User login
Traditional Chinese medicine improves outcomes in HFrEF
When added to guideline-directed therapies for heart failure with reduced ejection fraction (HFrEF), a traditional Chinese medicine called qiliqiangxin reduced the composite endpoint of cardiovascular death and heart failure hospitalization by more than 20%, results of a large placebo-controlled trial show.
reported Xinli Li, MD, PhD, First Affiliated Hospital, Nanjing Medical University, China.
Qiliqiangxin, a commonly used therapy in China for cardiovascular disease, is not a single chemical entity but a treatment composed of 11 plant-based substances that together are associated with diuretic effects, vasodilation, and “cardiotonic” activity, Dr. Li said. He also cited studies showing an upregulation effect on peroxisome proliferator-activated receptor gamma coactivator 1-beta (PGC1-beta).
The results were presented at the annual congress of the European Society of Cardiology.
Hard endpoints pursued in rigorous design
There have been numerous studies of qiliqiangxin for cardiovascular diseases, including a double-blind study that associated this agent with a greater than 30% reduction in the surrogate endpoint of N-terminal pro–B-type natriuretic peptide (NT-proBNP).
In the newly completed multicenter trial, called QUEST, the goal was to determine whether this therapy could reduce hard endpoints relative to placebo in a rigorously conducted trial enrolling patients receiving an optimized triple-therapy heart failure regimen.
Few patients in the study received a sodium glucose cotransporter-2 (SGLT-2 inhibitor), which was not a standard at the time the study was designed but is now part of the quadruple guideline-directed medical therapy in most European and North American guidelines.
In this trial, 3,119 patients were randomly assigned at 133 centers in China to take four capsules of qiliqiangxin or placebo three times per day. At a median follow-up of 18.3 months, outcomes were evaluable in nearly all 1,561 patients randomly assigned to the experimental therapy and 1,555 patients randomly assigned to placebo.
The key inclusion criteria were a left ventricular ejection fraction of 40% or less and a serum NT-proBNP level of at least 450 pg/mL. Patients in New York Heart Association class IV heart failure were excluded.
At enrollment, more than 80% of patients in both arms were receiving a renin-angiotensin system (RAS) inhibitor (angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or angiotensin receptor neprilysin inhibitor), more than 80% were receiving a mineralocorticoid receptor antagonist, and more than 85% were receiving a beta-blocker.
Death and hospitalization reduced 22%
By hazard ratio, the primary composite endpoint of CV death and heart failure hospitalization was reduced by 22% relative to placebo (HR, 0.78; P < .001). When evaluated separately, the relative reductions in these respective endpoints were 17% (HR, 0.83; P = .045) and 24% (HR, 0.76; P = .002).
The risk reduction was robust (HR, 0.76; P < .001) in patients with an ischemic cause but nonsignificant in those without (HR, 0.92; P = .575). A significant benefit was sustained in patients receiving an angiotensin receptor neprilysin inhibitor (HR, 0.84; P = .041), as well as those who did not receive this class of drug (HR, 0.77; P = .012).
However, the benefit of qiliqiangxin among patients receiving all components of guideline-directed triple therapy (RAS inhibitor, beta-blocker, and mineralocorticoid antagonist) was only a trend (HR, 0.86; P = .079).
All-cause mortality, a secondary endpoint, was lower among patients randomly assigned to qiliqiangxin than to those assigned to placebo, but this difference fell just short of statistical significance (14.21% vs. 16.85%; P = .058).
Qiliqiangxin was well tolerated. The proportion of patients with a serious adverse event was numerically lower with qiliqiangxin than with placebo (17.43% vs. 19.74%), whereas discontinuations associated with an adverse event were numerically higher in the qiliqiangxin group (1.03% vs. 0.58%), albeit still very low in both study arms.
Overlap of drug benefits suspected
Given the safety of this drug and its highly significant reduction in a composite endpoint used in other major HFrEF trials, the ESC-invited discussant, Carolyn S.P. Lam, MBBS, PhD, National Heart Centre, Singapore, called the outcome “remarkable” and a validation for “the millions of people” who are already taking qiliqiangxin in China and other Asian countries.
Using the DAPA-HF trial as a point of reference, Dr. Lam noted that relative reduction in the composite endpoint of cardiovascular death for the SGLT-2 inhibitor dapagliflozin relative to placebo on top of triple guideline-directed medical therapy was lower (17% vs. 24%), but there were significant reductions in each of the components, as well as a nonsignificant signal of a mortality benefit.
However, Dr. Lam pointed out that there does seem to be more of an overlap for the benefits of qiliqiangxin than dapagliflozin relative to other components of triple therapy based on the lower rate of benefit when patients were optimized on triple therapy.
“The subgroup analysis [of this study] is very important,” Dr. Lam said. Qiliqiangxin may be best in patients who cannot take one or more of the components of triple therapy, she suggested, even though she called for further studies to test this theory. She also cautioned that the pill burden of four capsules taken three times per day might be onerous for some patients.
Of the many questions still to be answered, Dr. Lam noted that the low rate of enrollment for patients (< 10%) taking SGLT-2 inhibitors makes the contribution of qiliqiangxin unclear among those receiving the current standard of quadruple guideline-directed medical therapy.
She also suggested that it will be important to dissect the relative contribution of the different active ingredients of qiliqiangxin.
“This is not a purified compound that we are used to in Western medicine,” Dr. Lam said. While she praised the study as “scientifically rigorous” and indicated that the results support a clinical benefit from qiliqiangxin, she thinks an exploration of the mechanism or mechanisms of benefit is a next step in understanding where this therapy fits in HFrEF management.
Dr. Li reports financial relationships with AstraZeneca, Bayer, Novartis, Roche, and Yiling. Dr. Lam reports financial relationships with more than 25 pharmaceutical or device manufacturers, many of which produce therapies for heart failure, as well as with Medscape/WebMD Global LLC. The study was supported by the Chinese National Key Research and Development Project and Yiling Pharmaceuticals.
A version of this article appeared on Medscape.com.
When added to guideline-directed therapies for heart failure with reduced ejection fraction (HFrEF), a traditional Chinese medicine called qiliqiangxin reduced the composite endpoint of cardiovascular death and heart failure hospitalization by more than 20%, results of a large placebo-controlled trial show.
reported Xinli Li, MD, PhD, First Affiliated Hospital, Nanjing Medical University, China.
Qiliqiangxin, a commonly used therapy in China for cardiovascular disease, is not a single chemical entity but a treatment composed of 11 plant-based substances that together are associated with diuretic effects, vasodilation, and “cardiotonic” activity, Dr. Li said. He also cited studies showing an upregulation effect on peroxisome proliferator-activated receptor gamma coactivator 1-beta (PGC1-beta).
The results were presented at the annual congress of the European Society of Cardiology.
Hard endpoints pursued in rigorous design
There have been numerous studies of qiliqiangxin for cardiovascular diseases, including a double-blind study that associated this agent with a greater than 30% reduction in the surrogate endpoint of N-terminal pro–B-type natriuretic peptide (NT-proBNP).
In the newly completed multicenter trial, called QUEST, the goal was to determine whether this therapy could reduce hard endpoints relative to placebo in a rigorously conducted trial enrolling patients receiving an optimized triple-therapy heart failure regimen.
Few patients in the study received a sodium glucose cotransporter-2 (SGLT-2 inhibitor), which was not a standard at the time the study was designed but is now part of the quadruple guideline-directed medical therapy in most European and North American guidelines.
In this trial, 3,119 patients were randomly assigned at 133 centers in China to take four capsules of qiliqiangxin or placebo three times per day. At a median follow-up of 18.3 months, outcomes were evaluable in nearly all 1,561 patients randomly assigned to the experimental therapy and 1,555 patients randomly assigned to placebo.
The key inclusion criteria were a left ventricular ejection fraction of 40% or less and a serum NT-proBNP level of at least 450 pg/mL. Patients in New York Heart Association class IV heart failure were excluded.
At enrollment, more than 80% of patients in both arms were receiving a renin-angiotensin system (RAS) inhibitor (angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or angiotensin receptor neprilysin inhibitor), more than 80% were receiving a mineralocorticoid receptor antagonist, and more than 85% were receiving a beta-blocker.
Death and hospitalization reduced 22%
By hazard ratio, the primary composite endpoint of CV death and heart failure hospitalization was reduced by 22% relative to placebo (HR, 0.78; P < .001). When evaluated separately, the relative reductions in these respective endpoints were 17% (HR, 0.83; P = .045) and 24% (HR, 0.76; P = .002).
The risk reduction was robust (HR, 0.76; P < .001) in patients with an ischemic cause but nonsignificant in those without (HR, 0.92; P = .575). A significant benefit was sustained in patients receiving an angiotensin receptor neprilysin inhibitor (HR, 0.84; P = .041), as well as those who did not receive this class of drug (HR, 0.77; P = .012).
However, the benefit of qiliqiangxin among patients receiving all components of guideline-directed triple therapy (RAS inhibitor, beta-blocker, and mineralocorticoid antagonist) was only a trend (HR, 0.86; P = .079).
All-cause mortality, a secondary endpoint, was lower among patients randomly assigned to qiliqiangxin than to those assigned to placebo, but this difference fell just short of statistical significance (14.21% vs. 16.85%; P = .058).
Qiliqiangxin was well tolerated. The proportion of patients with a serious adverse event was numerically lower with qiliqiangxin than with placebo (17.43% vs. 19.74%), whereas discontinuations associated with an adverse event were numerically higher in the qiliqiangxin group (1.03% vs. 0.58%), albeit still very low in both study arms.
Overlap of drug benefits suspected
Given the safety of this drug and its highly significant reduction in a composite endpoint used in other major HFrEF trials, the ESC-invited discussant, Carolyn S.P. Lam, MBBS, PhD, National Heart Centre, Singapore, called the outcome “remarkable” and a validation for “the millions of people” who are already taking qiliqiangxin in China and other Asian countries.
Using the DAPA-HF trial as a point of reference, Dr. Lam noted that relative reduction in the composite endpoint of cardiovascular death for the SGLT-2 inhibitor dapagliflozin relative to placebo on top of triple guideline-directed medical therapy was lower (17% vs. 24%), but there were significant reductions in each of the components, as well as a nonsignificant signal of a mortality benefit.
However, Dr. Lam pointed out that there does seem to be more of an overlap for the benefits of qiliqiangxin than dapagliflozin relative to other components of triple therapy based on the lower rate of benefit when patients were optimized on triple therapy.
“The subgroup analysis [of this study] is very important,” Dr. Lam said. Qiliqiangxin may be best in patients who cannot take one or more of the components of triple therapy, she suggested, even though she called for further studies to test this theory. She also cautioned that the pill burden of four capsules taken three times per day might be onerous for some patients.
Of the many questions still to be answered, Dr. Lam noted that the low rate of enrollment for patients (< 10%) taking SGLT-2 inhibitors makes the contribution of qiliqiangxin unclear among those receiving the current standard of quadruple guideline-directed medical therapy.
She also suggested that it will be important to dissect the relative contribution of the different active ingredients of qiliqiangxin.
“This is not a purified compound that we are used to in Western medicine,” Dr. Lam said. While she praised the study as “scientifically rigorous” and indicated that the results support a clinical benefit from qiliqiangxin, she thinks an exploration of the mechanism or mechanisms of benefit is a next step in understanding where this therapy fits in HFrEF management.
Dr. Li reports financial relationships with AstraZeneca, Bayer, Novartis, Roche, and Yiling. Dr. Lam reports financial relationships with more than 25 pharmaceutical or device manufacturers, many of which produce therapies for heart failure, as well as with Medscape/WebMD Global LLC. The study was supported by the Chinese National Key Research and Development Project and Yiling Pharmaceuticals.
A version of this article appeared on Medscape.com.
When added to guideline-directed therapies for heart failure with reduced ejection fraction (HFrEF), a traditional Chinese medicine called qiliqiangxin reduced the composite endpoint of cardiovascular death and heart failure hospitalization by more than 20%, results of a large placebo-controlled trial show.
reported Xinli Li, MD, PhD, First Affiliated Hospital, Nanjing Medical University, China.
Qiliqiangxin, a commonly used therapy in China for cardiovascular disease, is not a single chemical entity but a treatment composed of 11 plant-based substances that together are associated with diuretic effects, vasodilation, and “cardiotonic” activity, Dr. Li said. He also cited studies showing an upregulation effect on peroxisome proliferator-activated receptor gamma coactivator 1-beta (PGC1-beta).
The results were presented at the annual congress of the European Society of Cardiology.
Hard endpoints pursued in rigorous design
There have been numerous studies of qiliqiangxin for cardiovascular diseases, including a double-blind study that associated this agent with a greater than 30% reduction in the surrogate endpoint of N-terminal pro–B-type natriuretic peptide (NT-proBNP).
In the newly completed multicenter trial, called QUEST, the goal was to determine whether this therapy could reduce hard endpoints relative to placebo in a rigorously conducted trial enrolling patients receiving an optimized triple-therapy heart failure regimen.
Few patients in the study received a sodium glucose cotransporter-2 (SGLT-2 inhibitor), which was not a standard at the time the study was designed but is now part of the quadruple guideline-directed medical therapy in most European and North American guidelines.
In this trial, 3,119 patients were randomly assigned at 133 centers in China to take four capsules of qiliqiangxin or placebo three times per day. At a median follow-up of 18.3 months, outcomes were evaluable in nearly all 1,561 patients randomly assigned to the experimental therapy and 1,555 patients randomly assigned to placebo.
The key inclusion criteria were a left ventricular ejection fraction of 40% or less and a serum NT-proBNP level of at least 450 pg/mL. Patients in New York Heart Association class IV heart failure were excluded.
At enrollment, more than 80% of patients in both arms were receiving a renin-angiotensin system (RAS) inhibitor (angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or angiotensin receptor neprilysin inhibitor), more than 80% were receiving a mineralocorticoid receptor antagonist, and more than 85% were receiving a beta-blocker.
Death and hospitalization reduced 22%
By hazard ratio, the primary composite endpoint of CV death and heart failure hospitalization was reduced by 22% relative to placebo (HR, 0.78; P < .001). When evaluated separately, the relative reductions in these respective endpoints were 17% (HR, 0.83; P = .045) and 24% (HR, 0.76; P = .002).
The risk reduction was robust (HR, 0.76; P < .001) in patients with an ischemic cause but nonsignificant in those without (HR, 0.92; P = .575). A significant benefit was sustained in patients receiving an angiotensin receptor neprilysin inhibitor (HR, 0.84; P = .041), as well as those who did not receive this class of drug (HR, 0.77; P = .012).
However, the benefit of qiliqiangxin among patients receiving all components of guideline-directed triple therapy (RAS inhibitor, beta-blocker, and mineralocorticoid antagonist) was only a trend (HR, 0.86; P = .079).
All-cause mortality, a secondary endpoint, was lower among patients randomly assigned to qiliqiangxin than to those assigned to placebo, but this difference fell just short of statistical significance (14.21% vs. 16.85%; P = .058).
Qiliqiangxin was well tolerated. The proportion of patients with a serious adverse event was numerically lower with qiliqiangxin than with placebo (17.43% vs. 19.74%), whereas discontinuations associated with an adverse event were numerically higher in the qiliqiangxin group (1.03% vs. 0.58%), albeit still very low in both study arms.
Overlap of drug benefits suspected
Given the safety of this drug and its highly significant reduction in a composite endpoint used in other major HFrEF trials, the ESC-invited discussant, Carolyn S.P. Lam, MBBS, PhD, National Heart Centre, Singapore, called the outcome “remarkable” and a validation for “the millions of people” who are already taking qiliqiangxin in China and other Asian countries.
Using the DAPA-HF trial as a point of reference, Dr. Lam noted that relative reduction in the composite endpoint of cardiovascular death for the SGLT-2 inhibitor dapagliflozin relative to placebo on top of triple guideline-directed medical therapy was lower (17% vs. 24%), but there were significant reductions in each of the components, as well as a nonsignificant signal of a mortality benefit.
However, Dr. Lam pointed out that there does seem to be more of an overlap for the benefits of qiliqiangxin than dapagliflozin relative to other components of triple therapy based on the lower rate of benefit when patients were optimized on triple therapy.
“The subgroup analysis [of this study] is very important,” Dr. Lam said. Qiliqiangxin may be best in patients who cannot take one or more of the components of triple therapy, she suggested, even though she called for further studies to test this theory. She also cautioned that the pill burden of four capsules taken three times per day might be onerous for some patients.
Of the many questions still to be answered, Dr. Lam noted that the low rate of enrollment for patients (< 10%) taking SGLT-2 inhibitors makes the contribution of qiliqiangxin unclear among those receiving the current standard of quadruple guideline-directed medical therapy.
She also suggested that it will be important to dissect the relative contribution of the different active ingredients of qiliqiangxin.
“This is not a purified compound that we are used to in Western medicine,” Dr. Lam said. While she praised the study as “scientifically rigorous” and indicated that the results support a clinical benefit from qiliqiangxin, she thinks an exploration of the mechanism or mechanisms of benefit is a next step in understanding where this therapy fits in HFrEF management.
Dr. Li reports financial relationships with AstraZeneca, Bayer, Novartis, Roche, and Yiling. Dr. Lam reports financial relationships with more than 25 pharmaceutical or device manufacturers, many of which produce therapies for heart failure, as well as with Medscape/WebMD Global LLC. The study was supported by the Chinese National Key Research and Development Project and Yiling Pharmaceuticals.
A version of this article appeared on Medscape.com.
FROM ESC CONGRESS 2023
No reduction in AFib after noncardiac surgery with colchicine: COP-AF
Trends were seen with reductions in events, but these did not reach significance. However, benefit was seen in a post-hoc analysis looking at a composite of both of those endpoints, the researchers note, as well as a composite of vascular death, nonfatal MINS, nonfatal stroke, and clinically important perioperative AFib, the researchers report.
“We interpret that as there is a trend that is promising, a trend that needs to be further explored,” lead author David Conen, MD, Population Health Research Institute, Hamilton, Ont., said in an interview. “We think that further studies are needed to tease out which patients can benefit from colchicine and in what setting it can be used.”
Treatment was safe, with no effect on the risk for sepsis or infection, but it did cause an increase in noninfectious diarrhea. “These events were mostly benign and did not increase length of stay, and only one patient was readmitted because of diarrhea,” Dr. Conen noted.
Results of the COP-AF trial were presented at the annual congress of the European Society of Cardiology, Amsterdam, and published online in The Lancet .
Inflammation and perioperative AFib
AFib and MINS are common complications in patients undergoing major thoracic surgery, Dr. Conen explained. The literature suggests AFib occurs in about 10% and MINS in about 20% of these patients, “and patients with these complications have a much higher risk of additional complications, such as stroke or MI [myocardial infarction],” Dr. Conen said.
Both disorders are associated with high levels of inflammatory biomarkers, so they set out to test colchicine, a well-known anti-inflammatory drug used in higher doses to treat common clinical disorders, such as gout and pericarditis. Small, randomized trials had shown it reduced the incidence of perioperative AFib after cardiac surgery, he noted.
Low-dose colchicine (LoDoCo, Agepha Pharma) was recently approved by the U.S. Food and Drug Administration to reduce the risk for MI, stroke, coronary revascularization, or death in patients with established atherosclerotic disease or multiple risk factors for cardiovascular disease. It was approved on the basis of the LoDoCo 2 trial in patients with stable coronary artery disease and the COLCOT trial in patients with recent MI.
COP-AF was a randomized trial, conducted at 45 sites in 11 countries, and enrolled 3,209 patients aged 55 years or older (51.6% male) undergoing major noncardiac thoracic surgery. Patients were excluded if they had previous AFib, had any contraindications to colchicine, or required colchicine on a clinical basis.
Patients were randomly assigned 1:1 to receive oral colchicine at a dose of 0.5 mg twice daily (1,608 patients) or placebo (1,601 patients). Treatment was begun within 4 hours before surgery and continued for 10 days. Health care providers and patients, as well as data collectors and adjudicators, were blinded to treatment assignment.
The co-primary outcomes were clinically important perioperative AFib or MINS over 14 days of follow-up. The trial was originally looking only at clinically important AFib, Dr. Conen noted, but after the publication of LoDoCo 2 and COLCOT, “MINS was added as an independent co-primary outcome,” requiring more patients to achieve adequate power.
The main safety outcomes were a composite of sepsis or infection, along with noninfectious diarrhea.
Clinically important AFib was defined as AFib that results in angina, heart failure, or symptomatic hypotension or required treatment with a rate-controlling drug, antiarrhythmic drug, or electrical cardioversion. “This definition was chosen because of its prognostic relevance, and to avoid adding short, asymptomatic AFib episodes of uncertain clinical relevance to the primary outcome,” Dr. Conen said during his presentation.
MINS was defined as an MI or any postoperative troponin elevation that was judged by an adjudication panel to be of ischemic origin.
At 14 days, there was no significant difference between groups on either of the co-primary end points.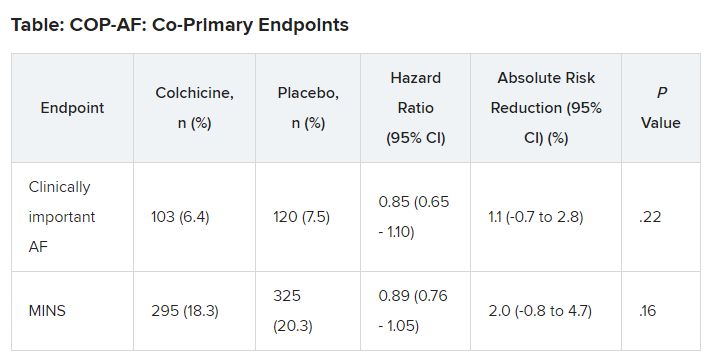
No significant differences but positive trends were similarly seen in secondary outcomes of a composite of all-cause death, nonfatal MINS, and nonfatal stroke; the composite of all-cause death, nonfatal MI, and nonfatal stroke; MINS not fulfilling the fourth universal definition of MI; or MI.
There were no differences in time to chest tube removal, days in hospital, nights in the step-down unit, or nights in the intensive care unit.
In terms of safety, there was no difference between groups on sepsis or infection, which occurred in 6.4% of patients in the colchicine group and 5.2% of those in the placebo group (hazard ratio, 1.24; 95% confidence interval, 0.93-1.66).
Noninfectious diarrhea was more common with colchicine, with 134 events (8.3%) versus 38 with placebo (2.4%), for an HR of 3.64 (95% CI, 2.54-5.22).
“In two post hoc analyses, colchicine significantly reduced the composite of the two co-primary outcomes,” Dr. Conen noted in his presentation. Clinically important perioperative AFib or MINS occurred in 22.4% in the colchicine group and 25.9% in the placebo group (HR, 0.84; 95% CI, 0.73-0.97; P = .02).
“Colchicine also significantly reduced the composite of vascular mortality, nonfatal MINS, nonfatal stroke, and clinically important AFib,” he said; 22.6% of patients in the colchicine group had one of these events versus 26.4% of those in the placebo group (HR, 0.83; 95% CI, 0.72-0.96; P = .01).
The researchers also reported significant interactions on both co-primary outcomes for the type of incision, “suggesting that stronger and statistically significant effects among patients undergoing thoracoscopic surgery as opposed to nonthoracoscopic surgery,” Dr. Conen said.
Patients undergoing thoracoscopic surgery treated with colchicine had a reduced risk for clinically important AFib (n = 2,397; HR, 0.53; 95% CI, 0.36-0.77), but colchicine treatment increased the risk in patients having open surgery (n = 784; HR, 1.59; 95% CI, 1.07-2.35; P for interaction < .0001).
There was a beneficial effect on MINS with colchicine among patients undergoing thoracoscopic surgery (HR, 0.80; 95% CI, 0.66-0.98), but no effect was seen among those having open surgery (HR, 1.15; 95% CI, 0.87-1.53; P for interaction = .041).
Low-risk patients
Jean-Claude Tardif, MD, Montreal Heart Institute and Université de Montréal, was the invited discussant for the COP-AF presentation and congratulated the researchers on “a job well done.”
He made the point that the risk for perioperative AFib has decreased substantially with the greater use of thoracoscopic rather than open surgical approaches. The population of this trial was relatively young, with an average age of 68 years; the presence of concomitant CVD was low, at about 9%; by design, patients with previous AFib were excluded; and only about 20% of patients had surgery with an open approach.
“So that population of patients were probably at relatively low risk of atrial fibrillation, and sure enough, the incidence of perioperative AFib in that population at 7.5% was lower than the assumed rate in the statistical powering of the study at 9%,” Dr. Tardif noted.
The post-hoc analyses showed a “nominally significant effect on the composite of MINS and AFib; however, that combination is fairly difficult to justify given the different pathophysiology and clinical consequences of both outcomes,” he pointed out.
The incidence of postoperative MI as a secondary outcome was low, less than 1%, as was the incidence of postoperative stroke in that study, Dr. Tardif added. “Given the link between presence of blood in the pericardium as a trigger for AFib, it would be interesting to know the incidence of perioperative pericarditis in COP-AF.”
In conclusion, he said, “when trying to put these results into the bigger picture of colchicine in cardiovascular disease, I believe we need large, well-powered clinical trials to determine the value of colchicine to reduce the risk of AFib after cardiac surgery and after catheter ablation,” Dr. Tardif said.
“We all know that colchicine represents the first line of therapy for the treatment of acute and recurrent pericarditis, and finally, low-dose colchicine, at a lower dose than was used in COP-AF, 0.5 mg once daily, is the first anti-inflammatory agent approved by both U.S. FDA and Health Canada to reduce the risk of atherothombotic events in patients with ASCVD [atherosclerotic cardiovascular disease], I believe offering a new pillar of treatment for the prevention of ischemic events in such patients.”
Session co-moderator Franz Weidinger, MD, Landstrasse Clinic, Vienna, Austria, called the COP-AF results “very important” but also noted that they show “the challenge of doing well-powered randomized trials these days when we have patients so well treated for a wide array of cardiovascular disease.”
The study was supported by the Canadian Institutes of Health Research (CIHR); Accelerating Clinical Trials Consortium; Innovation Fund of the Alternative Funding Plan for the Academic Health Sciences Centres of Ontario; Population Health Research Institute; Hamilton Health Sciences; Division of Cardiology at McMaster University, Canada; Hanela Foundation, Switzerland; and General Research Fund, Research Grants Council, Hong Kong. Dr. Conen reports receiving research grants from CIHR, speaker fees from Servier outside the current study, and advisory board fees from Roche Diagnostics and Trimedics outside the current study.
A version of this article appeared on Medscape.com.
Trends were seen with reductions in events, but these did not reach significance. However, benefit was seen in a post-hoc analysis looking at a composite of both of those endpoints, the researchers note, as well as a composite of vascular death, nonfatal MINS, nonfatal stroke, and clinically important perioperative AFib, the researchers report.
“We interpret that as there is a trend that is promising, a trend that needs to be further explored,” lead author David Conen, MD, Population Health Research Institute, Hamilton, Ont., said in an interview. “We think that further studies are needed to tease out which patients can benefit from colchicine and in what setting it can be used.”
Treatment was safe, with no effect on the risk for sepsis or infection, but it did cause an increase in noninfectious diarrhea. “These events were mostly benign and did not increase length of stay, and only one patient was readmitted because of diarrhea,” Dr. Conen noted.
Results of the COP-AF trial were presented at the annual congress of the European Society of Cardiology, Amsterdam, and published online in The Lancet .
Inflammation and perioperative AFib
AFib and MINS are common complications in patients undergoing major thoracic surgery, Dr. Conen explained. The literature suggests AFib occurs in about 10% and MINS in about 20% of these patients, “and patients with these complications have a much higher risk of additional complications, such as stroke or MI [myocardial infarction],” Dr. Conen said.
Both disorders are associated with high levels of inflammatory biomarkers, so they set out to test colchicine, a well-known anti-inflammatory drug used in higher doses to treat common clinical disorders, such as gout and pericarditis. Small, randomized trials had shown it reduced the incidence of perioperative AFib after cardiac surgery, he noted.
Low-dose colchicine (LoDoCo, Agepha Pharma) was recently approved by the U.S. Food and Drug Administration to reduce the risk for MI, stroke, coronary revascularization, or death in patients with established atherosclerotic disease or multiple risk factors for cardiovascular disease. It was approved on the basis of the LoDoCo 2 trial in patients with stable coronary artery disease and the COLCOT trial in patients with recent MI.
COP-AF was a randomized trial, conducted at 45 sites in 11 countries, and enrolled 3,209 patients aged 55 years or older (51.6% male) undergoing major noncardiac thoracic surgery. Patients were excluded if they had previous AFib, had any contraindications to colchicine, or required colchicine on a clinical basis.
Patients were randomly assigned 1:1 to receive oral colchicine at a dose of 0.5 mg twice daily (1,608 patients) or placebo (1,601 patients). Treatment was begun within 4 hours before surgery and continued for 10 days. Health care providers and patients, as well as data collectors and adjudicators, were blinded to treatment assignment.
The co-primary outcomes were clinically important perioperative AFib or MINS over 14 days of follow-up. The trial was originally looking only at clinically important AFib, Dr. Conen noted, but after the publication of LoDoCo 2 and COLCOT, “MINS was added as an independent co-primary outcome,” requiring more patients to achieve adequate power.
The main safety outcomes were a composite of sepsis or infection, along with noninfectious diarrhea.
Clinically important AFib was defined as AFib that results in angina, heart failure, or symptomatic hypotension or required treatment with a rate-controlling drug, antiarrhythmic drug, or electrical cardioversion. “This definition was chosen because of its prognostic relevance, and to avoid adding short, asymptomatic AFib episodes of uncertain clinical relevance to the primary outcome,” Dr. Conen said during his presentation.
MINS was defined as an MI or any postoperative troponin elevation that was judged by an adjudication panel to be of ischemic origin.
At 14 days, there was no significant difference between groups on either of the co-primary end points.
No significant differences but positive trends were similarly seen in secondary outcomes of a composite of all-cause death, nonfatal MINS, and nonfatal stroke; the composite of all-cause death, nonfatal MI, and nonfatal stroke; MINS not fulfilling the fourth universal definition of MI; or MI.
There were no differences in time to chest tube removal, days in hospital, nights in the step-down unit, or nights in the intensive care unit.
In terms of safety, there was no difference between groups on sepsis or infection, which occurred in 6.4% of patients in the colchicine group and 5.2% of those in the placebo group (hazard ratio, 1.24; 95% confidence interval, 0.93-1.66).
Noninfectious diarrhea was more common with colchicine, with 134 events (8.3%) versus 38 with placebo (2.4%), for an HR of 3.64 (95% CI, 2.54-5.22).
“In two post hoc analyses, colchicine significantly reduced the composite of the two co-primary outcomes,” Dr. Conen noted in his presentation. Clinically important perioperative AFib or MINS occurred in 22.4% in the colchicine group and 25.9% in the placebo group (HR, 0.84; 95% CI, 0.73-0.97; P = .02).
“Colchicine also significantly reduced the composite of vascular mortality, nonfatal MINS, nonfatal stroke, and clinically important AFib,” he said; 22.6% of patients in the colchicine group had one of these events versus 26.4% of those in the placebo group (HR, 0.83; 95% CI, 0.72-0.96; P = .01).
The researchers also reported significant interactions on both co-primary outcomes for the type of incision, “suggesting that stronger and statistically significant effects among patients undergoing thoracoscopic surgery as opposed to nonthoracoscopic surgery,” Dr. Conen said.
Patients undergoing thoracoscopic surgery treated with colchicine had a reduced risk for clinically important AFib (n = 2,397; HR, 0.53; 95% CI, 0.36-0.77), but colchicine treatment increased the risk in patients having open surgery (n = 784; HR, 1.59; 95% CI, 1.07-2.35; P for interaction < .0001).
There was a beneficial effect on MINS with colchicine among patients undergoing thoracoscopic surgery (HR, 0.80; 95% CI, 0.66-0.98), but no effect was seen among those having open surgery (HR, 1.15; 95% CI, 0.87-1.53; P for interaction = .041).
Low-risk patients
Jean-Claude Tardif, MD, Montreal Heart Institute and Université de Montréal, was the invited discussant for the COP-AF presentation and congratulated the researchers on “a job well done.”
He made the point that the risk for perioperative AFib has decreased substantially with the greater use of thoracoscopic rather than open surgical approaches. The population of this trial was relatively young, with an average age of 68 years; the presence of concomitant CVD was low, at about 9%; by design, patients with previous AFib were excluded; and only about 20% of patients had surgery with an open approach.
“So that population of patients were probably at relatively low risk of atrial fibrillation, and sure enough, the incidence of perioperative AFib in that population at 7.5% was lower than the assumed rate in the statistical powering of the study at 9%,” Dr. Tardif noted.
The post-hoc analyses showed a “nominally significant effect on the composite of MINS and AFib; however, that combination is fairly difficult to justify given the different pathophysiology and clinical consequences of both outcomes,” he pointed out.
The incidence of postoperative MI as a secondary outcome was low, less than 1%, as was the incidence of postoperative stroke in that study, Dr. Tardif added. “Given the link between presence of blood in the pericardium as a trigger for AFib, it would be interesting to know the incidence of perioperative pericarditis in COP-AF.”
In conclusion, he said, “when trying to put these results into the bigger picture of colchicine in cardiovascular disease, I believe we need large, well-powered clinical trials to determine the value of colchicine to reduce the risk of AFib after cardiac surgery and after catheter ablation,” Dr. Tardif said.
“We all know that colchicine represents the first line of therapy for the treatment of acute and recurrent pericarditis, and finally, low-dose colchicine, at a lower dose than was used in COP-AF, 0.5 mg once daily, is the first anti-inflammatory agent approved by both U.S. FDA and Health Canada to reduce the risk of atherothombotic events in patients with ASCVD [atherosclerotic cardiovascular disease], I believe offering a new pillar of treatment for the prevention of ischemic events in such patients.”
Session co-moderator Franz Weidinger, MD, Landstrasse Clinic, Vienna, Austria, called the COP-AF results “very important” but also noted that they show “the challenge of doing well-powered randomized trials these days when we have patients so well treated for a wide array of cardiovascular disease.”
The study was supported by the Canadian Institutes of Health Research (CIHR); Accelerating Clinical Trials Consortium; Innovation Fund of the Alternative Funding Plan for the Academic Health Sciences Centres of Ontario; Population Health Research Institute; Hamilton Health Sciences; Division of Cardiology at McMaster University, Canada; Hanela Foundation, Switzerland; and General Research Fund, Research Grants Council, Hong Kong. Dr. Conen reports receiving research grants from CIHR, speaker fees from Servier outside the current study, and advisory board fees from Roche Diagnostics and Trimedics outside the current study.
A version of this article appeared on Medscape.com.
Trends were seen with reductions in events, but these did not reach significance. However, benefit was seen in a post-hoc analysis looking at a composite of both of those endpoints, the researchers note, as well as a composite of vascular death, nonfatal MINS, nonfatal stroke, and clinically important perioperative AFib, the researchers report.
“We interpret that as there is a trend that is promising, a trend that needs to be further explored,” lead author David Conen, MD, Population Health Research Institute, Hamilton, Ont., said in an interview. “We think that further studies are needed to tease out which patients can benefit from colchicine and in what setting it can be used.”
Treatment was safe, with no effect on the risk for sepsis or infection, but it did cause an increase in noninfectious diarrhea. “These events were mostly benign and did not increase length of stay, and only one patient was readmitted because of diarrhea,” Dr. Conen noted.
Results of the COP-AF trial were presented at the annual congress of the European Society of Cardiology, Amsterdam, and published online in The Lancet .
Inflammation and perioperative AFib
AFib and MINS are common complications in patients undergoing major thoracic surgery, Dr. Conen explained. The literature suggests AFib occurs in about 10% and MINS in about 20% of these patients, “and patients with these complications have a much higher risk of additional complications, such as stroke or MI [myocardial infarction],” Dr. Conen said.
Both disorders are associated with high levels of inflammatory biomarkers, so they set out to test colchicine, a well-known anti-inflammatory drug used in higher doses to treat common clinical disorders, such as gout and pericarditis. Small, randomized trials had shown it reduced the incidence of perioperative AFib after cardiac surgery, he noted.
Low-dose colchicine (LoDoCo, Agepha Pharma) was recently approved by the U.S. Food and Drug Administration to reduce the risk for MI, stroke, coronary revascularization, or death in patients with established atherosclerotic disease or multiple risk factors for cardiovascular disease. It was approved on the basis of the LoDoCo 2 trial in patients with stable coronary artery disease and the COLCOT trial in patients with recent MI.
COP-AF was a randomized trial, conducted at 45 sites in 11 countries, and enrolled 3,209 patients aged 55 years or older (51.6% male) undergoing major noncardiac thoracic surgery. Patients were excluded if they had previous AFib, had any contraindications to colchicine, or required colchicine on a clinical basis.
Patients were randomly assigned 1:1 to receive oral colchicine at a dose of 0.5 mg twice daily (1,608 patients) or placebo (1,601 patients). Treatment was begun within 4 hours before surgery and continued for 10 days. Health care providers and patients, as well as data collectors and adjudicators, were blinded to treatment assignment.
The co-primary outcomes were clinically important perioperative AFib or MINS over 14 days of follow-up. The trial was originally looking only at clinically important AFib, Dr. Conen noted, but after the publication of LoDoCo 2 and COLCOT, “MINS was added as an independent co-primary outcome,” requiring more patients to achieve adequate power.
The main safety outcomes were a composite of sepsis or infection, along with noninfectious diarrhea.
Clinically important AFib was defined as AFib that results in angina, heart failure, or symptomatic hypotension or required treatment with a rate-controlling drug, antiarrhythmic drug, or electrical cardioversion. “This definition was chosen because of its prognostic relevance, and to avoid adding short, asymptomatic AFib episodes of uncertain clinical relevance to the primary outcome,” Dr. Conen said during his presentation.
MINS was defined as an MI or any postoperative troponin elevation that was judged by an adjudication panel to be of ischemic origin.
At 14 days, there was no significant difference between groups on either of the co-primary end points.
No significant differences but positive trends were similarly seen in secondary outcomes of a composite of all-cause death, nonfatal MINS, and nonfatal stroke; the composite of all-cause death, nonfatal MI, and nonfatal stroke; MINS not fulfilling the fourth universal definition of MI; or MI.
There were no differences in time to chest tube removal, days in hospital, nights in the step-down unit, or nights in the intensive care unit.
In terms of safety, there was no difference between groups on sepsis or infection, which occurred in 6.4% of patients in the colchicine group and 5.2% of those in the placebo group (hazard ratio, 1.24; 95% confidence interval, 0.93-1.66).
Noninfectious diarrhea was more common with colchicine, with 134 events (8.3%) versus 38 with placebo (2.4%), for an HR of 3.64 (95% CI, 2.54-5.22).
“In two post hoc analyses, colchicine significantly reduced the composite of the two co-primary outcomes,” Dr. Conen noted in his presentation. Clinically important perioperative AFib or MINS occurred in 22.4% in the colchicine group and 25.9% in the placebo group (HR, 0.84; 95% CI, 0.73-0.97; P = .02).
“Colchicine also significantly reduced the composite of vascular mortality, nonfatal MINS, nonfatal stroke, and clinically important AFib,” he said; 22.6% of patients in the colchicine group had one of these events versus 26.4% of those in the placebo group (HR, 0.83; 95% CI, 0.72-0.96; P = .01).
The researchers also reported significant interactions on both co-primary outcomes for the type of incision, “suggesting that stronger and statistically significant effects among patients undergoing thoracoscopic surgery as opposed to nonthoracoscopic surgery,” Dr. Conen said.
Patients undergoing thoracoscopic surgery treated with colchicine had a reduced risk for clinically important AFib (n = 2,397; HR, 0.53; 95% CI, 0.36-0.77), but colchicine treatment increased the risk in patients having open surgery (n = 784; HR, 1.59; 95% CI, 1.07-2.35; P for interaction < .0001).
There was a beneficial effect on MINS with colchicine among patients undergoing thoracoscopic surgery (HR, 0.80; 95% CI, 0.66-0.98), but no effect was seen among those having open surgery (HR, 1.15; 95% CI, 0.87-1.53; P for interaction = .041).
Low-risk patients
Jean-Claude Tardif, MD, Montreal Heart Institute and Université de Montréal, was the invited discussant for the COP-AF presentation and congratulated the researchers on “a job well done.”
He made the point that the risk for perioperative AFib has decreased substantially with the greater use of thoracoscopic rather than open surgical approaches. The population of this trial was relatively young, with an average age of 68 years; the presence of concomitant CVD was low, at about 9%; by design, patients with previous AFib were excluded; and only about 20% of patients had surgery with an open approach.
“So that population of patients were probably at relatively low risk of atrial fibrillation, and sure enough, the incidence of perioperative AFib in that population at 7.5% was lower than the assumed rate in the statistical powering of the study at 9%,” Dr. Tardif noted.
The post-hoc analyses showed a “nominally significant effect on the composite of MINS and AFib; however, that combination is fairly difficult to justify given the different pathophysiology and clinical consequences of both outcomes,” he pointed out.
The incidence of postoperative MI as a secondary outcome was low, less than 1%, as was the incidence of postoperative stroke in that study, Dr. Tardif added. “Given the link between presence of blood in the pericardium as a trigger for AFib, it would be interesting to know the incidence of perioperative pericarditis in COP-AF.”
In conclusion, he said, “when trying to put these results into the bigger picture of colchicine in cardiovascular disease, I believe we need large, well-powered clinical trials to determine the value of colchicine to reduce the risk of AFib after cardiac surgery and after catheter ablation,” Dr. Tardif said.
“We all know that colchicine represents the first line of therapy for the treatment of acute and recurrent pericarditis, and finally, low-dose colchicine, at a lower dose than was used in COP-AF, 0.5 mg once daily, is the first anti-inflammatory agent approved by both U.S. FDA and Health Canada to reduce the risk of atherothombotic events in patients with ASCVD [atherosclerotic cardiovascular disease], I believe offering a new pillar of treatment for the prevention of ischemic events in such patients.”
Session co-moderator Franz Weidinger, MD, Landstrasse Clinic, Vienna, Austria, called the COP-AF results “very important” but also noted that they show “the challenge of doing well-powered randomized trials these days when we have patients so well treated for a wide array of cardiovascular disease.”
The study was supported by the Canadian Institutes of Health Research (CIHR); Accelerating Clinical Trials Consortium; Innovation Fund of the Alternative Funding Plan for the Academic Health Sciences Centres of Ontario; Population Health Research Institute; Hamilton Health Sciences; Division of Cardiology at McMaster University, Canada; Hanela Foundation, Switzerland; and General Research Fund, Research Grants Council, Hong Kong. Dr. Conen reports receiving research grants from CIHR, speaker fees from Servier outside the current study, and advisory board fees from Roche Diagnostics and Trimedics outside the current study.
A version of this article appeared on Medscape.com.
FROM ESC CONGRESS 2023
ESC backs SGLT2 inhibitor plus GLP-1 in diabetes with high CVD risk
AMSTERDAM – The era of guidelines that recommended treatment with either a sodium-glucose cotransporter-2 (SGLT-2) inhibitor or a glucagonlike peptide-1 (GLP-1) receptor agonist in people with type 2 diabetes mellitus and established cardiovascular disease (CVD) ended with new recommendations from the European Society of Cardiology that call for starting both classes simultaneously.
said Darren K. McGuire, MD, at the annual congress of the European Society of Cardiology.
The society’s new guidelines for managing CVD in patients with diabetes, released on Aug. 25 and presented in several sessions at the Congress, also break with the past by calling for starting treatment with both an SGLT-2 inhibitor and a GLP-1 receptor agonist without regard to a person’s existing level of glucose control, including their current and target hemoglobin A1c levels, and regardless of background therapy, added Dr. McGuire, a cardiologist and professor at the UT Southwestern Medical Center in Dallas and a member of the ESC panel that wrote the new guidelines.
Instead, the new guidance calls for starting both drug classes promptly in people diagnosed with type 2 diabetes and established atherosclerotic CVD.
Both the previous ESC guidelines from 2019 as well as the current Standards of Care for 2023 document from the American Diabetes Association call for using one class or the other, but they hedge on combined treatment as discretionary.
Different mechanisms mean additive benefits
“With increasing numbers of patients with type 2 diabetes in trials for SGLT-2 inhibitors or GLP-1 receptor agonists who were also on the other drug class, we’ve done large, stratified analyses that suggest no treatment-effect modification” when people received agents from both drug classes, Dr. McGuire explained in an interview. “While we don’t understand the mechanisms of action of these drugs for CVD, we’ve become very confident that they use different mechanisms” that appear to have at least partially additive effects.
“Their benefits for CVD risk reduction are completely independent of their glucose effects. They are cardiology drugs,” Dr. McGuire added.
The new ESC guidelines highlight two other clinical settings where people with type 2 diabetes should receive an SGLT-2 inhibitor regardless of their existing level of glucose control and any other medical treatment: people with heart failure and people with chronic kidney disease (CKD) based on a depressed estimated glomerular filtration rate and an elevated urine albumin-to-creatinine ratio.
Nephropathy was considered by the ESC’s guideline panel to confer risk that is similar to that of established atherosclerotic CVD, Dr. McGuire said.
The guidelines also, for the first time for ESC recommendations, made treatment with finerenone (Kerendia, Bayer) a class 1 level A recommendation for people with type 2 diabetes and CKD.
SCORE2-Diabetes risk estimator
Another major change in the new ESC guideline revision is introduction of a CVD risk calculator intended to estimate the risk among people with type 2 diabetes but without established CVD, heart failure, or CKD.
Called the SCORE2-Diabetes risk estimator, it calculates a person’s 10-year risk for CVD and includes adjustment based on the European region where a person lives; it also tallies different risk levels for women and for men.
The researchers who developed the SCORE2-Diabetes calculator used data from nearly 230,000 people to devise the tool and then validated it with data from an additional 217,000 Europeans with type 2 diabetes.
Key features of the calculator include its use of routinely collected clinical values, such as age, sex, systolic blood pressure, smoking status, serum cholesterol levels, age at diabetes diagnosis, hemoglobin A1c level, and estimated glomerular filtration rate.
“For the first time we have a clear score to categorize risk” in people with type 2 diabetes and identify who needs more aggressive treatment to prevent CVD development,” said Emanuele Di Angelantonio, MD, PhD, deputy director of the cardiovascular epidemiology unit at the University of Cambridge (England).
The guidelines say that people who have a low (< 5%) or moderate (5%-9%) 10-year risk for CVD are possible candidates for metformin treatment. Those with high (10%-19%) or very high (≥ 20%) risk are possible candidates for treatment with metformin and/or an SGLT-2 inhibitor and/or a GLP-1 receptor agonist, said Dr. Di Angelantonio during his talk at the congress on the new risk score.
“The risk score is a good addition” because it estimates future CVD risk better and more systematically than usual practice, which generally relies on no systematic tool, said Naveed Sattar, PhD, professor of metabolic medicine at the University of Glasgow (Scotland) and also a member of the guideline-writing panel.
The new risk score “is a reasonable way” to identify people without CVD but at elevated risk who might benefit from treatment with a relatively expensive drug, such as an SGLT-2 inhibitor, Dr. Sattar said in an interview. “It doesn’t rely on any fancy biomarkers or imaging, and it takes about 30 seconds to calculate. It’s not perfect, but it gets the job done,” and it will increase the number of people with type 2 diabetes who will receive an SGLT-2 inhibitor, he predicted.
Dr. McGuire has been a consultant to Altimmune, Applied Therapeutics, AstraZeneca, Bayer, Boehringer-Ingelheim, Intercept, Lexion, Lilly, Merck, New Amsterdam, and Pfizer. Dr. Di Angelantonio had no disclosures. Dr. Sattar has been a consultant to Abbott, Amgen, AstraZeneca, Boehringer Ingelheim, Lilly, Novartis, Novo Nordisk, Pfizer, and Roche Diagnostics.
A version of this article appeared on Medscape.com.
AMSTERDAM – The era of guidelines that recommended treatment with either a sodium-glucose cotransporter-2 (SGLT-2) inhibitor or a glucagonlike peptide-1 (GLP-1) receptor agonist in people with type 2 diabetes mellitus and established cardiovascular disease (CVD) ended with new recommendations from the European Society of Cardiology that call for starting both classes simultaneously.
said Darren K. McGuire, MD, at the annual congress of the European Society of Cardiology.
The society’s new guidelines for managing CVD in patients with diabetes, released on Aug. 25 and presented in several sessions at the Congress, also break with the past by calling for starting treatment with both an SGLT-2 inhibitor and a GLP-1 receptor agonist without regard to a person’s existing level of glucose control, including their current and target hemoglobin A1c levels, and regardless of background therapy, added Dr. McGuire, a cardiologist and professor at the UT Southwestern Medical Center in Dallas and a member of the ESC panel that wrote the new guidelines.
Instead, the new guidance calls for starting both drug classes promptly in people diagnosed with type 2 diabetes and established atherosclerotic CVD.
Both the previous ESC guidelines from 2019 as well as the current Standards of Care for 2023 document from the American Diabetes Association call for using one class or the other, but they hedge on combined treatment as discretionary.
Different mechanisms mean additive benefits
“With increasing numbers of patients with type 2 diabetes in trials for SGLT-2 inhibitors or GLP-1 receptor agonists who were also on the other drug class, we’ve done large, stratified analyses that suggest no treatment-effect modification” when people received agents from both drug classes, Dr. McGuire explained in an interview. “While we don’t understand the mechanisms of action of these drugs for CVD, we’ve become very confident that they use different mechanisms” that appear to have at least partially additive effects.
“Their benefits for CVD risk reduction are completely independent of their glucose effects. They are cardiology drugs,” Dr. McGuire added.
The new ESC guidelines highlight two other clinical settings where people with type 2 diabetes should receive an SGLT-2 inhibitor regardless of their existing level of glucose control and any other medical treatment: people with heart failure and people with chronic kidney disease (CKD) based on a depressed estimated glomerular filtration rate and an elevated urine albumin-to-creatinine ratio.
Nephropathy was considered by the ESC’s guideline panel to confer risk that is similar to that of established atherosclerotic CVD, Dr. McGuire said.
The guidelines also, for the first time for ESC recommendations, made treatment with finerenone (Kerendia, Bayer) a class 1 level A recommendation for people with type 2 diabetes and CKD.
SCORE2-Diabetes risk estimator
Another major change in the new ESC guideline revision is introduction of a CVD risk calculator intended to estimate the risk among people with type 2 diabetes but without established CVD, heart failure, or CKD.
Called the SCORE2-Diabetes risk estimator, it calculates a person’s 10-year risk for CVD and includes adjustment based on the European region where a person lives; it also tallies different risk levels for women and for men.
The researchers who developed the SCORE2-Diabetes calculator used data from nearly 230,000 people to devise the tool and then validated it with data from an additional 217,000 Europeans with type 2 diabetes.
Key features of the calculator include its use of routinely collected clinical values, such as age, sex, systolic blood pressure, smoking status, serum cholesterol levels, age at diabetes diagnosis, hemoglobin A1c level, and estimated glomerular filtration rate.
“For the first time we have a clear score to categorize risk” in people with type 2 diabetes and identify who needs more aggressive treatment to prevent CVD development,” said Emanuele Di Angelantonio, MD, PhD, deputy director of the cardiovascular epidemiology unit at the University of Cambridge (England).
The guidelines say that people who have a low (< 5%) or moderate (5%-9%) 10-year risk for CVD are possible candidates for metformin treatment. Those with high (10%-19%) or very high (≥ 20%) risk are possible candidates for treatment with metformin and/or an SGLT-2 inhibitor and/or a GLP-1 receptor agonist, said Dr. Di Angelantonio during his talk at the congress on the new risk score.
“The risk score is a good addition” because it estimates future CVD risk better and more systematically than usual practice, which generally relies on no systematic tool, said Naveed Sattar, PhD, professor of metabolic medicine at the University of Glasgow (Scotland) and also a member of the guideline-writing panel.
The new risk score “is a reasonable way” to identify people without CVD but at elevated risk who might benefit from treatment with a relatively expensive drug, such as an SGLT-2 inhibitor, Dr. Sattar said in an interview. “It doesn’t rely on any fancy biomarkers or imaging, and it takes about 30 seconds to calculate. It’s not perfect, but it gets the job done,” and it will increase the number of people with type 2 diabetes who will receive an SGLT-2 inhibitor, he predicted.
Dr. McGuire has been a consultant to Altimmune, Applied Therapeutics, AstraZeneca, Bayer, Boehringer-Ingelheim, Intercept, Lexion, Lilly, Merck, New Amsterdam, and Pfizer. Dr. Di Angelantonio had no disclosures. Dr. Sattar has been a consultant to Abbott, Amgen, AstraZeneca, Boehringer Ingelheim, Lilly, Novartis, Novo Nordisk, Pfizer, and Roche Diagnostics.
A version of this article appeared on Medscape.com.
AMSTERDAM – The era of guidelines that recommended treatment with either a sodium-glucose cotransporter-2 (SGLT-2) inhibitor or a glucagonlike peptide-1 (GLP-1) receptor agonist in people with type 2 diabetes mellitus and established cardiovascular disease (CVD) ended with new recommendations from the European Society of Cardiology that call for starting both classes simultaneously.
said Darren K. McGuire, MD, at the annual congress of the European Society of Cardiology.
The society’s new guidelines for managing CVD in patients with diabetes, released on Aug. 25 and presented in several sessions at the Congress, also break with the past by calling for starting treatment with both an SGLT-2 inhibitor and a GLP-1 receptor agonist without regard to a person’s existing level of glucose control, including their current and target hemoglobin A1c levels, and regardless of background therapy, added Dr. McGuire, a cardiologist and professor at the UT Southwestern Medical Center in Dallas and a member of the ESC panel that wrote the new guidelines.
Instead, the new guidance calls for starting both drug classes promptly in people diagnosed with type 2 diabetes and established atherosclerotic CVD.
Both the previous ESC guidelines from 2019 as well as the current Standards of Care for 2023 document from the American Diabetes Association call for using one class or the other, but they hedge on combined treatment as discretionary.
Different mechanisms mean additive benefits
“With increasing numbers of patients with type 2 diabetes in trials for SGLT-2 inhibitors or GLP-1 receptor agonists who were also on the other drug class, we’ve done large, stratified analyses that suggest no treatment-effect modification” when people received agents from both drug classes, Dr. McGuire explained in an interview. “While we don’t understand the mechanisms of action of these drugs for CVD, we’ve become very confident that they use different mechanisms” that appear to have at least partially additive effects.
“Their benefits for CVD risk reduction are completely independent of their glucose effects. They are cardiology drugs,” Dr. McGuire added.
The new ESC guidelines highlight two other clinical settings where people with type 2 diabetes should receive an SGLT-2 inhibitor regardless of their existing level of glucose control and any other medical treatment: people with heart failure and people with chronic kidney disease (CKD) based on a depressed estimated glomerular filtration rate and an elevated urine albumin-to-creatinine ratio.
Nephropathy was considered by the ESC’s guideline panel to confer risk that is similar to that of established atherosclerotic CVD, Dr. McGuire said.
The guidelines also, for the first time for ESC recommendations, made treatment with finerenone (Kerendia, Bayer) a class 1 level A recommendation for people with type 2 diabetes and CKD.
SCORE2-Diabetes risk estimator
Another major change in the new ESC guideline revision is introduction of a CVD risk calculator intended to estimate the risk among people with type 2 diabetes but without established CVD, heart failure, or CKD.
Called the SCORE2-Diabetes risk estimator, it calculates a person’s 10-year risk for CVD and includes adjustment based on the European region where a person lives; it also tallies different risk levels for women and for men.
The researchers who developed the SCORE2-Diabetes calculator used data from nearly 230,000 people to devise the tool and then validated it with data from an additional 217,000 Europeans with type 2 diabetes.
Key features of the calculator include its use of routinely collected clinical values, such as age, sex, systolic blood pressure, smoking status, serum cholesterol levels, age at diabetes diagnosis, hemoglobin A1c level, and estimated glomerular filtration rate.
“For the first time we have a clear score to categorize risk” in people with type 2 diabetes and identify who needs more aggressive treatment to prevent CVD development,” said Emanuele Di Angelantonio, MD, PhD, deputy director of the cardiovascular epidemiology unit at the University of Cambridge (England).
The guidelines say that people who have a low (< 5%) or moderate (5%-9%) 10-year risk for CVD are possible candidates for metformin treatment. Those with high (10%-19%) or very high (≥ 20%) risk are possible candidates for treatment with metformin and/or an SGLT-2 inhibitor and/or a GLP-1 receptor agonist, said Dr. Di Angelantonio during his talk at the congress on the new risk score.
“The risk score is a good addition” because it estimates future CVD risk better and more systematically than usual practice, which generally relies on no systematic tool, said Naveed Sattar, PhD, professor of metabolic medicine at the University of Glasgow (Scotland) and also a member of the guideline-writing panel.
The new risk score “is a reasonable way” to identify people without CVD but at elevated risk who might benefit from treatment with a relatively expensive drug, such as an SGLT-2 inhibitor, Dr. Sattar said in an interview. “It doesn’t rely on any fancy biomarkers or imaging, and it takes about 30 seconds to calculate. It’s not perfect, but it gets the job done,” and it will increase the number of people with type 2 diabetes who will receive an SGLT-2 inhibitor, he predicted.
Dr. McGuire has been a consultant to Altimmune, Applied Therapeutics, AstraZeneca, Bayer, Boehringer-Ingelheim, Intercept, Lexion, Lilly, Merck, New Amsterdam, and Pfizer. Dr. Di Angelantonio had no disclosures. Dr. Sattar has been a consultant to Abbott, Amgen, AstraZeneca, Boehringer Ingelheim, Lilly, Novartis, Novo Nordisk, Pfizer, and Roche Diagnostics.
A version of this article appeared on Medscape.com.
AT ESC CONGRESS 2023
Liraglutide fixes learning limit tied to insulin resistance
A single injection of the GLP-1 receptor agonist liraglutide led to short-term normalization of associative learning in people with obesity and insulin resistance, a finding that suggests say the authors of a recent report in Nature Metabolism.
“We demonstrated that dopamine-driven associative learning about external sensory cues crucially depends on metabolic signaling,” said Marc Tittgemeyer, PhD, professor at the Max Planck Institute for Metabolism Research in Cologne, Germany, and senior author of the study. Study participants with impaired insulin sensitivity “exhibited a reduced amplitude of behavioral updating that was normalized” by a single subcutaneous injection of 0.6 mg of liraglutide (the starting daily dose for liraglutide for weight loss, available as Saxenda, Novo Nordisk) given the evening before testing.
The findings, from 30 adults with normal insulin sensitivity and normal weight and 24 adults with impaired insulin sensitivity and obesity, suggest that metabolic signals, particularly ones that promote energy restoration in a setting of energy deprivation caused by insulin or a glucagon-like peptide-1 (GLP-1) receptor agonist, “profoundly influence neuronal processing,” said Dr. Tittgemeyer. The findings suggest that impaired metabolic signaling such as occurs with insulin resistance in people with obesity can cause deficiencies in associative learning.
‘Liraglutide can normalize learning of associations’
“We show that in people with obesity, disrupted circuit mechanisms lead to impaired learning about sensory associations,” Dr. Tittgemeyer said in an interview. “The information provided by sensory systems that the brain must interpret to select a behavioral response are ‘off tune’ ” in these individuals.
“This is rather consequential for understanding food-intake behaviors. Modern obesity treatments, such as liraglutide, can normalize learning of associations and thereby render people susceptible again for sensory signals and make them more prone to react to subliminal interactions, such as weight-normalizing diets and conscious eating,” he added.
The normalization in associative learning that one dose of liraglutide produced in people with obesity “fits with studies showing that these drugs restore a normal feeling of satiety, causing people to eat less and therefore lose weight,” he explained.
Dr. Tittgemeyer noted that this effect is likely shared by other agents in the GLP-1 receptor agonist class, such as semaglutide (Ozempic, Wegovy, Novo Nordisk) but is likely not an effect when agents agonize receptors to other nutrient-stimulated hormones such as glucagon and the glucose-dependent insulinotropic polypeptide.
The findings “show that liraglutide restores associative learning in participants with greater insulin resistance,” a “highly relevant” discovery, commented Nils B. Kroemer, PhD, head of the section of medical psychology at the University of Bonn, Germany, who was not involved with this research, in a written statement.
The study run by Dr. Tittgemeyer and his associates included 54 healthy adult volunteers whom they assessed for insulin sensitivity with their homeostasis model assessment of insulin resistance. The researchers divided the cohort into groups; one group included 24 people with impaired insulin sensitivity, and one included 30 with normal insulin sensitivity. The average body mass index (BMI) of the normal sensitivity group was about 24 kg/m2; in the insulin-resistant subgroup, BMI averaged about 33 kg/m2.
The associative learning task tested the ability of participants to learn associations between auditory cues (a high or low tone) and a subsequent visual outcome (a picture of a face or a house). During each associative learning session, participants also underwent functional MRI of the brain.
Liraglutide treatment leveled learning
The results showed that the learning rate was significantly lower in the subgroup with impaired insulin sensitivity, compared with those with normal insulin sensitivity following treatment with a placebo injection. This indicates a decreased adaptation of learning to predictability variations in individuals with impaired insulin sensitivity.
In contrast, treatment with a single dose of liraglutide significantly enhanced the learning rate in the group with impaired insulin sensitivity but significantly reduced the learning rate in the group with normal insulin sensitivity. Liraglutide’s effect was twice as large in the group with impaired insulin sensitivity than in the group with normal insulin sensitivity, and these opposing effects of liraglutide resulted in a convergence of the two groups’ adaptive learning rates so that there wasn’t any significant between-group difference following liraglutide treatment.
After analyzing the functional MRI data along with the learning results, the researchers concluded that liraglutide normalized learning in individuals with impaired insulin sensitivity by enhancing adaptive prediction error encoding in the brain’s ventral striatum and mesocortical projection sites.
This apparent ability of GLP-1 analogues to correct this learning deficit in people with impaired insulin sensitivity and obesity has implications regarding potential benefit for people with other pathologies characterized by impaired dopaminergic function and associated with metabolic impairments, such as psychosis, Parkinson’s disease, and depression, the researchers say.
“The fascinating thing about GLP-1 receptor agonists is that they have an additional mechanism that relates to anti-inflammatory effects, especially for alleviating cell stress,” said Dr. Tittgemeyer. “Many ongoing clinical trials are assessing their effects in neuropsychiatric diseases,” he noted.
The study received no commercial funding. Dr. Tittgemyer and most of his coauthors had no disclosures. One coauthor had several disclosures, which are detailed in the report. Dr. Kroemer had no disclosures.
A version of this article first appeared on Medscape.com.
A single injection of the GLP-1 receptor agonist liraglutide led to short-term normalization of associative learning in people with obesity and insulin resistance, a finding that suggests say the authors of a recent report in Nature Metabolism.
“We demonstrated that dopamine-driven associative learning about external sensory cues crucially depends on metabolic signaling,” said Marc Tittgemeyer, PhD, professor at the Max Planck Institute for Metabolism Research in Cologne, Germany, and senior author of the study. Study participants with impaired insulin sensitivity “exhibited a reduced amplitude of behavioral updating that was normalized” by a single subcutaneous injection of 0.6 mg of liraglutide (the starting daily dose for liraglutide for weight loss, available as Saxenda, Novo Nordisk) given the evening before testing.
The findings, from 30 adults with normal insulin sensitivity and normal weight and 24 adults with impaired insulin sensitivity and obesity, suggest that metabolic signals, particularly ones that promote energy restoration in a setting of energy deprivation caused by insulin or a glucagon-like peptide-1 (GLP-1) receptor agonist, “profoundly influence neuronal processing,” said Dr. Tittgemeyer. The findings suggest that impaired metabolic signaling such as occurs with insulin resistance in people with obesity can cause deficiencies in associative learning.
‘Liraglutide can normalize learning of associations’
“We show that in people with obesity, disrupted circuit mechanisms lead to impaired learning about sensory associations,” Dr. Tittgemeyer said in an interview. “The information provided by sensory systems that the brain must interpret to select a behavioral response are ‘off tune’ ” in these individuals.
“This is rather consequential for understanding food-intake behaviors. Modern obesity treatments, such as liraglutide, can normalize learning of associations and thereby render people susceptible again for sensory signals and make them more prone to react to subliminal interactions, such as weight-normalizing diets and conscious eating,” he added.
The normalization in associative learning that one dose of liraglutide produced in people with obesity “fits with studies showing that these drugs restore a normal feeling of satiety, causing people to eat less and therefore lose weight,” he explained.
Dr. Tittgemeyer noted that this effect is likely shared by other agents in the GLP-1 receptor agonist class, such as semaglutide (Ozempic, Wegovy, Novo Nordisk) but is likely not an effect when agents agonize receptors to other nutrient-stimulated hormones such as glucagon and the glucose-dependent insulinotropic polypeptide.
The findings “show that liraglutide restores associative learning in participants with greater insulin resistance,” a “highly relevant” discovery, commented Nils B. Kroemer, PhD, head of the section of medical psychology at the University of Bonn, Germany, who was not involved with this research, in a written statement.
The study run by Dr. Tittgemeyer and his associates included 54 healthy adult volunteers whom they assessed for insulin sensitivity with their homeostasis model assessment of insulin resistance. The researchers divided the cohort into groups; one group included 24 people with impaired insulin sensitivity, and one included 30 with normal insulin sensitivity. The average body mass index (BMI) of the normal sensitivity group was about 24 kg/m2; in the insulin-resistant subgroup, BMI averaged about 33 kg/m2.
The associative learning task tested the ability of participants to learn associations between auditory cues (a high or low tone) and a subsequent visual outcome (a picture of a face or a house). During each associative learning session, participants also underwent functional MRI of the brain.
Liraglutide treatment leveled learning
The results showed that the learning rate was significantly lower in the subgroup with impaired insulin sensitivity, compared with those with normal insulin sensitivity following treatment with a placebo injection. This indicates a decreased adaptation of learning to predictability variations in individuals with impaired insulin sensitivity.
In contrast, treatment with a single dose of liraglutide significantly enhanced the learning rate in the group with impaired insulin sensitivity but significantly reduced the learning rate in the group with normal insulin sensitivity. Liraglutide’s effect was twice as large in the group with impaired insulin sensitivity than in the group with normal insulin sensitivity, and these opposing effects of liraglutide resulted in a convergence of the two groups’ adaptive learning rates so that there wasn’t any significant between-group difference following liraglutide treatment.
After analyzing the functional MRI data along with the learning results, the researchers concluded that liraglutide normalized learning in individuals with impaired insulin sensitivity by enhancing adaptive prediction error encoding in the brain’s ventral striatum and mesocortical projection sites.
This apparent ability of GLP-1 analogues to correct this learning deficit in people with impaired insulin sensitivity and obesity has implications regarding potential benefit for people with other pathologies characterized by impaired dopaminergic function and associated with metabolic impairments, such as psychosis, Parkinson’s disease, and depression, the researchers say.
“The fascinating thing about GLP-1 receptor agonists is that they have an additional mechanism that relates to anti-inflammatory effects, especially for alleviating cell stress,” said Dr. Tittgemeyer. “Many ongoing clinical trials are assessing their effects in neuropsychiatric diseases,” he noted.
The study received no commercial funding. Dr. Tittgemyer and most of his coauthors had no disclosures. One coauthor had several disclosures, which are detailed in the report. Dr. Kroemer had no disclosures.
A version of this article first appeared on Medscape.com.
A single injection of the GLP-1 receptor agonist liraglutide led to short-term normalization of associative learning in people with obesity and insulin resistance, a finding that suggests say the authors of a recent report in Nature Metabolism.
“We demonstrated that dopamine-driven associative learning about external sensory cues crucially depends on metabolic signaling,” said Marc Tittgemeyer, PhD, professor at the Max Planck Institute for Metabolism Research in Cologne, Germany, and senior author of the study. Study participants with impaired insulin sensitivity “exhibited a reduced amplitude of behavioral updating that was normalized” by a single subcutaneous injection of 0.6 mg of liraglutide (the starting daily dose for liraglutide for weight loss, available as Saxenda, Novo Nordisk) given the evening before testing.
The findings, from 30 adults with normal insulin sensitivity and normal weight and 24 adults with impaired insulin sensitivity and obesity, suggest that metabolic signals, particularly ones that promote energy restoration in a setting of energy deprivation caused by insulin or a glucagon-like peptide-1 (GLP-1) receptor agonist, “profoundly influence neuronal processing,” said Dr. Tittgemeyer. The findings suggest that impaired metabolic signaling such as occurs with insulin resistance in people with obesity can cause deficiencies in associative learning.
‘Liraglutide can normalize learning of associations’
“We show that in people with obesity, disrupted circuit mechanisms lead to impaired learning about sensory associations,” Dr. Tittgemeyer said in an interview. “The information provided by sensory systems that the brain must interpret to select a behavioral response are ‘off tune’ ” in these individuals.
“This is rather consequential for understanding food-intake behaviors. Modern obesity treatments, such as liraglutide, can normalize learning of associations and thereby render people susceptible again for sensory signals and make them more prone to react to subliminal interactions, such as weight-normalizing diets and conscious eating,” he added.
The normalization in associative learning that one dose of liraglutide produced in people with obesity “fits with studies showing that these drugs restore a normal feeling of satiety, causing people to eat less and therefore lose weight,” he explained.
Dr. Tittgemeyer noted that this effect is likely shared by other agents in the GLP-1 receptor agonist class, such as semaglutide (Ozempic, Wegovy, Novo Nordisk) but is likely not an effect when agents agonize receptors to other nutrient-stimulated hormones such as glucagon and the glucose-dependent insulinotropic polypeptide.
The findings “show that liraglutide restores associative learning in participants with greater insulin resistance,” a “highly relevant” discovery, commented Nils B. Kroemer, PhD, head of the section of medical psychology at the University of Bonn, Germany, who was not involved with this research, in a written statement.
The study run by Dr. Tittgemeyer and his associates included 54 healthy adult volunteers whom they assessed for insulin sensitivity with their homeostasis model assessment of insulin resistance. The researchers divided the cohort into groups; one group included 24 people with impaired insulin sensitivity, and one included 30 with normal insulin sensitivity. The average body mass index (BMI) of the normal sensitivity group was about 24 kg/m2; in the insulin-resistant subgroup, BMI averaged about 33 kg/m2.
The associative learning task tested the ability of participants to learn associations between auditory cues (a high or low tone) and a subsequent visual outcome (a picture of a face or a house). During each associative learning session, participants also underwent functional MRI of the brain.
Liraglutide treatment leveled learning
The results showed that the learning rate was significantly lower in the subgroup with impaired insulin sensitivity, compared with those with normal insulin sensitivity following treatment with a placebo injection. This indicates a decreased adaptation of learning to predictability variations in individuals with impaired insulin sensitivity.
In contrast, treatment with a single dose of liraglutide significantly enhanced the learning rate in the group with impaired insulin sensitivity but significantly reduced the learning rate in the group with normal insulin sensitivity. Liraglutide’s effect was twice as large in the group with impaired insulin sensitivity than in the group with normal insulin sensitivity, and these opposing effects of liraglutide resulted in a convergence of the two groups’ adaptive learning rates so that there wasn’t any significant between-group difference following liraglutide treatment.
After analyzing the functional MRI data along with the learning results, the researchers concluded that liraglutide normalized learning in individuals with impaired insulin sensitivity by enhancing adaptive prediction error encoding in the brain’s ventral striatum and mesocortical projection sites.
This apparent ability of GLP-1 analogues to correct this learning deficit in people with impaired insulin sensitivity and obesity has implications regarding potential benefit for people with other pathologies characterized by impaired dopaminergic function and associated with metabolic impairments, such as psychosis, Parkinson’s disease, and depression, the researchers say.
“The fascinating thing about GLP-1 receptor agonists is that they have an additional mechanism that relates to anti-inflammatory effects, especially for alleviating cell stress,” said Dr. Tittgemeyer. “Many ongoing clinical trials are assessing their effects in neuropsychiatric diseases,” he noted.
The study received no commercial funding. Dr. Tittgemyer and most of his coauthors had no disclosures. One coauthor had several disclosures, which are detailed in the report. Dr. Kroemer had no disclosures.
A version of this article first appeared on Medscape.com.
FROM NATURE METABOLISM
Wegovy scores HFpEF benefits in people with obesity
AMSTERDAM – Adults with heart failure with preserved ejection fraction (HFpEF) but without diabetes showed significant improvements in their heart failure-related symptoms and physical limitations, exercise function, and weight loss when treated with a weight-reducing dose of semaglutide for 52 weeks, compared with placebo, in the randomized STEP-HFpEF trial.
The results, which also showed the treatment’s safety in these patients, “indicate that treatment with semaglutide is a valuable therapeutic approach in the management of patients with HFpEF and obesity,” Mikhail Kosiborod, MD, said at the annual congress of the European Society of Cardiology.

The findings establish semaglutide, a glucagonlike peptide–1 (GLP-1) receptor agonist, as a second class of medication with proven efficacy and safety for people with HFpEF, joining two agents also proven beneficial for people with HFpEF, dapagliflozin (Farxiga) and empagliflozin (Jardiance), both from the class of sodium-glucose cotransporter 2 (SGLT2) inhibitors.
When administered at the approved dose for weight loss of 2.4 mg, injected subcutaneously weekly for 52 weeks, semaglutide (Wegovy) produced an average 7.8-point incremental improvement in patients’ scores on the Kansas City Cardiomyopathy Questionnaire (KCCQ), a validated measure of symptoms and functional limitations, compared with controls who received placebo injections, as well as an average incremental weight loss from baseline, compared with placebo, of 10.7%. Both were significant effects, compared with placebo, and clinically meaningful benefits for the study’s two primary endpoints.
Simultaneously with Kosiborod’s report the results also appeared in a report posted online in the New England Journal of Medicine.
A ‘paradigm shift’ for medical weight loss in cardiology
The findings from this study with 529 randomized patients immediately propelled the weight loss formulation of semaglutide into the ranks of agents used to treat and prevent cardiovascular disease events. This evolution in the indications for semaglutide will be driven not only by the STEP-HFpEF results but also by findings from the SELECT trial, which tested the same semaglutide weight-loss dose in people with obesity, established cardiovascular disease, and had positive top-line results for prevention of major cardiovascular adverse events, according to a press release from Novo Nordisk on Aug. 8.
The STEP-HFpEF and SELECT results will trigger “a paradigm shift” for cardiologists, who will now need to consider prescribing a weight-loss medication to many of their patients, agents that until now were not part of the usual pharmacologic toolbox for cardiologists, said Dr. Kosiborod, a cardiologist and codirector of the Haverty Cardiometabolic Center of Excellence at Saint Luke’s Mid America Heart Institute in Kansas City, Mo. This shift will require education to bring the clinical cardiology community on board, he added in an interview.
Given that semaglutide administered at this dose already has a Food and Drug Administration–approved indication for weight loss in people with obesity or overweight plus at least one comorbidity, clinicians could immediately start using the treatment in people with obesity and HFpEF, said Dr. Kosiborod and other cardiologists.
Weekly semaglutide injections “could be considered a treatment option right now” for people with obesity and HFpEF, Dr. Kosiborod said during a press briefing.
Other experts agreed, especially because the STEP-HFpEF results confirmed that weight loss treatment with semaglutide was safe in this population.
‘A terrific win for patients’
The new findings are “a terrific win and game changer for patients with HFpEF,” commented Gregg C. Fonarow, MD, professor and cochief of cardiology at the University of California, Los Angeles, who was not involved with the study.
“The magnitude of improvement in the patient-reported health status scores are large and impressive. These data support clinical use of this agent for individuals with HFpEF with a body mass index of 30 kg/m2, patients who already fall within existing indications,” Dr. Fonarow said in an interview.
“Given the improvements in clinical outcomes in the STEP-HFpEF and SELECT trials, cardiologists should be prescribing these medications to eligible patients without conditions,” he added. “The perception of [semaglutide] needs to shift and be viewed as a component of the comprehensive medical therapies provided to individuals with established cardiovascular disease or HFpEF who also have elevated body mass index to improve their clinical outcomes.”

Historically, cardiologists have had a concern that weight loss was potentially harmful in people with heart failure and that obesity was protective, a phenomenon known as the “obesity paradox,” but the STEP-HFpEF data help disprove that notion, commented Nancy K. Sweitzer, MD, PhD, a heart failure specialist and vice chair of clinical research in the department of medicine at Washington University in St. Louis, who also was not involved in the study.
No signal of an obesity paradox
“There’s been a concern in the heart failure community to use weight-loss strategies in people with heart failure because of this, but this evidence provides a lot of confidence that it’s safe to use this weight loss treatment. The results show that patients feel better and lose weight with no signal of harm,” Dr. Sweitzer said in an interview.
The “encouraging findings” for semaglutide in patients with HFpEF “potentially add a much needed extra option for these patients and provide another upstream treatment for patients with signs of this condition plus a high body mass index,” commented Yigal M. Pinto, MD, PhD, in an editorial that accompanied the published report.
“How these findings translate to hard end points remains to be established and will be important in determining the role of GLP-1 agonism,” wrote Dr. Pinto, a professor and heart failure specialist at Amsterdam University Medical Center.
But Dr. Kosiborod said that the improvement seen in the KCCQ score was itself an important benefit for patients. “Heart failure is defined clinically based on symptoms,” he noted, and results in prior studies documented that patients value improvements in symptoms and physical limitations even more than they value “hard endpoints” such as survival.
The new findings, which indicate that two different and expensive classes of medications are now standard of care for many people with HFpEF and obesity – the SGLT2 inhibitors and the GLP-1 receptor agonist semaglutide – also raise concerns over patient access and affordability, as many U.S. insurers have a history of requiring prior authorization, high copays, or coverage denials for these two medical classes.
But Dr. Sweitzer and Dr. Kosiborod both said that the insurance-coverage climate seems, in just the past couple of years or so, to have dramatically improved, although it’s still not ideal.
Prior authorization hoops have decreased
“We still have prior-authorization hoops to jump through, but I expect these will continue to decrease over time as evidence for clinical benefits [from weight loss] continues to accumulate,” said Dr. Sweitzer.
And “the SELECT data mean that cardiologists will need to become comfortable prescribing GLP-1 receptor agonists,” she added.
“It’s not okay for insurers to say we are not going to cover weight loss medications because it’s a cosmetic indication,” said Dr. Kosiborod. “Obesity appears to be very important in the pathogenesis and progression of heart failure, and if patients derive substantial benefit, they should have access to this treatment.”
The improvements in KCCQ score, as well as in several secondary and exploratory endpoints including a significant reduction in C-reactive protein (an indication of a potent anti-inflammatory effect), an average 20 m increase in 6-minute walk distance, a significant average drop in N-terminal pro-brain natriuretic peptide, and a drop in heart failure hospitalizations or urgent heart failure visits (although the trial was not powered to show differences in clinical events), “were the largest benefits in these outcomes we’ve seen,” compared with any other medical intervention in people with HFpEF, he noted.
“About 80% of U.S. patients with HFpEF have obesity or overweight,” Dr. Kosiborod noted. Using semaglutide on these patients “is an issue of access and insurance coverage. My hope is that these and other data will favorably change this.”
A related trial with a similar design, STEP-HFpEF DM, is still in progress and testing the same semaglutide treatment in adults with HFpEF, obesity, and type 2 diabetes, noted Dr. Kosiborod, who is also lead investigator for that study. He said those results will likely become available before the end of 2023.
The study was funded by Novo Nordisk, the company that markets semaglutide (Wegovy). Dr. Kosiborod has been a consultant and adviser to and has received honoraria from Novo Nordisk. He has also been a consultant to numerous other companies, received research grants from AstraZeneca, Boehringer Ingelheim, and Pfizer, honoraria from AstraZeneca, and is a stockholder in Artera Health and Saghmos Therapeutics. Dr. Fonarow has been a consultant to Abbott, Amgen, AstraZeneca, CHF Solutions, Cytokinetics, Edwards, Janssen, Medtronic, Merck, Novartis, and Regeneron. Dr. Sweitzer reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AMSTERDAM – Adults with heart failure with preserved ejection fraction (HFpEF) but without diabetes showed significant improvements in their heart failure-related symptoms and physical limitations, exercise function, and weight loss when treated with a weight-reducing dose of semaglutide for 52 weeks, compared with placebo, in the randomized STEP-HFpEF trial.
The results, which also showed the treatment’s safety in these patients, “indicate that treatment with semaglutide is a valuable therapeutic approach in the management of patients with HFpEF and obesity,” Mikhail Kosiborod, MD, said at the annual congress of the European Society of Cardiology.
The findings establish semaglutide, a glucagonlike peptide–1 (GLP-1) receptor agonist, as a second class of medication with proven efficacy and safety for people with HFpEF, joining two agents also proven beneficial for people with HFpEF, dapagliflozin (Farxiga) and empagliflozin (Jardiance), both from the class of sodium-glucose cotransporter 2 (SGLT2) inhibitors.
When administered at the approved dose for weight loss of 2.4 mg, injected subcutaneously weekly for 52 weeks, semaglutide (Wegovy) produced an average 7.8-point incremental improvement in patients’ scores on the Kansas City Cardiomyopathy Questionnaire (KCCQ), a validated measure of symptoms and functional limitations, compared with controls who received placebo injections, as well as an average incremental weight loss from baseline, compared with placebo, of 10.7%. Both were significant effects, compared with placebo, and clinically meaningful benefits for the study’s two primary endpoints.
Simultaneously with Kosiborod’s report the results also appeared in a report posted online in the New England Journal of Medicine.
A ‘paradigm shift’ for medical weight loss in cardiology
The findings from this study with 529 randomized patients immediately propelled the weight loss formulation of semaglutide into the ranks of agents used to treat and prevent cardiovascular disease events. This evolution in the indications for semaglutide will be driven not only by the STEP-HFpEF results but also by findings from the SELECT trial, which tested the same semaglutide weight-loss dose in people with obesity, established cardiovascular disease, and had positive top-line results for prevention of major cardiovascular adverse events, according to a press release from Novo Nordisk on Aug. 8.
The STEP-HFpEF and SELECT results will trigger “a paradigm shift” for cardiologists, who will now need to consider prescribing a weight-loss medication to many of their patients, agents that until now were not part of the usual pharmacologic toolbox for cardiologists, said Dr. Kosiborod, a cardiologist and codirector of the Haverty Cardiometabolic Center of Excellence at Saint Luke’s Mid America Heart Institute in Kansas City, Mo. This shift will require education to bring the clinical cardiology community on board, he added in an interview.
Given that semaglutide administered at this dose already has a Food and Drug Administration–approved indication for weight loss in people with obesity or overweight plus at least one comorbidity, clinicians could immediately start using the treatment in people with obesity and HFpEF, said Dr. Kosiborod and other cardiologists.
Weekly semaglutide injections “could be considered a treatment option right now” for people with obesity and HFpEF, Dr. Kosiborod said during a press briefing.
Other experts agreed, especially because the STEP-HFpEF results confirmed that weight loss treatment with semaglutide was safe in this population.
‘A terrific win for patients’
The new findings are “a terrific win and game changer for patients with HFpEF,” commented Gregg C. Fonarow, MD, professor and cochief of cardiology at the University of California, Los Angeles, who was not involved with the study.
“The magnitude of improvement in the patient-reported health status scores are large and impressive. These data support clinical use of this agent for individuals with HFpEF with a body mass index of 30 kg/m2, patients who already fall within existing indications,” Dr. Fonarow said in an interview.
“Given the improvements in clinical outcomes in the STEP-HFpEF and SELECT trials, cardiologists should be prescribing these medications to eligible patients without conditions,” he added. “The perception of [semaglutide] needs to shift and be viewed as a component of the comprehensive medical therapies provided to individuals with established cardiovascular disease or HFpEF who also have elevated body mass index to improve their clinical outcomes.”
Historically, cardiologists have had a concern that weight loss was potentially harmful in people with heart failure and that obesity was protective, a phenomenon known as the “obesity paradox,” but the STEP-HFpEF data help disprove that notion, commented Nancy K. Sweitzer, MD, PhD, a heart failure specialist and vice chair of clinical research in the department of medicine at Washington University in St. Louis, who also was not involved in the study.
No signal of an obesity paradox
“There’s been a concern in the heart failure community to use weight-loss strategies in people with heart failure because of this, but this evidence provides a lot of confidence that it’s safe to use this weight loss treatment. The results show that patients feel better and lose weight with no signal of harm,” Dr. Sweitzer said in an interview.
The “encouraging findings” for semaglutide in patients with HFpEF “potentially add a much needed extra option for these patients and provide another upstream treatment for patients with signs of this condition plus a high body mass index,” commented Yigal M. Pinto, MD, PhD, in an editorial that accompanied the published report.
“How these findings translate to hard end points remains to be established and will be important in determining the role of GLP-1 agonism,” wrote Dr. Pinto, a professor and heart failure specialist at Amsterdam University Medical Center.
But Dr. Kosiborod said that the improvement seen in the KCCQ score was itself an important benefit for patients. “Heart failure is defined clinically based on symptoms,” he noted, and results in prior studies documented that patients value improvements in symptoms and physical limitations even more than they value “hard endpoints” such as survival.
The new findings, which indicate that two different and expensive classes of medications are now standard of care for many people with HFpEF and obesity – the SGLT2 inhibitors and the GLP-1 receptor agonist semaglutide – also raise concerns over patient access and affordability, as many U.S. insurers have a history of requiring prior authorization, high copays, or coverage denials for these two medical classes.
But Dr. Sweitzer and Dr. Kosiborod both said that the insurance-coverage climate seems, in just the past couple of years or so, to have dramatically improved, although it’s still not ideal.
Prior authorization hoops have decreased
“We still have prior-authorization hoops to jump through, but I expect these will continue to decrease over time as evidence for clinical benefits [from weight loss] continues to accumulate,” said Dr. Sweitzer.
And “the SELECT data mean that cardiologists will need to become comfortable prescribing GLP-1 receptor agonists,” she added.
“It’s not okay for insurers to say we are not going to cover weight loss medications because it’s a cosmetic indication,” said Dr. Kosiborod. “Obesity appears to be very important in the pathogenesis and progression of heart failure, and if patients derive substantial benefit, they should have access to this treatment.”
The improvements in KCCQ score, as well as in several secondary and exploratory endpoints including a significant reduction in C-reactive protein (an indication of a potent anti-inflammatory effect), an average 20 m increase in 6-minute walk distance, a significant average drop in N-terminal pro-brain natriuretic peptide, and a drop in heart failure hospitalizations or urgent heart failure visits (although the trial was not powered to show differences in clinical events), “were the largest benefits in these outcomes we’ve seen,” compared with any other medical intervention in people with HFpEF, he noted.
“About 80% of U.S. patients with HFpEF have obesity or overweight,” Dr. Kosiborod noted. Using semaglutide on these patients “is an issue of access and insurance coverage. My hope is that these and other data will favorably change this.”
A related trial with a similar design, STEP-HFpEF DM, is still in progress and testing the same semaglutide treatment in adults with HFpEF, obesity, and type 2 diabetes, noted Dr. Kosiborod, who is also lead investigator for that study. He said those results will likely become available before the end of 2023.
The study was funded by Novo Nordisk, the company that markets semaglutide (Wegovy). Dr. Kosiborod has been a consultant and adviser to and has received honoraria from Novo Nordisk. He has also been a consultant to numerous other companies, received research grants from AstraZeneca, Boehringer Ingelheim, and Pfizer, honoraria from AstraZeneca, and is a stockholder in Artera Health and Saghmos Therapeutics. Dr. Fonarow has been a consultant to Abbott, Amgen, AstraZeneca, CHF Solutions, Cytokinetics, Edwards, Janssen, Medtronic, Merck, Novartis, and Regeneron. Dr. Sweitzer reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AMSTERDAM – Adults with heart failure with preserved ejection fraction (HFpEF) but without diabetes showed significant improvements in their heart failure-related symptoms and physical limitations, exercise function, and weight loss when treated with a weight-reducing dose of semaglutide for 52 weeks, compared with placebo, in the randomized STEP-HFpEF trial.
The results, which also showed the treatment’s safety in these patients, “indicate that treatment with semaglutide is a valuable therapeutic approach in the management of patients with HFpEF and obesity,” Mikhail Kosiborod, MD, said at the annual congress of the European Society of Cardiology.
The findings establish semaglutide, a glucagonlike peptide–1 (GLP-1) receptor agonist, as a second class of medication with proven efficacy and safety for people with HFpEF, joining two agents also proven beneficial for people with HFpEF, dapagliflozin (Farxiga) and empagliflozin (Jardiance), both from the class of sodium-glucose cotransporter 2 (SGLT2) inhibitors.
When administered at the approved dose for weight loss of 2.4 mg, injected subcutaneously weekly for 52 weeks, semaglutide (Wegovy) produced an average 7.8-point incremental improvement in patients’ scores on the Kansas City Cardiomyopathy Questionnaire (KCCQ), a validated measure of symptoms and functional limitations, compared with controls who received placebo injections, as well as an average incremental weight loss from baseline, compared with placebo, of 10.7%. Both were significant effects, compared with placebo, and clinically meaningful benefits for the study’s two primary endpoints.
Simultaneously with Kosiborod’s report the results also appeared in a report posted online in the New England Journal of Medicine.
A ‘paradigm shift’ for medical weight loss in cardiology
The findings from this study with 529 randomized patients immediately propelled the weight loss formulation of semaglutide into the ranks of agents used to treat and prevent cardiovascular disease events. This evolution in the indications for semaglutide will be driven not only by the STEP-HFpEF results but also by findings from the SELECT trial, which tested the same semaglutide weight-loss dose in people with obesity, established cardiovascular disease, and had positive top-line results for prevention of major cardiovascular adverse events, according to a press release from Novo Nordisk on Aug. 8.
The STEP-HFpEF and SELECT results will trigger “a paradigm shift” for cardiologists, who will now need to consider prescribing a weight-loss medication to many of their patients, agents that until now were not part of the usual pharmacologic toolbox for cardiologists, said Dr. Kosiborod, a cardiologist and codirector of the Haverty Cardiometabolic Center of Excellence at Saint Luke’s Mid America Heart Institute in Kansas City, Mo. This shift will require education to bring the clinical cardiology community on board, he added in an interview.
Given that semaglutide administered at this dose already has a Food and Drug Administration–approved indication for weight loss in people with obesity or overweight plus at least one comorbidity, clinicians could immediately start using the treatment in people with obesity and HFpEF, said Dr. Kosiborod and other cardiologists.
Weekly semaglutide injections “could be considered a treatment option right now” for people with obesity and HFpEF, Dr. Kosiborod said during a press briefing.
Other experts agreed, especially because the STEP-HFpEF results confirmed that weight loss treatment with semaglutide was safe in this population.
‘A terrific win for patients’
The new findings are “a terrific win and game changer for patients with HFpEF,” commented Gregg C. Fonarow, MD, professor and cochief of cardiology at the University of California, Los Angeles, who was not involved with the study.
“The magnitude of improvement in the patient-reported health status scores are large and impressive. These data support clinical use of this agent for individuals with HFpEF with a body mass index of 30 kg/m2, patients who already fall within existing indications,” Dr. Fonarow said in an interview.
“Given the improvements in clinical outcomes in the STEP-HFpEF and SELECT trials, cardiologists should be prescribing these medications to eligible patients without conditions,” he added. “The perception of [semaglutide] needs to shift and be viewed as a component of the comprehensive medical therapies provided to individuals with established cardiovascular disease or HFpEF who also have elevated body mass index to improve their clinical outcomes.”
Historically, cardiologists have had a concern that weight loss was potentially harmful in people with heart failure and that obesity was protective, a phenomenon known as the “obesity paradox,” but the STEP-HFpEF data help disprove that notion, commented Nancy K. Sweitzer, MD, PhD, a heart failure specialist and vice chair of clinical research in the department of medicine at Washington University in St. Louis, who also was not involved in the study.
No signal of an obesity paradox
“There’s been a concern in the heart failure community to use weight-loss strategies in people with heart failure because of this, but this evidence provides a lot of confidence that it’s safe to use this weight loss treatment. The results show that patients feel better and lose weight with no signal of harm,” Dr. Sweitzer said in an interview.
The “encouraging findings” for semaglutide in patients with HFpEF “potentially add a much needed extra option for these patients and provide another upstream treatment for patients with signs of this condition plus a high body mass index,” commented Yigal M. Pinto, MD, PhD, in an editorial that accompanied the published report.
“How these findings translate to hard end points remains to be established and will be important in determining the role of GLP-1 agonism,” wrote Dr. Pinto, a professor and heart failure specialist at Amsterdam University Medical Center.
But Dr. Kosiborod said that the improvement seen in the KCCQ score was itself an important benefit for patients. “Heart failure is defined clinically based on symptoms,” he noted, and results in prior studies documented that patients value improvements in symptoms and physical limitations even more than they value “hard endpoints” such as survival.
The new findings, which indicate that two different and expensive classes of medications are now standard of care for many people with HFpEF and obesity – the SGLT2 inhibitors and the GLP-1 receptor agonist semaglutide – also raise concerns over patient access and affordability, as many U.S. insurers have a history of requiring prior authorization, high copays, or coverage denials for these two medical classes.
But Dr. Sweitzer and Dr. Kosiborod both said that the insurance-coverage climate seems, in just the past couple of years or so, to have dramatically improved, although it’s still not ideal.
Prior authorization hoops have decreased
“We still have prior-authorization hoops to jump through, but I expect these will continue to decrease over time as evidence for clinical benefits [from weight loss] continues to accumulate,” said Dr. Sweitzer.
And “the SELECT data mean that cardiologists will need to become comfortable prescribing GLP-1 receptor agonists,” she added.
“It’s not okay for insurers to say we are not going to cover weight loss medications because it’s a cosmetic indication,” said Dr. Kosiborod. “Obesity appears to be very important in the pathogenesis and progression of heart failure, and if patients derive substantial benefit, they should have access to this treatment.”
The improvements in KCCQ score, as well as in several secondary and exploratory endpoints including a significant reduction in C-reactive protein (an indication of a potent anti-inflammatory effect), an average 20 m increase in 6-minute walk distance, a significant average drop in N-terminal pro-brain natriuretic peptide, and a drop in heart failure hospitalizations or urgent heart failure visits (although the trial was not powered to show differences in clinical events), “were the largest benefits in these outcomes we’ve seen,” compared with any other medical intervention in people with HFpEF, he noted.
“About 80% of U.S. patients with HFpEF have obesity or overweight,” Dr. Kosiborod noted. Using semaglutide on these patients “is an issue of access and insurance coverage. My hope is that these and other data will favorably change this.”
A related trial with a similar design, STEP-HFpEF DM, is still in progress and testing the same semaglutide treatment in adults with HFpEF, obesity, and type 2 diabetes, noted Dr. Kosiborod, who is also lead investigator for that study. He said those results will likely become available before the end of 2023.
The study was funded by Novo Nordisk, the company that markets semaglutide (Wegovy). Dr. Kosiborod has been a consultant and adviser to and has received honoraria from Novo Nordisk. He has also been a consultant to numerous other companies, received research grants from AstraZeneca, Boehringer Ingelheim, and Pfizer, honoraria from AstraZeneca, and is a stockholder in Artera Health and Saghmos Therapeutics. Dr. Fonarow has been a consultant to Abbott, Amgen, AstraZeneca, CHF Solutions, Cytokinetics, Edwards, Janssen, Medtronic, Merck, Novartis, and Regeneron. Dr. Sweitzer reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT THE ESC CONGRESS 2023
FDA okays first biosimilar for multiple sclerosis
including clinically isolated syndrome, relapsing remitting MS, and active secondary progressive disease.
“Biosimilar medications offer additional effective treatment options that have the potential to increase access for people living with relapsing forms of multiple sclerosis. [This] approval could have a meaningful impact for patients managing their disease,” Paul R. Lee, MD, PhD, director of the division of neurology II, FDA Center for Drug Evaluation and Research, said in a statement.
The natalizumab biosimilar is given using the same dosing and administration schedule. Like the reference product, it is indicated for adults with moderately to severely active Crohn’s disease unresponsive to other medications.
The approval of the natalizumab biosimilar is based on results of the phase 3 Antelope trial, which showed no clinically meaningful differences between it and the reference product.
The trial included 264 adults (mean age, 36 years; 61% women) with relapsing remitting MS from 48 centers in seven Eastern European countries.
All were randomly assigned to receive intravenous infusions every 4 weeks of 300 mg of the natalizumab biosimilar or the reference product for a total of 12 infusions.
At 24 and 48 weeks, there were no between-group differences in annualized relapse rates or Expanded Disability Status Scale scores, which were similar between treatment groups at baseline. There were also no significant differences in safety, tolerability, or immunogenicity.
The prescribing information for both natalizumab products includes a boxed warning about the increased risk of progressive multifocal leukoencephalopathy (PML), a viral infection of the brain that usually leads to death or severe disability.
Risk factors for the development of PML include the presence of antibodies to the JC virus, longer duration of therapy, and prior use of immunosuppressants.
“These factors should be considered in the context of expected benefit when initiating and continuing treatment with natalizumab products, and health care providers should monitor patients and withhold treatment immediately at the first sign or symptom suggestive of PML,” the FDA advises.
Because of the risks of PML, natalizumab products are available only through a restricted drug distribution program under a risk evaluation and mitigation strategy.
In a statement, Sandoz said it’s committed to having the product available in the United States “as soon as possible.”
A version of this article appeared on Medscape.com.
including clinically isolated syndrome, relapsing remitting MS, and active secondary progressive disease.
“Biosimilar medications offer additional effective treatment options that have the potential to increase access for people living with relapsing forms of multiple sclerosis. [This] approval could have a meaningful impact for patients managing their disease,” Paul R. Lee, MD, PhD, director of the division of neurology II, FDA Center for Drug Evaluation and Research, said in a statement.
The natalizumab biosimilar is given using the same dosing and administration schedule. Like the reference product, it is indicated for adults with moderately to severely active Crohn’s disease unresponsive to other medications.
The approval of the natalizumab biosimilar is based on results of the phase 3 Antelope trial, which showed no clinically meaningful differences between it and the reference product.
The trial included 264 adults (mean age, 36 years; 61% women) with relapsing remitting MS from 48 centers in seven Eastern European countries.
All were randomly assigned to receive intravenous infusions every 4 weeks of 300 mg of the natalizumab biosimilar or the reference product for a total of 12 infusions.
At 24 and 48 weeks, there were no between-group differences in annualized relapse rates or Expanded Disability Status Scale scores, which were similar between treatment groups at baseline. There were also no significant differences in safety, tolerability, or immunogenicity.
The prescribing information for both natalizumab products includes a boxed warning about the increased risk of progressive multifocal leukoencephalopathy (PML), a viral infection of the brain that usually leads to death or severe disability.
Risk factors for the development of PML include the presence of antibodies to the JC virus, longer duration of therapy, and prior use of immunosuppressants.
“These factors should be considered in the context of expected benefit when initiating and continuing treatment with natalizumab products, and health care providers should monitor patients and withhold treatment immediately at the first sign or symptom suggestive of PML,” the FDA advises.
Because of the risks of PML, natalizumab products are available only through a restricted drug distribution program under a risk evaluation and mitigation strategy.
In a statement, Sandoz said it’s committed to having the product available in the United States “as soon as possible.”
A version of this article appeared on Medscape.com.
including clinically isolated syndrome, relapsing remitting MS, and active secondary progressive disease.
“Biosimilar medications offer additional effective treatment options that have the potential to increase access for people living with relapsing forms of multiple sclerosis. [This] approval could have a meaningful impact for patients managing their disease,” Paul R. Lee, MD, PhD, director of the division of neurology II, FDA Center for Drug Evaluation and Research, said in a statement.
The natalizumab biosimilar is given using the same dosing and administration schedule. Like the reference product, it is indicated for adults with moderately to severely active Crohn’s disease unresponsive to other medications.
The approval of the natalizumab biosimilar is based on results of the phase 3 Antelope trial, which showed no clinically meaningful differences between it and the reference product.
The trial included 264 adults (mean age, 36 years; 61% women) with relapsing remitting MS from 48 centers in seven Eastern European countries.
All were randomly assigned to receive intravenous infusions every 4 weeks of 300 mg of the natalizumab biosimilar or the reference product for a total of 12 infusions.
At 24 and 48 weeks, there were no between-group differences in annualized relapse rates or Expanded Disability Status Scale scores, which were similar between treatment groups at baseline. There were also no significant differences in safety, tolerability, or immunogenicity.
The prescribing information for both natalizumab products includes a boxed warning about the increased risk of progressive multifocal leukoencephalopathy (PML), a viral infection of the brain that usually leads to death or severe disability.
Risk factors for the development of PML include the presence of antibodies to the JC virus, longer duration of therapy, and prior use of immunosuppressants.
“These factors should be considered in the context of expected benefit when initiating and continuing treatment with natalizumab products, and health care providers should monitor patients and withhold treatment immediately at the first sign or symptom suggestive of PML,” the FDA advises.
Because of the risks of PML, natalizumab products are available only through a restricted drug distribution program under a risk evaluation and mitigation strategy.
In a statement, Sandoz said it’s committed to having the product available in the United States “as soon as possible.”
A version of this article appeared on Medscape.com.
Breast cancer: Hope in sight for improved tamoxifen therapy?
A team at Lyon’s Cancer Research Center (CRCL) has revealed the role of an enzyme, PRMT5, in the response to tamoxifen, a drug used to prevent relapse in premenopausal women with breast cancer.
Muriel Le Romancer, MD, director of research at France’s Institute of Health and Medical Research, explained the issues involved in this discovery in an interview. She jointly led this research along with Olivier Trédan, MD, PhD, oncologist at Lyon’s Léon Bérard Clinic. The research concluded with the publication of a study in EMBO Molecular Medicine. The researchers both head up the CRCL’s hormone resistance, methylation, and breast cancer team.
Although the enzyme’s involvement in the mode of action of tamoxifen has been observed in close to 900 patients with breast cancer, these results need to be validated in other at-risk patient cohorts before the biomarker can be considered for routine use, said Dr. Le Romancer. She estimated that 2 more years of research are needed.
Can you tell us which cases involve the use of tamoxifen and what its mode of action is?
Dr. Le Romancer: Tamoxifen is a hormone therapy used to reduce the risk of breast cancer relapse. It is prescribed to premenopausal women with hormone-sensitive cancer, which equates to roughly 25% of women with breast cancer: 15,000 women each year. The drug, which is taken every day via oral administration, is an estrogen antagonist. By binding to these receptors, it blocks estrogen from mediating its biological effect in the breasts. Aromatase inhibitors are the preferred choice in postmenopausal women, as they have been shown to be more effective. These also have an antiestrogenic effect, but by inhibiting estrogen production.
Tamoxifen therapy is prescribed for a minimum period of 5 years. Despite this, 25% of women treated with tamoxifen relapse. Tamoxifen resistance is unique in that it occurs very late on, generally 10-15 years after starting treatment. This means that it’s really important for us to identify predictive markers of the response to hormone therapy to adapt treatment as best we can. For the moment, the only criteria used to prescribe tamoxifen are patient age and the presence of estrogen receptors within the tumor.
Exactly how would treatment be improved if a decisive predictive marker of response to tamoxifen could be identified?
Dr. Le Romancer: Currently, when a patient’s breast cancer relapses after several years of treatment with tamoxifen, we don’t know if the relapse is linked to tamoxifen resistance or not. This makes it difficult to choose the right treatment to manage such relapses, which remain complicated to treat. Lots of patients die because of metastases.
By predicting the response to tamoxifen using a marker, we will be able to either use another hormone therapy to prevent the relapse or prescribe tamoxifen alongside a molecule that stops resistance from developing. We hope that this will significantly reduce the rate of relapse.
You put forward PRMT5 as a potential predictive marker of response to tamoxifen. What makes you think it could be used in this way?
Dr. Le Romancer: Our research has allowed us to demonstrate that PRMT5, when present in the nuclei of tumor cells, is involved in the mechanisms of action of tamoxifen. Remember that estrogen receptors are located in cell nuclei. For tamoxifen to exert its antitumoral action, PRMT5, an enzyme, needs to enter the nucleus to modify the estrogen receptor. It’s this modification that allows tamoxifen to inhibit tumor growth. The proliferative effect induced by the estrogens is also blocked.
The results of our study showed that high nuclear expression of PRMT5, specifically in the nuclei of breast cancer cells, is associated with a prolonged survival of tamoxifen-treated patients. Until now, we thought this enzyme had an oncogenic role when present in the cytoplasm. It turns out that it also has the opposite effect when acting within the nucleus, at least in this patient cohort: women with hormone-sensitive breast cancer treated with tamoxifen.
What are the next steps in your research before we can begin to think about its use in clinical practice?
Dr. Le Romancer: Our next research will focus on understanding the circumstances surrounding PRMT5 entering and leaving the nucleus. We have also shown that in some patients, tamoxifen causes PRMT5 to enter the cell nucleus. This translocation is only seen in women who respond to tamoxifen, not in those who are resistant to treatment with the drug. All that remains is for us to work out how tamoxifen facilitates this translocation.
Once the elements promoting this translocation have been identified, we will be able to propose a treatment aimed at forcing the enzyme to enter the nucleus and stay there. Eventually, the idea is to combine treatment with antiestrogens with a medicinal product that promotes localization of PRMT5 in the nucleus to guarantee response to tamoxifen. It will be a few years of research before we can apply our findings to clinical practice.
Could we use this biomarker as is just to identify tamoxifen resistance?
Dr. Le Romancer: In the short term, yes, we could use this biomarker to better guide treatment choices at time of diagnosis. We have demonstrated the role of PRMT5 in response to tamoxifen by studying two cohorts of 900 patients with breast cancer receiving treatment at the Léon Bérard Center, Lyon. Before moving on to routine testing, we need to replicate these results in other cohorts, especially in high-risk patients with, for example, greater cell proliferation or those who experience relapse.
The use of this biomarker is based on histological examination of cancer tissue. Single antibody tissue staining targeting PRMT5 reveals the localization of the enzyme in the cells and provides a score evaluating its presence in the nucleus. Using this score, it would be possible to determine the level of response to tamoxifen and decide whether the treatment should be used. This biomarker is the first of its kind undergoing validation as part of the examination of resistance to hormone therapy. We should be able to confirm the findings within the next 2 years.
If clinical tests using this biomarker predict tamoxifen resistance, what alternative treatments are available to these patients?
Dr. Le Romancer: We could give them an aromatase inhibitor or one of the new estrogen antagonists that are currently in development. In a phase 3 study, fulvestrant (Faslodex), for example, demonstrated a significant benefit in treating women with hormone-sensitive advanced breast cancer when administered via injection. The same goes for oral treatment, elacestrant (Orserdu), which has recently been approved by the Food and Drug Administration. These treatments are usually deemed second line after tamoxifen, but they could certainly be used as first-line therapy in resistant patients.
The results obtained from research into novel estrogen antagonists are certainly encouraging. Can tamoxifen retain its prominent position while still ensuring its efficacy?
Dr. Le Romancer: Keeping in mind the current trend for personalized medicine, we should keep as many treatment options open as possible. When a patient relapses, there need to be other treatments available to them. Tamoxifen has been ousted in favor of aromatase inhibitors for postmenopausal women, but it’s still the gold standard for premenopausal women and has been for over 20 years. Despite having been replaced by a novel estrogen antagonist, it will still have a prominent place in the therapeutic arsenal of premenopausal women with breast cancer.
With the development of PRMT5 as a predictive biomarker, we could even see tamoxifen being proposed as first-line therapy for postmenopausal women in whom high levels of PRMT5 are found in the nuclei of their cancer cells. By predicting their response, we could achieve greater efficacy of tamoxifen, compared with aromatase inhibitors. For now, this remains a hypothesis and must be verified in further clinical studies.
This article was translated from the Medscape French Edition. A version appeared on Medscape.com.
A team at Lyon’s Cancer Research Center (CRCL) has revealed the role of an enzyme, PRMT5, in the response to tamoxifen, a drug used to prevent relapse in premenopausal women with breast cancer.
Muriel Le Romancer, MD, director of research at France’s Institute of Health and Medical Research, explained the issues involved in this discovery in an interview. She jointly led this research along with Olivier Trédan, MD, PhD, oncologist at Lyon’s Léon Bérard Clinic. The research concluded with the publication of a study in EMBO Molecular Medicine. The researchers both head up the CRCL’s hormone resistance, methylation, and breast cancer team.
Although the enzyme’s involvement in the mode of action of tamoxifen has been observed in close to 900 patients with breast cancer, these results need to be validated in other at-risk patient cohorts before the biomarker can be considered for routine use, said Dr. Le Romancer. She estimated that 2 more years of research are needed.
Can you tell us which cases involve the use of tamoxifen and what its mode of action is?
Dr. Le Romancer: Tamoxifen is a hormone therapy used to reduce the risk of breast cancer relapse. It is prescribed to premenopausal women with hormone-sensitive cancer, which equates to roughly 25% of women with breast cancer: 15,000 women each year. The drug, which is taken every day via oral administration, is an estrogen antagonist. By binding to these receptors, it blocks estrogen from mediating its biological effect in the breasts. Aromatase inhibitors are the preferred choice in postmenopausal women, as they have been shown to be more effective. These also have an antiestrogenic effect, but by inhibiting estrogen production.
Tamoxifen therapy is prescribed for a minimum period of 5 years. Despite this, 25% of women treated with tamoxifen relapse. Tamoxifen resistance is unique in that it occurs very late on, generally 10-15 years after starting treatment. This means that it’s really important for us to identify predictive markers of the response to hormone therapy to adapt treatment as best we can. For the moment, the only criteria used to prescribe tamoxifen are patient age and the presence of estrogen receptors within the tumor.
Exactly how would treatment be improved if a decisive predictive marker of response to tamoxifen could be identified?
Dr. Le Romancer: Currently, when a patient’s breast cancer relapses after several years of treatment with tamoxifen, we don’t know if the relapse is linked to tamoxifen resistance or not. This makes it difficult to choose the right treatment to manage such relapses, which remain complicated to treat. Lots of patients die because of metastases.
By predicting the response to tamoxifen using a marker, we will be able to either use another hormone therapy to prevent the relapse or prescribe tamoxifen alongside a molecule that stops resistance from developing. We hope that this will significantly reduce the rate of relapse.
You put forward PRMT5 as a potential predictive marker of response to tamoxifen. What makes you think it could be used in this way?
Dr. Le Romancer: Our research has allowed us to demonstrate that PRMT5, when present in the nuclei of tumor cells, is involved in the mechanisms of action of tamoxifen. Remember that estrogen receptors are located in cell nuclei. For tamoxifen to exert its antitumoral action, PRMT5, an enzyme, needs to enter the nucleus to modify the estrogen receptor. It’s this modification that allows tamoxifen to inhibit tumor growth. The proliferative effect induced by the estrogens is also blocked.
The results of our study showed that high nuclear expression of PRMT5, specifically in the nuclei of breast cancer cells, is associated with a prolonged survival of tamoxifen-treated patients. Until now, we thought this enzyme had an oncogenic role when present in the cytoplasm. It turns out that it also has the opposite effect when acting within the nucleus, at least in this patient cohort: women with hormone-sensitive breast cancer treated with tamoxifen.
What are the next steps in your research before we can begin to think about its use in clinical practice?
Dr. Le Romancer: Our next research will focus on understanding the circumstances surrounding PRMT5 entering and leaving the nucleus. We have also shown that in some patients, tamoxifen causes PRMT5 to enter the cell nucleus. This translocation is only seen in women who respond to tamoxifen, not in those who are resistant to treatment with the drug. All that remains is for us to work out how tamoxifen facilitates this translocation.
Once the elements promoting this translocation have been identified, we will be able to propose a treatment aimed at forcing the enzyme to enter the nucleus and stay there. Eventually, the idea is to combine treatment with antiestrogens with a medicinal product that promotes localization of PRMT5 in the nucleus to guarantee response to tamoxifen. It will be a few years of research before we can apply our findings to clinical practice.
Could we use this biomarker as is just to identify tamoxifen resistance?
Dr. Le Romancer: In the short term, yes, we could use this biomarker to better guide treatment choices at time of diagnosis. We have demonstrated the role of PRMT5 in response to tamoxifen by studying two cohorts of 900 patients with breast cancer receiving treatment at the Léon Bérard Center, Lyon. Before moving on to routine testing, we need to replicate these results in other cohorts, especially in high-risk patients with, for example, greater cell proliferation or those who experience relapse.
The use of this biomarker is based on histological examination of cancer tissue. Single antibody tissue staining targeting PRMT5 reveals the localization of the enzyme in the cells and provides a score evaluating its presence in the nucleus. Using this score, it would be possible to determine the level of response to tamoxifen and decide whether the treatment should be used. This biomarker is the first of its kind undergoing validation as part of the examination of resistance to hormone therapy. We should be able to confirm the findings within the next 2 years.
If clinical tests using this biomarker predict tamoxifen resistance, what alternative treatments are available to these patients?
Dr. Le Romancer: We could give them an aromatase inhibitor or one of the new estrogen antagonists that are currently in development. In a phase 3 study, fulvestrant (Faslodex), for example, demonstrated a significant benefit in treating women with hormone-sensitive advanced breast cancer when administered via injection. The same goes for oral treatment, elacestrant (Orserdu), which has recently been approved by the Food and Drug Administration. These treatments are usually deemed second line after tamoxifen, but they could certainly be used as first-line therapy in resistant patients.
The results obtained from research into novel estrogen antagonists are certainly encouraging. Can tamoxifen retain its prominent position while still ensuring its efficacy?
Dr. Le Romancer: Keeping in mind the current trend for personalized medicine, we should keep as many treatment options open as possible. When a patient relapses, there need to be other treatments available to them. Tamoxifen has been ousted in favor of aromatase inhibitors for postmenopausal women, but it’s still the gold standard for premenopausal women and has been for over 20 years. Despite having been replaced by a novel estrogen antagonist, it will still have a prominent place in the therapeutic arsenal of premenopausal women with breast cancer.
With the development of PRMT5 as a predictive biomarker, we could even see tamoxifen being proposed as first-line therapy for postmenopausal women in whom high levels of PRMT5 are found in the nuclei of their cancer cells. By predicting their response, we could achieve greater efficacy of tamoxifen, compared with aromatase inhibitors. For now, this remains a hypothesis and must be verified in further clinical studies.
This article was translated from the Medscape French Edition. A version appeared on Medscape.com.
A team at Lyon’s Cancer Research Center (CRCL) has revealed the role of an enzyme, PRMT5, in the response to tamoxifen, a drug used to prevent relapse in premenopausal women with breast cancer.
Muriel Le Romancer, MD, director of research at France’s Institute of Health and Medical Research, explained the issues involved in this discovery in an interview. She jointly led this research along with Olivier Trédan, MD, PhD, oncologist at Lyon’s Léon Bérard Clinic. The research concluded with the publication of a study in EMBO Molecular Medicine. The researchers both head up the CRCL’s hormone resistance, methylation, and breast cancer team.
Although the enzyme’s involvement in the mode of action of tamoxifen has been observed in close to 900 patients with breast cancer, these results need to be validated in other at-risk patient cohorts before the biomarker can be considered for routine use, said Dr. Le Romancer. She estimated that 2 more years of research are needed.
Can you tell us which cases involve the use of tamoxifen and what its mode of action is?
Dr. Le Romancer: Tamoxifen is a hormone therapy used to reduce the risk of breast cancer relapse. It is prescribed to premenopausal women with hormone-sensitive cancer, which equates to roughly 25% of women with breast cancer: 15,000 women each year. The drug, which is taken every day via oral administration, is an estrogen antagonist. By binding to these receptors, it blocks estrogen from mediating its biological effect in the breasts. Aromatase inhibitors are the preferred choice in postmenopausal women, as they have been shown to be more effective. These also have an antiestrogenic effect, but by inhibiting estrogen production.
Tamoxifen therapy is prescribed for a minimum period of 5 years. Despite this, 25% of women treated with tamoxifen relapse. Tamoxifen resistance is unique in that it occurs very late on, generally 10-15 years after starting treatment. This means that it’s really important for us to identify predictive markers of the response to hormone therapy to adapt treatment as best we can. For the moment, the only criteria used to prescribe tamoxifen are patient age and the presence of estrogen receptors within the tumor.
Exactly how would treatment be improved if a decisive predictive marker of response to tamoxifen could be identified?
Dr. Le Romancer: Currently, when a patient’s breast cancer relapses after several years of treatment with tamoxifen, we don’t know if the relapse is linked to tamoxifen resistance or not. This makes it difficult to choose the right treatment to manage such relapses, which remain complicated to treat. Lots of patients die because of metastases.
By predicting the response to tamoxifen using a marker, we will be able to either use another hormone therapy to prevent the relapse or prescribe tamoxifen alongside a molecule that stops resistance from developing. We hope that this will significantly reduce the rate of relapse.
You put forward PRMT5 as a potential predictive marker of response to tamoxifen. What makes you think it could be used in this way?
Dr. Le Romancer: Our research has allowed us to demonstrate that PRMT5, when present in the nuclei of tumor cells, is involved in the mechanisms of action of tamoxifen. Remember that estrogen receptors are located in cell nuclei. For tamoxifen to exert its antitumoral action, PRMT5, an enzyme, needs to enter the nucleus to modify the estrogen receptor. It’s this modification that allows tamoxifen to inhibit tumor growth. The proliferative effect induced by the estrogens is also blocked.
The results of our study showed that high nuclear expression of PRMT5, specifically in the nuclei of breast cancer cells, is associated with a prolonged survival of tamoxifen-treated patients. Until now, we thought this enzyme had an oncogenic role when present in the cytoplasm. It turns out that it also has the opposite effect when acting within the nucleus, at least in this patient cohort: women with hormone-sensitive breast cancer treated with tamoxifen.
What are the next steps in your research before we can begin to think about its use in clinical practice?
Dr. Le Romancer: Our next research will focus on understanding the circumstances surrounding PRMT5 entering and leaving the nucleus. We have also shown that in some patients, tamoxifen causes PRMT5 to enter the cell nucleus. This translocation is only seen in women who respond to tamoxifen, not in those who are resistant to treatment with the drug. All that remains is for us to work out how tamoxifen facilitates this translocation.
Once the elements promoting this translocation have been identified, we will be able to propose a treatment aimed at forcing the enzyme to enter the nucleus and stay there. Eventually, the idea is to combine treatment with antiestrogens with a medicinal product that promotes localization of PRMT5 in the nucleus to guarantee response to tamoxifen. It will be a few years of research before we can apply our findings to clinical practice.
Could we use this biomarker as is just to identify tamoxifen resistance?
Dr. Le Romancer: In the short term, yes, we could use this biomarker to better guide treatment choices at time of diagnosis. We have demonstrated the role of PRMT5 in response to tamoxifen by studying two cohorts of 900 patients with breast cancer receiving treatment at the Léon Bérard Center, Lyon. Before moving on to routine testing, we need to replicate these results in other cohorts, especially in high-risk patients with, for example, greater cell proliferation or those who experience relapse.
The use of this biomarker is based on histological examination of cancer tissue. Single antibody tissue staining targeting PRMT5 reveals the localization of the enzyme in the cells and provides a score evaluating its presence in the nucleus. Using this score, it would be possible to determine the level of response to tamoxifen and decide whether the treatment should be used. This biomarker is the first of its kind undergoing validation as part of the examination of resistance to hormone therapy. We should be able to confirm the findings within the next 2 years.
If clinical tests using this biomarker predict tamoxifen resistance, what alternative treatments are available to these patients?
Dr. Le Romancer: We could give them an aromatase inhibitor or one of the new estrogen antagonists that are currently in development. In a phase 3 study, fulvestrant (Faslodex), for example, demonstrated a significant benefit in treating women with hormone-sensitive advanced breast cancer when administered via injection. The same goes for oral treatment, elacestrant (Orserdu), which has recently been approved by the Food and Drug Administration. These treatments are usually deemed second line after tamoxifen, but they could certainly be used as first-line therapy in resistant patients.
The results obtained from research into novel estrogen antagonists are certainly encouraging. Can tamoxifen retain its prominent position while still ensuring its efficacy?
Dr. Le Romancer: Keeping in mind the current trend for personalized medicine, we should keep as many treatment options open as possible. When a patient relapses, there need to be other treatments available to them. Tamoxifen has been ousted in favor of aromatase inhibitors for postmenopausal women, but it’s still the gold standard for premenopausal women and has been for over 20 years. Despite having been replaced by a novel estrogen antagonist, it will still have a prominent place in the therapeutic arsenal of premenopausal women with breast cancer.
With the development of PRMT5 as a predictive biomarker, we could even see tamoxifen being proposed as first-line therapy for postmenopausal women in whom high levels of PRMT5 are found in the nuclei of their cancer cells. By predicting their response, we could achieve greater efficacy of tamoxifen, compared with aromatase inhibitors. For now, this remains a hypothesis and must be verified in further clinical studies.
This article was translated from the Medscape French Edition. A version appeared on Medscape.com.
New recommendation expands antiretroviral guidance for HIV
.
“With these new options we could potentially extend pre-exposure prophylaxis (PrEP) to a wider population,” says James Stevermer, MD, a member of the task force and a professor of family and community medicine at the University of Missouri–Columbia.
The guidance, published in JAMA, updates the group’s previous recommendation from 2019 to take into account the new options that have become available since the U.S. Food and Drug Administration approvals that included a long-acting injectable form.
In the original report, daily oral tenofovir disoproxil fumarate with emtricitabine was the only approved medication available and the task force recommended it. Since then, two new regimens have been approved: daily oral tenofovir alafenamide with emtricitabine and the long-acting injectable cabotegravir.
The task force is backing all three options and is recommending that clinicians use whichever formulation is most appropriate for their patients at risk for HIV infection.
Task force in primary and preventive care
The USPSTF is a volunteer group of experts in primary and preventive care who make recommendations on the best preventative interventions clinicians should take on everything from cancer screening, to preventive aspirin use, to behavioral counseling. The group is convened and supported by the Agency for Healthcare Research and Quality.
Recommendations from this group are particularly helpful for clinicians who may not see HIV as their area of expertise, says Carolyn Chu, MD, chief medical officer of the American Academy of HIV Medicine. “Hopefully, this will catch the eye of people who are not tracking all of the HIV updates,” she says.
A person’s risk for infection is mostly based on their behavior, Dr. Stevermer says. Those who use injectable drugs, particularly if they share needles, those who use condoms inconsistently and do not know their partner’s HIV status, and those who have recently had bacterial sexually transmitted infections like gonorrhea and syphilis are all at higher risk.
The efficacy of each of the three options is close enough to equal that it doesn’t usually matter which is prescribed, according to the task force. However, daily oral tenofovir alafenamide with emtricitabine is not approved for use by people engaging in receptive vaginal sex. For most people, the best medication option is the one they are most likely able to integrate into their routine. Cabotegravir, for example, which requires injections every 2 months, is an easier method for some people, particularly those who don’t think they could successfully take a daily pill.
Reducing risk
“The evidence is very clear that being able to adhere to taking the medication daily was very closely associated with the effectiveness of PrEP,” Dr. Stevermer says. “So, everything that we can do to make sure that the person who wants to prevent HIV is getting their PrEP as it is supposed to be taken makes it that much more effective.”
Expanding access to antiretrovirals among at-risk groups is an important part of the Ending the HIV Epidemic in the United States initiative that aims to reduce new HIV cases by 90% by 2030.
But an editorial published alongside the recommendation in JAMA notes that uptake of PrEP has been disproportionately low among populations most heavily affected by HIV.
In 2021, 78% of White people expected to benefit from PrEP received it, compared with just 11% of Black people and 21% of Hispanic people, despite both of those populations having a higher incidence of HIV than Whites. PrEP use is also substantially lower among cisgender and transgender women, youth, and people who inject drugs.
“We have an intervention that can markedly reduce people’s risk of getting HIV and so we want to make sure we get this out to all those populations at increased risk,” Dr. Stevermer says.
Having multiple options when it comes to PrEP is a big part of expanding access to the treatment for underserved groups, Dr. Chu says. “Even though oral tenofovir disoproxil fumarate with emtricitabine has been out for a while, we know it’s not getting to everyone, and there may be clinical circumstances that means it’s not the right option,” she says. “Making sure we are supporting choices so people can make the decision for themselves is important.”
But doctors also need to be willing to have an open conversation with their patients and bring up the topic of PrEP in a way that doesn’t feel judgmental or stigmatizing, Dr. Chu says.
It is also important not to make assumptions about who would want to talk about medication, she adds. “How can we change the narrative around PrEP?” she asks. “The evidence is there, these medications are effective and safe; weave PrEP into your preventive care portfolio to at least start the conversation.”
A version of this article appeared on Medscape.com.
.
“With these new options we could potentially extend pre-exposure prophylaxis (PrEP) to a wider population,” says James Stevermer, MD, a member of the task force and a professor of family and community medicine at the University of Missouri–Columbia.
The guidance, published in JAMA, updates the group’s previous recommendation from 2019 to take into account the new options that have become available since the U.S. Food and Drug Administration approvals that included a long-acting injectable form.
In the original report, daily oral tenofovir disoproxil fumarate with emtricitabine was the only approved medication available and the task force recommended it. Since then, two new regimens have been approved: daily oral tenofovir alafenamide with emtricitabine and the long-acting injectable cabotegravir.
The task force is backing all three options and is recommending that clinicians use whichever formulation is most appropriate for their patients at risk for HIV infection.
Task force in primary and preventive care
The USPSTF is a volunteer group of experts in primary and preventive care who make recommendations on the best preventative interventions clinicians should take on everything from cancer screening, to preventive aspirin use, to behavioral counseling. The group is convened and supported by the Agency for Healthcare Research and Quality.
Recommendations from this group are particularly helpful for clinicians who may not see HIV as their area of expertise, says Carolyn Chu, MD, chief medical officer of the American Academy of HIV Medicine. “Hopefully, this will catch the eye of people who are not tracking all of the HIV updates,” she says.
A person’s risk for infection is mostly based on their behavior, Dr. Stevermer says. Those who use injectable drugs, particularly if they share needles, those who use condoms inconsistently and do not know their partner’s HIV status, and those who have recently had bacterial sexually transmitted infections like gonorrhea and syphilis are all at higher risk.
The efficacy of each of the three options is close enough to equal that it doesn’t usually matter which is prescribed, according to the task force. However, daily oral tenofovir alafenamide with emtricitabine is not approved for use by people engaging in receptive vaginal sex. For most people, the best medication option is the one they are most likely able to integrate into their routine. Cabotegravir, for example, which requires injections every 2 months, is an easier method for some people, particularly those who don’t think they could successfully take a daily pill.
Reducing risk
“The evidence is very clear that being able to adhere to taking the medication daily was very closely associated with the effectiveness of PrEP,” Dr. Stevermer says. “So, everything that we can do to make sure that the person who wants to prevent HIV is getting their PrEP as it is supposed to be taken makes it that much more effective.”
Expanding access to antiretrovirals among at-risk groups is an important part of the Ending the HIV Epidemic in the United States initiative that aims to reduce new HIV cases by 90% by 2030.
But an editorial published alongside the recommendation in JAMA notes that uptake of PrEP has been disproportionately low among populations most heavily affected by HIV.
In 2021, 78% of White people expected to benefit from PrEP received it, compared with just 11% of Black people and 21% of Hispanic people, despite both of those populations having a higher incidence of HIV than Whites. PrEP use is also substantially lower among cisgender and transgender women, youth, and people who inject drugs.
“We have an intervention that can markedly reduce people’s risk of getting HIV and so we want to make sure we get this out to all those populations at increased risk,” Dr. Stevermer says.
Having multiple options when it comes to PrEP is a big part of expanding access to the treatment for underserved groups, Dr. Chu says. “Even though oral tenofovir disoproxil fumarate with emtricitabine has been out for a while, we know it’s not getting to everyone, and there may be clinical circumstances that means it’s not the right option,” she says. “Making sure we are supporting choices so people can make the decision for themselves is important.”
But doctors also need to be willing to have an open conversation with their patients and bring up the topic of PrEP in a way that doesn’t feel judgmental or stigmatizing, Dr. Chu says.
It is also important not to make assumptions about who would want to talk about medication, she adds. “How can we change the narrative around PrEP?” she asks. “The evidence is there, these medications are effective and safe; weave PrEP into your preventive care portfolio to at least start the conversation.”
A version of this article appeared on Medscape.com.
.
“With these new options we could potentially extend pre-exposure prophylaxis (PrEP) to a wider population,” says James Stevermer, MD, a member of the task force and a professor of family and community medicine at the University of Missouri–Columbia.
The guidance, published in JAMA, updates the group’s previous recommendation from 2019 to take into account the new options that have become available since the U.S. Food and Drug Administration approvals that included a long-acting injectable form.
In the original report, daily oral tenofovir disoproxil fumarate with emtricitabine was the only approved medication available and the task force recommended it. Since then, two new regimens have been approved: daily oral tenofovir alafenamide with emtricitabine and the long-acting injectable cabotegravir.
The task force is backing all three options and is recommending that clinicians use whichever formulation is most appropriate for their patients at risk for HIV infection.
Task force in primary and preventive care
The USPSTF is a volunteer group of experts in primary and preventive care who make recommendations on the best preventative interventions clinicians should take on everything from cancer screening, to preventive aspirin use, to behavioral counseling. The group is convened and supported by the Agency for Healthcare Research and Quality.
Recommendations from this group are particularly helpful for clinicians who may not see HIV as their area of expertise, says Carolyn Chu, MD, chief medical officer of the American Academy of HIV Medicine. “Hopefully, this will catch the eye of people who are not tracking all of the HIV updates,” she says.
A person’s risk for infection is mostly based on their behavior, Dr. Stevermer says. Those who use injectable drugs, particularly if they share needles, those who use condoms inconsistently and do not know their partner’s HIV status, and those who have recently had bacterial sexually transmitted infections like gonorrhea and syphilis are all at higher risk.
The efficacy of each of the three options is close enough to equal that it doesn’t usually matter which is prescribed, according to the task force. However, daily oral tenofovir alafenamide with emtricitabine is not approved for use by people engaging in receptive vaginal sex. For most people, the best medication option is the one they are most likely able to integrate into their routine. Cabotegravir, for example, which requires injections every 2 months, is an easier method for some people, particularly those who don’t think they could successfully take a daily pill.
Reducing risk
“The evidence is very clear that being able to adhere to taking the medication daily was very closely associated with the effectiveness of PrEP,” Dr. Stevermer says. “So, everything that we can do to make sure that the person who wants to prevent HIV is getting their PrEP as it is supposed to be taken makes it that much more effective.”
Expanding access to antiretrovirals among at-risk groups is an important part of the Ending the HIV Epidemic in the United States initiative that aims to reduce new HIV cases by 90% by 2030.
But an editorial published alongside the recommendation in JAMA notes that uptake of PrEP has been disproportionately low among populations most heavily affected by HIV.
In 2021, 78% of White people expected to benefit from PrEP received it, compared with just 11% of Black people and 21% of Hispanic people, despite both of those populations having a higher incidence of HIV than Whites. PrEP use is also substantially lower among cisgender and transgender women, youth, and people who inject drugs.
“We have an intervention that can markedly reduce people’s risk of getting HIV and so we want to make sure we get this out to all those populations at increased risk,” Dr. Stevermer says.
Having multiple options when it comes to PrEP is a big part of expanding access to the treatment for underserved groups, Dr. Chu says. “Even though oral tenofovir disoproxil fumarate with emtricitabine has been out for a while, we know it’s not getting to everyone, and there may be clinical circumstances that means it’s not the right option,” she says. “Making sure we are supporting choices so people can make the decision for themselves is important.”
But doctors also need to be willing to have an open conversation with their patients and bring up the topic of PrEP in a way that doesn’t feel judgmental or stigmatizing, Dr. Chu says.
It is also important not to make assumptions about who would want to talk about medication, she adds. “How can we change the narrative around PrEP?” she asks. “The evidence is there, these medications are effective and safe; weave PrEP into your preventive care portfolio to at least start the conversation.”
A version of this article appeared on Medscape.com.
New RSV shot is a monoclonal antibody, not a vaccine
For the first time in the fall of 2023, families will be offered season-long protection for infants and some children against respiratory syncytial virus (RSV).
The Food and Drug Administration in July approved a prevention called nirsevimab (Beyfortus, AstraZeneca/Sanofi) and it is expected to be widely rolled out in the coming weeks as the RSV season begins.
And monoclonal antibodies are often used for treatment rather than prevention.
Adding to potential confusion is the fact the Centers for Disease Control and Prevention has included nirsevimab in the Vaccines for Children program, which covers the costs for uninsured kids and makes it more accessible.
Nirsevimab is approved for infants (up to 8 months old) born during or entering their first RSV season, and in children up to 2 years of age who are still vulnerable to severe RSV through their second season.
It’s recommended that all infants get one injection in their first 8 months for prevention instead of the previous monthly shots used to help prevent kids at high risk from getting severe RSV.
If monoclonal antibodies can be used for preventing disease in infants, could they become a viable vaccine alternative for adults?
Specialists say no.
That’s partly because of the difference in body size. Although an injection is an option for a newborn, pediatricians suggest, it would take far too much of the treatment to work as a shot for adults.
Ruth Karron, MD, an expert in pediatric infectious diseases at Johns Hopkins Medicine, Baltimore, said that, while vaccines come in small amounts and activate immune cells, monoclonal antibodies are more like a drug, with the dose based on weight.
“You’d have to give it intravenously,” for larger doses, she explained, which has never been studied before and would also be very expensive. “It really couldn’t be an option for adults.”
What’s the difference between vaccines and antibodies?
Monoclonal antibodies are proteins made in a lab to mimic the immune system’s ability to fight pathogens such as viruses.
Dr. Karron explained that a wide variety of monoclonal antibodies have long been used to treat diseases such as cancers and autoimmune disease. In recent years, the antibodies have been used to treat COVID.
Monoclonal antibodies have also been used to treat RSV in children, but the effects don’t last long – they confer passive immunity and “when it’s gone, it’s gone,” Dr. Karron said.
That means kids at high risk for severe RSV have had to get monthly injections.
But with nirsevimab, the mutated antibodies stay in circulation longer so they can last 5 or 6 months, enough to cover the RSV season, Dr. Karron explained. “It’s highly, highly effective.”
Vaccines train the body
“The idea with vaccines is that you engage the individual’s immune system. You teach it to make antibodies,” Dr. Karron said. Conversely, “you give an antibody and it’s good for as long as the antibody lasts. It’s not teaching your body anything.”
Frank Esper, MD, a pediatric infectious disease specialist at Cleveland Clinic Children’s Hospital, said monoclonal antibody protection for RSV is particularly welcome. “We’ve been trying to make an RSV vaccine since the 1960s and have done nothing but fail miserably.”
“The best thing is always a vaccine,” Dr. Esper said, explaining that vaccines teach the body to make its own antibodies and confer long-term protection and are “probably more efficacious than anything that’s ever manmade.
“But since we’ve really not done very well for pediatric RSV vaccines, nirsevimab is certainly something I’m looking forward to,” he said.
Fast-acting monoclonal antibodies
An advantage for monoclonal antibodies is that they start working almost immediately.
Children can get sick with RSV in the first few months of life so the speed of the monoclonal antibodies to begin protection is important, Dr. Esper said, adding that RSV “is the worst during the first year of life.”
The peak age for babies getting infected enough to require hospitalization is about 2 months, he said.
By 14 months, he said, kids’ immune systems and airways have matured enough “that it’s not nearly as bad.”
To get protection from a vaccine, he added, “usually takes 2-4 weeks from the time you get your shot to the time you see some benefit. With an antibody, you’re bypassing the processing that the body has to do, and it goes straight to ‘protection’ mode,” Dr. Esper said. “You get protected pretty much as soon as you get the antibody.”
A version of this article first appeared on Medscape.com.
For the first time in the fall of 2023, families will be offered season-long protection for infants and some children against respiratory syncytial virus (RSV).
The Food and Drug Administration in July approved a prevention called nirsevimab (Beyfortus, AstraZeneca/Sanofi) and it is expected to be widely rolled out in the coming weeks as the RSV season begins.
And monoclonal antibodies are often used for treatment rather than prevention.
Adding to potential confusion is the fact the Centers for Disease Control and Prevention has included nirsevimab in the Vaccines for Children program, which covers the costs for uninsured kids and makes it more accessible.
Nirsevimab is approved for infants (up to 8 months old) born during or entering their first RSV season, and in children up to 2 years of age who are still vulnerable to severe RSV through their second season.
It’s recommended that all infants get one injection in their first 8 months for prevention instead of the previous monthly shots used to help prevent kids at high risk from getting severe RSV.
If monoclonal antibodies can be used for preventing disease in infants, could they become a viable vaccine alternative for adults?
Specialists say no.
That’s partly because of the difference in body size. Although an injection is an option for a newborn, pediatricians suggest, it would take far too much of the treatment to work as a shot for adults.
Ruth Karron, MD, an expert in pediatric infectious diseases at Johns Hopkins Medicine, Baltimore, said that, while vaccines come in small amounts and activate immune cells, monoclonal antibodies are more like a drug, with the dose based on weight.
“You’d have to give it intravenously,” for larger doses, she explained, which has never been studied before and would also be very expensive. “It really couldn’t be an option for adults.”
What’s the difference between vaccines and antibodies?
Monoclonal antibodies are proteins made in a lab to mimic the immune system’s ability to fight pathogens such as viruses.
Dr. Karron explained that a wide variety of monoclonal antibodies have long been used to treat diseases such as cancers and autoimmune disease. In recent years, the antibodies have been used to treat COVID.
Monoclonal antibodies have also been used to treat RSV in children, but the effects don’t last long – they confer passive immunity and “when it’s gone, it’s gone,” Dr. Karron said.
That means kids at high risk for severe RSV have had to get monthly injections.
But with nirsevimab, the mutated antibodies stay in circulation longer so they can last 5 or 6 months, enough to cover the RSV season, Dr. Karron explained. “It’s highly, highly effective.”
Vaccines train the body
“The idea with vaccines is that you engage the individual’s immune system. You teach it to make antibodies,” Dr. Karron said. Conversely, “you give an antibody and it’s good for as long as the antibody lasts. It’s not teaching your body anything.”
Frank Esper, MD, a pediatric infectious disease specialist at Cleveland Clinic Children’s Hospital, said monoclonal antibody protection for RSV is particularly welcome. “We’ve been trying to make an RSV vaccine since the 1960s and have done nothing but fail miserably.”
“The best thing is always a vaccine,” Dr. Esper said, explaining that vaccines teach the body to make its own antibodies and confer long-term protection and are “probably more efficacious than anything that’s ever manmade.
“But since we’ve really not done very well for pediatric RSV vaccines, nirsevimab is certainly something I’m looking forward to,” he said.
Fast-acting monoclonal antibodies
An advantage for monoclonal antibodies is that they start working almost immediately.
Children can get sick with RSV in the first few months of life so the speed of the monoclonal antibodies to begin protection is important, Dr. Esper said, adding that RSV “is the worst during the first year of life.”
The peak age for babies getting infected enough to require hospitalization is about 2 months, he said.
By 14 months, he said, kids’ immune systems and airways have matured enough “that it’s not nearly as bad.”
To get protection from a vaccine, he added, “usually takes 2-4 weeks from the time you get your shot to the time you see some benefit. With an antibody, you’re bypassing the processing that the body has to do, and it goes straight to ‘protection’ mode,” Dr. Esper said. “You get protected pretty much as soon as you get the antibody.”
A version of this article first appeared on Medscape.com.
For the first time in the fall of 2023, families will be offered season-long protection for infants and some children against respiratory syncytial virus (RSV).
The Food and Drug Administration in July approved a prevention called nirsevimab (Beyfortus, AstraZeneca/Sanofi) and it is expected to be widely rolled out in the coming weeks as the RSV season begins.
And monoclonal antibodies are often used for treatment rather than prevention.
Adding to potential confusion is the fact the Centers for Disease Control and Prevention has included nirsevimab in the Vaccines for Children program, which covers the costs for uninsured kids and makes it more accessible.
Nirsevimab is approved for infants (up to 8 months old) born during or entering their first RSV season, and in children up to 2 years of age who are still vulnerable to severe RSV through their second season.
It’s recommended that all infants get one injection in their first 8 months for prevention instead of the previous monthly shots used to help prevent kids at high risk from getting severe RSV.
If monoclonal antibodies can be used for preventing disease in infants, could they become a viable vaccine alternative for adults?
Specialists say no.
That’s partly because of the difference in body size. Although an injection is an option for a newborn, pediatricians suggest, it would take far too much of the treatment to work as a shot for adults.
Ruth Karron, MD, an expert in pediatric infectious diseases at Johns Hopkins Medicine, Baltimore, said that, while vaccines come in small amounts and activate immune cells, monoclonal antibodies are more like a drug, with the dose based on weight.
“You’d have to give it intravenously,” for larger doses, she explained, which has never been studied before and would also be very expensive. “It really couldn’t be an option for adults.”
What’s the difference between vaccines and antibodies?
Monoclonal antibodies are proteins made in a lab to mimic the immune system’s ability to fight pathogens such as viruses.
Dr. Karron explained that a wide variety of monoclonal antibodies have long been used to treat diseases such as cancers and autoimmune disease. In recent years, the antibodies have been used to treat COVID.
Monoclonal antibodies have also been used to treat RSV in children, but the effects don’t last long – they confer passive immunity and “when it’s gone, it’s gone,” Dr. Karron said.
That means kids at high risk for severe RSV have had to get monthly injections.
But with nirsevimab, the mutated antibodies stay in circulation longer so they can last 5 or 6 months, enough to cover the RSV season, Dr. Karron explained. “It’s highly, highly effective.”
Vaccines train the body
“The idea with vaccines is that you engage the individual’s immune system. You teach it to make antibodies,” Dr. Karron said. Conversely, “you give an antibody and it’s good for as long as the antibody lasts. It’s not teaching your body anything.”
Frank Esper, MD, a pediatric infectious disease specialist at Cleveland Clinic Children’s Hospital, said monoclonal antibody protection for RSV is particularly welcome. “We’ve been trying to make an RSV vaccine since the 1960s and have done nothing but fail miserably.”
“The best thing is always a vaccine,” Dr. Esper said, explaining that vaccines teach the body to make its own antibodies and confer long-term protection and are “probably more efficacious than anything that’s ever manmade.
“But since we’ve really not done very well for pediatric RSV vaccines, nirsevimab is certainly something I’m looking forward to,” he said.
Fast-acting monoclonal antibodies
An advantage for monoclonal antibodies is that they start working almost immediately.
Children can get sick with RSV in the first few months of life so the speed of the monoclonal antibodies to begin protection is important, Dr. Esper said, adding that RSV “is the worst during the first year of life.”
The peak age for babies getting infected enough to require hospitalization is about 2 months, he said.
By 14 months, he said, kids’ immune systems and airways have matured enough “that it’s not nearly as bad.”
To get protection from a vaccine, he added, “usually takes 2-4 weeks from the time you get your shot to the time you see some benefit. With an antibody, you’re bypassing the processing that the body has to do, and it goes straight to ‘protection’ mode,” Dr. Esper said. “You get protected pretty much as soon as you get the antibody.”
A version of this article first appeared on Medscape.com.
Expert calls for sparing oxygen use for dyspnea in the emergency department
PARIS – , as per the current guidelines. Florian Negrello, MD, an emergency medicine specialist at University Hospital of Martinique in Fort-de-France, reiterated this message at the 2023 conference held by France’s emergency medicine society (Urgences 2023). The recommendation is intended to prevent hyperoxia; increasing evidence indicates the harmful effects of such a state on the body.
“This is a real problem. Oxygen therapy is given all too readily despite studies now showing that excess oxygen is harmful, especially in patients with head trauma, ischemic stroke, or cardiac arrest,” stated the session’s moderator, Patrick Plaisance, MD, PhD, a doctor at Lariboisière Hospital in Paris.
No proven hypoxia
Described as difficulty breathing or shortness of breath, dyspnea is common in the emergency department, occurring in 5%-9% of patients. Close to 20% of intensive care unit admissions involve patients with dyspnea. “Since this is a very subjective symptom, it’s possible it’s being underdiagnosed,” said Dr. Negrello.
Lower respiratory tract infection, acute heart failure, chronic obstructive pulmonary disease, and exacerbation of asthma are the four main diagnoses linked to dyspnea, but this symptom is also seen in several medical conditions (gastrointestinal, metabolic, neurologic, etc.), he noted.
Often seen as a harmless treatment option, oxygen therapy is commonly administered to patients with breathing difficulties even when no hypoxemia is documented. This is particularly the case for patients brought into hospital via ambulance who are treated with oxygen without even having had their blood oxygen levels, SpO2, and partial pressure of oxygen checked.
In the United States, one of the few studies published on the topic showed that one-third of patients transported via ambulance are put on oxygen, with SpO2 being measured in just 5% of these cases. Finally, just 17% of patients receiving oxygen were experiencing hypoxia, defined as SpO2 < 94%.
Oxidative stress
Recently, several research studies have revealed the potential dangers of unjustified use of oxygen, which can lead to hyperoxia and increased mortality in hospitalized patients.
A meta-analysis reported a linear relationship between severe hyperoxia, in-hospital mortality, and length of stay in intensive care. Another study revealed a greater mortality rate in patients with acute respiratory distress syndrome (ARDS) experiencing an episode of hyperoxia, regardless of the severity of ARDS.
Oxygen toxicity in intensive care is said to be linked to oxidative stress caused by increased growth of reactive oxygen species but also to the systemic inflammation caused by hyperoxia, explained Dr. Negrello. Excess oxygen may also cause lung lesions with necrosis, the severity of which is proportional to the fraction of inspired oxygen and the length of exposure.
According to the most up-to-date international recommendations published in 2018 on the use of oxygen therapy in treating acute conditions, oxygen should not be used when SpO2 ≥ 93%. When treatment has been started, it must be stopped when SpO2 reaches 96%. SpO2 cannot be maintained above 96%, according to experts.
These threshold values can be found in the COVID-19 treatment guidelines produced by the French-Language Society of Respiratory Medicine, with oxygen therapy being recommended when SpO2 < 92%, added Dr. Negrello. The aim is to maintain normal oxygen levels, with SpO2 between 92% and 96%.
Use sparingly
For patients with COPD, the target levels are lower, due to the risk of hypercapnia (higher than normal carbon dioxide levels in the blood). Oxygen saturation levels should then be kept between 88% and 92%, “by using the minimum amount of oxygen necessary,” per the guidelines.
“Oxygen should be used sparingly,” concluded Dr. Negrello. “To treat our patients without harming them, we must be able to use it at the right time, meaning when a patient really has low blood oxygen, by focusing on normal saturation levels as the end goal.”
SpO2 measurement is the first step to be taken to determine oxygen requirements, followed by, if necessary, blood gas analysis once the patient has been admitted, he explained.
Questioned at the end of his session on how long oxygen therapy can be given for, Dr. Negrello reiterated that the risk for death is correlated with the length of time spent in a state of hyperoxia but that it is difficult to establish a maximum timeframe to be adhered to strictly.
Given that excess oxygen is harmful to patients in intensive care, “it would be better, when in doubt, to focus on physiological levels” and simply stop treatment when target saturation levels are reached.
This article was translated from the Medscape French Edition. A version appeared on Medscape.com.
PARIS – , as per the current guidelines. Florian Negrello, MD, an emergency medicine specialist at University Hospital of Martinique in Fort-de-France, reiterated this message at the 2023 conference held by France’s emergency medicine society (Urgences 2023). The recommendation is intended to prevent hyperoxia; increasing evidence indicates the harmful effects of such a state on the body.
“This is a real problem. Oxygen therapy is given all too readily despite studies now showing that excess oxygen is harmful, especially in patients with head trauma, ischemic stroke, or cardiac arrest,” stated the session’s moderator, Patrick Plaisance, MD, PhD, a doctor at Lariboisière Hospital in Paris.
No proven hypoxia
Described as difficulty breathing or shortness of breath, dyspnea is common in the emergency department, occurring in 5%-9% of patients. Close to 20% of intensive care unit admissions involve patients with dyspnea. “Since this is a very subjective symptom, it’s possible it’s being underdiagnosed,” said Dr. Negrello.
Lower respiratory tract infection, acute heart failure, chronic obstructive pulmonary disease, and exacerbation of asthma are the four main diagnoses linked to dyspnea, but this symptom is also seen in several medical conditions (gastrointestinal, metabolic, neurologic, etc.), he noted.
Often seen as a harmless treatment option, oxygen therapy is commonly administered to patients with breathing difficulties even when no hypoxemia is documented. This is particularly the case for patients brought into hospital via ambulance who are treated with oxygen without even having had their blood oxygen levels, SpO2, and partial pressure of oxygen checked.
In the United States, one of the few studies published on the topic showed that one-third of patients transported via ambulance are put on oxygen, with SpO2 being measured in just 5% of these cases. Finally, just 17% of patients receiving oxygen were experiencing hypoxia, defined as SpO2 < 94%.
Oxidative stress
Recently, several research studies have revealed the potential dangers of unjustified use of oxygen, which can lead to hyperoxia and increased mortality in hospitalized patients.
A meta-analysis reported a linear relationship between severe hyperoxia, in-hospital mortality, and length of stay in intensive care. Another study revealed a greater mortality rate in patients with acute respiratory distress syndrome (ARDS) experiencing an episode of hyperoxia, regardless of the severity of ARDS.
Oxygen toxicity in intensive care is said to be linked to oxidative stress caused by increased growth of reactive oxygen species but also to the systemic inflammation caused by hyperoxia, explained Dr. Negrello. Excess oxygen may also cause lung lesions with necrosis, the severity of which is proportional to the fraction of inspired oxygen and the length of exposure.
According to the most up-to-date international recommendations published in 2018 on the use of oxygen therapy in treating acute conditions, oxygen should not be used when SpO2 ≥ 93%. When treatment has been started, it must be stopped when SpO2 reaches 96%. SpO2 cannot be maintained above 96%, according to experts.
These threshold values can be found in the COVID-19 treatment guidelines produced by the French-Language Society of Respiratory Medicine, with oxygen therapy being recommended when SpO2 < 92%, added Dr. Negrello. The aim is to maintain normal oxygen levels, with SpO2 between 92% and 96%.
Use sparingly
For patients with COPD, the target levels are lower, due to the risk of hypercapnia (higher than normal carbon dioxide levels in the blood). Oxygen saturation levels should then be kept between 88% and 92%, “by using the minimum amount of oxygen necessary,” per the guidelines.
“Oxygen should be used sparingly,” concluded Dr. Negrello. “To treat our patients without harming them, we must be able to use it at the right time, meaning when a patient really has low blood oxygen, by focusing on normal saturation levels as the end goal.”
SpO2 measurement is the first step to be taken to determine oxygen requirements, followed by, if necessary, blood gas analysis once the patient has been admitted, he explained.
Questioned at the end of his session on how long oxygen therapy can be given for, Dr. Negrello reiterated that the risk for death is correlated with the length of time spent in a state of hyperoxia but that it is difficult to establish a maximum timeframe to be adhered to strictly.
Given that excess oxygen is harmful to patients in intensive care, “it would be better, when in doubt, to focus on physiological levels” and simply stop treatment when target saturation levels are reached.
This article was translated from the Medscape French Edition. A version appeared on Medscape.com.
PARIS – , as per the current guidelines. Florian Negrello, MD, an emergency medicine specialist at University Hospital of Martinique in Fort-de-France, reiterated this message at the 2023 conference held by France’s emergency medicine society (Urgences 2023). The recommendation is intended to prevent hyperoxia; increasing evidence indicates the harmful effects of such a state on the body.
“This is a real problem. Oxygen therapy is given all too readily despite studies now showing that excess oxygen is harmful, especially in patients with head trauma, ischemic stroke, or cardiac arrest,” stated the session’s moderator, Patrick Plaisance, MD, PhD, a doctor at Lariboisière Hospital in Paris.
No proven hypoxia
Described as difficulty breathing or shortness of breath, dyspnea is common in the emergency department, occurring in 5%-9% of patients. Close to 20% of intensive care unit admissions involve patients with dyspnea. “Since this is a very subjective symptom, it’s possible it’s being underdiagnosed,” said Dr. Negrello.
Lower respiratory tract infection, acute heart failure, chronic obstructive pulmonary disease, and exacerbation of asthma are the four main diagnoses linked to dyspnea, but this symptom is also seen in several medical conditions (gastrointestinal, metabolic, neurologic, etc.), he noted.
Often seen as a harmless treatment option, oxygen therapy is commonly administered to patients with breathing difficulties even when no hypoxemia is documented. This is particularly the case for patients brought into hospital via ambulance who are treated with oxygen without even having had their blood oxygen levels, SpO2, and partial pressure of oxygen checked.
In the United States, one of the few studies published on the topic showed that one-third of patients transported via ambulance are put on oxygen, with SpO2 being measured in just 5% of these cases. Finally, just 17% of patients receiving oxygen were experiencing hypoxia, defined as SpO2 < 94%.
Oxidative stress
Recently, several research studies have revealed the potential dangers of unjustified use of oxygen, which can lead to hyperoxia and increased mortality in hospitalized patients.
A meta-analysis reported a linear relationship between severe hyperoxia, in-hospital mortality, and length of stay in intensive care. Another study revealed a greater mortality rate in patients with acute respiratory distress syndrome (ARDS) experiencing an episode of hyperoxia, regardless of the severity of ARDS.
Oxygen toxicity in intensive care is said to be linked to oxidative stress caused by increased growth of reactive oxygen species but also to the systemic inflammation caused by hyperoxia, explained Dr. Negrello. Excess oxygen may also cause lung lesions with necrosis, the severity of which is proportional to the fraction of inspired oxygen and the length of exposure.
According to the most up-to-date international recommendations published in 2018 on the use of oxygen therapy in treating acute conditions, oxygen should not be used when SpO2 ≥ 93%. When treatment has been started, it must be stopped when SpO2 reaches 96%. SpO2 cannot be maintained above 96%, according to experts.
These threshold values can be found in the COVID-19 treatment guidelines produced by the French-Language Society of Respiratory Medicine, with oxygen therapy being recommended when SpO2 < 92%, added Dr. Negrello. The aim is to maintain normal oxygen levels, with SpO2 between 92% and 96%.
Use sparingly
For patients with COPD, the target levels are lower, due to the risk of hypercapnia (higher than normal carbon dioxide levels in the blood). Oxygen saturation levels should then be kept between 88% and 92%, “by using the minimum amount of oxygen necessary,” per the guidelines.
“Oxygen should be used sparingly,” concluded Dr. Negrello. “To treat our patients without harming them, we must be able to use it at the right time, meaning when a patient really has low blood oxygen, by focusing on normal saturation levels as the end goal.”
SpO2 measurement is the first step to be taken to determine oxygen requirements, followed by, if necessary, blood gas analysis once the patient has been admitted, he explained.
Questioned at the end of his session on how long oxygen therapy can be given for, Dr. Negrello reiterated that the risk for death is correlated with the length of time spent in a state of hyperoxia but that it is difficult to establish a maximum timeframe to be adhered to strictly.
Given that excess oxygen is harmful to patients in intensive care, “it would be better, when in doubt, to focus on physiological levels” and simply stop treatment when target saturation levels are reached.
This article was translated from the Medscape French Edition. A version appeared on Medscape.com.