User login
Substance abuse among older adults: A growing problem
Baby Boomers—a term used to refer to individuals born in the United States between 1946 and 1964—are now approaching old age. Surprisingly, these older adults are using illicit substances in a pattern not seen in prior generations of older adults, including developing substance use disorders (SUDs) at increasingly higher rates; in previous generations, the prevalence of such disorders typically lowered with advancing age.
This article discusses how to recognize and treat SUDs in older adults. Alcohol is the most commonly used substance among older adults,1 and there is a largebody of literature describing the identification and treatment of alcohol-related disorders in these patients. Therefore, this article will instead focus on older adults’ use of illicit substances, including marijuana, cocaine, and heroin.
Epidemiology
Prior clinical data regarding substance abuse in older adults focused on alcohol, prescription drugs, nicotine, and caffeine.2 In the past, compared with younger adults, older adults had lower rates of alcohol and other illicit drug use.3,4 Baby Boomers appear to be defying this trend.
A 2013 Substance Abuse and Mental Health Services Administration survey found that the percentage of adults ages 50 to 64 who used illicit substances increased from 2.7% in 2002 to 6.0% in 2013.5 Specifically, during that time, past-month illicit substance use increased from 3.4% to 7.9% among those ages 50 to 54, from 1.9% to 5.7% among those ages 55 to 59, and from 2.5% to 3.9% among those ages 60 to 64.5
More recently, a 2014 study of geriatric patients found that of the 1,302 patients age ≥65 admitted to a Level 1 trauma center, 48.3% had a positive urine drug screen.6 Someresearchers have estimated that 5.7 million older adults will require treatment for a substance use disorder in 2020, which is roughly double the 2.8 million who had an SUD in 2002 to 2006.7
Risk factors and patterns of substance abuse
Individual, social, and familial factors can contribute to substance use and abuse in late life. The Table1 outlines some of the potential risk factors for older adults associated with the use of illicit substances. Substance abuse among older adults can be divided into 2 broad categories: early onset (starting before age 50) and late onset (starting after age 50).8 While data are limited, in general, early-onset use is a more common pattern; late-onset use represents an estimated <10% of substance use among older adults. The factors that lead some adults to continue substance use in late life, or to begin substance use later in life, have not been thoroughly evaluated.

Although older adults may abuse a wide variety of illicit substances, here we describe their use of marijuana, cocaine, and heroin.
Marijuana use has changed substantially in the last decade. While marijuana is illegal under federal law, as of November 2017, 29 states had legalized marijuana for medicinal purposes and 7 states and the District of Columbia had legalized it for recreational use. The increased legal and social acceptance of marijuana has led to new businesses and methods of use beyond smoking. New types of marijuana products include edible substances, tinctures, and oils that can be vaporized and inhaled.
In addition to euphoria and relaxation, the effects of marijuana use include increased latency time and decreased ability to respond to stimuli.2 Nonpsychiatric effects of marijuana include shallow breathing, weakened immune system, and increasing cardiac workload.2 The latter effect is especially important for older adults, many of whom may have preexisting cardiac illness and may be more likely to experience an adverse cardiac event as a result of marijuana use.2 Older adults who begin to use marijuana in late life may do so not primarily as a social activity, but more likely to experience the drug’s potentially beneficial effects on pain or appetite.2 For more on theuse of marijuana for these reasons, see “Medical marijuana: Do the benefits outweigh the risks?” in
Cocaine. Although cocaine is a CNS stimulant that causes a short-lived euphoria, its adverse effects impact many body systems.9 Myocardial infarction (MI) secondary to coronary artery vasospasms, stroke (hemorrhagic and ischemic), seizures, psychosis, aortic dissection, and acute renal injury are some of the most severe complications. Acute MI is the most frequent and severe cardiovascular complication seen among abusers.10 Cocaine use can cause dizziness, restlessness, headache, mydriasis, and anxiety.
In a pilot study, Kalapatapu et al11 compared the effects of cocaine abuse in younger vs older users. They found that older users had similar patterns of cocaine abuse in terms of the amount of cocaine used and frequency of use.11 They also found that specific cognitive functions, including psychomotor speed, attention, and short-term memory, are particularly sensitive to the combined effects of aging and cocaine abuse.11
Heroin is an opioid and a CNS depressant. Common effects include slowed heart rate, decreased blood pressure, and decreased respiration rate. Chronic heroin users show an overall decrease in immune system functioning12; this deficit might be particularly pronounced in an older person whose immune system functioning has already begun to decline as a result of aging. In recent years, as is the case with younger substance users, prescription opioids have replaced heroin as the opioid of choice among older users. However, for some early-onset heroin users, the use of this particular drug becomes well entrenched and unlikely to change, even in late life. Each year of heroin use increases the likelihood of continued use the next year by approximately 3%.2 Some research suggests that older heroin users do not decrease their use over time, and face many of the same risks as younger users, including poorer physical and mental health, severe physical disability, and mortality.13
Challenges to recognizing the problem
There are no screening protocols in the clinical setting that are designed specifically for detecting illicit substance abuse among older adults. Furthermore, diagnosis can be easily overlooked because the signs and symptoms of illicit substance use can be mistaken for other illnesses. To complicate matters further, older adults often do not disclose their substance use, understate it, or even try to explain away their symptoms.1 Many older adults live alone, which may increase their risk of receiving no treatment.14
Older adults generally experience reduced tolerance to the effects of illicit substances because of age-related physiologic changes, such as decreases in renal functioning, motor functioning, and cardiac output; altered liver metabolism of certain drugs; and elevated blood glucose levels.15 As a result, symptoms of illicit substance use could be mistaken for dementia or other forms of cognitive impairment.1,16
Although not designed specifically for older adults, an evidence-based screening instrument, such as the CAGE Questionnaire Adapted to Include Drugs, may be helpful in identifying substance abuse in these patients. Urine and/or serum drug screening, along with obtaining a comprehensive history from a trustworthy source, is useful for diagnosis.
Pharmacologic treatments
Research evaluating the use of medication for treating substance abuse specifically in older adults is extremely limited; studies have focused primarily on younger patients or mixed-age populations. Treatments that have been shown to be effective for younger patients may or may not be effective for older adults.
Marijuana. There are no FDA-approved treatments for marijuana abuse. An open-label study found that N-acetylcysteine, 1,200 mg twice a day, resulted in a significant reduction in marijuana craving as measured by the 12-item version of the Marijuana Craving Questionnaire.17 In a double-blinded placebo-controlled study, adolescents who were dependent on marijuana who received N-acetylcysteine, 1,200 mg twice a day, were more than twice likely to stop marijuana use compared with those who received placebo.18 Some researchers have proposed that N-acetylcysteine may prevent continued use of marijuana via glutamate modulation in the nucleus accumbens. Animal models have demonstrated that chronic drug self-administration downregulates the cystine-glutamate exchanger in the nucleus accumbens, and that N-acetylcysteine upregulates this exchanger, which reduces reinstatement of drug seeking.Further studies are needed to verify this speculation.
Cocaine. There are no FDA-approved treatments for cocaine abuse. No specific treatment approach has been found to be consistently effective.
A potential “cocaine vaccine” called TA-CD, which is made from succinyl norcocaine conjugated to cholera toxin, is being evaluated. An initial study had promising results, finding a significant reduction in cocaine use among those who received TA-CD.19 A later double-blinded placebo-controlled study only partially replicated the efficacy found in the initial study.20
Currently, other cocaine treatments are also being investigated. An enzyme to rapidly metabolize cocaine is being evaluated.21 So far, none of these treatments have targeted older adults, and there may be age-specific issues to consider if these approaches eventually receive FDA approval.
Heroin. Several FDA-approved medications are available for treating dependency to heroin and other opioids, including naltrexone, buprenorphine, and methadone, but none have been studied specifically in older adults. Some studies of transdermal buprenorphine for treating chronic pain in older adults have concluded that this formulation may offer advantages for older patients.22,23 Compared with oral or sublingual buprenorphine, the transdermal formulation avoids the first-pass effect in the liver, thus greatly increasing bioavailability of the drug; avoids renal metabolism; and offers greater tolerability in patients with mild to moderate hepatic impairment.22,23 However, transdermal buprenorphine has been approved only for the treatment of pain. These beneficial aspects of transdermal buprenorphine may be applicable to older opioid users, but no age-specific studies of buprenorphine for treating opioid abuse have been conducted.
Nonpharmacologic treatments
The same psychotherapeutic treatments used to treat younger patients with SUDs may be appropriate for older adults. Older patients may experience feelings of isolation and shame related to needing treatment for substance abuse. These factors in treatment of older patients often are overcome by group psychotherapy. Self-help programs, such as Narcotics Anonymous or Alcoholics Anonymous, and group therapy also may be options.
On the other hand, individual psychotherapy, such as cognitive-behavioral therapy (CBT), interpersonal therapy, and psychodynamic therapy, can provide a private and confidential environment for older adults who are less social.24
The highly structured nature of CBT may be well suited to older adults who have memory difficulties.1 A study of 110 older veterans with substance abuse problems found evidence for the effectiveness of group CBT among these patients.25 All but 8 participants in this study were age ≥65. The intervention consisted of 16 weekly group sessions that began with analysis of substance use behavior to determine high-risk situations for use, followed by a series of modules to teach skills for coping with social pressure, being at home and alone, feelings of depression and loneliness, anxiety and tension, anger and frustration, cues for substance use, and other factors. Approximately 44% (49 of 110) completed treatment (≥13 sessions). Approximately 55% of those who completed the treatment were abstinent at 6-month follow-up.25
Don’t assume your older patient is not using illicit substances
It is a myth that older adults do not use and abuse illicit substances. Illicit drug use among older adults is increasing. Older adults with SUDs may not present with the same symptoms as their younger counterparts, and thus it may be difficult to identify the problem. Maintain a high index of suspicion regarding the use of illicit substances in these patients.
Treatment options are generally limited and health care settings offer few interventions designed specifically for older adults. In general, proper identification of SUDs and targeted treatment can highly improve outcomes.
1. Kuerbis A, Sacco P, Blazer DG, et al. Substance abuse among older adults. Clin Geriatr Med. 2014;30(3):629-654.
2. Taylor MH, Grossberg GT. (2012). The growing problem of illicit substance abuse in the elderly: a review. Prim Care Companion CNS Disord. 2012;14(4):PCC.11r01320. doi: 10.4088/PCC.11r01320.
3. Cummings SM, Bride B, Rawlings-Shaw AM. Alcohol abuse treatment for older adults: a review of recent empirical research. J Evid Based Soc Work. 2006;3(1):79-99.
4. Substance Abuse and Mental Health Services Administration. Results from the 2012 national survey on drug use and health: summary of national findings, NSDUH Series H-46, HHS Publication No (SMA) 13-4795. Rockville, MD: Substance Abuse and Mental Health Service Administration; 2013.
5. Substance Abuse and Mental Health Services Administration. Results from the 2013 national survey on drug use and health: summary of national findings. NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2014.
6. Ekeh AP, Parikh P, Walusimbi MS, et al. The prevalence of positive drug and alcohol screens in elderly trauma patients. Subst Abus. 2014;35(1):51-55.
7. Wu LT, Blazer DG. Illicit and nonmedical drug use among older adults: a review. J Aging Health. 2011;23(3):481-504.
8. Roe B, Beynon C, Pickering L, et al. Experiences of drug use and ageing: health, quality of life, relationship and service implications. J Adv Nurs. 2010;66(9):1968-1979.
9. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28(4):517-526.
10. Weber JE, Chudnofsky CR, Boczar M, et al. Cocaine-associated chest pain: how common is myocardial infarction? Acad Emerg Med. 2000;7(8):873-877.
11. Kalapatapu RK, Vadhan NP, Rubin E, et al. A pilot study of neurocognitive function in older and younger cocaine abusers and controls. Am J Addict. 2011;20(3):228-239.
12. Edelman EJ, Cheng DM, Krupitsky EM, et al. Heroin use and HIV disease progression: results from a pilot study of a Russian cohort. AIDS Behav. 2015;19(6):1089-1097.
13. Darke S, Mills KL, Ross J, et al. The ageing heroin user: career length, clinical profile and outcomes across 36 months. Drug Alcohol Rev. 2009;28(3):243-249.
14. West LA, Cole S, Goodkind D, et al. U.S. Census Bureau, P23-212. 65+ in the United States: 2010. Washington, DC: United States Census Bureau; 2014.
15. Boss GR, Seegmiller JE. Age-related physiological changes and their clinical significance. West J Med. 1981;135(6):434-440.
16. Ruiz P, Strain EC, Langrod JG. The substance abuse handbook. Philadelphia, PA: Wolters Kluwer Health; 2007.
17. Gray KM, Watson NL, Carpenter MJ, et al. N-acetylcysteine (NAC) in young marijuana users: an open-label pilot study. Am J Addict. 2010;19(2):187-189.
18. Gray KM, Carpenter MJ, Baker NL, et al. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169(8):805-812.
19. Martell BA, Orson FM, Poling J, et al. Cocaine vaccine for the treatment of cocaine dependence in methadone-maintained patients: a randomized, double-blind, placebo-controlled efficacy trial. Arch Gen Psychiatry. 2009;66(10):1116-1123
20. Kosten TR, Domingo CB, Shorter D, et al. Vaccine for cocaine dependence: a randomized double-blind placebo-controlled efficacy trial. Drug Alcohol Depend. 2014;140:42-47.
21. Gao Y, Brimijoin S. An engineered cocaine hydrolase blunts and reverses cardiovascular responses to cocaine in rats. J Pharmacol Exp Ther. 2004;310(3):1046-1052.
22. Vadivelu N, Hines RL. Management of chronic pain in the elderly: focus on transdermal buprenorphine. Clin Interv Aging. 2008;3(3):421-430.
23. Al-Tawil N, Odar-Cederlöf I, Berggren AC, et al. Pharmacokinetics of transdermal buprenorphine patch in the elderly. Eur J Clin Pharmacol. 2013;69(2):143-149.
24. Schultz SK, Arndt S, Liesveld J. Locations of facilities with special programs for older substance abuse clients in the US. Int J Geriatr Psychiatry. 2003;18(9):839-843.
25. Schonfeld L, Dupree LW, Dickson-Fuhrman E, et al. Cognitive-behavioral treatment of older veterans with substance abuse problems. J Geriatr Psychiatry Neurol. 2000;13(3):124-129.
Baby Boomers—a term used to refer to individuals born in the United States between 1946 and 1964—are now approaching old age. Surprisingly, these older adults are using illicit substances in a pattern not seen in prior generations of older adults, including developing substance use disorders (SUDs) at increasingly higher rates; in previous generations, the prevalence of such disorders typically lowered with advancing age.
This article discusses how to recognize and treat SUDs in older adults. Alcohol is the most commonly used substance among older adults,1 and there is a largebody of literature describing the identification and treatment of alcohol-related disorders in these patients. Therefore, this article will instead focus on older adults’ use of illicit substances, including marijuana, cocaine, and heroin.
Epidemiology
Prior clinical data regarding substance abuse in older adults focused on alcohol, prescription drugs, nicotine, and caffeine.2 In the past, compared with younger adults, older adults had lower rates of alcohol and other illicit drug use.3,4 Baby Boomers appear to be defying this trend.
A 2013 Substance Abuse and Mental Health Services Administration survey found that the percentage of adults ages 50 to 64 who used illicit substances increased from 2.7% in 2002 to 6.0% in 2013.5 Specifically, during that time, past-month illicit substance use increased from 3.4% to 7.9% among those ages 50 to 54, from 1.9% to 5.7% among those ages 55 to 59, and from 2.5% to 3.9% among those ages 60 to 64.5
More recently, a 2014 study of geriatric patients found that of the 1,302 patients age ≥65 admitted to a Level 1 trauma center, 48.3% had a positive urine drug screen.6 Someresearchers have estimated that 5.7 million older adults will require treatment for a substance use disorder in 2020, which is roughly double the 2.8 million who had an SUD in 2002 to 2006.7
Risk factors and patterns of substance abuse
Individual, social, and familial factors can contribute to substance use and abuse in late life. The Table1 outlines some of the potential risk factors for older adults associated with the use of illicit substances. Substance abuse among older adults can be divided into 2 broad categories: early onset (starting before age 50) and late onset (starting after age 50).8 While data are limited, in general, early-onset use is a more common pattern; late-onset use represents an estimated <10% of substance use among older adults. The factors that lead some adults to continue substance use in late life, or to begin substance use later in life, have not been thoroughly evaluated.
Although older adults may abuse a wide variety of illicit substances, here we describe their use of marijuana, cocaine, and heroin.
Marijuana use has changed substantially in the last decade. While marijuana is illegal under federal law, as of November 2017, 29 states had legalized marijuana for medicinal purposes and 7 states and the District of Columbia had legalized it for recreational use. The increased legal and social acceptance of marijuana has led to new businesses and methods of use beyond smoking. New types of marijuana products include edible substances, tinctures, and oils that can be vaporized and inhaled.
In addition to euphoria and relaxation, the effects of marijuana use include increased latency time and decreased ability to respond to stimuli.2 Nonpsychiatric effects of marijuana include shallow breathing, weakened immune system, and increasing cardiac workload.2 The latter effect is especially important for older adults, many of whom may have preexisting cardiac illness and may be more likely to experience an adverse cardiac event as a result of marijuana use.2 Older adults who begin to use marijuana in late life may do so not primarily as a social activity, but more likely to experience the drug’s potentially beneficial effects on pain or appetite.2 For more on theuse of marijuana for these reasons, see “Medical marijuana: Do the benefits outweigh the risks?” in
Cocaine. Although cocaine is a CNS stimulant that causes a short-lived euphoria, its adverse effects impact many body systems.9 Myocardial infarction (MI) secondary to coronary artery vasospasms, stroke (hemorrhagic and ischemic), seizures, psychosis, aortic dissection, and acute renal injury are some of the most severe complications. Acute MI is the most frequent and severe cardiovascular complication seen among abusers.10 Cocaine use can cause dizziness, restlessness, headache, mydriasis, and anxiety.
In a pilot study, Kalapatapu et al11 compared the effects of cocaine abuse in younger vs older users. They found that older users had similar patterns of cocaine abuse in terms of the amount of cocaine used and frequency of use.11 They also found that specific cognitive functions, including psychomotor speed, attention, and short-term memory, are particularly sensitive to the combined effects of aging and cocaine abuse.11
Heroin is an opioid and a CNS depressant. Common effects include slowed heart rate, decreased blood pressure, and decreased respiration rate. Chronic heroin users show an overall decrease in immune system functioning12; this deficit might be particularly pronounced in an older person whose immune system functioning has already begun to decline as a result of aging. In recent years, as is the case with younger substance users, prescription opioids have replaced heroin as the opioid of choice among older users. However, for some early-onset heroin users, the use of this particular drug becomes well entrenched and unlikely to change, even in late life. Each year of heroin use increases the likelihood of continued use the next year by approximately 3%.2 Some research suggests that older heroin users do not decrease their use over time, and face many of the same risks as younger users, including poorer physical and mental health, severe physical disability, and mortality.13
Challenges to recognizing the problem
There are no screening protocols in the clinical setting that are designed specifically for detecting illicit substance abuse among older adults. Furthermore, diagnosis can be easily overlooked because the signs and symptoms of illicit substance use can be mistaken for other illnesses. To complicate matters further, older adults often do not disclose their substance use, understate it, or even try to explain away their symptoms.1 Many older adults live alone, which may increase their risk of receiving no treatment.14
Older adults generally experience reduced tolerance to the effects of illicit substances because of age-related physiologic changes, such as decreases in renal functioning, motor functioning, and cardiac output; altered liver metabolism of certain drugs; and elevated blood glucose levels.15 As a result, symptoms of illicit substance use could be mistaken for dementia or other forms of cognitive impairment.1,16
Although not designed specifically for older adults, an evidence-based screening instrument, such as the CAGE Questionnaire Adapted to Include Drugs, may be helpful in identifying substance abuse in these patients. Urine and/or serum drug screening, along with obtaining a comprehensive history from a trustworthy source, is useful for diagnosis.
Pharmacologic treatments
Research evaluating the use of medication for treating substance abuse specifically in older adults is extremely limited; studies have focused primarily on younger patients or mixed-age populations. Treatments that have been shown to be effective for younger patients may or may not be effective for older adults.
Marijuana. There are no FDA-approved treatments for marijuana abuse. An open-label study found that N-acetylcysteine, 1,200 mg twice a day, resulted in a significant reduction in marijuana craving as measured by the 12-item version of the Marijuana Craving Questionnaire.17 In a double-blinded placebo-controlled study, adolescents who were dependent on marijuana who received N-acetylcysteine, 1,200 mg twice a day, were more than twice likely to stop marijuana use compared with those who received placebo.18 Some researchers have proposed that N-acetylcysteine may prevent continued use of marijuana via glutamate modulation in the nucleus accumbens. Animal models have demonstrated that chronic drug self-administration downregulates the cystine-glutamate exchanger in the nucleus accumbens, and that N-acetylcysteine upregulates this exchanger, which reduces reinstatement of drug seeking.Further studies are needed to verify this speculation.
Cocaine. There are no FDA-approved treatments for cocaine abuse. No specific treatment approach has been found to be consistently effective.
A potential “cocaine vaccine” called TA-CD, which is made from succinyl norcocaine conjugated to cholera toxin, is being evaluated. An initial study had promising results, finding a significant reduction in cocaine use among those who received TA-CD.19 A later double-blinded placebo-controlled study only partially replicated the efficacy found in the initial study.20
Currently, other cocaine treatments are also being investigated. An enzyme to rapidly metabolize cocaine is being evaluated.21 So far, none of these treatments have targeted older adults, and there may be age-specific issues to consider if these approaches eventually receive FDA approval.
Heroin. Several FDA-approved medications are available for treating dependency to heroin and other opioids, including naltrexone, buprenorphine, and methadone, but none have been studied specifically in older adults. Some studies of transdermal buprenorphine for treating chronic pain in older adults have concluded that this formulation may offer advantages for older patients.22,23 Compared with oral or sublingual buprenorphine, the transdermal formulation avoids the first-pass effect in the liver, thus greatly increasing bioavailability of the drug; avoids renal metabolism; and offers greater tolerability in patients with mild to moderate hepatic impairment.22,23 However, transdermal buprenorphine has been approved only for the treatment of pain. These beneficial aspects of transdermal buprenorphine may be applicable to older opioid users, but no age-specific studies of buprenorphine for treating opioid abuse have been conducted.
Nonpharmacologic treatments
The same psychotherapeutic treatments used to treat younger patients with SUDs may be appropriate for older adults. Older patients may experience feelings of isolation and shame related to needing treatment for substance abuse. These factors in treatment of older patients often are overcome by group psychotherapy. Self-help programs, such as Narcotics Anonymous or Alcoholics Anonymous, and group therapy also may be options.
On the other hand, individual psychotherapy, such as cognitive-behavioral therapy (CBT), interpersonal therapy, and psychodynamic therapy, can provide a private and confidential environment for older adults who are less social.24
The highly structured nature of CBT may be well suited to older adults who have memory difficulties.1 A study of 110 older veterans with substance abuse problems found evidence for the effectiveness of group CBT among these patients.25 All but 8 participants in this study were age ≥65. The intervention consisted of 16 weekly group sessions that began with analysis of substance use behavior to determine high-risk situations for use, followed by a series of modules to teach skills for coping with social pressure, being at home and alone, feelings of depression and loneliness, anxiety and tension, anger and frustration, cues for substance use, and other factors. Approximately 44% (49 of 110) completed treatment (≥13 sessions). Approximately 55% of those who completed the treatment were abstinent at 6-month follow-up.25
Don’t assume your older patient is not using illicit substances
It is a myth that older adults do not use and abuse illicit substances. Illicit drug use among older adults is increasing. Older adults with SUDs may not present with the same symptoms as their younger counterparts, and thus it may be difficult to identify the problem. Maintain a high index of suspicion regarding the use of illicit substances in these patients.
Treatment options are generally limited and health care settings offer few interventions designed specifically for older adults. In general, proper identification of SUDs and targeted treatment can highly improve outcomes.
Baby Boomers—a term used to refer to individuals born in the United States between 1946 and 1964—are now approaching old age. Surprisingly, these older adults are using illicit substances in a pattern not seen in prior generations of older adults, including developing substance use disorders (SUDs) at increasingly higher rates; in previous generations, the prevalence of such disorders typically lowered with advancing age.
This article discusses how to recognize and treat SUDs in older adults. Alcohol is the most commonly used substance among older adults,1 and there is a largebody of literature describing the identification and treatment of alcohol-related disorders in these patients. Therefore, this article will instead focus on older adults’ use of illicit substances, including marijuana, cocaine, and heroin.
Epidemiology
Prior clinical data regarding substance abuse in older adults focused on alcohol, prescription drugs, nicotine, and caffeine.2 In the past, compared with younger adults, older adults had lower rates of alcohol and other illicit drug use.3,4 Baby Boomers appear to be defying this trend.
A 2013 Substance Abuse and Mental Health Services Administration survey found that the percentage of adults ages 50 to 64 who used illicit substances increased from 2.7% in 2002 to 6.0% in 2013.5 Specifically, during that time, past-month illicit substance use increased from 3.4% to 7.9% among those ages 50 to 54, from 1.9% to 5.7% among those ages 55 to 59, and from 2.5% to 3.9% among those ages 60 to 64.5
More recently, a 2014 study of geriatric patients found that of the 1,302 patients age ≥65 admitted to a Level 1 trauma center, 48.3% had a positive urine drug screen.6 Someresearchers have estimated that 5.7 million older adults will require treatment for a substance use disorder in 2020, which is roughly double the 2.8 million who had an SUD in 2002 to 2006.7
Risk factors and patterns of substance abuse
Individual, social, and familial factors can contribute to substance use and abuse in late life. The Table1 outlines some of the potential risk factors for older adults associated with the use of illicit substances. Substance abuse among older adults can be divided into 2 broad categories: early onset (starting before age 50) and late onset (starting after age 50).8 While data are limited, in general, early-onset use is a more common pattern; late-onset use represents an estimated <10% of substance use among older adults. The factors that lead some adults to continue substance use in late life, or to begin substance use later in life, have not been thoroughly evaluated.
Although older adults may abuse a wide variety of illicit substances, here we describe their use of marijuana, cocaine, and heroin.
Marijuana use has changed substantially in the last decade. While marijuana is illegal under federal law, as of November 2017, 29 states had legalized marijuana for medicinal purposes and 7 states and the District of Columbia had legalized it for recreational use. The increased legal and social acceptance of marijuana has led to new businesses and methods of use beyond smoking. New types of marijuana products include edible substances, tinctures, and oils that can be vaporized and inhaled.
In addition to euphoria and relaxation, the effects of marijuana use include increased latency time and decreased ability to respond to stimuli.2 Nonpsychiatric effects of marijuana include shallow breathing, weakened immune system, and increasing cardiac workload.2 The latter effect is especially important for older adults, many of whom may have preexisting cardiac illness and may be more likely to experience an adverse cardiac event as a result of marijuana use.2 Older adults who begin to use marijuana in late life may do so not primarily as a social activity, but more likely to experience the drug’s potentially beneficial effects on pain or appetite.2 For more on theuse of marijuana for these reasons, see “Medical marijuana: Do the benefits outweigh the risks?” in
Cocaine. Although cocaine is a CNS stimulant that causes a short-lived euphoria, its adverse effects impact many body systems.9 Myocardial infarction (MI) secondary to coronary artery vasospasms, stroke (hemorrhagic and ischemic), seizures, psychosis, aortic dissection, and acute renal injury are some of the most severe complications. Acute MI is the most frequent and severe cardiovascular complication seen among abusers.10 Cocaine use can cause dizziness, restlessness, headache, mydriasis, and anxiety.
In a pilot study, Kalapatapu et al11 compared the effects of cocaine abuse in younger vs older users. They found that older users had similar patterns of cocaine abuse in terms of the amount of cocaine used and frequency of use.11 They also found that specific cognitive functions, including psychomotor speed, attention, and short-term memory, are particularly sensitive to the combined effects of aging and cocaine abuse.11
Heroin is an opioid and a CNS depressant. Common effects include slowed heart rate, decreased blood pressure, and decreased respiration rate. Chronic heroin users show an overall decrease in immune system functioning12; this deficit might be particularly pronounced in an older person whose immune system functioning has already begun to decline as a result of aging. In recent years, as is the case with younger substance users, prescription opioids have replaced heroin as the opioid of choice among older users. However, for some early-onset heroin users, the use of this particular drug becomes well entrenched and unlikely to change, even in late life. Each year of heroin use increases the likelihood of continued use the next year by approximately 3%.2 Some research suggests that older heroin users do not decrease their use over time, and face many of the same risks as younger users, including poorer physical and mental health, severe physical disability, and mortality.13
Challenges to recognizing the problem
There are no screening protocols in the clinical setting that are designed specifically for detecting illicit substance abuse among older adults. Furthermore, diagnosis can be easily overlooked because the signs and symptoms of illicit substance use can be mistaken for other illnesses. To complicate matters further, older adults often do not disclose their substance use, understate it, or even try to explain away their symptoms.1 Many older adults live alone, which may increase their risk of receiving no treatment.14
Older adults generally experience reduced tolerance to the effects of illicit substances because of age-related physiologic changes, such as decreases in renal functioning, motor functioning, and cardiac output; altered liver metabolism of certain drugs; and elevated blood glucose levels.15 As a result, symptoms of illicit substance use could be mistaken for dementia or other forms of cognitive impairment.1,16
Although not designed specifically for older adults, an evidence-based screening instrument, such as the CAGE Questionnaire Adapted to Include Drugs, may be helpful in identifying substance abuse in these patients. Urine and/or serum drug screening, along with obtaining a comprehensive history from a trustworthy source, is useful for diagnosis.
Pharmacologic treatments
Research evaluating the use of medication for treating substance abuse specifically in older adults is extremely limited; studies have focused primarily on younger patients or mixed-age populations. Treatments that have been shown to be effective for younger patients may or may not be effective for older adults.
Marijuana. There are no FDA-approved treatments for marijuana abuse. An open-label study found that N-acetylcysteine, 1,200 mg twice a day, resulted in a significant reduction in marijuana craving as measured by the 12-item version of the Marijuana Craving Questionnaire.17 In a double-blinded placebo-controlled study, adolescents who were dependent on marijuana who received N-acetylcysteine, 1,200 mg twice a day, were more than twice likely to stop marijuana use compared with those who received placebo.18 Some researchers have proposed that N-acetylcysteine may prevent continued use of marijuana via glutamate modulation in the nucleus accumbens. Animal models have demonstrated that chronic drug self-administration downregulates the cystine-glutamate exchanger in the nucleus accumbens, and that N-acetylcysteine upregulates this exchanger, which reduces reinstatement of drug seeking.Further studies are needed to verify this speculation.
Cocaine. There are no FDA-approved treatments for cocaine abuse. No specific treatment approach has been found to be consistently effective.
A potential “cocaine vaccine” called TA-CD, which is made from succinyl norcocaine conjugated to cholera toxin, is being evaluated. An initial study had promising results, finding a significant reduction in cocaine use among those who received TA-CD.19 A later double-blinded placebo-controlled study only partially replicated the efficacy found in the initial study.20
Currently, other cocaine treatments are also being investigated. An enzyme to rapidly metabolize cocaine is being evaluated.21 So far, none of these treatments have targeted older adults, and there may be age-specific issues to consider if these approaches eventually receive FDA approval.
Heroin. Several FDA-approved medications are available for treating dependency to heroin and other opioids, including naltrexone, buprenorphine, and methadone, but none have been studied specifically in older adults. Some studies of transdermal buprenorphine for treating chronic pain in older adults have concluded that this formulation may offer advantages for older patients.22,23 Compared with oral or sublingual buprenorphine, the transdermal formulation avoids the first-pass effect in the liver, thus greatly increasing bioavailability of the drug; avoids renal metabolism; and offers greater tolerability in patients with mild to moderate hepatic impairment.22,23 However, transdermal buprenorphine has been approved only for the treatment of pain. These beneficial aspects of transdermal buprenorphine may be applicable to older opioid users, but no age-specific studies of buprenorphine for treating opioid abuse have been conducted.
Nonpharmacologic treatments
The same psychotherapeutic treatments used to treat younger patients with SUDs may be appropriate for older adults. Older patients may experience feelings of isolation and shame related to needing treatment for substance abuse. These factors in treatment of older patients often are overcome by group psychotherapy. Self-help programs, such as Narcotics Anonymous or Alcoholics Anonymous, and group therapy also may be options.
On the other hand, individual psychotherapy, such as cognitive-behavioral therapy (CBT), interpersonal therapy, and psychodynamic therapy, can provide a private and confidential environment for older adults who are less social.24
The highly structured nature of CBT may be well suited to older adults who have memory difficulties.1 A study of 110 older veterans with substance abuse problems found evidence for the effectiveness of group CBT among these patients.25 All but 8 participants in this study were age ≥65. The intervention consisted of 16 weekly group sessions that began with analysis of substance use behavior to determine high-risk situations for use, followed by a series of modules to teach skills for coping with social pressure, being at home and alone, feelings of depression and loneliness, anxiety and tension, anger and frustration, cues for substance use, and other factors. Approximately 44% (49 of 110) completed treatment (≥13 sessions). Approximately 55% of those who completed the treatment were abstinent at 6-month follow-up.25
Don’t assume your older patient is not using illicit substances
It is a myth that older adults do not use and abuse illicit substances. Illicit drug use among older adults is increasing. Older adults with SUDs may not present with the same symptoms as their younger counterparts, and thus it may be difficult to identify the problem. Maintain a high index of suspicion regarding the use of illicit substances in these patients.
Treatment options are generally limited and health care settings offer few interventions designed specifically for older adults. In general, proper identification of SUDs and targeted treatment can highly improve outcomes.
1. Kuerbis A, Sacco P, Blazer DG, et al. Substance abuse among older adults. Clin Geriatr Med. 2014;30(3):629-654.
2. Taylor MH, Grossberg GT. (2012). The growing problem of illicit substance abuse in the elderly: a review. Prim Care Companion CNS Disord. 2012;14(4):PCC.11r01320. doi: 10.4088/PCC.11r01320.
3. Cummings SM, Bride B, Rawlings-Shaw AM. Alcohol abuse treatment for older adults: a review of recent empirical research. J Evid Based Soc Work. 2006;3(1):79-99.
4. Substance Abuse and Mental Health Services Administration. Results from the 2012 national survey on drug use and health: summary of national findings, NSDUH Series H-46, HHS Publication No (SMA) 13-4795. Rockville, MD: Substance Abuse and Mental Health Service Administration; 2013.
5. Substance Abuse and Mental Health Services Administration. Results from the 2013 national survey on drug use and health: summary of national findings. NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2014.
6. Ekeh AP, Parikh P, Walusimbi MS, et al. The prevalence of positive drug and alcohol screens in elderly trauma patients. Subst Abus. 2014;35(1):51-55.
7. Wu LT, Blazer DG. Illicit and nonmedical drug use among older adults: a review. J Aging Health. 2011;23(3):481-504.
8. Roe B, Beynon C, Pickering L, et al. Experiences of drug use and ageing: health, quality of life, relationship and service implications. J Adv Nurs. 2010;66(9):1968-1979.
9. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28(4):517-526.
10. Weber JE, Chudnofsky CR, Boczar M, et al. Cocaine-associated chest pain: how common is myocardial infarction? Acad Emerg Med. 2000;7(8):873-877.
11. Kalapatapu RK, Vadhan NP, Rubin E, et al. A pilot study of neurocognitive function in older and younger cocaine abusers and controls. Am J Addict. 2011;20(3):228-239.
12. Edelman EJ, Cheng DM, Krupitsky EM, et al. Heroin use and HIV disease progression: results from a pilot study of a Russian cohort. AIDS Behav. 2015;19(6):1089-1097.
13. Darke S, Mills KL, Ross J, et al. The ageing heroin user: career length, clinical profile and outcomes across 36 months. Drug Alcohol Rev. 2009;28(3):243-249.
14. West LA, Cole S, Goodkind D, et al. U.S. Census Bureau, P23-212. 65+ in the United States: 2010. Washington, DC: United States Census Bureau; 2014.
15. Boss GR, Seegmiller JE. Age-related physiological changes and their clinical significance. West J Med. 1981;135(6):434-440.
16. Ruiz P, Strain EC, Langrod JG. The substance abuse handbook. Philadelphia, PA: Wolters Kluwer Health; 2007.
17. Gray KM, Watson NL, Carpenter MJ, et al. N-acetylcysteine (NAC) in young marijuana users: an open-label pilot study. Am J Addict. 2010;19(2):187-189.
18. Gray KM, Carpenter MJ, Baker NL, et al. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169(8):805-812.
19. Martell BA, Orson FM, Poling J, et al. Cocaine vaccine for the treatment of cocaine dependence in methadone-maintained patients: a randomized, double-blind, placebo-controlled efficacy trial. Arch Gen Psychiatry. 2009;66(10):1116-1123
20. Kosten TR, Domingo CB, Shorter D, et al. Vaccine for cocaine dependence: a randomized double-blind placebo-controlled efficacy trial. Drug Alcohol Depend. 2014;140:42-47.
21. Gao Y, Brimijoin S. An engineered cocaine hydrolase blunts and reverses cardiovascular responses to cocaine in rats. J Pharmacol Exp Ther. 2004;310(3):1046-1052.
22. Vadivelu N, Hines RL. Management of chronic pain in the elderly: focus on transdermal buprenorphine. Clin Interv Aging. 2008;3(3):421-430.
23. Al-Tawil N, Odar-Cederlöf I, Berggren AC, et al. Pharmacokinetics of transdermal buprenorphine patch in the elderly. Eur J Clin Pharmacol. 2013;69(2):143-149.
24. Schultz SK, Arndt S, Liesveld J. Locations of facilities with special programs for older substance abuse clients in the US. Int J Geriatr Psychiatry. 2003;18(9):839-843.
25. Schonfeld L, Dupree LW, Dickson-Fuhrman E, et al. Cognitive-behavioral treatment of older veterans with substance abuse problems. J Geriatr Psychiatry Neurol. 2000;13(3):124-129.
1. Kuerbis A, Sacco P, Blazer DG, et al. Substance abuse among older adults. Clin Geriatr Med. 2014;30(3):629-654.
2. Taylor MH, Grossberg GT. (2012). The growing problem of illicit substance abuse in the elderly: a review. Prim Care Companion CNS Disord. 2012;14(4):PCC.11r01320. doi: 10.4088/PCC.11r01320.
3. Cummings SM, Bride B, Rawlings-Shaw AM. Alcohol abuse treatment for older adults: a review of recent empirical research. J Evid Based Soc Work. 2006;3(1):79-99.
4. Substance Abuse and Mental Health Services Administration. Results from the 2012 national survey on drug use and health: summary of national findings, NSDUH Series H-46, HHS Publication No (SMA) 13-4795. Rockville, MD: Substance Abuse and Mental Health Service Administration; 2013.
5. Substance Abuse and Mental Health Services Administration. Results from the 2013 national survey on drug use and health: summary of national findings. NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2014.
6. Ekeh AP, Parikh P, Walusimbi MS, et al. The prevalence of positive drug and alcohol screens in elderly trauma patients. Subst Abus. 2014;35(1):51-55.
7. Wu LT, Blazer DG. Illicit and nonmedical drug use among older adults: a review. J Aging Health. 2011;23(3):481-504.
8. Roe B, Beynon C, Pickering L, et al. Experiences of drug use and ageing: health, quality of life, relationship and service implications. J Adv Nurs. 2010;66(9):1968-1979.
9. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28(4):517-526.
10. Weber JE, Chudnofsky CR, Boczar M, et al. Cocaine-associated chest pain: how common is myocardial infarction? Acad Emerg Med. 2000;7(8):873-877.
11. Kalapatapu RK, Vadhan NP, Rubin E, et al. A pilot study of neurocognitive function in older and younger cocaine abusers and controls. Am J Addict. 2011;20(3):228-239.
12. Edelman EJ, Cheng DM, Krupitsky EM, et al. Heroin use and HIV disease progression: results from a pilot study of a Russian cohort. AIDS Behav. 2015;19(6):1089-1097.
13. Darke S, Mills KL, Ross J, et al. The ageing heroin user: career length, clinical profile and outcomes across 36 months. Drug Alcohol Rev. 2009;28(3):243-249.
14. West LA, Cole S, Goodkind D, et al. U.S. Census Bureau, P23-212. 65+ in the United States: 2010. Washington, DC: United States Census Bureau; 2014.
15. Boss GR, Seegmiller JE. Age-related physiological changes and their clinical significance. West J Med. 1981;135(6):434-440.
16. Ruiz P, Strain EC, Langrod JG. The substance abuse handbook. Philadelphia, PA: Wolters Kluwer Health; 2007.
17. Gray KM, Watson NL, Carpenter MJ, et al. N-acetylcysteine (NAC) in young marijuana users: an open-label pilot study. Am J Addict. 2010;19(2):187-189.
18. Gray KM, Carpenter MJ, Baker NL, et al. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169(8):805-812.
19. Martell BA, Orson FM, Poling J, et al. Cocaine vaccine for the treatment of cocaine dependence in methadone-maintained patients: a randomized, double-blind, placebo-controlled efficacy trial. Arch Gen Psychiatry. 2009;66(10):1116-1123
20. Kosten TR, Domingo CB, Shorter D, et al. Vaccine for cocaine dependence: a randomized double-blind placebo-controlled efficacy trial. Drug Alcohol Depend. 2014;140:42-47.
21. Gao Y, Brimijoin S. An engineered cocaine hydrolase blunts and reverses cardiovascular responses to cocaine in rats. J Pharmacol Exp Ther. 2004;310(3):1046-1052.
22. Vadivelu N, Hines RL. Management of chronic pain in the elderly: focus on transdermal buprenorphine. Clin Interv Aging. 2008;3(3):421-430.
23. Al-Tawil N, Odar-Cederlöf I, Berggren AC, et al. Pharmacokinetics of transdermal buprenorphine patch in the elderly. Eur J Clin Pharmacol. 2013;69(2):143-149.
24. Schultz SK, Arndt S, Liesveld J. Locations of facilities with special programs for older substance abuse clients in the US. Int J Geriatr Psychiatry. 2003;18(9):839-843.
25. Schonfeld L, Dupree LW, Dickson-Fuhrman E, et al. Cognitive-behavioral treatment of older veterans with substance abuse problems. J Geriatr Psychiatry Neurol. 2000;13(3):124-129.

Mental health apps: What to tell patients
Have your patients asked you about smartphone apps? If they haven’t yet, they may soon, as interest in apps for mental health continues to expand. There are now >10,000 mental health–related smartphone apps.1 The rapid rise of these apps is partly due to their potential to transform a patient’s smartphone into a monitoring and therapeutic platform, capable of capturing mental health symptoms in real time and delivering on-the-go therapy. Setting aside questions about the potential of mobile health, 2 urgent questions remain for the busy psychiatrist in clinical practice: What is the current evidence base for mental health apps, and what should you tell your patients about them?
For most apps, evidence of efficacy is limited
While the evidence base for mental health smartphone apps continues to expand, for many of these apps, there is no evidence of effectiveness. The growing consensus is that most commercially available apps are not evidence-based and some are even dangerous. For example, researchers who examined >700 mindfulness apps on the iTunes and Google Play stores found that only 4% provided acceptable mindfulness training and education.2 Another study of 58 apps that claimed to offer sobriety assessments found that none had ever been formally evaluated.3 Evidence-based reviews of suicide prevention apps have identified potentially harmful apps,4 and studies evaluating apps for bipolar disorder5 and depression6 have yielded similar results—few have any evidence supporting their use, and some offer dangerous and harmful advice. For example, researchers found that one app for bipolar disorder advised patients who are experiencing a manic episode to drink alcohol.5 Currently, the vast majority of commercially available apps are not appropriate for clinical care. This finding is not unique to mental health; similar findings have been reported for apps for cancer.7 The bottom line is that the apps that your patients are finding, and perhaps already using, may not be useful or effective.
However, early studies have demonstrated efficacy of some apps for several conditions, including schizophrenia,8 depression,9 anxiety disorders,10 and suicidal ideation.11 Although many of the apps evaluated in these studies are not available to the public, or still require large-scale assessment before they are ready for mainstream clinical care, this research demonstrates that mental health apps can help improve treatment outcomes. As this research develops, a wave of evidence-based and effective mental health apps may be available in the near future.
Although it is unknown how many patients are presently using mental health apps, there is strong anecdotal evidence that an increasing number of patients who use these apps and other forms of digital technology are finding some benefits. In many cases, patients may actually be ahead of the research. For example, one study that conducted an online survey of patients with schizophrenia noted that some patients are using their smartphones to play music to help block auditory hallucinations.12
Why online reviews are of limited use
As this evidence continues to mature, and with an ever-growing number of mental health apps available on commercial marketplaces, busy psychiatrists need to navigate this complex space. Even psychiatrists who decide to not use apps as part of care still need to be knowledgeable about them, because patients are likely to ask about the benefits of using apps, and they will expect an informed response. How would you reply if your patient asked you about a new mood-tracking app he or she recently heard about? On what would you base your recommendation and opinion?
Reading online app reviews for guidance is not a good solution. A recent study found little relationship between the star ratings of health apps and the quality of those apps,13 which suggests that a 5-star rating on the app store is of limited use.
Unlike medications whose ingredients do not change over time, or manualized psychotherapies that use specific protocols, mental health apps are dynamic and constantly changing.14 Think of how often the apps on your smartphone update. Thus, the version of a mental health app that your patient downloads today may be very different from the version that received a favorable user review last month. And just as there is no single medication or therapy that is ideal for every patient, neither is there a single “best” app for all patients with the same disorder. Picking an app is a personal decision that cannot be made based on a single score or numeric rating. Furthermore, the validity of app rating systems is unclear. One study found a wide variation in the interrater reliability of measures used to evaluate apps from sources that included PsyberGuide, the Anxiety and Depression Association of America, and the research literature. Quality measures such as effectiveness, ease of use, and performance had relatively poor interrater reliability.15 This means that, for example, an app that one patient finds “easy to use” may be difficult to use for another. Thus, providing patients with suggestions based on an app’s ratings may result in providing information that sounds useful, but often is misleading.
A model for evaluating apps
One possible solution is a risk-based and personalized assessment approach to evaluating mental health apps. Although it does not offer scoring or recommendations of specific apps, the American Psychiatric Association (APA) App Evaluation Model (Figure) provides a framework to guide discussion and informed decision-making about apps. (The authors of this article helped create this model, but receive no compensation for that volunteer work.) The pyramid shape reflects the hierarchical nature of the model. To begin the process, start at the base of the pyramid and work upward.
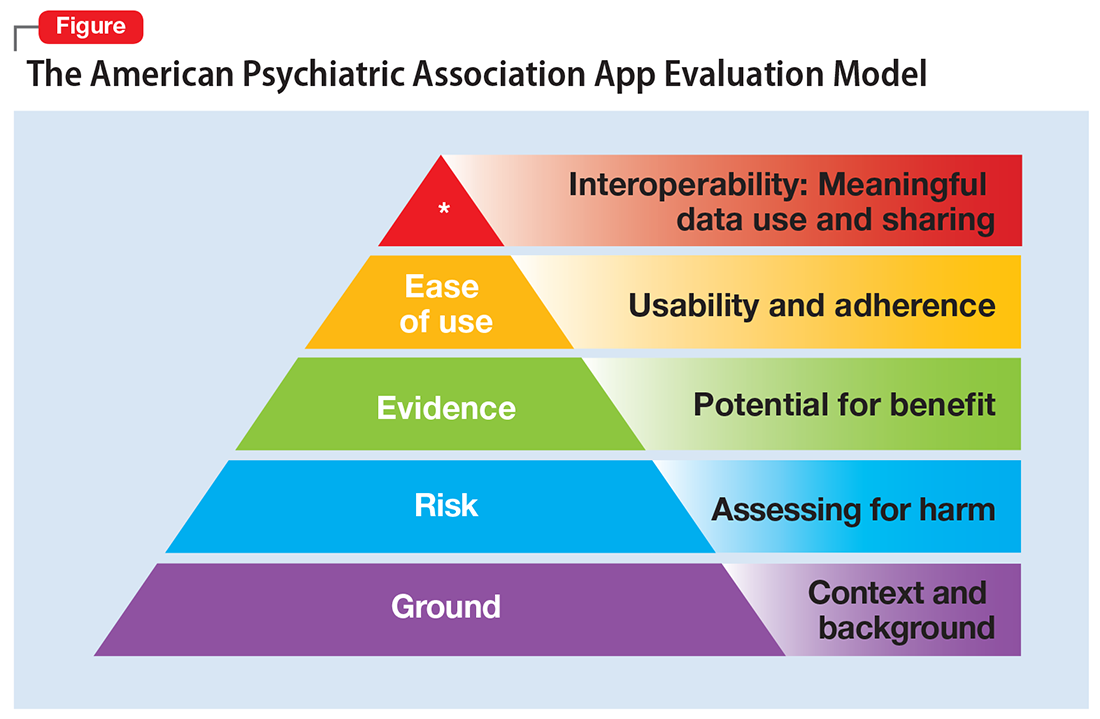
Ground. First, consider the context of the app by determining basic facts, such as who made it, how much it costs, and its technology requirements. This ground layer establishes the credibility of the app’s creator by questioning his or her reputation, ability to update the app, and funding sources. Understanding the app’s business model also will help you determine whether the app will stand the test of time: Will it continue to exist next month or next year, or will a lack of reliable funding lead the vendor to abandon it?
Risk. The next layer assesses the risk, privacy, and security features of the app. Many mental health apps actively aim to avoid falling under the jurisdiction of U.S. federal health care privacy rules, such as the Health Insurance Portability and Accountability Act of 1996, so there is no guarantee that sensitive data supplied to an app will be protected. The true cost of a “free” app often is your patient’s personal mental health information, which the app’s developer may accumulate and sell for profit. Thus, it is wise to check the privacy policy to learn where your patient’s data goes. Furthermore, patients and psychiatrists must be vigilant that malware-infected apps can be uploaded to the app store, which can further compromise privacy.16 You may be surprised to learn that many apps lack a privacy policy, which means there are no protections for personal information or safeguards against the misuse of mental health data.17 Checking that an app at least promises to digitally protect mental health data through encryption and secure storage also is a good step.
The goal of considering these factors is not to create a score, but rather to be aware of them and consider them in the context of the specific app, patient, and clinical situation. Doing so helps determine whether the app meets the appropriate risk, privacy, and security standards for your patient.
Evidence. The next layer of the evaluation framework is evidence. The goal is to seek an app with clinical evidence of effectiveness. Simply put, if a patient is going to use an app, he should use one that works. An app without formal evidence may be effective, but it is important to make sure the patient is aware that these claims have not been verified. Many apps claim that they offer cognitive-behavioral therapy or mindfulness therapy, but few deliver on such claims.18 It is wise to try an app before recommending it to a patient to ensure that it does what it claims it does, and does not offer dangerous or harmful recommendations.
Ease of use. Across all health apps, there is growing recognition that most downloaded apps are never used. Patient engagement with mental health apps appears to rapidly decline over the first week of use.19 There also is emerging evidence that many apps are not user-friendly. A recent study of several common mood-tracking apps found that patients with depression had difficulty entering and accessing their data.20 Because many psychiatric disorders are chronic or last at least several months, it is especially important to consider how engaging and usable the app will be for your patient. Usability varies from patient to patient, so it is best to check directly with your patient regarding his comfort with apps and mobile technology. Offering check-ins and support to help patients keep on track with apps may be critical for successful outcomes.
Interoperability. The final layer of the model is data sharing and interoperability. It is important to determine if the data collected or generated by the app are available to you, the patient, the treatment team, and others involved in the patient’s care. As mental health treatment moves toward integrated care, apps that fragment care (by not sharing information) impede care. Check if the app can share data with an electronic medical record, or if there is a plan to review and act on data from the app as part of your patient’s treatment plan.

More information about the APA App Evaluation Model, including additional factors to consider within each layer, is available from the APA for free at https://www.psychiatry.org/psychiatrists/practice/mental-health-apps/app-evaluation-model. For a sample of factors to consider when evaluating a mental health app, see the Table.
A reasonable strategy
Although the APA App Evaluation Model does not endorse any particular app, it can help guide more informed decision-making. As the evidence on mental health apps continues to evolve, it will become easier to make definitive statements on what constitutes a useful app. For now, the best strategy when discussing mental health apps with patients is to combine the use of this model with your clinical judgment.
1. Torous J, Roberts LW. Needed innovation in digital health and smartphone applications for mental health: transparency and trust. JAMA Psychiatry. 2017;74(5):437-438.
2. Mani M, Kavanagh DJ, Hides L, et al. Review and evaluation of mindfulness-based iPhone apps. JMIR Mhealth Uhealth. 2015;3(3):e82. doi: 10.2196/mhealth.4328.
3. Wilson H, Stoyanov SR, Gandabhai S, et al. The quality and accuracy of mobile apps to prevent driving after drinking alcohol. JMIR Mhealth Uhealth. 2016;4(3):e98. doi: 10.2196/mhealth.5961.
4. Larsen ME, Nicholas J, Christensen H. A systematic assessment of smartphone tools for suicide prevention. PLoS One. 2016;11(4):e0152285. doi: 10.1371/journal.pone.0152285.
5. Nicholas J, Larsen ME, Proudfoot J, et al. Mobile apps for bipolar disorder: a systematic review of features and content quality. J Med Internet Res. 2015;17(8):e198. doi: 10.2196/jmir.4581.
6. Shen N, Levitan MJ, Johnson A, et al. Finding a depression app: a review and content analysis of the depression app marketplace. JMIR Mhealth Uhealth. 2015;3(1):e16. doi: 10.2196/mhealth.3713.
7. Davis SW, Oakley-Girvan I. Achieving value in mobile health applications for cancer survivors. J Cancer Surviv. 2017;11(4):498-504.
8. Ben-Zeev D, Brenner CJ, Begale M, et al. Feasibility, acceptability, and preliminary efficacy of a smartphone intervention for schizophrenia. Schizophr Bull. 2014;40(6):1244-1253.
9. Mohr DC, Tomasino KN, Lattie EG, et al. IntelliCare: an eclectic, skills-based app suite for the treatment of depression and anxiety. J Med Internet Res. 2017;19(1):e10. doi: 10.2196/jmir.6645.
10. Tighe J, Shand F, Ridani R, et al. Ibobbly mobile health intervention for suicide prevention in Australian Indigenous youth: a pilot randomised controlled trial. BMJ Open. 2017;7(1):e013518. doi: 10.1136/bmjopen-2016-013518.
11. Firth J, Torous J, Nicholas J, et al. Can smartphone mental health interventions reduce symptoms of anxiety? A meta-analysis of randomized controlled trials. J Affect Disord. 2017;218:15-22.
12. Gay K, Torous J, Joseph A, et al. Digital technology use among individuals with schizophrenia: results of an online survey. JMIR Mental Health. 2016;3(2):e15. doi: 10.2196/mental.5379.
13. Singh K, Drouin K, Newmark LP, et al. Many mobile health apps target high-need, high-cost populations, but gaps remain. Health Aff (Millwood). 2016;35(12):2310-2318.
14. Larsen ME, Nicholas J, Christensen H. Quantifying app store dynamics: longitudinal tracking of mental health apps. JMIR Mhealth Uhealth. 2016;4(3):e96. doi: 10.2196/mhealth.6020.
15. Powell AC, Torous J, Chan S, et al. Interrater reliability of mHealth app rating measures: analysis of top depression and smoking cessation apps. JMIR Mhealth Uhealth. 2016;4(1):e15. doi: 10.2196/mhealth.5176.
16. Ducklin P. Apple’s XcodeGhost malware still in the machine…. https://nakedsecurity.sophos.com/2015/11/09/apples-xcodeghost-malware-still-in-the-machine. Published November 9, 2015. Accessed May 11, 2017.
17. Rosenfeld L, Torous J, Vahia IV. Data security and privacy in apps for dementia: an analysis of existing privacy policies. Am J Geriatr Psychiatry. 2017;25(8):873-877.
18. Torous J, Levin ME, Ahern DK, et al. Cognitive behavioral mobile applications: clinical studies, marketplace overview, and research agenda. Cogn Behav Pract. 2017;24(2):215-225.
19. Owen JE, Jaworski BK, Kuhn E, et al. mHealth in the wild: using novel data to examine the reach, use, and impact of PTSD coach. JMIR Ment Health. 2015;2(1):e7. doi: 10.2196/mental.3935.
20. Sarkar U, Gourley GI, Lyles CR, et al. Usability of commercially available mobile applications for diverse patients. J Gen Intern Med. 2016;31(12):1417-1426.
Have your patients asked you about smartphone apps? If they haven’t yet, they may soon, as interest in apps for mental health continues to expand. There are now >10,000 mental health–related smartphone apps.1 The rapid rise of these apps is partly due to their potential to transform a patient’s smartphone into a monitoring and therapeutic platform, capable of capturing mental health symptoms in real time and delivering on-the-go therapy. Setting aside questions about the potential of mobile health, 2 urgent questions remain for the busy psychiatrist in clinical practice: What is the current evidence base for mental health apps, and what should you tell your patients about them?
For most apps, evidence of efficacy is limited
While the evidence base for mental health smartphone apps continues to expand, for many of these apps, there is no evidence of effectiveness. The growing consensus is that most commercially available apps are not evidence-based and some are even dangerous. For example, researchers who examined >700 mindfulness apps on the iTunes and Google Play stores found that only 4% provided acceptable mindfulness training and education.2 Another study of 58 apps that claimed to offer sobriety assessments found that none had ever been formally evaluated.3 Evidence-based reviews of suicide prevention apps have identified potentially harmful apps,4 and studies evaluating apps for bipolar disorder5 and depression6 have yielded similar results—few have any evidence supporting their use, and some offer dangerous and harmful advice. For example, researchers found that one app for bipolar disorder advised patients who are experiencing a manic episode to drink alcohol.5 Currently, the vast majority of commercially available apps are not appropriate for clinical care. This finding is not unique to mental health; similar findings have been reported for apps for cancer.7 The bottom line is that the apps that your patients are finding, and perhaps already using, may not be useful or effective.
However, early studies have demonstrated efficacy of some apps for several conditions, including schizophrenia,8 depression,9 anxiety disorders,10 and suicidal ideation.11 Although many of the apps evaluated in these studies are not available to the public, or still require large-scale assessment before they are ready for mainstream clinical care, this research demonstrates that mental health apps can help improve treatment outcomes. As this research develops, a wave of evidence-based and effective mental health apps may be available in the near future.
Although it is unknown how many patients are presently using mental health apps, there is strong anecdotal evidence that an increasing number of patients who use these apps and other forms of digital technology are finding some benefits. In many cases, patients may actually be ahead of the research. For example, one study that conducted an online survey of patients with schizophrenia noted that some patients are using their smartphones to play music to help block auditory hallucinations.12
Why online reviews are of limited use
As this evidence continues to mature, and with an ever-growing number of mental health apps available on commercial marketplaces, busy psychiatrists need to navigate this complex space. Even psychiatrists who decide to not use apps as part of care still need to be knowledgeable about them, because patients are likely to ask about the benefits of using apps, and they will expect an informed response. How would you reply if your patient asked you about a new mood-tracking app he or she recently heard about? On what would you base your recommendation and opinion?
Reading online app reviews for guidance is not a good solution. A recent study found little relationship between the star ratings of health apps and the quality of those apps,13 which suggests that a 5-star rating on the app store is of limited use.
Unlike medications whose ingredients do not change over time, or manualized psychotherapies that use specific protocols, mental health apps are dynamic and constantly changing.14 Think of how often the apps on your smartphone update. Thus, the version of a mental health app that your patient downloads today may be very different from the version that received a favorable user review last month. And just as there is no single medication or therapy that is ideal for every patient, neither is there a single “best” app for all patients with the same disorder. Picking an app is a personal decision that cannot be made based on a single score or numeric rating. Furthermore, the validity of app rating systems is unclear. One study found a wide variation in the interrater reliability of measures used to evaluate apps from sources that included PsyberGuide, the Anxiety and Depression Association of America, and the research literature. Quality measures such as effectiveness, ease of use, and performance had relatively poor interrater reliability.15 This means that, for example, an app that one patient finds “easy to use” may be difficult to use for another. Thus, providing patients with suggestions based on an app’s ratings may result in providing information that sounds useful, but often is misleading.
A model for evaluating apps
One possible solution is a risk-based and personalized assessment approach to evaluating mental health apps. Although it does not offer scoring or recommendations of specific apps, the American Psychiatric Association (APA) App Evaluation Model (Figure) provides a framework to guide discussion and informed decision-making about apps. (The authors of this article helped create this model, but receive no compensation for that volunteer work.) The pyramid shape reflects the hierarchical nature of the model. To begin the process, start at the base of the pyramid and work upward.
Ground. First, consider the context of the app by determining basic facts, such as who made it, how much it costs, and its technology requirements. This ground layer establishes the credibility of the app’s creator by questioning his or her reputation, ability to update the app, and funding sources. Understanding the app’s business model also will help you determine whether the app will stand the test of time: Will it continue to exist next month or next year, or will a lack of reliable funding lead the vendor to abandon it?
Risk. The next layer assesses the risk, privacy, and security features of the app. Many mental health apps actively aim to avoid falling under the jurisdiction of U.S. federal health care privacy rules, such as the Health Insurance Portability and Accountability Act of 1996, so there is no guarantee that sensitive data supplied to an app will be protected. The true cost of a “free” app often is your patient’s personal mental health information, which the app’s developer may accumulate and sell for profit. Thus, it is wise to check the privacy policy to learn where your patient’s data goes. Furthermore, patients and psychiatrists must be vigilant that malware-infected apps can be uploaded to the app store, which can further compromise privacy.16 You may be surprised to learn that many apps lack a privacy policy, which means there are no protections for personal information or safeguards against the misuse of mental health data.17 Checking that an app at least promises to digitally protect mental health data through encryption and secure storage also is a good step.
The goal of considering these factors is not to create a score, but rather to be aware of them and consider them in the context of the specific app, patient, and clinical situation. Doing so helps determine whether the app meets the appropriate risk, privacy, and security standards for your patient.
Evidence. The next layer of the evaluation framework is evidence. The goal is to seek an app with clinical evidence of effectiveness. Simply put, if a patient is going to use an app, he should use one that works. An app without formal evidence may be effective, but it is important to make sure the patient is aware that these claims have not been verified. Many apps claim that they offer cognitive-behavioral therapy or mindfulness therapy, but few deliver on such claims.18 It is wise to try an app before recommending it to a patient to ensure that it does what it claims it does, and does not offer dangerous or harmful recommendations.
Ease of use. Across all health apps, there is growing recognition that most downloaded apps are never used. Patient engagement with mental health apps appears to rapidly decline over the first week of use.19 There also is emerging evidence that many apps are not user-friendly. A recent study of several common mood-tracking apps found that patients with depression had difficulty entering and accessing their data.20 Because many psychiatric disorders are chronic or last at least several months, it is especially important to consider how engaging and usable the app will be for your patient. Usability varies from patient to patient, so it is best to check directly with your patient regarding his comfort with apps and mobile technology. Offering check-ins and support to help patients keep on track with apps may be critical for successful outcomes.
Interoperability. The final layer of the model is data sharing and interoperability. It is important to determine if the data collected or generated by the app are available to you, the patient, the treatment team, and others involved in the patient’s care. As mental health treatment moves toward integrated care, apps that fragment care (by not sharing information) impede care. Check if the app can share data with an electronic medical record, or if there is a plan to review and act on data from the app as part of your patient’s treatment plan.
More information about the APA App Evaluation Model, including additional factors to consider within each layer, is available from the APA for free at https://www.psychiatry.org/psychiatrists/practice/mental-health-apps/app-evaluation-model. For a sample of factors to consider when evaluating a mental health app, see the Table.
A reasonable strategy
Although the APA App Evaluation Model does not endorse any particular app, it can help guide more informed decision-making. As the evidence on mental health apps continues to evolve, it will become easier to make definitive statements on what constitutes a useful app. For now, the best strategy when discussing mental health apps with patients is to combine the use of this model with your clinical judgment.
Have your patients asked you about smartphone apps? If they haven’t yet, they may soon, as interest in apps for mental health continues to expand. There are now >10,000 mental health–related smartphone apps.1 The rapid rise of these apps is partly due to their potential to transform a patient’s smartphone into a monitoring and therapeutic platform, capable of capturing mental health symptoms in real time and delivering on-the-go therapy. Setting aside questions about the potential of mobile health, 2 urgent questions remain for the busy psychiatrist in clinical practice: What is the current evidence base for mental health apps, and what should you tell your patients about them?
For most apps, evidence of efficacy is limited
While the evidence base for mental health smartphone apps continues to expand, for many of these apps, there is no evidence of effectiveness. The growing consensus is that most commercially available apps are not evidence-based and some are even dangerous. For example, researchers who examined >700 mindfulness apps on the iTunes and Google Play stores found that only 4% provided acceptable mindfulness training and education.2 Another study of 58 apps that claimed to offer sobriety assessments found that none had ever been formally evaluated.3 Evidence-based reviews of suicide prevention apps have identified potentially harmful apps,4 and studies evaluating apps for bipolar disorder5 and depression6 have yielded similar results—few have any evidence supporting their use, and some offer dangerous and harmful advice. For example, researchers found that one app for bipolar disorder advised patients who are experiencing a manic episode to drink alcohol.5 Currently, the vast majority of commercially available apps are not appropriate for clinical care. This finding is not unique to mental health; similar findings have been reported for apps for cancer.7 The bottom line is that the apps that your patients are finding, and perhaps already using, may not be useful or effective.
However, early studies have demonstrated efficacy of some apps for several conditions, including schizophrenia,8 depression,9 anxiety disorders,10 and suicidal ideation.11 Although many of the apps evaluated in these studies are not available to the public, or still require large-scale assessment before they are ready for mainstream clinical care, this research demonstrates that mental health apps can help improve treatment outcomes. As this research develops, a wave of evidence-based and effective mental health apps may be available in the near future.
Although it is unknown how many patients are presently using mental health apps, there is strong anecdotal evidence that an increasing number of patients who use these apps and other forms of digital technology are finding some benefits. In many cases, patients may actually be ahead of the research. For example, one study that conducted an online survey of patients with schizophrenia noted that some patients are using their smartphones to play music to help block auditory hallucinations.12
Why online reviews are of limited use
As this evidence continues to mature, and with an ever-growing number of mental health apps available on commercial marketplaces, busy psychiatrists need to navigate this complex space. Even psychiatrists who decide to not use apps as part of care still need to be knowledgeable about them, because patients are likely to ask about the benefits of using apps, and they will expect an informed response. How would you reply if your patient asked you about a new mood-tracking app he or she recently heard about? On what would you base your recommendation and opinion?
Reading online app reviews for guidance is not a good solution. A recent study found little relationship between the star ratings of health apps and the quality of those apps,13 which suggests that a 5-star rating on the app store is of limited use.
Unlike medications whose ingredients do not change over time, or manualized psychotherapies that use specific protocols, mental health apps are dynamic and constantly changing.14 Think of how often the apps on your smartphone update. Thus, the version of a mental health app that your patient downloads today may be very different from the version that received a favorable user review last month. And just as there is no single medication or therapy that is ideal for every patient, neither is there a single “best” app for all patients with the same disorder. Picking an app is a personal decision that cannot be made based on a single score or numeric rating. Furthermore, the validity of app rating systems is unclear. One study found a wide variation in the interrater reliability of measures used to evaluate apps from sources that included PsyberGuide, the Anxiety and Depression Association of America, and the research literature. Quality measures such as effectiveness, ease of use, and performance had relatively poor interrater reliability.15 This means that, for example, an app that one patient finds “easy to use” may be difficult to use for another. Thus, providing patients with suggestions based on an app’s ratings may result in providing information that sounds useful, but often is misleading.
A model for evaluating apps
One possible solution is a risk-based and personalized assessment approach to evaluating mental health apps. Although it does not offer scoring or recommendations of specific apps, the American Psychiatric Association (APA) App Evaluation Model (Figure) provides a framework to guide discussion and informed decision-making about apps. (The authors of this article helped create this model, but receive no compensation for that volunteer work.) The pyramid shape reflects the hierarchical nature of the model. To begin the process, start at the base of the pyramid and work upward.
Ground. First, consider the context of the app by determining basic facts, such as who made it, how much it costs, and its technology requirements. This ground layer establishes the credibility of the app’s creator by questioning his or her reputation, ability to update the app, and funding sources. Understanding the app’s business model also will help you determine whether the app will stand the test of time: Will it continue to exist next month or next year, or will a lack of reliable funding lead the vendor to abandon it?
Risk. The next layer assesses the risk, privacy, and security features of the app. Many mental health apps actively aim to avoid falling under the jurisdiction of U.S. federal health care privacy rules, such as the Health Insurance Portability and Accountability Act of 1996, so there is no guarantee that sensitive data supplied to an app will be protected. The true cost of a “free” app often is your patient’s personal mental health information, which the app’s developer may accumulate and sell for profit. Thus, it is wise to check the privacy policy to learn where your patient’s data goes. Furthermore, patients and psychiatrists must be vigilant that malware-infected apps can be uploaded to the app store, which can further compromise privacy.16 You may be surprised to learn that many apps lack a privacy policy, which means there are no protections for personal information or safeguards against the misuse of mental health data.17 Checking that an app at least promises to digitally protect mental health data through encryption and secure storage also is a good step.
The goal of considering these factors is not to create a score, but rather to be aware of them and consider them in the context of the specific app, patient, and clinical situation. Doing so helps determine whether the app meets the appropriate risk, privacy, and security standards for your patient.
Evidence. The next layer of the evaluation framework is evidence. The goal is to seek an app with clinical evidence of effectiveness. Simply put, if a patient is going to use an app, he should use one that works. An app without formal evidence may be effective, but it is important to make sure the patient is aware that these claims have not been verified. Many apps claim that they offer cognitive-behavioral therapy or mindfulness therapy, but few deliver on such claims.18 It is wise to try an app before recommending it to a patient to ensure that it does what it claims it does, and does not offer dangerous or harmful recommendations.
Ease of use. Across all health apps, there is growing recognition that most downloaded apps are never used. Patient engagement with mental health apps appears to rapidly decline over the first week of use.19 There also is emerging evidence that many apps are not user-friendly. A recent study of several common mood-tracking apps found that patients with depression had difficulty entering and accessing their data.20 Because many psychiatric disorders are chronic or last at least several months, it is especially important to consider how engaging and usable the app will be for your patient. Usability varies from patient to patient, so it is best to check directly with your patient regarding his comfort with apps and mobile technology. Offering check-ins and support to help patients keep on track with apps may be critical for successful outcomes.
Interoperability. The final layer of the model is data sharing and interoperability. It is important to determine if the data collected or generated by the app are available to you, the patient, the treatment team, and others involved in the patient’s care. As mental health treatment moves toward integrated care, apps that fragment care (by not sharing information) impede care. Check if the app can share data with an electronic medical record, or if there is a plan to review and act on data from the app as part of your patient’s treatment plan.
More information about the APA App Evaluation Model, including additional factors to consider within each layer, is available from the APA for free at https://www.psychiatry.org/psychiatrists/practice/mental-health-apps/app-evaluation-model. For a sample of factors to consider when evaluating a mental health app, see the Table.
A reasonable strategy
Although the APA App Evaluation Model does not endorse any particular app, it can help guide more informed decision-making. As the evidence on mental health apps continues to evolve, it will become easier to make definitive statements on what constitutes a useful app. For now, the best strategy when discussing mental health apps with patients is to combine the use of this model with your clinical judgment.
1. Torous J, Roberts LW. Needed innovation in digital health and smartphone applications for mental health: transparency and trust. JAMA Psychiatry. 2017;74(5):437-438.
2. Mani M, Kavanagh DJ, Hides L, et al. Review and evaluation of mindfulness-based iPhone apps. JMIR Mhealth Uhealth. 2015;3(3):e82. doi: 10.2196/mhealth.4328.
3. Wilson H, Stoyanov SR, Gandabhai S, et al. The quality and accuracy of mobile apps to prevent driving after drinking alcohol. JMIR Mhealth Uhealth. 2016;4(3):e98. doi: 10.2196/mhealth.5961.
4. Larsen ME, Nicholas J, Christensen H. A systematic assessment of smartphone tools for suicide prevention. PLoS One. 2016;11(4):e0152285. doi: 10.1371/journal.pone.0152285.
5. Nicholas J, Larsen ME, Proudfoot J, et al. Mobile apps for bipolar disorder: a systematic review of features and content quality. J Med Internet Res. 2015;17(8):e198. doi: 10.2196/jmir.4581.
6. Shen N, Levitan MJ, Johnson A, et al. Finding a depression app: a review and content analysis of the depression app marketplace. JMIR Mhealth Uhealth. 2015;3(1):e16. doi: 10.2196/mhealth.3713.
7. Davis SW, Oakley-Girvan I. Achieving value in mobile health applications for cancer survivors. J Cancer Surviv. 2017;11(4):498-504.
8. Ben-Zeev D, Brenner CJ, Begale M, et al. Feasibility, acceptability, and preliminary efficacy of a smartphone intervention for schizophrenia. Schizophr Bull. 2014;40(6):1244-1253.
9. Mohr DC, Tomasino KN, Lattie EG, et al. IntelliCare: an eclectic, skills-based app suite for the treatment of depression and anxiety. J Med Internet Res. 2017;19(1):e10. doi: 10.2196/jmir.6645.
10. Tighe J, Shand F, Ridani R, et al. Ibobbly mobile health intervention for suicide prevention in Australian Indigenous youth: a pilot randomised controlled trial. BMJ Open. 2017;7(1):e013518. doi: 10.1136/bmjopen-2016-013518.
11. Firth J, Torous J, Nicholas J, et al. Can smartphone mental health interventions reduce symptoms of anxiety? A meta-analysis of randomized controlled trials. J Affect Disord. 2017;218:15-22.
12. Gay K, Torous J, Joseph A, et al. Digital technology use among individuals with schizophrenia: results of an online survey. JMIR Mental Health. 2016;3(2):e15. doi: 10.2196/mental.5379.
13. Singh K, Drouin K, Newmark LP, et al. Many mobile health apps target high-need, high-cost populations, but gaps remain. Health Aff (Millwood). 2016;35(12):2310-2318.
14. Larsen ME, Nicholas J, Christensen H. Quantifying app store dynamics: longitudinal tracking of mental health apps. JMIR Mhealth Uhealth. 2016;4(3):e96. doi: 10.2196/mhealth.6020.
15. Powell AC, Torous J, Chan S, et al. Interrater reliability of mHealth app rating measures: analysis of top depression and smoking cessation apps. JMIR Mhealth Uhealth. 2016;4(1):e15. doi: 10.2196/mhealth.5176.
16. Ducklin P. Apple’s XcodeGhost malware still in the machine…. https://nakedsecurity.sophos.com/2015/11/09/apples-xcodeghost-malware-still-in-the-machine. Published November 9, 2015. Accessed May 11, 2017.
17. Rosenfeld L, Torous J, Vahia IV. Data security and privacy in apps for dementia: an analysis of existing privacy policies. Am J Geriatr Psychiatry. 2017;25(8):873-877.
18. Torous J, Levin ME, Ahern DK, et al. Cognitive behavioral mobile applications: clinical studies, marketplace overview, and research agenda. Cogn Behav Pract. 2017;24(2):215-225.
19. Owen JE, Jaworski BK, Kuhn E, et al. mHealth in the wild: using novel data to examine the reach, use, and impact of PTSD coach. JMIR Ment Health. 2015;2(1):e7. doi: 10.2196/mental.3935.
20. Sarkar U, Gourley GI, Lyles CR, et al. Usability of commercially available mobile applications for diverse patients. J Gen Intern Med. 2016;31(12):1417-1426.
1. Torous J, Roberts LW. Needed innovation in digital health and smartphone applications for mental health: transparency and trust. JAMA Psychiatry. 2017;74(5):437-438.
2. Mani M, Kavanagh DJ, Hides L, et al. Review and evaluation of mindfulness-based iPhone apps. JMIR Mhealth Uhealth. 2015;3(3):e82. doi: 10.2196/mhealth.4328.
3. Wilson H, Stoyanov SR, Gandabhai S, et al. The quality and accuracy of mobile apps to prevent driving after drinking alcohol. JMIR Mhealth Uhealth. 2016;4(3):e98. doi: 10.2196/mhealth.5961.
4. Larsen ME, Nicholas J, Christensen H. A systematic assessment of smartphone tools for suicide prevention. PLoS One. 2016;11(4):e0152285. doi: 10.1371/journal.pone.0152285.
5. Nicholas J, Larsen ME, Proudfoot J, et al. Mobile apps for bipolar disorder: a systematic review of features and content quality. J Med Internet Res. 2015;17(8):e198. doi: 10.2196/jmir.4581.
6. Shen N, Levitan MJ, Johnson A, et al. Finding a depression app: a review and content analysis of the depression app marketplace. JMIR Mhealth Uhealth. 2015;3(1):e16. doi: 10.2196/mhealth.3713.
7. Davis SW, Oakley-Girvan I. Achieving value in mobile health applications for cancer survivors. J Cancer Surviv. 2017;11(4):498-504.
8. Ben-Zeev D, Brenner CJ, Begale M, et al. Feasibility, acceptability, and preliminary efficacy of a smartphone intervention for schizophrenia. Schizophr Bull. 2014;40(6):1244-1253.
9. Mohr DC, Tomasino KN, Lattie EG, et al. IntelliCare: an eclectic, skills-based app suite for the treatment of depression and anxiety. J Med Internet Res. 2017;19(1):e10. doi: 10.2196/jmir.6645.
10. Tighe J, Shand F, Ridani R, et al. Ibobbly mobile health intervention for suicide prevention in Australian Indigenous youth: a pilot randomised controlled trial. BMJ Open. 2017;7(1):e013518. doi: 10.1136/bmjopen-2016-013518.
11. Firth J, Torous J, Nicholas J, et al. Can smartphone mental health interventions reduce symptoms of anxiety? A meta-analysis of randomized controlled trials. J Affect Disord. 2017;218:15-22.
12. Gay K, Torous J, Joseph A, et al. Digital technology use among individuals with schizophrenia: results of an online survey. JMIR Mental Health. 2016;3(2):e15. doi: 10.2196/mental.5379.
13. Singh K, Drouin K, Newmark LP, et al. Many mobile health apps target high-need, high-cost populations, but gaps remain. Health Aff (Millwood). 2016;35(12):2310-2318.
14. Larsen ME, Nicholas J, Christensen H. Quantifying app store dynamics: longitudinal tracking of mental health apps. JMIR Mhealth Uhealth. 2016;4(3):e96. doi: 10.2196/mhealth.6020.
15. Powell AC, Torous J, Chan S, et al. Interrater reliability of mHealth app rating measures: analysis of top depression and smoking cessation apps. JMIR Mhealth Uhealth. 2016;4(1):e15. doi: 10.2196/mhealth.5176.
16. Ducklin P. Apple’s XcodeGhost malware still in the machine…. https://nakedsecurity.sophos.com/2015/11/09/apples-xcodeghost-malware-still-in-the-machine. Published November 9, 2015. Accessed May 11, 2017.
17. Rosenfeld L, Torous J, Vahia IV. Data security and privacy in apps for dementia: an analysis of existing privacy policies. Am J Geriatr Psychiatry. 2017;25(8):873-877.
18. Torous J, Levin ME, Ahern DK, et al. Cognitive behavioral mobile applications: clinical studies, marketplace overview, and research agenda. Cogn Behav Pract. 2017;24(2):215-225.
19. Owen JE, Jaworski BK, Kuhn E, et al. mHealth in the wild: using novel data to examine the reach, use, and impact of PTSD coach. JMIR Ment Health. 2015;2(1):e7. doi: 10.2196/mental.3935.
20. Sarkar U, Gourley GI, Lyles CR, et al. Usability of commercially available mobile applications for diverse patients. J Gen Intern Med. 2016;31(12):1417-1426.

Neuromodulatory options for treatment-resistant depression
The emergence of treatment-resistant depression (TRD) poses a great clinical and public health challenge. There is no clear consensus on criteria to define TRD. The criteria range from failure to respond to 4 weeks of a single antidepressant to failure to respond to a single trial of electroconvulsive therapy (ECT).1
Neuromodulatory treatments for depression involve electrical stimulation of the brain through invasive or noninvasive methods. In this article, we discuss criteria for defining TRD, and compare the advantages and disadvantages of 4 neuromodulatory treatment options—ECT, vagus nerve stimulation (VNS), repetitive transcranial magnetic stimulation (rTMS), and deep brain stimulation (DBS)—for patients with depression who fail to respond to appropriate pharmacologic interventions (Table 1). Most of the studies we discuss selected patients who had severe depression and had not responded to numerous treatment trials.
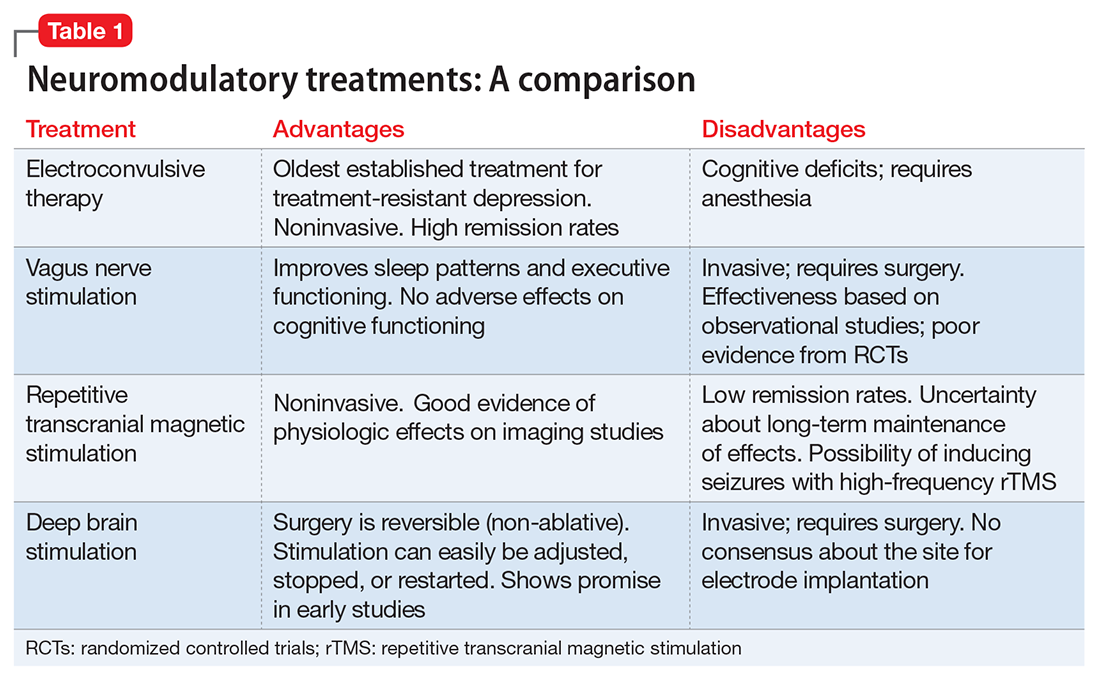
Defining treatment resistance
Thase and Rush2 suggested progressive stages for categorizing TRD, ranging from Stage I (failure of at least 1 adequate trial of antidepressants) to Stage V (failure of adequate treatment with 2 selective serotonin reuptake inhibitors [SSRIs], a tricyclic antidepressant, a monoamine oxidase inhibitor, and a course of bilateral ECT). The Massachusetts General Hospital Staging Model suggested a quantitative scale to help characterize the degree of treatment resistance in which a higher score corresponds to a higher level of resistance.3 For every failed 6-week trial with adequate dose of an antidepressant, the patient is given a score of 1. The patient receives an extra .5 point for failure to respond to optimization of the dosage and augmentation with another medication. The patient also is given 3 points for failure to respond to ECT. Souery et al4,5 proposed a model in which they defined TRD as a failure to respond after ≥1 adequate antidepressant trials of ≥12 weeks.
Treatment resistance often is the result of inadequate treatment of depressive symptoms. Inadequate treatment includes an inadequate dose of antidepressants and/or an inadequate duration of treatment. Treatment of depression also is often complicated by medical (cardiovascular, neurologic, endocrine disorders) and psychiatric (substance abuse disorders, personality disorders) comorbidities (Table 2). Patients with such comorbidities are at increased risk of mortality, and have lower response rates and increased morbidity.6
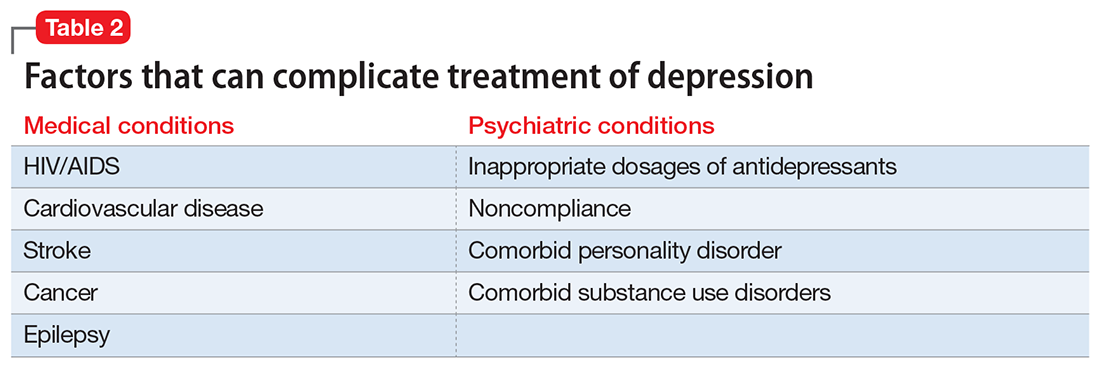
Electroconvulsive therapy
ECT involves the application of electric current to induce a self-limiting seizure. It affects multiple brain functions to produce its antidepressant effects. Patients with depression have a reduced concentration of γ-aminobutyric acid (GABA) in their plasma, CSF, and cortex. ECT increases GABAergic transmission in cortical circuits as demonstrated by increased levels of GABA in the occipital cortex, which may be responsible for ECT’s antidepressant effects.7 Sensitization of the 5-HT1A receptors and increased dopamine receptor binding in the striatum also have been associated with the antidepressant action of ECT.8 The antidepressant effects of ECT also can be attributed to increased neuroplasticity, as evidenced by increased neurotrophic factors and cell proliferation in animal models.9 Dysfunction of the HPA axis has long been associated with depressive disorders; ECT improves this dysfunction, as evidenced by normalization of the dexamethasone suppression test in patients who receive ECT.7
The results of neuroimaging studies exploring the effects of ECT vary widely based on the specific neuroimaging method, population, and statistical methods used to assess the changes. Some of the most consistent findings include reduced glucose metabolism in the frontal brain regions; reduced glucose metabolism in the hippocampus and medial temporal lobes; and reduction in functional connectivity in the anterior cingulate, parietal, medical frontal, and dorsolateral prefrontal cortex (DLPFC).10
Randomized control trials (RCTs) have established the superiority of ECT over pharmacotherapy and sham ECT. Compared with other neuromodulatory treatments, ECT has higher remission rates. On average, the remission rate among patients receiving ECT whose depression did not respond to pharmacotherapy is approximately 48%; this increases to 64.9% among patients who previously had responded to a medication.11
Some earlier trials found bilateral ECT to be more effective than unilateral ECT.12 Recent studies suggest that high-dose unilateral ECT (6 times the seizure threshold) is as effective as bilateral ECT.13 Studies have shown no significant differences in efficacy or treatment outcomes between twice- and thrice-weekly ECT regimens. Some studies suggest that twice-weekly ECT may be associated with a lower risk of short-term cognitive impairment compared with thrice-weekly ECT.14
In highly refractory cases, the effects of ECT can be augmented by using pre-treatment strategies such as hyperventilation, which may increase the duration of the seizure, and remifentanil, which helps reduce the anticonvulsant effect of agents used for anesthesia.15 Advanced age, psychotic features, resistance to pharmacotherapy, and comorbid personality disorders predict poor response to ECT.16
Adverse effects. Concerns about cognitive deficits secondary to ECT may curtail its use. Retrograde and anterograde amnesia are the most common deficits observed acutely after ECT.12 Other commonly affected cognitive functions include processing speed, attention/working memory, verbal and visual episodic memory, spatial problem solving, and executive functioning. The specific patterns of these deficits (in terms of duration and severity) vary between studies. In general, high-dose, thrice-weekly ECT and bilateral ECT are associated with greater cognitive deficits, whereas twice-weekly ECT and unilateral ECT are associated with a lower risk of cognitive adverse effects.12 A recent meta-analysis by Semkovska and McLoughlin17 found that most cognitive deficits seen after ECT are limited to the first 3 days after treatment. The authors of this meta-analysis concluded that these impairments improve over time and approach baseline 2 weeks after treatment. In fact, some of these impairments (processing speed, working memory, anterograde memory, and some aspects of executive function) improved beyond baseline after 15 days of treatment.17 The need for anesthesia and associated potential adverse effects also are a cause of concern with ECT.
Combining ECT with medication. Several patient-specific factors, including medication regimen and comorbid medical conditions, need to be considered before using ECT in combination with pharmacotherapy. Although most antipsychotics are safe to use with ECT, concomitant use of agents with higher antihistaminic properties may increase the risk of delirium. The risk of delirium also is increased with the use of anticonvulsants and mood stabilizers (eg, lithium) because these agents increase the seizure threshold. The potential for drug interactions may affect the choice of the anesthetic agents. Also, SSRIs and serotonin-norepinephrine reuptake inhibitors can increase the duration of induced seizures.18
Vagus nerve stimulation
VNS, in which an implanted device stimulates the vagus nerve with electrical impulses, initially was used to reduce the frequency of seizures in patients with epilepsy and treatment-resistant partial onset seizures.19 VNS was FDA-approved for TRD in July 2005.20 One VNS system, the NCP System, consists of an implantable, multi-programmable generator, known as a pulse generator, that is subcutaneously placed in the anterior chest wall during an outpatient surgical procedure. Separate bipolar nerve-stimulating electrodes are surgically wrapped around the left cervical vagus nerve, and then connected to the generator via a tunneling procedure. A telemetric wand is subsequently linked to a portable computer and used to adjust stimulation parameters.21,22
Support for using VNS for TRD came from a multitude of investigations and observations. Harden et al23 and Elger et al24 prospectively evaluated epileptic patients with standard depression symptom severity rating scales. They found that VNS was associated with statistically significant improvements in mood that were not related to reductions in seizures.23,24
The mechanism of action of VNS is not clear. Earlier researchers had found evidence that VNS affected brain regions associated with norepinephrine25 and serotonin systems26; both of these neurotransmitters have been implicated in the pathophysiology of depression. Positron emission tomography studies conducted during VNS treatment of epilepsy showed metabolic changes in cortical and subcortical areas of the brain, including the amygdala, hippocampus, and cingulate gyrus, all structures implicated in the pathophysiology of mood disorders.27
Most studies conducted to evaluate the efficacy of VNS have been observational, looking at depression ratings before and after treatment with VNS. The short-term studies measured the difference in depression rating scales at baseline and after 10 weeks of treatment. In most of these studies, treatment with VNS resulted in a statistically significant drop in depression rating scales scores, such as on the Hamilton Depression Rating Scale (HAM-D). Based on the study design and number of study participants, response rates have varied from 13%28 to 40%,29 whereas remission rates have varied from 15.3%30 to 28%.31 More than one-half of the reduction in symptoms occurred after 6 weeks of treatment.30 In longer-term follow-up studies, the antidepressant effect generally was sustained over time. Response rates remained essentially unchanged, but the remission rates increased to approximately 29%.29 Only 1 RCT has compared patients with controls; it found no significant differences in the response or remission rates between active VNS and sham VNS.32 In this study, all patients had VNS implanted, but in the control group, the VNS was never turned on.32 In a meta-analysis conducted by Martin and Martín-Sánchez,33 31.8% (95% confidence interval [CI], 23.2% to 41.8%; P < .001) of patients treated with VNS had a significant reduction in HAM-D scores. The response rate in patients with TRD ranged from 27% to 37% and the remission rate was approximately 13%. In studies that followed patients over longer periods, both the remission and response rates increased over time.34
Recent evidence suggests that the effectiveness of VNS may depend on the stimulation level. A multi-center double-blind study randomized patients to receive either a low (0.25 mA current, 130-millisecond pulse width), medium (0.5e1.0 mA, 250 millisecond), or high (1.25e1.5 mA, 250 millisecond) dose of VNS.35 Although all dose levels were associated with improvement in symptoms, a statistically significant durability in response was associated with the medium- and high-dose treatments.
Adverse effects. VNS has no major adverse effects on cognitive functioning, and some studies have found improvement in executive functioning that corresponded to improvement in depressive symptoms.30 VNS also may result in improved sleep patterns as evidenced by EEG changes.31 The most commonly reported adverse effects include pain in the incision site, hoarseness of voice, throat pain, and neck pain.36
Repetitive transcranial magnetic stimulation
rTMS is a noninvasive technique that uses high-intensity magnetic impulses to stimulate cortical neurons. A magnetic field is produced when current passes through a coil, which in turn causes electrical stimulation in the cortical neurons that results in transient changes in the excitability of the cortical neurons.37 Although many stimulation parameters exist for TMS, high-frequency stimulation to the left prefrontal cortex (HFL-rTMS) and low-frequency stimulation to the right prefrontal cortex (LFR-rTMS) have been shown most efficacious for treating depression.38 High-frequency (5 Hz to 20 Hz) stimulation using rTMS increases cortical neuron excitability, whereas low-frequency (approximately 1 Hz) is associated with reduced cortical neuron excitability.39 The choice of targeting the DLPFC stems from a large body of functional neuroimaging studies that have shown reduction in activity/blood flow in the left DLPFC and abnormal activity/blood flow in the right DLPFC.40
There is no dearth of RCTs evaluating the efficacy of rTMS vs sham rTMS (where no magnetic stimulation was provided). In a meta-analysis of 8 RCTs, low-frequency rTMS applied to the right DLPFC was associated with a remission rate of approximately 34.6%, compared with a 9.7% remission rate with sham rTMS.41 A response rate of approximately 38.2% was observed with HFL-rTMS, compared with a response rate of 15.1% for sham rTMS.41
Gaynes et al42 conducted a meta-analysis to determine the efficacy of rTMS in TRD. They found that for patients with TRD, rTMs produced a response rate of 29% and a remission rate of 30%. In long-term, naturalistic, observational studies, the response rates and remission rates were much higher (58% and 37.1%, respectively).43 Over a 1-year follow-up, almost two-thirds of patients continued to meet criteria for response to treatment.44 Trials comparing HFL-rTMS and LFR-rTMS have found no significant differences in efficacy.45
Advanced age, psychotic symptoms, and a longer duration of the current depressive episode predict poor response to rTMS. Also, imaging studies have shown that a lower metabolism in cerebellar, temporal, anterior cingulate, and occipital parts of the brain correlate with better response to HFL-rTMS.46,47
Adverse effects. The major adverse effect associated with rTMS is the risk of inducing seizures, which is more commonly associated with high-frequency rTMS. Other common adverse effects include headache, facial muscle twitching, and tinnitus.37
Deep brain stimulation
DBS is an invasive stereotactic surgical procedure. It involves unilateral or bilateral placement of electrodes at neuroanatomical locations to deliver continuous stimulation from a subcutaneously implanted pulse generator.48 In the past, destructive surgical procedures were used to treat intractable depression. Surgeries such as anterior cingulotomy, anterior capsulotomy, subcaudate tractotomy, and limbic leucotomy have been shown to effectively reduce depressive symptoms.49 The advantages of DBS over destructive procedures include the fact that DBS is reversible and that the stimulation levels can easily be adjusted, and the treatment can easily be stopped or restarted.
There is no consensus on the optimal anatomic locations for the electrode implantation in DBS. Electrodes have been implanted in the subcallosal cingulate gyrus, inferior thalamic peduncle, ventral capsule/ventral striatum, superolateral branch of the medial forebrain bundle (MFB), and nucleus accumbens.
The choice of anatomic locations stems from the large body of neuroimaging literature characterizing functional changes associated with acute depression and response to treatment. The electrode placement targets “nodes” that form an integral part of the affected neural circuits that are responsible for regulating depressive symptoms.50 Increased metabolic activity and blood flow to the subgenual cingulate gyrus and reduction in the blood flow to the DLPFC and the striatum have been associated with active depressed states. Response to antidepressant treatment has been associated with reversal of these findings.51 Functional magnetic resonance imaging studies have consistently shown increased activity in the amygdala in response to negative stimuli among patients with depression.
Regardless of the site of electrode placement, studies have reported symptomatic improvement among patients with depression who are treated with DBS. In 2 case reports, the electrode was implanted in the inferior thalamic peduncle.52,53 Each study had 1 participant, and each patient remitted.52,53
Placement of the electrodes in the nucleus accumbens resulted in a response rate of 45% in 1 study,54 whereas in a different study, all patients reported improvement in anhedonia.55 A response rate of 71% and a remission rate of 35% were observed in a study in which the electrode was implanted in the ventral capsule/ventral striatum area.56
Berlim et al57 published a systematic review and exploratory meta-analysis of studies in which the electrode had been implanted in the subgenual cingulate cortex. At 12 months, the response rate was 39.9% (95% CI, 28.4% to 52.8%), and 26.3% (95% CI, 13% to 45.9%) of patients achieved remission. The most significant drop in depression scores was observed 3 to 6 months after the surgery. No significant change in scores was observed between 6 to 12 months after surgery.57
The MFB, specifically the superolateral branch, is emerging as an exciting new target for electrode placement in DBS. Schlaepfer et al58 studied the effects of electrodes implanted bilaterally in the superolateral branch of the MFB. They observed an almost 50% reduction in symptoms by Day 7, and at the last follow-up visit (12 to 33 weeks) 4 of the 6 patients had achieved remission.58 In a recent systematic review, Gálvez et al59 found most studies had high response/remission rates without any significant adverse effects. In a recent study of DBS targeting the MFB, 3 of 4 patients had a >50% reduction in Montgomery-Åsberg Depression Rating Scale scores at the end of first week. Although 1 patient withdrew, 2 of the other 3 patients continued to report a >80% reduction in depressive symptoms, even at Week 26.60
Accurate localization of target areas (white matter tracts) and subsequent electrode placement might be an important factor governing treatment response. Riva-Posse et al61 found that clinical response was seen when the electrodes stimulated 3 specific white matter bundles. Interestingly, nonresponders were converted to responders simply by changing the position of the electrodes to include these white matter tracts.61
Adverse effects. The most common adverse effects noted during studies of DBS include pain at the site of implantation and wound infection. Other adverse effects include lead fracture, transient dysphagia, and other hardware-related problems.49
Sorting out the evidence
In the absence of head-to-head trials, it is difficult to establish a hierarchal algorithm for use of the 4 neuromodulatory treatments discussed in the article. If we were to base our decision solely on the current literature, ECT by far has the most evidence and highest remission rates.11 We can reduce the risk of cognitive deficits by using twice-weekly instead of thrice-weekly ECT, or by using unilateral instead of bilateral ECT.12 Another strategy for reducing adverse effects associated with long-term maintenance ECT is by using it in combination with VNS. ECT and VNS can be used safely concomitantly; ECT can be used to treat acutely worsening depression, and VNS for maintaining the antidepressant effect.62
Aside from ECT, rTMS is the only other treatment that has evidence from RCTs. Although the remission rates are not as high as ECT, its preferable adverse effects profile, noninvasive nature, and comparative low cost (compared with surgical procedures) make it a favorable choice. The Canadian Network for Mood and Anxiety Treatment guidelines suggest rTMS as the first-line treatment for patients who do not respond to pharmacologic treatments.63 ECT can be considered second-line treatment unless the patient has acute suicidal ideation, catatonia, psychotic features, greater treatment resistance, or physical deterioration, in which case ECT should be tried before TMS.63
Among the invasive options, VNS has more evidence and is FDA-approved for TRD. However, DBS has shown great promise in early studies, with remission rates as high as 35%.56 DBS has the advantage of being reversible, and the amount of stimulation can be adjusted easily. Despite early promise, more research is needed before DBS can be widely used in clinical settings.
1. Berlim MT, Turecki G. What is the meaning of treatment resistant/refractory major depression (TRD)? A systematic review of current randomized trials. Eur Neuropsychopharmacol. 2007;17(11):696-707.
2. Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(suppl 13):23-29.
3. Petersen T, Papakostas GI, Posternak MA, et al. Empirical testing of two models for staging antidepressant treatment resistance. J Clin Psychopharmacol. 2005;25(4):336-341.
4. Souery D, Papakostas GI, Trivedi MH. Treatment-resistant depression. J Clin Psychiatry. 2006;67(suppl 6):16-22.
5. Souery D, Amsterdam J, de Montigny C, et al. Treatment resistant depression: methodological overview and operational criteria. Eur Neuropsychopharmacol. 1999;9(1-2):83-91.
6. Evans DL, Charney DS. Mood disorders and medical illness: a major public health problem. Biol. Psychiatry. 2003;54(3):177-180.
7. Sanacora G, Mason GF, Rothman DL, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160(3):577-579.
8. Merkl A, Heuser I, Bajbouj M. Antidepressant electroconvulsive therapy: mechanism of action, recent advances and limitations. Exp Neurol. 2009;219(1):20-26.
9. Perera TD, Coplan JD, Lisanby SH, et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 2007;27(18):4894-4901.
10. Abbott CC, Gallegos P, Rediske N et al. A review of longitudinal electroconvulsive therapy: neuroimaging investigations. J Geriatr Psychiatry Neurol. 2014;27(1):33-46.
11. Heijnen WT, Birkenhäger TK, Wierdsma AI, et al. Antidepressant pharmacotherapy failure and response to subsequent electroconvulsive therapy: a meta-analysis. J Clin Psychopharmacol. 2010;30(5):616-619.
12. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
13. Semkovska M, Landau S, Dunne R et al. Bitemporal versus high-dose unilateral twice-weekly electroconvulsive therapy for depression (EFFECT-Dep): a pragmatic, randomized, non-inferiority trial. Am J Psychiatry. 2016;173(4):408-417.
14. Charlson F, Siskind D, Doi SA, et al. ECT efficacy and treatment course: a systematic review and meta-analysis of twice vs thrice weekly schedules. J Affect Disord. 2012;138(1-2):1-8.
15. Loo CK, Kaill A, Paton P, et al. The difficult-to-treat electroconvulsive therapy patient—strategies for augmenting outcomes. J Affect Disord. 2010;124(3):219-227.
16. de Vreede IM, Burger H, van Vliet IM. Prediction of response to ECT with routinely collected data in major depression. J Affect Disord. 2005;86(2-3):323-327.
17. Semkovska M, McLoughlin DM. Objective cognitive performance associated with electroconvulsive therapy for depression: a systematic review and meta-analysis. Biol Psychiatry. 2010;68(6):568-577.
18. Baghai TC, Marcuse A, Brosch M, et al. The influence of concomitant antidepressant medication on safety, tolerability and clinical effectiveness of electroconvulsive therapy. World J Biol Psychiatry. 2006;7(2):82-90.
19. Ben-Menachem E, Mañon-Espaillat R, Ristanovic R, et al. Vagus nerve stimulation for treatment of partial seizures: 1. A controlled study of effect on seizures. First International Vagus Nerve Stimulation Study Group. Epilepsia. 1994;35(3):616-626.
20. Nemeroff CB, Mayberg HS, Krahl SE, et al. VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology. 2006;31(7):1345-1355.
21. Matthews K, Eljamel MS. Vagus nerve stimulation and refractory depression: please can you switch me on doctor? Br J Psychiatry. 2003;183:181-183.
22. George MS, Rush AJ, Sackeim HA, et al. Vagus nerve stimulation (VNS): utility in neuropsychiatric disorders. Int J Neuropsychopharmacol. 2003;6(1):73-83.
23. Harden CL, Pulver MC, Ravdin LD, et al. A pilot study of mood in epilepsy patients treated with vagus nerve stimulation. Epilepsy Behav. 2000;1(2):93-99.
24. Elger G, Hoppe C, Falkai P, et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res. 2000;42(2-3):203-210.
25. Krahl SE, Clark KB, Smith DC, et al. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39(7):709-714.
26. Ben-Menachem E, Hamberger A, Hedner T, et al. Effects of vagus nerve stimulation on amino acids and other metabolites in the CSF of patients with partial seizures. Epilepsy Res. 1995;20(3):221-227.
27. Henry TR, Bakay RA, Votaw JR, et al. Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy: I. Acute effects at high and low levels of stimulation. Epilepsia. 1998;39(9):983-990.
28. O’Keane V, Dinan TG, Scott L, et al. Changes in hypothalamic-pituitary-adrenal axis measures after vagus nerve stimulation therapy in chronic depression. Biol Psychiatry. 2005;58(12):963-968.
29. Rush AJ, George MS, Sackeim HA, et al. Vagus nerve stimulation (VNS) for treatment-resistant depressions: a multicenter study. Biol Psychiatry. 2000;47(4):276-286.
30. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology. 2001;25(5):713-728.
31. Armitage R, Husain M, Hoffmann R, et al. The effects of vagus nerve stimulation on sleep EEG in depression: a preliminary report. J Psychosom Res. 2003;54(5):475-482.
32. Rush AJ, Marangell LB, Sackeim HA, et al. Vagus nerve stimulation for treatment-resistant depression: a randomized, controlled acute phase trial. Biol Psychiatry. 2005;58(5):347-354.
33. Martin JL, Martín-Sánchez E. Systematic review and meta-analysis of vagus nerve stimulation in the treatment of depression: variable results based on study designs. Eur Psychiatry. 2012;27(3):147-155.
34. Shah A, Carreno FR, Frazer A. Therapeutic modalities for treatment resistant depression: focus on vagal nerve stimulation and ketamine. Clin Psychopharmacol Neurosci. 2014;12(2):83-93.
35. Aaronson ST, Carpenter LL, Conway CR, et al. Vagus nerve stimulation therapy randomized to different amounts of electrical charge for treatment-resistant depression: acute and chronic effects. Brain Stimul. 2013;6(4):631-640.
36. Daban C, Martinez-Aran A, Cruz N, et al. Safety and efficacy of vagus nerve stimulation in treatment-resistant depression. A systematic review. J Affect Disord. 2008;110(1-2):1-15.
37. Eitan R, Lerer B. Nonpharmacological, somatic treatments of depression: electroconvulsive therapy and novel brain stimulation modalities. Dialogues Clin Neurosci. 2006;8(2):241-258.
38. Lam RW, Chan P, Wilkins-Ho M, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and metaanalysis. Can J Psychiatry. 2008;53(9):621-631.
39. Fitzgerald PB, Fountain S, Daskalakis ZJ. A comprehensive review of the effects of rTMS on motor cortical excitability and inhibition. Clin Neurophysiol. 2006;117(12):2584-2596.
40. Fitzgerald PB, Oxley TJ, Laird AR, et al. An analysis of functional neuroimaging studies of dorsolateral prefrontal cortical activity in depression. Psychiatry Res. 2006;148(1):33-45.
41. Berlim MT, Van den Eynde F, Daskalakis ZJ. Clinically meaningful efficacy and acceptability of low-frequency repetitive transcranial magnetic stimulation (rTMS) for treating primary major depression: a meta-analysis of randomized, double-blind and sham-controlled trials. Neuropsychopharmacology. 2013;38(4):543-551.
42. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
43. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
44. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder. J Clin Psychiatry. 2014;75(12):1394-1401.
45. Fitzgerald PB, Hoy K, Daskalakis ZJ, et al. A randomized trial of the anti-depressant effects of low- and high-frequency transcranial magnetic stimulation in treatment-resistant depression. Depress Anxiety. 2009;26(3):229-234.
46. Dumas R, Padovani R, Richieri R, et al. Repetitive transcranial magnetic stimulation in major depression: response factor [in French]. Encephale. 2012;38(4):360-368.
47. Fregni F, Marcolin MA, Myczkowski M, et al. Predictors of antidepressant response in clinical trials of transcranial magnetic stimulation. Int. J. Neuropsychopharmacol. 2006;9(6):641-654.
48. Kennedy SH, Giacobbe P, Rizvi SJ, et al. Deep brain stimulation for treatment-resistant depression: follow-up after 3 to 6 years. Am J Psychiatry. 2011;168(5):502-510.
49. Taghva AS, Malone DA, Rezai AR. Deep brain stimulation for treatment-resistant depression. World Neurosurg. 2013;80(3-4):S27.e17-S27.e24.
50. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 2003;65:193-207.
51. Mayberg HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
52. Jiménez F, Velasco F, Salín-Pascual R, et al. Neuromodulation of the inferior thalamic peduncle for major depression and obsessive compulsive disorder. Acta Neurochir Suppl. 2007;97(pt 2):393-398.
53. Jiménez F, Velasco F, Salin-Pascual R, et al. A patient with a resistant major depression disorder treated with deep brain stimulation in the inferior thalamic peduncle. Neurosurgery. 2005;57(3):585-593; discussion 585-593.
54. Bewernick BH, Hurlemann R, Matusch A, et al. Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry. 2010;67(2):110-116.
55. Schlaepfer TE, Bewernick BH, Kayser S, et al. Deep brain stimulation of the human reward system for major depression—rationale, outcomes and outlook. Neuropsychopharmacology. 2014;39(6):1303-1314.
56. Malone DA Jr, Dougherty DD, Rezai AR, et al. Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol Psychiatry. 2009;65(4):267-275.
57. Berlim MT, McGirr A, Van den Eynde F, et al. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
58. Schlaepfer TE, Bewernick BH, Kayser S, et al. Rapid effects of deep brain stimulation for treatment-resistant major depression. Biol Psychiatry. 2013;73(12):1204-1212.
59. Gálvez JF, Keser Z, Mwangi B, et al. The medial forebrain bundle as a deep brain stimulation target for treatment resistant depression: a review of published data. Prog Neuropsychopharmacol Biol Psychiatry. 2015;58:59-70.
60. Fenoy AJ, Schulz P, Selvaraj. Deep brain stimulation of the medial forebrain bundle: distinctive responses in resistant depression. J Affect Disord. 2016;203:143-151.
61. Riva-Posse P, Choi KS, Holtzheimer PE, et al. Defining critical white matter pathways mediating successful subcallosal cingulate deep brain stimulation for treatment-resistant depression. Biol Psychiatry. 2014;76(12):963-969.
62. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
63. Milev R V, Giacobbe P, Kennedy SH, et al; CANMAT Depression Work Group. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder: section 4. Neurostimulation treatments. Can J Psychiatry. 2016;61:561-575.
The emergence of treatment-resistant depression (TRD) poses a great clinical and public health challenge. There is no clear consensus on criteria to define TRD. The criteria range from failure to respond to 4 weeks of a single antidepressant to failure to respond to a single trial of electroconvulsive therapy (ECT).1
Neuromodulatory treatments for depression involve electrical stimulation of the brain through invasive or noninvasive methods. In this article, we discuss criteria for defining TRD, and compare the advantages and disadvantages of 4 neuromodulatory treatment options—ECT, vagus nerve stimulation (VNS), repetitive transcranial magnetic stimulation (rTMS), and deep brain stimulation (DBS)—for patients with depression who fail to respond to appropriate pharmacologic interventions (Table 1). Most of the studies we discuss selected patients who had severe depression and had not responded to numerous treatment trials.
Defining treatment resistance
Thase and Rush2 suggested progressive stages for categorizing TRD, ranging from Stage I (failure of at least 1 adequate trial of antidepressants) to Stage V (failure of adequate treatment with 2 selective serotonin reuptake inhibitors [SSRIs], a tricyclic antidepressant, a monoamine oxidase inhibitor, and a course of bilateral ECT). The Massachusetts General Hospital Staging Model suggested a quantitative scale to help characterize the degree of treatment resistance in which a higher score corresponds to a higher level of resistance.3 For every failed 6-week trial with adequate dose of an antidepressant, the patient is given a score of 1. The patient receives an extra .5 point for failure to respond to optimization of the dosage and augmentation with another medication. The patient also is given 3 points for failure to respond to ECT. Souery et al4,5 proposed a model in which they defined TRD as a failure to respond after ≥1 adequate antidepressant trials of ≥12 weeks.
Treatment resistance often is the result of inadequate treatment of depressive symptoms. Inadequate treatment includes an inadequate dose of antidepressants and/or an inadequate duration of treatment. Treatment of depression also is often complicated by medical (cardiovascular, neurologic, endocrine disorders) and psychiatric (substance abuse disorders, personality disorders) comorbidities (Table 2). Patients with such comorbidities are at increased risk of mortality, and have lower response rates and increased morbidity.6
Electroconvulsive therapy
ECT involves the application of electric current to induce a self-limiting seizure. It affects multiple brain functions to produce its antidepressant effects. Patients with depression have a reduced concentration of γ-aminobutyric acid (GABA) in their plasma, CSF, and cortex. ECT increases GABAergic transmission in cortical circuits as demonstrated by increased levels of GABA in the occipital cortex, which may be responsible for ECT’s antidepressant effects.7 Sensitization of the 5-HT1A receptors and increased dopamine receptor binding in the striatum also have been associated with the antidepressant action of ECT.8 The antidepressant effects of ECT also can be attributed to increased neuroplasticity, as evidenced by increased neurotrophic factors and cell proliferation in animal models.9 Dysfunction of the HPA axis has long been associated with depressive disorders; ECT improves this dysfunction, as evidenced by normalization of the dexamethasone suppression test in patients who receive ECT.7
The results of neuroimaging studies exploring the effects of ECT vary widely based on the specific neuroimaging method, population, and statistical methods used to assess the changes. Some of the most consistent findings include reduced glucose metabolism in the frontal brain regions; reduced glucose metabolism in the hippocampus and medial temporal lobes; and reduction in functional connectivity in the anterior cingulate, parietal, medical frontal, and dorsolateral prefrontal cortex (DLPFC).10
Randomized control trials (RCTs) have established the superiority of ECT over pharmacotherapy and sham ECT. Compared with other neuromodulatory treatments, ECT has higher remission rates. On average, the remission rate among patients receiving ECT whose depression did not respond to pharmacotherapy is approximately 48%; this increases to 64.9% among patients who previously had responded to a medication.11
Some earlier trials found bilateral ECT to be more effective than unilateral ECT.12 Recent studies suggest that high-dose unilateral ECT (6 times the seizure threshold) is as effective as bilateral ECT.13 Studies have shown no significant differences in efficacy or treatment outcomes between twice- and thrice-weekly ECT regimens. Some studies suggest that twice-weekly ECT may be associated with a lower risk of short-term cognitive impairment compared with thrice-weekly ECT.14
In highly refractory cases, the effects of ECT can be augmented by using pre-treatment strategies such as hyperventilation, which may increase the duration of the seizure, and remifentanil, which helps reduce the anticonvulsant effect of agents used for anesthesia.15 Advanced age, psychotic features, resistance to pharmacotherapy, and comorbid personality disorders predict poor response to ECT.16
Adverse effects. Concerns about cognitive deficits secondary to ECT may curtail its use. Retrograde and anterograde amnesia are the most common deficits observed acutely after ECT.12 Other commonly affected cognitive functions include processing speed, attention/working memory, verbal and visual episodic memory, spatial problem solving, and executive functioning. The specific patterns of these deficits (in terms of duration and severity) vary between studies. In general, high-dose, thrice-weekly ECT and bilateral ECT are associated with greater cognitive deficits, whereas twice-weekly ECT and unilateral ECT are associated with a lower risk of cognitive adverse effects.12 A recent meta-analysis by Semkovska and McLoughlin17 found that most cognitive deficits seen after ECT are limited to the first 3 days after treatment. The authors of this meta-analysis concluded that these impairments improve over time and approach baseline 2 weeks after treatment. In fact, some of these impairments (processing speed, working memory, anterograde memory, and some aspects of executive function) improved beyond baseline after 15 days of treatment.17 The need for anesthesia and associated potential adverse effects also are a cause of concern with ECT.
Combining ECT with medication. Several patient-specific factors, including medication regimen and comorbid medical conditions, need to be considered before using ECT in combination with pharmacotherapy. Although most antipsychotics are safe to use with ECT, concomitant use of agents with higher antihistaminic properties may increase the risk of delirium. The risk of delirium also is increased with the use of anticonvulsants and mood stabilizers (eg, lithium) because these agents increase the seizure threshold. The potential for drug interactions may affect the choice of the anesthetic agents. Also, SSRIs and serotonin-norepinephrine reuptake inhibitors can increase the duration of induced seizures.18
Vagus nerve stimulation
VNS, in which an implanted device stimulates the vagus nerve with electrical impulses, initially was used to reduce the frequency of seizures in patients with epilepsy and treatment-resistant partial onset seizures.19 VNS was FDA-approved for TRD in July 2005.20 One VNS system, the NCP System, consists of an implantable, multi-programmable generator, known as a pulse generator, that is subcutaneously placed in the anterior chest wall during an outpatient surgical procedure. Separate bipolar nerve-stimulating electrodes are surgically wrapped around the left cervical vagus nerve, and then connected to the generator via a tunneling procedure. A telemetric wand is subsequently linked to a portable computer and used to adjust stimulation parameters.21,22
Support for using VNS for TRD came from a multitude of investigations and observations. Harden et al23 and Elger et al24 prospectively evaluated epileptic patients with standard depression symptom severity rating scales. They found that VNS was associated with statistically significant improvements in mood that were not related to reductions in seizures.23,24
The mechanism of action of VNS is not clear. Earlier researchers had found evidence that VNS affected brain regions associated with norepinephrine25 and serotonin systems26; both of these neurotransmitters have been implicated in the pathophysiology of depression. Positron emission tomography studies conducted during VNS treatment of epilepsy showed metabolic changes in cortical and subcortical areas of the brain, including the amygdala, hippocampus, and cingulate gyrus, all structures implicated in the pathophysiology of mood disorders.27
Most studies conducted to evaluate the efficacy of VNS have been observational, looking at depression ratings before and after treatment with VNS. The short-term studies measured the difference in depression rating scales at baseline and after 10 weeks of treatment. In most of these studies, treatment with VNS resulted in a statistically significant drop in depression rating scales scores, such as on the Hamilton Depression Rating Scale (HAM-D). Based on the study design and number of study participants, response rates have varied from 13%28 to 40%,29 whereas remission rates have varied from 15.3%30 to 28%.31 More than one-half of the reduction in symptoms occurred after 6 weeks of treatment.30 In longer-term follow-up studies, the antidepressant effect generally was sustained over time. Response rates remained essentially unchanged, but the remission rates increased to approximately 29%.29 Only 1 RCT has compared patients with controls; it found no significant differences in the response or remission rates between active VNS and sham VNS.32 In this study, all patients had VNS implanted, but in the control group, the VNS was never turned on.32 In a meta-analysis conducted by Martin and Martín-Sánchez,33 31.8% (95% confidence interval [CI], 23.2% to 41.8%; P < .001) of patients treated with VNS had a significant reduction in HAM-D scores. The response rate in patients with TRD ranged from 27% to 37% and the remission rate was approximately 13%. In studies that followed patients over longer periods, both the remission and response rates increased over time.34
Recent evidence suggests that the effectiveness of VNS may depend on the stimulation level. A multi-center double-blind study randomized patients to receive either a low (0.25 mA current, 130-millisecond pulse width), medium (0.5e1.0 mA, 250 millisecond), or high (1.25e1.5 mA, 250 millisecond) dose of VNS.35 Although all dose levels were associated with improvement in symptoms, a statistically significant durability in response was associated with the medium- and high-dose treatments.
Adverse effects. VNS has no major adverse effects on cognitive functioning, and some studies have found improvement in executive functioning that corresponded to improvement in depressive symptoms.30 VNS also may result in improved sleep patterns as evidenced by EEG changes.31 The most commonly reported adverse effects include pain in the incision site, hoarseness of voice, throat pain, and neck pain.36
Repetitive transcranial magnetic stimulation
rTMS is a noninvasive technique that uses high-intensity magnetic impulses to stimulate cortical neurons. A magnetic field is produced when current passes through a coil, which in turn causes electrical stimulation in the cortical neurons that results in transient changes in the excitability of the cortical neurons.37 Although many stimulation parameters exist for TMS, high-frequency stimulation to the left prefrontal cortex (HFL-rTMS) and low-frequency stimulation to the right prefrontal cortex (LFR-rTMS) have been shown most efficacious for treating depression.38 High-frequency (5 Hz to 20 Hz) stimulation using rTMS increases cortical neuron excitability, whereas low-frequency (approximately 1 Hz) is associated with reduced cortical neuron excitability.39 The choice of targeting the DLPFC stems from a large body of functional neuroimaging studies that have shown reduction in activity/blood flow in the left DLPFC and abnormal activity/blood flow in the right DLPFC.40
There is no dearth of RCTs evaluating the efficacy of rTMS vs sham rTMS (where no magnetic stimulation was provided). In a meta-analysis of 8 RCTs, low-frequency rTMS applied to the right DLPFC was associated with a remission rate of approximately 34.6%, compared with a 9.7% remission rate with sham rTMS.41 A response rate of approximately 38.2% was observed with HFL-rTMS, compared with a response rate of 15.1% for sham rTMS.41
Gaynes et al42 conducted a meta-analysis to determine the efficacy of rTMS in TRD. They found that for patients with TRD, rTMs produced a response rate of 29% and a remission rate of 30%. In long-term, naturalistic, observational studies, the response rates and remission rates were much higher (58% and 37.1%, respectively).43 Over a 1-year follow-up, almost two-thirds of patients continued to meet criteria for response to treatment.44 Trials comparing HFL-rTMS and LFR-rTMS have found no significant differences in efficacy.45
Advanced age, psychotic symptoms, and a longer duration of the current depressive episode predict poor response to rTMS. Also, imaging studies have shown that a lower metabolism in cerebellar, temporal, anterior cingulate, and occipital parts of the brain correlate with better response to HFL-rTMS.46,47
Adverse effects. The major adverse effect associated with rTMS is the risk of inducing seizures, which is more commonly associated with high-frequency rTMS. Other common adverse effects include headache, facial muscle twitching, and tinnitus.37
Deep brain stimulation
DBS is an invasive stereotactic surgical procedure. It involves unilateral or bilateral placement of electrodes at neuroanatomical locations to deliver continuous stimulation from a subcutaneously implanted pulse generator.48 In the past, destructive surgical procedures were used to treat intractable depression. Surgeries such as anterior cingulotomy, anterior capsulotomy, subcaudate tractotomy, and limbic leucotomy have been shown to effectively reduce depressive symptoms.49 The advantages of DBS over destructive procedures include the fact that DBS is reversible and that the stimulation levels can easily be adjusted, and the treatment can easily be stopped or restarted.
There is no consensus on the optimal anatomic locations for the electrode implantation in DBS. Electrodes have been implanted in the subcallosal cingulate gyrus, inferior thalamic peduncle, ventral capsule/ventral striatum, superolateral branch of the medial forebrain bundle (MFB), and nucleus accumbens.
The choice of anatomic locations stems from the large body of neuroimaging literature characterizing functional changes associated with acute depression and response to treatment. The electrode placement targets “nodes” that form an integral part of the affected neural circuits that are responsible for regulating depressive symptoms.50 Increased metabolic activity and blood flow to the subgenual cingulate gyrus and reduction in the blood flow to the DLPFC and the striatum have been associated with active depressed states. Response to antidepressant treatment has been associated with reversal of these findings.51 Functional magnetic resonance imaging studies have consistently shown increased activity in the amygdala in response to negative stimuli among patients with depression.
Regardless of the site of electrode placement, studies have reported symptomatic improvement among patients with depression who are treated with DBS. In 2 case reports, the electrode was implanted in the inferior thalamic peduncle.52,53 Each study had 1 participant, and each patient remitted.52,53
Placement of the electrodes in the nucleus accumbens resulted in a response rate of 45% in 1 study,54 whereas in a different study, all patients reported improvement in anhedonia.55 A response rate of 71% and a remission rate of 35% were observed in a study in which the electrode was implanted in the ventral capsule/ventral striatum area.56
Berlim et al57 published a systematic review and exploratory meta-analysis of studies in which the electrode had been implanted in the subgenual cingulate cortex. At 12 months, the response rate was 39.9% (95% CI, 28.4% to 52.8%), and 26.3% (95% CI, 13% to 45.9%) of patients achieved remission. The most significant drop in depression scores was observed 3 to 6 months after the surgery. No significant change in scores was observed between 6 to 12 months after surgery.57
The MFB, specifically the superolateral branch, is emerging as an exciting new target for electrode placement in DBS. Schlaepfer et al58 studied the effects of electrodes implanted bilaterally in the superolateral branch of the MFB. They observed an almost 50% reduction in symptoms by Day 7, and at the last follow-up visit (12 to 33 weeks) 4 of the 6 patients had achieved remission.58 In a recent systematic review, Gálvez et al59 found most studies had high response/remission rates without any significant adverse effects. In a recent study of DBS targeting the MFB, 3 of 4 patients had a >50% reduction in Montgomery-Åsberg Depression Rating Scale scores at the end of first week. Although 1 patient withdrew, 2 of the other 3 patients continued to report a >80% reduction in depressive symptoms, even at Week 26.60
Accurate localization of target areas (white matter tracts) and subsequent electrode placement might be an important factor governing treatment response. Riva-Posse et al61 found that clinical response was seen when the electrodes stimulated 3 specific white matter bundles. Interestingly, nonresponders were converted to responders simply by changing the position of the electrodes to include these white matter tracts.61
Adverse effects. The most common adverse effects noted during studies of DBS include pain at the site of implantation and wound infection. Other adverse effects include lead fracture, transient dysphagia, and other hardware-related problems.49
Sorting out the evidence
In the absence of head-to-head trials, it is difficult to establish a hierarchal algorithm for use of the 4 neuromodulatory treatments discussed in the article. If we were to base our decision solely on the current literature, ECT by far has the most evidence and highest remission rates.11 We can reduce the risk of cognitive deficits by using twice-weekly instead of thrice-weekly ECT, or by using unilateral instead of bilateral ECT.12 Another strategy for reducing adverse effects associated with long-term maintenance ECT is by using it in combination with VNS. ECT and VNS can be used safely concomitantly; ECT can be used to treat acutely worsening depression, and VNS for maintaining the antidepressant effect.62
Aside from ECT, rTMS is the only other treatment that has evidence from RCTs. Although the remission rates are not as high as ECT, its preferable adverse effects profile, noninvasive nature, and comparative low cost (compared with surgical procedures) make it a favorable choice. The Canadian Network for Mood and Anxiety Treatment guidelines suggest rTMS as the first-line treatment for patients who do not respond to pharmacologic treatments.63 ECT can be considered second-line treatment unless the patient has acute suicidal ideation, catatonia, psychotic features, greater treatment resistance, or physical deterioration, in which case ECT should be tried before TMS.63
Among the invasive options, VNS has more evidence and is FDA-approved for TRD. However, DBS has shown great promise in early studies, with remission rates as high as 35%.56 DBS has the advantage of being reversible, and the amount of stimulation can be adjusted easily. Despite early promise, more research is needed before DBS can be widely used in clinical settings.
The emergence of treatment-resistant depression (TRD) poses a great clinical and public health challenge. There is no clear consensus on criteria to define TRD. The criteria range from failure to respond to 4 weeks of a single antidepressant to failure to respond to a single trial of electroconvulsive therapy (ECT).1
Neuromodulatory treatments for depression involve electrical stimulation of the brain through invasive or noninvasive methods. In this article, we discuss criteria for defining TRD, and compare the advantages and disadvantages of 4 neuromodulatory treatment options—ECT, vagus nerve stimulation (VNS), repetitive transcranial magnetic stimulation (rTMS), and deep brain stimulation (DBS)—for patients with depression who fail to respond to appropriate pharmacologic interventions (Table 1). Most of the studies we discuss selected patients who had severe depression and had not responded to numerous treatment trials.
Defining treatment resistance
Thase and Rush2 suggested progressive stages for categorizing TRD, ranging from Stage I (failure of at least 1 adequate trial of antidepressants) to Stage V (failure of adequate treatment with 2 selective serotonin reuptake inhibitors [SSRIs], a tricyclic antidepressant, a monoamine oxidase inhibitor, and a course of bilateral ECT). The Massachusetts General Hospital Staging Model suggested a quantitative scale to help characterize the degree of treatment resistance in which a higher score corresponds to a higher level of resistance.3 For every failed 6-week trial with adequate dose of an antidepressant, the patient is given a score of 1. The patient receives an extra .5 point for failure to respond to optimization of the dosage and augmentation with another medication. The patient also is given 3 points for failure to respond to ECT. Souery et al4,5 proposed a model in which they defined TRD as a failure to respond after ≥1 adequate antidepressant trials of ≥12 weeks.
Treatment resistance often is the result of inadequate treatment of depressive symptoms. Inadequate treatment includes an inadequate dose of antidepressants and/or an inadequate duration of treatment. Treatment of depression also is often complicated by medical (cardiovascular, neurologic, endocrine disorders) and psychiatric (substance abuse disorders, personality disorders) comorbidities (Table 2). Patients with such comorbidities are at increased risk of mortality, and have lower response rates and increased morbidity.6
Electroconvulsive therapy
ECT involves the application of electric current to induce a self-limiting seizure. It affects multiple brain functions to produce its antidepressant effects. Patients with depression have a reduced concentration of γ-aminobutyric acid (GABA) in their plasma, CSF, and cortex. ECT increases GABAergic transmission in cortical circuits as demonstrated by increased levels of GABA in the occipital cortex, which may be responsible for ECT’s antidepressant effects.7 Sensitization of the 5-HT1A receptors and increased dopamine receptor binding in the striatum also have been associated with the antidepressant action of ECT.8 The antidepressant effects of ECT also can be attributed to increased neuroplasticity, as evidenced by increased neurotrophic factors and cell proliferation in animal models.9 Dysfunction of the HPA axis has long been associated with depressive disorders; ECT improves this dysfunction, as evidenced by normalization of the dexamethasone suppression test in patients who receive ECT.7
The results of neuroimaging studies exploring the effects of ECT vary widely based on the specific neuroimaging method, population, and statistical methods used to assess the changes. Some of the most consistent findings include reduced glucose metabolism in the frontal brain regions; reduced glucose metabolism in the hippocampus and medial temporal lobes; and reduction in functional connectivity in the anterior cingulate, parietal, medical frontal, and dorsolateral prefrontal cortex (DLPFC).10
Randomized control trials (RCTs) have established the superiority of ECT over pharmacotherapy and sham ECT. Compared with other neuromodulatory treatments, ECT has higher remission rates. On average, the remission rate among patients receiving ECT whose depression did not respond to pharmacotherapy is approximately 48%; this increases to 64.9% among patients who previously had responded to a medication.11
Some earlier trials found bilateral ECT to be more effective than unilateral ECT.12 Recent studies suggest that high-dose unilateral ECT (6 times the seizure threshold) is as effective as bilateral ECT.13 Studies have shown no significant differences in efficacy or treatment outcomes between twice- and thrice-weekly ECT regimens. Some studies suggest that twice-weekly ECT may be associated with a lower risk of short-term cognitive impairment compared with thrice-weekly ECT.14
In highly refractory cases, the effects of ECT can be augmented by using pre-treatment strategies such as hyperventilation, which may increase the duration of the seizure, and remifentanil, which helps reduce the anticonvulsant effect of agents used for anesthesia.15 Advanced age, psychotic features, resistance to pharmacotherapy, and comorbid personality disorders predict poor response to ECT.16
Adverse effects. Concerns about cognitive deficits secondary to ECT may curtail its use. Retrograde and anterograde amnesia are the most common deficits observed acutely after ECT.12 Other commonly affected cognitive functions include processing speed, attention/working memory, verbal and visual episodic memory, spatial problem solving, and executive functioning. The specific patterns of these deficits (in terms of duration and severity) vary between studies. In general, high-dose, thrice-weekly ECT and bilateral ECT are associated with greater cognitive deficits, whereas twice-weekly ECT and unilateral ECT are associated with a lower risk of cognitive adverse effects.12 A recent meta-analysis by Semkovska and McLoughlin17 found that most cognitive deficits seen after ECT are limited to the first 3 days after treatment. The authors of this meta-analysis concluded that these impairments improve over time and approach baseline 2 weeks after treatment. In fact, some of these impairments (processing speed, working memory, anterograde memory, and some aspects of executive function) improved beyond baseline after 15 days of treatment.17 The need for anesthesia and associated potential adverse effects also are a cause of concern with ECT.
Combining ECT with medication. Several patient-specific factors, including medication regimen and comorbid medical conditions, need to be considered before using ECT in combination with pharmacotherapy. Although most antipsychotics are safe to use with ECT, concomitant use of agents with higher antihistaminic properties may increase the risk of delirium. The risk of delirium also is increased with the use of anticonvulsants and mood stabilizers (eg, lithium) because these agents increase the seizure threshold. The potential for drug interactions may affect the choice of the anesthetic agents. Also, SSRIs and serotonin-norepinephrine reuptake inhibitors can increase the duration of induced seizures.18
Vagus nerve stimulation
VNS, in which an implanted device stimulates the vagus nerve with electrical impulses, initially was used to reduce the frequency of seizures in patients with epilepsy and treatment-resistant partial onset seizures.19 VNS was FDA-approved for TRD in July 2005.20 One VNS system, the NCP System, consists of an implantable, multi-programmable generator, known as a pulse generator, that is subcutaneously placed in the anterior chest wall during an outpatient surgical procedure. Separate bipolar nerve-stimulating electrodes are surgically wrapped around the left cervical vagus nerve, and then connected to the generator via a tunneling procedure. A telemetric wand is subsequently linked to a portable computer and used to adjust stimulation parameters.21,22
Support for using VNS for TRD came from a multitude of investigations and observations. Harden et al23 and Elger et al24 prospectively evaluated epileptic patients with standard depression symptom severity rating scales. They found that VNS was associated with statistically significant improvements in mood that were not related to reductions in seizures.23,24
The mechanism of action of VNS is not clear. Earlier researchers had found evidence that VNS affected brain regions associated with norepinephrine25 and serotonin systems26; both of these neurotransmitters have been implicated in the pathophysiology of depression. Positron emission tomography studies conducted during VNS treatment of epilepsy showed metabolic changes in cortical and subcortical areas of the brain, including the amygdala, hippocampus, and cingulate gyrus, all structures implicated in the pathophysiology of mood disorders.27
Most studies conducted to evaluate the efficacy of VNS have been observational, looking at depression ratings before and after treatment with VNS. The short-term studies measured the difference in depression rating scales at baseline and after 10 weeks of treatment. In most of these studies, treatment with VNS resulted in a statistically significant drop in depression rating scales scores, such as on the Hamilton Depression Rating Scale (HAM-D). Based on the study design and number of study participants, response rates have varied from 13%28 to 40%,29 whereas remission rates have varied from 15.3%30 to 28%.31 More than one-half of the reduction in symptoms occurred after 6 weeks of treatment.30 In longer-term follow-up studies, the antidepressant effect generally was sustained over time. Response rates remained essentially unchanged, but the remission rates increased to approximately 29%.29 Only 1 RCT has compared patients with controls; it found no significant differences in the response or remission rates between active VNS and sham VNS.32 In this study, all patients had VNS implanted, but in the control group, the VNS was never turned on.32 In a meta-analysis conducted by Martin and Martín-Sánchez,33 31.8% (95% confidence interval [CI], 23.2% to 41.8%; P < .001) of patients treated with VNS had a significant reduction in HAM-D scores. The response rate in patients with TRD ranged from 27% to 37% and the remission rate was approximately 13%. In studies that followed patients over longer periods, both the remission and response rates increased over time.34
Recent evidence suggests that the effectiveness of VNS may depend on the stimulation level. A multi-center double-blind study randomized patients to receive either a low (0.25 mA current, 130-millisecond pulse width), medium (0.5e1.0 mA, 250 millisecond), or high (1.25e1.5 mA, 250 millisecond) dose of VNS.35 Although all dose levels were associated with improvement in symptoms, a statistically significant durability in response was associated with the medium- and high-dose treatments.
Adverse effects. VNS has no major adverse effects on cognitive functioning, and some studies have found improvement in executive functioning that corresponded to improvement in depressive symptoms.30 VNS also may result in improved sleep patterns as evidenced by EEG changes.31 The most commonly reported adverse effects include pain in the incision site, hoarseness of voice, throat pain, and neck pain.36
Repetitive transcranial magnetic stimulation
rTMS is a noninvasive technique that uses high-intensity magnetic impulses to stimulate cortical neurons. A magnetic field is produced when current passes through a coil, which in turn causes electrical stimulation in the cortical neurons that results in transient changes in the excitability of the cortical neurons.37 Although many stimulation parameters exist for TMS, high-frequency stimulation to the left prefrontal cortex (HFL-rTMS) and low-frequency stimulation to the right prefrontal cortex (LFR-rTMS) have been shown most efficacious for treating depression.38 High-frequency (5 Hz to 20 Hz) stimulation using rTMS increases cortical neuron excitability, whereas low-frequency (approximately 1 Hz) is associated with reduced cortical neuron excitability.39 The choice of targeting the DLPFC stems from a large body of functional neuroimaging studies that have shown reduction in activity/blood flow in the left DLPFC and abnormal activity/blood flow in the right DLPFC.40
There is no dearth of RCTs evaluating the efficacy of rTMS vs sham rTMS (where no magnetic stimulation was provided). In a meta-analysis of 8 RCTs, low-frequency rTMS applied to the right DLPFC was associated with a remission rate of approximately 34.6%, compared with a 9.7% remission rate with sham rTMS.41 A response rate of approximately 38.2% was observed with HFL-rTMS, compared with a response rate of 15.1% for sham rTMS.41
Gaynes et al42 conducted a meta-analysis to determine the efficacy of rTMS in TRD. They found that for patients with TRD, rTMs produced a response rate of 29% and a remission rate of 30%. In long-term, naturalistic, observational studies, the response rates and remission rates were much higher (58% and 37.1%, respectively).43 Over a 1-year follow-up, almost two-thirds of patients continued to meet criteria for response to treatment.44 Trials comparing HFL-rTMS and LFR-rTMS have found no significant differences in efficacy.45
Advanced age, psychotic symptoms, and a longer duration of the current depressive episode predict poor response to rTMS. Also, imaging studies have shown that a lower metabolism in cerebellar, temporal, anterior cingulate, and occipital parts of the brain correlate with better response to HFL-rTMS.46,47
Adverse effects. The major adverse effect associated with rTMS is the risk of inducing seizures, which is more commonly associated with high-frequency rTMS. Other common adverse effects include headache, facial muscle twitching, and tinnitus.37
Deep brain stimulation
DBS is an invasive stereotactic surgical procedure. It involves unilateral or bilateral placement of electrodes at neuroanatomical locations to deliver continuous stimulation from a subcutaneously implanted pulse generator.48 In the past, destructive surgical procedures were used to treat intractable depression. Surgeries such as anterior cingulotomy, anterior capsulotomy, subcaudate tractotomy, and limbic leucotomy have been shown to effectively reduce depressive symptoms.49 The advantages of DBS over destructive procedures include the fact that DBS is reversible and that the stimulation levels can easily be adjusted, and the treatment can easily be stopped or restarted.
There is no consensus on the optimal anatomic locations for the electrode implantation in DBS. Electrodes have been implanted in the subcallosal cingulate gyrus, inferior thalamic peduncle, ventral capsule/ventral striatum, superolateral branch of the medial forebrain bundle (MFB), and nucleus accumbens.
The choice of anatomic locations stems from the large body of neuroimaging literature characterizing functional changes associated with acute depression and response to treatment. The electrode placement targets “nodes” that form an integral part of the affected neural circuits that are responsible for regulating depressive symptoms.50 Increased metabolic activity and blood flow to the subgenual cingulate gyrus and reduction in the blood flow to the DLPFC and the striatum have been associated with active depressed states. Response to antidepressant treatment has been associated with reversal of these findings.51 Functional magnetic resonance imaging studies have consistently shown increased activity in the amygdala in response to negative stimuli among patients with depression.
Regardless of the site of electrode placement, studies have reported symptomatic improvement among patients with depression who are treated with DBS. In 2 case reports, the electrode was implanted in the inferior thalamic peduncle.52,53 Each study had 1 participant, and each patient remitted.52,53
Placement of the electrodes in the nucleus accumbens resulted in a response rate of 45% in 1 study,54 whereas in a different study, all patients reported improvement in anhedonia.55 A response rate of 71% and a remission rate of 35% were observed in a study in which the electrode was implanted in the ventral capsule/ventral striatum area.56
Berlim et al57 published a systematic review and exploratory meta-analysis of studies in which the electrode had been implanted in the subgenual cingulate cortex. At 12 months, the response rate was 39.9% (95% CI, 28.4% to 52.8%), and 26.3% (95% CI, 13% to 45.9%) of patients achieved remission. The most significant drop in depression scores was observed 3 to 6 months after the surgery. No significant change in scores was observed between 6 to 12 months after surgery.57
The MFB, specifically the superolateral branch, is emerging as an exciting new target for electrode placement in DBS. Schlaepfer et al58 studied the effects of electrodes implanted bilaterally in the superolateral branch of the MFB. They observed an almost 50% reduction in symptoms by Day 7, and at the last follow-up visit (12 to 33 weeks) 4 of the 6 patients had achieved remission.58 In a recent systematic review, Gálvez et al59 found most studies had high response/remission rates without any significant adverse effects. In a recent study of DBS targeting the MFB, 3 of 4 patients had a >50% reduction in Montgomery-Åsberg Depression Rating Scale scores at the end of first week. Although 1 patient withdrew, 2 of the other 3 patients continued to report a >80% reduction in depressive symptoms, even at Week 26.60
Accurate localization of target areas (white matter tracts) and subsequent electrode placement might be an important factor governing treatment response. Riva-Posse et al61 found that clinical response was seen when the electrodes stimulated 3 specific white matter bundles. Interestingly, nonresponders were converted to responders simply by changing the position of the electrodes to include these white matter tracts.61
Adverse effects. The most common adverse effects noted during studies of DBS include pain at the site of implantation and wound infection. Other adverse effects include lead fracture, transient dysphagia, and other hardware-related problems.49
Sorting out the evidence
In the absence of head-to-head trials, it is difficult to establish a hierarchal algorithm for use of the 4 neuromodulatory treatments discussed in the article. If we were to base our decision solely on the current literature, ECT by far has the most evidence and highest remission rates.11 We can reduce the risk of cognitive deficits by using twice-weekly instead of thrice-weekly ECT, or by using unilateral instead of bilateral ECT.12 Another strategy for reducing adverse effects associated with long-term maintenance ECT is by using it in combination with VNS. ECT and VNS can be used safely concomitantly; ECT can be used to treat acutely worsening depression, and VNS for maintaining the antidepressant effect.62
Aside from ECT, rTMS is the only other treatment that has evidence from RCTs. Although the remission rates are not as high as ECT, its preferable adverse effects profile, noninvasive nature, and comparative low cost (compared with surgical procedures) make it a favorable choice. The Canadian Network for Mood and Anxiety Treatment guidelines suggest rTMS as the first-line treatment for patients who do not respond to pharmacologic treatments.63 ECT can be considered second-line treatment unless the patient has acute suicidal ideation, catatonia, psychotic features, greater treatment resistance, or physical deterioration, in which case ECT should be tried before TMS.63
Among the invasive options, VNS has more evidence and is FDA-approved for TRD. However, DBS has shown great promise in early studies, with remission rates as high as 35%.56 DBS has the advantage of being reversible, and the amount of stimulation can be adjusted easily. Despite early promise, more research is needed before DBS can be widely used in clinical settings.
1. Berlim MT, Turecki G. What is the meaning of treatment resistant/refractory major depression (TRD)? A systematic review of current randomized trials. Eur Neuropsychopharmacol. 2007;17(11):696-707.
2. Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(suppl 13):23-29.
3. Petersen T, Papakostas GI, Posternak MA, et al. Empirical testing of two models for staging antidepressant treatment resistance. J Clin Psychopharmacol. 2005;25(4):336-341.
4. Souery D, Papakostas GI, Trivedi MH. Treatment-resistant depression. J Clin Psychiatry. 2006;67(suppl 6):16-22.
5. Souery D, Amsterdam J, de Montigny C, et al. Treatment resistant depression: methodological overview and operational criteria. Eur Neuropsychopharmacol. 1999;9(1-2):83-91.
6. Evans DL, Charney DS. Mood disorders and medical illness: a major public health problem. Biol. Psychiatry. 2003;54(3):177-180.
7. Sanacora G, Mason GF, Rothman DL, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160(3):577-579.
8. Merkl A, Heuser I, Bajbouj M. Antidepressant electroconvulsive therapy: mechanism of action, recent advances and limitations. Exp Neurol. 2009;219(1):20-26.
9. Perera TD, Coplan JD, Lisanby SH, et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 2007;27(18):4894-4901.
10. Abbott CC, Gallegos P, Rediske N et al. A review of longitudinal electroconvulsive therapy: neuroimaging investigations. J Geriatr Psychiatry Neurol. 2014;27(1):33-46.
11. Heijnen WT, Birkenhäger TK, Wierdsma AI, et al. Antidepressant pharmacotherapy failure and response to subsequent electroconvulsive therapy: a meta-analysis. J Clin Psychopharmacol. 2010;30(5):616-619.
12. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
13. Semkovska M, Landau S, Dunne R et al. Bitemporal versus high-dose unilateral twice-weekly electroconvulsive therapy for depression (EFFECT-Dep): a pragmatic, randomized, non-inferiority trial. Am J Psychiatry. 2016;173(4):408-417.
14. Charlson F, Siskind D, Doi SA, et al. ECT efficacy and treatment course: a systematic review and meta-analysis of twice vs thrice weekly schedules. J Affect Disord. 2012;138(1-2):1-8.
15. Loo CK, Kaill A, Paton P, et al. The difficult-to-treat electroconvulsive therapy patient—strategies for augmenting outcomes. J Affect Disord. 2010;124(3):219-227.
16. de Vreede IM, Burger H, van Vliet IM. Prediction of response to ECT with routinely collected data in major depression. J Affect Disord. 2005;86(2-3):323-327.
17. Semkovska M, McLoughlin DM. Objective cognitive performance associated with electroconvulsive therapy for depression: a systematic review and meta-analysis. Biol Psychiatry. 2010;68(6):568-577.
18. Baghai TC, Marcuse A, Brosch M, et al. The influence of concomitant antidepressant medication on safety, tolerability and clinical effectiveness of electroconvulsive therapy. World J Biol Psychiatry. 2006;7(2):82-90.
19. Ben-Menachem E, Mañon-Espaillat R, Ristanovic R, et al. Vagus nerve stimulation for treatment of partial seizures: 1. A controlled study of effect on seizures. First International Vagus Nerve Stimulation Study Group. Epilepsia. 1994;35(3):616-626.
20. Nemeroff CB, Mayberg HS, Krahl SE, et al. VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology. 2006;31(7):1345-1355.
21. Matthews K, Eljamel MS. Vagus nerve stimulation and refractory depression: please can you switch me on doctor? Br J Psychiatry. 2003;183:181-183.
22. George MS, Rush AJ, Sackeim HA, et al. Vagus nerve stimulation (VNS): utility in neuropsychiatric disorders. Int J Neuropsychopharmacol. 2003;6(1):73-83.
23. Harden CL, Pulver MC, Ravdin LD, et al. A pilot study of mood in epilepsy patients treated with vagus nerve stimulation. Epilepsy Behav. 2000;1(2):93-99.
24. Elger G, Hoppe C, Falkai P, et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res. 2000;42(2-3):203-210.
25. Krahl SE, Clark KB, Smith DC, et al. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39(7):709-714.
26. Ben-Menachem E, Hamberger A, Hedner T, et al. Effects of vagus nerve stimulation on amino acids and other metabolites in the CSF of patients with partial seizures. Epilepsy Res. 1995;20(3):221-227.
27. Henry TR, Bakay RA, Votaw JR, et al. Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy: I. Acute effects at high and low levels of stimulation. Epilepsia. 1998;39(9):983-990.
28. O’Keane V, Dinan TG, Scott L, et al. Changes in hypothalamic-pituitary-adrenal axis measures after vagus nerve stimulation therapy in chronic depression. Biol Psychiatry. 2005;58(12):963-968.
29. Rush AJ, George MS, Sackeim HA, et al. Vagus nerve stimulation (VNS) for treatment-resistant depressions: a multicenter study. Biol Psychiatry. 2000;47(4):276-286.
30. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology. 2001;25(5):713-728.
31. Armitage R, Husain M, Hoffmann R, et al. The effects of vagus nerve stimulation on sleep EEG in depression: a preliminary report. J Psychosom Res. 2003;54(5):475-482.
32. Rush AJ, Marangell LB, Sackeim HA, et al. Vagus nerve stimulation for treatment-resistant depression: a randomized, controlled acute phase trial. Biol Psychiatry. 2005;58(5):347-354.
33. Martin JL, Martín-Sánchez E. Systematic review and meta-analysis of vagus nerve stimulation in the treatment of depression: variable results based on study designs. Eur Psychiatry. 2012;27(3):147-155.
34. Shah A, Carreno FR, Frazer A. Therapeutic modalities for treatment resistant depression: focus on vagal nerve stimulation and ketamine. Clin Psychopharmacol Neurosci. 2014;12(2):83-93.
35. Aaronson ST, Carpenter LL, Conway CR, et al. Vagus nerve stimulation therapy randomized to different amounts of electrical charge for treatment-resistant depression: acute and chronic effects. Brain Stimul. 2013;6(4):631-640.
36. Daban C, Martinez-Aran A, Cruz N, et al. Safety and efficacy of vagus nerve stimulation in treatment-resistant depression. A systematic review. J Affect Disord. 2008;110(1-2):1-15.
37. Eitan R, Lerer B. Nonpharmacological, somatic treatments of depression: electroconvulsive therapy and novel brain stimulation modalities. Dialogues Clin Neurosci. 2006;8(2):241-258.
38. Lam RW, Chan P, Wilkins-Ho M, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and metaanalysis. Can J Psychiatry. 2008;53(9):621-631.
39. Fitzgerald PB, Fountain S, Daskalakis ZJ. A comprehensive review of the effects of rTMS on motor cortical excitability and inhibition. Clin Neurophysiol. 2006;117(12):2584-2596.
40. Fitzgerald PB, Oxley TJ, Laird AR, et al. An analysis of functional neuroimaging studies of dorsolateral prefrontal cortical activity in depression. Psychiatry Res. 2006;148(1):33-45.
41. Berlim MT, Van den Eynde F, Daskalakis ZJ. Clinically meaningful efficacy and acceptability of low-frequency repetitive transcranial magnetic stimulation (rTMS) for treating primary major depression: a meta-analysis of randomized, double-blind and sham-controlled trials. Neuropsychopharmacology. 2013;38(4):543-551.
42. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
43. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
44. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder. J Clin Psychiatry. 2014;75(12):1394-1401.
45. Fitzgerald PB, Hoy K, Daskalakis ZJ, et al. A randomized trial of the anti-depressant effects of low- and high-frequency transcranial magnetic stimulation in treatment-resistant depression. Depress Anxiety. 2009;26(3):229-234.
46. Dumas R, Padovani R, Richieri R, et al. Repetitive transcranial magnetic stimulation in major depression: response factor [in French]. Encephale. 2012;38(4):360-368.
47. Fregni F, Marcolin MA, Myczkowski M, et al. Predictors of antidepressant response in clinical trials of transcranial magnetic stimulation. Int. J. Neuropsychopharmacol. 2006;9(6):641-654.
48. Kennedy SH, Giacobbe P, Rizvi SJ, et al. Deep brain stimulation for treatment-resistant depression: follow-up after 3 to 6 years. Am J Psychiatry. 2011;168(5):502-510.
49. Taghva AS, Malone DA, Rezai AR. Deep brain stimulation for treatment-resistant depression. World Neurosurg. 2013;80(3-4):S27.e17-S27.e24.
50. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 2003;65:193-207.
51. Mayberg HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
52. Jiménez F, Velasco F, Salín-Pascual R, et al. Neuromodulation of the inferior thalamic peduncle for major depression and obsessive compulsive disorder. Acta Neurochir Suppl. 2007;97(pt 2):393-398.
53. Jiménez F, Velasco F, Salin-Pascual R, et al. A patient with a resistant major depression disorder treated with deep brain stimulation in the inferior thalamic peduncle. Neurosurgery. 2005;57(3):585-593; discussion 585-593.
54. Bewernick BH, Hurlemann R, Matusch A, et al. Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry. 2010;67(2):110-116.
55. Schlaepfer TE, Bewernick BH, Kayser S, et al. Deep brain stimulation of the human reward system for major depression—rationale, outcomes and outlook. Neuropsychopharmacology. 2014;39(6):1303-1314.
56. Malone DA Jr, Dougherty DD, Rezai AR, et al. Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol Psychiatry. 2009;65(4):267-275.
57. Berlim MT, McGirr A, Van den Eynde F, et al. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
58. Schlaepfer TE, Bewernick BH, Kayser S, et al. Rapid effects of deep brain stimulation for treatment-resistant major depression. Biol Psychiatry. 2013;73(12):1204-1212.
59. Gálvez JF, Keser Z, Mwangi B, et al. The medial forebrain bundle as a deep brain stimulation target for treatment resistant depression: a review of published data. Prog Neuropsychopharmacol Biol Psychiatry. 2015;58:59-70.
60. Fenoy AJ, Schulz P, Selvaraj. Deep brain stimulation of the medial forebrain bundle: distinctive responses in resistant depression. J Affect Disord. 2016;203:143-151.
61. Riva-Posse P, Choi KS, Holtzheimer PE, et al. Defining critical white matter pathways mediating successful subcallosal cingulate deep brain stimulation for treatment-resistant depression. Biol Psychiatry. 2014;76(12):963-969.
62. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
63. Milev R V, Giacobbe P, Kennedy SH, et al; CANMAT Depression Work Group. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder: section 4. Neurostimulation treatments. Can J Psychiatry. 2016;61:561-575.
1. Berlim MT, Turecki G. What is the meaning of treatment resistant/refractory major depression (TRD)? A systematic review of current randomized trials. Eur Neuropsychopharmacol. 2007;17(11):696-707.
2. Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(suppl 13):23-29.
3. Petersen T, Papakostas GI, Posternak MA, et al. Empirical testing of two models for staging antidepressant treatment resistance. J Clin Psychopharmacol. 2005;25(4):336-341.
4. Souery D, Papakostas GI, Trivedi MH. Treatment-resistant depression. J Clin Psychiatry. 2006;67(suppl 6):16-22.
5. Souery D, Amsterdam J, de Montigny C, et al. Treatment resistant depression: methodological overview and operational criteria. Eur Neuropsychopharmacol. 1999;9(1-2):83-91.
6. Evans DL, Charney DS. Mood disorders and medical illness: a major public health problem. Biol. Psychiatry. 2003;54(3):177-180.
7. Sanacora G, Mason GF, Rothman DL, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160(3):577-579.
8. Merkl A, Heuser I, Bajbouj M. Antidepressant electroconvulsive therapy: mechanism of action, recent advances and limitations. Exp Neurol. 2009;219(1):20-26.
9. Perera TD, Coplan JD, Lisanby SH, et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 2007;27(18):4894-4901.
10. Abbott CC, Gallegos P, Rediske N et al. A review of longitudinal electroconvulsive therapy: neuroimaging investigations. J Geriatr Psychiatry Neurol. 2014;27(1):33-46.
11. Heijnen WT, Birkenhäger TK, Wierdsma AI, et al. Antidepressant pharmacotherapy failure and response to subsequent electroconvulsive therapy: a meta-analysis. J Clin Psychopharmacol. 2010;30(5):616-619.
12. UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361(9360):799-808.
13. Semkovska M, Landau S, Dunne R et al. Bitemporal versus high-dose unilateral twice-weekly electroconvulsive therapy for depression (EFFECT-Dep): a pragmatic, randomized, non-inferiority trial. Am J Psychiatry. 2016;173(4):408-417.
14. Charlson F, Siskind D, Doi SA, et al. ECT efficacy and treatment course: a systematic review and meta-analysis of twice vs thrice weekly schedules. J Affect Disord. 2012;138(1-2):1-8.
15. Loo CK, Kaill A, Paton P, et al. The difficult-to-treat electroconvulsive therapy patient—strategies for augmenting outcomes. J Affect Disord. 2010;124(3):219-227.
16. de Vreede IM, Burger H, van Vliet IM. Prediction of response to ECT with routinely collected data in major depression. J Affect Disord. 2005;86(2-3):323-327.
17. Semkovska M, McLoughlin DM. Objective cognitive performance associated with electroconvulsive therapy for depression: a systematic review and meta-analysis. Biol Psychiatry. 2010;68(6):568-577.
18. Baghai TC, Marcuse A, Brosch M, et al. The influence of concomitant antidepressant medication on safety, tolerability and clinical effectiveness of electroconvulsive therapy. World J Biol Psychiatry. 2006;7(2):82-90.
19. Ben-Menachem E, Mañon-Espaillat R, Ristanovic R, et al. Vagus nerve stimulation for treatment of partial seizures: 1. A controlled study of effect on seizures. First International Vagus Nerve Stimulation Study Group. Epilepsia. 1994;35(3):616-626.
20. Nemeroff CB, Mayberg HS, Krahl SE, et al. VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology. 2006;31(7):1345-1355.
21. Matthews K, Eljamel MS. Vagus nerve stimulation and refractory depression: please can you switch me on doctor? Br J Psychiatry. 2003;183:181-183.
22. George MS, Rush AJ, Sackeim HA, et al. Vagus nerve stimulation (VNS): utility in neuropsychiatric disorders. Int J Neuropsychopharmacol. 2003;6(1):73-83.
23. Harden CL, Pulver MC, Ravdin LD, et al. A pilot study of mood in epilepsy patients treated with vagus nerve stimulation. Epilepsy Behav. 2000;1(2):93-99.
24. Elger G, Hoppe C, Falkai P, et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res. 2000;42(2-3):203-210.
25. Krahl SE, Clark KB, Smith DC, et al. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39(7):709-714.
26. Ben-Menachem E, Hamberger A, Hedner T, et al. Effects of vagus nerve stimulation on amino acids and other metabolites in the CSF of patients with partial seizures. Epilepsy Res. 1995;20(3):221-227.
27. Henry TR, Bakay RA, Votaw JR, et al. Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy: I. Acute effects at high and low levels of stimulation. Epilepsia. 1998;39(9):983-990.
28. O’Keane V, Dinan TG, Scott L, et al. Changes in hypothalamic-pituitary-adrenal axis measures after vagus nerve stimulation therapy in chronic depression. Biol Psychiatry. 2005;58(12):963-968.
29. Rush AJ, George MS, Sackeim HA, et al. Vagus nerve stimulation (VNS) for treatment-resistant depressions: a multicenter study. Biol Psychiatry. 2000;47(4):276-286.
30. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology. 2001;25(5):713-728.
31. Armitage R, Husain M, Hoffmann R, et al. The effects of vagus nerve stimulation on sleep EEG in depression: a preliminary report. J Psychosom Res. 2003;54(5):475-482.
32. Rush AJ, Marangell LB, Sackeim HA, et al. Vagus nerve stimulation for treatment-resistant depression: a randomized, controlled acute phase trial. Biol Psychiatry. 2005;58(5):347-354.
33. Martin JL, Martín-Sánchez E. Systematic review and meta-analysis of vagus nerve stimulation in the treatment of depression: variable results based on study designs. Eur Psychiatry. 2012;27(3):147-155.
34. Shah A, Carreno FR, Frazer A. Therapeutic modalities for treatment resistant depression: focus on vagal nerve stimulation and ketamine. Clin Psychopharmacol Neurosci. 2014;12(2):83-93.
35. Aaronson ST, Carpenter LL, Conway CR, et al. Vagus nerve stimulation therapy randomized to different amounts of electrical charge for treatment-resistant depression: acute and chronic effects. Brain Stimul. 2013;6(4):631-640.
36. Daban C, Martinez-Aran A, Cruz N, et al. Safety and efficacy of vagus nerve stimulation in treatment-resistant depression. A systematic review. J Affect Disord. 2008;110(1-2):1-15.
37. Eitan R, Lerer B. Nonpharmacological, somatic treatments of depression: electroconvulsive therapy and novel brain stimulation modalities. Dialogues Clin Neurosci. 2006;8(2):241-258.
38. Lam RW, Chan P, Wilkins-Ho M, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and metaanalysis. Can J Psychiatry. 2008;53(9):621-631.
39. Fitzgerald PB, Fountain S, Daskalakis ZJ. A comprehensive review of the effects of rTMS on motor cortical excitability and inhibition. Clin Neurophysiol. 2006;117(12):2584-2596.
40. Fitzgerald PB, Oxley TJ, Laird AR, et al. An analysis of functional neuroimaging studies of dorsolateral prefrontal cortical activity in depression. Psychiatry Res. 2006;148(1):33-45.
41. Berlim MT, Van den Eynde F, Daskalakis ZJ. Clinically meaningful efficacy and acceptability of low-frequency repetitive transcranial magnetic stimulation (rTMS) for treating primary major depression: a meta-analysis of randomized, double-blind and sham-controlled trials. Neuropsychopharmacology. 2013;38(4):543-551.
42. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
43. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
44. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder. J Clin Psychiatry. 2014;75(12):1394-1401.
45. Fitzgerald PB, Hoy K, Daskalakis ZJ, et al. A randomized trial of the anti-depressant effects of low- and high-frequency transcranial magnetic stimulation in treatment-resistant depression. Depress Anxiety. 2009;26(3):229-234.
46. Dumas R, Padovani R, Richieri R, et al. Repetitive transcranial magnetic stimulation in major depression: response factor [in French]. Encephale. 2012;38(4):360-368.
47. Fregni F, Marcolin MA, Myczkowski M, et al. Predictors of antidepressant response in clinical trials of transcranial magnetic stimulation. Int. J. Neuropsychopharmacol. 2006;9(6):641-654.
48. Kennedy SH, Giacobbe P, Rizvi SJ, et al. Deep brain stimulation for treatment-resistant depression: follow-up after 3 to 6 years. Am J Psychiatry. 2011;168(5):502-510.
49. Taghva AS, Malone DA, Rezai AR. Deep brain stimulation for treatment-resistant depression. World Neurosurg. 2013;80(3-4):S27.e17-S27.e24.
50. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 2003;65:193-207.
51. Mayberg HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
52. Jiménez F, Velasco F, Salín-Pascual R, et al. Neuromodulation of the inferior thalamic peduncle for major depression and obsessive compulsive disorder. Acta Neurochir Suppl. 2007;97(pt 2):393-398.
53. Jiménez F, Velasco F, Salin-Pascual R, et al. A patient with a resistant major depression disorder treated with deep brain stimulation in the inferior thalamic peduncle. Neurosurgery. 2005;57(3):585-593; discussion 585-593.
54. Bewernick BH, Hurlemann R, Matusch A, et al. Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry. 2010;67(2):110-116.
55. Schlaepfer TE, Bewernick BH, Kayser S, et al. Deep brain stimulation of the human reward system for major depression—rationale, outcomes and outlook. Neuropsychopharmacology. 2014;39(6):1303-1314.
56. Malone DA Jr, Dougherty DD, Rezai AR, et al. Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol Psychiatry. 2009;65(4):267-275.
57. Berlim MT, McGirr A, Van den Eynde F, et al. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
58. Schlaepfer TE, Bewernick BH, Kayser S, et al. Rapid effects of deep brain stimulation for treatment-resistant major depression. Biol Psychiatry. 2013;73(12):1204-1212.
59. Gálvez JF, Keser Z, Mwangi B, et al. The medial forebrain bundle as a deep brain stimulation target for treatment resistant depression: a review of published data. Prog Neuropsychopharmacol Biol Psychiatry. 2015;58:59-70.
60. Fenoy AJ, Schulz P, Selvaraj. Deep brain stimulation of the medial forebrain bundle: distinctive responses in resistant depression. J Affect Disord. 2016;203:143-151.
61. Riva-Posse P, Choi KS, Holtzheimer PE, et al. Defining critical white matter pathways mediating successful subcallosal cingulate deep brain stimulation for treatment-resistant depression. Biol Psychiatry. 2014;76(12):963-969.
62. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
63. Milev R V, Giacobbe P, Kennedy SH, et al; CANMAT Depression Work Group. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder: section 4. Neurostimulation treatments. Can J Psychiatry. 2016;61:561-575.
Benzodiazepines: Sensible prescribing in light of the risks
As a group, anxiety disorders are the most common mental illness in the Unites States, affecting 40 million adults. There is a nearly 30% lifetime prevalence of anxiety disorders in the general population.1 DSM-5 anxiety disorders include generalized anxiety disorder, social anxiety disorder (social phobia), panic disorder, specific phobia, and separation anxiety disorder. Although DSM-IV-TR also classified obsessive-compulsive disorder (OCD) and posttraumatic stress disorder (PTSD) as anxiety disorders, these diagnoses were reclassified in DSM-5. Anxiety also is a frequent symptom of many other psychiatric disorders, especially major depressive disorder.

Although benzodiazepines have many potential uses, they also carry risks that prescribers should recognize. This article reviews some of the risks of benzodiazepine use, identifies patients with higher risks of adverse effects, and presents a practical approach to prescribing these medications.
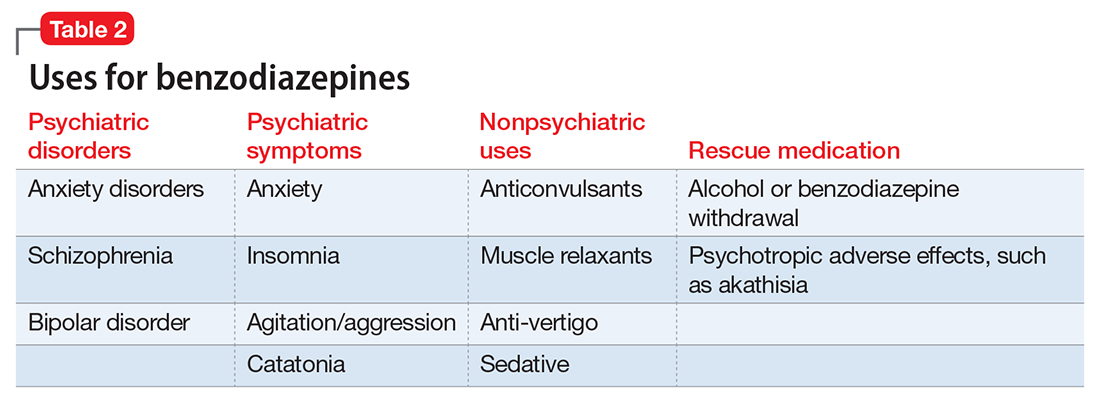
A wide range of risks
Abuse and addiction. Perhaps the most commonly recognized risk associated with benzodiazepine use is the potential for abuse and addiction.4 Prolonged benzodiazepine use typically results in physiologic tolerance, requiring higher dosing to achieve the same initial effect.5 American Psychiatric Association practice guidelines recognize the potential for benzodiazepine use to result in symptoms of dependence, including cravings and withdrawal, stating that “with ongoing use, all benzodiazepines will produce physiological dependence in most patients.”6 High-potency, short-acting compounds such as alprazolam have a higher risk for dependence, toxicity, and abuse.7 However, long-acting benzodiazepines (such as clonazepam) also can be habit-forming.8 Because of these properties, it is generally advisable to avoid prescribing benzodiazepines (and short-acting compounds in particular) when treating patients with current or past substance use disorders, except when treating withdrawal.9
Limited efficacy for other disorders. Although benzodiazepines can help reduce anxiety in patients with anxiety disorders, they have shown less promise in treating other disorders in which anxiety is a common symptom. Treating PTSD with benzodiazepines does not appear to offer any advantage over placebo, and may even result in increased symptoms over time.10,11 There is limited evidence supporting the use of benzodiazepines to treat OCD.12,13 Patients with borderline personality disorder who are treated with benzodiazepines may experience an increase in behavioral dysregulation.14
Physical ailments. Benzodiazepines can affect comorbid physical ailments. One study found that long-term benzodiazepine use among patients with comorbid pain disorders was correlated with high utilization of medical services and high disability levels.15 Benzodiazepine use also has been associated with an increased risk of exacerbating respiratory conditions, such as chronic obstructive pulmonary disease,16 and increased risk of pneumonia.17,18
Pregnancy and breastfeeding. Benzodiazepines carry risks for women who are pregnant or breastfeeding. Benzodiazepine use during pregnancy may increase the relative risk of major malformations and oral clefts. It also may result in neonatal lethargy, sedation, and weight loss. Benzodiazepine withdrawal symptoms can occur in the neonate.19 Benzodiazepines are secreted in breast milk and can result in sedation among breastfed infants.20
Geriatric patients. Older adults may be particularly vulnerable to the adverse effects of benzodiazepines. The Beers Criteria for Potentially Inappropriate Medication Use in Older Adults recommends against prescribing benzodiazepines to geriatric patients.21 Benzodiazepine use has been associated with an increased risk for falls among older adults,22,23 with an increased risk of fractures24 that can be fatal.25 Benzodiazepines also have been associated with an increased risk of cognitive dysfunction and dementia.26,27 Despite the documented risks of using benzodiazepines in geriatric patients, benzodiazepines continue to be frequently prescribed to this age group.28,29 One study found that the rate of prescribing benzodiazepines by primary care physicians increased from 2003 to 2012, primarily among older adults with no diagnosis of pain or a psychiatric disorder.30
Mortality. Benzodiazepine use also carries an increased risk of mortality. Benzodiazepine users are at increased risk of motor vehicle accidents because of difficulty maintaining road position.31 Some research has shown that patients with schizophrenia treated with benzodiazepines have an increased risk of death compared with those who are prescribed antipsychotics or antidepressants.32 Another study showed that patients with schizophrenia who were prescribed benzodiazepines had a greater risk of death by suicide and accidental poisoning.33 Benzodiazepine use has been associated with suicidal ideation and an increased risk of suicide.34 Prescription opioids and benzodiazepines are the top 2 causes of overdose-related deaths (benzodiazepines are involved in approximately 31% of fatal overdoses35), and from 2002 to 2015 there was a 4.3-fold increase in deaths from benzodiazepine overdose in the United States.36 CDC guidelines recommend against co-prescribing opioids and benzodiazepines because of the risk of death by respiratory depression.37 As of August 2016, the FDA required black-box warnings for opioids and benzodiazepines regarding the risk of respiratory depression and death when these agents are used in combination, noting that “If these medicines are prescribed together, limit the dosages and duration of each drug to the minimum possible while achieving the desired clinical effect.”38,39
A sensible approach to prescribing
Given the risks posed by benzodiazepines, what would constitute a sensible approach to their use? Clearly, there are some patients for whom benzodiazepine use should be minimized or avoided (Table 3). In a patient who is deemed a good candidate for benzodiazepines, a long-acting agent may be preferable because of the increased risk of dependence associated with short-acting compounds. Start with a low dose, and use the lowest dose that adequately treats the patient’s symptoms.40 Using scheduled rather than “as-needed” dosing may help reduce behavioral escape patterns that reinforce anxiety and dependence in the long term.
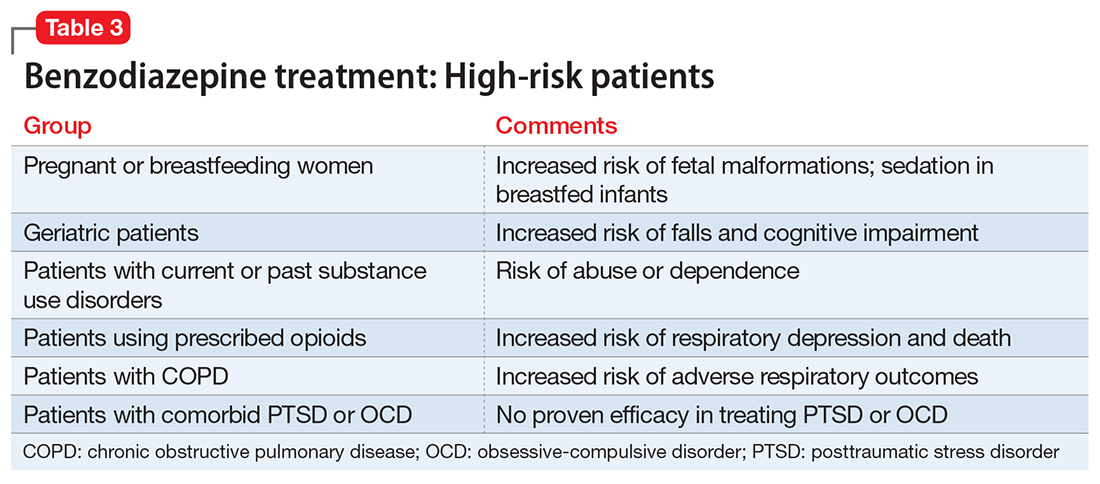
Before starting a patient on a benzodiazepine, discuss with him (her) the risks of use and an exit plan to discontinue the medication. For example, a benzodiazepine may be prescribed at the same time as a selective serotonin reuptake inhibitor (SSRI), with the goal of weaning off the benzodiazepine once the SSRI has achieved efficacy.6 Inform the patient that prescribing or treatment may be terminated if it is discovered that the patient is abusing or diverting the medication (regularly reviewing the state prescription monitoring program database can help determine if this has occurred). Strongly consider using non-benzodiazepine treatments for anxiety with (or eventually in place of) benzodiazepines (Table 441).
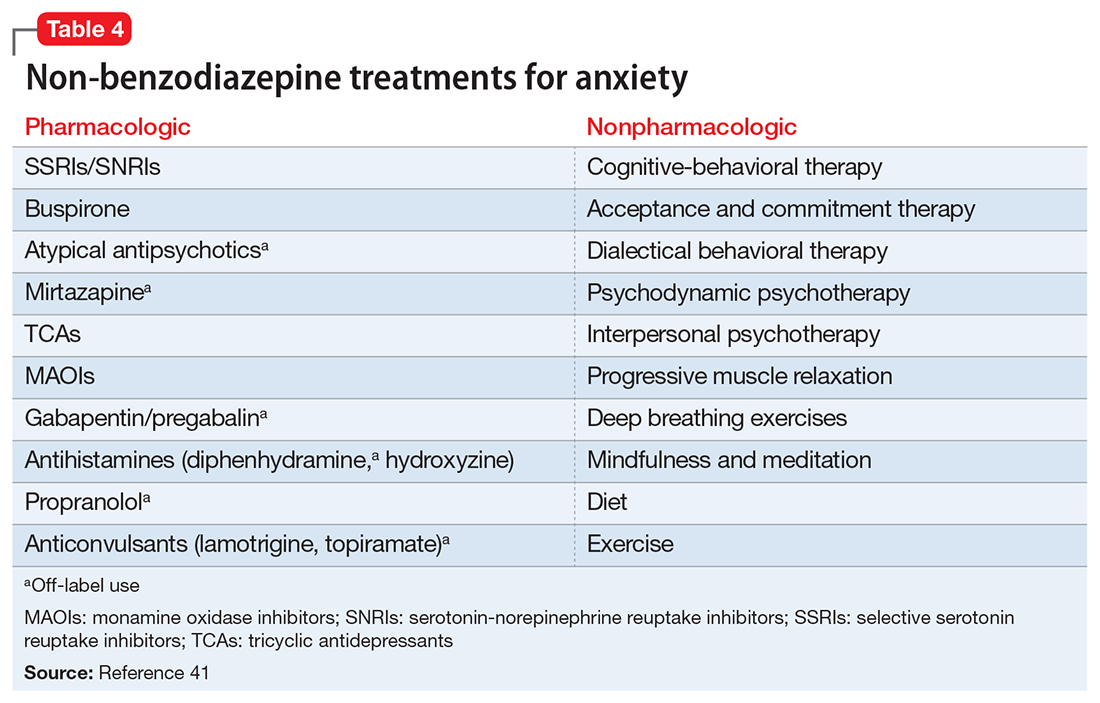
Reducing or stopping benzodiazepines can be challenging.42 Patients often are reluctant to stop such medications, and abrupt cessation can cause severe withdrawal. Benzodiazepine withdrawal symptoms can be severe or even fatal. Therefore, a safe and collaborative approach to reducing or stopping benzodiazepines is necessary. A starting point might be to review the risks associated with benzodiazepine use with the patient and ask about the frequency of use. Discuss with the patient a slow taper, perhaps reducing the dose by 10% to 25% increments weekly to biweekly.43,44 Less motivated patients may require a slower taper, more time, or repeated discussions. When starting a dose reduction, notify the patient that some rebound anxiety or insomnia are to be expected. With any progress the patient makes toward reducing his usage, congratulate him on such progress.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Balon R, Fava GA, Rickels K. Need for a realistic appraisal of benzodiazepines. World Psychiatry. 2015;14(2):243-244.
3. Ashton CH. Benzodiazepine equivalence table. http://www.benzo.org.uk/bzequiv.htm. Revised April 2007. Accessed May 3, 2017.
4. National Institute on Drug Abuse. Commonly abused drugs. https://d14rmgtrwzf5a.cloudfront.net/sites/default/files/commonly_abused_drugs_3.pdf. Revised January 2016. Accessed January 9, 2018.
5. Licata SC, Rowlett JK. Abuse and dependence liability of benzodiazepine-type drugs: GABA(A) receptor modulation and beyond. Pharmacol Biochem Behav. 2008;90(1):74-89.
6. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Published January 2009. Accessed May 3, 2017.
7. Salzman C. The APA Task Force report on benzodiazepine dependence, toxicity, and abuse. Am J Psychiatry. 1991;148(2):151-152.
8. Bushnell GA, Stürmer T, Gaynes BN, et al. Simultaneous antidepressant and benzodiazepine new use and subsequent long-term benzodiazepine use in adults with depression, United States, 2001-2014. JAMA Psychiatry. 2017;74(7):747-755.
9. O’Brien PL, Karnell LH, Gokhale M, et al. Prescribing of benzodiazepines and opioids to individuals with substance use disorders. Drug Alcohol Depend. 2017;178:223-230.
10. Mellman TA, Bustamante V, David D, et al. Hypnotic medication in the aftermath of trauma. J Clin Psychiatry. 2002;63(12):1183-1184.
11. Gelpin E, Bonne O, Peri T, et al. Treatment of recent trauma survivors with benzodiazepines: a prospective study. J Clin Psychiatry. 1996;57(9):390-394.
12. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed May 3, 2017.
13. Abdel-Ahad P, Kazour F. Non-antidepressant pharmacological treatment of obsessive compulsive disorder: a comprehensive review. Curr Clin Pharmacol. 2015;10(2):97-111.
14. Gardner DL, Cowdry RW. Alprazolam-induced dyscontrol in borderline personality disorder. Am J Psychiatry. 1985;142(1):98-100.
15. Ciccone DS, Just N, Bandilla EB, et al. Psychological correlates of opioid use in patients with chronic nonmalignant pain: a preliminary test of the downhill spiral hypothesis. J Pain Symptom Manage. 2000;20(3):180-192.
16. Vozoris NT, Fischer HD, Wang X, et al. Benzodiazepine drug use and adverse respiratory outcomes among older adults with COPD. Eur Respir J. 2014;44(2):332-340.
17. Obiora E, Hubbard R, Sanders RD, et al. The impact of benzodiazepines on occurrence of pneumonia and mortality from pneumonia: a nested case-control and survival analysis in a population-based cohort. Thorax. 2013;68(2):163-170.
18. Taipale H, Tolppanen AM, Koponen M, et al. Risk of pneumonia associated with incident benzodiazepine use among community-dwelling adults with Alzheimer disease. CMAJ. 2017;189(14):E519-E529.
19. Iqbal MM, Sobhan T, Ryals T. Effects of commonly used benzodiazepines on the fetus, the neonate, and the nursing infant. Psychiatric Serv. 2002;53:39-49.
20. U.S. National Library of Medicine, TOXNET Toxicology Data Network. Lactmed: alprazolam. http://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+lactmed:@term+@DOCNO+335. Accessed May 3, 2017.
21. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
22. Ray WA, Thapa PB, Gideon P. Benzodiazepines and the risk of falls in nursing home residents. J Am Geriatr Soc. 2000;48(6):682-685.
23. Woolcott JC, Richardson KJ, Wiens MO, et al. Meta-analysis of the impact of 9 medication classes on falls in elderly persons. Arch Intern Med. 2009;169(21):1952-1960.
24. Bolton JM, Morin SN, Majumdar SR, et al. Association of mental disorders and related medication use with risk for major osteoporotic fractures. JAMA Psychiatry. 2017;74(6):641-648.
25. Pariente A, Dartiques JF, Benichou J, et al. Benzodiazepines and injurious falls in community dwelling elders. Drugs Aging. 2008;25(1):61-70.
26. Lagnaoui R, Tournier M, Moride Y, et al. The risk of cognitive impairment in older community-dwelling women after benzodiazepine use. Age Ageing. 2009;38(2):226-228.
27. Billioti de Gage S, Bégaud B, Bazin F, et al. Benzodiazepine use and risk of dementia: prospective population based study. BMJ. 2012;345:e6231. doi: 10.1136/bmj.e6231.
28. Olfson M, King M, Schoenbaum M. Benzodiazepine use in the United States. JAMA Psychiatry. 2015;72(2):136-142.
29. Maust DT, Kales HC, Wiechers IR, et al. No end in sight: benzodiazepine use in older adults in the United States. J Am Geriatr Soc. 2016;64(12):2546-2553.
30. Maust DT, Blow FC, Wiechers IR, et al. National trends in antidepressant, benzodiazepine, and other sedative-hypnotic treatment of older adults in psychiatric and primary care. J Clin Psychiatry. 2017;78(4):e363-e371.
31. Rapoport MJ, Lanctôt KL, Streiner DL, et al. Benzodiazepine use and driving: a meta-analysis. J Clin Psychiatry. 2009;70(5):663-673.
32. Tiihonen J, Mittendorfer-Rutz E, Torniainen M, et al. Mortality and cumulative exposure to antipsychotics, antidepressants, and benzodiazepines in patients with schizophrenia: an observational follow-up study. Am J Psychiatry. 2016;173(6):600-606.
33. Fontanella CA, Campo JV, Phillips GS, et al. Benzodiazepine use and risk of mortality among patients with schizophrenia: a retrospective longitudinal study. J Clin Psychiatry. 2016;77(5):661-667.
34. McCall WV, Benca RM, Rosenguist PB, et al. Hypnotic medications and suicide: risk, mechanisms, mitigation, and the FDA. Am J Psychiatry. 2017;174(1):18-25.
35. Bachhuber MA, Hennessy S, Cunningham CO, et al. Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013. Am J Public Health. 2016;106(4):686-688.
36. National Institute on Drug Abuse. Overdose death rates. https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates. Updated September 2017. Accessed January 8, 2018.
37. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recomm Rep 2016;65(1):1-49.
38. U.S. Food and Drug Administration. FDA requires strong warnings for opioid analgesics, prescription opioid cough products, and benzodiazepine labeling related to serious risks and death from combined use [press release]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518697.htm. Published August 31, 2016. Accessed May 3, 2017.
39. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about serious risks and death when combining opioid pain or cough medicines with benzodiazepines; requires its strongest warning. http://www.fda.gov/Drugs/DrugSafety/ucm518473.htm. Published August 31, 2016. Accessed May 3, 2017.
40. National Institute for Health and Care Excellence. Controlled drugs: safe use and management. https://www.nice.org.uk/guidance/ng46/evidence/full-guideline-pdf-2427186353. Published April 2016. Accessed July 25, 2017.
41. Stahl SM. Anxiety disorders and anxiolytics. In: Stahl’s essential psychopharmacology. 3rd ed. New York, NY: Cambridge University Press; 2008:721-772.
42. Paquin AM, Zimmerman K, Rudolph JL. Risk versus risk: a review of benzodiazepine reduction in older adults. Expert Opin Drug Saf. 2014;13(7):919-934.
43. Nardi AE, Freire RC, Valença AM, et al. Tapering clonazepam in patients with panic disorder after at least 3 years of treatment. J Clin Psychopharmacol. 2010;30(3):290-293.
44. Tampi R. How to wean geriatric patients off benzodiazepines. Psychiatric News. http://psychnews.psychiatryonline.org/doi/full/10.1176/appi.pn.2016.PP3b6. Published March 18, 2016. Accessed May 3, 2017.
As a group, anxiety disorders are the most common mental illness in the Unites States, affecting 40 million adults. There is a nearly 30% lifetime prevalence of anxiety disorders in the general population.1 DSM-5 anxiety disorders include generalized anxiety disorder, social anxiety disorder (social phobia), panic disorder, specific phobia, and separation anxiety disorder. Although DSM-IV-TR also classified obsessive-compulsive disorder (OCD) and posttraumatic stress disorder (PTSD) as anxiety disorders, these diagnoses were reclassified in DSM-5. Anxiety also is a frequent symptom of many other psychiatric disorders, especially major depressive disorder.
Although benzodiazepines have many potential uses, they also carry risks that prescribers should recognize. This article reviews some of the risks of benzodiazepine use, identifies patients with higher risks of adverse effects, and presents a practical approach to prescribing these medications.
A wide range of risks
Abuse and addiction. Perhaps the most commonly recognized risk associated with benzodiazepine use is the potential for abuse and addiction.4 Prolonged benzodiazepine use typically results in physiologic tolerance, requiring higher dosing to achieve the same initial effect.5 American Psychiatric Association practice guidelines recognize the potential for benzodiazepine use to result in symptoms of dependence, including cravings and withdrawal, stating that “with ongoing use, all benzodiazepines will produce physiological dependence in most patients.”6 High-potency, short-acting compounds such as alprazolam have a higher risk for dependence, toxicity, and abuse.7 However, long-acting benzodiazepines (such as clonazepam) also can be habit-forming.8 Because of these properties, it is generally advisable to avoid prescribing benzodiazepines (and short-acting compounds in particular) when treating patients with current or past substance use disorders, except when treating withdrawal.9
Limited efficacy for other disorders. Although benzodiazepines can help reduce anxiety in patients with anxiety disorders, they have shown less promise in treating other disorders in which anxiety is a common symptom. Treating PTSD with benzodiazepines does not appear to offer any advantage over placebo, and may even result in increased symptoms over time.10,11 There is limited evidence supporting the use of benzodiazepines to treat OCD.12,13 Patients with borderline personality disorder who are treated with benzodiazepines may experience an increase in behavioral dysregulation.14
Physical ailments. Benzodiazepines can affect comorbid physical ailments. One study found that long-term benzodiazepine use among patients with comorbid pain disorders was correlated with high utilization of medical services and high disability levels.15 Benzodiazepine use also has been associated with an increased risk of exacerbating respiratory conditions, such as chronic obstructive pulmonary disease,16 and increased risk of pneumonia.17,18
Pregnancy and breastfeeding. Benzodiazepines carry risks for women who are pregnant or breastfeeding. Benzodiazepine use during pregnancy may increase the relative risk of major malformations and oral clefts. It also may result in neonatal lethargy, sedation, and weight loss. Benzodiazepine withdrawal symptoms can occur in the neonate.19 Benzodiazepines are secreted in breast milk and can result in sedation among breastfed infants.20
Geriatric patients. Older adults may be particularly vulnerable to the adverse effects of benzodiazepines. The Beers Criteria for Potentially Inappropriate Medication Use in Older Adults recommends against prescribing benzodiazepines to geriatric patients.21 Benzodiazepine use has been associated with an increased risk for falls among older adults,22,23 with an increased risk of fractures24 that can be fatal.25 Benzodiazepines also have been associated with an increased risk of cognitive dysfunction and dementia.26,27 Despite the documented risks of using benzodiazepines in geriatric patients, benzodiazepines continue to be frequently prescribed to this age group.28,29 One study found that the rate of prescribing benzodiazepines by primary care physicians increased from 2003 to 2012, primarily among older adults with no diagnosis of pain or a psychiatric disorder.30
Mortality. Benzodiazepine use also carries an increased risk of mortality. Benzodiazepine users are at increased risk of motor vehicle accidents because of difficulty maintaining road position.31 Some research has shown that patients with schizophrenia treated with benzodiazepines have an increased risk of death compared with those who are prescribed antipsychotics or antidepressants.32 Another study showed that patients with schizophrenia who were prescribed benzodiazepines had a greater risk of death by suicide and accidental poisoning.33 Benzodiazepine use has been associated with suicidal ideation and an increased risk of suicide.34 Prescription opioids and benzodiazepines are the top 2 causes of overdose-related deaths (benzodiazepines are involved in approximately 31% of fatal overdoses35), and from 2002 to 2015 there was a 4.3-fold increase in deaths from benzodiazepine overdose in the United States.36 CDC guidelines recommend against co-prescribing opioids and benzodiazepines because of the risk of death by respiratory depression.37 As of August 2016, the FDA required black-box warnings for opioids and benzodiazepines regarding the risk of respiratory depression and death when these agents are used in combination, noting that “If these medicines are prescribed together, limit the dosages and duration of each drug to the minimum possible while achieving the desired clinical effect.”38,39
A sensible approach to prescribing
Given the risks posed by benzodiazepines, what would constitute a sensible approach to their use? Clearly, there are some patients for whom benzodiazepine use should be minimized or avoided (Table 3). In a patient who is deemed a good candidate for benzodiazepines, a long-acting agent may be preferable because of the increased risk of dependence associated with short-acting compounds. Start with a low dose, and use the lowest dose that adequately treats the patient’s symptoms.40 Using scheduled rather than “as-needed” dosing may help reduce behavioral escape patterns that reinforce anxiety and dependence in the long term.
Before starting a patient on a benzodiazepine, discuss with him (her) the risks of use and an exit plan to discontinue the medication. For example, a benzodiazepine may be prescribed at the same time as a selective serotonin reuptake inhibitor (SSRI), with the goal of weaning off the benzodiazepine once the SSRI has achieved efficacy.6 Inform the patient that prescribing or treatment may be terminated if it is discovered that the patient is abusing or diverting the medication (regularly reviewing the state prescription monitoring program database can help determine if this has occurred). Strongly consider using non-benzodiazepine treatments for anxiety with (or eventually in place of) benzodiazepines (Table 441).
Reducing or stopping benzodiazepines can be challenging.42 Patients often are reluctant to stop such medications, and abrupt cessation can cause severe withdrawal. Benzodiazepine withdrawal symptoms can be severe or even fatal. Therefore, a safe and collaborative approach to reducing or stopping benzodiazepines is necessary. A starting point might be to review the risks associated with benzodiazepine use with the patient and ask about the frequency of use. Discuss with the patient a slow taper, perhaps reducing the dose by 10% to 25% increments weekly to biweekly.43,44 Less motivated patients may require a slower taper, more time, or repeated discussions. When starting a dose reduction, notify the patient that some rebound anxiety or insomnia are to be expected. With any progress the patient makes toward reducing his usage, congratulate him on such progress.
As a group, anxiety disorders are the most common mental illness in the Unites States, affecting 40 million adults. There is a nearly 30% lifetime prevalence of anxiety disorders in the general population.1 DSM-5 anxiety disorders include generalized anxiety disorder, social anxiety disorder (social phobia), panic disorder, specific phobia, and separation anxiety disorder. Although DSM-IV-TR also classified obsessive-compulsive disorder (OCD) and posttraumatic stress disorder (PTSD) as anxiety disorders, these diagnoses were reclassified in DSM-5. Anxiety also is a frequent symptom of many other psychiatric disorders, especially major depressive disorder.
Although benzodiazepines have many potential uses, they also carry risks that prescribers should recognize. This article reviews some of the risks of benzodiazepine use, identifies patients with higher risks of adverse effects, and presents a practical approach to prescribing these medications.
A wide range of risks
Abuse and addiction. Perhaps the most commonly recognized risk associated with benzodiazepine use is the potential for abuse and addiction.4 Prolonged benzodiazepine use typically results in physiologic tolerance, requiring higher dosing to achieve the same initial effect.5 American Psychiatric Association practice guidelines recognize the potential for benzodiazepine use to result in symptoms of dependence, including cravings and withdrawal, stating that “with ongoing use, all benzodiazepines will produce physiological dependence in most patients.”6 High-potency, short-acting compounds such as alprazolam have a higher risk for dependence, toxicity, and abuse.7 However, long-acting benzodiazepines (such as clonazepam) also can be habit-forming.8 Because of these properties, it is generally advisable to avoid prescribing benzodiazepines (and short-acting compounds in particular) when treating patients with current or past substance use disorders, except when treating withdrawal.9
Limited efficacy for other disorders. Although benzodiazepines can help reduce anxiety in patients with anxiety disorders, they have shown less promise in treating other disorders in which anxiety is a common symptom. Treating PTSD with benzodiazepines does not appear to offer any advantage over placebo, and may even result in increased symptoms over time.10,11 There is limited evidence supporting the use of benzodiazepines to treat OCD.12,13 Patients with borderline personality disorder who are treated with benzodiazepines may experience an increase in behavioral dysregulation.14
Physical ailments. Benzodiazepines can affect comorbid physical ailments. One study found that long-term benzodiazepine use among patients with comorbid pain disorders was correlated with high utilization of medical services and high disability levels.15 Benzodiazepine use also has been associated with an increased risk of exacerbating respiratory conditions, such as chronic obstructive pulmonary disease,16 and increased risk of pneumonia.17,18
Pregnancy and breastfeeding. Benzodiazepines carry risks for women who are pregnant or breastfeeding. Benzodiazepine use during pregnancy may increase the relative risk of major malformations and oral clefts. It also may result in neonatal lethargy, sedation, and weight loss. Benzodiazepine withdrawal symptoms can occur in the neonate.19 Benzodiazepines are secreted in breast milk and can result in sedation among breastfed infants.20
Geriatric patients. Older adults may be particularly vulnerable to the adverse effects of benzodiazepines. The Beers Criteria for Potentially Inappropriate Medication Use in Older Adults recommends against prescribing benzodiazepines to geriatric patients.21 Benzodiazepine use has been associated with an increased risk for falls among older adults,22,23 with an increased risk of fractures24 that can be fatal.25 Benzodiazepines also have been associated with an increased risk of cognitive dysfunction and dementia.26,27 Despite the documented risks of using benzodiazepines in geriatric patients, benzodiazepines continue to be frequently prescribed to this age group.28,29 One study found that the rate of prescribing benzodiazepines by primary care physicians increased from 2003 to 2012, primarily among older adults with no diagnosis of pain or a psychiatric disorder.30
Mortality. Benzodiazepine use also carries an increased risk of mortality. Benzodiazepine users are at increased risk of motor vehicle accidents because of difficulty maintaining road position.31 Some research has shown that patients with schizophrenia treated with benzodiazepines have an increased risk of death compared with those who are prescribed antipsychotics or antidepressants.32 Another study showed that patients with schizophrenia who were prescribed benzodiazepines had a greater risk of death by suicide and accidental poisoning.33 Benzodiazepine use has been associated with suicidal ideation and an increased risk of suicide.34 Prescription opioids and benzodiazepines are the top 2 causes of overdose-related deaths (benzodiazepines are involved in approximately 31% of fatal overdoses35), and from 2002 to 2015 there was a 4.3-fold increase in deaths from benzodiazepine overdose in the United States.36 CDC guidelines recommend against co-prescribing opioids and benzodiazepines because of the risk of death by respiratory depression.37 As of August 2016, the FDA required black-box warnings for opioids and benzodiazepines regarding the risk of respiratory depression and death when these agents are used in combination, noting that “If these medicines are prescribed together, limit the dosages and duration of each drug to the minimum possible while achieving the desired clinical effect.”38,39
A sensible approach to prescribing
Given the risks posed by benzodiazepines, what would constitute a sensible approach to their use? Clearly, there are some patients for whom benzodiazepine use should be minimized or avoided (Table 3). In a patient who is deemed a good candidate for benzodiazepines, a long-acting agent may be preferable because of the increased risk of dependence associated with short-acting compounds. Start with a low dose, and use the lowest dose that adequately treats the patient’s symptoms.40 Using scheduled rather than “as-needed” dosing may help reduce behavioral escape patterns that reinforce anxiety and dependence in the long term.
Before starting a patient on a benzodiazepine, discuss with him (her) the risks of use and an exit plan to discontinue the medication. For example, a benzodiazepine may be prescribed at the same time as a selective serotonin reuptake inhibitor (SSRI), with the goal of weaning off the benzodiazepine once the SSRI has achieved efficacy.6 Inform the patient that prescribing or treatment may be terminated if it is discovered that the patient is abusing or diverting the medication (regularly reviewing the state prescription monitoring program database can help determine if this has occurred). Strongly consider using non-benzodiazepine treatments for anxiety with (or eventually in place of) benzodiazepines (Table 441).
Reducing or stopping benzodiazepines can be challenging.42 Patients often are reluctant to stop such medications, and abrupt cessation can cause severe withdrawal. Benzodiazepine withdrawal symptoms can be severe or even fatal. Therefore, a safe and collaborative approach to reducing or stopping benzodiazepines is necessary. A starting point might be to review the risks associated with benzodiazepine use with the patient and ask about the frequency of use. Discuss with the patient a slow taper, perhaps reducing the dose by 10% to 25% increments weekly to biweekly.43,44 Less motivated patients may require a slower taper, more time, or repeated discussions. When starting a dose reduction, notify the patient that some rebound anxiety or insomnia are to be expected. With any progress the patient makes toward reducing his usage, congratulate him on such progress.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Balon R, Fava GA, Rickels K. Need for a realistic appraisal of benzodiazepines. World Psychiatry. 2015;14(2):243-244.
3. Ashton CH. Benzodiazepine equivalence table. http://www.benzo.org.uk/bzequiv.htm. Revised April 2007. Accessed May 3, 2017.
4. National Institute on Drug Abuse. Commonly abused drugs. https://d14rmgtrwzf5a.cloudfront.net/sites/default/files/commonly_abused_drugs_3.pdf. Revised January 2016. Accessed January 9, 2018.
5. Licata SC, Rowlett JK. Abuse and dependence liability of benzodiazepine-type drugs: GABA(A) receptor modulation and beyond. Pharmacol Biochem Behav. 2008;90(1):74-89.
6. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Published January 2009. Accessed May 3, 2017.
7. Salzman C. The APA Task Force report on benzodiazepine dependence, toxicity, and abuse. Am J Psychiatry. 1991;148(2):151-152.
8. Bushnell GA, Stürmer T, Gaynes BN, et al. Simultaneous antidepressant and benzodiazepine new use and subsequent long-term benzodiazepine use in adults with depression, United States, 2001-2014. JAMA Psychiatry. 2017;74(7):747-755.
9. O’Brien PL, Karnell LH, Gokhale M, et al. Prescribing of benzodiazepines and opioids to individuals with substance use disorders. Drug Alcohol Depend. 2017;178:223-230.
10. Mellman TA, Bustamante V, David D, et al. Hypnotic medication in the aftermath of trauma. J Clin Psychiatry. 2002;63(12):1183-1184.
11. Gelpin E, Bonne O, Peri T, et al. Treatment of recent trauma survivors with benzodiazepines: a prospective study. J Clin Psychiatry. 1996;57(9):390-394.
12. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed May 3, 2017.
13. Abdel-Ahad P, Kazour F. Non-antidepressant pharmacological treatment of obsessive compulsive disorder: a comprehensive review. Curr Clin Pharmacol. 2015;10(2):97-111.
14. Gardner DL, Cowdry RW. Alprazolam-induced dyscontrol in borderline personality disorder. Am J Psychiatry. 1985;142(1):98-100.
15. Ciccone DS, Just N, Bandilla EB, et al. Psychological correlates of opioid use in patients with chronic nonmalignant pain: a preliminary test of the downhill spiral hypothesis. J Pain Symptom Manage. 2000;20(3):180-192.
16. Vozoris NT, Fischer HD, Wang X, et al. Benzodiazepine drug use and adverse respiratory outcomes among older adults with COPD. Eur Respir J. 2014;44(2):332-340.
17. Obiora E, Hubbard R, Sanders RD, et al. The impact of benzodiazepines on occurrence of pneumonia and mortality from pneumonia: a nested case-control and survival analysis in a population-based cohort. Thorax. 2013;68(2):163-170.
18. Taipale H, Tolppanen AM, Koponen M, et al. Risk of pneumonia associated with incident benzodiazepine use among community-dwelling adults with Alzheimer disease. CMAJ. 2017;189(14):E519-E529.
19. Iqbal MM, Sobhan T, Ryals T. Effects of commonly used benzodiazepines on the fetus, the neonate, and the nursing infant. Psychiatric Serv. 2002;53:39-49.
20. U.S. National Library of Medicine, TOXNET Toxicology Data Network. Lactmed: alprazolam. http://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+lactmed:@term+@DOCNO+335. Accessed May 3, 2017.
21. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
22. Ray WA, Thapa PB, Gideon P. Benzodiazepines and the risk of falls in nursing home residents. J Am Geriatr Soc. 2000;48(6):682-685.
23. Woolcott JC, Richardson KJ, Wiens MO, et al. Meta-analysis of the impact of 9 medication classes on falls in elderly persons. Arch Intern Med. 2009;169(21):1952-1960.
24. Bolton JM, Morin SN, Majumdar SR, et al. Association of mental disorders and related medication use with risk for major osteoporotic fractures. JAMA Psychiatry. 2017;74(6):641-648.
25. Pariente A, Dartiques JF, Benichou J, et al. Benzodiazepines and injurious falls in community dwelling elders. Drugs Aging. 2008;25(1):61-70.
26. Lagnaoui R, Tournier M, Moride Y, et al. The risk of cognitive impairment in older community-dwelling women after benzodiazepine use. Age Ageing. 2009;38(2):226-228.
27. Billioti de Gage S, Bégaud B, Bazin F, et al. Benzodiazepine use and risk of dementia: prospective population based study. BMJ. 2012;345:e6231. doi: 10.1136/bmj.e6231.
28. Olfson M, King M, Schoenbaum M. Benzodiazepine use in the United States. JAMA Psychiatry. 2015;72(2):136-142.
29. Maust DT, Kales HC, Wiechers IR, et al. No end in sight: benzodiazepine use in older adults in the United States. J Am Geriatr Soc. 2016;64(12):2546-2553.
30. Maust DT, Blow FC, Wiechers IR, et al. National trends in antidepressant, benzodiazepine, and other sedative-hypnotic treatment of older adults in psychiatric and primary care. J Clin Psychiatry. 2017;78(4):e363-e371.
31. Rapoport MJ, Lanctôt KL, Streiner DL, et al. Benzodiazepine use and driving: a meta-analysis. J Clin Psychiatry. 2009;70(5):663-673.
32. Tiihonen J, Mittendorfer-Rutz E, Torniainen M, et al. Mortality and cumulative exposure to antipsychotics, antidepressants, and benzodiazepines in patients with schizophrenia: an observational follow-up study. Am J Psychiatry. 2016;173(6):600-606.
33. Fontanella CA, Campo JV, Phillips GS, et al. Benzodiazepine use and risk of mortality among patients with schizophrenia: a retrospective longitudinal study. J Clin Psychiatry. 2016;77(5):661-667.
34. McCall WV, Benca RM, Rosenguist PB, et al. Hypnotic medications and suicide: risk, mechanisms, mitigation, and the FDA. Am J Psychiatry. 2017;174(1):18-25.
35. Bachhuber MA, Hennessy S, Cunningham CO, et al. Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013. Am J Public Health. 2016;106(4):686-688.
36. National Institute on Drug Abuse. Overdose death rates. https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates. Updated September 2017. Accessed January 8, 2018.
37. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recomm Rep 2016;65(1):1-49.
38. U.S. Food and Drug Administration. FDA requires strong warnings for opioid analgesics, prescription opioid cough products, and benzodiazepine labeling related to serious risks and death from combined use [press release]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518697.htm. Published August 31, 2016. Accessed May 3, 2017.
39. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about serious risks and death when combining opioid pain or cough medicines with benzodiazepines; requires its strongest warning. http://www.fda.gov/Drugs/DrugSafety/ucm518473.htm. Published August 31, 2016. Accessed May 3, 2017.
40. National Institute for Health and Care Excellence. Controlled drugs: safe use and management. https://www.nice.org.uk/guidance/ng46/evidence/full-guideline-pdf-2427186353. Published April 2016. Accessed July 25, 2017.
41. Stahl SM. Anxiety disorders and anxiolytics. In: Stahl’s essential psychopharmacology. 3rd ed. New York, NY: Cambridge University Press; 2008:721-772.
42. Paquin AM, Zimmerman K, Rudolph JL. Risk versus risk: a review of benzodiazepine reduction in older adults. Expert Opin Drug Saf. 2014;13(7):919-934.
43. Nardi AE, Freire RC, Valença AM, et al. Tapering clonazepam in patients with panic disorder after at least 3 years of treatment. J Clin Psychopharmacol. 2010;30(3):290-293.
44. Tampi R. How to wean geriatric patients off benzodiazepines. Psychiatric News. http://psychnews.psychiatryonline.org/doi/full/10.1176/appi.pn.2016.PP3b6. Published March 18, 2016. Accessed May 3, 2017.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Balon R, Fava GA, Rickels K. Need for a realistic appraisal of benzodiazepines. World Psychiatry. 2015;14(2):243-244.
3. Ashton CH. Benzodiazepine equivalence table. http://www.benzo.org.uk/bzequiv.htm. Revised April 2007. Accessed May 3, 2017.
4. National Institute on Drug Abuse. Commonly abused drugs. https://d14rmgtrwzf5a.cloudfront.net/sites/default/files/commonly_abused_drugs_3.pdf. Revised January 2016. Accessed January 9, 2018.
5. Licata SC, Rowlett JK. Abuse and dependence liability of benzodiazepine-type drugs: GABA(A) receptor modulation and beyond. Pharmacol Biochem Behav. 2008;90(1):74-89.
6. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Published January 2009. Accessed May 3, 2017.
7. Salzman C. The APA Task Force report on benzodiazepine dependence, toxicity, and abuse. Am J Psychiatry. 1991;148(2):151-152.
8. Bushnell GA, Stürmer T, Gaynes BN, et al. Simultaneous antidepressant and benzodiazepine new use and subsequent long-term benzodiazepine use in adults with depression, United States, 2001-2014. JAMA Psychiatry. 2017;74(7):747-755.
9. O’Brien PL, Karnell LH, Gokhale M, et al. Prescribing of benzodiazepines and opioids to individuals with substance use disorders. Drug Alcohol Depend. 2017;178:223-230.
10. Mellman TA, Bustamante V, David D, et al. Hypnotic medication in the aftermath of trauma. J Clin Psychiatry. 2002;63(12):1183-1184.
11. Gelpin E, Bonne O, Peri T, et al. Treatment of recent trauma survivors with benzodiazepines: a prospective study. J Clin Psychiatry. 1996;57(9):390-394.
12. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed May 3, 2017.
13. Abdel-Ahad P, Kazour F. Non-antidepressant pharmacological treatment of obsessive compulsive disorder: a comprehensive review. Curr Clin Pharmacol. 2015;10(2):97-111.
14. Gardner DL, Cowdry RW. Alprazolam-induced dyscontrol in borderline personality disorder. Am J Psychiatry. 1985;142(1):98-100.
15. Ciccone DS, Just N, Bandilla EB, et al. Psychological correlates of opioid use in patients with chronic nonmalignant pain: a preliminary test of the downhill spiral hypothesis. J Pain Symptom Manage. 2000;20(3):180-192.
16. Vozoris NT, Fischer HD, Wang X, et al. Benzodiazepine drug use and adverse respiratory outcomes among older adults with COPD. Eur Respir J. 2014;44(2):332-340.
17. Obiora E, Hubbard R, Sanders RD, et al. The impact of benzodiazepines on occurrence of pneumonia and mortality from pneumonia: a nested case-control and survival analysis in a population-based cohort. Thorax. 2013;68(2):163-170.
18. Taipale H, Tolppanen AM, Koponen M, et al. Risk of pneumonia associated with incident benzodiazepine use among community-dwelling adults with Alzheimer disease. CMAJ. 2017;189(14):E519-E529.
19. Iqbal MM, Sobhan T, Ryals T. Effects of commonly used benzodiazepines on the fetus, the neonate, and the nursing infant. Psychiatric Serv. 2002;53:39-49.
20. U.S. National Library of Medicine, TOXNET Toxicology Data Network. Lactmed: alprazolam. http://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+lactmed:@term+@DOCNO+335. Accessed May 3, 2017.
21. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
22. Ray WA, Thapa PB, Gideon P. Benzodiazepines and the risk of falls in nursing home residents. J Am Geriatr Soc. 2000;48(6):682-685.
23. Woolcott JC, Richardson KJ, Wiens MO, et al. Meta-analysis of the impact of 9 medication classes on falls in elderly persons. Arch Intern Med. 2009;169(21):1952-1960.
24. Bolton JM, Morin SN, Majumdar SR, et al. Association of mental disorders and related medication use with risk for major osteoporotic fractures. JAMA Psychiatry. 2017;74(6):641-648.
25. Pariente A, Dartiques JF, Benichou J, et al. Benzodiazepines and injurious falls in community dwelling elders. Drugs Aging. 2008;25(1):61-70.
26. Lagnaoui R, Tournier M, Moride Y, et al. The risk of cognitive impairment in older community-dwelling women after benzodiazepine use. Age Ageing. 2009;38(2):226-228.
27. Billioti de Gage S, Bégaud B, Bazin F, et al. Benzodiazepine use and risk of dementia: prospective population based study. BMJ. 2012;345:e6231. doi: 10.1136/bmj.e6231.
28. Olfson M, King M, Schoenbaum M. Benzodiazepine use in the United States. JAMA Psychiatry. 2015;72(2):136-142.
29. Maust DT, Kales HC, Wiechers IR, et al. No end in sight: benzodiazepine use in older adults in the United States. J Am Geriatr Soc. 2016;64(12):2546-2553.
30. Maust DT, Blow FC, Wiechers IR, et al. National trends in antidepressant, benzodiazepine, and other sedative-hypnotic treatment of older adults in psychiatric and primary care. J Clin Psychiatry. 2017;78(4):e363-e371.
31. Rapoport MJ, Lanctôt KL, Streiner DL, et al. Benzodiazepine use and driving: a meta-analysis. J Clin Psychiatry. 2009;70(5):663-673.
32. Tiihonen J, Mittendorfer-Rutz E, Torniainen M, et al. Mortality and cumulative exposure to antipsychotics, antidepressants, and benzodiazepines in patients with schizophrenia: an observational follow-up study. Am J Psychiatry. 2016;173(6):600-606.
33. Fontanella CA, Campo JV, Phillips GS, et al. Benzodiazepine use and risk of mortality among patients with schizophrenia: a retrospective longitudinal study. J Clin Psychiatry. 2016;77(5):661-667.
34. McCall WV, Benca RM, Rosenguist PB, et al. Hypnotic medications and suicide: risk, mechanisms, mitigation, and the FDA. Am J Psychiatry. 2017;174(1):18-25.
35. Bachhuber MA, Hennessy S, Cunningham CO, et al. Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013. Am J Public Health. 2016;106(4):686-688.
36. National Institute on Drug Abuse. Overdose death rates. https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates. Updated September 2017. Accessed January 8, 2018.
37. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recomm Rep 2016;65(1):1-49.
38. U.S. Food and Drug Administration. FDA requires strong warnings for opioid analgesics, prescription opioid cough products, and benzodiazepine labeling related to serious risks and death from combined use [press release]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518697.htm. Published August 31, 2016. Accessed May 3, 2017.
39. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about serious risks and death when combining opioid pain or cough medicines with benzodiazepines; requires its strongest warning. http://www.fda.gov/Drugs/DrugSafety/ucm518473.htm. Published August 31, 2016. Accessed May 3, 2017.
40. National Institute for Health and Care Excellence. Controlled drugs: safe use and management. https://www.nice.org.uk/guidance/ng46/evidence/full-guideline-pdf-2427186353. Published April 2016. Accessed July 25, 2017.
41. Stahl SM. Anxiety disorders and anxiolytics. In: Stahl’s essential psychopharmacology. 3rd ed. New York, NY: Cambridge University Press; 2008:721-772.
42. Paquin AM, Zimmerman K, Rudolph JL. Risk versus risk: a review of benzodiazepine reduction in older adults. Expert Opin Drug Saf. 2014;13(7):919-934.
43. Nardi AE, Freire RC, Valença AM, et al. Tapering clonazepam in patients with panic disorder after at least 3 years of treatment. J Clin Psychopharmacol. 2010;30(3):290-293.
44. Tampi R. How to wean geriatric patients off benzodiazepines. Psychiatric News. http://psychnews.psychiatryonline.org/doi/full/10.1176/appi.pn.2016.PP3b6. Published March 18, 2016. Accessed May 3, 2017.

Compulsive sexual behavior: A nonjudgmental approach
Compulsive sexual behavior (CSB), also referred to as sexual addiction or hypersexuality, is characterized by repetitive and intense preoccupations with sexual fantasies, urges, and behaviors that are distressing to the individual and/or result in psychosocial impairment. Individuals with CSB often perceive their sexual behavior to be excessive but are unable to control it. CSB can involve fantasies and urges in addition to or in place of the behavior but must cause clinically significant distress and interference in daily life to qualify as a disorder.
Because of the lack of large-scale, population-based epidemiological studies assessing CSB, its true prevalence among adults is unknown. A study of 204 psychiatric inpatients found a current prevalence of 4.4%,1 while a university-based survey estimated the prevalence of CSB at approximately 2%.2 Others have estimated that the prevalence is between 3% to 6% of adults in the United States,3,4 with males comprising the majority (≥80%) of affected individuals.5
CSB usually develops during late adolescence/early adulthood, and most who present for treatment are male.5 Mood states, including depression, happiness, and loneliness, may trigger CSB.6 Many individuals report feelings of dissociation while engaging in CSB-related behaviors, whereas others report feeling important, powerful, excited, or gratified.
Why CSB is difficult to diagnose
Although CSB may be common, it usually goes undiagnosed. This potentially problematic behavior often is not diagnosed because of:
- Shame and secrecy. Embarrassment and shame, which are fundamental to CSB, appear to explain, in part, why few patients volunteer information regarding this behavior unless specifically asked.1
- Patient lack of knowledge. Patients often do not know that their behavior can be successfully treated.
- Clinician lack of knowledge. Few health care professionals have education or training in CSB. A lack of recognition of CSB also may be due to our limited understanding regarding the limits of sexual normality. In addition, the classification of CSB is unclear and not agreed upon (Box7-9), and moral judgments often are involved in understanding sexual behaviors.10
Box
Classifying compulsive sexual behavior
No consensus on diagnostic criteria
Accurately diagnosing CSB is difficult because of a lack of consensus about the diagnostic criteria for the disorder. Christenson et al11 developed an early set of criteria for CSB as part of a larger survey of impulse control disorders. They used the following 2 criteria to diagnose CSB: (1) excessive or uncontrolled sexual behavior(s) or sexual thoughts/urges to engage in behavior, and (2) these behaviors or thoughts/urges lead to significant distress, social or occupational impairment, or legal and financial consequences.11,12
During the DSM-5 revision process, a second approach to the diagnostic criteria was proposed for hypersexuality disorder. Under the proposed criteria for hypersexuality, a person would meet the diagnosis if ≥3 of the following were endorsed over a 6-month period: (a) time consumed by sexual fantasies, urges, or behaviors repetitively interferes with other important (non-sexual) goals, activities, and obligations; (b) repetitively engaging in sexual fantasies, urges, or behaviors in response to dysphoric mood states; (c) repetitively engaging in sexual fantasies, urges, or behaviors in response to stressful life events; (d) repetitive but unsuccessful efforts to control or significantly reduce these sexual fantasies, urges, or behaviors; and (e) repetitively engaging in sexual behaviors while disregarding the risk for physical or emotional harm to self or others.9
These 2 proposed approaches to diagnosis are somewhat similar. Both suggest that the core underlying issues involve sexual urges or behaviors that are difficult to control and that lead to psychosocial dysfunction. Differences in the criteria, however, could result in different rates of CSB diagnosis; therefore, further research will need to determine which diagnostic approach reflects the neurobiology underlying CSB.
Avoid misdiagnosis
Before making a diagnosis of CSB, it is important for clinicians to consider whether they are stigmatizing “negative consequences,” distress, or social impairment based on unconscious bias toward certain sexual behaviors. In addition, we need to ensure that we are not holding sex to different standards than other behaviors (for example, there are many things in life we do that result in negative consequences and yet do not classify as a mental disorder, such as indulging in less healthy food choices). Furthermore, excessive sexual behaviors might be associated with the normal coming out process for LGBTQ individuals, partner relationship problems, or sexual/gender identity. Therefore, the behavior needs to be assessed in the context of these psychosocial environmental factors.
Differential diagnosis
Various psychiatric disorders also may include excessive sexual behavior as part of their clinical presentation, and it is important to differentiate that behavior from CSB.
Bipolar disorder. Excessive sexual behavior can occur as part of a manic episode in bipolar disorder. If the problematic sexual behavior also occurs when the person’s mood is stable, the individual may have CSB and bipolar disorder. This distinction is important because the treatment for bipolar disorder is often different for CSB, because anticonvulsants have only case reports attesting to their use in CSB.
Substance abuse. Excessive sexual behavior can occur when a person is abusing substances, particularly stimulants such as cocaine and amphetamines.13 If the sexual behavior does not occur when the person is not using drugs, then the appropriate diagnosis would not likely be CSB.
Obsessive-compulsive disorder (OCD). Individuals with OCD often are preoccupied with sexual themes and feel that they think about sex excessively.14 Although patients with OCD may be preoccupied with thoughts of sex, the key difference is that persons with CSB report feeling excited by these thoughts and derive pleasure from the behavior, whereas the sexual thoughts of OCD are perceived as unpleasant.
Other disorders that may give rise to hypersexual behavior include neurocognitive disorders, attention-deficit/hyperactivity disorder, autism spectrum disorders, and depressive disorders.
Adverse effects of medication. It is important to ask the patient whether he (she)developed CSB after starting a medication. Certain medications (eg, medications for Parkinson’s disease or restless leg syndrome, or aripiprazole to treat depression or psychosis) may cause patients to engage in problematic sexual behavior.15,16 If the sexual behavior decreases or stops when the medication dosage is reduced or the medication is stopped, a diagnosis of CSB would not be appropriate.
Comorbidity is common
Research suggests that approximately one-half of adults with CSB meet criteria for at least 1 other psychiatric disorder, such as mood, anxiety, substance use, impulse control, or personality disorders. A study of men with CSB (N = 103) found that 71% met criteria for a mood disorder, 40% for an anxiety disorder, 41% for a substance use disorder, and 24% for an impulse control disorder such as gambling disorder.17 Therefore, to successfully treat CSB, clinicians also may need to focus on how and to what extent these co-occurring disorders drive the sexual behavior.
Co-occurring medical conditions also are common among individuals with CSB. Medical concerns may include unwanted pregnancy, sexually transmitted infections, and HIV/AIDS. Thus, treating psychiatric comorbidities and providing education about sexual health, with referrals to primary care specialists, often are part of CSB treatment.
Neuroimaging and cognition
One imaging study that compared participants with and without CSB found that participants with CSB had higher activity in the ventral striatum, anterior cingulate cortex, and amygdala relative to controls during a cue-reactivity functional MRI task.18 These findings show notable similarities to the patterns of activation seen in patients addicted to drugs when assessed using drug-craving paradigms. An additional neuroimaging study assessing patients with hypersexuality using diffusion tensor imaging noted that diffusivity in a prefrontal white matter tract within a superior frontal region was greater in patients with CSB.18 This study also indicated that there was a negative correlation between observed diffusion in the noted location and overall severity score for CSB symptoms such as frequency of urges or behaviors.
In terms of cognition, a preliminary assessment of young adults with CSB compared with healthy controls did not find any differences between groups across several tasks, although the previously mentioned diffusion tensor imaging study reported elevated impulsivity in CSB.18
Approaches to treatment
Most people with CSB are reluctant to mention it to their health care providers, and most physicians are generally uncomfortable talking about sex with their patients, in part, because of a lack of training.19 Patients are more likely to bring up the topic when they are receiving treatment for anxiety, depression, or substance abuse. Therefore, clinicians must consider that sexual behavior might be associated with a coping mechanism, distressing outcome, or comorbid condition in these patients.
Pharmacologic treatment
Evidence for the pharmacologic treatment of CSB consists primarily of small, open-label studies, case series, or retrospective analyses, except for 1 double-blind, placebo-controlled study. Based on this evidence, there may be several pharmacologic treatment options for patients with CSB; however, there are no FDA-approved medications for CSB.
Antidepressants. One of the most thoroughly documented categories of pharmacologic treatment for CSB is selective serotonin reuptake inhibitors (SSRIs). Several retrospective analyses and case series have reported on the general efficacy of SSRIs in reducing symptoms of CSB.20-23 Citalopram, the only treatment for CSB that has been assessed using a double-blind, placebo-controlled methodology, was associated with significant decreases in CSB symptoms, including sexual desire/drive, frequency of masturbation, and pornography use.24
In addition to SSRIs, several additional case reports have suggested that other classes of antidepressants, such as serotonin-norepinephrine reuptake inhibitors and tricyclic antidepressants, or stimulants may be beneficial when treating CSB.25 Several case reports have indicated significant improvement of CSB symptoms using clomipramine.22 A retrospective study of nefazodone also has suggested that it may be an option for treating CSB. Patients reported notable reductions in the frequency of sexual obsessions/compulsions while taking nefazodone and reported no notable sexual adverse effects.26 One branded version of nefazodone, Serzone, was associated with rare but severe liver problems and was withdrawn from the U.S. market in 2004.
Although some initial evidence regarding antidepressant use, particularly SSRIs, to treat CSB has suggested that these medications may be potentially beneficial, the findings are far from conclusive, with only 1 controlled trial and only single-subject case reports for many of the medications studied.
Naltrexone, an opioid antagonist, has received support from available cases, open-label studies, and retrospective analyses.17,27 Although evidence for the use of naltrexone in CSB is limited to case reports and retrospective analyses, results have been positive. Naltrexone has shown notable decreases in CSB symptom severity when used as monotherapy and when used in combination with other treatments.
Anticonvulsants. Several case reports have suggested that certain anticonvulsants may be beneficial for treating CSB.
Psychotherapy
Evidence supporting specific types of psychotherapy for CSB is limited and largely drawn from uncontrolled studies and case reports.
Cognitive-behavioral therapy (CBT) is one of the more common psychotherapeutic options used for CSB. Several uncontrolled studies and case reports have found that CBT is beneficial for CSB, although methodologies have varied.
Several cases found that combining CBT with motivational interviewing was associated with significant reductions in sexual behaviors, such as frequency of sexual partners and amount of time spent online during work hours.29,30 Group CBT also has been shown to be effective for CSB.31
Acceptance and commitment therapy (ACT) has received some initial support, with 1 uncontrolled study and 1 controlled study.32,33 The controlled study used 12 sessions of individual ACT compared with a wait-list condition.32 Improvements in CSB symptoms were maintained for 3 months. The overall reduction in problematic Internet pornography use was reported as 92% immediately after the study ended, and 86% after 3 months.
Marital/relationship therapy has been used successfully in several case series and case reports, although no studies have assessed its efficacy in treating CSB using a randomized protocol. In 1 case report, the researcher found that participation in marital sex therapy elicited notable improvements over the course of 1 year and 20 sessions.34
1. Grant JE, Levine L, Kim D, et al. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162(11):2184-2188.
2. Odlaug BL, Lust K, Schreiber LR, et al. Compulsive sexual behavior in young adults. Ann Clin Psychiatry. 2013;25(3):193-200.
3. Black DW. Compulsive sexual behavior: a review. J Psychiatr Pract. 1998;4(4):219-229.
4. Coleman E. Is your patient suffering from compulsive sexual behavior? Psychiatr Ann. 1992;22(6):320-325.
5. Kaplan MS, Krueger RB. Diagnosis, assessment, and treatment of hypersexuality. J Sex Res. 2010;47(2):181-198.
6. Black DW, Kehrberg LL, Flumerfelt DL, et al. Characteristics of 36 subjects reporting compulsive sexual behavior. Am J Psychiatry. 1997;154(2):243-249.
7. McElroy SL, Phillips KA, Keck PE Jr. Obsessive compulsive spectrum disorder. J Clin Psychiatry. 1994;(suppl 55):33-51; discussion 52-53.
8. McElroy SL, Pope HG Jr, Keck PE Jr, et al. Are impulse-control disorders related to bipolar disorder? Compr Psychiatry. 1996;37(4):229-240.
9. Kafka MP. Hypersexual disorder: a proposed diagnosis for DSM-V. Arch Sex Behav. 2010;39(2):377-400.
10. Levine SB. What is sexual addiction? J Sex Marital Ther. 2010;36(3):261-275.
11. Christenson GA, Faber RJ, de Zwaan M, et al. Compulsive buying: descriptive characteristics and psychiatric comorbidity. J Clin Psychiatry. 1994;55(1):5-11.
12. Grant JE. Impulse control disorders: a clinician’s guide to understanding and treating behavioral addictions. New York, NY: W.W. Norton & Company, Inc.; 2008.
13. Frohmader KS, Lehman MN, Laviolette SR, et al. Concurrent exposure to methamphetamine and sexual behavior enhances subsequent drug reward and causes compulsive sexual behavior in male rats. J Neurosci. 2011;31(45):16473-16482.
14. Grant JE, Pinto A, Gunnip M, et al. Sexual obsessions and clinical correlates in adults with obsessive-compulsive disorder. Compr Psychiatry. 2006;47(5):325-329.
15. Mété D, Dafreville C, Paitel V, et al. Aripiprazole, gambling disorder and compulsive sexuality [in French]. Encephale. 2016;42(3):281-283.
16. Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol. 2010;67(5):589-595.
17. Kraus SW, Meshberg-Cohen S, Martino S, et al. Treatment of compulsive pornography use with naltrexone: a case report. Am J Psychiatry. 2015;172(12):1260-1261.
18. Derbyshire KL, Grant JE. Compulsive sexual behavior: a review of the literature. J Behav Addict. 2015;4(2):37-43.
19. Levine SB, Scott DL. Sexual education for psychiatric residents. Acad Psychiatry. 2010;34(5):349-352.
20. Alsughier N. Compulsive masturbation treated with selective serotonin reuptake inhibitors. African J Psychiatry (Johannesbg). 2015;18:299.
21. Elmore JL. SSRI reduction of nonparaphilic sexual addiction. CNS Spectr. 2000;5(11);53-56.
22. Stein DJ, Hollander E, Anthony DT, et al. Serotonergic medications for sexual obsessions, sexual addictions, and paraphilias. J Clinical Psychiatry. 1992;53(8):267-271.
23. Kafka M. Psychopharmacologic treatments for nonparaphilic compulsive sexual behaviors. CNS Spectr. 200;5(1):49-59.
24. Wainberg ML, Muench F, Morgenstern J, et al. A double-blind study of citalopram versus placebo in the treatment of compulsive sexual behaviors in gay and bisexual men. J Clin Psychiatry. 2006;67(12):1968-1973.
25. Kafka MP, Hennen J. Psychostimulant augmentation during treatment with selective serotonin reuptake inhibitors in men with paraphilias and paraphilia-related disorders: a case series. J Clin Psychiatry. 2000;61(9):664-670.
26. Coleman E, Raymond N, McBean A. Assessment and treatment of compulsive sexual behavior. Minn Med. 2003;86(7):42-47.
27. Raymond NC, Grant JE, Coleman E. Augmentation with naltrexone to treat compulsive sexual behavior: a case series. Ann Clin Psychiatry. 2010;22(1):56-62.
28. Fong TW, De La Garza R 2nd, Newton TF. A case report of topiramate in the treatment of nonparaphilic sexual addiction. J Clin Psychopharmacol. 2005;25(5):512-514.
29. Del Giudice MJ, Kutinsky J. Applying motivational interviewing to the treatment of sexual compulsivity and addiction. Sex Addict Comp. 2007;14(4):303-319.
30. Shepherd L. Cognitive behavior therapy for sexually addictive behavior. Clin Case Stud. 2010;9(1):18-27.
31. Sadiza J, Varma R, Jena SPK, et al. Group cognitive behaviour therapy in the management of compulsive sex behaviour. International Journal of Criminal Justice Sciences. 2011;6(1-2):309-325.
32. Crosby JM, Twohig MP. Acceptance and commitment therapy for problematic Internet pornography use: a randomized trial. Behav Ther. 2016;47(3):355-366.
33. Twohig MP, Crosby JM. Acceptance and commitment therapy as a treatment for problematic internet pornography viewing. Behav Ther. 2010;41(3):285-295.
34. Sprenkle DH. Treating a sex addict through marital sex therapy. Fam Relat. 1987;36(1):11-14.
Compulsive sexual behavior (CSB), also referred to as sexual addiction or hypersexuality, is characterized by repetitive and intense preoccupations with sexual fantasies, urges, and behaviors that are distressing to the individual and/or result in psychosocial impairment. Individuals with CSB often perceive their sexual behavior to be excessive but are unable to control it. CSB can involve fantasies and urges in addition to or in place of the behavior but must cause clinically significant distress and interference in daily life to qualify as a disorder.
Because of the lack of large-scale, population-based epidemiological studies assessing CSB, its true prevalence among adults is unknown. A study of 204 psychiatric inpatients found a current prevalence of 4.4%,1 while a university-based survey estimated the prevalence of CSB at approximately 2%.2 Others have estimated that the prevalence is between 3% to 6% of adults in the United States,3,4 with males comprising the majority (≥80%) of affected individuals.5
CSB usually develops during late adolescence/early adulthood, and most who present for treatment are male.5 Mood states, including depression, happiness, and loneliness, may trigger CSB.6 Many individuals report feelings of dissociation while engaging in CSB-related behaviors, whereas others report feeling important, powerful, excited, or gratified.
Why CSB is difficult to diagnose
Although CSB may be common, it usually goes undiagnosed. This potentially problematic behavior often is not diagnosed because of:
- Shame and secrecy. Embarrassment and shame, which are fundamental to CSB, appear to explain, in part, why few patients volunteer information regarding this behavior unless specifically asked.1
- Patient lack of knowledge. Patients often do not know that their behavior can be successfully treated.
- Clinician lack of knowledge. Few health care professionals have education or training in CSB. A lack of recognition of CSB also may be due to our limited understanding regarding the limits of sexual normality. In addition, the classification of CSB is unclear and not agreed upon (Box7-9), and moral judgments often are involved in understanding sexual behaviors.10
Box
Classifying compulsive sexual behavior
No consensus on diagnostic criteria
Accurately diagnosing CSB is difficult because of a lack of consensus about the diagnostic criteria for the disorder. Christenson et al11 developed an early set of criteria for CSB as part of a larger survey of impulse control disorders. They used the following 2 criteria to diagnose CSB: (1) excessive or uncontrolled sexual behavior(s) or sexual thoughts/urges to engage in behavior, and (2) these behaviors or thoughts/urges lead to significant distress, social or occupational impairment, or legal and financial consequences.11,12
During the DSM-5 revision process, a second approach to the diagnostic criteria was proposed for hypersexuality disorder. Under the proposed criteria for hypersexuality, a person would meet the diagnosis if ≥3 of the following were endorsed over a 6-month period: (a) time consumed by sexual fantasies, urges, or behaviors repetitively interferes with other important (non-sexual) goals, activities, and obligations; (b) repetitively engaging in sexual fantasies, urges, or behaviors in response to dysphoric mood states; (c) repetitively engaging in sexual fantasies, urges, or behaviors in response to stressful life events; (d) repetitive but unsuccessful efforts to control or significantly reduce these sexual fantasies, urges, or behaviors; and (e) repetitively engaging in sexual behaviors while disregarding the risk for physical or emotional harm to self or others.9
These 2 proposed approaches to diagnosis are somewhat similar. Both suggest that the core underlying issues involve sexual urges or behaviors that are difficult to control and that lead to psychosocial dysfunction. Differences in the criteria, however, could result in different rates of CSB diagnosis; therefore, further research will need to determine which diagnostic approach reflects the neurobiology underlying CSB.
Avoid misdiagnosis
Before making a diagnosis of CSB, it is important for clinicians to consider whether they are stigmatizing “negative consequences,” distress, or social impairment based on unconscious bias toward certain sexual behaviors. In addition, we need to ensure that we are not holding sex to different standards than other behaviors (for example, there are many things in life we do that result in negative consequences and yet do not classify as a mental disorder, such as indulging in less healthy food choices). Furthermore, excessive sexual behaviors might be associated with the normal coming out process for LGBTQ individuals, partner relationship problems, or sexual/gender identity. Therefore, the behavior needs to be assessed in the context of these psychosocial environmental factors.
Differential diagnosis
Various psychiatric disorders also may include excessive sexual behavior as part of their clinical presentation, and it is important to differentiate that behavior from CSB.
Bipolar disorder. Excessive sexual behavior can occur as part of a manic episode in bipolar disorder. If the problematic sexual behavior also occurs when the person’s mood is stable, the individual may have CSB and bipolar disorder. This distinction is important because the treatment for bipolar disorder is often different for CSB, because anticonvulsants have only case reports attesting to their use in CSB.
Substance abuse. Excessive sexual behavior can occur when a person is abusing substances, particularly stimulants such as cocaine and amphetamines.13 If the sexual behavior does not occur when the person is not using drugs, then the appropriate diagnosis would not likely be CSB.
Obsessive-compulsive disorder (OCD). Individuals with OCD often are preoccupied with sexual themes and feel that they think about sex excessively.14 Although patients with OCD may be preoccupied with thoughts of sex, the key difference is that persons with CSB report feeling excited by these thoughts and derive pleasure from the behavior, whereas the sexual thoughts of OCD are perceived as unpleasant.
Other disorders that may give rise to hypersexual behavior include neurocognitive disorders, attention-deficit/hyperactivity disorder, autism spectrum disorders, and depressive disorders.
Adverse effects of medication. It is important to ask the patient whether he (she)developed CSB after starting a medication. Certain medications (eg, medications for Parkinson’s disease or restless leg syndrome, or aripiprazole to treat depression or psychosis) may cause patients to engage in problematic sexual behavior.15,16 If the sexual behavior decreases or stops when the medication dosage is reduced or the medication is stopped, a diagnosis of CSB would not be appropriate.
Comorbidity is common
Research suggests that approximately one-half of adults with CSB meet criteria for at least 1 other psychiatric disorder, such as mood, anxiety, substance use, impulse control, or personality disorders. A study of men with CSB (N = 103) found that 71% met criteria for a mood disorder, 40% for an anxiety disorder, 41% for a substance use disorder, and 24% for an impulse control disorder such as gambling disorder.17 Therefore, to successfully treat CSB, clinicians also may need to focus on how and to what extent these co-occurring disorders drive the sexual behavior.
Co-occurring medical conditions also are common among individuals with CSB. Medical concerns may include unwanted pregnancy, sexually transmitted infections, and HIV/AIDS. Thus, treating psychiatric comorbidities and providing education about sexual health, with referrals to primary care specialists, often are part of CSB treatment.
Neuroimaging and cognition
One imaging study that compared participants with and without CSB found that participants with CSB had higher activity in the ventral striatum, anterior cingulate cortex, and amygdala relative to controls during a cue-reactivity functional MRI task.18 These findings show notable similarities to the patterns of activation seen in patients addicted to drugs when assessed using drug-craving paradigms. An additional neuroimaging study assessing patients with hypersexuality using diffusion tensor imaging noted that diffusivity in a prefrontal white matter tract within a superior frontal region was greater in patients with CSB.18 This study also indicated that there was a negative correlation between observed diffusion in the noted location and overall severity score for CSB symptoms such as frequency of urges or behaviors.
In terms of cognition, a preliminary assessment of young adults with CSB compared with healthy controls did not find any differences between groups across several tasks, although the previously mentioned diffusion tensor imaging study reported elevated impulsivity in CSB.18
Approaches to treatment
Most people with CSB are reluctant to mention it to their health care providers, and most physicians are generally uncomfortable talking about sex with their patients, in part, because of a lack of training.19 Patients are more likely to bring up the topic when they are receiving treatment for anxiety, depression, or substance abuse. Therefore, clinicians must consider that sexual behavior might be associated with a coping mechanism, distressing outcome, or comorbid condition in these patients.
Pharmacologic treatment
Evidence for the pharmacologic treatment of CSB consists primarily of small, open-label studies, case series, or retrospective analyses, except for 1 double-blind, placebo-controlled study. Based on this evidence, there may be several pharmacologic treatment options for patients with CSB; however, there are no FDA-approved medications for CSB.
Antidepressants. One of the most thoroughly documented categories of pharmacologic treatment for CSB is selective serotonin reuptake inhibitors (SSRIs). Several retrospective analyses and case series have reported on the general efficacy of SSRIs in reducing symptoms of CSB.20-23 Citalopram, the only treatment for CSB that has been assessed using a double-blind, placebo-controlled methodology, was associated with significant decreases in CSB symptoms, including sexual desire/drive, frequency of masturbation, and pornography use.24
In addition to SSRIs, several additional case reports have suggested that other classes of antidepressants, such as serotonin-norepinephrine reuptake inhibitors and tricyclic antidepressants, or stimulants may be beneficial when treating CSB.25 Several case reports have indicated significant improvement of CSB symptoms using clomipramine.22 A retrospective study of nefazodone also has suggested that it may be an option for treating CSB. Patients reported notable reductions in the frequency of sexual obsessions/compulsions while taking nefazodone and reported no notable sexual adverse effects.26 One branded version of nefazodone, Serzone, was associated with rare but severe liver problems and was withdrawn from the U.S. market in 2004.
Although some initial evidence regarding antidepressant use, particularly SSRIs, to treat CSB has suggested that these medications may be potentially beneficial, the findings are far from conclusive, with only 1 controlled trial and only single-subject case reports for many of the medications studied.
Naltrexone, an opioid antagonist, has received support from available cases, open-label studies, and retrospective analyses.17,27 Although evidence for the use of naltrexone in CSB is limited to case reports and retrospective analyses, results have been positive. Naltrexone has shown notable decreases in CSB symptom severity when used as monotherapy and when used in combination with other treatments.
Anticonvulsants. Several case reports have suggested that certain anticonvulsants may be beneficial for treating CSB.
Psychotherapy
Evidence supporting specific types of psychotherapy for CSB is limited and largely drawn from uncontrolled studies and case reports.
Cognitive-behavioral therapy (CBT) is one of the more common psychotherapeutic options used for CSB. Several uncontrolled studies and case reports have found that CBT is beneficial for CSB, although methodologies have varied.
Several cases found that combining CBT with motivational interviewing was associated with significant reductions in sexual behaviors, such as frequency of sexual partners and amount of time spent online during work hours.29,30 Group CBT also has been shown to be effective for CSB.31
Acceptance and commitment therapy (ACT) has received some initial support, with 1 uncontrolled study and 1 controlled study.32,33 The controlled study used 12 sessions of individual ACT compared with a wait-list condition.32 Improvements in CSB symptoms were maintained for 3 months. The overall reduction in problematic Internet pornography use was reported as 92% immediately after the study ended, and 86% after 3 months.
Marital/relationship therapy has been used successfully in several case series and case reports, although no studies have assessed its efficacy in treating CSB using a randomized protocol. In 1 case report, the researcher found that participation in marital sex therapy elicited notable improvements over the course of 1 year and 20 sessions.34
Compulsive sexual behavior (CSB), also referred to as sexual addiction or hypersexuality, is characterized by repetitive and intense preoccupations with sexual fantasies, urges, and behaviors that are distressing to the individual and/or result in psychosocial impairment. Individuals with CSB often perceive their sexual behavior to be excessive but are unable to control it. CSB can involve fantasies and urges in addition to or in place of the behavior but must cause clinically significant distress and interference in daily life to qualify as a disorder.
Because of the lack of large-scale, population-based epidemiological studies assessing CSB, its true prevalence among adults is unknown. A study of 204 psychiatric inpatients found a current prevalence of 4.4%,1 while a university-based survey estimated the prevalence of CSB at approximately 2%.2 Others have estimated that the prevalence is between 3% to 6% of adults in the United States,3,4 with males comprising the majority (≥80%) of affected individuals.5
CSB usually develops during late adolescence/early adulthood, and most who present for treatment are male.5 Mood states, including depression, happiness, and loneliness, may trigger CSB.6 Many individuals report feelings of dissociation while engaging in CSB-related behaviors, whereas others report feeling important, powerful, excited, or gratified.
Why CSB is difficult to diagnose
Although CSB may be common, it usually goes undiagnosed. This potentially problematic behavior often is not diagnosed because of:
- Shame and secrecy. Embarrassment and shame, which are fundamental to CSB, appear to explain, in part, why few patients volunteer information regarding this behavior unless specifically asked.1
- Patient lack of knowledge. Patients often do not know that their behavior can be successfully treated.
- Clinician lack of knowledge. Few health care professionals have education or training in CSB. A lack of recognition of CSB also may be due to our limited understanding regarding the limits of sexual normality. In addition, the classification of CSB is unclear and not agreed upon (Box7-9), and moral judgments often are involved in understanding sexual behaviors.10
Box
Classifying compulsive sexual behavior
No consensus on diagnostic criteria
Accurately diagnosing CSB is difficult because of a lack of consensus about the diagnostic criteria for the disorder. Christenson et al11 developed an early set of criteria for CSB as part of a larger survey of impulse control disorders. They used the following 2 criteria to diagnose CSB: (1) excessive or uncontrolled sexual behavior(s) or sexual thoughts/urges to engage in behavior, and (2) these behaviors or thoughts/urges lead to significant distress, social or occupational impairment, or legal and financial consequences.11,12
During the DSM-5 revision process, a second approach to the diagnostic criteria was proposed for hypersexuality disorder. Under the proposed criteria for hypersexuality, a person would meet the diagnosis if ≥3 of the following were endorsed over a 6-month period: (a) time consumed by sexual fantasies, urges, or behaviors repetitively interferes with other important (non-sexual) goals, activities, and obligations; (b) repetitively engaging in sexual fantasies, urges, or behaviors in response to dysphoric mood states; (c) repetitively engaging in sexual fantasies, urges, or behaviors in response to stressful life events; (d) repetitive but unsuccessful efforts to control or significantly reduce these sexual fantasies, urges, or behaviors; and (e) repetitively engaging in sexual behaviors while disregarding the risk for physical or emotional harm to self or others.9
These 2 proposed approaches to diagnosis are somewhat similar. Both suggest that the core underlying issues involve sexual urges or behaviors that are difficult to control and that lead to psychosocial dysfunction. Differences in the criteria, however, could result in different rates of CSB diagnosis; therefore, further research will need to determine which diagnostic approach reflects the neurobiology underlying CSB.
Avoid misdiagnosis
Before making a diagnosis of CSB, it is important for clinicians to consider whether they are stigmatizing “negative consequences,” distress, or social impairment based on unconscious bias toward certain sexual behaviors. In addition, we need to ensure that we are not holding sex to different standards than other behaviors (for example, there are many things in life we do that result in negative consequences and yet do not classify as a mental disorder, such as indulging in less healthy food choices). Furthermore, excessive sexual behaviors might be associated with the normal coming out process for LGBTQ individuals, partner relationship problems, or sexual/gender identity. Therefore, the behavior needs to be assessed in the context of these psychosocial environmental factors.
Differential diagnosis
Various psychiatric disorders also may include excessive sexual behavior as part of their clinical presentation, and it is important to differentiate that behavior from CSB.
Bipolar disorder. Excessive sexual behavior can occur as part of a manic episode in bipolar disorder. If the problematic sexual behavior also occurs when the person’s mood is stable, the individual may have CSB and bipolar disorder. This distinction is important because the treatment for bipolar disorder is often different for CSB, because anticonvulsants have only case reports attesting to their use in CSB.
Substance abuse. Excessive sexual behavior can occur when a person is abusing substances, particularly stimulants such as cocaine and amphetamines.13 If the sexual behavior does not occur when the person is not using drugs, then the appropriate diagnosis would not likely be CSB.
Obsessive-compulsive disorder (OCD). Individuals with OCD often are preoccupied with sexual themes and feel that they think about sex excessively.14 Although patients with OCD may be preoccupied with thoughts of sex, the key difference is that persons with CSB report feeling excited by these thoughts and derive pleasure from the behavior, whereas the sexual thoughts of OCD are perceived as unpleasant.
Other disorders that may give rise to hypersexual behavior include neurocognitive disorders, attention-deficit/hyperactivity disorder, autism spectrum disorders, and depressive disorders.
Adverse effects of medication. It is important to ask the patient whether he (she)developed CSB after starting a medication. Certain medications (eg, medications for Parkinson’s disease or restless leg syndrome, or aripiprazole to treat depression or psychosis) may cause patients to engage in problematic sexual behavior.15,16 If the sexual behavior decreases or stops when the medication dosage is reduced or the medication is stopped, a diagnosis of CSB would not be appropriate.
Comorbidity is common
Research suggests that approximately one-half of adults with CSB meet criteria for at least 1 other psychiatric disorder, such as mood, anxiety, substance use, impulse control, or personality disorders. A study of men with CSB (N = 103) found that 71% met criteria for a mood disorder, 40% for an anxiety disorder, 41% for a substance use disorder, and 24% for an impulse control disorder such as gambling disorder.17 Therefore, to successfully treat CSB, clinicians also may need to focus on how and to what extent these co-occurring disorders drive the sexual behavior.
Co-occurring medical conditions also are common among individuals with CSB. Medical concerns may include unwanted pregnancy, sexually transmitted infections, and HIV/AIDS. Thus, treating psychiatric comorbidities and providing education about sexual health, with referrals to primary care specialists, often are part of CSB treatment.
Neuroimaging and cognition
One imaging study that compared participants with and without CSB found that participants with CSB had higher activity in the ventral striatum, anterior cingulate cortex, and amygdala relative to controls during a cue-reactivity functional MRI task.18 These findings show notable similarities to the patterns of activation seen in patients addicted to drugs when assessed using drug-craving paradigms. An additional neuroimaging study assessing patients with hypersexuality using diffusion tensor imaging noted that diffusivity in a prefrontal white matter tract within a superior frontal region was greater in patients with CSB.18 This study also indicated that there was a negative correlation between observed diffusion in the noted location and overall severity score for CSB symptoms such as frequency of urges or behaviors.
In terms of cognition, a preliminary assessment of young adults with CSB compared with healthy controls did not find any differences between groups across several tasks, although the previously mentioned diffusion tensor imaging study reported elevated impulsivity in CSB.18
Approaches to treatment
Most people with CSB are reluctant to mention it to their health care providers, and most physicians are generally uncomfortable talking about sex with their patients, in part, because of a lack of training.19 Patients are more likely to bring up the topic when they are receiving treatment for anxiety, depression, or substance abuse. Therefore, clinicians must consider that sexual behavior might be associated with a coping mechanism, distressing outcome, or comorbid condition in these patients.
Pharmacologic treatment
Evidence for the pharmacologic treatment of CSB consists primarily of small, open-label studies, case series, or retrospective analyses, except for 1 double-blind, placebo-controlled study. Based on this evidence, there may be several pharmacologic treatment options for patients with CSB; however, there are no FDA-approved medications for CSB.
Antidepressants. One of the most thoroughly documented categories of pharmacologic treatment for CSB is selective serotonin reuptake inhibitors (SSRIs). Several retrospective analyses and case series have reported on the general efficacy of SSRIs in reducing symptoms of CSB.20-23 Citalopram, the only treatment for CSB that has been assessed using a double-blind, placebo-controlled methodology, was associated with significant decreases in CSB symptoms, including sexual desire/drive, frequency of masturbation, and pornography use.24
In addition to SSRIs, several additional case reports have suggested that other classes of antidepressants, such as serotonin-norepinephrine reuptake inhibitors and tricyclic antidepressants, or stimulants may be beneficial when treating CSB.25 Several case reports have indicated significant improvement of CSB symptoms using clomipramine.22 A retrospective study of nefazodone also has suggested that it may be an option for treating CSB. Patients reported notable reductions in the frequency of sexual obsessions/compulsions while taking nefazodone and reported no notable sexual adverse effects.26 One branded version of nefazodone, Serzone, was associated with rare but severe liver problems and was withdrawn from the U.S. market in 2004.
Although some initial evidence regarding antidepressant use, particularly SSRIs, to treat CSB has suggested that these medications may be potentially beneficial, the findings are far from conclusive, with only 1 controlled trial and only single-subject case reports for many of the medications studied.
Naltrexone, an opioid antagonist, has received support from available cases, open-label studies, and retrospective analyses.17,27 Although evidence for the use of naltrexone in CSB is limited to case reports and retrospective analyses, results have been positive. Naltrexone has shown notable decreases in CSB symptom severity when used as monotherapy and when used in combination with other treatments.
Anticonvulsants. Several case reports have suggested that certain anticonvulsants may be beneficial for treating CSB.
Psychotherapy
Evidence supporting specific types of psychotherapy for CSB is limited and largely drawn from uncontrolled studies and case reports.
Cognitive-behavioral therapy (CBT) is one of the more common psychotherapeutic options used for CSB. Several uncontrolled studies and case reports have found that CBT is beneficial for CSB, although methodologies have varied.
Several cases found that combining CBT with motivational interviewing was associated with significant reductions in sexual behaviors, such as frequency of sexual partners and amount of time spent online during work hours.29,30 Group CBT also has been shown to be effective for CSB.31
Acceptance and commitment therapy (ACT) has received some initial support, with 1 uncontrolled study and 1 controlled study.32,33 The controlled study used 12 sessions of individual ACT compared with a wait-list condition.32 Improvements in CSB symptoms were maintained for 3 months. The overall reduction in problematic Internet pornography use was reported as 92% immediately after the study ended, and 86% after 3 months.
Marital/relationship therapy has been used successfully in several case series and case reports, although no studies have assessed its efficacy in treating CSB using a randomized protocol. In 1 case report, the researcher found that participation in marital sex therapy elicited notable improvements over the course of 1 year and 20 sessions.34
1. Grant JE, Levine L, Kim D, et al. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162(11):2184-2188.
2. Odlaug BL, Lust K, Schreiber LR, et al. Compulsive sexual behavior in young adults. Ann Clin Psychiatry. 2013;25(3):193-200.
3. Black DW. Compulsive sexual behavior: a review. J Psychiatr Pract. 1998;4(4):219-229.
4. Coleman E. Is your patient suffering from compulsive sexual behavior? Psychiatr Ann. 1992;22(6):320-325.
5. Kaplan MS, Krueger RB. Diagnosis, assessment, and treatment of hypersexuality. J Sex Res. 2010;47(2):181-198.
6. Black DW, Kehrberg LL, Flumerfelt DL, et al. Characteristics of 36 subjects reporting compulsive sexual behavior. Am J Psychiatry. 1997;154(2):243-249.
7. McElroy SL, Phillips KA, Keck PE Jr. Obsessive compulsive spectrum disorder. J Clin Psychiatry. 1994;(suppl 55):33-51; discussion 52-53.
8. McElroy SL, Pope HG Jr, Keck PE Jr, et al. Are impulse-control disorders related to bipolar disorder? Compr Psychiatry. 1996;37(4):229-240.
9. Kafka MP. Hypersexual disorder: a proposed diagnosis for DSM-V. Arch Sex Behav. 2010;39(2):377-400.
10. Levine SB. What is sexual addiction? J Sex Marital Ther. 2010;36(3):261-275.
11. Christenson GA, Faber RJ, de Zwaan M, et al. Compulsive buying: descriptive characteristics and psychiatric comorbidity. J Clin Psychiatry. 1994;55(1):5-11.
12. Grant JE. Impulse control disorders: a clinician’s guide to understanding and treating behavioral addictions. New York, NY: W.W. Norton & Company, Inc.; 2008.
13. Frohmader KS, Lehman MN, Laviolette SR, et al. Concurrent exposure to methamphetamine and sexual behavior enhances subsequent drug reward and causes compulsive sexual behavior in male rats. J Neurosci. 2011;31(45):16473-16482.
14. Grant JE, Pinto A, Gunnip M, et al. Sexual obsessions and clinical correlates in adults with obsessive-compulsive disorder. Compr Psychiatry. 2006;47(5):325-329.
15. Mété D, Dafreville C, Paitel V, et al. Aripiprazole, gambling disorder and compulsive sexuality [in French]. Encephale. 2016;42(3):281-283.
16. Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol. 2010;67(5):589-595.
17. Kraus SW, Meshberg-Cohen S, Martino S, et al. Treatment of compulsive pornography use with naltrexone: a case report. Am J Psychiatry. 2015;172(12):1260-1261.
18. Derbyshire KL, Grant JE. Compulsive sexual behavior: a review of the literature. J Behav Addict. 2015;4(2):37-43.
19. Levine SB, Scott DL. Sexual education for psychiatric residents. Acad Psychiatry. 2010;34(5):349-352.
20. Alsughier N. Compulsive masturbation treated with selective serotonin reuptake inhibitors. African J Psychiatry (Johannesbg). 2015;18:299.
21. Elmore JL. SSRI reduction of nonparaphilic sexual addiction. CNS Spectr. 2000;5(11);53-56.
22. Stein DJ, Hollander E, Anthony DT, et al. Serotonergic medications for sexual obsessions, sexual addictions, and paraphilias. J Clinical Psychiatry. 1992;53(8):267-271.
23. Kafka M. Psychopharmacologic treatments for nonparaphilic compulsive sexual behaviors. CNS Spectr. 200;5(1):49-59.
24. Wainberg ML, Muench F, Morgenstern J, et al. A double-blind study of citalopram versus placebo in the treatment of compulsive sexual behaviors in gay and bisexual men. J Clin Psychiatry. 2006;67(12):1968-1973.
25. Kafka MP, Hennen J. Psychostimulant augmentation during treatment with selective serotonin reuptake inhibitors in men with paraphilias and paraphilia-related disorders: a case series. J Clin Psychiatry. 2000;61(9):664-670.
26. Coleman E, Raymond N, McBean A. Assessment and treatment of compulsive sexual behavior. Minn Med. 2003;86(7):42-47.
27. Raymond NC, Grant JE, Coleman E. Augmentation with naltrexone to treat compulsive sexual behavior: a case series. Ann Clin Psychiatry. 2010;22(1):56-62.
28. Fong TW, De La Garza R 2nd, Newton TF. A case report of topiramate in the treatment of nonparaphilic sexual addiction. J Clin Psychopharmacol. 2005;25(5):512-514.
29. Del Giudice MJ, Kutinsky J. Applying motivational interviewing to the treatment of sexual compulsivity and addiction. Sex Addict Comp. 2007;14(4):303-319.
30. Shepherd L. Cognitive behavior therapy for sexually addictive behavior. Clin Case Stud. 2010;9(1):18-27.
31. Sadiza J, Varma R, Jena SPK, et al. Group cognitive behaviour therapy in the management of compulsive sex behaviour. International Journal of Criminal Justice Sciences. 2011;6(1-2):309-325.
32. Crosby JM, Twohig MP. Acceptance and commitment therapy for problematic Internet pornography use: a randomized trial. Behav Ther. 2016;47(3):355-366.
33. Twohig MP, Crosby JM. Acceptance and commitment therapy as a treatment for problematic internet pornography viewing. Behav Ther. 2010;41(3):285-295.
34. Sprenkle DH. Treating a sex addict through marital sex therapy. Fam Relat. 1987;36(1):11-14.
1. Grant JE, Levine L, Kim D, et al. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162(11):2184-2188.
2. Odlaug BL, Lust K, Schreiber LR, et al. Compulsive sexual behavior in young adults. Ann Clin Psychiatry. 2013;25(3):193-200.
3. Black DW. Compulsive sexual behavior: a review. J Psychiatr Pract. 1998;4(4):219-229.
4. Coleman E. Is your patient suffering from compulsive sexual behavior? Psychiatr Ann. 1992;22(6):320-325.
5. Kaplan MS, Krueger RB. Diagnosis, assessment, and treatment of hypersexuality. J Sex Res. 2010;47(2):181-198.
6. Black DW, Kehrberg LL, Flumerfelt DL, et al. Characteristics of 36 subjects reporting compulsive sexual behavior. Am J Psychiatry. 1997;154(2):243-249.
7. McElroy SL, Phillips KA, Keck PE Jr. Obsessive compulsive spectrum disorder. J Clin Psychiatry. 1994;(suppl 55):33-51; discussion 52-53.
8. McElroy SL, Pope HG Jr, Keck PE Jr, et al. Are impulse-control disorders related to bipolar disorder? Compr Psychiatry. 1996;37(4):229-240.
9. Kafka MP. Hypersexual disorder: a proposed diagnosis for DSM-V. Arch Sex Behav. 2010;39(2):377-400.
10. Levine SB. What is sexual addiction? J Sex Marital Ther. 2010;36(3):261-275.
11. Christenson GA, Faber RJ, de Zwaan M, et al. Compulsive buying: descriptive characteristics and psychiatric comorbidity. J Clin Psychiatry. 1994;55(1):5-11.
12. Grant JE. Impulse control disorders: a clinician’s guide to understanding and treating behavioral addictions. New York, NY: W.W. Norton & Company, Inc.; 2008.
13. Frohmader KS, Lehman MN, Laviolette SR, et al. Concurrent exposure to methamphetamine and sexual behavior enhances subsequent drug reward and causes compulsive sexual behavior in male rats. J Neurosci. 2011;31(45):16473-16482.
14. Grant JE, Pinto A, Gunnip M, et al. Sexual obsessions and clinical correlates in adults with obsessive-compulsive disorder. Compr Psychiatry. 2006;47(5):325-329.
15. Mété D, Dafreville C, Paitel V, et al. Aripiprazole, gambling disorder and compulsive sexuality [in French]. Encephale. 2016;42(3):281-283.
16. Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol. 2010;67(5):589-595.
17. Kraus SW, Meshberg-Cohen S, Martino S, et al. Treatment of compulsive pornography use with naltrexone: a case report. Am J Psychiatry. 2015;172(12):1260-1261.
18. Derbyshire KL, Grant JE. Compulsive sexual behavior: a review of the literature. J Behav Addict. 2015;4(2):37-43.
19. Levine SB, Scott DL. Sexual education for psychiatric residents. Acad Psychiatry. 2010;34(5):349-352.
20. Alsughier N. Compulsive masturbation treated with selective serotonin reuptake inhibitors. African J Psychiatry (Johannesbg). 2015;18:299.
21. Elmore JL. SSRI reduction of nonparaphilic sexual addiction. CNS Spectr. 2000;5(11);53-56.
22. Stein DJ, Hollander E, Anthony DT, et al. Serotonergic medications for sexual obsessions, sexual addictions, and paraphilias. J Clinical Psychiatry. 1992;53(8):267-271.
23. Kafka M. Psychopharmacologic treatments for nonparaphilic compulsive sexual behaviors. CNS Spectr. 200;5(1):49-59.
24. Wainberg ML, Muench F, Morgenstern J, et al. A double-blind study of citalopram versus placebo in the treatment of compulsive sexual behaviors in gay and bisexual men. J Clin Psychiatry. 2006;67(12):1968-1973.
25. Kafka MP, Hennen J. Psychostimulant augmentation during treatment with selective serotonin reuptake inhibitors in men with paraphilias and paraphilia-related disorders: a case series. J Clin Psychiatry. 2000;61(9):664-670.
26. Coleman E, Raymond N, McBean A. Assessment and treatment of compulsive sexual behavior. Minn Med. 2003;86(7):42-47.
27. Raymond NC, Grant JE, Coleman E. Augmentation with naltrexone to treat compulsive sexual behavior: a case series. Ann Clin Psychiatry. 2010;22(1):56-62.
28. Fong TW, De La Garza R 2nd, Newton TF. A case report of topiramate in the treatment of nonparaphilic sexual addiction. J Clin Psychopharmacol. 2005;25(5):512-514.
29. Del Giudice MJ, Kutinsky J. Applying motivational interviewing to the treatment of sexual compulsivity and addiction. Sex Addict Comp. 2007;14(4):303-319.
30. Shepherd L. Cognitive behavior therapy for sexually addictive behavior. Clin Case Stud. 2010;9(1):18-27.
31. Sadiza J, Varma R, Jena SPK, et al. Group cognitive behaviour therapy in the management of compulsive sex behaviour. International Journal of Criminal Justice Sciences. 2011;6(1-2):309-325.
32. Crosby JM, Twohig MP. Acceptance and commitment therapy for problematic Internet pornography use: a randomized trial. Behav Ther. 2016;47(3):355-366.
33. Twohig MP, Crosby JM. Acceptance and commitment therapy as a treatment for problematic internet pornography viewing. Behav Ther. 2010;41(3):285-295.
34. Sprenkle DH. Treating a sex addict through marital sex therapy. Fam Relat. 1987;36(1):11-14.

Disorders of diminished motivation: What they are, and how to treat them
Disorders of diminished motivation (DDM)—including apathy, abulia, and akinetic mutism—are characterized by impairment in goal-directed behavior, thought, and emotion.1 These disorders can be observed clinically as a gross underproduction of speech, movement, and emotional response.
DDM are not classified as disorders within DSM-5, and it remains unclear if they are distinct disorders or symptoms that overlap in other conditions. Some sources support distinct diagnoses, while the traditional position is that DDM are variations along a spectrum, with apathy as the mildest form and akinetic mutism as the most severe form (Figure).1-3 DDM can result from various neurologic, medical, psychiatric, socioeconomic, and drug-induced pathologies, and may represent differing severity of the same underlying pathology.1,4 It is postulated that DDM arise from disruptions in the dopaminergic frontal-subcortical-mesolimbic networks.1,4
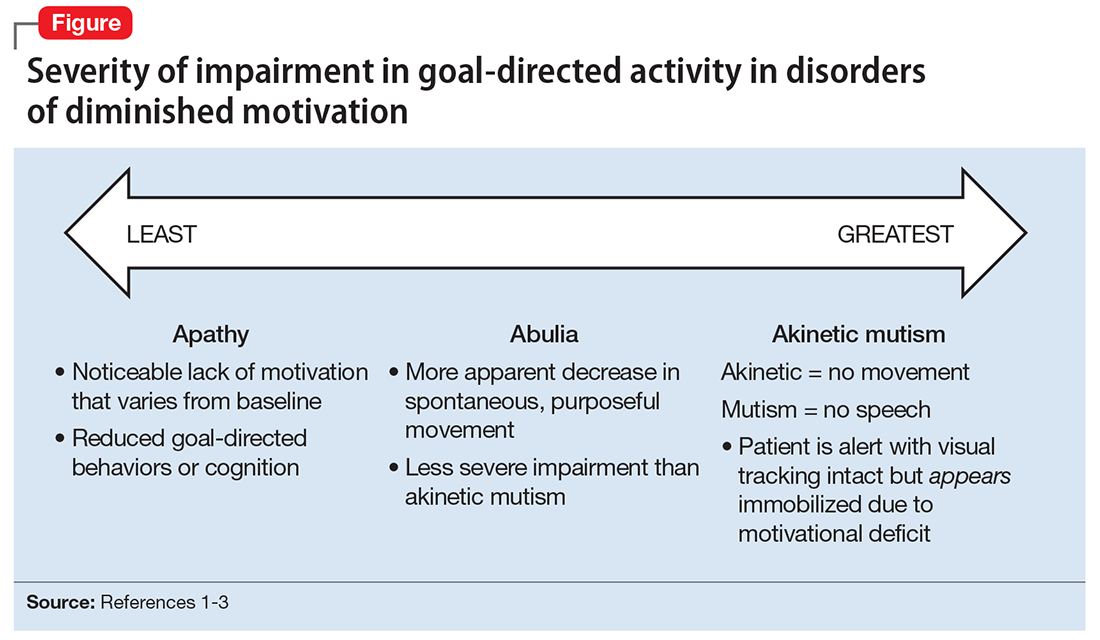
We present 2 cases of patients who developed distinct phenotypes within DDM. Despite differences in presentation and symptom severity, both patients showed clinical improvement on methylphenidate (not the only treatment option) as assessed by the Neuropsychiatric Inventory (NPI),5 a scale used to measure dementia-related behavioral symptoms that includes an Apathy/Indifference (A/I) subscale.
CASE 1
Apathy secondary to glioblastoma multiforme
Ms. E, age 59, presents with wound drainage 3 weeks after a repeat right craniotomy for recurrent glioblastoma multiforme (GBM) of the temporal lobe. Her medical history is not believed to have contributed to her current presentation.
On hospital day 2, Ms. E undergoes debridement and reclosure at the craniotomy site. Prior to the procedure, the patient was noted to have anhedonia and flat affect. Her family reports that she seems to get little enjoyment from life and “only slept and ate.” Psychiatry is consulted on hospital day 3 for evaluation and management of a perceived depressed mood.
On initial psychiatric evaluation, Ms. E continues to have a constricted affect with delayed psychomotor processing speed. However, she denies dysphoria or anhedonia. Richmond Agitation-Sedation Scale6 score is 0 (alert and calm) and test of sustained attention (‘Vigilant A’) is intact (ie, based on the Confusion Assessment Method for the Intensive Care Unit [CAM-ICU],7 Ms. E does not have delirium). The NPI A/I frequency score is 15, with a severity score of 3, for a total score of 45, indicating moderate behavioral disturbance on the NPI A/I subsection. A diagnosis of neuropsychiatric apathy due to recurrent GBM or craniotomy is made, although substance-induced mood disorder due to concurrent dexamethasone and opiate use is considered. Methylphenidate, 2.5 mg/d, is started, and Ms. E’s blood pressure remains stable with the initial dose.
Methylphenidate is titrated to 5 mg, twice daily, over a 1-week period. Ms. E’s NPI A/I subscale score improves to 3 (mild behavioral problem), with 3 points for frequency and a multiplier of 1 for mild severity, reflecting an improvement in neuropsychiatric apathy, and she is transferred to a long-term care rehabilitation center.
CASE 2
Akinetic mutism secondary to subarachnoid hemorrhage
Ms. G, age 47, is brought to an outside hospital with syncope and a severe headache radiating to her neck. Upon arrival, she is unconscious and requires intubation. A non-contrast head CT scan shows diffuse subarachnoid hemorrhage, 6 mm right midline shift, and a small left frontal subdural hematoma. A CT angiography of her head and neck reveals a 0.7 cm anterior paraclinoid left internal carotid artery aneurysm with ophthalmic involvement. Evidence of underlying left and right carotid fibromuscular dysplasia is also seen. Ms. G is transferred to our facility for neurosurgical intervention.
Neurosurgery proceeds with aneurysm coiling, followed by left craniotomy with subdural evacuation and ventriculostomy placement. Her postoperative course is complicated by prolonged nasogastric hyperalimentation, mild hypernatremia and hyperglycemia, tracheostomy, and recurrent central fever. She also develops persistent vasospasm, which requires balloon angioplasty of the left middle cerebral artery.
The psychiatry team is consulted on postoperative day 29 to assess for delirium. The CAM-ICU is positive for delirium, with nocturnal accentuation of agitation. Ms. G demonstrates paucity of speech and minimal verbal comprehension. She starts oral ziprasidone, 5 mg/d at bedtime. In addition to her original CNS insult, scopolamine patch, 1.5 mg, to decrease respiratory secretions, and IV metronidazole, 500 mg every 8 hours, for skin-site infection, may have been contributing to her delirium.
Ms. G’s delirium quickly resolves; however, on day 32 she continues to demonstrate behavioral and cognitive slowing; The NPI A/I frequency score is 28, with a severity score of 3, for a total score of 84, indicating severe behavioral disturbance on the NPI A/I subsection. Methylphenidate, 2.5 mg/d, is started and the next day is increased to 5 mg twice a day to treat severe akinetic mutism. Ms. G also is switched from ziprasidone to olanzapine, 2.5 mg/d at night.
By day 37, the tracheostomy is decannulated, and Ms. G demonstrates a full level of alertness, awareness, and attention. Her affect is full range and appropriate; however, she demonstrates residual language deficits, including dysnomia. On day 38, Ms. G is discharged with an NPI A/I subscale score of 5, indicating a mild behavioral problem.
What these cases demonstrate about DDM
These 2 cases are part of a larger, emerging conversation about the role of dopamine in DDM. Although not fully elucidated, the pathophysiology of abulia, apathy, and akinetic mutism is thought to be related to multiple neurotransmitters—especially dopamine—involved in the cortico-striatal-pallidal-thalamic network.1,8 This position has been supported by reports of clinical improvement in patients with DDM who are given dopaminergic agonists (Table 1).3,9-32
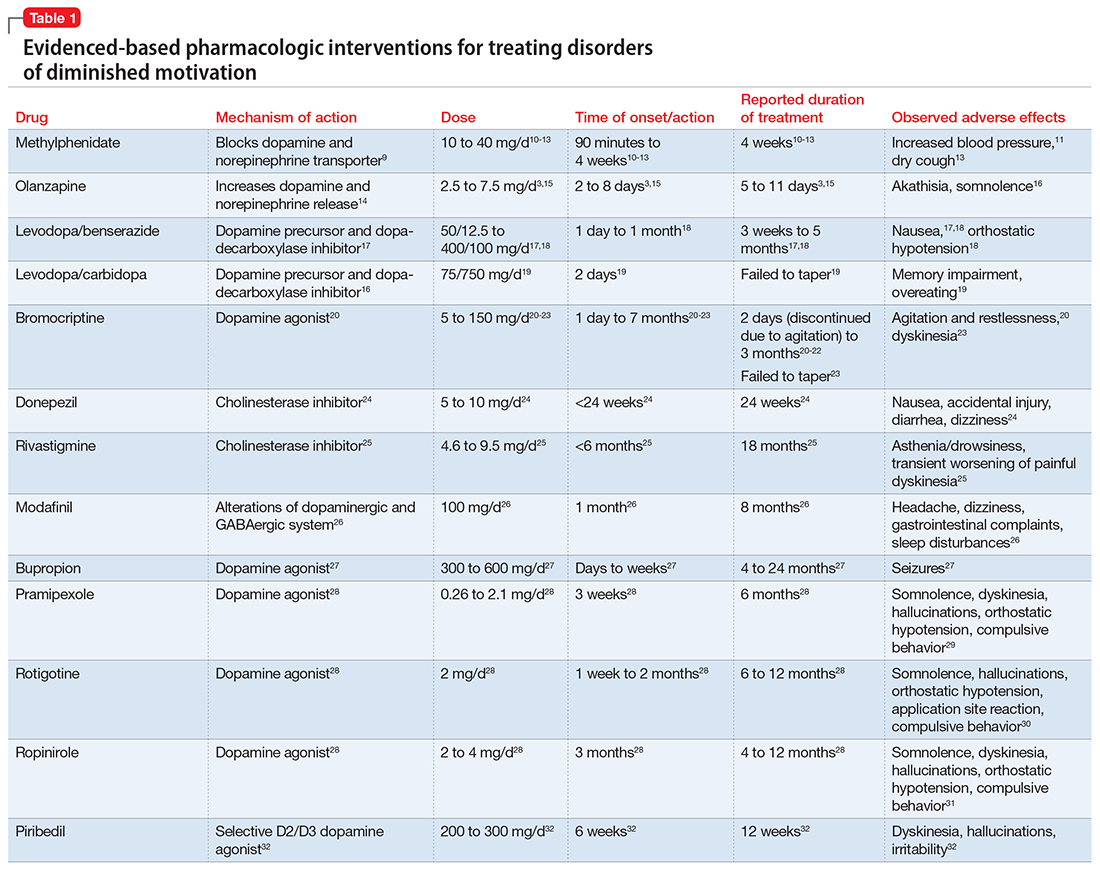
The clinical improvement seen in both of our patients after initiating methylphenidate is consistent with previous reports.10-13 Methylphenidate was selected because of its favorable adverse effect profile and potentially rapid onset of action in DDM.10-13 In cases where oral medication cannot be administered, such as in patients with akinetic mutism, short-term adjunctive IM olanzapine may be helpful, although it is not a first-line treatment.3,15
Interestingly, both of our patients showed improvement with low doses of methylphenidate. Ms. E showed rapid improvement at 2.5 mg/d, but eventually was increased to 10 mg/d. For Ms. G, who demonstrated severe akinetic mutism, rapid improvement was noted after the initial 2.5 mg/d dose; however, because of reports of efficacy of olanzapine in treating akinetic mutism, it is possible that these medications worked synergistically. The proposed mechanism of action of olanzapine in akinetic mutism is through increased dopamine transmission in the medial prefrontal cortex.3,15 Ms. G’s methylphenidate dose was increased to 5 mg/d, which was still “subtherapeutic,” because most reports have used dosages ranging from 10 to 40 mg/d.10-13 Although there were favorable acute results in both patients, their long-term requirements are unknown because of a lack of follow-up. Our findings are also limited by the fact that both patients were recovering from neurosurgical procedures, which could lead to natural improvement in symptoms over time.
Prevalence of DDM in psychiatric disorders
The successful treatment of DDM with dopaminergic drugs is meaningful because of the coexistence of DDM in various neuropsychiatric conditions. In Alzheimer’s disease (AD), disturbances in the dopaminergic system may explain the high comorbidity of apathy, which ranges from 47% in mild AD to 80% in moderate AD.33 In the dopamine-reduced states of cocaine and amphetamine withdrawal, 67% of patients report apathy and lack of motivation.8,34 Additionally, the prevalence of apathy is reported at 27% in Parkinson’s disease, 43% in mild cognitive impairment, 70% in mixed dementia, 94% in a major depressive episode, and 53% in schizophrenia.35 In schizophrenia with predominately negative symptoms, in vivo and postmortem studies have found reduced dopamine receptors.8 Meanwhile, the high rate of akinetic mutism in Creutzfeldt-Jakob disease allows for its use as a reliable diagnostic criteria in this disorder.36
However, the prevalence of DDM is best documented as it relates to stroke and traumatic brain injury (TBI). For instance, after experiencing a stroke, 20% to 25% of patients suffer from apathy.37 Many case reports describe abulia and akinetic mutism after cerebral infarction or hemorrhage, although the incidence of these disorders is unknown.2,38-40 Apathy following TBI is common, especially in younger patients who have sustained a severe injury.41 One study evaluated the prevalence of apathy after TBI among 83 consecutive patients in a neuropsychiatric clinic. Of the 83 patients, 10.84% had apathy without depression, and an equal number were depressed without apathy; another 60% of patients exhibited both apathy and depression. Younger patients (mean age, 29 years) were more likely to be apathetic than older patients, who were more likely to be depressed or depressed and apathetic (mean age, 42 and 38 years, respectively).41 Interestingly, DDM often are associated with cerebral lesions in distinct and distant anatomical locations that are not clearly connected to the neural circuits of motivational pathways. This phenomenon may be explained by the concept of diaschisis, which states that injury to one part of an interconnected neural network can affect other, separate parts of that network.2 If this concept is accurate, it may broaden the impact of DDM, especially as it relates to stroke and TBI.
The differential diagnosis of DDM includes depression and hypokinetic delirium (Table 21,3,42-50). A potential overlapping but confounding condition is stuporous catatonia, with symptoms that include psychomotor slowing such as immobility, staring, and stupor.47 It is important to differentiate these disorders because the treatment for each differs. As previously discussed, there is a clear role for dopamine receptor agonists in the treatment of DDM.

Although major depressive disorder can be treated with medications that increase dopaminergic transmission, selective serotonin reuptake inhibitors (SSRIs) are more commonly used as first-line agents.44 However, an SSRI would theoretically be contraindicated in DDM, because increased serotonin transmission decreases dopamine release from the midbrain, and therefore an SSRI may not only result in a lack of improvement but may worsen DDM.48 Finally, although delirium is treated with atypical or conventional antipsychotics vis-a-vis dopamine type 2 receptor antagonism,45 stuporous catatonia is preferentially treated with gamma-aminobutyric acid-A receptor agonists such as lorazepam.50
What to do when your patient’s presentation suggests DDM
Assessment of DDM should be structured, with input from the patient and the caregiver, and should incorporate the physician’s perspective. A history should be obtained applying recent criteria of apathy. The 3 core domains of apathy—behavior, cognition, and emotion—need to be evaluated. The revised criteria are based on the premise that change in motivation can be measured by examining a patient’s responsiveness to internal or external stimuli. Therefore, each of the 3 domains includes 2 symptoms: (1) self-initiated or “internal” behaviors, cognitions, and emotions (initiation symptom), and (2) the patient’s responsiveness to “external” stimuli (responsiveness symptom).51
One of the main diagnostic dilemmas is how to separate DDM from depression. The differentiation is difficult because of substantial overlap in the manifestation of key symptoms, such as a lack of interest, anergia, psychomotor slowing, and fatigue. Caregivers often mistakenly describe DDM as a depressive state, even though a lack of sadness, desperation, crying, and a depressive mood distinguish DDM from depression. Usually, DDM patients lack negative thoughts, emotional distress, sadness, vegetative symptoms, and somatic concerns, which are frequently observed in mood disorders.51
Several instruments have been developed for assessing neuropsychiatric symptoms. Some were specifically designed to measure apathy, whereas others were designed to provide a broader neuropsychiatric assessment. The NPI is the most widely used multidimensional instrument for assessing neuropsychiatric functioning in patients with neurocognitive disorders (NCDs). It is a valid, reliable instrument that consists of an interview of the patient’s caregiver. It is designed to assess the presence and severity of 10 symptoms, including apathy. The NPI includes both apathy and depression items, which can help clinicians distinguish the 2 conditions. Although beyond the scope of this article, more recent standardized instruments that can assess DDM include the Apathy Inventory, the Dementia Apathy Interview and Rating, and the Structured Clinical Interview for Apathy.52
As previously mentioned, researchers have proposed that DDM are simply a continuum of severity of reduced behavior, and akinetic mutism may be the extreme form. The dilemma is how to formally diagnose states of abulia and akinetic mutism, given the lack of diagnostic criteria and paucity of standardized instruments. Thus, distinguishing between abulia and akinetic mutism (and apathy) is more of a quantitative than qualitative exercise. One could hypothesize that higher scores on a standardized scale to measure apathy (ie, NPI) could imply abulia or akinetic mutism, although to the best of our knowledge, no formal “cut-off scores” exist.53
Treatment of apathy. The duration of pharmacotherapy to treat apathy is unknown and their usage is off-label. Further studies, including double-blind, randomized controlled trials (RCTs), are needed. Nonetheless, the 2 classes of medications that have the most evidence for treating apathy/DDM are psychostimulants and acetylcholinesterase inhibitors (AChEIs).
AChEIs are primarily used for treating cognitive symptoms in NCDs, but recent findings indicate that they have beneficial effects on noncognitive symptoms such as apathy. Of all medications used to treat apathy in NCDs, AChEIs have been used to treat the largest number of patients. Of 26 studies, 24 demonstrated improvement in apathy, with 21 demonstrating statistical significance. These studies ranged in duration from 8 weeks to 1 year, and most were open-label.54
Five studies (3 RCTs and 2 open-label studies) assessed the efficacy of methylphenidate for treating apathy due to AD. All the studies demonstrated at least some benefit in apathy scores after treatment with methylphenidate. These studies ranged from 5 to 12 weeks in duration. Notably, some patients reported adverse effects, including delusions and irritability.54
Although available evidence suggests AChEIs may be the most effective medications for treating apathy in NCDs, methylphenidate has been demonstrated to work faster.55 Thus, in cases where apathy can significantly affect activities of daily living or instrumental activities of daily living, a quicker response may dictate treatment with methylphenidate. It is imperative to note that safety studies and more large-scale double-blind RCTs are needed to further demonstrate the effectiveness and safety of methylphenidate.
Published in 2007, the American Psychiatric Association (APA) guidelines56 state that psychostimulants are a possible treatment option for patients with severe apathy. At the same time, clinicians are reminded that these agents—especially at higher doses—can produce various problematic adverse effects, including tachycardia, hypertension, restlessness, dyskinesia, agitation, sleep disturbances, psychosis, confusion, and decreased appetite. The APA guidelines recommend using low initial doses, with slow and careful titration. For example, methylphenidate should be started at 2.5 to 5 mg once in the morning, with daily doses not to exceed 30 to 40 mg. In our clinical experience, doses >20 mg/d have not been necessary.57
Treatment of akinetic mutism and abulia. In patients with akinetic mutism and possible abulia, for whom oral medication administration is either impossible or contraindicated (ie, due to the potential risk of aspiration pneumonia), atypical antipsychotics, such as IM olanazapine, have produced a therapeutic response in apathetic patients with NCD. However, extensive use of antipsychotics in NCD is not recommended because this class of medications has been associated with serious adverse effects, including an increased risk of death.55
Bottom Line
Apathy, abulia, and akinetic mutism have been categorized as disorders of diminished motivation (DDM). They commonly present after a stroke or traumatic brain injury, and should be differentiated from depression, hypokinetic delirium, and stuporous catatonia. DDM can be successfully treated with dopamine agonists.
Related Resources
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract. 2004;10(3):196-199.
- Dell’Osso B, Benatti B, Altamura AC, et al. Prevalence of selective serotonin reuptake inhibitor-related apathy in patients with obsessive compulsive disorder. J Clin Psychopharmacol. 2016;36(6):725-726.
- D’Souza G, Kakoullis A, Hegde N, et al. Recognition and management of abulia in the elderly. Prog Neurol Psychiatry. 2010;14(6):24-28.
Drug Brand Names
Bromocriptine • Parlodel
Bupropion • Wellbutrin XL, Zyban
Carbidopa • Lodosyn
Dexamethasone • DexPak, Ozurde
Donepezil • Aricept
Levodopa/benserazide • Prolopa
Levodopa/carbidopa • Pacopa Rytary Sinemet
Lorazepam • Ativan
Methylphenidate • Concerta, Methylin
Metronidazole • Flagyl, Metrogel
Modafinil • Provigil
Olanzapine • Zyprexa
Pramipexole • Mirapex
Rivastigmine • Exelon
Ropinirole • Requip
Rotigotine • Neurpro
Scopolamine • Transderm Scop
Ziprasidone • Geodon
1. Marin RS, Wilkosz PA. Disorders of diminished motivation. J Head Trauma Rehabil. 2005;20(4):377-388.
2. Ghoshal S, Gokhale S, Rebovich G, et al. The neurology of decreased activity: abulia. Rev Neurol Dis. 2011;8(3-4):e55-e67.
3. Spiegel DR, Chatterjee A. A case of abulia, status/post right middle cerebral artery territory infarct, treated successfully with olanzapine. Clin Neuropharmacol. 2014;37(6):186-189.
4. Marin RS. Differential diagnosis and classification of apathy. Am J Psychiatry. 1990;147(1):22-30.
5. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314.
6. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338-1344.
7. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370-1379.
8. Al-Adawi S, Dawe GS, Al-Hussaini AA. Aboulia: neurobehavioural dysfunction of dopaminergic system? Med Hypotheses. 2000;54(4):523-530.
9. Volkow ND, Fowler JS, Wang G, et al. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(suppl 1):S31-S43.
10. Chatterjee A, Fahn S. Methylphenidate treats apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2002;14(4):461-462.
11. Keenan S, Mavaddat N, Iddon J, et al. Effects of methylphenidate on cognition and apathy in normal pressure hydrocephalus: a case study and review. Br J Neurosurg. 2005;19(1):46-50.
12. Padala PR, Petty F, Bhatia SC. Methylphenidate may treat apathy independent of depression. Ann Pharmacother. 2005;39(11):1947-1949.
13. Padala PR, Burke WJ, Bhatia SC, et al. Treatment of apathy with methylphenidate. J Neuropsychiatry Clin Neurosci. 2007;19(1):81-83.
14. Li XM, Perry KW, Wong DT, et al. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl). 1998;136(2):153-161.
15. Spiegel DR, Casella DP, Callender DM, et al. Treatment of akinetic mutism with intramuscular olanzapine: a case series. J Neuropsychiatry Clin Neurosci. 2008;20(1):93-95.
16. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
17. Bakheit AM, Fletcher K, Brennan A. Successful treatment of severe abulia with co-beneldopa. NeuroRehabilitation. 2011;29(4):347-351.
18. Debette S, Kozlowski O, Steinling M, et al. Levodopa and bromocriptine in hypoxic brain injury. J Neurol. 2002;249(12):1678-1682.
19. Combarros O, Infante J, Berciano J. Akinetic mutism from frontal lobe damage responding to levodopa. J Neurol. 2000;247(7):568-569.
20. Echiverri HC, Tatum WO, Merens TA, et al. Akinetic mutism: pharmacologic probe of the dopaminergic mesencephalofrontal activating system. Pediatr Neurol. 1988;4(4):228-230.
21. Psarros T, Zouros A, Coimbra C. Bromocriptine-responsive akinetic mutism following endoscopy for ventricular neurocysticercosis. Case report and review of the literature. J Neurosurg. 2003;99(2):397-401.
22. Naik VD. Abulia following an episode of cardiac arrest [published online July 1, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-209357.
23. Kim MS, Rhee JJ, Lee SJ, et al. Akinetic mutism responsive to bromocriptine following subdural hematoma evacuation in a patient with hydrocephalus. Neurol Med Chir (Tokyo). 2007;47(9):419-423.
24. Rockwood K, Black S, Bedard MA; TOPS Study Investigators. Specific symptomatic changes following donepezil treatment of Alzheimer’s disease: a multi-centre, primary care, open-label study. Int J Geriatr Psychiatry. 2007;22(4):312-319.
25. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry. 2014;85(6):668-674.
26. Camargos EF, Quintas JL. Apathy syndrome treated successfully with modafinil [published online November 15, 2011]. BMJ Case Rep. doi: 10.1136/bcr.08.2011.4652.
27. Corcoran C, Wong ML, O’Keane V. Bupropion in the management of apathy. J Psychopharmacol. 2004;18(1):133-135.
28. Blundo C, Gerace C. Dopamine agonists can improve pure apathy associated with lesions of the prefrontal-basal ganglia functional system. Neurol Sci. 2015;36(7):1197-1201.
29. Mirapex [package insert]. Ridgefield, CT: Boehringer Ingelheim International GmbH; 2016.
30. Neupro [package insert]. Smyrna, GA: UBC, Inc.; 2012.
31. Requip [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
32. Thobois S, Lhommée E, Klinger H, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136(pt 5):1568-1577.
33. Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther. 2011;17(5):411-427.
34. Brower KJ, Maddahian E, Blow FC, et al. A comparison of self-reported symptoms and DSM-III-R criteria for cocaine withdrawal. Am J Drug Alcohol Abuse. 1988;14(3):347-356.
35. Mulin E, Leone E, Dujardin K, et al. Diagnostic criteria for apathy in clinical practice. Int J Geriatr Psychiatry. 2011;26(2):158-165.
36. Otto A, Zerr I, Lantsch M, et al. Akinetic mutism as a classification criterion for the diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1998;64(4):524-528.
37. Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350-354.
38. Hastak SM, Gorawara PS, Mishra NK. Abulia: no will, no way. J Assoc Physicians India. 2005;53:814-818.
39. Nagaratnam N, Nagaratnam K, Ng K, et al. Akinetic mutism following stroke. J Clin Neurosci. 2004;11(1):25-30.
40. Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34(6):693-698.
41. Schwarzbold M, Diaz A, Martins ET, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797-816.
42. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
43. Levy ML, Cummings JL, Fairbanks LA, et al. Apathy is not depression. J Neuropsychiatry Clin Neurosci. 1998;10(3):314-319.
44. Snow V, Lascher S, Mottur-Pilson C. Pharmacologic treatment of acute major depression and dysthymia. American College of Physicians-American Society of Internal Medicine. Ann Intern Med. 2000;132(9):738-742.
45. Schwartz AC, Fisher TJ, Greenspan HN, et al. Pharmacologic and nonpharmacologic approaches to the prevention and management of delirium. Int J Psychiatry Med. 2016;51(2):160-170.
46. Kang H, Zhao F, You L, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17(2):147-154.
47. Fricchione GL, Beach SR, Huffman J, et al. Life-threatening conditions in psychiatry: catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fava M, Wilens TE, eds. Massachusetts General Hospital comprehensive clinical psychiatry. London, United Kingdom: Elsevier; 2016:608-617.
48. Rogers RD. The roles of dopamine and serotonin in decision making: evidence from pharmacological experiments in humans. Neuropsychopharmacology. 2011;36(1):114-132.
49. Stransky M, Schmidt C, Ganslmeier P, et al. Hypoactive delirium after cardiac surgery as an independent risk factor for prolonged mechanical ventilation. J Cardiothorac Vasc Anesth. 2011;25(6):968-974.
50. Wilcox JA, Reid Duffy P. The syndrome of catatonia. Behav Sci (Basel). 2015;5(4):576-588.
51. Robert PH, Mulin E, Malléa P, et al. REVIEW: apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):263-271.
52. Cipriani G, Lucetti C, Danti S, et al. Apathy and dementia. Nosology, assessment and management. J Nerv Ment Dis. 2014;202(10):718-724.
53. Starkstein SE, Leentjens AF. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79(10):1088-1092.54. Berman K, Brodaty H, Withall A, et al. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry. 2012;20(2):104-122.
55. Theleritis C, Siarkos K, Katirtzoglou E, et al. Pharmacological and nonpharmacological treatment for apathy in Alzheimer disease: a systematic review across modalities. J Geriatr Psychiatry Neurol. 2017;30(1):26-49.
56. APA Work Group on Alzheimer’s Disease and other Dementias; Rabins PV, Blacker D, Rovner BW, et al. American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry. 2007;164(suppl 12):5-56.
57. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624-1632.
Disorders of diminished motivation (DDM)—including apathy, abulia, and akinetic mutism—are characterized by impairment in goal-directed behavior, thought, and emotion.1 These disorders can be observed clinically as a gross underproduction of speech, movement, and emotional response.
DDM are not classified as disorders within DSM-5, and it remains unclear if they are distinct disorders or symptoms that overlap in other conditions. Some sources support distinct diagnoses, while the traditional position is that DDM are variations along a spectrum, with apathy as the mildest form and akinetic mutism as the most severe form (Figure).1-3 DDM can result from various neurologic, medical, psychiatric, socioeconomic, and drug-induced pathologies, and may represent differing severity of the same underlying pathology.1,4 It is postulated that DDM arise from disruptions in the dopaminergic frontal-subcortical-mesolimbic networks.1,4
We present 2 cases of patients who developed distinct phenotypes within DDM. Despite differences in presentation and symptom severity, both patients showed clinical improvement on methylphenidate (not the only treatment option) as assessed by the Neuropsychiatric Inventory (NPI),5 a scale used to measure dementia-related behavioral symptoms that includes an Apathy/Indifference (A/I) subscale.
CASE 1
Apathy secondary to glioblastoma multiforme
Ms. E, age 59, presents with wound drainage 3 weeks after a repeat right craniotomy for recurrent glioblastoma multiforme (GBM) of the temporal lobe. Her medical history is not believed to have contributed to her current presentation.
On hospital day 2, Ms. E undergoes debridement and reclosure at the craniotomy site. Prior to the procedure, the patient was noted to have anhedonia and flat affect. Her family reports that she seems to get little enjoyment from life and “only slept and ate.” Psychiatry is consulted on hospital day 3 for evaluation and management of a perceived depressed mood.
On initial psychiatric evaluation, Ms. E continues to have a constricted affect with delayed psychomotor processing speed. However, she denies dysphoria or anhedonia. Richmond Agitation-Sedation Scale6 score is 0 (alert and calm) and test of sustained attention (‘Vigilant A’) is intact (ie, based on the Confusion Assessment Method for the Intensive Care Unit [CAM-ICU],7 Ms. E does not have delirium). The NPI A/I frequency score is 15, with a severity score of 3, for a total score of 45, indicating moderate behavioral disturbance on the NPI A/I subsection. A diagnosis of neuropsychiatric apathy due to recurrent GBM or craniotomy is made, although substance-induced mood disorder due to concurrent dexamethasone and opiate use is considered. Methylphenidate, 2.5 mg/d, is started, and Ms. E’s blood pressure remains stable with the initial dose.
Methylphenidate is titrated to 5 mg, twice daily, over a 1-week period. Ms. E’s NPI A/I subscale score improves to 3 (mild behavioral problem), with 3 points for frequency and a multiplier of 1 for mild severity, reflecting an improvement in neuropsychiatric apathy, and she is transferred to a long-term care rehabilitation center.
CASE 2
Akinetic mutism secondary to subarachnoid hemorrhage
Ms. G, age 47, is brought to an outside hospital with syncope and a severe headache radiating to her neck. Upon arrival, she is unconscious and requires intubation. A non-contrast head CT scan shows diffuse subarachnoid hemorrhage, 6 mm right midline shift, and a small left frontal subdural hematoma. A CT angiography of her head and neck reveals a 0.7 cm anterior paraclinoid left internal carotid artery aneurysm with ophthalmic involvement. Evidence of underlying left and right carotid fibromuscular dysplasia is also seen. Ms. G is transferred to our facility for neurosurgical intervention.
Neurosurgery proceeds with aneurysm coiling, followed by left craniotomy with subdural evacuation and ventriculostomy placement. Her postoperative course is complicated by prolonged nasogastric hyperalimentation, mild hypernatremia and hyperglycemia, tracheostomy, and recurrent central fever. She also develops persistent vasospasm, which requires balloon angioplasty of the left middle cerebral artery.
The psychiatry team is consulted on postoperative day 29 to assess for delirium. The CAM-ICU is positive for delirium, with nocturnal accentuation of agitation. Ms. G demonstrates paucity of speech and minimal verbal comprehension. She starts oral ziprasidone, 5 mg/d at bedtime. In addition to her original CNS insult, scopolamine patch, 1.5 mg, to decrease respiratory secretions, and IV metronidazole, 500 mg every 8 hours, for skin-site infection, may have been contributing to her delirium.
Ms. G’s delirium quickly resolves; however, on day 32 she continues to demonstrate behavioral and cognitive slowing; The NPI A/I frequency score is 28, with a severity score of 3, for a total score of 84, indicating severe behavioral disturbance on the NPI A/I subsection. Methylphenidate, 2.5 mg/d, is started and the next day is increased to 5 mg twice a day to treat severe akinetic mutism. Ms. G also is switched from ziprasidone to olanzapine, 2.5 mg/d at night.
By day 37, the tracheostomy is decannulated, and Ms. G demonstrates a full level of alertness, awareness, and attention. Her affect is full range and appropriate; however, she demonstrates residual language deficits, including dysnomia. On day 38, Ms. G is discharged with an NPI A/I subscale score of 5, indicating a mild behavioral problem.
What these cases demonstrate about DDM
These 2 cases are part of a larger, emerging conversation about the role of dopamine in DDM. Although not fully elucidated, the pathophysiology of abulia, apathy, and akinetic mutism is thought to be related to multiple neurotransmitters—especially dopamine—involved in the cortico-striatal-pallidal-thalamic network.1,8 This position has been supported by reports of clinical improvement in patients with DDM who are given dopaminergic agonists (Table 1).3,9-32
The clinical improvement seen in both of our patients after initiating methylphenidate is consistent with previous reports.10-13 Methylphenidate was selected because of its favorable adverse effect profile and potentially rapid onset of action in DDM.10-13 In cases where oral medication cannot be administered, such as in patients with akinetic mutism, short-term adjunctive IM olanzapine may be helpful, although it is not a first-line treatment.3,15
Interestingly, both of our patients showed improvement with low doses of methylphenidate. Ms. E showed rapid improvement at 2.5 mg/d, but eventually was increased to 10 mg/d. For Ms. G, who demonstrated severe akinetic mutism, rapid improvement was noted after the initial 2.5 mg/d dose; however, because of reports of efficacy of olanzapine in treating akinetic mutism, it is possible that these medications worked synergistically. The proposed mechanism of action of olanzapine in akinetic mutism is through increased dopamine transmission in the medial prefrontal cortex.3,15 Ms. G’s methylphenidate dose was increased to 5 mg/d, which was still “subtherapeutic,” because most reports have used dosages ranging from 10 to 40 mg/d.10-13 Although there were favorable acute results in both patients, their long-term requirements are unknown because of a lack of follow-up. Our findings are also limited by the fact that both patients were recovering from neurosurgical procedures, which could lead to natural improvement in symptoms over time.
Prevalence of DDM in psychiatric disorders
The successful treatment of DDM with dopaminergic drugs is meaningful because of the coexistence of DDM in various neuropsychiatric conditions. In Alzheimer’s disease (AD), disturbances in the dopaminergic system may explain the high comorbidity of apathy, which ranges from 47% in mild AD to 80% in moderate AD.33 In the dopamine-reduced states of cocaine and amphetamine withdrawal, 67% of patients report apathy and lack of motivation.8,34 Additionally, the prevalence of apathy is reported at 27% in Parkinson’s disease, 43% in mild cognitive impairment, 70% in mixed dementia, 94% in a major depressive episode, and 53% in schizophrenia.35 In schizophrenia with predominately negative symptoms, in vivo and postmortem studies have found reduced dopamine receptors.8 Meanwhile, the high rate of akinetic mutism in Creutzfeldt-Jakob disease allows for its use as a reliable diagnostic criteria in this disorder.36
However, the prevalence of DDM is best documented as it relates to stroke and traumatic brain injury (TBI). For instance, after experiencing a stroke, 20% to 25% of patients suffer from apathy.37 Many case reports describe abulia and akinetic mutism after cerebral infarction or hemorrhage, although the incidence of these disorders is unknown.2,38-40 Apathy following TBI is common, especially in younger patients who have sustained a severe injury.41 One study evaluated the prevalence of apathy after TBI among 83 consecutive patients in a neuropsychiatric clinic. Of the 83 patients, 10.84% had apathy without depression, and an equal number were depressed without apathy; another 60% of patients exhibited both apathy and depression. Younger patients (mean age, 29 years) were more likely to be apathetic than older patients, who were more likely to be depressed or depressed and apathetic (mean age, 42 and 38 years, respectively).41 Interestingly, DDM often are associated with cerebral lesions in distinct and distant anatomical locations that are not clearly connected to the neural circuits of motivational pathways. This phenomenon may be explained by the concept of diaschisis, which states that injury to one part of an interconnected neural network can affect other, separate parts of that network.2 If this concept is accurate, it may broaden the impact of DDM, especially as it relates to stroke and TBI.
The differential diagnosis of DDM includes depression and hypokinetic delirium (Table 21,3,42-50). A potential overlapping but confounding condition is stuporous catatonia, with symptoms that include psychomotor slowing such as immobility, staring, and stupor.47 It is important to differentiate these disorders because the treatment for each differs. As previously discussed, there is a clear role for dopamine receptor agonists in the treatment of DDM.
Although major depressive disorder can be treated with medications that increase dopaminergic transmission, selective serotonin reuptake inhibitors (SSRIs) are more commonly used as first-line agents.44 However, an SSRI would theoretically be contraindicated in DDM, because increased serotonin transmission decreases dopamine release from the midbrain, and therefore an SSRI may not only result in a lack of improvement but may worsen DDM.48 Finally, although delirium is treated with atypical or conventional antipsychotics vis-a-vis dopamine type 2 receptor antagonism,45 stuporous catatonia is preferentially treated with gamma-aminobutyric acid-A receptor agonists such as lorazepam.50
What to do when your patient’s presentation suggests DDM
Assessment of DDM should be structured, with input from the patient and the caregiver, and should incorporate the physician’s perspective. A history should be obtained applying recent criteria of apathy. The 3 core domains of apathy—behavior, cognition, and emotion—need to be evaluated. The revised criteria are based on the premise that change in motivation can be measured by examining a patient’s responsiveness to internal or external stimuli. Therefore, each of the 3 domains includes 2 symptoms: (1) self-initiated or “internal” behaviors, cognitions, and emotions (initiation symptom), and (2) the patient’s responsiveness to “external” stimuli (responsiveness symptom).51
One of the main diagnostic dilemmas is how to separate DDM from depression. The differentiation is difficult because of substantial overlap in the manifestation of key symptoms, such as a lack of interest, anergia, psychomotor slowing, and fatigue. Caregivers often mistakenly describe DDM as a depressive state, even though a lack of sadness, desperation, crying, and a depressive mood distinguish DDM from depression. Usually, DDM patients lack negative thoughts, emotional distress, sadness, vegetative symptoms, and somatic concerns, which are frequently observed in mood disorders.51
Several instruments have been developed for assessing neuropsychiatric symptoms. Some were specifically designed to measure apathy, whereas others were designed to provide a broader neuropsychiatric assessment. The NPI is the most widely used multidimensional instrument for assessing neuropsychiatric functioning in patients with neurocognitive disorders (NCDs). It is a valid, reliable instrument that consists of an interview of the patient’s caregiver. It is designed to assess the presence and severity of 10 symptoms, including apathy. The NPI includes both apathy and depression items, which can help clinicians distinguish the 2 conditions. Although beyond the scope of this article, more recent standardized instruments that can assess DDM include the Apathy Inventory, the Dementia Apathy Interview and Rating, and the Structured Clinical Interview for Apathy.52
As previously mentioned, researchers have proposed that DDM are simply a continuum of severity of reduced behavior, and akinetic mutism may be the extreme form. The dilemma is how to formally diagnose states of abulia and akinetic mutism, given the lack of diagnostic criteria and paucity of standardized instruments. Thus, distinguishing between abulia and akinetic mutism (and apathy) is more of a quantitative than qualitative exercise. One could hypothesize that higher scores on a standardized scale to measure apathy (ie, NPI) could imply abulia or akinetic mutism, although to the best of our knowledge, no formal “cut-off scores” exist.53
Treatment of apathy. The duration of pharmacotherapy to treat apathy is unknown and their usage is off-label. Further studies, including double-blind, randomized controlled trials (RCTs), are needed. Nonetheless, the 2 classes of medications that have the most evidence for treating apathy/DDM are psychostimulants and acetylcholinesterase inhibitors (AChEIs).
AChEIs are primarily used for treating cognitive symptoms in NCDs, but recent findings indicate that they have beneficial effects on noncognitive symptoms such as apathy. Of all medications used to treat apathy in NCDs, AChEIs have been used to treat the largest number of patients. Of 26 studies, 24 demonstrated improvement in apathy, with 21 demonstrating statistical significance. These studies ranged in duration from 8 weeks to 1 year, and most were open-label.54
Five studies (3 RCTs and 2 open-label studies) assessed the efficacy of methylphenidate for treating apathy due to AD. All the studies demonstrated at least some benefit in apathy scores after treatment with methylphenidate. These studies ranged from 5 to 12 weeks in duration. Notably, some patients reported adverse effects, including delusions and irritability.54
Although available evidence suggests AChEIs may be the most effective medications for treating apathy in NCDs, methylphenidate has been demonstrated to work faster.55 Thus, in cases where apathy can significantly affect activities of daily living or instrumental activities of daily living, a quicker response may dictate treatment with methylphenidate. It is imperative to note that safety studies and more large-scale double-blind RCTs are needed to further demonstrate the effectiveness and safety of methylphenidate.
Published in 2007, the American Psychiatric Association (APA) guidelines56 state that psychostimulants are a possible treatment option for patients with severe apathy. At the same time, clinicians are reminded that these agents—especially at higher doses—can produce various problematic adverse effects, including tachycardia, hypertension, restlessness, dyskinesia, agitation, sleep disturbances, psychosis, confusion, and decreased appetite. The APA guidelines recommend using low initial doses, with slow and careful titration. For example, methylphenidate should be started at 2.5 to 5 mg once in the morning, with daily doses not to exceed 30 to 40 mg. In our clinical experience, doses >20 mg/d have not been necessary.57
Treatment of akinetic mutism and abulia. In patients with akinetic mutism and possible abulia, for whom oral medication administration is either impossible or contraindicated (ie, due to the potential risk of aspiration pneumonia), atypical antipsychotics, such as IM olanazapine, have produced a therapeutic response in apathetic patients with NCD. However, extensive use of antipsychotics in NCD is not recommended because this class of medications has been associated with serious adverse effects, including an increased risk of death.55
Bottom Line
Apathy, abulia, and akinetic mutism have been categorized as disorders of diminished motivation (DDM). They commonly present after a stroke or traumatic brain injury, and should be differentiated from depression, hypokinetic delirium, and stuporous catatonia. DDM can be successfully treated with dopamine agonists.
Related Resources
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract. 2004;10(3):196-199.
- Dell’Osso B, Benatti B, Altamura AC, et al. Prevalence of selective serotonin reuptake inhibitor-related apathy in patients with obsessive compulsive disorder. J Clin Psychopharmacol. 2016;36(6):725-726.
- D’Souza G, Kakoullis A, Hegde N, et al. Recognition and management of abulia in the elderly. Prog Neurol Psychiatry. 2010;14(6):24-28.
Drug Brand Names
Bromocriptine • Parlodel
Bupropion • Wellbutrin XL, Zyban
Carbidopa • Lodosyn
Dexamethasone • DexPak, Ozurde
Donepezil • Aricept
Levodopa/benserazide • Prolopa
Levodopa/carbidopa • Pacopa Rytary Sinemet
Lorazepam • Ativan
Methylphenidate • Concerta, Methylin
Metronidazole • Flagyl, Metrogel
Modafinil • Provigil
Olanzapine • Zyprexa
Pramipexole • Mirapex
Rivastigmine • Exelon
Ropinirole • Requip
Rotigotine • Neurpro
Scopolamine • Transderm Scop
Ziprasidone • Geodon
Disorders of diminished motivation (DDM)—including apathy, abulia, and akinetic mutism—are characterized by impairment in goal-directed behavior, thought, and emotion.1 These disorders can be observed clinically as a gross underproduction of speech, movement, and emotional response.
DDM are not classified as disorders within DSM-5, and it remains unclear if they are distinct disorders or symptoms that overlap in other conditions. Some sources support distinct diagnoses, while the traditional position is that DDM are variations along a spectrum, with apathy as the mildest form and akinetic mutism as the most severe form (Figure).1-3 DDM can result from various neurologic, medical, psychiatric, socioeconomic, and drug-induced pathologies, and may represent differing severity of the same underlying pathology.1,4 It is postulated that DDM arise from disruptions in the dopaminergic frontal-subcortical-mesolimbic networks.1,4
We present 2 cases of patients who developed distinct phenotypes within DDM. Despite differences in presentation and symptom severity, both patients showed clinical improvement on methylphenidate (not the only treatment option) as assessed by the Neuropsychiatric Inventory (NPI),5 a scale used to measure dementia-related behavioral symptoms that includes an Apathy/Indifference (A/I) subscale.
CASE 1
Apathy secondary to glioblastoma multiforme
Ms. E, age 59, presents with wound drainage 3 weeks after a repeat right craniotomy for recurrent glioblastoma multiforme (GBM) of the temporal lobe. Her medical history is not believed to have contributed to her current presentation.
On hospital day 2, Ms. E undergoes debridement and reclosure at the craniotomy site. Prior to the procedure, the patient was noted to have anhedonia and flat affect. Her family reports that she seems to get little enjoyment from life and “only slept and ate.” Psychiatry is consulted on hospital day 3 for evaluation and management of a perceived depressed mood.
On initial psychiatric evaluation, Ms. E continues to have a constricted affect with delayed psychomotor processing speed. However, she denies dysphoria or anhedonia. Richmond Agitation-Sedation Scale6 score is 0 (alert and calm) and test of sustained attention (‘Vigilant A’) is intact (ie, based on the Confusion Assessment Method for the Intensive Care Unit [CAM-ICU],7 Ms. E does not have delirium). The NPI A/I frequency score is 15, with a severity score of 3, for a total score of 45, indicating moderate behavioral disturbance on the NPI A/I subsection. A diagnosis of neuropsychiatric apathy due to recurrent GBM or craniotomy is made, although substance-induced mood disorder due to concurrent dexamethasone and opiate use is considered. Methylphenidate, 2.5 mg/d, is started, and Ms. E’s blood pressure remains stable with the initial dose.
Methylphenidate is titrated to 5 mg, twice daily, over a 1-week period. Ms. E’s NPI A/I subscale score improves to 3 (mild behavioral problem), with 3 points for frequency and a multiplier of 1 for mild severity, reflecting an improvement in neuropsychiatric apathy, and she is transferred to a long-term care rehabilitation center.
CASE 2
Akinetic mutism secondary to subarachnoid hemorrhage
Ms. G, age 47, is brought to an outside hospital with syncope and a severe headache radiating to her neck. Upon arrival, she is unconscious and requires intubation. A non-contrast head CT scan shows diffuse subarachnoid hemorrhage, 6 mm right midline shift, and a small left frontal subdural hematoma. A CT angiography of her head and neck reveals a 0.7 cm anterior paraclinoid left internal carotid artery aneurysm with ophthalmic involvement. Evidence of underlying left and right carotid fibromuscular dysplasia is also seen. Ms. G is transferred to our facility for neurosurgical intervention.
Neurosurgery proceeds with aneurysm coiling, followed by left craniotomy with subdural evacuation and ventriculostomy placement. Her postoperative course is complicated by prolonged nasogastric hyperalimentation, mild hypernatremia and hyperglycemia, tracheostomy, and recurrent central fever. She also develops persistent vasospasm, which requires balloon angioplasty of the left middle cerebral artery.
The psychiatry team is consulted on postoperative day 29 to assess for delirium. The CAM-ICU is positive for delirium, with nocturnal accentuation of agitation. Ms. G demonstrates paucity of speech and minimal verbal comprehension. She starts oral ziprasidone, 5 mg/d at bedtime. In addition to her original CNS insult, scopolamine patch, 1.5 mg, to decrease respiratory secretions, and IV metronidazole, 500 mg every 8 hours, for skin-site infection, may have been contributing to her delirium.
Ms. G’s delirium quickly resolves; however, on day 32 she continues to demonstrate behavioral and cognitive slowing; The NPI A/I frequency score is 28, with a severity score of 3, for a total score of 84, indicating severe behavioral disturbance on the NPI A/I subsection. Methylphenidate, 2.5 mg/d, is started and the next day is increased to 5 mg twice a day to treat severe akinetic mutism. Ms. G also is switched from ziprasidone to olanzapine, 2.5 mg/d at night.
By day 37, the tracheostomy is decannulated, and Ms. G demonstrates a full level of alertness, awareness, and attention. Her affect is full range and appropriate; however, she demonstrates residual language deficits, including dysnomia. On day 38, Ms. G is discharged with an NPI A/I subscale score of 5, indicating a mild behavioral problem.
What these cases demonstrate about DDM
These 2 cases are part of a larger, emerging conversation about the role of dopamine in DDM. Although not fully elucidated, the pathophysiology of abulia, apathy, and akinetic mutism is thought to be related to multiple neurotransmitters—especially dopamine—involved in the cortico-striatal-pallidal-thalamic network.1,8 This position has been supported by reports of clinical improvement in patients with DDM who are given dopaminergic agonists (Table 1).3,9-32
The clinical improvement seen in both of our patients after initiating methylphenidate is consistent with previous reports.10-13 Methylphenidate was selected because of its favorable adverse effect profile and potentially rapid onset of action in DDM.10-13 In cases where oral medication cannot be administered, such as in patients with akinetic mutism, short-term adjunctive IM olanzapine may be helpful, although it is not a first-line treatment.3,15
Interestingly, both of our patients showed improvement with low doses of methylphenidate. Ms. E showed rapid improvement at 2.5 mg/d, but eventually was increased to 10 mg/d. For Ms. G, who demonstrated severe akinetic mutism, rapid improvement was noted after the initial 2.5 mg/d dose; however, because of reports of efficacy of olanzapine in treating akinetic mutism, it is possible that these medications worked synergistically. The proposed mechanism of action of olanzapine in akinetic mutism is through increased dopamine transmission in the medial prefrontal cortex.3,15 Ms. G’s methylphenidate dose was increased to 5 mg/d, which was still “subtherapeutic,” because most reports have used dosages ranging from 10 to 40 mg/d.10-13 Although there were favorable acute results in both patients, their long-term requirements are unknown because of a lack of follow-up. Our findings are also limited by the fact that both patients were recovering from neurosurgical procedures, which could lead to natural improvement in symptoms over time.
Prevalence of DDM in psychiatric disorders
The successful treatment of DDM with dopaminergic drugs is meaningful because of the coexistence of DDM in various neuropsychiatric conditions. In Alzheimer’s disease (AD), disturbances in the dopaminergic system may explain the high comorbidity of apathy, which ranges from 47% in mild AD to 80% in moderate AD.33 In the dopamine-reduced states of cocaine and amphetamine withdrawal, 67% of patients report apathy and lack of motivation.8,34 Additionally, the prevalence of apathy is reported at 27% in Parkinson’s disease, 43% in mild cognitive impairment, 70% in mixed dementia, 94% in a major depressive episode, and 53% in schizophrenia.35 In schizophrenia with predominately negative symptoms, in vivo and postmortem studies have found reduced dopamine receptors.8 Meanwhile, the high rate of akinetic mutism in Creutzfeldt-Jakob disease allows for its use as a reliable diagnostic criteria in this disorder.36
However, the prevalence of DDM is best documented as it relates to stroke and traumatic brain injury (TBI). For instance, after experiencing a stroke, 20% to 25% of patients suffer from apathy.37 Many case reports describe abulia and akinetic mutism after cerebral infarction or hemorrhage, although the incidence of these disorders is unknown.2,38-40 Apathy following TBI is common, especially in younger patients who have sustained a severe injury.41 One study evaluated the prevalence of apathy after TBI among 83 consecutive patients in a neuropsychiatric clinic. Of the 83 patients, 10.84% had apathy without depression, and an equal number were depressed without apathy; another 60% of patients exhibited both apathy and depression. Younger patients (mean age, 29 years) were more likely to be apathetic than older patients, who were more likely to be depressed or depressed and apathetic (mean age, 42 and 38 years, respectively).41 Interestingly, DDM often are associated with cerebral lesions in distinct and distant anatomical locations that are not clearly connected to the neural circuits of motivational pathways. This phenomenon may be explained by the concept of diaschisis, which states that injury to one part of an interconnected neural network can affect other, separate parts of that network.2 If this concept is accurate, it may broaden the impact of DDM, especially as it relates to stroke and TBI.
The differential diagnosis of DDM includes depression and hypokinetic delirium (Table 21,3,42-50). A potential overlapping but confounding condition is stuporous catatonia, with symptoms that include psychomotor slowing such as immobility, staring, and stupor.47 It is important to differentiate these disorders because the treatment for each differs. As previously discussed, there is a clear role for dopamine receptor agonists in the treatment of DDM.
Although major depressive disorder can be treated with medications that increase dopaminergic transmission, selective serotonin reuptake inhibitors (SSRIs) are more commonly used as first-line agents.44 However, an SSRI would theoretically be contraindicated in DDM, because increased serotonin transmission decreases dopamine release from the midbrain, and therefore an SSRI may not only result in a lack of improvement but may worsen DDM.48 Finally, although delirium is treated with atypical or conventional antipsychotics vis-a-vis dopamine type 2 receptor antagonism,45 stuporous catatonia is preferentially treated with gamma-aminobutyric acid-A receptor agonists such as lorazepam.50
What to do when your patient’s presentation suggests DDM
Assessment of DDM should be structured, with input from the patient and the caregiver, and should incorporate the physician’s perspective. A history should be obtained applying recent criteria of apathy. The 3 core domains of apathy—behavior, cognition, and emotion—need to be evaluated. The revised criteria are based on the premise that change in motivation can be measured by examining a patient’s responsiveness to internal or external stimuli. Therefore, each of the 3 domains includes 2 symptoms: (1) self-initiated or “internal” behaviors, cognitions, and emotions (initiation symptom), and (2) the patient’s responsiveness to “external” stimuli (responsiveness symptom).51
One of the main diagnostic dilemmas is how to separate DDM from depression. The differentiation is difficult because of substantial overlap in the manifestation of key symptoms, such as a lack of interest, anergia, psychomotor slowing, and fatigue. Caregivers often mistakenly describe DDM as a depressive state, even though a lack of sadness, desperation, crying, and a depressive mood distinguish DDM from depression. Usually, DDM patients lack negative thoughts, emotional distress, sadness, vegetative symptoms, and somatic concerns, which are frequently observed in mood disorders.51
Several instruments have been developed for assessing neuropsychiatric symptoms. Some were specifically designed to measure apathy, whereas others were designed to provide a broader neuropsychiatric assessment. The NPI is the most widely used multidimensional instrument for assessing neuropsychiatric functioning in patients with neurocognitive disorders (NCDs). It is a valid, reliable instrument that consists of an interview of the patient’s caregiver. It is designed to assess the presence and severity of 10 symptoms, including apathy. The NPI includes both apathy and depression items, which can help clinicians distinguish the 2 conditions. Although beyond the scope of this article, more recent standardized instruments that can assess DDM include the Apathy Inventory, the Dementia Apathy Interview and Rating, and the Structured Clinical Interview for Apathy.52
As previously mentioned, researchers have proposed that DDM are simply a continuum of severity of reduced behavior, and akinetic mutism may be the extreme form. The dilemma is how to formally diagnose states of abulia and akinetic mutism, given the lack of diagnostic criteria and paucity of standardized instruments. Thus, distinguishing between abulia and akinetic mutism (and apathy) is more of a quantitative than qualitative exercise. One could hypothesize that higher scores on a standardized scale to measure apathy (ie, NPI) could imply abulia or akinetic mutism, although to the best of our knowledge, no formal “cut-off scores” exist.53
Treatment of apathy. The duration of pharmacotherapy to treat apathy is unknown and their usage is off-label. Further studies, including double-blind, randomized controlled trials (RCTs), are needed. Nonetheless, the 2 classes of medications that have the most evidence for treating apathy/DDM are psychostimulants and acetylcholinesterase inhibitors (AChEIs).
AChEIs are primarily used for treating cognitive symptoms in NCDs, but recent findings indicate that they have beneficial effects on noncognitive symptoms such as apathy. Of all medications used to treat apathy in NCDs, AChEIs have been used to treat the largest number of patients. Of 26 studies, 24 demonstrated improvement in apathy, with 21 demonstrating statistical significance. These studies ranged in duration from 8 weeks to 1 year, and most were open-label.54
Five studies (3 RCTs and 2 open-label studies) assessed the efficacy of methylphenidate for treating apathy due to AD. All the studies demonstrated at least some benefit in apathy scores after treatment with methylphenidate. These studies ranged from 5 to 12 weeks in duration. Notably, some patients reported adverse effects, including delusions and irritability.54
Although available evidence suggests AChEIs may be the most effective medications for treating apathy in NCDs, methylphenidate has been demonstrated to work faster.55 Thus, in cases where apathy can significantly affect activities of daily living or instrumental activities of daily living, a quicker response may dictate treatment with methylphenidate. It is imperative to note that safety studies and more large-scale double-blind RCTs are needed to further demonstrate the effectiveness and safety of methylphenidate.
Published in 2007, the American Psychiatric Association (APA) guidelines56 state that psychostimulants are a possible treatment option for patients with severe apathy. At the same time, clinicians are reminded that these agents—especially at higher doses—can produce various problematic adverse effects, including tachycardia, hypertension, restlessness, dyskinesia, agitation, sleep disturbances, psychosis, confusion, and decreased appetite. The APA guidelines recommend using low initial doses, with slow and careful titration. For example, methylphenidate should be started at 2.5 to 5 mg once in the morning, with daily doses not to exceed 30 to 40 mg. In our clinical experience, doses >20 mg/d have not been necessary.57
Treatment of akinetic mutism and abulia. In patients with akinetic mutism and possible abulia, for whom oral medication administration is either impossible or contraindicated (ie, due to the potential risk of aspiration pneumonia), atypical antipsychotics, such as IM olanazapine, have produced a therapeutic response in apathetic patients with NCD. However, extensive use of antipsychotics in NCD is not recommended because this class of medications has been associated with serious adverse effects, including an increased risk of death.55
Bottom Line
Apathy, abulia, and akinetic mutism have been categorized as disorders of diminished motivation (DDM). They commonly present after a stroke or traumatic brain injury, and should be differentiated from depression, hypokinetic delirium, and stuporous catatonia. DDM can be successfully treated with dopamine agonists.
Related Resources
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract. 2004;10(3):196-199.
- Dell’Osso B, Benatti B, Altamura AC, et al. Prevalence of selective serotonin reuptake inhibitor-related apathy in patients with obsessive compulsive disorder. J Clin Psychopharmacol. 2016;36(6):725-726.
- D’Souza G, Kakoullis A, Hegde N, et al. Recognition and management of abulia in the elderly. Prog Neurol Psychiatry. 2010;14(6):24-28.
Drug Brand Names
Bromocriptine • Parlodel
Bupropion • Wellbutrin XL, Zyban
Carbidopa • Lodosyn
Dexamethasone • DexPak, Ozurde
Donepezil • Aricept
Levodopa/benserazide • Prolopa
Levodopa/carbidopa • Pacopa Rytary Sinemet
Lorazepam • Ativan
Methylphenidate • Concerta, Methylin
Metronidazole • Flagyl, Metrogel
Modafinil • Provigil
Olanzapine • Zyprexa
Pramipexole • Mirapex
Rivastigmine • Exelon
Ropinirole • Requip
Rotigotine • Neurpro
Scopolamine • Transderm Scop
Ziprasidone • Geodon
1. Marin RS, Wilkosz PA. Disorders of diminished motivation. J Head Trauma Rehabil. 2005;20(4):377-388.
2. Ghoshal S, Gokhale S, Rebovich G, et al. The neurology of decreased activity: abulia. Rev Neurol Dis. 2011;8(3-4):e55-e67.
3. Spiegel DR, Chatterjee A. A case of abulia, status/post right middle cerebral artery territory infarct, treated successfully with olanzapine. Clin Neuropharmacol. 2014;37(6):186-189.
4. Marin RS. Differential diagnosis and classification of apathy. Am J Psychiatry. 1990;147(1):22-30.
5. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314.
6. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338-1344.
7. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370-1379.
8. Al-Adawi S, Dawe GS, Al-Hussaini AA. Aboulia: neurobehavioural dysfunction of dopaminergic system? Med Hypotheses. 2000;54(4):523-530.
9. Volkow ND, Fowler JS, Wang G, et al. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(suppl 1):S31-S43.
10. Chatterjee A, Fahn S. Methylphenidate treats apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2002;14(4):461-462.
11. Keenan S, Mavaddat N, Iddon J, et al. Effects of methylphenidate on cognition and apathy in normal pressure hydrocephalus: a case study and review. Br J Neurosurg. 2005;19(1):46-50.
12. Padala PR, Petty F, Bhatia SC. Methylphenidate may treat apathy independent of depression. Ann Pharmacother. 2005;39(11):1947-1949.
13. Padala PR, Burke WJ, Bhatia SC, et al. Treatment of apathy with methylphenidate. J Neuropsychiatry Clin Neurosci. 2007;19(1):81-83.
14. Li XM, Perry KW, Wong DT, et al. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl). 1998;136(2):153-161.
15. Spiegel DR, Casella DP, Callender DM, et al. Treatment of akinetic mutism with intramuscular olanzapine: a case series. J Neuropsychiatry Clin Neurosci. 2008;20(1):93-95.
16. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
17. Bakheit AM, Fletcher K, Brennan A. Successful treatment of severe abulia with co-beneldopa. NeuroRehabilitation. 2011;29(4):347-351.
18. Debette S, Kozlowski O, Steinling M, et al. Levodopa and bromocriptine in hypoxic brain injury. J Neurol. 2002;249(12):1678-1682.
19. Combarros O, Infante J, Berciano J. Akinetic mutism from frontal lobe damage responding to levodopa. J Neurol. 2000;247(7):568-569.
20. Echiverri HC, Tatum WO, Merens TA, et al. Akinetic mutism: pharmacologic probe of the dopaminergic mesencephalofrontal activating system. Pediatr Neurol. 1988;4(4):228-230.
21. Psarros T, Zouros A, Coimbra C. Bromocriptine-responsive akinetic mutism following endoscopy for ventricular neurocysticercosis. Case report and review of the literature. J Neurosurg. 2003;99(2):397-401.
22. Naik VD. Abulia following an episode of cardiac arrest [published online July 1, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-209357.
23. Kim MS, Rhee JJ, Lee SJ, et al. Akinetic mutism responsive to bromocriptine following subdural hematoma evacuation in a patient with hydrocephalus. Neurol Med Chir (Tokyo). 2007;47(9):419-423.
24. Rockwood K, Black S, Bedard MA; TOPS Study Investigators. Specific symptomatic changes following donepezil treatment of Alzheimer’s disease: a multi-centre, primary care, open-label study. Int J Geriatr Psychiatry. 2007;22(4):312-319.
25. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry. 2014;85(6):668-674.
26. Camargos EF, Quintas JL. Apathy syndrome treated successfully with modafinil [published online November 15, 2011]. BMJ Case Rep. doi: 10.1136/bcr.08.2011.4652.
27. Corcoran C, Wong ML, O’Keane V. Bupropion in the management of apathy. J Psychopharmacol. 2004;18(1):133-135.
28. Blundo C, Gerace C. Dopamine agonists can improve pure apathy associated with lesions of the prefrontal-basal ganglia functional system. Neurol Sci. 2015;36(7):1197-1201.
29. Mirapex [package insert]. Ridgefield, CT: Boehringer Ingelheim International GmbH; 2016.
30. Neupro [package insert]. Smyrna, GA: UBC, Inc.; 2012.
31. Requip [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
32. Thobois S, Lhommée E, Klinger H, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136(pt 5):1568-1577.
33. Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther. 2011;17(5):411-427.
34. Brower KJ, Maddahian E, Blow FC, et al. A comparison of self-reported symptoms and DSM-III-R criteria for cocaine withdrawal. Am J Drug Alcohol Abuse. 1988;14(3):347-356.
35. Mulin E, Leone E, Dujardin K, et al. Diagnostic criteria for apathy in clinical practice. Int J Geriatr Psychiatry. 2011;26(2):158-165.
36. Otto A, Zerr I, Lantsch M, et al. Akinetic mutism as a classification criterion for the diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1998;64(4):524-528.
37. Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350-354.
38. Hastak SM, Gorawara PS, Mishra NK. Abulia: no will, no way. J Assoc Physicians India. 2005;53:814-818.
39. Nagaratnam N, Nagaratnam K, Ng K, et al. Akinetic mutism following stroke. J Clin Neurosci. 2004;11(1):25-30.
40. Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34(6):693-698.
41. Schwarzbold M, Diaz A, Martins ET, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797-816.
42. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
43. Levy ML, Cummings JL, Fairbanks LA, et al. Apathy is not depression. J Neuropsychiatry Clin Neurosci. 1998;10(3):314-319.
44. Snow V, Lascher S, Mottur-Pilson C. Pharmacologic treatment of acute major depression and dysthymia. American College of Physicians-American Society of Internal Medicine. Ann Intern Med. 2000;132(9):738-742.
45. Schwartz AC, Fisher TJ, Greenspan HN, et al. Pharmacologic and nonpharmacologic approaches to the prevention and management of delirium. Int J Psychiatry Med. 2016;51(2):160-170.
46. Kang H, Zhao F, You L, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17(2):147-154.
47. Fricchione GL, Beach SR, Huffman J, et al. Life-threatening conditions in psychiatry: catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fava M, Wilens TE, eds. Massachusetts General Hospital comprehensive clinical psychiatry. London, United Kingdom: Elsevier; 2016:608-617.
48. Rogers RD. The roles of dopamine and serotonin in decision making: evidence from pharmacological experiments in humans. Neuropsychopharmacology. 2011;36(1):114-132.
49. Stransky M, Schmidt C, Ganslmeier P, et al. Hypoactive delirium after cardiac surgery as an independent risk factor for prolonged mechanical ventilation. J Cardiothorac Vasc Anesth. 2011;25(6):968-974.
50. Wilcox JA, Reid Duffy P. The syndrome of catatonia. Behav Sci (Basel). 2015;5(4):576-588.
51. Robert PH, Mulin E, Malléa P, et al. REVIEW: apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):263-271.
52. Cipriani G, Lucetti C, Danti S, et al. Apathy and dementia. Nosology, assessment and management. J Nerv Ment Dis. 2014;202(10):718-724.
53. Starkstein SE, Leentjens AF. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79(10):1088-1092.54. Berman K, Brodaty H, Withall A, et al. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry. 2012;20(2):104-122.
55. Theleritis C, Siarkos K, Katirtzoglou E, et al. Pharmacological and nonpharmacological treatment for apathy in Alzheimer disease: a systematic review across modalities. J Geriatr Psychiatry Neurol. 2017;30(1):26-49.
56. APA Work Group on Alzheimer’s Disease and other Dementias; Rabins PV, Blacker D, Rovner BW, et al. American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry. 2007;164(suppl 12):5-56.
57. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624-1632.
1. Marin RS, Wilkosz PA. Disorders of diminished motivation. J Head Trauma Rehabil. 2005;20(4):377-388.
2. Ghoshal S, Gokhale S, Rebovich G, et al. The neurology of decreased activity: abulia. Rev Neurol Dis. 2011;8(3-4):e55-e67.
3. Spiegel DR, Chatterjee A. A case of abulia, status/post right middle cerebral artery territory infarct, treated successfully with olanzapine. Clin Neuropharmacol. 2014;37(6):186-189.
4. Marin RS. Differential diagnosis and classification of apathy. Am J Psychiatry. 1990;147(1):22-30.
5. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314.
6. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338-1344.
7. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370-1379.
8. Al-Adawi S, Dawe GS, Al-Hussaini AA. Aboulia: neurobehavioural dysfunction of dopaminergic system? Med Hypotheses. 2000;54(4):523-530.
9. Volkow ND, Fowler JS, Wang G, et al. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(suppl 1):S31-S43.
10. Chatterjee A, Fahn S. Methylphenidate treats apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2002;14(4):461-462.
11. Keenan S, Mavaddat N, Iddon J, et al. Effects of methylphenidate on cognition and apathy in normal pressure hydrocephalus: a case study and review. Br J Neurosurg. 2005;19(1):46-50.
12. Padala PR, Petty F, Bhatia SC. Methylphenidate may treat apathy independent of depression. Ann Pharmacother. 2005;39(11):1947-1949.
13. Padala PR, Burke WJ, Bhatia SC, et al. Treatment of apathy with methylphenidate. J Neuropsychiatry Clin Neurosci. 2007;19(1):81-83.
14. Li XM, Perry KW, Wong DT, et al. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl). 1998;136(2):153-161.
15. Spiegel DR, Casella DP, Callender DM, et al. Treatment of akinetic mutism with intramuscular olanzapine: a case series. J Neuropsychiatry Clin Neurosci. 2008;20(1):93-95.
16. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
17. Bakheit AM, Fletcher K, Brennan A. Successful treatment of severe abulia with co-beneldopa. NeuroRehabilitation. 2011;29(4):347-351.
18. Debette S, Kozlowski O, Steinling M, et al. Levodopa and bromocriptine in hypoxic brain injury. J Neurol. 2002;249(12):1678-1682.
19. Combarros O, Infante J, Berciano J. Akinetic mutism from frontal lobe damage responding to levodopa. J Neurol. 2000;247(7):568-569.
20. Echiverri HC, Tatum WO, Merens TA, et al. Akinetic mutism: pharmacologic probe of the dopaminergic mesencephalofrontal activating system. Pediatr Neurol. 1988;4(4):228-230.
21. Psarros T, Zouros A, Coimbra C. Bromocriptine-responsive akinetic mutism following endoscopy for ventricular neurocysticercosis. Case report and review of the literature. J Neurosurg. 2003;99(2):397-401.
22. Naik VD. Abulia following an episode of cardiac arrest [published online July 1, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-209357.
23. Kim MS, Rhee JJ, Lee SJ, et al. Akinetic mutism responsive to bromocriptine following subdural hematoma evacuation in a patient with hydrocephalus. Neurol Med Chir (Tokyo). 2007;47(9):419-423.
24. Rockwood K, Black S, Bedard MA; TOPS Study Investigators. Specific symptomatic changes following donepezil treatment of Alzheimer’s disease: a multi-centre, primary care, open-label study. Int J Geriatr Psychiatry. 2007;22(4):312-319.
25. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry. 2014;85(6):668-674.
26. Camargos EF, Quintas JL. Apathy syndrome treated successfully with modafinil [published online November 15, 2011]. BMJ Case Rep. doi: 10.1136/bcr.08.2011.4652.
27. Corcoran C, Wong ML, O’Keane V. Bupropion in the management of apathy. J Psychopharmacol. 2004;18(1):133-135.
28. Blundo C, Gerace C. Dopamine agonists can improve pure apathy associated with lesions of the prefrontal-basal ganglia functional system. Neurol Sci. 2015;36(7):1197-1201.
29. Mirapex [package insert]. Ridgefield, CT: Boehringer Ingelheim International GmbH; 2016.
30. Neupro [package insert]. Smyrna, GA: UBC, Inc.; 2012.
31. Requip [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
32. Thobois S, Lhommée E, Klinger H, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136(pt 5):1568-1577.
33. Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther. 2011;17(5):411-427.
34. Brower KJ, Maddahian E, Blow FC, et al. A comparison of self-reported symptoms and DSM-III-R criteria for cocaine withdrawal. Am J Drug Alcohol Abuse. 1988;14(3):347-356.
35. Mulin E, Leone E, Dujardin K, et al. Diagnostic criteria for apathy in clinical practice. Int J Geriatr Psychiatry. 2011;26(2):158-165.
36. Otto A, Zerr I, Lantsch M, et al. Akinetic mutism as a classification criterion for the diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1998;64(4):524-528.
37. Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350-354.
38. Hastak SM, Gorawara PS, Mishra NK. Abulia: no will, no way. J Assoc Physicians India. 2005;53:814-818.
39. Nagaratnam N, Nagaratnam K, Ng K, et al. Akinetic mutism following stroke. J Clin Neurosci. 2004;11(1):25-30.
40. Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34(6):693-698.
41. Schwarzbold M, Diaz A, Martins ET, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797-816.
42. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
43. Levy ML, Cummings JL, Fairbanks LA, et al. Apathy is not depression. J Neuropsychiatry Clin Neurosci. 1998;10(3):314-319.
44. Snow V, Lascher S, Mottur-Pilson C. Pharmacologic treatment of acute major depression and dysthymia. American College of Physicians-American Society of Internal Medicine. Ann Intern Med. 2000;132(9):738-742.
45. Schwartz AC, Fisher TJ, Greenspan HN, et al. Pharmacologic and nonpharmacologic approaches to the prevention and management of delirium. Int J Psychiatry Med. 2016;51(2):160-170.
46. Kang H, Zhao F, You L, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17(2):147-154.
47. Fricchione GL, Beach SR, Huffman J, et al. Life-threatening conditions in psychiatry: catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fava M, Wilens TE, eds. Massachusetts General Hospital comprehensive clinical psychiatry. London, United Kingdom: Elsevier; 2016:608-617.
48. Rogers RD. The roles of dopamine and serotonin in decision making: evidence from pharmacological experiments in humans. Neuropsychopharmacology. 2011;36(1):114-132.
49. Stransky M, Schmidt C, Ganslmeier P, et al. Hypoactive delirium after cardiac surgery as an independent risk factor for prolonged mechanical ventilation. J Cardiothorac Vasc Anesth. 2011;25(6):968-974.
50. Wilcox JA, Reid Duffy P. The syndrome of catatonia. Behav Sci (Basel). 2015;5(4):576-588.
51. Robert PH, Mulin E, Malléa P, et al. REVIEW: apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):263-271.
52. Cipriani G, Lucetti C, Danti S, et al. Apathy and dementia. Nosology, assessment and management. J Nerv Ment Dis. 2014;202(10):718-724.
53. Starkstein SE, Leentjens AF. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79(10):1088-1092.54. Berman K, Brodaty H, Withall A, et al. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry. 2012;20(2):104-122.
55. Theleritis C, Siarkos K, Katirtzoglou E, et al. Pharmacological and nonpharmacological treatment for apathy in Alzheimer disease: a systematic review across modalities. J Geriatr Psychiatry Neurol. 2017;30(1):26-49.
56. APA Work Group on Alzheimer’s Disease and other Dementias; Rabins PV, Blacker D, Rovner BW, et al. American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry. 2007;164(suppl 12):5-56.
57. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624-1632.
