User login
Complementary treatments for anxiety: Beyond pharmacotherapy and psychotherapy
Anxiety disorders are the most common psychiatric illnesses in the United States, with a prevalence of nearly 29%.1 These disorders typically are treated with pharmacotherapy, psychotherapy, or a combination of both. Pharmacotherapy for anxiety has evolved considerably during the last 30 years, but medications are not efficacious for or tolerated by all patients. For example, selective serotonin reuptake inhibitors, which are frequently used for treating anxiety, can cause sexual dysfunction,2 weight gain,2 drug interactions,2 coagulopathies,3 and gastrointestinal disturbances.4 Psychotherapeutic techniques, such as cognitive behavioral therapy (CBT) and interpersonal therapy (IPT), are efficacious for mild to moderate anxiety.5-7
In addition to standard pharmacotherapy and psychotherapy, some evidence suggests that complementary therapies, such as yoga, massage, and relaxation techniques, may be beneficial as adjunctive treatments for anxiety. In placebo-controlled trials, several of these complementary therapies have been shown to decrease serum levels of the inflammatory biomarker cortisol. Anxiety is associated with inflammation,8 so therapies that reduce inflammation may help reduce symptoms of anxiety. Here, we describe the results of select positive randomized controlled trials (RCTs) of several complementary interventions for anxiety that might be useful as adjunctive treatments to psychotherapy or pharmacotherapy.
A look at RCTs that measured both anxiety and cortisol
We searched PubMed, Google Scholar, and Scopus to identify RCTs of complementary nonpharmacologic and nonpsychotherapeutic therapies for anxiety published from January 2010 to May 2017. We included only studies that:
- blindly assessed anxiety levels through a validated instrument (the State-Trait Anxiety Inventory [STAI])9
- measured cortisol concentrations before and after treatment.
Evaluating both STAI scores and cortisol levels is useful because doing so gives insight into both the clinical and biological efficacy of the therapies. Studies were excluded if they employed a pharmacologic agent in addition to the approach being evaluated.
We identified 26 studies, of which 14 met the inclusion/exclusion criteria. These studies found beneficial effects for yoga, massage therapy, aromatherapy massage, pet therapy, Qigong, auricular acupressure, reiki touch therapy, acupuncture, music therapy, and relaxation techniques.
Yoga
Yoga has become increasingly popular in the Western world during the last 2 decades.10 There are a variety of yoga practices; common forms include hatha yoga, power yoga, kripalu yoga, and forrest yoga.11
A study of 92 depressed pregnant women monito
Hatha yoga consists of a combination of postural exercises, breathing techniques, relaxation, and meditation. In a 12-week study of 88 postmenopausal women, those who practiced hatha yoga for 75 minutes a day had significantly lower STAI scores compared with women who exercised for 75 minutes a day and those who performed no physical activity.13
Continue to: Massage therapy
Massage therapy
Receiving as little as 15 minutes of back massage has proven to be beneficial for individuals with anxiety. In an RCT conducted in Turkey, 44 caregivers of patients with cancer were assigned to receive a back massage or to rest quietly in a room for 15 minutes once each day for 1 week.14 By the end of the week, compared with those who quietly rested, those who received the back massage had a statistically significant reduction in serum cortisol levels and STAI scores.14
Aromatherapy massage
Aromatherapy is the use of essential oils from plants through distillation.15 The scent of the oils is purported to provide medical benefits. More than 60 essential oils are used therapeutically, including rose, lavender, lemon, and orange.16 These essential oils are frequently used in combination with a massage.
In South Korea, researchers investigated the effects of aromatherapy massage on 25 women who had children diagnosed with attention-deficit/hyperactivity disorder.17 Women assigned to the treatment group received a 40-minute aromatherapy massage using mixed essential oils that contained lavender and geranium twice a week for 4 weeks. Women in the control group received no treatment. Compared with those in the control group, women who received the aromatherapy massages had a statistically significant decrease in STAI scores and salivary cortisol levels. Plasma cortisol was not significantly different between groups.17
Pet therapy
The psychological benefits of animal-assisted therapy were not evident until World War II, when dogs were used to cheer up injured soldiers.18 Today, pet therapy has been used on many inpatient units.19
In a U.S. study, 48 healthy undergraduate students were assigned to a room with a dog, a room with a friend, or a room by themselves.20 All participants were given the Trier Social Stress Test (TSST), a protocol that measures stress by having participants give a speech and perform mental arithmetic in front of an audience.The TSST is known to induce increases in cortisol levels. Although no differences in STAI scores were found among groups, students in the room with the dog had a lower spike in salivary cortisol after the TSST compared with participants who were in a room with a friend or in a room alone.20
Continue to: Qigong
Qigong
In Chinese medicine, Qi is known as a vital life force that flows through the body. The disruption of Qi is hypothesized to contribute to disease.21
Qigong is a medical therapy that focuses on uniting the body, breath, and mind to improve health.21 It consists of rhythmic, choreographed movements used to position the body into postures believed to help direct Qi to specific areas in the body. Qigong also uses sound exercises, in which an individual creates certain syllables while breathing. Six syllables are used, each of which is believed to affect a certain organ.21
Korean researchers randomly assigned 32 healthy men to a Qigong training group or a sham Qigong control group.22 Individuals in the training group performed 25 minutes of sound exercises, 20 minutes of meditation, and 15 minutes of movements. The control group learned the same movements as the experimental group, but without the conscious effort of moving Qi. After 3 sessions, those in the Qigong training group had significantly decreased STAI scores and serum cortisol levels compared with those in the sham group.22
In a different Korean study, researchers randomly assigned 50 participants with elevated distress levels to a Qigong training group or a waitlist control group in which participants called a trainer to describe stressful events.23 After 4 weeks, participants in the Qigong group had significant decreases in STAI scores compared with the control group. However, there were no changes in salivary cortisol levels.23
Auricular acupressure
Auricular acupressure involves applying pressure on certain portions of the auricle (outer ear) to alleviate pain and disease.24 Similar to Qigong, auricular acupressure focuses on reestablishing Qi in the body. Researchers randomly assigned 80 post-caesarean section women in Taiwan to 5 days of auricular acupressure or usual care.25 The women who received auricular acupressure had significantly lower STAI scores and serum cortisol levels compared with women who received routine care.25
Continue to: Reiki touch therapy
Reiki touch therapy
Reiki touch therapy originated in Japan. In this therapy, healers apply a light touch or hover their hands above an individual’s body to help direct energy.26
The effects of reiki touch therapy were recently evaluated in a U.S. study.27 Researchers randomly assigned 37 patients with human immunodeficiency virus to an experimental group that received 30 minutes of reiki touch therapy plus music therapy 6 times a week for 10 weeks, or to a music therapy–only control group. Patients who received reiki touch therapy had a significant decrease in STAI scores. Patients in this group also had a statistically significant drop in salivary cortisol levels after the first week.27
Acupuncture
Acupuncture is the application of needles to specific areas on the body. Acupuncture has been proposed to activate pain receptors, thereby producing an analgesic response.28
Researchers in Brazil randomly assigned 57 lactating women with preterm infants to an experimental group that received acupuncture or to a control group that received sham acupuncture.29 Treatment was administered at 5 points on the ear unilaterally for 5 minutes once a week for 16 months. Custom-made needles that did not actually puncture the skin were used in the sham group; a toothpick was used to create the sensation of needle perforations. STAI scores were reduced in both groups, although there was no statistically significant difference in scores between the acupuncture and sham groups.29
Music therapy
Music has been long believed to have beneficial psychological effects. In Turkey, researchers evaluated the effects of music therapy in 100 oncology patients who received port catheters.30 Patients were randomly assigned to an experimental group that received music therapy throughout the procedure or to a control group that received normal care. Patients who listened to music during port catheter placement had significantly reduced STAI scores and serum cortisol levels compared with those in the control group.30
Continue to: Relaxation techniques
Relaxation techniques
A wide range of relaxation techniques are used for therapeutic purposes. In Switzerland, researchers evaluated the anxiolytic effects of 10 minutes of progressive muscle relaxation and guided imagery in 39 pregnant women.31 Women randomly assigned to progressive muscle relaxation were instructed to systematically tense and then release muscle groups throughout their body in sequential order. Women assigned to the guided imagery intervention were told to imagine a safe place and to think of someone who could confer security and reassurance. The remainder of the women were assigned to a control group, where they sat quietly without any formal instructions. Researchers found that each group had a decrease in STAI scores and salivary cortisol levels immediately after the intervention.31
The relaxation response was first described in 1975 by Herbert Benson, MD, as a deep meditative state characterized by a decrease in tension, heart rate, and breathing rate. Several techniques can induce this state, including hypnosis, progressive muscle relaxation, yoga, and transcendental meditation.32 In a study of 15 healthy older adults (age 65 to 80), researchers randomly assigned participants to a relaxation response training group or to a control group.33 The relaxation response training included meditation, imagery, and relaxation techniques. After 5 weeks, participants who received the relaxation response training had marginally significant decreases in STAI scores compared with those in the control group.33
Consider these therapies as adjuncts
Our review of select positive RCTs (Table12-14,17,20,22,23,25,27,29-31,33) suggests that some nonpharmacologic/nonpsychotherapeutic adjunctive interventions may have beneficial effects for patients who have anxiety. Several of the controlled studies we reviewed demonstrated that these interventions are superior to placebo. The reductions in both anxiety severity as measured by the STAI and cortisol levels suggests that some of these complementary therapies deserve a second look as useful adjuncts to established anxiety treatments.

Bottom Line
A review of select randomized controlled trials suggests that some complementary therapies may be helpful as adjunctive therapy in patients with anxiety. These include yoga, massage therapy, aromatherapy massage, pet therapy, Qigong, auricular acupressure, reiki touch therapy, acupuncture, music therapy, and relaxation techniques.
Related Resources
- Bandelow B, Baldwin D, Abelli M, et al. Biological markers for anxiety disorders, OCD and PTSD: a consensus statement. Part II: neurochemistry, neurophysiology and neurocognition. World J Biol Psychiatry. 2017;18(3):162-214.
- National Institute of Mental Health. Anxiety disorders. https://www.nimh.nih.gov/health/topics/anxiety-disorders/index.shtml.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Masand PS, Gupta S. Long-term side effects of newer-generation antidepressants: SSRIs, venlafaxine, nefazodone, bupropion, and mirtazapine. Ann Clin Psychiatry. 2002;14(3):175-182.
3. Siddiqui R, Gawande S, Shende T, et al. SSRI-induced coagulopathy: is it reality? Therapeutic Advances in Psychopharmacology. 2011;1(6):169-174.
4. Brambilla P, Cipriani A, Hotopf M, et al. Side-effect profile of fluoxetine in comparison with other SSRIs, tricyclic and newer antidepressants: a meta-analysis of clinical trial data. Pharmacopsychiatry. 2005;38(2):69-77.
5. Slomski A. Blended CBT controls anxiety in cancer survivors. JAMA. 2017;318(4):323.
6. Forsell E, Bendix M, Holländare F, et al. Internet delivered cognitive behavior therapy for antenatal depression: a randomised controlled trial. J Affect Disord. 2017;221:56-64.
7. Lilliengren P, Johansson R, Town JM, et al. Intensive Short-Term Dynamic Psychotherapy for generalized anxiety disorder: A pilot effectiveness and process-outcome study. Clin Psychol Psychother. 2017;24(6):1313-1321.
8. Furtado M, Katzman MA. Neuroinflammatory pathways in anxiety, posttraumatic stress, and obsessive compulsive disorders. Psychiatry Res. 2015;229(1-2):37-48.
9. Spielberger CD, Gorsuch RL, Lushene R, et al. Manual for the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press; 1983.
10. Saper RB, Eisenberg DM, Davis RB, et al. Prevalence and patterns of adult yoga use in the United States: results of a national survey. Altern Ther Health Med. 2004;10(2):44-49.
11. Farmer J. Americanasana. Reviews in American history. 2012;40(1):145-158.
12. Field T, Diego M, Delgado J, et al. Yoga and social support reduce prenatal depression, anxiety and cortisol. J Bodyw Mov Ther: 2013;17(4):397-403.
13. Jorge MP, Santaella DF, Pontes IM, et al. Hatha Yoga practice decreases menopause symptoms and improves quality of life: a randomized controlled trial. Complement Ther Med. 2016;26:128-135.
14. Pinar R, Afsar F. Back massage to decrease state anxiety, cortisol level, blood pressure, heart rate and increase sleep quality in family caregivers of patients with cancer: a randomised controlled trial. Asian Pac J Cancer Prev. 2015;16(18):8127-8133.
15. Kuriyama H, Watanabe S, Nakaya, et al. Immunological and psychological benefits of aromatherapy massage. Evid Based Complement Alternat Med. 2005;2(2):179-184.
16. Setzer WN. Essential oils and anxiolytic aromatherapy. Nat Prod Commun. 2009;4(9):1305-1316.
17. Wu JJ, Cui Y, Yang YS, et al. Modulatory effects of aromatherapy massage intervention on electroencephalogram, psychological assessments, salivary cortisol and plasma brain-derived neurotrophic factor. Complement Ther Med. 2014;22(3):456-462.
18. Fine A. Forward. In: Fine A, ed. Handbook on animal-assisted therapy-theoretical foundations and guidelines for practice. 3rd ed. Academic Press; 2010:xvii-xviii.
19. Snipelisky D, Burton MC. Canine-assisted therapy in the inpatient setting. South Med J. 2014;107(4):265-273.
20. Polheber JP, Matchock RL. The presence of a dog attenuates cortisol and heart rate in the Trier Social Stress Test compared to human friends. J Behav Med. 2014;37(5):860-867.
21. Liu T, Qiang X, eds. Chinese medical Qigong. Philadelphia, PA: Singing Dragon; 2013:1-100,192,238,511.
22. Lee MS, Kang CW, Lim HJ, et al. Effects of Qi-training on anxiety and plasma concentrations of cortisol, ACTH, and aldosterone: a randomized placebo-controlled pilot study. Stress Health. 2004;20(5):243-248.
23. Hwang EY, Chung SY, Cho JH, et al. Effects of a brief Qigong-based stress reduction program (BQSRP) in a distressed Korean population: a randomized trial. BMC Complement Altern Med. 2013;13:113.
24. Oleson, T. Overview and history of auriculotherapy. In: Auriculotherapy manual: Chinese and Western systems of ear acupuncture. 4th ed. London: Churchill Livingstone; 2014:1.
25. Kuo SY, Tsai SH, Chen SL, et al. Auricular acupressure relieves anxiety and fatigue, and reduces cortisol levels in post-caesarean section women: a single-blind, randomised controlled study. Int J Nurs Stud. 2016;53:17-26.
26. Horan P. Introduction. In: Horan P. Empowerment through reiki: the path to personal and global transformation. 8th ed. Twin Lakes, WI: Lotus Press; 1998:13-15.
27. Bremner MN, Blake BJ, Wagner VD, et al. Effects of reiki with music compared to music only among people living with HIV. J Assoc Nurses AIDS Care. 2016;27(5):635-647.
28. Helmes JM. The basic, clinical, and speculative science of acupuncture. In: Acupuncture energetics: a clinical approach for physicians. Volume 1. Berkeley, CA: Medical Acupuncture Publishers; 1995:19-32.
29. Haddad-Rodrigues M, Spanó Nakano A, Stefanello J, et al. Acupuncture for anxiety in lactating mothers with preterm infants: a randomized controlled trial. Evid Based Complement Alternat Med. 2013;2013:169184. doi: 10.1155/2013/169184.
30. Zengin S, Kabul S, Al B, et al. Effects of music therapy on pain and anxiety in patients undergoing port catheter placement procedure. Complement Ther Med. 2013;21(6):689-696.
31. Urech C, Fink NS, Hoesli I, et al. Effects of relaxation on psychobiological wellbeing during pregnancy: a randomized controlled trial. Psychoneuroendocrinology. 2010;35(9):1348-1355.
32. Goleman D. The relaxation response. In: Mind body medicine: how to use your mind for better health. Yonkers, NY: Consumer Reports; 1993:125-149.
33. Galvin JA, Benson H, Deckro GR, et al. The relaxation response: reducing stress and improving cognition in healthy aging adults. Complement Ther Clin Pract. 2006;12(3):186-191.
Anxiety disorders are the most common psychiatric illnesses in the United States, with a prevalence of nearly 29%.1 These disorders typically are treated with pharmacotherapy, psychotherapy, or a combination of both. Pharmacotherapy for anxiety has evolved considerably during the last 30 years, but medications are not efficacious for or tolerated by all patients. For example, selective serotonin reuptake inhibitors, which are frequently used for treating anxiety, can cause sexual dysfunction,2 weight gain,2 drug interactions,2 coagulopathies,3 and gastrointestinal disturbances.4 Psychotherapeutic techniques, such as cognitive behavioral therapy (CBT) and interpersonal therapy (IPT), are efficacious for mild to moderate anxiety.5-7
In addition to standard pharmacotherapy and psychotherapy, some evidence suggests that complementary therapies, such as yoga, massage, and relaxation techniques, may be beneficial as adjunctive treatments for anxiety. In placebo-controlled trials, several of these complementary therapies have been shown to decrease serum levels of the inflammatory biomarker cortisol. Anxiety is associated with inflammation,8 so therapies that reduce inflammation may help reduce symptoms of anxiety. Here, we describe the results of select positive randomized controlled trials (RCTs) of several complementary interventions for anxiety that might be useful as adjunctive treatments to psychotherapy or pharmacotherapy.
A look at RCTs that measured both anxiety and cortisol
We searched PubMed, Google Scholar, and Scopus to identify RCTs of complementary nonpharmacologic and nonpsychotherapeutic therapies for anxiety published from January 2010 to May 2017. We included only studies that:
- blindly assessed anxiety levels through a validated instrument (the State-Trait Anxiety Inventory [STAI])9
- measured cortisol concentrations before and after treatment.
Evaluating both STAI scores and cortisol levels is useful because doing so gives insight into both the clinical and biological efficacy of the therapies. Studies were excluded if they employed a pharmacologic agent in addition to the approach being evaluated.
We identified 26 studies, of which 14 met the inclusion/exclusion criteria. These studies found beneficial effects for yoga, massage therapy, aromatherapy massage, pet therapy, Qigong, auricular acupressure, reiki touch therapy, acupuncture, music therapy, and relaxation techniques.
Yoga
Yoga has become increasingly popular in the Western world during the last 2 decades.10 There are a variety of yoga practices; common forms include hatha yoga, power yoga, kripalu yoga, and forrest yoga.11
A study of 92 depressed pregnant women monito
Hatha yoga consists of a combination of postural exercises, breathing techniques, relaxation, and meditation. In a 12-week study of 88 postmenopausal women, those who practiced hatha yoga for 75 minutes a day had significantly lower STAI scores compared with women who exercised for 75 minutes a day and those who performed no physical activity.13
Continue to: Massage therapy
Massage therapy
Receiving as little as 15 minutes of back massage has proven to be beneficial for individuals with anxiety. In an RCT conducted in Turkey, 44 caregivers of patients with cancer were assigned to receive a back massage or to rest quietly in a room for 15 minutes once each day for 1 week.14 By the end of the week, compared with those who quietly rested, those who received the back massage had a statistically significant reduction in serum cortisol levels and STAI scores.14
Aromatherapy massage
Aromatherapy is the use of essential oils from plants through distillation.15 The scent of the oils is purported to provide medical benefits. More than 60 essential oils are used therapeutically, including rose, lavender, lemon, and orange.16 These essential oils are frequently used in combination with a massage.
In South Korea, researchers investigated the effects of aromatherapy massage on 25 women who had children diagnosed with attention-deficit/hyperactivity disorder.17 Women assigned to the treatment group received a 40-minute aromatherapy massage using mixed essential oils that contained lavender and geranium twice a week for 4 weeks. Women in the control group received no treatment. Compared with those in the control group, women who received the aromatherapy massages had a statistically significant decrease in STAI scores and salivary cortisol levels. Plasma cortisol was not significantly different between groups.17
Pet therapy
The psychological benefits of animal-assisted therapy were not evident until World War II, when dogs were used to cheer up injured soldiers.18 Today, pet therapy has been used on many inpatient units.19
In a U.S. study, 48 healthy undergraduate students were assigned to a room with a dog, a room with a friend, or a room by themselves.20 All participants were given the Trier Social Stress Test (TSST), a protocol that measures stress by having participants give a speech and perform mental arithmetic in front of an audience.The TSST is known to induce increases in cortisol levels. Although no differences in STAI scores were found among groups, students in the room with the dog had a lower spike in salivary cortisol after the TSST compared with participants who were in a room with a friend or in a room alone.20
Continue to: Qigong
Qigong
In Chinese medicine, Qi is known as a vital life force that flows through the body. The disruption of Qi is hypothesized to contribute to disease.21
Qigong is a medical therapy that focuses on uniting the body, breath, and mind to improve health.21 It consists of rhythmic, choreographed movements used to position the body into postures believed to help direct Qi to specific areas in the body. Qigong also uses sound exercises, in which an individual creates certain syllables while breathing. Six syllables are used, each of which is believed to affect a certain organ.21
Korean researchers randomly assigned 32 healthy men to a Qigong training group or a sham Qigong control group.22 Individuals in the training group performed 25 minutes of sound exercises, 20 minutes of meditation, and 15 minutes of movements. The control group learned the same movements as the experimental group, but without the conscious effort of moving Qi. After 3 sessions, those in the Qigong training group had significantly decreased STAI scores and serum cortisol levels compared with those in the sham group.22
In a different Korean study, researchers randomly assigned 50 participants with elevated distress levels to a Qigong training group or a waitlist control group in which participants called a trainer to describe stressful events.23 After 4 weeks, participants in the Qigong group had significant decreases in STAI scores compared with the control group. However, there were no changes in salivary cortisol levels.23
Auricular acupressure
Auricular acupressure involves applying pressure on certain portions of the auricle (outer ear) to alleviate pain and disease.24 Similar to Qigong, auricular acupressure focuses on reestablishing Qi in the body. Researchers randomly assigned 80 post-caesarean section women in Taiwan to 5 days of auricular acupressure or usual care.25 The women who received auricular acupressure had significantly lower STAI scores and serum cortisol levels compared with women who received routine care.25
Continue to: Reiki touch therapy
Reiki touch therapy
Reiki touch therapy originated in Japan. In this therapy, healers apply a light touch or hover their hands above an individual’s body to help direct energy.26
The effects of reiki touch therapy were recently evaluated in a U.S. study.27 Researchers randomly assigned 37 patients with human immunodeficiency virus to an experimental group that received 30 minutes of reiki touch therapy plus music therapy 6 times a week for 10 weeks, or to a music therapy–only control group. Patients who received reiki touch therapy had a significant decrease in STAI scores. Patients in this group also had a statistically significant drop in salivary cortisol levels after the first week.27
Acupuncture
Acupuncture is the application of needles to specific areas on the body. Acupuncture has been proposed to activate pain receptors, thereby producing an analgesic response.28
Researchers in Brazil randomly assigned 57 lactating women with preterm infants to an experimental group that received acupuncture or to a control group that received sham acupuncture.29 Treatment was administered at 5 points on the ear unilaterally for 5 minutes once a week for 16 months. Custom-made needles that did not actually puncture the skin were used in the sham group; a toothpick was used to create the sensation of needle perforations. STAI scores were reduced in both groups, although there was no statistically significant difference in scores between the acupuncture and sham groups.29
Music therapy
Music has been long believed to have beneficial psychological effects. In Turkey, researchers evaluated the effects of music therapy in 100 oncology patients who received port catheters.30 Patients were randomly assigned to an experimental group that received music therapy throughout the procedure or to a control group that received normal care. Patients who listened to music during port catheter placement had significantly reduced STAI scores and serum cortisol levels compared with those in the control group.30
Continue to: Relaxation techniques
Relaxation techniques
A wide range of relaxation techniques are used for therapeutic purposes. In Switzerland, researchers evaluated the anxiolytic effects of 10 minutes of progressive muscle relaxation and guided imagery in 39 pregnant women.31 Women randomly assigned to progressive muscle relaxation were instructed to systematically tense and then release muscle groups throughout their body in sequential order. Women assigned to the guided imagery intervention were told to imagine a safe place and to think of someone who could confer security and reassurance. The remainder of the women were assigned to a control group, where they sat quietly without any formal instructions. Researchers found that each group had a decrease in STAI scores and salivary cortisol levels immediately after the intervention.31
The relaxation response was first described in 1975 by Herbert Benson, MD, as a deep meditative state characterized by a decrease in tension, heart rate, and breathing rate. Several techniques can induce this state, including hypnosis, progressive muscle relaxation, yoga, and transcendental meditation.32 In a study of 15 healthy older adults (age 65 to 80), researchers randomly assigned participants to a relaxation response training group or to a control group.33 The relaxation response training included meditation, imagery, and relaxation techniques. After 5 weeks, participants who received the relaxation response training had marginally significant decreases in STAI scores compared with those in the control group.33
Consider these therapies as adjuncts
Our review of select positive RCTs (Table12-14,17,20,22,23,25,27,29-31,33) suggests that some nonpharmacologic/nonpsychotherapeutic adjunctive interventions may have beneficial effects for patients who have anxiety. Several of the controlled studies we reviewed demonstrated that these interventions are superior to placebo. The reductions in both anxiety severity as measured by the STAI and cortisol levels suggests that some of these complementary therapies deserve a second look as useful adjuncts to established anxiety treatments.
Bottom Line
A review of select randomized controlled trials suggests that some complementary therapies may be helpful as adjunctive therapy in patients with anxiety. These include yoga, massage therapy, aromatherapy massage, pet therapy, Qigong, auricular acupressure, reiki touch therapy, acupuncture, music therapy, and relaxation techniques.
Related Resources
- Bandelow B, Baldwin D, Abelli M, et al. Biological markers for anxiety disorders, OCD and PTSD: a consensus statement. Part II: neurochemistry, neurophysiology and neurocognition. World J Biol Psychiatry. 2017;18(3):162-214.
- National Institute of Mental Health. Anxiety disorders. https://www.nimh.nih.gov/health/topics/anxiety-disorders/index.shtml.
Anxiety disorders are the most common psychiatric illnesses in the United States, with a prevalence of nearly 29%.1 These disorders typically are treated with pharmacotherapy, psychotherapy, or a combination of both. Pharmacotherapy for anxiety has evolved considerably during the last 30 years, but medications are not efficacious for or tolerated by all patients. For example, selective serotonin reuptake inhibitors, which are frequently used for treating anxiety, can cause sexual dysfunction,2 weight gain,2 drug interactions,2 coagulopathies,3 and gastrointestinal disturbances.4 Psychotherapeutic techniques, such as cognitive behavioral therapy (CBT) and interpersonal therapy (IPT), are efficacious for mild to moderate anxiety.5-7
In addition to standard pharmacotherapy and psychotherapy, some evidence suggests that complementary therapies, such as yoga, massage, and relaxation techniques, may be beneficial as adjunctive treatments for anxiety. In placebo-controlled trials, several of these complementary therapies have been shown to decrease serum levels of the inflammatory biomarker cortisol. Anxiety is associated with inflammation,8 so therapies that reduce inflammation may help reduce symptoms of anxiety. Here, we describe the results of select positive randomized controlled trials (RCTs) of several complementary interventions for anxiety that might be useful as adjunctive treatments to psychotherapy or pharmacotherapy.
A look at RCTs that measured both anxiety and cortisol
We searched PubMed, Google Scholar, and Scopus to identify RCTs of complementary nonpharmacologic and nonpsychotherapeutic therapies for anxiety published from January 2010 to May 2017. We included only studies that:
- blindly assessed anxiety levels through a validated instrument (the State-Trait Anxiety Inventory [STAI])9
- measured cortisol concentrations before and after treatment.
Evaluating both STAI scores and cortisol levels is useful because doing so gives insight into both the clinical and biological efficacy of the therapies. Studies were excluded if they employed a pharmacologic agent in addition to the approach being evaluated.
We identified 26 studies, of which 14 met the inclusion/exclusion criteria. These studies found beneficial effects for yoga, massage therapy, aromatherapy massage, pet therapy, Qigong, auricular acupressure, reiki touch therapy, acupuncture, music therapy, and relaxation techniques.
Yoga
Yoga has become increasingly popular in the Western world during the last 2 decades.10 There are a variety of yoga practices; common forms include hatha yoga, power yoga, kripalu yoga, and forrest yoga.11
A study of 92 depressed pregnant women monito
Hatha yoga consists of a combination of postural exercises, breathing techniques, relaxation, and meditation. In a 12-week study of 88 postmenopausal women, those who practiced hatha yoga for 75 minutes a day had significantly lower STAI scores compared with women who exercised for 75 minutes a day and those who performed no physical activity.13
Continue to: Massage therapy
Massage therapy
Receiving as little as 15 minutes of back massage has proven to be beneficial for individuals with anxiety. In an RCT conducted in Turkey, 44 caregivers of patients with cancer were assigned to receive a back massage or to rest quietly in a room for 15 minutes once each day for 1 week.14 By the end of the week, compared with those who quietly rested, those who received the back massage had a statistically significant reduction in serum cortisol levels and STAI scores.14
Aromatherapy massage
Aromatherapy is the use of essential oils from plants through distillation.15 The scent of the oils is purported to provide medical benefits. More than 60 essential oils are used therapeutically, including rose, lavender, lemon, and orange.16 These essential oils are frequently used in combination with a massage.
In South Korea, researchers investigated the effects of aromatherapy massage on 25 women who had children diagnosed with attention-deficit/hyperactivity disorder.17 Women assigned to the treatment group received a 40-minute aromatherapy massage using mixed essential oils that contained lavender and geranium twice a week for 4 weeks. Women in the control group received no treatment. Compared with those in the control group, women who received the aromatherapy massages had a statistically significant decrease in STAI scores and salivary cortisol levels. Plasma cortisol was not significantly different between groups.17
Pet therapy
The psychological benefits of animal-assisted therapy were not evident until World War II, when dogs were used to cheer up injured soldiers.18 Today, pet therapy has been used on many inpatient units.19
In a U.S. study, 48 healthy undergraduate students were assigned to a room with a dog, a room with a friend, or a room by themselves.20 All participants were given the Trier Social Stress Test (TSST), a protocol that measures stress by having participants give a speech and perform mental arithmetic in front of an audience.The TSST is known to induce increases in cortisol levels. Although no differences in STAI scores were found among groups, students in the room with the dog had a lower spike in salivary cortisol after the TSST compared with participants who were in a room with a friend or in a room alone.20
Continue to: Qigong
Qigong
In Chinese medicine, Qi is known as a vital life force that flows through the body. The disruption of Qi is hypothesized to contribute to disease.21
Qigong is a medical therapy that focuses on uniting the body, breath, and mind to improve health.21 It consists of rhythmic, choreographed movements used to position the body into postures believed to help direct Qi to specific areas in the body. Qigong also uses sound exercises, in which an individual creates certain syllables while breathing. Six syllables are used, each of which is believed to affect a certain organ.21
Korean researchers randomly assigned 32 healthy men to a Qigong training group or a sham Qigong control group.22 Individuals in the training group performed 25 minutes of sound exercises, 20 minutes of meditation, and 15 minutes of movements. The control group learned the same movements as the experimental group, but without the conscious effort of moving Qi. After 3 sessions, those in the Qigong training group had significantly decreased STAI scores and serum cortisol levels compared with those in the sham group.22
In a different Korean study, researchers randomly assigned 50 participants with elevated distress levels to a Qigong training group or a waitlist control group in which participants called a trainer to describe stressful events.23 After 4 weeks, participants in the Qigong group had significant decreases in STAI scores compared with the control group. However, there were no changes in salivary cortisol levels.23
Auricular acupressure
Auricular acupressure involves applying pressure on certain portions of the auricle (outer ear) to alleviate pain and disease.24 Similar to Qigong, auricular acupressure focuses on reestablishing Qi in the body. Researchers randomly assigned 80 post-caesarean section women in Taiwan to 5 days of auricular acupressure or usual care.25 The women who received auricular acupressure had significantly lower STAI scores and serum cortisol levels compared with women who received routine care.25
Continue to: Reiki touch therapy
Reiki touch therapy
Reiki touch therapy originated in Japan. In this therapy, healers apply a light touch or hover their hands above an individual’s body to help direct energy.26
The effects of reiki touch therapy were recently evaluated in a U.S. study.27 Researchers randomly assigned 37 patients with human immunodeficiency virus to an experimental group that received 30 minutes of reiki touch therapy plus music therapy 6 times a week for 10 weeks, or to a music therapy–only control group. Patients who received reiki touch therapy had a significant decrease in STAI scores. Patients in this group also had a statistically significant drop in salivary cortisol levels after the first week.27
Acupuncture
Acupuncture is the application of needles to specific areas on the body. Acupuncture has been proposed to activate pain receptors, thereby producing an analgesic response.28
Researchers in Brazil randomly assigned 57 lactating women with preterm infants to an experimental group that received acupuncture or to a control group that received sham acupuncture.29 Treatment was administered at 5 points on the ear unilaterally for 5 minutes once a week for 16 months. Custom-made needles that did not actually puncture the skin were used in the sham group; a toothpick was used to create the sensation of needle perforations. STAI scores were reduced in both groups, although there was no statistically significant difference in scores between the acupuncture and sham groups.29
Music therapy
Music has been long believed to have beneficial psychological effects. In Turkey, researchers evaluated the effects of music therapy in 100 oncology patients who received port catheters.30 Patients were randomly assigned to an experimental group that received music therapy throughout the procedure or to a control group that received normal care. Patients who listened to music during port catheter placement had significantly reduced STAI scores and serum cortisol levels compared with those in the control group.30
Continue to: Relaxation techniques
Relaxation techniques
A wide range of relaxation techniques are used for therapeutic purposes. In Switzerland, researchers evaluated the anxiolytic effects of 10 minutes of progressive muscle relaxation and guided imagery in 39 pregnant women.31 Women randomly assigned to progressive muscle relaxation were instructed to systematically tense and then release muscle groups throughout their body in sequential order. Women assigned to the guided imagery intervention were told to imagine a safe place and to think of someone who could confer security and reassurance. The remainder of the women were assigned to a control group, where they sat quietly without any formal instructions. Researchers found that each group had a decrease in STAI scores and salivary cortisol levels immediately after the intervention.31
The relaxation response was first described in 1975 by Herbert Benson, MD, as a deep meditative state characterized by a decrease in tension, heart rate, and breathing rate. Several techniques can induce this state, including hypnosis, progressive muscle relaxation, yoga, and transcendental meditation.32 In a study of 15 healthy older adults (age 65 to 80), researchers randomly assigned participants to a relaxation response training group or to a control group.33 The relaxation response training included meditation, imagery, and relaxation techniques. After 5 weeks, participants who received the relaxation response training had marginally significant decreases in STAI scores compared with those in the control group.33
Consider these therapies as adjuncts
Our review of select positive RCTs (Table12-14,17,20,22,23,25,27,29-31,33) suggests that some nonpharmacologic/nonpsychotherapeutic adjunctive interventions may have beneficial effects for patients who have anxiety. Several of the controlled studies we reviewed demonstrated that these interventions are superior to placebo. The reductions in both anxiety severity as measured by the STAI and cortisol levels suggests that some of these complementary therapies deserve a second look as useful adjuncts to established anxiety treatments.
Bottom Line
A review of select randomized controlled trials suggests that some complementary therapies may be helpful as adjunctive therapy in patients with anxiety. These include yoga, massage therapy, aromatherapy massage, pet therapy, Qigong, auricular acupressure, reiki touch therapy, acupuncture, music therapy, and relaxation techniques.
Related Resources
- Bandelow B, Baldwin D, Abelli M, et al. Biological markers for anxiety disorders, OCD and PTSD: a consensus statement. Part II: neurochemistry, neurophysiology and neurocognition. World J Biol Psychiatry. 2017;18(3):162-214.
- National Institute of Mental Health. Anxiety disorders. https://www.nimh.nih.gov/health/topics/anxiety-disorders/index.shtml.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Masand PS, Gupta S. Long-term side effects of newer-generation antidepressants: SSRIs, venlafaxine, nefazodone, bupropion, and mirtazapine. Ann Clin Psychiatry. 2002;14(3):175-182.
3. Siddiqui R, Gawande S, Shende T, et al. SSRI-induced coagulopathy: is it reality? Therapeutic Advances in Psychopharmacology. 2011;1(6):169-174.
4. Brambilla P, Cipriani A, Hotopf M, et al. Side-effect profile of fluoxetine in comparison with other SSRIs, tricyclic and newer antidepressants: a meta-analysis of clinical trial data. Pharmacopsychiatry. 2005;38(2):69-77.
5. Slomski A. Blended CBT controls anxiety in cancer survivors. JAMA. 2017;318(4):323.
6. Forsell E, Bendix M, Holländare F, et al. Internet delivered cognitive behavior therapy for antenatal depression: a randomised controlled trial. J Affect Disord. 2017;221:56-64.
7. Lilliengren P, Johansson R, Town JM, et al. Intensive Short-Term Dynamic Psychotherapy for generalized anxiety disorder: A pilot effectiveness and process-outcome study. Clin Psychol Psychother. 2017;24(6):1313-1321.
8. Furtado M, Katzman MA. Neuroinflammatory pathways in anxiety, posttraumatic stress, and obsessive compulsive disorders. Psychiatry Res. 2015;229(1-2):37-48.
9. Spielberger CD, Gorsuch RL, Lushene R, et al. Manual for the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press; 1983.
10. Saper RB, Eisenberg DM, Davis RB, et al. Prevalence and patterns of adult yoga use in the United States: results of a national survey. Altern Ther Health Med. 2004;10(2):44-49.
11. Farmer J. Americanasana. Reviews in American history. 2012;40(1):145-158.
12. Field T, Diego M, Delgado J, et al. Yoga and social support reduce prenatal depression, anxiety and cortisol. J Bodyw Mov Ther: 2013;17(4):397-403.
13. Jorge MP, Santaella DF, Pontes IM, et al. Hatha Yoga practice decreases menopause symptoms and improves quality of life: a randomized controlled trial. Complement Ther Med. 2016;26:128-135.
14. Pinar R, Afsar F. Back massage to decrease state anxiety, cortisol level, blood pressure, heart rate and increase sleep quality in family caregivers of patients with cancer: a randomised controlled trial. Asian Pac J Cancer Prev. 2015;16(18):8127-8133.
15. Kuriyama H, Watanabe S, Nakaya, et al. Immunological and psychological benefits of aromatherapy massage. Evid Based Complement Alternat Med. 2005;2(2):179-184.
16. Setzer WN. Essential oils and anxiolytic aromatherapy. Nat Prod Commun. 2009;4(9):1305-1316.
17. Wu JJ, Cui Y, Yang YS, et al. Modulatory effects of aromatherapy massage intervention on electroencephalogram, psychological assessments, salivary cortisol and plasma brain-derived neurotrophic factor. Complement Ther Med. 2014;22(3):456-462.
18. Fine A. Forward. In: Fine A, ed. Handbook on animal-assisted therapy-theoretical foundations and guidelines for practice. 3rd ed. Academic Press; 2010:xvii-xviii.
19. Snipelisky D, Burton MC. Canine-assisted therapy in the inpatient setting. South Med J. 2014;107(4):265-273.
20. Polheber JP, Matchock RL. The presence of a dog attenuates cortisol and heart rate in the Trier Social Stress Test compared to human friends. J Behav Med. 2014;37(5):860-867.
21. Liu T, Qiang X, eds. Chinese medical Qigong. Philadelphia, PA: Singing Dragon; 2013:1-100,192,238,511.
22. Lee MS, Kang CW, Lim HJ, et al. Effects of Qi-training on anxiety and plasma concentrations of cortisol, ACTH, and aldosterone: a randomized placebo-controlled pilot study. Stress Health. 2004;20(5):243-248.
23. Hwang EY, Chung SY, Cho JH, et al. Effects of a brief Qigong-based stress reduction program (BQSRP) in a distressed Korean population: a randomized trial. BMC Complement Altern Med. 2013;13:113.
24. Oleson, T. Overview and history of auriculotherapy. In: Auriculotherapy manual: Chinese and Western systems of ear acupuncture. 4th ed. London: Churchill Livingstone; 2014:1.
25. Kuo SY, Tsai SH, Chen SL, et al. Auricular acupressure relieves anxiety and fatigue, and reduces cortisol levels in post-caesarean section women: a single-blind, randomised controlled study. Int J Nurs Stud. 2016;53:17-26.
26. Horan P. Introduction. In: Horan P. Empowerment through reiki: the path to personal and global transformation. 8th ed. Twin Lakes, WI: Lotus Press; 1998:13-15.
27. Bremner MN, Blake BJ, Wagner VD, et al. Effects of reiki with music compared to music only among people living with HIV. J Assoc Nurses AIDS Care. 2016;27(5):635-647.
28. Helmes JM. The basic, clinical, and speculative science of acupuncture. In: Acupuncture energetics: a clinical approach for physicians. Volume 1. Berkeley, CA: Medical Acupuncture Publishers; 1995:19-32.
29. Haddad-Rodrigues M, Spanó Nakano A, Stefanello J, et al. Acupuncture for anxiety in lactating mothers with preterm infants: a randomized controlled trial. Evid Based Complement Alternat Med. 2013;2013:169184. doi: 10.1155/2013/169184.
30. Zengin S, Kabul S, Al B, et al. Effects of music therapy on pain and anxiety in patients undergoing port catheter placement procedure. Complement Ther Med. 2013;21(6):689-696.
31. Urech C, Fink NS, Hoesli I, et al. Effects of relaxation on psychobiological wellbeing during pregnancy: a randomized controlled trial. Psychoneuroendocrinology. 2010;35(9):1348-1355.
32. Goleman D. The relaxation response. In: Mind body medicine: how to use your mind for better health. Yonkers, NY: Consumer Reports; 1993:125-149.
33. Galvin JA, Benson H, Deckro GR, et al. The relaxation response: reducing stress and improving cognition in healthy aging adults. Complement Ther Clin Pract. 2006;12(3):186-191.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Masand PS, Gupta S. Long-term side effects of newer-generation antidepressants: SSRIs, venlafaxine, nefazodone, bupropion, and mirtazapine. Ann Clin Psychiatry. 2002;14(3):175-182.
3. Siddiqui R, Gawande S, Shende T, et al. SSRI-induced coagulopathy: is it reality? Therapeutic Advances in Psychopharmacology. 2011;1(6):169-174.
4. Brambilla P, Cipriani A, Hotopf M, et al. Side-effect profile of fluoxetine in comparison with other SSRIs, tricyclic and newer antidepressants: a meta-analysis of clinical trial data. Pharmacopsychiatry. 2005;38(2):69-77.
5. Slomski A. Blended CBT controls anxiety in cancer survivors. JAMA. 2017;318(4):323.
6. Forsell E, Bendix M, Holländare F, et al. Internet delivered cognitive behavior therapy for antenatal depression: a randomised controlled trial. J Affect Disord. 2017;221:56-64.
7. Lilliengren P, Johansson R, Town JM, et al. Intensive Short-Term Dynamic Psychotherapy for generalized anxiety disorder: A pilot effectiveness and process-outcome study. Clin Psychol Psychother. 2017;24(6):1313-1321.
8. Furtado M, Katzman MA. Neuroinflammatory pathways in anxiety, posttraumatic stress, and obsessive compulsive disorders. Psychiatry Res. 2015;229(1-2):37-48.
9. Spielberger CD, Gorsuch RL, Lushene R, et al. Manual for the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press; 1983.
10. Saper RB, Eisenberg DM, Davis RB, et al. Prevalence and patterns of adult yoga use in the United States: results of a national survey. Altern Ther Health Med. 2004;10(2):44-49.
11. Farmer J. Americanasana. Reviews in American history. 2012;40(1):145-158.
12. Field T, Diego M, Delgado J, et al. Yoga and social support reduce prenatal depression, anxiety and cortisol. J Bodyw Mov Ther: 2013;17(4):397-403.
13. Jorge MP, Santaella DF, Pontes IM, et al. Hatha Yoga practice decreases menopause symptoms and improves quality of life: a randomized controlled trial. Complement Ther Med. 2016;26:128-135.
14. Pinar R, Afsar F. Back massage to decrease state anxiety, cortisol level, blood pressure, heart rate and increase sleep quality in family caregivers of patients with cancer: a randomised controlled trial. Asian Pac J Cancer Prev. 2015;16(18):8127-8133.
15. Kuriyama H, Watanabe S, Nakaya, et al. Immunological and psychological benefits of aromatherapy massage. Evid Based Complement Alternat Med. 2005;2(2):179-184.
16. Setzer WN. Essential oils and anxiolytic aromatherapy. Nat Prod Commun. 2009;4(9):1305-1316.
17. Wu JJ, Cui Y, Yang YS, et al. Modulatory effects of aromatherapy massage intervention on electroencephalogram, psychological assessments, salivary cortisol and plasma brain-derived neurotrophic factor. Complement Ther Med. 2014;22(3):456-462.
18. Fine A. Forward. In: Fine A, ed. Handbook on animal-assisted therapy-theoretical foundations and guidelines for practice. 3rd ed. Academic Press; 2010:xvii-xviii.
19. Snipelisky D, Burton MC. Canine-assisted therapy in the inpatient setting. South Med J. 2014;107(4):265-273.
20. Polheber JP, Matchock RL. The presence of a dog attenuates cortisol and heart rate in the Trier Social Stress Test compared to human friends. J Behav Med. 2014;37(5):860-867.
21. Liu T, Qiang X, eds. Chinese medical Qigong. Philadelphia, PA: Singing Dragon; 2013:1-100,192,238,511.
22. Lee MS, Kang CW, Lim HJ, et al. Effects of Qi-training on anxiety and plasma concentrations of cortisol, ACTH, and aldosterone: a randomized placebo-controlled pilot study. Stress Health. 2004;20(5):243-248.
23. Hwang EY, Chung SY, Cho JH, et al. Effects of a brief Qigong-based stress reduction program (BQSRP) in a distressed Korean population: a randomized trial. BMC Complement Altern Med. 2013;13:113.
24. Oleson, T. Overview and history of auriculotherapy. In: Auriculotherapy manual: Chinese and Western systems of ear acupuncture. 4th ed. London: Churchill Livingstone; 2014:1.
25. Kuo SY, Tsai SH, Chen SL, et al. Auricular acupressure relieves anxiety and fatigue, and reduces cortisol levels in post-caesarean section women: a single-blind, randomised controlled study. Int J Nurs Stud. 2016;53:17-26.
26. Horan P. Introduction. In: Horan P. Empowerment through reiki: the path to personal and global transformation. 8th ed. Twin Lakes, WI: Lotus Press; 1998:13-15.
27. Bremner MN, Blake BJ, Wagner VD, et al. Effects of reiki with music compared to music only among people living with HIV. J Assoc Nurses AIDS Care. 2016;27(5):635-647.
28. Helmes JM. The basic, clinical, and speculative science of acupuncture. In: Acupuncture energetics: a clinical approach for physicians. Volume 1. Berkeley, CA: Medical Acupuncture Publishers; 1995:19-32.
29. Haddad-Rodrigues M, Spanó Nakano A, Stefanello J, et al. Acupuncture for anxiety in lactating mothers with preterm infants: a randomized controlled trial. Evid Based Complement Alternat Med. 2013;2013:169184. doi: 10.1155/2013/169184.
30. Zengin S, Kabul S, Al B, et al. Effects of music therapy on pain and anxiety in patients undergoing port catheter placement procedure. Complement Ther Med. 2013;21(6):689-696.
31. Urech C, Fink NS, Hoesli I, et al. Effects of relaxation on psychobiological wellbeing during pregnancy: a randomized controlled trial. Psychoneuroendocrinology. 2010;35(9):1348-1355.
32. Goleman D. The relaxation response. In: Mind body medicine: how to use your mind for better health. Yonkers, NY: Consumer Reports; 1993:125-149.
33. Galvin JA, Benson H, Deckro GR, et al. The relaxation response: reducing stress and improving cognition in healthy aging adults. Complement Ther Clin Pract. 2006;12(3):186-191.

N-acetylcysteine: A potential treatment for substance use disorders
Pharmacologic treatment options for many substance use disorders (SUDs) are limited. This is especially true for cocaine use disorder and cannabis use disorder, for which there are no FDA-approved medications. FDA-approved medications for other SUDs often take the form of replacement or agonist therapies (eg, nicotine replacement therapy) that substitute the effects of the substance to aid in cessation. Other pharmacotherapies treat symptoms of withdrawal, reduce craving, or provide aversive counter-conditioning if the patient consumes the substance while on the medication (eg, disulfiram).
The over-the-counter (OTC) antioxidant N-acetylcysteine (NAC) may be a potential treatment for SUDs. Although NAC is not approved by the FDA for treating SUDs, its proposed mechanism of action differs from that of current FDA-approved medications for SUDs. NAC’s potential for broad applicability, favorable adverse-effect profile, accessibility, and low cost make it an intriguing option for patients with multiple comorbidities, and potentially for individuals with polysubstance use. This article reviews the current evidence supporting NAC for treating SUDs, to provide insight about which patients may benefit from NAC and under which circumstances they are most likely to benefit.
NAC may correct glutamate dysregulation
Approximately 85% of individuals with an SUD do not seek treatment for it, and those who do are older, have a longer history of use, have more severe dependence, and have sought treatment numerous times before.1 By the time most people seek treatment, years of chronic substance use have likely led to significant brain-related adaptations. Individuals with SUDs often indicate that their substance use began as a pleasurable activity—the effects of the drug were enjoyable and they were motivated to use it again. With repeated substance use, they may begin to develop a stronger urge to use the drug, driven not necessarily by a desire for pleasure, but by compulsion.2
Numerous neural adaptations underlie the transition from “liking” a substance to engaging in the compulsive use that is characteristic of an SUD.2 For example, repeated use of an addictive substance may result in excess glutamate in the nucleus accumbens,3,4 an area of the brain that plays a critical role in motivation and learning. As a result, it has been proposed that pharmacotherapies that help correct glutamate dysregulation may be effective in promoting abstinence or preventing relapse to a substance.5,6
NAC may reverse the neural dysfunction seen in SUDs. As an OTC antioxidant that impacts glutamatergic functioning in the brain, NAC has long been used to treat acetaminophen overdose; however, in recent years, researchers have begun to tap its potential for treating substance use and psychiatric disorders. NAC is thought to upregulate the glutamate transporter (GLT-1) that removes excess glutamate from the nucleus accumbens.6 Several published reviews provide more in-depth information about the neurobiology of NAC.6-10
The adverse-effect profile of NAC is relatively benign. Nausea, vomiting, diarrhea, and sleepiness are relatively infrequent and mild.11,12 The bioavailability of NAC is about 4% to 9%, with an approximate half-life of 6.25 hours when orally administered.13 Because NAC is classified as an OTC supplement, the potency and preparation may vary by supplier. To maximize consistency, NAC should be obtained from a supplier that meets United States Pharmacopeia (USP) standards.
NAC for SUDs: Emerging evidence
Several recent reviews have described the efficacy of NAC for SUDs and other psychiatric disorders. Here we summarize the current research examining the efficacy of NAC for stimulant (ie, cocaine and methamphetamine), cannabis, tobacco, and alcohol use disorders.
Continue to: Stimulant use disorders
Stimulant use disorders. The United Nations Office for Drugs and Crime estimates that worldwide, more than 18 million people use cocaine and more than 35 million use amphetamines.14 There are currently no FDA-approved treatments for stimulant use disorders, and clinicians treating patients with cocaine or amphetamine dependence often are at a loss for how best to promote abstinence. Recent studies suggest that NAC may decrease drug-seeking behavior and cravings in adults who seek treatment. The results of studies examining NAC for treating cocaine use and methamphetamine use are summarized in Table 115-17 and Table 2,18,19 respectively.
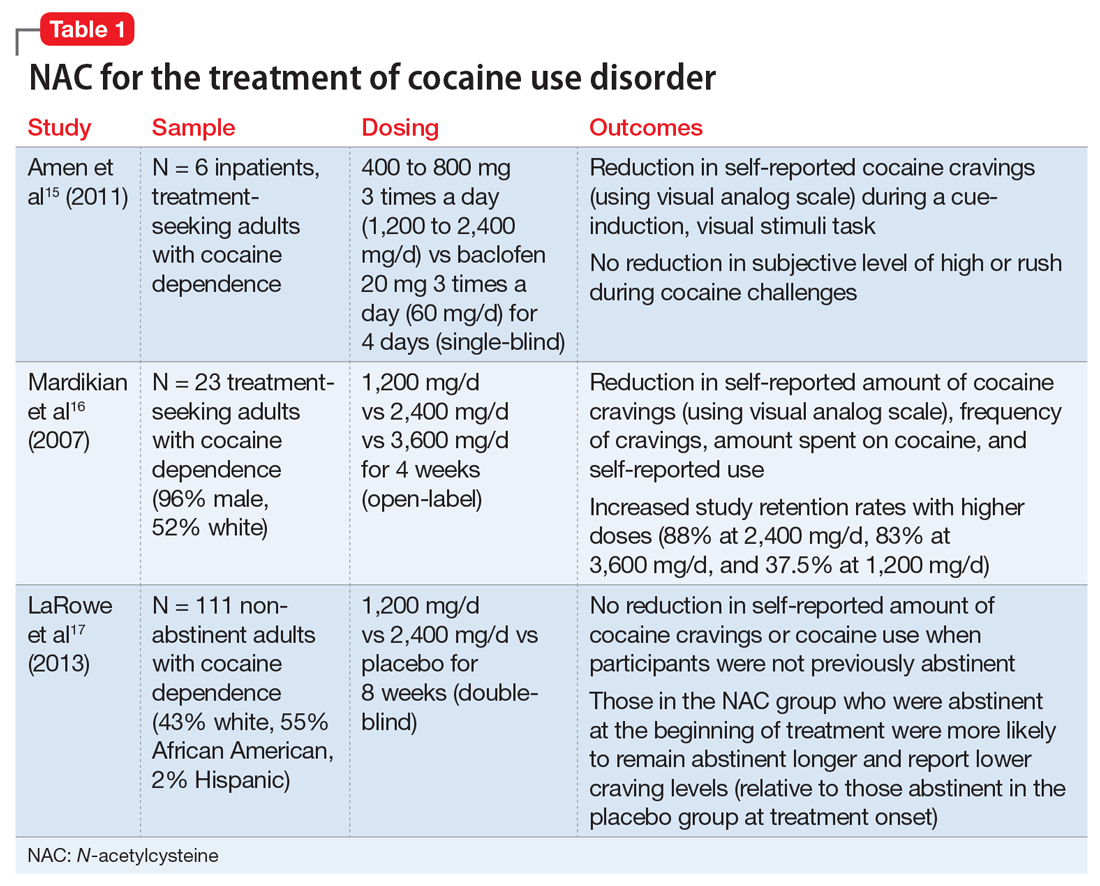
Cocaine cessation and relapse prevention. Several small pilot projects15,16 found that compared with placebo, various doses of NAC reduced craving (as measured with a visual analog scale). However, in a double-blind, placebo-controlled study, NAC did not decrease cravings or use after 8 weeks of treatment in individuals with cocaine use disorder who were still using cocaine (ie, they had not yet become abstinent). Interestingly, those who were abstinent when treatment began reported lower craving and remained abstinent longer if they received NAC (vs placebo), which suggests that NAC may be useful for preventing relapse.17

Methamphetamine cessation and relapse prevention. One study (N = 32) that evaluated the use of NAC, 1,200 mg/d for 4 weeks, vs placebo found reduced cravings among methamphetamine users who were seeking treatment.18 In contrast, a study of 31 methamphetamine users who were not seeking treatment evaluated the use of NAC, 2,400 mg/d, plus naltrexone, 200 mg/d, vs placebo for 8 weeks.19 It found no significant differences in craving or use patterns. Further research is needed to optimize the use of NAC for stimulant use disorders, and to better understand the role that abstinence plays.
Appropriate populations. The most support for use of NAC has been as an anti-relapse agent in treatment-seeking adults.
Continue to: Safety and dosing
Safety and dosing. Suggested dosages for the treatment of cocaine use disorder range from 1,200 to 3,600 mg/d (typically 600 to 1,800 mg twice daily, due to NAC’s short half-life), with higher retention rates noted in individuals who received 2,400 mg/d and 3,600 mg/d.16
Clinical implications. NAC is thought to act as an anti-relapse agent, rather than an agent that can help someone who is actively using stimulants to stop. Consequently, NAC will likely be most helpful for patients who are motivated to quit and are abstinent when they start taking NAC; however, this hypothesis needs further testing.
Cannabis use disorder
There are no FDA-approved treatments for cannabis use disorder. Individuals who use marijuana or other forms of cannabis may be less likely to report negative consequences or seek treatment compared with those who use other substances. Approximately 9% of individuals who use marijuana develop cannabis use disorder20; those who begin using marijuana earlier in adolescence are at increased risk.21 Commonly reported reasons for wanting to stop using marijuana include being concerned about health consequences, regaining or demonstrating self-control, saving money, avoiding legal consequences, obtaining or keeping employment, and reducing interpersonal conflict.22,23 Table 324-27 summarizes initial evidence that suggests NAC may be particularly useful in reducing marijuana use among adolescents (age 15 to 21).24,25

Cessation. An open-label, pilot clinical trial found significant reductions in self-reported marijuana use and craving—but not in biomarkers of use—among 24 adolescents after 4 weeks of NAC, 1,200 mg twice daily.24 In an 8-week, double-blind, randomized controlled trial of 116 adolescents, NAC, 1,200 mg twice daily, plus contingency management doubled the odds of abstinence, but had no effect on self-reported craving or use.25,26 In a sample of 302 adults, a 12-week trial of NAC, 1,200 mg twice daily, plus contingency management was no more effective than contingency management alone in promoting abstinence.27
Continue to: Appropriate populations
Appropriate populations. Evidence is stronger for use of NAC among adolescents (age 15 to 21) than for individuals older than age 21.25,27 Further research is needed to explore potential reasons for age-specific effects.
Safety and dosing. A safe and potentially efficacious dosage for the treatment of cannabis use disorder is 2,400 mg/d (1,200 mg twice daily).24,25,27
Clinical implications. Combined with contingency management, NAC might be efficacious for adolescents with cannabis use disorder, with treatment gains evident by the fourth week of treatment.24,25 To date, no clinical trials have examined the efficacy of NAC for treating cannabis use disorder without adjunctive contingency management, and research is needed to isolate the clinical effect of NAC among adolescents.
Tobacco use disorder
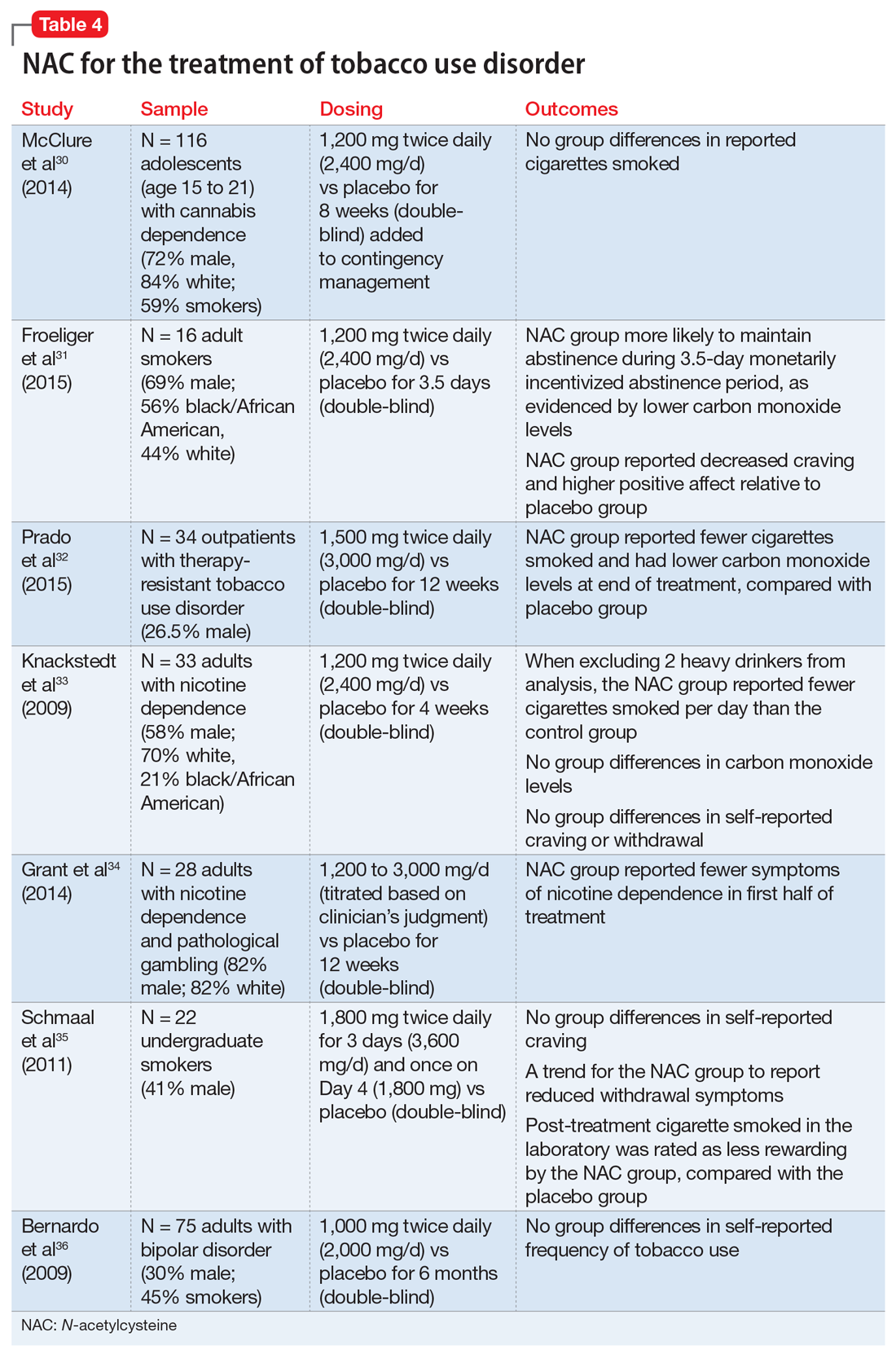
Cigarette smoking remains a leading cause of preventable death in the United States,28 and nearly 70% of people who start using tobacco become dependent.20 Existing FDA-approved treatments include nicotine replacement products, varenicline, and bupropion. Even though efficacious treatments exist, successful and sustained quit attempts are infrequent.29 NAC may exert a complementary effect to existing tobacco cessation interventions, such as varenicline.30 While these medications promote abstinence, NAC may be particularly beneficial in preventing relapse after abstinence has been achieved (Table 430-36).
Continue to: Cessation and relapse prevention
Cessation and relapse prevention. Several pilot studies found that adult smokers who received NAC (alone or in combination with another treatment) had lower carbon monoxide levels,31,32 smoked fewer cigarettes,32,33 and had fewer self-reported symptoms of nicotine dependence34 and/or less craving for cigarettes.31 However, one study of 33 smokers did not find a reduction in craving or carbon monoxide for NAC compared with placebo.33 Another pilot study of 22 young adult smokers found that those who received NAC rated their first cigarette after treatment (smoked in the laboratory) as less rewarding, relative to smokers who received a placebo.35
Secondary analyses of adults with bipolar disorder36 and adolescents with cannabis use disorder37 found no decreases in tobacco use among those who received NAC compared with placebo. However, the studies in these analyses did not specifically recruit tobacco users, and participants who were tobacco users were not necessarily interested in quitting. This may partially explain discrepant findings.
Appropriate populations. NAC has been studied mostly in adult cigarette smokers.
Safety and dosing. Suggested dosages for treating tobacco use disorder range from 1,200 to 3,600 mg/d (600 to 1,800 mg twice daily).
Continue to: Clinical implications
Clinical implications. Data on NAC’s efficacy for tobacco use disorder come from small, pilot trials. Although initial evidence is promising, it is premature to suggest NAC for smoking cessation until a fully powered, randomized clinical trial provides evidence of efficacy.
Alcohol use disorder
Alcohol use disorders are widely prevalent; 13.9% of U.S. adults met criteria in the past year, and 29.1% of U.S. adults meet criteria in their lifetime.38 Alcohol use disorders can result in significant negative consequences, including relationship problems, violent behavior, medical problems, and death. Existing FDA-approved medications for alcohol use disorder include naltrexone, acamprosate, and disulfiram.
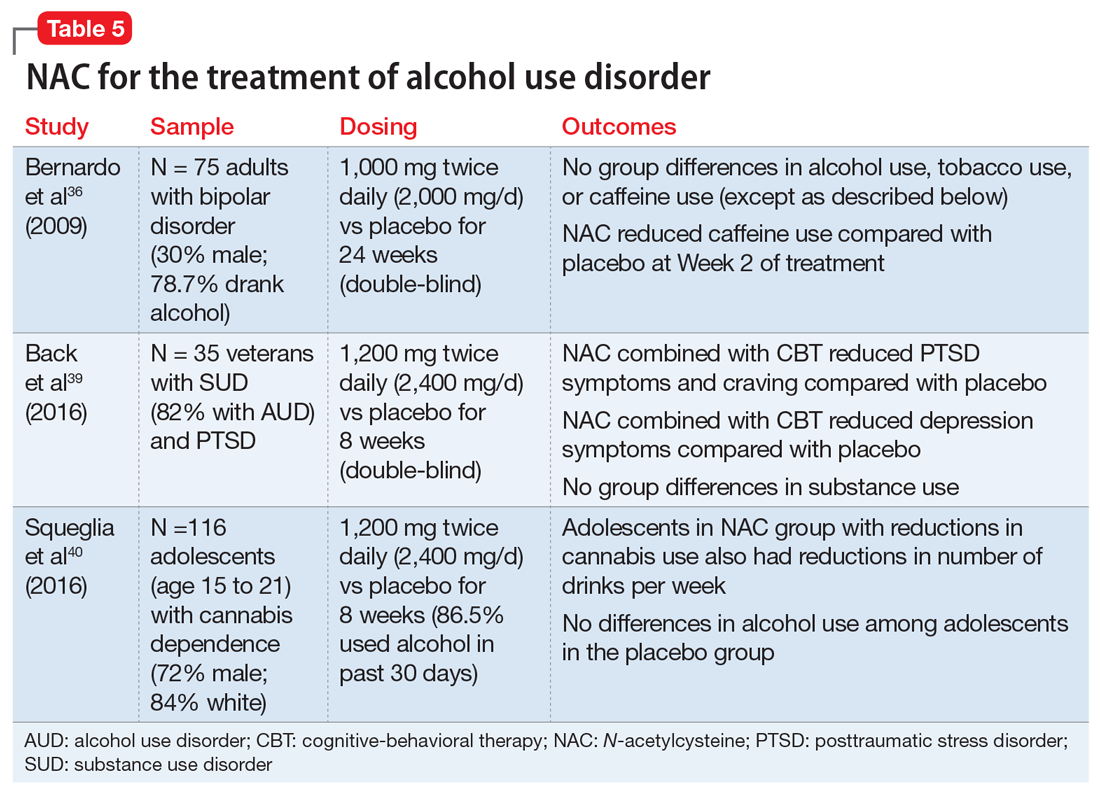
Due to the severe potential health consequences of alcohol, NAC has been examined as a possible aid in preventing relapse. However, most studies have been conducted using animals. Three studies have examined alcohol use in humans (Table 536,39,40). One was a pilot study,39 and the other 2 were secondary data analyses.36,40 None of them specifically focused on alcohol use disorders. A pilot study of 35 veterans with co-occurring posttraumautic stress disorder (PTSD) and SUDs (82% of whom had an alcohol use disorder) found that compared with placebo, NAC significantly decreased PTSD symptoms, craving, and depression.39 In a study of 75 adults with bipolar disorder, secondary alcohol use was not significantly reduced.36 However, one study suggested that NAC may decrease adolescent alcohol and marijuana co-use.40 Future work is needed to examine the potential clinical utility of NAC in individuals with alcohol use disorders.
Findings from animal studies indicate that NAC may:
- reduce alcohol-seeking41
- reduce withdrawal symptoms42
- reduce the teratogenic effects of alcohol43
- prevent alcohol toxicity44
- reduce health-related consequences of alcohol (eg, myocardial oxidative stress45 and alcohol-related steatohepatitis46).
Continue to: Appropriate populations
Appropriate populations. Pilot studies have suggested that appropriate populations may include veterans with SUD and PTSD39 and adolescents with marijuana dependence who use alcohol.40
Safety and dosing. Suggested dosages for the treatment of alcohol use disorder based on these studies range from 1,000 to 2,400 mg/d (500 to 1,200 mg twice daily).
Clinical implications. Future work is needed to determine if NAC is effective for treating alcohol use disorders. Ongoing randomized clinical trials are examining the efficacy of NAC in reducing alcohol use among individuals with alcohol use disorder. It is premature to recommend NAC for treatment of alcohol use disorders.
Other psychiatric uses
Although we have highlighted NAC’s effect on glutamatergic transmission, evidence suggests that NAC may have multiple mechanisms of action that could impact psychiatric functioning. For example, NAC may also reverse oxidative stress, which is frequently observed in psychiatric disorders such as schizophrenia and bipolar disorder.10,12 NAC also has anti-inflammatory properties. When inflammatory pathways of the CNS are dysregulated, production of neurotransmitters may be impaired, resulting in depression-like symptoms.10,12,47 Preliminary evidence suggests that NAC may be effective in treating mood-related symptoms (eg, irritability, depression) in individuals with psychiatric disorders (eg, bipolar and depressive disorders, PTSD, and SUDs) and general symptoms of schizophrenia, obsessive-compulsive disorder, and trichotillomania, although mixed findings in controlled studies suggest a need for further research.12,39
Continue to: NAC: A promising candidate
NAC: A promising candidate
Initial evidence suggests NAC may be helpful for treating patients with SUDs. A patient seeking SUD treatment who is treated with NAC may experience a decreased drive, craving, or compulsion to use. Notably, NAC may be particularly useful in preventing relapse after an individual has achieved abstinence. Evidence suggests that NAC may be useful in the treatment of adults with cocaine use disorders who have achieved abstinence, and adolescents with cannabis use disorders. Preliminary results for adult tobacco use disorder are also promising. Human data examining the efficacy of NAC for alcohol use disorder is limited. Researchers’ ongoing challenge is to identify which patients with which SUDs are most likely to benefit from NAC, and to create clear clinical guidelines for the provider.
Bottom Line
N-acetylcysteine is likely to have modest effects for some patients who have a substance use disorder, particularly adults who use cocaine and adolescents who use marijuana. It may be useful in preventing relapse to substance use after an individual has achieved abstinence.
Related Resources
- Deepmala, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294-321.
- Roberts-Wolfe D, Kalivas P. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745-756.
- National Institute on Drug Abuse. Treatment approaches for drug addiction. https://www.drugabuse.gov/publications/drugfacts/treatment-approaches-drug-addiction.
Drug Brand Names
Acamprosate • Campral
Acetaminophen • Tylenol
Baclofen • Lioresal
Bupropion • Zyban
Disulfiram • Antabuse
Naltrexone • Revia,Vivitrol
Varenicline • Chantix
1. Grella CE, Karno MP, Warda US, et al. Perceptions of need and help received for substance dependence in a national probability survey. Psychiatr Serv. 2009;60(8):1068-1074.
2. Everitt BJ, Robbins TW. Drug addiction: updating actions to habits to compulsions ten years on. Annu Rev Psychol. 2016;67:23-50.
3. McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23(8):3531-3537.
4. LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28(12):3170-3177.
5. Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16(10):974-986.
6. Roberts-Wolfe D, Kalivas PW. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745-756.
7. Berk M, Malhi GS, Gray LJ, et al. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34(3):167-177.
8. McClure EA, Gipson CD, Malcolm RJ, et al. Potential role of N-acetylcysteine in the management of substance use disorders. CNS drugs. 2014;28(2):95-106.
9. Deepmala, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294-321.
10. Minarini A, Ferrari S, Galletti M, et al. N-acetylcysteine in the treatment of psychiatric disorders: current status and future prospects. Expert Opin Drug Metab Toxicol. 2017;13(3):279-292.
11. Grandjean EM, Berthet P, Ruffman R, et al. Efficacy of oral long-term N‑acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22(2):209‑221.
12. Rhodes K, Braakhuis A. Performance and side effects of supplementation with N-acetylcysteine: a systematic review and meta-analysis. Sports Med. 2017;47(8):1619-1636.
13. Olsson B, Johansson M, Gabrielsson J, et al. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol. 1988;34(1):77-82.
14. United Nations Office on Drugs and Crime. World Drug Report 2016 (United Nations publication, Sales No. E.16.XI.7). https://www.unodc.org/doc/wdr2016/WORLD_DRUG_REPORT_2016_web.pdf. Published May 2016. Accessed April 26, 2018.
15. Amen SL, Piacentine LB, Ahmad ME, et al. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology. 2011;36(4):871-878.
16. Mardikian PN, LaRowe SD, Hedden S, et al. An open-label trial of N-acetylcysteine for the treatment of cocaine dependence: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(2):389-394.
17. LaRowe SD, Kalivas PW, Nicholas JS, et al. A double‐blind placebo‐controlled trial of N‐acetylcysteine in the treatment of cocaine dependence. Am J Addict. 2013;22(5):443-452.
18. Mousavi SG, Sharbafchi MR, Salehi M, et al. The efficacy of N-acetylcysteine in the treatment of methamphetamine dependence: a double-blind controlled, crossover study. Arch Iran Med. 2015;18(1):28-33.
19. Grant JE, Odlaug BL, Kim SW. A double-blind, placebo-controlled study of N-acetyl cysteine plus naltrexone for methamphetamine dependence. Eur Neuropsychopharmacol. 2010;20(11):823-828.
20. Lopez-Quintero C, Pérez de los Cobos J, Hasin DS, et al. Probability and predictors of transition from first use to dependence on nicotine, alcohol, cannabis, and cocaine: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Drug Alcohol Depend. 2011;115(1-2):120-130.
21. Chen CY, O’Brien MS, Anthony JC. Who becomes cannabis dependent soon after onset of use? Epidemiological evidence from the United States: 2000-2001. Drug Alcohol Depend. 2005;79(1):11-22.
22. Copersino ML, Boyd SJ, Tashkin DP, et al. Quitting among non-treatment-seeking marijuana users: reasons and changes in other substance use. Am J Addict. 2006;15(4):297-302.
23. Weiner MD, Sussman S, McCuller WJ, et al. Factors in marijuana cessation among high-risk youth. J Drug Educ. 1999;29(4):337-357.
24. Gray KM, Watson NL, Carpenter MJ, et al. N-acetylcysteine (NAC) in young marijuana users: an open-label pilot study. Am J Addict. 2010;19(2):187-189.
25. Gray KM, Carpenter MJ, Baker NL, et al. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169(8):805-812.
26. Roten AT, Baker NL, Gray KM. Marijuana craving trajectories in an adolescent marijuana cessation pharmacotherapy trial. Addict Behav. 2013;38(3):1788-1791.
27. Gray KM, Sonne SC, McClure EA, et al. A randomized placebo-controlled trial of N-acetylcysteine for cannabis use disorder in adults. Drug Alcohol Depend. 2017;177:249-257.
28. Rostron B. Mortality risks associated with environmental tobacco smoke exposure in the United States. Nicotine Tob Res. 2013;15(10):1722-1728.
29. Centers for Disease Control and Prevention. Quitting smoking among adults – United States, 2001–2010. MMWR. 2011;60(44):1513-1519.
30. McClure EA, Baker NL, Gipson CD, et al. An open-label pilot trial of N-acetylcysteine and varenicline in adult cigarette smokers. Am J Drug Alcohol Abuse. 2015;41(1):52-56.
31. Froeliger B, McConnell P, Stankeviciute N, et al. The effects of N-acetylcysteine on frontostriatal resting-state functional connectivity, withdrawal symptoms and smoking abstinence: a double-blind, placebo-controlled fMRI pilot study. Drug Alcohol Depend. 2015;156:234-242.
32. Prado E, Maes M, Piccoli LG, et al. N-acetylcysteine for therapy-resistant tobacco use disorder: a pilot study. Redox Rep. 2015;20(5):215-222.
33. Knackstedt LA, LaRowe S, Mardikian P, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65(10):841-845.
34. Grant JE, Odlaug BL, Chamberlain SR, et al. A randomized, placebo-controlled trial of N-acetylcysteine plus imaginal desensitization for nicotine-dependent pathological gamblers. J Clin Psychiatry. 2014;75(1):39-45.
35. Schmaal L, Berk L, Hulstijn KP, et al. Efficacy of N-acetylcysteine in the treatment of nicotine dependence: a double-blind placebo-controlled pilot study. Eur Addiction Res. 2011;17(4):211-216.
36. Bernardo M, Dodd S, Gama CS, et al. Effects of N‐acetylcysteine on substance use in bipolar disorder: a randomised placebo‐controlled clinical trial. Acta Neuropsychiatr. 2009;21(5):239-245.
37. McClure EA, Baker NL, Gray KM. Cigarette smoking during an N-acetylcysteine-assisted cannabis cessation trial in adolescents. Am J Drug Alcohol Abuse. 2014;40(4):285-291.
38. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM-5 alcohol use disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72(8):757-766.
39. Back SE, McCauley JL, Korte KJ, et al. A double-blind randomized controlled pilot trial of N-acetylcysteine in veterans with PTSD and substance use disorders. J Clin Psychiatry. 2016;77(11):e1439-e1446.
40. Squeglia LM, Baker NL, McClure EA, et al. Alcohol use during a trial of N-acetylcysteine for adolescent marijuana cessation. Addict Behav. 2016;63:172-177.
41. Lebourgeois S, González-Marín MC, Jeanblanc J, et al. Effect of N-acetylcysteine on motivation, seeking and relapse to ethanol self-administration. Addict Biol. 2018;23(2):643-652.
42. Schneider R Jr, Santos CF, Clarimundo V, et al. N-acetylcysteine prevents behavioral and biochemical changes induced by alcohol cessation in rats. Alcohol. 2015;49(3):259-263.
43. Parnell SE, Sulik KK, Dehart DB, et al. Reduction of ethanol-induced ocular abnormalities in mice via dietary administration of N-acetylcysteine. Alcohol. 2010;44(7-8):699-705.
44. Ozkol H, Bulut G, Balahoroglu R, et al. Protective effects of Selenium, N-acetylcysteine and Vitamin E against acute ethanol intoxication in rats. Biol Trace Elem Res. 2017;175(1):177-185.
45. Seiva FR, Amauchi JF, Rocha KK, et al. Alcoholism and alcohol abstinence: N-acetylcysteine to improve energy expenditure, myocardial oxidative stress, and energy metabolism in alcoholic heart disease. Alcohol. 2009;43(8):649-656.
46. Setshedi M, Longato L, Petersen DR, et al. Limited therapeutic effect of N‐acetylcysteine on hepatic insulin resistance in an experimental model of alcohol‐induced steatohepatitis. Alcohol Clin Exp Res. 2011;35(12):2139-2151.
47. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
Pharmacologic treatment options for many substance use disorders (SUDs) are limited. This is especially true for cocaine use disorder and cannabis use disorder, for which there are no FDA-approved medications. FDA-approved medications for other SUDs often take the form of replacement or agonist therapies (eg, nicotine replacement therapy) that substitute the effects of the substance to aid in cessation. Other pharmacotherapies treat symptoms of withdrawal, reduce craving, or provide aversive counter-conditioning if the patient consumes the substance while on the medication (eg, disulfiram).
The over-the-counter (OTC) antioxidant N-acetylcysteine (NAC) may be a potential treatment for SUDs. Although NAC is not approved by the FDA for treating SUDs, its proposed mechanism of action differs from that of current FDA-approved medications for SUDs. NAC’s potential for broad applicability, favorable adverse-effect profile, accessibility, and low cost make it an intriguing option for patients with multiple comorbidities, and potentially for individuals with polysubstance use. This article reviews the current evidence supporting NAC for treating SUDs, to provide insight about which patients may benefit from NAC and under which circumstances they are most likely to benefit.
NAC may correct glutamate dysregulation
Approximately 85% of individuals with an SUD do not seek treatment for it, and those who do are older, have a longer history of use, have more severe dependence, and have sought treatment numerous times before.1 By the time most people seek treatment, years of chronic substance use have likely led to significant brain-related adaptations. Individuals with SUDs often indicate that their substance use began as a pleasurable activity—the effects of the drug were enjoyable and they were motivated to use it again. With repeated substance use, they may begin to develop a stronger urge to use the drug, driven not necessarily by a desire for pleasure, but by compulsion.2
Numerous neural adaptations underlie the transition from “liking” a substance to engaging in the compulsive use that is characteristic of an SUD.2 For example, repeated use of an addictive substance may result in excess glutamate in the nucleus accumbens,3,4 an area of the brain that plays a critical role in motivation and learning. As a result, it has been proposed that pharmacotherapies that help correct glutamate dysregulation may be effective in promoting abstinence or preventing relapse to a substance.5,6
NAC may reverse the neural dysfunction seen in SUDs. As an OTC antioxidant that impacts glutamatergic functioning in the brain, NAC has long been used to treat acetaminophen overdose; however, in recent years, researchers have begun to tap its potential for treating substance use and psychiatric disorders. NAC is thought to upregulate the glutamate transporter (GLT-1) that removes excess glutamate from the nucleus accumbens.6 Several published reviews provide more in-depth information about the neurobiology of NAC.6-10
The adverse-effect profile of NAC is relatively benign. Nausea, vomiting, diarrhea, and sleepiness are relatively infrequent and mild.11,12 The bioavailability of NAC is about 4% to 9%, with an approximate half-life of 6.25 hours when orally administered.13 Because NAC is classified as an OTC supplement, the potency and preparation may vary by supplier. To maximize consistency, NAC should be obtained from a supplier that meets United States Pharmacopeia (USP) standards.
NAC for SUDs: Emerging evidence
Several recent reviews have described the efficacy of NAC for SUDs and other psychiatric disorders. Here we summarize the current research examining the efficacy of NAC for stimulant (ie, cocaine and methamphetamine), cannabis, tobacco, and alcohol use disorders.
Continue to: Stimulant use disorders
Stimulant use disorders. The United Nations Office for Drugs and Crime estimates that worldwide, more than 18 million people use cocaine and more than 35 million use amphetamines.14 There are currently no FDA-approved treatments for stimulant use disorders, and clinicians treating patients with cocaine or amphetamine dependence often are at a loss for how best to promote abstinence. Recent studies suggest that NAC may decrease drug-seeking behavior and cravings in adults who seek treatment. The results of studies examining NAC for treating cocaine use and methamphetamine use are summarized in Table 115-17 and Table 2,18,19 respectively.
Cocaine cessation and relapse prevention. Several small pilot projects15,16 found that compared with placebo, various doses of NAC reduced craving (as measured with a visual analog scale). However, in a double-blind, placebo-controlled study, NAC did not decrease cravings or use after 8 weeks of treatment in individuals with cocaine use disorder who were still using cocaine (ie, they had not yet become abstinent). Interestingly, those who were abstinent when treatment began reported lower craving and remained abstinent longer if they received NAC (vs placebo), which suggests that NAC may be useful for preventing relapse.17
Methamphetamine cessation and relapse prevention. One study (N = 32) that evaluated the use of NAC, 1,200 mg/d for 4 weeks, vs placebo found reduced cravings among methamphetamine users who were seeking treatment.18 In contrast, a study of 31 methamphetamine users who were not seeking treatment evaluated the use of NAC, 2,400 mg/d, plus naltrexone, 200 mg/d, vs placebo for 8 weeks.19 It found no significant differences in craving or use patterns. Further research is needed to optimize the use of NAC for stimulant use disorders, and to better understand the role that abstinence plays.
Appropriate populations. The most support for use of NAC has been as an anti-relapse agent in treatment-seeking adults.
Continue to: Safety and dosing
Safety and dosing. Suggested dosages for the treatment of cocaine use disorder range from 1,200 to 3,600 mg/d (typically 600 to 1,800 mg twice daily, due to NAC’s short half-life), with higher retention rates noted in individuals who received 2,400 mg/d and 3,600 mg/d.16
Clinical implications. NAC is thought to act as an anti-relapse agent, rather than an agent that can help someone who is actively using stimulants to stop. Consequently, NAC will likely be most helpful for patients who are motivated to quit and are abstinent when they start taking NAC; however, this hypothesis needs further testing.
Cannabis use disorder
There are no FDA-approved treatments for cannabis use disorder. Individuals who use marijuana or other forms of cannabis may be less likely to report negative consequences or seek treatment compared with those who use other substances. Approximately 9% of individuals who use marijuana develop cannabis use disorder20; those who begin using marijuana earlier in adolescence are at increased risk.21 Commonly reported reasons for wanting to stop using marijuana include being concerned about health consequences, regaining or demonstrating self-control, saving money, avoiding legal consequences, obtaining or keeping employment, and reducing interpersonal conflict.22,23 Table 324-27 summarizes initial evidence that suggests NAC may be particularly useful in reducing marijuana use among adolescents (age 15 to 21).24,25
Cessation. An open-label, pilot clinical trial found significant reductions in self-reported marijuana use and craving—but not in biomarkers of use—among 24 adolescents after 4 weeks of NAC, 1,200 mg twice daily.24 In an 8-week, double-blind, randomized controlled trial of 116 adolescents, NAC, 1,200 mg twice daily, plus contingency management doubled the odds of abstinence, but had no effect on self-reported craving or use.25,26 In a sample of 302 adults, a 12-week trial of NAC, 1,200 mg twice daily, plus contingency management was no more effective than contingency management alone in promoting abstinence.27
Continue to: Appropriate populations
Appropriate populations. Evidence is stronger for use of NAC among adolescents (age 15 to 21) than for individuals older than age 21.25,27 Further research is needed to explore potential reasons for age-specific effects.
Safety and dosing. A safe and potentially efficacious dosage for the treatment of cannabis use disorder is 2,400 mg/d (1,200 mg twice daily).24,25,27
Clinical implications. Combined with contingency management, NAC might be efficacious for adolescents with cannabis use disorder, with treatment gains evident by the fourth week of treatment.24,25 To date, no clinical trials have examined the efficacy of NAC for treating cannabis use disorder without adjunctive contingency management, and research is needed to isolate the clinical effect of NAC among adolescents.
Tobacco use disorder
Cigarette smoking remains a leading cause of preventable death in the United States,28 and nearly 70% of people who start using tobacco become dependent.20 Existing FDA-approved treatments include nicotine replacement products, varenicline, and bupropion. Even though efficacious treatments exist, successful and sustained quit attempts are infrequent.29 NAC may exert a complementary effect to existing tobacco cessation interventions, such as varenicline.30 While these medications promote abstinence, NAC may be particularly beneficial in preventing relapse after abstinence has been achieved (Table 430-36).
Continue to: Cessation and relapse prevention
Cessation and relapse prevention. Several pilot studies found that adult smokers who received NAC (alone or in combination with another treatment) had lower carbon monoxide levels,31,32 smoked fewer cigarettes,32,33 and had fewer self-reported symptoms of nicotine dependence34 and/or less craving for cigarettes.31 However, one study of 33 smokers did not find a reduction in craving or carbon monoxide for NAC compared with placebo.33 Another pilot study of 22 young adult smokers found that those who received NAC rated their first cigarette after treatment (smoked in the laboratory) as less rewarding, relative to smokers who received a placebo.35
Secondary analyses of adults with bipolar disorder36 and adolescents with cannabis use disorder37 found no decreases in tobacco use among those who received NAC compared with placebo. However, the studies in these analyses did not specifically recruit tobacco users, and participants who were tobacco users were not necessarily interested in quitting. This may partially explain discrepant findings.
Appropriate populations. NAC has been studied mostly in adult cigarette smokers.
Safety and dosing. Suggested dosages for treating tobacco use disorder range from 1,200 to 3,600 mg/d (600 to 1,800 mg twice daily).
Continue to: Clinical implications
Clinical implications. Data on NAC’s efficacy for tobacco use disorder come from small, pilot trials. Although initial evidence is promising, it is premature to suggest NAC for smoking cessation until a fully powered, randomized clinical trial provides evidence of efficacy.
Alcohol use disorder
Alcohol use disorders are widely prevalent; 13.9% of U.S. adults met criteria in the past year, and 29.1% of U.S. adults meet criteria in their lifetime.38 Alcohol use disorders can result in significant negative consequences, including relationship problems, violent behavior, medical problems, and death. Existing FDA-approved medications for alcohol use disorder include naltrexone, acamprosate, and disulfiram.
Due to the severe potential health consequences of alcohol, NAC has been examined as a possible aid in preventing relapse. However, most studies have been conducted using animals. Three studies have examined alcohol use in humans (Table 536,39,40). One was a pilot study,39 and the other 2 were secondary data analyses.36,40 None of them specifically focused on alcohol use disorders. A pilot study of 35 veterans with co-occurring posttraumautic stress disorder (PTSD) and SUDs (82% of whom had an alcohol use disorder) found that compared with placebo, NAC significantly decreased PTSD symptoms, craving, and depression.39 In a study of 75 adults with bipolar disorder, secondary alcohol use was not significantly reduced.36 However, one study suggested that NAC may decrease adolescent alcohol and marijuana co-use.40 Future work is needed to examine the potential clinical utility of NAC in individuals with alcohol use disorders.
Findings from animal studies indicate that NAC may:
- reduce alcohol-seeking41
- reduce withdrawal symptoms42
- reduce the teratogenic effects of alcohol43
- prevent alcohol toxicity44
- reduce health-related consequences of alcohol (eg, myocardial oxidative stress45 and alcohol-related steatohepatitis46).
Continue to: Appropriate populations
Appropriate populations. Pilot studies have suggested that appropriate populations may include veterans with SUD and PTSD39 and adolescents with marijuana dependence who use alcohol.40
Safety and dosing. Suggested dosages for the treatment of alcohol use disorder based on these studies range from 1,000 to 2,400 mg/d (500 to 1,200 mg twice daily).
Clinical implications. Future work is needed to determine if NAC is effective for treating alcohol use disorders. Ongoing randomized clinical trials are examining the efficacy of NAC in reducing alcohol use among individuals with alcohol use disorder. It is premature to recommend NAC for treatment of alcohol use disorders.
Other psychiatric uses
Although we have highlighted NAC’s effect on glutamatergic transmission, evidence suggests that NAC may have multiple mechanisms of action that could impact psychiatric functioning. For example, NAC may also reverse oxidative stress, which is frequently observed in psychiatric disorders such as schizophrenia and bipolar disorder.10,12 NAC also has anti-inflammatory properties. When inflammatory pathways of the CNS are dysregulated, production of neurotransmitters may be impaired, resulting in depression-like symptoms.10,12,47 Preliminary evidence suggests that NAC may be effective in treating mood-related symptoms (eg, irritability, depression) in individuals with psychiatric disorders (eg, bipolar and depressive disorders, PTSD, and SUDs) and general symptoms of schizophrenia, obsessive-compulsive disorder, and trichotillomania, although mixed findings in controlled studies suggest a need for further research.12,39
Continue to: NAC: A promising candidate
NAC: A promising candidate
Initial evidence suggests NAC may be helpful for treating patients with SUDs. A patient seeking SUD treatment who is treated with NAC may experience a decreased drive, craving, or compulsion to use. Notably, NAC may be particularly useful in preventing relapse after an individual has achieved abstinence. Evidence suggests that NAC may be useful in the treatment of adults with cocaine use disorders who have achieved abstinence, and adolescents with cannabis use disorders. Preliminary results for adult tobacco use disorder are also promising. Human data examining the efficacy of NAC for alcohol use disorder is limited. Researchers’ ongoing challenge is to identify which patients with which SUDs are most likely to benefit from NAC, and to create clear clinical guidelines for the provider.
Bottom Line
N-acetylcysteine is likely to have modest effects for some patients who have a substance use disorder, particularly adults who use cocaine and adolescents who use marijuana. It may be useful in preventing relapse to substance use after an individual has achieved abstinence.
Related Resources
- Deepmala, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294-321.
- Roberts-Wolfe D, Kalivas P. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745-756.
- National Institute on Drug Abuse. Treatment approaches for drug addiction. https://www.drugabuse.gov/publications/drugfacts/treatment-approaches-drug-addiction.
Drug Brand Names
Acamprosate • Campral
Acetaminophen • Tylenol
Baclofen • Lioresal
Bupropion • Zyban
Disulfiram • Antabuse
Naltrexone • Revia,Vivitrol
Varenicline • Chantix
Pharmacologic treatment options for many substance use disorders (SUDs) are limited. This is especially true for cocaine use disorder and cannabis use disorder, for which there are no FDA-approved medications. FDA-approved medications for other SUDs often take the form of replacement or agonist therapies (eg, nicotine replacement therapy) that substitute the effects of the substance to aid in cessation. Other pharmacotherapies treat symptoms of withdrawal, reduce craving, or provide aversive counter-conditioning if the patient consumes the substance while on the medication (eg, disulfiram).
The over-the-counter (OTC) antioxidant N-acetylcysteine (NAC) may be a potential treatment for SUDs. Although NAC is not approved by the FDA for treating SUDs, its proposed mechanism of action differs from that of current FDA-approved medications for SUDs. NAC’s potential for broad applicability, favorable adverse-effect profile, accessibility, and low cost make it an intriguing option for patients with multiple comorbidities, and potentially for individuals with polysubstance use. This article reviews the current evidence supporting NAC for treating SUDs, to provide insight about which patients may benefit from NAC and under which circumstances they are most likely to benefit.
NAC may correct glutamate dysregulation
Approximately 85% of individuals with an SUD do not seek treatment for it, and those who do are older, have a longer history of use, have more severe dependence, and have sought treatment numerous times before.1 By the time most people seek treatment, years of chronic substance use have likely led to significant brain-related adaptations. Individuals with SUDs often indicate that their substance use began as a pleasurable activity—the effects of the drug were enjoyable and they were motivated to use it again. With repeated substance use, they may begin to develop a stronger urge to use the drug, driven not necessarily by a desire for pleasure, but by compulsion.2
Numerous neural adaptations underlie the transition from “liking” a substance to engaging in the compulsive use that is characteristic of an SUD.2 For example, repeated use of an addictive substance may result in excess glutamate in the nucleus accumbens,3,4 an area of the brain that plays a critical role in motivation and learning. As a result, it has been proposed that pharmacotherapies that help correct glutamate dysregulation may be effective in promoting abstinence or preventing relapse to a substance.5,6
NAC may reverse the neural dysfunction seen in SUDs. As an OTC antioxidant that impacts glutamatergic functioning in the brain, NAC has long been used to treat acetaminophen overdose; however, in recent years, researchers have begun to tap its potential for treating substance use and psychiatric disorders. NAC is thought to upregulate the glutamate transporter (GLT-1) that removes excess glutamate from the nucleus accumbens.6 Several published reviews provide more in-depth information about the neurobiology of NAC.6-10
The adverse-effect profile of NAC is relatively benign. Nausea, vomiting, diarrhea, and sleepiness are relatively infrequent and mild.11,12 The bioavailability of NAC is about 4% to 9%, with an approximate half-life of 6.25 hours when orally administered.13 Because NAC is classified as an OTC supplement, the potency and preparation may vary by supplier. To maximize consistency, NAC should be obtained from a supplier that meets United States Pharmacopeia (USP) standards.
NAC for SUDs: Emerging evidence
Several recent reviews have described the efficacy of NAC for SUDs and other psychiatric disorders. Here we summarize the current research examining the efficacy of NAC for stimulant (ie, cocaine and methamphetamine), cannabis, tobacco, and alcohol use disorders.
Continue to: Stimulant use disorders
Stimulant use disorders. The United Nations Office for Drugs and Crime estimates that worldwide, more than 18 million people use cocaine and more than 35 million use amphetamines.14 There are currently no FDA-approved treatments for stimulant use disorders, and clinicians treating patients with cocaine or amphetamine dependence often are at a loss for how best to promote abstinence. Recent studies suggest that NAC may decrease drug-seeking behavior and cravings in adults who seek treatment. The results of studies examining NAC for treating cocaine use and methamphetamine use are summarized in Table 115-17 and Table 2,18,19 respectively.
Cocaine cessation and relapse prevention. Several small pilot projects15,16 found that compared with placebo, various doses of NAC reduced craving (as measured with a visual analog scale). However, in a double-blind, placebo-controlled study, NAC did not decrease cravings or use after 8 weeks of treatment in individuals with cocaine use disorder who were still using cocaine (ie, they had not yet become abstinent). Interestingly, those who were abstinent when treatment began reported lower craving and remained abstinent longer if they received NAC (vs placebo), which suggests that NAC may be useful for preventing relapse.17
Methamphetamine cessation and relapse prevention. One study (N = 32) that evaluated the use of NAC, 1,200 mg/d for 4 weeks, vs placebo found reduced cravings among methamphetamine users who were seeking treatment.18 In contrast, a study of 31 methamphetamine users who were not seeking treatment evaluated the use of NAC, 2,400 mg/d, plus naltrexone, 200 mg/d, vs placebo for 8 weeks.19 It found no significant differences in craving or use patterns. Further research is needed to optimize the use of NAC for stimulant use disorders, and to better understand the role that abstinence plays.
Appropriate populations. The most support for use of NAC has been as an anti-relapse agent in treatment-seeking adults.
Continue to: Safety and dosing
Safety and dosing. Suggested dosages for the treatment of cocaine use disorder range from 1,200 to 3,600 mg/d (typically 600 to 1,800 mg twice daily, due to NAC’s short half-life), with higher retention rates noted in individuals who received 2,400 mg/d and 3,600 mg/d.16
Clinical implications. NAC is thought to act as an anti-relapse agent, rather than an agent that can help someone who is actively using stimulants to stop. Consequently, NAC will likely be most helpful for patients who are motivated to quit and are abstinent when they start taking NAC; however, this hypothesis needs further testing.
Cannabis use disorder
There are no FDA-approved treatments for cannabis use disorder. Individuals who use marijuana or other forms of cannabis may be less likely to report negative consequences or seek treatment compared with those who use other substances. Approximately 9% of individuals who use marijuana develop cannabis use disorder20; those who begin using marijuana earlier in adolescence are at increased risk.21 Commonly reported reasons for wanting to stop using marijuana include being concerned about health consequences, regaining or demonstrating self-control, saving money, avoiding legal consequences, obtaining or keeping employment, and reducing interpersonal conflict.22,23 Table 324-27 summarizes initial evidence that suggests NAC may be particularly useful in reducing marijuana use among adolescents (age 15 to 21).24,25
Cessation. An open-label, pilot clinical trial found significant reductions in self-reported marijuana use and craving—but not in biomarkers of use—among 24 adolescents after 4 weeks of NAC, 1,200 mg twice daily.24 In an 8-week, double-blind, randomized controlled trial of 116 adolescents, NAC, 1,200 mg twice daily, plus contingency management doubled the odds of abstinence, but had no effect on self-reported craving or use.25,26 In a sample of 302 adults, a 12-week trial of NAC, 1,200 mg twice daily, plus contingency management was no more effective than contingency management alone in promoting abstinence.27
Continue to: Appropriate populations
Appropriate populations. Evidence is stronger for use of NAC among adolescents (age 15 to 21) than for individuals older than age 21.25,27 Further research is needed to explore potential reasons for age-specific effects.
Safety and dosing. A safe and potentially efficacious dosage for the treatment of cannabis use disorder is 2,400 mg/d (1,200 mg twice daily).24,25,27
Clinical implications. Combined with contingency management, NAC might be efficacious for adolescents with cannabis use disorder, with treatment gains evident by the fourth week of treatment.24,25 To date, no clinical trials have examined the efficacy of NAC for treating cannabis use disorder without adjunctive contingency management, and research is needed to isolate the clinical effect of NAC among adolescents.
Tobacco use disorder
Cigarette smoking remains a leading cause of preventable death in the United States,28 and nearly 70% of people who start using tobacco become dependent.20 Existing FDA-approved treatments include nicotine replacement products, varenicline, and bupropion. Even though efficacious treatments exist, successful and sustained quit attempts are infrequent.29 NAC may exert a complementary effect to existing tobacco cessation interventions, such as varenicline.30 While these medications promote abstinence, NAC may be particularly beneficial in preventing relapse after abstinence has been achieved (Table 430-36).
Continue to: Cessation and relapse prevention
Cessation and relapse prevention. Several pilot studies found that adult smokers who received NAC (alone or in combination with another treatment) had lower carbon monoxide levels,31,32 smoked fewer cigarettes,32,33 and had fewer self-reported symptoms of nicotine dependence34 and/or less craving for cigarettes.31 However, one study of 33 smokers did not find a reduction in craving or carbon monoxide for NAC compared with placebo.33 Another pilot study of 22 young adult smokers found that those who received NAC rated their first cigarette after treatment (smoked in the laboratory) as less rewarding, relative to smokers who received a placebo.35
Secondary analyses of adults with bipolar disorder36 and adolescents with cannabis use disorder37 found no decreases in tobacco use among those who received NAC compared with placebo. However, the studies in these analyses did not specifically recruit tobacco users, and participants who were tobacco users were not necessarily interested in quitting. This may partially explain discrepant findings.
Appropriate populations. NAC has been studied mostly in adult cigarette smokers.
Safety and dosing. Suggested dosages for treating tobacco use disorder range from 1,200 to 3,600 mg/d (600 to 1,800 mg twice daily).
Continue to: Clinical implications
Clinical implications. Data on NAC’s efficacy for tobacco use disorder come from small, pilot trials. Although initial evidence is promising, it is premature to suggest NAC for smoking cessation until a fully powered, randomized clinical trial provides evidence of efficacy.
Alcohol use disorder
Alcohol use disorders are widely prevalent; 13.9% of U.S. adults met criteria in the past year, and 29.1% of U.S. adults meet criteria in their lifetime.38 Alcohol use disorders can result in significant negative consequences, including relationship problems, violent behavior, medical problems, and death. Existing FDA-approved medications for alcohol use disorder include naltrexone, acamprosate, and disulfiram.
Due to the severe potential health consequences of alcohol, NAC has been examined as a possible aid in preventing relapse. However, most studies have been conducted using animals. Three studies have examined alcohol use in humans (Table 536,39,40). One was a pilot study,39 and the other 2 were secondary data analyses.36,40 None of them specifically focused on alcohol use disorders. A pilot study of 35 veterans with co-occurring posttraumautic stress disorder (PTSD) and SUDs (82% of whom had an alcohol use disorder) found that compared with placebo, NAC significantly decreased PTSD symptoms, craving, and depression.39 In a study of 75 adults with bipolar disorder, secondary alcohol use was not significantly reduced.36 However, one study suggested that NAC may decrease adolescent alcohol and marijuana co-use.40 Future work is needed to examine the potential clinical utility of NAC in individuals with alcohol use disorders.
Findings from animal studies indicate that NAC may:
- reduce alcohol-seeking41
- reduce withdrawal symptoms42
- reduce the teratogenic effects of alcohol43
- prevent alcohol toxicity44
- reduce health-related consequences of alcohol (eg, myocardial oxidative stress45 and alcohol-related steatohepatitis46).
Continue to: Appropriate populations
Appropriate populations. Pilot studies have suggested that appropriate populations may include veterans with SUD and PTSD39 and adolescents with marijuana dependence who use alcohol.40
Safety and dosing. Suggested dosages for the treatment of alcohol use disorder based on these studies range from 1,000 to 2,400 mg/d (500 to 1,200 mg twice daily).
Clinical implications. Future work is needed to determine if NAC is effective for treating alcohol use disorders. Ongoing randomized clinical trials are examining the efficacy of NAC in reducing alcohol use among individuals with alcohol use disorder. It is premature to recommend NAC for treatment of alcohol use disorders.
Other psychiatric uses
Although we have highlighted NAC’s effect on glutamatergic transmission, evidence suggests that NAC may have multiple mechanisms of action that could impact psychiatric functioning. For example, NAC may also reverse oxidative stress, which is frequently observed in psychiatric disorders such as schizophrenia and bipolar disorder.10,12 NAC also has anti-inflammatory properties. When inflammatory pathways of the CNS are dysregulated, production of neurotransmitters may be impaired, resulting in depression-like symptoms.10,12,47 Preliminary evidence suggests that NAC may be effective in treating mood-related symptoms (eg, irritability, depression) in individuals with psychiatric disorders (eg, bipolar and depressive disorders, PTSD, and SUDs) and general symptoms of schizophrenia, obsessive-compulsive disorder, and trichotillomania, although mixed findings in controlled studies suggest a need for further research.12,39
Continue to: NAC: A promising candidate
NAC: A promising candidate
Initial evidence suggests NAC may be helpful for treating patients with SUDs. A patient seeking SUD treatment who is treated with NAC may experience a decreased drive, craving, or compulsion to use. Notably, NAC may be particularly useful in preventing relapse after an individual has achieved abstinence. Evidence suggests that NAC may be useful in the treatment of adults with cocaine use disorders who have achieved abstinence, and adolescents with cannabis use disorders. Preliminary results for adult tobacco use disorder are also promising. Human data examining the efficacy of NAC for alcohol use disorder is limited. Researchers’ ongoing challenge is to identify which patients with which SUDs are most likely to benefit from NAC, and to create clear clinical guidelines for the provider.
Bottom Line
N-acetylcysteine is likely to have modest effects for some patients who have a substance use disorder, particularly adults who use cocaine and adolescents who use marijuana. It may be useful in preventing relapse to substance use after an individual has achieved abstinence.
Related Resources
- Deepmala, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294-321.
- Roberts-Wolfe D, Kalivas P. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745-756.
- National Institute on Drug Abuse. Treatment approaches for drug addiction. https://www.drugabuse.gov/publications/drugfacts/treatment-approaches-drug-addiction.
Drug Brand Names
Acamprosate • Campral
Acetaminophen • Tylenol
Baclofen • Lioresal
Bupropion • Zyban
Disulfiram • Antabuse
Naltrexone • Revia,Vivitrol
Varenicline • Chantix
1. Grella CE, Karno MP, Warda US, et al. Perceptions of need and help received for substance dependence in a national probability survey. Psychiatr Serv. 2009;60(8):1068-1074.
2. Everitt BJ, Robbins TW. Drug addiction: updating actions to habits to compulsions ten years on. Annu Rev Psychol. 2016;67:23-50.
3. McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23(8):3531-3537.
4. LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28(12):3170-3177.
5. Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16(10):974-986.
6. Roberts-Wolfe D, Kalivas PW. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745-756.
7. Berk M, Malhi GS, Gray LJ, et al. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34(3):167-177.
8. McClure EA, Gipson CD, Malcolm RJ, et al. Potential role of N-acetylcysteine in the management of substance use disorders. CNS drugs. 2014;28(2):95-106.
9. Deepmala, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294-321.
10. Minarini A, Ferrari S, Galletti M, et al. N-acetylcysteine in the treatment of psychiatric disorders: current status and future prospects. Expert Opin Drug Metab Toxicol. 2017;13(3):279-292.
11. Grandjean EM, Berthet P, Ruffman R, et al. Efficacy of oral long-term N‑acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22(2):209‑221.
12. Rhodes K, Braakhuis A. Performance and side effects of supplementation with N-acetylcysteine: a systematic review and meta-analysis. Sports Med. 2017;47(8):1619-1636.
13. Olsson B, Johansson M, Gabrielsson J, et al. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol. 1988;34(1):77-82.
14. United Nations Office on Drugs and Crime. World Drug Report 2016 (United Nations publication, Sales No. E.16.XI.7). https://www.unodc.org/doc/wdr2016/WORLD_DRUG_REPORT_2016_web.pdf. Published May 2016. Accessed April 26, 2018.
15. Amen SL, Piacentine LB, Ahmad ME, et al. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology. 2011;36(4):871-878.
16. Mardikian PN, LaRowe SD, Hedden S, et al. An open-label trial of N-acetylcysteine for the treatment of cocaine dependence: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(2):389-394.
17. LaRowe SD, Kalivas PW, Nicholas JS, et al. A double‐blind placebo‐controlled trial of N‐acetylcysteine in the treatment of cocaine dependence. Am J Addict. 2013;22(5):443-452.
18. Mousavi SG, Sharbafchi MR, Salehi M, et al. The efficacy of N-acetylcysteine in the treatment of methamphetamine dependence: a double-blind controlled, crossover study. Arch Iran Med. 2015;18(1):28-33.
19. Grant JE, Odlaug BL, Kim SW. A double-blind, placebo-controlled study of N-acetyl cysteine plus naltrexone for methamphetamine dependence. Eur Neuropsychopharmacol. 2010;20(11):823-828.
20. Lopez-Quintero C, Pérez de los Cobos J, Hasin DS, et al. Probability and predictors of transition from first use to dependence on nicotine, alcohol, cannabis, and cocaine: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Drug Alcohol Depend. 2011;115(1-2):120-130.
21. Chen CY, O’Brien MS, Anthony JC. Who becomes cannabis dependent soon after onset of use? Epidemiological evidence from the United States: 2000-2001. Drug Alcohol Depend. 2005;79(1):11-22.
22. Copersino ML, Boyd SJ, Tashkin DP, et al. Quitting among non-treatment-seeking marijuana users: reasons and changes in other substance use. Am J Addict. 2006;15(4):297-302.
23. Weiner MD, Sussman S, McCuller WJ, et al. Factors in marijuana cessation among high-risk youth. J Drug Educ. 1999;29(4):337-357.
24. Gray KM, Watson NL, Carpenter MJ, et al. N-acetylcysteine (NAC) in young marijuana users: an open-label pilot study. Am J Addict. 2010;19(2):187-189.
25. Gray KM, Carpenter MJ, Baker NL, et al. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169(8):805-812.
26. Roten AT, Baker NL, Gray KM. Marijuana craving trajectories in an adolescent marijuana cessation pharmacotherapy trial. Addict Behav. 2013;38(3):1788-1791.
27. Gray KM, Sonne SC, McClure EA, et al. A randomized placebo-controlled trial of N-acetylcysteine for cannabis use disorder in adults. Drug Alcohol Depend. 2017;177:249-257.
28. Rostron B. Mortality risks associated with environmental tobacco smoke exposure in the United States. Nicotine Tob Res. 2013;15(10):1722-1728.
29. Centers for Disease Control and Prevention. Quitting smoking among adults – United States, 2001–2010. MMWR. 2011;60(44):1513-1519.
30. McClure EA, Baker NL, Gipson CD, et al. An open-label pilot trial of N-acetylcysteine and varenicline in adult cigarette smokers. Am J Drug Alcohol Abuse. 2015;41(1):52-56.
31. Froeliger B, McConnell P, Stankeviciute N, et al. The effects of N-acetylcysteine on frontostriatal resting-state functional connectivity, withdrawal symptoms and smoking abstinence: a double-blind, placebo-controlled fMRI pilot study. Drug Alcohol Depend. 2015;156:234-242.
32. Prado E, Maes M, Piccoli LG, et al. N-acetylcysteine for therapy-resistant tobacco use disorder: a pilot study. Redox Rep. 2015;20(5):215-222.
33. Knackstedt LA, LaRowe S, Mardikian P, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65(10):841-845.
34. Grant JE, Odlaug BL, Chamberlain SR, et al. A randomized, placebo-controlled trial of N-acetylcysteine plus imaginal desensitization for nicotine-dependent pathological gamblers. J Clin Psychiatry. 2014;75(1):39-45.
35. Schmaal L, Berk L, Hulstijn KP, et al. Efficacy of N-acetylcysteine in the treatment of nicotine dependence: a double-blind placebo-controlled pilot study. Eur Addiction Res. 2011;17(4):211-216.
36. Bernardo M, Dodd S, Gama CS, et al. Effects of N‐acetylcysteine on substance use in bipolar disorder: a randomised placebo‐controlled clinical trial. Acta Neuropsychiatr. 2009;21(5):239-245.
37. McClure EA, Baker NL, Gray KM. Cigarette smoking during an N-acetylcysteine-assisted cannabis cessation trial in adolescents. Am J Drug Alcohol Abuse. 2014;40(4):285-291.
38. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM-5 alcohol use disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72(8):757-766.
39. Back SE, McCauley JL, Korte KJ, et al. A double-blind randomized controlled pilot trial of N-acetylcysteine in veterans with PTSD and substance use disorders. J Clin Psychiatry. 2016;77(11):e1439-e1446.
40. Squeglia LM, Baker NL, McClure EA, et al. Alcohol use during a trial of N-acetylcysteine for adolescent marijuana cessation. Addict Behav. 2016;63:172-177.
41. Lebourgeois S, González-Marín MC, Jeanblanc J, et al. Effect of N-acetylcysteine on motivation, seeking and relapse to ethanol self-administration. Addict Biol. 2018;23(2):643-652.
42. Schneider R Jr, Santos CF, Clarimundo V, et al. N-acetylcysteine prevents behavioral and biochemical changes induced by alcohol cessation in rats. Alcohol. 2015;49(3):259-263.
43. Parnell SE, Sulik KK, Dehart DB, et al. Reduction of ethanol-induced ocular abnormalities in mice via dietary administration of N-acetylcysteine. Alcohol. 2010;44(7-8):699-705.
44. Ozkol H, Bulut G, Balahoroglu R, et al. Protective effects of Selenium, N-acetylcysteine and Vitamin E against acute ethanol intoxication in rats. Biol Trace Elem Res. 2017;175(1):177-185.
45. Seiva FR, Amauchi JF, Rocha KK, et al. Alcoholism and alcohol abstinence: N-acetylcysteine to improve energy expenditure, myocardial oxidative stress, and energy metabolism in alcoholic heart disease. Alcohol. 2009;43(8):649-656.
46. Setshedi M, Longato L, Petersen DR, et al. Limited therapeutic effect of N‐acetylcysteine on hepatic insulin resistance in an experimental model of alcohol‐induced steatohepatitis. Alcohol Clin Exp Res. 2011;35(12):2139-2151.
47. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
1. Grella CE, Karno MP, Warda US, et al. Perceptions of need and help received for substance dependence in a national probability survey. Psychiatr Serv. 2009;60(8):1068-1074.
2. Everitt BJ, Robbins TW. Drug addiction: updating actions to habits to compulsions ten years on. Annu Rev Psychol. 2016;67:23-50.
3. McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23(8):3531-3537.
4. LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28(12):3170-3177.
5. Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16(10):974-986.
6. Roberts-Wolfe D, Kalivas PW. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745-756.
7. Berk M, Malhi GS, Gray LJ, et al. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34(3):167-177.
8. McClure EA, Gipson CD, Malcolm RJ, et al. Potential role of N-acetylcysteine in the management of substance use disorders. CNS drugs. 2014;28(2):95-106.
9. Deepmala, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294-321.
10. Minarini A, Ferrari S, Galletti M, et al. N-acetylcysteine in the treatment of psychiatric disorders: current status and future prospects. Expert Opin Drug Metab Toxicol. 2017;13(3):279-292.
11. Grandjean EM, Berthet P, Ruffman R, et al. Efficacy of oral long-term N‑acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22(2):209‑221.
12. Rhodes K, Braakhuis A. Performance and side effects of supplementation with N-acetylcysteine: a systematic review and meta-analysis. Sports Med. 2017;47(8):1619-1636.
13. Olsson B, Johansson M, Gabrielsson J, et al. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol. 1988;34(1):77-82.
14. United Nations Office on Drugs and Crime. World Drug Report 2016 (United Nations publication, Sales No. E.16.XI.7). https://www.unodc.org/doc/wdr2016/WORLD_DRUG_REPORT_2016_web.pdf. Published May 2016. Accessed April 26, 2018.
15. Amen SL, Piacentine LB, Ahmad ME, et al. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology. 2011;36(4):871-878.
16. Mardikian PN, LaRowe SD, Hedden S, et al. An open-label trial of N-acetylcysteine for the treatment of cocaine dependence: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(2):389-394.
17. LaRowe SD, Kalivas PW, Nicholas JS, et al. A double‐blind placebo‐controlled trial of N‐acetylcysteine in the treatment of cocaine dependence. Am J Addict. 2013;22(5):443-452.
18. Mousavi SG, Sharbafchi MR, Salehi M, et al. The efficacy of N-acetylcysteine in the treatment of methamphetamine dependence: a double-blind controlled, crossover study. Arch Iran Med. 2015;18(1):28-33.
19. Grant JE, Odlaug BL, Kim SW. A double-blind, placebo-controlled study of N-acetyl cysteine plus naltrexone for methamphetamine dependence. Eur Neuropsychopharmacol. 2010;20(11):823-828.
20. Lopez-Quintero C, Pérez de los Cobos J, Hasin DS, et al. Probability and predictors of transition from first use to dependence on nicotine, alcohol, cannabis, and cocaine: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Drug Alcohol Depend. 2011;115(1-2):120-130.
21. Chen CY, O’Brien MS, Anthony JC. Who becomes cannabis dependent soon after onset of use? Epidemiological evidence from the United States: 2000-2001. Drug Alcohol Depend. 2005;79(1):11-22.
22. Copersino ML, Boyd SJ, Tashkin DP, et al. Quitting among non-treatment-seeking marijuana users: reasons and changes in other substance use. Am J Addict. 2006;15(4):297-302.
23. Weiner MD, Sussman S, McCuller WJ, et al. Factors in marijuana cessation among high-risk youth. J Drug Educ. 1999;29(4):337-357.
24. Gray KM, Watson NL, Carpenter MJ, et al. N-acetylcysteine (NAC) in young marijuana users: an open-label pilot study. Am J Addict. 2010;19(2):187-189.
25. Gray KM, Carpenter MJ, Baker NL, et al. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169(8):805-812.
26. Roten AT, Baker NL, Gray KM. Marijuana craving trajectories in an adolescent marijuana cessation pharmacotherapy trial. Addict Behav. 2013;38(3):1788-1791.
27. Gray KM, Sonne SC, McClure EA, et al. A randomized placebo-controlled trial of N-acetylcysteine for cannabis use disorder in adults. Drug Alcohol Depend. 2017;177:249-257.
28. Rostron B. Mortality risks associated with environmental tobacco smoke exposure in the United States. Nicotine Tob Res. 2013;15(10):1722-1728.
29. Centers for Disease Control and Prevention. Quitting smoking among adults – United States, 2001–2010. MMWR. 2011;60(44):1513-1519.
30. McClure EA, Baker NL, Gipson CD, et al. An open-label pilot trial of N-acetylcysteine and varenicline in adult cigarette smokers. Am J Drug Alcohol Abuse. 2015;41(1):52-56.
31. Froeliger B, McConnell P, Stankeviciute N, et al. The effects of N-acetylcysteine on frontostriatal resting-state functional connectivity, withdrawal symptoms and smoking abstinence: a double-blind, placebo-controlled fMRI pilot study. Drug Alcohol Depend. 2015;156:234-242.
32. Prado E, Maes M, Piccoli LG, et al. N-acetylcysteine for therapy-resistant tobacco use disorder: a pilot study. Redox Rep. 2015;20(5):215-222.
33. Knackstedt LA, LaRowe S, Mardikian P, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65(10):841-845.
34. Grant JE, Odlaug BL, Chamberlain SR, et al. A randomized, placebo-controlled trial of N-acetylcysteine plus imaginal desensitization for nicotine-dependent pathological gamblers. J Clin Psychiatry. 2014;75(1):39-45.
35. Schmaal L, Berk L, Hulstijn KP, et al. Efficacy of N-acetylcysteine in the treatment of nicotine dependence: a double-blind placebo-controlled pilot study. Eur Addiction Res. 2011;17(4):211-216.
36. Bernardo M, Dodd S, Gama CS, et al. Effects of N‐acetylcysteine on substance use in bipolar disorder: a randomised placebo‐controlled clinical trial. Acta Neuropsychiatr. 2009;21(5):239-245.
37. McClure EA, Baker NL, Gray KM. Cigarette smoking during an N-acetylcysteine-assisted cannabis cessation trial in adolescents. Am J Drug Alcohol Abuse. 2014;40(4):285-291.
38. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM-5 alcohol use disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72(8):757-766.
39. Back SE, McCauley JL, Korte KJ, et al. A double-blind randomized controlled pilot trial of N-acetylcysteine in veterans with PTSD and substance use disorders. J Clin Psychiatry. 2016;77(11):e1439-e1446.
40. Squeglia LM, Baker NL, McClure EA, et al. Alcohol use during a trial of N-acetylcysteine for adolescent marijuana cessation. Addict Behav. 2016;63:172-177.
41. Lebourgeois S, González-Marín MC, Jeanblanc J, et al. Effect of N-acetylcysteine on motivation, seeking and relapse to ethanol self-administration. Addict Biol. 2018;23(2):643-652.
42. Schneider R Jr, Santos CF, Clarimundo V, et al. N-acetylcysteine prevents behavioral and biochemical changes induced by alcohol cessation in rats. Alcohol. 2015;49(3):259-263.
43. Parnell SE, Sulik KK, Dehart DB, et al. Reduction of ethanol-induced ocular abnormalities in mice via dietary administration of N-acetylcysteine. Alcohol. 2010;44(7-8):699-705.
44. Ozkol H, Bulut G, Balahoroglu R, et al. Protective effects of Selenium, N-acetylcysteine and Vitamin E against acute ethanol intoxication in rats. Biol Trace Elem Res. 2017;175(1):177-185.
45. Seiva FR, Amauchi JF, Rocha KK, et al. Alcoholism and alcohol abstinence: N-acetylcysteine to improve energy expenditure, myocardial oxidative stress, and energy metabolism in alcoholic heart disease. Alcohol. 2009;43(8):649-656.
46. Setshedi M, Longato L, Petersen DR, et al. Limited therapeutic effect of N‐acetylcysteine on hepatic insulin resistance in an experimental model of alcohol‐induced steatohepatitis. Alcohol Clin Exp Res. 2011;35(12):2139-2151.
47. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.

Repetitive transcranial magnetic stimulation for tic disorders
Tourette syndrome (TS) is a chronic neuropsychiatric disorder occurring in early childhood or adolescence that’s characterized by multiple motor and vocal tics that are usually preceded by premonitory urges.1,2 Usually, the tics are repetitive, sudden, stereotypical, non-rhythmic movements and/or vocalizations.3,4 Individuals with TS and other tic disorders often experience impulsivity, aggression, obsessive-compulsive disorder (OCD), attention-deficit/hyperactivity disorder, and various mood and anxiety disorders.3 Psychosocial issues may include having low self-esteem, increased family conflict, and poor social skills. Males are affected 3 to 5 times more often than females.3
There is no definitive treatment for TS. Commonly used interventions are pharmacotherapy and/or behavioral therapy, which includes supportive psychotherapy, habit reversal training, exposure with response prevention, relaxation therapy, cognitive-behavioral therapy, and self-monitoring. Pharmacotherapy for TS and other tic disorders consists mainly of antipsychotics such as haloperidol, pimozide, and aripiprazole, and alpha-2 agonists (guanfacine and clonidine).4,8-10 Unfortunately, not all children respond to these medications, and these agents are associated with multiple adverse effects.11 Therefore, there is a need for additional treatment options for patients with TS and other tic disorders, especially those who are not helped by conventional treatments.
Repetitive transcranial magnetic stimulation (rTMS) is a non-invasive therapeutic technique in which high-intensity magnetic impulses are delivered through an electromagnetic coil placed on the patient’s scalp to stimulate cortical neurons. The effect is determined by various parameters, including the intensity, frequency, pulse number, duration, coil location, and type of coil.3,8
rTMS is FDA-approved for treating depression, and has been used to treat anxiety disorders, Parkinson’s disease, chronic pain syndromes, and dystonia.12,13 Researchers have begun to evaluate the usefulness of rTMS for patients with TS or other tic disorders. In this article, we review the findings of 11 studies—9 clinical trials and 2 case studies—that evaluated rTMS as a treatment option for patients with tic disorders.
A proposed mechanism of action
TS is believed to be caused by multiple factors, including neurotransmitter imbalances and genetic, environmental, and psychosocial factors.14 Evidence strongly suggests the involvement of the motor cortex, basal ganglia, and reticular activating system in the expression of TS.2,15-17
Researchers have consistently identified networks of regions in the brain, including the supplementary motor area (SMA), that are active in the seconds before tics occur in patients with these disorders.6,18-22 The SMA modulates the way information is channeled between motor circuits, the limbic system, and the cognitive processes.3,23-26 The SMA can be used as a target for focal brain stimulation to modulate activity in those circuits and improve symptoms in resistant patients. Recent rTMS studies that targeted the SMA have found that stimulation to this area may be an effective way to treat TS.19,20,23,27
Continue to: rTMS for tics: Mixed evidence
rTMS for tics: Mixed evidence
We reviewed the results of 11 studies that described the use of rTMS for TS and other tic disorders (Table 11,24-26,28,29 and Table 23,8,23,27,30,31). They included:
- 2 double-blind, randomized controlled trials28,29
- 2 single-blind trials24-26
- 1 double-blind trial with an open-label extension1
- 4 open-label studies3,8,23,30
- 1 case series27 and 1 case report.31

Study characteristics. In the 11 studies we reviewed, the duration of rTMS treatment varied from 2 days to 4 weeks. The pulses used were 900, 1,200, 1,800, and 2,400 per day, and the frequencies were 1 Hz, 4 Hz, 15 Hz and 30 Hz. Seven studies did not use placebo- or sham-controlled arms.1,3,8,23,27,30,31

Efficacy. Two double-blind trials28,29 found no significant improvement in tic severity in patients treated with rTMS (P = .066 and P = .43, respectively). In addition, the 2 single-blind studies showed no beneficial effects of rTMS for patients with tics (P > .05).24-26 However, 3 of the 4 open-label studies found a significant improvement in tics.3,23,30 In one of the double-blind trials, researchers added an open-label extension phase.1 They found no significant results in the double-blind phase of the study (P = .27), but in the open-label phase, patients experienced a significant improvement in tic severity (P = .04).1 Lastly, the case series and case report found an improvement in tic severity and improvement in TS symptoms, respectively, with rTMS treatment.
rTMS might also improve symptoms of OCD that may co-occur with TS.8,23,28 Two studies found significant improvement in tic severity in a subgroup of patients suffering from comorbid OCD.8,28
Continue to: Safety profile and adverse effects
Safety profile and adverse effects. In the studies we reviewed, the adverse effects associated with rTMS included headache (45%),1,8,24,26,28,29 scalp pain (18%),8,30 self-injurious crisis (9%),31 abdominal pain (9%),29 red eyes (9%),29 neck pain (9%),1 muscle sprain (9%),1 tiredness (9%),24,26 and increase in motor excitability (9%).28 There were no severe adverse effects reported in any of the studies. The self-injurious crisis reported by a patient early in one study as a seizure was later ruled out after careful clinical and electroencephalographic evaluation. This patient demonstrated self-injurious behaviors prior to the treatment, and overall there was a reduction in frequency and intensity of self-injurious behavior as well as an improvement in tics.31
Dissimilar studies
There was great heterogeneity among the 11 studies we reviewed. One case series27 and one case report31 found significant improvement in tics, but these studies did not have control groups. Both studies employed rTMS with a frequency of 1 Hz and between 900 to 1,200 pulses per day. Three open-label studies that found significant improvement in tic severity used the same frequency of stimulation (1 Hz with 1,200 pulses per day).3,23,30 All studies we analyzed differed in the total number of rTMS sessions and number of trains per stimulation.
The studies also differed in terms of the age of the participants. Some studies focused primarily on pediatric patients,3,30 but many of them also included adults. The main limitations of the 11 studies included a small sample size,1,3,8,23-25,28-30 no placebo or controlled arm,1,3,8,23,27,30,31 concomitant psychiatric comorbidities8,28,29 or medications,1,3,23,29,30 low stimulation intensity,24-26 and use of short trains24,26 or unilateral cerebral stimulation.24,26 Among the blinded studies, limitations included a small sample size, prior medications used, comorbidities, low stimulation intensity, and high rTMS dose.1,24-26,28,29
A possible option for treatment-resistant tics
We cannot offer a definitive conclusion on the safety and effectiveness of rTMS for the treatment of TS and other tic disorders because of the inconsistent results, heterogeneity, and small sample sizes of the studies we analyzed. Higher-quality studies failed to find evidence supporting the use of rTMS for treating TS and other tics disorders, but open-label studies and case reports found significant improvements. In light of this evidence and the treatment’s relatively favorable adverse-effects profile, rTMS might be an option for certain patients with treatment-resistant tics, particularly those with comorbid OCD symptoms.
Continue to: Bottom Line
Bottom Line
The evidence for using repetitive transcranial stimulation (rTMS) to treat patients with Tourette syndrome and other tic disorders is mixed. Higher-quality studies have found no significant improvements, whereas open-label studies and case studies have. Although not recommended for the routine treatment of tic disorders, rTMS may be an option for patients with treatment-resistant tics, particularly those with comorbid obsessive-compulsive symptoms.
Related Resources
- Tourette Association of America. https://www.tourette.org/.
- Harris E. Children with tic disorders: How to match treatment with symptoms. Current Psychiatry. 2010;9(3):29-36.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres, Duraclon
Guanfacine • Intuniv, Tenex
Haloperidol • Haldol
Pimozide • Orap
1. Landeros-Weisenberger A, Mantovani A, Motlagh MG, et al. Randomized sham controlled double-blind trial of repetitive transcranial magnetic stimulation for adults with severe Tourette syndrome. Brain Stimulat. 2015;8(3):574-581.
2. Kamble N, Netravathi M, Pal PK. Therapeutic applications of repetitive transcranial magnetic stimulation (rTMS) in movement disorders: a review. Parkinsonism Relat Disord. 2014;20(7):695-707.
3. Le K, Liu L, Sun M, et al. Transcranial magnetic stimulation at 1 Hertz improves clinical symptoms in children with Tourette syndrome for at least 6 months. J Clin Neurosci. 2013;20(2):257-262.
4. Cavanna AE, Seri S. Tourette’s syndrome. BMJ. 2013;347:f4964. doi:10.1136/bmj.f4964.
5. Leckman JF, Bloch MH, Scahill L, et al. Tourette syndrome: the self under siege. J Child Neurol. 2006;21(8):642-649.
6. Bloch MH, Peterson BS, Scahill L, et al. Adulthood outcome of tic and obsessive-compulsive symptom severity in children with Tourette syndrome. Arch Pediatr Adolesc Med. 2006;160(1):65-69.
7. Bloch M, State M, Pittenger C. Recent advances in Tourette syndrome. Curr Opin Neurol. 2011;24(2):119-125.
8. Bloch Y, Arad S, Levkovitz Y. Deep TMS add-on treatment for intractable Tourette syndrome: a feasibility study. World J Biol Psychiatry. 2016;17(7):557-561.
9. Robertson MM. The Gilles de la Tourette syndrome: the current status. Arch Dis Child Educ Pract Ed. 2012;97(5):166-175.
10. Párraga HC, Harris KM, Párraga KL, et al. An overview of the treatment of Tourette’s disorder and tics. J Child Adolesc Psychopharmacol. 2010;20(4):249-262.
11. Du JC, Chiu TF, Lee KM, et al. Tourette syndrome in children: an updated review. Pediatr Neonatol. 2010;51(5):255-264.
12. Malizia AL. What do brain imaging studies tell us about anxiety disorders? J Psychopharmacol. 1999;13(4):372-378.
13. Di Lazzaro V, Oliviero A, Berardelli A, et al. Direct demonstration of the effects of repetitive transcranial magnetic stimulation on the excitability of the human motor cortex. Exp Brain Res. 2002;144(4):549-553.
14. Olson LL, Singer HS, Goodman WK, et al. Tourette syndrome: diagnosis, strategies, therapies, pathogenesis, and future research directions. J Child Neurol. 2006;21(8):630-641.
15. Gerard E, Peterson BS. Developmental processes and brain imaging studies in Tourette syndrome. J Psychosom Res. 2003;55(1):13-22.
16. Kurlan R. Hypothesis II: Tourette’s syndrome is part of a clinical spectrum that includes normal brain development. Arch Neurol. 1994;51(11):1145-1150.
17. Peterson BS. Neuroimaging in child and adolescent neuropsychiatric disorders. J Am Acad Child Adolesc Psychiatry. 1995;34(12):1560-1576.
18. Sheppard DM, Bradshaw JL, Purcell R, et al. Tourette’s and comorbid syndromes: obsessive compulsive and attention deficit hyperactivity disorder. A common etiology? Clin Psychol Rev. 1999;19(5):531-552.
19. Bohlhalter S, Goldfine A, Matteson S, et al. Neural correlates of tic generation in Tourette syndrome: an event-related functional MRI study. Brain. 2006;129(pt 8):2029-2037.
20. Hampson M, Tokoglu F, King RA, et al. Brain areas coactivating with motor cortex during chronic motor tics and intentional movements. Biol Psychiatry. 2009;65(7):594-599.
21. Eichele H, Plessen KJ. Neural plasticity in functional and anatomical MRI studies of children with Tourette syndrome. Behav Neurol. 2013;27(1):33-45.
22. Neuner I, Schneider F, Shah NJ. Functional neuroanatomy of tics. Int Rev Neurobiol. 2013;112:35-71.
23. Mantovani A, Lisanby SH, Pieraccini F, et al. Repetitive transcranial magnetic stimulation (rTMS) in the treatment of obsessive-compulsive disorder (OCD) and Tourette’s syndrome (TS). Int J Neuropsychopharmacol. 2006;9(1):95-100.
24. Münchau A, Bloem BR, Thilo KV, et al. Repetitive transcranial magnetic stimulation for Tourette syndrome. Neurology. 2002;59(11):1789-1791.
25. Orth M, Kirby R, Richardson MP, et al. Subthreshold rTMS over pre-motor cortex has no effect on tics in patients with Gilles de la Tourette syndrome. Clin Neurophysiol. 2005;116(4):764-768.
26. Snijders AH, Bloem BR, Orth M, et al. Video assessment of rTMS for Tourette syndrome. J Neurol Neurosurg Psychiatry. 2005;76(12):1743-1744.
27. Mantovani A, Leckman JF, Grantz H, et al. Repetitive transcranial magnetic stimulation of the supplementary motor area in the treatment of Tourette syndrome: report of two cases. Clin Neurophysiol. 2007;118(10):2314-2315.
28. Chae JH, Nahas Z, Wassermann E, et al. A pilot safety study of repetitive transcranial magnetic stimulation (rTMS) in Tourette’s syndrome. Cogn Behav Neurol. 2004;17(2):109-117.
29. Wu SW, Maloney T, Gilbert DL, et al. Functional MRI-navigated repetitive transcranial magnetic stimulation over supplementary motor area in chronic tic disorders. Brain Stimul. 2014;7(2):212-218.
30. Kwon HJ, Lim WS, Lim MH, et al. 1-Hz low frequency repetitive transcranial magnetic stimulation in children with Tourette’s syndrome. Neurosci Lett. 2011;492(1):1-4.
31. Salatino A, Momo E, Nobili M, et al. Awareness of symptoms amelioration following low-frequency repetitive transcranial magnetic stimulation in a patient with Tourette syndrome and comorbid obsessive-compulsive disorder. Brain Stimulat. 2014;7(2):341-343.
Tourette syndrome (TS) is a chronic neuropsychiatric disorder occurring in early childhood or adolescence that’s characterized by multiple motor and vocal tics that are usually preceded by premonitory urges.1,2 Usually, the tics are repetitive, sudden, stereotypical, non-rhythmic movements and/or vocalizations.3,4 Individuals with TS and other tic disorders often experience impulsivity, aggression, obsessive-compulsive disorder (OCD), attention-deficit/hyperactivity disorder, and various mood and anxiety disorders.3 Psychosocial issues may include having low self-esteem, increased family conflict, and poor social skills. Males are affected 3 to 5 times more often than females.3
There is no definitive treatment for TS. Commonly used interventions are pharmacotherapy and/or behavioral therapy, which includes supportive psychotherapy, habit reversal training, exposure with response prevention, relaxation therapy, cognitive-behavioral therapy, and self-monitoring. Pharmacotherapy for TS and other tic disorders consists mainly of antipsychotics such as haloperidol, pimozide, and aripiprazole, and alpha-2 agonists (guanfacine and clonidine).4,8-10 Unfortunately, not all children respond to these medications, and these agents are associated with multiple adverse effects.11 Therefore, there is a need for additional treatment options for patients with TS and other tic disorders, especially those who are not helped by conventional treatments.
Repetitive transcranial magnetic stimulation (rTMS) is a non-invasive therapeutic technique in which high-intensity magnetic impulses are delivered through an electromagnetic coil placed on the patient’s scalp to stimulate cortical neurons. The effect is determined by various parameters, including the intensity, frequency, pulse number, duration, coil location, and type of coil.3,8
rTMS is FDA-approved for treating depression, and has been used to treat anxiety disorders, Parkinson’s disease, chronic pain syndromes, and dystonia.12,13 Researchers have begun to evaluate the usefulness of rTMS for patients with TS or other tic disorders. In this article, we review the findings of 11 studies—9 clinical trials and 2 case studies—that evaluated rTMS as a treatment option for patients with tic disorders.
A proposed mechanism of action
TS is believed to be caused by multiple factors, including neurotransmitter imbalances and genetic, environmental, and psychosocial factors.14 Evidence strongly suggests the involvement of the motor cortex, basal ganglia, and reticular activating system in the expression of TS.2,15-17
Researchers have consistently identified networks of regions in the brain, including the supplementary motor area (SMA), that are active in the seconds before tics occur in patients with these disorders.6,18-22 The SMA modulates the way information is channeled between motor circuits, the limbic system, and the cognitive processes.3,23-26 The SMA can be used as a target for focal brain stimulation to modulate activity in those circuits and improve symptoms in resistant patients. Recent rTMS studies that targeted the SMA have found that stimulation to this area may be an effective way to treat TS.19,20,23,27
Continue to: rTMS for tics: Mixed evidence
rTMS for tics: Mixed evidence
We reviewed the results of 11 studies that described the use of rTMS for TS and other tic disorders (Table 11,24-26,28,29 and Table 23,8,23,27,30,31). They included:
- 2 double-blind, randomized controlled trials28,29
- 2 single-blind trials24-26
- 1 double-blind trial with an open-label extension1
- 4 open-label studies3,8,23,30
- 1 case series27 and 1 case report.31
Study characteristics. In the 11 studies we reviewed, the duration of rTMS treatment varied from 2 days to 4 weeks. The pulses used were 900, 1,200, 1,800, and 2,400 per day, and the frequencies were 1 Hz, 4 Hz, 15 Hz and 30 Hz. Seven studies did not use placebo- or sham-controlled arms.1,3,8,23,27,30,31
Efficacy. Two double-blind trials28,29 found no significant improvement in tic severity in patients treated with rTMS (P = .066 and P = .43, respectively). In addition, the 2 single-blind studies showed no beneficial effects of rTMS for patients with tics (P > .05).24-26 However, 3 of the 4 open-label studies found a significant improvement in tics.3,23,30 In one of the double-blind trials, researchers added an open-label extension phase.1 They found no significant results in the double-blind phase of the study (P = .27), but in the open-label phase, patients experienced a significant improvement in tic severity (P = .04).1 Lastly, the case series and case report found an improvement in tic severity and improvement in TS symptoms, respectively, with rTMS treatment.
rTMS might also improve symptoms of OCD that may co-occur with TS.8,23,28 Two studies found significant improvement in tic severity in a subgroup of patients suffering from comorbid OCD.8,28
Continue to: Safety profile and adverse effects
Safety profile and adverse effects. In the studies we reviewed, the adverse effects associated with rTMS included headache (45%),1,8,24,26,28,29 scalp pain (18%),8,30 self-injurious crisis (9%),31 abdominal pain (9%),29 red eyes (9%),29 neck pain (9%),1 muscle sprain (9%),1 tiredness (9%),24,26 and increase in motor excitability (9%).28 There were no severe adverse effects reported in any of the studies. The self-injurious crisis reported by a patient early in one study as a seizure was later ruled out after careful clinical and electroencephalographic evaluation. This patient demonstrated self-injurious behaviors prior to the treatment, and overall there was a reduction in frequency and intensity of self-injurious behavior as well as an improvement in tics.31
Dissimilar studies
There was great heterogeneity among the 11 studies we reviewed. One case series27 and one case report31 found significant improvement in tics, but these studies did not have control groups. Both studies employed rTMS with a frequency of 1 Hz and between 900 to 1,200 pulses per day. Three open-label studies that found significant improvement in tic severity used the same frequency of stimulation (1 Hz with 1,200 pulses per day).3,23,30 All studies we analyzed differed in the total number of rTMS sessions and number of trains per stimulation.
The studies also differed in terms of the age of the participants. Some studies focused primarily on pediatric patients,3,30 but many of them also included adults. The main limitations of the 11 studies included a small sample size,1,3,8,23-25,28-30 no placebo or controlled arm,1,3,8,23,27,30,31 concomitant psychiatric comorbidities8,28,29 or medications,1,3,23,29,30 low stimulation intensity,24-26 and use of short trains24,26 or unilateral cerebral stimulation.24,26 Among the blinded studies, limitations included a small sample size, prior medications used, comorbidities, low stimulation intensity, and high rTMS dose.1,24-26,28,29
A possible option for treatment-resistant tics
We cannot offer a definitive conclusion on the safety and effectiveness of rTMS for the treatment of TS and other tic disorders because of the inconsistent results, heterogeneity, and small sample sizes of the studies we analyzed. Higher-quality studies failed to find evidence supporting the use of rTMS for treating TS and other tics disorders, but open-label studies and case reports found significant improvements. In light of this evidence and the treatment’s relatively favorable adverse-effects profile, rTMS might be an option for certain patients with treatment-resistant tics, particularly those with comorbid OCD symptoms.
Continue to: Bottom Line
Bottom Line
The evidence for using repetitive transcranial stimulation (rTMS) to treat patients with Tourette syndrome and other tic disorders is mixed. Higher-quality studies have found no significant improvements, whereas open-label studies and case studies have. Although not recommended for the routine treatment of tic disorders, rTMS may be an option for patients with treatment-resistant tics, particularly those with comorbid obsessive-compulsive symptoms.
Related Resources
- Tourette Association of America. https://www.tourette.org/.
- Harris E. Children with tic disorders: How to match treatment with symptoms. Current Psychiatry. 2010;9(3):29-36.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres, Duraclon
Guanfacine • Intuniv, Tenex
Haloperidol • Haldol
Pimozide • Orap
Tourette syndrome (TS) is a chronic neuropsychiatric disorder occurring in early childhood or adolescence that’s characterized by multiple motor and vocal tics that are usually preceded by premonitory urges.1,2 Usually, the tics are repetitive, sudden, stereotypical, non-rhythmic movements and/or vocalizations.3,4 Individuals with TS and other tic disorders often experience impulsivity, aggression, obsessive-compulsive disorder (OCD), attention-deficit/hyperactivity disorder, and various mood and anxiety disorders.3 Psychosocial issues may include having low self-esteem, increased family conflict, and poor social skills. Males are affected 3 to 5 times more often than females.3
There is no definitive treatment for TS. Commonly used interventions are pharmacotherapy and/or behavioral therapy, which includes supportive psychotherapy, habit reversal training, exposure with response prevention, relaxation therapy, cognitive-behavioral therapy, and self-monitoring. Pharmacotherapy for TS and other tic disorders consists mainly of antipsychotics such as haloperidol, pimozide, and aripiprazole, and alpha-2 agonists (guanfacine and clonidine).4,8-10 Unfortunately, not all children respond to these medications, and these agents are associated with multiple adverse effects.11 Therefore, there is a need for additional treatment options for patients with TS and other tic disorders, especially those who are not helped by conventional treatments.
Repetitive transcranial magnetic stimulation (rTMS) is a non-invasive therapeutic technique in which high-intensity magnetic impulses are delivered through an electromagnetic coil placed on the patient’s scalp to stimulate cortical neurons. The effect is determined by various parameters, including the intensity, frequency, pulse number, duration, coil location, and type of coil.3,8
rTMS is FDA-approved for treating depression, and has been used to treat anxiety disorders, Parkinson’s disease, chronic pain syndromes, and dystonia.12,13 Researchers have begun to evaluate the usefulness of rTMS for patients with TS or other tic disorders. In this article, we review the findings of 11 studies—9 clinical trials and 2 case studies—that evaluated rTMS as a treatment option for patients with tic disorders.
A proposed mechanism of action
TS is believed to be caused by multiple factors, including neurotransmitter imbalances and genetic, environmental, and psychosocial factors.14 Evidence strongly suggests the involvement of the motor cortex, basal ganglia, and reticular activating system in the expression of TS.2,15-17
Researchers have consistently identified networks of regions in the brain, including the supplementary motor area (SMA), that are active in the seconds before tics occur in patients with these disorders.6,18-22 The SMA modulates the way information is channeled between motor circuits, the limbic system, and the cognitive processes.3,23-26 The SMA can be used as a target for focal brain stimulation to modulate activity in those circuits and improve symptoms in resistant patients. Recent rTMS studies that targeted the SMA have found that stimulation to this area may be an effective way to treat TS.19,20,23,27
Continue to: rTMS for tics: Mixed evidence
rTMS for tics: Mixed evidence
We reviewed the results of 11 studies that described the use of rTMS for TS and other tic disorders (Table 11,24-26,28,29 and Table 23,8,23,27,30,31). They included:
- 2 double-blind, randomized controlled trials28,29
- 2 single-blind trials24-26
- 1 double-blind trial with an open-label extension1
- 4 open-label studies3,8,23,30
- 1 case series27 and 1 case report.31
Study characteristics. In the 11 studies we reviewed, the duration of rTMS treatment varied from 2 days to 4 weeks. The pulses used were 900, 1,200, 1,800, and 2,400 per day, and the frequencies were 1 Hz, 4 Hz, 15 Hz and 30 Hz. Seven studies did not use placebo- or sham-controlled arms.1,3,8,23,27,30,31
Efficacy. Two double-blind trials28,29 found no significant improvement in tic severity in patients treated with rTMS (P = .066 and P = .43, respectively). In addition, the 2 single-blind studies showed no beneficial effects of rTMS for patients with tics (P > .05).24-26 However, 3 of the 4 open-label studies found a significant improvement in tics.3,23,30 In one of the double-blind trials, researchers added an open-label extension phase.1 They found no significant results in the double-blind phase of the study (P = .27), but in the open-label phase, patients experienced a significant improvement in tic severity (P = .04).1 Lastly, the case series and case report found an improvement in tic severity and improvement in TS symptoms, respectively, with rTMS treatment.
rTMS might also improve symptoms of OCD that may co-occur with TS.8,23,28 Two studies found significant improvement in tic severity in a subgroup of patients suffering from comorbid OCD.8,28
Continue to: Safety profile and adverse effects
Safety profile and adverse effects. In the studies we reviewed, the adverse effects associated with rTMS included headache (45%),1,8,24,26,28,29 scalp pain (18%),8,30 self-injurious crisis (9%),31 abdominal pain (9%),29 red eyes (9%),29 neck pain (9%),1 muscle sprain (9%),1 tiredness (9%),24,26 and increase in motor excitability (9%).28 There were no severe adverse effects reported in any of the studies. The self-injurious crisis reported by a patient early in one study as a seizure was later ruled out after careful clinical and electroencephalographic evaluation. This patient demonstrated self-injurious behaviors prior to the treatment, and overall there was a reduction in frequency and intensity of self-injurious behavior as well as an improvement in tics.31
Dissimilar studies
There was great heterogeneity among the 11 studies we reviewed. One case series27 and one case report31 found significant improvement in tics, but these studies did not have control groups. Both studies employed rTMS with a frequency of 1 Hz and between 900 to 1,200 pulses per day. Three open-label studies that found significant improvement in tic severity used the same frequency of stimulation (1 Hz with 1,200 pulses per day).3,23,30 All studies we analyzed differed in the total number of rTMS sessions and number of trains per stimulation.
The studies also differed in terms of the age of the participants. Some studies focused primarily on pediatric patients,3,30 but many of them also included adults. The main limitations of the 11 studies included a small sample size,1,3,8,23-25,28-30 no placebo or controlled arm,1,3,8,23,27,30,31 concomitant psychiatric comorbidities8,28,29 or medications,1,3,23,29,30 low stimulation intensity,24-26 and use of short trains24,26 or unilateral cerebral stimulation.24,26 Among the blinded studies, limitations included a small sample size, prior medications used, comorbidities, low stimulation intensity, and high rTMS dose.1,24-26,28,29
A possible option for treatment-resistant tics
We cannot offer a definitive conclusion on the safety and effectiveness of rTMS for the treatment of TS and other tic disorders because of the inconsistent results, heterogeneity, and small sample sizes of the studies we analyzed. Higher-quality studies failed to find evidence supporting the use of rTMS for treating TS and other tics disorders, but open-label studies and case reports found significant improvements. In light of this evidence and the treatment’s relatively favorable adverse-effects profile, rTMS might be an option for certain patients with treatment-resistant tics, particularly those with comorbid OCD symptoms.
Continue to: Bottom Line
Bottom Line
The evidence for using repetitive transcranial stimulation (rTMS) to treat patients with Tourette syndrome and other tic disorders is mixed. Higher-quality studies have found no significant improvements, whereas open-label studies and case studies have. Although not recommended for the routine treatment of tic disorders, rTMS may be an option for patients with treatment-resistant tics, particularly those with comorbid obsessive-compulsive symptoms.
Related Resources
- Tourette Association of America. https://www.tourette.org/.
- Harris E. Children with tic disorders: How to match treatment with symptoms. Current Psychiatry. 2010;9(3):29-36.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres, Duraclon
Guanfacine • Intuniv, Tenex
Haloperidol • Haldol
Pimozide • Orap
1. Landeros-Weisenberger A, Mantovani A, Motlagh MG, et al. Randomized sham controlled double-blind trial of repetitive transcranial magnetic stimulation for adults with severe Tourette syndrome. Brain Stimulat. 2015;8(3):574-581.
2. Kamble N, Netravathi M, Pal PK. Therapeutic applications of repetitive transcranial magnetic stimulation (rTMS) in movement disorders: a review. Parkinsonism Relat Disord. 2014;20(7):695-707.
3. Le K, Liu L, Sun M, et al. Transcranial magnetic stimulation at 1 Hertz improves clinical symptoms in children with Tourette syndrome for at least 6 months. J Clin Neurosci. 2013;20(2):257-262.
4. Cavanna AE, Seri S. Tourette’s syndrome. BMJ. 2013;347:f4964. doi:10.1136/bmj.f4964.
5. Leckman JF, Bloch MH, Scahill L, et al. Tourette syndrome: the self under siege. J Child Neurol. 2006;21(8):642-649.
6. Bloch MH, Peterson BS, Scahill L, et al. Adulthood outcome of tic and obsessive-compulsive symptom severity in children with Tourette syndrome. Arch Pediatr Adolesc Med. 2006;160(1):65-69.
7. Bloch M, State M, Pittenger C. Recent advances in Tourette syndrome. Curr Opin Neurol. 2011;24(2):119-125.
8. Bloch Y, Arad S, Levkovitz Y. Deep TMS add-on treatment for intractable Tourette syndrome: a feasibility study. World J Biol Psychiatry. 2016;17(7):557-561.
9. Robertson MM. The Gilles de la Tourette syndrome: the current status. Arch Dis Child Educ Pract Ed. 2012;97(5):166-175.
10. Párraga HC, Harris KM, Párraga KL, et al. An overview of the treatment of Tourette’s disorder and tics. J Child Adolesc Psychopharmacol. 2010;20(4):249-262.
11. Du JC, Chiu TF, Lee KM, et al. Tourette syndrome in children: an updated review. Pediatr Neonatol. 2010;51(5):255-264.
12. Malizia AL. What do brain imaging studies tell us about anxiety disorders? J Psychopharmacol. 1999;13(4):372-378.
13. Di Lazzaro V, Oliviero A, Berardelli A, et al. Direct demonstration of the effects of repetitive transcranial magnetic stimulation on the excitability of the human motor cortex. Exp Brain Res. 2002;144(4):549-553.
14. Olson LL, Singer HS, Goodman WK, et al. Tourette syndrome: diagnosis, strategies, therapies, pathogenesis, and future research directions. J Child Neurol. 2006;21(8):630-641.
15. Gerard E, Peterson BS. Developmental processes and brain imaging studies in Tourette syndrome. J Psychosom Res. 2003;55(1):13-22.
16. Kurlan R. Hypothesis II: Tourette’s syndrome is part of a clinical spectrum that includes normal brain development. Arch Neurol. 1994;51(11):1145-1150.
17. Peterson BS. Neuroimaging in child and adolescent neuropsychiatric disorders. J Am Acad Child Adolesc Psychiatry. 1995;34(12):1560-1576.
18. Sheppard DM, Bradshaw JL, Purcell R, et al. Tourette’s and comorbid syndromes: obsessive compulsive and attention deficit hyperactivity disorder. A common etiology? Clin Psychol Rev. 1999;19(5):531-552.
19. Bohlhalter S, Goldfine A, Matteson S, et al. Neural correlates of tic generation in Tourette syndrome: an event-related functional MRI study. Brain. 2006;129(pt 8):2029-2037.
20. Hampson M, Tokoglu F, King RA, et al. Brain areas coactivating with motor cortex during chronic motor tics and intentional movements. Biol Psychiatry. 2009;65(7):594-599.
21. Eichele H, Plessen KJ. Neural plasticity in functional and anatomical MRI studies of children with Tourette syndrome. Behav Neurol. 2013;27(1):33-45.
22. Neuner I, Schneider F, Shah NJ. Functional neuroanatomy of tics. Int Rev Neurobiol. 2013;112:35-71.
23. Mantovani A, Lisanby SH, Pieraccini F, et al. Repetitive transcranial magnetic stimulation (rTMS) in the treatment of obsessive-compulsive disorder (OCD) and Tourette’s syndrome (TS). Int J Neuropsychopharmacol. 2006;9(1):95-100.
24. Münchau A, Bloem BR, Thilo KV, et al. Repetitive transcranial magnetic stimulation for Tourette syndrome. Neurology. 2002;59(11):1789-1791.
25. Orth M, Kirby R, Richardson MP, et al. Subthreshold rTMS over pre-motor cortex has no effect on tics in patients with Gilles de la Tourette syndrome. Clin Neurophysiol. 2005;116(4):764-768.
26. Snijders AH, Bloem BR, Orth M, et al. Video assessment of rTMS for Tourette syndrome. J Neurol Neurosurg Psychiatry. 2005;76(12):1743-1744.
27. Mantovani A, Leckman JF, Grantz H, et al. Repetitive transcranial magnetic stimulation of the supplementary motor area in the treatment of Tourette syndrome: report of two cases. Clin Neurophysiol. 2007;118(10):2314-2315.
28. Chae JH, Nahas Z, Wassermann E, et al. A pilot safety study of repetitive transcranial magnetic stimulation (rTMS) in Tourette’s syndrome. Cogn Behav Neurol. 2004;17(2):109-117.
29. Wu SW, Maloney T, Gilbert DL, et al. Functional MRI-navigated repetitive transcranial magnetic stimulation over supplementary motor area in chronic tic disorders. Brain Stimul. 2014;7(2):212-218.
30. Kwon HJ, Lim WS, Lim MH, et al. 1-Hz low frequency repetitive transcranial magnetic stimulation in children with Tourette’s syndrome. Neurosci Lett. 2011;492(1):1-4.
31. Salatino A, Momo E, Nobili M, et al. Awareness of symptoms amelioration following low-frequency repetitive transcranial magnetic stimulation in a patient with Tourette syndrome and comorbid obsessive-compulsive disorder. Brain Stimulat. 2014;7(2):341-343.
1. Landeros-Weisenberger A, Mantovani A, Motlagh MG, et al. Randomized sham controlled double-blind trial of repetitive transcranial magnetic stimulation for adults with severe Tourette syndrome. Brain Stimulat. 2015;8(3):574-581.
2. Kamble N, Netravathi M, Pal PK. Therapeutic applications of repetitive transcranial magnetic stimulation (rTMS) in movement disorders: a review. Parkinsonism Relat Disord. 2014;20(7):695-707.
3. Le K, Liu L, Sun M, et al. Transcranial magnetic stimulation at 1 Hertz improves clinical symptoms in children with Tourette syndrome for at least 6 months. J Clin Neurosci. 2013;20(2):257-262.
4. Cavanna AE, Seri S. Tourette’s syndrome. BMJ. 2013;347:f4964. doi:10.1136/bmj.f4964.
5. Leckman JF, Bloch MH, Scahill L, et al. Tourette syndrome: the self under siege. J Child Neurol. 2006;21(8):642-649.
6. Bloch MH, Peterson BS, Scahill L, et al. Adulthood outcome of tic and obsessive-compulsive symptom severity in children with Tourette syndrome. Arch Pediatr Adolesc Med. 2006;160(1):65-69.
7. Bloch M, State M, Pittenger C. Recent advances in Tourette syndrome. Curr Opin Neurol. 2011;24(2):119-125.
8. Bloch Y, Arad S, Levkovitz Y. Deep TMS add-on treatment for intractable Tourette syndrome: a feasibility study. World J Biol Psychiatry. 2016;17(7):557-561.
9. Robertson MM. The Gilles de la Tourette syndrome: the current status. Arch Dis Child Educ Pract Ed. 2012;97(5):166-175.
10. Párraga HC, Harris KM, Párraga KL, et al. An overview of the treatment of Tourette’s disorder and tics. J Child Adolesc Psychopharmacol. 2010;20(4):249-262.
11. Du JC, Chiu TF, Lee KM, et al. Tourette syndrome in children: an updated review. Pediatr Neonatol. 2010;51(5):255-264.
12. Malizia AL. What do brain imaging studies tell us about anxiety disorders? J Psychopharmacol. 1999;13(4):372-378.
13. Di Lazzaro V, Oliviero A, Berardelli A, et al. Direct demonstration of the effects of repetitive transcranial magnetic stimulation on the excitability of the human motor cortex. Exp Brain Res. 2002;144(4):549-553.
14. Olson LL, Singer HS, Goodman WK, et al. Tourette syndrome: diagnosis, strategies, therapies, pathogenesis, and future research directions. J Child Neurol. 2006;21(8):630-641.
15. Gerard E, Peterson BS. Developmental processes and brain imaging studies in Tourette syndrome. J Psychosom Res. 2003;55(1):13-22.
16. Kurlan R. Hypothesis II: Tourette’s syndrome is part of a clinical spectrum that includes normal brain development. Arch Neurol. 1994;51(11):1145-1150.
17. Peterson BS. Neuroimaging in child and adolescent neuropsychiatric disorders. J Am Acad Child Adolesc Psychiatry. 1995;34(12):1560-1576.
18. Sheppard DM, Bradshaw JL, Purcell R, et al. Tourette’s and comorbid syndromes: obsessive compulsive and attention deficit hyperactivity disorder. A common etiology? Clin Psychol Rev. 1999;19(5):531-552.
19. Bohlhalter S, Goldfine A, Matteson S, et al. Neural correlates of tic generation in Tourette syndrome: an event-related functional MRI study. Brain. 2006;129(pt 8):2029-2037.
20. Hampson M, Tokoglu F, King RA, et al. Brain areas coactivating with motor cortex during chronic motor tics and intentional movements. Biol Psychiatry. 2009;65(7):594-599.
21. Eichele H, Plessen KJ. Neural plasticity in functional and anatomical MRI studies of children with Tourette syndrome. Behav Neurol. 2013;27(1):33-45.
22. Neuner I, Schneider F, Shah NJ. Functional neuroanatomy of tics. Int Rev Neurobiol. 2013;112:35-71.
23. Mantovani A, Lisanby SH, Pieraccini F, et al. Repetitive transcranial magnetic stimulation (rTMS) in the treatment of obsessive-compulsive disorder (OCD) and Tourette’s syndrome (TS). Int J Neuropsychopharmacol. 2006;9(1):95-100.
24. Münchau A, Bloem BR, Thilo KV, et al. Repetitive transcranial magnetic stimulation for Tourette syndrome. Neurology. 2002;59(11):1789-1791.
25. Orth M, Kirby R, Richardson MP, et al. Subthreshold rTMS over pre-motor cortex has no effect on tics in patients with Gilles de la Tourette syndrome. Clin Neurophysiol. 2005;116(4):764-768.
26. Snijders AH, Bloem BR, Orth M, et al. Video assessment of rTMS for Tourette syndrome. J Neurol Neurosurg Psychiatry. 2005;76(12):1743-1744.
27. Mantovani A, Leckman JF, Grantz H, et al. Repetitive transcranial magnetic stimulation of the supplementary motor area in the treatment of Tourette syndrome: report of two cases. Clin Neurophysiol. 2007;118(10):2314-2315.
28. Chae JH, Nahas Z, Wassermann E, et al. A pilot safety study of repetitive transcranial magnetic stimulation (rTMS) in Tourette’s syndrome. Cogn Behav Neurol. 2004;17(2):109-117.
29. Wu SW, Maloney T, Gilbert DL, et al. Functional MRI-navigated repetitive transcranial magnetic stimulation over supplementary motor area in chronic tic disorders. Brain Stimul. 2014;7(2):212-218.
30. Kwon HJ, Lim WS, Lim MH, et al. 1-Hz low frequency repetitive transcranial magnetic stimulation in children with Tourette’s syndrome. Neurosci Lett. 2011;492(1):1-4.
31. Salatino A, Momo E, Nobili M, et al. Awareness of symptoms amelioration following low-frequency repetitive transcranial magnetic stimulation in a patient with Tourette syndrome and comorbid obsessive-compulsive disorder. Brain Stimulat. 2014;7(2):341-343.

Chief complaint: Homicidal. Assessing violence risk
Mr. F, age 35, is homeless and has a history of cocaine and alcohol use disorders. He is admitted voluntarily to the psychiatric unit because he has homicidal thoughts toward Ms. S, who works in the shelter where he has been staying. Mr. F reports that he is thinking of killing Ms. S if he is discharged because she has been rude to him. He states that he has access to several firearms, but he will not disclose the location. He has been diagnosed with unspecified depressive disorder and exhibited antisocial personality disorder traits. He is being treated with sertraline. However, his mood appears to be relatively stable, except for occasional angry verbal outbursts. The outbursts have been related to intrusive peers or staff turning the television off for group meetings. Mr. F has been joking with peers, eating well, and sleeping appropriately. He reports no suicidal thoughts and has not been physically violent on the unit. However, Mr. F has had a history of violence since his teenage years. He has been incarcerated twice for assault and once for drug possession.
How would you approach assessing and managing Mr. F’s risk for violence?
We all have encountered a patient similar to Mr. F on the psychiatric unit or in the emergency department—a patient who makes violent threats and appears angry, intimidating, manipulative, and/or demanding, despite exhibiting no evidence of mania or psychosis. This patient often has a history of substance abuse and a lifelong pattern of viewing violence as an acceptable way of addressing life’s problems. Many psychiatrists suspect that more time on the inpatient unit is unlikely to reduce this patient’s risk of violence. Why? Because the violence risk does not stem from a treatable mental illness. Further, psychiatrists may be apprehensive about this patient’s potential for violence after discharge and their liability in the event of a bad outcome. No one wants their name associated with a headline that reads “Psychiatrist discharged man less than 24 hours before he killed 3 people.”
The purported relationship between mental illness and violence often is sensationalized in the media. However, research reveals that the vast majority of violence is in fact not due to symptoms of mental illness.1,2 A common clinical challenge in psychiatry involves evaluating individuals at elevated risk of violence and determining how to address their risk factors for violence. When the risk is primarily due to psychosis and can be reduced with antipsychotic medication, the job is easy. But how should we proceed when the risk stems from factors other than mental illness?
This article
Violence and mental illness: A tenuous link
Violence is a major public health concern in the United States. Although in recent years the rates of homicide and aggravated assault have decreased dramatically, there are approximately 16,000 homicides annually in the United States, and more than 1.6 million injuries from assaults treated in emergency departments each year.3 Homicide continues to be one of the leading causes of death among teenagers and young adults.4
The most effective methods of preventing widespread violence are public health approaches, such as parent- and family-focused programs, early childhood education, programs in school, and public policy changes.3 However, as psychiatrists, we are routinely asked to assess the risk of violence for an individual patient and devise strategies to mitigate violence risk.
Continue to: Although certain mental illnesses...
Although certain mental illnesses increase the relative risk of violence (compared with people without mental illness),5,6 recent studies suggest that mental illness plays only a “minor role in explaining violence in populations.”7 It is estimated that as little as 4% of the violence in the United States can be attributed to mental illness.1 According to a 1998 meta-analysis of 48 studies of criminal recidivism, the risk factors for violent recidivism were “almost identical” among offenders who had a mental disorder and those who did not.8
Approaches to assessing violence risk
Psychiatrists can assess the risk of future violence via 3 broad approaches.9,10
Unaided clinical judgment is when a mental health professional estimates violence risk based on his or her own experience and intuition, with knowledge of violence risk factors, but without the use of structured tools.
Actuarial tools are statistical models that use formulae to show relationships between data (risk factors) and outcomes (violence).10,11
Continue to: Structured professional judgment
Structured professional judgment is a hybrid of unaided clinical judgment and actuarial methods. Structured professional judgment tools help the evaluator identify empirically established risk factors. Once the information is collected, it is combined with clinical judgment in decision making.9,10 There are now more than 200 structured tools available for assessing violence risk in criminal justice and forensic mental health populations.12
Clinical judgment, although commonly used in practice, is less accurate than actuarial tools or structured professional judgment.10,11 In general, risk assessment tools offer moderate levels of accuracy in categorizing people at low risk vs high risk.5,13 The tools have better ability to accurately categorize individuals at low risk, compared with high risk, where false positives are common.12,14
Two types of risk factors
Risk factors for violence are commonly categorized as static or dynamic factors. Static factors are historical factors that cannot be changed with intervention (eg, age, sex, history of abuse). Dynamic factors can be changed with intervention (eg, substance abuse).15
Static risk factors. The best predictor of future violence is past violent behavior.5,16,17 Violence risk increases with each prior episode of violence.5 Prior arrests for any crime, especially if the individual was a juvenile at the time of arrest for his or her first violent offense, increase future violence risk.5 Other important static violence risk factors include demographic factors such as age, sex, and socioeconomic status. Swanson et al6 reviewed a large pool of data (approximately 10,000 respondents) from the Epidemiologic Catchment Area survey. Being young, male, and of low socioeconomic status were all associated with violence in the community.6 The highest-risk age group for violence is age 15 to 24.5 Males perpetrate violence in the community at a rate 10 times that of females.18 However, among individuals with severe mental illness, men and women have similar rates of violence.19,20 Unstable employment,21 less education,22 low intelligence,16 and a history of a significant head injury5 also are risk factors for violence.5
Continue to: Being abused as a child...
Being abused as a child, witnessing violence in the home,5,16 and growing up with an unstable parental situation (eg, parental loss or separation) has been linked to violence.16,23,24 Early disruptive behavior in childhood (eg, fighting, lying and stealing, truancy, and school problems) increases violence risk.21,23
Personality factors are important static risk factors for violence. Antisocial personality disorder is the most common personality disorder linked with violence.17 Several studies consistently show psychopathy to be a strong predictor of both violence and criminal behavior.5,25 A psychopath is a person who lacks empathy and close relationships, behaves impulsively, has superficially charming qualities, and is primarily interested in self-gratification.26 Harris et al27 studied 169 released forensic patients and found that 77% of the psychopaths (according to Psychopathy Checklist-Revised [PCL-R] scores) violently recidivated. In contrast, only 21% of the non-psychopaths violently recidivated.27
Other personality factors associated with violence include a predisposition toward feelings of anger and hatred (as opposed to empathy, anxiety, or guilt, which may reduce risk), hostile attributional biases (a tendency to interpret benign behavior of others as intentionally antagonistic), violent fantasies, poor anger control, and impulsivity.5 Although personality factors tend to be longstanding and more difficult to modify, in the outpatient setting, therapeutic efforts can be made to modify hostile attribution biases, poor anger control, and impulsive behavior.
Dynamic risk factors. Substance abuse is strongly associated with violence.6,17 The prevalence of violence is 12 times greater among individuals with alcohol use disorder and 16 times greater among individuals with other substance use disorders, compared with those with no such diagnoses.5,6
Continue to: Steadman et al...
Steadman et al28 compared 1,136 adult patients with mental disorders discharged from psychiatric hospitals with 519 individuals living in the same neighborhoods as the hospitalized patients. They found that the prevalence of violence among discharged patients without substance abuse was “statistically indistinguishable” from the prevalence of violence among community members, in the same neighborhood, who did not have symptoms of substance abuse.28 Swanson et al6 found that the combination of a mental disorder plus an alcohol or substance use disorder substantially increased the risk of violence.
Other dynamic risk factors for violence include mental illness symptoms such as psychosis, especially threat/control-override delusions, where the individual believes that they are being threatened or controlled by an external force.17
Contextual factors to consider in violence risk assessments include current stressors, lack of social support, availability of weapons, access to drugs and alcohol, and the presence of similar circumstances that led to violent behavior in the past.5
How to assess the risk of targeted violence
Targeted violence is a predatory act of violence intentionally committed against a preselected person, group of people, or place.29 Due to the low base rates of these incidents, targeted violence is difficult to study.7,30 These risk assessments require a more specialized approach.
Continue to: In their 1999 article...
In their 1999 article, Borum et al30 discussed threat assessment strategies utilized by the U.S. Secret Service and recommended investigating “pathways of ideas and behaviors that may lead to violent action.” Borum et al30 summarized 3 fundamental principles of threat assessment (Table 130).

What to do when violence risk is not due to mental illness
Based on the information in Mr. F’s case scenario, it is likely that his homicidal ideation is not due to mental illness. Despite this, several risk factors for violence are present. Where do we go from here?
Scott and Resnick17 recommend considering the concept of dangerousness as 5 components (Table 217). When this model of dangerousness is applied to Mr. F’s case, one can see that the magnitude of the harm is great because of threatened homicide. With regard to the imminence of the harm, it would help to clarify whether Mr. F plans to kill Ms. S immediately after discharge, or sometime in the next few months. Is his threat contingent on further provocations by Ms. S? Alternatively, does he intend to kill her for past grievances, regardless of further perceived insults?

Next, the frequency of a behavior relates to how often Mr. F has been aggressive in the past. The severity of his past aggression is also important. What is the most violent act he has ever done? Situational factors in this case include Mr. F’s access to weapons, financial problems, housing problems, and access to drugs and alcohol.17 Mr. F should be asked about what situations previously provoked his violent behavior. Consider how similar the present conditions are to past conditions to which Mr. F responded violently.5 The likelihood that a homicide will occur should take into account Mr. F’s risk factors for violence, as well as the seriousness of his intent to cause harm.
Continue to: Consider using a structured tool...
Consider using a structured tool, such as the Classification of Violence Risk, to help identify Mr. F’s risk factors for violence, or some other formal method to ensure that the proper data are collected. Violence risk assessments are more accurate when structured risk assessment tools are used, compared with clinical judgment alone.
It is important to review collateral sources of information. In Mr. F’s case, useful collateral sources may include his criminal docket (usually available online), past medical records, information from the shelter where he lives, and, potentially, friends or family.
Because Mr. F is making threats of targeted violence, be sure to ask about attack-related behaviors (Table 130).
Regarding the seriousness of Mr. F’s intent to cause harm, it may be helpful to ask him the following questions:
- How likely are you to carry out this act of violence?
- Do you have a plan? Have you taken any steps toward this plan?
- Do you see other, nonviolent solutions to this problem?
- What do you hope that we can do for you to help with this problem?
Continue to: Mr. F's answers...
Mr. F’s answers may suggest the possibility of a hidden agenda. Some patients express homicidal thoughts in order to stay in the hospital. If Mr. F expresses threats that are contingent on discharge and declines to engage in problem-solving discussions, this would cast doubt on the genuineness of his threat. However, doubt about the genuineness of the threat alone is not sufficient to simply discharge Mr. F. Assessment of his intent needs to be considered with other relevant risk factors, risk reduction strategies, and any Tarasoff duties that may apply.
In addition to risk factors, consider mitigating factors. For example, does Mr. F express concern over prison time as a reason to not engage in violence? It would be more ominous if Mr. F says that he does not care if he goes to prison because life is lousy being homeless and unemployed. At this point, an estimation can be made regarding whether Mr. F is a low-, moderate-, or high-risk of violence.
The next step is to organize Mr. F’s risk factors into static (historical) and dynamic (subject to intervention) factors. This will be helpful in formulating a strategy to manage risk because continued hospitalization can only address dynamic risk factors. Often in these cases, the static risk factors are far more numerous than the dynamic risk factors.
Once the data are collected and organized, the final step is to devise a risk management strategy. Some interventions, such as substance use treatment, will be straightforward. A mood-stabilizing medication could be considered, if clinically appropriate, to help reduce aggression and irritability.31 Efforts should be made to eliminate Mr. F’s access to firearms; however, in this case, it sounds unlikely that he will cooperate with those efforts. Ultimately, you may find yourself with a list of risk factors that are unlikely to be altered with further hospitalization, particularly if Mr. F’s homicidal thoughts and intent are due to antisocial personality traits.
Continue to: In that case...
In that case, the most important step will be to carry out your duty to warn/protect others prior to Mr. F’s discharge. Most states either require or permit mental health professionals to take reasonable steps to protect victims from violence when certain conditions are present, such as an explicit threat or identifiable victim (see Related Resources).
Once dynamic risk factors have been addressed, and duty to warn/protect is carried out, if there is no further clinical indication for hospitalization, it would be appropriate to discharge Mr. F. Continued homicidal threats stemming from antisocial personality traits, in the absence of a treatable mental illness (or other modifiable risk factors for violence that can be actively addressed), is not a reason for continued hospitalization. It may be useful to obtain a second opinion from a colleague in such scenarios. A second opinion may offer additional risk management ideas. In the event of a bad outcome, this will also help to show that the decision to discharge the patient was not taken lightly.
The psychiatrist should document a thoughtful risk assessment, the strategies that were implemented to reduce risk, the details of the warning, and the reasoning why continued hospitalization was not indicated (Table 3).

CASE CONTINUED
Decision to discharge
In Mr. F’s case, the treating psychiatrist determined that Mr. F’s risk of violence toward Ms. S was moderate. The psychiatrist identified several static risk factors for violence that raised Mr. F’s risk, but also noted that Mr. F’s threats were likely a manipulative effort to prolong his hospital stay. The psychiatrist carried out his duty to protect by notifying police and Ms. S of the nature of the threat prior to Mr. F’s discharge. The unit social worker helped Mr. F schedule an intake appointment for a substance use disorder treatment facility. Mr. F ultimately stated that he no longer experienced homicidal ideas once a bed was secured for him in a substance use treatment program. The psychiatrist carefully documented Mr. F’s risk assessment and the reasons why Mr. F’s risk would not be significantly altered by further inpatient hospitalization. Mr. F was discharged, and Ms. S remained unharmed.
Continue to: Bottom Line
Bottom Line
Use a structured approach to identify risk factors for violence. Address dynamic risk factors, including access to weapons. Carry out the duty to warn/protect if applicable. Document your decisions and actions carefully, and then discharge the patient if clinically indicated. Do not be “held hostage” by a patient’s homicidal ideation.
Related Resources
- Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
- Douglas KS, Hart SD, Webster CD, et al. HCR-20V3: Assessing risk of violence–user guide. Burnaby, Canada: Mental Health, Law, and Policy Institute, Simon Fraser University; 2013.
- National Conference of State Legislatures. Mental health professionals’ duty to warn. http://www.ncsl.org/research/health/mental-health-professionals-duty-to-warn.aspx. Published September 28, 2015.
Drug Brand Names
Sertraline • Zoloft
1. Skeem J, Kennealy P, Monahan J, et al. Psychosis uncommonly and inconsistently precedes violence among high-risk individuals. Clin Psychol Sci. 2016;4(1):40-49.
2. McGinty E, Frattaroli S, Appelbaum PS, et al. Using research evidence to reframe the policy debate around mental illness and guns: process and recommendations. Am J Public Health. 2014;104(11):e22-e26.
3. Sumner SA, Mercy JA, Dahlberg LL, et al. Violence in the United States: status, challenges, and opportunities. JAMA. 2015;314(5):478-488.
4. Heron M. Deaths: leading causes for 2014. Natl Vital Stat Rep. 2016;65(5):1-96.
5. Borum R, Swartz M, Swanson J. Assessing and managing violence risk in clinical practice. J Prac Psychiatry Behav Health. 1996;2(4):205-215.
6. Swanson JW, Holzer CE 3rd, Ganju VK, et al. Violence and psychiatric disorder in the community: Evidence from the epidemiologic catchment area surveys. Hosp Community Psychiatry. 1990;41(7):761-770.
7. Swanson JW. Explaining rare acts of violence: the limits of evidence from population research. Psychiatr Serv. 2011;62(11):1369-1371.
8. Bonta J, Law M, Hanson K. The prediction of criminal and violent recidivism among mentally disordered offenders: a meta-analysis. Psychol Bull. 1998;123(2):123-142.
9. Monahan J. The inclusion of biological risk factors in violence risk assessments. In: Singh I, Sinnott-Armstrong W, Savulescu J, eds. Bioprediction, biomarkers, and bad behavior: scientific, legal, and ethical implications. New York, NY: Oxford University Press; 2014:57-76.
10. Murray J, Thomson ME. Clinical judgement in violence risk assessment. Eur J Psychol. 2010;6(1):128-149.
11. Mossman D. Violence risk: is clinical judgment enough? Current Psychiatry. 2008;7(6):66-72.
12. Douglas T, Pugh J, Singh I, et al. Risk assessment tools in criminal justice and forensic psychiatry: the need for better data. Eur Psychiatry. 2017;42:134-137.
13. Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
14. Fazel S, Singh J, Doll H, et al. Use of risk assessment instruments to predict violence and antisocial behaviour in 73 samples involving 24 827 people: systematic review and meta-analysis. BMJ. 2012;345:e4692. doi: 10.1136/bmj.e4692.
15. National Collaborating Centre for Mental Health (UK). Violence and aggression: short- term management in mental health, health, and community settings: updated edition. London: British Psychological Society; 2015. NICE Guideline, No 10.
16. Klassen D, O’Connor WA. Predicting violence in schizophrenic and non-schizophrenic patients: a prospective study. J Community Psychol. 1988;16(2):217-227.
17. Scott C, Resnick P. Clinical assessment of aggression and violence. In: Rosner R, Scott C, eds. Principles and practice of forensic psychiatry, 3rd ed. Boca Raton, FL: CRC Press; 2017:623-631.
18. Tardiff K, Sweillam A. Assault, suicide, and mental illness. Arch Gen Psychiatry. 1980;37(2):164-169.
19. Lidz CW, Mulvey EP, Gardner W. The accuracy of predictions of violence to others. JAMA. 1993;269(8):1007-1011.
20. Newhill CE, Mulvey EP, Lidz CW. Characteristics of violence in the community by female patients seen in a psychiatric emergency service. Psychiatric Serv. 1995;46(8):785-789.
21. Mulvey E, Lidz C. Clinical considerations in the prediction of dangerousness in mental patients. Clin Psychol Rev. 1984;4(4):379-401.
22. Link BG, Andrews H, Cullen FT. The violent and illegal behavior of mental patients reconsidered. Am Sociol Rev. 1992;57(3):275-292.
23. Harris GT, Rice ME, Quinsey VL. Violent recidivism of mentally disordered offenders: the development of a statistical prediction instrument. Crim Justice and Behav. 1993;20(4):315-335.
24. Klassen D, O’Connor W. Demographic and case history variables in risk assessment. In: Monahan J, Steadman H, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:229-257.
25. Hart SD, Hare RD, Forth AE. Psychopathy as a risk marker for violence: development and validation of a screening version of the revised Psychopathy Checklist. In: Monahan J, Steadman HJ, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:81-98.
26. Cleckley H. The mask of sanity. St. Louis, MO: Mosby; 1941.
27. Harris GT, Rice ME, Cormier CA. Psychopathy and violent recidivism. Law Hum Behav. 1991;15(6):625-637.
28. Steadman HJ, Mulvey EP, Monahan J. Violence by people discharged from acute psychiatric inpatient facilities and by others in the same neighborhoods. Arch Gen Psychiatry. 1998;55:393-401.
29. Meloy JR, White SG, Hart S. Workplace assessment of targeted violence risk: the development and reliability of the WAVR-21. J Forensic Sci. 2013;58(5):1353-1358.
30. Borum R, Fein R, Vossekuil B, et al. Threat assessment: defining an approach for evaluating risk of targeted violence. Behav Sci Law. 1999;17(3):323-337.
31. Tyrer P, Bateman AW. Drug treatment for personality disorders. Adv Psychiatr Treat. 2004;10(5):389-398.
Mr. F, age 35, is homeless and has a history of cocaine and alcohol use disorders. He is admitted voluntarily to the psychiatric unit because he has homicidal thoughts toward Ms. S, who works in the shelter where he has been staying. Mr. F reports that he is thinking of killing Ms. S if he is discharged because she has been rude to him. He states that he has access to several firearms, but he will not disclose the location. He has been diagnosed with unspecified depressive disorder and exhibited antisocial personality disorder traits. He is being treated with sertraline. However, his mood appears to be relatively stable, except for occasional angry verbal outbursts. The outbursts have been related to intrusive peers or staff turning the television off for group meetings. Mr. F has been joking with peers, eating well, and sleeping appropriately. He reports no suicidal thoughts and has not been physically violent on the unit. However, Mr. F has had a history of violence since his teenage years. He has been incarcerated twice for assault and once for drug possession.
How would you approach assessing and managing Mr. F’s risk for violence?
We all have encountered a patient similar to Mr. F on the psychiatric unit or in the emergency department—a patient who makes violent threats and appears angry, intimidating, manipulative, and/or demanding, despite exhibiting no evidence of mania or psychosis. This patient often has a history of substance abuse and a lifelong pattern of viewing violence as an acceptable way of addressing life’s problems. Many psychiatrists suspect that more time on the inpatient unit is unlikely to reduce this patient’s risk of violence. Why? Because the violence risk does not stem from a treatable mental illness. Further, psychiatrists may be apprehensive about this patient’s potential for violence after discharge and their liability in the event of a bad outcome. No one wants their name associated with a headline that reads “Psychiatrist discharged man less than 24 hours before he killed 3 people.”
The purported relationship between mental illness and violence often is sensationalized in the media. However, research reveals that the vast majority of violence is in fact not due to symptoms of mental illness.1,2 A common clinical challenge in psychiatry involves evaluating individuals at elevated risk of violence and determining how to address their risk factors for violence. When the risk is primarily due to psychosis and can be reduced with antipsychotic medication, the job is easy. But how should we proceed when the risk stems from factors other than mental illness?
This article
Violence and mental illness: A tenuous link
Violence is a major public health concern in the United States. Although in recent years the rates of homicide and aggravated assault have decreased dramatically, there are approximately 16,000 homicides annually in the United States, and more than 1.6 million injuries from assaults treated in emergency departments each year.3 Homicide continues to be one of the leading causes of death among teenagers and young adults.4
The most effective methods of preventing widespread violence are public health approaches, such as parent- and family-focused programs, early childhood education, programs in school, and public policy changes.3 However, as psychiatrists, we are routinely asked to assess the risk of violence for an individual patient and devise strategies to mitigate violence risk.
Continue to: Although certain mental illnesses...
Although certain mental illnesses increase the relative risk of violence (compared with people without mental illness),5,6 recent studies suggest that mental illness plays only a “minor role in explaining violence in populations.”7 It is estimated that as little as 4% of the violence in the United States can be attributed to mental illness.1 According to a 1998 meta-analysis of 48 studies of criminal recidivism, the risk factors for violent recidivism were “almost identical” among offenders who had a mental disorder and those who did not.8
Approaches to assessing violence risk
Psychiatrists can assess the risk of future violence via 3 broad approaches.9,10
Unaided clinical judgment is when a mental health professional estimates violence risk based on his or her own experience and intuition, with knowledge of violence risk factors, but without the use of structured tools.
Actuarial tools are statistical models that use formulae to show relationships between data (risk factors) and outcomes (violence).10,11
Continue to: Structured professional judgment
Structured professional judgment is a hybrid of unaided clinical judgment and actuarial methods. Structured professional judgment tools help the evaluator identify empirically established risk factors. Once the information is collected, it is combined with clinical judgment in decision making.9,10 There are now more than 200 structured tools available for assessing violence risk in criminal justice and forensic mental health populations.12
Clinical judgment, although commonly used in practice, is less accurate than actuarial tools or structured professional judgment.10,11 In general, risk assessment tools offer moderate levels of accuracy in categorizing people at low risk vs high risk.5,13 The tools have better ability to accurately categorize individuals at low risk, compared with high risk, where false positives are common.12,14
Two types of risk factors
Risk factors for violence are commonly categorized as static or dynamic factors. Static factors are historical factors that cannot be changed with intervention (eg, age, sex, history of abuse). Dynamic factors can be changed with intervention (eg, substance abuse).15
Static risk factors. The best predictor of future violence is past violent behavior.5,16,17 Violence risk increases with each prior episode of violence.5 Prior arrests for any crime, especially if the individual was a juvenile at the time of arrest for his or her first violent offense, increase future violence risk.5 Other important static violence risk factors include demographic factors such as age, sex, and socioeconomic status. Swanson et al6 reviewed a large pool of data (approximately 10,000 respondents) from the Epidemiologic Catchment Area survey. Being young, male, and of low socioeconomic status were all associated with violence in the community.6 The highest-risk age group for violence is age 15 to 24.5 Males perpetrate violence in the community at a rate 10 times that of females.18 However, among individuals with severe mental illness, men and women have similar rates of violence.19,20 Unstable employment,21 less education,22 low intelligence,16 and a history of a significant head injury5 also are risk factors for violence.5
Continue to: Being abused as a child...
Being abused as a child, witnessing violence in the home,5,16 and growing up with an unstable parental situation (eg, parental loss or separation) has been linked to violence.16,23,24 Early disruptive behavior in childhood (eg, fighting, lying and stealing, truancy, and school problems) increases violence risk.21,23
Personality factors are important static risk factors for violence. Antisocial personality disorder is the most common personality disorder linked with violence.17 Several studies consistently show psychopathy to be a strong predictor of both violence and criminal behavior.5,25 A psychopath is a person who lacks empathy and close relationships, behaves impulsively, has superficially charming qualities, and is primarily interested in self-gratification.26 Harris et al27 studied 169 released forensic patients and found that 77% of the psychopaths (according to Psychopathy Checklist-Revised [PCL-R] scores) violently recidivated. In contrast, only 21% of the non-psychopaths violently recidivated.27
Other personality factors associated with violence include a predisposition toward feelings of anger and hatred (as opposed to empathy, anxiety, or guilt, which may reduce risk), hostile attributional biases (a tendency to interpret benign behavior of others as intentionally antagonistic), violent fantasies, poor anger control, and impulsivity.5 Although personality factors tend to be longstanding and more difficult to modify, in the outpatient setting, therapeutic efforts can be made to modify hostile attribution biases, poor anger control, and impulsive behavior.
Dynamic risk factors. Substance abuse is strongly associated with violence.6,17 The prevalence of violence is 12 times greater among individuals with alcohol use disorder and 16 times greater among individuals with other substance use disorders, compared with those with no such diagnoses.5,6
Continue to: Steadman et al...
Steadman et al28 compared 1,136 adult patients with mental disorders discharged from psychiatric hospitals with 519 individuals living in the same neighborhoods as the hospitalized patients. They found that the prevalence of violence among discharged patients without substance abuse was “statistically indistinguishable” from the prevalence of violence among community members, in the same neighborhood, who did not have symptoms of substance abuse.28 Swanson et al6 found that the combination of a mental disorder plus an alcohol or substance use disorder substantially increased the risk of violence.
Other dynamic risk factors for violence include mental illness symptoms such as psychosis, especially threat/control-override delusions, where the individual believes that they are being threatened or controlled by an external force.17
Contextual factors to consider in violence risk assessments include current stressors, lack of social support, availability of weapons, access to drugs and alcohol, and the presence of similar circumstances that led to violent behavior in the past.5
How to assess the risk of targeted violence
Targeted violence is a predatory act of violence intentionally committed against a preselected person, group of people, or place.29 Due to the low base rates of these incidents, targeted violence is difficult to study.7,30 These risk assessments require a more specialized approach.
Continue to: In their 1999 article...
In their 1999 article, Borum et al30 discussed threat assessment strategies utilized by the U.S. Secret Service and recommended investigating “pathways of ideas and behaviors that may lead to violent action.” Borum et al30 summarized 3 fundamental principles of threat assessment (Table 130).
What to do when violence risk is not due to mental illness
Based on the information in Mr. F’s case scenario, it is likely that his homicidal ideation is not due to mental illness. Despite this, several risk factors for violence are present. Where do we go from here?
Scott and Resnick17 recommend considering the concept of dangerousness as 5 components (Table 217). When this model of dangerousness is applied to Mr. F’s case, one can see that the magnitude of the harm is great because of threatened homicide. With regard to the imminence of the harm, it would help to clarify whether Mr. F plans to kill Ms. S immediately after discharge, or sometime in the next few months. Is his threat contingent on further provocations by Ms. S? Alternatively, does he intend to kill her for past grievances, regardless of further perceived insults?
Next, the frequency of a behavior relates to how often Mr. F has been aggressive in the past. The severity of his past aggression is also important. What is the most violent act he has ever done? Situational factors in this case include Mr. F’s access to weapons, financial problems, housing problems, and access to drugs and alcohol.17 Mr. F should be asked about what situations previously provoked his violent behavior. Consider how similar the present conditions are to past conditions to which Mr. F responded violently.5 The likelihood that a homicide will occur should take into account Mr. F’s risk factors for violence, as well as the seriousness of his intent to cause harm.
Continue to: Consider using a structured tool...
Consider using a structured tool, such as the Classification of Violence Risk, to help identify Mr. F’s risk factors for violence, or some other formal method to ensure that the proper data are collected. Violence risk assessments are more accurate when structured risk assessment tools are used, compared with clinical judgment alone.
It is important to review collateral sources of information. In Mr. F’s case, useful collateral sources may include his criminal docket (usually available online), past medical records, information from the shelter where he lives, and, potentially, friends or family.
Because Mr. F is making threats of targeted violence, be sure to ask about attack-related behaviors (Table 130).
Regarding the seriousness of Mr. F’s intent to cause harm, it may be helpful to ask him the following questions:
- How likely are you to carry out this act of violence?
- Do you have a plan? Have you taken any steps toward this plan?
- Do you see other, nonviolent solutions to this problem?
- What do you hope that we can do for you to help with this problem?
Continue to: Mr. F's answers...
Mr. F’s answers may suggest the possibility of a hidden agenda. Some patients express homicidal thoughts in order to stay in the hospital. If Mr. F expresses threats that are contingent on discharge and declines to engage in problem-solving discussions, this would cast doubt on the genuineness of his threat. However, doubt about the genuineness of the threat alone is not sufficient to simply discharge Mr. F. Assessment of his intent needs to be considered with other relevant risk factors, risk reduction strategies, and any Tarasoff duties that may apply.
In addition to risk factors, consider mitigating factors. For example, does Mr. F express concern over prison time as a reason to not engage in violence? It would be more ominous if Mr. F says that he does not care if he goes to prison because life is lousy being homeless and unemployed. At this point, an estimation can be made regarding whether Mr. F is a low-, moderate-, or high-risk of violence.
The next step is to organize Mr. F’s risk factors into static (historical) and dynamic (subject to intervention) factors. This will be helpful in formulating a strategy to manage risk because continued hospitalization can only address dynamic risk factors. Often in these cases, the static risk factors are far more numerous than the dynamic risk factors.
Once the data are collected and organized, the final step is to devise a risk management strategy. Some interventions, such as substance use treatment, will be straightforward. A mood-stabilizing medication could be considered, if clinically appropriate, to help reduce aggression and irritability.31 Efforts should be made to eliminate Mr. F’s access to firearms; however, in this case, it sounds unlikely that he will cooperate with those efforts. Ultimately, you may find yourself with a list of risk factors that are unlikely to be altered with further hospitalization, particularly if Mr. F’s homicidal thoughts and intent are due to antisocial personality traits.
Continue to: In that case...
In that case, the most important step will be to carry out your duty to warn/protect others prior to Mr. F’s discharge. Most states either require or permit mental health professionals to take reasonable steps to protect victims from violence when certain conditions are present, such as an explicit threat or identifiable victim (see Related Resources).
Once dynamic risk factors have been addressed, and duty to warn/protect is carried out, if there is no further clinical indication for hospitalization, it would be appropriate to discharge Mr. F. Continued homicidal threats stemming from antisocial personality traits, in the absence of a treatable mental illness (or other modifiable risk factors for violence that can be actively addressed), is not a reason for continued hospitalization. It may be useful to obtain a second opinion from a colleague in such scenarios. A second opinion may offer additional risk management ideas. In the event of a bad outcome, this will also help to show that the decision to discharge the patient was not taken lightly.
The psychiatrist should document a thoughtful risk assessment, the strategies that were implemented to reduce risk, the details of the warning, and the reasoning why continued hospitalization was not indicated (Table 3).
CASE CONTINUED
Decision to discharge
In Mr. F’s case, the treating psychiatrist determined that Mr. F’s risk of violence toward Ms. S was moderate. The psychiatrist identified several static risk factors for violence that raised Mr. F’s risk, but also noted that Mr. F’s threats were likely a manipulative effort to prolong his hospital stay. The psychiatrist carried out his duty to protect by notifying police and Ms. S of the nature of the threat prior to Mr. F’s discharge. The unit social worker helped Mr. F schedule an intake appointment for a substance use disorder treatment facility. Mr. F ultimately stated that he no longer experienced homicidal ideas once a bed was secured for him in a substance use treatment program. The psychiatrist carefully documented Mr. F’s risk assessment and the reasons why Mr. F’s risk would not be significantly altered by further inpatient hospitalization. Mr. F was discharged, and Ms. S remained unharmed.
Continue to: Bottom Line
Bottom Line
Use a structured approach to identify risk factors for violence. Address dynamic risk factors, including access to weapons. Carry out the duty to warn/protect if applicable. Document your decisions and actions carefully, and then discharge the patient if clinically indicated. Do not be “held hostage” by a patient’s homicidal ideation.
Related Resources
- Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
- Douglas KS, Hart SD, Webster CD, et al. HCR-20V3: Assessing risk of violence–user guide. Burnaby, Canada: Mental Health, Law, and Policy Institute, Simon Fraser University; 2013.
- National Conference of State Legislatures. Mental health professionals’ duty to warn. http://www.ncsl.org/research/health/mental-health-professionals-duty-to-warn.aspx. Published September 28, 2015.
Drug Brand Names
Sertraline • Zoloft
Mr. F, age 35, is homeless and has a history of cocaine and alcohol use disorders. He is admitted voluntarily to the psychiatric unit because he has homicidal thoughts toward Ms. S, who works in the shelter where he has been staying. Mr. F reports that he is thinking of killing Ms. S if he is discharged because she has been rude to him. He states that he has access to several firearms, but he will not disclose the location. He has been diagnosed with unspecified depressive disorder and exhibited antisocial personality disorder traits. He is being treated with sertraline. However, his mood appears to be relatively stable, except for occasional angry verbal outbursts. The outbursts have been related to intrusive peers or staff turning the television off for group meetings. Mr. F has been joking with peers, eating well, and sleeping appropriately. He reports no suicidal thoughts and has not been physically violent on the unit. However, Mr. F has had a history of violence since his teenage years. He has been incarcerated twice for assault and once for drug possession.
How would you approach assessing and managing Mr. F’s risk for violence?
We all have encountered a patient similar to Mr. F on the psychiatric unit or in the emergency department—a patient who makes violent threats and appears angry, intimidating, manipulative, and/or demanding, despite exhibiting no evidence of mania or psychosis. This patient often has a history of substance abuse and a lifelong pattern of viewing violence as an acceptable way of addressing life’s problems. Many psychiatrists suspect that more time on the inpatient unit is unlikely to reduce this patient’s risk of violence. Why? Because the violence risk does not stem from a treatable mental illness. Further, psychiatrists may be apprehensive about this patient’s potential for violence after discharge and their liability in the event of a bad outcome. No one wants their name associated with a headline that reads “Psychiatrist discharged man less than 24 hours before he killed 3 people.”
The purported relationship between mental illness and violence often is sensationalized in the media. However, research reveals that the vast majority of violence is in fact not due to symptoms of mental illness.1,2 A common clinical challenge in psychiatry involves evaluating individuals at elevated risk of violence and determining how to address their risk factors for violence. When the risk is primarily due to psychosis and can be reduced with antipsychotic medication, the job is easy. But how should we proceed when the risk stems from factors other than mental illness?
This article
Violence and mental illness: A tenuous link
Violence is a major public health concern in the United States. Although in recent years the rates of homicide and aggravated assault have decreased dramatically, there are approximately 16,000 homicides annually in the United States, and more than 1.6 million injuries from assaults treated in emergency departments each year.3 Homicide continues to be one of the leading causes of death among teenagers and young adults.4
The most effective methods of preventing widespread violence are public health approaches, such as parent- and family-focused programs, early childhood education, programs in school, and public policy changes.3 However, as psychiatrists, we are routinely asked to assess the risk of violence for an individual patient and devise strategies to mitigate violence risk.
Continue to: Although certain mental illnesses...
Although certain mental illnesses increase the relative risk of violence (compared with people without mental illness),5,6 recent studies suggest that mental illness plays only a “minor role in explaining violence in populations.”7 It is estimated that as little as 4% of the violence in the United States can be attributed to mental illness.1 According to a 1998 meta-analysis of 48 studies of criminal recidivism, the risk factors for violent recidivism were “almost identical” among offenders who had a mental disorder and those who did not.8
Approaches to assessing violence risk
Psychiatrists can assess the risk of future violence via 3 broad approaches.9,10
Unaided clinical judgment is when a mental health professional estimates violence risk based on his or her own experience and intuition, with knowledge of violence risk factors, but without the use of structured tools.
Actuarial tools are statistical models that use formulae to show relationships between data (risk factors) and outcomes (violence).10,11
Continue to: Structured professional judgment
Structured professional judgment is a hybrid of unaided clinical judgment and actuarial methods. Structured professional judgment tools help the evaluator identify empirically established risk factors. Once the information is collected, it is combined with clinical judgment in decision making.9,10 There are now more than 200 structured tools available for assessing violence risk in criminal justice and forensic mental health populations.12
Clinical judgment, although commonly used in practice, is less accurate than actuarial tools or structured professional judgment.10,11 In general, risk assessment tools offer moderate levels of accuracy in categorizing people at low risk vs high risk.5,13 The tools have better ability to accurately categorize individuals at low risk, compared with high risk, where false positives are common.12,14
Two types of risk factors
Risk factors for violence are commonly categorized as static or dynamic factors. Static factors are historical factors that cannot be changed with intervention (eg, age, sex, history of abuse). Dynamic factors can be changed with intervention (eg, substance abuse).15
Static risk factors. The best predictor of future violence is past violent behavior.5,16,17 Violence risk increases with each prior episode of violence.5 Prior arrests for any crime, especially if the individual was a juvenile at the time of arrest for his or her first violent offense, increase future violence risk.5 Other important static violence risk factors include demographic factors such as age, sex, and socioeconomic status. Swanson et al6 reviewed a large pool of data (approximately 10,000 respondents) from the Epidemiologic Catchment Area survey. Being young, male, and of low socioeconomic status were all associated with violence in the community.6 The highest-risk age group for violence is age 15 to 24.5 Males perpetrate violence in the community at a rate 10 times that of females.18 However, among individuals with severe mental illness, men and women have similar rates of violence.19,20 Unstable employment,21 less education,22 low intelligence,16 and a history of a significant head injury5 also are risk factors for violence.5
Continue to: Being abused as a child...
Being abused as a child, witnessing violence in the home,5,16 and growing up with an unstable parental situation (eg, parental loss or separation) has been linked to violence.16,23,24 Early disruptive behavior in childhood (eg, fighting, lying and stealing, truancy, and school problems) increases violence risk.21,23
Personality factors are important static risk factors for violence. Antisocial personality disorder is the most common personality disorder linked with violence.17 Several studies consistently show psychopathy to be a strong predictor of both violence and criminal behavior.5,25 A psychopath is a person who lacks empathy and close relationships, behaves impulsively, has superficially charming qualities, and is primarily interested in self-gratification.26 Harris et al27 studied 169 released forensic patients and found that 77% of the psychopaths (according to Psychopathy Checklist-Revised [PCL-R] scores) violently recidivated. In contrast, only 21% of the non-psychopaths violently recidivated.27
Other personality factors associated with violence include a predisposition toward feelings of anger and hatred (as opposed to empathy, anxiety, or guilt, which may reduce risk), hostile attributional biases (a tendency to interpret benign behavior of others as intentionally antagonistic), violent fantasies, poor anger control, and impulsivity.5 Although personality factors tend to be longstanding and more difficult to modify, in the outpatient setting, therapeutic efforts can be made to modify hostile attribution biases, poor anger control, and impulsive behavior.
Dynamic risk factors. Substance abuse is strongly associated with violence.6,17 The prevalence of violence is 12 times greater among individuals with alcohol use disorder and 16 times greater among individuals with other substance use disorders, compared with those with no such diagnoses.5,6
Continue to: Steadman et al...
Steadman et al28 compared 1,136 adult patients with mental disorders discharged from psychiatric hospitals with 519 individuals living in the same neighborhoods as the hospitalized patients. They found that the prevalence of violence among discharged patients without substance abuse was “statistically indistinguishable” from the prevalence of violence among community members, in the same neighborhood, who did not have symptoms of substance abuse.28 Swanson et al6 found that the combination of a mental disorder plus an alcohol or substance use disorder substantially increased the risk of violence.
Other dynamic risk factors for violence include mental illness symptoms such as psychosis, especially threat/control-override delusions, where the individual believes that they are being threatened or controlled by an external force.17
Contextual factors to consider in violence risk assessments include current stressors, lack of social support, availability of weapons, access to drugs and alcohol, and the presence of similar circumstances that led to violent behavior in the past.5
How to assess the risk of targeted violence
Targeted violence is a predatory act of violence intentionally committed against a preselected person, group of people, or place.29 Due to the low base rates of these incidents, targeted violence is difficult to study.7,30 These risk assessments require a more specialized approach.
Continue to: In their 1999 article...
In their 1999 article, Borum et al30 discussed threat assessment strategies utilized by the U.S. Secret Service and recommended investigating “pathways of ideas and behaviors that may lead to violent action.” Borum et al30 summarized 3 fundamental principles of threat assessment (Table 130).
What to do when violence risk is not due to mental illness
Based on the information in Mr. F’s case scenario, it is likely that his homicidal ideation is not due to mental illness. Despite this, several risk factors for violence are present. Where do we go from here?
Scott and Resnick17 recommend considering the concept of dangerousness as 5 components (Table 217). When this model of dangerousness is applied to Mr. F’s case, one can see that the magnitude of the harm is great because of threatened homicide. With regard to the imminence of the harm, it would help to clarify whether Mr. F plans to kill Ms. S immediately after discharge, or sometime in the next few months. Is his threat contingent on further provocations by Ms. S? Alternatively, does he intend to kill her for past grievances, regardless of further perceived insults?
Next, the frequency of a behavior relates to how often Mr. F has been aggressive in the past. The severity of his past aggression is also important. What is the most violent act he has ever done? Situational factors in this case include Mr. F’s access to weapons, financial problems, housing problems, and access to drugs and alcohol.17 Mr. F should be asked about what situations previously provoked his violent behavior. Consider how similar the present conditions are to past conditions to which Mr. F responded violently.5 The likelihood that a homicide will occur should take into account Mr. F’s risk factors for violence, as well as the seriousness of his intent to cause harm.
Continue to: Consider using a structured tool...
Consider using a structured tool, such as the Classification of Violence Risk, to help identify Mr. F’s risk factors for violence, or some other formal method to ensure that the proper data are collected. Violence risk assessments are more accurate when structured risk assessment tools are used, compared with clinical judgment alone.
It is important to review collateral sources of information. In Mr. F’s case, useful collateral sources may include his criminal docket (usually available online), past medical records, information from the shelter where he lives, and, potentially, friends or family.
Because Mr. F is making threats of targeted violence, be sure to ask about attack-related behaviors (Table 130).
Regarding the seriousness of Mr. F’s intent to cause harm, it may be helpful to ask him the following questions:
- How likely are you to carry out this act of violence?
- Do you have a plan? Have you taken any steps toward this plan?
- Do you see other, nonviolent solutions to this problem?
- What do you hope that we can do for you to help with this problem?
Continue to: Mr. F's answers...
Mr. F’s answers may suggest the possibility of a hidden agenda. Some patients express homicidal thoughts in order to stay in the hospital. If Mr. F expresses threats that are contingent on discharge and declines to engage in problem-solving discussions, this would cast doubt on the genuineness of his threat. However, doubt about the genuineness of the threat alone is not sufficient to simply discharge Mr. F. Assessment of his intent needs to be considered with other relevant risk factors, risk reduction strategies, and any Tarasoff duties that may apply.
In addition to risk factors, consider mitigating factors. For example, does Mr. F express concern over prison time as a reason to not engage in violence? It would be more ominous if Mr. F says that he does not care if he goes to prison because life is lousy being homeless and unemployed. At this point, an estimation can be made regarding whether Mr. F is a low-, moderate-, or high-risk of violence.
The next step is to organize Mr. F’s risk factors into static (historical) and dynamic (subject to intervention) factors. This will be helpful in formulating a strategy to manage risk because continued hospitalization can only address dynamic risk factors. Often in these cases, the static risk factors are far more numerous than the dynamic risk factors.
Once the data are collected and organized, the final step is to devise a risk management strategy. Some interventions, such as substance use treatment, will be straightforward. A mood-stabilizing medication could be considered, if clinically appropriate, to help reduce aggression and irritability.31 Efforts should be made to eliminate Mr. F’s access to firearms; however, in this case, it sounds unlikely that he will cooperate with those efforts. Ultimately, you may find yourself with a list of risk factors that are unlikely to be altered with further hospitalization, particularly if Mr. F’s homicidal thoughts and intent are due to antisocial personality traits.
Continue to: In that case...
In that case, the most important step will be to carry out your duty to warn/protect others prior to Mr. F’s discharge. Most states either require or permit mental health professionals to take reasonable steps to protect victims from violence when certain conditions are present, such as an explicit threat or identifiable victim (see Related Resources).
Once dynamic risk factors have been addressed, and duty to warn/protect is carried out, if there is no further clinical indication for hospitalization, it would be appropriate to discharge Mr. F. Continued homicidal threats stemming from antisocial personality traits, in the absence of a treatable mental illness (or other modifiable risk factors for violence that can be actively addressed), is not a reason for continued hospitalization. It may be useful to obtain a second opinion from a colleague in such scenarios. A second opinion may offer additional risk management ideas. In the event of a bad outcome, this will also help to show that the decision to discharge the patient was not taken lightly.
The psychiatrist should document a thoughtful risk assessment, the strategies that were implemented to reduce risk, the details of the warning, and the reasoning why continued hospitalization was not indicated (Table 3).
CASE CONTINUED
Decision to discharge
In Mr. F’s case, the treating psychiatrist determined that Mr. F’s risk of violence toward Ms. S was moderate. The psychiatrist identified several static risk factors for violence that raised Mr. F’s risk, but also noted that Mr. F’s threats were likely a manipulative effort to prolong his hospital stay. The psychiatrist carried out his duty to protect by notifying police and Ms. S of the nature of the threat prior to Mr. F’s discharge. The unit social worker helped Mr. F schedule an intake appointment for a substance use disorder treatment facility. Mr. F ultimately stated that he no longer experienced homicidal ideas once a bed was secured for him in a substance use treatment program. The psychiatrist carefully documented Mr. F’s risk assessment and the reasons why Mr. F’s risk would not be significantly altered by further inpatient hospitalization. Mr. F was discharged, and Ms. S remained unharmed.
Continue to: Bottom Line
Bottom Line
Use a structured approach to identify risk factors for violence. Address dynamic risk factors, including access to weapons. Carry out the duty to warn/protect if applicable. Document your decisions and actions carefully, and then discharge the patient if clinically indicated. Do not be “held hostage” by a patient’s homicidal ideation.
Related Resources
- Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
- Douglas KS, Hart SD, Webster CD, et al. HCR-20V3: Assessing risk of violence–user guide. Burnaby, Canada: Mental Health, Law, and Policy Institute, Simon Fraser University; 2013.
- National Conference of State Legislatures. Mental health professionals’ duty to warn. http://www.ncsl.org/research/health/mental-health-professionals-duty-to-warn.aspx. Published September 28, 2015.
Drug Brand Names
Sertraline • Zoloft
1. Skeem J, Kennealy P, Monahan J, et al. Psychosis uncommonly and inconsistently precedes violence among high-risk individuals. Clin Psychol Sci. 2016;4(1):40-49.
2. McGinty E, Frattaroli S, Appelbaum PS, et al. Using research evidence to reframe the policy debate around mental illness and guns: process and recommendations. Am J Public Health. 2014;104(11):e22-e26.
3. Sumner SA, Mercy JA, Dahlberg LL, et al. Violence in the United States: status, challenges, and opportunities. JAMA. 2015;314(5):478-488.
4. Heron M. Deaths: leading causes for 2014. Natl Vital Stat Rep. 2016;65(5):1-96.
5. Borum R, Swartz M, Swanson J. Assessing and managing violence risk in clinical practice. J Prac Psychiatry Behav Health. 1996;2(4):205-215.
6. Swanson JW, Holzer CE 3rd, Ganju VK, et al. Violence and psychiatric disorder in the community: Evidence from the epidemiologic catchment area surveys. Hosp Community Psychiatry. 1990;41(7):761-770.
7. Swanson JW. Explaining rare acts of violence: the limits of evidence from population research. Psychiatr Serv. 2011;62(11):1369-1371.
8. Bonta J, Law M, Hanson K. The prediction of criminal and violent recidivism among mentally disordered offenders: a meta-analysis. Psychol Bull. 1998;123(2):123-142.
9. Monahan J. The inclusion of biological risk factors in violence risk assessments. In: Singh I, Sinnott-Armstrong W, Savulescu J, eds. Bioprediction, biomarkers, and bad behavior: scientific, legal, and ethical implications. New York, NY: Oxford University Press; 2014:57-76.
10. Murray J, Thomson ME. Clinical judgement in violence risk assessment. Eur J Psychol. 2010;6(1):128-149.
11. Mossman D. Violence risk: is clinical judgment enough? Current Psychiatry. 2008;7(6):66-72.
12. Douglas T, Pugh J, Singh I, et al. Risk assessment tools in criminal justice and forensic psychiatry: the need for better data. Eur Psychiatry. 2017;42:134-137.
13. Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
14. Fazel S, Singh J, Doll H, et al. Use of risk assessment instruments to predict violence and antisocial behaviour in 73 samples involving 24 827 people: systematic review and meta-analysis. BMJ. 2012;345:e4692. doi: 10.1136/bmj.e4692.
15. National Collaborating Centre for Mental Health (UK). Violence and aggression: short- term management in mental health, health, and community settings: updated edition. London: British Psychological Society; 2015. NICE Guideline, No 10.
16. Klassen D, O’Connor WA. Predicting violence in schizophrenic and non-schizophrenic patients: a prospective study. J Community Psychol. 1988;16(2):217-227.
17. Scott C, Resnick P. Clinical assessment of aggression and violence. In: Rosner R, Scott C, eds. Principles and practice of forensic psychiatry, 3rd ed. Boca Raton, FL: CRC Press; 2017:623-631.
18. Tardiff K, Sweillam A. Assault, suicide, and mental illness. Arch Gen Psychiatry. 1980;37(2):164-169.
19. Lidz CW, Mulvey EP, Gardner W. The accuracy of predictions of violence to others. JAMA. 1993;269(8):1007-1011.
20. Newhill CE, Mulvey EP, Lidz CW. Characteristics of violence in the community by female patients seen in a psychiatric emergency service. Psychiatric Serv. 1995;46(8):785-789.
21. Mulvey E, Lidz C. Clinical considerations in the prediction of dangerousness in mental patients. Clin Psychol Rev. 1984;4(4):379-401.
22. Link BG, Andrews H, Cullen FT. The violent and illegal behavior of mental patients reconsidered. Am Sociol Rev. 1992;57(3):275-292.
23. Harris GT, Rice ME, Quinsey VL. Violent recidivism of mentally disordered offenders: the development of a statistical prediction instrument. Crim Justice and Behav. 1993;20(4):315-335.
24. Klassen D, O’Connor W. Demographic and case history variables in risk assessment. In: Monahan J, Steadman H, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:229-257.
25. Hart SD, Hare RD, Forth AE. Psychopathy as a risk marker for violence: development and validation of a screening version of the revised Psychopathy Checklist. In: Monahan J, Steadman HJ, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:81-98.
26. Cleckley H. The mask of sanity. St. Louis, MO: Mosby; 1941.
27. Harris GT, Rice ME, Cormier CA. Psychopathy and violent recidivism. Law Hum Behav. 1991;15(6):625-637.
28. Steadman HJ, Mulvey EP, Monahan J. Violence by people discharged from acute psychiatric inpatient facilities and by others in the same neighborhoods. Arch Gen Psychiatry. 1998;55:393-401.
29. Meloy JR, White SG, Hart S. Workplace assessment of targeted violence risk: the development and reliability of the WAVR-21. J Forensic Sci. 2013;58(5):1353-1358.
30. Borum R, Fein R, Vossekuil B, et al. Threat assessment: defining an approach for evaluating risk of targeted violence. Behav Sci Law. 1999;17(3):323-337.
31. Tyrer P, Bateman AW. Drug treatment for personality disorders. Adv Psychiatr Treat. 2004;10(5):389-398.
1. Skeem J, Kennealy P, Monahan J, et al. Psychosis uncommonly and inconsistently precedes violence among high-risk individuals. Clin Psychol Sci. 2016;4(1):40-49.
2. McGinty E, Frattaroli S, Appelbaum PS, et al. Using research evidence to reframe the policy debate around mental illness and guns: process and recommendations. Am J Public Health. 2014;104(11):e22-e26.
3. Sumner SA, Mercy JA, Dahlberg LL, et al. Violence in the United States: status, challenges, and opportunities. JAMA. 2015;314(5):478-488.
4. Heron M. Deaths: leading causes for 2014. Natl Vital Stat Rep. 2016;65(5):1-96.
5. Borum R, Swartz M, Swanson J. Assessing and managing violence risk in clinical practice. J Prac Psychiatry Behav Health. 1996;2(4):205-215.
6. Swanson JW, Holzer CE 3rd, Ganju VK, et al. Violence and psychiatric disorder in the community: Evidence from the epidemiologic catchment area surveys. Hosp Community Psychiatry. 1990;41(7):761-770.
7. Swanson JW. Explaining rare acts of violence: the limits of evidence from population research. Psychiatr Serv. 2011;62(11):1369-1371.
8. Bonta J, Law M, Hanson K. The prediction of criminal and violent recidivism among mentally disordered offenders: a meta-analysis. Psychol Bull. 1998;123(2):123-142.
9. Monahan J. The inclusion of biological risk factors in violence risk assessments. In: Singh I, Sinnott-Armstrong W, Savulescu J, eds. Bioprediction, biomarkers, and bad behavior: scientific, legal, and ethical implications. New York, NY: Oxford University Press; 2014:57-76.
10. Murray J, Thomson ME. Clinical judgement in violence risk assessment. Eur J Psychol. 2010;6(1):128-149.
11. Mossman D. Violence risk: is clinical judgment enough? Current Psychiatry. 2008;7(6):66-72.
12. Douglas T, Pugh J, Singh I, et al. Risk assessment tools in criminal justice and forensic psychiatry: the need for better data. Eur Psychiatry. 2017;42:134-137.
13. Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
14. Fazel S, Singh J, Doll H, et al. Use of risk assessment instruments to predict violence and antisocial behaviour in 73 samples involving 24 827 people: systematic review and meta-analysis. BMJ. 2012;345:e4692. doi: 10.1136/bmj.e4692.
15. National Collaborating Centre for Mental Health (UK). Violence and aggression: short- term management in mental health, health, and community settings: updated edition. London: British Psychological Society; 2015. NICE Guideline, No 10.
16. Klassen D, O’Connor WA. Predicting violence in schizophrenic and non-schizophrenic patients: a prospective study. J Community Psychol. 1988;16(2):217-227.
17. Scott C, Resnick P. Clinical assessment of aggression and violence. In: Rosner R, Scott C, eds. Principles and practice of forensic psychiatry, 3rd ed. Boca Raton, FL: CRC Press; 2017:623-631.
18. Tardiff K, Sweillam A. Assault, suicide, and mental illness. Arch Gen Psychiatry. 1980;37(2):164-169.
19. Lidz CW, Mulvey EP, Gardner W. The accuracy of predictions of violence to others. JAMA. 1993;269(8):1007-1011.
20. Newhill CE, Mulvey EP, Lidz CW. Characteristics of violence in the community by female patients seen in a psychiatric emergency service. Psychiatric Serv. 1995;46(8):785-789.
21. Mulvey E, Lidz C. Clinical considerations in the prediction of dangerousness in mental patients. Clin Psychol Rev. 1984;4(4):379-401.
22. Link BG, Andrews H, Cullen FT. The violent and illegal behavior of mental patients reconsidered. Am Sociol Rev. 1992;57(3):275-292.
23. Harris GT, Rice ME, Quinsey VL. Violent recidivism of mentally disordered offenders: the development of a statistical prediction instrument. Crim Justice and Behav. 1993;20(4):315-335.
24. Klassen D, O’Connor W. Demographic and case history variables in risk assessment. In: Monahan J, Steadman H, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:229-257.
25. Hart SD, Hare RD, Forth AE. Psychopathy as a risk marker for violence: development and validation of a screening version of the revised Psychopathy Checklist. In: Monahan J, Steadman HJ, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:81-98.
26. Cleckley H. The mask of sanity. St. Louis, MO: Mosby; 1941.
27. Harris GT, Rice ME, Cormier CA. Psychopathy and violent recidivism. Law Hum Behav. 1991;15(6):625-637.
28. Steadman HJ, Mulvey EP, Monahan J. Violence by people discharged from acute psychiatric inpatient facilities and by others in the same neighborhoods. Arch Gen Psychiatry. 1998;55:393-401.
29. Meloy JR, White SG, Hart S. Workplace assessment of targeted violence risk: the development and reliability of the WAVR-21. J Forensic Sci. 2013;58(5):1353-1358.
30. Borum R, Fein R, Vossekuil B, et al. Threat assessment: defining an approach for evaluating risk of targeted violence. Behav Sci Law. 1999;17(3):323-337.
31. Tyrer P, Bateman AW. Drug treatment for personality disorders. Adv Psychiatr Treat. 2004;10(5):389-398.

Aripiprazole, brexpiprazole, and cariprazine: Not all the same
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.

Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
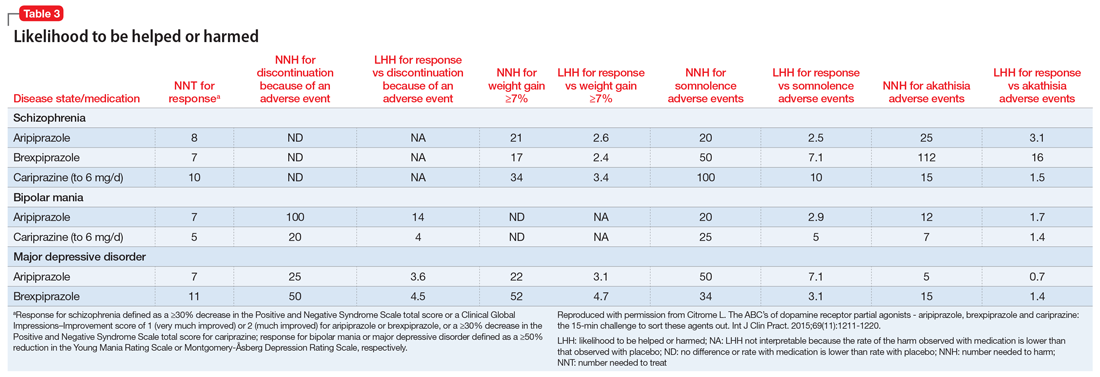
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.

Anxiety and joint hypermobility: An unexpected association
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.

The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
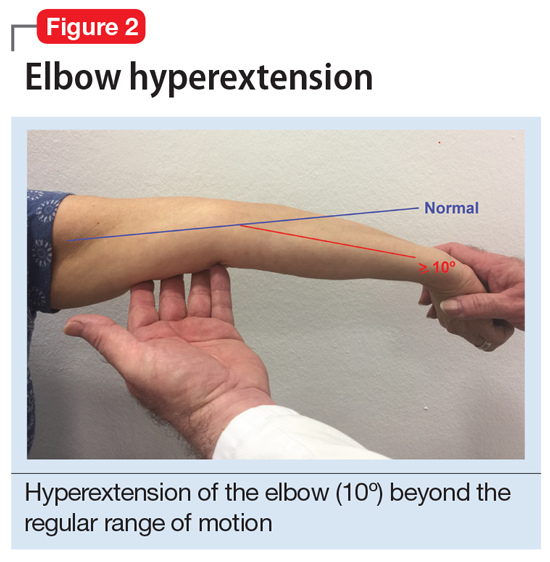
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.
The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.
The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
PTSD: A systematic approach to diagnosis and treatment
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.

Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
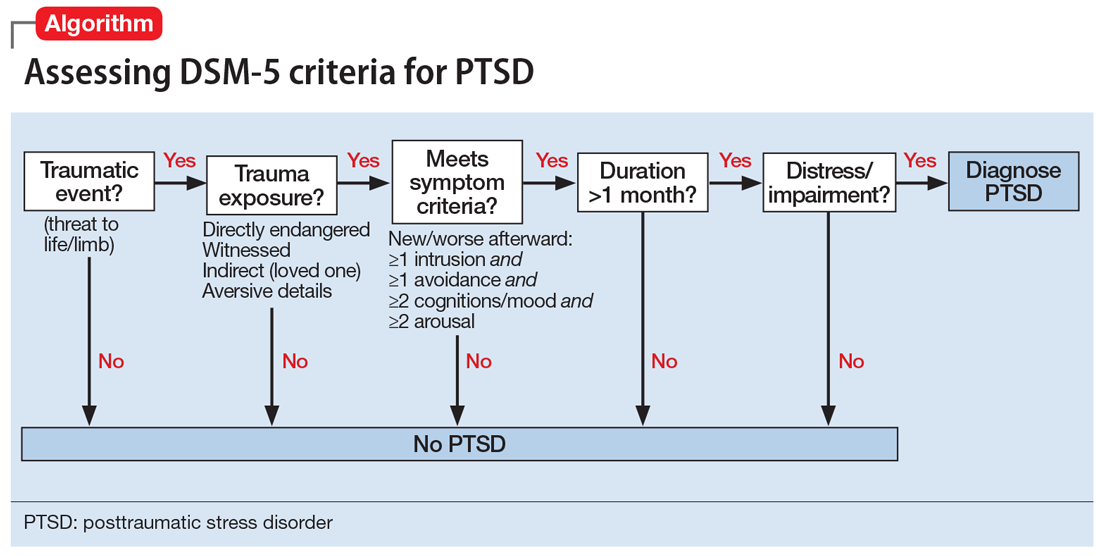
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
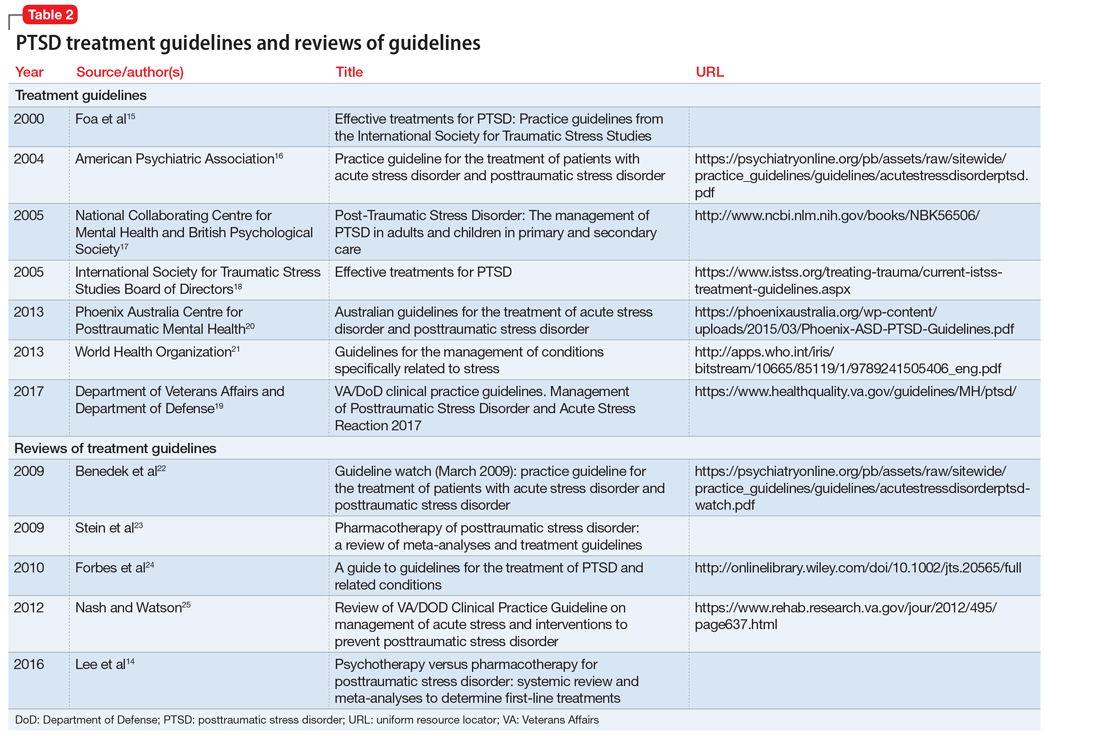
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
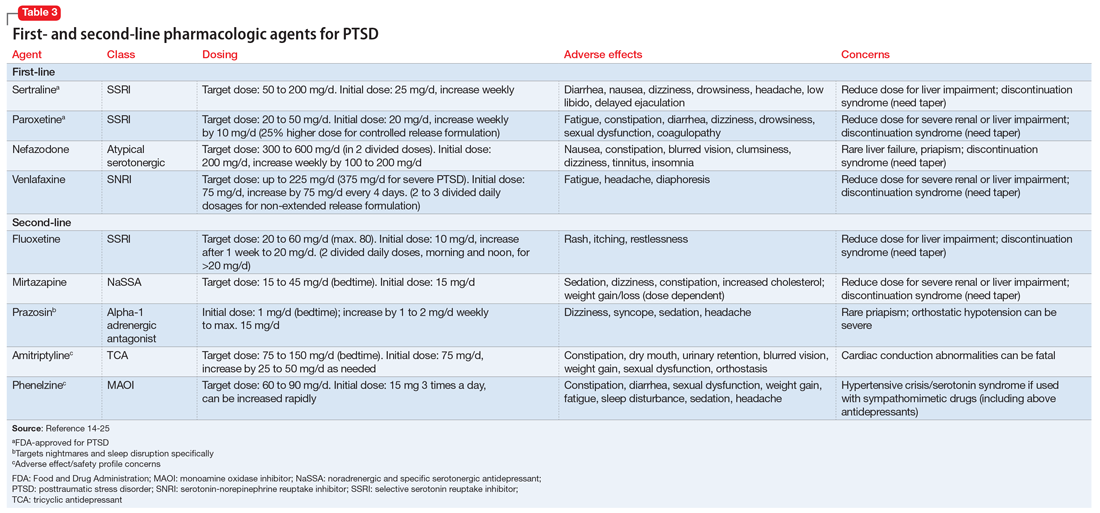
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.
Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.
Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
