User login
Medical marijuana: Do the benefits outweigh the risks?
There is a need for additional treatment options to improve symptoms, enhance the quality of life (QOL), and reduce suffering among patients who have chronic medical illness. Medical marijuana (MM) has the potential to help patients who have certain medical conditions in states where it is legal for prescription by a licensed medical provider.
Cannabis has a long history of medicinal use (Box 11-12). Two derivatives of the Cannabis plant—cannabinoid delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD)—are responsible for most of its effects. Some of these effects, including analgesia, decreased muscle spasticity, and reduced eye pressure, have been harnessed for their potential therapeutic effects (Box 213-19). As of November 2017, 29 states had legalized Cannabis for medical use, and several had legalized its recreational use.12
With the increasing availability of MM, psychiatrists are likely to encounter patients who are using it or who will ask them about it. This article reviews evidence related to using MM to treat patients with neuropathic pain; chemotherapyinduced nausea and vomiting (CINV); epilepsy; multiple sclerosis (MS); glaucoma; Crohn’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; dementia-related behavioral disturbances; posttraumatic stress disorder (PTSD); and anxiety.
Box 1
Cannabis: A history of medicinal use
Cannabis has been cultivated since ancient times, beginning in China and India. The earliest reference of its use for healing purposes may have been in the Chinese Pharmacopeia, circa 1500 BC.1 In 1839, Dr. William Brooke O’Shaughnessy introduced Cannabis Indica, or “Indian hemp,” to the western world after a professorship in Calcutta, India.2 In the early 1840s, an English physician, Dr. John Clendinning, prescribed Cannabis for migraine headache.3 In the 19th and early 20th centuries, several prominent physicians advocated using Cannabis for migraines; Sir William Osler did so in his textbook, The principles and practice of medicine.4 It was listed in the U.S. Pharmacopeia in 1850 but removed in 1942.5,6
Until 1937, Cannabis was used in the United States for medicinal purposes, such as for treating inflamed skin, incontinence, and sexually transmitted diseases.7 In 1937, the Marihuana Tax Act, which prohibited the production, importation, possession, use, and dispersal of Cannabis, was passed.8 Cannabis became a Schedule I drug under the Controlled Substance Act of 1970.9
In 1999, based on available evidence, the Institute of Medicine (IOM) concluded Cannabis had less likelihood of dependence than benzodiazepines, opiates, cocaine, or nicotine. The IOM also concluded that the symptoms of withdrawal were mild in comparison with benzodiazepines or opiates. Finally, the IOM stated that Cannabis was not a “gateway” drug.10
In 1996, California was the first state to reimplement medicinal use of Cannabis under the Compassionate Use Act, also known as Proposition 215.11 This act allowed individuals to retain or produce Cannabis for personal consumption with a physician’s approval. Many states eventually followed California’s lead. As of November 2017, 29 states, the District of Columbia, Guam, and Puerto Rico had regulated Cannabis use for medical purposes,12 and recreational use had been approved in 7 states and the District of Columbia.
Medical illnesses
Neuropathic pain. Chronic neuropathic pain affects an estimated 7% to 8% of adults.20 Patients with neuropathic pain are often treated with anticonvulsants, antidepressants, opioids, and local anesthetics21; however, these medications may not provide substantial relief. Research has revealed that THC and CBD can improve central and peripheral neuropathic pain, as well as pain associated with rheumatoid arthritis and fibromyalgia.22
Wilsey et al23 evaluated the analgesic effects of smoked MM for neuropathic pain in a small (N = 38) double-blind, randomized controlled trial (RCT). Patients in this study had a preexisting diagnosis of complex regional pain syndrome, spinal cord injury, peripheral neuropathy, or nerve injury. To prevent any unforeseen adverse outcomes related to Cannabis use, participants were required to have previous exposure to Cannabis. Patients were excluded if they had major mental illness, substance abuse, or other major medical ailments.
Participants smoked high-dose Cannabis cigarettes (7% THC), low-dose Cannabis cigarettes (3.5% THC), or placebo cigarettes. Pain was measured on a visual analog scale (VAS) that ranged from 0 (no pain) to 100 (worst possible pain). Compared with the placebo group, significant analgesia was achieved in both Cannabis groups (P = .016). The high-dose group had greater neurocognitive impairment.
Ware et al24 conducted a crossover RCT (N = 23) to determine the efficacy of smoked MM for neuropathic pain. Participants had neuropathic pain for at least 3 months that was caused by trauma or surgery, with an average weekly pain intensity score >4 on scale of 0 to 10. Patients with pain due to cancer, nociceptive causes, unstable medical conditions, current substance abuse, history of a psychotic disorder, or suicidal ideation were excluded. Participants were assigned to a 9.4% THC group or a 0% THC group. Pain intensity was evaluated daily via telephone. Participants in the 9.4% THC group had statistically lower pain intensity compared with the 0% THC group (P = .023). Common adverse effects reported by those in the 9.4% group included headache, dry eyes, burning sensation, dizziness, numbness, and cough.
Box 2
The effects of Cannabis
Marijuana is harvested from the plant Cannabis sativa and composed of 400 lipophilic chemical compounds, including phytocannabinoids, terpenoids, and flavonoids.13 The plant contains compounds termed “cannabinoids.” Two of these derivatives in particular are responsible for most of the effects of marijuana: cannabinoid delta-9- tetrahydrocannabinol (THC) and cannabidiol (CBD). THC has a comparable structure and binding mechanism to anandamide, a naturally occurring fatty acid neurotransmitter present within the human brain.14-16 The endogenous endocannabinoid system and its receptors are found throughout the entire body (brain, organs, glands, immune cells, and connective tissues).
THC binds to cannabinoid receptors CB1 and CB2. CB1 is found predominantly in the CNS. CB2 is found predominantly outside the CNS and is associated with the immune system.14-16 The effects of THC include euphoria, relaxation, appetite stimulation, improvement of nausea and vomiting, analgesia, decreased muscle spasticity, and reduced eye pressure.14,15 CBD may have anxiolytic, antipsychotic, anticonvulsive, and analgesic effects.
The rate of absorption of THC and CBD depends both on the potency of the cannabinoid as well as the mechanism of consumption. Cannabis can be administered by multiple routes, including via smoking, oral ingestion, or IV.16 When Cannabis is smoked (the route for the most rapid delivery), THC is transported from the lungs to the bloodstream and reaches peak concentrations in 3 to 10 minutes. Oral ingestion (capsules, tinctures, sprays, and edibles) has a more flexible onset of action, usually occurring in 30 to 120 minutes, with effects lasting 5 to 6 hours. IV administration has rapid effects; the onset can occur within seconds to minutes, and effects can last 2 to 3 hours. The IV form allows 90% of THC to be distributed in plasma and can rapidly penetrate highly vascularized tissues, such as the liver, heart, fat, lungs, and muscles.
Pharmaceutical manufacturers have used cannabinoid derivatives to produce Cannabis-based medications for treating medical conditions. Nabilone, a potent agonist of the CB1 receptor, became available as a Schedule II medication in 1981 and was approved for patients with chemotherapy-induced nausea and vomiting (CINV).17 In 1985, dronabinol was introduced as an antiemetic for CINV as well as an appetite stimulant for patients with conditions associated with excessive weight loss.18 Another option, nabiximols, is an oral mucosal spray that consists of THC and CBD in a 1:1 ratio.19 Nabiximols is approved in Canada for pain relief in end-stage cancer patients and pain associated with multiple sclerosis.19
In an RCT of vaporized Cannabis, 39 patients with a diagnosis of complex regional pain syndrome, thalamic pain, spinal cord injury, peripheral neuropathy, radiculopathy, or nerve injury were assigned to a medium-dose (3.53% THC), low-dose (1.29% THC), or placebo group.25 Serious mental illness, substance abuse, and medical conditions were cause for exclusion. Participants received vaporized marijuana (average 8 to 12 puffs per visit) over 3 sessions. A 30% pain reduction was achieved by 26% of those in the placebo group, 57% of those in the low-dose group, and 61% of individuals in the high-dose group; the difference between placebo and each Cannabis group was statistically significant.
Chemotherapy-induced nausea and vomiting. Up to 80% of patients who receive chemotherapy experience CINV, which occurs from 24 hours to 7 days after receiving such therapy.26 CINV negatively influences a patient’s QOL and may impact the decision to continue with chemotherapy. Use of MM can help to diminish vomiting by binding to central CB1 receptors and averting the proemetic effects of dopamine and serotonin.27 Two synthetically derived cannabinoids, dronabinol and nabilone, are FDA-approved for treating CINV.
In a small (N = 64) parallel-group RCT, Meiri et al27 compared dronabinol with the commonly used antiemetic ondansetron and with a combination of dronabinol and ondansetron for treating CINV in adults. The primary outcome was prevention of delayed-onset CINV. Patients were eligible for this study if they had a malignancy that did not involve bone marrow, were receiving treatment with a moderately to highly emetogenic regimen, were not pregnant, and had an estimated life expectancy of at least 6 weeks after chemotherapy. The patients were randomized to 1 of 4 treatment groups: dronabinol alone, ondansetron alone, dronabinol plus ondansetron, or placebo. Overall, 47% to 58% of the active treatment groups improved, compared with 20% of the placebo group. Combination therapy did not provide any benefit beyond any single agent alone. All active treatments reduced nausea compared with placebo; there was no difference between active treatment groups. This study was limited by low enrollment.
Tramèr et al28 conducted a systematic review of 30 randomized comparisons of MM with placebo or antiemetics. The reviewed studies were completed between 1975 to 1997 and analyzed a total of 1,366 patients. Nabilone was evaluated in 16 trials; dronabinol was utilized in 13 trials; and IM levonantradol, a synthetic cannabinoid analog of dronabinol, was used in 1 trial. These agents were found to be more effective as an antiemetic compared with prochlorperazine, metoclopramide, chlorpromazine, thiethylperazine, haloperidol, domperidone, or alizapride. In addition, 38% to 90% of patients in these studies preferred MM over the traditional antiemetics.
A Cochrane review29 suggested that MM may be a viable option for treatment-resistant CINV; however, further studies are needed because current studies have methodological limitations.
Epilepsy. Maa and Figi30 reported a case of a 5-year-old girl who had Dravet syndrome, which resulted in 50 generalized tonic-clonic seizures daily; multiple anticonvulsants did not alleviate these seizures. Because of her recurring seizures, the patient had multiple cognitive and motor delays and needed a feeding tube. In addition to her existing antiepileptic drug regimen, she was started on adjunctive therapy with a sublingual Cannabis extract containing a high concentration of CBD. Her seizures decreased from 50 per day to 2 to 3 nocturnal convulsions per month. The treatment enabled her to stop using a feeding tube, resume walking and talking, and sleep soundly.
dos Santos et al31 reviewed studies of MM for treating epilepsy. One was a double-blind, placebo-controlled trial that included 15 patients ages 14 to 49 who had secondary generalized epilepsy with a temporal lobe focus. Eight patients received 200 to 300 mg/d of oral CBD for 8 to 18 weeks, and 7 received placebo. Seven patients had fewer seizures and 4 had no seizures. Only 1 patient in the placebo group demonstrated any improvement. Another study in this review included 19 children with treatment-resistant epilepsy: Dravet syndrome (n = 13), Doose syndrome (n = 4), Lennox-Gastaut syndrome (n = 1), or idiopathic epilepsy (n = 1). These patients experienced various types of seizures with a frequency ranging from 2 per week to 250 per day. Overall, 84% of children treated with CBD had fewer seizures: 11% were seizure-free, 42% had a >80% reduction in seizures, and 32% had a 25% to 60% reduction in seizures. Parents also noted additional benefits, including increased attention, improved mood, and improved sleep. CBD was well tolerated in most patients in both studies.
Despite these results, a Cochrane review32 found that no reliable conclusions can be drawn regarding the efficacy of MM for treating epilepsy.
Multiple sclerosis. According to American Academy of Neurology guidelines, physicians may provide MM as an alternative treatment for patients with MS-related spasticity.33 Multiple studies have tested MM and MM-related extracts for treating spasticity related to MS.34,35 In a placebo-controlled crossover study, Corey-Bloom et al34 reported a significant reduction in spasticity, measured using the modified Ashworth scale, in MS patients receiving Cannabis cigarettes vs placebo cigarettes (P < .0001). However, compared with the placebo group, patients who received MM had significant adverse effects, primarily cognitive impairment (P = .003).
In a multicenter RCT (N = 572 patients with refractory MS spasticity), Novotna et al36 evaluated nabiximols, an oral mucosal spray of a formulated extract of Cannabis that contains THC and CBD in a 1:1 ratio. They assessed spasticity using the Numerical Spasticity Rating Scale (NRS). Results were confirmed by measuring the number of daily spasms, self-report of sleep quality, and activities of daily living. After 4 weeks of single-blind treatment, patients who responded to nabiximols (≥20% improvement in spasticity) were randomized to a placebo group or nabiximols group for 12 additional weeks. After 12 weeks, compared with those who received placebo, those in the nabiximols group experienced a statistically significant reduction in spasticity based on NRS score (P = .0002).
For a summary of evidence on MM for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, see Box 3.37-43
Box 3
Cannabis for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
Glaucoma. In a placebo-controlled study, oromucosal administration of medical marijuana (MM) reduced intraocular pressure from 28 mm Hg to 22 mm Hg, with a duration of action of 3.5 hours.37However, the American Academy of Ophthalmologists does not recommend treating glaucoma with MM because the effect is short-lasting, and MM causes significant cognitive impairment compared with other standardized treatments.38 MM also leads to decreased blood pressure, which lowers blood flow to the optic nerve, thus increasing the risk of blindness.
Crohn’s disease. A randomized controlled trial (RCT) of MM for Crohn’s disease was conducted using the Crohn’s Disease Activity Index (CDAI) to assess for remission. In this 8-week study,21 individuals with Crohn’s disease were administered smoked MM (115 mg of delta-9-tetrahydrocannabinol [THC]) or placebo.39 Eligible patients were at least 20 years old, had active Crohn’s disease (CDAI >200), and had not responded to medical treatment for the illness. Compared with those who received placebo, patients who received MM experienced a statistically significant reduction in CDAI scores (P < .05). However, at follow-up 2 weeks after the study, when MM was no longer administered, there was no difference in mean CDAI scores between the 2 groups. Five of the 11 patients in the MM group achieved clinical remission, compared with 1 of 10 in the placebo group, but this difference was not statistically significant.
Parkinson’s disease (PD). According to the American Academy of Neurology, oral Cannabis extracts are “probably ineffective” for levodopa-induced dyskinesia in patients with PD.40 Reported benefits have come mainly from self-report studies. A 2014 survey (22 patients) found a significant reduction in PD symptoms—mainly relief from drug-induced tremor and pain—when measured using the Unified Parkinson’s Disease Rating Scale (UPDRS). Patients also reported better sleep and reduced pain (measured with a visual analog scale [VAS]). An exploratory double-blind placebo trial (N = 119) found no difference in mean UPDRS and no difference in any neuroprotective measures.41 However, the experimental group had a significantly higher quality of life (QOL; P = .05). A similar double-blind crossover study that included 19 patients found no significant difference in dyskinesia, as measured with the UPDRS, in the group receiving oral Cannabis extract compared with the placebo group.42
Amyotrophic lateral sclerosis (ALS). A randomized double-blind crossover trial of 27 ALS patients found that an oral THC extract (dronabinol, 5 mg, twice daily) had no significant effects on spasticity, as measured with the VAS.43 There was also no significant difference between the experimental and placebo groups on number of spasms (also measured with a VAS), quality of sleep (measured with the Sleep Disorders Questionnaire), or QOL (measured with the Amyotrophic Lateral Sclerosis Assessment questionnaire).
Psychiatric illnesses
Dementia-related behavioral disturbances. A few clinical trials with small sample sizes have found evidence supporting the use of MM compounds for alleviating neuropsychiatric symptoms of patients with dementia. An open-label pilot study of 6 individuals with late-stage dementia who received dronabinol, 2.5 mg/d, for 2 weeks, found a significant reduction (compared with baseline) in nighttime motor activity as measured with an actometer (P < .0028).44 The secondary Neuropsychiatric Inventory (NPI) assessment found reductions in aberrant motor behavior (P = .042), agitation (P = .042), and nighttime behaviors (P = .42).
A 2014 retrospective analysis of 40 inpatients with dementia-related agitation and appetite loss who were treated with dronabinol (mean dosage: 7.03 mg/d) found reductions in all aspects of agitation, including aberrant vocalization, motor agitation, aggressiveness, and treatment resistance, as measured with the Pittsburgh Agitation Scale (P < .0001).45 The study found no significant improvements in appetite, Global Assessment of Functioning mean score, or number of times patients awoke during the night. Adverse effects included sedation and delirium.
A RCT of 50 dementia patients with clinically relevant neuropsychiatric symptoms found no significant difference in mean NPI scores between patients given placebo and those who received nabiximols, 1.5 mg, 3 times daily.46 There were no significant differences found in agitation, QOL, life activities, or caregiver-scored Caregiver Global Impression of Change scale.
In a small RCT, THC was safe and well tolerated in 10 older patients with dementia.47 A 2009 Cochrane review48 concluded that there was no evidence for the efficacy of MM in treating the neuropsychiatric symptoms related to dementia.
PTSD. Preclinical evidence shows that the endocannabinoid system is involved in regulating emotional memory. Evidence also suggests that cannabinoids may facilitate the extinction of aversive memories.49,50
In 2009, New Mexico became the first state to authorize the use of MM for patients with PTSD. In a study of patients applying for the New Mexico Medical Cannabis Program, researchers used the Clinician Administered Posttraumatic Scale (CAPS) to assess PTSD symptoms.51 A retrospective chart review of the first 80 patients evaluated found significant (P < .0001) reductions of several PTSD symptoms, including intrusive memories, distressing dreams, flashbacks, numbing and avoidance, and hyperarousal, in the group using MM vs those not using MM. There also was a significant difference in CAPS total score (P < .0001). Patients reported a 75% reduction in PTSD symptoms while using MM. This study has several limitations: It was a retrospective review, not an RCT, and patients were prescreened and knew before the study began that MM helped their PTSD symptoms.
In another retrospective study, researchers evaluated treatment with nabilone, 0.5 to 6 mg/d, in 104 incarcerated men with various major mental illnesses; most (91%) met criteria for Cannabis dependence.52 They found significant improvements in sleep and PTSD symptoms.
A double-blind RCT evaluated MM in 10 Canadian male soldiers with PTSD who experienced nightmares despite standard medication treatment. Adjunctive nabilone (maximum dose: 3 mg/d) resulted in a reduction in nightmares as measured by the CAPS recurrent distressing dream of the event item score.53
Currently, there are no adequately powered RCTs of MM in a diverse group of PTSD patients. Most studies are open-label, enriched design, and included white male veterans. No well-conducted trials have evaluated patients with noncombat-related PTSD. Most of the relevant literature consists of case reports of Cannabis use by patients with PTSD.
Anxiety disorders.Patients frequently indicate that smoking Cannabis helps relieve their anxiety, although there is no replicated evidence based on double-blind RCTs to support this. However, in rat models CBD has been shown to facilitate extinction of conditioned fear via the endocannabinoid system.54-56 The mechanism of action is not completely understood. CBD has been shown to have antagonistic action at CB1 and CB2 receptors. It may have similar effects on memory extinction and may be an adjunct to exposure therapies for anxiety disorders.
Das et al57 studied the effects of CBD (32 mg) on extinction and consolidation of memory related to contextual fear in 48 individuals. They found that CBD can enhance extinction learning, and suggested it may have potential as an adjunct to extinction-based therapies for anxiety disorders.
Caveats: Adverse effects, lack of RCTs
Cannabis use causes impairment of learning, memory, attention, and working memory. Adolescents are particularly vulnerable to the effects of Cannabis on brain development at a time when synaptic pruning and increased myelination occur. Normal brain development could be disrupted. Some studies have linked Cannabis use to abnormalities in the amygdala, hippocampus, frontal lobe, and cerebellum. From 1995 to 2014, the potency of Cannabis (THC concentration) increased from 4% to 12%.58 This has substantial implications for increased abuse among adolescents and the deleterious effects of Cannabis on the brain.
Heavy Cannabis use impairs motivation and could precipitate psychosis in vulnerable individuals. Cannabis use may be linked to the development of schizophrenia.59
There are no well-conducted RCTs on the efficacy of MM, and adequate safety data are lacking. There is also lack of consensus among qualified experts. There is soft evidence that MM may be helpful in some medical conditions, including but not limited to CINV, neuropathic pain, epilepsy, and MS-related spasticity. Currently, the benefits of using MM do not appear to outweigh the risks.
Bottom Line
Limited evidence suggests medical marijuana (MM) may be beneficial for treating a few medical conditions, including neuropathic pain and chemotherapy-induced nausea and vomiting. There is no clear and convincing evidence MM is beneficial for psychiatric disorders, and Cannabis can impair cognition and attention and may precipitate psychosis. The risk of deleterious effects are greater in adolescents.
Related Resources
- Nguyen DH, Thant TM. Caring for medical marijuana patients who request controlled prescriptions. Current Psychiatry. 2017;16(8):50-51.
- National Institute on Drug Abuse. Marijuana as medicine. https://www.drugabuse.gov/publications/drugfacts/ marijuana-medicine.
Drug Brand Names
Alizapride • Litican, Superan
Chlorpromazine • Thorazine
Domperidone • Motilium
Dronabinol • Marinol, Syndros
Haloperidol • Haldol
Metoclopramide • Reglan
Nabilone • Cesamet
Nabiximols • Sativex
Ondansetron • Zofran, Zuplenz
Prochlorperazine • Compazine
Thiethylperazine • Torecan
1. National Institute on Drug Abuse (NIDA). Marijuana Research Findings 1976. NIDA research monograph 14. https://archives.drugabuse.gov/sites/default/files/monograph14.pdf. Published July 1977. Accessed November 15, 2017.
2. O’Shaughnessy WB. On the preparations of the Indian hemp, or gunjah- cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363-369.
3. Clendinning J. Observations on the medical properties of the Cannabis Sativa of India. Med Chir Trans. 1843;26:188-210.
4. Osler W, McCrae T. The principles and practice of medicine. 9th ed. New York, NY: D. Appleton and Company; 1921.
5. The pharmacopoeia of the United States of America. 3rd ed. Philadelphia, PA: Lippincott; 1851.
6. The pharmacopoeia of the United States of America. 12th ed. Easton, PA: Mack Printing Company; 1942.
7. Philipsen N, Butler RD, Simon C, et al. Medical marijuana: a primer on ethics, evidence, and politics. Journal Nurse Pract. 2014;10(9):633-640.
8. Marihuana Tax Act of 1937, Pub L No. 75-238, 75th Cong, 50 Stat 551 (1937).
9. Controlled Substances Act, 21 USC §812.
10. Watson SJ, Benson JA, Joy JE, eds. Marijuana and medicine: assessing the science base. Washington, DC: National Academy Press; 1999.
11. California Proposition 215, the medical marijuana initiative (1996). https://ballotpedia.org/California_Proposition_215,_the_Medical_Marijuana_Initiative_(1996). Accessed November 16, 2017.
12. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated September 14, 2017. Accessed November 16, 2017.
13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344-1364.
14. Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013:14. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3997295. Accessed December 5, 2017.
15. Galal AM, Slade D, Gul W, et al. Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications. Recent Pat CNS Drug Discov. 2009;4(2):112-136.
16. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804.
17. Cesamet [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2013.
18. Marinol [package insert]. Chicago, IL: AbbVie Inc.; 2017.
19. Sativex [package insert]. Mississauga, Ontario: Bayer Inc.; 2015.
20. Torrance N, Ferguson JA, Afolabi E, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690-699.
21. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162-173.
22. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313(24):2456-2473.
23. Wilsey B, Marcotte, T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9(6):506-521.
24. Ware MA, Wang T, Shapiro S, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ. 2010;182(14):E694-E701.
25. Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14(2):136-148.
26. National Cancer Institute. Treatment-related nausea and vomiting (PDQ®)-health professional version. https://www.cancer.gov/about-cancer/treatment/side-effects/nausea/nausea-hp-pdq. Updated May 10, 2017. Accessed November 7, 2017.
27. Meiri E, Jhangiani H, Vrendenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533-543.
28. Tramèr MR, Carroll D, Campbell FA, et al. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ. 2001;323(7303):16-21.
29. Smith LA, Azariah F, Lavender VT, et al. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev. 2015;(11):CD009464.
30. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783-786.
31. dos Santos RG, Hallak JE, Leite JP, et al. Phytocannabinoids and epilepsy. J Clin Pharm Ther. 2015;40(2):135-143.
32. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;(3):CD009270.
33. Yadav V, Bever C Jr, Bowen J, et al. Summary of evidence-based guideline: complementary and alternative medicine in multiple sclerosis: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2014;82(12):1083-1092.
34. Corey-Bloom J, Wolfson T, Gamst A, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. CMAJ. 2012;184(10):1143-1150.
35. Zajicek J, Ball S, Wright D, et al; CUPID investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
36. Novotna A, Mares J, Ratcliffe S, et al; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122-1131.
37. Merritt JC, Crawford WJ, Alexander PC, et al. Effect of marihuana on intraocular and blood pressure in glaucoma. Ophthalmology. 1980;87(3):222-228.
38. American Academy of Ophthalmology. American Academy of Ophthalmology reiterates position that marijuana is not a proven treatment for glaucoma. https://www.aao.org/newsroom/news-releases/detail/american-academy-of-ophthalmology-reiterates-posit. Published June 27, 2014. Accessed May 29, 2017.
39. Naftali T, Bar-Lev Schleider L, Dotan I, et al. Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11(10):1276.e1-1280.e1.
40. Koppel BS Brust JC, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in certain neurological disorders. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82(17):1556-1563.
41. Chagas MH, Zuardi AW, Tumas V, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. 2014;28(11):1088-1098.
42. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology. 2004;63(7):1245-1250.
43. Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135-1140.
44. Walther S, Mahlberg R, Eichmann U, et al. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006;185(4):524-528.
45. Woodward MR, Harper DG, Stolyar A, et al. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am J Geriatr Psychiatry. 2014;22(4):415-419.
46. van den Elsen GA, Ahmed A, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: a randomized controlled trial. Neurology. 2015;84(23):2338-2346.
47. Ahmed AI, van den Elsen GA, Colbers A, et al. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology (Berl). 2015;232(14):25872595.
48. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev. 2009;(2):CD007204.
49. de Bitencourt RM, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389-395.
50. Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci Ther. 2009;15(1):84-88.
51. Greer GR, Grob CS, Halberstadt AL. PTSD symptom reports of patients evaluated for the New Mexico Medical Cannabis Program. J Psychoactive Drugs. 2014;46(1):73-77.
52. Cameron C, Watson D, Robinson J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. J Clin Psychopharmacol. 2014;34(5):559-564.
53. Jetly R, Heber A, Fraser G, et al. The efficacy of nabilone, a synthetic cannabinoid, in the treatment of PTSD-associated nightmares: a preliminary randomized, double-blind, placebo-controlled cross-over design study. Psychoneuroendocrinology. 2015;51:585-588.
54. Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear, memory extinction, and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18(12):849-859.
55. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199-215.
56. Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613-623.
57. Das RK, Kamboj SK, Ramadas M, et al. Cannabidiol enhances consolidation of explicit fear extinction in humans. Psychopharmacology (Berl). 2013;226(4):781-792.
58. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613-619.
59. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73(3):292297.
There is a need for additional treatment options to improve symptoms, enhance the quality of life (QOL), and reduce suffering among patients who have chronic medical illness. Medical marijuana (MM) has the potential to help patients who have certain medical conditions in states where it is legal for prescription by a licensed medical provider.
Cannabis has a long history of medicinal use (Box 11-12). Two derivatives of the Cannabis plant—cannabinoid delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD)—are responsible for most of its effects. Some of these effects, including analgesia, decreased muscle spasticity, and reduced eye pressure, have been harnessed for their potential therapeutic effects (Box 213-19). As of November 2017, 29 states had legalized Cannabis for medical use, and several had legalized its recreational use.12
With the increasing availability of MM, psychiatrists are likely to encounter patients who are using it or who will ask them about it. This article reviews evidence related to using MM to treat patients with neuropathic pain; chemotherapyinduced nausea and vomiting (CINV); epilepsy; multiple sclerosis (MS); glaucoma; Crohn’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; dementia-related behavioral disturbances; posttraumatic stress disorder (PTSD); and anxiety.
Box 1
Cannabis: A history of medicinal use
Cannabis has been cultivated since ancient times, beginning in China and India. The earliest reference of its use for healing purposes may have been in the Chinese Pharmacopeia, circa 1500 BC.1 In 1839, Dr. William Brooke O’Shaughnessy introduced Cannabis Indica, or “Indian hemp,” to the western world after a professorship in Calcutta, India.2 In the early 1840s, an English physician, Dr. John Clendinning, prescribed Cannabis for migraine headache.3 In the 19th and early 20th centuries, several prominent physicians advocated using Cannabis for migraines; Sir William Osler did so in his textbook, The principles and practice of medicine.4 It was listed in the U.S. Pharmacopeia in 1850 but removed in 1942.5,6
Until 1937, Cannabis was used in the United States for medicinal purposes, such as for treating inflamed skin, incontinence, and sexually transmitted diseases.7 In 1937, the Marihuana Tax Act, which prohibited the production, importation, possession, use, and dispersal of Cannabis, was passed.8 Cannabis became a Schedule I drug under the Controlled Substance Act of 1970.9
In 1999, based on available evidence, the Institute of Medicine (IOM) concluded Cannabis had less likelihood of dependence than benzodiazepines, opiates, cocaine, or nicotine. The IOM also concluded that the symptoms of withdrawal were mild in comparison with benzodiazepines or opiates. Finally, the IOM stated that Cannabis was not a “gateway” drug.10
In 1996, California was the first state to reimplement medicinal use of Cannabis under the Compassionate Use Act, also known as Proposition 215.11 This act allowed individuals to retain or produce Cannabis for personal consumption with a physician’s approval. Many states eventually followed California’s lead. As of November 2017, 29 states, the District of Columbia, Guam, and Puerto Rico had regulated Cannabis use for medical purposes,12 and recreational use had been approved in 7 states and the District of Columbia.
Medical illnesses
Neuropathic pain. Chronic neuropathic pain affects an estimated 7% to 8% of adults.20 Patients with neuropathic pain are often treated with anticonvulsants, antidepressants, opioids, and local anesthetics21; however, these medications may not provide substantial relief. Research has revealed that THC and CBD can improve central and peripheral neuropathic pain, as well as pain associated with rheumatoid arthritis and fibromyalgia.22
Wilsey et al23 evaluated the analgesic effects of smoked MM for neuropathic pain in a small (N = 38) double-blind, randomized controlled trial (RCT). Patients in this study had a preexisting diagnosis of complex regional pain syndrome, spinal cord injury, peripheral neuropathy, or nerve injury. To prevent any unforeseen adverse outcomes related to Cannabis use, participants were required to have previous exposure to Cannabis. Patients were excluded if they had major mental illness, substance abuse, or other major medical ailments.
Participants smoked high-dose Cannabis cigarettes (7% THC), low-dose Cannabis cigarettes (3.5% THC), or placebo cigarettes. Pain was measured on a visual analog scale (VAS) that ranged from 0 (no pain) to 100 (worst possible pain). Compared with the placebo group, significant analgesia was achieved in both Cannabis groups (P = .016). The high-dose group had greater neurocognitive impairment.
Ware et al24 conducted a crossover RCT (N = 23) to determine the efficacy of smoked MM for neuropathic pain. Participants had neuropathic pain for at least 3 months that was caused by trauma or surgery, with an average weekly pain intensity score >4 on scale of 0 to 10. Patients with pain due to cancer, nociceptive causes, unstable medical conditions, current substance abuse, history of a psychotic disorder, or suicidal ideation were excluded. Participants were assigned to a 9.4% THC group or a 0% THC group. Pain intensity was evaluated daily via telephone. Participants in the 9.4% THC group had statistically lower pain intensity compared with the 0% THC group (P = .023). Common adverse effects reported by those in the 9.4% group included headache, dry eyes, burning sensation, dizziness, numbness, and cough.
Box 2
The effects of Cannabis
Marijuana is harvested from the plant Cannabis sativa and composed of 400 lipophilic chemical compounds, including phytocannabinoids, terpenoids, and flavonoids.13 The plant contains compounds termed “cannabinoids.” Two of these derivatives in particular are responsible for most of the effects of marijuana: cannabinoid delta-9- tetrahydrocannabinol (THC) and cannabidiol (CBD). THC has a comparable structure and binding mechanism to anandamide, a naturally occurring fatty acid neurotransmitter present within the human brain.14-16 The endogenous endocannabinoid system and its receptors are found throughout the entire body (brain, organs, glands, immune cells, and connective tissues).
THC binds to cannabinoid receptors CB1 and CB2. CB1 is found predominantly in the CNS. CB2 is found predominantly outside the CNS and is associated with the immune system.14-16 The effects of THC include euphoria, relaxation, appetite stimulation, improvement of nausea and vomiting, analgesia, decreased muscle spasticity, and reduced eye pressure.14,15 CBD may have anxiolytic, antipsychotic, anticonvulsive, and analgesic effects.
The rate of absorption of THC and CBD depends both on the potency of the cannabinoid as well as the mechanism of consumption. Cannabis can be administered by multiple routes, including via smoking, oral ingestion, or IV.16 When Cannabis is smoked (the route for the most rapid delivery), THC is transported from the lungs to the bloodstream and reaches peak concentrations in 3 to 10 minutes. Oral ingestion (capsules, tinctures, sprays, and edibles) has a more flexible onset of action, usually occurring in 30 to 120 minutes, with effects lasting 5 to 6 hours. IV administration has rapid effects; the onset can occur within seconds to minutes, and effects can last 2 to 3 hours. The IV form allows 90% of THC to be distributed in plasma and can rapidly penetrate highly vascularized tissues, such as the liver, heart, fat, lungs, and muscles.
Pharmaceutical manufacturers have used cannabinoid derivatives to produce Cannabis-based medications for treating medical conditions. Nabilone, a potent agonist of the CB1 receptor, became available as a Schedule II medication in 1981 and was approved for patients with chemotherapy-induced nausea and vomiting (CINV).17 In 1985, dronabinol was introduced as an antiemetic for CINV as well as an appetite stimulant for patients with conditions associated with excessive weight loss.18 Another option, nabiximols, is an oral mucosal spray that consists of THC and CBD in a 1:1 ratio.19 Nabiximols is approved in Canada for pain relief in end-stage cancer patients and pain associated with multiple sclerosis.19
In an RCT of vaporized Cannabis, 39 patients with a diagnosis of complex regional pain syndrome, thalamic pain, spinal cord injury, peripheral neuropathy, radiculopathy, or nerve injury were assigned to a medium-dose (3.53% THC), low-dose (1.29% THC), or placebo group.25 Serious mental illness, substance abuse, and medical conditions were cause for exclusion. Participants received vaporized marijuana (average 8 to 12 puffs per visit) over 3 sessions. A 30% pain reduction was achieved by 26% of those in the placebo group, 57% of those in the low-dose group, and 61% of individuals in the high-dose group; the difference between placebo and each Cannabis group was statistically significant.
Chemotherapy-induced nausea and vomiting. Up to 80% of patients who receive chemotherapy experience CINV, which occurs from 24 hours to 7 days after receiving such therapy.26 CINV negatively influences a patient’s QOL and may impact the decision to continue with chemotherapy. Use of MM can help to diminish vomiting by binding to central CB1 receptors and averting the proemetic effects of dopamine and serotonin.27 Two synthetically derived cannabinoids, dronabinol and nabilone, are FDA-approved for treating CINV.
In a small (N = 64) parallel-group RCT, Meiri et al27 compared dronabinol with the commonly used antiemetic ondansetron and with a combination of dronabinol and ondansetron for treating CINV in adults. The primary outcome was prevention of delayed-onset CINV. Patients were eligible for this study if they had a malignancy that did not involve bone marrow, were receiving treatment with a moderately to highly emetogenic regimen, were not pregnant, and had an estimated life expectancy of at least 6 weeks after chemotherapy. The patients were randomized to 1 of 4 treatment groups: dronabinol alone, ondansetron alone, dronabinol plus ondansetron, or placebo. Overall, 47% to 58% of the active treatment groups improved, compared with 20% of the placebo group. Combination therapy did not provide any benefit beyond any single agent alone. All active treatments reduced nausea compared with placebo; there was no difference between active treatment groups. This study was limited by low enrollment.
Tramèr et al28 conducted a systematic review of 30 randomized comparisons of MM with placebo or antiemetics. The reviewed studies were completed between 1975 to 1997 and analyzed a total of 1,366 patients. Nabilone was evaluated in 16 trials; dronabinol was utilized in 13 trials; and IM levonantradol, a synthetic cannabinoid analog of dronabinol, was used in 1 trial. These agents were found to be more effective as an antiemetic compared with prochlorperazine, metoclopramide, chlorpromazine, thiethylperazine, haloperidol, domperidone, or alizapride. In addition, 38% to 90% of patients in these studies preferred MM over the traditional antiemetics.
A Cochrane review29 suggested that MM may be a viable option for treatment-resistant CINV; however, further studies are needed because current studies have methodological limitations.
Epilepsy. Maa and Figi30 reported a case of a 5-year-old girl who had Dravet syndrome, which resulted in 50 generalized tonic-clonic seizures daily; multiple anticonvulsants did not alleviate these seizures. Because of her recurring seizures, the patient had multiple cognitive and motor delays and needed a feeding tube. In addition to her existing antiepileptic drug regimen, she was started on adjunctive therapy with a sublingual Cannabis extract containing a high concentration of CBD. Her seizures decreased from 50 per day to 2 to 3 nocturnal convulsions per month. The treatment enabled her to stop using a feeding tube, resume walking and talking, and sleep soundly.
dos Santos et al31 reviewed studies of MM for treating epilepsy. One was a double-blind, placebo-controlled trial that included 15 patients ages 14 to 49 who had secondary generalized epilepsy with a temporal lobe focus. Eight patients received 200 to 300 mg/d of oral CBD for 8 to 18 weeks, and 7 received placebo. Seven patients had fewer seizures and 4 had no seizures. Only 1 patient in the placebo group demonstrated any improvement. Another study in this review included 19 children with treatment-resistant epilepsy: Dravet syndrome (n = 13), Doose syndrome (n = 4), Lennox-Gastaut syndrome (n = 1), or idiopathic epilepsy (n = 1). These patients experienced various types of seizures with a frequency ranging from 2 per week to 250 per day. Overall, 84% of children treated with CBD had fewer seizures: 11% were seizure-free, 42% had a >80% reduction in seizures, and 32% had a 25% to 60% reduction in seizures. Parents also noted additional benefits, including increased attention, improved mood, and improved sleep. CBD was well tolerated in most patients in both studies.
Despite these results, a Cochrane review32 found that no reliable conclusions can be drawn regarding the efficacy of MM for treating epilepsy.
Multiple sclerosis. According to American Academy of Neurology guidelines, physicians may provide MM as an alternative treatment for patients with MS-related spasticity.33 Multiple studies have tested MM and MM-related extracts for treating spasticity related to MS.34,35 In a placebo-controlled crossover study, Corey-Bloom et al34 reported a significant reduction in spasticity, measured using the modified Ashworth scale, in MS patients receiving Cannabis cigarettes vs placebo cigarettes (P < .0001). However, compared with the placebo group, patients who received MM had significant adverse effects, primarily cognitive impairment (P = .003).
In a multicenter RCT (N = 572 patients with refractory MS spasticity), Novotna et al36 evaluated nabiximols, an oral mucosal spray of a formulated extract of Cannabis that contains THC and CBD in a 1:1 ratio. They assessed spasticity using the Numerical Spasticity Rating Scale (NRS). Results were confirmed by measuring the number of daily spasms, self-report of sleep quality, and activities of daily living. After 4 weeks of single-blind treatment, patients who responded to nabiximols (≥20% improvement in spasticity) were randomized to a placebo group or nabiximols group for 12 additional weeks. After 12 weeks, compared with those who received placebo, those in the nabiximols group experienced a statistically significant reduction in spasticity based on NRS score (P = .0002).
For a summary of evidence on MM for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, see Box 3.37-43
Box 3
Cannabis for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
Glaucoma. In a placebo-controlled study, oromucosal administration of medical marijuana (MM) reduced intraocular pressure from 28 mm Hg to 22 mm Hg, with a duration of action of 3.5 hours.37However, the American Academy of Ophthalmologists does not recommend treating glaucoma with MM because the effect is short-lasting, and MM causes significant cognitive impairment compared with other standardized treatments.38 MM also leads to decreased blood pressure, which lowers blood flow to the optic nerve, thus increasing the risk of blindness.
Crohn’s disease. A randomized controlled trial (RCT) of MM for Crohn’s disease was conducted using the Crohn’s Disease Activity Index (CDAI) to assess for remission. In this 8-week study,21 individuals with Crohn’s disease were administered smoked MM (115 mg of delta-9-tetrahydrocannabinol [THC]) or placebo.39 Eligible patients were at least 20 years old, had active Crohn’s disease (CDAI >200), and had not responded to medical treatment for the illness. Compared with those who received placebo, patients who received MM experienced a statistically significant reduction in CDAI scores (P < .05). However, at follow-up 2 weeks after the study, when MM was no longer administered, there was no difference in mean CDAI scores between the 2 groups. Five of the 11 patients in the MM group achieved clinical remission, compared with 1 of 10 in the placebo group, but this difference was not statistically significant.
Parkinson’s disease (PD). According to the American Academy of Neurology, oral Cannabis extracts are “probably ineffective” for levodopa-induced dyskinesia in patients with PD.40 Reported benefits have come mainly from self-report studies. A 2014 survey (22 patients) found a significant reduction in PD symptoms—mainly relief from drug-induced tremor and pain—when measured using the Unified Parkinson’s Disease Rating Scale (UPDRS). Patients also reported better sleep and reduced pain (measured with a visual analog scale [VAS]). An exploratory double-blind placebo trial (N = 119) found no difference in mean UPDRS and no difference in any neuroprotective measures.41 However, the experimental group had a significantly higher quality of life (QOL; P = .05). A similar double-blind crossover study that included 19 patients found no significant difference in dyskinesia, as measured with the UPDRS, in the group receiving oral Cannabis extract compared with the placebo group.42
Amyotrophic lateral sclerosis (ALS). A randomized double-blind crossover trial of 27 ALS patients found that an oral THC extract (dronabinol, 5 mg, twice daily) had no significant effects on spasticity, as measured with the VAS.43 There was also no significant difference between the experimental and placebo groups on number of spasms (also measured with a VAS), quality of sleep (measured with the Sleep Disorders Questionnaire), or QOL (measured with the Amyotrophic Lateral Sclerosis Assessment questionnaire).
Psychiatric illnesses
Dementia-related behavioral disturbances. A few clinical trials with small sample sizes have found evidence supporting the use of MM compounds for alleviating neuropsychiatric symptoms of patients with dementia. An open-label pilot study of 6 individuals with late-stage dementia who received dronabinol, 2.5 mg/d, for 2 weeks, found a significant reduction (compared with baseline) in nighttime motor activity as measured with an actometer (P < .0028).44 The secondary Neuropsychiatric Inventory (NPI) assessment found reductions in aberrant motor behavior (P = .042), agitation (P = .042), and nighttime behaviors (P = .42).
A 2014 retrospective analysis of 40 inpatients with dementia-related agitation and appetite loss who were treated with dronabinol (mean dosage: 7.03 mg/d) found reductions in all aspects of agitation, including aberrant vocalization, motor agitation, aggressiveness, and treatment resistance, as measured with the Pittsburgh Agitation Scale (P < .0001).45 The study found no significant improvements in appetite, Global Assessment of Functioning mean score, or number of times patients awoke during the night. Adverse effects included sedation and delirium.
A RCT of 50 dementia patients with clinically relevant neuropsychiatric symptoms found no significant difference in mean NPI scores between patients given placebo and those who received nabiximols, 1.5 mg, 3 times daily.46 There were no significant differences found in agitation, QOL, life activities, or caregiver-scored Caregiver Global Impression of Change scale.
In a small RCT, THC was safe and well tolerated in 10 older patients with dementia.47 A 2009 Cochrane review48 concluded that there was no evidence for the efficacy of MM in treating the neuropsychiatric symptoms related to dementia.
PTSD. Preclinical evidence shows that the endocannabinoid system is involved in regulating emotional memory. Evidence also suggests that cannabinoids may facilitate the extinction of aversive memories.49,50
In 2009, New Mexico became the first state to authorize the use of MM for patients with PTSD. In a study of patients applying for the New Mexico Medical Cannabis Program, researchers used the Clinician Administered Posttraumatic Scale (CAPS) to assess PTSD symptoms.51 A retrospective chart review of the first 80 patients evaluated found significant (P < .0001) reductions of several PTSD symptoms, including intrusive memories, distressing dreams, flashbacks, numbing and avoidance, and hyperarousal, in the group using MM vs those not using MM. There also was a significant difference in CAPS total score (P < .0001). Patients reported a 75% reduction in PTSD symptoms while using MM. This study has several limitations: It was a retrospective review, not an RCT, and patients were prescreened and knew before the study began that MM helped their PTSD symptoms.
In another retrospective study, researchers evaluated treatment with nabilone, 0.5 to 6 mg/d, in 104 incarcerated men with various major mental illnesses; most (91%) met criteria for Cannabis dependence.52 They found significant improvements in sleep and PTSD symptoms.
A double-blind RCT evaluated MM in 10 Canadian male soldiers with PTSD who experienced nightmares despite standard medication treatment. Adjunctive nabilone (maximum dose: 3 mg/d) resulted in a reduction in nightmares as measured by the CAPS recurrent distressing dream of the event item score.53
Currently, there are no adequately powered RCTs of MM in a diverse group of PTSD patients. Most studies are open-label, enriched design, and included white male veterans. No well-conducted trials have evaluated patients with noncombat-related PTSD. Most of the relevant literature consists of case reports of Cannabis use by patients with PTSD.
Anxiety disorders.Patients frequently indicate that smoking Cannabis helps relieve their anxiety, although there is no replicated evidence based on double-blind RCTs to support this. However, in rat models CBD has been shown to facilitate extinction of conditioned fear via the endocannabinoid system.54-56 The mechanism of action is not completely understood. CBD has been shown to have antagonistic action at CB1 and CB2 receptors. It may have similar effects on memory extinction and may be an adjunct to exposure therapies for anxiety disorders.
Das et al57 studied the effects of CBD (32 mg) on extinction and consolidation of memory related to contextual fear in 48 individuals. They found that CBD can enhance extinction learning, and suggested it may have potential as an adjunct to extinction-based therapies for anxiety disorders.
Caveats: Adverse effects, lack of RCTs
Cannabis use causes impairment of learning, memory, attention, and working memory. Adolescents are particularly vulnerable to the effects of Cannabis on brain development at a time when synaptic pruning and increased myelination occur. Normal brain development could be disrupted. Some studies have linked Cannabis use to abnormalities in the amygdala, hippocampus, frontal lobe, and cerebellum. From 1995 to 2014, the potency of Cannabis (THC concentration) increased from 4% to 12%.58 This has substantial implications for increased abuse among adolescents and the deleterious effects of Cannabis on the brain.
Heavy Cannabis use impairs motivation and could precipitate psychosis in vulnerable individuals. Cannabis use may be linked to the development of schizophrenia.59
There are no well-conducted RCTs on the efficacy of MM, and adequate safety data are lacking. There is also lack of consensus among qualified experts. There is soft evidence that MM may be helpful in some medical conditions, including but not limited to CINV, neuropathic pain, epilepsy, and MS-related spasticity. Currently, the benefits of using MM do not appear to outweigh the risks.
Bottom Line
Limited evidence suggests medical marijuana (MM) may be beneficial for treating a few medical conditions, including neuropathic pain and chemotherapy-induced nausea and vomiting. There is no clear and convincing evidence MM is beneficial for psychiatric disorders, and Cannabis can impair cognition and attention and may precipitate psychosis. The risk of deleterious effects are greater in adolescents.
Related Resources
- Nguyen DH, Thant TM. Caring for medical marijuana patients who request controlled prescriptions. Current Psychiatry. 2017;16(8):50-51.
- National Institute on Drug Abuse. Marijuana as medicine. https://www.drugabuse.gov/publications/drugfacts/ marijuana-medicine.
Drug Brand Names
Alizapride • Litican, Superan
Chlorpromazine • Thorazine
Domperidone • Motilium
Dronabinol • Marinol, Syndros
Haloperidol • Haldol
Metoclopramide • Reglan
Nabilone • Cesamet
Nabiximols • Sativex
Ondansetron • Zofran, Zuplenz
Prochlorperazine • Compazine
Thiethylperazine • Torecan
There is a need for additional treatment options to improve symptoms, enhance the quality of life (QOL), and reduce suffering among patients who have chronic medical illness. Medical marijuana (MM) has the potential to help patients who have certain medical conditions in states where it is legal for prescription by a licensed medical provider.
Cannabis has a long history of medicinal use (Box 11-12). Two derivatives of the Cannabis plant—cannabinoid delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD)—are responsible for most of its effects. Some of these effects, including analgesia, decreased muscle spasticity, and reduced eye pressure, have been harnessed for their potential therapeutic effects (Box 213-19). As of November 2017, 29 states had legalized Cannabis for medical use, and several had legalized its recreational use.12
With the increasing availability of MM, psychiatrists are likely to encounter patients who are using it or who will ask them about it. This article reviews evidence related to using MM to treat patients with neuropathic pain; chemotherapyinduced nausea and vomiting (CINV); epilepsy; multiple sclerosis (MS); glaucoma; Crohn’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; dementia-related behavioral disturbances; posttraumatic stress disorder (PTSD); and anxiety.
Box 1
Cannabis: A history of medicinal use
Cannabis has been cultivated since ancient times, beginning in China and India. The earliest reference of its use for healing purposes may have been in the Chinese Pharmacopeia, circa 1500 BC.1 In 1839, Dr. William Brooke O’Shaughnessy introduced Cannabis Indica, or “Indian hemp,” to the western world after a professorship in Calcutta, India.2 In the early 1840s, an English physician, Dr. John Clendinning, prescribed Cannabis for migraine headache.3 In the 19th and early 20th centuries, several prominent physicians advocated using Cannabis for migraines; Sir William Osler did so in his textbook, The principles and practice of medicine.4 It was listed in the U.S. Pharmacopeia in 1850 but removed in 1942.5,6
Until 1937, Cannabis was used in the United States for medicinal purposes, such as for treating inflamed skin, incontinence, and sexually transmitted diseases.7 In 1937, the Marihuana Tax Act, which prohibited the production, importation, possession, use, and dispersal of Cannabis, was passed.8 Cannabis became a Schedule I drug under the Controlled Substance Act of 1970.9
In 1999, based on available evidence, the Institute of Medicine (IOM) concluded Cannabis had less likelihood of dependence than benzodiazepines, opiates, cocaine, or nicotine. The IOM also concluded that the symptoms of withdrawal were mild in comparison with benzodiazepines or opiates. Finally, the IOM stated that Cannabis was not a “gateway” drug.10
In 1996, California was the first state to reimplement medicinal use of Cannabis under the Compassionate Use Act, also known as Proposition 215.11 This act allowed individuals to retain or produce Cannabis for personal consumption with a physician’s approval. Many states eventually followed California’s lead. As of November 2017, 29 states, the District of Columbia, Guam, and Puerto Rico had regulated Cannabis use for medical purposes,12 and recreational use had been approved in 7 states and the District of Columbia.
Medical illnesses
Neuropathic pain. Chronic neuropathic pain affects an estimated 7% to 8% of adults.20 Patients with neuropathic pain are often treated with anticonvulsants, antidepressants, opioids, and local anesthetics21; however, these medications may not provide substantial relief. Research has revealed that THC and CBD can improve central and peripheral neuropathic pain, as well as pain associated with rheumatoid arthritis and fibromyalgia.22
Wilsey et al23 evaluated the analgesic effects of smoked MM for neuropathic pain in a small (N = 38) double-blind, randomized controlled trial (RCT). Patients in this study had a preexisting diagnosis of complex regional pain syndrome, spinal cord injury, peripheral neuropathy, or nerve injury. To prevent any unforeseen adverse outcomes related to Cannabis use, participants were required to have previous exposure to Cannabis. Patients were excluded if they had major mental illness, substance abuse, or other major medical ailments.
Participants smoked high-dose Cannabis cigarettes (7% THC), low-dose Cannabis cigarettes (3.5% THC), or placebo cigarettes. Pain was measured on a visual analog scale (VAS) that ranged from 0 (no pain) to 100 (worst possible pain). Compared with the placebo group, significant analgesia was achieved in both Cannabis groups (P = .016). The high-dose group had greater neurocognitive impairment.
Ware et al24 conducted a crossover RCT (N = 23) to determine the efficacy of smoked MM for neuropathic pain. Participants had neuropathic pain for at least 3 months that was caused by trauma or surgery, with an average weekly pain intensity score >4 on scale of 0 to 10. Patients with pain due to cancer, nociceptive causes, unstable medical conditions, current substance abuse, history of a psychotic disorder, or suicidal ideation were excluded. Participants were assigned to a 9.4% THC group or a 0% THC group. Pain intensity was evaluated daily via telephone. Participants in the 9.4% THC group had statistically lower pain intensity compared with the 0% THC group (P = .023). Common adverse effects reported by those in the 9.4% group included headache, dry eyes, burning sensation, dizziness, numbness, and cough.
Box 2
The effects of Cannabis
Marijuana is harvested from the plant Cannabis sativa and composed of 400 lipophilic chemical compounds, including phytocannabinoids, terpenoids, and flavonoids.13 The plant contains compounds termed “cannabinoids.” Two of these derivatives in particular are responsible for most of the effects of marijuana: cannabinoid delta-9- tetrahydrocannabinol (THC) and cannabidiol (CBD). THC has a comparable structure and binding mechanism to anandamide, a naturally occurring fatty acid neurotransmitter present within the human brain.14-16 The endogenous endocannabinoid system and its receptors are found throughout the entire body (brain, organs, glands, immune cells, and connective tissues).
THC binds to cannabinoid receptors CB1 and CB2. CB1 is found predominantly in the CNS. CB2 is found predominantly outside the CNS and is associated with the immune system.14-16 The effects of THC include euphoria, relaxation, appetite stimulation, improvement of nausea and vomiting, analgesia, decreased muscle spasticity, and reduced eye pressure.14,15 CBD may have anxiolytic, antipsychotic, anticonvulsive, and analgesic effects.
The rate of absorption of THC and CBD depends both on the potency of the cannabinoid as well as the mechanism of consumption. Cannabis can be administered by multiple routes, including via smoking, oral ingestion, or IV.16 When Cannabis is smoked (the route for the most rapid delivery), THC is transported from the lungs to the bloodstream and reaches peak concentrations in 3 to 10 minutes. Oral ingestion (capsules, tinctures, sprays, and edibles) has a more flexible onset of action, usually occurring in 30 to 120 minutes, with effects lasting 5 to 6 hours. IV administration has rapid effects; the onset can occur within seconds to minutes, and effects can last 2 to 3 hours. The IV form allows 90% of THC to be distributed in plasma and can rapidly penetrate highly vascularized tissues, such as the liver, heart, fat, lungs, and muscles.
Pharmaceutical manufacturers have used cannabinoid derivatives to produce Cannabis-based medications for treating medical conditions. Nabilone, a potent agonist of the CB1 receptor, became available as a Schedule II medication in 1981 and was approved for patients with chemotherapy-induced nausea and vomiting (CINV).17 In 1985, dronabinol was introduced as an antiemetic for CINV as well as an appetite stimulant for patients with conditions associated with excessive weight loss.18 Another option, nabiximols, is an oral mucosal spray that consists of THC and CBD in a 1:1 ratio.19 Nabiximols is approved in Canada for pain relief in end-stage cancer patients and pain associated with multiple sclerosis.19
In an RCT of vaporized Cannabis, 39 patients with a diagnosis of complex regional pain syndrome, thalamic pain, spinal cord injury, peripheral neuropathy, radiculopathy, or nerve injury were assigned to a medium-dose (3.53% THC), low-dose (1.29% THC), or placebo group.25 Serious mental illness, substance abuse, and medical conditions were cause for exclusion. Participants received vaporized marijuana (average 8 to 12 puffs per visit) over 3 sessions. A 30% pain reduction was achieved by 26% of those in the placebo group, 57% of those in the low-dose group, and 61% of individuals in the high-dose group; the difference between placebo and each Cannabis group was statistically significant.
Chemotherapy-induced nausea and vomiting. Up to 80% of patients who receive chemotherapy experience CINV, which occurs from 24 hours to 7 days after receiving such therapy.26 CINV negatively influences a patient’s QOL and may impact the decision to continue with chemotherapy. Use of MM can help to diminish vomiting by binding to central CB1 receptors and averting the proemetic effects of dopamine and serotonin.27 Two synthetically derived cannabinoids, dronabinol and nabilone, are FDA-approved for treating CINV.
In a small (N = 64) parallel-group RCT, Meiri et al27 compared dronabinol with the commonly used antiemetic ondansetron and with a combination of dronabinol and ondansetron for treating CINV in adults. The primary outcome was prevention of delayed-onset CINV. Patients were eligible for this study if they had a malignancy that did not involve bone marrow, were receiving treatment with a moderately to highly emetogenic regimen, were not pregnant, and had an estimated life expectancy of at least 6 weeks after chemotherapy. The patients were randomized to 1 of 4 treatment groups: dronabinol alone, ondansetron alone, dronabinol plus ondansetron, or placebo. Overall, 47% to 58% of the active treatment groups improved, compared with 20% of the placebo group. Combination therapy did not provide any benefit beyond any single agent alone. All active treatments reduced nausea compared with placebo; there was no difference between active treatment groups. This study was limited by low enrollment.
Tramèr et al28 conducted a systematic review of 30 randomized comparisons of MM with placebo or antiemetics. The reviewed studies were completed between 1975 to 1997 and analyzed a total of 1,366 patients. Nabilone was evaluated in 16 trials; dronabinol was utilized in 13 trials; and IM levonantradol, a synthetic cannabinoid analog of dronabinol, was used in 1 trial. These agents were found to be more effective as an antiemetic compared with prochlorperazine, metoclopramide, chlorpromazine, thiethylperazine, haloperidol, domperidone, or alizapride. In addition, 38% to 90% of patients in these studies preferred MM over the traditional antiemetics.
A Cochrane review29 suggested that MM may be a viable option for treatment-resistant CINV; however, further studies are needed because current studies have methodological limitations.
Epilepsy. Maa and Figi30 reported a case of a 5-year-old girl who had Dravet syndrome, which resulted in 50 generalized tonic-clonic seizures daily; multiple anticonvulsants did not alleviate these seizures. Because of her recurring seizures, the patient had multiple cognitive and motor delays and needed a feeding tube. In addition to her existing antiepileptic drug regimen, she was started on adjunctive therapy with a sublingual Cannabis extract containing a high concentration of CBD. Her seizures decreased from 50 per day to 2 to 3 nocturnal convulsions per month. The treatment enabled her to stop using a feeding tube, resume walking and talking, and sleep soundly.
dos Santos et al31 reviewed studies of MM for treating epilepsy. One was a double-blind, placebo-controlled trial that included 15 patients ages 14 to 49 who had secondary generalized epilepsy with a temporal lobe focus. Eight patients received 200 to 300 mg/d of oral CBD for 8 to 18 weeks, and 7 received placebo. Seven patients had fewer seizures and 4 had no seizures. Only 1 patient in the placebo group demonstrated any improvement. Another study in this review included 19 children with treatment-resistant epilepsy: Dravet syndrome (n = 13), Doose syndrome (n = 4), Lennox-Gastaut syndrome (n = 1), or idiopathic epilepsy (n = 1). These patients experienced various types of seizures with a frequency ranging from 2 per week to 250 per day. Overall, 84% of children treated with CBD had fewer seizures: 11% were seizure-free, 42% had a >80% reduction in seizures, and 32% had a 25% to 60% reduction in seizures. Parents also noted additional benefits, including increased attention, improved mood, and improved sleep. CBD was well tolerated in most patients in both studies.
Despite these results, a Cochrane review32 found that no reliable conclusions can be drawn regarding the efficacy of MM for treating epilepsy.
Multiple sclerosis. According to American Academy of Neurology guidelines, physicians may provide MM as an alternative treatment for patients with MS-related spasticity.33 Multiple studies have tested MM and MM-related extracts for treating spasticity related to MS.34,35 In a placebo-controlled crossover study, Corey-Bloom et al34 reported a significant reduction in spasticity, measured using the modified Ashworth scale, in MS patients receiving Cannabis cigarettes vs placebo cigarettes (P < .0001). However, compared with the placebo group, patients who received MM had significant adverse effects, primarily cognitive impairment (P = .003).
In a multicenter RCT (N = 572 patients with refractory MS spasticity), Novotna et al36 evaluated nabiximols, an oral mucosal spray of a formulated extract of Cannabis that contains THC and CBD in a 1:1 ratio. They assessed spasticity using the Numerical Spasticity Rating Scale (NRS). Results were confirmed by measuring the number of daily spasms, self-report of sleep quality, and activities of daily living. After 4 weeks of single-blind treatment, patients who responded to nabiximols (≥20% improvement in spasticity) were randomized to a placebo group or nabiximols group for 12 additional weeks. After 12 weeks, compared with those who received placebo, those in the nabiximols group experienced a statistically significant reduction in spasticity based on NRS score (P = .0002).
For a summary of evidence on MM for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, see Box 3.37-43
Box 3
Cannabis for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
Glaucoma. In a placebo-controlled study, oromucosal administration of medical marijuana (MM) reduced intraocular pressure from 28 mm Hg to 22 mm Hg, with a duration of action of 3.5 hours.37However, the American Academy of Ophthalmologists does not recommend treating glaucoma with MM because the effect is short-lasting, and MM causes significant cognitive impairment compared with other standardized treatments.38 MM also leads to decreased blood pressure, which lowers blood flow to the optic nerve, thus increasing the risk of blindness.
Crohn’s disease. A randomized controlled trial (RCT) of MM for Crohn’s disease was conducted using the Crohn’s Disease Activity Index (CDAI) to assess for remission. In this 8-week study,21 individuals with Crohn’s disease were administered smoked MM (115 mg of delta-9-tetrahydrocannabinol [THC]) or placebo.39 Eligible patients were at least 20 years old, had active Crohn’s disease (CDAI >200), and had not responded to medical treatment for the illness. Compared with those who received placebo, patients who received MM experienced a statistically significant reduction in CDAI scores (P < .05). However, at follow-up 2 weeks after the study, when MM was no longer administered, there was no difference in mean CDAI scores between the 2 groups. Five of the 11 patients in the MM group achieved clinical remission, compared with 1 of 10 in the placebo group, but this difference was not statistically significant.
Parkinson’s disease (PD). According to the American Academy of Neurology, oral Cannabis extracts are “probably ineffective” for levodopa-induced dyskinesia in patients with PD.40 Reported benefits have come mainly from self-report studies. A 2014 survey (22 patients) found a significant reduction in PD symptoms—mainly relief from drug-induced tremor and pain—when measured using the Unified Parkinson’s Disease Rating Scale (UPDRS). Patients also reported better sleep and reduced pain (measured with a visual analog scale [VAS]). An exploratory double-blind placebo trial (N = 119) found no difference in mean UPDRS and no difference in any neuroprotective measures.41 However, the experimental group had a significantly higher quality of life (QOL; P = .05). A similar double-blind crossover study that included 19 patients found no significant difference in dyskinesia, as measured with the UPDRS, in the group receiving oral Cannabis extract compared with the placebo group.42
Amyotrophic lateral sclerosis (ALS). A randomized double-blind crossover trial of 27 ALS patients found that an oral THC extract (dronabinol, 5 mg, twice daily) had no significant effects on spasticity, as measured with the VAS.43 There was also no significant difference between the experimental and placebo groups on number of spasms (also measured with a VAS), quality of sleep (measured with the Sleep Disorders Questionnaire), or QOL (measured with the Amyotrophic Lateral Sclerosis Assessment questionnaire).
Psychiatric illnesses
Dementia-related behavioral disturbances. A few clinical trials with small sample sizes have found evidence supporting the use of MM compounds for alleviating neuropsychiatric symptoms of patients with dementia. An open-label pilot study of 6 individuals with late-stage dementia who received dronabinol, 2.5 mg/d, for 2 weeks, found a significant reduction (compared with baseline) in nighttime motor activity as measured with an actometer (P < .0028).44 The secondary Neuropsychiatric Inventory (NPI) assessment found reductions in aberrant motor behavior (P = .042), agitation (P = .042), and nighttime behaviors (P = .42).
A 2014 retrospective analysis of 40 inpatients with dementia-related agitation and appetite loss who were treated with dronabinol (mean dosage: 7.03 mg/d) found reductions in all aspects of agitation, including aberrant vocalization, motor agitation, aggressiveness, and treatment resistance, as measured with the Pittsburgh Agitation Scale (P < .0001).45 The study found no significant improvements in appetite, Global Assessment of Functioning mean score, or number of times patients awoke during the night. Adverse effects included sedation and delirium.
A RCT of 50 dementia patients with clinically relevant neuropsychiatric symptoms found no significant difference in mean NPI scores between patients given placebo and those who received nabiximols, 1.5 mg, 3 times daily.46 There were no significant differences found in agitation, QOL, life activities, or caregiver-scored Caregiver Global Impression of Change scale.
In a small RCT, THC was safe and well tolerated in 10 older patients with dementia.47 A 2009 Cochrane review48 concluded that there was no evidence for the efficacy of MM in treating the neuropsychiatric symptoms related to dementia.
PTSD. Preclinical evidence shows that the endocannabinoid system is involved in regulating emotional memory. Evidence also suggests that cannabinoids may facilitate the extinction of aversive memories.49,50
In 2009, New Mexico became the first state to authorize the use of MM for patients with PTSD. In a study of patients applying for the New Mexico Medical Cannabis Program, researchers used the Clinician Administered Posttraumatic Scale (CAPS) to assess PTSD symptoms.51 A retrospective chart review of the first 80 patients evaluated found significant (P < .0001) reductions of several PTSD symptoms, including intrusive memories, distressing dreams, flashbacks, numbing and avoidance, and hyperarousal, in the group using MM vs those not using MM. There also was a significant difference in CAPS total score (P < .0001). Patients reported a 75% reduction in PTSD symptoms while using MM. This study has several limitations: It was a retrospective review, not an RCT, and patients were prescreened and knew before the study began that MM helped their PTSD symptoms.
In another retrospective study, researchers evaluated treatment with nabilone, 0.5 to 6 mg/d, in 104 incarcerated men with various major mental illnesses; most (91%) met criteria for Cannabis dependence.52 They found significant improvements in sleep and PTSD symptoms.
A double-blind RCT evaluated MM in 10 Canadian male soldiers with PTSD who experienced nightmares despite standard medication treatment. Adjunctive nabilone (maximum dose: 3 mg/d) resulted in a reduction in nightmares as measured by the CAPS recurrent distressing dream of the event item score.53
Currently, there are no adequately powered RCTs of MM in a diverse group of PTSD patients. Most studies are open-label, enriched design, and included white male veterans. No well-conducted trials have evaluated patients with noncombat-related PTSD. Most of the relevant literature consists of case reports of Cannabis use by patients with PTSD.
Anxiety disorders.Patients frequently indicate that smoking Cannabis helps relieve their anxiety, although there is no replicated evidence based on double-blind RCTs to support this. However, in rat models CBD has been shown to facilitate extinction of conditioned fear via the endocannabinoid system.54-56 The mechanism of action is not completely understood. CBD has been shown to have antagonistic action at CB1 and CB2 receptors. It may have similar effects on memory extinction and may be an adjunct to exposure therapies for anxiety disorders.
Das et al57 studied the effects of CBD (32 mg) on extinction and consolidation of memory related to contextual fear in 48 individuals. They found that CBD can enhance extinction learning, and suggested it may have potential as an adjunct to extinction-based therapies for anxiety disorders.
Caveats: Adverse effects, lack of RCTs
Cannabis use causes impairment of learning, memory, attention, and working memory. Adolescents are particularly vulnerable to the effects of Cannabis on brain development at a time when synaptic pruning and increased myelination occur. Normal brain development could be disrupted. Some studies have linked Cannabis use to abnormalities in the amygdala, hippocampus, frontal lobe, and cerebellum. From 1995 to 2014, the potency of Cannabis (THC concentration) increased from 4% to 12%.58 This has substantial implications for increased abuse among adolescents and the deleterious effects of Cannabis on the brain.
Heavy Cannabis use impairs motivation and could precipitate psychosis in vulnerable individuals. Cannabis use may be linked to the development of schizophrenia.59
There are no well-conducted RCTs on the efficacy of MM, and adequate safety data are lacking. There is also lack of consensus among qualified experts. There is soft evidence that MM may be helpful in some medical conditions, including but not limited to CINV, neuropathic pain, epilepsy, and MS-related spasticity. Currently, the benefits of using MM do not appear to outweigh the risks.
Bottom Line
Limited evidence suggests medical marijuana (MM) may be beneficial for treating a few medical conditions, including neuropathic pain and chemotherapy-induced nausea and vomiting. There is no clear and convincing evidence MM is beneficial for psychiatric disorders, and Cannabis can impair cognition and attention and may precipitate psychosis. The risk of deleterious effects are greater in adolescents.
Related Resources
- Nguyen DH, Thant TM. Caring for medical marijuana patients who request controlled prescriptions. Current Psychiatry. 2017;16(8):50-51.
- National Institute on Drug Abuse. Marijuana as medicine. https://www.drugabuse.gov/publications/drugfacts/ marijuana-medicine.
Drug Brand Names
Alizapride • Litican, Superan
Chlorpromazine • Thorazine
Domperidone • Motilium
Dronabinol • Marinol, Syndros
Haloperidol • Haldol
Metoclopramide • Reglan
Nabilone • Cesamet
Nabiximols • Sativex
Ondansetron • Zofran, Zuplenz
Prochlorperazine • Compazine
Thiethylperazine • Torecan
1. National Institute on Drug Abuse (NIDA). Marijuana Research Findings 1976. NIDA research monograph 14. https://archives.drugabuse.gov/sites/default/files/monograph14.pdf. Published July 1977. Accessed November 15, 2017.
2. O’Shaughnessy WB. On the preparations of the Indian hemp, or gunjah- cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363-369.
3. Clendinning J. Observations on the medical properties of the Cannabis Sativa of India. Med Chir Trans. 1843;26:188-210.
4. Osler W, McCrae T. The principles and practice of medicine. 9th ed. New York, NY: D. Appleton and Company; 1921.
5. The pharmacopoeia of the United States of America. 3rd ed. Philadelphia, PA: Lippincott; 1851.
6. The pharmacopoeia of the United States of America. 12th ed. Easton, PA: Mack Printing Company; 1942.
7. Philipsen N, Butler RD, Simon C, et al. Medical marijuana: a primer on ethics, evidence, and politics. Journal Nurse Pract. 2014;10(9):633-640.
8. Marihuana Tax Act of 1937, Pub L No. 75-238, 75th Cong, 50 Stat 551 (1937).
9. Controlled Substances Act, 21 USC §812.
10. Watson SJ, Benson JA, Joy JE, eds. Marijuana and medicine: assessing the science base. Washington, DC: National Academy Press; 1999.
11. California Proposition 215, the medical marijuana initiative (1996). https://ballotpedia.org/California_Proposition_215,_the_Medical_Marijuana_Initiative_(1996). Accessed November 16, 2017.
12. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated September 14, 2017. Accessed November 16, 2017.
13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344-1364.
14. Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013:14. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3997295. Accessed December 5, 2017.
15. Galal AM, Slade D, Gul W, et al. Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications. Recent Pat CNS Drug Discov. 2009;4(2):112-136.
16. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804.
17. Cesamet [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2013.
18. Marinol [package insert]. Chicago, IL: AbbVie Inc.; 2017.
19. Sativex [package insert]. Mississauga, Ontario: Bayer Inc.; 2015.
20. Torrance N, Ferguson JA, Afolabi E, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690-699.
21. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162-173.
22. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313(24):2456-2473.
23. Wilsey B, Marcotte, T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9(6):506-521.
24. Ware MA, Wang T, Shapiro S, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ. 2010;182(14):E694-E701.
25. Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14(2):136-148.
26. National Cancer Institute. Treatment-related nausea and vomiting (PDQ®)-health professional version. https://www.cancer.gov/about-cancer/treatment/side-effects/nausea/nausea-hp-pdq. Updated May 10, 2017. Accessed November 7, 2017.
27. Meiri E, Jhangiani H, Vrendenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533-543.
28. Tramèr MR, Carroll D, Campbell FA, et al. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ. 2001;323(7303):16-21.
29. Smith LA, Azariah F, Lavender VT, et al. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev. 2015;(11):CD009464.
30. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783-786.
31. dos Santos RG, Hallak JE, Leite JP, et al. Phytocannabinoids and epilepsy. J Clin Pharm Ther. 2015;40(2):135-143.
32. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;(3):CD009270.
33. Yadav V, Bever C Jr, Bowen J, et al. Summary of evidence-based guideline: complementary and alternative medicine in multiple sclerosis: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2014;82(12):1083-1092.
34. Corey-Bloom J, Wolfson T, Gamst A, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. CMAJ. 2012;184(10):1143-1150.
35. Zajicek J, Ball S, Wright D, et al; CUPID investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
36. Novotna A, Mares J, Ratcliffe S, et al; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122-1131.
37. Merritt JC, Crawford WJ, Alexander PC, et al. Effect of marihuana on intraocular and blood pressure in glaucoma. Ophthalmology. 1980;87(3):222-228.
38. American Academy of Ophthalmology. American Academy of Ophthalmology reiterates position that marijuana is not a proven treatment for glaucoma. https://www.aao.org/newsroom/news-releases/detail/american-academy-of-ophthalmology-reiterates-posit. Published June 27, 2014. Accessed May 29, 2017.
39. Naftali T, Bar-Lev Schleider L, Dotan I, et al. Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11(10):1276.e1-1280.e1.
40. Koppel BS Brust JC, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in certain neurological disorders. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82(17):1556-1563.
41. Chagas MH, Zuardi AW, Tumas V, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. 2014;28(11):1088-1098.
42. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology. 2004;63(7):1245-1250.
43. Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135-1140.
44. Walther S, Mahlberg R, Eichmann U, et al. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006;185(4):524-528.
45. Woodward MR, Harper DG, Stolyar A, et al. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am J Geriatr Psychiatry. 2014;22(4):415-419.
46. van den Elsen GA, Ahmed A, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: a randomized controlled trial. Neurology. 2015;84(23):2338-2346.
47. Ahmed AI, van den Elsen GA, Colbers A, et al. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology (Berl). 2015;232(14):25872595.
48. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev. 2009;(2):CD007204.
49. de Bitencourt RM, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389-395.
50. Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci Ther. 2009;15(1):84-88.
51. Greer GR, Grob CS, Halberstadt AL. PTSD symptom reports of patients evaluated for the New Mexico Medical Cannabis Program. J Psychoactive Drugs. 2014;46(1):73-77.
52. Cameron C, Watson D, Robinson J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. J Clin Psychopharmacol. 2014;34(5):559-564.
53. Jetly R, Heber A, Fraser G, et al. The efficacy of nabilone, a synthetic cannabinoid, in the treatment of PTSD-associated nightmares: a preliminary randomized, double-blind, placebo-controlled cross-over design study. Psychoneuroendocrinology. 2015;51:585-588.
54. Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear, memory extinction, and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18(12):849-859.
55. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199-215.
56. Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613-623.
57. Das RK, Kamboj SK, Ramadas M, et al. Cannabidiol enhances consolidation of explicit fear extinction in humans. Psychopharmacology (Berl). 2013;226(4):781-792.
58. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613-619.
59. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73(3):292297.
1. National Institute on Drug Abuse (NIDA). Marijuana Research Findings 1976. NIDA research monograph 14. https://archives.drugabuse.gov/sites/default/files/monograph14.pdf. Published July 1977. Accessed November 15, 2017.
2. O’Shaughnessy WB. On the preparations of the Indian hemp, or gunjah- cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363-369.
3. Clendinning J. Observations on the medical properties of the Cannabis Sativa of India. Med Chir Trans. 1843;26:188-210.
4. Osler W, McCrae T. The principles and practice of medicine. 9th ed. New York, NY: D. Appleton and Company; 1921.
5. The pharmacopoeia of the United States of America. 3rd ed. Philadelphia, PA: Lippincott; 1851.
6. The pharmacopoeia of the United States of America. 12th ed. Easton, PA: Mack Printing Company; 1942.
7. Philipsen N, Butler RD, Simon C, et al. Medical marijuana: a primer on ethics, evidence, and politics. Journal Nurse Pract. 2014;10(9):633-640.
8. Marihuana Tax Act of 1937, Pub L No. 75-238, 75th Cong, 50 Stat 551 (1937).
9. Controlled Substances Act, 21 USC §812.
10. Watson SJ, Benson JA, Joy JE, eds. Marijuana and medicine: assessing the science base. Washington, DC: National Academy Press; 1999.
11. California Proposition 215, the medical marijuana initiative (1996). https://ballotpedia.org/California_Proposition_215,_the_Medical_Marijuana_Initiative_(1996). Accessed November 16, 2017.
12. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated September 14, 2017. Accessed November 16, 2017.
13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344-1364.
14. Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013:14. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3997295. Accessed December 5, 2017.
15. Galal AM, Slade D, Gul W, et al. Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications. Recent Pat CNS Drug Discov. 2009;4(2):112-136.
16. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804.
17. Cesamet [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2013.
18. Marinol [package insert]. Chicago, IL: AbbVie Inc.; 2017.
19. Sativex [package insert]. Mississauga, Ontario: Bayer Inc.; 2015.
20. Torrance N, Ferguson JA, Afolabi E, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690-699.
21. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162-173.
22. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313(24):2456-2473.
23. Wilsey B, Marcotte, T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9(6):506-521.
24. Ware MA, Wang T, Shapiro S, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ. 2010;182(14):E694-E701.
25. Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14(2):136-148.
26. National Cancer Institute. Treatment-related nausea and vomiting (PDQ®)-health professional version. https://www.cancer.gov/about-cancer/treatment/side-effects/nausea/nausea-hp-pdq. Updated May 10, 2017. Accessed November 7, 2017.
27. Meiri E, Jhangiani H, Vrendenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533-543.
28. Tramèr MR, Carroll D, Campbell FA, et al. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ. 2001;323(7303):16-21.
29. Smith LA, Azariah F, Lavender VT, et al. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev. 2015;(11):CD009464.
30. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783-786.
31. dos Santos RG, Hallak JE, Leite JP, et al. Phytocannabinoids and epilepsy. J Clin Pharm Ther. 2015;40(2):135-143.
32. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;(3):CD009270.
33. Yadav V, Bever C Jr, Bowen J, et al. Summary of evidence-based guideline: complementary and alternative medicine in multiple sclerosis: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2014;82(12):1083-1092.
34. Corey-Bloom J, Wolfson T, Gamst A, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. CMAJ. 2012;184(10):1143-1150.
35. Zajicek J, Ball S, Wright D, et al; CUPID investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
36. Novotna A, Mares J, Ratcliffe S, et al; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122-1131.
37. Merritt JC, Crawford WJ, Alexander PC, et al. Effect of marihuana on intraocular and blood pressure in glaucoma. Ophthalmology. 1980;87(3):222-228.
38. American Academy of Ophthalmology. American Academy of Ophthalmology reiterates position that marijuana is not a proven treatment for glaucoma. https://www.aao.org/newsroom/news-releases/detail/american-academy-of-ophthalmology-reiterates-posit. Published June 27, 2014. Accessed May 29, 2017.
39. Naftali T, Bar-Lev Schleider L, Dotan I, et al. Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11(10):1276.e1-1280.e1.
40. Koppel BS Brust JC, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in certain neurological disorders. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82(17):1556-1563.
41. Chagas MH, Zuardi AW, Tumas V, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. 2014;28(11):1088-1098.
42. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology. 2004;63(7):1245-1250.
43. Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135-1140.
44. Walther S, Mahlberg R, Eichmann U, et al. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006;185(4):524-528.
45. Woodward MR, Harper DG, Stolyar A, et al. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am J Geriatr Psychiatry. 2014;22(4):415-419.
46. van den Elsen GA, Ahmed A, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: a randomized controlled trial. Neurology. 2015;84(23):2338-2346.
47. Ahmed AI, van den Elsen GA, Colbers A, et al. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology (Berl). 2015;232(14):25872595.
48. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev. 2009;(2):CD007204.
49. de Bitencourt RM, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389-395.
50. Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci Ther. 2009;15(1):84-88.
51. Greer GR, Grob CS, Halberstadt AL. PTSD symptom reports of patients evaluated for the New Mexico Medical Cannabis Program. J Psychoactive Drugs. 2014;46(1):73-77.
52. Cameron C, Watson D, Robinson J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. J Clin Psychopharmacol. 2014;34(5):559-564.
53. Jetly R, Heber A, Fraser G, et al. The efficacy of nabilone, a synthetic cannabinoid, in the treatment of PTSD-associated nightmares: a preliminary randomized, double-blind, placebo-controlled cross-over design study. Psychoneuroendocrinology. 2015;51:585-588.
54. Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear, memory extinction, and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18(12):849-859.
55. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199-215.
56. Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613-623.
57. Das RK, Kamboj SK, Ramadas M, et al. Cannabidiol enhances consolidation of explicit fear extinction in humans. Psychopharmacology (Berl). 2013;226(4):781-792.
58. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613-619.
59. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73(3):292297.

A concise guide to monoamine oxidase inhibitors: How to avoid drug interactions
Monoamine oxidase inhibitors (MAOIs) have well-established efficacy for treating depression, panic disorder, and social phobia. However, a lack of familiarity with these agents and misconceptions about the risks associated with their use have led to MAOIs being substantially underutilized. The goal of this 2-part guide to MAOIs is to educate clinicians about this often-overlooked class of medications. Part 1 (“A concise guide to monoamine inhibitors,”
MAOIs and potential drug interactions
One source of concern in patients receiving irreversible nonselective MAOIs is the development of excessive serotonergic neurotransmission resulting in SS. In the 1960s, researchers noted that administering large doses of
- mild symptoms: tremor, akathisia, inducible clonus
- moderate symptoms: spontaneous or sustained clonus, muscular hypertonicity
- severe symptoms: hyperthermia, diaphoresis.2
Although SS can be induced by significant exposure to individual agents that promote excess synaptic serotonin (eg, overdose of selective serotonin reuptake inhibitors [SSRIs]), the majority of fatal cases have occurred among patients taking MAOIs who were coadministered an agent that inhibited serotonin reuptake (Table 13). Animal studies have determined that excessive stimulation of the 5HT2A receptor is primarily responsible for SS,4 and that 5HT2A antagonists, such as mirtazapine, can block the development of SS in a mouse coadministered

Risk for SS. Most medications that promote serotonergic activity are well known for their use as antidepressants, but other agents that have 5HT reuptake properties (eg, the antihistamine chlorpheniramine) must be avoided. Although older literature suggests that the use of lower doses of certain tricyclic antidepressants concurrently with MAOIs may not be as dangerous as once believed,6 there are sufficient reports of serious outcomes that tricyclics should be avoided in patients taking MAOIs because of the risk of SS, and also because, in general, tricyclics are poorly tolerated.7
Desipramine, a potent norepinephrine transporter (NET) inhibitor, blocks the entry of tyramine into cells by NET, thereby preventing hypertensive events in animal models of tyramine overexposure. However, in some assays, the affinity for the serotonin transporter is not insignificant, so at higher doses desipramine may pose the same theoretical risk for SS as seen with other tricyclics.3
Lastly
Astute clinicians will recognize that antidepressants that lack 5HT reuptake (eg, bupropion, mirtazapine) are not on this list of agents that may increase SS risk when taken with an MAOI. Older papers often list mirtazapine, but as a 5HT2A antagonist, it does not possess a plausible mechanism by which it can induce 5HT toxicity.9,10 Most atypical antipsychotics have significant 5HT2A antagonism and can be combined with MAOIs, but ziprasidone is an exception: as a moderate SNRI, it has been associated with SS when administered with an MAOI.11
Pressor reactions. The only theoretical sources of concern for pressor effects are medications that act as norepinephrine releasers through interactions at the trace amine-associated receptor 1 (TAAR1) (for more information on TAAR1, see
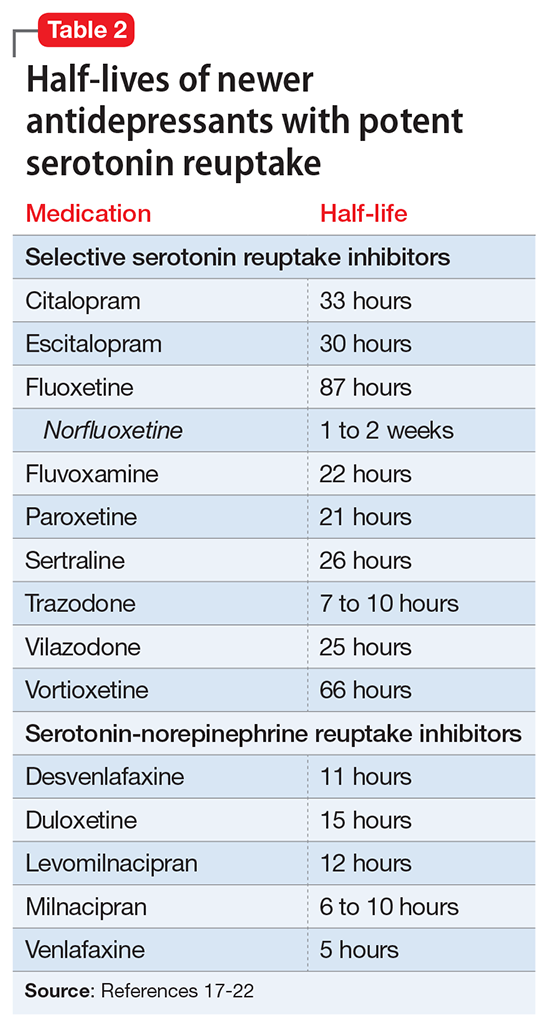
Starting a patient on an MAOI
Contraindicated medications need to be tapered before beginning MAOI treatment. The duration of the washout period depends on the half-life of the medication and any active metabolites. Antidepressants with half-lives of approximately ≤24 hours should be tapered over 7 to 14 days (depending on the dose) to minimize the risk of withdrawal syndromes, while those with long half-lives (eg, fluoxetine,
Initiation of an MAOI is always based on whether the patient can reliably follow the basic dietary advice (see “A concise guide to monoamine inhibitors,”
The orthostasis management strategy is similar to that employed for
Augmentation options for patients taking MAOIs
For depressed patients who do not achieve remission of symptoms from MAOI therapy, augmentation options should be sought, as patients who respond but fail to remit are at increased risk of relapse.26 Lithium augmentation is one of the more common strategies, with abundant data supporting its use.27,28 Case reports dating back >12 years describe the concurrent use o
1. Krishnamoorthy S, Ma Z, Zhang G, et al. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107(4):830-841.
2. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-713.
3. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-90.
4. Haberzettl R, Fink H, Bert B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J Pharmacol Toxicol Methods. 2014;70(2):129-133.
5. Shioda K, Nisijima K, Yoshino T, et al. Mirtazapine abolishes hyperthermia in an animal model of serotonin syndrome. Neurosci Lett. 2010;482(3):216-219.
6. White K, Simpson G. Combined MAOI-tricyclic antidepressant treatment: a reevaluation. J Clin Psychopharmacol. 1981;1(5):264-282.
7. Otte W, Birkenhager TK, van den Broek WW. Fatal interaction between tranylcypromine and imipramine. Eur Psychiatry. 2003;18(5):264-265.
8. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
9. Gillman PK. Mirtazapine: unable to induce serotonin toxicity? Clin Neuropharmacol. 2003;26(6):288-289; author reply 289-290.
10. Gillman PK. A systematic review of the serotonergic effects of mirtazapine in humans: implications for its dual action status. Hum Psychopharmacol. 2006;21(2):117-125.
11. Meyer JM, Cummings MA, Proctor G. Augmentation of phenelzine with aripiprazole and quetiapine in a treatment resistant patient with psychotic unipolar depression: case report and literature review. CNS Spectr. 2017;22(5):391-396.
12. Feinberg SS. Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication. J Clin Psychiatry. 2004;65(11):1520-1524.
13. Israel JA. Combining stimulants and monoamine oxidase inhibitors: a reexamination of the literature and a report of a new treatment combination. Prim Care Companion CNS Disord. 2015;17(6). doi: 10.4088/PCC.15br01836.
14. Simmler LD, Buchy D, Chaboz S, et al. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J Pharmacol Exp Ther. 2016;357(1):134-144.
15. Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with enhanced selectivity for the dopamine transporter. J Med Chem. 2007;50(2):219-232.
16. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
17. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85(1):11-28.
18. Pristiq [package insert]. New York, NY: Pfizer Inc; 2016.
19. Savella [package insert]. Irvine, CA: Allergan USA Inc; 2016.
20. Viibryd [package insert]. Irvine, CA: Allergan USA Inc; 2016.
21. Trintellix [package insert]. Deerfield, IL: Takeda Pharmaceuticals America Inc; 2016.
22. Fetzima [package insert]. Irvine, CA: Allergan USA Inc; 2017.
23. Nardil [package insert]. New York, NY: Pfizer Inc; 2009.
24. Testani M Jr. Clozapine-induced orthostatic hypotension treated with fludrocortisone. J Clin Psychiatry. 1994;55(11):497-498.
25. Emsam [package insert]. Morgantown, WV: Somerset Pharmaceuticals Inc; 2015.
26. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
27. Tariot PN, Murphy DL, Sunderland T, et al. Rapid antidepressant effect of addition of lithium to tranylcypromine. J Clin Psychopharmacol. 1986;6(3):165-167.
28. Kok RM, Vink D, Heeren TJ, et al. Lithium augmentation compared with phenelzine in treatment-resistant depression in the elderly: an open, randomized, controlled trial. J Clin Psychiatry. 2007;68(8):1177-1185.
29. Quante A, Zeugmann S. Tranylcypromine and bupropion combination therapy in treatment-resistant major depression: a report of 2 cases. J Clin Psychopharmacol. 2012;32(4):572-574.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry. 1988;49(10):409-410.
31. Hullett FJ, Bidder TG. Phenelzine plus triiodothyronine combination in a case of refractory depression. J Nerv Ment Dis. 1983;171(5):318-320.
Monoamine oxidase inhibitors (MAOIs) have well-established efficacy for treating depression, panic disorder, and social phobia. However, a lack of familiarity with these agents and misconceptions about the risks associated with their use have led to MAOIs being substantially underutilized. The goal of this 2-part guide to MAOIs is to educate clinicians about this often-overlooked class of medications. Part 1 (“A concise guide to monoamine inhibitors,”
MAOIs and potential drug interactions
One source of concern in patients receiving irreversible nonselective MAOIs is the development of excessive serotonergic neurotransmission resulting in SS. In the 1960s, researchers noted that administering large doses of
- mild symptoms: tremor, akathisia, inducible clonus
- moderate symptoms: spontaneous or sustained clonus, muscular hypertonicity
- severe symptoms: hyperthermia, diaphoresis.2
Although SS can be induced by significant exposure to individual agents that promote excess synaptic serotonin (eg, overdose of selective serotonin reuptake inhibitors [SSRIs]), the majority of fatal cases have occurred among patients taking MAOIs who were coadministered an agent that inhibited serotonin reuptake (Table 13). Animal studies have determined that excessive stimulation of the 5HT2A receptor is primarily responsible for SS,4 and that 5HT2A antagonists, such as mirtazapine, can block the development of SS in a mouse coadministered
Risk for SS. Most medications that promote serotonergic activity are well known for their use as antidepressants, but other agents that have 5HT reuptake properties (eg, the antihistamine chlorpheniramine) must be avoided. Although older literature suggests that the use of lower doses of certain tricyclic antidepressants concurrently with MAOIs may not be as dangerous as once believed,6 there are sufficient reports of serious outcomes that tricyclics should be avoided in patients taking MAOIs because of the risk of SS, and also because, in general, tricyclics are poorly tolerated.7
Desipramine, a potent norepinephrine transporter (NET) inhibitor, blocks the entry of tyramine into cells by NET, thereby preventing hypertensive events in animal models of tyramine overexposure. However, in some assays, the affinity for the serotonin transporter is not insignificant, so at higher doses desipramine may pose the same theoretical risk for SS as seen with other tricyclics.3
Lastly
Astute clinicians will recognize that antidepressants that lack 5HT reuptake (eg, bupropion, mirtazapine) are not on this list of agents that may increase SS risk when taken with an MAOI. Older papers often list mirtazapine, but as a 5HT2A antagonist, it does not possess a plausible mechanism by which it can induce 5HT toxicity.9,10 Most atypical antipsychotics have significant 5HT2A antagonism and can be combined with MAOIs, but ziprasidone is an exception: as a moderate SNRI, it has been associated with SS when administered with an MAOI.11
Pressor reactions. The only theoretical sources of concern for pressor effects are medications that act as norepinephrine releasers through interactions at the trace amine-associated receptor 1 (TAAR1) (for more information on TAAR1, see
Starting a patient on an MAOI
Contraindicated medications need to be tapered before beginning MAOI treatment. The duration of the washout period depends on the half-life of the medication and any active metabolites. Antidepressants with half-lives of approximately ≤24 hours should be tapered over 7 to 14 days (depending on the dose) to minimize the risk of withdrawal syndromes, while those with long half-lives (eg, fluoxetine,
Initiation of an MAOI is always based on whether the patient can reliably follow the basic dietary advice (see “A concise guide to monoamine inhibitors,”
The orthostasis management strategy is similar to that employed for
Augmentation options for patients taking MAOIs
For depressed patients who do not achieve remission of symptoms from MAOI therapy, augmentation options should be sought, as patients who respond but fail to remit are at increased risk of relapse.26 Lithium augmentation is one of the more common strategies, with abundant data supporting its use.27,28 Case reports dating back >12 years describe the concurrent use o
Monoamine oxidase inhibitors (MAOIs) have well-established efficacy for treating depression, panic disorder, and social phobia. However, a lack of familiarity with these agents and misconceptions about the risks associated with their use have led to MAOIs being substantially underutilized. The goal of this 2-part guide to MAOIs is to educate clinicians about this often-overlooked class of medications. Part 1 (“A concise guide to monoamine inhibitors,”
MAOIs and potential drug interactions
One source of concern in patients receiving irreversible nonselective MAOIs is the development of excessive serotonergic neurotransmission resulting in SS. In the 1960s, researchers noted that administering large doses of
- mild symptoms: tremor, akathisia, inducible clonus
- moderate symptoms: spontaneous or sustained clonus, muscular hypertonicity
- severe symptoms: hyperthermia, diaphoresis.2
Although SS can be induced by significant exposure to individual agents that promote excess synaptic serotonin (eg, overdose of selective serotonin reuptake inhibitors [SSRIs]), the majority of fatal cases have occurred among patients taking MAOIs who were coadministered an agent that inhibited serotonin reuptake (Table 13). Animal studies have determined that excessive stimulation of the 5HT2A receptor is primarily responsible for SS,4 and that 5HT2A antagonists, such as mirtazapine, can block the development of SS in a mouse coadministered
Risk for SS. Most medications that promote serotonergic activity are well known for their use as antidepressants, but other agents that have 5HT reuptake properties (eg, the antihistamine chlorpheniramine) must be avoided. Although older literature suggests that the use of lower doses of certain tricyclic antidepressants concurrently with MAOIs may not be as dangerous as once believed,6 there are sufficient reports of serious outcomes that tricyclics should be avoided in patients taking MAOIs because of the risk of SS, and also because, in general, tricyclics are poorly tolerated.7
Desipramine, a potent norepinephrine transporter (NET) inhibitor, blocks the entry of tyramine into cells by NET, thereby preventing hypertensive events in animal models of tyramine overexposure. However, in some assays, the affinity for the serotonin transporter is not insignificant, so at higher doses desipramine may pose the same theoretical risk for SS as seen with other tricyclics.3
Lastly
Astute clinicians will recognize that antidepressants that lack 5HT reuptake (eg, bupropion, mirtazapine) are not on this list of agents that may increase SS risk when taken with an MAOI. Older papers often list mirtazapine, but as a 5HT2A antagonist, it does not possess a plausible mechanism by which it can induce 5HT toxicity.9,10 Most atypical antipsychotics have significant 5HT2A antagonism and can be combined with MAOIs, but ziprasidone is an exception: as a moderate SNRI, it has been associated with SS when administered with an MAOI.11
Pressor reactions. The only theoretical sources of concern for pressor effects are medications that act as norepinephrine releasers through interactions at the trace amine-associated receptor 1 (TAAR1) (for more information on TAAR1, see
Starting a patient on an MAOI
Contraindicated medications need to be tapered before beginning MAOI treatment. The duration of the washout period depends on the half-life of the medication and any active metabolites. Antidepressants with half-lives of approximately ≤24 hours should be tapered over 7 to 14 days (depending on the dose) to minimize the risk of withdrawal syndromes, while those with long half-lives (eg, fluoxetine,
Initiation of an MAOI is always based on whether the patient can reliably follow the basic dietary advice (see “A concise guide to monoamine inhibitors,”
The orthostasis management strategy is similar to that employed for
Augmentation options for patients taking MAOIs
For depressed patients who do not achieve remission of symptoms from MAOI therapy, augmentation options should be sought, as patients who respond but fail to remit are at increased risk of relapse.26 Lithium augmentation is one of the more common strategies, with abundant data supporting its use.27,28 Case reports dating back >12 years describe the concurrent use o
1. Krishnamoorthy S, Ma Z, Zhang G, et al. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107(4):830-841.
2. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-713.
3. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-90.
4. Haberzettl R, Fink H, Bert B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J Pharmacol Toxicol Methods. 2014;70(2):129-133.
5. Shioda K, Nisijima K, Yoshino T, et al. Mirtazapine abolishes hyperthermia in an animal model of serotonin syndrome. Neurosci Lett. 2010;482(3):216-219.
6. White K, Simpson G. Combined MAOI-tricyclic antidepressant treatment: a reevaluation. J Clin Psychopharmacol. 1981;1(5):264-282.
7. Otte W, Birkenhager TK, van den Broek WW. Fatal interaction between tranylcypromine and imipramine. Eur Psychiatry. 2003;18(5):264-265.
8. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
9. Gillman PK. Mirtazapine: unable to induce serotonin toxicity? Clin Neuropharmacol. 2003;26(6):288-289; author reply 289-290.
10. Gillman PK. A systematic review of the serotonergic effects of mirtazapine in humans: implications for its dual action status. Hum Psychopharmacol. 2006;21(2):117-125.
11. Meyer JM, Cummings MA, Proctor G. Augmentation of phenelzine with aripiprazole and quetiapine in a treatment resistant patient with psychotic unipolar depression: case report and literature review. CNS Spectr. 2017;22(5):391-396.
12. Feinberg SS. Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication. J Clin Psychiatry. 2004;65(11):1520-1524.
13. Israel JA. Combining stimulants and monoamine oxidase inhibitors: a reexamination of the literature and a report of a new treatment combination. Prim Care Companion CNS Disord. 2015;17(6). doi: 10.4088/PCC.15br01836.
14. Simmler LD, Buchy D, Chaboz S, et al. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J Pharmacol Exp Ther. 2016;357(1):134-144.
15. Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with enhanced selectivity for the dopamine transporter. J Med Chem. 2007;50(2):219-232.
16. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
17. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85(1):11-28.
18. Pristiq [package insert]. New York, NY: Pfizer Inc; 2016.
19. Savella [package insert]. Irvine, CA: Allergan USA Inc; 2016.
20. Viibryd [package insert]. Irvine, CA: Allergan USA Inc; 2016.
21. Trintellix [package insert]. Deerfield, IL: Takeda Pharmaceuticals America Inc; 2016.
22. Fetzima [package insert]. Irvine, CA: Allergan USA Inc; 2017.
23. Nardil [package insert]. New York, NY: Pfizer Inc; 2009.
24. Testani M Jr. Clozapine-induced orthostatic hypotension treated with fludrocortisone. J Clin Psychiatry. 1994;55(11):497-498.
25. Emsam [package insert]. Morgantown, WV: Somerset Pharmaceuticals Inc; 2015.
26. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
27. Tariot PN, Murphy DL, Sunderland T, et al. Rapid antidepressant effect of addition of lithium to tranylcypromine. J Clin Psychopharmacol. 1986;6(3):165-167.
28. Kok RM, Vink D, Heeren TJ, et al. Lithium augmentation compared with phenelzine in treatment-resistant depression in the elderly: an open, randomized, controlled trial. J Clin Psychiatry. 2007;68(8):1177-1185.
29. Quante A, Zeugmann S. Tranylcypromine and bupropion combination therapy in treatment-resistant major depression: a report of 2 cases. J Clin Psychopharmacol. 2012;32(4):572-574.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry. 1988;49(10):409-410.
31. Hullett FJ, Bidder TG. Phenelzine plus triiodothyronine combination in a case of refractory depression. J Nerv Ment Dis. 1983;171(5):318-320.
1. Krishnamoorthy S, Ma Z, Zhang G, et al. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107(4):830-841.
2. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-713.
3. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-90.
4. Haberzettl R, Fink H, Bert B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J Pharmacol Toxicol Methods. 2014;70(2):129-133.
5. Shioda K, Nisijima K, Yoshino T, et al. Mirtazapine abolishes hyperthermia in an animal model of serotonin syndrome. Neurosci Lett. 2010;482(3):216-219.
6. White K, Simpson G. Combined MAOI-tricyclic antidepressant treatment: a reevaluation. J Clin Psychopharmacol. 1981;1(5):264-282.
7. Otte W, Birkenhager TK, van den Broek WW. Fatal interaction between tranylcypromine and imipramine. Eur Psychiatry. 2003;18(5):264-265.
8. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
9. Gillman PK. Mirtazapine: unable to induce serotonin toxicity? Clin Neuropharmacol. 2003;26(6):288-289; author reply 289-290.
10. Gillman PK. A systematic review of the serotonergic effects of mirtazapine in humans: implications for its dual action status. Hum Psychopharmacol. 2006;21(2):117-125.
11. Meyer JM, Cummings MA, Proctor G. Augmentation of phenelzine with aripiprazole and quetiapine in a treatment resistant patient with psychotic unipolar depression: case report and literature review. CNS Spectr. 2017;22(5):391-396.
12. Feinberg SS. Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication. J Clin Psychiatry. 2004;65(11):1520-1524.
13. Israel JA. Combining stimulants and monoamine oxidase inhibitors: a reexamination of the literature and a report of a new treatment combination. Prim Care Companion CNS Disord. 2015;17(6). doi: 10.4088/PCC.15br01836.
14. Simmler LD, Buchy D, Chaboz S, et al. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J Pharmacol Exp Ther. 2016;357(1):134-144.
15. Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with enhanced selectivity for the dopamine transporter. J Med Chem. 2007;50(2):219-232.
16. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
17. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85(1):11-28.
18. Pristiq [package insert]. New York, NY: Pfizer Inc; 2016.
19. Savella [package insert]. Irvine, CA: Allergan USA Inc; 2016.
20. Viibryd [package insert]. Irvine, CA: Allergan USA Inc; 2016.
21. Trintellix [package insert]. Deerfield, IL: Takeda Pharmaceuticals America Inc; 2016.
22. Fetzima [package insert]. Irvine, CA: Allergan USA Inc; 2017.
23. Nardil [package insert]. New York, NY: Pfizer Inc; 2009.
24. Testani M Jr. Clozapine-induced orthostatic hypotension treated with fludrocortisone. J Clin Psychiatry. 1994;55(11):497-498.
25. Emsam [package insert]. Morgantown, WV: Somerset Pharmaceuticals Inc; 2015.
26. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
27. Tariot PN, Murphy DL, Sunderland T, et al. Rapid antidepressant effect of addition of lithium to tranylcypromine. J Clin Psychopharmacol. 1986;6(3):165-167.
28. Kok RM, Vink D, Heeren TJ, et al. Lithium augmentation compared with phenelzine in treatment-resistant depression in the elderly: an open, randomized, controlled trial. J Clin Psychiatry. 2007;68(8):1177-1185.
29. Quante A, Zeugmann S. Tranylcypromine and bupropion combination therapy in treatment-resistant major depression: a report of 2 cases. J Clin Psychopharmacol. 2012;32(4):572-574.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry. 1988;49(10):409-410.
31. Hullett FJ, Bidder TG. Phenelzine plus triiodothyronine combination in a case of refractory depression. J Nerv Ment Dis. 1983;171(5):318-320.

A concise guide to monoamine oxidase inhibitors
Despite an abundance of evidenced-based literature supporting monoamine oxidase inhibitors (MAOIs) as an effective treatment for depression, use of these agents has decreased drastically in the past 3 decades. A lack of industry support and the ease of use of other agents are contributing factors, but the biggest impediments to routine use of MAOIs are unfamiliarity with their efficacy advantages and concerns about adverse effects, particularly the risk of hypertensive crises and serotonin syndrome. Many misconceptions regarding these medications are based on outdated data and studies that are no longer reliable.
The goal of this 2-part review is to provide clinicians with updated information regarding MAOIs. Part 1 provides a brief description of:
- the pharmacology of nonselective irreversible MAOIs
- the mechanism by which tyramine induces hypertension
- sources of clinically significant tyramine exposure
- what to tell patients about dietary restrictions and MAOIs.
Part 2 of this guide will cover the risk of serotonin syndrome when MAOIs are combined with inhibitors of serotonin reuptake, how to initiate MAOI therapy, and augmenting MAOIs with other agents.
The pharmacology of MAOIs
First used clinically in the 1950s to treat tuberculosis, MAOIs have a long and interesting history (see the Box “A brief history of monoamine oxidase inhibitors”). Table 11 lists MAOIs currently available in the United States, including the MAO-B–specific agent rasagiline, which is used for Parkinson’s disease.

Manipulation of the monoamines serotonin, norepinephrine, and dopamine is fundamental to managing major depressive disorder (MDD), yet only nonselective MAOIs directly promote neurotransmission of all 3 by inhibiting MAO-A and MAO-B.2 The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study demonstrated that <50% of MDD patients achieve remission in monotherapy trials of selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, mirtazapine, or bupropion, necessitating consideration of antidepressant combinations, augmentation options, and eventually irreversible, nonselective MAOIs such as phenelzine, tranylcypromine, or isocarboxazid.3,4 Nonselective MAOIs thus offer a therapeutic opportunity for patients who do not respond to single or dual-mechanism strategies; moreover, nonselective MAOIs have compelling effectiveness data for other conditions, including panic disorder and social phobia.5 Although MAOIs are among the most effective pharmacologic agents for MDD,6 they are underutilized because of an inadequate understanding of risk mechanisms and resultant fear of catastrophic outcomes. Because of the difficulties encountered in achieving clinical remission for MDD, the nonselective MAOIs deserve a second look.
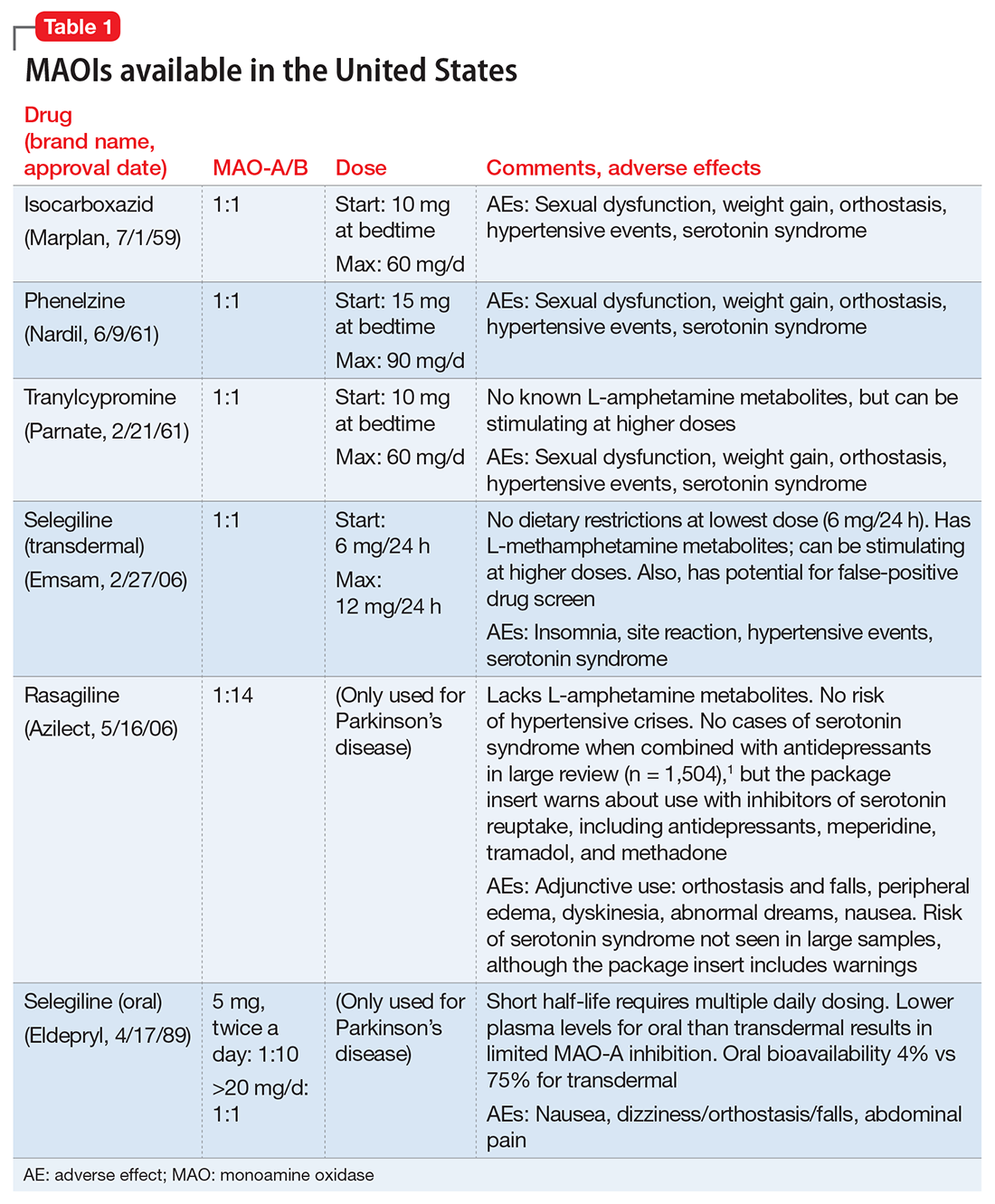
Differentiation of MAO-A from MAO-B. It is essential to understand the mechanism of action of MAOIs, specifically the impact of MAO-A inhibition. Although the enzyme MAO was known in the 1950s, it wasn’t until 1968 that Johnston7 postulated the existence of >1 form. In 1971, Goridis and Neff8 used clorgyline to examine the deamination rate by MAO of tyramine and norepinephrine. They found that tyramine appeared to be a substrate of both MAO isoforms, but only 1 of the MAO types was sensitive to the inhibitory effects of clorgyline. They also discerned that norepinephrine was only a substrate for MAO-A, and that this form of MAO was sensitive to clorgyline inhibition. Thus, the forms of MAO were characterized by their preferred substrates (Table 29,10), and then later by their tissue distribution. Phenylethylamine is a naturally occurring compound found in foods, such as chocolate, and has an in vitro pharmacology similar to amphetamine but with 1 important difference: it has a short half-life of 5 to 10 minutes after oral ingestion, and therefore no appreciable CNS impact.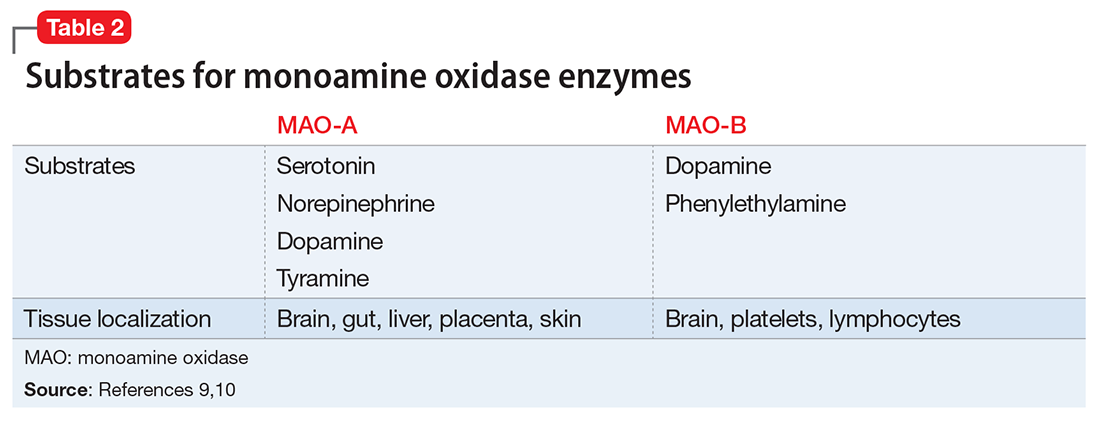
Within the CNS, norepinephrine and dopamine neurons possess both MAO forms, with the MAO-A content greater than MAO-B. Serotonergic neurons only contain MAO-B.11 Outside of the CNS, MAO-A predominates, with only platelets and lymphocytes possessing MAO-B activity.11 The overall relative tissue proportions of MAO-A to MAO-B activity are: brain, 25% MAO-A, 75% MAO-B; liver, 50% MAO-A, 50% MAO-B; intestine, 80% MAO-A, 20% MAO-B; and peripheral adrenergic neurons, 90% MAO-A, 10% MAO-B.
Because of its specificity for serotonin and norepinephrine, CNS MAO-A inhibition is necessary for antidepressant effects. MAO-B inhibition by itself does not appear to raise CNS dopamine levels unless exogenous dopamine is supplied.11 All MAOIs used in the United States to treat depression are irreversible, nonselective inhibitors of MAO-A and MAO-B.
Selegiline in oral form generates low plasma levels and primarily inhibits MAO-B. The transdermal form of selegiline achieves significantly greater systemic exposure, and at these higher plasma levels selegiline is a nonselective, irreversible MAOI effective for MDD (Figure 112). Administering selegiline systemically via a transdermal patch avoids clinically significant MAOI effects in the gut, so no dietary warnings exist for the lowest dose (6 mg/24 hours), although there are warnings for the higher dosages (9 mg/24 hours and 12 mg/24 hours).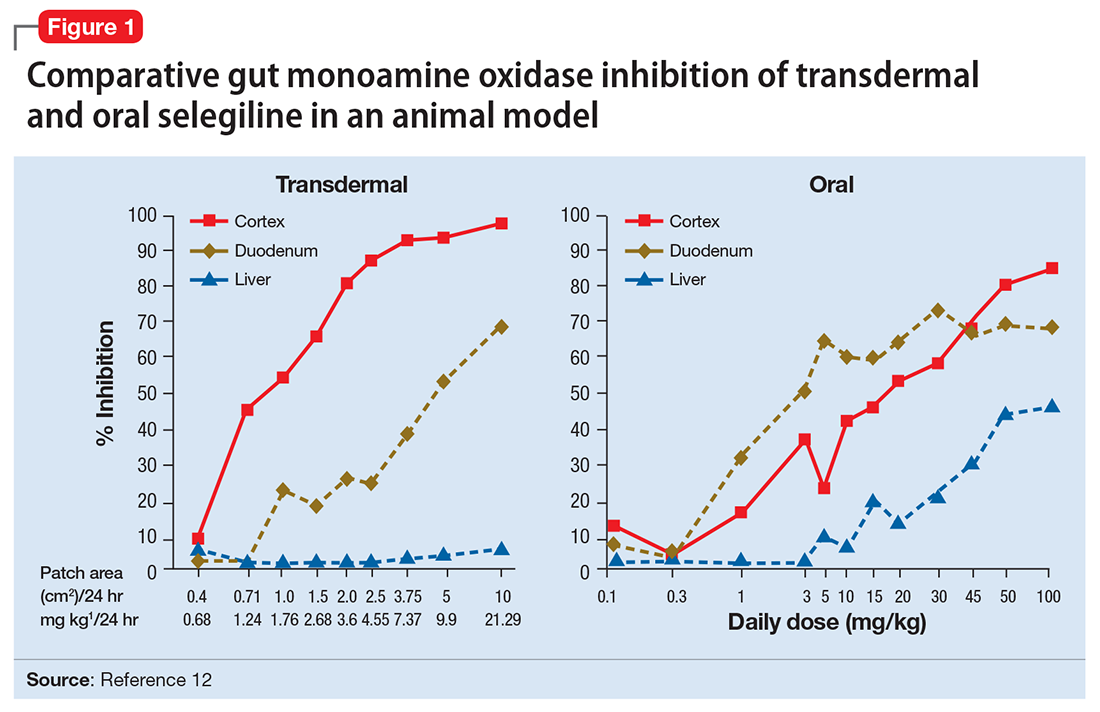
Differentiation of MAOIs by chemical class. The earliest MAOI, iproniazid, was a hydrazine derivative and exhibited hepatotoxicity,13 as did certain other hydrazine MAOIs. This lead to a search for safer hydrazine and nonhydrazine alternatives. Isocarboxazid and phenelzine are the 2 hydrazine MAOIs available in the United States, while tranylcypromine and selegiline transdermal are nonhydrazines (Figure 2).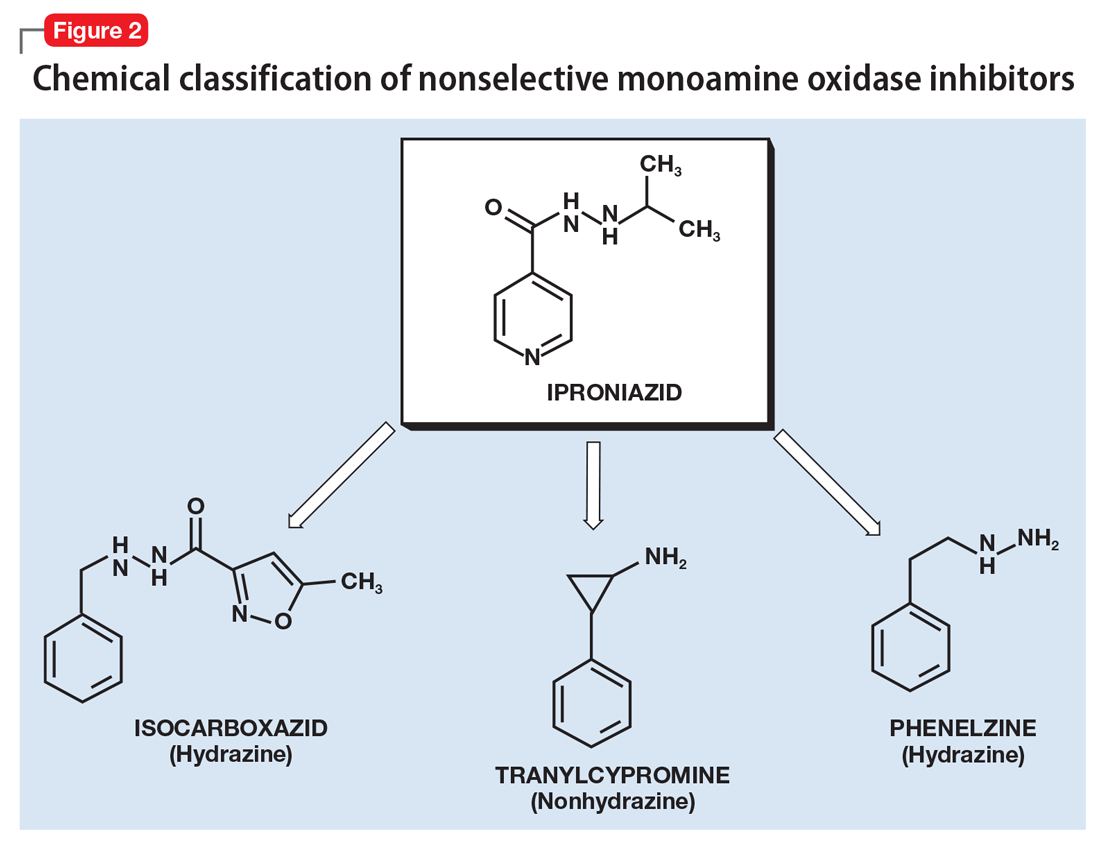
What distinguishes the nonhydrazine medication selegiline is that its metabolism generates L-amphetamine metabolites (Figure 314). This property was thought to be shared by other nonhydrazines, but recent studies indicate than neither tranylcypromine15 nor the MAO-B–selective rasagiline possess amphetamine metabolites.16 Unlike the dextro isomers, L-amphetamine structures do not inhibit dopamine reuptake or cause euphoria, but can cause stimulation (eg, sleep disturbance) by inhibiting norepinephrine reuptake, and also by interacting with the trace amine-associated receptor 1 (TAAR1), an intracellular receptor expressed within the presynaptic terminal of monoamine neurons. Activation of TAAR1 by tyramine is an important part of the hypertensive effects related to excessive tyramine exposure.17 (The importance of TAAR1 and the interaction with tyramine is discussed in the next section.) Importantly, patients taking selegiline must be warned that certain drug screens may not discriminate between levo and dextro isomers of amphetamines, and that the use of selegiline should be disclosed prior to drug testing procedures.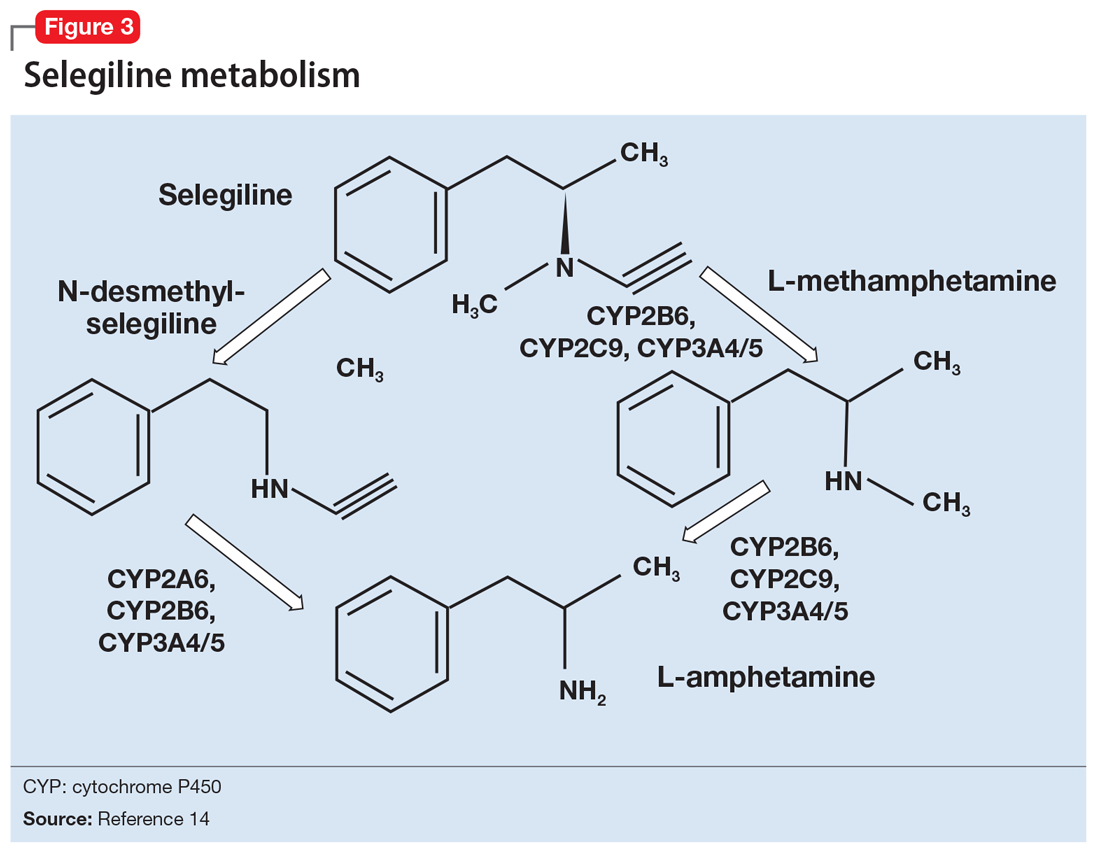
MAOIs and tyramine: Dietary requirements
Clinicians who are familiar with MAOIs recognize that there are dietary restrictions to minimize patients’ exposure to tyramine. As most clinicians know, significant tyramine ingestion may cause an increase in blood pressure (BP) in patients taking an MAOI, but many overestimate the prevalence of foods high in tyramine content since the original reports emerged in the early 1960s.18 In a recent monograph, one of the leading experts on MAOIs, Professor Ken Gillman, stated:
Very few foods now contain problematically high tyramine levels, that is a result of great changes in international food production methods and hygiene regulations. Cheese is the only food that, in the past, has been associated with documented fatalities resulting from hypertension. Nowadays most cheeses are quite safe, and even ‘matured’ cheeses are usually safe in healthy-sized portions. The variability of sensitivity to tyramine between individuals, and the sometimes unpredictable amount of tyramine content in foods, means a little knowledge and care are still required.19
What is tyramine? Tyramine is a biogenic amine that is virtually absent in fresh animal protein sources but is enriched after decay or fermentation.20 Modern food processing and handling methods have significantly limited the tyramine content in processed foods, with the exception of certain cheeses and sauces, as discussed below. Moreover, modern assaying techniques using high-performance liquid chromatography have generated extremely accurate assessments of the tyramine content of specific foods.21 Data published prior to 2000 are not reliable, because many of these publications employed outdated methods.17
When ingested, tyramine is metabolized by gut MAO-A, with doses up to 400 mg causing no known effects, although most people rarely ingest >25 mg during a meal.22 In addition to being a substrate for MAO-A, tyramine is also a substrate for the dopamine transporter, norepinephrine transporter (NET), the vesicular monoamine transporter 2, and TAAR1.23 Tyramine enters the cell via NET, where it interacts with TAAR1, a G protein-coupled receptor that is responsive to trace amines, such as tyramine, as well as amphetamines.20 The agonist properties at TAAR1 are the presumed site of action for the BP effects of tyramine, because binding results in potent release of norepinephrine.20,24 When tyramine is supplied to an animal in which MAO-A is inhibited, the decreased peripheral catabolism of tyramine results in markedly increased norepinephrine release by peripheral adrenergic neurons. Moreover, the absence of MAO-A activity in those neurons prevents any norepinephrine breakdown, resulting in robust synaptic norepinephrine delivery and peripheral effects.
All orally administered irreversible MAOIs potently inhibit gut and systemic MAO-A, and are susceptible to the impact of significant tyramine ingestion. The exception is selegiline transdermal (Figure 112), as appreciable gut MAO-A inhibition does not occur until doses >6 mg/24 hours are reached.22 No significant pressor response was seen in participants taking selegiline transdermal, 6 mg/24 hours for 13 days, who consumed a meal that provided 400 mg of tyramine.22 Conversely, for oral agents that produce gut MAO-A inhibition, tyramine doses as low as 8 to 10 mg (when administered as tyramine capsules) may increase systolic pressure by 30 mm Hg.25 The dietary warnings do not apply to rasagiline, which is a selective MAO-B inhibitor, although rasagiline may have an impact on resting BP; the prescribing information for rasagiline includes warnings about hypotension and hypertension.26
What to tell patients about tyramine. Although administering pure tyramine capsules can induce a measurable change in systolic BP, when ingested as food, tyramine doses <50 mg are unlikely to cause an increase in BP sufficient to warrant clinical intervention, although some individuals can be sensitive to 10 to 25 mg.19 When discussing with patients safety issues related to diet, there are a few important concepts to remember19:
- In an era when the tyramine content of foods was much higher (1960 to 1964) and MAOI users received no dietary guidance, only 14 deaths were reported among an estimated 1.5 million patients who took MAOIs.
- MAOIs do not raise BP, and their use is associated with orthostasis in some patients.
- Routine exercise or other vigorous activities (eg, weightlifting) can raise systolic pressure well above 200 mm Hg, and routine baseline systolic pressures, ranging from 180 to 220 mm Hg, do not increase the risk of subarachnoid hemorrhage.
- Hospital evaluation is needed only if a substantial amount of tyramine is ingested (eg, estimated ≥100 mg), and self-monitoring shows a systolic BP ≥220 mm Hg over a prolonged period (eg, 2 hours). Ingestion of 100 mg of tyramine would almost certainly have to be intentional, as it would require one to consume 3.5 oz of the most highly tyramine-laden cheeses.
Emphasize to patients that only a small number of highly aged cheeses, foods, and sauces contain high quantities of tyramine, and that even these foods can be enjoyed in small amounts. All patients who are prescribed an MAOI also should purchase a portable BP cuff for those rare instances when a dietary indiscretion may have occurred and the person experiences a headache within 1 to 2 hours after tyramine ingestion. Most reactions are self-limited and resolve over 2 to 4 hours.
Patients who ingest ≥100 mg of tyramine should be evaluated by a physician. Under no circumstances should a patient be given a prescription for nifedipine or other medications that can abruptly lower BP, because this may result in complications, including myocardial infarction.27,28 Counsel patients to remain calm. Some clinicians endorse the use of low doses of benzodiazepines (the equivalent of alprazolam 0.5 mg) to facilitate this, because anxiety elevates BP. A recent emergency room study of patients with an initial systolic BP ≥160 mm Hg or diastolic BP ≥100 mm Hg without end organ damage demonstrated that alprazolam, 0.5 mg, was as effective as captopril, 25 mg, in lowering BP.29
Also, tell patients that if a food is unfamiliar and highly aged or fermented, they should avoid it until they can further inquire about it. In a review, Gillman19 provides the tyramine content of an exhaustive list of cheeses, aged meats, and sauces (see Related Resources). For other products, patients often can obtain information directly from the manufacturer. In many parts of the world, assays for tyramine content are required as a demonstration of adequate product safety procedures. Even the most highly aged cheeses with a tyramine content of 1,000 g/kg can be enjoyed in small amounts (<1 oz), and most products would require heroic intake to achieve clinically significant tyramine ingestion (Table 319).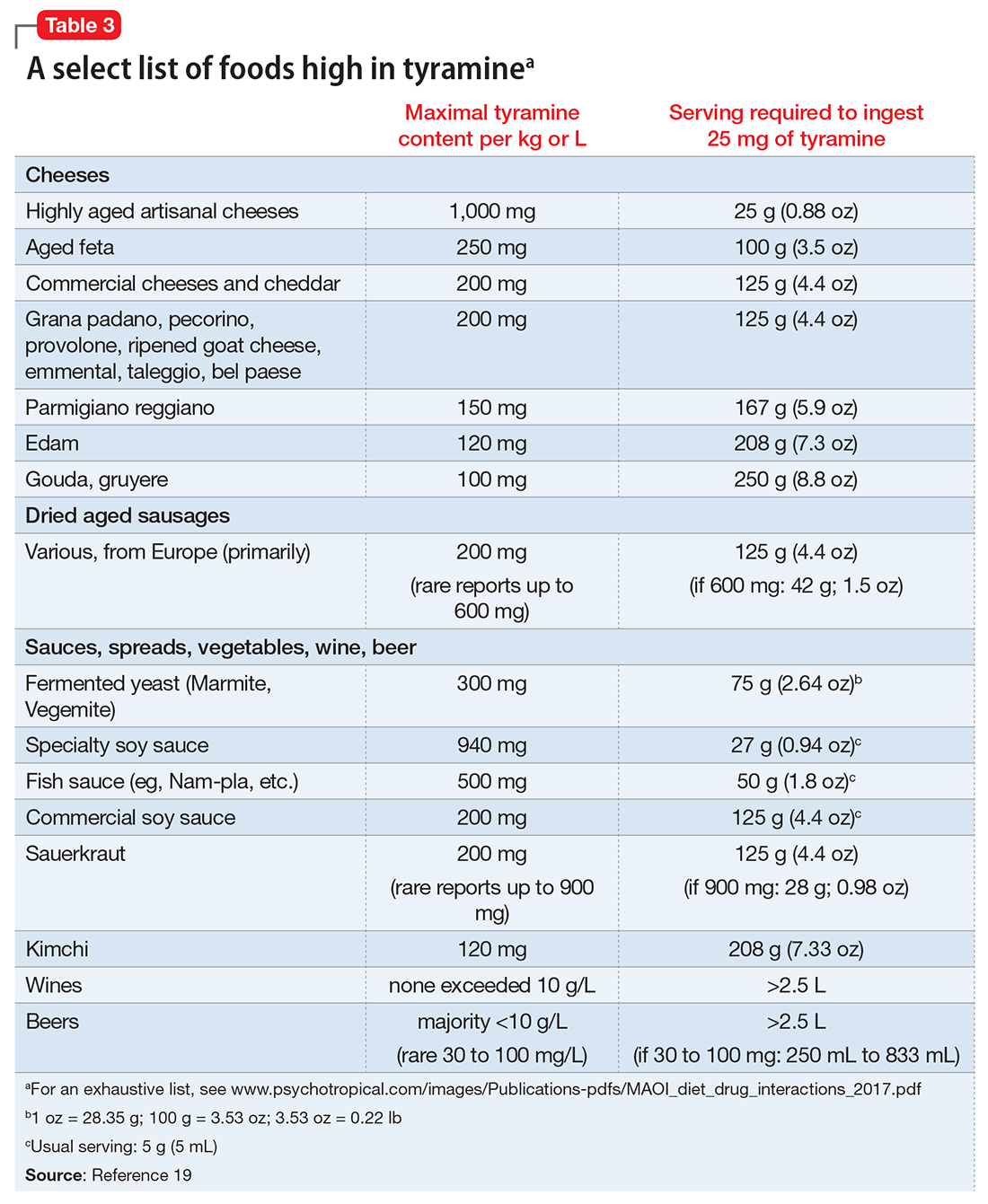
Improved education can clarify the risks
Medications such as lithium, clozapine, and MAOIs have a proven record of efficacy, yet often are underused due to fears engendered by lack of systematic training. A recent initiative in New York thus aimed to increase rates of
1. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
2. López-Muñoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 2009;15(14):1563-1586.
3. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
4. Trivedi MH, Fava M, Wisniewski SR, et al; STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. New Engl J Med. 2006;354(12):1243-1252.
5. Bandelow B, Zohar J, Hollander E, et al; World Federation of Societies of Biological Psychiatry Task Force on Treatment Guidelines for Anxiety, Obsessive-Compulsive and Posttraumatic Stress Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and posttraumatic stress disorders. World J Biol Psychiatry. 2002;3(4):171-199.
6. Shulman KI, Herrmann N, Walker SE. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013;27(10):789-797.
7. Johnston JP. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem Pharmacol. 1968;17(7):1285-1297.
8. Goridis C, Neff NH. Monoamine oxidase in sympathetic nerves: a transmitter specific enzyme type. Br J Pharmacol. 1971;43(4):814-818.
9. Geha RM, Rebrin I, Chen K, et al. Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J Biol Chem. 2001;276(13):9877-9882.
10. O’Carroll AM, Fowler CJ, Phillips JP, et al. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch Pharmacol. 1983;322(3):198-202.
11. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
12. Mawhinney M, Cole D, Azzaro AJ. Daily transdermal administration of selegiline to guinea-pigs preferentially inhibits monoamine oxidase activity in brain when compared with intestinal and hepatic tissues. J Pharm Pharmacol. 2003;55(1):27-34.
13. Maille F, Duvoux C, Cherqui D, et al. Auxiliary hepatic transplantation in iproniazid-induced subfulminant hepatitis. Should iproniazid still be sold in France? [in French]. Gastroenterol Clin Biol. 1999;23(10):1083-1085.
14. Salonen JS, Nyman L, Boobis AR, et al. Comparative studies on the cytochrome p450-associated metabolism and interaction potential of selegiline between human liver-derived in vitro systems. Drug Metab Dispos. 2003;31(9):1093-1102.
15. Iwersen S, Schmoldt A. One fatal and one nonfatal intoxication with tranylcypromine. Absence of amphetamines as metabolites. J Anal Toxicol. 1996;20(5):301-304.
16. Müller T, Hoffmann JA, Dimpfel W, et al. Switch from selegiline to rasagiline is beneficial in patients with Parkinson’s disease. J Neural Transm (Vienna). 2013;120(5):761-765.
17. Lewin AH, Miller GM, Gilmour B. Trace amine-associated receptor 1 is a stereoselective binding site for compounds in the amphetamine class. Bioorg Med Chem. 2011;19(23):7044-7048.
18. Blackwell B. Hypertensive crisis due to monoamine-oxidase inhibitors. Lancet. 1963;2(7313):849-850.
19. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-97.
20. Pei Y, Asif-Malik A, Canales JJ. Trace amines and the trace amine-associated receptor 1: pharmacology, neurochemistry, and clinical implications. Front Neurosci. 2016;10:148.
21. Fiechter G, Sivec G, Mayer HK. Application of UHPLC for the simultaneous analysis of free amino acids and biogenic amines in ripened acid-curd cheeses. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;927:191-200.
22. Blob LF, Sharoky M, Campbell BJ, et al. Effects of a tyramine-enriched meal on blood pressure response in healthy male volunteers treated with selegiline transdermal system 6 mg/24 hour. CNS Spectr. 2007;12(1):25-34.
23. Partilla JS, Dempsey AG, Nagpal AS, et al. Interaction of amphetamines and related compounds at the vesicular monoamine transporter. J Pharmacol Exp Ther. 2006;319(1):237-246.
24. Borowsky B, Adham N, Jones KA, et al. Trace amines: identification of a family of mammalian G protein-coupled receptors. Proc Natl Acad Sci U S A. 2001;98(16):8966-8971.
25. Azzaro AJ, Vandenberg CM, Blob LF, et al. Tyramine pressor sensitivity during treatment with the selegiline transdermal system 6 mg/24 h in healthy subjects. J Clin Pharmacol. 2006;46(8):933-944.
26. Azilect [package insert]. Overland Park, KS: Teva Neuroscience, Inc.; 2014.
27. Marik PE, Varon J. Hypertensive crises: challenges and management. Chest. 2007;131(6):1949-1962.
28. Burton TJ, Wilkinson IB. The dangers of immediate-release nifedipine in the emergency treatment of hypertension. J Hum Hypertens. 2008;22(4):301-302.
29. Yilmaz S, Pekdemir M, Tural U, et al. Comparison of alprazolam versus captopril in high blood pressure: a randomized controlled trial. Blood Press. 2011;20(4):239-243.
30. Carruthers J, Radigan M, Erlich MD, et al. An initiative to improve clozapine prescribing in New York State. Psychiatr Serv. 2016;67(4):369-371.
Despite an abundance of evidenced-based literature supporting monoamine oxidase inhibitors (MAOIs) as an effective treatment for depression, use of these agents has decreased drastically in the past 3 decades. A lack of industry support and the ease of use of other agents are contributing factors, but the biggest impediments to routine use of MAOIs are unfamiliarity with their efficacy advantages and concerns about adverse effects, particularly the risk of hypertensive crises and serotonin syndrome. Many misconceptions regarding these medications are based on outdated data and studies that are no longer reliable.
The goal of this 2-part review is to provide clinicians with updated information regarding MAOIs. Part 1 provides a brief description of:
- the pharmacology of nonselective irreversible MAOIs
- the mechanism by which tyramine induces hypertension
- sources of clinically significant tyramine exposure
- what to tell patients about dietary restrictions and MAOIs.
Part 2 of this guide will cover the risk of serotonin syndrome when MAOIs are combined with inhibitors of serotonin reuptake, how to initiate MAOI therapy, and augmenting MAOIs with other agents.
The pharmacology of MAOIs
First used clinically in the 1950s to treat tuberculosis, MAOIs have a long and interesting history (see the Box “A brief history of monoamine oxidase inhibitors”). Table 11 lists MAOIs currently available in the United States, including the MAO-B–specific agent rasagiline, which is used for Parkinson’s disease.
Manipulation of the monoamines serotonin, norepinephrine, and dopamine is fundamental to managing major depressive disorder (MDD), yet only nonselective MAOIs directly promote neurotransmission of all 3 by inhibiting MAO-A and MAO-B.2 The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study demonstrated that <50% of MDD patients achieve remission in monotherapy trials of selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, mirtazapine, or bupropion, necessitating consideration of antidepressant combinations, augmentation options, and eventually irreversible, nonselective MAOIs such as phenelzine, tranylcypromine, or isocarboxazid.3,4 Nonselective MAOIs thus offer a therapeutic opportunity for patients who do not respond to single or dual-mechanism strategies; moreover, nonselective MAOIs have compelling effectiveness data for other conditions, including panic disorder and social phobia.5 Although MAOIs are among the most effective pharmacologic agents for MDD,6 they are underutilized because of an inadequate understanding of risk mechanisms and resultant fear of catastrophic outcomes. Because of the difficulties encountered in achieving clinical remission for MDD, the nonselective MAOIs deserve a second look.
Differentiation of MAO-A from MAO-B. It is essential to understand the mechanism of action of MAOIs, specifically the impact of MAO-A inhibition. Although the enzyme MAO was known in the 1950s, it wasn’t until 1968 that Johnston7 postulated the existence of >1 form. In 1971, Goridis and Neff8 used clorgyline to examine the deamination rate by MAO of tyramine and norepinephrine. They found that tyramine appeared to be a substrate of both MAO isoforms, but only 1 of the MAO types was sensitive to the inhibitory effects of clorgyline. They also discerned that norepinephrine was only a substrate for MAO-A, and that this form of MAO was sensitive to clorgyline inhibition. Thus, the forms of MAO were characterized by their preferred substrates (Table 29,10), and then later by their tissue distribution. Phenylethylamine is a naturally occurring compound found in foods, such as chocolate, and has an in vitro pharmacology similar to amphetamine but with 1 important difference: it has a short half-life of 5 to 10 minutes after oral ingestion, and therefore no appreciable CNS impact.
Within the CNS, norepinephrine and dopamine neurons possess both MAO forms, with the MAO-A content greater than MAO-B. Serotonergic neurons only contain MAO-B.11 Outside of the CNS, MAO-A predominates, with only platelets and lymphocytes possessing MAO-B activity.11 The overall relative tissue proportions of MAO-A to MAO-B activity are: brain, 25% MAO-A, 75% MAO-B; liver, 50% MAO-A, 50% MAO-B; intestine, 80% MAO-A, 20% MAO-B; and peripheral adrenergic neurons, 90% MAO-A, 10% MAO-B.
Because of its specificity for serotonin and norepinephrine, CNS MAO-A inhibition is necessary for antidepressant effects. MAO-B inhibition by itself does not appear to raise CNS dopamine levels unless exogenous dopamine is supplied.11 All MAOIs used in the United States to treat depression are irreversible, nonselective inhibitors of MAO-A and MAO-B.
Selegiline in oral form generates low plasma levels and primarily inhibits MAO-B. The transdermal form of selegiline achieves significantly greater systemic exposure, and at these higher plasma levels selegiline is a nonselective, irreversible MAOI effective for MDD (Figure 112). Administering selegiline systemically via a transdermal patch avoids clinically significant MAOI effects in the gut, so no dietary warnings exist for the lowest dose (6 mg/24 hours), although there are warnings for the higher dosages (9 mg/24 hours and 12 mg/24 hours).
Differentiation of MAOIs by chemical class. The earliest MAOI, iproniazid, was a hydrazine derivative and exhibited hepatotoxicity,13 as did certain other hydrazine MAOIs. This lead to a search for safer hydrazine and nonhydrazine alternatives. Isocarboxazid and phenelzine are the 2 hydrazine MAOIs available in the United States, while tranylcypromine and selegiline transdermal are nonhydrazines (Figure 2).
What distinguishes the nonhydrazine medication selegiline is that its metabolism generates L-amphetamine metabolites (Figure 314). This property was thought to be shared by other nonhydrazines, but recent studies indicate than neither tranylcypromine15 nor the MAO-B–selective rasagiline possess amphetamine metabolites.16 Unlike the dextro isomers, L-amphetamine structures do not inhibit dopamine reuptake or cause euphoria, but can cause stimulation (eg, sleep disturbance) by inhibiting norepinephrine reuptake, and also by interacting with the trace amine-associated receptor 1 (TAAR1), an intracellular receptor expressed within the presynaptic terminal of monoamine neurons. Activation of TAAR1 by tyramine is an important part of the hypertensive effects related to excessive tyramine exposure.17 (The importance of TAAR1 and the interaction with tyramine is discussed in the next section.) Importantly, patients taking selegiline must be warned that certain drug screens may not discriminate between levo and dextro isomers of amphetamines, and that the use of selegiline should be disclosed prior to drug testing procedures.
MAOIs and tyramine: Dietary requirements
Clinicians who are familiar with MAOIs recognize that there are dietary restrictions to minimize patients’ exposure to tyramine. As most clinicians know, significant tyramine ingestion may cause an increase in blood pressure (BP) in patients taking an MAOI, but many overestimate the prevalence of foods high in tyramine content since the original reports emerged in the early 1960s.18 In a recent monograph, one of the leading experts on MAOIs, Professor Ken Gillman, stated:
Very few foods now contain problematically high tyramine levels, that is a result of great changes in international food production methods and hygiene regulations. Cheese is the only food that, in the past, has been associated with documented fatalities resulting from hypertension. Nowadays most cheeses are quite safe, and even ‘matured’ cheeses are usually safe in healthy-sized portions. The variability of sensitivity to tyramine between individuals, and the sometimes unpredictable amount of tyramine content in foods, means a little knowledge and care are still required.19
What is tyramine? Tyramine is a biogenic amine that is virtually absent in fresh animal protein sources but is enriched after decay or fermentation.20 Modern food processing and handling methods have significantly limited the tyramine content in processed foods, with the exception of certain cheeses and sauces, as discussed below. Moreover, modern assaying techniques using high-performance liquid chromatography have generated extremely accurate assessments of the tyramine content of specific foods.21 Data published prior to 2000 are not reliable, because many of these publications employed outdated methods.17
When ingested, tyramine is metabolized by gut MAO-A, with doses up to 400 mg causing no known effects, although most people rarely ingest >25 mg during a meal.22 In addition to being a substrate for MAO-A, tyramine is also a substrate for the dopamine transporter, norepinephrine transporter (NET), the vesicular monoamine transporter 2, and TAAR1.23 Tyramine enters the cell via NET, where it interacts with TAAR1, a G protein-coupled receptor that is responsive to trace amines, such as tyramine, as well as amphetamines.20 The agonist properties at TAAR1 are the presumed site of action for the BP effects of tyramine, because binding results in potent release of norepinephrine.20,24 When tyramine is supplied to an animal in which MAO-A is inhibited, the decreased peripheral catabolism of tyramine results in markedly increased norepinephrine release by peripheral adrenergic neurons. Moreover, the absence of MAO-A activity in those neurons prevents any norepinephrine breakdown, resulting in robust synaptic norepinephrine delivery and peripheral effects.
All orally administered irreversible MAOIs potently inhibit gut and systemic MAO-A, and are susceptible to the impact of significant tyramine ingestion. The exception is selegiline transdermal (Figure 112), as appreciable gut MAO-A inhibition does not occur until doses >6 mg/24 hours are reached.22 No significant pressor response was seen in participants taking selegiline transdermal, 6 mg/24 hours for 13 days, who consumed a meal that provided 400 mg of tyramine.22 Conversely, for oral agents that produce gut MAO-A inhibition, tyramine doses as low as 8 to 10 mg (when administered as tyramine capsules) may increase systolic pressure by 30 mm Hg.25 The dietary warnings do not apply to rasagiline, which is a selective MAO-B inhibitor, although rasagiline may have an impact on resting BP; the prescribing information for rasagiline includes warnings about hypotension and hypertension.26
What to tell patients about tyramine. Although administering pure tyramine capsules can induce a measurable change in systolic BP, when ingested as food, tyramine doses <50 mg are unlikely to cause an increase in BP sufficient to warrant clinical intervention, although some individuals can be sensitive to 10 to 25 mg.19 When discussing with patients safety issues related to diet, there are a few important concepts to remember19:
- In an era when the tyramine content of foods was much higher (1960 to 1964) and MAOI users received no dietary guidance, only 14 deaths were reported among an estimated 1.5 million patients who took MAOIs.
- MAOIs do not raise BP, and their use is associated with orthostasis in some patients.
- Routine exercise or other vigorous activities (eg, weightlifting) can raise systolic pressure well above 200 mm Hg, and routine baseline systolic pressures, ranging from 180 to 220 mm Hg, do not increase the risk of subarachnoid hemorrhage.
- Hospital evaluation is needed only if a substantial amount of tyramine is ingested (eg, estimated ≥100 mg), and self-monitoring shows a systolic BP ≥220 mm Hg over a prolonged period (eg, 2 hours). Ingestion of 100 mg of tyramine would almost certainly have to be intentional, as it would require one to consume 3.5 oz of the most highly tyramine-laden cheeses.
Emphasize to patients that only a small number of highly aged cheeses, foods, and sauces contain high quantities of tyramine, and that even these foods can be enjoyed in small amounts. All patients who are prescribed an MAOI also should purchase a portable BP cuff for those rare instances when a dietary indiscretion may have occurred and the person experiences a headache within 1 to 2 hours after tyramine ingestion. Most reactions are self-limited and resolve over 2 to 4 hours.
Patients who ingest ≥100 mg of tyramine should be evaluated by a physician. Under no circumstances should a patient be given a prescription for nifedipine or other medications that can abruptly lower BP, because this may result in complications, including myocardial infarction.27,28 Counsel patients to remain calm. Some clinicians endorse the use of low doses of benzodiazepines (the equivalent of alprazolam 0.5 mg) to facilitate this, because anxiety elevates BP. A recent emergency room study of patients with an initial systolic BP ≥160 mm Hg or diastolic BP ≥100 mm Hg without end organ damage demonstrated that alprazolam, 0.5 mg, was as effective as captopril, 25 mg, in lowering BP.29
Also, tell patients that if a food is unfamiliar and highly aged or fermented, they should avoid it until they can further inquire about it. In a review, Gillman19 provides the tyramine content of an exhaustive list of cheeses, aged meats, and sauces (see Related Resources). For other products, patients often can obtain information directly from the manufacturer. In many parts of the world, assays for tyramine content are required as a demonstration of adequate product safety procedures. Even the most highly aged cheeses with a tyramine content of 1,000 g/kg can be enjoyed in small amounts (<1 oz), and most products would require heroic intake to achieve clinically significant tyramine ingestion (Table 319).
Improved education can clarify the risks
Medications such as lithium, clozapine, and MAOIs have a proven record of efficacy, yet often are underused due to fears engendered by lack of systematic training. A recent initiative in New York thus aimed to increase rates of
Despite an abundance of evidenced-based literature supporting monoamine oxidase inhibitors (MAOIs) as an effective treatment for depression, use of these agents has decreased drastically in the past 3 decades. A lack of industry support and the ease of use of other agents are contributing factors, but the biggest impediments to routine use of MAOIs are unfamiliarity with their efficacy advantages and concerns about adverse effects, particularly the risk of hypertensive crises and serotonin syndrome. Many misconceptions regarding these medications are based on outdated data and studies that are no longer reliable.
The goal of this 2-part review is to provide clinicians with updated information regarding MAOIs. Part 1 provides a brief description of:
- the pharmacology of nonselective irreversible MAOIs
- the mechanism by which tyramine induces hypertension
- sources of clinically significant tyramine exposure
- what to tell patients about dietary restrictions and MAOIs.
Part 2 of this guide will cover the risk of serotonin syndrome when MAOIs are combined with inhibitors of serotonin reuptake, how to initiate MAOI therapy, and augmenting MAOIs with other agents.
The pharmacology of MAOIs
First used clinically in the 1950s to treat tuberculosis, MAOIs have a long and interesting history (see the Box “A brief history of monoamine oxidase inhibitors”). Table 11 lists MAOIs currently available in the United States, including the MAO-B–specific agent rasagiline, which is used for Parkinson’s disease.
Manipulation of the monoamines serotonin, norepinephrine, and dopamine is fundamental to managing major depressive disorder (MDD), yet only nonselective MAOIs directly promote neurotransmission of all 3 by inhibiting MAO-A and MAO-B.2 The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study demonstrated that <50% of MDD patients achieve remission in monotherapy trials of selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, mirtazapine, or bupropion, necessitating consideration of antidepressant combinations, augmentation options, and eventually irreversible, nonselective MAOIs such as phenelzine, tranylcypromine, or isocarboxazid.3,4 Nonselective MAOIs thus offer a therapeutic opportunity for patients who do not respond to single or dual-mechanism strategies; moreover, nonselective MAOIs have compelling effectiveness data for other conditions, including panic disorder and social phobia.5 Although MAOIs are among the most effective pharmacologic agents for MDD,6 they are underutilized because of an inadequate understanding of risk mechanisms and resultant fear of catastrophic outcomes. Because of the difficulties encountered in achieving clinical remission for MDD, the nonselective MAOIs deserve a second look.
Differentiation of MAO-A from MAO-B. It is essential to understand the mechanism of action of MAOIs, specifically the impact of MAO-A inhibition. Although the enzyme MAO was known in the 1950s, it wasn’t until 1968 that Johnston7 postulated the existence of >1 form. In 1971, Goridis and Neff8 used clorgyline to examine the deamination rate by MAO of tyramine and norepinephrine. They found that tyramine appeared to be a substrate of both MAO isoforms, but only 1 of the MAO types was sensitive to the inhibitory effects of clorgyline. They also discerned that norepinephrine was only a substrate for MAO-A, and that this form of MAO was sensitive to clorgyline inhibition. Thus, the forms of MAO were characterized by their preferred substrates (Table 29,10), and then later by their tissue distribution. Phenylethylamine is a naturally occurring compound found in foods, such as chocolate, and has an in vitro pharmacology similar to amphetamine but with 1 important difference: it has a short half-life of 5 to 10 minutes after oral ingestion, and therefore no appreciable CNS impact.
Within the CNS, norepinephrine and dopamine neurons possess both MAO forms, with the MAO-A content greater than MAO-B. Serotonergic neurons only contain MAO-B.11 Outside of the CNS, MAO-A predominates, with only platelets and lymphocytes possessing MAO-B activity.11 The overall relative tissue proportions of MAO-A to MAO-B activity are: brain, 25% MAO-A, 75% MAO-B; liver, 50% MAO-A, 50% MAO-B; intestine, 80% MAO-A, 20% MAO-B; and peripheral adrenergic neurons, 90% MAO-A, 10% MAO-B.
Because of its specificity for serotonin and norepinephrine, CNS MAO-A inhibition is necessary for antidepressant effects. MAO-B inhibition by itself does not appear to raise CNS dopamine levels unless exogenous dopamine is supplied.11 All MAOIs used in the United States to treat depression are irreversible, nonselective inhibitors of MAO-A and MAO-B.
Selegiline in oral form generates low plasma levels and primarily inhibits MAO-B. The transdermal form of selegiline achieves significantly greater systemic exposure, and at these higher plasma levels selegiline is a nonselective, irreversible MAOI effective for MDD (Figure 112). Administering selegiline systemically via a transdermal patch avoids clinically significant MAOI effects in the gut, so no dietary warnings exist for the lowest dose (6 mg/24 hours), although there are warnings for the higher dosages (9 mg/24 hours and 12 mg/24 hours).
Differentiation of MAOIs by chemical class. The earliest MAOI, iproniazid, was a hydrazine derivative and exhibited hepatotoxicity,13 as did certain other hydrazine MAOIs. This lead to a search for safer hydrazine and nonhydrazine alternatives. Isocarboxazid and phenelzine are the 2 hydrazine MAOIs available in the United States, while tranylcypromine and selegiline transdermal are nonhydrazines (Figure 2).
What distinguishes the nonhydrazine medication selegiline is that its metabolism generates L-amphetamine metabolites (Figure 314). This property was thought to be shared by other nonhydrazines, but recent studies indicate than neither tranylcypromine15 nor the MAO-B–selective rasagiline possess amphetamine metabolites.16 Unlike the dextro isomers, L-amphetamine structures do not inhibit dopamine reuptake or cause euphoria, but can cause stimulation (eg, sleep disturbance) by inhibiting norepinephrine reuptake, and also by interacting with the trace amine-associated receptor 1 (TAAR1), an intracellular receptor expressed within the presynaptic terminal of monoamine neurons. Activation of TAAR1 by tyramine is an important part of the hypertensive effects related to excessive tyramine exposure.17 (The importance of TAAR1 and the interaction with tyramine is discussed in the next section.) Importantly, patients taking selegiline must be warned that certain drug screens may not discriminate between levo and dextro isomers of amphetamines, and that the use of selegiline should be disclosed prior to drug testing procedures.
MAOIs and tyramine: Dietary requirements
Clinicians who are familiar with MAOIs recognize that there are dietary restrictions to minimize patients’ exposure to tyramine. As most clinicians know, significant tyramine ingestion may cause an increase in blood pressure (BP) in patients taking an MAOI, but many overestimate the prevalence of foods high in tyramine content since the original reports emerged in the early 1960s.18 In a recent monograph, one of the leading experts on MAOIs, Professor Ken Gillman, stated:
Very few foods now contain problematically high tyramine levels, that is a result of great changes in international food production methods and hygiene regulations. Cheese is the only food that, in the past, has been associated with documented fatalities resulting from hypertension. Nowadays most cheeses are quite safe, and even ‘matured’ cheeses are usually safe in healthy-sized portions. The variability of sensitivity to tyramine between individuals, and the sometimes unpredictable amount of tyramine content in foods, means a little knowledge and care are still required.19
What is tyramine? Tyramine is a biogenic amine that is virtually absent in fresh animal protein sources but is enriched after decay or fermentation.20 Modern food processing and handling methods have significantly limited the tyramine content in processed foods, with the exception of certain cheeses and sauces, as discussed below. Moreover, modern assaying techniques using high-performance liquid chromatography have generated extremely accurate assessments of the tyramine content of specific foods.21 Data published prior to 2000 are not reliable, because many of these publications employed outdated methods.17
When ingested, tyramine is metabolized by gut MAO-A, with doses up to 400 mg causing no known effects, although most people rarely ingest >25 mg during a meal.22 In addition to being a substrate for MAO-A, tyramine is also a substrate for the dopamine transporter, norepinephrine transporter (NET), the vesicular monoamine transporter 2, and TAAR1.23 Tyramine enters the cell via NET, where it interacts with TAAR1, a G protein-coupled receptor that is responsive to trace amines, such as tyramine, as well as amphetamines.20 The agonist properties at TAAR1 are the presumed site of action for the BP effects of tyramine, because binding results in potent release of norepinephrine.20,24 When tyramine is supplied to an animal in which MAO-A is inhibited, the decreased peripheral catabolism of tyramine results in markedly increased norepinephrine release by peripheral adrenergic neurons. Moreover, the absence of MAO-A activity in those neurons prevents any norepinephrine breakdown, resulting in robust synaptic norepinephrine delivery and peripheral effects.
All orally administered irreversible MAOIs potently inhibit gut and systemic MAO-A, and are susceptible to the impact of significant tyramine ingestion. The exception is selegiline transdermal (Figure 112), as appreciable gut MAO-A inhibition does not occur until doses >6 mg/24 hours are reached.22 No significant pressor response was seen in participants taking selegiline transdermal, 6 mg/24 hours for 13 days, who consumed a meal that provided 400 mg of tyramine.22 Conversely, for oral agents that produce gut MAO-A inhibition, tyramine doses as low as 8 to 10 mg (when administered as tyramine capsules) may increase systolic pressure by 30 mm Hg.25 The dietary warnings do not apply to rasagiline, which is a selective MAO-B inhibitor, although rasagiline may have an impact on resting BP; the prescribing information for rasagiline includes warnings about hypotension and hypertension.26
What to tell patients about tyramine. Although administering pure tyramine capsules can induce a measurable change in systolic BP, when ingested as food, tyramine doses <50 mg are unlikely to cause an increase in BP sufficient to warrant clinical intervention, although some individuals can be sensitive to 10 to 25 mg.19 When discussing with patients safety issues related to diet, there are a few important concepts to remember19:
- In an era when the tyramine content of foods was much higher (1960 to 1964) and MAOI users received no dietary guidance, only 14 deaths were reported among an estimated 1.5 million patients who took MAOIs.
- MAOIs do not raise BP, and their use is associated with orthostasis in some patients.
- Routine exercise or other vigorous activities (eg, weightlifting) can raise systolic pressure well above 200 mm Hg, and routine baseline systolic pressures, ranging from 180 to 220 mm Hg, do not increase the risk of subarachnoid hemorrhage.
- Hospital evaluation is needed only if a substantial amount of tyramine is ingested (eg, estimated ≥100 mg), and self-monitoring shows a systolic BP ≥220 mm Hg over a prolonged period (eg, 2 hours). Ingestion of 100 mg of tyramine would almost certainly have to be intentional, as it would require one to consume 3.5 oz of the most highly tyramine-laden cheeses.
Emphasize to patients that only a small number of highly aged cheeses, foods, and sauces contain high quantities of tyramine, and that even these foods can be enjoyed in small amounts. All patients who are prescribed an MAOI also should purchase a portable BP cuff for those rare instances when a dietary indiscretion may have occurred and the person experiences a headache within 1 to 2 hours after tyramine ingestion. Most reactions are self-limited and resolve over 2 to 4 hours.
Patients who ingest ≥100 mg of tyramine should be evaluated by a physician. Under no circumstances should a patient be given a prescription for nifedipine or other medications that can abruptly lower BP, because this may result in complications, including myocardial infarction.27,28 Counsel patients to remain calm. Some clinicians endorse the use of low doses of benzodiazepines (the equivalent of alprazolam 0.5 mg) to facilitate this, because anxiety elevates BP. A recent emergency room study of patients with an initial systolic BP ≥160 mm Hg or diastolic BP ≥100 mm Hg without end organ damage demonstrated that alprazolam, 0.5 mg, was as effective as captopril, 25 mg, in lowering BP.29
Also, tell patients that if a food is unfamiliar and highly aged or fermented, they should avoid it until they can further inquire about it. In a review, Gillman19 provides the tyramine content of an exhaustive list of cheeses, aged meats, and sauces (see Related Resources). For other products, patients often can obtain information directly from the manufacturer. In many parts of the world, assays for tyramine content are required as a demonstration of adequate product safety procedures. Even the most highly aged cheeses with a tyramine content of 1,000 g/kg can be enjoyed in small amounts (<1 oz), and most products would require heroic intake to achieve clinically significant tyramine ingestion (Table 319).
Improved education can clarify the risks
Medications such as lithium, clozapine, and MAOIs have a proven record of efficacy, yet often are underused due to fears engendered by lack of systematic training. A recent initiative in New York thus aimed to increase rates of
1. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
2. López-Muñoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 2009;15(14):1563-1586.
3. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
4. Trivedi MH, Fava M, Wisniewski SR, et al; STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. New Engl J Med. 2006;354(12):1243-1252.
5. Bandelow B, Zohar J, Hollander E, et al; World Federation of Societies of Biological Psychiatry Task Force on Treatment Guidelines for Anxiety, Obsessive-Compulsive and Posttraumatic Stress Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and posttraumatic stress disorders. World J Biol Psychiatry. 2002;3(4):171-199.
6. Shulman KI, Herrmann N, Walker SE. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013;27(10):789-797.
7. Johnston JP. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem Pharmacol. 1968;17(7):1285-1297.
8. Goridis C, Neff NH. Monoamine oxidase in sympathetic nerves: a transmitter specific enzyme type. Br J Pharmacol. 1971;43(4):814-818.
9. Geha RM, Rebrin I, Chen K, et al. Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J Biol Chem. 2001;276(13):9877-9882.
10. O’Carroll AM, Fowler CJ, Phillips JP, et al. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch Pharmacol. 1983;322(3):198-202.
11. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
12. Mawhinney M, Cole D, Azzaro AJ. Daily transdermal administration of selegiline to guinea-pigs preferentially inhibits monoamine oxidase activity in brain when compared with intestinal and hepatic tissues. J Pharm Pharmacol. 2003;55(1):27-34.
13. Maille F, Duvoux C, Cherqui D, et al. Auxiliary hepatic transplantation in iproniazid-induced subfulminant hepatitis. Should iproniazid still be sold in France? [in French]. Gastroenterol Clin Biol. 1999;23(10):1083-1085.
14. Salonen JS, Nyman L, Boobis AR, et al. Comparative studies on the cytochrome p450-associated metabolism and interaction potential of selegiline between human liver-derived in vitro systems. Drug Metab Dispos. 2003;31(9):1093-1102.
15. Iwersen S, Schmoldt A. One fatal and one nonfatal intoxication with tranylcypromine. Absence of amphetamines as metabolites. J Anal Toxicol. 1996;20(5):301-304.
16. Müller T, Hoffmann JA, Dimpfel W, et al. Switch from selegiline to rasagiline is beneficial in patients with Parkinson’s disease. J Neural Transm (Vienna). 2013;120(5):761-765.
17. Lewin AH, Miller GM, Gilmour B. Trace amine-associated receptor 1 is a stereoselective binding site for compounds in the amphetamine class. Bioorg Med Chem. 2011;19(23):7044-7048.
18. Blackwell B. Hypertensive crisis due to monoamine-oxidase inhibitors. Lancet. 1963;2(7313):849-850.
19. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-97.
20. Pei Y, Asif-Malik A, Canales JJ. Trace amines and the trace amine-associated receptor 1: pharmacology, neurochemistry, and clinical implications. Front Neurosci. 2016;10:148.
21. Fiechter G, Sivec G, Mayer HK. Application of UHPLC for the simultaneous analysis of free amino acids and biogenic amines in ripened acid-curd cheeses. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;927:191-200.
22. Blob LF, Sharoky M, Campbell BJ, et al. Effects of a tyramine-enriched meal on blood pressure response in healthy male volunteers treated with selegiline transdermal system 6 mg/24 hour. CNS Spectr. 2007;12(1):25-34.
23. Partilla JS, Dempsey AG, Nagpal AS, et al. Interaction of amphetamines and related compounds at the vesicular monoamine transporter. J Pharmacol Exp Ther. 2006;319(1):237-246.
24. Borowsky B, Adham N, Jones KA, et al. Trace amines: identification of a family of mammalian G protein-coupled receptors. Proc Natl Acad Sci U S A. 2001;98(16):8966-8971.
25. Azzaro AJ, Vandenberg CM, Blob LF, et al. Tyramine pressor sensitivity during treatment with the selegiline transdermal system 6 mg/24 h in healthy subjects. J Clin Pharmacol. 2006;46(8):933-944.
26. Azilect [package insert]. Overland Park, KS: Teva Neuroscience, Inc.; 2014.
27. Marik PE, Varon J. Hypertensive crises: challenges and management. Chest. 2007;131(6):1949-1962.
28. Burton TJ, Wilkinson IB. The dangers of immediate-release nifedipine in the emergency treatment of hypertension. J Hum Hypertens. 2008;22(4):301-302.
29. Yilmaz S, Pekdemir M, Tural U, et al. Comparison of alprazolam versus captopril in high blood pressure: a randomized controlled trial. Blood Press. 2011;20(4):239-243.
30. Carruthers J, Radigan M, Erlich MD, et al. An initiative to improve clozapine prescribing in New York State. Psychiatr Serv. 2016;67(4):369-371.
1. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
2. López-Muñoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 2009;15(14):1563-1586.
3. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
4. Trivedi MH, Fava M, Wisniewski SR, et al; STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. New Engl J Med. 2006;354(12):1243-1252.
5. Bandelow B, Zohar J, Hollander E, et al; World Federation of Societies of Biological Psychiatry Task Force on Treatment Guidelines for Anxiety, Obsessive-Compulsive and Posttraumatic Stress Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and posttraumatic stress disorders. World J Biol Psychiatry. 2002;3(4):171-199.
6. Shulman KI, Herrmann N, Walker SE. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013;27(10):789-797.
7. Johnston JP. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem Pharmacol. 1968;17(7):1285-1297.
8. Goridis C, Neff NH. Monoamine oxidase in sympathetic nerves: a transmitter specific enzyme type. Br J Pharmacol. 1971;43(4):814-818.
9. Geha RM, Rebrin I, Chen K, et al. Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J Biol Chem. 2001;276(13):9877-9882.
10. O’Carroll AM, Fowler CJ, Phillips JP, et al. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch Pharmacol. 1983;322(3):198-202.
11. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
12. Mawhinney M, Cole D, Azzaro AJ. Daily transdermal administration of selegiline to guinea-pigs preferentially inhibits monoamine oxidase activity in brain when compared with intestinal and hepatic tissues. J Pharm Pharmacol. 2003;55(1):27-34.
13. Maille F, Duvoux C, Cherqui D, et al. Auxiliary hepatic transplantation in iproniazid-induced subfulminant hepatitis. Should iproniazid still be sold in France? [in French]. Gastroenterol Clin Biol. 1999;23(10):1083-1085.
14. Salonen JS, Nyman L, Boobis AR, et al. Comparative studies on the cytochrome p450-associated metabolism and interaction potential of selegiline between human liver-derived in vitro systems. Drug Metab Dispos. 2003;31(9):1093-1102.
15. Iwersen S, Schmoldt A. One fatal and one nonfatal intoxication with tranylcypromine. Absence of amphetamines as metabolites. J Anal Toxicol. 1996;20(5):301-304.
16. Müller T, Hoffmann JA, Dimpfel W, et al. Switch from selegiline to rasagiline is beneficial in patients with Parkinson’s disease. J Neural Transm (Vienna). 2013;120(5):761-765.
17. Lewin AH, Miller GM, Gilmour B. Trace amine-associated receptor 1 is a stereoselective binding site for compounds in the amphetamine class. Bioorg Med Chem. 2011;19(23):7044-7048.
18. Blackwell B. Hypertensive crisis due to monoamine-oxidase inhibitors. Lancet. 1963;2(7313):849-850.
19. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-97.
20. Pei Y, Asif-Malik A, Canales JJ. Trace amines and the trace amine-associated receptor 1: pharmacology, neurochemistry, and clinical implications. Front Neurosci. 2016;10:148.
21. Fiechter G, Sivec G, Mayer HK. Application of UHPLC for the simultaneous analysis of free amino acids and biogenic amines in ripened acid-curd cheeses. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;927:191-200.
22. Blob LF, Sharoky M, Campbell BJ, et al. Effects of a tyramine-enriched meal on blood pressure response in healthy male volunteers treated with selegiline transdermal system 6 mg/24 hour. CNS Spectr. 2007;12(1):25-34.
23. Partilla JS, Dempsey AG, Nagpal AS, et al. Interaction of amphetamines and related compounds at the vesicular monoamine transporter. J Pharmacol Exp Ther. 2006;319(1):237-246.
24. Borowsky B, Adham N, Jones KA, et al. Trace amines: identification of a family of mammalian G protein-coupled receptors. Proc Natl Acad Sci U S A. 2001;98(16):8966-8971.
25. Azzaro AJ, Vandenberg CM, Blob LF, et al. Tyramine pressor sensitivity during treatment with the selegiline transdermal system 6 mg/24 h in healthy subjects. J Clin Pharmacol. 2006;46(8):933-944.
26. Azilect [package insert]. Overland Park, KS: Teva Neuroscience, Inc.; 2014.
27. Marik PE, Varon J. Hypertensive crises: challenges and management. Chest. 2007;131(6):1949-1962.
28. Burton TJ, Wilkinson IB. The dangers of immediate-release nifedipine in the emergency treatment of hypertension. J Hum Hypertens. 2008;22(4):301-302.
29. Yilmaz S, Pekdemir M, Tural U, et al. Comparison of alprazolam versus captopril in high blood pressure: a randomized controlled trial. Blood Press. 2011;20(4):239-243.
30. Carruthers J, Radigan M, Erlich MD, et al. An initiative to improve clozapine prescribing in New York State. Psychiatr Serv. 2016;67(4):369-371.

Prescribing antipsychotics in geriatric patients: Focus on dementia
According to the U.S. Department of Health and Human Services, in 2007, 88% of 1.4 million Medicare claims for
Because of the aging population and widespread prescription of antipsychotics to older patients, clinicians need information on the relative risks of using these medications in this population. In the United States, all antipsychotics carry a FDA “black-box” warning of the increased risk of death in older adults with dementia. In addition, the risk of death is increased when prescribing antipsychotics to older adults with other conditions, such as Parkinson’s disease,6 and other safety and tolerability concerns, including falls and fractures, sedation, metabolic abnormalities, and extrapyramidal effects, are highly relevant to geriatric patients.
This 3-part review summarizes findings and recommendations on prescribing antipsychotics to older individuals with schizophrenia, bipolar disorder, depression, and dementia. This third and final installment:
- briefly summarizes the major studies and analyses relevant to prescribing antipsychotics to older patients with dementia
- provides a summative opinion on safety and tolerability issues in these older patients
- highlights the gaps in the evidence base and areas that need additional research.
Summary of benefits, place in treatment armamentarium
Behavioral and psychological symptoms of dementia (BPSD) include agitation, delusional beliefs, repetitive questioning, hallucinations, aggression, wandering, and various socially inappropriate behaviors.7 These occur almost universally in all types and stages of dementia.7 BPSD are among the most complex, stressful, and costly aspects of dementia care, and lead to a myriad of poor health outcomes, including excess morbidity, mortality, hospital stays, and early nursing home placement.8-11 Because BPSD usually occur across all types and stages of dementia,7,12-16 the prevalence of BPSD mirrors the overall prevalence of dementia.
Although all expert organizations, including the American Psychiatric Association,17 recommend nonpharmacologic strategies as first-line treatment for BPSD, for the most part, these recommendations have not been translated into standard clinical management or routine care.18 Because of a perceived lack of other options, the current mainstay of treatment is the off-label use of psychotropics such as antipsychotics. Of all the agents currently used for BPSD, SGAs have the strongest evidence base, although benefits are modest at best (standardized effect size 0.13 to 0.16).19,20 In terms of individual SGAs, only risperidone is indicated for aggression in Canada and in Europe (not in the United States);
Clinical Trials
Adverse effects. A meta-analysis of RCTs of SGAs found that, compared with placebo, SGAs have increased rates of several adverse effects. These include somnolence (17% drug vs 7% placebo;
In the 42-site Clinical Antipsychotic Trials of Intervention Effectiveness Alzheimer’s disease RCT, 421 outpatients with Alzheimer’s disease and BPSD were randomized to an SGA (risperidone,
In the 2005 FDA black-box warning, pneumonia and cardiac adverse effects were cited as primary causes of death for patients with dementia taking SGAs. A subsequent observational study confirmed that use of either FGAs or SGAs in geriatric patients was associated with an increased risk of pneumonia, in a dose-dependent manner.27 Although there is limited data on cardiac adverse effects in older adults, especially those with dementia taking antipsychotics,28 1 observational study of nursing home residents29 found that those taking FGAs had a significantly higher risk of hospitalization for ventricular arrhythmia or cardiac arrest compared with those who were not taking FGAs. In contrast, there was no increased risk with SGAs.
Mortality.
In 2005, the FDA announced that based on a reanalysis of 17 placebo-controlled trials (many of which were unpublished) that SGAs were associated with a 1.7-fold increase in mortality compared with placebo.30 As a result, the FDA issued a black-box warning for using SGAs in patients with dementia. The overall OR in a published meta-analysis of mortality with SGAs was 1.54 (1.06 to 2.23; z = 2.28; P = .02), with pooled events of 3.5% mortality vs 2.3% (drug vs placebo).21 This meta-analysis21 also included ad hoc analyses of haloperidol; using combined data from 2 contrasts of haloperidol (with risperidone and quetiapine; 243 patients receiving haloperidol and 239 receiving placebo) they also found 15 deaths (6.2%) with haloperidol and 9 (3.8%) with placebo, resulting in an OR of 1.68.
Other clinical data
Observational studies. Most observational studies have confirmed concerns regarding increased mortality in patients with BPSD who take antipsychotics, with FGAs having a higher risk than SGAs18,31 and SGAs having a higher risk compared with most other psychotropics.32 Three studies that found no increase in mortality with antipsychotics in patients with dementia had methodological issues, including examining prevalence as opposed to new users,33,34 not controlling for exposure,10,33,34 power issues,10,34 not controlling for other psychiatric medications,10 and varying lengths of follow-up.10 An FDA black-box warning for FGAs was announced in 200830 based on 2 observational studies that showed an increased risk of mortality in older adults taking FGAs vs SGAs.35,36
In terms of specific SGAs, Kales et al37 examined the mortality risk associated with individual antipsychotics using various methods to control for confounding. Among a national sample of >33,000 older veterans with dementia newly started on haloperidol, risperidone, olanzapine, quetiapine, or
Most recently, a retrospective case-control study (90,786 patients age ≥65 with dementia) examined the number needed to harm (NNH; ie, number of patients needed to receive treatment that would result in 1 death) over 180 days following initiation of an FGA or SGA.38 This study found the following NNHs: haloperidol, 26 (95% CI, 15 to 99); risperidone, 27 (95% CI, 19 to 46); olanzapine, 40 (95% CI, 21 to 312); and quetiapine, 50 (95% CI, 30 to 150).38 These results are congruent with a review of observational studies that found the highest risk of mortality was associated with haloperidol and
Patterns of antipsychotic use in older dementia patients
There are high rates of antipsychotic use in patients with dementia. Before the FDA issued the black-box warning, the Aging Demographics and Memory study found that the rate of antipsychotic use in community (outpatient) older adults with dementia was approximately 19% between 2002 and 2004 in a representative sample of 307 older adults.39 Another study examining trends in community antipsychotic use in the U.S. Department of Veterans Affairs (VA) found that in the 1990s, SGA use was increasing; approximately 18% of outpatients with dementia were taking these agents.40 Use of SGAs began to decline in 2003, ahead of the 2005 black-box warning, in tandem with other advisories (eg, diabetes, metabolic syndrome,41 and stroke risk).42,43 Olanzapine and risperidone showed declining rates between 2003 and 2005, whereas quetiapine use significantly increased during this period. All 3 SGAs declined after the black-box warning. However, by the end of 2007, the use of SGAs had leveled off to approximately 12% of VA patients with dementia. A recent U.S. Government Accountability Office (GAO) report found that in 2012, 14% of older adult Medicare Part D enrollees with dementia living in the community were prescribed an antipsychotic.44
Use in nursing home residents. Because BPSD are one of the main reasons people with dementia are placed in nursing homes, it is not surprising that rates of antipsychotic use are higher in these settings than in the community. Prior to the black-box warning, studies found that 24% to 32% of nursing home residents were treated with antipsychotics.45-47 A study examining VA nursing homes (n = 133 facilities, n = 3,692 veterans) found that approximately 26% of residents were prescribed antipsychotics in 2004 to 2005.48 The Center for Medicare and Medicaid Services (CMS) National Partnership to Improve Dementia Care in Nursing Homes has appeared to lower antipsychotic medication use in nursing homes; the rate decreased from 24% in long-stay nursing home residents nationwide in 2011 to 19% by the end of 2014. Specific to dementia, a 2010 CMS report49 indicated that approximately 40% of nursing home residents with cognitive impairment and behavioral issues, without psychosis, received antipsychotics. The GAO data indicated that approximately 33% of older Medicare Part D enrollees with dementia who spent >100 days in a nursing home were prescribed an antipsychotic in 2012.44 A recent Canadian study using drug claims data found that overall psychotropic use in patients with dementia remains high, finding that three-fourths of all patients with dementia in long-term care are given at least 1 psychotropic, and up to one-third are prescribed SGAs.50 European data similarly show that antipsychotics continue to be prescribed to up to one-third of long-term care residents with dementia, with 7 out of 10 receiving an SGA.1

Conclusions
The Table provides a summary of the evidence regarding the use of antipsychotics in patients with dementia. Expert consensus is that among BPSD, aggression and psychosis are the primary indications for using antipsychotics.51 Based on multiple RCTs and meta-analyses, the evidence for using SGAs to treat these symptoms is moderate at best. However, in real-world practice settings, SGAs are widely used for symptoms, such as wandering, inappropriate behaviors, resistance to care, etc., for which there is no evidence for efficacy other than sedation. Furthermore, even when there is a potential for benefit, this must be balanced against the risk of adverse effects, including somnolence, worsened cognition, extrapyramidal symptoms, stroke, and mortality.
Clinicians who care for older adults with BPSD should strive to increase the use of first-line nonpharmacologic strategies, by using structured approaches such as DICE (Describe, Investigate, Create, Evaluate) described in the Box.51 Antipsychotics should be reserved for situations in which nonpharmacologic approaches are unsuccessful, or there is concern for serious or imminent risk to the patient or others.

In the future, observational studies using biomarkers, such as neuroimaging markers, of brain health in older patients taking antipsychotics for various durations may give us a better understanding of long-term antipsychotic safety and tolerability and the monitoring required to assess long-term burden of specific antipsychotics in real-world samples.52 However, because of various biases, observational data may not provide answers to all questions,53 and a major challenge is that the number of published RCTs specific to geriatric patients is not growing substantially. Pharmacotherapy evidence is not keeping up with demographic trends. Key developments in RCTs will be the inclusion of biomarkers via neuroimaging, drug serum or brain levels, and genetic profiling. Because of the modest findings of benefits of antipsychotics in dementia and safety concerns addressing brain health in preclinical or early stages, identification of effective non-drug interventions and identifying true disease-modifying agents will be the next challenges of dementia research.
1. Foebel AD, Liperoti R, Onder G, et al; SHELTER Study Investigators. Use of antipsychotic drugs among residents with dementia in European long-term care facilities: results from the SHELTER study. J Am Med Dir Assoc. 2014;15(12):911-917.
2. Foebel A, Ballokova A, Wellens NI, et al. A retrospective, longitudinal study of factors associated with new antipsychotic medication use among recently admitted long-term care residents. BMC Geriatr. 2015;15:128.
3. Parsons C, Johnston S, Mathie E, et al. Potentially inappropriate prescribing in older people with dementia in care homes: a retrospective analysis. Drugs Aging. 2012;29(2):143-155.
4. Vidal X, Agustí A, Vallano A, et al; Potentially Inappropriate Prescription in Older Patients in Spain (PIPOPS) Investigators’ project. Elderly patients treated with psychotropic medicines admitted to hospital: associated characteristics and inappropriate use. Eur J Clin Pharmacol. 2016;72(6):755-764.
5. Caron L, Cottencin O, Lapeyre-Mestre M, et al. Off-label prescribing of antipsychotics in adults, children and elderly individuals: a systematic review of recent prescription trends. Curr Pharm Des. 2015;21(23):3280-3297.
6. Weintraub D, Chiang C, Kim HM, et al. Association of antipsychotic use with mortality risk in patients with parkinson disease. JAMA Neurol. 2016;73(5):535-541.
7. Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 2011;7(5):532-539.
8. Kales HC, Chen P, Blow FC, et al. Rates of clinical depression diagnosis, functional impairment, and nursing home placement in coexisting dementia and depression. Am J Geriatr Psychiatry. 2005;13(6):441-449.
9. Yaffe K, Fox P, Newcomer R, et al. Patient and caregiver characteristics and nursing home placement in patients with dementia. JAMA. 2002;287(16):2090-2097.
10. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
11. Vilalta-Franch J, López-Pousa S, Calvó-Perxas L, et al. Psychosis of Alzheimer disease: prevalence, incidence, persistence, risk factors, and mortality. Am J Geriatr Psychiatry. 2013;21(11):1135-1143.
12. Spalletta G, Musicco M, Padovani A, et al. Neuropsychiatric symptoms and syndromes in a large cohort of newly diagnosed, untreated patients with Alzheimer disease. Am J Geriatr Psychiatry. 2010;18(11):1026-1035.
13. Steinberg M, Shao H, Zandi P, et al; Cache County Investigators. Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: the Cache County Study. Int J Geriatr Psychiatry. 2008;23(2):170-177.
14. Finkel SI, Burns A. Behavioral and psychological symptoms of dementia (BPSD): a clinical and research update-introduction. International Psychogeriatrics. 2000;12:9-12.
15. Lyketsos CG. Neuropsychiatric symptoms (behavioral and psychological symptoms of dementia) and the development of dementia treatments. Int Psychogeriatr. 2007;19(3):409-420.
16. Kunik ME, Snow AL, Davila JA, et al. Causes of aggressive behavior in patients with dementia. J Clin Psychiatry. 2010;71(9):1145-1152.
17. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
18. Kales HC, Gitlin LN, Lyketsos CG. Assessment and management of behavioral and psychological symptoms of dementia. BMJ. 2015;350:h369. doi: 10.1136/bmj.h369.
19. Schneider LS, Pollock VE, Lyness SA. A metaanalysis of controlled trials of neuroleptic treatment in dementia. J Am Geriatr Soc. 1990;38(5):553-563.
20. Yury CA, Fisher JE. Meta-analysis of the effectiveness of atypical antipsychotics for the treatment of behavioural problems in persons with dementia. Psychother Psychosom. 2007;76(4):213-218.
21. Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. 2006;14(3):191-210.
22. Ballard CG, Waite J. The effectiveness of atypical antipsychotics for aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst Rev. 2006:1:CD003476.
23. Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA. 2005;293(5):596-608.
24. Aisen PS, Cummings J, Schneider LS. Symptomatic and nonamyloid/tau based pharmacologic treatment for Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006395. doi: 10.1101/cshperspect.a006395.
25. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
26. Trifirò G, Sultana J, Spina E. Are the safety profiles of antipsychotic drugs used in dementia the same? An updated review of observational studies. Drug Saf. 2014;37(7):501-520.
27. Trifirò G, Gambassi G, Sen EF, et al. Association of community-acquired pneumonia with antipsychotic drug use in elderly patients: a nested case-control study. Ann Intern Med. 2010;152(7):418-425, W139-W140.
28. Sultana J, Trifirò G. Drug safety warnings: a message in a bottle. Analysis. 2008;179:438-446.
29. Liperoti R, Gambassi G, Lapane KL, et al. Cerebrovascular events among elderly nursing home patients treated with conventional or atypical antipsychotics. J Clin Psychiatry. 2005;66(9):1090-1096.
30. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafety information forpatientsandproviders/ucm053171. Updated August 16, 2013. Accessed October 20, 2017.
31. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353(22):2335-2341.
32. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
33. Simoni-Wastila L, Ryder PT, Qian J, et al. Association of antipsychotic use with hospital events and mortality among medicare beneficiaries residing in long-term care facilities. Am J Geriatr Psychiatry. 2009;17(5):417-427.
34. Raivio MM, Laurila JV, Strandberg TE, et al. Neither atypical nor conventional antipsychotics increase mortality or hospital admissions among elderly patients with dementia: a two-year prospective study. Am J Geriatr Psychiatry. 2007;15(5):416-424.
35. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
36. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ. 2007;176(5):627-632.
37. Kales HC, Kim HM, Zivin K, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. 2012;169(1):71-79.
38. Maust DT, Kim HM, Seyfried LS, et al. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry. 2015;72(5):438-445.
39. Rhee Y, Csernansky JG, Emanuel LL, et al. Psychotropic medication burden and factors associated with antipsychotic use: an analysis of a population-based sample of community-dwelling older persons with dementia. J Am Geriatr Soc. 2011;59(11):2100-2107.
40. Kales HC, Zivin K, Kim HM, et al. Trends in antipsychotic use in dementia 1999-2007. Arch Gen Psychiatry. 2011;68(2):190-197.
41. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
42. Brodaty H, Ames D, Snowdon J, et al. A randomized placebo-controlled trial of risperidone for the treatment of aggression, agitation, and psychosis of dementia. J Clin Psychiatry. 2003;64(2):134-143.
43. Wooltorton E. Risperidone (Risperdal): increased rate of cerebrovascular events in dementia trials. CMAJ. 2002;167(11):1269-1270.
44. United States Government Accountability Office. Antipsychotic drug use: HHS has initiatives to reduce use among older adults in nursing homes, but should expand efforts to other settings. http://www.gao.gov/assets/670/668221.pdf. Published January 2015. Accessed October 20, 2017.
45. Chen Y, Briesacher BA, Field TS, et al. Unexplained variation across US nursing homes in antipsychotic prescribing rates. Arch Intern Med. 2010;170(1):89-95.
46. Feng Z, Hirdes JP, Smith TF, et al. Use of physical restraints and antipsychotic medications in nursing homes: a cross-national study. Int J Geriatr Psychiatry. 2009;24(10):1110-1118.
47. Kamble P, Chen H, Sherer J, et al. Antipsychotic drug use among elderly nursing home residents in the United States. Am J Geriatr Pharmacother. 2008;6(4):187-197.
48. Gellad WF, Aspinall SL, Handler SM, et al. Use of antipsychotics among older residents in VA nursing homes. Med Care. 2012;50(11):954-960.
49. Bonner A. Improving dementia care and reducing unnecessary use of antipsychotic medications in nursing homes. Center for Medicare and Medicaid Services. http://ltcombudsman.org/uploads/files/support/alice-bonner-slides.pdf. Published April 28, 2013. Accessed October 20, 2017.
50. Vasudev A, Shariff SZ, Liu K, et al. Trends in psychotropic dispensing among older adults with dementia living in long-term care facilities: 2004-2013. Am J Geriatr Psychiatry. 2015;23(12):1259-1269.
51. Kales HC, Gitlin LN, Lyketsos CG, et al; Detroit Expert Panel on Assessment and Management of Neuropsychiatric Symptoms of Dementia. Management of neuropsychiatric symptoms of dementia in clinical settings: recommendations from a multidisciplinary expert panel. J Am Geriatr Soc. 2014;62(4):762-769.
52. Andreasen NC, Liu D, Ziebell S, et al. Relapse duration, treatment intensity, and brain tissue loss in schizophrenia: a prospective longitudinal MRI study. Am J Psychiatry. 2013;170(6):609-615.
53. Mulsant BH. Challenges of the treatment of neuropsychiatric symptoms associated with dementia. Am J Geriatr Psychiatry. 2014;22(4):317-320.
According to the U.S. Department of Health and Human Services, in 2007, 88% of 1.4 million Medicare claims for
Because of the aging population and widespread prescription of antipsychotics to older patients, clinicians need information on the relative risks of using these medications in this population. In the United States, all antipsychotics carry a FDA “black-box” warning of the increased risk of death in older adults with dementia. In addition, the risk of death is increased when prescribing antipsychotics to older adults with other conditions, such as Parkinson’s disease,6 and other safety and tolerability concerns, including falls and fractures, sedation, metabolic abnormalities, and extrapyramidal effects, are highly relevant to geriatric patients.
This 3-part review summarizes findings and recommendations on prescribing antipsychotics to older individuals with schizophrenia, bipolar disorder, depression, and dementia. This third and final installment:
- briefly summarizes the major studies and analyses relevant to prescribing antipsychotics to older patients with dementia
- provides a summative opinion on safety and tolerability issues in these older patients
- highlights the gaps in the evidence base and areas that need additional research.
Summary of benefits, place in treatment armamentarium
Behavioral and psychological symptoms of dementia (BPSD) include agitation, delusional beliefs, repetitive questioning, hallucinations, aggression, wandering, and various socially inappropriate behaviors.7 These occur almost universally in all types and stages of dementia.7 BPSD are among the most complex, stressful, and costly aspects of dementia care, and lead to a myriad of poor health outcomes, including excess morbidity, mortality, hospital stays, and early nursing home placement.8-11 Because BPSD usually occur across all types and stages of dementia,7,12-16 the prevalence of BPSD mirrors the overall prevalence of dementia.
Although all expert organizations, including the American Psychiatric Association,17 recommend nonpharmacologic strategies as first-line treatment for BPSD, for the most part, these recommendations have not been translated into standard clinical management or routine care.18 Because of a perceived lack of other options, the current mainstay of treatment is the off-label use of psychotropics such as antipsychotics. Of all the agents currently used for BPSD, SGAs have the strongest evidence base, although benefits are modest at best (standardized effect size 0.13 to 0.16).19,20 In terms of individual SGAs, only risperidone is indicated for aggression in Canada and in Europe (not in the United States);
Clinical Trials
Adverse effects. A meta-analysis of RCTs of SGAs found that, compared with placebo, SGAs have increased rates of several adverse effects. These include somnolence (17% drug vs 7% placebo;
In the 42-site Clinical Antipsychotic Trials of Intervention Effectiveness Alzheimer’s disease RCT, 421 outpatients with Alzheimer’s disease and BPSD were randomized to an SGA (risperidone,
In the 2005 FDA black-box warning, pneumonia and cardiac adverse effects were cited as primary causes of death for patients with dementia taking SGAs. A subsequent observational study confirmed that use of either FGAs or SGAs in geriatric patients was associated with an increased risk of pneumonia, in a dose-dependent manner.27 Although there is limited data on cardiac adverse effects in older adults, especially those with dementia taking antipsychotics,28 1 observational study of nursing home residents29 found that those taking FGAs had a significantly higher risk of hospitalization for ventricular arrhythmia or cardiac arrest compared with those who were not taking FGAs. In contrast, there was no increased risk with SGAs.
Mortality.
In 2005, the FDA announced that based on a reanalysis of 17 placebo-controlled trials (many of which were unpublished) that SGAs were associated with a 1.7-fold increase in mortality compared with placebo.30 As a result, the FDA issued a black-box warning for using SGAs in patients with dementia. The overall OR in a published meta-analysis of mortality with SGAs was 1.54 (1.06 to 2.23; z = 2.28; P = .02), with pooled events of 3.5% mortality vs 2.3% (drug vs placebo).21 This meta-analysis21 also included ad hoc analyses of haloperidol; using combined data from 2 contrasts of haloperidol (with risperidone and quetiapine; 243 patients receiving haloperidol and 239 receiving placebo) they also found 15 deaths (6.2%) with haloperidol and 9 (3.8%) with placebo, resulting in an OR of 1.68.
Other clinical data
Observational studies. Most observational studies have confirmed concerns regarding increased mortality in patients with BPSD who take antipsychotics, with FGAs having a higher risk than SGAs18,31 and SGAs having a higher risk compared with most other psychotropics.32 Three studies that found no increase in mortality with antipsychotics in patients with dementia had methodological issues, including examining prevalence as opposed to new users,33,34 not controlling for exposure,10,33,34 power issues,10,34 not controlling for other psychiatric medications,10 and varying lengths of follow-up.10 An FDA black-box warning for FGAs was announced in 200830 based on 2 observational studies that showed an increased risk of mortality in older adults taking FGAs vs SGAs.35,36
In terms of specific SGAs, Kales et al37 examined the mortality risk associated with individual antipsychotics using various methods to control for confounding. Among a national sample of >33,000 older veterans with dementia newly started on haloperidol, risperidone, olanzapine, quetiapine, or
Most recently, a retrospective case-control study (90,786 patients age ≥65 with dementia) examined the number needed to harm (NNH; ie, number of patients needed to receive treatment that would result in 1 death) over 180 days following initiation of an FGA or SGA.38 This study found the following NNHs: haloperidol, 26 (95% CI, 15 to 99); risperidone, 27 (95% CI, 19 to 46); olanzapine, 40 (95% CI, 21 to 312); and quetiapine, 50 (95% CI, 30 to 150).38 These results are congruent with a review of observational studies that found the highest risk of mortality was associated with haloperidol and
Patterns of antipsychotic use in older dementia patients
There are high rates of antipsychotic use in patients with dementia. Before the FDA issued the black-box warning, the Aging Demographics and Memory study found that the rate of antipsychotic use in community (outpatient) older adults with dementia was approximately 19% between 2002 and 2004 in a representative sample of 307 older adults.39 Another study examining trends in community antipsychotic use in the U.S. Department of Veterans Affairs (VA) found that in the 1990s, SGA use was increasing; approximately 18% of outpatients with dementia were taking these agents.40 Use of SGAs began to decline in 2003, ahead of the 2005 black-box warning, in tandem with other advisories (eg, diabetes, metabolic syndrome,41 and stroke risk).42,43 Olanzapine and risperidone showed declining rates between 2003 and 2005, whereas quetiapine use significantly increased during this period. All 3 SGAs declined after the black-box warning. However, by the end of 2007, the use of SGAs had leveled off to approximately 12% of VA patients with dementia. A recent U.S. Government Accountability Office (GAO) report found that in 2012, 14% of older adult Medicare Part D enrollees with dementia living in the community were prescribed an antipsychotic.44
Use in nursing home residents. Because BPSD are one of the main reasons people with dementia are placed in nursing homes, it is not surprising that rates of antipsychotic use are higher in these settings than in the community. Prior to the black-box warning, studies found that 24% to 32% of nursing home residents were treated with antipsychotics.45-47 A study examining VA nursing homes (n = 133 facilities, n = 3,692 veterans) found that approximately 26% of residents were prescribed antipsychotics in 2004 to 2005.48 The Center for Medicare and Medicaid Services (CMS) National Partnership to Improve Dementia Care in Nursing Homes has appeared to lower antipsychotic medication use in nursing homes; the rate decreased from 24% in long-stay nursing home residents nationwide in 2011 to 19% by the end of 2014. Specific to dementia, a 2010 CMS report49 indicated that approximately 40% of nursing home residents with cognitive impairment and behavioral issues, without psychosis, received antipsychotics. The GAO data indicated that approximately 33% of older Medicare Part D enrollees with dementia who spent >100 days in a nursing home were prescribed an antipsychotic in 2012.44 A recent Canadian study using drug claims data found that overall psychotropic use in patients with dementia remains high, finding that three-fourths of all patients with dementia in long-term care are given at least 1 psychotropic, and up to one-third are prescribed SGAs.50 European data similarly show that antipsychotics continue to be prescribed to up to one-third of long-term care residents with dementia, with 7 out of 10 receiving an SGA.1
Conclusions
The Table provides a summary of the evidence regarding the use of antipsychotics in patients with dementia. Expert consensus is that among BPSD, aggression and psychosis are the primary indications for using antipsychotics.51 Based on multiple RCTs and meta-analyses, the evidence for using SGAs to treat these symptoms is moderate at best. However, in real-world practice settings, SGAs are widely used for symptoms, such as wandering, inappropriate behaviors, resistance to care, etc., for which there is no evidence for efficacy other than sedation. Furthermore, even when there is a potential for benefit, this must be balanced against the risk of adverse effects, including somnolence, worsened cognition, extrapyramidal symptoms, stroke, and mortality.
Clinicians who care for older adults with BPSD should strive to increase the use of first-line nonpharmacologic strategies, by using structured approaches such as DICE (Describe, Investigate, Create, Evaluate) described in the Box.51 Antipsychotics should be reserved for situations in which nonpharmacologic approaches are unsuccessful, or there is concern for serious or imminent risk to the patient or others.
In the future, observational studies using biomarkers, such as neuroimaging markers, of brain health in older patients taking antipsychotics for various durations may give us a better understanding of long-term antipsychotic safety and tolerability and the monitoring required to assess long-term burden of specific antipsychotics in real-world samples.52 However, because of various biases, observational data may not provide answers to all questions,53 and a major challenge is that the number of published RCTs specific to geriatric patients is not growing substantially. Pharmacotherapy evidence is not keeping up with demographic trends. Key developments in RCTs will be the inclusion of biomarkers via neuroimaging, drug serum or brain levels, and genetic profiling. Because of the modest findings of benefits of antipsychotics in dementia and safety concerns addressing brain health in preclinical or early stages, identification of effective non-drug interventions and identifying true disease-modifying agents will be the next challenges of dementia research.
According to the U.S. Department of Health and Human Services, in 2007, 88% of 1.4 million Medicare claims for
Because of the aging population and widespread prescription of antipsychotics to older patients, clinicians need information on the relative risks of using these medications in this population. In the United States, all antipsychotics carry a FDA “black-box” warning of the increased risk of death in older adults with dementia. In addition, the risk of death is increased when prescribing antipsychotics to older adults with other conditions, such as Parkinson’s disease,6 and other safety and tolerability concerns, including falls and fractures, sedation, metabolic abnormalities, and extrapyramidal effects, are highly relevant to geriatric patients.
This 3-part review summarizes findings and recommendations on prescribing antipsychotics to older individuals with schizophrenia, bipolar disorder, depression, and dementia. This third and final installment:
- briefly summarizes the major studies and analyses relevant to prescribing antipsychotics to older patients with dementia
- provides a summative opinion on safety and tolerability issues in these older patients
- highlights the gaps in the evidence base and areas that need additional research.
Summary of benefits, place in treatment armamentarium
Behavioral and psychological symptoms of dementia (BPSD) include agitation, delusional beliefs, repetitive questioning, hallucinations, aggression, wandering, and various socially inappropriate behaviors.7 These occur almost universally in all types and stages of dementia.7 BPSD are among the most complex, stressful, and costly aspects of dementia care, and lead to a myriad of poor health outcomes, including excess morbidity, mortality, hospital stays, and early nursing home placement.8-11 Because BPSD usually occur across all types and stages of dementia,7,12-16 the prevalence of BPSD mirrors the overall prevalence of dementia.
Although all expert organizations, including the American Psychiatric Association,17 recommend nonpharmacologic strategies as first-line treatment for BPSD, for the most part, these recommendations have not been translated into standard clinical management or routine care.18 Because of a perceived lack of other options, the current mainstay of treatment is the off-label use of psychotropics such as antipsychotics. Of all the agents currently used for BPSD, SGAs have the strongest evidence base, although benefits are modest at best (standardized effect size 0.13 to 0.16).19,20 In terms of individual SGAs, only risperidone is indicated for aggression in Canada and in Europe (not in the United States);
Clinical Trials
Adverse effects. A meta-analysis of RCTs of SGAs found that, compared with placebo, SGAs have increased rates of several adverse effects. These include somnolence (17% drug vs 7% placebo;
In the 42-site Clinical Antipsychotic Trials of Intervention Effectiveness Alzheimer’s disease RCT, 421 outpatients with Alzheimer’s disease and BPSD were randomized to an SGA (risperidone,
In the 2005 FDA black-box warning, pneumonia and cardiac adverse effects were cited as primary causes of death for patients with dementia taking SGAs. A subsequent observational study confirmed that use of either FGAs or SGAs in geriatric patients was associated with an increased risk of pneumonia, in a dose-dependent manner.27 Although there is limited data on cardiac adverse effects in older adults, especially those with dementia taking antipsychotics,28 1 observational study of nursing home residents29 found that those taking FGAs had a significantly higher risk of hospitalization for ventricular arrhythmia or cardiac arrest compared with those who were not taking FGAs. In contrast, there was no increased risk with SGAs.
Mortality.
In 2005, the FDA announced that based on a reanalysis of 17 placebo-controlled trials (many of which were unpublished) that SGAs were associated with a 1.7-fold increase in mortality compared with placebo.30 As a result, the FDA issued a black-box warning for using SGAs in patients with dementia. The overall OR in a published meta-analysis of mortality with SGAs was 1.54 (1.06 to 2.23; z = 2.28; P = .02), with pooled events of 3.5% mortality vs 2.3% (drug vs placebo).21 This meta-analysis21 also included ad hoc analyses of haloperidol; using combined data from 2 contrasts of haloperidol (with risperidone and quetiapine; 243 patients receiving haloperidol and 239 receiving placebo) they also found 15 deaths (6.2%) with haloperidol and 9 (3.8%) with placebo, resulting in an OR of 1.68.
Other clinical data
Observational studies. Most observational studies have confirmed concerns regarding increased mortality in patients with BPSD who take antipsychotics, with FGAs having a higher risk than SGAs18,31 and SGAs having a higher risk compared with most other psychotropics.32 Three studies that found no increase in mortality with antipsychotics in patients with dementia had methodological issues, including examining prevalence as opposed to new users,33,34 not controlling for exposure,10,33,34 power issues,10,34 not controlling for other psychiatric medications,10 and varying lengths of follow-up.10 An FDA black-box warning for FGAs was announced in 200830 based on 2 observational studies that showed an increased risk of mortality in older adults taking FGAs vs SGAs.35,36
In terms of specific SGAs, Kales et al37 examined the mortality risk associated with individual antipsychotics using various methods to control for confounding. Among a national sample of >33,000 older veterans with dementia newly started on haloperidol, risperidone, olanzapine, quetiapine, or
Most recently, a retrospective case-control study (90,786 patients age ≥65 with dementia) examined the number needed to harm (NNH; ie, number of patients needed to receive treatment that would result in 1 death) over 180 days following initiation of an FGA or SGA.38 This study found the following NNHs: haloperidol, 26 (95% CI, 15 to 99); risperidone, 27 (95% CI, 19 to 46); olanzapine, 40 (95% CI, 21 to 312); and quetiapine, 50 (95% CI, 30 to 150).38 These results are congruent with a review of observational studies that found the highest risk of mortality was associated with haloperidol and
Patterns of antipsychotic use in older dementia patients
There are high rates of antipsychotic use in patients with dementia. Before the FDA issued the black-box warning, the Aging Demographics and Memory study found that the rate of antipsychotic use in community (outpatient) older adults with dementia was approximately 19% between 2002 and 2004 in a representative sample of 307 older adults.39 Another study examining trends in community antipsychotic use in the U.S. Department of Veterans Affairs (VA) found that in the 1990s, SGA use was increasing; approximately 18% of outpatients with dementia were taking these agents.40 Use of SGAs began to decline in 2003, ahead of the 2005 black-box warning, in tandem with other advisories (eg, diabetes, metabolic syndrome,41 and stroke risk).42,43 Olanzapine and risperidone showed declining rates between 2003 and 2005, whereas quetiapine use significantly increased during this period. All 3 SGAs declined after the black-box warning. However, by the end of 2007, the use of SGAs had leveled off to approximately 12% of VA patients with dementia. A recent U.S. Government Accountability Office (GAO) report found that in 2012, 14% of older adult Medicare Part D enrollees with dementia living in the community were prescribed an antipsychotic.44
Use in nursing home residents. Because BPSD are one of the main reasons people with dementia are placed in nursing homes, it is not surprising that rates of antipsychotic use are higher in these settings than in the community. Prior to the black-box warning, studies found that 24% to 32% of nursing home residents were treated with antipsychotics.45-47 A study examining VA nursing homes (n = 133 facilities, n = 3,692 veterans) found that approximately 26% of residents were prescribed antipsychotics in 2004 to 2005.48 The Center for Medicare and Medicaid Services (CMS) National Partnership to Improve Dementia Care in Nursing Homes has appeared to lower antipsychotic medication use in nursing homes; the rate decreased from 24% in long-stay nursing home residents nationwide in 2011 to 19% by the end of 2014. Specific to dementia, a 2010 CMS report49 indicated that approximately 40% of nursing home residents with cognitive impairment and behavioral issues, without psychosis, received antipsychotics. The GAO data indicated that approximately 33% of older Medicare Part D enrollees with dementia who spent >100 days in a nursing home were prescribed an antipsychotic in 2012.44 A recent Canadian study using drug claims data found that overall psychotropic use in patients with dementia remains high, finding that three-fourths of all patients with dementia in long-term care are given at least 1 psychotropic, and up to one-third are prescribed SGAs.50 European data similarly show that antipsychotics continue to be prescribed to up to one-third of long-term care residents with dementia, with 7 out of 10 receiving an SGA.1
Conclusions
The Table provides a summary of the evidence regarding the use of antipsychotics in patients with dementia. Expert consensus is that among BPSD, aggression and psychosis are the primary indications for using antipsychotics.51 Based on multiple RCTs and meta-analyses, the evidence for using SGAs to treat these symptoms is moderate at best. However, in real-world practice settings, SGAs are widely used for symptoms, such as wandering, inappropriate behaviors, resistance to care, etc., for which there is no evidence for efficacy other than sedation. Furthermore, even when there is a potential for benefit, this must be balanced against the risk of adverse effects, including somnolence, worsened cognition, extrapyramidal symptoms, stroke, and mortality.
Clinicians who care for older adults with BPSD should strive to increase the use of first-line nonpharmacologic strategies, by using structured approaches such as DICE (Describe, Investigate, Create, Evaluate) described in the Box.51 Antipsychotics should be reserved for situations in which nonpharmacologic approaches are unsuccessful, or there is concern for serious or imminent risk to the patient or others.
In the future, observational studies using biomarkers, such as neuroimaging markers, of brain health in older patients taking antipsychotics for various durations may give us a better understanding of long-term antipsychotic safety and tolerability and the monitoring required to assess long-term burden of specific antipsychotics in real-world samples.52 However, because of various biases, observational data may not provide answers to all questions,53 and a major challenge is that the number of published RCTs specific to geriatric patients is not growing substantially. Pharmacotherapy evidence is not keeping up with demographic trends. Key developments in RCTs will be the inclusion of biomarkers via neuroimaging, drug serum or brain levels, and genetic profiling. Because of the modest findings of benefits of antipsychotics in dementia and safety concerns addressing brain health in preclinical or early stages, identification of effective non-drug interventions and identifying true disease-modifying agents will be the next challenges of dementia research.
1. Foebel AD, Liperoti R, Onder G, et al; SHELTER Study Investigators. Use of antipsychotic drugs among residents with dementia in European long-term care facilities: results from the SHELTER study. J Am Med Dir Assoc. 2014;15(12):911-917.
2. Foebel A, Ballokova A, Wellens NI, et al. A retrospective, longitudinal study of factors associated with new antipsychotic medication use among recently admitted long-term care residents. BMC Geriatr. 2015;15:128.
3. Parsons C, Johnston S, Mathie E, et al. Potentially inappropriate prescribing in older people with dementia in care homes: a retrospective analysis. Drugs Aging. 2012;29(2):143-155.
4. Vidal X, Agustí A, Vallano A, et al; Potentially Inappropriate Prescription in Older Patients in Spain (PIPOPS) Investigators’ project. Elderly patients treated with psychotropic medicines admitted to hospital: associated characteristics and inappropriate use. Eur J Clin Pharmacol. 2016;72(6):755-764.
5. Caron L, Cottencin O, Lapeyre-Mestre M, et al. Off-label prescribing of antipsychotics in adults, children and elderly individuals: a systematic review of recent prescription trends. Curr Pharm Des. 2015;21(23):3280-3297.
6. Weintraub D, Chiang C, Kim HM, et al. Association of antipsychotic use with mortality risk in patients with parkinson disease. JAMA Neurol. 2016;73(5):535-541.
7. Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 2011;7(5):532-539.
8. Kales HC, Chen P, Blow FC, et al. Rates of clinical depression diagnosis, functional impairment, and nursing home placement in coexisting dementia and depression. Am J Geriatr Psychiatry. 2005;13(6):441-449.
9. Yaffe K, Fox P, Newcomer R, et al. Patient and caregiver characteristics and nursing home placement in patients with dementia. JAMA. 2002;287(16):2090-2097.
10. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
11. Vilalta-Franch J, López-Pousa S, Calvó-Perxas L, et al. Psychosis of Alzheimer disease: prevalence, incidence, persistence, risk factors, and mortality. Am J Geriatr Psychiatry. 2013;21(11):1135-1143.
12. Spalletta G, Musicco M, Padovani A, et al. Neuropsychiatric symptoms and syndromes in a large cohort of newly diagnosed, untreated patients with Alzheimer disease. Am J Geriatr Psychiatry. 2010;18(11):1026-1035.
13. Steinberg M, Shao H, Zandi P, et al; Cache County Investigators. Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: the Cache County Study. Int J Geriatr Psychiatry. 2008;23(2):170-177.
14. Finkel SI, Burns A. Behavioral and psychological symptoms of dementia (BPSD): a clinical and research update-introduction. International Psychogeriatrics. 2000;12:9-12.
15. Lyketsos CG. Neuropsychiatric symptoms (behavioral and psychological symptoms of dementia) and the development of dementia treatments. Int Psychogeriatr. 2007;19(3):409-420.
16. Kunik ME, Snow AL, Davila JA, et al. Causes of aggressive behavior in patients with dementia. J Clin Psychiatry. 2010;71(9):1145-1152.
17. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
18. Kales HC, Gitlin LN, Lyketsos CG. Assessment and management of behavioral and psychological symptoms of dementia. BMJ. 2015;350:h369. doi: 10.1136/bmj.h369.
19. Schneider LS, Pollock VE, Lyness SA. A metaanalysis of controlled trials of neuroleptic treatment in dementia. J Am Geriatr Soc. 1990;38(5):553-563.
20. Yury CA, Fisher JE. Meta-analysis of the effectiveness of atypical antipsychotics for the treatment of behavioural problems in persons with dementia. Psychother Psychosom. 2007;76(4):213-218.
21. Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. 2006;14(3):191-210.
22. Ballard CG, Waite J. The effectiveness of atypical antipsychotics for aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst Rev. 2006:1:CD003476.
23. Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA. 2005;293(5):596-608.
24. Aisen PS, Cummings J, Schneider LS. Symptomatic and nonamyloid/tau based pharmacologic treatment for Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006395. doi: 10.1101/cshperspect.a006395.
25. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
26. Trifirò G, Sultana J, Spina E. Are the safety profiles of antipsychotic drugs used in dementia the same? An updated review of observational studies. Drug Saf. 2014;37(7):501-520.
27. Trifirò G, Gambassi G, Sen EF, et al. Association of community-acquired pneumonia with antipsychotic drug use in elderly patients: a nested case-control study. Ann Intern Med. 2010;152(7):418-425, W139-W140.
28. Sultana J, Trifirò G. Drug safety warnings: a message in a bottle. Analysis. 2008;179:438-446.
29. Liperoti R, Gambassi G, Lapane KL, et al. Cerebrovascular events among elderly nursing home patients treated with conventional or atypical antipsychotics. J Clin Psychiatry. 2005;66(9):1090-1096.
30. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafety information forpatientsandproviders/ucm053171. Updated August 16, 2013. Accessed October 20, 2017.
31. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353(22):2335-2341.
32. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
33. Simoni-Wastila L, Ryder PT, Qian J, et al. Association of antipsychotic use with hospital events and mortality among medicare beneficiaries residing in long-term care facilities. Am J Geriatr Psychiatry. 2009;17(5):417-427.
34. Raivio MM, Laurila JV, Strandberg TE, et al. Neither atypical nor conventional antipsychotics increase mortality or hospital admissions among elderly patients with dementia: a two-year prospective study. Am J Geriatr Psychiatry. 2007;15(5):416-424.
35. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
36. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ. 2007;176(5):627-632.
37. Kales HC, Kim HM, Zivin K, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. 2012;169(1):71-79.
38. Maust DT, Kim HM, Seyfried LS, et al. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry. 2015;72(5):438-445.
39. Rhee Y, Csernansky JG, Emanuel LL, et al. Psychotropic medication burden and factors associated with antipsychotic use: an analysis of a population-based sample of community-dwelling older persons with dementia. J Am Geriatr Soc. 2011;59(11):2100-2107.
40. Kales HC, Zivin K, Kim HM, et al. Trends in antipsychotic use in dementia 1999-2007. Arch Gen Psychiatry. 2011;68(2):190-197.
41. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
42. Brodaty H, Ames D, Snowdon J, et al. A randomized placebo-controlled trial of risperidone for the treatment of aggression, agitation, and psychosis of dementia. J Clin Psychiatry. 2003;64(2):134-143.
43. Wooltorton E. Risperidone (Risperdal): increased rate of cerebrovascular events in dementia trials. CMAJ. 2002;167(11):1269-1270.
44. United States Government Accountability Office. Antipsychotic drug use: HHS has initiatives to reduce use among older adults in nursing homes, but should expand efforts to other settings. http://www.gao.gov/assets/670/668221.pdf. Published January 2015. Accessed October 20, 2017.
45. Chen Y, Briesacher BA, Field TS, et al. Unexplained variation across US nursing homes in antipsychotic prescribing rates. Arch Intern Med. 2010;170(1):89-95.
46. Feng Z, Hirdes JP, Smith TF, et al. Use of physical restraints and antipsychotic medications in nursing homes: a cross-national study. Int J Geriatr Psychiatry. 2009;24(10):1110-1118.
47. Kamble P, Chen H, Sherer J, et al. Antipsychotic drug use among elderly nursing home residents in the United States. Am J Geriatr Pharmacother. 2008;6(4):187-197.
48. Gellad WF, Aspinall SL, Handler SM, et al. Use of antipsychotics among older residents in VA nursing homes. Med Care. 2012;50(11):954-960.
49. Bonner A. Improving dementia care and reducing unnecessary use of antipsychotic medications in nursing homes. Center for Medicare and Medicaid Services. http://ltcombudsman.org/uploads/files/support/alice-bonner-slides.pdf. Published April 28, 2013. Accessed October 20, 2017.
50. Vasudev A, Shariff SZ, Liu K, et al. Trends in psychotropic dispensing among older adults with dementia living in long-term care facilities: 2004-2013. Am J Geriatr Psychiatry. 2015;23(12):1259-1269.
51. Kales HC, Gitlin LN, Lyketsos CG, et al; Detroit Expert Panel on Assessment and Management of Neuropsychiatric Symptoms of Dementia. Management of neuropsychiatric symptoms of dementia in clinical settings: recommendations from a multidisciplinary expert panel. J Am Geriatr Soc. 2014;62(4):762-769.
52. Andreasen NC, Liu D, Ziebell S, et al. Relapse duration, treatment intensity, and brain tissue loss in schizophrenia: a prospective longitudinal MRI study. Am J Psychiatry. 2013;170(6):609-615.
53. Mulsant BH. Challenges of the treatment of neuropsychiatric symptoms associated with dementia. Am J Geriatr Psychiatry. 2014;22(4):317-320.
1. Foebel AD, Liperoti R, Onder G, et al; SHELTER Study Investigators. Use of antipsychotic drugs among residents with dementia in European long-term care facilities: results from the SHELTER study. J Am Med Dir Assoc. 2014;15(12):911-917.
2. Foebel A, Ballokova A, Wellens NI, et al. A retrospective, longitudinal study of factors associated with new antipsychotic medication use among recently admitted long-term care residents. BMC Geriatr. 2015;15:128.
3. Parsons C, Johnston S, Mathie E, et al. Potentially inappropriate prescribing in older people with dementia in care homes: a retrospective analysis. Drugs Aging. 2012;29(2):143-155.
4. Vidal X, Agustí A, Vallano A, et al; Potentially Inappropriate Prescription in Older Patients in Spain (PIPOPS) Investigators’ project. Elderly patients treated with psychotropic medicines admitted to hospital: associated characteristics and inappropriate use. Eur J Clin Pharmacol. 2016;72(6):755-764.
5. Caron L, Cottencin O, Lapeyre-Mestre M, et al. Off-label prescribing of antipsychotics in adults, children and elderly individuals: a systematic review of recent prescription trends. Curr Pharm Des. 2015;21(23):3280-3297.
6. Weintraub D, Chiang C, Kim HM, et al. Association of antipsychotic use with mortality risk in patients with parkinson disease. JAMA Neurol. 2016;73(5):535-541.
7. Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 2011;7(5):532-539.
8. Kales HC, Chen P, Blow FC, et al. Rates of clinical depression diagnosis, functional impairment, and nursing home placement in coexisting dementia and depression. Am J Geriatr Psychiatry. 2005;13(6):441-449.
9. Yaffe K, Fox P, Newcomer R, et al. Patient and caregiver characteristics and nursing home placement in patients with dementia. JAMA. 2002;287(16):2090-2097.
10. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
11. Vilalta-Franch J, López-Pousa S, Calvó-Perxas L, et al. Psychosis of Alzheimer disease: prevalence, incidence, persistence, risk factors, and mortality. Am J Geriatr Psychiatry. 2013;21(11):1135-1143.
12. Spalletta G, Musicco M, Padovani A, et al. Neuropsychiatric symptoms and syndromes in a large cohort of newly diagnosed, untreated patients with Alzheimer disease. Am J Geriatr Psychiatry. 2010;18(11):1026-1035.
13. Steinberg M, Shao H, Zandi P, et al; Cache County Investigators. Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: the Cache County Study. Int J Geriatr Psychiatry. 2008;23(2):170-177.
14. Finkel SI, Burns A. Behavioral and psychological symptoms of dementia (BPSD): a clinical and research update-introduction. International Psychogeriatrics. 2000;12:9-12.
15. Lyketsos CG. Neuropsychiatric symptoms (behavioral and psychological symptoms of dementia) and the development of dementia treatments. Int Psychogeriatr. 2007;19(3):409-420.
16. Kunik ME, Snow AL, Davila JA, et al. Causes of aggressive behavior in patients with dementia. J Clin Psychiatry. 2010;71(9):1145-1152.
17. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
18. Kales HC, Gitlin LN, Lyketsos CG. Assessment and management of behavioral and psychological symptoms of dementia. BMJ. 2015;350:h369. doi: 10.1136/bmj.h369.
19. Schneider LS, Pollock VE, Lyness SA. A metaanalysis of controlled trials of neuroleptic treatment in dementia. J Am Geriatr Soc. 1990;38(5):553-563.
20. Yury CA, Fisher JE. Meta-analysis of the effectiveness of atypical antipsychotics for the treatment of behavioural problems in persons with dementia. Psychother Psychosom. 2007;76(4):213-218.
21. Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. 2006;14(3):191-210.
22. Ballard CG, Waite J. The effectiveness of atypical antipsychotics for aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst Rev. 2006:1:CD003476.
23. Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA. 2005;293(5):596-608.
24. Aisen PS, Cummings J, Schneider LS. Symptomatic and nonamyloid/tau based pharmacologic treatment for Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006395. doi: 10.1101/cshperspect.a006395.
25. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
26. Trifirò G, Sultana J, Spina E. Are the safety profiles of antipsychotic drugs used in dementia the same? An updated review of observational studies. Drug Saf. 2014;37(7):501-520.
27. Trifirò G, Gambassi G, Sen EF, et al. Association of community-acquired pneumonia with antipsychotic drug use in elderly patients: a nested case-control study. Ann Intern Med. 2010;152(7):418-425, W139-W140.
28. Sultana J, Trifirò G. Drug safety warnings: a message in a bottle. Analysis. 2008;179:438-446.
29. Liperoti R, Gambassi G, Lapane KL, et al. Cerebrovascular events among elderly nursing home patients treated with conventional or atypical antipsychotics. J Clin Psychiatry. 2005;66(9):1090-1096.
30. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafety information forpatientsandproviders/ucm053171. Updated August 16, 2013. Accessed October 20, 2017.
31. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353(22):2335-2341.
32. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
33. Simoni-Wastila L, Ryder PT, Qian J, et al. Association of antipsychotic use with hospital events and mortality among medicare beneficiaries residing in long-term care facilities. Am J Geriatr Psychiatry. 2009;17(5):417-427.
34. Raivio MM, Laurila JV, Strandberg TE, et al. Neither atypical nor conventional antipsychotics increase mortality or hospital admissions among elderly patients with dementia: a two-year prospective study. Am J Geriatr Psychiatry. 2007;15(5):416-424.
35. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
36. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ. 2007;176(5):627-632.
37. Kales HC, Kim HM, Zivin K, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. 2012;169(1):71-79.
38. Maust DT, Kim HM, Seyfried LS, et al. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry. 2015;72(5):438-445.
39. Rhee Y, Csernansky JG, Emanuel LL, et al. Psychotropic medication burden and factors associated with antipsychotic use: an analysis of a population-based sample of community-dwelling older persons with dementia. J Am Geriatr Soc. 2011;59(11):2100-2107.
40. Kales HC, Zivin K, Kim HM, et al. Trends in antipsychotic use in dementia 1999-2007. Arch Gen Psychiatry. 2011;68(2):190-197.
41. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
42. Brodaty H, Ames D, Snowdon J, et al. A randomized placebo-controlled trial of risperidone for the treatment of aggression, agitation, and psychosis of dementia. J Clin Psychiatry. 2003;64(2):134-143.
43. Wooltorton E. Risperidone (Risperdal): increased rate of cerebrovascular events in dementia trials. CMAJ. 2002;167(11):1269-1270.
44. United States Government Accountability Office. Antipsychotic drug use: HHS has initiatives to reduce use among older adults in nursing homes, but should expand efforts to other settings. http://www.gao.gov/assets/670/668221.pdf. Published January 2015. Accessed October 20, 2017.
45. Chen Y, Briesacher BA, Field TS, et al. Unexplained variation across US nursing homes in antipsychotic prescribing rates. Arch Intern Med. 2010;170(1):89-95.
46. Feng Z, Hirdes JP, Smith TF, et al. Use of physical restraints and antipsychotic medications in nursing homes: a cross-national study. Int J Geriatr Psychiatry. 2009;24(10):1110-1118.
47. Kamble P, Chen H, Sherer J, et al. Antipsychotic drug use among elderly nursing home residents in the United States. Am J Geriatr Pharmacother. 2008;6(4):187-197.
48. Gellad WF, Aspinall SL, Handler SM, et al. Use of antipsychotics among older residents in VA nursing homes. Med Care. 2012;50(11):954-960.
49. Bonner A. Improving dementia care and reducing unnecessary use of antipsychotic medications in nursing homes. Center for Medicare and Medicaid Services. http://ltcombudsman.org/uploads/files/support/alice-bonner-slides.pdf. Published April 28, 2013. Accessed October 20, 2017.
50. Vasudev A, Shariff SZ, Liu K, et al. Trends in psychotropic dispensing among older adults with dementia living in long-term care facilities: 2004-2013. Am J Geriatr Psychiatry. 2015;23(12):1259-1269.
51. Kales HC, Gitlin LN, Lyketsos CG, et al; Detroit Expert Panel on Assessment and Management of Neuropsychiatric Symptoms of Dementia. Management of neuropsychiatric symptoms of dementia in clinical settings: recommendations from a multidisciplinary expert panel. J Am Geriatr Soc. 2014;62(4):762-769.
52. Andreasen NC, Liu D, Ziebell S, et al. Relapse duration, treatment intensity, and brain tissue loss in schizophrenia: a prospective longitudinal MRI study. Am J Psychiatry. 2013;170(6):609-615.
53. Mulsant BH. Challenges of the treatment of neuropsychiatric symptoms associated with dementia. Am J Geriatr Psychiatry. 2014;22(4):317-320.

Sexting: What are the clinical and legal implications?
Sexting includes sending sexually explicit (or sexually suggestive) text messages and photos, usually by cell phone. This article focuses on sexts involving photos. Cell phones are almost ubiquitous among American teens, and with technological advances, sexts are getting easier to send. Sexting may occur to initiate a relationship or sustain one. Some teenagers are coerced into sexting. Many people do not realize the potential long-term consequences of sexting—particularly because of the impulsive nature of sexting and the belief that the behavior is harmless.
Media attention has recently focused on teens who face legal charges related to sexting. Sexting photos may be considered child pornography—even though the teens made it themselves. There are also social consequences to sexting. Photos meant to be private are sometimes forwarded to others. Cyberbullying is not uncommon with teen sexting, and suicides after experiencing this behavior have been reported.
Sexting may be a form of modern flirtation, but in some cases, it may be a marker of other risk behaviors, such as substance abuse. Psychiatrists must be aware of the frequency and meaning of this potentially dangerous behavior. Clinicians should feel comfortable asking their patients about it and provide education and counseling.
CASE
Private photos get shared
K, age 14, a freshman with no psychiatric history, is referred to you by her school psychologist for evaluation of suicidal ideation. K reports depressed mood, poor sleep, inattention, loss of appetite, anhedonia, and feelings of guilt for the past month. She recently ended a relationship with her boyfriend of 1 year after she learned that he had shared with his friends naked photos of her that she had sent him. The school administration learned of the photos when a student posted them on one of the school computers.
K’s boyfriend, age 16, was suspended after the school learned that he had shared the photos without K’s consent. K, who is a good student, missed several days of school, including cheerleading practice; previously she had never missed a day of school.
On evaluation, K is tearful, stating that her life is “over.” She says that her ex-boyfriend’s friends are harassing her, calling her “slut” and making sexual comments. She also feels guilty, because she learned that the police interviewed her ex-boyfriend in connection with posting her photos on the Internet. In a text, he said he “might get charged with child pornography.” On further questioning, K confides that she had naked photos of her ex-boyfriend on her phone. She admits to sharing the pictures with her best friend, because she was “angry and wanted to get back” at her ex-boyfriend. She also reports a several-month history of sexting with her ex-boyfriend. K deleted the photos and texts after learning that her ex-boyfriend “was in trouble with the police.”
K has no prior sexual experience. She dated 1 boy her age prior to her ex-boyfriend. She had never been evaluated by a mental health clinician. She is dysphoric and reports feeling “hopeless … Unless this can be erased, I can’t go back to school.”
Sexting: What is the extent of the problem?
The true prevalence of sexting is difficult to ascertain, because different studies have used different definitions and methodologies. However, the rates are far from negligible. Sexting rates increase with age, over the teen years.1-3 Among American minors, 2.5% to 28% of middle school and high school students report that they have sent a sext (Table 11-9). Studies of American young adults (age ≥18) and university students have found 30% to 60% have sent sexts, and >40% have received a sext.4,5
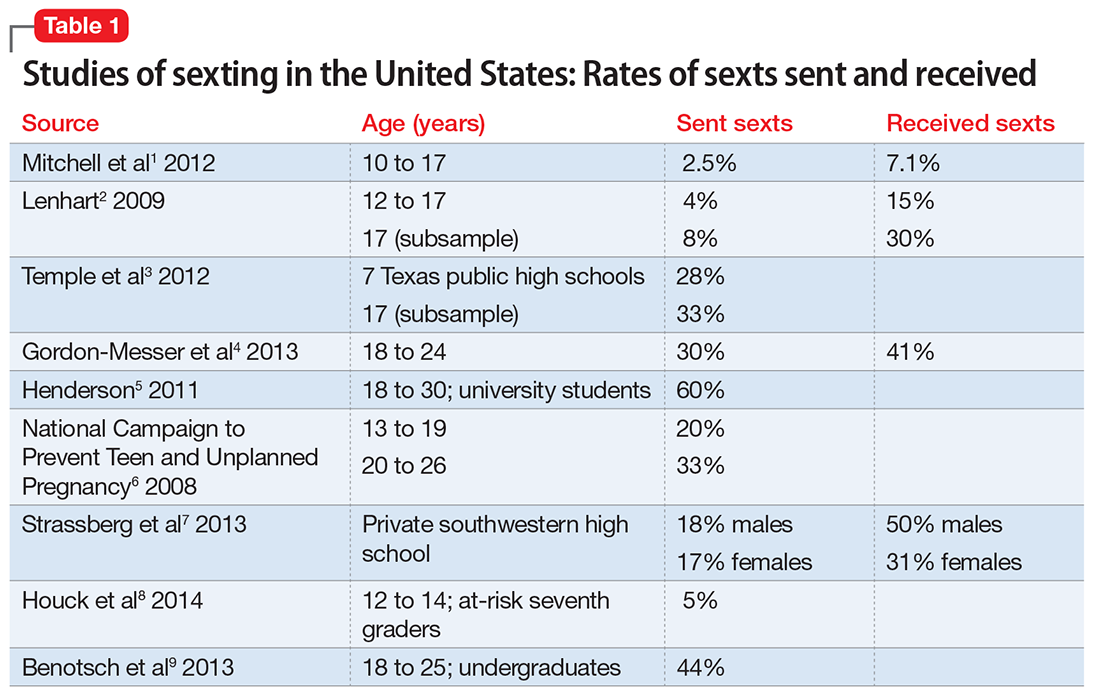
Many people receive sexts—including individuals who are not the intended recipient. In 1 study, although most teens intended to share sexts only with their boyfriend/girlfriend, 25% to 33% had received sext photos intended for other people.6 In another recent study, 25% of teens had forwarded a sext that they received.7 Moreover, 12% of teenage boys and 5% of teenage girls had sent a sexually explicit photo that they took of another teen to a third person.7 Forwarding sexts can add exponentially to the psychosocial risks of the photographed teenager.
Who sexts? Current research indicates that the likelihood of sexting is related to age, personality, and social situation. Teens are approaching the peak age of their sex drive, and often are curious and feel invincible. Teens are more impulsive than adults. When it takes less than a minute to send a sext, irreversible poor choices can be made quickly. Teens who send sexts often engage in more text messaging than other teens.7
Teens may use sexting to initiate or sustain a relationship. Sexts also may be sent because of coercion. More than one-half of girls cited pressure to sext from a boy.6 Temple et al3 found that more than one-half of their study sample had been asked to send a sext. Girls were more likely than boys to be asked to send a sext; most were troubled by this.
One study that assessed knowledge of potential legal consequences of sexting found that many teens who sent sexts were aware of the potential consequences.7 Regarding personality traits, sexting among undergraduates was predicted by neuroticism and low agreeableness.10 Conversely, sending text messages with sexually suggestive writing was predicted by extraversion and problematic cell phone use.
Comorbidities. There are mixed findings about whether sexting is simply a modern dating strategy or a marker of other risk behaviors; age may play an important discriminating role. Sexual activity appears to be correlated with sexting. According to Temple and Choi,11 “Sexting fits within the context of adolescent sexual development and may be a viable indicator of adolescent sexual activity.”11
Some authors have suggested that sexting is a contemporary risk behavior that is likely to correlate with other risk behaviors. Among young teens—seventh graders who were referred to a risk prevention trial because of behavioral/emotional difficulties—those who sexted were more likely to engage in early sexual behaviors.8 These younger at-risk teens also had less understanding of their emotions and greater difficulty in regulating their emotions.
Among the general population of high school students, teens who sext are more likely to be sexually active.3 High school girls who engaged in sexting were noted to engage in other risk behaviors, including having multiple partners in the past year and using alcohol or drugs before sex.3 Teens who had sent a sext were more likely to be sexually active 1 year later than teens who had not.11Studies of sexting among university students also have had mixed findings. One study found that among undergraduates, sexting was associated with substance use and other risk behaviors.9 Another young adult study found sexting was not related to sexual risk or psychological well-being.4
Legal issues affect psychiatrists as well as patients
As a psychiatrist evaluating K, what are your duties as a mandated reporter? Psychiatrists are legally required to report suspected maltreatment or abuse of children.12 The circumstances under which psychiatrists may have a mandate to report include when a psychiatrist:
- evaluates a child and suspects abuse
- suspects child abuse based on an adult patient’s report
- learns from a third party that a child may have been/is being abused.
Psychiatrists usually are not mandated to report other types of potentially criminal behavior. As such, reporting sexting might be considered a breach of confidentiality. Psychiatrists should be familiar with the specific reporting guidelines for the jurisdiction in which they practice. Psychiatrists who work with individuals who commit crimes should focus on changing the potentially dangerous behaviors rather than reporting them.
Does the transmission of naked photos of a minor in a sexual pose or act constitute child pornography or another criminal offense? The legal answer varies, but the role of the psychiatrist does not. Psychiatrists should educate their patients about potentially dangerous behaviors.
With regards to the legal consequences, some states classify underage sexting photos as child pornography. Others have less rigid definitions of child pornography and take into account the age of the participants and their intent. Such jurisdictions point out that sexting naked photos among adolescents is “age appropriate.” Some have enacted specific sexting laws to address the transmission of obscene material to a child through the Internet. In some jurisdictions, sexting laws are categorized to refer to behavior of individuals under or over age 18. The term “revenge porn” is used to refer to nonconsensual pornography with its dissemination motivated by spite.13 Some states have defined specific revenge porn laws to address the behavior. Currently, 20 states have sexting laws and 26 states have revenge porn laws.14 Twenty states address a minor age <18 sending the photo, while only 18 address the recipient. The law in this area can be complex and detailed, taking into account the age of the sender, the intentions of the sender, and the nature of the relationship between the sender and the recipient and the behavior of the recipient.
Laws regarding sexting vary greatly. Sexting may be a misdemeanor or a felony, depending on the state, the specific behavior, and the frequency. In the United States, 11 state laws include a diversion remedy—an option to pursue the case outside of the criminal juvenile system; 10 laws require counseling or another informal sanction; 11 states laws have the potential for misdemeanor punishment; and 4 state laws have the potential for felony punishment.14 Depending on the criminal charge, the perpetrator may have to register as a sex offender. For example, in some jurisdictions, a conviction for possession of child pornography requires sex offender registration. Thirty-eight states include juvenile sex offenders in their sex offender registries. Other states require juveniles to register if they are age ≥15 years or have been tried as an adult.15
The frequency of police involvement in sexting cases also greatly varies. A national study examining the characteristics of youth sexting cases revealed that law enforcement agencies handled approximately 3,477 cases of youth-produced sexual photos in 2008 and 2009.16 Situations that involved an adult or a minor engaged in malicious, nonconsensual, or abusive behavior comprised two-thirds of cases. Arrests occurred in 62% of the adult-involved cases and 36% of the aggravated youth-only cases. Arrests occurred in only 18% of investigated non-aggravated youth-only cases. Table 2 describes recent American sexting legal cases and their outcomes.
In K’s case, depending on the jurisdiction, K or her ex-boyfriend may be subject to arrest for child pornography, revenge pornography, or sexting.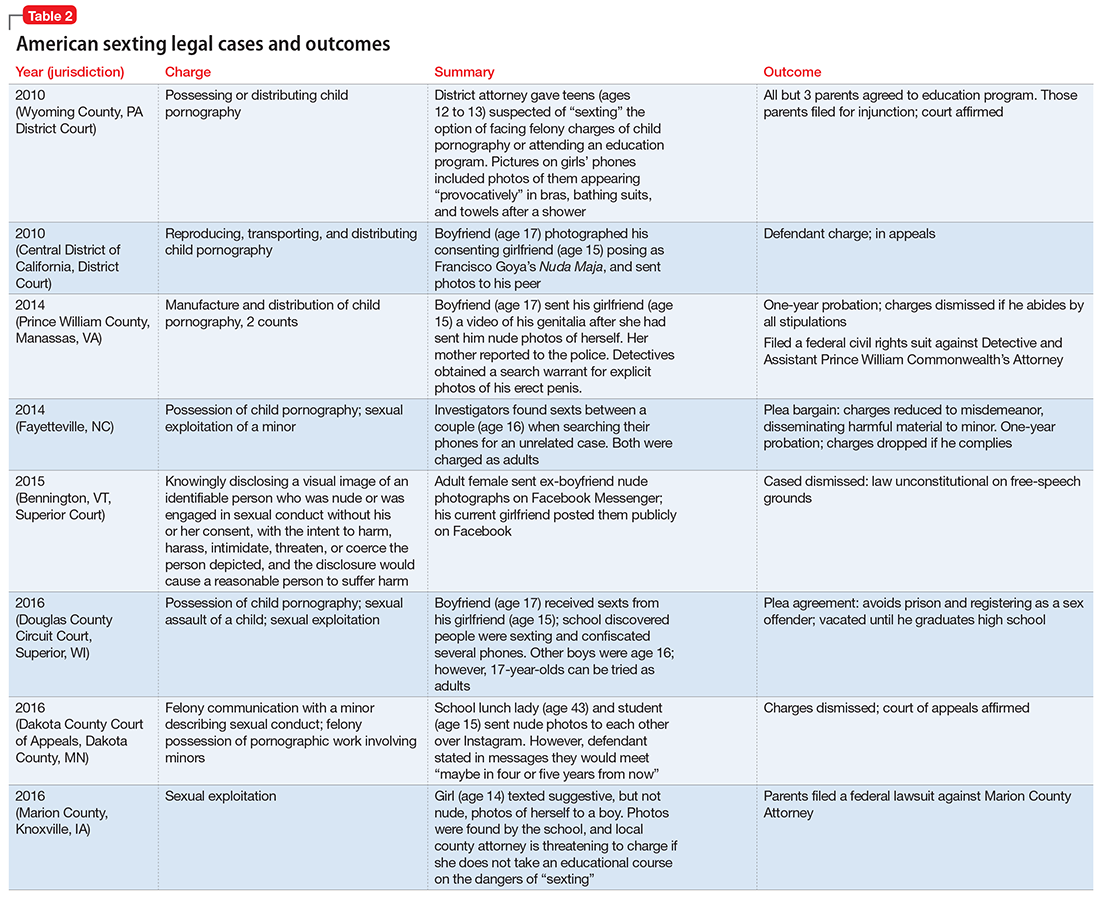
Potential social and psychiatric consequences
What are the social and psychiatric ramifications for K? Aside from potential legal consequences of sexting, K is experiencing psychological and social consequences. She has developed depressive symptoms and suicidal ideation. Her ex-boyfriend’s dissemination of her nude photos on the school computer could be interpreted as cyberbullying. (The National Center for Missing and Exploited Children defines cyberbullying as “bullying through the use of technology or electronic devices such as telephones, cell phones, computers, and the Internet.”17 All 50 states have enacted laws against bullying; 48 states have electronic harassment in their bullying laws; and 22 states have laws specifically referencing “cyberbullying.”)
Her depressive symptoms developed in response to her feelings of guilt and shame related to sexting as well as the subsequent peer harassment. She is refusing to return to school because of her concerns about bullying. A careful inquiry into suicidality should be part of the evaluation when sexting has led to psychiatric symptoms. Several cases of sexting and cyberbullying have ended in suicide (Table 3).

How to ask patients about sexting
To screen patients for sexting, clinicians need to develop a new skill set, which at first may be uncomfortable. However, the questions to ask are not all that different from other questions about adolescent and young adult sexuality. The importance of patients seeing that we as physicians are comfortable with the topic and approachable about their sexual health cannot be overemphasized. When discussing sexting with patients, it is essential to:
- explain that you are asking questions about their sexual health because they are important to overall health
- engage patients in discussion in a nonthreatening and nonjudgmental way
- develop rapport so patients feel comfortable disclosing behavior that may be embarrassing
- listen to their stories and build a context for understanding their experiences. As you listen, ask questions when needed to help move the story along.
Sometimes when asking about topics that are uncomfortable, clinicians revert from open-ended to closed-ended questions, but when asking about a patient’s sexual life, it is especially important to be open-ended and ask questions in a nonjudgmental way. Contextualizing sexual questions by (for example) asking them while discussing the teen’s relationships will make them seem more natural.18 To best understand, inquire explicitly about specific behaviors, but do so without appearing voyeuristic.18
Sexting may precede sexual intercourse. Keep in mind that a patient may report that she (he) is not sexually active but still may be involved in sexting. Therefore, discuss sexting even if your patient reports not being sexually active. By understanding the prevalence of sexting among teens, you can ask questions in a normalizing way. Clinicians can inquire about sexting while discussing relationships and dating or online risk behaviors.
Also consider whether any of your patient’s sexual behaviors, including sexting, are the result of coercion: “Some of my patients tell me they feel pressured or coerced into having sex. Have you ever felt this way?”19 and “Have you ever been picked on or bullied? Is that still a problem?” are suggested safety screening questions about bullying,18 and one can also ask about specific cyberbullying behaviors.
1. Mitchell KJ, Finkelhor D, Jones LM, et al. Prevalence and characteristics of youth sexting: a national study. Pediatrics. 2012;129(1):13-20.
2. Lenhart A. Teens and sexting. The Pew Research Center. http://www.pewinternet.org/2009/12/15/teens-and-sexting. Published December 15, 2009. Accessed October 31, 2017.
3. Temple JR, Paul JA, van den Berg P, et al. Teen sexting and its association with sexual behaviors. Arch Pediatr Adolesc Med. 2012;166(9):828-833.
4. Gordon-Messer D, Bauermeister JA, Grodzinski A, et al. Sexting among young adults. J Adolesc Health. 2013;52(3):301-306.
5. Henderson L. Sexting and sexual relationships among teens and young adults. McNair Scholars Research Journal. 2011;7(1):31-39.
6. The National Campaign to Prevent Teen and Unplanned Pregnancy. Sex and tech: results from a survey of teens and young adults. https://thenationalcampaign.org/sites/default/files/resource-primary-download/sex_and_tech_summary.pdf. Published December 2008. Accessed October 31, 2017.
7. Strassberg DS, McKinnon RK, Sustaíta MA, et al. Sexting by high school students: an exploratory and descriptive study. Arch Sex Behav. 2013;42(1):15-21.
8. Houck CD, Barker D, Rizzo C, et al. Sexting and sexual behavior in at-risk adolescents. Pediatrics. 2014;133(2):e276-e282.
9. Benotsch EG, Snipes DJ, Martin AM, et al. Sexting, substance use, and sexual risk behavior in young adults. J Adolesc Health. 2013;52(3):307-313.
10. Delevi R, Weisskirch RS. Personality factors as predictors of sexting. Comput Human Behav. 2013;29(6):2589-2594.
11. Temple JR, Choi H. Longitudinal association between teen sexting and sexual behavior. Pediatrics. 2014;134(5):1287-1292.
12. McEwan M, Friedman SH. Violence by parents against their children: reporting of maltreatment suspicions, child protection, and risk in mental illness. Psych Clin North Am. 2016;39(4):691-700.
13. Citron DK, Franks MA. Criminalizing revenge porn. Wake Forest Law Review. 2014;49:345-391.
14. Hinduja S, Patchin JW. State cyberbullying laws: a brief review of state cyberbullying laws and policies. Cyberbullying Research Center. https://cyberbullying.org/Bullying-and-Cyberbullying-Laws.pdf. Updated 2016. Accessed October 31, 2017.
15. Beitsch R. States slowly scale back juvenile sex offender registries. The Pew Charitable Trusts. http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2015/11/19/states-slowly-scale-back-juvenile-sex-offender-registries. Published November 19, 2015. Accessed October 31, 2017.
16. Wolak J, Finkelhor D, Mitchell KJ. How often are teens arrested for sexting? Data from a national sample of police cases. Pediatrics. 2012;129(1):4-12.
17. The Campus School at Boston College. Bullying prevention policy. https://www.bc.edu/bc-web/schools/lsoe/sites/campus-school/who-we-are/policies-and-procedures/bullying-prevention-policy.html. Accessed October 31, 2017.
18. Goldenring JM, Rosen DS. Getting into adolescent heads: an essential update. Contemporary Pediatrics. 2004;21(1):64.
19. Klein DA, Goldenring JM, Adelman WP. HEEADSSS 3.0: the psychosocial interview for adolescents updated for a new century fueled by media. Contemporary Pediatrics. http://contemporarypediatrics.modernmedicine.com/contemporary-pediatrics/content/tags/adolescent-medicine/heeadsss-30-psychosocial-interview-adolesce?page=full. Published January 1, 2014. Accessed October 31, 2017.
Sexting includes sending sexually explicit (or sexually suggestive) text messages and photos, usually by cell phone. This article focuses on sexts involving photos. Cell phones are almost ubiquitous among American teens, and with technological advances, sexts are getting easier to send. Sexting may occur to initiate a relationship or sustain one. Some teenagers are coerced into sexting. Many people do not realize the potential long-term consequences of sexting—particularly because of the impulsive nature of sexting and the belief that the behavior is harmless.
Media attention has recently focused on teens who face legal charges related to sexting. Sexting photos may be considered child pornography—even though the teens made it themselves. There are also social consequences to sexting. Photos meant to be private are sometimes forwarded to others. Cyberbullying is not uncommon with teen sexting, and suicides after experiencing this behavior have been reported.
Sexting may be a form of modern flirtation, but in some cases, it may be a marker of other risk behaviors, such as substance abuse. Psychiatrists must be aware of the frequency and meaning of this potentially dangerous behavior. Clinicians should feel comfortable asking their patients about it and provide education and counseling.
CASE
Private photos get shared
K, age 14, a freshman with no psychiatric history, is referred to you by her school psychologist for evaluation of suicidal ideation. K reports depressed mood, poor sleep, inattention, loss of appetite, anhedonia, and feelings of guilt for the past month. She recently ended a relationship with her boyfriend of 1 year after she learned that he had shared with his friends naked photos of her that she had sent him. The school administration learned of the photos when a student posted them on one of the school computers.
K’s boyfriend, age 16, was suspended after the school learned that he had shared the photos without K’s consent. K, who is a good student, missed several days of school, including cheerleading practice; previously she had never missed a day of school.
On evaluation, K is tearful, stating that her life is “over.” She says that her ex-boyfriend’s friends are harassing her, calling her “slut” and making sexual comments. She also feels guilty, because she learned that the police interviewed her ex-boyfriend in connection with posting her photos on the Internet. In a text, he said he “might get charged with child pornography.” On further questioning, K confides that she had naked photos of her ex-boyfriend on her phone. She admits to sharing the pictures with her best friend, because she was “angry and wanted to get back” at her ex-boyfriend. She also reports a several-month history of sexting with her ex-boyfriend. K deleted the photos and texts after learning that her ex-boyfriend “was in trouble with the police.”
K has no prior sexual experience. She dated 1 boy her age prior to her ex-boyfriend. She had never been evaluated by a mental health clinician. She is dysphoric and reports feeling “hopeless … Unless this can be erased, I can’t go back to school.”
Sexting: What is the extent of the problem?
The true prevalence of sexting is difficult to ascertain, because different studies have used different definitions and methodologies. However, the rates are far from negligible. Sexting rates increase with age, over the teen years.1-3 Among American minors, 2.5% to 28% of middle school and high school students report that they have sent a sext (Table 11-9). Studies of American young adults (age ≥18) and university students have found 30% to 60% have sent sexts, and >40% have received a sext.4,5
Many people receive sexts—including individuals who are not the intended recipient. In 1 study, although most teens intended to share sexts only with their boyfriend/girlfriend, 25% to 33% had received sext photos intended for other people.6 In another recent study, 25% of teens had forwarded a sext that they received.7 Moreover, 12% of teenage boys and 5% of teenage girls had sent a sexually explicit photo that they took of another teen to a third person.7 Forwarding sexts can add exponentially to the psychosocial risks of the photographed teenager.
Who sexts? Current research indicates that the likelihood of sexting is related to age, personality, and social situation. Teens are approaching the peak age of their sex drive, and often are curious and feel invincible. Teens are more impulsive than adults. When it takes less than a minute to send a sext, irreversible poor choices can be made quickly. Teens who send sexts often engage in more text messaging than other teens.7
Teens may use sexting to initiate or sustain a relationship. Sexts also may be sent because of coercion. More than one-half of girls cited pressure to sext from a boy.6 Temple et al3 found that more than one-half of their study sample had been asked to send a sext. Girls were more likely than boys to be asked to send a sext; most were troubled by this.
One study that assessed knowledge of potential legal consequences of sexting found that many teens who sent sexts were aware of the potential consequences.7 Regarding personality traits, sexting among undergraduates was predicted by neuroticism and low agreeableness.10 Conversely, sending text messages with sexually suggestive writing was predicted by extraversion and problematic cell phone use.
Comorbidities. There are mixed findings about whether sexting is simply a modern dating strategy or a marker of other risk behaviors; age may play an important discriminating role. Sexual activity appears to be correlated with sexting. According to Temple and Choi,11 “Sexting fits within the context of adolescent sexual development and may be a viable indicator of adolescent sexual activity.”11
Some authors have suggested that sexting is a contemporary risk behavior that is likely to correlate with other risk behaviors. Among young teens—seventh graders who were referred to a risk prevention trial because of behavioral/emotional difficulties—those who sexted were more likely to engage in early sexual behaviors.8 These younger at-risk teens also had less understanding of their emotions and greater difficulty in regulating their emotions.
Among the general population of high school students, teens who sext are more likely to be sexually active.3 High school girls who engaged in sexting were noted to engage in other risk behaviors, including having multiple partners in the past year and using alcohol or drugs before sex.3 Teens who had sent a sext were more likely to be sexually active 1 year later than teens who had not.11Studies of sexting among university students also have had mixed findings. One study found that among undergraduates, sexting was associated with substance use and other risk behaviors.9 Another young adult study found sexting was not related to sexual risk or psychological well-being.4
Legal issues affect psychiatrists as well as patients
As a psychiatrist evaluating K, what are your duties as a mandated reporter? Psychiatrists are legally required to report suspected maltreatment or abuse of children.12 The circumstances under which psychiatrists may have a mandate to report include when a psychiatrist:
- evaluates a child and suspects abuse
- suspects child abuse based on an adult patient’s report
- learns from a third party that a child may have been/is being abused.
Psychiatrists usually are not mandated to report other types of potentially criminal behavior. As such, reporting sexting might be considered a breach of confidentiality. Psychiatrists should be familiar with the specific reporting guidelines for the jurisdiction in which they practice. Psychiatrists who work with individuals who commit crimes should focus on changing the potentially dangerous behaviors rather than reporting them.
Does the transmission of naked photos of a minor in a sexual pose or act constitute child pornography or another criminal offense? The legal answer varies, but the role of the psychiatrist does not. Psychiatrists should educate their patients about potentially dangerous behaviors.
With regards to the legal consequences, some states classify underage sexting photos as child pornography. Others have less rigid definitions of child pornography and take into account the age of the participants and their intent. Such jurisdictions point out that sexting naked photos among adolescents is “age appropriate.” Some have enacted specific sexting laws to address the transmission of obscene material to a child through the Internet. In some jurisdictions, sexting laws are categorized to refer to behavior of individuals under or over age 18. The term “revenge porn” is used to refer to nonconsensual pornography with its dissemination motivated by spite.13 Some states have defined specific revenge porn laws to address the behavior. Currently, 20 states have sexting laws and 26 states have revenge porn laws.14 Twenty states address a minor age <18 sending the photo, while only 18 address the recipient. The law in this area can be complex and detailed, taking into account the age of the sender, the intentions of the sender, and the nature of the relationship between the sender and the recipient and the behavior of the recipient.
Laws regarding sexting vary greatly. Sexting may be a misdemeanor or a felony, depending on the state, the specific behavior, and the frequency. In the United States, 11 state laws include a diversion remedy—an option to pursue the case outside of the criminal juvenile system; 10 laws require counseling or another informal sanction; 11 states laws have the potential for misdemeanor punishment; and 4 state laws have the potential for felony punishment.14 Depending on the criminal charge, the perpetrator may have to register as a sex offender. For example, in some jurisdictions, a conviction for possession of child pornography requires sex offender registration. Thirty-eight states include juvenile sex offenders in their sex offender registries. Other states require juveniles to register if they are age ≥15 years or have been tried as an adult.15
The frequency of police involvement in sexting cases also greatly varies. A national study examining the characteristics of youth sexting cases revealed that law enforcement agencies handled approximately 3,477 cases of youth-produced sexual photos in 2008 and 2009.16 Situations that involved an adult or a minor engaged in malicious, nonconsensual, or abusive behavior comprised two-thirds of cases. Arrests occurred in 62% of the adult-involved cases and 36% of the aggravated youth-only cases. Arrests occurred in only 18% of investigated non-aggravated youth-only cases. Table 2 describes recent American sexting legal cases and their outcomes.
In K’s case, depending on the jurisdiction, K or her ex-boyfriend may be subject to arrest for child pornography, revenge pornography, or sexting.
Potential social and psychiatric consequences
What are the social and psychiatric ramifications for K? Aside from potential legal consequences of sexting, K is experiencing psychological and social consequences. She has developed depressive symptoms and suicidal ideation. Her ex-boyfriend’s dissemination of her nude photos on the school computer could be interpreted as cyberbullying. (The National Center for Missing and Exploited Children defines cyberbullying as “bullying through the use of technology or electronic devices such as telephones, cell phones, computers, and the Internet.”17 All 50 states have enacted laws against bullying; 48 states have electronic harassment in their bullying laws; and 22 states have laws specifically referencing “cyberbullying.”)
Her depressive symptoms developed in response to her feelings of guilt and shame related to sexting as well as the subsequent peer harassment. She is refusing to return to school because of her concerns about bullying. A careful inquiry into suicidality should be part of the evaluation when sexting has led to psychiatric symptoms. Several cases of sexting and cyberbullying have ended in suicide (Table 3).
How to ask patients about sexting
To screen patients for sexting, clinicians need to develop a new skill set, which at first may be uncomfortable. However, the questions to ask are not all that different from other questions about adolescent and young adult sexuality. The importance of patients seeing that we as physicians are comfortable with the topic and approachable about their sexual health cannot be overemphasized. When discussing sexting with patients, it is essential to:
- explain that you are asking questions about their sexual health because they are important to overall health
- engage patients in discussion in a nonthreatening and nonjudgmental way
- develop rapport so patients feel comfortable disclosing behavior that may be embarrassing
- listen to their stories and build a context for understanding their experiences. As you listen, ask questions when needed to help move the story along.
Sometimes when asking about topics that are uncomfortable, clinicians revert from open-ended to closed-ended questions, but when asking about a patient’s sexual life, it is especially important to be open-ended and ask questions in a nonjudgmental way. Contextualizing sexual questions by (for example) asking them while discussing the teen’s relationships will make them seem more natural.18 To best understand, inquire explicitly about specific behaviors, but do so without appearing voyeuristic.18
Sexting may precede sexual intercourse. Keep in mind that a patient may report that she (he) is not sexually active but still may be involved in sexting. Therefore, discuss sexting even if your patient reports not being sexually active. By understanding the prevalence of sexting among teens, you can ask questions in a normalizing way. Clinicians can inquire about sexting while discussing relationships and dating or online risk behaviors.
Also consider whether any of your patient’s sexual behaviors, including sexting, are the result of coercion: “Some of my patients tell me they feel pressured or coerced into having sex. Have you ever felt this way?”19 and “Have you ever been picked on or bullied? Is that still a problem?” are suggested safety screening questions about bullying,18 and one can also ask about specific cyberbullying behaviors.
Sexting includes sending sexually explicit (or sexually suggestive) text messages and photos, usually by cell phone. This article focuses on sexts involving photos. Cell phones are almost ubiquitous among American teens, and with technological advances, sexts are getting easier to send. Sexting may occur to initiate a relationship or sustain one. Some teenagers are coerced into sexting. Many people do not realize the potential long-term consequences of sexting—particularly because of the impulsive nature of sexting and the belief that the behavior is harmless.
Media attention has recently focused on teens who face legal charges related to sexting. Sexting photos may be considered child pornography—even though the teens made it themselves. There are also social consequences to sexting. Photos meant to be private are sometimes forwarded to others. Cyberbullying is not uncommon with teen sexting, and suicides after experiencing this behavior have been reported.
Sexting may be a form of modern flirtation, but in some cases, it may be a marker of other risk behaviors, such as substance abuse. Psychiatrists must be aware of the frequency and meaning of this potentially dangerous behavior. Clinicians should feel comfortable asking their patients about it and provide education and counseling.
CASE
Private photos get shared
K, age 14, a freshman with no psychiatric history, is referred to you by her school psychologist for evaluation of suicidal ideation. K reports depressed mood, poor sleep, inattention, loss of appetite, anhedonia, and feelings of guilt for the past month. She recently ended a relationship with her boyfriend of 1 year after she learned that he had shared with his friends naked photos of her that she had sent him. The school administration learned of the photos when a student posted them on one of the school computers.
K’s boyfriend, age 16, was suspended after the school learned that he had shared the photos without K’s consent. K, who is a good student, missed several days of school, including cheerleading practice; previously she had never missed a day of school.
On evaluation, K is tearful, stating that her life is “over.” She says that her ex-boyfriend’s friends are harassing her, calling her “slut” and making sexual comments. She also feels guilty, because she learned that the police interviewed her ex-boyfriend in connection with posting her photos on the Internet. In a text, he said he “might get charged with child pornography.” On further questioning, K confides that she had naked photos of her ex-boyfriend on her phone. She admits to sharing the pictures with her best friend, because she was “angry and wanted to get back” at her ex-boyfriend. She also reports a several-month history of sexting with her ex-boyfriend. K deleted the photos and texts after learning that her ex-boyfriend “was in trouble with the police.”
K has no prior sexual experience. She dated 1 boy her age prior to her ex-boyfriend. She had never been evaluated by a mental health clinician. She is dysphoric and reports feeling “hopeless … Unless this can be erased, I can’t go back to school.”
Sexting: What is the extent of the problem?
The true prevalence of sexting is difficult to ascertain, because different studies have used different definitions and methodologies. However, the rates are far from negligible. Sexting rates increase with age, over the teen years.1-3 Among American minors, 2.5% to 28% of middle school and high school students report that they have sent a sext (Table 11-9). Studies of American young adults (age ≥18) and university students have found 30% to 60% have sent sexts, and >40% have received a sext.4,5
Many people receive sexts—including individuals who are not the intended recipient. In 1 study, although most teens intended to share sexts only with their boyfriend/girlfriend, 25% to 33% had received sext photos intended for other people.6 In another recent study, 25% of teens had forwarded a sext that they received.7 Moreover, 12% of teenage boys and 5% of teenage girls had sent a sexually explicit photo that they took of another teen to a third person.7 Forwarding sexts can add exponentially to the psychosocial risks of the photographed teenager.
Who sexts? Current research indicates that the likelihood of sexting is related to age, personality, and social situation. Teens are approaching the peak age of their sex drive, and often are curious and feel invincible. Teens are more impulsive than adults. When it takes less than a minute to send a sext, irreversible poor choices can be made quickly. Teens who send sexts often engage in more text messaging than other teens.7
Teens may use sexting to initiate or sustain a relationship. Sexts also may be sent because of coercion. More than one-half of girls cited pressure to sext from a boy.6 Temple et al3 found that more than one-half of their study sample had been asked to send a sext. Girls were more likely than boys to be asked to send a sext; most were troubled by this.
One study that assessed knowledge of potential legal consequences of sexting found that many teens who sent sexts were aware of the potential consequences.7 Regarding personality traits, sexting among undergraduates was predicted by neuroticism and low agreeableness.10 Conversely, sending text messages with sexually suggestive writing was predicted by extraversion and problematic cell phone use.
Comorbidities. There are mixed findings about whether sexting is simply a modern dating strategy or a marker of other risk behaviors; age may play an important discriminating role. Sexual activity appears to be correlated with sexting. According to Temple and Choi,11 “Sexting fits within the context of adolescent sexual development and may be a viable indicator of adolescent sexual activity.”11
Some authors have suggested that sexting is a contemporary risk behavior that is likely to correlate with other risk behaviors. Among young teens—seventh graders who were referred to a risk prevention trial because of behavioral/emotional difficulties—those who sexted were more likely to engage in early sexual behaviors.8 These younger at-risk teens also had less understanding of their emotions and greater difficulty in regulating their emotions.
Among the general population of high school students, teens who sext are more likely to be sexually active.3 High school girls who engaged in sexting were noted to engage in other risk behaviors, including having multiple partners in the past year and using alcohol or drugs before sex.3 Teens who had sent a sext were more likely to be sexually active 1 year later than teens who had not.11Studies of sexting among university students also have had mixed findings. One study found that among undergraduates, sexting was associated with substance use and other risk behaviors.9 Another young adult study found sexting was not related to sexual risk or psychological well-being.4
Legal issues affect psychiatrists as well as patients
As a psychiatrist evaluating K, what are your duties as a mandated reporter? Psychiatrists are legally required to report suspected maltreatment or abuse of children.12 The circumstances under which psychiatrists may have a mandate to report include when a psychiatrist:
- evaluates a child and suspects abuse
- suspects child abuse based on an adult patient’s report
- learns from a third party that a child may have been/is being abused.
Psychiatrists usually are not mandated to report other types of potentially criminal behavior. As such, reporting sexting might be considered a breach of confidentiality. Psychiatrists should be familiar with the specific reporting guidelines for the jurisdiction in which they practice. Psychiatrists who work with individuals who commit crimes should focus on changing the potentially dangerous behaviors rather than reporting them.
Does the transmission of naked photos of a minor in a sexual pose or act constitute child pornography or another criminal offense? The legal answer varies, but the role of the psychiatrist does not. Psychiatrists should educate their patients about potentially dangerous behaviors.
With regards to the legal consequences, some states classify underage sexting photos as child pornography. Others have less rigid definitions of child pornography and take into account the age of the participants and their intent. Such jurisdictions point out that sexting naked photos among adolescents is “age appropriate.” Some have enacted specific sexting laws to address the transmission of obscene material to a child through the Internet. In some jurisdictions, sexting laws are categorized to refer to behavior of individuals under or over age 18. The term “revenge porn” is used to refer to nonconsensual pornography with its dissemination motivated by spite.13 Some states have defined specific revenge porn laws to address the behavior. Currently, 20 states have sexting laws and 26 states have revenge porn laws.14 Twenty states address a minor age <18 sending the photo, while only 18 address the recipient. The law in this area can be complex and detailed, taking into account the age of the sender, the intentions of the sender, and the nature of the relationship between the sender and the recipient and the behavior of the recipient.
Laws regarding sexting vary greatly. Sexting may be a misdemeanor or a felony, depending on the state, the specific behavior, and the frequency. In the United States, 11 state laws include a diversion remedy—an option to pursue the case outside of the criminal juvenile system; 10 laws require counseling or another informal sanction; 11 states laws have the potential for misdemeanor punishment; and 4 state laws have the potential for felony punishment.14 Depending on the criminal charge, the perpetrator may have to register as a sex offender. For example, in some jurisdictions, a conviction for possession of child pornography requires sex offender registration. Thirty-eight states include juvenile sex offenders in their sex offender registries. Other states require juveniles to register if they are age ≥15 years or have been tried as an adult.15
The frequency of police involvement in sexting cases also greatly varies. A national study examining the characteristics of youth sexting cases revealed that law enforcement agencies handled approximately 3,477 cases of youth-produced sexual photos in 2008 and 2009.16 Situations that involved an adult or a minor engaged in malicious, nonconsensual, or abusive behavior comprised two-thirds of cases. Arrests occurred in 62% of the adult-involved cases and 36% of the aggravated youth-only cases. Arrests occurred in only 18% of investigated non-aggravated youth-only cases. Table 2 describes recent American sexting legal cases and their outcomes.
In K’s case, depending on the jurisdiction, K or her ex-boyfriend may be subject to arrest for child pornography, revenge pornography, or sexting.
Potential social and psychiatric consequences
What are the social and psychiatric ramifications for K? Aside from potential legal consequences of sexting, K is experiencing psychological and social consequences. She has developed depressive symptoms and suicidal ideation. Her ex-boyfriend’s dissemination of her nude photos on the school computer could be interpreted as cyberbullying. (The National Center for Missing and Exploited Children defines cyberbullying as “bullying through the use of technology or electronic devices such as telephones, cell phones, computers, and the Internet.”17 All 50 states have enacted laws against bullying; 48 states have electronic harassment in their bullying laws; and 22 states have laws specifically referencing “cyberbullying.”)
Her depressive symptoms developed in response to her feelings of guilt and shame related to sexting as well as the subsequent peer harassment. She is refusing to return to school because of her concerns about bullying. A careful inquiry into suicidality should be part of the evaluation when sexting has led to psychiatric symptoms. Several cases of sexting and cyberbullying have ended in suicide (Table 3).
How to ask patients about sexting
To screen patients for sexting, clinicians need to develop a new skill set, which at first may be uncomfortable. However, the questions to ask are not all that different from other questions about adolescent and young adult sexuality. The importance of patients seeing that we as physicians are comfortable with the topic and approachable about their sexual health cannot be overemphasized. When discussing sexting with patients, it is essential to:
- explain that you are asking questions about their sexual health because they are important to overall health
- engage patients in discussion in a nonthreatening and nonjudgmental way
- develop rapport so patients feel comfortable disclosing behavior that may be embarrassing
- listen to their stories and build a context for understanding their experiences. As you listen, ask questions when needed to help move the story along.
Sometimes when asking about topics that are uncomfortable, clinicians revert from open-ended to closed-ended questions, but when asking about a patient’s sexual life, it is especially important to be open-ended and ask questions in a nonjudgmental way. Contextualizing sexual questions by (for example) asking them while discussing the teen’s relationships will make them seem more natural.18 To best understand, inquire explicitly about specific behaviors, but do so without appearing voyeuristic.18
Sexting may precede sexual intercourse. Keep in mind that a patient may report that she (he) is not sexually active but still may be involved in sexting. Therefore, discuss sexting even if your patient reports not being sexually active. By understanding the prevalence of sexting among teens, you can ask questions in a normalizing way. Clinicians can inquire about sexting while discussing relationships and dating or online risk behaviors.
Also consider whether any of your patient’s sexual behaviors, including sexting, are the result of coercion: “Some of my patients tell me they feel pressured or coerced into having sex. Have you ever felt this way?”19 and “Have you ever been picked on or bullied? Is that still a problem?” are suggested safety screening questions about bullying,18 and one can also ask about specific cyberbullying behaviors.
1. Mitchell KJ, Finkelhor D, Jones LM, et al. Prevalence and characteristics of youth sexting: a national study. Pediatrics. 2012;129(1):13-20.
2. Lenhart A. Teens and sexting. The Pew Research Center. http://www.pewinternet.org/2009/12/15/teens-and-sexting. Published December 15, 2009. Accessed October 31, 2017.
3. Temple JR, Paul JA, van den Berg P, et al. Teen sexting and its association with sexual behaviors. Arch Pediatr Adolesc Med. 2012;166(9):828-833.
4. Gordon-Messer D, Bauermeister JA, Grodzinski A, et al. Sexting among young adults. J Adolesc Health. 2013;52(3):301-306.
5. Henderson L. Sexting and sexual relationships among teens and young adults. McNair Scholars Research Journal. 2011;7(1):31-39.
6. The National Campaign to Prevent Teen and Unplanned Pregnancy. Sex and tech: results from a survey of teens and young adults. https://thenationalcampaign.org/sites/default/files/resource-primary-download/sex_and_tech_summary.pdf. Published December 2008. Accessed October 31, 2017.
7. Strassberg DS, McKinnon RK, Sustaíta MA, et al. Sexting by high school students: an exploratory and descriptive study. Arch Sex Behav. 2013;42(1):15-21.
8. Houck CD, Barker D, Rizzo C, et al. Sexting and sexual behavior in at-risk adolescents. Pediatrics. 2014;133(2):e276-e282.
9. Benotsch EG, Snipes DJ, Martin AM, et al. Sexting, substance use, and sexual risk behavior in young adults. J Adolesc Health. 2013;52(3):307-313.
10. Delevi R, Weisskirch RS. Personality factors as predictors of sexting. Comput Human Behav. 2013;29(6):2589-2594.
11. Temple JR, Choi H. Longitudinal association between teen sexting and sexual behavior. Pediatrics. 2014;134(5):1287-1292.
12. McEwan M, Friedman SH. Violence by parents against their children: reporting of maltreatment suspicions, child protection, and risk in mental illness. Psych Clin North Am. 2016;39(4):691-700.
13. Citron DK, Franks MA. Criminalizing revenge porn. Wake Forest Law Review. 2014;49:345-391.
14. Hinduja S, Patchin JW. State cyberbullying laws: a brief review of state cyberbullying laws and policies. Cyberbullying Research Center. https://cyberbullying.org/Bullying-and-Cyberbullying-Laws.pdf. Updated 2016. Accessed October 31, 2017.
15. Beitsch R. States slowly scale back juvenile sex offender registries. The Pew Charitable Trusts. http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2015/11/19/states-slowly-scale-back-juvenile-sex-offender-registries. Published November 19, 2015. Accessed October 31, 2017.
16. Wolak J, Finkelhor D, Mitchell KJ. How often are teens arrested for sexting? Data from a national sample of police cases. Pediatrics. 2012;129(1):4-12.
17. The Campus School at Boston College. Bullying prevention policy. https://www.bc.edu/bc-web/schools/lsoe/sites/campus-school/who-we-are/policies-and-procedures/bullying-prevention-policy.html. Accessed October 31, 2017.
18. Goldenring JM, Rosen DS. Getting into adolescent heads: an essential update. Contemporary Pediatrics. 2004;21(1):64.
19. Klein DA, Goldenring JM, Adelman WP. HEEADSSS 3.0: the psychosocial interview for adolescents updated for a new century fueled by media. Contemporary Pediatrics. http://contemporarypediatrics.modernmedicine.com/contemporary-pediatrics/content/tags/adolescent-medicine/heeadsss-30-psychosocial-interview-adolesce?page=full. Published January 1, 2014. Accessed October 31, 2017.
1. Mitchell KJ, Finkelhor D, Jones LM, et al. Prevalence and characteristics of youth sexting: a national study. Pediatrics. 2012;129(1):13-20.
2. Lenhart A. Teens and sexting. The Pew Research Center. http://www.pewinternet.org/2009/12/15/teens-and-sexting. Published December 15, 2009. Accessed October 31, 2017.
3. Temple JR, Paul JA, van den Berg P, et al. Teen sexting and its association with sexual behaviors. Arch Pediatr Adolesc Med. 2012;166(9):828-833.
4. Gordon-Messer D, Bauermeister JA, Grodzinski A, et al. Sexting among young adults. J Adolesc Health. 2013;52(3):301-306.
5. Henderson L. Sexting and sexual relationships among teens and young adults. McNair Scholars Research Journal. 2011;7(1):31-39.
6. The National Campaign to Prevent Teen and Unplanned Pregnancy. Sex and tech: results from a survey of teens and young adults. https://thenationalcampaign.org/sites/default/files/resource-primary-download/sex_and_tech_summary.pdf. Published December 2008. Accessed October 31, 2017.
7. Strassberg DS, McKinnon RK, Sustaíta MA, et al. Sexting by high school students: an exploratory and descriptive study. Arch Sex Behav. 2013;42(1):15-21.
8. Houck CD, Barker D, Rizzo C, et al. Sexting and sexual behavior in at-risk adolescents. Pediatrics. 2014;133(2):e276-e282.
9. Benotsch EG, Snipes DJ, Martin AM, et al. Sexting, substance use, and sexual risk behavior in young adults. J Adolesc Health. 2013;52(3):307-313.
10. Delevi R, Weisskirch RS. Personality factors as predictors of sexting. Comput Human Behav. 2013;29(6):2589-2594.
11. Temple JR, Choi H. Longitudinal association between teen sexting and sexual behavior. Pediatrics. 2014;134(5):1287-1292.
12. McEwan M, Friedman SH. Violence by parents against their children: reporting of maltreatment suspicions, child protection, and risk in mental illness. Psych Clin North Am. 2016;39(4):691-700.
13. Citron DK, Franks MA. Criminalizing revenge porn. Wake Forest Law Review. 2014;49:345-391.
14. Hinduja S, Patchin JW. State cyberbullying laws: a brief review of state cyberbullying laws and policies. Cyberbullying Research Center. https://cyberbullying.org/Bullying-and-Cyberbullying-Laws.pdf. Updated 2016. Accessed October 31, 2017.
15. Beitsch R. States slowly scale back juvenile sex offender registries. The Pew Charitable Trusts. http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2015/11/19/states-slowly-scale-back-juvenile-sex-offender-registries. Published November 19, 2015. Accessed October 31, 2017.
16. Wolak J, Finkelhor D, Mitchell KJ. How often are teens arrested for sexting? Data from a national sample of police cases. Pediatrics. 2012;129(1):4-12.
17. The Campus School at Boston College. Bullying prevention policy. https://www.bc.edu/bc-web/schools/lsoe/sites/campus-school/who-we-are/policies-and-procedures/bullying-prevention-policy.html. Accessed October 31, 2017.
18. Goldenring JM, Rosen DS. Getting into adolescent heads: an essential update. Contemporary Pediatrics. 2004;21(1):64.
19. Klein DA, Goldenring JM, Adelman WP. HEEADSSS 3.0: the psychosocial interview for adolescents updated for a new century fueled by media. Contemporary Pediatrics. http://contemporarypediatrics.modernmedicine.com/contemporary-pediatrics/content/tags/adolescent-medicine/heeadsss-30-psychosocial-interview-adolesce?page=full. Published January 1, 2014. Accessed October 31, 2017.

Methamphetamine-induced psychosis: Who says all drug use is reversible?
Use of methamphetamine, an N-methyl analog of amphetamine, is a serious public health problem; throughout the world an estimated 35.7 million people use the drug recreationally.1 Methamphetamine is easy to obtain because it is cheap to produce and can be synthesized anywhere. In the United States, methamphetamine is commonly manufactured in small-scale laboratories using relatively inexpensive, legally available ingredients. Large-scale manufacturing in clandestine laboratories also contributes to methamphetamine abuse. The drug, known as meth, crystal meth, ice, and other names, is available as a powder, tablet, or crystalline salt, and is used by various routes of administration (Table).

Although FDA-approved for treating attention-deficit/hyperactivity disorder, methamphetamine is taken recreationally for its euphoric effects; however, it also produces anhedonia, paranoia, and a host of cognitive deficits and other adverse effects.
Methamphetamine causes psychiatric diseases that resemble naturally occurring illnesses but are more difficult to treat. Dependence occurs over a period of escalating use (Figure). Long-term exposure to the drug has been shown to cause severe neurotoxic and neuropathological effects with consequent disturbances in several cognitive domains.4
Despite advances in understanding the basic neurobiology of methamphetamine-induced effects on the brain, much remains to be done to translate this knowledge to treating patients and the complications that result from chronic abuse of this stimulant. In this review, we:
- provide a brief synopsis of the clinical presentation of patients who use methamphetamine
- describe some of the complications of methamphetamine abuse/dependence, focusing on methamphetamine-induced psychosis
- suggest ways to approach the treatment of these patients, including those with methamphetamine-induced psychosis.
Acute effects of methamphetamine use
Psychiatric symptoms. Patients under the influence of methamphetamine may present with clinical symptoms that mimic psychiatric disorders. For example, the drug can cause marked euphoria, hyperactivity, and disturbed speech patterns, thus mimicking a manic state. Patients also may present with anxiety, agitation, and irritability or aggressiveness. Although an individual may take methamphetamine for sexual enhancement, the drug can cause hypersexuality, which often is associated with unintended and unsafe sexual activities. These signs and symptoms are exacerbated during drug binges that can last for days, during which time large quantities of the drug are consumed.
Methamphetamine users may become preoccupied with their own thought patterns, and their actions can become compulsive and nonsensical. For example, a patient may become obsessed with an object of no specific value in his (her) environment, such as a doorknob or a cloud. Patients also may become suspicious of their friends and family members or think that police officers are after them. Less commonly, a patient also may suffer from poverty of speech, psychomotor retardation, and diminished social engagement similar to that reported in some patients with schizophrenia with deficit syndrome. Usually, acute symptoms will last 4 to 7 days after drug cessation, and then resolve completely with protracted abstinence from the drug.
Neurologic signs of methamphetamine use include hemorrhagic strokes in young people without any evidence of previous neurologic impairments. Studies have documented similarities between methamphetamine-induced neurotoxicity and traumatic brain injury.5 Postmortem studies have reported the presence of arteriovenous malformation in some patients with hemorrhagic strokes.
Hyperthermia is a dangerous acute effect of methamphetamine use. High body temperatures can cause both peripheral and central abnormalities, including muscular and cardiovascular dysfunction, renal failure secondary to rhabdomyolysis, heat stroke, and other heat-induced malignant syndromes. Some of the central dysfunctions may be related to heat-induced production of free radicals in various brain regions. There are no pharmacologic treatments for methamphetamine-induced thermal dysregulation.6 Therefore, clinicians need to focus on reducing body temperature by using cooling fans or cold water baths. Efforts should be made to avoid overhydrating patients because of the risk of developing the syndrome of inappropriate antidiuretic hormone secretion.
Chronic methamphetamine abuse
Psychosis is a long-term complication of chronic abuse of the drug.7 Although psychosis has been a reported complication of methamphetamine use since the 1950s,8 most of the subsequent literature is from Japan, where methamphetamine use was highly prevalent after World War II.9,10 The prevalence of methamphetamine-induced psychosis in methamphetamine-dependent patients varies from 13% (in the United States11) to 50% (in Asia12). This difference might be related to variability in the purity of methamphetamine used in different locations.
Methamphetamine users may experience a pre-psychotic state that consists of ideas of reference and delusional moods. This is followed by a psychotic state that includes hallucinations and delusions. The time it takes to develop these symptoms can vary from a few months up to >20 years after starting to use methamphetamine.10,13 Psychosis can occur in patients who do not have a history of psychiatric illness.10
The clinical presentation of methamphetamine-induced psychosis includes delusions of reference and persecutions.8-10 Paranoid delusions may be accompanied by violent behavior. Some patients may present with grandiose or jealousy delusions. Patients may experience auditory, tactile, or visual hallucinations. They may exhibit mania and logorrheic verbal outputs, symptoms consistent with a diagnosis of methamphetamine-induced mood disorder with manic features. Patients who use large daily doses of the drug also may report that there are ants or other parasites crawling under their skin (eg, formication, “meth mites”) and might present with infected excoriations of their skin as a result of attempting to remove insects. This is clinically important because penicillin-resistant bacteria are common in patients who use methamphetamine, and strains tend to be virulent.
Psychotic symptoms can last from a few days to several weeks after stopping methamphetamine use, although methamphetamine-induced psychosis can persist after long periods of abstinence.14 Psychotic symptoms may recur with re-exposure to the drug9 or repeated stressful life events.15 Patients with recurrent psychosis in the absence of a drug trigger appear to have high levels of peripheral norepinephrine.15 Patients with psychosis caused by long-term methamphetamine use will not necessarily show signs of sympathomimetic dysfunction because they may not have any methamphetamine in the body when they first present for clinical evaluation. Importantly, patients with methamphetamine-induced psychosis have been reported to have poor outcomes at follow-up.16 They have an increased risk of suicide, recurrent drug-induced psychosis, and comorbid alcohol abuse.16
Doses required to induce psychosis vary from patient to patient and may depend on the patient’s genetic background and/or environmental conditions. Methamphetamine can increase the severity of many psychiatric symptoms17 and may expedite the development of schizophrenia in first-degree relatives of patients with schizophrenia.18
The diagnosis of methamphetamine-induced psychosis should focus on differentiating it from schizophrenia. Wang et al19 found similar patterns of delusions in patients with schizophrenia and those with methamphetamine-induced psychosis. However, compared with patients with schizophrenia, patients with methamphetamine-induced psychosis have a higher prevalence of visual and tactile hallucinations, and less disorganization, blunted affect, and motor retardation. Some patients may present with depression and suicidal ideation; these features may be more prominent during withdrawal, but also may be obvious during periods of active use.16
Although these clinical features may be helpful initially, more comparative neurobiologic investigations are needed to identify potential biologic differences between schizophrenia and methamphetamine-induced psychosis because these differences will impact therapeutic approaches to these diverse population groups.
Neurologic complications. Chronic methamphetamine users may develop various neurologic disorders.20 They may present with stereotypies involving finger movements or repeated rubbing of mouth or face, orofacial dyskinesia, and choreoathetoid movements reminiscent of classical neurologic disorders. These movement disorders can persist after cessation of methamphetamine use. In some cases, these movement abnormalities may respond to dopamine receptor antagonists such as haloperidol.
Neuropsychological findings. Chronic methamphetamine users show mild signs of cognitive decline that affects a broad range of neuropsychological functions.21-23 There are deficits in several cognitive processes that are dependent on the function of frontostriatal and limbic circuits.24-26 Specifically, episodic memory, executive functions, complex information processing speed, and psychomotor functions all have been reported to be negatively impacted.
Methamphetamine use often results in psychiatric distress that impacts users’ interpersonal relationships.27 Additionally, impulsivity may exacerbate their psychosocial difficulties and promote maintenance of drug-seeking behaviors.28 Cognitive deficits lead to poor health outcomes, high-risk behaviors, employment difficulties, and repeated relapse.29,30
Partial recovery of neuropsychological functioning and improvement in affective distress can be achieved after sustained abstinence from methamphetamine, but recovery may not be complete. Because cognitive dysfunction can influence treatment outcomes, clinicians need to be fully aware of the cognitive status of those patients, and a thorough neuropsychological evaluation is necessary before initiating treatment.
Treatment
Methamphetamine abuse. Because patients who abuse methamphetamine are at high risk of developing psychosis, neurologic complications, and neuropsychological disorders, initiating treatment early in the course of their addiction is of paramount importance. Treatment of methamphetamine addiction is complicated by the fact that these patients have a high prevalence of comorbid psychiatric disorders, which clinicians need to keep in mind when selecting therapeutic interventions.
There are no FDA-approved agents for treating methamphetamine abuse.31 Several drugs have been tried with varying degrees of success, including bupropion, modafinil, and naltrexone. A study of modafinil found no clinically significant effects for treating methamphetamine abuse; however, only approximately one-half of participants in this study took modafinil as instructed.32 Certain selective serotonin reuptake inhibitors, including fluoxetine and paroxetine, have not been shown to be effective in treating these patients. Naltrexone may be a reasonable medication to consider because of the high prevalence of comorbid alcohol abuse among methamphetamine users.
Other treatments for methamphetamine addiction consist of behavioral interventions such as cognitive-behavioral therapy. Clinical experience has shown that the risk of relapse depends on how long the patient has been abstinent prior to entering a treatment program, the presence of attention and memory deficits, and findings of poor decision-making on neuropsychological tests.
The presence of cognitive abnormalities has been reported to impact methamphetamine abusers’ response to treatment.33 These findings suggest the need to develop approaches that might improve cognition in patients who are undergoing treatment for methamphetamine abuse. The monoaminergic agent modafinil and similar drugs need to be evaluated in large populations to increase the possibility of identifying characteristics of patients who might respond to cognitive enhancement.34
Methamphetamine-induced psychosis. First-generation antipsychotics, such as haloperidol or fluphenazine, need to be used sparingly in patients with methamphetamine-induced psychosis because of the risk of developing extrapyramidal symptoms (EPS) and because these patients are prone to develop motor complications as a result of methamphetamine abuse. Second-generation antipsychotics, such as risperidone and olanzapine, may be more appropriate because of the lower risks of EPS.35 The presence of high norepinephrine levels in some patients with recurrent methamphetamine psychosis suggests that drugs that block norepinephrine receptors, such as prazosin or propranolol, might be of therapeutic benefit if they are shown to be effective in controlled clinical trials.
1. United Nations Office on Drugs and Crime. World Drug Report 2016. United Nations publication, Sales No. E.16.XI.7. http://www.unodc.org/wdr2016. Published 2016. Accessed September 28, 2017.
2. Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60(2):379-407.
3. Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760-773.
4. Cadet JL, Bisagno V, Milroy CM. Neuropathology of substance use disorders. Acta Neuropathol. 2014;127(1):91-107.
5. Gold MS, Kobeissy FH, Wang KK, et al. Methamphetamine- and trauma-induced brain injuries: comparative cellular and molecular neurobiological substrates. Biol Psychiatry. 2009;66(2):118-127.
6. Gold MS, Graham NA, Kobeissy FH, et al. Speed, cocaine, and other psychostimulants death rates. Am J Cardiol. 2007;100(7):1184.
7. Shelly J, Uhlmann A, Sinclair H, et al. First-rank symptoms in methamphetamine psychosis and schizophrenia. Psychopathology. 2016;49(6):429-435.
8. Connell PH. Amphetamine psychosis. In: Connell PH. Maudsley monographs. No. 5. London, United Kingdom: Oxford Press; 1958:5.
9. Sato M. A lasting vulnerability to psychosis in patients with previous methamphetamine psychosis. Ann N Y Acad Sci. 1992;654(1):160-170.
10. Ujike H, Sato M. Clinical features of sensitization to methamphetamine observed in patients with methamphetamine dependence and psychosis. Ann N Y Acad Sci. 2004;1025(1):279-287.
11. Glasner-Edwards S, Mooney LJ, Marinelli-Casey P, et al; Methamphetamine Treatment Project Corporate Authors. Psychopathology in methamphetamine-dependent adults 3 years after treatment. Drug Alcohol Rev. 2010;29(1):12-20.
12. Sulaiman AH, Said MA, Habil MH, et al. The risk and associated factors of methamphetamine psychosis in methamphetamine-dependent patients in Malaysia. Compr Psychiatry. 2014;55(suppl 1):S89-S94.
13. Fasihpour B, Molavi S, Shariat SV. Clinical features of inpatients with methamphetamine-induced psychosis. J Ment Health. 2013;22(4):341-349.
14. Akiyama K, Saito A, Shimoda K. Chronic methamphetamine psychosis after long-term abstinence in Japanese incarcerated patients. Am J Addict. 2011;20(3):240-249.
15. Yui K, Goto K, Ikemoto S, et al. Methamphetamine psychosis: spontaneous recurrence of paranoid-hallucinatory states and monoamine neurotransmitter function. J Clin Psychopharmacol. 1997;17(1):34-43.
16. Kittirattanapaiboon P, Mahatnirunkul S, Booncharoen H, et al. Long-term outcomes in methamphetamine psychosis patients after first hospitalisation. Drug Alcohol Rev. 2010;29(4):456-461.
17. McKetin R, Dawe S, Burns RA, et al. The profile of psychiatric symptoms exacerbated by methamphetamine use. Drug Alcohol Depend. 2016;161:104-109.
18. Li H, Lu Q, Xiao E, et al. Methamphetamine enhances the development of schizophrenia in first-degree relatives of patients with schizophrenia. Can J Psychiatry. 2014;59(2):107-113.
19. Wang LJ, Lin SK, Chen YC, et al. Differences in clinical features of methamphetamine users with persistent psychosis and patients with schizophrenia. Psychopathology. 2016;49(2):108-115.
20. Rusyniak DE. Neurologic manifestations of chronic methamphetamine abuse. Psychiatr Clin North Am. 2013;36(2):261-275.
21. Simon SL, Domier C, Carnell J, et al. Cognitive impairment in individuals currently using methamphetamine. Am J Addict. 2000;9(3):222-231.
22. Paulus MP, Hozack NE, Zauscher BE, et al. Behavioral and functional neuroimaging evidence for prefrontal dysfunction in methamphetamine-dependent subjects. Neuropsychopharmacology. 2002;26(1):53-63.
23. Rendell PG, Mazur M, Henry JD. Prospective memory impairment in former users of methamphetamine. Psychopharmacology (Berl). 2009;203(3):609-616.
24. Monterosso JR, Ainslie G, Xu J, et al. Frontoparietal cortical activity of methamphetamine-dependent and comparison subjects performing a delay discounting task. Hum Brain Mapp. 2007;28(5):383-393.
25. Nestor LJ, Ghahremani DG, Monterosso J, et al. Prefrontal hypoactivation during cognitive control in early abstinent methamphetamine-dependent subjects. Psychiatry Res. 2011;194(3):287-295.
26. Scott JC, Woods SP, Matt GE, et al. Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev. 2007;17(3):275-297.
27. Cretzmeyer M, Sarrazin MV, Huber DL, et al. Treatment of methamphetamine abuse: research findings and clinical directions. J Subst Abuse Treat. 2003;24(3):267-277.
28. Semple SJ, Zians J, Grant I, et al. Impulsivity and methamphetamine use. J Subst Abuse Treat. 2005;29(2):85-93.
29. Hester R, Lee N, Pennay A, et al. The effects of modafinil treatment on neuropsychological and attentional bias performance during 7-day inpatient withdrawal from methamphetamine dependence. Exp Clin Psychopharmacol. 2010;18(6):489-497.
30. Weber E, Blackstone K, Iudicello JE, et al; Translational Methamphetamine AIDS Research Center (TMARC) Group. Neurocognitive deficits are associated with unemployment in chronic methamphetamine users. Drug Alcohol Depend. 2012;125(1-2):146-153.
31. Ballester J, Valentine G, Sofuoglu M. Pharmacological treatments for methamphetamine addiction: current status and future directions. Expert Rev Clin Pharmacol. 2017;10(3):305-314.
32. Anderson AL, Li SH, Biswas K, et al. Modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2012;120(1-3):135-141.
33. Cadet JL, Bisagno V. Neuropsychological consequences of chronic drug use: relevance to treatment approaches. Front Psychiatry. 2016;6:189.
34. Loland CJ, Mereu M, Okunola OM, et al. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol Psychiatry. 2012;72(5):405-413.
35. Farnia V, Shakeri J, Tatari F, et al. Randomized controlled trial of aripiprazole versus risperidone for the treatment of amphetamine-induced psychosis. Am J Drug Alcohol Abuse. 2014;40(1):10-15.
Use of methamphetamine, an N-methyl analog of amphetamine, is a serious public health problem; throughout the world an estimated 35.7 million people use the drug recreationally.1 Methamphetamine is easy to obtain because it is cheap to produce and can be synthesized anywhere. In the United States, methamphetamine is commonly manufactured in small-scale laboratories using relatively inexpensive, legally available ingredients. Large-scale manufacturing in clandestine laboratories also contributes to methamphetamine abuse. The drug, known as meth, crystal meth, ice, and other names, is available as a powder, tablet, or crystalline salt, and is used by various routes of administration (Table).
Although FDA-approved for treating attention-deficit/hyperactivity disorder, methamphetamine is taken recreationally for its euphoric effects; however, it also produces anhedonia, paranoia, and a host of cognitive deficits and other adverse effects.
Methamphetamine causes psychiatric diseases that resemble naturally occurring illnesses but are more difficult to treat. Dependence occurs over a period of escalating use (Figure). Long-term exposure to the drug has been shown to cause severe neurotoxic and neuropathological effects with consequent disturbances in several cognitive domains.4
Despite advances in understanding the basic neurobiology of methamphetamine-induced effects on the brain, much remains to be done to translate this knowledge to treating patients and the complications that result from chronic abuse of this stimulant. In this review, we:
- provide a brief synopsis of the clinical presentation of patients who use methamphetamine
- describe some of the complications of methamphetamine abuse/dependence, focusing on methamphetamine-induced psychosis
- suggest ways to approach the treatment of these patients, including those with methamphetamine-induced psychosis.
Acute effects of methamphetamine use
Psychiatric symptoms. Patients under the influence of methamphetamine may present with clinical symptoms that mimic psychiatric disorders. For example, the drug can cause marked euphoria, hyperactivity, and disturbed speech patterns, thus mimicking a manic state. Patients also may present with anxiety, agitation, and irritability or aggressiveness. Although an individual may take methamphetamine for sexual enhancement, the drug can cause hypersexuality, which often is associated with unintended and unsafe sexual activities. These signs and symptoms are exacerbated during drug binges that can last for days, during which time large quantities of the drug are consumed.
Methamphetamine users may become preoccupied with their own thought patterns, and their actions can become compulsive and nonsensical. For example, a patient may become obsessed with an object of no specific value in his (her) environment, such as a doorknob or a cloud. Patients also may become suspicious of their friends and family members or think that police officers are after them. Less commonly, a patient also may suffer from poverty of speech, psychomotor retardation, and diminished social engagement similar to that reported in some patients with schizophrenia with deficit syndrome. Usually, acute symptoms will last 4 to 7 days after drug cessation, and then resolve completely with protracted abstinence from the drug.
Neurologic signs of methamphetamine use include hemorrhagic strokes in young people without any evidence of previous neurologic impairments. Studies have documented similarities between methamphetamine-induced neurotoxicity and traumatic brain injury.5 Postmortem studies have reported the presence of arteriovenous malformation in some patients with hemorrhagic strokes.
Hyperthermia is a dangerous acute effect of methamphetamine use. High body temperatures can cause both peripheral and central abnormalities, including muscular and cardiovascular dysfunction, renal failure secondary to rhabdomyolysis, heat stroke, and other heat-induced malignant syndromes. Some of the central dysfunctions may be related to heat-induced production of free radicals in various brain regions. There are no pharmacologic treatments for methamphetamine-induced thermal dysregulation.6 Therefore, clinicians need to focus on reducing body temperature by using cooling fans or cold water baths. Efforts should be made to avoid overhydrating patients because of the risk of developing the syndrome of inappropriate antidiuretic hormone secretion.
Chronic methamphetamine abuse
Psychosis is a long-term complication of chronic abuse of the drug.7 Although psychosis has been a reported complication of methamphetamine use since the 1950s,8 most of the subsequent literature is from Japan, where methamphetamine use was highly prevalent after World War II.9,10 The prevalence of methamphetamine-induced psychosis in methamphetamine-dependent patients varies from 13% (in the United States11) to 50% (in Asia12). This difference might be related to variability in the purity of methamphetamine used in different locations.
Methamphetamine users may experience a pre-psychotic state that consists of ideas of reference and delusional moods. This is followed by a psychotic state that includes hallucinations and delusions. The time it takes to develop these symptoms can vary from a few months up to >20 years after starting to use methamphetamine.10,13 Psychosis can occur in patients who do not have a history of psychiatric illness.10
The clinical presentation of methamphetamine-induced psychosis includes delusions of reference and persecutions.8-10 Paranoid delusions may be accompanied by violent behavior. Some patients may present with grandiose or jealousy delusions. Patients may experience auditory, tactile, or visual hallucinations. They may exhibit mania and logorrheic verbal outputs, symptoms consistent with a diagnosis of methamphetamine-induced mood disorder with manic features. Patients who use large daily doses of the drug also may report that there are ants or other parasites crawling under their skin (eg, formication, “meth mites”) and might present with infected excoriations of their skin as a result of attempting to remove insects. This is clinically important because penicillin-resistant bacteria are common in patients who use methamphetamine, and strains tend to be virulent.
Psychotic symptoms can last from a few days to several weeks after stopping methamphetamine use, although methamphetamine-induced psychosis can persist after long periods of abstinence.14 Psychotic symptoms may recur with re-exposure to the drug9 or repeated stressful life events.15 Patients with recurrent psychosis in the absence of a drug trigger appear to have high levels of peripheral norepinephrine.15 Patients with psychosis caused by long-term methamphetamine use will not necessarily show signs of sympathomimetic dysfunction because they may not have any methamphetamine in the body when they first present for clinical evaluation. Importantly, patients with methamphetamine-induced psychosis have been reported to have poor outcomes at follow-up.16 They have an increased risk of suicide, recurrent drug-induced psychosis, and comorbid alcohol abuse.16
Doses required to induce psychosis vary from patient to patient and may depend on the patient’s genetic background and/or environmental conditions. Methamphetamine can increase the severity of many psychiatric symptoms17 and may expedite the development of schizophrenia in first-degree relatives of patients with schizophrenia.18
The diagnosis of methamphetamine-induced psychosis should focus on differentiating it from schizophrenia. Wang et al19 found similar patterns of delusions in patients with schizophrenia and those with methamphetamine-induced psychosis. However, compared with patients with schizophrenia, patients with methamphetamine-induced psychosis have a higher prevalence of visual and tactile hallucinations, and less disorganization, blunted affect, and motor retardation. Some patients may present with depression and suicidal ideation; these features may be more prominent during withdrawal, but also may be obvious during periods of active use.16
Although these clinical features may be helpful initially, more comparative neurobiologic investigations are needed to identify potential biologic differences between schizophrenia and methamphetamine-induced psychosis because these differences will impact therapeutic approaches to these diverse population groups.
Neurologic complications. Chronic methamphetamine users may develop various neurologic disorders.20 They may present with stereotypies involving finger movements or repeated rubbing of mouth or face, orofacial dyskinesia, and choreoathetoid movements reminiscent of classical neurologic disorders. These movement disorders can persist after cessation of methamphetamine use. In some cases, these movement abnormalities may respond to dopamine receptor antagonists such as haloperidol.
Neuropsychological findings. Chronic methamphetamine users show mild signs of cognitive decline that affects a broad range of neuropsychological functions.21-23 There are deficits in several cognitive processes that are dependent on the function of frontostriatal and limbic circuits.24-26 Specifically, episodic memory, executive functions, complex information processing speed, and psychomotor functions all have been reported to be negatively impacted.
Methamphetamine use often results in psychiatric distress that impacts users’ interpersonal relationships.27 Additionally, impulsivity may exacerbate their psychosocial difficulties and promote maintenance of drug-seeking behaviors.28 Cognitive deficits lead to poor health outcomes, high-risk behaviors, employment difficulties, and repeated relapse.29,30
Partial recovery of neuropsychological functioning and improvement in affective distress can be achieved after sustained abstinence from methamphetamine, but recovery may not be complete. Because cognitive dysfunction can influence treatment outcomes, clinicians need to be fully aware of the cognitive status of those patients, and a thorough neuropsychological evaluation is necessary before initiating treatment.
Treatment
Methamphetamine abuse. Because patients who abuse methamphetamine are at high risk of developing psychosis, neurologic complications, and neuropsychological disorders, initiating treatment early in the course of their addiction is of paramount importance. Treatment of methamphetamine addiction is complicated by the fact that these patients have a high prevalence of comorbid psychiatric disorders, which clinicians need to keep in mind when selecting therapeutic interventions.
There are no FDA-approved agents for treating methamphetamine abuse.31 Several drugs have been tried with varying degrees of success, including bupropion, modafinil, and naltrexone. A study of modafinil found no clinically significant effects for treating methamphetamine abuse; however, only approximately one-half of participants in this study took modafinil as instructed.32 Certain selective serotonin reuptake inhibitors, including fluoxetine and paroxetine, have not been shown to be effective in treating these patients. Naltrexone may be a reasonable medication to consider because of the high prevalence of comorbid alcohol abuse among methamphetamine users.
Other treatments for methamphetamine addiction consist of behavioral interventions such as cognitive-behavioral therapy. Clinical experience has shown that the risk of relapse depends on how long the patient has been abstinent prior to entering a treatment program, the presence of attention and memory deficits, and findings of poor decision-making on neuropsychological tests.
The presence of cognitive abnormalities has been reported to impact methamphetamine abusers’ response to treatment.33 These findings suggest the need to develop approaches that might improve cognition in patients who are undergoing treatment for methamphetamine abuse. The monoaminergic agent modafinil and similar drugs need to be evaluated in large populations to increase the possibility of identifying characteristics of patients who might respond to cognitive enhancement.34
Methamphetamine-induced psychosis. First-generation antipsychotics, such as haloperidol or fluphenazine, need to be used sparingly in patients with methamphetamine-induced psychosis because of the risk of developing extrapyramidal symptoms (EPS) and because these patients are prone to develop motor complications as a result of methamphetamine abuse. Second-generation antipsychotics, such as risperidone and olanzapine, may be more appropriate because of the lower risks of EPS.35 The presence of high norepinephrine levels in some patients with recurrent methamphetamine psychosis suggests that drugs that block norepinephrine receptors, such as prazosin or propranolol, might be of therapeutic benefit if they are shown to be effective in controlled clinical trials.
Use of methamphetamine, an N-methyl analog of amphetamine, is a serious public health problem; throughout the world an estimated 35.7 million people use the drug recreationally.1 Methamphetamine is easy to obtain because it is cheap to produce and can be synthesized anywhere. In the United States, methamphetamine is commonly manufactured in small-scale laboratories using relatively inexpensive, legally available ingredients. Large-scale manufacturing in clandestine laboratories also contributes to methamphetamine abuse. The drug, known as meth, crystal meth, ice, and other names, is available as a powder, tablet, or crystalline salt, and is used by various routes of administration (Table).
Although FDA-approved for treating attention-deficit/hyperactivity disorder, methamphetamine is taken recreationally for its euphoric effects; however, it also produces anhedonia, paranoia, and a host of cognitive deficits and other adverse effects.
Methamphetamine causes psychiatric diseases that resemble naturally occurring illnesses but are more difficult to treat. Dependence occurs over a period of escalating use (Figure). Long-term exposure to the drug has been shown to cause severe neurotoxic and neuropathological effects with consequent disturbances in several cognitive domains.4
Despite advances in understanding the basic neurobiology of methamphetamine-induced effects on the brain, much remains to be done to translate this knowledge to treating patients and the complications that result from chronic abuse of this stimulant. In this review, we:
- provide a brief synopsis of the clinical presentation of patients who use methamphetamine
- describe some of the complications of methamphetamine abuse/dependence, focusing on methamphetamine-induced psychosis
- suggest ways to approach the treatment of these patients, including those with methamphetamine-induced psychosis.
Acute effects of methamphetamine use
Psychiatric symptoms. Patients under the influence of methamphetamine may present with clinical symptoms that mimic psychiatric disorders. For example, the drug can cause marked euphoria, hyperactivity, and disturbed speech patterns, thus mimicking a manic state. Patients also may present with anxiety, agitation, and irritability or aggressiveness. Although an individual may take methamphetamine for sexual enhancement, the drug can cause hypersexuality, which often is associated with unintended and unsafe sexual activities. These signs and symptoms are exacerbated during drug binges that can last for days, during which time large quantities of the drug are consumed.
Methamphetamine users may become preoccupied with their own thought patterns, and their actions can become compulsive and nonsensical. For example, a patient may become obsessed with an object of no specific value in his (her) environment, such as a doorknob or a cloud. Patients also may become suspicious of their friends and family members or think that police officers are after them. Less commonly, a patient also may suffer from poverty of speech, psychomotor retardation, and diminished social engagement similar to that reported in some patients with schizophrenia with deficit syndrome. Usually, acute symptoms will last 4 to 7 days after drug cessation, and then resolve completely with protracted abstinence from the drug.
Neurologic signs of methamphetamine use include hemorrhagic strokes in young people without any evidence of previous neurologic impairments. Studies have documented similarities between methamphetamine-induced neurotoxicity and traumatic brain injury.5 Postmortem studies have reported the presence of arteriovenous malformation in some patients with hemorrhagic strokes.
Hyperthermia is a dangerous acute effect of methamphetamine use. High body temperatures can cause both peripheral and central abnormalities, including muscular and cardiovascular dysfunction, renal failure secondary to rhabdomyolysis, heat stroke, and other heat-induced malignant syndromes. Some of the central dysfunctions may be related to heat-induced production of free radicals in various brain regions. There are no pharmacologic treatments for methamphetamine-induced thermal dysregulation.6 Therefore, clinicians need to focus on reducing body temperature by using cooling fans or cold water baths. Efforts should be made to avoid overhydrating patients because of the risk of developing the syndrome of inappropriate antidiuretic hormone secretion.
Chronic methamphetamine abuse
Psychosis is a long-term complication of chronic abuse of the drug.7 Although psychosis has been a reported complication of methamphetamine use since the 1950s,8 most of the subsequent literature is from Japan, where methamphetamine use was highly prevalent after World War II.9,10 The prevalence of methamphetamine-induced psychosis in methamphetamine-dependent patients varies from 13% (in the United States11) to 50% (in Asia12). This difference might be related to variability in the purity of methamphetamine used in different locations.
Methamphetamine users may experience a pre-psychotic state that consists of ideas of reference and delusional moods. This is followed by a psychotic state that includes hallucinations and delusions. The time it takes to develop these symptoms can vary from a few months up to >20 years after starting to use methamphetamine.10,13 Psychosis can occur in patients who do not have a history of psychiatric illness.10
The clinical presentation of methamphetamine-induced psychosis includes delusions of reference and persecutions.8-10 Paranoid delusions may be accompanied by violent behavior. Some patients may present with grandiose or jealousy delusions. Patients may experience auditory, tactile, or visual hallucinations. They may exhibit mania and logorrheic verbal outputs, symptoms consistent with a diagnosis of methamphetamine-induced mood disorder with manic features. Patients who use large daily doses of the drug also may report that there are ants or other parasites crawling under their skin (eg, formication, “meth mites”) and might present with infected excoriations of their skin as a result of attempting to remove insects. This is clinically important because penicillin-resistant bacteria are common in patients who use methamphetamine, and strains tend to be virulent.
Psychotic symptoms can last from a few days to several weeks after stopping methamphetamine use, although methamphetamine-induced psychosis can persist after long periods of abstinence.14 Psychotic symptoms may recur with re-exposure to the drug9 or repeated stressful life events.15 Patients with recurrent psychosis in the absence of a drug trigger appear to have high levels of peripheral norepinephrine.15 Patients with psychosis caused by long-term methamphetamine use will not necessarily show signs of sympathomimetic dysfunction because they may not have any methamphetamine in the body when they first present for clinical evaluation. Importantly, patients with methamphetamine-induced psychosis have been reported to have poor outcomes at follow-up.16 They have an increased risk of suicide, recurrent drug-induced psychosis, and comorbid alcohol abuse.16
Doses required to induce psychosis vary from patient to patient and may depend on the patient’s genetic background and/or environmental conditions. Methamphetamine can increase the severity of many psychiatric symptoms17 and may expedite the development of schizophrenia in first-degree relatives of patients with schizophrenia.18
The diagnosis of methamphetamine-induced psychosis should focus on differentiating it from schizophrenia. Wang et al19 found similar patterns of delusions in patients with schizophrenia and those with methamphetamine-induced psychosis. However, compared with patients with schizophrenia, patients with methamphetamine-induced psychosis have a higher prevalence of visual and tactile hallucinations, and less disorganization, blunted affect, and motor retardation. Some patients may present with depression and suicidal ideation; these features may be more prominent during withdrawal, but also may be obvious during periods of active use.16
Although these clinical features may be helpful initially, more comparative neurobiologic investigations are needed to identify potential biologic differences between schizophrenia and methamphetamine-induced psychosis because these differences will impact therapeutic approaches to these diverse population groups.
Neurologic complications. Chronic methamphetamine users may develop various neurologic disorders.20 They may present with stereotypies involving finger movements or repeated rubbing of mouth or face, orofacial dyskinesia, and choreoathetoid movements reminiscent of classical neurologic disorders. These movement disorders can persist after cessation of methamphetamine use. In some cases, these movement abnormalities may respond to dopamine receptor antagonists such as haloperidol.
Neuropsychological findings. Chronic methamphetamine users show mild signs of cognitive decline that affects a broad range of neuropsychological functions.21-23 There are deficits in several cognitive processes that are dependent on the function of frontostriatal and limbic circuits.24-26 Specifically, episodic memory, executive functions, complex information processing speed, and psychomotor functions all have been reported to be negatively impacted.
Methamphetamine use often results in psychiatric distress that impacts users’ interpersonal relationships.27 Additionally, impulsivity may exacerbate their psychosocial difficulties and promote maintenance of drug-seeking behaviors.28 Cognitive deficits lead to poor health outcomes, high-risk behaviors, employment difficulties, and repeated relapse.29,30
Partial recovery of neuropsychological functioning and improvement in affective distress can be achieved after sustained abstinence from methamphetamine, but recovery may not be complete. Because cognitive dysfunction can influence treatment outcomes, clinicians need to be fully aware of the cognitive status of those patients, and a thorough neuropsychological evaluation is necessary before initiating treatment.
Treatment
Methamphetamine abuse. Because patients who abuse methamphetamine are at high risk of developing psychosis, neurologic complications, and neuropsychological disorders, initiating treatment early in the course of their addiction is of paramount importance. Treatment of methamphetamine addiction is complicated by the fact that these patients have a high prevalence of comorbid psychiatric disorders, which clinicians need to keep in mind when selecting therapeutic interventions.
There are no FDA-approved agents for treating methamphetamine abuse.31 Several drugs have been tried with varying degrees of success, including bupropion, modafinil, and naltrexone. A study of modafinil found no clinically significant effects for treating methamphetamine abuse; however, only approximately one-half of participants in this study took modafinil as instructed.32 Certain selective serotonin reuptake inhibitors, including fluoxetine and paroxetine, have not been shown to be effective in treating these patients. Naltrexone may be a reasonable medication to consider because of the high prevalence of comorbid alcohol abuse among methamphetamine users.
Other treatments for methamphetamine addiction consist of behavioral interventions such as cognitive-behavioral therapy. Clinical experience has shown that the risk of relapse depends on how long the patient has been abstinent prior to entering a treatment program, the presence of attention and memory deficits, and findings of poor decision-making on neuropsychological tests.
The presence of cognitive abnormalities has been reported to impact methamphetamine abusers’ response to treatment.33 These findings suggest the need to develop approaches that might improve cognition in patients who are undergoing treatment for methamphetamine abuse. The monoaminergic agent modafinil and similar drugs need to be evaluated in large populations to increase the possibility of identifying characteristics of patients who might respond to cognitive enhancement.34
Methamphetamine-induced psychosis. First-generation antipsychotics, such as haloperidol or fluphenazine, need to be used sparingly in patients with methamphetamine-induced psychosis because of the risk of developing extrapyramidal symptoms (EPS) and because these patients are prone to develop motor complications as a result of methamphetamine abuse. Second-generation antipsychotics, such as risperidone and olanzapine, may be more appropriate because of the lower risks of EPS.35 The presence of high norepinephrine levels in some patients with recurrent methamphetamine psychosis suggests that drugs that block norepinephrine receptors, such as prazosin or propranolol, might be of therapeutic benefit if they are shown to be effective in controlled clinical trials.
1. United Nations Office on Drugs and Crime. World Drug Report 2016. United Nations publication, Sales No. E.16.XI.7. http://www.unodc.org/wdr2016. Published 2016. Accessed September 28, 2017.
2. Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60(2):379-407.
3. Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760-773.
4. Cadet JL, Bisagno V, Milroy CM. Neuropathology of substance use disorders. Acta Neuropathol. 2014;127(1):91-107.
5. Gold MS, Kobeissy FH, Wang KK, et al. Methamphetamine- and trauma-induced brain injuries: comparative cellular and molecular neurobiological substrates. Biol Psychiatry. 2009;66(2):118-127.
6. Gold MS, Graham NA, Kobeissy FH, et al. Speed, cocaine, and other psychostimulants death rates. Am J Cardiol. 2007;100(7):1184.
7. Shelly J, Uhlmann A, Sinclair H, et al. First-rank symptoms in methamphetamine psychosis and schizophrenia. Psychopathology. 2016;49(6):429-435.
8. Connell PH. Amphetamine psychosis. In: Connell PH. Maudsley monographs. No. 5. London, United Kingdom: Oxford Press; 1958:5.
9. Sato M. A lasting vulnerability to psychosis in patients with previous methamphetamine psychosis. Ann N Y Acad Sci. 1992;654(1):160-170.
10. Ujike H, Sato M. Clinical features of sensitization to methamphetamine observed in patients with methamphetamine dependence and psychosis. Ann N Y Acad Sci. 2004;1025(1):279-287.
11. Glasner-Edwards S, Mooney LJ, Marinelli-Casey P, et al; Methamphetamine Treatment Project Corporate Authors. Psychopathology in methamphetamine-dependent adults 3 years after treatment. Drug Alcohol Rev. 2010;29(1):12-20.
12. Sulaiman AH, Said MA, Habil MH, et al. The risk and associated factors of methamphetamine psychosis in methamphetamine-dependent patients in Malaysia. Compr Psychiatry. 2014;55(suppl 1):S89-S94.
13. Fasihpour B, Molavi S, Shariat SV. Clinical features of inpatients with methamphetamine-induced psychosis. J Ment Health. 2013;22(4):341-349.
14. Akiyama K, Saito A, Shimoda K. Chronic methamphetamine psychosis after long-term abstinence in Japanese incarcerated patients. Am J Addict. 2011;20(3):240-249.
15. Yui K, Goto K, Ikemoto S, et al. Methamphetamine psychosis: spontaneous recurrence of paranoid-hallucinatory states and monoamine neurotransmitter function. J Clin Psychopharmacol. 1997;17(1):34-43.
16. Kittirattanapaiboon P, Mahatnirunkul S, Booncharoen H, et al. Long-term outcomes in methamphetamine psychosis patients after first hospitalisation. Drug Alcohol Rev. 2010;29(4):456-461.
17. McKetin R, Dawe S, Burns RA, et al. The profile of psychiatric symptoms exacerbated by methamphetamine use. Drug Alcohol Depend. 2016;161:104-109.
18. Li H, Lu Q, Xiao E, et al. Methamphetamine enhances the development of schizophrenia in first-degree relatives of patients with schizophrenia. Can J Psychiatry. 2014;59(2):107-113.
19. Wang LJ, Lin SK, Chen YC, et al. Differences in clinical features of methamphetamine users with persistent psychosis and patients with schizophrenia. Psychopathology. 2016;49(2):108-115.
20. Rusyniak DE. Neurologic manifestations of chronic methamphetamine abuse. Psychiatr Clin North Am. 2013;36(2):261-275.
21. Simon SL, Domier C, Carnell J, et al. Cognitive impairment in individuals currently using methamphetamine. Am J Addict. 2000;9(3):222-231.
22. Paulus MP, Hozack NE, Zauscher BE, et al. Behavioral and functional neuroimaging evidence for prefrontal dysfunction in methamphetamine-dependent subjects. Neuropsychopharmacology. 2002;26(1):53-63.
23. Rendell PG, Mazur M, Henry JD. Prospective memory impairment in former users of methamphetamine. Psychopharmacology (Berl). 2009;203(3):609-616.
24. Monterosso JR, Ainslie G, Xu J, et al. Frontoparietal cortical activity of methamphetamine-dependent and comparison subjects performing a delay discounting task. Hum Brain Mapp. 2007;28(5):383-393.
25. Nestor LJ, Ghahremani DG, Monterosso J, et al. Prefrontal hypoactivation during cognitive control in early abstinent methamphetamine-dependent subjects. Psychiatry Res. 2011;194(3):287-295.
26. Scott JC, Woods SP, Matt GE, et al. Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev. 2007;17(3):275-297.
27. Cretzmeyer M, Sarrazin MV, Huber DL, et al. Treatment of methamphetamine abuse: research findings and clinical directions. J Subst Abuse Treat. 2003;24(3):267-277.
28. Semple SJ, Zians J, Grant I, et al. Impulsivity and methamphetamine use. J Subst Abuse Treat. 2005;29(2):85-93.
29. Hester R, Lee N, Pennay A, et al. The effects of modafinil treatment on neuropsychological and attentional bias performance during 7-day inpatient withdrawal from methamphetamine dependence. Exp Clin Psychopharmacol. 2010;18(6):489-497.
30. Weber E, Blackstone K, Iudicello JE, et al; Translational Methamphetamine AIDS Research Center (TMARC) Group. Neurocognitive deficits are associated with unemployment in chronic methamphetamine users. Drug Alcohol Depend. 2012;125(1-2):146-153.
31. Ballester J, Valentine G, Sofuoglu M. Pharmacological treatments for methamphetamine addiction: current status and future directions. Expert Rev Clin Pharmacol. 2017;10(3):305-314.
32. Anderson AL, Li SH, Biswas K, et al. Modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2012;120(1-3):135-141.
33. Cadet JL, Bisagno V. Neuropsychological consequences of chronic drug use: relevance to treatment approaches. Front Psychiatry. 2016;6:189.
34. Loland CJ, Mereu M, Okunola OM, et al. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol Psychiatry. 2012;72(5):405-413.
35. Farnia V, Shakeri J, Tatari F, et al. Randomized controlled trial of aripiprazole versus risperidone for the treatment of amphetamine-induced psychosis. Am J Drug Alcohol Abuse. 2014;40(1):10-15.
1. United Nations Office on Drugs and Crime. World Drug Report 2016. United Nations publication, Sales No. E.16.XI.7. http://www.unodc.org/wdr2016. Published 2016. Accessed September 28, 2017.
2. Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60(2):379-407.
3. Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760-773.
4. Cadet JL, Bisagno V, Milroy CM. Neuropathology of substance use disorders. Acta Neuropathol. 2014;127(1):91-107.
5. Gold MS, Kobeissy FH, Wang KK, et al. Methamphetamine- and trauma-induced brain injuries: comparative cellular and molecular neurobiological substrates. Biol Psychiatry. 2009;66(2):118-127.
6. Gold MS, Graham NA, Kobeissy FH, et al. Speed, cocaine, and other psychostimulants death rates. Am J Cardiol. 2007;100(7):1184.
7. Shelly J, Uhlmann A, Sinclair H, et al. First-rank symptoms in methamphetamine psychosis and schizophrenia. Psychopathology. 2016;49(6):429-435.
8. Connell PH. Amphetamine psychosis. In: Connell PH. Maudsley monographs. No. 5. London, United Kingdom: Oxford Press; 1958:5.
9. Sato M. A lasting vulnerability to psychosis in patients with previous methamphetamine psychosis. Ann N Y Acad Sci. 1992;654(1):160-170.
10. Ujike H, Sato M. Clinical features of sensitization to methamphetamine observed in patients with methamphetamine dependence and psychosis. Ann N Y Acad Sci. 2004;1025(1):279-287.
11. Glasner-Edwards S, Mooney LJ, Marinelli-Casey P, et al; Methamphetamine Treatment Project Corporate Authors. Psychopathology in methamphetamine-dependent adults 3 years after treatment. Drug Alcohol Rev. 2010;29(1):12-20.
12. Sulaiman AH, Said MA, Habil MH, et al. The risk and associated factors of methamphetamine psychosis in methamphetamine-dependent patients in Malaysia. Compr Psychiatry. 2014;55(suppl 1):S89-S94.
13. Fasihpour B, Molavi S, Shariat SV. Clinical features of inpatients with methamphetamine-induced psychosis. J Ment Health. 2013;22(4):341-349.
14. Akiyama K, Saito A, Shimoda K. Chronic methamphetamine psychosis after long-term abstinence in Japanese incarcerated patients. Am J Addict. 2011;20(3):240-249.
15. Yui K, Goto K, Ikemoto S, et al. Methamphetamine psychosis: spontaneous recurrence of paranoid-hallucinatory states and monoamine neurotransmitter function. J Clin Psychopharmacol. 1997;17(1):34-43.
16. Kittirattanapaiboon P, Mahatnirunkul S, Booncharoen H, et al. Long-term outcomes in methamphetamine psychosis patients after first hospitalisation. Drug Alcohol Rev. 2010;29(4):456-461.
17. McKetin R, Dawe S, Burns RA, et al. The profile of psychiatric symptoms exacerbated by methamphetamine use. Drug Alcohol Depend. 2016;161:104-109.
18. Li H, Lu Q, Xiao E, et al. Methamphetamine enhances the development of schizophrenia in first-degree relatives of patients with schizophrenia. Can J Psychiatry. 2014;59(2):107-113.
19. Wang LJ, Lin SK, Chen YC, et al. Differences in clinical features of methamphetamine users with persistent psychosis and patients with schizophrenia. Psychopathology. 2016;49(2):108-115.
20. Rusyniak DE. Neurologic manifestations of chronic methamphetamine abuse. Psychiatr Clin North Am. 2013;36(2):261-275.
21. Simon SL, Domier C, Carnell J, et al. Cognitive impairment in individuals currently using methamphetamine. Am J Addict. 2000;9(3):222-231.
22. Paulus MP, Hozack NE, Zauscher BE, et al. Behavioral and functional neuroimaging evidence for prefrontal dysfunction in methamphetamine-dependent subjects. Neuropsychopharmacology. 2002;26(1):53-63.
23. Rendell PG, Mazur M, Henry JD. Prospective memory impairment in former users of methamphetamine. Psychopharmacology (Berl). 2009;203(3):609-616.
24. Monterosso JR, Ainslie G, Xu J, et al. Frontoparietal cortical activity of methamphetamine-dependent and comparison subjects performing a delay discounting task. Hum Brain Mapp. 2007;28(5):383-393.
25. Nestor LJ, Ghahremani DG, Monterosso J, et al. Prefrontal hypoactivation during cognitive control in early abstinent methamphetamine-dependent subjects. Psychiatry Res. 2011;194(3):287-295.
26. Scott JC, Woods SP, Matt GE, et al. Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev. 2007;17(3):275-297.
27. Cretzmeyer M, Sarrazin MV, Huber DL, et al. Treatment of methamphetamine abuse: research findings and clinical directions. J Subst Abuse Treat. 2003;24(3):267-277.
28. Semple SJ, Zians J, Grant I, et al. Impulsivity and methamphetamine use. J Subst Abuse Treat. 2005;29(2):85-93.
29. Hester R, Lee N, Pennay A, et al. The effects of modafinil treatment on neuropsychological and attentional bias performance during 7-day inpatient withdrawal from methamphetamine dependence. Exp Clin Psychopharmacol. 2010;18(6):489-497.
30. Weber E, Blackstone K, Iudicello JE, et al; Translational Methamphetamine AIDS Research Center (TMARC) Group. Neurocognitive deficits are associated with unemployment in chronic methamphetamine users. Drug Alcohol Depend. 2012;125(1-2):146-153.
31. Ballester J, Valentine G, Sofuoglu M. Pharmacological treatments for methamphetamine addiction: current status and future directions. Expert Rev Clin Pharmacol. 2017;10(3):305-314.
32. Anderson AL, Li SH, Biswas K, et al. Modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2012;120(1-3):135-141.
33. Cadet JL, Bisagno V. Neuropsychological consequences of chronic drug use: relevance to treatment approaches. Front Psychiatry. 2016;6:189.
34. Loland CJ, Mereu M, Okunola OM, et al. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol Psychiatry. 2012;72(5):405-413.
35. Farnia V, Shakeri J, Tatari F, et al. Randomized controlled trial of aripiprazole versus risperidone for the treatment of amphetamine-induced psychosis. Am J Drug Alcohol Abuse. 2014;40(1):10-15.

Prescribing antipsychotics in geriatric patients: Focus on major depressive disorder
The proportion of older adults in the world population is growing rapidly. In the next 10 to 15 years, the population age >60 will grow 3.5 times more rapidly than the general population.1 As a result, there is an increased urgency in examining benefits vs risks of antipsychotics in older individuals. In a 2010 U.S. nationally representative observational study, antipsychotic use was observed to rise slowly during early and middle adulthood, peaking at approximately age 55, declining slightly between ages 55 and 65, and then rising again after age 65, with >2% of individuals ages 80 to 84 receiving an antipsychotic.2 This is likely due to the chronology of psychotic, mood, and neurocognitive disorders across the life span. In this large national study, long-term antipsychotic treatment was common, and older patients were more likely to receive their prescriptions from non-psychiatrist physicians than from psychiatrists.2 Among patients receiving an antipsychotic, the proportion of those receiving it for >120 days was 54% for individuals ages 70 to 74; 49% for individuals ages 75 to 79; and 46% for individuals ages 80 to 84.
This 3-part review summarizes findings and risk–benefit considerations when prescribing antipsychotics to older individuals. Part 1 focused on those with chronic psychotic disorders, such as schizophrenia or bipolar disorder,3 and part 3 will cover patients with dementia. This review (part 2) aims to:
- briefly summarize the results of randomized controlled trials (RCTs) of second-generation antipsychotics (SGAs) and other major studies and analyses in older patients with major depressive disorder (MDD)
- provide a summative opinion on the relative risks and benefits associated with using antipsychotics in older adults with MDD
- highlight the gaps in the evidence base and areas that need additional research.
Summary of benefits, place in treatment armamentarium
The prevalence of MDD and clinically significant depressive symptoms in communitydwelling older adults is 3% to 4% and 15%, respectively, and as high as 16% and 50%, respectively, in nursing home residents.4 Because late-life depression is associated with suffering, disability, and excessive mortality, it needs to be recognized and treated aggressively.5 Antidepressants are the mainstay of pharmacotherapy for late-life depression. Guidelines and expert opinion informed by the current evidence recommend using selective serotonin reuptake inhibitors, such as
Over the past decade, several antipsychotics have been FDA-approved for treating MDD:

Data from several rigorously conducted RCTs support using an antidepressant plus an FGA or SGA as first-line pharmacotherapy in younger and older patients with “psychotic depression.”8-12 SGAs also can be used as augmenting agents when there is only a partial response to antidepressants.13-15 In this situation, guidelines and experts favor an augmentation strategy over switching to another antidepressant.5,9,10,16 Until recently, most published pharmacologic trials for late-life treatment-resistant depression supported using lithium to augment antidepressants.14,17 However, because several antipsychotics are now FDA-approved for treating MDD, and in light of positive findings from several studies relevant to older patients,18-21 many experts now support using SGAs to augment antidepressants in older patients with nonpsychotic depression.5,15
Clinical trials
Olanzapine plus sertraline as first-line pharmacotherapy for MDD with psychotic features. Meyers et al11 reported on a double-blind randomized comparison of olanzapine plus placebo vs olanzapine plus sertraline in 259 patients with MDD with psychotic features. An unusual feature of this trial is that it included a similar number of younger and older participants (ages 18 to 93): 117 participants were age <60 (mean age [standard deviation (SD)]: 41.3 [10.8]) and 142 were age ≥60 (mean age [SD]: 71.7 [7.8]). The same dose titration schedules based on efficacy and tolerability were used in both younger and older participants. At the end of the study, the mean dose (SD) of sertraline (or placebo) did not differ significantly in younger (174.3 mg/d [34.1]) and older participants (165.7 mg/d [43.4]). However, the mean dose (SD) of olanzapine was significantly higher in younger patients (15.7 mg/d [4.7]) than in older participants (13.4 mg/d [5.1]).
In both age groups, olanzapine plus sertraline was more efficacious than olanzapine plus placebo, and there was no statistical interaction between age, time, and treatment group (ie, the trajectories of improvement were similar in older and younger patients receiving either olanzapine or olanzapine plus sertraline). Similarly, drop-out rates because of poor tolerability did not differ significantly in younger (4.3%) and older participants (5.6%). However, in a multinomial regression, older participants were more likely to discontinue treatment because of poor tolerability.22 Older participants were significantly less likely to experience weight gain (mean [SD]: +3.3 [4.9] vs +6.5 [6.6] kg) or an increase in fasting glucose and more likely to experience a fall, pedal edema, or extrapyramidal symptoms.11,22-24 Cholesterol and triglyceride increased significantly and similarly in both age groups. The incidence of symptoms of tardive dyskinesia (TD) over the 12-week trial was low (<5%) in both younger and older participants, and clinically diagnosed TD was reported in only 1 (older) participant.25
Venlafaxine plus aripiprazole for treatment-resistant MDD. In the largest double-blind randomized study of augmentation pharmacotherapy for late-life treatment-resistant depression published to date, Lenze et al21 compared venlafaxine plus aripiprazole vs venlafaxine plus placebo in 181 patients age >60 (mean age 66, with 49 participants age >70) with MDD who did not remit after 12 weeks of treatment with venlafaxine (up to 300 mg/d). After 12 weeks of augmentation, remission rates were significantly higher with aripiprazole than with placebo: 40 (44%) vs 26 (29%); odds ratio (95% confidence interval [CI]): 2.0 (1.1 to 3.7). The median final aripiprazole dose was 7 mg/d (range 2 to 15 mg/d) in remitters and 10 mg/d (range 2 to 15 mg/d) in nonremitters.
Five of 90 participants (5%) discontinued aripiprazole (1 each: suicide, jitteriness/akathisia, worsening parkinsonism; and 2 withdrew consent); 8 of 90 (9%) discontinued placebo (2 each: lack of efficacy, headache; 1: worsening parkinsonism; and 3 withdrew consent). The completed suicide occurred after 5 weeks of treatment with aripiprazole and was judged to be “neither due to emergent suicidal ideation nor to aripiprazole side-effects, but was concluded by investigators to be a result of the individual’s persisting and long-standing suicidal ideation.”21 Including the suicide, there were 4 serious adverse events (5%) in those receiving aripiprazole (1 each: suicide, congestive heart failure, mild stroke, and diverticulitis) and 2 (2%) in those receiving placebo (1 each: myocardial infarction, hospitalized for vomiting due to accidentally taking extra venlafaxine). In 86 participants receiving aripiprazole and 87 receiving placebo, the most frequently reported adverse effects were increased dream activity (aripiprazole: 23 [27%] vs placebo: 12 [14%]), weight gain (17 [20%] vs 8 [9%]), and tremor (5 [6%] vs 0). Akathisia and parkinsonism were observed more frequently with aripiprazole than with placebo (akathisia: 24 [26%] of 91 vs 11 [12%] of 90; parkinsonism: 15 [17%] of 86 vs 2 [2%] of 81). Akathisia was generally mild and resolved with dose adjustment; however, it was associated with a transient increase in suicidality in 3 (3%) participants receiving aripiprazole vs 0 receiving placebo and persisted at the end of the trial in 5 (5%) participants receiving aripiprazole vs 2 (2%) receiving placebo. Participants receiving aripiprazole had a significantly larger increase in weight (mean [SD]: +1.93 [3.00] vs +0.01 [3.15] kg), but there were no differences between aripiprazole and placebo in changes in body fat, total cholesterol, high-density lipoprotein, low-density lipoprotein, triglycerides, glucose, insulin concentration, or QTc.
Citalopram plus risperidone for treatment-resistant MDD. Alexopoulos et al26 reported an analysis of data from 110 patients age ≥55 years (mean age [SD]: 63.4 [4.8]), among 489 mixed-age patients with MDD. Participants (n = 110) who did not respond to 1 to 3 antidepressants (venlafaxine, sertraline, mirtazapine, fluoxetine, paroxetine, or bupropion in >90%) during their current depressive episode completed 4 to 6 weeks of treatment with citalopram up to 40 mg/d; 93 did not respond and were treated with open-label risperidone (0.25 to 1 mg/d) augmentation for 4 to 6 weeks. Sixty-three (68%) of these 93 patients remitted and were randomized to 24 weeks of double-blind continuation treatment with citalopram plus risperidone vs citalopram plus placebo. Neither the median times to relapse (105 vs 57 days) nor the relapse rates (risperidone: 18 of 32 [56%] vs placebo: 20 of 31 [65%]) differed significantly. During the open-label risperidone augmentation, the most common adverse events were dizziness and dry mouth (n = 9 each, 9.7% of 93). During the continuation phase, headache (n = 3; 9.1% of 32) was observed with risperidone but not with placebo (n = 0). There was no incident parkinsonism or abnormal movements noted, but risperidone was associated with weight gain during both the open-label risperidone augmentation phase (mean [SD]: +0.9 [2.1] kg) and the continuation phase (risperidone: +0.8 [3.5] vs placebo: −0.3 [2.8] kg).
Quetiapine XR monotherapy for MDD. Katila et al27 reported on a placebo-controlled RCT of quetiapine XR (median dose, 158.7 mg/d; range, 50 to 300 mg/d) in 338 patients age ≥66 years (mean age [SD], 71.3 [7.5]) presenting with MDD and a major depressive episode with a duration <1 year and no history of failed antidepressants trials from 2 classes (more than two-thirds of participants had not received treatment). After 9 weeks, the reduction in depressive symptoms on the Montgomery-Åsberg Depression Rating Scale was significantly larger with quetiapine XR than with placebo (mean [SD]: −16.0 [9.3] vs −9.0 [9.9]). There were congruent, significant differences between quetiapine and placebo in terms of response rate (quetiapine XR: 105 of 164 [64%] vs placebo: 52 of 171 [30.4%]) and remission rate (92 of 164 [56.1%] vs placebo: 40 of 171 [23.4%]). The drop-out rates for all causes were similar, but the drop-out rate attributed to adverse events was higher with quetiapine than placebo (16 of 166 [9.6%] vs 7 of 172 [4.1%]). Most quetiapine drop-outs were attributable to dizziness, headache, and somnolence (n = 4 each), and placebo drop-outs were because of headache (n = 2). Consistent with the profile of quetiapine, adverse events with a rate that was at least 5% higher with quetiapine than with placebo included somnolence (64 of 166 [38.6%] vs 16 of 172 [9.3%]), dry mouth (34 [20.5%] vs 18 [10.5%]), and extrapyramidal symptoms (12 [7.2%] vs 4 [2.3%]). Changes in weight and laboratory test results (eg, glucose, lipid profile) were minimal and not clinically meaningful.
Other clinical data. The efficacy and relatively good tolerability of aripiprazole in older patients with treatment-resistant depression observed in the RCT by Lenze et al21 is congruent with the earlier results of 2 small (N = 20 and 24) pilot studies.18,19 In both studies, the remission rate was 50%, and the most prevalent adverse effects were agitation/restlessness/akathisia or drowsiness/sedation. Similarly, in a post hoc pooled analysis of 409 participants ages 50 to 67 from 3 placebo-controlled randomized trials, the remission rate was significantly higher with aripiprazole than with placebo (32.5% vs 17.1%), and the most common adverse effects were akathisia or restlessness (64 of 210 [30.4%]), somnolence (18 [8.6%]), and insomnia (17 [8.1%]).20
Clinical considerations
When assessing the relative benefits and risks of antipsychotics in older patients, it is important to remember that conclusions and summative opinions are necessarily influenced by the source of the data. Because much of what we know about the use of antipsychotics in geriatric adults is from clinical trials, we know more about their acute efficacy and tolerability than their long-term effectiveness and safety.28 There are similar issues regarding the role of antipsychotics in treating MDD in late life. Based on the results of several RCTs,8,11 a combination of an antidepressant plus an antipsychotic is the recommended pharmacotherapy for the acute treatment of MDD with psychotic features (Table 2).8,11,12,19-21,23-27 However, there are no published data to guide how long the antipsychotic should be continued.29

In older patients with MDD without psychotic features, 1 relatively large placebo-controlled RCT,21 2 smaller open studies,18,19 and a post hoc analysis of a large placebo-controlled RCT in mixed-age adults20 support the efficacy and relatively good tolerability of aripiprazole augmentation of an antidepressant for treatment-resistant MDD. Similarly, 1 large placebo-controlled RCT supports the efficacy and relatively good tolerability of quetiapine for non–treatment-resistant MDD. However, there are no comparative data assessing the relative merits of using these antipsychotics vs other pharmacologic strategies (eg, switching to another antidepressant, lithium augmentation, or combination of 2 antidepressants). Because older patients are more likely to experience adverse effects that may have more serious consequences (Table 3), many prudent clinicians reserve using antipsychotics as a third-line treatment in older patients with MDD without psychotic features and limit the duration of their use to a few months.30
Although some data have accumulated in recent years, there are significant gaps in knowledge on the safety and tolerability of antipsychotics in older adults. The era of “big data” may provide important answers to questions such as the relative place of antipsychotics vs lithium in preserving brain health among people with bipolar disorder or treatment-resistant MDD31; whether there are true ethnic differences in terms of drugs response and adverse effect prevalence in antipsychotics32,33; or the role of pharmacogenetic evaluation in establishing individual risk–benefit ratios of antipsychotics.34
1. United Nations. Department of Economic and Social Affairs Population Division. World Population Ageing: 1950-2050. http://www.un.org/esa/population/publications/worldageing19502050. Published 2001. Accessed September 27, 2017.
2. Olfson M, King M, Schoenbaum M. Antipsychotic treatment of adults in the United States. J Clin Psychiatry. 2015;76(10):1346-1353.
3. Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
4. Hybels CF, Blazer DG. Epidemiology of late-life mental disorders. Clin Geriatr Med. 2003;19(4):663-696,v.
5. Mulsant BH, Blumberger DM, Ismail Z, et al. A systematic approach to pharmacotherapy for geriatric major depression. Clin Geriatr Med. 2014;30(3):517-534.
6. Mulsant BH, Pollock BG. Psychopharmacology. In: Steffens DC, Blazer DG, Thakur ME, eds. The American Psychiatric Publishing textbook of geriatric psychiatry. 5th ed. Arlington, VA: American Psychiatric Publishing; 2015:527-587.
7. U.S. Food and Drug Administration. Public health advisory. deaths with antipsychotics in elderly patients with behavioral disturbances. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Updated August 16, 2013. Accessed September 27, 2017.
8. Andreescu C, Mulsant BH, Rothschild AJ, et al. Pharmacotherapy of major depression with psychotic features: what is the evidence? Psychiatric Annals. 2006;35(1):31-38.
9. Buchanan D, Tourigny-Rivard MF, Cappeliez P, et al. National guidelines for seniors’ mental health: the assessment and treatment of depression. Canadian Journal of Geriatrics. 2006;9(suppl 2):S52-S58.
10. Canadian Coalition for Senior’s Mental Health. National guidelines for senior’s mental health. The assessment and treatment of depression 2006. http://www.ccsmh.ca/projects/depression. Accessed February 28, 2016.
11. Meyers BS, Flint AJ, Rothschild AJ, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy of psychotic depression (STOP-PD). Arch Gen Psychiatry. 2009;66(8):838-847.
12. Mulsant BH, Sweet RA, Rosen J, et al. A double-blind randomized comparison of nortriptyline plus perphenazine versus nortriptyline plus placebo in the treatment of psychotic depression in late life. J Clin Psychiatry. 2001;62(8):597-604.
13. Cakir S, Senkal Z. Atypical antipsychotics as add-on treatment in late-life depression. Clin Interv Aging. 2016;11:1193-1198.
14. Maust DT, Oslin DW, Thase ME. Going beyond antidepressant monotherapy for incomplete response in nonpsychotic late-life depression: a critical review. Am J Geriatr Psychiatry. 2013;21(10):973-986.
15. Patel K, Abdool PS, Rajji TK, et al. Pharmacotherapy of major depression in late life: what is the role of new agents? Expert Opin Pharmacother. 2017;18(6):599-609.
16. Alexopoulos GS, Katz IR, Reynolds CF 3rd, et al. Pharmacotherapy of depression in older patients: a summary of the expert consensus guidelines. J Psychiatr Pract. 2001;7(6):361-376.
17. Cooper C, Katona C, Lyketsos K, et al. A systematic review of treatments for refractory depression in older people. Am J Psychiatry. 2011;168(7):681-688.
18. Rutherford B, Sneed J, Miyazaki M, et al. An open trial of aripiprazole augmentation for SSRI non-remitters with late-life depression. Int J Geriatr Psychiatry. 2007;22(10):986-991.
19. Sheffrin M, Driscoll HC, Lenze EJ, et al. Pilot study of augmentation with aripiprazole for incomplete response in late-life depression: getting to remission. J Clin Psychiatry. 2009;70(2):208-213.
20. Steffens DC, Nelson JC, Eudicone JM, et al. Efficacy and safety of adjunctive aripiprazole in major depressive disorder in older patients: a pooled subpopulation analysis. Int J Geriatr Psychiatry. 2011;26(6):564-572.
21. Lenze EJ, Mulsant BH, Blumberger DM, et al. Efficacy, safety, and tolerability of augmentation pharmacotherapy with aripiprazole for treatment-resistant depression in late life: a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(10011):2404-2412.
22. Deligiannidis KM, Rothschild AJ, Barton BA, et al. A gender analysis of the study of pharmacotherapy of psychotic depression (STOP-PD): gender and age as predictors of response and treatment-associated changes in body mass index and metabolic measures. J Clin Psychiatry. 2013;74(10):1003-1009.
23. Flint AJ, Iaboni A, Mulsant BH, et al. Effect of sertraline on risk of falling in older adults with psychotic depression on olanzapine: results of a randomized placebo-controlled trial. Am J Geriatr Psychiatry. 2014;22(4):332-336.
24. Smith E, Rothschild AJ, Heo M, et al. Weight gain during olanzapine treatment for psychotic depression: effects of dose and age. Int Clin Psychopharmacol. 2008;23(3):130-137.
25. Blumberger DM, Mulsant BH, Kanellopoulos D, et al. The incidence of tardive dyskinesia in the study of pharmacotherapy for psychotic depression. J Clin Psychopharmacol. 2013;33(3):391-397.
26. Alexopoulos GS, Canuso CM, Gharabawi GM, et al. Placebo-controlled study of relapse prevention with risperidone augmentation in older patients with resistant depression. Am J Geriatr Psychiatry. 2008;16(1):21-30.
27. Katila H, Mezhebovsky I, Mulroy A, et al. Randomized, double-blind study of the efficacy and tolerability of extended release quetiapine fumarate (quetiapine XR) monotherapy in elderly patients with major depressive disorder. Am J Geriatr Psychiatry. 2013;21(8):769-784.
28. Sultana J, Trifiro G. Drug safety warnings: a message in a bottle. J Drug Des Res. 2014;1(1):1004.
29. Flint A, Meyers BS, Rothschild AR, et al; STOP-PD II Study Group. Sustaining remission of psychotic depression: rationale, design and methodology of STOP-PD II. BMC Psychiatry. 2013;13:38.
30. Alexopoulos GS; PROSPECT Group. Interventions for depressed elderly primary care patients. Int J Geriatr Psychiatry. 2001;16(6):553-559.
31. Sajatovic M, Forester BP, Gildengers A, et al. Aging changes and medical complexity in late-life bipolar disorder: emerging research findings that may help advance care. Neuropsychiatry (London). 2013;3(6):621-633.
32. Bigos KL, Bies RR, Pollock BG, et al. Genetic variation in CYP3A43 explains racial difference in olanzapine clearance. Mol Psychiatry. 2011;16(6):620-625.
33. Jin Y, Pollock BG, Coley K, et al. Population pharmacokinetics of perphenazine in schizophrenia patients from CATIE: impact of race and smoking. J Clin Pharmacol. 2010;50(1):73-80.
34. Mulsant BH. Is there a role for antidepressant and antipsychotic pharmacogenetics in clinical practice in 2014? Can J Psychiatry. 2014;59(2):59-61.
The proportion of older adults in the world population is growing rapidly. In the next 10 to 15 years, the population age >60 will grow 3.5 times more rapidly than the general population.1 As a result, there is an increased urgency in examining benefits vs risks of antipsychotics in older individuals. In a 2010 U.S. nationally representative observational study, antipsychotic use was observed to rise slowly during early and middle adulthood, peaking at approximately age 55, declining slightly between ages 55 and 65, and then rising again after age 65, with >2% of individuals ages 80 to 84 receiving an antipsychotic.2 This is likely due to the chronology of psychotic, mood, and neurocognitive disorders across the life span. In this large national study, long-term antipsychotic treatment was common, and older patients were more likely to receive their prescriptions from non-psychiatrist physicians than from psychiatrists.2 Among patients receiving an antipsychotic, the proportion of those receiving it for >120 days was 54% for individuals ages 70 to 74; 49% for individuals ages 75 to 79; and 46% for individuals ages 80 to 84.
This 3-part review summarizes findings and risk–benefit considerations when prescribing antipsychotics to older individuals. Part 1 focused on those with chronic psychotic disorders, such as schizophrenia or bipolar disorder,3 and part 3 will cover patients with dementia. This review (part 2) aims to:
- briefly summarize the results of randomized controlled trials (RCTs) of second-generation antipsychotics (SGAs) and other major studies and analyses in older patients with major depressive disorder (MDD)
- provide a summative opinion on the relative risks and benefits associated with using antipsychotics in older adults with MDD
- highlight the gaps in the evidence base and areas that need additional research.
Summary of benefits, place in treatment armamentarium
The prevalence of MDD and clinically significant depressive symptoms in communitydwelling older adults is 3% to 4% and 15%, respectively, and as high as 16% and 50%, respectively, in nursing home residents.4 Because late-life depression is associated with suffering, disability, and excessive mortality, it needs to be recognized and treated aggressively.5 Antidepressants are the mainstay of pharmacotherapy for late-life depression. Guidelines and expert opinion informed by the current evidence recommend using selective serotonin reuptake inhibitors, such as
Over the past decade, several antipsychotics have been FDA-approved for treating MDD:
Data from several rigorously conducted RCTs support using an antidepressant plus an FGA or SGA as first-line pharmacotherapy in younger and older patients with “psychotic depression.”8-12 SGAs also can be used as augmenting agents when there is only a partial response to antidepressants.13-15 In this situation, guidelines and experts favor an augmentation strategy over switching to another antidepressant.5,9,10,16 Until recently, most published pharmacologic trials for late-life treatment-resistant depression supported using lithium to augment antidepressants.14,17 However, because several antipsychotics are now FDA-approved for treating MDD, and in light of positive findings from several studies relevant to older patients,18-21 many experts now support using SGAs to augment antidepressants in older patients with nonpsychotic depression.5,15
Clinical trials
Olanzapine plus sertraline as first-line pharmacotherapy for MDD with psychotic features. Meyers et al11 reported on a double-blind randomized comparison of olanzapine plus placebo vs olanzapine plus sertraline in 259 patients with MDD with psychotic features. An unusual feature of this trial is that it included a similar number of younger and older participants (ages 18 to 93): 117 participants were age <60 (mean age [standard deviation (SD)]: 41.3 [10.8]) and 142 were age ≥60 (mean age [SD]: 71.7 [7.8]). The same dose titration schedules based on efficacy and tolerability were used in both younger and older participants. At the end of the study, the mean dose (SD) of sertraline (or placebo) did not differ significantly in younger (174.3 mg/d [34.1]) and older participants (165.7 mg/d [43.4]). However, the mean dose (SD) of olanzapine was significantly higher in younger patients (15.7 mg/d [4.7]) than in older participants (13.4 mg/d [5.1]).
In both age groups, olanzapine plus sertraline was more efficacious than olanzapine plus placebo, and there was no statistical interaction between age, time, and treatment group (ie, the trajectories of improvement were similar in older and younger patients receiving either olanzapine or olanzapine plus sertraline). Similarly, drop-out rates because of poor tolerability did not differ significantly in younger (4.3%) and older participants (5.6%). However, in a multinomial regression, older participants were more likely to discontinue treatment because of poor tolerability.22 Older participants were significantly less likely to experience weight gain (mean [SD]: +3.3 [4.9] vs +6.5 [6.6] kg) or an increase in fasting glucose and more likely to experience a fall, pedal edema, or extrapyramidal symptoms.11,22-24 Cholesterol and triglyceride increased significantly and similarly in both age groups. The incidence of symptoms of tardive dyskinesia (TD) over the 12-week trial was low (<5%) in both younger and older participants, and clinically diagnosed TD was reported in only 1 (older) participant.25
Venlafaxine plus aripiprazole for treatment-resistant MDD. In the largest double-blind randomized study of augmentation pharmacotherapy for late-life treatment-resistant depression published to date, Lenze et al21 compared venlafaxine plus aripiprazole vs venlafaxine plus placebo in 181 patients age >60 (mean age 66, with 49 participants age >70) with MDD who did not remit after 12 weeks of treatment with venlafaxine (up to 300 mg/d). After 12 weeks of augmentation, remission rates were significantly higher with aripiprazole than with placebo: 40 (44%) vs 26 (29%); odds ratio (95% confidence interval [CI]): 2.0 (1.1 to 3.7). The median final aripiprazole dose was 7 mg/d (range 2 to 15 mg/d) in remitters and 10 mg/d (range 2 to 15 mg/d) in nonremitters.
Five of 90 participants (5%) discontinued aripiprazole (1 each: suicide, jitteriness/akathisia, worsening parkinsonism; and 2 withdrew consent); 8 of 90 (9%) discontinued placebo (2 each: lack of efficacy, headache; 1: worsening parkinsonism; and 3 withdrew consent). The completed suicide occurred after 5 weeks of treatment with aripiprazole and was judged to be “neither due to emergent suicidal ideation nor to aripiprazole side-effects, but was concluded by investigators to be a result of the individual’s persisting and long-standing suicidal ideation.”21 Including the suicide, there were 4 serious adverse events (5%) in those receiving aripiprazole (1 each: suicide, congestive heart failure, mild stroke, and diverticulitis) and 2 (2%) in those receiving placebo (1 each: myocardial infarction, hospitalized for vomiting due to accidentally taking extra venlafaxine). In 86 participants receiving aripiprazole and 87 receiving placebo, the most frequently reported adverse effects were increased dream activity (aripiprazole: 23 [27%] vs placebo: 12 [14%]), weight gain (17 [20%] vs 8 [9%]), and tremor (5 [6%] vs 0). Akathisia and parkinsonism were observed more frequently with aripiprazole than with placebo (akathisia: 24 [26%] of 91 vs 11 [12%] of 90; parkinsonism: 15 [17%] of 86 vs 2 [2%] of 81). Akathisia was generally mild and resolved with dose adjustment; however, it was associated with a transient increase in suicidality in 3 (3%) participants receiving aripiprazole vs 0 receiving placebo and persisted at the end of the trial in 5 (5%) participants receiving aripiprazole vs 2 (2%) receiving placebo. Participants receiving aripiprazole had a significantly larger increase in weight (mean [SD]: +1.93 [3.00] vs +0.01 [3.15] kg), but there were no differences between aripiprazole and placebo in changes in body fat, total cholesterol, high-density lipoprotein, low-density lipoprotein, triglycerides, glucose, insulin concentration, or QTc.
Citalopram plus risperidone for treatment-resistant MDD. Alexopoulos et al26 reported an analysis of data from 110 patients age ≥55 years (mean age [SD]: 63.4 [4.8]), among 489 mixed-age patients with MDD. Participants (n = 110) who did not respond to 1 to 3 antidepressants (venlafaxine, sertraline, mirtazapine, fluoxetine, paroxetine, or bupropion in >90%) during their current depressive episode completed 4 to 6 weeks of treatment with citalopram up to 40 mg/d; 93 did not respond and were treated with open-label risperidone (0.25 to 1 mg/d) augmentation for 4 to 6 weeks. Sixty-three (68%) of these 93 patients remitted and were randomized to 24 weeks of double-blind continuation treatment with citalopram plus risperidone vs citalopram plus placebo. Neither the median times to relapse (105 vs 57 days) nor the relapse rates (risperidone: 18 of 32 [56%] vs placebo: 20 of 31 [65%]) differed significantly. During the open-label risperidone augmentation, the most common adverse events were dizziness and dry mouth (n = 9 each, 9.7% of 93). During the continuation phase, headache (n = 3; 9.1% of 32) was observed with risperidone but not with placebo (n = 0). There was no incident parkinsonism or abnormal movements noted, but risperidone was associated with weight gain during both the open-label risperidone augmentation phase (mean [SD]: +0.9 [2.1] kg) and the continuation phase (risperidone: +0.8 [3.5] vs placebo: −0.3 [2.8] kg).
Quetiapine XR monotherapy for MDD. Katila et al27 reported on a placebo-controlled RCT of quetiapine XR (median dose, 158.7 mg/d; range, 50 to 300 mg/d) in 338 patients age ≥66 years (mean age [SD], 71.3 [7.5]) presenting with MDD and a major depressive episode with a duration <1 year and no history of failed antidepressants trials from 2 classes (more than two-thirds of participants had not received treatment). After 9 weeks, the reduction in depressive symptoms on the Montgomery-Åsberg Depression Rating Scale was significantly larger with quetiapine XR than with placebo (mean [SD]: −16.0 [9.3] vs −9.0 [9.9]). There were congruent, significant differences between quetiapine and placebo in terms of response rate (quetiapine XR: 105 of 164 [64%] vs placebo: 52 of 171 [30.4%]) and remission rate (92 of 164 [56.1%] vs placebo: 40 of 171 [23.4%]). The drop-out rates for all causes were similar, but the drop-out rate attributed to adverse events was higher with quetiapine than placebo (16 of 166 [9.6%] vs 7 of 172 [4.1%]). Most quetiapine drop-outs were attributable to dizziness, headache, and somnolence (n = 4 each), and placebo drop-outs were because of headache (n = 2). Consistent with the profile of quetiapine, adverse events with a rate that was at least 5% higher with quetiapine than with placebo included somnolence (64 of 166 [38.6%] vs 16 of 172 [9.3%]), dry mouth (34 [20.5%] vs 18 [10.5%]), and extrapyramidal symptoms (12 [7.2%] vs 4 [2.3%]). Changes in weight and laboratory test results (eg, glucose, lipid profile) were minimal and not clinically meaningful.
Other clinical data. The efficacy and relatively good tolerability of aripiprazole in older patients with treatment-resistant depression observed in the RCT by Lenze et al21 is congruent with the earlier results of 2 small (N = 20 and 24) pilot studies.18,19 In both studies, the remission rate was 50%, and the most prevalent adverse effects were agitation/restlessness/akathisia or drowsiness/sedation. Similarly, in a post hoc pooled analysis of 409 participants ages 50 to 67 from 3 placebo-controlled randomized trials, the remission rate was significantly higher with aripiprazole than with placebo (32.5% vs 17.1%), and the most common adverse effects were akathisia or restlessness (64 of 210 [30.4%]), somnolence (18 [8.6%]), and insomnia (17 [8.1%]).20
Clinical considerations
When assessing the relative benefits and risks of antipsychotics in older patients, it is important to remember that conclusions and summative opinions are necessarily influenced by the source of the data. Because much of what we know about the use of antipsychotics in geriatric adults is from clinical trials, we know more about their acute efficacy and tolerability than their long-term effectiveness and safety.28 There are similar issues regarding the role of antipsychotics in treating MDD in late life. Based on the results of several RCTs,8,11 a combination of an antidepressant plus an antipsychotic is the recommended pharmacotherapy for the acute treatment of MDD with psychotic features (Table 2).8,11,12,19-21,23-27 However, there are no published data to guide how long the antipsychotic should be continued.29
In older patients with MDD without psychotic features, 1 relatively large placebo-controlled RCT,21 2 smaller open studies,18,19 and a post hoc analysis of a large placebo-controlled RCT in mixed-age adults20 support the efficacy and relatively good tolerability of aripiprazole augmentation of an antidepressant for treatment-resistant MDD. Similarly, 1 large placebo-controlled RCT supports the efficacy and relatively good tolerability of quetiapine for non–treatment-resistant MDD. However, there are no comparative data assessing the relative merits of using these antipsychotics vs other pharmacologic strategies (eg, switching to another antidepressant, lithium augmentation, or combination of 2 antidepressants). Because older patients are more likely to experience adverse effects that may have more serious consequences (Table 3), many prudent clinicians reserve using antipsychotics as a third-line treatment in older patients with MDD without psychotic features and limit the duration of their use to a few months.30
Although some data have accumulated in recent years, there are significant gaps in knowledge on the safety and tolerability of antipsychotics in older adults. The era of “big data” may provide important answers to questions such as the relative place of antipsychotics vs lithium in preserving brain health among people with bipolar disorder or treatment-resistant MDD31; whether there are true ethnic differences in terms of drugs response and adverse effect prevalence in antipsychotics32,33; or the role of pharmacogenetic evaluation in establishing individual risk–benefit ratios of antipsychotics.34
The proportion of older adults in the world population is growing rapidly. In the next 10 to 15 years, the population age >60 will grow 3.5 times more rapidly than the general population.1 As a result, there is an increased urgency in examining benefits vs risks of antipsychotics in older individuals. In a 2010 U.S. nationally representative observational study, antipsychotic use was observed to rise slowly during early and middle adulthood, peaking at approximately age 55, declining slightly between ages 55 and 65, and then rising again after age 65, with >2% of individuals ages 80 to 84 receiving an antipsychotic.2 This is likely due to the chronology of psychotic, mood, and neurocognitive disorders across the life span. In this large national study, long-term antipsychotic treatment was common, and older patients were more likely to receive their prescriptions from non-psychiatrist physicians than from psychiatrists.2 Among patients receiving an antipsychotic, the proportion of those receiving it for >120 days was 54% for individuals ages 70 to 74; 49% for individuals ages 75 to 79; and 46% for individuals ages 80 to 84.
This 3-part review summarizes findings and risk–benefit considerations when prescribing antipsychotics to older individuals. Part 1 focused on those with chronic psychotic disorders, such as schizophrenia or bipolar disorder,3 and part 3 will cover patients with dementia. This review (part 2) aims to:
- briefly summarize the results of randomized controlled trials (RCTs) of second-generation antipsychotics (SGAs) and other major studies and analyses in older patients with major depressive disorder (MDD)
- provide a summative opinion on the relative risks and benefits associated with using antipsychotics in older adults with MDD
- highlight the gaps in the evidence base and areas that need additional research.
Summary of benefits, place in treatment armamentarium
The prevalence of MDD and clinically significant depressive symptoms in communitydwelling older adults is 3% to 4% and 15%, respectively, and as high as 16% and 50%, respectively, in nursing home residents.4 Because late-life depression is associated with suffering, disability, and excessive mortality, it needs to be recognized and treated aggressively.5 Antidepressants are the mainstay of pharmacotherapy for late-life depression. Guidelines and expert opinion informed by the current evidence recommend using selective serotonin reuptake inhibitors, such as
Over the past decade, several antipsychotics have been FDA-approved for treating MDD:
Data from several rigorously conducted RCTs support using an antidepressant plus an FGA or SGA as first-line pharmacotherapy in younger and older patients with “psychotic depression.”8-12 SGAs also can be used as augmenting agents when there is only a partial response to antidepressants.13-15 In this situation, guidelines and experts favor an augmentation strategy over switching to another antidepressant.5,9,10,16 Until recently, most published pharmacologic trials for late-life treatment-resistant depression supported using lithium to augment antidepressants.14,17 However, because several antipsychotics are now FDA-approved for treating MDD, and in light of positive findings from several studies relevant to older patients,18-21 many experts now support using SGAs to augment antidepressants in older patients with nonpsychotic depression.5,15
Clinical trials
Olanzapine plus sertraline as first-line pharmacotherapy for MDD with psychotic features. Meyers et al11 reported on a double-blind randomized comparison of olanzapine plus placebo vs olanzapine plus sertraline in 259 patients with MDD with psychotic features. An unusual feature of this trial is that it included a similar number of younger and older participants (ages 18 to 93): 117 participants were age <60 (mean age [standard deviation (SD)]: 41.3 [10.8]) and 142 were age ≥60 (mean age [SD]: 71.7 [7.8]). The same dose titration schedules based on efficacy and tolerability were used in both younger and older participants. At the end of the study, the mean dose (SD) of sertraline (or placebo) did not differ significantly in younger (174.3 mg/d [34.1]) and older participants (165.7 mg/d [43.4]). However, the mean dose (SD) of olanzapine was significantly higher in younger patients (15.7 mg/d [4.7]) than in older participants (13.4 mg/d [5.1]).
In both age groups, olanzapine plus sertraline was more efficacious than olanzapine plus placebo, and there was no statistical interaction between age, time, and treatment group (ie, the trajectories of improvement were similar in older and younger patients receiving either olanzapine or olanzapine plus sertraline). Similarly, drop-out rates because of poor tolerability did not differ significantly in younger (4.3%) and older participants (5.6%). However, in a multinomial regression, older participants were more likely to discontinue treatment because of poor tolerability.22 Older participants were significantly less likely to experience weight gain (mean [SD]: +3.3 [4.9] vs +6.5 [6.6] kg) or an increase in fasting glucose and more likely to experience a fall, pedal edema, or extrapyramidal symptoms.11,22-24 Cholesterol and triglyceride increased significantly and similarly in both age groups. The incidence of symptoms of tardive dyskinesia (TD) over the 12-week trial was low (<5%) in both younger and older participants, and clinically diagnosed TD was reported in only 1 (older) participant.25
Venlafaxine plus aripiprazole for treatment-resistant MDD. In the largest double-blind randomized study of augmentation pharmacotherapy for late-life treatment-resistant depression published to date, Lenze et al21 compared venlafaxine plus aripiprazole vs venlafaxine plus placebo in 181 patients age >60 (mean age 66, with 49 participants age >70) with MDD who did not remit after 12 weeks of treatment with venlafaxine (up to 300 mg/d). After 12 weeks of augmentation, remission rates were significantly higher with aripiprazole than with placebo: 40 (44%) vs 26 (29%); odds ratio (95% confidence interval [CI]): 2.0 (1.1 to 3.7). The median final aripiprazole dose was 7 mg/d (range 2 to 15 mg/d) in remitters and 10 mg/d (range 2 to 15 mg/d) in nonremitters.
Five of 90 participants (5%) discontinued aripiprazole (1 each: suicide, jitteriness/akathisia, worsening parkinsonism; and 2 withdrew consent); 8 of 90 (9%) discontinued placebo (2 each: lack of efficacy, headache; 1: worsening parkinsonism; and 3 withdrew consent). The completed suicide occurred after 5 weeks of treatment with aripiprazole and was judged to be “neither due to emergent suicidal ideation nor to aripiprazole side-effects, but was concluded by investigators to be a result of the individual’s persisting and long-standing suicidal ideation.”21 Including the suicide, there were 4 serious adverse events (5%) in those receiving aripiprazole (1 each: suicide, congestive heart failure, mild stroke, and diverticulitis) and 2 (2%) in those receiving placebo (1 each: myocardial infarction, hospitalized for vomiting due to accidentally taking extra venlafaxine). In 86 participants receiving aripiprazole and 87 receiving placebo, the most frequently reported adverse effects were increased dream activity (aripiprazole: 23 [27%] vs placebo: 12 [14%]), weight gain (17 [20%] vs 8 [9%]), and tremor (5 [6%] vs 0). Akathisia and parkinsonism were observed more frequently with aripiprazole than with placebo (akathisia: 24 [26%] of 91 vs 11 [12%] of 90; parkinsonism: 15 [17%] of 86 vs 2 [2%] of 81). Akathisia was generally mild and resolved with dose adjustment; however, it was associated with a transient increase in suicidality in 3 (3%) participants receiving aripiprazole vs 0 receiving placebo and persisted at the end of the trial in 5 (5%) participants receiving aripiprazole vs 2 (2%) receiving placebo. Participants receiving aripiprazole had a significantly larger increase in weight (mean [SD]: +1.93 [3.00] vs +0.01 [3.15] kg), but there were no differences between aripiprazole and placebo in changes in body fat, total cholesterol, high-density lipoprotein, low-density lipoprotein, triglycerides, glucose, insulin concentration, or QTc.
Citalopram plus risperidone for treatment-resistant MDD. Alexopoulos et al26 reported an analysis of data from 110 patients age ≥55 years (mean age [SD]: 63.4 [4.8]), among 489 mixed-age patients with MDD. Participants (n = 110) who did not respond to 1 to 3 antidepressants (venlafaxine, sertraline, mirtazapine, fluoxetine, paroxetine, or bupropion in >90%) during their current depressive episode completed 4 to 6 weeks of treatment with citalopram up to 40 mg/d; 93 did not respond and were treated with open-label risperidone (0.25 to 1 mg/d) augmentation for 4 to 6 weeks. Sixty-three (68%) of these 93 patients remitted and were randomized to 24 weeks of double-blind continuation treatment with citalopram plus risperidone vs citalopram plus placebo. Neither the median times to relapse (105 vs 57 days) nor the relapse rates (risperidone: 18 of 32 [56%] vs placebo: 20 of 31 [65%]) differed significantly. During the open-label risperidone augmentation, the most common adverse events were dizziness and dry mouth (n = 9 each, 9.7% of 93). During the continuation phase, headache (n = 3; 9.1% of 32) was observed with risperidone but not with placebo (n = 0). There was no incident parkinsonism or abnormal movements noted, but risperidone was associated with weight gain during both the open-label risperidone augmentation phase (mean [SD]: +0.9 [2.1] kg) and the continuation phase (risperidone: +0.8 [3.5] vs placebo: −0.3 [2.8] kg).
Quetiapine XR monotherapy for MDD. Katila et al27 reported on a placebo-controlled RCT of quetiapine XR (median dose, 158.7 mg/d; range, 50 to 300 mg/d) in 338 patients age ≥66 years (mean age [SD], 71.3 [7.5]) presenting with MDD and a major depressive episode with a duration <1 year and no history of failed antidepressants trials from 2 classes (more than two-thirds of participants had not received treatment). After 9 weeks, the reduction in depressive symptoms on the Montgomery-Åsberg Depression Rating Scale was significantly larger with quetiapine XR than with placebo (mean [SD]: −16.0 [9.3] vs −9.0 [9.9]). There were congruent, significant differences between quetiapine and placebo in terms of response rate (quetiapine XR: 105 of 164 [64%] vs placebo: 52 of 171 [30.4%]) and remission rate (92 of 164 [56.1%] vs placebo: 40 of 171 [23.4%]). The drop-out rates for all causes were similar, but the drop-out rate attributed to adverse events was higher with quetiapine than placebo (16 of 166 [9.6%] vs 7 of 172 [4.1%]). Most quetiapine drop-outs were attributable to dizziness, headache, and somnolence (n = 4 each), and placebo drop-outs were because of headache (n = 2). Consistent with the profile of quetiapine, adverse events with a rate that was at least 5% higher with quetiapine than with placebo included somnolence (64 of 166 [38.6%] vs 16 of 172 [9.3%]), dry mouth (34 [20.5%] vs 18 [10.5%]), and extrapyramidal symptoms (12 [7.2%] vs 4 [2.3%]). Changes in weight and laboratory test results (eg, glucose, lipid profile) were minimal and not clinically meaningful.
Other clinical data. The efficacy and relatively good tolerability of aripiprazole in older patients with treatment-resistant depression observed in the RCT by Lenze et al21 is congruent with the earlier results of 2 small (N = 20 and 24) pilot studies.18,19 In both studies, the remission rate was 50%, and the most prevalent adverse effects were agitation/restlessness/akathisia or drowsiness/sedation. Similarly, in a post hoc pooled analysis of 409 participants ages 50 to 67 from 3 placebo-controlled randomized trials, the remission rate was significantly higher with aripiprazole than with placebo (32.5% vs 17.1%), and the most common adverse effects were akathisia or restlessness (64 of 210 [30.4%]), somnolence (18 [8.6%]), and insomnia (17 [8.1%]).20
Clinical considerations
When assessing the relative benefits and risks of antipsychotics in older patients, it is important to remember that conclusions and summative opinions are necessarily influenced by the source of the data. Because much of what we know about the use of antipsychotics in geriatric adults is from clinical trials, we know more about their acute efficacy and tolerability than their long-term effectiveness and safety.28 There are similar issues regarding the role of antipsychotics in treating MDD in late life. Based on the results of several RCTs,8,11 a combination of an antidepressant plus an antipsychotic is the recommended pharmacotherapy for the acute treatment of MDD with psychotic features (Table 2).8,11,12,19-21,23-27 However, there are no published data to guide how long the antipsychotic should be continued.29
In older patients with MDD without psychotic features, 1 relatively large placebo-controlled RCT,21 2 smaller open studies,18,19 and a post hoc analysis of a large placebo-controlled RCT in mixed-age adults20 support the efficacy and relatively good tolerability of aripiprazole augmentation of an antidepressant for treatment-resistant MDD. Similarly, 1 large placebo-controlled RCT supports the efficacy and relatively good tolerability of quetiapine for non–treatment-resistant MDD. However, there are no comparative data assessing the relative merits of using these antipsychotics vs other pharmacologic strategies (eg, switching to another antidepressant, lithium augmentation, or combination of 2 antidepressants). Because older patients are more likely to experience adverse effects that may have more serious consequences (Table 3), many prudent clinicians reserve using antipsychotics as a third-line treatment in older patients with MDD without psychotic features and limit the duration of their use to a few months.30
Although some data have accumulated in recent years, there are significant gaps in knowledge on the safety and tolerability of antipsychotics in older adults. The era of “big data” may provide important answers to questions such as the relative place of antipsychotics vs lithium in preserving brain health among people with bipolar disorder or treatment-resistant MDD31; whether there are true ethnic differences in terms of drugs response and adverse effect prevalence in antipsychotics32,33; or the role of pharmacogenetic evaluation in establishing individual risk–benefit ratios of antipsychotics.34
1. United Nations. Department of Economic and Social Affairs Population Division. World Population Ageing: 1950-2050. http://www.un.org/esa/population/publications/worldageing19502050. Published 2001. Accessed September 27, 2017.
2. Olfson M, King M, Schoenbaum M. Antipsychotic treatment of adults in the United States. J Clin Psychiatry. 2015;76(10):1346-1353.
3. Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
4. Hybels CF, Blazer DG. Epidemiology of late-life mental disorders. Clin Geriatr Med. 2003;19(4):663-696,v.
5. Mulsant BH, Blumberger DM, Ismail Z, et al. A systematic approach to pharmacotherapy for geriatric major depression. Clin Geriatr Med. 2014;30(3):517-534.
6. Mulsant BH, Pollock BG. Psychopharmacology. In: Steffens DC, Blazer DG, Thakur ME, eds. The American Psychiatric Publishing textbook of geriatric psychiatry. 5th ed. Arlington, VA: American Psychiatric Publishing; 2015:527-587.
7. U.S. Food and Drug Administration. Public health advisory. deaths with antipsychotics in elderly patients with behavioral disturbances. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Updated August 16, 2013. Accessed September 27, 2017.
8. Andreescu C, Mulsant BH, Rothschild AJ, et al. Pharmacotherapy of major depression with psychotic features: what is the evidence? Psychiatric Annals. 2006;35(1):31-38.
9. Buchanan D, Tourigny-Rivard MF, Cappeliez P, et al. National guidelines for seniors’ mental health: the assessment and treatment of depression. Canadian Journal of Geriatrics. 2006;9(suppl 2):S52-S58.
10. Canadian Coalition for Senior’s Mental Health. National guidelines for senior’s mental health. The assessment and treatment of depression 2006. http://www.ccsmh.ca/projects/depression. Accessed February 28, 2016.
11. Meyers BS, Flint AJ, Rothschild AJ, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy of psychotic depression (STOP-PD). Arch Gen Psychiatry. 2009;66(8):838-847.
12. Mulsant BH, Sweet RA, Rosen J, et al. A double-blind randomized comparison of nortriptyline plus perphenazine versus nortriptyline plus placebo in the treatment of psychotic depression in late life. J Clin Psychiatry. 2001;62(8):597-604.
13. Cakir S, Senkal Z. Atypical antipsychotics as add-on treatment in late-life depression. Clin Interv Aging. 2016;11:1193-1198.
14. Maust DT, Oslin DW, Thase ME. Going beyond antidepressant monotherapy for incomplete response in nonpsychotic late-life depression: a critical review. Am J Geriatr Psychiatry. 2013;21(10):973-986.
15. Patel K, Abdool PS, Rajji TK, et al. Pharmacotherapy of major depression in late life: what is the role of new agents? Expert Opin Pharmacother. 2017;18(6):599-609.
16. Alexopoulos GS, Katz IR, Reynolds CF 3rd, et al. Pharmacotherapy of depression in older patients: a summary of the expert consensus guidelines. J Psychiatr Pract. 2001;7(6):361-376.
17. Cooper C, Katona C, Lyketsos K, et al. A systematic review of treatments for refractory depression in older people. Am J Psychiatry. 2011;168(7):681-688.
18. Rutherford B, Sneed J, Miyazaki M, et al. An open trial of aripiprazole augmentation for SSRI non-remitters with late-life depression. Int J Geriatr Psychiatry. 2007;22(10):986-991.
19. Sheffrin M, Driscoll HC, Lenze EJ, et al. Pilot study of augmentation with aripiprazole for incomplete response in late-life depression: getting to remission. J Clin Psychiatry. 2009;70(2):208-213.
20. Steffens DC, Nelson JC, Eudicone JM, et al. Efficacy and safety of adjunctive aripiprazole in major depressive disorder in older patients: a pooled subpopulation analysis. Int J Geriatr Psychiatry. 2011;26(6):564-572.
21. Lenze EJ, Mulsant BH, Blumberger DM, et al. Efficacy, safety, and tolerability of augmentation pharmacotherapy with aripiprazole for treatment-resistant depression in late life: a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(10011):2404-2412.
22. Deligiannidis KM, Rothschild AJ, Barton BA, et al. A gender analysis of the study of pharmacotherapy of psychotic depression (STOP-PD): gender and age as predictors of response and treatment-associated changes in body mass index and metabolic measures. J Clin Psychiatry. 2013;74(10):1003-1009.
23. Flint AJ, Iaboni A, Mulsant BH, et al. Effect of sertraline on risk of falling in older adults with psychotic depression on olanzapine: results of a randomized placebo-controlled trial. Am J Geriatr Psychiatry. 2014;22(4):332-336.
24. Smith E, Rothschild AJ, Heo M, et al. Weight gain during olanzapine treatment for psychotic depression: effects of dose and age. Int Clin Psychopharmacol. 2008;23(3):130-137.
25. Blumberger DM, Mulsant BH, Kanellopoulos D, et al. The incidence of tardive dyskinesia in the study of pharmacotherapy for psychotic depression. J Clin Psychopharmacol. 2013;33(3):391-397.
26. Alexopoulos GS, Canuso CM, Gharabawi GM, et al. Placebo-controlled study of relapse prevention with risperidone augmentation in older patients with resistant depression. Am J Geriatr Psychiatry. 2008;16(1):21-30.
27. Katila H, Mezhebovsky I, Mulroy A, et al. Randomized, double-blind study of the efficacy and tolerability of extended release quetiapine fumarate (quetiapine XR) monotherapy in elderly patients with major depressive disorder. Am J Geriatr Psychiatry. 2013;21(8):769-784.
28. Sultana J, Trifiro G. Drug safety warnings: a message in a bottle. J Drug Des Res. 2014;1(1):1004.
29. Flint A, Meyers BS, Rothschild AR, et al; STOP-PD II Study Group. Sustaining remission of psychotic depression: rationale, design and methodology of STOP-PD II. BMC Psychiatry. 2013;13:38.
30. Alexopoulos GS; PROSPECT Group. Interventions for depressed elderly primary care patients. Int J Geriatr Psychiatry. 2001;16(6):553-559.
31. Sajatovic M, Forester BP, Gildengers A, et al. Aging changes and medical complexity in late-life bipolar disorder: emerging research findings that may help advance care. Neuropsychiatry (London). 2013;3(6):621-633.
32. Bigos KL, Bies RR, Pollock BG, et al. Genetic variation in CYP3A43 explains racial difference in olanzapine clearance. Mol Psychiatry. 2011;16(6):620-625.
33. Jin Y, Pollock BG, Coley K, et al. Population pharmacokinetics of perphenazine in schizophrenia patients from CATIE: impact of race and smoking. J Clin Pharmacol. 2010;50(1):73-80.
34. Mulsant BH. Is there a role for antidepressant and antipsychotic pharmacogenetics in clinical practice in 2014? Can J Psychiatry. 2014;59(2):59-61.
1. United Nations. Department of Economic and Social Affairs Population Division. World Population Ageing: 1950-2050. http://www.un.org/esa/population/publications/worldageing19502050. Published 2001. Accessed September 27, 2017.
2. Olfson M, King M, Schoenbaum M. Antipsychotic treatment of adults in the United States. J Clin Psychiatry. 2015;76(10):1346-1353.
3. Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
4. Hybels CF, Blazer DG. Epidemiology of late-life mental disorders. Clin Geriatr Med. 2003;19(4):663-696,v.
5. Mulsant BH, Blumberger DM, Ismail Z, et al. A systematic approach to pharmacotherapy for geriatric major depression. Clin Geriatr Med. 2014;30(3):517-534.
6. Mulsant BH, Pollock BG. Psychopharmacology. In: Steffens DC, Blazer DG, Thakur ME, eds. The American Psychiatric Publishing textbook of geriatric psychiatry. 5th ed. Arlington, VA: American Psychiatric Publishing; 2015:527-587.
7. U.S. Food and Drug Administration. Public health advisory. deaths with antipsychotics in elderly patients with behavioral disturbances. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Updated August 16, 2013. Accessed September 27, 2017.
8. Andreescu C, Mulsant BH, Rothschild AJ, et al. Pharmacotherapy of major depression with psychotic features: what is the evidence? Psychiatric Annals. 2006;35(1):31-38.
9. Buchanan D, Tourigny-Rivard MF, Cappeliez P, et al. National guidelines for seniors’ mental health: the assessment and treatment of depression. Canadian Journal of Geriatrics. 2006;9(suppl 2):S52-S58.
10. Canadian Coalition for Senior’s Mental Health. National guidelines for senior’s mental health. The assessment and treatment of depression 2006. http://www.ccsmh.ca/projects/depression. Accessed February 28, 2016.
11. Meyers BS, Flint AJ, Rothschild AJ, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy of psychotic depression (STOP-PD). Arch Gen Psychiatry. 2009;66(8):838-847.
12. Mulsant BH, Sweet RA, Rosen J, et al. A double-blind randomized comparison of nortriptyline plus perphenazine versus nortriptyline plus placebo in the treatment of psychotic depression in late life. J Clin Psychiatry. 2001;62(8):597-604.
13. Cakir S, Senkal Z. Atypical antipsychotics as add-on treatment in late-life depression. Clin Interv Aging. 2016;11:1193-1198.
14. Maust DT, Oslin DW, Thase ME. Going beyond antidepressant monotherapy for incomplete response in nonpsychotic late-life depression: a critical review. Am J Geriatr Psychiatry. 2013;21(10):973-986.
15. Patel K, Abdool PS, Rajji TK, et al. Pharmacotherapy of major depression in late life: what is the role of new agents? Expert Opin Pharmacother. 2017;18(6):599-609.
16. Alexopoulos GS, Katz IR, Reynolds CF 3rd, et al. Pharmacotherapy of depression in older patients: a summary of the expert consensus guidelines. J Psychiatr Pract. 2001;7(6):361-376.
17. Cooper C, Katona C, Lyketsos K, et al. A systematic review of treatments for refractory depression in older people. Am J Psychiatry. 2011;168(7):681-688.
18. Rutherford B, Sneed J, Miyazaki M, et al. An open trial of aripiprazole augmentation for SSRI non-remitters with late-life depression. Int J Geriatr Psychiatry. 2007;22(10):986-991.
19. Sheffrin M, Driscoll HC, Lenze EJ, et al. Pilot study of augmentation with aripiprazole for incomplete response in late-life depression: getting to remission. J Clin Psychiatry. 2009;70(2):208-213.
20. Steffens DC, Nelson JC, Eudicone JM, et al. Efficacy and safety of adjunctive aripiprazole in major depressive disorder in older patients: a pooled subpopulation analysis. Int J Geriatr Psychiatry. 2011;26(6):564-572.
21. Lenze EJ, Mulsant BH, Blumberger DM, et al. Efficacy, safety, and tolerability of augmentation pharmacotherapy with aripiprazole for treatment-resistant depression in late life: a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(10011):2404-2412.
22. Deligiannidis KM, Rothschild AJ, Barton BA, et al. A gender analysis of the study of pharmacotherapy of psychotic depression (STOP-PD): gender and age as predictors of response and treatment-associated changes in body mass index and metabolic measures. J Clin Psychiatry. 2013;74(10):1003-1009.
23. Flint AJ, Iaboni A, Mulsant BH, et al. Effect of sertraline on risk of falling in older adults with psychotic depression on olanzapine: results of a randomized placebo-controlled trial. Am J Geriatr Psychiatry. 2014;22(4):332-336.
24. Smith E, Rothschild AJ, Heo M, et al. Weight gain during olanzapine treatment for psychotic depression: effects of dose and age. Int Clin Psychopharmacol. 2008;23(3):130-137.
25. Blumberger DM, Mulsant BH, Kanellopoulos D, et al. The incidence of tardive dyskinesia in the study of pharmacotherapy for psychotic depression. J Clin Psychopharmacol. 2013;33(3):391-397.
26. Alexopoulos GS, Canuso CM, Gharabawi GM, et al. Placebo-controlled study of relapse prevention with risperidone augmentation in older patients with resistant depression. Am J Geriatr Psychiatry. 2008;16(1):21-30.
27. Katila H, Mezhebovsky I, Mulroy A, et al. Randomized, double-blind study of the efficacy and tolerability of extended release quetiapine fumarate (quetiapine XR) monotherapy in elderly patients with major depressive disorder. Am J Geriatr Psychiatry. 2013;21(8):769-784.
28. Sultana J, Trifiro G. Drug safety warnings: a message in a bottle. J Drug Des Res. 2014;1(1):1004.
29. Flint A, Meyers BS, Rothschild AR, et al; STOP-PD II Study Group. Sustaining remission of psychotic depression: rationale, design and methodology of STOP-PD II. BMC Psychiatry. 2013;13:38.
30. Alexopoulos GS; PROSPECT Group. Interventions for depressed elderly primary care patients. Int J Geriatr Psychiatry. 2001;16(6):553-559.
31. Sajatovic M, Forester BP, Gildengers A, et al. Aging changes and medical complexity in late-life bipolar disorder: emerging research findings that may help advance care. Neuropsychiatry (London). 2013;3(6):621-633.
32. Bigos KL, Bies RR, Pollock BG, et al. Genetic variation in CYP3A43 explains racial difference in olanzapine clearance. Mol Psychiatry. 2011;16(6):620-625.
33. Jin Y, Pollock BG, Coley K, et al. Population pharmacokinetics of perphenazine in schizophrenia patients from CATIE: impact of race and smoking. J Clin Pharmacol. 2010;50(1):73-80.
34. Mulsant BH. Is there a role for antidepressant and antipsychotic pharmacogenetics in clinical practice in 2014? Can J Psychiatry. 2014;59(2):59-61.

The placebo effect in psychiatric practice
“It is a mystery how a ubiquitous treatment used since antiquity was unknown, unnamed, and unidentified until recently. It is even more remarkable because this is the only treatment common to all societies and cultures.”1
The treatment discussed above is not a specific pill, surgery, plant, or herb. Rather, the authors are referring to placebo. Indeed, the history of medical treatment is largely a chronicle of placebos. When subjected to scientific scrutiny, the overwhelming majority of treatments have turned out to be devoid of intrinsic therapeutic value; they derived their benefits from the placebo effect. Despite these benefits, the term “placebo” comes with unfortunate baggage. Latin for “I shall please,” it is the first word of the Christian vespers for the dead. In the 12th century these vespers were commonly referred to as placebos. By the 1300s, the term had become secular and pejorative, suggesting a flatterer or sycophant. When the word entered medical terminology in the late 18th century, the negative connotation stuck. A placebo was defined as a medicine given to please patients rather than to benefit them. In the modern era, the lack of pharmacologic activity became part of the definition as well.
The word placebo brings with it connotations of deception, fakery, and ineffectiveness. But one of the things about placebos that contribute mightily to the health care community’s aversion toward them is, in fact, their effectiveness. They bring relief across a wide range of medical conditions.2 In doing so, placebos impugn the value of our most cherished remedies, hamper the development of new therapeutics, and threaten our livelihoods as health professionals.3
Placebos often are conceptualized as any treatment that lacks intrinsic therapeutic value, such as sugar pills. But looking at what placebo treatment actually entails, both in placebo-controlled treatment trials and in clinical settings, suggests a more comprehensive definition. Placebos encompass all the elements common to any treatment or healing situation. These include a recognized healer, evaluation, diagnosis, prognosis, plausible treatment, and most importantly, the expectation that one will recover. Along these lines, the placebo response can be thought of as the response to the common elements of the treatment or healing situation.3
Research regarding the placebo effect has mushroomed in the past 2 decades. Over this time, we have learned a good deal about both the mechanisms underlying the placebo effect and how the placebo effect can be applied to enhance the benefit of conventional treatment. Brain imaging technology has revealed that when placebo treatment alleviates pain, Parkinson’s disease, and depression, brain changes occur that are similar to those observed with active pharmacologic treatment.4,5 Recent studies also show that deliberate, open (nondeceptive) use of placebo can improve the symptoms of several conditions, including depression, pain, and irritable bowel syndrome.6 Furthermore, intermittent substitution of placebo pills for pharmacologically active treatment in a conditioning paradigm can be as effective as the “real” treatment.7 Also, research over the past decade has verified that certain common features of the treatment situation, particularly the quality of the doctor–patient encounter, contribute to the placebo response and have a demonstrable impact on the outcome of treatment.8 Clearly, the placebo effect has gone from being simply a nuisance that interferes with the evaluation of new treatments to a variable worthy of study and application in its own right. Although, for the most part, clinical practice has not kept up with these advances.
Placebos seem to have their greatest impact on the subjective symptoms of disease—pain, distress, and discouragement. It should come as no surprise, then, that placebos are particularly effective in certain psychiatric conditions. In some forms of anxiety and depressive disorders, for example, distress is the illness, and placebos reliably bring relief. Patients with panic disorder, mild to moderate depression, or generalized anxiety disorder get almost as much relief with placebo as they do with conventional treatment (about one-half improve with placebo).9-11 But <20% of those with obsessive-compulsive disorder improve with placebo, and placebo response rates are also low in patients with schizophrenia or dementia. Mania, attention-deficit/hyperactivity disorder (ADHD), and severe depression fall somewhere in the middle.3
Harnessing the placebo response
There may be a few circumstances in psychiatric practice when it makes sense to intentionally prescribe a placebo as treatment, and we discuss those below. But far more frequently, what we know about the elements that contribute to the placebo effect can be applied to enhance the benefits of any treatment. Patients might be best served if deliberate mobilization of the placebo effect was a standard adjunct to conventional clinical care.
Various components of the treatment situation, collectively referred to as placebo, are a powerful antidote for illness, and some of these healing components exert their influence without special activity on the clinician’s part:
- Simply seeking psychiatric care can bring relief by providing some sense of control over distressing symptoms. The standard trappings of the office or clinic and customary office procedures—from the presentation of one’s insurance card to taking a history—offer reassurance and evoke the expectation that improvement or recovery is around the corner.
- The comfort provided by the psychiatrist’s presence is enhanced when patients feel that they are in the hands of a recognized healer. Psychiatrists inspire confidence when they look like a psychiatrist, or more precisely, like the patient’s idea of what a psychiatrist should look like. In our culture, that means a white coat or business attire.
A thorough evaluation is one of the common treatment elements that does the most to reduce distress and inspire confidence. The quality of an evaluation bears a strong relationship to patients’ satisfaction with the medical encounter, and can influence the amount of disability they suffer.3,12-15
Although guidelines for conducting effective psychiatric interviews have been around for almost 100 years, psychiatrists vary considerably in the extent to which they elicit complete and accurate information, build rapport, give patients the sense that they are listened to, and provide a thorough assessment. The degree to which patients feel that the clinician is responsive to their concerns depends as much on the style of the interview as on the amount of time devoted to it. Nonverbal behavior can carry the message that the clinician is paying full attention. Something as simple as not answering the phone during an interview (this seems obvious, but a surprising and troubling number of mental health professionals take phone calls during interviews and treatment sessions) conveys an important message about the importance that the clinician places on the patient’s problems.3
The idea that the treatment situation itself provides reassurance and reduces distress, and in doing so, powers a good bit of the placebo effect, is enshrined in such concepts as the importance of good bedside manner. Many feel that the doctor’s thoughtful attention, positive regard, and optimism—so valued by patients—are justified on humanitarian grounds alone; actual evidence that this caring behavior contributes to healing isn’t required. To many, the healing properties of the treatment situation are self-evident. But as the costs of health care snowball and the demands for efficiency and cost-effectiveness rise, the time that psychiatrists can devote to patients has dwindled. Third-party payors demand evidence, beyond intuition and common sense, that diagnostic procedures and treatments have some usefulness, and rightly so.
Is there any evidence that the common components of the treatment situation provide benefit?3 More specifically, does the quality of the doctor–patient relationship and the patient’s feelings about a therapeutic encounter promote healing? Several studies suggest that the doctor–patient relationship has a demonstrable impact on symptom relief.16 In 1 study, oncologists were randomly assigned to receive a Communication Skills Training (CST) program or not. CST included a 1.5-day face-to-face workshop and 6 hours of monthly videoconferencing that focused on improving communication skills with patients.17 Lessons included building rapport, engaging in appropriate eye contact, and normalizing difficult experiences. One week after initially consulting with their physician, patients who saw an oncologist in the CST group experienced less anxiety and depression than those who saw an oncologist who did not receive CST. The benefit of CST for patient anxiety mostly persisted at a 3-month follow-up.
A recent meta-analysis pooled the results of 47 studies to examine the relationship between how much trust patients have for their doctors and health outcomes. There was a small to medium association: More trust was associated with greater improvement.18 It is possible that a good doctor–patient relationship enhances expectancies. However, it is also likely that a positive therapeutic relationship is inherently soothing and reduces distress or dysfunction independent of expectation. Regardless of the precise mechanism, these studies warrant attention. We all understand that it is important on ethical grounds to treat patients with respect and kindness. Research shows that this type of behavior also promotes recovery.
Patient expectations. The idea that expectation of improvement has a major impact on treatment outcome is firmly grounded in research on the placebo effect. Studies have shown that what people expect to experience as an outcome of treatment has a substantial impact on what they actually experience. In a classic study, a doctor told some patients with symptoms of minor illness that they would feel better soon and another group with the same symptoms that he didn’t know what ailed them.19 Two weeks later, 64% of patients in the “positive expectation” group were improved, compared with only 39% of patients in the “negative” group. In another study, adults were exposed to an allergen that caused a skin reaction.20 Hand lotion (ie, a therapeutically inert substance) was then spread on the skin. Patients were led to believe that the cream would either alleviate or exacerbate the itching. The experimentally-induced wheal-and-flare was measured in both groups a few minutes after the allergen and cream were applied. The wheal-and-flare were worse for participants in the group that expected exacerbation.
Not uncommonly, expectation can have more impact on clinical outcome than a drug’s pharmacologic activity. In a double-blind placebo-controlled study, patients with depression were treated with St. John’s wort, sertraline, or placebo.21 They improved to the same extent with all 3 treatments. But when patients were asked to guess the treatment to which they had been assigned, those who thought they had received placebo showed little improvement, irrespective of which intervention they actually received, and those who guessed they had been given St. John’s wort or sertraline showed uniformly large improvement, irrespective of which intervention they actually received (including placebo). The researchers concluded that “Patient beliefs regarding treatment may have a stronger association with clinical outcome than the actual medication received.”
Psychiatrists who wish to use all the therapeutic tools at their disposal must attend to and manage patient expectations. One part of channeling a patient’s expectation is to thoroughly assess the patient’s beliefs regarding the efficacy of various treatments. If a patient’s uncle said that a certain drug is a miracle cure for anxiety, and the patient believes it to be true, then that expectation must be taken into consideration. Many patients prefer alternative treatments to conventional therapies. As long as there is no reason to think an alternative treatment will cause harm, a compromise might be reasonable. For example, if a patient with schizophrenia wants to treat her symptoms with herbal tea, the psychiatrist could say, “In addition to the tea, I recommend that you also take clozapine. The combination is likely to improve your symptoms.”3 More than anything else, the words a psychiatrist uses when recommending treatment shape the patient’s expectations. “You should be feeling a lot less anxious soon after you start taking this” has a different effect than “Try this. It may help.”
Prescribing ‘open-label’ placebo
There may be some limited circumstances where an actual placebo (eg, a sugar pill) might be suitable as a treatment. These include when placebo and conventional treatment provide similar results and a patient is reluctant to take conventional medicine, or when there is no effective conventional treatment. The deceptive prescription of placebo (providing placebo and calling it a drug) has a long history and was considered ethical—and recommended by medical authorities—until the latter half of the 20th century. This practice was deemed unethical in the 1980s, because it was dishonest and violated patient autonomy. Because it was widely believed that placebos given openly would be ineffective, the end of placebo treatment seemed at hand. An intriguing body of evidence, however, suggests that placebos can be effective even when patients know they are taking a placebo. Patients given an “open-label” placebo are told something along the lines of “the pill being prescribed contains no medicine, but some people improve with it, perhaps because the pill stimulates the body’s self-healing.” Open-label placebo has been evaluated for depression,22 low back pain,23 irritable bowel syndrome,24 neurosis,25 allergic rhinitis,26 and anxiety.27 Most of these studies are small, and some were uncontrolled. Yet they consistently have shown that symptoms improve with a nondeceptive placebo, and improve to a greater extent than with no treatment.
The most recent trial is a promising example of the potential of open-label placebos. In this study, 96 patients with chronic low back pain were randomly assigned to 3 weeks of treatment as usual (TAU) or 3 weeks of TAU plus open-label placebo.23 Patients who received open-label placebo were educated about the placebo effect and shown a film clip describing promising results of a prior open-label placebo study. They were then given placebo pills to be take once daily, and clearly told the pills contained no active medication. After 3 weeks, patients in the TAU plus placebo group reported less pain and less disability than patients who received TAU without a placebo. Some patients even requested a placebo prescription at the end of the study.
The placebo response provides a rational basis for prescribing innocuous alternative therapies with no intrinsic therapeutic value. Patients who prefer and believe in the effectiveness of alternative remedies—herbal compounds, massage, magnets, homeopathic solutions, etc.—can be recommended these treatments to mobilize a placebo response.
Using a conditioning model. Prescribing a placebo to obtain a conditioned drug response has enormous but untapped clinical potential. Both animal and human research indicates that a wide range of drug responses, from immune suppression to motor stimulation, can be conditioned (a neutral stimulus, such as a pill or injection, associated with drug administration can in itself evoke the drug effect). In many conditioning or dose-extending models, a particular response to real medication (such as pain relief after analgesics) first becomes conditioned due to repeated exposure to the drug given in a particular vehicle. Then, the treatment shifts to some doses comprising of real medicine and some doses comprising of placebo. Because the drug response has been conditioned, it is thought that the response to an identically appearing placebo will mirror the drug response. The active drug often is only replaced by placebo for certain doses under a schedule of partial reinforcement, given the ubiquity of extinction (the conditioned response lessens when the conditioned stimulus is presented alone on repeated trials).
In 1 version of a conditioning study, children with ADHD were randomized to 1 of 3 groups.28 One group (full dose) took the standard dose of medication for 2 months, a second group (reduced dose) took a standard dose during 1 month followed by a half dose during the second month, and children in the third group (reduced dose with placebo) took the standard dose plus a visually distinctive placebo during the first month, followed by a half dose plus the visually distinctive placebo during the second month. Not surprisingly, ADHD symptoms were worse among children in the reduced-dose group. However, there was no difference between those in the reduced-dose with placebo group and those in the full-dose group. It appears as though the symptom reduction associated with a 100% dose was an unconditioned response that could be mimicked with the addition of a placebo pill.
In another study, patients with psoriasis were randomly assigned to receive a full dose of active medication (0.1% triamcinolone cream) twice a day, or a full dose of active medication for 25% to 50% of the doses, with a placebo (moisturizing cream) given for the other 50% to 75% of the doses.29 Relapse rates were not statistically different between groups.
1. Shapiro AK, Shapiro E. The powerful placebo: from ancient priest to modern physician. Baltimore, MD: Johns Hopkins University Press; 1997.
2. Beecher HK. The powerful placebo. J Am Med Assoc. 1955;159(17):1602-1606.
3. Brown WA. The placebo effect in clinical practice. New York, NY: Oxford University Press; 2013.
4. Mayberg HS, Silva JA, Brannan SK, et al. The functional neuroanatomy of the placebo effect. Am J Psychiatry. 2002;159(5):728-737.
5. de la Fuente-Fernández R, Ruth TJ, Sossi V, et al. Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science. 2001;293(5532):1164-1166.
6. Charlesworth JEG, Petkovic G, Kelley JM, et al. Effects of placebos without deception compared with no treatment: a systematic review and meta‐analysis. J Evid Based Med. 2017;10(2):97-107.
7. Colloca L, Enck P, DeGrazia D. Relieving pain using dose-extending placebos: a scoping review. Pain. 2016;157(8):1590-1598.
8. Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336(7651):999-1003.
9. Khan A, Kolts RL, Rapaport MH, et al. Magnitude of placebo response and drug-placebo differences across psychiatric disorders. Psychol Med. 2005;35(5):743-749.
10. Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840-1847.
11. Kirsch I, Deacon BJ, Huedo-Medina TB, et al. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008;5(2):e45.
12. Kaptchuk TJ. Acupuncture: theory, efficacy, and practice. Ann Intern Med. 2002;136(5):374-383.
13. Kelley JM, Kraft-Todd G, Schapira L, et al. The influence of the patient-clinician relationship on healthcare outcomes: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2014;9(4):e94207.
14. Olsson B, Olsson B, Tibblin G. Effect of patients’ expectations on recovery from acute tonsillitis. Fam Pract. 1989;6(3):188-192.
15. Sox HC, Margulies I, Sox CH. Psychologically mediated effects of diagnostic tests. Ann Intern Med. 1981;95(6):680-685.
16. Stewart MA. Effective physician-patient communication and health outcomes: a review. CMAJ. 1995;152(9):1423-1433.
17. Girgis A, Cockburn J, Butow P, et al. Improving patient emotional functioning and psychological morbidity: evaluation of a consultation skills training program for oncologists. Patient Educ Couns. 2009;77(3):456-462.
18. Birkhäuer J, Gaab J, Kossowsky J, et al. Trust in the health care professional and health outcome: a meta-analysis. PLoS One. 2017;12(2):e0170988.
19. Thomas KB. General practice consultations: is there any point in being positive? Br Med J (Clin Res Ed). 1987;294(6581):1200-1202.
20. Howe LC, Goyer JP, Crum AJ. Harnessing the placebo effect: exploring the influence of physician characteristics on placebo response [published online May 9, 2017]. Health Psychol. doi: 10.1037/hea0000499.
21. Chen JA, Papakostas GI, Youn SJ, et al. Association between patient beliefs regarding assigned treatment and clinical response: reanalysis of data from the Hypericum Depression Trial Study Group. J Clin Psychiatry. 2011;72(12):1669-1676.
22. Kelley JM, Kaptchuk TJ, Cusin C, et al. Open-label placebo for major depressive disorder: a pilot randomized controlled trial. Psychother Psychosom. 2012;81(5):312-314.
23. Carvalho C, Caetano JM, Cunha L, et al. Open-label placebo treatment in chronic low back pain: a randomized controlled trial. Pain. 2016;157(12):2766-2772.
24. Kaptchuk TJ, Friedlander E, Kelley JM, et al. Placebos without deception: a randomized controlled trial in irritable bowel syndrome. PLoS One. 2010;5(12):e15591.
25. Park LC, Covi L. Nonblind placebo trial: an exploration of neurotic patients’ responses to placebo when its inert content is disclosed. Arch Gen Psychiatry. 1965;12(4):336-345.
26. Schaefer M, Harke R, Denke C. Open-label placebos improve symptoms in allergic rhinitis: a randomized controlled trial. Psychother Psychosom. 2016;85(6):373-374.
27. Aulas JJ, Rosner I. Efficacy of a non blind placebo prescription [in French]. Encephale. 2003;29(1):68-71.
28. Sandler AD, Glesne CE, Bodfish JW. Conditioned placebo dose reduction: a new treatment in attention deficit hyperactivity disorder? J Dev Behav Pediatr. 2010;31(5):369-375.
29. Ader R, Mercurio MG, Walton J, et al. Conditioned pharmacotherapeutic effects: a preliminary study. Psychosom Med. 2010;72(2):192-197.
30. Weiss RD, O’Malley SS, Hosking JD, et al; COMBINE Study Research Group. Do patients with alcohol dependence respond to placebo? Results from the COMBINE Study. J Stud Alcohol Drugs. 2008;69(6):878-884.
31. Mattick RP, Breen C, Kimber J, et al. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev. 2014;(2):CD002207.
“It is a mystery how a ubiquitous treatment used since antiquity was unknown, unnamed, and unidentified until recently. It is even more remarkable because this is the only treatment common to all societies and cultures.”1
The treatment discussed above is not a specific pill, surgery, plant, or herb. Rather, the authors are referring to placebo. Indeed, the history of medical treatment is largely a chronicle of placebos. When subjected to scientific scrutiny, the overwhelming majority of treatments have turned out to be devoid of intrinsic therapeutic value; they derived their benefits from the placebo effect. Despite these benefits, the term “placebo” comes with unfortunate baggage. Latin for “I shall please,” it is the first word of the Christian vespers for the dead. In the 12th century these vespers were commonly referred to as placebos. By the 1300s, the term had become secular and pejorative, suggesting a flatterer or sycophant. When the word entered medical terminology in the late 18th century, the negative connotation stuck. A placebo was defined as a medicine given to please patients rather than to benefit them. In the modern era, the lack of pharmacologic activity became part of the definition as well.
The word placebo brings with it connotations of deception, fakery, and ineffectiveness. But one of the things about placebos that contribute mightily to the health care community’s aversion toward them is, in fact, their effectiveness. They bring relief across a wide range of medical conditions.2 In doing so, placebos impugn the value of our most cherished remedies, hamper the development of new therapeutics, and threaten our livelihoods as health professionals.3
Placebos often are conceptualized as any treatment that lacks intrinsic therapeutic value, such as sugar pills. But looking at what placebo treatment actually entails, both in placebo-controlled treatment trials and in clinical settings, suggests a more comprehensive definition. Placebos encompass all the elements common to any treatment or healing situation. These include a recognized healer, evaluation, diagnosis, prognosis, plausible treatment, and most importantly, the expectation that one will recover. Along these lines, the placebo response can be thought of as the response to the common elements of the treatment or healing situation.3
Research regarding the placebo effect has mushroomed in the past 2 decades. Over this time, we have learned a good deal about both the mechanisms underlying the placebo effect and how the placebo effect can be applied to enhance the benefit of conventional treatment. Brain imaging technology has revealed that when placebo treatment alleviates pain, Parkinson’s disease, and depression, brain changes occur that are similar to those observed with active pharmacologic treatment.4,5 Recent studies also show that deliberate, open (nondeceptive) use of placebo can improve the symptoms of several conditions, including depression, pain, and irritable bowel syndrome.6 Furthermore, intermittent substitution of placebo pills for pharmacologically active treatment in a conditioning paradigm can be as effective as the “real” treatment.7 Also, research over the past decade has verified that certain common features of the treatment situation, particularly the quality of the doctor–patient encounter, contribute to the placebo response and have a demonstrable impact on the outcome of treatment.8 Clearly, the placebo effect has gone from being simply a nuisance that interferes with the evaluation of new treatments to a variable worthy of study and application in its own right. Although, for the most part, clinical practice has not kept up with these advances.
Placebos seem to have their greatest impact on the subjective symptoms of disease—pain, distress, and discouragement. It should come as no surprise, then, that placebos are particularly effective in certain psychiatric conditions. In some forms of anxiety and depressive disorders, for example, distress is the illness, and placebos reliably bring relief. Patients with panic disorder, mild to moderate depression, or generalized anxiety disorder get almost as much relief with placebo as they do with conventional treatment (about one-half improve with placebo).9-11 But <20% of those with obsessive-compulsive disorder improve with placebo, and placebo response rates are also low in patients with schizophrenia or dementia. Mania, attention-deficit/hyperactivity disorder (ADHD), and severe depression fall somewhere in the middle.3
Harnessing the placebo response
There may be a few circumstances in psychiatric practice when it makes sense to intentionally prescribe a placebo as treatment, and we discuss those below. But far more frequently, what we know about the elements that contribute to the placebo effect can be applied to enhance the benefits of any treatment. Patients might be best served if deliberate mobilization of the placebo effect was a standard adjunct to conventional clinical care.
Various components of the treatment situation, collectively referred to as placebo, are a powerful antidote for illness, and some of these healing components exert their influence without special activity on the clinician’s part:
- Simply seeking psychiatric care can bring relief by providing some sense of control over distressing symptoms. The standard trappings of the office or clinic and customary office procedures—from the presentation of one’s insurance card to taking a history—offer reassurance and evoke the expectation that improvement or recovery is around the corner.
- The comfort provided by the psychiatrist’s presence is enhanced when patients feel that they are in the hands of a recognized healer. Psychiatrists inspire confidence when they look like a psychiatrist, or more precisely, like the patient’s idea of what a psychiatrist should look like. In our culture, that means a white coat or business attire.
A thorough evaluation is one of the common treatment elements that does the most to reduce distress and inspire confidence. The quality of an evaluation bears a strong relationship to patients’ satisfaction with the medical encounter, and can influence the amount of disability they suffer.3,12-15
Although guidelines for conducting effective psychiatric interviews have been around for almost 100 years, psychiatrists vary considerably in the extent to which they elicit complete and accurate information, build rapport, give patients the sense that they are listened to, and provide a thorough assessment. The degree to which patients feel that the clinician is responsive to their concerns depends as much on the style of the interview as on the amount of time devoted to it. Nonverbal behavior can carry the message that the clinician is paying full attention. Something as simple as not answering the phone during an interview (this seems obvious, but a surprising and troubling number of mental health professionals take phone calls during interviews and treatment sessions) conveys an important message about the importance that the clinician places on the patient’s problems.3
The idea that the treatment situation itself provides reassurance and reduces distress, and in doing so, powers a good bit of the placebo effect, is enshrined in such concepts as the importance of good bedside manner. Many feel that the doctor’s thoughtful attention, positive regard, and optimism—so valued by patients—are justified on humanitarian grounds alone; actual evidence that this caring behavior contributes to healing isn’t required. To many, the healing properties of the treatment situation are self-evident. But as the costs of health care snowball and the demands for efficiency and cost-effectiveness rise, the time that psychiatrists can devote to patients has dwindled. Third-party payors demand evidence, beyond intuition and common sense, that diagnostic procedures and treatments have some usefulness, and rightly so.
Is there any evidence that the common components of the treatment situation provide benefit?3 More specifically, does the quality of the doctor–patient relationship and the patient’s feelings about a therapeutic encounter promote healing? Several studies suggest that the doctor–patient relationship has a demonstrable impact on symptom relief.16 In 1 study, oncologists were randomly assigned to receive a Communication Skills Training (CST) program or not. CST included a 1.5-day face-to-face workshop and 6 hours of monthly videoconferencing that focused on improving communication skills with patients.17 Lessons included building rapport, engaging in appropriate eye contact, and normalizing difficult experiences. One week after initially consulting with their physician, patients who saw an oncologist in the CST group experienced less anxiety and depression than those who saw an oncologist who did not receive CST. The benefit of CST for patient anxiety mostly persisted at a 3-month follow-up.
A recent meta-analysis pooled the results of 47 studies to examine the relationship between how much trust patients have for their doctors and health outcomes. There was a small to medium association: More trust was associated with greater improvement.18 It is possible that a good doctor–patient relationship enhances expectancies. However, it is also likely that a positive therapeutic relationship is inherently soothing and reduces distress or dysfunction independent of expectation. Regardless of the precise mechanism, these studies warrant attention. We all understand that it is important on ethical grounds to treat patients with respect and kindness. Research shows that this type of behavior also promotes recovery.
Patient expectations. The idea that expectation of improvement has a major impact on treatment outcome is firmly grounded in research on the placebo effect. Studies have shown that what people expect to experience as an outcome of treatment has a substantial impact on what they actually experience. In a classic study, a doctor told some patients with symptoms of minor illness that they would feel better soon and another group with the same symptoms that he didn’t know what ailed them.19 Two weeks later, 64% of patients in the “positive expectation” group were improved, compared with only 39% of patients in the “negative” group. In another study, adults were exposed to an allergen that caused a skin reaction.20 Hand lotion (ie, a therapeutically inert substance) was then spread on the skin. Patients were led to believe that the cream would either alleviate or exacerbate the itching. The experimentally-induced wheal-and-flare was measured in both groups a few minutes after the allergen and cream were applied. The wheal-and-flare were worse for participants in the group that expected exacerbation.
Not uncommonly, expectation can have more impact on clinical outcome than a drug’s pharmacologic activity. In a double-blind placebo-controlled study, patients with depression were treated with St. John’s wort, sertraline, or placebo.21 They improved to the same extent with all 3 treatments. But when patients were asked to guess the treatment to which they had been assigned, those who thought they had received placebo showed little improvement, irrespective of which intervention they actually received, and those who guessed they had been given St. John’s wort or sertraline showed uniformly large improvement, irrespective of which intervention they actually received (including placebo). The researchers concluded that “Patient beliefs regarding treatment may have a stronger association with clinical outcome than the actual medication received.”
Psychiatrists who wish to use all the therapeutic tools at their disposal must attend to and manage patient expectations. One part of channeling a patient’s expectation is to thoroughly assess the patient’s beliefs regarding the efficacy of various treatments. If a patient’s uncle said that a certain drug is a miracle cure for anxiety, and the patient believes it to be true, then that expectation must be taken into consideration. Many patients prefer alternative treatments to conventional therapies. As long as there is no reason to think an alternative treatment will cause harm, a compromise might be reasonable. For example, if a patient with schizophrenia wants to treat her symptoms with herbal tea, the psychiatrist could say, “In addition to the tea, I recommend that you also take clozapine. The combination is likely to improve your symptoms.”3 More than anything else, the words a psychiatrist uses when recommending treatment shape the patient’s expectations. “You should be feeling a lot less anxious soon after you start taking this” has a different effect than “Try this. It may help.”
Prescribing ‘open-label’ placebo
There may be some limited circumstances where an actual placebo (eg, a sugar pill) might be suitable as a treatment. These include when placebo and conventional treatment provide similar results and a patient is reluctant to take conventional medicine, or when there is no effective conventional treatment. The deceptive prescription of placebo (providing placebo and calling it a drug) has a long history and was considered ethical—and recommended by medical authorities—until the latter half of the 20th century. This practice was deemed unethical in the 1980s, because it was dishonest and violated patient autonomy. Because it was widely believed that placebos given openly would be ineffective, the end of placebo treatment seemed at hand. An intriguing body of evidence, however, suggests that placebos can be effective even when patients know they are taking a placebo. Patients given an “open-label” placebo are told something along the lines of “the pill being prescribed contains no medicine, but some people improve with it, perhaps because the pill stimulates the body’s self-healing.” Open-label placebo has been evaluated for depression,22 low back pain,23 irritable bowel syndrome,24 neurosis,25 allergic rhinitis,26 and anxiety.27 Most of these studies are small, and some were uncontrolled. Yet they consistently have shown that symptoms improve with a nondeceptive placebo, and improve to a greater extent than with no treatment.
The most recent trial is a promising example of the potential of open-label placebos. In this study, 96 patients with chronic low back pain were randomly assigned to 3 weeks of treatment as usual (TAU) or 3 weeks of TAU plus open-label placebo.23 Patients who received open-label placebo were educated about the placebo effect and shown a film clip describing promising results of a prior open-label placebo study. They were then given placebo pills to be take once daily, and clearly told the pills contained no active medication. After 3 weeks, patients in the TAU plus placebo group reported less pain and less disability than patients who received TAU without a placebo. Some patients even requested a placebo prescription at the end of the study.
The placebo response provides a rational basis for prescribing innocuous alternative therapies with no intrinsic therapeutic value. Patients who prefer and believe in the effectiveness of alternative remedies—herbal compounds, massage, magnets, homeopathic solutions, etc.—can be recommended these treatments to mobilize a placebo response.
Using a conditioning model. Prescribing a placebo to obtain a conditioned drug response has enormous but untapped clinical potential. Both animal and human research indicates that a wide range of drug responses, from immune suppression to motor stimulation, can be conditioned (a neutral stimulus, such as a pill or injection, associated with drug administration can in itself evoke the drug effect). In many conditioning or dose-extending models, a particular response to real medication (such as pain relief after analgesics) first becomes conditioned due to repeated exposure to the drug given in a particular vehicle. Then, the treatment shifts to some doses comprising of real medicine and some doses comprising of placebo. Because the drug response has been conditioned, it is thought that the response to an identically appearing placebo will mirror the drug response. The active drug often is only replaced by placebo for certain doses under a schedule of partial reinforcement, given the ubiquity of extinction (the conditioned response lessens when the conditioned stimulus is presented alone on repeated trials).
In 1 version of a conditioning study, children with ADHD were randomized to 1 of 3 groups.28 One group (full dose) took the standard dose of medication for 2 months, a second group (reduced dose) took a standard dose during 1 month followed by a half dose during the second month, and children in the third group (reduced dose with placebo) took the standard dose plus a visually distinctive placebo during the first month, followed by a half dose plus the visually distinctive placebo during the second month. Not surprisingly, ADHD symptoms were worse among children in the reduced-dose group. However, there was no difference between those in the reduced-dose with placebo group and those in the full-dose group. It appears as though the symptom reduction associated with a 100% dose was an unconditioned response that could be mimicked with the addition of a placebo pill.
In another study, patients with psoriasis were randomly assigned to receive a full dose of active medication (0.1% triamcinolone cream) twice a day, or a full dose of active medication for 25% to 50% of the doses, with a placebo (moisturizing cream) given for the other 50% to 75% of the doses.29 Relapse rates were not statistically different between groups.
“It is a mystery how a ubiquitous treatment used since antiquity was unknown, unnamed, and unidentified until recently. It is even more remarkable because this is the only treatment common to all societies and cultures.”1
The treatment discussed above is not a specific pill, surgery, plant, or herb. Rather, the authors are referring to placebo. Indeed, the history of medical treatment is largely a chronicle of placebos. When subjected to scientific scrutiny, the overwhelming majority of treatments have turned out to be devoid of intrinsic therapeutic value; they derived their benefits from the placebo effect. Despite these benefits, the term “placebo” comes with unfortunate baggage. Latin for “I shall please,” it is the first word of the Christian vespers for the dead. In the 12th century these vespers were commonly referred to as placebos. By the 1300s, the term had become secular and pejorative, suggesting a flatterer or sycophant. When the word entered medical terminology in the late 18th century, the negative connotation stuck. A placebo was defined as a medicine given to please patients rather than to benefit them. In the modern era, the lack of pharmacologic activity became part of the definition as well.
The word placebo brings with it connotations of deception, fakery, and ineffectiveness. But one of the things about placebos that contribute mightily to the health care community’s aversion toward them is, in fact, their effectiveness. They bring relief across a wide range of medical conditions.2 In doing so, placebos impugn the value of our most cherished remedies, hamper the development of new therapeutics, and threaten our livelihoods as health professionals.3
Placebos often are conceptualized as any treatment that lacks intrinsic therapeutic value, such as sugar pills. But looking at what placebo treatment actually entails, both in placebo-controlled treatment trials and in clinical settings, suggests a more comprehensive definition. Placebos encompass all the elements common to any treatment or healing situation. These include a recognized healer, evaluation, diagnosis, prognosis, plausible treatment, and most importantly, the expectation that one will recover. Along these lines, the placebo response can be thought of as the response to the common elements of the treatment or healing situation.3
Research regarding the placebo effect has mushroomed in the past 2 decades. Over this time, we have learned a good deal about both the mechanisms underlying the placebo effect and how the placebo effect can be applied to enhance the benefit of conventional treatment. Brain imaging technology has revealed that when placebo treatment alleviates pain, Parkinson’s disease, and depression, brain changes occur that are similar to those observed with active pharmacologic treatment.4,5 Recent studies also show that deliberate, open (nondeceptive) use of placebo can improve the symptoms of several conditions, including depression, pain, and irritable bowel syndrome.6 Furthermore, intermittent substitution of placebo pills for pharmacologically active treatment in a conditioning paradigm can be as effective as the “real” treatment.7 Also, research over the past decade has verified that certain common features of the treatment situation, particularly the quality of the doctor–patient encounter, contribute to the placebo response and have a demonstrable impact on the outcome of treatment.8 Clearly, the placebo effect has gone from being simply a nuisance that interferes with the evaluation of new treatments to a variable worthy of study and application in its own right. Although, for the most part, clinical practice has not kept up with these advances.
Placebos seem to have their greatest impact on the subjective symptoms of disease—pain, distress, and discouragement. It should come as no surprise, then, that placebos are particularly effective in certain psychiatric conditions. In some forms of anxiety and depressive disorders, for example, distress is the illness, and placebos reliably bring relief. Patients with panic disorder, mild to moderate depression, or generalized anxiety disorder get almost as much relief with placebo as they do with conventional treatment (about one-half improve with placebo).9-11 But <20% of those with obsessive-compulsive disorder improve with placebo, and placebo response rates are also low in patients with schizophrenia or dementia. Mania, attention-deficit/hyperactivity disorder (ADHD), and severe depression fall somewhere in the middle.3
Harnessing the placebo response
There may be a few circumstances in psychiatric practice when it makes sense to intentionally prescribe a placebo as treatment, and we discuss those below. But far more frequently, what we know about the elements that contribute to the placebo effect can be applied to enhance the benefits of any treatment. Patients might be best served if deliberate mobilization of the placebo effect was a standard adjunct to conventional clinical care.
Various components of the treatment situation, collectively referred to as placebo, are a powerful antidote for illness, and some of these healing components exert their influence without special activity on the clinician’s part:
- Simply seeking psychiatric care can bring relief by providing some sense of control over distressing symptoms. The standard trappings of the office or clinic and customary office procedures—from the presentation of one’s insurance card to taking a history—offer reassurance and evoke the expectation that improvement or recovery is around the corner.
- The comfort provided by the psychiatrist’s presence is enhanced when patients feel that they are in the hands of a recognized healer. Psychiatrists inspire confidence when they look like a psychiatrist, or more precisely, like the patient’s idea of what a psychiatrist should look like. In our culture, that means a white coat or business attire.
A thorough evaluation is one of the common treatment elements that does the most to reduce distress and inspire confidence. The quality of an evaluation bears a strong relationship to patients’ satisfaction with the medical encounter, and can influence the amount of disability they suffer.3,12-15
Although guidelines for conducting effective psychiatric interviews have been around for almost 100 years, psychiatrists vary considerably in the extent to which they elicit complete and accurate information, build rapport, give patients the sense that they are listened to, and provide a thorough assessment. The degree to which patients feel that the clinician is responsive to their concerns depends as much on the style of the interview as on the amount of time devoted to it. Nonverbal behavior can carry the message that the clinician is paying full attention. Something as simple as not answering the phone during an interview (this seems obvious, but a surprising and troubling number of mental health professionals take phone calls during interviews and treatment sessions) conveys an important message about the importance that the clinician places on the patient’s problems.3
The idea that the treatment situation itself provides reassurance and reduces distress, and in doing so, powers a good bit of the placebo effect, is enshrined in such concepts as the importance of good bedside manner. Many feel that the doctor’s thoughtful attention, positive regard, and optimism—so valued by patients—are justified on humanitarian grounds alone; actual evidence that this caring behavior contributes to healing isn’t required. To many, the healing properties of the treatment situation are self-evident. But as the costs of health care snowball and the demands for efficiency and cost-effectiveness rise, the time that psychiatrists can devote to patients has dwindled. Third-party payors demand evidence, beyond intuition and common sense, that diagnostic procedures and treatments have some usefulness, and rightly so.
Is there any evidence that the common components of the treatment situation provide benefit?3 More specifically, does the quality of the doctor–patient relationship and the patient’s feelings about a therapeutic encounter promote healing? Several studies suggest that the doctor–patient relationship has a demonstrable impact on symptom relief.16 In 1 study, oncologists were randomly assigned to receive a Communication Skills Training (CST) program or not. CST included a 1.5-day face-to-face workshop and 6 hours of monthly videoconferencing that focused on improving communication skills with patients.17 Lessons included building rapport, engaging in appropriate eye contact, and normalizing difficult experiences. One week after initially consulting with their physician, patients who saw an oncologist in the CST group experienced less anxiety and depression than those who saw an oncologist who did not receive CST. The benefit of CST for patient anxiety mostly persisted at a 3-month follow-up.
A recent meta-analysis pooled the results of 47 studies to examine the relationship between how much trust patients have for their doctors and health outcomes. There was a small to medium association: More trust was associated with greater improvement.18 It is possible that a good doctor–patient relationship enhances expectancies. However, it is also likely that a positive therapeutic relationship is inherently soothing and reduces distress or dysfunction independent of expectation. Regardless of the precise mechanism, these studies warrant attention. We all understand that it is important on ethical grounds to treat patients with respect and kindness. Research shows that this type of behavior also promotes recovery.
Patient expectations. The idea that expectation of improvement has a major impact on treatment outcome is firmly grounded in research on the placebo effect. Studies have shown that what people expect to experience as an outcome of treatment has a substantial impact on what they actually experience. In a classic study, a doctor told some patients with symptoms of minor illness that they would feel better soon and another group with the same symptoms that he didn’t know what ailed them.19 Two weeks later, 64% of patients in the “positive expectation” group were improved, compared with only 39% of patients in the “negative” group. In another study, adults were exposed to an allergen that caused a skin reaction.20 Hand lotion (ie, a therapeutically inert substance) was then spread on the skin. Patients were led to believe that the cream would either alleviate or exacerbate the itching. The experimentally-induced wheal-and-flare was measured in both groups a few minutes after the allergen and cream were applied. The wheal-and-flare were worse for participants in the group that expected exacerbation.
Not uncommonly, expectation can have more impact on clinical outcome than a drug’s pharmacologic activity. In a double-blind placebo-controlled study, patients with depression were treated with St. John’s wort, sertraline, or placebo.21 They improved to the same extent with all 3 treatments. But when patients were asked to guess the treatment to which they had been assigned, those who thought they had received placebo showed little improvement, irrespective of which intervention they actually received, and those who guessed they had been given St. John’s wort or sertraline showed uniformly large improvement, irrespective of which intervention they actually received (including placebo). The researchers concluded that “Patient beliefs regarding treatment may have a stronger association with clinical outcome than the actual medication received.”
Psychiatrists who wish to use all the therapeutic tools at their disposal must attend to and manage patient expectations. One part of channeling a patient’s expectation is to thoroughly assess the patient’s beliefs regarding the efficacy of various treatments. If a patient’s uncle said that a certain drug is a miracle cure for anxiety, and the patient believes it to be true, then that expectation must be taken into consideration. Many patients prefer alternative treatments to conventional therapies. As long as there is no reason to think an alternative treatment will cause harm, a compromise might be reasonable. For example, if a patient with schizophrenia wants to treat her symptoms with herbal tea, the psychiatrist could say, “In addition to the tea, I recommend that you also take clozapine. The combination is likely to improve your symptoms.”3 More than anything else, the words a psychiatrist uses when recommending treatment shape the patient’s expectations. “You should be feeling a lot less anxious soon after you start taking this” has a different effect than “Try this. It may help.”
Prescribing ‘open-label’ placebo
There may be some limited circumstances where an actual placebo (eg, a sugar pill) might be suitable as a treatment. These include when placebo and conventional treatment provide similar results and a patient is reluctant to take conventional medicine, or when there is no effective conventional treatment. The deceptive prescription of placebo (providing placebo and calling it a drug) has a long history and was considered ethical—and recommended by medical authorities—until the latter half of the 20th century. This practice was deemed unethical in the 1980s, because it was dishonest and violated patient autonomy. Because it was widely believed that placebos given openly would be ineffective, the end of placebo treatment seemed at hand. An intriguing body of evidence, however, suggests that placebos can be effective even when patients know they are taking a placebo. Patients given an “open-label” placebo are told something along the lines of “the pill being prescribed contains no medicine, but some people improve with it, perhaps because the pill stimulates the body’s self-healing.” Open-label placebo has been evaluated for depression,22 low back pain,23 irritable bowel syndrome,24 neurosis,25 allergic rhinitis,26 and anxiety.27 Most of these studies are small, and some were uncontrolled. Yet they consistently have shown that symptoms improve with a nondeceptive placebo, and improve to a greater extent than with no treatment.
The most recent trial is a promising example of the potential of open-label placebos. In this study, 96 patients with chronic low back pain were randomly assigned to 3 weeks of treatment as usual (TAU) or 3 weeks of TAU plus open-label placebo.23 Patients who received open-label placebo were educated about the placebo effect and shown a film clip describing promising results of a prior open-label placebo study. They were then given placebo pills to be take once daily, and clearly told the pills contained no active medication. After 3 weeks, patients in the TAU plus placebo group reported less pain and less disability than patients who received TAU without a placebo. Some patients even requested a placebo prescription at the end of the study.
The placebo response provides a rational basis for prescribing innocuous alternative therapies with no intrinsic therapeutic value. Patients who prefer and believe in the effectiveness of alternative remedies—herbal compounds, massage, magnets, homeopathic solutions, etc.—can be recommended these treatments to mobilize a placebo response.
Using a conditioning model. Prescribing a placebo to obtain a conditioned drug response has enormous but untapped clinical potential. Both animal and human research indicates that a wide range of drug responses, from immune suppression to motor stimulation, can be conditioned (a neutral stimulus, such as a pill or injection, associated with drug administration can in itself evoke the drug effect). In many conditioning or dose-extending models, a particular response to real medication (such as pain relief after analgesics) first becomes conditioned due to repeated exposure to the drug given in a particular vehicle. Then, the treatment shifts to some doses comprising of real medicine and some doses comprising of placebo. Because the drug response has been conditioned, it is thought that the response to an identically appearing placebo will mirror the drug response. The active drug often is only replaced by placebo for certain doses under a schedule of partial reinforcement, given the ubiquity of extinction (the conditioned response lessens when the conditioned stimulus is presented alone on repeated trials).
In 1 version of a conditioning study, children with ADHD were randomized to 1 of 3 groups.28 One group (full dose) took the standard dose of medication for 2 months, a second group (reduced dose) took a standard dose during 1 month followed by a half dose during the second month, and children in the third group (reduced dose with placebo) took the standard dose plus a visually distinctive placebo during the first month, followed by a half dose plus the visually distinctive placebo during the second month. Not surprisingly, ADHD symptoms were worse among children in the reduced-dose group. However, there was no difference between those in the reduced-dose with placebo group and those in the full-dose group. It appears as though the symptom reduction associated with a 100% dose was an unconditioned response that could be mimicked with the addition of a placebo pill.
In another study, patients with psoriasis were randomly assigned to receive a full dose of active medication (0.1% triamcinolone cream) twice a day, or a full dose of active medication for 25% to 50% of the doses, with a placebo (moisturizing cream) given for the other 50% to 75% of the doses.29 Relapse rates were not statistically different between groups.
1. Shapiro AK, Shapiro E. The powerful placebo: from ancient priest to modern physician. Baltimore, MD: Johns Hopkins University Press; 1997.
2. Beecher HK. The powerful placebo. J Am Med Assoc. 1955;159(17):1602-1606.
3. Brown WA. The placebo effect in clinical practice. New York, NY: Oxford University Press; 2013.
4. Mayberg HS, Silva JA, Brannan SK, et al. The functional neuroanatomy of the placebo effect. Am J Psychiatry. 2002;159(5):728-737.
5. de la Fuente-Fernández R, Ruth TJ, Sossi V, et al. Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science. 2001;293(5532):1164-1166.
6. Charlesworth JEG, Petkovic G, Kelley JM, et al. Effects of placebos without deception compared with no treatment: a systematic review and meta‐analysis. J Evid Based Med. 2017;10(2):97-107.
7. Colloca L, Enck P, DeGrazia D. Relieving pain using dose-extending placebos: a scoping review. Pain. 2016;157(8):1590-1598.
8. Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336(7651):999-1003.
9. Khan A, Kolts RL, Rapaport MH, et al. Magnitude of placebo response and drug-placebo differences across psychiatric disorders. Psychol Med. 2005;35(5):743-749.
10. Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840-1847.
11. Kirsch I, Deacon BJ, Huedo-Medina TB, et al. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008;5(2):e45.
12. Kaptchuk TJ. Acupuncture: theory, efficacy, and practice. Ann Intern Med. 2002;136(5):374-383.
13. Kelley JM, Kraft-Todd G, Schapira L, et al. The influence of the patient-clinician relationship on healthcare outcomes: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2014;9(4):e94207.
14. Olsson B, Olsson B, Tibblin G. Effect of patients’ expectations on recovery from acute tonsillitis. Fam Pract. 1989;6(3):188-192.
15. Sox HC, Margulies I, Sox CH. Psychologically mediated effects of diagnostic tests. Ann Intern Med. 1981;95(6):680-685.
16. Stewart MA. Effective physician-patient communication and health outcomes: a review. CMAJ. 1995;152(9):1423-1433.
17. Girgis A, Cockburn J, Butow P, et al. Improving patient emotional functioning and psychological morbidity: evaluation of a consultation skills training program for oncologists. Patient Educ Couns. 2009;77(3):456-462.
18. Birkhäuer J, Gaab J, Kossowsky J, et al. Trust in the health care professional and health outcome: a meta-analysis. PLoS One. 2017;12(2):e0170988.
19. Thomas KB. General practice consultations: is there any point in being positive? Br Med J (Clin Res Ed). 1987;294(6581):1200-1202.
20. Howe LC, Goyer JP, Crum AJ. Harnessing the placebo effect: exploring the influence of physician characteristics on placebo response [published online May 9, 2017]. Health Psychol. doi: 10.1037/hea0000499.
21. Chen JA, Papakostas GI, Youn SJ, et al. Association between patient beliefs regarding assigned treatment and clinical response: reanalysis of data from the Hypericum Depression Trial Study Group. J Clin Psychiatry. 2011;72(12):1669-1676.
22. Kelley JM, Kaptchuk TJ, Cusin C, et al. Open-label placebo for major depressive disorder: a pilot randomized controlled trial. Psychother Psychosom. 2012;81(5):312-314.
23. Carvalho C, Caetano JM, Cunha L, et al. Open-label placebo treatment in chronic low back pain: a randomized controlled trial. Pain. 2016;157(12):2766-2772.
24. Kaptchuk TJ, Friedlander E, Kelley JM, et al. Placebos without deception: a randomized controlled trial in irritable bowel syndrome. PLoS One. 2010;5(12):e15591.
25. Park LC, Covi L. Nonblind placebo trial: an exploration of neurotic patients’ responses to placebo when its inert content is disclosed. Arch Gen Psychiatry. 1965;12(4):336-345.
26. Schaefer M, Harke R, Denke C. Open-label placebos improve symptoms in allergic rhinitis: a randomized controlled trial. Psychother Psychosom. 2016;85(6):373-374.
27. Aulas JJ, Rosner I. Efficacy of a non blind placebo prescription [in French]. Encephale. 2003;29(1):68-71.
28. Sandler AD, Glesne CE, Bodfish JW. Conditioned placebo dose reduction: a new treatment in attention deficit hyperactivity disorder? J Dev Behav Pediatr. 2010;31(5):369-375.
29. Ader R, Mercurio MG, Walton J, et al. Conditioned pharmacotherapeutic effects: a preliminary study. Psychosom Med. 2010;72(2):192-197.
30. Weiss RD, O’Malley SS, Hosking JD, et al; COMBINE Study Research Group. Do patients with alcohol dependence respond to placebo? Results from the COMBINE Study. J Stud Alcohol Drugs. 2008;69(6):878-884.
31. Mattick RP, Breen C, Kimber J, et al. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev. 2014;(2):CD002207.
1. Shapiro AK, Shapiro E. The powerful placebo: from ancient priest to modern physician. Baltimore, MD: Johns Hopkins University Press; 1997.
2. Beecher HK. The powerful placebo. J Am Med Assoc. 1955;159(17):1602-1606.
3. Brown WA. The placebo effect in clinical practice. New York, NY: Oxford University Press; 2013.
4. Mayberg HS, Silva JA, Brannan SK, et al. The functional neuroanatomy of the placebo effect. Am J Psychiatry. 2002;159(5):728-737.
5. de la Fuente-Fernández R, Ruth TJ, Sossi V, et al. Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science. 2001;293(5532):1164-1166.
6. Charlesworth JEG, Petkovic G, Kelley JM, et al. Effects of placebos without deception compared with no treatment: a systematic review and meta‐analysis. J Evid Based Med. 2017;10(2):97-107.
7. Colloca L, Enck P, DeGrazia D. Relieving pain using dose-extending placebos: a scoping review. Pain. 2016;157(8):1590-1598.
8. Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336(7651):999-1003.
9. Khan A, Kolts RL, Rapaport MH, et al. Magnitude of placebo response and drug-placebo differences across psychiatric disorders. Psychol Med. 2005;35(5):743-749.
10. Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840-1847.
11. Kirsch I, Deacon BJ, Huedo-Medina TB, et al. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008;5(2):e45.
12. Kaptchuk TJ. Acupuncture: theory, efficacy, and practice. Ann Intern Med. 2002;136(5):374-383.
13. Kelley JM, Kraft-Todd G, Schapira L, et al. The influence of the patient-clinician relationship on healthcare outcomes: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2014;9(4):e94207.
14. Olsson B, Olsson B, Tibblin G. Effect of patients’ expectations on recovery from acute tonsillitis. Fam Pract. 1989;6(3):188-192.
15. Sox HC, Margulies I, Sox CH. Psychologically mediated effects of diagnostic tests. Ann Intern Med. 1981;95(6):680-685.
16. Stewart MA. Effective physician-patient communication and health outcomes: a review. CMAJ. 1995;152(9):1423-1433.
17. Girgis A, Cockburn J, Butow P, et al. Improving patient emotional functioning and psychological morbidity: evaluation of a consultation skills training program for oncologists. Patient Educ Couns. 2009;77(3):456-462.
18. Birkhäuer J, Gaab J, Kossowsky J, et al. Trust in the health care professional and health outcome: a meta-analysis. PLoS One. 2017;12(2):e0170988.
19. Thomas KB. General practice consultations: is there any point in being positive? Br Med J (Clin Res Ed). 1987;294(6581):1200-1202.
20. Howe LC, Goyer JP, Crum AJ. Harnessing the placebo effect: exploring the influence of physician characteristics on placebo response [published online May 9, 2017]. Health Psychol. doi: 10.1037/hea0000499.
21. Chen JA, Papakostas GI, Youn SJ, et al. Association between patient beliefs regarding assigned treatment and clinical response: reanalysis of data from the Hypericum Depression Trial Study Group. J Clin Psychiatry. 2011;72(12):1669-1676.
22. Kelley JM, Kaptchuk TJ, Cusin C, et al. Open-label placebo for major depressive disorder: a pilot randomized controlled trial. Psychother Psychosom. 2012;81(5):312-314.
23. Carvalho C, Caetano JM, Cunha L, et al. Open-label placebo treatment in chronic low back pain: a randomized controlled trial. Pain. 2016;157(12):2766-2772.
24. Kaptchuk TJ, Friedlander E, Kelley JM, et al. Placebos without deception: a randomized controlled trial in irritable bowel syndrome. PLoS One. 2010;5(12):e15591.
25. Park LC, Covi L. Nonblind placebo trial: an exploration of neurotic patients’ responses to placebo when its inert content is disclosed. Arch Gen Psychiatry. 1965;12(4):336-345.
26. Schaefer M, Harke R, Denke C. Open-label placebos improve symptoms in allergic rhinitis: a randomized controlled trial. Psychother Psychosom. 2016;85(6):373-374.
27. Aulas JJ, Rosner I. Efficacy of a non blind placebo prescription [in French]. Encephale. 2003;29(1):68-71.
28. Sandler AD, Glesne CE, Bodfish JW. Conditioned placebo dose reduction: a new treatment in attention deficit hyperactivity disorder? J Dev Behav Pediatr. 2010;31(5):369-375.
29. Ader R, Mercurio MG, Walton J, et al. Conditioned pharmacotherapeutic effects: a preliminary study. Psychosom Med. 2010;72(2):192-197.
30. Weiss RD, O’Malley SS, Hosking JD, et al; COMBINE Study Research Group. Do patients with alcohol dependence respond to placebo? Results from the COMBINE Study. J Stud Alcohol Drugs. 2008;69(6):878-884.
31. Mattick RP, Breen C, Kimber J, et al. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev. 2014;(2):CD002207.
