User login
Oral Bowenoid Papulosis
To the Editor:
A 22-year-old Somali woman presented to our institution with oral lesions of 2 years’ duration. The lesions started as small papules in the corners of the mouth that gradually continued to spread to the mucosal lips and gums. The lesions did not drain any material. The patient reported that they were not painful and had not regressed. She was concerned about the cosmetic appearance of the lesions. The patient believed the lesions had developed from working in a chicken factory and was concerned that they appeared possibly due to contact with a substance in the factory. Additionally, she noted that her voice had become hoarse. She was otherwise healthy and denied any sexual contact or ever having a blood transfusion.
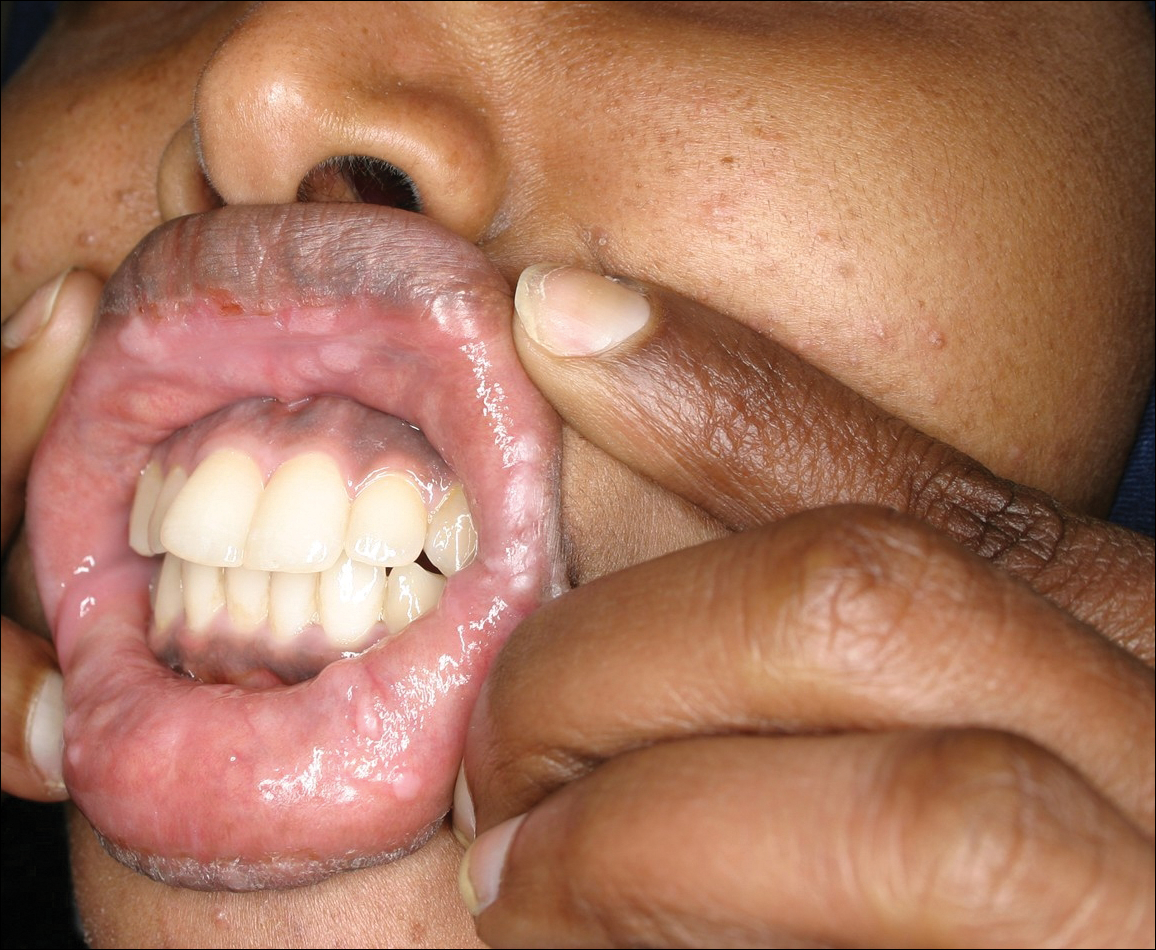
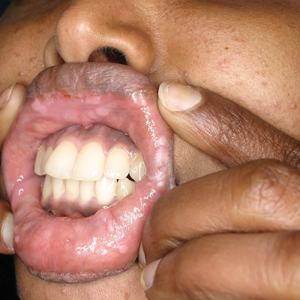
Physical examination revealed 10 to 15 flesh-colored papules measuring 2 to 3 mm in diameter on the vermilion, mucosal surfaces of the lips, and upper and lower gingivae (Figure 1). No lesions were seen on the hard and soft palate, tongue, buccal mucosa, or posterior pharynx.
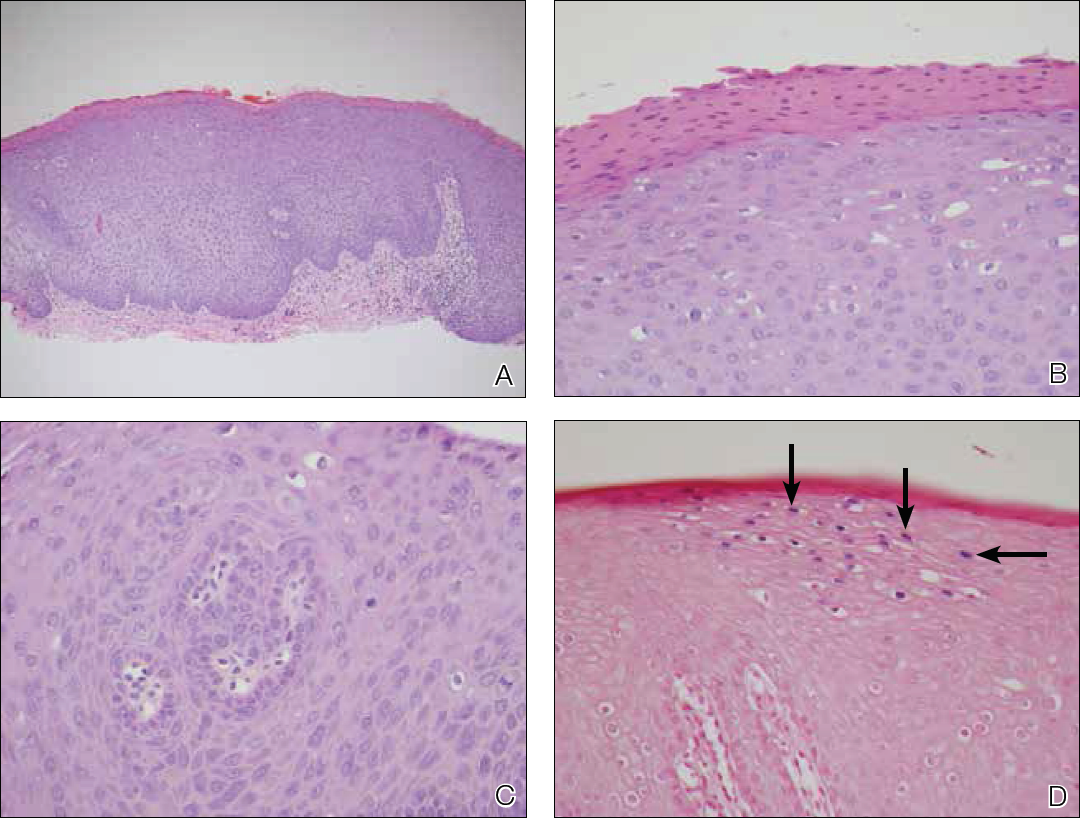
Skin biopsy of the left lower mucosal lip revealed parakeratosis, acanthosis, superficial koilocytes, and atypical keratinocytes with frequent mitoses (Figures 2A–2C). In situ hybridization testing for human papillomavirus (HPV) was negative for low-risk types 6 and 11 but positive for high-risk types 16 and 18 (Figure 2D). Laboratory investigations including complete blood cell count, electrolyte panel, and liver function studies were normal, and serum was negative for syphilis and human immunodeficiency virus antibodies.
The combined clinical and histologic findings were diagnostic of oral bowenoid papulosis. Gynecologic evaluation showed that the patient had undergone female circumcision, and she had a normal Papanicolaou test. The patient was referred to both the ear, nose, and throat clinic as well as the dermatologic surgery department to discuss treatment options, but she was lost to follow-up.
Bowenoid papulosis is triggered by HPV infection and manifests clinically as solitary or multiple verrucous papules and plaques that are usually located on the genitalia.1 Only a few cases of bowenoid papulosis have been reported in the oral cavity.1-5 Because this disease is sexually transmitted, the mean age of onset of bowenoid papulosis is 31 years.2 There is a small risk (2%–3%) of developing invasive carcinoma in bowenoid papulosis.1-3,6 Most lesions are associated with HPV type 16; however, bowenoid papulosis also has been associated with HPV types 18, 31, 32, 35, and 39.2
Some investigators consider bowenoid papulosis and Bowen disease (a type of squamous cell carcinoma [SCC] in situ) to be histologically identical1,6; however, some histologic differences have been reported.1-3,6 Bowenoid papulosis has more dilated and tortuous dermal capillaries and less atypia and dyskeratosis than Bowen disease.1,6 In contrast to bowenoid papulosis, Bowen disease is characterized clinically as well-defined scaly plaques on sun-exposed areas of the skin in older adults. Invasive SCC can be seen in 5% of skin lesions and 30% of penile lesions associated with Bowen disease.2 Risk factors for Bowen disease include sun exposure; arsenic poisoning; and infection with HPV types 2, 16, 18, 31, 33, 52, and 67.1,6
Oral bowenoid papulosis is rare. A PubMed search of articles indexed for MEDLINE using the term oral bowenoid papulosis yielded 7 additional cases, which are summarized in the Table. In 1987 Lookingbill et al2 described one of the first reported cases of oral disease in a 33-year-old immunosuppressed man receiving prednisone therapy for systemic lupus erythematosus who had both mouth and genital lesions. All lesions were positive for HPV type 16. The patient subsequently developed SCC of the tongue.2

The risk for progression of oral bowenoid papulosis to invasive SCC is not known. Our search yielded only 1 case of this occurrence.2
Two of 3 cases of solitary lip lesions in oral bowenoid papulosis were treated with surgical excision.1 Other treatment options include CO2 laser therapy, cryotherapy, 5-fluorouracil, bleomycin, intralesional interferon alfa, and imiquimod.1-3,5,6
Our case represents a rare report of oral bowenoid papulosis. Recognition of this unusual presentation is important for the diagnosis and management of this disease.
- Daley T, Birek C, Wysocki GP. Oral bowenoid lesions: differential diagnosis and pathogenetic insights. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:466-473.
- Lookingbill DP, Kreider JW, Howett MK, et al. Human papillomavirus type 16 in bowenoid papulosis, intraoral papillomas, and squamous cell carcinoma of the tongue. Arch Dermatol. 1987;123:363-368.
- Kratochvil FJ, Cioffi GA, Auclair PL, et al. Virus-associated dysplasia (bowenoid papulosis?) of the oral cavity. Oral Surg Oral Med Oral Pathol. 1989;68:312-316.
- Degener AM, Latino L, Pierangeli A, et al. Human papilloma virus-32-positive extragenital bowenoid papulosis in a HIV patient with typical genital bowenoid papulosis localization. Sex Transm Dis. 2004;31:619-622.
- Rinaggio J, Glick M, Lambert WC. Oral bowenoid papulosis in an HIV-positive male [published online October 14, 2005]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:328-332.
- Regezi JA, Dekker NP, Ramos DM, et al. Proliferation and invasion factors in HIV-associated dysplastic and nondysplastic oral warts and in oral squamous cell carcinoma: an immunohistochemical and RT-PCR evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:724-731.
To the Editor:
A 22-year-old Somali woman presented to our institution with oral lesions of 2 years’ duration. The lesions started as small papules in the corners of the mouth that gradually continued to spread to the mucosal lips and gums. The lesions did not drain any material. The patient reported that they were not painful and had not regressed. She was concerned about the cosmetic appearance of the lesions. The patient believed the lesions had developed from working in a chicken factory and was concerned that they appeared possibly due to contact with a substance in the factory. Additionally, she noted that her voice had become hoarse. She was otherwise healthy and denied any sexual contact or ever having a blood transfusion.
Physical examination revealed 10 to 15 flesh-colored papules measuring 2 to 3 mm in diameter on the vermilion, mucosal surfaces of the lips, and upper and lower gingivae (Figure 1). No lesions were seen on the hard and soft palate, tongue, buccal mucosa, or posterior pharynx.
Skin biopsy of the left lower mucosal lip revealed parakeratosis, acanthosis, superficial koilocytes, and atypical keratinocytes with frequent mitoses (Figures 2A–2C). In situ hybridization testing for human papillomavirus (HPV) was negative for low-risk types 6 and 11 but positive for high-risk types 16 and 18 (Figure 2D). Laboratory investigations including complete blood cell count, electrolyte panel, and liver function studies were normal, and serum was negative for syphilis and human immunodeficiency virus antibodies.
The combined clinical and histologic findings were diagnostic of oral bowenoid papulosis. Gynecologic evaluation showed that the patient had undergone female circumcision, and she had a normal Papanicolaou test. The patient was referred to both the ear, nose, and throat clinic as well as the dermatologic surgery department to discuss treatment options, but she was lost to follow-up.
Bowenoid papulosis is triggered by HPV infection and manifests clinically as solitary or multiple verrucous papules and plaques that are usually located on the genitalia.1 Only a few cases of bowenoid papulosis have been reported in the oral cavity.1-5 Because this disease is sexually transmitted, the mean age of onset of bowenoid papulosis is 31 years.2 There is a small risk (2%–3%) of developing invasive carcinoma in bowenoid papulosis.1-3,6 Most lesions are associated with HPV type 16; however, bowenoid papulosis also has been associated with HPV types 18, 31, 32, 35, and 39.2
Some investigators consider bowenoid papulosis and Bowen disease (a type of squamous cell carcinoma [SCC] in situ) to be histologically identical1,6; however, some histologic differences have been reported.1-3,6 Bowenoid papulosis has more dilated and tortuous dermal capillaries and less atypia and dyskeratosis than Bowen disease.1,6 In contrast to bowenoid papulosis, Bowen disease is characterized clinically as well-defined scaly plaques on sun-exposed areas of the skin in older adults. Invasive SCC can be seen in 5% of skin lesions and 30% of penile lesions associated with Bowen disease.2 Risk factors for Bowen disease include sun exposure; arsenic poisoning; and infection with HPV types 2, 16, 18, 31, 33, 52, and 67.1,6
Oral bowenoid papulosis is rare. A PubMed search of articles indexed for MEDLINE using the term oral bowenoid papulosis yielded 7 additional cases, which are summarized in the Table. In 1987 Lookingbill et al2 described one of the first reported cases of oral disease in a 33-year-old immunosuppressed man receiving prednisone therapy for systemic lupus erythematosus who had both mouth and genital lesions. All lesions were positive for HPV type 16. The patient subsequently developed SCC of the tongue.2
The risk for progression of oral bowenoid papulosis to invasive SCC is not known. Our search yielded only 1 case of this occurrence.2
Two of 3 cases of solitary lip lesions in oral bowenoid papulosis were treated with surgical excision.1 Other treatment options include CO2 laser therapy, cryotherapy, 5-fluorouracil, bleomycin, intralesional interferon alfa, and imiquimod.1-3,5,6
Our case represents a rare report of oral bowenoid papulosis. Recognition of this unusual presentation is important for the diagnosis and management of this disease.
To the Editor:
A 22-year-old Somali woman presented to our institution with oral lesions of 2 years’ duration. The lesions started as small papules in the corners of the mouth that gradually continued to spread to the mucosal lips and gums. The lesions did not drain any material. The patient reported that they were not painful and had not regressed. She was concerned about the cosmetic appearance of the lesions. The patient believed the lesions had developed from working in a chicken factory and was concerned that they appeared possibly due to contact with a substance in the factory. Additionally, she noted that her voice had become hoarse. She was otherwise healthy and denied any sexual contact or ever having a blood transfusion.
Physical examination revealed 10 to 15 flesh-colored papules measuring 2 to 3 mm in diameter on the vermilion, mucosal surfaces of the lips, and upper and lower gingivae (Figure 1). No lesions were seen on the hard and soft palate, tongue, buccal mucosa, or posterior pharynx.
Skin biopsy of the left lower mucosal lip revealed parakeratosis, acanthosis, superficial koilocytes, and atypical keratinocytes with frequent mitoses (Figures 2A–2C). In situ hybridization testing for human papillomavirus (HPV) was negative for low-risk types 6 and 11 but positive for high-risk types 16 and 18 (Figure 2D). Laboratory investigations including complete blood cell count, electrolyte panel, and liver function studies were normal, and serum was negative for syphilis and human immunodeficiency virus antibodies.
The combined clinical and histologic findings were diagnostic of oral bowenoid papulosis. Gynecologic evaluation showed that the patient had undergone female circumcision, and she had a normal Papanicolaou test. The patient was referred to both the ear, nose, and throat clinic as well as the dermatologic surgery department to discuss treatment options, but she was lost to follow-up.
Bowenoid papulosis is triggered by HPV infection and manifests clinically as solitary or multiple verrucous papules and plaques that are usually located on the genitalia.1 Only a few cases of bowenoid papulosis have been reported in the oral cavity.1-5 Because this disease is sexually transmitted, the mean age of onset of bowenoid papulosis is 31 years.2 There is a small risk (2%–3%) of developing invasive carcinoma in bowenoid papulosis.1-3,6 Most lesions are associated with HPV type 16; however, bowenoid papulosis also has been associated with HPV types 18, 31, 32, 35, and 39.2
Some investigators consider bowenoid papulosis and Bowen disease (a type of squamous cell carcinoma [SCC] in situ) to be histologically identical1,6; however, some histologic differences have been reported.1-3,6 Bowenoid papulosis has more dilated and tortuous dermal capillaries and less atypia and dyskeratosis than Bowen disease.1,6 In contrast to bowenoid papulosis, Bowen disease is characterized clinically as well-defined scaly plaques on sun-exposed areas of the skin in older adults. Invasive SCC can be seen in 5% of skin lesions and 30% of penile lesions associated with Bowen disease.2 Risk factors for Bowen disease include sun exposure; arsenic poisoning; and infection with HPV types 2, 16, 18, 31, 33, 52, and 67.1,6
Oral bowenoid papulosis is rare. A PubMed search of articles indexed for MEDLINE using the term oral bowenoid papulosis yielded 7 additional cases, which are summarized in the Table. In 1987 Lookingbill et al2 described one of the first reported cases of oral disease in a 33-year-old immunosuppressed man receiving prednisone therapy for systemic lupus erythematosus who had both mouth and genital lesions. All lesions were positive for HPV type 16. The patient subsequently developed SCC of the tongue.2
The risk for progression of oral bowenoid papulosis to invasive SCC is not known. Our search yielded only 1 case of this occurrence.2
Two of 3 cases of solitary lip lesions in oral bowenoid papulosis were treated with surgical excision.1 Other treatment options include CO2 laser therapy, cryotherapy, 5-fluorouracil, bleomycin, intralesional interferon alfa, and imiquimod.1-3,5,6
Our case represents a rare report of oral bowenoid papulosis. Recognition of this unusual presentation is important for the diagnosis and management of this disease.
- Daley T, Birek C, Wysocki GP. Oral bowenoid lesions: differential diagnosis and pathogenetic insights. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:466-473.
- Lookingbill DP, Kreider JW, Howett MK, et al. Human papillomavirus type 16 in bowenoid papulosis, intraoral papillomas, and squamous cell carcinoma of the tongue. Arch Dermatol. 1987;123:363-368.
- Kratochvil FJ, Cioffi GA, Auclair PL, et al. Virus-associated dysplasia (bowenoid papulosis?) of the oral cavity. Oral Surg Oral Med Oral Pathol. 1989;68:312-316.
- Degener AM, Latino L, Pierangeli A, et al. Human papilloma virus-32-positive extragenital bowenoid papulosis in a HIV patient with typical genital bowenoid papulosis localization. Sex Transm Dis. 2004;31:619-622.
- Rinaggio J, Glick M, Lambert WC. Oral bowenoid papulosis in an HIV-positive male [published online October 14, 2005]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:328-332.
- Regezi JA, Dekker NP, Ramos DM, et al. Proliferation and invasion factors in HIV-associated dysplastic and nondysplastic oral warts and in oral squamous cell carcinoma: an immunohistochemical and RT-PCR evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:724-731.
- Daley T, Birek C, Wysocki GP. Oral bowenoid lesions: differential diagnosis and pathogenetic insights. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:466-473.
- Lookingbill DP, Kreider JW, Howett MK, et al. Human papillomavirus type 16 in bowenoid papulosis, intraoral papillomas, and squamous cell carcinoma of the tongue. Arch Dermatol. 1987;123:363-368.
- Kratochvil FJ, Cioffi GA, Auclair PL, et al. Virus-associated dysplasia (bowenoid papulosis?) of the oral cavity. Oral Surg Oral Med Oral Pathol. 1989;68:312-316.
- Degener AM, Latino L, Pierangeli A, et al. Human papilloma virus-32-positive extragenital bowenoid papulosis in a HIV patient with typical genital bowenoid papulosis localization. Sex Transm Dis. 2004;31:619-622.
- Rinaggio J, Glick M, Lambert WC. Oral bowenoid papulosis in an HIV-positive male [published online October 14, 2005]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:328-332.
- Regezi JA, Dekker NP, Ramos DM, et al. Proliferation and invasion factors in HIV-associated dysplastic and nondysplastic oral warts and in oral squamous cell carcinoma: an immunohistochemical and RT-PCR evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:724-731.
Practice Points
- Bowenoid papulosis is triggered by human papillomavirus infection and manifests clinically as solitary or multiple verrucous papules and plaques that usually are located on the genitalia.
- Oral bowenoid papulosis is rare, and recognition of this unusual presentation is important for the diagnosis and management of this disease.
Gemcitabine-Induced Pseudocellulitis
To the Editor:
Gemcitabine is a nucleoside analogue used to treat a variety of solid and hematologic malignancies. Cutaneous toxicities include radiation recall dermatitis and erysipelaslike reactions that occur in areas not previously treated with radiation. Often referred to as pseudocellulitis, these reactions generally have been reported in areas of lymphedema in patients with solid malignancies.1-6 Herein, we report a rare case of gemcitabine-induced pseudocellulitis on the legs in a patient with a history of hematologic malignancy and total body irradiation (TBI).
A 61-year-old woman with history of peripheral T-cell lymphoma presented to the emergency department at our institution with acute-onset redness, tenderness, and swelling of the legs that was concerning for cellulitis. The patient’s history was notable for receiving gemcitabine 1000 mg/m2 for treatment of refractory lymphoma (12 and 4 days prior to presentation) as well as lymphedema of the legs. Her complete treatment course included multiple rounds of chemotherapy and matched unrelated donor nonmyeloablative allogeneic stem cell transplantation with a single dose of TBI at 200 cGy at our institution. Her transplant was complicated only by mild cutaneous graft-versus-host disease, which resolved with prednisone and tacrolimus.
On physical examination, the patient was afebrile with symmetric erythema and induration extending from the bilateral knees to the dorsal feet. A complete blood cell count was notable for a white blood cell count of 5400/µL (reference range, 4500–11,000/µL) and a platelet count of 96,000/µL (reference range, 150,000–400,000/µL). Plain film radiographs of the bilateral ankles were remarkable only for moderate subcutaneous edema. She received vancomycin in the emergency department and was admitted to the oncology service. Blood cultures drawn on admission were negative. Dermatology was consulted on admission, and a diagnosis of pseudocellulitis was made in conjunction with oncology (Figure). Antibiotics were held, and the patient was treated symptomatically with ibuprofen and was discharged 1 day after admission. The reaction resolved after 1 week with the use of diphenhydramine, nonsteroidal anti-inflammatory drugs, and compression. The patient was not rechallenged with gemcitabine.
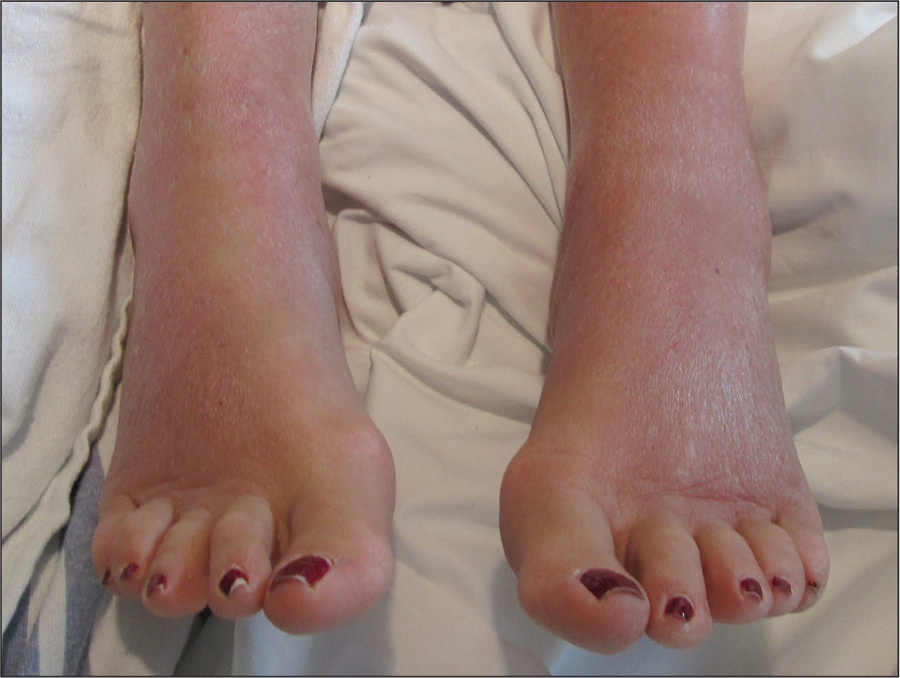
Gemcitabine-induced pseudocellulitis is a rare cutaneous side effect of gemcitabine therapy. Reported cases have suggested key characteristics of pseudocellulitis (Table). The reaction is characterized by localized erythema, edema, and tenderness of the skin, with onset generally 48 hours to 1 week after receiving gemcitabine.1-6 Lymphedema appears to be a risk factor.1,3-5 Six cases (including the current case) demonstrated confinement of these findings to areas of prior lymphedema.1,4,6 Infectious workup is negative, and rechallenging with gemcitabine likely will reproduce the reaction. Unlike radiation recall dermatitis, there is no prior localized radiation exposure.
Our patient had a history of hematologic malignancy and a one-time low-dose TBI of 200 cGy, unlike the other reported cases described in the Table. It is difficult to attribute our patient’s localized eruption to radiation recall given the history of TBI. The clinical examination, laboratory findings, and time frame of the reaction were consistent with gemcitabine-induced pseudocellulitis.
It is important to be aware of pseudocellulitis as a possible complication of gemcitabine therapy in patients without history of localized radiation. Early recognition of pseudocellulitis may prevent unnecessary exposure to broad-spectrum antibiotics. Patients’ temperature, white blood cell count, clinical examination, and potentially ancillary studies (eg, vascular studies, inflammatory markers) should be reviewed carefully to determine whether there is an infectious or alternate etiology. In patients with known prior lymphedema, it may be beneficial to educate clinicians and patients alike about this potential adverse effect of gemcitabine and the high likelihood of recurrence on re-exposure.
- Brandes A, Reichmann U, Plasswilm L, et al. Time- and dose-limiting erysipeloid rash confined to areas of lymphedema following treatment with gemcitabine—a report of three cases. Anticancer Drugs. 2000;11:15-17.
- Kuku I, Kaya E, Sevinc A, et al. Gemcitabine-induced erysipeloid skin lesions in a patient with malignant mesothelioma. J Eur Acad Dermatol Venereol. 2002;16:271-272.
- Zustovich F, Pavei P, Cartei G. Erysipeloid skin toxicity induced by gemcitabine. J Eur Acad Dermatol Venereol. 2006;20:757-758.
- Korniyenko A, Lozada J, Ranade A, et al. Recurrent lower extremity pseudocellulitis. Am J Ther. 2012;19:e141-e142.
- Singh A, Hampole H. Gemcitabine associated pseudocellulitis [published online June 14, 2012]. J Gen Intern Med. 2012;27:1721.
- Curtis S, Hong S, Gucalp R, et al. Gemcitabine-induced pseudocellulitis in a patient with recurrent lymphedema: a case report and review of the current literature. Am J Ther. 2016;23:e321-323.
To the Editor:
Gemcitabine is a nucleoside analogue used to treat a variety of solid and hematologic malignancies. Cutaneous toxicities include radiation recall dermatitis and erysipelaslike reactions that occur in areas not previously treated with radiation. Often referred to as pseudocellulitis, these reactions generally have been reported in areas of lymphedema in patients with solid malignancies.1-6 Herein, we report a rare case of gemcitabine-induced pseudocellulitis on the legs in a patient with a history of hematologic malignancy and total body irradiation (TBI).
A 61-year-old woman with history of peripheral T-cell lymphoma presented to the emergency department at our institution with acute-onset redness, tenderness, and swelling of the legs that was concerning for cellulitis. The patient’s history was notable for receiving gemcitabine 1000 mg/m2 for treatment of refractory lymphoma (12 and 4 days prior to presentation) as well as lymphedema of the legs. Her complete treatment course included multiple rounds of chemotherapy and matched unrelated donor nonmyeloablative allogeneic stem cell transplantation with a single dose of TBI at 200 cGy at our institution. Her transplant was complicated only by mild cutaneous graft-versus-host disease, which resolved with prednisone and tacrolimus.
On physical examination, the patient was afebrile with symmetric erythema and induration extending from the bilateral knees to the dorsal feet. A complete blood cell count was notable for a white blood cell count of 5400/µL (reference range, 4500–11,000/µL) and a platelet count of 96,000/µL (reference range, 150,000–400,000/µL). Plain film radiographs of the bilateral ankles were remarkable only for moderate subcutaneous edema. She received vancomycin in the emergency department and was admitted to the oncology service. Blood cultures drawn on admission were negative. Dermatology was consulted on admission, and a diagnosis of pseudocellulitis was made in conjunction with oncology (Figure). Antibiotics were held, and the patient was treated symptomatically with ibuprofen and was discharged 1 day after admission. The reaction resolved after 1 week with the use of diphenhydramine, nonsteroidal anti-inflammatory drugs, and compression. The patient was not rechallenged with gemcitabine.
Gemcitabine-induced pseudocellulitis is a rare cutaneous side effect of gemcitabine therapy. Reported cases have suggested key characteristics of pseudocellulitis (Table). The reaction is characterized by localized erythema, edema, and tenderness of the skin, with onset generally 48 hours to 1 week after receiving gemcitabine.1-6 Lymphedema appears to be a risk factor.1,3-5 Six cases (including the current case) demonstrated confinement of these findings to areas of prior lymphedema.1,4,6 Infectious workup is negative, and rechallenging with gemcitabine likely will reproduce the reaction. Unlike radiation recall dermatitis, there is no prior localized radiation exposure.
Our patient had a history of hematologic malignancy and a one-time low-dose TBI of 200 cGy, unlike the other reported cases described in the Table. It is difficult to attribute our patient’s localized eruption to radiation recall given the history of TBI. The clinical examination, laboratory findings, and time frame of the reaction were consistent with gemcitabine-induced pseudocellulitis.
It is important to be aware of pseudocellulitis as a possible complication of gemcitabine therapy in patients without history of localized radiation. Early recognition of pseudocellulitis may prevent unnecessary exposure to broad-spectrum antibiotics. Patients’ temperature, white blood cell count, clinical examination, and potentially ancillary studies (eg, vascular studies, inflammatory markers) should be reviewed carefully to determine whether there is an infectious or alternate etiology. In patients with known prior lymphedema, it may be beneficial to educate clinicians and patients alike about this potential adverse effect of gemcitabine and the high likelihood of recurrence on re-exposure.
To the Editor:
Gemcitabine is a nucleoside analogue used to treat a variety of solid and hematologic malignancies. Cutaneous toxicities include radiation recall dermatitis and erysipelaslike reactions that occur in areas not previously treated with radiation. Often referred to as pseudocellulitis, these reactions generally have been reported in areas of lymphedema in patients with solid malignancies.1-6 Herein, we report a rare case of gemcitabine-induced pseudocellulitis on the legs in a patient with a history of hematologic malignancy and total body irradiation (TBI).
A 61-year-old woman with history of peripheral T-cell lymphoma presented to the emergency department at our institution with acute-onset redness, tenderness, and swelling of the legs that was concerning for cellulitis. The patient’s history was notable for receiving gemcitabine 1000 mg/m2 for treatment of refractory lymphoma (12 and 4 days prior to presentation) as well as lymphedema of the legs. Her complete treatment course included multiple rounds of chemotherapy and matched unrelated donor nonmyeloablative allogeneic stem cell transplantation with a single dose of TBI at 200 cGy at our institution. Her transplant was complicated only by mild cutaneous graft-versus-host disease, which resolved with prednisone and tacrolimus.
On physical examination, the patient was afebrile with symmetric erythema and induration extending from the bilateral knees to the dorsal feet. A complete blood cell count was notable for a white blood cell count of 5400/µL (reference range, 4500–11,000/µL) and a platelet count of 96,000/µL (reference range, 150,000–400,000/µL). Plain film radiographs of the bilateral ankles were remarkable only for moderate subcutaneous edema. She received vancomycin in the emergency department and was admitted to the oncology service. Blood cultures drawn on admission were negative. Dermatology was consulted on admission, and a diagnosis of pseudocellulitis was made in conjunction with oncology (Figure). Antibiotics were held, and the patient was treated symptomatically with ibuprofen and was discharged 1 day after admission. The reaction resolved after 1 week with the use of diphenhydramine, nonsteroidal anti-inflammatory drugs, and compression. The patient was not rechallenged with gemcitabine.
Gemcitabine-induced pseudocellulitis is a rare cutaneous side effect of gemcitabine therapy. Reported cases have suggested key characteristics of pseudocellulitis (Table). The reaction is characterized by localized erythema, edema, and tenderness of the skin, with onset generally 48 hours to 1 week after receiving gemcitabine.1-6 Lymphedema appears to be a risk factor.1,3-5 Six cases (including the current case) demonstrated confinement of these findings to areas of prior lymphedema.1,4,6 Infectious workup is negative, and rechallenging with gemcitabine likely will reproduce the reaction. Unlike radiation recall dermatitis, there is no prior localized radiation exposure.
Our patient had a history of hematologic malignancy and a one-time low-dose TBI of 200 cGy, unlike the other reported cases described in the Table. It is difficult to attribute our patient’s localized eruption to radiation recall given the history of TBI. The clinical examination, laboratory findings, and time frame of the reaction were consistent with gemcitabine-induced pseudocellulitis.
It is important to be aware of pseudocellulitis as a possible complication of gemcitabine therapy in patients without history of localized radiation. Early recognition of pseudocellulitis may prevent unnecessary exposure to broad-spectrum antibiotics. Patients’ temperature, white blood cell count, clinical examination, and potentially ancillary studies (eg, vascular studies, inflammatory markers) should be reviewed carefully to determine whether there is an infectious or alternate etiology. In patients with known prior lymphedema, it may be beneficial to educate clinicians and patients alike about this potential adverse effect of gemcitabine and the high likelihood of recurrence on re-exposure.
- Brandes A, Reichmann U, Plasswilm L, et al. Time- and dose-limiting erysipeloid rash confined to areas of lymphedema following treatment with gemcitabine—a report of three cases. Anticancer Drugs. 2000;11:15-17.
- Kuku I, Kaya E, Sevinc A, et al. Gemcitabine-induced erysipeloid skin lesions in a patient with malignant mesothelioma. J Eur Acad Dermatol Venereol. 2002;16:271-272.
- Zustovich F, Pavei P, Cartei G. Erysipeloid skin toxicity induced by gemcitabine. J Eur Acad Dermatol Venereol. 2006;20:757-758.
- Korniyenko A, Lozada J, Ranade A, et al. Recurrent lower extremity pseudocellulitis. Am J Ther. 2012;19:e141-e142.
- Singh A, Hampole H. Gemcitabine associated pseudocellulitis [published online June 14, 2012]. J Gen Intern Med. 2012;27:1721.
- Curtis S, Hong S, Gucalp R, et al. Gemcitabine-induced pseudocellulitis in a patient with recurrent lymphedema: a case report and review of the current literature. Am J Ther. 2016;23:e321-323.
- Brandes A, Reichmann U, Plasswilm L, et al. Time- and dose-limiting erysipeloid rash confined to areas of lymphedema following treatment with gemcitabine—a report of three cases. Anticancer Drugs. 2000;11:15-17.
- Kuku I, Kaya E, Sevinc A, et al. Gemcitabine-induced erysipeloid skin lesions in a patient with malignant mesothelioma. J Eur Acad Dermatol Venereol. 2002;16:271-272.
- Zustovich F, Pavei P, Cartei G. Erysipeloid skin toxicity induced by gemcitabine. J Eur Acad Dermatol Venereol. 2006;20:757-758.
- Korniyenko A, Lozada J, Ranade A, et al. Recurrent lower extremity pseudocellulitis. Am J Ther. 2012;19:e141-e142.
- Singh A, Hampole H. Gemcitabine associated pseudocellulitis [published online June 14, 2012]. J Gen Intern Med. 2012;27:1721.
- Curtis S, Hong S, Gucalp R, et al. Gemcitabine-induced pseudocellulitis in a patient with recurrent lymphedema: a case report and review of the current literature. Am J Ther. 2016;23:e321-323.
Practice Points
- Gemcitabine is a nucleoside analogue used to treat a variety of solid and hematologic malignancies.
- Gemcitabine-induced pseudocellulitis is a rare cutaneous side effect of gemcitabine therapy.
- Early recognition of pseudocellulitis may prevent unnecessary exposure to broad-spectrum antibiotics.
DRESS Syndrome Induced by Telaprevir: A Potentially Fatal Adverse Event in Chronic Hepatitis C Therapy
To the Editor:
A 58-year-old woman with a history of hyperprolactinemia and gastrointestinal angiodysplasia presented to the dermatology department with a generalized skin rash of 3 weeks’ duration. She did not have a history of toxic habits. She had a history of chronic hepatitis C virus (HCV) genotype 1b (IL-28B locus) with severe hepatic fibrosis (stage 4) as assessed by ultrasound-based elastography. Due to lack of response, plasma HCV RNA was still detectable at week 12 of pegylated interferon and ribavirin (RIB) therapy, and triple therapy with pegylated interferon, RIB, and telaprevir was initiated.
Two months later, she was admitted to the hospital after developing a generalized cutaneous rash that covered 90% of the body surface area (BSA) along with fever (temperature, 38.5°C). Laboratory blood tests showed an elevated absolute eosinophil count (2000 cells/µL [reference range, 0–500 cells/µL]), anemia (hemoglobin, 6.5 g/dL [reference range, 12–16 g/dL]), thrombocytopenia (26×103/µL [reference range, 150–400×103/µL]), and altered liver function tests (serum alanine aminotransferase, 60 U/L [reference range, 0–45 U/L]; aspartate aminotransferase, 80 U/L [reference range, 0–40 U/L]). Plasma HCV RNA was undetectable at this visit. On physical examination a generalized exanthema with coalescing plaques was observed, as well as crusted vesicles covering the arms, legs, chest, abdomen, and back. Palmoplantar papules (Figure, A) and facial swelling (Figure, B) also were present. A skin biopsy specimen taken from a papule on the left arm showed superficial perivascular lymphocytic infiltration with dermal edema. These findings were consistent with a diagnosis of DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome. Application of the Adverse Drug Reaction Probability Scale1 in our patient (total score of 5) suggested that DRESS syndrome was a moderate adverse event likely related to the use of telaprevir.
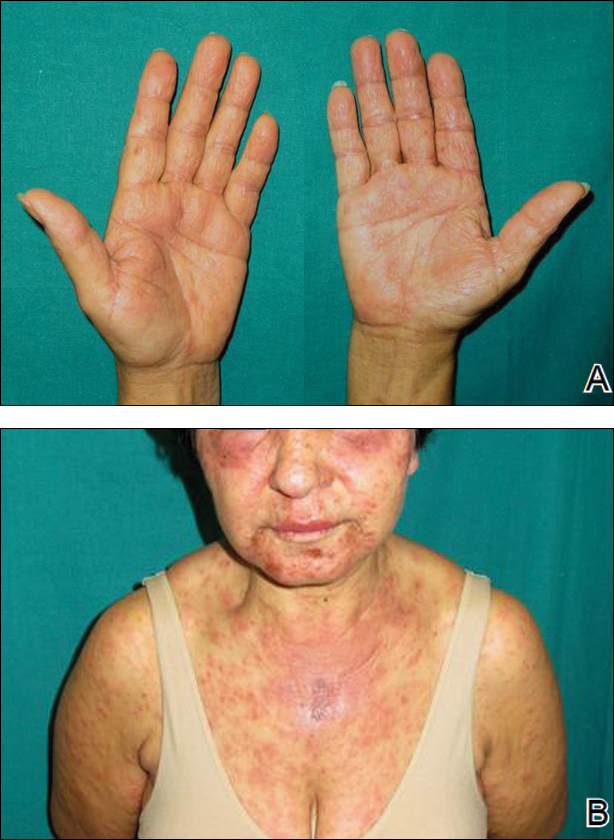
After diagnosis of DRESS syndrome, telaprevir was discontinued, and the doses of RIB and pegylated interferon were reduced to 200 mg and 180 µg weekly, respectively. Laboratory test values including liver function tests normalized within 3 weeks and remained normal on follow-up. Plasma HCV RNA continued to be undetectable.
Hepatitis C virus is relatively common with an incidence of 3% worldwide.2 It may present as an acute hepatitis or, more frequently, as asymptomatic chronic hepatitis. The acute process is self-limited and rarely causes hepatic failure. It usually leads to a chronic infection, which can result in cirrhosis, hepatocellular carcinoma, and the need for liver transplantation. The aim of treatment is eradication of HCV RNA, which is predicted by the attainment of a sustained virologic response. The latter is defined by the absence of HCV RNA by a polymerase chain reaction within 3 to 6 months after cessation of treatment.
Treatment of chronic HCV was based on the combination of pegylated interferon alfa-2a or -2b with RIB until 2015. Guidelines for the diagnosis and management of HCV infection have been published by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.2 These guidelines include new protease inhibitors, telaprevir and boceprevir, in the therapeutic approach of these patients. The main limitation of both drugs is the cutaneous toxicity.
Factors to be considered when treating HCV include viral genotype, if the patient is naïve or pretreated, the degree of fibrosis, established cirrhosis, and the treatment response. For patients with genotype 1,2 as in our case, combination therapy with 3 drugs is recommended: pegylated interferon 180 µg subcutaneous injection weekly, RIB 15 mg/kg daily, and telaprevir 2250 mg or boceprevir 2400 mg daily. Triple therapy has been shown to achieve a successful response in 75% of naïve patients and in 50% of patients refractory to standard therapy.3
Telaprevir is an NS3/4A protease inhibitor approved by the US Food and Drug Administration and the European Medicines Agency for treatment of chronic HCV infection in naïve patients and in those unresponsive to double therapy. In phase 2 clinical trials, 41% to 61% of patients treated with telaprevir developed cutaneous reactions, of which 5% to 8% required cessation of treatment.4 The predicting risk factors for developing a secondary rash to telaprevir include age older than 45 years, body mass index less than 30, Caucasian ethnicity, and receiving HCV therapy for the first time.4
This cutaneous side effect is managed depending on the extension of the lesions, the presence of systemic symptoms, and laboratory abnormalities.5 Therefore, the severity of the skin reaction can be divided into 4 stages4,5: (1) grade I or mild, defined as a localized rash with no systemic signs or mucosal involvement; (2) grade II or moderate, a maximum of 50% BSA involvement without epidermal detachment, and inflammation of the mucous membranes may be present without ulcers, as well as systemic symptoms such as fever, arthralgia, or eosinophilia; (3) grade III or severe, skin lesions affecting more than 50% BSA or less if any of the following lesions are present: vesicles or blisters, ulcers, epidermal detachment, palpable purpura, or erythema that does not blanch under pressure; (4) grade IV or life-threatening, when the clinical picture is consistent with acute generalized exanthematous pustulosis, DRESS syndrome, toxic epidermal necrolysis, or Stevens-Johnson syndrome.
DRESS syndrome is a condition clinically characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests. Cutaneous histopathologic examination may be unspecific, though atypical lymphocytes with a marked epidermotropism mimicking fungoid mycosis also have been described.6 In addition, human herpesvirus 6 serology may be negative, despite infection with this herpesvirus subtype having been associated with the development of DRESS syndrome. The pathophysiologic mechanism of DRESS syndrome is not completely understood; however, one theory ascribes an immunologic activation due to drug metabolite formation as the main mechanism.1
Eleven patients7 with possible DRESS syndrome have been reported in clinical trials (less than 5% of the total of patients), with an addition of 1 more by Montaudié et al.8 No notable differences were found between telaprevir levels in these patients with respect to those of the control group.
For the management of DRESS syndrome, the occurrence of early signs of a severe acute skin reaction requires the immediate cessation of the drug, telaprevir in this case. The withdrawal of the dual therapy will depend on the short-term clinical course, according to the general condition of the patient, as well as the analytical abnormalities observed.9
In conclusion, telaprevir is a promising novel therapy for the treatment of HCV infection, but its cutaneous side effects still need to be properly established.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharacol Ther. 1981;30:239-245.
- HCV guidance: recommendations for testing, managing, and treating hepatitis C. HCV Guidelines website. http://www.hcvguidelines.org. Accessed August 11, 2018.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405-2416.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Roujeau JC, Mockenhaupt M, Tahan SR, et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013;149:152-158.
- De Vriese AS, Philippe J, Van Renterghem DM, et al. Carbamazepine hypersensitivity syndrome: report of 4 cases and review of the literature. Medicine (Baltimore). 1995;74:144-151.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review [published online May 17, 2011]. Am J Med. 2011;124:588-597.
- Montaudié H, Passeron T, Cardot-Leccia N, et al. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303-305.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
To the Editor:
A 58-year-old woman with a history of hyperprolactinemia and gastrointestinal angiodysplasia presented to the dermatology department with a generalized skin rash of 3 weeks’ duration. She did not have a history of toxic habits. She had a history of chronic hepatitis C virus (HCV) genotype 1b (IL-28B locus) with severe hepatic fibrosis (stage 4) as assessed by ultrasound-based elastography. Due to lack of response, plasma HCV RNA was still detectable at week 12 of pegylated interferon and ribavirin (RIB) therapy, and triple therapy with pegylated interferon, RIB, and telaprevir was initiated.
Two months later, she was admitted to the hospital after developing a generalized cutaneous rash that covered 90% of the body surface area (BSA) along with fever (temperature, 38.5°C). Laboratory blood tests showed an elevated absolute eosinophil count (2000 cells/µL [reference range, 0–500 cells/µL]), anemia (hemoglobin, 6.5 g/dL [reference range, 12–16 g/dL]), thrombocytopenia (26×103/µL [reference range, 150–400×103/µL]), and altered liver function tests (serum alanine aminotransferase, 60 U/L [reference range, 0–45 U/L]; aspartate aminotransferase, 80 U/L [reference range, 0–40 U/L]). Plasma HCV RNA was undetectable at this visit. On physical examination a generalized exanthema with coalescing plaques was observed, as well as crusted vesicles covering the arms, legs, chest, abdomen, and back. Palmoplantar papules (Figure, A) and facial swelling (Figure, B) also were present. A skin biopsy specimen taken from a papule on the left arm showed superficial perivascular lymphocytic infiltration with dermal edema. These findings were consistent with a diagnosis of DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome. Application of the Adverse Drug Reaction Probability Scale1 in our patient (total score of 5) suggested that DRESS syndrome was a moderate adverse event likely related to the use of telaprevir.
After diagnosis of DRESS syndrome, telaprevir was discontinued, and the doses of RIB and pegylated interferon were reduced to 200 mg and 180 µg weekly, respectively. Laboratory test values including liver function tests normalized within 3 weeks and remained normal on follow-up. Plasma HCV RNA continued to be undetectable.
Hepatitis C virus is relatively common with an incidence of 3% worldwide.2 It may present as an acute hepatitis or, more frequently, as asymptomatic chronic hepatitis. The acute process is self-limited and rarely causes hepatic failure. It usually leads to a chronic infection, which can result in cirrhosis, hepatocellular carcinoma, and the need for liver transplantation. The aim of treatment is eradication of HCV RNA, which is predicted by the attainment of a sustained virologic response. The latter is defined by the absence of HCV RNA by a polymerase chain reaction within 3 to 6 months after cessation of treatment.
Treatment of chronic HCV was based on the combination of pegylated interferon alfa-2a or -2b with RIB until 2015. Guidelines for the diagnosis and management of HCV infection have been published by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.2 These guidelines include new protease inhibitors, telaprevir and boceprevir, in the therapeutic approach of these patients. The main limitation of both drugs is the cutaneous toxicity.
Factors to be considered when treating HCV include viral genotype, if the patient is naïve or pretreated, the degree of fibrosis, established cirrhosis, and the treatment response. For patients with genotype 1,2 as in our case, combination therapy with 3 drugs is recommended: pegylated interferon 180 µg subcutaneous injection weekly, RIB 15 mg/kg daily, and telaprevir 2250 mg or boceprevir 2400 mg daily. Triple therapy has been shown to achieve a successful response in 75% of naïve patients and in 50% of patients refractory to standard therapy.3
Telaprevir is an NS3/4A protease inhibitor approved by the US Food and Drug Administration and the European Medicines Agency for treatment of chronic HCV infection in naïve patients and in those unresponsive to double therapy. In phase 2 clinical trials, 41% to 61% of patients treated with telaprevir developed cutaneous reactions, of which 5% to 8% required cessation of treatment.4 The predicting risk factors for developing a secondary rash to telaprevir include age older than 45 years, body mass index less than 30, Caucasian ethnicity, and receiving HCV therapy for the first time.4
This cutaneous side effect is managed depending on the extension of the lesions, the presence of systemic symptoms, and laboratory abnormalities.5 Therefore, the severity of the skin reaction can be divided into 4 stages4,5: (1) grade I or mild, defined as a localized rash with no systemic signs or mucosal involvement; (2) grade II or moderate, a maximum of 50% BSA involvement without epidermal detachment, and inflammation of the mucous membranes may be present without ulcers, as well as systemic symptoms such as fever, arthralgia, or eosinophilia; (3) grade III or severe, skin lesions affecting more than 50% BSA or less if any of the following lesions are present: vesicles or blisters, ulcers, epidermal detachment, palpable purpura, or erythema that does not blanch under pressure; (4) grade IV or life-threatening, when the clinical picture is consistent with acute generalized exanthematous pustulosis, DRESS syndrome, toxic epidermal necrolysis, or Stevens-Johnson syndrome.
DRESS syndrome is a condition clinically characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests. Cutaneous histopathologic examination may be unspecific, though atypical lymphocytes with a marked epidermotropism mimicking fungoid mycosis also have been described.6 In addition, human herpesvirus 6 serology may be negative, despite infection with this herpesvirus subtype having been associated with the development of DRESS syndrome. The pathophysiologic mechanism of DRESS syndrome is not completely understood; however, one theory ascribes an immunologic activation due to drug metabolite formation as the main mechanism.1
Eleven patients7 with possible DRESS syndrome have been reported in clinical trials (less than 5% of the total of patients), with an addition of 1 more by Montaudié et al.8 No notable differences were found between telaprevir levels in these patients with respect to those of the control group.
For the management of DRESS syndrome, the occurrence of early signs of a severe acute skin reaction requires the immediate cessation of the drug, telaprevir in this case. The withdrawal of the dual therapy will depend on the short-term clinical course, according to the general condition of the patient, as well as the analytical abnormalities observed.9
In conclusion, telaprevir is a promising novel therapy for the treatment of HCV infection, but its cutaneous side effects still need to be properly established.
To the Editor:
A 58-year-old woman with a history of hyperprolactinemia and gastrointestinal angiodysplasia presented to the dermatology department with a generalized skin rash of 3 weeks’ duration. She did not have a history of toxic habits. She had a history of chronic hepatitis C virus (HCV) genotype 1b (IL-28B locus) with severe hepatic fibrosis (stage 4) as assessed by ultrasound-based elastography. Due to lack of response, plasma HCV RNA was still detectable at week 12 of pegylated interferon and ribavirin (RIB) therapy, and triple therapy with pegylated interferon, RIB, and telaprevir was initiated.
Two months later, she was admitted to the hospital after developing a generalized cutaneous rash that covered 90% of the body surface area (BSA) along with fever (temperature, 38.5°C). Laboratory blood tests showed an elevated absolute eosinophil count (2000 cells/µL [reference range, 0–500 cells/µL]), anemia (hemoglobin, 6.5 g/dL [reference range, 12–16 g/dL]), thrombocytopenia (26×103/µL [reference range, 150–400×103/µL]), and altered liver function tests (serum alanine aminotransferase, 60 U/L [reference range, 0–45 U/L]; aspartate aminotransferase, 80 U/L [reference range, 0–40 U/L]). Plasma HCV RNA was undetectable at this visit. On physical examination a generalized exanthema with coalescing plaques was observed, as well as crusted vesicles covering the arms, legs, chest, abdomen, and back. Palmoplantar papules (Figure, A) and facial swelling (Figure, B) also were present. A skin biopsy specimen taken from a papule on the left arm showed superficial perivascular lymphocytic infiltration with dermal edema. These findings were consistent with a diagnosis of DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome. Application of the Adverse Drug Reaction Probability Scale1 in our patient (total score of 5) suggested that DRESS syndrome was a moderate adverse event likely related to the use of telaprevir.
After diagnosis of DRESS syndrome, telaprevir was discontinued, and the doses of RIB and pegylated interferon were reduced to 200 mg and 180 µg weekly, respectively. Laboratory test values including liver function tests normalized within 3 weeks and remained normal on follow-up. Plasma HCV RNA continued to be undetectable.
Hepatitis C virus is relatively common with an incidence of 3% worldwide.2 It may present as an acute hepatitis or, more frequently, as asymptomatic chronic hepatitis. The acute process is self-limited and rarely causes hepatic failure. It usually leads to a chronic infection, which can result in cirrhosis, hepatocellular carcinoma, and the need for liver transplantation. The aim of treatment is eradication of HCV RNA, which is predicted by the attainment of a sustained virologic response. The latter is defined by the absence of HCV RNA by a polymerase chain reaction within 3 to 6 months after cessation of treatment.
Treatment of chronic HCV was based on the combination of pegylated interferon alfa-2a or -2b with RIB until 2015. Guidelines for the diagnosis and management of HCV infection have been published by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.2 These guidelines include new protease inhibitors, telaprevir and boceprevir, in the therapeutic approach of these patients. The main limitation of both drugs is the cutaneous toxicity.
Factors to be considered when treating HCV include viral genotype, if the patient is naïve or pretreated, the degree of fibrosis, established cirrhosis, and the treatment response. For patients with genotype 1,2 as in our case, combination therapy with 3 drugs is recommended: pegylated interferon 180 µg subcutaneous injection weekly, RIB 15 mg/kg daily, and telaprevir 2250 mg or boceprevir 2400 mg daily. Triple therapy has been shown to achieve a successful response in 75% of naïve patients and in 50% of patients refractory to standard therapy.3
Telaprevir is an NS3/4A protease inhibitor approved by the US Food and Drug Administration and the European Medicines Agency for treatment of chronic HCV infection in naïve patients and in those unresponsive to double therapy. In phase 2 clinical trials, 41% to 61% of patients treated with telaprevir developed cutaneous reactions, of which 5% to 8% required cessation of treatment.4 The predicting risk factors for developing a secondary rash to telaprevir include age older than 45 years, body mass index less than 30, Caucasian ethnicity, and receiving HCV therapy for the first time.4
This cutaneous side effect is managed depending on the extension of the lesions, the presence of systemic symptoms, and laboratory abnormalities.5 Therefore, the severity of the skin reaction can be divided into 4 stages4,5: (1) grade I or mild, defined as a localized rash with no systemic signs or mucosal involvement; (2) grade II or moderate, a maximum of 50% BSA involvement without epidermal detachment, and inflammation of the mucous membranes may be present without ulcers, as well as systemic symptoms such as fever, arthralgia, or eosinophilia; (3) grade III or severe, skin lesions affecting more than 50% BSA or less if any of the following lesions are present: vesicles or blisters, ulcers, epidermal detachment, palpable purpura, or erythema that does not blanch under pressure; (4) grade IV or life-threatening, when the clinical picture is consistent with acute generalized exanthematous pustulosis, DRESS syndrome, toxic epidermal necrolysis, or Stevens-Johnson syndrome.
DRESS syndrome is a condition clinically characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests. Cutaneous histopathologic examination may be unspecific, though atypical lymphocytes with a marked epidermotropism mimicking fungoid mycosis also have been described.6 In addition, human herpesvirus 6 serology may be negative, despite infection with this herpesvirus subtype having been associated with the development of DRESS syndrome. The pathophysiologic mechanism of DRESS syndrome is not completely understood; however, one theory ascribes an immunologic activation due to drug metabolite formation as the main mechanism.1
Eleven patients7 with possible DRESS syndrome have been reported in clinical trials (less than 5% of the total of patients), with an addition of 1 more by Montaudié et al.8 No notable differences were found between telaprevir levels in these patients with respect to those of the control group.
For the management of DRESS syndrome, the occurrence of early signs of a severe acute skin reaction requires the immediate cessation of the drug, telaprevir in this case. The withdrawal of the dual therapy will depend on the short-term clinical course, according to the general condition of the patient, as well as the analytical abnormalities observed.9
In conclusion, telaprevir is a promising novel therapy for the treatment of HCV infection, but its cutaneous side effects still need to be properly established.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharacol Ther. 1981;30:239-245.
- HCV guidance: recommendations for testing, managing, and treating hepatitis C. HCV Guidelines website. http://www.hcvguidelines.org. Accessed August 11, 2018.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405-2416.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Roujeau JC, Mockenhaupt M, Tahan SR, et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013;149:152-158.
- De Vriese AS, Philippe J, Van Renterghem DM, et al. Carbamazepine hypersensitivity syndrome: report of 4 cases and review of the literature. Medicine (Baltimore). 1995;74:144-151.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review [published online May 17, 2011]. Am J Med. 2011;124:588-597.
- Montaudié H, Passeron T, Cardot-Leccia N, et al. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303-305.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharacol Ther. 1981;30:239-245.
- HCV guidance: recommendations for testing, managing, and treating hepatitis C. HCV Guidelines website. http://www.hcvguidelines.org. Accessed August 11, 2018.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405-2416.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Roujeau JC, Mockenhaupt M, Tahan SR, et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013;149:152-158.
- De Vriese AS, Philippe J, Van Renterghem DM, et al. Carbamazepine hypersensitivity syndrome: report of 4 cases and review of the literature. Medicine (Baltimore). 1995;74:144-151.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review [published online May 17, 2011]. Am J Med. 2011;124:588-597.
- Montaudié H, Passeron T, Cardot-Leccia N, et al. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303-305.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
Practice Points
- DRESS syndrome is characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests.
- Severity of the skin reaction can be divided into 4 stages; in the third and fourth stages, adequate patient monitoring is necessary.
- Telaprevir is an NS3/4A protease inhibitor approved for treatment of chronic hepatitis C virus infection in naïve patients and in those unresponsive to double therapy. Its cutaneous side effects still need to be properly established.
Eumycetoma Pedis in an Albanian Farmer
To the Editor:
Mycetoma is a noncontagious chronic infection of the skin and subcutaneous tissue caused by exogenous fungi or bacteria that can involve deeper structures such as the fasciae, muscles, and bones. Clinically it is characterized by increased swelling of the affected area, fibrosis, nodules, tumefaction, formation of draining sinuses, and abscesses that drain pus-containing grains through fistulae.1 The initiation of the infection is related to local trauma and can involve muscle, underlying bone, and adjacent organs. The feet are the most commonly affected region, and the incubation period is variable. Patients rarely report prior trauma to the affected area and only seek medical consultation when the nodules and draining sinuses become evident. The etiopathogenesis of mycetoma is associated with aerobic actinomycetes (ie, Nocardia, Actinomadura, Streptomyces), known as actinomycetoma, and fungal infections, known as eumycetomas.1
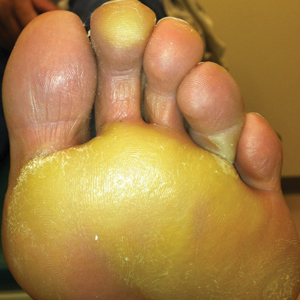
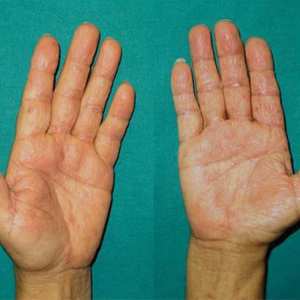
We report the case of a 57-year-old Albanian man who was referred to the outpatient clinic of our dermatology department for diagnosis and treatment of a chronic, suppurative, subcutaneous infection on the right foot presenting as abscesses and draining sinuses. The patient was a farmer and reported that the condition appeared 4 years prior following a laceration he sustained while at work. Dermatologic examination revealed local tumefaction, fistulated nodules, and abscesses discharging a serohemorrhagic fluid on the right foot (Figure 1). Perilesional erythema and subcutaneous swelling were evident. There was no regional lymphadenopathy. Standard laboratory examination was normal. Radiography of the right foot showed no osteolytic lesions or evidence of osteomyelitis.
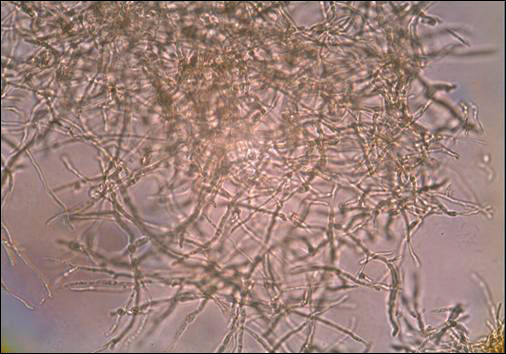
A skin biopsy from a lesion on the right foot was performed, and identification of the possible etiologic agent was based on direct microscopic examination of the granules, culture isolation of the agent, and fungal microscopic morphology.2 Granules were studied under direct examination with potassium hydroxide solution 20% and showed septate branching hyphae (Figure 2). The culture produced colonies that were white, yellow, and brown. Colonies were comprised of dense mycelium with melanin pigment and were grown at 37°C. A lactose tolerance test was positive.2 Therefore, the strain was identified as Madurella mycetomatis, and a diagnosis of eumycetoma pedis was made.
The patient was hospitalized for 2 weeks and treated with intravenous fluconazole, then treatment with oral itraconazole 200 mg once daily was initiated. At 4-month follow-up, he had self-discontinued treatment but demonstrated partial improvement of the tumefaction, healing of sinus tracts, and functional recovery of the right foot.
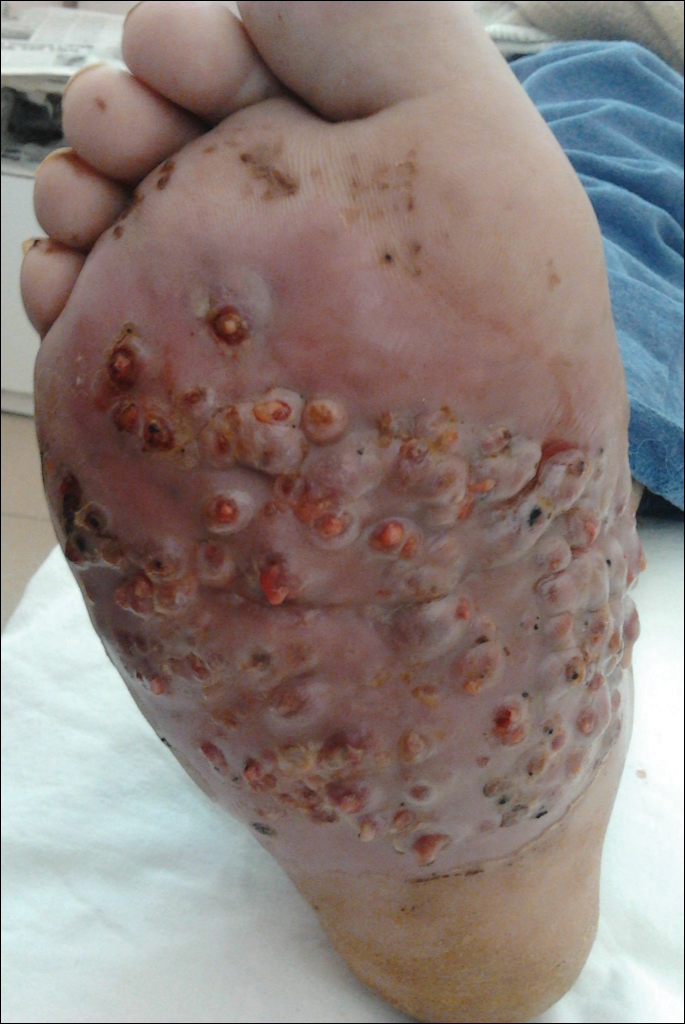
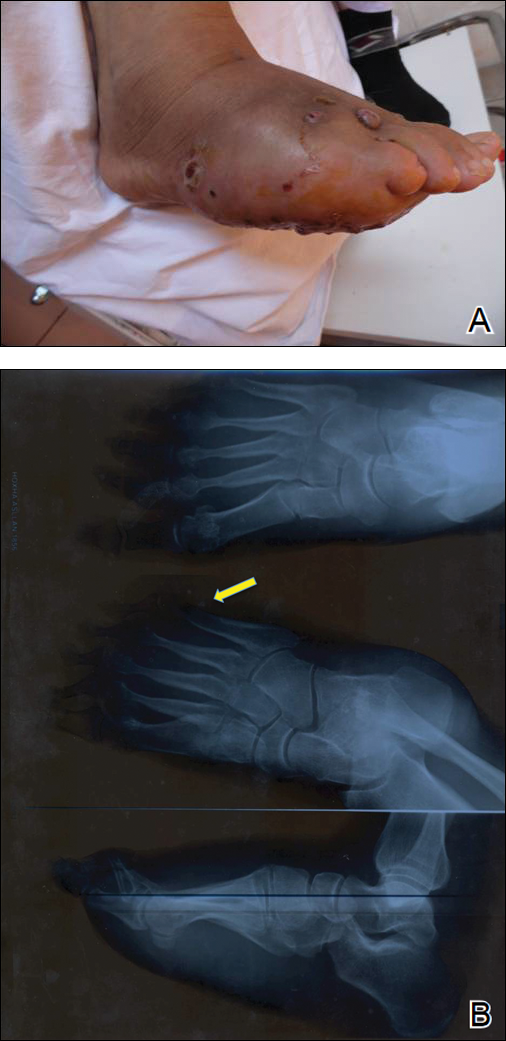
One year following the initial presentation, the patient’s clinical condition worsened (Figure 3A). Radiography of the right foot showed osteolytic lesions on bones in the right foot (Figure 3B), and a repeat culture showed the presence of Staphylococcus aureus; thus, treatment with itraconazole 200 mg once daily along with antibiotics (cefuroxime and gentamicin) was started immediately. Surgical treatment was recommended, but the patient refused treatment.
Mycetomas are rare in Albania but are common in countries of tropical and subtropical regions. K
Clinical features of eumycetoma include lesions with clear margins, few sinuses, black grains, slow progression, and long-term involvement of bone. The grains represent an aggregate of hyphae produced by fungi; thus, the characteristic feature of eumycetoma is the formation of large granules that can involve bone.1 A critical diagnostic step is to distinguish between eumycetoma and actinomycetoma. If possible, it is important to culture the organism because treatment varies depending on the cause of the infection.
Fungal identification is crucial in the diagnosis of mycetoma. In our case, diagnosis of eumycetoma pedis was based on clinical examination and detection of fungal species by microscopic examination and culture. The color of small granules (black grains) is a parameter used to identify different pathogens on histology but is not sufficient for diagnosis.5 The examination by potassium hydroxide preparation is helpful to identify the hyphae; however, culture is necessary.2
Therapeutic management of eumycetoma needs a combined strategy that includes systemic treatment and surgical therapy. Eumycetomas generally are more difficult to treat then actinomycetomas. Some authors recommend a high dose of amphotericin B as the treatment of choice for eumycetoma,6,7 but there are some that emphasize that amphotericin B is partially effective.8,9 There also is evidence in the literature of resistance of eumycetoma to ketoconazole treatment10,11 and successful treatment with fluconazole and itraconazole.10-13 For this reason, we treated our patient with the latter agents. In cases of osteolysis, amputation often is required.
In conclusion, eumycetoma pedis is a rare deep fungal infection that can cause considerable morbidity. P
- Rook A, Burns T. Rook’s Textbook of Dermatology. 8th ed. West Sussex, UK; Hoboken, NJ: Wiley-Blackwell; 2010.
- Balows A, Hausler WJ, eds. Manual of Clinical Microbiology. 5th ed. Washington, DC: American Society for Microbiology; 1991.
- Carter HV. On a new striking form of fungus disease principally affecting the foot and prevailing endemically in many parts of India. Trans Med Phys Soc Bombay. 1860;6:104-142.
- Kwon-Chung KJ, Bennet JE. Medical Mycology. Philadelphia, PA: Lea & Febiger; 1992.
- Venugopal PV, Venugopal TV. Pale grain eumycetomas in Madras. Australas J Dermatol. 1995;36:149-151.
- Guarro J, Gams W, Pujol I, et al. Acremonium species: new emerging fungal opportunists—in vitro antifungal susceptibilities and review. Clin Infec Dis. 1997;25:1222-1229.
- Lau YL, Yuen KY, Lee CW, et al. Invasive Acremonium falciforme infection in a patient with severe combined immunodeficiency. Clin Infect Dis. 1995;20:197-198.
- Fincher RM, Fisher JF, Lovell RD, et al. Infection due to the fungus Acremonium (cephalosporium). Medicine (Baltimore). 1991;70:398-409.
- Milburn PB, Papayanopulos DM, Pomerantz BM. Mycetoma due to Acremonium falciforme. Int J Dermatol. 1988;27:408-410.
- Welsh O, Salinas MC, Rodriguez MA. Treatment of eumycetoma and actinomycetoma. Cur Top Med Mycol. 1995;6:47-71.
- Restrepo A. Treatment of tropical mycoses. J Am Acad Dermatol. 1994;31:S91-S102.
- Gugnani HC, Ezeanolue BC, Khalil M, et al. Fluconazole in the therapy of tropical deep mycoses. Mycoses. 1995;38:485-488.
- Welsh O. Mycetoma. current concepts in treatment. Int J Dermatol. 1991;30:387-398.
To the Editor:
Mycetoma is a noncontagious chronic infection of the skin and subcutaneous tissue caused by exogenous fungi or bacteria that can involve deeper structures such as the fasciae, muscles, and bones. Clinically it is characterized by increased swelling of the affected area, fibrosis, nodules, tumefaction, formation of draining sinuses, and abscesses that drain pus-containing grains through fistulae.1 The initiation of the infection is related to local trauma and can involve muscle, underlying bone, and adjacent organs. The feet are the most commonly affected region, and the incubation period is variable. Patients rarely report prior trauma to the affected area and only seek medical consultation when the nodules and draining sinuses become evident. The etiopathogenesis of mycetoma is associated with aerobic actinomycetes (ie, Nocardia, Actinomadura, Streptomyces), known as actinomycetoma, and fungal infections, known as eumycetomas.1
We report the case of a 57-year-old Albanian man who was referred to the outpatient clinic of our dermatology department for diagnosis and treatment of a chronic, suppurative, subcutaneous infection on the right foot presenting as abscesses and draining sinuses. The patient was a farmer and reported that the condition appeared 4 years prior following a laceration he sustained while at work. Dermatologic examination revealed local tumefaction, fistulated nodules, and abscesses discharging a serohemorrhagic fluid on the right foot (Figure 1). Perilesional erythema and subcutaneous swelling were evident. There was no regional lymphadenopathy. Standard laboratory examination was normal. Radiography of the right foot showed no osteolytic lesions or evidence of osteomyelitis.
A skin biopsy from a lesion on the right foot was performed, and identification of the possible etiologic agent was based on direct microscopic examination of the granules, culture isolation of the agent, and fungal microscopic morphology.2 Granules were studied under direct examination with potassium hydroxide solution 20% and showed septate branching hyphae (Figure 2). The culture produced colonies that were white, yellow, and brown. Colonies were comprised of dense mycelium with melanin pigment and were grown at 37°C. A lactose tolerance test was positive.2 Therefore, the strain was identified as Madurella mycetomatis, and a diagnosis of eumycetoma pedis was made.
The patient was hospitalized for 2 weeks and treated with intravenous fluconazole, then treatment with oral itraconazole 200 mg once daily was initiated. At 4-month follow-up, he had self-discontinued treatment but demonstrated partial improvement of the tumefaction, healing of sinus tracts, and functional recovery of the right foot.
One year following the initial presentation, the patient’s clinical condition worsened (Figure 3A). Radiography of the right foot showed osteolytic lesions on bones in the right foot (Figure 3B), and a repeat culture showed the presence of Staphylococcus aureus; thus, treatment with itraconazole 200 mg once daily along with antibiotics (cefuroxime and gentamicin) was started immediately. Surgical treatment was recommended, but the patient refused treatment.
Mycetomas are rare in Albania but are common in countries of tropical and subtropical regions. K
Clinical features of eumycetoma include lesions with clear margins, few sinuses, black grains, slow progression, and long-term involvement of bone. The grains represent an aggregate of hyphae produced by fungi; thus, the characteristic feature of eumycetoma is the formation of large granules that can involve bone.1 A critical diagnostic step is to distinguish between eumycetoma and actinomycetoma. If possible, it is important to culture the organism because treatment varies depending on the cause of the infection.
Fungal identification is crucial in the diagnosis of mycetoma. In our case, diagnosis of eumycetoma pedis was based on clinical examination and detection of fungal species by microscopic examination and culture. The color of small granules (black grains) is a parameter used to identify different pathogens on histology but is not sufficient for diagnosis.5 The examination by potassium hydroxide preparation is helpful to identify the hyphae; however, culture is necessary.2
Therapeutic management of eumycetoma needs a combined strategy that includes systemic treatment and surgical therapy. Eumycetomas generally are more difficult to treat then actinomycetomas. Some authors recommend a high dose of amphotericin B as the treatment of choice for eumycetoma,6,7 but there are some that emphasize that amphotericin B is partially effective.8,9 There also is evidence in the literature of resistance of eumycetoma to ketoconazole treatment10,11 and successful treatment with fluconazole and itraconazole.10-13 For this reason, we treated our patient with the latter agents. In cases of osteolysis, amputation often is required.
In conclusion, eumycetoma pedis is a rare deep fungal infection that can cause considerable morbidity. P
To the Editor:
Mycetoma is a noncontagious chronic infection of the skin and subcutaneous tissue caused by exogenous fungi or bacteria that can involve deeper structures such as the fasciae, muscles, and bones. Clinically it is characterized by increased swelling of the affected area, fibrosis, nodules, tumefaction, formation of draining sinuses, and abscesses that drain pus-containing grains through fistulae.1 The initiation of the infection is related to local trauma and can involve muscle, underlying bone, and adjacent organs. The feet are the most commonly affected region, and the incubation period is variable. Patients rarely report prior trauma to the affected area and only seek medical consultation when the nodules and draining sinuses become evident. The etiopathogenesis of mycetoma is associated with aerobic actinomycetes (ie, Nocardia, Actinomadura, Streptomyces), known as actinomycetoma, and fungal infections, known as eumycetomas.1
We report the case of a 57-year-old Albanian man who was referred to the outpatient clinic of our dermatology department for diagnosis and treatment of a chronic, suppurative, subcutaneous infection on the right foot presenting as abscesses and draining sinuses. The patient was a farmer and reported that the condition appeared 4 years prior following a laceration he sustained while at work. Dermatologic examination revealed local tumefaction, fistulated nodules, and abscesses discharging a serohemorrhagic fluid on the right foot (Figure 1). Perilesional erythema and subcutaneous swelling were evident. There was no regional lymphadenopathy. Standard laboratory examination was normal. Radiography of the right foot showed no osteolytic lesions or evidence of osteomyelitis.
A skin biopsy from a lesion on the right foot was performed, and identification of the possible etiologic agent was based on direct microscopic examination of the granules, culture isolation of the agent, and fungal microscopic morphology.2 Granules were studied under direct examination with potassium hydroxide solution 20% and showed septate branching hyphae (Figure 2). The culture produced colonies that were white, yellow, and brown. Colonies were comprised of dense mycelium with melanin pigment and were grown at 37°C. A lactose tolerance test was positive.2 Therefore, the strain was identified as Madurella mycetomatis, and a diagnosis of eumycetoma pedis was made.
The patient was hospitalized for 2 weeks and treated with intravenous fluconazole, then treatment with oral itraconazole 200 mg once daily was initiated. At 4-month follow-up, he had self-discontinued treatment but demonstrated partial improvement of the tumefaction, healing of sinus tracts, and functional recovery of the right foot.
One year following the initial presentation, the patient’s clinical condition worsened (Figure 3A). Radiography of the right foot showed osteolytic lesions on bones in the right foot (Figure 3B), and a repeat culture showed the presence of Staphylococcus aureus; thus, treatment with itraconazole 200 mg once daily along with antibiotics (cefuroxime and gentamicin) was started immediately. Surgical treatment was recommended, but the patient refused treatment.
Mycetomas are rare in Albania but are common in countries of tropical and subtropical regions. K
Clinical features of eumycetoma include lesions with clear margins, few sinuses, black grains, slow progression, and long-term involvement of bone. The grains represent an aggregate of hyphae produced by fungi; thus, the characteristic feature of eumycetoma is the formation of large granules that can involve bone.1 A critical diagnostic step is to distinguish between eumycetoma and actinomycetoma. If possible, it is important to culture the organism because treatment varies depending on the cause of the infection.
Fungal identification is crucial in the diagnosis of mycetoma. In our case, diagnosis of eumycetoma pedis was based on clinical examination and detection of fungal species by microscopic examination and culture. The color of small granules (black grains) is a parameter used to identify different pathogens on histology but is not sufficient for diagnosis.5 The examination by potassium hydroxide preparation is helpful to identify the hyphae; however, culture is necessary.2
Therapeutic management of eumycetoma needs a combined strategy that includes systemic treatment and surgical therapy. Eumycetomas generally are more difficult to treat then actinomycetomas. Some authors recommend a high dose of amphotericin B as the treatment of choice for eumycetoma,6,7 but there are some that emphasize that amphotericin B is partially effective.8,9 There also is evidence in the literature of resistance of eumycetoma to ketoconazole treatment10,11 and successful treatment with fluconazole and itraconazole.10-13 For this reason, we treated our patient with the latter agents. In cases of osteolysis, amputation often is required.
In conclusion, eumycetoma pedis is a rare deep fungal infection that can cause considerable morbidity. P
- Rook A, Burns T. Rook’s Textbook of Dermatology. 8th ed. West Sussex, UK; Hoboken, NJ: Wiley-Blackwell; 2010.
- Balows A, Hausler WJ, eds. Manual of Clinical Microbiology. 5th ed. Washington, DC: American Society for Microbiology; 1991.
- Carter HV. On a new striking form of fungus disease principally affecting the foot and prevailing endemically in many parts of India. Trans Med Phys Soc Bombay. 1860;6:104-142.
- Kwon-Chung KJ, Bennet JE. Medical Mycology. Philadelphia, PA: Lea & Febiger; 1992.
- Venugopal PV, Venugopal TV. Pale grain eumycetomas in Madras. Australas J Dermatol. 1995;36:149-151.
- Guarro J, Gams W, Pujol I, et al. Acremonium species: new emerging fungal opportunists—in vitro antifungal susceptibilities and review. Clin Infec Dis. 1997;25:1222-1229.
- Lau YL, Yuen KY, Lee CW, et al. Invasive Acremonium falciforme infection in a patient with severe combined immunodeficiency. Clin Infect Dis. 1995;20:197-198.
- Fincher RM, Fisher JF, Lovell RD, et al. Infection due to the fungus Acremonium (cephalosporium). Medicine (Baltimore). 1991;70:398-409.
- Milburn PB, Papayanopulos DM, Pomerantz BM. Mycetoma due to Acremonium falciforme. Int J Dermatol. 1988;27:408-410.
- Welsh O, Salinas MC, Rodriguez MA. Treatment of eumycetoma and actinomycetoma. Cur Top Med Mycol. 1995;6:47-71.
- Restrepo A. Treatment of tropical mycoses. J Am Acad Dermatol. 1994;31:S91-S102.
- Gugnani HC, Ezeanolue BC, Khalil M, et al. Fluconazole in the therapy of tropical deep mycoses. Mycoses. 1995;38:485-488.
- Welsh O. Mycetoma. current concepts in treatment. Int J Dermatol. 1991;30:387-398.
- Rook A, Burns T. Rook’s Textbook of Dermatology. 8th ed. West Sussex, UK; Hoboken, NJ: Wiley-Blackwell; 2010.
- Balows A, Hausler WJ, eds. Manual of Clinical Microbiology. 5th ed. Washington, DC: American Society for Microbiology; 1991.
- Carter HV. On a new striking form of fungus disease principally affecting the foot and prevailing endemically in many parts of India. Trans Med Phys Soc Bombay. 1860;6:104-142.
- Kwon-Chung KJ, Bennet JE. Medical Mycology. Philadelphia, PA: Lea & Febiger; 1992.
- Venugopal PV, Venugopal TV. Pale grain eumycetomas in Madras. Australas J Dermatol. 1995;36:149-151.
- Guarro J, Gams W, Pujol I, et al. Acremonium species: new emerging fungal opportunists—in vitro antifungal susceptibilities and review. Clin Infec Dis. 1997;25:1222-1229.
- Lau YL, Yuen KY, Lee CW, et al. Invasive Acremonium falciforme infection in a patient with severe combined immunodeficiency. Clin Infect Dis. 1995;20:197-198.
- Fincher RM, Fisher JF, Lovell RD, et al. Infection due to the fungus Acremonium (cephalosporium). Medicine (Baltimore). 1991;70:398-409.
- Milburn PB, Papayanopulos DM, Pomerantz BM. Mycetoma due to Acremonium falciforme. Int J Dermatol. 1988;27:408-410.
- Welsh O, Salinas MC, Rodriguez MA. Treatment of eumycetoma and actinomycetoma. Cur Top Med Mycol. 1995;6:47-71.
- Restrepo A. Treatment of tropical mycoses. J Am Acad Dermatol. 1994;31:S91-S102.
- Gugnani HC, Ezeanolue BC, Khalil M, et al. Fluconazole in the therapy of tropical deep mycoses. Mycoses. 1995;38:485-488.
- Welsh O. Mycetoma. current concepts in treatment. Int J Dermatol. 1991;30:387-398.
Practice Points
- A critical step in the diagnosis of mycetomas is to distinguish between eumycetoma and actinomycetoma.
- Potassium hydroxide preparation is helpful to identify fungal infection.
- Eumycetomas generally are more difficult to treat and require a combined strategy including systemic treatment and surgical therapy.
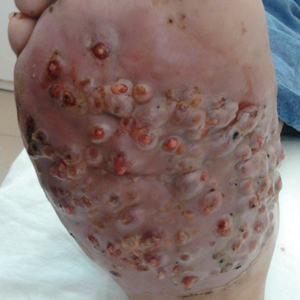
Postirradiation Morphea: Unique Presentation on the Breast
To the Editor:
Postirradiation morphea (PIM) is a rare but well-documented phenomenon that primarily occurs in breast cancer patients who have received radiation therapy; however, it also has been reported in patients who have received radiation therapy for lymphoma as well as endocervical, endometrial, and gastric carcinomas.1 Importantly, clinicians must be able to recognize and differentiate this condition from other causes of new-onset induration and erythema of the breast, such as cancer recurrence, a new primary malignancy, or inflammatory etiologies (eg, radiation or contact dermatitis). Typically, PIM presents months to years after radiation therapy as an erythematous patch within the irradiated area that progressively becomes indurated. We report an unusual case of PIM with a reticulated appearance occurring 3 weeks after radiotherapy, chemotherapy, and surgery for an infiltrating ductal carcinoma of the left breast.
A 62-year-old woman presented to the dermatology department with a stage IIA, lymph node–negative, estrogen and progesterone receptor–negative, human epidermal growth factor receptor 2–negative infiltrating ductal carcinoma of the left breast. She was treated with a partial mastectomy of the left breast followed by external beam radiotherapy to the entire left breast in combination with chemotherapy (doxorubicin, cyclophosphamide, paclitaxel). The patient received 15 fractions of 270 cGy (4050 cGy total) with a weekly 600-cGy boost over 21 days without any complications.
Three weeks after finishing radiation therapy, the patient developed redness and swelling of the left breast that did not encompass the entire radiation field. There was no associated pain or pruritus. She was treated by her surgical oncologist with topical calendula and 3 courses of cephalexin for suspected mastitis with only modest improvement, then was referred to dermatology 3 months later.
At the initial dermatology evaluation, the patient reported little improvement after antibiotics and topical calendula. On physical examination, there were erythematous, reticulated, dusky, indurated patches on the entire left breast. The area of most pronounced induration surrounded the surgical scar on the left superior breast. Punch biopsy for hematoxylin and eosin staining and tissue cultures was obtained at this appointment. The patient was started on doxycycline 100 mg twice daily and was instructed to apply triamcinolone ointment 0.1% twice daily to the affected area. After 1 month of therapy, she reported slight improvement in the degree of erythema with this regimen, but the involved area continued to extend outside of the radiation field to the central chest wall and medial right breast (Figure 1). Two additional biopsies—one from the central chest and another from the right breast—were then taken over the course of 4 months, given the consistently inconclusive clinicopathologic nature and failure of the eruption to respond to antibiotics plus topical corticosteroids.
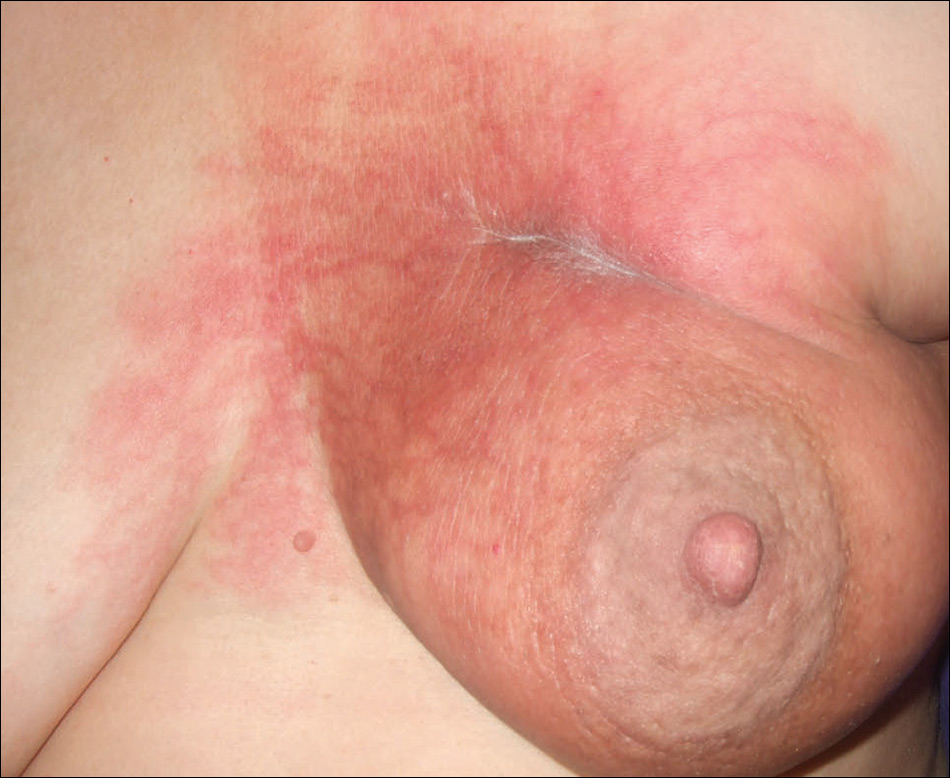
Punch biopsy from the central chest revealed a sparse perivascular infiltrate comprised predominantly of lymphocytes with occasional eosinophils (Figure 2). There were foci suggestive of early dermal sclerosis, an increased number of small blood vessels in the dermis, and scattered enlarged fibroblasts. Metastatic carcinoma was not identified. Although the histologic findings were not entirely specific, the changes were most suggestive of PIM, for which the patient was started on pentoxifylline (400 mg 3 times daily) and oral vitamin E supplementation (400 IU daily). At subsequent follow-up appointments, she showed markedly decreased skin erythema and induration.
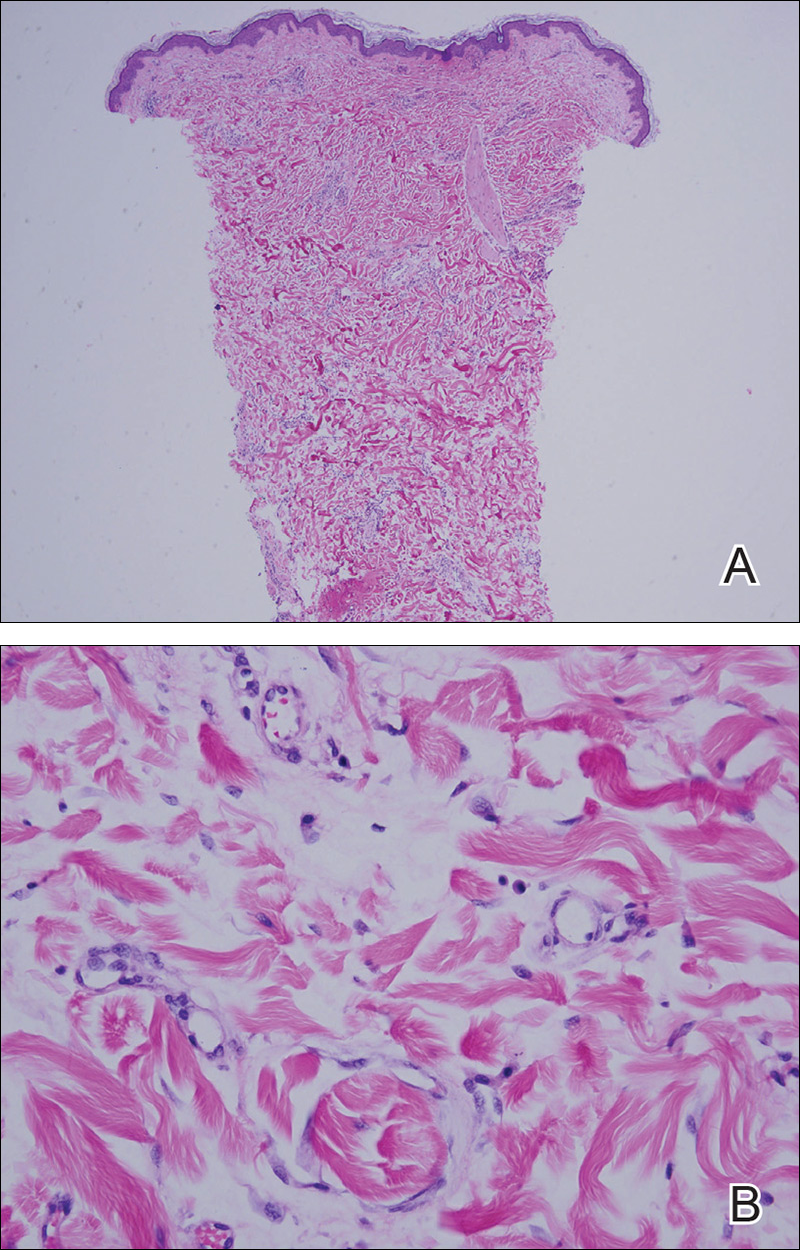
Morphea, also known as localized scleroderma, is an inflammatory skin condition characterized by sclerosis of the dermis and subcutis leading to scarlike tissue formation. Worldwide incidence ranges from 0.4 to 2.7 cases per 100,000 individuals with a predilection for white women.2 Unlike systemic scleroderma, morphea patients lack Raynaud phenomenon and visceral involvement.3,4
There are several clinical subtypes of morphea, including plaque, linear, generalized, and pansclerotic morphea. Lesions may vary in appearance based on configuration, stage of development, and depth of involvement.4 During the earliest phases, morphea lesions are asymptomatic, asymmetrically distributed, erythematous to violaceous patches or subtly indurated plaques expanding centrifugally with a lilac ring. Central sclerosis with loss of follicles and sweat glands is a later finding associated with advanced disease. Moreover, some reports of early-stage morphea have suggested a reticulated or geographic vascular morphology that may be misdiagnosed for other conditions such as a port-wine stain.5
Local skin exposures have long been hypothesized to contribute to development of morphea, including infection, especially Borrelia burgdorferi; trauma; chronic venous insufficiency; cosmetic surgery; medications; and exposure to toxic cooking oils, silicones, silica, pesticides, organic solvents, and vinyl chloride.2,6,7
Radiation therapy is an often overlooked cause of morphea. It was first described in 1905 but then rarely discussed until a 1989 case series of 9 patients, 7 of whom had received irradiation for breast cancer.8,9 Today, the increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in PIM incidence.10
In contrast to other radiation-induced skin conditions, development of PIM is independent of the presence or absence of adjuvant chemotherapy, type of radiation therapy, or the total radiation dose or fractionation number, with reported doses ranging from less than 20.0 Gy to up to 59.4 Gy and dose fractions ranging from 10 to 30. In 20% to 30% of cases, PIM extends beyond the radiation field, sometimes involving distant sites never exposed to high-energy rays.1,10,11 This observation suggests a mechanism reliant on more widespread cascade rather than solely local tissue damage.
Prominent culture-negative, lymphoplasmacytic inflammation is another important diagnostic clue. Radiation dermatitis and fibrosis do not have the marked erythematous to violaceous hue seen in early morphea plaques. This color seen in early morphea plaques may be intense enough and in a geographic pattern, emulating a vascular lesion.
There is no standardized treatment of PIM, but traditional therapies for morphea may provide some benefit. Several randomized controlled clinical trials have shown success with pentoxifylline and oral vitamin E supplementation to treat or prevent radiation-induced breast fibrosis.12 Extrapolating from this data, our patient was started on this combination therapy and showed marked improvement in skin color and texture.
- Morganroth PA, Dehoratius D, Curry H, et al. Postirradiation morphea: a case report with a review of the literature and summary of the clinicopathologic differential diagnosis [published online October 4, 2013]. Am J
Dermatopathol. doi:10.1097/DAD.0b013e3181cb3fdd. - Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228; quiz 229-230.
- Noh JW, Kim J, Kim JW. Localized scleroderma: a clinical study at a single center in Korea. Int J Rheum Dis. 2013;16:437-441.
- Vasquez R, Sendejo C, Jacobe H. Morphea and other localized forms of scleroderma. Curr Opin Rheumatol. 2012;24:685-693.
- Nijhawan RI, Bard S, Blyumin M, et al. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64:779-782.
- Haustein UF, Ziegler V. Environmentally induced systemic sclerosis-like disorders. Int J Dermatol. 1985;24:147-151.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol. 2009;36:2383-2396.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol. 1989;120:831-835.
- Crocker HR. Diseases of the Skin: Their Description, Pathology, Diagnosis, and Treatment. Philadelphia, PA: P. Blakiston Son & Co; 1905.
- Laetsch B, Hofer T, Lombriser N, et al. Irradiation-induced morphea: x-rays as triggers of autoimmunity. Dermatology. 2011;223:9-12.
- Shetty G, Lewis F, Thrush S. Morphea of the breast: case reports and review of literature. Breast J. 2007;13:302-304.
- Jacobson G, Bhatia S, Smith BJ, et al. Randomized trial of pentoxifylline and vitamin E vs standard follow-up after breast irradiation to prevent breast fibrosis, evaluated by tissue compliance meter. Int J Radiat Oncol Biol Phys. 2013;85:604-608.
To the Editor:
Postirradiation morphea (PIM) is a rare but well-documented phenomenon that primarily occurs in breast cancer patients who have received radiation therapy; however, it also has been reported in patients who have received radiation therapy for lymphoma as well as endocervical, endometrial, and gastric carcinomas.1 Importantly, clinicians must be able to recognize and differentiate this condition from other causes of new-onset induration and erythema of the breast, such as cancer recurrence, a new primary malignancy, or inflammatory etiologies (eg, radiation or contact dermatitis). Typically, PIM presents months to years after radiation therapy as an erythematous patch within the irradiated area that progressively becomes indurated. We report an unusual case of PIM with a reticulated appearance occurring 3 weeks after radiotherapy, chemotherapy, and surgery for an infiltrating ductal carcinoma of the left breast.
A 62-year-old woman presented to the dermatology department with a stage IIA, lymph node–negative, estrogen and progesterone receptor–negative, human epidermal growth factor receptor 2–negative infiltrating ductal carcinoma of the left breast. She was treated with a partial mastectomy of the left breast followed by external beam radiotherapy to the entire left breast in combination with chemotherapy (doxorubicin, cyclophosphamide, paclitaxel). The patient received 15 fractions of 270 cGy (4050 cGy total) with a weekly 600-cGy boost over 21 days without any complications.
Three weeks after finishing radiation therapy, the patient developed redness and swelling of the left breast that did not encompass the entire radiation field. There was no associated pain or pruritus. She was treated by her surgical oncologist with topical calendula and 3 courses of cephalexin for suspected mastitis with only modest improvement, then was referred to dermatology 3 months later.
At the initial dermatology evaluation, the patient reported little improvement after antibiotics and topical calendula. On physical examination, there were erythematous, reticulated, dusky, indurated patches on the entire left breast. The area of most pronounced induration surrounded the surgical scar on the left superior breast. Punch biopsy for hematoxylin and eosin staining and tissue cultures was obtained at this appointment. The patient was started on doxycycline 100 mg twice daily and was instructed to apply triamcinolone ointment 0.1% twice daily to the affected area. After 1 month of therapy, she reported slight improvement in the degree of erythema with this regimen, but the involved area continued to extend outside of the radiation field to the central chest wall and medial right breast (Figure 1). Two additional biopsies—one from the central chest and another from the right breast—were then taken over the course of 4 months, given the consistently inconclusive clinicopathologic nature and failure of the eruption to respond to antibiotics plus topical corticosteroids.
Punch biopsy from the central chest revealed a sparse perivascular infiltrate comprised predominantly of lymphocytes with occasional eosinophils (Figure 2). There were foci suggestive of early dermal sclerosis, an increased number of small blood vessels in the dermis, and scattered enlarged fibroblasts. Metastatic carcinoma was not identified. Although the histologic findings were not entirely specific, the changes were most suggestive of PIM, for which the patient was started on pentoxifylline (400 mg 3 times daily) and oral vitamin E supplementation (400 IU daily). At subsequent follow-up appointments, she showed markedly decreased skin erythema and induration.
Morphea, also known as localized scleroderma, is an inflammatory skin condition characterized by sclerosis of the dermis and subcutis leading to scarlike tissue formation. Worldwide incidence ranges from 0.4 to 2.7 cases per 100,000 individuals with a predilection for white women.2 Unlike systemic scleroderma, morphea patients lack Raynaud phenomenon and visceral involvement.3,4
There are several clinical subtypes of morphea, including plaque, linear, generalized, and pansclerotic morphea. Lesions may vary in appearance based on configuration, stage of development, and depth of involvement.4 During the earliest phases, morphea lesions are asymptomatic, asymmetrically distributed, erythematous to violaceous patches or subtly indurated plaques expanding centrifugally with a lilac ring. Central sclerosis with loss of follicles and sweat glands is a later finding associated with advanced disease. Moreover, some reports of early-stage morphea have suggested a reticulated or geographic vascular morphology that may be misdiagnosed for other conditions such as a port-wine stain.5
Local skin exposures have long been hypothesized to contribute to development of morphea, including infection, especially Borrelia burgdorferi; trauma; chronic venous insufficiency; cosmetic surgery; medications; and exposure to toxic cooking oils, silicones, silica, pesticides, organic solvents, and vinyl chloride.2,6,7
Radiation therapy is an often overlooked cause of morphea. It was first described in 1905 but then rarely discussed until a 1989 case series of 9 patients, 7 of whom had received irradiation for breast cancer.8,9 Today, the increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in PIM incidence.10
In contrast to other radiation-induced skin conditions, development of PIM is independent of the presence or absence of adjuvant chemotherapy, type of radiation therapy, or the total radiation dose or fractionation number, with reported doses ranging from less than 20.0 Gy to up to 59.4 Gy and dose fractions ranging from 10 to 30. In 20% to 30% of cases, PIM extends beyond the radiation field, sometimes involving distant sites never exposed to high-energy rays.1,10,11 This observation suggests a mechanism reliant on more widespread cascade rather than solely local tissue damage.
Prominent culture-negative, lymphoplasmacytic inflammation is another important diagnostic clue. Radiation dermatitis and fibrosis do not have the marked erythematous to violaceous hue seen in early morphea plaques. This color seen in early morphea plaques may be intense enough and in a geographic pattern, emulating a vascular lesion.
There is no standardized treatment of PIM, but traditional therapies for morphea may provide some benefit. Several randomized controlled clinical trials have shown success with pentoxifylline and oral vitamin E supplementation to treat or prevent radiation-induced breast fibrosis.12 Extrapolating from this data, our patient was started on this combination therapy and showed marked improvement in skin color and texture.
To the Editor:
Postirradiation morphea (PIM) is a rare but well-documented phenomenon that primarily occurs in breast cancer patients who have received radiation therapy; however, it also has been reported in patients who have received radiation therapy for lymphoma as well as endocervical, endometrial, and gastric carcinomas.1 Importantly, clinicians must be able to recognize and differentiate this condition from other causes of new-onset induration and erythema of the breast, such as cancer recurrence, a new primary malignancy, or inflammatory etiologies (eg, radiation or contact dermatitis). Typically, PIM presents months to years after radiation therapy as an erythematous patch within the irradiated area that progressively becomes indurated. We report an unusual case of PIM with a reticulated appearance occurring 3 weeks after radiotherapy, chemotherapy, and surgery for an infiltrating ductal carcinoma of the left breast.
A 62-year-old woman presented to the dermatology department with a stage IIA, lymph node–negative, estrogen and progesterone receptor–negative, human epidermal growth factor receptor 2–negative infiltrating ductal carcinoma of the left breast. She was treated with a partial mastectomy of the left breast followed by external beam radiotherapy to the entire left breast in combination with chemotherapy (doxorubicin, cyclophosphamide, paclitaxel). The patient received 15 fractions of 270 cGy (4050 cGy total) with a weekly 600-cGy boost over 21 days without any complications.
Three weeks after finishing radiation therapy, the patient developed redness and swelling of the left breast that did not encompass the entire radiation field. There was no associated pain or pruritus. She was treated by her surgical oncologist with topical calendula and 3 courses of cephalexin for suspected mastitis with only modest improvement, then was referred to dermatology 3 months later.
At the initial dermatology evaluation, the patient reported little improvement after antibiotics and topical calendula. On physical examination, there were erythematous, reticulated, dusky, indurated patches on the entire left breast. The area of most pronounced induration surrounded the surgical scar on the left superior breast. Punch biopsy for hematoxylin and eosin staining and tissue cultures was obtained at this appointment. The patient was started on doxycycline 100 mg twice daily and was instructed to apply triamcinolone ointment 0.1% twice daily to the affected area. After 1 month of therapy, she reported slight improvement in the degree of erythema with this regimen, but the involved area continued to extend outside of the radiation field to the central chest wall and medial right breast (Figure 1). Two additional biopsies—one from the central chest and another from the right breast—were then taken over the course of 4 months, given the consistently inconclusive clinicopathologic nature and failure of the eruption to respond to antibiotics plus topical corticosteroids.
Punch biopsy from the central chest revealed a sparse perivascular infiltrate comprised predominantly of lymphocytes with occasional eosinophils (Figure 2). There were foci suggestive of early dermal sclerosis, an increased number of small blood vessels in the dermis, and scattered enlarged fibroblasts. Metastatic carcinoma was not identified. Although the histologic findings were not entirely specific, the changes were most suggestive of PIM, for which the patient was started on pentoxifylline (400 mg 3 times daily) and oral vitamin E supplementation (400 IU daily). At subsequent follow-up appointments, she showed markedly decreased skin erythema and induration.
Morphea, also known as localized scleroderma, is an inflammatory skin condition characterized by sclerosis of the dermis and subcutis leading to scarlike tissue formation. Worldwide incidence ranges from 0.4 to 2.7 cases per 100,000 individuals with a predilection for white women.2 Unlike systemic scleroderma, morphea patients lack Raynaud phenomenon and visceral involvement.3,4
There are several clinical subtypes of morphea, including plaque, linear, generalized, and pansclerotic morphea. Lesions may vary in appearance based on configuration, stage of development, and depth of involvement.4 During the earliest phases, morphea lesions are asymptomatic, asymmetrically distributed, erythematous to violaceous patches or subtly indurated plaques expanding centrifugally with a lilac ring. Central sclerosis with loss of follicles and sweat glands is a later finding associated with advanced disease. Moreover, some reports of early-stage morphea have suggested a reticulated or geographic vascular morphology that may be misdiagnosed for other conditions such as a port-wine stain.5
Local skin exposures have long been hypothesized to contribute to development of morphea, including infection, especially Borrelia burgdorferi; trauma; chronic venous insufficiency; cosmetic surgery; medications; and exposure to toxic cooking oils, silicones, silica, pesticides, organic solvents, and vinyl chloride.2,6,7
Radiation therapy is an often overlooked cause of morphea. It was first described in 1905 but then rarely discussed until a 1989 case series of 9 patients, 7 of whom had received irradiation for breast cancer.8,9 Today, the increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in PIM incidence.10
In contrast to other radiation-induced skin conditions, development of PIM is independent of the presence or absence of adjuvant chemotherapy, type of radiation therapy, or the total radiation dose or fractionation number, with reported doses ranging from less than 20.0 Gy to up to 59.4 Gy and dose fractions ranging from 10 to 30. In 20% to 30% of cases, PIM extends beyond the radiation field, sometimes involving distant sites never exposed to high-energy rays.1,10,11 This observation suggests a mechanism reliant on more widespread cascade rather than solely local tissue damage.
Prominent culture-negative, lymphoplasmacytic inflammation is another important diagnostic clue. Radiation dermatitis and fibrosis do not have the marked erythematous to violaceous hue seen in early morphea plaques. This color seen in early morphea plaques may be intense enough and in a geographic pattern, emulating a vascular lesion.
There is no standardized treatment of PIM, but traditional therapies for morphea may provide some benefit. Several randomized controlled clinical trials have shown success with pentoxifylline and oral vitamin E supplementation to treat or prevent radiation-induced breast fibrosis.12 Extrapolating from this data, our patient was started on this combination therapy and showed marked improvement in skin color and texture.
- Morganroth PA, Dehoratius D, Curry H, et al. Postirradiation morphea: a case report with a review of the literature and summary of the clinicopathologic differential diagnosis [published online October 4, 2013]. Am J
Dermatopathol. doi:10.1097/DAD.0b013e3181cb3fdd. - Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228; quiz 229-230.
- Noh JW, Kim J, Kim JW. Localized scleroderma: a clinical study at a single center in Korea. Int J Rheum Dis. 2013;16:437-441.
- Vasquez R, Sendejo C, Jacobe H. Morphea and other localized forms of scleroderma. Curr Opin Rheumatol. 2012;24:685-693.
- Nijhawan RI, Bard S, Blyumin M, et al. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64:779-782.
- Haustein UF, Ziegler V. Environmentally induced systemic sclerosis-like disorders. Int J Dermatol. 1985;24:147-151.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol. 2009;36:2383-2396.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol. 1989;120:831-835.
- Crocker HR. Diseases of the Skin: Their Description, Pathology, Diagnosis, and Treatment. Philadelphia, PA: P. Blakiston Son & Co; 1905.
- Laetsch B, Hofer T, Lombriser N, et al. Irradiation-induced morphea: x-rays as triggers of autoimmunity. Dermatology. 2011;223:9-12.
- Shetty G, Lewis F, Thrush S. Morphea of the breast: case reports and review of literature. Breast J. 2007;13:302-304.
- Jacobson G, Bhatia S, Smith BJ, et al. Randomized trial of pentoxifylline and vitamin E vs standard follow-up after breast irradiation to prevent breast fibrosis, evaluated by tissue compliance meter. Int J Radiat Oncol Biol Phys. 2013;85:604-608.
- Morganroth PA, Dehoratius D, Curry H, et al. Postirradiation morphea: a case report with a review of the literature and summary of the clinicopathologic differential diagnosis [published online October 4, 2013]. Am J
Dermatopathol. doi:10.1097/DAD.0b013e3181cb3fdd. - Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228; quiz 229-230.
- Noh JW, Kim J, Kim JW. Localized scleroderma: a clinical study at a single center in Korea. Int J Rheum Dis. 2013;16:437-441.
- Vasquez R, Sendejo C, Jacobe H. Morphea and other localized forms of scleroderma. Curr Opin Rheumatol. 2012;24:685-693.
- Nijhawan RI, Bard S, Blyumin M, et al. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64:779-782.
- Haustein UF, Ziegler V. Environmentally induced systemic sclerosis-like disorders. Int J Dermatol. 1985;24:147-151.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol. 2009;36:2383-2396.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol. 1989;120:831-835.
- Crocker HR. Diseases of the Skin: Their Description, Pathology, Diagnosis, and Treatment. Philadelphia, PA: P. Blakiston Son & Co; 1905.
- Laetsch B, Hofer T, Lombriser N, et al. Irradiation-induced morphea: x-rays as triggers of autoimmunity. Dermatology. 2011;223:9-12.
- Shetty G, Lewis F, Thrush S. Morphea of the breast: case reports and review of literature. Breast J. 2007;13:302-304.
- Jacobson G, Bhatia S, Smith BJ, et al. Randomized trial of pentoxifylline and vitamin E vs standard follow-up after breast irradiation to prevent breast fibrosis, evaluated by tissue compliance meter. Int J Radiat Oncol Biol Phys. 2013;85:604-608.
Practice Points
- Radiation therapy is an often overlooked cause of morphea.
- The increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in postirradiation morphea incidence.
- Tissue changes occur as early as weeks or as late as 32 years after radiation treatment.
- Postirradiation morphea may extend beyond the radiation field.
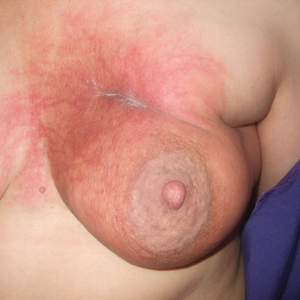
Autoimmune Progesterone Dermatitis
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
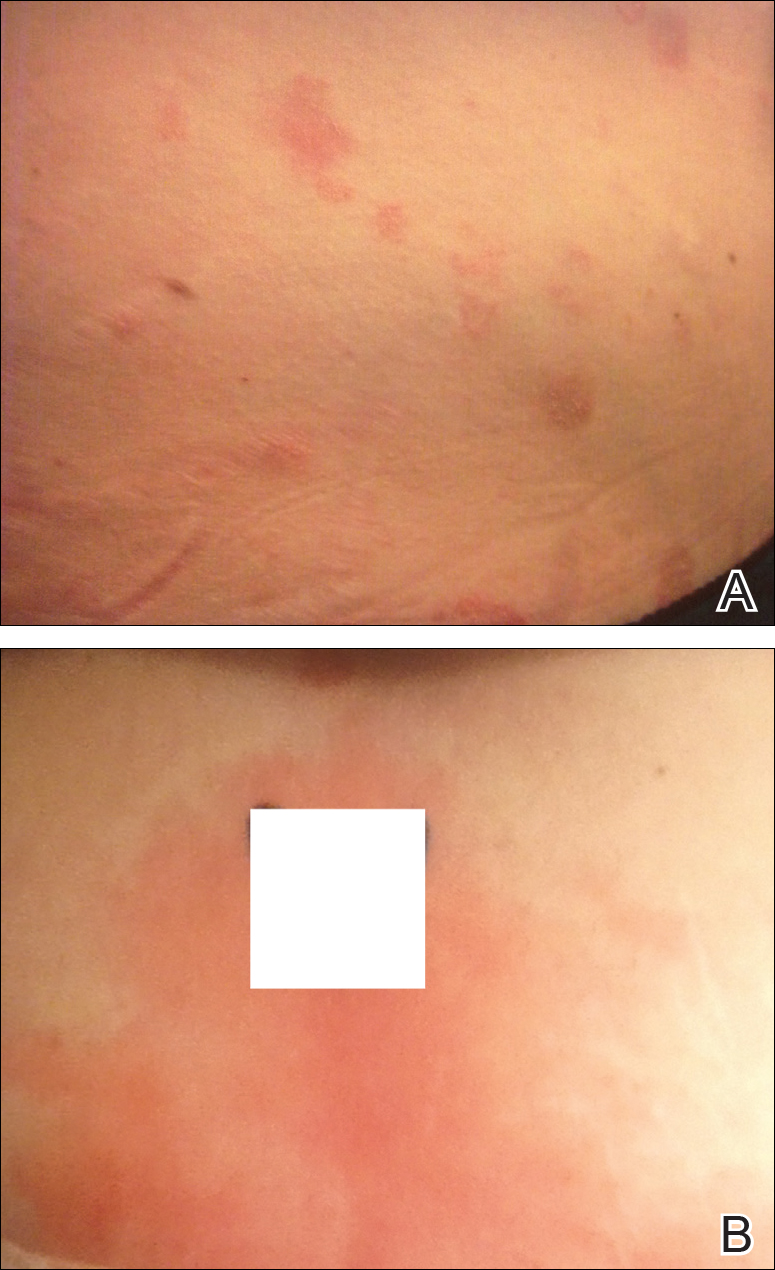
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
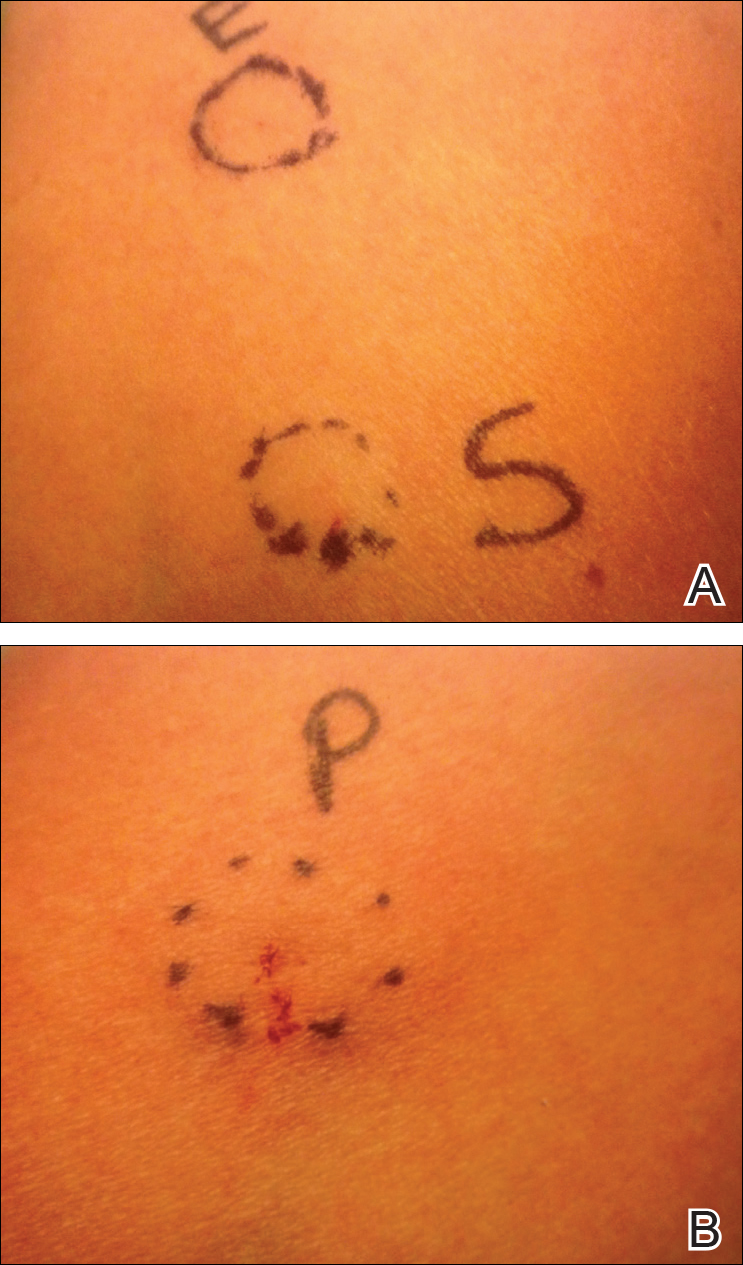
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
Practice Points
- Autoimmune progesterone dermatitis (APD) is a hypersensitivity reaction to the progesterone surge during a woman’s menstrual cycle.
- Patients with APD often are misdiagnosed for years due to the variability of each woman’s menstrual cycle, making the correlation difficult.
- It is important to keep APD in mind for any recalcitrant or recurrent rash in females. A thorough history is critical when formulating a diagnosis.
Mohs Micrographic Surgery Overlying a Pacemaker
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.

Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
Practice Points
- Surgical treatment of a cutaneous lesion overlying a cardiac device requires caution, both in avoidance of the device itself as well as electrocautery interference.
- Local anesthesia infiltrates can be used to create a temporary edematous thickening to minimize potential exposure of the device during the procedure.
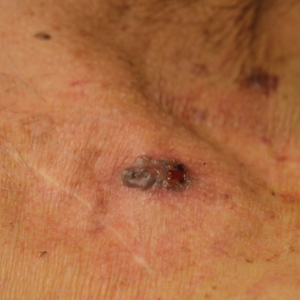
Intrahepatic Cholestasis of Pregnancy
To the Editor:
A 28-year-old primigravid woman at 32 weeks’ gestation presented to an outpatient dermatology clinic with a generalized rash and itch of 3 months’ duration. She was distressed with the itch and had tried antihistamines (eg, chlorpheniramine, cetirizine) without relief. She had no notable medical history. Physical examination revealed generalized erythematous papules and nodules on the chest, back, periumbilical region, arms, and legs (Figure). A few pustules were noted on the upper back. No wheals, plaques, vesicles, or bullae were seen.
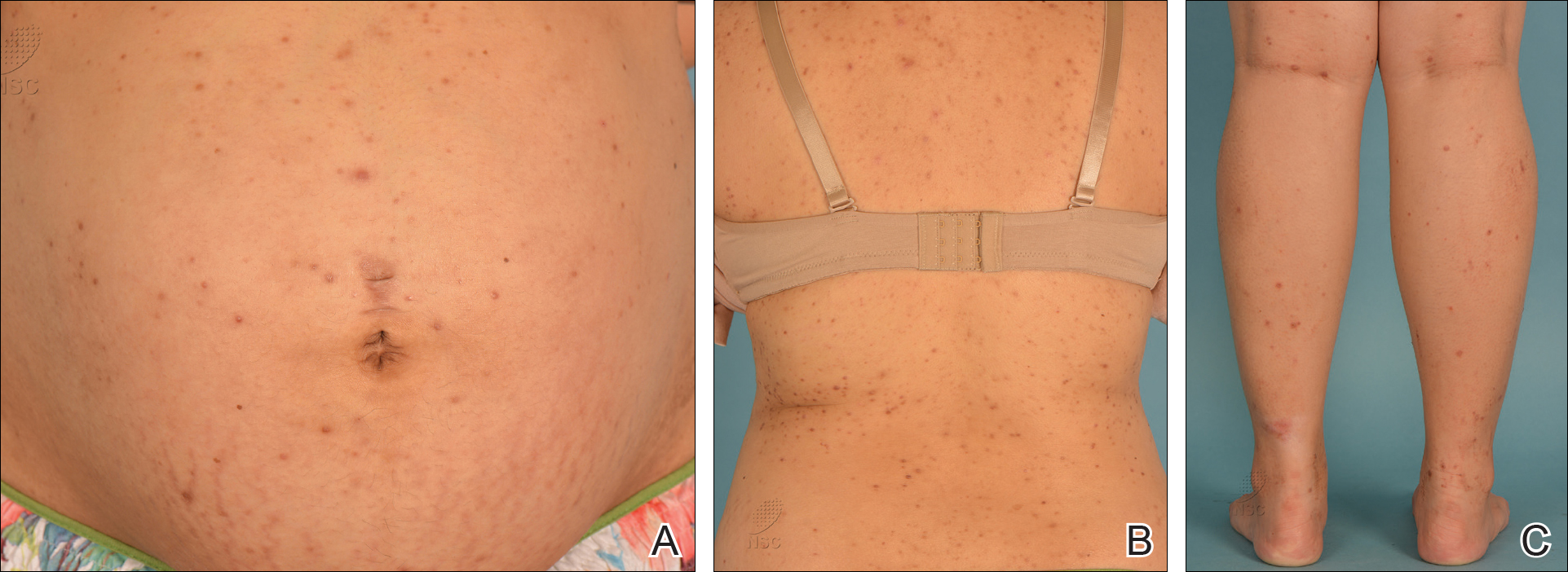
Laboratory investigations revealed elevated alkaline phosphatase (187 U/L [reference range, 30–120 U/L]), aspartate aminotransferase (45 U/L [reference range, 10–30 U/L]), alanine aminotransferase (120 U/L [reference range, 10–40 U/L]), and γ-glutamyltransferase (48 U/L [reference range, 9–40 U/L]) levels. A fungal scrape of the papules on the upper back demonstrated spores. Subsequent tests included ultrasonography of the liver, which showed fatty changes, as well as rising levels of alkaline phosphatase. Fasting glucose and 2-hour oral glucose tolerance tests showed poorly controlled gestational diabetes mellitus (DM) as well as raised triglycerides.
Based on the patient’s reports of itch, signs of erythematous papules and nodules, and laboratory results of cholestasis, a diagnosis of intrahepatic cholestasis of pregnancy (ICP) was made. The finding of Pityrosporum folliculitis also prompted screening for gestational DM, which was positive.
Treatment with ursodeoxycholic acid (UDCA) 250 mg twice daily was prescribed, which led to some relief of the skin symptoms. Her cutaneous symptoms were discussed with her obstetrician, and a decision was made for emergency cesarean delivery at 37 weeks’ gestation in light of nonreassuring fetal status during her follow-up antenatal ultrasonograph. Her pruritus and poor liver function resolved within 2 weeks after delivery.
Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy. The incidence varies with geographical location and ethnicity.1 It is one of the specific dermatoses of pregnancy and usually presents in the third trimester. It is characterized by pruritus, elevation of serum total bile acids and mild elevations of other liver function tests, and increased rates of adverse fetal outcomes. A positive diagnosis is made by the elevation of the serum total bile acid levels (>11.0 μmol/L [reference range, 0.73–5.63 μmol/L]). It is important for clinicians to recognize ICP because it is associated with fetal prematurity, intrapartal fetal distress, and stillbirths.2
The pathogenesis of ICP is not fully understood. During pregnancy, estrogens interfere with bile acid secretion, and progestins inhibit hepatic glucuronyltransferase. Increased IFN-γ, natural killer cells, and natural killer T cells, as well as decreased T cells in decidua parietalis, also have been reported.3
Mutations in the ATP binding cassette subfamily B member 4 gene, ABCB4, which encodes the multidrug resistance protein 3, a canalicular phosphatidylcholine translocase, have been found in several women with ICP.4 Clinically, patients usually present with pruritus that may precede or follow laboratory abnormalities. The pruritus worsens as the pregnancy advances and can resolve within 48 hours of delivery. Pruritus usually affects the palms and soles but may extend to the legs and abdomen or become generalized.4
Generally, there are no cutaneous signs other than excoriation marks, contrary to primary skin lesions found in other specific dermatoses of pregnancy. Mild jaundice can develop 2 to 4 weeks after the onset of pruritus, which may be associated with subclinical steatorrhea and increased risk of intrapartum and postpartum hemorrhage.5 Of note, ICP may be associated with increased risk for gestational DM, as illustrated in our case.6
Ursodeoxycholic acid currently is the most effective pharmacologic treatment of ICP. It reduces bile acids in cord blood, colostrum, and amniotic fluid.7 A meta-analysis of randomized controlled trials demonstrated that UDCA (450–1200 mg daily) is highly effective in alleviating pruritus and normalizing laboratory abnormalities associated with ICP.8 No severe adverse events have been reported related to UDCA.9,10
Intrahepatic cholestasis of pregnancy has been associated with increased risk for preterm delivery (19%–60%), meconium staining of amniotic fluid (≤27%), fetal bradycardia (≤14%), fetal distress (22%–41%), and fetal loss (0.4%–4.1%).11 The risk for serious fetal complications in ICP makes intensive fetal surveillance mandatory, including weekly nonstress cardiotocography or biophysical assessment from 34 weeks’ gestation. Delivery at 36 weeks or earlier (if lung maturity is achieved and cervix favorable) should be considered for severe cases with jaundice, progressive elevations in serum total bile acids, and suspected fetal distress. At more than 36 weeks’ gestation, amniocentesis and delivery should be considered if cervix is favorable and fetal lung maturity satisfactory.12-14
Our case highlights the importance of diagnosing ICP when a pregnant patient presents with generalized itch associated with elevated liver function tests. Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
- Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40:467-474.
- Ling B, Yao F, Zhou Y, et al. Cell-mediated immunity imbalance in patients with intrahepatic cholestasis of pregnancy. Cell Mol Immunol. 2007;4:71-75.
- Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genetics. 2000;9:1209-1217.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Martineau M, Raker C, Powrie R, et al. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur J Obstet Gynecol Reprod Biol. 2014;176:80-85.
- Laifer SA, Stiller RJ, Siddiqui DS, et al. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J Matern Fetal Med. 2001;10:131-135.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
- Tan LK. Obstetric cholestasis: current opinions and management. Ann Acad Med Singapore. 2003;32:294-298.
- Ghosh S, Chaudhuri S. Intra-hepatic cholestasis of pregnancy: a comprehensive review. Indian J Dermatol. 2013;58:327.
- Rioseco AJ, Ivankovic MB, Manzur A, et al. Intrahepatic cholestasis of pregnancy: retrospective case-control study of perinatal outcome. Am J Obstet Gynecol. 1994;170:890-895.
- Saleh MM, Abdo KR. Intrahepatic cholestasis of pregnancy: review of the literature and evaluation of current evidence. J Womens Health (Larchmt). 2007;16:833-841.
- Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management. Eur J Obstet Gynecol Reprod Biol. 2002;100:167-170.
To the Editor:
A 28-year-old primigravid woman at 32 weeks’ gestation presented to an outpatient dermatology clinic with a generalized rash and itch of 3 months’ duration. She was distressed with the itch and had tried antihistamines (eg, chlorpheniramine, cetirizine) without relief. She had no notable medical history. Physical examination revealed generalized erythematous papules and nodules on the chest, back, periumbilical region, arms, and legs (Figure). A few pustules were noted on the upper back. No wheals, plaques, vesicles, or bullae were seen.
Laboratory investigations revealed elevated alkaline phosphatase (187 U/L [reference range, 30–120 U/L]), aspartate aminotransferase (45 U/L [reference range, 10–30 U/L]), alanine aminotransferase (120 U/L [reference range, 10–40 U/L]), and γ-glutamyltransferase (48 U/L [reference range, 9–40 U/L]) levels. A fungal scrape of the papules on the upper back demonstrated spores. Subsequent tests included ultrasonography of the liver, which showed fatty changes, as well as rising levels of alkaline phosphatase. Fasting glucose and 2-hour oral glucose tolerance tests showed poorly controlled gestational diabetes mellitus (DM) as well as raised triglycerides.
Based on the patient’s reports of itch, signs of erythematous papules and nodules, and laboratory results of cholestasis, a diagnosis of intrahepatic cholestasis of pregnancy (ICP) was made. The finding of Pityrosporum folliculitis also prompted screening for gestational DM, which was positive.
Treatment with ursodeoxycholic acid (UDCA) 250 mg twice daily was prescribed, which led to some relief of the skin symptoms. Her cutaneous symptoms were discussed with her obstetrician, and a decision was made for emergency cesarean delivery at 37 weeks’ gestation in light of nonreassuring fetal status during her follow-up antenatal ultrasonograph. Her pruritus and poor liver function resolved within 2 weeks after delivery.
Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy. The incidence varies with geographical location and ethnicity.1 It is one of the specific dermatoses of pregnancy and usually presents in the third trimester. It is characterized by pruritus, elevation of serum total bile acids and mild elevations of other liver function tests, and increased rates of adverse fetal outcomes. A positive diagnosis is made by the elevation of the serum total bile acid levels (>11.0 μmol/L [reference range, 0.73–5.63 μmol/L]). It is important for clinicians to recognize ICP because it is associated with fetal prematurity, intrapartal fetal distress, and stillbirths.2
The pathogenesis of ICP is not fully understood. During pregnancy, estrogens interfere with bile acid secretion, and progestins inhibit hepatic glucuronyltransferase. Increased IFN-γ, natural killer cells, and natural killer T cells, as well as decreased T cells in decidua parietalis, also have been reported.3
Mutations in the ATP binding cassette subfamily B member 4 gene, ABCB4, which encodes the multidrug resistance protein 3, a canalicular phosphatidylcholine translocase, have been found in several women with ICP.4 Clinically, patients usually present with pruritus that may precede or follow laboratory abnormalities. The pruritus worsens as the pregnancy advances and can resolve within 48 hours of delivery. Pruritus usually affects the palms and soles but may extend to the legs and abdomen or become generalized.4
Generally, there are no cutaneous signs other than excoriation marks, contrary to primary skin lesions found in other specific dermatoses of pregnancy. Mild jaundice can develop 2 to 4 weeks after the onset of pruritus, which may be associated with subclinical steatorrhea and increased risk of intrapartum and postpartum hemorrhage.5 Of note, ICP may be associated with increased risk for gestational DM, as illustrated in our case.6
Ursodeoxycholic acid currently is the most effective pharmacologic treatment of ICP. It reduces bile acids in cord blood, colostrum, and amniotic fluid.7 A meta-analysis of randomized controlled trials demonstrated that UDCA (450–1200 mg daily) is highly effective in alleviating pruritus and normalizing laboratory abnormalities associated with ICP.8 No severe adverse events have been reported related to UDCA.9,10
Intrahepatic cholestasis of pregnancy has been associated with increased risk for preterm delivery (19%–60%), meconium staining of amniotic fluid (≤27%), fetal bradycardia (≤14%), fetal distress (22%–41%), and fetal loss (0.4%–4.1%).11 The risk for serious fetal complications in ICP makes intensive fetal surveillance mandatory, including weekly nonstress cardiotocography or biophysical assessment from 34 weeks’ gestation. Delivery at 36 weeks or earlier (if lung maturity is achieved and cervix favorable) should be considered for severe cases with jaundice, progressive elevations in serum total bile acids, and suspected fetal distress. At more than 36 weeks’ gestation, amniocentesis and delivery should be considered if cervix is favorable and fetal lung maturity satisfactory.12-14
Our case highlights the importance of diagnosing ICP when a pregnant patient presents with generalized itch associated with elevated liver function tests. Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
To the Editor:
A 28-year-old primigravid woman at 32 weeks’ gestation presented to an outpatient dermatology clinic with a generalized rash and itch of 3 months’ duration. She was distressed with the itch and had tried antihistamines (eg, chlorpheniramine, cetirizine) without relief. She had no notable medical history. Physical examination revealed generalized erythematous papules and nodules on the chest, back, periumbilical region, arms, and legs (Figure). A few pustules were noted on the upper back. No wheals, plaques, vesicles, or bullae were seen.
Laboratory investigations revealed elevated alkaline phosphatase (187 U/L [reference range, 30–120 U/L]), aspartate aminotransferase (45 U/L [reference range, 10–30 U/L]), alanine aminotransferase (120 U/L [reference range, 10–40 U/L]), and γ-glutamyltransferase (48 U/L [reference range, 9–40 U/L]) levels. A fungal scrape of the papules on the upper back demonstrated spores. Subsequent tests included ultrasonography of the liver, which showed fatty changes, as well as rising levels of alkaline phosphatase. Fasting glucose and 2-hour oral glucose tolerance tests showed poorly controlled gestational diabetes mellitus (DM) as well as raised triglycerides.
Based on the patient’s reports of itch, signs of erythematous papules and nodules, and laboratory results of cholestasis, a diagnosis of intrahepatic cholestasis of pregnancy (ICP) was made. The finding of Pityrosporum folliculitis also prompted screening for gestational DM, which was positive.
Treatment with ursodeoxycholic acid (UDCA) 250 mg twice daily was prescribed, which led to some relief of the skin symptoms. Her cutaneous symptoms were discussed with her obstetrician, and a decision was made for emergency cesarean delivery at 37 weeks’ gestation in light of nonreassuring fetal status during her follow-up antenatal ultrasonograph. Her pruritus and poor liver function resolved within 2 weeks after delivery.
Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy. The incidence varies with geographical location and ethnicity.1 It is one of the specific dermatoses of pregnancy and usually presents in the third trimester. It is characterized by pruritus, elevation of serum total bile acids and mild elevations of other liver function tests, and increased rates of adverse fetal outcomes. A positive diagnosis is made by the elevation of the serum total bile acid levels (>11.0 μmol/L [reference range, 0.73–5.63 μmol/L]). It is important for clinicians to recognize ICP because it is associated with fetal prematurity, intrapartal fetal distress, and stillbirths.2
The pathogenesis of ICP is not fully understood. During pregnancy, estrogens interfere with bile acid secretion, and progestins inhibit hepatic glucuronyltransferase. Increased IFN-γ, natural killer cells, and natural killer T cells, as well as decreased T cells in decidua parietalis, also have been reported.3
Mutations in the ATP binding cassette subfamily B member 4 gene, ABCB4, which encodes the multidrug resistance protein 3, a canalicular phosphatidylcholine translocase, have been found in several women with ICP.4 Clinically, patients usually present with pruritus that may precede or follow laboratory abnormalities. The pruritus worsens as the pregnancy advances and can resolve within 48 hours of delivery. Pruritus usually affects the palms and soles but may extend to the legs and abdomen or become generalized.4
Generally, there are no cutaneous signs other than excoriation marks, contrary to primary skin lesions found in other specific dermatoses of pregnancy. Mild jaundice can develop 2 to 4 weeks after the onset of pruritus, which may be associated with subclinical steatorrhea and increased risk of intrapartum and postpartum hemorrhage.5 Of note, ICP may be associated with increased risk for gestational DM, as illustrated in our case.6
Ursodeoxycholic acid currently is the most effective pharmacologic treatment of ICP. It reduces bile acids in cord blood, colostrum, and amniotic fluid.7 A meta-analysis of randomized controlled trials demonstrated that UDCA (450–1200 mg daily) is highly effective in alleviating pruritus and normalizing laboratory abnormalities associated with ICP.8 No severe adverse events have been reported related to UDCA.9,10
Intrahepatic cholestasis of pregnancy has been associated with increased risk for preterm delivery (19%–60%), meconium staining of amniotic fluid (≤27%), fetal bradycardia (≤14%), fetal distress (22%–41%), and fetal loss (0.4%–4.1%).11 The risk for serious fetal complications in ICP makes intensive fetal surveillance mandatory, including weekly nonstress cardiotocography or biophysical assessment from 34 weeks’ gestation. Delivery at 36 weeks or earlier (if lung maturity is achieved and cervix favorable) should be considered for severe cases with jaundice, progressive elevations in serum total bile acids, and suspected fetal distress. At more than 36 weeks’ gestation, amniocentesis and delivery should be considered if cervix is favorable and fetal lung maturity satisfactory.12-14
Our case highlights the importance of diagnosing ICP when a pregnant patient presents with generalized itch associated with elevated liver function tests. Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
- Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40:467-474.
- Ling B, Yao F, Zhou Y, et al. Cell-mediated immunity imbalance in patients with intrahepatic cholestasis of pregnancy. Cell Mol Immunol. 2007;4:71-75.
- Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genetics. 2000;9:1209-1217.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Martineau M, Raker C, Powrie R, et al. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur J Obstet Gynecol Reprod Biol. 2014;176:80-85.
- Laifer SA, Stiller RJ, Siddiqui DS, et al. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J Matern Fetal Med. 2001;10:131-135.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
- Tan LK. Obstetric cholestasis: current opinions and management. Ann Acad Med Singapore. 2003;32:294-298.
- Ghosh S, Chaudhuri S. Intra-hepatic cholestasis of pregnancy: a comprehensive review. Indian J Dermatol. 2013;58:327.
- Rioseco AJ, Ivankovic MB, Manzur A, et al. Intrahepatic cholestasis of pregnancy: retrospective case-control study of perinatal outcome. Am J Obstet Gynecol. 1994;170:890-895.
- Saleh MM, Abdo KR. Intrahepatic cholestasis of pregnancy: review of the literature and evaluation of current evidence. J Womens Health (Larchmt). 2007;16:833-841.
- Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management. Eur J Obstet Gynecol Reprod Biol. 2002;100:167-170.
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
- Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40:467-474.
- Ling B, Yao F, Zhou Y, et al. Cell-mediated immunity imbalance in patients with intrahepatic cholestasis of pregnancy. Cell Mol Immunol. 2007;4:71-75.
- Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genetics. 2000;9:1209-1217.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Martineau M, Raker C, Powrie R, et al. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur J Obstet Gynecol Reprod Biol. 2014;176:80-85.
- Laifer SA, Stiller RJ, Siddiqui DS, et al. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J Matern Fetal Med. 2001;10:131-135.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
- Tan LK. Obstetric cholestasis: current opinions and management. Ann Acad Med Singapore. 2003;32:294-298.
- Ghosh S, Chaudhuri S. Intra-hepatic cholestasis of pregnancy: a comprehensive review. Indian J Dermatol. 2013;58:327.
- Rioseco AJ, Ivankovic MB, Manzur A, et al. Intrahepatic cholestasis of pregnancy: retrospective case-control study of perinatal outcome. Am J Obstet Gynecol. 1994;170:890-895.
- Saleh MM, Abdo KR. Intrahepatic cholestasis of pregnancy: review of the literature and evaluation of current evidence. J Womens Health (Larchmt). 2007;16:833-841.
- Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management. Eur J Obstet Gynecol Reprod Biol. 2002;100:167-170.
Practice Points
- Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy.
- Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
Sweat Regeneration Following CO2 Fractionated Laser Therapy
To the Editor:
It is not uncommon for patients with extensive dermal scarring to overheat due to the inability to regulate body temperature through evaporative heat loss, as lack of perspiration in areas of prior full-thickness skin injury is well known. One of the authors (C.M.H.) previously reported a case of a patient with considerable hypertrophic scarring after surviving an episode of toxic epidermal necrolysis that was likely precipitated by lamotrigine.1 The patient initially presented to our clinic in consultation for laser therapy to improve the pliability and cosmetic appearance of the scars; however, approximately 3 weeks after initiating treatment with a fractional CO2 laser, the patient noticed perspiration in areas where she once lacked the ability to perspire as well as improved functionality.1 It was speculated that scar remodeling stimulated by the CO2 fractional laser allowed new connections to form between eccrine ducts in the dermis and epidermis.2
These findings are even more notable in light of a study by Rittié et al3 that suggested the primary appendages of the skin involved in human wound healing are the eccrine sweat glands. The investigators were able to demonstrate that eccrine sweat glands are major contributors in reepithelialization and wound healing in humans; therefore, it is possible that stimulating these glands with the CO2 laser may promote enhanced reepithelialization in addition to the reestablishment of perspiration and wound healing.3 Considering inadequate wound repair represents a substantial disturbance to the patient and health care system, this finding offers promise as a potential means to decrease morbidity in patients with dermal scarring from burns and traumatic injuries. We have since evaluated and treated 3 patients who demonstrated sweat regeneration following treatment with the fractional CO2 laser (Table).
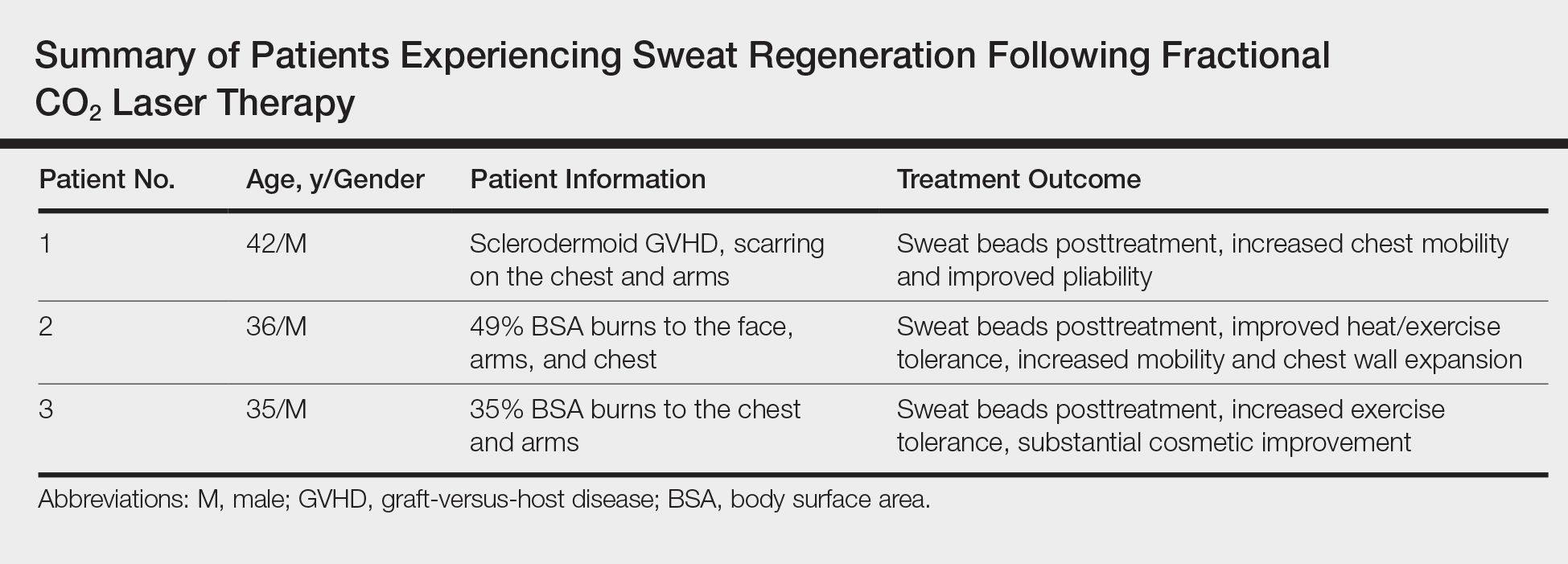
A 42-year-old man was our first patient to demonstrate functional scar improvement following bone marrow transplant for acute lymphoblastic leukemia complicated by chronic sclerodermoid graft-versus-host disease and subsequent extensive scarring on the chest and arms. Approximately 2 weeks after the first treatment with the fractional CO2 laser, the patient began to notice the presence of sweat beads in the treated areas. In addition to the reestablishment of perspiration, the patient had perceived increased mobility with improved pliability and “softness” (as described by family members) in treated areas likely related to scar remodeling.
A 36-year-old wounded army veteran presented with burns to the face, arms, and chest affecting 49% of the body surface area. After only 1 treatment, the patient reported that he could subjectively tolerate 10°F more ambient temperature and work all day outside in south Texas when heat intolerance previously would allow him to work only 2 to 3 hours. Additionally, he noted increased mobility and chest wall expansion, which in combination contributed to overall increased exercise tolerance and enhanced quality of life.
A 35-year-old US Marine and firefighter with burns primarily on the chest and arms involving 35% body surface area experienced increased exercise tolerance and sweat regeneration, particularly on the chest after a single treatment with the fractional CO2 laser but continued to experience improvement after a total of 3 treatments. Additionally, the cosmetic improvement was so substantial that the physician (C.M.H) had to review older photographs to verify the location of the scars.
We have now treated 3 patients with various mechanisms of injury and extensive scarring who noticed improved heat tolerance from sweat regeneration following fractional CO2 laser therapy. At this point, we only have anecdotal evidence of subjective functional improvement, and further research is warranted to elucidate the exact mechanism of action to support our findings.
- Neiner J, Whittemore D, Hivnor C. Buried alive: functional eccrine coils buried under scar tissue? J Am Acad Dermatol. 2011;65:661-663.
- Waibel J, Beer K, Narurkar V, et al. Preliminary observations on fractional ablative resurfacing devices: clinical impressions. J Drugs Dermatol. 2009;8:481-485.
- Rittié L, Sachs D, Orringer J, et al. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am J Pathol. 2013;1:163-171.
To the Editor:
It is not uncommon for patients with extensive dermal scarring to overheat due to the inability to regulate body temperature through evaporative heat loss, as lack of perspiration in areas of prior full-thickness skin injury is well known. One of the authors (C.M.H.) previously reported a case of a patient with considerable hypertrophic scarring after surviving an episode of toxic epidermal necrolysis that was likely precipitated by lamotrigine.1 The patient initially presented to our clinic in consultation for laser therapy to improve the pliability and cosmetic appearance of the scars; however, approximately 3 weeks after initiating treatment with a fractional CO2 laser, the patient noticed perspiration in areas where she once lacked the ability to perspire as well as improved functionality.1 It was speculated that scar remodeling stimulated by the CO2 fractional laser allowed new connections to form between eccrine ducts in the dermis and epidermis.2
These findings are even more notable in light of a study by Rittié et al3 that suggested the primary appendages of the skin involved in human wound healing are the eccrine sweat glands. The investigators were able to demonstrate that eccrine sweat glands are major contributors in reepithelialization and wound healing in humans; therefore, it is possible that stimulating these glands with the CO2 laser may promote enhanced reepithelialization in addition to the reestablishment of perspiration and wound healing.3 Considering inadequate wound repair represents a substantial disturbance to the patient and health care system, this finding offers promise as a potential means to decrease morbidity in patients with dermal scarring from burns and traumatic injuries. We have since evaluated and treated 3 patients who demonstrated sweat regeneration following treatment with the fractional CO2 laser (Table).
A 42-year-old man was our first patient to demonstrate functional scar improvement following bone marrow transplant for acute lymphoblastic leukemia complicated by chronic sclerodermoid graft-versus-host disease and subsequent extensive scarring on the chest and arms. Approximately 2 weeks after the first treatment with the fractional CO2 laser, the patient began to notice the presence of sweat beads in the treated areas. In addition to the reestablishment of perspiration, the patient had perceived increased mobility with improved pliability and “softness” (as described by family members) in treated areas likely related to scar remodeling.
A 36-year-old wounded army veteran presented with burns to the face, arms, and chest affecting 49% of the body surface area. After only 1 treatment, the patient reported that he could subjectively tolerate 10°F more ambient temperature and work all day outside in south Texas when heat intolerance previously would allow him to work only 2 to 3 hours. Additionally, he noted increased mobility and chest wall expansion, which in combination contributed to overall increased exercise tolerance and enhanced quality of life.
A 35-year-old US Marine and firefighter with burns primarily on the chest and arms involving 35% body surface area experienced increased exercise tolerance and sweat regeneration, particularly on the chest after a single treatment with the fractional CO2 laser but continued to experience improvement after a total of 3 treatments. Additionally, the cosmetic improvement was so substantial that the physician (C.M.H) had to review older photographs to verify the location of the scars.
We have now treated 3 patients with various mechanisms of injury and extensive scarring who noticed improved heat tolerance from sweat regeneration following fractional CO2 laser therapy. At this point, we only have anecdotal evidence of subjective functional improvement, and further research is warranted to elucidate the exact mechanism of action to support our findings.
To the Editor:
It is not uncommon for patients with extensive dermal scarring to overheat due to the inability to regulate body temperature through evaporative heat loss, as lack of perspiration in areas of prior full-thickness skin injury is well known. One of the authors (C.M.H.) previously reported a case of a patient with considerable hypertrophic scarring after surviving an episode of toxic epidermal necrolysis that was likely precipitated by lamotrigine.1 The patient initially presented to our clinic in consultation for laser therapy to improve the pliability and cosmetic appearance of the scars; however, approximately 3 weeks after initiating treatment with a fractional CO2 laser, the patient noticed perspiration in areas where she once lacked the ability to perspire as well as improved functionality.1 It was speculated that scar remodeling stimulated by the CO2 fractional laser allowed new connections to form between eccrine ducts in the dermis and epidermis.2
These findings are even more notable in light of a study by Rittié et al3 that suggested the primary appendages of the skin involved in human wound healing are the eccrine sweat glands. The investigators were able to demonstrate that eccrine sweat glands are major contributors in reepithelialization and wound healing in humans; therefore, it is possible that stimulating these glands with the CO2 laser may promote enhanced reepithelialization in addition to the reestablishment of perspiration and wound healing.3 Considering inadequate wound repair represents a substantial disturbance to the patient and health care system, this finding offers promise as a potential means to decrease morbidity in patients with dermal scarring from burns and traumatic injuries. We have since evaluated and treated 3 patients who demonstrated sweat regeneration following treatment with the fractional CO2 laser (Table).
A 42-year-old man was our first patient to demonstrate functional scar improvement following bone marrow transplant for acute lymphoblastic leukemia complicated by chronic sclerodermoid graft-versus-host disease and subsequent extensive scarring on the chest and arms. Approximately 2 weeks after the first treatment with the fractional CO2 laser, the patient began to notice the presence of sweat beads in the treated areas. In addition to the reestablishment of perspiration, the patient had perceived increased mobility with improved pliability and “softness” (as described by family members) in treated areas likely related to scar remodeling.
A 36-year-old wounded army veteran presented with burns to the face, arms, and chest affecting 49% of the body surface area. After only 1 treatment, the patient reported that he could subjectively tolerate 10°F more ambient temperature and work all day outside in south Texas when heat intolerance previously would allow him to work only 2 to 3 hours. Additionally, he noted increased mobility and chest wall expansion, which in combination contributed to overall increased exercise tolerance and enhanced quality of life.
A 35-year-old US Marine and firefighter with burns primarily on the chest and arms involving 35% body surface area experienced increased exercise tolerance and sweat regeneration, particularly on the chest after a single treatment with the fractional CO2 laser but continued to experience improvement after a total of 3 treatments. Additionally, the cosmetic improvement was so substantial that the physician (C.M.H) had to review older photographs to verify the location of the scars.
We have now treated 3 patients with various mechanisms of injury and extensive scarring who noticed improved heat tolerance from sweat regeneration following fractional CO2 laser therapy. At this point, we only have anecdotal evidence of subjective functional improvement, and further research is warranted to elucidate the exact mechanism of action to support our findings.
- Neiner J, Whittemore D, Hivnor C. Buried alive: functional eccrine coils buried under scar tissue? J Am Acad Dermatol. 2011;65:661-663.
- Waibel J, Beer K, Narurkar V, et al. Preliminary observations on fractional ablative resurfacing devices: clinical impressions. J Drugs Dermatol. 2009;8:481-485.
- Rittié L, Sachs D, Orringer J, et al. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am J Pathol. 2013;1:163-171.
- Neiner J, Whittemore D, Hivnor C. Buried alive: functional eccrine coils buried under scar tissue? J Am Acad Dermatol. 2011;65:661-663.
- Waibel J, Beer K, Narurkar V, et al. Preliminary observations on fractional ablative resurfacing devices: clinical impressions. J Drugs Dermatol. 2009;8:481-485.
- Rittié L, Sachs D, Orringer J, et al. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am J Pathol. 2013;1:163-171.
Practice Points
- Treatment of dermal scarring with fractional CO2 laser may contribute to eccrine sweat gland regeneration during the remodeling process in addition to increased skin pliability.
- Sweat regeneration has been demonstrated following treatment with fractional CO2 laser in patients with extensive scarring; this case shows sweat regeneration secondary to burns and chronic sclerodermoid graft-versus-host disease.
Vemurafenib-Induced Plantar Hyperkeratosis
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
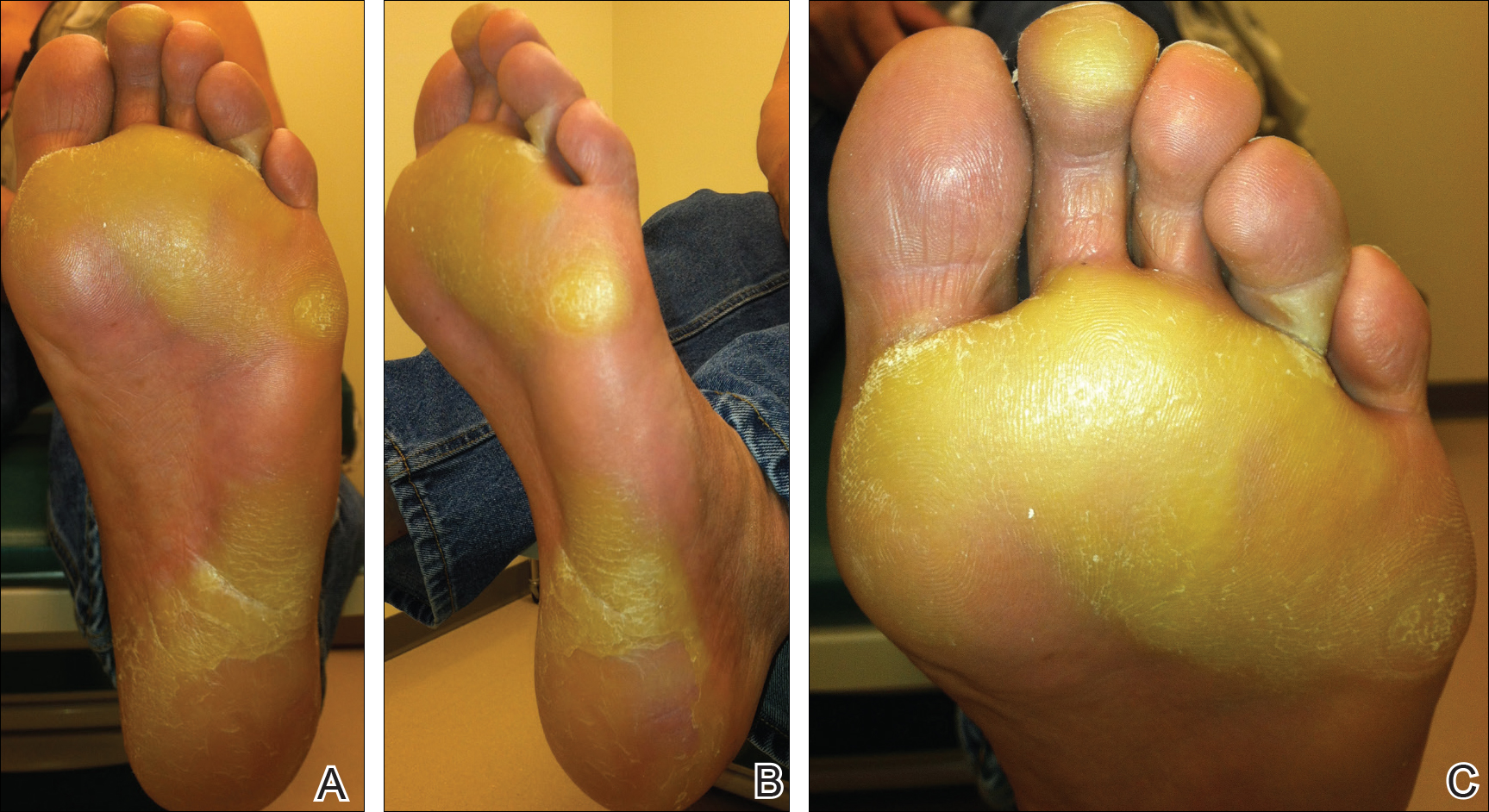
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
Practice Points
- BRAF inhibitors such as vemurafenib are associated with a high incidence of cutaneous side effects, including rash, hyperkeratosis, and cutaneous squamous cell carcinoma.
- Practitioners should be aware of these side effects and their management to avoid discontinuation or interruption of therapy.