User login
Severe Acne Fulminans Following Low-Dose Isotretinoin and Testosterone Use
To the Editor:
Acne fulminans (AF), the most severe form of acne, is a rare condition with an incidence of less than 1% of total acne cases.1 Adolescent boys are the most susceptible group of patients.2 Painful inflammatory pustules that transform into deep ulcerations covered by abundant hemorrhagic crust are typical of AF. Commonly affected areas include the face, back, neck, and chest. Additionally, fever and polyarthralgia may be present, and there often is myopathy due to rapid weight loss.3,4 Less often, erythema nodosum and splenomegaly may be observed.5 Laboratory testing also may reveal markers of systemic inflammation such as leukocytosis with neutrophilia, elevated C-reactive protein levels, increased erythrocyte sedimentation rate, and thrombocytosis. Anemia and elevated hepatic enzyme levels also may be present in AF.2 It is suspected that AF may be induced by low doses of isotretinoin therapy with concomitant inherited susceptibility.6
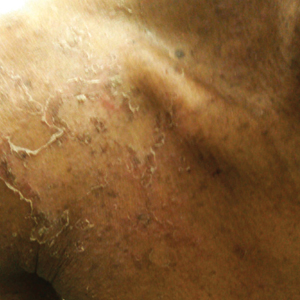
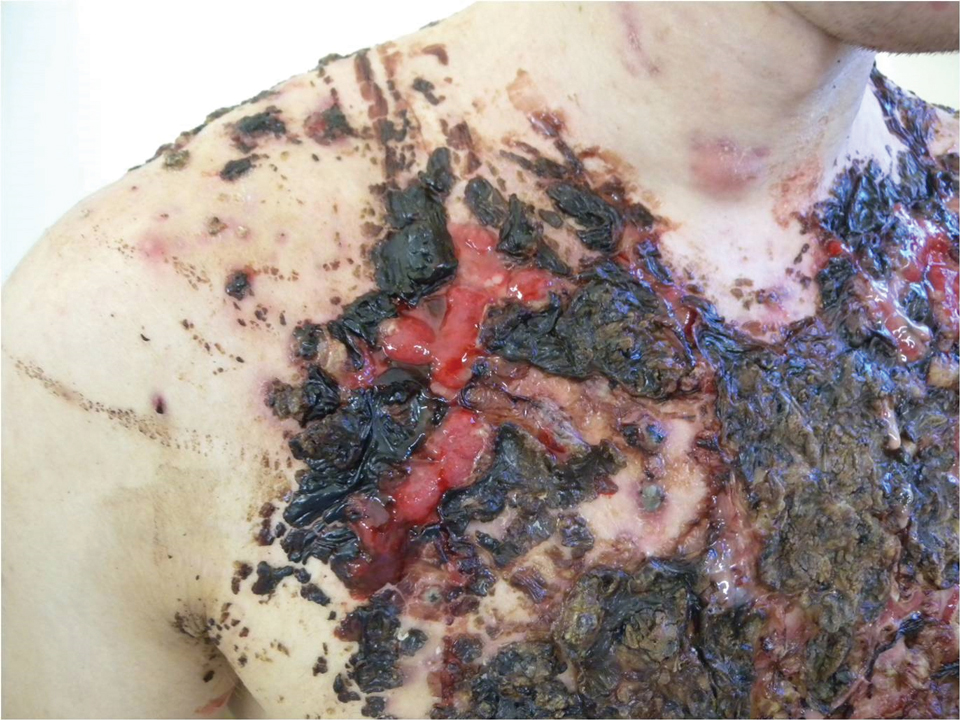
We report the case of a 21-year-old man who was referred to the Department of Dermatology by his primary care physician for evaluation of severe hemorrhagic lesions on the trunk following use of oral isotretinoin (Figure 1). Prior to development of the lesions, the patient had started weekly intramuscular injections of testosterone 500 mg, which he purchased online without consulting a physician, to address muscle mass reduction associated with sudden weight loss from intense physical training. After 8 months of testosterone supplementation along with continued physical training, the patient presented to his primary care physician for treatment of acne vulgaris on the back and trunk of 2 months’ duration. Oral isotretinoin 20 mg once daily was initiated; however, the patient reported that the acne lesions showed progression after 1 month of treatment. Isotretinoin was increased to a more weight-appropriate dosage of 60 mg once daily 2 weeks before admission to our dermatology clinic.
At the current presentation, dermatologic examination revealed numerous inflamed ulcerations covered by a hemorrhagic crust on the back and trunk. The patient also reported knee, elbow, and inguinal pain, especially at night. No fever or loss of appetite was reported. The patient was otherwise healthy and had no remarkable family history of acne or other dermatologic diseases.
Laboratory testing showed leukocytosis (11,000/µL [reference range, 4500–11,000/µL]), an elevated C-reactive protein level (66 mg/L [reference range, 0.08–3.1 mg/L]), and an elevated erythrocyte sedimentation rate (46 mm/h [reference range, 0–20 mm/h]). There were laboratory and clinical signs of a secondary bacterial infection in the affected areas, and a culture of secretions collected from lesions on the back grew Staphylococcus aureus with sensitivity to erythromycin, clindamycin, doxycycline, and trimethoprim-sulfamethoxazole and resistance to penicillin. A diagnosis of AF was made based on the clinical presentation and systemic symptoms, and anabolic-androgenic steroids and low-dose isotretinoin were identified as etiologic factors.
Treatment initially included cessation of isotretinoin and administration of prednisone, omeprazole, clindamycin, and doxycycline. Prednisone was given at a dosage of 40 mg once daily for 1 week, then decreased by 5 mg every 7 days. Omeprazole was given concurrently as prophylaxis for the gastrointestinal tract side effects of long-term prednisone use. Clindamycin was given at a dosage of 300 mg 3 times daily. Doxycycline was given for 6 weeks at a dosage of 100 mg twice daily. Topical octenidine dihydrochloride also was given.
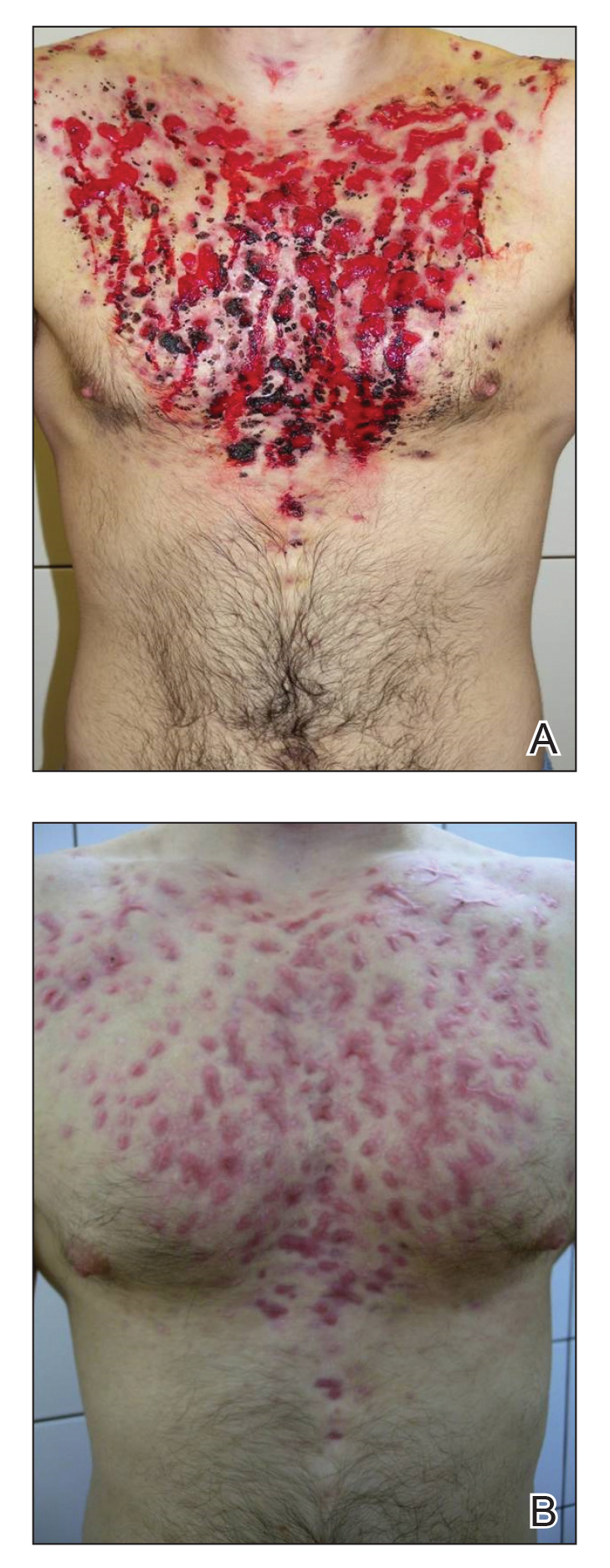
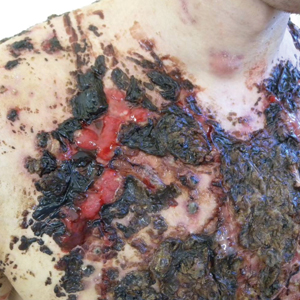
Marked improvement was noted after 24 hours (Figure 2) as well as on the third day of treatment (Figure 3A). After 6 weeks, only disfiguring scars were visible (Figure 3B). Oral isotretinoin was reincorporated after 8 weeks and was subsequently discontinued after 5 months of therapy with a cumulative dose of 150 mg/kg.

It is important to differentiate AF from exacerbation of acne vulgaris because patients typically have mild or moderate acne vulgaris before the onset of acute symptoms.1 Acne fulminans is characterized by systemic symptoms such as myalgia, polyarthralgia, fatigue, and osteolytic bone lesions.1,7 Additionally, hematologic symptoms such as fever, leukocytosis, anemia, splenomegaly, and hepatomegaly may be present.1,5,7 Our patient demonstrated the polysymptomatic form of AF. The patient had severe acne with a tendency to scar. There also were some systemic manifestations such as polyarthralgia, weight loss, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level.
The clinical diagnosis in our patient also was supported by the hypothesis that heredity, overactive immune reactions, bacterial infections, and use of some drugs (eg, isotretinoin, tetracycline, testosterone) can trigger AF.8 The most well-known theory is that low doses of isotretinoin induce AF.6 The majority of cases are caused by doses of less than 20 mg/kg once daily, but there have been reports of patients using full doses and developing this condition.9 The fact that the use of low- and high-dose isotretinoin can provoke AF suggests an idiosyncratic reaction that is not clearly dose related. The most dangerous triggering factor of AF is concomitant usage of testosterone and isotretinoin.10 Our patient used testosterone injections to increase muscle mass and underwent treatment with isotretinoin for acne.
Treatment of AF is controversial, as there is no standard therapy. Currently, steroids and isotretinoin are the treatments of choice. Antibiotic use is controversial because of a lack of randomized trials.11
In the first stage of treatment, prednisone 0.5 to 1 mg/kg once daily is recommended as an initial anti-inflammatory therapy, with gradual dose reduction. According to evidence-based recommendations, a low dose of isotretinoin can be introduced after crusted lesions have healed. The overlapping therapy with steroids and isotretinoin should be provided for at least 4 weeks. High-potency topical corticosteroids can be used on granulation tissue, which can shorten the systemic treatment with prednisone or can be an alternative treatment for patients with contraindications to systemic corticosteroids.11
Additionally, local care of the lesions including compresses and topical emollients is crucial. There are some case reports in which there is introduction of high doses of isotretinoin, subsequently with systemic steroids.7,8,12 Seukeran and Cunliffe5 proved that it is beneficial to give acne prophylaxis to prevent further episodes. Our patient was similarly treated with systemic steroids and isotretinoin. Treatment guidelines for AF do not recommend oral antibiotics,11 but data are limited in the case of isotretinoin-induced AF. Our patient was given doxycycline concomitant with systemic steroids, which was necessary due to signs of secondary infection from a lesion culture. Doxycycline was stopped when isotretinoin treatment was initiated to prevent pseudotumor cerebri. The patient achieved good clinical improvement with no relapse.
Using isotretinoin to treat acne vulgaris has many benefits, despite the possibility of developing AF as an extremely rare complication. Clinicians should be aware of the risk of this complication to make the diagnosis and provide appropriate care, especially in young men. It is particularly important to consider the possibility of concomitant testosterone and isotretinoin when documenting the patient’s medical history.
- Romiti R, Jansen T, Plewig G. Acne fulminans. An Bras Dermatol. 2000;75:611-617.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Kelly AP, Burns RE. Acute febrile ulcerative conglobate acne with polyarthralgia. Arch Dermatol. 1971;104:182-187.
- Plewig G, Kligman AM. Vitamin A acid in acneiform dermatoses. Acta Derm Venereol Suppl. 1975;74:119-127.
- Seukeran DC, Cunliffe WJ. The treatment of acne fulminans: a review of 25 cases. Br J Dermatol. 1999;141:307-309.
- Kraus SL, Emmert S, Schön MP, et al. The dark side of beauty: acne fulminans induced by anabolic steroids in a male bodybuilder. Arch Dermatol. 2012;148:1210-1212.
- Jansen T, Plewig G. Acne fulminans. Int J Dermatol. 1998;37:254-257.
- Zanelato TP, Gontijo GM, Alves CA, et al. Disabling acne fulminans. An Bras Dermatol. 2011;86:9-12.
- Azulay DR, Abulafia LA, Costa JAN, et al. Tecido de granulação exuberante. efeito colateral da terapêutica com isotretinoína. An Bras Dermatol. 1985;60:179-182.
- Traupe H, von Mühlendahl KE, Brämswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol. 1988;124:414-417.
- Greywal T, Zaenglein AL, Baldwin HE, et al. Evidence-based recommendations for the management of acne fulminans and its variants. J Am Acad Dermatol. 2017;77:109-117.
- Honma M, Murakami M, Iinuma S, et al. Acne fulminans following measles infection. J Dermatol. 2009;36:471-473.
To the Editor:
Acne fulminans (AF), the most severe form of acne, is a rare condition with an incidence of less than 1% of total acne cases.1 Adolescent boys are the most susceptible group of patients.2 Painful inflammatory pustules that transform into deep ulcerations covered by abundant hemorrhagic crust are typical of AF. Commonly affected areas include the face, back, neck, and chest. Additionally, fever and polyarthralgia may be present, and there often is myopathy due to rapid weight loss.3,4 Less often, erythema nodosum and splenomegaly may be observed.5 Laboratory testing also may reveal markers of systemic inflammation such as leukocytosis with neutrophilia, elevated C-reactive protein levels, increased erythrocyte sedimentation rate, and thrombocytosis. Anemia and elevated hepatic enzyme levels also may be present in AF.2 It is suspected that AF may be induced by low doses of isotretinoin therapy with concomitant inherited susceptibility.6
We report the case of a 21-year-old man who was referred to the Department of Dermatology by his primary care physician for evaluation of severe hemorrhagic lesions on the trunk following use of oral isotretinoin (Figure 1). Prior to development of the lesions, the patient had started weekly intramuscular injections of testosterone 500 mg, which he purchased online without consulting a physician, to address muscle mass reduction associated with sudden weight loss from intense physical training. After 8 months of testosterone supplementation along with continued physical training, the patient presented to his primary care physician for treatment of acne vulgaris on the back and trunk of 2 months’ duration. Oral isotretinoin 20 mg once daily was initiated; however, the patient reported that the acne lesions showed progression after 1 month of treatment. Isotretinoin was increased to a more weight-appropriate dosage of 60 mg once daily 2 weeks before admission to our dermatology clinic.
At the current presentation, dermatologic examination revealed numerous inflamed ulcerations covered by a hemorrhagic crust on the back and trunk. The patient also reported knee, elbow, and inguinal pain, especially at night. No fever or loss of appetite was reported. The patient was otherwise healthy and had no remarkable family history of acne or other dermatologic diseases.
Laboratory testing showed leukocytosis (11,000/µL [reference range, 4500–11,000/µL]), an elevated C-reactive protein level (66 mg/L [reference range, 0.08–3.1 mg/L]), and an elevated erythrocyte sedimentation rate (46 mm/h [reference range, 0–20 mm/h]). There were laboratory and clinical signs of a secondary bacterial infection in the affected areas, and a culture of secretions collected from lesions on the back grew Staphylococcus aureus with sensitivity to erythromycin, clindamycin, doxycycline, and trimethoprim-sulfamethoxazole and resistance to penicillin. A diagnosis of AF was made based on the clinical presentation and systemic symptoms, and anabolic-androgenic steroids and low-dose isotretinoin were identified as etiologic factors.
Treatment initially included cessation of isotretinoin and administration of prednisone, omeprazole, clindamycin, and doxycycline. Prednisone was given at a dosage of 40 mg once daily for 1 week, then decreased by 5 mg every 7 days. Omeprazole was given concurrently as prophylaxis for the gastrointestinal tract side effects of long-term prednisone use. Clindamycin was given at a dosage of 300 mg 3 times daily. Doxycycline was given for 6 weeks at a dosage of 100 mg twice daily. Topical octenidine dihydrochloride also was given.
Marked improvement was noted after 24 hours (Figure 2) as well as on the third day of treatment (Figure 3A). After 6 weeks, only disfiguring scars were visible (Figure 3B). Oral isotretinoin was reincorporated after 8 weeks and was subsequently discontinued after 5 months of therapy with a cumulative dose of 150 mg/kg.
It is important to differentiate AF from exacerbation of acne vulgaris because patients typically have mild or moderate acne vulgaris before the onset of acute symptoms.1 Acne fulminans is characterized by systemic symptoms such as myalgia, polyarthralgia, fatigue, and osteolytic bone lesions.1,7 Additionally, hematologic symptoms such as fever, leukocytosis, anemia, splenomegaly, and hepatomegaly may be present.1,5,7 Our patient demonstrated the polysymptomatic form of AF. The patient had severe acne with a tendency to scar. There also were some systemic manifestations such as polyarthralgia, weight loss, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level.
The clinical diagnosis in our patient also was supported by the hypothesis that heredity, overactive immune reactions, bacterial infections, and use of some drugs (eg, isotretinoin, tetracycline, testosterone) can trigger AF.8 The most well-known theory is that low doses of isotretinoin induce AF.6 The majority of cases are caused by doses of less than 20 mg/kg once daily, but there have been reports of patients using full doses and developing this condition.9 The fact that the use of low- and high-dose isotretinoin can provoke AF suggests an idiosyncratic reaction that is not clearly dose related. The most dangerous triggering factor of AF is concomitant usage of testosterone and isotretinoin.10 Our patient used testosterone injections to increase muscle mass and underwent treatment with isotretinoin for acne.
Treatment of AF is controversial, as there is no standard therapy. Currently, steroids and isotretinoin are the treatments of choice. Antibiotic use is controversial because of a lack of randomized trials.11
In the first stage of treatment, prednisone 0.5 to 1 mg/kg once daily is recommended as an initial anti-inflammatory therapy, with gradual dose reduction. According to evidence-based recommendations, a low dose of isotretinoin can be introduced after crusted lesions have healed. The overlapping therapy with steroids and isotretinoin should be provided for at least 4 weeks. High-potency topical corticosteroids can be used on granulation tissue, which can shorten the systemic treatment with prednisone or can be an alternative treatment for patients with contraindications to systemic corticosteroids.11
Additionally, local care of the lesions including compresses and topical emollients is crucial. There are some case reports in which there is introduction of high doses of isotretinoin, subsequently with systemic steroids.7,8,12 Seukeran and Cunliffe5 proved that it is beneficial to give acne prophylaxis to prevent further episodes. Our patient was similarly treated with systemic steroids and isotretinoin. Treatment guidelines for AF do not recommend oral antibiotics,11 but data are limited in the case of isotretinoin-induced AF. Our patient was given doxycycline concomitant with systemic steroids, which was necessary due to signs of secondary infection from a lesion culture. Doxycycline was stopped when isotretinoin treatment was initiated to prevent pseudotumor cerebri. The patient achieved good clinical improvement with no relapse.
Using isotretinoin to treat acne vulgaris has many benefits, despite the possibility of developing AF as an extremely rare complication. Clinicians should be aware of the risk of this complication to make the diagnosis and provide appropriate care, especially in young men. It is particularly important to consider the possibility of concomitant testosterone and isotretinoin when documenting the patient’s medical history.
To the Editor:
Acne fulminans (AF), the most severe form of acne, is a rare condition with an incidence of less than 1% of total acne cases.1 Adolescent boys are the most susceptible group of patients.2 Painful inflammatory pustules that transform into deep ulcerations covered by abundant hemorrhagic crust are typical of AF. Commonly affected areas include the face, back, neck, and chest. Additionally, fever and polyarthralgia may be present, and there often is myopathy due to rapid weight loss.3,4 Less often, erythema nodosum and splenomegaly may be observed.5 Laboratory testing also may reveal markers of systemic inflammation such as leukocytosis with neutrophilia, elevated C-reactive protein levels, increased erythrocyte sedimentation rate, and thrombocytosis. Anemia and elevated hepatic enzyme levels also may be present in AF.2 It is suspected that AF may be induced by low doses of isotretinoin therapy with concomitant inherited susceptibility.6
We report the case of a 21-year-old man who was referred to the Department of Dermatology by his primary care physician for evaluation of severe hemorrhagic lesions on the trunk following use of oral isotretinoin (Figure 1). Prior to development of the lesions, the patient had started weekly intramuscular injections of testosterone 500 mg, which he purchased online without consulting a physician, to address muscle mass reduction associated with sudden weight loss from intense physical training. After 8 months of testosterone supplementation along with continued physical training, the patient presented to his primary care physician for treatment of acne vulgaris on the back and trunk of 2 months’ duration. Oral isotretinoin 20 mg once daily was initiated; however, the patient reported that the acne lesions showed progression after 1 month of treatment. Isotretinoin was increased to a more weight-appropriate dosage of 60 mg once daily 2 weeks before admission to our dermatology clinic.
At the current presentation, dermatologic examination revealed numerous inflamed ulcerations covered by a hemorrhagic crust on the back and trunk. The patient also reported knee, elbow, and inguinal pain, especially at night. No fever or loss of appetite was reported. The patient was otherwise healthy and had no remarkable family history of acne or other dermatologic diseases.
Laboratory testing showed leukocytosis (11,000/µL [reference range, 4500–11,000/µL]), an elevated C-reactive protein level (66 mg/L [reference range, 0.08–3.1 mg/L]), and an elevated erythrocyte sedimentation rate (46 mm/h [reference range, 0–20 mm/h]). There were laboratory and clinical signs of a secondary bacterial infection in the affected areas, and a culture of secretions collected from lesions on the back grew Staphylococcus aureus with sensitivity to erythromycin, clindamycin, doxycycline, and trimethoprim-sulfamethoxazole and resistance to penicillin. A diagnosis of AF was made based on the clinical presentation and systemic symptoms, and anabolic-androgenic steroids and low-dose isotretinoin were identified as etiologic factors.
Treatment initially included cessation of isotretinoin and administration of prednisone, omeprazole, clindamycin, and doxycycline. Prednisone was given at a dosage of 40 mg once daily for 1 week, then decreased by 5 mg every 7 days. Omeprazole was given concurrently as prophylaxis for the gastrointestinal tract side effects of long-term prednisone use. Clindamycin was given at a dosage of 300 mg 3 times daily. Doxycycline was given for 6 weeks at a dosage of 100 mg twice daily. Topical octenidine dihydrochloride also was given.
Marked improvement was noted after 24 hours (Figure 2) as well as on the third day of treatment (Figure 3A). After 6 weeks, only disfiguring scars were visible (Figure 3B). Oral isotretinoin was reincorporated after 8 weeks and was subsequently discontinued after 5 months of therapy with a cumulative dose of 150 mg/kg.
It is important to differentiate AF from exacerbation of acne vulgaris because patients typically have mild or moderate acne vulgaris before the onset of acute symptoms.1 Acne fulminans is characterized by systemic symptoms such as myalgia, polyarthralgia, fatigue, and osteolytic bone lesions.1,7 Additionally, hematologic symptoms such as fever, leukocytosis, anemia, splenomegaly, and hepatomegaly may be present.1,5,7 Our patient demonstrated the polysymptomatic form of AF. The patient had severe acne with a tendency to scar. There also were some systemic manifestations such as polyarthralgia, weight loss, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level.
The clinical diagnosis in our patient also was supported by the hypothesis that heredity, overactive immune reactions, bacterial infections, and use of some drugs (eg, isotretinoin, tetracycline, testosterone) can trigger AF.8 The most well-known theory is that low doses of isotretinoin induce AF.6 The majority of cases are caused by doses of less than 20 mg/kg once daily, but there have been reports of patients using full doses and developing this condition.9 The fact that the use of low- and high-dose isotretinoin can provoke AF suggests an idiosyncratic reaction that is not clearly dose related. The most dangerous triggering factor of AF is concomitant usage of testosterone and isotretinoin.10 Our patient used testosterone injections to increase muscle mass and underwent treatment with isotretinoin for acne.
Treatment of AF is controversial, as there is no standard therapy. Currently, steroids and isotretinoin are the treatments of choice. Antibiotic use is controversial because of a lack of randomized trials.11
In the first stage of treatment, prednisone 0.5 to 1 mg/kg once daily is recommended as an initial anti-inflammatory therapy, with gradual dose reduction. According to evidence-based recommendations, a low dose of isotretinoin can be introduced after crusted lesions have healed. The overlapping therapy with steroids and isotretinoin should be provided for at least 4 weeks. High-potency topical corticosteroids can be used on granulation tissue, which can shorten the systemic treatment with prednisone or can be an alternative treatment for patients with contraindications to systemic corticosteroids.11
Additionally, local care of the lesions including compresses and topical emollients is crucial. There are some case reports in which there is introduction of high doses of isotretinoin, subsequently with systemic steroids.7,8,12 Seukeran and Cunliffe5 proved that it is beneficial to give acne prophylaxis to prevent further episodes. Our patient was similarly treated with systemic steroids and isotretinoin. Treatment guidelines for AF do not recommend oral antibiotics,11 but data are limited in the case of isotretinoin-induced AF. Our patient was given doxycycline concomitant with systemic steroids, which was necessary due to signs of secondary infection from a lesion culture. Doxycycline was stopped when isotretinoin treatment was initiated to prevent pseudotumor cerebri. The patient achieved good clinical improvement with no relapse.
Using isotretinoin to treat acne vulgaris has many benefits, despite the possibility of developing AF as an extremely rare complication. Clinicians should be aware of the risk of this complication to make the diagnosis and provide appropriate care, especially in young men. It is particularly important to consider the possibility of concomitant testosterone and isotretinoin when documenting the patient’s medical history.
- Romiti R, Jansen T, Plewig G. Acne fulminans. An Bras Dermatol. 2000;75:611-617.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Kelly AP, Burns RE. Acute febrile ulcerative conglobate acne with polyarthralgia. Arch Dermatol. 1971;104:182-187.
- Plewig G, Kligman AM. Vitamin A acid in acneiform dermatoses. Acta Derm Venereol Suppl. 1975;74:119-127.
- Seukeran DC, Cunliffe WJ. The treatment of acne fulminans: a review of 25 cases. Br J Dermatol. 1999;141:307-309.
- Kraus SL, Emmert S, Schön MP, et al. The dark side of beauty: acne fulminans induced by anabolic steroids in a male bodybuilder. Arch Dermatol. 2012;148:1210-1212.
- Jansen T, Plewig G. Acne fulminans. Int J Dermatol. 1998;37:254-257.
- Zanelato TP, Gontijo GM, Alves CA, et al. Disabling acne fulminans. An Bras Dermatol. 2011;86:9-12.
- Azulay DR, Abulafia LA, Costa JAN, et al. Tecido de granulação exuberante. efeito colateral da terapêutica com isotretinoína. An Bras Dermatol. 1985;60:179-182.
- Traupe H, von Mühlendahl KE, Brämswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol. 1988;124:414-417.
- Greywal T, Zaenglein AL, Baldwin HE, et al. Evidence-based recommendations for the management of acne fulminans and its variants. J Am Acad Dermatol. 2017;77:109-117.
- Honma M, Murakami M, Iinuma S, et al. Acne fulminans following measles infection. J Dermatol. 2009;36:471-473.
- Romiti R, Jansen T, Plewig G. Acne fulminans. An Bras Dermatol. 2000;75:611-617.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Kelly AP, Burns RE. Acute febrile ulcerative conglobate acne with polyarthralgia. Arch Dermatol. 1971;104:182-187.
- Plewig G, Kligman AM. Vitamin A acid in acneiform dermatoses. Acta Derm Venereol Suppl. 1975;74:119-127.
- Seukeran DC, Cunliffe WJ. The treatment of acne fulminans: a review of 25 cases. Br J Dermatol. 1999;141:307-309.
- Kraus SL, Emmert S, Schön MP, et al. The dark side of beauty: acne fulminans induced by anabolic steroids in a male bodybuilder. Arch Dermatol. 2012;148:1210-1212.
- Jansen T, Plewig G. Acne fulminans. Int J Dermatol. 1998;37:254-257.
- Zanelato TP, Gontijo GM, Alves CA, et al. Disabling acne fulminans. An Bras Dermatol. 2011;86:9-12.
- Azulay DR, Abulafia LA, Costa JAN, et al. Tecido de granulação exuberante. efeito colateral da terapêutica com isotretinoína. An Bras Dermatol. 1985;60:179-182.
- Traupe H, von Mühlendahl KE, Brämswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol. 1988;124:414-417.
- Greywal T, Zaenglein AL, Baldwin HE, et al. Evidence-based recommendations for the management of acne fulminans and its variants. J Am Acad Dermatol. 2017;77:109-117.
- Honma M, Murakami M, Iinuma S, et al. Acne fulminans following measles infection. J Dermatol. 2009;36:471-473.
Practice Points
- Acne fulminans, the most severe form of acne, is characterized by deep ulcerations covered by a hemorrhagic crust. It is commonly associated with fever, polyarthralgia, and myopathy caused by rapid weight loss.
- This rare condition is recognized as a potential complication of oral isotretinoin therapy.
Allergic Contact Dermatitis With Sparing of Exposed Psoriasis Plaques
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
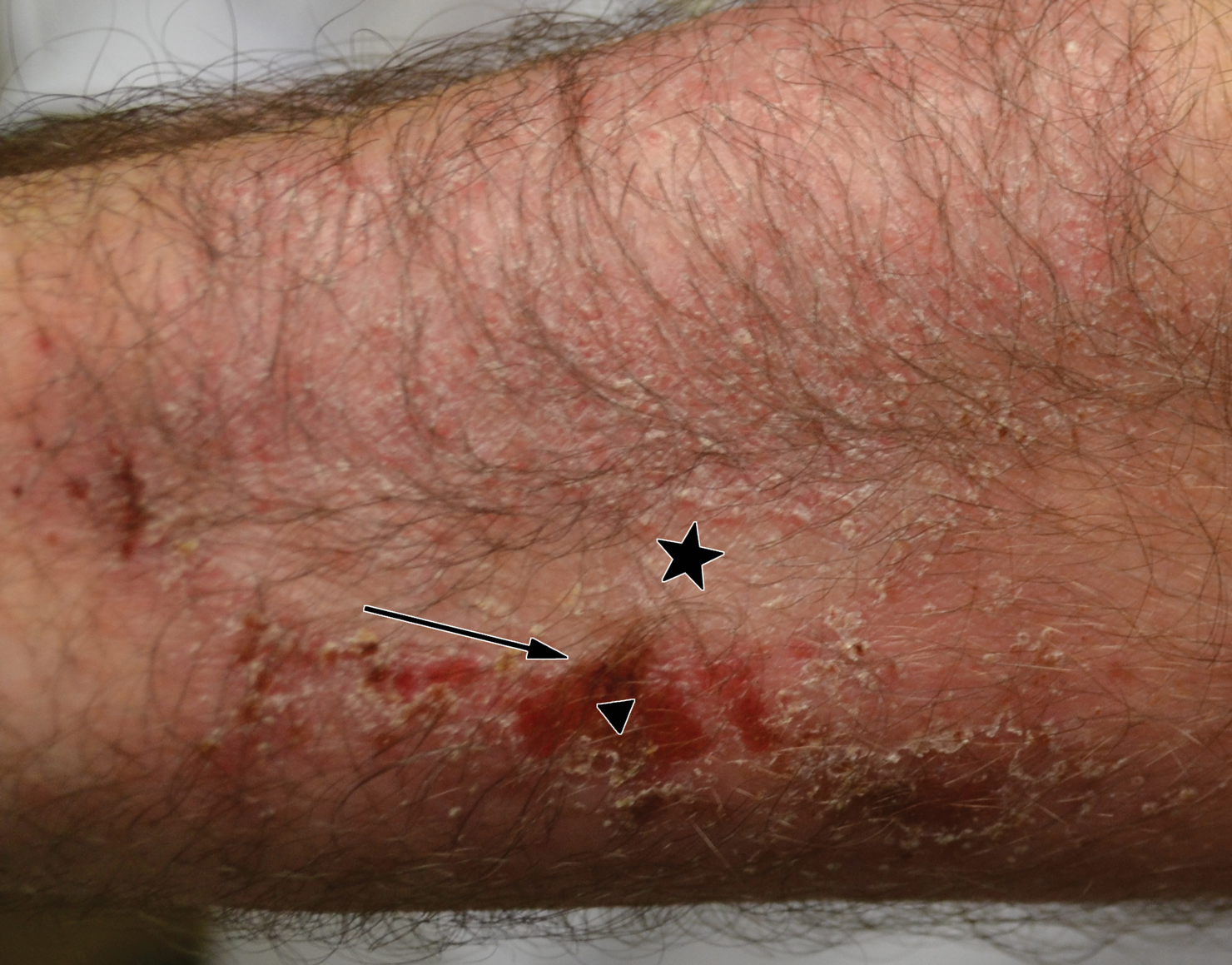
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
Practice Points
- Patients with plaque-type psoriasis who experience allergic contact dermatitis (ACD) may present with sparing of exposed psoriatic plaques.
- The divergent immunologic milieus present in ACD and psoriasis likely underly the decreased incidence of ACD in patients with psoriasis.
Generalized Granuloma Annulare Responsive to Narrowband UVB
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.

Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
Practice Points
- The generalized variant of granuloma annulare (GA) can be persistent, sometimes lasting years to decades; treatment is not always effective.
- The safety profile and tolerability of narrowband UVB phototherapy make it a suitable treatment option for generalized GA.
Pustular Tinea Id Reaction
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
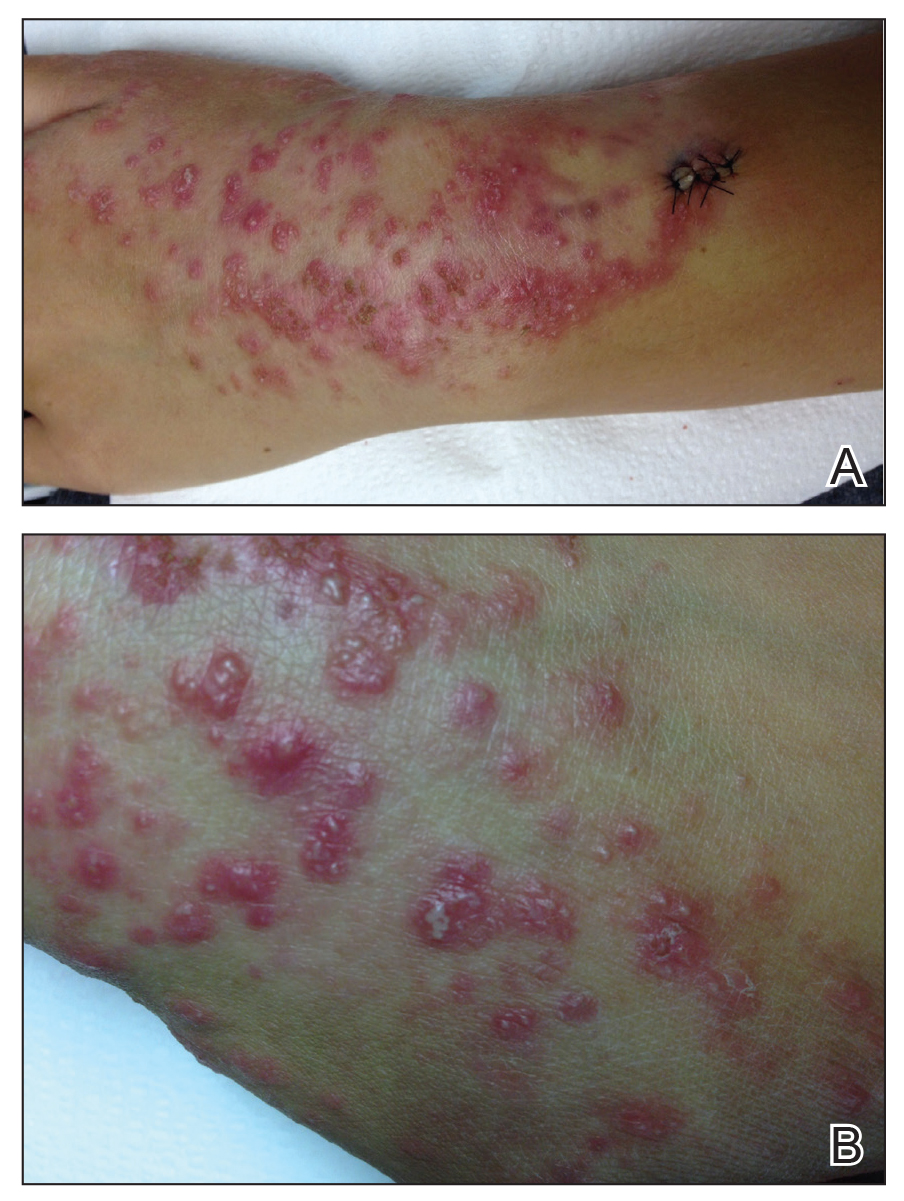
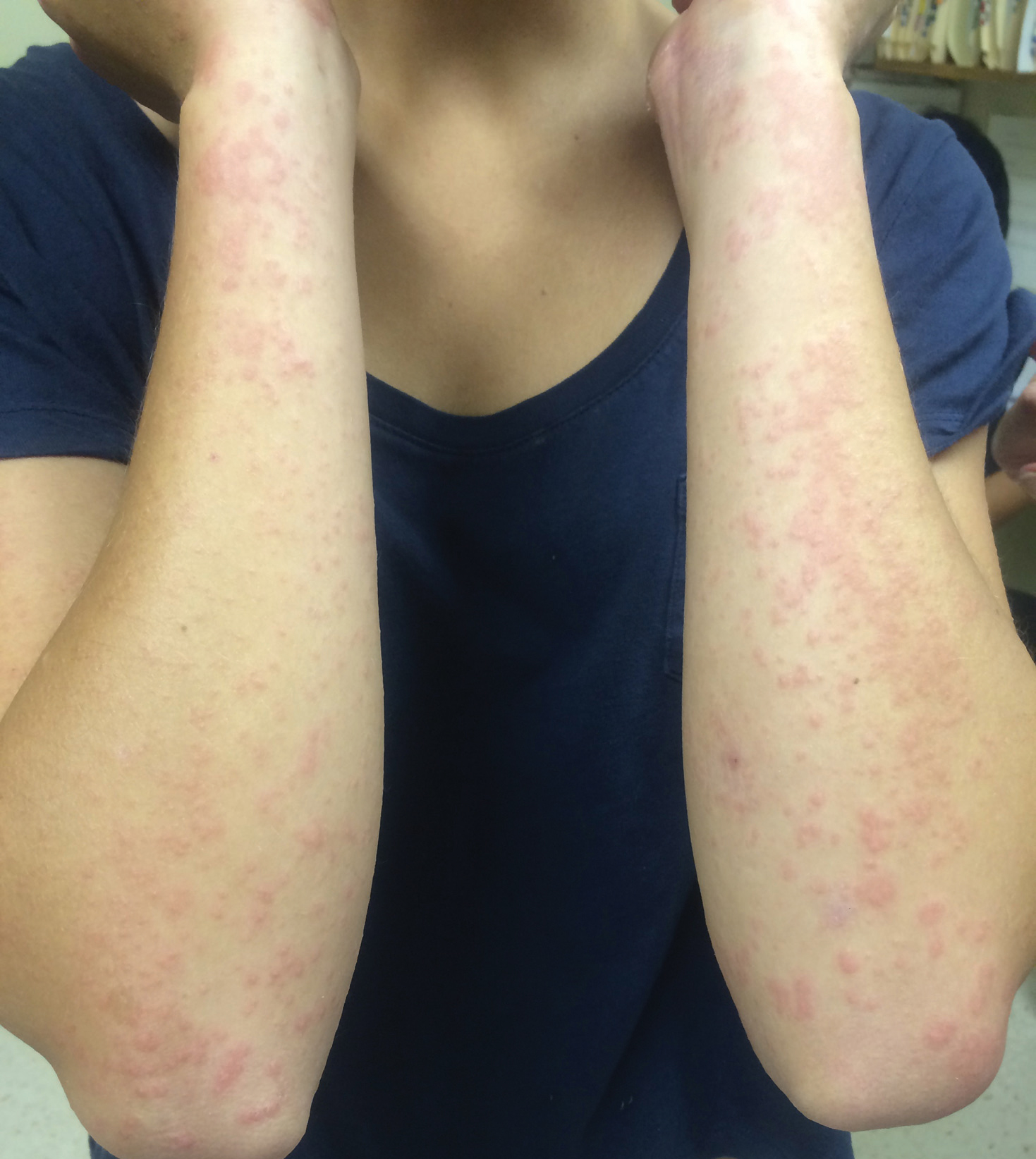
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
Practice Points
• Id reactions, or autoeczematization, can occur secondary to dermatophyte infections, possibly due to a hypersensitivity reaction to the fungus. These eruptions can occur in many forms of tinea and in a variety of clinical presentations.
• Treatment is based on clearance of the original dermatophyte infection.
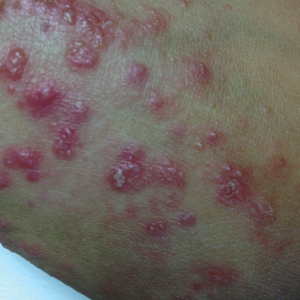
Dupilumab for Treatment of Severe Atopic Dermatitis in a Heart Transplant Recipient
To the Editor:
Solid-organ transplant recipients can develop a range of dermatologic consequences due to chronic immunosuppression, including frequent skin infections and malignancies. Atopic dermatitis (AD) and psoriasis are relatively rare in this population because many immunosuppressive therapies, such as mycophenolate mofetil and tacrolimus, also are used to treat inflammatory dermatoses.1 In a large renal transplant population, the prevalence of AD was 1.3%.2 The pathogenesis of posttransplantation AD is poorly understood, and standard treatment regimens have not been defined. Dupilumab is a novel biologic medication that has demonstrated efficacy in the treatment of AD.3 Reports of dupilumab use for AD management in solid-organ transplant recipients are limited in the literature.
A 29-year-old woman with a history of a heart transplant 4 years prior presented to our dermatology clinic with an itchy rash over the entire body. Since the transplant, she had been on long-term immunosuppression with prednisone, mycophenolate mofetil, and tacrolimus. The rash appeared after she switched from brand-name to generic versions of the medications. Physical examination revealed erythematous scaly plaques on the lateral face, back, chest, arms, and legs covering approximately 10% of the body surface area. The patient’s total serum IgE level was elevated at 711,500 µg/L (reference range, 0–1500 µg/L). Outside biopsies revealed changes consistent with spongiotic dermatitis, and patch testing performed by an outside physician was positive for sensitivity to the preservative bronopol.
The patient was switched back to brand-name tacrolimus, but the rash did not improve. Topical steroids, phototherapy, and omalizumab were ineffective. The itching was primarily managed with desoximetasone spray, mometasone cream, and loratidine. With approval from the patient’s transplant team outside of our hospital system, she was started on dupilumab 300 mg once every 14 days. Complete clearance of the rash was noted within 3 months of treatment. Besides bilateral conjunctivitis, which was treated with ophthalmic prednisolone and moxifloxacin solutions, dupilumab was well tolerated. No issues related to immunosuppressant levels or graft-related issues, including rejection, were reported at 6-, 12-, and 18-month follow-up visits.
Atopic dermatitis is characterized by activation of type 2 immune responses, skin barrier defects, and increased Staphylococcus aureus colonization.4 A potential mechanism for the development of AD in transplant recipients relates to their use of tacrolimus for chronic immunosuppression. Tacrolimus increases intestinal permeability and therefore allows greater absorption of allergens. This influx of allergens promotes hypersensitivity reactions, resulting in elevated IgE levels and eosinophilia. Tacrolimus also facilitates predominance of helper T cells (TH2 cytokines) through selective inhibition of the TH1 cytokine IL-2.5
Dupilumab is a human monoclonal antibody that blocks IL-4 and IL-13, which are key drivers of TH2-mediated inflammation. In addition to downregulation of inflammatory mediators, dupilumab also increases production of epidermal barrier proteins, resulting in skin repair. It has demonstrated rapid, dose-dependent efficacy in patients with moderate to severe AD.6 Dupilumab boasts a good safety profile with no increase in risk for skin infections compared to placebo6; however, its safety has not yet been verified in transplant recipients.
Our case is notable for the severity of the patient’s AD despite considerable immunosuppression with transplant medications. Development of AD was associated with a switch from brand-name to generic drugs, which is not commonly reported. Her condition was refractory to a litany of treatments prior to a trial of dupilumab. The rapid clearance observed with this novel biologic medication highlights its potential to provide relief to patients who have particularly tenacious cases of AD. Prior to starting dupilumab, we do recommend more extensive laboratory testing in immunosuppressed patients including transplant recipients and patients with human immunodeficiency virus. We illustrate that a history of solid-organ transplant need not exclude patients from consideration for dupilumab therapy.
- Savoia P, Cavaliere G, Zavattaro E, et al. Inflammatory cutaneous diseases in renal transplant recipients [published online August 19, 2016]. Int J Mol Sci. doi:10.3390/ijms17081362.
- Lally A, Casabonne D, Imko-Walczuk B, et al. Prevalence of benign cutaneous disease among Oxford renal transplant recipients. J Eur Acad Dermatol Venereol. 2011;25:462-470.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bieber T, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Machura E, Chodór B, Kleszyk M, et al. Atopic allergy and chronic inflammation of the oral mucosa in a 3-year-old boy after heart transplantation—diagnostic and therapeutic difficulties. Kardiochir Torakochirurgia Pol. 2015;12:176-180.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
To the Editor:
Solid-organ transplant recipients can develop a range of dermatologic consequences due to chronic immunosuppression, including frequent skin infections and malignancies. Atopic dermatitis (AD) and psoriasis are relatively rare in this population because many immunosuppressive therapies, such as mycophenolate mofetil and tacrolimus, also are used to treat inflammatory dermatoses.1 In a large renal transplant population, the prevalence of AD was 1.3%.2 The pathogenesis of posttransplantation AD is poorly understood, and standard treatment regimens have not been defined. Dupilumab is a novel biologic medication that has demonstrated efficacy in the treatment of AD.3 Reports of dupilumab use for AD management in solid-organ transplant recipients are limited in the literature.
A 29-year-old woman with a history of a heart transplant 4 years prior presented to our dermatology clinic with an itchy rash over the entire body. Since the transplant, she had been on long-term immunosuppression with prednisone, mycophenolate mofetil, and tacrolimus. The rash appeared after she switched from brand-name to generic versions of the medications. Physical examination revealed erythematous scaly plaques on the lateral face, back, chest, arms, and legs covering approximately 10% of the body surface area. The patient’s total serum IgE level was elevated at 711,500 µg/L (reference range, 0–1500 µg/L). Outside biopsies revealed changes consistent with spongiotic dermatitis, and patch testing performed by an outside physician was positive for sensitivity to the preservative bronopol.
The patient was switched back to brand-name tacrolimus, but the rash did not improve. Topical steroids, phototherapy, and omalizumab were ineffective. The itching was primarily managed with desoximetasone spray, mometasone cream, and loratidine. With approval from the patient’s transplant team outside of our hospital system, she was started on dupilumab 300 mg once every 14 days. Complete clearance of the rash was noted within 3 months of treatment. Besides bilateral conjunctivitis, which was treated with ophthalmic prednisolone and moxifloxacin solutions, dupilumab was well tolerated. No issues related to immunosuppressant levels or graft-related issues, including rejection, were reported at 6-, 12-, and 18-month follow-up visits.
Atopic dermatitis is characterized by activation of type 2 immune responses, skin barrier defects, and increased Staphylococcus aureus colonization.4 A potential mechanism for the development of AD in transplant recipients relates to their use of tacrolimus for chronic immunosuppression. Tacrolimus increases intestinal permeability and therefore allows greater absorption of allergens. This influx of allergens promotes hypersensitivity reactions, resulting in elevated IgE levels and eosinophilia. Tacrolimus also facilitates predominance of helper T cells (TH2 cytokines) through selective inhibition of the TH1 cytokine IL-2.5
Dupilumab is a human monoclonal antibody that blocks IL-4 and IL-13, which are key drivers of TH2-mediated inflammation. In addition to downregulation of inflammatory mediators, dupilumab also increases production of epidermal barrier proteins, resulting in skin repair. It has demonstrated rapid, dose-dependent efficacy in patients with moderate to severe AD.6 Dupilumab boasts a good safety profile with no increase in risk for skin infections compared to placebo6; however, its safety has not yet been verified in transplant recipients.
Our case is notable for the severity of the patient’s AD despite considerable immunosuppression with transplant medications. Development of AD was associated with a switch from brand-name to generic drugs, which is not commonly reported. Her condition was refractory to a litany of treatments prior to a trial of dupilumab. The rapid clearance observed with this novel biologic medication highlights its potential to provide relief to patients who have particularly tenacious cases of AD. Prior to starting dupilumab, we do recommend more extensive laboratory testing in immunosuppressed patients including transplant recipients and patients with human immunodeficiency virus. We illustrate that a history of solid-organ transplant need not exclude patients from consideration for dupilumab therapy.
To the Editor:
Solid-organ transplant recipients can develop a range of dermatologic consequences due to chronic immunosuppression, including frequent skin infections and malignancies. Atopic dermatitis (AD) and psoriasis are relatively rare in this population because many immunosuppressive therapies, such as mycophenolate mofetil and tacrolimus, also are used to treat inflammatory dermatoses.1 In a large renal transplant population, the prevalence of AD was 1.3%.2 The pathogenesis of posttransplantation AD is poorly understood, and standard treatment regimens have not been defined. Dupilumab is a novel biologic medication that has demonstrated efficacy in the treatment of AD.3 Reports of dupilumab use for AD management in solid-organ transplant recipients are limited in the literature.
A 29-year-old woman with a history of a heart transplant 4 years prior presented to our dermatology clinic with an itchy rash over the entire body. Since the transplant, she had been on long-term immunosuppression with prednisone, mycophenolate mofetil, and tacrolimus. The rash appeared after she switched from brand-name to generic versions of the medications. Physical examination revealed erythematous scaly plaques on the lateral face, back, chest, arms, and legs covering approximately 10% of the body surface area. The patient’s total serum IgE level was elevated at 711,500 µg/L (reference range, 0–1500 µg/L). Outside biopsies revealed changes consistent with spongiotic dermatitis, and patch testing performed by an outside physician was positive for sensitivity to the preservative bronopol.
The patient was switched back to brand-name tacrolimus, but the rash did not improve. Topical steroids, phototherapy, and omalizumab were ineffective. The itching was primarily managed with desoximetasone spray, mometasone cream, and loratidine. With approval from the patient’s transplant team outside of our hospital system, she was started on dupilumab 300 mg once every 14 days. Complete clearance of the rash was noted within 3 months of treatment. Besides bilateral conjunctivitis, which was treated with ophthalmic prednisolone and moxifloxacin solutions, dupilumab was well tolerated. No issues related to immunosuppressant levels or graft-related issues, including rejection, were reported at 6-, 12-, and 18-month follow-up visits.
Atopic dermatitis is characterized by activation of type 2 immune responses, skin barrier defects, and increased Staphylococcus aureus colonization.4 A potential mechanism for the development of AD in transplant recipients relates to their use of tacrolimus for chronic immunosuppression. Tacrolimus increases intestinal permeability and therefore allows greater absorption of allergens. This influx of allergens promotes hypersensitivity reactions, resulting in elevated IgE levels and eosinophilia. Tacrolimus also facilitates predominance of helper T cells (TH2 cytokines) through selective inhibition of the TH1 cytokine IL-2.5
Dupilumab is a human monoclonal antibody that blocks IL-4 and IL-13, which are key drivers of TH2-mediated inflammation. In addition to downregulation of inflammatory mediators, dupilumab also increases production of epidermal barrier proteins, resulting in skin repair. It has demonstrated rapid, dose-dependent efficacy in patients with moderate to severe AD.6 Dupilumab boasts a good safety profile with no increase in risk for skin infections compared to placebo6; however, its safety has not yet been verified in transplant recipients.
Our case is notable for the severity of the patient’s AD despite considerable immunosuppression with transplant medications. Development of AD was associated with a switch from brand-name to generic drugs, which is not commonly reported. Her condition was refractory to a litany of treatments prior to a trial of dupilumab. The rapid clearance observed with this novel biologic medication highlights its potential to provide relief to patients who have particularly tenacious cases of AD. Prior to starting dupilumab, we do recommend more extensive laboratory testing in immunosuppressed patients including transplant recipients and patients with human immunodeficiency virus. We illustrate that a history of solid-organ transplant need not exclude patients from consideration for dupilumab therapy.
- Savoia P, Cavaliere G, Zavattaro E, et al. Inflammatory cutaneous diseases in renal transplant recipients [published online August 19, 2016]. Int J Mol Sci. doi:10.3390/ijms17081362.
- Lally A, Casabonne D, Imko-Walczuk B, et al. Prevalence of benign cutaneous disease among Oxford renal transplant recipients. J Eur Acad Dermatol Venereol. 2011;25:462-470.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bieber T, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Machura E, Chodór B, Kleszyk M, et al. Atopic allergy and chronic inflammation of the oral mucosa in a 3-year-old boy after heart transplantation—diagnostic and therapeutic difficulties. Kardiochir Torakochirurgia Pol. 2015;12:176-180.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Savoia P, Cavaliere G, Zavattaro E, et al. Inflammatory cutaneous diseases in renal transplant recipients [published online August 19, 2016]. Int J Mol Sci. doi:10.3390/ijms17081362.
- Lally A, Casabonne D, Imko-Walczuk B, et al. Prevalence of benign cutaneous disease among Oxford renal transplant recipients. J Eur Acad Dermatol Venereol. 2011;25:462-470.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bieber T, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Machura E, Chodór B, Kleszyk M, et al. Atopic allergy and chronic inflammation of the oral mucosa in a 3-year-old boy after heart transplantation—diagnostic and therapeutic difficulties. Kardiochir Torakochirurgia Pol. 2015;12:176-180.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
Practice Points
- Chronic tacrolimus use in solid-organ transplant recipients may increase intestinal permeability to allergens and is a potential cause for development of atopic dermatitis (AD).
- Dupilumab has the potential to provide relief from particularly tenacious cases of AD.
- History of solid-organ transplant should not be cause for exclusion from consideration for dupilumab therapy.
Graham-Little-Piccardi-Lassueur Syndrome
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.

The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.
The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.
The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
Practice Points
- Graham-Little-Piccardi-Lassueur syndrome (GLPL) is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp.
- Graham-Little-Piccardi-Lassueur syndrome is considered one of the 3 clinical variants of lichen planopilaris.
- Potential therapies for GLPL include hydroxychloroquine, cyclosporine, tetracyclines, and pioglitazone.

Multiple Subcutaneous Dermoid Cysts
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
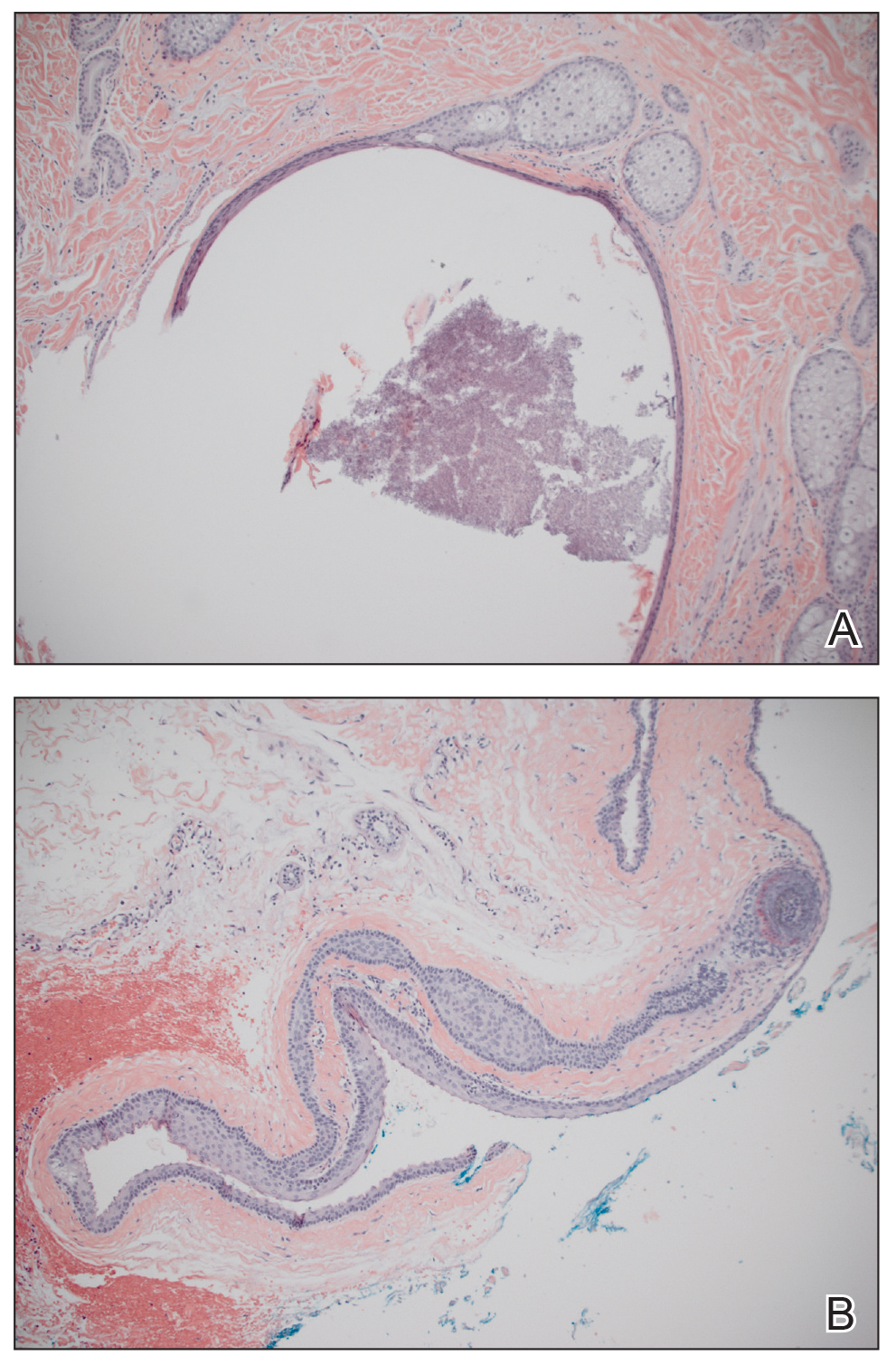
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
Practice Points
- The majority of dermoid cysts are congenital; however, they may be acquired from traumatic implantation of epidermal elements into the dermis.
- The most common location for dermoid cysts is the lateral third of the eyebrows; however, they also may occur on the mid forehead, scalp, nose, anterior neck, and trunk.
- Imaging studies may be needed to rule out intracranial or intraspinal extension of dermoid cysts, particularly for those presenting in the midline.
Multiple Eruptive Syringomas on the Penis
To the Editor:
Syringomas are small, benign, asymptomatic eccrine or apocrine tumors that present as multiple discrete flesh-colored papules. They are more common in females than males.1 The etiology of eruptive syringomas is unclear, though an inflammatory process has been implicated in the abnormal proliferation of sweat glands.2 However, a minority of tumors have been known to have an autosomal-dominant mode of transmission. Multiple or eruptive syringomas are associated with Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, and Blau syndrome.3 The clear cell variant has been found to be associated with diabetes mellitus.4 Syringomas most commonly appear on the lower eyelids, upper cheeks, neck, and upper chest; presentation on the penis is rare.5 We report a case of multiple eruptive syringomas located exclusively on the penis mimicking a sexually transmitted condition.
A 53-year-old man who was otherwise healthy presented with multiple flesh-colored papules on the penis that initially began to develop 30 years prior, but increased crops of lesions appeared 4 to 6 weeks prior to presentation. The patient described the lesions as rashlike, nonpruritic, and sensitive to the touch. He denied any discharge, oozing, crusting, or bleeding from the lesions. He did not report any high-risk sexual behaviors and stated that he was in a monogamous relationship with his wife. He had a medical history of molluscum contagiosum that was diagnosed and treated with cryotherapy 30 years prior; however, he did not have a history of any other sexually transmitted diseases. He also did not have a history of diabetes mellitus or thyroid disease.
Physical examination revealed multiple pink papules on the dorsal and ventral shaft of the penis, measuring 2 to 4 mm in diameter, with koebnerization (Figure 1). Based on clinical examination, the differential included condyloma, inflamed seborrheic keratosis, bowenoid papulosis, atypical molluscum contagiosum, or lichen planus. Consequently, a punch biopsy of the penile shaft was performed and histopathologic examination revealed proliferation of ducts focally that were tadpole shaped and embedded in a sclerotic stroma. The lining of the ducts was composed of cuboidal cells, some with clear cell change. The microscopic findings were consistent with penile syringomas (Figure 2). Laboratory results revealed the patient was negative for human immunodeficiency virus, hepatitis B, hepatitis C, and syphilis. The patient was given topical hydrocortisone butyrate and tacrolimus for symptomatic treatment. He declined further aggressive treatment.

Due to the rarity of syringomas on the penis, presentation of these benign eccrine tumors can be commonly mistaken for lichen planus, molluscum contagiosum, genital warts, or bowenoid papulosis.5 The characteristic histopathology of syringomas consists of multiple, small, tadpole or paisley tie–shaped ducts within an eosinophilic stroma. Often, the findings can be histologically confused with desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma. Although the histopathology of our patient’s biopsy showed clear cell change, the patient did not report a history of diabetes mellitus, which is a disease that can be associated with the clear cell variant of syringoma. Because syringomas are benign tumors, treatment is not medically necessary unless the lesions are symptomatic. Treatment often is regarded as challenging, as lesions often recur and scarring is a consideration. Possible treatments for removal of the benign papules include surgical excision, electrodesiccation and curettage, shave removal, chemical peels, liquid nitrogen cryotherapy, and CO2 laser vaporization.6
To prevent misdiagnosis and unnecessary treatment, it is important to have syringomas as part of the differential diagnosis when patients present with multiple small flesh-colored papules on the penis. The lesions should be biopsied for accurate diagnosis and to provide reassurance to patients who usually come in for evaluation for fear of having acquired a sexually transmitted disease.
- Yalisove B, Stolar EEH, Williams CM. Multiple penile papules. syringoma. Arch Dermatol. 1987;123:1391-1396.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Yoshimi N, Kurokawa I, Kakuno A, et al. Case of generalized eruptive clear cell syringoma with diabetes mellitus. J Dermatol. 2012;39:744-745.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Wu CY. Multifocal penile syringoma masquerading as genital warts. Clin Exp Dermatol. 2009;34:e290-e291.
- Lipshutz RL, Kantor GR, Vonderheid EC. Multiple penile syringomas mimicking verrucae. Int J Dermatol. 1991;30:69.
To the Editor:
Syringomas are small, benign, asymptomatic eccrine or apocrine tumors that present as multiple discrete flesh-colored papules. They are more common in females than males.1 The etiology of eruptive syringomas is unclear, though an inflammatory process has been implicated in the abnormal proliferation of sweat glands.2 However, a minority of tumors have been known to have an autosomal-dominant mode of transmission. Multiple or eruptive syringomas are associated with Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, and Blau syndrome.3 The clear cell variant has been found to be associated with diabetes mellitus.4 Syringomas most commonly appear on the lower eyelids, upper cheeks, neck, and upper chest; presentation on the penis is rare.5 We report a case of multiple eruptive syringomas located exclusively on the penis mimicking a sexually transmitted condition.
A 53-year-old man who was otherwise healthy presented with multiple flesh-colored papules on the penis that initially began to develop 30 years prior, but increased crops of lesions appeared 4 to 6 weeks prior to presentation. The patient described the lesions as rashlike, nonpruritic, and sensitive to the touch. He denied any discharge, oozing, crusting, or bleeding from the lesions. He did not report any high-risk sexual behaviors and stated that he was in a monogamous relationship with his wife. He had a medical history of molluscum contagiosum that was diagnosed and treated with cryotherapy 30 years prior; however, he did not have a history of any other sexually transmitted diseases. He also did not have a history of diabetes mellitus or thyroid disease.
Physical examination revealed multiple pink papules on the dorsal and ventral shaft of the penis, measuring 2 to 4 mm in diameter, with koebnerization (Figure 1). Based on clinical examination, the differential included condyloma, inflamed seborrheic keratosis, bowenoid papulosis, atypical molluscum contagiosum, or lichen planus. Consequently, a punch biopsy of the penile shaft was performed and histopathologic examination revealed proliferation of ducts focally that were tadpole shaped and embedded in a sclerotic stroma. The lining of the ducts was composed of cuboidal cells, some with clear cell change. The microscopic findings were consistent with penile syringomas (Figure 2). Laboratory results revealed the patient was negative for human immunodeficiency virus, hepatitis B, hepatitis C, and syphilis. The patient was given topical hydrocortisone butyrate and tacrolimus for symptomatic treatment. He declined further aggressive treatment.
Due to the rarity of syringomas on the penis, presentation of these benign eccrine tumors can be commonly mistaken for lichen planus, molluscum contagiosum, genital warts, or bowenoid papulosis.5 The characteristic histopathology of syringomas consists of multiple, small, tadpole or paisley tie–shaped ducts within an eosinophilic stroma. Often, the findings can be histologically confused with desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma. Although the histopathology of our patient’s biopsy showed clear cell change, the patient did not report a history of diabetes mellitus, which is a disease that can be associated with the clear cell variant of syringoma. Because syringomas are benign tumors, treatment is not medically necessary unless the lesions are symptomatic. Treatment often is regarded as challenging, as lesions often recur and scarring is a consideration. Possible treatments for removal of the benign papules include surgical excision, electrodesiccation and curettage, shave removal, chemical peels, liquid nitrogen cryotherapy, and CO2 laser vaporization.6
To prevent misdiagnosis and unnecessary treatment, it is important to have syringomas as part of the differential diagnosis when patients present with multiple small flesh-colored papules on the penis. The lesions should be biopsied for accurate diagnosis and to provide reassurance to patients who usually come in for evaluation for fear of having acquired a sexually transmitted disease.
To the Editor:
Syringomas are small, benign, asymptomatic eccrine or apocrine tumors that present as multiple discrete flesh-colored papules. They are more common in females than males.1 The etiology of eruptive syringomas is unclear, though an inflammatory process has been implicated in the abnormal proliferation of sweat glands.2 However, a minority of tumors have been known to have an autosomal-dominant mode of transmission. Multiple or eruptive syringomas are associated with Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, and Blau syndrome.3 The clear cell variant has been found to be associated with diabetes mellitus.4 Syringomas most commonly appear on the lower eyelids, upper cheeks, neck, and upper chest; presentation on the penis is rare.5 We report a case of multiple eruptive syringomas located exclusively on the penis mimicking a sexually transmitted condition.
A 53-year-old man who was otherwise healthy presented with multiple flesh-colored papules on the penis that initially began to develop 30 years prior, but increased crops of lesions appeared 4 to 6 weeks prior to presentation. The patient described the lesions as rashlike, nonpruritic, and sensitive to the touch. He denied any discharge, oozing, crusting, or bleeding from the lesions. He did not report any high-risk sexual behaviors and stated that he was in a monogamous relationship with his wife. He had a medical history of molluscum contagiosum that was diagnosed and treated with cryotherapy 30 years prior; however, he did not have a history of any other sexually transmitted diseases. He also did not have a history of diabetes mellitus or thyroid disease.
Physical examination revealed multiple pink papules on the dorsal and ventral shaft of the penis, measuring 2 to 4 mm in diameter, with koebnerization (Figure 1). Based on clinical examination, the differential included condyloma, inflamed seborrheic keratosis, bowenoid papulosis, atypical molluscum contagiosum, or lichen planus. Consequently, a punch biopsy of the penile shaft was performed and histopathologic examination revealed proliferation of ducts focally that were tadpole shaped and embedded in a sclerotic stroma. The lining of the ducts was composed of cuboidal cells, some with clear cell change. The microscopic findings were consistent with penile syringomas (Figure 2). Laboratory results revealed the patient was negative for human immunodeficiency virus, hepatitis B, hepatitis C, and syphilis. The patient was given topical hydrocortisone butyrate and tacrolimus for symptomatic treatment. He declined further aggressive treatment.
Due to the rarity of syringomas on the penis, presentation of these benign eccrine tumors can be commonly mistaken for lichen planus, molluscum contagiosum, genital warts, or bowenoid papulosis.5 The characteristic histopathology of syringomas consists of multiple, small, tadpole or paisley tie–shaped ducts within an eosinophilic stroma. Often, the findings can be histologically confused with desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma. Although the histopathology of our patient’s biopsy showed clear cell change, the patient did not report a history of diabetes mellitus, which is a disease that can be associated with the clear cell variant of syringoma. Because syringomas are benign tumors, treatment is not medically necessary unless the lesions are symptomatic. Treatment often is regarded as challenging, as lesions often recur and scarring is a consideration. Possible treatments for removal of the benign papules include surgical excision, electrodesiccation and curettage, shave removal, chemical peels, liquid nitrogen cryotherapy, and CO2 laser vaporization.6
To prevent misdiagnosis and unnecessary treatment, it is important to have syringomas as part of the differential diagnosis when patients present with multiple small flesh-colored papules on the penis. The lesions should be biopsied for accurate diagnosis and to provide reassurance to patients who usually come in for evaluation for fear of having acquired a sexually transmitted disease.
- Yalisove B, Stolar EEH, Williams CM. Multiple penile papules. syringoma. Arch Dermatol. 1987;123:1391-1396.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Yoshimi N, Kurokawa I, Kakuno A, et al. Case of generalized eruptive clear cell syringoma with diabetes mellitus. J Dermatol. 2012;39:744-745.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Wu CY. Multifocal penile syringoma masquerading as genital warts. Clin Exp Dermatol. 2009;34:e290-e291.
- Lipshutz RL, Kantor GR, Vonderheid EC. Multiple penile syringomas mimicking verrucae. Int J Dermatol. 1991;30:69.
- Yalisove B, Stolar EEH, Williams CM. Multiple penile papules. syringoma. Arch Dermatol. 1987;123:1391-1396.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Yoshimi N, Kurokawa I, Kakuno A, et al. Case of generalized eruptive clear cell syringoma with diabetes mellitus. J Dermatol. 2012;39:744-745.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Wu CY. Multifocal penile syringoma masquerading as genital warts. Clin Exp Dermatol. 2009;34:e290-e291.
- Lipshutz RL, Kantor GR, Vonderheid EC. Multiple penile syringomas mimicking verrucae. Int J Dermatol. 1991;30:69.
Practice Points
- Penile syringoma can mimic sexually transmitted disease such as condyloma acuminatum or molluscum contagiosum.
- Penile syringomas can be long-standing and require biopsy to differentiate from other conditions.

Apremilast and Phototherapy for Treatment of Psoriasis in a Patient With Human Immunodeficiency Virus
To the Editor:
A 50-year old man with Fitzpatrick skin type IV, human immunodeficiency virus (HIV), fatty liver disease, and moderate psoriasis (10% body surface area [BSA] affected) currently treated with clobetasol spray and calcitriol ointment presented with persistent psoriatic lesions on the trunk, arms, legs, and buttocks. His CD4 count was 460 and his HIV RNA count was 48 copies/mL on polymerase chain reaction 2 months prior to the current presentation. He had been undergoing phototherapy 3 times weekly for the last 5 months for treatment of psoriasis.
At the current presentation, he was started on an apremilast starter pack with the dosage titrated from 10 mg to 30 mg over the course of 1 week. He was maintained on a dose of 30 mg twice daily after 1 week and continued clobetasol spray, calcitriol ointment, and phototherapy 3 times weekly with the intent to reduce the frequency after adequate control of psoriasis was achieved. After 3 months of treatment, the affected BSA was 0%. He continued apremilast, and phototherapy was reduced to once weekly. Phototherapy was discontinued after 7 months of concomitant treatment with apremilast after clearance was maintained. It was reinitiated twice weekly after a mild flare (3% BSA affected). After 20 total months of treatment, the patient was no longer able to afford apremilast treatment and presented with a severe psoriasis flare (40% BSA affected). He was switched to acitretin with a plan to apply for apremilast financial assistance programs.
Psoriasis treatment in the HIV population poses a challenge given the immunosuppressed state of these patients, the risk of reactivation of latent infections, and the refractory nature of psoriasis in the setting of HIV. Two of the authors (S.P.R. and J.J.W.) previously reported a case of moderate to severe psoriasis in a patient with HIV and hepatitis C who demonstrated treatment success with apremilast until it was discontinued due to financial implications.1 Currently, apremilast is not widely used to treat psoriasis in the HIV population. The National Psoriasis Foundation 2010 guidelines recommended UV light therapy for treatment of moderate to severe psoriasis in HIV-positive patients, with oral retinoids as the second-line treatment.2 There remains a need for updated guidelines on the use of systemic agents for psoriasis treatment in the HIV population.
Apremilast, a phosphodiesterase 4 inhibitor, is an oral therapy that restores the balance of proinflammatory and anti-inflammatory cytokines by inhibiting inflammatory cytokine (eg, tumor necrosis factor α, IFN-γ, IL-2, IL-12, IL-23) secretion and stimulating anti-inflammatory cytokine (eg, IL-6, IL-10) production. In 2015, the phase 3 ESTEEM 13 and ESTEEM 24 trials demonstrated the efficacy of apremilast 30 mg twice daily for treatment of psoriasis. In both trials, the psoriasis area and severity index 75 response rate at week 16 was significantly
Use of other systemic agents such as tumor necrosis factor α inhibitors and ustekinumab has been reported in HIV-positive patients.5-7 There is no current data on IL-17 and IL-23 inhibitors. Acitretin generally is recommended as a second-line agent in HIV patients given its lack of immunosuppression2; however, methotrexate and cyclosporine should be avoided given the risk of opportunistic infections.8
Apremilast is a promising therapy with a favorable safety profile that should be considered as an adjuvant treatment to first-line agents such as phototherapy in HIV-positive patients. Apremilast has been successfully used in an HIV patient with a concomitant chronic hepatitis C infection.1 Systemic medications such as apremilast should be managed in coordination with infectious disease specialists with close monitoring of CD4 levels and viral loads as well as prophylactic agents.
- Reddy SP, Shah VV, Wu JJ. Apremilast for a psoriasis patient with HIV and hepatitis C. J Eur Acad Dermatol Venereol. 2017;31:e481-e482.
- Menon K, Van Voorhees AS, Bebo BF Jr, et al. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation [published online July 31, 2009]. J Am Acad Dermatol. 2010;62:291-299.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871.
- Saeki H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatolog Treat. 2012;23:398-399.
- Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections [published online July 11, 2018]. J Am Acad Dermatol. 2019;80:43-53.
To the Editor:
A 50-year old man with Fitzpatrick skin type IV, human immunodeficiency virus (HIV), fatty liver disease, and moderate psoriasis (10% body surface area [BSA] affected) currently treated with clobetasol spray and calcitriol ointment presented with persistent psoriatic lesions on the trunk, arms, legs, and buttocks. His CD4 count was 460 and his HIV RNA count was 48 copies/mL on polymerase chain reaction 2 months prior to the current presentation. He had been undergoing phototherapy 3 times weekly for the last 5 months for treatment of psoriasis.
At the current presentation, he was started on an apremilast starter pack with the dosage titrated from 10 mg to 30 mg over the course of 1 week. He was maintained on a dose of 30 mg twice daily after 1 week and continued clobetasol spray, calcitriol ointment, and phototherapy 3 times weekly with the intent to reduce the frequency after adequate control of psoriasis was achieved. After 3 months of treatment, the affected BSA was 0%. He continued apremilast, and phototherapy was reduced to once weekly. Phototherapy was discontinued after 7 months of concomitant treatment with apremilast after clearance was maintained. It was reinitiated twice weekly after a mild flare (3% BSA affected). After 20 total months of treatment, the patient was no longer able to afford apremilast treatment and presented with a severe psoriasis flare (40% BSA affected). He was switched to acitretin with a plan to apply for apremilast financial assistance programs.
Psoriasis treatment in the HIV population poses a challenge given the immunosuppressed state of these patients, the risk of reactivation of latent infections, and the refractory nature of psoriasis in the setting of HIV. Two of the authors (S.P.R. and J.J.W.) previously reported a case of moderate to severe psoriasis in a patient with HIV and hepatitis C who demonstrated treatment success with apremilast until it was discontinued due to financial implications.1 Currently, apremilast is not widely used to treat psoriasis in the HIV population. The National Psoriasis Foundation 2010 guidelines recommended UV light therapy for treatment of moderate to severe psoriasis in HIV-positive patients, with oral retinoids as the second-line treatment.2 There remains a need for updated guidelines on the use of systemic agents for psoriasis treatment in the HIV population.
Apremilast, a phosphodiesterase 4 inhibitor, is an oral therapy that restores the balance of proinflammatory and anti-inflammatory cytokines by inhibiting inflammatory cytokine (eg, tumor necrosis factor α, IFN-γ, IL-2, IL-12, IL-23) secretion and stimulating anti-inflammatory cytokine (eg, IL-6, IL-10) production. In 2015, the phase 3 ESTEEM 13 and ESTEEM 24 trials demonstrated the efficacy of apremilast 30 mg twice daily for treatment of psoriasis. In both trials, the psoriasis area and severity index 75 response rate at week 16 was significantly
Use of other systemic agents such as tumor necrosis factor α inhibitors and ustekinumab has been reported in HIV-positive patients.5-7 There is no current data on IL-17 and IL-23 inhibitors. Acitretin generally is recommended as a second-line agent in HIV patients given its lack of immunosuppression2; however, methotrexate and cyclosporine should be avoided given the risk of opportunistic infections.8
Apremilast is a promising therapy with a favorable safety profile that should be considered as an adjuvant treatment to first-line agents such as phototherapy in HIV-positive patients. Apremilast has been successfully used in an HIV patient with a concomitant chronic hepatitis C infection.1 Systemic medications such as apremilast should be managed in coordination with infectious disease specialists with close monitoring of CD4 levels and viral loads as well as prophylactic agents.
To the Editor:
A 50-year old man with Fitzpatrick skin type IV, human immunodeficiency virus (HIV), fatty liver disease, and moderate psoriasis (10% body surface area [BSA] affected) currently treated with clobetasol spray and calcitriol ointment presented with persistent psoriatic lesions on the trunk, arms, legs, and buttocks. His CD4 count was 460 and his HIV RNA count was 48 copies/mL on polymerase chain reaction 2 months prior to the current presentation. He had been undergoing phototherapy 3 times weekly for the last 5 months for treatment of psoriasis.
At the current presentation, he was started on an apremilast starter pack with the dosage titrated from 10 mg to 30 mg over the course of 1 week. He was maintained on a dose of 30 mg twice daily after 1 week and continued clobetasol spray, calcitriol ointment, and phototherapy 3 times weekly with the intent to reduce the frequency after adequate control of psoriasis was achieved. After 3 months of treatment, the affected BSA was 0%. He continued apremilast, and phototherapy was reduced to once weekly. Phototherapy was discontinued after 7 months of concomitant treatment with apremilast after clearance was maintained. It was reinitiated twice weekly after a mild flare (3% BSA affected). After 20 total months of treatment, the patient was no longer able to afford apremilast treatment and presented with a severe psoriasis flare (40% BSA affected). He was switched to acitretin with a plan to apply for apremilast financial assistance programs.
Psoriasis treatment in the HIV population poses a challenge given the immunosuppressed state of these patients, the risk of reactivation of latent infections, and the refractory nature of psoriasis in the setting of HIV. Two of the authors (S.P.R. and J.J.W.) previously reported a case of moderate to severe psoriasis in a patient with HIV and hepatitis C who demonstrated treatment success with apremilast until it was discontinued due to financial implications.1 Currently, apremilast is not widely used to treat psoriasis in the HIV population. The National Psoriasis Foundation 2010 guidelines recommended UV light therapy for treatment of moderate to severe psoriasis in HIV-positive patients, with oral retinoids as the second-line treatment.2 There remains a need for updated guidelines on the use of systemic agents for psoriasis treatment in the HIV population.
Apremilast, a phosphodiesterase 4 inhibitor, is an oral therapy that restores the balance of proinflammatory and anti-inflammatory cytokines by inhibiting inflammatory cytokine (eg, tumor necrosis factor α, IFN-γ, IL-2, IL-12, IL-23) secretion and stimulating anti-inflammatory cytokine (eg, IL-6, IL-10) production. In 2015, the phase 3 ESTEEM 13 and ESTEEM 24 trials demonstrated the efficacy of apremilast 30 mg twice daily for treatment of psoriasis. In both trials, the psoriasis area and severity index 75 response rate at week 16 was significantly
Use of other systemic agents such as tumor necrosis factor α inhibitors and ustekinumab has been reported in HIV-positive patients.5-7 There is no current data on IL-17 and IL-23 inhibitors. Acitretin generally is recommended as a second-line agent in HIV patients given its lack of immunosuppression2; however, methotrexate and cyclosporine should be avoided given the risk of opportunistic infections.8
Apremilast is a promising therapy with a favorable safety profile that should be considered as an adjuvant treatment to first-line agents such as phototherapy in HIV-positive patients. Apremilast has been successfully used in an HIV patient with a concomitant chronic hepatitis C infection.1 Systemic medications such as apremilast should be managed in coordination with infectious disease specialists with close monitoring of CD4 levels and viral loads as well as prophylactic agents.
- Reddy SP, Shah VV, Wu JJ. Apremilast for a psoriasis patient with HIV and hepatitis C. J Eur Acad Dermatol Venereol. 2017;31:e481-e482.
- Menon K, Van Voorhees AS, Bebo BF Jr, et al. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation [published online July 31, 2009]. J Am Acad Dermatol. 2010;62:291-299.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871.
- Saeki H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatolog Treat. 2012;23:398-399.
- Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections [published online July 11, 2018]. J Am Acad Dermatol. 2019;80:43-53.
- Reddy SP, Shah VV, Wu JJ. Apremilast for a psoriasis patient with HIV and hepatitis C. J Eur Acad Dermatol Venereol. 2017;31:e481-e482.
- Menon K, Van Voorhees AS, Bebo BF Jr, et al. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation [published online July 31, 2009]. J Am Acad Dermatol. 2010;62:291-299.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871.
- Saeki H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatolog Treat. 2012;23:398-399.
- Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections [published online July 11, 2018]. J Am Acad Dermatol. 2019;80:43-53.
Practice Point
- Apremilast may be considered as a first-line therapy in the human immunodeficiency virus population due to decreased immunosuppression.
Netherton Syndrome: An Atypical Presentation
To the Editor:
Netherton syndrome (NS) is a rare autosomal-recessive ichthyosiform disease.1 The incidence is estimated to be 1 in 200,000 individuals.2 Netherton syndrome presents with generalized erythroderma and scaling, characteristic hair shaft abnormalities, and dysregulation of the immune system. Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination. We report a case of NS in a man with congenital erythroderma, pili torti, and elevated IgE levels.
A 23-year-old man presented with generalized scaly skin that was present since birth. He was the first child born of nonconsanguineous parents. His medical history was suggestive of atopic diatheses such as allergic rhinitis and recurrent urticaria. The patient was of thin build and had widespread erythematous, annular, and polycyclic scaly lesions (Figure 1A), some with characteristic double-edged scale (Figure 1B). The skin was dry due to anhidrosis that was present since birth. Flexural lichenification was present at the cubital fossa of both arms. Scalp hairs were easily pluckable and had generalized thinning of hair density. Hair mount examination showed characteristic features of both trichorrhexis invaginata (Figure 2A) and pili torti (Figure 2B).
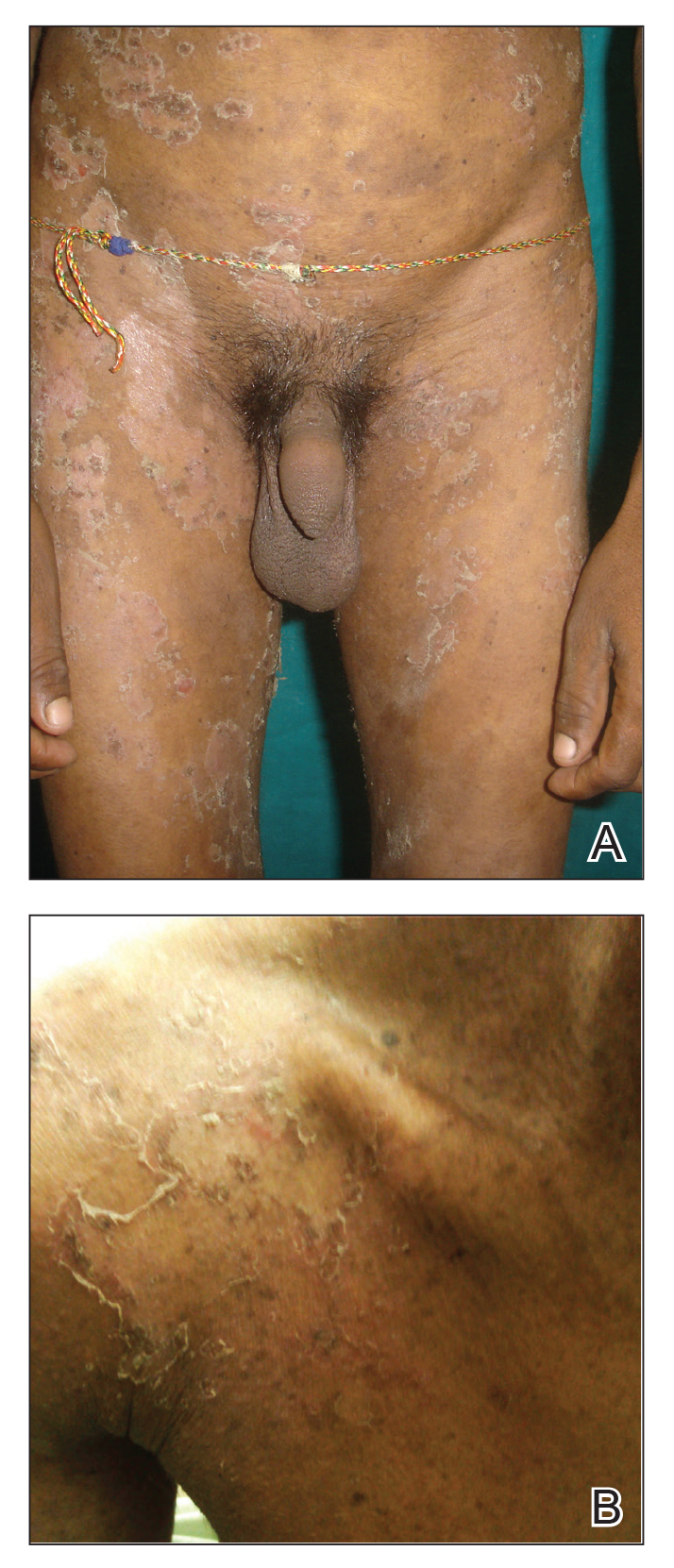

hair shaft known as bamboo hair or trichorrhexis invaginata. B, Features of pili torti; the hair
shaft twisted at irregular intervals.
Potassium hydroxide mount from a lesion was negative for fungal elements. Complete hematologic workup showed moderate anemia at 8.0 g/dL (reference range, 8.0–10.9 g/dL) and peripheral eosinophilia at 12% (reference range, 0%–6%). His IgE level was markedly elevated at6331 IU/mL (reference range, 150–1000 IU/mL) when tested with fully automated bidirectionally interfaced chemiluminescent immunoassay. Histopathologic examination of a lesion biopsy showed psoriasiform epidermal hyperplasia, papillomatosis, and acanthosis, consistent with ichthyosis linearis circumflexa (ILC)(Figure 3). Clinicopathologic correlation led to a diagnosis of ILC, trichorrhexis invaginata/pili torti, and atopic diathesis, which is a constellation of disorders related to NS.
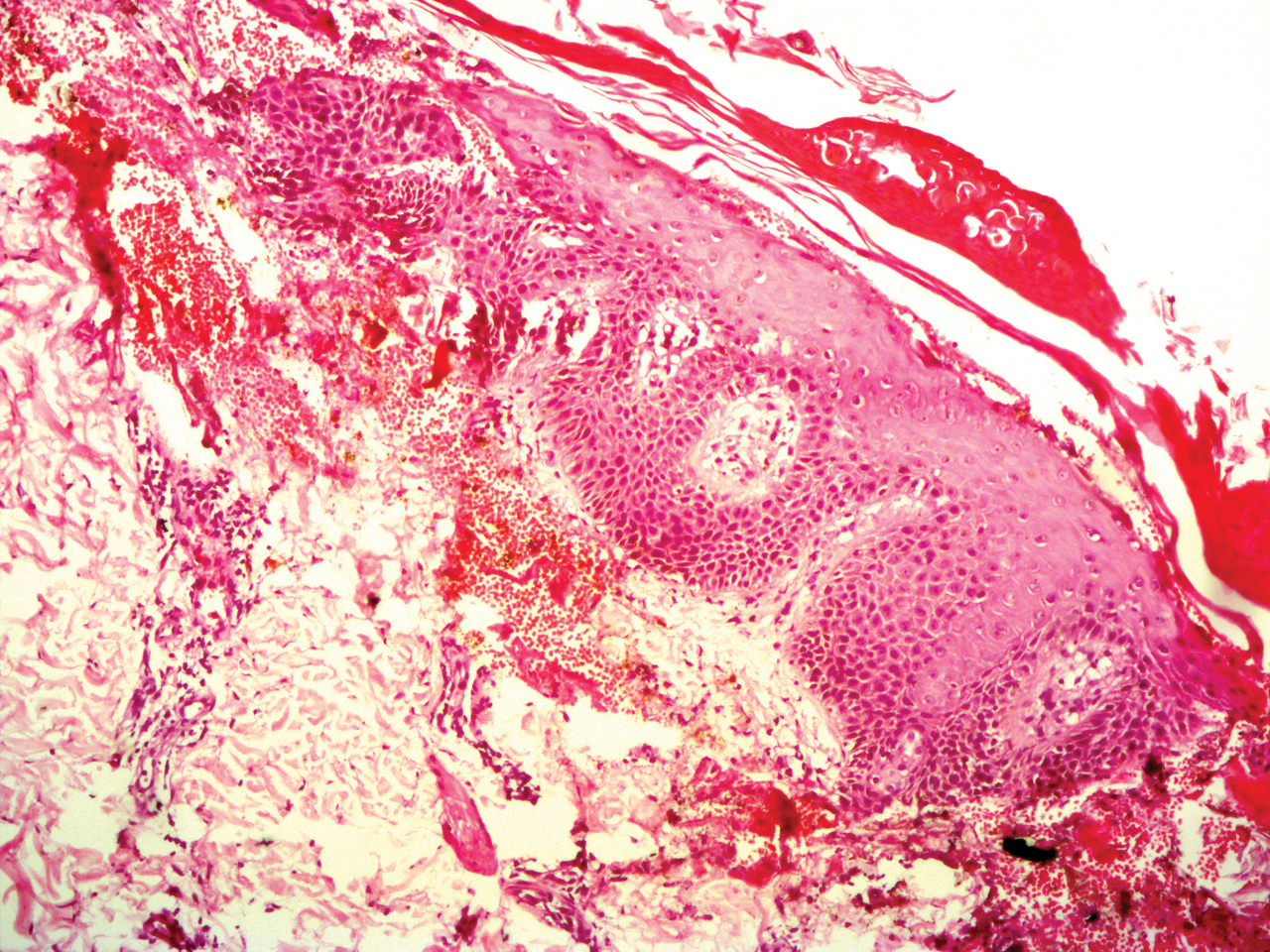
We prescribed oral acitretin 25 mg once daily and instructed the patient to apply petroleum jelly; however, the patient returned after 2 weeks due to aggravation of the skin condition with increased scaling and redness. Because the patient showed signs of acute skin failure and erythroderma, we stopped acitretin treatment and managed his condition conservatively with the application of petroleum jelly.
Netherton syndrome is caused by mutation of the SPINK5 gene, serine protease inhibitor Kazal type 5; the corresponding gene is located on the long arm of chromosome 5.3 The gene encodes a serine protease inhibitor proprotein LEKTI (lymphoepithelial Kazal type inhibitor).4 The product of the gene is thought to be necessary for epidermal cell growth and differentiation. The classic clinical triad of NS includes ichthyosiform dermatosis with double-edged scale, hair shaft abnormalities, and atopy or elevated IgE levels.5 Generalized (congenital) erythroderma usually becomes evident at birth or shortly thereafter. Half of patients develop lesions of ILC on the trunk and limbs during childhood.6 A typical ILC lesion is characterized by an erythematous scaly patch that may be annular or polycyclic with double-edged scale at the advancing border. The ability to sweat is impaired, which may cause episodes of hyperpyrexia, especially during humid weather. Patients with hyperpyrexia may be incorrectly diagnosed with bacterial infection and treated with antipyretic drugs or a prolonged course of antibiotics. Trichorrhexis invaginata, also referred to as bamboo hair or ball-and-socket defect, is the pathognomonic hair shaft abnormality seen in NS.7 Other hair shaft abnormalities in this syndrome include trichorrhexis nodosa and pili torti.8 Our patient had hair shaft abnormalities of trichorrhexis invaginata and pili torti, which are rare findings. The third component of this syndrome is atopy, which generally manifests as angioedema, urticaria, allergic rhinitis, peripheral eosinophilia, atopic dermatitis–like skin lesions, asthma, and elevated IgE levels.9
Treatment with emollients, topical steroids, tacrolimus, and psoralen plus UVA does not elicit a satisfactory response. The Table highlights the clinical features and management of NS.
Generally, systemic retinoid therapy is helpful in cases of erythrodermic ichthyosis, but a unique feature of NS is that erythroderma may worsen with systemic retinoid therapy, as retinoids aggravate atopic dermatitis by worsening existing xerosis.4 Our case highlights the rare association of trichorrhexis invaginata with pili torti as well as acitretin treatment worsening our patient’s condition. This paradoxical effect of retinoid therapy further confirmed the diagnosis of NS.
- Suhaila O, Muzhirah A. Netherton syndrome: a case report. Malaysian J Pediatr Child Health. 2010;16:26.
- Emre S, Metin A, Demirseren D, et al. Two siblings with Netherton syndrome. Turk J Med Sci. 2010;40:819-823.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141-142.
- Judge MR, Mclean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010:19.1-19.122.
- Greene SL, Muller SA. Netherton’s syndrome. report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329-337.
- Khan I-U, Chaudhary R. Netherton’s syndrome, an uncommon genodermatosis. J Pakistan Assoc Dermatol. 2006;16.
- Boskabadi H, Maamouri G, Mafinejad S. Netherton syndrome, a case report and review of literature. Iran J Pediatr. 2013;23:611-612.
- Hurwitz S. Hereditary skin disorders: the genodermatoses. In: Hurwitz, ed. Clinical Pediatric Dermatology. Philadelphia, PA: WB Saunders; 1993:173.
- Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. Vol 2. Oxford, England: Blackwell Science; 2004:34.35.
To the Editor:
Netherton syndrome (NS) is a rare autosomal-recessive ichthyosiform disease.1 The incidence is estimated to be 1 in 200,000 individuals.2 Netherton syndrome presents with generalized erythroderma and scaling, characteristic hair shaft abnormalities, and dysregulation of the immune system. Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination. We report a case of NS in a man with congenital erythroderma, pili torti, and elevated IgE levels.
A 23-year-old man presented with generalized scaly skin that was present since birth. He was the first child born of nonconsanguineous parents. His medical history was suggestive of atopic diatheses such as allergic rhinitis and recurrent urticaria. The patient was of thin build and had widespread erythematous, annular, and polycyclic scaly lesions (Figure 1A), some with characteristic double-edged scale (Figure 1B). The skin was dry due to anhidrosis that was present since birth. Flexural lichenification was present at the cubital fossa of both arms. Scalp hairs were easily pluckable and had generalized thinning of hair density. Hair mount examination showed characteristic features of both trichorrhexis invaginata (Figure 2A) and pili torti (Figure 2B).
hair shaft known as bamboo hair or trichorrhexis invaginata. B, Features of pili torti; the hair
shaft twisted at irregular intervals.
Potassium hydroxide mount from a lesion was negative for fungal elements. Complete hematologic workup showed moderate anemia at 8.0 g/dL (reference range, 8.0–10.9 g/dL) and peripheral eosinophilia at 12% (reference range, 0%–6%). His IgE level was markedly elevated at6331 IU/mL (reference range, 150–1000 IU/mL) when tested with fully automated bidirectionally interfaced chemiluminescent immunoassay. Histopathologic examination of a lesion biopsy showed psoriasiform epidermal hyperplasia, papillomatosis, and acanthosis, consistent with ichthyosis linearis circumflexa (ILC)(Figure 3). Clinicopathologic correlation led to a diagnosis of ILC, trichorrhexis invaginata/pili torti, and atopic diathesis, which is a constellation of disorders related to NS.
We prescribed oral acitretin 25 mg once daily and instructed the patient to apply petroleum jelly; however, the patient returned after 2 weeks due to aggravation of the skin condition with increased scaling and redness. Because the patient showed signs of acute skin failure and erythroderma, we stopped acitretin treatment and managed his condition conservatively with the application of petroleum jelly.
Netherton syndrome is caused by mutation of the SPINK5 gene, serine protease inhibitor Kazal type 5; the corresponding gene is located on the long arm of chromosome 5.3 The gene encodes a serine protease inhibitor proprotein LEKTI (lymphoepithelial Kazal type inhibitor).4 The product of the gene is thought to be necessary for epidermal cell growth and differentiation. The classic clinical triad of NS includes ichthyosiform dermatosis with double-edged scale, hair shaft abnormalities, and atopy or elevated IgE levels.5 Generalized (congenital) erythroderma usually becomes evident at birth or shortly thereafter. Half of patients develop lesions of ILC on the trunk and limbs during childhood.6 A typical ILC lesion is characterized by an erythematous scaly patch that may be annular or polycyclic with double-edged scale at the advancing border. The ability to sweat is impaired, which may cause episodes of hyperpyrexia, especially during humid weather. Patients with hyperpyrexia may be incorrectly diagnosed with bacterial infection and treated with antipyretic drugs or a prolonged course of antibiotics. Trichorrhexis invaginata, also referred to as bamboo hair or ball-and-socket defect, is the pathognomonic hair shaft abnormality seen in NS.7 Other hair shaft abnormalities in this syndrome include trichorrhexis nodosa and pili torti.8 Our patient had hair shaft abnormalities of trichorrhexis invaginata and pili torti, which are rare findings. The third component of this syndrome is atopy, which generally manifests as angioedema, urticaria, allergic rhinitis, peripheral eosinophilia, atopic dermatitis–like skin lesions, asthma, and elevated IgE levels.9
Treatment with emollients, topical steroids, tacrolimus, and psoralen plus UVA does not elicit a satisfactory response. The Table highlights the clinical features and management of NS.
Generally, systemic retinoid therapy is helpful in cases of erythrodermic ichthyosis, but a unique feature of NS is that erythroderma may worsen with systemic retinoid therapy, as retinoids aggravate atopic dermatitis by worsening existing xerosis.4 Our case highlights the rare association of trichorrhexis invaginata with pili torti as well as acitretin treatment worsening our patient’s condition. This paradoxical effect of retinoid therapy further confirmed the diagnosis of NS.
To the Editor:
Netherton syndrome (NS) is a rare autosomal-recessive ichthyosiform disease.1 The incidence is estimated to be 1 in 200,000 individuals.2 Netherton syndrome presents with generalized erythroderma and scaling, characteristic hair shaft abnormalities, and dysregulation of the immune system. Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination. We report a case of NS in a man with congenital erythroderma, pili torti, and elevated IgE levels.
A 23-year-old man presented with generalized scaly skin that was present since birth. He was the first child born of nonconsanguineous parents. His medical history was suggestive of atopic diatheses such as allergic rhinitis and recurrent urticaria. The patient was of thin build and had widespread erythematous, annular, and polycyclic scaly lesions (Figure 1A), some with characteristic double-edged scale (Figure 1B). The skin was dry due to anhidrosis that was present since birth. Flexural lichenification was present at the cubital fossa of both arms. Scalp hairs were easily pluckable and had generalized thinning of hair density. Hair mount examination showed characteristic features of both trichorrhexis invaginata (Figure 2A) and pili torti (Figure 2B).
hair shaft known as bamboo hair or trichorrhexis invaginata. B, Features of pili torti; the hair
shaft twisted at irregular intervals.
Potassium hydroxide mount from a lesion was negative for fungal elements. Complete hematologic workup showed moderate anemia at 8.0 g/dL (reference range, 8.0–10.9 g/dL) and peripheral eosinophilia at 12% (reference range, 0%–6%). His IgE level was markedly elevated at6331 IU/mL (reference range, 150–1000 IU/mL) when tested with fully automated bidirectionally interfaced chemiluminescent immunoassay. Histopathologic examination of a lesion biopsy showed psoriasiform epidermal hyperplasia, papillomatosis, and acanthosis, consistent with ichthyosis linearis circumflexa (ILC)(Figure 3). Clinicopathologic correlation led to a diagnosis of ILC, trichorrhexis invaginata/pili torti, and atopic diathesis, which is a constellation of disorders related to NS.
We prescribed oral acitretin 25 mg once daily and instructed the patient to apply petroleum jelly; however, the patient returned after 2 weeks due to aggravation of the skin condition with increased scaling and redness. Because the patient showed signs of acute skin failure and erythroderma, we stopped acitretin treatment and managed his condition conservatively with the application of petroleum jelly.
Netherton syndrome is caused by mutation of the SPINK5 gene, serine protease inhibitor Kazal type 5; the corresponding gene is located on the long arm of chromosome 5.3 The gene encodes a serine protease inhibitor proprotein LEKTI (lymphoepithelial Kazal type inhibitor).4 The product of the gene is thought to be necessary for epidermal cell growth and differentiation. The classic clinical triad of NS includes ichthyosiform dermatosis with double-edged scale, hair shaft abnormalities, and atopy or elevated IgE levels.5 Generalized (congenital) erythroderma usually becomes evident at birth or shortly thereafter. Half of patients develop lesions of ILC on the trunk and limbs during childhood.6 A typical ILC lesion is characterized by an erythematous scaly patch that may be annular or polycyclic with double-edged scale at the advancing border. The ability to sweat is impaired, which may cause episodes of hyperpyrexia, especially during humid weather. Patients with hyperpyrexia may be incorrectly diagnosed with bacterial infection and treated with antipyretic drugs or a prolonged course of antibiotics. Trichorrhexis invaginata, also referred to as bamboo hair or ball-and-socket defect, is the pathognomonic hair shaft abnormality seen in NS.7 Other hair shaft abnormalities in this syndrome include trichorrhexis nodosa and pili torti.8 Our patient had hair shaft abnormalities of trichorrhexis invaginata and pili torti, which are rare findings. The third component of this syndrome is atopy, which generally manifests as angioedema, urticaria, allergic rhinitis, peripheral eosinophilia, atopic dermatitis–like skin lesions, asthma, and elevated IgE levels.9
Treatment with emollients, topical steroids, tacrolimus, and psoralen plus UVA does not elicit a satisfactory response. The Table highlights the clinical features and management of NS.
Generally, systemic retinoid therapy is helpful in cases of erythrodermic ichthyosis, but a unique feature of NS is that erythroderma may worsen with systemic retinoid therapy, as retinoids aggravate atopic dermatitis by worsening existing xerosis.4 Our case highlights the rare association of trichorrhexis invaginata with pili torti as well as acitretin treatment worsening our patient’s condition. This paradoxical effect of retinoid therapy further confirmed the diagnosis of NS.
- Suhaila O, Muzhirah A. Netherton syndrome: a case report. Malaysian J Pediatr Child Health. 2010;16:26.
- Emre S, Metin A, Demirseren D, et al. Two siblings with Netherton syndrome. Turk J Med Sci. 2010;40:819-823.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141-142.
- Judge MR, Mclean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010:19.1-19.122.
- Greene SL, Muller SA. Netherton’s syndrome. report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329-337.
- Khan I-U, Chaudhary R. Netherton’s syndrome, an uncommon genodermatosis. J Pakistan Assoc Dermatol. 2006;16.
- Boskabadi H, Maamouri G, Mafinejad S. Netherton syndrome, a case report and review of literature. Iran J Pediatr. 2013;23:611-612.
- Hurwitz S. Hereditary skin disorders: the genodermatoses. In: Hurwitz, ed. Clinical Pediatric Dermatology. Philadelphia, PA: WB Saunders; 1993:173.
- Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. Vol 2. Oxford, England: Blackwell Science; 2004:34.35.
- Suhaila O, Muzhirah A. Netherton syndrome: a case report. Malaysian J Pediatr Child Health. 2010;16:26.
- Emre S, Metin A, Demirseren D, et al. Two siblings with Netherton syndrome. Turk J Med Sci. 2010;40:819-823.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141-142.
- Judge MR, Mclean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010:19.1-19.122.
- Greene SL, Muller SA. Netherton’s syndrome. report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329-337.
- Khan I-U, Chaudhary R. Netherton’s syndrome, an uncommon genodermatosis. J Pakistan Assoc Dermatol. 2006;16.
- Boskabadi H, Maamouri G, Mafinejad S. Netherton syndrome, a case report and review of literature. Iran J Pediatr. 2013;23:611-612.
- Hurwitz S. Hereditary skin disorders: the genodermatoses. In: Hurwitz, ed. Clinical Pediatric Dermatology. Philadelphia, PA: WB Saunders; 1993:173.
- Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. Vol 2. Oxford, England: Blackwell Science; 2004:34.35.
Practice Points
- Netherton syndrome is characterized by generalized erythroderma and scaling, hair shaft abnormalities, and dysregulation of the immune system.
- Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination.