User login
Acantholytic Anaplastic Extramammary Paget Disease
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
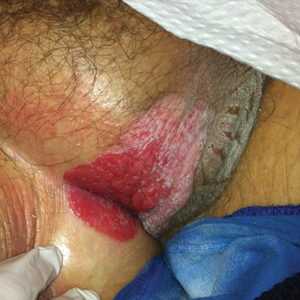
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
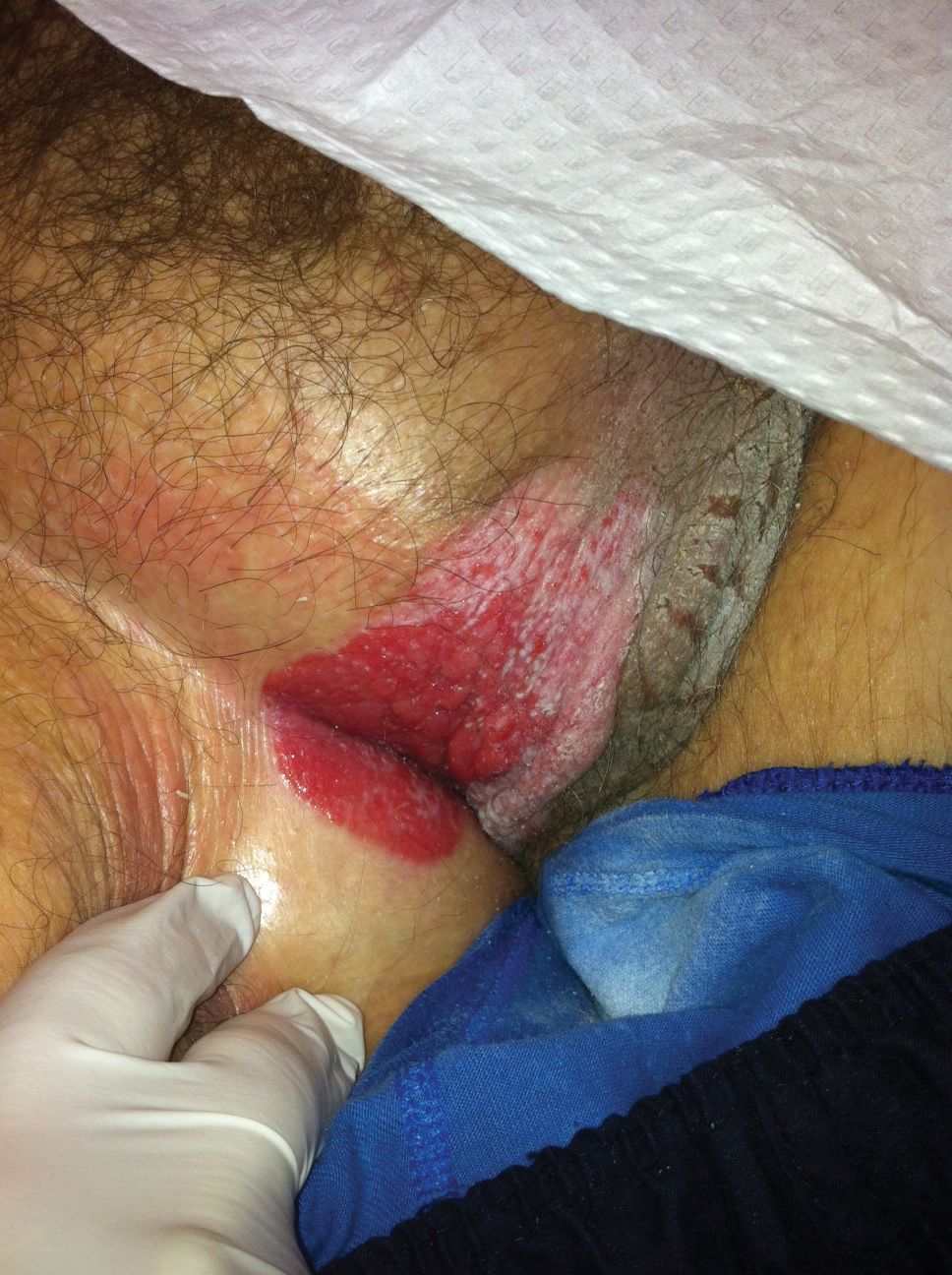
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
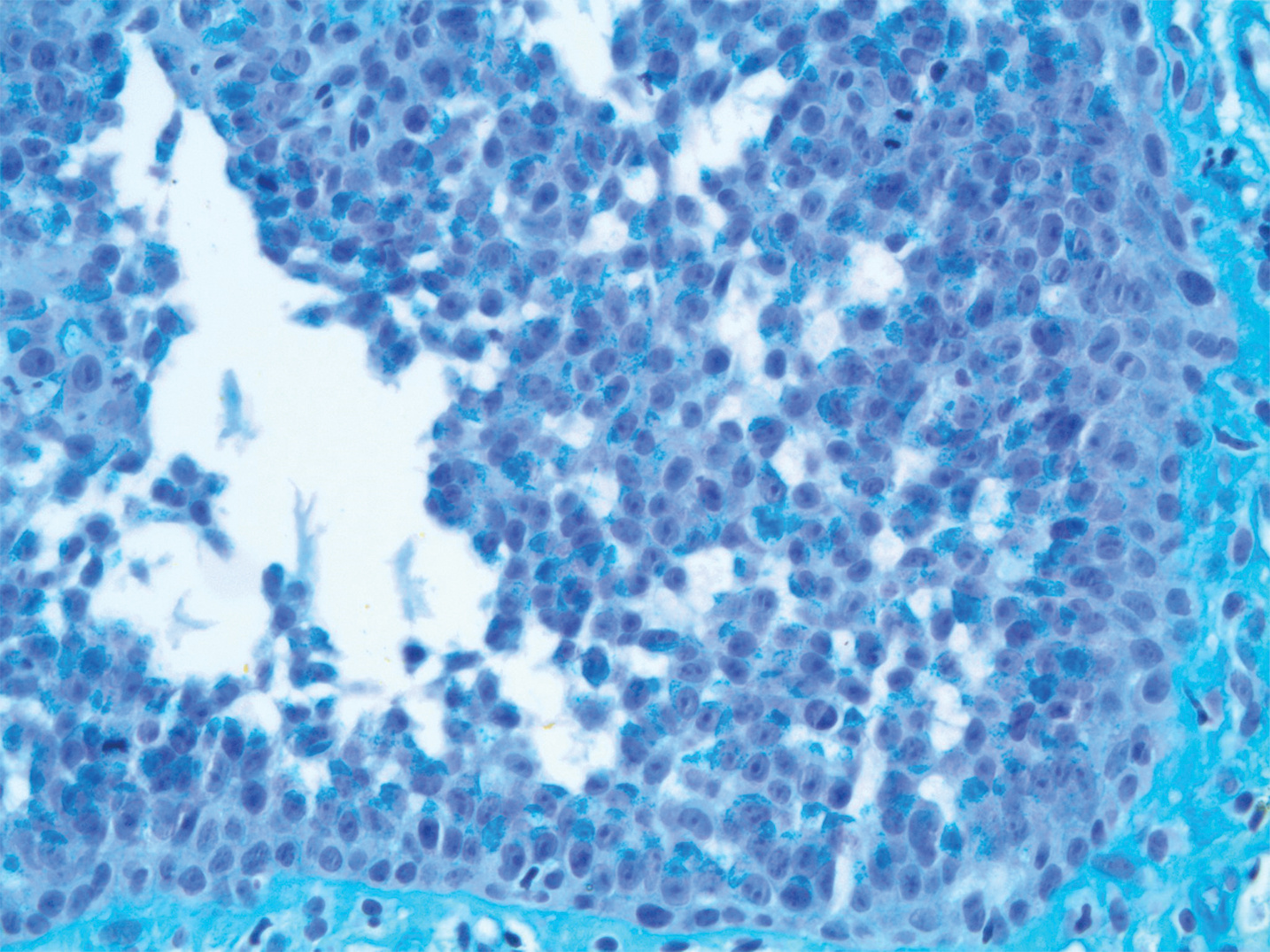
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
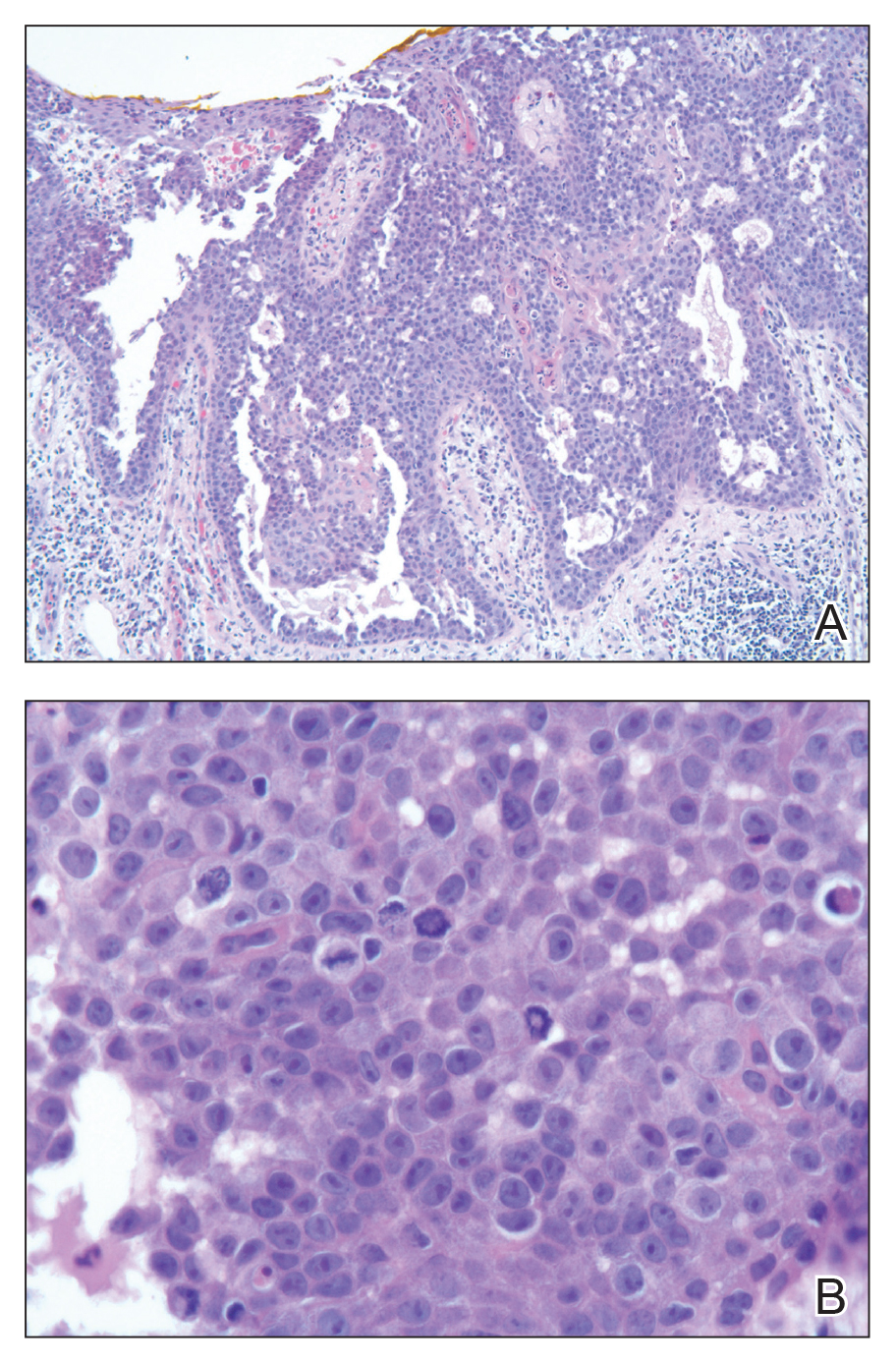
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
Practice Points
- The acantholytic anaplastic variant of extramammary Paget disease (EMPD) can be mimicked by many other entities including Bowen disease, acantholytic dyskeratosis of the genitocrural area, and pemphigus vulgaris.
- A good immunohistochemical panel to evaluate for EMPD includes cytokeratin (CK) 7, pancytokeratin (CKAE1/AE3), CK20, and carcinoembryonic antigen.
Barber’s Sinus Between the Toes of a Female Hairdresser
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
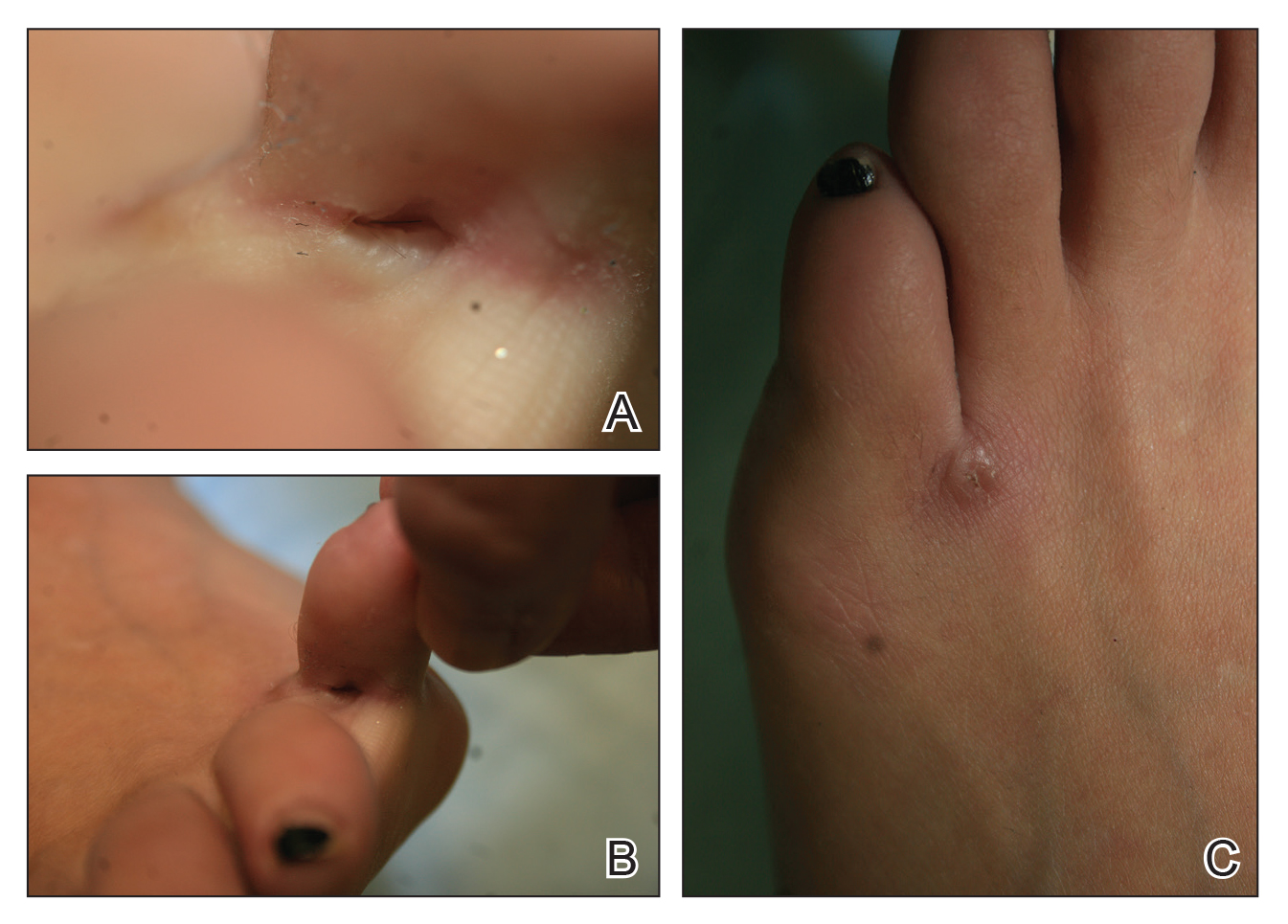
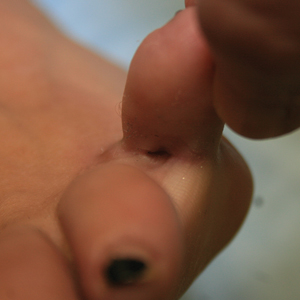
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
Practice Points
- This case illustrates a disease in which a medical history and simple clinical examination can lead to the diagnosis.
- Patients may value a diagnosis without treatment. A patient with barber’s sinus may be satisfied with watchful waiting.
Management of Refractory Pain From Hereditary Cutaneous Leiomyomas With Nifedipine and Gabapentin
To the Editor:
Leiomyomas are benign smooth muscle tumors. There are 3 types of cutaneous leiomyomas: (1) piloleiomyomas, arising from the arrector pili muscles; (2) angioleiomyomas, arising from the muscles surrounding dermal blood vessels; and (3) leiomyomas of the external genitalia, arising from the dartoic, vulvar, or mammary smooth muscles.1 There is no gender predilection for cutaneous leiomyomas, and lesions present on average at approximately 40 to 45 years of age.2
Piloleiomyomas are the most common type of cutaneous leiomyomas and typically present as red-brown papules and nodules on the trunk, arms, and legs.3 Piloleiomyomas often are associated with spontaneous or induced pain (eg, with cold exposure). The pain associated with piloleiomyomas can be severely debilitating to patients and may have a considerable impact on their quality of life.
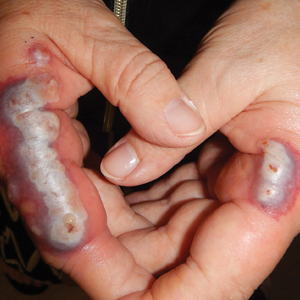
A 40-year-old woman presented to our clinic with numerous widespread, painful, red-brown papules and nodules on the head, neck, chest, abdomen, back, arms, and legs of 6 years’ duration that were increasing in number (Figure 1). She had a history of uterine leiomyomas and type 2 renal papillary carcinoma following a left nephrectomy at 38 years of age. The patient’s mother had a history of similar skin lesions as well as uterine cancer. Multiple excisional biopsies were performed, all of which showed piloleiomyomas on histopathology (Figure 2). The pain associated with the patient’s extensive cutaneous leiomyomas considerably impaired her quality of life. Although she experienced pain in all affected areas of the body, the pain was the worst in the upper arms. She reported having requested a nerve ablation procedure from an outside pain management clinic, which was denied for unknown reasons.


Two years prior to the current presentation, the patient had been treated by a pain management specialist with gabapentin 300 mg twice daily as needed for pain associated with leiomyomas. The patient followed this regimen approximately 3 times weekly for the preceding 1 to 2 years with reduction in her pain symptoms; however, the painful episodes became more frequent and severe over time. The patient reported being unable to further increase the gabapentin dosing frequency because it made her too drowsy and impacted her ability to work a job that required heavy lifting. Thus, the patient requested additional therapy and was subsequently treated at our clinic with numerous excisional biopsies of the most painful lesions during the 2 years prior to her current presentation.
When the patient re-presented to our clinic, she requested additional lesion excisions given that she had experienced some pain relief from this treatment modality in the past; however, these prior excisions only resulted in local pain relief limited to the site of the excision. Because of the extent of the lesions and the patient’s inability to tolerate pain from the lidocaine injections, we did not feel multiple excisions were a practical treatment option. The patient subsequently was offered a trial of cryotherapy for symptom relief based on a reported case in which this modality was successfully used.4 After discussing the risks and benefits associated with this treatment, cryotherapy was attempted on a few of the leiomyomas on the patient’s right shoulder; however, she experienced severe pain during cryotherapy treatment, and the procedure had to be aborted.
We then increased the patient’s gabapentin regimen to 300 mg in the morning and 600 mg in the evening, as tolerated. The patient reported that she was better able to tolerate the sedating side effects of the increased dose of gabapentin because she had stopped working due to her severe pain episodes. We also added oral nifedipine 10 mg 3 times daily, as needed. Within 30 minutes of starting this treatment regimen, the pain associated with the lesions remarkably improved (10/10 severity before starting treatment vs 3/10 after starting treatment). Her pain levels remained stable (3/10 severity) during 3 weeks of treatment with this combination regimen, but unfortunately she developed headaches and malaise, which she associated with the nifedipine at the 3 times daily dose.
The patient was able to better tolerate the nifedipine after reducing the dose to once daily on an as-needed basis. On average, the patient took nifedipine once every 3 days; however, she reported that she had to periodically increase the frequency of the nifedipine to once daily for up to 2 weeks at a time for periods of more frequent pain flares. The patient reported a consistent pattern of the breakthrough symptoms rapidly improving with each dose of nifedipine, though she did feel that taking consistent gabapentin enhanced baseline symptom control. The patient also noticed on a few occasions when she did not have access to her nifedipine that her pain would flare to 10/10 severity and would decrease to 4/10 severity 30 minutes after restarting nifedipine at 10 mg once daily. She experienced breakthrough pain due to exacerbating factors including her menstrual cycle; exposure to the sun and cold temperatures or water; excessive physical activity; and mild trauma. Due to exacerbations from sun exposure, the patient often wore long-sleeved shirts, which helped reduce the severity of the pain episodes while she was outdoors.
The exact mechanism for the pain associated with cutaneous leiomyomas is unknown but is thought to be due to infringement of the lesion on the surrounding cutaneous nerves. In addition, norepinephrine activates alpha receptors on the smooth muscle to contract through an influx of ions such as calcium. When smooth muscle contracts, the compression of nerves likely is worsened.
There are a limited number of case reports in the literature that have demonstrated successful treatment of the pain associated with cutaneous leiomyomas. Previously reported treatment modalities have included phenoxybenzamine, an alpha-blocking agent that may reduce pain through its antiadrenergic effects2; nitroglycerin, a venous and arterial dilator that may reduce pain by decreasing muscle oxygen requirements2; gabapentin, an antiepileptic and analgesic medication with structural similarity to the gamma-aminobutyric acid neurotransmitter for which the exact mechanism of action is unknown3; botulinum toxin, a neuromuscular blocker that prevents the release of presynaptic acetylcholine and may decrease neuropathic pain by reducing hyperactive nerves5,6; hyoscine butylbromide and topical hyoscine hydrobromide, both antispasmodics that may reduce pain through their anticholinergic effects, which relax smooth muscle7,8; and the CO2 laser, a treatment that has been utilized for its resurfacing, excisional, and ablative properties.9,10
Calcium channel blockers such as amlodipine, verapamil, and nifedipine also have been used to treat the pain associated with piloleiomyomas.11 Calcium ion channel antagonists inhibit the influx of calcium ions across the cell membrane; therefore, nifedipine and other calcium channel blockers may prevent the smooth muscle contraction that is hypothesized to cause pain in patients with cutaneous leiomyomas.12
Mean plasma concentration of nifedipine has been shown to reach maximum values of 160 +/− 49 µg/L after 30 to 60 minutes following oral administration of 10 mg of nifedipine.13 After 8 hours, the mean concentration drops to 3.4 +/− 1.2 µg/L. The clinical response in our patient appeared consistent with the reported pharmacokinetics of the drug, as she was able to consistently obtain considerable reduction in her pain symptoms within 30 minutes of starting nifedipine, coinciding with the period of time it takes for the nifedipine to reach maximum plasma concentrations.13
Interestingly, our patient had worsening pain episodes associated with sun exposure, which typically is not reported as one of the usual triggers for cutaneous leiomyomas. We are not aware of any described mechanisms that would explain this phenomenon.
Importantly, any patient presenting with multiple cutaneous and uterine (if female) leiomyomas should be screened for hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC), an autosomal-dominant disorder linked to a mutation in the fumarate hydratase tumor suppressor gene. Clinically, HLRCC patients typically present with multiple cutaneous leiomyomas, uterine leiomyomas, and renal cell cancer (most often type 2 papillary renal cell carcinoma).14 Hereditary leiomyomatosis and renal cell carcinoma syndrome (also known as multiple cutaneous and uterine leiomyomatosis syndrome) previously was thought to be a separate disease entity from Reed syndrome; however, after the same mutation in the fumarate hydratase tumor suppressor gene was found to be responsible for both Reed syndrome and HLRCC, they are now thought to be the same disease process.15
Diagnosis of HLRCC is likely when the patient meets the major criterion of multiple cutaneous piloleiomyomas confirmed histopathologically. Clinical diagnosis of HLRCC is suspected if 2 or more of the following minor criteria are present: type 2 papillary renal cell carcinoma before 40 years of age; onset of severely symptomatic (requiring surgery) uterine fibroids before 40 years of age in females; and first-degree family member who meets 1 or more of these criteria.15 At the time of presentation, the patient met clinical criteria for HLRCC, including multiple cutaneous leiomyomas (major criterion) and type 2 papillary renal cell carcinoma before 40 years of age (minor criterion). The patient also had a history of uterine leiomyomas, but these lesions did not fulfill the criterion of being severely symptomatic requiring surgery. Furthermore, the patient’s mother had similar cutaneous leiomyomas and a history of uterine cancer, which fulfilled additional minor criterion, consistent with an autosomal-dominant inheritance pattern (with variable penetrance) seen in HLRCC. An important issue for counseling and monitoring patients is that premenopausal women with HLRCC are at an increased risk of developing uterine leiomyosarcoma.15 Our patient followed up with an oncologist for tumor surveillance and subsequently underwent genetic testing, which revealed a mutation in the fumarate hydratase gene.
Treatment of painful cutaneous leiomyomas, particularly in patients with HLRCC, remains a therapeutic challenge. Although surgical and/or destructive treatments can provide pain relief for patients who have a limited number of lesions, these options are impracticable when a patient has numerous widespread leiomyomas; therefore, systemic therapies may be more beneficial. Clinicians should be aware of nifedipine, which may be used in combination with gabapentin as a viable treatment option in the management of acute and breakthrough pain associated with cutaneous leiomyomas.
Acknowledgment
The a
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477-494.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Alam M, Rabinowitz AD, Engler DE. Gabapentin treatment of multiple piloleiomyoma-related pain. J Am Acad Dermatol. 2002;46:S27-S29.
- Basendwh MA, Fatani M, Baltow B. Reed’s syndrome: a case of multiple cutaneous leiomyomas treated with liquid nitrogen cryotherapy. Case Rep Dermatol. 2016;8:65-70.
- Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A–treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2008;218:44-47.
- Onder M, Adıs¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Kaliyadan F, Manoj J, Dharmaratnam AD. Multiple cutaneous leiomyomas: pain relief with pulsed hysocine butyl bromide. Indian J Dermatol. 2009;54:72.
- Archer CB, Whittaker S, Greaves MW. Pharmacological modulation of cold‐induced pain in cutaneous leiomyomata. Br J Dermatol. 1988;118:255-260.
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322.
- Michajłowski I, Błaz˙ewicz I, Karpinsky G, et al. Successful treatment of multiple cutaneous leiomyomas with carbon dioxide laser ablation. Postepy Dermatol Alergol. 2015;32:480-482.
- Archer CB, Greaves MW. Assessment of treatment for painful cutaneous leiomyomas. J Am Acad Dermatol. 1987;17:141-142.
- Thompson JA. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865-867.
- Raemsch KD, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5(4 pt 2):II18-II24.
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95-106.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49-59.
To the Editor:
Leiomyomas are benign smooth muscle tumors. There are 3 types of cutaneous leiomyomas: (1) piloleiomyomas, arising from the arrector pili muscles; (2) angioleiomyomas, arising from the muscles surrounding dermal blood vessels; and (3) leiomyomas of the external genitalia, arising from the dartoic, vulvar, or mammary smooth muscles.1 There is no gender predilection for cutaneous leiomyomas, and lesions present on average at approximately 40 to 45 years of age.2
Piloleiomyomas are the most common type of cutaneous leiomyomas and typically present as red-brown papules and nodules on the trunk, arms, and legs.3 Piloleiomyomas often are associated with spontaneous or induced pain (eg, with cold exposure). The pain associated with piloleiomyomas can be severely debilitating to patients and may have a considerable impact on their quality of life.
A 40-year-old woman presented to our clinic with numerous widespread, painful, red-brown papules and nodules on the head, neck, chest, abdomen, back, arms, and legs of 6 years’ duration that were increasing in number (Figure 1). She had a history of uterine leiomyomas and type 2 renal papillary carcinoma following a left nephrectomy at 38 years of age. The patient’s mother had a history of similar skin lesions as well as uterine cancer. Multiple excisional biopsies were performed, all of which showed piloleiomyomas on histopathology (Figure 2). The pain associated with the patient’s extensive cutaneous leiomyomas considerably impaired her quality of life. Although she experienced pain in all affected areas of the body, the pain was the worst in the upper arms. She reported having requested a nerve ablation procedure from an outside pain management clinic, which was denied for unknown reasons.
Two years prior to the current presentation, the patient had been treated by a pain management specialist with gabapentin 300 mg twice daily as needed for pain associated with leiomyomas. The patient followed this regimen approximately 3 times weekly for the preceding 1 to 2 years with reduction in her pain symptoms; however, the painful episodes became more frequent and severe over time. The patient reported being unable to further increase the gabapentin dosing frequency because it made her too drowsy and impacted her ability to work a job that required heavy lifting. Thus, the patient requested additional therapy and was subsequently treated at our clinic with numerous excisional biopsies of the most painful lesions during the 2 years prior to her current presentation.
When the patient re-presented to our clinic, she requested additional lesion excisions given that she had experienced some pain relief from this treatment modality in the past; however, these prior excisions only resulted in local pain relief limited to the site of the excision. Because of the extent of the lesions and the patient’s inability to tolerate pain from the lidocaine injections, we did not feel multiple excisions were a practical treatment option. The patient subsequently was offered a trial of cryotherapy for symptom relief based on a reported case in which this modality was successfully used.4 After discussing the risks and benefits associated with this treatment, cryotherapy was attempted on a few of the leiomyomas on the patient’s right shoulder; however, she experienced severe pain during cryotherapy treatment, and the procedure had to be aborted.
We then increased the patient’s gabapentin regimen to 300 mg in the morning and 600 mg in the evening, as tolerated. The patient reported that she was better able to tolerate the sedating side effects of the increased dose of gabapentin because she had stopped working due to her severe pain episodes. We also added oral nifedipine 10 mg 3 times daily, as needed. Within 30 minutes of starting this treatment regimen, the pain associated with the lesions remarkably improved (10/10 severity before starting treatment vs 3/10 after starting treatment). Her pain levels remained stable (3/10 severity) during 3 weeks of treatment with this combination regimen, but unfortunately she developed headaches and malaise, which she associated with the nifedipine at the 3 times daily dose.
The patient was able to better tolerate the nifedipine after reducing the dose to once daily on an as-needed basis. On average, the patient took nifedipine once every 3 days; however, she reported that she had to periodically increase the frequency of the nifedipine to once daily for up to 2 weeks at a time for periods of more frequent pain flares. The patient reported a consistent pattern of the breakthrough symptoms rapidly improving with each dose of nifedipine, though she did feel that taking consistent gabapentin enhanced baseline symptom control. The patient also noticed on a few occasions when she did not have access to her nifedipine that her pain would flare to 10/10 severity and would decrease to 4/10 severity 30 minutes after restarting nifedipine at 10 mg once daily. She experienced breakthrough pain due to exacerbating factors including her menstrual cycle; exposure to the sun and cold temperatures or water; excessive physical activity; and mild trauma. Due to exacerbations from sun exposure, the patient often wore long-sleeved shirts, which helped reduce the severity of the pain episodes while she was outdoors.
The exact mechanism for the pain associated with cutaneous leiomyomas is unknown but is thought to be due to infringement of the lesion on the surrounding cutaneous nerves. In addition, norepinephrine activates alpha receptors on the smooth muscle to contract through an influx of ions such as calcium. When smooth muscle contracts, the compression of nerves likely is worsened.
There are a limited number of case reports in the literature that have demonstrated successful treatment of the pain associated with cutaneous leiomyomas. Previously reported treatment modalities have included phenoxybenzamine, an alpha-blocking agent that may reduce pain through its antiadrenergic effects2; nitroglycerin, a venous and arterial dilator that may reduce pain by decreasing muscle oxygen requirements2; gabapentin, an antiepileptic and analgesic medication with structural similarity to the gamma-aminobutyric acid neurotransmitter for which the exact mechanism of action is unknown3; botulinum toxin, a neuromuscular blocker that prevents the release of presynaptic acetylcholine and may decrease neuropathic pain by reducing hyperactive nerves5,6; hyoscine butylbromide and topical hyoscine hydrobromide, both antispasmodics that may reduce pain through their anticholinergic effects, which relax smooth muscle7,8; and the CO2 laser, a treatment that has been utilized for its resurfacing, excisional, and ablative properties.9,10
Calcium channel blockers such as amlodipine, verapamil, and nifedipine also have been used to treat the pain associated with piloleiomyomas.11 Calcium ion channel antagonists inhibit the influx of calcium ions across the cell membrane; therefore, nifedipine and other calcium channel blockers may prevent the smooth muscle contraction that is hypothesized to cause pain in patients with cutaneous leiomyomas.12
Mean plasma concentration of nifedipine has been shown to reach maximum values of 160 +/− 49 µg/L after 30 to 60 minutes following oral administration of 10 mg of nifedipine.13 After 8 hours, the mean concentration drops to 3.4 +/− 1.2 µg/L. The clinical response in our patient appeared consistent with the reported pharmacokinetics of the drug, as she was able to consistently obtain considerable reduction in her pain symptoms within 30 minutes of starting nifedipine, coinciding with the period of time it takes for the nifedipine to reach maximum plasma concentrations.13
Interestingly, our patient had worsening pain episodes associated with sun exposure, which typically is not reported as one of the usual triggers for cutaneous leiomyomas. We are not aware of any described mechanisms that would explain this phenomenon.
Importantly, any patient presenting with multiple cutaneous and uterine (if female) leiomyomas should be screened for hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC), an autosomal-dominant disorder linked to a mutation in the fumarate hydratase tumor suppressor gene. Clinically, HLRCC patients typically present with multiple cutaneous leiomyomas, uterine leiomyomas, and renal cell cancer (most often type 2 papillary renal cell carcinoma).14 Hereditary leiomyomatosis and renal cell carcinoma syndrome (also known as multiple cutaneous and uterine leiomyomatosis syndrome) previously was thought to be a separate disease entity from Reed syndrome; however, after the same mutation in the fumarate hydratase tumor suppressor gene was found to be responsible for both Reed syndrome and HLRCC, they are now thought to be the same disease process.15
Diagnosis of HLRCC is likely when the patient meets the major criterion of multiple cutaneous piloleiomyomas confirmed histopathologically. Clinical diagnosis of HLRCC is suspected if 2 or more of the following minor criteria are present: type 2 papillary renal cell carcinoma before 40 years of age; onset of severely symptomatic (requiring surgery) uterine fibroids before 40 years of age in females; and first-degree family member who meets 1 or more of these criteria.15 At the time of presentation, the patient met clinical criteria for HLRCC, including multiple cutaneous leiomyomas (major criterion) and type 2 papillary renal cell carcinoma before 40 years of age (minor criterion). The patient also had a history of uterine leiomyomas, but these lesions did not fulfill the criterion of being severely symptomatic requiring surgery. Furthermore, the patient’s mother had similar cutaneous leiomyomas and a history of uterine cancer, which fulfilled additional minor criterion, consistent with an autosomal-dominant inheritance pattern (with variable penetrance) seen in HLRCC. An important issue for counseling and monitoring patients is that premenopausal women with HLRCC are at an increased risk of developing uterine leiomyosarcoma.15 Our patient followed up with an oncologist for tumor surveillance and subsequently underwent genetic testing, which revealed a mutation in the fumarate hydratase gene.
Treatment of painful cutaneous leiomyomas, particularly in patients with HLRCC, remains a therapeutic challenge. Although surgical and/or destructive treatments can provide pain relief for patients who have a limited number of lesions, these options are impracticable when a patient has numerous widespread leiomyomas; therefore, systemic therapies may be more beneficial. Clinicians should be aware of nifedipine, which may be used in combination with gabapentin as a viable treatment option in the management of acute and breakthrough pain associated with cutaneous leiomyomas.
Acknowledgment
The a
To the Editor:
Leiomyomas are benign smooth muscle tumors. There are 3 types of cutaneous leiomyomas: (1) piloleiomyomas, arising from the arrector pili muscles; (2) angioleiomyomas, arising from the muscles surrounding dermal blood vessels; and (3) leiomyomas of the external genitalia, arising from the dartoic, vulvar, or mammary smooth muscles.1 There is no gender predilection for cutaneous leiomyomas, and lesions present on average at approximately 40 to 45 years of age.2
Piloleiomyomas are the most common type of cutaneous leiomyomas and typically present as red-brown papules and nodules on the trunk, arms, and legs.3 Piloleiomyomas often are associated with spontaneous or induced pain (eg, with cold exposure). The pain associated with piloleiomyomas can be severely debilitating to patients and may have a considerable impact on their quality of life.
A 40-year-old woman presented to our clinic with numerous widespread, painful, red-brown papules and nodules on the head, neck, chest, abdomen, back, arms, and legs of 6 years’ duration that were increasing in number (Figure 1). She had a history of uterine leiomyomas and type 2 renal papillary carcinoma following a left nephrectomy at 38 years of age. The patient’s mother had a history of similar skin lesions as well as uterine cancer. Multiple excisional biopsies were performed, all of which showed piloleiomyomas on histopathology (Figure 2). The pain associated with the patient’s extensive cutaneous leiomyomas considerably impaired her quality of life. Although she experienced pain in all affected areas of the body, the pain was the worst in the upper arms. She reported having requested a nerve ablation procedure from an outside pain management clinic, which was denied for unknown reasons.
Two years prior to the current presentation, the patient had been treated by a pain management specialist with gabapentin 300 mg twice daily as needed for pain associated with leiomyomas. The patient followed this regimen approximately 3 times weekly for the preceding 1 to 2 years with reduction in her pain symptoms; however, the painful episodes became more frequent and severe over time. The patient reported being unable to further increase the gabapentin dosing frequency because it made her too drowsy and impacted her ability to work a job that required heavy lifting. Thus, the patient requested additional therapy and was subsequently treated at our clinic with numerous excisional biopsies of the most painful lesions during the 2 years prior to her current presentation.
When the patient re-presented to our clinic, she requested additional lesion excisions given that she had experienced some pain relief from this treatment modality in the past; however, these prior excisions only resulted in local pain relief limited to the site of the excision. Because of the extent of the lesions and the patient’s inability to tolerate pain from the lidocaine injections, we did not feel multiple excisions were a practical treatment option. The patient subsequently was offered a trial of cryotherapy for symptom relief based on a reported case in which this modality was successfully used.4 After discussing the risks and benefits associated with this treatment, cryotherapy was attempted on a few of the leiomyomas on the patient’s right shoulder; however, she experienced severe pain during cryotherapy treatment, and the procedure had to be aborted.
We then increased the patient’s gabapentin regimen to 300 mg in the morning and 600 mg in the evening, as tolerated. The patient reported that she was better able to tolerate the sedating side effects of the increased dose of gabapentin because she had stopped working due to her severe pain episodes. We also added oral nifedipine 10 mg 3 times daily, as needed. Within 30 minutes of starting this treatment regimen, the pain associated with the lesions remarkably improved (10/10 severity before starting treatment vs 3/10 after starting treatment). Her pain levels remained stable (3/10 severity) during 3 weeks of treatment with this combination regimen, but unfortunately she developed headaches and malaise, which she associated with the nifedipine at the 3 times daily dose.
The patient was able to better tolerate the nifedipine after reducing the dose to once daily on an as-needed basis. On average, the patient took nifedipine once every 3 days; however, she reported that she had to periodically increase the frequency of the nifedipine to once daily for up to 2 weeks at a time for periods of more frequent pain flares. The patient reported a consistent pattern of the breakthrough symptoms rapidly improving with each dose of nifedipine, though she did feel that taking consistent gabapentin enhanced baseline symptom control. The patient also noticed on a few occasions when she did not have access to her nifedipine that her pain would flare to 10/10 severity and would decrease to 4/10 severity 30 minutes after restarting nifedipine at 10 mg once daily. She experienced breakthrough pain due to exacerbating factors including her menstrual cycle; exposure to the sun and cold temperatures or water; excessive physical activity; and mild trauma. Due to exacerbations from sun exposure, the patient often wore long-sleeved shirts, which helped reduce the severity of the pain episodes while she was outdoors.
The exact mechanism for the pain associated with cutaneous leiomyomas is unknown but is thought to be due to infringement of the lesion on the surrounding cutaneous nerves. In addition, norepinephrine activates alpha receptors on the smooth muscle to contract through an influx of ions such as calcium. When smooth muscle contracts, the compression of nerves likely is worsened.
There are a limited number of case reports in the literature that have demonstrated successful treatment of the pain associated with cutaneous leiomyomas. Previously reported treatment modalities have included phenoxybenzamine, an alpha-blocking agent that may reduce pain through its antiadrenergic effects2; nitroglycerin, a venous and arterial dilator that may reduce pain by decreasing muscle oxygen requirements2; gabapentin, an antiepileptic and analgesic medication with structural similarity to the gamma-aminobutyric acid neurotransmitter for which the exact mechanism of action is unknown3; botulinum toxin, a neuromuscular blocker that prevents the release of presynaptic acetylcholine and may decrease neuropathic pain by reducing hyperactive nerves5,6; hyoscine butylbromide and topical hyoscine hydrobromide, both antispasmodics that may reduce pain through their anticholinergic effects, which relax smooth muscle7,8; and the CO2 laser, a treatment that has been utilized for its resurfacing, excisional, and ablative properties.9,10
Calcium channel blockers such as amlodipine, verapamil, and nifedipine also have been used to treat the pain associated with piloleiomyomas.11 Calcium ion channel antagonists inhibit the influx of calcium ions across the cell membrane; therefore, nifedipine and other calcium channel blockers may prevent the smooth muscle contraction that is hypothesized to cause pain in patients with cutaneous leiomyomas.12
Mean plasma concentration of nifedipine has been shown to reach maximum values of 160 +/− 49 µg/L after 30 to 60 minutes following oral administration of 10 mg of nifedipine.13 After 8 hours, the mean concentration drops to 3.4 +/− 1.2 µg/L. The clinical response in our patient appeared consistent with the reported pharmacokinetics of the drug, as she was able to consistently obtain considerable reduction in her pain symptoms within 30 minutes of starting nifedipine, coinciding with the period of time it takes for the nifedipine to reach maximum plasma concentrations.13
Interestingly, our patient had worsening pain episodes associated with sun exposure, which typically is not reported as one of the usual triggers for cutaneous leiomyomas. We are not aware of any described mechanisms that would explain this phenomenon.
Importantly, any patient presenting with multiple cutaneous and uterine (if female) leiomyomas should be screened for hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC), an autosomal-dominant disorder linked to a mutation in the fumarate hydratase tumor suppressor gene. Clinically, HLRCC patients typically present with multiple cutaneous leiomyomas, uterine leiomyomas, and renal cell cancer (most often type 2 papillary renal cell carcinoma).14 Hereditary leiomyomatosis and renal cell carcinoma syndrome (also known as multiple cutaneous and uterine leiomyomatosis syndrome) previously was thought to be a separate disease entity from Reed syndrome; however, after the same mutation in the fumarate hydratase tumor suppressor gene was found to be responsible for both Reed syndrome and HLRCC, they are now thought to be the same disease process.15
Diagnosis of HLRCC is likely when the patient meets the major criterion of multiple cutaneous piloleiomyomas confirmed histopathologically. Clinical diagnosis of HLRCC is suspected if 2 or more of the following minor criteria are present: type 2 papillary renal cell carcinoma before 40 years of age; onset of severely symptomatic (requiring surgery) uterine fibroids before 40 years of age in females; and first-degree family member who meets 1 or more of these criteria.15 At the time of presentation, the patient met clinical criteria for HLRCC, including multiple cutaneous leiomyomas (major criterion) and type 2 papillary renal cell carcinoma before 40 years of age (minor criterion). The patient also had a history of uterine leiomyomas, but these lesions did not fulfill the criterion of being severely symptomatic requiring surgery. Furthermore, the patient’s mother had similar cutaneous leiomyomas and a history of uterine cancer, which fulfilled additional minor criterion, consistent with an autosomal-dominant inheritance pattern (with variable penetrance) seen in HLRCC. An important issue for counseling and monitoring patients is that premenopausal women with HLRCC are at an increased risk of developing uterine leiomyosarcoma.15 Our patient followed up with an oncologist for tumor surveillance and subsequently underwent genetic testing, which revealed a mutation in the fumarate hydratase gene.
Treatment of painful cutaneous leiomyomas, particularly in patients with HLRCC, remains a therapeutic challenge. Although surgical and/or destructive treatments can provide pain relief for patients who have a limited number of lesions, these options are impracticable when a patient has numerous widespread leiomyomas; therefore, systemic therapies may be more beneficial. Clinicians should be aware of nifedipine, which may be used in combination with gabapentin as a viable treatment option in the management of acute and breakthrough pain associated with cutaneous leiomyomas.
Acknowledgment
The a
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477-494.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Alam M, Rabinowitz AD, Engler DE. Gabapentin treatment of multiple piloleiomyoma-related pain. J Am Acad Dermatol. 2002;46:S27-S29.
- Basendwh MA, Fatani M, Baltow B. Reed’s syndrome: a case of multiple cutaneous leiomyomas treated with liquid nitrogen cryotherapy. Case Rep Dermatol. 2016;8:65-70.
- Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A–treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2008;218:44-47.
- Onder M, Adıs¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Kaliyadan F, Manoj J, Dharmaratnam AD. Multiple cutaneous leiomyomas: pain relief with pulsed hysocine butyl bromide. Indian J Dermatol. 2009;54:72.
- Archer CB, Whittaker S, Greaves MW. Pharmacological modulation of cold‐induced pain in cutaneous leiomyomata. Br J Dermatol. 1988;118:255-260.
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322.
- Michajłowski I, Błaz˙ewicz I, Karpinsky G, et al. Successful treatment of multiple cutaneous leiomyomas with carbon dioxide laser ablation. Postepy Dermatol Alergol. 2015;32:480-482.
- Archer CB, Greaves MW. Assessment of treatment for painful cutaneous leiomyomas. J Am Acad Dermatol. 1987;17:141-142.
- Thompson JA. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865-867.
- Raemsch KD, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5(4 pt 2):II18-II24.
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95-106.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49-59.
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477-494.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Alam M, Rabinowitz AD, Engler DE. Gabapentin treatment of multiple piloleiomyoma-related pain. J Am Acad Dermatol. 2002;46:S27-S29.
- Basendwh MA, Fatani M, Baltow B. Reed’s syndrome: a case of multiple cutaneous leiomyomas treated with liquid nitrogen cryotherapy. Case Rep Dermatol. 2016;8:65-70.
- Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A–treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2008;218:44-47.
- Onder M, Adıs¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Kaliyadan F, Manoj J, Dharmaratnam AD. Multiple cutaneous leiomyomas: pain relief with pulsed hysocine butyl bromide. Indian J Dermatol. 2009;54:72.
- Archer CB, Whittaker S, Greaves MW. Pharmacological modulation of cold‐induced pain in cutaneous leiomyomata. Br J Dermatol. 1988;118:255-260.
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322.
- Michajłowski I, Błaz˙ewicz I, Karpinsky G, et al. Successful treatment of multiple cutaneous leiomyomas with carbon dioxide laser ablation. Postepy Dermatol Alergol. 2015;32:480-482.
- Archer CB, Greaves MW. Assessment of treatment for painful cutaneous leiomyomas. J Am Acad Dermatol. 1987;17:141-142.
- Thompson JA. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865-867.
- Raemsch KD, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5(4 pt 2):II18-II24.
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95-106.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49-59.
Practice Points
- Cutaneous leiomyomas (piloleiomyomas) are benign smooth muscle tumors derived from the arrector pili muscle.
- Patients presenting with multiple cutaneous leiomyomas should be evaluated for hereditary leiomyomatosis and renal cell carcinoma syndrome, an autosomal-dominant disorder, which also predisposes to the development of symptomatic uterine fibroids and uterine leiomyosarcoma.
- Cutaneous leiomyomas may be a source of considerable pain, which may respond to treatment with nifedipine in combination with gabapentin.

Mycobacterium haemophilum: A Challenging Treatment Dilemma in an Immunocompromised Patient
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
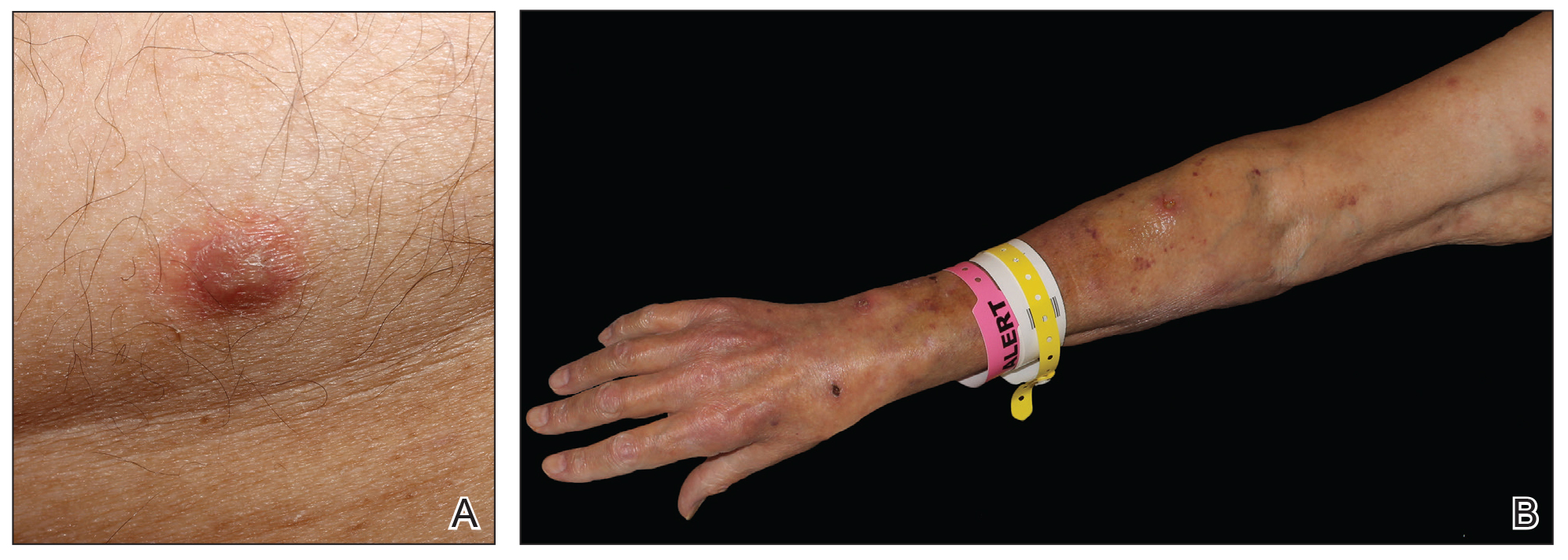

The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
Practice Points
- Mycobacterium haemophilum is a slow-growing acid-fast bacillus that requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. Because these requirements for growth are not standard for acid-fast bacteria cultures, M haemophilum infection may be underrecognized and underreported.
- There are no species-specific treatment guidelines, but extended course of treatment with multiple active antibacterials typically is recommended.
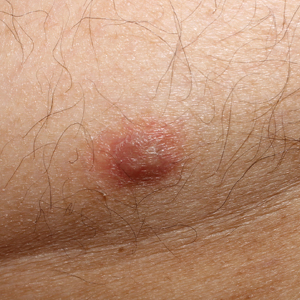
Severe Pretibial Myxedema Refractory to Systemic Immunosuppressants
To the Editor:
A 55-year-old man with a history of Graves disease treated with radioactive iodine and Graves ophthalmopathy was referred to our dermatology clinic by his endocrinologist with a 2-year history of severe pretibial myxedema (PM) that had failed treatment with systemic immunosuppressants after diagnosis by an outside dermatologist in the United Kingdom approximately 2 years prior. In addition to burning pain and difficulty walking associated with progressive “enlarging” of the lower legs and feet (Figure, A and B), the patient reported that he consistently had to find larger shoes (size 13 at the current presentation). His medications included gabapentin for foot pain and levothyroxine for hypothyroidism.
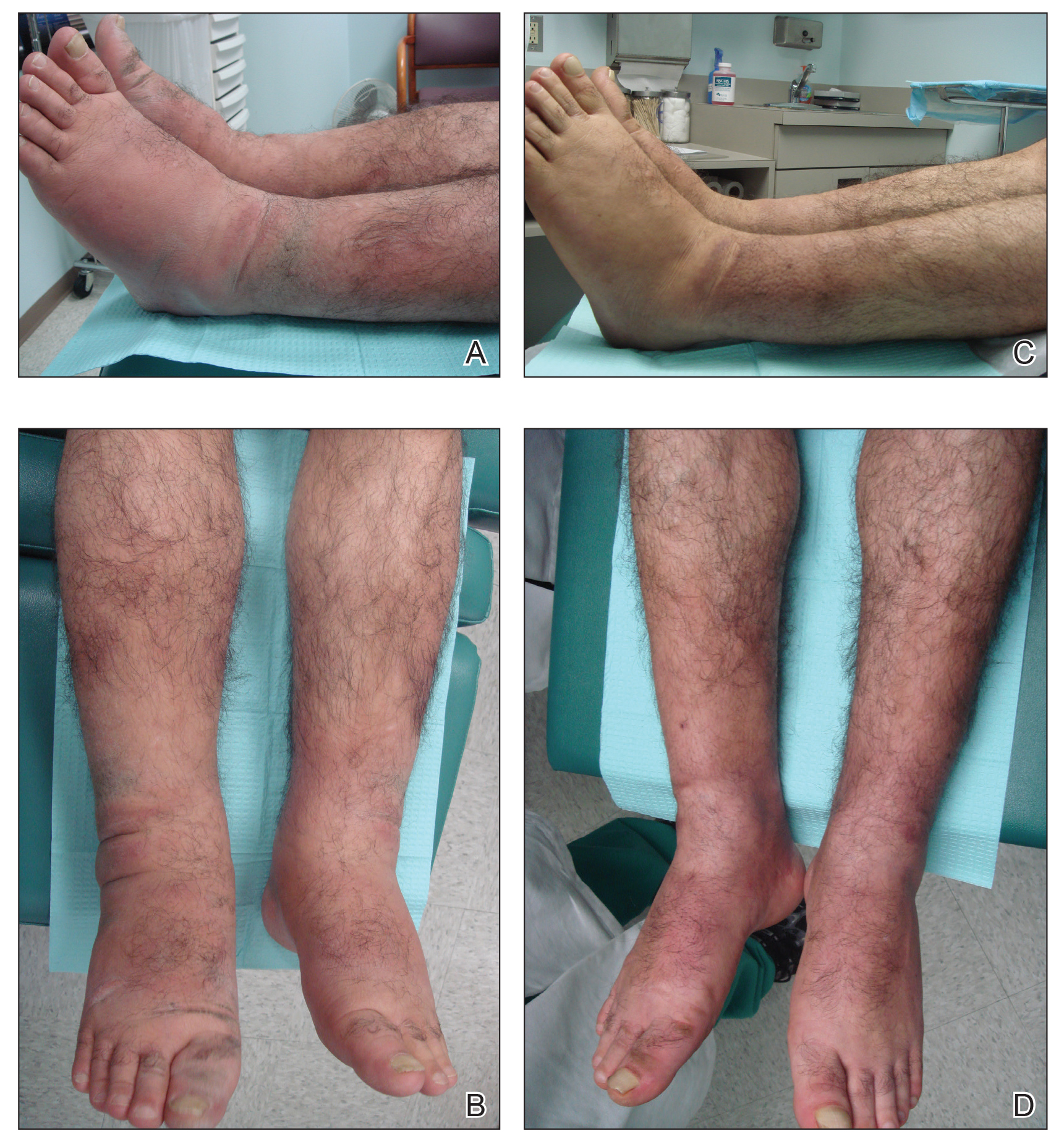
Physical examination revealed diffuse, waxy, indurated, flesh-colored and erythematous plaques and nodules with a peau d’orange appearance on the dorsal feet, ankles, and lower legs. Laboratory evaluation revealed a thyroid stimulating immunoglobulin level of 617% (reference range, <140%) and mild anemia. His thyroid stimulating hormone and free T4 levels, a comprehensive metabolic panel, and lipid panel were all within reference range.
Treatment with oral, intravenous, and intralesional steroids; cyclosporine; and azathioprine were tried prior to presentation to our clinic with no improvement. The patient was started on pentoxifylline (400 mg 3 times daily), intralesional triamcinolone acetonide (5 mg/mL every 3–4 weeks), clobetasol propionate ointment 0.05% under occlusion twice daily, short-stretch bandages, and compression stockings (20–30 mm Hg). The baseline circumference of the extremities also were measured (right ankle, 12 in; left ankle, 11.5 in; right and left mid-plantar feet, 12 in).
At 3-week follow-up, the lesions had flattened with softening of the skin. The patient reported his legs were smaller and he had bought a new pair of shoes at size 8.5 (Figure, C). He noted less pain and difficulty with walking. The circumference of the extremities was measured again (right ankle, 10.2 in; left ankle, 10 in; right and left mid-plantar feet, 10.5 in). The patient continued treatment and was followed for 3 months. At each visit, clinical improvement was noted as well as report of decreased pain while walking (Figure, D).
Pretibial myxedema is a known manifestation of Graves disease that almost always occurs in the presence of Graves ophthalmopathy. Pretibial myxedema occurs in 0.5% to 4.3% of patients with Graves disease and variably manifests as diffuse nonpitting edema or localized, waxy, indurated plaques or nodules.1,2
The proposed pathogenesis of PM is that autoantibodies directed against the thyroid receptors cross-react with the fibroblasts of the skin,2,3 which stimulates the fibroblasts to produce high amounts of glycosaminoglycans, especially hyaluronic acid, in the dermis and subcutis of the pretibial area. It is not known why there is a predilection for the anterior shins, but mechanical factors and dependent position (ie, leg position is lower than the level of the heart) may be involved.4
The mainstay of treatment for PM is topical and intralesional corticosteroids, which may have a benefit in mild to moderate disease; however, in cases of severe disease that is refractory to intralesional and topical corticosteroids under occlusion, more aggressive treatment is required. Systemic immunosuppressants such as cyclosporine, azathioprine, and corticosteroids have proven useful in some but not all cases.5,6
Our patient did not respond to treatment with systemic and intralesional corticosteroids, cyclosporine, or azathioprine before he presented to our clinic; however, the lesions were dramatically improved after 3 weeks of treatment with pentoxifylline, intralesional and topical corticosteroids under occlusion, short-stretch bandages, and compression stockings.
Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves ophthalmology and PM.7 It has been shown to reduce thickness of skin lesions when used in combination with topical or intralesional steroids.3,8 Corticosteroids are thought to block fibroblast-mediated glycosaminoglycan production.3,9 The deposition of mucin, which is comprised of glycosaminoglycans, expands the dermal tissue and causes fluid to accumulate; it also causes compression of dermal lymphatics, worsening the dermal edema. Because fluid accumulates, the use of short-stretch bandages and compression stockings may provide additional benefit, as was seen in our patient, whose shoe size decreased from a 13 to an 8.5 within 3 weeks of treatment.
In conclusion, the combination of pentoxifylline, intralesional and topical corticosteroids under occlusion, short-stretch bandages, and compression garments can cause substantial improvement in severe PM refractory to systemic immunosuppressants.
- Susser WS, Heermans AG, Chapman MS, et al. Elephantiasic pretibial myxedema: a novel treatment for an uncommon disorder. J Am Acad Dermatol. 2002;46:723-726.
- Kriss J. Pathogenesis and treatment of pretibial myxedema. Endocrinol Metab Clin North Am. 1987;16:409-415.
- Pineda AM, Tianco EA, Tan JB, et al. Oral pentoxifylline and topical clobetasol propionate ointment in the treatment of pretibial myxoedema, with concomitant improvement of Graves’ ophthalmopathy. J Eur Acad Dermatol Venereol. 2007; 21:1441-1443.
- Fatourechi V. Pretibial myxedema. Am J Clin Dermatol. 2005;6:295-309.
- Benoit FL, Greenspan FS. Corticoid therapy for pretibial myxedema: observations on the long-acting thyroid stimulator. Ann Intern Med. 1967;66:711-720.
- Hanke CW, Bergfeld WF, Guirguis MN, et al. Pretibial myxedema (elephantiasis form): treatment with cytotoxic therapy. Cleve Clin Q. 1983;50:183-188.
- Chang CC, Chang TC, Kao SC, et al. Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves’ ophthalmopathy and pretibial myxoedema. Acta Endocrinol (Copenh). 1993;129:322-327.
- Engin B, Gümüs¸el M, Ozdemir M, et al. Successful combined pentoxifylline and intralesional triamcinolone acetonide treatment of severe pretibial myxedema. Dermatol Online J. 2007;13:16.
- Lang PG, Sisson JC, Lynch PJ. Intralesional triamcinolone therapy for pretibial myxedema. Arch Dermatol. 1975;111:197-202.
To the Editor:
A 55-year-old man with a history of Graves disease treated with radioactive iodine and Graves ophthalmopathy was referred to our dermatology clinic by his endocrinologist with a 2-year history of severe pretibial myxedema (PM) that had failed treatment with systemic immunosuppressants after diagnosis by an outside dermatologist in the United Kingdom approximately 2 years prior. In addition to burning pain and difficulty walking associated with progressive “enlarging” of the lower legs and feet (Figure, A and B), the patient reported that he consistently had to find larger shoes (size 13 at the current presentation). His medications included gabapentin for foot pain and levothyroxine for hypothyroidism.
Physical examination revealed diffuse, waxy, indurated, flesh-colored and erythematous plaques and nodules with a peau d’orange appearance on the dorsal feet, ankles, and lower legs. Laboratory evaluation revealed a thyroid stimulating immunoglobulin level of 617% (reference range, <140%) and mild anemia. His thyroid stimulating hormone and free T4 levels, a comprehensive metabolic panel, and lipid panel were all within reference range.
Treatment with oral, intravenous, and intralesional steroids; cyclosporine; and azathioprine were tried prior to presentation to our clinic with no improvement. The patient was started on pentoxifylline (400 mg 3 times daily), intralesional triamcinolone acetonide (5 mg/mL every 3–4 weeks), clobetasol propionate ointment 0.05% under occlusion twice daily, short-stretch bandages, and compression stockings (20–30 mm Hg). The baseline circumference of the extremities also were measured (right ankle, 12 in; left ankle, 11.5 in; right and left mid-plantar feet, 12 in).
At 3-week follow-up, the lesions had flattened with softening of the skin. The patient reported his legs were smaller and he had bought a new pair of shoes at size 8.5 (Figure, C). He noted less pain and difficulty with walking. The circumference of the extremities was measured again (right ankle, 10.2 in; left ankle, 10 in; right and left mid-plantar feet, 10.5 in). The patient continued treatment and was followed for 3 months. At each visit, clinical improvement was noted as well as report of decreased pain while walking (Figure, D).
Pretibial myxedema is a known manifestation of Graves disease that almost always occurs in the presence of Graves ophthalmopathy. Pretibial myxedema occurs in 0.5% to 4.3% of patients with Graves disease and variably manifests as diffuse nonpitting edema or localized, waxy, indurated plaques or nodules.1,2
The proposed pathogenesis of PM is that autoantibodies directed against the thyroid receptors cross-react with the fibroblasts of the skin,2,3 which stimulates the fibroblasts to produce high amounts of glycosaminoglycans, especially hyaluronic acid, in the dermis and subcutis of the pretibial area. It is not known why there is a predilection for the anterior shins, but mechanical factors and dependent position (ie, leg position is lower than the level of the heart) may be involved.4
The mainstay of treatment for PM is topical and intralesional corticosteroids, which may have a benefit in mild to moderate disease; however, in cases of severe disease that is refractory to intralesional and topical corticosteroids under occlusion, more aggressive treatment is required. Systemic immunosuppressants such as cyclosporine, azathioprine, and corticosteroids have proven useful in some but not all cases.5,6
Our patient did not respond to treatment with systemic and intralesional corticosteroids, cyclosporine, or azathioprine before he presented to our clinic; however, the lesions were dramatically improved after 3 weeks of treatment with pentoxifylline, intralesional and topical corticosteroids under occlusion, short-stretch bandages, and compression stockings.
Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves ophthalmology and PM.7 It has been shown to reduce thickness of skin lesions when used in combination with topical or intralesional steroids.3,8 Corticosteroids are thought to block fibroblast-mediated glycosaminoglycan production.3,9 The deposition of mucin, which is comprised of glycosaminoglycans, expands the dermal tissue and causes fluid to accumulate; it also causes compression of dermal lymphatics, worsening the dermal edema. Because fluid accumulates, the use of short-stretch bandages and compression stockings may provide additional benefit, as was seen in our patient, whose shoe size decreased from a 13 to an 8.5 within 3 weeks of treatment.
In conclusion, the combination of pentoxifylline, intralesional and topical corticosteroids under occlusion, short-stretch bandages, and compression garments can cause substantial improvement in severe PM refractory to systemic immunosuppressants.
To the Editor:
A 55-year-old man with a history of Graves disease treated with radioactive iodine and Graves ophthalmopathy was referred to our dermatology clinic by his endocrinologist with a 2-year history of severe pretibial myxedema (PM) that had failed treatment with systemic immunosuppressants after diagnosis by an outside dermatologist in the United Kingdom approximately 2 years prior. In addition to burning pain and difficulty walking associated with progressive “enlarging” of the lower legs and feet (Figure, A and B), the patient reported that he consistently had to find larger shoes (size 13 at the current presentation). His medications included gabapentin for foot pain and levothyroxine for hypothyroidism.
Physical examination revealed diffuse, waxy, indurated, flesh-colored and erythematous plaques and nodules with a peau d’orange appearance on the dorsal feet, ankles, and lower legs. Laboratory evaluation revealed a thyroid stimulating immunoglobulin level of 617% (reference range, <140%) and mild anemia. His thyroid stimulating hormone and free T4 levels, a comprehensive metabolic panel, and lipid panel were all within reference range.
Treatment with oral, intravenous, and intralesional steroids; cyclosporine; and azathioprine were tried prior to presentation to our clinic with no improvement. The patient was started on pentoxifylline (400 mg 3 times daily), intralesional triamcinolone acetonide (5 mg/mL every 3–4 weeks), clobetasol propionate ointment 0.05% under occlusion twice daily, short-stretch bandages, and compression stockings (20–30 mm Hg). The baseline circumference of the extremities also were measured (right ankle, 12 in; left ankle, 11.5 in; right and left mid-plantar feet, 12 in).
At 3-week follow-up, the lesions had flattened with softening of the skin. The patient reported his legs were smaller and he had bought a new pair of shoes at size 8.5 (Figure, C). He noted less pain and difficulty with walking. The circumference of the extremities was measured again (right ankle, 10.2 in; left ankle, 10 in; right and left mid-plantar feet, 10.5 in). The patient continued treatment and was followed for 3 months. At each visit, clinical improvement was noted as well as report of decreased pain while walking (Figure, D).
Pretibial myxedema is a known manifestation of Graves disease that almost always occurs in the presence of Graves ophthalmopathy. Pretibial myxedema occurs in 0.5% to 4.3% of patients with Graves disease and variably manifests as diffuse nonpitting edema or localized, waxy, indurated plaques or nodules.1,2
The proposed pathogenesis of PM is that autoantibodies directed against the thyroid receptors cross-react with the fibroblasts of the skin,2,3 which stimulates the fibroblasts to produce high amounts of glycosaminoglycans, especially hyaluronic acid, in the dermis and subcutis of the pretibial area. It is not known why there is a predilection for the anterior shins, but mechanical factors and dependent position (ie, leg position is lower than the level of the heart) may be involved.4
The mainstay of treatment for PM is topical and intralesional corticosteroids, which may have a benefit in mild to moderate disease; however, in cases of severe disease that is refractory to intralesional and topical corticosteroids under occlusion, more aggressive treatment is required. Systemic immunosuppressants such as cyclosporine, azathioprine, and corticosteroids have proven useful in some but not all cases.5,6
Our patient did not respond to treatment with systemic and intralesional corticosteroids, cyclosporine, or azathioprine before he presented to our clinic; however, the lesions were dramatically improved after 3 weeks of treatment with pentoxifylline, intralesional and topical corticosteroids under occlusion, short-stretch bandages, and compression stockings.
Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves ophthalmology and PM.7 It has been shown to reduce thickness of skin lesions when used in combination with topical or intralesional steroids.3,8 Corticosteroids are thought to block fibroblast-mediated glycosaminoglycan production.3,9 The deposition of mucin, which is comprised of glycosaminoglycans, expands the dermal tissue and causes fluid to accumulate; it also causes compression of dermal lymphatics, worsening the dermal edema. Because fluid accumulates, the use of short-stretch bandages and compression stockings may provide additional benefit, as was seen in our patient, whose shoe size decreased from a 13 to an 8.5 within 3 weeks of treatment.
In conclusion, the combination of pentoxifylline, intralesional and topical corticosteroids under occlusion, short-stretch bandages, and compression garments can cause substantial improvement in severe PM refractory to systemic immunosuppressants.
- Susser WS, Heermans AG, Chapman MS, et al. Elephantiasic pretibial myxedema: a novel treatment for an uncommon disorder. J Am Acad Dermatol. 2002;46:723-726.
- Kriss J. Pathogenesis and treatment of pretibial myxedema. Endocrinol Metab Clin North Am. 1987;16:409-415.
- Pineda AM, Tianco EA, Tan JB, et al. Oral pentoxifylline and topical clobetasol propionate ointment in the treatment of pretibial myxoedema, with concomitant improvement of Graves’ ophthalmopathy. J Eur Acad Dermatol Venereol. 2007; 21:1441-1443.
- Fatourechi V. Pretibial myxedema. Am J Clin Dermatol. 2005;6:295-309.
- Benoit FL, Greenspan FS. Corticoid therapy for pretibial myxedema: observations on the long-acting thyroid stimulator. Ann Intern Med. 1967;66:711-720.
- Hanke CW, Bergfeld WF, Guirguis MN, et al. Pretibial myxedema (elephantiasis form): treatment with cytotoxic therapy. Cleve Clin Q. 1983;50:183-188.
- Chang CC, Chang TC, Kao SC, et al. Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves’ ophthalmopathy and pretibial myxoedema. Acta Endocrinol (Copenh). 1993;129:322-327.
- Engin B, Gümüs¸el M, Ozdemir M, et al. Successful combined pentoxifylline and intralesional triamcinolone acetonide treatment of severe pretibial myxedema. Dermatol Online J. 2007;13:16.
- Lang PG, Sisson JC, Lynch PJ. Intralesional triamcinolone therapy for pretibial myxedema. Arch Dermatol. 1975;111:197-202.
- Susser WS, Heermans AG, Chapman MS, et al. Elephantiasic pretibial myxedema: a novel treatment for an uncommon disorder. J Am Acad Dermatol. 2002;46:723-726.
- Kriss J. Pathogenesis and treatment of pretibial myxedema. Endocrinol Metab Clin North Am. 1987;16:409-415.
- Pineda AM, Tianco EA, Tan JB, et al. Oral pentoxifylline and topical clobetasol propionate ointment in the treatment of pretibial myxoedema, with concomitant improvement of Graves’ ophthalmopathy. J Eur Acad Dermatol Venereol. 2007; 21:1441-1443.
- Fatourechi V. Pretibial myxedema. Am J Clin Dermatol. 2005;6:295-309.
- Benoit FL, Greenspan FS. Corticoid therapy for pretibial myxedema: observations on the long-acting thyroid stimulator. Ann Intern Med. 1967;66:711-720.
- Hanke CW, Bergfeld WF, Guirguis MN, et al. Pretibial myxedema (elephantiasis form): treatment with cytotoxic therapy. Cleve Clin Q. 1983;50:183-188.
- Chang CC, Chang TC, Kao SC, et al. Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves’ ophthalmopathy and pretibial myxoedema. Acta Endocrinol (Copenh). 1993;129:322-327.
- Engin B, Gümüs¸el M, Ozdemir M, et al. Successful combined pentoxifylline and intralesional triamcinolone acetonide treatment of severe pretibial myxedema. Dermatol Online J. 2007;13:16.
- Lang PG, Sisson JC, Lynch PJ. Intralesional triamcinolone therapy for pretibial myxedema. Arch Dermatol. 1975;111:197-202.
Practice Points
- Pretibial myxedema (PM) is a known manifestation of Graves disease that almost always occurs in the presence of Graves ophthalmopathy.
- The proposed pathogenesis of PM is cross-reaction of autoantibodies directed against the thyroid receptors with the fibroblasts of the skin. It is not known why there is a predilection for the anterior shins, but mechanical factors and dependent position may be involved.
- The mainstay of treatment for PM is topical and intralesional corticosteroids, which may have a benefit in mild to moderate disease; however, in cases of severe disease that is refractory to intralesional and topical corticosteroids under occlusion, more aggressive treatment is required.
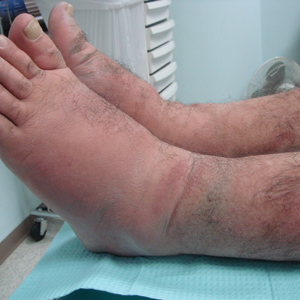
Sniffing Out Malignant Melanoma: A Case of Canine Olfactory Detection
To the Editor:
A 43-year-old woman presented with a mole on the central back that had been present since childhood and had changed and grown over the last few years. The patient reported that her 2-year-old rescue dog frequently sniffed the mole and would subsequently get agitated and try to scratch and bite the lesion. This behavior prompted the patient to visit a dermatologist.
She reported no personal history of melanoma or nonmelanoma skin cancer, tanning booth exposure, blistering sunburns, or use of immunosuppressant medications. Her family history was remarkable for basal cell carcinoma in her father but no family history of melanoma. Physical examination revealed a 1.2×1.5-cm brown patch along with a 1×1-cm ulcerated nodule on the lower aspect of the lesion (Figure 1). Dermoscopy showed a blue-white veil and an irregular vascular pattern (Figure 2). No cervical, axillary, or inguinal lymphadenopathy was appreciated on physical examination. Reflectance confocal microscopy showed pagetoid spread of atypical round melanocytes as well as melanocytes in the stratum corneum (Figure 3).
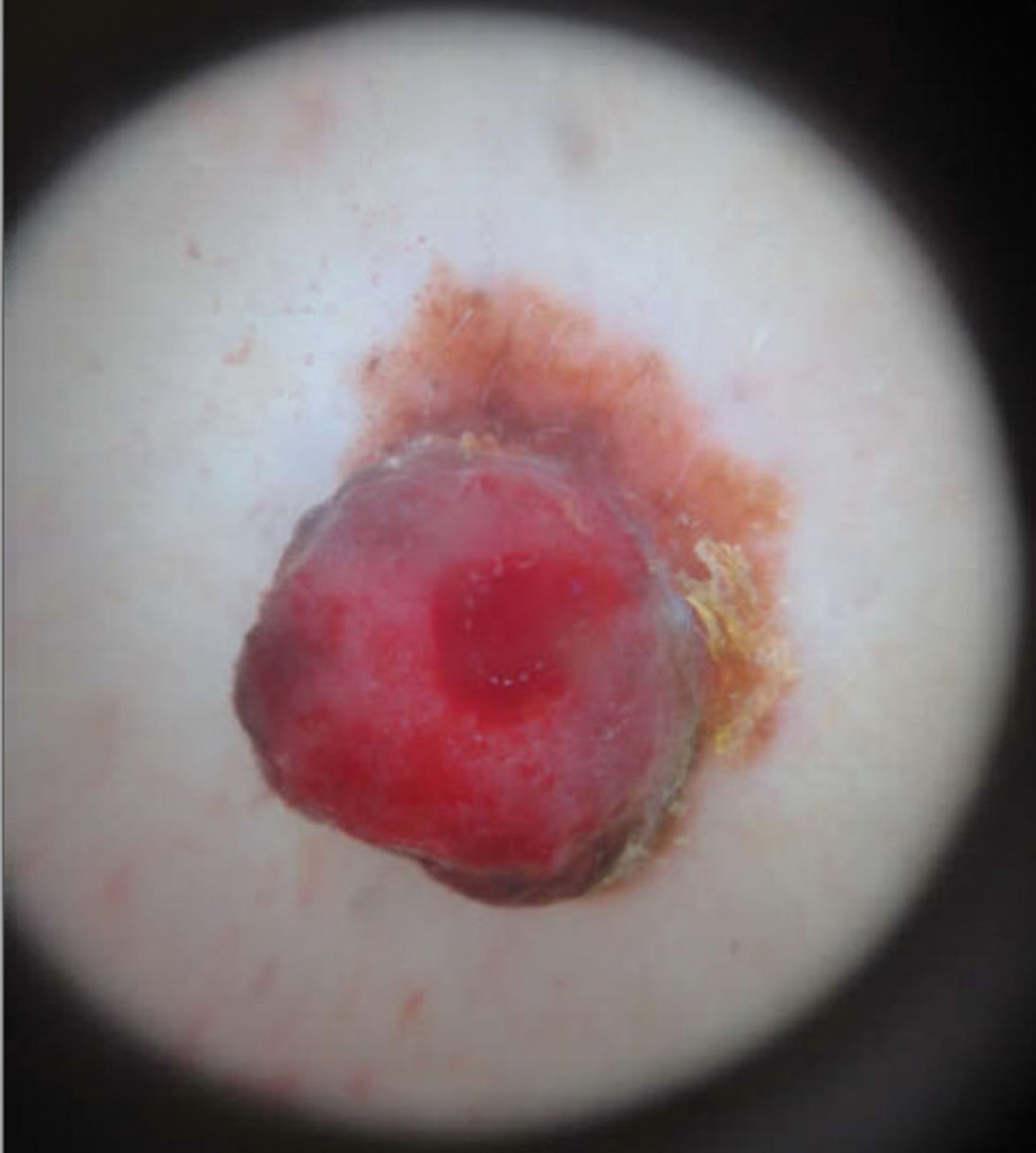

The patient was referred to a surgical oncologist for wide local excision and sentinel lymph node biopsy. Pathology showed a 4-mm-thick melanoma with numerous positive lymph nodes (Figure 4). The patient subsequently underwent a right axillary lymphadenectomy and was diagnosed with stage IIIB malignant melanoma. After surgery, the patient reported that her dog would now sniff her back and calmly rest his head in her lap.
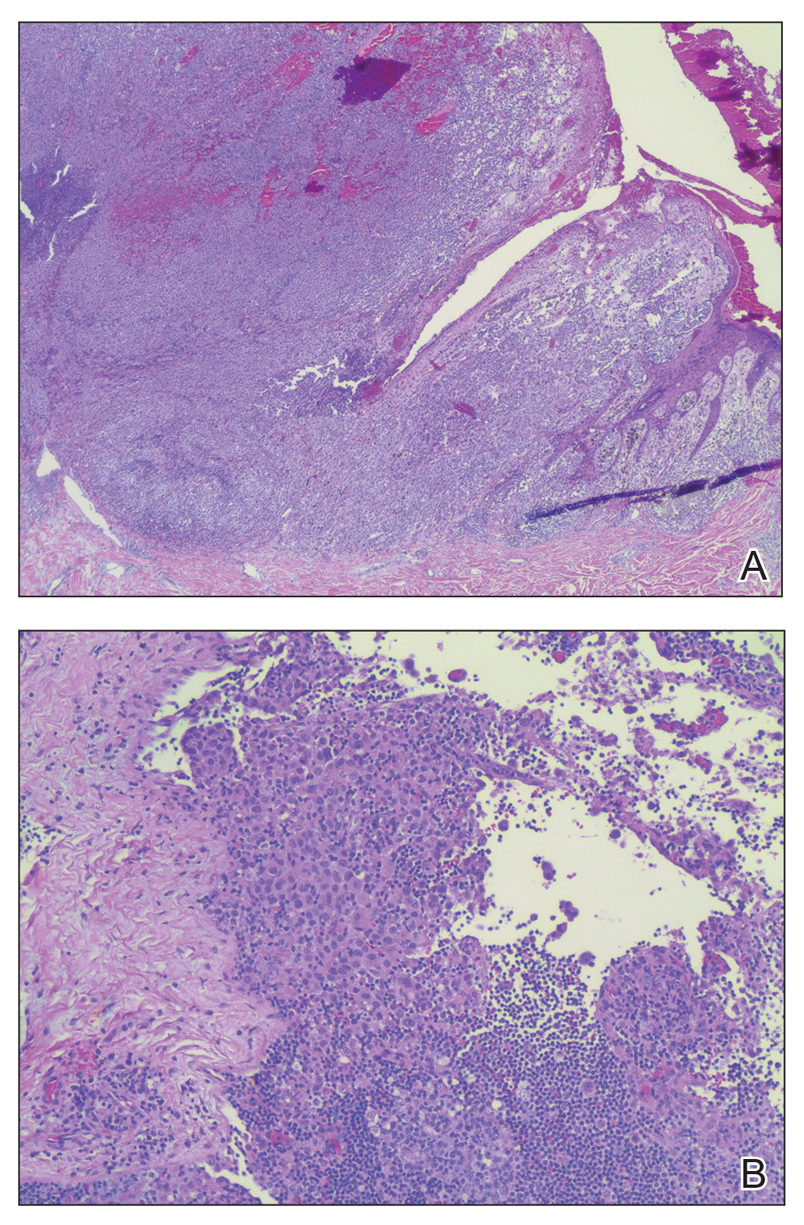
She was treated with ipilimumab but subsequently developed panhypopituitarism, so she was taken off the ipilimumab. Currently, the patient is doing well. She follows up annually for full-body skin examinations and has not had any recurrence in the last 7 years. The patient credits her dog for prompting her to see a dermatologist and saving her life.
Both anecdotal and systematic evidence have emerged on the role of canine olfaction in the detection of lung, breast, colorectal, ovarian, prostate, and skin cancers, including malignant melanoma.1-6 A 1989 case report described a woman who was prompted to seek dermatologic evaluation of a pigmented lesion because her dog consistently targeted the lesion. Excision and subsequent histopathologic examination of the lesion revealed that it was malignant melanoma.5 Another case report described a patient whose dog, which was not trained to detect cancers in humans, persistently licked a lesion behind the patient’s ear that eventually was found to be malignant melanoma.6 These reports have inspired considerable research interest regarding canine olfaction as a potential method to noninvasively screen for and even diagnose malignant melanomas in humans.
Both physiologic and pathologic metabolic processes result in the production of volatile organic compounds (VOCs), or small odorant molecules that evaporate at normal temperatures and pressures.1 Individual cells release VOCs in extremely low concentrations into the blood, urine, feces, and breath, as well as onto the skin’s surface, but there are methods for detecting these VOCs, including gas chromatography–mass spectrometry and canine olfaction.7,8 Pathologic processes, such as infection and malignancy, result in irregular protein synthesis and metabolism, producing new VOCs or differing concentrations of VOCs as compared to normal processes.1
Dimethyl disulfide and dimethyl trisulfide compounds have been identified in malignant melanoma, and these compounds are not produced by normal melanocytes.7 Furthermore, malignant melanoma produces differing quantities of these compounds as compared to normal melanocytes, including isovaleric acid, 2-methylbutyric acid, isoamyl alcohol (3-methyl-1-butanol), and 2-methyl-1-butanol, resulting in a distinct odorant profile that previously has been detected via canine olfaction.7 Canine olfaction can identify odorant molecules at up to 1 part per trillion (a magnitude more sensitive than the currently available gas chromatography–mass spectrometry technologies) and can detect the production of new VOCs or altered VOC ratios due to pathologic processes.1 Systematic studies with dogs that are trained to detect cancers in humans have shown that canine olfaction correctly identified malignant melanomas against healthy skin, benign nevi, and even basal cell carcinomas at higher rates than what would have been expected by chance alone.2,3
Canine olfaction can identify new or altered ratios of odorant VOCs associated with pathologic metabolic processes, and canines can be trained to target odor profiles associated with specific diseases.1 Canine olfaction for melanoma screening and diagnosis may seem appealing, as it provides an easily transportable, real-time, low-cost method compared to other techniques such as gas chromatography–mass spectrometry.1 Although preliminary results have shown that canine olfaction detects melanoma at higher rates than would be expected by chance alone, these findings have not approached clinical utility for the widespread use of canine olfaction as a screening method for melanoma.2,3,9 Further studies are needed to understand the role of canine olfaction in melanoma screening and diagnosis as well as to explore methods to optimize sensitivity and specificity. Until then, patients and dermatologists should not ignore the behavior of dogs toward skin lesions. Dogs may be beneficial in the detection of melanoma and help save lives, as was seen in our case.
- Angle C, Waggoner LP, Ferrando A, et al. Canine detection of the volatilome: a review of implications for pathogen and disease detection. Front Vet Sci. 2016;3:47.
- Pickel D, Mauncy GP, Walker DB, et al. Evidence for canine olfactory detection of melanoma. Applied Animal Behaviour Science. 2004;89:107-116.
- Willis CM, Britton LE, Swindells MA, et al. Invasive melanoma in vivo can be distinguished from basal cell carcinoma, benign naevi and healthy skin by canine olfaction: a proof‐of‐principle study of differential volatile organic compound emission. Br J Dermatol. 2016;175:1020-1029.
- Jezierski T, Walczak M, Ligor T, et al. Study of the art: canine olfaction used for cancer detection on the basis of breath odour. perspectives and limitations. J Breath Res. 2015;9:027001.
- Williams H, Pembroke A. Sniffer dogs in the melanoma clinic? Lancet. 1989;1:734.
- Campbell LF, Farmery L, George SM, et al. Canine olfactory detection of malignant melanoma. BMJ Case Rep. 2013. doi:10.1136/bcr-2013-008566.
- Kwak J, Gallagher M, Ozdener MH, et al. Volatile biomarkers from human melanoma cells. J Chromotogr B Analyt Technol Biomed Life Sci. 2013;931:90-96.
- D’Amico A, Bono R, Pennazza G, et al. Identification of melanoma with a gas sensor array. Skin Res Technol. 2008;14:226-236.
- Elliker KR, Williams HC. Detection of skin cancer odours using dogs: a step forward in melanoma detection training and research methodologies. Br J Dermatol. 2016;175:851-852.
To the Editor:
A 43-year-old woman presented with a mole on the central back that had been present since childhood and had changed and grown over the last few years. The patient reported that her 2-year-old rescue dog frequently sniffed the mole and would subsequently get agitated and try to scratch and bite the lesion. This behavior prompted the patient to visit a dermatologist.
She reported no personal history of melanoma or nonmelanoma skin cancer, tanning booth exposure, blistering sunburns, or use of immunosuppressant medications. Her family history was remarkable for basal cell carcinoma in her father but no family history of melanoma. Physical examination revealed a 1.2×1.5-cm brown patch along with a 1×1-cm ulcerated nodule on the lower aspect of the lesion (Figure 1). Dermoscopy showed a blue-white veil and an irregular vascular pattern (Figure 2). No cervical, axillary, or inguinal lymphadenopathy was appreciated on physical examination. Reflectance confocal microscopy showed pagetoid spread of atypical round melanocytes as well as melanocytes in the stratum corneum (Figure 3).
The patient was referred to a surgical oncologist for wide local excision and sentinel lymph node biopsy. Pathology showed a 4-mm-thick melanoma with numerous positive lymph nodes (Figure 4). The patient subsequently underwent a right axillary lymphadenectomy and was diagnosed with stage IIIB malignant melanoma. After surgery, the patient reported that her dog would now sniff her back and calmly rest his head in her lap.
She was treated with ipilimumab but subsequently developed panhypopituitarism, so she was taken off the ipilimumab. Currently, the patient is doing well. She follows up annually for full-body skin examinations and has not had any recurrence in the last 7 years. The patient credits her dog for prompting her to see a dermatologist and saving her life.
Both anecdotal and systematic evidence have emerged on the role of canine olfaction in the detection of lung, breast, colorectal, ovarian, prostate, and skin cancers, including malignant melanoma.1-6 A 1989 case report described a woman who was prompted to seek dermatologic evaluation of a pigmented lesion because her dog consistently targeted the lesion. Excision and subsequent histopathologic examination of the lesion revealed that it was malignant melanoma.5 Another case report described a patient whose dog, which was not trained to detect cancers in humans, persistently licked a lesion behind the patient’s ear that eventually was found to be malignant melanoma.6 These reports have inspired considerable research interest regarding canine olfaction as a potential method to noninvasively screen for and even diagnose malignant melanomas in humans.
Both physiologic and pathologic metabolic processes result in the production of volatile organic compounds (VOCs), or small odorant molecules that evaporate at normal temperatures and pressures.1 Individual cells release VOCs in extremely low concentrations into the blood, urine, feces, and breath, as well as onto the skin’s surface, but there are methods for detecting these VOCs, including gas chromatography–mass spectrometry and canine olfaction.7,8 Pathologic processes, such as infection and malignancy, result in irregular protein synthesis and metabolism, producing new VOCs or differing concentrations of VOCs as compared to normal processes.1
Dimethyl disulfide and dimethyl trisulfide compounds have been identified in malignant melanoma, and these compounds are not produced by normal melanocytes.7 Furthermore, malignant melanoma produces differing quantities of these compounds as compared to normal melanocytes, including isovaleric acid, 2-methylbutyric acid, isoamyl alcohol (3-methyl-1-butanol), and 2-methyl-1-butanol, resulting in a distinct odorant profile that previously has been detected via canine olfaction.7 Canine olfaction can identify odorant molecules at up to 1 part per trillion (a magnitude more sensitive than the currently available gas chromatography–mass spectrometry technologies) and can detect the production of new VOCs or altered VOC ratios due to pathologic processes.1 Systematic studies with dogs that are trained to detect cancers in humans have shown that canine olfaction correctly identified malignant melanomas against healthy skin, benign nevi, and even basal cell carcinomas at higher rates than what would have been expected by chance alone.2,3
Canine olfaction can identify new or altered ratios of odorant VOCs associated with pathologic metabolic processes, and canines can be trained to target odor profiles associated with specific diseases.1 Canine olfaction for melanoma screening and diagnosis may seem appealing, as it provides an easily transportable, real-time, low-cost method compared to other techniques such as gas chromatography–mass spectrometry.1 Although preliminary results have shown that canine olfaction detects melanoma at higher rates than would be expected by chance alone, these findings have not approached clinical utility for the widespread use of canine olfaction as a screening method for melanoma.2,3,9 Further studies are needed to understand the role of canine olfaction in melanoma screening and diagnosis as well as to explore methods to optimize sensitivity and specificity. Until then, patients and dermatologists should not ignore the behavior of dogs toward skin lesions. Dogs may be beneficial in the detection of melanoma and help save lives, as was seen in our case.
To the Editor:
A 43-year-old woman presented with a mole on the central back that had been present since childhood and had changed and grown over the last few years. The patient reported that her 2-year-old rescue dog frequently sniffed the mole and would subsequently get agitated and try to scratch and bite the lesion. This behavior prompted the patient to visit a dermatologist.
She reported no personal history of melanoma or nonmelanoma skin cancer, tanning booth exposure, blistering sunburns, or use of immunosuppressant medications. Her family history was remarkable for basal cell carcinoma in her father but no family history of melanoma. Physical examination revealed a 1.2×1.5-cm brown patch along with a 1×1-cm ulcerated nodule on the lower aspect of the lesion (Figure 1). Dermoscopy showed a blue-white veil and an irregular vascular pattern (Figure 2). No cervical, axillary, or inguinal lymphadenopathy was appreciated on physical examination. Reflectance confocal microscopy showed pagetoid spread of atypical round melanocytes as well as melanocytes in the stratum corneum (Figure 3).
The patient was referred to a surgical oncologist for wide local excision and sentinel lymph node biopsy. Pathology showed a 4-mm-thick melanoma with numerous positive lymph nodes (Figure 4). The patient subsequently underwent a right axillary lymphadenectomy and was diagnosed with stage IIIB malignant melanoma. After surgery, the patient reported that her dog would now sniff her back and calmly rest his head in her lap.
She was treated with ipilimumab but subsequently developed panhypopituitarism, so she was taken off the ipilimumab. Currently, the patient is doing well. She follows up annually for full-body skin examinations and has not had any recurrence in the last 7 years. The patient credits her dog for prompting her to see a dermatologist and saving her life.
Both anecdotal and systematic evidence have emerged on the role of canine olfaction in the detection of lung, breast, colorectal, ovarian, prostate, and skin cancers, including malignant melanoma.1-6 A 1989 case report described a woman who was prompted to seek dermatologic evaluation of a pigmented lesion because her dog consistently targeted the lesion. Excision and subsequent histopathologic examination of the lesion revealed that it was malignant melanoma.5 Another case report described a patient whose dog, which was not trained to detect cancers in humans, persistently licked a lesion behind the patient’s ear that eventually was found to be malignant melanoma.6 These reports have inspired considerable research interest regarding canine olfaction as a potential method to noninvasively screen for and even diagnose malignant melanomas in humans.
Both physiologic and pathologic metabolic processes result in the production of volatile organic compounds (VOCs), or small odorant molecules that evaporate at normal temperatures and pressures.1 Individual cells release VOCs in extremely low concentrations into the blood, urine, feces, and breath, as well as onto the skin’s surface, but there are methods for detecting these VOCs, including gas chromatography–mass spectrometry and canine olfaction.7,8 Pathologic processes, such as infection and malignancy, result in irregular protein synthesis and metabolism, producing new VOCs or differing concentrations of VOCs as compared to normal processes.1
Dimethyl disulfide and dimethyl trisulfide compounds have been identified in malignant melanoma, and these compounds are not produced by normal melanocytes.7 Furthermore, malignant melanoma produces differing quantities of these compounds as compared to normal melanocytes, including isovaleric acid, 2-methylbutyric acid, isoamyl alcohol (3-methyl-1-butanol), and 2-methyl-1-butanol, resulting in a distinct odorant profile that previously has been detected via canine olfaction.7 Canine olfaction can identify odorant molecules at up to 1 part per trillion (a magnitude more sensitive than the currently available gas chromatography–mass spectrometry technologies) and can detect the production of new VOCs or altered VOC ratios due to pathologic processes.1 Systematic studies with dogs that are trained to detect cancers in humans have shown that canine olfaction correctly identified malignant melanomas against healthy skin, benign nevi, and even basal cell carcinomas at higher rates than what would have been expected by chance alone.2,3
Canine olfaction can identify new or altered ratios of odorant VOCs associated with pathologic metabolic processes, and canines can be trained to target odor profiles associated with specific diseases.1 Canine olfaction for melanoma screening and diagnosis may seem appealing, as it provides an easily transportable, real-time, low-cost method compared to other techniques such as gas chromatography–mass spectrometry.1 Although preliminary results have shown that canine olfaction detects melanoma at higher rates than would be expected by chance alone, these findings have not approached clinical utility for the widespread use of canine olfaction as a screening method for melanoma.2,3,9 Further studies are needed to understand the role of canine olfaction in melanoma screening and diagnosis as well as to explore methods to optimize sensitivity and specificity. Until then, patients and dermatologists should not ignore the behavior of dogs toward skin lesions. Dogs may be beneficial in the detection of melanoma and help save lives, as was seen in our case.
- Angle C, Waggoner LP, Ferrando A, et al. Canine detection of the volatilome: a review of implications for pathogen and disease detection. Front Vet Sci. 2016;3:47.
- Pickel D, Mauncy GP, Walker DB, et al. Evidence for canine olfactory detection of melanoma. Applied Animal Behaviour Science. 2004;89:107-116.
- Willis CM, Britton LE, Swindells MA, et al. Invasive melanoma in vivo can be distinguished from basal cell carcinoma, benign naevi and healthy skin by canine olfaction: a proof‐of‐principle study of differential volatile organic compound emission. Br J Dermatol. 2016;175:1020-1029.
- Jezierski T, Walczak M, Ligor T, et al. Study of the art: canine olfaction used for cancer detection on the basis of breath odour. perspectives and limitations. J Breath Res. 2015;9:027001.
- Williams H, Pembroke A. Sniffer dogs in the melanoma clinic? Lancet. 1989;1:734.
- Campbell LF, Farmery L, George SM, et al. Canine olfactory detection of malignant melanoma. BMJ Case Rep. 2013. doi:10.1136/bcr-2013-008566.
- Kwak J, Gallagher M, Ozdener MH, et al. Volatile biomarkers from human melanoma cells. J Chromotogr B Analyt Technol Biomed Life Sci. 2013;931:90-96.
- D’Amico A, Bono R, Pennazza G, et al. Identification of melanoma with a gas sensor array. Skin Res Technol. 2008;14:226-236.
- Elliker KR, Williams HC. Detection of skin cancer odours using dogs: a step forward in melanoma detection training and research methodologies. Br J Dermatol. 2016;175:851-852.
- Angle C, Waggoner LP, Ferrando A, et al. Canine detection of the volatilome: a review of implications for pathogen and disease detection. Front Vet Sci. 2016;3:47.
- Pickel D, Mauncy GP, Walker DB, et al. Evidence for canine olfactory detection of melanoma. Applied Animal Behaviour Science. 2004;89:107-116.
- Willis CM, Britton LE, Swindells MA, et al. Invasive melanoma in vivo can be distinguished from basal cell carcinoma, benign naevi and healthy skin by canine olfaction: a proof‐of‐principle study of differential volatile organic compound emission. Br J Dermatol. 2016;175:1020-1029.
- Jezierski T, Walczak M, Ligor T, et al. Study of the art: canine olfaction used for cancer detection on the basis of breath odour. perspectives and limitations. J Breath Res. 2015;9:027001.
- Williams H, Pembroke A. Sniffer dogs in the melanoma clinic? Lancet. 1989;1:734.
- Campbell LF, Farmery L, George SM, et al. Canine olfactory detection of malignant melanoma. BMJ Case Rep. 2013. doi:10.1136/bcr-2013-008566.
- Kwak J, Gallagher M, Ozdener MH, et al. Volatile biomarkers from human melanoma cells. J Chromotogr B Analyt Technol Biomed Life Sci. 2013;931:90-96.
- D’Amico A, Bono R, Pennazza G, et al. Identification of melanoma with a gas sensor array. Skin Res Technol. 2008;14:226-236.
- Elliker KR, Williams HC. Detection of skin cancer odours using dogs: a step forward in melanoma detection training and research methodologies. Br J Dermatol. 2016;175:851-852.
Practice Points
- Physiologic and pathologic processes produce volatile organic compounds in the skin and other tissues.
- Malignant melanocytes release unique volatile organic compounds (VOCs) as well as differing combinations and quantities of VOCs as compared to normal melanocytes.
- Volatile organic compounds released at the skin’s surface can be detected by various methods, including canine olfaction; therefore, unusual canine behavior toward skin lesions should not be ignored.
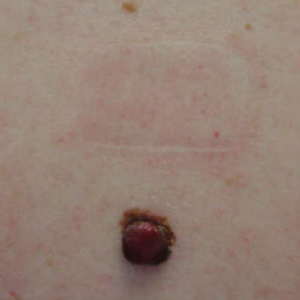
Cutaneous Sarcoidosis Presenting as a Cutaneous Horn
To the Editor:
A 53-year-old woman presented to our dermatology clinic with a painful growth on the right ear of 2 months’ duration. A complete review of systems was negative except for an isolated episode of shortness of breath prior to presentation that resolved without intervention. During this episode, her primary care physician made a diagnosis of chronic obstructive pulmonary disease based on a chest radiograph. The patient reported minimal tobacco use, specifically that she had smoked a few cigarettes daily for several years but had quit 6 months prior to the current presentation.
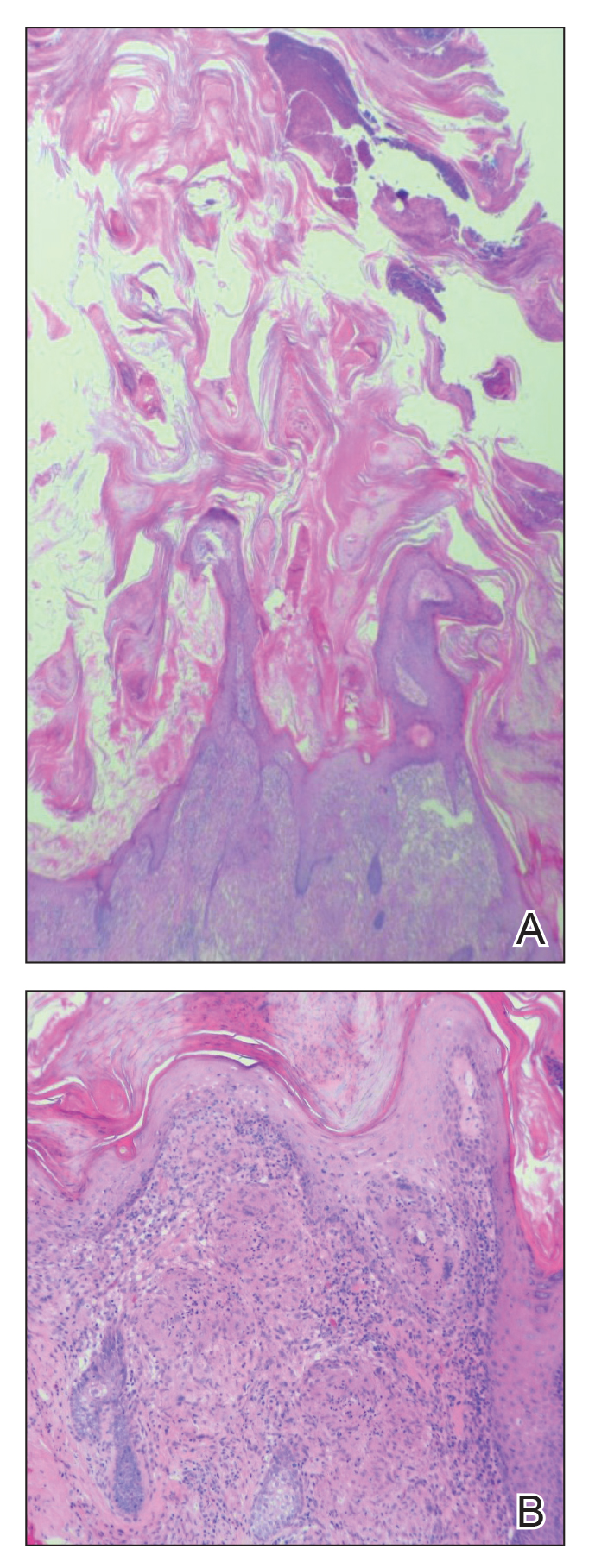
Cutaneous horn is a clinical term used to describe hyperkeratotic horn-shaped growths of highly variable shapes and sizes. Although the pathogenesis and incidence of cutaneous horns remain unknown, these lesions most often are the result of a neoplastic rather than an inflammatory process. The differential diagnosis typically includes entities characterized by marked hyperkeratosis, including hypertrophic actinic keratosis, squamous cell carcinoma (SCC), seborrheic keratosis, and verruca vulgaris. The base of the horn must be biopsied to determine the underlying etiology, paying careful attention to avoid a superficial biopsy, as it may be nondiagnostic.
Studies analyzing the underlying diagnoses and clinical features of cutaneous horns are limited. In a large retrospective study of 643 cutaneous horns, 61% were benign, 23% were premalignant, and 16% were malignant. In this study, 4 features were associated with premalignant or malignant pathology: (1) older age (mid- 60s to 70s); (2) male sex; (3) location on the nose, pinnae, dorsal hands, scalp, forearms, or face; and (4) a wide base (4.4 mm or larger) and a lower height-to-base ratio than benign lesions.1 Two additional studies of more than 200 horns each showed higher rates of premalignant horns (42% and 38%, respectively) with malignancy found in 7% and 20% of horns, respectively.2,3 One prospective study sought to identify clinical and dermatoscopic features of SCCs underlying cutaneous horns, concluding that SCC diagnosis was more likely if a horn had (1) a height less than the diameter of its base, (2) a lack of terrace morphology (a dermatoscopic feature defined as horizontal parallel layers of keratin), (3) erythema at the base, and (4) the presence of pain.4
Our patient had a cutaneous horn on the pinna that was painful, wider than it was tall, and erythematous at the base, suggesting a malignant process; however, a complete cutaneous physical examination revealed other skin lesions that were concerning for sarcoidosis and raised suspicion that the horn also was a manifestation of the same inflammatory process.
Although unusual, cutaneous sarcoidosis presenting as a cutaneous horn is not unexpected. In a histopathologic study of 62 cases of cutaneous sarcoidosis, 79% (49/62) showed epidermal changes and 13% (8/62) demonstrated hyperkeratosis. Other epidermal changes included parakeratosis (16% [10/62]), acanthosis (10% [6/62]), and epidermal atrophy (57% [35/62]).5 The spectrum of epidermal pathology in cutaneous sarcoidosis is evident in its well-documented verrucous, psoriasiform, and ichthyosiform presentations. For completeness, cutaneous horn is added to the list of clinical morphologies for this “great imitator” of cutaneous diseases.
- Yu RC, Pryce DW, Macfarlane AW, et al. A histopathological study of 643 cutaneous horns. Br J Dermatol. 1991;124:449-452.
- Schosser RH, Hodge SJ, Gaba CR, et al. Cutaneous horns: a histopathologic study. South Med J. 1979;72:1129-1131.
- Mantese SA, Diogo PM, Rocha A, et al. Cutaneous horn: a retrospective histopathological study of 222 cases. An Bras Dermatol. 2010;85:157-163.
- Pyne J, Sapkota D, Wong JC. Cutaneous horns: clues to invasive squamous cell carcinoma being present in the horn base. Dermatol Pract Concept. 2013;3:3-7.
- Hiroyuki O. Epidermal changes in cutaneous lesions of sarcoidosis. Am J Dermatopathol. 1999;21:229-233.
To the Editor:
A 53-year-old woman presented to our dermatology clinic with a painful growth on the right ear of 2 months’ duration. A complete review of systems was negative except for an isolated episode of shortness of breath prior to presentation that resolved without intervention. During this episode, her primary care physician made a diagnosis of chronic obstructive pulmonary disease based on a chest radiograph. The patient reported minimal tobacco use, specifically that she had smoked a few cigarettes daily for several years but had quit 6 months prior to the current presentation.
Cutaneous horn is a clinical term used to describe hyperkeratotic horn-shaped growths of highly variable shapes and sizes. Although the pathogenesis and incidence of cutaneous horns remain unknown, these lesions most often are the result of a neoplastic rather than an inflammatory process. The differential diagnosis typically includes entities characterized by marked hyperkeratosis, including hypertrophic actinic keratosis, squamous cell carcinoma (SCC), seborrheic keratosis, and verruca vulgaris. The base of the horn must be biopsied to determine the underlying etiology, paying careful attention to avoid a superficial biopsy, as it may be nondiagnostic.
Studies analyzing the underlying diagnoses and clinical features of cutaneous horns are limited. In a large retrospective study of 643 cutaneous horns, 61% were benign, 23% were premalignant, and 16% were malignant. In this study, 4 features were associated with premalignant or malignant pathology: (1) older age (mid- 60s to 70s); (2) male sex; (3) location on the nose, pinnae, dorsal hands, scalp, forearms, or face; and (4) a wide base (4.4 mm or larger) and a lower height-to-base ratio than benign lesions.1 Two additional studies of more than 200 horns each showed higher rates of premalignant horns (42% and 38%, respectively) with malignancy found in 7% and 20% of horns, respectively.2,3 One prospective study sought to identify clinical and dermatoscopic features of SCCs underlying cutaneous horns, concluding that SCC diagnosis was more likely if a horn had (1) a height less than the diameter of its base, (2) a lack of terrace morphology (a dermatoscopic feature defined as horizontal parallel layers of keratin), (3) erythema at the base, and (4) the presence of pain.4
Our patient had a cutaneous horn on the pinna that was painful, wider than it was tall, and erythematous at the base, suggesting a malignant process; however, a complete cutaneous physical examination revealed other skin lesions that were concerning for sarcoidosis and raised suspicion that the horn also was a manifestation of the same inflammatory process.
Although unusual, cutaneous sarcoidosis presenting as a cutaneous horn is not unexpected. In a histopathologic study of 62 cases of cutaneous sarcoidosis, 79% (49/62) showed epidermal changes and 13% (8/62) demonstrated hyperkeratosis. Other epidermal changes included parakeratosis (16% [10/62]), acanthosis (10% [6/62]), and epidermal atrophy (57% [35/62]).5 The spectrum of epidermal pathology in cutaneous sarcoidosis is evident in its well-documented verrucous, psoriasiform, and ichthyosiform presentations. For completeness, cutaneous horn is added to the list of clinical morphologies for this “great imitator” of cutaneous diseases.
To the Editor:
A 53-year-old woman presented to our dermatology clinic with a painful growth on the right ear of 2 months’ duration. A complete review of systems was negative except for an isolated episode of shortness of breath prior to presentation that resolved without intervention. During this episode, her primary care physician made a diagnosis of chronic obstructive pulmonary disease based on a chest radiograph. The patient reported minimal tobacco use, specifically that she had smoked a few cigarettes daily for several years but had quit 6 months prior to the current presentation.
Cutaneous horn is a clinical term used to describe hyperkeratotic horn-shaped growths of highly variable shapes and sizes. Although the pathogenesis and incidence of cutaneous horns remain unknown, these lesions most often are the result of a neoplastic rather than an inflammatory process. The differential diagnosis typically includes entities characterized by marked hyperkeratosis, including hypertrophic actinic keratosis, squamous cell carcinoma (SCC), seborrheic keratosis, and verruca vulgaris. The base of the horn must be biopsied to determine the underlying etiology, paying careful attention to avoid a superficial biopsy, as it may be nondiagnostic.
Studies analyzing the underlying diagnoses and clinical features of cutaneous horns are limited. In a large retrospective study of 643 cutaneous horns, 61% were benign, 23% were premalignant, and 16% were malignant. In this study, 4 features were associated with premalignant or malignant pathology: (1) older age (mid- 60s to 70s); (2) male sex; (3) location on the nose, pinnae, dorsal hands, scalp, forearms, or face; and (4) a wide base (4.4 mm or larger) and a lower height-to-base ratio than benign lesions.1 Two additional studies of more than 200 horns each showed higher rates of premalignant horns (42% and 38%, respectively) with malignancy found in 7% and 20% of horns, respectively.2,3 One prospective study sought to identify clinical and dermatoscopic features of SCCs underlying cutaneous horns, concluding that SCC diagnosis was more likely if a horn had (1) a height less than the diameter of its base, (2) a lack of terrace morphology (a dermatoscopic feature defined as horizontal parallel layers of keratin), (3) erythema at the base, and (4) the presence of pain.4
Our patient had a cutaneous horn on the pinna that was painful, wider than it was tall, and erythematous at the base, suggesting a malignant process; however, a complete cutaneous physical examination revealed other skin lesions that were concerning for sarcoidosis and raised suspicion that the horn also was a manifestation of the same inflammatory process.
Although unusual, cutaneous sarcoidosis presenting as a cutaneous horn is not unexpected. In a histopathologic study of 62 cases of cutaneous sarcoidosis, 79% (49/62) showed epidermal changes and 13% (8/62) demonstrated hyperkeratosis. Other epidermal changes included parakeratosis (16% [10/62]), acanthosis (10% [6/62]), and epidermal atrophy (57% [35/62]).5 The spectrum of epidermal pathology in cutaneous sarcoidosis is evident in its well-documented verrucous, psoriasiform, and ichthyosiform presentations. For completeness, cutaneous horn is added to the list of clinical morphologies for this “great imitator” of cutaneous diseases.
- Yu RC, Pryce DW, Macfarlane AW, et al. A histopathological study of 643 cutaneous horns. Br J Dermatol. 1991;124:449-452.
- Schosser RH, Hodge SJ, Gaba CR, et al. Cutaneous horns: a histopathologic study. South Med J. 1979;72:1129-1131.
- Mantese SA, Diogo PM, Rocha A, et al. Cutaneous horn: a retrospective histopathological study of 222 cases. An Bras Dermatol. 2010;85:157-163.
- Pyne J, Sapkota D, Wong JC. Cutaneous horns: clues to invasive squamous cell carcinoma being present in the horn base. Dermatol Pract Concept. 2013;3:3-7.
- Hiroyuki O. Epidermal changes in cutaneous lesions of sarcoidosis. Am J Dermatopathol. 1999;21:229-233.
- Yu RC, Pryce DW, Macfarlane AW, et al. A histopathological study of 643 cutaneous horns. Br J Dermatol. 1991;124:449-452.
- Schosser RH, Hodge SJ, Gaba CR, et al. Cutaneous horns: a histopathologic study. South Med J. 1979;72:1129-1131.
- Mantese SA, Diogo PM, Rocha A, et al. Cutaneous horn: a retrospective histopathological study of 222 cases. An Bras Dermatol. 2010;85:157-163.
- Pyne J, Sapkota D, Wong JC. Cutaneous horns: clues to invasive squamous cell carcinoma being present in the horn base. Dermatol Pract Concept. 2013;3:3-7.
- Hiroyuki O. Epidermal changes in cutaneous lesions of sarcoidosis. Am J Dermatopathol. 1999;21:229-233.
Practice Points
- Biopsy of a cutaneous horn should be deep enough to capture the neoplastic or inflammatory process at the base of the lesion.
- Cutaneous sarcoidosis can present with variable morphologies including the epidermal changes of a cutaneous horn.
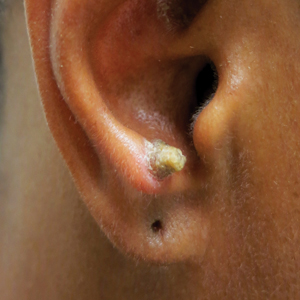
Unusually Early-Onset Plantar Verrucous Carcinoma
To the Editor:
Verrucous carcinoma (VC) is a rare type of squamous cell carcinoma characterized by a well-differentiated low-grade tumor with a high degree of keratinization. First described by Ackerman1 in 1948, VC presents on the skin or oral and genital mucosae with minimal atypical cytologic findings.1-3 It most commonly is seen in late middle-aged men (85% of cases) and presents as a slow-growing mass, often of more than 10 years’ duration.2,3 Verrucous carcinoma frequently is observed at 3 particular anatomic sites: the oral cavity, known as oral florid papillomatosis; the anogenital area, known as Buschke-Löwenstein tumor; and on the plantar surface, known as epithelioma cuniculatum.2-13
A 19-year-old man presented with an ulcerous lesion on the right big toe of 2 years’ duration. He reported that the lesion had gradually increased in size and was painful when walking. Physical examination revealed an ulcerated lesion on the right big toe with purulent inflammation and necrosis, unclear edges, and border nodules containing a fatty, yellowish, foul-smelling material (Figure 1). Histologic examination of purulent material from deep within the primary lesion revealed gram-negative rods and gram-positive diplococci. Erlich-Ziehl-Neelsen staining and culture in Lowenstein-Jensen medium were negative for mycobacteria. Histologic examination and fungal culture were not diagnostic for fungal infection.
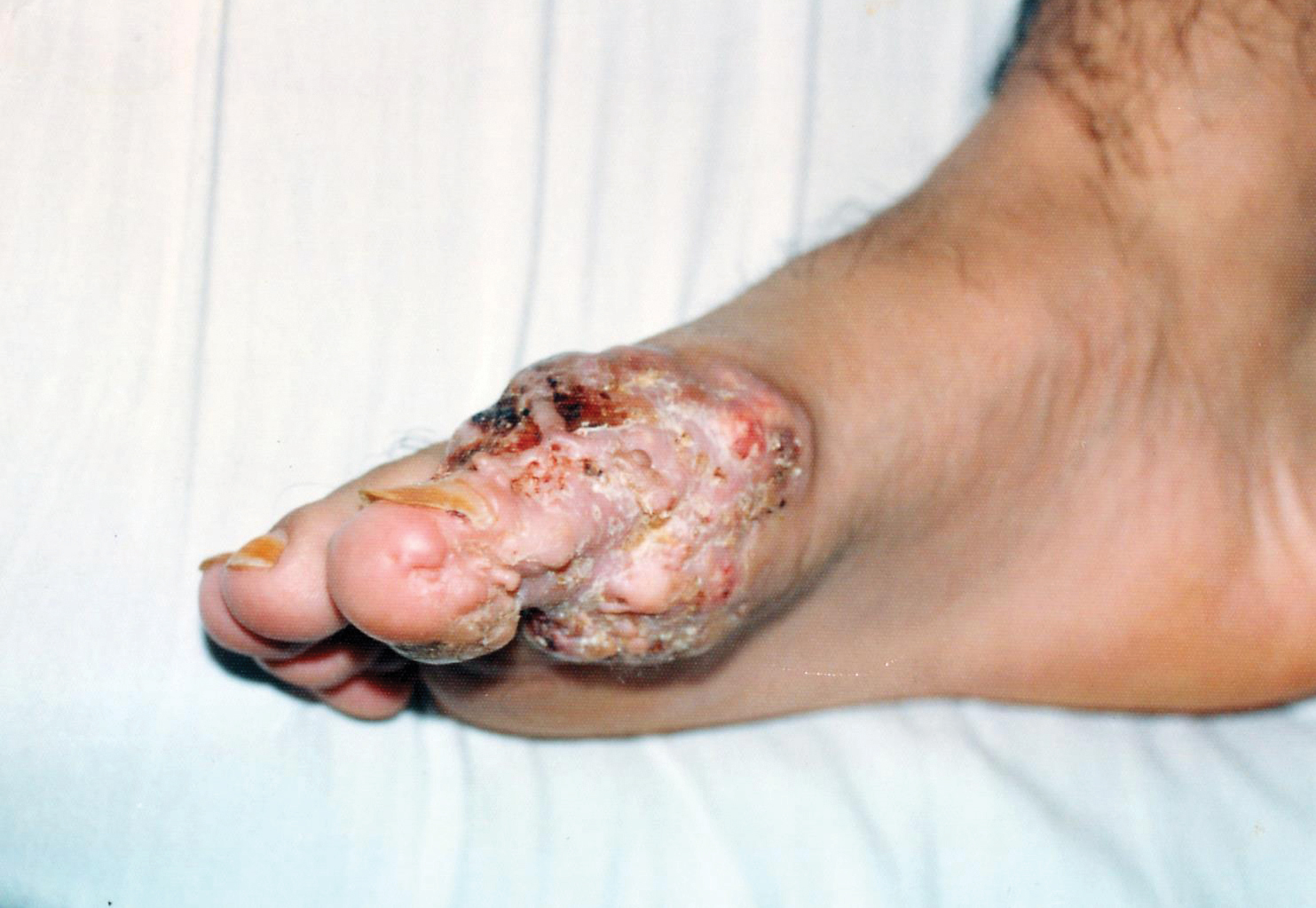
The differential diagnosis included tuberculosis cutis verrucosa, subcutaneous mycoses, swimming pool granuloma, leishmania cutis, chronic pyoderma vegetans, and VC. A punch biopsy of the lesion showed chronic nonspecific inflammation, hyperkeratosis, parakeratosis, and pseudoepitheliomatous hyperplasia. A repeat biopsy performed 15 days later also showed a nonspecific inflammation. At the initial presentation, an anti–human immunodeficiency virus test was negative. A purified protein derivative (PPD) skin test was positive and showed a 17-mm induration, and a sputum test was negative for Mycobacterium tuberculosis. A chest radiograph was normal. We considered the positive PPD skin test to be clinically insignificant; we did not find an accompanying tuberculosis infection, and the high exposure to atypical tuberculosis in developing countries such as Turkey, which is where the patient resided, often explains a positive PPD test.
At the initial presentation, radiography of the right big toe revealed porotic signs and cortical irregularity of the distal phalanx. A deep incisional biopsy of the lesion was performed for pathologic and microbiologic analysis. Erlich-Ziehl-Neelsen staining was negative, fungal elements could not be observed, and there was no growth in Lowenstein-Jensen medium or Sabouraud dextrose agar. Polymerase chain reaction for human papillomavirus, M tuberculosis, and atypical mycobacterium was negative. Periodic acid–Schiff staining was negative for fungal elements. Histopathologic examination revealed an exophytic as well as endophytic squamous cell proliferation infiltrating deeper layers of the dermis with a desmoplastic stroma (Figure 2). Slight cytologic atypia was noted. A diagnosis of VC was made based on the clinical and histopathologic findings. The patient’s right big toe was amputated by plastic surgery 6 months after the initial presentation.
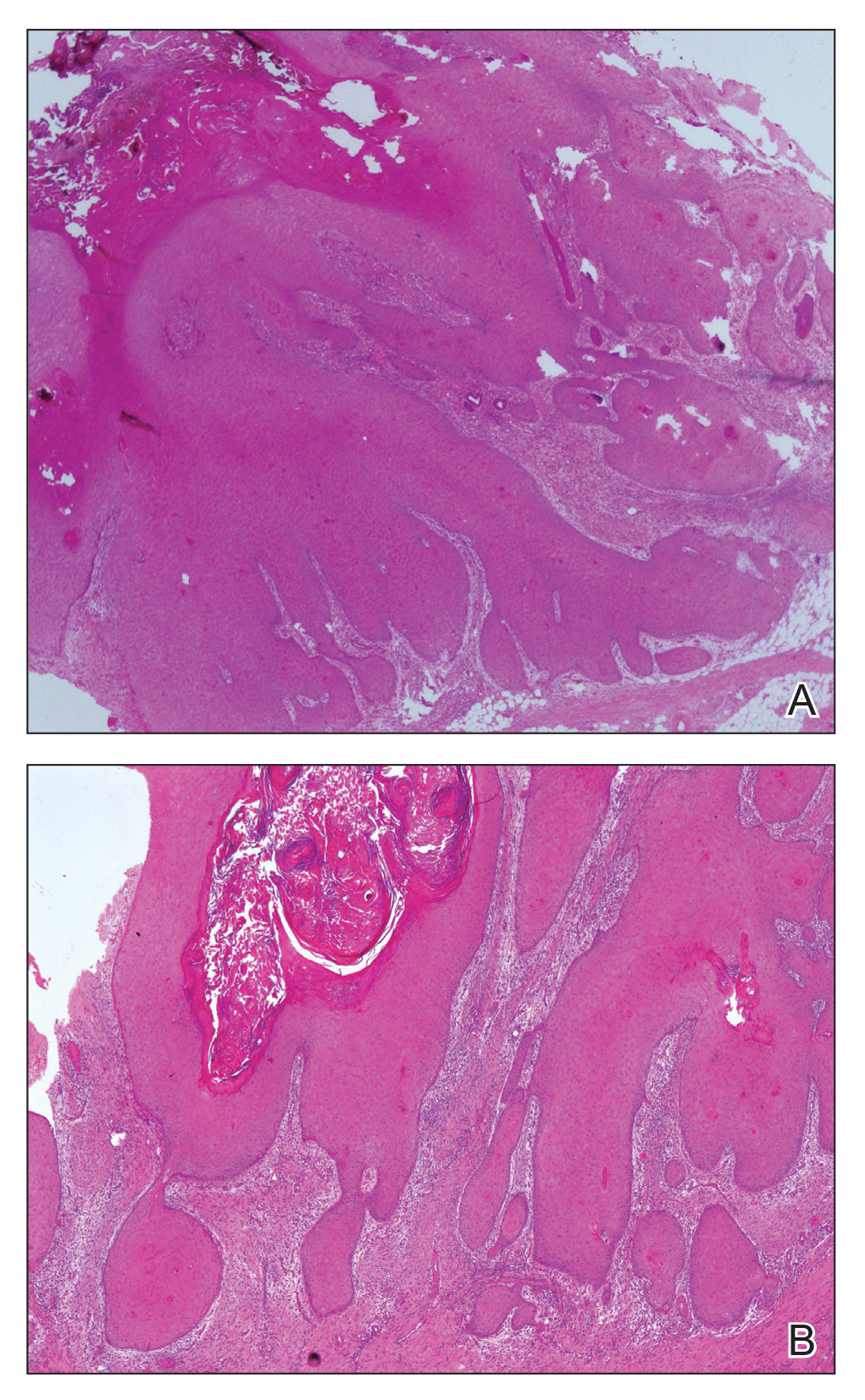
The term epithelioma cuniculatum was first used in 1954 to describe plantar VC. The term cuniculus is Latin for rabbit nest.3 At the distal part of the plantar surface of the foot, VC presents as an exophytic funguslike mass with abundant keratin-filled sinuses.14 When pressure is applied to the lesion, a greasy, yellowish, foul-smelling material with the consistency of toothpaste emerges from the sinuses. The lesion resembles pyoderma vegetans and may present with secondary infections (eg, Staphylococcus aureus, gram-negative bacteria, fungal infection) and/or ulcerations. Its appearance resembles an inflammatory lesion more than a neoplasm.6 Sometimes the skin surrounding the lesion may be a yellowish color, giving the impression of a plantar wart.3,4 In most cases, in situ hybridization demonstrates a human papillomavirus genome.2-5,10 Other factors implicated in the etiopathogenesis of VC include chronic inflammation; a cicatrice associated with a condition such as chronic cutaneous tuberculosis, ulcerative leprosy, dystrophic epidermolysis bullosa, or chronic osteomyelitis4; recurrent trauma3; and/or lichen planus.2,4 In spite of its slow development and benign appearance, VC may cause severe destruction affecting surrounding bony structures and may ultimately require amputation.2,4 In its early stages, VC can be mistaken for a benign tumor or other benign lesion, such as giant seborrheic keratosis, giant keratoacanthoma, eccrine poroma, or verruciform xanthoma, potentially leading to an incorrect diagnosis.5
Histopathologic examination, especially of superficial biopsies, generally reveals squamous cell proliferation demonstrating minimal pleomorphism and cytologic atypia with sparse mitotic figures.4-6 Diagnosis of VC can be challenging if the endophytic proliferation, which characteristically pushes into the dermis and even deeper tissues at the base of the lesion, is not seen. This feature is uncommon in squamous cell carcinomas.3,4,6 Histopathologic detection of koilocytes can lead to difficulty in distinguishing VC from warts.5 The growth of lesions is exophytic in plantar verrucae, whereas in VC it may be either exophytic or endophytic.4 At early stages, it is too difficult to distinguish VC from pseudoepitheliomatous hyperplasia caused by chronic inflammation, as well as from tuberculosis and subcutaneous mycoses.3,6 In these situations, possible responsible microorganisms must be sought out. Amelanotic malignant melanoma and eccrine poroma also should be considered in the differential diagnosis.3,5 If the biopsy specimen is obtained superficially and is fragmented, the diagnosis is more difficult, making deep biopsies essential in suspicious cases.4 Excision is the best treatment, and Mohs micrographic surgery may be required in some cases.2,3,11 It is important to consider that radiotherapy may lead to anaplastic transformation and metastasis.2 Metastasis to lymph nodes is very rare, and the prognosis is excellent when complete excision is performed.2 Recurrence may be observed.4
Our case of plantar VC is notable because of the patient’s young age, which is uncommon, as the typical age for developing VC is late middle age (ie, fifth and sixth decades of life). A long-standing lesion that is therapy resistant and without a detectable microorganism should be investigated for malignancy by repetitive deep biopsy regardless of the patient’s age, as demonstrated in our case.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Schwartz RA. Verrucous carcinoma of the skin and mucosal. J Am Acad Dermatol. 1995;32:1-21.
- Kao GF, Graham JH, Helwig EB. Carcinoma cuniculatum (verrucous carcinoma of the skin): a clinicopathologic study of 46 cases with ultrastructural observations. Cancer. 1982;49:2395-2403.
- Mc Kee PH, ed. Pathology of the Skin. 2nd ed. London, England: Mosby-Wolfe; 1996.
- Schwartz RA, Stoll HL. Squamous cell carcinoma. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York, NY: Mc-Graw Hill; 1999:840-856.
- MacKie RM. Epidermal skin tumours. In: Rook A, Wilkinson DS, Ebling FJG, et al, eds. Textbook of Dermatology. 5th ed. Oxford, United Kingdom: Blackwell Scientific; 1992:1500-1556.
- Yoshtatsu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Van Geertruyden JP, Olemans C, Laporte M, et al. Verrucous carcinoma of the nail bed. Foot Ankle Int. 1998;19:327-328.
- Sanchez-Yus E, Velasco E, Robledo A. Verrucous carcinoma of the back. J Am Acad Dermatol. 1986;14(5 pt 2):947-950.
- Noel JC, Peny MO, Detremmerie O, et al. Demonstration of human papillomavirus type 2 in a verrucous carcinoma of the foot. Dermatology. 1993;187:58-61.
- Mora RG. Microscopically controlled surgery (Mohs’ chemosurgery) for treatment of verrucous squamous cell carcinoma of the foot (epithelioma cuniculatum). J Am Acad Dermatol. 1983;8:354-362.
- Kathuria S, Rieker J, Jablokow VR, et al. Plantar verrucous carcinoma (epithelioma cuniculatum): case report with review of the literature. J Surg Oncol. 1986;31:71-75.
- Brownstein MH, Shapiro L. Verrucous carcinoma of skin: epithelioma cuniculatum plantare. Cancer. 1976;38:1710-1716.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. verrucous carcinoma (epithelioma cuniculatum). Arch Dermatol. 2000;136:547-548, 550-551.
To the Editor:
Verrucous carcinoma (VC) is a rare type of squamous cell carcinoma characterized by a well-differentiated low-grade tumor with a high degree of keratinization. First described by Ackerman1 in 1948, VC presents on the skin or oral and genital mucosae with minimal atypical cytologic findings.1-3 It most commonly is seen in late middle-aged men (85% of cases) and presents as a slow-growing mass, often of more than 10 years’ duration.2,3 Verrucous carcinoma frequently is observed at 3 particular anatomic sites: the oral cavity, known as oral florid papillomatosis; the anogenital area, known as Buschke-Löwenstein tumor; and on the plantar surface, known as epithelioma cuniculatum.2-13
A 19-year-old man presented with an ulcerous lesion on the right big toe of 2 years’ duration. He reported that the lesion had gradually increased in size and was painful when walking. Physical examination revealed an ulcerated lesion on the right big toe with purulent inflammation and necrosis, unclear edges, and border nodules containing a fatty, yellowish, foul-smelling material (Figure 1). Histologic examination of purulent material from deep within the primary lesion revealed gram-negative rods and gram-positive diplococci. Erlich-Ziehl-Neelsen staining and culture in Lowenstein-Jensen medium were negative for mycobacteria. Histologic examination and fungal culture were not diagnostic for fungal infection.
The differential diagnosis included tuberculosis cutis verrucosa, subcutaneous mycoses, swimming pool granuloma, leishmania cutis, chronic pyoderma vegetans, and VC. A punch biopsy of the lesion showed chronic nonspecific inflammation, hyperkeratosis, parakeratosis, and pseudoepitheliomatous hyperplasia. A repeat biopsy performed 15 days later also showed a nonspecific inflammation. At the initial presentation, an anti–human immunodeficiency virus test was negative. A purified protein derivative (PPD) skin test was positive and showed a 17-mm induration, and a sputum test was negative for Mycobacterium tuberculosis. A chest radiograph was normal. We considered the positive PPD skin test to be clinically insignificant; we did not find an accompanying tuberculosis infection, and the high exposure to atypical tuberculosis in developing countries such as Turkey, which is where the patient resided, often explains a positive PPD test.
At the initial presentation, radiography of the right big toe revealed porotic signs and cortical irregularity of the distal phalanx. A deep incisional biopsy of the lesion was performed for pathologic and microbiologic analysis. Erlich-Ziehl-Neelsen staining was negative, fungal elements could not be observed, and there was no growth in Lowenstein-Jensen medium or Sabouraud dextrose agar. Polymerase chain reaction for human papillomavirus, M tuberculosis, and atypical mycobacterium was negative. Periodic acid–Schiff staining was negative for fungal elements. Histopathologic examination revealed an exophytic as well as endophytic squamous cell proliferation infiltrating deeper layers of the dermis with a desmoplastic stroma (Figure 2). Slight cytologic atypia was noted. A diagnosis of VC was made based on the clinical and histopathologic findings. The patient’s right big toe was amputated by plastic surgery 6 months after the initial presentation.
The term epithelioma cuniculatum was first used in 1954 to describe plantar VC. The term cuniculus is Latin for rabbit nest.3 At the distal part of the plantar surface of the foot, VC presents as an exophytic funguslike mass with abundant keratin-filled sinuses.14 When pressure is applied to the lesion, a greasy, yellowish, foul-smelling material with the consistency of toothpaste emerges from the sinuses. The lesion resembles pyoderma vegetans and may present with secondary infections (eg, Staphylococcus aureus, gram-negative bacteria, fungal infection) and/or ulcerations. Its appearance resembles an inflammatory lesion more than a neoplasm.6 Sometimes the skin surrounding the lesion may be a yellowish color, giving the impression of a plantar wart.3,4 In most cases, in situ hybridization demonstrates a human papillomavirus genome.2-5,10 Other factors implicated in the etiopathogenesis of VC include chronic inflammation; a cicatrice associated with a condition such as chronic cutaneous tuberculosis, ulcerative leprosy, dystrophic epidermolysis bullosa, or chronic osteomyelitis4; recurrent trauma3; and/or lichen planus.2,4 In spite of its slow development and benign appearance, VC may cause severe destruction affecting surrounding bony structures and may ultimately require amputation.2,4 In its early stages, VC can be mistaken for a benign tumor or other benign lesion, such as giant seborrheic keratosis, giant keratoacanthoma, eccrine poroma, or verruciform xanthoma, potentially leading to an incorrect diagnosis.5
Histopathologic examination, especially of superficial biopsies, generally reveals squamous cell proliferation demonstrating minimal pleomorphism and cytologic atypia with sparse mitotic figures.4-6 Diagnosis of VC can be challenging if the endophytic proliferation, which characteristically pushes into the dermis and even deeper tissues at the base of the lesion, is not seen. This feature is uncommon in squamous cell carcinomas.3,4,6 Histopathologic detection of koilocytes can lead to difficulty in distinguishing VC from warts.5 The growth of lesions is exophytic in plantar verrucae, whereas in VC it may be either exophytic or endophytic.4 At early stages, it is too difficult to distinguish VC from pseudoepitheliomatous hyperplasia caused by chronic inflammation, as well as from tuberculosis and subcutaneous mycoses.3,6 In these situations, possible responsible microorganisms must be sought out. Amelanotic malignant melanoma and eccrine poroma also should be considered in the differential diagnosis.3,5 If the biopsy specimen is obtained superficially and is fragmented, the diagnosis is more difficult, making deep biopsies essential in suspicious cases.4 Excision is the best treatment, and Mohs micrographic surgery may be required in some cases.2,3,11 It is important to consider that radiotherapy may lead to anaplastic transformation and metastasis.2 Metastasis to lymph nodes is very rare, and the prognosis is excellent when complete excision is performed.2 Recurrence may be observed.4
Our case of plantar VC is notable because of the patient’s young age, which is uncommon, as the typical age for developing VC is late middle age (ie, fifth and sixth decades of life). A long-standing lesion that is therapy resistant and without a detectable microorganism should be investigated for malignancy by repetitive deep biopsy regardless of the patient’s age, as demonstrated in our case.
To the Editor:
Verrucous carcinoma (VC) is a rare type of squamous cell carcinoma characterized by a well-differentiated low-grade tumor with a high degree of keratinization. First described by Ackerman1 in 1948, VC presents on the skin or oral and genital mucosae with minimal atypical cytologic findings.1-3 It most commonly is seen in late middle-aged men (85% of cases) and presents as a slow-growing mass, often of more than 10 years’ duration.2,3 Verrucous carcinoma frequently is observed at 3 particular anatomic sites: the oral cavity, known as oral florid papillomatosis; the anogenital area, known as Buschke-Löwenstein tumor; and on the plantar surface, known as epithelioma cuniculatum.2-13
A 19-year-old man presented with an ulcerous lesion on the right big toe of 2 years’ duration. He reported that the lesion had gradually increased in size and was painful when walking. Physical examination revealed an ulcerated lesion on the right big toe with purulent inflammation and necrosis, unclear edges, and border nodules containing a fatty, yellowish, foul-smelling material (Figure 1). Histologic examination of purulent material from deep within the primary lesion revealed gram-negative rods and gram-positive diplococci. Erlich-Ziehl-Neelsen staining and culture in Lowenstein-Jensen medium were negative for mycobacteria. Histologic examination and fungal culture were not diagnostic for fungal infection.
The differential diagnosis included tuberculosis cutis verrucosa, subcutaneous mycoses, swimming pool granuloma, leishmania cutis, chronic pyoderma vegetans, and VC. A punch biopsy of the lesion showed chronic nonspecific inflammation, hyperkeratosis, parakeratosis, and pseudoepitheliomatous hyperplasia. A repeat biopsy performed 15 days later also showed a nonspecific inflammation. At the initial presentation, an anti–human immunodeficiency virus test was negative. A purified protein derivative (PPD) skin test was positive and showed a 17-mm induration, and a sputum test was negative for Mycobacterium tuberculosis. A chest radiograph was normal. We considered the positive PPD skin test to be clinically insignificant; we did not find an accompanying tuberculosis infection, and the high exposure to atypical tuberculosis in developing countries such as Turkey, which is where the patient resided, often explains a positive PPD test.
At the initial presentation, radiography of the right big toe revealed porotic signs and cortical irregularity of the distal phalanx. A deep incisional biopsy of the lesion was performed for pathologic and microbiologic analysis. Erlich-Ziehl-Neelsen staining was negative, fungal elements could not be observed, and there was no growth in Lowenstein-Jensen medium or Sabouraud dextrose agar. Polymerase chain reaction for human papillomavirus, M tuberculosis, and atypical mycobacterium was negative. Periodic acid–Schiff staining was negative for fungal elements. Histopathologic examination revealed an exophytic as well as endophytic squamous cell proliferation infiltrating deeper layers of the dermis with a desmoplastic stroma (Figure 2). Slight cytologic atypia was noted. A diagnosis of VC was made based on the clinical and histopathologic findings. The patient’s right big toe was amputated by plastic surgery 6 months after the initial presentation.
The term epithelioma cuniculatum was first used in 1954 to describe plantar VC. The term cuniculus is Latin for rabbit nest.3 At the distal part of the plantar surface of the foot, VC presents as an exophytic funguslike mass with abundant keratin-filled sinuses.14 When pressure is applied to the lesion, a greasy, yellowish, foul-smelling material with the consistency of toothpaste emerges from the sinuses. The lesion resembles pyoderma vegetans and may present with secondary infections (eg, Staphylococcus aureus, gram-negative bacteria, fungal infection) and/or ulcerations. Its appearance resembles an inflammatory lesion more than a neoplasm.6 Sometimes the skin surrounding the lesion may be a yellowish color, giving the impression of a plantar wart.3,4 In most cases, in situ hybridization demonstrates a human papillomavirus genome.2-5,10 Other factors implicated in the etiopathogenesis of VC include chronic inflammation; a cicatrice associated with a condition such as chronic cutaneous tuberculosis, ulcerative leprosy, dystrophic epidermolysis bullosa, or chronic osteomyelitis4; recurrent trauma3; and/or lichen planus.2,4 In spite of its slow development and benign appearance, VC may cause severe destruction affecting surrounding bony structures and may ultimately require amputation.2,4 In its early stages, VC can be mistaken for a benign tumor or other benign lesion, such as giant seborrheic keratosis, giant keratoacanthoma, eccrine poroma, or verruciform xanthoma, potentially leading to an incorrect diagnosis.5
Histopathologic examination, especially of superficial biopsies, generally reveals squamous cell proliferation demonstrating minimal pleomorphism and cytologic atypia with sparse mitotic figures.4-6 Diagnosis of VC can be challenging if the endophytic proliferation, which characteristically pushes into the dermis and even deeper tissues at the base of the lesion, is not seen. This feature is uncommon in squamous cell carcinomas.3,4,6 Histopathologic detection of koilocytes can lead to difficulty in distinguishing VC from warts.5 The growth of lesions is exophytic in plantar verrucae, whereas in VC it may be either exophytic or endophytic.4 At early stages, it is too difficult to distinguish VC from pseudoepitheliomatous hyperplasia caused by chronic inflammation, as well as from tuberculosis and subcutaneous mycoses.3,6 In these situations, possible responsible microorganisms must be sought out. Amelanotic malignant melanoma and eccrine poroma also should be considered in the differential diagnosis.3,5 If the biopsy specimen is obtained superficially and is fragmented, the diagnosis is more difficult, making deep biopsies essential in suspicious cases.4 Excision is the best treatment, and Mohs micrographic surgery may be required in some cases.2,3,11 It is important to consider that radiotherapy may lead to anaplastic transformation and metastasis.2 Metastasis to lymph nodes is very rare, and the prognosis is excellent when complete excision is performed.2 Recurrence may be observed.4
Our case of plantar VC is notable because of the patient’s young age, which is uncommon, as the typical age for developing VC is late middle age (ie, fifth and sixth decades of life). A long-standing lesion that is therapy resistant and without a detectable microorganism should be investigated for malignancy by repetitive deep biopsy regardless of the patient’s age, as demonstrated in our case.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Schwartz RA. Verrucous carcinoma of the skin and mucosal. J Am Acad Dermatol. 1995;32:1-21.
- Kao GF, Graham JH, Helwig EB. Carcinoma cuniculatum (verrucous carcinoma of the skin): a clinicopathologic study of 46 cases with ultrastructural observations. Cancer. 1982;49:2395-2403.
- Mc Kee PH, ed. Pathology of the Skin. 2nd ed. London, England: Mosby-Wolfe; 1996.
- Schwartz RA, Stoll HL. Squamous cell carcinoma. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York, NY: Mc-Graw Hill; 1999:840-856.
- MacKie RM. Epidermal skin tumours. In: Rook A, Wilkinson DS, Ebling FJG, et al, eds. Textbook of Dermatology. 5th ed. Oxford, United Kingdom: Blackwell Scientific; 1992:1500-1556.
- Yoshtatsu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Van Geertruyden JP, Olemans C, Laporte M, et al. Verrucous carcinoma of the nail bed. Foot Ankle Int. 1998;19:327-328.
- Sanchez-Yus E, Velasco E, Robledo A. Verrucous carcinoma of the back. J Am Acad Dermatol. 1986;14(5 pt 2):947-950.
- Noel JC, Peny MO, Detremmerie O, et al. Demonstration of human papillomavirus type 2 in a verrucous carcinoma of the foot. Dermatology. 1993;187:58-61.
- Mora RG. Microscopically controlled surgery (Mohs’ chemosurgery) for treatment of verrucous squamous cell carcinoma of the foot (epithelioma cuniculatum). J Am Acad Dermatol. 1983;8:354-362.
- Kathuria S, Rieker J, Jablokow VR, et al. Plantar verrucous carcinoma (epithelioma cuniculatum): case report with review of the literature. J Surg Oncol. 1986;31:71-75.
- Brownstein MH, Shapiro L. Verrucous carcinoma of skin: epithelioma cuniculatum plantare. Cancer. 1976;38:1710-1716.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. verrucous carcinoma (epithelioma cuniculatum). Arch Dermatol. 2000;136:547-548, 550-551.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Schwartz RA. Verrucous carcinoma of the skin and mucosal. J Am Acad Dermatol. 1995;32:1-21.
- Kao GF, Graham JH, Helwig EB. Carcinoma cuniculatum (verrucous carcinoma of the skin): a clinicopathologic study of 46 cases with ultrastructural observations. Cancer. 1982;49:2395-2403.
- Mc Kee PH, ed. Pathology of the Skin. 2nd ed. London, England: Mosby-Wolfe; 1996.
- Schwartz RA, Stoll HL. Squamous cell carcinoma. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York, NY: Mc-Graw Hill; 1999:840-856.
- MacKie RM. Epidermal skin tumours. In: Rook A, Wilkinson DS, Ebling FJG, et al, eds. Textbook of Dermatology. 5th ed. Oxford, United Kingdom: Blackwell Scientific; 1992:1500-1556.
- Yoshtatsu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Van Geertruyden JP, Olemans C, Laporte M, et al. Verrucous carcinoma of the nail bed. Foot Ankle Int. 1998;19:327-328.
- Sanchez-Yus E, Velasco E, Robledo A. Verrucous carcinoma of the back. J Am Acad Dermatol. 1986;14(5 pt 2):947-950.
- Noel JC, Peny MO, Detremmerie O, et al. Demonstration of human papillomavirus type 2 in a verrucous carcinoma of the foot. Dermatology. 1993;187:58-61.
- Mora RG. Microscopically controlled surgery (Mohs’ chemosurgery) for treatment of verrucous squamous cell carcinoma of the foot (epithelioma cuniculatum). J Am Acad Dermatol. 1983;8:354-362.
- Kathuria S, Rieker J, Jablokow VR, et al. Plantar verrucous carcinoma (epithelioma cuniculatum): case report with review of the literature. J Surg Oncol. 1986;31:71-75.
- Brownstein MH, Shapiro L. Verrucous carcinoma of skin: epithelioma cuniculatum plantare. Cancer. 1976;38:1710-1716.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. verrucous carcinoma (epithelioma cuniculatum). Arch Dermatol. 2000;136:547-548, 550-551.
Practice Points
- Verrucous carcinoma (VC) frequently is observed at 3 particular anatomic sites: the oral cavity, the anogenital area, and on the plantar surface.
- Plantar VC is rare, with a male predominance and most patients presenting in the fifth to sixth decades of life.
- Differentiating VS from benign tumors may be difficult, especially if only superficial biopsies are taken. Multiple biopsies and a close clinical correlation are required before a definite diagnosis is possible.
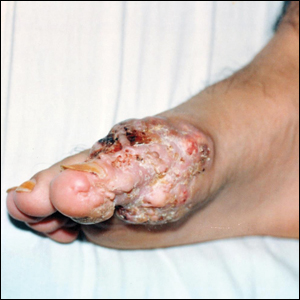
Postinflammatory Hyperpigmentation Following Treatment of Hyperkeratosis Lenticularis Perstans With Tazarotene Cream 0.1%
To the Editor:
Hyperkeratosis lenticularis perstans (HLP), or Flegel disease, is a rare keratinization disorder characterized by asymptomatic, red-brown, 1- to 5-mm papules with irregular horny scales commonly seen on the dorsal feet and lower legs.1 Hyperkeratosis lenticularis perstans is notorious for being difficult to treat. Various treatment options, including 5-fluorouracil, topical and oral retinoids, vitamin D3 derivatives, psoralen plus UVA therapy, and dermabrasion, have been explored but none have proven to be consistently effective.
A woman in her 50s presented with an asymptomatic eruption on the legs and thighs that had been present for the last 20 years. She had been misdiagnosed by multiple outside providers with atopic dermatitis and was treated with topical steroids without considerable improvement. Upon initial presentation to our clinic , physical examination revealed a woman with Fitzpatrick skin type II with multiple hyperpigmented, red-brown, 2- to 6-mm papules on the extensor surfaces of the lower legs and upper thighs (Figure, A). A 3-mm punch biopsy of a lesion on the right upper thigh revealed hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate. The clinical and histopathologic findings were consistent with HLP.
The patient was started on treatment with 5-fluorouracil cream on the right leg and tazarotene cream 0.1% on the left leg to determine which agent would work best. After 9 weeks of treatment, slight improvement was observed on both legs, but the lesions were still erythematous (Figure, B). Treatment was continued, and after 14 weeks complete resolution of the lesions was noted on both legs; however, postinflammatory hyperpigmentation (PIH) was observed on the left leg, which had been treated with tazarotene (Figure, C). The patient was lost to follow-up prior to treatment of the PIH.
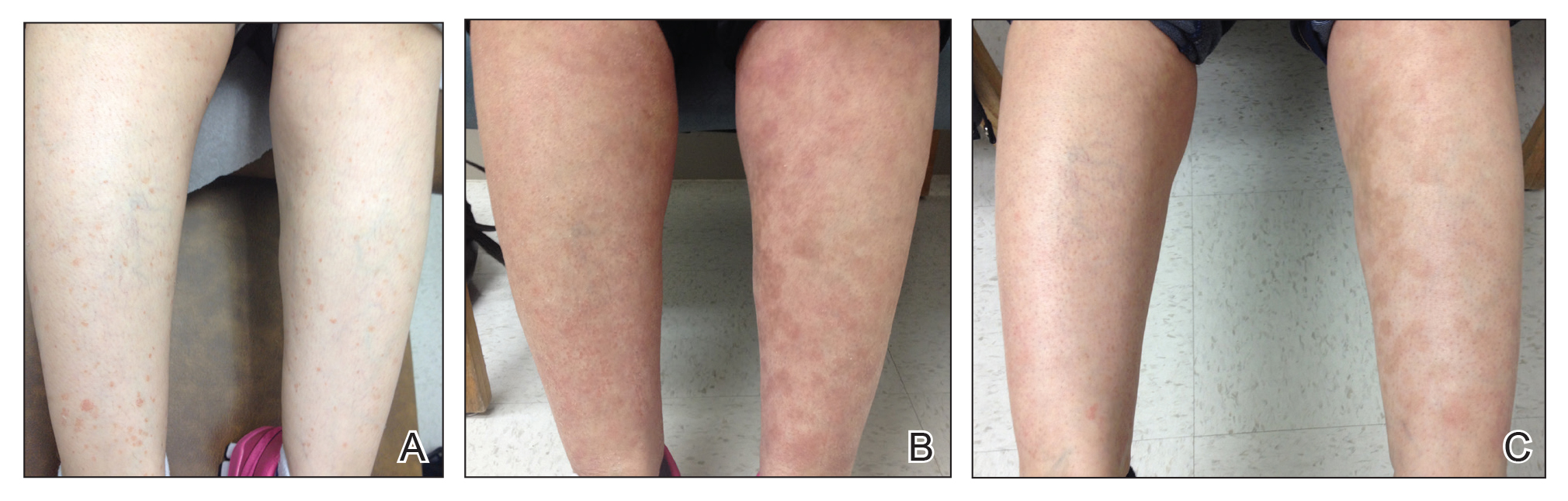
Postinflammatory hyperpigmentation is an acquired excess of pigment due to a prior disease process such as an infection, allergic reaction, trauma, inflammatory disease, or drug reaction. In our patient, this finding was unusual because tazarotene has been shown to be an effective treatment of PIH.2,3
In PIH, there is either abnormal production or distribution of melanin pigment in the epidermis and/or dermis. Several mechanisms for PIH have been suggested. One potential mechanism is disruption of the basal cell layer due to dermal lymphocytic inflammation, causing melanin to be released and trapped by macrophages present in the dermal papillae. Another possible mechanism is epidermal hypermelanosis, in which the release and oxidation of arachidonic acid to prostaglandins and leukotrienes alters immune cells and melanocytes, causing an increase in melanin and increased transfer of melanin to keratinocytes in the surrounding epidermis.4
Treatment of PIH can be a difficult and prolonged process, especially when a dermal rather than epidermal melanosis is observed. Topical retinoids, topical hydroquinone, azelaic acid, corticosteroids, tretinoin cream, glycolic acid, and trichloroacetic acid have been shown to be effective in treating epidermal PIH. Tazarotene is a synthetic retinoid that has been proven to be an effective treatment of PIH3; however, in our patient the PIH progressed with treatment. One plausible explanation is that irritation caused by the medication led to further PIH.2,5
It is uncommon for tazarotene to cause PIH. Hyperpigmentation is listed as an adverse effect observed during the postmarketing experience according to one manufacturer6 and the US Food and Drug Administration; however, details about prior incidents of hyperpigmentation have not been reported in the literature. Our case is unique because both treatments showed considerable improvement in HLP, but more PIH was observed on the tazarotene-treated leg.
- Bean SF. Hyperkeratosis lenticularis perstans. a clinical, histopathologic, and genetic study. Arch Dermatol. 1969;99:705-709.
- Callender V, St. Surin-Lord S, Davis E, et al. Postinflammatory hyperpigmentation: etiologic and therapeutic considerations. Am J Clin Dermatol. 2011;12:87-99.
- McEvoy G. Tazarotene (topical). In: AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2014:84-92.
- Lacz N, Vafaie J, Kihiczak N, et al. Postinflammatory hyperpigmentation: a common but troubling condition. Int J Dermatol. 2004;43:362-365.
- Tazorac (tazarotene) cream [package insert]. Irvine, CA: Allergan, Inc; 2013.
- Tazorac (tazarotene) gel [package insert]. Irvine, CA: Allergan, Inc; 2014.
To the Editor:
Hyperkeratosis lenticularis perstans (HLP), or Flegel disease, is a rare keratinization disorder characterized by asymptomatic, red-brown, 1- to 5-mm papules with irregular horny scales commonly seen on the dorsal feet and lower legs.1 Hyperkeratosis lenticularis perstans is notorious for being difficult to treat. Various treatment options, including 5-fluorouracil, topical and oral retinoids, vitamin D3 derivatives, psoralen plus UVA therapy, and dermabrasion, have been explored but none have proven to be consistently effective.
A woman in her 50s presented with an asymptomatic eruption on the legs and thighs that had been present for the last 20 years. She had been misdiagnosed by multiple outside providers with atopic dermatitis and was treated with topical steroids without considerable improvement. Upon initial presentation to our clinic , physical examination revealed a woman with Fitzpatrick skin type II with multiple hyperpigmented, red-brown, 2- to 6-mm papules on the extensor surfaces of the lower legs and upper thighs (Figure, A). A 3-mm punch biopsy of a lesion on the right upper thigh revealed hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate. The clinical and histopathologic findings were consistent with HLP.
The patient was started on treatment with 5-fluorouracil cream on the right leg and tazarotene cream 0.1% on the left leg to determine which agent would work best. After 9 weeks of treatment, slight improvement was observed on both legs, but the lesions were still erythematous (Figure, B). Treatment was continued, and after 14 weeks complete resolution of the lesions was noted on both legs; however, postinflammatory hyperpigmentation (PIH) was observed on the left leg, which had been treated with tazarotene (Figure, C). The patient was lost to follow-up prior to treatment of the PIH.
Postinflammatory hyperpigmentation is an acquired excess of pigment due to a prior disease process such as an infection, allergic reaction, trauma, inflammatory disease, or drug reaction. In our patient, this finding was unusual because tazarotene has been shown to be an effective treatment of PIH.2,3
In PIH, there is either abnormal production or distribution of melanin pigment in the epidermis and/or dermis. Several mechanisms for PIH have been suggested. One potential mechanism is disruption of the basal cell layer due to dermal lymphocytic inflammation, causing melanin to be released and trapped by macrophages present in the dermal papillae. Another possible mechanism is epidermal hypermelanosis, in which the release and oxidation of arachidonic acid to prostaglandins and leukotrienes alters immune cells and melanocytes, causing an increase in melanin and increased transfer of melanin to keratinocytes in the surrounding epidermis.4
Treatment of PIH can be a difficult and prolonged process, especially when a dermal rather than epidermal melanosis is observed. Topical retinoids, topical hydroquinone, azelaic acid, corticosteroids, tretinoin cream, glycolic acid, and trichloroacetic acid have been shown to be effective in treating epidermal PIH. Tazarotene is a synthetic retinoid that has been proven to be an effective treatment of PIH3; however, in our patient the PIH progressed with treatment. One plausible explanation is that irritation caused by the medication led to further PIH.2,5
It is uncommon for tazarotene to cause PIH. Hyperpigmentation is listed as an adverse effect observed during the postmarketing experience according to one manufacturer6 and the US Food and Drug Administration; however, details about prior incidents of hyperpigmentation have not been reported in the literature. Our case is unique because both treatments showed considerable improvement in HLP, but more PIH was observed on the tazarotene-treated leg.
To the Editor:
Hyperkeratosis lenticularis perstans (HLP), or Flegel disease, is a rare keratinization disorder characterized by asymptomatic, red-brown, 1- to 5-mm papules with irregular horny scales commonly seen on the dorsal feet and lower legs.1 Hyperkeratosis lenticularis perstans is notorious for being difficult to treat. Various treatment options, including 5-fluorouracil, topical and oral retinoids, vitamin D3 derivatives, psoralen plus UVA therapy, and dermabrasion, have been explored but none have proven to be consistently effective.
A woman in her 50s presented with an asymptomatic eruption on the legs and thighs that had been present for the last 20 years. She had been misdiagnosed by multiple outside providers with atopic dermatitis and was treated with topical steroids without considerable improvement. Upon initial presentation to our clinic , physical examination revealed a woman with Fitzpatrick skin type II with multiple hyperpigmented, red-brown, 2- to 6-mm papules on the extensor surfaces of the lower legs and upper thighs (Figure, A). A 3-mm punch biopsy of a lesion on the right upper thigh revealed hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate. The clinical and histopathologic findings were consistent with HLP.
The patient was started on treatment with 5-fluorouracil cream on the right leg and tazarotene cream 0.1% on the left leg to determine which agent would work best. After 9 weeks of treatment, slight improvement was observed on both legs, but the lesions were still erythematous (Figure, B). Treatment was continued, and after 14 weeks complete resolution of the lesions was noted on both legs; however, postinflammatory hyperpigmentation (PIH) was observed on the left leg, which had been treated with tazarotene (Figure, C). The patient was lost to follow-up prior to treatment of the PIH.
Postinflammatory hyperpigmentation is an acquired excess of pigment due to a prior disease process such as an infection, allergic reaction, trauma, inflammatory disease, or drug reaction. In our patient, this finding was unusual because tazarotene has been shown to be an effective treatment of PIH.2,3
In PIH, there is either abnormal production or distribution of melanin pigment in the epidermis and/or dermis. Several mechanisms for PIH have been suggested. One potential mechanism is disruption of the basal cell layer due to dermal lymphocytic inflammation, causing melanin to be released and trapped by macrophages present in the dermal papillae. Another possible mechanism is epidermal hypermelanosis, in which the release and oxidation of arachidonic acid to prostaglandins and leukotrienes alters immune cells and melanocytes, causing an increase in melanin and increased transfer of melanin to keratinocytes in the surrounding epidermis.4
Treatment of PIH can be a difficult and prolonged process, especially when a dermal rather than epidermal melanosis is observed. Topical retinoids, topical hydroquinone, azelaic acid, corticosteroids, tretinoin cream, glycolic acid, and trichloroacetic acid have been shown to be effective in treating epidermal PIH. Tazarotene is a synthetic retinoid that has been proven to be an effective treatment of PIH3; however, in our patient the PIH progressed with treatment. One plausible explanation is that irritation caused by the medication led to further PIH.2,5
It is uncommon for tazarotene to cause PIH. Hyperpigmentation is listed as an adverse effect observed during the postmarketing experience according to one manufacturer6 and the US Food and Drug Administration; however, details about prior incidents of hyperpigmentation have not been reported in the literature. Our case is unique because both treatments showed considerable improvement in HLP, but more PIH was observed on the tazarotene-treated leg.
- Bean SF. Hyperkeratosis lenticularis perstans. a clinical, histopathologic, and genetic study. Arch Dermatol. 1969;99:705-709.
- Callender V, St. Surin-Lord S, Davis E, et al. Postinflammatory hyperpigmentation: etiologic and therapeutic considerations. Am J Clin Dermatol. 2011;12:87-99.
- McEvoy G. Tazarotene (topical). In: AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2014:84-92.
- Lacz N, Vafaie J, Kihiczak N, et al. Postinflammatory hyperpigmentation: a common but troubling condition. Int J Dermatol. 2004;43:362-365.
- Tazorac (tazarotene) cream [package insert]. Irvine, CA: Allergan, Inc; 2013.
- Tazorac (tazarotene) gel [package insert]. Irvine, CA: Allergan, Inc; 2014.
- Bean SF. Hyperkeratosis lenticularis perstans. a clinical, histopathologic, and genetic study. Arch Dermatol. 1969;99:705-709.
- Callender V, St. Surin-Lord S, Davis E, et al. Postinflammatory hyperpigmentation: etiologic and therapeutic considerations. Am J Clin Dermatol. 2011;12:87-99.
- McEvoy G. Tazarotene (topical). In: AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2014:84-92.
- Lacz N, Vafaie J, Kihiczak N, et al. Postinflammatory hyperpigmentation: a common but troubling condition. Int J Dermatol. 2004;43:362-365.
- Tazorac (tazarotene) cream [package insert]. Irvine, CA: Allergan, Inc; 2013.
- Tazorac (tazarotene) gel [package insert]. Irvine, CA: Allergan, Inc; 2014.
Practice Points
- Hyperkeratosis lenticularis perstans is a rare keratinization disorder that presents with asymptomatic red-brown papules with irregular horny scales on the lower extremities.
- Hyperkeratosis lenticularis perstans can be difficult to diagnose and treat. Hematoxylin and eosin staining generally will show hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate.
- Tazarotene cream 0.1% is a synthetic retinoid sometimes used for treatment of hyperpigmentation, but it also can cause postinflammatory hyperpigmentation.
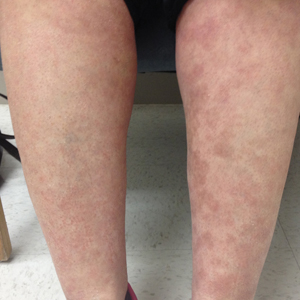
Dapagliflozin-Induced Sweet Syndrome
To the Editor:
A 75-year-old woman with a history of hypertension, gout, and adult-onset diabetes mellitus was started on dapagliflozin (5 mg) for glycemic control (hemoglobin A1c, 7.9 [reference range, 4–7]). Within 1 week of starting the medication, she developed a fine papular eruption in a photodistributed area on the neck and chest with associated malaise. The rash progressed over the next 2 weeks, evolving into edematous papules and plaques, some with vesicles involving the neck, chest, postauricular areas, and nose. Approximately 3 weeks after starting dapagliflozin, the patient also developed bilateral painful, hemorrhagic, bullous plaques (10×3 cm overall) without satellite lesions on the dorsal aspects of the hands. The borders of the bullae had rapidly expanding geographic margins and were extremely painful. The patient’s primary care physician advised to discontinue dapagliflozin, as this medication was thought to be triggering the eruption. She was administered triamcinolone (40 mg intramuscularly) and advised to take ibuprofen as needed. She had malaise and reported that she felt hot but had no known fever. No laboratory tests were ordered.
The lesions on the neck and chest began to fade within 1 week of stopping the medication and administering corticosteroids; however, the hand lesions continued to progress and began to involve the base of the digits (Figure 1). The patient was then seen by a dermatologist who biopsied the hand lesions. Histopathology was characteristic of Sweet syndrome, also known as acute febrile neutrophilic dermatosis and Gomm-Button disease. Notably, there was a dense nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris without leukocytoclastic vasculitis (Figure 2).
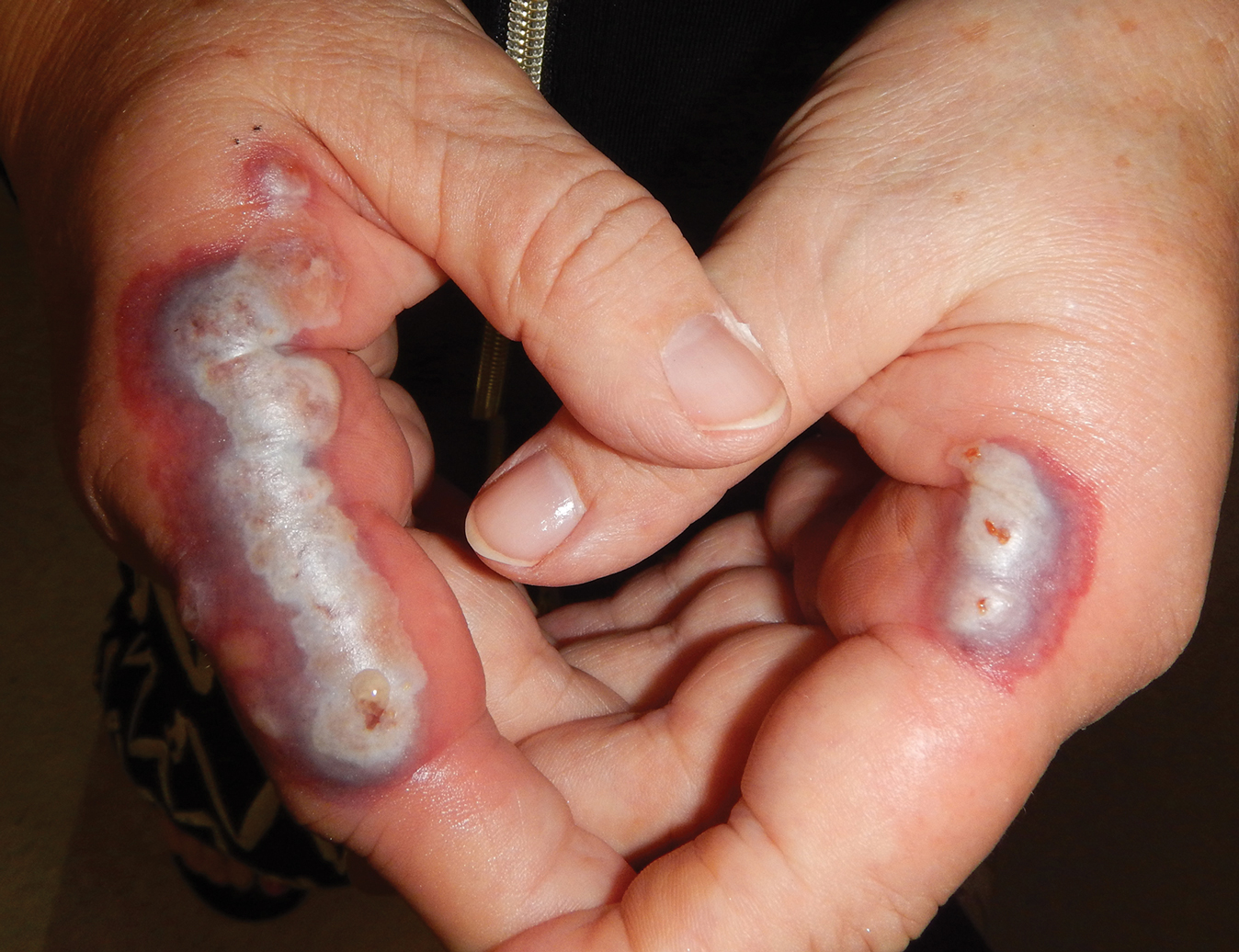
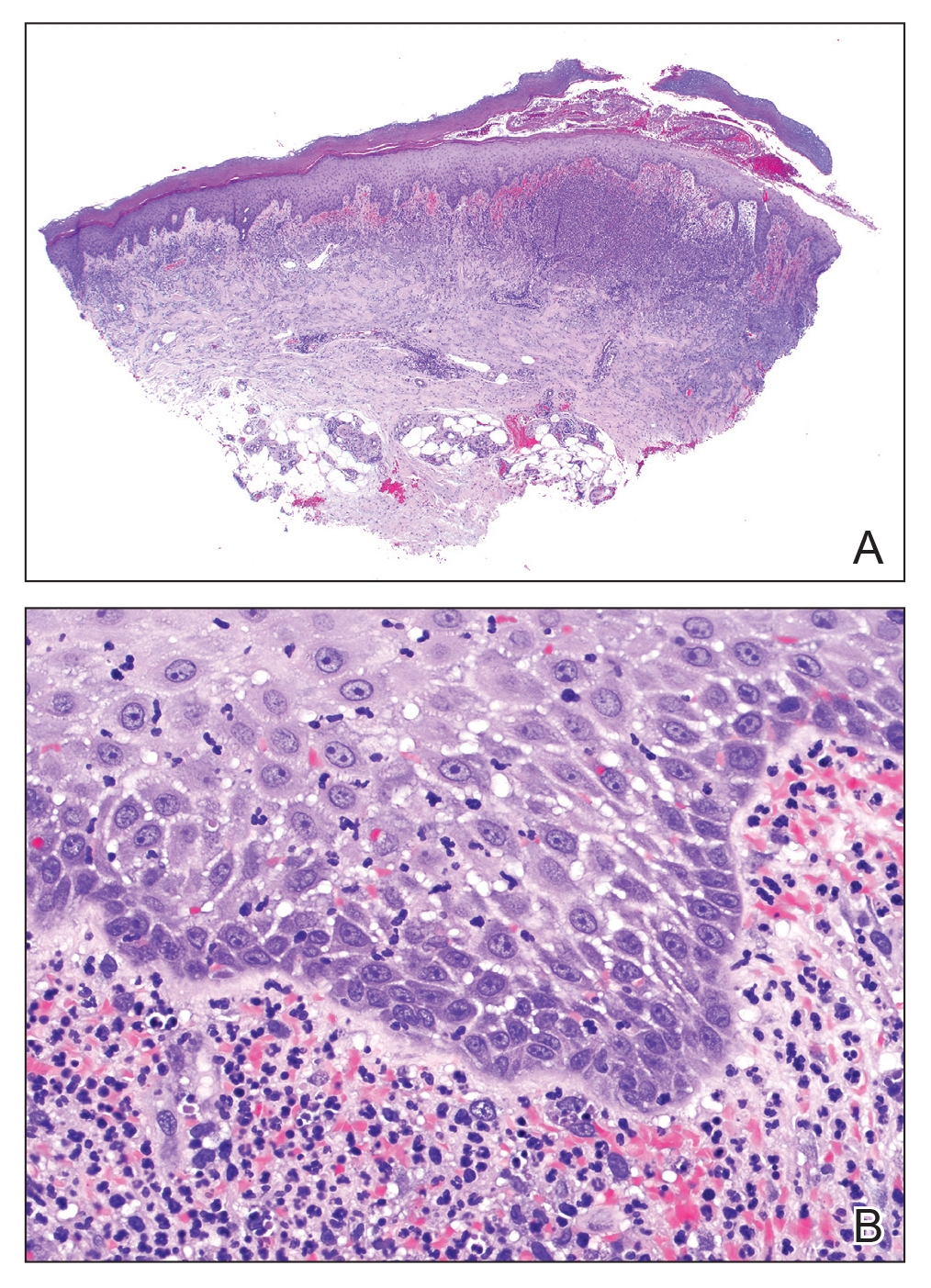
The following therapies in addition to gentle wound care were prescribed: betamethasone injectable suspension (9 mg intramuscularly), oral prednisone (60 mg daily for 5 days, tapering off over 2 months), clobetasol ointment 0.05% twice daily, and tacrolimus ointment 0.1% twice daily. The patient responded well to therapy, with complete resolution and healing of the skin lesions except for mild postinflammatory pigment alteration. The systemic steroids were slowly tapered over 2 months, and the patient remained free of symptoms or recurrences more than 3 years after discontinuation of the medication.
Dapagliflozin is a member of a new class of medications (gliflozins) used for the treatment of type 2 diabetes mellitus.3,4 The medication lowers blood glucose by inhibiting the sodium-glucose cotransport protein 2, thus lowering the blood glucose levels by increasing urinary excretion of glucose. Because many patients with type 2 diabetes mellitus are overweight, these medications are poised to gain popularity for weight loss and decreased blood pressure side effects. Three other medications in this class also have been approved by US Food and Drug Administration: empagliflozin, canagliflozin, and ertugliflozin.
Sweet syndrome consists of 4 cardinal features that were first described in 1964: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.5 Since then, Su and Liu6 proposed guidelines consisting of major and minor criteria. In 1996, Walker and Cohen7 suggested a set of diagnostic criteria specifically for drug-induced Sweet syndrome, including painful erythematous plaques, histopathologic neutrophilic infiltrate, and fever. Additional criteria included a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.7 The lesions of drug-induced Sweet syndrome often are described as painful red papules that can form plaques, may appear vesicular, and are more common in women. These lesions classically appear on the upper extremities, as well as the head, neck, trunk, and back.8 Clinically, symptoms most commonly include fever and musculoskeletal involvement, both of which were experienced by the patient who described herself as feeling feverish when the lesions first appeared and reported malaise. Our patient experienced all of these features, and although a fever was not measured in the acute stage of presentation, she reported feeling hot. Other symptoms that may occur include arthralgia, headache, and myalgia.9 Microscopically, there is a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris. The pathogenesis of Sweet syndrome remains unclear but can be associated with malignancy, pregnancy, autoimmune disorders, and drug reactions.10 Many different classes of medications have been reported to cause drug-induced Sweet syndrome and are listed in the Table.1,8,11 The recommended treatment of Sweet syndrome is systemic corticosteroids.12
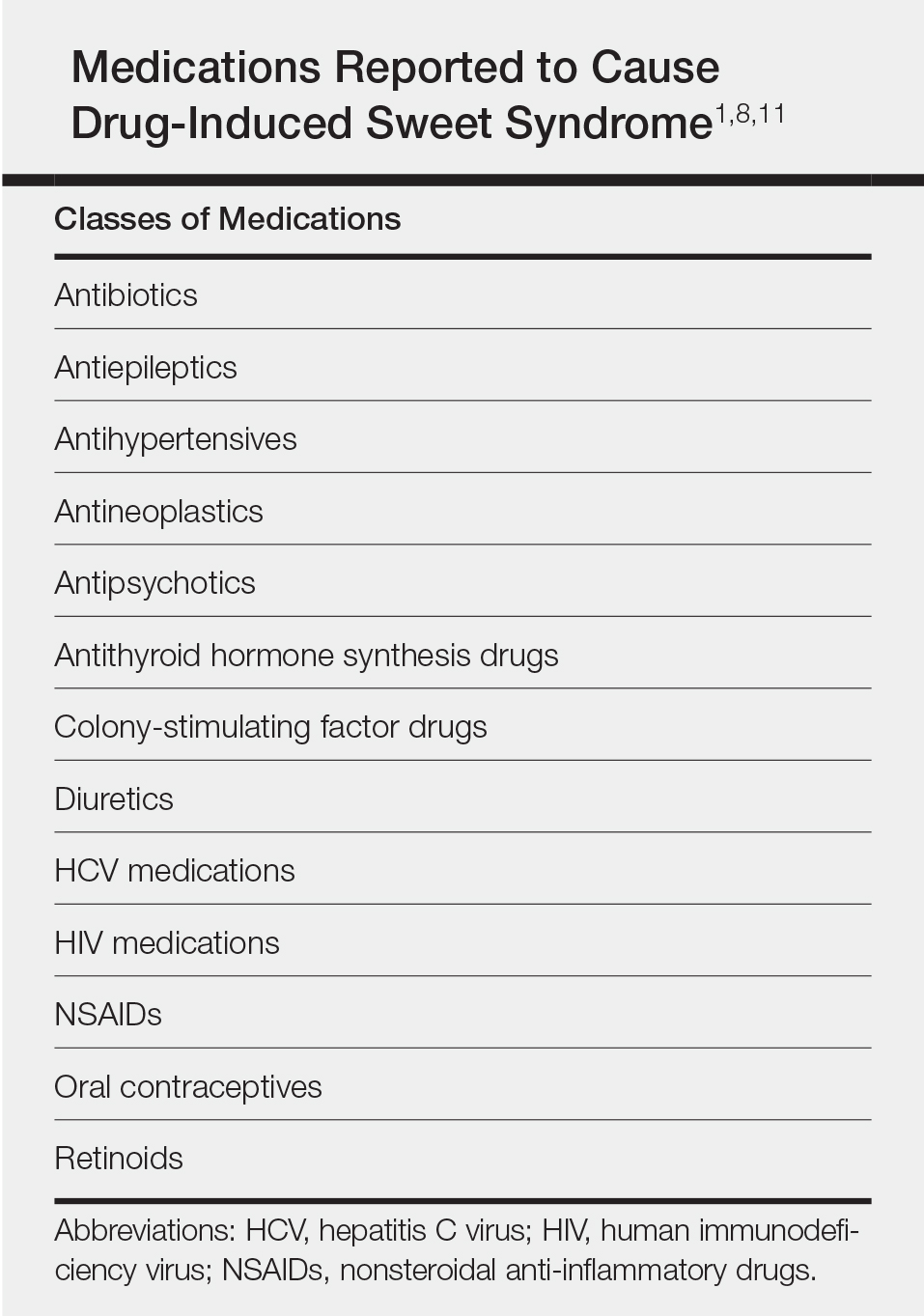
The temporal use of dapagliflozin and appearance of the hand lesions, along with the histology, favored drug-induced Sweet syndrome to dapagliflozin as the cause of the plaques. Our patient did not seek medical attention at the onset of the initial chest and neck rash but did so several weeks after the painful hand lesions that were consistent with Sweet syndrome had appeared. Discontinuation of dapagliflozin and treatment with immunosuppressive medications resulted in resolution of the skin lesions on the hands. This scenario raises the question whether or not she would have developed the inflammatory hand lesions if she had stopped the medication earlier. Because dapagliflozin is a relatively new medication and boasts the potentially beneficial side effects of reducing body weight and blood pressure in addition to glucose control, we expect additional cases may occur, especially if use of this medication notably increases. Furthermore, this reaction may be a drug-class side effect and not one specific to dapagliflozin.
- Weedon D. The vasculopathic reaction pattern. Weedon’s Skin Pathology. 3rd ed. Oxford, UK: Churchill Livingstone; 2010:218-225.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Dapagliflozin (Farxiga) for type 2 diabetes. Med Lett Drugs Ther. 2014;56:13-15.
- Aylsworth A, Dean Z, VanNorman C, et al. Dapagliflozin for the treatment of type 2 diabetes mellitus [published online June 20, 2014]. Ann Pharmacother. 2014;48:1202-1208.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 pt 2):918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Cohen PR, Kurzrock R. Sweet’s syndrome. a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18:265-282.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
To the Editor:
A 75-year-old woman with a history of hypertension, gout, and adult-onset diabetes mellitus was started on dapagliflozin (5 mg) for glycemic control (hemoglobin A1c, 7.9 [reference range, 4–7]). Within 1 week of starting the medication, she developed a fine papular eruption in a photodistributed area on the neck and chest with associated malaise. The rash progressed over the next 2 weeks, evolving into edematous papules and plaques, some with vesicles involving the neck, chest, postauricular areas, and nose. Approximately 3 weeks after starting dapagliflozin, the patient also developed bilateral painful, hemorrhagic, bullous plaques (10×3 cm overall) without satellite lesions on the dorsal aspects of the hands. The borders of the bullae had rapidly expanding geographic margins and were extremely painful. The patient’s primary care physician advised to discontinue dapagliflozin, as this medication was thought to be triggering the eruption. She was administered triamcinolone (40 mg intramuscularly) and advised to take ibuprofen as needed. She had malaise and reported that she felt hot but had no known fever. No laboratory tests were ordered.
The lesions on the neck and chest began to fade within 1 week of stopping the medication and administering corticosteroids; however, the hand lesions continued to progress and began to involve the base of the digits (Figure 1). The patient was then seen by a dermatologist who biopsied the hand lesions. Histopathology was characteristic of Sweet syndrome, also known as acute febrile neutrophilic dermatosis and Gomm-Button disease. Notably, there was a dense nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris without leukocytoclastic vasculitis (Figure 2).
The following therapies in addition to gentle wound care were prescribed: betamethasone injectable suspension (9 mg intramuscularly), oral prednisone (60 mg daily for 5 days, tapering off over 2 months), clobetasol ointment 0.05% twice daily, and tacrolimus ointment 0.1% twice daily. The patient responded well to therapy, with complete resolution and healing of the skin lesions except for mild postinflammatory pigment alteration. The systemic steroids were slowly tapered over 2 months, and the patient remained free of symptoms or recurrences more than 3 years after discontinuation of the medication.
Dapagliflozin is a member of a new class of medications (gliflozins) used for the treatment of type 2 diabetes mellitus.3,4 The medication lowers blood glucose by inhibiting the sodium-glucose cotransport protein 2, thus lowering the blood glucose levels by increasing urinary excretion of glucose. Because many patients with type 2 diabetes mellitus are overweight, these medications are poised to gain popularity for weight loss and decreased blood pressure side effects. Three other medications in this class also have been approved by US Food and Drug Administration: empagliflozin, canagliflozin, and ertugliflozin.
Sweet syndrome consists of 4 cardinal features that were first described in 1964: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.5 Since then, Su and Liu6 proposed guidelines consisting of major and minor criteria. In 1996, Walker and Cohen7 suggested a set of diagnostic criteria specifically for drug-induced Sweet syndrome, including painful erythematous plaques, histopathologic neutrophilic infiltrate, and fever. Additional criteria included a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.7 The lesions of drug-induced Sweet syndrome often are described as painful red papules that can form plaques, may appear vesicular, and are more common in women. These lesions classically appear on the upper extremities, as well as the head, neck, trunk, and back.8 Clinically, symptoms most commonly include fever and musculoskeletal involvement, both of which were experienced by the patient who described herself as feeling feverish when the lesions first appeared and reported malaise. Our patient experienced all of these features, and although a fever was not measured in the acute stage of presentation, she reported feeling hot. Other symptoms that may occur include arthralgia, headache, and myalgia.9 Microscopically, there is a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris. The pathogenesis of Sweet syndrome remains unclear but can be associated with malignancy, pregnancy, autoimmune disorders, and drug reactions.10 Many different classes of medications have been reported to cause drug-induced Sweet syndrome and are listed in the Table.1,8,11 The recommended treatment of Sweet syndrome is systemic corticosteroids.12
The temporal use of dapagliflozin and appearance of the hand lesions, along with the histology, favored drug-induced Sweet syndrome to dapagliflozin as the cause of the plaques. Our patient did not seek medical attention at the onset of the initial chest and neck rash but did so several weeks after the painful hand lesions that were consistent with Sweet syndrome had appeared. Discontinuation of dapagliflozin and treatment with immunosuppressive medications resulted in resolution of the skin lesions on the hands. This scenario raises the question whether or not she would have developed the inflammatory hand lesions if she had stopped the medication earlier. Because dapagliflozin is a relatively new medication and boasts the potentially beneficial side effects of reducing body weight and blood pressure in addition to glucose control, we expect additional cases may occur, especially if use of this medication notably increases. Furthermore, this reaction may be a drug-class side effect and not one specific to dapagliflozin.
To the Editor:
A 75-year-old woman with a history of hypertension, gout, and adult-onset diabetes mellitus was started on dapagliflozin (5 mg) for glycemic control (hemoglobin A1c, 7.9 [reference range, 4–7]). Within 1 week of starting the medication, she developed a fine papular eruption in a photodistributed area on the neck and chest with associated malaise. The rash progressed over the next 2 weeks, evolving into edematous papules and plaques, some with vesicles involving the neck, chest, postauricular areas, and nose. Approximately 3 weeks after starting dapagliflozin, the patient also developed bilateral painful, hemorrhagic, bullous plaques (10×3 cm overall) without satellite lesions on the dorsal aspects of the hands. The borders of the bullae had rapidly expanding geographic margins and were extremely painful. The patient’s primary care physician advised to discontinue dapagliflozin, as this medication was thought to be triggering the eruption. She was administered triamcinolone (40 mg intramuscularly) and advised to take ibuprofen as needed. She had malaise and reported that she felt hot but had no known fever. No laboratory tests were ordered.
The lesions on the neck and chest began to fade within 1 week of stopping the medication and administering corticosteroids; however, the hand lesions continued to progress and began to involve the base of the digits (Figure 1). The patient was then seen by a dermatologist who biopsied the hand lesions. Histopathology was characteristic of Sweet syndrome, also known as acute febrile neutrophilic dermatosis and Gomm-Button disease. Notably, there was a dense nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris without leukocytoclastic vasculitis (Figure 2).
The following therapies in addition to gentle wound care were prescribed: betamethasone injectable suspension (9 mg intramuscularly), oral prednisone (60 mg daily for 5 days, tapering off over 2 months), clobetasol ointment 0.05% twice daily, and tacrolimus ointment 0.1% twice daily. The patient responded well to therapy, with complete resolution and healing of the skin lesions except for mild postinflammatory pigment alteration. The systemic steroids were slowly tapered over 2 months, and the patient remained free of symptoms or recurrences more than 3 years after discontinuation of the medication.
Dapagliflozin is a member of a new class of medications (gliflozins) used for the treatment of type 2 diabetes mellitus.3,4 The medication lowers blood glucose by inhibiting the sodium-glucose cotransport protein 2, thus lowering the blood glucose levels by increasing urinary excretion of glucose. Because many patients with type 2 diabetes mellitus are overweight, these medications are poised to gain popularity for weight loss and decreased blood pressure side effects. Three other medications in this class also have been approved by US Food and Drug Administration: empagliflozin, canagliflozin, and ertugliflozin.
Sweet syndrome consists of 4 cardinal features that were first described in 1964: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.5 Since then, Su and Liu6 proposed guidelines consisting of major and minor criteria. In 1996, Walker and Cohen7 suggested a set of diagnostic criteria specifically for drug-induced Sweet syndrome, including painful erythematous plaques, histopathologic neutrophilic infiltrate, and fever. Additional criteria included a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.7 The lesions of drug-induced Sweet syndrome often are described as painful red papules that can form plaques, may appear vesicular, and are more common in women. These lesions classically appear on the upper extremities, as well as the head, neck, trunk, and back.8 Clinically, symptoms most commonly include fever and musculoskeletal involvement, both of which were experienced by the patient who described herself as feeling feverish when the lesions first appeared and reported malaise. Our patient experienced all of these features, and although a fever was not measured in the acute stage of presentation, she reported feeling hot. Other symptoms that may occur include arthralgia, headache, and myalgia.9 Microscopically, there is a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris. The pathogenesis of Sweet syndrome remains unclear but can be associated with malignancy, pregnancy, autoimmune disorders, and drug reactions.10 Many different classes of medications have been reported to cause drug-induced Sweet syndrome and are listed in the Table.1,8,11 The recommended treatment of Sweet syndrome is systemic corticosteroids.12
The temporal use of dapagliflozin and appearance of the hand lesions, along with the histology, favored drug-induced Sweet syndrome to dapagliflozin as the cause of the plaques. Our patient did not seek medical attention at the onset of the initial chest and neck rash but did so several weeks after the painful hand lesions that were consistent with Sweet syndrome had appeared. Discontinuation of dapagliflozin and treatment with immunosuppressive medications resulted in resolution of the skin lesions on the hands. This scenario raises the question whether or not she would have developed the inflammatory hand lesions if she had stopped the medication earlier. Because dapagliflozin is a relatively new medication and boasts the potentially beneficial side effects of reducing body weight and blood pressure in addition to glucose control, we expect additional cases may occur, especially if use of this medication notably increases. Furthermore, this reaction may be a drug-class side effect and not one specific to dapagliflozin.
- Weedon D. The vasculopathic reaction pattern. Weedon’s Skin Pathology. 3rd ed. Oxford, UK: Churchill Livingstone; 2010:218-225.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Dapagliflozin (Farxiga) for type 2 diabetes. Med Lett Drugs Ther. 2014;56:13-15.
- Aylsworth A, Dean Z, VanNorman C, et al. Dapagliflozin for the treatment of type 2 diabetes mellitus [published online June 20, 2014]. Ann Pharmacother. 2014;48:1202-1208.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 pt 2):918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Cohen PR, Kurzrock R. Sweet’s syndrome. a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18:265-282.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Weedon D. The vasculopathic reaction pattern. Weedon’s Skin Pathology. 3rd ed. Oxford, UK: Churchill Livingstone; 2010:218-225.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Dapagliflozin (Farxiga) for type 2 diabetes. Med Lett Drugs Ther. 2014;56:13-15.
- Aylsworth A, Dean Z, VanNorman C, et al. Dapagliflozin for the treatment of type 2 diabetes mellitus [published online June 20, 2014]. Ann Pharmacother. 2014;48:1202-1208.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 pt 2):918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Cohen PR, Kurzrock R. Sweet’s syndrome. a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18:265-282.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
Practice Points
- Sweet syndrome consists of 4 cardinal features: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.
- In drug-induced Sweet syndrome, there is a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.
- Microscopic findings of Sweet syndrome include a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris.
- Dapagliflozin is a member of a new class of medications (gliflozins) used for treatment of type 2 diabetes mellitus, which may cause drug-induced Sweet syndrome.