User login
Lipid instigates leukemic cell death

Scientists say they’ve identified a tumor-associated lipid that successfully stimulates T cells to kill leukemia cells in vitro and in vivo.
The team noted that certain T cells can recognize the presentation of self-derived lipids on the CD1c protein.
These T cells are commonly found in healthy individuals and are known to kill transformed hematopoietic cells.
However, their antigen specificity and potential antileukemic effects have not been well characterized.
So Gennaro De Libero, MD, PhD, of the University Hospital Basel in Switzerland, and his colleagues analyzed these cells and reported their findings in The Journal of Experimental Medicine.
The researchers discovered that CD1c self-reactive T cells recognize a novel class of self-lipids called methyl-lysophosphatidic acids (mLPAs), which were abundant in several human leukemias.
These mLPAs are the first example of lipid tumor-associated antigens (TAAs). TAAs are produced by tumors and stimulate T cells that can kill leukemia cells.
However, protein TAA accumulation can be drastically reduced by variant leukemia cells. And some TAAs may change their structure, which prevents recognition by T cells and facilitates tumor evasion.
mLPAs, on the other hand, do not change their structure and remain abundant in leukemia cells.
Furthermore, Dr De Libero and his colleagues found they could isolate the T cells that recognize and kill mLPA-expressing leukemia cells in in vitro tests.
And when the team transplanted the T cells into mice, the cells displayed therapeutic efficacy against leukemia cells.
The researchers noted that this type of immunotherapy, if proven effective in humans, could be used to help prevent leukemia relapse after chemotherapy or stem cell transplant. ![]()
Scientists say they’ve identified a tumor-associated lipid that successfully stimulates T cells to kill leukemia cells in vitro and in vivo.
The team noted that certain T cells can recognize the presentation of self-derived lipids on the CD1c protein.
These T cells are commonly found in healthy individuals and are known to kill transformed hematopoietic cells.
However, their antigen specificity and potential antileukemic effects have not been well characterized.
So Gennaro De Libero, MD, PhD, of the University Hospital Basel in Switzerland, and his colleagues analyzed these cells and reported their findings in The Journal of Experimental Medicine.
The researchers discovered that CD1c self-reactive T cells recognize a novel class of self-lipids called methyl-lysophosphatidic acids (mLPAs), which were abundant in several human leukemias.
These mLPAs are the first example of lipid tumor-associated antigens (TAAs). TAAs are produced by tumors and stimulate T cells that can kill leukemia cells.
However, protein TAA accumulation can be drastically reduced by variant leukemia cells. And some TAAs may change their structure, which prevents recognition by T cells and facilitates tumor evasion.
mLPAs, on the other hand, do not change their structure and remain abundant in leukemia cells.
Furthermore, Dr De Libero and his colleagues found they could isolate the T cells that recognize and kill mLPA-expressing leukemia cells in in vitro tests.
And when the team transplanted the T cells into mice, the cells displayed therapeutic efficacy against leukemia cells.
The researchers noted that this type of immunotherapy, if proven effective in humans, could be used to help prevent leukemia relapse after chemotherapy or stem cell transplant. ![]()
Scientists say they’ve identified a tumor-associated lipid that successfully stimulates T cells to kill leukemia cells in vitro and in vivo.
The team noted that certain T cells can recognize the presentation of self-derived lipids on the CD1c protein.
These T cells are commonly found in healthy individuals and are known to kill transformed hematopoietic cells.
However, their antigen specificity and potential antileukemic effects have not been well characterized.
So Gennaro De Libero, MD, PhD, of the University Hospital Basel in Switzerland, and his colleagues analyzed these cells and reported their findings in The Journal of Experimental Medicine.
The researchers discovered that CD1c self-reactive T cells recognize a novel class of self-lipids called methyl-lysophosphatidic acids (mLPAs), which were abundant in several human leukemias.
These mLPAs are the first example of lipid tumor-associated antigens (TAAs). TAAs are produced by tumors and stimulate T cells that can kill leukemia cells.
However, protein TAA accumulation can be drastically reduced by variant leukemia cells. And some TAAs may change their structure, which prevents recognition by T cells and facilitates tumor evasion.
mLPAs, on the other hand, do not change their structure and remain abundant in leukemia cells.
Furthermore, Dr De Libero and his colleagues found they could isolate the T cells that recognize and kill mLPA-expressing leukemia cells in in vitro tests.
And when the team transplanted the T cells into mice, the cells displayed therapeutic efficacy against leukemia cells.
The researchers noted that this type of immunotherapy, if proven effective in humans, could be used to help prevent leukemia relapse after chemotherapy or stem cell transplant. ![]()
Later-life PTSD boosts vascular risk, study finds
ORLANDO – Military veterans aged 55 years or older with current posttraumatic stress disorder are at significantly higher risk of developing new-onset vascular disease than are those without PTSD, according to a very large national longitudinal study.
"This study suggests the need for greater monitoring and treatment of PTSD in older veterans to assist in the prevention of vascular disorders," Amy L. Byers, Ph.D., said at the annual meeting of the American Association for Geriatric Psychiatry.
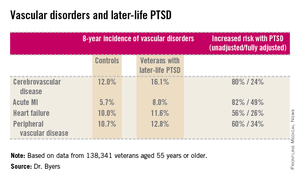
She reported on 138,341 veterans aged 55 years or older who were free of known vascular disease at baseline. During 8 years of follow-up, those with PTSD had significantly higher rates of incident cerebrovascular disease, acute MI, heart failure, and peripheral vascular disease than did those without PTSD, even after adjustment for demographics, comorbid diabetes, hypertension, cancer, chronic obstructive pulmonary disease, renal disease, traumatic brain injury, dementia, substance use disorders, and psychiatric diagnoses. The fully adjusted increased risk of each of the forms of vascular disease under study still remained significant at P less than .001, noted Dr. Byers, an epidemiologist in the psychiatry department at the University of California, San Francisco.
In a separate study led by Dr. Byers, PTSD in the general population with onset prior to and persistence beyond age 55 was a powerful independent predictor of global disability.
Dr. Byers’ study of older veterans was funded by the Department of Defense. She had no disclosures.
This paper continues to strengthen a link between PTSD and inflammatory markers. Dewleen Baker of the VA health care system in San Diego reported that there was a 10-fold increase in C-reactive protein (CRP) post deployment as compared with these same soldiers predeployment CRP levels. After adjustment for battlefield experience scores and combat exposures, those patients with PTSD symptoms had elevated CRP levels of 1.0 ng/mL versus 0.7 ng/mL without postdeployment symptoms (JAMA Psychiatry. 2014;71:423-31). So it seems that there may be a link between PTSD negative cardiovascular outcomes. And there may be a link between PTSD and elevated CRP. So, this leaves us with at least two questions: Is elevated CRP related to increased incidence of negative cardiovascular outcomes? And, which came first, the chicken (PTSD) or the egg (elevated CRP)?
Dr. Mark A. Adelman is chief of vascular and endovascular surgery at NYU Langone Medical Center, New York, and an associate medical editor for Vascular Specialist.
This paper continues to strengthen a link between PTSD and inflammatory markers. Dewleen Baker of the VA health care system in San Diego reported that there was a 10-fold increase in C-reactive protein (CRP) post deployment as compared with these same soldiers predeployment CRP levels. After adjustment for battlefield experience scores and combat exposures, those patients with PTSD symptoms had elevated CRP levels of 1.0 ng/mL versus 0.7 ng/mL without postdeployment symptoms (JAMA Psychiatry. 2014;71:423-31). So it seems that there may be a link between PTSD negative cardiovascular outcomes. And there may be a link between PTSD and elevated CRP. So, this leaves us with at least two questions: Is elevated CRP related to increased incidence of negative cardiovascular outcomes? And, which came first, the chicken (PTSD) or the egg (elevated CRP)?
Dr. Mark A. Adelman is chief of vascular and endovascular surgery at NYU Langone Medical Center, New York, and an associate medical editor for Vascular Specialist.
This paper continues to strengthen a link between PTSD and inflammatory markers. Dewleen Baker of the VA health care system in San Diego reported that there was a 10-fold increase in C-reactive protein (CRP) post deployment as compared with these same soldiers predeployment CRP levels. After adjustment for battlefield experience scores and combat exposures, those patients with PTSD symptoms had elevated CRP levels of 1.0 ng/mL versus 0.7 ng/mL without postdeployment symptoms (JAMA Psychiatry. 2014;71:423-31). So it seems that there may be a link between PTSD negative cardiovascular outcomes. And there may be a link between PTSD and elevated CRP. So, this leaves us with at least two questions: Is elevated CRP related to increased incidence of negative cardiovascular outcomes? And, which came first, the chicken (PTSD) or the egg (elevated CRP)?
Dr. Mark A. Adelman is chief of vascular and endovascular surgery at NYU Langone Medical Center, New York, and an associate medical editor for Vascular Specialist.
ORLANDO – Military veterans aged 55 years or older with current posttraumatic stress disorder are at significantly higher risk of developing new-onset vascular disease than are those without PTSD, according to a very large national longitudinal study.
"This study suggests the need for greater monitoring and treatment of PTSD in older veterans to assist in the prevention of vascular disorders," Amy L. Byers, Ph.D., said at the annual meeting of the American Association for Geriatric Psychiatry.
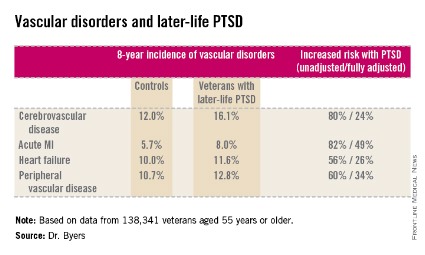
She reported on 138,341 veterans aged 55 years or older who were free of known vascular disease at baseline. During 8 years of follow-up, those with PTSD had significantly higher rates of incident cerebrovascular disease, acute MI, heart failure, and peripheral vascular disease than did those without PTSD, even after adjustment for demographics, comorbid diabetes, hypertension, cancer, chronic obstructive pulmonary disease, renal disease, traumatic brain injury, dementia, substance use disorders, and psychiatric diagnoses. The fully adjusted increased risk of each of the forms of vascular disease under study still remained significant at P less than .001, noted Dr. Byers, an epidemiologist in the psychiatry department at the University of California, San Francisco.
In a separate study led by Dr. Byers, PTSD in the general population with onset prior to and persistence beyond age 55 was a powerful independent predictor of global disability.
Dr. Byers’ study of older veterans was funded by the Department of Defense. She had no disclosures.
ORLANDO – Military veterans aged 55 years or older with current posttraumatic stress disorder are at significantly higher risk of developing new-onset vascular disease than are those without PTSD, according to a very large national longitudinal study.
"This study suggests the need for greater monitoring and treatment of PTSD in older veterans to assist in the prevention of vascular disorders," Amy L. Byers, Ph.D., said at the annual meeting of the American Association for Geriatric Psychiatry.
She reported on 138,341 veterans aged 55 years or older who were free of known vascular disease at baseline. During 8 years of follow-up, those with PTSD had significantly higher rates of incident cerebrovascular disease, acute MI, heart failure, and peripheral vascular disease than did those without PTSD, even after adjustment for demographics, comorbid diabetes, hypertension, cancer, chronic obstructive pulmonary disease, renal disease, traumatic brain injury, dementia, substance use disorders, and psychiatric diagnoses. The fully adjusted increased risk of each of the forms of vascular disease under study still remained significant at P less than .001, noted Dr. Byers, an epidemiologist in the psychiatry department at the University of California, San Francisco.
In a separate study led by Dr. Byers, PTSD in the general population with onset prior to and persistence beyond age 55 was a powerful independent predictor of global disability.
Dr. Byers’ study of older veterans was funded by the Department of Defense. She had no disclosures.
Major finding: Military veterans with late-life posttraumatic stress disorder were 80% more likely to develop new-onset cerebrovascular disease during 8 years of follow-up than were those without PTSD. They were also 82% more likely to have a first acute myocardial infarction, 56% more likely to develop heart failure, and 60% more likely to be diagnosed with peripheral vascular disease.
Data source: This was a longitudinal observational study in 138,341 veterans aged 55 years or older who were free of known vascular disease at baseline and were followed for 8 years.
Disclosures: Dr. Byers’ study of older veterans was funded by the Department of Defense. She reported having no financial conflicts.
Another surgeon’s error – Must you tell the patient?
YES: Surgeons have a duty to tell patients when a medical error has been made by a physician colleague.
Surgeons have a moral and ethical obligation to inform a patient when a medical error has occurred, including cases when the error was made by another surgeon.

Principles that support complete and honest disclosure to the patient and/or the patient’s family in such cases include professional obligation on the part of both the surgeon who made the error and the surgeon who discovered the error, the integrity of both surgeons, the patient’s right to informed care throughout the continuum of care, and the patient’s right to informed consent.
With respect to the first, the American Medical Association’s code of ethics provides a framework for disclosure; it clearly states that situations occur in which a patient experiences significant complications that may have resulted from a physician’s mistake or judgment and that the physician is ethically required to inform the patient of all facts necessary to ensure understanding of the error that occurred.
The American College of Physicians’ ethics manual also states that physicians should disclose to patients information about procedural or judgment errors made during the course of care, as long as that information is pertinent and material to the patient’s well-being.
Errors do not necessarily imply negligence or unethical behavior, but failure to disclose may.

As for patients’ rights, I think that patients are entitled to honest information. They shouldn’t bear the burden of determining how they came to be in another surgeon’s care.
Patients with complications may have impactful financial burdens that result from the additional treatment that is needed, and without all pertinent information, they may have difficulty understanding the benefits, such as deferment of payments, to which they are entitled.
The patient must also be kept informed as to the long-term care plan, and honest and timely disclosure will facilitate moving beyond blame and toward patient advocacy.
The patient is entitled to informed consent, and this requires an understanding of the conditions under which they arrived in another surgeon’s care. If a second procedure is required, the patient must be made aware of potential complications – including how the effects of the initial error might impact outcomes.
Although surgeons have an ethical obligation to disclose errors made by another surgeon, this is a difficult task. Pressures from society and medical professionals can make disclosure difficult, but the benefits of disclosure are real; studies show that open, honest communication improves patient satisfaction, strengthens the physician-patient relationship, and can improve outcomes.
Disclosure also has the potential to improve the well-being of the surgeons involved, through relieving feelings of guilt, and satisfying the need to fulfill one’s obligations. Furthermore, data suggest that error disclosure reduces long-term litigation and costs. Admittedly, however, there are little data on how disclosure of another surgeon’s errors ultimately reduces litigation and costs.
Ultimately, supporting a just culture allows us to emphasize the importance of disclosing errors and to be accountable in setting a standard that involves exploring errors rather than ignoring them; it must be remembered, though, that this process of disclosure involves obtaining facts to help both surgeons and patients understand what truly happened.
Surgeon-to-surgeon discussions can be productive and can facilitate disclosure. However, if the doctor who made the error declines to be part of the disclosure process, one still has an obligation to disclose the error and to answer the patient’s questions honestly.
This approach requires a commitment to support surgeons in their efforts to promote transparency, and it requires a clear understanding of our obligations and the role of disclosure during training; we need to engage medical students and residents.
Dr. Susan D. Moffatt-Bruce is an associate professor at the Ohio State University, Columbus.
NO: Surgeons are not required to inform a patient of another physician’s possible error.
Surgeons do not have an obligation to disclose to a patient another surgeon’s possible medical error.

A consensus has been reached in medicine about our ethical duty to inform patients about our own medical errors. Although nondisclosure has previously been rationalized by concerns about invoking anxiety or confusion in the patient, this approach has largely been discredited; disclosure preserves patient trust and bolsters the physician-patient relationship.
However, it is an entirely different story when it comes to disclosing another surgeon’s mistake – a situation that is quite common. A recent survey showed that two-thirds of respondents had encountered a similar dilemma in the past 6 months (Qual. Saf. Health Care 2009;18:209-12).
The approach that physicians have previously taken when faced with this dilemma is an important measure of what they believe represents an ethical or just response. A poll of many of my colleagues across the country and at my own institution suggests that the preferred approach is to provide appropriate care for the patient and to answer their questions honestly, but to not proactively disclose the perceived medical error.
In fact, this was the preferred approach of every surgeon who responded.
A recent article in the New England Journal of Medicine addressed this very topic. The authors noted that there is little guidance available regarding the reporting of another physician’s error (2013;369:1752-7).
Among the challenges inherent in disclosing another’s mistake is the difficulty in determining exactly what happened. Uncertainty inevitably exists regarding the conversations that took place between the patient and the surgeon, and also about what actually defines a medical error. Incidents regarded as medical errors may comprise a large spectrum, ranging from "not what I would have done – but within the standard of care," to "blatant negligence."
Several studies suggest that highly trained physicians and surgeons routinely disagree about whether negligence has occurred in a given case. In one study, two reviewers disagreed 38% of the time as to whether appropriate care was provided.
Physicians have difficulty judging if the standard of care has been met. Therefore, it is not acceptable for each of us to assume we are the medical expert capable of rendering an opinion of whether previous care was appropriate and informing patients of our opinion.
Physicians overwhelmingly report that in the event they are responsible for a medical error discovered by another physician, they would prefer that the physician come to them first to discuss the matter. In fact, 93% of 400 respondents in one survey reported this preference.
The most acceptable approach when dealing with a peer’s medical error is to discuss the error with the responsible physician and to encourage the physician to disclose any error with the patient.
If there is disagreement as to whether an error occurred, institutional guidance should be applied. Only a collaborative approach can appropriately meet the needs of the patient and family after harmful medical errors.
Dr. Chadrick E. Denlinger is an associate professor at the Medical University of South Carolina, Charleston.
YES: Surgeons have a duty to tell patients when a medical error has been made by a physician colleague.
Surgeons have a moral and ethical obligation to inform a patient when a medical error has occurred, including cases when the error was made by another surgeon.
Principles that support complete and honest disclosure to the patient and/or the patient’s family in such cases include professional obligation on the part of both the surgeon who made the error and the surgeon who discovered the error, the integrity of both surgeons, the patient’s right to informed care throughout the continuum of care, and the patient’s right to informed consent.
With respect to the first, the American Medical Association’s code of ethics provides a framework for disclosure; it clearly states that situations occur in which a patient experiences significant complications that may have resulted from a physician’s mistake or judgment and that the physician is ethically required to inform the patient of all facts necessary to ensure understanding of the error that occurred.
The American College of Physicians’ ethics manual also states that physicians should disclose to patients information about procedural or judgment errors made during the course of care, as long as that information is pertinent and material to the patient’s well-being.
Errors do not necessarily imply negligence or unethical behavior, but failure to disclose may.
As for patients’ rights, I think that patients are entitled to honest information. They shouldn’t bear the burden of determining how they came to be in another surgeon’s care.
Patients with complications may have impactful financial burdens that result from the additional treatment that is needed, and without all pertinent information, they may have difficulty understanding the benefits, such as deferment of payments, to which they are entitled.
The patient must also be kept informed as to the long-term care plan, and honest and timely disclosure will facilitate moving beyond blame and toward patient advocacy.
The patient is entitled to informed consent, and this requires an understanding of the conditions under which they arrived in another surgeon’s care. If a second procedure is required, the patient must be made aware of potential complications – including how the effects of the initial error might impact outcomes.
Although surgeons have an ethical obligation to disclose errors made by another surgeon, this is a difficult task. Pressures from society and medical professionals can make disclosure difficult, but the benefits of disclosure are real; studies show that open, honest communication improves patient satisfaction, strengthens the physician-patient relationship, and can improve outcomes.
Disclosure also has the potential to improve the well-being of the surgeons involved, through relieving feelings of guilt, and satisfying the need to fulfill one’s obligations. Furthermore, data suggest that error disclosure reduces long-term litigation and costs. Admittedly, however, there are little data on how disclosure of another surgeon’s errors ultimately reduces litigation and costs.
Ultimately, supporting a just culture allows us to emphasize the importance of disclosing errors and to be accountable in setting a standard that involves exploring errors rather than ignoring them; it must be remembered, though, that this process of disclosure involves obtaining facts to help both surgeons and patients understand what truly happened.
Surgeon-to-surgeon discussions can be productive and can facilitate disclosure. However, if the doctor who made the error declines to be part of the disclosure process, one still has an obligation to disclose the error and to answer the patient’s questions honestly.
This approach requires a commitment to support surgeons in their efforts to promote transparency, and it requires a clear understanding of our obligations and the role of disclosure during training; we need to engage medical students and residents.
Dr. Susan D. Moffatt-Bruce is an associate professor at the Ohio State University, Columbus.
NO: Surgeons are not required to inform a patient of another physician’s possible error.
Surgeons do not have an obligation to disclose to a patient another surgeon’s possible medical error.
A consensus has been reached in medicine about our ethical duty to inform patients about our own medical errors. Although nondisclosure has previously been rationalized by concerns about invoking anxiety or confusion in the patient, this approach has largely been discredited; disclosure preserves patient trust and bolsters the physician-patient relationship.
However, it is an entirely different story when it comes to disclosing another surgeon’s mistake – a situation that is quite common. A recent survey showed that two-thirds of respondents had encountered a similar dilemma in the past 6 months (Qual. Saf. Health Care 2009;18:209-12).
The approach that physicians have previously taken when faced with this dilemma is an important measure of what they believe represents an ethical or just response. A poll of many of my colleagues across the country and at my own institution suggests that the preferred approach is to provide appropriate care for the patient and to answer their questions honestly, but to not proactively disclose the perceived medical error.
In fact, this was the preferred approach of every surgeon who responded.
A recent article in the New England Journal of Medicine addressed this very topic. The authors noted that there is little guidance available regarding the reporting of another physician’s error (2013;369:1752-7).
Among the challenges inherent in disclosing another’s mistake is the difficulty in determining exactly what happened. Uncertainty inevitably exists regarding the conversations that took place between the patient and the surgeon, and also about what actually defines a medical error. Incidents regarded as medical errors may comprise a large spectrum, ranging from "not what I would have done – but within the standard of care," to "blatant negligence."
Several studies suggest that highly trained physicians and surgeons routinely disagree about whether negligence has occurred in a given case. In one study, two reviewers disagreed 38% of the time as to whether appropriate care was provided.
Physicians have difficulty judging if the standard of care has been met. Therefore, it is not acceptable for each of us to assume we are the medical expert capable of rendering an opinion of whether previous care was appropriate and informing patients of our opinion.
Physicians overwhelmingly report that in the event they are responsible for a medical error discovered by another physician, they would prefer that the physician come to them first to discuss the matter. In fact, 93% of 400 respondents in one survey reported this preference.
The most acceptable approach when dealing with a peer’s medical error is to discuss the error with the responsible physician and to encourage the physician to disclose any error with the patient.
If there is disagreement as to whether an error occurred, institutional guidance should be applied. Only a collaborative approach can appropriately meet the needs of the patient and family after harmful medical errors.
Dr. Chadrick E. Denlinger is an associate professor at the Medical University of South Carolina, Charleston.
YES: Surgeons have a duty to tell patients when a medical error has been made by a physician colleague.
Surgeons have a moral and ethical obligation to inform a patient when a medical error has occurred, including cases when the error was made by another surgeon.
Principles that support complete and honest disclosure to the patient and/or the patient’s family in such cases include professional obligation on the part of both the surgeon who made the error and the surgeon who discovered the error, the integrity of both surgeons, the patient’s right to informed care throughout the continuum of care, and the patient’s right to informed consent.
With respect to the first, the American Medical Association’s code of ethics provides a framework for disclosure; it clearly states that situations occur in which a patient experiences significant complications that may have resulted from a physician’s mistake or judgment and that the physician is ethically required to inform the patient of all facts necessary to ensure understanding of the error that occurred.
The American College of Physicians’ ethics manual also states that physicians should disclose to patients information about procedural or judgment errors made during the course of care, as long as that information is pertinent and material to the patient’s well-being.
Errors do not necessarily imply negligence or unethical behavior, but failure to disclose may.
As for patients’ rights, I think that patients are entitled to honest information. They shouldn’t bear the burden of determining how they came to be in another surgeon’s care.
Patients with complications may have impactful financial burdens that result from the additional treatment that is needed, and without all pertinent information, they may have difficulty understanding the benefits, such as deferment of payments, to which they are entitled.
The patient must also be kept informed as to the long-term care plan, and honest and timely disclosure will facilitate moving beyond blame and toward patient advocacy.
The patient is entitled to informed consent, and this requires an understanding of the conditions under which they arrived in another surgeon’s care. If a second procedure is required, the patient must be made aware of potential complications – including how the effects of the initial error might impact outcomes.
Although surgeons have an ethical obligation to disclose errors made by another surgeon, this is a difficult task. Pressures from society and medical professionals can make disclosure difficult, but the benefits of disclosure are real; studies show that open, honest communication improves patient satisfaction, strengthens the physician-patient relationship, and can improve outcomes.
Disclosure also has the potential to improve the well-being of the surgeons involved, through relieving feelings of guilt, and satisfying the need to fulfill one’s obligations. Furthermore, data suggest that error disclosure reduces long-term litigation and costs. Admittedly, however, there are little data on how disclosure of another surgeon’s errors ultimately reduces litigation and costs.
Ultimately, supporting a just culture allows us to emphasize the importance of disclosing errors and to be accountable in setting a standard that involves exploring errors rather than ignoring them; it must be remembered, though, that this process of disclosure involves obtaining facts to help both surgeons and patients understand what truly happened.
Surgeon-to-surgeon discussions can be productive and can facilitate disclosure. However, if the doctor who made the error declines to be part of the disclosure process, one still has an obligation to disclose the error and to answer the patient’s questions honestly.
This approach requires a commitment to support surgeons in their efforts to promote transparency, and it requires a clear understanding of our obligations and the role of disclosure during training; we need to engage medical students and residents.
Dr. Susan D. Moffatt-Bruce is an associate professor at the Ohio State University, Columbus.
NO: Surgeons are not required to inform a patient of another physician’s possible error.
Surgeons do not have an obligation to disclose to a patient another surgeon’s possible medical error.
A consensus has been reached in medicine about our ethical duty to inform patients about our own medical errors. Although nondisclosure has previously been rationalized by concerns about invoking anxiety or confusion in the patient, this approach has largely been discredited; disclosure preserves patient trust and bolsters the physician-patient relationship.
However, it is an entirely different story when it comes to disclosing another surgeon’s mistake – a situation that is quite common. A recent survey showed that two-thirds of respondents had encountered a similar dilemma in the past 6 months (Qual. Saf. Health Care 2009;18:209-12).
The approach that physicians have previously taken when faced with this dilemma is an important measure of what they believe represents an ethical or just response. A poll of many of my colleagues across the country and at my own institution suggests that the preferred approach is to provide appropriate care for the patient and to answer their questions honestly, but to not proactively disclose the perceived medical error.
In fact, this was the preferred approach of every surgeon who responded.
A recent article in the New England Journal of Medicine addressed this very topic. The authors noted that there is little guidance available regarding the reporting of another physician’s error (2013;369:1752-7).
Among the challenges inherent in disclosing another’s mistake is the difficulty in determining exactly what happened. Uncertainty inevitably exists regarding the conversations that took place between the patient and the surgeon, and also about what actually defines a medical error. Incidents regarded as medical errors may comprise a large spectrum, ranging from "not what I would have done – but within the standard of care," to "blatant negligence."
Several studies suggest that highly trained physicians and surgeons routinely disagree about whether negligence has occurred in a given case. In one study, two reviewers disagreed 38% of the time as to whether appropriate care was provided.
Physicians have difficulty judging if the standard of care has been met. Therefore, it is not acceptable for each of us to assume we are the medical expert capable of rendering an opinion of whether previous care was appropriate and informing patients of our opinion.
Physicians overwhelmingly report that in the event they are responsible for a medical error discovered by another physician, they would prefer that the physician come to them first to discuss the matter. In fact, 93% of 400 respondents in one survey reported this preference.
The most acceptable approach when dealing with a peer’s medical error is to discuss the error with the responsible physician and to encourage the physician to disclose any error with the patient.
If there is disagreement as to whether an error occurred, institutional guidance should be applied. Only a collaborative approach can appropriately meet the needs of the patient and family after harmful medical errors.
Dr. Chadrick E. Denlinger is an associate professor at the Medical University of South Carolina, Charleston.

Paclitaxel-eluting balloon shows high claudication efficacy
PARIS – A drug-eluting balloon produced a stentlike rate of primary patency and need for target vessel revascularization in a multinational, controlled trial with 331 patients with claudication.
The results showed that the paclitaxel-eluting angioplasty balloon used in the study, the IN.PACT model made by Medtronic, has the "potential to become the standard of care" for treating stenoses in the superficial femoral and popliteal arteries, Dr. Marianne Brodmann said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions. After 1 year, the rate of clinically driven target-vessel revascularizations was 2% in the 220 patients treated with the drug-eluting balloon and 21% in 111 control patients treated with plain balloon angioplasty, a statistically significant difference, reported Dr. Brodmann, professor of angiology at the Medical University of Graz (Austria).*

The results seen in this trial contrast with results from studies of other types of drug-eluting balloons in these arteries, said Dr. Marc Bosiers, head of the department of vascular surgery at St. Blasius Hospital in Dendermonde, Belgium, and a coinvestigator in the study. "What we’ve learned from this trial, if you look at the results from other trials [of drug-eluting balloons], is that not all drug-eluting balloons are equal, just as not all stents are equal," Dr. Bosiers said.
The IN.PACT SFA Trial enrolled 150 patients at 13 centers in Europe and 181 patients at 44 U.S. centers. All patients were adults with Rutherford stage 2, 3, or 4 disease; claudication and rest pain; and a single or closely tandem lesion in the superficial femoral or popliteal arteries with a total length of no more than 18 cm. Their average age was 68, and about 40% had diabetes. The trial protocol allowed provisional stenting, which occurred in 7% of the patients treated with a paclitaxel-eluting balloon and in 13% of those treated with a plain balloon. The average lesion length treated was about 9 cm in both arms of the study.
The study’s primary endpoint was the rate of primary patency at 12 months, defined as freedom from clinically driven target-vessel revascularization and freedom from restenosis assessed by Doppler ultrasound at 12 months, which was 82% in patients treated with the drug-eluting balloon and 52% among patients in the control arm, a statistically significant difference.
The study’s primary safety endpoint was the combined rate of procedure- and device-related death at 30 days, freedom from target-limb major amputation at 1 year, and freedom from clinically driven target-vessel revascularization at 1 year, which occurred in 96% of patients treated with the paclitaxel-eluting balloon and in 77% of the control patients, a statistically significant difference.
These outcomes included "the lowest target-vessel revascularization rates and the highest patency rates ever reported" in this setting, and provide "robust, level 1 evidence" for the safety and efficacy of the paclitaxel-eluting balloon for this indication, Dr. Brodmann concluded.
If restenosis were to occur in the target vessel following treatment with the paclitaxel-eluting balloon, it would be possible to retreat the same vessel with a second paclitaxel-eluting balloon, although that scenario was not tested in the trial, Dr. Brodmann said in an interview. The paclitaxel essentially disappears within a few months of treatment, which should allow safe retreatment.
A written statement from Medtronic said that the company has an application pending with the Food and Drug Administration for U.S. marketing approval of the IN.PACT balloon for this indication. The balloon has been available in Europe since 2009.
The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said that he had no disclosures.
On Twitter @mitchelzoler
*Correction, 6/24/2014: A previous version of this article misstated the number of patients in the control arm, the number of study centers, the name of the study, and two references to the device.

|
Mitchel L. Zoler/Frontline Medical News
|
The results from this trial change the way we think about treating stenoses in the superficial femoral and popliteal arteries. These results are probably the first to show with such robust, level 1 evidence that a drug-eluting balloon works at least as well as the best stent available today.
The results mean that the concept of "leave nothing behind" when treating vascular disease in the superficial femoral artery will be the best approach going forward. The 82% 1-year patency rate and the 2.4% rate of clinically driven target-vessel revascularizations were absolutely outstanding results.
Dr. Alberto Cremonesi, director of the interventional cardioangiology unit at Villa Maria Cecilia Hospital in Cotignola-Ravenna, Italy, made these comments in an interview. He said he had no relevant financial disclosures.
|
|
Mitchel L. Zoler/Frontline Medical News
|
The results from this trial change the way we think about treating stenoses in the superficial femoral and popliteal arteries. These results are probably the first to show with such robust, level 1 evidence that a drug-eluting balloon works at least as well as the best stent available today.
The results mean that the concept of "leave nothing behind" when treating vascular disease in the superficial femoral artery will be the best approach going forward. The 82% 1-year patency rate and the 2.4% rate of clinically driven target-vessel revascularizations were absolutely outstanding results.
Dr. Alberto Cremonesi, director of the interventional cardioangiology unit at Villa Maria Cecilia Hospital in Cotignola-Ravenna, Italy, made these comments in an interview. He said he had no relevant financial disclosures.
|
|
Mitchel L. Zoler/Frontline Medical News
|
The results from this trial change the way we think about treating stenoses in the superficial femoral and popliteal arteries. These results are probably the first to show with such robust, level 1 evidence that a drug-eluting balloon works at least as well as the best stent available today.
The results mean that the concept of "leave nothing behind" when treating vascular disease in the superficial femoral artery will be the best approach going forward. The 82% 1-year patency rate and the 2.4% rate of clinically driven target-vessel revascularizations were absolutely outstanding results.
Dr. Alberto Cremonesi, director of the interventional cardioangiology unit at Villa Maria Cecilia Hospital in Cotignola-Ravenna, Italy, made these comments in an interview. He said he had no relevant financial disclosures.
PARIS – A drug-eluting balloon produced a stentlike rate of primary patency and need for target vessel revascularization in a multinational, controlled trial with 331 patients with claudication.
The results showed that the paclitaxel-eluting angioplasty balloon used in the study, the IN.PACT model made by Medtronic, has the "potential to become the standard of care" for treating stenoses in the superficial femoral and popliteal arteries, Dr. Marianne Brodmann said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions. After 1 year, the rate of clinically driven target-vessel revascularizations was 2% in the 220 patients treated with the drug-eluting balloon and 21% in 111 control patients treated with plain balloon angioplasty, a statistically significant difference, reported Dr. Brodmann, professor of angiology at the Medical University of Graz (Austria).*
The results seen in this trial contrast with results from studies of other types of drug-eluting balloons in these arteries, said Dr. Marc Bosiers, head of the department of vascular surgery at St. Blasius Hospital in Dendermonde, Belgium, and a coinvestigator in the study. "What we’ve learned from this trial, if you look at the results from other trials [of drug-eluting balloons], is that not all drug-eluting balloons are equal, just as not all stents are equal," Dr. Bosiers said.
The IN.PACT SFA Trial enrolled 150 patients at 13 centers in Europe and 181 patients at 44 U.S. centers. All patients were adults with Rutherford stage 2, 3, or 4 disease; claudication and rest pain; and a single or closely tandem lesion in the superficial femoral or popliteal arteries with a total length of no more than 18 cm. Their average age was 68, and about 40% had diabetes. The trial protocol allowed provisional stenting, which occurred in 7% of the patients treated with a paclitaxel-eluting balloon and in 13% of those treated with a plain balloon. The average lesion length treated was about 9 cm in both arms of the study.
The study’s primary endpoint was the rate of primary patency at 12 months, defined as freedom from clinically driven target-vessel revascularization and freedom from restenosis assessed by Doppler ultrasound at 12 months, which was 82% in patients treated with the drug-eluting balloon and 52% among patients in the control arm, a statistically significant difference.
The study’s primary safety endpoint was the combined rate of procedure- and device-related death at 30 days, freedom from target-limb major amputation at 1 year, and freedom from clinically driven target-vessel revascularization at 1 year, which occurred in 96% of patients treated with the paclitaxel-eluting balloon and in 77% of the control patients, a statistically significant difference.
These outcomes included "the lowest target-vessel revascularization rates and the highest patency rates ever reported" in this setting, and provide "robust, level 1 evidence" for the safety and efficacy of the paclitaxel-eluting balloon for this indication, Dr. Brodmann concluded.
If restenosis were to occur in the target vessel following treatment with the paclitaxel-eluting balloon, it would be possible to retreat the same vessel with a second paclitaxel-eluting balloon, although that scenario was not tested in the trial, Dr. Brodmann said in an interview. The paclitaxel essentially disappears within a few months of treatment, which should allow safe retreatment.
A written statement from Medtronic said that the company has an application pending with the Food and Drug Administration for U.S. marketing approval of the IN.PACT balloon for this indication. The balloon has been available in Europe since 2009.
The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said that he had no disclosures.
On Twitter @mitchelzoler
*Correction, 6/24/2014: A previous version of this article misstated the number of patients in the control arm, the number of study centers, the name of the study, and two references to the device.
PARIS – A drug-eluting balloon produced a stentlike rate of primary patency and need for target vessel revascularization in a multinational, controlled trial with 331 patients with claudication.
The results showed that the paclitaxel-eluting angioplasty balloon used in the study, the IN.PACT model made by Medtronic, has the "potential to become the standard of care" for treating stenoses in the superficial femoral and popliteal arteries, Dr. Marianne Brodmann said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions. After 1 year, the rate of clinically driven target-vessel revascularizations was 2% in the 220 patients treated with the drug-eluting balloon and 21% in 111 control patients treated with plain balloon angioplasty, a statistically significant difference, reported Dr. Brodmann, professor of angiology at the Medical University of Graz (Austria).*
The results seen in this trial contrast with results from studies of other types of drug-eluting balloons in these arteries, said Dr. Marc Bosiers, head of the department of vascular surgery at St. Blasius Hospital in Dendermonde, Belgium, and a coinvestigator in the study. "What we’ve learned from this trial, if you look at the results from other trials [of drug-eluting balloons], is that not all drug-eluting balloons are equal, just as not all stents are equal," Dr. Bosiers said.
The IN.PACT SFA Trial enrolled 150 patients at 13 centers in Europe and 181 patients at 44 U.S. centers. All patients were adults with Rutherford stage 2, 3, or 4 disease; claudication and rest pain; and a single or closely tandem lesion in the superficial femoral or popliteal arteries with a total length of no more than 18 cm. Their average age was 68, and about 40% had diabetes. The trial protocol allowed provisional stenting, which occurred in 7% of the patients treated with a paclitaxel-eluting balloon and in 13% of those treated with a plain balloon. The average lesion length treated was about 9 cm in both arms of the study.
The study’s primary endpoint was the rate of primary patency at 12 months, defined as freedom from clinically driven target-vessel revascularization and freedom from restenosis assessed by Doppler ultrasound at 12 months, which was 82% in patients treated with the drug-eluting balloon and 52% among patients in the control arm, a statistically significant difference.
The study’s primary safety endpoint was the combined rate of procedure- and device-related death at 30 days, freedom from target-limb major amputation at 1 year, and freedom from clinically driven target-vessel revascularization at 1 year, which occurred in 96% of patients treated with the paclitaxel-eluting balloon and in 77% of the control patients, a statistically significant difference.
These outcomes included "the lowest target-vessel revascularization rates and the highest patency rates ever reported" in this setting, and provide "robust, level 1 evidence" for the safety and efficacy of the paclitaxel-eluting balloon for this indication, Dr. Brodmann concluded.
If restenosis were to occur in the target vessel following treatment with the paclitaxel-eluting balloon, it would be possible to retreat the same vessel with a second paclitaxel-eluting balloon, although that scenario was not tested in the trial, Dr. Brodmann said in an interview. The paclitaxel essentially disappears within a few months of treatment, which should allow safe retreatment.
A written statement from Medtronic said that the company has an application pending with the Food and Drug Administration for U.S. marketing approval of the IN.PACT balloon for this indication. The balloon has been available in Europe since 2009.
The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said that he had no disclosures.
On Twitter @mitchelzoler
*Correction, 6/24/2014: A previous version of this article misstated the number of patients in the control arm, the number of study centers, the name of the study, and two references to the device.
AT EUROPCR 2014
Key clinical point: A drug-eluting balloon produced stentlike patency after 1 year in superficial femoral and popliteal arteries.
Major finding: Angioplasty with a paclitaxel-eluting balloon produced a 1-year 82% primary patency rate, compared with 52% in controls.
Data source: A multicenter, randomized controlled trial with 331 patients with claudication and rest pain treated at 57 international sites.
Disclosures: The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said he had no disclosures.

Repeat biopsy and long-term surveillance key for rare Hodgkin’s lymphoma subtype
Time to progression was inferior in patients with advanced-stage nodular lymphocyte-predominant Hodgkin’s lymphoma, compared with patients with classical Hodgkin’s lymphoma, in a study that compared outcomes between the two groups of Hodgkin’s lymphoma patients enrolled in the British Columbia Cancer Agency database.
Over 10 years, time to progression was 63% in the nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) group, vs. 73% in the classical Hodgkin’s lymphoma (CHL) group (P =.040), reported Dr. Katharine Xing of the Centre for Lymphoid Cancer at the BCCA and the University of British Columbia, Vancouver, and her associates.
Transformation to an aggressive non–Hodgkin’s lymphoma (NHL) over 15 years occurred in 24% of those with NLPHL, but in none of those with CHL (P = .00018), and the median time to transformation among those with NLPHL was 5.45 years (Blood 2014;123:3567-73).
The study compared 42 patients with advanced-stage NLPHL to 84 controls with advanced CHL, matched for age, sex, decade of diagnosis, stage, and chemotherapy type; all had been diagnosed between 1970 and 2011. Their mean age was 37 years, about two-thirds were men, most in both groups had stage III disease, and they were followed up for a median of about 11 years. Treatments included standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and most received standard ABVD or ABVD-equivalent chemotherapy. The study was conducted to "highlight the distinct natural history of this rare HL subtype," which accounts for 5% of HL cases, the authors noted.
Over 10 years, "HL freedom from treatment failure," which reflects only relapses from HL, was 75% among those with NLPHL and 73% among those with CHL. Overall survival was also similar between the two groups (83.5% among those with NLPHL and 81% among those with CHL at 10 years).
Among their other findings was a significantly higher incidence of transformation over 10 years among those who had splenic involvement at the time of NLPHL diagnosis, compared with those who did not have splenic involvement (29% vs. 7.8%). When they looked at only those NLPHL patients who had received ABVD-like treatment, the incidence of transformation over 10 years was 34% among those with splenic involvement at diagnosis, vs. 9% among those who did not have splenic involvement (P = .014).
Since NLPHL is rare, information on the optimal treatment is limited, particularly for those with advanced disease, the authors pointed out. The analysis "highlights the distinct disease behavior of NLPHL, compared with CHL, and the need for repeat biopsy at relapse as well as long-term surveillance," the authors concluded. "Given the strong expression of CD20" on the lymphocyte predominant cells that distinguishes NLPHL from CHL, the results also provide "a rationale for further evaluation" of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with rituximab, they added.
Time to progression was inferior in patients with advanced-stage nodular lymphocyte-predominant Hodgkin’s lymphoma, compared with patients with classical Hodgkin’s lymphoma, in a study that compared outcomes between the two groups of Hodgkin’s lymphoma patients enrolled in the British Columbia Cancer Agency database.
Over 10 years, time to progression was 63% in the nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) group, vs. 73% in the classical Hodgkin’s lymphoma (CHL) group (P =.040), reported Dr. Katharine Xing of the Centre for Lymphoid Cancer at the BCCA and the University of British Columbia, Vancouver, and her associates.
Transformation to an aggressive non–Hodgkin’s lymphoma (NHL) over 15 years occurred in 24% of those with NLPHL, but in none of those with CHL (P = .00018), and the median time to transformation among those with NLPHL was 5.45 years (Blood 2014;123:3567-73).
The study compared 42 patients with advanced-stage NLPHL to 84 controls with advanced CHL, matched for age, sex, decade of diagnosis, stage, and chemotherapy type; all had been diagnosed between 1970 and 2011. Their mean age was 37 years, about two-thirds were men, most in both groups had stage III disease, and they were followed up for a median of about 11 years. Treatments included standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and most received standard ABVD or ABVD-equivalent chemotherapy. The study was conducted to "highlight the distinct natural history of this rare HL subtype," which accounts for 5% of HL cases, the authors noted.
Over 10 years, "HL freedom from treatment failure," which reflects only relapses from HL, was 75% among those with NLPHL and 73% among those with CHL. Overall survival was also similar between the two groups (83.5% among those with NLPHL and 81% among those with CHL at 10 years).
Among their other findings was a significantly higher incidence of transformation over 10 years among those who had splenic involvement at the time of NLPHL diagnosis, compared with those who did not have splenic involvement (29% vs. 7.8%). When they looked at only those NLPHL patients who had received ABVD-like treatment, the incidence of transformation over 10 years was 34% among those with splenic involvement at diagnosis, vs. 9% among those who did not have splenic involvement (P = .014).
Since NLPHL is rare, information on the optimal treatment is limited, particularly for those with advanced disease, the authors pointed out. The analysis "highlights the distinct disease behavior of NLPHL, compared with CHL, and the need for repeat biopsy at relapse as well as long-term surveillance," the authors concluded. "Given the strong expression of CD20" on the lymphocyte predominant cells that distinguishes NLPHL from CHL, the results also provide "a rationale for further evaluation" of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with rituximab, they added.
Time to progression was inferior in patients with advanced-stage nodular lymphocyte-predominant Hodgkin’s lymphoma, compared with patients with classical Hodgkin’s lymphoma, in a study that compared outcomes between the two groups of Hodgkin’s lymphoma patients enrolled in the British Columbia Cancer Agency database.
Over 10 years, time to progression was 63% in the nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) group, vs. 73% in the classical Hodgkin’s lymphoma (CHL) group (P =.040), reported Dr. Katharine Xing of the Centre for Lymphoid Cancer at the BCCA and the University of British Columbia, Vancouver, and her associates.
Transformation to an aggressive non–Hodgkin’s lymphoma (NHL) over 15 years occurred in 24% of those with NLPHL, but in none of those with CHL (P = .00018), and the median time to transformation among those with NLPHL was 5.45 years (Blood 2014;123:3567-73).
The study compared 42 patients with advanced-stage NLPHL to 84 controls with advanced CHL, matched for age, sex, decade of diagnosis, stage, and chemotherapy type; all had been diagnosed between 1970 and 2011. Their mean age was 37 years, about two-thirds were men, most in both groups had stage III disease, and they were followed up for a median of about 11 years. Treatments included standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and most received standard ABVD or ABVD-equivalent chemotherapy. The study was conducted to "highlight the distinct natural history of this rare HL subtype," which accounts for 5% of HL cases, the authors noted.
Over 10 years, "HL freedom from treatment failure," which reflects only relapses from HL, was 75% among those with NLPHL and 73% among those with CHL. Overall survival was also similar between the two groups (83.5% among those with NLPHL and 81% among those with CHL at 10 years).
Among their other findings was a significantly higher incidence of transformation over 10 years among those who had splenic involvement at the time of NLPHL diagnosis, compared with those who did not have splenic involvement (29% vs. 7.8%). When they looked at only those NLPHL patients who had received ABVD-like treatment, the incidence of transformation over 10 years was 34% among those with splenic involvement at diagnosis, vs. 9% among those who did not have splenic involvement (P = .014).
Since NLPHL is rare, information on the optimal treatment is limited, particularly for those with advanced disease, the authors pointed out. The analysis "highlights the distinct disease behavior of NLPHL, compared with CHL, and the need for repeat biopsy at relapse as well as long-term surveillance," the authors concluded. "Given the strong expression of CD20" on the lymphocyte predominant cells that distinguishes NLPHL from CHL, the results also provide "a rationale for further evaluation" of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with rituximab, they added.
FROM BLOOD
Key clinical point: Repeat biopsy and long-term surveillance are necessary in nodular lymphocyte-predominant Hodgkin’s lymphoma.
Major finding: Overall survival was similar between patients with advanced-stage NLPHL and those with advanced-stage CHL, but differences between the two groups included an inferior time to progression among those with NLPHL over 10 years (63% vs 73%).
Data source: The study compared outcomes in 42 patients with advanced-stage NLPHL and 84 matched controls with advanced CHL, who were diagnosed between 1970 and 2011 and were enrolled in a Canadian cancer database.
Disclosures: Fourauthors received research funding from Roche; the remaining seven authors, including the lead author, had no relevant disclosures.
Studies confirm importance of CALR mutation in PMF
MILAN—Two new studies appear to confirm the prognostic significance of CALR mutations in patients with primary myelofibrosis (PMF).
One study showed that PMF patients with mutated CALR had prolonged overall survival (OS) compared to patients with wild-type CALR. And additional subclonal mutations did not impair the positive impact of CALR mutations.
Another study suggested that indels in exon 9 of CALR are founding driver mutations in PMF. These mutations are independent predictors of clinical course, disease progression, and OS.
Both studies were presented at the 19th Congress of the European Hematology Association (EHA).
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented data on CALR mutations in the context of additional mutations (abstract S1355).
And Elisa Rumi, MD, of the University of Pavia in Italy, presented information on mutated CALR and other founding driver mutations in PMF (abstract S1356).
CALR & other subclonal mutations in PMF
To investigate the prognostic role of CALR mutations in relation to additional subclonal mutations, Dr Guglielmelli and her colleagues analyzed 274 samples from PMF patients.
The team genotyped the samples for mutations in 11 genes: JAK2, CALR, MPL, EZH2, ASXL1, SRSF2, IDH1, IDH2, CBL, TET2, and DNMT3A.
Two hundred and fifty-six patients (93.4%) presented with at least 1 somatic mutation, and 104 (38%) presented with at least 2.
The median follow-up was 3.8 years (range, 0.52-29.20 years). Among all patients, the median OS was 12.2 years (range, 5.6-18.8 years). Eighty-four patients died (30.7%), 44 (16.1%) of them due to leukemia.
The presence of CALR mutations was associated with better OS, independent of IPSS and molecular risk categories (hazard ratio [HR] 0.51, P=0.03). But CALR mutations did not impact the risk of progression to acute leukemia.
Among patients with a low- to intermediate-1-risk IPSS score, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR 0.4, P=0.02). Among patients with intermediate-2 to high risk, those with CALR mutations lived a median of 4.2 years, and those without lived a median of 2.6 years (HR=0.5, P=0.09).
Among patients with high molecular risk, those with CALR mutations lived a median of 17.7 years, and those without lived a median of 4.3 years, (HR=0.3, P=0.008). High molecular risk was defined as at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2.
Among patients in the low-molecular-risk group, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR=0.6, P=0.048).
Dr Guglielmelli said these results confirm the association between CALR mutation and favorable outcomes in PMF. They also show that additional subclonal mutations do not impair the positive impact of CALR mutation, thereby reinforcing the idea that CALR-mutated PMF is a distinct entity in terms of prognosis.
Founding driver mutations in PMF
In another presentation at the EHA Congress, Dr Rumi presented data on founding driver mutations in PMF. She and her colleagues analyzed 617 PMF patients, screening them for JAK2 V617F mutations, indels of CALR exon 9, and MPL exon 10 mutations.
The researchers assessed the impact of these mutations on thrombosis, progression to leukemia, and OS.
Their analysis suggested CALR-mutated PMF patients have a lower risk of thrombosis than JAK2-mutated patients (P=0.021). And this difference retained significance after adjusting for age.
Triple-negative PMF patients had a higher risk of leukemic evolution than CALR-mutated patients (P=0.016) and JAK2-mutated patients (P=0.043).
After adjusting for age, the risk remained significantly higher in triple-negative patients compared to JAK2-mutated patients (P=0.04) and retained borderline significance compared to CALR-mutated patients (P=0.052).
Patients with CALR-mutated PMF had a better OS than JAK2-mutated patients (P<0.001), MPL-mutated patients (P=0.009), and triple-negative patients (P<0.001).
In a multivariate analysis, CALR-mutated patients maintained a better OS than JAK2-mutated patients (P=0.019) and triple-negative patients (P<0.001).
Based on these results, Dr Rumi concluded that mutations in JAK2, CALR, and MPL are independent predictors of clinical course, disease progression, and OS in PMF. So screening patients for these mutations can likely improve upon the risk stratification provided by IPSS. ![]()
MILAN—Two new studies appear to confirm the prognostic significance of CALR mutations in patients with primary myelofibrosis (PMF).
One study showed that PMF patients with mutated CALR had prolonged overall survival (OS) compared to patients with wild-type CALR. And additional subclonal mutations did not impair the positive impact of CALR mutations.
Another study suggested that indels in exon 9 of CALR are founding driver mutations in PMF. These mutations are independent predictors of clinical course, disease progression, and OS.
Both studies were presented at the 19th Congress of the European Hematology Association (EHA).
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented data on CALR mutations in the context of additional mutations (abstract S1355).
And Elisa Rumi, MD, of the University of Pavia in Italy, presented information on mutated CALR and other founding driver mutations in PMF (abstract S1356).
CALR & other subclonal mutations in PMF
To investigate the prognostic role of CALR mutations in relation to additional subclonal mutations, Dr Guglielmelli and her colleagues analyzed 274 samples from PMF patients.
The team genotyped the samples for mutations in 11 genes: JAK2, CALR, MPL, EZH2, ASXL1, SRSF2, IDH1, IDH2, CBL, TET2, and DNMT3A.
Two hundred and fifty-six patients (93.4%) presented with at least 1 somatic mutation, and 104 (38%) presented with at least 2.
The median follow-up was 3.8 years (range, 0.52-29.20 years). Among all patients, the median OS was 12.2 years (range, 5.6-18.8 years). Eighty-four patients died (30.7%), 44 (16.1%) of them due to leukemia.
The presence of CALR mutations was associated with better OS, independent of IPSS and molecular risk categories (hazard ratio [HR] 0.51, P=0.03). But CALR mutations did not impact the risk of progression to acute leukemia.
Among patients with a low- to intermediate-1-risk IPSS score, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR 0.4, P=0.02). Among patients with intermediate-2 to high risk, those with CALR mutations lived a median of 4.2 years, and those without lived a median of 2.6 years (HR=0.5, P=0.09).
Among patients with high molecular risk, those with CALR mutations lived a median of 17.7 years, and those without lived a median of 4.3 years, (HR=0.3, P=0.008). High molecular risk was defined as at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2.
Among patients in the low-molecular-risk group, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR=0.6, P=0.048).
Dr Guglielmelli said these results confirm the association between CALR mutation and favorable outcomes in PMF. They also show that additional subclonal mutations do not impair the positive impact of CALR mutation, thereby reinforcing the idea that CALR-mutated PMF is a distinct entity in terms of prognosis.
Founding driver mutations in PMF
In another presentation at the EHA Congress, Dr Rumi presented data on founding driver mutations in PMF. She and her colleagues analyzed 617 PMF patients, screening them for JAK2 V617F mutations, indels of CALR exon 9, and MPL exon 10 mutations.
The researchers assessed the impact of these mutations on thrombosis, progression to leukemia, and OS.
Their analysis suggested CALR-mutated PMF patients have a lower risk of thrombosis than JAK2-mutated patients (P=0.021). And this difference retained significance after adjusting for age.
Triple-negative PMF patients had a higher risk of leukemic evolution than CALR-mutated patients (P=0.016) and JAK2-mutated patients (P=0.043).
After adjusting for age, the risk remained significantly higher in triple-negative patients compared to JAK2-mutated patients (P=0.04) and retained borderline significance compared to CALR-mutated patients (P=0.052).
Patients with CALR-mutated PMF had a better OS than JAK2-mutated patients (P<0.001), MPL-mutated patients (P=0.009), and triple-negative patients (P<0.001).
In a multivariate analysis, CALR-mutated patients maintained a better OS than JAK2-mutated patients (P=0.019) and triple-negative patients (P<0.001).
Based on these results, Dr Rumi concluded that mutations in JAK2, CALR, and MPL are independent predictors of clinical course, disease progression, and OS in PMF. So screening patients for these mutations can likely improve upon the risk stratification provided by IPSS. ![]()
MILAN—Two new studies appear to confirm the prognostic significance of CALR mutations in patients with primary myelofibrosis (PMF).
One study showed that PMF patients with mutated CALR had prolonged overall survival (OS) compared to patients with wild-type CALR. And additional subclonal mutations did not impair the positive impact of CALR mutations.
Another study suggested that indels in exon 9 of CALR are founding driver mutations in PMF. These mutations are independent predictors of clinical course, disease progression, and OS.
Both studies were presented at the 19th Congress of the European Hematology Association (EHA).
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented data on CALR mutations in the context of additional mutations (abstract S1355).
And Elisa Rumi, MD, of the University of Pavia in Italy, presented information on mutated CALR and other founding driver mutations in PMF (abstract S1356).
CALR & other subclonal mutations in PMF
To investigate the prognostic role of CALR mutations in relation to additional subclonal mutations, Dr Guglielmelli and her colleagues analyzed 274 samples from PMF patients.
The team genotyped the samples for mutations in 11 genes: JAK2, CALR, MPL, EZH2, ASXL1, SRSF2, IDH1, IDH2, CBL, TET2, and DNMT3A.
Two hundred and fifty-six patients (93.4%) presented with at least 1 somatic mutation, and 104 (38%) presented with at least 2.
The median follow-up was 3.8 years (range, 0.52-29.20 years). Among all patients, the median OS was 12.2 years (range, 5.6-18.8 years). Eighty-four patients died (30.7%), 44 (16.1%) of them due to leukemia.
The presence of CALR mutations was associated with better OS, independent of IPSS and molecular risk categories (hazard ratio [HR] 0.51, P=0.03). But CALR mutations did not impact the risk of progression to acute leukemia.
Among patients with a low- to intermediate-1-risk IPSS score, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR 0.4, P=0.02). Among patients with intermediate-2 to high risk, those with CALR mutations lived a median of 4.2 years, and those without lived a median of 2.6 years (HR=0.5, P=0.09).
Among patients with high molecular risk, those with CALR mutations lived a median of 17.7 years, and those without lived a median of 4.3 years, (HR=0.3, P=0.008). High molecular risk was defined as at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2.
Among patients in the low-molecular-risk group, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR=0.6, P=0.048).
Dr Guglielmelli said these results confirm the association between CALR mutation and favorable outcomes in PMF. They also show that additional subclonal mutations do not impair the positive impact of CALR mutation, thereby reinforcing the idea that CALR-mutated PMF is a distinct entity in terms of prognosis.
Founding driver mutations in PMF
In another presentation at the EHA Congress, Dr Rumi presented data on founding driver mutations in PMF. She and her colleagues analyzed 617 PMF patients, screening them for JAK2 V617F mutations, indels of CALR exon 9, and MPL exon 10 mutations.
The researchers assessed the impact of these mutations on thrombosis, progression to leukemia, and OS.
Their analysis suggested CALR-mutated PMF patients have a lower risk of thrombosis than JAK2-mutated patients (P=0.021). And this difference retained significance after adjusting for age.
Triple-negative PMF patients had a higher risk of leukemic evolution than CALR-mutated patients (P=0.016) and JAK2-mutated patients (P=0.043).
After adjusting for age, the risk remained significantly higher in triple-negative patients compared to JAK2-mutated patients (P=0.04) and retained borderline significance compared to CALR-mutated patients (P=0.052).
Patients with CALR-mutated PMF had a better OS than JAK2-mutated patients (P<0.001), MPL-mutated patients (P=0.009), and triple-negative patients (P<0.001).
In a multivariate analysis, CALR-mutated patients maintained a better OS than JAK2-mutated patients (P=0.019) and triple-negative patients (P<0.001).
Based on these results, Dr Rumi concluded that mutations in JAK2, CALR, and MPL are independent predictors of clinical course, disease progression, and OS in PMF. So screening patients for these mutations can likely improve upon the risk stratification provided by IPSS. ![]()
How genetics, race affect clopidogrel outcomes

Credit: Robert Boston
Washington University
New research helps explain the higher risk of death observed among some patients taking the anticoagulant clopidogrel after a heart attack.
Researchers identified genetic variants that increased the risk of dying in the year following a first heart attack, but they appeared to do so for different reasons depending on a patient’s race.
Two DNA variants common in African Americans were associated with an increased risk of both bleeding and death.
And in Caucasians, a different variant was linked to additional heart attacks and a higher risk of death.
The variations influence the way patients metabolize clopidogrel and can alter its effectiveness.
These findings were published in Circulation: Cardiovascular Genetics.
“The research is provocative,” said the study’s first author Sharon Cresci, MD, of the Washington University School of Medicine in St Louis.
“Knowing about potential genetic differences based on race can help physicians tailor drugs to patients based on their genetic makeup.”
Clopidogrel is metabolized in the liver, where it is turned into its active form via a group of enzymes called cytochrome P450 (CYP). Although clopidogrel is effective in many patients, earlier studies showed that some patients metabolize the drug better than others.
Indeed, in 2010, the US Food and Drug Administration added a black box warning to labels of clopidogrel after research (which primarily involved Caucasians) showed that people with a particular CYP genetic variant metabolized the drug poorly, which reduced the amount of the drug circulating in the blood. These patients had a higher risk of heart attack and stroke.
Additional studies showed that other CYP gene variants are linked to the rapid metabolism of clopidogrel, and patients with those variants had a higher risk of bleeding. But it has not been clear, until now, that the effects of these particular gene variations can vary by race.
To uncover this finding, Dr Cresci and her colleagues analyzed CYP variants among 2062 Caucasians and 670 African Americans who suffered heart attacks. Nearly 80% of the Caucasians and 65% of the African Americans were prescribed clopidogrel.
The patients were enrolled in a major study known as TRIUMPH (Translational Research Investigating Underlying disparities in acute Myocardial infarction Patients’ Health), which was conducted from 2005 to 2008 at 24 US hospitals.
Among patients taking clopidogrel, the 1-year mortality rate for African Americans was 7.2%, compared with 3.6% for Caucasians.
Caucasians who carried the CYP2C19*2 variant, which has been linked to poor metabolism of the drug, had a higher rate of repeat heart attacks and death. However, among African Americans treated with clopidogrel, the CYP2C19*2 variant was not associated with a higher rate of death.
African Americans had higher rates of bleeding and death if they carried either of 2 other variants: CYP1A2*1C or CYP2C19*17, the latter of which has been associated with the rapid metabolism of clopidogrel. Among Caucasians on clopidogrel, neither variant increased the risk of death.
“This is very novel information that begs for more research,” said study author Richard G. Bach, MD, also of Washington University.
Although genetic testing is available to identify CYP variants in a patient’s DNA, these tests are not widely used by cardiologists. Results of the current study suggest this practice may need to be reconsidered.
“This research is an important addition to the field because previous studies looking at CYP gene variants and their effects on risks of repeat heart attacks, bleeding, and death have included predominantly Caucasian patients of European ancestry,” Dr Cresci noted. “There is almost no data, until now, about these variants in African Americans.”
Research examining how genetic variants alter the effectiveness of clopidogrel remains somewhat controversial, Dr Bach said. Many physicians feel that before they can tailor therapy for heart attack patients, more data is needed to prove a clear link between genetic variants and negative health consequences and that tailoring therapy will improve patients’ outcomes, he explained.
His hope is that additional research would provide more definitive conclusions to help physicians choose the best medications for patients after a heart attack and, ultimately, “to reduce the too-high rate of death and disability for patients after a heart attack,” Dr Bach said.
Dr Cresci agreed, adding, “By focusing on genetic differences, we may be able to individualize therapies after heart attacks and achieve the best treatment for each patient.” ![]()
Credit: Robert Boston
Washington University
New research helps explain the higher risk of death observed among some patients taking the anticoagulant clopidogrel after a heart attack.
Researchers identified genetic variants that increased the risk of dying in the year following a first heart attack, but they appeared to do so for different reasons depending on a patient’s race.
Two DNA variants common in African Americans were associated with an increased risk of both bleeding and death.
And in Caucasians, a different variant was linked to additional heart attacks and a higher risk of death.
The variations influence the way patients metabolize clopidogrel and can alter its effectiveness.
These findings were published in Circulation: Cardiovascular Genetics.
“The research is provocative,” said the study’s first author Sharon Cresci, MD, of the Washington University School of Medicine in St Louis.
“Knowing about potential genetic differences based on race can help physicians tailor drugs to patients based on their genetic makeup.”
Clopidogrel is metabolized in the liver, where it is turned into its active form via a group of enzymes called cytochrome P450 (CYP). Although clopidogrel is effective in many patients, earlier studies showed that some patients metabolize the drug better than others.
Indeed, in 2010, the US Food and Drug Administration added a black box warning to labels of clopidogrel after research (which primarily involved Caucasians) showed that people with a particular CYP genetic variant metabolized the drug poorly, which reduced the amount of the drug circulating in the blood. These patients had a higher risk of heart attack and stroke.
Additional studies showed that other CYP gene variants are linked to the rapid metabolism of clopidogrel, and patients with those variants had a higher risk of bleeding. But it has not been clear, until now, that the effects of these particular gene variations can vary by race.
To uncover this finding, Dr Cresci and her colleagues analyzed CYP variants among 2062 Caucasians and 670 African Americans who suffered heart attacks. Nearly 80% of the Caucasians and 65% of the African Americans were prescribed clopidogrel.
The patients were enrolled in a major study known as TRIUMPH (Translational Research Investigating Underlying disparities in acute Myocardial infarction Patients’ Health), which was conducted from 2005 to 2008 at 24 US hospitals.
Among patients taking clopidogrel, the 1-year mortality rate for African Americans was 7.2%, compared with 3.6% for Caucasians.
Caucasians who carried the CYP2C19*2 variant, which has been linked to poor metabolism of the drug, had a higher rate of repeat heart attacks and death. However, among African Americans treated with clopidogrel, the CYP2C19*2 variant was not associated with a higher rate of death.
African Americans had higher rates of bleeding and death if they carried either of 2 other variants: CYP1A2*1C or CYP2C19*17, the latter of which has been associated with the rapid metabolism of clopidogrel. Among Caucasians on clopidogrel, neither variant increased the risk of death.
“This is very novel information that begs for more research,” said study author Richard G. Bach, MD, also of Washington University.
Although genetic testing is available to identify CYP variants in a patient’s DNA, these tests are not widely used by cardiologists. Results of the current study suggest this practice may need to be reconsidered.
“This research is an important addition to the field because previous studies looking at CYP gene variants and their effects on risks of repeat heart attacks, bleeding, and death have included predominantly Caucasian patients of European ancestry,” Dr Cresci noted. “There is almost no data, until now, about these variants in African Americans.”
Research examining how genetic variants alter the effectiveness of clopidogrel remains somewhat controversial, Dr Bach said. Many physicians feel that before they can tailor therapy for heart attack patients, more data is needed to prove a clear link between genetic variants and negative health consequences and that tailoring therapy will improve patients’ outcomes, he explained.
His hope is that additional research would provide more definitive conclusions to help physicians choose the best medications for patients after a heart attack and, ultimately, “to reduce the too-high rate of death and disability for patients after a heart attack,” Dr Bach said.
Dr Cresci agreed, adding, “By focusing on genetic differences, we may be able to individualize therapies after heart attacks and achieve the best treatment for each patient.” ![]()
Credit: Robert Boston
Washington University
New research helps explain the higher risk of death observed among some patients taking the anticoagulant clopidogrel after a heart attack.
Researchers identified genetic variants that increased the risk of dying in the year following a first heart attack, but they appeared to do so for different reasons depending on a patient’s race.
Two DNA variants common in African Americans were associated with an increased risk of both bleeding and death.
And in Caucasians, a different variant was linked to additional heart attacks and a higher risk of death.
The variations influence the way patients metabolize clopidogrel and can alter its effectiveness.
These findings were published in Circulation: Cardiovascular Genetics.
“The research is provocative,” said the study’s first author Sharon Cresci, MD, of the Washington University School of Medicine in St Louis.
“Knowing about potential genetic differences based on race can help physicians tailor drugs to patients based on their genetic makeup.”
Clopidogrel is metabolized in the liver, where it is turned into its active form via a group of enzymes called cytochrome P450 (CYP). Although clopidogrel is effective in many patients, earlier studies showed that some patients metabolize the drug better than others.
Indeed, in 2010, the US Food and Drug Administration added a black box warning to labels of clopidogrel after research (which primarily involved Caucasians) showed that people with a particular CYP genetic variant metabolized the drug poorly, which reduced the amount of the drug circulating in the blood. These patients had a higher risk of heart attack and stroke.
Additional studies showed that other CYP gene variants are linked to the rapid metabolism of clopidogrel, and patients with those variants had a higher risk of bleeding. But it has not been clear, until now, that the effects of these particular gene variations can vary by race.
To uncover this finding, Dr Cresci and her colleagues analyzed CYP variants among 2062 Caucasians and 670 African Americans who suffered heart attacks. Nearly 80% of the Caucasians and 65% of the African Americans were prescribed clopidogrel.
The patients were enrolled in a major study known as TRIUMPH (Translational Research Investigating Underlying disparities in acute Myocardial infarction Patients’ Health), which was conducted from 2005 to 2008 at 24 US hospitals.
Among patients taking clopidogrel, the 1-year mortality rate for African Americans was 7.2%, compared with 3.6% for Caucasians.
Caucasians who carried the CYP2C19*2 variant, which has been linked to poor metabolism of the drug, had a higher rate of repeat heart attacks and death. However, among African Americans treated with clopidogrel, the CYP2C19*2 variant was not associated with a higher rate of death.
African Americans had higher rates of bleeding and death if they carried either of 2 other variants: CYP1A2*1C or CYP2C19*17, the latter of which has been associated with the rapid metabolism of clopidogrel. Among Caucasians on clopidogrel, neither variant increased the risk of death.
“This is very novel information that begs for more research,” said study author Richard G. Bach, MD, also of Washington University.
Although genetic testing is available to identify CYP variants in a patient’s DNA, these tests are not widely used by cardiologists. Results of the current study suggest this practice may need to be reconsidered.
“This research is an important addition to the field because previous studies looking at CYP gene variants and their effects on risks of repeat heart attacks, bleeding, and death have included predominantly Caucasian patients of European ancestry,” Dr Cresci noted. “There is almost no data, until now, about these variants in African Americans.”
Research examining how genetic variants alter the effectiveness of clopidogrel remains somewhat controversial, Dr Bach said. Many physicians feel that before they can tailor therapy for heart attack patients, more data is needed to prove a clear link between genetic variants and negative health consequences and that tailoring therapy will improve patients’ outcomes, he explained.
His hope is that additional research would provide more definitive conclusions to help physicians choose the best medications for patients after a heart attack and, ultimately, “to reduce the too-high rate of death and disability for patients after a heart attack,” Dr Bach said.
Dr Cresci agreed, adding, “By focusing on genetic differences, we may be able to individualize therapies after heart attacks and achieve the best treatment for each patient.” ![]()
Restoring gene function can ‘reverse’ B-ALL

and Ross Dickins, PhD
Credit: Walter and Eliza Hall
Institute of Medical Research
Results of preclinical research suggest B-progenitor acute lymphoblastic leukemia (B-ALL) can be “reversed” by coaxing leukemic cells back into normal development.
Researchers found that switching off the gene Pax5 could induce B-ALL in mice, but restoring Pax5 function could prompt disease remission.
Grace Liu, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia, and her colleagues detailed this research in Genes & Development.
“Pax5 is essential for normal development of [B cells],” Liu said. “When Pax5 function is compromised, developing B cells can get trapped in an immature state and become cancerous.”
The researchers used transgenic RNAi to suppress endogenous Pax5 expression in the hematopoietic compartment of mice, and this induced B-ALL.
“Along with other genetic changes, deactivating Pax5 drives normal blood cells to turn into leukemia cells, which has been shown before,” Liu said. “However, we showed, for the first time, that reactivating Pax5 enabled the cells to resume their normal development and lose their cancer-like qualities, effectively curing the leukemia.”
The team found that restoring endogenous Pax5 expression triggered immunophenotypic maturation and durable disease remission.
Even brief Pax5 restoration disabled B-ALL cells’ leukemia-initiating capacity in mice. And the researchers observed similar results in human B-ALL cell lines.
“This work shows how inactivating the tumor suppressor gene Pax5 contributes to B-ALL development and how leukemia cells become addicted to low Pax5 levels to continue proliferating,” said study author Ross Dickins, PhD, also of the Walter and Eliza Hall Institute of Medical Research.
“Even though the B-ALL cells have multiple genetic mutations, simply reactivating Pax5 causes tumor cells to resume normal development and lose their cancerous properties.”
Dr Dickins added that forcing B-ALL cells to resume their normal development could provide a new strategy for treating leukemia. However, genes lost in tumor cells are not traditionally considered suitable drug targets.
“It is very difficult to develop drugs that restore the function of genes that are lost during cancer development,” Dr Dickins said. “However, by understanding the mechanisms by which Pax5 loss causes leukemia, we can begin to look at ways of developing drugs that could have the same effect as restoring Pax5 function.” ![]()
and Ross Dickins, PhD
Credit: Walter and Eliza Hall
Institute of Medical Research
Results of preclinical research suggest B-progenitor acute lymphoblastic leukemia (B-ALL) can be “reversed” by coaxing leukemic cells back into normal development.
Researchers found that switching off the gene Pax5 could induce B-ALL in mice, but restoring Pax5 function could prompt disease remission.
Grace Liu, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia, and her colleagues detailed this research in Genes & Development.
“Pax5 is essential for normal development of [B cells],” Liu said. “When Pax5 function is compromised, developing B cells can get trapped in an immature state and become cancerous.”
The researchers used transgenic RNAi to suppress endogenous Pax5 expression in the hematopoietic compartment of mice, and this induced B-ALL.
“Along with other genetic changes, deactivating Pax5 drives normal blood cells to turn into leukemia cells, which has been shown before,” Liu said. “However, we showed, for the first time, that reactivating Pax5 enabled the cells to resume their normal development and lose their cancer-like qualities, effectively curing the leukemia.”
The team found that restoring endogenous Pax5 expression triggered immunophenotypic maturation and durable disease remission.
Even brief Pax5 restoration disabled B-ALL cells’ leukemia-initiating capacity in mice. And the researchers observed similar results in human B-ALL cell lines.
“This work shows how inactivating the tumor suppressor gene Pax5 contributes to B-ALL development and how leukemia cells become addicted to low Pax5 levels to continue proliferating,” said study author Ross Dickins, PhD, also of the Walter and Eliza Hall Institute of Medical Research.
“Even though the B-ALL cells have multiple genetic mutations, simply reactivating Pax5 causes tumor cells to resume normal development and lose their cancerous properties.”
Dr Dickins added that forcing B-ALL cells to resume their normal development could provide a new strategy for treating leukemia. However, genes lost in tumor cells are not traditionally considered suitable drug targets.
“It is very difficult to develop drugs that restore the function of genes that are lost during cancer development,” Dr Dickins said. “However, by understanding the mechanisms by which Pax5 loss causes leukemia, we can begin to look at ways of developing drugs that could have the same effect as restoring Pax5 function.” ![]()
and Ross Dickins, PhD
Credit: Walter and Eliza Hall
Institute of Medical Research
Results of preclinical research suggest B-progenitor acute lymphoblastic leukemia (B-ALL) can be “reversed” by coaxing leukemic cells back into normal development.
Researchers found that switching off the gene Pax5 could induce B-ALL in mice, but restoring Pax5 function could prompt disease remission.
Grace Liu, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia, and her colleagues detailed this research in Genes & Development.
“Pax5 is essential for normal development of [B cells],” Liu said. “When Pax5 function is compromised, developing B cells can get trapped in an immature state and become cancerous.”
The researchers used transgenic RNAi to suppress endogenous Pax5 expression in the hematopoietic compartment of mice, and this induced B-ALL.
“Along with other genetic changes, deactivating Pax5 drives normal blood cells to turn into leukemia cells, which has been shown before,” Liu said. “However, we showed, for the first time, that reactivating Pax5 enabled the cells to resume their normal development and lose their cancer-like qualities, effectively curing the leukemia.”
The team found that restoring endogenous Pax5 expression triggered immunophenotypic maturation and durable disease remission.
Even brief Pax5 restoration disabled B-ALL cells’ leukemia-initiating capacity in mice. And the researchers observed similar results in human B-ALL cell lines.
“This work shows how inactivating the tumor suppressor gene Pax5 contributes to B-ALL development and how leukemia cells become addicted to low Pax5 levels to continue proliferating,” said study author Ross Dickins, PhD, also of the Walter and Eliza Hall Institute of Medical Research.
“Even though the B-ALL cells have multiple genetic mutations, simply reactivating Pax5 causes tumor cells to resume normal development and lose their cancerous properties.”
Dr Dickins added that forcing B-ALL cells to resume their normal development could provide a new strategy for treating leukemia. However, genes lost in tumor cells are not traditionally considered suitable drug targets.
“It is very difficult to develop drugs that restore the function of genes that are lost during cancer development,” Dr Dickins said. “However, by understanding the mechanisms by which Pax5 loss causes leukemia, we can begin to look at ways of developing drugs that could have the same effect as restoring Pax5 function.” ![]()
Elderly males with DLBCL require increased rituximab dosing

©ASCO/Phil McCarten
CHICAGO—Elderly males with non-Hodgkin lymphoma (NHL) may require one-third higher doses of rituximab than the current standard to attain optimal responses to rituximab-containing chemotherapy, a new study suggests.
Increasing the rituximab dose eliminated any gender-related differences in survival among elderly patients with aggressive, CD20+, B-cell lymphomas, said investigator Michael Pfreundschuh, MD, of Saarland University Medical Center in Germany.
He presented this finding at the 2014 ASCO Annual Meeting (abstract 8501).
Although rituximab has been used in NHL for nearly 2 decades, standard dosing of the drug is still largely empiric. Only recently have researchers examined whether responses to the drug may vary by gender and age.
New data show that rituximab clears the body more rapidly in elderly males than elderly females, suggesting that rituximab dosing may need to be increased in older men to maintain adequate drug exposure.
Dr Pfreundschuh noted that the RICOVER-60 trial established 6 cycles of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone every 14 days (R-CHOP-14), followed by 2 additional cycles of rituximab, as the new standard of care for elderly patients with NHL in Germany.
Further analysis of the trial’s results indicates that rituximab dosing may be inadequate in males older than 60, since they fare much worse than elderly females in terms of progression-free survival (PFS).
To test this gender difference, the German High-grade Non-Hodgkin Lymphoma Study Group designed the SEXIE-R-CHOP-14 trial.
The group tested 2 different rituximab doses in patients aged 61 to 80 with stage I to IV, CD20+, diffuse large B-cell lymphoma (DLBCL). A group of 120 females received rituximab at 375 mg/m2 per treatment cycle, and a group of 148 males received 500 mg/m2 per treatment cycle.
The increased rituximab dose in males resulted in slightly higher trough serum levels than in females. However, rituximab levels dropped faster in males, resulting in nearly identical serum levels thereafter and a very similar overall rituximab exposure time, Dr Pfreundschuh said.
The higher dose of rituximab given to elderly males did not result in increased toxicity.
Survival rates were similar between the male and female groups. The 3-year PFS was 74% in males and 68% in females, and 3-year overall survival was 82% in males and 72% in females.
A multivariate analysis that evaluated gender as a risk factor revealed that the male-vs-female hazard ratio for PFS was 0.8 in SEXIE-R-CHOP-14, compared to 1.6 in RICOVER-60. Similarly, the male-vs-female hazard ratio for overall survival was 0.7 in SEXIE-R-CHOP-14 and 1.4 in RICOVER-60.
“Increasing the rituximab dose by one-third eliminated the increased risk [of death and progression in] elderly males,” Dr Pfreundschuh concluded.
He also noted that younger males and females have faster rituximab clearance than elderly females. So increasing the dose in these populations should also result in a better outcome. ![]()
©ASCO/Phil McCarten
CHICAGO—Elderly males with non-Hodgkin lymphoma (NHL) may require one-third higher doses of rituximab than the current standard to attain optimal responses to rituximab-containing chemotherapy, a new study suggests.
Increasing the rituximab dose eliminated any gender-related differences in survival among elderly patients with aggressive, CD20+, B-cell lymphomas, said investigator Michael Pfreundschuh, MD, of Saarland University Medical Center in Germany.
He presented this finding at the 2014 ASCO Annual Meeting (abstract 8501).
Although rituximab has been used in NHL for nearly 2 decades, standard dosing of the drug is still largely empiric. Only recently have researchers examined whether responses to the drug may vary by gender and age.
New data show that rituximab clears the body more rapidly in elderly males than elderly females, suggesting that rituximab dosing may need to be increased in older men to maintain adequate drug exposure.
Dr Pfreundschuh noted that the RICOVER-60 trial established 6 cycles of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone every 14 days (R-CHOP-14), followed by 2 additional cycles of rituximab, as the new standard of care for elderly patients with NHL in Germany.
Further analysis of the trial’s results indicates that rituximab dosing may be inadequate in males older than 60, since they fare much worse than elderly females in terms of progression-free survival (PFS).
To test this gender difference, the German High-grade Non-Hodgkin Lymphoma Study Group designed the SEXIE-R-CHOP-14 trial.
The group tested 2 different rituximab doses in patients aged 61 to 80 with stage I to IV, CD20+, diffuse large B-cell lymphoma (DLBCL). A group of 120 females received rituximab at 375 mg/m2 per treatment cycle, and a group of 148 males received 500 mg/m2 per treatment cycle.
The increased rituximab dose in males resulted in slightly higher trough serum levels than in females. However, rituximab levels dropped faster in males, resulting in nearly identical serum levels thereafter and a very similar overall rituximab exposure time, Dr Pfreundschuh said.
The higher dose of rituximab given to elderly males did not result in increased toxicity.
Survival rates were similar between the male and female groups. The 3-year PFS was 74% in males and 68% in females, and 3-year overall survival was 82% in males and 72% in females.
A multivariate analysis that evaluated gender as a risk factor revealed that the male-vs-female hazard ratio for PFS was 0.8 in SEXIE-R-CHOP-14, compared to 1.6 in RICOVER-60. Similarly, the male-vs-female hazard ratio for overall survival was 0.7 in SEXIE-R-CHOP-14 and 1.4 in RICOVER-60.
“Increasing the rituximab dose by one-third eliminated the increased risk [of death and progression in] elderly males,” Dr Pfreundschuh concluded.
He also noted that younger males and females have faster rituximab clearance than elderly females. So increasing the dose in these populations should also result in a better outcome. ![]()
©ASCO/Phil McCarten
CHICAGO—Elderly males with non-Hodgkin lymphoma (NHL) may require one-third higher doses of rituximab than the current standard to attain optimal responses to rituximab-containing chemotherapy, a new study suggests.
Increasing the rituximab dose eliminated any gender-related differences in survival among elderly patients with aggressive, CD20+, B-cell lymphomas, said investigator Michael Pfreundschuh, MD, of Saarland University Medical Center in Germany.
He presented this finding at the 2014 ASCO Annual Meeting (abstract 8501).
Although rituximab has been used in NHL for nearly 2 decades, standard dosing of the drug is still largely empiric. Only recently have researchers examined whether responses to the drug may vary by gender and age.
New data show that rituximab clears the body more rapidly in elderly males than elderly females, suggesting that rituximab dosing may need to be increased in older men to maintain adequate drug exposure.
Dr Pfreundschuh noted that the RICOVER-60 trial established 6 cycles of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone every 14 days (R-CHOP-14), followed by 2 additional cycles of rituximab, as the new standard of care for elderly patients with NHL in Germany.
Further analysis of the trial’s results indicates that rituximab dosing may be inadequate in males older than 60, since they fare much worse than elderly females in terms of progression-free survival (PFS).
To test this gender difference, the German High-grade Non-Hodgkin Lymphoma Study Group designed the SEXIE-R-CHOP-14 trial.
The group tested 2 different rituximab doses in patients aged 61 to 80 with stage I to IV, CD20+, diffuse large B-cell lymphoma (DLBCL). A group of 120 females received rituximab at 375 mg/m2 per treatment cycle, and a group of 148 males received 500 mg/m2 per treatment cycle.
The increased rituximab dose in males resulted in slightly higher trough serum levels than in females. However, rituximab levels dropped faster in males, resulting in nearly identical serum levels thereafter and a very similar overall rituximab exposure time, Dr Pfreundschuh said.
The higher dose of rituximab given to elderly males did not result in increased toxicity.
Survival rates were similar between the male and female groups. The 3-year PFS was 74% in males and 68% in females, and 3-year overall survival was 82% in males and 72% in females.
A multivariate analysis that evaluated gender as a risk factor revealed that the male-vs-female hazard ratio for PFS was 0.8 in SEXIE-R-CHOP-14, compared to 1.6 in RICOVER-60. Similarly, the male-vs-female hazard ratio for overall survival was 0.7 in SEXIE-R-CHOP-14 and 1.4 in RICOVER-60.
“Increasing the rituximab dose by one-third eliminated the increased risk [of death and progression in] elderly males,” Dr Pfreundschuh concluded.
He also noted that younger males and females have faster rituximab clearance than elderly females. So increasing the dose in these populations should also result in a better outcome. ![]()
Passing stones with PDE5 inhibitors
Now that we have made the most likely diagnosis of a kidney stone, we can potentially avoid a urologic procedure if we expel it. Of the 22% of kidney stones that wind up in the ureter, two-thirds will be lodged in the distal ureter.
Tamsulosin works, and prednisolone may also be helpful. Anything else?
Dr. Kumar Jayant of Sudha Hospital and Medical Research Center, Kota, India, and associates investigated the efficacy of a phosphodiesterase type 5 (PDE5) inhibitor (tadalafil) to facilitate kidney stone expulsion. PDE5 inhibitors increase levels of cyclic guanosine monophosphate and cause ureteric relaxation (Int. J. Urol. 2014 June 3 [doi:10.1111/iju.12496]).
In this study, 244 patients with distal ureteral stones between 5 and 10 mm (about a 50% chance of passing) quantitated with noncontrast CT were randomized to two groups: tamsulosin (0.4 mg daily) or tamsulosin (0.4 mg daily) plus tadalafil (10 mg daily). Medications were given for 4 weeks.
The average patient age was about 37 years, and the mean stone size was 7 mm. Participants were included only if their pain was relieved within a day by diclofenac injection. Potential participants were excluded if they had fever, hydronephrosis, multiple kidney stones, a history of surgery or endoscopic procedures, or diabetes; were taking calcium channel blockers or nitrates; were pregnant or lactating; or required immediate treatment. If stones were not passed in 4 weeks, ureterorenoscopy was used to remove them.
The tamsulosin/tadalafil combination was associated with a statistically significantly higher rate of expulsion (83.6% vs. 65.5%; P = .031) and a shorter time to expulsion (14.9 days vs. 16.7 days; P =.003). Tamsulosin/tadalafil was associated with significantly fewer hospital visits and less need for pain medications. Not surprisingly, tamsulosin/tadalafil improved erectile function. However, patients taking the tamsulosin/tadalafil combination also had more headaches, dizziness, orthostatic hypotension, and backaches.
In certain patients, hypotension may be a concern with this combination. However, the researchers highlighted a study whose authors concluded, "in subjects on tamsulosin, tadalafil 10 and 20 mg produced mean maximal decreases in standing [systolic blood pressure] that were similar to placebo" (J. Urol. 2004;172(5, pt. 1):1935-40).
Out-of-pocket cost may be a barrier for some patients. But given the significance of these findings (an almost 20% difference in expulsion rate), total cost of care may be significantly reduced for these patients.
Dr. Ebbert is a professor of medicine and general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. He reports no disclosures.
Now that we have made the most likely diagnosis of a kidney stone, we can potentially avoid a urologic procedure if we expel it. Of the 22% of kidney stones that wind up in the ureter, two-thirds will be lodged in the distal ureter.
Tamsulosin works, and prednisolone may also be helpful. Anything else?
Dr. Kumar Jayant of Sudha Hospital and Medical Research Center, Kota, India, and associates investigated the efficacy of a phosphodiesterase type 5 (PDE5) inhibitor (tadalafil) to facilitate kidney stone expulsion. PDE5 inhibitors increase levels of cyclic guanosine monophosphate and cause ureteric relaxation (Int. J. Urol. 2014 June 3 [doi:10.1111/iju.12496]).
In this study, 244 patients with distal ureteral stones between 5 and 10 mm (about a 50% chance of passing) quantitated with noncontrast CT were randomized to two groups: tamsulosin (0.4 mg daily) or tamsulosin (0.4 mg daily) plus tadalafil (10 mg daily). Medications were given for 4 weeks.
The average patient age was about 37 years, and the mean stone size was 7 mm. Participants were included only if their pain was relieved within a day by diclofenac injection. Potential participants were excluded if they had fever, hydronephrosis, multiple kidney stones, a history of surgery or endoscopic procedures, or diabetes; were taking calcium channel blockers or nitrates; were pregnant or lactating; or required immediate treatment. If stones were not passed in 4 weeks, ureterorenoscopy was used to remove them.
The tamsulosin/tadalafil combination was associated with a statistically significantly higher rate of expulsion (83.6% vs. 65.5%; P = .031) and a shorter time to expulsion (14.9 days vs. 16.7 days; P =.003). Tamsulosin/tadalafil was associated with significantly fewer hospital visits and less need for pain medications. Not surprisingly, tamsulosin/tadalafil improved erectile function. However, patients taking the tamsulosin/tadalafil combination also had more headaches, dizziness, orthostatic hypotension, and backaches.
In certain patients, hypotension may be a concern with this combination. However, the researchers highlighted a study whose authors concluded, "in subjects on tamsulosin, tadalafil 10 and 20 mg produced mean maximal decreases in standing [systolic blood pressure] that were similar to placebo" (J. Urol. 2004;172(5, pt. 1):1935-40).
Out-of-pocket cost may be a barrier for some patients. But given the significance of these findings (an almost 20% difference in expulsion rate), total cost of care may be significantly reduced for these patients.
Dr. Ebbert is a professor of medicine and general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. He reports no disclosures.
Now that we have made the most likely diagnosis of a kidney stone, we can potentially avoid a urologic procedure if we expel it. Of the 22% of kidney stones that wind up in the ureter, two-thirds will be lodged in the distal ureter.
Tamsulosin works, and prednisolone may also be helpful. Anything else?
Dr. Kumar Jayant of Sudha Hospital and Medical Research Center, Kota, India, and associates investigated the efficacy of a phosphodiesterase type 5 (PDE5) inhibitor (tadalafil) to facilitate kidney stone expulsion. PDE5 inhibitors increase levels of cyclic guanosine monophosphate and cause ureteric relaxation (Int. J. Urol. 2014 June 3 [doi:10.1111/iju.12496]).
In this study, 244 patients with distal ureteral stones between 5 and 10 mm (about a 50% chance of passing) quantitated with noncontrast CT were randomized to two groups: tamsulosin (0.4 mg daily) or tamsulosin (0.4 mg daily) plus tadalafil (10 mg daily). Medications were given for 4 weeks.
The average patient age was about 37 years, and the mean stone size was 7 mm. Participants were included only if their pain was relieved within a day by diclofenac injection. Potential participants were excluded if they had fever, hydronephrosis, multiple kidney stones, a history of surgery or endoscopic procedures, or diabetes; were taking calcium channel blockers or nitrates; were pregnant or lactating; or required immediate treatment. If stones were not passed in 4 weeks, ureterorenoscopy was used to remove them.
The tamsulosin/tadalafil combination was associated with a statistically significantly higher rate of expulsion (83.6% vs. 65.5%; P = .031) and a shorter time to expulsion (14.9 days vs. 16.7 days; P =.003). Tamsulosin/tadalafil was associated with significantly fewer hospital visits and less need for pain medications. Not surprisingly, tamsulosin/tadalafil improved erectile function. However, patients taking the tamsulosin/tadalafil combination also had more headaches, dizziness, orthostatic hypotension, and backaches.
In certain patients, hypotension may be a concern with this combination. However, the researchers highlighted a study whose authors concluded, "in subjects on tamsulosin, tadalafil 10 and 20 mg produced mean maximal decreases in standing [systolic blood pressure] that were similar to placebo" (J. Urol. 2004;172(5, pt. 1):1935-40).
Out-of-pocket cost may be a barrier for some patients. But given the significance of these findings (an almost 20% difference in expulsion rate), total cost of care may be significantly reduced for these patients.
Dr. Ebbert is a professor of medicine and general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. He reports no disclosures.
