User login
ICD-10 Will Allow Dermatologists to Effectively Communicate With Payors About Patient Visits
It is important that dermatologists do not overlook the changes associated with the transition to International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM). Some physicians believe that providing this level of specificity is not important because at the end of the day, who is watching? You will see with several of the codes highlighted in this column that specificity may be required before a claim can be submitted to the payor for processing. In addition, some older catchall codes will go away, forcing us to provide additional specificity.1
Scabies coding is a good example of an added specificity requirement. Currently, with the International Classification of Diseases, Ninth Revision (ICD-9), we have a code for scabies but no code to address the postscabetic pruritus that patients often develop. The ICD-9 code 133.0 applies to scabies, and if we would like to code for the itch, we must enter a second code for unspecified pruritic disorder (698.9).2 With ICD-10-CM, we can be more specific and actually code for the cause of the itch. We will be able to mark the initial visit as B86 (scabies), but subsequent visits for the itch related to scabies can be coded as pruritus using the primary code L29.8 (other pruritus) and a new sequelae or late effect code of B94 (sequelae of other and unspecified infectious and parasitic diseases).3 You will still be able to submit the second visit with scabies as the primary diagnosis, but this practice should be avoided to prevent claim rejection on the backend. In addition, there will be codes to allow for evaluation of a family member or close contact for this condition. Although coding these patients as an initial visit for scabies may be easier, for epidemiologic purposes a more complete code to provide would be the ICD-10-CM code of Z11.8 (encounter for screening for other infectious and parasitic diseases), particularly if they are not found to have scabies.3 This code also would be useful when screening for head lice when a close contact is not found to have it. What do we currently code when someone comes for screening of head lice because a classmate or sibling has it? With ICD-10-CM we will have a more accurate way to communicate with the payor.
The increase in the number of codes with ICD-10-CM permits us to give a description on a situation or circumstance when a person who may not be sick comes into the office for a specific reason or when circumstances influence a person’s health status but those circumstances are not an actual illness or injury. It is frustrating that I currently am not able to code for a skin examination appropriately using ICD-9. Let me be clear, I am not saying that this new code will be reimbursable. We will have to wait and see how we are reimbursed for all of these codes, but at least we will have the option of coding for the reason the patient presents to me, which is often a skin examination, and then the secondary code could be benign nevi or seborrheic keratosis. The code Z12.83 (encounter for screening for malignant neoplasm of skin) will now be the best code for these purposes.3 In addition, codes that address postoperative nursing visits have not been available. With ICD-10-CM we will have a new code for encounter for surgical aftercare following surgery on the skin and subcutaneous tissue that does not include a standard suture removal but is a necessary visit (Z48.817). In addition, we will have a specific code to document encounters for allergy testing (Z01.82).3 With ICD-9, rash not otherwise specified or eczema would have to be the primary code (782.1) and you were forced to place a code for the patient’s allergy, regardless of whether or not the allergy was known.
The reimbursement of these codes has not been addressed, so ideally during the testing period there can be a head-to-head comparison. By adhering to some of the new rules, hopefully we will be able to more completely communicate about what is occurring during the patient visit.
1. Lamb A. Dermatology coding changes with ICD-10. Cutis. 2014;93:284-285.
2. ICD-9 code lookup. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/medicare-coverage-database/staticpages/icd-9-code-lookup.aspx. Accessed July 17, 2014.
3. Centers for Medicare & Medicaid Services. ICD-10-CM Tabular List of Diseases and Injuries. http://www.cms.gov/Medicare/Coding/ICD10/downloads/6_I10tab2010.pdf. Published 2010. Accessed July 17, 2014.
It is important that dermatologists do not overlook the changes associated with the transition to International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM). Some physicians believe that providing this level of specificity is not important because at the end of the day, who is watching? You will see with several of the codes highlighted in this column that specificity may be required before a claim can be submitted to the payor for processing. In addition, some older catchall codes will go away, forcing us to provide additional specificity.1
Scabies coding is a good example of an added specificity requirement. Currently, with the International Classification of Diseases, Ninth Revision (ICD-9), we have a code for scabies but no code to address the postscabetic pruritus that patients often develop. The ICD-9 code 133.0 applies to scabies, and if we would like to code for the itch, we must enter a second code for unspecified pruritic disorder (698.9).2 With ICD-10-CM, we can be more specific and actually code for the cause of the itch. We will be able to mark the initial visit as B86 (scabies), but subsequent visits for the itch related to scabies can be coded as pruritus using the primary code L29.8 (other pruritus) and a new sequelae or late effect code of B94 (sequelae of other and unspecified infectious and parasitic diseases).3 You will still be able to submit the second visit with scabies as the primary diagnosis, but this practice should be avoided to prevent claim rejection on the backend. In addition, there will be codes to allow for evaluation of a family member or close contact for this condition. Although coding these patients as an initial visit for scabies may be easier, for epidemiologic purposes a more complete code to provide would be the ICD-10-CM code of Z11.8 (encounter for screening for other infectious and parasitic diseases), particularly if they are not found to have scabies.3 This code also would be useful when screening for head lice when a close contact is not found to have it. What do we currently code when someone comes for screening of head lice because a classmate or sibling has it? With ICD-10-CM we will have a more accurate way to communicate with the payor.
The increase in the number of codes with ICD-10-CM permits us to give a description on a situation or circumstance when a person who may not be sick comes into the office for a specific reason or when circumstances influence a person’s health status but those circumstances are not an actual illness or injury. It is frustrating that I currently am not able to code for a skin examination appropriately using ICD-9. Let me be clear, I am not saying that this new code will be reimbursable. We will have to wait and see how we are reimbursed for all of these codes, but at least we will have the option of coding for the reason the patient presents to me, which is often a skin examination, and then the secondary code could be benign nevi or seborrheic keratosis. The code Z12.83 (encounter for screening for malignant neoplasm of skin) will now be the best code for these purposes.3 In addition, codes that address postoperative nursing visits have not been available. With ICD-10-CM we will have a new code for encounter for surgical aftercare following surgery on the skin and subcutaneous tissue that does not include a standard suture removal but is a necessary visit (Z48.817). In addition, we will have a specific code to document encounters for allergy testing (Z01.82).3 With ICD-9, rash not otherwise specified or eczema would have to be the primary code (782.1) and you were forced to place a code for the patient’s allergy, regardless of whether or not the allergy was known.
The reimbursement of these codes has not been addressed, so ideally during the testing period there can be a head-to-head comparison. By adhering to some of the new rules, hopefully we will be able to more completely communicate about what is occurring during the patient visit.
It is important that dermatologists do not overlook the changes associated with the transition to International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM). Some physicians believe that providing this level of specificity is not important because at the end of the day, who is watching? You will see with several of the codes highlighted in this column that specificity may be required before a claim can be submitted to the payor for processing. In addition, some older catchall codes will go away, forcing us to provide additional specificity.1
Scabies coding is a good example of an added specificity requirement. Currently, with the International Classification of Diseases, Ninth Revision (ICD-9), we have a code for scabies but no code to address the postscabetic pruritus that patients often develop. The ICD-9 code 133.0 applies to scabies, and if we would like to code for the itch, we must enter a second code for unspecified pruritic disorder (698.9).2 With ICD-10-CM, we can be more specific and actually code for the cause of the itch. We will be able to mark the initial visit as B86 (scabies), but subsequent visits for the itch related to scabies can be coded as pruritus using the primary code L29.8 (other pruritus) and a new sequelae or late effect code of B94 (sequelae of other and unspecified infectious and parasitic diseases).3 You will still be able to submit the second visit with scabies as the primary diagnosis, but this practice should be avoided to prevent claim rejection on the backend. In addition, there will be codes to allow for evaluation of a family member or close contact for this condition. Although coding these patients as an initial visit for scabies may be easier, for epidemiologic purposes a more complete code to provide would be the ICD-10-CM code of Z11.8 (encounter for screening for other infectious and parasitic diseases), particularly if they are not found to have scabies.3 This code also would be useful when screening for head lice when a close contact is not found to have it. What do we currently code when someone comes for screening of head lice because a classmate or sibling has it? With ICD-10-CM we will have a more accurate way to communicate with the payor.
The increase in the number of codes with ICD-10-CM permits us to give a description on a situation or circumstance when a person who may not be sick comes into the office for a specific reason or when circumstances influence a person’s health status but those circumstances are not an actual illness or injury. It is frustrating that I currently am not able to code for a skin examination appropriately using ICD-9. Let me be clear, I am not saying that this new code will be reimbursable. We will have to wait and see how we are reimbursed for all of these codes, but at least we will have the option of coding for the reason the patient presents to me, which is often a skin examination, and then the secondary code could be benign nevi or seborrheic keratosis. The code Z12.83 (encounter for screening for malignant neoplasm of skin) will now be the best code for these purposes.3 In addition, codes that address postoperative nursing visits have not been available. With ICD-10-CM we will have a new code for encounter for surgical aftercare following surgery on the skin and subcutaneous tissue that does not include a standard suture removal but is a necessary visit (Z48.817). In addition, we will have a specific code to document encounters for allergy testing (Z01.82).3 With ICD-9, rash not otherwise specified or eczema would have to be the primary code (782.1) and you were forced to place a code for the patient’s allergy, regardless of whether or not the allergy was known.
The reimbursement of these codes has not been addressed, so ideally during the testing period there can be a head-to-head comparison. By adhering to some of the new rules, hopefully we will be able to more completely communicate about what is occurring during the patient visit.
1. Lamb A. Dermatology coding changes with ICD-10. Cutis. 2014;93:284-285.
2. ICD-9 code lookup. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/medicare-coverage-database/staticpages/icd-9-code-lookup.aspx. Accessed July 17, 2014.
3. Centers for Medicare & Medicaid Services. ICD-10-CM Tabular List of Diseases and Injuries. http://www.cms.gov/Medicare/Coding/ICD10/downloads/6_I10tab2010.pdf. Published 2010. Accessed July 17, 2014.
1. Lamb A. Dermatology coding changes with ICD-10. Cutis. 2014;93:284-285.
2. ICD-9 code lookup. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/medicare-coverage-database/staticpages/icd-9-code-lookup.aspx. Accessed July 17, 2014.
3. Centers for Medicare & Medicaid Services. ICD-10-CM Tabular List of Diseases and Injuries. http://www.cms.gov/Medicare/Coding/ICD10/downloads/6_I10tab2010.pdf. Published 2010. Accessed July 17, 2014.
Practice Points
- With International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM), dermatologists will have a more accurate way to communicate with the payor. For example, physicians will be able to code for scabies as well as the cause of postscabetic pruritus.
- Physicians will have the option of coding for the reason the patient presented. For example, dermatologists may code for a skin examination to screen for a malignant neoplasm.
- The reimbursement of the new codes has not been addressed; a head-to-head comparison will be needed during the testing period.
Can private practice survive?
I ran into Peter the other day. We weren’t close back in high school 50 years ago – Peter was a jock and I wasn’t – but both of us ended up in medicine.
"How’s the radiology business?" I asked him.
"Two more years," he said. "I should be able to hang on."
"That bad?"
"We were taken over by the academic department of a big teaching hospital," Peter said.
"What’s the problem? They want you to publish papers?" I asked.
"They’re not crazy," he said. "They know what kind of papers we’d write. They just want to measure things."
"Measure what?"
"Anything they can," he said. "Productivity, quality. They micromanage everything. We’re not a private practice anymore. We don’t call our own shots."
You hear a lot of talk these days about whether private practice can survive. Judging by anecdotal chats with colleagues in other specialties, I’d say prospects are iffy. A lot of the future is already here.
"I’m going crazy," a local community rheumatologist told me. "There was just one other guy in town. The local hospital knocked on his door and said, ‘Either sell out and work for us on our terms, or you’ll never get another referral.’ So he just closed up and moved out of state."
"How about you?" I asked him. "Have they made you an offer you can’t refuse?"
"Not yet," he said.
Then I met an oncologist from my wife’s family. He is a couple of years younger than I am. "I never thought I’d be working harder than I ever did at my age," he said. "And earning less.
"But we just sold our practice. No choice, really. Reimbursements don’t cover the cost of chemo drugs, so we lose money on every patient we give them to. The hospital that’s buying us can bill for drugs at a higher rate. So I’ll work for them for 5 years, then I’m out."
Then I heard about a neurologist, whose practice his hospital covets. He orders scans and other diagnostic procedures that generate hundreds of thousands of dollars annually, but that of course he can’t bill for. The hospital can. I don’t get how that works, but obviously the hospital does.
The trend toward consolidation proceeds. Doctors become salaried employees of large organizations. Maybe the main reason we dermatologists aren’t being swallowed yet by the big fish is that we’re just not tasty enough. We don’t generate admissions or enough costly procedures to make gulping us down worthwhile.
Switching from being an independent practitioner to a salaried employee means more than a change of location or even lower income; it signals a loss of status. I got a glimpse of how that plays out when I met another friend, an internist who used to be out on his own, but who now works for a big health care organization. "Have you got a quota of patients you have to meet?" I asked him.
"Yes," he said. "They just set that up recently."
"How did you find out?" I asked him.
"They sent out a memo," he said.
In other words, management notified doctors of productivity guidelines the same way that they would tell clerks or auxiliary personnel by which quality metrics their performance and salary would be judged.
"What if you don’t meet your quota because patients cancel or don’t keep their appointments?" I asked him. "You have no control over how patients are booked or reminded to come?"
"They blame the doctors anyway," he said. "Anyway, I’m not full time anymore. I’ll be retiring soon."
To say that hanging around colleagues who talk this way is dispiriting would be an understatement.
Peter the radiologist spoke in similar downbeat tones. "I’m adapting to the new reality fairly well," he said, "because I see the end in sight. But my younger colleagues are having a harder time. They thought they were joining a private practice, and none of them would have signed on for what they’re getting now: harder work, longer hours, no autonomy. They show up every morning cranky, muttering under their breath."
"I can see why," I said. "Whom do they take it out on?"
"Not the administrators," said Peter. "They don’t care. And certainly not the patients. They take it out on their families, I guess."
We agreed that seemed the best strategy.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Skin & Allergy News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years.
I ran into Peter the other day. We weren’t close back in high school 50 years ago – Peter was a jock and I wasn’t – but both of us ended up in medicine.
"How’s the radiology business?" I asked him.
"Two more years," he said. "I should be able to hang on."
"That bad?"
"We were taken over by the academic department of a big teaching hospital," Peter said.
"What’s the problem? They want you to publish papers?" I asked.
"They’re not crazy," he said. "They know what kind of papers we’d write. They just want to measure things."
"Measure what?"
"Anything they can," he said. "Productivity, quality. They micromanage everything. We’re not a private practice anymore. We don’t call our own shots."
You hear a lot of talk these days about whether private practice can survive. Judging by anecdotal chats with colleagues in other specialties, I’d say prospects are iffy. A lot of the future is already here.
"I’m going crazy," a local community rheumatologist told me. "There was just one other guy in town. The local hospital knocked on his door and said, ‘Either sell out and work for us on our terms, or you’ll never get another referral.’ So he just closed up and moved out of state."
"How about you?" I asked him. "Have they made you an offer you can’t refuse?"
"Not yet," he said.
Then I met an oncologist from my wife’s family. He is a couple of years younger than I am. "I never thought I’d be working harder than I ever did at my age," he said. "And earning less.
"But we just sold our practice. No choice, really. Reimbursements don’t cover the cost of chemo drugs, so we lose money on every patient we give them to. The hospital that’s buying us can bill for drugs at a higher rate. So I’ll work for them for 5 years, then I’m out."
Then I heard about a neurologist, whose practice his hospital covets. He orders scans and other diagnostic procedures that generate hundreds of thousands of dollars annually, but that of course he can’t bill for. The hospital can. I don’t get how that works, but obviously the hospital does.
The trend toward consolidation proceeds. Doctors become salaried employees of large organizations. Maybe the main reason we dermatologists aren’t being swallowed yet by the big fish is that we’re just not tasty enough. We don’t generate admissions or enough costly procedures to make gulping us down worthwhile.
Switching from being an independent practitioner to a salaried employee means more than a change of location or even lower income; it signals a loss of status. I got a glimpse of how that plays out when I met another friend, an internist who used to be out on his own, but who now works for a big health care organization. "Have you got a quota of patients you have to meet?" I asked him.
"Yes," he said. "They just set that up recently."
"How did you find out?" I asked him.
"They sent out a memo," he said.
In other words, management notified doctors of productivity guidelines the same way that they would tell clerks or auxiliary personnel by which quality metrics their performance and salary would be judged.
"What if you don’t meet your quota because patients cancel or don’t keep their appointments?" I asked him. "You have no control over how patients are booked or reminded to come?"
"They blame the doctors anyway," he said. "Anyway, I’m not full time anymore. I’ll be retiring soon."
To say that hanging around colleagues who talk this way is dispiriting would be an understatement.
Peter the radiologist spoke in similar downbeat tones. "I’m adapting to the new reality fairly well," he said, "because I see the end in sight. But my younger colleagues are having a harder time. They thought they were joining a private practice, and none of them would have signed on for what they’re getting now: harder work, longer hours, no autonomy. They show up every morning cranky, muttering under their breath."
"I can see why," I said. "Whom do they take it out on?"
"Not the administrators," said Peter. "They don’t care. And certainly not the patients. They take it out on their families, I guess."
We agreed that seemed the best strategy.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Skin & Allergy News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years.
I ran into Peter the other day. We weren’t close back in high school 50 years ago – Peter was a jock and I wasn’t – but both of us ended up in medicine.
"How’s the radiology business?" I asked him.
"Two more years," he said. "I should be able to hang on."
"That bad?"
"We were taken over by the academic department of a big teaching hospital," Peter said.
"What’s the problem? They want you to publish papers?" I asked.
"They’re not crazy," he said. "They know what kind of papers we’d write. They just want to measure things."
"Measure what?"
"Anything they can," he said. "Productivity, quality. They micromanage everything. We’re not a private practice anymore. We don’t call our own shots."
You hear a lot of talk these days about whether private practice can survive. Judging by anecdotal chats with colleagues in other specialties, I’d say prospects are iffy. A lot of the future is already here.
"I’m going crazy," a local community rheumatologist told me. "There was just one other guy in town. The local hospital knocked on his door and said, ‘Either sell out and work for us on our terms, or you’ll never get another referral.’ So he just closed up and moved out of state."
"How about you?" I asked him. "Have they made you an offer you can’t refuse?"
"Not yet," he said.
Then I met an oncologist from my wife’s family. He is a couple of years younger than I am. "I never thought I’d be working harder than I ever did at my age," he said. "And earning less.
"But we just sold our practice. No choice, really. Reimbursements don’t cover the cost of chemo drugs, so we lose money on every patient we give them to. The hospital that’s buying us can bill for drugs at a higher rate. So I’ll work for them for 5 years, then I’m out."
Then I heard about a neurologist, whose practice his hospital covets. He orders scans and other diagnostic procedures that generate hundreds of thousands of dollars annually, but that of course he can’t bill for. The hospital can. I don’t get how that works, but obviously the hospital does.
The trend toward consolidation proceeds. Doctors become salaried employees of large organizations. Maybe the main reason we dermatologists aren’t being swallowed yet by the big fish is that we’re just not tasty enough. We don’t generate admissions or enough costly procedures to make gulping us down worthwhile.
Switching from being an independent practitioner to a salaried employee means more than a change of location or even lower income; it signals a loss of status. I got a glimpse of how that plays out when I met another friend, an internist who used to be out on his own, but who now works for a big health care organization. "Have you got a quota of patients you have to meet?" I asked him.
"Yes," he said. "They just set that up recently."
"How did you find out?" I asked him.
"They sent out a memo," he said.
In other words, management notified doctors of productivity guidelines the same way that they would tell clerks or auxiliary personnel by which quality metrics their performance and salary would be judged.
"What if you don’t meet your quota because patients cancel or don’t keep their appointments?" I asked him. "You have no control over how patients are booked or reminded to come?"
"They blame the doctors anyway," he said. "Anyway, I’m not full time anymore. I’ll be retiring soon."
To say that hanging around colleagues who talk this way is dispiriting would be an understatement.
Peter the radiologist spoke in similar downbeat tones. "I’m adapting to the new reality fairly well," he said, "because I see the end in sight. But my younger colleagues are having a harder time. They thought they were joining a private practice, and none of them would have signed on for what they’re getting now: harder work, longer hours, no autonomy. They show up every morning cranky, muttering under their breath."
"I can see why," I said. "Whom do they take it out on?"
"Not the administrators," said Peter. "They don’t care. And certainly not the patients. They take it out on their families, I guess."
We agreed that seemed the best strategy.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Skin & Allergy News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years.

Laparoscopic power morcellation coverage will end as of September 1 for Blue Cross Blue Shield affiliate
About 5.2 million people insured by Highmark Inc, a Blue Cross and Blue Shield (BCBS)–affiliated company in Pennsylvania, Delaware, and West Virginia will be denied coverage for laparoscopic power morcellation used in gynecologic surgery. Highmark is the fourth-largest BCBS affiliate.1
On August 2, spokesman Aaron Billger announced by email that, in the best interest of their members, Highmark is halting coverage as of September 1, 2014.2 This decision comes in response to the US Food and Drug Administration (FDA) Safety Communication of April 17, 2014, that discouraged the use of power morcellation during hysterectomy and myomectomy because of the increased risk of dispersion of benign myoma tissue and occult malignant tissue through the abdominal cavity.2,3
University of Pittsburgh Medical Center (UPMC), the largest hospital group in western Pennsylvania, publicized that it will stop performing laparoscopic power morcellation as of September 1. UPMC spokesperson Gloria Kreps said the decision was an “appropriate and prudent course of action,” and that the hospital was “looking to the FDA for further guidance.”4 Other medical centers and hospitals have banned the use of power morcellation, beginning with Brigham and Women’s and Massachusetts General in April.5
In April 2014, Ethicon, a unit of Johnson & Johnson, suspended sales of all its morcellation devices. In an urgent Medical Device Market Withdrawal dated July 30, 2014, Ethicon initiated a “worldwide voluntary market withdrawal of all Ethicon Morcellation Devices that currently remain on the market.”6 Ethicon will issue prorated credit for morcellation devices returned by December 30, 2014, with full credit issued for unopened, unexpired disposable products.6
The FDA Obstetrics and Gynecology Devices Panel Advisory Committee held a 2-day hearing in July 2014 to weigh the risks and benefits of power morcellation. The panel will send recommendations to the FDA, and a final decision on the use of laparoscopic power morcellation will be forthcoming.7
1.
Reuters. U.S. insurer to stop coverage of gynecological procedure. http://in.reuters.com/article/2014/08/02/highmark-procedure-idINL2N0Q80IY20140802. Published August 3, 2014. Accessed August 6, 2014.
2. Kreiter M. Highmark Inc. says it won’t cover laparoscopic power morcellation because of cancer risk. International Business Times. http://www.ibtimes.com/highmark-inc-says-it-wont-cover-laparoscopic-power-morcellation-because-cancer-risk-1647160. Published August 2, 2014. Accessed August 6, 2014.
3. Yates J. FDA, hospitals caution against laparoscopic power morcellation during hysterectomy and myomectomy. OBG Manag. 2014;26(5):68, 67. http://www.obgmanagement.com/topic-collections/morcellation/article/fda-hospitals-caution-against-laparoscopic-power-morcellation-during-hysterectomy-and-myomectomy.html.
4. Mamula KB. UPMC drops controversial surgical procedure. Pittsburgh Business Times. http://www.bizjournals.com/pittsburgh/news/2014/08/01/upmc-drops-controversial-surgical-procedure.html. Published August 1, 2014. Accessed August 6, 2014.
5. Yates J. Open power morcellation of uterine tumors during hysterectomy banned at two Boston hospitals. OBG Manag. 2014;26(4). http://www.obgmanagement.com/topic-collections/morcellation/article/open-power-morcellation-of-uterine-tumors-during-hysterectomy-banned-at-two-boston-hospitals.html
6. Ethicon. Urgent: Medical device market withdrawal. http://www.ethicon.com/sites/default/files/managed-documents/Ethicon%20Morcellation%20Devices%20Customer%20Letter%20Final.pdf. Published July 30, 2014. Accessed August 6, 2014.
7. Iglesia C, Yates J. Why FDA hearing on morcellation safety could drive innovation [audiocast]. OBG Manag. 2014;26(7). http://www.obgmanagement.com/home/article/why-fda-hearing-on-morcellation-safety-could-drive-innovation/d1071c5e8326e8a2de76a30f0446b1ab.html. Published July 17, 2014. Accessed August 6, 2014.
About 5.2 million people insured by Highmark Inc, a Blue Cross and Blue Shield (BCBS)–affiliated company in Pennsylvania, Delaware, and West Virginia will be denied coverage for laparoscopic power morcellation used in gynecologic surgery. Highmark is the fourth-largest BCBS affiliate.1
On August 2, spokesman Aaron Billger announced by email that, in the best interest of their members, Highmark is halting coverage as of September 1, 2014.2 This decision comes in response to the US Food and Drug Administration (FDA) Safety Communication of April 17, 2014, that discouraged the use of power morcellation during hysterectomy and myomectomy because of the increased risk of dispersion of benign myoma tissue and occult malignant tissue through the abdominal cavity.2,3
University of Pittsburgh Medical Center (UPMC), the largest hospital group in western Pennsylvania, publicized that it will stop performing laparoscopic power morcellation as of September 1. UPMC spokesperson Gloria Kreps said the decision was an “appropriate and prudent course of action,” and that the hospital was “looking to the FDA for further guidance.”4 Other medical centers and hospitals have banned the use of power morcellation, beginning with Brigham and Women’s and Massachusetts General in April.5
In April 2014, Ethicon, a unit of Johnson & Johnson, suspended sales of all its morcellation devices. In an urgent Medical Device Market Withdrawal dated July 30, 2014, Ethicon initiated a “worldwide voluntary market withdrawal of all Ethicon Morcellation Devices that currently remain on the market.”6 Ethicon will issue prorated credit for morcellation devices returned by December 30, 2014, with full credit issued for unopened, unexpired disposable products.6
The FDA Obstetrics and Gynecology Devices Panel Advisory Committee held a 2-day hearing in July 2014 to weigh the risks and benefits of power morcellation. The panel will send recommendations to the FDA, and a final decision on the use of laparoscopic power morcellation will be forthcoming.7
About 5.2 million people insured by Highmark Inc, a Blue Cross and Blue Shield (BCBS)–affiliated company in Pennsylvania, Delaware, and West Virginia will be denied coverage for laparoscopic power morcellation used in gynecologic surgery. Highmark is the fourth-largest BCBS affiliate.1
On August 2, spokesman Aaron Billger announced by email that, in the best interest of their members, Highmark is halting coverage as of September 1, 2014.2 This decision comes in response to the US Food and Drug Administration (FDA) Safety Communication of April 17, 2014, that discouraged the use of power morcellation during hysterectomy and myomectomy because of the increased risk of dispersion of benign myoma tissue and occult malignant tissue through the abdominal cavity.2,3
University of Pittsburgh Medical Center (UPMC), the largest hospital group in western Pennsylvania, publicized that it will stop performing laparoscopic power morcellation as of September 1. UPMC spokesperson Gloria Kreps said the decision was an “appropriate and prudent course of action,” and that the hospital was “looking to the FDA for further guidance.”4 Other medical centers and hospitals have banned the use of power morcellation, beginning with Brigham and Women’s and Massachusetts General in April.5
In April 2014, Ethicon, a unit of Johnson & Johnson, suspended sales of all its morcellation devices. In an urgent Medical Device Market Withdrawal dated July 30, 2014, Ethicon initiated a “worldwide voluntary market withdrawal of all Ethicon Morcellation Devices that currently remain on the market.”6 Ethicon will issue prorated credit for morcellation devices returned by December 30, 2014, with full credit issued for unopened, unexpired disposable products.6
The FDA Obstetrics and Gynecology Devices Panel Advisory Committee held a 2-day hearing in July 2014 to weigh the risks and benefits of power morcellation. The panel will send recommendations to the FDA, and a final decision on the use of laparoscopic power morcellation will be forthcoming.7
1.
Reuters. U.S. insurer to stop coverage of gynecological procedure. http://in.reuters.com/article/2014/08/02/highmark-procedure-idINL2N0Q80IY20140802. Published August 3, 2014. Accessed August 6, 2014.
2. Kreiter M. Highmark Inc. says it won’t cover laparoscopic power morcellation because of cancer risk. International Business Times. http://www.ibtimes.com/highmark-inc-says-it-wont-cover-laparoscopic-power-morcellation-because-cancer-risk-1647160. Published August 2, 2014. Accessed August 6, 2014.
3. Yates J. FDA, hospitals caution against laparoscopic power morcellation during hysterectomy and myomectomy. OBG Manag. 2014;26(5):68, 67. http://www.obgmanagement.com/topic-collections/morcellation/article/fda-hospitals-caution-against-laparoscopic-power-morcellation-during-hysterectomy-and-myomectomy.html.
4. Mamula KB. UPMC drops controversial surgical procedure. Pittsburgh Business Times. http://www.bizjournals.com/pittsburgh/news/2014/08/01/upmc-drops-controversial-surgical-procedure.html. Published August 1, 2014. Accessed August 6, 2014.
5. Yates J. Open power morcellation of uterine tumors during hysterectomy banned at two Boston hospitals. OBG Manag. 2014;26(4). http://www.obgmanagement.com/topic-collections/morcellation/article/open-power-morcellation-of-uterine-tumors-during-hysterectomy-banned-at-two-boston-hospitals.html
6. Ethicon. Urgent: Medical device market withdrawal. http://www.ethicon.com/sites/default/files/managed-documents/Ethicon%20Morcellation%20Devices%20Customer%20Letter%20Final.pdf. Published July 30, 2014. Accessed August 6, 2014.
7. Iglesia C, Yates J. Why FDA hearing on morcellation safety could drive innovation [audiocast]. OBG Manag. 2014;26(7). http://www.obgmanagement.com/home/article/why-fda-hearing-on-morcellation-safety-could-drive-innovation/d1071c5e8326e8a2de76a30f0446b1ab.html. Published July 17, 2014. Accessed August 6, 2014.
1.
Reuters. U.S. insurer to stop coverage of gynecological procedure. http://in.reuters.com/article/2014/08/02/highmark-procedure-idINL2N0Q80IY20140802. Published August 3, 2014. Accessed August 6, 2014.
2. Kreiter M. Highmark Inc. says it won’t cover laparoscopic power morcellation because of cancer risk. International Business Times. http://www.ibtimes.com/highmark-inc-says-it-wont-cover-laparoscopic-power-morcellation-because-cancer-risk-1647160. Published August 2, 2014. Accessed August 6, 2014.
3. Yates J. FDA, hospitals caution against laparoscopic power morcellation during hysterectomy and myomectomy. OBG Manag. 2014;26(5):68, 67. http://www.obgmanagement.com/topic-collections/morcellation/article/fda-hospitals-caution-against-laparoscopic-power-morcellation-during-hysterectomy-and-myomectomy.html.
4. Mamula KB. UPMC drops controversial surgical procedure. Pittsburgh Business Times. http://www.bizjournals.com/pittsburgh/news/2014/08/01/upmc-drops-controversial-surgical-procedure.html. Published August 1, 2014. Accessed August 6, 2014.
5. Yates J. Open power morcellation of uterine tumors during hysterectomy banned at two Boston hospitals. OBG Manag. 2014;26(4). http://www.obgmanagement.com/topic-collections/morcellation/article/open-power-morcellation-of-uterine-tumors-during-hysterectomy-banned-at-two-boston-hospitals.html
6. Ethicon. Urgent: Medical device market withdrawal. http://www.ethicon.com/sites/default/files/managed-documents/Ethicon%20Morcellation%20Devices%20Customer%20Letter%20Final.pdf. Published July 30, 2014. Accessed August 6, 2014.
7. Iglesia C, Yates J. Why FDA hearing on morcellation safety could drive innovation [audiocast]. OBG Manag. 2014;26(7). http://www.obgmanagement.com/home/article/why-fda-hearing-on-morcellation-safety-could-drive-innovation/d1071c5e8326e8a2de76a30f0446b1ab.html. Published July 17, 2014. Accessed August 6, 2014.
Ebola, fear, and us
Recently, two American missionary workers in Liberia made headlines when they became infected with the Ebola virus, which causes a highly fatal hemorrhagic fever. They both received an experimental "secret serum" called ZMapp while in Africa and have since been flown back to the United States to be cared for at Emory University Hospital in Atlanta.
The response to their return has been mixed. While some hail their incredible selflessness and the mission they set out to accomplish, others are lukewarm, indifferent, or even very opposed to their return – so opposed that security was heightened in response to threats. I, of course, am among the former.
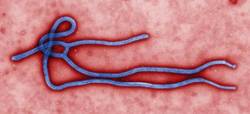
I once went on a 5-day missionary trip to Nicaragua. Optimistic about the potential to help the poor and hurting, my biggest challenges were the long plane ride and the hotel accommodations, which were not quite to my liking. The thought of contracting a potentially fatal disease and never returning home to my family never, ever crossed my mind.
Dr. Kent Brantly and Nancy Writebol, both missionary workers, were of a completely different mindset; they had different goals, they faced different challenges. They willingly boarded a plane to fly thousands of miles away from the comfort of their homes, the love of their families, the security of their close friends. They risked everything to care for complete strangers, strangers who posed a real threat to their lives. They never expected to receive any earthly thing in return. Interestingly, after contracting Ebola, Dr. Brantley did receive a unit of blood from a 14-year-old who had survived his fight with the Ebola virus while under his care.
Personally, I can only hope that one day I will have even half of the compassion and servant’s heart that these brave, incredibly selfless individuals possess.
I have to believe that those who object to their presence on American soil are afraid. They do not know what to expect and fear the epidemic spreading to the United States. I remember when I first started treating HIV/AIDS patients in the early ’90s. It was new and it was very, very scary. In those days, we donned protective garments above and beyond what we now know is necessary. Still, at times I, along with countless other health care workers, was overwhelmed by fear.
Like the human immunodeficiency virus, the Ebola virus is spread through direct contact with the blood or other body fluids of an infected person or through exposure to infected objects such as needles. It is not airborne, nor is it spread by contaminated food and water. Unlike with HIV, however, Ebola can be fatal within days. Another difference is that it is believed to be transmissible only if the individual is symptomatic, while countless cases of HIV were the result of exposure to seemingly healthy individuals.
While some in the American public still decry that Dr. Brantley and Mrs. Writebol were brought to a hospital in the United States, I have to believe that the great majority of Americans keep these two incredible individuals in their thoughts and prayers.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected].
Recently, two American missionary workers in Liberia made headlines when they became infected with the Ebola virus, which causes a highly fatal hemorrhagic fever. They both received an experimental "secret serum" called ZMapp while in Africa and have since been flown back to the United States to be cared for at Emory University Hospital in Atlanta.
The response to their return has been mixed. While some hail their incredible selflessness and the mission they set out to accomplish, others are lukewarm, indifferent, or even very opposed to their return – so opposed that security was heightened in response to threats. I, of course, am among the former.
I once went on a 5-day missionary trip to Nicaragua. Optimistic about the potential to help the poor and hurting, my biggest challenges were the long plane ride and the hotel accommodations, which were not quite to my liking. The thought of contracting a potentially fatal disease and never returning home to my family never, ever crossed my mind.
Dr. Kent Brantly and Nancy Writebol, both missionary workers, were of a completely different mindset; they had different goals, they faced different challenges. They willingly boarded a plane to fly thousands of miles away from the comfort of their homes, the love of their families, the security of their close friends. They risked everything to care for complete strangers, strangers who posed a real threat to their lives. They never expected to receive any earthly thing in return. Interestingly, after contracting Ebola, Dr. Brantley did receive a unit of blood from a 14-year-old who had survived his fight with the Ebola virus while under his care.
Personally, I can only hope that one day I will have even half of the compassion and servant’s heart that these brave, incredibly selfless individuals possess.
I have to believe that those who object to their presence on American soil are afraid. They do not know what to expect and fear the epidemic spreading to the United States. I remember when I first started treating HIV/AIDS patients in the early ’90s. It was new and it was very, very scary. In those days, we donned protective garments above and beyond what we now know is necessary. Still, at times I, along with countless other health care workers, was overwhelmed by fear.
Like the human immunodeficiency virus, the Ebola virus is spread through direct contact with the blood or other body fluids of an infected person or through exposure to infected objects such as needles. It is not airborne, nor is it spread by contaminated food and water. Unlike with HIV, however, Ebola can be fatal within days. Another difference is that it is believed to be transmissible only if the individual is symptomatic, while countless cases of HIV were the result of exposure to seemingly healthy individuals.
While some in the American public still decry that Dr. Brantley and Mrs. Writebol were brought to a hospital in the United States, I have to believe that the great majority of Americans keep these two incredible individuals in their thoughts and prayers.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected].
Recently, two American missionary workers in Liberia made headlines when they became infected with the Ebola virus, which causes a highly fatal hemorrhagic fever. They both received an experimental "secret serum" called ZMapp while in Africa and have since been flown back to the United States to be cared for at Emory University Hospital in Atlanta.
The response to their return has been mixed. While some hail their incredible selflessness and the mission they set out to accomplish, others are lukewarm, indifferent, or even very opposed to their return – so opposed that security was heightened in response to threats. I, of course, am among the former.
I once went on a 5-day missionary trip to Nicaragua. Optimistic about the potential to help the poor and hurting, my biggest challenges were the long plane ride and the hotel accommodations, which were not quite to my liking. The thought of contracting a potentially fatal disease and never returning home to my family never, ever crossed my mind.
Dr. Kent Brantly and Nancy Writebol, both missionary workers, were of a completely different mindset; they had different goals, they faced different challenges. They willingly boarded a plane to fly thousands of miles away from the comfort of their homes, the love of their families, the security of their close friends. They risked everything to care for complete strangers, strangers who posed a real threat to their lives. They never expected to receive any earthly thing in return. Interestingly, after contracting Ebola, Dr. Brantley did receive a unit of blood from a 14-year-old who had survived his fight with the Ebola virus while under his care.
Personally, I can only hope that one day I will have even half of the compassion and servant’s heart that these brave, incredibly selfless individuals possess.
I have to believe that those who object to their presence on American soil are afraid. They do not know what to expect and fear the epidemic spreading to the United States. I remember when I first started treating HIV/AIDS patients in the early ’90s. It was new and it was very, very scary. In those days, we donned protective garments above and beyond what we now know is necessary. Still, at times I, along with countless other health care workers, was overwhelmed by fear.
Like the human immunodeficiency virus, the Ebola virus is spread through direct contact with the blood or other body fluids of an infected person or through exposure to infected objects such as needles. It is not airborne, nor is it spread by contaminated food and water. Unlike with HIV, however, Ebola can be fatal within days. Another difference is that it is believed to be transmissible only if the individual is symptomatic, while countless cases of HIV were the result of exposure to seemingly healthy individuals.
While some in the American public still decry that Dr. Brantley and Mrs. Writebol were brought to a hospital in the United States, I have to believe that the great majority of Americans keep these two incredible individuals in their thoughts and prayers.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected].

When ‘normal’ just isn’t normal
As pediatricians, we are constantly evaluating children of all ages. We make determinations of normal and abnormal all the time. But, sometimes determining normal can be a challenge because children come in all shapes, sizes, and complexions, so "normal" can appear in a variety of ways.
When it comes to the adolescent, this is an even greater challenge because the onset of puberty is so varied that children of the same age can look vastly different, and pubertal changes only widen the variety. Obesity also has impacted the appearance of normal because it makes children look older, pubertal changes more advanced, and a thorough exam more difficult. So when is it okay to say, "They will just grow out of it?" Well, the best answer is when all the serious illnesses have been considered and ruled out.

Gynecomastia is a common finding in the adolescent wellness exam; 50%-60% of adolescent males experience some degree of breast enlargement starting at the age of 10 years. This peaks at ages 13-14, then regresses over a period of 18 months (N. Engl. J. Med. 2007;357:1229-37). For approximately 25% of children, the breast tissue persists, which leads to significant anxiety and insecurities among adolescent males. Even when asked if they have concerns, few will admit to it because the thought of the evaluation is more than they can handle.
Gynecomastia is caused from the increased ratio of estrogen to androgen. Antiandrogens, drugs, and weight gain have all been implicated. But in the evaluation of increased breast tissue, normal as well as abnormal causes have to be considered.
Exogenous causes include herbal products, such as tea tree oil, or medications. The most common drugs are cimetidine, ranitidine, and omeprazole, as well spirolactone and ketoconazole. With the exception of spirolactone, these are all drugs that are used commonly for minor illness in children, but have been identified as a cause for gynecomastia. Discontinuation of these products usually resolves the issue within a few months (Pharmacotherapy 1993;13:37-45).
Obesity can cause a pseudogynecomastia as well as a true gynecomastia because aromatase enzyme increases with the increase in fat tissue, which converts testosterone to estradiol. Clinically, pseudogynecomastia can be distinguished from true gynecomastia by doing a breast exam. True gynecomastia is a concentric, rubbery firm mass greater than 0.5cm, and directly below the areola, where pseudogynecomastia has diffuse enlargement and no discernable glandular tissue.
Abnormal causes of gynecomastia are much less common, but do occur. A careful physical examination and a detailed review of systems can be very helpful in ruling in or out serious causes.
An imbalance of estrogen and testosterone can result from estrogen or testosterone going up or down. These changes can be caused by other hormonal stimulation. Human chorionic gonadotropin (HGC) is increased with germ cell tumors, which can be found in abdominal or testicular masses, resulting in secondary hypogonadism. Elevated estradiol is found with testicular tumors and adrenal tumors.
Hyperthyroidism can cause gynecomastia. Additional symptoms include palpitations, weight loss, and anxiety. Physical findings include a goiter, exophthalmoses, and tremors.
Klinefelter’s syndrome, a condition that occurs in men who have an extra X chromosome, includes gynecomastia and hypogonadism. There is a 20%-60% increased risk of breast cancer in these patients, who tend to have less facial and body hair, reduced muscle tone, and narrower shoulders and wider hips (N. Engl. J. Med. 2007;357:1229-37). Suspicion of breast cancer should increase if the mass is unilateral, nontender, and eccentric to the areola.
Although the vast majority of patients with gynecomastia will resolve spontaneously, careful evaluation and consideration of abnormal causes can lead to early diagnosis and treatment.
Experienced pediatricians know it’s never "nothing" unless all the possible "somethings" have been ruled out!
Dr. Pearce is a pediatrician in Frankfort, Ill. E-mail her at [email protected].
As pediatricians, we are constantly evaluating children of all ages. We make determinations of normal and abnormal all the time. But, sometimes determining normal can be a challenge because children come in all shapes, sizes, and complexions, so "normal" can appear in a variety of ways.
When it comes to the adolescent, this is an even greater challenge because the onset of puberty is so varied that children of the same age can look vastly different, and pubertal changes only widen the variety. Obesity also has impacted the appearance of normal because it makes children look older, pubertal changes more advanced, and a thorough exam more difficult. So when is it okay to say, "They will just grow out of it?" Well, the best answer is when all the serious illnesses have been considered and ruled out.
Gynecomastia is a common finding in the adolescent wellness exam; 50%-60% of adolescent males experience some degree of breast enlargement starting at the age of 10 years. This peaks at ages 13-14, then regresses over a period of 18 months (N. Engl. J. Med. 2007;357:1229-37). For approximately 25% of children, the breast tissue persists, which leads to significant anxiety and insecurities among adolescent males. Even when asked if they have concerns, few will admit to it because the thought of the evaluation is more than they can handle.
Gynecomastia is caused from the increased ratio of estrogen to androgen. Antiandrogens, drugs, and weight gain have all been implicated. But in the evaluation of increased breast tissue, normal as well as abnormal causes have to be considered.
Exogenous causes include herbal products, such as tea tree oil, or medications. The most common drugs are cimetidine, ranitidine, and omeprazole, as well spirolactone and ketoconazole. With the exception of spirolactone, these are all drugs that are used commonly for minor illness in children, but have been identified as a cause for gynecomastia. Discontinuation of these products usually resolves the issue within a few months (Pharmacotherapy 1993;13:37-45).
Obesity can cause a pseudogynecomastia as well as a true gynecomastia because aromatase enzyme increases with the increase in fat tissue, which converts testosterone to estradiol. Clinically, pseudogynecomastia can be distinguished from true gynecomastia by doing a breast exam. True gynecomastia is a concentric, rubbery firm mass greater than 0.5cm, and directly below the areola, where pseudogynecomastia has diffuse enlargement and no discernable glandular tissue.
Abnormal causes of gynecomastia are much less common, but do occur. A careful physical examination and a detailed review of systems can be very helpful in ruling in or out serious causes.
An imbalance of estrogen and testosterone can result from estrogen or testosterone going up or down. These changes can be caused by other hormonal stimulation. Human chorionic gonadotropin (HGC) is increased with germ cell tumors, which can be found in abdominal or testicular masses, resulting in secondary hypogonadism. Elevated estradiol is found with testicular tumors and adrenal tumors.
Hyperthyroidism can cause gynecomastia. Additional symptoms include palpitations, weight loss, and anxiety. Physical findings include a goiter, exophthalmoses, and tremors.
Klinefelter’s syndrome, a condition that occurs in men who have an extra X chromosome, includes gynecomastia and hypogonadism. There is a 20%-60% increased risk of breast cancer in these patients, who tend to have less facial and body hair, reduced muscle tone, and narrower shoulders and wider hips (N. Engl. J. Med. 2007;357:1229-37). Suspicion of breast cancer should increase if the mass is unilateral, nontender, and eccentric to the areola.
Although the vast majority of patients with gynecomastia will resolve spontaneously, careful evaluation and consideration of abnormal causes can lead to early diagnosis and treatment.
Experienced pediatricians know it’s never "nothing" unless all the possible "somethings" have been ruled out!
Dr. Pearce is a pediatrician in Frankfort, Ill. E-mail her at [email protected].
As pediatricians, we are constantly evaluating children of all ages. We make determinations of normal and abnormal all the time. But, sometimes determining normal can be a challenge because children come in all shapes, sizes, and complexions, so "normal" can appear in a variety of ways.
When it comes to the adolescent, this is an even greater challenge because the onset of puberty is so varied that children of the same age can look vastly different, and pubertal changes only widen the variety. Obesity also has impacted the appearance of normal because it makes children look older, pubertal changes more advanced, and a thorough exam more difficult. So when is it okay to say, "They will just grow out of it?" Well, the best answer is when all the serious illnesses have been considered and ruled out.
Gynecomastia is a common finding in the adolescent wellness exam; 50%-60% of adolescent males experience some degree of breast enlargement starting at the age of 10 years. This peaks at ages 13-14, then regresses over a period of 18 months (N. Engl. J. Med. 2007;357:1229-37). For approximately 25% of children, the breast tissue persists, which leads to significant anxiety and insecurities among adolescent males. Even when asked if they have concerns, few will admit to it because the thought of the evaluation is more than they can handle.
Gynecomastia is caused from the increased ratio of estrogen to androgen. Antiandrogens, drugs, and weight gain have all been implicated. But in the evaluation of increased breast tissue, normal as well as abnormal causes have to be considered.
Exogenous causes include herbal products, such as tea tree oil, or medications. The most common drugs are cimetidine, ranitidine, and omeprazole, as well spirolactone and ketoconazole. With the exception of spirolactone, these are all drugs that are used commonly for minor illness in children, but have been identified as a cause for gynecomastia. Discontinuation of these products usually resolves the issue within a few months (Pharmacotherapy 1993;13:37-45).
Obesity can cause a pseudogynecomastia as well as a true gynecomastia because aromatase enzyme increases with the increase in fat tissue, which converts testosterone to estradiol. Clinically, pseudogynecomastia can be distinguished from true gynecomastia by doing a breast exam. True gynecomastia is a concentric, rubbery firm mass greater than 0.5cm, and directly below the areola, where pseudogynecomastia has diffuse enlargement and no discernable glandular tissue.
Abnormal causes of gynecomastia are much less common, but do occur. A careful physical examination and a detailed review of systems can be very helpful in ruling in or out serious causes.
An imbalance of estrogen and testosterone can result from estrogen or testosterone going up or down. These changes can be caused by other hormonal stimulation. Human chorionic gonadotropin (HGC) is increased with germ cell tumors, which can be found in abdominal or testicular masses, resulting in secondary hypogonadism. Elevated estradiol is found with testicular tumors and adrenal tumors.
Hyperthyroidism can cause gynecomastia. Additional symptoms include palpitations, weight loss, and anxiety. Physical findings include a goiter, exophthalmoses, and tremors.
Klinefelter’s syndrome, a condition that occurs in men who have an extra X chromosome, includes gynecomastia and hypogonadism. There is a 20%-60% increased risk of breast cancer in these patients, who tend to have less facial and body hair, reduced muscle tone, and narrower shoulders and wider hips (N. Engl. J. Med. 2007;357:1229-37). Suspicion of breast cancer should increase if the mass is unilateral, nontender, and eccentric to the areola.
Although the vast majority of patients with gynecomastia will resolve spontaneously, careful evaluation and consideration of abnormal causes can lead to early diagnosis and treatment.
Experienced pediatricians know it’s never "nothing" unless all the possible "somethings" have been ruled out!
Dr. Pearce is a pediatrician in Frankfort, Ill. E-mail her at [email protected].

Method corrects β-thalassemia mutations

Credit: Salk Institute
Genome editing technology allows for seamless correction of disease-causing mutations in cells from patients with β-thalassemia, investigators have reported in Genome Research.
The team noted that β-thalassemia results from inherited mutations in the hemoglobin beta (HBB) gene, which prompt reduced HBB expression in red blood cells, as well as anemia.
The only established curative treatment is hematopoietic stem cell transplant, but this requires a matched donor.
Gene therapy could eliminate this need.
To correct HBB mutations directly in a patient’s genome, Yuet Wai Kan, MD, of the University of California, San Francisco, and his colleagues first generated induced pluripotent stem cells (iPSCs) from patients’ skin cells.
The team then used CRISPR/Cas9 technology to precisely engineer a double-strand DNA break at the HBB locus in the iPSCs, allowing a donor plasmid with the corrected sites to be efficiently integrated, thus replacing the mutated sites.
The donor plasmid also contained selectable markers to identify cells with corrected copies of the gene. These selectable markers were subsequently removed with transposase and a second round of selection, generating a seamless, corrected version of HBB in the patient’s genome.
The investigators found the corrected iPSCs could differentiate into mature blood cells, and these blood cells showed restored expression of hemoglobin.
However, the team said a lot more work is needed before these cells could be transplanted to treat a patient with β-thalassemia.
“Although we and others are able to differentiate iPSCs into blood cell progenitors as well as mature blood cells, the transplantation of the progenitors into mouse models to test them has, so far, proven very difficult,” Dr Kan said. “I believe it will take quite a few more years before we can apply it in a clinical setting.” ![]()
Credit: Salk Institute
Genome editing technology allows for seamless correction of disease-causing mutations in cells from patients with β-thalassemia, investigators have reported in Genome Research.
The team noted that β-thalassemia results from inherited mutations in the hemoglobin beta (HBB) gene, which prompt reduced HBB expression in red blood cells, as well as anemia.
The only established curative treatment is hematopoietic stem cell transplant, but this requires a matched donor.
Gene therapy could eliminate this need.
To correct HBB mutations directly in a patient’s genome, Yuet Wai Kan, MD, of the University of California, San Francisco, and his colleagues first generated induced pluripotent stem cells (iPSCs) from patients’ skin cells.
The team then used CRISPR/Cas9 technology to precisely engineer a double-strand DNA break at the HBB locus in the iPSCs, allowing a donor plasmid with the corrected sites to be efficiently integrated, thus replacing the mutated sites.
The donor plasmid also contained selectable markers to identify cells with corrected copies of the gene. These selectable markers were subsequently removed with transposase and a second round of selection, generating a seamless, corrected version of HBB in the patient’s genome.
The investigators found the corrected iPSCs could differentiate into mature blood cells, and these blood cells showed restored expression of hemoglobin.
However, the team said a lot more work is needed before these cells could be transplanted to treat a patient with β-thalassemia.
“Although we and others are able to differentiate iPSCs into blood cell progenitors as well as mature blood cells, the transplantation of the progenitors into mouse models to test them has, so far, proven very difficult,” Dr Kan said. “I believe it will take quite a few more years before we can apply it in a clinical setting.” ![]()
Credit: Salk Institute
Genome editing technology allows for seamless correction of disease-causing mutations in cells from patients with β-thalassemia, investigators have reported in Genome Research.
The team noted that β-thalassemia results from inherited mutations in the hemoglobin beta (HBB) gene, which prompt reduced HBB expression in red blood cells, as well as anemia.
The only established curative treatment is hematopoietic stem cell transplant, but this requires a matched donor.
Gene therapy could eliminate this need.
To correct HBB mutations directly in a patient’s genome, Yuet Wai Kan, MD, of the University of California, San Francisco, and his colleagues first generated induced pluripotent stem cells (iPSCs) from patients’ skin cells.
The team then used CRISPR/Cas9 technology to precisely engineer a double-strand DNA break at the HBB locus in the iPSCs, allowing a donor plasmid with the corrected sites to be efficiently integrated, thus replacing the mutated sites.
The donor plasmid also contained selectable markers to identify cells with corrected copies of the gene. These selectable markers were subsequently removed with transposase and a second round of selection, generating a seamless, corrected version of HBB in the patient’s genome.
The investigators found the corrected iPSCs could differentiate into mature blood cells, and these blood cells showed restored expression of hemoglobin.
However, the team said a lot more work is needed before these cells could be transplanted to treat a patient with β-thalassemia.
“Although we and others are able to differentiate iPSCs into blood cell progenitors as well as mature blood cells, the transplantation of the progenitors into mouse models to test them has, so far, proven very difficult,” Dr Kan said. “I believe it will take quite a few more years before we can apply it in a clinical setting.” ![]()
Battling Multidrug Resistant UTIs With Methenamine Hippurate
Recently, Federal Practitioner talked with Rebecca McAllister, MS, FNP-BC, about her role in treating complicated urinary tract infections (UTIs) in elderly patients at the Community Living Center of the Bay Pines VA Healthcare System in Florida. The original July 2014 Case in Point, “Recurrent Multidrug Resistant Urinary Tract Infections in Geriatric Patients,” discussed 4 case studies, which suggested the safety and efficacy of treatment with methenamine hippurate.
Federal Practitioner: Much of the focus in your article was on the use of methenamine in Norway and Sweden and a lack thereof in the U.S. How are multidrug resistant UTIs generally treated in the U.S., and how could this be handled differently?
Rebecca McAllister, MS, FNP-BC: Because of the increased rates of bacterial resistance, treating recurrent UTIs prophylactically with low-dose antibiotics is no longer the standard of care. Currently, multidrug resistant UTIs are treated with broad-spectrum antibiotics, that organisms are susceptible to. The promise of methenamine relies on the bacteria not developing resistance to it; in turn, long-term use in patients does not contribute to developing resistance.
FP: Is methenamine hippurate readily available within the VA, and what are the guidelines surrounding its use?
RM: Methenamine is on the VA formulary available in 1-gm doses. Standard guidelines per Micromedex are for prophylaxis of recurrent UTIs, as mentioned in the article, and contraindicates use in patients with impaired renal function, although specific parameters are not identified, because testing was never done with geriatric patients.
FP: Of the 4 patients discussed in the article, 3 were aged > 89 years and the fourth was aged exactly 89 years. Was this a coincidence, or does the success of methenamine in this oldest-old cohort highlight UTI recurrence rate late in life, the failed efficacy of other drugs over time, or both?
RM: The success of methenamine highlights both UTI recurrence rate late in life and the failed efficacy of other drugs over time. The primary patient group in these case studies was composed of homebound veterans.
FP: At the beginning of the discussion portion of your article, following 4 case studies, you mention, “Patients with similar profiles to those discussed in this report were treated with less dramatic results.” How do you, as a family nurse practitioner, consider treatment a success, and how might this differ from expectations set by a medical facility?
RM: As is always the challenge with preventive interventions, it is difficult to measure what does not happen. Successful treatment for recurrent UTIs is lack of recurrence, also asymptomatic colonization vs a symptomatic UTI, which can be measured by a urinalysis is a success. In the oldest of the old, delayed hospitalization for urosepsis, reduced risk of falls, and increased mortality are also successes. Cost savings to the health care system by administering an inexpensive preventive medication vs very expensive IV antibiotic therapy, another success. The observed changes in bacterial resistance in patients treated with methenamine offers great hope in the battle against bacteria.
Ms. McAllister coauthored the July 2014 article, “Recurrent Multidrug Resistant Urinary Tract Infections in Geriatric Patients,” with Janice Allwood, MS, ARNP, CUNP.
Ms. McAllister is a Community Living Center family nurse practitioner and Ms. Allwood is an advanced registered nurse practitioner in Urology Surgery, both at the Bay Pines VA Healthcare System in Florida.
Recently, Federal Practitioner talked with Rebecca McAllister, MS, FNP-BC, about her role in treating complicated urinary tract infections (UTIs) in elderly patients at the Community Living Center of the Bay Pines VA Healthcare System in Florida. The original July 2014 Case in Point, “Recurrent Multidrug Resistant Urinary Tract Infections in Geriatric Patients,” discussed 4 case studies, which suggested the safety and efficacy of treatment with methenamine hippurate.
Federal Practitioner: Much of the focus in your article was on the use of methenamine in Norway and Sweden and a lack thereof in the U.S. How are multidrug resistant UTIs generally treated in the U.S., and how could this be handled differently?
Rebecca McAllister, MS, FNP-BC: Because of the increased rates of bacterial resistance, treating recurrent UTIs prophylactically with low-dose antibiotics is no longer the standard of care. Currently, multidrug resistant UTIs are treated with broad-spectrum antibiotics, that organisms are susceptible to. The promise of methenamine relies on the bacteria not developing resistance to it; in turn, long-term use in patients does not contribute to developing resistance.
FP: Is methenamine hippurate readily available within the VA, and what are the guidelines surrounding its use?
RM: Methenamine is on the VA formulary available in 1-gm doses. Standard guidelines per Micromedex are for prophylaxis of recurrent UTIs, as mentioned in the article, and contraindicates use in patients with impaired renal function, although specific parameters are not identified, because testing was never done with geriatric patients.
FP: Of the 4 patients discussed in the article, 3 were aged > 89 years and the fourth was aged exactly 89 years. Was this a coincidence, or does the success of methenamine in this oldest-old cohort highlight UTI recurrence rate late in life, the failed efficacy of other drugs over time, or both?
RM: The success of methenamine highlights both UTI recurrence rate late in life and the failed efficacy of other drugs over time. The primary patient group in these case studies was composed of homebound veterans.
FP: At the beginning of the discussion portion of your article, following 4 case studies, you mention, “Patients with similar profiles to those discussed in this report were treated with less dramatic results.” How do you, as a family nurse practitioner, consider treatment a success, and how might this differ from expectations set by a medical facility?
RM: As is always the challenge with preventive interventions, it is difficult to measure what does not happen. Successful treatment for recurrent UTIs is lack of recurrence, also asymptomatic colonization vs a symptomatic UTI, which can be measured by a urinalysis is a success. In the oldest of the old, delayed hospitalization for urosepsis, reduced risk of falls, and increased mortality are also successes. Cost savings to the health care system by administering an inexpensive preventive medication vs very expensive IV antibiotic therapy, another success. The observed changes in bacterial resistance in patients treated with methenamine offers great hope in the battle against bacteria.
Ms. McAllister coauthored the July 2014 article, “Recurrent Multidrug Resistant Urinary Tract Infections in Geriatric Patients,” with Janice Allwood, MS, ARNP, CUNP.
Ms. McAllister is a Community Living Center family nurse practitioner and Ms. Allwood is an advanced registered nurse practitioner in Urology Surgery, both at the Bay Pines VA Healthcare System in Florida.
Recently, Federal Practitioner talked with Rebecca McAllister, MS, FNP-BC, about her role in treating complicated urinary tract infections (UTIs) in elderly patients at the Community Living Center of the Bay Pines VA Healthcare System in Florida. The original July 2014 Case in Point, “Recurrent Multidrug Resistant Urinary Tract Infections in Geriatric Patients,” discussed 4 case studies, which suggested the safety and efficacy of treatment with methenamine hippurate.
Federal Practitioner: Much of the focus in your article was on the use of methenamine in Norway and Sweden and a lack thereof in the U.S. How are multidrug resistant UTIs generally treated in the U.S., and how could this be handled differently?
Rebecca McAllister, MS, FNP-BC: Because of the increased rates of bacterial resistance, treating recurrent UTIs prophylactically with low-dose antibiotics is no longer the standard of care. Currently, multidrug resistant UTIs are treated with broad-spectrum antibiotics, that organisms are susceptible to. The promise of methenamine relies on the bacteria not developing resistance to it; in turn, long-term use in patients does not contribute to developing resistance.
FP: Is methenamine hippurate readily available within the VA, and what are the guidelines surrounding its use?
RM: Methenamine is on the VA formulary available in 1-gm doses. Standard guidelines per Micromedex are for prophylaxis of recurrent UTIs, as mentioned in the article, and contraindicates use in patients with impaired renal function, although specific parameters are not identified, because testing was never done with geriatric patients.
FP: Of the 4 patients discussed in the article, 3 were aged > 89 years and the fourth was aged exactly 89 years. Was this a coincidence, or does the success of methenamine in this oldest-old cohort highlight UTI recurrence rate late in life, the failed efficacy of other drugs over time, or both?
RM: The success of methenamine highlights both UTI recurrence rate late in life and the failed efficacy of other drugs over time. The primary patient group in these case studies was composed of homebound veterans.
FP: At the beginning of the discussion portion of your article, following 4 case studies, you mention, “Patients with similar profiles to those discussed in this report were treated with less dramatic results.” How do you, as a family nurse practitioner, consider treatment a success, and how might this differ from expectations set by a medical facility?
RM: As is always the challenge with preventive interventions, it is difficult to measure what does not happen. Successful treatment for recurrent UTIs is lack of recurrence, also asymptomatic colonization vs a symptomatic UTI, which can be measured by a urinalysis is a success. In the oldest of the old, delayed hospitalization for urosepsis, reduced risk of falls, and increased mortality are also successes. Cost savings to the health care system by administering an inexpensive preventive medication vs very expensive IV antibiotic therapy, another success. The observed changes in bacterial resistance in patients treated with methenamine offers great hope in the battle against bacteria.
Ms. McAllister coauthored the July 2014 article, “Recurrent Multidrug Resistant Urinary Tract Infections in Geriatric Patients,” with Janice Allwood, MS, ARNP, CUNP.
Ms. McAllister is a Community Living Center family nurse practitioner and Ms. Allwood is an advanced registered nurse practitioner in Urology Surgery, both at the Bay Pines VA Healthcare System in Florida.

Malaria prophylaxis appears safe, effective in kids

Credit: Malayaka house
Year-round prophylaxis with a newer antimalaria treatment can reduce the risk of malaria in young children without posing a risk of serious adverse events, according to a study published in PLOS Medicine.
The researchers found that dihydroartemisinin-piperaquine (DP), an artemisinin-based combination therapy, was the most effective of 3 treatments at reducing malaria risk in children aged 6 months to 24 months.
The other 2 treatments, the antifolates sulfadoxine-pyrimethamine (SP) and trimethoprim-sulfamethoxazole (TS), have been in use longer than DP. And, in many locations, the malaria parasite has developed a resistance to them.
The researchers conducted this study to determine if the benefits of malaria prophylaxis outweighed the potential risk of anemia and other side effects from the drugs.
And they found the benefits did outweigh the risks. There was no significant increase in grade 3/4 adverse events with any of the treatments when compared to a control group.
The researchers also wanted to look specifically at the effects of year-round treatment. They noted that most previous studies of malaria prophylaxis have been limited to areas where there is only a seasonal risk of the disease. But this study took place in Uganda, where the risk persists throughout the year.
“Our study showed that preventive drug treatment can greatly reduce malaria in young children in areas where there are year-round high rates of transmission,” said study author Grant Dorsey, MD, of the University of California, San Francisco.
To make this discovery, the researchers studied 393 children from Tororo, Uganda. Beginning at 6 months of age, the children were randomized to 1 of 4 groups: monthly DP, monthly SP, daily TS, or a group that didn’t receive any prophylaxis, which is the standard medical practice in the area.
Treatments were given at home without supervision. Piperaquine levels were used as a measure of compliance in the DP arm.
All of the families involved in the study received insecticide-treated bed nets to put over the children when they slept. By 24 months of age, 352 children were still taking part in the study.
There were 3.02 malaria episodes per person-year in the DP group, 5.21 in the TS group, 6.73 in the SP group, and 6.95 in the control group. Protective efficacy measured 58% for the DP group, 28% for the TS group, and 7% for the SP group.
Piperaquine levels were below the detection limit 52% of the time when malaria was diagnosed in the DP group, which suggests non-adherence to treatment.
Between the groups, there was no significant difference in the rate of grade 3/4 adverse events related to treatment. There were 8 such events in the SP group, 8 in the TS group, and 3 in the DP group. Events included elevated temperature, anemia, neutropenia, thrombocytopenia, and elevated ALT/AST.
Considering all grade 3/4 adverse events regardless of their relationship to treatment, the researchers found the overall incidence was significantly lower in the DP group, but not the SP or TS groups, compared to the control group. The same was true for the incidence of elevated temperature, anemia, and thrombocytopenia.
After discontinuing the children’s treatment at 24 months of age, the researchers followed the children until age 3 and found no difference in malaria rates between the groups.
The team said these results suggest monthly administration of DP is a safe and effective option for reducing malaria among infants in regions with year-round transmission and high resistance to antifolates.
The findings also help to allay any concerns that continuous treatment might interfere with the children’s ability to develop an immune response against malaria, thereby making them more likely to contract the disease after treatment stops.
The researchers noted, however, that additional research is needed to evaluate the preventive efficacy of DP in other areas, maintain surveillance for potential selection of drug-resistant parasites, and evaluate the role of preventative treatment in the context of other malaria control interventions. ![]()
Credit: Malayaka house
Year-round prophylaxis with a newer antimalaria treatment can reduce the risk of malaria in young children without posing a risk of serious adverse events, according to a study published in PLOS Medicine.
The researchers found that dihydroartemisinin-piperaquine (DP), an artemisinin-based combination therapy, was the most effective of 3 treatments at reducing malaria risk in children aged 6 months to 24 months.
The other 2 treatments, the antifolates sulfadoxine-pyrimethamine (SP) and trimethoprim-sulfamethoxazole (TS), have been in use longer than DP. And, in many locations, the malaria parasite has developed a resistance to them.
The researchers conducted this study to determine if the benefits of malaria prophylaxis outweighed the potential risk of anemia and other side effects from the drugs.
And they found the benefits did outweigh the risks. There was no significant increase in grade 3/4 adverse events with any of the treatments when compared to a control group.
The researchers also wanted to look specifically at the effects of year-round treatment. They noted that most previous studies of malaria prophylaxis have been limited to areas where there is only a seasonal risk of the disease. But this study took place in Uganda, where the risk persists throughout the year.
“Our study showed that preventive drug treatment can greatly reduce malaria in young children in areas where there are year-round high rates of transmission,” said study author Grant Dorsey, MD, of the University of California, San Francisco.
To make this discovery, the researchers studied 393 children from Tororo, Uganda. Beginning at 6 months of age, the children were randomized to 1 of 4 groups: monthly DP, monthly SP, daily TS, or a group that didn’t receive any prophylaxis, which is the standard medical practice in the area.
Treatments were given at home without supervision. Piperaquine levels were used as a measure of compliance in the DP arm.
All of the families involved in the study received insecticide-treated bed nets to put over the children when they slept. By 24 months of age, 352 children were still taking part in the study.
There were 3.02 malaria episodes per person-year in the DP group, 5.21 in the TS group, 6.73 in the SP group, and 6.95 in the control group. Protective efficacy measured 58% for the DP group, 28% for the TS group, and 7% for the SP group.
Piperaquine levels were below the detection limit 52% of the time when malaria was diagnosed in the DP group, which suggests non-adherence to treatment.
Between the groups, there was no significant difference in the rate of grade 3/4 adverse events related to treatment. There were 8 such events in the SP group, 8 in the TS group, and 3 in the DP group. Events included elevated temperature, anemia, neutropenia, thrombocytopenia, and elevated ALT/AST.
Considering all grade 3/4 adverse events regardless of their relationship to treatment, the researchers found the overall incidence was significantly lower in the DP group, but not the SP or TS groups, compared to the control group. The same was true for the incidence of elevated temperature, anemia, and thrombocytopenia.
After discontinuing the children’s treatment at 24 months of age, the researchers followed the children until age 3 and found no difference in malaria rates between the groups.
The team said these results suggest monthly administration of DP is a safe and effective option for reducing malaria among infants in regions with year-round transmission and high resistance to antifolates.
The findings also help to allay any concerns that continuous treatment might interfere with the children’s ability to develop an immune response against malaria, thereby making them more likely to contract the disease after treatment stops.
The researchers noted, however, that additional research is needed to evaluate the preventive efficacy of DP in other areas, maintain surveillance for potential selection of drug-resistant parasites, and evaluate the role of preventative treatment in the context of other malaria control interventions. ![]()
Credit: Malayaka house
Year-round prophylaxis with a newer antimalaria treatment can reduce the risk of malaria in young children without posing a risk of serious adverse events, according to a study published in PLOS Medicine.
The researchers found that dihydroartemisinin-piperaquine (DP), an artemisinin-based combination therapy, was the most effective of 3 treatments at reducing malaria risk in children aged 6 months to 24 months.
The other 2 treatments, the antifolates sulfadoxine-pyrimethamine (SP) and trimethoprim-sulfamethoxazole (TS), have been in use longer than DP. And, in many locations, the malaria parasite has developed a resistance to them.
The researchers conducted this study to determine if the benefits of malaria prophylaxis outweighed the potential risk of anemia and other side effects from the drugs.
And they found the benefits did outweigh the risks. There was no significant increase in grade 3/4 adverse events with any of the treatments when compared to a control group.
The researchers also wanted to look specifically at the effects of year-round treatment. They noted that most previous studies of malaria prophylaxis have been limited to areas where there is only a seasonal risk of the disease. But this study took place in Uganda, where the risk persists throughout the year.
“Our study showed that preventive drug treatment can greatly reduce malaria in young children in areas where there are year-round high rates of transmission,” said study author Grant Dorsey, MD, of the University of California, San Francisco.
To make this discovery, the researchers studied 393 children from Tororo, Uganda. Beginning at 6 months of age, the children were randomized to 1 of 4 groups: monthly DP, monthly SP, daily TS, or a group that didn’t receive any prophylaxis, which is the standard medical practice in the area.
Treatments were given at home without supervision. Piperaquine levels were used as a measure of compliance in the DP arm.
All of the families involved in the study received insecticide-treated bed nets to put over the children when they slept. By 24 months of age, 352 children were still taking part in the study.
There were 3.02 malaria episodes per person-year in the DP group, 5.21 in the TS group, 6.73 in the SP group, and 6.95 in the control group. Protective efficacy measured 58% for the DP group, 28% for the TS group, and 7% for the SP group.
Piperaquine levels were below the detection limit 52% of the time when malaria was diagnosed in the DP group, which suggests non-adherence to treatment.
Between the groups, there was no significant difference in the rate of grade 3/4 adverse events related to treatment. There were 8 such events in the SP group, 8 in the TS group, and 3 in the DP group. Events included elevated temperature, anemia, neutropenia, thrombocytopenia, and elevated ALT/AST.
Considering all grade 3/4 adverse events regardless of their relationship to treatment, the researchers found the overall incidence was significantly lower in the DP group, but not the SP or TS groups, compared to the control group. The same was true for the incidence of elevated temperature, anemia, and thrombocytopenia.
After discontinuing the children’s treatment at 24 months of age, the researchers followed the children until age 3 and found no difference in malaria rates between the groups.
The team said these results suggest monthly administration of DP is a safe and effective option for reducing malaria among infants in regions with year-round transmission and high resistance to antifolates.
The findings also help to allay any concerns that continuous treatment might interfere with the children’s ability to develop an immune response against malaria, thereby making them more likely to contract the disease after treatment stops.
The researchers noted, however, that additional research is needed to evaluate the preventive efficacy of DP in other areas, maintain surveillance for potential selection of drug-resistant parasites, and evaluate the role of preventative treatment in the context of other malaria control interventions. ![]()
Protein-targeting drug could treat cancers, other diseases

surrounding fused cells
Credit: IRB Barcelona
Experiments in mice suggest the mitochondrial chaperone TRAP-1 is involved in the development of cancers and age-related diseases.
Previous research showed that TRAP-1 is overexpressed in leukemia, lymphoma, and many other cancers.
The new research, published in Cell Reports, clarifies TRAP-1’s role in cancers and age-related conditions. It also suggests gamitrinib, a novel agent targeting TRAP-1, could prove useful in treating these diseases.
TRAP-1 is a member of the heat shock protein 90 (HSP90) family, chaperone proteins that guide the physical formation of other proteins and serve a regulatory function within mitochondria. Tumors use HSP90 proteins like TRAP-1 to help survive therapeutic attack.
To further investigate the effects of TRAP-1, researchers bred TRAP-1 knockout mice. The team found the mice compensate for losing the protein by switching to alternative cellular mechanisms for making energy.
“We see this astounding change in TRAP-1 knockout mice, where they show fewer signs of aging and are less likely to develop cancers,” said Dario C. Altieri, MD, of The Wistar Institute in Philadelphia, Pennsylvania.
“Our findings provide an unexpected explanation for how TRAP-1 and related proteins regulate metabolism within our cells. We usually link the reprogramming of metabolic pathways with human diseases, such as cancer. What we didn’t expect to see were healthier mice with fewer tumors.”
Dr Altieri and his colleagues created the TRAP-1 knockout mice as part of their ongoing investigation into their novel drug, gamitrinib, which targets TRAP-1 in the mitochondria of tumor cells.
“In tumors, the loss of TRAP-1 is devastating, triggering a host of catastrophic defects, including metabolic problems that ultimately result in the death of the tumor cells,” Dr Altieri said. ”Mice that lack TRAP-1 from the start, however, have 3 weeks in the womb to compensate for the loss of the protein.”
The researchers found that, in the knockout mice, the loss of TRAP-1 causes mitochondrial proteins to misfold, which triggers a compensatory response that causes cells to consume more oxygen and metabolize more sugar. This prompts the mitochondria to produce deregulated levels of ATP.
This increased mitochondrial activity actually creates a moderate boost in oxidative stress (free radical damage) and the associated DNA damage. While DNA damage may seem counterproductive to longevity and good health, the low level of DNA damage actually reduces cell proliferation, slowing growth to allow the cell’s natural repair mechanisms to take effect.
According to Dr Altieri, his group’s observations provide a mechanistic foundation for the role of chaperone molecules like HSP90 in the regulation of bioenergetics in mitochondria—how cells produce and use the chemical energy they need to survive and grow.
Their results explain some contradictory findings in the scientific literature regarding the regulation of bioenergetics and show how compensatory mechanisms can arise when these chaperone molecules are taken out of the equation.
“Our findings strengthen the case for targeting HSP90 in tumor cells, but they also open up a fascinating array of questions that may have implications for metabolism and longevity,” Dr Altieri said. “I predict that the TRAP-1 knockout mouse will be a valuable tool for answering these questions.” ![]()
surrounding fused cells
Credit: IRB Barcelona
Experiments in mice suggest the mitochondrial chaperone TRAP-1 is involved in the development of cancers and age-related diseases.
Previous research showed that TRAP-1 is overexpressed in leukemia, lymphoma, and many other cancers.
The new research, published in Cell Reports, clarifies TRAP-1’s role in cancers and age-related conditions. It also suggests gamitrinib, a novel agent targeting TRAP-1, could prove useful in treating these diseases.
TRAP-1 is a member of the heat shock protein 90 (HSP90) family, chaperone proteins that guide the physical formation of other proteins and serve a regulatory function within mitochondria. Tumors use HSP90 proteins like TRAP-1 to help survive therapeutic attack.
To further investigate the effects of TRAP-1, researchers bred TRAP-1 knockout mice. The team found the mice compensate for losing the protein by switching to alternative cellular mechanisms for making energy.
“We see this astounding change in TRAP-1 knockout mice, where they show fewer signs of aging and are less likely to develop cancers,” said Dario C. Altieri, MD, of The Wistar Institute in Philadelphia, Pennsylvania.
“Our findings provide an unexpected explanation for how TRAP-1 and related proteins regulate metabolism within our cells. We usually link the reprogramming of metabolic pathways with human diseases, such as cancer. What we didn’t expect to see were healthier mice with fewer tumors.”
Dr Altieri and his colleagues created the TRAP-1 knockout mice as part of their ongoing investigation into their novel drug, gamitrinib, which targets TRAP-1 in the mitochondria of tumor cells.
“In tumors, the loss of TRAP-1 is devastating, triggering a host of catastrophic defects, including metabolic problems that ultimately result in the death of the tumor cells,” Dr Altieri said. ”Mice that lack TRAP-1 from the start, however, have 3 weeks in the womb to compensate for the loss of the protein.”
The researchers found that, in the knockout mice, the loss of TRAP-1 causes mitochondrial proteins to misfold, which triggers a compensatory response that causes cells to consume more oxygen and metabolize more sugar. This prompts the mitochondria to produce deregulated levels of ATP.
This increased mitochondrial activity actually creates a moderate boost in oxidative stress (free radical damage) and the associated DNA damage. While DNA damage may seem counterproductive to longevity and good health, the low level of DNA damage actually reduces cell proliferation, slowing growth to allow the cell’s natural repair mechanisms to take effect.
According to Dr Altieri, his group’s observations provide a mechanistic foundation for the role of chaperone molecules like HSP90 in the regulation of bioenergetics in mitochondria—how cells produce and use the chemical energy they need to survive and grow.
Their results explain some contradictory findings in the scientific literature regarding the regulation of bioenergetics and show how compensatory mechanisms can arise when these chaperone molecules are taken out of the equation.
“Our findings strengthen the case for targeting HSP90 in tumor cells, but they also open up a fascinating array of questions that may have implications for metabolism and longevity,” Dr Altieri said. “I predict that the TRAP-1 knockout mouse will be a valuable tool for answering these questions.” ![]()
surrounding fused cells
Credit: IRB Barcelona
Experiments in mice suggest the mitochondrial chaperone TRAP-1 is involved in the development of cancers and age-related diseases.
Previous research showed that TRAP-1 is overexpressed in leukemia, lymphoma, and many other cancers.
The new research, published in Cell Reports, clarifies TRAP-1’s role in cancers and age-related conditions. It also suggests gamitrinib, a novel agent targeting TRAP-1, could prove useful in treating these diseases.
TRAP-1 is a member of the heat shock protein 90 (HSP90) family, chaperone proteins that guide the physical formation of other proteins and serve a regulatory function within mitochondria. Tumors use HSP90 proteins like TRAP-1 to help survive therapeutic attack.
To further investigate the effects of TRAP-1, researchers bred TRAP-1 knockout mice. The team found the mice compensate for losing the protein by switching to alternative cellular mechanisms for making energy.
“We see this astounding change in TRAP-1 knockout mice, where they show fewer signs of aging and are less likely to develop cancers,” said Dario C. Altieri, MD, of The Wistar Institute in Philadelphia, Pennsylvania.
“Our findings provide an unexpected explanation for how TRAP-1 and related proteins regulate metabolism within our cells. We usually link the reprogramming of metabolic pathways with human diseases, such as cancer. What we didn’t expect to see were healthier mice with fewer tumors.”
Dr Altieri and his colleagues created the TRAP-1 knockout mice as part of their ongoing investigation into their novel drug, gamitrinib, which targets TRAP-1 in the mitochondria of tumor cells.
“In tumors, the loss of TRAP-1 is devastating, triggering a host of catastrophic defects, including metabolic problems that ultimately result in the death of the tumor cells,” Dr Altieri said. ”Mice that lack TRAP-1 from the start, however, have 3 weeks in the womb to compensate for the loss of the protein.”
The researchers found that, in the knockout mice, the loss of TRAP-1 causes mitochondrial proteins to misfold, which triggers a compensatory response that causes cells to consume more oxygen and metabolize more sugar. This prompts the mitochondria to produce deregulated levels of ATP.
This increased mitochondrial activity actually creates a moderate boost in oxidative stress (free radical damage) and the associated DNA damage. While DNA damage may seem counterproductive to longevity and good health, the low level of DNA damage actually reduces cell proliferation, slowing growth to allow the cell’s natural repair mechanisms to take effect.
According to Dr Altieri, his group’s observations provide a mechanistic foundation for the role of chaperone molecules like HSP90 in the regulation of bioenergetics in mitochondria—how cells produce and use the chemical energy they need to survive and grow.
Their results explain some contradictory findings in the scientific literature regarding the regulation of bioenergetics and show how compensatory mechanisms can arise when these chaperone molecules are taken out of the equation.
“Our findings strengthen the case for targeting HSP90 in tumor cells, but they also open up a fascinating array of questions that may have implications for metabolism and longevity,” Dr Altieri said. “I predict that the TRAP-1 knockout mouse will be a valuable tool for answering these questions.” ![]()
STAP cell researcher commits suicide

Credit: NIH
An author of the retracted Nature papers on STAP cells (stimulus-triggered acquisition of pluripotency cells) has committed suicide at the age of 52.
Yoshiki Sasai, MD, PhD, was found dead at the RIKEN Center for Developmental Biology in Kobe, Japan, where he was deputy director.
Dr Sasai reportedly hanged himself and left several suicide notes.
Members of the scientific community have expressed shock and sadness upon learning of Dr Sasai’s death.
RIKEN President Ryoji Noyori, PhD, said he was “overcome with grief” when he heard the unfortunate news.
“The scientific world has lost a talented and dedicated researcher, who earned our deep respect for the advanced research he carried out over many years,” Dr Noyori said.
Nature’s editor-in-chief, Phil Campbell, PhD, echoed that sentiment, saying, “Yoshiki Sasai was an exceptional scientist, and he has left an extraordinary legacy of pioneering work across many fields within stem cell and developmental biology.”
Dr Sasai was a respected expert on embryonic stem cells, but the STAP cell scandal damaged his reputation and reportedly took a toll on his health. According to a spokesperson at RIKEN, Dr Sasai was hospitalized for stress and required counseling in the wake of the scandal.
Dr Sasai had worked closely with the lead author of the STAP cell papers, Haruko Obokata, PhD, although he said his main duty was editing the papers.
The papers, an article and a letter, were published in Nature in January. They recounted the creation of STAP cells—inducing pluripotency in somatic cells by exposing them to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation. In April, the investigative committee concluded that Dr Obokata was guilty of research misconduct, while Dr Sasai and another author from RIKEN, Teruhiko Wakayama, PhD, were guilty of negligence.
RIKEN also said the researchers would be disciplined, although details were not released.
At a subsequent news conference, Dr Sasai said the Nature papers should be retracted because of the errors and inconsistencies, but the data do indicate the STAP cell phenomenon is real.
Likewise, Dr Obokata insisted the phenomenon is real and appealed the findings of RIKEN’s investigation. But RIKEN said another investigation was not warranted and called for a retraction of the papers. In July, Nature published retractions.
A RIKEN group is still attempting to recreate the STAP cell phenomenon, with Dr Obokata’s help. RIKEN plans to release an interim report on this attempt soon.
Other researchers said they have tried and failed to replicate the STAP cell experiments. One group reported their failed attempt in F1000Research. ![]()
Credit: NIH
An author of the retracted Nature papers on STAP cells (stimulus-triggered acquisition of pluripotency cells) has committed suicide at the age of 52.
Yoshiki Sasai, MD, PhD, was found dead at the RIKEN Center for Developmental Biology in Kobe, Japan, where he was deputy director.
Dr Sasai reportedly hanged himself and left several suicide notes.
Members of the scientific community have expressed shock and sadness upon learning of Dr Sasai’s death.
RIKEN President Ryoji Noyori, PhD, said he was “overcome with grief” when he heard the unfortunate news.
“The scientific world has lost a talented and dedicated researcher, who earned our deep respect for the advanced research he carried out over many years,” Dr Noyori said.
Nature’s editor-in-chief, Phil Campbell, PhD, echoed that sentiment, saying, “Yoshiki Sasai was an exceptional scientist, and he has left an extraordinary legacy of pioneering work across many fields within stem cell and developmental biology.”
Dr Sasai was a respected expert on embryonic stem cells, but the STAP cell scandal damaged his reputation and reportedly took a toll on his health. According to a spokesperson at RIKEN, Dr Sasai was hospitalized for stress and required counseling in the wake of the scandal.
Dr Sasai had worked closely with the lead author of the STAP cell papers, Haruko Obokata, PhD, although he said his main duty was editing the papers.
The papers, an article and a letter, were published in Nature in January. They recounted the creation of STAP cells—inducing pluripotency in somatic cells by exposing them to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation. In April, the investigative committee concluded that Dr Obokata was guilty of research misconduct, while Dr Sasai and another author from RIKEN, Teruhiko Wakayama, PhD, were guilty of negligence.
RIKEN also said the researchers would be disciplined, although details were not released.
At a subsequent news conference, Dr Sasai said the Nature papers should be retracted because of the errors and inconsistencies, but the data do indicate the STAP cell phenomenon is real.
Likewise, Dr Obokata insisted the phenomenon is real and appealed the findings of RIKEN’s investigation. But RIKEN said another investigation was not warranted and called for a retraction of the papers. In July, Nature published retractions.
A RIKEN group is still attempting to recreate the STAP cell phenomenon, with Dr Obokata’s help. RIKEN plans to release an interim report on this attempt soon.
Other researchers said they have tried and failed to replicate the STAP cell experiments. One group reported their failed attempt in F1000Research. ![]()
Credit: NIH
An author of the retracted Nature papers on STAP cells (stimulus-triggered acquisition of pluripotency cells) has committed suicide at the age of 52.
Yoshiki Sasai, MD, PhD, was found dead at the RIKEN Center for Developmental Biology in Kobe, Japan, where he was deputy director.
Dr Sasai reportedly hanged himself and left several suicide notes.
Members of the scientific community have expressed shock and sadness upon learning of Dr Sasai’s death.
RIKEN President Ryoji Noyori, PhD, said he was “overcome with grief” when he heard the unfortunate news.
“The scientific world has lost a talented and dedicated researcher, who earned our deep respect for the advanced research he carried out over many years,” Dr Noyori said.
Nature’s editor-in-chief, Phil Campbell, PhD, echoed that sentiment, saying, “Yoshiki Sasai was an exceptional scientist, and he has left an extraordinary legacy of pioneering work across many fields within stem cell and developmental biology.”
Dr Sasai was a respected expert on embryonic stem cells, but the STAP cell scandal damaged his reputation and reportedly took a toll on his health. According to a spokesperson at RIKEN, Dr Sasai was hospitalized for stress and required counseling in the wake of the scandal.
Dr Sasai had worked closely with the lead author of the STAP cell papers, Haruko Obokata, PhD, although he said his main duty was editing the papers.
The papers, an article and a letter, were published in Nature in January. They recounted the creation of STAP cells—inducing pluripotency in somatic cells by exposing them to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation. In April, the investigative committee concluded that Dr Obokata was guilty of research misconduct, while Dr Sasai and another author from RIKEN, Teruhiko Wakayama, PhD, were guilty of negligence.
RIKEN also said the researchers would be disciplined, although details were not released.
At a subsequent news conference, Dr Sasai said the Nature papers should be retracted because of the errors and inconsistencies, but the data do indicate the STAP cell phenomenon is real.
Likewise, Dr Obokata insisted the phenomenon is real and appealed the findings of RIKEN’s investigation. But RIKEN said another investigation was not warranted and called for a retraction of the papers. In July, Nature published retractions.
A RIKEN group is still attempting to recreate the STAP cell phenomenon, with Dr Obokata’s help. RIKEN plans to release an interim report on this attempt soon.
Other researchers said they have tried and failed to replicate the STAP cell experiments. One group reported their failed attempt in F1000Research. ![]()