User login
Painful rash on feet
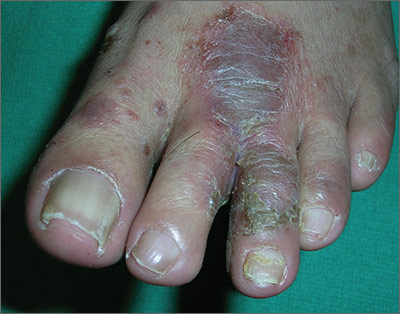
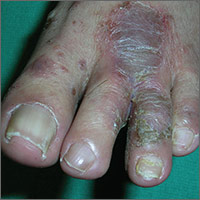
The FP told the patient that he had ulcerative tinea pedis related to a bacterial superinfection. The FP performed a potassium hydroxide (KOH) preparation, which was positive for branching septate hyphae. (See a video on how to perform a KOH preparation.) The small ulcerations and crusts were due to a bacterial superinfection on top of the standard tinea pedis.
Ulcerative tinea pedis is uncommon and, thus, is not always mentioned when describing the standard types of tinea pedis (interdigital, moccasin distribution, and vesicular [vesiculobullous]). Ulcerative tinea pedis does not have to have large ulcers, but there is usually disruption in the skin barrier along with signs of bacterial superinfection (such as crusting and exudate).
This patient demonstrated these signs and the FP chose to treat the patient with an antibiotic that would cover typical skin organisms. There was no need for a culture, as it was likely to grow out many types of skin flora found on the foot. This case was unlikely to be the result of methicillin-resistant Staphylococcus aureus, so a first-generation cephalosporin was adequate.
The FP treated the patient with oral terbinafine 250 mg/d for 2 weeks, along with a course of oral cephalexin 500 mg 3 times daily for one week. Three weeks later, the skin on the patient’s feet was clear, except for some postinflammatory hyperpigmentation that was likely to fade over time.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Reppa R. Tinea pedis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:799-804.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP told the patient that he had ulcerative tinea pedis related to a bacterial superinfection. The FP performed a potassium hydroxide (KOH) preparation, which was positive for branching septate hyphae. (See a video on how to perform a KOH preparation.) The small ulcerations and crusts were due to a bacterial superinfection on top of the standard tinea pedis.
Ulcerative tinea pedis is uncommon and, thus, is not always mentioned when describing the standard types of tinea pedis (interdigital, moccasin distribution, and vesicular [vesiculobullous]). Ulcerative tinea pedis does not have to have large ulcers, but there is usually disruption in the skin barrier along with signs of bacterial superinfection (such as crusting and exudate).
This patient demonstrated these signs and the FP chose to treat the patient with an antibiotic that would cover typical skin organisms. There was no need for a culture, as it was likely to grow out many types of skin flora found on the foot. This case was unlikely to be the result of methicillin-resistant Staphylococcus aureus, so a first-generation cephalosporin was adequate.
The FP treated the patient with oral terbinafine 250 mg/d for 2 weeks, along with a course of oral cephalexin 500 mg 3 times daily for one week. Three weeks later, the skin on the patient’s feet was clear, except for some postinflammatory hyperpigmentation that was likely to fade over time.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Reppa R. Tinea pedis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:799-804.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP told the patient that he had ulcerative tinea pedis related to a bacterial superinfection. The FP performed a potassium hydroxide (KOH) preparation, which was positive for branching septate hyphae. (See a video on how to perform a KOH preparation.) The small ulcerations and crusts were due to a bacterial superinfection on top of the standard tinea pedis.
Ulcerative tinea pedis is uncommon and, thus, is not always mentioned when describing the standard types of tinea pedis (interdigital, moccasin distribution, and vesicular [vesiculobullous]). Ulcerative tinea pedis does not have to have large ulcers, but there is usually disruption in the skin barrier along with signs of bacterial superinfection (such as crusting and exudate).
This patient demonstrated these signs and the FP chose to treat the patient with an antibiotic that would cover typical skin organisms. There was no need for a culture, as it was likely to grow out many types of skin flora found on the foot. This case was unlikely to be the result of methicillin-resistant Staphylococcus aureus, so a first-generation cephalosporin was adequate.
The FP treated the patient with oral terbinafine 250 mg/d for 2 weeks, along with a course of oral cephalexin 500 mg 3 times daily for one week. Three weeks later, the skin on the patient’s feet was clear, except for some postinflammatory hyperpigmentation that was likely to fade over time.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Reppa R. Tinea pedis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:799-804.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Into the Wild: PA Edition
The island of Adak is Alaska’s last frontier. There are no stores, doctors, streetlights, traffic lights, or even a need for license plates. Just the lonely cry of a bald eagle and the howl of the arctic wind accompany you along the dark streets of this ghost town on the edge of the earth, where the North Pacific and the Bering Sea collide. The frigid Arctic front travels down the west coast, while the warmer Japanese current comes up the Pacific side. The low tundra does little to block the winds generated by that confluence, which often whip at 90 mph or more for days on end. Otherwise, the weather is generally gray and misty, and temperatures range from about 20°F to 65°F.
The weather matches the ambience and surroundings—decaying shambles of a once grand Navy base with facilities designed to make life as pleasant as possible for service members. But the Navy left in the late 1990s. By the time I arrived to practice here in 2013, only a few hundred people inhabited the island. There was virtually no economy, and almost all the roads and buildings were deserted. Man’s creation had fallen victim to vandals and nature.
My practice claimed a converted high school principal’s office and a two-bed emergency department (ED) that had some nice equipment—but with no medical aid, x-ray technician, phlebotomist, or lab technician, there was only me to operate it.
One of the oddities of the Alaskan bush is that medical providers often perform as All Species Providers; my first patient, Sadie, was a very well-behaved black Labrador retriever. Unable to anesthetize her, I was thankful for her even-tempered, patient breed (and for my experience as a veterinarian). Sadie lay calmly on the ED bed, her head in her owner’s hands, while I sutured her forepaw. In hindsight, she was one of my easier cases.
Adak’s isolated location (450 miles from the nearest settled outpost), rugged terrain, and vast wildlife come with an elevated risk for injuries and no shortage of challenges in treating them. During my first week, an autistic child presented with a foot laceration. There was no electricity when he arrived, as was often the case due to the wind; the only available light came through the open door. It was dusk and snow was gently falling, but we huddled in the doorway, as the patient’s mother and my husband held the boy while I sutured him. I just managed to get the last stitch in before the child entered his incoherent world.

Not surprisingly for an island, the surrounding seas brought many of my patients. Workers on commercial fishing vessels frequently sustained injuries that the conditions at sea would exacerbate. Hand injuries were common, as were slip-and-fall injuries such as dislocations and broken bones. It could take hours to get them into port. The most memorable was a fisherman who had lost the end of his thumb when it was crushed between the gunwale and a swinging, loaded crab pot being hoisted aboard. The electricity (surprise) was out, so I was forced to treat him by flashlight. He was a young man (early 30s) but especially nice. What I remember most is that he declined a digital block. I debrided the devitalized tissue via sharp dissection, but he never once cursed. So much for all those jokes about cussing like a sailor.
On a seemingly calm Saturday in my third week, one of the supervisors from the fish processing plant came in with two of his workers. One had bilateral ocular exposure to a cleaning solvent, while the other was stumbling and disoriented. My triage skills were put to the test as I anesthetized Worker 1’s eyes with proparacaine, then inserted Morgan lenses (which, thankfully, came with instructions in the packaging!). The supervisor, who was keeping Worker 2 calm simultaneously, held a basin below the patient’s eyes to catch runoff as I quickly attached a liter of saline to each lens and opened the valve full bore. With that situation under control for the moment, I switched gears to assess Worker 2, who was markedly incoherent and unable to give a history. What I did know was that he was ataxic and nauseous, with a temperature of 104°F. I flashed my penlight into his eyes, and he reacted like Dracula faced with a crucifix. In addition to his exquisite photophobia, he had marked neck stiffness. The diagnosis was my first case of human meningitis. I inserted an IV providing ceftriaxone, acyclovir, and ondansetron. He required 2.5 L of IV fluid, but he gradually recovered over the next two days.

Why, with all these challenges, did I go to Alaska? Because I felt that there I could be of most service; if ever there was an underserved community, Adak was it. Ironically, the other job offer I considered was on the Hawaiian island of Kauai. As idyllic as that might have been, it wasn’t quite the adventure that Adak turned out to be—and it wasn’t as desperately in need of a provider.
When I took the Adak post, I didn’t realize how much I would learn. Above all, I realized that a complete and meticulous physical exam is king. If I missed any life-threatening conditions, the ramifications would be extreme. There were no referrals or second opinions, just me with the patient, and I had to make the right decisions.
The bonds I formed with the people on this remote island, and the paths I trod through this unusual land, made for a unique experience. It was a thrill to look out toward the horizon and know that everything I could see had remained unchanged since the birth of these volcanic islands. In time, I came to recognize that I was not in charge. The planes carrying supplies in or patients out would come if the weather allowed. What was on the island was what I had to work with, and if I didn’t have what I wanted, it was only because I didn’t want what I had. The waves would come as they will. I could either surf them or be tossed by them. I chose to dive in.
The island of Adak is Alaska’s last frontier. There are no stores, doctors, streetlights, traffic lights, or even a need for license plates. Just the lonely cry of a bald eagle and the howl of the arctic wind accompany you along the dark streets of this ghost town on the edge of the earth, where the North Pacific and the Bering Sea collide. The frigid Arctic front travels down the west coast, while the warmer Japanese current comes up the Pacific side. The low tundra does little to block the winds generated by that confluence, which often whip at 90 mph or more for days on end. Otherwise, the weather is generally gray and misty, and temperatures range from about 20°F to 65°F.
The weather matches the ambience and surroundings—decaying shambles of a once grand Navy base with facilities designed to make life as pleasant as possible for service members. But the Navy left in the late 1990s. By the time I arrived to practice here in 2013, only a few hundred people inhabited the island. There was virtually no economy, and almost all the roads and buildings were deserted. Man’s creation had fallen victim to vandals and nature.
My practice claimed a converted high school principal’s office and a two-bed emergency department (ED) that had some nice equipment—but with no medical aid, x-ray technician, phlebotomist, or lab technician, there was only me to operate it.
One of the oddities of the Alaskan bush is that medical providers often perform as All Species Providers; my first patient, Sadie, was a very well-behaved black Labrador retriever. Unable to anesthetize her, I was thankful for her even-tempered, patient breed (and for my experience as a veterinarian). Sadie lay calmly on the ED bed, her head in her owner’s hands, while I sutured her forepaw. In hindsight, she was one of my easier cases.
Adak’s isolated location (450 miles from the nearest settled outpost), rugged terrain, and vast wildlife come with an elevated risk for injuries and no shortage of challenges in treating them. During my first week, an autistic child presented with a foot laceration. There was no electricity when he arrived, as was often the case due to the wind; the only available light came through the open door. It was dusk and snow was gently falling, but we huddled in the doorway, as the patient’s mother and my husband held the boy while I sutured him. I just managed to get the last stitch in before the child entered his incoherent world.
Not surprisingly for an island, the surrounding seas brought many of my patients. Workers on commercial fishing vessels frequently sustained injuries that the conditions at sea would exacerbate. Hand injuries were common, as were slip-and-fall injuries such as dislocations and broken bones. It could take hours to get them into port. The most memorable was a fisherman who had lost the end of his thumb when it was crushed between the gunwale and a swinging, loaded crab pot being hoisted aboard. The electricity (surprise) was out, so I was forced to treat him by flashlight. He was a young man (early 30s) but especially nice. What I remember most is that he declined a digital block. I debrided the devitalized tissue via sharp dissection, but he never once cursed. So much for all those jokes about cussing like a sailor.
On a seemingly calm Saturday in my third week, one of the supervisors from the fish processing plant came in with two of his workers. One had bilateral ocular exposure to a cleaning solvent, while the other was stumbling and disoriented. My triage skills were put to the test as I anesthetized Worker 1’s eyes with proparacaine, then inserted Morgan lenses (which, thankfully, came with instructions in the packaging!). The supervisor, who was keeping Worker 2 calm simultaneously, held a basin below the patient’s eyes to catch runoff as I quickly attached a liter of saline to each lens and opened the valve full bore. With that situation under control for the moment, I switched gears to assess Worker 2, who was markedly incoherent and unable to give a history. What I did know was that he was ataxic and nauseous, with a temperature of 104°F. I flashed my penlight into his eyes, and he reacted like Dracula faced with a crucifix. In addition to his exquisite photophobia, he had marked neck stiffness. The diagnosis was my first case of human meningitis. I inserted an IV providing ceftriaxone, acyclovir, and ondansetron. He required 2.5 L of IV fluid, but he gradually recovered over the next two days.
Why, with all these challenges, did I go to Alaska? Because I felt that there I could be of most service; if ever there was an underserved community, Adak was it. Ironically, the other job offer I considered was on the Hawaiian island of Kauai. As idyllic as that might have been, it wasn’t quite the adventure that Adak turned out to be—and it wasn’t as desperately in need of a provider.
When I took the Adak post, I didn’t realize how much I would learn. Above all, I realized that a complete and meticulous physical exam is king. If I missed any life-threatening conditions, the ramifications would be extreme. There were no referrals or second opinions, just me with the patient, and I had to make the right decisions.
The bonds I formed with the people on this remote island, and the paths I trod through this unusual land, made for a unique experience. It was a thrill to look out toward the horizon and know that everything I could see had remained unchanged since the birth of these volcanic islands. In time, I came to recognize that I was not in charge. The planes carrying supplies in or patients out would come if the weather allowed. What was on the island was what I had to work with, and if I didn’t have what I wanted, it was only because I didn’t want what I had. The waves would come as they will. I could either surf them or be tossed by them. I chose to dive in.
The island of Adak is Alaska’s last frontier. There are no stores, doctors, streetlights, traffic lights, or even a need for license plates. Just the lonely cry of a bald eagle and the howl of the arctic wind accompany you along the dark streets of this ghost town on the edge of the earth, where the North Pacific and the Bering Sea collide. The frigid Arctic front travels down the west coast, while the warmer Japanese current comes up the Pacific side. The low tundra does little to block the winds generated by that confluence, which often whip at 90 mph or more for days on end. Otherwise, the weather is generally gray and misty, and temperatures range from about 20°F to 65°F.
The weather matches the ambience and surroundings—decaying shambles of a once grand Navy base with facilities designed to make life as pleasant as possible for service members. But the Navy left in the late 1990s. By the time I arrived to practice here in 2013, only a few hundred people inhabited the island. There was virtually no economy, and almost all the roads and buildings were deserted. Man’s creation had fallen victim to vandals and nature.
My practice claimed a converted high school principal’s office and a two-bed emergency department (ED) that had some nice equipment—but with no medical aid, x-ray technician, phlebotomist, or lab technician, there was only me to operate it.
One of the oddities of the Alaskan bush is that medical providers often perform as All Species Providers; my first patient, Sadie, was a very well-behaved black Labrador retriever. Unable to anesthetize her, I was thankful for her even-tempered, patient breed (and for my experience as a veterinarian). Sadie lay calmly on the ED bed, her head in her owner’s hands, while I sutured her forepaw. In hindsight, she was one of my easier cases.
Adak’s isolated location (450 miles from the nearest settled outpost), rugged terrain, and vast wildlife come with an elevated risk for injuries and no shortage of challenges in treating them. During my first week, an autistic child presented with a foot laceration. There was no electricity when he arrived, as was often the case due to the wind; the only available light came through the open door. It was dusk and snow was gently falling, but we huddled in the doorway, as the patient’s mother and my husband held the boy while I sutured him. I just managed to get the last stitch in before the child entered his incoherent world.
Not surprisingly for an island, the surrounding seas brought many of my patients. Workers on commercial fishing vessels frequently sustained injuries that the conditions at sea would exacerbate. Hand injuries were common, as were slip-and-fall injuries such as dislocations and broken bones. It could take hours to get them into port. The most memorable was a fisherman who had lost the end of his thumb when it was crushed between the gunwale and a swinging, loaded crab pot being hoisted aboard. The electricity (surprise) was out, so I was forced to treat him by flashlight. He was a young man (early 30s) but especially nice. What I remember most is that he declined a digital block. I debrided the devitalized tissue via sharp dissection, but he never once cursed. So much for all those jokes about cussing like a sailor.
On a seemingly calm Saturday in my third week, one of the supervisors from the fish processing plant came in with two of his workers. One had bilateral ocular exposure to a cleaning solvent, while the other was stumbling and disoriented. My triage skills were put to the test as I anesthetized Worker 1’s eyes with proparacaine, then inserted Morgan lenses (which, thankfully, came with instructions in the packaging!). The supervisor, who was keeping Worker 2 calm simultaneously, held a basin below the patient’s eyes to catch runoff as I quickly attached a liter of saline to each lens and opened the valve full bore. With that situation under control for the moment, I switched gears to assess Worker 2, who was markedly incoherent and unable to give a history. What I did know was that he was ataxic and nauseous, with a temperature of 104°F. I flashed my penlight into his eyes, and he reacted like Dracula faced with a crucifix. In addition to his exquisite photophobia, he had marked neck stiffness. The diagnosis was my first case of human meningitis. I inserted an IV providing ceftriaxone, acyclovir, and ondansetron. He required 2.5 L of IV fluid, but he gradually recovered over the next two days.
Why, with all these challenges, did I go to Alaska? Because I felt that there I could be of most service; if ever there was an underserved community, Adak was it. Ironically, the other job offer I considered was on the Hawaiian island of Kauai. As idyllic as that might have been, it wasn’t quite the adventure that Adak turned out to be—and it wasn’t as desperately in need of a provider.
When I took the Adak post, I didn’t realize how much I would learn. Above all, I realized that a complete and meticulous physical exam is king. If I missed any life-threatening conditions, the ramifications would be extreme. There were no referrals or second opinions, just me with the patient, and I had to make the right decisions.
The bonds I formed with the people on this remote island, and the paths I trod through this unusual land, made for a unique experience. It was a thrill to look out toward the horizon and know that everything I could see had remained unchanged since the birth of these volcanic islands. In time, I came to recognize that I was not in charge. The planes carrying supplies in or patients out would come if the weather allowed. What was on the island was what I had to work with, and if I didn’t have what I wanted, it was only because I didn’t want what I had. The waves would come as they will. I could either surf them or be tossed by them. I chose to dive in.
Lack of natalizumab concentration increase before PML argues against extending dosing interval
Serum concentrations of natalizumab do not appear to rise before patients with relapsing-remitting multiple sclerosis are diagnosed with progressive multifocal leukoencephalopathy, contradicting the hypothesis that exposure to elevated concentrations of the drug is a risk factor for the disease, according to findings from a prospective, observational cohort study.
Zoé L.E. van Kempen, MD, and her colleagues at VU Medical Center Amsterdam noted that a small but increasing number of “neurologists are extending dose intervals of natalizumab [Tysabri] with the primary aim of reducing the risk of PML [progressive multifocal leukoencephalopathy] by lowering the natalizumab exposure per patient,” which also potentially has the benefit of decreasing drug costs, as well as clinic or hospital visits. However, there have been no prospective studies that confirm that extending natalizumab dose intervals does not affect efficacy.
Dr. van Kempen and her coinvestigators investigated this hypothesis by comparing serum concentrations of natalizumab in 5 patients with PML and 10 age- and sex-matched controls from a cohort of 219 relapsing-remitting multiple sclerosis patients taking natalizumab who had blood samples routinely drawn every 12 weeks before the infusion of natalizumab (Mult Scler. 2016 Dec 13. doi: 10.1177/1352458516684023). These 10 controls had a mean concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed before the diagnosis of PML in the 5 cases from the cohort. The five cases also did not show a rise in concentration before the diagnosis of PML and did not have concentrations fluctuate more than 11 mcg/mL during treatment. The patients with PML received a mean of 43 infusions, whereas controls received an average of 106 infusions. A median number of 5 pre-PML samples underwent testing, compared with yearly concentration measurements in the 10 controls who had at least 7 years’ treatment duration.
The increased risk of PML that has been documented in other studies with more than 24 months of natalizumab treatment also was not explained in this study by a rise in serum concentration over long-term follow-up.
“Although this cohort is too small to draw definite conclusions, our results do not support the hypothesis of high serum concentrations as a risk factor for developing PML,” the investigators wrote.
“With our small cohort in mind and insufficient data on this subject so far, neurologists should be careful in extending dose intervals and not overstate a possible decrease in the risk of developing PML. Obviously, if neurologists choose to extend dosing intervals of natalizumab in [John Cunningham virus]–positive patients, these patients should still receive stringent PML monitoring according to current recommendations,” they advised.
The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
Serum concentrations of natalizumab do not appear to rise before patients with relapsing-remitting multiple sclerosis are diagnosed with progressive multifocal leukoencephalopathy, contradicting the hypothesis that exposure to elevated concentrations of the drug is a risk factor for the disease, according to findings from a prospective, observational cohort study.
Zoé L.E. van Kempen, MD, and her colleagues at VU Medical Center Amsterdam noted that a small but increasing number of “neurologists are extending dose intervals of natalizumab [Tysabri] with the primary aim of reducing the risk of PML [progressive multifocal leukoencephalopathy] by lowering the natalizumab exposure per patient,” which also potentially has the benefit of decreasing drug costs, as well as clinic or hospital visits. However, there have been no prospective studies that confirm that extending natalizumab dose intervals does not affect efficacy.
Dr. van Kempen and her coinvestigators investigated this hypothesis by comparing serum concentrations of natalizumab in 5 patients with PML and 10 age- and sex-matched controls from a cohort of 219 relapsing-remitting multiple sclerosis patients taking natalizumab who had blood samples routinely drawn every 12 weeks before the infusion of natalizumab (Mult Scler. 2016 Dec 13. doi: 10.1177/1352458516684023). These 10 controls had a mean concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed before the diagnosis of PML in the 5 cases from the cohort. The five cases also did not show a rise in concentration before the diagnosis of PML and did not have concentrations fluctuate more than 11 mcg/mL during treatment. The patients with PML received a mean of 43 infusions, whereas controls received an average of 106 infusions. A median number of 5 pre-PML samples underwent testing, compared with yearly concentration measurements in the 10 controls who had at least 7 years’ treatment duration.
The increased risk of PML that has been documented in other studies with more than 24 months of natalizumab treatment also was not explained in this study by a rise in serum concentration over long-term follow-up.
“Although this cohort is too small to draw definite conclusions, our results do not support the hypothesis of high serum concentrations as a risk factor for developing PML,” the investigators wrote.
“With our small cohort in mind and insufficient data on this subject so far, neurologists should be careful in extending dose intervals and not overstate a possible decrease in the risk of developing PML. Obviously, if neurologists choose to extend dosing intervals of natalizumab in [John Cunningham virus]–positive patients, these patients should still receive stringent PML monitoring according to current recommendations,” they advised.
The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
Serum concentrations of natalizumab do not appear to rise before patients with relapsing-remitting multiple sclerosis are diagnosed with progressive multifocal leukoencephalopathy, contradicting the hypothesis that exposure to elevated concentrations of the drug is a risk factor for the disease, according to findings from a prospective, observational cohort study.
Zoé L.E. van Kempen, MD, and her colleagues at VU Medical Center Amsterdam noted that a small but increasing number of “neurologists are extending dose intervals of natalizumab [Tysabri] with the primary aim of reducing the risk of PML [progressive multifocal leukoencephalopathy] by lowering the natalizumab exposure per patient,” which also potentially has the benefit of decreasing drug costs, as well as clinic or hospital visits. However, there have been no prospective studies that confirm that extending natalizumab dose intervals does not affect efficacy.
Dr. van Kempen and her coinvestigators investigated this hypothesis by comparing serum concentrations of natalizumab in 5 patients with PML and 10 age- and sex-matched controls from a cohort of 219 relapsing-remitting multiple sclerosis patients taking natalizumab who had blood samples routinely drawn every 12 weeks before the infusion of natalizumab (Mult Scler. 2016 Dec 13. doi: 10.1177/1352458516684023). These 10 controls had a mean concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed before the diagnosis of PML in the 5 cases from the cohort. The five cases also did not show a rise in concentration before the diagnosis of PML and did not have concentrations fluctuate more than 11 mcg/mL during treatment. The patients with PML received a mean of 43 infusions, whereas controls received an average of 106 infusions. A median number of 5 pre-PML samples underwent testing, compared with yearly concentration measurements in the 10 controls who had at least 7 years’ treatment duration.
The increased risk of PML that has been documented in other studies with more than 24 months of natalizumab treatment also was not explained in this study by a rise in serum concentration over long-term follow-up.
“Although this cohort is too small to draw definite conclusions, our results do not support the hypothesis of high serum concentrations as a risk factor for developing PML,” the investigators wrote.
“With our small cohort in mind and insufficient data on this subject so far, neurologists should be careful in extending dose intervals and not overstate a possible decrease in the risk of developing PML. Obviously, if neurologists choose to extend dosing intervals of natalizumab in [John Cunningham virus]–positive patients, these patients should still receive stringent PML monitoring according to current recommendations,” they advised.
The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
FROM MULTIPLE SCLEROSIS JOURNAL
Key clinical point:
Major finding: A total of 10 control patients had a mean natalizumab serum concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed in 5 patients before a diagnosis of PML.
Data source: A comparison of 5 patients with PML and 10 age- and sex-matched controls from a prospective cohort of 219 patients with relapsing-remitting MS who were treated with natalizumab at one center.
Disclosures: The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
FDA gives nod to crisaborole for atopic dermatitis
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.

At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.
At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.
At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
Ohio governor vetoes 6-week abortion ban in favor of 20 weeks
Ohio Gov. John Kasich (R) has rejected a controversial bill that would have barred women in the state from having abortions as early as 6 weeks, but he signed another measure banning the procedure at 20 weeks.
The legislation, known as “the heartbeat bill,” would have prohibited abortions if a fetal heartbeat could be detected – usually around 6 weeks – and jailed physicians who performed the procedure without checking for a heartbeat or who provided an abortion after a heartbeat was found. The Ohio House approved the measure on Dec. 6 after earlier passage by the state senate. In his veto of the 6-week ban, Gov. Kasich noted that federal courts have struck down similar legislation in Arkansas and North Dakota, and that he did not want Ohio taxpayers to fund a losing legal challenge.
“Because the federal courts are bound to follow the Supreme Court’s rulings on abortion, the amendment ... will be struck down,” Gov. Kasich said in a statement. “The state of Ohio will be the losing party in that lawsuit and ... be forced to pay hundreds of thousands of taxpayer dollars to cover the legal fees of the pro-choice activists’ lawyers. Furthermore, such a defeat invites additional challenges to Ohio’s strong legal protections for unborn life. Therefore, this veto is in the public interest.”
Abortion rights advocates were quick to criticize the 20-week ban (SB 127), saying that Ohio lawmakers still intend to effectively end abortion in the state.
“The 20-week ban will force women to travel long distances and cross state lines in order to access safe, legal abortion – a barrier that many women simply cannot afford,” Dawn Laguens, Planned Parenthood Action Fund executive vice president said in a statement.
The American Congress of Obstetricians and Gynecologists Ohio Section also opposes the 20-week ban. In a Dec. 9 letter to Gov. Kasich, Wayne Trout, MD, chair of ACOG’s Ohio Section, said these types of laws come between physicians and patients and create unnecessary medical risks.
“A great number of pregnancy terminations beyond 20 weeks are due to identification of serious and fatal birth defects in otherwise highly desired pregnancies,” Dr. Trout wrote. “Continuation of these pregnancies would result in certain death of the baby and expose the newborn to needless pain. Meanwhile the mother is forced to carry the fetus to term and may be exposed to a myriad of medical and reproductive risks.”
The Ohio law also places doctors in a “precarious situation,” he noted, by criminalizing a medical procedure that is recognized by ACOG as a standard of care when women have a medical indication for termination of pregnancy.
Abortion opponents meanwhile, praised the 20-week ban, calling it a vehicle to end abortion. “It challenges the current national abortion standard and properly moves the legal needle from viability to the baby’s ability to feel pain,” Mike Gonidakis, President of Ohio Right to Life said in a statement.
[email protected]
On Twitter @legal_med
Ohio Gov. John Kasich (R) has rejected a controversial bill that would have barred women in the state from having abortions as early as 6 weeks, but he signed another measure banning the procedure at 20 weeks.
The legislation, known as “the heartbeat bill,” would have prohibited abortions if a fetal heartbeat could be detected – usually around 6 weeks – and jailed physicians who performed the procedure without checking for a heartbeat or who provided an abortion after a heartbeat was found. The Ohio House approved the measure on Dec. 6 after earlier passage by the state senate. In his veto of the 6-week ban, Gov. Kasich noted that federal courts have struck down similar legislation in Arkansas and North Dakota, and that he did not want Ohio taxpayers to fund a losing legal challenge.
“Because the federal courts are bound to follow the Supreme Court’s rulings on abortion, the amendment ... will be struck down,” Gov. Kasich said in a statement. “The state of Ohio will be the losing party in that lawsuit and ... be forced to pay hundreds of thousands of taxpayer dollars to cover the legal fees of the pro-choice activists’ lawyers. Furthermore, such a defeat invites additional challenges to Ohio’s strong legal protections for unborn life. Therefore, this veto is in the public interest.”
Abortion rights advocates were quick to criticize the 20-week ban (SB 127), saying that Ohio lawmakers still intend to effectively end abortion in the state.
“The 20-week ban will force women to travel long distances and cross state lines in order to access safe, legal abortion – a barrier that many women simply cannot afford,” Dawn Laguens, Planned Parenthood Action Fund executive vice president said in a statement.
The American Congress of Obstetricians and Gynecologists Ohio Section also opposes the 20-week ban. In a Dec. 9 letter to Gov. Kasich, Wayne Trout, MD, chair of ACOG’s Ohio Section, said these types of laws come between physicians and patients and create unnecessary medical risks.
“A great number of pregnancy terminations beyond 20 weeks are due to identification of serious and fatal birth defects in otherwise highly desired pregnancies,” Dr. Trout wrote. “Continuation of these pregnancies would result in certain death of the baby and expose the newborn to needless pain. Meanwhile the mother is forced to carry the fetus to term and may be exposed to a myriad of medical and reproductive risks.”
The Ohio law also places doctors in a “precarious situation,” he noted, by criminalizing a medical procedure that is recognized by ACOG as a standard of care when women have a medical indication for termination of pregnancy.
Abortion opponents meanwhile, praised the 20-week ban, calling it a vehicle to end abortion. “It challenges the current national abortion standard and properly moves the legal needle from viability to the baby’s ability to feel pain,” Mike Gonidakis, President of Ohio Right to Life said in a statement.
[email protected]
On Twitter @legal_med
Ohio Gov. John Kasich (R) has rejected a controversial bill that would have barred women in the state from having abortions as early as 6 weeks, but he signed another measure banning the procedure at 20 weeks.
The legislation, known as “the heartbeat bill,” would have prohibited abortions if a fetal heartbeat could be detected – usually around 6 weeks – and jailed physicians who performed the procedure without checking for a heartbeat or who provided an abortion after a heartbeat was found. The Ohio House approved the measure on Dec. 6 after earlier passage by the state senate. In his veto of the 6-week ban, Gov. Kasich noted that federal courts have struck down similar legislation in Arkansas and North Dakota, and that he did not want Ohio taxpayers to fund a losing legal challenge.
“Because the federal courts are bound to follow the Supreme Court’s rulings on abortion, the amendment ... will be struck down,” Gov. Kasich said in a statement. “The state of Ohio will be the losing party in that lawsuit and ... be forced to pay hundreds of thousands of taxpayer dollars to cover the legal fees of the pro-choice activists’ lawyers. Furthermore, such a defeat invites additional challenges to Ohio’s strong legal protections for unborn life. Therefore, this veto is in the public interest.”
Abortion rights advocates were quick to criticize the 20-week ban (SB 127), saying that Ohio lawmakers still intend to effectively end abortion in the state.
“The 20-week ban will force women to travel long distances and cross state lines in order to access safe, legal abortion – a barrier that many women simply cannot afford,” Dawn Laguens, Planned Parenthood Action Fund executive vice president said in a statement.
The American Congress of Obstetricians and Gynecologists Ohio Section also opposes the 20-week ban. In a Dec. 9 letter to Gov. Kasich, Wayne Trout, MD, chair of ACOG’s Ohio Section, said these types of laws come between physicians and patients and create unnecessary medical risks.
“A great number of pregnancy terminations beyond 20 weeks are due to identification of serious and fatal birth defects in otherwise highly desired pregnancies,” Dr. Trout wrote. “Continuation of these pregnancies would result in certain death of the baby and expose the newborn to needless pain. Meanwhile the mother is forced to carry the fetus to term and may be exposed to a myriad of medical and reproductive risks.”
The Ohio law also places doctors in a “precarious situation,” he noted, by criminalizing a medical procedure that is recognized by ACOG as a standard of care when women have a medical indication for termination of pregnancy.
Abortion opponents meanwhile, praised the 20-week ban, calling it a vehicle to end abortion. “It challenges the current national abortion standard and properly moves the legal needle from viability to the baby’s ability to feel pain,” Mike Gonidakis, President of Ohio Right to Life said in a statement.
[email protected]
On Twitter @legal_med
Ask service members and veterans about sexual health
SILVER SPRING, MD. – Tending to the psychiatric and physical needs of military servicemen, servicewomen, and veterans must include attention to their sexual functioning, according to Col. (Ret.) Elspeth Cameron Ritchie, MD, MPH.
“Think of sexual activity as an activity of daily living,” Dr. Ritchie said at the Trauma Treatment from the Trenches meeting at the Forest Glen Annex in Silver Spring, Md., organized by Sawsan Ghurani, MD, a psychiatrist and Navy captain, and sponsored by the Walter Reed National Military Medical Center in Bethesda, Md.

Despite the deployment of 2.7 million service members over more than 15 years of war in the United States, research on the sexual health of this population has been scant, Dr. Ritchie said. Research has been similarly limited in the civilian population, except for the work that has been done among civilians on the impact of spinal cord injuries on sexual dysfunction, she said (J Spinal Cord Med. 2016 Aug 31:1-12).
Clinicians who work with service members and veterans find that wives complain about the impact of medical interventions on their partners, said Dr. Ritchie, a former Army psychiatry consultant and current chief of Community-Based Outpatient Clinics at the Washington (D.C.) Veterans Affairs Medical Center. “As part of the discussion on sexual health, I remind them that sexual activity is broader than penetration.”
Another issue to be aware of among service members is anger. “Often our patients are angry, and often that anger comes out to us as therapists,” she said. “We need to tell our colleagues about this and get them to expect it.”
Treating post-traumatic stress disorder can be a tricky proposition because of the sexual side effects caused by selective serotonin reuptake inhibitors, Dr. Ritchie said. She prefers bupropion because it is not linked to sexual side effects. Mood stabilizers cause weight gain, as do antipsychotics. Meanwhile, drug holidays lead to problems with adherence, she said. To mitigate side effects, Dr. Ritchie advised “trying one thing at a time and adding trazodone in low doses for sleep.” However, trazodone has been linked to priapism (Gen Hosp Psychiatry. 2015 Jan-Feb;37[1]:40-5), and so must be used with care.
Another treatment for PTSD that is showing promise is stellate ganglion block, which has proven effective for treating hot flashes in postmenopausal women and in addressing estrogen depletion tied to breast cancer treatment in small numbers of patients (Med Hypotheses. 2009 Jun;72[6]:657-61).
“Studies have found a reduction in PTSD symptoms as well as pain,” Dr. Ritchie said. “I’m pushing the VA to do more research in this area.”
In effort to treat sexual dysfunction, Dr. Ritchie said her family practice colleagues prescribe a lot of Viagra and other phosphodiesterase inhibitors. She speculated that flibanserin, a selective agonist for 5-HT1A and an antagonist for 5-HT2A receptors approved in 2015 by the Food and Drug Administration for premenopausal women with hypoactive sexual desire disorder, “will start making its way into the general population,” said Dr. Ritchie, whose comments about using phosphodiesterase inhibitors pertain to VA patients.
Many service members who participated in Operation Iraqi Freedom and Operation Enduring Freedom were aged 18-25 years. Partly because the Department of Defense (DOD) and the VA work with very young families, clinicians are tasked with teaching them about their needs as couples, including the need to discuss intimacy and sexual health. For political reasons, Dr. Ritchie said, in vitro fertilization is not covered by the VA, even though injuries from bomb blasts can make it impossible for couples to conceive naturally.
Toxic and infectious substances faced by troops that are not commonly found in the United States, such as chemicals, pesticides, and motor oils, also need to be acknowledged and addressed by clinicians. “We don’t do a good job about what a veteran thinks about what exposure does to their reproductive systems,” Dr. Ritchie said.
In an interview, Dr. Ghurani said that she came up with the title of the meeting to illustrate the extent to which providers treat PTSD in the military every day. Conference speakers examined the latest treatments on sexual trauma, and the state of the art therapy and research taking place at Walter Reed.
At the meeting, she discussed the distinction between military sexual trauma (MST) and sexual trauma within the civilian population. Research shows that veterans who screen positive for MST “are more likely to have a history of a suicide attempt documented in their VA medical record” (Psychiatry Res. 2016;244:257-65).
A 2008 study found that women are far more likely to screen positive for MST than men. Specifically, the study found that 25% of women and 1.3% of men who are screened for MST within the Veterans Health Administration screen positive (Trauma Violence Abuse. 2008 Oct;9[4]250-69).
Walter Reed’s Interpersonal Recovery Program at the Psychiatry Continuity Service is the only intensive outpatient program within the DOD that provides ongoing treatment for active duty service members with PTSD from a sexual trauma, Dr. Ghurani said.
Earlier during the meeting, Capt. (Ret.) William P. Nash, MD, director of psychological health at the Marine Corps and a 30-year veteran of the Navy, discussed moral injury, and contrasted it with PTSD and other sequelae of psychological trauma. He also described his work on the Moral Injury Events Scale as a tool for recognizing potentially morally injurious events in clinical and research settings.
Dr. Ritchie is the editor of “Intimacy Post-Injury: Combat Trauma and Sexual Health,” (New York: Oxford University Press, 2016). Dr. Ghurani contributed to a chapter in “Intimacy Post Injury,” and Dr. Nash has written extensively about PTSD, particularly among deployed Marines.
[email protected]
On Twitter @ginalhenderson
SILVER SPRING, MD. – Tending to the psychiatric and physical needs of military servicemen, servicewomen, and veterans must include attention to their sexual functioning, according to Col. (Ret.) Elspeth Cameron Ritchie, MD, MPH.
“Think of sexual activity as an activity of daily living,” Dr. Ritchie said at the Trauma Treatment from the Trenches meeting at the Forest Glen Annex in Silver Spring, Md., organized by Sawsan Ghurani, MD, a psychiatrist and Navy captain, and sponsored by the Walter Reed National Military Medical Center in Bethesda, Md.
Despite the deployment of 2.7 million service members over more than 15 years of war in the United States, research on the sexual health of this population has been scant, Dr. Ritchie said. Research has been similarly limited in the civilian population, except for the work that has been done among civilians on the impact of spinal cord injuries on sexual dysfunction, she said (J Spinal Cord Med. 2016 Aug 31:1-12).
Clinicians who work with service members and veterans find that wives complain about the impact of medical interventions on their partners, said Dr. Ritchie, a former Army psychiatry consultant and current chief of Community-Based Outpatient Clinics at the Washington (D.C.) Veterans Affairs Medical Center. “As part of the discussion on sexual health, I remind them that sexual activity is broader than penetration.”
Another issue to be aware of among service members is anger. “Often our patients are angry, and often that anger comes out to us as therapists,” she said. “We need to tell our colleagues about this and get them to expect it.”
Treating post-traumatic stress disorder can be a tricky proposition because of the sexual side effects caused by selective serotonin reuptake inhibitors, Dr. Ritchie said. She prefers bupropion because it is not linked to sexual side effects. Mood stabilizers cause weight gain, as do antipsychotics. Meanwhile, drug holidays lead to problems with adherence, she said. To mitigate side effects, Dr. Ritchie advised “trying one thing at a time and adding trazodone in low doses for sleep.” However, trazodone has been linked to priapism (Gen Hosp Psychiatry. 2015 Jan-Feb;37[1]:40-5), and so must be used with care.
Another treatment for PTSD that is showing promise is stellate ganglion block, which has proven effective for treating hot flashes in postmenopausal women and in addressing estrogen depletion tied to breast cancer treatment in small numbers of patients (Med Hypotheses. 2009 Jun;72[6]:657-61).
“Studies have found a reduction in PTSD symptoms as well as pain,” Dr. Ritchie said. “I’m pushing the VA to do more research in this area.”
In effort to treat sexual dysfunction, Dr. Ritchie said her family practice colleagues prescribe a lot of Viagra and other phosphodiesterase inhibitors. She speculated that flibanserin, a selective agonist for 5-HT1A and an antagonist for 5-HT2A receptors approved in 2015 by the Food and Drug Administration for premenopausal women with hypoactive sexual desire disorder, “will start making its way into the general population,” said Dr. Ritchie, whose comments about using phosphodiesterase inhibitors pertain to VA patients.
Many service members who participated in Operation Iraqi Freedom and Operation Enduring Freedom were aged 18-25 years. Partly because the Department of Defense (DOD) and the VA work with very young families, clinicians are tasked with teaching them about their needs as couples, including the need to discuss intimacy and sexual health. For political reasons, Dr. Ritchie said, in vitro fertilization is not covered by the VA, even though injuries from bomb blasts can make it impossible for couples to conceive naturally.
Toxic and infectious substances faced by troops that are not commonly found in the United States, such as chemicals, pesticides, and motor oils, also need to be acknowledged and addressed by clinicians. “We don’t do a good job about what a veteran thinks about what exposure does to their reproductive systems,” Dr. Ritchie said.
In an interview, Dr. Ghurani said that she came up with the title of the meeting to illustrate the extent to which providers treat PTSD in the military every day. Conference speakers examined the latest treatments on sexual trauma, and the state of the art therapy and research taking place at Walter Reed.
At the meeting, she discussed the distinction between military sexual trauma (MST) and sexual trauma within the civilian population. Research shows that veterans who screen positive for MST “are more likely to have a history of a suicide attempt documented in their VA medical record” (Psychiatry Res. 2016;244:257-65).
A 2008 study found that women are far more likely to screen positive for MST than men. Specifically, the study found that 25% of women and 1.3% of men who are screened for MST within the Veterans Health Administration screen positive (Trauma Violence Abuse. 2008 Oct;9[4]250-69).
Walter Reed’s Interpersonal Recovery Program at the Psychiatry Continuity Service is the only intensive outpatient program within the DOD that provides ongoing treatment for active duty service members with PTSD from a sexual trauma, Dr. Ghurani said.
Earlier during the meeting, Capt. (Ret.) William P. Nash, MD, director of psychological health at the Marine Corps and a 30-year veteran of the Navy, discussed moral injury, and contrasted it with PTSD and other sequelae of psychological trauma. He also described his work on the Moral Injury Events Scale as a tool for recognizing potentially morally injurious events in clinical and research settings.
Dr. Ritchie is the editor of “Intimacy Post-Injury: Combat Trauma and Sexual Health,” (New York: Oxford University Press, 2016). Dr. Ghurani contributed to a chapter in “Intimacy Post Injury,” and Dr. Nash has written extensively about PTSD, particularly among deployed Marines.
[email protected]
On Twitter @ginalhenderson
SILVER SPRING, MD. – Tending to the psychiatric and physical needs of military servicemen, servicewomen, and veterans must include attention to their sexual functioning, according to Col. (Ret.) Elspeth Cameron Ritchie, MD, MPH.
“Think of sexual activity as an activity of daily living,” Dr. Ritchie said at the Trauma Treatment from the Trenches meeting at the Forest Glen Annex in Silver Spring, Md., organized by Sawsan Ghurani, MD, a psychiatrist and Navy captain, and sponsored by the Walter Reed National Military Medical Center in Bethesda, Md.
Despite the deployment of 2.7 million service members over more than 15 years of war in the United States, research on the sexual health of this population has been scant, Dr. Ritchie said. Research has been similarly limited in the civilian population, except for the work that has been done among civilians on the impact of spinal cord injuries on sexual dysfunction, she said (J Spinal Cord Med. 2016 Aug 31:1-12).
Clinicians who work with service members and veterans find that wives complain about the impact of medical interventions on their partners, said Dr. Ritchie, a former Army psychiatry consultant and current chief of Community-Based Outpatient Clinics at the Washington (D.C.) Veterans Affairs Medical Center. “As part of the discussion on sexual health, I remind them that sexual activity is broader than penetration.”
Another issue to be aware of among service members is anger. “Often our patients are angry, and often that anger comes out to us as therapists,” she said. “We need to tell our colleagues about this and get them to expect it.”
Treating post-traumatic stress disorder can be a tricky proposition because of the sexual side effects caused by selective serotonin reuptake inhibitors, Dr. Ritchie said. She prefers bupropion because it is not linked to sexual side effects. Mood stabilizers cause weight gain, as do antipsychotics. Meanwhile, drug holidays lead to problems with adherence, she said. To mitigate side effects, Dr. Ritchie advised “trying one thing at a time and adding trazodone in low doses for sleep.” However, trazodone has been linked to priapism (Gen Hosp Psychiatry. 2015 Jan-Feb;37[1]:40-5), and so must be used with care.
Another treatment for PTSD that is showing promise is stellate ganglion block, which has proven effective for treating hot flashes in postmenopausal women and in addressing estrogen depletion tied to breast cancer treatment in small numbers of patients (Med Hypotheses. 2009 Jun;72[6]:657-61).
“Studies have found a reduction in PTSD symptoms as well as pain,” Dr. Ritchie said. “I’m pushing the VA to do more research in this area.”
In effort to treat sexual dysfunction, Dr. Ritchie said her family practice colleagues prescribe a lot of Viagra and other phosphodiesterase inhibitors. She speculated that flibanserin, a selective agonist for 5-HT1A and an antagonist for 5-HT2A receptors approved in 2015 by the Food and Drug Administration for premenopausal women with hypoactive sexual desire disorder, “will start making its way into the general population,” said Dr. Ritchie, whose comments about using phosphodiesterase inhibitors pertain to VA patients.
Many service members who participated in Operation Iraqi Freedom and Operation Enduring Freedom were aged 18-25 years. Partly because the Department of Defense (DOD) and the VA work with very young families, clinicians are tasked with teaching them about their needs as couples, including the need to discuss intimacy and sexual health. For political reasons, Dr. Ritchie said, in vitro fertilization is not covered by the VA, even though injuries from bomb blasts can make it impossible for couples to conceive naturally.
Toxic and infectious substances faced by troops that are not commonly found in the United States, such as chemicals, pesticides, and motor oils, also need to be acknowledged and addressed by clinicians. “We don’t do a good job about what a veteran thinks about what exposure does to their reproductive systems,” Dr. Ritchie said.
In an interview, Dr. Ghurani said that she came up with the title of the meeting to illustrate the extent to which providers treat PTSD in the military every day. Conference speakers examined the latest treatments on sexual trauma, and the state of the art therapy and research taking place at Walter Reed.
At the meeting, she discussed the distinction between military sexual trauma (MST) and sexual trauma within the civilian population. Research shows that veterans who screen positive for MST “are more likely to have a history of a suicide attempt documented in their VA medical record” (Psychiatry Res. 2016;244:257-65).
A 2008 study found that women are far more likely to screen positive for MST than men. Specifically, the study found that 25% of women and 1.3% of men who are screened for MST within the Veterans Health Administration screen positive (Trauma Violence Abuse. 2008 Oct;9[4]250-69).
Walter Reed’s Interpersonal Recovery Program at the Psychiatry Continuity Service is the only intensive outpatient program within the DOD that provides ongoing treatment for active duty service members with PTSD from a sexual trauma, Dr. Ghurani said.
Earlier during the meeting, Capt. (Ret.) William P. Nash, MD, director of psychological health at the Marine Corps and a 30-year veteran of the Navy, discussed moral injury, and contrasted it with PTSD and other sequelae of psychological trauma. He also described his work on the Moral Injury Events Scale as a tool for recognizing potentially morally injurious events in clinical and research settings.
Dr. Ritchie is the editor of “Intimacy Post-Injury: Combat Trauma and Sexual Health,” (New York: Oxford University Press, 2016). Dr. Ghurani contributed to a chapter in “Intimacy Post Injury,” and Dr. Nash has written extensively about PTSD, particularly among deployed Marines.
[email protected]
On Twitter @ginalhenderson
FROM TRAUMA TREATMENT FROM THE TRENCHES
Kimford J. Meador, MD
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

What can administrators and ObGyns do together to reduce physician burnout?
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Related content on burnout:
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Related content on burnout:
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Related content on burnout:

Lower-risk approach for aortic arch repair
In newborns with a borderline hypoplastic aortic arch, the type of operation and surgical approach can be critical in determining the risk of recurrent obstruction, but aortic arch reconstruction through a median sternotomy on bypass may carry a lower risk of recurrence than use of a thoracotomy.
In a study of 183 newborns and infants (median age of 15 days) who had surgery for coarctation and hypoplastic aortic arch over a 17-year period, researchers led by Andreas Tulzer, MD, of Children’s Heart Center, Linz, Austria, found that resection and extended end-to-end anastomosis (REEEA) as well as end-to-side anastomosis (ESA) yielded low mortality. The findings were published in the December 2016 issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1506-13).
In the study, 72 patients had a median sternotomy – 71 with cardiopulmonary bypass (CPB) – and the remaining 111 had a lateral thoracotomy. Fifty-two patients (28.4%) had an additional ventricular septal defect closure. In the 71 patients who had median sternotomy with CPB, 41 had REEEA and 30 had ESA.
One patient who had median sternotomy with CPB had complications whereas 10 who had undergone primary repair with REEEA through a lateral thoracotomy had complications, for complications rates of 1.4% and 9%, respectively.
“Access through a median sternotomy with the use of CPB was superior to a lateral thoracotomy in terms of necessary reinterventions,” noted Dr. Tulzer and coauthors.
Of the 131 patients who had isolated repair of coarctation of the aorta with associated hypoplastic aortic arch, 116 had REEEA and 15 had ESA. There were no in-hospital deaths in this group and one patient needed an early reintervention. One patient had a severe neurologic complication.
On long-term follow-up of 139 patients at a median duration of 6.3 years, no late deaths were reported. “The calculated freedom from mortality (early and late) at 10 years for the entire group was 99.27%,” the researchers said. “In none of the patients of the follow-up population did we notice any signs of permanent left laryngeal nerve injury, bronchial compression, or left pulmonary artery stenosis.”
Lateral thoracotomy as access was a significant risk factor for recurrent obstruction at P = .03.
In the study, an experienced pediatric cardiologist and a pediatric cardiac surgeon determined which of three procedures to use – ESA on bypass, REEEA with a median sternotomy on CPB, or REEEA with a lateral thoracotomy without CPB – based on the size and anatomy of the proximal transverse aortic arch. In the early study period, cut-off values were proximal transverse arch diameters of 4 mm or less in newborns and young infants, but in the later study period the cut-off was z scores of –4.5 or less.
Adverse outcomes were minimal. There was one death within 30 days of surgery in the overall population (0.54%). The one severe complication consisted of paraplegia and cerebral hypoxemia after REEEA. At 10 years, 99.27% of all patients survived and 90.12% were free from intervention.
Either approach with REEEA and ESA is safe and effective, Dr. Tulzer and colleagues said, but they did determine a suitable population for the median sternotomy using CPB. “In patients with proximal transverse aortic arch, z scores of less than –4.59, arch repair should be performed through a median sternotomy using CPB, rather than through a lateral thoracotomy to reduce the risk for recurrent arch obstructions,” the researchers concluded.
Dr. Tulzer and coauthors had no financial relationships to disclose.
While the outcomes that Dr. Tulzer and colleagues reported are “remarkable,” the findings raise a question if mortality and risk of interventions are the sole determinants in selecting a surgical strategy, Petros V. Anagnostopoulos, MD, said in his invited commentary (J Thorac Cardiovasc Surg. 2016;152:1475-6). “The mortality should be low irrespective of approach,” Dr. Anagnostopoulos said.
He said that even if the surgeon pursues repair for coarctation and hypoplastic aortic arch through a thoracotomy instead of the median sternotomy the Austrian authors advocate and coarctation should recur, most of these cases can be treated with catheterization at low risk. “Will such a suboptimal outcome prove to be superior to that of a patient who has perfect anatomic repair but potentially faces adverse neurodevelopmental consequences of a neonatal cardiopulmonary bypass run?” he asked.
But the long-term outcomes are “poorly defined” as clinical investigators continue to “push the limits” to avoid deep hypothermia by using perfusion modifications to perform arch reconstruction, said Dr. Anagnostopoulos of the division of pediatric cardiothoracic surgery, American Family Children’s Hospital, University of Wisconsin, Madison. “It may be time to start taking into account not only survival and accuracy of repair, but also long-term sequelae of our therapies,” he concluded.
Dr. Anagnostopoulos had no financial relationships to disclose.
While the outcomes that Dr. Tulzer and colleagues reported are “remarkable,” the findings raise a question if mortality and risk of interventions are the sole determinants in selecting a surgical strategy, Petros V. Anagnostopoulos, MD, said in his invited commentary (J Thorac Cardiovasc Surg. 2016;152:1475-6). “The mortality should be low irrespective of approach,” Dr. Anagnostopoulos said.
He said that even if the surgeon pursues repair for coarctation and hypoplastic aortic arch through a thoracotomy instead of the median sternotomy the Austrian authors advocate and coarctation should recur, most of these cases can be treated with catheterization at low risk. “Will such a suboptimal outcome prove to be superior to that of a patient who has perfect anatomic repair but potentially faces adverse neurodevelopmental consequences of a neonatal cardiopulmonary bypass run?” he asked.
But the long-term outcomes are “poorly defined” as clinical investigators continue to “push the limits” to avoid deep hypothermia by using perfusion modifications to perform arch reconstruction, said Dr. Anagnostopoulos of the division of pediatric cardiothoracic surgery, American Family Children’s Hospital, University of Wisconsin, Madison. “It may be time to start taking into account not only survival and accuracy of repair, but also long-term sequelae of our therapies,” he concluded.
Dr. Anagnostopoulos had no financial relationships to disclose.
While the outcomes that Dr. Tulzer and colleagues reported are “remarkable,” the findings raise a question if mortality and risk of interventions are the sole determinants in selecting a surgical strategy, Petros V. Anagnostopoulos, MD, said in his invited commentary (J Thorac Cardiovasc Surg. 2016;152:1475-6). “The mortality should be low irrespective of approach,” Dr. Anagnostopoulos said.
He said that even if the surgeon pursues repair for coarctation and hypoplastic aortic arch through a thoracotomy instead of the median sternotomy the Austrian authors advocate and coarctation should recur, most of these cases can be treated with catheterization at low risk. “Will such a suboptimal outcome prove to be superior to that of a patient who has perfect anatomic repair but potentially faces adverse neurodevelopmental consequences of a neonatal cardiopulmonary bypass run?” he asked.
But the long-term outcomes are “poorly defined” as clinical investigators continue to “push the limits” to avoid deep hypothermia by using perfusion modifications to perform arch reconstruction, said Dr. Anagnostopoulos of the division of pediatric cardiothoracic surgery, American Family Children’s Hospital, University of Wisconsin, Madison. “It may be time to start taking into account not only survival and accuracy of repair, but also long-term sequelae of our therapies,” he concluded.
Dr. Anagnostopoulos had no financial relationships to disclose.
In newborns with a borderline hypoplastic aortic arch, the type of operation and surgical approach can be critical in determining the risk of recurrent obstruction, but aortic arch reconstruction through a median sternotomy on bypass may carry a lower risk of recurrence than use of a thoracotomy.
In a study of 183 newborns and infants (median age of 15 days) who had surgery for coarctation and hypoplastic aortic arch over a 17-year period, researchers led by Andreas Tulzer, MD, of Children’s Heart Center, Linz, Austria, found that resection and extended end-to-end anastomosis (REEEA) as well as end-to-side anastomosis (ESA) yielded low mortality. The findings were published in the December 2016 issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1506-13).
In the study, 72 patients had a median sternotomy – 71 with cardiopulmonary bypass (CPB) – and the remaining 111 had a lateral thoracotomy. Fifty-two patients (28.4%) had an additional ventricular septal defect closure. In the 71 patients who had median sternotomy with CPB, 41 had REEEA and 30 had ESA.
One patient who had median sternotomy with CPB had complications whereas 10 who had undergone primary repair with REEEA through a lateral thoracotomy had complications, for complications rates of 1.4% and 9%, respectively.
“Access through a median sternotomy with the use of CPB was superior to a lateral thoracotomy in terms of necessary reinterventions,” noted Dr. Tulzer and coauthors.
Of the 131 patients who had isolated repair of coarctation of the aorta with associated hypoplastic aortic arch, 116 had REEEA and 15 had ESA. There were no in-hospital deaths in this group and one patient needed an early reintervention. One patient had a severe neurologic complication.
On long-term follow-up of 139 patients at a median duration of 6.3 years, no late deaths were reported. “The calculated freedom from mortality (early and late) at 10 years for the entire group was 99.27%,” the researchers said. “In none of the patients of the follow-up population did we notice any signs of permanent left laryngeal nerve injury, bronchial compression, or left pulmonary artery stenosis.”
Lateral thoracotomy as access was a significant risk factor for recurrent obstruction at P = .03.
In the study, an experienced pediatric cardiologist and a pediatric cardiac surgeon determined which of three procedures to use – ESA on bypass, REEEA with a median sternotomy on CPB, or REEEA with a lateral thoracotomy without CPB – based on the size and anatomy of the proximal transverse aortic arch. In the early study period, cut-off values were proximal transverse arch diameters of 4 mm or less in newborns and young infants, but in the later study period the cut-off was z scores of –4.5 or less.
Adverse outcomes were minimal. There was one death within 30 days of surgery in the overall population (0.54%). The one severe complication consisted of paraplegia and cerebral hypoxemia after REEEA. At 10 years, 99.27% of all patients survived and 90.12% were free from intervention.
Either approach with REEEA and ESA is safe and effective, Dr. Tulzer and colleagues said, but they did determine a suitable population for the median sternotomy using CPB. “In patients with proximal transverse aortic arch, z scores of less than –4.59, arch repair should be performed through a median sternotomy using CPB, rather than through a lateral thoracotomy to reduce the risk for recurrent arch obstructions,” the researchers concluded.
Dr. Tulzer and coauthors had no financial relationships to disclose.
In newborns with a borderline hypoplastic aortic arch, the type of operation and surgical approach can be critical in determining the risk of recurrent obstruction, but aortic arch reconstruction through a median sternotomy on bypass may carry a lower risk of recurrence than use of a thoracotomy.
In a study of 183 newborns and infants (median age of 15 days) who had surgery for coarctation and hypoplastic aortic arch over a 17-year period, researchers led by Andreas Tulzer, MD, of Children’s Heart Center, Linz, Austria, found that resection and extended end-to-end anastomosis (REEEA) as well as end-to-side anastomosis (ESA) yielded low mortality. The findings were published in the December 2016 issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1506-13).
In the study, 72 patients had a median sternotomy – 71 with cardiopulmonary bypass (CPB) – and the remaining 111 had a lateral thoracotomy. Fifty-two patients (28.4%) had an additional ventricular septal defect closure. In the 71 patients who had median sternotomy with CPB, 41 had REEEA and 30 had ESA.
One patient who had median sternotomy with CPB had complications whereas 10 who had undergone primary repair with REEEA through a lateral thoracotomy had complications, for complications rates of 1.4% and 9%, respectively.
“Access through a median sternotomy with the use of CPB was superior to a lateral thoracotomy in terms of necessary reinterventions,” noted Dr. Tulzer and coauthors.
Of the 131 patients who had isolated repair of coarctation of the aorta with associated hypoplastic aortic arch, 116 had REEEA and 15 had ESA. There were no in-hospital deaths in this group and one patient needed an early reintervention. One patient had a severe neurologic complication.
On long-term follow-up of 139 patients at a median duration of 6.3 years, no late deaths were reported. “The calculated freedom from mortality (early and late) at 10 years for the entire group was 99.27%,” the researchers said. “In none of the patients of the follow-up population did we notice any signs of permanent left laryngeal nerve injury, bronchial compression, or left pulmonary artery stenosis.”
Lateral thoracotomy as access was a significant risk factor for recurrent obstruction at P = .03.
In the study, an experienced pediatric cardiologist and a pediatric cardiac surgeon determined which of three procedures to use – ESA on bypass, REEEA with a median sternotomy on CPB, or REEEA with a lateral thoracotomy without CPB – based on the size and anatomy of the proximal transverse aortic arch. In the early study period, cut-off values were proximal transverse arch diameters of 4 mm or less in newborns and young infants, but in the later study period the cut-off was z scores of –4.5 or less.
Adverse outcomes were minimal. There was one death within 30 days of surgery in the overall population (0.54%). The one severe complication consisted of paraplegia and cerebral hypoxemia after REEEA. At 10 years, 99.27% of all patients survived and 90.12% were free from intervention.
Either approach with REEEA and ESA is safe and effective, Dr. Tulzer and colleagues said, but they did determine a suitable population for the median sternotomy using CPB. “In patients with proximal transverse aortic arch, z scores of less than –4.59, arch repair should be performed through a median sternotomy using CPB, rather than through a lateral thoracotomy to reduce the risk for recurrent arch obstructions,” the researchers concluded.
Dr. Tulzer and coauthors had no financial relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Aortic arch reconstruction through a median sternotomy on bypass in newborns and infants had lower rates for recurrent obstruction than did a thoracotomy approach.
Major finding: Of 11 patients who required reintervention, one had a median sternotomy and 10 had a lateral thoracotomy.
Data source: Retrospective review of 183 consecutive newborns and infants with coarctation and hypoplastic aortic arch from 1996 to 2013.
Disclosures: Dr. Tulzer and coauthors had no financial relationships to disclose.
Novel trial aims to BEAT AML
SAN DIEGO – A multi-arm clinical trial aims to transform the treatment of acute myeloid leukemia, a deadly blood cancer whose standard of care has remained essentially unchanged for 4 decades.
Launched in October 2016, the multicenter BEAT AML Master Trial is based on a simple but radical goal – turn around genomic tests of bone marrow biopsies within 7 days to allow targeted therapy, said lead investigator Brian Druker, MD, of Oregon Health and Science University Knight Cancer Institute in Portland.

Speaking at a press conference at the annual meeting of the American Society of Hematology, he emphasized that rapid, accurate genomic testing is the only way to prescribe targeted agents for AML in time for them to help patients. “It really is about matching the right patient with the right drug,” he said. He also spoke about AML in a video interview at the conference.
That is a major departure from the current approach to treating AML, in which patients receive standard chemotherapy regimens that are toxic and largely ineffective. “Patients themselves call this barbaric therapy,” said John Byrd, MD, who is co-leading the trial on behalf of the Ohio State University Wexner Medical Center in Columbus. “In this trial, we’re going to move away from toxic therapy that is not potentially curative to give more targeted medicine instead.”
In addition to Dr. Druker’s and Dr. Byrd’s centers, Memorial Sloan Kettering Cancer Center, New York, and Dana-Farber Cancer Institute and Massachusetts General Hospital, both in Boston, are onboard for the study. The lead investigators hope to add another six centers to the study group and to have 10 arms of the study underway by mid-2017.
Older patients with AML find chemotherapy especially hard to tolerate and typically respond poorly. Accordingly, the trial will enroll those aged 60 years and up regardless of their genomic profile, the researchers said. Patients lacking targetable markers will be offered investigational therapies showing broad activity in AML.
Another complexity of AML is that any patient can have a variety of mutations, including some affecting only a small subset of leukemia cells, Dr. Byrd noted. Targeting those mutations cannot eradicate disease, but past trials did not rank or choose therapies based on mutation prevalence. Thus, this trial is the first to ask “which mutation is in all of the cells, which gives you the opportunity to get rid of all the disease,” he emphasized. Again, patients – not individual markers or agents – are the priority.
The study also is meant to be nimble – arms can be quickly opened or closed if bench or clinical data are promising or lackluster. This design does not preclude FDA approvals, said Louis J. DeGennaro, PhD, of the Leukemia and Lymphoma Society, which is sponsoring the trial. “We have worked closely with FDA to design a unique protocol that we believe will change the paradigm of AML treatment and future clinical trials,” he added. “This is an unprecedented collaboration.”
Dr. Druker agreed. “If we do this correctly, we can potentially see large effects, and that can become the impetus for rapid FDA approval of these drugs for the right patients,” he said. “That one of the things this trial is designed to do.”
Dr. DeGennaro is president and chief executive officer of the Leukemia and Lymphoma Society, which is sponsoring the BEAT AML Master trial. Dr. Druker disclosed ties to a number of pharmaceutical companies. Dr. Byrd had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – A multi-arm clinical trial aims to transform the treatment of acute myeloid leukemia, a deadly blood cancer whose standard of care has remained essentially unchanged for 4 decades.
Launched in October 2016, the multicenter BEAT AML Master Trial is based on a simple but radical goal – turn around genomic tests of bone marrow biopsies within 7 days to allow targeted therapy, said lead investigator Brian Druker, MD, of Oregon Health and Science University Knight Cancer Institute in Portland.
Speaking at a press conference at the annual meeting of the American Society of Hematology, he emphasized that rapid, accurate genomic testing is the only way to prescribe targeted agents for AML in time for them to help patients. “It really is about matching the right patient with the right drug,” he said. He also spoke about AML in a video interview at the conference.
That is a major departure from the current approach to treating AML, in which patients receive standard chemotherapy regimens that are toxic and largely ineffective. “Patients themselves call this barbaric therapy,” said John Byrd, MD, who is co-leading the trial on behalf of the Ohio State University Wexner Medical Center in Columbus. “In this trial, we’re going to move away from toxic therapy that is not potentially curative to give more targeted medicine instead.”
In addition to Dr. Druker’s and Dr. Byrd’s centers, Memorial Sloan Kettering Cancer Center, New York, and Dana-Farber Cancer Institute and Massachusetts General Hospital, both in Boston, are onboard for the study. The lead investigators hope to add another six centers to the study group and to have 10 arms of the study underway by mid-2017.
Older patients with AML find chemotherapy especially hard to tolerate and typically respond poorly. Accordingly, the trial will enroll those aged 60 years and up regardless of their genomic profile, the researchers said. Patients lacking targetable markers will be offered investigational therapies showing broad activity in AML.
Another complexity of AML is that any patient can have a variety of mutations, including some affecting only a small subset of leukemia cells, Dr. Byrd noted. Targeting those mutations cannot eradicate disease, but past trials did not rank or choose therapies based on mutation prevalence. Thus, this trial is the first to ask “which mutation is in all of the cells, which gives you the opportunity to get rid of all the disease,” he emphasized. Again, patients – not individual markers or agents – are the priority.
The study also is meant to be nimble – arms can be quickly opened or closed if bench or clinical data are promising or lackluster. This design does not preclude FDA approvals, said Louis J. DeGennaro, PhD, of the Leukemia and Lymphoma Society, which is sponsoring the trial. “We have worked closely with FDA to design a unique protocol that we believe will change the paradigm of AML treatment and future clinical trials,” he added. “This is an unprecedented collaboration.”
Dr. Druker agreed. “If we do this correctly, we can potentially see large effects, and that can become the impetus for rapid FDA approval of these drugs for the right patients,” he said. “That one of the things this trial is designed to do.”
Dr. DeGennaro is president and chief executive officer of the Leukemia and Lymphoma Society, which is sponsoring the BEAT AML Master trial. Dr. Druker disclosed ties to a number of pharmaceutical companies. Dr. Byrd had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – A multi-arm clinical trial aims to transform the treatment of acute myeloid leukemia, a deadly blood cancer whose standard of care has remained essentially unchanged for 4 decades.
Launched in October 2016, the multicenter BEAT AML Master Trial is based on a simple but radical goal – turn around genomic tests of bone marrow biopsies within 7 days to allow targeted therapy, said lead investigator Brian Druker, MD, of Oregon Health and Science University Knight Cancer Institute in Portland.
Speaking at a press conference at the annual meeting of the American Society of Hematology, he emphasized that rapid, accurate genomic testing is the only way to prescribe targeted agents for AML in time for them to help patients. “It really is about matching the right patient with the right drug,” he said. He also spoke about AML in a video interview at the conference.
That is a major departure from the current approach to treating AML, in which patients receive standard chemotherapy regimens that are toxic and largely ineffective. “Patients themselves call this barbaric therapy,” said John Byrd, MD, who is co-leading the trial on behalf of the Ohio State University Wexner Medical Center in Columbus. “In this trial, we’re going to move away from toxic therapy that is not potentially curative to give more targeted medicine instead.”
In addition to Dr. Druker’s and Dr. Byrd’s centers, Memorial Sloan Kettering Cancer Center, New York, and Dana-Farber Cancer Institute and Massachusetts General Hospital, both in Boston, are onboard for the study. The lead investigators hope to add another six centers to the study group and to have 10 arms of the study underway by mid-2017.
Older patients with AML find chemotherapy especially hard to tolerate and typically respond poorly. Accordingly, the trial will enroll those aged 60 years and up regardless of their genomic profile, the researchers said. Patients lacking targetable markers will be offered investigational therapies showing broad activity in AML.
Another complexity of AML is that any patient can have a variety of mutations, including some affecting only a small subset of leukemia cells, Dr. Byrd noted. Targeting those mutations cannot eradicate disease, but past trials did not rank or choose therapies based on mutation prevalence. Thus, this trial is the first to ask “which mutation is in all of the cells, which gives you the opportunity to get rid of all the disease,” he emphasized. Again, patients – not individual markers or agents – are the priority.
The study also is meant to be nimble – arms can be quickly opened or closed if bench or clinical data are promising or lackluster. This design does not preclude FDA approvals, said Louis J. DeGennaro, PhD, of the Leukemia and Lymphoma Society, which is sponsoring the trial. “We have worked closely with FDA to design a unique protocol that we believe will change the paradigm of AML treatment and future clinical trials,” he added. “This is an unprecedented collaboration.”
Dr. Druker agreed. “If we do this correctly, we can potentially see large effects, and that can become the impetus for rapid FDA approval of these drugs for the right patients,” he said. “That one of the things this trial is designed to do.”
Dr. DeGennaro is president and chief executive officer of the Leukemia and Lymphoma Society, which is sponsoring the BEAT AML Master trial. Dr. Druker disclosed ties to a number of pharmaceutical companies. Dr. Byrd had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2016
