User login
Symmetric hair loss across frontal and temporal scalp

The FP referred the patient to a dermatology clinic that specialized in hair loss. Based on the clinical findings, physicians at the clinic suspected that this was a case of frontal fibrosing alopecia (FFA), a primary lymphocytic cicatricial (scarring) alopecia. A dermatopathologist confirmed the diagnosis via histologic review.
FFA is characterized by symmetric band-like hair loss with evidence of scarring in the frontal and temporal regions of the scalp. (The extent of hair loss can be assessed by retracting the patient’s hair and having the patient raise his or her eyebrows and wrinkle the forehead in a surprised look.) FFA is accompanied by eyebrow loss in 73% to 95% of patients. Mild to severe perifollicular (and possibly more generalized) erythema and scale are usually present. In addition, erythematous or skin-colored papules may appear on the face, and marked exaggeration of the temporal veins is a common finding.
Most patients with FFA (83%) are postmenopausal women and nearly all (98.6%) have Fitzpatrick skin type 1 or 2 (white skin that burns easily and doesn’t readily tan). Other pertinent findings include the absence of oral lesions, nail changes, or other skin diseases.
A punch biopsy taken from the leading edge of the hair loss confirms the diagnosis of FFA and, ideally, should be reviewed by a dermatopathologist. Histologic examination will reveal a lichenoid lymphocytic infiltrate (predominantly around the hair follicle where the follicular stem cells reside), resulting in fibrosis and scarring. In addition to confirming the diagnosis with histologic examination, the following conditions must be ruled out in the differential: alopecia areata, female pattern hair loss, discoid lupus erythematosus, central centrifugal cicatricial alopecia, and traction alopecia.
Early detection is key. In general, physicians should initiate treatment as soon as possible to prevent disease progression and reduce permanent scarring and hair loss. Intralesional steroids such as triamcinolone acetonide (5-10 mg/cc), as well as high-potency topical steroids, are generally helpful to stabilize the disease. There is also some evidence of benefit from oral dutasteride or finasteride at variable doses. Immunosuppressants such as hydroxychloroquine also may be used as second-line treatments, although the benefit-to-risk ratio needs to be taken into consideration.
This patient was started on a regimen of topical high-potency steroids (clobetasol foam, 0.05%), with targeted, intralesional injection of steroids (10 mg/cc of triamcinolone acetonide) to areas with the most inflammation. She also was advised to use ketoconazole 2% shampoo while showering. These interventions decreased the patient’s symptoms dramatically. Her scalp erythema and scale improved, but the hair did not regrow. One year later, her hairline was clinically stable with no evidence of disease progression. She continued to see a dermatologist annually.
This case was adapted from: Power DV, Disse M, Hordinsky M. Progressive hair loss. J Fam Pract. 2017;66:521-523.
The FP referred the patient to a dermatology clinic that specialized in hair loss. Based on the clinical findings, physicians at the clinic suspected that this was a case of frontal fibrosing alopecia (FFA), a primary lymphocytic cicatricial (scarring) alopecia. A dermatopathologist confirmed the diagnosis via histologic review.
FFA is characterized by symmetric band-like hair loss with evidence of scarring in the frontal and temporal regions of the scalp. (The extent of hair loss can be assessed by retracting the patient’s hair and having the patient raise his or her eyebrows and wrinkle the forehead in a surprised look.) FFA is accompanied by eyebrow loss in 73% to 95% of patients. Mild to severe perifollicular (and possibly more generalized) erythema and scale are usually present. In addition, erythematous or skin-colored papules may appear on the face, and marked exaggeration of the temporal veins is a common finding.
Most patients with FFA (83%) are postmenopausal women and nearly all (98.6%) have Fitzpatrick skin type 1 or 2 (white skin that burns easily and doesn’t readily tan). Other pertinent findings include the absence of oral lesions, nail changes, or other skin diseases.
A punch biopsy taken from the leading edge of the hair loss confirms the diagnosis of FFA and, ideally, should be reviewed by a dermatopathologist. Histologic examination will reveal a lichenoid lymphocytic infiltrate (predominantly around the hair follicle where the follicular stem cells reside), resulting in fibrosis and scarring. In addition to confirming the diagnosis with histologic examination, the following conditions must be ruled out in the differential: alopecia areata, female pattern hair loss, discoid lupus erythematosus, central centrifugal cicatricial alopecia, and traction alopecia.
Early detection is key. In general, physicians should initiate treatment as soon as possible to prevent disease progression and reduce permanent scarring and hair loss. Intralesional steroids such as triamcinolone acetonide (5-10 mg/cc), as well as high-potency topical steroids, are generally helpful to stabilize the disease. There is also some evidence of benefit from oral dutasteride or finasteride at variable doses. Immunosuppressants such as hydroxychloroquine also may be used as second-line treatments, although the benefit-to-risk ratio needs to be taken into consideration.
This patient was started on a regimen of topical high-potency steroids (clobetasol foam, 0.05%), with targeted, intralesional injection of steroids (10 mg/cc of triamcinolone acetonide) to areas with the most inflammation. She also was advised to use ketoconazole 2% shampoo while showering. These interventions decreased the patient’s symptoms dramatically. Her scalp erythema and scale improved, but the hair did not regrow. One year later, her hairline was clinically stable with no evidence of disease progression. She continued to see a dermatologist annually.
This case was adapted from: Power DV, Disse M, Hordinsky M. Progressive hair loss. J Fam Pract. 2017;66:521-523.
The FP referred the patient to a dermatology clinic that specialized in hair loss. Based on the clinical findings, physicians at the clinic suspected that this was a case of frontal fibrosing alopecia (FFA), a primary lymphocytic cicatricial (scarring) alopecia. A dermatopathologist confirmed the diagnosis via histologic review.
FFA is characterized by symmetric band-like hair loss with evidence of scarring in the frontal and temporal regions of the scalp. (The extent of hair loss can be assessed by retracting the patient’s hair and having the patient raise his or her eyebrows and wrinkle the forehead in a surprised look.) FFA is accompanied by eyebrow loss in 73% to 95% of patients. Mild to severe perifollicular (and possibly more generalized) erythema and scale are usually present. In addition, erythematous or skin-colored papules may appear on the face, and marked exaggeration of the temporal veins is a common finding.
Most patients with FFA (83%) are postmenopausal women and nearly all (98.6%) have Fitzpatrick skin type 1 or 2 (white skin that burns easily and doesn’t readily tan). Other pertinent findings include the absence of oral lesions, nail changes, or other skin diseases.
A punch biopsy taken from the leading edge of the hair loss confirms the diagnosis of FFA and, ideally, should be reviewed by a dermatopathologist. Histologic examination will reveal a lichenoid lymphocytic infiltrate (predominantly around the hair follicle where the follicular stem cells reside), resulting in fibrosis and scarring. In addition to confirming the diagnosis with histologic examination, the following conditions must be ruled out in the differential: alopecia areata, female pattern hair loss, discoid lupus erythematosus, central centrifugal cicatricial alopecia, and traction alopecia.
Early detection is key. In general, physicians should initiate treatment as soon as possible to prevent disease progression and reduce permanent scarring and hair loss. Intralesional steroids such as triamcinolone acetonide (5-10 mg/cc), as well as high-potency topical steroids, are generally helpful to stabilize the disease. There is also some evidence of benefit from oral dutasteride or finasteride at variable doses. Immunosuppressants such as hydroxychloroquine also may be used as second-line treatments, although the benefit-to-risk ratio needs to be taken into consideration.
This patient was started on a regimen of topical high-potency steroids (clobetasol foam, 0.05%), with targeted, intralesional injection of steroids (10 mg/cc of triamcinolone acetonide) to areas with the most inflammation. She also was advised to use ketoconazole 2% shampoo while showering. These interventions decreased the patient’s symptoms dramatically. Her scalp erythema and scale improved, but the hair did not regrow. One year later, her hairline was clinically stable with no evidence of disease progression. She continued to see a dermatologist annually.
This case was adapted from: Power DV, Disse M, Hordinsky M. Progressive hair loss. J Fam Pract. 2017;66:521-523.

Keeping Lesions at Arm’s Length
A 14-year-old boy presents to dermatology for evaluation of an asymptomatic “rash” present on his arms since age 6. The condition has caught the attention of family members and teachers over the years, particularly in regard to possible contagion.
The patient is otherwise reasonably healthy, although he has asthma and seasonal allergies.

EXAMINATION
The "rash" consists of uniformly distributed and sized planar papules. Although they are tiny, averaging only 1 mm wide, they are prominent enough to be noticeable and palpable. They appear slightly lighter than the surrounding skin. Distribution is from the lower deltoid to mid-dorsal forearm, affecting both arms identically. The volar aspects and triceps of both arms are totally spared.
The patient has type IV skin.
What’s the diagnosis?
DISCUSSION
This case is an almost perfect representation of lichen nitidus (LN), in terms of morphology, distribution, and configuration. Close examination of individual lesions revealed that the papules were somewhat planar (ie, flat-topped), giving their surfaces a reflective appearance that the eye interprets as white (particularly contrasted with darker skin).
LN can occur in anyone, but it is most often seen in those with darker skin. It is also frequently seen in children, many of whom are atopic, with dry, sensitive skin that is prone to eczema.
In terms of distribution, LN typically affects the extensor triceps, elbow, and forearms bilaterally. With its flat-topped and shiny appearance, LN is sometimes called "mini-lichen planus"—a condition that can demonstrate similar features. Fortunately, LN is seldom itchy and shares none of the distinct histologic characteristics of lichen planus.
LN is quite unusual, if not rare. It is also idiopathic and nearly always resolves on its own—although this can take months to years.
Emollients help to make the affected skin smoother and less visible. Class 4 steroid creams (eg, triamcinolone 0.05%) can help with itching.
TAKE-HOME LEARNING POINTS
- Lichen nitidus (LN) is a rare idiopathic skin condition manifesting with patches of tiny planar papules; it typically affects the elbow and dorsal forearm.
- LN has no pathologic implications and is asymptomatic and self-limited.
- The lesions of LN have a “lichenoid” appearance—ie, a shiny, flat-topped look similar to that seen with lichen planus.
- Fortunately, LN rarely requires treatment, aside from relief of mild itching.
A 14-year-old boy presents to dermatology for evaluation of an asymptomatic “rash” present on his arms since age 6. The condition has caught the attention of family members and teachers over the years, particularly in regard to possible contagion.
The patient is otherwise reasonably healthy, although he has asthma and seasonal allergies.
EXAMINATION
The "rash" consists of uniformly distributed and sized planar papules. Although they are tiny, averaging only 1 mm wide, they are prominent enough to be noticeable and palpable. They appear slightly lighter than the surrounding skin. Distribution is from the lower deltoid to mid-dorsal forearm, affecting both arms identically. The volar aspects and triceps of both arms are totally spared.
The patient has type IV skin.
What’s the diagnosis?
DISCUSSION
This case is an almost perfect representation of lichen nitidus (LN), in terms of morphology, distribution, and configuration. Close examination of individual lesions revealed that the papules were somewhat planar (ie, flat-topped), giving their surfaces a reflective appearance that the eye interprets as white (particularly contrasted with darker skin).
LN can occur in anyone, but it is most often seen in those with darker skin. It is also frequently seen in children, many of whom are atopic, with dry, sensitive skin that is prone to eczema.
In terms of distribution, LN typically affects the extensor triceps, elbow, and forearms bilaterally. With its flat-topped and shiny appearance, LN is sometimes called "mini-lichen planus"—a condition that can demonstrate similar features. Fortunately, LN is seldom itchy and shares none of the distinct histologic characteristics of lichen planus.
LN is quite unusual, if not rare. It is also idiopathic and nearly always resolves on its own—although this can take months to years.
Emollients help to make the affected skin smoother and less visible. Class 4 steroid creams (eg, triamcinolone 0.05%) can help with itching.
TAKE-HOME LEARNING POINTS
- Lichen nitidus (LN) is a rare idiopathic skin condition manifesting with patches of tiny planar papules; it typically affects the elbow and dorsal forearm.
- LN has no pathologic implications and is asymptomatic and self-limited.
- The lesions of LN have a “lichenoid” appearance—ie, a shiny, flat-topped look similar to that seen with lichen planus.
- Fortunately, LN rarely requires treatment, aside from relief of mild itching.
A 14-year-old boy presents to dermatology for evaluation of an asymptomatic “rash” present on his arms since age 6. The condition has caught the attention of family members and teachers over the years, particularly in regard to possible contagion.
The patient is otherwise reasonably healthy, although he has asthma and seasonal allergies.
EXAMINATION
The "rash" consists of uniformly distributed and sized planar papules. Although they are tiny, averaging only 1 mm wide, they are prominent enough to be noticeable and palpable. They appear slightly lighter than the surrounding skin. Distribution is from the lower deltoid to mid-dorsal forearm, affecting both arms identically. The volar aspects and triceps of both arms are totally spared.
The patient has type IV skin.
What’s the diagnosis?
DISCUSSION
This case is an almost perfect representation of lichen nitidus (LN), in terms of morphology, distribution, and configuration. Close examination of individual lesions revealed that the papules were somewhat planar (ie, flat-topped), giving their surfaces a reflective appearance that the eye interprets as white (particularly contrasted with darker skin).
LN can occur in anyone, but it is most often seen in those with darker skin. It is also frequently seen in children, many of whom are atopic, with dry, sensitive skin that is prone to eczema.
In terms of distribution, LN typically affects the extensor triceps, elbow, and forearms bilaterally. With its flat-topped and shiny appearance, LN is sometimes called "mini-lichen planus"—a condition that can demonstrate similar features. Fortunately, LN is seldom itchy and shares none of the distinct histologic characteristics of lichen planus.
LN is quite unusual, if not rare. It is also idiopathic and nearly always resolves on its own—although this can take months to years.
Emollients help to make the affected skin smoother and less visible. Class 4 steroid creams (eg, triamcinolone 0.05%) can help with itching.
TAKE-HOME LEARNING POINTS
- Lichen nitidus (LN) is a rare idiopathic skin condition manifesting with patches of tiny planar papules; it typically affects the elbow and dorsal forearm.
- LN has no pathologic implications and is asymptomatic and self-limited.
- The lesions of LN have a “lichenoid” appearance—ie, a shiny, flat-topped look similar to that seen with lichen planus.
- Fortunately, LN rarely requires treatment, aside from relief of mild itching.

Newborns’ maternal protection against measles wanes within 6 months
according to new research.

In fact, most of the 196 infants’ maternal measles antibodies had dropped below the protective threshold by 3 months of age – well before the recommended age of 12-15 months for the first dose of MMR vaccine.
The odds of inadequate protection doubled for each additional month of age, Michelle Science, MD, of the University of Toronto and associates reported in Pediatrics.
“The widening gap between loss of maternal antibodies and measles vaccination described in our study leaves infants vulnerable to measles for much of their infancy and highlights the need for further research to support public health policy,” Dr. Science and colleagues wrote.
The findings are not surprising for a setting in which measles has been eliminated and align with results from past research, Huong Q. McLean, PhD, MPH, of the Marshfield (Wis.) Clinic Research Institute and Walter A. Orenstein, MD, of Emory University in Atlanta wrote in an accompanying editorial (Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-2541).
However, this susceptibility prior to receiving the MMR has taken on a new significance more recently, Dr. McLean and Dr. Orenstein suggested.
“In light of increasing measles outbreaks during the past year reaching levels not recorded in the United States since 1992 and increased measles elsewhere, coupled with the risk of severe illness in infants, there is increased concern regarding the protection of infants against measles,” the editorialists wrote.
Dr. Science and colleagues tested serum samples from 196 term infants, all under 12 months old, for antibodies against measles. The sera had been previously collected at a single tertiary care center in Ontario for clinical testing and then stored. Measles has been eliminated in Canada since 1998.
The researchers randomly selected 25 samples for each of eight different age groups: up to 30 days old; 1 month (31-60 days); 2 months (61-89 days); 3 months (90-119 days); 4 months; 5 months; 6-9 months; and 9-11 months.
Just over half the babies (56%) were male, and 35% had an underlying condition, but none had conditions that might affect antibody levels. The conditions were primarily a developmental delay or otherwise affecting the central nervous system, liver, or gastrointestinal function. Mean maternal age was 32 years.
To ensure high test sensitivity, the researchers used the plaque-reduction neutralization test (PRNT) to test for measles-neutralizing antibodies instead of using enzyme-linked immunosorbent assay (ELISA) because “ELISA sensitivity decreases as antibody titers decrease,” Dr. Science and colleagues wrote. They used a neutralization titer of less than 192 mIU/mL as the threshold for protection against measles.
When the researchers calculated the predicted standardized mean antibody titer for infants with a mother aged 32 years, they determined their mean to be 541 mIU/mL at 1 month, 142 mIU/mL at 3 months (below the measles threshold of susceptibility of 192 mIU/mL) , and 64 mIU/mL at 6 months. None of the infants had measles antibodies above the protective threshold at 6 months old, the authors noted.
Children’s odds of susceptibility to measles doubled for each additional month of age, after adjustment for infant sex and maternal age (odds ratio, 2.13). Children’s likelihood of susceptibility to measles modestly increased as maternal age increased in 5-year increments from 25 to 40 years.
Children with an underlying conditions had greater susceptibility to measles (83%), compared with those without a comorbidity (68%, P = .03). No difference in susceptibility existed between males and females or based on gestational age at birth (ranging from 37 to 41 weeks).
The Advisory Committee on Immunization Practices permits measles vaccination “as early as 6 months for infants who plan to travel internationally, infants with ongoing risk for exposure during measles outbreaks and as postexposure prophylaxis,” Dr. McLean and Dr. Orenstein noted in their editorial.
They discussed the rationale for various changes in the recommended schedule for measles immunization, based on changes in epidemiology of the disease and improved understanding of the immune response to vaccination since the vaccine became available in 1963. Then they posed the question of whether the recommendation should be revised again.
“Ideally, the schedule should minimize the risk of measles and its complications and optimize vaccine-induced protection,” Dr. McLean and Dr. Orenstein wrote.
They argued that the evidence cannot currently support changing the first MMR dose to a younger age because measles incidence in the United States remains extremely low outside of the extraordinary outbreaks in 2014 and 2019. Further, infants under 12 months of age make up less than 15% of measles cases during outbreaks, and unvaccinated people make up more than 70% of cases.
Rather, they stated, this new study emphasizes the importance of following the current schedule, with consideration of an earlier schedule only warranted during outbreaks.
“Health care providers must work to maintain high levels of coverage with 2 doses of MMR among vaccine-eligible populations and minimize pockets of susceptibility to prevent transmission to infants and prevent reestablishment of endemic transmission,” they concluded.
The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures. The editorialists had no external funding and no relevant financial disclosures.
SOURCE: Science M et al. Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-0630.
according to new research.
In fact, most of the 196 infants’ maternal measles antibodies had dropped below the protective threshold by 3 months of age – well before the recommended age of 12-15 months for the first dose of MMR vaccine.
The odds of inadequate protection doubled for each additional month of age, Michelle Science, MD, of the University of Toronto and associates reported in Pediatrics.
“The widening gap between loss of maternal antibodies and measles vaccination described in our study leaves infants vulnerable to measles for much of their infancy and highlights the need for further research to support public health policy,” Dr. Science and colleagues wrote.
The findings are not surprising for a setting in which measles has been eliminated and align with results from past research, Huong Q. McLean, PhD, MPH, of the Marshfield (Wis.) Clinic Research Institute and Walter A. Orenstein, MD, of Emory University in Atlanta wrote in an accompanying editorial (Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-2541).
However, this susceptibility prior to receiving the MMR has taken on a new significance more recently, Dr. McLean and Dr. Orenstein suggested.
“In light of increasing measles outbreaks during the past year reaching levels not recorded in the United States since 1992 and increased measles elsewhere, coupled with the risk of severe illness in infants, there is increased concern regarding the protection of infants against measles,” the editorialists wrote.
Dr. Science and colleagues tested serum samples from 196 term infants, all under 12 months old, for antibodies against measles. The sera had been previously collected at a single tertiary care center in Ontario for clinical testing and then stored. Measles has been eliminated in Canada since 1998.
The researchers randomly selected 25 samples for each of eight different age groups: up to 30 days old; 1 month (31-60 days); 2 months (61-89 days); 3 months (90-119 days); 4 months; 5 months; 6-9 months; and 9-11 months.
Just over half the babies (56%) were male, and 35% had an underlying condition, but none had conditions that might affect antibody levels. The conditions were primarily a developmental delay or otherwise affecting the central nervous system, liver, or gastrointestinal function. Mean maternal age was 32 years.
To ensure high test sensitivity, the researchers used the plaque-reduction neutralization test (PRNT) to test for measles-neutralizing antibodies instead of using enzyme-linked immunosorbent assay (ELISA) because “ELISA sensitivity decreases as antibody titers decrease,” Dr. Science and colleagues wrote. They used a neutralization titer of less than 192 mIU/mL as the threshold for protection against measles.
When the researchers calculated the predicted standardized mean antibody titer for infants with a mother aged 32 years, they determined their mean to be 541 mIU/mL at 1 month, 142 mIU/mL at 3 months (below the measles threshold of susceptibility of 192 mIU/mL) , and 64 mIU/mL at 6 months. None of the infants had measles antibodies above the protective threshold at 6 months old, the authors noted.
Children’s odds of susceptibility to measles doubled for each additional month of age, after adjustment for infant sex and maternal age (odds ratio, 2.13). Children’s likelihood of susceptibility to measles modestly increased as maternal age increased in 5-year increments from 25 to 40 years.
Children with an underlying conditions had greater susceptibility to measles (83%), compared with those without a comorbidity (68%, P = .03). No difference in susceptibility existed between males and females or based on gestational age at birth (ranging from 37 to 41 weeks).
The Advisory Committee on Immunization Practices permits measles vaccination “as early as 6 months for infants who plan to travel internationally, infants with ongoing risk for exposure during measles outbreaks and as postexposure prophylaxis,” Dr. McLean and Dr. Orenstein noted in their editorial.
They discussed the rationale for various changes in the recommended schedule for measles immunization, based on changes in epidemiology of the disease and improved understanding of the immune response to vaccination since the vaccine became available in 1963. Then they posed the question of whether the recommendation should be revised again.
“Ideally, the schedule should minimize the risk of measles and its complications and optimize vaccine-induced protection,” Dr. McLean and Dr. Orenstein wrote.
They argued that the evidence cannot currently support changing the first MMR dose to a younger age because measles incidence in the United States remains extremely low outside of the extraordinary outbreaks in 2014 and 2019. Further, infants under 12 months of age make up less than 15% of measles cases during outbreaks, and unvaccinated people make up more than 70% of cases.
Rather, they stated, this new study emphasizes the importance of following the current schedule, with consideration of an earlier schedule only warranted during outbreaks.
“Health care providers must work to maintain high levels of coverage with 2 doses of MMR among vaccine-eligible populations and minimize pockets of susceptibility to prevent transmission to infants and prevent reestablishment of endemic transmission,” they concluded.
The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures. The editorialists had no external funding and no relevant financial disclosures.
SOURCE: Science M et al. Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-0630.
according to new research.
In fact, most of the 196 infants’ maternal measles antibodies had dropped below the protective threshold by 3 months of age – well before the recommended age of 12-15 months for the first dose of MMR vaccine.
The odds of inadequate protection doubled for each additional month of age, Michelle Science, MD, of the University of Toronto and associates reported in Pediatrics.
“The widening gap between loss of maternal antibodies and measles vaccination described in our study leaves infants vulnerable to measles for much of their infancy and highlights the need for further research to support public health policy,” Dr. Science and colleagues wrote.
The findings are not surprising for a setting in which measles has been eliminated and align with results from past research, Huong Q. McLean, PhD, MPH, of the Marshfield (Wis.) Clinic Research Institute and Walter A. Orenstein, MD, of Emory University in Atlanta wrote in an accompanying editorial (Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-2541).
However, this susceptibility prior to receiving the MMR has taken on a new significance more recently, Dr. McLean and Dr. Orenstein suggested.
“In light of increasing measles outbreaks during the past year reaching levels not recorded in the United States since 1992 and increased measles elsewhere, coupled with the risk of severe illness in infants, there is increased concern regarding the protection of infants against measles,” the editorialists wrote.
Dr. Science and colleagues tested serum samples from 196 term infants, all under 12 months old, for antibodies against measles. The sera had been previously collected at a single tertiary care center in Ontario for clinical testing and then stored. Measles has been eliminated in Canada since 1998.
The researchers randomly selected 25 samples for each of eight different age groups: up to 30 days old; 1 month (31-60 days); 2 months (61-89 days); 3 months (90-119 days); 4 months; 5 months; 6-9 months; and 9-11 months.
Just over half the babies (56%) were male, and 35% had an underlying condition, but none had conditions that might affect antibody levels. The conditions were primarily a developmental delay or otherwise affecting the central nervous system, liver, or gastrointestinal function. Mean maternal age was 32 years.
To ensure high test sensitivity, the researchers used the plaque-reduction neutralization test (PRNT) to test for measles-neutralizing antibodies instead of using enzyme-linked immunosorbent assay (ELISA) because “ELISA sensitivity decreases as antibody titers decrease,” Dr. Science and colleagues wrote. They used a neutralization titer of less than 192 mIU/mL as the threshold for protection against measles.
When the researchers calculated the predicted standardized mean antibody titer for infants with a mother aged 32 years, they determined their mean to be 541 mIU/mL at 1 month, 142 mIU/mL at 3 months (below the measles threshold of susceptibility of 192 mIU/mL) , and 64 mIU/mL at 6 months. None of the infants had measles antibodies above the protective threshold at 6 months old, the authors noted.
Children’s odds of susceptibility to measles doubled for each additional month of age, after adjustment for infant sex and maternal age (odds ratio, 2.13). Children’s likelihood of susceptibility to measles modestly increased as maternal age increased in 5-year increments from 25 to 40 years.
Children with an underlying conditions had greater susceptibility to measles (83%), compared with those without a comorbidity (68%, P = .03). No difference in susceptibility existed between males and females or based on gestational age at birth (ranging from 37 to 41 weeks).
The Advisory Committee on Immunization Practices permits measles vaccination “as early as 6 months for infants who plan to travel internationally, infants with ongoing risk for exposure during measles outbreaks and as postexposure prophylaxis,” Dr. McLean and Dr. Orenstein noted in their editorial.
They discussed the rationale for various changes in the recommended schedule for measles immunization, based on changes in epidemiology of the disease and improved understanding of the immune response to vaccination since the vaccine became available in 1963. Then they posed the question of whether the recommendation should be revised again.
“Ideally, the schedule should minimize the risk of measles and its complications and optimize vaccine-induced protection,” Dr. McLean and Dr. Orenstein wrote.
They argued that the evidence cannot currently support changing the first MMR dose to a younger age because measles incidence in the United States remains extremely low outside of the extraordinary outbreaks in 2014 and 2019. Further, infants under 12 months of age make up less than 15% of measles cases during outbreaks, and unvaccinated people make up more than 70% of cases.
Rather, they stated, this new study emphasizes the importance of following the current schedule, with consideration of an earlier schedule only warranted during outbreaks.
“Health care providers must work to maintain high levels of coverage with 2 doses of MMR among vaccine-eligible populations and minimize pockets of susceptibility to prevent transmission to infants and prevent reestablishment of endemic transmission,” they concluded.
The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures. The editorialists had no external funding and no relevant financial disclosures.
SOURCE: Science M et al. Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-0630.
FROM PEDIATRICS
Key clinical point: Infants’ maternal measles antibodies fell below protective levels by 6 months old.
Major finding: Infants were twice as likely not to have protective immunity against measles for each month of age after birth (odds ratio, 2.13).
Study details: The findings are based on measles antibody testing of 196 serum samples from infants born in a tertiary care center in Ontario.
Disclosures: The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures.
Source: Science M et al. Pediatrics. 2019 Nov 21. doi: 10.1542/peds.2019-0630.

Menopause

Firm and tender growth in right nostril

The clinical features were consistent with a filiform wart, which is caused by human papillomavirus and common on the face. Filiform warts may occur on mucosal surfaces, including the nasal mucosa, lips, or eyelids. Most are benign and resolve within 2 years without treatment, but others can be symptomatic. Larger filiform warts may develop the clinical features of a cutaneous horn and mimic a squamous cell carcinoma.
Patients often want the wart removed for functional or cosmetic reasons. Although, there are many treatments available for warts, none have success rates that exceed about 70%. The most common options for treatment include topical salicylic acid in a liquid or plaster, cryotherapy, intralesional immunotherapy with candida antigen, excision, and topical acid.
The location of this patient’s wart limited the treatment options to cryotherapy or snip excision and cautery. The patient opted for cryotherapy. In this process, a hemostat or heavy gauge tweezer is dipped in liquid nitrogen and allowed to cool. Then, without anesthesia, the clinician pinches the lesion gently with the instrument and holds it until the freeze horizon extends to the base. This is then repeated. Each cycle may take 10 and 15 seconds. A benefit of this technique (which is also useful for skin tags and lesions close to the eye) is the ability to avoid overspray of liquid nitrogen and thus, minimize collateral tissue damage. It is also quick and bloodless.
The FP performed cryotherapy on the wart and advised the patient to expect the lesion to become more inflamed over the next 2 to 3 days and then peel off within 1 to 2 weeks. The FP also instructed the patient to return in a week or 2 so that the FP could evaluate whether additional treatment would be necessary. In general, non-genital warts require 2 to 3 individual treatments to clear, but a smaller and pedunculated wart like the one seen in this case tend to clear more easily.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained).
The clinical features were consistent with a filiform wart, which is caused by human papillomavirus and common on the face. Filiform warts may occur on mucosal surfaces, including the nasal mucosa, lips, or eyelids. Most are benign and resolve within 2 years without treatment, but others can be symptomatic. Larger filiform warts may develop the clinical features of a cutaneous horn and mimic a squamous cell carcinoma.
Patients often want the wart removed for functional or cosmetic reasons. Although, there are many treatments available for warts, none have success rates that exceed about 70%. The most common options for treatment include topical salicylic acid in a liquid or plaster, cryotherapy, intralesional immunotherapy with candida antigen, excision, and topical acid.
The location of this patient’s wart limited the treatment options to cryotherapy or snip excision and cautery. The patient opted for cryotherapy. In this process, a hemostat or heavy gauge tweezer is dipped in liquid nitrogen and allowed to cool. Then, without anesthesia, the clinician pinches the lesion gently with the instrument and holds it until the freeze horizon extends to the base. This is then repeated. Each cycle may take 10 and 15 seconds. A benefit of this technique (which is also useful for skin tags and lesions close to the eye) is the ability to avoid overspray of liquid nitrogen and thus, minimize collateral tissue damage. It is also quick and bloodless.
The FP performed cryotherapy on the wart and advised the patient to expect the lesion to become more inflamed over the next 2 to 3 days and then peel off within 1 to 2 weeks. The FP also instructed the patient to return in a week or 2 so that the FP could evaluate whether additional treatment would be necessary. In general, non-genital warts require 2 to 3 individual treatments to clear, but a smaller and pedunculated wart like the one seen in this case tend to clear more easily.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained).
The clinical features were consistent with a filiform wart, which is caused by human papillomavirus and common on the face. Filiform warts may occur on mucosal surfaces, including the nasal mucosa, lips, or eyelids. Most are benign and resolve within 2 years without treatment, but others can be symptomatic. Larger filiform warts may develop the clinical features of a cutaneous horn and mimic a squamous cell carcinoma.
Patients often want the wart removed for functional or cosmetic reasons. Although, there are many treatments available for warts, none have success rates that exceed about 70%. The most common options for treatment include topical salicylic acid in a liquid or plaster, cryotherapy, intralesional immunotherapy with candida antigen, excision, and topical acid.
The location of this patient’s wart limited the treatment options to cryotherapy or snip excision and cautery. The patient opted for cryotherapy. In this process, a hemostat or heavy gauge tweezer is dipped in liquid nitrogen and allowed to cool. Then, without anesthesia, the clinician pinches the lesion gently with the instrument and holds it until the freeze horizon extends to the base. This is then repeated. Each cycle may take 10 and 15 seconds. A benefit of this technique (which is also useful for skin tags and lesions close to the eye) is the ability to avoid overspray of liquid nitrogen and thus, minimize collateral tissue damage. It is also quick and bloodless.
The FP performed cryotherapy on the wart and advised the patient to expect the lesion to become more inflamed over the next 2 to 3 days and then peel off within 1 to 2 weeks. The FP also instructed the patient to return in a week or 2 so that the FP could evaluate whether additional treatment would be necessary. In general, non-genital warts require 2 to 3 individual treatments to clear, but a smaller and pedunculated wart like the one seen in this case tend to clear more easily.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained).

Vaping front and center at Hahn’s first FDA confirmation hearing
Stephen Hahn, MD, President Trump’s pick to head the Food and Drug Administration, faced questions from both sides of the aisle on youth vaping, but came up short when asked to commit to taking action, particularly on banning flavored vaping products.

Speaking at a Nov. 20 confirmation hearing before the Senate Health, Education, Labor, and Pensions Committee, Dr. Hahn said that youth vaping and e-cigarette use is “an important, urgent crisis in this country. I do not want to see another generation of Americans become addicted to tobacco and nicotine and I believe that we need to take aggressive to stop that.”
Sen. Patty Murray (D-Wash), the committee’s ranking member, asked Dr. Hahn whether he would work to finalize a ban flavored e-cigarette products, first proposed but then backed away from, by the president in September.
“I understand that the final compliance policy is under consideration by the administration, and I look forward to their decision,” Dr. Hahn said. “I am not privy to those decision-making processes, but I very much agree and support that aggressive action needs to be taken to protect our children.”
When pressed by Sen. Murray as to whether he told President Trump that he disagrees with the decision to back away the proposed ban, Dr. Hahn revealed that he has “not had a conversation with the president.”
Dr. Hahn, a radiation oncologist who currently serves as chief medical executive at MD Anderson Cancer Center, Houston, held firm to just coming up short of making that commitment when questioned by senators from both parties.
Sen. Mitt Romney (R-Utah) warned Dr. Hahn that the playing of politics would be unlike anything he has seen and is already being played out in the lobbying of the administration to change its stance on flavored e-cigarette products, which can run counter to the science about the harmful effects of these products.
“The question is how you will balance those things in which you put forward,” Sen. Romney asked. “How you will deal with this issue is a pretty good test case for how you would deal with this issue on an ongoing basis on matters not just related to vaping.”
He also brought up President Trump’s September announcement on a flavor ban and the administration’s signaling they are moving away from a flavor ban. “Is the FDA, under your leadership, able and willing to take action which will protect our kids, whether or not the White House wants you to take that action?”
Dr. Hahn cited his pledge as a doctor to always put the patient first and reiterated that “I take that pledge very seriously and I think if you ask anyone who has worked with me, they will tell you that I have upheld that pledge.”
But he fell short of saying that he would take actions that would oppose the White House, saying only that “patients need to come first and the decisions that we make need to be guided by science and data, congruent with the law.”
When asked by Sen. Romney if he saw any reason for holding off on a flavor ban, given the evidence that suggests flavored e-cigarette products are the gateway to youths nicotine addiction, Dr. Hahn said that he has seen the same evidence and that it requires “bold action,” but did not commit to a flavor ban. “I will use science and data to guide the decisions if I am fortunate enough to be confirmed, and I won’t back away from that.”
Sen. Doug Jones (D-Ala.) expressed concern about Dr. Hahn’s answers.
“I was less than happy with many of the answers you gave to members of this committee with regard to vaping and the potential ban on flavored e-cigarettes,” Sen. Jones said. “I think you can tell from the questions of so many senators that is one of the biggest issues that the United States Senate and Congress is facing right now. It is with this committee.”
Outside of vaping, much of the senators’ questioning was nonconfrontational, with questions spanning a gamut of issues facing the FDA.
Dr. Hahn offered his commitment to working with Congress to address drug shortages, noting nonspecifically that, “there are things that we can do to help.”
He also pledged to work with Congress on addressing patent reform to get more biosimilars to market in an effort to help drive down drug prices.
Regarding opioids, Dr. Hahn was asked about balancing the needs of those who legitimately need access to opioids against abuse and diversion.
“When I first went to medical school and started taking care of cancer patients, the teaching was that cancer patients should be treated liberally with opioids and that they don’t become addicted to pain medications,” he said. “We found out that wasn’t the case – and in some instances – with tragic consequences.”
He noted that pain therapy has evolved and that his institution now takes a multidisciplinary approach employing both opioid and nonopioid medications.
“I am very much a supporter of the multidisciplinary approach to treating pain,” he said. “I think it is something that we need to more of and if I am fortunate enough to be confirmed as commissioner of [FDA], I look forward to furthering the education efforts for providers and patients.”
Other areas he committed to included helping to improve clinical trial design for psychiatric medications and improving development of therapies for rare diseases.
Committee Chairman Lamar Alexander (R-Tenn.) said he plans to schedule a Dec. 3 vote to advance Dr. Hahn’s nomination to the full Senate for its consideration.
Stephen Hahn, MD, President Trump’s pick to head the Food and Drug Administration, faced questions from both sides of the aisle on youth vaping, but came up short when asked to commit to taking action, particularly on banning flavored vaping products.
Speaking at a Nov. 20 confirmation hearing before the Senate Health, Education, Labor, and Pensions Committee, Dr. Hahn said that youth vaping and e-cigarette use is “an important, urgent crisis in this country. I do not want to see another generation of Americans become addicted to tobacco and nicotine and I believe that we need to take aggressive to stop that.”
Sen. Patty Murray (D-Wash), the committee’s ranking member, asked Dr. Hahn whether he would work to finalize a ban flavored e-cigarette products, first proposed but then backed away from, by the president in September.
“I understand that the final compliance policy is under consideration by the administration, and I look forward to their decision,” Dr. Hahn said. “I am not privy to those decision-making processes, but I very much agree and support that aggressive action needs to be taken to protect our children.”
When pressed by Sen. Murray as to whether he told President Trump that he disagrees with the decision to back away the proposed ban, Dr. Hahn revealed that he has “not had a conversation with the president.”
Dr. Hahn, a radiation oncologist who currently serves as chief medical executive at MD Anderson Cancer Center, Houston, held firm to just coming up short of making that commitment when questioned by senators from both parties.
Sen. Mitt Romney (R-Utah) warned Dr. Hahn that the playing of politics would be unlike anything he has seen and is already being played out in the lobbying of the administration to change its stance on flavored e-cigarette products, which can run counter to the science about the harmful effects of these products.
“The question is how you will balance those things in which you put forward,” Sen. Romney asked. “How you will deal with this issue is a pretty good test case for how you would deal with this issue on an ongoing basis on matters not just related to vaping.”
He also brought up President Trump’s September announcement on a flavor ban and the administration’s signaling they are moving away from a flavor ban. “Is the FDA, under your leadership, able and willing to take action which will protect our kids, whether or not the White House wants you to take that action?”
Dr. Hahn cited his pledge as a doctor to always put the patient first and reiterated that “I take that pledge very seriously and I think if you ask anyone who has worked with me, they will tell you that I have upheld that pledge.”
But he fell short of saying that he would take actions that would oppose the White House, saying only that “patients need to come first and the decisions that we make need to be guided by science and data, congruent with the law.”
When asked by Sen. Romney if he saw any reason for holding off on a flavor ban, given the evidence that suggests flavored e-cigarette products are the gateway to youths nicotine addiction, Dr. Hahn said that he has seen the same evidence and that it requires “bold action,” but did not commit to a flavor ban. “I will use science and data to guide the decisions if I am fortunate enough to be confirmed, and I won’t back away from that.”
Sen. Doug Jones (D-Ala.) expressed concern about Dr. Hahn’s answers.
“I was less than happy with many of the answers you gave to members of this committee with regard to vaping and the potential ban on flavored e-cigarettes,” Sen. Jones said. “I think you can tell from the questions of so many senators that is one of the biggest issues that the United States Senate and Congress is facing right now. It is with this committee.”
Outside of vaping, much of the senators’ questioning was nonconfrontational, with questions spanning a gamut of issues facing the FDA.
Dr. Hahn offered his commitment to working with Congress to address drug shortages, noting nonspecifically that, “there are things that we can do to help.”
He also pledged to work with Congress on addressing patent reform to get more biosimilars to market in an effort to help drive down drug prices.
Regarding opioids, Dr. Hahn was asked about balancing the needs of those who legitimately need access to opioids against abuse and diversion.
“When I first went to medical school and started taking care of cancer patients, the teaching was that cancer patients should be treated liberally with opioids and that they don’t become addicted to pain medications,” he said. “We found out that wasn’t the case – and in some instances – with tragic consequences.”
He noted that pain therapy has evolved and that his institution now takes a multidisciplinary approach employing both opioid and nonopioid medications.
“I am very much a supporter of the multidisciplinary approach to treating pain,” he said. “I think it is something that we need to more of and if I am fortunate enough to be confirmed as commissioner of [FDA], I look forward to furthering the education efforts for providers and patients.”
Other areas he committed to included helping to improve clinical trial design for psychiatric medications and improving development of therapies for rare diseases.
Committee Chairman Lamar Alexander (R-Tenn.) said he plans to schedule a Dec. 3 vote to advance Dr. Hahn’s nomination to the full Senate for its consideration.
Stephen Hahn, MD, President Trump’s pick to head the Food and Drug Administration, faced questions from both sides of the aisle on youth vaping, but came up short when asked to commit to taking action, particularly on banning flavored vaping products.
Speaking at a Nov. 20 confirmation hearing before the Senate Health, Education, Labor, and Pensions Committee, Dr. Hahn said that youth vaping and e-cigarette use is “an important, urgent crisis in this country. I do not want to see another generation of Americans become addicted to tobacco and nicotine and I believe that we need to take aggressive to stop that.”
Sen. Patty Murray (D-Wash), the committee’s ranking member, asked Dr. Hahn whether he would work to finalize a ban flavored e-cigarette products, first proposed but then backed away from, by the president in September.
“I understand that the final compliance policy is under consideration by the administration, and I look forward to their decision,” Dr. Hahn said. “I am not privy to those decision-making processes, but I very much agree and support that aggressive action needs to be taken to protect our children.”
When pressed by Sen. Murray as to whether he told President Trump that he disagrees with the decision to back away the proposed ban, Dr. Hahn revealed that he has “not had a conversation with the president.”
Dr. Hahn, a radiation oncologist who currently serves as chief medical executive at MD Anderson Cancer Center, Houston, held firm to just coming up short of making that commitment when questioned by senators from both parties.
Sen. Mitt Romney (R-Utah) warned Dr. Hahn that the playing of politics would be unlike anything he has seen and is already being played out in the lobbying of the administration to change its stance on flavored e-cigarette products, which can run counter to the science about the harmful effects of these products.
“The question is how you will balance those things in which you put forward,” Sen. Romney asked. “How you will deal with this issue is a pretty good test case for how you would deal with this issue on an ongoing basis on matters not just related to vaping.”
He also brought up President Trump’s September announcement on a flavor ban and the administration’s signaling they are moving away from a flavor ban. “Is the FDA, under your leadership, able and willing to take action which will protect our kids, whether or not the White House wants you to take that action?”
Dr. Hahn cited his pledge as a doctor to always put the patient first and reiterated that “I take that pledge very seriously and I think if you ask anyone who has worked with me, they will tell you that I have upheld that pledge.”
But he fell short of saying that he would take actions that would oppose the White House, saying only that “patients need to come first and the decisions that we make need to be guided by science and data, congruent with the law.”
When asked by Sen. Romney if he saw any reason for holding off on a flavor ban, given the evidence that suggests flavored e-cigarette products are the gateway to youths nicotine addiction, Dr. Hahn said that he has seen the same evidence and that it requires “bold action,” but did not commit to a flavor ban. “I will use science and data to guide the decisions if I am fortunate enough to be confirmed, and I won’t back away from that.”
Sen. Doug Jones (D-Ala.) expressed concern about Dr. Hahn’s answers.
“I was less than happy with many of the answers you gave to members of this committee with regard to vaping and the potential ban on flavored e-cigarettes,” Sen. Jones said. “I think you can tell from the questions of so many senators that is one of the biggest issues that the United States Senate and Congress is facing right now. It is with this committee.”
Outside of vaping, much of the senators’ questioning was nonconfrontational, with questions spanning a gamut of issues facing the FDA.
Dr. Hahn offered his commitment to working with Congress to address drug shortages, noting nonspecifically that, “there are things that we can do to help.”
He also pledged to work with Congress on addressing patent reform to get more biosimilars to market in an effort to help drive down drug prices.
Regarding opioids, Dr. Hahn was asked about balancing the needs of those who legitimately need access to opioids against abuse and diversion.
“When I first went to medical school and started taking care of cancer patients, the teaching was that cancer patients should be treated liberally with opioids and that they don’t become addicted to pain medications,” he said. “We found out that wasn’t the case – and in some instances – with tragic consequences.”
He noted that pain therapy has evolved and that his institution now takes a multidisciplinary approach employing both opioid and nonopioid medications.
“I am very much a supporter of the multidisciplinary approach to treating pain,” he said. “I think it is something that we need to more of and if I am fortunate enough to be confirmed as commissioner of [FDA], I look forward to furthering the education efforts for providers and patients.”
Other areas he committed to included helping to improve clinical trial design for psychiatric medications and improving development of therapies for rare diseases.
Committee Chairman Lamar Alexander (R-Tenn.) said he plans to schedule a Dec. 3 vote to advance Dr. Hahn’s nomination to the full Senate for its consideration.
REPORTING FROM A SENATE SUBCOMMITTEE HEARING
Scalp Psoriasis: A Challenge for Patients and Dermatologists
Prevalence of Scalp Psoriasis
Scalp psoriasis is a common and difficult-to-treat manifestation of psoriasis.1,2 The prevalence of scalp psoriasis in patients with psoriasis is estimated to be 45% to 56%.3 Other studies have shown 80% to 90% of patients with psoriasis have scalp involvement at some point during the course of their disease.2,4-6
Clinical Presentation
Scalp psoriasis typically presents as red thickened patches with silvery white scales that flake and may be mistaken for dandruff.7,8
The lesions may be limited to the hairline or may extend to the forehead, ears, and back of the neck.1,9 Patients often report intense itching, feelings of soreness, and burning.10,11 Patients with scalp psoriasis also are vulnerable to Koebner phenomenon because normal hair care along with scratching or picking at lesions can result in skin trauma and a cycle of exacerbating disease.11
Quality of Life Implications
Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.2,12,13 In one study, more than 70% of patients with scalp psoriasis reported difficulty with daily life.12 Patients frequently report feelings of shame, embarrassment, or self-consciousness about scalp psoriasis; many grow their hair long or wear hats to hide scalp lesions. Others report that flaking sometimes, often, or always affects their choice of clothing color.7,12
Psoriatic Alopecia
Alopecia is another common sequala in the setting of scalp psoriasis, though it is not well understood.14,15 First described by Shuster16 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.14,17 Clinically, it presents as pink scaly plaques consistent with psoriasis with overlying alopecia. In most patients, hair loss is usually reversible following effective treatment of psoriasis; however, instances of psoriatic alopecia have been reported as cicatricial (permanent) hair loss and generalized telogen effluvium. Cicatricial alopecia is increasingly being linked with chronic relapsing episodes of psoriasis.14,15 Patients with psoriatic alopecia are known to have a higher proportion of telogen and catagen hairs.14,18 Moreover, patients with psoriasis have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients.14
The patient described here had scalp psoriasis with increased and preserved hair density. The case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.14,15
Therapeutic Options
Although numerous treatment options for psoriasis are available, the scalp remains a difficult area to treat.1,14 Increased hair density can complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.14 The presence of hair also has been shown to strongly influence treatment adherence.1,8 Patients often discuss the greasy effect of medications in this area and difficulty removing products from the hair.1
Topical corticosteroids, with or without the addition of the vitamin D analogs calcipotriol or calcipotriene, remain the first-line treatment of mild scalp psoriasis. It is possible that the development of new formulations in recent years—foams, shampoos, and sprays—may improve adherence. Systemic treatment should be considered in severe or intractable cases.
Bottom Line
Although hair loss is more common, scalp psoriasis also may present with increased hair density, which may make topical medications more difficult to apply and can affect treatment adherence. Topical corticosteroids, with or without the addition of the vitamin D analog calcipotriol, remain the first-line treatment of mild scalp psoriasis. Systemic therapy should be considered in severe or recalcitrant cases.
- Blakely K, Gooderham M. Management of scalp psoriasis: current perspectives. Psoriasis (Auckl). 2016;6:33-40.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- Merola JF, Li T, Li WQ, et al. Prevalence of psoriasis phenotypes among men and women in the USA. Clin Exp Dermatol. 2016;41:486-489.
- Frez ML, Asawanonda P, Gunasekara C, et al. Recommendations for a patient-centered approach to the assessment and treatment of scalp psoriasis: a consensus statement from the Asia Scalp Psoriasis Study Group. J Dermatol Treat. 2014;25:38-45.
- van de Kerkhof PC, Franssen ME. Psoriasis of the scalp. diagnosis and management. Am J Clin Dermatol. 2001;2:159-165.
- Chan CS, Van Voorhees AS, Lebwohl MG, et al. Treatment of severe scalp psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2009;60:962-971.
- Aldredge LM, Higham RC. Manifestations and management of difficult-to-treat psoriasis. J Dermatol Nurses Assoc. 2018;10:189-197.
- Dopytalska K, Sobolewski P, Blaszczak A, et al. Psoriasis in special localizations. Reumatologia. 2018;56:392-398.
- Papp K, Berth-Jones J, Kragballe K, et al. Scalp psoriasis: a review of current topical treatment options. J Eur Acad Dermatol Venereol. 2007;21:1151-1160.
- Kircik LH, Kumar S. Scalp psoriasis. J Drugs Dermatol. 2010;9(8 suppl):S101-S105.
- Wozel G. Psoriasis treatment in difficult locations: scalp, nails, and intertriginous areas. Clin Dermatol. 2008;26:448-459.
- Sampogna F, Linder D, Piaserico S, et al. Quality of life assessment of patients with scalp dermatitis using the Italian version of the Scalpdex. Acta Dermato-Venereologica. 2014;94:411-414.
- Crowley J. Scalp psoriasis: an overview of the disease and available therapies. J Drugs Dermatol. 2010;9:912-918.
- Shah VV, Lee EB, Reddy SP, et al. Scalp psoriasis with increased hair density. Cutis. 2018;102:63-64.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
Prevalence of Scalp Psoriasis
Scalp psoriasis is a common and difficult-to-treat manifestation of psoriasis.1,2 The prevalence of scalp psoriasis in patients with psoriasis is estimated to be 45% to 56%.3 Other studies have shown 80% to 90% of patients with psoriasis have scalp involvement at some point during the course of their disease.2,4-6
Clinical Presentation
Scalp psoriasis typically presents as red thickened patches with silvery white scales that flake and may be mistaken for dandruff.7,8
The lesions may be limited to the hairline or may extend to the forehead, ears, and back of the neck.1,9 Patients often report intense itching, feelings of soreness, and burning.10,11 Patients with scalp psoriasis also are vulnerable to Koebner phenomenon because normal hair care along with scratching or picking at lesions can result in skin trauma and a cycle of exacerbating disease.11
Quality of Life Implications
Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.2,12,13 In one study, more than 70% of patients with scalp psoriasis reported difficulty with daily life.12 Patients frequently report feelings of shame, embarrassment, or self-consciousness about scalp psoriasis; many grow their hair long or wear hats to hide scalp lesions. Others report that flaking sometimes, often, or always affects their choice of clothing color.7,12
Psoriatic Alopecia
Alopecia is another common sequala in the setting of scalp psoriasis, though it is not well understood.14,15 First described by Shuster16 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.14,17 Clinically, it presents as pink scaly plaques consistent with psoriasis with overlying alopecia. In most patients, hair loss is usually reversible following effective treatment of psoriasis; however, instances of psoriatic alopecia have been reported as cicatricial (permanent) hair loss and generalized telogen effluvium. Cicatricial alopecia is increasingly being linked with chronic relapsing episodes of psoriasis.14,15 Patients with psoriatic alopecia are known to have a higher proportion of telogen and catagen hairs.14,18 Moreover, patients with psoriasis have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients.14
The patient described here had scalp psoriasis with increased and preserved hair density. The case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.14,15
Therapeutic Options
Although numerous treatment options for psoriasis are available, the scalp remains a difficult area to treat.1,14 Increased hair density can complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.14 The presence of hair also has been shown to strongly influence treatment adherence.1,8 Patients often discuss the greasy effect of medications in this area and difficulty removing products from the hair.1
Topical corticosteroids, with or without the addition of the vitamin D analogs calcipotriol or calcipotriene, remain the first-line treatment of mild scalp psoriasis. It is possible that the development of new formulations in recent years—foams, shampoos, and sprays—may improve adherence. Systemic treatment should be considered in severe or intractable cases.
Bottom Line
Although hair loss is more common, scalp psoriasis also may present with increased hair density, which may make topical medications more difficult to apply and can affect treatment adherence. Topical corticosteroids, with or without the addition of the vitamin D analog calcipotriol, remain the first-line treatment of mild scalp psoriasis. Systemic therapy should be considered in severe or recalcitrant cases.
Prevalence of Scalp Psoriasis
Scalp psoriasis is a common and difficult-to-treat manifestation of psoriasis.1,2 The prevalence of scalp psoriasis in patients with psoriasis is estimated to be 45% to 56%.3 Other studies have shown 80% to 90% of patients with psoriasis have scalp involvement at some point during the course of their disease.2,4-6
Clinical Presentation
Scalp psoriasis typically presents as red thickened patches with silvery white scales that flake and may be mistaken for dandruff.7,8
The lesions may be limited to the hairline or may extend to the forehead, ears, and back of the neck.1,9 Patients often report intense itching, feelings of soreness, and burning.10,11 Patients with scalp psoriasis also are vulnerable to Koebner phenomenon because normal hair care along with scratching or picking at lesions can result in skin trauma and a cycle of exacerbating disease.11
Quality of Life Implications
Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.2,12,13 In one study, more than 70% of patients with scalp psoriasis reported difficulty with daily life.12 Patients frequently report feelings of shame, embarrassment, or self-consciousness about scalp psoriasis; many grow their hair long or wear hats to hide scalp lesions. Others report that flaking sometimes, often, or always affects their choice of clothing color.7,12
Psoriatic Alopecia
Alopecia is another common sequala in the setting of scalp psoriasis, though it is not well understood.14,15 First described by Shuster16 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.14,17 Clinically, it presents as pink scaly plaques consistent with psoriasis with overlying alopecia. In most patients, hair loss is usually reversible following effective treatment of psoriasis; however, instances of psoriatic alopecia have been reported as cicatricial (permanent) hair loss and generalized telogen effluvium. Cicatricial alopecia is increasingly being linked with chronic relapsing episodes of psoriasis.14,15 Patients with psoriatic alopecia are known to have a higher proportion of telogen and catagen hairs.14,18 Moreover, patients with psoriasis have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients.14
The patient described here had scalp psoriasis with increased and preserved hair density. The case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.14,15
Therapeutic Options
Although numerous treatment options for psoriasis are available, the scalp remains a difficult area to treat.1,14 Increased hair density can complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.14 The presence of hair also has been shown to strongly influence treatment adherence.1,8 Patients often discuss the greasy effect of medications in this area and difficulty removing products from the hair.1
Topical corticosteroids, with or without the addition of the vitamin D analogs calcipotriol or calcipotriene, remain the first-line treatment of mild scalp psoriasis. It is possible that the development of new formulations in recent years—foams, shampoos, and sprays—may improve adherence. Systemic treatment should be considered in severe or intractable cases.
Bottom Line
Although hair loss is more common, scalp psoriasis also may present with increased hair density, which may make topical medications more difficult to apply and can affect treatment adherence. Topical corticosteroids, with or without the addition of the vitamin D analog calcipotriol, remain the first-line treatment of mild scalp psoriasis. Systemic therapy should be considered in severe or recalcitrant cases.
- Blakely K, Gooderham M. Management of scalp psoriasis: current perspectives. Psoriasis (Auckl). 2016;6:33-40.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- Merola JF, Li T, Li WQ, et al. Prevalence of psoriasis phenotypes among men and women in the USA. Clin Exp Dermatol. 2016;41:486-489.
- Frez ML, Asawanonda P, Gunasekara C, et al. Recommendations for a patient-centered approach to the assessment and treatment of scalp psoriasis: a consensus statement from the Asia Scalp Psoriasis Study Group. J Dermatol Treat. 2014;25:38-45.
- van de Kerkhof PC, Franssen ME. Psoriasis of the scalp. diagnosis and management. Am J Clin Dermatol. 2001;2:159-165.
- Chan CS, Van Voorhees AS, Lebwohl MG, et al. Treatment of severe scalp psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2009;60:962-971.
- Aldredge LM, Higham RC. Manifestations and management of difficult-to-treat psoriasis. J Dermatol Nurses Assoc. 2018;10:189-197.
- Dopytalska K, Sobolewski P, Blaszczak A, et al. Psoriasis in special localizations. Reumatologia. 2018;56:392-398.
- Papp K, Berth-Jones J, Kragballe K, et al. Scalp psoriasis: a review of current topical treatment options. J Eur Acad Dermatol Venereol. 2007;21:1151-1160.
- Kircik LH, Kumar S. Scalp psoriasis. J Drugs Dermatol. 2010;9(8 suppl):S101-S105.
- Wozel G. Psoriasis treatment in difficult locations: scalp, nails, and intertriginous areas. Clin Dermatol. 2008;26:448-459.
- Sampogna F, Linder D, Piaserico S, et al. Quality of life assessment of patients with scalp dermatitis using the Italian version of the Scalpdex. Acta Dermato-Venereologica. 2014;94:411-414.
- Crowley J. Scalp psoriasis: an overview of the disease and available therapies. J Drugs Dermatol. 2010;9:912-918.
- Shah VV, Lee EB, Reddy SP, et al. Scalp psoriasis with increased hair density. Cutis. 2018;102:63-64.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
- Blakely K, Gooderham M. Management of scalp psoriasis: current perspectives. Psoriasis (Auckl). 2016;6:33-40.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- Merola JF, Li T, Li WQ, et al. Prevalence of psoriasis phenotypes among men and women in the USA. Clin Exp Dermatol. 2016;41:486-489.
- Frez ML, Asawanonda P, Gunasekara C, et al. Recommendations for a patient-centered approach to the assessment and treatment of scalp psoriasis: a consensus statement from the Asia Scalp Psoriasis Study Group. J Dermatol Treat. 2014;25:38-45.
- van de Kerkhof PC, Franssen ME. Psoriasis of the scalp. diagnosis and management. Am J Clin Dermatol. 2001;2:159-165.
- Chan CS, Van Voorhees AS, Lebwohl MG, et al. Treatment of severe scalp psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2009;60:962-971.
- Aldredge LM, Higham RC. Manifestations and management of difficult-to-treat psoriasis. J Dermatol Nurses Assoc. 2018;10:189-197.
- Dopytalska K, Sobolewski P, Blaszczak A, et al. Psoriasis in special localizations. Reumatologia. 2018;56:392-398.
- Papp K, Berth-Jones J, Kragballe K, et al. Scalp psoriasis: a review of current topical treatment options. J Eur Acad Dermatol Venereol. 2007;21:1151-1160.
- Kircik LH, Kumar S. Scalp psoriasis. J Drugs Dermatol. 2010;9(8 suppl):S101-S105.
- Wozel G. Psoriasis treatment in difficult locations: scalp, nails, and intertriginous areas. Clin Dermatol. 2008;26:448-459.
- Sampogna F, Linder D, Piaserico S, et al. Quality of life assessment of patients with scalp dermatitis using the Italian version of the Scalpdex. Acta Dermato-Venereologica. 2014;94:411-414.
- Crowley J. Scalp psoriasis: an overview of the disease and available therapies. J Drugs Dermatol. 2010;9:912-918.
- Shah VV, Lee EB, Reddy SP, et al. Scalp psoriasis with increased hair density. Cutis. 2018;102:63-64.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
The Case
A 19-year-old man initially presented for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. The patient’s medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made.
Treatment
The patient was prescribed topical corticosteroids for 2 weeks and as needed thereafter.
Patient Outcomes
The patient remained stable for 5 years before again presenting to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye.
Clobetasol solution 0.05% twice daily was prescribed for application to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.

This case was adapted from Shah VV, Lee EB, Reddy SP, et al. Scalp psoriasis with increased hair density. Cutis. 2018;102:63-64.
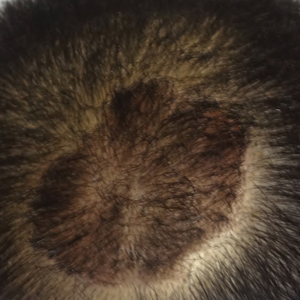
TARGET-NASH: One-third of NAFLD, NASH patients lost weight, but not all kept it off
BOSTON – While about one-third of overweight patients with nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH) slimmed down after a weight-loss intervention, about 20% gained weight back within the next few years, according to results from a large, long-term observational cohort study.
Effectively, that meant about one-quarter of overweight subjects enrolled in the TARGET-NASH study were able to lose weight and keep it off, said investigator Miguel H. Malespin, MD, a transplant hepatologist at Tampa General Hospital.
These findings illustrate how challenging lifestyle interventions can be for patients with NAFLD and NASH, and have clinical implications for identifying those individuals who are more or less likely to respond to weight-loss intervention as an initial treatment, Dr. Malespin said.
“I think we as providers need to better identify what are the barriers for weight loss for these patients, and try to use data, such as this data from TARGET-NASH, to help our clinical judgment and to learn strategies to be able to help these patients optimize their weight loss,” he added in an interview.
With no approved pharmacologic treatments for NAFLD and NASH, weight reduction remains one of the mainstays of treatment, Dr. Malespin said.
However, there are few studies evaluating structured programs designed to achieve such weight loss, and the long-term sustainability of this approach remains in question, according to Dr. Malespin, who presented findings of TARGET-NASH at the annual meeting of the American Association for the Study of Liver Diseases.
To date, more than 4,500 patients have been enrolled in TARGET-NASH at 59 U.S. centers. The present analysis, which Dr. Malespin described in an oral abstract presentation, included 2,037 patients with a body mass index of at least 25 kg/m2 and no decompensated cirrhosis, bariatric surgery, weight-loss medication use, or cancer diagnoses.
In sum, 34% of patients had 5% or more weight loss over about 3 years of follow-up, while about 10% had a 10% or greater weight loss, Dr. Malespin reported.
It took patients about 18 months, on average, to achieve 5% or greater weight loss. Those who achieved weight loss tended to be older, according to the investigator, and were about 1.5 times as likely to be obese class II or III, as opposed to just being overweight.
Patients were significantly more likely to lose weight, Dr. Malespin and coauthors found, if they had cirrhosis or comorbid medical conditions such as diabetes, osteoarthritis, or cardiovascular disease.
“All these factors essentially imply that these patients are hopefully being seen by multiple providers,” Dr. Malespin said, “so hopefully they’re getting the message [on the need for lifestyle modifications] from multiple different providers, and this is contributing to why we’re seeing some of this weight loss.”
However, weight loss did not persist in all patients, with 20.2% regaining weight back up to baseline or greater, according to the report. The median time to weight regain was 24-31 months, depending on initial weight class.
TARGET-NASH, sponsored by TARGET PharmaSolutions, is a 5-year longitudinal observational study with an enrollment goal of 15,000 patients with NAFLD or NASH and an estimated study completion date of 2021, according to a ClinicalTrials.gov listing.
Dr. Malespin reported disclosures with Gilead (speaker board), Intercept (speaker board, advisory committee), and TARGET PharmaSolutions (research grant paid to institution).
SOURCE: Malespin MH et al. The Liver Meeting 2019, Abstract 240.
BOSTON – While about one-third of overweight patients with nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH) slimmed down after a weight-loss intervention, about 20% gained weight back within the next few years, according to results from a large, long-term observational cohort study.
Effectively, that meant about one-quarter of overweight subjects enrolled in the TARGET-NASH study were able to lose weight and keep it off, said investigator Miguel H. Malespin, MD, a transplant hepatologist at Tampa General Hospital.
These findings illustrate how challenging lifestyle interventions can be for patients with NAFLD and NASH, and have clinical implications for identifying those individuals who are more or less likely to respond to weight-loss intervention as an initial treatment, Dr. Malespin said.
“I think we as providers need to better identify what are the barriers for weight loss for these patients, and try to use data, such as this data from TARGET-NASH, to help our clinical judgment and to learn strategies to be able to help these patients optimize their weight loss,” he added in an interview.
With no approved pharmacologic treatments for NAFLD and NASH, weight reduction remains one of the mainstays of treatment, Dr. Malespin said.
However, there are few studies evaluating structured programs designed to achieve such weight loss, and the long-term sustainability of this approach remains in question, according to Dr. Malespin, who presented findings of TARGET-NASH at the annual meeting of the American Association for the Study of Liver Diseases.
To date, more than 4,500 patients have been enrolled in TARGET-NASH at 59 U.S. centers. The present analysis, which Dr. Malespin described in an oral abstract presentation, included 2,037 patients with a body mass index of at least 25 kg/m2 and no decompensated cirrhosis, bariatric surgery, weight-loss medication use, or cancer diagnoses.
In sum, 34% of patients had 5% or more weight loss over about 3 years of follow-up, while about 10% had a 10% or greater weight loss, Dr. Malespin reported.
It took patients about 18 months, on average, to achieve 5% or greater weight loss. Those who achieved weight loss tended to be older, according to the investigator, and were about 1.5 times as likely to be obese class II or III, as opposed to just being overweight.
Patients were significantly more likely to lose weight, Dr. Malespin and coauthors found, if they had cirrhosis or comorbid medical conditions such as diabetes, osteoarthritis, or cardiovascular disease.
“All these factors essentially imply that these patients are hopefully being seen by multiple providers,” Dr. Malespin said, “so hopefully they’re getting the message [on the need for lifestyle modifications] from multiple different providers, and this is contributing to why we’re seeing some of this weight loss.”
However, weight loss did not persist in all patients, with 20.2% regaining weight back up to baseline or greater, according to the report. The median time to weight regain was 24-31 months, depending on initial weight class.
TARGET-NASH, sponsored by TARGET PharmaSolutions, is a 5-year longitudinal observational study with an enrollment goal of 15,000 patients with NAFLD or NASH and an estimated study completion date of 2021, according to a ClinicalTrials.gov listing.
Dr. Malespin reported disclosures with Gilead (speaker board), Intercept (speaker board, advisory committee), and TARGET PharmaSolutions (research grant paid to institution).
SOURCE: Malespin MH et al. The Liver Meeting 2019, Abstract 240.
BOSTON – While about one-third of overweight patients with nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH) slimmed down after a weight-loss intervention, about 20% gained weight back within the next few years, according to results from a large, long-term observational cohort study.
Effectively, that meant about one-quarter of overweight subjects enrolled in the TARGET-NASH study were able to lose weight and keep it off, said investigator Miguel H. Malespin, MD, a transplant hepatologist at Tampa General Hospital.
These findings illustrate how challenging lifestyle interventions can be for patients with NAFLD and NASH, and have clinical implications for identifying those individuals who are more or less likely to respond to weight-loss intervention as an initial treatment, Dr. Malespin said.
“I think we as providers need to better identify what are the barriers for weight loss for these patients, and try to use data, such as this data from TARGET-NASH, to help our clinical judgment and to learn strategies to be able to help these patients optimize their weight loss,” he added in an interview.
With no approved pharmacologic treatments for NAFLD and NASH, weight reduction remains one of the mainstays of treatment, Dr. Malespin said.
However, there are few studies evaluating structured programs designed to achieve such weight loss, and the long-term sustainability of this approach remains in question, according to Dr. Malespin, who presented findings of TARGET-NASH at the annual meeting of the American Association for the Study of Liver Diseases.
To date, more than 4,500 patients have been enrolled in TARGET-NASH at 59 U.S. centers. The present analysis, which Dr. Malespin described in an oral abstract presentation, included 2,037 patients with a body mass index of at least 25 kg/m2 and no decompensated cirrhosis, bariatric surgery, weight-loss medication use, or cancer diagnoses.
In sum, 34% of patients had 5% or more weight loss over about 3 years of follow-up, while about 10% had a 10% or greater weight loss, Dr. Malespin reported.
It took patients about 18 months, on average, to achieve 5% or greater weight loss. Those who achieved weight loss tended to be older, according to the investigator, and were about 1.5 times as likely to be obese class II or III, as opposed to just being overweight.
Patients were significantly more likely to lose weight, Dr. Malespin and coauthors found, if they had cirrhosis or comorbid medical conditions such as diabetes, osteoarthritis, or cardiovascular disease.
“All these factors essentially imply that these patients are hopefully being seen by multiple providers,” Dr. Malespin said, “so hopefully they’re getting the message [on the need for lifestyle modifications] from multiple different providers, and this is contributing to why we’re seeing some of this weight loss.”
However, weight loss did not persist in all patients, with 20.2% regaining weight back up to baseline or greater, according to the report. The median time to weight regain was 24-31 months, depending on initial weight class.
TARGET-NASH, sponsored by TARGET PharmaSolutions, is a 5-year longitudinal observational study with an enrollment goal of 15,000 patients with NAFLD or NASH and an estimated study completion date of 2021, according to a ClinicalTrials.gov listing.
Dr. Malespin reported disclosures with Gilead (speaker board), Intercept (speaker board, advisory committee), and TARGET PharmaSolutions (research grant paid to institution).
SOURCE: Malespin MH et al. The Liver Meeting 2019, Abstract 240.
REPORTING FROM THE LIVER MEETING 2019
Time to Disability Milestones in MS Is Lengthening
Key clinical point: Disability milestones are not being met as quickly for patients with multiple sclerosis (MS), as disease progression has slowed.
Major finding: In patients with relapsing-onset MS there was a decreased risk of reaching disability milestones by 3%, 6%, and 7% in Expanded Disability Status Score (EDSS) scores of 3.0, 4.0, and 6.0, respectively. In patients with progressive-onset MS there was no significant decrease.
Study details: This was a nationwide population-based retrospective cohort study of 12,512 patients in Sweden. Of those patients 7,331 (5,196 females) were diagnosed with MS at a mean age of 38.3 years between January 1995 and December 2010 and had 2 recorded EDSS scores.
Disclosures: Research was funded by the Swedish Research Council and the Swedish Brain Foundation.
Citation: Beiki O, et al. JAMA Neurol. 2019;76(6):665-671. doi: 10.1001/jamaneurol.2019.0330.
Key clinical point: Disability milestones are not being met as quickly for patients with multiple sclerosis (MS), as disease progression has slowed.
Major finding: In patients with relapsing-onset MS there was a decreased risk of reaching disability milestones by 3%, 6%, and 7% in Expanded Disability Status Score (EDSS) scores of 3.0, 4.0, and 6.0, respectively. In patients with progressive-onset MS there was no significant decrease.
Study details: This was a nationwide population-based retrospective cohort study of 12,512 patients in Sweden. Of those patients 7,331 (5,196 females) were diagnosed with MS at a mean age of 38.3 years between January 1995 and December 2010 and had 2 recorded EDSS scores.
Disclosures: Research was funded by the Swedish Research Council and the Swedish Brain Foundation.
Citation: Beiki O, et al. JAMA Neurol. 2019;76(6):665-671. doi: 10.1001/jamaneurol.2019.0330.
Key clinical point: Disability milestones are not being met as quickly for patients with multiple sclerosis (MS), as disease progression has slowed.
Major finding: In patients with relapsing-onset MS there was a decreased risk of reaching disability milestones by 3%, 6%, and 7% in Expanded Disability Status Score (EDSS) scores of 3.0, 4.0, and 6.0, respectively. In patients with progressive-onset MS there was no significant decrease.
Study details: This was a nationwide population-based retrospective cohort study of 12,512 patients in Sweden. Of those patients 7,331 (5,196 females) were diagnosed with MS at a mean age of 38.3 years between January 1995 and December 2010 and had 2 recorded EDSS scores.
Disclosures: Research was funded by the Swedish Research Council and the Swedish Brain Foundation.
Citation: Beiki O, et al. JAMA Neurol. 2019;76(6):665-671. doi: 10.1001/jamaneurol.2019.0330.
From DMTs to Secondary Progressive MS
Key clinical point: The use, type, and timing of disease-modifying therapies (DMTs) in relapsing-remitting multiple sclerosis (RRMS) patients may lead to secondary progressive MS.
Major finding: RRMS patients beginning DMTs of fingolimod, alemtuzumab, or natalizumab had a lower chance of secondary progressive MS, whereas patients taking glatiramer acetate or interferon beta had a higher chance.
Study details: This was a cohort study that examined 1,555 RRMS patients (1,123 females) across 21 countries that began DMTs (interferon beta, glatiramer acetate, fingolimod, natalizumab, or alemtuzumab) between 1988 and 2012.
Disclosures: This study was financially supported by the National Health and Medical Research Council of Australia, the University of Melbourne, a Next Generation Fellowship funded by the Grand Charity of the Freemason’s, and the MSBase 2017 Fellowship. Alemtuzumab studies done in Cambridge were supported by the NIHR Cambridge Biomedical Research Centre and the MS Society UK.
Citation: Brown JWL, et al. JAMA. 2019;321(2):175-187. doi: 10.1001/jama.2018.20588.
Key clinical point: The use, type, and timing of disease-modifying therapies (DMTs) in relapsing-remitting multiple sclerosis (RRMS) patients may lead to secondary progressive MS.
Major finding: RRMS patients beginning DMTs of fingolimod, alemtuzumab, or natalizumab had a lower chance of secondary progressive MS, whereas patients taking glatiramer acetate or interferon beta had a higher chance.
Study details: This was a cohort study that examined 1,555 RRMS patients (1,123 females) across 21 countries that began DMTs (interferon beta, glatiramer acetate, fingolimod, natalizumab, or alemtuzumab) between 1988 and 2012.
Disclosures: This study was financially supported by the National Health and Medical Research Council of Australia, the University of Melbourne, a Next Generation Fellowship funded by the Grand Charity of the Freemason’s, and the MSBase 2017 Fellowship. Alemtuzumab studies done in Cambridge were supported by the NIHR Cambridge Biomedical Research Centre and the MS Society UK.
Citation: Brown JWL, et al. JAMA. 2019;321(2):175-187. doi: 10.1001/jama.2018.20588.
Key clinical point: The use, type, and timing of disease-modifying therapies (DMTs) in relapsing-remitting multiple sclerosis (RRMS) patients may lead to secondary progressive MS.
Major finding: RRMS patients beginning DMTs of fingolimod, alemtuzumab, or natalizumab had a lower chance of secondary progressive MS, whereas patients taking glatiramer acetate or interferon beta had a higher chance.
Study details: This was a cohort study that examined 1,555 RRMS patients (1,123 females) across 21 countries that began DMTs (interferon beta, glatiramer acetate, fingolimod, natalizumab, or alemtuzumab) between 1988 and 2012.
Disclosures: This study was financially supported by the National Health and Medical Research Council of Australia, the University of Melbourne, a Next Generation Fellowship funded by the Grand Charity of the Freemason’s, and the MSBase 2017 Fellowship. Alemtuzumab studies done in Cambridge were supported by the NIHR Cambridge Biomedical Research Centre and the MS Society UK.
Citation: Brown JWL, et al. JAMA. 2019;321(2):175-187. doi: 10.1001/jama.2018.20588.