User login
In high-risk first relapse ALL, blinatumomab seen superior to consolidation chemo
Blinatumomab was superior to high-risk consolidation (HC) 3 chemotherapy in a phase 3 clinical trial among children with high-risk first-relapse acute lymphoblastic leukemia (ALL), according to Franco Locatelli, MD, PhD, Ospedale Pediatrico Bambino Gesú and Sapienza, Rome.
Blinatumomab constitutes a new standard of care because of superior event-free survival (EFS) and other comparative benefits, including fewer and less severe toxicities, he said in a presentation at theannual meeting of the American Society of Hematology, which was held virtually.
About 15% of children with B-cell precursor (BCP) ALL relapse after standard treatment. Prognosis depends largely on time from diagnosis to relapse and the site of relapse. After relapse, when a second morphological complete remission (M1 marrow) is achieved, most are candidates for allogeneic hematopoietic stem cell transplant (alloHSCT), Dr. Locatelli noted. Immuno-oncotherapy with blinatumomab, a bispecific T-cell–engager molecule, has been shown to be efficacious in children with relapsed/refractory BCP-ALL.
In the open-label, controlled trial, investigators randomized children with M1 (<5% blasts) or M2 (<25% and 5% or greater blasts) marrow 1:1 after induction therapy and cycles of HC1 and HC2 chemotherapy to a third consolidation course with blinatumomab (15 µg/m2/day for 4 weeks) or HC3 (dexamethasone, vincristine, daunorubicin, methotrexate, ifosfamide, PEG-asparaginase); intrathecal chemotherapy (methotrexate/cytarabine/prednisolone) was administered before treatment. Patients achieving a second complete morphological remission (M1 marrow) after blinatumomab or HC3 proceeded to alloHSCT. EFS was the primary endpoint (from randomization until relapse date or M2 marrow after a complete response [CR], failure to achieve CR at end of treatment, second malignancy, or death from any cause).
Investigators had enrolled 108 (54 received HC3; 54 received blinatumomab) out of a target of about 202 patients when the data-monitoring committee recommended termination because of blinatumomab benefit observed at the first interim analysis. Median age was around 5.5 years (1-17), with the mean time from first diagnosis to relapse at approximately 22 months.
Dr. Locatelli reported events for 18/54 (33.3%) in the blinatumomab arm and 31/54 (57.4%) in the HC3 arm, with a median EFS of “not reached” and 7.4 months, respectively. The risk of relapse with blinatumomab was reduced by 64% versus HC3 (hazard ratio, 0.36; 95% confidence interval, 0.19-0.66, P < .001). Overall survival (OS) favored blinatumomab over HC3, as well, with a hazard ratio of 0.43 (95% CI, 0.18-1.01). Minimal residual disease (MRD) remission (MRD < 10-4) was seen in 43/46 (93.5%) blinatumomab-randomized and 25/46 (54.3%) HC3-randomized patients.
Relapses occurred more often in the HC3 group (blinatumomab 13, 24%; HC3 29, 54%) overall, and at each of the assessments at 6 months, 12 months, and 24 months. Also, MRD remissions by PCR (polymerase chain reaction) were superior in the blinatumomab arm overall (90% versus 54%) and according to baseline MRD status with strikingly divergent rates in those with MRD greater than or equal to 104 at baseline (93% blinatumomab/24% HC3). Rates were relatively similar in patients with MRD less than 104 at baseline (85% blinatumomab/87% HC3).
Grade 3 or greater treatment-emergent adverse events were reported by 30/53 (57%) and 41/51 (80%) patients in the blinatumomab and HC3 groups, respectively, with several markedly lower in the blinatumomab group (neutropenia/neutrophil count decrease 17 versus 31; anemia 15 versus 41; febrile neutropenia 4 versus 26). As expected, grade 3 or greater neurologic events occurred more frequently with blinatumomab than with HC3 (48% versus 29%); no grade 3 or greater cytokine release syndrome events were reported.
Tallying the blinatumomab benefits (superior EFS and MRD negativity prior to alloHSCT, improved OS, fewer relapses, fewer and less severe toxicities), Dr. Locatelli concluded, “Blinatumomab constitutes a new standard of care in children with high-risk first-relapse ALL.”
In the postpresentation discussion, Dr. Locatelli underscored the blinatumomab benefit versus a third course of chemotherapy: “Monotherapy with blinatumomab was able to present a higher proportion of patients in CR2 who could proceed to transplant.”
Dr. Locatelli disclosed relationships with multiple companies.
SOURCE: Locatelli F et al. ASH 2020, Abstract 268.
Blinatumomab was superior to high-risk consolidation (HC) 3 chemotherapy in a phase 3 clinical trial among children with high-risk first-relapse acute lymphoblastic leukemia (ALL), according to Franco Locatelli, MD, PhD, Ospedale Pediatrico Bambino Gesú and Sapienza, Rome.
Blinatumomab constitutes a new standard of care because of superior event-free survival (EFS) and other comparative benefits, including fewer and less severe toxicities, he said in a presentation at theannual meeting of the American Society of Hematology, which was held virtually.
About 15% of children with B-cell precursor (BCP) ALL relapse after standard treatment. Prognosis depends largely on time from diagnosis to relapse and the site of relapse. After relapse, when a second morphological complete remission (M1 marrow) is achieved, most are candidates for allogeneic hematopoietic stem cell transplant (alloHSCT), Dr. Locatelli noted. Immuno-oncotherapy with blinatumomab, a bispecific T-cell–engager molecule, has been shown to be efficacious in children with relapsed/refractory BCP-ALL.
In the open-label, controlled trial, investigators randomized children with M1 (<5% blasts) or M2 (<25% and 5% or greater blasts) marrow 1:1 after induction therapy and cycles of HC1 and HC2 chemotherapy to a third consolidation course with blinatumomab (15 µg/m2/day for 4 weeks) or HC3 (dexamethasone, vincristine, daunorubicin, methotrexate, ifosfamide, PEG-asparaginase); intrathecal chemotherapy (methotrexate/cytarabine/prednisolone) was administered before treatment. Patients achieving a second complete morphological remission (M1 marrow) after blinatumomab or HC3 proceeded to alloHSCT. EFS was the primary endpoint (from randomization until relapse date or M2 marrow after a complete response [CR], failure to achieve CR at end of treatment, second malignancy, or death from any cause).
Investigators had enrolled 108 (54 received HC3; 54 received blinatumomab) out of a target of about 202 patients when the data-monitoring committee recommended termination because of blinatumomab benefit observed at the first interim analysis. Median age was around 5.5 years (1-17), with the mean time from first diagnosis to relapse at approximately 22 months.
Dr. Locatelli reported events for 18/54 (33.3%) in the blinatumomab arm and 31/54 (57.4%) in the HC3 arm, with a median EFS of “not reached” and 7.4 months, respectively. The risk of relapse with blinatumomab was reduced by 64% versus HC3 (hazard ratio, 0.36; 95% confidence interval, 0.19-0.66, P < .001). Overall survival (OS) favored blinatumomab over HC3, as well, with a hazard ratio of 0.43 (95% CI, 0.18-1.01). Minimal residual disease (MRD) remission (MRD < 10-4) was seen in 43/46 (93.5%) blinatumomab-randomized and 25/46 (54.3%) HC3-randomized patients.
Relapses occurred more often in the HC3 group (blinatumomab 13, 24%; HC3 29, 54%) overall, and at each of the assessments at 6 months, 12 months, and 24 months. Also, MRD remissions by PCR (polymerase chain reaction) were superior in the blinatumomab arm overall (90% versus 54%) and according to baseline MRD status with strikingly divergent rates in those with MRD greater than or equal to 104 at baseline (93% blinatumomab/24% HC3). Rates were relatively similar in patients with MRD less than 104 at baseline (85% blinatumomab/87% HC3).
Grade 3 or greater treatment-emergent adverse events were reported by 30/53 (57%) and 41/51 (80%) patients in the blinatumomab and HC3 groups, respectively, with several markedly lower in the blinatumomab group (neutropenia/neutrophil count decrease 17 versus 31; anemia 15 versus 41; febrile neutropenia 4 versus 26). As expected, grade 3 or greater neurologic events occurred more frequently with blinatumomab than with HC3 (48% versus 29%); no grade 3 or greater cytokine release syndrome events were reported.
Tallying the blinatumomab benefits (superior EFS and MRD negativity prior to alloHSCT, improved OS, fewer relapses, fewer and less severe toxicities), Dr. Locatelli concluded, “Blinatumomab constitutes a new standard of care in children with high-risk first-relapse ALL.”
In the postpresentation discussion, Dr. Locatelli underscored the blinatumomab benefit versus a third course of chemotherapy: “Monotherapy with blinatumomab was able to present a higher proportion of patients in CR2 who could proceed to transplant.”
Dr. Locatelli disclosed relationships with multiple companies.
SOURCE: Locatelli F et al. ASH 2020, Abstract 268.
Blinatumomab was superior to high-risk consolidation (HC) 3 chemotherapy in a phase 3 clinical trial among children with high-risk first-relapse acute lymphoblastic leukemia (ALL), according to Franco Locatelli, MD, PhD, Ospedale Pediatrico Bambino Gesú and Sapienza, Rome.
Blinatumomab constitutes a new standard of care because of superior event-free survival (EFS) and other comparative benefits, including fewer and less severe toxicities, he said in a presentation at theannual meeting of the American Society of Hematology, which was held virtually.
About 15% of children with B-cell precursor (BCP) ALL relapse after standard treatment. Prognosis depends largely on time from diagnosis to relapse and the site of relapse. After relapse, when a second morphological complete remission (M1 marrow) is achieved, most are candidates for allogeneic hematopoietic stem cell transplant (alloHSCT), Dr. Locatelli noted. Immuno-oncotherapy with blinatumomab, a bispecific T-cell–engager molecule, has been shown to be efficacious in children with relapsed/refractory BCP-ALL.
In the open-label, controlled trial, investigators randomized children with M1 (<5% blasts) or M2 (<25% and 5% or greater blasts) marrow 1:1 after induction therapy and cycles of HC1 and HC2 chemotherapy to a third consolidation course with blinatumomab (15 µg/m2/day for 4 weeks) or HC3 (dexamethasone, vincristine, daunorubicin, methotrexate, ifosfamide, PEG-asparaginase); intrathecal chemotherapy (methotrexate/cytarabine/prednisolone) was administered before treatment. Patients achieving a second complete morphological remission (M1 marrow) after blinatumomab or HC3 proceeded to alloHSCT. EFS was the primary endpoint (from randomization until relapse date or M2 marrow after a complete response [CR], failure to achieve CR at end of treatment, second malignancy, or death from any cause).
Investigators had enrolled 108 (54 received HC3; 54 received blinatumomab) out of a target of about 202 patients when the data-monitoring committee recommended termination because of blinatumomab benefit observed at the first interim analysis. Median age was around 5.5 years (1-17), with the mean time from first diagnosis to relapse at approximately 22 months.
Dr. Locatelli reported events for 18/54 (33.3%) in the blinatumomab arm and 31/54 (57.4%) in the HC3 arm, with a median EFS of “not reached” and 7.4 months, respectively. The risk of relapse with blinatumomab was reduced by 64% versus HC3 (hazard ratio, 0.36; 95% confidence interval, 0.19-0.66, P < .001). Overall survival (OS) favored blinatumomab over HC3, as well, with a hazard ratio of 0.43 (95% CI, 0.18-1.01). Minimal residual disease (MRD) remission (MRD < 10-4) was seen in 43/46 (93.5%) blinatumomab-randomized and 25/46 (54.3%) HC3-randomized patients.
Relapses occurred more often in the HC3 group (blinatumomab 13, 24%; HC3 29, 54%) overall, and at each of the assessments at 6 months, 12 months, and 24 months. Also, MRD remissions by PCR (polymerase chain reaction) were superior in the blinatumomab arm overall (90% versus 54%) and according to baseline MRD status with strikingly divergent rates in those with MRD greater than or equal to 104 at baseline (93% blinatumomab/24% HC3). Rates were relatively similar in patients with MRD less than 104 at baseline (85% blinatumomab/87% HC3).
Grade 3 or greater treatment-emergent adverse events were reported by 30/53 (57%) and 41/51 (80%) patients in the blinatumomab and HC3 groups, respectively, with several markedly lower in the blinatumomab group (neutropenia/neutrophil count decrease 17 versus 31; anemia 15 versus 41; febrile neutropenia 4 versus 26). As expected, grade 3 or greater neurologic events occurred more frequently with blinatumomab than with HC3 (48% versus 29%); no grade 3 or greater cytokine release syndrome events were reported.
Tallying the blinatumomab benefits (superior EFS and MRD negativity prior to alloHSCT, improved OS, fewer relapses, fewer and less severe toxicities), Dr. Locatelli concluded, “Blinatumomab constitutes a new standard of care in children with high-risk first-relapse ALL.”
In the postpresentation discussion, Dr. Locatelli underscored the blinatumomab benefit versus a third course of chemotherapy: “Monotherapy with blinatumomab was able to present a higher proportion of patients in CR2 who could proceed to transplant.”
Dr. Locatelli disclosed relationships with multiple companies.
SOURCE: Locatelli F et al. ASH 2020, Abstract 268.
FROM ASH 2020
IBD patients more likely to stick with vedolizumab than anti-TNF drugs
Adults with inflammatory bowel disease were more likely to continue using vedolizumab, compared with anti–tumor necrosis factor (TNF) drugs over 3 years, based on data from a retrospective study of nearly 16,000 patients.
Patient persistence with prescribed therapy is essential to managing chronic inflammatory bowel disease (IBD), but data on the persistence of patients with treatments are limited, wrote Ulf Helwig, MD, of the Practice for Internal Medicine, Oldenburg, Germany, and colleagues. “With the advent of vedolizumab, physicians for the first time had the choice between biologicals with different modes of action,” they wrote.
In a study published in the Journal of Clinical Gastroenterology, the researchers used a national prescription database to identify 15,984 adults aged 18 years and older who were treatment-naive to biologics and received prescriptions between July 2014 and March 2017. Treatment persistence was defined as continuous treatment time of at least 90 days without prescription.
A total of 2,076 vedolizumab patients were matched with 2,076 adalimumab patients; 716 vedolizumab patients were matched with 716 golimumab patients; and 2,055 vedolizumab patients were matched with 2,055 infliximab patients.
Within 3 years after the first prescription, the overall persistence rates were 35.9% for vedolizumab, 27.8% for adalimumab, 20.7% for golimumab, and 29.8% for infliximab.
In matched-pair analysis, 35.2% of vedolizumab patients were persistent, compared with 28.9% of adalimumab patients over a 3-year period; the difference was statistically significant. In addition, 30.5% of vedolizumab patients persisted, compared with 25.4% of golimumab patients, also statistically significant. A matched-pair comparison between vedolizumab and infliximab (35.7% vs. 30.2%) was not statistically significant (P = 0.119).
In addition, vedolizumab patients were significantly less likely to discontinue therapy, compared with both adalimumab and golimumab patients, with hazard ratios of 0.86 and 0.60, respectively, in the matched pair analysis; discontinuation, compared with infliximab, was not statistically significant.
“Several reasons may account for significant rates of discontinuation reported for all biological treatments in IBD,” the researchers noted. “These comprise differences in health care systems in the concerned countries, including differences in availability of biologicals, access to reimbursed drugs, or different patient care settings,” they wrote.
The study findings were limited by several factors including the lack of data on specific IBD diagnoses, IBD severity, disease course, and dose escalation, they noted.
However, the study was strengthened by the large sample size and use of a real-world setting, they said.
“Further studies are needed to identify the reasons for persistence differences between vedolizumab and anti-TNF drugs,” they concluded.
Comparisons inform choices
“There are multiple biologic options for therapy of inflammatory bowel disease, and response to therapy tends to drop off over time in many patients for a variety of reasons including development of antibodies and escape from the mechanism of the action of the drug,” said Kim L. Isaacs, MD, of the University of North Carolina at Chapel Hill, in an interview.
“Intolerance or side effects of medication also may lead to discontinuation of therapy,” said Dr. Isaacs. “This trial looks at therapy discontinuation among four biologics used for inflammatory bowel disease over a 3-year period after initiation of therapy in patients who were previous biologically naive. Reasons for discontinuation cannot be assessed with this data set,” she noted. “There are very few comparative trials with the different biologic therapies in IBD. This trial is important because it compares the two distinct biologic mechanisms of action and continuation of therapy in biologically naive patients,” she said.
Dr. Isaacs said she was not surprised by the study findings. “Discontinuation of anti-TNF therapy was more common, compared to vedolizumab and golimumab. There was no statistical difference in terms of therapy discontinuation with infliximab,” she said. “In general, vedolizumab is felt to be less systemically immunosuppressant with targeting of white blood cell trafficking to the gut, whereas anti-TNF therapy is more systemically immunosuppressant and may be associated with more systemic side effects,” she explained.
The study design does not allow for comment on comparative efficacy, “although the findings are intriguing,” said Dr. Isaacs. “If the discontinuations were caused by lack of efficacy, the findings in this study may help in positioning biologic therapy in the biologic-naive patients,” she said.
The study is “a ‘real-world’ experiment that suggests there is a difference between different biologic therapies for inflammatory bowel disease,” said Dr. Isaacs. “More controlled comparative efficacy trials are needed that can look at reasons for drug discontinuation between different populations. To date, the VARSITY trial comparing vedolizumab to adalimumab in ulcerative colitis is the only published trial to do this,” she added.
The study received no outside funding. Lead author Dr. Helwig disclosed lecture and consulting fees from AbbVie, Amgen, Biogen, Celltrion, Hexal, MSD, Ferring, Falk Foundation, Takeda, Mundipharma, Pfizer, Hospira, and Vifor Pharma. Dr. Isaacs disclosed serving on the Data and Safety Monitoring Board (DSMB) for Janssen.
Help your patients better understand their IBD treatment options by sharing AGA’s patient education, “Living with IBD,” in the AGA GI Patient Center at www.gastro.org/IBD.
SOURCE: Helwig U et al. J Clin Gastroenterol. 2021 Jan. doi: 10.1097/MCG.0000000000001323
Story updated Jan. 5, 2021.
Adults with inflammatory bowel disease were more likely to continue using vedolizumab, compared with anti–tumor necrosis factor (TNF) drugs over 3 years, based on data from a retrospective study of nearly 16,000 patients.
Patient persistence with prescribed therapy is essential to managing chronic inflammatory bowel disease (IBD), but data on the persistence of patients with treatments are limited, wrote Ulf Helwig, MD, of the Practice for Internal Medicine, Oldenburg, Germany, and colleagues. “With the advent of vedolizumab, physicians for the first time had the choice between biologicals with different modes of action,” they wrote.
In a study published in the Journal of Clinical Gastroenterology, the researchers used a national prescription database to identify 15,984 adults aged 18 years and older who were treatment-naive to biologics and received prescriptions between July 2014 and March 2017. Treatment persistence was defined as continuous treatment time of at least 90 days without prescription.
A total of 2,076 vedolizumab patients were matched with 2,076 adalimumab patients; 716 vedolizumab patients were matched with 716 golimumab patients; and 2,055 vedolizumab patients were matched with 2,055 infliximab patients.
Within 3 years after the first prescription, the overall persistence rates were 35.9% for vedolizumab, 27.8% for adalimumab, 20.7% for golimumab, and 29.8% for infliximab.
In matched-pair analysis, 35.2% of vedolizumab patients were persistent, compared with 28.9% of adalimumab patients over a 3-year period; the difference was statistically significant. In addition, 30.5% of vedolizumab patients persisted, compared with 25.4% of golimumab patients, also statistically significant. A matched-pair comparison between vedolizumab and infliximab (35.7% vs. 30.2%) was not statistically significant (P = 0.119).
In addition, vedolizumab patients were significantly less likely to discontinue therapy, compared with both adalimumab and golimumab patients, with hazard ratios of 0.86 and 0.60, respectively, in the matched pair analysis; discontinuation, compared with infliximab, was not statistically significant.
“Several reasons may account for significant rates of discontinuation reported for all biological treatments in IBD,” the researchers noted. “These comprise differences in health care systems in the concerned countries, including differences in availability of biologicals, access to reimbursed drugs, or different patient care settings,” they wrote.
The study findings were limited by several factors including the lack of data on specific IBD diagnoses, IBD severity, disease course, and dose escalation, they noted.
However, the study was strengthened by the large sample size and use of a real-world setting, they said.
“Further studies are needed to identify the reasons for persistence differences between vedolizumab and anti-TNF drugs,” they concluded.
Comparisons inform choices
“There are multiple biologic options for therapy of inflammatory bowel disease, and response to therapy tends to drop off over time in many patients for a variety of reasons including development of antibodies and escape from the mechanism of the action of the drug,” said Kim L. Isaacs, MD, of the University of North Carolina at Chapel Hill, in an interview.
“Intolerance or side effects of medication also may lead to discontinuation of therapy,” said Dr. Isaacs. “This trial looks at therapy discontinuation among four biologics used for inflammatory bowel disease over a 3-year period after initiation of therapy in patients who were previous biologically naive. Reasons for discontinuation cannot be assessed with this data set,” she noted. “There are very few comparative trials with the different biologic therapies in IBD. This trial is important because it compares the two distinct biologic mechanisms of action and continuation of therapy in biologically naive patients,” she said.
Dr. Isaacs said she was not surprised by the study findings. “Discontinuation of anti-TNF therapy was more common, compared to vedolizumab and golimumab. There was no statistical difference in terms of therapy discontinuation with infliximab,” she said. “In general, vedolizumab is felt to be less systemically immunosuppressant with targeting of white blood cell trafficking to the gut, whereas anti-TNF therapy is more systemically immunosuppressant and may be associated with more systemic side effects,” she explained.
The study design does not allow for comment on comparative efficacy, “although the findings are intriguing,” said Dr. Isaacs. “If the discontinuations were caused by lack of efficacy, the findings in this study may help in positioning biologic therapy in the biologic-naive patients,” she said.
The study is “a ‘real-world’ experiment that suggests there is a difference between different biologic therapies for inflammatory bowel disease,” said Dr. Isaacs. “More controlled comparative efficacy trials are needed that can look at reasons for drug discontinuation between different populations. To date, the VARSITY trial comparing vedolizumab to adalimumab in ulcerative colitis is the only published trial to do this,” she added.
The study received no outside funding. Lead author Dr. Helwig disclosed lecture and consulting fees from AbbVie, Amgen, Biogen, Celltrion, Hexal, MSD, Ferring, Falk Foundation, Takeda, Mundipharma, Pfizer, Hospira, and Vifor Pharma. Dr. Isaacs disclosed serving on the Data and Safety Monitoring Board (DSMB) for Janssen.
Help your patients better understand their IBD treatment options by sharing AGA’s patient education, “Living with IBD,” in the AGA GI Patient Center at www.gastro.org/IBD.
SOURCE: Helwig U et al. J Clin Gastroenterol. 2021 Jan. doi: 10.1097/MCG.0000000000001323
Story updated Jan. 5, 2021.
Adults with inflammatory bowel disease were more likely to continue using vedolizumab, compared with anti–tumor necrosis factor (TNF) drugs over 3 years, based on data from a retrospective study of nearly 16,000 patients.
Patient persistence with prescribed therapy is essential to managing chronic inflammatory bowel disease (IBD), but data on the persistence of patients with treatments are limited, wrote Ulf Helwig, MD, of the Practice for Internal Medicine, Oldenburg, Germany, and colleagues. “With the advent of vedolizumab, physicians for the first time had the choice between biologicals with different modes of action,” they wrote.
In a study published in the Journal of Clinical Gastroenterology, the researchers used a national prescription database to identify 15,984 adults aged 18 years and older who were treatment-naive to biologics and received prescriptions between July 2014 and March 2017. Treatment persistence was defined as continuous treatment time of at least 90 days without prescription.
A total of 2,076 vedolizumab patients were matched with 2,076 adalimumab patients; 716 vedolizumab patients were matched with 716 golimumab patients; and 2,055 vedolizumab patients were matched with 2,055 infliximab patients.
Within 3 years after the first prescription, the overall persistence rates were 35.9% for vedolizumab, 27.8% for adalimumab, 20.7% for golimumab, and 29.8% for infliximab.
In matched-pair analysis, 35.2% of vedolizumab patients were persistent, compared with 28.9% of adalimumab patients over a 3-year period; the difference was statistically significant. In addition, 30.5% of vedolizumab patients persisted, compared with 25.4% of golimumab patients, also statistically significant. A matched-pair comparison between vedolizumab and infliximab (35.7% vs. 30.2%) was not statistically significant (P = 0.119).
In addition, vedolizumab patients were significantly less likely to discontinue therapy, compared with both adalimumab and golimumab patients, with hazard ratios of 0.86 and 0.60, respectively, in the matched pair analysis; discontinuation, compared with infliximab, was not statistically significant.
“Several reasons may account for significant rates of discontinuation reported for all biological treatments in IBD,” the researchers noted. “These comprise differences in health care systems in the concerned countries, including differences in availability of biologicals, access to reimbursed drugs, or different patient care settings,” they wrote.
The study findings were limited by several factors including the lack of data on specific IBD diagnoses, IBD severity, disease course, and dose escalation, they noted.
However, the study was strengthened by the large sample size and use of a real-world setting, they said.
“Further studies are needed to identify the reasons for persistence differences between vedolizumab and anti-TNF drugs,” they concluded.
Comparisons inform choices
“There are multiple biologic options for therapy of inflammatory bowel disease, and response to therapy tends to drop off over time in many patients for a variety of reasons including development of antibodies and escape from the mechanism of the action of the drug,” said Kim L. Isaacs, MD, of the University of North Carolina at Chapel Hill, in an interview.
“Intolerance or side effects of medication also may lead to discontinuation of therapy,” said Dr. Isaacs. “This trial looks at therapy discontinuation among four biologics used for inflammatory bowel disease over a 3-year period after initiation of therapy in patients who were previous biologically naive. Reasons for discontinuation cannot be assessed with this data set,” she noted. “There are very few comparative trials with the different biologic therapies in IBD. This trial is important because it compares the two distinct biologic mechanisms of action and continuation of therapy in biologically naive patients,” she said.
Dr. Isaacs said she was not surprised by the study findings. “Discontinuation of anti-TNF therapy was more common, compared to vedolizumab and golimumab. There was no statistical difference in terms of therapy discontinuation with infliximab,” she said. “In general, vedolizumab is felt to be less systemically immunosuppressant with targeting of white blood cell trafficking to the gut, whereas anti-TNF therapy is more systemically immunosuppressant and may be associated with more systemic side effects,” she explained.
The study design does not allow for comment on comparative efficacy, “although the findings are intriguing,” said Dr. Isaacs. “If the discontinuations were caused by lack of efficacy, the findings in this study may help in positioning biologic therapy in the biologic-naive patients,” she said.
The study is “a ‘real-world’ experiment that suggests there is a difference between different biologic therapies for inflammatory bowel disease,” said Dr. Isaacs. “More controlled comparative efficacy trials are needed that can look at reasons for drug discontinuation between different populations. To date, the VARSITY trial comparing vedolizumab to adalimumab in ulcerative colitis is the only published trial to do this,” she added.
The study received no outside funding. Lead author Dr. Helwig disclosed lecture and consulting fees from AbbVie, Amgen, Biogen, Celltrion, Hexal, MSD, Ferring, Falk Foundation, Takeda, Mundipharma, Pfizer, Hospira, and Vifor Pharma. Dr. Isaacs disclosed serving on the Data and Safety Monitoring Board (DSMB) for Janssen.
Help your patients better understand their IBD treatment options by sharing AGA’s patient education, “Living with IBD,” in the AGA GI Patient Center at www.gastro.org/IBD.
SOURCE: Helwig U et al. J Clin Gastroenterol. 2021 Jan. doi: 10.1097/MCG.0000000000001323
Story updated Jan. 5, 2021.
FROM THE JOURNAL OF CLINICAL GASTROENTEROLOGY
COVID-19–induced drop in first measles vaccinations sparks resurgence concerns
Widespread use of the MMR vaccine is not only crucial for protecting the community against infectious outbreaks, but also serves as the overall pacesetter for preventive services, said Sara M. Bode, MD and colleagues at Nationwide Children’s Hospital in Columbus.
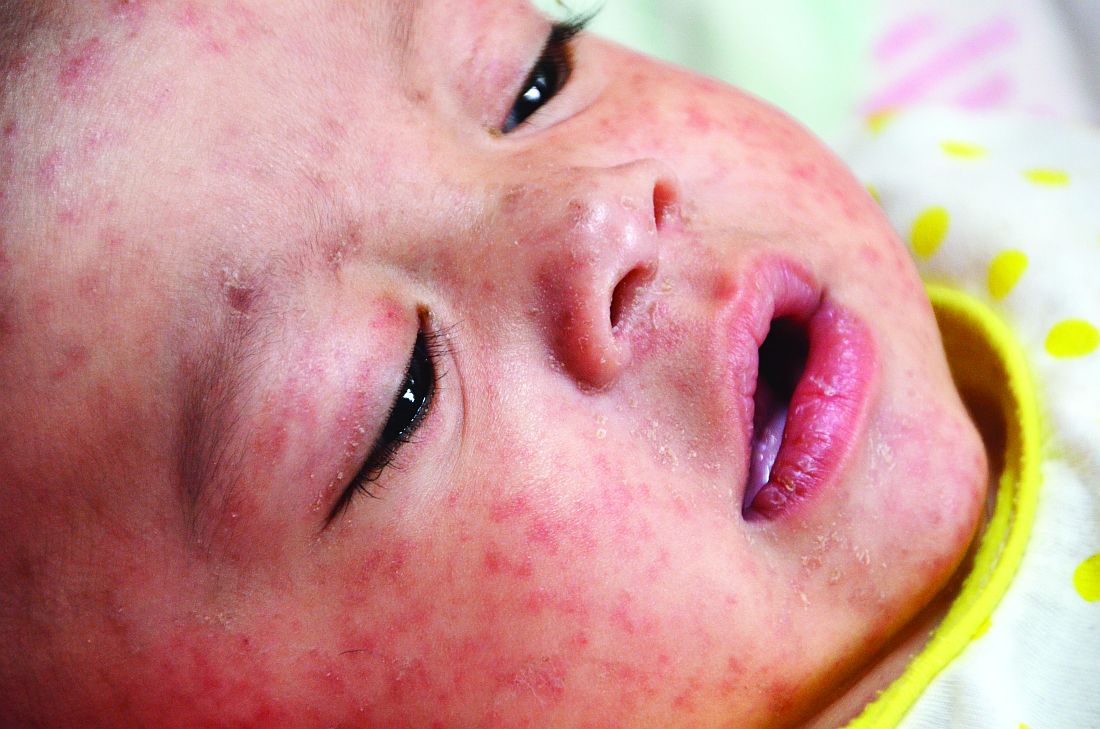
As part of a bivariate logistic regression analysis, Dr. Bode and colleagues sought to evaluate changes in measles vaccination rates across 12 clinic sites of the Nationwide Children’s Hospital pediatric primary care network in Columbus among 23,534 children aged 16 months. The study period targeted the time between April and May 2020, when clinic access and appointment attendance declined following the start of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, until the June-to-August 2020 time period, when clinical care was allowed to return.
The need for the study was prompted by Centers for Disease Control and Prevention reporting on a state-specific precipitous decline in MMR vaccination rates shortly after the onset of COVID-19 in May 2020. Citing the results of one study, such reductions in vaccination have raised concerns over the possibility of a measles resurgence, noted Dr. Bode and associates.
MMR vaccination rates begin to drop with onset of COVID-19 pandemic.
From March 2017 to March 2020, the average rate of MMR vaccination in 16-month-olds was 72%. It subsequently decreased to 67% from April to May 2020, and then dropped further to 62% during the period June to August, 2020 (P = .001). Those without insurance were less likely to be vaccinated than were those carrying private insurance or Medicaid.
Among patients who had not attended a preventive care visit after 12 months of age, the proportion who received vaccines declined during the same time periods, from 10% before the pandemic to 6% at the start of the pandemic and 3% during the summer months of 2020.
“Given the baseline low vaccination rates even before the pandemic and the subsequent decline, we face a critical need to improve timely vaccination and provide catch-up opportunities” in areas with the highest incidence of COVID-19, observed Dr. Bode and colleagues.
Innovative approaches are needed to encourage families to seek preventive care.
In response, the researchers announced the implementation of new community-based vaccination approaches in Ohio, including pop-up vaccine clinics, mobile clinics, and school-based clinics to provide families, who are reluctant to visit health care facilities over COVID-19 related concerns, with safe alternatives. “We believe that it is critical to develop innovative approaches to have families return for preventive care,” they added.
In a separate interview, Herschel Lessin, MD, a private practice pediatrician in Poughkeepsie, N.Y., noted: “This study confirms the anecdotal experience of pediatricians around the country, and our greatest fear that the pandemic will interfere with herd immunity of children for vaccine-preventable illness. Although the study was of urban offices with a primarily Medicaid population, I believe the results to be very worrisome should they prove to be generalizable to the country, as a whole. The significant reduction of well-child visits due to COVID-19 (and fear of COVID-19) seriously impaired the vaccination status of a standard required vaccine in a large population. What is even more worrisome is that the rates continued to fall even after the initial closure of many offices and well into their reopening, despite concerted efforts to try to catch up these missed visits and immunizations.”
Measles is an intensely contagious illness that has not been eradicated, as evidenced by the enormous measles outbreak stemming from Disneyland in 2014-2015, and again with the possible exposure of hundreds to an infected Disneyland visitor last fall, where coverage rates were even higher than in this study, added Dr. Lessin. “This phenomenon, unless forcefully remedied, could easily result in large outbreaks of other vaccine-preventable illness besides COVID-19,” he cautioned.
Dr. Bode and colleagues as well as Dr. Lessin had no conflicts of interest and no relevant financial disclosures.
SOURCE: Bode SM et al. Pediatrics. 2021. doi: 10.1542/peds.2020-035576.
Widespread use of the MMR vaccine is not only crucial for protecting the community against infectious outbreaks, but also serves as the overall pacesetter for preventive services, said Sara M. Bode, MD and colleagues at Nationwide Children’s Hospital in Columbus.
As part of a bivariate logistic regression analysis, Dr. Bode and colleagues sought to evaluate changes in measles vaccination rates across 12 clinic sites of the Nationwide Children’s Hospital pediatric primary care network in Columbus among 23,534 children aged 16 months. The study period targeted the time between April and May 2020, when clinic access and appointment attendance declined following the start of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, until the June-to-August 2020 time period, when clinical care was allowed to return.
The need for the study was prompted by Centers for Disease Control and Prevention reporting on a state-specific precipitous decline in MMR vaccination rates shortly after the onset of COVID-19 in May 2020. Citing the results of one study, such reductions in vaccination have raised concerns over the possibility of a measles resurgence, noted Dr. Bode and associates.
MMR vaccination rates begin to drop with onset of COVID-19 pandemic.
From March 2017 to March 2020, the average rate of MMR vaccination in 16-month-olds was 72%. It subsequently decreased to 67% from April to May 2020, and then dropped further to 62% during the period June to August, 2020 (P = .001). Those without insurance were less likely to be vaccinated than were those carrying private insurance or Medicaid.
Among patients who had not attended a preventive care visit after 12 months of age, the proportion who received vaccines declined during the same time periods, from 10% before the pandemic to 6% at the start of the pandemic and 3% during the summer months of 2020.
“Given the baseline low vaccination rates even before the pandemic and the subsequent decline, we face a critical need to improve timely vaccination and provide catch-up opportunities” in areas with the highest incidence of COVID-19, observed Dr. Bode and colleagues.
Innovative approaches are needed to encourage families to seek preventive care.
In response, the researchers announced the implementation of new community-based vaccination approaches in Ohio, including pop-up vaccine clinics, mobile clinics, and school-based clinics to provide families, who are reluctant to visit health care facilities over COVID-19 related concerns, with safe alternatives. “We believe that it is critical to develop innovative approaches to have families return for preventive care,” they added.
In a separate interview, Herschel Lessin, MD, a private practice pediatrician in Poughkeepsie, N.Y., noted: “This study confirms the anecdotal experience of pediatricians around the country, and our greatest fear that the pandemic will interfere with herd immunity of children for vaccine-preventable illness. Although the study was of urban offices with a primarily Medicaid population, I believe the results to be very worrisome should they prove to be generalizable to the country, as a whole. The significant reduction of well-child visits due to COVID-19 (and fear of COVID-19) seriously impaired the vaccination status of a standard required vaccine in a large population. What is even more worrisome is that the rates continued to fall even after the initial closure of many offices and well into their reopening, despite concerted efforts to try to catch up these missed visits and immunizations.”
Measles is an intensely contagious illness that has not been eradicated, as evidenced by the enormous measles outbreak stemming from Disneyland in 2014-2015, and again with the possible exposure of hundreds to an infected Disneyland visitor last fall, where coverage rates were even higher than in this study, added Dr. Lessin. “This phenomenon, unless forcefully remedied, could easily result in large outbreaks of other vaccine-preventable illness besides COVID-19,” he cautioned.
Dr. Bode and colleagues as well as Dr. Lessin had no conflicts of interest and no relevant financial disclosures.
SOURCE: Bode SM et al. Pediatrics. 2021. doi: 10.1542/peds.2020-035576.
Widespread use of the MMR vaccine is not only crucial for protecting the community against infectious outbreaks, but also serves as the overall pacesetter for preventive services, said Sara M. Bode, MD and colleagues at Nationwide Children’s Hospital in Columbus.
As part of a bivariate logistic regression analysis, Dr. Bode and colleagues sought to evaluate changes in measles vaccination rates across 12 clinic sites of the Nationwide Children’s Hospital pediatric primary care network in Columbus among 23,534 children aged 16 months. The study period targeted the time between April and May 2020, when clinic access and appointment attendance declined following the start of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, until the June-to-August 2020 time period, when clinical care was allowed to return.
The need for the study was prompted by Centers for Disease Control and Prevention reporting on a state-specific precipitous decline in MMR vaccination rates shortly after the onset of COVID-19 in May 2020. Citing the results of one study, such reductions in vaccination have raised concerns over the possibility of a measles resurgence, noted Dr. Bode and associates.
MMR vaccination rates begin to drop with onset of COVID-19 pandemic.
From March 2017 to March 2020, the average rate of MMR vaccination in 16-month-olds was 72%. It subsequently decreased to 67% from April to May 2020, and then dropped further to 62% during the period June to August, 2020 (P = .001). Those without insurance were less likely to be vaccinated than were those carrying private insurance or Medicaid.
Among patients who had not attended a preventive care visit after 12 months of age, the proportion who received vaccines declined during the same time periods, from 10% before the pandemic to 6% at the start of the pandemic and 3% during the summer months of 2020.
“Given the baseline low vaccination rates even before the pandemic and the subsequent decline, we face a critical need to improve timely vaccination and provide catch-up opportunities” in areas with the highest incidence of COVID-19, observed Dr. Bode and colleagues.
Innovative approaches are needed to encourage families to seek preventive care.
In response, the researchers announced the implementation of new community-based vaccination approaches in Ohio, including pop-up vaccine clinics, mobile clinics, and school-based clinics to provide families, who are reluctant to visit health care facilities over COVID-19 related concerns, with safe alternatives. “We believe that it is critical to develop innovative approaches to have families return for preventive care,” they added.
In a separate interview, Herschel Lessin, MD, a private practice pediatrician in Poughkeepsie, N.Y., noted: “This study confirms the anecdotal experience of pediatricians around the country, and our greatest fear that the pandemic will interfere with herd immunity of children for vaccine-preventable illness. Although the study was of urban offices with a primarily Medicaid population, I believe the results to be very worrisome should they prove to be generalizable to the country, as a whole. The significant reduction of well-child visits due to COVID-19 (and fear of COVID-19) seriously impaired the vaccination status of a standard required vaccine in a large population. What is even more worrisome is that the rates continued to fall even after the initial closure of many offices and well into their reopening, despite concerted efforts to try to catch up these missed visits and immunizations.”
Measles is an intensely contagious illness that has not been eradicated, as evidenced by the enormous measles outbreak stemming from Disneyland in 2014-2015, and again with the possible exposure of hundreds to an infected Disneyland visitor last fall, where coverage rates were even higher than in this study, added Dr. Lessin. “This phenomenon, unless forcefully remedied, could easily result in large outbreaks of other vaccine-preventable illness besides COVID-19,” he cautioned.
Dr. Bode and colleagues as well as Dr. Lessin had no conflicts of interest and no relevant financial disclosures.
SOURCE: Bode SM et al. Pediatrics. 2021. doi: 10.1542/peds.2020-035576.
FROM PEDIATRICS
Latest rise in child COVID-19 cases is relatively small
For the seventh week out of the last eight, more new cases of COVID-19 in children were reported in the United States than any week before, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
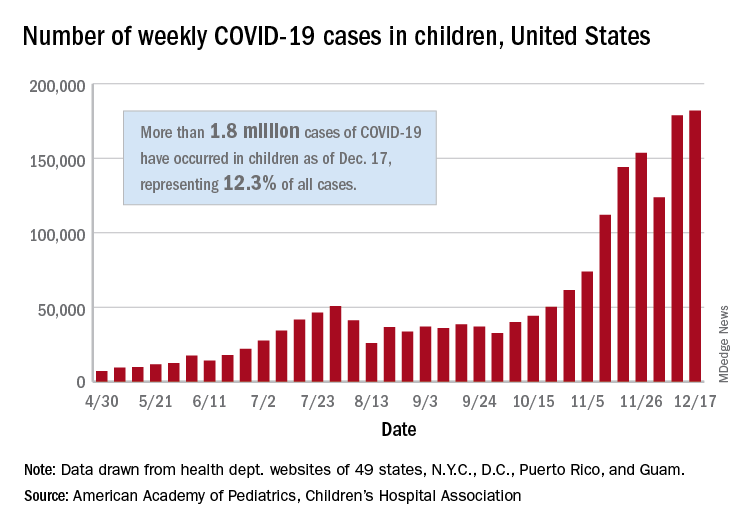
There were just over 182,000 new cases of COVID-19 in children during the week ending Dec. 17, topping the previous high of almost 179,000 set the previous week. – a stretch of 11 weeks that has produced only one decline, based on data from the latest AAP/CHA weekly report.
As of Dec. 17, there had been over 1.8 million cases of COVID-19 in children, which represents 12.3% of all U.S. cases. For the week, 14% of all cases occurred in children, which was up slightly from 13.8% the week before (Dec. 10). The overall rate of coronavirus infection is now 2,420 cases per 100,000 children in the population, the AAP and CHA said.
A total of 30 states are above that national rate, with North Dakota the highest at 7,515 cases per 100,000 children, followed by South Dakota (5,618), Wyoming (5,157), Wisconsin (5,106), and Tennessee (4,994). Wyoming has the highest proportion of cases occurring in children at 20.8%, but that is down from 23.4% in mid-November, based on data collected by the AAP and CHA from the health department websites of 49 states (New York does not provide age distributions), the District of Columbia, New York City, Puerto Rico, and Guam.
In the last 2 weeks, however, the largest percent increases in new cases came in states with low-to-average rates of cumulative child infection. California, Connecticut, Delaware, Maine, Maryland, New Hampshire, and Vermont all saw increases of over 35% from Dec. 3 to Dec. 17, while the smallest increases occurred in Hawaii, North Dakota, and Wyoming, the AAP and CHA reported.
For the seventh week out of the last eight, more new cases of COVID-19 in children were reported in the United States than any week before, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
There were just over 182,000 new cases of COVID-19 in children during the week ending Dec. 17, topping the previous high of almost 179,000 set the previous week. – a stretch of 11 weeks that has produced only one decline, based on data from the latest AAP/CHA weekly report.
As of Dec. 17, there had been over 1.8 million cases of COVID-19 in children, which represents 12.3% of all U.S. cases. For the week, 14% of all cases occurred in children, which was up slightly from 13.8% the week before (Dec. 10). The overall rate of coronavirus infection is now 2,420 cases per 100,000 children in the population, the AAP and CHA said.
A total of 30 states are above that national rate, with North Dakota the highest at 7,515 cases per 100,000 children, followed by South Dakota (5,618), Wyoming (5,157), Wisconsin (5,106), and Tennessee (4,994). Wyoming has the highest proportion of cases occurring in children at 20.8%, but that is down from 23.4% in mid-November, based on data collected by the AAP and CHA from the health department websites of 49 states (New York does not provide age distributions), the District of Columbia, New York City, Puerto Rico, and Guam.
In the last 2 weeks, however, the largest percent increases in new cases came in states with low-to-average rates of cumulative child infection. California, Connecticut, Delaware, Maine, Maryland, New Hampshire, and Vermont all saw increases of over 35% from Dec. 3 to Dec. 17, while the smallest increases occurred in Hawaii, North Dakota, and Wyoming, the AAP and CHA reported.
For the seventh week out of the last eight, more new cases of COVID-19 in children were reported in the United States than any week before, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
There were just over 182,000 new cases of COVID-19 in children during the week ending Dec. 17, topping the previous high of almost 179,000 set the previous week. – a stretch of 11 weeks that has produced only one decline, based on data from the latest AAP/CHA weekly report.
As of Dec. 17, there had been over 1.8 million cases of COVID-19 in children, which represents 12.3% of all U.S. cases. For the week, 14% of all cases occurred in children, which was up slightly from 13.8% the week before (Dec. 10). The overall rate of coronavirus infection is now 2,420 cases per 100,000 children in the population, the AAP and CHA said.
A total of 30 states are above that national rate, with North Dakota the highest at 7,515 cases per 100,000 children, followed by South Dakota (5,618), Wyoming (5,157), Wisconsin (5,106), and Tennessee (4,994). Wyoming has the highest proportion of cases occurring in children at 20.8%, but that is down from 23.4% in mid-November, based on data collected by the AAP and CHA from the health department websites of 49 states (New York does not provide age distributions), the District of Columbia, New York City, Puerto Rico, and Guam.
In the last 2 weeks, however, the largest percent increases in new cases came in states with low-to-average rates of cumulative child infection. California, Connecticut, Delaware, Maine, Maryland, New Hampshire, and Vermont all saw increases of over 35% from Dec. 3 to Dec. 17, while the smallest increases occurred in Hawaii, North Dakota, and Wyoming, the AAP and CHA reported.
Shortcomings identified in study of acne videos on TikTok
, according to an analysis of the top 100 videos using a consumer health validation tool.
The popularity of TikTok among adolescents in particular has implications for the dissemination of acne information, as some teens become “skinfluencers” and receive sponsorship from skin care brands in exchange for social media promotion, wrote David X. Zheng, BA, of the department of dermatology, Case Western Reserve University, Cleveland, and colleagues.
“However, the quality of dermatologic information found on TikTok is largely unknown,” they said.
In a brief report published in Pediatric Dermatology, the researchers identified the top 100 videos on TikTok on May 1, 2020, that were tagged with “#acne.” The information on each video included date of upload, type and gender of the individual uploading the video, physician specialty if applicable, and video category. These top 100 videos had 13,470,501 likes and 64,775 comments over a 7.6-month time period.
The researchers used the DISCERN criteria, a validated 1-5 scale designed to assess consumer health information, to evaluate the video content, with 1 (having “serious” or “extensive shortcomings”) and 5 (having “minimal shortcomings.”)
Overall, the average quality rating of the TikTok acne videos was 2.03. A total of 9 videos were produced by board-certified physicians in the United States, with an average DISCERN score of 2.41.
“Analysis of the DISCERN criteria dimensions suggested that major shortcomings common to both physician and nonphysician uploaders included failure to cite information sources, discuss treatment risks, and provide support for shared decision-making,” the researchers said.
Approximately one-third (34%) of the videos fell into the treatment-product advertisement category, while 26% were personal anecdotes, 20% presented information related to acne, 13% featured home remedy treatments, and 7% were classified as “other.” The researchers also identified the top 200 “#acne” videos on TikTok once a week from May 8, 2020 to June 5, 2020, to determine the evolution of acne content on the app and found a turnover rate of 10.9% per week.
Based on the high turnover and low quality based on DISCERN ratings, the authors suggested that patients seeking acne information should “view acne-related TikTok videos with caution and consult evidence-based resources whenever possible.”
The study findings were limited by several factors including the small sample size of physicians uploading videos, lack of information about the number of nonphysician medical professionals who uploaded videos, and lack of information about the number of video views and country of origin, the researchers noted. However, the results highlight the need for dermatologists to be aware that patients, especially teens, may be using TikTok for acne information that may be of poor quality, they said.
“Conversely, we understand that social media can be a powerful tool for advancing health literacy,” the researchers noted. “Therefore, we also recommend that health care professionals engaging on TikTok create thorough and perhaps standardized educational videos regarding acne, as well as correct any acne-related misinformation that may be present,” they concluded.
The other authors of the study were from the departments of dermatology at Case Western Reserve, University Hospitals Cleveland, and Johns Hopkins University, Baltimore.
The study received no outside funding. The researchers had no financial conflicts to disclose.
SOURCE: Zheng DX et al. Pediatr Dermatol. 2020 Nov 28. doi: 10.1111/pde.14471.
, according to an analysis of the top 100 videos using a consumer health validation tool.
The popularity of TikTok among adolescents in particular has implications for the dissemination of acne information, as some teens become “skinfluencers” and receive sponsorship from skin care brands in exchange for social media promotion, wrote David X. Zheng, BA, of the department of dermatology, Case Western Reserve University, Cleveland, and colleagues.
“However, the quality of dermatologic information found on TikTok is largely unknown,” they said.
In a brief report published in Pediatric Dermatology, the researchers identified the top 100 videos on TikTok on May 1, 2020, that were tagged with “#acne.” The information on each video included date of upload, type and gender of the individual uploading the video, physician specialty if applicable, and video category. These top 100 videos had 13,470,501 likes and 64,775 comments over a 7.6-month time period.
The researchers used the DISCERN criteria, a validated 1-5 scale designed to assess consumer health information, to evaluate the video content, with 1 (having “serious” or “extensive shortcomings”) and 5 (having “minimal shortcomings.”)
Overall, the average quality rating of the TikTok acne videos was 2.03. A total of 9 videos were produced by board-certified physicians in the United States, with an average DISCERN score of 2.41.
“Analysis of the DISCERN criteria dimensions suggested that major shortcomings common to both physician and nonphysician uploaders included failure to cite information sources, discuss treatment risks, and provide support for shared decision-making,” the researchers said.
Approximately one-third (34%) of the videos fell into the treatment-product advertisement category, while 26% were personal anecdotes, 20% presented information related to acne, 13% featured home remedy treatments, and 7% were classified as “other.” The researchers also identified the top 200 “#acne” videos on TikTok once a week from May 8, 2020 to June 5, 2020, to determine the evolution of acne content on the app and found a turnover rate of 10.9% per week.
Based on the high turnover and low quality based on DISCERN ratings, the authors suggested that patients seeking acne information should “view acne-related TikTok videos with caution and consult evidence-based resources whenever possible.”
The study findings were limited by several factors including the small sample size of physicians uploading videos, lack of information about the number of nonphysician medical professionals who uploaded videos, and lack of information about the number of video views and country of origin, the researchers noted. However, the results highlight the need for dermatologists to be aware that patients, especially teens, may be using TikTok for acne information that may be of poor quality, they said.
“Conversely, we understand that social media can be a powerful tool for advancing health literacy,” the researchers noted. “Therefore, we also recommend that health care professionals engaging on TikTok create thorough and perhaps standardized educational videos regarding acne, as well as correct any acne-related misinformation that may be present,” they concluded.
The other authors of the study were from the departments of dermatology at Case Western Reserve, University Hospitals Cleveland, and Johns Hopkins University, Baltimore.
The study received no outside funding. The researchers had no financial conflicts to disclose.
SOURCE: Zheng DX et al. Pediatr Dermatol. 2020 Nov 28. doi: 10.1111/pde.14471.
, according to an analysis of the top 100 videos using a consumer health validation tool.
The popularity of TikTok among adolescents in particular has implications for the dissemination of acne information, as some teens become “skinfluencers” and receive sponsorship from skin care brands in exchange for social media promotion, wrote David X. Zheng, BA, of the department of dermatology, Case Western Reserve University, Cleveland, and colleagues.
“However, the quality of dermatologic information found on TikTok is largely unknown,” they said.
In a brief report published in Pediatric Dermatology, the researchers identified the top 100 videos on TikTok on May 1, 2020, that were tagged with “#acne.” The information on each video included date of upload, type and gender of the individual uploading the video, physician specialty if applicable, and video category. These top 100 videos had 13,470,501 likes and 64,775 comments over a 7.6-month time period.
The researchers used the DISCERN criteria, a validated 1-5 scale designed to assess consumer health information, to evaluate the video content, with 1 (having “serious” or “extensive shortcomings”) and 5 (having “minimal shortcomings.”)
Overall, the average quality rating of the TikTok acne videos was 2.03. A total of 9 videos were produced by board-certified physicians in the United States, with an average DISCERN score of 2.41.
“Analysis of the DISCERN criteria dimensions suggested that major shortcomings common to both physician and nonphysician uploaders included failure to cite information sources, discuss treatment risks, and provide support for shared decision-making,” the researchers said.
Approximately one-third (34%) of the videos fell into the treatment-product advertisement category, while 26% were personal anecdotes, 20% presented information related to acne, 13% featured home remedy treatments, and 7% were classified as “other.” The researchers also identified the top 200 “#acne” videos on TikTok once a week from May 8, 2020 to June 5, 2020, to determine the evolution of acne content on the app and found a turnover rate of 10.9% per week.
Based on the high turnover and low quality based on DISCERN ratings, the authors suggested that patients seeking acne information should “view acne-related TikTok videos with caution and consult evidence-based resources whenever possible.”
The study findings were limited by several factors including the small sample size of physicians uploading videos, lack of information about the number of nonphysician medical professionals who uploaded videos, and lack of information about the number of video views and country of origin, the researchers noted. However, the results highlight the need for dermatologists to be aware that patients, especially teens, may be using TikTok for acne information that may be of poor quality, they said.
“Conversely, we understand that social media can be a powerful tool for advancing health literacy,” the researchers noted. “Therefore, we also recommend that health care professionals engaging on TikTok create thorough and perhaps standardized educational videos regarding acne, as well as correct any acne-related misinformation that may be present,” they concluded.
The other authors of the study were from the departments of dermatology at Case Western Reserve, University Hospitals Cleveland, and Johns Hopkins University, Baltimore.
The study received no outside funding. The researchers had no financial conflicts to disclose.
SOURCE: Zheng DX et al. Pediatr Dermatol. 2020 Nov 28. doi: 10.1111/pde.14471.
FROM PEDIATRIC DERMATOLOGY
ASH guidelines for venous thromboembolism: What family physicians need to know
Each year in the United States, approximately one to two out of every thousand people suffer from venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism. .

These guidelines, which were recently published in Blood Advances (Ortel T L et al. Blood Adv 2020 doi: 10.1182/bloodadvances.2020001830), include 28 recommendations.
How to treat uncomplicated patients
For uncomplicated deep vein thrombosis (DVT) and/or pulmonary embolism (PE), the guidelines suggest treating patients at home rather than in the hospital. This is especially important for family physicians to note as many of these patients will now be the responsibility of the primary care doctor to treat and follow. Patients treated at home can avoid the risk of nosocomial infections, especially in the days of COVID-19. Evidence also suggests that being treated at home was shown to reduce the risk of PE versus being treated in the hospital. It is, therefore, crucial that family physicians know which patients are low versus high risk.
Further, the guidelines suggest that these patients with low risk of complications are better treated with direct oral anticoagulants (DOACs) instead of vitamin K antagonists, such as Coumadin.
Medication-related suggestions
The guidelines also suggest that no DOAC is preferred over another. Since DOACs are relatively newer agents, family doctors need to become comfortable with their use. For proximal DVTs, anticoagulation alone can be used without thrombolytics.
Family physicians are often tasked with the decision on when to stop anticoagulation. The authors recommend against using diagnostic tests such as D-Dimer or ultrasound to decide when to stop these medications in low-risk patients. In patients at risk of recurrent VTE due to chronic medical conditions, it is suggested to continue anti-coagulants indefinitely. While anticoagulant therapy effectively reduces risk of VTE, it does increase the risk of bleeding events.
The guidelines are quite extensive and specific in their recommendations and family physicians need to understand them. We are often the first ones in the medical system to diagnose VTE, and it is quite possible to keep these patients home, thereby eliminating risks they may encounter by being hospitalized. In addition, the recommendation regarding the use of DOACs may ease some of the burden of monitoring patients on long-term Coumadin. These medications do not come without risks, and we must be comfortable evaluating for any complications. In our current health care system, different insurance companies have different formularies making it necessary for us to know all these medications.
In the past, the diagnosis of PE and even a DVT would mean a hospital stay. We now know, and these guidelines reaffirm, that this is not necessary in uncomplicated cases.
In addition to diagnosing VTE, family physicians are also tasked with following up with patients who were hospitalized or started on treatment by other physicians. We need to know the plan on when to stop the medication or when to reevaluate its use. Patients often bring this question to us, and these guidelines will help us answer that question.
Many patients who have more complicated medical conditions often see multiple specialists. The ASH recommendations help standardize the care of these patients across specialties.
What the recommendations are missing
As family doctors, we often treat patients with multiple comorbidities. These guidelines do not make recommendations for patients with cancer, who are at high risk of VTE events. Some patients also have conditions that increase their risk of bleeding or have contraindications to the use of anticoagulants. It would be helpful to have more recommendations for both of these types of patients in addition to the use of inferior vena cava filter in patients with proximal DVT. The document is also missing recommendations for pregnant patients, which would be useful.
Overall, these guidelines include much of what we already do in our practices while doing a great job of incorporating the newer DOACs. These guidelines are easy for family physicians to put into practice.
Dr. Girgis practices family medicine in South River, N.J., and is a clinical assistant professor of family medicine at Robert Wood Johnson Medical School, New Brunswick, N.J. You can contact her at [email protected].
Each year in the United States, approximately one to two out of every thousand people suffer from venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism. .
These guidelines, which were recently published in Blood Advances (Ortel T L et al. Blood Adv 2020 doi: 10.1182/bloodadvances.2020001830), include 28 recommendations.
How to treat uncomplicated patients
For uncomplicated deep vein thrombosis (DVT) and/or pulmonary embolism (PE), the guidelines suggest treating patients at home rather than in the hospital. This is especially important for family physicians to note as many of these patients will now be the responsibility of the primary care doctor to treat and follow. Patients treated at home can avoid the risk of nosocomial infections, especially in the days of COVID-19. Evidence also suggests that being treated at home was shown to reduce the risk of PE versus being treated in the hospital. It is, therefore, crucial that family physicians know which patients are low versus high risk.
Further, the guidelines suggest that these patients with low risk of complications are better treated with direct oral anticoagulants (DOACs) instead of vitamin K antagonists, such as Coumadin.
Medication-related suggestions
The guidelines also suggest that no DOAC is preferred over another. Since DOACs are relatively newer agents, family doctors need to become comfortable with their use. For proximal DVTs, anticoagulation alone can be used without thrombolytics.
Family physicians are often tasked with the decision on when to stop anticoagulation. The authors recommend against using diagnostic tests such as D-Dimer or ultrasound to decide when to stop these medications in low-risk patients. In patients at risk of recurrent VTE due to chronic medical conditions, it is suggested to continue anti-coagulants indefinitely. While anticoagulant therapy effectively reduces risk of VTE, it does increase the risk of bleeding events.
The guidelines are quite extensive and specific in their recommendations and family physicians need to understand them. We are often the first ones in the medical system to diagnose VTE, and it is quite possible to keep these patients home, thereby eliminating risks they may encounter by being hospitalized. In addition, the recommendation regarding the use of DOACs may ease some of the burden of monitoring patients on long-term Coumadin. These medications do not come without risks, and we must be comfortable evaluating for any complications. In our current health care system, different insurance companies have different formularies making it necessary for us to know all these medications.
In the past, the diagnosis of PE and even a DVT would mean a hospital stay. We now know, and these guidelines reaffirm, that this is not necessary in uncomplicated cases.
In addition to diagnosing VTE, family physicians are also tasked with following up with patients who were hospitalized or started on treatment by other physicians. We need to know the plan on when to stop the medication or when to reevaluate its use. Patients often bring this question to us, and these guidelines will help us answer that question.
Many patients who have more complicated medical conditions often see multiple specialists. The ASH recommendations help standardize the care of these patients across specialties.
What the recommendations are missing
As family doctors, we often treat patients with multiple comorbidities. These guidelines do not make recommendations for patients with cancer, who are at high risk of VTE events. Some patients also have conditions that increase their risk of bleeding or have contraindications to the use of anticoagulants. It would be helpful to have more recommendations for both of these types of patients in addition to the use of inferior vena cava filter in patients with proximal DVT. The document is also missing recommendations for pregnant patients, which would be useful.
Overall, these guidelines include much of what we already do in our practices while doing a great job of incorporating the newer DOACs. These guidelines are easy for family physicians to put into practice.
Dr. Girgis practices family medicine in South River, N.J., and is a clinical assistant professor of family medicine at Robert Wood Johnson Medical School, New Brunswick, N.J. You can contact her at [email protected].
Each year in the United States, approximately one to two out of every thousand people suffer from venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism. .
These guidelines, which were recently published in Blood Advances (Ortel T L et al. Blood Adv 2020 doi: 10.1182/bloodadvances.2020001830), include 28 recommendations.
How to treat uncomplicated patients
For uncomplicated deep vein thrombosis (DVT) and/or pulmonary embolism (PE), the guidelines suggest treating patients at home rather than in the hospital. This is especially important for family physicians to note as many of these patients will now be the responsibility of the primary care doctor to treat and follow. Patients treated at home can avoid the risk of nosocomial infections, especially in the days of COVID-19. Evidence also suggests that being treated at home was shown to reduce the risk of PE versus being treated in the hospital. It is, therefore, crucial that family physicians know which patients are low versus high risk.
Further, the guidelines suggest that these patients with low risk of complications are better treated with direct oral anticoagulants (DOACs) instead of vitamin K antagonists, such as Coumadin.
Medication-related suggestions
The guidelines also suggest that no DOAC is preferred over another. Since DOACs are relatively newer agents, family doctors need to become comfortable with their use. For proximal DVTs, anticoagulation alone can be used without thrombolytics.
Family physicians are often tasked with the decision on when to stop anticoagulation. The authors recommend against using diagnostic tests such as D-Dimer or ultrasound to decide when to stop these medications in low-risk patients. In patients at risk of recurrent VTE due to chronic medical conditions, it is suggested to continue anti-coagulants indefinitely. While anticoagulant therapy effectively reduces risk of VTE, it does increase the risk of bleeding events.
The guidelines are quite extensive and specific in their recommendations and family physicians need to understand them. We are often the first ones in the medical system to diagnose VTE, and it is quite possible to keep these patients home, thereby eliminating risks they may encounter by being hospitalized. In addition, the recommendation regarding the use of DOACs may ease some of the burden of monitoring patients on long-term Coumadin. These medications do not come without risks, and we must be comfortable evaluating for any complications. In our current health care system, different insurance companies have different formularies making it necessary for us to know all these medications.
In the past, the diagnosis of PE and even a DVT would mean a hospital stay. We now know, and these guidelines reaffirm, that this is not necessary in uncomplicated cases.
In addition to diagnosing VTE, family physicians are also tasked with following up with patients who were hospitalized or started on treatment by other physicians. We need to know the plan on when to stop the medication or when to reevaluate its use. Patients often bring this question to us, and these guidelines will help us answer that question.
Many patients who have more complicated medical conditions often see multiple specialists. The ASH recommendations help standardize the care of these patients across specialties.
What the recommendations are missing
As family doctors, we often treat patients with multiple comorbidities. These guidelines do not make recommendations for patients with cancer, who are at high risk of VTE events. Some patients also have conditions that increase their risk of bleeding or have contraindications to the use of anticoagulants. It would be helpful to have more recommendations for both of these types of patients in addition to the use of inferior vena cava filter in patients with proximal DVT. The document is also missing recommendations for pregnant patients, which would be useful.
Overall, these guidelines include much of what we already do in our practices while doing a great job of incorporating the newer DOACs. These guidelines are easy for family physicians to put into practice.
Dr. Girgis practices family medicine in South River, N.J., and is a clinical assistant professor of family medicine at Robert Wood Johnson Medical School, New Brunswick, N.J. You can contact her at [email protected].
Labor induction at 39 weeks may improve neonatal outcomes
Aaron B. Caughey, MD, PhD, said at the 2020 virtual meeting of the American College of Obstetricians and Gynecologists.
For much of the 20th century, term gestation has been defined as 37 weeks and beyond, said Dr. Caughey, of Oregon Health & Science University, Portland. He noted several studies showing a U-shaped distribution in neonatal outcomes during the period from 37 weeks to 41 weeks for some outcomes, including Apgar scores. However, respiratory outcomes in a study from 2008 showed an increase, with meconium stained amniotic fluid increasing from 2.27% at 37 weeks to 10.33% at 41 weeks, and meconium aspiration increasing from 0.07% at 37 weeks to 0.27% at 41 weeks.
Late-term induction may carry more risk
The study “that really got everyone’s attention” in terms of neonatal outcomes was published in 2009 in the New England Journal of Medicine. The cohort study included 24,077 elective cesarean deliveries between 37 and 42 weeks and reviewed a range of neonatal outcomes based on gestational age.
The rate of any adverse outcome decreased from 37 weeks to 39 weeks, “but then started going back up again,” Dr. Caughey said. He reviewed data from another study that factored in stillbirth and the risk of expectant management based on gestational age. A composite risk of perinatal death with expectant management was 15.4 deaths per 10,000 cases at 37 weeks and 39 weeks, but increased to 19.9 at 42 weeks.
“The morbidity appears to have a U-shaped distribution and the mortality seems to favor delivery at 39 weeks,” he said.
When it comes to induction of labor, medically indicated vs. nonmedically indicated does matter, Dr. Caughey said. Factors not considered a medical indication include impending macrosomia, increased risk for developing preeclampsia or intrauterine growth retardation, and a favorable cervix, he noted.
“For indicated induction of labor, the risks and benefits of induction of labor vs. expectant management have been considered and weighed in by the field of experts that care for pregnant women,” he said. With nonmedically indicated induction, experts “either decided that risks and benefits don’t favor induction of labor, or we haven’t come down hard on what the protocol might be.
“It is important to consider the risks and benefits,” said Dr. Caughey. The factors you want to include are neonatal outcomes, maternal preferences, and doctor preferences. However, “we want to be thoughtful about this intervention,” because of the association of higher costs and increased risk of cesarean with induction of labor.
As for timing of induction of labor, certain conditions favoring early-term induction include preeclampsia and gestational hypertension, chronic hypertension, diabetes, intrauterine growth restriction, nonreassuring fetal testing, cholestasis, placenta previa or accreta, and twins.
Data support value of 39 weeks
As for late-term induction of labor, “at 41 weeks it is pretty clear that neonatal outcomes would be improved by delivery,” he said. Historically, clinicians have raised concerns about the increased risk of cesarean delivery following induction of labor, but this risk has not been borne out in recent studies. Dr. Caughey said. However, in the findings from the ARRIVE trial, a large study of 6,106 women who were randomized to induction or labor or expectant management at 39 weeks, “they found a reduction in their risk of cesarean delivery compared to expectant management (18.6% vs. 22.2%). Rates of preeclampsia also were lower among induced women, while rate of chorioamnionitis, postpartum hemorrhage, and intensive care were similar between the groups. The researchers did not find significant differences in perinatal outcomes.
Dr. Caughey and colleagues conducted a systematic review of cesarean risk and induction of labor, and found a risk ratio of 0.83, similar to the ARRIVE trial. “The data suggest a consistently reduced risk for cesarean delivery with the induction of labor.”
However, “I would caution us to be thoughtful about research protocols vs. actual practice,” he said. “You must think about the environment.” The latent phase of labor can continue for a long time after induction, and patience is called for, he emphasized.
Dr. Caughey said that despite the ARRIVE trial and other studies, 39 weeks should not necessarily be the new standard for induction of labor. “The proportion of women impacted is dramatically different, if you would be inducing every woman at 39 weeks, that would be 60% to 70%,” which could have a great impact on resources.
Based on current research, early-term induction of labor at 37 weeks “is a bad idea without indication,” said Dr. Caughey. Induction at 41 weeks (sometimes considered post term) is the current ACOG recommendation and is associated with improved outcomes.
Induction of labor at full term (39-40 weeks) depends in part on the environment, and is not a violation of standard of care, he said. “Evidence is evolving, and individual hospitals are trying to figure this out.”
Cesarean data are convincing, at least in some settings, he said. However, “we need more global trials and different medical settings” to determine the optimal time for induction of labor.
Consider maternal preferences and characteristics
During a question-and-answer session, Dr. Caughey was asked whether all women should be offered induction of labor at 39 weeks.
“I think it is OK if your entire health system has agreed to offering, to have that shared medical decision making, but you need to have careful conversation to make sure you have the resources,” he noted. Also, he said he believed clinicians should respond to women as they request labor induction at 39 weeks.
In response to a question about induction of labor in obese women, he noted that women with a body mass index greater than 35 kg/m2 are not equally successful with induction of labor. “We know they have a higher risk of cesarean delivery,” however, “it has been demonstrated that they have the same potential benefits of reduced risk of cesarean.”
As for factoring in the Bishop score to determine a favorable or unfavorable cervix, Dr. Caughey noted that women with a favorable cervix are more likely to go into labor on their own, while those with an unfavorable cervix may benefit from cervical ripening.
Dr. Caughey had no financial conflicts relevant to this talk, but disclosed serving as a medical adviser to Celmatix and Mindchild, as well as an endowment to his academic department from Bob’s Red Mill, an Oregon-based whole grain foods manufacturer.
Aaron B. Caughey, MD, PhD, said at the 2020 virtual meeting of the American College of Obstetricians and Gynecologists.
For much of the 20th century, term gestation has been defined as 37 weeks and beyond, said Dr. Caughey, of Oregon Health & Science University, Portland. He noted several studies showing a U-shaped distribution in neonatal outcomes during the period from 37 weeks to 41 weeks for some outcomes, including Apgar scores. However, respiratory outcomes in a study from 2008 showed an increase, with meconium stained amniotic fluid increasing from 2.27% at 37 weeks to 10.33% at 41 weeks, and meconium aspiration increasing from 0.07% at 37 weeks to 0.27% at 41 weeks.
Late-term induction may carry more risk
The study “that really got everyone’s attention” in terms of neonatal outcomes was published in 2009 in the New England Journal of Medicine. The cohort study included 24,077 elective cesarean deliveries between 37 and 42 weeks and reviewed a range of neonatal outcomes based on gestational age.
The rate of any adverse outcome decreased from 37 weeks to 39 weeks, “but then started going back up again,” Dr. Caughey said. He reviewed data from another study that factored in stillbirth and the risk of expectant management based on gestational age. A composite risk of perinatal death with expectant management was 15.4 deaths per 10,000 cases at 37 weeks and 39 weeks, but increased to 19.9 at 42 weeks.
“The morbidity appears to have a U-shaped distribution and the mortality seems to favor delivery at 39 weeks,” he said.
When it comes to induction of labor, medically indicated vs. nonmedically indicated does matter, Dr. Caughey said. Factors not considered a medical indication include impending macrosomia, increased risk for developing preeclampsia or intrauterine growth retardation, and a favorable cervix, he noted.
“For indicated induction of labor, the risks and benefits of induction of labor vs. expectant management have been considered and weighed in by the field of experts that care for pregnant women,” he said. With nonmedically indicated induction, experts “either decided that risks and benefits don’t favor induction of labor, or we haven’t come down hard on what the protocol might be.
“It is important to consider the risks and benefits,” said Dr. Caughey. The factors you want to include are neonatal outcomes, maternal preferences, and doctor preferences. However, “we want to be thoughtful about this intervention,” because of the association of higher costs and increased risk of cesarean with induction of labor.
As for timing of induction of labor, certain conditions favoring early-term induction include preeclampsia and gestational hypertension, chronic hypertension, diabetes, intrauterine growth restriction, nonreassuring fetal testing, cholestasis, placenta previa or accreta, and twins.
Data support value of 39 weeks
As for late-term induction of labor, “at 41 weeks it is pretty clear that neonatal outcomes would be improved by delivery,” he said. Historically, clinicians have raised concerns about the increased risk of cesarean delivery following induction of labor, but this risk has not been borne out in recent studies. Dr. Caughey said. However, in the findings from the ARRIVE trial, a large study of 6,106 women who were randomized to induction or labor or expectant management at 39 weeks, “they found a reduction in their risk of cesarean delivery compared to expectant management (18.6% vs. 22.2%). Rates of preeclampsia also were lower among induced women, while rate of chorioamnionitis, postpartum hemorrhage, and intensive care were similar between the groups. The researchers did not find significant differences in perinatal outcomes.
Dr. Caughey and colleagues conducted a systematic review of cesarean risk and induction of labor, and found a risk ratio of 0.83, similar to the ARRIVE trial. “The data suggest a consistently reduced risk for cesarean delivery with the induction of labor.”
However, “I would caution us to be thoughtful about research protocols vs. actual practice,” he said. “You must think about the environment.” The latent phase of labor can continue for a long time after induction, and patience is called for, he emphasized.
Dr. Caughey said that despite the ARRIVE trial and other studies, 39 weeks should not necessarily be the new standard for induction of labor. “The proportion of women impacted is dramatically different, if you would be inducing every woman at 39 weeks, that would be 60% to 70%,” which could have a great impact on resources.
Based on current research, early-term induction of labor at 37 weeks “is a bad idea without indication,” said Dr. Caughey. Induction at 41 weeks (sometimes considered post term) is the current ACOG recommendation and is associated with improved outcomes.
Induction of labor at full term (39-40 weeks) depends in part on the environment, and is not a violation of standard of care, he said. “Evidence is evolving, and individual hospitals are trying to figure this out.”
Cesarean data are convincing, at least in some settings, he said. However, “we need more global trials and different medical settings” to determine the optimal time for induction of labor.
Consider maternal preferences and characteristics
During a question-and-answer session, Dr. Caughey was asked whether all women should be offered induction of labor at 39 weeks.
“I think it is OK if your entire health system has agreed to offering, to have that shared medical decision making, but you need to have careful conversation to make sure you have the resources,” he noted. Also, he said he believed clinicians should respond to women as they request labor induction at 39 weeks.
In response to a question about induction of labor in obese women, he noted that women with a body mass index greater than 35 kg/m2 are not equally successful with induction of labor. “We know they have a higher risk of cesarean delivery,” however, “it has been demonstrated that they have the same potential benefits of reduced risk of cesarean.”
As for factoring in the Bishop score to determine a favorable or unfavorable cervix, Dr. Caughey noted that women with a favorable cervix are more likely to go into labor on their own, while those with an unfavorable cervix may benefit from cervical ripening.
Dr. Caughey had no financial conflicts relevant to this talk, but disclosed serving as a medical adviser to Celmatix and Mindchild, as well as an endowment to his academic department from Bob’s Red Mill, an Oregon-based whole grain foods manufacturer.
Aaron B. Caughey, MD, PhD, said at the 2020 virtual meeting of the American College of Obstetricians and Gynecologists.
For much of the 20th century, term gestation has been defined as 37 weeks and beyond, said Dr. Caughey, of Oregon Health & Science University, Portland. He noted several studies showing a U-shaped distribution in neonatal outcomes during the period from 37 weeks to 41 weeks for some outcomes, including Apgar scores. However, respiratory outcomes in a study from 2008 showed an increase, with meconium stained amniotic fluid increasing from 2.27% at 37 weeks to 10.33% at 41 weeks, and meconium aspiration increasing from 0.07% at 37 weeks to 0.27% at 41 weeks.
Late-term induction may carry more risk
The study “that really got everyone’s attention” in terms of neonatal outcomes was published in 2009 in the New England Journal of Medicine. The cohort study included 24,077 elective cesarean deliveries between 37 and 42 weeks and reviewed a range of neonatal outcomes based on gestational age.
The rate of any adverse outcome decreased from 37 weeks to 39 weeks, “but then started going back up again,” Dr. Caughey said. He reviewed data from another study that factored in stillbirth and the risk of expectant management based on gestational age. A composite risk of perinatal death with expectant management was 15.4 deaths per 10,000 cases at 37 weeks and 39 weeks, but increased to 19.9 at 42 weeks.
“The morbidity appears to have a U-shaped distribution and the mortality seems to favor delivery at 39 weeks,” he said.
When it comes to induction of labor, medically indicated vs. nonmedically indicated does matter, Dr. Caughey said. Factors not considered a medical indication include impending macrosomia, increased risk for developing preeclampsia or intrauterine growth retardation, and a favorable cervix, he noted.
“For indicated induction of labor, the risks and benefits of induction of labor vs. expectant management have been considered and weighed in by the field of experts that care for pregnant women,” he said. With nonmedically indicated induction, experts “either decided that risks and benefits don’t favor induction of labor, or we haven’t come down hard on what the protocol might be.
“It is important to consider the risks and benefits,” said Dr. Caughey. The factors you want to include are neonatal outcomes, maternal preferences, and doctor preferences. However, “we want to be thoughtful about this intervention,” because of the association of higher costs and increased risk of cesarean with induction of labor.
As for timing of induction of labor, certain conditions favoring early-term induction include preeclampsia and gestational hypertension, chronic hypertension, diabetes, intrauterine growth restriction, nonreassuring fetal testing, cholestasis, placenta previa or accreta, and twins.
Data support value of 39 weeks
As for late-term induction of labor, “at 41 weeks it is pretty clear that neonatal outcomes would be improved by delivery,” he said. Historically, clinicians have raised concerns about the increased risk of cesarean delivery following induction of labor, but this risk has not been borne out in recent studies. Dr. Caughey said. However, in the findings from the ARRIVE trial, a large study of 6,106 women who were randomized to induction or labor or expectant management at 39 weeks, “they found a reduction in their risk of cesarean delivery compared to expectant management (18.6% vs. 22.2%). Rates of preeclampsia also were lower among induced women, while rate of chorioamnionitis, postpartum hemorrhage, and intensive care were similar between the groups. The researchers did not find significant differences in perinatal outcomes.
Dr. Caughey and colleagues conducted a systematic review of cesarean risk and induction of labor, and found a risk ratio of 0.83, similar to the ARRIVE trial. “The data suggest a consistently reduced risk for cesarean delivery with the induction of labor.”
However, “I would caution us to be thoughtful about research protocols vs. actual practice,” he said. “You must think about the environment.” The latent phase of labor can continue for a long time after induction, and patience is called for, he emphasized.
Dr. Caughey said that despite the ARRIVE trial and other studies, 39 weeks should not necessarily be the new standard for induction of labor. “The proportion of women impacted is dramatically different, if you would be inducing every woman at 39 weeks, that would be 60% to 70%,” which could have a great impact on resources.
Based on current research, early-term induction of labor at 37 weeks “is a bad idea without indication,” said Dr. Caughey. Induction at 41 weeks (sometimes considered post term) is the current ACOG recommendation and is associated with improved outcomes.
Induction of labor at full term (39-40 weeks) depends in part on the environment, and is not a violation of standard of care, he said. “Evidence is evolving, and individual hospitals are trying to figure this out.”
Cesarean data are convincing, at least in some settings, he said. However, “we need more global trials and different medical settings” to determine the optimal time for induction of labor.
Consider maternal preferences and characteristics
During a question-and-answer session, Dr. Caughey was asked whether all women should be offered induction of labor at 39 weeks.
“I think it is OK if your entire health system has agreed to offering, to have that shared medical decision making, but you need to have careful conversation to make sure you have the resources,” he noted. Also, he said he believed clinicians should respond to women as they request labor induction at 39 weeks.
In response to a question about induction of labor in obese women, he noted that women with a body mass index greater than 35 kg/m2 are not equally successful with induction of labor. “We know they have a higher risk of cesarean delivery,” however, “it has been demonstrated that they have the same potential benefits of reduced risk of cesarean.”
As for factoring in the Bishop score to determine a favorable or unfavorable cervix, Dr. Caughey noted that women with a favorable cervix are more likely to go into labor on their own, while those with an unfavorable cervix may benefit from cervical ripening.
Dr. Caughey had no financial conflicts relevant to this talk, but disclosed serving as a medical adviser to Celmatix and Mindchild, as well as an endowment to his academic department from Bob’s Red Mill, an Oregon-based whole grain foods manufacturer.
EXPERT ANALYSIS FROM ACOG 2020
Home phototherapy never looked better, expert says
Kenneth B. Gordon, MD, asserted at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.

“In my practice, I’m using more and more home UVB, and there are a number of reasons for that. It’s more convenient and easier for the patient, as it’s getting more difficult for patients to give up time from work to come to the office. And I might add that, in this time of COVID-19, people don’t want to come to the office. It’s generally less expensive for patients because of copays, which increase the cost of UVB. And believe it or not, I believe it’s easier for the clinician as well. I write a prescription, the patient gets a number of treatments, and I don’t lose any sleep because I think it’s very difficult for patients to get into trouble with narrow-band UVB at home,” explained Dr. Gordon, professor and chair of the department of dermatology at the Medical College of Wisconsin, Milwaukee.
“There’s all sorts of insurance company silliness in getting this paid for, but if you do get it paid for, I think it’s a really effective way to treat psoriasis,” the dermatologist added.
A Dutch multicenter randomized trial demonstrated that home UVB phototherapy for psoriasis was equally safe and effective as outpatient UVB phototherapy, and with greater patient satisfaction.
Surveys show most dermatologists consider phototherapy their preferred treatment for patients with extensive psoriasis because its side effect profile is so benign, compared with that of systemic therapies, be they biologic agents or older drugs such as methotrexate or acitretin. Phototherapy is particularly popular for use in women of childbearing potential, since it’s a nonsystemic therapy.
And speaking of side effects, Dr. Gordon declared, “The risks of narrow-band UVB are sometimes, I believe, exaggerated.” Indeed, he considers the No. 1 side effect of office-based phototherapy to be the loss of productive time.
“Simply put, phototherapy in the office is very easy for me. I write a prescription, the tech takes care of it, and if there’s a problem I’m handy to see the patient. But for the patient, it’s very difficult. Whereas it might take only a few minutes to get the treatment in-office, it takes a lot of time to get to the office, and many patients don’t have transportation. So I think the loss of productive time with phototherapy has to be considered a side effect,” Dr. Gordon said.
Turning to the therapy’s other side effects, he said that although there is some degree of photoaging associated with narrow-band UVB – which is far and away the most commonly used form of phototherapy in the United States – it’s nothing close to the photoaging caused by PUVA.
“I don’t believe that PUVA, with all the destruction of the skin that you see with it, is a significant part of our treatment modalities today,” Dr. Gordon said.
Sunburn is a risk with narrow-band UVB, especially if the dose is ramped up too quickly. Reactivation of herpes simplex virus infection is a frequent problem, and one patients find especially concerning when it manifests as eruptions of cold sores on the face.
The side effect of narrow-band UVB of greatest interest to most patients and physicians is skin cancer. “This is an extremely controversial area,” the dermatologist observed.
Unlike with PUVA, there has never been a convincing study to show that narrow-band UVB is associated with significantly increased risks of keratinocyte carcinomas or melanoma. A large Scottish study found no significantly increased risk, but a modestly increased trend for more squamous cell carcinomas. How modest? The investigators calculated that it would require 50,000 psoriasis patients with a minimum of 100 narrow-band UVB treatments to be followed for 5 years in order to demonstrate a twofold increased risk of the malignancy.
“In other words, it takes an incredible number of patients to be able to see a difference in a skin cancer that we can relatively easily treat. That’s why when I see patients, I don’t emphasize the risk of skin cancer,” Dr. Gordon said.
Similarly reassuring was a Swedish study, which showed the skin cancer rate in UVB-treated psoriasis patients was no different than in the general population.
Guideline recommendations regarding UVB phototherapy and skin cancer risk are all over the map. French guidelines advise a maximum of 230 narrow-band UVB treatments. British guidelines recommend reducing narrow-band UVB exposure to skin areas with significant sun exposure. American guidelines leave the topic untouched, Dr. Gordon noted.
He reported having no financial conflicts of interest regarding his presentation, as neither he, the Medical College of Wisconsin, or its department of dermatology receive any payment for phototherapy services he prescribes. Those payments go to the hospital system where he works. MedscapeLive and this news organization are owned by the same parent company.
Kenneth B. Gordon, MD, asserted at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
“In my practice, I’m using more and more home UVB, and there are a number of reasons for that. It’s more convenient and easier for the patient, as it’s getting more difficult for patients to give up time from work to come to the office. And I might add that, in this time of COVID-19, people don’t want to come to the office. It’s generally less expensive for patients because of copays, which increase the cost of UVB. And believe it or not, I believe it’s easier for the clinician as well. I write a prescription, the patient gets a number of treatments, and I don’t lose any sleep because I think it’s very difficult for patients to get into trouble with narrow-band UVB at home,” explained Dr. Gordon, professor and chair of the department of dermatology at the Medical College of Wisconsin, Milwaukee.
“There’s all sorts of insurance company silliness in getting this paid for, but if you do get it paid for, I think it’s a really effective way to treat psoriasis,” the dermatologist added.
A Dutch multicenter randomized trial demonstrated that home UVB phototherapy for psoriasis was equally safe and effective as outpatient UVB phototherapy, and with greater patient satisfaction.
Surveys show most dermatologists consider phototherapy their preferred treatment for patients with extensive psoriasis because its side effect profile is so benign, compared with that of systemic therapies, be they biologic agents or older drugs such as methotrexate or acitretin. Phototherapy is particularly popular for use in women of childbearing potential, since it’s a nonsystemic therapy.
And speaking of side effects, Dr. Gordon declared, “The risks of narrow-band UVB are sometimes, I believe, exaggerated.” Indeed, he considers the No. 1 side effect of office-based phototherapy to be the loss of productive time.
“Simply put, phototherapy in the office is very easy for me. I write a prescription, the tech takes care of it, and if there’s a problem I’m handy to see the patient. But for the patient, it’s very difficult. Whereas it might take only a few minutes to get the treatment in-office, it takes a lot of time to get to the office, and many patients don’t have transportation. So I think the loss of productive time with phototherapy has to be considered a side effect,” Dr. Gordon said.
Turning to the therapy’s other side effects, he said that although there is some degree of photoaging associated with narrow-band UVB – which is far and away the most commonly used form of phototherapy in the United States – it’s nothing close to the photoaging caused by PUVA.
“I don’t believe that PUVA, with all the destruction of the skin that you see with it, is a significant part of our treatment modalities today,” Dr. Gordon said.
Sunburn is a risk with narrow-band UVB, especially if the dose is ramped up too quickly. Reactivation of herpes simplex virus infection is a frequent problem, and one patients find especially concerning when it manifests as eruptions of cold sores on the face.
The side effect of narrow-band UVB of greatest interest to most patients and physicians is skin cancer. “This is an extremely controversial area,” the dermatologist observed.
Unlike with PUVA, there has never been a convincing study to show that narrow-band UVB is associated with significantly increased risks of keratinocyte carcinomas or melanoma. A large Scottish study found no significantly increased risk, but a modestly increased trend for more squamous cell carcinomas. How modest? The investigators calculated that it would require 50,000 psoriasis patients with a minimum of 100 narrow-band UVB treatments to be followed for 5 years in order to demonstrate a twofold increased risk of the malignancy.
“In other words, it takes an incredible number of patients to be able to see a difference in a skin cancer that we can relatively easily treat. That’s why when I see patients, I don’t emphasize the risk of skin cancer,” Dr. Gordon said.
Similarly reassuring was a Swedish study, which showed the skin cancer rate in UVB-treated psoriasis patients was no different than in the general population.
Guideline recommendations regarding UVB phototherapy and skin cancer risk are all over the map. French guidelines advise a maximum of 230 narrow-band UVB treatments. British guidelines recommend reducing narrow-band UVB exposure to skin areas with significant sun exposure. American guidelines leave the topic untouched, Dr. Gordon noted.
He reported having no financial conflicts of interest regarding his presentation, as neither he, the Medical College of Wisconsin, or its department of dermatology receive any payment for phototherapy services he prescribes. Those payments go to the hospital system where he works. MedscapeLive and this news organization are owned by the same parent company.
Kenneth B. Gordon, MD, asserted at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
“In my practice, I’m using more and more home UVB, and there are a number of reasons for that. It’s more convenient and easier for the patient, as it’s getting more difficult for patients to give up time from work to come to the office. And I might add that, in this time of COVID-19, people don’t want to come to the office. It’s generally less expensive for patients because of copays, which increase the cost of UVB. And believe it or not, I believe it’s easier for the clinician as well. I write a prescription, the patient gets a number of treatments, and I don’t lose any sleep because I think it’s very difficult for patients to get into trouble with narrow-band UVB at home,” explained Dr. Gordon, professor and chair of the department of dermatology at the Medical College of Wisconsin, Milwaukee.
“There’s all sorts of insurance company silliness in getting this paid for, but if you do get it paid for, I think it’s a really effective way to treat psoriasis,” the dermatologist added.
A Dutch multicenter randomized trial demonstrated that home UVB phototherapy for psoriasis was equally safe and effective as outpatient UVB phototherapy, and with greater patient satisfaction.
Surveys show most dermatologists consider phototherapy their preferred treatment for patients with extensive psoriasis because its side effect profile is so benign, compared with that of systemic therapies, be they biologic agents or older drugs such as methotrexate or acitretin. Phototherapy is particularly popular for use in women of childbearing potential, since it’s a nonsystemic therapy.
And speaking of side effects, Dr. Gordon declared, “The risks of narrow-band UVB are sometimes, I believe, exaggerated.” Indeed, he considers the No. 1 side effect of office-based phototherapy to be the loss of productive time.
“Simply put, phototherapy in the office is very easy for me. I write a prescription, the tech takes care of it, and if there’s a problem I’m handy to see the patient. But for the patient, it’s very difficult. Whereas it might take only a few minutes to get the treatment in-office, it takes a lot of time to get to the office, and many patients don’t have transportation. So I think the loss of productive time with phototherapy has to be considered a side effect,” Dr. Gordon said.
Turning to the therapy’s other side effects, he said that although there is some degree of photoaging associated with narrow-band UVB – which is far and away the most commonly used form of phototherapy in the United States – it’s nothing close to the photoaging caused by PUVA.
“I don’t believe that PUVA, with all the destruction of the skin that you see with it, is a significant part of our treatment modalities today,” Dr. Gordon said.
Sunburn is a risk with narrow-band UVB, especially if the dose is ramped up too quickly. Reactivation of herpes simplex virus infection is a frequent problem, and one patients find especially concerning when it manifests as eruptions of cold sores on the face.
The side effect of narrow-band UVB of greatest interest to most patients and physicians is skin cancer. “This is an extremely controversial area,” the dermatologist observed.
Unlike with PUVA, there has never been a convincing study to show that narrow-band UVB is associated with significantly increased risks of keratinocyte carcinomas or melanoma. A large Scottish study found no significantly increased risk, but a modestly increased trend for more squamous cell carcinomas. How modest? The investigators calculated that it would require 50,000 psoriasis patients with a minimum of 100 narrow-band UVB treatments to be followed for 5 years in order to demonstrate a twofold increased risk of the malignancy.
“In other words, it takes an incredible number of patients to be able to see a difference in a skin cancer that we can relatively easily treat. That’s why when I see patients, I don’t emphasize the risk of skin cancer,” Dr. Gordon said.
Similarly reassuring was a Swedish study, which showed the skin cancer rate in UVB-treated psoriasis patients was no different than in the general population.
Guideline recommendations regarding UVB phototherapy and skin cancer risk are all over the map. French guidelines advise a maximum of 230 narrow-band UVB treatments. British guidelines recommend reducing narrow-band UVB exposure to skin areas with significant sun exposure. American guidelines leave the topic untouched, Dr. Gordon noted.
He reported having no financial conflicts of interest regarding his presentation, as neither he, the Medical College of Wisconsin, or its department of dermatology receive any payment for phototherapy services he prescribes. Those payments go to the hospital system where he works. MedscapeLive and this news organization are owned by the same parent company.
FROM MEDSCAPELIVE LAS VEGAS DERMATOLOGY SEMINAR
Tackling screen time from birth
In this day and age, a new question should make its way to the top of the list for new parents and their pediatricians: How will the family approach the issue of household technology use – for their children as well as themselves?

Even the most vigilant among them will find much available guidance conflicting – and a good deal of it may feel oblivious to their day-to-day realities. This is especially true now, as many families face the daunting demands of pandemic parenting – juggling full-time jobs with childcare and distance learning.
While the American Academy of Pediatrics still specifies daily time limits by age (difficult for many families to achieve even in “normal” times), pediatricians and other child development experts now recognize that a host of factors should inform decisions about healthy screen use. There’s no one “magic number” for acceptable daily use, as not all screen time is the same (particularly during the pandemic) – and different children will experience the effects of technology usage differently.
The quality of content the child engages with; the degree of parental involvement; and the individual cognitive and behavioral considerations of a child are just some of the factors that should be considered. Given the complexities, it’s unfortunate that many parents will not have discussed screen use with their pediatrician or even their partner before their child first reaches for that glowing screen – and certainly not before they are using technology to multitask work, social connections, and day-to-day child rearing. Screen habits form early for both children and families.
An early well-visit priority
Pediatricians have an important role to play in helping families develop a more purposeful approach to household technology by providing them with trusted, evidence-based information on screens and the potential impact on children’s development. This is not information parents should receive when a child is 2 or 3 years of age. By then, toddlers are often already avid tablet users and parents may have come to rely on technology as a parenting aid.

Parents also need to know that, from birth, they can influence their child’s brain and communication development through undistracted time for talking, reading, singing, and interacting with their baby. Though the way children develop speech and language skills has not changed, the level of distraction posed by technology has grown immensely. It is much more difficult for parents to notice a baby’s subtle communication attempts – a coo or a smile, for instance, and respond accordingly – with a smartphone in hand. Rather than admonish parents for overusing their devices, we can focus them on what supports healthy child development instead.
By talking to parents earlier, pediatricians can help prevent premature or excessive exposure to technology – as well as encourage parents to dedicate “unplugged” times of day with their baby, such as during feeding time or before bedtime. By accenting the positive – educating parents about how they can encourage development and growth, how they can bond with their baby, and how they can establish a loving and nurturing relationship – pediatricians can motivate them towards simple steps that limit technology’s disruption of daily developmental opportunities. They can also help them lay a solid foundation for healthy screen time habits in the long term for their family.
The critical period for speech and language development is early
The impact of screen use on a child’s communication development is a significant concern. The critical time window for the development of speech, language, and social skills is between birth and 3 years. What happens then is the precursor for later reading and writing skills that are key to academic and vocational success.

A meta-analysis published in JAMA Pediatrics in July 2020 found that greater quantity of screen use was associated with poorer language skills – while screen-use onset at later ages was also associated with stronger child language skills. A November 2019 JAMA Pediatrics study using brain imaging in pre–school age children found an association between increased screen-based media use, compared with the AAP guidelines, and lower integrity of white matter tracts supporting language and emergent literacy skills. That study also used several language and literacy tests, finding that children with higher screen exposure had poorer expressive language and worse language processing speed. A 2017 study of 900 children between 18 and 24 months found that every 30-minute increase in daily screen time was linked to a 49% increased risk of expressive speech delay. And on the parental side, numerous studies have documented decreased verbal and nonverbal interactions initiated by parents when they are using a device.
Be tech wise with baby
AAP offers many resources in its Media and Children Communication Toolkit that serve as helpful starting points for conversations, including its family media use plan templates. However, more resources tailored to parents of newborns and very young children are needed.
One new option recently developed by the American Speech-Language-Hearing Association (ASHA) and the Children’s Screen Time Action Network is Be Tech Wise With Baby! Aimed at prospective and new parents, this simple handout, available for free in English and Spanish, is an easy takeaway from early well-child visits that pediatricians and pediatric nurses can distribute.
Pediatricians have a vital role to play championing healthy, balanced screen time use for children and adults – starting from baby’s first moments of life. By guiding new parents towards simple steps, such as carving out tech-free times of day and delaying introduction of screens, they can positively influence the screen-time habits of the next generation of connected kids.
Mark Bertin, MD, is a developmental pediatrician and author of numerous parenting books, including “How Children Thrive: The Practical Science of Raising Independent, Resilient, and Happy Kids” and “Mindful Parenting for ADHD.” Diane Paul, PhD, CCC-SLP, is the director of clinical issues in speech-language pathology for the American Speech-Language-Hearing Association (ASHA) and author of numerous books, including “Talking on the Go: Everyday Activities to Enhance Speech and Language Development.” Email them at [email protected].
In this day and age, a new question should make its way to the top of the list for new parents and their pediatricians: How will the family approach the issue of household technology use – for their children as well as themselves?
Even the most vigilant among them will find much available guidance conflicting – and a good deal of it may feel oblivious to their day-to-day realities. This is especially true now, as many families face the daunting demands of pandemic parenting – juggling full-time jobs with childcare and distance learning.
While the American Academy of Pediatrics still specifies daily time limits by age (difficult for many families to achieve even in “normal” times), pediatricians and other child development experts now recognize that a host of factors should inform decisions about healthy screen use. There’s no one “magic number” for acceptable daily use, as not all screen time is the same (particularly during the pandemic) – and different children will experience the effects of technology usage differently.
The quality of content the child engages with; the degree of parental involvement; and the individual cognitive and behavioral considerations of a child are just some of the factors that should be considered. Given the complexities, it’s unfortunate that many parents will not have discussed screen use with their pediatrician or even their partner before their child first reaches for that glowing screen – and certainly not before they are using technology to multitask work, social connections, and day-to-day child rearing. Screen habits form early for both children and families.
An early well-visit priority
Pediatricians have an important role to play in helping families develop a more purposeful approach to household technology by providing them with trusted, evidence-based information on screens and the potential impact on children’s development. This is not information parents should receive when a child is 2 or 3 years of age. By then, toddlers are often already avid tablet users and parents may have come to rely on technology as a parenting aid.
Parents also need to know that, from birth, they can influence their child’s brain and communication development through undistracted time for talking, reading, singing, and interacting with their baby. Though the way children develop speech and language skills has not changed, the level of distraction posed by technology has grown immensely. It is much more difficult for parents to notice a baby’s subtle communication attempts – a coo or a smile, for instance, and respond accordingly – with a smartphone in hand. Rather than admonish parents for overusing their devices, we can focus them on what supports healthy child development instead.
By talking to parents earlier, pediatricians can help prevent premature or excessive exposure to technology – as well as encourage parents to dedicate “unplugged” times of day with their baby, such as during feeding time or before bedtime. By accenting the positive – educating parents about how they can encourage development and growth, how they can bond with their baby, and how they can establish a loving and nurturing relationship – pediatricians can motivate them towards simple steps that limit technology’s disruption of daily developmental opportunities. They can also help them lay a solid foundation for healthy screen time habits in the long term for their family.
The critical period for speech and language development is early
The impact of screen use on a child’s communication development is a significant concern. The critical time window for the development of speech, language, and social skills is between birth and 3 years. What happens then is the precursor for later reading and writing skills that are key to academic and vocational success.
A meta-analysis published in JAMA Pediatrics in July 2020 found that greater quantity of screen use was associated with poorer language skills – while screen-use onset at later ages was also associated with stronger child language skills. A November 2019 JAMA Pediatrics study using brain imaging in pre–school age children found an association between increased screen-based media use, compared with the AAP guidelines, and lower integrity of white matter tracts supporting language and emergent literacy skills. That study also used several language and literacy tests, finding that children with higher screen exposure had poorer expressive language and worse language processing speed. A 2017 study of 900 children between 18 and 24 months found that every 30-minute increase in daily screen time was linked to a 49% increased risk of expressive speech delay. And on the parental side, numerous studies have documented decreased verbal and nonverbal interactions initiated by parents when they are using a device.
Be tech wise with baby
AAP offers many resources in its Media and Children Communication Toolkit that serve as helpful starting points for conversations, including its family media use plan templates. However, more resources tailored to parents of newborns and very young children are needed.
One new option recently developed by the American Speech-Language-Hearing Association (ASHA) and the Children’s Screen Time Action Network is Be Tech Wise With Baby! Aimed at prospective and new parents, this simple handout, available for free in English and Spanish, is an easy takeaway from early well-child visits that pediatricians and pediatric nurses can distribute.
Pediatricians have a vital role to play championing healthy, balanced screen time use for children and adults – starting from baby’s first moments of life. By guiding new parents towards simple steps, such as carving out tech-free times of day and delaying introduction of screens, they can positively influence the screen-time habits of the next generation of connected kids.
Mark Bertin, MD, is a developmental pediatrician and author of numerous parenting books, including “How Children Thrive: The Practical Science of Raising Independent, Resilient, and Happy Kids” and “Mindful Parenting for ADHD.” Diane Paul, PhD, CCC-SLP, is the director of clinical issues in speech-language pathology for the American Speech-Language-Hearing Association (ASHA) and author of numerous books, including “Talking on the Go: Everyday Activities to Enhance Speech and Language Development.” Email them at [email protected].
In this day and age, a new question should make its way to the top of the list for new parents and their pediatricians: How will the family approach the issue of household technology use – for their children as well as themselves?
Even the most vigilant among them will find much available guidance conflicting – and a good deal of it may feel oblivious to their day-to-day realities. This is especially true now, as many families face the daunting demands of pandemic parenting – juggling full-time jobs with childcare and distance learning.
While the American Academy of Pediatrics still specifies daily time limits by age (difficult for many families to achieve even in “normal” times), pediatricians and other child development experts now recognize that a host of factors should inform decisions about healthy screen use. There’s no one “magic number” for acceptable daily use, as not all screen time is the same (particularly during the pandemic) – and different children will experience the effects of technology usage differently.
The quality of content the child engages with; the degree of parental involvement; and the individual cognitive and behavioral considerations of a child are just some of the factors that should be considered. Given the complexities, it’s unfortunate that many parents will not have discussed screen use with their pediatrician or even their partner before their child first reaches for that glowing screen – and certainly not before they are using technology to multitask work, social connections, and day-to-day child rearing. Screen habits form early for both children and families.
An early well-visit priority
Pediatricians have an important role to play in helping families develop a more purposeful approach to household technology by providing them with trusted, evidence-based information on screens and the potential impact on children’s development. This is not information parents should receive when a child is 2 or 3 years of age. By then, toddlers are often already avid tablet users and parents may have come to rely on technology as a parenting aid.
Parents also need to know that, from birth, they can influence their child’s brain and communication development through undistracted time for talking, reading, singing, and interacting with their baby. Though the way children develop speech and language skills has not changed, the level of distraction posed by technology has grown immensely. It is much more difficult for parents to notice a baby’s subtle communication attempts – a coo or a smile, for instance, and respond accordingly – with a smartphone in hand. Rather than admonish parents for overusing their devices, we can focus them on what supports healthy child development instead.
By talking to parents earlier, pediatricians can help prevent premature or excessive exposure to technology – as well as encourage parents to dedicate “unplugged” times of day with their baby, such as during feeding time or before bedtime. By accenting the positive – educating parents about how they can encourage development and growth, how they can bond with their baby, and how they can establish a loving and nurturing relationship – pediatricians can motivate them towards simple steps that limit technology’s disruption of daily developmental opportunities. They can also help them lay a solid foundation for healthy screen time habits in the long term for their family.
The critical period for speech and language development is early
The impact of screen use on a child’s communication development is a significant concern. The critical time window for the development of speech, language, and social skills is between birth and 3 years. What happens then is the precursor for later reading and writing skills that are key to academic and vocational success.
A meta-analysis published in JAMA Pediatrics in July 2020 found that greater quantity of screen use was associated with poorer language skills – while screen-use onset at later ages was also associated with stronger child language skills. A November 2019 JAMA Pediatrics study using brain imaging in pre–school age children found an association between increased screen-based media use, compared with the AAP guidelines, and lower integrity of white matter tracts supporting language and emergent literacy skills. That study also used several language and literacy tests, finding that children with higher screen exposure had poorer expressive language and worse language processing speed. A 2017 study of 900 children between 18 and 24 months found that every 30-minute increase in daily screen time was linked to a 49% increased risk of expressive speech delay. And on the parental side, numerous studies have documented decreased verbal and nonverbal interactions initiated by parents when they are using a device.
Be tech wise with baby
AAP offers many resources in its Media and Children Communication Toolkit that serve as helpful starting points for conversations, including its family media use plan templates. However, more resources tailored to parents of newborns and very young children are needed.
One new option recently developed by the American Speech-Language-Hearing Association (ASHA) and the Children’s Screen Time Action Network is Be Tech Wise With Baby! Aimed at prospective and new parents, this simple handout, available for free in English and Spanish, is an easy takeaway from early well-child visits that pediatricians and pediatric nurses can distribute.
Pediatricians have a vital role to play championing healthy, balanced screen time use for children and adults – starting from baby’s first moments of life. By guiding new parents towards simple steps, such as carving out tech-free times of day and delaying introduction of screens, they can positively influence the screen-time habits of the next generation of connected kids.
Mark Bertin, MD, is a developmental pediatrician and author of numerous parenting books, including “How Children Thrive: The Practical Science of Raising Independent, Resilient, and Happy Kids” and “Mindful Parenting for ADHD.” Diane Paul, PhD, CCC-SLP, is the director of clinical issues in speech-language pathology for the American Speech-Language-Hearing Association (ASHA) and author of numerous books, including “Talking on the Go: Everyday Activities to Enhance Speech and Language Development.” Email them at [email protected].
On being an elite
Regardless of who received the most electoral votes it is pretty clear that each candidate has millions of supporters, and that they are separated by only a few percentage points. I guess one could argue that so many people being able to express their opinions is healthy. However, from my side of the divide I have difficulty understanding how so many of my fellow citizens could arrive at an opinion so diametrically opposed to my own.

Since the 2016 election I have tried to read as many articles as I could find in search of an explanation for that outcome and continuing partisan support. I have never had much interest in political science because it always sounded like an oxymoron. But I am willing to listen to anyone who claims to understand how so many other citizens can see the world so differently from the way I do. It simply may be that for whatever reason one person, in this case one man, has such charismatic power that his supporters willingly abandon the moral skeleton on which their lives had been draped. Or is this us versus them primarily a chasm between the elites and the nonelites?
I don’t know much about you but the fact that you are reading this column means that, like me, you are an elite. Even if you are a woman of color and the daughter of immigrants you have taken advantage of what opportunities you have been offered, stayed in school long enough to adopt a reverence for the scientific method, and have a job that pays well because you have acquired some expertise.
Tom Nichols, a political scientist teaching at Harvard Extension School, says that “expertise is a very exclusionary idea because it’s supposed to be: Not everybody gets a vote on how to fly the plane” (Why isn’t the right more afraid of COVID-19? by Christina Pazzanese, Harvard Gazette, Oct 30, 2020) This exclusivity may in part explain the cultural trend that has eroded faith in experts in general, but particularly around issues such as climate change. Ironically, although science continues to be held in esteem in our culture, many scientists have become targets for those citizens who wish to attack authority figures.
How is it that you and I as pediatricians have avoided those attacks and the derogatory label as “so-called experts”?
You may live and practice in a community where many of your patients’ families don’t share your political views. But you have probably been successful at maintaining a trusting relationship with them in large part because you have cast yourself in the role of an adviser and not a dictator. And, although at times it has been difficult, you have been careful to avoid sharing your advice in a manner that sounds condescending. You have succeeded in functioning as an expert while carefully disguising yourself as a nonelite.
However, you are skating on thin ice if you venture into topics that run counter to your patients’ religious beliefs. Theda Skocpol, professor of government and psychology at Harvard University, Cambridge, Mass., has observed that studies have shown that while religious conservatives are aware of the science and don’t reject the finding, “they resent the use of experts as political authorities.” This may explain why all across this diverse country, our patients are eager for and accepting of our advice on all manners of health-related issues until we step into a swampy area that threatens their political views – such as vaccination or gun control.
With one misstep in the wrong direction, you can go from being a compassionate adviser to an elitist “so-called expert.”
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Regardless of who received the most electoral votes it is pretty clear that each candidate has millions of supporters, and that they are separated by only a few percentage points. I guess one could argue that so many people being able to express their opinions is healthy. However, from my side of the divide I have difficulty understanding how so many of my fellow citizens could arrive at an opinion so diametrically opposed to my own.
Since the 2016 election I have tried to read as many articles as I could find in search of an explanation for that outcome and continuing partisan support. I have never had much interest in political science because it always sounded like an oxymoron. But I am willing to listen to anyone who claims to understand how so many other citizens can see the world so differently from the way I do. It simply may be that for whatever reason one person, in this case one man, has such charismatic power that his supporters willingly abandon the moral skeleton on which their lives had been draped. Or is this us versus them primarily a chasm between the elites and the nonelites?
I don’t know much about you but the fact that you are reading this column means that, like me, you are an elite. Even if you are a woman of color and the daughter of immigrants you have taken advantage of what opportunities you have been offered, stayed in school long enough to adopt a reverence for the scientific method, and have a job that pays well because you have acquired some expertise.
Tom Nichols, a political scientist teaching at Harvard Extension School, says that “expertise is a very exclusionary idea because it’s supposed to be: Not everybody gets a vote on how to fly the plane” (Why isn’t the right more afraid of COVID-19? by Christina Pazzanese, Harvard Gazette, Oct 30, 2020) This exclusivity may in part explain the cultural trend that has eroded faith in experts in general, but particularly around issues such as climate change. Ironically, although science continues to be held in esteem in our culture, many scientists have become targets for those citizens who wish to attack authority figures.
How is it that you and I as pediatricians have avoided those attacks and the derogatory label as “so-called experts”?
You may live and practice in a community where many of your patients’ families don’t share your political views. But you have probably been successful at maintaining a trusting relationship with them in large part because you have cast yourself in the role of an adviser and not a dictator. And, although at times it has been difficult, you have been careful to avoid sharing your advice in a manner that sounds condescending. You have succeeded in functioning as an expert while carefully disguising yourself as a nonelite.
However, you are skating on thin ice if you venture into topics that run counter to your patients’ religious beliefs. Theda Skocpol, professor of government and psychology at Harvard University, Cambridge, Mass., has observed that studies have shown that while religious conservatives are aware of the science and don’t reject the finding, “they resent the use of experts as political authorities.” This may explain why all across this diverse country, our patients are eager for and accepting of our advice on all manners of health-related issues until we step into a swampy area that threatens their political views – such as vaccination or gun control.
With one misstep in the wrong direction, you can go from being a compassionate adviser to an elitist “so-called expert.”
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Regardless of who received the most electoral votes it is pretty clear that each candidate has millions of supporters, and that they are separated by only a few percentage points. I guess one could argue that so many people being able to express their opinions is healthy. However, from my side of the divide I have difficulty understanding how so many of my fellow citizens could arrive at an opinion so diametrically opposed to my own.
Since the 2016 election I have tried to read as many articles as I could find in search of an explanation for that outcome and continuing partisan support. I have never had much interest in political science because it always sounded like an oxymoron. But I am willing to listen to anyone who claims to understand how so many other citizens can see the world so differently from the way I do. It simply may be that for whatever reason one person, in this case one man, has such charismatic power that his supporters willingly abandon the moral skeleton on which their lives had been draped. Or is this us versus them primarily a chasm between the elites and the nonelites?
I don’t know much about you but the fact that you are reading this column means that, like me, you are an elite. Even if you are a woman of color and the daughter of immigrants you have taken advantage of what opportunities you have been offered, stayed in school long enough to adopt a reverence for the scientific method, and have a job that pays well because you have acquired some expertise.
Tom Nichols, a political scientist teaching at Harvard Extension School, says that “expertise is a very exclusionary idea because it’s supposed to be: Not everybody gets a vote on how to fly the plane” (Why isn’t the right more afraid of COVID-19? by Christina Pazzanese, Harvard Gazette, Oct 30, 2020) This exclusivity may in part explain the cultural trend that has eroded faith in experts in general, but particularly around issues such as climate change. Ironically, although science continues to be held in esteem in our culture, many scientists have become targets for those citizens who wish to attack authority figures.
How is it that you and I as pediatricians have avoided those attacks and the derogatory label as “so-called experts”?
You may live and practice in a community where many of your patients’ families don’t share your political views. But you have probably been successful at maintaining a trusting relationship with them in large part because you have cast yourself in the role of an adviser and not a dictator. And, although at times it has been difficult, you have been careful to avoid sharing your advice in a manner that sounds condescending. You have succeeded in functioning as an expert while carefully disguising yourself as a nonelite.
However, you are skating on thin ice if you venture into topics that run counter to your patients’ religious beliefs. Theda Skocpol, professor of government and psychology at Harvard University, Cambridge, Mass., has observed that studies have shown that while religious conservatives are aware of the science and don’t reject the finding, “they resent the use of experts as political authorities.” This may explain why all across this diverse country, our patients are eager for and accepting of our advice on all manners of health-related issues until we step into a swampy area that threatens their political views – such as vaccination or gun control.
With one misstep in the wrong direction, you can go from being a compassionate adviser to an elitist “so-called expert.”
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].