User login
Guidelines vary on what age to begin screening for cervical cancer. What age do you typically recommend for patients?
[polldaddy:11135441]
[polldaddy:11135441]
[polldaddy:11135441]
Alcohol, degraded sleep related in young adults
CHARLOTTE, N.C. – Sleep and alcohol consumption in young adults seems to follow a “vicious cycle,” as one observer called it. and those who went to bed earlier and slept longer tended to drink less the next day, a study of drinking and sleeping habits in 21- to 29-year-olds found.
“Sleep is a potential factor that we could intervene on to really identify how to improve drinking behaviors among young adults,” David Reichenberger, a graduate student at Penn State University, University Park, said in an interview after he presented his findings at the annual meeting of the Associated Professional Sleep Societies.

This is one of the few studies of alcohol consumption and sleep patterns that used an objective measure of alcohol consumption, Mr. Reichenberger said. The study evaluated sleep and alcohol consumption patterns in 222 regularly drinking young adults over 6 consecutive days. Study participants completed morning smartphone-based questionnaires, reporting their previous night’s bedtime, sleep duration, sleep quality, and number of drinks consumed. They also wore an alcohol monitor that continuously measured their transdermal alcohol consumption (TAC).
The study analyzed the data using two sets of multilevel models: A linear model that looked at how each drinking predictor was associated with each sleep variable and a Poisson model to determine how sleep predicted next-day alcohol use.
“We found that higher average peak TAC – that is, how intoxicated they got – was associated with a 19-minute later bedtime among young adults,” Mr. Reichenberger said. “Later bedtimes were then associated with a 26% greater TAC among those adults” (P < .02).
Patterns of alcohol consumption and sleep
On days when participants recorded a higher peak TAC, bedtime was delayed, sleep duration was shorter, and subjective sleep quality was worse, he said. However, none of the sleep variables predicted next-day peak TAC.
“We found an association between the duration of the drinking episode and later bedtimes among young adults,” he added. “And on days when the drinking episodes were longer, subsequent sleep was delayed and sleep quality was worse. But we also found that after nights when they had a later bedtime, next-day drinking episodes were about 7% longer.”
Conversely, young adults who had earlier bedtimes and longer sleep durations tended to consume fewer drinks and they achieved lower intoxication levels the next day, Mr. Reichenberger said.
Between-person results showed that young adults who tended to go to bed later drank on average 24% more the next day (P < .01). Also, each extra hour of sleep was associated with a 14% decrease in drinking the next day (P < .03).
Participants who drank more went to bed on average 12-19 minutes later (P < .01) and slept 5 fewer minutes (P < .01). Within-person results showed that on nights when participants drank more than usual they went to bed 8-13 minutes later (P < .01), slept 2-4 fewer minutes (P < .03), and had worse sleep quality (P < .01).
Mr. Reichenberger acknowledged one limitation of the study: Measuring sleep and alcohol consumption patterns over 6 days might not be long enough. Future studies should address that.
A ‘vicious cycle’
Hans P.A. Van Dongen, PhD, director of the Sleep and Performance Research Center at Washington State University, Spokane, said in an interview that the findings imply a “vicious cycle” between sleep and alcohol consumption. “You create a problem and then it perpetuates itself or reinforces itself.”

In older adults, alcohol tends to act as a “sleep aid,” Dr. Van Dongen noted. “Then it disrupts their sleep later on and then the next night they need to use the sleep aid again because they had a really poor night and they’re tired and they want to fall asleep.”
He added: “I think what is new here is that’s not very likely the mechanism that they’re using alcohol as a sleep aid in younger adults that we see in older adults, so I think there is a new element to it. Now does anybody know how that works exactly? No, that’s the next thing.”
The Penn State study identifies “a signal there that needs to be followed up on,” Dr. Van Dongen said. “There’s something nature’s trying to tell us but it’s not exactly clear what it’s trying to tell us.”
The National Institute on Drug Abuse provided funding for the study. Mr. Reichenberger has no relevant disclosures. Dr. Van Dongen has no disclosures to report.
CHARLOTTE, N.C. – Sleep and alcohol consumption in young adults seems to follow a “vicious cycle,” as one observer called it. and those who went to bed earlier and slept longer tended to drink less the next day, a study of drinking and sleeping habits in 21- to 29-year-olds found.
“Sleep is a potential factor that we could intervene on to really identify how to improve drinking behaviors among young adults,” David Reichenberger, a graduate student at Penn State University, University Park, said in an interview after he presented his findings at the annual meeting of the Associated Professional Sleep Societies.
This is one of the few studies of alcohol consumption and sleep patterns that used an objective measure of alcohol consumption, Mr. Reichenberger said. The study evaluated sleep and alcohol consumption patterns in 222 regularly drinking young adults over 6 consecutive days. Study participants completed morning smartphone-based questionnaires, reporting their previous night’s bedtime, sleep duration, sleep quality, and number of drinks consumed. They also wore an alcohol monitor that continuously measured their transdermal alcohol consumption (TAC).
The study analyzed the data using two sets of multilevel models: A linear model that looked at how each drinking predictor was associated with each sleep variable and a Poisson model to determine how sleep predicted next-day alcohol use.
“We found that higher average peak TAC – that is, how intoxicated they got – was associated with a 19-minute later bedtime among young adults,” Mr. Reichenberger said. “Later bedtimes were then associated with a 26% greater TAC among those adults” (P < .02).
Patterns of alcohol consumption and sleep
On days when participants recorded a higher peak TAC, bedtime was delayed, sleep duration was shorter, and subjective sleep quality was worse, he said. However, none of the sleep variables predicted next-day peak TAC.
“We found an association between the duration of the drinking episode and later bedtimes among young adults,” he added. “And on days when the drinking episodes were longer, subsequent sleep was delayed and sleep quality was worse. But we also found that after nights when they had a later bedtime, next-day drinking episodes were about 7% longer.”
Conversely, young adults who had earlier bedtimes and longer sleep durations tended to consume fewer drinks and they achieved lower intoxication levels the next day, Mr. Reichenberger said.
Between-person results showed that young adults who tended to go to bed later drank on average 24% more the next day (P < .01). Also, each extra hour of sleep was associated with a 14% decrease in drinking the next day (P < .03).
Participants who drank more went to bed on average 12-19 minutes later (P < .01) and slept 5 fewer minutes (P < .01). Within-person results showed that on nights when participants drank more than usual they went to bed 8-13 minutes later (P < .01), slept 2-4 fewer minutes (P < .03), and had worse sleep quality (P < .01).
Mr. Reichenberger acknowledged one limitation of the study: Measuring sleep and alcohol consumption patterns over 6 days might not be long enough. Future studies should address that.
A ‘vicious cycle’
Hans P.A. Van Dongen, PhD, director of the Sleep and Performance Research Center at Washington State University, Spokane, said in an interview that the findings imply a “vicious cycle” between sleep and alcohol consumption. “You create a problem and then it perpetuates itself or reinforces itself.”
In older adults, alcohol tends to act as a “sleep aid,” Dr. Van Dongen noted. “Then it disrupts their sleep later on and then the next night they need to use the sleep aid again because they had a really poor night and they’re tired and they want to fall asleep.”
He added: “I think what is new here is that’s not very likely the mechanism that they’re using alcohol as a sleep aid in younger adults that we see in older adults, so I think there is a new element to it. Now does anybody know how that works exactly? No, that’s the next thing.”
The Penn State study identifies “a signal there that needs to be followed up on,” Dr. Van Dongen said. “There’s something nature’s trying to tell us but it’s not exactly clear what it’s trying to tell us.”
The National Institute on Drug Abuse provided funding for the study. Mr. Reichenberger has no relevant disclosures. Dr. Van Dongen has no disclosures to report.
CHARLOTTE, N.C. – Sleep and alcohol consumption in young adults seems to follow a “vicious cycle,” as one observer called it. and those who went to bed earlier and slept longer tended to drink less the next day, a study of drinking and sleeping habits in 21- to 29-year-olds found.
“Sleep is a potential factor that we could intervene on to really identify how to improve drinking behaviors among young adults,” David Reichenberger, a graduate student at Penn State University, University Park, said in an interview after he presented his findings at the annual meeting of the Associated Professional Sleep Societies.
This is one of the few studies of alcohol consumption and sleep patterns that used an objective measure of alcohol consumption, Mr. Reichenberger said. The study evaluated sleep and alcohol consumption patterns in 222 regularly drinking young adults over 6 consecutive days. Study participants completed morning smartphone-based questionnaires, reporting their previous night’s bedtime, sleep duration, sleep quality, and number of drinks consumed. They also wore an alcohol monitor that continuously measured their transdermal alcohol consumption (TAC).
The study analyzed the data using two sets of multilevel models: A linear model that looked at how each drinking predictor was associated with each sleep variable and a Poisson model to determine how sleep predicted next-day alcohol use.
“We found that higher average peak TAC – that is, how intoxicated they got – was associated with a 19-minute later bedtime among young adults,” Mr. Reichenberger said. “Later bedtimes were then associated with a 26% greater TAC among those adults” (P < .02).
Patterns of alcohol consumption and sleep
On days when participants recorded a higher peak TAC, bedtime was delayed, sleep duration was shorter, and subjective sleep quality was worse, he said. However, none of the sleep variables predicted next-day peak TAC.
“We found an association between the duration of the drinking episode and later bedtimes among young adults,” he added. “And on days when the drinking episodes were longer, subsequent sleep was delayed and sleep quality was worse. But we also found that after nights when they had a later bedtime, next-day drinking episodes were about 7% longer.”
Conversely, young adults who had earlier bedtimes and longer sleep durations tended to consume fewer drinks and they achieved lower intoxication levels the next day, Mr. Reichenberger said.
Between-person results showed that young adults who tended to go to bed later drank on average 24% more the next day (P < .01). Also, each extra hour of sleep was associated with a 14% decrease in drinking the next day (P < .03).
Participants who drank more went to bed on average 12-19 minutes later (P < .01) and slept 5 fewer minutes (P < .01). Within-person results showed that on nights when participants drank more than usual they went to bed 8-13 minutes later (P < .01), slept 2-4 fewer minutes (P < .03), and had worse sleep quality (P < .01).
Mr. Reichenberger acknowledged one limitation of the study: Measuring sleep and alcohol consumption patterns over 6 days might not be long enough. Future studies should address that.
A ‘vicious cycle’
Hans P.A. Van Dongen, PhD, director of the Sleep and Performance Research Center at Washington State University, Spokane, said in an interview that the findings imply a “vicious cycle” between sleep and alcohol consumption. “You create a problem and then it perpetuates itself or reinforces itself.”
In older adults, alcohol tends to act as a “sleep aid,” Dr. Van Dongen noted. “Then it disrupts their sleep later on and then the next night they need to use the sleep aid again because they had a really poor night and they’re tired and they want to fall asleep.”
He added: “I think what is new here is that’s not very likely the mechanism that they’re using alcohol as a sleep aid in younger adults that we see in older adults, so I think there is a new element to it. Now does anybody know how that works exactly? No, that’s the next thing.”
The Penn State study identifies “a signal there that needs to be followed up on,” Dr. Van Dongen said. “There’s something nature’s trying to tell us but it’s not exactly clear what it’s trying to tell us.”
The National Institute on Drug Abuse provided funding for the study. Mr. Reichenberger has no relevant disclosures. Dr. Van Dongen has no disclosures to report.
At SLEEP 2022
Insurer told to pay $5.2 million to woman who caught STD in a car
A Missouri lawsuit adds a new twist to the kind of “bodily harm” in a car that’s covered by insurance.
On June 7,
The woman, identified in court documents as M.O., said she contracted human papillomavirus from her boyfriend. She said he knew he had the disease but didn’t tell her.
An arbitrator found in May 2021 that the in-car sex had “directly caused, or directly contributed to cause” the STD transmission. The man was found liable. The woman was awarded $5.2 million to be paid by GEICO, which insured the man’s vehicle.
GEICO filed for the award to be overturned, alleging it had been denied due process and that the arbitration deal was unenforceable.
Court documents show that GEICO claimed the man’s policy covered only injuries that came “out of the ownership, maintenance or use of the ... auto” and that the woman’s “injuries arose from an intervening cause – namely, her failure to prevent transmission of STDs by having unprotected sex.”
The state appellate panel ruled that the lower court made no mistake in the case and upheld the decision.
The Kansas City Star reported that one of the judges concurred but said GEICO was offered “no meaningful opportunity to participate” in the lawsuit and existing law “relegat(es) the insurer to the status of a bystander.”
“This case presents novel and potentially important issues about whether an insurance carrier can be held liable under such policies for the consequences of two adults voluntarily having unprotected sex in the insured’s automobile,” noted U.S. Magistrate Judge Angel D. Mitchell in court documents. “Interpretation of these policies could have far-reaching implications for other policies with similar terms.”
A version of this article first appeared on WebMD.com.
A Missouri lawsuit adds a new twist to the kind of “bodily harm” in a car that’s covered by insurance.
On June 7,
The woman, identified in court documents as M.O., said she contracted human papillomavirus from her boyfriend. She said he knew he had the disease but didn’t tell her.
An arbitrator found in May 2021 that the in-car sex had “directly caused, or directly contributed to cause” the STD transmission. The man was found liable. The woman was awarded $5.2 million to be paid by GEICO, which insured the man’s vehicle.
GEICO filed for the award to be overturned, alleging it had been denied due process and that the arbitration deal was unenforceable.
Court documents show that GEICO claimed the man’s policy covered only injuries that came “out of the ownership, maintenance or use of the ... auto” and that the woman’s “injuries arose from an intervening cause – namely, her failure to prevent transmission of STDs by having unprotected sex.”
The state appellate panel ruled that the lower court made no mistake in the case and upheld the decision.
The Kansas City Star reported that one of the judges concurred but said GEICO was offered “no meaningful opportunity to participate” in the lawsuit and existing law “relegat(es) the insurer to the status of a bystander.”
“This case presents novel and potentially important issues about whether an insurance carrier can be held liable under such policies for the consequences of two adults voluntarily having unprotected sex in the insured’s automobile,” noted U.S. Magistrate Judge Angel D. Mitchell in court documents. “Interpretation of these policies could have far-reaching implications for other policies with similar terms.”
A version of this article first appeared on WebMD.com.
A Missouri lawsuit adds a new twist to the kind of “bodily harm” in a car that’s covered by insurance.
On June 7,
The woman, identified in court documents as M.O., said she contracted human papillomavirus from her boyfriend. She said he knew he had the disease but didn’t tell her.
An arbitrator found in May 2021 that the in-car sex had “directly caused, or directly contributed to cause” the STD transmission. The man was found liable. The woman was awarded $5.2 million to be paid by GEICO, which insured the man’s vehicle.
GEICO filed for the award to be overturned, alleging it had been denied due process and that the arbitration deal was unenforceable.
Court documents show that GEICO claimed the man’s policy covered only injuries that came “out of the ownership, maintenance or use of the ... auto” and that the woman’s “injuries arose from an intervening cause – namely, her failure to prevent transmission of STDs by having unprotected sex.”
The state appellate panel ruled that the lower court made no mistake in the case and upheld the decision.
The Kansas City Star reported that one of the judges concurred but said GEICO was offered “no meaningful opportunity to participate” in the lawsuit and existing law “relegat(es) the insurer to the status of a bystander.”
“This case presents novel and potentially important issues about whether an insurance carrier can be held liable under such policies for the consequences of two adults voluntarily having unprotected sex in the insured’s automobile,” noted U.S. Magistrate Judge Angel D. Mitchell in court documents. “Interpretation of these policies could have far-reaching implications for other policies with similar terms.”
A version of this article first appeared on WebMD.com.
Harmony pulmonary valve update: Regurgitation resolved 1 year out
The 1-year results of the Harmony transcatheter pulmonary valve to treat severe pulmonary regurgitation have shown a high rate of eliminating or reducing the degree of symptoms as well as freedom from endocarditis, sustained ventricular tachycardia, and the need for further interventions.
“Simply put, the good news is no endocarditis,” said Daniel S. Levi, MD, in presenting results from three different studies with 108 patients who received three different iterations of the device at the Society for Cardiovascular Angiography & Interventions annual scientific sessions.
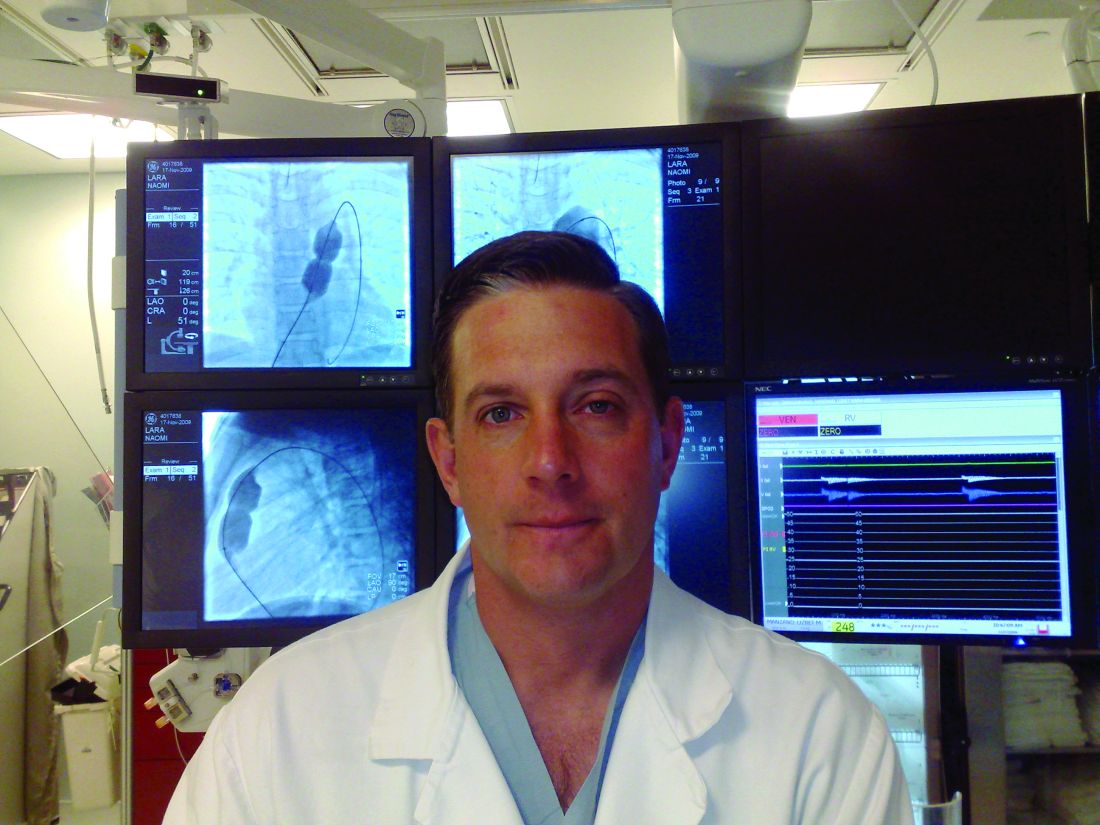
“Endocarditis has been an issue for us in the pulmonary position; we have yet to have an endocarditis in these patients in 1 year,” he stressed.
The studies evaluated three different versions of the Harmony valve: TPV22 (42 patients), the first version with a 22-mm diameter; the Clinical TPV25 (17 patients), the first iteration of a 25 mm–wide device that has since been discontinued; and the modified TPV25 (45 patients), the second version of the 25-mm valve. The three studies are the early feasibility study of the TPV22, the continued-access study of the TPV22 and the mTPV25, and the pivotal study that included all three versions.
At baseline, 89% of patients had severe and 11% had moderate pulmonary regurgitation (PR). At 1 year, 92% had none or trace PR, 3% had mild PR, and 4% moderate disease.
Dr. Levi said the device “speaks for itself” in the results he presented. They include no deaths, no heart attacks, and no pulmonary thromboembolism. Other key outcomes include:
- One major stent fracture in one of the early feasibility study patients at 1-month follow-up.
- Four explants, with two in the discontinued cTPV25 and two with the TPV22 in the early-feasibility study.
- Four reinterventions, two with the discontinued cTPV25 and two valve-in-valve procedures with the mTPV25 in the continued-access study, one with stent placement in the right ventricular outflow tract.
Dr. Levi and coinvestigators also performed a breakdown of 1-year outcomes – freedom from PR, stenosis, and interventions – by device: 95.1% for TPV22; 89.7% for mTPV25; and 73.3% for the discontinued cTPV25.
Although the valve is indicated for adolescents and adults, most of the patients in the three studies were adults, with an average weight of 165 pounds (75 kg) who have had PR for decades, said Dr. Levi, an interventional pediatric cardiologist at the University of California, Los Angeles. “With a device like this we are hopefully shifting to treating that a little bit earlier, but fortunately we don’t usually need to treat it before puberty.” The 25-mm TPV gives “a really nice landing zone” for future valve placement. “The goal is to keep patients out of the operating room for at least a few decades if not their whole lives,” he said.
Dr. Levi said the Harmony investigators will follow outcomes with the 22- and modified 25-mm Harmony valves, both of which remain commercially available, out to 10 years.

The study represents the first collective cohort evaluating the Harmony device across the early feasibility, continued access and pivotal studies, said Brian Morray, MD. “It’s important that people understand that evolution and how that impacts the way we look at outcomes, because when you aggregate the data, particularly for the TPV25, some of the procedural outcomes and the adverse events are no longer really reflective in the current time frame.”
These Harmony results “represent another big step in the evolution of interventional cardiology and will be up there with development of the Melody valve and the utility and the use of the Sapien valve in the pulmonary position,” said Dr. Morray, an associate professor of pediatrics at the University of Washington, Seattle, and an interventional cardiologist at Seattle Children’s Hospital.
Dr. Levi disclosed he is a consultant to Medtronic and Edwards Lifesciences. Dr. Morray disclosed he is a clinical proctor for Abbott and a consultant to Medtronic, but not for the Harmony device.
The 1-year results of the Harmony transcatheter pulmonary valve to treat severe pulmonary regurgitation have shown a high rate of eliminating or reducing the degree of symptoms as well as freedom from endocarditis, sustained ventricular tachycardia, and the need for further interventions.
“Simply put, the good news is no endocarditis,” said Daniel S. Levi, MD, in presenting results from three different studies with 108 patients who received three different iterations of the device at the Society for Cardiovascular Angiography & Interventions annual scientific sessions.
“Endocarditis has been an issue for us in the pulmonary position; we have yet to have an endocarditis in these patients in 1 year,” he stressed.
The studies evaluated three different versions of the Harmony valve: TPV22 (42 patients), the first version with a 22-mm diameter; the Clinical TPV25 (17 patients), the first iteration of a 25 mm–wide device that has since been discontinued; and the modified TPV25 (45 patients), the second version of the 25-mm valve. The three studies are the early feasibility study of the TPV22, the continued-access study of the TPV22 and the mTPV25, and the pivotal study that included all three versions.
At baseline, 89% of patients had severe and 11% had moderate pulmonary regurgitation (PR). At 1 year, 92% had none or trace PR, 3% had mild PR, and 4% moderate disease.
Dr. Levi said the device “speaks for itself” in the results he presented. They include no deaths, no heart attacks, and no pulmonary thromboembolism. Other key outcomes include:
- One major stent fracture in one of the early feasibility study patients at 1-month follow-up.
- Four explants, with two in the discontinued cTPV25 and two with the TPV22 in the early-feasibility study.
- Four reinterventions, two with the discontinued cTPV25 and two valve-in-valve procedures with the mTPV25 in the continued-access study, one with stent placement in the right ventricular outflow tract.
Dr. Levi and coinvestigators also performed a breakdown of 1-year outcomes – freedom from PR, stenosis, and interventions – by device: 95.1% for TPV22; 89.7% for mTPV25; and 73.3% for the discontinued cTPV25.
Although the valve is indicated for adolescents and adults, most of the patients in the three studies were adults, with an average weight of 165 pounds (75 kg) who have had PR for decades, said Dr. Levi, an interventional pediatric cardiologist at the University of California, Los Angeles. “With a device like this we are hopefully shifting to treating that a little bit earlier, but fortunately we don’t usually need to treat it before puberty.” The 25-mm TPV gives “a really nice landing zone” for future valve placement. “The goal is to keep patients out of the operating room for at least a few decades if not their whole lives,” he said.
Dr. Levi said the Harmony investigators will follow outcomes with the 22- and modified 25-mm Harmony valves, both of which remain commercially available, out to 10 years.
The study represents the first collective cohort evaluating the Harmony device across the early feasibility, continued access and pivotal studies, said Brian Morray, MD. “It’s important that people understand that evolution and how that impacts the way we look at outcomes, because when you aggregate the data, particularly for the TPV25, some of the procedural outcomes and the adverse events are no longer really reflective in the current time frame.”
These Harmony results “represent another big step in the evolution of interventional cardiology and will be up there with development of the Melody valve and the utility and the use of the Sapien valve in the pulmonary position,” said Dr. Morray, an associate professor of pediatrics at the University of Washington, Seattle, and an interventional cardiologist at Seattle Children’s Hospital.
Dr. Levi disclosed he is a consultant to Medtronic and Edwards Lifesciences. Dr. Morray disclosed he is a clinical proctor for Abbott and a consultant to Medtronic, but not for the Harmony device.
The 1-year results of the Harmony transcatheter pulmonary valve to treat severe pulmonary regurgitation have shown a high rate of eliminating or reducing the degree of symptoms as well as freedom from endocarditis, sustained ventricular tachycardia, and the need for further interventions.
“Simply put, the good news is no endocarditis,” said Daniel S. Levi, MD, in presenting results from three different studies with 108 patients who received three different iterations of the device at the Society for Cardiovascular Angiography & Interventions annual scientific sessions.
“Endocarditis has been an issue for us in the pulmonary position; we have yet to have an endocarditis in these patients in 1 year,” he stressed.
The studies evaluated three different versions of the Harmony valve: TPV22 (42 patients), the first version with a 22-mm diameter; the Clinical TPV25 (17 patients), the first iteration of a 25 mm–wide device that has since been discontinued; and the modified TPV25 (45 patients), the second version of the 25-mm valve. The three studies are the early feasibility study of the TPV22, the continued-access study of the TPV22 and the mTPV25, and the pivotal study that included all three versions.
At baseline, 89% of patients had severe and 11% had moderate pulmonary regurgitation (PR). At 1 year, 92% had none or trace PR, 3% had mild PR, and 4% moderate disease.
Dr. Levi said the device “speaks for itself” in the results he presented. They include no deaths, no heart attacks, and no pulmonary thromboembolism. Other key outcomes include:
- One major stent fracture in one of the early feasibility study patients at 1-month follow-up.
- Four explants, with two in the discontinued cTPV25 and two with the TPV22 in the early-feasibility study.
- Four reinterventions, two with the discontinued cTPV25 and two valve-in-valve procedures with the mTPV25 in the continued-access study, one with stent placement in the right ventricular outflow tract.
Dr. Levi and coinvestigators also performed a breakdown of 1-year outcomes – freedom from PR, stenosis, and interventions – by device: 95.1% for TPV22; 89.7% for mTPV25; and 73.3% for the discontinued cTPV25.
Although the valve is indicated for adolescents and adults, most of the patients in the three studies were adults, with an average weight of 165 pounds (75 kg) who have had PR for decades, said Dr. Levi, an interventional pediatric cardiologist at the University of California, Los Angeles. “With a device like this we are hopefully shifting to treating that a little bit earlier, but fortunately we don’t usually need to treat it before puberty.” The 25-mm TPV gives “a really nice landing zone” for future valve placement. “The goal is to keep patients out of the operating room for at least a few decades if not their whole lives,” he said.
Dr. Levi said the Harmony investigators will follow outcomes with the 22- and modified 25-mm Harmony valves, both of which remain commercially available, out to 10 years.
The study represents the first collective cohort evaluating the Harmony device across the early feasibility, continued access and pivotal studies, said Brian Morray, MD. “It’s important that people understand that evolution and how that impacts the way we look at outcomes, because when you aggregate the data, particularly for the TPV25, some of the procedural outcomes and the adverse events are no longer really reflective in the current time frame.”
These Harmony results “represent another big step in the evolution of interventional cardiology and will be up there with development of the Melody valve and the utility and the use of the Sapien valve in the pulmonary position,” said Dr. Morray, an associate professor of pediatrics at the University of Washington, Seattle, and an interventional cardiologist at Seattle Children’s Hospital.
Dr. Levi disclosed he is a consultant to Medtronic and Edwards Lifesciences. Dr. Morray disclosed he is a clinical proctor for Abbott and a consultant to Medtronic, but not for the Harmony device.
FROM SCAI 2022
Therapeutic patient education can help with adherence to treatment
, Andreas Wollenberg, MD, said at the Revolutionizing Atopic Dermatitis symposium.
A major goal of patient education is increasing medication adherence, noted Dr. Wollenberg, professor in the department of dermatology and allergy at Ludwig Maximilian University of Munich. Quoting former U.S. Surgeon General C. Everett Koop, MD, he said, “drugs don’t work in patients who don’t take them.”
While this is a simple message, it is important, Dr. Wollenberg said, noting that there can be a gap between a physician’s well-intentioned message and how it is interpreted by the patient. “Our messages may not be heard, not understood, not accepted, and even if they are put into place, how long will they last?” he asked. “We need to find a way [to] place sticky messages in the brains of our patients who are sitting and interacting with us.”
One way to improve treatment adherence is through patient education, such as using a written action plan or graphics; simplifying treatment regimens; minimizing treatment costs; setting up reminder programs, early follow-up visits, and short-term treatment goals; and minimizing nocebo effects. “This is more than providing just leaflets to patients. It is a complete program. It is a holistic approach. It should be structured and should be interdisciplinary, and it should contain a psychological component,” Dr. Wollenberg said.
Therapeutic patient education is recommended at baseline for children and adults with moderate to severe AD in the 2020 European Task Force on Atopic Dermatitis (ETFAD) and European Academy of Dermatology and Venereology (EADV) position paper on the diagnosis and treatment of AD in adults and children, alongside other interventions, such as emollients, bath oils, and avoidance of clinically relevant allergens, noted Dr. Wollenberg, the first author . “Therapeutic patient education is an extremely helpful tool to address patient beliefs and questions regarding disease and treatment,” he and his coauthors wrote in the paper.
When considering a therapeutic patient education program for AD, content is key, but just as important is consideration of legal and cultural conditions in the local area, Dr. Wollenberg explained. Every country will need some degree of standardization of content, he noted. Clinicians interested in adopting a patient education program need to consider who will pay for it – patients, foundations, or insurance companies – as well as the time commitment needed.
Dr. Wollenberg said that his team uses an evidence-based education program for AD in Germany that works across patients with different personalities, with a multidisciplinary team that includes a dermatologist, a specialist nurse, a nutrition expert, and a psychologist. “Sometimes we replace the specialized nurse with the dermatology resident because, in Germany, it’s difficult to find any type of specialized nurse,” although this is not an issue in many other countries, he said.
The model for children involves six 90-minute sessions, which cover topics that include emollients and basic care, food allergies and diet, medical treatment, and psychology of itch. The program for adults involves six 2-hour sessions, which cover topics that include psychology, skin care/nutrition, and medical treatment.
While this education program improves adherence in patients with AD, he acknowledged it is time consuming, and may not work for people who live far away from a clinic or who have other time commitments, making an alternative format necessary.
In terms of improving patient adherence to a doctor’s recommendations regarding chronic skin disease, “we cannot change our patients, we cannot change the disease, but we can strongly influence the treatment that we choose and how we interact as physicians with our patients,” said Dr. Wollenberg.
“Therapeutic patient education is virtually free of side effects, but evidence based. Have a look [at] it and adapt it to your own practice,” he added.
Dr. Wollenberg is a consultant, speaker and receives fees from numerous pharmaceutical companies.
, Andreas Wollenberg, MD, said at the Revolutionizing Atopic Dermatitis symposium.
A major goal of patient education is increasing medication adherence, noted Dr. Wollenberg, professor in the department of dermatology and allergy at Ludwig Maximilian University of Munich. Quoting former U.S. Surgeon General C. Everett Koop, MD, he said, “drugs don’t work in patients who don’t take them.”
While this is a simple message, it is important, Dr. Wollenberg said, noting that there can be a gap between a physician’s well-intentioned message and how it is interpreted by the patient. “Our messages may not be heard, not understood, not accepted, and even if they are put into place, how long will they last?” he asked. “We need to find a way [to] place sticky messages in the brains of our patients who are sitting and interacting with us.”
One way to improve treatment adherence is through patient education, such as using a written action plan or graphics; simplifying treatment regimens; minimizing treatment costs; setting up reminder programs, early follow-up visits, and short-term treatment goals; and minimizing nocebo effects. “This is more than providing just leaflets to patients. It is a complete program. It is a holistic approach. It should be structured and should be interdisciplinary, and it should contain a psychological component,” Dr. Wollenberg said.
Therapeutic patient education is recommended at baseline for children and adults with moderate to severe AD in the 2020 European Task Force on Atopic Dermatitis (ETFAD) and European Academy of Dermatology and Venereology (EADV) position paper on the diagnosis and treatment of AD in adults and children, alongside other interventions, such as emollients, bath oils, and avoidance of clinically relevant allergens, noted Dr. Wollenberg, the first author . “Therapeutic patient education is an extremely helpful tool to address patient beliefs and questions regarding disease and treatment,” he and his coauthors wrote in the paper.
When considering a therapeutic patient education program for AD, content is key, but just as important is consideration of legal and cultural conditions in the local area, Dr. Wollenberg explained. Every country will need some degree of standardization of content, he noted. Clinicians interested in adopting a patient education program need to consider who will pay for it – patients, foundations, or insurance companies – as well as the time commitment needed.
Dr. Wollenberg said that his team uses an evidence-based education program for AD in Germany that works across patients with different personalities, with a multidisciplinary team that includes a dermatologist, a specialist nurse, a nutrition expert, and a psychologist. “Sometimes we replace the specialized nurse with the dermatology resident because, in Germany, it’s difficult to find any type of specialized nurse,” although this is not an issue in many other countries, he said.
The model for children involves six 90-minute sessions, which cover topics that include emollients and basic care, food allergies and diet, medical treatment, and psychology of itch. The program for adults involves six 2-hour sessions, which cover topics that include psychology, skin care/nutrition, and medical treatment.
While this education program improves adherence in patients with AD, he acknowledged it is time consuming, and may not work for people who live far away from a clinic or who have other time commitments, making an alternative format necessary.
In terms of improving patient adherence to a doctor’s recommendations regarding chronic skin disease, “we cannot change our patients, we cannot change the disease, but we can strongly influence the treatment that we choose and how we interact as physicians with our patients,” said Dr. Wollenberg.
“Therapeutic patient education is virtually free of side effects, but evidence based. Have a look [at] it and adapt it to your own practice,” he added.
Dr. Wollenberg is a consultant, speaker and receives fees from numerous pharmaceutical companies.
, Andreas Wollenberg, MD, said at the Revolutionizing Atopic Dermatitis symposium.
A major goal of patient education is increasing medication adherence, noted Dr. Wollenberg, professor in the department of dermatology and allergy at Ludwig Maximilian University of Munich. Quoting former U.S. Surgeon General C. Everett Koop, MD, he said, “drugs don’t work in patients who don’t take them.”
While this is a simple message, it is important, Dr. Wollenberg said, noting that there can be a gap between a physician’s well-intentioned message and how it is interpreted by the patient. “Our messages may not be heard, not understood, not accepted, and even if they are put into place, how long will they last?” he asked. “We need to find a way [to] place sticky messages in the brains of our patients who are sitting and interacting with us.”
One way to improve treatment adherence is through patient education, such as using a written action plan or graphics; simplifying treatment regimens; minimizing treatment costs; setting up reminder programs, early follow-up visits, and short-term treatment goals; and minimizing nocebo effects. “This is more than providing just leaflets to patients. It is a complete program. It is a holistic approach. It should be structured and should be interdisciplinary, and it should contain a psychological component,” Dr. Wollenberg said.
Therapeutic patient education is recommended at baseline for children and adults with moderate to severe AD in the 2020 European Task Force on Atopic Dermatitis (ETFAD) and European Academy of Dermatology and Venereology (EADV) position paper on the diagnosis and treatment of AD in adults and children, alongside other interventions, such as emollients, bath oils, and avoidance of clinically relevant allergens, noted Dr. Wollenberg, the first author . “Therapeutic patient education is an extremely helpful tool to address patient beliefs and questions regarding disease and treatment,” he and his coauthors wrote in the paper.
When considering a therapeutic patient education program for AD, content is key, but just as important is consideration of legal and cultural conditions in the local area, Dr. Wollenberg explained. Every country will need some degree of standardization of content, he noted. Clinicians interested in adopting a patient education program need to consider who will pay for it – patients, foundations, or insurance companies – as well as the time commitment needed.
Dr. Wollenberg said that his team uses an evidence-based education program for AD in Germany that works across patients with different personalities, with a multidisciplinary team that includes a dermatologist, a specialist nurse, a nutrition expert, and a psychologist. “Sometimes we replace the specialized nurse with the dermatology resident because, in Germany, it’s difficult to find any type of specialized nurse,” although this is not an issue in many other countries, he said.
The model for children involves six 90-minute sessions, which cover topics that include emollients and basic care, food allergies and diet, medical treatment, and psychology of itch. The program for adults involves six 2-hour sessions, which cover topics that include psychology, skin care/nutrition, and medical treatment.
While this education program improves adherence in patients with AD, he acknowledged it is time consuming, and may not work for people who live far away from a clinic or who have other time commitments, making an alternative format necessary.
In terms of improving patient adherence to a doctor’s recommendations regarding chronic skin disease, “we cannot change our patients, we cannot change the disease, but we can strongly influence the treatment that we choose and how we interact as physicians with our patients,” said Dr. Wollenberg.
“Therapeutic patient education is virtually free of side effects, but evidence based. Have a look [at] it and adapt it to your own practice,” he added.
Dr. Wollenberg is a consultant, speaker and receives fees from numerous pharmaceutical companies.
FROM RAD 2022
2022 Update: Beyond prenatal exome sequencing
Last year, our Update focused on the expansion of sequencing in prenatal diagnosis. This year, we are taking a step sideways to remember the many diagnoses we may miss if we rely on exome sequencing alone. A recent case report in Prenatal Diagnosis describes a pregnancy affected by fetal akinesia sequence and polyhydramnios in which sequencing did not reveal a diagnosis. Expansion of the differential to include congenital myotonic dystrophy and subsequent triplet repeat testing led the clinicians to the diagnosis and identification of a triplet repeat expansion in the DMPK gene. This case serves as our first example of how complementary testing and technologies should continue to help us make critical diagnoses.
What is the yield of exome sequencing vs panels in nonimmune hydrops?
Rogers R, Moyer K, Moise KJ Jr. Congenital myotonic dystrophy: an overlooked diagnosis not amenable to detection by sequencing. Prenat Diagn. 2022;42:233-235. doi:10.1002/pd.6105.
Norton ME, Ziffle JV, Lianoglou BR, et al. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2021;28:S0002-9378(21)00828-0. doi:10.1016/j.ajog.2021.07.014.
We have had several illuminating discussions with our colleagues about the merits of exome sequencing (ES) versus panels and other modalities for fetal diagnosis. Many obstetricians practicing at the leading edge may feel like ES should be utilized uniformly for fetal anomalies with nondiagnostic karyotype or microarray. However, for well-defined phenotypes with clear and narrow lists of implicated genes (eg, skeletal dysplasias) or patients without insurance coverage, panel sequencing still has utility in prenatal diagnosis. The question of which phenotypes most benefit from ES versus panel sequencing is an area of interesting, ongoing research for several investigators.
Secondary analysis of nonimmune hydrops cohort
Norton and colleagues tackled one such cohort in a study presented in the American Journal of Obstetrics and Gynecology. They compared the proportion of diagnoses that would have been identified in commercial lab panels with their research of phenotype-driven ES in a cohort of 127 fetuses with features of nonimmune hydrops fetalis (NIHF). NIHF can be caused by a variety of single-gene disorders in addition to chromosomal disorders and copy number variants on chromosomal microarray. Patients were eligible for inclusion in the cohort if they had a nondiagnostic karyotype or microarray and any of the following features: nuchal translucency of 3.5 mm or greater, cystic hygroma, pleural effusion, pericardial effusion, ascites, or skin edema. Standard sequencing methods and variant analysis were performed. They assumed 100% analytical sensitivity and specificity of the panels for variant detection and collected cost information on the targeted gene panels.
Study outcomes
In the ES analysis of cases, 37 of 127 cases (29%) had a pathogenic or likely pathogenic variant in 1 of 29 genes, and another 12 of 127 cases (9%) had variants of uncertain significance that were strongly suspected to be the etiology during clinical analysis. The types of disorders that were identified are listed in the TABLE. In addition to a feature of NIHF, 50% of the cases had a structural anomaly.

There were 10 identified clinical panels from 7 clinical laboratories. These panels ranged in size from 11 to 128 genes. The highest simulated yield of any commercial panel was only 62% of the pathogenic variants identified by ES. The other commercial laboratory panels detection yield ranged from 11% to 62% of pathogenic variants detected by ES. For overall yield, the largest panel would have a diagnostic yield of 18% of diagnoses relative to the 29% diagnostic yield from ES.
The largest panel included 128 genes prior to the publication of the original cohort and was updated after publication to include 148 genes. The larger updated panel would have identified all of the patients in the ES cohort. However, many of the other panels listed would have identified a smaller fraction of the variants identified by ES (range, 11%-62%). At the time of publication, the cost of the panels ranged from $640 to $3,500, and the cost of prenatal ES ranged from $2,458 to $7,500.
Continue to: Strengths and limitations...
Strengths and limitations
Twenty-three percent of the patients who were sequenced had an increased fetal nuchal translucency or cystic hygroma, and another 17% had a single fetal effusion. This inclusivity makes this study more applicable to broader fetal anomaly populations. However, it is worth noting that only 61% of patients had NIHF by the definition of 2 or more fluid collections or skin thickening.
The authors assumed 100% sensitivity and specificity for the panel tests relative to diagnostic ES results, but this may not reflect real-life analysis. There is inherent subjectivity and subsequent differences in variant calling (deciding which genetic changes are pathogenic) between institutions and companies despite efforts to standardize this process. Due to the simulated nature of this study, these differences are not captured. Additionally, although the authors note that the research ES had at least 30 times the coverage (an adequate number of sequence reads for accurate testing) than did the commercial lab panels, some gene panels have additional sequencing of intronic regions, copy number analysis, and up to 10 times more coverage than ES, which could lead to more diagnoses.
This study illustrates that there is nuance involved in selecting which type of gene sequencing and which clinical laboratory to use for prenatal diagnosis. Labs with more updated literature searches and more inclusive gene panels may be excellent options for patients in whom ES is not covered by insurance or with phenotypes with a narrow range of suspected causative genes. However, there is a lag time in updating the genes offered on each panel, and new genedisease associations will not be captured by existing panels.
From a cost, speed-of-analysis, and depth-of-sequencing perspective, panel sequencing can have advantages that should be considered in some patients, particularly if the panels are large and regularly updated. However, the authors summarize our sentiments and their findings with the following:
“For disorders, such as NIHF with marked genetic heterogeneity and less clear in utero phenotypes of underlying genetic diseases, the broader coverage of exome sequencing makes it a superior option to targeted panel testing.”
We look forward to the publication of further anomaly-specific cohorts and secondary analyses of the utility of current panels and ES that may follow.
Frequency of Beckwith-Widemann syndrome in prenatally diagnosed omphaloceles
Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41:798-816. doi:10.1002/pd.5930.
An omphalocele is diagnosed prenatally on ultrasound when an anterior midline mass, often containing abdominal contents, is seen herniating into the base of the umbilical cord. Omphaloceles are often associated with additional structural abnormalities and underlying genetic syndromes, thus a thorough fetal assessment is required for accurate prenatal counseling and neonatal care.
Identification of Beckwith-Widemann syndrome (BWS) in the setting of a prenatally diagnosed omphalocele is difficult because of its wide range of clinical features and its unique genetic basis. Unlike many genetic disorders that are caused by specific genetic variants, or spelling changes in the genes, BWS results from a change in the expression of one or more of the genes in a specific region of chromosome 11. A high index of clinical suspicion as well as an understanding of the various genetic and epigenetics alterations that cause BWS is required for prenatal diagnosis.
Retrospective cohort at a single center
The authors in this study reviewed all pregnancies in which an omphalocele was diagnosed prenatally at a single center between 2010 and 2015. They describe a standard prenatal evaluation following identification of an omphalocele including echocardiogram, detailed anatomic survey, and availability of an amniocentesis to facilitate aneuploidy screening and testing for BWS. This review also includes an overview of perinatal and long-term outcomes for cases of BWS diagnosed at their center between 2000 and 2015.
Study outcomes
Results of prenatal genetic testing in this cohort were divided between cases of an isolated omphalocele (without other structural changes) and cases of nonisolated omphaloceles. In the group of pregnancies with an isolated omphalocele, 2 of 27 pregnancies (7.4%) were found to have an abnormal karyotype, and 6 of 16 of the remaining pregnancies (37.5%) were diagnosed with BWS. Among the group of pregnancies with a nonisolated omphalocele, 23 of 59 pregnancies (39%) were found to have an abnormal karyotype, and 1 of 20 pregnancies (5%) were diagnosed with BWS.
Prenatal sonographic features associated with cases of BWS included polyhydramnios in 12 of 19 cases (63%) and macrosomia in 8 of 19 cases (42%). Macroglossia is another characteristic feature of the disorder, which was identified in 4 of 19 cases (21%) prenatally and in an additional 10 of 19 cases (52.6%) postnatally. Interestingly, only 1 of the cases of BWS was caused by a microdeletion at 11p15.4—a change that was identified on microarray. The additional 6 cases of BWS were caused by imprinting changes in the region, which are only detectable with a specific methylation-analysis technique.
Among the 19 cases of BWS identified over a 15-year period, there was 1 intrauterine demise. Preterm birth occurred in 10 of 19 cases (52.6%), including 8 of 19 cases (42.1%) of spontaneous preterm labor. Respiratory distress (27.8%), hypoglycemia (61%), and gastrointestinal reflux (59%) were common neonatal complications. Embryonal tumors were diagnosed in 2 of 16 patients (12.5%). Although neurodevelopmental outcomes were incomplete, their data suggested normal development in 75% of children. There were 2 neonatal deaths in this cohort and 1 childhood death at age 2 years.
Study strengths and limitations
As with many studies investigating a rare disorder, this study is limited by its retrospective nature and small sample size. Nevertheless, it adds an important cohort of patients with a prenatally diagnosed omphalocele to the literature and illuminates the utility of a standardized approach to testing for BWS in this population.
In this cohort with prenatally diagnosed omphaloceles with standardized testing for BWS, the prevalence of the disorder was approximately 8% and more common in cases of an isolated omphalocele. The most common supporting sonographic features of BWS may not be detected until later in gestation, including polyhydramnios and macrosomia. This demonstrates the importance of both sonographic follow-up as well as universal testing for BWS in euploid cases of a prenatally diagnosed omphalocele. Almost all cases of BWS in this cohort required specialized molecular techniques for diagnosis, and the diagnosis would have been missed on karyotype, microarray, and ES.
Continue to: Genetic diagnoses that could have been identified by expanded carrier screening...
Genetic diagnoses that could have been identified by expanded carrier screening
Stevens BK, Nunley PB, Wagner C, et al. Utility of expanded carrier screening in pregnancies with ultrasound abnormalities. Prenat Diagn. 2022;42:60-78. doi:10.1002/pd.6069.
This series is a thorough retrospective review of patients evaluated in a pediatric genetics clinic from 2014 through 2017. Patients were included if they were evaluated in the first 6 months of life and had a structural abnormality that might be detected on prenatal ultrasonography. The genetic testing results were analyzed and categorized according to types of genetic disorders, with the goal of identifying how many patients might have been identified by expanded carrier screening (ECS) panels.
Study outcomes
A total of 931 charts were reviewed, and 85% (791 of 931) of patients evaluated in the first 6 months of life were determined to have a structural anomaly that might be detected on prenatal ultrasonography. Of those patients, 691 went on to have genetic testing and 32.1% (222 of 691) of them had a diagnostic (pathogenic) genetic testing result related to the phenotype. The types of diagnostic testing results are shown in the FIGURE. Notably, 42 single-gene disorders were detected.
FIGURE Diagnostic test result in pediatric patients evaluated under age 6 months
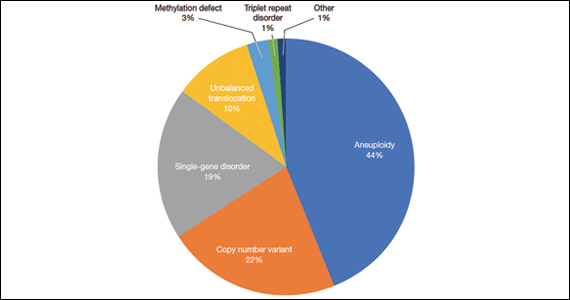
Of those 222 patients with diagnostic results, there were 8 patients with autosomal recessive and X-linked conditions that could be detected using a 500-gene ECS panel. Five patients could be detected with a 271-gene panel. After nondiagnostic microarray, 11.3% of patients had a condition that could be detected by using a 500-gene ECS panel. The identified conditions included cystic fibrosis, CYP21‐related congenital adrenal hyperplasia, autosomal recessive polycystic kidney disease, Antley‐Bixler syndrome, and Morquio syndrome type A.
Furthermore, the authors conducted a literature review of 271 conditions and found that 32% (88 of 271) of conditions may be associated with ultrasound findings.
Study strengths and limitations
When applying these data to prenatal populations, the authors acknowledge several notable limitations. There is a selection bias toward less-severe phenotypes for many patients choosing to continue rather than to interrupt a pregnancy. Additionally, only 23% of the patients in the study had a microarray and ES, which may lead to an underrepresentation of single-gene disorders and an underestimation of the utility of ECS. Finally, a retrospective classification of structural abnormalities that may be detectable by ultrasonography may not always reflect what is actually reported in prenatal imaging.
However, the work that the authors put forth to evaluate and categorize 931 participants by the results of genetic testing and structural anomalies is appreciated, and the level of detail is impressive for this retrospective chart review. Additionally, the tables itemizing the authors’ review of 271 ECS disorders that may have ultrasonography findings categorized by disorder and system are helpful and quick diagnostic references for clinicians providing prenatal care. ●
This study of potentially detectable prenatal findings from the lens of a pediatric genetics clinic lends an interesting perspective: Exome sequencing is not the primary route to establish a diagnosis; karyotype, microarray, methylation disorders, and triplet repeat disorders all have an established role in the diagnostic toolkit. Keeping in mind the contribution of these modalities to pediatric testing may shorten the diagnostic odyssey to continue pregnancies or help to fully counsel patients on expectations and decision-making after birth.
Carrier screening is not a substitute for diagnostic testing in pregnancy. However, in appropriately selected patients, a broad carrier screening panel may have added utility. ECS can be conducted while awaiting microarray results to help target testing and may be particularly useful for patients who decline diagnostic testing until the postnatal period. It is important to counsel patients that carrier screening is not a diagnostic test, and results will only report likely pathogenic or pathogenic variants, not variants of uncertain significance that may be of clinical relevance. However, our practice has had several insightful diagnoses reached through ECS, in conjunction with microarray testing that allowed for faster and more targeted sequencing and precise fetal diagnosis. This readily available molecular tool (often covered by insurance) deserves a spot in your fetal diagnosis tool belt based on available evidence.

Rebecca Reimers, MD
Dr. Reimers is a Clinical Fellow, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital and Boston Children’s Hospital, Massachusetts.
Stephanie Guseh, MD

Dr. Guseh is a Clinical Instructor, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital.
The authors report no financial relationships relevant to this article.
Rebecca Reimers, MD
Dr. Reimers is a Clinical Fellow, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital and Boston Children’s Hospital, Massachusetts.
Stephanie Guseh, MD
Dr. Guseh is a Clinical Instructor, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital.
The authors report no financial relationships relevant to this article.
Rebecca Reimers, MD
Dr. Reimers is a Clinical Fellow, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital and Boston Children’s Hospital, Massachusetts.
Stephanie Guseh, MD
Dr. Guseh is a Clinical Instructor, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital.
The authors report no financial relationships relevant to this article.
Last year, our Update focused on the expansion of sequencing in prenatal diagnosis. This year, we are taking a step sideways to remember the many diagnoses we may miss if we rely on exome sequencing alone. A recent case report in Prenatal Diagnosis describes a pregnancy affected by fetal akinesia sequence and polyhydramnios in which sequencing did not reveal a diagnosis. Expansion of the differential to include congenital myotonic dystrophy and subsequent triplet repeat testing led the clinicians to the diagnosis and identification of a triplet repeat expansion in the DMPK gene. This case serves as our first example of how complementary testing and technologies should continue to help us make critical diagnoses.
What is the yield of exome sequencing vs panels in nonimmune hydrops?
Rogers R, Moyer K, Moise KJ Jr. Congenital myotonic dystrophy: an overlooked diagnosis not amenable to detection by sequencing. Prenat Diagn. 2022;42:233-235. doi:10.1002/pd.6105.
Norton ME, Ziffle JV, Lianoglou BR, et al. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2021;28:S0002-9378(21)00828-0. doi:10.1016/j.ajog.2021.07.014.
We have had several illuminating discussions with our colleagues about the merits of exome sequencing (ES) versus panels and other modalities for fetal diagnosis. Many obstetricians practicing at the leading edge may feel like ES should be utilized uniformly for fetal anomalies with nondiagnostic karyotype or microarray. However, for well-defined phenotypes with clear and narrow lists of implicated genes (eg, skeletal dysplasias) or patients without insurance coverage, panel sequencing still has utility in prenatal diagnosis. The question of which phenotypes most benefit from ES versus panel sequencing is an area of interesting, ongoing research for several investigators.
Secondary analysis of nonimmune hydrops cohort
Norton and colleagues tackled one such cohort in a study presented in the American Journal of Obstetrics and Gynecology. They compared the proportion of diagnoses that would have been identified in commercial lab panels with their research of phenotype-driven ES in a cohort of 127 fetuses with features of nonimmune hydrops fetalis (NIHF). NIHF can be caused by a variety of single-gene disorders in addition to chromosomal disorders and copy number variants on chromosomal microarray. Patients were eligible for inclusion in the cohort if they had a nondiagnostic karyotype or microarray and any of the following features: nuchal translucency of 3.5 mm or greater, cystic hygroma, pleural effusion, pericardial effusion, ascites, or skin edema. Standard sequencing methods and variant analysis were performed. They assumed 100% analytical sensitivity and specificity of the panels for variant detection and collected cost information on the targeted gene panels.
Study outcomes
In the ES analysis of cases, 37 of 127 cases (29%) had a pathogenic or likely pathogenic variant in 1 of 29 genes, and another 12 of 127 cases (9%) had variants of uncertain significance that were strongly suspected to be the etiology during clinical analysis. The types of disorders that were identified are listed in the TABLE. In addition to a feature of NIHF, 50% of the cases had a structural anomaly.
There were 10 identified clinical panels from 7 clinical laboratories. These panels ranged in size from 11 to 128 genes. The highest simulated yield of any commercial panel was only 62% of the pathogenic variants identified by ES. The other commercial laboratory panels detection yield ranged from 11% to 62% of pathogenic variants detected by ES. For overall yield, the largest panel would have a diagnostic yield of 18% of diagnoses relative to the 29% diagnostic yield from ES.
The largest panel included 128 genes prior to the publication of the original cohort and was updated after publication to include 148 genes. The larger updated panel would have identified all of the patients in the ES cohort. However, many of the other panels listed would have identified a smaller fraction of the variants identified by ES (range, 11%-62%). At the time of publication, the cost of the panels ranged from $640 to $3,500, and the cost of prenatal ES ranged from $2,458 to $7,500.
Continue to: Strengths and limitations...
Strengths and limitations
Twenty-three percent of the patients who were sequenced had an increased fetal nuchal translucency or cystic hygroma, and another 17% had a single fetal effusion. This inclusivity makes this study more applicable to broader fetal anomaly populations. However, it is worth noting that only 61% of patients had NIHF by the definition of 2 or more fluid collections or skin thickening.
The authors assumed 100% sensitivity and specificity for the panel tests relative to diagnostic ES results, but this may not reflect real-life analysis. There is inherent subjectivity and subsequent differences in variant calling (deciding which genetic changes are pathogenic) between institutions and companies despite efforts to standardize this process. Due to the simulated nature of this study, these differences are not captured. Additionally, although the authors note that the research ES had at least 30 times the coverage (an adequate number of sequence reads for accurate testing) than did the commercial lab panels, some gene panels have additional sequencing of intronic regions, copy number analysis, and up to 10 times more coverage than ES, which could lead to more diagnoses.
This study illustrates that there is nuance involved in selecting which type of gene sequencing and which clinical laboratory to use for prenatal diagnosis. Labs with more updated literature searches and more inclusive gene panels may be excellent options for patients in whom ES is not covered by insurance or with phenotypes with a narrow range of suspected causative genes. However, there is a lag time in updating the genes offered on each panel, and new genedisease associations will not be captured by existing panels.
From a cost, speed-of-analysis, and depth-of-sequencing perspective, panel sequencing can have advantages that should be considered in some patients, particularly if the panels are large and regularly updated. However, the authors summarize our sentiments and their findings with the following:
“For disorders, such as NIHF with marked genetic heterogeneity and less clear in utero phenotypes of underlying genetic diseases, the broader coverage of exome sequencing makes it a superior option to targeted panel testing.”
We look forward to the publication of further anomaly-specific cohorts and secondary analyses of the utility of current panels and ES that may follow.
Frequency of Beckwith-Widemann syndrome in prenatally diagnosed omphaloceles
Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41:798-816. doi:10.1002/pd.5930.
An omphalocele is diagnosed prenatally on ultrasound when an anterior midline mass, often containing abdominal contents, is seen herniating into the base of the umbilical cord. Omphaloceles are often associated with additional structural abnormalities and underlying genetic syndromes, thus a thorough fetal assessment is required for accurate prenatal counseling and neonatal care.
Identification of Beckwith-Widemann syndrome (BWS) in the setting of a prenatally diagnosed omphalocele is difficult because of its wide range of clinical features and its unique genetic basis. Unlike many genetic disorders that are caused by specific genetic variants, or spelling changes in the genes, BWS results from a change in the expression of one or more of the genes in a specific region of chromosome 11. A high index of clinical suspicion as well as an understanding of the various genetic and epigenetics alterations that cause BWS is required for prenatal diagnosis.
Retrospective cohort at a single center
The authors in this study reviewed all pregnancies in which an omphalocele was diagnosed prenatally at a single center between 2010 and 2015. They describe a standard prenatal evaluation following identification of an omphalocele including echocardiogram, detailed anatomic survey, and availability of an amniocentesis to facilitate aneuploidy screening and testing for BWS. This review also includes an overview of perinatal and long-term outcomes for cases of BWS diagnosed at their center between 2000 and 2015.
Study outcomes
Results of prenatal genetic testing in this cohort were divided between cases of an isolated omphalocele (without other structural changes) and cases of nonisolated omphaloceles. In the group of pregnancies with an isolated omphalocele, 2 of 27 pregnancies (7.4%) were found to have an abnormal karyotype, and 6 of 16 of the remaining pregnancies (37.5%) were diagnosed with BWS. Among the group of pregnancies with a nonisolated omphalocele, 23 of 59 pregnancies (39%) were found to have an abnormal karyotype, and 1 of 20 pregnancies (5%) were diagnosed with BWS.
Prenatal sonographic features associated with cases of BWS included polyhydramnios in 12 of 19 cases (63%) and macrosomia in 8 of 19 cases (42%). Macroglossia is another characteristic feature of the disorder, which was identified in 4 of 19 cases (21%) prenatally and in an additional 10 of 19 cases (52.6%) postnatally. Interestingly, only 1 of the cases of BWS was caused by a microdeletion at 11p15.4—a change that was identified on microarray. The additional 6 cases of BWS were caused by imprinting changes in the region, which are only detectable with a specific methylation-analysis technique.
Among the 19 cases of BWS identified over a 15-year period, there was 1 intrauterine demise. Preterm birth occurred in 10 of 19 cases (52.6%), including 8 of 19 cases (42.1%) of spontaneous preterm labor. Respiratory distress (27.8%), hypoglycemia (61%), and gastrointestinal reflux (59%) were common neonatal complications. Embryonal tumors were diagnosed in 2 of 16 patients (12.5%). Although neurodevelopmental outcomes were incomplete, their data suggested normal development in 75% of children. There were 2 neonatal deaths in this cohort and 1 childhood death at age 2 years.
Study strengths and limitations
As with many studies investigating a rare disorder, this study is limited by its retrospective nature and small sample size. Nevertheless, it adds an important cohort of patients with a prenatally diagnosed omphalocele to the literature and illuminates the utility of a standardized approach to testing for BWS in this population.
In this cohort with prenatally diagnosed omphaloceles with standardized testing for BWS, the prevalence of the disorder was approximately 8% and more common in cases of an isolated omphalocele. The most common supporting sonographic features of BWS may not be detected until later in gestation, including polyhydramnios and macrosomia. This demonstrates the importance of both sonographic follow-up as well as universal testing for BWS in euploid cases of a prenatally diagnosed omphalocele. Almost all cases of BWS in this cohort required specialized molecular techniques for diagnosis, and the diagnosis would have been missed on karyotype, microarray, and ES.
Continue to: Genetic diagnoses that could have been identified by expanded carrier screening...
Genetic diagnoses that could have been identified by expanded carrier screening
Stevens BK, Nunley PB, Wagner C, et al. Utility of expanded carrier screening in pregnancies with ultrasound abnormalities. Prenat Diagn. 2022;42:60-78. doi:10.1002/pd.6069.
This series is a thorough retrospective review of patients evaluated in a pediatric genetics clinic from 2014 through 2017. Patients were included if they were evaluated in the first 6 months of life and had a structural abnormality that might be detected on prenatal ultrasonography. The genetic testing results were analyzed and categorized according to types of genetic disorders, with the goal of identifying how many patients might have been identified by expanded carrier screening (ECS) panels.
Study outcomes
A total of 931 charts were reviewed, and 85% (791 of 931) of patients evaluated in the first 6 months of life were determined to have a structural anomaly that might be detected on prenatal ultrasonography. Of those patients, 691 went on to have genetic testing and 32.1% (222 of 691) of them had a diagnostic (pathogenic) genetic testing result related to the phenotype. The types of diagnostic testing results are shown in the FIGURE. Notably, 42 single-gene disorders were detected.
FIGURE Diagnostic test result in pediatric patients evaluated under age 6 months
Of those 222 patients with diagnostic results, there were 8 patients with autosomal recessive and X-linked conditions that could be detected using a 500-gene ECS panel. Five patients could be detected with a 271-gene panel. After nondiagnostic microarray, 11.3% of patients had a condition that could be detected by using a 500-gene ECS panel. The identified conditions included cystic fibrosis, CYP21‐related congenital adrenal hyperplasia, autosomal recessive polycystic kidney disease, Antley‐Bixler syndrome, and Morquio syndrome type A.
Furthermore, the authors conducted a literature review of 271 conditions and found that 32% (88 of 271) of conditions may be associated with ultrasound findings.
Study strengths and limitations
When applying these data to prenatal populations, the authors acknowledge several notable limitations. There is a selection bias toward less-severe phenotypes for many patients choosing to continue rather than to interrupt a pregnancy. Additionally, only 23% of the patients in the study had a microarray and ES, which may lead to an underrepresentation of single-gene disorders and an underestimation of the utility of ECS. Finally, a retrospective classification of structural abnormalities that may be detectable by ultrasonography may not always reflect what is actually reported in prenatal imaging.
However, the work that the authors put forth to evaluate and categorize 931 participants by the results of genetic testing and structural anomalies is appreciated, and the level of detail is impressive for this retrospective chart review. Additionally, the tables itemizing the authors’ review of 271 ECS disorders that may have ultrasonography findings categorized by disorder and system are helpful and quick diagnostic references for clinicians providing prenatal care. ●
This study of potentially detectable prenatal findings from the lens of a pediatric genetics clinic lends an interesting perspective: Exome sequencing is not the primary route to establish a diagnosis; karyotype, microarray, methylation disorders, and triplet repeat disorders all have an established role in the diagnostic toolkit. Keeping in mind the contribution of these modalities to pediatric testing may shorten the diagnostic odyssey to continue pregnancies or help to fully counsel patients on expectations and decision-making after birth.
Carrier screening is not a substitute for diagnostic testing in pregnancy. However, in appropriately selected patients, a broad carrier screening panel may have added utility. ECS can be conducted while awaiting microarray results to help target testing and may be particularly useful for patients who decline diagnostic testing until the postnatal period. It is important to counsel patients that carrier screening is not a diagnostic test, and results will only report likely pathogenic or pathogenic variants, not variants of uncertain significance that may be of clinical relevance. However, our practice has had several insightful diagnoses reached through ECS, in conjunction with microarray testing that allowed for faster and more targeted sequencing and precise fetal diagnosis. This readily available molecular tool (often covered by insurance) deserves a spot in your fetal diagnosis tool belt based on available evidence.
Last year, our Update focused on the expansion of sequencing in prenatal diagnosis. This year, we are taking a step sideways to remember the many diagnoses we may miss if we rely on exome sequencing alone. A recent case report in Prenatal Diagnosis describes a pregnancy affected by fetal akinesia sequence and polyhydramnios in which sequencing did not reveal a diagnosis. Expansion of the differential to include congenital myotonic dystrophy and subsequent triplet repeat testing led the clinicians to the diagnosis and identification of a triplet repeat expansion in the DMPK gene. This case serves as our first example of how complementary testing and technologies should continue to help us make critical diagnoses.
What is the yield of exome sequencing vs panels in nonimmune hydrops?
Rogers R, Moyer K, Moise KJ Jr. Congenital myotonic dystrophy: an overlooked diagnosis not amenable to detection by sequencing. Prenat Diagn. 2022;42:233-235. doi:10.1002/pd.6105.
Norton ME, Ziffle JV, Lianoglou BR, et al. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2021;28:S0002-9378(21)00828-0. doi:10.1016/j.ajog.2021.07.014.
We have had several illuminating discussions with our colleagues about the merits of exome sequencing (ES) versus panels and other modalities for fetal diagnosis. Many obstetricians practicing at the leading edge may feel like ES should be utilized uniformly for fetal anomalies with nondiagnostic karyotype or microarray. However, for well-defined phenotypes with clear and narrow lists of implicated genes (eg, skeletal dysplasias) or patients without insurance coverage, panel sequencing still has utility in prenatal diagnosis. The question of which phenotypes most benefit from ES versus panel sequencing is an area of interesting, ongoing research for several investigators.
Secondary analysis of nonimmune hydrops cohort
Norton and colleagues tackled one such cohort in a study presented in the American Journal of Obstetrics and Gynecology. They compared the proportion of diagnoses that would have been identified in commercial lab panels with their research of phenotype-driven ES in a cohort of 127 fetuses with features of nonimmune hydrops fetalis (NIHF). NIHF can be caused by a variety of single-gene disorders in addition to chromosomal disorders and copy number variants on chromosomal microarray. Patients were eligible for inclusion in the cohort if they had a nondiagnostic karyotype or microarray and any of the following features: nuchal translucency of 3.5 mm or greater, cystic hygroma, pleural effusion, pericardial effusion, ascites, or skin edema. Standard sequencing methods and variant analysis were performed. They assumed 100% analytical sensitivity and specificity of the panels for variant detection and collected cost information on the targeted gene panels.
Study outcomes
In the ES analysis of cases, 37 of 127 cases (29%) had a pathogenic or likely pathogenic variant in 1 of 29 genes, and another 12 of 127 cases (9%) had variants of uncertain significance that were strongly suspected to be the etiology during clinical analysis. The types of disorders that were identified are listed in the TABLE. In addition to a feature of NIHF, 50% of the cases had a structural anomaly.
There were 10 identified clinical panels from 7 clinical laboratories. These panels ranged in size from 11 to 128 genes. The highest simulated yield of any commercial panel was only 62% of the pathogenic variants identified by ES. The other commercial laboratory panels detection yield ranged from 11% to 62% of pathogenic variants detected by ES. For overall yield, the largest panel would have a diagnostic yield of 18% of diagnoses relative to the 29% diagnostic yield from ES.
The largest panel included 128 genes prior to the publication of the original cohort and was updated after publication to include 148 genes. The larger updated panel would have identified all of the patients in the ES cohort. However, many of the other panels listed would have identified a smaller fraction of the variants identified by ES (range, 11%-62%). At the time of publication, the cost of the panels ranged from $640 to $3,500, and the cost of prenatal ES ranged from $2,458 to $7,500.
Continue to: Strengths and limitations...
Strengths and limitations
Twenty-three percent of the patients who were sequenced had an increased fetal nuchal translucency or cystic hygroma, and another 17% had a single fetal effusion. This inclusivity makes this study more applicable to broader fetal anomaly populations. However, it is worth noting that only 61% of patients had NIHF by the definition of 2 or more fluid collections or skin thickening.
The authors assumed 100% sensitivity and specificity for the panel tests relative to diagnostic ES results, but this may not reflect real-life analysis. There is inherent subjectivity and subsequent differences in variant calling (deciding which genetic changes are pathogenic) between institutions and companies despite efforts to standardize this process. Due to the simulated nature of this study, these differences are not captured. Additionally, although the authors note that the research ES had at least 30 times the coverage (an adequate number of sequence reads for accurate testing) than did the commercial lab panels, some gene panels have additional sequencing of intronic regions, copy number analysis, and up to 10 times more coverage than ES, which could lead to more diagnoses.
This study illustrates that there is nuance involved in selecting which type of gene sequencing and which clinical laboratory to use for prenatal diagnosis. Labs with more updated literature searches and more inclusive gene panels may be excellent options for patients in whom ES is not covered by insurance or with phenotypes with a narrow range of suspected causative genes. However, there is a lag time in updating the genes offered on each panel, and new genedisease associations will not be captured by existing panels.
From a cost, speed-of-analysis, and depth-of-sequencing perspective, panel sequencing can have advantages that should be considered in some patients, particularly if the panels are large and regularly updated. However, the authors summarize our sentiments and their findings with the following:
“For disorders, such as NIHF with marked genetic heterogeneity and less clear in utero phenotypes of underlying genetic diseases, the broader coverage of exome sequencing makes it a superior option to targeted panel testing.”
We look forward to the publication of further anomaly-specific cohorts and secondary analyses of the utility of current panels and ES that may follow.
Frequency of Beckwith-Widemann syndrome in prenatally diagnosed omphaloceles
Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41:798-816. doi:10.1002/pd.5930.
An omphalocele is diagnosed prenatally on ultrasound when an anterior midline mass, often containing abdominal contents, is seen herniating into the base of the umbilical cord. Omphaloceles are often associated with additional structural abnormalities and underlying genetic syndromes, thus a thorough fetal assessment is required for accurate prenatal counseling and neonatal care.
Identification of Beckwith-Widemann syndrome (BWS) in the setting of a prenatally diagnosed omphalocele is difficult because of its wide range of clinical features and its unique genetic basis. Unlike many genetic disorders that are caused by specific genetic variants, or spelling changes in the genes, BWS results from a change in the expression of one or more of the genes in a specific region of chromosome 11. A high index of clinical suspicion as well as an understanding of the various genetic and epigenetics alterations that cause BWS is required for prenatal diagnosis.
Retrospective cohort at a single center
The authors in this study reviewed all pregnancies in which an omphalocele was diagnosed prenatally at a single center between 2010 and 2015. They describe a standard prenatal evaluation following identification of an omphalocele including echocardiogram, detailed anatomic survey, and availability of an amniocentesis to facilitate aneuploidy screening and testing for BWS. This review also includes an overview of perinatal and long-term outcomes for cases of BWS diagnosed at their center between 2000 and 2015.
Study outcomes
Results of prenatal genetic testing in this cohort were divided between cases of an isolated omphalocele (without other structural changes) and cases of nonisolated omphaloceles. In the group of pregnancies with an isolated omphalocele, 2 of 27 pregnancies (7.4%) were found to have an abnormal karyotype, and 6 of 16 of the remaining pregnancies (37.5%) were diagnosed with BWS. Among the group of pregnancies with a nonisolated omphalocele, 23 of 59 pregnancies (39%) were found to have an abnormal karyotype, and 1 of 20 pregnancies (5%) were diagnosed with BWS.
Prenatal sonographic features associated with cases of BWS included polyhydramnios in 12 of 19 cases (63%) and macrosomia in 8 of 19 cases (42%). Macroglossia is another characteristic feature of the disorder, which was identified in 4 of 19 cases (21%) prenatally and in an additional 10 of 19 cases (52.6%) postnatally. Interestingly, only 1 of the cases of BWS was caused by a microdeletion at 11p15.4—a change that was identified on microarray. The additional 6 cases of BWS were caused by imprinting changes in the region, which are only detectable with a specific methylation-analysis technique.
Among the 19 cases of BWS identified over a 15-year period, there was 1 intrauterine demise. Preterm birth occurred in 10 of 19 cases (52.6%), including 8 of 19 cases (42.1%) of spontaneous preterm labor. Respiratory distress (27.8%), hypoglycemia (61%), and gastrointestinal reflux (59%) were common neonatal complications. Embryonal tumors were diagnosed in 2 of 16 patients (12.5%). Although neurodevelopmental outcomes were incomplete, their data suggested normal development in 75% of children. There were 2 neonatal deaths in this cohort and 1 childhood death at age 2 years.
Study strengths and limitations
As with many studies investigating a rare disorder, this study is limited by its retrospective nature and small sample size. Nevertheless, it adds an important cohort of patients with a prenatally diagnosed omphalocele to the literature and illuminates the utility of a standardized approach to testing for BWS in this population.
In this cohort with prenatally diagnosed omphaloceles with standardized testing for BWS, the prevalence of the disorder was approximately 8% and more common in cases of an isolated omphalocele. The most common supporting sonographic features of BWS may not be detected until later in gestation, including polyhydramnios and macrosomia. This demonstrates the importance of both sonographic follow-up as well as universal testing for BWS in euploid cases of a prenatally diagnosed omphalocele. Almost all cases of BWS in this cohort required specialized molecular techniques for diagnosis, and the diagnosis would have been missed on karyotype, microarray, and ES.
Continue to: Genetic diagnoses that could have been identified by expanded carrier screening...
Genetic diagnoses that could have been identified by expanded carrier screening
Stevens BK, Nunley PB, Wagner C, et al. Utility of expanded carrier screening in pregnancies with ultrasound abnormalities. Prenat Diagn. 2022;42:60-78. doi:10.1002/pd.6069.
This series is a thorough retrospective review of patients evaluated in a pediatric genetics clinic from 2014 through 2017. Patients were included if they were evaluated in the first 6 months of life and had a structural abnormality that might be detected on prenatal ultrasonography. The genetic testing results were analyzed and categorized according to types of genetic disorders, with the goal of identifying how many patients might have been identified by expanded carrier screening (ECS) panels.
Study outcomes
A total of 931 charts were reviewed, and 85% (791 of 931) of patients evaluated in the first 6 months of life were determined to have a structural anomaly that might be detected on prenatal ultrasonography. Of those patients, 691 went on to have genetic testing and 32.1% (222 of 691) of them had a diagnostic (pathogenic) genetic testing result related to the phenotype. The types of diagnostic testing results are shown in the FIGURE. Notably, 42 single-gene disorders were detected.
FIGURE Diagnostic test result in pediatric patients evaluated under age 6 months
Of those 222 patients with diagnostic results, there were 8 patients with autosomal recessive and X-linked conditions that could be detected using a 500-gene ECS panel. Five patients could be detected with a 271-gene panel. After nondiagnostic microarray, 11.3% of patients had a condition that could be detected by using a 500-gene ECS panel. The identified conditions included cystic fibrosis, CYP21‐related congenital adrenal hyperplasia, autosomal recessive polycystic kidney disease, Antley‐Bixler syndrome, and Morquio syndrome type A.
Furthermore, the authors conducted a literature review of 271 conditions and found that 32% (88 of 271) of conditions may be associated with ultrasound findings.
Study strengths and limitations
When applying these data to prenatal populations, the authors acknowledge several notable limitations. There is a selection bias toward less-severe phenotypes for many patients choosing to continue rather than to interrupt a pregnancy. Additionally, only 23% of the patients in the study had a microarray and ES, which may lead to an underrepresentation of single-gene disorders and an underestimation of the utility of ECS. Finally, a retrospective classification of structural abnormalities that may be detectable by ultrasonography may not always reflect what is actually reported in prenatal imaging.
However, the work that the authors put forth to evaluate and categorize 931 participants by the results of genetic testing and structural anomalies is appreciated, and the level of detail is impressive for this retrospective chart review. Additionally, the tables itemizing the authors’ review of 271 ECS disorders that may have ultrasonography findings categorized by disorder and system are helpful and quick diagnostic references for clinicians providing prenatal care. ●
This study of potentially detectable prenatal findings from the lens of a pediatric genetics clinic lends an interesting perspective: Exome sequencing is not the primary route to establish a diagnosis; karyotype, microarray, methylation disorders, and triplet repeat disorders all have an established role in the diagnostic toolkit. Keeping in mind the contribution of these modalities to pediatric testing may shorten the diagnostic odyssey to continue pregnancies or help to fully counsel patients on expectations and decision-making after birth.
Carrier screening is not a substitute for diagnostic testing in pregnancy. However, in appropriately selected patients, a broad carrier screening panel may have added utility. ECS can be conducted while awaiting microarray results to help target testing and may be particularly useful for patients who decline diagnostic testing until the postnatal period. It is important to counsel patients that carrier screening is not a diagnostic test, and results will only report likely pathogenic or pathogenic variants, not variants of uncertain significance that may be of clinical relevance. However, our practice has had several insightful diagnoses reached through ECS, in conjunction with microarray testing that allowed for faster and more targeted sequencing and precise fetal diagnosis. This readily available molecular tool (often covered by insurance) deserves a spot in your fetal diagnosis tool belt based on available evidence.
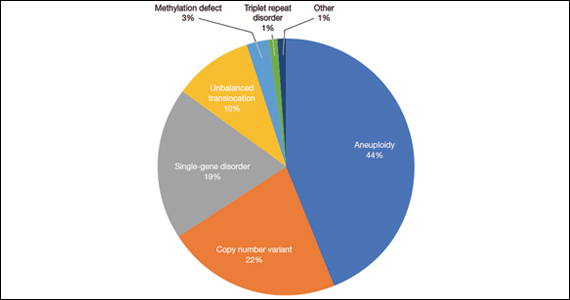
Challenges and innovations in training gyn surgeons
Obstetrics and gynecology (ObGyn) is a surgical specialty, yet the training of ObGyn residents differs significantly from that of residents in other surgical specialties. In addition to attaining competency in both the distinct but related fields of obstetrics and gynecology, ObGyn residents have their training condensed into 4 years rather than the 5 years’ training of many other surgical specialties. This limits the time dedicated to gynecologic surgery, currently 18 to 20 months in most programs, and has been exacerbated by tighter duty-hour restrictions.1
Additionally, with increasing demand for minimally invasive procedures, residents are expected to attain competency in a growing breadth of gynecologic procedures in a patient population with increasing morbidity, and they may have less autonomy to do so in an increasingly litigious environment.2 Furthermore, annual hysterectomy cases are declining, from about 680,000 in 2002 to 430,000 in 2010,3 and these declining rates are seen in the low case numbers of recent graduates.4
Training time, procedure complexity
With less time to master a growing body of increasingly complex procedures, is the profession adequately training gynecologic surgeons? Many gynecologic surgeons are concerned that the answer is no and that significant shifts in resident training are needed to generate safe and competent gynecologic surgeons. These training deficits represent a deficiency in the quality of care for women specifically, and thus the inattention to training gynecologic surgeons should be considered a health care disparity.
The concern over insufficient attention to gynecologic surgical training is not new, nor are proposed solutions, with many physicians citing the above concerns.5-9 In 2018, the Accreditation Council for Graduate Medical Education (ACGME) case minimums for hysterectomy increased to 85 from 70 hysterectomies, with a shift toward minimally invasive hysterectomy.10 Otherwise, minimal national changes have been made in this century to training gynecologic surgeons.
Tracking as an option
Many critics of current ObGyn training argue that obstetrics and gynecology, while related, have significantly different pathologies, surgical approaches, and skill sets and thus warrant the option to track toward obstetrics or gynecology after attaining limited core skill set in residency. In 2010, the Carnegie Foundation for the Advancement of Teaching called for the need for increased individualization opportunities in graduate medical education, citing that minimal changes have been made to medical education since the Flexner Report a century prior.11
Notably, tracking has been implemented with success at Cleveland Clinic, where residents are given 5 to 10 weeks of time allotted to their specific fields of interest, while still meeting minimum ACGME requirements and, in some cases, exceeding hysterectomy minimums by as much as 500%.12 Tracking is viewed positively by a majority of program directors.13 See the box below for Dr. Ferrando’s experience on tracking at the Cleveland Clinic.
Simulation training
Other educators advocate for maximizing preparedness for the operating room by using high-fidelity simulation.14,15 Simulation allows for the acquisition of basic technical skills needed for surgery as well as for repetition not easily achieved in the current surgical environment. Additionally, it provides lower-level learners the opportunity to acquire basic skills in a safe setting, thereby enhancing the ability to participate meaningfully on arrival in the operating room.16
In 2018, the American Board of Obstetrics and Gynecology added the Fundamentals of Laparoscopic Surgery certification as a new requirement for board certification.17 Laparoscopic and robotic surgery simulators allow trainees to develop coordination and specific skills, like knot tying and suturing. Additionally, models are available with varying levels of fidelity for vaginal and abdominal hysterectomy.18-20 See the box below for Dr. Miyazaki’s experience in developing the Miya Model trainer for vaginal surgery simulation.
Structured feedback
Finally, if a resident has limited exposure to a specific procedure, maximizing the preparation and feedback for each procedure is paramount. However, surgeons receive minimal formal training in teaching trainees, which leads to inconsistent and underutilized feedback.21 Specific structured feedback models have been implemented with success in the general surgery literature, including the SHARP (Set learning objectives, How did it go, Address concerns, Review learning points, Plan ahead) and BID (Briefing, Intraoperative, Debriefing) models.22,23
Reimbursement reform
While surgical reimbursement is not directly tied to resident education, decreased reimbursement to women’s health pathology and procedures has the downstream effect of decreasing the funds available for ObGyn departments to invest in research and education. Additionally, “suboptimal mastery or maintenance of appropriate surgical skills results in procedural inefficiencies that compound surgical cost.”5 Providers and payors alike should therefore be motivated to improve funding in order to improve adequate training of gynecologic surgeons. Payment reform is necessary to equally value women’s health procedures but also can ensure that gynecologic surgeons have the funds needed to train a competent next generation of ObGyn physicians. ●
- Residents and fellows have significant constraints that limit adequate training in gynecologic surgery. In a panel discussion at the 48th annual meeting of the Society of Gynecologic Surgeons, Drs. Zimmerman, Ferrando, and Miyazaki spoke about potential solutions.
- Allowing residents to track toward obstetric or gynecologic subspecialties may improve surgical volume of trainees who aim for a future career in gynecologic surgery.
- Simulation has demonstrated efficacy in enabling residents to prepare and improve their technical skills for specific procedures prior to entering the operating room.
Cecile A. Ferrando, MD, MPH
In his 2013 presidential address at the opening ceremony of the 42nd AAGL Global Congress on Minimally Invasive Gynecology, Javier Magrina, MD, asked the audience, “Isn’t it time to separate the O from the G?”7 Since that address, this catchy question has been posed several times, and it continues to be a topic of interest to many ObGyn educators seeking to innovate the curriculum and to better train our next generation’s gynecologic surgeons.
Several concerns have been raised about the current traditional 4-year residency training program, which has been impacted by the reduction of training hours due to duty-hour rules in the setting of decreased surgical volume and new technologies used to perform surgery. While other surgical specialties have begun to innovate their pathways for trainees, ObGyn has been a little slower to make a significant transition in its approach to training.
In 2012, Cleveland Clinic decided to lead the way in innovation regarding residency training. At its inception, the curriculum was designed to allow “tracking blocks” through each academic year to allow residents to gain additional experience in their specialty of choice. The program was carefully designed to assure that residents would achieve all 28 of the core obstetrics and gynecology milestones while still allowing for curricular flexibility.
Currently, residents are given autonomy to design their own tracking blocks with an assigned mentor for the rotation. Allowing residents to spend more time in their specialty of choice permits them to fine-tune skills that a standard curriculum may not have afforded the opportunity to home in on. It also allows residents to gain exposure to specialties that are not part of the core program, such as vulvar health, breast health and surgery, and gender affirmation surgery.
The Cleveland Clinic experience has been successful thus far. Importantly, preliminary data show that the tracking program does not interfere with the overall case number necessary for graduation. Residents also have succeeded in their postgraduation pursuits, including those who chose to specialize in general obstetrics and gynecology.
Cleveland Clinic is no longer the only program to incorporate tracking into its curriculum. This innovation is likely to become more standard as medical education in ObGyn evolves. We have not yet “separated the O from the G” completely in our specialty. However, thought leaders in our field are recognizing the need to better prepare our trainees, and this flexibility in mindset is bound to lead to a paradigm that may become the new standard for our specialty.
Acknowledgments: John E. Jelovsek, MD, the first Program Director of the Cleveland Clinic Residency in Obstetrics & Gynecology, who was responsible for creating the tracking program; and Vicki Reed, MD, the current Program Director, who has continued to innovate the program.
The Miya Model (Miyazaki Enterprises LLC) is a multiprocedural vaginal surgery simulator born from the need for standardized, scalable training in response to reductions in the average surgical case volume per resident. The Miya Model supports various basic procedures, such as pelvic exams and dilation and curettage, as well as full surgical procedures, including anterior and posterior colporrhaphy, midurethral and retropubic slings, cystoscopy, and vaginal hysterectomy. Training with the Miya Model moves resident surgical education from the operating room to any simulation lab or office-based setting. With rapidly declining resident surgical case volumes, there is an even stronger need to provide additional training outside of the operating room theater. Creation and development of the Miya Model were fueled by a desire to create a safer and more efficient method to educate residents without the risk of patient harm.
Miyazaki Enterprises has taken the Miya Model from a vision on paper to a standardized, commercially available product to help support resident and physician education. The Miya Model has undergone numerous rounds of waterfall and agile development, validity testing, and the creation of internal and external processes to achieve this vision. It serves as an example that ideas originating from significant demonstrated market need can be successfully created and deployed by a physician.
- Espey E, Ogburn T, Puscheck E. Impact of duty hour limitations on resident and student education in obstetrics and gynecology. J Reprod Med. 2007;52:345-348.
- Pulliam SJ, Berkowitz LR. Smaller pieces of the hysterectomy pie: current challenges in resident surgical education. Obstet Gynecol. 2009;113(2 pt 1):395-398. doi: 10.1097/AOG.0b013e3181955011.
- Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241. doi: 10.1097/AOG.0b013e318299a6cf.
- Cadish LA, Kropat G, Muffly TM. Hysterectomy volume among recent obstetrics and gynecology residency graduates. Female Pelvic Med Reconstr Surg. 2021;27:382-387. doi: 10.1097/SPV.0000000000000879.
- Podratz KC. Gynecologic surgery: an imperiled ballet. Presidential address. Am J Obstet Gynecol. 1998;178:1229-1234. doi: 10.1016/ s0002-9378(98)70327-8.
- Bissonnette JM, Gabbe SG, Hammond CB, et al. Restructuring residency training in obstetrics and gynecology. Am J Obstet Gynecol. 1999;180(3 pt 1):516-518. doi: 10.1016/s0002-9378(99)70246-2.
- Magrina JF. Isn’t it time to separate the O from the G? J Minim Invasive Gynecol. 2014;21:501-503. doi: 10.1016/j.jmig.2014.01.022.
- Merrill JA. Needed changes in obstetric-gynecologic training. Obstet Gynecol Surv. 1994;49:1-2.
- Lauer JK, Advincula AP. The future of the gynecologic surgeon: rationale for and steps toward subspecialization of complex gynecologic surgery. J Minim Invasive Gynecol. 2021;28:726-729. doi: 10.1016/j.jmig.2020.12.031.
- Hall EF, Raker CA, Hampton BS. Variability in gynecologic case volume of obstetrician-gynecologist residents graduating from 2009 to 2017. Am J Obstet Gynecol. 2020;222:617.e1-617.e8. doi: 10.1016/j .ajog.2019.11.1258.
- Irby DM, Cooke M, O’Brien BC. Calls for reform of medical education by the Carnegie Foundation for the Advancement of Teaching: 1910 and 2010. Acad Med. 2010;85:220-227. doi: 10.1097 /ACM.0b013e3181c88449.
- Reed VR, Emery J, Farrell RM, et al. Tracking—a flexible obstetrics and gynecology residency curriculum. Obstet Gynecol. 2019;134(suppl 1):29s-33s. doi: 10.1097/AOG.0000000000003464.
- Hariton E, Freret TS, Nitecki R, et al. Program director perceptions of subspecialty tracking in obstetrics and gynecology residency. J Grad Med Educ. 2018;10:665-670. doi: 10.4300/JGME-D-18-00096.1.
- Azadi S, Green IC, Arnold A, et al. Robotic surgery: the impact of simulation and other innovative platforms on performance and training. J Minim Invasive Gynecol. 2021;28:490-495. doi: 10.1016/j .jmig.2020.12.001.
- Wohlrab K, Jelovsek JE, Myers D. Incorporating simulation into gynecologic surgical training. Am J Obstet Gynecol. 2017;217:522-526. doi: 10.1016/j.ajog.2017.05.017.
- Chen CC, Green IC, Colbert-Getz JM, et al. Warm-up on a simulator improves residents’ performance in laparoscopic surgery: a randomized trial. Int Urogynecol J. 2013;24:1615-1622. doi: 10.1007 /s00192-013-2066-2.
- Fundamentals of Laparoscopic Surgery. ABOG announces new eligibility requirement for board certification. January 23, 2018. Accessed May 12, 2022. https://www.flsprogram.org/news/abog -announces-new-eligibility-requirement-board-certification/.
- Zoorob D, Frenn R, Moffitt M, et al. Multi-institutional validation of a vaginal hysterectomy simulation model for resident training. J Minim Invasive Gynecol. 2021;28:1490-1496.e1. doi: 10.1016/j .jmig.2020.12.006.
- Barrier BF, Thompson AB, McCullough MW, et al. A novel and inexpensive vaginal hysterectomy simulator. Simul Healthc. 2012;7:374-379. doi: 10.1097/SIH.0b013e318266d0c6.
- Stickrath E, Alston M. A novel abdominal hysterectomy simulator and its impact on obstetrics and gynecology residents’ surgical confidence. MedEdPORTAL. 2017;13:10636. doi: 10.15766/mep_2374-8265.10636.
- McKendy KM, Watanabe Y, Lee L, et al. Perioperative feedback in surgical training: a systematic review. Am J Surg. 2017;214:117-126. doi: 10.1016/j.amjsurg.2016.12.014.
- Ahmed M, Arora S, Russ S, et al. Operation debrief: a SHARP improvement in performance feedback in the operating room. Ann Surg. 2013;258:958-963. doi: 10.1097/SLA.0b013e31828c88fc.
- Anderson CI, Gupta RN, Larson JR, et al. Impact of objectively assessing surgeons’ teaching on effective perioperative instructional behaviors. JAMA Surg. 2013;148:915-922. doi: 10.1001/jamasurg.2013.2144.
Obstetrics and gynecology (ObGyn) is a surgical specialty, yet the training of ObGyn residents differs significantly from that of residents in other surgical specialties. In addition to attaining competency in both the distinct but related fields of obstetrics and gynecology, ObGyn residents have their training condensed into 4 years rather than the 5 years’ training of many other surgical specialties. This limits the time dedicated to gynecologic surgery, currently 18 to 20 months in most programs, and has been exacerbated by tighter duty-hour restrictions.1
Additionally, with increasing demand for minimally invasive procedures, residents are expected to attain competency in a growing breadth of gynecologic procedures in a patient population with increasing morbidity, and they may have less autonomy to do so in an increasingly litigious environment.2 Furthermore, annual hysterectomy cases are declining, from about 680,000 in 2002 to 430,000 in 2010,3 and these declining rates are seen in the low case numbers of recent graduates.4
Training time, procedure complexity
With less time to master a growing body of increasingly complex procedures, is the profession adequately training gynecologic surgeons? Many gynecologic surgeons are concerned that the answer is no and that significant shifts in resident training are needed to generate safe and competent gynecologic surgeons. These training deficits represent a deficiency in the quality of care for women specifically, and thus the inattention to training gynecologic surgeons should be considered a health care disparity.
The concern over insufficient attention to gynecologic surgical training is not new, nor are proposed solutions, with many physicians citing the above concerns.5-9 In 2018, the Accreditation Council for Graduate Medical Education (ACGME) case minimums for hysterectomy increased to 85 from 70 hysterectomies, with a shift toward minimally invasive hysterectomy.10 Otherwise, minimal national changes have been made in this century to training gynecologic surgeons.
Tracking as an option
Many critics of current ObGyn training argue that obstetrics and gynecology, while related, have significantly different pathologies, surgical approaches, and skill sets and thus warrant the option to track toward obstetrics or gynecology after attaining limited core skill set in residency. In 2010, the Carnegie Foundation for the Advancement of Teaching called for the need for increased individualization opportunities in graduate medical education, citing that minimal changes have been made to medical education since the Flexner Report a century prior.11
Notably, tracking has been implemented with success at Cleveland Clinic, where residents are given 5 to 10 weeks of time allotted to their specific fields of interest, while still meeting minimum ACGME requirements and, in some cases, exceeding hysterectomy minimums by as much as 500%.12 Tracking is viewed positively by a majority of program directors.13 See the box below for Dr. Ferrando’s experience on tracking at the Cleveland Clinic.
Simulation training
Other educators advocate for maximizing preparedness for the operating room by using high-fidelity simulation.14,15 Simulation allows for the acquisition of basic technical skills needed for surgery as well as for repetition not easily achieved in the current surgical environment. Additionally, it provides lower-level learners the opportunity to acquire basic skills in a safe setting, thereby enhancing the ability to participate meaningfully on arrival in the operating room.16
In 2018, the American Board of Obstetrics and Gynecology added the Fundamentals of Laparoscopic Surgery certification as a new requirement for board certification.17 Laparoscopic and robotic surgery simulators allow trainees to develop coordination and specific skills, like knot tying and suturing. Additionally, models are available with varying levels of fidelity for vaginal and abdominal hysterectomy.18-20 See the box below for Dr. Miyazaki’s experience in developing the Miya Model trainer for vaginal surgery simulation.
Structured feedback
Finally, if a resident has limited exposure to a specific procedure, maximizing the preparation and feedback for each procedure is paramount. However, surgeons receive minimal formal training in teaching trainees, which leads to inconsistent and underutilized feedback.21 Specific structured feedback models have been implemented with success in the general surgery literature, including the SHARP (Set learning objectives, How did it go, Address concerns, Review learning points, Plan ahead) and BID (Briefing, Intraoperative, Debriefing) models.22,23
Reimbursement reform
While surgical reimbursement is not directly tied to resident education, decreased reimbursement to women’s health pathology and procedures has the downstream effect of decreasing the funds available for ObGyn departments to invest in research and education. Additionally, “suboptimal mastery or maintenance of appropriate surgical skills results in procedural inefficiencies that compound surgical cost.”5 Providers and payors alike should therefore be motivated to improve funding in order to improve adequate training of gynecologic surgeons. Payment reform is necessary to equally value women’s health procedures but also can ensure that gynecologic surgeons have the funds needed to train a competent next generation of ObGyn physicians. ●
- Residents and fellows have significant constraints that limit adequate training in gynecologic surgery. In a panel discussion at the 48th annual meeting of the Society of Gynecologic Surgeons, Drs. Zimmerman, Ferrando, and Miyazaki spoke about potential solutions.
- Allowing residents to track toward obstetric or gynecologic subspecialties may improve surgical volume of trainees who aim for a future career in gynecologic surgery.
- Simulation has demonstrated efficacy in enabling residents to prepare and improve their technical skills for specific procedures prior to entering the operating room.
Cecile A. Ferrando, MD, MPH
In his 2013 presidential address at the opening ceremony of the 42nd AAGL Global Congress on Minimally Invasive Gynecology, Javier Magrina, MD, asked the audience, “Isn’t it time to separate the O from the G?”7 Since that address, this catchy question has been posed several times, and it continues to be a topic of interest to many ObGyn educators seeking to innovate the curriculum and to better train our next generation’s gynecologic surgeons.
Several concerns have been raised about the current traditional 4-year residency training program, which has been impacted by the reduction of training hours due to duty-hour rules in the setting of decreased surgical volume and new technologies used to perform surgery. While other surgical specialties have begun to innovate their pathways for trainees, ObGyn has been a little slower to make a significant transition in its approach to training.
In 2012, Cleveland Clinic decided to lead the way in innovation regarding residency training. At its inception, the curriculum was designed to allow “tracking blocks” through each academic year to allow residents to gain additional experience in their specialty of choice. The program was carefully designed to assure that residents would achieve all 28 of the core obstetrics and gynecology milestones while still allowing for curricular flexibility.
Currently, residents are given autonomy to design their own tracking blocks with an assigned mentor for the rotation. Allowing residents to spend more time in their specialty of choice permits them to fine-tune skills that a standard curriculum may not have afforded the opportunity to home in on. It also allows residents to gain exposure to specialties that are not part of the core program, such as vulvar health, breast health and surgery, and gender affirmation surgery.
The Cleveland Clinic experience has been successful thus far. Importantly, preliminary data show that the tracking program does not interfere with the overall case number necessary for graduation. Residents also have succeeded in their postgraduation pursuits, including those who chose to specialize in general obstetrics and gynecology.
Cleveland Clinic is no longer the only program to incorporate tracking into its curriculum. This innovation is likely to become more standard as medical education in ObGyn evolves. We have not yet “separated the O from the G” completely in our specialty. However, thought leaders in our field are recognizing the need to better prepare our trainees, and this flexibility in mindset is bound to lead to a paradigm that may become the new standard for our specialty.
Acknowledgments: John E. Jelovsek, MD, the first Program Director of the Cleveland Clinic Residency in Obstetrics & Gynecology, who was responsible for creating the tracking program; and Vicki Reed, MD, the current Program Director, who has continued to innovate the program.
The Miya Model (Miyazaki Enterprises LLC) is a multiprocedural vaginal surgery simulator born from the need for standardized, scalable training in response to reductions in the average surgical case volume per resident. The Miya Model supports various basic procedures, such as pelvic exams and dilation and curettage, as well as full surgical procedures, including anterior and posterior colporrhaphy, midurethral and retropubic slings, cystoscopy, and vaginal hysterectomy. Training with the Miya Model moves resident surgical education from the operating room to any simulation lab or office-based setting. With rapidly declining resident surgical case volumes, there is an even stronger need to provide additional training outside of the operating room theater. Creation and development of the Miya Model were fueled by a desire to create a safer and more efficient method to educate residents without the risk of patient harm.
Miyazaki Enterprises has taken the Miya Model from a vision on paper to a standardized, commercially available product to help support resident and physician education. The Miya Model has undergone numerous rounds of waterfall and agile development, validity testing, and the creation of internal and external processes to achieve this vision. It serves as an example that ideas originating from significant demonstrated market need can be successfully created and deployed by a physician.
Obstetrics and gynecology (ObGyn) is a surgical specialty, yet the training of ObGyn residents differs significantly from that of residents in other surgical specialties. In addition to attaining competency in both the distinct but related fields of obstetrics and gynecology, ObGyn residents have their training condensed into 4 years rather than the 5 years’ training of many other surgical specialties. This limits the time dedicated to gynecologic surgery, currently 18 to 20 months in most programs, and has been exacerbated by tighter duty-hour restrictions.1
Additionally, with increasing demand for minimally invasive procedures, residents are expected to attain competency in a growing breadth of gynecologic procedures in a patient population with increasing morbidity, and they may have less autonomy to do so in an increasingly litigious environment.2 Furthermore, annual hysterectomy cases are declining, from about 680,000 in 2002 to 430,000 in 2010,3 and these declining rates are seen in the low case numbers of recent graduates.4
Training time, procedure complexity
With less time to master a growing body of increasingly complex procedures, is the profession adequately training gynecologic surgeons? Many gynecologic surgeons are concerned that the answer is no and that significant shifts in resident training are needed to generate safe and competent gynecologic surgeons. These training deficits represent a deficiency in the quality of care for women specifically, and thus the inattention to training gynecologic surgeons should be considered a health care disparity.
The concern over insufficient attention to gynecologic surgical training is not new, nor are proposed solutions, with many physicians citing the above concerns.5-9 In 2018, the Accreditation Council for Graduate Medical Education (ACGME) case minimums for hysterectomy increased to 85 from 70 hysterectomies, with a shift toward minimally invasive hysterectomy.10 Otherwise, minimal national changes have been made in this century to training gynecologic surgeons.
Tracking as an option
Many critics of current ObGyn training argue that obstetrics and gynecology, while related, have significantly different pathologies, surgical approaches, and skill sets and thus warrant the option to track toward obstetrics or gynecology after attaining limited core skill set in residency. In 2010, the Carnegie Foundation for the Advancement of Teaching called for the need for increased individualization opportunities in graduate medical education, citing that minimal changes have been made to medical education since the Flexner Report a century prior.11
Notably, tracking has been implemented with success at Cleveland Clinic, where residents are given 5 to 10 weeks of time allotted to their specific fields of interest, while still meeting minimum ACGME requirements and, in some cases, exceeding hysterectomy minimums by as much as 500%.12 Tracking is viewed positively by a majority of program directors.13 See the box below for Dr. Ferrando’s experience on tracking at the Cleveland Clinic.
Simulation training
Other educators advocate for maximizing preparedness for the operating room by using high-fidelity simulation.14,15 Simulation allows for the acquisition of basic technical skills needed for surgery as well as for repetition not easily achieved in the current surgical environment. Additionally, it provides lower-level learners the opportunity to acquire basic skills in a safe setting, thereby enhancing the ability to participate meaningfully on arrival in the operating room.16
In 2018, the American Board of Obstetrics and Gynecology added the Fundamentals of Laparoscopic Surgery certification as a new requirement for board certification.17 Laparoscopic and robotic surgery simulators allow trainees to develop coordination and specific skills, like knot tying and suturing. Additionally, models are available with varying levels of fidelity for vaginal and abdominal hysterectomy.18-20 See the box below for Dr. Miyazaki’s experience in developing the Miya Model trainer for vaginal surgery simulation.
Structured feedback
Finally, if a resident has limited exposure to a specific procedure, maximizing the preparation and feedback for each procedure is paramount. However, surgeons receive minimal formal training in teaching trainees, which leads to inconsistent and underutilized feedback.21 Specific structured feedback models have been implemented with success in the general surgery literature, including the SHARP (Set learning objectives, How did it go, Address concerns, Review learning points, Plan ahead) and BID (Briefing, Intraoperative, Debriefing) models.22,23
Reimbursement reform
While surgical reimbursement is not directly tied to resident education, decreased reimbursement to women’s health pathology and procedures has the downstream effect of decreasing the funds available for ObGyn departments to invest in research and education. Additionally, “suboptimal mastery or maintenance of appropriate surgical skills results in procedural inefficiencies that compound surgical cost.”5 Providers and payors alike should therefore be motivated to improve funding in order to improve adequate training of gynecologic surgeons. Payment reform is necessary to equally value women’s health procedures but also can ensure that gynecologic surgeons have the funds needed to train a competent next generation of ObGyn physicians. ●
- Residents and fellows have significant constraints that limit adequate training in gynecologic surgery. In a panel discussion at the 48th annual meeting of the Society of Gynecologic Surgeons, Drs. Zimmerman, Ferrando, and Miyazaki spoke about potential solutions.
- Allowing residents to track toward obstetric or gynecologic subspecialties may improve surgical volume of trainees who aim for a future career in gynecologic surgery.
- Simulation has demonstrated efficacy in enabling residents to prepare and improve their technical skills for specific procedures prior to entering the operating room.
Cecile A. Ferrando, MD, MPH
In his 2013 presidential address at the opening ceremony of the 42nd AAGL Global Congress on Minimally Invasive Gynecology, Javier Magrina, MD, asked the audience, “Isn’t it time to separate the O from the G?”7 Since that address, this catchy question has been posed several times, and it continues to be a topic of interest to many ObGyn educators seeking to innovate the curriculum and to better train our next generation’s gynecologic surgeons.
Several concerns have been raised about the current traditional 4-year residency training program, which has been impacted by the reduction of training hours due to duty-hour rules in the setting of decreased surgical volume and new technologies used to perform surgery. While other surgical specialties have begun to innovate their pathways for trainees, ObGyn has been a little slower to make a significant transition in its approach to training.
In 2012, Cleveland Clinic decided to lead the way in innovation regarding residency training. At its inception, the curriculum was designed to allow “tracking blocks” through each academic year to allow residents to gain additional experience in their specialty of choice. The program was carefully designed to assure that residents would achieve all 28 of the core obstetrics and gynecology milestones while still allowing for curricular flexibility.
Currently, residents are given autonomy to design their own tracking blocks with an assigned mentor for the rotation. Allowing residents to spend more time in their specialty of choice permits them to fine-tune skills that a standard curriculum may not have afforded the opportunity to home in on. It also allows residents to gain exposure to specialties that are not part of the core program, such as vulvar health, breast health and surgery, and gender affirmation surgery.
The Cleveland Clinic experience has been successful thus far. Importantly, preliminary data show that the tracking program does not interfere with the overall case number necessary for graduation. Residents also have succeeded in their postgraduation pursuits, including those who chose to specialize in general obstetrics and gynecology.
Cleveland Clinic is no longer the only program to incorporate tracking into its curriculum. This innovation is likely to become more standard as medical education in ObGyn evolves. We have not yet “separated the O from the G” completely in our specialty. However, thought leaders in our field are recognizing the need to better prepare our trainees, and this flexibility in mindset is bound to lead to a paradigm that may become the new standard for our specialty.
Acknowledgments: John E. Jelovsek, MD, the first Program Director of the Cleveland Clinic Residency in Obstetrics & Gynecology, who was responsible for creating the tracking program; and Vicki Reed, MD, the current Program Director, who has continued to innovate the program.
The Miya Model (Miyazaki Enterprises LLC) is a multiprocedural vaginal surgery simulator born from the need for standardized, scalable training in response to reductions in the average surgical case volume per resident. The Miya Model supports various basic procedures, such as pelvic exams and dilation and curettage, as well as full surgical procedures, including anterior and posterior colporrhaphy, midurethral and retropubic slings, cystoscopy, and vaginal hysterectomy. Training with the Miya Model moves resident surgical education from the operating room to any simulation lab or office-based setting. With rapidly declining resident surgical case volumes, there is an even stronger need to provide additional training outside of the operating room theater. Creation and development of the Miya Model were fueled by a desire to create a safer and more efficient method to educate residents without the risk of patient harm.
Miyazaki Enterprises has taken the Miya Model from a vision on paper to a standardized, commercially available product to help support resident and physician education. The Miya Model has undergone numerous rounds of waterfall and agile development, validity testing, and the creation of internal and external processes to achieve this vision. It serves as an example that ideas originating from significant demonstrated market need can be successfully created and deployed by a physician.
- Espey E, Ogburn T, Puscheck E. Impact of duty hour limitations on resident and student education in obstetrics and gynecology. J Reprod Med. 2007;52:345-348.
- Pulliam SJ, Berkowitz LR. Smaller pieces of the hysterectomy pie: current challenges in resident surgical education. Obstet Gynecol. 2009;113(2 pt 1):395-398. doi: 10.1097/AOG.0b013e3181955011.
- Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241. doi: 10.1097/AOG.0b013e318299a6cf.
- Cadish LA, Kropat G, Muffly TM. Hysterectomy volume among recent obstetrics and gynecology residency graduates. Female Pelvic Med Reconstr Surg. 2021;27:382-387. doi: 10.1097/SPV.0000000000000879.
- Podratz KC. Gynecologic surgery: an imperiled ballet. Presidential address. Am J Obstet Gynecol. 1998;178:1229-1234. doi: 10.1016/ s0002-9378(98)70327-8.
- Bissonnette JM, Gabbe SG, Hammond CB, et al. Restructuring residency training in obstetrics and gynecology. Am J Obstet Gynecol. 1999;180(3 pt 1):516-518. doi: 10.1016/s0002-9378(99)70246-2.
- Magrina JF. Isn’t it time to separate the O from the G? J Minim Invasive Gynecol. 2014;21:501-503. doi: 10.1016/j.jmig.2014.01.022.
- Merrill JA. Needed changes in obstetric-gynecologic training. Obstet Gynecol Surv. 1994;49:1-2.
- Lauer JK, Advincula AP. The future of the gynecologic surgeon: rationale for and steps toward subspecialization of complex gynecologic surgery. J Minim Invasive Gynecol. 2021;28:726-729. doi: 10.1016/j.jmig.2020.12.031.
- Hall EF, Raker CA, Hampton BS. Variability in gynecologic case volume of obstetrician-gynecologist residents graduating from 2009 to 2017. Am J Obstet Gynecol. 2020;222:617.e1-617.e8. doi: 10.1016/j .ajog.2019.11.1258.
- Irby DM, Cooke M, O’Brien BC. Calls for reform of medical education by the Carnegie Foundation for the Advancement of Teaching: 1910 and 2010. Acad Med. 2010;85:220-227. doi: 10.1097 /ACM.0b013e3181c88449.
- Reed VR, Emery J, Farrell RM, et al. Tracking—a flexible obstetrics and gynecology residency curriculum. Obstet Gynecol. 2019;134(suppl 1):29s-33s. doi: 10.1097/AOG.0000000000003464.
- Hariton E, Freret TS, Nitecki R, et al. Program director perceptions of subspecialty tracking in obstetrics and gynecology residency. J Grad Med Educ. 2018;10:665-670. doi: 10.4300/JGME-D-18-00096.1.
- Azadi S, Green IC, Arnold A, et al. Robotic surgery: the impact of simulation and other innovative platforms on performance and training. J Minim Invasive Gynecol. 2021;28:490-495. doi: 10.1016/j .jmig.2020.12.001.
- Wohlrab K, Jelovsek JE, Myers D. Incorporating simulation into gynecologic surgical training. Am J Obstet Gynecol. 2017;217:522-526. doi: 10.1016/j.ajog.2017.05.017.
- Chen CC, Green IC, Colbert-Getz JM, et al. Warm-up on a simulator improves residents’ performance in laparoscopic surgery: a randomized trial. Int Urogynecol J. 2013;24:1615-1622. doi: 10.1007 /s00192-013-2066-2.
- Fundamentals of Laparoscopic Surgery. ABOG announces new eligibility requirement for board certification. January 23, 2018. Accessed May 12, 2022. https://www.flsprogram.org/news/abog -announces-new-eligibility-requirement-board-certification/.
- Zoorob D, Frenn R, Moffitt M, et al. Multi-institutional validation of a vaginal hysterectomy simulation model for resident training. J Minim Invasive Gynecol. 2021;28:1490-1496.e1. doi: 10.1016/j .jmig.2020.12.006.
- Barrier BF, Thompson AB, McCullough MW, et al. A novel and inexpensive vaginal hysterectomy simulator. Simul Healthc. 2012;7:374-379. doi: 10.1097/SIH.0b013e318266d0c6.
- Stickrath E, Alston M. A novel abdominal hysterectomy simulator and its impact on obstetrics and gynecology residents’ surgical confidence. MedEdPORTAL. 2017;13:10636. doi: 10.15766/mep_2374-8265.10636.
- McKendy KM, Watanabe Y, Lee L, et al. Perioperative feedback in surgical training: a systematic review. Am J Surg. 2017;214:117-126. doi: 10.1016/j.amjsurg.2016.12.014.
- Ahmed M, Arora S, Russ S, et al. Operation debrief: a SHARP improvement in performance feedback in the operating room. Ann Surg. 2013;258:958-963. doi: 10.1097/SLA.0b013e31828c88fc.
- Anderson CI, Gupta RN, Larson JR, et al. Impact of objectively assessing surgeons’ teaching on effective perioperative instructional behaviors. JAMA Surg. 2013;148:915-922. doi: 10.1001/jamasurg.2013.2144.
- Espey E, Ogburn T, Puscheck E. Impact of duty hour limitations on resident and student education in obstetrics and gynecology. J Reprod Med. 2007;52:345-348.
- Pulliam SJ, Berkowitz LR. Smaller pieces of the hysterectomy pie: current challenges in resident surgical education. Obstet Gynecol. 2009;113(2 pt 1):395-398. doi: 10.1097/AOG.0b013e3181955011.
- Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241. doi: 10.1097/AOG.0b013e318299a6cf.
- Cadish LA, Kropat G, Muffly TM. Hysterectomy volume among recent obstetrics and gynecology residency graduates. Female Pelvic Med Reconstr Surg. 2021;27:382-387. doi: 10.1097/SPV.0000000000000879.
- Podratz KC. Gynecologic surgery: an imperiled ballet. Presidential address. Am J Obstet Gynecol. 1998;178:1229-1234. doi: 10.1016/ s0002-9378(98)70327-8.
- Bissonnette JM, Gabbe SG, Hammond CB, et al. Restructuring residency training in obstetrics and gynecology. Am J Obstet Gynecol. 1999;180(3 pt 1):516-518. doi: 10.1016/s0002-9378(99)70246-2.
- Magrina JF. Isn’t it time to separate the O from the G? J Minim Invasive Gynecol. 2014;21:501-503. doi: 10.1016/j.jmig.2014.01.022.
- Merrill JA. Needed changes in obstetric-gynecologic training. Obstet Gynecol Surv. 1994;49:1-2.
- Lauer JK, Advincula AP. The future of the gynecologic surgeon: rationale for and steps toward subspecialization of complex gynecologic surgery. J Minim Invasive Gynecol. 2021;28:726-729. doi: 10.1016/j.jmig.2020.12.031.
- Hall EF, Raker CA, Hampton BS. Variability in gynecologic case volume of obstetrician-gynecologist residents graduating from 2009 to 2017. Am J Obstet Gynecol. 2020;222:617.e1-617.e8. doi: 10.1016/j .ajog.2019.11.1258.
- Irby DM, Cooke M, O’Brien BC. Calls for reform of medical education by the Carnegie Foundation for the Advancement of Teaching: 1910 and 2010. Acad Med. 2010;85:220-227. doi: 10.1097 /ACM.0b013e3181c88449.
- Reed VR, Emery J, Farrell RM, et al. Tracking—a flexible obstetrics and gynecology residency curriculum. Obstet Gynecol. 2019;134(suppl 1):29s-33s. doi: 10.1097/AOG.0000000000003464.
- Hariton E, Freret TS, Nitecki R, et al. Program director perceptions of subspecialty tracking in obstetrics and gynecology residency. J Grad Med Educ. 2018;10:665-670. doi: 10.4300/JGME-D-18-00096.1.
- Azadi S, Green IC, Arnold A, et al. Robotic surgery: the impact of simulation and other innovative platforms on performance and training. J Minim Invasive Gynecol. 2021;28:490-495. doi: 10.1016/j .jmig.2020.12.001.
- Wohlrab K, Jelovsek JE, Myers D. Incorporating simulation into gynecologic surgical training. Am J Obstet Gynecol. 2017;217:522-526. doi: 10.1016/j.ajog.2017.05.017.
- Chen CC, Green IC, Colbert-Getz JM, et al. Warm-up on a simulator improves residents’ performance in laparoscopic surgery: a randomized trial. Int Urogynecol J. 2013;24:1615-1622. doi: 10.1007 /s00192-013-2066-2.
- Fundamentals of Laparoscopic Surgery. ABOG announces new eligibility requirement for board certification. January 23, 2018. Accessed May 12, 2022. https://www.flsprogram.org/news/abog -announces-new-eligibility-requirement-board-certification/.
- Zoorob D, Frenn R, Moffitt M, et al. Multi-institutional validation of a vaginal hysterectomy simulation model for resident training. J Minim Invasive Gynecol. 2021;28:1490-1496.e1. doi: 10.1016/j .jmig.2020.12.006.
- Barrier BF, Thompson AB, McCullough MW, et al. A novel and inexpensive vaginal hysterectomy simulator. Simul Healthc. 2012;7:374-379. doi: 10.1097/SIH.0b013e318266d0c6.
- Stickrath E, Alston M. A novel abdominal hysterectomy simulator and its impact on obstetrics and gynecology residents’ surgical confidence. MedEdPORTAL. 2017;13:10636. doi: 10.15766/mep_2374-8265.10636.
- McKendy KM, Watanabe Y, Lee L, et al. Perioperative feedback in surgical training: a systematic review. Am J Surg. 2017;214:117-126. doi: 10.1016/j.amjsurg.2016.12.014.
- Ahmed M, Arora S, Russ S, et al. Operation debrief: a SHARP improvement in performance feedback in the operating room. Ann Surg. 2013;258:958-963. doi: 10.1097/SLA.0b013e31828c88fc.
- Anderson CI, Gupta RN, Larson JR, et al. Impact of objectively assessing surgeons’ teaching on effective perioperative instructional behaviors. JAMA Surg. 2013;148:915-922. doi: 10.1001/jamasurg.2013.2144.
Vesicovaginal and rectovaginal fistulas from obstetric-related causes: Diagnosis and management
Although rare in the United States and more common in low-resource countries, fistulas due to obstructed labor do occur. In developed countries, other obstetric causes for fistula are usually surgery, trauma, or infection related. An abnormal communication between organs—be it the urethra, bladder, ureter, uterus, cervix, or rectum—can develop1 and lead to vesicovaginal fistula (VVF), urethrovaginal fistula (FIGURE 1), vesicocervical fistula, vesicouterine fistula, ureterovaginal fistula (FIGURE 2), and rectovaginal fistula (RVF). Other nonobstetric causes include gynecologic surgery, radiation, malignancy, and congenital malformations.
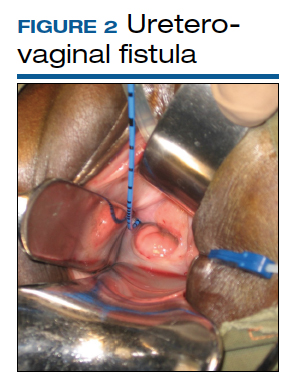
During labor, hypoxia, subsequent ischemia, and pressure necrosis contribute to fistula formation. Injury sustained during a cesarean delivery (CD) or cesarean hysterectomy can lead to fistula formation; at times, however, complications are unavoidable given the nature of the pathologic condition that the patient presents with.
VVF and RVF have a devastating impact on a woman’s quality of life as they lead to significant morbidity and short- and long-term psychological distress. The fistula may not be recognized at the time of injury. The presenting signs and symptoms may be intermittent and confusing. Immediate surgical intervention may not be possible due to ongoing inflammation or infection. Recovery often is prolonged. As there is significant concomitant postpartum anxiety and depression, patients with fistula often require psychosocial support and counseling. After repair, there is still a risk for recurrence and voiding dysfunction.
Fistula signs and symptoms and evaluation
In cases of VVF, patients present with continuing large or small volume urinary incontinence. Depending on the time to diagnosis, patients may have calculi formation, prolapse, scarring, external perineal dermatitis, perineal nerve injury, and even motor weakness. Cyclic hematuria may be seen in vesicouterine fistulas.2
Multiple classification systems for diagnosis and staging of VVF have been suggested.3,4 A classification system for RVF was published by Tsang and colleagues.5 All these classification systems have attempted to characterize fistulas in terms of level of surgical complexity for repair, providing a guideline for preoperative assessment. These classification systems do not translate into prediction regarding outcomes.
Evaluation of pelvic fistula from the urinary tract starts with a thorough history that includes onset, duration, and description of leakage (continuous, intermittent, or positional) and whether there is concomitant stress and urge incontinence. A detailed obstetric history, including circumstances around the mode of delivery, underlying risk factors, and psychosocial history, should be obtained.
The pelvic examination with a plastic speculum and adequate lighting should assess the external perineum for dermatitis; bulbocavernosus and anal reflexes; and the vagina for length, caliber, level of scarring, and any prolapse. For VVFs, the location, size, and number of the fistula tracts can be visualized and confirmed with a retrograde fill of the bladder via a Foley catheter with saline or water mixed with methylene blue or any other blue dye (FIGURE 3). If a ureterovaginal fistula is suspected, the patient can simultaneously be given oral phenazopyridine and a tampon inserted within the vagina; the patient can then ambulate, and re-examination of the end of the tampon can reveal orange staining. The bladder meanwhile is retrograde filled with blue dye, with no blue staining of the tampon.
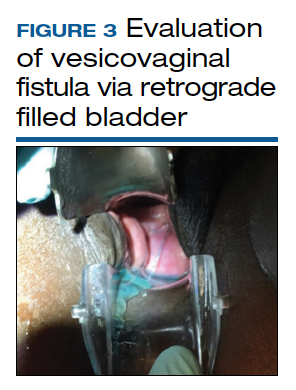
For RVF, history taking should include the onset, duration, and description of leakage, and the external anal sphincter should be assessed, with careful examination of the distal vagina at the vestibule as this is the most common location for RVF (fistula in ano). Patients may describe vaginal flatus and sometimes only brownish discharge, which can be intermittent, leading to an incorrect diagnosis of vaginitis that is treated repeatedly without success.
There is no consensus regarding optimal imaging for the assessment of VVF. Imaging used for diagnosis of VVF includes a voiding cystogram with opacification of the vagina after filling the bladder with contrast if there is a fistula. A cystoscopy can evaluate for calculi, retained suture, level of inflammation, and location of the ureters in relation to the fistula. Renal ultrasonography is of limited use. Intravenous pyelography can miss lesions by the trigone. In general, a computed tomography (CT) urogram and magnetic resonance imaging (MRI) with bladder contrast are more sensitive.
In the diagnosis of RVF, contrast vaginoscopy, double contrast barium enema, CT scan with contrast, and MRI can be used. MRI is more sensitive.6 A high index of suspicion is required based on the patient’s history as these imaging modalities do not always confirm RVF despite patient’s clear history of leakage. When the history is convincing, a thorough rectovaginal exam under anesthesia may be imperative. If rectal trauma is present, endoanal ultrasonography can delineate external and internal anal sphincter defects.
Prolonged Foley catheter placement after obstetric injury can lead to successful closure of a VVF. Prior to surgical intervention, assessing if there is possible ureteral involvement and use of intraoperative ureteral stents is a consideration. The route of surgery can be vaginal, abdominal, combined abdominal-vaginal, laparoscopic, or robotic.7 The robotic approach is increasingly utilized.8,9 However, the general consensus among fistula surgeons is that the vaginal approach should be considered first.
Continue to: Surgical repair...
Surgical repair
VVF repair. Factors that influence successful repair of VVF include the size and number of fistula, location, degree of scarring, bladder capacity, and urethral length.
Surgical technique requires wide mobilization and adequate exposure. The fistula tract can be delineated and manipulated with a pediatric Foley catheter, ureteral stent, or even a ureteral guidewire to aid in dissection (FIGURE 4). Intraoperative visualization of the ureters, including stenting, often is needed. The fistulous track is excised depending on the level of scarring. Closure of the bladder uroepithelium for the first layer is with absorbable interrupted 3-0 or 2-0 sutures in a tension-free closure. The bladder is then evaluated with a retrograde fill with saline and methylene blue to ensure a watertight closure for the first layer. If the first layer is not watertight, the second layer closure will not compensate and the fistula will persist. Particular attention is paid to the angles of the fistula at the first layer closure to prevent recurrence of the fistula at the angles. A running second layer with absorbable 2-0 suture is done. At times, a Martius flap or an omental J flap can be used to provide an additional layer for support and to increase vascularity.10 The patient is sent home with a Foley catheter for drainage for 10 to 14 days.11 Antibiotics are not needed postoperatively for VVF surgery.12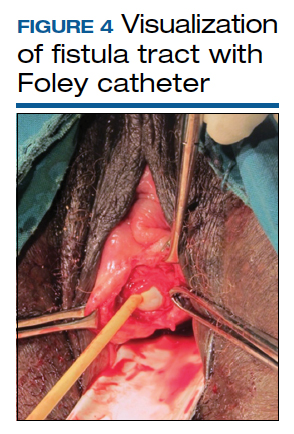
CT cystogram or retrograde cystogram is usually done to evaluate closure of the fistula prior to removal of the Foley catheter; retrograde fill with contrast directly into the bladder with 300 mL is sufficient (FIGURE 5). Patients are advised to refrain from sexual activity for a minimum of 6 weeks, but depending on the level of complexity and scarring, this can be up to 12 weeks.
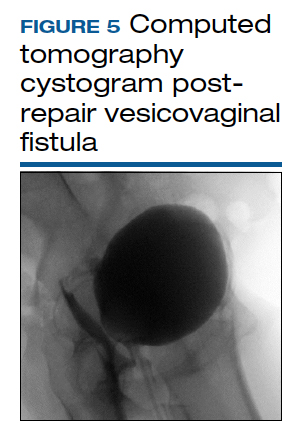
The success rate in general is in the 95% range. Patients with successful closure of VVF are at risk for urge incontinence due to decreased bladder capacity, stress incontinence especially if the continence mechanism or urethra is involved, vaginal scarring, dyspareunia, and infertility.13 In general, sexual function improves after surgical repair.
RVF repair. Prior to surgical repair of RVF, the integrity of the external anal sphincter must be determined. If it is not involved, a vertical incision is made in the posterior vaginal wall, the vaginal epithelium is separated from the vaginal muscularis, and the fistula tract is identified. After complete wide mobilization of the tissue surrounding the tract, it is excised. The rectal wall is repaired with 3-0 or 4-0 absorbable interrupted sutures; a second layer and if possible even a third layer and finally the vaginal epithelium are all closed with 2-0 absorbable interrupted sutures.
If the sphincter complex is involved, the dissection involves an inverted U incision separating the vaginal wall from the rectum. The fistula tract is excised, the rectal wall is closed, and the internal anal sphincter is identified and reapproximated with interrupted absorbable 2-0 or 0 sutures. The disrupted external sphincter is then reapproximated with 2-0 or 0 sutures, and finally the transverse perineal and bulbocavernosus muscles are brought together with Lembert 0 sutures prior to closure of the external skin. Perioperative antibiotics have been shown to improve success rates in the correction of RVF.5 In patients with sphincter trauma and known RVF, outcomes with a sphincteroplasty are better, compared with endorectal advancement flaps. The patient is discharged with a bowel regimen and dietary precautions that aim for daily soft bowel movements.
After surgical treatment of fistulas, patients benefit from pelvic floor physical therapy that focuses on pelvic floor strengthening. Incorporating the habit of Kegel exercises after every void, timed (scheduled) bladder voiding, and avoidance of straining with urination or defecation should be emphasized.
Continue to: CASE 1 Pregnant woman with rectal bleeding...
CASE 1 Pregnant woman with rectal bleeding
A 37-year-old woman at 36 3/7 weeks’ gestation presented with acute rectal bleeding and pain. This was found to result from a catastrophic rupture of a pelvic arteriovenous malformation that caused an 11 x 7 x 9.5 cm size inferior pelvic hematoma and a full-thickness rectal tear at the dentate line. During examination under anesthesia, the baby was delivered by a stat CD due to breech presentation and a prolonged fetal heart rate deceleration. The patient underwent embolization of the right middle rectal artery and right internal iliac artery by a radiologic intervention. Further bleeding required surgical intervention for evacuation of about 1,000 mL of hematoma, repair of the rectal tear, and laparoscopic diverting loop ileostomy. In total, the patient received 8 U of packed red blood cells, 6 U of fresh frozen plasma, 5 L of crystalloid solution, and 2 g of tranexamic acid. The patient reported increased foul-smelling vaginal discharge, bedside exam suggested possible fistulous tract, and on postoperative day 16, an exam under anesthesia by Urogynecology confirmed a rectovaginal fistula in the right mid vagina. After 2 months of observation to allow resolution of inflammation, successful excision of the fistula tract and repair of RVF using the above-mentioned technique was accomplished.
CASE 2 Patient with VVF after cesarean hysterectomy
A 40-year-old (G6P2222) patient underwent cesarean hysterectomy for placenta percreta and uterine rupture at 24 weeks’ gestation. Intraoperatively, there were right ureteral ligation and posterior bladder wall cystotomies. The right ureter was reimplanted in the right upper posterior wall and the cystostomies were closed. As the patient had continuous urinary leakage postoperatively, a CT urogram was obtained, which showed left ureteral obstruction and VVF. Urinary incontinence persisted despite bilateral robotic ureteral reimplantation with omental flap by the urology team. Percutaneous nephrostomy tubes were placed bilaterally. The patient underwent additional imaging studies, including MRI, with findings of VVF and possible ureterovaginal fistula.
On referral to Urogynecology, the patient underwent cystoscopy with antegrade pyelogram, and the bilateral ureteroneocystostomy orifices had 5 French open-ended ureteral stents placed. A 10 French pediatric Foley catheter was inserted intravaginally into the bladder through the VVF. Via the vaginal approach, cervical remnant and skin bridges overlying the VVF were excised. The scarred fistula tract was excised with a circumferential incision. Horizontal interrupted Lembert sutures with 3-0 absorbable suture were used to reapproximate the first layer, which was confirmed to be watertight on testing with retrograde fill. Second-layer closure was completed with horizontal mattress 2-0 absorbable sutures, followed by a third-layer closure done in similar fashion. Fibrin glue was then placed. The vaginal epithelium was closed with 2-0 absorbable suture. Percutaneous nephrostomy tubes were removed. Postoperatively, the patient had a CT cystogram with no leak and no incontinence, but she developed urgency, which was controlled with timed voids and oxybutynin.
- Adler AJ, Ronsmans C, Calvert C, et al. Estimating the presence of obstetric fistula: a systematic review and meta-analysis BMC Pregnancy Childbirth. 2013;13:246.
- Battacharjee S, Kohli UA, Sood A, et al. Vesicouterine fistula: Youssef’s syndrome. Med J Armed Forces India. 2015;71(suppl 1):S175-S177. doi: 10.1016/j.mjafi.2013.11.006.
- Waaldijk K. Step-by-Step Surgery of Vesicovaginal Fistulas. Campion Press; 1994.
- Goh, JTW. A new classification for female genital tract fistula. Aust N Z J Ob Gynecol. 2004:44:502-504.
- Tsang CB, Rothenberger DA. Rectovaginal fistulas: therapeutic options. Surg Clin North Am. 1997;77:95-114.
- Champagne BJ, McGee MF. Rectovaginal fistula. Surg Clin North Am. 2010;90:69-82.
- Bodner-Adler B, Hanzal E, Pablik E, et al. Management of vesicovaginal fistulas in women following benign gynecologic surgery: a systematic review and meta-analysis. PLoS One. 2017;12:e0171554.
- Randazzo M, Lengauer L, Rochat CH, et al. Best practices in robotic-assisted repair of vesicovaginal fistula: a consensus report from the European Association of Urology Robotic Urology Section Scientific Working Group for Reconstructive Urology. Eur Urol. 2020;78: 432-442.
- Miklos JR, Moore RD, Chinthakanan O. Laparoscopic and robotic assisted vesicovaginal fistula repair: a systematic review of the literature. J Minim Invasive Gynecol. 2015:22:727-736.
- Hancock B. Practical Obstetric Fistula Surgery. Royal Society of Medicine Press; 2009.
- Nardos R, Menber B, Browning A. Outcome of obstetric fistula repair after 10-day versus 14-day Foley catheterization. Int J Gynaecol 0bstet. 2012;118:21-23.
- Tomlinson AJ, Thornton JG. A randomized controlled trial of antibiotic prophylaxis for vesico-vaginal fistula repair. Br J Obstet Gynaecol. 2005;105:397-399.
- Bengtson AM, Kopp D, Tang JH, et al. Identifying patients with vesicovaginal fistula at high risk of urinary incontinence after surgery. Obstet Gynecol. 2016;128:945-953.
Although rare in the United States and more common in low-resource countries, fistulas due to obstructed labor do occur. In developed countries, other obstetric causes for fistula are usually surgery, trauma, or infection related. An abnormal communication between organs—be it the urethra, bladder, ureter, uterus, cervix, or rectum—can develop1 and lead to vesicovaginal fistula (VVF), urethrovaginal fistula (FIGURE 1), vesicocervical fistula, vesicouterine fistula, ureterovaginal fistula (FIGURE 2), and rectovaginal fistula (RVF). Other nonobstetric causes include gynecologic surgery, radiation, malignancy, and congenital malformations.
During labor, hypoxia, subsequent ischemia, and pressure necrosis contribute to fistula formation. Injury sustained during a cesarean delivery (CD) or cesarean hysterectomy can lead to fistula formation; at times, however, complications are unavoidable given the nature of the pathologic condition that the patient presents with.
VVF and RVF have a devastating impact on a woman’s quality of life as they lead to significant morbidity and short- and long-term psychological distress. The fistula may not be recognized at the time of injury. The presenting signs and symptoms may be intermittent and confusing. Immediate surgical intervention may not be possible due to ongoing inflammation or infection. Recovery often is prolonged. As there is significant concomitant postpartum anxiety and depression, patients with fistula often require psychosocial support and counseling. After repair, there is still a risk for recurrence and voiding dysfunction.
Fistula signs and symptoms and evaluation
In cases of VVF, patients present with continuing large or small volume urinary incontinence. Depending on the time to diagnosis, patients may have calculi formation, prolapse, scarring, external perineal dermatitis, perineal nerve injury, and even motor weakness. Cyclic hematuria may be seen in vesicouterine fistulas.2
Multiple classification systems for diagnosis and staging of VVF have been suggested.3,4 A classification system for RVF was published by Tsang and colleagues.5 All these classification systems have attempted to characterize fistulas in terms of level of surgical complexity for repair, providing a guideline for preoperative assessment. These classification systems do not translate into prediction regarding outcomes.
Evaluation of pelvic fistula from the urinary tract starts with a thorough history that includes onset, duration, and description of leakage (continuous, intermittent, or positional) and whether there is concomitant stress and urge incontinence. A detailed obstetric history, including circumstances around the mode of delivery, underlying risk factors, and psychosocial history, should be obtained.
The pelvic examination with a plastic speculum and adequate lighting should assess the external perineum for dermatitis; bulbocavernosus and anal reflexes; and the vagina for length, caliber, level of scarring, and any prolapse. For VVFs, the location, size, and number of the fistula tracts can be visualized and confirmed with a retrograde fill of the bladder via a Foley catheter with saline or water mixed with methylene blue or any other blue dye (FIGURE 3). If a ureterovaginal fistula is suspected, the patient can simultaneously be given oral phenazopyridine and a tampon inserted within the vagina; the patient can then ambulate, and re-examination of the end of the tampon can reveal orange staining. The bladder meanwhile is retrograde filled with blue dye, with no blue staining of the tampon.
For RVF, history taking should include the onset, duration, and description of leakage, and the external anal sphincter should be assessed, with careful examination of the distal vagina at the vestibule as this is the most common location for RVF (fistula in ano). Patients may describe vaginal flatus and sometimes only brownish discharge, which can be intermittent, leading to an incorrect diagnosis of vaginitis that is treated repeatedly without success.
There is no consensus regarding optimal imaging for the assessment of VVF. Imaging used for diagnosis of VVF includes a voiding cystogram with opacification of the vagina after filling the bladder with contrast if there is a fistula. A cystoscopy can evaluate for calculi, retained suture, level of inflammation, and location of the ureters in relation to the fistula. Renal ultrasonography is of limited use. Intravenous pyelography can miss lesions by the trigone. In general, a computed tomography (CT) urogram and magnetic resonance imaging (MRI) with bladder contrast are more sensitive.
In the diagnosis of RVF, contrast vaginoscopy, double contrast barium enema, CT scan with contrast, and MRI can be used. MRI is more sensitive.6 A high index of suspicion is required based on the patient’s history as these imaging modalities do not always confirm RVF despite patient’s clear history of leakage. When the history is convincing, a thorough rectovaginal exam under anesthesia may be imperative. If rectal trauma is present, endoanal ultrasonography can delineate external and internal anal sphincter defects.
Prolonged Foley catheter placement after obstetric injury can lead to successful closure of a VVF. Prior to surgical intervention, assessing if there is possible ureteral involvement and use of intraoperative ureteral stents is a consideration. The route of surgery can be vaginal, abdominal, combined abdominal-vaginal, laparoscopic, or robotic.7 The robotic approach is increasingly utilized.8,9 However, the general consensus among fistula surgeons is that the vaginal approach should be considered first.
Continue to: Surgical repair...
Surgical repair
VVF repair. Factors that influence successful repair of VVF include the size and number of fistula, location, degree of scarring, bladder capacity, and urethral length.
Surgical technique requires wide mobilization and adequate exposure. The fistula tract can be delineated and manipulated with a pediatric Foley catheter, ureteral stent, or even a ureteral guidewire to aid in dissection (FIGURE 4). Intraoperative visualization of the ureters, including stenting, often is needed. The fistulous track is excised depending on the level of scarring. Closure of the bladder uroepithelium for the first layer is with absorbable interrupted 3-0 or 2-0 sutures in a tension-free closure. The bladder is then evaluated with a retrograde fill with saline and methylene blue to ensure a watertight closure for the first layer. If the first layer is not watertight, the second layer closure will not compensate and the fistula will persist. Particular attention is paid to the angles of the fistula at the first layer closure to prevent recurrence of the fistula at the angles. A running second layer with absorbable 2-0 suture is done. At times, a Martius flap or an omental J flap can be used to provide an additional layer for support and to increase vascularity.10 The patient is sent home with a Foley catheter for drainage for 10 to 14 days.11 Antibiotics are not needed postoperatively for VVF surgery.12
CT cystogram or retrograde cystogram is usually done to evaluate closure of the fistula prior to removal of the Foley catheter; retrograde fill with contrast directly into the bladder with 300 mL is sufficient (FIGURE 5). Patients are advised to refrain from sexual activity for a minimum of 6 weeks, but depending on the level of complexity and scarring, this can be up to 12 weeks.
The success rate in general is in the 95% range. Patients with successful closure of VVF are at risk for urge incontinence due to decreased bladder capacity, stress incontinence especially if the continence mechanism or urethra is involved, vaginal scarring, dyspareunia, and infertility.13 In general, sexual function improves after surgical repair.
RVF repair. Prior to surgical repair of RVF, the integrity of the external anal sphincter must be determined. If it is not involved, a vertical incision is made in the posterior vaginal wall, the vaginal epithelium is separated from the vaginal muscularis, and the fistula tract is identified. After complete wide mobilization of the tissue surrounding the tract, it is excised. The rectal wall is repaired with 3-0 or 4-0 absorbable interrupted sutures; a second layer and if possible even a third layer and finally the vaginal epithelium are all closed with 2-0 absorbable interrupted sutures.
If the sphincter complex is involved, the dissection involves an inverted U incision separating the vaginal wall from the rectum. The fistula tract is excised, the rectal wall is closed, and the internal anal sphincter is identified and reapproximated with interrupted absorbable 2-0 or 0 sutures. The disrupted external sphincter is then reapproximated with 2-0 or 0 sutures, and finally the transverse perineal and bulbocavernosus muscles are brought together with Lembert 0 sutures prior to closure of the external skin. Perioperative antibiotics have been shown to improve success rates in the correction of RVF.5 In patients with sphincter trauma and known RVF, outcomes with a sphincteroplasty are better, compared with endorectal advancement flaps. The patient is discharged with a bowel regimen and dietary precautions that aim for daily soft bowel movements.
After surgical treatment of fistulas, patients benefit from pelvic floor physical therapy that focuses on pelvic floor strengthening. Incorporating the habit of Kegel exercises after every void, timed (scheduled) bladder voiding, and avoidance of straining with urination or defecation should be emphasized.
Continue to: CASE 1 Pregnant woman with rectal bleeding...
CASE 1 Pregnant woman with rectal bleeding
A 37-year-old woman at 36 3/7 weeks’ gestation presented with acute rectal bleeding and pain. This was found to result from a catastrophic rupture of a pelvic arteriovenous malformation that caused an 11 x 7 x 9.5 cm size inferior pelvic hematoma and a full-thickness rectal tear at the dentate line. During examination under anesthesia, the baby was delivered by a stat CD due to breech presentation and a prolonged fetal heart rate deceleration. The patient underwent embolization of the right middle rectal artery and right internal iliac artery by a radiologic intervention. Further bleeding required surgical intervention for evacuation of about 1,000 mL of hematoma, repair of the rectal tear, and laparoscopic diverting loop ileostomy. In total, the patient received 8 U of packed red blood cells, 6 U of fresh frozen plasma, 5 L of crystalloid solution, and 2 g of tranexamic acid. The patient reported increased foul-smelling vaginal discharge, bedside exam suggested possible fistulous tract, and on postoperative day 16, an exam under anesthesia by Urogynecology confirmed a rectovaginal fistula in the right mid vagina. After 2 months of observation to allow resolution of inflammation, successful excision of the fistula tract and repair of RVF using the above-mentioned technique was accomplished.
CASE 2 Patient with VVF after cesarean hysterectomy
A 40-year-old (G6P2222) patient underwent cesarean hysterectomy for placenta percreta and uterine rupture at 24 weeks’ gestation. Intraoperatively, there were right ureteral ligation and posterior bladder wall cystotomies. The right ureter was reimplanted in the right upper posterior wall and the cystostomies were closed. As the patient had continuous urinary leakage postoperatively, a CT urogram was obtained, which showed left ureteral obstruction and VVF. Urinary incontinence persisted despite bilateral robotic ureteral reimplantation with omental flap by the urology team. Percutaneous nephrostomy tubes were placed bilaterally. The patient underwent additional imaging studies, including MRI, with findings of VVF and possible ureterovaginal fistula.
On referral to Urogynecology, the patient underwent cystoscopy with antegrade pyelogram, and the bilateral ureteroneocystostomy orifices had 5 French open-ended ureteral stents placed. A 10 French pediatric Foley catheter was inserted intravaginally into the bladder through the VVF. Via the vaginal approach, cervical remnant and skin bridges overlying the VVF were excised. The scarred fistula tract was excised with a circumferential incision. Horizontal interrupted Lembert sutures with 3-0 absorbable suture were used to reapproximate the first layer, which was confirmed to be watertight on testing with retrograde fill. Second-layer closure was completed with horizontal mattress 2-0 absorbable sutures, followed by a third-layer closure done in similar fashion. Fibrin glue was then placed. The vaginal epithelium was closed with 2-0 absorbable suture. Percutaneous nephrostomy tubes were removed. Postoperatively, the patient had a CT cystogram with no leak and no incontinence, but she developed urgency, which was controlled with timed voids and oxybutynin.
Although rare in the United States and more common in low-resource countries, fistulas due to obstructed labor do occur. In developed countries, other obstetric causes for fistula are usually surgery, trauma, or infection related. An abnormal communication between organs—be it the urethra, bladder, ureter, uterus, cervix, or rectum—can develop1 and lead to vesicovaginal fistula (VVF), urethrovaginal fistula (FIGURE 1), vesicocervical fistula, vesicouterine fistula, ureterovaginal fistula (FIGURE 2), and rectovaginal fistula (RVF). Other nonobstetric causes include gynecologic surgery, radiation, malignancy, and congenital malformations.
During labor, hypoxia, subsequent ischemia, and pressure necrosis contribute to fistula formation. Injury sustained during a cesarean delivery (CD) or cesarean hysterectomy can lead to fistula formation; at times, however, complications are unavoidable given the nature of the pathologic condition that the patient presents with.
VVF and RVF have a devastating impact on a woman’s quality of life as they lead to significant morbidity and short- and long-term psychological distress. The fistula may not be recognized at the time of injury. The presenting signs and symptoms may be intermittent and confusing. Immediate surgical intervention may not be possible due to ongoing inflammation or infection. Recovery often is prolonged. As there is significant concomitant postpartum anxiety and depression, patients with fistula often require psychosocial support and counseling. After repair, there is still a risk for recurrence and voiding dysfunction.
Fistula signs and symptoms and evaluation
In cases of VVF, patients present with continuing large or small volume urinary incontinence. Depending on the time to diagnosis, patients may have calculi formation, prolapse, scarring, external perineal dermatitis, perineal nerve injury, and even motor weakness. Cyclic hematuria may be seen in vesicouterine fistulas.2
Multiple classification systems for diagnosis and staging of VVF have been suggested.3,4 A classification system for RVF was published by Tsang and colleagues.5 All these classification systems have attempted to characterize fistulas in terms of level of surgical complexity for repair, providing a guideline for preoperative assessment. These classification systems do not translate into prediction regarding outcomes.
Evaluation of pelvic fistula from the urinary tract starts with a thorough history that includes onset, duration, and description of leakage (continuous, intermittent, or positional) and whether there is concomitant stress and urge incontinence. A detailed obstetric history, including circumstances around the mode of delivery, underlying risk factors, and psychosocial history, should be obtained.
The pelvic examination with a plastic speculum and adequate lighting should assess the external perineum for dermatitis; bulbocavernosus and anal reflexes; and the vagina for length, caliber, level of scarring, and any prolapse. For VVFs, the location, size, and number of the fistula tracts can be visualized and confirmed with a retrograde fill of the bladder via a Foley catheter with saline or water mixed with methylene blue or any other blue dye (FIGURE 3). If a ureterovaginal fistula is suspected, the patient can simultaneously be given oral phenazopyridine and a tampon inserted within the vagina; the patient can then ambulate, and re-examination of the end of the tampon can reveal orange staining. The bladder meanwhile is retrograde filled with blue dye, with no blue staining of the tampon.
For RVF, history taking should include the onset, duration, and description of leakage, and the external anal sphincter should be assessed, with careful examination of the distal vagina at the vestibule as this is the most common location for RVF (fistula in ano). Patients may describe vaginal flatus and sometimes only brownish discharge, which can be intermittent, leading to an incorrect diagnosis of vaginitis that is treated repeatedly without success.
There is no consensus regarding optimal imaging for the assessment of VVF. Imaging used for diagnosis of VVF includes a voiding cystogram with opacification of the vagina after filling the bladder with contrast if there is a fistula. A cystoscopy can evaluate for calculi, retained suture, level of inflammation, and location of the ureters in relation to the fistula. Renal ultrasonography is of limited use. Intravenous pyelography can miss lesions by the trigone. In general, a computed tomography (CT) urogram and magnetic resonance imaging (MRI) with bladder contrast are more sensitive.
In the diagnosis of RVF, contrast vaginoscopy, double contrast barium enema, CT scan with contrast, and MRI can be used. MRI is more sensitive.6 A high index of suspicion is required based on the patient’s history as these imaging modalities do not always confirm RVF despite patient’s clear history of leakage. When the history is convincing, a thorough rectovaginal exam under anesthesia may be imperative. If rectal trauma is present, endoanal ultrasonography can delineate external and internal anal sphincter defects.
Prolonged Foley catheter placement after obstetric injury can lead to successful closure of a VVF. Prior to surgical intervention, assessing if there is possible ureteral involvement and use of intraoperative ureteral stents is a consideration. The route of surgery can be vaginal, abdominal, combined abdominal-vaginal, laparoscopic, or robotic.7 The robotic approach is increasingly utilized.8,9 However, the general consensus among fistula surgeons is that the vaginal approach should be considered first.
Continue to: Surgical repair...
Surgical repair
VVF repair. Factors that influence successful repair of VVF include the size and number of fistula, location, degree of scarring, bladder capacity, and urethral length.
Surgical technique requires wide mobilization and adequate exposure. The fistula tract can be delineated and manipulated with a pediatric Foley catheter, ureteral stent, or even a ureteral guidewire to aid in dissection (FIGURE 4). Intraoperative visualization of the ureters, including stenting, often is needed. The fistulous track is excised depending on the level of scarring. Closure of the bladder uroepithelium for the first layer is with absorbable interrupted 3-0 or 2-0 sutures in a tension-free closure. The bladder is then evaluated with a retrograde fill with saline and methylene blue to ensure a watertight closure for the first layer. If the first layer is not watertight, the second layer closure will not compensate and the fistula will persist. Particular attention is paid to the angles of the fistula at the first layer closure to prevent recurrence of the fistula at the angles. A running second layer with absorbable 2-0 suture is done. At times, a Martius flap or an omental J flap can be used to provide an additional layer for support and to increase vascularity.10 The patient is sent home with a Foley catheter for drainage for 10 to 14 days.11 Antibiotics are not needed postoperatively for VVF surgery.12
CT cystogram or retrograde cystogram is usually done to evaluate closure of the fistula prior to removal of the Foley catheter; retrograde fill with contrast directly into the bladder with 300 mL is sufficient (FIGURE 5). Patients are advised to refrain from sexual activity for a minimum of 6 weeks, but depending on the level of complexity and scarring, this can be up to 12 weeks.
The success rate in general is in the 95% range. Patients with successful closure of VVF are at risk for urge incontinence due to decreased bladder capacity, stress incontinence especially if the continence mechanism or urethra is involved, vaginal scarring, dyspareunia, and infertility.13 In general, sexual function improves after surgical repair.
RVF repair. Prior to surgical repair of RVF, the integrity of the external anal sphincter must be determined. If it is not involved, a vertical incision is made in the posterior vaginal wall, the vaginal epithelium is separated from the vaginal muscularis, and the fistula tract is identified. After complete wide mobilization of the tissue surrounding the tract, it is excised. The rectal wall is repaired with 3-0 or 4-0 absorbable interrupted sutures; a second layer and if possible even a third layer and finally the vaginal epithelium are all closed with 2-0 absorbable interrupted sutures.
If the sphincter complex is involved, the dissection involves an inverted U incision separating the vaginal wall from the rectum. The fistula tract is excised, the rectal wall is closed, and the internal anal sphincter is identified and reapproximated with interrupted absorbable 2-0 or 0 sutures. The disrupted external sphincter is then reapproximated with 2-0 or 0 sutures, and finally the transverse perineal and bulbocavernosus muscles are brought together with Lembert 0 sutures prior to closure of the external skin. Perioperative antibiotics have been shown to improve success rates in the correction of RVF.5 In patients with sphincter trauma and known RVF, outcomes with a sphincteroplasty are better, compared with endorectal advancement flaps. The patient is discharged with a bowel regimen and dietary precautions that aim for daily soft bowel movements.
After surgical treatment of fistulas, patients benefit from pelvic floor physical therapy that focuses on pelvic floor strengthening. Incorporating the habit of Kegel exercises after every void, timed (scheduled) bladder voiding, and avoidance of straining with urination or defecation should be emphasized.
Continue to: CASE 1 Pregnant woman with rectal bleeding...
CASE 1 Pregnant woman with rectal bleeding
A 37-year-old woman at 36 3/7 weeks’ gestation presented with acute rectal bleeding and pain. This was found to result from a catastrophic rupture of a pelvic arteriovenous malformation that caused an 11 x 7 x 9.5 cm size inferior pelvic hematoma and a full-thickness rectal tear at the dentate line. During examination under anesthesia, the baby was delivered by a stat CD due to breech presentation and a prolonged fetal heart rate deceleration. The patient underwent embolization of the right middle rectal artery and right internal iliac artery by a radiologic intervention. Further bleeding required surgical intervention for evacuation of about 1,000 mL of hematoma, repair of the rectal tear, and laparoscopic diverting loop ileostomy. In total, the patient received 8 U of packed red blood cells, 6 U of fresh frozen plasma, 5 L of crystalloid solution, and 2 g of tranexamic acid. The patient reported increased foul-smelling vaginal discharge, bedside exam suggested possible fistulous tract, and on postoperative day 16, an exam under anesthesia by Urogynecology confirmed a rectovaginal fistula in the right mid vagina. After 2 months of observation to allow resolution of inflammation, successful excision of the fistula tract and repair of RVF using the above-mentioned technique was accomplished.
CASE 2 Patient with VVF after cesarean hysterectomy
A 40-year-old (G6P2222) patient underwent cesarean hysterectomy for placenta percreta and uterine rupture at 24 weeks’ gestation. Intraoperatively, there were right ureteral ligation and posterior bladder wall cystotomies. The right ureter was reimplanted in the right upper posterior wall and the cystostomies were closed. As the patient had continuous urinary leakage postoperatively, a CT urogram was obtained, which showed left ureteral obstruction and VVF. Urinary incontinence persisted despite bilateral robotic ureteral reimplantation with omental flap by the urology team. Percutaneous nephrostomy tubes were placed bilaterally. The patient underwent additional imaging studies, including MRI, with findings of VVF and possible ureterovaginal fistula.
On referral to Urogynecology, the patient underwent cystoscopy with antegrade pyelogram, and the bilateral ureteroneocystostomy orifices had 5 French open-ended ureteral stents placed. A 10 French pediatric Foley catheter was inserted intravaginally into the bladder through the VVF. Via the vaginal approach, cervical remnant and skin bridges overlying the VVF were excised. The scarred fistula tract was excised with a circumferential incision. Horizontal interrupted Lembert sutures with 3-0 absorbable suture were used to reapproximate the first layer, which was confirmed to be watertight on testing with retrograde fill. Second-layer closure was completed with horizontal mattress 2-0 absorbable sutures, followed by a third-layer closure done in similar fashion. Fibrin glue was then placed. The vaginal epithelium was closed with 2-0 absorbable suture. Percutaneous nephrostomy tubes were removed. Postoperatively, the patient had a CT cystogram with no leak and no incontinence, but she developed urgency, which was controlled with timed voids and oxybutynin.
- Adler AJ, Ronsmans C, Calvert C, et al. Estimating the presence of obstetric fistula: a systematic review and meta-analysis BMC Pregnancy Childbirth. 2013;13:246.
- Battacharjee S, Kohli UA, Sood A, et al. Vesicouterine fistula: Youssef’s syndrome. Med J Armed Forces India. 2015;71(suppl 1):S175-S177. doi: 10.1016/j.mjafi.2013.11.006.
- Waaldijk K. Step-by-Step Surgery of Vesicovaginal Fistulas. Campion Press; 1994.
- Goh, JTW. A new classification for female genital tract fistula. Aust N Z J Ob Gynecol. 2004:44:502-504.
- Tsang CB, Rothenberger DA. Rectovaginal fistulas: therapeutic options. Surg Clin North Am. 1997;77:95-114.
- Champagne BJ, McGee MF. Rectovaginal fistula. Surg Clin North Am. 2010;90:69-82.
- Bodner-Adler B, Hanzal E, Pablik E, et al. Management of vesicovaginal fistulas in women following benign gynecologic surgery: a systematic review and meta-analysis. PLoS One. 2017;12:e0171554.
- Randazzo M, Lengauer L, Rochat CH, et al. Best practices in robotic-assisted repair of vesicovaginal fistula: a consensus report from the European Association of Urology Robotic Urology Section Scientific Working Group for Reconstructive Urology. Eur Urol. 2020;78: 432-442.
- Miklos JR, Moore RD, Chinthakanan O. Laparoscopic and robotic assisted vesicovaginal fistula repair: a systematic review of the literature. J Minim Invasive Gynecol. 2015:22:727-736.
- Hancock B. Practical Obstetric Fistula Surgery. Royal Society of Medicine Press; 2009.
- Nardos R, Menber B, Browning A. Outcome of obstetric fistula repair after 10-day versus 14-day Foley catheterization. Int J Gynaecol 0bstet. 2012;118:21-23.
- Tomlinson AJ, Thornton JG. A randomized controlled trial of antibiotic prophylaxis for vesico-vaginal fistula repair. Br J Obstet Gynaecol. 2005;105:397-399.
- Bengtson AM, Kopp D, Tang JH, et al. Identifying patients with vesicovaginal fistula at high risk of urinary incontinence after surgery. Obstet Gynecol. 2016;128:945-953.
- Adler AJ, Ronsmans C, Calvert C, et al. Estimating the presence of obstetric fistula: a systematic review and meta-analysis BMC Pregnancy Childbirth. 2013;13:246.
- Battacharjee S, Kohli UA, Sood A, et al. Vesicouterine fistula: Youssef’s syndrome. Med J Armed Forces India. 2015;71(suppl 1):S175-S177. doi: 10.1016/j.mjafi.2013.11.006.
- Waaldijk K. Step-by-Step Surgery of Vesicovaginal Fistulas. Campion Press; 1994.
- Goh, JTW. A new classification for female genital tract fistula. Aust N Z J Ob Gynecol. 2004:44:502-504.
- Tsang CB, Rothenberger DA. Rectovaginal fistulas: therapeutic options. Surg Clin North Am. 1997;77:95-114.
- Champagne BJ, McGee MF. Rectovaginal fistula. Surg Clin North Am. 2010;90:69-82.
- Bodner-Adler B, Hanzal E, Pablik E, et al. Management of vesicovaginal fistulas in women following benign gynecologic surgery: a systematic review and meta-analysis. PLoS One. 2017;12:e0171554.
- Randazzo M, Lengauer L, Rochat CH, et al. Best practices in robotic-assisted repair of vesicovaginal fistula: a consensus report from the European Association of Urology Robotic Urology Section Scientific Working Group for Reconstructive Urology. Eur Urol. 2020;78: 432-442.
- Miklos JR, Moore RD, Chinthakanan O. Laparoscopic and robotic assisted vesicovaginal fistula repair: a systematic review of the literature. J Minim Invasive Gynecol. 2015:22:727-736.
- Hancock B. Practical Obstetric Fistula Surgery. Royal Society of Medicine Press; 2009.
- Nardos R, Menber B, Browning A. Outcome of obstetric fistula repair after 10-day versus 14-day Foley catheterization. Int J Gynaecol 0bstet. 2012;118:21-23.
- Tomlinson AJ, Thornton JG. A randomized controlled trial of antibiotic prophylaxis for vesico-vaginal fistula repair. Br J Obstet Gynaecol. 2005;105:397-399.
- Bengtson AM, Kopp D, Tang JH, et al. Identifying patients with vesicovaginal fistula at high risk of urinary incontinence after surgery. Obstet Gynecol. 2016;128:945-953.
OR safety and efficiency: Measuring and monitoring all factors—including surgical volume
The operating room (OR) is a key contributor to a hospital’s profitability. It is a complex environment with ever-advancing technology. A successful surgery completed without complications within an optimal time depends not only on the surgeon’s experience, skills, and knowledge but also on numerous other structural, human, and nontechnical factors over which the surgeon has limited control.
As in any setting that deals with human life, in the OR, team dynamics, communication, and environment play a major role. Research has indicated the benefits of dedicated teams, reduced handoffs, and innovative modalities that continuously and systematically monitor potential breakdowns and propose solutions for the detected problems.
Finally, who should perform your loved one’s hysterectomy? This article also attempts to address the impact of surgeons’ and hospitals’ volume on operative outcomes with a diminishing number of hysterectomies but an increasing number of approaches.
Human factors in the OR
Human factors research was born as a product of the industrial revolution and mass production. It aims to optimize human experience and improve system performance by studying how humans interact with system. The aviation industry, for example, minimized errors significantly by using methods developed by human factors scientists. As another industry with no tolerance for mistakes, the health care sector followed suit. Ultimately, the goal of human factors research in health care is to improve patient safety, optimize work and environment, reduce costs, and enhance employees’ physical and mental health, engagement, comfort, and quality of life (FIGURE 1).1
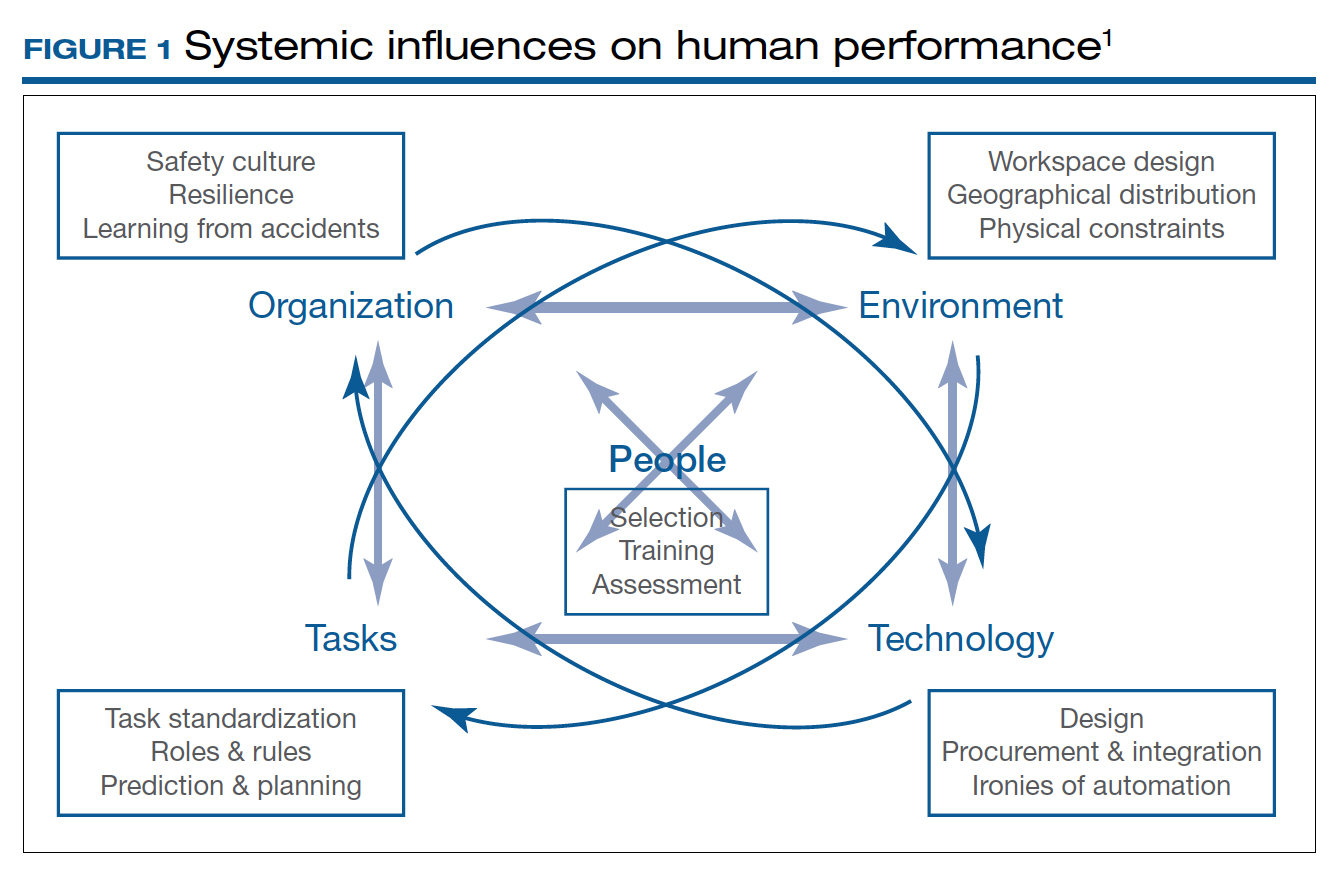
Today’s OR is so complex that it is hard to understand its dynamics without human factors research. Every new OR technology is first tested in controlled and simulated environments to determine “work as imagined.” However, it is necessary to study “work as done” in the real world via direct observation, video recording, questionnaires, and semistructured interviews by an on-site multidisciplinary team. This process not only focuses on surgical skills, process efficiency, and outcomes but also monitors the entire process according to Human Factors and Ergonomics Engineering principles to explore otherwise hidden complexities and latent safety concerns. The Systems Engineering Initiative for Patient Safety (SEIPS) framework is used to study the impact of interactions between people, tasks, technologies, environment, and organization.1
Robot-assisted surgery (RAS), an increasingly popular surgical approach among gynecologic surgeons, recently has been the focus of human factors science. A robotic OR poses unique challenges: the surgeon is not scrubbed, is removed from the operating table, and controls a complex highly technologic device in a crowded and darkened room. These are ideal conditions waiting to be optimized by human factor experts. To demonstrate the importance of human factors in the OR, we review the evidence for RAS.
Continue to: Impact of flow disruptions...
Impact of flow disruptions
Flow disruptions (FDs) were found to be more common in RAS. Catchpole and colleagues identified a mean of 9.62 FDs per hour in 89 robotic procedures, including hysterectomies and sacrocolpopexies, from a variety of fields; FDs occurred more often during the docking stage, followed by the console time, and they mostly were caused by communication breakdown and lack of team familiarity.2
Surgeon experience significantly reduced FDs. Surgeons who had done more than 700 RAS cases experienced 60% fewer FDs than those who had done less than 250 cases (13 vs 8 per hour).2 A study focusing on residents’ impact on RAS outcomes found that each FD increased the total operative time by an average 2.4 minutes, with the number significantly higher when a resident was involved.3 About one-quarter of the training-related FDs were procedure-specific instructions, while one-third were related to instrument and robotic instruction. However, pauses to teach residents did not appear to create significant intraoperative delays. Expectedly, experienced surgeons could anticipate and reduce these disruptions by supporting the whole team.
Human ergonomics, turnover time, and robot-specific skills
In a study of human ergonomics in RAS, Yu and colleagues noted that bedside assistants could experience neck posture problems. Surprisingly, the console could constrain the surgeon’s neck-shoulder region.4 Studies that reported on communication problems in a robotic OR suggest that innovative forms of verbal and nonverbal communication may support successful team communication.5
On the learning curve for RAS, OR turnover time, a key value metric, has been longer. However, turnover time was reduced almost by half from 99.2 to 53.2 minutes over 3 months after concepts from motor racing pit stops were employed, including briefings, leadership, role definition, task allocation, and task sequencing. Average room-ready time also was lowered from 42.2 to 27.2 minutes.6 RAS presents new challenges with sterile instrument processing as well. A successful RAS program, therefore, has organizational needs that include the training of OR and sterile processing staff and appropriate shift management.1
In a robotic OR, not only the surgeon but also the whole team requires robot-specific skills. New training approaches to teamwork, communication, and situation awareness skills are necessary. Robotic equipment, with its data and power cables, 2 consoles, and changing movement paths, necessitate larger rooms with a specific layout.7
In a review of recordings of RAS that used a multidimensional assessment tool to measure team effectiveness and cognitive load, Sexton and colleagues identified anticipation, active team engagement, and higher familiarity scores as the best predictors of team efficiency.8 Several studies emphasized the need for a stable team, especially in the learning phase of robotic surgery.5,9,10 A dedicated robotic team reduced the operative time by 18% during robot-assisted sacrocolpopexy (RASCP).10 RASCP procedures that extended into the afternoon took significantly longer time.9 A dedicated anesthesiologist improved the preoperative time.9 Surgical team handoffs also have reduced OR efficiency.11,12
Studying the impact of human factors is paramount for safe and efficient surgery. It is especially necessary in ORs that are equipped with high technologic instruments such as those used in RAS.
Surgical Black Box: Using data for OR safety and efficiency
Surgical procedures account for more than 50% of medical errors in a hospital setting, many of which are preventable. Postevent analysis with traditional methods, such as “Morbidity and Mortality” meetings held many days later, misses many adverse events in the OR.13 Another challenge with ever-changing and fast-multiplying surgical approaches is the development of effective surgical skill. Reviewing video recording of surgical procedures has been proposed as an instrument for recognizing adverse events and perfecting surgical skills.Recently, an innovative data-capture platform called the OR Black Box, developed by Teodor Grantcharov, MD, PhD, and colleagues, went beyond simple audiovisual recording.14 This high technologic platform not only video records the actual surgical procedure with laparoscopic camera capture (and wearable cameras for open cases) but also monitors the entire OR environment via wide-angle cameras, utilizes sensors, and records both the patient’s and the surgeon’s physiologic parameters.
The OR Black Box generates a holistic view of the OR after synchronization, encryption, and secure storage of all inputs for further analysis by experts and software-based algorithms (FIGURE 2). Computer vision algorithms can recognize improper dissection techniques and complications, such as bleeding. Adverse events are flagged with an automated software on a procedural timeline to facilitate review of procedural steps, disruptive environmental and organizational factors, OR team technical and nontechnical skills, surgeon physiologic stress, and intraoperative errors, events, and rectification processes using validated instruments.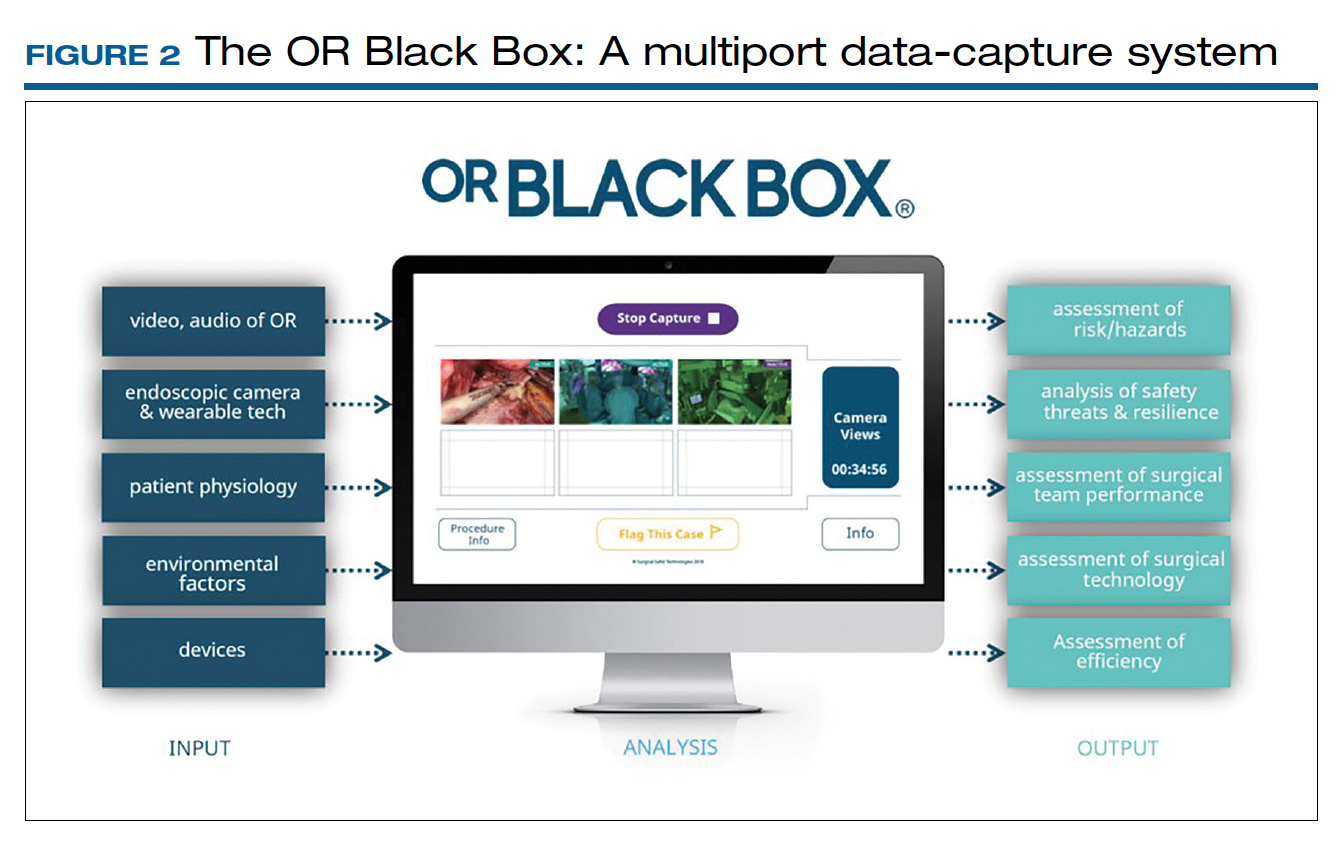
Artificial intelligence built into this platform can automatically extract objective, high-quality, and structured data to generate explainable insights by recognizing adverse events and procedural segments of interest for training and quality improvement and provide a foundation with objective measurements of technical and nontechnical performance for formative and summative assessment. This system, a major step up compared with retrospective review of likely biased medical records and labor-intensive multidisciplinary human observers, has the potential to increase efficiency and reduce costs by studying human factors that include clinical design, technology, and organization. OR efficiency, measured in real time objectively and thoroughly, may save time and resources.
OR Black Box platforms have already started to generate meaningful data. It is not surprising that auditory disruptions—OR doors opening, loud noises, pagers beeping, telephones ringing—were recorded almost every minute during laparoscopic procedures.15 Most technical errors occurred during dissection, resection, and reconstruction and most commonly were associated with improper estimations of force applied to tissue and distance to the target tissue during operative steps of a laparoscopic procedure.16 Another study based on this system showed that technical performance was an independent predictor of postoperative outcomes.17 The OR Black Box identified a device-related interruption in 30% of elective laparoscopic general surgery cases, most commonly in sleeve gastrectomy and oncologic gastrectomy procedures. This sophisticated surgical data recording system also demonstrated a significantly better ability to detect Veress needle injuries (12 vs 3) and near misses (47 vs 0) when compared with traditional chart review.18
Data from the OR Black Box also have been applied to better analyze nontechnical performance, including teamwork and interpersonal dynamics.19 Surgeons most commonly exhibited adept situational awareness and leadership, while the nurse team excelled at task management and situational awareness.19 Of the total care provider team studied, the surgeon and scrub nurse demonstrated the most favorable nontechnical behavior.19 Of note, continuous physiologic monitoring of the surgeon with this system revealed that surgeons under stress had 66% higher adverse events.
The OR Black Box is currently utilized at 20 institutions in North America and Europe. The data compiled from all these institutions revealed that there was a 10% decrease in intraoperative adverse events for each 10-point increase in technical skill score on a scale of 0 to 100 (unpublished data). This centralized data indicated that turnover time ranged widely between 7 and 91 minutes, with variation of cleanup time from 1 to 25 minutes and setup time from 22 to 43 minutes. Institutions can learn from each other using this platform. For example, the information about block time utilization (20%–99%) across institutions provides opportunities for system improvements.
With any revolutionary technology, it is imperative to study its effects on outcomes, training, costs, and privacy before it is widely implemented. We, obstetricians and gynecologists, are very familiar with the impact of electronic fetal monitoring, a great example of a technologic advance that did not improve perinatal outcomes but led to unintended consequences, such as higher rates of cesarean deliveries and lawsuits. Such a tool may lead to potential misrepresentation of intraoperative events unless legal aspects are clearly delineated. As exciting as it is, this disruptive technology requires further exploration with scientific vigor.
Continue to: Surgeon and hospital volume: Surgical outcomes paradigm...
Surgeon and hospital volume: Surgical outcomes paradigm
A landmark study in 1979 that showed decreased mortality in high-volume centers underscored the need for regionalization for certain surgical procedures.20 This association was further substantiated by 2 reports on 2.5 million Medicare beneficiaries that demonstrated significantly lower mortality for all 14 cardiovascular and oncologic procedures for hospitals with larger surgical volume (16% vs 4%) and high-volume surgeons for certain procedures, for example, 15% versus 5% for pancreatic resections for cancer.21,22
A similar association was found for all routes of hysterectomies performed for benign indications. Boyd and colleagues showed that gynecologists who performed fewer than 10 hysterectomies per year had a higher perioperative morbidity rate (16.5%) compared with those who did more (11.7%).23 Specific to vaginal hysterectomy, in a study of more than 6,000 women, surgeons who performed 13 procedures per year had 31% less risk of operative injury than those who did 5.5 procedures per year (2.5% vs 1.7%).24 Overall perioperative complications (5.0% vs 4.0%) and medical complications (5.7% vs 3.9%) were also reduced for higher-volume surgeons. In a cohort of approximately 8,000 women who underwent a laparoscopic hysterectomy, high-volume surgeons had a considerably lower complication rate (4.2% vs 6.2%).25
As expected, lower complication rates of high-volume surgeons led to lower resource utilization, including lower transfusion rates, less intensive care unit utilization, and shorter operative times and, in several studies, length of stay.24,25 Of note, low-volume surgeons were less likely to offer minimally invasive routes and were more likely to convert to laparotomy.26 In addition, significant cost savings have been associated with high surgical volume, which one study showed was 16% ($6,500 vs $5,600) for high-volume surgeons.26 With regard to mortality, a study of 7,800 women found that perioperative mortality increased more than 10-fold for surgeons who performed an average 1 case per year compared with all other surgeons (2.5% vs 0.2%).27
When gynecologic cancers are concerned, arguably, long-term survival outcomes may be more critical than perioperative morbidity and mortality. Higher surgeon and hospital volume are associated with improved perioperative outcomes for endometrial and cervical cancers.28 Importantly, minimally invasive hysterectomy was offered for endometrial cancer significantly more often by surgeons with high volume.28 Survival outcomes were not affected by surgeon or hospital volume, likely due to overall more favorable prognosis for endometrial cancer after treatment.
Although it is intuitive to assume that a surgeon’s skills and experience would make the most impact in procedures for ovarian cancer due to the complexity of ovarian cancer surgery, evidence on short-term outcomes has been mixed. Intriguingly, some studies reported that high-volume institutions had higher complication and readmission rates. However, evidence supports that the surgeon’s volume, and especially hospital volume, improves long-term survival for ovarian cancer, with a negative impact on immediate postoperative morbidity.29 This may suggest that a more aggressive surgical effort improves long-term survival but also can cause more perioperative complications. Further, longer survival may result not only from operative skills but also because of better care by a structured multidisciplinary team at more established high-volume cancer centers.
The association of improved outcomes with higher volume led to public reporting of hospital outcomes. Policy efforts toward regionalization have impacted surgical practice. Based on their analysis of 3.2 million Medicare patients who underwent 1 of 8 different cancer surgeries or cardiovascular operations from 1999 to 2008, Finks and colleagues demonstrated that care was concentrated to fewer hospitals over time for many of these procedures.29 This trend was noted for gynecologic cancer surgery but not for benign gynecologic surgery.
Regionalization of care limits access particularly for minority and underserved communities because of longer travel distances, logistic challenges, and financial strain. An alternative to regionalization of care is targeted quality improvement by rigorous adherence to quality guidelines at low-volume hospitals.
Is there a critical minimum volume that may be used as a requirement for surgeons to maintain their privileges and for hospitals to offer certain procedures? In 2015, minimum volume standards for a number of common procedures were proposed by Johns Hopkins Medicine and Dartmouth-Hitchcock Medical Center, such as 50 hip replacement surgeries per hospital and 25 per physician per year, and 20 pancreatectomies per hospital and 5 per surgeon per year.30 A modeling study for hysterectomy showed that a volume cut point of >1 procedure in the prior year would restrict privileges for a substantial number of surgeons performing abdominal (17.5%), robot-assisted (12.5%), laparoscopic (16.8%), and vaginal (27.6%) hysterectomies.27 This study concluded that minimum-volume standards for hysterectomy for even the lowest volume physicians would restrict a significant number of gynecologic surgeons, including many with outcomes that are better than predicted.
Therefore, while there is good evidence that favors better outcomes in the hands of high-volume surgeons in gynecology, the impact of such policies on gynecologic practice clearly warrants careful monitoring and further study.
- What factors besides the surgeon’s skills influence surgical safety and efficiency?
- Are you ready to have audio, video, and sensor-based recording of everything in the OR?
- Who should perform your loved one’s hysterectomy? Do the surgeon’s and hospital’s volume matter?
- Catchpole K, Bisantz A, Hallbeck MS, et al. Human factors in robotic assisted surgery: lessons from studies ‘in the wild’. Appl Ergon. 2019;78:270-276.
- Catchpole K, Perkins C, Bresee C, et al. Safety, efficiency and learning curves in robotic surgery: a human factors analysis. Surg Endosc. 2016;30:3749-3761.
- Jain M, Fry BT, Hess LW, et al. Barriers to efficiency in robotic surgery: the resident effect. J Surg. Res. 2016;205:296-304.
- Yu D, Dural C, Morrow MM, et al. Intraoperative workload in robotic surgery assessed by wearable motion tracking sensors and questionnaires. Surg Endosc. 2017;31:877-886.
- Randell R, Honey S, Alvarado N, et al. Embedding robotic surgery into routine practice and impacts on communication and decision making: a review of the experience of surgical teams. Cognit Technol Work. 2016;18:423-437.
- Souders CP, Catchpole KR, Wood LN, et al. Reducing operating room turnover time for robotic surgery using a motor racing pit stop model. World J Surg. 2017;4:1943–1949.
- Ahmad N, Hussein AA, Cavuoto L, et al. Ambulatory movements, team dynamics and interactions during robot-assisted surgery. BJU Int. 2016;118:132-139.
- Sexton K, Johnson A, Gotsch A, et al. Anticipation, teamwork, and cognitive load: chasing efficiency during robot-assisted surgery. BMJ Qual Saf. 2018;27:148-154.
- Harmanli O, Solak S, Bayram A, et al. Optimizing the robotic surgery team: an operations management perspective. Int Urogynecol J. 2021;32:1379-1385.
- Carter-Brooks CM, Du AL, Bonidie MJ, et al. The impact of a dedicated robotic team on robotic-assisted sacrocolpopexy outcomes. Female Pelvic Med Reconstr Surg. 2018;24:13-16.
- Giugale LE, Sears S, Lavelle ES, et al. Evaluating the impact of intraoperative surgical team handoffs on patient outcomes. Female Pelvic Med Reconstr Surg. 2017;23:288-292.
- Geynisman-Tan J, Brown O, Mueller M, et al. Operating room efficiency: examining the impact of personnel handoffs. Female Pelvic Med Reconstr Surg. 2018;24:87-89.
- Alsubaie H, Goldenberg M, Grantcharov T. Quantifying recall bias in surgical safety: a need for a modern approach to morbidity and mortality reviews. Can J Surg. 2019;62:39-43.
- Goldenberg MG, Jung J, Grantcharov TP. Using data to enhance performance and improve quality and safety in surgery. JAMA Surg. 2017;152:972-973.
- Jung JJ, Grantcharov TP. The operating room black box: a prospective observational study of the operating room. J Am Coll Surg. 2017;225:S127-S128.
- Jung JJ, Jüni P, Lebovic G, et al. First-year analysis of the operating room black box study. Ann Surg. 2020;271:122-127.
- Jung JJ, Kashfi A, Sharma S, et al. Characterization of device-related interruptions in minimally invasive surgery: need for intraoperative data and effective mitigation strategies. Surg Endosc. 2019;33:717-723.
- Jung JJ, Adams-McGavin RC, Grantcharov TP. Underreporting of Veress needle injuries: comparing direct observation and chart review methods. J Surg Res. 2019;236:266-270.
- Fesco AB, Kuzulugil SS, Babaoglu C, et al. Relationship between intraoperative nontechnical performance and technical events in bariatric surgery. Br J Surg. 2018;105:1044-1050.
- Luft HS, Bunker JP, Enthoven AC. Should operations be regionalized? The empirical relation between surgical volume and mortality. N Engl J Med. 1979;301:1364-1369.
- Birkmeyer JD, Siewers AE, Finlayson EV, et al. Hospital volume and surgical mortality in the United States. N Engl J Med. 2002;346:1128-1137.
- Birkmeyer JD, Stukel TA, Siewers AE, et al. Surgeon volume and operative mortality in the United States. N Engl J Med. 2003;349:21172127.
- Boyd LR, Novetsky AP, Curtin JP. Effect of surgical volume on route of hysterectomy and short-term morbidity. Obstet Gynecol. 2010;116:909-915.
- Rogo-Gupta LJ, Lewin SN, Kim JH, et al. The effect of surgeon volume on outcomes and resource use for vaginal hysterectomy. Obstet Gynecol. 2010;116:1341-1347.
- Wallenstein MR, Ananth CV, Kim JH, et al. Effect of surgical volume on outcomes for laparoscopic hysterectomy for benign indications. Obstet Gynecol. 2012;119:709-716.
- Bretschneider CE, Frazzini Padilla P, Das D, et al. The impact of surgeon volume on perioperative adverse events in women undergoing minimally invasive hysterectomy for the large uterus. Am J Obstet Gynecol. 2018;219:490.e1-490.e8.
- Ruiz MP, Chen L, Hou JY, et al. Outcomes of hysterectomy performed by very low-volume surgeons. Obstet Gynecol. 2018;131:981-990.
- Wright JD. The volume-outcome paradigm for gynecologic surgery: clinical and policy implications. Clin Obstet Gynecol. 2020;63:252-265.
- Finks JF, Osborne NH, Birkmeyer JD. Trends in hospital volume and operative mortality for high risk surgery. N Engl J Med. 2011;364:21282137.
- Sternberg S. Hospitals move to limit low-volume surgeries. US News & World Report. May 19, 2015. www.usnews.com/news /articles/2015/05/19/hospitals-move-to-limit-low-volume-surgeries. Accessed April 19, 2022.
The operating room (OR) is a key contributor to a hospital’s profitability. It is a complex environment with ever-advancing technology. A successful surgery completed without complications within an optimal time depends not only on the surgeon’s experience, skills, and knowledge but also on numerous other structural, human, and nontechnical factors over which the surgeon has limited control.
As in any setting that deals with human life, in the OR, team dynamics, communication, and environment play a major role. Research has indicated the benefits of dedicated teams, reduced handoffs, and innovative modalities that continuously and systematically monitor potential breakdowns and propose solutions for the detected problems.
Finally, who should perform your loved one’s hysterectomy? This article also attempts to address the impact of surgeons’ and hospitals’ volume on operative outcomes with a diminishing number of hysterectomies but an increasing number of approaches.
Human factors in the OR
Human factors research was born as a product of the industrial revolution and mass production. It aims to optimize human experience and improve system performance by studying how humans interact with system. The aviation industry, for example, minimized errors significantly by using methods developed by human factors scientists. As another industry with no tolerance for mistakes, the health care sector followed suit. Ultimately, the goal of human factors research in health care is to improve patient safety, optimize work and environment, reduce costs, and enhance employees’ physical and mental health, engagement, comfort, and quality of life (FIGURE 1).1
Today’s OR is so complex that it is hard to understand its dynamics without human factors research. Every new OR technology is first tested in controlled and simulated environments to determine “work as imagined.” However, it is necessary to study “work as done” in the real world via direct observation, video recording, questionnaires, and semistructured interviews by an on-site multidisciplinary team. This process not only focuses on surgical skills, process efficiency, and outcomes but also monitors the entire process according to Human Factors and Ergonomics Engineering principles to explore otherwise hidden complexities and latent safety concerns. The Systems Engineering Initiative for Patient Safety (SEIPS) framework is used to study the impact of interactions between people, tasks, technologies, environment, and organization.1
Robot-assisted surgery (RAS), an increasingly popular surgical approach among gynecologic surgeons, recently has been the focus of human factors science. A robotic OR poses unique challenges: the surgeon is not scrubbed, is removed from the operating table, and controls a complex highly technologic device in a crowded and darkened room. These are ideal conditions waiting to be optimized by human factor experts. To demonstrate the importance of human factors in the OR, we review the evidence for RAS.
Continue to: Impact of flow disruptions...
Impact of flow disruptions
Flow disruptions (FDs) were found to be more common in RAS. Catchpole and colleagues identified a mean of 9.62 FDs per hour in 89 robotic procedures, including hysterectomies and sacrocolpopexies, from a variety of fields; FDs occurred more often during the docking stage, followed by the console time, and they mostly were caused by communication breakdown and lack of team familiarity.2
Surgeon experience significantly reduced FDs. Surgeons who had done more than 700 RAS cases experienced 60% fewer FDs than those who had done less than 250 cases (13 vs 8 per hour).2 A study focusing on residents’ impact on RAS outcomes found that each FD increased the total operative time by an average 2.4 minutes, with the number significantly higher when a resident was involved.3 About one-quarter of the training-related FDs were procedure-specific instructions, while one-third were related to instrument and robotic instruction. However, pauses to teach residents did not appear to create significant intraoperative delays. Expectedly, experienced surgeons could anticipate and reduce these disruptions by supporting the whole team.
Human ergonomics, turnover time, and robot-specific skills
In a study of human ergonomics in RAS, Yu and colleagues noted that bedside assistants could experience neck posture problems. Surprisingly, the console could constrain the surgeon’s neck-shoulder region.4 Studies that reported on communication problems in a robotic OR suggest that innovative forms of verbal and nonverbal communication may support successful team communication.5
On the learning curve for RAS, OR turnover time, a key value metric, has been longer. However, turnover time was reduced almost by half from 99.2 to 53.2 minutes over 3 months after concepts from motor racing pit stops were employed, including briefings, leadership, role definition, task allocation, and task sequencing. Average room-ready time also was lowered from 42.2 to 27.2 minutes.6 RAS presents new challenges with sterile instrument processing as well. A successful RAS program, therefore, has organizational needs that include the training of OR and sterile processing staff and appropriate shift management.1
In a robotic OR, not only the surgeon but also the whole team requires robot-specific skills. New training approaches to teamwork, communication, and situation awareness skills are necessary. Robotic equipment, with its data and power cables, 2 consoles, and changing movement paths, necessitate larger rooms with a specific layout.7
In a review of recordings of RAS that used a multidimensional assessment tool to measure team effectiveness and cognitive load, Sexton and colleagues identified anticipation, active team engagement, and higher familiarity scores as the best predictors of team efficiency.8 Several studies emphasized the need for a stable team, especially in the learning phase of robotic surgery.5,9,10 A dedicated robotic team reduced the operative time by 18% during robot-assisted sacrocolpopexy (RASCP).10 RASCP procedures that extended into the afternoon took significantly longer time.9 A dedicated anesthesiologist improved the preoperative time.9 Surgical team handoffs also have reduced OR efficiency.11,12
Studying the impact of human factors is paramount for safe and efficient surgery. It is especially necessary in ORs that are equipped with high technologic instruments such as those used in RAS.
Surgical Black Box: Using data for OR safety and efficiency
Surgical procedures account for more than 50% of medical errors in a hospital setting, many of which are preventable. Postevent analysis with traditional methods, such as “Morbidity and Mortality” meetings held many days later, misses many adverse events in the OR.13 Another challenge with ever-changing and fast-multiplying surgical approaches is the development of effective surgical skill. Reviewing video recording of surgical procedures has been proposed as an instrument for recognizing adverse events and perfecting surgical skills.Recently, an innovative data-capture platform called the OR Black Box, developed by Teodor Grantcharov, MD, PhD, and colleagues, went beyond simple audiovisual recording.14 This high technologic platform not only video records the actual surgical procedure with laparoscopic camera capture (and wearable cameras for open cases) but also monitors the entire OR environment via wide-angle cameras, utilizes sensors, and records both the patient’s and the surgeon’s physiologic parameters.
The OR Black Box generates a holistic view of the OR after synchronization, encryption, and secure storage of all inputs for further analysis by experts and software-based algorithms (FIGURE 2). Computer vision algorithms can recognize improper dissection techniques and complications, such as bleeding. Adverse events are flagged with an automated software on a procedural timeline to facilitate review of procedural steps, disruptive environmental and organizational factors, OR team technical and nontechnical skills, surgeon physiologic stress, and intraoperative errors, events, and rectification processes using validated instruments.
Artificial intelligence built into this platform can automatically extract objective, high-quality, and structured data to generate explainable insights by recognizing adverse events and procedural segments of interest for training and quality improvement and provide a foundation with objective measurements of technical and nontechnical performance for formative and summative assessment. This system, a major step up compared with retrospective review of likely biased medical records and labor-intensive multidisciplinary human observers, has the potential to increase efficiency and reduce costs by studying human factors that include clinical design, technology, and organization. OR efficiency, measured in real time objectively and thoroughly, may save time and resources.
OR Black Box platforms have already started to generate meaningful data. It is not surprising that auditory disruptions—OR doors opening, loud noises, pagers beeping, telephones ringing—were recorded almost every minute during laparoscopic procedures.15 Most technical errors occurred during dissection, resection, and reconstruction and most commonly were associated with improper estimations of force applied to tissue and distance to the target tissue during operative steps of a laparoscopic procedure.16 Another study based on this system showed that technical performance was an independent predictor of postoperative outcomes.17 The OR Black Box identified a device-related interruption in 30% of elective laparoscopic general surgery cases, most commonly in sleeve gastrectomy and oncologic gastrectomy procedures. This sophisticated surgical data recording system also demonstrated a significantly better ability to detect Veress needle injuries (12 vs 3) and near misses (47 vs 0) when compared with traditional chart review.18
Data from the OR Black Box also have been applied to better analyze nontechnical performance, including teamwork and interpersonal dynamics.19 Surgeons most commonly exhibited adept situational awareness and leadership, while the nurse team excelled at task management and situational awareness.19 Of the total care provider team studied, the surgeon and scrub nurse demonstrated the most favorable nontechnical behavior.19 Of note, continuous physiologic monitoring of the surgeon with this system revealed that surgeons under stress had 66% higher adverse events.
The OR Black Box is currently utilized at 20 institutions in North America and Europe. The data compiled from all these institutions revealed that there was a 10% decrease in intraoperative adverse events for each 10-point increase in technical skill score on a scale of 0 to 100 (unpublished data). This centralized data indicated that turnover time ranged widely between 7 and 91 minutes, with variation of cleanup time from 1 to 25 minutes and setup time from 22 to 43 minutes. Institutions can learn from each other using this platform. For example, the information about block time utilization (20%–99%) across institutions provides opportunities for system improvements.
With any revolutionary technology, it is imperative to study its effects on outcomes, training, costs, and privacy before it is widely implemented. We, obstetricians and gynecologists, are very familiar with the impact of electronic fetal monitoring, a great example of a technologic advance that did not improve perinatal outcomes but led to unintended consequences, such as higher rates of cesarean deliveries and lawsuits. Such a tool may lead to potential misrepresentation of intraoperative events unless legal aspects are clearly delineated. As exciting as it is, this disruptive technology requires further exploration with scientific vigor.
Continue to: Surgeon and hospital volume: Surgical outcomes paradigm...
Surgeon and hospital volume: Surgical outcomes paradigm
A landmark study in 1979 that showed decreased mortality in high-volume centers underscored the need for regionalization for certain surgical procedures.20 This association was further substantiated by 2 reports on 2.5 million Medicare beneficiaries that demonstrated significantly lower mortality for all 14 cardiovascular and oncologic procedures for hospitals with larger surgical volume (16% vs 4%) and high-volume surgeons for certain procedures, for example, 15% versus 5% for pancreatic resections for cancer.21,22
A similar association was found for all routes of hysterectomies performed for benign indications. Boyd and colleagues showed that gynecologists who performed fewer than 10 hysterectomies per year had a higher perioperative morbidity rate (16.5%) compared with those who did more (11.7%).23 Specific to vaginal hysterectomy, in a study of more than 6,000 women, surgeons who performed 13 procedures per year had 31% less risk of operative injury than those who did 5.5 procedures per year (2.5% vs 1.7%).24 Overall perioperative complications (5.0% vs 4.0%) and medical complications (5.7% vs 3.9%) were also reduced for higher-volume surgeons. In a cohort of approximately 8,000 women who underwent a laparoscopic hysterectomy, high-volume surgeons had a considerably lower complication rate (4.2% vs 6.2%).25
As expected, lower complication rates of high-volume surgeons led to lower resource utilization, including lower transfusion rates, less intensive care unit utilization, and shorter operative times and, in several studies, length of stay.24,25 Of note, low-volume surgeons were less likely to offer minimally invasive routes and were more likely to convert to laparotomy.26 In addition, significant cost savings have been associated with high surgical volume, which one study showed was 16% ($6,500 vs $5,600) for high-volume surgeons.26 With regard to mortality, a study of 7,800 women found that perioperative mortality increased more than 10-fold for surgeons who performed an average 1 case per year compared with all other surgeons (2.5% vs 0.2%).27
When gynecologic cancers are concerned, arguably, long-term survival outcomes may be more critical than perioperative morbidity and mortality. Higher surgeon and hospital volume are associated with improved perioperative outcomes for endometrial and cervical cancers.28 Importantly, minimally invasive hysterectomy was offered for endometrial cancer significantly more often by surgeons with high volume.28 Survival outcomes were not affected by surgeon or hospital volume, likely due to overall more favorable prognosis for endometrial cancer after treatment.
Although it is intuitive to assume that a surgeon’s skills and experience would make the most impact in procedures for ovarian cancer due to the complexity of ovarian cancer surgery, evidence on short-term outcomes has been mixed. Intriguingly, some studies reported that high-volume institutions had higher complication and readmission rates. However, evidence supports that the surgeon’s volume, and especially hospital volume, improves long-term survival for ovarian cancer, with a negative impact on immediate postoperative morbidity.29 This may suggest that a more aggressive surgical effort improves long-term survival but also can cause more perioperative complications. Further, longer survival may result not only from operative skills but also because of better care by a structured multidisciplinary team at more established high-volume cancer centers.
The association of improved outcomes with higher volume led to public reporting of hospital outcomes. Policy efforts toward regionalization have impacted surgical practice. Based on their analysis of 3.2 million Medicare patients who underwent 1 of 8 different cancer surgeries or cardiovascular operations from 1999 to 2008, Finks and colleagues demonstrated that care was concentrated to fewer hospitals over time for many of these procedures.29 This trend was noted for gynecologic cancer surgery but not for benign gynecologic surgery.
Regionalization of care limits access particularly for minority and underserved communities because of longer travel distances, logistic challenges, and financial strain. An alternative to regionalization of care is targeted quality improvement by rigorous adherence to quality guidelines at low-volume hospitals.
Is there a critical minimum volume that may be used as a requirement for surgeons to maintain their privileges and for hospitals to offer certain procedures? In 2015, minimum volume standards for a number of common procedures were proposed by Johns Hopkins Medicine and Dartmouth-Hitchcock Medical Center, such as 50 hip replacement surgeries per hospital and 25 per physician per year, and 20 pancreatectomies per hospital and 5 per surgeon per year.30 A modeling study for hysterectomy showed that a volume cut point of >1 procedure in the prior year would restrict privileges for a substantial number of surgeons performing abdominal (17.5%), robot-assisted (12.5%), laparoscopic (16.8%), and vaginal (27.6%) hysterectomies.27 This study concluded that minimum-volume standards for hysterectomy for even the lowest volume physicians would restrict a significant number of gynecologic surgeons, including many with outcomes that are better than predicted.
Therefore, while there is good evidence that favors better outcomes in the hands of high-volume surgeons in gynecology, the impact of such policies on gynecologic practice clearly warrants careful monitoring and further study.
- What factors besides the surgeon’s skills influence surgical safety and efficiency?
- Are you ready to have audio, video, and sensor-based recording of everything in the OR?
- Who should perform your loved one’s hysterectomy? Do the surgeon’s and hospital’s volume matter?
The operating room (OR) is a key contributor to a hospital’s profitability. It is a complex environment with ever-advancing technology. A successful surgery completed without complications within an optimal time depends not only on the surgeon’s experience, skills, and knowledge but also on numerous other structural, human, and nontechnical factors over which the surgeon has limited control.
As in any setting that deals with human life, in the OR, team dynamics, communication, and environment play a major role. Research has indicated the benefits of dedicated teams, reduced handoffs, and innovative modalities that continuously and systematically monitor potential breakdowns and propose solutions for the detected problems.
Finally, who should perform your loved one’s hysterectomy? This article also attempts to address the impact of surgeons’ and hospitals’ volume on operative outcomes with a diminishing number of hysterectomies but an increasing number of approaches.
Human factors in the OR
Human factors research was born as a product of the industrial revolution and mass production. It aims to optimize human experience and improve system performance by studying how humans interact with system. The aviation industry, for example, minimized errors significantly by using methods developed by human factors scientists. As another industry with no tolerance for mistakes, the health care sector followed suit. Ultimately, the goal of human factors research in health care is to improve patient safety, optimize work and environment, reduce costs, and enhance employees’ physical and mental health, engagement, comfort, and quality of life (FIGURE 1).1
Today’s OR is so complex that it is hard to understand its dynamics without human factors research. Every new OR technology is first tested in controlled and simulated environments to determine “work as imagined.” However, it is necessary to study “work as done” in the real world via direct observation, video recording, questionnaires, and semistructured interviews by an on-site multidisciplinary team. This process not only focuses on surgical skills, process efficiency, and outcomes but also monitors the entire process according to Human Factors and Ergonomics Engineering principles to explore otherwise hidden complexities and latent safety concerns. The Systems Engineering Initiative for Patient Safety (SEIPS) framework is used to study the impact of interactions between people, tasks, technologies, environment, and organization.1
Robot-assisted surgery (RAS), an increasingly popular surgical approach among gynecologic surgeons, recently has been the focus of human factors science. A robotic OR poses unique challenges: the surgeon is not scrubbed, is removed from the operating table, and controls a complex highly technologic device in a crowded and darkened room. These are ideal conditions waiting to be optimized by human factor experts. To demonstrate the importance of human factors in the OR, we review the evidence for RAS.
Continue to: Impact of flow disruptions...
Impact of flow disruptions
Flow disruptions (FDs) were found to be more common in RAS. Catchpole and colleagues identified a mean of 9.62 FDs per hour in 89 robotic procedures, including hysterectomies and sacrocolpopexies, from a variety of fields; FDs occurred more often during the docking stage, followed by the console time, and they mostly were caused by communication breakdown and lack of team familiarity.2
Surgeon experience significantly reduced FDs. Surgeons who had done more than 700 RAS cases experienced 60% fewer FDs than those who had done less than 250 cases (13 vs 8 per hour).2 A study focusing on residents’ impact on RAS outcomes found that each FD increased the total operative time by an average 2.4 minutes, with the number significantly higher when a resident was involved.3 About one-quarter of the training-related FDs were procedure-specific instructions, while one-third were related to instrument and robotic instruction. However, pauses to teach residents did not appear to create significant intraoperative delays. Expectedly, experienced surgeons could anticipate and reduce these disruptions by supporting the whole team.
Human ergonomics, turnover time, and robot-specific skills
In a study of human ergonomics in RAS, Yu and colleagues noted that bedside assistants could experience neck posture problems. Surprisingly, the console could constrain the surgeon’s neck-shoulder region.4 Studies that reported on communication problems in a robotic OR suggest that innovative forms of verbal and nonverbal communication may support successful team communication.5
On the learning curve for RAS, OR turnover time, a key value metric, has been longer. However, turnover time was reduced almost by half from 99.2 to 53.2 minutes over 3 months after concepts from motor racing pit stops were employed, including briefings, leadership, role definition, task allocation, and task sequencing. Average room-ready time also was lowered from 42.2 to 27.2 minutes.6 RAS presents new challenges with sterile instrument processing as well. A successful RAS program, therefore, has organizational needs that include the training of OR and sterile processing staff and appropriate shift management.1
In a robotic OR, not only the surgeon but also the whole team requires robot-specific skills. New training approaches to teamwork, communication, and situation awareness skills are necessary. Robotic equipment, with its data and power cables, 2 consoles, and changing movement paths, necessitate larger rooms with a specific layout.7
In a review of recordings of RAS that used a multidimensional assessment tool to measure team effectiveness and cognitive load, Sexton and colleagues identified anticipation, active team engagement, and higher familiarity scores as the best predictors of team efficiency.8 Several studies emphasized the need for a stable team, especially in the learning phase of robotic surgery.5,9,10 A dedicated robotic team reduced the operative time by 18% during robot-assisted sacrocolpopexy (RASCP).10 RASCP procedures that extended into the afternoon took significantly longer time.9 A dedicated anesthesiologist improved the preoperative time.9 Surgical team handoffs also have reduced OR efficiency.11,12
Studying the impact of human factors is paramount for safe and efficient surgery. It is especially necessary in ORs that are equipped with high technologic instruments such as those used in RAS.
Surgical Black Box: Using data for OR safety and efficiency
Surgical procedures account for more than 50% of medical errors in a hospital setting, many of which are preventable. Postevent analysis with traditional methods, such as “Morbidity and Mortality” meetings held many days later, misses many adverse events in the OR.13 Another challenge with ever-changing and fast-multiplying surgical approaches is the development of effective surgical skill. Reviewing video recording of surgical procedures has been proposed as an instrument for recognizing adverse events and perfecting surgical skills.Recently, an innovative data-capture platform called the OR Black Box, developed by Teodor Grantcharov, MD, PhD, and colleagues, went beyond simple audiovisual recording.14 This high technologic platform not only video records the actual surgical procedure with laparoscopic camera capture (and wearable cameras for open cases) but also monitors the entire OR environment via wide-angle cameras, utilizes sensors, and records both the patient’s and the surgeon’s physiologic parameters.
The OR Black Box generates a holistic view of the OR after synchronization, encryption, and secure storage of all inputs for further analysis by experts and software-based algorithms (FIGURE 2). Computer vision algorithms can recognize improper dissection techniques and complications, such as bleeding. Adverse events are flagged with an automated software on a procedural timeline to facilitate review of procedural steps, disruptive environmental and organizational factors, OR team technical and nontechnical skills, surgeon physiologic stress, and intraoperative errors, events, and rectification processes using validated instruments.
Artificial intelligence built into this platform can automatically extract objective, high-quality, and structured data to generate explainable insights by recognizing adverse events and procedural segments of interest for training and quality improvement and provide a foundation with objective measurements of technical and nontechnical performance for formative and summative assessment. This system, a major step up compared with retrospective review of likely biased medical records and labor-intensive multidisciplinary human observers, has the potential to increase efficiency and reduce costs by studying human factors that include clinical design, technology, and organization. OR efficiency, measured in real time objectively and thoroughly, may save time and resources.
OR Black Box platforms have already started to generate meaningful data. It is not surprising that auditory disruptions—OR doors opening, loud noises, pagers beeping, telephones ringing—were recorded almost every minute during laparoscopic procedures.15 Most technical errors occurred during dissection, resection, and reconstruction and most commonly were associated with improper estimations of force applied to tissue and distance to the target tissue during operative steps of a laparoscopic procedure.16 Another study based on this system showed that technical performance was an independent predictor of postoperative outcomes.17 The OR Black Box identified a device-related interruption in 30% of elective laparoscopic general surgery cases, most commonly in sleeve gastrectomy and oncologic gastrectomy procedures. This sophisticated surgical data recording system also demonstrated a significantly better ability to detect Veress needle injuries (12 vs 3) and near misses (47 vs 0) when compared with traditional chart review.18
Data from the OR Black Box also have been applied to better analyze nontechnical performance, including teamwork and interpersonal dynamics.19 Surgeons most commonly exhibited adept situational awareness and leadership, while the nurse team excelled at task management and situational awareness.19 Of the total care provider team studied, the surgeon and scrub nurse demonstrated the most favorable nontechnical behavior.19 Of note, continuous physiologic monitoring of the surgeon with this system revealed that surgeons under stress had 66% higher adverse events.
The OR Black Box is currently utilized at 20 institutions in North America and Europe. The data compiled from all these institutions revealed that there was a 10% decrease in intraoperative adverse events for each 10-point increase in technical skill score on a scale of 0 to 100 (unpublished data). This centralized data indicated that turnover time ranged widely between 7 and 91 minutes, with variation of cleanup time from 1 to 25 minutes and setup time from 22 to 43 minutes. Institutions can learn from each other using this platform. For example, the information about block time utilization (20%–99%) across institutions provides opportunities for system improvements.
With any revolutionary technology, it is imperative to study its effects on outcomes, training, costs, and privacy before it is widely implemented. We, obstetricians and gynecologists, are very familiar with the impact of electronic fetal monitoring, a great example of a technologic advance that did not improve perinatal outcomes but led to unintended consequences, such as higher rates of cesarean deliveries and lawsuits. Such a tool may lead to potential misrepresentation of intraoperative events unless legal aspects are clearly delineated. As exciting as it is, this disruptive technology requires further exploration with scientific vigor.
Continue to: Surgeon and hospital volume: Surgical outcomes paradigm...
Surgeon and hospital volume: Surgical outcomes paradigm
A landmark study in 1979 that showed decreased mortality in high-volume centers underscored the need for regionalization for certain surgical procedures.20 This association was further substantiated by 2 reports on 2.5 million Medicare beneficiaries that demonstrated significantly lower mortality for all 14 cardiovascular and oncologic procedures for hospitals with larger surgical volume (16% vs 4%) and high-volume surgeons for certain procedures, for example, 15% versus 5% for pancreatic resections for cancer.21,22
A similar association was found for all routes of hysterectomies performed for benign indications. Boyd and colleagues showed that gynecologists who performed fewer than 10 hysterectomies per year had a higher perioperative morbidity rate (16.5%) compared with those who did more (11.7%).23 Specific to vaginal hysterectomy, in a study of more than 6,000 women, surgeons who performed 13 procedures per year had 31% less risk of operative injury than those who did 5.5 procedures per year (2.5% vs 1.7%).24 Overall perioperative complications (5.0% vs 4.0%) and medical complications (5.7% vs 3.9%) were also reduced for higher-volume surgeons. In a cohort of approximately 8,000 women who underwent a laparoscopic hysterectomy, high-volume surgeons had a considerably lower complication rate (4.2% vs 6.2%).25
As expected, lower complication rates of high-volume surgeons led to lower resource utilization, including lower transfusion rates, less intensive care unit utilization, and shorter operative times and, in several studies, length of stay.24,25 Of note, low-volume surgeons were less likely to offer minimally invasive routes and were more likely to convert to laparotomy.26 In addition, significant cost savings have been associated with high surgical volume, which one study showed was 16% ($6,500 vs $5,600) for high-volume surgeons.26 With regard to mortality, a study of 7,800 women found that perioperative mortality increased more than 10-fold for surgeons who performed an average 1 case per year compared with all other surgeons (2.5% vs 0.2%).27
When gynecologic cancers are concerned, arguably, long-term survival outcomes may be more critical than perioperative morbidity and mortality. Higher surgeon and hospital volume are associated with improved perioperative outcomes for endometrial and cervical cancers.28 Importantly, minimally invasive hysterectomy was offered for endometrial cancer significantly more often by surgeons with high volume.28 Survival outcomes were not affected by surgeon or hospital volume, likely due to overall more favorable prognosis for endometrial cancer after treatment.
Although it is intuitive to assume that a surgeon’s skills and experience would make the most impact in procedures for ovarian cancer due to the complexity of ovarian cancer surgery, evidence on short-term outcomes has been mixed. Intriguingly, some studies reported that high-volume institutions had higher complication and readmission rates. However, evidence supports that the surgeon’s volume, and especially hospital volume, improves long-term survival for ovarian cancer, with a negative impact on immediate postoperative morbidity.29 This may suggest that a more aggressive surgical effort improves long-term survival but also can cause more perioperative complications. Further, longer survival may result not only from operative skills but also because of better care by a structured multidisciplinary team at more established high-volume cancer centers.
The association of improved outcomes with higher volume led to public reporting of hospital outcomes. Policy efforts toward regionalization have impacted surgical practice. Based on their analysis of 3.2 million Medicare patients who underwent 1 of 8 different cancer surgeries or cardiovascular operations from 1999 to 2008, Finks and colleagues demonstrated that care was concentrated to fewer hospitals over time for many of these procedures.29 This trend was noted for gynecologic cancer surgery but not for benign gynecologic surgery.
Regionalization of care limits access particularly for minority and underserved communities because of longer travel distances, logistic challenges, and financial strain. An alternative to regionalization of care is targeted quality improvement by rigorous adherence to quality guidelines at low-volume hospitals.
Is there a critical minimum volume that may be used as a requirement for surgeons to maintain their privileges and for hospitals to offer certain procedures? In 2015, minimum volume standards for a number of common procedures were proposed by Johns Hopkins Medicine and Dartmouth-Hitchcock Medical Center, such as 50 hip replacement surgeries per hospital and 25 per physician per year, and 20 pancreatectomies per hospital and 5 per surgeon per year.30 A modeling study for hysterectomy showed that a volume cut point of >1 procedure in the prior year would restrict privileges for a substantial number of surgeons performing abdominal (17.5%), robot-assisted (12.5%), laparoscopic (16.8%), and vaginal (27.6%) hysterectomies.27 This study concluded that minimum-volume standards for hysterectomy for even the lowest volume physicians would restrict a significant number of gynecologic surgeons, including many with outcomes that are better than predicted.
Therefore, while there is good evidence that favors better outcomes in the hands of high-volume surgeons in gynecology, the impact of such policies on gynecologic practice clearly warrants careful monitoring and further study.
- What factors besides the surgeon’s skills influence surgical safety and efficiency?
- Are you ready to have audio, video, and sensor-based recording of everything in the OR?
- Who should perform your loved one’s hysterectomy? Do the surgeon’s and hospital’s volume matter?
- Catchpole K, Bisantz A, Hallbeck MS, et al. Human factors in robotic assisted surgery: lessons from studies ‘in the wild’. Appl Ergon. 2019;78:270-276.
- Catchpole K, Perkins C, Bresee C, et al. Safety, efficiency and learning curves in robotic surgery: a human factors analysis. Surg Endosc. 2016;30:3749-3761.
- Jain M, Fry BT, Hess LW, et al. Barriers to efficiency in robotic surgery: the resident effect. J Surg. Res. 2016;205:296-304.
- Yu D, Dural C, Morrow MM, et al. Intraoperative workload in robotic surgery assessed by wearable motion tracking sensors and questionnaires. Surg Endosc. 2017;31:877-886.
- Randell R, Honey S, Alvarado N, et al. Embedding robotic surgery into routine practice and impacts on communication and decision making: a review of the experience of surgical teams. Cognit Technol Work. 2016;18:423-437.
- Souders CP, Catchpole KR, Wood LN, et al. Reducing operating room turnover time for robotic surgery using a motor racing pit stop model. World J Surg. 2017;4:1943–1949.
- Ahmad N, Hussein AA, Cavuoto L, et al. Ambulatory movements, team dynamics and interactions during robot-assisted surgery. BJU Int. 2016;118:132-139.
- Sexton K, Johnson A, Gotsch A, et al. Anticipation, teamwork, and cognitive load: chasing efficiency during robot-assisted surgery. BMJ Qual Saf. 2018;27:148-154.
- Harmanli O, Solak S, Bayram A, et al. Optimizing the robotic surgery team: an operations management perspective. Int Urogynecol J. 2021;32:1379-1385.
- Carter-Brooks CM, Du AL, Bonidie MJ, et al. The impact of a dedicated robotic team on robotic-assisted sacrocolpopexy outcomes. Female Pelvic Med Reconstr Surg. 2018;24:13-16.
- Giugale LE, Sears S, Lavelle ES, et al. Evaluating the impact of intraoperative surgical team handoffs on patient outcomes. Female Pelvic Med Reconstr Surg. 2017;23:288-292.
- Geynisman-Tan J, Brown O, Mueller M, et al. Operating room efficiency: examining the impact of personnel handoffs. Female Pelvic Med Reconstr Surg. 2018;24:87-89.
- Alsubaie H, Goldenberg M, Grantcharov T. Quantifying recall bias in surgical safety: a need for a modern approach to morbidity and mortality reviews. Can J Surg. 2019;62:39-43.
- Goldenberg MG, Jung J, Grantcharov TP. Using data to enhance performance and improve quality and safety in surgery. JAMA Surg. 2017;152:972-973.
- Jung JJ, Grantcharov TP. The operating room black box: a prospective observational study of the operating room. J Am Coll Surg. 2017;225:S127-S128.
- Jung JJ, Jüni P, Lebovic G, et al. First-year analysis of the operating room black box study. Ann Surg. 2020;271:122-127.
- Jung JJ, Kashfi A, Sharma S, et al. Characterization of device-related interruptions in minimally invasive surgery: need for intraoperative data and effective mitigation strategies. Surg Endosc. 2019;33:717-723.
- Jung JJ, Adams-McGavin RC, Grantcharov TP. Underreporting of Veress needle injuries: comparing direct observation and chart review methods. J Surg Res. 2019;236:266-270.
- Fesco AB, Kuzulugil SS, Babaoglu C, et al. Relationship between intraoperative nontechnical performance and technical events in bariatric surgery. Br J Surg. 2018;105:1044-1050.
- Luft HS, Bunker JP, Enthoven AC. Should operations be regionalized? The empirical relation between surgical volume and mortality. N Engl J Med. 1979;301:1364-1369.
- Birkmeyer JD, Siewers AE, Finlayson EV, et al. Hospital volume and surgical mortality in the United States. N Engl J Med. 2002;346:1128-1137.
- Birkmeyer JD, Stukel TA, Siewers AE, et al. Surgeon volume and operative mortality in the United States. N Engl J Med. 2003;349:21172127.
- Boyd LR, Novetsky AP, Curtin JP. Effect of surgical volume on route of hysterectomy and short-term morbidity. Obstet Gynecol. 2010;116:909-915.
- Rogo-Gupta LJ, Lewin SN, Kim JH, et al. The effect of surgeon volume on outcomes and resource use for vaginal hysterectomy. Obstet Gynecol. 2010;116:1341-1347.
- Wallenstein MR, Ananth CV, Kim JH, et al. Effect of surgical volume on outcomes for laparoscopic hysterectomy for benign indications. Obstet Gynecol. 2012;119:709-716.
- Bretschneider CE, Frazzini Padilla P, Das D, et al. The impact of surgeon volume on perioperative adverse events in women undergoing minimally invasive hysterectomy for the large uterus. Am J Obstet Gynecol. 2018;219:490.e1-490.e8.
- Ruiz MP, Chen L, Hou JY, et al. Outcomes of hysterectomy performed by very low-volume surgeons. Obstet Gynecol. 2018;131:981-990.
- Wright JD. The volume-outcome paradigm for gynecologic surgery: clinical and policy implications. Clin Obstet Gynecol. 2020;63:252-265.
- Finks JF, Osborne NH, Birkmeyer JD. Trends in hospital volume and operative mortality for high risk surgery. N Engl J Med. 2011;364:21282137.
- Sternberg S. Hospitals move to limit low-volume surgeries. US News & World Report. May 19, 2015. www.usnews.com/news /articles/2015/05/19/hospitals-move-to-limit-low-volume-surgeries. Accessed April 19, 2022.
- Catchpole K, Bisantz A, Hallbeck MS, et al. Human factors in robotic assisted surgery: lessons from studies ‘in the wild’. Appl Ergon. 2019;78:270-276.
- Catchpole K, Perkins C, Bresee C, et al. Safety, efficiency and learning curves in robotic surgery: a human factors analysis. Surg Endosc. 2016;30:3749-3761.
- Jain M, Fry BT, Hess LW, et al. Barriers to efficiency in robotic surgery: the resident effect. J Surg. Res. 2016;205:296-304.
- Yu D, Dural C, Morrow MM, et al. Intraoperative workload in robotic surgery assessed by wearable motion tracking sensors and questionnaires. Surg Endosc. 2017;31:877-886.
- Randell R, Honey S, Alvarado N, et al. Embedding robotic surgery into routine practice and impacts on communication and decision making: a review of the experience of surgical teams. Cognit Technol Work. 2016;18:423-437.
- Souders CP, Catchpole KR, Wood LN, et al. Reducing operating room turnover time for robotic surgery using a motor racing pit stop model. World J Surg. 2017;4:1943–1949.
- Ahmad N, Hussein AA, Cavuoto L, et al. Ambulatory movements, team dynamics and interactions during robot-assisted surgery. BJU Int. 2016;118:132-139.
- Sexton K, Johnson A, Gotsch A, et al. Anticipation, teamwork, and cognitive load: chasing efficiency during robot-assisted surgery. BMJ Qual Saf. 2018;27:148-154.
- Harmanli O, Solak S, Bayram A, et al. Optimizing the robotic surgery team: an operations management perspective. Int Urogynecol J. 2021;32:1379-1385.
- Carter-Brooks CM, Du AL, Bonidie MJ, et al. The impact of a dedicated robotic team on robotic-assisted sacrocolpopexy outcomes. Female Pelvic Med Reconstr Surg. 2018;24:13-16.
- Giugale LE, Sears S, Lavelle ES, et al. Evaluating the impact of intraoperative surgical team handoffs on patient outcomes. Female Pelvic Med Reconstr Surg. 2017;23:288-292.
- Geynisman-Tan J, Brown O, Mueller M, et al. Operating room efficiency: examining the impact of personnel handoffs. Female Pelvic Med Reconstr Surg. 2018;24:87-89.
- Alsubaie H, Goldenberg M, Grantcharov T. Quantifying recall bias in surgical safety: a need for a modern approach to morbidity and mortality reviews. Can J Surg. 2019;62:39-43.
- Goldenberg MG, Jung J, Grantcharov TP. Using data to enhance performance and improve quality and safety in surgery. JAMA Surg. 2017;152:972-973.
- Jung JJ, Grantcharov TP. The operating room black box: a prospective observational study of the operating room. J Am Coll Surg. 2017;225:S127-S128.
- Jung JJ, Jüni P, Lebovic G, et al. First-year analysis of the operating room black box study. Ann Surg. 2020;271:122-127.
- Jung JJ, Kashfi A, Sharma S, et al. Characterization of device-related interruptions in minimally invasive surgery: need for intraoperative data and effective mitigation strategies. Surg Endosc. 2019;33:717-723.
- Jung JJ, Adams-McGavin RC, Grantcharov TP. Underreporting of Veress needle injuries: comparing direct observation and chart review methods. J Surg Res. 2019;236:266-270.
- Fesco AB, Kuzulugil SS, Babaoglu C, et al. Relationship between intraoperative nontechnical performance and technical events in bariatric surgery. Br J Surg. 2018;105:1044-1050.
- Luft HS, Bunker JP, Enthoven AC. Should operations be regionalized? The empirical relation between surgical volume and mortality. N Engl J Med. 1979;301:1364-1369.
- Birkmeyer JD, Siewers AE, Finlayson EV, et al. Hospital volume and surgical mortality in the United States. N Engl J Med. 2002;346:1128-1137.
- Birkmeyer JD, Stukel TA, Siewers AE, et al. Surgeon volume and operative mortality in the United States. N Engl J Med. 2003;349:21172127.
- Boyd LR, Novetsky AP, Curtin JP. Effect of surgical volume on route of hysterectomy and short-term morbidity. Obstet Gynecol. 2010;116:909-915.
- Rogo-Gupta LJ, Lewin SN, Kim JH, et al. The effect of surgeon volume on outcomes and resource use for vaginal hysterectomy. Obstet Gynecol. 2010;116:1341-1347.
- Wallenstein MR, Ananth CV, Kim JH, et al. Effect of surgical volume on outcomes for laparoscopic hysterectomy for benign indications. Obstet Gynecol. 2012;119:709-716.
- Bretschneider CE, Frazzini Padilla P, Das D, et al. The impact of surgeon volume on perioperative adverse events in women undergoing minimally invasive hysterectomy for the large uterus. Am J Obstet Gynecol. 2018;219:490.e1-490.e8.
- Ruiz MP, Chen L, Hou JY, et al. Outcomes of hysterectomy performed by very low-volume surgeons. Obstet Gynecol. 2018;131:981-990.
- Wright JD. The volume-outcome paradigm for gynecologic surgery: clinical and policy implications. Clin Obstet Gynecol. 2020;63:252-265.
- Finks JF, Osborne NH, Birkmeyer JD. Trends in hospital volume and operative mortality for high risk surgery. N Engl J Med. 2011;364:21282137.
- Sternberg S. Hospitals move to limit low-volume surgeries. US News & World Report. May 19, 2015. www.usnews.com/news /articles/2015/05/19/hospitals-move-to-limit-low-volume-surgeries. Accessed April 19, 2022.
Knowledge gaps and challenges in care for menopausal women
The transition to menopause begins with ovarian fluctuation and hormonal changes, often beginning before significant changes in menstruation. Reproductive aging with loss of follicular activity progresses over a wide age range (42 to 58 years) with an average onset at approximately age 47, ranging from 4 to 8 years. Although most women have heard about menopause, defined as 12 months after the last period, they often lack understanding about perimenopause or that the menopausal transition usually begins 5 years before menopause.1
Perimenopause, defined as early and late menopause transition stages, may be viewed as a window of potential vulnerability for women who develop or have worsening menstrual-related mood disorders. Over time, hormonal fluctuations often lead to menstrual cycle irregularity (either shorter or longer). Changes occurring during perimenopause may be confusing as it may not be clear whether symptoms are related to menopause, aging, or stress. Often not recognized or treated adequately, perimenopausal symptoms may be challenging to navigate for both women and clinicians.
The perimenopausal process is often even more confusing for women with early menopause—whether due to bilateral oophorectomy, chemotherapy or radiation therapy, genetics, or an autoimmune process—because of lack of recognition that an early menopausal transition is occurring or what solutions are available for symptoms. While there is support in the workplace for women during pregnancy and breastfeeding, there remains little support or recognition for the oft challenging perimenopausal transition leading to menopause.
Perimenopause: Common symptoms and treatments
Symptoms may be related to either estrogen level deficiency or excess during perimenopause, and these level changes may even occur within the same cycle.
Cyclic breast tenderness may develop, worsened by caffeine or high salt intake (which can be potentially improved, although without clinical trial evidence, with decreased caffeine or a trial of evening primrose oil or vitamin E).
Changes in menstrual flow and frequency of menses are typical. Flow may be lighter or heavier, longer or shorter, and there may be cycle variability, missed menses, or midcycle spotting.2 Bleeding may be heavy, with or without cramping. In addition to imaging with vaginal ultrasonography or hysteroscopy to identify structural issues, symptoms may be managed with nonsteroidal anti-inflammatory drugs (NSAIDs), hormonal therapy (HT) with short hormone-free interval contraceptives, oral progestogens, or progestin intrauterine systems. Newer medical treatments include antifibrinolytic drugs and selective progesterone-receptor modulators. Uterine ablation to decrease or stop bleeding is effective if there are no structural abnormalities, such as fibroids or polyps or the presence of adenomyosis, where glands will regrow into the endometrium after ablation. Endometrial biopsy is indicated for persistent abnormal uterine bleeding or those with risk factors such as chronic anovulation.
Worsening headaches or menstrual migraines may be triggered by hormonal changes, which may respond to NSAIDs; dihydroergotamine; triptans; the combination of aspirin, acetaminophen, and caffeine; or estrogen the week before menses. For women taking oral contraceptives (OCPs), adding estradiol the week before menses, or using the OCP continuously, may decrease headache frequency. These short-term prophylactic strategies during the perimenstrual time are often effective. If not, preventive therapy is available for women with frequent, severe headaches.
Mood complaints and poor sleep are independently associated with menstrual irregularity, and can lead to fatigue or anxiety, worsening premenstrual syndrome, or depressive moods. Sleep is disrupted premenstrually for up to one-third of women, and sleep disruption is particularly prevalent in those with premenstrual mood disorders and worsens during perimenopause.3
Reproductive hormones act on the neurotransmitter systems in the brain involved in mood regulation and emotion. The fluctuating hormones occurring during perimenopause may exacerbate pre-existing menstrual-related mood disorders. A subset of women experience depressive moods due to perimenopausal elevations in ovarian hormones.4 Others may exhibit increased mood sensitivity with the ovarian hormone withdrawal accompanying late menopause transition and early postmenopausal phase.5 There is significant comorbidity between premenstrual mood disorder (PMDD) and postpartum depression.6 During perimenopause and early menopause, clinicians should ask about prior hormonally-related depression (puberty, postpartum) and recognize that current or past premenstrual syndrome may worsen into a more severe premenstrual dysphoric disorder. Evidence-based treatments for PMDD include selective serotonin reuptake inhibitors (SSRIs); either taken continuously or only during the luteal phase; drospirenone-containing oral contraceptives, often with shorter pill-free intervals; GnRH analogues with or without hormone add-back; and cognitive behavioral therapy.7 For women whose perimenopausal moods improve with HT or develop worsened mood sensitivity with ovarian hormone withdrawal, clinicians should recognize that mood may worsen when treatment is ceased.5

Continue to: Menopausal symptoms...
Menopausal symptoms
Vasomotor symptoms (VMS), hot flashes, or night sweats occur in up to 75% of women as they develop more menstrual irregularity and move closer to their final period and menopause.
Hot flashes are transient episodes of flushing with the sensation of warmth (up to intense heat) on the upper body and face or head, often associated with sweating, chills or flushing, an increase in heart rate, and lowered blood pressure. Hot flashes can sometimes be preceded by an intense feeling of dread, followed by rapid heat dissipation. The etiology of hot flashes is still not clear, but the neurokinin receptors are involved. They are related to small fluctuations in core body temperature superimposed on a narrow thermoneutral zone in symptomatic women. Hot flashes are triggered when core body temperature rises above the upper (sweating) threshold. Shivering occurs if the core body temperature falls below the lower threshold. Sleep may be disrupted, with less rapid eye movement (REM) sleep, and associated with throwing covers on and off or changing sheets or nightclothes. On average, hot flashes last 7.2 years,8 and they are more bothersome if night sweats interfere with sleep or disrupt performance during the day.
In the Stages of Reproductive Aging Workshop (STRAW + 10), women reported VMS within 1-3 years after the menopausal transition.8 Four trajectories of hot flashes were identified in the Study of Women’s Health Across the Nation (SWAN) trial,9 including low levels throughout the menopause transition, early onset, late onset, and a group which had frequent hot flashes, starting early and lasting longer. Serum estrogen levels were not predictive of hot flash frequency or severity.
Hot flashes have been associated with low levels of exercise, cigarette smoking, high follicle-stimulating hormone levels and low estradiol levels, increasing body mass index, ethnicity (with hot flashes more common among Black and Hispanic women), low socioeconomic status, prior PMDD, anxiety, perceived stress, and depression.8 Women with a history of premenstrual syndrome, stress, sexual dysfunction, physical inactivity, or hot flashes are more vulnerable to depressive symptoms during perimenopause and early menopause.5
Depression may co-occur or overlap with menopause symptoms. Diagnosis involves menopausal stage, co-occurring psychiatric and menopause symptoms, psychosocial stressors, and a validated screening tool such as PQ9. Treatments for perimenopausal depression, such as antidepressants, psychotherapy, or cognitive behavioral therapy, are recommended first line for perimenopausal depression. Estrogen therapy has not been approved to treat perimenopausal depression but appears to have antidepressant effects in perimenopausal women, particularly those with bothersome vasomotor symptoms.5
Anxiety can worsen during menopause, and may respond to calming apps, meditation, cognitive behavioral therapy, hypnosis, yoga or tai chi, HT, or antianxiety medications.
Weight gain around the abdomen (ie, belly fat) is a common complaint during the menopausal transition, despite women reporting not changing their eating or exercise patterns. Increasing exercise or bursts of higher intensity, decreasing portion sizes or limiting carbohydrates and alcohol may help.
Memory and concentration problems, described as brain fog, tend to be more of an issue in perimenopause and level out after menopause. Counsel midlife women that these changes are not due to dementia but are related to normal aging, hormonal changes, mood, stress, or other life circumstances. Identifying and addressing sleep issues and mood disorders may help mitigate brain fog, as can advising women to avoid excess caffeine, alcohol, nicotine, and eating before bed. Improvements in memory, cognition, and health have been found with the Mediterranean diet, regular exercise, avoiding multitasking, and engaging in mentally stimulating activities.
Sleeping concerns in peri- and postmenopausal women include sleeping less and more frequent insomnia. Women are more likely to use prescription sleeping aids during these times of their lives. The data from SWAN8 show that the menopausal transition is related to self-reported difficulty sleeping, independent of age. Sleep latency interval is increased while REM sleep decreases. Night sweats can trigger awakenings in the first half of the night. The perceived decline in sleep quality also may be attributed to general aging effects, nocturnal urination, sleep-related disorders such as sleep apnea or restless legs, or chronic pain, stress, or depression.10 Suggestions for management include sleep apps, cognitive behavioral therapy, low-dose antidepressant therapy, addressing sleep routines, and HT. Hypnotics should be avoided.
Sexuality issues are common complaints during the menopausal transition. Cross-sectional data reported from a longitudinal, population-based Australian cohort of women aged 45 to 55 years, found a decrease in sexual responsivity, sexual frequency, libido, vaginal dyspareunia, and more partner problems.11 Low libido may be related to relationship issues, dyspareunia with vaginal narrowing, loss of lubrication, levator spasm, stress, anxiety, exhaustion or mood disorder, lowered hormone levels, excess alcohol intake, underlying health concerns, or a side effect of medications for depression or pain. There is no direct correlation between testosterone levels and libido.
When HT at menopause may be helpful
For healthy symptomatic women without contraindications who are younger than age 60, or within 10 years of menopause onset, the benefits of initiating HT most likely outweigh the risks to relieve bothersome hot flashes and night sweats.12-17 For older women, or for those further from menopause, the greater absolute risks of coronary heart disease, stroke, venous thromboembolism, and dementia, in general, outweigh the potential benefits.12-17 Extended durations of HT have less safety and efficacy data and should be considered primarily for those with persistent menopausal symptoms, with periodic re-evaluation.13,14 For bothersome genitourinary syndrome of menopause symptoms that do not respond to vaginal moisturizers or lubricants, low-dose vaginal HTs are encouraged.13-17

Continue to: Early-onset menopause...
Early-onset menopause
According to observational studies,18 early menopause is associated with a higher risk of osteoporosis, coronary heart disease, cognitive changes, vaginal dryness, loss of libido, and mood changes. Studies have shown that women with early menopause who take HT, without contraindications, to the average age of menopause (age 52) decrease the health risks of early menopause (bone loss, heart disease, mood, and cognition changes).13,14,18
Women with early menopause, whether spontaneous or following bilateral oophorectomy or cancer treatment, should be counseled to get adequate calcium (dietary recommended over supplementation) and vitamin D intake, eat a healthy diet, and exercise regularly. Evaluation should include risk for bone loss, heart disease, mood changes, and vaginal changes.
Extended use of HT
Up to 8% of women have hot flashes for 20 years or more after menopause.19 The decision to continue or to stop HT is not always clear for women:
- with persistent hot flashes after a trial period of HT discontinuation
- with bone loss that cannot be treated with bone-specific medications
- who request continuation for quality of life.
Extended use of HT should include an ongoing assessment of its risks and benefits, periodic trials off of HT, and documentation of rationale and informed discussions about continuing. Lower doses and transdermal therapies appear safer, as does micronized progesterone instead of more potent synthetic progestins.13-17
Genitourinary syndrome of menopause
Once women are further into menopause, they may notice vaginal dryness, vulvar itching or burning, bothersome vaginal discharge, or urinary urgency or frequency. The development of painful intercourse frequently occurs, a combination of the loss of estrogen with thinning of the vaginal mucosa, a loss of the acidic vaginal milieu with less elasticity, and spasm of the levator muscles. Some women develop urinary tract infections after intercourse or have more frequent reoccurrences. First-line therapy is often vaginal moisturizers and lubricants. Vaginal therapies (estradiol, conjugated estrogen, or dehydroepiandrosterone) or oral selective estrogen-receptor modulators (SERMs; ospemifene) improve vaginal dryness and dyspareunia.13,14 Pelvic therapy has also proved valuable for incontinence, pelvic floor dysfunction, and levator spasms.20
Where are there gaps in clinician knowledge?
Studies on emotional health, mood, and sleep need to incorporate measures of menstrual timing into data collection and analyses. Does the sleep disruption occurring premenstrually during perimenopause disproportionately contribute to a woman’s vulnerability to depressive disorders? The risk of clinically significant depressive symptoms increases 1.5- to 2.9-fold in the menopause transition.5 Research into premenstrual dysphoria during the menopause transition may identify different trajectories in the timing of symptoms related to either cycle itself or the ovarian hormone fluctuations or both.21 Gamma-aminobutyric acid (GABA)-modulating drugs, such as sepranolone, which blocks allopregnanolone’s actions at the GABAA receptor, may allow treatment of menstrual-related mood disorders without the need for hormonal interventions.21
Despite extended observational trial data, more data are needed to inform us about the long-term risks and benefits of using menopausal HT, particularly when initiated at menopause and to help address the timing of HT discontinuation. Furthermore, there are many unanswered questions. For instance:
- How much safer are lower dose and transdermal therapies?
- Do untreated hot flashes increase the risk of cardiovascular disease or dementia?
- Will newer non-HT options, such as the neurokinin receptor antagonists that are in testing but are not yet available, lower cardiovascular or dementia risks?
- What will be the risks and benefits for the newer estrogen in testing (estetrol, or E4), considered a natural estrogen and which appears to have lower thrombotic risks?
- What will be the role of intravaginal energy-based therapies, such as vaginal laser or radiofrequency devices?
- How do we address diverse populations and the effects of menopause on race, gender, culture, prior trauma, and socioeconomic status?
Lack of recognition of menopausal symptoms, particularly in the workplace
Clinicians need to understand the varied physical and emotional symptoms that may occur with hormonal changes as women traverse perimenopause and early menopause. We need to recognize that the lack of discussion about women’s health during this time may make women feel ashamed and fearful of bringing up their symptoms due to fear of being dismissed or stigmatized.22 Women may not seek help until a crisis at home or work occurs, as they may fear that admitting symptoms or a need for help or time away from work will threaten how they are viewed at work or affect their chances of promotion. Although there are economic costs around menopause for appointments, tests, therapies, and missed time at work, not addressing menopausal health leads to poorer performance, workplace absences, and additional medical costs.22

Conclusion
Menopause occurs naturally as a part of a woman’s life cycle. However, women need assistance navigating perimenopausal hormonal fluctuations and decisions about HT once in menopause. Increased awareness and education about perimenopause and menopause will allow compassionate, individualized, informed care, including lifestyle changes, behavioral or complementary strategies, or medical therapies, hormonal or nonhormonal.27 As a medical society, we need to challenge the stigma associated with aging and menopause and educate ourselves and our patients to help women navigate this challenging time. ●
Myth 1: All hot flashes are the same
The truth: Seventy-five percent of women will have hot flashes, but only 25% are severe enough to cause women to seek treatment. Duration varies with identified patterns, including starting early or late, being mild or starting early, and going late. Ethnicity affects the duration of hot flashes, with longer durations seen in Black and Hispanic women. About 15% of women have had hot flashes for more than 15 or 20 years.1,2
Myth 2: There is no help for hot flashes
The truth: For some women, lifestyle changes are helpful, such as dressing in layers, turning down the thermostat at night, avoiding hot beverages or alcohol, or using technology (Femtech) for cooling devices. Over-the-counter products that are available, but are not clearly proven to help more than placebo, include soy (which may be estrogenic), black cohosh supplements, and nutritional supplements. Cognitive behavioral therapy, hypnosis, weight loss, or mindfulness may help.3 Nonhormone medications such as low-dose antidepressants or gabapentin have shown benefit. Newer treatments in testing, including neurokinin receptor antagonists, appear to work quickly and as effectively as HT. When initiating HT, healthy women with bothersome hot flashes under age 60 or within 10 years of menopause are the best candidates for HT; many lower doses and oral and non-oral therapies are available.
Myth 3: Compounded bioidentical hormones made by a compounding pharmacy are safer and more effective than FDA-approved ones
The truth: Compounded bioidentical hormones are touted as safer or more effective, but there is no good evidence to back up those claims. Whether US Food and Drug Administration (FDA)-approved or compounded, hormones come from the same precursors and have potential risks. With custom compounded HT, there is additional concern about precisely what is in the compounded product, whether levels are similar batch to batch, and the degree of absorption of progesterone, which is better absorbed oral.4-6 FDA-approved bioidentical HTs have been tested for safety, proven to contain consistent, effective levels of hormones, and are monitored by the FDA. For menopausal symptoms, FDA-approved therapies are available as estradiol (oral, patch, spray, gel, lotion, and vaginal ring) and progesterone (as an oral compound or combined with estradiol). Pellets made of compounded hormones have shown higher serum levels and more adverse events.5,7
Myth 4: Menopause causes weight gain
The truth is that fluctuating and declining hormones and the slowing of metabolism affect weight. Weight gain is not inevitable, just harder to prevent. Many women gain an average of 5 lb (2.27 kg) at midlife, which is mainly related to aging and lifestyle and not to menopause or HT. However, menopause may be related to body composition and fat distribution changes. Counsel women to decrease portion sizes, limit carbs, and increase exercise intensity, including strength training. The goal is 30 minutes of moderate aerobic activity per day, all at once or through smaller time increments, to improve their energy, mood, and sleep.
References
1. The NAMS 2017 HT Position Statement Advisory Panel. The 2017 HT position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
2. Pinkerton JV. HT for postmenopausal women. N Engl J Med. 2020;382:446-455.
3. Paramsothy P. Duration of the menopausal transition is longer in women with young age at onset: the multiethnic Study of Women’s Health Across the Nation. Menopause. 2017;24:142-149.
4. Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
5. Eisenlohr-Moul TA, Kaiser G, Weise C, et al. Are there temporal subtypes of premenstrual dysphoric disorder? Using group-based trajectory modeling to identify individual differences in symptom change. Psychol Med. 2020;50:964-972.
6. Seibel M, Seibel S. Working through Menopause: The Impact on Women, Businesses and the Bottom Line. Bookbaby. March 8, 2022.
7. Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
- Paramsothy P. Duration of the menopausal transition is longer in women with young age at onset: the multiethnic Study of Women’s Health Across the Nation. Menopause. 2017;24:142–149.
- Harlow SD, Gass M, Hall JE, et al. STRAW 10 Collaborative Group. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. Menopause. 2012;19:387-95.
- Meers JM, Nowakowski S. Sleep, premenstrual mood disorder, and women’s health. Curr Opin Psychol. 2020;34:43-49.
- Sander B, Gordon JL. Premenstrual mood symptoms in the perimenopause. Curr Psychiatry Rep. 2021;23:73.
- Maki PM, Kornstein SG, Joffe H, et al. Guidelines for the evaluation and treatment of perimenopausal depression: summary and recommendations. J Women’s Health. 2019;28:117–134.
- Cao S, Jones M, Tooth L, et al. History of premenstrual syndrome and development of postpartum depression: a systematic review and meta-analysis. J Psychiatr Res. 2020;121:82–90.
- Rapkin AJ, Korotkaya Y, Taylor KC. Contraception counseling for women with premenstrual dysphoric disorder (PMDD): current perspectives. Open Access J Contracept. 2019;10:27–39.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women's Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med. 2015;175:531.
- Tepper PG, Brooks MM, Randolph JF Jr, et al. Characterizing the trajectories of vasomotor symptoms across the menopausal transition. Menopause. 2016;23:1067-1074.
- Kravitz HM, Ganz PA, Bromberger J, et al. Sleep difficulty in women at midlife: a community survey of sleep and the menopausal transition. Menopause. 2003;10:19-28.
- Dennerstein L, Dudley EC, Hopper JL, et al. A prospective population-based study of menopausal symptoms. Obstet Gynecol. 2000;96:351-358.
- Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal HT and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA. 2013;310:1353-1368.
- The NAMS 2017 HT Position Statement Advisory Panel. T he 2017 HT position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
- Pinkerton JV. HT for postmenopausal women. N Engl J Med. 2020;382:446-455.
- Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:39754011.
- Manson JE, Kaunitz AM. Menopause management—getting clinical care back on track. N Engl J Med. 2016;374:803–806.
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 141: Management of menopausal symptoms. Obstet Gynecol. 2014;123:202-216.
- Shuster LT, Rhodes DJ, Gostout BS, et al. Premature menopause or early menopause: long-term health consequences. Maturitas. 2010;65:161-166.
- Zeleke BM, Davis SR, Fradkin P, et al. Vasomotor symptoms and urogenital atrophy in older women: a systematic review. Climacteric. 2015;18:112-120.
- Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
- Eisenlohr-Moul TA, Kaiser G, Weise C, et al. Are there temporal subtypes of pre- menstrual dysphoric disorder? Using group-based trajectory modeling to identify individual differences in symptom change. Psychol Med. 2020;50: 964-972.
- Seibel M, Seibel S. Working through Menopause: The Impact on Women, Businesses and the Bottom Line. Bookbaby. March 8, 2022.
- Jackson LM, Parker RM, Mattison DR, eds. The Clinical Utility of Compounded Bioidentical HT: A Review of Safety, Effectiveness, and Use. Washington, DC: National Academies Press; 2020.
- Pinkerton JV. Concerns about safety and efficacy of compounded bioidentical HT. Menopause. 2021;28:847-849.
- Liu JH, Pinkerton JV. Prescription therapies. In: CJ Crandall, ed. Menopause Practice: A Clinician’s Guide, 6th ed. Pepper Pike, OH: The North American Menopause Society; 2019: 277-309.
- Jiang X, Bossert A, Parthasarathy KN, et al. Safety assessment of compounded non-FDA-approved hormonal therapy versus FDA-approved hormonal therapy in treating postmenopausal women. Menopause. 2021;28:867-874.
- Aninye IO, Laitner MH, Chinnappan S; Society for Women’s Health Research Menopause Working Group. Menopause preparedness: perspectives for patient, provider, and policymaker consideration. Menopause. 2021;28:1186-1191.

Dr. Pinkerton is Division Director, Midlife Health Center, and Professor, Department of Obstetrics and Gynecology, University of Virginia Health, Charlottesville, Virginia.
The author reports participating in a multicenter clinical trial on nonhormone therapy for hot flashes, for which the University of Virgina received fees from Bayer.
Dr. Pinkerton is Division Director, Midlife Health Center, and Professor, Department of Obstetrics and Gynecology, University of Virginia Health, Charlottesville, Virginia.
The author reports participating in a multicenter clinical trial on nonhormone therapy for hot flashes, for which the University of Virgina received fees from Bayer.
Dr. Pinkerton is Division Director, Midlife Health Center, and Professor, Department of Obstetrics and Gynecology, University of Virginia Health, Charlottesville, Virginia.
The author reports participating in a multicenter clinical trial on nonhormone therapy for hot flashes, for which the University of Virgina received fees from Bayer.
The transition to menopause begins with ovarian fluctuation and hormonal changes, often beginning before significant changes in menstruation. Reproductive aging with loss of follicular activity progresses over a wide age range (42 to 58 years) with an average onset at approximately age 47, ranging from 4 to 8 years. Although most women have heard about menopause, defined as 12 months after the last period, they often lack understanding about perimenopause or that the menopausal transition usually begins 5 years before menopause.1
Perimenopause, defined as early and late menopause transition stages, may be viewed as a window of potential vulnerability for women who develop or have worsening menstrual-related mood disorders. Over time, hormonal fluctuations often lead to menstrual cycle irregularity (either shorter or longer). Changes occurring during perimenopause may be confusing as it may not be clear whether symptoms are related to menopause, aging, or stress. Often not recognized or treated adequately, perimenopausal symptoms may be challenging to navigate for both women and clinicians.
The perimenopausal process is often even more confusing for women with early menopause—whether due to bilateral oophorectomy, chemotherapy or radiation therapy, genetics, or an autoimmune process—because of lack of recognition that an early menopausal transition is occurring or what solutions are available for symptoms. While there is support in the workplace for women during pregnancy and breastfeeding, there remains little support or recognition for the oft challenging perimenopausal transition leading to menopause.
Perimenopause: Common symptoms and treatments
Symptoms may be related to either estrogen level deficiency or excess during perimenopause, and these level changes may even occur within the same cycle.
Cyclic breast tenderness may develop, worsened by caffeine or high salt intake (which can be potentially improved, although without clinical trial evidence, with decreased caffeine or a trial of evening primrose oil or vitamin E).
Changes in menstrual flow and frequency of menses are typical. Flow may be lighter or heavier, longer or shorter, and there may be cycle variability, missed menses, or midcycle spotting.2 Bleeding may be heavy, with or without cramping. In addition to imaging with vaginal ultrasonography or hysteroscopy to identify structural issues, symptoms may be managed with nonsteroidal anti-inflammatory drugs (NSAIDs), hormonal therapy (HT) with short hormone-free interval contraceptives, oral progestogens, or progestin intrauterine systems. Newer medical treatments include antifibrinolytic drugs and selective progesterone-receptor modulators. Uterine ablation to decrease or stop bleeding is effective if there are no structural abnormalities, such as fibroids or polyps or the presence of adenomyosis, where glands will regrow into the endometrium after ablation. Endometrial biopsy is indicated for persistent abnormal uterine bleeding or those with risk factors such as chronic anovulation.
Worsening headaches or menstrual migraines may be triggered by hormonal changes, which may respond to NSAIDs; dihydroergotamine; triptans; the combination of aspirin, acetaminophen, and caffeine; or estrogen the week before menses. For women taking oral contraceptives (OCPs), adding estradiol the week before menses, or using the OCP continuously, may decrease headache frequency. These short-term prophylactic strategies during the perimenstrual time are often effective. If not, preventive therapy is available for women with frequent, severe headaches.
Mood complaints and poor sleep are independently associated with menstrual irregularity, and can lead to fatigue or anxiety, worsening premenstrual syndrome, or depressive moods. Sleep is disrupted premenstrually for up to one-third of women, and sleep disruption is particularly prevalent in those with premenstrual mood disorders and worsens during perimenopause.3
Reproductive hormones act on the neurotransmitter systems in the brain involved in mood regulation and emotion. The fluctuating hormones occurring during perimenopause may exacerbate pre-existing menstrual-related mood disorders. A subset of women experience depressive moods due to perimenopausal elevations in ovarian hormones.4 Others may exhibit increased mood sensitivity with the ovarian hormone withdrawal accompanying late menopause transition and early postmenopausal phase.5 There is significant comorbidity between premenstrual mood disorder (PMDD) and postpartum depression.6 During perimenopause and early menopause, clinicians should ask about prior hormonally-related depression (puberty, postpartum) and recognize that current or past premenstrual syndrome may worsen into a more severe premenstrual dysphoric disorder. Evidence-based treatments for PMDD include selective serotonin reuptake inhibitors (SSRIs); either taken continuously or only during the luteal phase; drospirenone-containing oral contraceptives, often with shorter pill-free intervals; GnRH analogues with or without hormone add-back; and cognitive behavioral therapy.7 For women whose perimenopausal moods improve with HT or develop worsened mood sensitivity with ovarian hormone withdrawal, clinicians should recognize that mood may worsen when treatment is ceased.5
Continue to: Menopausal symptoms...
Menopausal symptoms
Vasomotor symptoms (VMS), hot flashes, or night sweats occur in up to 75% of women as they develop more menstrual irregularity and move closer to their final period and menopause.
Hot flashes are transient episodes of flushing with the sensation of warmth (up to intense heat) on the upper body and face or head, often associated with sweating, chills or flushing, an increase in heart rate, and lowered blood pressure. Hot flashes can sometimes be preceded by an intense feeling of dread, followed by rapid heat dissipation. The etiology of hot flashes is still not clear, but the neurokinin receptors are involved. They are related to small fluctuations in core body temperature superimposed on a narrow thermoneutral zone in symptomatic women. Hot flashes are triggered when core body temperature rises above the upper (sweating) threshold. Shivering occurs if the core body temperature falls below the lower threshold. Sleep may be disrupted, with less rapid eye movement (REM) sleep, and associated with throwing covers on and off or changing sheets or nightclothes. On average, hot flashes last 7.2 years,8 and they are more bothersome if night sweats interfere with sleep or disrupt performance during the day.
In the Stages of Reproductive Aging Workshop (STRAW + 10), women reported VMS within 1-3 years after the menopausal transition.8 Four trajectories of hot flashes were identified in the Study of Women’s Health Across the Nation (SWAN) trial,9 including low levels throughout the menopause transition, early onset, late onset, and a group which had frequent hot flashes, starting early and lasting longer. Serum estrogen levels were not predictive of hot flash frequency or severity.
Hot flashes have been associated with low levels of exercise, cigarette smoking, high follicle-stimulating hormone levels and low estradiol levels, increasing body mass index, ethnicity (with hot flashes more common among Black and Hispanic women), low socioeconomic status, prior PMDD, anxiety, perceived stress, and depression.8 Women with a history of premenstrual syndrome, stress, sexual dysfunction, physical inactivity, or hot flashes are more vulnerable to depressive symptoms during perimenopause and early menopause.5
Depression may co-occur or overlap with menopause symptoms. Diagnosis involves menopausal stage, co-occurring psychiatric and menopause symptoms, psychosocial stressors, and a validated screening tool such as PQ9. Treatments for perimenopausal depression, such as antidepressants, psychotherapy, or cognitive behavioral therapy, are recommended first line for perimenopausal depression. Estrogen therapy has not been approved to treat perimenopausal depression but appears to have antidepressant effects in perimenopausal women, particularly those with bothersome vasomotor symptoms.5
Anxiety can worsen during menopause, and may respond to calming apps, meditation, cognitive behavioral therapy, hypnosis, yoga or tai chi, HT, or antianxiety medications.
Weight gain around the abdomen (ie, belly fat) is a common complaint during the menopausal transition, despite women reporting not changing their eating or exercise patterns. Increasing exercise or bursts of higher intensity, decreasing portion sizes or limiting carbohydrates and alcohol may help.
Memory and concentration problems, described as brain fog, tend to be more of an issue in perimenopause and level out after menopause. Counsel midlife women that these changes are not due to dementia but are related to normal aging, hormonal changes, mood, stress, or other life circumstances. Identifying and addressing sleep issues and mood disorders may help mitigate brain fog, as can advising women to avoid excess caffeine, alcohol, nicotine, and eating before bed. Improvements in memory, cognition, and health have been found with the Mediterranean diet, regular exercise, avoiding multitasking, and engaging in mentally stimulating activities.
Sleeping concerns in peri- and postmenopausal women include sleeping less and more frequent insomnia. Women are more likely to use prescription sleeping aids during these times of their lives. The data from SWAN8 show that the menopausal transition is related to self-reported difficulty sleeping, independent of age. Sleep latency interval is increased while REM sleep decreases. Night sweats can trigger awakenings in the first half of the night. The perceived decline in sleep quality also may be attributed to general aging effects, nocturnal urination, sleep-related disorders such as sleep apnea or restless legs, or chronic pain, stress, or depression.10 Suggestions for management include sleep apps, cognitive behavioral therapy, low-dose antidepressant therapy, addressing sleep routines, and HT. Hypnotics should be avoided.
Sexuality issues are common complaints during the menopausal transition. Cross-sectional data reported from a longitudinal, population-based Australian cohort of women aged 45 to 55 years, found a decrease in sexual responsivity, sexual frequency, libido, vaginal dyspareunia, and more partner problems.11 Low libido may be related to relationship issues, dyspareunia with vaginal narrowing, loss of lubrication, levator spasm, stress, anxiety, exhaustion or mood disorder, lowered hormone levels, excess alcohol intake, underlying health concerns, or a side effect of medications for depression or pain. There is no direct correlation between testosterone levels and libido.
When HT at menopause may be helpful
For healthy symptomatic women without contraindications who are younger than age 60, or within 10 years of menopause onset, the benefits of initiating HT most likely outweigh the risks to relieve bothersome hot flashes and night sweats.12-17 For older women, or for those further from menopause, the greater absolute risks of coronary heart disease, stroke, venous thromboembolism, and dementia, in general, outweigh the potential benefits.12-17 Extended durations of HT have less safety and efficacy data and should be considered primarily for those with persistent menopausal symptoms, with periodic re-evaluation.13,14 For bothersome genitourinary syndrome of menopause symptoms that do not respond to vaginal moisturizers or lubricants, low-dose vaginal HTs are encouraged.13-17
Continue to: Early-onset menopause...
Early-onset menopause
According to observational studies,18 early menopause is associated with a higher risk of osteoporosis, coronary heart disease, cognitive changes, vaginal dryness, loss of libido, and mood changes. Studies have shown that women with early menopause who take HT, without contraindications, to the average age of menopause (age 52) decrease the health risks of early menopause (bone loss, heart disease, mood, and cognition changes).13,14,18
Women with early menopause, whether spontaneous or following bilateral oophorectomy or cancer treatment, should be counseled to get adequate calcium (dietary recommended over supplementation) and vitamin D intake, eat a healthy diet, and exercise regularly. Evaluation should include risk for bone loss, heart disease, mood changes, and vaginal changes.
Extended use of HT
Up to 8% of women have hot flashes for 20 years or more after menopause.19 The decision to continue or to stop HT is not always clear for women:
- with persistent hot flashes after a trial period of HT discontinuation
- with bone loss that cannot be treated with bone-specific medications
- who request continuation for quality of life.
Extended use of HT should include an ongoing assessment of its risks and benefits, periodic trials off of HT, and documentation of rationale and informed discussions about continuing. Lower doses and transdermal therapies appear safer, as does micronized progesterone instead of more potent synthetic progestins.13-17
Genitourinary syndrome of menopause
Once women are further into menopause, they may notice vaginal dryness, vulvar itching or burning, bothersome vaginal discharge, or urinary urgency or frequency. The development of painful intercourse frequently occurs, a combination of the loss of estrogen with thinning of the vaginal mucosa, a loss of the acidic vaginal milieu with less elasticity, and spasm of the levator muscles. Some women develop urinary tract infections after intercourse or have more frequent reoccurrences. First-line therapy is often vaginal moisturizers and lubricants. Vaginal therapies (estradiol, conjugated estrogen, or dehydroepiandrosterone) or oral selective estrogen-receptor modulators (SERMs; ospemifene) improve vaginal dryness and dyspareunia.13,14 Pelvic therapy has also proved valuable for incontinence, pelvic floor dysfunction, and levator spasms.20
Where are there gaps in clinician knowledge?
Studies on emotional health, mood, and sleep need to incorporate measures of menstrual timing into data collection and analyses. Does the sleep disruption occurring premenstrually during perimenopause disproportionately contribute to a woman’s vulnerability to depressive disorders? The risk of clinically significant depressive symptoms increases 1.5- to 2.9-fold in the menopause transition.5 Research into premenstrual dysphoria during the menopause transition may identify different trajectories in the timing of symptoms related to either cycle itself or the ovarian hormone fluctuations or both.21 Gamma-aminobutyric acid (GABA)-modulating drugs, such as sepranolone, which blocks allopregnanolone’s actions at the GABAA receptor, may allow treatment of menstrual-related mood disorders without the need for hormonal interventions.21
Despite extended observational trial data, more data are needed to inform us about the long-term risks and benefits of using menopausal HT, particularly when initiated at menopause and to help address the timing of HT discontinuation. Furthermore, there are many unanswered questions. For instance:
- How much safer are lower dose and transdermal therapies?
- Do untreated hot flashes increase the risk of cardiovascular disease or dementia?
- Will newer non-HT options, such as the neurokinin receptor antagonists that are in testing but are not yet available, lower cardiovascular or dementia risks?
- What will be the risks and benefits for the newer estrogen in testing (estetrol, or E4), considered a natural estrogen and which appears to have lower thrombotic risks?
- What will be the role of intravaginal energy-based therapies, such as vaginal laser or radiofrequency devices?
- How do we address diverse populations and the effects of menopause on race, gender, culture, prior trauma, and socioeconomic status?
Lack of recognition of menopausal symptoms, particularly in the workplace
Clinicians need to understand the varied physical and emotional symptoms that may occur with hormonal changes as women traverse perimenopause and early menopause. We need to recognize that the lack of discussion about women’s health during this time may make women feel ashamed and fearful of bringing up their symptoms due to fear of being dismissed or stigmatized.22 Women may not seek help until a crisis at home or work occurs, as they may fear that admitting symptoms or a need for help or time away from work will threaten how they are viewed at work or affect their chances of promotion. Although there are economic costs around menopause for appointments, tests, therapies, and missed time at work, not addressing menopausal health leads to poorer performance, workplace absences, and additional medical costs.22
Conclusion
Menopause occurs naturally as a part of a woman’s life cycle. However, women need assistance navigating perimenopausal hormonal fluctuations and decisions about HT once in menopause. Increased awareness and education about perimenopause and menopause will allow compassionate, individualized, informed care, including lifestyle changes, behavioral or complementary strategies, or medical therapies, hormonal or nonhormonal.27 As a medical society, we need to challenge the stigma associated with aging and menopause and educate ourselves and our patients to help women navigate this challenging time. ●
Myth 1: All hot flashes are the same
The truth: Seventy-five percent of women will have hot flashes, but only 25% are severe enough to cause women to seek treatment. Duration varies with identified patterns, including starting early or late, being mild or starting early, and going late. Ethnicity affects the duration of hot flashes, with longer durations seen in Black and Hispanic women. About 15% of women have had hot flashes for more than 15 or 20 years.1,2
Myth 2: There is no help for hot flashes
The truth: For some women, lifestyle changes are helpful, such as dressing in layers, turning down the thermostat at night, avoiding hot beverages or alcohol, or using technology (Femtech) for cooling devices. Over-the-counter products that are available, but are not clearly proven to help more than placebo, include soy (which may be estrogenic), black cohosh supplements, and nutritional supplements. Cognitive behavioral therapy, hypnosis, weight loss, or mindfulness may help.3 Nonhormone medications such as low-dose antidepressants or gabapentin have shown benefit. Newer treatments in testing, including neurokinin receptor antagonists, appear to work quickly and as effectively as HT. When initiating HT, healthy women with bothersome hot flashes under age 60 or within 10 years of menopause are the best candidates for HT; many lower doses and oral and non-oral therapies are available.
Myth 3: Compounded bioidentical hormones made by a compounding pharmacy are safer and more effective than FDA-approved ones
The truth: Compounded bioidentical hormones are touted as safer or more effective, but there is no good evidence to back up those claims. Whether US Food and Drug Administration (FDA)-approved or compounded, hormones come from the same precursors and have potential risks. With custom compounded HT, there is additional concern about precisely what is in the compounded product, whether levels are similar batch to batch, and the degree of absorption of progesterone, which is better absorbed oral.4-6 FDA-approved bioidentical HTs have been tested for safety, proven to contain consistent, effective levels of hormones, and are monitored by the FDA. For menopausal symptoms, FDA-approved therapies are available as estradiol (oral, patch, spray, gel, lotion, and vaginal ring) and progesterone (as an oral compound or combined with estradiol). Pellets made of compounded hormones have shown higher serum levels and more adverse events.5,7
Myth 4: Menopause causes weight gain
The truth is that fluctuating and declining hormones and the slowing of metabolism affect weight. Weight gain is not inevitable, just harder to prevent. Many women gain an average of 5 lb (2.27 kg) at midlife, which is mainly related to aging and lifestyle and not to menopause or HT. However, menopause may be related to body composition and fat distribution changes. Counsel women to decrease portion sizes, limit carbs, and increase exercise intensity, including strength training. The goal is 30 minutes of moderate aerobic activity per day, all at once or through smaller time increments, to improve their energy, mood, and sleep.
References
1. The NAMS 2017 HT Position Statement Advisory Panel. The 2017 HT position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
2. Pinkerton JV. HT for postmenopausal women. N Engl J Med. 2020;382:446-455.
3. Paramsothy P. Duration of the menopausal transition is longer in women with young age at onset: the multiethnic Study of Women’s Health Across the Nation. Menopause. 2017;24:142-149.
4. Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
5. Eisenlohr-Moul TA, Kaiser G, Weise C, et al. Are there temporal subtypes of premenstrual dysphoric disorder? Using group-based trajectory modeling to identify individual differences in symptom change. Psychol Med. 2020;50:964-972.
6. Seibel M, Seibel S. Working through Menopause: The Impact on Women, Businesses and the Bottom Line. Bookbaby. March 8, 2022.
7. Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
The transition to menopause begins with ovarian fluctuation and hormonal changes, often beginning before significant changes in menstruation. Reproductive aging with loss of follicular activity progresses over a wide age range (42 to 58 years) with an average onset at approximately age 47, ranging from 4 to 8 years. Although most women have heard about menopause, defined as 12 months after the last period, they often lack understanding about perimenopause or that the menopausal transition usually begins 5 years before menopause.1
Perimenopause, defined as early and late menopause transition stages, may be viewed as a window of potential vulnerability for women who develop or have worsening menstrual-related mood disorders. Over time, hormonal fluctuations often lead to menstrual cycle irregularity (either shorter or longer). Changes occurring during perimenopause may be confusing as it may not be clear whether symptoms are related to menopause, aging, or stress. Often not recognized or treated adequately, perimenopausal symptoms may be challenging to navigate for both women and clinicians.
The perimenopausal process is often even more confusing for women with early menopause—whether due to bilateral oophorectomy, chemotherapy or radiation therapy, genetics, or an autoimmune process—because of lack of recognition that an early menopausal transition is occurring or what solutions are available for symptoms. While there is support in the workplace for women during pregnancy and breastfeeding, there remains little support or recognition for the oft challenging perimenopausal transition leading to menopause.
Perimenopause: Common symptoms and treatments
Symptoms may be related to either estrogen level deficiency or excess during perimenopause, and these level changes may even occur within the same cycle.
Cyclic breast tenderness may develop, worsened by caffeine or high salt intake (which can be potentially improved, although without clinical trial evidence, with decreased caffeine or a trial of evening primrose oil or vitamin E).
Changes in menstrual flow and frequency of menses are typical. Flow may be lighter or heavier, longer or shorter, and there may be cycle variability, missed menses, or midcycle spotting.2 Bleeding may be heavy, with or without cramping. In addition to imaging with vaginal ultrasonography or hysteroscopy to identify structural issues, symptoms may be managed with nonsteroidal anti-inflammatory drugs (NSAIDs), hormonal therapy (HT) with short hormone-free interval contraceptives, oral progestogens, or progestin intrauterine systems. Newer medical treatments include antifibrinolytic drugs and selective progesterone-receptor modulators. Uterine ablation to decrease or stop bleeding is effective if there are no structural abnormalities, such as fibroids or polyps or the presence of adenomyosis, where glands will regrow into the endometrium after ablation. Endometrial biopsy is indicated for persistent abnormal uterine bleeding or those with risk factors such as chronic anovulation.
Worsening headaches or menstrual migraines may be triggered by hormonal changes, which may respond to NSAIDs; dihydroergotamine; triptans; the combination of aspirin, acetaminophen, and caffeine; or estrogen the week before menses. For women taking oral contraceptives (OCPs), adding estradiol the week before menses, or using the OCP continuously, may decrease headache frequency. These short-term prophylactic strategies during the perimenstrual time are often effective. If not, preventive therapy is available for women with frequent, severe headaches.
Mood complaints and poor sleep are independently associated with menstrual irregularity, and can lead to fatigue or anxiety, worsening premenstrual syndrome, or depressive moods. Sleep is disrupted premenstrually for up to one-third of women, and sleep disruption is particularly prevalent in those with premenstrual mood disorders and worsens during perimenopause.3
Reproductive hormones act on the neurotransmitter systems in the brain involved in mood regulation and emotion. The fluctuating hormones occurring during perimenopause may exacerbate pre-existing menstrual-related mood disorders. A subset of women experience depressive moods due to perimenopausal elevations in ovarian hormones.4 Others may exhibit increased mood sensitivity with the ovarian hormone withdrawal accompanying late menopause transition and early postmenopausal phase.5 There is significant comorbidity between premenstrual mood disorder (PMDD) and postpartum depression.6 During perimenopause and early menopause, clinicians should ask about prior hormonally-related depression (puberty, postpartum) and recognize that current or past premenstrual syndrome may worsen into a more severe premenstrual dysphoric disorder. Evidence-based treatments for PMDD include selective serotonin reuptake inhibitors (SSRIs); either taken continuously or only during the luteal phase; drospirenone-containing oral contraceptives, often with shorter pill-free intervals; GnRH analogues with or without hormone add-back; and cognitive behavioral therapy.7 For women whose perimenopausal moods improve with HT or develop worsened mood sensitivity with ovarian hormone withdrawal, clinicians should recognize that mood may worsen when treatment is ceased.5
Continue to: Menopausal symptoms...
Menopausal symptoms
Vasomotor symptoms (VMS), hot flashes, or night sweats occur in up to 75% of women as they develop more menstrual irregularity and move closer to their final period and menopause.
Hot flashes are transient episodes of flushing with the sensation of warmth (up to intense heat) on the upper body and face or head, often associated with sweating, chills or flushing, an increase in heart rate, and lowered blood pressure. Hot flashes can sometimes be preceded by an intense feeling of dread, followed by rapid heat dissipation. The etiology of hot flashes is still not clear, but the neurokinin receptors are involved. They are related to small fluctuations in core body temperature superimposed on a narrow thermoneutral zone in symptomatic women. Hot flashes are triggered when core body temperature rises above the upper (sweating) threshold. Shivering occurs if the core body temperature falls below the lower threshold. Sleep may be disrupted, with less rapid eye movement (REM) sleep, and associated with throwing covers on and off or changing sheets or nightclothes. On average, hot flashes last 7.2 years,8 and they are more bothersome if night sweats interfere with sleep or disrupt performance during the day.
In the Stages of Reproductive Aging Workshop (STRAW + 10), women reported VMS within 1-3 years after the menopausal transition.8 Four trajectories of hot flashes were identified in the Study of Women’s Health Across the Nation (SWAN) trial,9 including low levels throughout the menopause transition, early onset, late onset, and a group which had frequent hot flashes, starting early and lasting longer. Serum estrogen levels were not predictive of hot flash frequency or severity.
Hot flashes have been associated with low levels of exercise, cigarette smoking, high follicle-stimulating hormone levels and low estradiol levels, increasing body mass index, ethnicity (with hot flashes more common among Black and Hispanic women), low socioeconomic status, prior PMDD, anxiety, perceived stress, and depression.8 Women with a history of premenstrual syndrome, stress, sexual dysfunction, physical inactivity, or hot flashes are more vulnerable to depressive symptoms during perimenopause and early menopause.5
Depression may co-occur or overlap with menopause symptoms. Diagnosis involves menopausal stage, co-occurring psychiatric and menopause symptoms, psychosocial stressors, and a validated screening tool such as PQ9. Treatments for perimenopausal depression, such as antidepressants, psychotherapy, or cognitive behavioral therapy, are recommended first line for perimenopausal depression. Estrogen therapy has not been approved to treat perimenopausal depression but appears to have antidepressant effects in perimenopausal women, particularly those with bothersome vasomotor symptoms.5
Anxiety can worsen during menopause, and may respond to calming apps, meditation, cognitive behavioral therapy, hypnosis, yoga or tai chi, HT, or antianxiety medications.
Weight gain around the abdomen (ie, belly fat) is a common complaint during the menopausal transition, despite women reporting not changing their eating or exercise patterns. Increasing exercise or bursts of higher intensity, decreasing portion sizes or limiting carbohydrates and alcohol may help.
Memory and concentration problems, described as brain fog, tend to be more of an issue in perimenopause and level out after menopause. Counsel midlife women that these changes are not due to dementia but are related to normal aging, hormonal changes, mood, stress, or other life circumstances. Identifying and addressing sleep issues and mood disorders may help mitigate brain fog, as can advising women to avoid excess caffeine, alcohol, nicotine, and eating before bed. Improvements in memory, cognition, and health have been found with the Mediterranean diet, regular exercise, avoiding multitasking, and engaging in mentally stimulating activities.
Sleeping concerns in peri- and postmenopausal women include sleeping less and more frequent insomnia. Women are more likely to use prescription sleeping aids during these times of their lives. The data from SWAN8 show that the menopausal transition is related to self-reported difficulty sleeping, independent of age. Sleep latency interval is increased while REM sleep decreases. Night sweats can trigger awakenings in the first half of the night. The perceived decline in sleep quality also may be attributed to general aging effects, nocturnal urination, sleep-related disorders such as sleep apnea or restless legs, or chronic pain, stress, or depression.10 Suggestions for management include sleep apps, cognitive behavioral therapy, low-dose antidepressant therapy, addressing sleep routines, and HT. Hypnotics should be avoided.
Sexuality issues are common complaints during the menopausal transition. Cross-sectional data reported from a longitudinal, population-based Australian cohort of women aged 45 to 55 years, found a decrease in sexual responsivity, sexual frequency, libido, vaginal dyspareunia, and more partner problems.11 Low libido may be related to relationship issues, dyspareunia with vaginal narrowing, loss of lubrication, levator spasm, stress, anxiety, exhaustion or mood disorder, lowered hormone levels, excess alcohol intake, underlying health concerns, or a side effect of medications for depression or pain. There is no direct correlation between testosterone levels and libido.
When HT at menopause may be helpful
For healthy symptomatic women without contraindications who are younger than age 60, or within 10 years of menopause onset, the benefits of initiating HT most likely outweigh the risks to relieve bothersome hot flashes and night sweats.12-17 For older women, or for those further from menopause, the greater absolute risks of coronary heart disease, stroke, venous thromboembolism, and dementia, in general, outweigh the potential benefits.12-17 Extended durations of HT have less safety and efficacy data and should be considered primarily for those with persistent menopausal symptoms, with periodic re-evaluation.13,14 For bothersome genitourinary syndrome of menopause symptoms that do not respond to vaginal moisturizers or lubricants, low-dose vaginal HTs are encouraged.13-17
Continue to: Early-onset menopause...
Early-onset menopause
According to observational studies,18 early menopause is associated with a higher risk of osteoporosis, coronary heart disease, cognitive changes, vaginal dryness, loss of libido, and mood changes. Studies have shown that women with early menopause who take HT, without contraindications, to the average age of menopause (age 52) decrease the health risks of early menopause (bone loss, heart disease, mood, and cognition changes).13,14,18
Women with early menopause, whether spontaneous or following bilateral oophorectomy or cancer treatment, should be counseled to get adequate calcium (dietary recommended over supplementation) and vitamin D intake, eat a healthy diet, and exercise regularly. Evaluation should include risk for bone loss, heart disease, mood changes, and vaginal changes.
Extended use of HT
Up to 8% of women have hot flashes for 20 years or more after menopause.19 The decision to continue or to stop HT is not always clear for women:
- with persistent hot flashes after a trial period of HT discontinuation
- with bone loss that cannot be treated with bone-specific medications
- who request continuation for quality of life.
Extended use of HT should include an ongoing assessment of its risks and benefits, periodic trials off of HT, and documentation of rationale and informed discussions about continuing. Lower doses and transdermal therapies appear safer, as does micronized progesterone instead of more potent synthetic progestins.13-17
Genitourinary syndrome of menopause
Once women are further into menopause, they may notice vaginal dryness, vulvar itching or burning, bothersome vaginal discharge, or urinary urgency or frequency. The development of painful intercourse frequently occurs, a combination of the loss of estrogen with thinning of the vaginal mucosa, a loss of the acidic vaginal milieu with less elasticity, and spasm of the levator muscles. Some women develop urinary tract infections after intercourse or have more frequent reoccurrences. First-line therapy is often vaginal moisturizers and lubricants. Vaginal therapies (estradiol, conjugated estrogen, or dehydroepiandrosterone) or oral selective estrogen-receptor modulators (SERMs; ospemifene) improve vaginal dryness and dyspareunia.13,14 Pelvic therapy has also proved valuable for incontinence, pelvic floor dysfunction, and levator spasms.20
Where are there gaps in clinician knowledge?
Studies on emotional health, mood, and sleep need to incorporate measures of menstrual timing into data collection and analyses. Does the sleep disruption occurring premenstrually during perimenopause disproportionately contribute to a woman’s vulnerability to depressive disorders? The risk of clinically significant depressive symptoms increases 1.5- to 2.9-fold in the menopause transition.5 Research into premenstrual dysphoria during the menopause transition may identify different trajectories in the timing of symptoms related to either cycle itself or the ovarian hormone fluctuations or both.21 Gamma-aminobutyric acid (GABA)-modulating drugs, such as sepranolone, which blocks allopregnanolone’s actions at the GABAA receptor, may allow treatment of menstrual-related mood disorders without the need for hormonal interventions.21
Despite extended observational trial data, more data are needed to inform us about the long-term risks and benefits of using menopausal HT, particularly when initiated at menopause and to help address the timing of HT discontinuation. Furthermore, there are many unanswered questions. For instance:
- How much safer are lower dose and transdermal therapies?
- Do untreated hot flashes increase the risk of cardiovascular disease or dementia?
- Will newer non-HT options, such as the neurokinin receptor antagonists that are in testing but are not yet available, lower cardiovascular or dementia risks?
- What will be the risks and benefits for the newer estrogen in testing (estetrol, or E4), considered a natural estrogen and which appears to have lower thrombotic risks?
- What will be the role of intravaginal energy-based therapies, such as vaginal laser or radiofrequency devices?
- How do we address diverse populations and the effects of menopause on race, gender, culture, prior trauma, and socioeconomic status?
Lack of recognition of menopausal symptoms, particularly in the workplace
Clinicians need to understand the varied physical and emotional symptoms that may occur with hormonal changes as women traverse perimenopause and early menopause. We need to recognize that the lack of discussion about women’s health during this time may make women feel ashamed and fearful of bringing up their symptoms due to fear of being dismissed or stigmatized.22 Women may not seek help until a crisis at home or work occurs, as they may fear that admitting symptoms or a need for help or time away from work will threaten how they are viewed at work or affect their chances of promotion. Although there are economic costs around menopause for appointments, tests, therapies, and missed time at work, not addressing menopausal health leads to poorer performance, workplace absences, and additional medical costs.22
Conclusion
Menopause occurs naturally as a part of a woman’s life cycle. However, women need assistance navigating perimenopausal hormonal fluctuations and decisions about HT once in menopause. Increased awareness and education about perimenopause and menopause will allow compassionate, individualized, informed care, including lifestyle changes, behavioral or complementary strategies, or medical therapies, hormonal or nonhormonal.27 As a medical society, we need to challenge the stigma associated with aging and menopause and educate ourselves and our patients to help women navigate this challenging time. ●
Myth 1: All hot flashes are the same
The truth: Seventy-five percent of women will have hot flashes, but only 25% are severe enough to cause women to seek treatment. Duration varies with identified patterns, including starting early or late, being mild or starting early, and going late. Ethnicity affects the duration of hot flashes, with longer durations seen in Black and Hispanic women. About 15% of women have had hot flashes for more than 15 or 20 years.1,2
Myth 2: There is no help for hot flashes
The truth: For some women, lifestyle changes are helpful, such as dressing in layers, turning down the thermostat at night, avoiding hot beverages or alcohol, or using technology (Femtech) for cooling devices. Over-the-counter products that are available, but are not clearly proven to help more than placebo, include soy (which may be estrogenic), black cohosh supplements, and nutritional supplements. Cognitive behavioral therapy, hypnosis, weight loss, or mindfulness may help.3 Nonhormone medications such as low-dose antidepressants or gabapentin have shown benefit. Newer treatments in testing, including neurokinin receptor antagonists, appear to work quickly and as effectively as HT. When initiating HT, healthy women with bothersome hot flashes under age 60 or within 10 years of menopause are the best candidates for HT; many lower doses and oral and non-oral therapies are available.
Myth 3: Compounded bioidentical hormones made by a compounding pharmacy are safer and more effective than FDA-approved ones
The truth: Compounded bioidentical hormones are touted as safer or more effective, but there is no good evidence to back up those claims. Whether US Food and Drug Administration (FDA)-approved or compounded, hormones come from the same precursors and have potential risks. With custom compounded HT, there is additional concern about precisely what is in the compounded product, whether levels are similar batch to batch, and the degree of absorption of progesterone, which is better absorbed oral.4-6 FDA-approved bioidentical HTs have been tested for safety, proven to contain consistent, effective levels of hormones, and are monitored by the FDA. For menopausal symptoms, FDA-approved therapies are available as estradiol (oral, patch, spray, gel, lotion, and vaginal ring) and progesterone (as an oral compound or combined with estradiol). Pellets made of compounded hormones have shown higher serum levels and more adverse events.5,7
Myth 4: Menopause causes weight gain
The truth is that fluctuating and declining hormones and the slowing of metabolism affect weight. Weight gain is not inevitable, just harder to prevent. Many women gain an average of 5 lb (2.27 kg) at midlife, which is mainly related to aging and lifestyle and not to menopause or HT. However, menopause may be related to body composition and fat distribution changes. Counsel women to decrease portion sizes, limit carbs, and increase exercise intensity, including strength training. The goal is 30 minutes of moderate aerobic activity per day, all at once or through smaller time increments, to improve their energy, mood, and sleep.
References
1. The NAMS 2017 HT Position Statement Advisory Panel. The 2017 HT position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
2. Pinkerton JV. HT for postmenopausal women. N Engl J Med. 2020;382:446-455.
3. Paramsothy P. Duration of the menopausal transition is longer in women with young age at onset: the multiethnic Study of Women’s Health Across the Nation. Menopause. 2017;24:142-149.
4. Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
5. Eisenlohr-Moul TA, Kaiser G, Weise C, et al. Are there temporal subtypes of premenstrual dysphoric disorder? Using group-based trajectory modeling to identify individual differences in symptom change. Psychol Med. 2020;50:964-972.
6. Seibel M, Seibel S. Working through Menopause: The Impact on Women, Businesses and the Bottom Line. Bookbaby. March 8, 2022.
7. Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
- Paramsothy P. Duration of the menopausal transition is longer in women with young age at onset: the multiethnic Study of Women’s Health Across the Nation. Menopause. 2017;24:142–149.
- Harlow SD, Gass M, Hall JE, et al. STRAW 10 Collaborative Group. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. Menopause. 2012;19:387-95.
- Meers JM, Nowakowski S. Sleep, premenstrual mood disorder, and women’s health. Curr Opin Psychol. 2020;34:43-49.
- Sander B, Gordon JL. Premenstrual mood symptoms in the perimenopause. Curr Psychiatry Rep. 2021;23:73.
- Maki PM, Kornstein SG, Joffe H, et al. Guidelines for the evaluation and treatment of perimenopausal depression: summary and recommendations. J Women’s Health. 2019;28:117–134.
- Cao S, Jones M, Tooth L, et al. History of premenstrual syndrome and development of postpartum depression: a systematic review and meta-analysis. J Psychiatr Res. 2020;121:82–90.
- Rapkin AJ, Korotkaya Y, Taylor KC. Contraception counseling for women with premenstrual dysphoric disorder (PMDD): current perspectives. Open Access J Contracept. 2019;10:27–39.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women's Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med. 2015;175:531.
- Tepper PG, Brooks MM, Randolph JF Jr, et al. Characterizing the trajectories of vasomotor symptoms across the menopausal transition. Menopause. 2016;23:1067-1074.
- Kravitz HM, Ganz PA, Bromberger J, et al. Sleep difficulty in women at midlife: a community survey of sleep and the menopausal transition. Menopause. 2003;10:19-28.
- Dennerstein L, Dudley EC, Hopper JL, et al. A prospective population-based study of menopausal symptoms. Obstet Gynecol. 2000;96:351-358.
- Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal HT and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA. 2013;310:1353-1368.
- The NAMS 2017 HT Position Statement Advisory Panel. T he 2017 HT position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
- Pinkerton JV. HT for postmenopausal women. N Engl J Med. 2020;382:446-455.
- Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:39754011.
- Manson JE, Kaunitz AM. Menopause management—getting clinical care back on track. N Engl J Med. 2016;374:803–806.
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 141: Management of menopausal symptoms. Obstet Gynecol. 2014;123:202-216.
- Shuster LT, Rhodes DJ, Gostout BS, et al. Premature menopause or early menopause: long-term health consequences. Maturitas. 2010;65:161-166.
- Zeleke BM, Davis SR, Fradkin P, et al. Vasomotor symptoms and urogenital atrophy in older women: a systematic review. Climacteric. 2015;18:112-120.
- Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
- Eisenlohr-Moul TA, Kaiser G, Weise C, et al. Are there temporal subtypes of pre- menstrual dysphoric disorder? Using group-based trajectory modeling to identify individual differences in symptom change. Psychol Med. 2020;50: 964-972.
- Seibel M, Seibel S. Working through Menopause: The Impact on Women, Businesses and the Bottom Line. Bookbaby. March 8, 2022.
- Jackson LM, Parker RM, Mattison DR, eds. The Clinical Utility of Compounded Bioidentical HT: A Review of Safety, Effectiveness, and Use. Washington, DC: National Academies Press; 2020.
- Pinkerton JV. Concerns about safety and efficacy of compounded bioidentical HT. Menopause. 2021;28:847-849.
- Liu JH, Pinkerton JV. Prescription therapies. In: CJ Crandall, ed. Menopause Practice: A Clinician’s Guide, 6th ed. Pepper Pike, OH: The North American Menopause Society; 2019: 277-309.
- Jiang X, Bossert A, Parthasarathy KN, et al. Safety assessment of compounded non-FDA-approved hormonal therapy versus FDA-approved hormonal therapy in treating postmenopausal women. Menopause. 2021;28:867-874.
- Aninye IO, Laitner MH, Chinnappan S; Society for Women’s Health Research Menopause Working Group. Menopause preparedness: perspectives for patient, provider, and policymaker consideration. Menopause. 2021;28:1186-1191.
- Paramsothy P. Duration of the menopausal transition is longer in women with young age at onset: the multiethnic Study of Women’s Health Across the Nation. Menopause. 2017;24:142–149.
- Harlow SD, Gass M, Hall JE, et al. STRAW 10 Collaborative Group. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. Menopause. 2012;19:387-95.
- Meers JM, Nowakowski S. Sleep, premenstrual mood disorder, and women’s health. Curr Opin Psychol. 2020;34:43-49.
- Sander B, Gordon JL. Premenstrual mood symptoms in the perimenopause. Curr Psychiatry Rep. 2021;23:73.
- Maki PM, Kornstein SG, Joffe H, et al. Guidelines for the evaluation and treatment of perimenopausal depression: summary and recommendations. J Women’s Health. 2019;28:117–134.
- Cao S, Jones M, Tooth L, et al. History of premenstrual syndrome and development of postpartum depression: a systematic review and meta-analysis. J Psychiatr Res. 2020;121:82–90.
- Rapkin AJ, Korotkaya Y, Taylor KC. Contraception counseling for women with premenstrual dysphoric disorder (PMDD): current perspectives. Open Access J Contracept. 2019;10:27–39.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women's Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med. 2015;175:531.
- Tepper PG, Brooks MM, Randolph JF Jr, et al. Characterizing the trajectories of vasomotor symptoms across the menopausal transition. Menopause. 2016;23:1067-1074.
- Kravitz HM, Ganz PA, Bromberger J, et al. Sleep difficulty in women at midlife: a community survey of sleep and the menopausal transition. Menopause. 2003;10:19-28.
- Dennerstein L, Dudley EC, Hopper JL, et al. A prospective population-based study of menopausal symptoms. Obstet Gynecol. 2000;96:351-358.
- Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal HT and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA. 2013;310:1353-1368.
- The NAMS 2017 HT Position Statement Advisory Panel. T he 2017 HT position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
- Pinkerton JV. HT for postmenopausal women. N Engl J Med. 2020;382:446-455.
- Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:39754011.
- Manson JE, Kaunitz AM. Menopause management—getting clinical care back on track. N Engl J Med. 2016;374:803–806.
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 141: Management of menopausal symptoms. Obstet Gynecol. 2014;123:202-216.
- Shuster LT, Rhodes DJ, Gostout BS, et al. Premature menopause or early menopause: long-term health consequences. Maturitas. 2010;65:161-166.
- Zeleke BM, Davis SR, Fradkin P, et al. Vasomotor symptoms and urogenital atrophy in older women: a systematic review. Climacteric. 2015;18:112-120.
- Kingsberg SA, Schaffir J, Faught BM, et al. Female sexual health: barriers to optimal outcomes and a roadmap for improved patient-clinician communications. J Womens Health (Larchmt). 2019;28:432-443.
- Eisenlohr-Moul TA, Kaiser G, Weise C, et al. Are there temporal subtypes of pre- menstrual dysphoric disorder? Using group-based trajectory modeling to identify individual differences in symptom change. Psychol Med. 2020;50: 964-972.
- Seibel M, Seibel S. Working through Menopause: The Impact on Women, Businesses and the Bottom Line. Bookbaby. March 8, 2022.
- Jackson LM, Parker RM, Mattison DR, eds. The Clinical Utility of Compounded Bioidentical HT: A Review of Safety, Effectiveness, and Use. Washington, DC: National Academies Press; 2020.
- Pinkerton JV. Concerns about safety and efficacy of compounded bioidentical HT. Menopause. 2021;28:847-849.
- Liu JH, Pinkerton JV. Prescription therapies. In: CJ Crandall, ed. Menopause Practice: A Clinician’s Guide, 6th ed. Pepper Pike, OH: The North American Menopause Society; 2019: 277-309.
- Jiang X, Bossert A, Parthasarathy KN, et al. Safety assessment of compounded non-FDA-approved hormonal therapy versus FDA-approved hormonal therapy in treating postmenopausal women. Menopause. 2021;28:867-874.
- Aninye IO, Laitner MH, Chinnappan S; Society for Women’s Health Research Menopause Working Group. Menopause preparedness: perspectives for patient, provider, and policymaker consideration. Menopause. 2021;28:1186-1191.
