User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
What’s Eating You? Ixodes Tick and Related Diseases, Part 3: Coinfection and Tick-Bite Prevention
Tick-borne diseases are increasing in prevalence, likely due to climate change in combination with human movement into tick habitats.1-3 The Ixodes genus of hard ticks is a common vector for the transmission of pathogenic viruses, bacteria, parasites, and toxins. Among these, Lyme disease, which is caused by Borrelia burgdorferi, is the most prevalent, followed by babesiosis and human granulocytic anaplasmosis (HGA), respectively.4 In Europe, tick-borne encephalitis is commonly encountered. More recently identified diseases transmitted by Ixodes ticks include Powassan virus and Borrelia miyamotoi infection; however, these diseases are less frequently encountered than other tick-borne diseases.5,6
As tick-borne diseases become more prevalent, the likelihood of coinfection with more than one Ixodes-transmitted pathogen is increasing.7 Therefore, it is important for physicians who practice in endemic areas to be aware of the possibility of coinfection, which can alter clinical presentation, disease severity, and treatment response in tick-borne diseases. Additionally, public education on tick-bite prevention and prompt tick removal is necessary to combat the rising prevalence of these diseases.
Coinfection
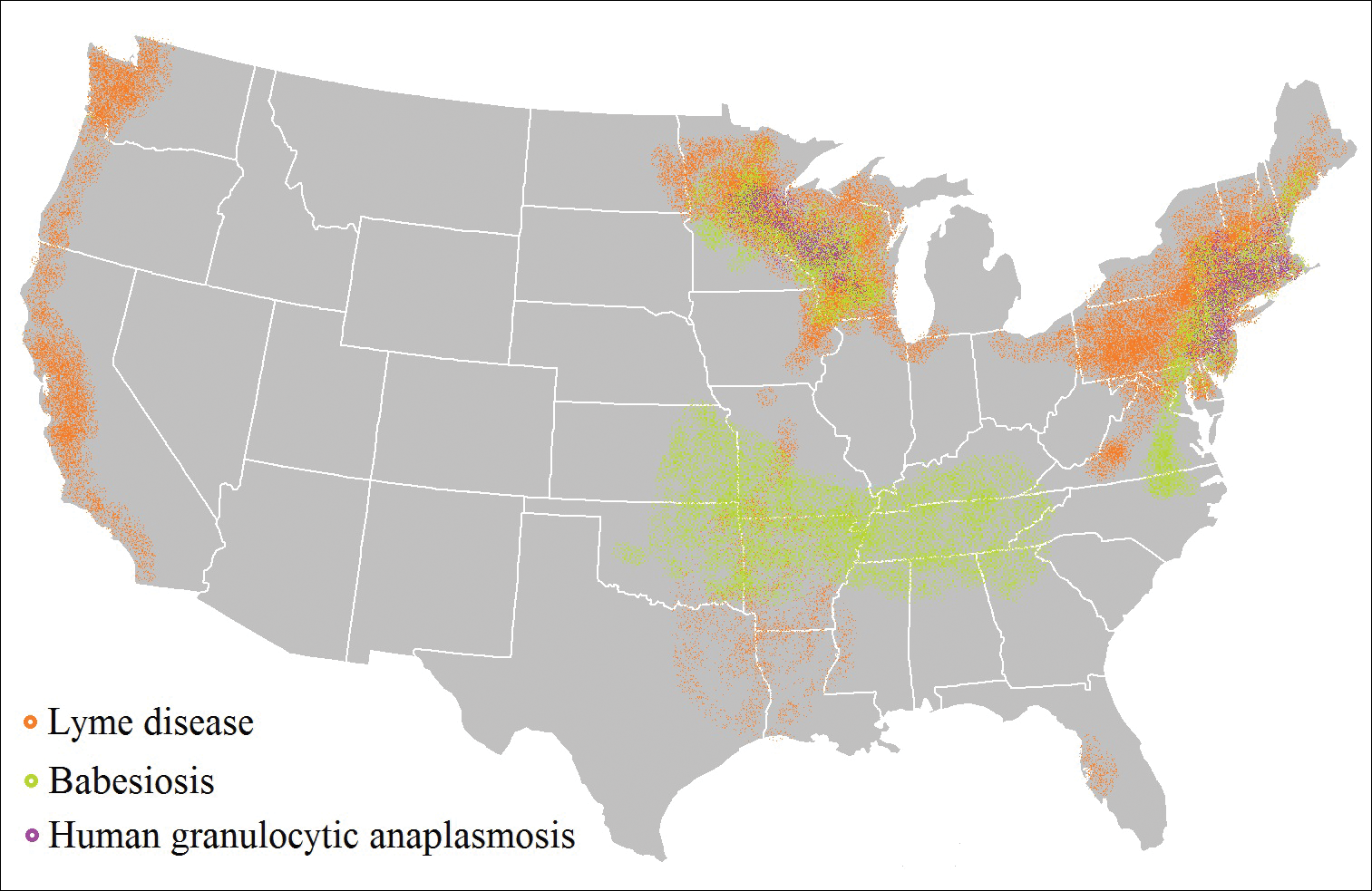
Risk of coinfection with more than one tick-borne disease is contingent on the geographic distribution of the tick species as well as the particular pathogen’s prevalence within reservoir hosts in a given area (Figure). Most coinfections occur with B. burgdorferi and an additional pathogen, usually Anaplasma phagocytophilum (which causes human granulocytic anaplasmosis [HGA]) or Babesia microti (which causes babesiosis). In Europe, coinfection with tick-borne encephalitis virus may occur. There is limited evidence of human coinfection with B miyamotoi or Powassan virus, as isolated infection with either of these pathogens is rare.
In patients with Lyme disease, as many as 35% may have concurrent babesiosis, and as many as 12% may have concurrent HGA in endemic areas (eg, northeast and northern central United States).7-9 Concurrent HGA and babesiosis in the absence of Lyme disease also has been documented.7-9 Coinfection generally increases the diversity of presenting symptoms, often obscuring the primary diagnosis. In addition, these patients may have more severe and prolonged illness.8,10,11
In endemic areas, coinfection with B burgdorferi and an additional pathogen should be suspected if a patient presents with typical symptoms of early Lyme disease, especially erythema migrans, along with (1) combination of fever, chills, and headache; (2) prolonged viral-like illness, particularly 48 hours after appropriate antibiotic treatment; and (3) unexplained blood dyscrasia.7,11,12 When a patient presents with erythema migrans, it is unnecessary to test for HGA, as treatment of Lyme disease with doxycycline also is adequate for treating HGA; however, if systemic symptoms persist despite treatment, testing for babesiosis and other tick-borne illnesses should be considered, as babesiosis requires treatment with atovaquone plus azithromycin or clindamycin plus quinine.13
A complete blood count and peripheral blood smear can aid in the diagnosis of coinfection. The complete blood count may reveal leukopenia, anemia, or thrombocytopenia associated with HGA or babesiosis. The peripheral blood smear can reveal inclusions of intra-erythrocytic ring forms and tetrads (the “Maltese cross” appearance) in babesiosis and intragranulocytic morulae in HGA.12 The most sensitive diagnostic tests for tick-borne diseases are organism-specific IgM and IgG serology for Lyme disease, babesiosis, and HGA and polymerase chain reaction for babesiosis and HGA.7
Prevention Strategies
The most effective means of controlling tick-borne disease is avoiding tick bites altogether. One method is to avoid spending time in high-risk areas that may be infested with ticks, particularly low-lying brush, where ticks are likely to hide.14 For individuals traveling in environments with a high risk of tick exposure, behavioral methods of avoidance are indicated, including wearing long pants and a shirt with long sleeves, tucking the shirt into the pants, and wearing closed-toe shoes. Wearing light-colored clothing may aid in tick identification and prompt removal prior to attachment. Permethrin-impregnated clothing has been proven to decrease the likelihood of tick bites in adults working outdoors.15-17
Topical repellents also play a role in the prevention of tick-borne diseases. The most effective and safe synthetic repellents are N,N-diethyl-meta-toluamide (DEET); picaridin; p-menthane-3,8-diol; and insect repellent 3535 (IR3535)(ethyl butylacetylaminopropionate).16-19 Plant-based repellents also are available, but their efficacy is strongly influenced by the surrounding environment (eg, temperature, humidity, organic matter).20-22 Individuals also may be exposed to ticks following contact with domesticated animals and pets.23,24 Tick prevention in pets with the use of ectoparasiticides should be directed by a qualified veterinarian.25
Tick Removal
Following a bite, the tick should be removed promptly to avoid transmission of pathogens. Numerous commercial and in-home methods of tick removal are available, but not all are equally effective. Detachment techniques include removal with a card or commercially available radiofrequency device, lassoing, or freezing.26,27 However, the most effective method is simple removal with tweezers. The tick should be grasped close to the skin surface and pulled upward with an even pressure. Commercially available tick-removal devices have not been shown to produce better outcomes than removal of the tick with tweezers.28
Conclusion
When patients do not respond to therapy for presumed tick-borne infection, the diagnosis should be reconsidered. One important consideration is coinfection with a second organism. Prompt identification and removal of ticks can prevent disease transmission.
- McMichael C, Barnett J, McMichael AJ. An ill wind? climate change, migration, and health. Environ Health Perspect. 2012;120:646-654.
- Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140051.
- Ogden NH, Bigras-Poulin M, O’Callaghan CJ, et al. Vector seasonality, host infection dynamics and fitness of pathogens transmitted by the tick Ixodes scapularis. Parasitology. 2007;134(pt 2):209-227.
- Tickborne diseases of the United States. Centers for Disease Control and Prevention website. http://www.cdc.gov/ticks/diseases/index.html. Updated July 25, 2017. Accessed April 10, 2018.
- Hinten SR, Beckett GA, Gensheimer KF, et al. Increased recognition of Powassan encephalitis in the United States, 1999-2005. Vector Borne Zoonotic Dis. 2008;8:733-740.
- Platonov AE, Karan LS, Kolyasnikova NM, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816-1823.
- Krause PJ, McKay K, Thompson CA, et al; Deer-Associated Infection Study Group. Disease-specific diagnosis of coinfecting tickborne zoonoses: babesiosis, human granulocytic ehrlichiosis, and Lyme disease. Clin Infect Dis. 2002;34:1184-1191.
- Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent Lyme disease and babesiosis. evidence for increased severity and duration of illness. JAMA. 1996;275:1657-1660.
- Belongia EA, Reed KD, Mitchell PD, et al. Clinical and epidemiological features of early Lyme disease and human granulocytic ehrlichiosis in Wisconsin. Clin Infect Dis. 1999;29:1472-1477.
- Sweeny CJ, Ghassemi M, Agger WA, et al. Coinfection with Babesia microti and Borrelia burgdorferi in a western Wisconsin resident. Mayo Clin Proc.1998;73:338-341.
- Nadelman RB, Horowitz HW, Hsieh TC, et al. Simultaneous human granulocytic ehrlichiosis and Lyme borreliosis. N Engl J Med. 1997;337:27-30.
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-1134.
- Swanson SJ, Neitzel D, Reed DK, et al. Coinfections acquired from Ixodes ticks. Clin Microbiol Rev. 2006;19:708-727.
- Hayes EB, Piesman J. How can we prevent Lyme disease? N Engl J Med. 2003;348:2424-2430.
- Vaughn MF, Funkhouser SW, Lin FC, et al. Long-lasting permethrin impregnated uniforms: a randomized-controlled trial for tick bite prevention. Am J Prev Med. 2014;46:473-480.
- Miller NJ, Rainone EE, Dyer MC, et al. Tick bite protection with permethrin-treated summer-weight clothing. J Med Entomol. 2011;48:327-333.
- Richards SL, Balanay JAG, Harris JW. Effectiveness of permethrin-treated clothing to prevent tick exposure in foresters in the central Appalachian region of the USA. Int J Environ Health Res. 2015;25:453-462.
- Pages F, Dautel H, Duvallet G, et al. Tick repellents for human use: prevention of tick bites and tick-borne diseases. Vector Borne Zoonotic Dis. 2014;14:85-93.
- Büchel K, Bendin J, Gharbi A, et al. Repellent efficacy of DEET, icaridin, and EBAAP against Ixodes ricinus and Ixodes scapularis nymphs (Acari, Ixodidae). Ticks Tick Borne Dis. 2015;6:494-498.
- Schwantes U, Dautel H, Jung G. Prevention of infectious tick-borne diseases in humans: comparative studies of the repellency of different dodecanoic acid-formulations against Ixodes ricinus ticks (Acari: Ixodidae). Parasit Vectors. 2008;8:1-8.
- Bissinger BW, Apperson CS, Sonenshine DE, et al. Efficacy of the new repellent BioUD against three species of ixodid ticks. Exp Appl Acarol. 2009;48:239-250.
- Feaster JE, Scialdone MA, Todd RG, et al. Dihydronepetalactones deter feeding activity by mosquitoes, stable flies, and deer ticks. J Med Entomol. 2009;46:832-840.
- Jennett AL, Smith FD, Wall R. Tick infestation risk for dogs in a peri-urban park. Parasit Vectors. 2013;6:358.
- Rand PW, Smith RP Jr, Lacombe EH. Canine seroprevalence and the distribution of Ixodes dammini in an area of emerging Lyme disease. Am J Public Health. 1991;81:1331-1334.
- Baneth G, Bourdeau P, Bourdoiseau G, et al; CVBD World Forum. Vector-borne diseases—constant challenge for practicing veterinarians: recommendations from the CVBD World Forum. Parasit Vectors. 2012;5:55.
- Akin Belli A, Dervis E, Kar S, et al. Revisiting detachment techniques in human-biting ticks. J Am Acad Dermatol. 2016;75:393-397.
- Ashique KT, Kaliyadan F. Radiofrequency device for tick removal. J Am Acad Dermatol. 2015;72:155-156.
- Due C, Fox W, Medlock JM, et al. Tick bite prevention and tick removal. BMJ. 2013;347:f7123.
Tick-borne diseases are increasing in prevalence, likely due to climate change in combination with human movement into tick habitats.1-3 The Ixodes genus of hard ticks is a common vector for the transmission of pathogenic viruses, bacteria, parasites, and toxins. Among these, Lyme disease, which is caused by Borrelia burgdorferi, is the most prevalent, followed by babesiosis and human granulocytic anaplasmosis (HGA), respectively.4 In Europe, tick-borne encephalitis is commonly encountered. More recently identified diseases transmitted by Ixodes ticks include Powassan virus and Borrelia miyamotoi infection; however, these diseases are less frequently encountered than other tick-borne diseases.5,6
As tick-borne diseases become more prevalent, the likelihood of coinfection with more than one Ixodes-transmitted pathogen is increasing.7 Therefore, it is important for physicians who practice in endemic areas to be aware of the possibility of coinfection, which can alter clinical presentation, disease severity, and treatment response in tick-borne diseases. Additionally, public education on tick-bite prevention and prompt tick removal is necessary to combat the rising prevalence of these diseases.
Coinfection
Risk of coinfection with more than one tick-borne disease is contingent on the geographic distribution of the tick species as well as the particular pathogen’s prevalence within reservoir hosts in a given area (Figure). Most coinfections occur with B. burgdorferi and an additional pathogen, usually Anaplasma phagocytophilum (which causes human granulocytic anaplasmosis [HGA]) or Babesia microti (which causes babesiosis). In Europe, coinfection with tick-borne encephalitis virus may occur. There is limited evidence of human coinfection with B miyamotoi or Powassan virus, as isolated infection with either of these pathogens is rare.
In patients with Lyme disease, as many as 35% may have concurrent babesiosis, and as many as 12% may have concurrent HGA in endemic areas (eg, northeast and northern central United States).7-9 Concurrent HGA and babesiosis in the absence of Lyme disease also has been documented.7-9 Coinfection generally increases the diversity of presenting symptoms, often obscuring the primary diagnosis. In addition, these patients may have more severe and prolonged illness.8,10,11
In endemic areas, coinfection with B burgdorferi and an additional pathogen should be suspected if a patient presents with typical symptoms of early Lyme disease, especially erythema migrans, along with (1) combination of fever, chills, and headache; (2) prolonged viral-like illness, particularly 48 hours after appropriate antibiotic treatment; and (3) unexplained blood dyscrasia.7,11,12 When a patient presents with erythema migrans, it is unnecessary to test for HGA, as treatment of Lyme disease with doxycycline also is adequate for treating HGA; however, if systemic symptoms persist despite treatment, testing for babesiosis and other tick-borne illnesses should be considered, as babesiosis requires treatment with atovaquone plus azithromycin or clindamycin plus quinine.13
A complete blood count and peripheral blood smear can aid in the diagnosis of coinfection. The complete blood count may reveal leukopenia, anemia, or thrombocytopenia associated with HGA or babesiosis. The peripheral blood smear can reveal inclusions of intra-erythrocytic ring forms and tetrads (the “Maltese cross” appearance) in babesiosis and intragranulocytic morulae in HGA.12 The most sensitive diagnostic tests for tick-borne diseases are organism-specific IgM and IgG serology for Lyme disease, babesiosis, and HGA and polymerase chain reaction for babesiosis and HGA.7
Prevention Strategies
The most effective means of controlling tick-borne disease is avoiding tick bites altogether. One method is to avoid spending time in high-risk areas that may be infested with ticks, particularly low-lying brush, where ticks are likely to hide.14 For individuals traveling in environments with a high risk of tick exposure, behavioral methods of avoidance are indicated, including wearing long pants and a shirt with long sleeves, tucking the shirt into the pants, and wearing closed-toe shoes. Wearing light-colored clothing may aid in tick identification and prompt removal prior to attachment. Permethrin-impregnated clothing has been proven to decrease the likelihood of tick bites in adults working outdoors.15-17
Topical repellents also play a role in the prevention of tick-borne diseases. The most effective and safe synthetic repellents are N,N-diethyl-meta-toluamide (DEET); picaridin; p-menthane-3,8-diol; and insect repellent 3535 (IR3535)(ethyl butylacetylaminopropionate).16-19 Plant-based repellents also are available, but their efficacy is strongly influenced by the surrounding environment (eg, temperature, humidity, organic matter).20-22 Individuals also may be exposed to ticks following contact with domesticated animals and pets.23,24 Tick prevention in pets with the use of ectoparasiticides should be directed by a qualified veterinarian.25
Tick Removal
Following a bite, the tick should be removed promptly to avoid transmission of pathogens. Numerous commercial and in-home methods of tick removal are available, but not all are equally effective. Detachment techniques include removal with a card or commercially available radiofrequency device, lassoing, or freezing.26,27 However, the most effective method is simple removal with tweezers. The tick should be grasped close to the skin surface and pulled upward with an even pressure. Commercially available tick-removal devices have not been shown to produce better outcomes than removal of the tick with tweezers.28
Conclusion
When patients do not respond to therapy for presumed tick-borne infection, the diagnosis should be reconsidered. One important consideration is coinfection with a second organism. Prompt identification and removal of ticks can prevent disease transmission.
Tick-borne diseases are increasing in prevalence, likely due to climate change in combination with human movement into tick habitats.1-3 The Ixodes genus of hard ticks is a common vector for the transmission of pathogenic viruses, bacteria, parasites, and toxins. Among these, Lyme disease, which is caused by Borrelia burgdorferi, is the most prevalent, followed by babesiosis and human granulocytic anaplasmosis (HGA), respectively.4 In Europe, tick-borne encephalitis is commonly encountered. More recently identified diseases transmitted by Ixodes ticks include Powassan virus and Borrelia miyamotoi infection; however, these diseases are less frequently encountered than other tick-borne diseases.5,6
As tick-borne diseases become more prevalent, the likelihood of coinfection with more than one Ixodes-transmitted pathogen is increasing.7 Therefore, it is important for physicians who practice in endemic areas to be aware of the possibility of coinfection, which can alter clinical presentation, disease severity, and treatment response in tick-borne diseases. Additionally, public education on tick-bite prevention and prompt tick removal is necessary to combat the rising prevalence of these diseases.
Coinfection
Risk of coinfection with more than one tick-borne disease is contingent on the geographic distribution of the tick species as well as the particular pathogen’s prevalence within reservoir hosts in a given area (Figure). Most coinfections occur with B. burgdorferi and an additional pathogen, usually Anaplasma phagocytophilum (which causes human granulocytic anaplasmosis [HGA]) or Babesia microti (which causes babesiosis). In Europe, coinfection with tick-borne encephalitis virus may occur. There is limited evidence of human coinfection with B miyamotoi or Powassan virus, as isolated infection with either of these pathogens is rare.
In patients with Lyme disease, as many as 35% may have concurrent babesiosis, and as many as 12% may have concurrent HGA in endemic areas (eg, northeast and northern central United States).7-9 Concurrent HGA and babesiosis in the absence of Lyme disease also has been documented.7-9 Coinfection generally increases the diversity of presenting symptoms, often obscuring the primary diagnosis. In addition, these patients may have more severe and prolonged illness.8,10,11
In endemic areas, coinfection with B burgdorferi and an additional pathogen should be suspected if a patient presents with typical symptoms of early Lyme disease, especially erythema migrans, along with (1) combination of fever, chills, and headache; (2) prolonged viral-like illness, particularly 48 hours after appropriate antibiotic treatment; and (3) unexplained blood dyscrasia.7,11,12 When a patient presents with erythema migrans, it is unnecessary to test for HGA, as treatment of Lyme disease with doxycycline also is adequate for treating HGA; however, if systemic symptoms persist despite treatment, testing for babesiosis and other tick-borne illnesses should be considered, as babesiosis requires treatment with atovaquone plus azithromycin or clindamycin plus quinine.13
A complete blood count and peripheral blood smear can aid in the diagnosis of coinfection. The complete blood count may reveal leukopenia, anemia, or thrombocytopenia associated with HGA or babesiosis. The peripheral blood smear can reveal inclusions of intra-erythrocytic ring forms and tetrads (the “Maltese cross” appearance) in babesiosis and intragranulocytic morulae in HGA.12 The most sensitive diagnostic tests for tick-borne diseases are organism-specific IgM and IgG serology for Lyme disease, babesiosis, and HGA and polymerase chain reaction for babesiosis and HGA.7
Prevention Strategies
The most effective means of controlling tick-borne disease is avoiding tick bites altogether. One method is to avoid spending time in high-risk areas that may be infested with ticks, particularly low-lying brush, where ticks are likely to hide.14 For individuals traveling in environments with a high risk of tick exposure, behavioral methods of avoidance are indicated, including wearing long pants and a shirt with long sleeves, tucking the shirt into the pants, and wearing closed-toe shoes. Wearing light-colored clothing may aid in tick identification and prompt removal prior to attachment. Permethrin-impregnated clothing has been proven to decrease the likelihood of tick bites in adults working outdoors.15-17
Topical repellents also play a role in the prevention of tick-borne diseases. The most effective and safe synthetic repellents are N,N-diethyl-meta-toluamide (DEET); picaridin; p-menthane-3,8-diol; and insect repellent 3535 (IR3535)(ethyl butylacetylaminopropionate).16-19 Plant-based repellents also are available, but their efficacy is strongly influenced by the surrounding environment (eg, temperature, humidity, organic matter).20-22 Individuals also may be exposed to ticks following contact with domesticated animals and pets.23,24 Tick prevention in pets with the use of ectoparasiticides should be directed by a qualified veterinarian.25
Tick Removal
Following a bite, the tick should be removed promptly to avoid transmission of pathogens. Numerous commercial and in-home methods of tick removal are available, but not all are equally effective. Detachment techniques include removal with a card or commercially available radiofrequency device, lassoing, or freezing.26,27 However, the most effective method is simple removal with tweezers. The tick should be grasped close to the skin surface and pulled upward with an even pressure. Commercially available tick-removal devices have not been shown to produce better outcomes than removal of the tick with tweezers.28
Conclusion
When patients do not respond to therapy for presumed tick-borne infection, the diagnosis should be reconsidered. One important consideration is coinfection with a second organism. Prompt identification and removal of ticks can prevent disease transmission.
- McMichael C, Barnett J, McMichael AJ. An ill wind? climate change, migration, and health. Environ Health Perspect. 2012;120:646-654.
- Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140051.
- Ogden NH, Bigras-Poulin M, O’Callaghan CJ, et al. Vector seasonality, host infection dynamics and fitness of pathogens transmitted by the tick Ixodes scapularis. Parasitology. 2007;134(pt 2):209-227.
- Tickborne diseases of the United States. Centers for Disease Control and Prevention website. http://www.cdc.gov/ticks/diseases/index.html. Updated July 25, 2017. Accessed April 10, 2018.
- Hinten SR, Beckett GA, Gensheimer KF, et al. Increased recognition of Powassan encephalitis in the United States, 1999-2005. Vector Borne Zoonotic Dis. 2008;8:733-740.
- Platonov AE, Karan LS, Kolyasnikova NM, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816-1823.
- Krause PJ, McKay K, Thompson CA, et al; Deer-Associated Infection Study Group. Disease-specific diagnosis of coinfecting tickborne zoonoses: babesiosis, human granulocytic ehrlichiosis, and Lyme disease. Clin Infect Dis. 2002;34:1184-1191.
- Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent Lyme disease and babesiosis. evidence for increased severity and duration of illness. JAMA. 1996;275:1657-1660.
- Belongia EA, Reed KD, Mitchell PD, et al. Clinical and epidemiological features of early Lyme disease and human granulocytic ehrlichiosis in Wisconsin. Clin Infect Dis. 1999;29:1472-1477.
- Sweeny CJ, Ghassemi M, Agger WA, et al. Coinfection with Babesia microti and Borrelia burgdorferi in a western Wisconsin resident. Mayo Clin Proc.1998;73:338-341.
- Nadelman RB, Horowitz HW, Hsieh TC, et al. Simultaneous human granulocytic ehrlichiosis and Lyme borreliosis. N Engl J Med. 1997;337:27-30.
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-1134.
- Swanson SJ, Neitzel D, Reed DK, et al. Coinfections acquired from Ixodes ticks. Clin Microbiol Rev. 2006;19:708-727.
- Hayes EB, Piesman J. How can we prevent Lyme disease? N Engl J Med. 2003;348:2424-2430.
- Vaughn MF, Funkhouser SW, Lin FC, et al. Long-lasting permethrin impregnated uniforms: a randomized-controlled trial for tick bite prevention. Am J Prev Med. 2014;46:473-480.
- Miller NJ, Rainone EE, Dyer MC, et al. Tick bite protection with permethrin-treated summer-weight clothing. J Med Entomol. 2011;48:327-333.
- Richards SL, Balanay JAG, Harris JW. Effectiveness of permethrin-treated clothing to prevent tick exposure in foresters in the central Appalachian region of the USA. Int J Environ Health Res. 2015;25:453-462.
- Pages F, Dautel H, Duvallet G, et al. Tick repellents for human use: prevention of tick bites and tick-borne diseases. Vector Borne Zoonotic Dis. 2014;14:85-93.
- Büchel K, Bendin J, Gharbi A, et al. Repellent efficacy of DEET, icaridin, and EBAAP against Ixodes ricinus and Ixodes scapularis nymphs (Acari, Ixodidae). Ticks Tick Borne Dis. 2015;6:494-498.
- Schwantes U, Dautel H, Jung G. Prevention of infectious tick-borne diseases in humans: comparative studies of the repellency of different dodecanoic acid-formulations against Ixodes ricinus ticks (Acari: Ixodidae). Parasit Vectors. 2008;8:1-8.
- Bissinger BW, Apperson CS, Sonenshine DE, et al. Efficacy of the new repellent BioUD against three species of ixodid ticks. Exp Appl Acarol. 2009;48:239-250.
- Feaster JE, Scialdone MA, Todd RG, et al. Dihydronepetalactones deter feeding activity by mosquitoes, stable flies, and deer ticks. J Med Entomol. 2009;46:832-840.
- Jennett AL, Smith FD, Wall R. Tick infestation risk for dogs in a peri-urban park. Parasit Vectors. 2013;6:358.
- Rand PW, Smith RP Jr, Lacombe EH. Canine seroprevalence and the distribution of Ixodes dammini in an area of emerging Lyme disease. Am J Public Health. 1991;81:1331-1334.
- Baneth G, Bourdeau P, Bourdoiseau G, et al; CVBD World Forum. Vector-borne diseases—constant challenge for practicing veterinarians: recommendations from the CVBD World Forum. Parasit Vectors. 2012;5:55.
- Akin Belli A, Dervis E, Kar S, et al. Revisiting detachment techniques in human-biting ticks. J Am Acad Dermatol. 2016;75:393-397.
- Ashique KT, Kaliyadan F. Radiofrequency device for tick removal. J Am Acad Dermatol. 2015;72:155-156.
- Due C, Fox W, Medlock JM, et al. Tick bite prevention and tick removal. BMJ. 2013;347:f7123.
- McMichael C, Barnett J, McMichael AJ. An ill wind? climate change, migration, and health. Environ Health Perspect. 2012;120:646-654.
- Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140051.
- Ogden NH, Bigras-Poulin M, O’Callaghan CJ, et al. Vector seasonality, host infection dynamics and fitness of pathogens transmitted by the tick Ixodes scapularis. Parasitology. 2007;134(pt 2):209-227.
- Tickborne diseases of the United States. Centers for Disease Control and Prevention website. http://www.cdc.gov/ticks/diseases/index.html. Updated July 25, 2017. Accessed April 10, 2018.
- Hinten SR, Beckett GA, Gensheimer KF, et al. Increased recognition of Powassan encephalitis in the United States, 1999-2005. Vector Borne Zoonotic Dis. 2008;8:733-740.
- Platonov AE, Karan LS, Kolyasnikova NM, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816-1823.
- Krause PJ, McKay K, Thompson CA, et al; Deer-Associated Infection Study Group. Disease-specific diagnosis of coinfecting tickborne zoonoses: babesiosis, human granulocytic ehrlichiosis, and Lyme disease. Clin Infect Dis. 2002;34:1184-1191.
- Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent Lyme disease and babesiosis. evidence for increased severity and duration of illness. JAMA. 1996;275:1657-1660.
- Belongia EA, Reed KD, Mitchell PD, et al. Clinical and epidemiological features of early Lyme disease and human granulocytic ehrlichiosis in Wisconsin. Clin Infect Dis. 1999;29:1472-1477.
- Sweeny CJ, Ghassemi M, Agger WA, et al. Coinfection with Babesia microti and Borrelia burgdorferi in a western Wisconsin resident. Mayo Clin Proc.1998;73:338-341.
- Nadelman RB, Horowitz HW, Hsieh TC, et al. Simultaneous human granulocytic ehrlichiosis and Lyme borreliosis. N Engl J Med. 1997;337:27-30.
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-1134.
- Swanson SJ, Neitzel D, Reed DK, et al. Coinfections acquired from Ixodes ticks. Clin Microbiol Rev. 2006;19:708-727.
- Hayes EB, Piesman J. How can we prevent Lyme disease? N Engl J Med. 2003;348:2424-2430.
- Vaughn MF, Funkhouser SW, Lin FC, et al. Long-lasting permethrin impregnated uniforms: a randomized-controlled trial for tick bite prevention. Am J Prev Med. 2014;46:473-480.
- Miller NJ, Rainone EE, Dyer MC, et al. Tick bite protection with permethrin-treated summer-weight clothing. J Med Entomol. 2011;48:327-333.
- Richards SL, Balanay JAG, Harris JW. Effectiveness of permethrin-treated clothing to prevent tick exposure in foresters in the central Appalachian region of the USA. Int J Environ Health Res. 2015;25:453-462.
- Pages F, Dautel H, Duvallet G, et al. Tick repellents for human use: prevention of tick bites and tick-borne diseases. Vector Borne Zoonotic Dis. 2014;14:85-93.
- Büchel K, Bendin J, Gharbi A, et al. Repellent efficacy of DEET, icaridin, and EBAAP against Ixodes ricinus and Ixodes scapularis nymphs (Acari, Ixodidae). Ticks Tick Borne Dis. 2015;6:494-498.
- Schwantes U, Dautel H, Jung G. Prevention of infectious tick-borne diseases in humans: comparative studies of the repellency of different dodecanoic acid-formulations against Ixodes ricinus ticks (Acari: Ixodidae). Parasit Vectors. 2008;8:1-8.
- Bissinger BW, Apperson CS, Sonenshine DE, et al. Efficacy of the new repellent BioUD against three species of ixodid ticks. Exp Appl Acarol. 2009;48:239-250.
- Feaster JE, Scialdone MA, Todd RG, et al. Dihydronepetalactones deter feeding activity by mosquitoes, stable flies, and deer ticks. J Med Entomol. 2009;46:832-840.
- Jennett AL, Smith FD, Wall R. Tick infestation risk for dogs in a peri-urban park. Parasit Vectors. 2013;6:358.
- Rand PW, Smith RP Jr, Lacombe EH. Canine seroprevalence and the distribution of Ixodes dammini in an area of emerging Lyme disease. Am J Public Health. 1991;81:1331-1334.
- Baneth G, Bourdeau P, Bourdoiseau G, et al; CVBD World Forum. Vector-borne diseases—constant challenge for practicing veterinarians: recommendations from the CVBD World Forum. Parasit Vectors. 2012;5:55.
- Akin Belli A, Dervis E, Kar S, et al. Revisiting detachment techniques in human-biting ticks. J Am Acad Dermatol. 2016;75:393-397.
- Ashique KT, Kaliyadan F. Radiofrequency device for tick removal. J Am Acad Dermatol. 2015;72:155-156.
- Due C, Fox W, Medlock JM, et al. Tick bite prevention and tick removal. BMJ. 2013;347:f7123.
Practice Points
- As tick-borne diseases become more prevalent, the likelihood of coinfection with more than one Ixodes-transmitted pathogen is increasing, particularly in endemic areas.
- Coinfection generally increases the diversity of presenting symptoms, obscuring the primary diagnosis. The disease course also may be prolonged and more severe.
- Prevention of tick attachment and prompt tick removal are critical to combating the rising prevalence of tick-borne diseases.
Painful Mouth Ulcers
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
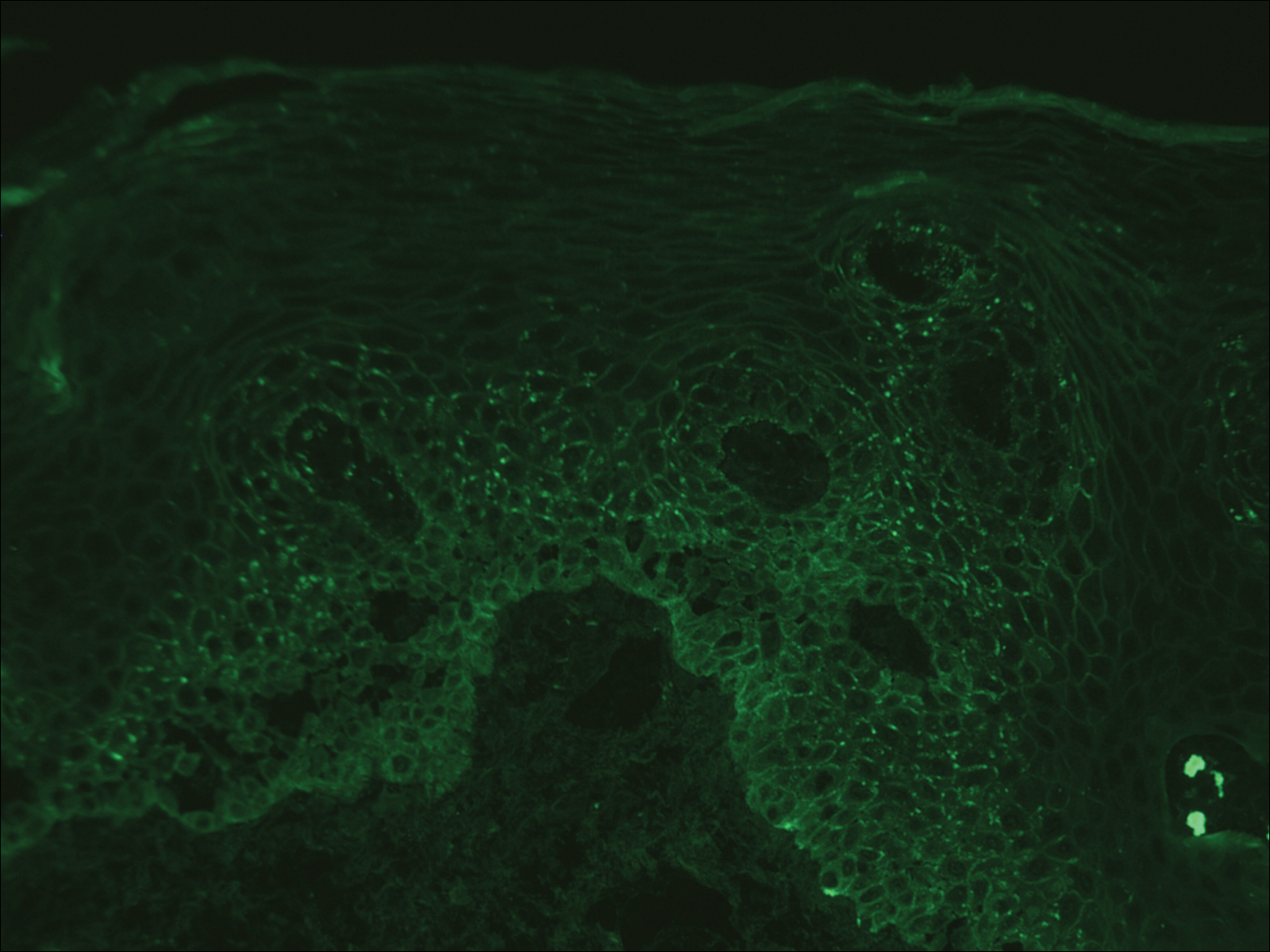
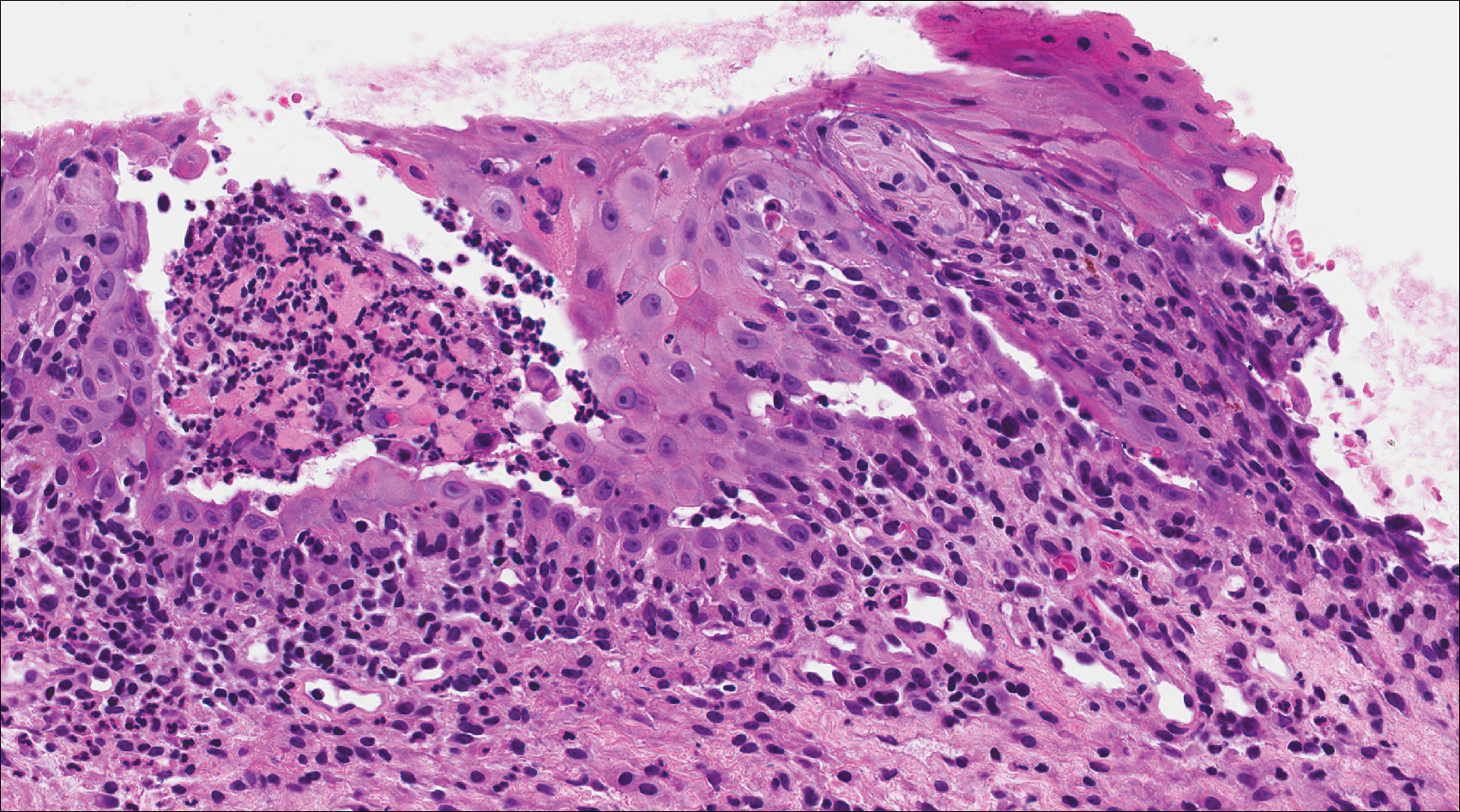
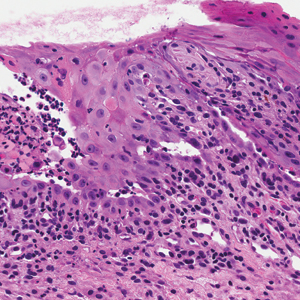
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6

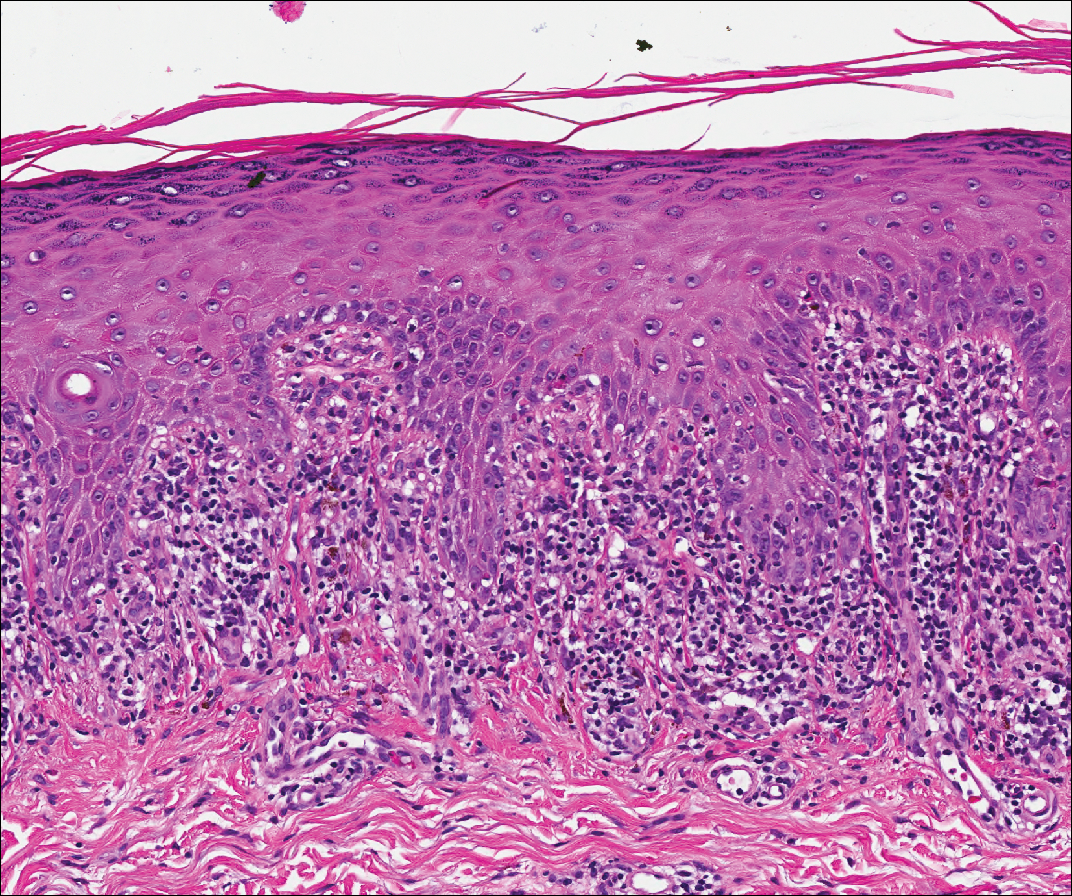
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
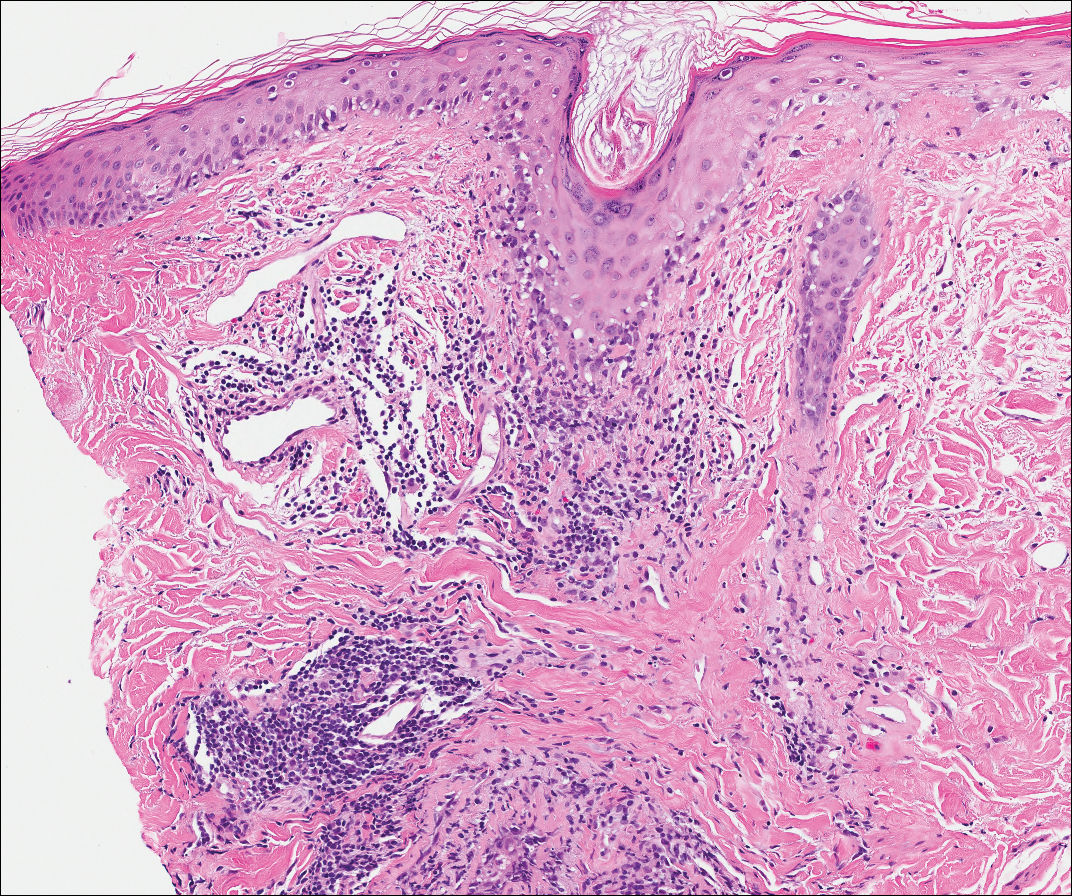
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
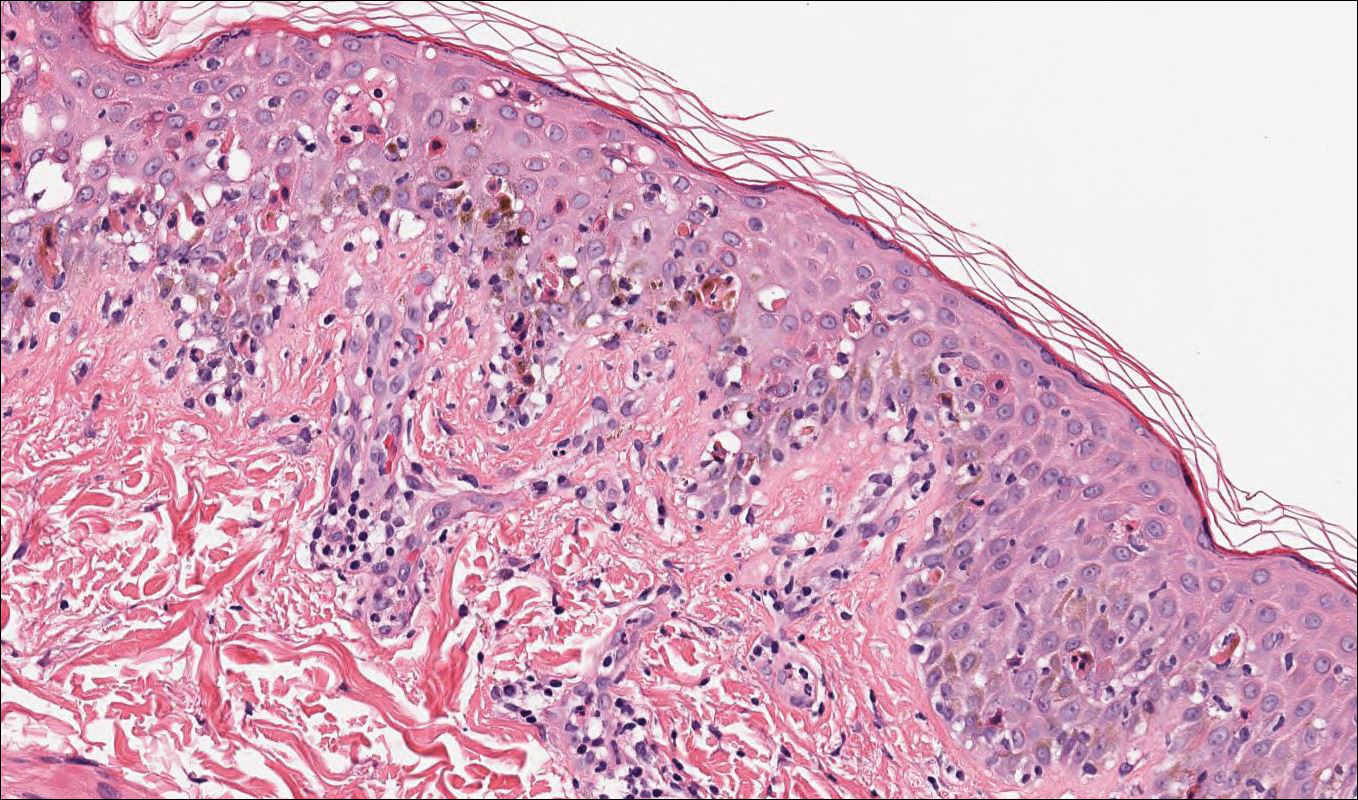
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
A 41-year-old woman presented with painful ulcers on the oral mucosa of 2 months' duration that were unresponsive to treatment with acyclovir. She had been diagnosed with a pelvic tumor a few weeks prior to the development of the mouth ulcers. Direct immunofluorescence of the perilesional mucosa showed positive IgG and complement C3 with an intercellular distribution. A biopsy of an oral lesion was performed.
Reticular Hyperpigmented Patches With Indurated Subcutaneous Plaques
The Diagnosis: Superficial Migratory Thrombophlebitis
On initial presentation, the differential diagnosis included livedoid vasculopathy, cutaneous polyarteritis nodosa, erythema ab igne, cholesterol embolism, and livedo reticularis. Laboratory investigation included antiphospholipid antibody syndrome (APS), antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibody, serum protein electrophoresis, and coagulation tests. Pertinent findings included transient low total complement activity but normal complement protein C2, C3, and C5 levels and negative cryoglobulins. Additional laboratory testing revealed elevated antiphosphatidylserine IgG, which remained elevated 12 weeks later.
New lesions continued to appear over the next several months as painful, erythematous, linear, pruritic nodules that resolved as hyperpigmented linear patches, which intersected to form a livedo reticularis-like pattern that covered the lower legs. Biopsy of an erythematous nodule on the right leg revealed fibrin occlusion of a medium-sized vein in the subcutaneous fat. Direct immunofluorescence was not specific. Venous duplex ultrasonography demonstrated chronic superficial thrombophlebitis and was crucial to the diagnosis. Ultimately, the patient's history, clinical presentation, laboratory results, venous studies, and histopathologic analysis were consistent with a diagnosis of superficial migratory thrombophlebitis (SMT) with resultant postinflammatory hyperpigmentation presenting in a reticular pattern that mimicked livedoid vasculopathy, livedo reticularis, or erythema ab igne.
Superficial migratory thrombophlebitis, also known as thrombophlebitis migrans, is defined as the recurrent formation of thrombi within superficial veins.1 The presence of a thrombus in a superficial vein evokes an inflammatory response, resulting in swelling, tenderness, erythema, and warmth in the affected area. Superficial migratory thrombophlebitis has been associated with several etiologies, including pregnancy, oral contraceptive use, APS, vasculitic disorders, and malignancies (eg, pancreas, lung, breast), as well as infections such as secondary syphilis.1
When SMT is associated with an occult malignancy, it is known as Trousseau syndrome. Common malignancies found in association with Trousseau syndrome include pancreatic, lung, and breast cancers.2 A systematic review from 2008 evaluated the utility of extensive cancer screening strategies in patients with newly diagnosed, unprovoked venous thromboembolic events.3 Using a wide screening strategy that included computed tomography of the abdomen and pelvis, the investigators detected a considerable number of formerly undiagnosed cancers, increasing detection rates from 49.4% to 69.7%. After the diagnosis of SMT was made in our patient, computed tomography of the chest, abdomen, and pelvis was performed, but the findings were unremarkable.
Because occult malignancy was excluded in our patient, the likely etiology of SMT was APS, an acquired autoimmune condition diagnosed based on the presence of a vascular thrombosis and/or pregnancy failure in women as well as elevation of at least one antiphospholipid antibody laboratory marker (eg, lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein I antibody) on 2 or more occasions at least 12 weeks apart.4 Other antibodies such as those directed against negatively charged phospholipids (eg, antiphosphatidylserine [which was elevated in our patient], phosphatidylinositol, phosphatidic acid) have been reported in patients with APS, although their diagnostic use is controversial.5 For example, the presence of antiphosphatidylserine antibodies has been considered common but not specific in patients with APS.4 However, a recent observational study demonstrated that antiphosphatidylserine antibodies are highly specific (87%) and useful in diagnosing clinical APS cases in the presence of other negative markers.6
In our patient, diagnosis of SMT with resultant postinflammatory hyperpigmentation in a reticular pattern was based on the patient's medical history, clinical examination, and histopathologic findings, as well as laboratory results and venous studies. However, it is important to note that a livedo reticularis-like pattern also is a very common finding in APS and must be included in the differential diagnosis of a reticular network on the skin.7 Moreover, differentiating livedo reticularis from SMT has prognostic importance since SMT may be associated with underlying malignancies while livedo reticularis may be associated with Sneddon syndrome, a disorder in which neurologic vascular events (eg, cerebrovascular accidents) are present.8 While this distinction is important, there are no pathognomonic histologic findings seen in livedo reticularis, and consideration of the clinical picture and additional testing is critical.4,8
Livedo vasculopathy was excluded in our patient due to the lack of diagnostic histopathologic findings, such as fibrin deposition and thrombus formation involving the upper- and mid-dermal capillaries.9 Furthermore, characteristic direct immunofluorescence findings of a homogenous or granular deposition in the vessel wall consisting of immune complexes, complement, and fibrin were absent in our patient.9 Our patient also lacked common clinical findings found in livedo vasculopathy such as small ulcerations or atrophic, porcelain-white scars on the lower legs. Erythema ab igne also was excluded in our patient due to the absence of heat exposure and presence of fibrin occlusion in the superficial leg veins. Physiologic livedo reticularis, defined as a livedoid pattern due to physiologic changes in the skin in response to cold exposure,10 also was excluded, as our patient's cutaneous changes included an alteration in pigmentation with a brown reticular pattern and no blanching, erythematous or violaceous hue, warmth, or tenderness.
In conclusion, SMT is a disorder with multiple associations that may clinically mimic livedo reticularis and livedoid vasculopathy when postinflammatory hyperpigmentation has a lacelike or livedoid pattern. While nontraditional antibodies may be useful in diagnosis in patients suspected of having APS with otherwise negative markers, standardized assays and further studies are needed to determine the specificity and value of these antibodies, particularly when used in isolation. Our patient's elevated antiphosphatidylserine IgG may have been the cause of her hypercoagulable state causing the SMT. A livedoid pattern is a common finding in APS and also was seen in our patient with SMT, but the differentiation of the brown pigmentary change and more active erythema was critical to the appropriate clinical workup of our patient.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. secondary hypercoagulable states. J Am Acad Dermatol. 1990;23:1-18.
- Rigdon EE. Trousseau's syndrome and acute arterial thrombosis. Cardiovasc Surg. 2000;8:214-218.
- Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149:323-333.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Bertolaccini ML, Amengual O, Atsumi T, et al. 'Non-criteria' aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus. 2011;20:191-205.
- Khogeer H, Alfattani A, Al Kaff M, et al. Antiphosphatidylserine antibodies as diagnostic indicators of antiphospholipid syndrome. Lupus. 2015;24:186-190.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6 pt 1):970-982.
- Francès C, Papo T, Wechsler B, et al. Sneddon syndrome with or without antiphospholipid antibodies. a comparative study in 46 patients. Medicine (Baltimore). 1999;78:209-219.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478-488.
- James WD, Berger TG, Elston DM. Andrews' Diseases Of The Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: Elsevier Saunders; 2006.
The Diagnosis: Superficial Migratory Thrombophlebitis
On initial presentation, the differential diagnosis included livedoid vasculopathy, cutaneous polyarteritis nodosa, erythema ab igne, cholesterol embolism, and livedo reticularis. Laboratory investigation included antiphospholipid antibody syndrome (APS), antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibody, serum protein electrophoresis, and coagulation tests. Pertinent findings included transient low total complement activity but normal complement protein C2, C3, and C5 levels and negative cryoglobulins. Additional laboratory testing revealed elevated antiphosphatidylserine IgG, which remained elevated 12 weeks later.
New lesions continued to appear over the next several months as painful, erythematous, linear, pruritic nodules that resolved as hyperpigmented linear patches, which intersected to form a livedo reticularis-like pattern that covered the lower legs. Biopsy of an erythematous nodule on the right leg revealed fibrin occlusion of a medium-sized vein in the subcutaneous fat. Direct immunofluorescence was not specific. Venous duplex ultrasonography demonstrated chronic superficial thrombophlebitis and was crucial to the diagnosis. Ultimately, the patient's history, clinical presentation, laboratory results, venous studies, and histopathologic analysis were consistent with a diagnosis of superficial migratory thrombophlebitis (SMT) with resultant postinflammatory hyperpigmentation presenting in a reticular pattern that mimicked livedoid vasculopathy, livedo reticularis, or erythema ab igne.
Superficial migratory thrombophlebitis, also known as thrombophlebitis migrans, is defined as the recurrent formation of thrombi within superficial veins.1 The presence of a thrombus in a superficial vein evokes an inflammatory response, resulting in swelling, tenderness, erythema, and warmth in the affected area. Superficial migratory thrombophlebitis has been associated with several etiologies, including pregnancy, oral contraceptive use, APS, vasculitic disorders, and malignancies (eg, pancreas, lung, breast), as well as infections such as secondary syphilis.1
When SMT is associated with an occult malignancy, it is known as Trousseau syndrome. Common malignancies found in association with Trousseau syndrome include pancreatic, lung, and breast cancers.2 A systematic review from 2008 evaluated the utility of extensive cancer screening strategies in patients with newly diagnosed, unprovoked venous thromboembolic events.3 Using a wide screening strategy that included computed tomography of the abdomen and pelvis, the investigators detected a considerable number of formerly undiagnosed cancers, increasing detection rates from 49.4% to 69.7%. After the diagnosis of SMT was made in our patient, computed tomography of the chest, abdomen, and pelvis was performed, but the findings were unremarkable.
Because occult malignancy was excluded in our patient, the likely etiology of SMT was APS, an acquired autoimmune condition diagnosed based on the presence of a vascular thrombosis and/or pregnancy failure in women as well as elevation of at least one antiphospholipid antibody laboratory marker (eg, lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein I antibody) on 2 or more occasions at least 12 weeks apart.4 Other antibodies such as those directed against negatively charged phospholipids (eg, antiphosphatidylserine [which was elevated in our patient], phosphatidylinositol, phosphatidic acid) have been reported in patients with APS, although their diagnostic use is controversial.5 For example, the presence of antiphosphatidylserine antibodies has been considered common but not specific in patients with APS.4 However, a recent observational study demonstrated that antiphosphatidylserine antibodies are highly specific (87%) and useful in diagnosing clinical APS cases in the presence of other negative markers.6
In our patient, diagnosis of SMT with resultant postinflammatory hyperpigmentation in a reticular pattern was based on the patient's medical history, clinical examination, and histopathologic findings, as well as laboratory results and venous studies. However, it is important to note that a livedo reticularis-like pattern also is a very common finding in APS and must be included in the differential diagnosis of a reticular network on the skin.7 Moreover, differentiating livedo reticularis from SMT has prognostic importance since SMT may be associated with underlying malignancies while livedo reticularis may be associated with Sneddon syndrome, a disorder in which neurologic vascular events (eg, cerebrovascular accidents) are present.8 While this distinction is important, there are no pathognomonic histologic findings seen in livedo reticularis, and consideration of the clinical picture and additional testing is critical.4,8
Livedo vasculopathy was excluded in our patient due to the lack of diagnostic histopathologic findings, such as fibrin deposition and thrombus formation involving the upper- and mid-dermal capillaries.9 Furthermore, characteristic direct immunofluorescence findings of a homogenous or granular deposition in the vessel wall consisting of immune complexes, complement, and fibrin were absent in our patient.9 Our patient also lacked common clinical findings found in livedo vasculopathy such as small ulcerations or atrophic, porcelain-white scars on the lower legs. Erythema ab igne also was excluded in our patient due to the absence of heat exposure and presence of fibrin occlusion in the superficial leg veins. Physiologic livedo reticularis, defined as a livedoid pattern due to physiologic changes in the skin in response to cold exposure,10 also was excluded, as our patient's cutaneous changes included an alteration in pigmentation with a brown reticular pattern and no blanching, erythematous or violaceous hue, warmth, or tenderness.
In conclusion, SMT is a disorder with multiple associations that may clinically mimic livedo reticularis and livedoid vasculopathy when postinflammatory hyperpigmentation has a lacelike or livedoid pattern. While nontraditional antibodies may be useful in diagnosis in patients suspected of having APS with otherwise negative markers, standardized assays and further studies are needed to determine the specificity and value of these antibodies, particularly when used in isolation. Our patient's elevated antiphosphatidylserine IgG may have been the cause of her hypercoagulable state causing the SMT. A livedoid pattern is a common finding in APS and also was seen in our patient with SMT, but the differentiation of the brown pigmentary change and more active erythema was critical to the appropriate clinical workup of our patient.
The Diagnosis: Superficial Migratory Thrombophlebitis
On initial presentation, the differential diagnosis included livedoid vasculopathy, cutaneous polyarteritis nodosa, erythema ab igne, cholesterol embolism, and livedo reticularis. Laboratory investigation included antiphospholipid antibody syndrome (APS), antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibody, serum protein electrophoresis, and coagulation tests. Pertinent findings included transient low total complement activity but normal complement protein C2, C3, and C5 levels and negative cryoglobulins. Additional laboratory testing revealed elevated antiphosphatidylserine IgG, which remained elevated 12 weeks later.
New lesions continued to appear over the next several months as painful, erythematous, linear, pruritic nodules that resolved as hyperpigmented linear patches, which intersected to form a livedo reticularis-like pattern that covered the lower legs. Biopsy of an erythematous nodule on the right leg revealed fibrin occlusion of a medium-sized vein in the subcutaneous fat. Direct immunofluorescence was not specific. Venous duplex ultrasonography demonstrated chronic superficial thrombophlebitis and was crucial to the diagnosis. Ultimately, the patient's history, clinical presentation, laboratory results, venous studies, and histopathologic analysis were consistent with a diagnosis of superficial migratory thrombophlebitis (SMT) with resultant postinflammatory hyperpigmentation presenting in a reticular pattern that mimicked livedoid vasculopathy, livedo reticularis, or erythema ab igne.
Superficial migratory thrombophlebitis, also known as thrombophlebitis migrans, is defined as the recurrent formation of thrombi within superficial veins.1 The presence of a thrombus in a superficial vein evokes an inflammatory response, resulting in swelling, tenderness, erythema, and warmth in the affected area. Superficial migratory thrombophlebitis has been associated with several etiologies, including pregnancy, oral contraceptive use, APS, vasculitic disorders, and malignancies (eg, pancreas, lung, breast), as well as infections such as secondary syphilis.1
When SMT is associated with an occult malignancy, it is known as Trousseau syndrome. Common malignancies found in association with Trousseau syndrome include pancreatic, lung, and breast cancers.2 A systematic review from 2008 evaluated the utility of extensive cancer screening strategies in patients with newly diagnosed, unprovoked venous thromboembolic events.3 Using a wide screening strategy that included computed tomography of the abdomen and pelvis, the investigators detected a considerable number of formerly undiagnosed cancers, increasing detection rates from 49.4% to 69.7%. After the diagnosis of SMT was made in our patient, computed tomography of the chest, abdomen, and pelvis was performed, but the findings were unremarkable.
Because occult malignancy was excluded in our patient, the likely etiology of SMT was APS, an acquired autoimmune condition diagnosed based on the presence of a vascular thrombosis and/or pregnancy failure in women as well as elevation of at least one antiphospholipid antibody laboratory marker (eg, lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein I antibody) on 2 or more occasions at least 12 weeks apart.4 Other antibodies such as those directed against negatively charged phospholipids (eg, antiphosphatidylserine [which was elevated in our patient], phosphatidylinositol, phosphatidic acid) have been reported in patients with APS, although their diagnostic use is controversial.5 For example, the presence of antiphosphatidylserine antibodies has been considered common but not specific in patients with APS.4 However, a recent observational study demonstrated that antiphosphatidylserine antibodies are highly specific (87%) and useful in diagnosing clinical APS cases in the presence of other negative markers.6
In our patient, diagnosis of SMT with resultant postinflammatory hyperpigmentation in a reticular pattern was based on the patient's medical history, clinical examination, and histopathologic findings, as well as laboratory results and venous studies. However, it is important to note that a livedo reticularis-like pattern also is a very common finding in APS and must be included in the differential diagnosis of a reticular network on the skin.7 Moreover, differentiating livedo reticularis from SMT has prognostic importance since SMT may be associated with underlying malignancies while livedo reticularis may be associated with Sneddon syndrome, a disorder in which neurologic vascular events (eg, cerebrovascular accidents) are present.8 While this distinction is important, there are no pathognomonic histologic findings seen in livedo reticularis, and consideration of the clinical picture and additional testing is critical.4,8
Livedo vasculopathy was excluded in our patient due to the lack of diagnostic histopathologic findings, such as fibrin deposition and thrombus formation involving the upper- and mid-dermal capillaries.9 Furthermore, characteristic direct immunofluorescence findings of a homogenous or granular deposition in the vessel wall consisting of immune complexes, complement, and fibrin were absent in our patient.9 Our patient also lacked common clinical findings found in livedo vasculopathy such as small ulcerations or atrophic, porcelain-white scars on the lower legs. Erythema ab igne also was excluded in our patient due to the absence of heat exposure and presence of fibrin occlusion in the superficial leg veins. Physiologic livedo reticularis, defined as a livedoid pattern due to physiologic changes in the skin in response to cold exposure,10 also was excluded, as our patient's cutaneous changes included an alteration in pigmentation with a brown reticular pattern and no blanching, erythematous or violaceous hue, warmth, or tenderness.
In conclusion, SMT is a disorder with multiple associations that may clinically mimic livedo reticularis and livedoid vasculopathy when postinflammatory hyperpigmentation has a lacelike or livedoid pattern. While nontraditional antibodies may be useful in diagnosis in patients suspected of having APS with otherwise negative markers, standardized assays and further studies are needed to determine the specificity and value of these antibodies, particularly when used in isolation. Our patient's elevated antiphosphatidylserine IgG may have been the cause of her hypercoagulable state causing the SMT. A livedoid pattern is a common finding in APS and also was seen in our patient with SMT, but the differentiation of the brown pigmentary change and more active erythema was critical to the appropriate clinical workup of our patient.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. secondary hypercoagulable states. J Am Acad Dermatol. 1990;23:1-18.
- Rigdon EE. Trousseau's syndrome and acute arterial thrombosis. Cardiovasc Surg. 2000;8:214-218.
- Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149:323-333.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Bertolaccini ML, Amengual O, Atsumi T, et al. 'Non-criteria' aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus. 2011;20:191-205.
- Khogeer H, Alfattani A, Al Kaff M, et al. Antiphosphatidylserine antibodies as diagnostic indicators of antiphospholipid syndrome. Lupus. 2015;24:186-190.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6 pt 1):970-982.
- Francès C, Papo T, Wechsler B, et al. Sneddon syndrome with or without antiphospholipid antibodies. a comparative study in 46 patients. Medicine (Baltimore). 1999;78:209-219.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478-488.
- James WD, Berger TG, Elston DM. Andrews' Diseases Of The Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: Elsevier Saunders; 2006.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. secondary hypercoagulable states. J Am Acad Dermatol. 1990;23:1-18.
- Rigdon EE. Trousseau's syndrome and acute arterial thrombosis. Cardiovasc Surg. 2000;8:214-218.
- Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149:323-333.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Bertolaccini ML, Amengual O, Atsumi T, et al. 'Non-criteria' aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus. 2011;20:191-205.
- Khogeer H, Alfattani A, Al Kaff M, et al. Antiphosphatidylserine antibodies as diagnostic indicators of antiphospholipid syndrome. Lupus. 2015;24:186-190.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6 pt 1):970-982.
- Francès C, Papo T, Wechsler B, et al. Sneddon syndrome with or without antiphospholipid antibodies. a comparative study in 46 patients. Medicine (Baltimore). 1999;78:209-219.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478-488.
- James WD, Berger TG, Elston DM. Andrews' Diseases Of The Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: Elsevier Saunders; 2006.

A 32-year-old woman presented for evaluation of small, tender, erythematous nodules on the lower legs of 1 year's duration that had started to spread to the thighs over several months prior to presentation. The patient reported no history of ulceration or other cutaneous findings. On physical examination, a hyperpigmented, linear to reticular pattern was noted on the lower legs with a few 1-cm, erythematous, mildly indurated and tender subcutaneous nodules. The patient denied any recent medical procedures, history of malignancy or cardiovascular disease, use of tobacco or illicit drugs, prolonged contact with a heat source, recent unintentional weight loss, fevers, or night sweats. Her medical history was notable for asthma and migraines, which were treated with albuterol, fluticasone, and topiramate.
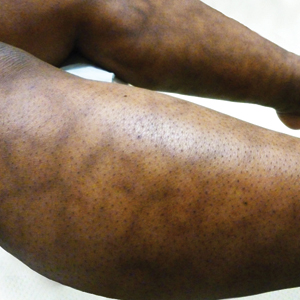
New Guidelines of Care for the Management of Nonmelanoma Skin Cancer
In January 2018, the American Academy of Dermatology (AAD) released its first guidelines of care for the management of nonmelanoma skin cancer (NMSC), which established official recommendations for the treatment of basal cell carcinoma (BCC)1 and cutaneous squamous cell carcinoma (cSCC).2 The guidelines will help dermatologists address the growing health concern of skin cancer, which remains the most common of any type of cancer in the United States.3 Affecting more than 3 million Americans every year, NMSC is the most common type of skin cancer, and its incidence has continued to increase every year over the past few decades.3,4 During the past 30 years, the incidence of both BCC and cSCC has more than doubled.5
Commonly used guidelines for the management of NMSC are available from the National Comprehensive Cancer Network (NCCN).6,7 Although the NCCN aimed to develop multidisciplinary guidelines, the new AAD guidelines were established primarily by dermatologists for dermatologists. The NCCN guidelines frequently are referenced throughout the new AAD guidelines, which also recognize the importance of multidisciplinary care. The authors of the AAD guidelines noted that, although many of the NCCN recommendations reiterated prevailing knowledge or current practice, some recommendations highlighted alternative tenets that were not as widely considered or were supported by insufficient evidence.
The AAD guidelines address the complete management of NMSC, which includes biopsy technique, staging, treatment, follow-up, metastatic disease, and prevention.1,2 Also included are evidence tables evaluating the current literature and available recommendations.
BCC Guidelines
For suspected BCCs, the recommended biopsy techniques are punch biopsy, shave biopsy, and excisional biopsy, all of which can detect the most aggressive histology subtypes.1 Rebiopsy is recommended if the initial specimen is inadequate. The pathology report should include histologic subtype, invasion beyond the reticular dermis, and perineural involvement. The AAD guidelines do not include a formal staging system for risk stratification but rather refer to the NCCN guidelines, which take both clinical and pathologic parameters into account. The AAD treatment recommendations are based on this stratification.1
Treatment of BCC includes a broad range of therapeutic modalities. Recurrence rate, preservation of function, patient expectations, and potential adverse effects should be considered in the treatment plan.1 Curettage and electrodessication may be considered for low-risk tumors in nonterminal hair-bearing locations. Surgical excision with 4-mm margins is recommended for low-risk primary tumors. For high-risk BCC, Mohs micrographic surgery is recommended, although standard excision along with attention to margin control may also be considered. Nonsurgical treatments also may be considered when more effective surgical therapies are contraindicated or impractical. If surgical therapy is not feasible or preferred, other treatment options for low-risk BCCs include cryotherapy, topical
Multidisciplinary consultation is recommended in patients with metastatic BCCs along with first-line treatment with a smoothened inhibitor.1 Alternative treatment options include platinum-based chemotherapy and/or supportive care. For locally advanced disease, surgery and radiation therapy remain the initial treatments, but smoothened inhibitors and supportive care are suitable alternative treatments.1
The AAD guidelines also offer recommendations for follow-up and reducing future risk of skin cancer. After the first diagnosis of BCC, a skin cancer screening should be performed at least annually, and patients should be counseled about self-examinations and sun protection.1 Topical and oral retinoids are not recommended for the prevention of additional skin cancers, nor is dietary supplementation with selenium or beta-carotene. There also is insufficient evidence regarding the use of oral nicotinamide, celecoxib, or α-difluoromethylornithine for chemoprevention of disease.1
cSCC Guidelines
For suspected cSCCs, no single optimal biopsy technique is recommended, but repeat biopsy may be considered if the initial biopsy is insufficient for diagnosis.2 The guidelines further recommend an extensive list of elements to be included in the final pathology report (eg, lesion size, immunosuppression, depth of invasion, degree of differentiation). There is no universally recognized stratification for localized cSCC; therefore, the AAD guidelines refer to the framework provided by the NCCN. Also mentioned is the recent release of the American Joint Committee on Cancer’s staging manual,8 which includes the management of cSCC in conjunction with all SCCs of the head and neck. The Brigham and Women’s system9 was considered as an alternative classification system; however, the NCCN guidelines were chosen because they primarily provide clinical guidance for treatment of cSCC rather than provide accurate prognostication or outcome assessment.
Considerations for surgical treatment of cSCC are similar to those for BCC.2 In low-risk tumors, surgical excision with 4- to 6-mm margins to the midsubcutaneous fat or curettage with electrodessication may be considered. Mohs micrographic surgery or standard excision with attention to margin control may be considered for high-risk tumors. Nonsurgical therapies generally are not recommended as a first-line treatment, particularly in cSCC, due to possible recurrence and metastasis. When nonsurgical therapies are preferred, options may include cryosurgery or radiation therapy, with the understanding that cure rates may be lower than with surgical options. Topical therapy with imiquimod or 5-fluorouracil as well as photodynamic or laser therapy are not recommended for cSCCs.2
For patients with metastatic cSCC or locally advanced disease, multidisciplinary consultation is recommended.2 In cSCCs with regional lymph node metastases, the recommended approach includes surgical resection with possible adjuvant radiation therapy and/or systemic therapy. For inoperable disease, combination chemoradiation may be considered. Epidermal growth factor inhibitors and cisplatin may be considered in metastatic disease, although there are limited data to support their efficacy. As with BCC, all patients with cSCCs should receive supportive and palliative care to optimize quality of life.2
Recommendations for follow-up after the first diagnosis of cSCC are the same as those for BCC.2 Additionally, acitretin is the only therapy that may be beneficial in the reduction of recurrent skin cancer in patients who are solid-organ transplant recipients.
Final Thoughts
A comprehensive understanding of the management of NMSC and the evidence on which recommendations are based is critically important for optimal patient care. These guidelines are an efficient way for dermatologists and their colleagues to understand the latest evidence and recommendations. The AAD guidelines provide support for clinical decision making with standardized approaches to the diagnosis, care, and prevention of NMSC that are consistent with established practice patterns.
With few exceptions, surgical therapy is the most effective approach for the treatment of BCC and cSCC; however, the AAD guidelines include an important review on nonsurgical management options.1,2 The AAD guidelines help to highlight where data on evidence-based outcomes exist and reveal where data remain insufficient. This is illustrated by the guideline recommendations for providing additional histopathologic characteristics in the pathology reports, which will likely produce future data to enhance the prognosis and eventual treatment of patients with NMSC.1,2 Future guidelines also may include newer technologies (eg, gene expression profiling).
The guidelines do not cover the management of premalignant and in situ lesions, nor do they provide details on the management of metastatic or locally advanced disease. These topics certainly will require a similar critical review and may be addressed separately. The guidelines are identifying unanswered questions about patient care and are concurrently establishing the collection of appropriate data to answer these questions in the future.
Official guidelines often become the primary source for the measured standard of both treatment and outcomes in patient care; therefore, it is critical that dermatologists and the AAD take the lead in creating these guidelines so that we can provide our patients with the best evidenced-based comprehensive care.
The AAD guidelines emphasize the importance of considering the patient perspective in determining how to treat BCCs and cSCCs.1,2 It is important for patients to understand the available treatment options and participate in their own medical care. The AAD work group for these guidelines included patient advocates to ensure that the guidelines would promote further dialogue between physicians and their patients.
The AAD guidelines for the management of NMSC were developed by board-certified dermatologists and other experts in the field. They allow dermatologists to work with patients diagnosed with NMSC to determine the treatment option that is best for each individual patient.
- Bichakjian C, Armstrong A, Baum C, et al. Guidelines of care for the management of basal cell carcinoma. J Am Acad Dermatol. 2018;78:540-559.
- Alam M, Armstrong A, Baum C, et al. Guidelines of care for the management of cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2018;78:560-578.
- Burden of skin disease. American Academy of Dermatology website. https://www.aad.org/about/burden-of-skin-disease. Accessed April 17, 2018.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population. JAMA Dermatol. 2015;151:1081-1086.
- Muzic JG, Schmitt AR, Wright AC, et al. Incidence and trends of basal cell carcinoma and cutaneous squamous cell carcinoma: a population-based study in Olmstead County, Minnnesota, 2000-2010. Mayo Clin Proc. 2017;92:890-898.
- Bichakjian CK, Olencki T, Aasi SZ, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Basal Cell Skin Cancer. National Comprehensive Cancer Network website. https://www.nccn.org/professionals/physician_gls/pdf/nmsc.pdf. Published September 18, 2017. Accessed April 17, 2018.
- Bichakjian CK, Olencki T, Aasi SZ, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Squamous Cell Skin Cancer. National Comprehensive Cancer Network website. Published October 5, 2017. Accessed April 17, 2018.
- Amin MB, Edge SB, Greene FL, et al. AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer International Publishing; 2016.
- Jambusaria-Pahlajani A, Kanetsky PA, Karia PS, et al. Evaluation of AJCC tumor staging for cutaneous squamous cell carcinoma and a proposed alternative tumor staging system. JAMA Dermatol. 2013;149:402-410.
In January 2018, the American Academy of Dermatology (AAD) released its first guidelines of care for the management of nonmelanoma skin cancer (NMSC), which established official recommendations for the treatment of basal cell carcinoma (BCC)1 and cutaneous squamous cell carcinoma (cSCC).2 The guidelines will help dermatologists address the growing health concern of skin cancer, which remains the most common of any type of cancer in the United States.3 Affecting more than 3 million Americans every year, NMSC is the most common type of skin cancer, and its incidence has continued to increase every year over the past few decades.3,4 During the past 30 years, the incidence of both BCC and cSCC has more than doubled.5
Commonly used guidelines for the management of NMSC are available from the National Comprehensive Cancer Network (NCCN).6,7 Although the NCCN aimed to develop multidisciplinary guidelines, the new AAD guidelines were established primarily by dermatologists for dermatologists. The NCCN guidelines frequently are referenced throughout the new AAD guidelines, which also recognize the importance of multidisciplinary care. The authors of the AAD guidelines noted that, although many of the NCCN recommendations reiterated prevailing knowledge or current practice, some recommendations highlighted alternative tenets that were not as widely considered or were supported by insufficient evidence.
The AAD guidelines address the complete management of NMSC, which includes biopsy technique, staging, treatment, follow-up, metastatic disease, and prevention.1,2 Also included are evidence tables evaluating the current literature and available recommendations.
BCC Guidelines
For suspected BCCs, the recommended biopsy techniques are punch biopsy, shave biopsy, and excisional biopsy, all of which can detect the most aggressive histology subtypes.1 Rebiopsy is recommended if the initial specimen is inadequate. The pathology report should include histologic subtype, invasion beyond the reticular dermis, and perineural involvement. The AAD guidelines do not include a formal staging system for risk stratification but rather refer to the NCCN guidelines, which take both clinical and pathologic parameters into account. The AAD treatment recommendations are based on this stratification.1
Treatment of BCC includes a broad range of therapeutic modalities. Recurrence rate, preservation of function, patient expectations, and potential adverse effects should be considered in the treatment plan.1 Curettage and electrodessication may be considered for low-risk tumors in nonterminal hair-bearing locations. Surgical excision with 4-mm margins is recommended for low-risk primary tumors. For high-risk BCC, Mohs micrographic surgery is recommended, although standard excision along with attention to margin control may also be considered. Nonsurgical treatments also may be considered when more effective surgical therapies are contraindicated or impractical. If surgical therapy is not feasible or preferred, other treatment options for low-risk BCCs include cryotherapy, topical
Multidisciplinary consultation is recommended in patients with metastatic BCCs along with first-line treatment with a smoothened inhibitor.1 Alternative treatment options include platinum-based chemotherapy and/or supportive care. For locally advanced disease, surgery and radiation therapy remain the initial treatments, but smoothened inhibitors and supportive care are suitable alternative treatments.1
The AAD guidelines also offer recommendations for follow-up and reducing future risk of skin cancer. After the first diagnosis of BCC, a skin cancer screening should be performed at least annually, and patients should be counseled about self-examinations and sun protection.1 Topical and oral retinoids are not recommended for the prevention of additional skin cancers, nor is dietary supplementation with selenium or beta-carotene. There also is insufficient evidence regarding the use of oral nicotinamide, celecoxib, or α-difluoromethylornithine for chemoprevention of disease.1
cSCC Guidelines
For suspected cSCCs, no single optimal biopsy technique is recommended, but repeat biopsy may be considered if the initial biopsy is insufficient for diagnosis.2 The guidelines further recommend an extensive list of elements to be included in the final pathology report (eg, lesion size, immunosuppression, depth of invasion, degree of differentiation). There is no universally recognized stratification for localized cSCC; therefore, the AAD guidelines refer to the framework provided by the NCCN. Also mentioned is the recent release of the American Joint Committee on Cancer’s staging manual,8 which includes the management of cSCC in conjunction with all SCCs of the head and neck. The Brigham and Women’s system9 was considered as an alternative classification system; however, the NCCN guidelines were chosen because they primarily provide clinical guidance for treatment of cSCC rather than provide accurate prognostication or outcome assessment.
Considerations for surgical treatment of cSCC are similar to those for BCC.2 In low-risk tumors, surgical excision with 4- to 6-mm margins to the midsubcutaneous fat or curettage with electrodessication may be considered. Mohs micrographic surgery or standard excision with attention to margin control may be considered for high-risk tumors. Nonsurgical therapies generally are not recommended as a first-line treatment, particularly in cSCC, due to possible recurrence and metastasis. When nonsurgical therapies are preferred, options may include cryosurgery or radiation therapy, with the understanding that cure rates may be lower than with surgical options. Topical therapy with imiquimod or 5-fluorouracil as well as photodynamic or laser therapy are not recommended for cSCCs.2
For patients with metastatic cSCC or locally advanced disease, multidisciplinary consultation is recommended.2 In cSCCs with regional lymph node metastases, the recommended approach includes surgical resection with possible adjuvant radiation therapy and/or systemic therapy. For inoperable disease, combination chemoradiation may be considered. Epidermal growth factor inhibitors and cisplatin may be considered in metastatic disease, although there are limited data to support their efficacy. As with BCC, all patients with cSCCs should receive supportive and palliative care to optimize quality of life.2
Recommendations for follow-up after the first diagnosis of cSCC are the same as those for BCC.2 Additionally, acitretin is the only therapy that may be beneficial in the reduction of recurrent skin cancer in patients who are solid-organ transplant recipients.
Final Thoughts
A comprehensive understanding of the management of NMSC and the evidence on which recommendations are based is critically important for optimal patient care. These guidelines are an efficient way for dermatologists and their colleagues to understand the latest evidence and recommendations. The AAD guidelines provide support for clinical decision making with standardized approaches to the diagnosis, care, and prevention of NMSC that are consistent with established practice patterns.
With few exceptions, surgical therapy is the most effective approach for the treatment of BCC and cSCC; however, the AAD guidelines include an important review on nonsurgical management options.1,2 The AAD guidelines help to highlight where data on evidence-based outcomes exist and reveal where data remain insufficient. This is illustrated by the guideline recommendations for providing additional histopathologic characteristics in the pathology reports, which will likely produce future data to enhance the prognosis and eventual treatment of patients with NMSC.1,2 Future guidelines also may include newer technologies (eg, gene expression profiling).
The guidelines do not cover the management of premalignant and in situ lesions, nor do they provide details on the management of metastatic or locally advanced disease. These topics certainly will require a similar critical review and may be addressed separately. The guidelines are identifying unanswered questions about patient care and are concurrently establishing the collection of appropriate data to answer these questions in the future.
Official guidelines often become the primary source for the measured standard of both treatment and outcomes in patient care; therefore, it is critical that dermatologists and the AAD take the lead in creating these guidelines so that we can provide our patients with the best evidenced-based comprehensive care.
The AAD guidelines emphasize the importance of considering the patient perspective in determining how to treat BCCs and cSCCs.1,2 It is important for patients to understand the available treatment options and participate in their own medical care. The AAD work group for these guidelines included patient advocates to ensure that the guidelines would promote further dialogue between physicians and their patients.
The AAD guidelines for the management of NMSC were developed by board-certified dermatologists and other experts in the field. They allow dermatologists to work with patients diagnosed with NMSC to determine the treatment option that is best for each individual patient.
In January 2018, the American Academy of Dermatology (AAD) released its first guidelines of care for the management of nonmelanoma skin cancer (NMSC), which established official recommendations for the treatment of basal cell carcinoma (BCC)1 and cutaneous squamous cell carcinoma (cSCC).2 The guidelines will help dermatologists address the growing health concern of skin cancer, which remains the most common of any type of cancer in the United States.3 Affecting more than 3 million Americans every year, NMSC is the most common type of skin cancer, and its incidence has continued to increase every year over the past few decades.3,4 During the past 30 years, the incidence of both BCC and cSCC has more than doubled.5
Commonly used guidelines for the management of NMSC are available from the National Comprehensive Cancer Network (NCCN).6,7 Although the NCCN aimed to develop multidisciplinary guidelines, the new AAD guidelines were established primarily by dermatologists for dermatologists. The NCCN guidelines frequently are referenced throughout the new AAD guidelines, which also recognize the importance of multidisciplinary care. The authors of the AAD guidelines noted that, although many of the NCCN recommendations reiterated prevailing knowledge or current practice, some recommendations highlighted alternative tenets that were not as widely considered or were supported by insufficient evidence.
The AAD guidelines address the complete management of NMSC, which includes biopsy technique, staging, treatment, follow-up, metastatic disease, and prevention.1,2 Also included are evidence tables evaluating the current literature and available recommendations.
BCC Guidelines
For suspected BCCs, the recommended biopsy techniques are punch biopsy, shave biopsy, and excisional biopsy, all of which can detect the most aggressive histology subtypes.1 Rebiopsy is recommended if the initial specimen is inadequate. The pathology report should include histologic subtype, invasion beyond the reticular dermis, and perineural involvement. The AAD guidelines do not include a formal staging system for risk stratification but rather refer to the NCCN guidelines, which take both clinical and pathologic parameters into account. The AAD treatment recommendations are based on this stratification.1
Treatment of BCC includes a broad range of therapeutic modalities. Recurrence rate, preservation of function, patient expectations, and potential adverse effects should be considered in the treatment plan.1 Curettage and electrodessication may be considered for low-risk tumors in nonterminal hair-bearing locations. Surgical excision with 4-mm margins is recommended for low-risk primary tumors. For high-risk BCC, Mohs micrographic surgery is recommended, although standard excision along with attention to margin control may also be considered. Nonsurgical treatments also may be considered when more effective surgical therapies are contraindicated or impractical. If surgical therapy is not feasible or preferred, other treatment options for low-risk BCCs include cryotherapy, topical
Multidisciplinary consultation is recommended in patients with metastatic BCCs along with first-line treatment with a smoothened inhibitor.1 Alternative treatment options include platinum-based chemotherapy and/or supportive care. For locally advanced disease, surgery and radiation therapy remain the initial treatments, but smoothened inhibitors and supportive care are suitable alternative treatments.1
The AAD guidelines also offer recommendations for follow-up and reducing future risk of skin cancer. After the first diagnosis of BCC, a skin cancer screening should be performed at least annually, and patients should be counseled about self-examinations and sun protection.1 Topical and oral retinoids are not recommended for the prevention of additional skin cancers, nor is dietary supplementation with selenium or beta-carotene. There also is insufficient evidence regarding the use of oral nicotinamide, celecoxib, or α-difluoromethylornithine for chemoprevention of disease.1
cSCC Guidelines
For suspected cSCCs, no single optimal biopsy technique is recommended, but repeat biopsy may be considered if the initial biopsy is insufficient for diagnosis.2 The guidelines further recommend an extensive list of elements to be included in the final pathology report (eg, lesion size, immunosuppression, depth of invasion, degree of differentiation). There is no universally recognized stratification for localized cSCC; therefore, the AAD guidelines refer to the framework provided by the NCCN. Also mentioned is the recent release of the American Joint Committee on Cancer’s staging manual,8 which includes the management of cSCC in conjunction with all SCCs of the head and neck. The Brigham and Women’s system9 was considered as an alternative classification system; however, the NCCN guidelines were chosen because they primarily provide clinical guidance for treatment of cSCC rather than provide accurate prognostication or outcome assessment.
Considerations for surgical treatment of cSCC are similar to those for BCC.2 In low-risk tumors, surgical excision with 4- to 6-mm margins to the midsubcutaneous fat or curettage with electrodessication may be considered. Mohs micrographic surgery or standard excision with attention to margin control may be considered for high-risk tumors. Nonsurgical therapies generally are not recommended as a first-line treatment, particularly in cSCC, due to possible recurrence and metastasis. When nonsurgical therapies are preferred, options may include cryosurgery or radiation therapy, with the understanding that cure rates may be lower than with surgical options. Topical therapy with imiquimod or 5-fluorouracil as well as photodynamic or laser therapy are not recommended for cSCCs.2
For patients with metastatic cSCC or locally advanced disease, multidisciplinary consultation is recommended.2 In cSCCs with regional lymph node metastases, the recommended approach includes surgical resection with possible adjuvant radiation therapy and/or systemic therapy. For inoperable disease, combination chemoradiation may be considered. Epidermal growth factor inhibitors and cisplatin may be considered in metastatic disease, although there are limited data to support their efficacy. As with BCC, all patients with cSCCs should receive supportive and palliative care to optimize quality of life.2
Recommendations for follow-up after the first diagnosis of cSCC are the same as those for BCC.2 Additionally, acitretin is the only therapy that may be beneficial in the reduction of recurrent skin cancer in patients who are solid-organ transplant recipients.
Final Thoughts
A comprehensive understanding of the management of NMSC and the evidence on which recommendations are based is critically important for optimal patient care. These guidelines are an efficient way for dermatologists and their colleagues to understand the latest evidence and recommendations. The AAD guidelines provide support for clinical decision making with standardized approaches to the diagnosis, care, and prevention of NMSC that are consistent with established practice patterns.
With few exceptions, surgical therapy is the most effective approach for the treatment of BCC and cSCC; however, the AAD guidelines include an important review on nonsurgical management options.1,2 The AAD guidelines help to highlight where data on evidence-based outcomes exist and reveal where data remain insufficient. This is illustrated by the guideline recommendations for providing additional histopathologic characteristics in the pathology reports, which will likely produce future data to enhance the prognosis and eventual treatment of patients with NMSC.1,2 Future guidelines also may include newer technologies (eg, gene expression profiling).
The guidelines do not cover the management of premalignant and in situ lesions, nor do they provide details on the management of metastatic or locally advanced disease. These topics certainly will require a similar critical review and may be addressed separately. The guidelines are identifying unanswered questions about patient care and are concurrently establishing the collection of appropriate data to answer these questions in the future.
Official guidelines often become the primary source for the measured standard of both treatment and outcomes in patient care; therefore, it is critical that dermatologists and the AAD take the lead in creating these guidelines so that we can provide our patients with the best evidenced-based comprehensive care.
The AAD guidelines emphasize the importance of considering the patient perspective in determining how to treat BCCs and cSCCs.1,2 It is important for patients to understand the available treatment options and participate in their own medical care. The AAD work group for these guidelines included patient advocates to ensure that the guidelines would promote further dialogue between physicians and their patients.
The AAD guidelines for the management of NMSC were developed by board-certified dermatologists and other experts in the field. They allow dermatologists to work with patients diagnosed with NMSC to determine the treatment option that is best for each individual patient.
- Bichakjian C, Armstrong A, Baum C, et al. Guidelines of care for the management of basal cell carcinoma. J Am Acad Dermatol. 2018;78:540-559.
- Alam M, Armstrong A, Baum C, et al. Guidelines of care for the management of cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2018;78:560-578.
- Burden of skin disease. American Academy of Dermatology website. https://www.aad.org/about/burden-of-skin-disease. Accessed April 17, 2018.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population. JAMA Dermatol. 2015;151:1081-1086.
- Muzic JG, Schmitt AR, Wright AC, et al. Incidence and trends of basal cell carcinoma and cutaneous squamous cell carcinoma: a population-based study in Olmstead County, Minnnesota, 2000-2010. Mayo Clin Proc. 2017;92:890-898.
- Bichakjian CK, Olencki T, Aasi SZ, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Basal Cell Skin Cancer. National Comprehensive Cancer Network website. https://www.nccn.org/professionals/physician_gls/pdf/nmsc.pdf. Published September 18, 2017. Accessed April 17, 2018.
- Bichakjian CK, Olencki T, Aasi SZ, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Squamous Cell Skin Cancer. National Comprehensive Cancer Network website. Published October 5, 2017. Accessed April 17, 2018.
- Amin MB, Edge SB, Greene FL, et al. AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer International Publishing; 2016.
- Jambusaria-Pahlajani A, Kanetsky PA, Karia PS, et al. Evaluation of AJCC tumor staging for cutaneous squamous cell carcinoma and a proposed alternative tumor staging system. JAMA Dermatol. 2013;149:402-410.
- Bichakjian C, Armstrong A, Baum C, et al. Guidelines of care for the management of basal cell carcinoma. J Am Acad Dermatol. 2018;78:540-559.
- Alam M, Armstrong A, Baum C, et al. Guidelines of care for the management of cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2018;78:560-578.
- Burden of skin disease. American Academy of Dermatology website. https://www.aad.org/about/burden-of-skin-disease. Accessed April 17, 2018.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population. JAMA Dermatol. 2015;151:1081-1086.
- Muzic JG, Schmitt AR, Wright AC, et al. Incidence and trends of basal cell carcinoma and cutaneous squamous cell carcinoma: a population-based study in Olmstead County, Minnnesota, 2000-2010. Mayo Clin Proc. 2017;92:890-898.
- Bichakjian CK, Olencki T, Aasi SZ, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Basal Cell Skin Cancer. National Comprehensive Cancer Network website. https://www.nccn.org/professionals/physician_gls/pdf/nmsc.pdf. Published September 18, 2017. Accessed April 17, 2018.
- Bichakjian CK, Olencki T, Aasi SZ, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Squamous Cell Skin Cancer. National Comprehensive Cancer Network website. Published October 5, 2017. Accessed April 17, 2018.
- Amin MB, Edge SB, Greene FL, et al. AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer International Publishing; 2016.
- Jambusaria-Pahlajani A, Kanetsky PA, Karia PS, et al. Evaluation of AJCC tumor staging for cutaneous squamous cell carcinoma and a proposed alternative tumor staging system. JAMA Dermatol. 2013;149:402-410.

Penile Squamous Cell Carcinoma With Urethral Extension Treated With Mohs Micrographic Surgery
Penile squamous cell carcinoma (SCC) with considerable urethral extension is uncommon and difficult to manage. It often is resistant to less invasive and nonsurgical treatments and frequently results in partial or total penectomy, which can lead to cosmetic disfigurement, functional issues, and psychological distress. We report a case of penile SCC in situ with considerable urethral extension with a focus of cells suspicious for moderately well-differentiated and invasive SCC that was treated with
Mohs micrographic surgery with distal urethrectomy and reconstruction is a valuable treatment technique for cases of SCC involving the glans penis and distal urethra. It offers equivalent or better overall cure rates compared to more radical interventions. Additionally, preservation of the penis with MMS spares patients from considerable physical and psychosocial morbidity. Our case, along with growing body of literature,1-4 calls on dermatologists and urologists to consider MMS as a treatment for penile SCC with or without urethral involvement.
Case Report
A 61-year-old man presented to the dermatology department with a pruritic lesion on the penis that had been present for 6 years. Shave biopsy demonstrated SCC in situ with a focus of cells suspicious for moderately well-differentiated and invasive SCC. Physical examination revealed an ill-defined, 2.2×1.9-cm, pink, eroded plaque involving the tip of the penis and surrounding the external urinary meatus (Figure 1). There was no palpable inguinal lymphadenopathy.
Distal penectomy and lymph node biopsy was recommended following evaluation by the urologic oncology department, but the patient declined these interventions and presented to our dermatology department (A.H.) for a second opinion. The tumor, including the invasive perineural portion, was removed using MMS several weeks after initially presenting to urologic oncology. Ventral meatotomy allowed access to the SCC in situ portion extending proximally up the pendulous urethra (Figure 2). Clear margins were obtained after the eighth stage of MMS, which required removal of 4 to 5 cm of the distal urethra (Figure 3). Reconstruction of the wound required urethral advancement, urethrostomy, and meatoplasty. A positive outcome was achieved with preservation of the length and shape of the penis as well as the cosmetic appearance of the glans penis (Figure 4). The patient was satisfied with the outcome. At 49 months’ follow-up, no evidence of local recurrence or disease progression was noted, and the distal urethrostomy remained intact and functional.


Comment
Penile SCC is a rare malignancy that represents between 0.4% and 0.6% of all malignant tumors in the United States and occurs most commonly in men aged 50 to 70 years.4 The incidence is higher in developing countries, approaching 10% of malignancies in men. It occurs most commonly on the glans penis, prepuce, and coronal sulcus, and has multiple possible appearances, including erythematous and indurated, warty and exophytic, or flat and ulcerated lesions.5 Some reports indicate that more than 40% of penile SCCs are attributable to human papilloma virus,6 while lack of circumcision, chronic inflammation, poor hygiene, balanitis xerotica obliterans, penile trauma, human immunodeficiency virus, UVA treatment of penile psoriasis, and tobacco use are known risk factors.5
Invasive penile SCC generally is treated with penectomy (partial or total), radiation therapy, or MMS; SCC in situ can be treated with topical chemotherapy, laser therapy, and wide local excision (2-cm margins) including circumcision, complete glansectomy, or MMS.5 Squamous cell carcinoma in situ with urethral involvement treated with nonsurgical therapies is associated with higher recurrence rates, ultimately necessitating more aggressive treatments, most commonly partial penectomy.7 The high local recurrence rate of SCC in situ with urethral involvement treated with nonsurgical therapies reflects the fact that determining the presence of urethral extension is difficult and, if present, is inherently inaccessible to these local therapies because the urethra is not an outward-facing tissue surface; MMS represents one possible solution to these issues.
Across all treatment modalities, the most prognostic factor of cancer-specific survival in patients with penile SCC is pelvic lymph node involvement. Some reports cite 5-year survival rates as low as 0% in the setting of pelvic lymph node involvement,5 whereas others had cited rates of 29% to 40%4; 5-year survival rates of higher than 85% have been reported in node-negative patients.4 Recurrence rates vary widely by treatment modality, ranging from less than 10% with partial penectomy and long-term follow-up8 and up to 50% within 2 years with penile-preserving approaches (eg, topical chemotherapy, laser therapy, radiotherapy).5 Multiple case series of penile cancer (the most common of which was SCC/SCC in situ) treated with MMS report comparable and at times superior survival and recurrence data (Table).1-4 Slightly higher recurrences of penile SCC treated with MMS compared to penectomy have been reported, along with considerably higher recurrence rates compared to nonpenile cutaneous SCC treated with MMS (reported to be less than 3%).4 The elastic and expansile nature of penile tissue may lead to distortion from swelling/local anesthesia when taking individual Mohs layers. Additionally, as a large percentage of penile SCCs are attributable to human papillomavirus, difficulty in detecting human papilloma virus–infected cells (which may have oncogenic potential) with the naked eye or histologically with typical staining techniques may help explain the higher recurrence rate of penile SCC treated with MMS compared to penectomy. Despite the higher recurrence rates, survival is comparable or higher in cases treated with MMS (Table).
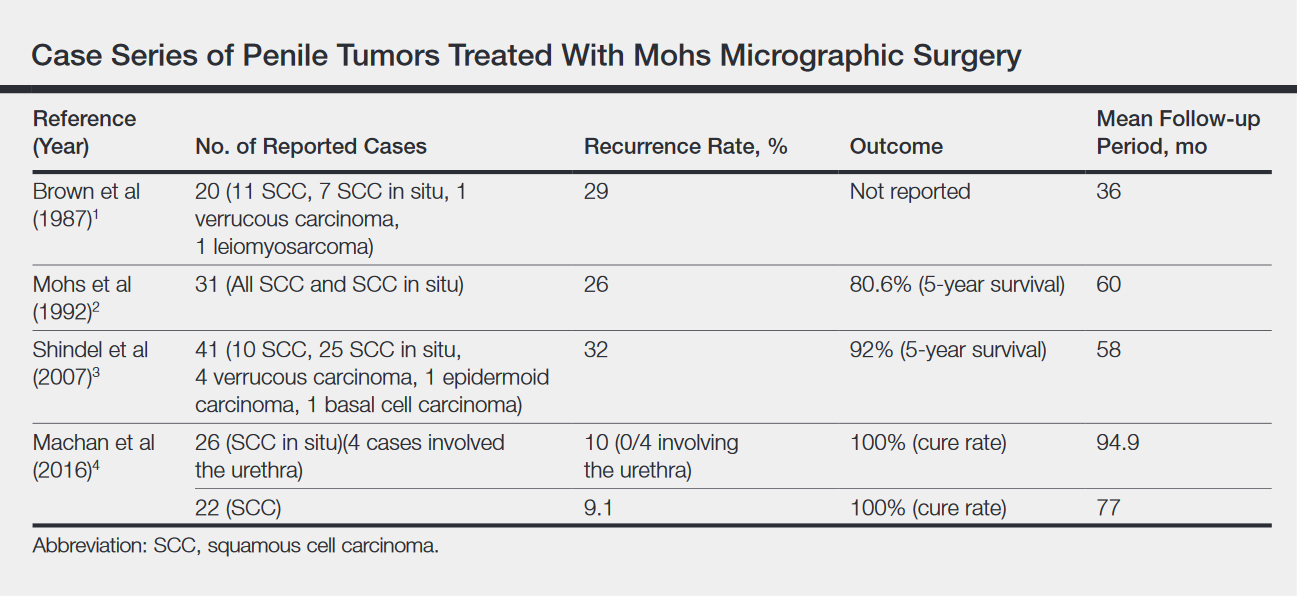
Partial penectomy also has a negative impact on health-related quality of life. Kieffer et al9 compared the impact of penile-sparing surgery (PSS)(including MMS) versus partial or total penectomy on sexual function and health-related quality of life in 90 patients with penile cancer. Although the association between the extent of surgery (partial penectomy/total penectomy/PSS) surgery type and extent and most outcome measures was not statistically significant, partial penectomy was associated with significantly more problems with orgasm (P=.031), concerns about appearance (P=.008), interference in daily life (P=.032), and urinary function (P<.0001) when compared to patients treated with PSS.9 Although this study included only laser/local excision with or without circumcision or glans penis amputation with or without reconstruction as PSSs and did not explicitly include MMS, MMS is clearly a tissue-sparing technique and the study results are generaliz
Conclusion
Penile SCC with considerable urethral extension is uncommon, difficult to manage, and often is resistant to less invasive and nonsurgical treatments. As a result, partial or total penectomy is sometimes necessary. Such cases benefit from MMS with distal urethrectomy and reconstruction because MMS provides equivalent or better overall cure rates compared to more radical interventions.1-4 Importantly, preservation of the penis with MMS can spare patients considerable physical and psychosocial morbidity. Partial penectomy is associated with more health-related quality-of-life problems with orgasm, concerns about appearance, interference in daily life, and urinary function compared to PSSs such as MMS.9 This case, and a growing body of literature, are a call to dermatologists and urologists to consider MMS as a treatment for penile SCC, even with involvement of the urethra.
- Brown MD, Zachary CB, Grekin RC, et al. Penile tumors: their management by Mohs micrographic surgery. J Dermatol Surg Oncol. 1987;13:1163-1167.
- Mohs FE, Snow SN, Larson PO. Mohs micrographic surgery for penile tumors. Urol Clin North Am. 1992;19:291-304.
- Shindel AW, Mann MW, Lev RY, et al. Mohs micrographic surgery for penile cancer: management and long-term followup. J Urol. 2007;178:1980-1985.
- Machan M, Brodland D, Zitelli J. Penile squamous cell carcinoma: penis-preserving treatment with Mohs micrographic surgery. Dermatol Surg. 2016;42:936-944.
- Spiess PE, Horenblas S, Pagliaro LC, et al. Current concepts in penile cancer. J Natl Compr Canc Netw. 2013;11:617-624.
- Hernandez BY, Barnholtz-Sloan J, German RR, et al. Burden of invasive squamous cell carcinoma of the penis in the United States, 1998-2003. Cancer. 2008;113(10 suppl):2883-2891.
- Nash PA, Bihrle R, Gleason PE, et al. Mohs micrographic surgery and distal urethrectomy with immediate urethral reconstruction for glanular carcinoma in situ with significant urethral extension. Urology. 1996;47:108-110.
- Djordjevic ML, Palminteri E, Martins F. Male genital reconstruction for the penile cancer survivor. Curr Opin Urol. 2014;24:427-433.
- Kieffer JM, Djajadiningrat RS, van Muilekom EA, et al. Quality of life for patients treated for penile cancer. J Urol. 2014;192:1105-1110.
Penile squamous cell carcinoma (SCC) with considerable urethral extension is uncommon and difficult to manage. It often is resistant to less invasive and nonsurgical treatments and frequently results in partial or total penectomy, which can lead to cosmetic disfigurement, functional issues, and psychological distress. We report a case of penile SCC in situ with considerable urethral extension with a focus of cells suspicious for moderately well-differentiated and invasive SCC that was treated with
Mohs micrographic surgery with distal urethrectomy and reconstruction is a valuable treatment technique for cases of SCC involving the glans penis and distal urethra. It offers equivalent or better overall cure rates compared to more radical interventions. Additionally, preservation of the penis with MMS spares patients from considerable physical and psychosocial morbidity. Our case, along with growing body of literature,1-4 calls on dermatologists and urologists to consider MMS as a treatment for penile SCC with or without urethral involvement.
Case Report
A 61-year-old man presented to the dermatology department with a pruritic lesion on the penis that had been present for 6 years. Shave biopsy demonstrated SCC in situ with a focus of cells suspicious for moderately well-differentiated and invasive SCC. Physical examination revealed an ill-defined, 2.2×1.9-cm, pink, eroded plaque involving the tip of the penis and surrounding the external urinary meatus (Figure 1). There was no palpable inguinal lymphadenopathy.
Distal penectomy and lymph node biopsy was recommended following evaluation by the urologic oncology department, but the patient declined these interventions and presented to our dermatology department (A.H.) for a second opinion. The tumor, including the invasive perineural portion, was removed using MMS several weeks after initially presenting to urologic oncology. Ventral meatotomy allowed access to the SCC in situ portion extending proximally up the pendulous urethra (Figure 2). Clear margins were obtained after the eighth stage of MMS, which required removal of 4 to 5 cm of the distal urethra (Figure 3). Reconstruction of the wound required urethral advancement, urethrostomy, and meatoplasty. A positive outcome was achieved with preservation of the length and shape of the penis as well as the cosmetic appearance of the glans penis (Figure 4). The patient was satisfied with the outcome. At 49 months’ follow-up, no evidence of local recurrence or disease progression was noted, and the distal urethrostomy remained intact and functional.
Comment
Penile SCC is a rare malignancy that represents between 0.4% and 0.6% of all malignant tumors in the United States and occurs most commonly in men aged 50 to 70 years.4 The incidence is higher in developing countries, approaching 10% of malignancies in men. It occurs most commonly on the glans penis, prepuce, and coronal sulcus, and has multiple possible appearances, including erythematous and indurated, warty and exophytic, or flat and ulcerated lesions.5 Some reports indicate that more than 40% of penile SCCs are attributable to human papilloma virus,6 while lack of circumcision, chronic inflammation, poor hygiene, balanitis xerotica obliterans, penile trauma, human immunodeficiency virus, UVA treatment of penile psoriasis, and tobacco use are known risk factors.5
Invasive penile SCC generally is treated with penectomy (partial or total), radiation therapy, or MMS; SCC in situ can be treated with topical chemotherapy, laser therapy, and wide local excision (2-cm margins) including circumcision, complete glansectomy, or MMS.5 Squamous cell carcinoma in situ with urethral involvement treated with nonsurgical therapies is associated with higher recurrence rates, ultimately necessitating more aggressive treatments, most commonly partial penectomy.7 The high local recurrence rate of SCC in situ with urethral involvement treated with nonsurgical therapies reflects the fact that determining the presence of urethral extension is difficult and, if present, is inherently inaccessible to these local therapies because the urethra is not an outward-facing tissue surface; MMS represents one possible solution to these issues.
Across all treatment modalities, the most prognostic factor of cancer-specific survival in patients with penile SCC is pelvic lymph node involvement. Some reports cite 5-year survival rates as low as 0% in the setting of pelvic lymph node involvement,5 whereas others had cited rates of 29% to 40%4; 5-year survival rates of higher than 85% have been reported in node-negative patients.4 Recurrence rates vary widely by treatment modality, ranging from less than 10% with partial penectomy and long-term follow-up8 and up to 50% within 2 years with penile-preserving approaches (eg, topical chemotherapy, laser therapy, radiotherapy).5 Multiple case series of penile cancer (the most common of which was SCC/SCC in situ) treated with MMS report comparable and at times superior survival and recurrence data (Table).1-4 Slightly higher recurrences of penile SCC treated with MMS compared to penectomy have been reported, along with considerably higher recurrence rates compared to nonpenile cutaneous SCC treated with MMS (reported to be less than 3%).4 The elastic and expansile nature of penile tissue may lead to distortion from swelling/local anesthesia when taking individual Mohs layers. Additionally, as a large percentage of penile SCCs are attributable to human papillomavirus, difficulty in detecting human papilloma virus–infected cells (which may have oncogenic potential) with the naked eye or histologically with typical staining techniques may help explain the higher recurrence rate of penile SCC treated with MMS compared to penectomy. Despite the higher recurrence rates, survival is comparable or higher in cases treated with MMS (Table).
Partial penectomy also has a negative impact on health-related quality of life. Kieffer et al9 compared the impact of penile-sparing surgery (PSS)(including MMS) versus partial or total penectomy on sexual function and health-related quality of life in 90 patients with penile cancer. Although the association between the extent of surgery (partial penectomy/total penectomy/PSS) surgery type and extent and most outcome measures was not statistically significant, partial penectomy was associated with significantly more problems with orgasm (P=.031), concerns about appearance (P=.008), interference in daily life (P=.032), and urinary function (P<.0001) when compared to patients treated with PSS.9 Although this study included only laser/local excision with or without circumcision or glans penis amputation with or without reconstruction as PSSs and did not explicitly include MMS, MMS is clearly a tissue-sparing technique and the study results are generaliz
Conclusion
Penile SCC with considerable urethral extension is uncommon, difficult to manage, and often is resistant to less invasive and nonsurgical treatments. As a result, partial or total penectomy is sometimes necessary. Such cases benefit from MMS with distal urethrectomy and reconstruction because MMS provides equivalent or better overall cure rates compared to more radical interventions.1-4 Importantly, preservation of the penis with MMS can spare patients considerable physical and psychosocial morbidity. Partial penectomy is associated with more health-related quality-of-life problems with orgasm, concerns about appearance, interference in daily life, and urinary function compared to PSSs such as MMS.9 This case, and a growing body of literature, are a call to dermatologists and urologists to consider MMS as a treatment for penile SCC, even with involvement of the urethra.
Penile squamous cell carcinoma (SCC) with considerable urethral extension is uncommon and difficult to manage. It often is resistant to less invasive and nonsurgical treatments and frequently results in partial or total penectomy, which can lead to cosmetic disfigurement, functional issues, and psychological distress. We report a case of penile SCC in situ with considerable urethral extension with a focus of cells suspicious for moderately well-differentiated and invasive SCC that was treated with
Mohs micrographic surgery with distal urethrectomy and reconstruction is a valuable treatment technique for cases of SCC involving the glans penis and distal urethra. It offers equivalent or better overall cure rates compared to more radical interventions. Additionally, preservation of the penis with MMS spares patients from considerable physical and psychosocial morbidity. Our case, along with growing body of literature,1-4 calls on dermatologists and urologists to consider MMS as a treatment for penile SCC with or without urethral involvement.
Case Report
A 61-year-old man presented to the dermatology department with a pruritic lesion on the penis that had been present for 6 years. Shave biopsy demonstrated SCC in situ with a focus of cells suspicious for moderately well-differentiated and invasive SCC. Physical examination revealed an ill-defined, 2.2×1.9-cm, pink, eroded plaque involving the tip of the penis and surrounding the external urinary meatus (Figure 1). There was no palpable inguinal lymphadenopathy.
Distal penectomy and lymph node biopsy was recommended following evaluation by the urologic oncology department, but the patient declined these interventions and presented to our dermatology department (A.H.) for a second opinion. The tumor, including the invasive perineural portion, was removed using MMS several weeks after initially presenting to urologic oncology. Ventral meatotomy allowed access to the SCC in situ portion extending proximally up the pendulous urethra (Figure 2). Clear margins were obtained after the eighth stage of MMS, which required removal of 4 to 5 cm of the distal urethra (Figure 3). Reconstruction of the wound required urethral advancement, urethrostomy, and meatoplasty. A positive outcome was achieved with preservation of the length and shape of the penis as well as the cosmetic appearance of the glans penis (Figure 4). The patient was satisfied with the outcome. At 49 months’ follow-up, no evidence of local recurrence or disease progression was noted, and the distal urethrostomy remained intact and functional.
Comment
Penile SCC is a rare malignancy that represents between 0.4% and 0.6% of all malignant tumors in the United States and occurs most commonly in men aged 50 to 70 years.4 The incidence is higher in developing countries, approaching 10% of malignancies in men. It occurs most commonly on the glans penis, prepuce, and coronal sulcus, and has multiple possible appearances, including erythematous and indurated, warty and exophytic, or flat and ulcerated lesions.5 Some reports indicate that more than 40% of penile SCCs are attributable to human papilloma virus,6 while lack of circumcision, chronic inflammation, poor hygiene, balanitis xerotica obliterans, penile trauma, human immunodeficiency virus, UVA treatment of penile psoriasis, and tobacco use are known risk factors.5
Invasive penile SCC generally is treated with penectomy (partial or total), radiation therapy, or MMS; SCC in situ can be treated with topical chemotherapy, laser therapy, and wide local excision (2-cm margins) including circumcision, complete glansectomy, or MMS.5 Squamous cell carcinoma in situ with urethral involvement treated with nonsurgical therapies is associated with higher recurrence rates, ultimately necessitating more aggressive treatments, most commonly partial penectomy.7 The high local recurrence rate of SCC in situ with urethral involvement treated with nonsurgical therapies reflects the fact that determining the presence of urethral extension is difficult and, if present, is inherently inaccessible to these local therapies because the urethra is not an outward-facing tissue surface; MMS represents one possible solution to these issues.
Across all treatment modalities, the most prognostic factor of cancer-specific survival in patients with penile SCC is pelvic lymph node involvement. Some reports cite 5-year survival rates as low as 0% in the setting of pelvic lymph node involvement,5 whereas others had cited rates of 29% to 40%4; 5-year survival rates of higher than 85% have been reported in node-negative patients.4 Recurrence rates vary widely by treatment modality, ranging from less than 10% with partial penectomy and long-term follow-up8 and up to 50% within 2 years with penile-preserving approaches (eg, topical chemotherapy, laser therapy, radiotherapy).5 Multiple case series of penile cancer (the most common of which was SCC/SCC in situ) treated with MMS report comparable and at times superior survival and recurrence data (Table).1-4 Slightly higher recurrences of penile SCC treated with MMS compared to penectomy have been reported, along with considerably higher recurrence rates compared to nonpenile cutaneous SCC treated with MMS (reported to be less than 3%).4 The elastic and expansile nature of penile tissue may lead to distortion from swelling/local anesthesia when taking individual Mohs layers. Additionally, as a large percentage of penile SCCs are attributable to human papillomavirus, difficulty in detecting human papilloma virus–infected cells (which may have oncogenic potential) with the naked eye or histologically with typical staining techniques may help explain the higher recurrence rate of penile SCC treated with MMS compared to penectomy. Despite the higher recurrence rates, survival is comparable or higher in cases treated with MMS (Table).
Partial penectomy also has a negative impact on health-related quality of life. Kieffer et al9 compared the impact of penile-sparing surgery (PSS)(including MMS) versus partial or total penectomy on sexual function and health-related quality of life in 90 patients with penile cancer. Although the association between the extent of surgery (partial penectomy/total penectomy/PSS) surgery type and extent and most outcome measures was not statistically significant, partial penectomy was associated with significantly more problems with orgasm (P=.031), concerns about appearance (P=.008), interference in daily life (P=.032), and urinary function (P<.0001) when compared to patients treated with PSS.9 Although this study included only laser/local excision with or without circumcision or glans penis amputation with or without reconstruction as PSSs and did not explicitly include MMS, MMS is clearly a tissue-sparing technique and the study results are generaliz
Conclusion
Penile SCC with considerable urethral extension is uncommon, difficult to manage, and often is resistant to less invasive and nonsurgical treatments. As a result, partial or total penectomy is sometimes necessary. Such cases benefit from MMS with distal urethrectomy and reconstruction because MMS provides equivalent or better overall cure rates compared to more radical interventions.1-4 Importantly, preservation of the penis with MMS can spare patients considerable physical and psychosocial morbidity. Partial penectomy is associated with more health-related quality-of-life problems with orgasm, concerns about appearance, interference in daily life, and urinary function compared to PSSs such as MMS.9 This case, and a growing body of literature, are a call to dermatologists and urologists to consider MMS as a treatment for penile SCC, even with involvement of the urethra.
- Brown MD, Zachary CB, Grekin RC, et al. Penile tumors: their management by Mohs micrographic surgery. J Dermatol Surg Oncol. 1987;13:1163-1167.
- Mohs FE, Snow SN, Larson PO. Mohs micrographic surgery for penile tumors. Urol Clin North Am. 1992;19:291-304.
- Shindel AW, Mann MW, Lev RY, et al. Mohs micrographic surgery for penile cancer: management and long-term followup. J Urol. 2007;178:1980-1985.
- Machan M, Brodland D, Zitelli J. Penile squamous cell carcinoma: penis-preserving treatment with Mohs micrographic surgery. Dermatol Surg. 2016;42:936-944.
- Spiess PE, Horenblas S, Pagliaro LC, et al. Current concepts in penile cancer. J Natl Compr Canc Netw. 2013;11:617-624.
- Hernandez BY, Barnholtz-Sloan J, German RR, et al. Burden of invasive squamous cell carcinoma of the penis in the United States, 1998-2003. Cancer. 2008;113(10 suppl):2883-2891.
- Nash PA, Bihrle R, Gleason PE, et al. Mohs micrographic surgery and distal urethrectomy with immediate urethral reconstruction for glanular carcinoma in situ with significant urethral extension. Urology. 1996;47:108-110.
- Djordjevic ML, Palminteri E, Martins F. Male genital reconstruction for the penile cancer survivor. Curr Opin Urol. 2014;24:427-433.
- Kieffer JM, Djajadiningrat RS, van Muilekom EA, et al. Quality of life for patients treated for penile cancer. J Urol. 2014;192:1105-1110.
- Brown MD, Zachary CB, Grekin RC, et al. Penile tumors: their management by Mohs micrographic surgery. J Dermatol Surg Oncol. 1987;13:1163-1167.
- Mohs FE, Snow SN, Larson PO. Mohs micrographic surgery for penile tumors. Urol Clin North Am. 1992;19:291-304.
- Shindel AW, Mann MW, Lev RY, et al. Mohs micrographic surgery for penile cancer: management and long-term followup. J Urol. 2007;178:1980-1985.
- Machan M, Brodland D, Zitelli J. Penile squamous cell carcinoma: penis-preserving treatment with Mohs micrographic surgery. Dermatol Surg. 2016;42:936-944.
- Spiess PE, Horenblas S, Pagliaro LC, et al. Current concepts in penile cancer. J Natl Compr Canc Netw. 2013;11:617-624.
- Hernandez BY, Barnholtz-Sloan J, German RR, et al. Burden of invasive squamous cell carcinoma of the penis in the United States, 1998-2003. Cancer. 2008;113(10 suppl):2883-2891.
- Nash PA, Bihrle R, Gleason PE, et al. Mohs micrographic surgery and distal urethrectomy with immediate urethral reconstruction for glanular carcinoma in situ with significant urethral extension. Urology. 1996;47:108-110.
- Djordjevic ML, Palminteri E, Martins F. Male genital reconstruction for the penile cancer survivor. Curr Opin Urol. 2014;24:427-433.
- Kieffer JM, Djajadiningrat RS, van Muilekom EA, et al. Quality of life for patients treated for penile cancer. J Urol. 2014;192:1105-1110.
Resident Pearl
- Penile squamous cell carcinoma (SCC) often is treated with partial or total penectomy, especially when there is urethral extension. Mohs micrographic surgery for penile SCC results in equivalent or better overall cure rates and decreases morbidity.

Merkel Cell Carcinoma: Presentation, Pathogenesis, and Spontaneous Regression


Intralymphatic Histiocytosis Treated With Intralesional Triamcinolone Acetonide and Pressure Bandage
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
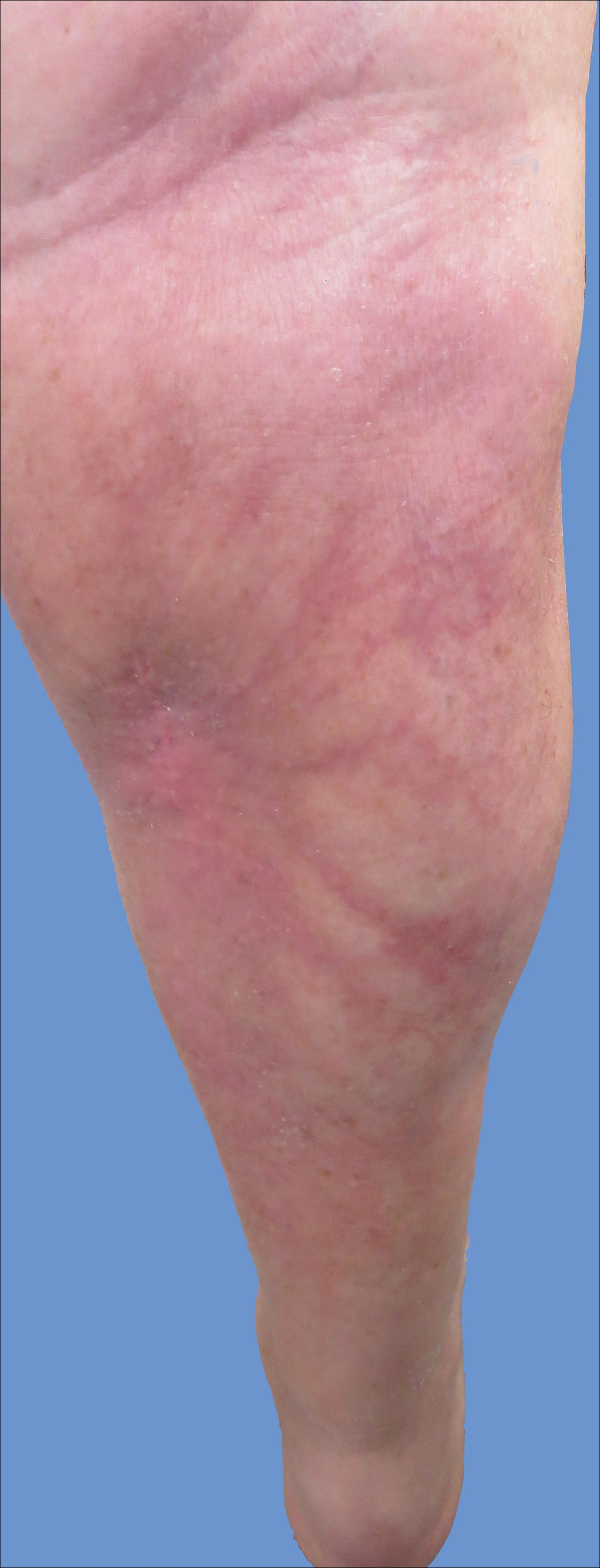
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
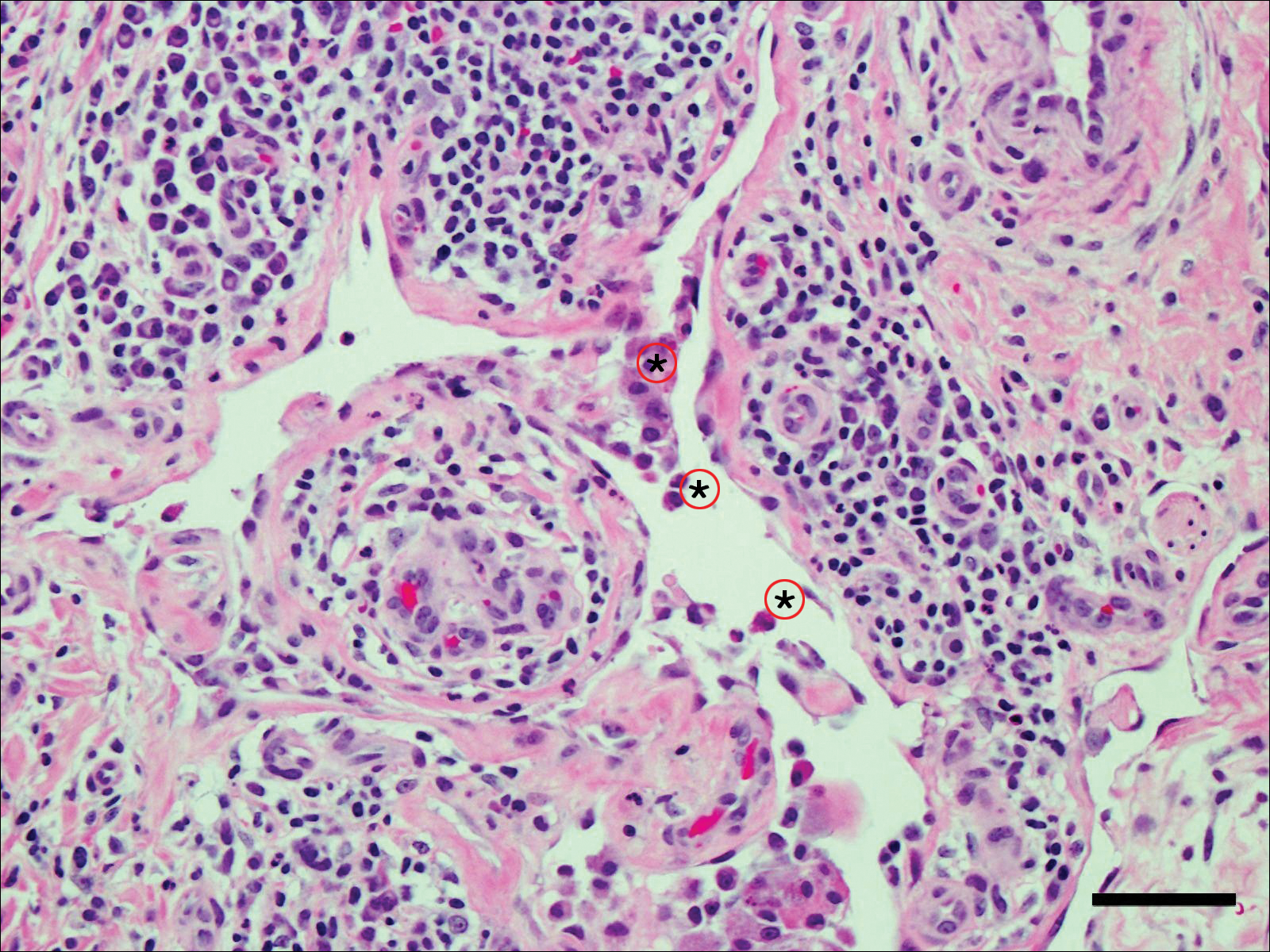
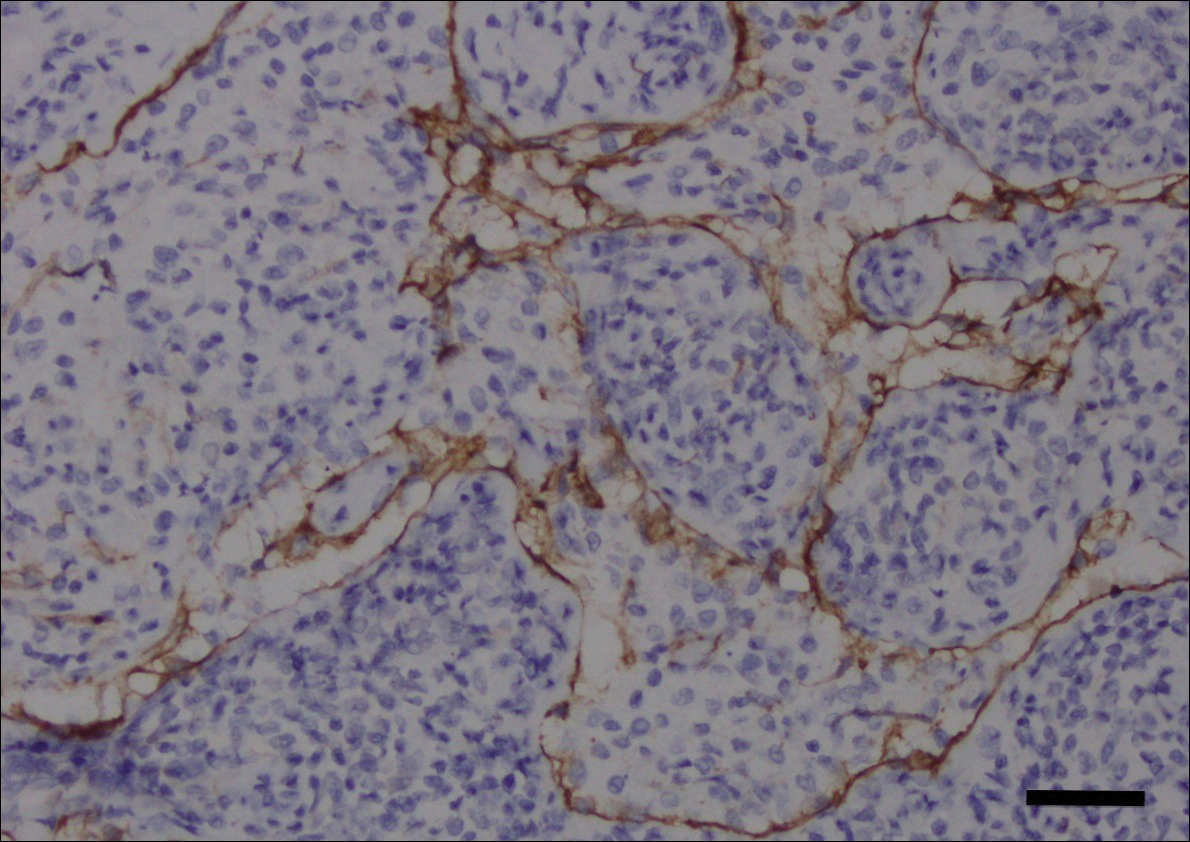
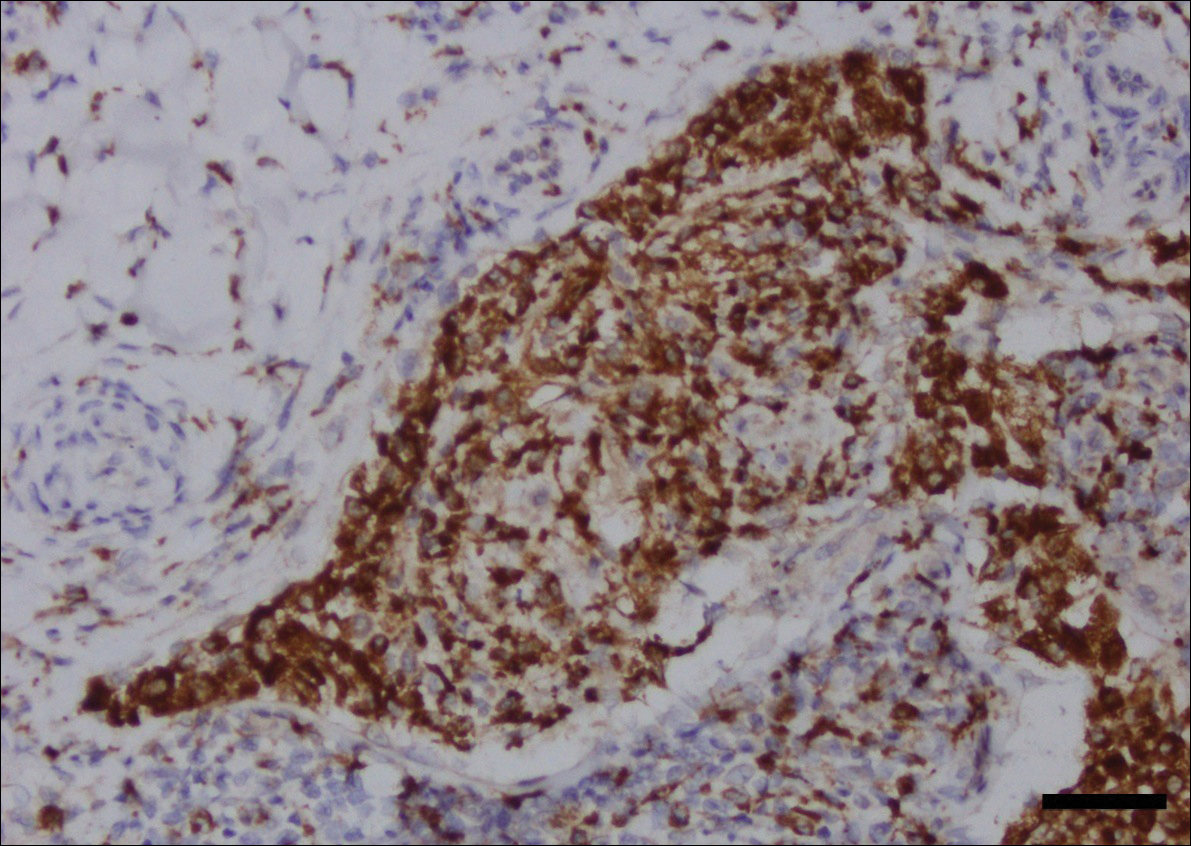
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
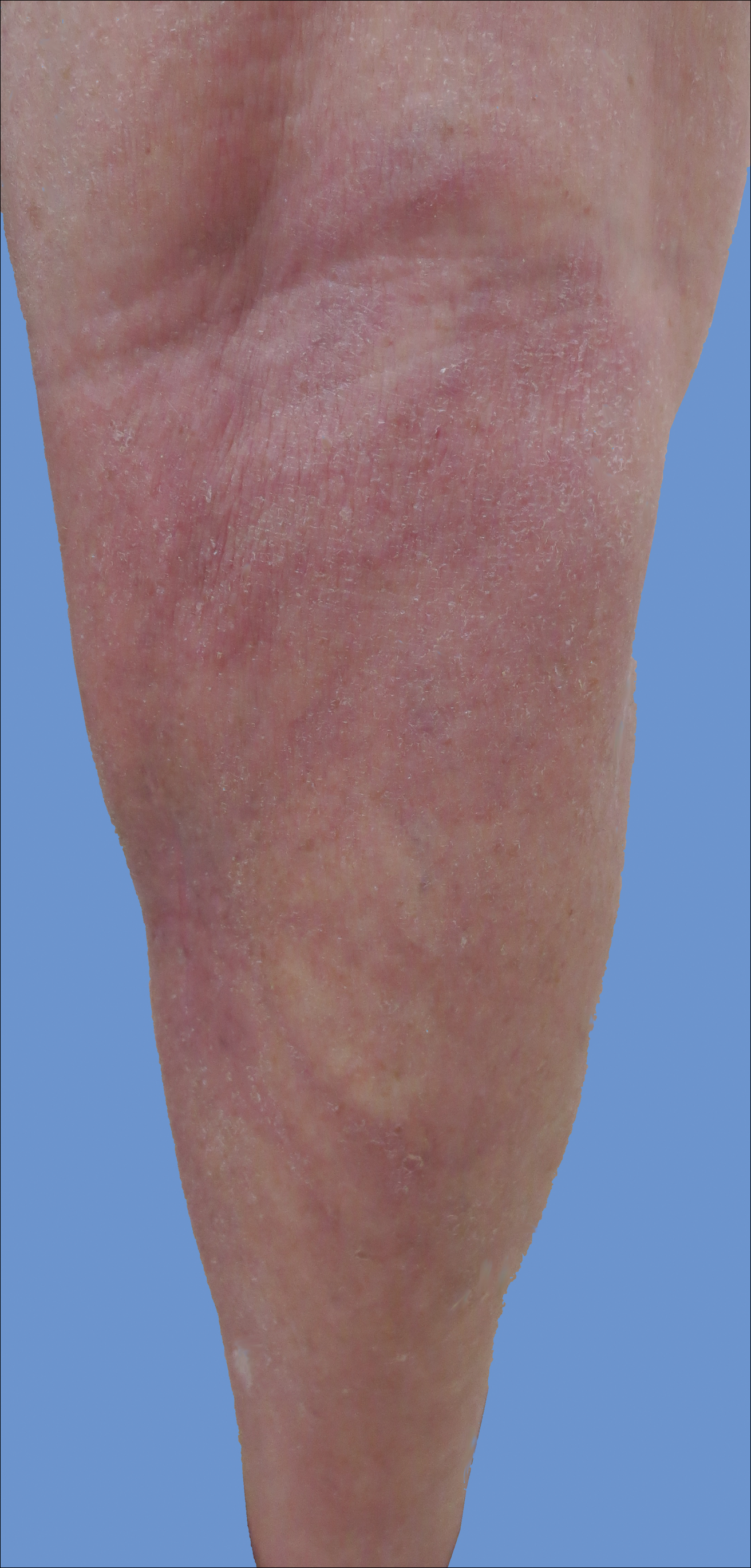
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
Practice Points
- Intralymphatic histiocytosis is a rare disorder often associated with rheumatic arthritis and joint prostheses.
- The diagnosis is made by histopathology as well as D2-40 and CD68 immunostaining.
- While there is no gold standard of treatment for intralymphatic histiocytosis, intralesional triamcinolone proved efficacious in this case with prolonged results.
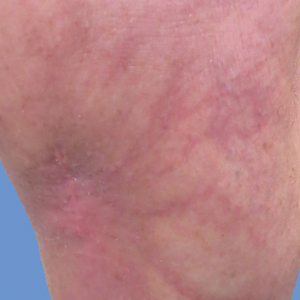
Climate Change and Skin Disease
The term climate refers to the average weather conditions of a specific geographic location measured over several decades.1 While a certain degree of variation in the Earth’s climate is expected, a persistent warming or cooling trend is not. The factors driving the Earth’s warming remain difficult to prove.2 We know the Earth previously has undergone dramatic climate changes and that natural factors driving these changes are varied (eg, the relationship between the Earth and the Sun, volcanic eruptions, solar irradiance).1,3 These factors ideally change over protracted periods of time in a way that allows organisms to adapt to new environments.
Anthropogenic climate change refers to human-caused climate change. This is thought to be a major driving factor in the Earth’s recent warming trend, partly due to the rapidity of warming in recent years.3 According to climate scientists, the Earth’s temperature has risen 4°C to 7°C over the past 5000 years, but it has risen 0.7°C in just the past 100 years alone.4 Greenhouse gases such as carbon dioxide are emitted by various natural processes and human activities and play a central role in current warming because they trap solar heat and increase ambient temperature.3
In a recent edition of the commonly cited textbook Dermatology, Bolognia et al5 referenced climate change only once in a figure legend regarding the expansion of dengue fever in the Americas. However, climate change may have the potential to cause outright skin disease epidemics worldwide, and the Climate Change Committee of the International Society of Dermatology has called upon dermatologists across the globe to help raise awareness of this issue.6
Much of the literature regarding the effects of climate change on human health focuses on insect-borne diseases, but over the past decade other areas of impact also have been investigated, such as increases in airborne diseases, zoonoses, newly endemic saprophytic and dimorphic fungal infections, fecal-oral diseases, and severe allergic disease.7,8 It is postulated that climate change leads to region-specific increases in human disease because it creates newly favorable habitats for infectious agents, their vectors. and their reservoirs, allowing expansion of their ranges and access to immunologically naïve populations.9 Furthermore, extreme weather events such as heat waves, hurricanes, and flooding, which are expected to increase in frequency as a result of climate change, have all been linked to infectious disease outbreaks.10
Lyme Disease
In the past 20 years, Lyme disease incidence has tripled in the United States.11 It has been hypothesized that the increase may be occurring as a result of the expanding geographic distribution of the Ixodes tick and its mammalian hosts (eg, white-tailed deer) under the influence of climate change.12 Lyme disease is a multisystem disease affecting the skin, joints, heart, and nervous system. Its most characteristic manifestation is cutaneous in the form of erythema migrans. Dermatologists may be called upon to play an increasingly important role in early detection and treatment of this potentially chronic and debilitating condition.
Arboviruses
Arboviruses are transmitted by arthropods and are an important category of climate change–related diseases due to the expansion of the mosquito habitat worldwide. The vectorial capacity for the transmission of dengue fever has increased worldwide by 9.4% via Aedes aegypti and 11.1% via Aedes albopictus since 1950.13 Dengue fever, also known as breakbone fever, presents with intense joint pain, fevers, headaches, and a transient morbilliform rash that desquamates with defervescence and in some cases will incite hemorrhagic skin lesions.14 Dengue fever previously was considered to be a tropical disease but locally acquired cases have been reported in the United States, including Texas, Hawaii, and Florida.15,16
Reports of local transmission of chikungunya, another arbovirus transmitted by A albopictus and A aegypti mosquitoes in Florida, the US Virgin Islands, and Puerto Rico, began in 2014.17 A higher prevalence of these diseases within the United States also may be related to increased globalization, with US travelers returning from endemic regions with infections. Prior to 2014, transmission occurred in traditional endemic regions, primarily in Asia, Africa, or island nations in the Indian Ocean. Like dengue fever, chikungunya causes high fevers, cutaneous manifestations (eg, urticarial papules, morbilliform eruption, hypermelanosis, intertriginous lesions, lymphedema),17 and intense joint pain. Unlike dengue fever, however, joint involvement can be chronic, erosive, and debilitating.
Lastly, New World leishmaniasis, an arboviral disease characterized by mucocutaneous ulcers and transmitted by phlebotomine sand flies, has been acquired locally in Oklahoma and Texas when it was previously considered to be endemic to Mexico and Central and South America.14,18 The habitats of New World Leishmania species are expected to expand northward, with an ecological niche model predicting that they reach southern Canada by the year 2080 due to the expanding habitats of sand fly and rodent vectors.19
Fungal Infections
In the Pacific Northwest, there have been reports of newly endemic Cryptococcus gattii and Coccidioides immitis, both of which previously had been confined to the southwestern United States.8 Endemic ranges of these mycotic pathogens may be expanding for a variety of reasons, with climate change creating new regions conducive to the colonization of these species.8,20,21Coccidioides immitis is a soil-dwelling fungus that usually presents with primary pulmonary disease that can disseminate acutely or even months later. Prompt recognition of disseminated disease may allow life-saving therapy to be initiated. Cryptococcus gattii is a fungus with multiple niches, including oil, trees, and birds.20 This fungus also is acquired via inhalation, with dissemination occurring most commonly in immunosuppressed patients to the central nervous system, bone, and skin. Primary or secondary infection with both of these fungi may present with cutaneous manifestations presenting as polymorphous lesions, including umbilicated or ulcerated papules, indurated nodules, and acneiform pustules.
Final Thoughts
Awareness of the shifting habitats of microorganisms and vectors locally is important in order for clinicians to make correct diagnoses in a timely fashion. Regional or endemic diseases are presenting outside their traditional boundaries due to changing habitats of microbes and vectors and may be easily overlooked, resulting in a delayed diagnosis. Being prepared to diagnose diseases with increasing incidence secondary to climate change and discussing this with patients is an important physician obligation, but it is not the only one. We cannot effectively advocate for the health of patients and the community while ignoring the destruction of the environment. Our additional responsibility is straightforward—being advocates for good stewardship of the Earth’s resources now on both a personal and a policy level.22
- Climate Central. Global Weirdness: Severe Storms, Deadly Heat Waves, Relentless Drought, Rising Seas, and the Weather of the Future. New York, NY: Pantheon Books; 2012.
- Cook J, Nuccitelli D, Green SA, et al. Quantifying the consensus on anthropogenic global warming in the scientific literature. Environ Res Lett. 2013;8:024024.
- A blanket around the Earth. NASA Climate website. https://climate.nasa.gov/causes/. Accessed February 5, 2018.
- How is today’s warming different from the past? NASA Earth Observatory website. https://earthobservatory.nasa.gov/Features/GlobalWarming/page3.php. Accessed February 5, 2018.
- Mancini AJ, Shani-Adir A, Sidbury R. Other viral diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2017:1425-1446.
- Andersen LK, Davis MDP. A wake-up call to dermatologists—climate change affects the skin. Int J Dermatol. 2017;56:E198-E199.
- Liang L, Gong P. Climate change and human infectious diseases: a synthesis of research findings from global and spatio-temporal perspectives [published online March 23, 2017]. Environ Int. 2017;103:99-108.
- Lockhart SR, McCotter OZ, Chiller TM. Emerging fungal infections in the Pacific Northwest: the unrecognized burden and geographic range of Cryptococcus gattii and Coccidioides immitis. Microbiol Spectr. 2016;4. doi:10.1128/microbiolspec.EI10-0016-2016.
- Kilpatrick AM, Randolph SE. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet. 2012;380:1946-1955.
- McMichael AJ. Extreme weather events and infectious disease outbreaks. Virulence. 2015;6:543-547.
- Lyme disease graphs. CDC website. https://www.cdc.gov/lyme/stats/graphs.html. Updated November 1, 2017. Accessed April 12, 2018.
- Stone BL, Tourand Y, Brissette CA. Brave new worlds: the expanding universe of Lyme disease. Vector Borne Zoonotic Dis. 2017;17:619-629.
- Watts N, Amann M, Ayeb-Karlsson S, et al. The Lancet Countdown on health and climate change: from 25 years of inaction to a global transformation for public health [published online October 30, 2017]. Lancet. doi:10.1016/S0140-6736(17)32464-9.
- Nawas ZY, Tong Y, Kollipara R, et al. Emerging infectious diseases with cutaneous manifestations: viral and bacterial infections. J Am Acad Dermatol. 2016;75:1-16.
- Kaffenberger BH, Shetlar D, Norton SA, et al. The effect of climate change on skin disease in North America. J Am Acad Dermatol. 2017;76:140-147.
- Dengue. CDC website. https://www.cdc.gov/dengue/epidemiology/index.html. Updated June 9, 2014. Accessed April 3, 2018.
- Chikungunya virus in the United States. CDC website. https://www.cdc.gov/chikungunya/geo/united-states.html. Updated October 30, 2017. Accessed April 4, 2018.
- Clarke CF, Bradley KK, Wright JH, et al. Emergence of autochthonous cutaneous leishmaniasis in northeastern Texas and southeastern Oklahoma. Am J Trop Med Hyg. 2013;88:157-61.
- González C, Wang O, Strutz SE, et al. Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl Trop Dis. 2010;4:E585.
- Chang CC, Chen SC. Colliding epidemics and the rise of cryptococcosis. J Fungi (Basel). 2015;2. doi: 10.3390/jof2010001.
- Marsden-Haug N, Goldoft M, Ralston C, et al. Coccidioidomycosis acquired in Washington state. Clin Infect Dis. 2013;56:847-850.
- Rosenbach M. Climate change & dermatology: what can you do? Paper presented at: American Academy of Dermatology Annual Meeting; March 3-7, 2017; Orlando, FL.
The term climate refers to the average weather conditions of a specific geographic location measured over several decades.1 While a certain degree of variation in the Earth’s climate is expected, a persistent warming or cooling trend is not. The factors driving the Earth’s warming remain difficult to prove.2 We know the Earth previously has undergone dramatic climate changes and that natural factors driving these changes are varied (eg, the relationship between the Earth and the Sun, volcanic eruptions, solar irradiance).1,3 These factors ideally change over protracted periods of time in a way that allows organisms to adapt to new environments.
Anthropogenic climate change refers to human-caused climate change. This is thought to be a major driving factor in the Earth’s recent warming trend, partly due to the rapidity of warming in recent years.3 According to climate scientists, the Earth’s temperature has risen 4°C to 7°C over the past 5000 years, but it has risen 0.7°C in just the past 100 years alone.4 Greenhouse gases such as carbon dioxide are emitted by various natural processes and human activities and play a central role in current warming because they trap solar heat and increase ambient temperature.3
In a recent edition of the commonly cited textbook Dermatology, Bolognia et al5 referenced climate change only once in a figure legend regarding the expansion of dengue fever in the Americas. However, climate change may have the potential to cause outright skin disease epidemics worldwide, and the Climate Change Committee of the International Society of Dermatology has called upon dermatologists across the globe to help raise awareness of this issue.6
Much of the literature regarding the effects of climate change on human health focuses on insect-borne diseases, but over the past decade other areas of impact also have been investigated, such as increases in airborne diseases, zoonoses, newly endemic saprophytic and dimorphic fungal infections, fecal-oral diseases, and severe allergic disease.7,8 It is postulated that climate change leads to region-specific increases in human disease because it creates newly favorable habitats for infectious agents, their vectors. and their reservoirs, allowing expansion of their ranges and access to immunologically naïve populations.9 Furthermore, extreme weather events such as heat waves, hurricanes, and flooding, which are expected to increase in frequency as a result of climate change, have all been linked to infectious disease outbreaks.10
Lyme Disease
In the past 20 years, Lyme disease incidence has tripled in the United States.11 It has been hypothesized that the increase may be occurring as a result of the expanding geographic distribution of the Ixodes tick and its mammalian hosts (eg, white-tailed deer) under the influence of climate change.12 Lyme disease is a multisystem disease affecting the skin, joints, heart, and nervous system. Its most characteristic manifestation is cutaneous in the form of erythema migrans. Dermatologists may be called upon to play an increasingly important role in early detection and treatment of this potentially chronic and debilitating condition.
Arboviruses
Arboviruses are transmitted by arthropods and are an important category of climate change–related diseases due to the expansion of the mosquito habitat worldwide. The vectorial capacity for the transmission of dengue fever has increased worldwide by 9.4% via Aedes aegypti and 11.1% via Aedes albopictus since 1950.13 Dengue fever, also known as breakbone fever, presents with intense joint pain, fevers, headaches, and a transient morbilliform rash that desquamates with defervescence and in some cases will incite hemorrhagic skin lesions.14 Dengue fever previously was considered to be a tropical disease but locally acquired cases have been reported in the United States, including Texas, Hawaii, and Florida.15,16
Reports of local transmission of chikungunya, another arbovirus transmitted by A albopictus and A aegypti mosquitoes in Florida, the US Virgin Islands, and Puerto Rico, began in 2014.17 A higher prevalence of these diseases within the United States also may be related to increased globalization, with US travelers returning from endemic regions with infections. Prior to 2014, transmission occurred in traditional endemic regions, primarily in Asia, Africa, or island nations in the Indian Ocean. Like dengue fever, chikungunya causes high fevers, cutaneous manifestations (eg, urticarial papules, morbilliform eruption, hypermelanosis, intertriginous lesions, lymphedema),17 and intense joint pain. Unlike dengue fever, however, joint involvement can be chronic, erosive, and debilitating.
Lastly, New World leishmaniasis, an arboviral disease characterized by mucocutaneous ulcers and transmitted by phlebotomine sand flies, has been acquired locally in Oklahoma and Texas when it was previously considered to be endemic to Mexico and Central and South America.14,18 The habitats of New World Leishmania species are expected to expand northward, with an ecological niche model predicting that they reach southern Canada by the year 2080 due to the expanding habitats of sand fly and rodent vectors.19
Fungal Infections
In the Pacific Northwest, there have been reports of newly endemic Cryptococcus gattii and Coccidioides immitis, both of which previously had been confined to the southwestern United States.8 Endemic ranges of these mycotic pathogens may be expanding for a variety of reasons, with climate change creating new regions conducive to the colonization of these species.8,20,21Coccidioides immitis is a soil-dwelling fungus that usually presents with primary pulmonary disease that can disseminate acutely or even months later. Prompt recognition of disseminated disease may allow life-saving therapy to be initiated. Cryptococcus gattii is a fungus with multiple niches, including oil, trees, and birds.20 This fungus also is acquired via inhalation, with dissemination occurring most commonly in immunosuppressed patients to the central nervous system, bone, and skin. Primary or secondary infection with both of these fungi may present with cutaneous manifestations presenting as polymorphous lesions, including umbilicated or ulcerated papules, indurated nodules, and acneiform pustules.
Final Thoughts
Awareness of the shifting habitats of microorganisms and vectors locally is important in order for clinicians to make correct diagnoses in a timely fashion. Regional or endemic diseases are presenting outside their traditional boundaries due to changing habitats of microbes and vectors and may be easily overlooked, resulting in a delayed diagnosis. Being prepared to diagnose diseases with increasing incidence secondary to climate change and discussing this with patients is an important physician obligation, but it is not the only one. We cannot effectively advocate for the health of patients and the community while ignoring the destruction of the environment. Our additional responsibility is straightforward—being advocates for good stewardship of the Earth’s resources now on both a personal and a policy level.22
The term climate refers to the average weather conditions of a specific geographic location measured over several decades.1 While a certain degree of variation in the Earth’s climate is expected, a persistent warming or cooling trend is not. The factors driving the Earth’s warming remain difficult to prove.2 We know the Earth previously has undergone dramatic climate changes and that natural factors driving these changes are varied (eg, the relationship between the Earth and the Sun, volcanic eruptions, solar irradiance).1,3 These factors ideally change over protracted periods of time in a way that allows organisms to adapt to new environments.
Anthropogenic climate change refers to human-caused climate change. This is thought to be a major driving factor in the Earth’s recent warming trend, partly due to the rapidity of warming in recent years.3 According to climate scientists, the Earth’s temperature has risen 4°C to 7°C over the past 5000 years, but it has risen 0.7°C in just the past 100 years alone.4 Greenhouse gases such as carbon dioxide are emitted by various natural processes and human activities and play a central role in current warming because they trap solar heat and increase ambient temperature.3
In a recent edition of the commonly cited textbook Dermatology, Bolognia et al5 referenced climate change only once in a figure legend regarding the expansion of dengue fever in the Americas. However, climate change may have the potential to cause outright skin disease epidemics worldwide, and the Climate Change Committee of the International Society of Dermatology has called upon dermatologists across the globe to help raise awareness of this issue.6
Much of the literature regarding the effects of climate change on human health focuses on insect-borne diseases, but over the past decade other areas of impact also have been investigated, such as increases in airborne diseases, zoonoses, newly endemic saprophytic and dimorphic fungal infections, fecal-oral diseases, and severe allergic disease.7,8 It is postulated that climate change leads to region-specific increases in human disease because it creates newly favorable habitats for infectious agents, their vectors. and their reservoirs, allowing expansion of their ranges and access to immunologically naïve populations.9 Furthermore, extreme weather events such as heat waves, hurricanes, and flooding, which are expected to increase in frequency as a result of climate change, have all been linked to infectious disease outbreaks.10
Lyme Disease
In the past 20 years, Lyme disease incidence has tripled in the United States.11 It has been hypothesized that the increase may be occurring as a result of the expanding geographic distribution of the Ixodes tick and its mammalian hosts (eg, white-tailed deer) under the influence of climate change.12 Lyme disease is a multisystem disease affecting the skin, joints, heart, and nervous system. Its most characteristic manifestation is cutaneous in the form of erythema migrans. Dermatologists may be called upon to play an increasingly important role in early detection and treatment of this potentially chronic and debilitating condition.
Arboviruses
Arboviruses are transmitted by arthropods and are an important category of climate change–related diseases due to the expansion of the mosquito habitat worldwide. The vectorial capacity for the transmission of dengue fever has increased worldwide by 9.4% via Aedes aegypti and 11.1% via Aedes albopictus since 1950.13 Dengue fever, also known as breakbone fever, presents with intense joint pain, fevers, headaches, and a transient morbilliform rash that desquamates with defervescence and in some cases will incite hemorrhagic skin lesions.14 Dengue fever previously was considered to be a tropical disease but locally acquired cases have been reported in the United States, including Texas, Hawaii, and Florida.15,16
Reports of local transmission of chikungunya, another arbovirus transmitted by A albopictus and A aegypti mosquitoes in Florida, the US Virgin Islands, and Puerto Rico, began in 2014.17 A higher prevalence of these diseases within the United States also may be related to increased globalization, with US travelers returning from endemic regions with infections. Prior to 2014, transmission occurred in traditional endemic regions, primarily in Asia, Africa, or island nations in the Indian Ocean. Like dengue fever, chikungunya causes high fevers, cutaneous manifestations (eg, urticarial papules, morbilliform eruption, hypermelanosis, intertriginous lesions, lymphedema),17 and intense joint pain. Unlike dengue fever, however, joint involvement can be chronic, erosive, and debilitating.
Lastly, New World leishmaniasis, an arboviral disease characterized by mucocutaneous ulcers and transmitted by phlebotomine sand flies, has been acquired locally in Oklahoma and Texas when it was previously considered to be endemic to Mexico and Central and South America.14,18 The habitats of New World Leishmania species are expected to expand northward, with an ecological niche model predicting that they reach southern Canada by the year 2080 due to the expanding habitats of sand fly and rodent vectors.19
Fungal Infections
In the Pacific Northwest, there have been reports of newly endemic Cryptococcus gattii and Coccidioides immitis, both of which previously had been confined to the southwestern United States.8 Endemic ranges of these mycotic pathogens may be expanding for a variety of reasons, with climate change creating new regions conducive to the colonization of these species.8,20,21Coccidioides immitis is a soil-dwelling fungus that usually presents with primary pulmonary disease that can disseminate acutely or even months later. Prompt recognition of disseminated disease may allow life-saving therapy to be initiated. Cryptococcus gattii is a fungus with multiple niches, including oil, trees, and birds.20 This fungus also is acquired via inhalation, with dissemination occurring most commonly in immunosuppressed patients to the central nervous system, bone, and skin. Primary or secondary infection with both of these fungi may present with cutaneous manifestations presenting as polymorphous lesions, including umbilicated or ulcerated papules, indurated nodules, and acneiform pustules.
Final Thoughts
Awareness of the shifting habitats of microorganisms and vectors locally is important in order for clinicians to make correct diagnoses in a timely fashion. Regional or endemic diseases are presenting outside their traditional boundaries due to changing habitats of microbes and vectors and may be easily overlooked, resulting in a delayed diagnosis. Being prepared to diagnose diseases with increasing incidence secondary to climate change and discussing this with patients is an important physician obligation, but it is not the only one. We cannot effectively advocate for the health of patients and the community while ignoring the destruction of the environment. Our additional responsibility is straightforward—being advocates for good stewardship of the Earth’s resources now on both a personal and a policy level.22
- Climate Central. Global Weirdness: Severe Storms, Deadly Heat Waves, Relentless Drought, Rising Seas, and the Weather of the Future. New York, NY: Pantheon Books; 2012.
- Cook J, Nuccitelli D, Green SA, et al. Quantifying the consensus on anthropogenic global warming in the scientific literature. Environ Res Lett. 2013;8:024024.
- A blanket around the Earth. NASA Climate website. https://climate.nasa.gov/causes/. Accessed February 5, 2018.
- How is today’s warming different from the past? NASA Earth Observatory website. https://earthobservatory.nasa.gov/Features/GlobalWarming/page3.php. Accessed February 5, 2018.
- Mancini AJ, Shani-Adir A, Sidbury R. Other viral diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2017:1425-1446.
- Andersen LK, Davis MDP. A wake-up call to dermatologists—climate change affects the skin. Int J Dermatol. 2017;56:E198-E199.
- Liang L, Gong P. Climate change and human infectious diseases: a synthesis of research findings from global and spatio-temporal perspectives [published online March 23, 2017]. Environ Int. 2017;103:99-108.
- Lockhart SR, McCotter OZ, Chiller TM. Emerging fungal infections in the Pacific Northwest: the unrecognized burden and geographic range of Cryptococcus gattii and Coccidioides immitis. Microbiol Spectr. 2016;4. doi:10.1128/microbiolspec.EI10-0016-2016.
- Kilpatrick AM, Randolph SE. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet. 2012;380:1946-1955.
- McMichael AJ. Extreme weather events and infectious disease outbreaks. Virulence. 2015;6:543-547.
- Lyme disease graphs. CDC website. https://www.cdc.gov/lyme/stats/graphs.html. Updated November 1, 2017. Accessed April 12, 2018.
- Stone BL, Tourand Y, Brissette CA. Brave new worlds: the expanding universe of Lyme disease. Vector Borne Zoonotic Dis. 2017;17:619-629.
- Watts N, Amann M, Ayeb-Karlsson S, et al. The Lancet Countdown on health and climate change: from 25 years of inaction to a global transformation for public health [published online October 30, 2017]. Lancet. doi:10.1016/S0140-6736(17)32464-9.
- Nawas ZY, Tong Y, Kollipara R, et al. Emerging infectious diseases with cutaneous manifestations: viral and bacterial infections. J Am Acad Dermatol. 2016;75:1-16.
- Kaffenberger BH, Shetlar D, Norton SA, et al. The effect of climate change on skin disease in North America. J Am Acad Dermatol. 2017;76:140-147.
- Dengue. CDC website. https://www.cdc.gov/dengue/epidemiology/index.html. Updated June 9, 2014. Accessed April 3, 2018.
- Chikungunya virus in the United States. CDC website. https://www.cdc.gov/chikungunya/geo/united-states.html. Updated October 30, 2017. Accessed April 4, 2018.
- Clarke CF, Bradley KK, Wright JH, et al. Emergence of autochthonous cutaneous leishmaniasis in northeastern Texas and southeastern Oklahoma. Am J Trop Med Hyg. 2013;88:157-61.
- González C, Wang O, Strutz SE, et al. Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl Trop Dis. 2010;4:E585.
- Chang CC, Chen SC. Colliding epidemics and the rise of cryptococcosis. J Fungi (Basel). 2015;2. doi: 10.3390/jof2010001.
- Marsden-Haug N, Goldoft M, Ralston C, et al. Coccidioidomycosis acquired in Washington state. Clin Infect Dis. 2013;56:847-850.
- Rosenbach M. Climate change & dermatology: what can you do? Paper presented at: American Academy of Dermatology Annual Meeting; March 3-7, 2017; Orlando, FL.
- Climate Central. Global Weirdness: Severe Storms, Deadly Heat Waves, Relentless Drought, Rising Seas, and the Weather of the Future. New York, NY: Pantheon Books; 2012.
- Cook J, Nuccitelli D, Green SA, et al. Quantifying the consensus on anthropogenic global warming in the scientific literature. Environ Res Lett. 2013;8:024024.
- A blanket around the Earth. NASA Climate website. https://climate.nasa.gov/causes/. Accessed February 5, 2018.
- How is today’s warming different from the past? NASA Earth Observatory website. https://earthobservatory.nasa.gov/Features/GlobalWarming/page3.php. Accessed February 5, 2018.
- Mancini AJ, Shani-Adir A, Sidbury R. Other viral diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2017:1425-1446.
- Andersen LK, Davis MDP. A wake-up call to dermatologists—climate change affects the skin. Int J Dermatol. 2017;56:E198-E199.
- Liang L, Gong P. Climate change and human infectious diseases: a synthesis of research findings from global and spatio-temporal perspectives [published online March 23, 2017]. Environ Int. 2017;103:99-108.
- Lockhart SR, McCotter OZ, Chiller TM. Emerging fungal infections in the Pacific Northwest: the unrecognized burden and geographic range of Cryptococcus gattii and Coccidioides immitis. Microbiol Spectr. 2016;4. doi:10.1128/microbiolspec.EI10-0016-2016.
- Kilpatrick AM, Randolph SE. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet. 2012;380:1946-1955.
- McMichael AJ. Extreme weather events and infectious disease outbreaks. Virulence. 2015;6:543-547.
- Lyme disease graphs. CDC website. https://www.cdc.gov/lyme/stats/graphs.html. Updated November 1, 2017. Accessed April 12, 2018.
- Stone BL, Tourand Y, Brissette CA. Brave new worlds: the expanding universe of Lyme disease. Vector Borne Zoonotic Dis. 2017;17:619-629.
- Watts N, Amann M, Ayeb-Karlsson S, et al. The Lancet Countdown on health and climate change: from 25 years of inaction to a global transformation for public health [published online October 30, 2017]. Lancet. doi:10.1016/S0140-6736(17)32464-9.
- Nawas ZY, Tong Y, Kollipara R, et al. Emerging infectious diseases with cutaneous manifestations: viral and bacterial infections. J Am Acad Dermatol. 2016;75:1-16.
- Kaffenberger BH, Shetlar D, Norton SA, et al. The effect of climate change on skin disease in North America. J Am Acad Dermatol. 2017;76:140-147.
- Dengue. CDC website. https://www.cdc.gov/dengue/epidemiology/index.html. Updated June 9, 2014. Accessed April 3, 2018.
- Chikungunya virus in the United States. CDC website. https://www.cdc.gov/chikungunya/geo/united-states.html. Updated October 30, 2017. Accessed April 4, 2018.
- Clarke CF, Bradley KK, Wright JH, et al. Emergence of autochthonous cutaneous leishmaniasis in northeastern Texas and southeastern Oklahoma. Am J Trop Med Hyg. 2013;88:157-61.
- González C, Wang O, Strutz SE, et al. Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl Trop Dis. 2010;4:E585.
- Chang CC, Chen SC. Colliding epidemics and the rise of cryptococcosis. J Fungi (Basel). 2015;2. doi: 10.3390/jof2010001.
- Marsden-Haug N, Goldoft M, Ralston C, et al. Coccidioidomycosis acquired in Washington state. Clin Infect Dis. 2013;56:847-850.
- Rosenbach M. Climate change & dermatology: what can you do? Paper presented at: American Academy of Dermatology Annual Meeting; March 3-7, 2017; Orlando, FL.

Pigmented Peduncule on the Leg
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
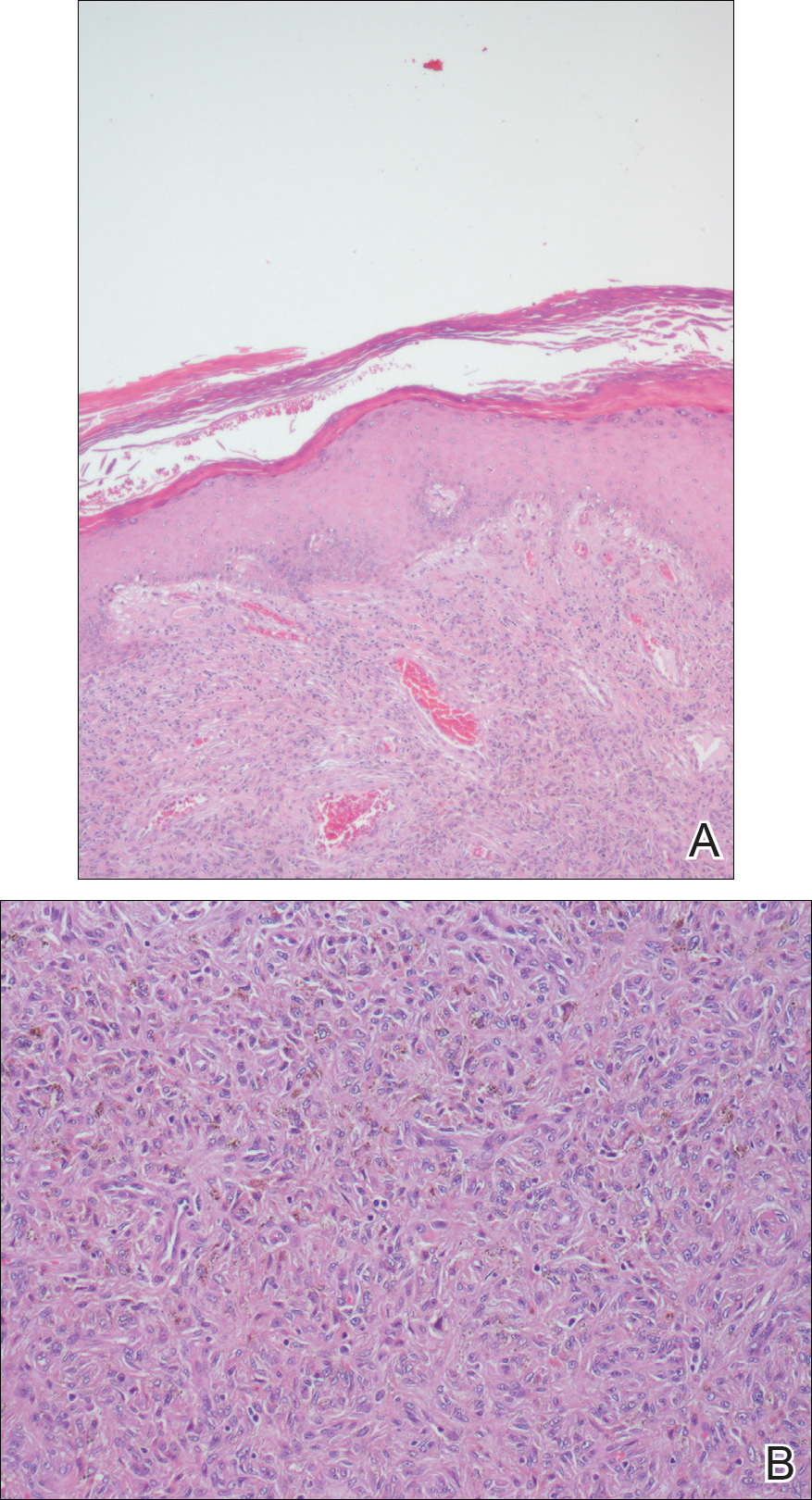
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
A 68-year-old man with a history of type 2 diabetes mellitus and hypercholesterolemia presented to the dermatology department with a cutaneous lesion on the posterior aspect of the right thigh of 2 years' duration. The lesion had become larger during the 4 months prior to presentation and was mostly asymptomatic but became tender when subjected to trauma. Physical examination revealed a firm, 2-cm, slightly pigmented peduncule on the posterior right thigh. No lymphadenopathies were noted. The lesion was completely excised for histologic examination.