User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Vemurafenib-Induced Plantar Hyperkeratosis
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
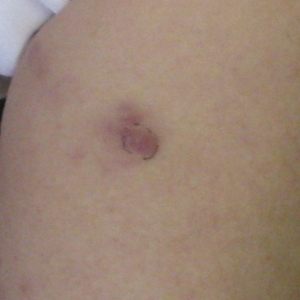
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
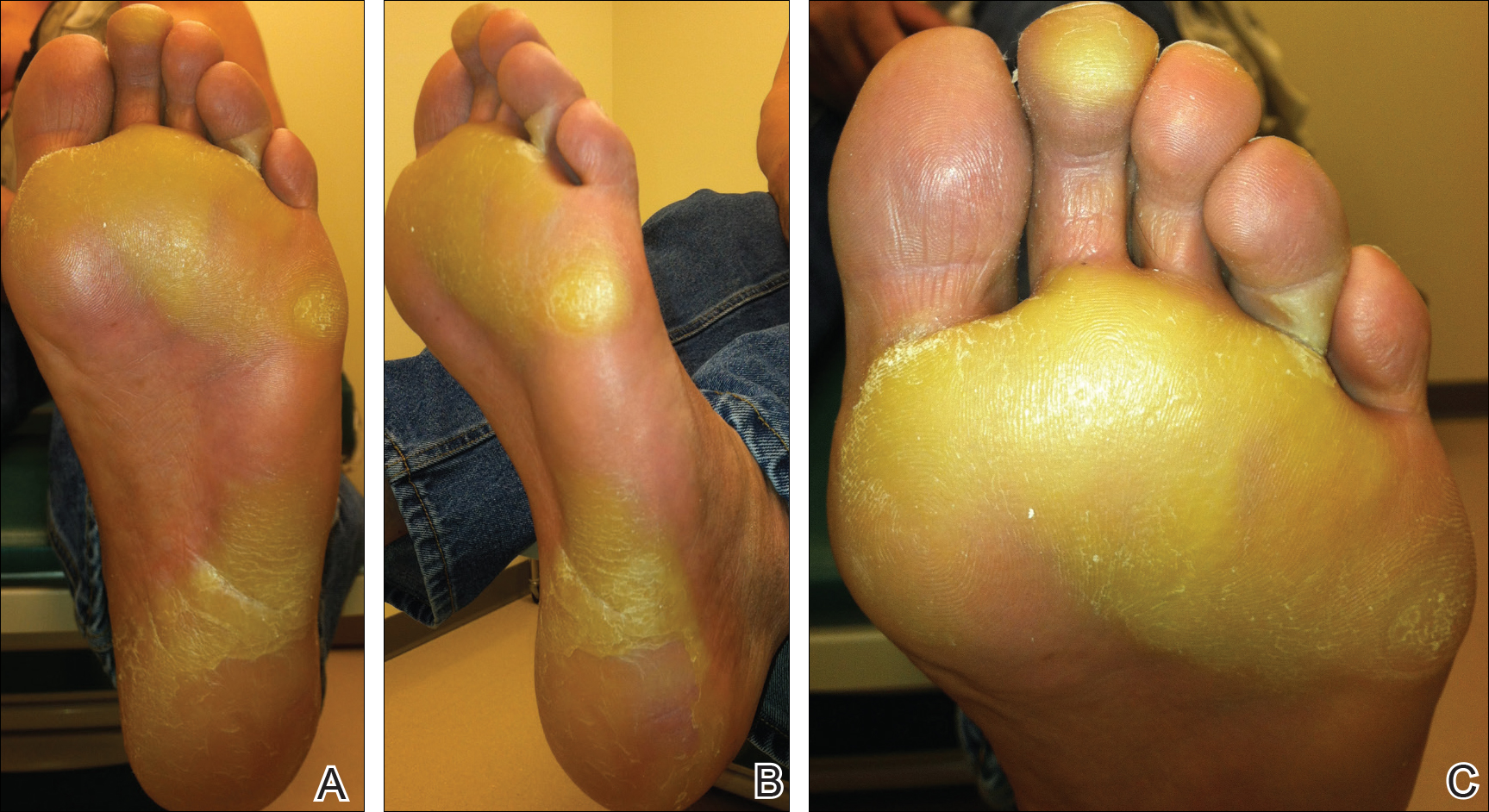
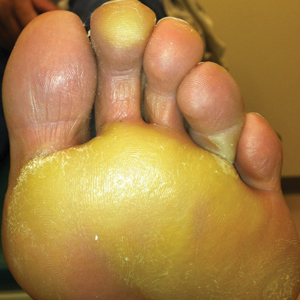
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
Practice Points
- BRAF inhibitors such as vemurafenib are associated with a high incidence of cutaneous side effects, including rash, hyperkeratosis, and cutaneous squamous cell carcinoma.
- Practitioners should be aware of these side effects and their management to avoid discontinuation or interruption of therapy.
Acral Cutaneous Metastasis From a Primary Breast Carcinoma Following Chemotherapy With Bevacizumab and Paclitaxel
Cutaneous metastasis of internal malignancy is a relatively uncommon phenomenon, with an overall incidence of 5.3% in cancer patients.1 Cutaneous involvement typically occurs late in the course of disease but can occasionally be the first extranodal sign of metastatic disease. Breast cancer has the highest rate of cutaneous metastasis, most often involving the chest wall1; however, cutaneous metastasis to the acral sites is exceedingly rare. The hand is the site of 0.1% of all metastatic lesions, with only 10% of these being cutaneous lesions and the remaining 90% being osseous metastases.2 Herein, we report a case of multiple cutaneous metastases to acral sites involving the palmar and plantar surfaces of the hands and feet.
Case Report
A 54-year-old black woman with a history of stage IV carcinoma of the breast was admitted to the university medical center with exquisitely painful cutaneous nodules on the hands and feet of 5 weeks’ duration that had started to cause difficulty with walking and daily activities. The patient reported that the breast carcinoma had initially been diagnosed in Nigeria 2 years prior, but she did not receive treatment until moving to the United States. She received a total of 4 cycles of chemotherapy with paclitaxel and bevacizumab, which was discontinued 6 weeks prior to admission due to pain in the lower extremities that was thought to be secondary to neuropathy. One week after discontinuation of chemotherapy, the patient reported increasing pain in the extremities and new-onset painful nodules on the hands and feet. Treatment with gabapentin as well as several courses of antibiotics failed to improve the condition.
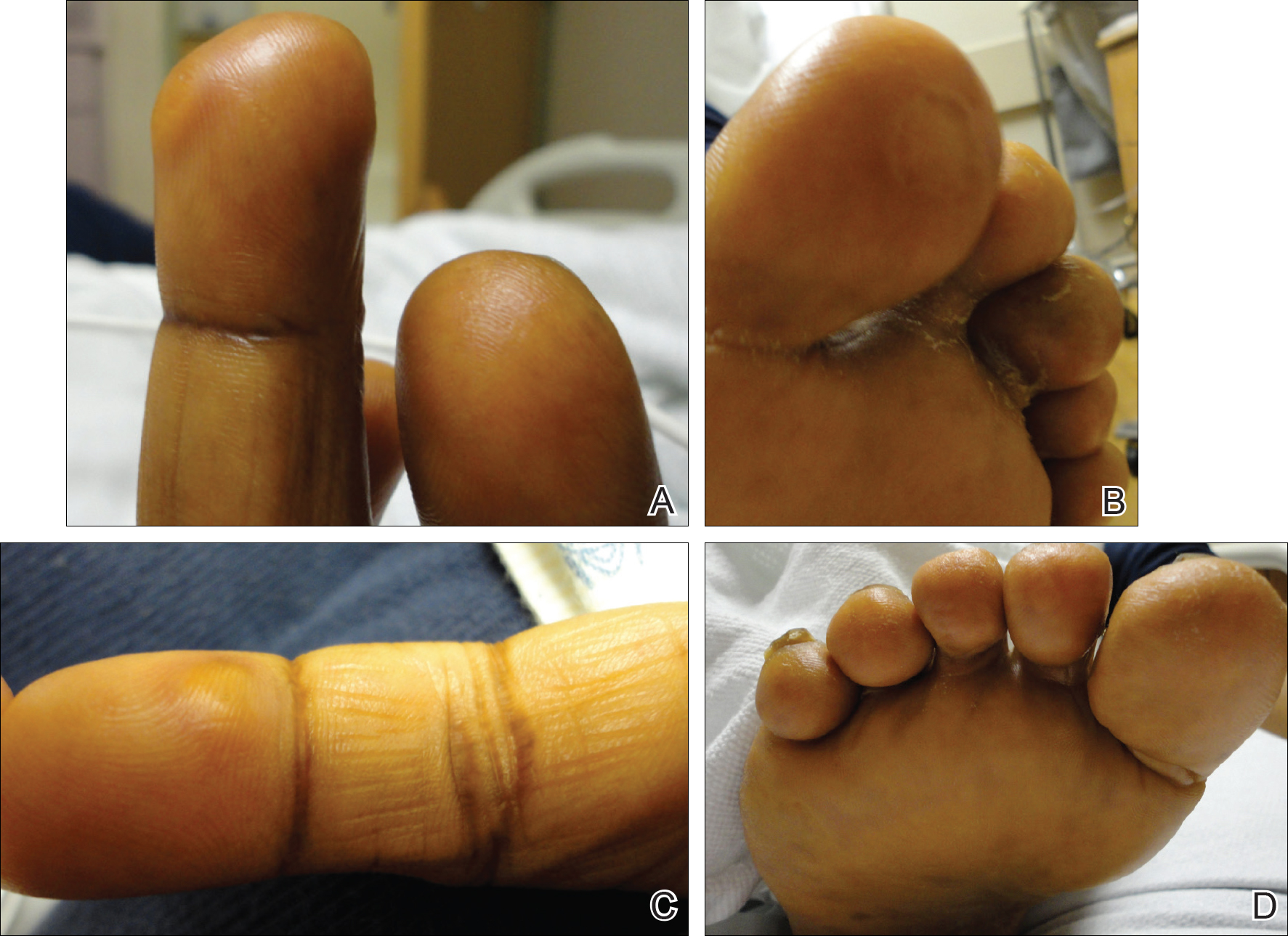
She was admitted for symptomatic pain control and a dermatology consultation. Physical examination revealed multiple firm, tender, subcutaneous nodules on the volar surfaces of the soles, toes, palms, and fingertips (Figure 1). A nodule also was noted on the scalp. A punch biopsy of a nodule on the right fourth finger revealed a dermal carcinoma (Figure 2). On immunohistochemistry, the tumor stained positive for cytokeratin 5/6, cytokeratin 7, and gross cystic disease fluid protein 15. It did not demonstrate connection to the epidermis or adnexal structures. Although the tumor did not express estrogen or progesterone receptors, the findings were compatible with metastasis from the patient’s primary breast carcinoma with poor differentiation. A biopsy of the primary breast carcinoma was not available for review from Nigeria.
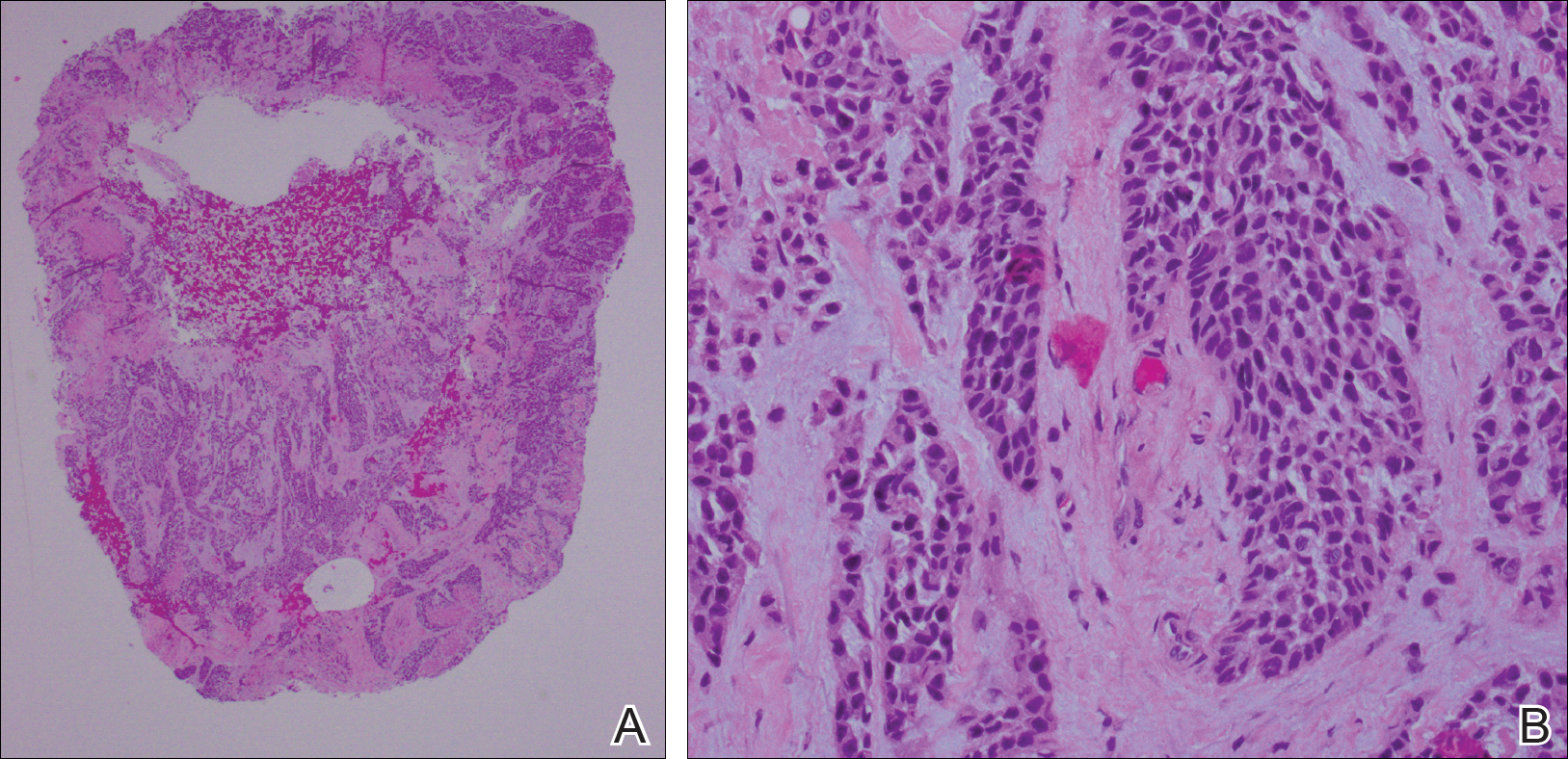
Comment
The majority of cases reporting acral cutaneous metastasis from internal malignancies are unilateral, involving only one extremity. Several hypotheses have been provided, including spread from localized trauma, which causes disruption of blood vessels and consequent extravasation and localization of tumor cells into the extravascular space.3 The distal extremities are particularly vulnerable to trauma, making this hypothesis plausible.
Considering the overall rarity of metastases to acral sites, it is interesting that our patient developed multiple distal nodules on both the hands and feet. The rapid onset of cutaneous nodules shortly after a course of chemotherapy led the team to consider the physiologic effects of paclitaxel and bevacizumab in the etiology of the acral cutaneous metastases. Karamouzis et al3 described a similar case of multiple cutaneous metastases with a bilateral acral distribution. This case also was associated with chemotherapy in the treatment of breast cancer. The authors proposed hand-foot syndrome, a chemotherapy-related eruption localized to acral skin, as a possible mechanism for hematogenous spread of malignant cells.3 The pathogenesis of hand-foot syndrome is not well understood, but the unique anatomy and physiology of acral skin including temperature gradients, rapidly dividing epidermal cells, absence of hair follicles and sebaceous glands, wide dermal papillae, and exposure to high pressures from carrying body weight and repetitive minor trauma may contribute to the localization of signs and symptoms.3,4 Our case supports a chemotherapy-related etiology of acral cutaneous metastasis of a primary breast cancer; however, our patient did not have apparent signs or symptoms of hand-foot syndrome during the course of treatment. We propose that effects of bevacizumab on acral skin may have contributed to the development of our patient’s metastatic pattern.
Bevacizumab, a monoclonal antibody to vascular endothelial growth factor A, has well-known vascular side effects. Unlike the inhibition of vascular endothelial growth factor A provided by the receptor tyrosine kinase inhibitors sorafenib and sunitinib, bevacizumab typically is not associated with hand-foot syndrome.5 However, several cases have been reported with chemotherapy-associated palmoplantar eruptions that resolved after withholding bevacizumab while continuing other chemotherapeutic agents, suggesting that bevacizumab-induced changes in acral skin contributed to the eruption.6 Specific factors that could contribute to acral metastasis in patients taking bevacizumab are endothelial dysfunction and capillary rarefaction of the acral skin, as well as hemorrhage, decreased wound healing, and changes in vascular permeability.5,7
We present a rare case of acral cutaneous metastasis associated with bevacizumab, one of few reported cases associated with a taxane chemotherapeutic agent.3 More cases need to be identified and reported to establish a causative association, if indeed existent, between acral cutaneous metastasis of breast carcinoma and the use of bevacizumab as well as other chemotherapeutic drugs.
- Krathen RA, Orengo IF, Rosen T. Cutaneous metastasis: a meta-analysis of data. South Med J. 2003;96:164-167.
- Wu CY, Gao HW, Huang WH, et al. Infection-like acral cutaneous metastasis as the presenting sign of an occult breast cancer. Clin Exp Dermatol. 2009;34:409-410.
- Karamouzis MV, Ardavanis A, Alexopoulos A, et al. Multiple cutaneous acral metastases in a woman with breast adenocarcinoma treated with pegylated liposomal doxorubicin: incidental or aetiological association? Eur J Cancer Care (Engl). 2005;14:267-271.
- Nagore E, Insa A, Sanmartin O. Antineoplastic therapy-induced palmar plantar erythrodysesthesia (‘hand-foot’) syndrome. incidence, recognition and management. Am J Clin Dermatol. 2000;1:225-234.
- Wozel G, Sticherling M, Schon MP. Cutaneous side effects of inhibition of VEGF signal transduction. J Dtsch Dermatol Ges. 2010;8:243-249.
- Munehiro A, Yoneda K, Nakai K, et al. Bevacizumab-induced hand-foot syndrome: circumscribed type. Br J Dermatol. 2010;162:1411-1413.
- Mourad JJ, des Guetz G, Debbabi H, et al. Blood pressure rise following angiogenesis inhibition by bevacizumab. a crucial role for microcirculation. Ann Oncol. 2008;19:927-934.
Cutaneous metastasis of internal malignancy is a relatively uncommon phenomenon, with an overall incidence of 5.3% in cancer patients.1 Cutaneous involvement typically occurs late in the course of disease but can occasionally be the first extranodal sign of metastatic disease. Breast cancer has the highest rate of cutaneous metastasis, most often involving the chest wall1; however, cutaneous metastasis to the acral sites is exceedingly rare. The hand is the site of 0.1% of all metastatic lesions, with only 10% of these being cutaneous lesions and the remaining 90% being osseous metastases.2 Herein, we report a case of multiple cutaneous metastases to acral sites involving the palmar and plantar surfaces of the hands and feet.
Case Report
A 54-year-old black woman with a history of stage IV carcinoma of the breast was admitted to the university medical center with exquisitely painful cutaneous nodules on the hands and feet of 5 weeks’ duration that had started to cause difficulty with walking and daily activities. The patient reported that the breast carcinoma had initially been diagnosed in Nigeria 2 years prior, but she did not receive treatment until moving to the United States. She received a total of 4 cycles of chemotherapy with paclitaxel and bevacizumab, which was discontinued 6 weeks prior to admission due to pain in the lower extremities that was thought to be secondary to neuropathy. One week after discontinuation of chemotherapy, the patient reported increasing pain in the extremities and new-onset painful nodules on the hands and feet. Treatment with gabapentin as well as several courses of antibiotics failed to improve the condition.
She was admitted for symptomatic pain control and a dermatology consultation. Physical examination revealed multiple firm, tender, subcutaneous nodules on the volar surfaces of the soles, toes, palms, and fingertips (Figure 1). A nodule also was noted on the scalp. A punch biopsy of a nodule on the right fourth finger revealed a dermal carcinoma (Figure 2). On immunohistochemistry, the tumor stained positive for cytokeratin 5/6, cytokeratin 7, and gross cystic disease fluid protein 15. It did not demonstrate connection to the epidermis or adnexal structures. Although the tumor did not express estrogen or progesterone receptors, the findings were compatible with metastasis from the patient’s primary breast carcinoma with poor differentiation. A biopsy of the primary breast carcinoma was not available for review from Nigeria.
Comment
The majority of cases reporting acral cutaneous metastasis from internal malignancies are unilateral, involving only one extremity. Several hypotheses have been provided, including spread from localized trauma, which causes disruption of blood vessels and consequent extravasation and localization of tumor cells into the extravascular space.3 The distal extremities are particularly vulnerable to trauma, making this hypothesis plausible.
Considering the overall rarity of metastases to acral sites, it is interesting that our patient developed multiple distal nodules on both the hands and feet. The rapid onset of cutaneous nodules shortly after a course of chemotherapy led the team to consider the physiologic effects of paclitaxel and bevacizumab in the etiology of the acral cutaneous metastases. Karamouzis et al3 described a similar case of multiple cutaneous metastases with a bilateral acral distribution. This case also was associated with chemotherapy in the treatment of breast cancer. The authors proposed hand-foot syndrome, a chemotherapy-related eruption localized to acral skin, as a possible mechanism for hematogenous spread of malignant cells.3 The pathogenesis of hand-foot syndrome is not well understood, but the unique anatomy and physiology of acral skin including temperature gradients, rapidly dividing epidermal cells, absence of hair follicles and sebaceous glands, wide dermal papillae, and exposure to high pressures from carrying body weight and repetitive minor trauma may contribute to the localization of signs and symptoms.3,4 Our case supports a chemotherapy-related etiology of acral cutaneous metastasis of a primary breast cancer; however, our patient did not have apparent signs or symptoms of hand-foot syndrome during the course of treatment. We propose that effects of bevacizumab on acral skin may have contributed to the development of our patient’s metastatic pattern.
Bevacizumab, a monoclonal antibody to vascular endothelial growth factor A, has well-known vascular side effects. Unlike the inhibition of vascular endothelial growth factor A provided by the receptor tyrosine kinase inhibitors sorafenib and sunitinib, bevacizumab typically is not associated with hand-foot syndrome.5 However, several cases have been reported with chemotherapy-associated palmoplantar eruptions that resolved after withholding bevacizumab while continuing other chemotherapeutic agents, suggesting that bevacizumab-induced changes in acral skin contributed to the eruption.6 Specific factors that could contribute to acral metastasis in patients taking bevacizumab are endothelial dysfunction and capillary rarefaction of the acral skin, as well as hemorrhage, decreased wound healing, and changes in vascular permeability.5,7
We present a rare case of acral cutaneous metastasis associated with bevacizumab, one of few reported cases associated with a taxane chemotherapeutic agent.3 More cases need to be identified and reported to establish a causative association, if indeed existent, between acral cutaneous metastasis of breast carcinoma and the use of bevacizumab as well as other chemotherapeutic drugs.
Cutaneous metastasis of internal malignancy is a relatively uncommon phenomenon, with an overall incidence of 5.3% in cancer patients.1 Cutaneous involvement typically occurs late in the course of disease but can occasionally be the first extranodal sign of metastatic disease. Breast cancer has the highest rate of cutaneous metastasis, most often involving the chest wall1; however, cutaneous metastasis to the acral sites is exceedingly rare. The hand is the site of 0.1% of all metastatic lesions, with only 10% of these being cutaneous lesions and the remaining 90% being osseous metastases.2 Herein, we report a case of multiple cutaneous metastases to acral sites involving the palmar and plantar surfaces of the hands and feet.
Case Report
A 54-year-old black woman with a history of stage IV carcinoma of the breast was admitted to the university medical center with exquisitely painful cutaneous nodules on the hands and feet of 5 weeks’ duration that had started to cause difficulty with walking and daily activities. The patient reported that the breast carcinoma had initially been diagnosed in Nigeria 2 years prior, but she did not receive treatment until moving to the United States. She received a total of 4 cycles of chemotherapy with paclitaxel and bevacizumab, which was discontinued 6 weeks prior to admission due to pain in the lower extremities that was thought to be secondary to neuropathy. One week after discontinuation of chemotherapy, the patient reported increasing pain in the extremities and new-onset painful nodules on the hands and feet. Treatment with gabapentin as well as several courses of antibiotics failed to improve the condition.
She was admitted for symptomatic pain control and a dermatology consultation. Physical examination revealed multiple firm, tender, subcutaneous nodules on the volar surfaces of the soles, toes, palms, and fingertips (Figure 1). A nodule also was noted on the scalp. A punch biopsy of a nodule on the right fourth finger revealed a dermal carcinoma (Figure 2). On immunohistochemistry, the tumor stained positive for cytokeratin 5/6, cytokeratin 7, and gross cystic disease fluid protein 15. It did not demonstrate connection to the epidermis or adnexal structures. Although the tumor did not express estrogen or progesterone receptors, the findings were compatible with metastasis from the patient’s primary breast carcinoma with poor differentiation. A biopsy of the primary breast carcinoma was not available for review from Nigeria.
Comment
The majority of cases reporting acral cutaneous metastasis from internal malignancies are unilateral, involving only one extremity. Several hypotheses have been provided, including spread from localized trauma, which causes disruption of blood vessels and consequent extravasation and localization of tumor cells into the extravascular space.3 The distal extremities are particularly vulnerable to trauma, making this hypothesis plausible.
Considering the overall rarity of metastases to acral sites, it is interesting that our patient developed multiple distal nodules on both the hands and feet. The rapid onset of cutaneous nodules shortly after a course of chemotherapy led the team to consider the physiologic effects of paclitaxel and bevacizumab in the etiology of the acral cutaneous metastases. Karamouzis et al3 described a similar case of multiple cutaneous metastases with a bilateral acral distribution. This case also was associated with chemotherapy in the treatment of breast cancer. The authors proposed hand-foot syndrome, a chemotherapy-related eruption localized to acral skin, as a possible mechanism for hematogenous spread of malignant cells.3 The pathogenesis of hand-foot syndrome is not well understood, but the unique anatomy and physiology of acral skin including temperature gradients, rapidly dividing epidermal cells, absence of hair follicles and sebaceous glands, wide dermal papillae, and exposure to high pressures from carrying body weight and repetitive minor trauma may contribute to the localization of signs and symptoms.3,4 Our case supports a chemotherapy-related etiology of acral cutaneous metastasis of a primary breast cancer; however, our patient did not have apparent signs or symptoms of hand-foot syndrome during the course of treatment. We propose that effects of bevacizumab on acral skin may have contributed to the development of our patient’s metastatic pattern.
Bevacizumab, a monoclonal antibody to vascular endothelial growth factor A, has well-known vascular side effects. Unlike the inhibition of vascular endothelial growth factor A provided by the receptor tyrosine kinase inhibitors sorafenib and sunitinib, bevacizumab typically is not associated with hand-foot syndrome.5 However, several cases have been reported with chemotherapy-associated palmoplantar eruptions that resolved after withholding bevacizumab while continuing other chemotherapeutic agents, suggesting that bevacizumab-induced changes in acral skin contributed to the eruption.6 Specific factors that could contribute to acral metastasis in patients taking bevacizumab are endothelial dysfunction and capillary rarefaction of the acral skin, as well as hemorrhage, decreased wound healing, and changes in vascular permeability.5,7
We present a rare case of acral cutaneous metastasis associated with bevacizumab, one of few reported cases associated with a taxane chemotherapeutic agent.3 More cases need to be identified and reported to establish a causative association, if indeed existent, between acral cutaneous metastasis of breast carcinoma and the use of bevacizumab as well as other chemotherapeutic drugs.
- Krathen RA, Orengo IF, Rosen T. Cutaneous metastasis: a meta-analysis of data. South Med J. 2003;96:164-167.
- Wu CY, Gao HW, Huang WH, et al. Infection-like acral cutaneous metastasis as the presenting sign of an occult breast cancer. Clin Exp Dermatol. 2009;34:409-410.
- Karamouzis MV, Ardavanis A, Alexopoulos A, et al. Multiple cutaneous acral metastases in a woman with breast adenocarcinoma treated with pegylated liposomal doxorubicin: incidental or aetiological association? Eur J Cancer Care (Engl). 2005;14:267-271.
- Nagore E, Insa A, Sanmartin O. Antineoplastic therapy-induced palmar plantar erythrodysesthesia (‘hand-foot’) syndrome. incidence, recognition and management. Am J Clin Dermatol. 2000;1:225-234.
- Wozel G, Sticherling M, Schon MP. Cutaneous side effects of inhibition of VEGF signal transduction. J Dtsch Dermatol Ges. 2010;8:243-249.
- Munehiro A, Yoneda K, Nakai K, et al. Bevacizumab-induced hand-foot syndrome: circumscribed type. Br J Dermatol. 2010;162:1411-1413.
- Mourad JJ, des Guetz G, Debbabi H, et al. Blood pressure rise following angiogenesis inhibition by bevacizumab. a crucial role for microcirculation. Ann Oncol. 2008;19:927-934.
- Krathen RA, Orengo IF, Rosen T. Cutaneous metastasis: a meta-analysis of data. South Med J. 2003;96:164-167.
- Wu CY, Gao HW, Huang WH, et al. Infection-like acral cutaneous metastasis as the presenting sign of an occult breast cancer. Clin Exp Dermatol. 2009;34:409-410.
- Karamouzis MV, Ardavanis A, Alexopoulos A, et al. Multiple cutaneous acral metastases in a woman with breast adenocarcinoma treated with pegylated liposomal doxorubicin: incidental or aetiological association? Eur J Cancer Care (Engl). 2005;14:267-271.
- Nagore E, Insa A, Sanmartin O. Antineoplastic therapy-induced palmar plantar erythrodysesthesia (‘hand-foot’) syndrome. incidence, recognition and management. Am J Clin Dermatol. 2000;1:225-234.
- Wozel G, Sticherling M, Schon MP. Cutaneous side effects of inhibition of VEGF signal transduction. J Dtsch Dermatol Ges. 2010;8:243-249.
- Munehiro A, Yoneda K, Nakai K, et al. Bevacizumab-induced hand-foot syndrome: circumscribed type. Br J Dermatol. 2010;162:1411-1413.
- Mourad JJ, des Guetz G, Debbabi H, et al. Blood pressure rise following angiogenesis inhibition by bevacizumab. a crucial role for microcirculation. Ann Oncol. 2008;19:927-934.
Practice Points
- Cutaneous involvement of internal malignancy typically occurs late in the disease course but can occasionally be the first extranodal sign of metastatic disease.
- Acral cutaneous metastasis from internal malignancies typically is unilateral, involving only one extremity; however, this case demonstrates involvement on both the hands and feet.
- This case support a chemotherapy-related etiology of acral cutaneous metastasis of a primary breast cancer.

Perianal Ulceration and Verrucous Papules
The Diagnosis: Herpes Simplex Virus Infection
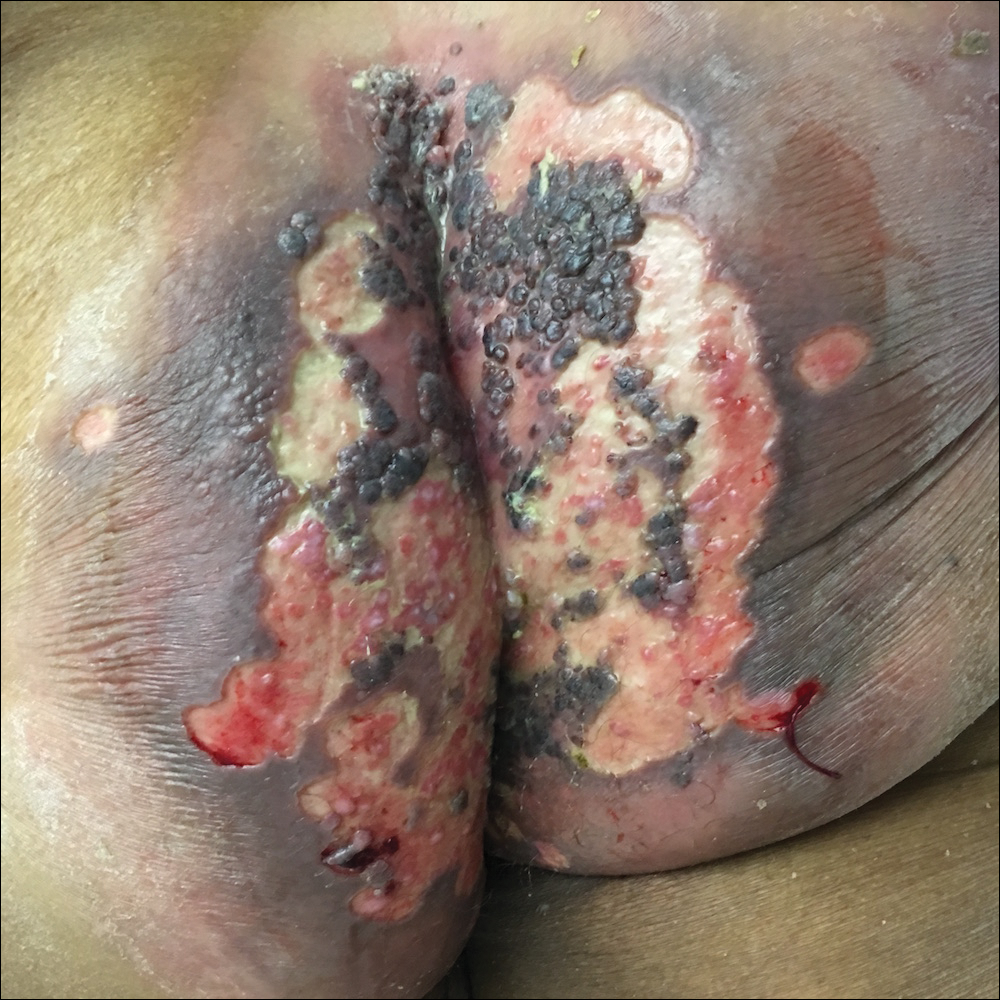
Viral culture of the ulcer was positive for herpes simplex virus type 2 (HHV-2). Bacterial culture grew enteric flora. The patient was started on intravenous acyclovir 5 mg/kg every 8 hours for 7 days and then transitioned to oral acyclovir for chronic suppressive therapy. One month later, there was near-complete reepithelialization with 2 remaining 1-cm shallow ulcers. The verrucous lesions had dried up and were flaking off (Figure). At 6-month follow-up, the ulcers and verrucous lesions had completely resolved.
Herpes simplex virus type 2 is the most common cause of genital and perianal ulcers in immunocompromised individuals.1 Patients classically present with painful grouped vesicles followed by painful superficial ulcers that may rapidly progress to extensive confluent ulceration. A hypertrophic variant of genital herpes characterized by anogenital verrucous lesions, similar to condyloma acuminata, also can be seen in immunocompromised individuals.2 This form has almost exclusively been observed in patients with human immunodeficiency virus and may occur in isolation or together with the ulcerative form.1-5 A case of vegetative HHV infection of the genital area in a patient with common variable immunodeficiency has been reported.6 Verrucous lesions of the mouth secondary to HHV have been observed in Hodgkin lymphoma, acute myelogenous leukemia, and individuals on immunosuppressive medications.7-10
Perianal involvement of Crohn disease typically presents with fistulas, ulcers, abscesses, strictures, and skin tags in some cases. Invasive squamous cell carcinoma may arise within a chronic ulcer of the anogenital area or may itself manifest as an ulcer or anal fissure. Perianal ulcerative skin tuberculosis has been reported in the literature as a rare manifestation of extrapulmonary tuberculosis and should be considered in a patient with an appropriate clinical history. Pyoderma gangrenosum classically pre-sents as a large ulcer with irregular rolled borders, though a rare variant of vegetative pyoderma gangrenosum may manifest as a nodular or verrucous plaque.
Studies to diagnose HHV include viral cell culture, HHV polymerase chain reaction testing, HHV serology, and direct fluorescent antibody testing. Skin biopsy may be necessary to rule out underlying malignancy. Treatment of perianal HHV infection includes acyclovir, valacyclovir, or famciclovir.1,5,6 Hypertrophic lesions often are refractory to first-line antiviral therapy and may require surgical resection or treatment with alternative medications such as imiquimod, a topical immunomodulator.3,5,6,11
- Ranu H, Lee J, Chio M, et al. Tumour-like presentations of anogenital herpes simplex in HIV-positive patients. Int J STD AIDS. 2011;22:181-186.
- Tong P, Mutasim DF. Herpes simplex virus infection masquerading as condylomata accuminata in a patient with HIV disease. Br J Dermatol. 1996;134:797-800.
- Mosunjac M, Park J, Wang W, et al. Genital and perianal herpes simplex simulating neoplasia in patients with AIDS. AIDS Patient Care STDS. 2009;23:153-158.
- Gubinelli E, Cocuroccia B, Lazzarotto T, et al. Nodular perianal herpes simplex with prominent plasma cell infiltration. Sexually Transm Dis. 2003;30:157-159.
- Nadal SR, Calore EE, Manzione CR, et al. Hypertrophic herpes simplex simulating anal neoplasia in AIDS patients: report of five cases. Dis Colon Rectum. 2005;48:2289-2293.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Burke EM, Karp DL, Wu TC, et al. Atypical oral presentation of herpes simplex virus infection in a patient after orthotopic liver transplantation. Eur Arch Otorhinolaryngol. 1994;251:301-303.
- Tabaee A, Saltman B, Shutter J, et al. Recurrent oral herpes simplex virus infection presenting as a tongue mass. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:376-380.
- Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin's disease. Cancer. 1984;54:3043-3047.
- Burgoyne M, Burke W. Atypical herpes simplex infection in patients with acute myelogenous leukemia recovering from chemotherapy. J Am Acad Dermatol. 1989;20:1125-1126.
- Deza G, Martin-Ezquerra G, Curto-Barredo L, et al. Successful treatment of hypertrophic herpes simplex genitalis in HIV-infected patient with topical imiquimod. J Dermatol. 2015;42:1176-1178.
The Diagnosis: Herpes Simplex Virus Infection
Viral culture of the ulcer was positive for herpes simplex virus type 2 (HHV-2). Bacterial culture grew enteric flora. The patient was started on intravenous acyclovir 5 mg/kg every 8 hours for 7 days and then transitioned to oral acyclovir for chronic suppressive therapy. One month later, there was near-complete reepithelialization with 2 remaining 1-cm shallow ulcers. The verrucous lesions had dried up and were flaking off (Figure). At 6-month follow-up, the ulcers and verrucous lesions had completely resolved.
Herpes simplex virus type 2 is the most common cause of genital and perianal ulcers in immunocompromised individuals.1 Patients classically present with painful grouped vesicles followed by painful superficial ulcers that may rapidly progress to extensive confluent ulceration. A hypertrophic variant of genital herpes characterized by anogenital verrucous lesions, similar to condyloma acuminata, also can be seen in immunocompromised individuals.2 This form has almost exclusively been observed in patients with human immunodeficiency virus and may occur in isolation or together with the ulcerative form.1-5 A case of vegetative HHV infection of the genital area in a patient with common variable immunodeficiency has been reported.6 Verrucous lesions of the mouth secondary to HHV have been observed in Hodgkin lymphoma, acute myelogenous leukemia, and individuals on immunosuppressive medications.7-10
Perianal involvement of Crohn disease typically presents with fistulas, ulcers, abscesses, strictures, and skin tags in some cases. Invasive squamous cell carcinoma may arise within a chronic ulcer of the anogenital area or may itself manifest as an ulcer or anal fissure. Perianal ulcerative skin tuberculosis has been reported in the literature as a rare manifestation of extrapulmonary tuberculosis and should be considered in a patient with an appropriate clinical history. Pyoderma gangrenosum classically pre-sents as a large ulcer with irregular rolled borders, though a rare variant of vegetative pyoderma gangrenosum may manifest as a nodular or verrucous plaque.
Studies to diagnose HHV include viral cell culture, HHV polymerase chain reaction testing, HHV serology, and direct fluorescent antibody testing. Skin biopsy may be necessary to rule out underlying malignancy. Treatment of perianal HHV infection includes acyclovir, valacyclovir, or famciclovir.1,5,6 Hypertrophic lesions often are refractory to first-line antiviral therapy and may require surgical resection or treatment with alternative medications such as imiquimod, a topical immunomodulator.3,5,6,11
The Diagnosis: Herpes Simplex Virus Infection
Viral culture of the ulcer was positive for herpes simplex virus type 2 (HHV-2). Bacterial culture grew enteric flora. The patient was started on intravenous acyclovir 5 mg/kg every 8 hours for 7 days and then transitioned to oral acyclovir for chronic suppressive therapy. One month later, there was near-complete reepithelialization with 2 remaining 1-cm shallow ulcers. The verrucous lesions had dried up and were flaking off (Figure). At 6-month follow-up, the ulcers and verrucous lesions had completely resolved.
Herpes simplex virus type 2 is the most common cause of genital and perianal ulcers in immunocompromised individuals.1 Patients classically present with painful grouped vesicles followed by painful superficial ulcers that may rapidly progress to extensive confluent ulceration. A hypertrophic variant of genital herpes characterized by anogenital verrucous lesions, similar to condyloma acuminata, also can be seen in immunocompromised individuals.2 This form has almost exclusively been observed in patients with human immunodeficiency virus and may occur in isolation or together with the ulcerative form.1-5 A case of vegetative HHV infection of the genital area in a patient with common variable immunodeficiency has been reported.6 Verrucous lesions of the mouth secondary to HHV have been observed in Hodgkin lymphoma, acute myelogenous leukemia, and individuals on immunosuppressive medications.7-10
Perianal involvement of Crohn disease typically presents with fistulas, ulcers, abscesses, strictures, and skin tags in some cases. Invasive squamous cell carcinoma may arise within a chronic ulcer of the anogenital area or may itself manifest as an ulcer or anal fissure. Perianal ulcerative skin tuberculosis has been reported in the literature as a rare manifestation of extrapulmonary tuberculosis and should be considered in a patient with an appropriate clinical history. Pyoderma gangrenosum classically pre-sents as a large ulcer with irregular rolled borders, though a rare variant of vegetative pyoderma gangrenosum may manifest as a nodular or verrucous plaque.
Studies to diagnose HHV include viral cell culture, HHV polymerase chain reaction testing, HHV serology, and direct fluorescent antibody testing. Skin biopsy may be necessary to rule out underlying malignancy. Treatment of perianal HHV infection includes acyclovir, valacyclovir, or famciclovir.1,5,6 Hypertrophic lesions often are refractory to first-line antiviral therapy and may require surgical resection or treatment with alternative medications such as imiquimod, a topical immunomodulator.3,5,6,11
- Ranu H, Lee J, Chio M, et al. Tumour-like presentations of anogenital herpes simplex in HIV-positive patients. Int J STD AIDS. 2011;22:181-186.
- Tong P, Mutasim DF. Herpes simplex virus infection masquerading as condylomata accuminata in a patient with HIV disease. Br J Dermatol. 1996;134:797-800.
- Mosunjac M, Park J, Wang W, et al. Genital and perianal herpes simplex simulating neoplasia in patients with AIDS. AIDS Patient Care STDS. 2009;23:153-158.
- Gubinelli E, Cocuroccia B, Lazzarotto T, et al. Nodular perianal herpes simplex with prominent plasma cell infiltration. Sexually Transm Dis. 2003;30:157-159.
- Nadal SR, Calore EE, Manzione CR, et al. Hypertrophic herpes simplex simulating anal neoplasia in AIDS patients: report of five cases. Dis Colon Rectum. 2005;48:2289-2293.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Burke EM, Karp DL, Wu TC, et al. Atypical oral presentation of herpes simplex virus infection in a patient after orthotopic liver transplantation. Eur Arch Otorhinolaryngol. 1994;251:301-303.
- Tabaee A, Saltman B, Shutter J, et al. Recurrent oral herpes simplex virus infection presenting as a tongue mass. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:376-380.
- Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin's disease. Cancer. 1984;54:3043-3047.
- Burgoyne M, Burke W. Atypical herpes simplex infection in patients with acute myelogenous leukemia recovering from chemotherapy. J Am Acad Dermatol. 1989;20:1125-1126.
- Deza G, Martin-Ezquerra G, Curto-Barredo L, et al. Successful treatment of hypertrophic herpes simplex genitalis in HIV-infected patient with topical imiquimod. J Dermatol. 2015;42:1176-1178.
- Ranu H, Lee J, Chio M, et al. Tumour-like presentations of anogenital herpes simplex in HIV-positive patients. Int J STD AIDS. 2011;22:181-186.
- Tong P, Mutasim DF. Herpes simplex virus infection masquerading as condylomata accuminata in a patient with HIV disease. Br J Dermatol. 1996;134:797-800.
- Mosunjac M, Park J, Wang W, et al. Genital and perianal herpes simplex simulating neoplasia in patients with AIDS. AIDS Patient Care STDS. 2009;23:153-158.
- Gubinelli E, Cocuroccia B, Lazzarotto T, et al. Nodular perianal herpes simplex with prominent plasma cell infiltration. Sexually Transm Dis. 2003;30:157-159.
- Nadal SR, Calore EE, Manzione CR, et al. Hypertrophic herpes simplex simulating anal neoplasia in AIDS patients: report of five cases. Dis Colon Rectum. 2005;48:2289-2293.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Burke EM, Karp DL, Wu TC, et al. Atypical oral presentation of herpes simplex virus infection in a patient after orthotopic liver transplantation. Eur Arch Otorhinolaryngol. 1994;251:301-303.
- Tabaee A, Saltman B, Shutter J, et al. Recurrent oral herpes simplex virus infection presenting as a tongue mass. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:376-380.
- Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin's disease. Cancer. 1984;54:3043-3047.
- Burgoyne M, Burke W. Atypical herpes simplex infection in patients with acute myelogenous leukemia recovering from chemotherapy. J Am Acad Dermatol. 1989;20:1125-1126.
- Deza G, Martin-Ezquerra G, Curto-Barredo L, et al. Successful treatment of hypertrophic herpes simplex genitalis in HIV-infected patient with topical imiquimod. J Dermatol. 2015;42:1176-1178.
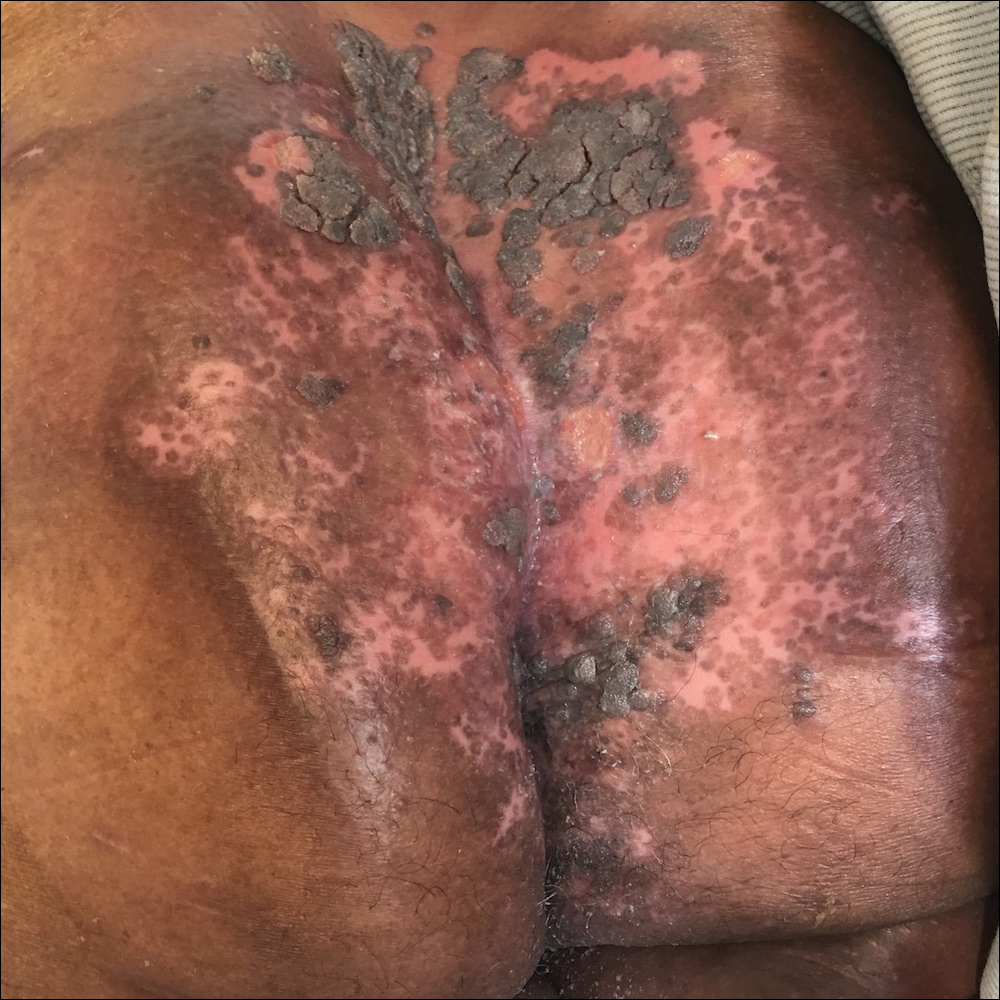
A 75-year-old woman with chronic lymphocytic leukemia undergoing ibrutinib targeted therapy presented to the emergency department with fever and perianal pain of 4 months' duration. The patient denied history of genital or perianal ulcers, warts, masses, bedsores, prolonged immobilization, anal surgeries, or recent travel. She had not been previously treated for the perianal pain. On physical examination there was an 18×15-cm shallow ulceration with rolled borders involving the intergluteal cleft and perianal area. There were numerous hyperpigmented verrucous papules clustered in the center of the ulceration. No vesicles or bullae were present. Laboratory results were pertinent for a white blood cell count of 3600/µL (reference range, 4500-11,000/µL) and absolute neutrophil count of 1300/µL (reference range, 1900-8000/µL). Human immunodeficiency virus testing was negative.
Amantadine-Induced Livedo Reticularis in a Child Treated Off Label for Neurobehavioral Disorders
Livedo reticularis (LR) is a common dermatologic finding consisting of diffuse, reticulated, violaceous patches. It often is a benign physical finding known as cutis marmorata; however, LR can be associated with other medical conditions as well as with the use of some medications.1,2 Amantadine is a common cause of LR in Parkinson disease patients.3,4 We present a rare case of amantadine-induced LR in a pediatric patient and highlight the off-label use of this medication in children.
Case Report
An 8-year-old boy presented with a diffuse rash on the trunk, arms, and legs of 9 months’ duration. The patient denied any associated symptoms as well as alleviating or exacerbating factors. He also denied any changes with temperature. He had no recent international travel and no prior drug allergies. His medical history was remarkable for attention deficit hyperactivity disorder (ADHD), bipolar disorder, and autism spectrum disorder. His previously prescribed medications included atomoxetine, quetiapine, and valproic acid. The only new medication that had been started within the last year was amantadine. Physical examination revealed a diffuse, reticulated, erythematous to violaceous, blanching rash that was most notable on the legs (Figure 1A) but also was present on the trunk (Figure 1B) and arms (Figure 1C). The clinical examination was consistent with LR, which was presumed to be secondary to amantadine use. Given the multiple psychiatric diagnoses and medication history in this young patient, a consultation with child psychiatry was facilitated. His medications and diagnosis were reviewed, and amantadine was discontinued. At a follow-up visit 5 months later, the patient’s LR had improved (Figure 2).
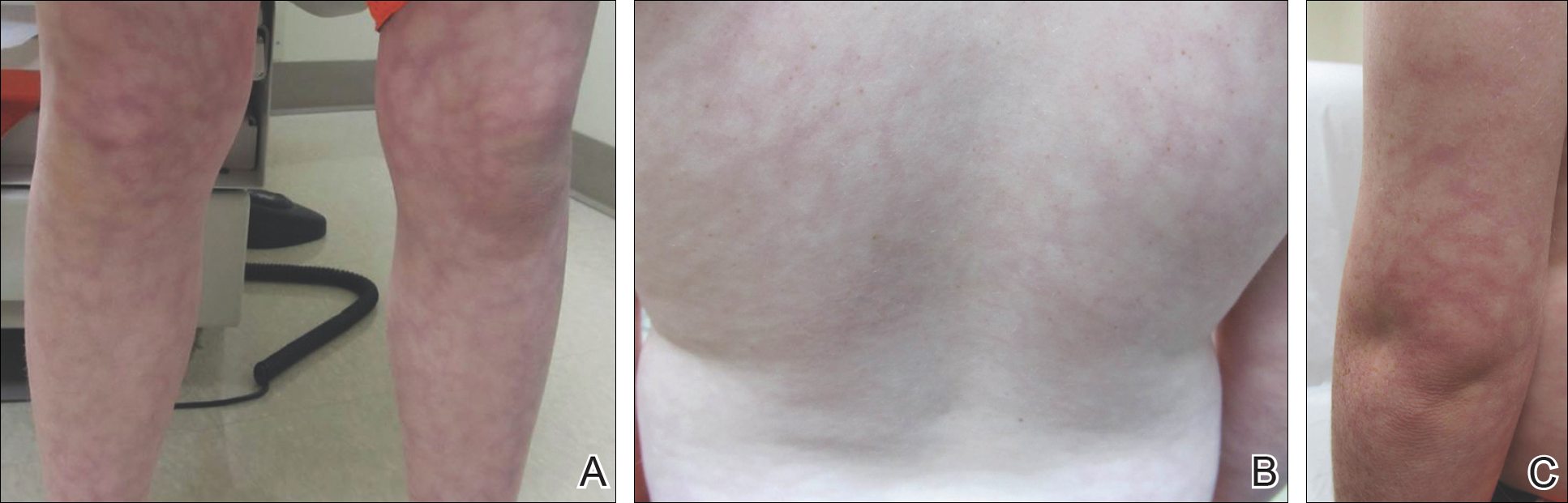
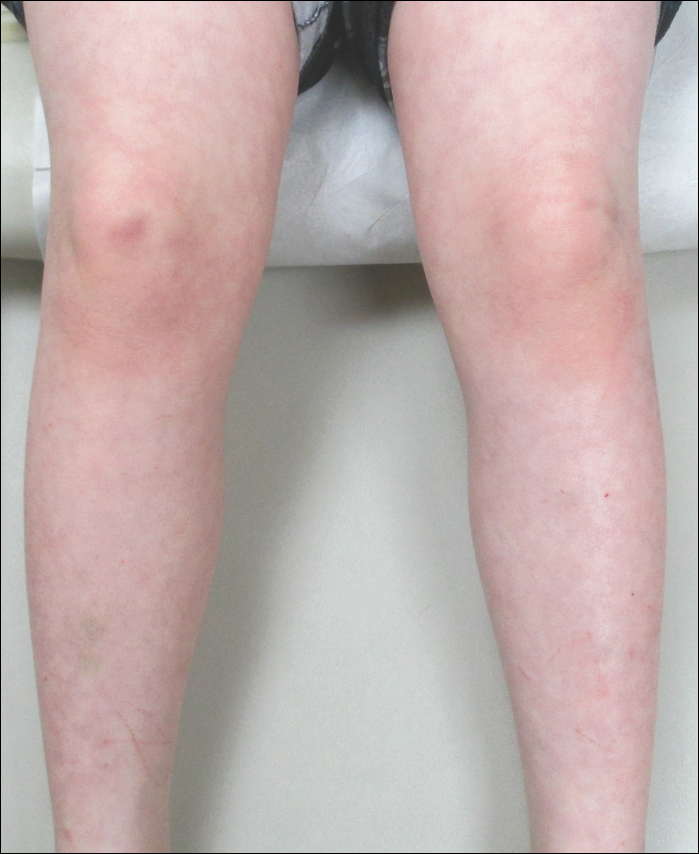
Comment
Amantadine has a well-documented association with LR in patients with Parkinson disease,3,4 which has been reported in up to 40% of those taking amantadine.2 More recently, amantadine has been used off label to treat neurobehavioral disorders in children due to beneficial effects including improvement in attention and concentration, distractibility, and fatigue.5 Our patient was being treated off label with amantadine for ADHD and bipolar disorder. Amantadine acts as a noncompetitive antagonist of the N-methyl-D-aspartate receptor, enhancing dopamine release to reduce symptoms of ADHD.5,6 Additionally, amantadine can cause a depletion of catecholamines in the peripheral nerve terminals, which may lead to dilatation of dermal vessels.4,6 This sequence of events has been proposed as a possible mechanism contributing to amantadine-induced LR, though the pathophysiology is not fully understood.1,3,4
Our case of LR likely was induced by amantadine given the temporal relationship between initiation of the medication, onset of the rash, and the considerable improvement of the rash upon discontinuation of amantadine. Barrera and Browning6 reported another case of amantadine-induced LR in a pediatric patient. Because amantadine is increasingly being used off label to treat childhood neurobehavioral disorders, amantadine-induced LR may become more prevalent in patients who do not have Parkinson disease; therefore, physicians who treat pediatric patients must be aware of this side effect.5
- Quaresma MV, Gomes-Dias AC, Serruya A, et al. Amantadine-induced livedo reticularis: a case report. An Bras Dermatol. 2015;90:745-747.
- Gibbs MB, English JC, Zirwas MJ. Livedo reticularis: an update. J Am Acad Dermatol. 2005;52:1009-1019.
- Silva SB, Miot HA. Case for diagnosis. amantadine-induced livedo reticularis. An Bras Dermatol. 2012;87:319-321.
- Vollum DI, Parkes JD, Doyle D. Livedo reticularis during amantadine treatment. Br Med J. 1971;2:627-628.
- Hosenbocus S, Chahal R. Amantadine: a review of use in child and adolescent psychiatry. J Can Acad Child Adolesc Psychiatry. 2013;22:55-60.
- Barrera F, Browning JC. Likely amantadine-induced livedo reticularis in a child. Pediatr Dermatol. 2012;29:329-330.
Livedo reticularis (LR) is a common dermatologic finding consisting of diffuse, reticulated, violaceous patches. It often is a benign physical finding known as cutis marmorata; however, LR can be associated with other medical conditions as well as with the use of some medications.1,2 Amantadine is a common cause of LR in Parkinson disease patients.3,4 We present a rare case of amantadine-induced LR in a pediatric patient and highlight the off-label use of this medication in children.
Case Report
An 8-year-old boy presented with a diffuse rash on the trunk, arms, and legs of 9 months’ duration. The patient denied any associated symptoms as well as alleviating or exacerbating factors. He also denied any changes with temperature. He had no recent international travel and no prior drug allergies. His medical history was remarkable for attention deficit hyperactivity disorder (ADHD), bipolar disorder, and autism spectrum disorder. His previously prescribed medications included atomoxetine, quetiapine, and valproic acid. The only new medication that had been started within the last year was amantadine. Physical examination revealed a diffuse, reticulated, erythematous to violaceous, blanching rash that was most notable on the legs (Figure 1A) but also was present on the trunk (Figure 1B) and arms (Figure 1C). The clinical examination was consistent with LR, which was presumed to be secondary to amantadine use. Given the multiple psychiatric diagnoses and medication history in this young patient, a consultation with child psychiatry was facilitated. His medications and diagnosis were reviewed, and amantadine was discontinued. At a follow-up visit 5 months later, the patient’s LR had improved (Figure 2).
Comment
Amantadine has a well-documented association with LR in patients with Parkinson disease,3,4 which has been reported in up to 40% of those taking amantadine.2 More recently, amantadine has been used off label to treat neurobehavioral disorders in children due to beneficial effects including improvement in attention and concentration, distractibility, and fatigue.5 Our patient was being treated off label with amantadine for ADHD and bipolar disorder. Amantadine acts as a noncompetitive antagonist of the N-methyl-D-aspartate receptor, enhancing dopamine release to reduce symptoms of ADHD.5,6 Additionally, amantadine can cause a depletion of catecholamines in the peripheral nerve terminals, which may lead to dilatation of dermal vessels.4,6 This sequence of events has been proposed as a possible mechanism contributing to amantadine-induced LR, though the pathophysiology is not fully understood.1,3,4
Our case of LR likely was induced by amantadine given the temporal relationship between initiation of the medication, onset of the rash, and the considerable improvement of the rash upon discontinuation of amantadine. Barrera and Browning6 reported another case of amantadine-induced LR in a pediatric patient. Because amantadine is increasingly being used off label to treat childhood neurobehavioral disorders, amantadine-induced LR may become more prevalent in patients who do not have Parkinson disease; therefore, physicians who treat pediatric patients must be aware of this side effect.5
Livedo reticularis (LR) is a common dermatologic finding consisting of diffuse, reticulated, violaceous patches. It often is a benign physical finding known as cutis marmorata; however, LR can be associated with other medical conditions as well as with the use of some medications.1,2 Amantadine is a common cause of LR in Parkinson disease patients.3,4 We present a rare case of amantadine-induced LR in a pediatric patient and highlight the off-label use of this medication in children.
Case Report
An 8-year-old boy presented with a diffuse rash on the trunk, arms, and legs of 9 months’ duration. The patient denied any associated symptoms as well as alleviating or exacerbating factors. He also denied any changes with temperature. He had no recent international travel and no prior drug allergies. His medical history was remarkable for attention deficit hyperactivity disorder (ADHD), bipolar disorder, and autism spectrum disorder. His previously prescribed medications included atomoxetine, quetiapine, and valproic acid. The only new medication that had been started within the last year was amantadine. Physical examination revealed a diffuse, reticulated, erythematous to violaceous, blanching rash that was most notable on the legs (Figure 1A) but also was present on the trunk (Figure 1B) and arms (Figure 1C). The clinical examination was consistent with LR, which was presumed to be secondary to amantadine use. Given the multiple psychiatric diagnoses and medication history in this young patient, a consultation with child psychiatry was facilitated. His medications and diagnosis were reviewed, and amantadine was discontinued. At a follow-up visit 5 months later, the patient’s LR had improved (Figure 2).
Comment
Amantadine has a well-documented association with LR in patients with Parkinson disease,3,4 which has been reported in up to 40% of those taking amantadine.2 More recently, amantadine has been used off label to treat neurobehavioral disorders in children due to beneficial effects including improvement in attention and concentration, distractibility, and fatigue.5 Our patient was being treated off label with amantadine for ADHD and bipolar disorder. Amantadine acts as a noncompetitive antagonist of the N-methyl-D-aspartate receptor, enhancing dopamine release to reduce symptoms of ADHD.5,6 Additionally, amantadine can cause a depletion of catecholamines in the peripheral nerve terminals, which may lead to dilatation of dermal vessels.4,6 This sequence of events has been proposed as a possible mechanism contributing to amantadine-induced LR, though the pathophysiology is not fully understood.1,3,4
Our case of LR likely was induced by amantadine given the temporal relationship between initiation of the medication, onset of the rash, and the considerable improvement of the rash upon discontinuation of amantadine. Barrera and Browning6 reported another case of amantadine-induced LR in a pediatric patient. Because amantadine is increasingly being used off label to treat childhood neurobehavioral disorders, amantadine-induced LR may become more prevalent in patients who do not have Parkinson disease; therefore, physicians who treat pediatric patients must be aware of this side effect.5
- Quaresma MV, Gomes-Dias AC, Serruya A, et al. Amantadine-induced livedo reticularis: a case report. An Bras Dermatol. 2015;90:745-747.
- Gibbs MB, English JC, Zirwas MJ. Livedo reticularis: an update. J Am Acad Dermatol. 2005;52:1009-1019.
- Silva SB, Miot HA. Case for diagnosis. amantadine-induced livedo reticularis. An Bras Dermatol. 2012;87:319-321.
- Vollum DI, Parkes JD, Doyle D. Livedo reticularis during amantadine treatment. Br Med J. 1971;2:627-628.
- Hosenbocus S, Chahal R. Amantadine: a review of use in child and adolescent psychiatry. J Can Acad Child Adolesc Psychiatry. 2013;22:55-60.
- Barrera F, Browning JC. Likely amantadine-induced livedo reticularis in a child. Pediatr Dermatol. 2012;29:329-330.
- Quaresma MV, Gomes-Dias AC, Serruya A, et al. Amantadine-induced livedo reticularis: a case report. An Bras Dermatol. 2015;90:745-747.
- Gibbs MB, English JC, Zirwas MJ. Livedo reticularis: an update. J Am Acad Dermatol. 2005;52:1009-1019.
- Silva SB, Miot HA. Case for diagnosis. amantadine-induced livedo reticularis. An Bras Dermatol. 2012;87:319-321.
- Vollum DI, Parkes JD, Doyle D. Livedo reticularis during amantadine treatment. Br Med J. 1971;2:627-628.
- Hosenbocus S, Chahal R. Amantadine: a review of use in child and adolescent psychiatry. J Can Acad Child Adolesc Psychiatry. 2013;22:55-60.
- Barrera F, Browning JC. Likely amantadine-induced livedo reticularis in a child. Pediatr Dermatol. 2012;29:329-330.
Practice Points
- Amantadine is a generally well-tolerated medication that is more commonly used for off-label treatment of several pediatric neurobehavioral conditions such as attention deficit hyperactivity disorder, autism spectrum disorders, obsessive compulsive disorder, depression, and others.
- Livedo reticularis has known associations with several medications and diseases; however, the most common presentation is cutis marmorata, a benign condition that typically affects newborns.

Inframammary Macerated Erosion
The Diagnosis: Hailey-Hailey Disease (Benign Familial Chronic Pemphigus)
Our patient had a long-standing history of Hailey-Hailey disease, as confirmed by multiple prior skin biopsies at outside institutions as well as our affiliated site. He began treatment with oral doxycycline 50 mg twice daily for 2 weeks, triamcinolone cream 0.1% twice daily to the affected region, and aluminum acetate solution soaks and chlorhexidine wash daily along with petroleum jelly, which resulted in good control of the disease. The differential diagnosis of eroded plaques, particularly in the axillary, crural, and inframammary folds, is broad and includes candidiasis, inverse psoriasis, contact dermatitis, dermatophyte infection, pemphigus vegetans or foliaceus, and granular parakeratosis.
Hailey-Hailey disease is a genetic disorder with a prevalence of 1 in 50,000 individuals. Most patients develop symptoms during the second or third decades of life.1 Hailey-Hailey disease exhibits an autosomal-dominant pattern of inheritance secondary to mutation in the human ATP2C1 gene, which codes for the ATPase secretory pathway of the Ca2+ transporting pump type 1 (SPCA1) localized in the Golgi apparatus.2 Altered SPCA1 protein reduces concentration of Ca2+ within the Golgi lumen, which in turn impairs the processing of junctional proteins needed for normal cell-to-cell adhesion.1
Clinically, Hailey-Hailey disease is characterized by vesicular or erosive plaques that have a predilection for intertriginous areas of the body.1 The primary lesions often are flaccid vesicles that easily rupture, leaving behind crusted erosions that spread peripherally. The lesions also can appear as macerated plaques resembling torn tissue paper, as in our case. Friction, heat, and sweat exacerbate the disease. Complications occur from secondary bacterial, fungal, and viral colonization. Malodor and vegetations can indicate bacterial or fungal infections and can lead to persistence of skin lesions. Herpes simplex virus infections can exacerbate preexisting lesions.3 Hailey-Hailey disease of the anogenital region also can be complicated by infection with oncogenic strains of human papillomavirus and lead to cutaneous squamous cell carcinoma.4
Hailey-Hailey disease histologically appears as suprabasal and intraepidermal keratinocyte acantholysis,5 which typically is widespread in the epidermis, with large areas of dyscohesion with a dilapidated brick wall-like appearance.1 In more chronic lesions, epidermal hyperplasia, parakeratosis, and focal crusts may be observed. A moderate perivascular lymphocytic infiltrate can be observed in the superficial dermis. Direct immunofluorescence typically is negative.
Topical corticosteroids and antimicrobials are first-line therapies that often only provide temporary suppression. When the disease is refractory to topical therapies, intralesional corticosteroids may be attempted. There is no strong evidence to support the use of systemic therapy, aside from antimicrobial agents (eg, doxycycline) for the use of superinfections. In severe cases, immunomodulating therapies such as prednisone, cyclosporine, methotrexate, dapsone, alefacept, and oral retinoids may be effective.6-8 Surgical therapy also can be considered for recalcitrant disease, including wide excision and grafting, though these techniques can be associated with morbidity.9
Superficial ablative techniques including dermabrasion, laser therapy with CO2 and erbium-doped YAG, photodynamic therapy, and electron beam radiation have been shown to be effective modalities in severe cases.5,9-11 It has been hypothesized that keratinocytes expressing the molecular defect are ablated, while the surrounding normal adnexal epithelium can regenerate normal epithelium. It also is thought that dermal fibrosis leads to better support of the diseased epidermis and decreases the risk for ulceration and fissuring.9
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:887-896.
- Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7:E2259.
- Peppiatt T, Keefe M,White JE. Hailey-Hailey disease--exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 1992;17:201-202.
- Chen MY, Chiu HC, Su LH, et al. Presence of human papillomavirus type 6 DNA in the perineal verrucoid lesions of Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2006;20:1356-1357.
- Graham PM, Melkonian A, Fivenson D. Familial benign chronic pemphigus (Hailey-Hailey disease) treated with electron beam radiation. JAAD Case Rep. 2016;2:159-161.
- Berth-Jones J, Smith SG, Graham-Brown RA, et al. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Sire DJ, Johnson BL. Benign familial chronic pemphigus treated with dapsone. Arch Dermatol. 1971;103:262-265.
- Hunt MJ, Salisbury EL, Painter DM. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996;37:196-198.
- Ortiz AE, Zachary CB. Laser therapy for Hailey-Hailey disease: review of the literature and a case report. Dermatol Reports. 2011;3:E28.
- Don PC, Carney PS, Lynch WS, et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease). J Dermatol Surg Oncol. 1987;13:1187-1194.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;135:423-427.
The Diagnosis: Hailey-Hailey Disease (Benign Familial Chronic Pemphigus)
Our patient had a long-standing history of Hailey-Hailey disease, as confirmed by multiple prior skin biopsies at outside institutions as well as our affiliated site. He began treatment with oral doxycycline 50 mg twice daily for 2 weeks, triamcinolone cream 0.1% twice daily to the affected region, and aluminum acetate solution soaks and chlorhexidine wash daily along with petroleum jelly, which resulted in good control of the disease. The differential diagnosis of eroded plaques, particularly in the axillary, crural, and inframammary folds, is broad and includes candidiasis, inverse psoriasis, contact dermatitis, dermatophyte infection, pemphigus vegetans or foliaceus, and granular parakeratosis.
Hailey-Hailey disease is a genetic disorder with a prevalence of 1 in 50,000 individuals. Most patients develop symptoms during the second or third decades of life.1 Hailey-Hailey disease exhibits an autosomal-dominant pattern of inheritance secondary to mutation in the human ATP2C1 gene, which codes for the ATPase secretory pathway of the Ca2+ transporting pump type 1 (SPCA1) localized in the Golgi apparatus.2 Altered SPCA1 protein reduces concentration of Ca2+ within the Golgi lumen, which in turn impairs the processing of junctional proteins needed for normal cell-to-cell adhesion.1
Clinically, Hailey-Hailey disease is characterized by vesicular or erosive plaques that have a predilection for intertriginous areas of the body.1 The primary lesions often are flaccid vesicles that easily rupture, leaving behind crusted erosions that spread peripherally. The lesions also can appear as macerated plaques resembling torn tissue paper, as in our case. Friction, heat, and sweat exacerbate the disease. Complications occur from secondary bacterial, fungal, and viral colonization. Malodor and vegetations can indicate bacterial or fungal infections and can lead to persistence of skin lesions. Herpes simplex virus infections can exacerbate preexisting lesions.3 Hailey-Hailey disease of the anogenital region also can be complicated by infection with oncogenic strains of human papillomavirus and lead to cutaneous squamous cell carcinoma.4
Hailey-Hailey disease histologically appears as suprabasal and intraepidermal keratinocyte acantholysis,5 which typically is widespread in the epidermis, with large areas of dyscohesion with a dilapidated brick wall-like appearance.1 In more chronic lesions, epidermal hyperplasia, parakeratosis, and focal crusts may be observed. A moderate perivascular lymphocytic infiltrate can be observed in the superficial dermis. Direct immunofluorescence typically is negative.
Topical corticosteroids and antimicrobials are first-line therapies that often only provide temporary suppression. When the disease is refractory to topical therapies, intralesional corticosteroids may be attempted. There is no strong evidence to support the use of systemic therapy, aside from antimicrobial agents (eg, doxycycline) for the use of superinfections. In severe cases, immunomodulating therapies such as prednisone, cyclosporine, methotrexate, dapsone, alefacept, and oral retinoids may be effective.6-8 Surgical therapy also can be considered for recalcitrant disease, including wide excision and grafting, though these techniques can be associated with morbidity.9
Superficial ablative techniques including dermabrasion, laser therapy with CO2 and erbium-doped YAG, photodynamic therapy, and electron beam radiation have been shown to be effective modalities in severe cases.5,9-11 It has been hypothesized that keratinocytes expressing the molecular defect are ablated, while the surrounding normal adnexal epithelium can regenerate normal epithelium. It also is thought that dermal fibrosis leads to better support of the diseased epidermis and decreases the risk for ulceration and fissuring.9
The Diagnosis: Hailey-Hailey Disease (Benign Familial Chronic Pemphigus)
Our patient had a long-standing history of Hailey-Hailey disease, as confirmed by multiple prior skin biopsies at outside institutions as well as our affiliated site. He began treatment with oral doxycycline 50 mg twice daily for 2 weeks, triamcinolone cream 0.1% twice daily to the affected region, and aluminum acetate solution soaks and chlorhexidine wash daily along with petroleum jelly, which resulted in good control of the disease. The differential diagnosis of eroded plaques, particularly in the axillary, crural, and inframammary folds, is broad and includes candidiasis, inverse psoriasis, contact dermatitis, dermatophyte infection, pemphigus vegetans or foliaceus, and granular parakeratosis.
Hailey-Hailey disease is a genetic disorder with a prevalence of 1 in 50,000 individuals. Most patients develop symptoms during the second or third decades of life.1 Hailey-Hailey disease exhibits an autosomal-dominant pattern of inheritance secondary to mutation in the human ATP2C1 gene, which codes for the ATPase secretory pathway of the Ca2+ transporting pump type 1 (SPCA1) localized in the Golgi apparatus.2 Altered SPCA1 protein reduces concentration of Ca2+ within the Golgi lumen, which in turn impairs the processing of junctional proteins needed for normal cell-to-cell adhesion.1
Clinically, Hailey-Hailey disease is characterized by vesicular or erosive plaques that have a predilection for intertriginous areas of the body.1 The primary lesions often are flaccid vesicles that easily rupture, leaving behind crusted erosions that spread peripherally. The lesions also can appear as macerated plaques resembling torn tissue paper, as in our case. Friction, heat, and sweat exacerbate the disease. Complications occur from secondary bacterial, fungal, and viral colonization. Malodor and vegetations can indicate bacterial or fungal infections and can lead to persistence of skin lesions. Herpes simplex virus infections can exacerbate preexisting lesions.3 Hailey-Hailey disease of the anogenital region also can be complicated by infection with oncogenic strains of human papillomavirus and lead to cutaneous squamous cell carcinoma.4
Hailey-Hailey disease histologically appears as suprabasal and intraepidermal keratinocyte acantholysis,5 which typically is widespread in the epidermis, with large areas of dyscohesion with a dilapidated brick wall-like appearance.1 In more chronic lesions, epidermal hyperplasia, parakeratosis, and focal crusts may be observed. A moderate perivascular lymphocytic infiltrate can be observed in the superficial dermis. Direct immunofluorescence typically is negative.
Topical corticosteroids and antimicrobials are first-line therapies that often only provide temporary suppression. When the disease is refractory to topical therapies, intralesional corticosteroids may be attempted. There is no strong evidence to support the use of systemic therapy, aside from antimicrobial agents (eg, doxycycline) for the use of superinfections. In severe cases, immunomodulating therapies such as prednisone, cyclosporine, methotrexate, dapsone, alefacept, and oral retinoids may be effective.6-8 Surgical therapy also can be considered for recalcitrant disease, including wide excision and grafting, though these techniques can be associated with morbidity.9
Superficial ablative techniques including dermabrasion, laser therapy with CO2 and erbium-doped YAG, photodynamic therapy, and electron beam radiation have been shown to be effective modalities in severe cases.5,9-11 It has been hypothesized that keratinocytes expressing the molecular defect are ablated, while the surrounding normal adnexal epithelium can regenerate normal epithelium. It also is thought that dermal fibrosis leads to better support of the diseased epidermis and decreases the risk for ulceration and fissuring.9
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:887-896.
- Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7:E2259.
- Peppiatt T, Keefe M,White JE. Hailey-Hailey disease--exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 1992;17:201-202.
- Chen MY, Chiu HC, Su LH, et al. Presence of human papillomavirus type 6 DNA in the perineal verrucoid lesions of Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2006;20:1356-1357.
- Graham PM, Melkonian A, Fivenson D. Familial benign chronic pemphigus (Hailey-Hailey disease) treated with electron beam radiation. JAAD Case Rep. 2016;2:159-161.
- Berth-Jones J, Smith SG, Graham-Brown RA, et al. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Sire DJ, Johnson BL. Benign familial chronic pemphigus treated with dapsone. Arch Dermatol. 1971;103:262-265.
- Hunt MJ, Salisbury EL, Painter DM. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996;37:196-198.
- Ortiz AE, Zachary CB. Laser therapy for Hailey-Hailey disease: review of the literature and a case report. Dermatol Reports. 2011;3:E28.
- Don PC, Carney PS, Lynch WS, et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease). J Dermatol Surg Oncol. 1987;13:1187-1194.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;135:423-427.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:887-896.
- Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7:E2259.
- Peppiatt T, Keefe M,White JE. Hailey-Hailey disease--exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 1992;17:201-202.
- Chen MY, Chiu HC, Su LH, et al. Presence of human papillomavirus type 6 DNA in the perineal verrucoid lesions of Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2006;20:1356-1357.
- Graham PM, Melkonian A, Fivenson D. Familial benign chronic pemphigus (Hailey-Hailey disease) treated with electron beam radiation. JAAD Case Rep. 2016;2:159-161.
- Berth-Jones J, Smith SG, Graham-Brown RA, et al. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Sire DJ, Johnson BL. Benign familial chronic pemphigus treated with dapsone. Arch Dermatol. 1971;103:262-265.
- Hunt MJ, Salisbury EL, Painter DM. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996;37:196-198.
- Ortiz AE, Zachary CB. Laser therapy for Hailey-Hailey disease: review of the literature and a case report. Dermatol Reports. 2011;3:E28.
- Don PC, Carney PS, Lynch WS, et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease). J Dermatol Surg Oncol. 1987;13:1187-1194.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;135:423-427.
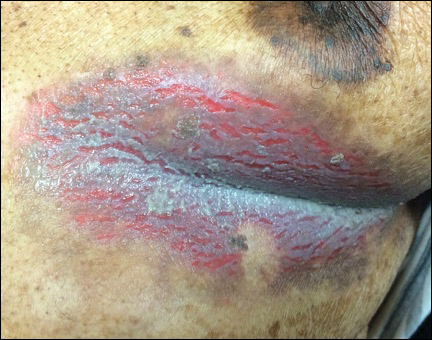
An 81-year-old man presented with a painful erosion in the left inframammary region of 2 weeks' duration. He described the lesion as pruritic and burning. He reported having prior similar episodes in the bilateral groin, axilla, and lower abdomen that often were malodorous. Use of triamcinolone cream 0.1% up to 4 times daily resulted in little relief of the erosion. Of note, the patient reported therapies for prior sites had included oral doxycycline 50 mg twice daily, clobetasol cream, and clindamycin solution, which provided limited relief but eventual resolution. Application of cold aluminum acetate solution compresses for 5 minutes daily irritated the skin even further and led to bleeding at the affected sites. The patient's father had a history of similar skin lesions.
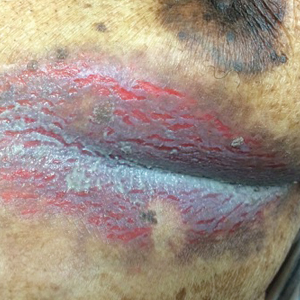
Sunscreens: Survey of the Cutis Editorial Board
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on sunscreens. Here’s what we found.
What sun protection factor (SPF) do you recommend for the majority of your patients?

Fifty percent of dermatologists we surveyed recommend SPF 30. SPF 50 was recommended by 26%, SPF 50+ by 21%, and SPF 15 by only 2%.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Half of our Editorial Board recommends sunscreen with SPF 30, with many recommending SPF 50 or higher. This trend toward sunscreens with higher SPF is consistent with a survey-based study with 97% of dermatologists stating they were comfortable recommending sunscreens with an SPF of 50 or higher and 83.3% stating that they believe that high SPF sunscreens provide an additional margin of safety (Farberg et al). These trends are supported by a randomized, double-blind, split-face clinical trial in which participants applied either SPF 50+ or SPF 100+ sunscreen after exposure to natural sunlight. The results showed that SPF 100+ sunscreen was remarkably more effective in protecting against sunburn than SPF 50+ sunscreen in actual use conditions (Williams et al).
Next page: Spray sunscreens
Which patient populations do you feel may benefit from spray sunscreens?

Two-thirds of dermatologists indicated that spray sunscreens may benefit patients traveling alone. Men with bald spots also may benefit (62%), as well as athletes, children, and older patients (57% each).
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
As dermatologists, we tell our patients that the best sunscreens are ones that are used consistently. Spray sunscreens are likely as effective as lotions (Ou-Yang et al). There has been a clear trend in consumer purchasing of spray sunscreens from 2011 to 2016 (Teplitz et al). Spray sunscreens may benefit those traveling alone, particularly for hard-to-reach areas.
Next page: Supplemental vitamin D
In patients who apply sunscreen regularly, do you recommend supplemental vitamin D3?

More than half (53%) of dermatologists recommend supplemental vitamin D3.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Because use of photoprotection results in decreased vitamin D levels in most individuals, it is good practice to recommend vitamin D supplementation in patients who are applying sunscreen regularly (Bogaczewicz et al).
Next page: Sunscreen compliance
What is the most often heard reason(s) for not using sunscreen in your patients?

Nearly three-quarters (72%) of dermatologists reported that patients do not use sunscreen because of cosmetic acceptance. Almost one-third (31%) said their patients prefer “natural” products. Price was a factor for 26%. Fewer dermatologists indicated risk of environmental damage (14%), allergy (12%), cancer induction (5%), and hormonal alteration (5%) were reasons patients are not compliant.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Cosmetic acceptance is paramount for patient compliance for sunscreen application. These results from our Editorial Board echo a study on sunscreen product performance and other determinants of consumer preferences, which cited “cosmetic elegance” as an important factor in choosing sunscreens (Xu et al). Dermatologists must stress to patients to find a sunscreen that they find acceptable in terms of vehicle and price to increase compliance.
Next page: Sunscreens in pregnant women
What sunscreens do you recommend to pregnant women and children?

Most dermatologists (86%) recommend physical blockers “chemical-free” only sunscreens to pregnant women and children.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
While absorption of sunscreen by human embryos is likely negligible, because there is limited data on sunscreen effects in embryos and children, it is reasonable to recommend physical blockers for pregnant women and children.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
As a dermatologist married to a pediatrician, I try to get my kids to embrace sun-protection strategies. For the little ones it’s hard, but as they have gotten older and been exposed to more derm journals sitting around with pretty graphic pictures, they seem to get on board, even when away at summer camp on their own. If only our patients knew what our kids do.—Joel L. Cohen, MD (Denver, Colorado)
The most important factor in getting patient compliance with sunscreen usage is “cosmetic acceptance.” If they or their children or their spouse don’t like the feel, they won’t use it.—Vincent A. DeLeo, MD (Los Angeles, California)
Not using photoprotection with sunscreen is like crossing a busy road without looking both ways first.—James Q. Del Rosso, DO (Las Vegas, Nevada)
I do not recommend spray sunscreens. At least half of the spray seems to go in the air rather than on the skin. And people often do not rub the spray into their skin well enough. Lotions are better!—Lawrence J. Green, MD (Washington, DC)
The most important factor in sunscreen is not SPF; educate patients on the important role vehicle and sweating play in the length of sun protection.—Orit Markowitz, MD (New York, New York)
Reapplying sunscreen in the appropriate amount is key to blocking the danger rays of the sun.—Vineet Mishra, MD (San Antonio, Texas)
A good sunscreen is the one you put on properly. Regardless of the formulation, make sure you apply the sunscreen evenly to all exposed skin and reapply according to directions on the container. Remember, a regular white T-shirt has minimal SPF 4-5. Either wear sun-protective clothing or wear sunscreen underneath!—Larisa Ravitskiy, MD (Gahanna, Ohio)
Sun protection and sunscreen application go hand-in-hand. We can still enjoy the outdoors without getting excessive UV exposure.—Anthony M. Rossi, MD (New York, New York)
Sunscreens are only part of sun protection. Make sure to reapply them regularly, try to avoid direct sun between about 10 AM and 2 PM if possible, and wear a hat with a wide brim (not a baseball cap, which, after all, is designed for catching baseballs, not sun protection).—Robert I. Rudolph, MD (Wyomissing, Pennsylvania)
Sunscreens keep you younger looking longer!—Richard K. Scher, MD (New York, New York)
The dentist says only floss the teeth you want to keep. I tell patients to only sun block the skin they want to keep.—Daniel M. Siegel, MD, MS (Brooklyn, New York)
The best sunscreen is the one that is used! If it's too greasy or drying, smells bad or stings, it won't be used. Stick to the one YOU like, but at least SPF 30 or better.—Stephen P. Stone, MD, (Springfield, Illinois)
Sunscreen can be a meaningful part of your sun-protection regimen used in conjunction with sun-protective clothing, sun safe behaviors, and a diet rich in natural antioxidants.—Michelle Tarbox, MD (Lubbock, Texas)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from August 2, 2018, to September 2, 2018. A total of 42 usable responses were received.
Bogaczewicz J, Karczmarewicz E, Pludowski P, et al. Requirement for vitamin D supplementation in patients using photoprotection: variations in vitamin D levels and bone formation markers. Int J Dermatol. 2016;55:e176-e183.
Farberg AS, Glazer AM, Rigel AC, et al. Dermatologists’ perceptions, recommendations, and use of sunscreen. JAMA Dermatol. 2017;153:99-101.
Ou-Yang H, Stanfield J, Cole C, et al. High-SPF sunscreens (SPF ≥ 70) may provide ultraviolet protection above minimal recommended levels by adequately compensating for lower sunscreen user application amounts. J Am Acad Dermatol. 2012;67:1220-1227.
Teplitz RW, Glazer AM, Svoboda RM, et al. Trends in US sunscreen formulations: impact of increasing spray usage. J Am Acad Dermatol. 2018;78:187-189.
Williams JD, Maitra P, Atillasoy E, et al. SPF 100+ sunscreen is more protective against sunburn than SPF 50+ in actual use: Results of a randomized, double-blind, split-face, natural sunlight exposure clinical trial. J Am Acad Dermatol. 2018;78:902.e2-910.e2.
Xu S, Kwa M, Agarwal A, et al. Sunscreen product performance and other determinants of consumer preferences. JAMA Dermatol. 2016;152:920-927.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on sunscreens. Here’s what we found.
What sun protection factor (SPF) do you recommend for the majority of your patients?
Fifty percent of dermatologists we surveyed recommend SPF 30. SPF 50 was recommended by 26%, SPF 50+ by 21%, and SPF 15 by only 2%.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Half of our Editorial Board recommends sunscreen with SPF 30, with many recommending SPF 50 or higher. This trend toward sunscreens with higher SPF is consistent with a survey-based study with 97% of dermatologists stating they were comfortable recommending sunscreens with an SPF of 50 or higher and 83.3% stating that they believe that high SPF sunscreens provide an additional margin of safety (Farberg et al). These trends are supported by a randomized, double-blind, split-face clinical trial in which participants applied either SPF 50+ or SPF 100+ sunscreen after exposure to natural sunlight. The results showed that SPF 100+ sunscreen was remarkably more effective in protecting against sunburn than SPF 50+ sunscreen in actual use conditions (Williams et al).
Next page: Spray sunscreens
Which patient populations do you feel may benefit from spray sunscreens?
Two-thirds of dermatologists indicated that spray sunscreens may benefit patients traveling alone. Men with bald spots also may benefit (62%), as well as athletes, children, and older patients (57% each).
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
As dermatologists, we tell our patients that the best sunscreens are ones that are used consistently. Spray sunscreens are likely as effective as lotions (Ou-Yang et al). There has been a clear trend in consumer purchasing of spray sunscreens from 2011 to 2016 (Teplitz et al). Spray sunscreens may benefit those traveling alone, particularly for hard-to-reach areas.
Next page: Supplemental vitamin D
In patients who apply sunscreen regularly, do you recommend supplemental vitamin D3?
More than half (53%) of dermatologists recommend supplemental vitamin D3.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Because use of photoprotection results in decreased vitamin D levels in most individuals, it is good practice to recommend vitamin D supplementation in patients who are applying sunscreen regularly (Bogaczewicz et al).
Next page: Sunscreen compliance
What is the most often heard reason(s) for not using sunscreen in your patients?
Nearly three-quarters (72%) of dermatologists reported that patients do not use sunscreen because of cosmetic acceptance. Almost one-third (31%) said their patients prefer “natural” products. Price was a factor for 26%. Fewer dermatologists indicated risk of environmental damage (14%), allergy (12%), cancer induction (5%), and hormonal alteration (5%) were reasons patients are not compliant.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Cosmetic acceptance is paramount for patient compliance for sunscreen application. These results from our Editorial Board echo a study on sunscreen product performance and other determinants of consumer preferences, which cited “cosmetic elegance” as an important factor in choosing sunscreens (Xu et al). Dermatologists must stress to patients to find a sunscreen that they find acceptable in terms of vehicle and price to increase compliance.
Next page: Sunscreens in pregnant women
What sunscreens do you recommend to pregnant women and children?
Most dermatologists (86%) recommend physical blockers “chemical-free” only sunscreens to pregnant women and children.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
While absorption of sunscreen by human embryos is likely negligible, because there is limited data on sunscreen effects in embryos and children, it is reasonable to recommend physical blockers for pregnant women and children.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
As a dermatologist married to a pediatrician, I try to get my kids to embrace sun-protection strategies. For the little ones it’s hard, but as they have gotten older and been exposed to more derm journals sitting around with pretty graphic pictures, they seem to get on board, even when away at summer camp on their own. If only our patients knew what our kids do.—Joel L. Cohen, MD (Denver, Colorado)
The most important factor in getting patient compliance with sunscreen usage is “cosmetic acceptance.” If they or their children or their spouse don’t like the feel, they won’t use it.—Vincent A. DeLeo, MD (Los Angeles, California)
Not using photoprotection with sunscreen is like crossing a busy road without looking both ways first.—James Q. Del Rosso, DO (Las Vegas, Nevada)
I do not recommend spray sunscreens. At least half of the spray seems to go in the air rather than on the skin. And people often do not rub the spray into their skin well enough. Lotions are better!—Lawrence J. Green, MD (Washington, DC)
The most important factor in sunscreen is not SPF; educate patients on the important role vehicle and sweating play in the length of sun protection.—Orit Markowitz, MD (New York, New York)
Reapplying sunscreen in the appropriate amount is key to blocking the danger rays of the sun.—Vineet Mishra, MD (San Antonio, Texas)
A good sunscreen is the one you put on properly. Regardless of the formulation, make sure you apply the sunscreen evenly to all exposed skin and reapply according to directions on the container. Remember, a regular white T-shirt has minimal SPF 4-5. Either wear sun-protective clothing or wear sunscreen underneath!—Larisa Ravitskiy, MD (Gahanna, Ohio)
Sun protection and sunscreen application go hand-in-hand. We can still enjoy the outdoors without getting excessive UV exposure.—Anthony M. Rossi, MD (New York, New York)
Sunscreens are only part of sun protection. Make sure to reapply them regularly, try to avoid direct sun between about 10 AM and 2 PM if possible, and wear a hat with a wide brim (not a baseball cap, which, after all, is designed for catching baseballs, not sun protection).—Robert I. Rudolph, MD (Wyomissing, Pennsylvania)
Sunscreens keep you younger looking longer!—Richard K. Scher, MD (New York, New York)
The dentist says only floss the teeth you want to keep. I tell patients to only sun block the skin they want to keep.—Daniel M. Siegel, MD, MS (Brooklyn, New York)
The best sunscreen is the one that is used! If it's too greasy or drying, smells bad or stings, it won't be used. Stick to the one YOU like, but at least SPF 30 or better.—Stephen P. Stone, MD, (Springfield, Illinois)
Sunscreen can be a meaningful part of your sun-protection regimen used in conjunction with sun-protective clothing, sun safe behaviors, and a diet rich in natural antioxidants.—Michelle Tarbox, MD (Lubbock, Texas)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from August 2, 2018, to September 2, 2018. A total of 42 usable responses were received.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on sunscreens. Here’s what we found.
What sun protection factor (SPF) do you recommend for the majority of your patients?
Fifty percent of dermatologists we surveyed recommend SPF 30. SPF 50 was recommended by 26%, SPF 50+ by 21%, and SPF 15 by only 2%.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Half of our Editorial Board recommends sunscreen with SPF 30, with many recommending SPF 50 or higher. This trend toward sunscreens with higher SPF is consistent with a survey-based study with 97% of dermatologists stating they were comfortable recommending sunscreens with an SPF of 50 or higher and 83.3% stating that they believe that high SPF sunscreens provide an additional margin of safety (Farberg et al). These trends are supported by a randomized, double-blind, split-face clinical trial in which participants applied either SPF 50+ or SPF 100+ sunscreen after exposure to natural sunlight. The results showed that SPF 100+ sunscreen was remarkably more effective in protecting against sunburn than SPF 50+ sunscreen in actual use conditions (Williams et al).
Next page: Spray sunscreens
Which patient populations do you feel may benefit from spray sunscreens?
Two-thirds of dermatologists indicated that spray sunscreens may benefit patients traveling alone. Men with bald spots also may benefit (62%), as well as athletes, children, and older patients (57% each).
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
As dermatologists, we tell our patients that the best sunscreens are ones that are used consistently. Spray sunscreens are likely as effective as lotions (Ou-Yang et al). There has been a clear trend in consumer purchasing of spray sunscreens from 2011 to 2016 (Teplitz et al). Spray sunscreens may benefit those traveling alone, particularly for hard-to-reach areas.
Next page: Supplemental vitamin D
In patients who apply sunscreen regularly, do you recommend supplemental vitamin D3?
More than half (53%) of dermatologists recommend supplemental vitamin D3.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Because use of photoprotection results in decreased vitamin D levels in most individuals, it is good practice to recommend vitamin D supplementation in patients who are applying sunscreen regularly (Bogaczewicz et al).
Next page: Sunscreen compliance
What is the most often heard reason(s) for not using sunscreen in your patients?
Nearly three-quarters (72%) of dermatologists reported that patients do not use sunscreen because of cosmetic acceptance. Almost one-third (31%) said their patients prefer “natural” products. Price was a factor for 26%. Fewer dermatologists indicated risk of environmental damage (14%), allergy (12%), cancer induction (5%), and hormonal alteration (5%) were reasons patients are not compliant.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Cosmetic acceptance is paramount for patient compliance for sunscreen application. These results from our Editorial Board echo a study on sunscreen product performance and other determinants of consumer preferences, which cited “cosmetic elegance” as an important factor in choosing sunscreens (Xu et al). Dermatologists must stress to patients to find a sunscreen that they find acceptable in terms of vehicle and price to increase compliance.
Next page: Sunscreens in pregnant women
What sunscreens do you recommend to pregnant women and children?
Most dermatologists (86%) recommend physical blockers “chemical-free” only sunscreens to pregnant women and children.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
While absorption of sunscreen by human embryos is likely negligible, because there is limited data on sunscreen effects in embryos and children, it is reasonable to recommend physical blockers for pregnant women and children.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
As a dermatologist married to a pediatrician, I try to get my kids to embrace sun-protection strategies. For the little ones it’s hard, but as they have gotten older and been exposed to more derm journals sitting around with pretty graphic pictures, they seem to get on board, even when away at summer camp on their own. If only our patients knew what our kids do.—Joel L. Cohen, MD (Denver, Colorado)
The most important factor in getting patient compliance with sunscreen usage is “cosmetic acceptance.” If they or their children or their spouse don’t like the feel, they won’t use it.—Vincent A. DeLeo, MD (Los Angeles, California)
Not using photoprotection with sunscreen is like crossing a busy road without looking both ways first.—James Q. Del Rosso, DO (Las Vegas, Nevada)
I do not recommend spray sunscreens. At least half of the spray seems to go in the air rather than on the skin. And people often do not rub the spray into their skin well enough. Lotions are better!—Lawrence J. Green, MD (Washington, DC)
The most important factor in sunscreen is not SPF; educate patients on the important role vehicle and sweating play in the length of sun protection.—Orit Markowitz, MD (New York, New York)
Reapplying sunscreen in the appropriate amount is key to blocking the danger rays of the sun.—Vineet Mishra, MD (San Antonio, Texas)
A good sunscreen is the one you put on properly. Regardless of the formulation, make sure you apply the sunscreen evenly to all exposed skin and reapply according to directions on the container. Remember, a regular white T-shirt has minimal SPF 4-5. Either wear sun-protective clothing or wear sunscreen underneath!—Larisa Ravitskiy, MD (Gahanna, Ohio)
Sun protection and sunscreen application go hand-in-hand. We can still enjoy the outdoors without getting excessive UV exposure.—Anthony M. Rossi, MD (New York, New York)
Sunscreens are only part of sun protection. Make sure to reapply them regularly, try to avoid direct sun between about 10 AM and 2 PM if possible, and wear a hat with a wide brim (not a baseball cap, which, after all, is designed for catching baseballs, not sun protection).—Robert I. Rudolph, MD (Wyomissing, Pennsylvania)
Sunscreens keep you younger looking longer!—Richard K. Scher, MD (New York, New York)
The dentist says only floss the teeth you want to keep. I tell patients to only sun block the skin they want to keep.—Daniel M. Siegel, MD, MS (Brooklyn, New York)
The best sunscreen is the one that is used! If it's too greasy or drying, smells bad or stings, it won't be used. Stick to the one YOU like, but at least SPF 30 or better.—Stephen P. Stone, MD, (Springfield, Illinois)
Sunscreen can be a meaningful part of your sun-protection regimen used in conjunction with sun-protective clothing, sun safe behaviors, and a diet rich in natural antioxidants.—Michelle Tarbox, MD (Lubbock, Texas)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from August 2, 2018, to September 2, 2018. A total of 42 usable responses were received.
Bogaczewicz J, Karczmarewicz E, Pludowski P, et al. Requirement for vitamin D supplementation in patients using photoprotection: variations in vitamin D levels and bone formation markers. Int J Dermatol. 2016;55:e176-e183.
Farberg AS, Glazer AM, Rigel AC, et al. Dermatologists’ perceptions, recommendations, and use of sunscreen. JAMA Dermatol. 2017;153:99-101.
Ou-Yang H, Stanfield J, Cole C, et al. High-SPF sunscreens (SPF ≥ 70) may provide ultraviolet protection above minimal recommended levels by adequately compensating for lower sunscreen user application amounts. J Am Acad Dermatol. 2012;67:1220-1227.
Teplitz RW, Glazer AM, Svoboda RM, et al. Trends in US sunscreen formulations: impact of increasing spray usage. J Am Acad Dermatol. 2018;78:187-189.
Williams JD, Maitra P, Atillasoy E, et al. SPF 100+ sunscreen is more protective against sunburn than SPF 50+ in actual use: Results of a randomized, double-blind, split-face, natural sunlight exposure clinical trial. J Am Acad Dermatol. 2018;78:902.e2-910.e2.
Xu S, Kwa M, Agarwal A, et al. Sunscreen product performance and other determinants of consumer preferences. JAMA Dermatol. 2016;152:920-927.
Bogaczewicz J, Karczmarewicz E, Pludowski P, et al. Requirement for vitamin D supplementation in patients using photoprotection: variations in vitamin D levels and bone formation markers. Int J Dermatol. 2016;55:e176-e183.
Farberg AS, Glazer AM, Rigel AC, et al. Dermatologists’ perceptions, recommendations, and use of sunscreen. JAMA Dermatol. 2017;153:99-101.
Ou-Yang H, Stanfield J, Cole C, et al. High-SPF sunscreens (SPF ≥ 70) may provide ultraviolet protection above minimal recommended levels by adequately compensating for lower sunscreen user application amounts. J Am Acad Dermatol. 2012;67:1220-1227.
Teplitz RW, Glazer AM, Svoboda RM, et al. Trends in US sunscreen formulations: impact of increasing spray usage. J Am Acad Dermatol. 2018;78:187-189.
Williams JD, Maitra P, Atillasoy E, et al. SPF 100+ sunscreen is more protective against sunburn than SPF 50+ in actual use: Results of a randomized, double-blind, split-face, natural sunlight exposure clinical trial. J Am Acad Dermatol. 2018;78:902.e2-910.e2.
Xu S, Kwa M, Agarwal A, et al. Sunscreen product performance and other determinants of consumer preferences. JAMA Dermatol. 2016;152:920-927.
Nevus of Ota Associated With a Primary Uveal Melanoma and Intracranial Melanoma Metastasis
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
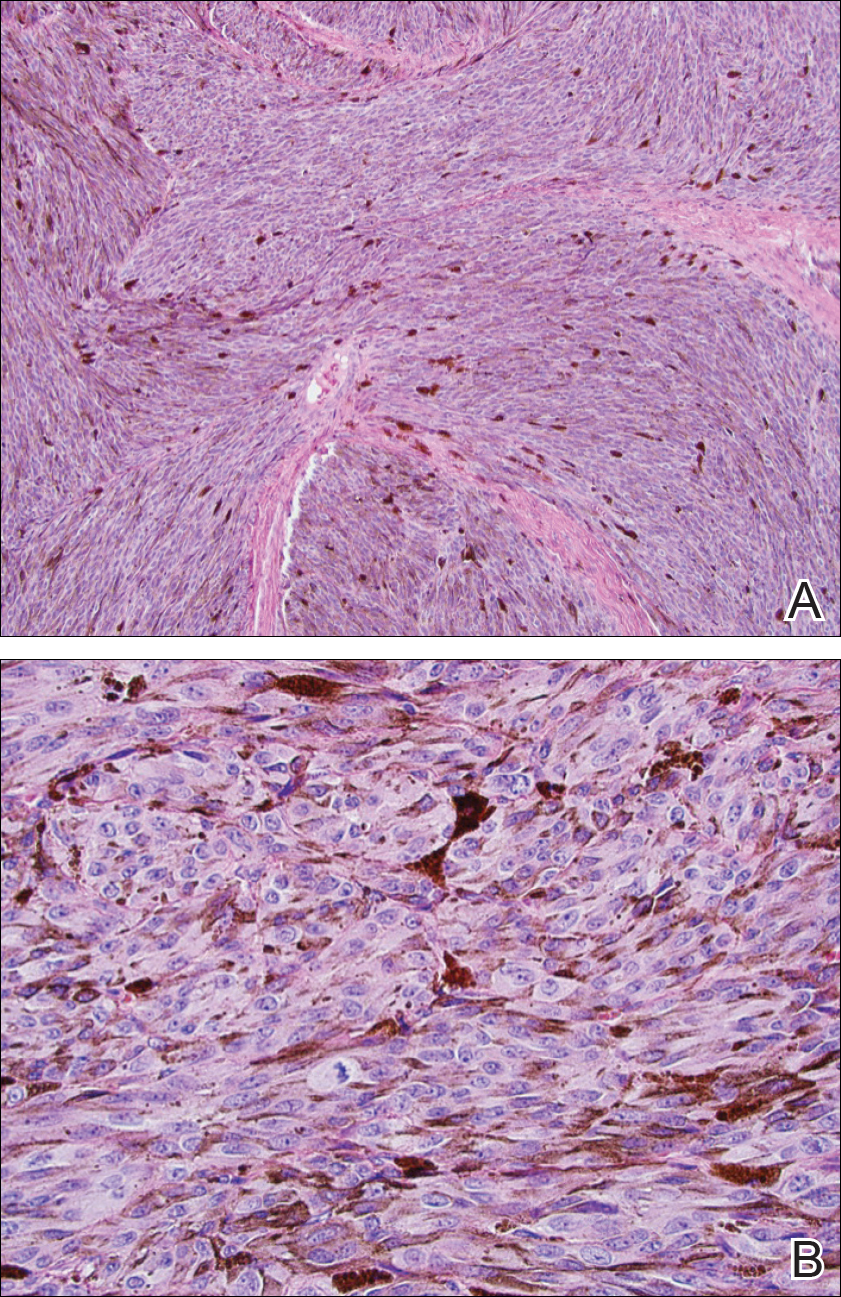
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
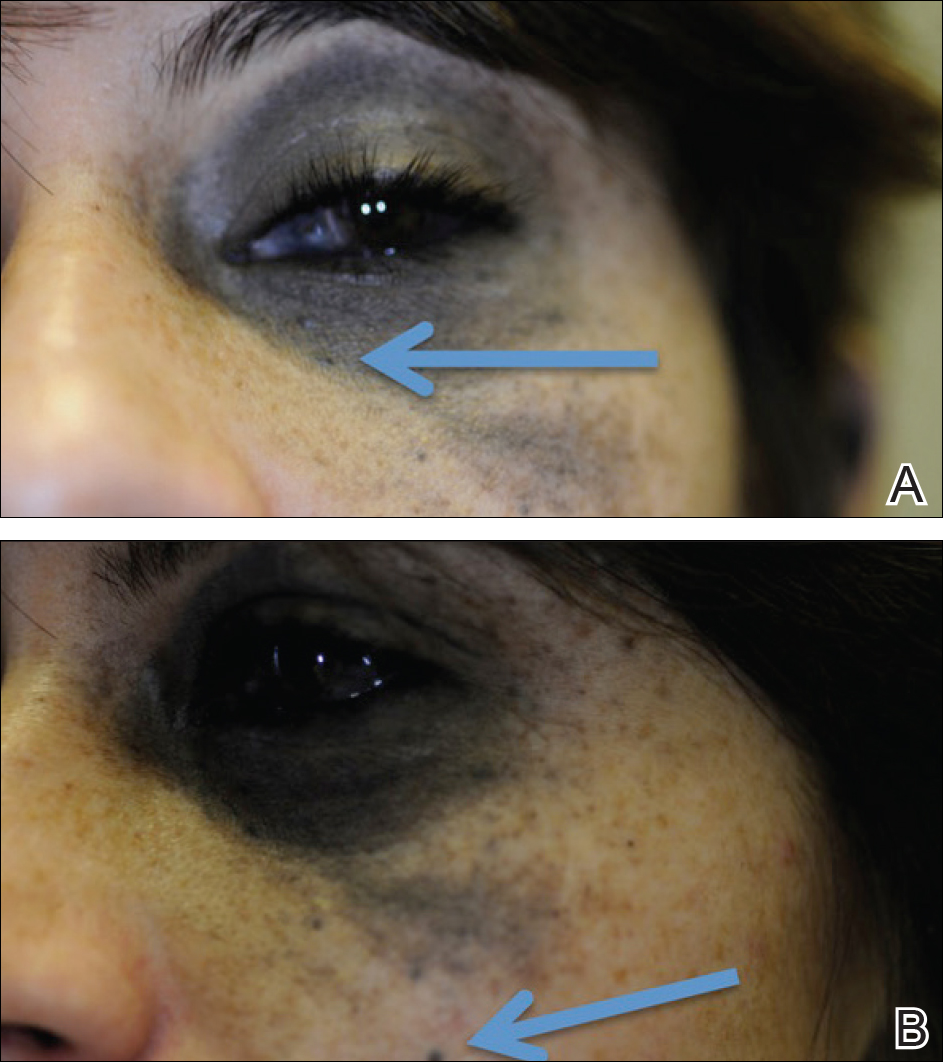

The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
Practice Points
- Nevus of Ota is a hyperpigmented dermatosis that typically is distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.
- GNAQ and BAP1 mutations in patients with nevus of Ota confer a greater risk for malignant melanoma and metastatic progression.
- Ongoing ophthalmologic screening is paramount in patients with nevus of Ota and may prevent devastating sequelae.
Terra Firma-Forme Dermatosis Mimicking Livedo Racemosa
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
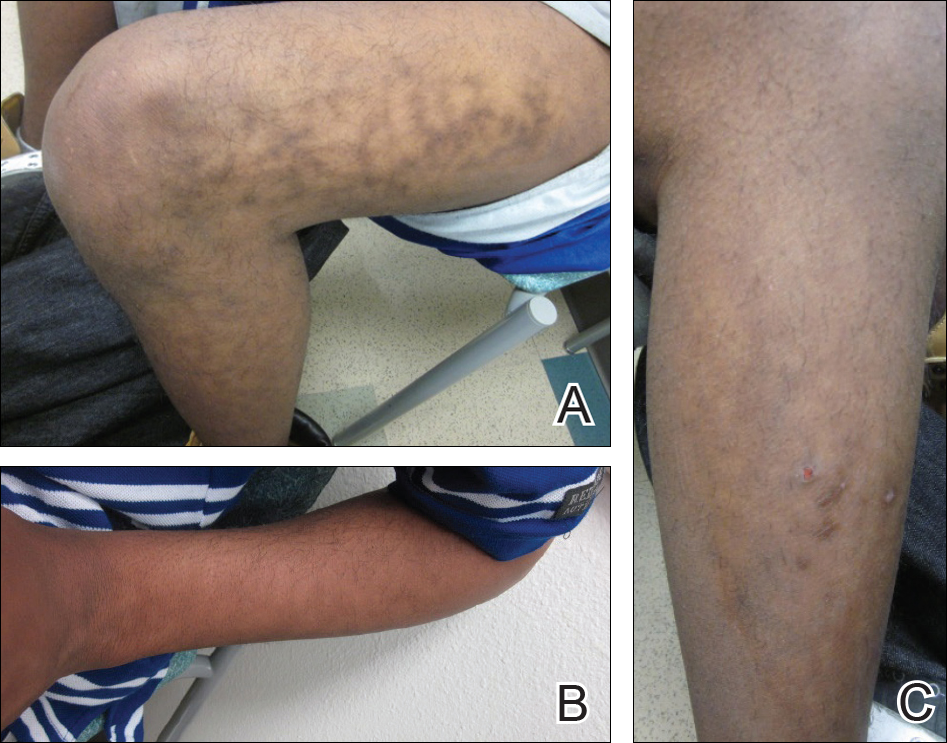
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
Practice Points
- Clinicians should include terra firma-forme dermatosis in the differential diagnosis of any hyperpigmented condition, regardless of pattern of presentation.
- Clean the skin with an alcohol wipe to rule out a diagnosis of terra firma-forme dermatosis.
Agminated Papules on the Neck
The Diagnosis: Pseudoxanthoma Elasticum
Histopathology showed abnormal curled frayed elastic fibers in the mid dermis (Figure, A); von Kossa stain was positive for calcified and fragmented elastic fibers (Figure, B). Based on clinical and histological findings, a diagnosis of pseudoxanthoma elasticum (PXE) was made.
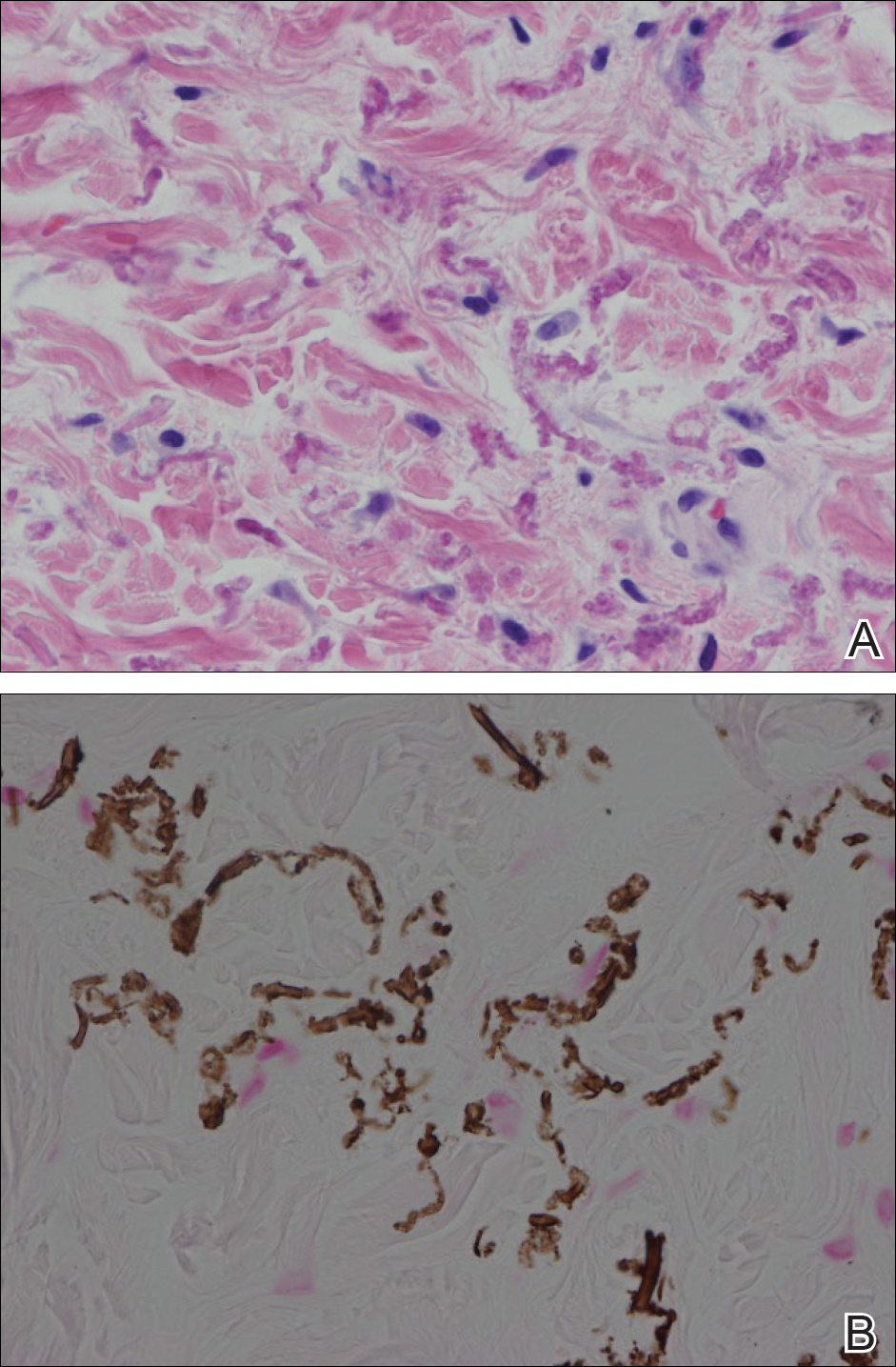
Pseudoxanthoma elasticum is a rare multisystem heterogeneous genetic disorder that causes abnormal mineralization and fragmentation of tissue elastin fibers. Clinically, accumulation of mineralized elastin fibers leads to soft tissue calcification and late-onset pathology in the dermis, retinal Bruch membrane, and medial layers of large- and medium-sized arterial walls.
Pseudoxanthoma elasticum is an autosomal-recessive disease associated with more than 300 loss mutations in the ATP-binding cassette subfamily C member 6 gene, ABCC6.1,2 However, PXE clinically is characterized by wide variability in clinical progression and outcome as well as phenotypic overlap with other disorders such as generalized arterial calcification of infancy. Pseudoxanthoma elasticum affects an estimated 1 in 25,000 to 100,000 individuals with a female preponderance (2:1 ratio).1-3 Age of onset typically is in the second to third decades of life, with 80% of cases demonstrating skin manifestations before 20 years of age.2,3
The first and most benign finding often is the appearance of small soft asymptomatic yellow papules with a plucked chicken skin-like appearance that occur on the flexural areas such as the neck, axilla, antecubital, popliteal, inguinal, and periumbilical areas. These papules may progress to irregularly shaped, yellowish plaques with a leathery appearance; mucous membranes, often occurring on the inner aspect of the lower lips, also may be involved. More severe abdominal striae also may affect some but not all women with PXE. Histologic examination demonstrates swollen, clumped, and fragmented elastin fibers with calcium deposits in the mid dermis. Elastin-specific stains such as orcein and calcium-specific stains such as the von Kossa stain aid in the diagnosis.
Vision impairment subsequently develops in 50% to 70% of patients, with severe vision loss in 3% to 8% of patients.4,5 Ophthalmologic examination identifies characteristic angioid streaks (ie, gray lines radiating from the optic disk) and subretinal hemorrhages caused by brittle new vessel formation.
Bleeding complications, especially from the gastrointestinal tract, caused by arterial wall fragility may affect 10% of PXE patients.5 Although bleeding complications also may affect the genitourinary system, the risk for fetal loss or adverse reproductive outcomes is considered low.6 More insidiously, progressive arterial calcification and peripheral arterial disease contribute to accelerated atherosclerosis, causing earlier presentations of claudication, angina pectoris, myocardial infarction, and hypertension by the third and fourth decades of life.
Management of PXE is limited. Primary care providers should be attentive to cardiovascular screening for coronary and peripheral arterial disease. Patients should receive regular eye examinations, and choroidal neovascularization should be aggressively treated with photocoagulation, photodynamic therapy, and vascular endothelial growth factor inhibitors.1,3
Collagenous fibromas are slow-growing tumors but are histologically distinct, showing fibrous or myxoid connective tissue arising within adipose tissue. Cutaneous leiomyomas may be solitary or grouped, often painful papules composed histologically of bundles of smooth muscle. Cutaneous sclerosis in sclerosing mesenteritis is a rare cutaneous manifestation of an internal disorder and presents as asymptomatic indurated subcutaneous nodules but histologically is distinctive, demonstrating sclerosis with fat necrosis. Xanthoma disseminatum is a rare form of histiocytosis that commonly presents as hundreds of small yellowish brown or reddish brown papules symmetrically distributed on the face, trunk, and intertriginous areas.
On follow-up within a year after initial presentation, our patient was found to have early subtle angioid streaks on ophthalmologic examination with no vision loss. A transthoracic echocardiogram was performed and showed no cardiac abnormalities. Her pregnancy was complicated by intrauterine growth retardation in the third trimester; however, the patient delivered a healthy-appearing 2835 g neonate (10th percentile for gestational age) at 39 weeks of gestations via an uncomplicated cesarean delivery.
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155A:1517-1526.
- Li Q, Jiang Q, Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1-11.
- Finger RP, Charbel Issa P, Ladewig MS, et al. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272-285.
- Li Y, Cui Y, Zhao H, et al. Pseudoxanthoma elasticum: a review of 86 cases in China. Intractable Rare Dis Res. 2014;3:75-78.
- Laube S, Moss C. Pseudoxanthoma elasticum. Arch Dis Child. 2005;90:754-756.
- Bercovitch L, Leroux T, Terry S, et al. Pregnancy and obstetrical outcomes in pseudoxanthoma elasticum. Br J Dermatol. 2004;151:1011-1018.
The Diagnosis: Pseudoxanthoma Elasticum
Histopathology showed abnormal curled frayed elastic fibers in the mid dermis (Figure, A); von Kossa stain was positive for calcified and fragmented elastic fibers (Figure, B). Based on clinical and histological findings, a diagnosis of pseudoxanthoma elasticum (PXE) was made.
Pseudoxanthoma elasticum is a rare multisystem heterogeneous genetic disorder that causes abnormal mineralization and fragmentation of tissue elastin fibers. Clinically, accumulation of mineralized elastin fibers leads to soft tissue calcification and late-onset pathology in the dermis, retinal Bruch membrane, and medial layers of large- and medium-sized arterial walls.
Pseudoxanthoma elasticum is an autosomal-recessive disease associated with more than 300 loss mutations in the ATP-binding cassette subfamily C member 6 gene, ABCC6.1,2 However, PXE clinically is characterized by wide variability in clinical progression and outcome as well as phenotypic overlap with other disorders such as generalized arterial calcification of infancy. Pseudoxanthoma elasticum affects an estimated 1 in 25,000 to 100,000 individuals with a female preponderance (2:1 ratio).1-3 Age of onset typically is in the second to third decades of life, with 80% of cases demonstrating skin manifestations before 20 years of age.2,3
The first and most benign finding often is the appearance of small soft asymptomatic yellow papules with a plucked chicken skin-like appearance that occur on the flexural areas such as the neck, axilla, antecubital, popliteal, inguinal, and periumbilical areas. These papules may progress to irregularly shaped, yellowish plaques with a leathery appearance; mucous membranes, often occurring on the inner aspect of the lower lips, also may be involved. More severe abdominal striae also may affect some but not all women with PXE. Histologic examination demonstrates swollen, clumped, and fragmented elastin fibers with calcium deposits in the mid dermis. Elastin-specific stains such as orcein and calcium-specific stains such as the von Kossa stain aid in the diagnosis.
Vision impairment subsequently develops in 50% to 70% of patients, with severe vision loss in 3% to 8% of patients.4,5 Ophthalmologic examination identifies characteristic angioid streaks (ie, gray lines radiating from the optic disk) and subretinal hemorrhages caused by brittle new vessel formation.
Bleeding complications, especially from the gastrointestinal tract, caused by arterial wall fragility may affect 10% of PXE patients.5 Although bleeding complications also may affect the genitourinary system, the risk for fetal loss or adverse reproductive outcomes is considered low.6 More insidiously, progressive arterial calcification and peripheral arterial disease contribute to accelerated atherosclerosis, causing earlier presentations of claudication, angina pectoris, myocardial infarction, and hypertension by the third and fourth decades of life.
Management of PXE is limited. Primary care providers should be attentive to cardiovascular screening for coronary and peripheral arterial disease. Patients should receive regular eye examinations, and choroidal neovascularization should be aggressively treated with photocoagulation, photodynamic therapy, and vascular endothelial growth factor inhibitors.1,3
Collagenous fibromas are slow-growing tumors but are histologically distinct, showing fibrous or myxoid connective tissue arising within adipose tissue. Cutaneous leiomyomas may be solitary or grouped, often painful papules composed histologically of bundles of smooth muscle. Cutaneous sclerosis in sclerosing mesenteritis is a rare cutaneous manifestation of an internal disorder and presents as asymptomatic indurated subcutaneous nodules but histologically is distinctive, demonstrating sclerosis with fat necrosis. Xanthoma disseminatum is a rare form of histiocytosis that commonly presents as hundreds of small yellowish brown or reddish brown papules symmetrically distributed on the face, trunk, and intertriginous areas.
On follow-up within a year after initial presentation, our patient was found to have early subtle angioid streaks on ophthalmologic examination with no vision loss. A transthoracic echocardiogram was performed and showed no cardiac abnormalities. Her pregnancy was complicated by intrauterine growth retardation in the third trimester; however, the patient delivered a healthy-appearing 2835 g neonate (10th percentile for gestational age) at 39 weeks of gestations via an uncomplicated cesarean delivery.
The Diagnosis: Pseudoxanthoma Elasticum
Histopathology showed abnormal curled frayed elastic fibers in the mid dermis (Figure, A); von Kossa stain was positive for calcified and fragmented elastic fibers (Figure, B). Based on clinical and histological findings, a diagnosis of pseudoxanthoma elasticum (PXE) was made.
Pseudoxanthoma elasticum is a rare multisystem heterogeneous genetic disorder that causes abnormal mineralization and fragmentation of tissue elastin fibers. Clinically, accumulation of mineralized elastin fibers leads to soft tissue calcification and late-onset pathology in the dermis, retinal Bruch membrane, and medial layers of large- and medium-sized arterial walls.
Pseudoxanthoma elasticum is an autosomal-recessive disease associated with more than 300 loss mutations in the ATP-binding cassette subfamily C member 6 gene, ABCC6.1,2 However, PXE clinically is characterized by wide variability in clinical progression and outcome as well as phenotypic overlap with other disorders such as generalized arterial calcification of infancy. Pseudoxanthoma elasticum affects an estimated 1 in 25,000 to 100,000 individuals with a female preponderance (2:1 ratio).1-3 Age of onset typically is in the second to third decades of life, with 80% of cases demonstrating skin manifestations before 20 years of age.2,3
The first and most benign finding often is the appearance of small soft asymptomatic yellow papules with a plucked chicken skin-like appearance that occur on the flexural areas such as the neck, axilla, antecubital, popliteal, inguinal, and periumbilical areas. These papules may progress to irregularly shaped, yellowish plaques with a leathery appearance; mucous membranes, often occurring on the inner aspect of the lower lips, also may be involved. More severe abdominal striae also may affect some but not all women with PXE. Histologic examination demonstrates swollen, clumped, and fragmented elastin fibers with calcium deposits in the mid dermis. Elastin-specific stains such as orcein and calcium-specific stains such as the von Kossa stain aid in the diagnosis.
Vision impairment subsequently develops in 50% to 70% of patients, with severe vision loss in 3% to 8% of patients.4,5 Ophthalmologic examination identifies characteristic angioid streaks (ie, gray lines radiating from the optic disk) and subretinal hemorrhages caused by brittle new vessel formation.
Bleeding complications, especially from the gastrointestinal tract, caused by arterial wall fragility may affect 10% of PXE patients.5 Although bleeding complications also may affect the genitourinary system, the risk for fetal loss or adverse reproductive outcomes is considered low.6 More insidiously, progressive arterial calcification and peripheral arterial disease contribute to accelerated atherosclerosis, causing earlier presentations of claudication, angina pectoris, myocardial infarction, and hypertension by the third and fourth decades of life.
Management of PXE is limited. Primary care providers should be attentive to cardiovascular screening for coronary and peripheral arterial disease. Patients should receive regular eye examinations, and choroidal neovascularization should be aggressively treated with photocoagulation, photodynamic therapy, and vascular endothelial growth factor inhibitors.1,3
Collagenous fibromas are slow-growing tumors but are histologically distinct, showing fibrous or myxoid connective tissue arising within adipose tissue. Cutaneous leiomyomas may be solitary or grouped, often painful papules composed histologically of bundles of smooth muscle. Cutaneous sclerosis in sclerosing mesenteritis is a rare cutaneous manifestation of an internal disorder and presents as asymptomatic indurated subcutaneous nodules but histologically is distinctive, demonstrating sclerosis with fat necrosis. Xanthoma disseminatum is a rare form of histiocytosis that commonly presents as hundreds of small yellowish brown or reddish brown papules symmetrically distributed on the face, trunk, and intertriginous areas.
On follow-up within a year after initial presentation, our patient was found to have early subtle angioid streaks on ophthalmologic examination with no vision loss. A transthoracic echocardiogram was performed and showed no cardiac abnormalities. Her pregnancy was complicated by intrauterine growth retardation in the third trimester; however, the patient delivered a healthy-appearing 2835 g neonate (10th percentile for gestational age) at 39 weeks of gestations via an uncomplicated cesarean delivery.
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155A:1517-1526.
- Li Q, Jiang Q, Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1-11.
- Finger RP, Charbel Issa P, Ladewig MS, et al. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272-285.
- Li Y, Cui Y, Zhao H, et al. Pseudoxanthoma elasticum: a review of 86 cases in China. Intractable Rare Dis Res. 2014;3:75-78.
- Laube S, Moss C. Pseudoxanthoma elasticum. Arch Dis Child. 2005;90:754-756.
- Bercovitch L, Leroux T, Terry S, et al. Pregnancy and obstetrical outcomes in pseudoxanthoma elasticum. Br J Dermatol. 2004;151:1011-1018.
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155A:1517-1526.
- Li Q, Jiang Q, Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1-11.
- Finger RP, Charbel Issa P, Ladewig MS, et al. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272-285.
- Li Y, Cui Y, Zhao H, et al. Pseudoxanthoma elasticum: a review of 86 cases in China. Intractable Rare Dis Res. 2014;3:75-78.
- Laube S, Moss C. Pseudoxanthoma elasticum. Arch Dis Child. 2005;90:754-756.
- Bercovitch L, Leroux T, Terry S, et al. Pregnancy and obstetrical outcomes in pseudoxanthoma elasticum. Br J Dermatol. 2004;151:1011-1018.
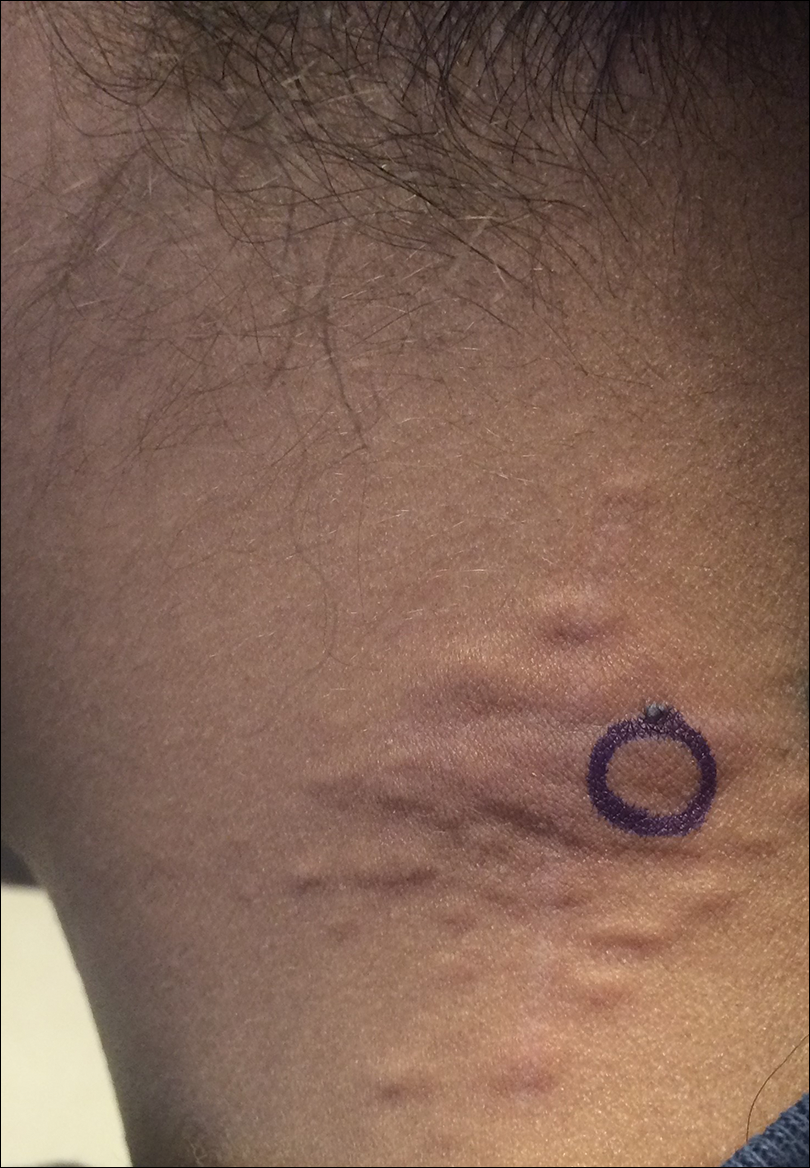
A 24-year-old woman presented with a lesion on the neck of 3 months' duration. She noted occasional mild pruritus at the site but no other symptoms or similar lesions elsewhere. At the time of presentation, she was at 17 weeks of gestation without any complications. Her medical history was notable for hypertension, unspecified chest pain with a normal electrocardiogram, and 2 spontaneous abortions. She denied a personal or family history of notable cardiovascular or gastrointestinal tract diseases. Examination of the skin showed indurated 3- to 5-mm papules coalescing into a 3- to 4-cm plaque on the left posterolateral neck.
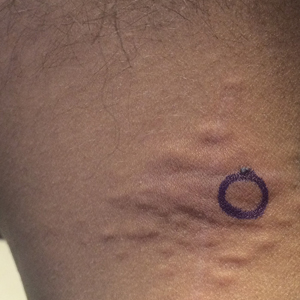
A Rare Case of Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
CASE REPORT
A 74-year-old woman presented with a painful lesion on the left lower leg that was getting larger and more edematous and erythematous over the last 5 months. She experienced numbness and burning of the left lower leg 1 year prior to the development of the lesion. A review of her medical history revealed an otherwise healthy woman with no constitutional symptoms of fever, chills, nausea, vomiting, diarrhea, or chest pain. The patient did not exhibit mucosal, genital, or nail involvement. Physical examination revealed a group of four 1-cm, ill-defined, irregularly bordered, violaceous plaques on the left anterior tibial leg with faint surrounding erythematous to violaceous patches (Figure 1). The plaques were tender to palpation with no bleeding or drainage.
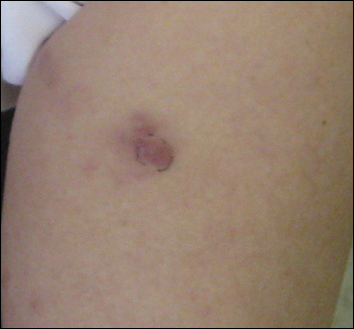
An 8.0-mm punch biopsy of the lesion was obtained. Hematoxylin and eosin staining on low-power magnification demonstrated a diffuse lymphocytic inflammatory infiltrate in the dermis and subcutis. Notable sparing of the subepidermal area (free grenz zone) was present (Figure 2A). On higher power, centroblasts and immunoblasts were visualized alongside extravasated red blood cells (Figure 2B). A diagnosis of primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) was made. Various immunohistochemical stains confirmed the diagnosis, including B-cell lymphoma 2 (BCL-2)(Figure 3A) and multiple myeloma oncogene 1 (MUM-1)(Figure 3B), which were highly positive in our patient. The patient had a negative bone marrow biopsy and positron emission tomography scan. She was started on rituximab infusions and multiple radiation treatments. At 2-year follow-up the lymphoma continued to recur despite radiation therapy.
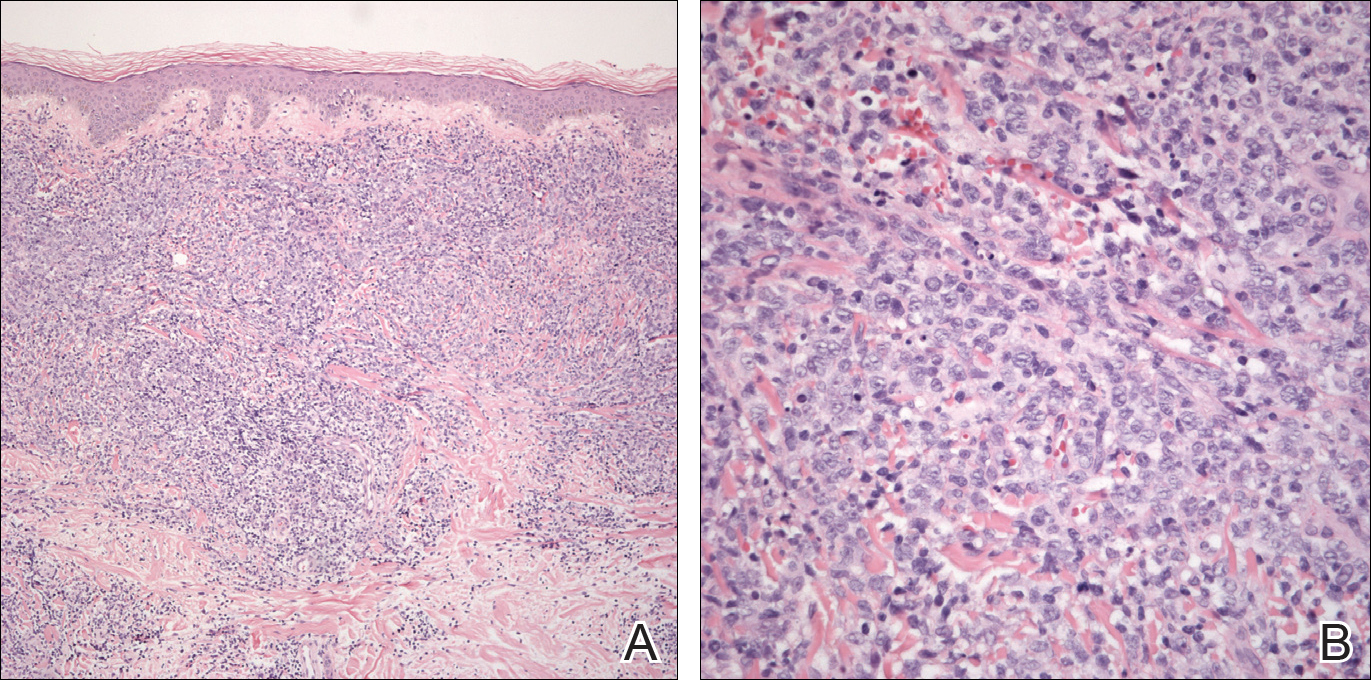
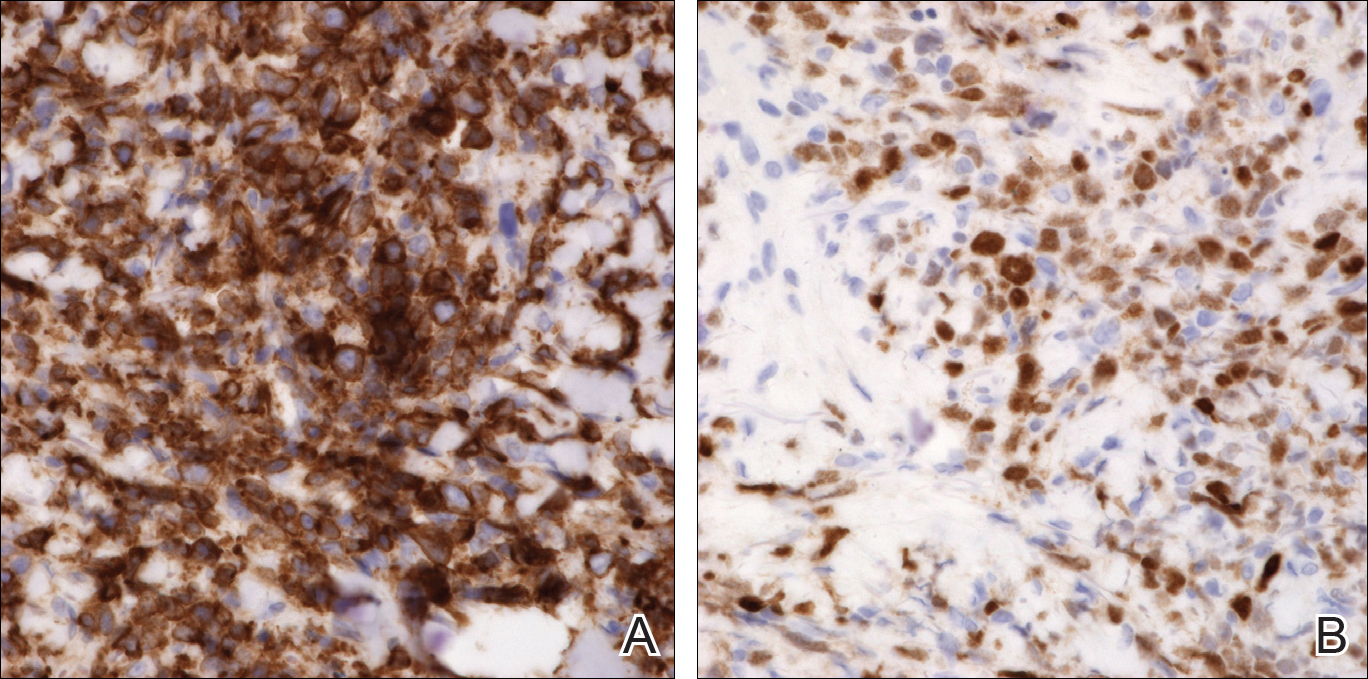
COMMENT
Incidence and Clinical Characteristics
Primary cutaneous DLBCLLT is an intermediately aggressive form of primary cutaneous B-cell lymphoma (CBCL) that accounts for approximately 10% to 20% of all primary CBCLs and 1% to 3% of all cutaneous lymphomas.1 Diffuse large B-cell lymphoma, leg type primarily affects elderly patients (median age, 70 years). Women are more commonly affected. Clinically, primary cutaneous DLBCLLT presents as red-brown to bluish nodules or tumors on one or both distal legs.
Histopathology
The diagnosis of DLBCLLT is best made histologically. There is a dense inflammatory infiltrate present in the dermis and subcutis that may extend upward into the dermoepidermal junction. Often a subepidermal free grenz zone may be seen, and adnexal structures may be destroyed. This infiltrate is composed of confluent sheets of large round cells including centroblasts and immunoblasts.2 Centroblasts are large cells that have nuclei with several small nucleoli adhering to the membrane, while immunoblasts are large round cells containing nuclei with large central nucleoli. Both centroblasts and immunoblasts stain positively for BCL-2. Centrocytes typically are absent. Staining for BCL-2 can be important in distinguishing DLBCLLT from other forms of CBCL. Diffuse large B-cell lymphoma, leg type also can demonstrate clusters of large atypical cells in the epidermis simulating epidermotropism and Pautrier microabscesses. Neoplastic cells in this condition may express monoclonal surface and cytoplasmic immunoglobulins. Primary cutaneous DLBCLLT typically is positive for B-cell markers CD20 and CD79a. Additionally, MUM-1/IRF4 (interferon regulatory factor 4) and forkhead box protein 1 (FOXP1) are strongly expressed by most patients, which helps distinguish it from other forms of CBCL.
Treatment
Diffuse large B-cell lymphoma, leg type is a relatively aggressive form of CBCL that requires more aggressive treatment than the conservative watchful waiting of some of the more indolent forms of primary CBCL. One regimen involves using cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Local chemotherapy or radiation with rituximab is another treatment option.1,2 In patients with severe comorbidities, rituximab alone may be administered. The prognosis for DLBCLLT is not as favorable as other types of primary CBCL, with an estimated 5-year survival rate of approximately 50%.2
Differential Diagnosis
Lymphomas are malignancies of the lymphocytes that may be subdivided depending on the organ of origin. Both primary nodal lymphomas and primary cutaneous lymphomas exist. Primary nodal lymphomas arise from the lymph nodes and are divided into Hodgkin and non-Hodgkin lymphomas. There are 2 major types of primary cutaneous lymphomas: cutaneous T-cell lymphoma (CTCL) and CBCL. Most primary cutaneous lymphomas are CTCLs, accounting for 75% to 80%.3
Pseudolymphoma
Pseudolymphoma is an inflammatory condition that may histologically mimic cutaneous lymphoma but has a benign clinical course. Pseudolymphoma is not a specific disease but rather is a reactive lymphoproliferative response to a known or unknown stimulus.4 Pseudolymphoma can be broken down into 2 or 3 major categories: cutaneous B-cell pseudolymphoma; cutaneous T-cell pseudolymphoma; and debatably lymphomatoid papulosis, a chronic, self-remitting, papulonecrotic condition that resembles lymphoma histologically but clinically appears benign. It is unknown if lymphomatoid papulosis represents a pseudolymphoma or a true lymphoma. Lymphomatoid papulosis may represent an early indolent form of CTCL.4
Pseudolymphomas can be triggered by a variety of causes. Most cases are idiopathic, and a causative stimulus is never identified. Drugs are known to cause many cases of pseudolymphoma, either by a causing a hypersensitivity reaction or by depressing immunosurveillance.5 Pseudolymphomas may result from exogenous stimuli such as jewelry, tattoo dyes, injectable fillers (eg, silicone), insect bites, vaccines, and trauma.6,7 Lastly, infections in the form of Borrelia, varicella, and molluscum contagiosum can potentially cause pseudolymphomas.4
Clinically, pseudolymphomas may demonstrate a B-cell or T-cell pattern. In cutaneous B-cell pseudolymphomas, asymptomatic solitary erythematous, violaceous, or flesh-colored nodules appear on the face, followed by the chest and arms. Cutaneous T-cell pseudolymphomas present with erythematous patches that are more likely to be symptomatic.4
Histologically, pseudolymphomas also are classified as demonstrating B-cell or T-cell patterns. The nodular inflammatory infiltrate of cutaneous B-cell pseudolymphoma corresponds with its clinically apparent nodules. It can be distinguished from lymphoma in that it is not solely a lymphocytic infiltrate but rather a mixed infiltrate including histiocytes, lymphocytes, eosinophils, and plasma cells. Additionally, cutaneous B-cell pseudolymphoma does not penetrate the dermis as deeply as CBCL.8 Cutaneous T-cell pseudolymphoma is more difficult to distinguish from CTCL because it also demonstrates a bandlike lymphocytic infiltrate in the papillary dermis with epidermotropism.9
Treatment must address the underlying cause of pseudolymphoma for resolution. Other treatment options include surgery, cryotherapy, local radiotherapy, topical steroids, and topical immunomodulators. Spontaneous resolution also can occur. The prognosis is better when a known trigger is eliminated, though idiopathic pseudolymphomas may be chronic in nature. It is important to rule out concurrent cutaneous lymphoma or rare transformation into cutaneous lymphoma.
Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphomas are a diverse group of neoplasms that account for most cutaneous lymphomas seen by dermatologists. In 1806, the first case of CTCL in the form of mycosis fungoides (MF) was described by Jean Louis Alibert. Mycosis fungoides represents the most common form of CTCL, accounting for approximately 50% of all primary cutaneous lymphomas.10 Mycosis fungoides was named after its morphological resemblance to mushrooms. Although not all cases exhibit a classic progression, MF is known for its stepwise progression from patch stage to tumor stage.
Clinically, lesions typically begin as patches that progress to plaques and finally tumors. This progression may not always occur and often can take years to decades to progress. Patches are characterized by erythematous, finely scaling lesions that may be easily confused with eczema or psoriasis. Lesions occur primarily in a swimming trunk distribution.
Mycosis fungoides histologically demonstrates a bandlike lymphocytic infiltrate with epidermotropism, which occurs when lymphocytes infiltrate the epidermis without spongiosis. These lymphocytes are larger, darker, and more angulated than normal lymphocytes. Intraepidermal nests of these atypical lymphocytes creating Pautrier microabscesses may be present. Tumor-stage lesions demonstrate diminished epidermotropism with dense sheets of lymphocytes in the dermis, and fat cells with cerebriform nuclei are present.
Therapies for MF may control the disease but may not prolong patients’ lives. Topical corticosteroids, phototherapy, and radiotherapy are options for skin-targeting therapies. Systemic chemotherapy and biological response modifiers also are viable treatment options. Prognosis for MF is poor.
There are a few notable variants of MF that are important to consider. Sézary syndrome is an erythrodermic variant of MF characterized by atypical Sézary cells. Clinically, it presents with generalized erythroderma with leonine facies, facial edema, and alopecia with associated symptoms of burning and pruritus. Histologically, Sézary syndrome is similar to MF with an increased CD4:CD8 ratio.10 Sézary syndrome may be treated with methotrexate or photopheresis, but the prognosis remains poor with an average survival of 5 years.
Cutaneous B-Cell Lymphoma
There are 5 types of primary CBCL: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone B-cell lymphoma; primary cutaneous diffuse large B-cell lymphoma, other; precursor B-cell lymphoblastic lymphoma; and primary cutaneous DLBCLLT, which was seen in our patient.11
Primary cutaneous follicle center lymphoma is an indolent neoplastic proliferation in the skin. Clinically, it presents with solitary or grouped pinkish purple papules, plaques, or nodules on the trunk with surrounding patches of erythema.3 Lesions located on the back are referred to as Crosti lymphoma. Histopathology reveals a lymphocytic infiltrate with a diffuse follicular pattern and large round centroblasts, centrocytes, and immunoblasts with epidermal sparing. Tumor cells stain positively for κ or λ light chains, as well as CD20, CD79a, and B-cell lymphoma 6 (BCL-6); however, staining for the protein product of BCL-2 may be negative, which differentiates this form of CBCL from primary nodal B-cell lymphoma. Staining for MUM-1 may be negative, which contrasts with the strong expression seen in DLBCLLT. The follicular pattern of follicle center lymphoma stains positive for CD10, but the diffuse pattern may be CD10 negative. The prognosis for primary cutaneous follicle center lymphoma is favorable, but the recurrence rate is up to 50%.3 Treatment includes local radiotherapy or surgical excision.
Primary cutaneous marginal zone B-cell lymphoma is another indolent primary CBCL subtype that is closely related to mucosa-associated lymphoid tissue lymphomas and arises in areas of acrodermatitis chronica atrophicans and Borrelia infection. Clinically, it presents with recurrent, asymptomatic, red-brown papules, plaques, and nodules of the arms and legs. Histologically, there is a patchy infiltrate in the dermis and subcutis with sparing of the epidermis with pale-staining cells with indented nuclei, along with plasma cells and eosinophils. Primary cutaneous marginal zone B-cell lymphoma typically does not demonstrate epidermotropism. Centrocyte cells stain positively for CD20, CD79a, and BCL-2. The prognosis of primary cutaneous marginal zone B-cell lymphoma is favorable. Treatment is similar to primary cutaneous follicle center lymphoma with surgical excision, radiotherapy, and surveillance being the main modalities.
Primary cutaneous diffuse large B-cell lymphoma, other is an intermediately aggressive form of primary CBCL that is thought to be related to primary cutaneous DLBCLLT. Clinically, it presents with indurated erythematous to violaceous plaques on the trunk and thighs that may resemble a vascular tumor or panniculitis.2,12 Histopathologically, this form of lymphoma presents with a round cell morphology without BCL-2 expression, which distinguishes it from DLBCLLT. If limited to skin, the prognosis is better than the systemic form but is still less favorable than other forms of CBCL.
Precursor B-cell lymphoblastic lymphoma is an extremely rare type of CBCL that potentially can occur in the skin. It primarily affects children and young adults. Clinically, it presents as a solitary large erythematous tumor of the head. Histol
CONCLUSION
We present a rare case of primary cutaneous DLBCLLT. Our case demonstrates the classic presentation of primary cutaneous DLBCLLT in a 74-year-old woman with a tumor on the lower left leg. Histologically, a dense dermal and subcutis infiltrate of centroblasts and immunoblasts with a grenz zone was present. Immunostaining in our patient was consistent with characteristic findings in the literature, staining highly positive for BCL-2 and MUM-1. Primary cutaneous DLBCLLT is an extremely rare and unique form of cutaneous lymphoma that can have potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.
- Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control. 2012;19:236-244.
- Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143:1144-1150.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:2768-3785.
- Brodell RT, Santa Cruz DJ. Cutaneous pseudolymphomas. Dermatol Clin. 1985;3:719-734.
- Albrecht J, Fine LA, Piette W. Drug-associated lymphoma and pseudolymphoma: recognition and management. Dermatol Clin. 2007;25:233-244; vii.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Kluger N, Vermeulen C, Moguelet P, et al. Cutaneous lymphoid hyperplasia (pseudolymphoma) in tattoos: a case series of seven patients. J Eur Acad Dermatol Venereol. 2010;24:208-213.
- Burg G, Kerl H, Schmoeckel C. Differentiation between malignant B-cell lymphomas and pseudolymphomas of the skin. J Dermatol Surg Oncol. 1984;10:271-275.
- Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38(6, pt 1):877-895; quiz 896-897.
- Diamandidou E, Cohen PR, Kurzrock R. Mycosis fungoides and Sézary syndrome. Blood. 1996;88:2385-2409.
- Kempf W, Ralfkiaer E, Duncan LM, et al. Cutaneous marginal zone B-cell lymphoma. In: LeBoit P, Burg G, Weedon D, et al, eds. Pathology and Genetics of Skin Tumors. Lyon, France: IARC Press; 2006:194-195.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in cutaneous large B-cell lymphomas: a European multicentric study. J Clin Oncol. 2001;19:3602-3610.
- Chimenti S, Fink-Puches R, Peris K, et al. Cutaneous involvement in lymphoblastic lymphoma. J Cutan Pathol. 1999;26:379-385.
CASE REPORT
A 74-year-old woman presented with a painful lesion on the left lower leg that was getting larger and more edematous and erythematous over the last 5 months. She experienced numbness and burning of the left lower leg 1 year prior to the development of the lesion. A review of her medical history revealed an otherwise healthy woman with no constitutional symptoms of fever, chills, nausea, vomiting, diarrhea, or chest pain. The patient did not exhibit mucosal, genital, or nail involvement. Physical examination revealed a group of four 1-cm, ill-defined, irregularly bordered, violaceous plaques on the left anterior tibial leg with faint surrounding erythematous to violaceous patches (Figure 1). The plaques were tender to palpation with no bleeding or drainage.
An 8.0-mm punch biopsy of the lesion was obtained. Hematoxylin and eosin staining on low-power magnification demonstrated a diffuse lymphocytic inflammatory infiltrate in the dermis and subcutis. Notable sparing of the subepidermal area (free grenz zone) was present (Figure 2A). On higher power, centroblasts and immunoblasts were visualized alongside extravasated red blood cells (Figure 2B). A diagnosis of primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) was made. Various immunohistochemical stains confirmed the diagnosis, including B-cell lymphoma 2 (BCL-2)(Figure 3A) and multiple myeloma oncogene 1 (MUM-1)(Figure 3B), which were highly positive in our patient. The patient had a negative bone marrow biopsy and positron emission tomography scan. She was started on rituximab infusions and multiple radiation treatments. At 2-year follow-up the lymphoma continued to recur despite radiation therapy.
COMMENT
Incidence and Clinical Characteristics
Primary cutaneous DLBCLLT is an intermediately aggressive form of primary cutaneous B-cell lymphoma (CBCL) that accounts for approximately 10% to 20% of all primary CBCLs and 1% to 3% of all cutaneous lymphomas.1 Diffuse large B-cell lymphoma, leg type primarily affects elderly patients (median age, 70 years). Women are more commonly affected. Clinically, primary cutaneous DLBCLLT presents as red-brown to bluish nodules or tumors on one or both distal legs.
Histopathology
The diagnosis of DLBCLLT is best made histologically. There is a dense inflammatory infiltrate present in the dermis and subcutis that may extend upward into the dermoepidermal junction. Often a subepidermal free grenz zone may be seen, and adnexal structures may be destroyed. This infiltrate is composed of confluent sheets of large round cells including centroblasts and immunoblasts.2 Centroblasts are large cells that have nuclei with several small nucleoli adhering to the membrane, while immunoblasts are large round cells containing nuclei with large central nucleoli. Both centroblasts and immunoblasts stain positively for BCL-2. Centrocytes typically are absent. Staining for BCL-2 can be important in distinguishing DLBCLLT from other forms of CBCL. Diffuse large B-cell lymphoma, leg type also can demonstrate clusters of large atypical cells in the epidermis simulating epidermotropism and Pautrier microabscesses. Neoplastic cells in this condition may express monoclonal surface and cytoplasmic immunoglobulins. Primary cutaneous DLBCLLT typically is positive for B-cell markers CD20 and CD79a. Additionally, MUM-1/IRF4 (interferon regulatory factor 4) and forkhead box protein 1 (FOXP1) are strongly expressed by most patients, which helps distinguish it from other forms of CBCL.
Treatment
Diffuse large B-cell lymphoma, leg type is a relatively aggressive form of CBCL that requires more aggressive treatment than the conservative watchful waiting of some of the more indolent forms of primary CBCL. One regimen involves using cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Local chemotherapy or radiation with rituximab is another treatment option.1,2 In patients with severe comorbidities, rituximab alone may be administered. The prognosis for DLBCLLT is not as favorable as other types of primary CBCL, with an estimated 5-year survival rate of approximately 50%.2
Differential Diagnosis
Lymphomas are malignancies of the lymphocytes that may be subdivided depending on the organ of origin. Both primary nodal lymphomas and primary cutaneous lymphomas exist. Primary nodal lymphomas arise from the lymph nodes and are divided into Hodgkin and non-Hodgkin lymphomas. There are 2 major types of primary cutaneous lymphomas: cutaneous T-cell lymphoma (CTCL) and CBCL. Most primary cutaneous lymphomas are CTCLs, accounting for 75% to 80%.3
Pseudolymphoma
Pseudolymphoma is an inflammatory condition that may histologically mimic cutaneous lymphoma but has a benign clinical course. Pseudolymphoma is not a specific disease but rather is a reactive lymphoproliferative response to a known or unknown stimulus.4 Pseudolymphoma can be broken down into 2 or 3 major categories: cutaneous B-cell pseudolymphoma; cutaneous T-cell pseudolymphoma; and debatably lymphomatoid papulosis, a chronic, self-remitting, papulonecrotic condition that resembles lymphoma histologically but clinically appears benign. It is unknown if lymphomatoid papulosis represents a pseudolymphoma or a true lymphoma. Lymphomatoid papulosis may represent an early indolent form of CTCL.4
Pseudolymphomas can be triggered by a variety of causes. Most cases are idiopathic, and a causative stimulus is never identified. Drugs are known to cause many cases of pseudolymphoma, either by a causing a hypersensitivity reaction or by depressing immunosurveillance.5 Pseudolymphomas may result from exogenous stimuli such as jewelry, tattoo dyes, injectable fillers (eg, silicone), insect bites, vaccines, and trauma.6,7 Lastly, infections in the form of Borrelia, varicella, and molluscum contagiosum can potentially cause pseudolymphomas.4
Clinically, pseudolymphomas may demonstrate a B-cell or T-cell pattern. In cutaneous B-cell pseudolymphomas, asymptomatic solitary erythematous, violaceous, or flesh-colored nodules appear on the face, followed by the chest and arms. Cutaneous T-cell pseudolymphomas present with erythematous patches that are more likely to be symptomatic.4
Histologically, pseudolymphomas also are classified as demonstrating B-cell or T-cell patterns. The nodular inflammatory infiltrate of cutaneous B-cell pseudolymphoma corresponds with its clinically apparent nodules. It can be distinguished from lymphoma in that it is not solely a lymphocytic infiltrate but rather a mixed infiltrate including histiocytes, lymphocytes, eosinophils, and plasma cells. Additionally, cutaneous B-cell pseudolymphoma does not penetrate the dermis as deeply as CBCL.8 Cutaneous T-cell pseudolymphoma is more difficult to distinguish from CTCL because it also demonstrates a bandlike lymphocytic infiltrate in the papillary dermis with epidermotropism.9
Treatment must address the underlying cause of pseudolymphoma for resolution. Other treatment options include surgery, cryotherapy, local radiotherapy, topical steroids, and topical immunomodulators. Spontaneous resolution also can occur. The prognosis is better when a known trigger is eliminated, though idiopathic pseudolymphomas may be chronic in nature. It is important to rule out concurrent cutaneous lymphoma or rare transformation into cutaneous lymphoma.
Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphomas are a diverse group of neoplasms that account for most cutaneous lymphomas seen by dermatologists. In 1806, the first case of CTCL in the form of mycosis fungoides (MF) was described by Jean Louis Alibert. Mycosis fungoides represents the most common form of CTCL, accounting for approximately 50% of all primary cutaneous lymphomas.10 Mycosis fungoides was named after its morphological resemblance to mushrooms. Although not all cases exhibit a classic progression, MF is known for its stepwise progression from patch stage to tumor stage.
Clinically, lesions typically begin as patches that progress to plaques and finally tumors. This progression may not always occur and often can take years to decades to progress. Patches are characterized by erythematous, finely scaling lesions that may be easily confused with eczema or psoriasis. Lesions occur primarily in a swimming trunk distribution.
Mycosis fungoides histologically demonstrates a bandlike lymphocytic infiltrate with epidermotropism, which occurs when lymphocytes infiltrate the epidermis without spongiosis. These lymphocytes are larger, darker, and more angulated than normal lymphocytes. Intraepidermal nests of these atypical lymphocytes creating Pautrier microabscesses may be present. Tumor-stage lesions demonstrate diminished epidermotropism with dense sheets of lymphocytes in the dermis, and fat cells with cerebriform nuclei are present.
Therapies for MF may control the disease but may not prolong patients’ lives. Topical corticosteroids, phototherapy, and radiotherapy are options for skin-targeting therapies. Systemic chemotherapy and biological response modifiers also are viable treatment options. Prognosis for MF is poor.
There are a few notable variants of MF that are important to consider. Sézary syndrome is an erythrodermic variant of MF characterized by atypical Sézary cells. Clinically, it presents with generalized erythroderma with leonine facies, facial edema, and alopecia with associated symptoms of burning and pruritus. Histologically, Sézary syndrome is similar to MF with an increased CD4:CD8 ratio.10 Sézary syndrome may be treated with methotrexate or photopheresis, but the prognosis remains poor with an average survival of 5 years.
Cutaneous B-Cell Lymphoma
There are 5 types of primary CBCL: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone B-cell lymphoma; primary cutaneous diffuse large B-cell lymphoma, other; precursor B-cell lymphoblastic lymphoma; and primary cutaneous DLBCLLT, which was seen in our patient.11
Primary cutaneous follicle center lymphoma is an indolent neoplastic proliferation in the skin. Clinically, it presents with solitary or grouped pinkish purple papules, plaques, or nodules on the trunk with surrounding patches of erythema.3 Lesions located on the back are referred to as Crosti lymphoma. Histopathology reveals a lymphocytic infiltrate with a diffuse follicular pattern and large round centroblasts, centrocytes, and immunoblasts with epidermal sparing. Tumor cells stain positively for κ or λ light chains, as well as CD20, CD79a, and B-cell lymphoma 6 (BCL-6); however, staining for the protein product of BCL-2 may be negative, which differentiates this form of CBCL from primary nodal B-cell lymphoma. Staining for MUM-1 may be negative, which contrasts with the strong expression seen in DLBCLLT. The follicular pattern of follicle center lymphoma stains positive for CD10, but the diffuse pattern may be CD10 negative. The prognosis for primary cutaneous follicle center lymphoma is favorable, but the recurrence rate is up to 50%.3 Treatment includes local radiotherapy or surgical excision.
Primary cutaneous marginal zone B-cell lymphoma is another indolent primary CBCL subtype that is closely related to mucosa-associated lymphoid tissue lymphomas and arises in areas of acrodermatitis chronica atrophicans and Borrelia infection. Clinically, it presents with recurrent, asymptomatic, red-brown papules, plaques, and nodules of the arms and legs. Histologically, there is a patchy infiltrate in the dermis and subcutis with sparing of the epidermis with pale-staining cells with indented nuclei, along with plasma cells and eosinophils. Primary cutaneous marginal zone B-cell lymphoma typically does not demonstrate epidermotropism. Centrocyte cells stain positively for CD20, CD79a, and BCL-2. The prognosis of primary cutaneous marginal zone B-cell lymphoma is favorable. Treatment is similar to primary cutaneous follicle center lymphoma with surgical excision, radiotherapy, and surveillance being the main modalities.
Primary cutaneous diffuse large B-cell lymphoma, other is an intermediately aggressive form of primary CBCL that is thought to be related to primary cutaneous DLBCLLT. Clinically, it presents with indurated erythematous to violaceous plaques on the trunk and thighs that may resemble a vascular tumor or panniculitis.2,12 Histopathologically, this form of lymphoma presents with a round cell morphology without BCL-2 expression, which distinguishes it from DLBCLLT. If limited to skin, the prognosis is better than the systemic form but is still less favorable than other forms of CBCL.
Precursor B-cell lymphoblastic lymphoma is an extremely rare type of CBCL that potentially can occur in the skin. It primarily affects children and young adults. Clinically, it presents as a solitary large erythematous tumor of the head. Histol
CONCLUSION
We present a rare case of primary cutaneous DLBCLLT. Our case demonstrates the classic presentation of primary cutaneous DLBCLLT in a 74-year-old woman with a tumor on the lower left leg. Histologically, a dense dermal and subcutis infiltrate of centroblasts and immunoblasts with a grenz zone was present. Immunostaining in our patient was consistent with characteristic findings in the literature, staining highly positive for BCL-2 and MUM-1. Primary cutaneous DLBCLLT is an extremely rare and unique form of cutaneous lymphoma that can have potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.
CASE REPORT
A 74-year-old woman presented with a painful lesion on the left lower leg that was getting larger and more edematous and erythematous over the last 5 months. She experienced numbness and burning of the left lower leg 1 year prior to the development of the lesion. A review of her medical history revealed an otherwise healthy woman with no constitutional symptoms of fever, chills, nausea, vomiting, diarrhea, or chest pain. The patient did not exhibit mucosal, genital, or nail involvement. Physical examination revealed a group of four 1-cm, ill-defined, irregularly bordered, violaceous plaques on the left anterior tibial leg with faint surrounding erythematous to violaceous patches (Figure 1). The plaques were tender to palpation with no bleeding or drainage.
An 8.0-mm punch biopsy of the lesion was obtained. Hematoxylin and eosin staining on low-power magnification demonstrated a diffuse lymphocytic inflammatory infiltrate in the dermis and subcutis. Notable sparing of the subepidermal area (free grenz zone) was present (Figure 2A). On higher power, centroblasts and immunoblasts were visualized alongside extravasated red blood cells (Figure 2B). A diagnosis of primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) was made. Various immunohistochemical stains confirmed the diagnosis, including B-cell lymphoma 2 (BCL-2)(Figure 3A) and multiple myeloma oncogene 1 (MUM-1)(Figure 3B), which were highly positive in our patient. The patient had a negative bone marrow biopsy and positron emission tomography scan. She was started on rituximab infusions and multiple radiation treatments. At 2-year follow-up the lymphoma continued to recur despite radiation therapy.
COMMENT
Incidence and Clinical Characteristics
Primary cutaneous DLBCLLT is an intermediately aggressive form of primary cutaneous B-cell lymphoma (CBCL) that accounts for approximately 10% to 20% of all primary CBCLs and 1% to 3% of all cutaneous lymphomas.1 Diffuse large B-cell lymphoma, leg type primarily affects elderly patients (median age, 70 years). Women are more commonly affected. Clinically, primary cutaneous DLBCLLT presents as red-brown to bluish nodules or tumors on one or both distal legs.
Histopathology
The diagnosis of DLBCLLT is best made histologically. There is a dense inflammatory infiltrate present in the dermis and subcutis that may extend upward into the dermoepidermal junction. Often a subepidermal free grenz zone may be seen, and adnexal structures may be destroyed. This infiltrate is composed of confluent sheets of large round cells including centroblasts and immunoblasts.2 Centroblasts are large cells that have nuclei with several small nucleoli adhering to the membrane, while immunoblasts are large round cells containing nuclei with large central nucleoli. Both centroblasts and immunoblasts stain positively for BCL-2. Centrocytes typically are absent. Staining for BCL-2 can be important in distinguishing DLBCLLT from other forms of CBCL. Diffuse large B-cell lymphoma, leg type also can demonstrate clusters of large atypical cells in the epidermis simulating epidermotropism and Pautrier microabscesses. Neoplastic cells in this condition may express monoclonal surface and cytoplasmic immunoglobulins. Primary cutaneous DLBCLLT typically is positive for B-cell markers CD20 and CD79a. Additionally, MUM-1/IRF4 (interferon regulatory factor 4) and forkhead box protein 1 (FOXP1) are strongly expressed by most patients, which helps distinguish it from other forms of CBCL.
Treatment
Diffuse large B-cell lymphoma, leg type is a relatively aggressive form of CBCL that requires more aggressive treatment than the conservative watchful waiting of some of the more indolent forms of primary CBCL. One regimen involves using cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Local chemotherapy or radiation with rituximab is another treatment option.1,2 In patients with severe comorbidities, rituximab alone may be administered. The prognosis for DLBCLLT is not as favorable as other types of primary CBCL, with an estimated 5-year survival rate of approximately 50%.2
Differential Diagnosis
Lymphomas are malignancies of the lymphocytes that may be subdivided depending on the organ of origin. Both primary nodal lymphomas and primary cutaneous lymphomas exist. Primary nodal lymphomas arise from the lymph nodes and are divided into Hodgkin and non-Hodgkin lymphomas. There are 2 major types of primary cutaneous lymphomas: cutaneous T-cell lymphoma (CTCL) and CBCL. Most primary cutaneous lymphomas are CTCLs, accounting for 75% to 80%.3
Pseudolymphoma
Pseudolymphoma is an inflammatory condition that may histologically mimic cutaneous lymphoma but has a benign clinical course. Pseudolymphoma is not a specific disease but rather is a reactive lymphoproliferative response to a known or unknown stimulus.4 Pseudolymphoma can be broken down into 2 or 3 major categories: cutaneous B-cell pseudolymphoma; cutaneous T-cell pseudolymphoma; and debatably lymphomatoid papulosis, a chronic, self-remitting, papulonecrotic condition that resembles lymphoma histologically but clinically appears benign. It is unknown if lymphomatoid papulosis represents a pseudolymphoma or a true lymphoma. Lymphomatoid papulosis may represent an early indolent form of CTCL.4
Pseudolymphomas can be triggered by a variety of causes. Most cases are idiopathic, and a causative stimulus is never identified. Drugs are known to cause many cases of pseudolymphoma, either by a causing a hypersensitivity reaction or by depressing immunosurveillance.5 Pseudolymphomas may result from exogenous stimuli such as jewelry, tattoo dyes, injectable fillers (eg, silicone), insect bites, vaccines, and trauma.6,7 Lastly, infections in the form of Borrelia, varicella, and molluscum contagiosum can potentially cause pseudolymphomas.4
Clinically, pseudolymphomas may demonstrate a B-cell or T-cell pattern. In cutaneous B-cell pseudolymphomas, asymptomatic solitary erythematous, violaceous, or flesh-colored nodules appear on the face, followed by the chest and arms. Cutaneous T-cell pseudolymphomas present with erythematous patches that are more likely to be symptomatic.4
Histologically, pseudolymphomas also are classified as demonstrating B-cell or T-cell patterns. The nodular inflammatory infiltrate of cutaneous B-cell pseudolymphoma corresponds with its clinically apparent nodules. It can be distinguished from lymphoma in that it is not solely a lymphocytic infiltrate but rather a mixed infiltrate including histiocytes, lymphocytes, eosinophils, and plasma cells. Additionally, cutaneous B-cell pseudolymphoma does not penetrate the dermis as deeply as CBCL.8 Cutaneous T-cell pseudolymphoma is more difficult to distinguish from CTCL because it also demonstrates a bandlike lymphocytic infiltrate in the papillary dermis with epidermotropism.9
Treatment must address the underlying cause of pseudolymphoma for resolution. Other treatment options include surgery, cryotherapy, local radiotherapy, topical steroids, and topical immunomodulators. Spontaneous resolution also can occur. The prognosis is better when a known trigger is eliminated, though idiopathic pseudolymphomas may be chronic in nature. It is important to rule out concurrent cutaneous lymphoma or rare transformation into cutaneous lymphoma.
Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphomas are a diverse group of neoplasms that account for most cutaneous lymphomas seen by dermatologists. In 1806, the first case of CTCL in the form of mycosis fungoides (MF) was described by Jean Louis Alibert. Mycosis fungoides represents the most common form of CTCL, accounting for approximately 50% of all primary cutaneous lymphomas.10 Mycosis fungoides was named after its morphological resemblance to mushrooms. Although not all cases exhibit a classic progression, MF is known for its stepwise progression from patch stage to tumor stage.
Clinically, lesions typically begin as patches that progress to plaques and finally tumors. This progression may not always occur and often can take years to decades to progress. Patches are characterized by erythematous, finely scaling lesions that may be easily confused with eczema or psoriasis. Lesions occur primarily in a swimming trunk distribution.
Mycosis fungoides histologically demonstrates a bandlike lymphocytic infiltrate with epidermotropism, which occurs when lymphocytes infiltrate the epidermis without spongiosis. These lymphocytes are larger, darker, and more angulated than normal lymphocytes. Intraepidermal nests of these atypical lymphocytes creating Pautrier microabscesses may be present. Tumor-stage lesions demonstrate diminished epidermotropism with dense sheets of lymphocytes in the dermis, and fat cells with cerebriform nuclei are present.
Therapies for MF may control the disease but may not prolong patients’ lives. Topical corticosteroids, phototherapy, and radiotherapy are options for skin-targeting therapies. Systemic chemotherapy and biological response modifiers also are viable treatment options. Prognosis for MF is poor.
There are a few notable variants of MF that are important to consider. Sézary syndrome is an erythrodermic variant of MF characterized by atypical Sézary cells. Clinically, it presents with generalized erythroderma with leonine facies, facial edema, and alopecia with associated symptoms of burning and pruritus. Histologically, Sézary syndrome is similar to MF with an increased CD4:CD8 ratio.10 Sézary syndrome may be treated with methotrexate or photopheresis, but the prognosis remains poor with an average survival of 5 years.
Cutaneous B-Cell Lymphoma
There are 5 types of primary CBCL: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone B-cell lymphoma; primary cutaneous diffuse large B-cell lymphoma, other; precursor B-cell lymphoblastic lymphoma; and primary cutaneous DLBCLLT, which was seen in our patient.11
Primary cutaneous follicle center lymphoma is an indolent neoplastic proliferation in the skin. Clinically, it presents with solitary or grouped pinkish purple papules, plaques, or nodules on the trunk with surrounding patches of erythema.3 Lesions located on the back are referred to as Crosti lymphoma. Histopathology reveals a lymphocytic infiltrate with a diffuse follicular pattern and large round centroblasts, centrocytes, and immunoblasts with epidermal sparing. Tumor cells stain positively for κ or λ light chains, as well as CD20, CD79a, and B-cell lymphoma 6 (BCL-6); however, staining for the protein product of BCL-2 may be negative, which differentiates this form of CBCL from primary nodal B-cell lymphoma. Staining for MUM-1 may be negative, which contrasts with the strong expression seen in DLBCLLT. The follicular pattern of follicle center lymphoma stains positive for CD10, but the diffuse pattern may be CD10 negative. The prognosis for primary cutaneous follicle center lymphoma is favorable, but the recurrence rate is up to 50%.3 Treatment includes local radiotherapy or surgical excision.
Primary cutaneous marginal zone B-cell lymphoma is another indolent primary CBCL subtype that is closely related to mucosa-associated lymphoid tissue lymphomas and arises in areas of acrodermatitis chronica atrophicans and Borrelia infection. Clinically, it presents with recurrent, asymptomatic, red-brown papules, plaques, and nodules of the arms and legs. Histologically, there is a patchy infiltrate in the dermis and subcutis with sparing of the epidermis with pale-staining cells with indented nuclei, along with plasma cells and eosinophils. Primary cutaneous marginal zone B-cell lymphoma typically does not demonstrate epidermotropism. Centrocyte cells stain positively for CD20, CD79a, and BCL-2. The prognosis of primary cutaneous marginal zone B-cell lymphoma is favorable. Treatment is similar to primary cutaneous follicle center lymphoma with surgical excision, radiotherapy, and surveillance being the main modalities.
Primary cutaneous diffuse large B-cell lymphoma, other is an intermediately aggressive form of primary CBCL that is thought to be related to primary cutaneous DLBCLLT. Clinically, it presents with indurated erythematous to violaceous plaques on the trunk and thighs that may resemble a vascular tumor or panniculitis.2,12 Histopathologically, this form of lymphoma presents with a round cell morphology without BCL-2 expression, which distinguishes it from DLBCLLT. If limited to skin, the prognosis is better than the systemic form but is still less favorable than other forms of CBCL.
Precursor B-cell lymphoblastic lymphoma is an extremely rare type of CBCL that potentially can occur in the skin. It primarily affects children and young adults. Clinically, it presents as a solitary large erythematous tumor of the head. Histol
CONCLUSION
We present a rare case of primary cutaneous DLBCLLT. Our case demonstrates the classic presentation of primary cutaneous DLBCLLT in a 74-year-old woman with a tumor on the lower left leg. Histologically, a dense dermal and subcutis infiltrate of centroblasts and immunoblasts with a grenz zone was present. Immunostaining in our patient was consistent with characteristic findings in the literature, staining highly positive for BCL-2 and MUM-1. Primary cutaneous DLBCLLT is an extremely rare and unique form of cutaneous lymphoma that can have potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.
- Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control. 2012;19:236-244.
- Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143:1144-1150.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:2768-3785.
- Brodell RT, Santa Cruz DJ. Cutaneous pseudolymphomas. Dermatol Clin. 1985;3:719-734.
- Albrecht J, Fine LA, Piette W. Drug-associated lymphoma and pseudolymphoma: recognition and management. Dermatol Clin. 2007;25:233-244; vii.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Kluger N, Vermeulen C, Moguelet P, et al. Cutaneous lymphoid hyperplasia (pseudolymphoma) in tattoos: a case series of seven patients. J Eur Acad Dermatol Venereol. 2010;24:208-213.
- Burg G, Kerl H, Schmoeckel C. Differentiation between malignant B-cell lymphomas and pseudolymphomas of the skin. J Dermatol Surg Oncol. 1984;10:271-275.
- Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38(6, pt 1):877-895; quiz 896-897.
- Diamandidou E, Cohen PR, Kurzrock R. Mycosis fungoides and Sézary syndrome. Blood. 1996;88:2385-2409.
- Kempf W, Ralfkiaer E, Duncan LM, et al. Cutaneous marginal zone B-cell lymphoma. In: LeBoit P, Burg G, Weedon D, et al, eds. Pathology and Genetics of Skin Tumors. Lyon, France: IARC Press; 2006:194-195.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in cutaneous large B-cell lymphomas: a European multicentric study. J Clin Oncol. 2001;19:3602-3610.
- Chimenti S, Fink-Puches R, Peris K, et al. Cutaneous involvement in lymphoblastic lymphoma. J Cutan Pathol. 1999;26:379-385.
- Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control. 2012;19:236-244.
- Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143:1144-1150.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:2768-3785.
- Brodell RT, Santa Cruz DJ. Cutaneous pseudolymphomas. Dermatol Clin. 1985;3:719-734.
- Albrecht J, Fine LA, Piette W. Drug-associated lymphoma and pseudolymphoma: recognition and management. Dermatol Clin. 2007;25:233-244; vii.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Kluger N, Vermeulen C, Moguelet P, et al. Cutaneous lymphoid hyperplasia (pseudolymphoma) in tattoos: a case series of seven patients. J Eur Acad Dermatol Venereol. 2010;24:208-213.
- Burg G, Kerl H, Schmoeckel C. Differentiation between malignant B-cell lymphomas and pseudolymphomas of the skin. J Dermatol Surg Oncol. 1984;10:271-275.
- Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38(6, pt 1):877-895; quiz 896-897.
- Diamandidou E, Cohen PR, Kurzrock R. Mycosis fungoides and Sézary syndrome. Blood. 1996;88:2385-2409.
- Kempf W, Ralfkiaer E, Duncan LM, et al. Cutaneous marginal zone B-cell lymphoma. In: LeBoit P, Burg G, Weedon D, et al, eds. Pathology and Genetics of Skin Tumors. Lyon, France: IARC Press; 2006:194-195.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in cutaneous large B-cell lymphomas: a European multicentric study. J Clin Oncol. 2001;19:3602-3610.
- Chimenti S, Fink-Puches R, Peris K, et al. Cutaneous involvement in lymphoblastic lymphoma. J Cutan Pathol. 1999;26:379-385.
Practice Points
- Primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) is characterized by the presence of large round cells on histopathology.
- There are potentially fatal consequences if undiagnosed; therefore, clinicians must take great care to make the correct diagnosis based on a knowledge of the clinical and immunohistochemical findings of DLBCLLT.