User login
-
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
FDA OKs emergency use of Merck pill for COVID-19
Similar to FDA authorization of another antiviral pill regimen – ritonavir plus nirmatrelvir, or Paxlovid – granted to Pfizer on Wednesday, molnupiravir (brand name Lagevrio) should be taken early in the course of COVID-19 illness.
Pfizer’s drug is authorized for anyone aged 12 and up. But Merck’s is only for adults aged 18 and older.
Merck filed an application for emergency use authorization with the FDA in October. The company included results of its phase 3 study showing the treatment could lead to a 50% reduction in COVID-19 hospitalizations. Data later showed this efficacy at closer to a 30% reduction. In November, an FDA advisory panel narrowly recommended the agency grant authorization by a 13-10 vote.
Animal studies found the drug may harm a fetus, so it is not recommended for pregnant people, the FDA says. It may be prescribed to a pregnant person only after their doctor determines the benefits outweigh the risks and the patient is told of those risks.
Women who may get pregnant should use a reliable method of birth control if being treated with molnupiravir and for 4 days after the final dose.
Two weapons against COVID
Two antiviral pills could be better than one, at least in terms of making more COVID-19 treatments available in early 2022. It is yet to be seen if the drugmakers will be able to keep up with demand, which could substantially increase with an expected surge in Omicron variant cases.
Ritonavir and molnupiravir join remdesivir (brand name Veklury) as available antivirals to treat COVID-19. Remdesivir is fully approved by the FDA but is given only through an IV to people in the hospital.
Officials point out that COVID-19 treatments in tablet form are more convenient for patients in the United States and across the globe, particularly where IV infusion services may be limited.
In March 2021, experts accurately predicted that the molnupiravir pill would be available by year’s end.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of SARS-CoV-2 because the agent does not target the virus’s spike protein.
Perhaps in part because of early promising results, the U.S. government announced in November intentions to purchase $1 billion worth of molnupiravir. That new order came on top of $1.2 billion worth of the pills the U.S. ordered in June.
A version of this article first appeared on WebMD.com.
Similar to FDA authorization of another antiviral pill regimen – ritonavir plus nirmatrelvir, or Paxlovid – granted to Pfizer on Wednesday, molnupiravir (brand name Lagevrio) should be taken early in the course of COVID-19 illness.
Pfizer’s drug is authorized for anyone aged 12 and up. But Merck’s is only for adults aged 18 and older.
Merck filed an application for emergency use authorization with the FDA in October. The company included results of its phase 3 study showing the treatment could lead to a 50% reduction in COVID-19 hospitalizations. Data later showed this efficacy at closer to a 30% reduction. In November, an FDA advisory panel narrowly recommended the agency grant authorization by a 13-10 vote.
Animal studies found the drug may harm a fetus, so it is not recommended for pregnant people, the FDA says. It may be prescribed to a pregnant person only after their doctor determines the benefits outweigh the risks and the patient is told of those risks.
Women who may get pregnant should use a reliable method of birth control if being treated with molnupiravir and for 4 days after the final dose.
Two weapons against COVID
Two antiviral pills could be better than one, at least in terms of making more COVID-19 treatments available in early 2022. It is yet to be seen if the drugmakers will be able to keep up with demand, which could substantially increase with an expected surge in Omicron variant cases.
Ritonavir and molnupiravir join remdesivir (brand name Veklury) as available antivirals to treat COVID-19. Remdesivir is fully approved by the FDA but is given only through an IV to people in the hospital.
Officials point out that COVID-19 treatments in tablet form are more convenient for patients in the United States and across the globe, particularly where IV infusion services may be limited.
In March 2021, experts accurately predicted that the molnupiravir pill would be available by year’s end.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of SARS-CoV-2 because the agent does not target the virus’s spike protein.
Perhaps in part because of early promising results, the U.S. government announced in November intentions to purchase $1 billion worth of molnupiravir. That new order came on top of $1.2 billion worth of the pills the U.S. ordered in June.
A version of this article first appeared on WebMD.com.
Similar to FDA authorization of another antiviral pill regimen – ritonavir plus nirmatrelvir, or Paxlovid – granted to Pfizer on Wednesday, molnupiravir (brand name Lagevrio) should be taken early in the course of COVID-19 illness.
Pfizer’s drug is authorized for anyone aged 12 and up. But Merck’s is only for adults aged 18 and older.
Merck filed an application for emergency use authorization with the FDA in October. The company included results of its phase 3 study showing the treatment could lead to a 50% reduction in COVID-19 hospitalizations. Data later showed this efficacy at closer to a 30% reduction. In November, an FDA advisory panel narrowly recommended the agency grant authorization by a 13-10 vote.
Animal studies found the drug may harm a fetus, so it is not recommended for pregnant people, the FDA says. It may be prescribed to a pregnant person only after their doctor determines the benefits outweigh the risks and the patient is told of those risks.
Women who may get pregnant should use a reliable method of birth control if being treated with molnupiravir and for 4 days after the final dose.
Two weapons against COVID
Two antiviral pills could be better than one, at least in terms of making more COVID-19 treatments available in early 2022. It is yet to be seen if the drugmakers will be able to keep up with demand, which could substantially increase with an expected surge in Omicron variant cases.
Ritonavir and molnupiravir join remdesivir (brand name Veklury) as available antivirals to treat COVID-19. Remdesivir is fully approved by the FDA but is given only through an IV to people in the hospital.
Officials point out that COVID-19 treatments in tablet form are more convenient for patients in the United States and across the globe, particularly where IV infusion services may be limited.
In March 2021, experts accurately predicted that the molnupiravir pill would be available by year’s end.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of SARS-CoV-2 because the agent does not target the virus’s spike protein.
Perhaps in part because of early promising results, the U.S. government announced in November intentions to purchase $1 billion worth of molnupiravir. That new order came on top of $1.2 billion worth of the pills the U.S. ordered in June.
A version of this article first appeared on WebMD.com.
New studies suggest Omicron infections are less severe than Delta ones
People who get COVID-19 infections caused by the Omicron variant are less likely to need hospital care, compared with those infected by the Delta variant, according to two large new studies from the U.K. and South Africa.
The findings, which were released ahead of peer review, add to previous glimmers of evidence suggesting that Omicron – while extremely contagious -– may result in less severe symptoms than its predecessors.
“This is helping us quantify how much less severe Omicron is than Delta, and it appears to be between 40 to 75% reduced risk of hospitalizations, adjusted for many factors, which is very good,” said Eric Topol, MD, the editor-in-chief of Medscape and a cardiologist at Scripps Research Translational Institute in La Jolla, CA.
The first analysis, which was done by the World Health Organization Collaborating Centre for Infectious Disease Modelling and Imperial College London, found that overall, people infected by Omicron had about a 20% reduced risk of needing any hospital care for their infections and a 40% lower risk of an overnight hospital stay, compared to those infected with Delta.
Meanwhile, people who were re-infected – meaning they caught Omicron after recovering from a previous COVID-19 infection – had a 50%-60% lower risk of needing hospital care, likely reflecting the benefits of having some prior immunity against the same family of viruses.
The study included everyone with polymerase chain reaction-confirmed COVID-19 in the U.K. during the first 2 weeks of December – roughly 56,000 Omicron cases and 269,000 Delta infections.
The second study, from researchers at the National Institute for Communicable Diseases in South Africa, included more than 29,000 COVID-19 cases that had lab results highly suggestive of Omicron infections. Compared to people infected with the Delta variant, those with presumed Omicron infections were about 70% less likely to have severe disease.
While the news is hopeful for individuals, on a population level, health care systems may still be stressed, the study authors warned.
“Given the high transmissibility of the Omicron virus, there remains the potential for health services to face increasing demand if Omicron cases continue to grow at the rate that has been seen in recent weeks,” said study author Neil Ferguson, PhD, who studies how infectious diseases spread at Imperial College London.
The study authors say their findings are specific to the U.K. and South Africa, where substantial portions of the population have some immune protection from past infection. In other words, they may not apply to countries where fewer people have been vaccinated or recovered from a bout with COVID-19.
A version of this article first appeared on WebMD.com.
People who get COVID-19 infections caused by the Omicron variant are less likely to need hospital care, compared with those infected by the Delta variant, according to two large new studies from the U.K. and South Africa.
The findings, which were released ahead of peer review, add to previous glimmers of evidence suggesting that Omicron – while extremely contagious -– may result in less severe symptoms than its predecessors.
“This is helping us quantify how much less severe Omicron is than Delta, and it appears to be between 40 to 75% reduced risk of hospitalizations, adjusted for many factors, which is very good,” said Eric Topol, MD, the editor-in-chief of Medscape and a cardiologist at Scripps Research Translational Institute in La Jolla, CA.
The first analysis, which was done by the World Health Organization Collaborating Centre for Infectious Disease Modelling and Imperial College London, found that overall, people infected by Omicron had about a 20% reduced risk of needing any hospital care for their infections and a 40% lower risk of an overnight hospital stay, compared to those infected with Delta.
Meanwhile, people who were re-infected – meaning they caught Omicron after recovering from a previous COVID-19 infection – had a 50%-60% lower risk of needing hospital care, likely reflecting the benefits of having some prior immunity against the same family of viruses.
The study included everyone with polymerase chain reaction-confirmed COVID-19 in the U.K. during the first 2 weeks of December – roughly 56,000 Omicron cases and 269,000 Delta infections.
The second study, from researchers at the National Institute for Communicable Diseases in South Africa, included more than 29,000 COVID-19 cases that had lab results highly suggestive of Omicron infections. Compared to people infected with the Delta variant, those with presumed Omicron infections were about 70% less likely to have severe disease.
While the news is hopeful for individuals, on a population level, health care systems may still be stressed, the study authors warned.
“Given the high transmissibility of the Omicron virus, there remains the potential for health services to face increasing demand if Omicron cases continue to grow at the rate that has been seen in recent weeks,” said study author Neil Ferguson, PhD, who studies how infectious diseases spread at Imperial College London.
The study authors say their findings are specific to the U.K. and South Africa, where substantial portions of the population have some immune protection from past infection. In other words, they may not apply to countries where fewer people have been vaccinated or recovered from a bout with COVID-19.
A version of this article first appeared on WebMD.com.
People who get COVID-19 infections caused by the Omicron variant are less likely to need hospital care, compared with those infected by the Delta variant, according to two large new studies from the U.K. and South Africa.
The findings, which were released ahead of peer review, add to previous glimmers of evidence suggesting that Omicron – while extremely contagious -– may result in less severe symptoms than its predecessors.
“This is helping us quantify how much less severe Omicron is than Delta, and it appears to be between 40 to 75% reduced risk of hospitalizations, adjusted for many factors, which is very good,” said Eric Topol, MD, the editor-in-chief of Medscape and a cardiologist at Scripps Research Translational Institute in La Jolla, CA.
The first analysis, which was done by the World Health Organization Collaborating Centre for Infectious Disease Modelling and Imperial College London, found that overall, people infected by Omicron had about a 20% reduced risk of needing any hospital care for their infections and a 40% lower risk of an overnight hospital stay, compared to those infected with Delta.
Meanwhile, people who were re-infected – meaning they caught Omicron after recovering from a previous COVID-19 infection – had a 50%-60% lower risk of needing hospital care, likely reflecting the benefits of having some prior immunity against the same family of viruses.
The study included everyone with polymerase chain reaction-confirmed COVID-19 in the U.K. during the first 2 weeks of December – roughly 56,000 Omicron cases and 269,000 Delta infections.
The second study, from researchers at the National Institute for Communicable Diseases in South Africa, included more than 29,000 COVID-19 cases that had lab results highly suggestive of Omicron infections. Compared to people infected with the Delta variant, those with presumed Omicron infections were about 70% less likely to have severe disease.
While the news is hopeful for individuals, on a population level, health care systems may still be stressed, the study authors warned.
“Given the high transmissibility of the Omicron virus, there remains the potential for health services to face increasing demand if Omicron cases continue to grow at the rate that has been seen in recent weeks,” said study author Neil Ferguson, PhD, who studies how infectious diseases spread at Imperial College London.
The study authors say their findings are specific to the U.K. and South Africa, where substantial portions of the population have some immune protection from past infection. In other words, they may not apply to countries where fewer people have been vaccinated or recovered from a bout with COVID-19.
A version of this article first appeared on WebMD.com.
FDA authorizes Pfizer antiviral pill for COVID-19
The Food and Drug Administration on Dec. 22, 2021, granted emergency use authorization (EUA) for a new antiviral pill to treat people with symptomatic COVID-19.
Pfizer’s ritonavir, name brand Paxlovid, can now be taken by patients ages 12 and up who weigh at least 88 pounds.
The antiviral is only for people who test positive for the coronavirus and who are at high risk for severe COVID-19, including hospitalization or death. It is available by prescription only and should be taken as soon as possible after diagnosis and within 5 days of the start of symptoms.
Paxlovid is taken as three tablets together orally twice a day for 5 days, for a total of 30 tablets.
Possible side effects include a reduced sense of taste, diarrhea, high blood pressure, and muscle aches.
The authorization arrives as U.S. cases of the Omicron variant are surging, some monoclonal antibody treatments are becoming less effective, and Americans struggle to maintain some sense of tradition and normalcy around the holidays.
Paxlovid joins remdesivir as an available antiviral to treat COVID-19. Remdesivir is fully approved by the FDA but is given only intravenously in a hospital.
The COVID-19 antiviral pills come with some obvious advantages, including greater convenience for consumers – such as home use – and the potential to expand treatment for people in low- and middle-income countries.
‘An exciting step forward’
The EUA for Pfizer’s new drug has been highly anticipated, and news of its impending authorization circulated on social media on Tuesday. Eric Topol, MD, called the development an “exciting step forward.” Dr. Topol is editor in chief of Medscape, the parent company of MDedge.
He and many others also expected the FDA to grant emergency use authorization for an antiviral from Merck. But there was no immediate word Wednesday if that was still going to happen.
An accelerated authorization?
The FDA’s authorization for Pfizer’s antiviral comes about 5 weeks after the company submitted an application to the agency. In its submission, the company said a study showed the pill reduced by 89% the rate of hospitalization and death for people with mild to moderate COVID-19 illness.
In April 2021, Pfizer announced its antiviral pill for COVID-19 could be available by year’s end. In September, an official at the National Institutes of Allergy and Infectious Diseases seconded the prediction.
Merck filed its EUA application with the FDA in October. The company included results of its phase 3 study showing the treatment was linked to a 50% reduction in COVID-19 hospitalizations.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of the coronavirus because the agent does not target the virus’s spike protein. At the time, Delta was the dominant variant in the United States.
Faith-based purchasing
The U.S. government has already recognized the potential of these oral therapies, at least in terms of preorders.
Last month, it announced intentions to purchase $1 billion worth of Merck’s molnupiravir, adding to the $1.2 billion worth of the pills the U.S. ordered in June 2021. Also in November, the government announced it would purchase 10 million courses of the Pfizer pill at an estimated cost of $5.3 billion.
The government preorders of the antiviral pills for COVID-19 are separate from the orders for COVID-19 vaccines. Most recently, the Biden administration announced it will make 500 million tests for coronavirus infection available to Americans for free in early 2022.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration on Dec. 22, 2021, granted emergency use authorization (EUA) for a new antiviral pill to treat people with symptomatic COVID-19.
Pfizer’s ritonavir, name brand Paxlovid, can now be taken by patients ages 12 and up who weigh at least 88 pounds.
The antiviral is only for people who test positive for the coronavirus and who are at high risk for severe COVID-19, including hospitalization or death. It is available by prescription only and should be taken as soon as possible after diagnosis and within 5 days of the start of symptoms.
Paxlovid is taken as three tablets together orally twice a day for 5 days, for a total of 30 tablets.
Possible side effects include a reduced sense of taste, diarrhea, high blood pressure, and muscle aches.
The authorization arrives as U.S. cases of the Omicron variant are surging, some monoclonal antibody treatments are becoming less effective, and Americans struggle to maintain some sense of tradition and normalcy around the holidays.
Paxlovid joins remdesivir as an available antiviral to treat COVID-19. Remdesivir is fully approved by the FDA but is given only intravenously in a hospital.
The COVID-19 antiviral pills come with some obvious advantages, including greater convenience for consumers – such as home use – and the potential to expand treatment for people in low- and middle-income countries.
‘An exciting step forward’
The EUA for Pfizer’s new drug has been highly anticipated, and news of its impending authorization circulated on social media on Tuesday. Eric Topol, MD, called the development an “exciting step forward.” Dr. Topol is editor in chief of Medscape, the parent company of MDedge.
He and many others also expected the FDA to grant emergency use authorization for an antiviral from Merck. But there was no immediate word Wednesday if that was still going to happen.
An accelerated authorization?
The FDA’s authorization for Pfizer’s antiviral comes about 5 weeks after the company submitted an application to the agency. In its submission, the company said a study showed the pill reduced by 89% the rate of hospitalization and death for people with mild to moderate COVID-19 illness.
In April 2021, Pfizer announced its antiviral pill for COVID-19 could be available by year’s end. In September, an official at the National Institutes of Allergy and Infectious Diseases seconded the prediction.
Merck filed its EUA application with the FDA in October. The company included results of its phase 3 study showing the treatment was linked to a 50% reduction in COVID-19 hospitalizations.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of the coronavirus because the agent does not target the virus’s spike protein. At the time, Delta was the dominant variant in the United States.
Faith-based purchasing
The U.S. government has already recognized the potential of these oral therapies, at least in terms of preorders.
Last month, it announced intentions to purchase $1 billion worth of Merck’s molnupiravir, adding to the $1.2 billion worth of the pills the U.S. ordered in June 2021. Also in November, the government announced it would purchase 10 million courses of the Pfizer pill at an estimated cost of $5.3 billion.
The government preorders of the antiviral pills for COVID-19 are separate from the orders for COVID-19 vaccines. Most recently, the Biden administration announced it will make 500 million tests for coronavirus infection available to Americans for free in early 2022.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration on Dec. 22, 2021, granted emergency use authorization (EUA) for a new antiviral pill to treat people with symptomatic COVID-19.
Pfizer’s ritonavir, name brand Paxlovid, can now be taken by patients ages 12 and up who weigh at least 88 pounds.
The antiviral is only for people who test positive for the coronavirus and who are at high risk for severe COVID-19, including hospitalization or death. It is available by prescription only and should be taken as soon as possible after diagnosis and within 5 days of the start of symptoms.
Paxlovid is taken as three tablets together orally twice a day for 5 days, for a total of 30 tablets.
Possible side effects include a reduced sense of taste, diarrhea, high blood pressure, and muscle aches.
The authorization arrives as U.S. cases of the Omicron variant are surging, some monoclonal antibody treatments are becoming less effective, and Americans struggle to maintain some sense of tradition and normalcy around the holidays.
Paxlovid joins remdesivir as an available antiviral to treat COVID-19. Remdesivir is fully approved by the FDA but is given only intravenously in a hospital.
The COVID-19 antiviral pills come with some obvious advantages, including greater convenience for consumers – such as home use – and the potential to expand treatment for people in low- and middle-income countries.
‘An exciting step forward’
The EUA for Pfizer’s new drug has been highly anticipated, and news of its impending authorization circulated on social media on Tuesday. Eric Topol, MD, called the development an “exciting step forward.” Dr. Topol is editor in chief of Medscape, the parent company of MDedge.
He and many others also expected the FDA to grant emergency use authorization for an antiviral from Merck. But there was no immediate word Wednesday if that was still going to happen.
An accelerated authorization?
The FDA’s authorization for Pfizer’s antiviral comes about 5 weeks after the company submitted an application to the agency. In its submission, the company said a study showed the pill reduced by 89% the rate of hospitalization and death for people with mild to moderate COVID-19 illness.
In April 2021, Pfizer announced its antiviral pill for COVID-19 could be available by year’s end. In September, an official at the National Institutes of Allergy and Infectious Diseases seconded the prediction.
Merck filed its EUA application with the FDA in October. The company included results of its phase 3 study showing the treatment was linked to a 50% reduction in COVID-19 hospitalizations.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of the coronavirus because the agent does not target the virus’s spike protein. At the time, Delta was the dominant variant in the United States.
Faith-based purchasing
The U.S. government has already recognized the potential of these oral therapies, at least in terms of preorders.
Last month, it announced intentions to purchase $1 billion worth of Merck’s molnupiravir, adding to the $1.2 billion worth of the pills the U.S. ordered in June 2021. Also in November, the government announced it would purchase 10 million courses of the Pfizer pill at an estimated cost of $5.3 billion.
The government preorders of the antiviral pills for COVID-19 are separate from the orders for COVID-19 vaccines. Most recently, the Biden administration announced it will make 500 million tests for coronavirus infection available to Americans for free in early 2022.
A version of this article first appeared on WebMD.com.
Convalescent plasma cuts COVID-19 hospitalizations in half: Study
A “definitive study” from Johns Hopkins University researchers and others shows that convalescent plasma can cut hospital admissions for COVID-19 by 54% if therapy is administered within 8 days of symptom onset.
In the study of 1,181 adults randomly assigned to high-titer convalescent plasma or placebo, 2.9% of people receiving the therapy were hospitalized, compared with 6.3% who received placebo control plasma.
This translates to a 54% risk reduction for hospitalization with convalescent plasma.
“We have a clear difference,” principal investigator David Sullivan, MD, a professor at Johns Hopkins University, Baltimore, said during a Dec. 21 media briefing.
“This is very good news since we are in the midst of the Omicron surge, which has defeated [some of] our major monocular antibody therapies,” said Arturo Casadevall, MD, chair of the department of molecular microbiology and immunology at Johns Hopkins.
“So we have a new tool to keep people from progressing in their disease and to reduce progression or hospitalization,” Dr. Casadevall said.
The findings were published as a preprint study on Dec. 21, 2021, on medRxiv. The study has not yet been peer reviewed.
Whereas many convalescent plasma studies were done in hospitalized patients, this is one of only a handful performed in outpatients, the researchers noted.
There is a regulatory catch. The Food and Drug Administration restricted emergency use authorization (EUA) for convalescent plasma in February 2021 to include only high-dose titer plasma and to limit the therapy to hospitalized patients with early disease or for immunocompromised people who cannot mount an adequate antibody response.
Dr. Sullivan and colleagues hoped their findings will prompt the FDA to expand the EUA to include outpatients.
“We have shared this data with both the World Health Organization and the FDA,” study coauthor Kelly Gebo, MD, MPH, said during the media briefing.
“We do believe that this could be scaled up quickly,” added Dr. Gebo, professor of medicine at Johns Hopkins University. Convalescent plasma “could be used as a potential treatment as variants continue to evolve, such as we’ve seen with Omicron.”
Pre-Omicron results
The study was conducted at Johns Hopkins University and 23 other sites nationwide between June 2020 and October 2021. This means researchers enrolled symptomatic adults during circulation of the SARS-CoV-2 ancestral strain and the Alpha and Delta variants.
However, Dr. Sullivan said, “we think that ... plasma with high levels of antibodies can adapt faster to Omicron, although it will take us longer to get an Omicron-specific supply.”
Because of the timing of the study, 80% of participants were unvaccinated. Mean age was 44 years and 57% were women. Black and Hispanic participants each accounted for more than 12% of the study population.
On average, participants received a transfusion within 6 days of the start of symptoms.
In the study, 37 people out of 589 control group participants were hospitalized, compared with 17 of the 592 who received the convalescent plasma.
“We know antibodies work against SARS-CoV-2. The vaccines have been spectacular – producing antibodies that reduce hospitalizations and prevent transmission,” Dr. Sullivan said. “Convalescent plasma provides much of the same antibodies instantly.”
Convalescent and controversial
Convalescent plasma has been one of the controversial treatments for people with COVID-19 – with studies going back and forth on the potential benefits and efficacy. A National Institutes of Health–funded study published in August 2021, for example, showed no significant benefit.
“As you know, convalescent plasma has had a rocky ride,” Dr. Casadevall said.
“It was deployed with great excitement in the terrible, early days of the pandemic. Unfortunately, the early excitement and optimism was dampened with some of the randomized control trials appearing to show no benefit in reducing mortality and hospitalized patients,” he added.
In contrast, the current study shows “where convalescent plasma works using the latest, most rigorous clinical investigation tools available: a double-blinded, randomized, placebo-control trial,” Dr. Casadevall said.
Why a preprint, and why now?
The researchers decided to release their data in recognition of the lag time between reporting of COVID-19 cases and hospitalizations, Dr. Sullivan said. “That’s part of the reason we decided to act now with this knowledge – that it does take a couple of weeks – with cases of Omicron going up.”
Furthermore, “we thought this was actionable data for decision-makers,” he added.
A reporter asked why the Johns Hopkins researchers chose to hold a media briefing for a preprint study.
A preprint is “not so unusual given the SARS-CoV-2 pandemic,” said study senior author Daniel Hanley, MD, division director of brain injury outcomes at Johns Hopkins University.
“The data are the data,” Dr. Casadevall added. “This is not going to change from peer review.”
Peer review may change some of the wording of the manuscript, but not the numbers, he added.
“Now with the Omicron crisis and the fact that we have lost some more main monoclonal antibodies, it is essential to get this information out,” Dr. Casadevall said.
Plasma therapy nothing new
Donation and transfusion of convalescent plasma is highly regulated with strict criteria, said Evan Bloch, MBChB, associate director of the transfusion medicine division at Johns Hopkins University.
If the FDA opts to expand the EUA based on this or other evidence, administration of convalescent plasma could be rolled out fairly quickly, the researchers noted.
Plasma transfusion takes place in hospitals and at infusion centers every day. The infrastructure is in place in many countries, even low- and middle-resource nations, around the world to provide convalescent plasma therapy. The major difference between traditional plasma and SARS-CoV-2 convalescent plasma is the indication, Dr. Bloch added.
In addition, convalescent plasma has a polyclonal composition – a benefit compared with monoclonal antibodies, he added. “It’s more durable or adaptive [compared with] some of the targeted therapies, such as monoclonal antibodies, where we’ve witnessed this diminished efficacy with viral evolution.”
A version of this article first appeared on Medscape.com.
A “definitive study” from Johns Hopkins University researchers and others shows that convalescent plasma can cut hospital admissions for COVID-19 by 54% if therapy is administered within 8 days of symptom onset.
In the study of 1,181 adults randomly assigned to high-titer convalescent plasma or placebo, 2.9% of people receiving the therapy were hospitalized, compared with 6.3% who received placebo control plasma.
This translates to a 54% risk reduction for hospitalization with convalescent plasma.
“We have a clear difference,” principal investigator David Sullivan, MD, a professor at Johns Hopkins University, Baltimore, said during a Dec. 21 media briefing.
“This is very good news since we are in the midst of the Omicron surge, which has defeated [some of] our major monocular antibody therapies,” said Arturo Casadevall, MD, chair of the department of molecular microbiology and immunology at Johns Hopkins.
“So we have a new tool to keep people from progressing in their disease and to reduce progression or hospitalization,” Dr. Casadevall said.
The findings were published as a preprint study on Dec. 21, 2021, on medRxiv. The study has not yet been peer reviewed.
Whereas many convalescent plasma studies were done in hospitalized patients, this is one of only a handful performed in outpatients, the researchers noted.
There is a regulatory catch. The Food and Drug Administration restricted emergency use authorization (EUA) for convalescent plasma in February 2021 to include only high-dose titer plasma and to limit the therapy to hospitalized patients with early disease or for immunocompromised people who cannot mount an adequate antibody response.
Dr. Sullivan and colleagues hoped their findings will prompt the FDA to expand the EUA to include outpatients.
“We have shared this data with both the World Health Organization and the FDA,” study coauthor Kelly Gebo, MD, MPH, said during the media briefing.
“We do believe that this could be scaled up quickly,” added Dr. Gebo, professor of medicine at Johns Hopkins University. Convalescent plasma “could be used as a potential treatment as variants continue to evolve, such as we’ve seen with Omicron.”
Pre-Omicron results
The study was conducted at Johns Hopkins University and 23 other sites nationwide between June 2020 and October 2021. This means researchers enrolled symptomatic adults during circulation of the SARS-CoV-2 ancestral strain and the Alpha and Delta variants.
However, Dr. Sullivan said, “we think that ... plasma with high levels of antibodies can adapt faster to Omicron, although it will take us longer to get an Omicron-specific supply.”
Because of the timing of the study, 80% of participants were unvaccinated. Mean age was 44 years and 57% were women. Black and Hispanic participants each accounted for more than 12% of the study population.
On average, participants received a transfusion within 6 days of the start of symptoms.
In the study, 37 people out of 589 control group participants were hospitalized, compared with 17 of the 592 who received the convalescent plasma.
“We know antibodies work against SARS-CoV-2. The vaccines have been spectacular – producing antibodies that reduce hospitalizations and prevent transmission,” Dr. Sullivan said. “Convalescent plasma provides much of the same antibodies instantly.”
Convalescent and controversial
Convalescent plasma has been one of the controversial treatments for people with COVID-19 – with studies going back and forth on the potential benefits and efficacy. A National Institutes of Health–funded study published in August 2021, for example, showed no significant benefit.
“As you know, convalescent plasma has had a rocky ride,” Dr. Casadevall said.
“It was deployed with great excitement in the terrible, early days of the pandemic. Unfortunately, the early excitement and optimism was dampened with some of the randomized control trials appearing to show no benefit in reducing mortality and hospitalized patients,” he added.
In contrast, the current study shows “where convalescent plasma works using the latest, most rigorous clinical investigation tools available: a double-blinded, randomized, placebo-control trial,” Dr. Casadevall said.
Why a preprint, and why now?
The researchers decided to release their data in recognition of the lag time between reporting of COVID-19 cases and hospitalizations, Dr. Sullivan said. “That’s part of the reason we decided to act now with this knowledge – that it does take a couple of weeks – with cases of Omicron going up.”
Furthermore, “we thought this was actionable data for decision-makers,” he added.
A reporter asked why the Johns Hopkins researchers chose to hold a media briefing for a preprint study.
A preprint is “not so unusual given the SARS-CoV-2 pandemic,” said study senior author Daniel Hanley, MD, division director of brain injury outcomes at Johns Hopkins University.
“The data are the data,” Dr. Casadevall added. “This is not going to change from peer review.”
Peer review may change some of the wording of the manuscript, but not the numbers, he added.
“Now with the Omicron crisis and the fact that we have lost some more main monoclonal antibodies, it is essential to get this information out,” Dr. Casadevall said.
Plasma therapy nothing new
Donation and transfusion of convalescent plasma is highly regulated with strict criteria, said Evan Bloch, MBChB, associate director of the transfusion medicine division at Johns Hopkins University.
If the FDA opts to expand the EUA based on this or other evidence, administration of convalescent plasma could be rolled out fairly quickly, the researchers noted.
Plasma transfusion takes place in hospitals and at infusion centers every day. The infrastructure is in place in many countries, even low- and middle-resource nations, around the world to provide convalescent plasma therapy. The major difference between traditional plasma and SARS-CoV-2 convalescent plasma is the indication, Dr. Bloch added.
In addition, convalescent plasma has a polyclonal composition – a benefit compared with monoclonal antibodies, he added. “It’s more durable or adaptive [compared with] some of the targeted therapies, such as monoclonal antibodies, where we’ve witnessed this diminished efficacy with viral evolution.”
A version of this article first appeared on Medscape.com.
A “definitive study” from Johns Hopkins University researchers and others shows that convalescent plasma can cut hospital admissions for COVID-19 by 54% if therapy is administered within 8 days of symptom onset.
In the study of 1,181 adults randomly assigned to high-titer convalescent plasma or placebo, 2.9% of people receiving the therapy were hospitalized, compared with 6.3% who received placebo control plasma.
This translates to a 54% risk reduction for hospitalization with convalescent plasma.
“We have a clear difference,” principal investigator David Sullivan, MD, a professor at Johns Hopkins University, Baltimore, said during a Dec. 21 media briefing.
“This is very good news since we are in the midst of the Omicron surge, which has defeated [some of] our major monocular antibody therapies,” said Arturo Casadevall, MD, chair of the department of molecular microbiology and immunology at Johns Hopkins.
“So we have a new tool to keep people from progressing in their disease and to reduce progression or hospitalization,” Dr. Casadevall said.
The findings were published as a preprint study on Dec. 21, 2021, on medRxiv. The study has not yet been peer reviewed.
Whereas many convalescent plasma studies were done in hospitalized patients, this is one of only a handful performed in outpatients, the researchers noted.
There is a regulatory catch. The Food and Drug Administration restricted emergency use authorization (EUA) for convalescent plasma in February 2021 to include only high-dose titer plasma and to limit the therapy to hospitalized patients with early disease or for immunocompromised people who cannot mount an adequate antibody response.
Dr. Sullivan and colleagues hoped their findings will prompt the FDA to expand the EUA to include outpatients.
“We have shared this data with both the World Health Organization and the FDA,” study coauthor Kelly Gebo, MD, MPH, said during the media briefing.
“We do believe that this could be scaled up quickly,” added Dr. Gebo, professor of medicine at Johns Hopkins University. Convalescent plasma “could be used as a potential treatment as variants continue to evolve, such as we’ve seen with Omicron.”
Pre-Omicron results
The study was conducted at Johns Hopkins University and 23 other sites nationwide between June 2020 and October 2021. This means researchers enrolled symptomatic adults during circulation of the SARS-CoV-2 ancestral strain and the Alpha and Delta variants.
However, Dr. Sullivan said, “we think that ... plasma with high levels of antibodies can adapt faster to Omicron, although it will take us longer to get an Omicron-specific supply.”
Because of the timing of the study, 80% of participants were unvaccinated. Mean age was 44 years and 57% were women. Black and Hispanic participants each accounted for more than 12% of the study population.
On average, participants received a transfusion within 6 days of the start of symptoms.
In the study, 37 people out of 589 control group participants were hospitalized, compared with 17 of the 592 who received the convalescent plasma.
“We know antibodies work against SARS-CoV-2. The vaccines have been spectacular – producing antibodies that reduce hospitalizations and prevent transmission,” Dr. Sullivan said. “Convalescent plasma provides much of the same antibodies instantly.”
Convalescent and controversial
Convalescent plasma has been one of the controversial treatments for people with COVID-19 – with studies going back and forth on the potential benefits and efficacy. A National Institutes of Health–funded study published in August 2021, for example, showed no significant benefit.
“As you know, convalescent plasma has had a rocky ride,” Dr. Casadevall said.
“It was deployed with great excitement in the terrible, early days of the pandemic. Unfortunately, the early excitement and optimism was dampened with some of the randomized control trials appearing to show no benefit in reducing mortality and hospitalized patients,” he added.
In contrast, the current study shows “where convalescent plasma works using the latest, most rigorous clinical investigation tools available: a double-blinded, randomized, placebo-control trial,” Dr. Casadevall said.
Why a preprint, and why now?
The researchers decided to release their data in recognition of the lag time between reporting of COVID-19 cases and hospitalizations, Dr. Sullivan said. “That’s part of the reason we decided to act now with this knowledge – that it does take a couple of weeks – with cases of Omicron going up.”
Furthermore, “we thought this was actionable data for decision-makers,” he added.
A reporter asked why the Johns Hopkins researchers chose to hold a media briefing for a preprint study.
A preprint is “not so unusual given the SARS-CoV-2 pandemic,” said study senior author Daniel Hanley, MD, division director of brain injury outcomes at Johns Hopkins University.
“The data are the data,” Dr. Casadevall added. “This is not going to change from peer review.”
Peer review may change some of the wording of the manuscript, but not the numbers, he added.
“Now with the Omicron crisis and the fact that we have lost some more main monoclonal antibodies, it is essential to get this information out,” Dr. Casadevall said.
Plasma therapy nothing new
Donation and transfusion of convalescent plasma is highly regulated with strict criteria, said Evan Bloch, MBChB, associate director of the transfusion medicine division at Johns Hopkins University.
If the FDA opts to expand the EUA based on this or other evidence, administration of convalescent plasma could be rolled out fairly quickly, the researchers noted.
Plasma transfusion takes place in hospitals and at infusion centers every day. The infrastructure is in place in many countries, even low- and middle-resource nations, around the world to provide convalescent plasma therapy. The major difference between traditional plasma and SARS-CoV-2 convalescent plasma is the indication, Dr. Bloch added.
In addition, convalescent plasma has a polyclonal composition – a benefit compared with monoclonal antibodies, he added. “It’s more durable or adaptive [compared with] some of the targeted therapies, such as monoclonal antibodies, where we’ve witnessed this diminished efficacy with viral evolution.”
A version of this article first appeared on Medscape.com.
FROM MEDRXIV
Bamlanivimab’s effects in COVID-19 depend on antibodies
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
FROM ANNALS OF INTERNAL MEDICINE
Children and COVID: New cases up slightly, vaccinations continue to slow
New COVID-19 vaccinations in children were down by almost 24% in the last week as new cases rose by just 3.5%, based on new data.
That fairly low number suggests the latest case count from the American Academy of Pediatrics and the Children’s Hospital Association has not caught up yet to the reality of the Omicron variant, which has sent new cases climbing among all ages and now represents the majority of COVID-19 infections nationwide, the Centers for Disease Control and Prevention said.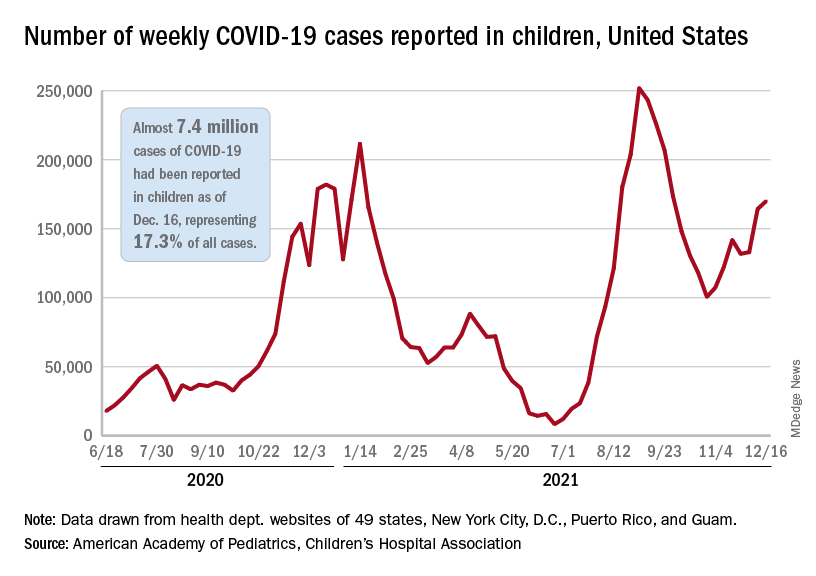
Meanwhile, in the midst of the latest surge, the United States just passed yet another sobering COVID milestone: 1,000 deaths in children aged 17 and under. The total as of Dec. 20 was 1,015, according to the CDC, with the largest share, almost 32%, occurring in children less than 5 years of age.
Regionally, the majority of that increase came in the Northeast, with a small rise in the South and decreases in the Midwest and West, the AAP and CHA said in their weekly COVID report.
At the state level, the largest percent increases in cases over the past 2 weeks were seen in Maine and New Hampshire, as well as Vermont, which has the nation’s highest vaccination rates for children aged 5-11 (51%) and 12-17 (84%), the AAP said in its vaccination trends report.
Nationally, new COVID vaccinations in children continue to trend downward. The number of children aged 5-17 years who had received at least one dose increased by about 498,000 for the week of Dec. 13-19, down from 654,000 (–23.9%) the previous week. Children aged 5-11 years still represented the largest share (22.7%) of all vaccine initiators in the last 2 weeks, but that proportion was 42.8% just before Thanksgiving, according to data from the CDC.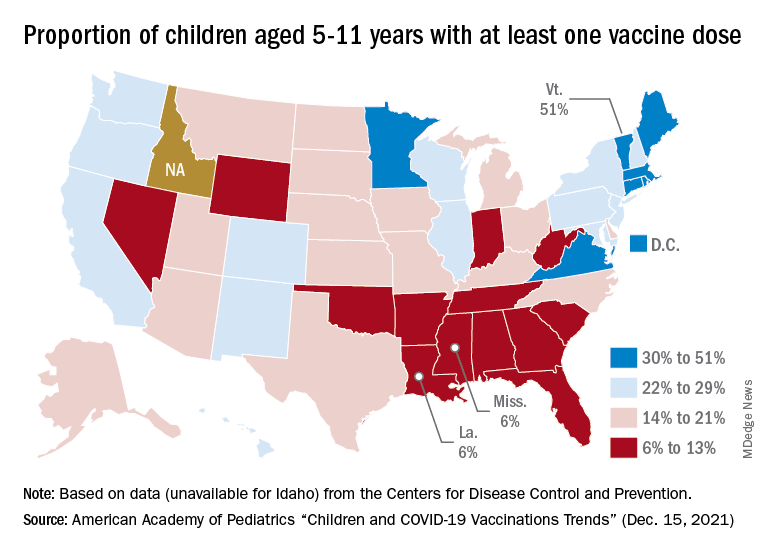
On a more positive note, children aged 5-11 made up 51% of all Americans who completed the vaccine regimen during the 2 weeks ending Dec. 20. The cumulative completion count is 3.6 million in that age group, along with almost 13.4 million children aged 12-17, and the CDC data show that 6.1 million children aged 5-11 and 15.9 million children aged 12-17 have received at least one dose.
On a less positive note, however, that means almost half (47%) of 12- to 17-year-olds still are not fully vaccinated and that over a third (37%) have received no vaccine at all, according to the COVID Data Tracker.
New COVID-19 vaccinations in children were down by almost 24% in the last week as new cases rose by just 3.5%, based on new data.
That fairly low number suggests the latest case count from the American Academy of Pediatrics and the Children’s Hospital Association has not caught up yet to the reality of the Omicron variant, which has sent new cases climbing among all ages and now represents the majority of COVID-19 infections nationwide, the Centers for Disease Control and Prevention said.
Meanwhile, in the midst of the latest surge, the United States just passed yet another sobering COVID milestone: 1,000 deaths in children aged 17 and under. The total as of Dec. 20 was 1,015, according to the CDC, with the largest share, almost 32%, occurring in children less than 5 years of age.
Regionally, the majority of that increase came in the Northeast, with a small rise in the South and decreases in the Midwest and West, the AAP and CHA said in their weekly COVID report.
At the state level, the largest percent increases in cases over the past 2 weeks were seen in Maine and New Hampshire, as well as Vermont, which has the nation’s highest vaccination rates for children aged 5-11 (51%) and 12-17 (84%), the AAP said in its vaccination trends report.
Nationally, new COVID vaccinations in children continue to trend downward. The number of children aged 5-17 years who had received at least one dose increased by about 498,000 for the week of Dec. 13-19, down from 654,000 (–23.9%) the previous week. Children aged 5-11 years still represented the largest share (22.7%) of all vaccine initiators in the last 2 weeks, but that proportion was 42.8% just before Thanksgiving, according to data from the CDC.
On a more positive note, children aged 5-11 made up 51% of all Americans who completed the vaccine regimen during the 2 weeks ending Dec. 20. The cumulative completion count is 3.6 million in that age group, along with almost 13.4 million children aged 12-17, and the CDC data show that 6.1 million children aged 5-11 and 15.9 million children aged 12-17 have received at least one dose.
On a less positive note, however, that means almost half (47%) of 12- to 17-year-olds still are not fully vaccinated and that over a third (37%) have received no vaccine at all, according to the COVID Data Tracker.
New COVID-19 vaccinations in children were down by almost 24% in the last week as new cases rose by just 3.5%, based on new data.
That fairly low number suggests the latest case count from the American Academy of Pediatrics and the Children’s Hospital Association has not caught up yet to the reality of the Omicron variant, which has sent new cases climbing among all ages and now represents the majority of COVID-19 infections nationwide, the Centers for Disease Control and Prevention said.
Meanwhile, in the midst of the latest surge, the United States just passed yet another sobering COVID milestone: 1,000 deaths in children aged 17 and under. The total as of Dec. 20 was 1,015, according to the CDC, with the largest share, almost 32%, occurring in children less than 5 years of age.
Regionally, the majority of that increase came in the Northeast, with a small rise in the South and decreases in the Midwest and West, the AAP and CHA said in their weekly COVID report.
At the state level, the largest percent increases in cases over the past 2 weeks were seen in Maine and New Hampshire, as well as Vermont, which has the nation’s highest vaccination rates for children aged 5-11 (51%) and 12-17 (84%), the AAP said in its vaccination trends report.
Nationally, new COVID vaccinations in children continue to trend downward. The number of children aged 5-17 years who had received at least one dose increased by about 498,000 for the week of Dec. 13-19, down from 654,000 (–23.9%) the previous week. Children aged 5-11 years still represented the largest share (22.7%) of all vaccine initiators in the last 2 weeks, but that proportion was 42.8% just before Thanksgiving, according to data from the CDC.
On a more positive note, children aged 5-11 made up 51% of all Americans who completed the vaccine regimen during the 2 weeks ending Dec. 20. The cumulative completion count is 3.6 million in that age group, along with almost 13.4 million children aged 12-17, and the CDC data show that 6.1 million children aged 5-11 and 15.9 million children aged 12-17 have received at least one dose.
On a less positive note, however, that means almost half (47%) of 12- to 17-year-olds still are not fully vaccinated and that over a third (37%) have received no vaccine at all, according to the COVID Data Tracker.
RSV resurgence likely in wake of COVID-19
The impact of respiratory syncytial virus (RSV)will likely be greater in 2021 and 2022 in the United States than in previous years as a result of the ongoing COVID-19 pandemic, based on data from a simulation-modeling study involving approximately 19 million individuals.
Although RSV usually follows consistent patterns of timing and duration, the disease all but disappeared starting in March 2020 after the introduction of measures to mitigate the spread of COVID-19, Zhe Zheng, MBBS, of Yale University, New Haven, Conn., and colleagues wrote.
However, lifting of mitigation measures has resulted in emergence of RSV in various parts of the world in early 2021, and trends may be similar in the United States, but data are needed to plan for prophylaxis and hospital use, they noted.
In a study published in JAMA Network Open, the researchers developed a simulation model for epidemics of RSV based on historical data. They acquired inpatient records from New York during 2005-2014 and from California during 2003-2011. The primary clinical outcome was the estimated monthly hospitalizations for RSV.
The simulated study population was 19.45 million individuals. After evaluating several scenarios including continued low transmission associated with social distancing and other mitigation measures, the researchers focused on the likely scenario that introduction of RSV from other regions would likely spark RSV epidemics in the United States.
They determined that spring and summer 2021 would show an increase in hospitalizations for RSV. Overall, higher rates of virus introduction from other regions were associated with more intense spring and summer RSV epidemics, with the trade-off of smaller winter epidemics. In the model, the expected RSV epidemic in spring and summer 2021 in New York was small, with a peak incidence of 419 hospitalizations per 100,000 people in April; by contrast, for states with less seasonal variability, such as Florida, the model predicted a larger summer epidemic.
In the model, the mean age of hospitalization for children younger than 5 years for January 2022 was expected to be 1.17 years, compared with 0.84 years in January 2019, the researchers noted.
Across all age groups, the greatest relative increase in the incidence of RSV infection was predicted for children aged 1-4 years (ranging from 82% to 86%), as were lower respiratory infections (87%-101%) and hospitalization (99%-119%), compared with prepandemic levels.
Hospitalizations for children aged 1 year were predicted to double compared with prepandemic seasons; 707 per 100,000 children per year for 2021 and 2022 versus 355 per 100,000 children per year in a typical prepandemic season. However, the largest incidence of lower respiratory infections (30,075 per 100,000) was predicted for infants aged 3-5 months, and the largest incidence of hospitalizations (3,116 per 100,000) was predicted for infants younger than 3 months.
“Without virus importation, the risk of RSV infections across all age groups in the winter of 2021 and 2022 would be greater, as more susceptible individuals were spared from infections in the absence of summer epidemics,” the researchers noted.
The older mean hospitalization age seen in the model was similar to the reported median patient age in Australia both before the pandemic and during the reemergent RSV epidemic.
“This makes intuitive sense, since many children born in 2020 were spared from RSV infection due to the low virus activity; these children will be older when they get infected for the first time during the reemergent epidemics,” the researchers wrote. “Consequently, stakeholders should consider modifying prophylaxis guidelines to include high-risk infants less than 2 years of age for the 2021-2022 season.”
The study findings were limited by several factors including the lack of data on level of virus introduction or on the impact of lack of boosting on infants with only transplacentally acquired RSV antibodies, the researchers noted. Other limitations include the use of historical data and the lack of data on values outside those included in the model, as well as the inability to control for other factors that could influence RSV, such as vaccines or long-lasting antibodies.
However, the results suggest that the rate of imported infections is associated with RSV hospitalizations, and the model effectively captured the RSV epidemics in the United States in spring and summer 2021.
Models can guide clinical preparations
“Health care simulation modeling is a growing field, with very exciting implications,” Lenore Jarvis, MD, of George Washington University, Washington, said in an interview. The field has the potential ability to influence health care in a data-driven way, including, but not limited to, staffing and other hospital operations, as well as patient care decision-making. “In short, accurate modeling and predictions can help us to make informed health care decisions that can lead to increased quality of care, potential cost savings, and even to help save lives,” she said.
Although the details of transmission modeling were not mentioned in the study, the authors evaluated the performances of several models and scenarios. “Scenario 4, for example, was focused on in particular because it best captured the observed dynamics [for RSV] that emerged during the spring and summer of 2021,” Dr. Jarvis said.
“Pediatricians can speak to these trends firsthand. A decrease in expected RSV infections and hospitalizations in 2020, followed by an unprecedented and early increase in RSV infections and severity in 2021, and the factors that the authors account for make sense, such as reintroduction of RSV from other regions and low immunity in the population,” she said. “It also makes sense that, in these transmission modeling scenarios, the expected mean age of hospitalization because of RSV increased with a temporary (hopefully) increase in RSV hospitalizations in the 2021 season, and potentially the 2022 RSV season.”
As for additional research, Dr. Jarvis said she would like to see follow-up data on the RSV transmission modeling. “For example, with scenario 4, does this scenario continue to perform well in other time periods, such as the winter? If the modeling continues to be accurate during other periods of evaluation and reevaluation, this modeling could be very useful in helping pediatric clinics and hospitals to prepare for RSV care and hospital capacity management.”
The study was supported by grants to various researchers from the National Institute of Allergy and Infectious Diseases/National Institutes of Health, the National Center for Advancing Translational Science at the National Institutes of Health, and NIH Roadmap for Medical Research. Lead author Ms. Zheng had no financial conflicts to disclose. Her study coauthors disclosed relationships with companies including AbbVie, Merck, Pfizer, GlaxoSmithKline, MedImmune, and Janssen. Dr. Jarvis had no financial conflicts to disclose and serves on the Pediatric News editorial advisory board.
The impact of respiratory syncytial virus (RSV)will likely be greater in 2021 and 2022 in the United States than in previous years as a result of the ongoing COVID-19 pandemic, based on data from a simulation-modeling study involving approximately 19 million individuals.
Although RSV usually follows consistent patterns of timing and duration, the disease all but disappeared starting in March 2020 after the introduction of measures to mitigate the spread of COVID-19, Zhe Zheng, MBBS, of Yale University, New Haven, Conn., and colleagues wrote.
However, lifting of mitigation measures has resulted in emergence of RSV in various parts of the world in early 2021, and trends may be similar in the United States, but data are needed to plan for prophylaxis and hospital use, they noted.
In a study published in JAMA Network Open, the researchers developed a simulation model for epidemics of RSV based on historical data. They acquired inpatient records from New York during 2005-2014 and from California during 2003-2011. The primary clinical outcome was the estimated monthly hospitalizations for RSV.
The simulated study population was 19.45 million individuals. After evaluating several scenarios including continued low transmission associated with social distancing and other mitigation measures, the researchers focused on the likely scenario that introduction of RSV from other regions would likely spark RSV epidemics in the United States.
They determined that spring and summer 2021 would show an increase in hospitalizations for RSV. Overall, higher rates of virus introduction from other regions were associated with more intense spring and summer RSV epidemics, with the trade-off of smaller winter epidemics. In the model, the expected RSV epidemic in spring and summer 2021 in New York was small, with a peak incidence of 419 hospitalizations per 100,000 people in April; by contrast, for states with less seasonal variability, such as Florida, the model predicted a larger summer epidemic.
In the model, the mean age of hospitalization for children younger than 5 years for January 2022 was expected to be 1.17 years, compared with 0.84 years in January 2019, the researchers noted.
Across all age groups, the greatest relative increase in the incidence of RSV infection was predicted for children aged 1-4 years (ranging from 82% to 86%), as were lower respiratory infections (87%-101%) and hospitalization (99%-119%), compared with prepandemic levels.
Hospitalizations for children aged 1 year were predicted to double compared with prepandemic seasons; 707 per 100,000 children per year for 2021 and 2022 versus 355 per 100,000 children per year in a typical prepandemic season. However, the largest incidence of lower respiratory infections (30,075 per 100,000) was predicted for infants aged 3-5 months, and the largest incidence of hospitalizations (3,116 per 100,000) was predicted for infants younger than 3 months.
“Without virus importation, the risk of RSV infections across all age groups in the winter of 2021 and 2022 would be greater, as more susceptible individuals were spared from infections in the absence of summer epidemics,” the researchers noted.
The older mean hospitalization age seen in the model was similar to the reported median patient age in Australia both before the pandemic and during the reemergent RSV epidemic.
“This makes intuitive sense, since many children born in 2020 were spared from RSV infection due to the low virus activity; these children will be older when they get infected for the first time during the reemergent epidemics,” the researchers wrote. “Consequently, stakeholders should consider modifying prophylaxis guidelines to include high-risk infants less than 2 years of age for the 2021-2022 season.”
The study findings were limited by several factors including the lack of data on level of virus introduction or on the impact of lack of boosting on infants with only transplacentally acquired RSV antibodies, the researchers noted. Other limitations include the use of historical data and the lack of data on values outside those included in the model, as well as the inability to control for other factors that could influence RSV, such as vaccines or long-lasting antibodies.
However, the results suggest that the rate of imported infections is associated with RSV hospitalizations, and the model effectively captured the RSV epidemics in the United States in spring and summer 2021.
Models can guide clinical preparations
“Health care simulation modeling is a growing field, with very exciting implications,” Lenore Jarvis, MD, of George Washington University, Washington, said in an interview. The field has the potential ability to influence health care in a data-driven way, including, but not limited to, staffing and other hospital operations, as well as patient care decision-making. “In short, accurate modeling and predictions can help us to make informed health care decisions that can lead to increased quality of care, potential cost savings, and even to help save lives,” she said.
Although the details of transmission modeling were not mentioned in the study, the authors evaluated the performances of several models and scenarios. “Scenario 4, for example, was focused on in particular because it best captured the observed dynamics [for RSV] that emerged during the spring and summer of 2021,” Dr. Jarvis said.
“Pediatricians can speak to these trends firsthand. A decrease in expected RSV infections and hospitalizations in 2020, followed by an unprecedented and early increase in RSV infections and severity in 2021, and the factors that the authors account for make sense, such as reintroduction of RSV from other regions and low immunity in the population,” she said. “It also makes sense that, in these transmission modeling scenarios, the expected mean age of hospitalization because of RSV increased with a temporary (hopefully) increase in RSV hospitalizations in the 2021 season, and potentially the 2022 RSV season.”
As for additional research, Dr. Jarvis said she would like to see follow-up data on the RSV transmission modeling. “For example, with scenario 4, does this scenario continue to perform well in other time periods, such as the winter? If the modeling continues to be accurate during other periods of evaluation and reevaluation, this modeling could be very useful in helping pediatric clinics and hospitals to prepare for RSV care and hospital capacity management.”
The study was supported by grants to various researchers from the National Institute of Allergy and Infectious Diseases/National Institutes of Health, the National Center for Advancing Translational Science at the National Institutes of Health, and NIH Roadmap for Medical Research. Lead author Ms. Zheng had no financial conflicts to disclose. Her study coauthors disclosed relationships with companies including AbbVie, Merck, Pfizer, GlaxoSmithKline, MedImmune, and Janssen. Dr. Jarvis had no financial conflicts to disclose and serves on the Pediatric News editorial advisory board.
The impact of respiratory syncytial virus (RSV)will likely be greater in 2021 and 2022 in the United States than in previous years as a result of the ongoing COVID-19 pandemic, based on data from a simulation-modeling study involving approximately 19 million individuals.
Although RSV usually follows consistent patterns of timing and duration, the disease all but disappeared starting in March 2020 after the introduction of measures to mitigate the spread of COVID-19, Zhe Zheng, MBBS, of Yale University, New Haven, Conn., and colleagues wrote.
However, lifting of mitigation measures has resulted in emergence of RSV in various parts of the world in early 2021, and trends may be similar in the United States, but data are needed to plan for prophylaxis and hospital use, they noted.
In a study published in JAMA Network Open, the researchers developed a simulation model for epidemics of RSV based on historical data. They acquired inpatient records from New York during 2005-2014 and from California during 2003-2011. The primary clinical outcome was the estimated monthly hospitalizations for RSV.
The simulated study population was 19.45 million individuals. After evaluating several scenarios including continued low transmission associated with social distancing and other mitigation measures, the researchers focused on the likely scenario that introduction of RSV from other regions would likely spark RSV epidemics in the United States.
They determined that spring and summer 2021 would show an increase in hospitalizations for RSV. Overall, higher rates of virus introduction from other regions were associated with more intense spring and summer RSV epidemics, with the trade-off of smaller winter epidemics. In the model, the expected RSV epidemic in spring and summer 2021 in New York was small, with a peak incidence of 419 hospitalizations per 100,000 people in April; by contrast, for states with less seasonal variability, such as Florida, the model predicted a larger summer epidemic.
In the model, the mean age of hospitalization for children younger than 5 years for January 2022 was expected to be 1.17 years, compared with 0.84 years in January 2019, the researchers noted.
Across all age groups, the greatest relative increase in the incidence of RSV infection was predicted for children aged 1-4 years (ranging from 82% to 86%), as were lower respiratory infections (87%-101%) and hospitalization (99%-119%), compared with prepandemic levels.
Hospitalizations for children aged 1 year were predicted to double compared with prepandemic seasons; 707 per 100,000 children per year for 2021 and 2022 versus 355 per 100,000 children per year in a typical prepandemic season. However, the largest incidence of lower respiratory infections (30,075 per 100,000) was predicted for infants aged 3-5 months, and the largest incidence of hospitalizations (3,116 per 100,000) was predicted for infants younger than 3 months.
“Without virus importation, the risk of RSV infections across all age groups in the winter of 2021 and 2022 would be greater, as more susceptible individuals were spared from infections in the absence of summer epidemics,” the researchers noted.
The older mean hospitalization age seen in the model was similar to the reported median patient age in Australia both before the pandemic and during the reemergent RSV epidemic.
“This makes intuitive sense, since many children born in 2020 were spared from RSV infection due to the low virus activity; these children will be older when they get infected for the first time during the reemergent epidemics,” the researchers wrote. “Consequently, stakeholders should consider modifying prophylaxis guidelines to include high-risk infants less than 2 years of age for the 2021-2022 season.”
The study findings were limited by several factors including the lack of data on level of virus introduction or on the impact of lack of boosting on infants with only transplacentally acquired RSV antibodies, the researchers noted. Other limitations include the use of historical data and the lack of data on values outside those included in the model, as well as the inability to control for other factors that could influence RSV, such as vaccines or long-lasting antibodies.
However, the results suggest that the rate of imported infections is associated with RSV hospitalizations, and the model effectively captured the RSV epidemics in the United States in spring and summer 2021.
Models can guide clinical preparations
“Health care simulation modeling is a growing field, with very exciting implications,” Lenore Jarvis, MD, of George Washington University, Washington, said in an interview. The field has the potential ability to influence health care in a data-driven way, including, but not limited to, staffing and other hospital operations, as well as patient care decision-making. “In short, accurate modeling and predictions can help us to make informed health care decisions that can lead to increased quality of care, potential cost savings, and even to help save lives,” she said.
Although the details of transmission modeling were not mentioned in the study, the authors evaluated the performances of several models and scenarios. “Scenario 4, for example, was focused on in particular because it best captured the observed dynamics [for RSV] that emerged during the spring and summer of 2021,” Dr. Jarvis said.
“Pediatricians can speak to these trends firsthand. A decrease in expected RSV infections and hospitalizations in 2020, followed by an unprecedented and early increase in RSV infections and severity in 2021, and the factors that the authors account for make sense, such as reintroduction of RSV from other regions and low immunity in the population,” she said. “It also makes sense that, in these transmission modeling scenarios, the expected mean age of hospitalization because of RSV increased with a temporary (hopefully) increase in RSV hospitalizations in the 2021 season, and potentially the 2022 RSV season.”
As for additional research, Dr. Jarvis said she would like to see follow-up data on the RSV transmission modeling. “For example, with scenario 4, does this scenario continue to perform well in other time periods, such as the winter? If the modeling continues to be accurate during other periods of evaluation and reevaluation, this modeling could be very useful in helping pediatric clinics and hospitals to prepare for RSV care and hospital capacity management.”
The study was supported by grants to various researchers from the National Institute of Allergy and Infectious Diseases/National Institutes of Health, the National Center for Advancing Translational Science at the National Institutes of Health, and NIH Roadmap for Medical Research. Lead author Ms. Zheng had no financial conflicts to disclose. Her study coauthors disclosed relationships with companies including AbbVie, Merck, Pfizer, GlaxoSmithKline, MedImmune, and Janssen. Dr. Jarvis had no financial conflicts to disclose and serves on the Pediatric News editorial advisory board.
FROM JAMA NETWORK OPEN
BMJ slams ‘incompetent’ Facebook fact-checking of vaccine article
According to an open letter written by outgoing BMJ editor-in-chief Fiona Godlee, MD, and incoming editor-in-chief Kamran Abbasi, MD, Facebook hired a third-party contractor to evaluate the article’s findings. This resulted in “inaccurate, incompetent, and irresponsible” conclusions that “should be of concern to anyone who values and relies on sources such as the BMJ for reliable medical information.”
The article in question investigated data integrity concerns at Pfizer vaccine clinical trial sites. In September 2020, the letter states, a former employee of the research group involved in Pfizer’s main vaccine trials, Ventavia, reached out to the BMJ and “began providing ... dozens of internal company documents, photos, audio recordings, and emails.” According to the company’s website, Ventavia “played a significant part in [COVID-19 clinical trial] recruitment” and “has received recognition by Pfizer for their contribution to vaccine trials.”
It was previously reported that the whistle-blower is a former regional director who was involved in Pfizer’s vaccine trials in Texas during the fall of 2020. She alleges “the company falsified data, unblinded patients, employed inadequately trained vaccinators, and was slow to follow up on adverse events reported in Pfizer’s pivotal phase 3 trial.”
The images provided to the BMJ “showed needles discarded in a plastic biohazard bag instead of a sharps container box” and another displayed “vaccine packaging materials with trial participants’ identification numbers written on them left out in the open, potentially unblinding participants.”
Despite informing Ventavia, the director’s concerns went unaddressed. She then filed a complaint with the Food and Drug Administration and was subsequently fired the same day. The FDA did not investigate the director’s allegations, said Dr. Godlee and Dr. Abbasi, even though the evidence “revealed a host of poor clinical trial research practices occurring at Ventavia that could impact data integrity and patient safety.”
Article labeled as ‘hoax,’ without pointing out errors
The BMJ hired an investigative reporter to follow up on the clinical trial claims. The findings were published in an article on Nov. 2, 2021, after the article “went through ... the usual high-level legal and editorial oversight and peer review,” according to the journal.
However, by Nov. 10, the journal began receiving complaints from readers unable to share the article on social media. Others had their posts flagged with warnings, such as “missing context ... independent fact-checkers say this information could mislead people.” Administrators of various Facebook groups were notified that posts containing the article were “partly false.”
Readers were informed that Facebook contractor Lead Stories performed the article’s “fact check.” Lead Stories is “an award-winning innovative fact checking and debunking website” and “an active part of Facebook’s partnership with third-party fact checkers” – with the latter granting them “access to listings of content that has been flagged as potentially false by Facebook’s systems or its users.” The company said they “decide independently if we want to fact check it or not.”
Lead Stories stated that they “can enter our fact checks into a tool provided by Facebook and Facebook then uses our data to help slow down the spread of false information on its platform.” Although the contractor is compensated, Lead Stories claims they have “no say or influence over what we fact check or what our conclusions are.”
Both editors question the validity of the fact check performed by Lead Stories, as it failed to provide any “assertions of fact” as to what the BMJ got wrong. Moreover, the editors take issue with Lead Stories referring to the journal as a “news blog” and using the phrase “hoax-alert” in the URL when publishing the story on its site.
The BMJ has reached out to Lead Stories and Facebook, said the letter, but Lead Stories refuses to “change anything about their article or actions that have led to Facebook flagging our article.” Requests for Facebook to remove the “fact-checking” label and allow “readers to freely share the article on [Facebook’s] platform” have been unfruitful.
Dr. Godlee and Dr. Abbasi expressed concern that other “high quality information provider[s] have been affected by the incompetence of Meta’s fact checking regime.” In November, Instagram censored Cochrane, an international provider of independent systematic medical reviews. Instagram, also owned by Meta, prohibited users from tagging Cochrane because the organization “repeatedly posted ... false content about COVID-19 or vaccines.” Cochrane refuted the allegations.
While “fact checking has been a staple of good journalism for decades,” said the editors, Meta has “apparently delegated responsibility to people incompetent in carrying out this crucial task.” They urged the company to reconsider its fact-checking strategy and review the issues that contributed to the error.
This news organization reached out to Meta for comment but did not receive a response at press time.
Lead Stories has posted a reply (Lead Stories’ Response To BMJ Open Letter Objecting To A Lead Stories Fact Check) to the BMJ’s complaint on its website.
A version of this article first appeared on Medscape.com.
According to an open letter written by outgoing BMJ editor-in-chief Fiona Godlee, MD, and incoming editor-in-chief Kamran Abbasi, MD, Facebook hired a third-party contractor to evaluate the article’s findings. This resulted in “inaccurate, incompetent, and irresponsible” conclusions that “should be of concern to anyone who values and relies on sources such as the BMJ for reliable medical information.”
The article in question investigated data integrity concerns at Pfizer vaccine clinical trial sites. In September 2020, the letter states, a former employee of the research group involved in Pfizer’s main vaccine trials, Ventavia, reached out to the BMJ and “began providing ... dozens of internal company documents, photos, audio recordings, and emails.” According to the company’s website, Ventavia “played a significant part in [COVID-19 clinical trial] recruitment” and “has received recognition by Pfizer for their contribution to vaccine trials.”
It was previously reported that the whistle-blower is a former regional director who was involved in Pfizer’s vaccine trials in Texas during the fall of 2020. She alleges “the company falsified data, unblinded patients, employed inadequately trained vaccinators, and was slow to follow up on adverse events reported in Pfizer’s pivotal phase 3 trial.”
The images provided to the BMJ “showed needles discarded in a plastic biohazard bag instead of a sharps container box” and another displayed “vaccine packaging materials with trial participants’ identification numbers written on them left out in the open, potentially unblinding participants.”
Despite informing Ventavia, the director’s concerns went unaddressed. She then filed a complaint with the Food and Drug Administration and was subsequently fired the same day. The FDA did not investigate the director’s allegations, said Dr. Godlee and Dr. Abbasi, even though the evidence “revealed a host of poor clinical trial research practices occurring at Ventavia that could impact data integrity and patient safety.”
Article labeled as ‘hoax,’ without pointing out errors
The BMJ hired an investigative reporter to follow up on the clinical trial claims. The findings were published in an article on Nov. 2, 2021, after the article “went through ... the usual high-level legal and editorial oversight and peer review,” according to the journal.
However, by Nov. 10, the journal began receiving complaints from readers unable to share the article on social media. Others had their posts flagged with warnings, such as “missing context ... independent fact-checkers say this information could mislead people.” Administrators of various Facebook groups were notified that posts containing the article were “partly false.”
Readers were informed that Facebook contractor Lead Stories performed the article’s “fact check.” Lead Stories is “an award-winning innovative fact checking and debunking website” and “an active part of Facebook’s partnership with third-party fact checkers” – with the latter granting them “access to listings of content that has been flagged as potentially false by Facebook’s systems or its users.” The company said they “decide independently if we want to fact check it or not.”
Lead Stories stated that they “can enter our fact checks into a tool provided by Facebook and Facebook then uses our data to help slow down the spread of false information on its platform.” Although the contractor is compensated, Lead Stories claims they have “no say or influence over what we fact check or what our conclusions are.”
Both editors question the validity of the fact check performed by Lead Stories, as it failed to provide any “assertions of fact” as to what the BMJ got wrong. Moreover, the editors take issue with Lead Stories referring to the journal as a “news blog” and using the phrase “hoax-alert” in the URL when publishing the story on its site.
The BMJ has reached out to Lead Stories and Facebook, said the letter, but Lead Stories refuses to “change anything about their article or actions that have led to Facebook flagging our article.” Requests for Facebook to remove the “fact-checking” label and allow “readers to freely share the article on [Facebook’s] platform” have been unfruitful.
Dr. Godlee and Dr. Abbasi expressed concern that other “high quality information provider[s] have been affected by the incompetence of Meta’s fact checking regime.” In November, Instagram censored Cochrane, an international provider of independent systematic medical reviews. Instagram, also owned by Meta, prohibited users from tagging Cochrane because the organization “repeatedly posted ... false content about COVID-19 or vaccines.” Cochrane refuted the allegations.
While “fact checking has been a staple of good journalism for decades,” said the editors, Meta has “apparently delegated responsibility to people incompetent in carrying out this crucial task.” They urged the company to reconsider its fact-checking strategy and review the issues that contributed to the error.
This news organization reached out to Meta for comment but did not receive a response at press time.
Lead Stories has posted a reply (Lead Stories’ Response To BMJ Open Letter Objecting To A Lead Stories Fact Check) to the BMJ’s complaint on its website.
A version of this article first appeared on Medscape.com.
According to an open letter written by outgoing BMJ editor-in-chief Fiona Godlee, MD, and incoming editor-in-chief Kamran Abbasi, MD, Facebook hired a third-party contractor to evaluate the article’s findings. This resulted in “inaccurate, incompetent, and irresponsible” conclusions that “should be of concern to anyone who values and relies on sources such as the BMJ for reliable medical information.”
The article in question investigated data integrity concerns at Pfizer vaccine clinical trial sites. In September 2020, the letter states, a former employee of the research group involved in Pfizer’s main vaccine trials, Ventavia, reached out to the BMJ and “began providing ... dozens of internal company documents, photos, audio recordings, and emails.” According to the company’s website, Ventavia “played a significant part in [COVID-19 clinical trial] recruitment” and “has received recognition by Pfizer for their contribution to vaccine trials.”
It was previously reported that the whistle-blower is a former regional director who was involved in Pfizer’s vaccine trials in Texas during the fall of 2020. She alleges “the company falsified data, unblinded patients, employed inadequately trained vaccinators, and was slow to follow up on adverse events reported in Pfizer’s pivotal phase 3 trial.”
The images provided to the BMJ “showed needles discarded in a plastic biohazard bag instead of a sharps container box” and another displayed “vaccine packaging materials with trial participants’ identification numbers written on them left out in the open, potentially unblinding participants.”
Despite informing Ventavia, the director’s concerns went unaddressed. She then filed a complaint with the Food and Drug Administration and was subsequently fired the same day. The FDA did not investigate the director’s allegations, said Dr. Godlee and Dr. Abbasi, even though the evidence “revealed a host of poor clinical trial research practices occurring at Ventavia that could impact data integrity and patient safety.”
Article labeled as ‘hoax,’ without pointing out errors
The BMJ hired an investigative reporter to follow up on the clinical trial claims. The findings were published in an article on Nov. 2, 2021, after the article “went through ... the usual high-level legal and editorial oversight and peer review,” according to the journal.
However, by Nov. 10, the journal began receiving complaints from readers unable to share the article on social media. Others had their posts flagged with warnings, such as “missing context ... independent fact-checkers say this information could mislead people.” Administrators of various Facebook groups were notified that posts containing the article were “partly false.”
Readers were informed that Facebook contractor Lead Stories performed the article’s “fact check.” Lead Stories is “an award-winning innovative fact checking and debunking website” and “an active part of Facebook’s partnership with third-party fact checkers” – with the latter granting them “access to listings of content that has been flagged as potentially false by Facebook’s systems or its users.” The company said they “decide independently if we want to fact check it or not.”
Lead Stories stated that they “can enter our fact checks into a tool provided by Facebook and Facebook then uses our data to help slow down the spread of false information on its platform.” Although the contractor is compensated, Lead Stories claims they have “no say or influence over what we fact check or what our conclusions are.”
Both editors question the validity of the fact check performed by Lead Stories, as it failed to provide any “assertions of fact” as to what the BMJ got wrong. Moreover, the editors take issue with Lead Stories referring to the journal as a “news blog” and using the phrase “hoax-alert” in the URL when publishing the story on its site.
The BMJ has reached out to Lead Stories and Facebook, said the letter, but Lead Stories refuses to “change anything about their article or actions that have led to Facebook flagging our article.” Requests for Facebook to remove the “fact-checking” label and allow “readers to freely share the article on [Facebook’s] platform” have been unfruitful.
Dr. Godlee and Dr. Abbasi expressed concern that other “high quality information provider[s] have been affected by the incompetence of Meta’s fact checking regime.” In November, Instagram censored Cochrane, an international provider of independent systematic medical reviews. Instagram, also owned by Meta, prohibited users from tagging Cochrane because the organization “repeatedly posted ... false content about COVID-19 or vaccines.” Cochrane refuted the allegations.
While “fact checking has been a staple of good journalism for decades,” said the editors, Meta has “apparently delegated responsibility to people incompetent in carrying out this crucial task.” They urged the company to reconsider its fact-checking strategy and review the issues that contributed to the error.
This news organization reached out to Meta for comment but did not receive a response at press time.
Lead Stories has posted a reply (Lead Stories’ Response To BMJ Open Letter Objecting To A Lead Stories Fact Check) to the BMJ’s complaint on its website.
A version of this article first appeared on Medscape.com.
CDC supports ‘test-to-stay’ for COVID- exposed students
The Centers for Disease Control and Prevention has announced that in the following days.
The new guidance, known as the “test-to-stay” protocol, would reduce the number of children who are expected to stay home as a close contact to someone who tested positive for the virus.
“Test-to-stay is an encouraging public health practice to keep our children in schools,” Rochelle Walensky, MD, director of the CDC, said during a White House press briefing.
When a COVID-19 case is identified in a school, the test-to-stay strategy allows schools to implement regular testing rather than quarantine close contacts. If the contacts don’t experience symptoms and test negative at least twice in a seven-day period, they can continue in-person learning. If they test positive, then they are required to isolate.
In recent months, the CDC has collaborated with several school districts across the United States to evaluate test-to-stay programs. On Dec. 17, the CDC published two studies in its Morbidity and Mortality Weekly Report that demonstrated the effectiveness of these programs in limiting the spread of the virus while also keeping students in class.
“CDC is updating our materials to help schools and parents know how to best implement this promising and now-proven practice, along with our multi-layer prevention strategies that will help keep our children in the classroom safely,” Dr. Walensky said. “These studies demonstrated that test-to-stay works to keep unvaccinated children in school safely.”
In one study, researchers analyzed data for public schools in Los Angeles County between Aug. 16 and Oct. 31, where 432 schools implemented test-to-stay and 1,635 did not.
The Los Angeles County Department of Public Health found that COVID-19 cases did not increase among the schools that used the protocol, as compared with schools that didn’t.
Before test-to-stay was implemented, the average daily number of cases was 10 cases per 100,000 students in districts that later adopted the protocol and 20 cases per 100,000 students in districts that didn’t. After the program was implemented, average daily case rates declined in all school districts but remained lower in test-to-stay districts, with 6 cases per 100,000 students as compared with 11 cases per 100,000 students in districts that didn’t do the protocol.
In addition, schools that didn’t use the test-to-stay program “lost substantial in-person school days,” researchers wrote. At the same time, implementing the program “requires resources that might be currently unavailable for some schools,” they added, noting that “a higher percentage of disadvantaged schools” didn’t do the protocol.
The program requires personnel who can track which students need to be tested, their results and when they can come off the list of close contacts, officials told CNN. This can be a challenge for overstretched school nursing staff.
In another study published last week, researchers analyzed data between Aug. 9 and Oct. 29 for 90 schools across 31 districts in Lake County, Ill., that implemented test-to-stay programs. During that time, the schools reported 258 COVID-19 cases and 1,664 close contacts.
The Lake County Health Department examined the number of close contacts that later tested positive and whether the virus further spread from the close contacts to other people. They found that 16 of the close contacts tested positive and that these were all students. No one appeared to transmit the virus to others at school, but nine cases were identified among household contacts.
Overall, study authors wrote, the test-to-stay protocol preserved in-person learning days for students. In addition, regular testing, masking, and physical distancing led to lower virus transmission in school.
“The test-to-stay-programs are really good at balancing the costs and benefits,” Zoe McLaren, a health policy expert at the University of Maryland at Baltimore, told The New York Times.
“What the test-to-stay program does is help us keep COVID cases down, while also trying to make sure we keep kids in school as much as possible, which I think is really important,” she said.
A version of this article first appeared on WebMD.com.
The Centers for Disease Control and Prevention has announced that in the following days.
The new guidance, known as the “test-to-stay” protocol, would reduce the number of children who are expected to stay home as a close contact to someone who tested positive for the virus.
“Test-to-stay is an encouraging public health practice to keep our children in schools,” Rochelle Walensky, MD, director of the CDC, said during a White House press briefing.
When a COVID-19 case is identified in a school, the test-to-stay strategy allows schools to implement regular testing rather than quarantine close contacts. If the contacts don’t experience symptoms and test negative at least twice in a seven-day period, they can continue in-person learning. If they test positive, then they are required to isolate.
In recent months, the CDC has collaborated with several school districts across the United States to evaluate test-to-stay programs. On Dec. 17, the CDC published two studies in its Morbidity and Mortality Weekly Report that demonstrated the effectiveness of these programs in limiting the spread of the virus while also keeping students in class.
“CDC is updating our materials to help schools and parents know how to best implement this promising and now-proven practice, along with our multi-layer prevention strategies that will help keep our children in the classroom safely,” Dr. Walensky said. “These studies demonstrated that test-to-stay works to keep unvaccinated children in school safely.”
In one study, researchers analyzed data for public schools in Los Angeles County between Aug. 16 and Oct. 31, where 432 schools implemented test-to-stay and 1,635 did not.
The Los Angeles County Department of Public Health found that COVID-19 cases did not increase among the schools that used the protocol, as compared with schools that didn’t.
Before test-to-stay was implemented, the average daily number of cases was 10 cases per 100,000 students in districts that later adopted the protocol and 20 cases per 100,000 students in districts that didn’t. After the program was implemented, average daily case rates declined in all school districts but remained lower in test-to-stay districts, with 6 cases per 100,000 students as compared with 11 cases per 100,000 students in districts that didn’t do the protocol.
In addition, schools that didn’t use the test-to-stay program “lost substantial in-person school days,” researchers wrote. At the same time, implementing the program “requires resources that might be currently unavailable for some schools,” they added, noting that “a higher percentage of disadvantaged schools” didn’t do the protocol.
The program requires personnel who can track which students need to be tested, their results and when they can come off the list of close contacts, officials told CNN. This can be a challenge for overstretched school nursing staff.
In another study published last week, researchers analyzed data between Aug. 9 and Oct. 29 for 90 schools across 31 districts in Lake County, Ill., that implemented test-to-stay programs. During that time, the schools reported 258 COVID-19 cases and 1,664 close contacts.
The Lake County Health Department examined the number of close contacts that later tested positive and whether the virus further spread from the close contacts to other people. They found that 16 of the close contacts tested positive and that these were all students. No one appeared to transmit the virus to others at school, but nine cases were identified among household contacts.
Overall, study authors wrote, the test-to-stay protocol preserved in-person learning days for students. In addition, regular testing, masking, and physical distancing led to lower virus transmission in school.
“The test-to-stay-programs are really good at balancing the costs and benefits,” Zoe McLaren, a health policy expert at the University of Maryland at Baltimore, told The New York Times.
“What the test-to-stay program does is help us keep COVID cases down, while also trying to make sure we keep kids in school as much as possible, which I think is really important,” she said.
A version of this article first appeared on WebMD.com.
The Centers for Disease Control and Prevention has announced that in the following days.
The new guidance, known as the “test-to-stay” protocol, would reduce the number of children who are expected to stay home as a close contact to someone who tested positive for the virus.
“Test-to-stay is an encouraging public health practice to keep our children in schools,” Rochelle Walensky, MD, director of the CDC, said during a White House press briefing.
When a COVID-19 case is identified in a school, the test-to-stay strategy allows schools to implement regular testing rather than quarantine close contacts. If the contacts don’t experience symptoms and test negative at least twice in a seven-day period, they can continue in-person learning. If they test positive, then they are required to isolate.
In recent months, the CDC has collaborated with several school districts across the United States to evaluate test-to-stay programs. On Dec. 17, the CDC published two studies in its Morbidity and Mortality Weekly Report that demonstrated the effectiveness of these programs in limiting the spread of the virus while also keeping students in class.
“CDC is updating our materials to help schools and parents know how to best implement this promising and now-proven practice, along with our multi-layer prevention strategies that will help keep our children in the classroom safely,” Dr. Walensky said. “These studies demonstrated that test-to-stay works to keep unvaccinated children in school safely.”
In one study, researchers analyzed data for public schools in Los Angeles County between Aug. 16 and Oct. 31, where 432 schools implemented test-to-stay and 1,635 did not.
The Los Angeles County Department of Public Health found that COVID-19 cases did not increase among the schools that used the protocol, as compared with schools that didn’t.
Before test-to-stay was implemented, the average daily number of cases was 10 cases per 100,000 students in districts that later adopted the protocol and 20 cases per 100,000 students in districts that didn’t. After the program was implemented, average daily case rates declined in all school districts but remained lower in test-to-stay districts, with 6 cases per 100,000 students as compared with 11 cases per 100,000 students in districts that didn’t do the protocol.
In addition, schools that didn’t use the test-to-stay program “lost substantial in-person school days,” researchers wrote. At the same time, implementing the program “requires resources that might be currently unavailable for some schools,” they added, noting that “a higher percentage of disadvantaged schools” didn’t do the protocol.
The program requires personnel who can track which students need to be tested, their results and when they can come off the list of close contacts, officials told CNN. This can be a challenge for overstretched school nursing staff.
In another study published last week, researchers analyzed data between Aug. 9 and Oct. 29 for 90 schools across 31 districts in Lake County, Ill., that implemented test-to-stay programs. During that time, the schools reported 258 COVID-19 cases and 1,664 close contacts.
The Lake County Health Department examined the number of close contacts that later tested positive and whether the virus further spread from the close contacts to other people. They found that 16 of the close contacts tested positive and that these were all students. No one appeared to transmit the virus to others at school, but nine cases were identified among household contacts.
Overall, study authors wrote, the test-to-stay protocol preserved in-person learning days for students. In addition, regular testing, masking, and physical distancing led to lower virus transmission in school.
“The test-to-stay-programs are really good at balancing the costs and benefits,” Zoe McLaren, a health policy expert at the University of Maryland at Baltimore, told The New York Times.
“What the test-to-stay program does is help us keep COVID cases down, while also trying to make sure we keep kids in school as much as possible, which I think is really important,” she said.
A version of this article first appeared on WebMD.com.
FDA approves tezepelumab-ekko (Tezspire) for severe asthma
The Food and Drug Administration has approved tezepelumab-ekko (Tezspire) as a first-in-class treatment for severe asthma in adults and pediatric patients aged 12 years and older. It is not recommended for the relief of acute bronchospasm or status asthmaticus.

Tezepelumab-ekko is a human monoclonal antibody that acts as a thymic stromal lymphopoietin (TSLP) blocker. TSLP is an epithelial cell–derived cytokine implicated in the pathogenesis of asthma. Tezepelumab-ekko is administered by subcutaneous injection at a recommended dosage of 210 mg given once every 4 weeks.
“Tezspire represents a much-needed new treatment for the many patients who remain underserved and continue to struggle with severe, uncontrolled asthma,” said professor Andrew Menzies-Gow, MD, PhD, director of the lung division, Royal Brompton Hospital, London, and the principal investigator of the pivotal NAVIGATOR trial, in a Dec. 17 Amgen press release.
Trial results
The early approval of the treatment was based on the results of various clinical trials, primarily the NAVIGATOR phase 3 trial, results of which were published in the New England Journal of Medicine in May 2021.
In the NAVIGATOR trial, a total of 1,061 patients were randomly assigned to receive tezepelumab (529 patients) or placebo (532 patients).
With tezepelumab, the annualized rate of asthma exacerbations was 0.93; with placebo, the rate was 2.10 (P < .001).
“Patients with severe, uncontrolled asthma who received tezepelumab had fewer exacerbations and better lung function, asthma control, and health-related quality of life than those who received placebo,” according to the report of NAVIGATOR trial, which was funded by AstraZeneca and Amgen.
Tezepelumab details
The full prescribing information for tezepelumab-ekko is available, including specific warnings and areas of concern where information is not available. The drug should not be administered to individuals with known hypersensitivity to tezepelumab-ekko or excipients, and hypersensitivity reactions (e.g., rash and allergic conjunctivitis), can occur within hours of administration, but in some instances have a delayed onset (i.e., days).
The drug should not be used to treat acute asthma symptoms, acute exacerbations, acute bronchospasm, or status asthmaticus, and the use of live-attenuated vaccines in patients receiving tezepelumab-ekko should be avoided.
There is no available data regarding the use of tezepelumab-ekko in patients who are pregnant, although placental transfer of monoclonal antibodies such as tezepelumab-ekko is greater during the third trimester of pregnancy; therefore, potential effects on a fetus are likely to be greater during the third trimester of pregnancy, according to the company.
The most common adverse reactions for the drug, with a reported incidence of at least 3%, are pharyngitis, arthralgia, and back pain.
“The approval of Tezspire is long-awaited positive news for the asthma community,” said Tonya Winders, president and CEO at the Allergy & Asthma Network and president of the Global Allergy and Airways Patient Platform in the Amgen press release. “For the first time, many people living with severe asthma have the opportunity to receive treatment regardless of the cause of their inflammation.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved tezepelumab-ekko (Tezspire) as a first-in-class treatment for severe asthma in adults and pediatric patients aged 12 years and older. It is not recommended for the relief of acute bronchospasm or status asthmaticus.
Tezepelumab-ekko is a human monoclonal antibody that acts as a thymic stromal lymphopoietin (TSLP) blocker. TSLP is an epithelial cell–derived cytokine implicated in the pathogenesis of asthma. Tezepelumab-ekko is administered by subcutaneous injection at a recommended dosage of 210 mg given once every 4 weeks.
“Tezspire represents a much-needed new treatment for the many patients who remain underserved and continue to struggle with severe, uncontrolled asthma,” said professor Andrew Menzies-Gow, MD, PhD, director of the lung division, Royal Brompton Hospital, London, and the principal investigator of the pivotal NAVIGATOR trial, in a Dec. 17 Amgen press release.
Trial results
The early approval of the treatment was based on the results of various clinical trials, primarily the NAVIGATOR phase 3 trial, results of which were published in the New England Journal of Medicine in May 2021.
In the NAVIGATOR trial, a total of 1,061 patients were randomly assigned to receive tezepelumab (529 patients) or placebo (532 patients).
With tezepelumab, the annualized rate of asthma exacerbations was 0.93; with placebo, the rate was 2.10 (P < .001).
“Patients with severe, uncontrolled asthma who received tezepelumab had fewer exacerbations and better lung function, asthma control, and health-related quality of life than those who received placebo,” according to the report of NAVIGATOR trial, which was funded by AstraZeneca and Amgen.
Tezepelumab details
The full prescribing information for tezepelumab-ekko is available, including specific warnings and areas of concern where information is not available. The drug should not be administered to individuals with known hypersensitivity to tezepelumab-ekko or excipients, and hypersensitivity reactions (e.g., rash and allergic conjunctivitis), can occur within hours of administration, but in some instances have a delayed onset (i.e., days).
The drug should not be used to treat acute asthma symptoms, acute exacerbations, acute bronchospasm, or status asthmaticus, and the use of live-attenuated vaccines in patients receiving tezepelumab-ekko should be avoided.
There is no available data regarding the use of tezepelumab-ekko in patients who are pregnant, although placental transfer of monoclonal antibodies such as tezepelumab-ekko is greater during the third trimester of pregnancy; therefore, potential effects on a fetus are likely to be greater during the third trimester of pregnancy, according to the company.
The most common adverse reactions for the drug, with a reported incidence of at least 3%, are pharyngitis, arthralgia, and back pain.
“The approval of Tezspire is long-awaited positive news for the asthma community,” said Tonya Winders, president and CEO at the Allergy & Asthma Network and president of the Global Allergy and Airways Patient Platform in the Amgen press release. “For the first time, many people living with severe asthma have the opportunity to receive treatment regardless of the cause of their inflammation.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved tezepelumab-ekko (Tezspire) as a first-in-class treatment for severe asthma in adults and pediatric patients aged 12 years and older. It is not recommended for the relief of acute bronchospasm or status asthmaticus.
Tezepelumab-ekko is a human monoclonal antibody that acts as a thymic stromal lymphopoietin (TSLP) blocker. TSLP is an epithelial cell–derived cytokine implicated in the pathogenesis of asthma. Tezepelumab-ekko is administered by subcutaneous injection at a recommended dosage of 210 mg given once every 4 weeks.
“Tezspire represents a much-needed new treatment for the many patients who remain underserved and continue to struggle with severe, uncontrolled asthma,” said professor Andrew Menzies-Gow, MD, PhD, director of the lung division, Royal Brompton Hospital, London, and the principal investigator of the pivotal NAVIGATOR trial, in a Dec. 17 Amgen press release.
Trial results
The early approval of the treatment was based on the results of various clinical trials, primarily the NAVIGATOR phase 3 trial, results of which were published in the New England Journal of Medicine in May 2021.
In the NAVIGATOR trial, a total of 1,061 patients were randomly assigned to receive tezepelumab (529 patients) or placebo (532 patients).
With tezepelumab, the annualized rate of asthma exacerbations was 0.93; with placebo, the rate was 2.10 (P < .001).
“Patients with severe, uncontrolled asthma who received tezepelumab had fewer exacerbations and better lung function, asthma control, and health-related quality of life than those who received placebo,” according to the report of NAVIGATOR trial, which was funded by AstraZeneca and Amgen.
Tezepelumab details
The full prescribing information for tezepelumab-ekko is available, including specific warnings and areas of concern where information is not available. The drug should not be administered to individuals with known hypersensitivity to tezepelumab-ekko or excipients, and hypersensitivity reactions (e.g., rash and allergic conjunctivitis), can occur within hours of administration, but in some instances have a delayed onset (i.e., days).
The drug should not be used to treat acute asthma symptoms, acute exacerbations, acute bronchospasm, or status asthmaticus, and the use of live-attenuated vaccines in patients receiving tezepelumab-ekko should be avoided.
There is no available data regarding the use of tezepelumab-ekko in patients who are pregnant, although placental transfer of monoclonal antibodies such as tezepelumab-ekko is greater during the third trimester of pregnancy; therefore, potential effects on a fetus are likely to be greater during the third trimester of pregnancy, according to the company.
The most common adverse reactions for the drug, with a reported incidence of at least 3%, are pharyngitis, arthralgia, and back pain.
“The approval of Tezspire is long-awaited positive news for the asthma community,” said Tonya Winders, president and CEO at the Allergy & Asthma Network and president of the Global Allergy and Airways Patient Platform in the Amgen press release. “For the first time, many people living with severe asthma have the opportunity to receive treatment regardless of the cause of their inflammation.”
A version of this article first appeared on Medscape.com.