User login
AAN publishes guideline on the treatment of sleep problems in children with autism
The guideline was published online ahead of print Feb. 12 in Neurology.
“While up to 40% of children and teens in the general population will have sleep problems at some point during their childhood, such problems usually lessen with age,” lead author Ashura Williams Buckley, MD, director of the Sleep and Neurodevelopment Service at the National Institute of Mental Health in Bethesda, Md., said in a press release. “For children and teens with autism, sleep problems are more common and more likely to persist, resulting in poor health and poor quality of life. Some sleep problems may be directly related to autism, but others are not. Regardless, autism symptoms may make sleep problems worse.”
Few evidence-based treatments are available
Dr. Williams Buckley and colleagues developed the current guideline to evaluate which pharmacologic, behavioral, and complementary and alternative medicine (CAM) interventions improve bedtime resistance, sleep onset latency, sleep continuity, total sleep time, and daytime behavior in children and adolescents with ASD. The panel evaluated 900 abstracts of articles that had been included in systematic reviews, as well as 1,087 additional abstracts. One hundred thirty-nine articles were potentially relevant, 12 met criteria for data extraction, and eight were rated class III or higher and were included in the panel’s review.
The authors observed what they called a dearth of evidence-based treatments for sleep dysregulation in ASD. Evidence indicates that melatonin, with or without cognitive–behavioral therapy (CBT), improves several sleep outcomes, compared with placebo. “Evidence for other interventions is largely lacking,” wrote Dr. Williams Buckley and colleagues. They observed a lack of long-term safety data for melatonin in children, which they considered concerning, because melatonin affects the hypothalamic–gonadal axis and can potentially influence pubertal development.
Screening for comorbid conditions and concomitant medications
The guideline recommends that clinicians assess children with ASD and sleep disturbances for coexisting conditions and concomitant medications that could be contributing to these sleep disturbances. They should ensure that children receive appropriate treatment for coexisting conditions and adjust or discontinue potentially problematic medications appropriately, according to the guideline.
Furthermore, clinicians should counsel parents or guardians about behavioral strategies as a first-line treatment for improving sleep function. These strategies could be administered alone or with pharmacologic or neutraceutical approaches as needed, according to the authors. Suggested behavioral approaches include unmodified extinction (i.e., imposing a bedtime and ignoring a child’s protests), graduated extinction (i.e., ignoring protests for a specified period before responding), positive routines (i.e., establishing pre-bedtime calming rituals), and bedtime fading (i.e., putting a child to bed close to the time he or she begins to fall asleep).
If a child’s contributing coexisting conditions and medications have been addressed and behavioral strategies have not been helpful, clinicians should offer melatonin, according to the guideline. Because over-the-counter formulations contain variable concentrations of melatonin, clinicians should write a prescription for it or recommend high-purity pharmaceutical grade melatonin. The initial dose should be 1-3 mg/day at 60-30 minutes before bedtime. The dose can be titrated to 10 mg/day. Clinicians also should counsel children and their parents about potential adverse events of melatonin and the lack of long-term safety data, according to the guideline.
In addition, clinicians should advise children and parents that no evidence supports the routine use of weighted blankets or specialized mattress technology for improving sleep. Parents who ask about weighted blankets should be told that the reviewed trial reported no serious adverse events with this intervention, and that blankets could be a reasonable nonpharmacologic approach for some patients, according to the guideline.
Optimal outcome measures are undefined
Dr. Williams Buckley and colleagues also suggested areas for future research. Investigators have not yet defined optimal outcome measures (e.g., questionnaires, polysomnography, and actigraphy) that balance tolerability and accuracy, they wrote. Clinically important differences for most measures also have yet to be determined. Researchers should investigate whether long-term adverse events are associated with chronic melatonin use and study patients with ASD and comorbid mood disorders, wrote the authors. “Research tying the underlying neurobiology in early-life sleep disruption to behavior might help clinicians and researchers understand which treatments might work for which people with ASD,” they concluded.
The AAN supported the development of the guideline. Dr. Williams Buckley had no conflicts of interest. Six authors had conflicts of interest that the AAN deemed not significant enough to prevent their participation in the development of the guideline.
SOURCE: Williams Buckley A et al. Neurology. 2020;94:393-405. doi: 10.1212/WNL0000000000009033.
The guideline was published online ahead of print Feb. 12 in Neurology.
“While up to 40% of children and teens in the general population will have sleep problems at some point during their childhood, such problems usually lessen with age,” lead author Ashura Williams Buckley, MD, director of the Sleep and Neurodevelopment Service at the National Institute of Mental Health in Bethesda, Md., said in a press release. “For children and teens with autism, sleep problems are more common and more likely to persist, resulting in poor health and poor quality of life. Some sleep problems may be directly related to autism, but others are not. Regardless, autism symptoms may make sleep problems worse.”
Few evidence-based treatments are available
Dr. Williams Buckley and colleagues developed the current guideline to evaluate which pharmacologic, behavioral, and complementary and alternative medicine (CAM) interventions improve bedtime resistance, sleep onset latency, sleep continuity, total sleep time, and daytime behavior in children and adolescents with ASD. The panel evaluated 900 abstracts of articles that had been included in systematic reviews, as well as 1,087 additional abstracts. One hundred thirty-nine articles were potentially relevant, 12 met criteria for data extraction, and eight were rated class III or higher and were included in the panel’s review.
The authors observed what they called a dearth of evidence-based treatments for sleep dysregulation in ASD. Evidence indicates that melatonin, with or without cognitive–behavioral therapy (CBT), improves several sleep outcomes, compared with placebo. “Evidence for other interventions is largely lacking,” wrote Dr. Williams Buckley and colleagues. They observed a lack of long-term safety data for melatonin in children, which they considered concerning, because melatonin affects the hypothalamic–gonadal axis and can potentially influence pubertal development.
Screening for comorbid conditions and concomitant medications
The guideline recommends that clinicians assess children with ASD and sleep disturbances for coexisting conditions and concomitant medications that could be contributing to these sleep disturbances. They should ensure that children receive appropriate treatment for coexisting conditions and adjust or discontinue potentially problematic medications appropriately, according to the guideline.
Furthermore, clinicians should counsel parents or guardians about behavioral strategies as a first-line treatment for improving sleep function. These strategies could be administered alone or with pharmacologic or neutraceutical approaches as needed, according to the authors. Suggested behavioral approaches include unmodified extinction (i.e., imposing a bedtime and ignoring a child’s protests), graduated extinction (i.e., ignoring protests for a specified period before responding), positive routines (i.e., establishing pre-bedtime calming rituals), and bedtime fading (i.e., putting a child to bed close to the time he or she begins to fall asleep).
If a child’s contributing coexisting conditions and medications have been addressed and behavioral strategies have not been helpful, clinicians should offer melatonin, according to the guideline. Because over-the-counter formulations contain variable concentrations of melatonin, clinicians should write a prescription for it or recommend high-purity pharmaceutical grade melatonin. The initial dose should be 1-3 mg/day at 60-30 minutes before bedtime. The dose can be titrated to 10 mg/day. Clinicians also should counsel children and their parents about potential adverse events of melatonin and the lack of long-term safety data, according to the guideline.
In addition, clinicians should advise children and parents that no evidence supports the routine use of weighted blankets or specialized mattress technology for improving sleep. Parents who ask about weighted blankets should be told that the reviewed trial reported no serious adverse events with this intervention, and that blankets could be a reasonable nonpharmacologic approach for some patients, according to the guideline.
Optimal outcome measures are undefined
Dr. Williams Buckley and colleagues also suggested areas for future research. Investigators have not yet defined optimal outcome measures (e.g., questionnaires, polysomnography, and actigraphy) that balance tolerability and accuracy, they wrote. Clinically important differences for most measures also have yet to be determined. Researchers should investigate whether long-term adverse events are associated with chronic melatonin use and study patients with ASD and comorbid mood disorders, wrote the authors. “Research tying the underlying neurobiology in early-life sleep disruption to behavior might help clinicians and researchers understand which treatments might work for which people with ASD,” they concluded.
The AAN supported the development of the guideline. Dr. Williams Buckley had no conflicts of interest. Six authors had conflicts of interest that the AAN deemed not significant enough to prevent their participation in the development of the guideline.
SOURCE: Williams Buckley A et al. Neurology. 2020;94:393-405. doi: 10.1212/WNL0000000000009033.
The guideline was published online ahead of print Feb. 12 in Neurology.
“While up to 40% of children and teens in the general population will have sleep problems at some point during their childhood, such problems usually lessen with age,” lead author Ashura Williams Buckley, MD, director of the Sleep and Neurodevelopment Service at the National Institute of Mental Health in Bethesda, Md., said in a press release. “For children and teens with autism, sleep problems are more common and more likely to persist, resulting in poor health and poor quality of life. Some sleep problems may be directly related to autism, but others are not. Regardless, autism symptoms may make sleep problems worse.”
Few evidence-based treatments are available
Dr. Williams Buckley and colleagues developed the current guideline to evaluate which pharmacologic, behavioral, and complementary and alternative medicine (CAM) interventions improve bedtime resistance, sleep onset latency, sleep continuity, total sleep time, and daytime behavior in children and adolescents with ASD. The panel evaluated 900 abstracts of articles that had been included in systematic reviews, as well as 1,087 additional abstracts. One hundred thirty-nine articles were potentially relevant, 12 met criteria for data extraction, and eight were rated class III or higher and were included in the panel’s review.
The authors observed what they called a dearth of evidence-based treatments for sleep dysregulation in ASD. Evidence indicates that melatonin, with or without cognitive–behavioral therapy (CBT), improves several sleep outcomes, compared with placebo. “Evidence for other interventions is largely lacking,” wrote Dr. Williams Buckley and colleagues. They observed a lack of long-term safety data for melatonin in children, which they considered concerning, because melatonin affects the hypothalamic–gonadal axis and can potentially influence pubertal development.
Screening for comorbid conditions and concomitant medications
The guideline recommends that clinicians assess children with ASD and sleep disturbances for coexisting conditions and concomitant medications that could be contributing to these sleep disturbances. They should ensure that children receive appropriate treatment for coexisting conditions and adjust or discontinue potentially problematic medications appropriately, according to the guideline.
Furthermore, clinicians should counsel parents or guardians about behavioral strategies as a first-line treatment for improving sleep function. These strategies could be administered alone or with pharmacologic or neutraceutical approaches as needed, according to the authors. Suggested behavioral approaches include unmodified extinction (i.e., imposing a bedtime and ignoring a child’s protests), graduated extinction (i.e., ignoring protests for a specified period before responding), positive routines (i.e., establishing pre-bedtime calming rituals), and bedtime fading (i.e., putting a child to bed close to the time he or she begins to fall asleep).
If a child’s contributing coexisting conditions and medications have been addressed and behavioral strategies have not been helpful, clinicians should offer melatonin, according to the guideline. Because over-the-counter formulations contain variable concentrations of melatonin, clinicians should write a prescription for it or recommend high-purity pharmaceutical grade melatonin. The initial dose should be 1-3 mg/day at 60-30 minutes before bedtime. The dose can be titrated to 10 mg/day. Clinicians also should counsel children and their parents about potential adverse events of melatonin and the lack of long-term safety data, according to the guideline.
In addition, clinicians should advise children and parents that no evidence supports the routine use of weighted blankets or specialized mattress technology for improving sleep. Parents who ask about weighted blankets should be told that the reviewed trial reported no serious adverse events with this intervention, and that blankets could be a reasonable nonpharmacologic approach for some patients, according to the guideline.
Optimal outcome measures are undefined
Dr. Williams Buckley and colleagues also suggested areas for future research. Investigators have not yet defined optimal outcome measures (e.g., questionnaires, polysomnography, and actigraphy) that balance tolerability and accuracy, they wrote. Clinically important differences for most measures also have yet to be determined. Researchers should investigate whether long-term adverse events are associated with chronic melatonin use and study patients with ASD and comorbid mood disorders, wrote the authors. “Research tying the underlying neurobiology in early-life sleep disruption to behavior might help clinicians and researchers understand which treatments might work for which people with ASD,” they concluded.
The AAN supported the development of the guideline. Dr. Williams Buckley had no conflicts of interest. Six authors had conflicts of interest that the AAN deemed not significant enough to prevent their participation in the development of the guideline.
SOURCE: Williams Buckley A et al. Neurology. 2020;94:393-405. doi: 10.1212/WNL0000000000009033.
FROM NEUROLOGY
Key clinical point: The AAN has published a guideline on the treatment of sleep problems in children with autism.
Major finding: The guideline recommends behavioral strategies as a first-line treatment.
Study details: A review of 1,987 peer-reviewed studies.
Disclosures: The AAN funded the development of the guideline. The first author had no conflicts of interest, and the other authors had no significant conflicts.
Source: Williams Buckley A et al. Neurology. 2020;94:393-405. doi: 10.1212/WNL0000000000009033.
Mobile stroke unit had clinical impact on EVT
In its first year of operation, a mobile stroke unit in Melbourne demonstrated substantial savings in time to commencement of both thrombolysis and endovascular thrombectomy (EVT), results from a prospective study showed.
“While previously published data from MSU [mobile stroke unit] services in Europe and North America show substantial reductions in time to thrombolysis of approximately 30-45 minutes, little is known about the clinical impact on EVT,” first author Henry Zhao, MBBS, and colleagues wrote in a study published in Stroke.
Launched in November 2017, the Melbourne MSU is based at a large comprehensive stroke center and operates with a 20-km radius, servicing about 1.7 million people within the city of Melbourne. It is staffed with an onboard neurologist or senior stroke fellow who provides primary assessment and treatment decisions, a stroke advanced practice nurse who provides clinical support and treatment administration, a clinician who provides CT imaging, and advanced life support and mobile intensive care paramedics who provide transport logistics and paramedicine support. For the current analysis, MSU patients who received reperfusion therapy were compared with control patients presenting to metropolitan Melbourne stroke units via standard ambulance within MSU operating hours. The primary outcome was median time difference in first ambulance dispatch to treatment, which the researchers used quantile regression analysis to determine. Time savings were subsequently converted to disability-adjusted life years (DALY) avoiding using published estimates.
Dr. Zhao of the Melbourne Brain Centre and department of neurology at Royal Melbourne Hospital and his colleagues reported that, in its first year of operation, the Melbourne MSU administered prehospital thrombolysis to 100 patients with a mean age of nearly 74 years. More than half of the patients (62%) were male. Compared with controls, the median time savings per MSU patient was 26 minutes for dispatch to hospital arrival and 15 minutes for hospital arrival to thrombolysis (P less than .0010 for both associations). The calculated overall time saving from dispatch to thrombolysis was 42.5 minutes.
Over the same time period, 41 MSU patients with a mean age of 76 years received EVT dispatch-to-treatment time saving of 51 minutes (P less than 0.001). This included a median time saving of 17 minutes for EVT hospital arrival to arterial puncture for MSU patients (P = .001). Overall estimated median DALYs saved through earlier provision of reperfusion therapies were 20.9 for thrombolysis and 24.6 for EVT.
“The benefit in EVT patients was primarily driven by prehospital MSU diagnosis of large vessel occlusion, which enabled bypass of a local non-EVT center directly to a comprehensive stroke center in almost 50% of patients with large vessel occlusion,” the researchers wrote. “Even when patients were located close to an EVT center, MSU pre-notification and facilitated workflows achieved a reduction in hospital arrival to arterial puncture by one-third. Furthermore, the time saving was seen despite the majority of EVT patients receiving repeat imaging in hospital to visualize the extracranial circulation.”
The study is scheduled to be presented at the International Stroke Conference on Feb. 20.
The Melbourne MSU received funding from the Australian Commonwealth Government, Victorian State Government, Royal Melbourne Hospital Neurosciences Foundation, Stroke Foundation, the Florey Institute of Neurosciences and Mental Health, the University of Melbourne, Boehringer Ingelheim, and private donation. Dr. Zhao disclosed that he has received grants from the Australian Commonwealth Government and the University of Melbourne and personal fees from Boehringer Ingelheim.
SOURCE: Zhao H et al. Stroke. 2020 Feb 12. doi: 10.1161/strokeaha.119.027843.
In its first year of operation, a mobile stroke unit in Melbourne demonstrated substantial savings in time to commencement of both thrombolysis and endovascular thrombectomy (EVT), results from a prospective study showed.
“While previously published data from MSU [mobile stroke unit] services in Europe and North America show substantial reductions in time to thrombolysis of approximately 30-45 minutes, little is known about the clinical impact on EVT,” first author Henry Zhao, MBBS, and colleagues wrote in a study published in Stroke.
Launched in November 2017, the Melbourne MSU is based at a large comprehensive stroke center and operates with a 20-km radius, servicing about 1.7 million people within the city of Melbourne. It is staffed with an onboard neurologist or senior stroke fellow who provides primary assessment and treatment decisions, a stroke advanced practice nurse who provides clinical support and treatment administration, a clinician who provides CT imaging, and advanced life support and mobile intensive care paramedics who provide transport logistics and paramedicine support. For the current analysis, MSU patients who received reperfusion therapy were compared with control patients presenting to metropolitan Melbourne stroke units via standard ambulance within MSU operating hours. The primary outcome was median time difference in first ambulance dispatch to treatment, which the researchers used quantile regression analysis to determine. Time savings were subsequently converted to disability-adjusted life years (DALY) avoiding using published estimates.
Dr. Zhao of the Melbourne Brain Centre and department of neurology at Royal Melbourne Hospital and his colleagues reported that, in its first year of operation, the Melbourne MSU administered prehospital thrombolysis to 100 patients with a mean age of nearly 74 years. More than half of the patients (62%) were male. Compared with controls, the median time savings per MSU patient was 26 minutes for dispatch to hospital arrival and 15 minutes for hospital arrival to thrombolysis (P less than .0010 for both associations). The calculated overall time saving from dispatch to thrombolysis was 42.5 minutes.
Over the same time period, 41 MSU patients with a mean age of 76 years received EVT dispatch-to-treatment time saving of 51 minutes (P less than 0.001). This included a median time saving of 17 minutes for EVT hospital arrival to arterial puncture for MSU patients (P = .001). Overall estimated median DALYs saved through earlier provision of reperfusion therapies were 20.9 for thrombolysis and 24.6 for EVT.
“The benefit in EVT patients was primarily driven by prehospital MSU diagnosis of large vessel occlusion, which enabled bypass of a local non-EVT center directly to a comprehensive stroke center in almost 50% of patients with large vessel occlusion,” the researchers wrote. “Even when patients were located close to an EVT center, MSU pre-notification and facilitated workflows achieved a reduction in hospital arrival to arterial puncture by one-third. Furthermore, the time saving was seen despite the majority of EVT patients receiving repeat imaging in hospital to visualize the extracranial circulation.”
The study is scheduled to be presented at the International Stroke Conference on Feb. 20.
The Melbourne MSU received funding from the Australian Commonwealth Government, Victorian State Government, Royal Melbourne Hospital Neurosciences Foundation, Stroke Foundation, the Florey Institute of Neurosciences and Mental Health, the University of Melbourne, Boehringer Ingelheim, and private donation. Dr. Zhao disclosed that he has received grants from the Australian Commonwealth Government and the University of Melbourne and personal fees from Boehringer Ingelheim.
SOURCE: Zhao H et al. Stroke. 2020 Feb 12. doi: 10.1161/strokeaha.119.027843.
In its first year of operation, a mobile stroke unit in Melbourne demonstrated substantial savings in time to commencement of both thrombolysis and endovascular thrombectomy (EVT), results from a prospective study showed.
“While previously published data from MSU [mobile stroke unit] services in Europe and North America show substantial reductions in time to thrombolysis of approximately 30-45 minutes, little is known about the clinical impact on EVT,” first author Henry Zhao, MBBS, and colleagues wrote in a study published in Stroke.
Launched in November 2017, the Melbourne MSU is based at a large comprehensive stroke center and operates with a 20-km radius, servicing about 1.7 million people within the city of Melbourne. It is staffed with an onboard neurologist or senior stroke fellow who provides primary assessment and treatment decisions, a stroke advanced practice nurse who provides clinical support and treatment administration, a clinician who provides CT imaging, and advanced life support and mobile intensive care paramedics who provide transport logistics and paramedicine support. For the current analysis, MSU patients who received reperfusion therapy were compared with control patients presenting to metropolitan Melbourne stroke units via standard ambulance within MSU operating hours. The primary outcome was median time difference in first ambulance dispatch to treatment, which the researchers used quantile regression analysis to determine. Time savings were subsequently converted to disability-adjusted life years (DALY) avoiding using published estimates.
Dr. Zhao of the Melbourne Brain Centre and department of neurology at Royal Melbourne Hospital and his colleagues reported that, in its first year of operation, the Melbourne MSU administered prehospital thrombolysis to 100 patients with a mean age of nearly 74 years. More than half of the patients (62%) were male. Compared with controls, the median time savings per MSU patient was 26 minutes for dispatch to hospital arrival and 15 minutes for hospital arrival to thrombolysis (P less than .0010 for both associations). The calculated overall time saving from dispatch to thrombolysis was 42.5 minutes.
Over the same time period, 41 MSU patients with a mean age of 76 years received EVT dispatch-to-treatment time saving of 51 minutes (P less than 0.001). This included a median time saving of 17 minutes for EVT hospital arrival to arterial puncture for MSU patients (P = .001). Overall estimated median DALYs saved through earlier provision of reperfusion therapies were 20.9 for thrombolysis and 24.6 for EVT.
“The benefit in EVT patients was primarily driven by prehospital MSU diagnosis of large vessel occlusion, which enabled bypass of a local non-EVT center directly to a comprehensive stroke center in almost 50% of patients with large vessel occlusion,” the researchers wrote. “Even when patients were located close to an EVT center, MSU pre-notification and facilitated workflows achieved a reduction in hospital arrival to arterial puncture by one-third. Furthermore, the time saving was seen despite the majority of EVT patients receiving repeat imaging in hospital to visualize the extracranial circulation.”
The study is scheduled to be presented at the International Stroke Conference on Feb. 20.
The Melbourne MSU received funding from the Australian Commonwealth Government, Victorian State Government, Royal Melbourne Hospital Neurosciences Foundation, Stroke Foundation, the Florey Institute of Neurosciences and Mental Health, the University of Melbourne, Boehringer Ingelheim, and private donation. Dr. Zhao disclosed that he has received grants from the Australian Commonwealth Government and the University of Melbourne and personal fees from Boehringer Ingelheim.
SOURCE: Zhao H et al. Stroke. 2020 Feb 12. doi: 10.1161/strokeaha.119.027843.
FROM STROKE
Key clinical point: A mobile stroke unit (MSU) substantially reduced time to reperfusion therapies.
Major finding: Compared with controls, the median time savings per MSU patient was 26 minutes for dispatch to hospital arrival and 15 minutes for hospital arrival to thrombolysis (P less than .0010 for both associations).
Study details: A prospective study of 100 stroke patients.
Disclosures: The Melbourne MSU received funding from the Australian Commonwealth Government, Victorian State Government, Royal Melbourne Hospital Neurosciences Foundation, Stroke Foundation, the Florey Institute of Neurosciences and Mental Health, the University of Melbourne, Boehringer Ingelheim, and private donation. Dr. Zhao disclosed that he has received grants from the Australian Commonwealth Government and the University of Melbourne and personal fees from Boehringer Ingelheim.
Source: Zhao H et al. Stroke. 2020 Feb 12. doi: 10.1161/strokeaha.119.027843.
Pharmacologic prophylaxis fails in pediatric migraine

Clinicians hoped that medications used in adults – such as antidepressants, antiepileptics, antihypertensive agents, calcium channel blockers, and food supplements – would find similar success in children. Unfortunately, researchers found only short-term signs of efficacy over placebo, with no benefit lasting more than 6 months.
The study, conducted by a team led by Cosima Locher, PhD, of Boston Children’s Hospital, included 23 double-blind, randomized, controlled trials with a total of 2,217 patients; the mean age was 11 years. They compared 12 pharmacologic agents with each other or with placebo in the study, published online in JAMA Pediatrics.
In a main efficacy analysis that included 19 studies, only two treatments outperformed placebo: propranolol (standardized mean difference, 0.60; 95% confidence interval, 0.03-1.17) and topiramate (SMD, 0.59; 95% CI, 0.03-1.15). There were no statistically significant between-treatment differences.
The results had an overall low to moderate certainty.
When propranolol was compared to placebo, the 95% prediction interval (–0.62 to 1.82) was wider than the significant confidence interval (0.03-1.17), and comprised both beneficial and detrimental effects. A similar result was found with topiramate, with a prediction interval of –0.62 to 1.80 extending into nonsignificant effects (95% CI, 0.03-1.15). In both cases, significant effects were found only when the prediction interval was 70%.
In a long-term analysis (greater than 6 months), no treatment outperformed placebo.
The treatments generally were acceptable. The researchers found no significant difference in tolerability between any of the treatments and each other or placebo. Safety data analyzed from 13 trials revealed no significant differences between treatments and placebo.
“Because specific effects of drugs are associated with the size of the placebo effect, the lack of drug efficacy in our NMA [network meta-analysis] could be owing to a comparatively high placebo effect in children. In fact, there is indirect evidence [from other studies] that the placebo effect is more pronounced in children and adolescents than in adults,” Dr. Locher and associates said. They suggested that studies were needed to quantify the placebo effect in pediatric migraine, and if it was large, to develop innovative therapies making use of this.
The findings should lead to some changes in practice, Boris Zernikow, MD, PhD, of Children’s and Adolescents’ Hospital Datteln (Germany) wrote in an accompanying editorial.
Pharmacological prophylactic treatment of childhood migraine should be an exception rather than the rule, and nonpharmacologic approaches should be emphasized, particularly because the placebo effect is magnified in children, he said.
Many who suffer migraines in childhood will continue to be affected in adulthood, so pediatric intervention is a good opportunity to instill effective strategies. These include: using abortive medication early in an attack and using antimigraine medications for only that specific type of headache; engaging in physical activity to reduce migraine attacks; getting sufficient sleep; and learning relaxation and other psychological approaches to counter migraines.
Dr. Zernikow had no relevant financial disclosures. One study author received grants from Amgen and other support from Grunenthal and Akelos. The study received funding from the Sara Page Mayo Endowment for Pediatric Pain Research, Education, and Treatment; the Swiss National Science Foundation; the Schweizer-Arau-Foundation; and the Theophrastus Foundation.
SOURCES: Locher C et al. JAMA Pediatrics. 2020 Feb 10. doi: 10.1001/jamapediatrics.2019.5856; Zernikow B. JAMA Pediatrics. 2020 Feb 10. doi: 10.1001/jamapediatrics.2019.5907.
Clinicians hoped that medications used in adults – such as antidepressants, antiepileptics, antihypertensive agents, calcium channel blockers, and food supplements – would find similar success in children. Unfortunately, researchers found only short-term signs of efficacy over placebo, with no benefit lasting more than 6 months.
The study, conducted by a team led by Cosima Locher, PhD, of Boston Children’s Hospital, included 23 double-blind, randomized, controlled trials with a total of 2,217 patients; the mean age was 11 years. They compared 12 pharmacologic agents with each other or with placebo in the study, published online in JAMA Pediatrics.
In a main efficacy analysis that included 19 studies, only two treatments outperformed placebo: propranolol (standardized mean difference, 0.60; 95% confidence interval, 0.03-1.17) and topiramate (SMD, 0.59; 95% CI, 0.03-1.15). There were no statistically significant between-treatment differences.
The results had an overall low to moderate certainty.
When propranolol was compared to placebo, the 95% prediction interval (–0.62 to 1.82) was wider than the significant confidence interval (0.03-1.17), and comprised both beneficial and detrimental effects. A similar result was found with topiramate, with a prediction interval of –0.62 to 1.80 extending into nonsignificant effects (95% CI, 0.03-1.15). In both cases, significant effects were found only when the prediction interval was 70%.
In a long-term analysis (greater than 6 months), no treatment outperformed placebo.
The treatments generally were acceptable. The researchers found no significant difference in tolerability between any of the treatments and each other or placebo. Safety data analyzed from 13 trials revealed no significant differences between treatments and placebo.
“Because specific effects of drugs are associated with the size of the placebo effect, the lack of drug efficacy in our NMA [network meta-analysis] could be owing to a comparatively high placebo effect in children. In fact, there is indirect evidence [from other studies] that the placebo effect is more pronounced in children and adolescents than in adults,” Dr. Locher and associates said. They suggested that studies were needed to quantify the placebo effect in pediatric migraine, and if it was large, to develop innovative therapies making use of this.
The findings should lead to some changes in practice, Boris Zernikow, MD, PhD, of Children’s and Adolescents’ Hospital Datteln (Germany) wrote in an accompanying editorial.
Pharmacological prophylactic treatment of childhood migraine should be an exception rather than the rule, and nonpharmacologic approaches should be emphasized, particularly because the placebo effect is magnified in children, he said.
Many who suffer migraines in childhood will continue to be affected in adulthood, so pediatric intervention is a good opportunity to instill effective strategies. These include: using abortive medication early in an attack and using antimigraine medications for only that specific type of headache; engaging in physical activity to reduce migraine attacks; getting sufficient sleep; and learning relaxation and other psychological approaches to counter migraines.
Dr. Zernikow had no relevant financial disclosures. One study author received grants from Amgen and other support from Grunenthal and Akelos. The study received funding from the Sara Page Mayo Endowment for Pediatric Pain Research, Education, and Treatment; the Swiss National Science Foundation; the Schweizer-Arau-Foundation; and the Theophrastus Foundation.
SOURCES: Locher C et al. JAMA Pediatrics. 2020 Feb 10. doi: 10.1001/jamapediatrics.2019.5856; Zernikow B. JAMA Pediatrics. 2020 Feb 10. doi: 10.1001/jamapediatrics.2019.5907.
Clinicians hoped that medications used in adults – such as antidepressants, antiepileptics, antihypertensive agents, calcium channel blockers, and food supplements – would find similar success in children. Unfortunately, researchers found only short-term signs of efficacy over placebo, with no benefit lasting more than 6 months.
The study, conducted by a team led by Cosima Locher, PhD, of Boston Children’s Hospital, included 23 double-blind, randomized, controlled trials with a total of 2,217 patients; the mean age was 11 years. They compared 12 pharmacologic agents with each other or with placebo in the study, published online in JAMA Pediatrics.
In a main efficacy analysis that included 19 studies, only two treatments outperformed placebo: propranolol (standardized mean difference, 0.60; 95% confidence interval, 0.03-1.17) and topiramate (SMD, 0.59; 95% CI, 0.03-1.15). There were no statistically significant between-treatment differences.
The results had an overall low to moderate certainty.
When propranolol was compared to placebo, the 95% prediction interval (–0.62 to 1.82) was wider than the significant confidence interval (0.03-1.17), and comprised both beneficial and detrimental effects. A similar result was found with topiramate, with a prediction interval of –0.62 to 1.80 extending into nonsignificant effects (95% CI, 0.03-1.15). In both cases, significant effects were found only when the prediction interval was 70%.
In a long-term analysis (greater than 6 months), no treatment outperformed placebo.
The treatments generally were acceptable. The researchers found no significant difference in tolerability between any of the treatments and each other or placebo. Safety data analyzed from 13 trials revealed no significant differences between treatments and placebo.
“Because specific effects of drugs are associated with the size of the placebo effect, the lack of drug efficacy in our NMA [network meta-analysis] could be owing to a comparatively high placebo effect in children. In fact, there is indirect evidence [from other studies] that the placebo effect is more pronounced in children and adolescents than in adults,” Dr. Locher and associates said. They suggested that studies were needed to quantify the placebo effect in pediatric migraine, and if it was large, to develop innovative therapies making use of this.
The findings should lead to some changes in practice, Boris Zernikow, MD, PhD, of Children’s and Adolescents’ Hospital Datteln (Germany) wrote in an accompanying editorial.
Pharmacological prophylactic treatment of childhood migraine should be an exception rather than the rule, and nonpharmacologic approaches should be emphasized, particularly because the placebo effect is magnified in children, he said.
Many who suffer migraines in childhood will continue to be affected in adulthood, so pediatric intervention is a good opportunity to instill effective strategies. These include: using abortive medication early in an attack and using antimigraine medications for only that specific type of headache; engaging in physical activity to reduce migraine attacks; getting sufficient sleep; and learning relaxation and other psychological approaches to counter migraines.
Dr. Zernikow had no relevant financial disclosures. One study author received grants from Amgen and other support from Grunenthal and Akelos. The study received funding from the Sara Page Mayo Endowment for Pediatric Pain Research, Education, and Treatment; the Swiss National Science Foundation; the Schweizer-Arau-Foundation; and the Theophrastus Foundation.
SOURCES: Locher C et al. JAMA Pediatrics. 2020 Feb 10. doi: 10.1001/jamapediatrics.2019.5856; Zernikow B. JAMA Pediatrics. 2020 Feb 10. doi: 10.1001/jamapediatrics.2019.5907.
FROM JAMA PEDIATRICS
DLBCL tops cases of HBV-associated NHL in Europe
The majority of hepatitis B virus (HBV)–associated non-Hodgkin lymphoma (NHL) cases in Western Europe were patients with advanced-stage diffuse large B-cell lymphoma (DLBCL), according to results of a retrospective study.
The findings suggest additional research is needed to better understand the nature of HBV-related lymphomas in nonendemic regions.
“Our aim was to describe the characteristics and outcomes of patients with NHL and active hepatitis B in France and Italy, where the prevalence of HBV is low,” wrote Marine Lemaitre of the Centre Hospitalier de Versailles in Le Chesnay, France, and colleagues. The findings were published in the Journal of Infection.
The researchers retrospectively studied a cohort of 39 patients with B-cell NHL and active HBV infection. Clinical data was collected from medical records at three hematology centers in France and Italy. The team evaluated clinical characteristics, including histologic subtype of the lymphoma, type of treatment, patient demographics, and prognostic outcomes. In addition, they compared these data with a separate cohort of patients with B-cell NHL and active HCV infection. Among study patients, the median age at lymphoma diagnosis was 59 years (range, 29-88 years), and most were men. The most common subtype of lymphoma was DLBCL (62%), followed by other subtypes (38%), including marginal zone lymphomas, follicular lymphomas, and mantle cell lymphomas. With respect to treatment, 92% of patients with DLBCL were treated with R-CHOP or a similar regimen, while 90% of patients received antivirals, resulting in a complete remission for 75% of patients. At 12-month follow-up, 88% and 87% of patients with DLBCL and other B-cell lymphomas were alive, respectively.
“Patients had predominantly advanced-stage DLBCL, with frequent liver involvement, and frequent long-term hematological responses when they received a combination of immuno-chemotherapy and antiviral treatment,” the researchers explained. They also noted that extra-nodal involvement was frequently seen in both HBV- and HCV-associated NHL.
“Additional studies are needed to explore the lymphomagenesis of [these] association[s],” they concluded.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Lemaitre M et al. J Infect. 2019 Dec 14. doi: 10.1016/j.jinf.2019.12.005.
The majority of hepatitis B virus (HBV)–associated non-Hodgkin lymphoma (NHL) cases in Western Europe were patients with advanced-stage diffuse large B-cell lymphoma (DLBCL), according to results of a retrospective study.
The findings suggest additional research is needed to better understand the nature of HBV-related lymphomas in nonendemic regions.
“Our aim was to describe the characteristics and outcomes of patients with NHL and active hepatitis B in France and Italy, where the prevalence of HBV is low,” wrote Marine Lemaitre of the Centre Hospitalier de Versailles in Le Chesnay, France, and colleagues. The findings were published in the Journal of Infection.
The researchers retrospectively studied a cohort of 39 patients with B-cell NHL and active HBV infection. Clinical data was collected from medical records at three hematology centers in France and Italy. The team evaluated clinical characteristics, including histologic subtype of the lymphoma, type of treatment, patient demographics, and prognostic outcomes. In addition, they compared these data with a separate cohort of patients with B-cell NHL and active HCV infection. Among study patients, the median age at lymphoma diagnosis was 59 years (range, 29-88 years), and most were men. The most common subtype of lymphoma was DLBCL (62%), followed by other subtypes (38%), including marginal zone lymphomas, follicular lymphomas, and mantle cell lymphomas. With respect to treatment, 92% of patients with DLBCL were treated with R-CHOP or a similar regimen, while 90% of patients received antivirals, resulting in a complete remission for 75% of patients. At 12-month follow-up, 88% and 87% of patients with DLBCL and other B-cell lymphomas were alive, respectively.
“Patients had predominantly advanced-stage DLBCL, with frequent liver involvement, and frequent long-term hematological responses when they received a combination of immuno-chemotherapy and antiviral treatment,” the researchers explained. They also noted that extra-nodal involvement was frequently seen in both HBV- and HCV-associated NHL.
“Additional studies are needed to explore the lymphomagenesis of [these] association[s],” they concluded.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Lemaitre M et al. J Infect. 2019 Dec 14. doi: 10.1016/j.jinf.2019.12.005.
The majority of hepatitis B virus (HBV)–associated non-Hodgkin lymphoma (NHL) cases in Western Europe were patients with advanced-stage diffuse large B-cell lymphoma (DLBCL), according to results of a retrospective study.
The findings suggest additional research is needed to better understand the nature of HBV-related lymphomas in nonendemic regions.
“Our aim was to describe the characteristics and outcomes of patients with NHL and active hepatitis B in France and Italy, where the prevalence of HBV is low,” wrote Marine Lemaitre of the Centre Hospitalier de Versailles in Le Chesnay, France, and colleagues. The findings were published in the Journal of Infection.
The researchers retrospectively studied a cohort of 39 patients with B-cell NHL and active HBV infection. Clinical data was collected from medical records at three hematology centers in France and Italy. The team evaluated clinical characteristics, including histologic subtype of the lymphoma, type of treatment, patient demographics, and prognostic outcomes. In addition, they compared these data with a separate cohort of patients with B-cell NHL and active HCV infection. Among study patients, the median age at lymphoma diagnosis was 59 years (range, 29-88 years), and most were men. The most common subtype of lymphoma was DLBCL (62%), followed by other subtypes (38%), including marginal zone lymphomas, follicular lymphomas, and mantle cell lymphomas. With respect to treatment, 92% of patients with DLBCL were treated with R-CHOP or a similar regimen, while 90% of patients received antivirals, resulting in a complete remission for 75% of patients. At 12-month follow-up, 88% and 87% of patients with DLBCL and other B-cell lymphomas were alive, respectively.
“Patients had predominantly advanced-stage DLBCL, with frequent liver involvement, and frequent long-term hematological responses when they received a combination of immuno-chemotherapy and antiviral treatment,” the researchers explained. They also noted that extra-nodal involvement was frequently seen in both HBV- and HCV-associated NHL.
“Additional studies are needed to explore the lymphomagenesis of [these] association[s],” they concluded.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Lemaitre M et al. J Infect. 2019 Dec 14. doi: 10.1016/j.jinf.2019.12.005.
FROM THE JOURNAL OF INFECTION
Palliative care improves QoL for patients with Parkinson’s disease and related disorders
, according to results from a randomized clinical trial in JAMA Neurology.
The benefits of palliative care even extended to patients’ caregivers, who also appeared to benefit from outpatient palliative care at the 12-month mark, according to lead author Benzi M. Kluger, MD, of the department of neurology, University of Colorado at Denver, Aurora, and colleagues.
Between November 2015 and September 2017, Dr. Kluger and colleagues included 210 patients into the trial from three participating academic tertiary care centers who had “moderate to high palliative care needs” as assessed by the Palliative Care Needs Assessment Tool, which the researchers said are “common reasons for referral” and “reflect a desire to meet patient-centered needs rather than disease-centered markers.” Patients were primarily non-Hispanic white men with a mean age of about 70 years. The researchers also included 175 caregivers in the trial, most of whom were women, spouses to the patients, and in their caregiver role for over 5.5 years.
Patients with PDRD were randomized to receive standard care – usual care through their primary care physician and a neurologist – or “integrated outpatient palliative care,” from a team consisting of a palliative neurologist, nurse, social worker, chaplain, and board-certified palliative medicine physician. The goal of palliative care was addressing “nonmotor symptoms, goals of care, anticipatory guidance, difficult emotions, and caregiver support,” which patients received every 3 months through an in-person outpatient visit or telemedicine.
Quality of life for patients was measured through the Quality of Life in Alzheimer’s Disease (QoL-AD) scale, and caregiver burden was assessed with the Zarit Burden Interview (ZBI-12). The researchers also measured symptom burden and other QoL measures using the Edmonton Symptom Assessment Scale–Revised for Parkinson’s Disease, Parkinson’s Disease Questionnaire, Hospital Anxiety and Depression Scale, Prolonged Grief Disorder questionnaire, and Functional Assessment of Chronic Illness Therapy–Spiritual Well-Being.
Overall, 87 of 105 (82.1%) of patients in the palliative care group went to all their outpatient palliative care visits, and 19 of 106 (17.9%) patients received palliative care through telemedicine. Patients in the palliative care group also attended more neurology visits (4.66 visits) than those in the standard care (3.16 visits) group.
Quality of life significantly improved for patients in the palliative care group, compared with patients receiving standard care only at 6 months (0.66 vs. –0.84; between-group difference, 1.87; 95% confidence interval, 0.47-3.27; P = .009) and at 12 months (0.68 vs. –0.42; between-group difference, 1.36; 95% CI, −0.01 to 2.73; P = .05). These results remained significant at 6 months and 12 months after researchers used multiple imputation to fill in missing data. While there was no significant difference in caregiver burden between groups at 6 months, there was a statistically significant difference at 12 months favoring the palliative care group (between-group difference, −2.60; 95% CI, −4.58 to −0.61; P = .01).
Patients receiving palliative care also had better nonmotor symptom burden, motor symptom severity, and were more likely to complete advance directives, compared with patients receiving standard care alone. “We hypothesize that motor improvements may have reflected an unanticipated benefit of our palliative care team’s general goal of encouraging activities that promoted joy, meaning, and connection,” Dr. Kluger and colleagues said. Researchers also noted that the intervention patients with greater need for palliative care tended to benefit more than patients with patients with lower palliative care needs.
“Because the palliative care intervention is time-intensive and resource-intensive, future studies should optimize triage tools and consider alternative models of care delivery, such as telemedicine or care navigators, to provide key aspects of the intervention at lower cost,” they said.
In a related editorial, Bastiaan R. Bloem, MD, PhD, from the Center of Expertise for Parkinson & Movement Disorders, at Radboud University Medical Center, in the Netherlands, and colleagues acknowledged that the study by Kluger et al. is “timely and practical” because it introduces a system for outpatient palliative care for patients with PDRD at a time when there is “growing awareness that palliative care may also benefit persons with neurodegenerative diseases like Parkinson’s disease.”
The study is also important because it highlights that patients at varying stages of disease, including mild disease, may benefit from an integrated outpatient palliative model, which is not usually considered when determining candidates for palliative care in other scenarios, such as in patients with cancer. Future studies are warranted to assess how palliative care models can be implemented in different disease states and health care settings, they said.
“These new studies should continue to highlight the fact that palliative care is not about terminal diseases and dying,” Dr. Bloem and colleagues concluded. “As Kluger and colleagues indicate in their important clinical trial, palliative care is about how to live well.”
Six authors reported receiving a grant from the Patient-Centered Outcomes Research Institute, which was the funding source for the study. Two authors reported receiving grants from the University Hospital Foundation during the study. One author reported receiving grants from Allergan and Merz Pharma and is a consultant for GE Pharmaceuticals and Sunovion Pharmaceuticals; another reported receiving grants from the Archstone Foundation, the California Health Care Foundation, the Cambia Health Foundation, the Gordon and Betty Moore Foundation, the National Institute of Nursing Research, the Stupski Foundation, and the UniHealth Foundation. Dr. Bloem and a colleague reported their institution received a center of excellence grant from the Parkinson’s Foundation.
SOURCE: Kluger B et al. JAMA Neurol. doi: 10.1001/jamaneurol.2019.4992.
, according to results from a randomized clinical trial in JAMA Neurology.
The benefits of palliative care even extended to patients’ caregivers, who also appeared to benefit from outpatient palliative care at the 12-month mark, according to lead author Benzi M. Kluger, MD, of the department of neurology, University of Colorado at Denver, Aurora, and colleagues.
Between November 2015 and September 2017, Dr. Kluger and colleagues included 210 patients into the trial from three participating academic tertiary care centers who had “moderate to high palliative care needs” as assessed by the Palliative Care Needs Assessment Tool, which the researchers said are “common reasons for referral” and “reflect a desire to meet patient-centered needs rather than disease-centered markers.” Patients were primarily non-Hispanic white men with a mean age of about 70 years. The researchers also included 175 caregivers in the trial, most of whom were women, spouses to the patients, and in their caregiver role for over 5.5 years.
Patients with PDRD were randomized to receive standard care – usual care through their primary care physician and a neurologist – or “integrated outpatient palliative care,” from a team consisting of a palliative neurologist, nurse, social worker, chaplain, and board-certified palliative medicine physician. The goal of palliative care was addressing “nonmotor symptoms, goals of care, anticipatory guidance, difficult emotions, and caregiver support,” which patients received every 3 months through an in-person outpatient visit or telemedicine.
Quality of life for patients was measured through the Quality of Life in Alzheimer’s Disease (QoL-AD) scale, and caregiver burden was assessed with the Zarit Burden Interview (ZBI-12). The researchers also measured symptom burden and other QoL measures using the Edmonton Symptom Assessment Scale–Revised for Parkinson’s Disease, Parkinson’s Disease Questionnaire, Hospital Anxiety and Depression Scale, Prolonged Grief Disorder questionnaire, and Functional Assessment of Chronic Illness Therapy–Spiritual Well-Being.
Overall, 87 of 105 (82.1%) of patients in the palliative care group went to all their outpatient palliative care visits, and 19 of 106 (17.9%) patients received palliative care through telemedicine. Patients in the palliative care group also attended more neurology visits (4.66 visits) than those in the standard care (3.16 visits) group.
Quality of life significantly improved for patients in the palliative care group, compared with patients receiving standard care only at 6 months (0.66 vs. –0.84; between-group difference, 1.87; 95% confidence interval, 0.47-3.27; P = .009) and at 12 months (0.68 vs. –0.42; between-group difference, 1.36; 95% CI, −0.01 to 2.73; P = .05). These results remained significant at 6 months and 12 months after researchers used multiple imputation to fill in missing data. While there was no significant difference in caregiver burden between groups at 6 months, there was a statistically significant difference at 12 months favoring the palliative care group (between-group difference, −2.60; 95% CI, −4.58 to −0.61; P = .01).
Patients receiving palliative care also had better nonmotor symptom burden, motor symptom severity, and were more likely to complete advance directives, compared with patients receiving standard care alone. “We hypothesize that motor improvements may have reflected an unanticipated benefit of our palliative care team’s general goal of encouraging activities that promoted joy, meaning, and connection,” Dr. Kluger and colleagues said. Researchers also noted that the intervention patients with greater need for palliative care tended to benefit more than patients with patients with lower palliative care needs.
“Because the palliative care intervention is time-intensive and resource-intensive, future studies should optimize triage tools and consider alternative models of care delivery, such as telemedicine or care navigators, to provide key aspects of the intervention at lower cost,” they said.
In a related editorial, Bastiaan R. Bloem, MD, PhD, from the Center of Expertise for Parkinson & Movement Disorders, at Radboud University Medical Center, in the Netherlands, and colleagues acknowledged that the study by Kluger et al. is “timely and practical” because it introduces a system for outpatient palliative care for patients with PDRD at a time when there is “growing awareness that palliative care may also benefit persons with neurodegenerative diseases like Parkinson’s disease.”
The study is also important because it highlights that patients at varying stages of disease, including mild disease, may benefit from an integrated outpatient palliative model, which is not usually considered when determining candidates for palliative care in other scenarios, such as in patients with cancer. Future studies are warranted to assess how palliative care models can be implemented in different disease states and health care settings, they said.
“These new studies should continue to highlight the fact that palliative care is not about terminal diseases and dying,” Dr. Bloem and colleagues concluded. “As Kluger and colleagues indicate in their important clinical trial, palliative care is about how to live well.”
Six authors reported receiving a grant from the Patient-Centered Outcomes Research Institute, which was the funding source for the study. Two authors reported receiving grants from the University Hospital Foundation during the study. One author reported receiving grants from Allergan and Merz Pharma and is a consultant for GE Pharmaceuticals and Sunovion Pharmaceuticals; another reported receiving grants from the Archstone Foundation, the California Health Care Foundation, the Cambia Health Foundation, the Gordon and Betty Moore Foundation, the National Institute of Nursing Research, the Stupski Foundation, and the UniHealth Foundation. Dr. Bloem and a colleague reported their institution received a center of excellence grant from the Parkinson’s Foundation.
SOURCE: Kluger B et al. JAMA Neurol. doi: 10.1001/jamaneurol.2019.4992.
, according to results from a randomized clinical trial in JAMA Neurology.
The benefits of palliative care even extended to patients’ caregivers, who also appeared to benefit from outpatient palliative care at the 12-month mark, according to lead author Benzi M. Kluger, MD, of the department of neurology, University of Colorado at Denver, Aurora, and colleagues.
Between November 2015 and September 2017, Dr. Kluger and colleagues included 210 patients into the trial from three participating academic tertiary care centers who had “moderate to high palliative care needs” as assessed by the Palliative Care Needs Assessment Tool, which the researchers said are “common reasons for referral” and “reflect a desire to meet patient-centered needs rather than disease-centered markers.” Patients were primarily non-Hispanic white men with a mean age of about 70 years. The researchers also included 175 caregivers in the trial, most of whom were women, spouses to the patients, and in their caregiver role for over 5.5 years.
Patients with PDRD were randomized to receive standard care – usual care through their primary care physician and a neurologist – or “integrated outpatient palliative care,” from a team consisting of a palliative neurologist, nurse, social worker, chaplain, and board-certified palliative medicine physician. The goal of palliative care was addressing “nonmotor symptoms, goals of care, anticipatory guidance, difficult emotions, and caregiver support,” which patients received every 3 months through an in-person outpatient visit or telemedicine.
Quality of life for patients was measured through the Quality of Life in Alzheimer’s Disease (QoL-AD) scale, and caregiver burden was assessed with the Zarit Burden Interview (ZBI-12). The researchers also measured symptom burden and other QoL measures using the Edmonton Symptom Assessment Scale–Revised for Parkinson’s Disease, Parkinson’s Disease Questionnaire, Hospital Anxiety and Depression Scale, Prolonged Grief Disorder questionnaire, and Functional Assessment of Chronic Illness Therapy–Spiritual Well-Being.
Overall, 87 of 105 (82.1%) of patients in the palliative care group went to all their outpatient palliative care visits, and 19 of 106 (17.9%) patients received palliative care through telemedicine. Patients in the palliative care group also attended more neurology visits (4.66 visits) than those in the standard care (3.16 visits) group.
Quality of life significantly improved for patients in the palliative care group, compared with patients receiving standard care only at 6 months (0.66 vs. –0.84; between-group difference, 1.87; 95% confidence interval, 0.47-3.27; P = .009) and at 12 months (0.68 vs. –0.42; between-group difference, 1.36; 95% CI, −0.01 to 2.73; P = .05). These results remained significant at 6 months and 12 months after researchers used multiple imputation to fill in missing data. While there was no significant difference in caregiver burden between groups at 6 months, there was a statistically significant difference at 12 months favoring the palliative care group (between-group difference, −2.60; 95% CI, −4.58 to −0.61; P = .01).
Patients receiving palliative care also had better nonmotor symptom burden, motor symptom severity, and were more likely to complete advance directives, compared with patients receiving standard care alone. “We hypothesize that motor improvements may have reflected an unanticipated benefit of our palliative care team’s general goal of encouraging activities that promoted joy, meaning, and connection,” Dr. Kluger and colleagues said. Researchers also noted that the intervention patients with greater need for palliative care tended to benefit more than patients with patients with lower palliative care needs.
“Because the palliative care intervention is time-intensive and resource-intensive, future studies should optimize triage tools and consider alternative models of care delivery, such as telemedicine or care navigators, to provide key aspects of the intervention at lower cost,” they said.
In a related editorial, Bastiaan R. Bloem, MD, PhD, from the Center of Expertise for Parkinson & Movement Disorders, at Radboud University Medical Center, in the Netherlands, and colleagues acknowledged that the study by Kluger et al. is “timely and practical” because it introduces a system for outpatient palliative care for patients with PDRD at a time when there is “growing awareness that palliative care may also benefit persons with neurodegenerative diseases like Parkinson’s disease.”
The study is also important because it highlights that patients at varying stages of disease, including mild disease, may benefit from an integrated outpatient palliative model, which is not usually considered when determining candidates for palliative care in other scenarios, such as in patients with cancer. Future studies are warranted to assess how palliative care models can be implemented in different disease states and health care settings, they said.
“These new studies should continue to highlight the fact that palliative care is not about terminal diseases and dying,” Dr. Bloem and colleagues concluded. “As Kluger and colleagues indicate in their important clinical trial, palliative care is about how to live well.”
Six authors reported receiving a grant from the Patient-Centered Outcomes Research Institute, which was the funding source for the study. Two authors reported receiving grants from the University Hospital Foundation during the study. One author reported receiving grants from Allergan and Merz Pharma and is a consultant for GE Pharmaceuticals and Sunovion Pharmaceuticals; another reported receiving grants from the Archstone Foundation, the California Health Care Foundation, the Cambia Health Foundation, the Gordon and Betty Moore Foundation, the National Institute of Nursing Research, the Stupski Foundation, and the UniHealth Foundation. Dr. Bloem and a colleague reported their institution received a center of excellence grant from the Parkinson’s Foundation.
SOURCE: Kluger B et al. JAMA Neurol. doi: 10.1001/jamaneurol.2019.4992.
FROM JAMA NEUROLOGY
APOE genotype directly regulates alpha-synuclein accumulation
Apolipoprotein E epsilon 4 (APOE4) directly and independently exacerbates accumulation of alpha-synuclein in patients with Lewy body dementia, whereas APOE2 may have a protective effect, based on two recent studies involving mouse models and human patients.

These insights confirm the importance of APOE in synucleinopathies, and may lead to new treatments, according to Eliezer Masliah, MD, director of the division of neuroscience at the National Institute on Aging.
“These [studies] definitely implicate a role of APOE4,” Dr. Masliah said in an interview.
According to Dr. Masliah, previous studies linked the APOE4 genotype with cognitive decline in synucleinopathies, but underlying molecular mechanisms remained unknown.
“We [now] have more direct confirmation [based on] different experimental animal models,” Dr. Masliah said. “It also means that APOE4 could be a therapeutic target for dementia with Lewy bodies.”
The two studies were published simultaneously in Science Translational Medicine. The first study was conducted by Albert A. Davis, MD, PhD, of Washington University, St. Louis, and colleagues; the second was led by Na Zhao, MD, PhD, of the Mayo Clinic in Jacksonville, Fla.
“The studies are very synergistic, but used different techniques,” said Dr. Masliah, who was not involved in the studies.
Both studies involved mice that expressed a human variant of APOE: APOE2, APOE3, or APOE4. Three independent techniques were used to concurrently overexpress alpha-synuclein; Dr. Davis and colleagues used a transgenic approach, as well as striatal injection of alpha-synuclein preformed fibrils, whereas Dr. Zhao and colleagues turned to a viral vector. Regardless of technique, each APOE variant had a distinct impact on the level of alpha-synuclein accumulation.
“In a nutshell, [Dr. Davis and colleagues] found that those mice that have synuclein and APOE4 have a much more rapid progression of the disease,” Dr. Masliah said. “They become Parkinsonian much faster, but also, they become cognitively impaired much faster, and they have more synuclein in the brain. Remarkably, on the opposite side, those that were expressing APOE2, which we know is a protective allele, actually were far less impaired. So that’s really a remarkable finding.”
The study at the Mayo Clinic echoed these findings.
“Essentially, [Dr. Zhao and colleagues] had very similar results,” Dr. Masliah said. “[In mice expressing] APOE4, synuclein accumulation was worse and pathology was worse, and with APOE2, there was relative protection.”
Both studies found that the exacerbating effect of APOE4 translated to human patients.
Dr. Davis and colleagues evaluated data from 251 patients in the Parkinson’s Progression Markers Initiative. A multivariate model showed that patients with the APOE4 genotype had faster cognitive decline, an impact that was independent of other variables, including cerebrospinal fluid concentrations of amyloid beta and tau protein (P = .0119). This finding was further supported by additional analyses involving 177 patients with Parkinson’s disease from the Washington University Movement Disorders Center, and another 1,030 patients enrolled in the NeuroGenetics Research Consortium study.
Dr. Zhao and colleagues evaluated postmortem samples from patients with Lewy body dementia who had minimal amyloid pathology. Comparing 22 APOE4 carriers versus 22 age- and sex-matched noncarriers, they found that carriers had significantly greater accumulations of alpha-synuclein (P less than .05).
According to the investigators, these findings could have both prognostic and therapeutic implications.
“[I]t is intriguing to speculate whether APOE and other potential genetic risk or resilience genes could be useful as screening tools to stratify risk for individual patients,” Dr. Davis and colleagues wrote in their paper. They went on to suggest that APOE genotyping may one day be used to personalize treatments for patients with neurodegenerative disease.
According to Dr. Masliah, several treatment strategies are under investigation.
“There are some pharmaceutical companies and also some academic groups that have been developing antibodies against APOE4 for Alzheimer’s disease, but certainly that could also be used for dementia with Lewy bodies,” he said. “There are other ways. One could [be] to suppress the expression of APOE4 with antisense or other technologies.
“There is also a very innovative technology that has been developed by the group at the Gladstone Institutes in San Francisco, which is to switch APOE4 to APOE3.” This technique, Dr. Masliah explained, is accomplished by breaking a disulfide bond in APOE4, which opens the structure into an isoform that mimics APOE3. “They have developed small molecules that actually can break that bond and essentially chemically switch APOE4 to APOE3,” he said.
Although multiple techniques are feasible, Dr. Masliah stressed that these therapeutic efforts are still in their infancy.
“We need to better understand the mechanisms as to how APOE4 and alpha-synuclein interact,” he said. “I think we need a lot more work in this area.”
The Davis study was funded by the American Academy of Neurology/American Brain Foundation, the BrightFocus Foundation, the Mary E. Groff Charitable Trust, and others; the investigators reported additional relationships with Biogen, Alector, Parabon, and others. The Zhao study was funded by the National Institutes of Health and the Lewy Body Dementia Center Without Walls; the investigators reported no competing interests. Dr. Masliah reported no conflicts of interest.
SOURCES: Davis AA et al. Sci Transl Med. 2020 Feb 5. doi: 10.1126/scitranslmed.aay3069; Zhao N et al. Sci Transl Med. 2020 Feb 5. doi: 10.1126/scitranslmed.aay1809.
Apolipoprotein E epsilon 4 (APOE4) directly and independently exacerbates accumulation of alpha-synuclein in patients with Lewy body dementia, whereas APOE2 may have a protective effect, based on two recent studies involving mouse models and human patients.
These insights confirm the importance of APOE in synucleinopathies, and may lead to new treatments, according to Eliezer Masliah, MD, director of the division of neuroscience at the National Institute on Aging.
“These [studies] definitely implicate a role of APOE4,” Dr. Masliah said in an interview.
According to Dr. Masliah, previous studies linked the APOE4 genotype with cognitive decline in synucleinopathies, but underlying molecular mechanisms remained unknown.
“We [now] have more direct confirmation [based on] different experimental animal models,” Dr. Masliah said. “It also means that APOE4 could be a therapeutic target for dementia with Lewy bodies.”
The two studies were published simultaneously in Science Translational Medicine. The first study was conducted by Albert A. Davis, MD, PhD, of Washington University, St. Louis, and colleagues; the second was led by Na Zhao, MD, PhD, of the Mayo Clinic in Jacksonville, Fla.
“The studies are very synergistic, but used different techniques,” said Dr. Masliah, who was not involved in the studies.
Both studies involved mice that expressed a human variant of APOE: APOE2, APOE3, or APOE4. Three independent techniques were used to concurrently overexpress alpha-synuclein; Dr. Davis and colleagues used a transgenic approach, as well as striatal injection of alpha-synuclein preformed fibrils, whereas Dr. Zhao and colleagues turned to a viral vector. Regardless of technique, each APOE variant had a distinct impact on the level of alpha-synuclein accumulation.
“In a nutshell, [Dr. Davis and colleagues] found that those mice that have synuclein and APOE4 have a much more rapid progression of the disease,” Dr. Masliah said. “They become Parkinsonian much faster, but also, they become cognitively impaired much faster, and they have more synuclein in the brain. Remarkably, on the opposite side, those that were expressing APOE2, which we know is a protective allele, actually were far less impaired. So that’s really a remarkable finding.”
The study at the Mayo Clinic echoed these findings.
“Essentially, [Dr. Zhao and colleagues] had very similar results,” Dr. Masliah said. “[In mice expressing] APOE4, synuclein accumulation was worse and pathology was worse, and with APOE2, there was relative protection.”
Both studies found that the exacerbating effect of APOE4 translated to human patients.
Dr. Davis and colleagues evaluated data from 251 patients in the Parkinson’s Progression Markers Initiative. A multivariate model showed that patients with the APOE4 genotype had faster cognitive decline, an impact that was independent of other variables, including cerebrospinal fluid concentrations of amyloid beta and tau protein (P = .0119). This finding was further supported by additional analyses involving 177 patients with Parkinson’s disease from the Washington University Movement Disorders Center, and another 1,030 patients enrolled in the NeuroGenetics Research Consortium study.
Dr. Zhao and colleagues evaluated postmortem samples from patients with Lewy body dementia who had minimal amyloid pathology. Comparing 22 APOE4 carriers versus 22 age- and sex-matched noncarriers, they found that carriers had significantly greater accumulations of alpha-synuclein (P less than .05).
According to the investigators, these findings could have both prognostic and therapeutic implications.
“[I]t is intriguing to speculate whether APOE and other potential genetic risk or resilience genes could be useful as screening tools to stratify risk for individual patients,” Dr. Davis and colleagues wrote in their paper. They went on to suggest that APOE genotyping may one day be used to personalize treatments for patients with neurodegenerative disease.
According to Dr. Masliah, several treatment strategies are under investigation.
“There are some pharmaceutical companies and also some academic groups that have been developing antibodies against APOE4 for Alzheimer’s disease, but certainly that could also be used for dementia with Lewy bodies,” he said. “There are other ways. One could [be] to suppress the expression of APOE4 with antisense or other technologies.
“There is also a very innovative technology that has been developed by the group at the Gladstone Institutes in San Francisco, which is to switch APOE4 to APOE3.” This technique, Dr. Masliah explained, is accomplished by breaking a disulfide bond in APOE4, which opens the structure into an isoform that mimics APOE3. “They have developed small molecules that actually can break that bond and essentially chemically switch APOE4 to APOE3,” he said.
Although multiple techniques are feasible, Dr. Masliah stressed that these therapeutic efforts are still in their infancy.
“We need to better understand the mechanisms as to how APOE4 and alpha-synuclein interact,” he said. “I think we need a lot more work in this area.”
The Davis study was funded by the American Academy of Neurology/American Brain Foundation, the BrightFocus Foundation, the Mary E. Groff Charitable Trust, and others; the investigators reported additional relationships with Biogen, Alector, Parabon, and others. The Zhao study was funded by the National Institutes of Health and the Lewy Body Dementia Center Without Walls; the investigators reported no competing interests. Dr. Masliah reported no conflicts of interest.
SOURCES: Davis AA et al. Sci Transl Med. 2020 Feb 5. doi: 10.1126/scitranslmed.aay3069; Zhao N et al. Sci Transl Med. 2020 Feb 5. doi: 10.1126/scitranslmed.aay1809.
Apolipoprotein E epsilon 4 (APOE4) directly and independently exacerbates accumulation of alpha-synuclein in patients with Lewy body dementia, whereas APOE2 may have a protective effect, based on two recent studies involving mouse models and human patients.
These insights confirm the importance of APOE in synucleinopathies, and may lead to new treatments, according to Eliezer Masliah, MD, director of the division of neuroscience at the National Institute on Aging.
“These [studies] definitely implicate a role of APOE4,” Dr. Masliah said in an interview.
According to Dr. Masliah, previous studies linked the APOE4 genotype with cognitive decline in synucleinopathies, but underlying molecular mechanisms remained unknown.
“We [now] have more direct confirmation [based on] different experimental animal models,” Dr. Masliah said. “It also means that APOE4 could be a therapeutic target for dementia with Lewy bodies.”
The two studies were published simultaneously in Science Translational Medicine. The first study was conducted by Albert A. Davis, MD, PhD, of Washington University, St. Louis, and colleagues; the second was led by Na Zhao, MD, PhD, of the Mayo Clinic in Jacksonville, Fla.
“The studies are very synergistic, but used different techniques,” said Dr. Masliah, who was not involved in the studies.
Both studies involved mice that expressed a human variant of APOE: APOE2, APOE3, or APOE4. Three independent techniques were used to concurrently overexpress alpha-synuclein; Dr. Davis and colleagues used a transgenic approach, as well as striatal injection of alpha-synuclein preformed fibrils, whereas Dr. Zhao and colleagues turned to a viral vector. Regardless of technique, each APOE variant had a distinct impact on the level of alpha-synuclein accumulation.
“In a nutshell, [Dr. Davis and colleagues] found that those mice that have synuclein and APOE4 have a much more rapid progression of the disease,” Dr. Masliah said. “They become Parkinsonian much faster, but also, they become cognitively impaired much faster, and they have more synuclein in the brain. Remarkably, on the opposite side, those that were expressing APOE2, which we know is a protective allele, actually were far less impaired. So that’s really a remarkable finding.”
The study at the Mayo Clinic echoed these findings.
“Essentially, [Dr. Zhao and colleagues] had very similar results,” Dr. Masliah said. “[In mice expressing] APOE4, synuclein accumulation was worse and pathology was worse, and with APOE2, there was relative protection.”
Both studies found that the exacerbating effect of APOE4 translated to human patients.
Dr. Davis and colleagues evaluated data from 251 patients in the Parkinson’s Progression Markers Initiative. A multivariate model showed that patients with the APOE4 genotype had faster cognitive decline, an impact that was independent of other variables, including cerebrospinal fluid concentrations of amyloid beta and tau protein (P = .0119). This finding was further supported by additional analyses involving 177 patients with Parkinson’s disease from the Washington University Movement Disorders Center, and another 1,030 patients enrolled in the NeuroGenetics Research Consortium study.
Dr. Zhao and colleagues evaluated postmortem samples from patients with Lewy body dementia who had minimal amyloid pathology. Comparing 22 APOE4 carriers versus 22 age- and sex-matched noncarriers, they found that carriers had significantly greater accumulations of alpha-synuclein (P less than .05).
According to the investigators, these findings could have both prognostic and therapeutic implications.
“[I]t is intriguing to speculate whether APOE and other potential genetic risk or resilience genes could be useful as screening tools to stratify risk for individual patients,” Dr. Davis and colleagues wrote in their paper. They went on to suggest that APOE genotyping may one day be used to personalize treatments for patients with neurodegenerative disease.
According to Dr. Masliah, several treatment strategies are under investigation.
“There are some pharmaceutical companies and also some academic groups that have been developing antibodies against APOE4 for Alzheimer’s disease, but certainly that could also be used for dementia with Lewy bodies,” he said. “There are other ways. One could [be] to suppress the expression of APOE4 with antisense or other technologies.
“There is also a very innovative technology that has been developed by the group at the Gladstone Institutes in San Francisco, which is to switch APOE4 to APOE3.” This technique, Dr. Masliah explained, is accomplished by breaking a disulfide bond in APOE4, which opens the structure into an isoform that mimics APOE3. “They have developed small molecules that actually can break that bond and essentially chemically switch APOE4 to APOE3,” he said.
Although multiple techniques are feasible, Dr. Masliah stressed that these therapeutic efforts are still in their infancy.
“We need to better understand the mechanisms as to how APOE4 and alpha-synuclein interact,” he said. “I think we need a lot more work in this area.”
The Davis study was funded by the American Academy of Neurology/American Brain Foundation, the BrightFocus Foundation, the Mary E. Groff Charitable Trust, and others; the investigators reported additional relationships with Biogen, Alector, Parabon, and others. The Zhao study was funded by the National Institutes of Health and the Lewy Body Dementia Center Without Walls; the investigators reported no competing interests. Dr. Masliah reported no conflicts of interest.
SOURCES: Davis AA et al. Sci Transl Med. 2020 Feb 5. doi: 10.1126/scitranslmed.aay3069; Zhao N et al. Sci Transl Med. 2020 Feb 5. doi: 10.1126/scitranslmed.aay1809.
FROM SCIENCE TRANSLATIONAL MEDICINE
Serum levels of neurofilament light are increased before clinical onset of MS
(MS), according to research published in the January issue of JAMA Neurology. These results lend weight to the idea that MS has a prodromal phase, and this phase appears to be associated with neurodegeneration, according to the authors.
Patients often have CNS lesions of various stages of development at the time of their first demyelinating event, and this finding was one basis for neurologists’ hypothesis of a prodromal phase of MS. The finding that one-third of patients with radiologically isolated syndrome develop MS within 5 years also lends credence to this idea. Diagnosing MS early would enable early treatment that could prevent demyelination and the progression of neurodegeneration.
Researchers compared presymptomatic and symptomatic samples
With this idea in mind, Kjetil Bjornevik, MD, PhD, a member of the neuroepidemiology research group at Harvard TH Chan School of Public Health in Boston, and colleagues evaluated whether serum levels of NfL, a marker of ongoing neuroaxonal degeneration, were increased in the years before and around the time of clinical onset of MS. For their study population, the investigators chose active-duty U.S. military personnel who have at least one serum sample stored in the U.S. Department of Defense Serum Repository. Samples are collected after routine HIV type 1 antibody testing.
Within this population, Dr. Bjornevik and colleagues identified patients with MS who had at least one presymptomatic serum sample. The date of clinical MS onset was defined as the date of the first neurologic symptoms attributable to MS documented in the medical record. The investigators randomly selected two control individuals from the population and matched them to each case by age, sex, race or ethnicity, and dates of sample collection. Eligible controls were on active duty on the date of onset of the matched case.
Dr. Bjornevik and colleagues identified 245 patients with MS. Among this sample, the researchers selected two groups that each included 30 cases and 30 controls. The first group included patients who had provided at least one serum sample before MS onset and one sample within 2 years after MS onset. The second group included cases with at least two presymptomatic serum samples, one of which was collected more than 5 years before MS diagnosis, and the other of which was collected between 2 and 5 years before diagnosis. The investigators handled pairs of serum samples in the same way and assayed them in the same batch. The order of the samples in each pair was arranged at random.
Levels were higher in cases than in controls
About 77% of the population was male. Sixty percent of participants were white, 28% were black, and 6.7% were Hispanic. The population’s mean age at first sample collection was approximately 27 years. Mean age at MS onset was approximately 31 years.
For patients who provided samples before and after the clinical onset of MS, serum NfL levels were higher than in matched controls at both points. Most patients who passed from the presymptomatic stage to the symptomatic stage had a significant increase in serum NfL level (i.e., from a median of 25.0 pg/mL to a median of 45.1 pg/mL). Serum NfL levels at the two time points in controls did not differ significantly. For any given patient, an increase in serum NfL level from the presymptomatic measurement to the symptomatic measurement was associated with an increased risk of MS.
In patients with two presymptomatic samples, serum NfL levels were significantly higher in both samples than in the corresponding samples from matched controls. In cases, the earlier sample was collected at a median of 6 years before clinical onset of MS, and the later sample was collected at a median of 1 year before clinical onset. The serum NfL levels increased significantly between the two points for cases (i.e., a median increase of 1.3 pg/mL per year), but there was no significant difference in serum NfL level between the two samples in controls. A within-patient increase in presymptomatic serum NfL level was associated with an increased risk of MS.
Population included few women
“Our study differs from previous studies on the prodromal phase of MS because these have used indirect markers of this phase, which included unspecific symptoms or disturbances occurring before the clinical onset, compared with a marker of neurodegeneration,” wrote Dr. Bjornevik and colleagues. Initiation of treatment with disease-modifying therapy is associated with reductions in serum NfL levels, and this association could explain why some patients in the current study had higher NfL levels before MS onset than afterward. Furthermore, serum NfL levels are highly associated with levels of NfL in cerebrospinal fluid. “Thus, our findings of a presymptomatic increase in serum NfL not only suggest the presence of a prodromal phase in MS, but also that this phase is associated with neurodegeneration,” wrote the investigators.
The study’s well-defined population helped to minimize selection bias, and the blinded, randomized method of analyzing the serum samples eliminated artifactual differences in serum NfL concentrations. But the small sample size precluded analyses that could have influenced clinical practice, wrote Dr. Bjornevik and colleagues. For example, the researchers could not evaluate distinct cutoffs in serum NfL level that could mark the beginning of the prodromal phase of MS. Nor could they determine whether presymptomatic serum NfL levels varied with age at clinical onset, sex, or race. The small number of women in the sample was another limitation of the study.
The Swiss National Research Foundation and the National Institute of Neurologic Disorders and Stroke funded the study. Several of the investigators received fees from various drug companies that were unrelated to the study, and one researcher received grants from the National Institutes of Health during the study.
SOURCE: Bjornevik K et al. JAMA Neurol. 2020;77(1):58-64.
(MS), according to research published in the January issue of JAMA Neurology. These results lend weight to the idea that MS has a prodromal phase, and this phase appears to be associated with neurodegeneration, according to the authors.
Patients often have CNS lesions of various stages of development at the time of their first demyelinating event, and this finding was one basis for neurologists’ hypothesis of a prodromal phase of MS. The finding that one-third of patients with radiologically isolated syndrome develop MS within 5 years also lends credence to this idea. Diagnosing MS early would enable early treatment that could prevent demyelination and the progression of neurodegeneration.
Researchers compared presymptomatic and symptomatic samples
With this idea in mind, Kjetil Bjornevik, MD, PhD, a member of the neuroepidemiology research group at Harvard TH Chan School of Public Health in Boston, and colleagues evaluated whether serum levels of NfL, a marker of ongoing neuroaxonal degeneration, were increased in the years before and around the time of clinical onset of MS. For their study population, the investigators chose active-duty U.S. military personnel who have at least one serum sample stored in the U.S. Department of Defense Serum Repository. Samples are collected after routine HIV type 1 antibody testing.
Within this population, Dr. Bjornevik and colleagues identified patients with MS who had at least one presymptomatic serum sample. The date of clinical MS onset was defined as the date of the first neurologic symptoms attributable to MS documented in the medical record. The investigators randomly selected two control individuals from the population and matched them to each case by age, sex, race or ethnicity, and dates of sample collection. Eligible controls were on active duty on the date of onset of the matched case.
Dr. Bjornevik and colleagues identified 245 patients with MS. Among this sample, the researchers selected two groups that each included 30 cases and 30 controls. The first group included patients who had provided at least one serum sample before MS onset and one sample within 2 years after MS onset. The second group included cases with at least two presymptomatic serum samples, one of which was collected more than 5 years before MS diagnosis, and the other of which was collected between 2 and 5 years before diagnosis. The investigators handled pairs of serum samples in the same way and assayed them in the same batch. The order of the samples in each pair was arranged at random.
Levels were higher in cases than in controls
About 77% of the population was male. Sixty percent of participants were white, 28% were black, and 6.7% were Hispanic. The population’s mean age at first sample collection was approximately 27 years. Mean age at MS onset was approximately 31 years.
For patients who provided samples before and after the clinical onset of MS, serum NfL levels were higher than in matched controls at both points. Most patients who passed from the presymptomatic stage to the symptomatic stage had a significant increase in serum NfL level (i.e., from a median of 25.0 pg/mL to a median of 45.1 pg/mL). Serum NfL levels at the two time points in controls did not differ significantly. For any given patient, an increase in serum NfL level from the presymptomatic measurement to the symptomatic measurement was associated with an increased risk of MS.
In patients with two presymptomatic samples, serum NfL levels were significantly higher in both samples than in the corresponding samples from matched controls. In cases, the earlier sample was collected at a median of 6 years before clinical onset of MS, and the later sample was collected at a median of 1 year before clinical onset. The serum NfL levels increased significantly between the two points for cases (i.e., a median increase of 1.3 pg/mL per year), but there was no significant difference in serum NfL level between the two samples in controls. A within-patient increase in presymptomatic serum NfL level was associated with an increased risk of MS.
Population included few women
“Our study differs from previous studies on the prodromal phase of MS because these have used indirect markers of this phase, which included unspecific symptoms or disturbances occurring before the clinical onset, compared with a marker of neurodegeneration,” wrote Dr. Bjornevik and colleagues. Initiation of treatment with disease-modifying therapy is associated with reductions in serum NfL levels, and this association could explain why some patients in the current study had higher NfL levels before MS onset than afterward. Furthermore, serum NfL levels are highly associated with levels of NfL in cerebrospinal fluid. “Thus, our findings of a presymptomatic increase in serum NfL not only suggest the presence of a prodromal phase in MS, but also that this phase is associated with neurodegeneration,” wrote the investigators.
The study’s well-defined population helped to minimize selection bias, and the blinded, randomized method of analyzing the serum samples eliminated artifactual differences in serum NfL concentrations. But the small sample size precluded analyses that could have influenced clinical practice, wrote Dr. Bjornevik and colleagues. For example, the researchers could not evaluate distinct cutoffs in serum NfL level that could mark the beginning of the prodromal phase of MS. Nor could they determine whether presymptomatic serum NfL levels varied with age at clinical onset, sex, or race. The small number of women in the sample was another limitation of the study.
The Swiss National Research Foundation and the National Institute of Neurologic Disorders and Stroke funded the study. Several of the investigators received fees from various drug companies that were unrelated to the study, and one researcher received grants from the National Institutes of Health during the study.
SOURCE: Bjornevik K et al. JAMA Neurol. 2020;77(1):58-64.
(MS), according to research published in the January issue of JAMA Neurology. These results lend weight to the idea that MS has a prodromal phase, and this phase appears to be associated with neurodegeneration, according to the authors.
Patients often have CNS lesions of various stages of development at the time of their first demyelinating event, and this finding was one basis for neurologists’ hypothesis of a prodromal phase of MS. The finding that one-third of patients with radiologically isolated syndrome develop MS within 5 years also lends credence to this idea. Diagnosing MS early would enable early treatment that could prevent demyelination and the progression of neurodegeneration.
Researchers compared presymptomatic and symptomatic samples
With this idea in mind, Kjetil Bjornevik, MD, PhD, a member of the neuroepidemiology research group at Harvard TH Chan School of Public Health in Boston, and colleagues evaluated whether serum levels of NfL, a marker of ongoing neuroaxonal degeneration, were increased in the years before and around the time of clinical onset of MS. For their study population, the investigators chose active-duty U.S. military personnel who have at least one serum sample stored in the U.S. Department of Defense Serum Repository. Samples are collected after routine HIV type 1 antibody testing.
Within this population, Dr. Bjornevik and colleagues identified patients with MS who had at least one presymptomatic serum sample. The date of clinical MS onset was defined as the date of the first neurologic symptoms attributable to MS documented in the medical record. The investigators randomly selected two control individuals from the population and matched them to each case by age, sex, race or ethnicity, and dates of sample collection. Eligible controls were on active duty on the date of onset of the matched case.
Dr. Bjornevik and colleagues identified 245 patients with MS. Among this sample, the researchers selected two groups that each included 30 cases and 30 controls. The first group included patients who had provided at least one serum sample before MS onset and one sample within 2 years after MS onset. The second group included cases with at least two presymptomatic serum samples, one of which was collected more than 5 years before MS diagnosis, and the other of which was collected between 2 and 5 years before diagnosis. The investigators handled pairs of serum samples in the same way and assayed them in the same batch. The order of the samples in each pair was arranged at random.
Levels were higher in cases than in controls
About 77% of the population was male. Sixty percent of participants were white, 28% were black, and 6.7% were Hispanic. The population’s mean age at first sample collection was approximately 27 years. Mean age at MS onset was approximately 31 years.
For patients who provided samples before and after the clinical onset of MS, serum NfL levels were higher than in matched controls at both points. Most patients who passed from the presymptomatic stage to the symptomatic stage had a significant increase in serum NfL level (i.e., from a median of 25.0 pg/mL to a median of 45.1 pg/mL). Serum NfL levels at the two time points in controls did not differ significantly. For any given patient, an increase in serum NfL level from the presymptomatic measurement to the symptomatic measurement was associated with an increased risk of MS.
In patients with two presymptomatic samples, serum NfL levels were significantly higher in both samples than in the corresponding samples from matched controls. In cases, the earlier sample was collected at a median of 6 years before clinical onset of MS, and the later sample was collected at a median of 1 year before clinical onset. The serum NfL levels increased significantly between the two points for cases (i.e., a median increase of 1.3 pg/mL per year), but there was no significant difference in serum NfL level between the two samples in controls. A within-patient increase in presymptomatic serum NfL level was associated with an increased risk of MS.
Population included few women
“Our study differs from previous studies on the prodromal phase of MS because these have used indirect markers of this phase, which included unspecific symptoms or disturbances occurring before the clinical onset, compared with a marker of neurodegeneration,” wrote Dr. Bjornevik and colleagues. Initiation of treatment with disease-modifying therapy is associated with reductions in serum NfL levels, and this association could explain why some patients in the current study had higher NfL levels before MS onset than afterward. Furthermore, serum NfL levels are highly associated with levels of NfL in cerebrospinal fluid. “Thus, our findings of a presymptomatic increase in serum NfL not only suggest the presence of a prodromal phase in MS, but also that this phase is associated with neurodegeneration,” wrote the investigators.
The study’s well-defined population helped to minimize selection bias, and the blinded, randomized method of analyzing the serum samples eliminated artifactual differences in serum NfL concentrations. But the small sample size precluded analyses that could have influenced clinical practice, wrote Dr. Bjornevik and colleagues. For example, the researchers could not evaluate distinct cutoffs in serum NfL level that could mark the beginning of the prodromal phase of MS. Nor could they determine whether presymptomatic serum NfL levels varied with age at clinical onset, sex, or race. The small number of women in the sample was another limitation of the study.
The Swiss National Research Foundation and the National Institute of Neurologic Disorders and Stroke funded the study. Several of the investigators received fees from various drug companies that were unrelated to the study, and one researcher received grants from the National Institutes of Health during the study.
SOURCE: Bjornevik K et al. JAMA Neurol. 2020;77(1):58-64.
FROM JAMA NEUROLOGY
Rate of suicide is higher in people with neurologic disorders
The absolute risk difference is small, but statistically significant. “These findings do not necessarily warrant changing the management of treatment for individual patients,” wrote Annette Erlangsen, PhD, a researcher at the Danish Research Institute for Suicide Prevention in Hellerup, and colleagues. “As with all patients, physicians should be aware of the potential for depression, demoralization, and suicide.”
In addition, dementia, Alzheimer’s disease, and intellectual disabilities may be associated with lower suicide rates, according to the study, which was published in JAMA.
“Plausible mechanisms” could underlie the association between neurologic disease and suicide, the authors wrote. A neurologic diagnosis “may constitute a distressing life event,” and the diseases may have psychological, physical, and psychiatric effects. Patients may see themselves as a burden or have less financial security. In addition, the diseases may entail “communication difficulties, poor sleep, and pain.” Neurologic diseases may alter brain circuitry and functioning and influence aggression and impulsivity. “People with neurologic disorders may also have easier access to toxic medication,” they added.
More than a dozen conditions examined
Prior studies have found associations between neurologic conditions and rates of suicide, but data have been inconclusive or inconsistent for some of the disorders. To examine whether people with neurologic disorders have higher suicide rates, relative to people without these disorders, the researchers conducted a retrospective study. They analyzed data from more than 7.3 million people aged 15 years or older who lived in Denmark between 1980 and 2016. The cohort included more than 1.2 million people with neurologic disorders. The investigators identified neurologic disorders using ICD codes for head injury, stroke, epilepsy, polyneuropathy, diseases of the myoneural junction, Parkinson’s disease, multiple sclerosis, CNS infections, meningitis, encephalitis, amyotrophic lateral sclerosis, Huntington’s disease, dementia, intellectual disability, and other brain disorders. They compared incidence rates using a Poisson regression model and adjusted for time period, sex, age, region, socioeconomic status, comorbidity, self-harm or psychiatric hospitalization prior to a neurologic diagnosis, and whether a person lived alone.
In all, 35,483 people in the cohort died by suicide at an average age of about 52 years; 77.4% were male. About 15% of those who died by suicide had a neurologic disorder. The suicide incidence rate among people with a neurologic disorder was 44.0 per 100,000 person-years, whereas the rate among people without a neurologic disorder was 20.1 per 100,000 person-years.
The adjusted incidence rate ratio for people with a neurologic disorder was 1.8. The rate ratio was highest during the 3 months after diagnosis, at 3.1. Huntington’s disease and amyotrophic lateral sclerosis were associated with “the largest excess adjusted [incidence rate ratios] of suicide mortality,” with a rate ratio of 4.9 for each condition, the researchers reported. The adjusted incidence rate ratio was 1.7 for head injury, 1.3 for stroke, 1.7 for epilepsy, 1.4 for intracerebral hemorrhage, 1.3 for cerebral infarction, 1.3 for subarachnoid hemorrhage, 1.7 for polyneuropathy and peripheral neuropathy, 2.2 for Guillain-Barré syndrome, 1.9 for diseases of myoneural junction and muscle, 1.8 for other brain disorders, 1.7 for Parkinson’s disease, 2.2 for multiple sclerosis, and 1.6 for CNS infection.
Compared with people without a neurologic condition, people with dementia, Alzheimer’s disease, and intellectual disabilities had lower suicide rates, with adjusted incidence rate ratios of 0.8, 0.2, and 0.6, respectively. “However, the adjusted [incidence rate ratio] for people with dementia during the first month after diagnosis was 3.0,” the researchers wrote.
In addition, the suicide rate increased with an increasing cumulative number of hospital contacts for neurologic conditions.
Overall incidence rates declined
“Over the study period, the suicide incidence rate for people with neurological disorders decreased from 78.6 per 100,000 person-years during the 1980-1999 years to 27.3 per 100,000 person-years during the 2000-2016 years,” wrote Dr. Erlangsen and colleagues. “The suicide incidence rate for those without a disorder decreased from 26.3 to 12.7 during the same time spans. ... The decline in the overall suicide rate over time did not affect the relative risk pattern.”
The decline in the general suicide rate in Denmark “has largely been attributed to means restriction, such as efforts to limit availability of firearms and particularly toxic medication,” the authors added.
In those time spans, the adjusted incidence rate ratio for suicide among those with dementia decreased from 2.4 to 1.0, and among those with multiple sclerosis from 2.0 to 1.0. “It is possible that the improvements observed for dementia and multiple sclerosis may be related to improvements in treatment and intensified community-based support,” Dr. Erlangsen and coauthors wrote.
When the researchers used people with rheumatoid arthritis as a reference group, those with a neurologic disorder had a higher suicide rate per 100,000 person-years, 30.2 versus 18.4. The adjusted incidence rate ratio for that comparison was 1.4.
In patients with Huntington’s disease, depression mediated by hyperactivity in the hypothalamic-pituitary-adrenal axis may contribute to the risk of suicide. “Witnessing the course of the disease in one’s parent” also may contribute the risk, the researchers wrote.
The analysis may have missed people with neurologic disorders diagnosed before 1977 if they did not have subsequent contact with a hospital, the investigators noted. In addition, diagnoses given in primary care were not included, suicide deaths may be underrecorded, and “adjusting for preexisting mental disorders could be viewed as overadjusting,” they wrote.
The study was supported by a grant from the Psychiatric Research Foundation in Denmark. The authors reported that they had no disclosures.
SOURCE: Erlangsen A et al. JAMA. 2020 Feb 4. doi: 10.1001/jama.2019.21834.
The absolute risk difference is small, but statistically significant. “These findings do not necessarily warrant changing the management of treatment for individual patients,” wrote Annette Erlangsen, PhD, a researcher at the Danish Research Institute for Suicide Prevention in Hellerup, and colleagues. “As with all patients, physicians should be aware of the potential for depression, demoralization, and suicide.”
In addition, dementia, Alzheimer’s disease, and intellectual disabilities may be associated with lower suicide rates, according to the study, which was published in JAMA.
“Plausible mechanisms” could underlie the association between neurologic disease and suicide, the authors wrote. A neurologic diagnosis “may constitute a distressing life event,” and the diseases may have psychological, physical, and psychiatric effects. Patients may see themselves as a burden or have less financial security. In addition, the diseases may entail “communication difficulties, poor sleep, and pain.” Neurologic diseases may alter brain circuitry and functioning and influence aggression and impulsivity. “People with neurologic disorders may also have easier access to toxic medication,” they added.
More than a dozen conditions examined
Prior studies have found associations between neurologic conditions and rates of suicide, but data have been inconclusive or inconsistent for some of the disorders. To examine whether people with neurologic disorders have higher suicide rates, relative to people without these disorders, the researchers conducted a retrospective study. They analyzed data from more than 7.3 million people aged 15 years or older who lived in Denmark between 1980 and 2016. The cohort included more than 1.2 million people with neurologic disorders. The investigators identified neurologic disorders using ICD codes for head injury, stroke, epilepsy, polyneuropathy, diseases of the myoneural junction, Parkinson’s disease, multiple sclerosis, CNS infections, meningitis, encephalitis, amyotrophic lateral sclerosis, Huntington’s disease, dementia, intellectual disability, and other brain disorders. They compared incidence rates using a Poisson regression model and adjusted for time period, sex, age, region, socioeconomic status, comorbidity, self-harm or psychiatric hospitalization prior to a neurologic diagnosis, and whether a person lived alone.
In all, 35,483 people in the cohort died by suicide at an average age of about 52 years; 77.4% were male. About 15% of those who died by suicide had a neurologic disorder. The suicide incidence rate among people with a neurologic disorder was 44.0 per 100,000 person-years, whereas the rate among people without a neurologic disorder was 20.1 per 100,000 person-years.
The adjusted incidence rate ratio for people with a neurologic disorder was 1.8. The rate ratio was highest during the 3 months after diagnosis, at 3.1. Huntington’s disease and amyotrophic lateral sclerosis were associated with “the largest excess adjusted [incidence rate ratios] of suicide mortality,” with a rate ratio of 4.9 for each condition, the researchers reported. The adjusted incidence rate ratio was 1.7 for head injury, 1.3 for stroke, 1.7 for epilepsy, 1.4 for intracerebral hemorrhage, 1.3 for cerebral infarction, 1.3 for subarachnoid hemorrhage, 1.7 for polyneuropathy and peripheral neuropathy, 2.2 for Guillain-Barré syndrome, 1.9 for diseases of myoneural junction and muscle, 1.8 for other brain disorders, 1.7 for Parkinson’s disease, 2.2 for multiple sclerosis, and 1.6 for CNS infection.
Compared with people without a neurologic condition, people with dementia, Alzheimer’s disease, and intellectual disabilities had lower suicide rates, with adjusted incidence rate ratios of 0.8, 0.2, and 0.6, respectively. “However, the adjusted [incidence rate ratio] for people with dementia during the first month after diagnosis was 3.0,” the researchers wrote.
In addition, the suicide rate increased with an increasing cumulative number of hospital contacts for neurologic conditions.
Overall incidence rates declined
“Over the study period, the suicide incidence rate for people with neurological disorders decreased from 78.6 per 100,000 person-years during the 1980-1999 years to 27.3 per 100,000 person-years during the 2000-2016 years,” wrote Dr. Erlangsen and colleagues. “The suicide incidence rate for those without a disorder decreased from 26.3 to 12.7 during the same time spans. ... The decline in the overall suicide rate over time did not affect the relative risk pattern.”
The decline in the general suicide rate in Denmark “has largely been attributed to means restriction, such as efforts to limit availability of firearms and particularly toxic medication,” the authors added.
In those time spans, the adjusted incidence rate ratio for suicide among those with dementia decreased from 2.4 to 1.0, and among those with multiple sclerosis from 2.0 to 1.0. “It is possible that the improvements observed for dementia and multiple sclerosis may be related to improvements in treatment and intensified community-based support,” Dr. Erlangsen and coauthors wrote.
When the researchers used people with rheumatoid arthritis as a reference group, those with a neurologic disorder had a higher suicide rate per 100,000 person-years, 30.2 versus 18.4. The adjusted incidence rate ratio for that comparison was 1.4.
In patients with Huntington’s disease, depression mediated by hyperactivity in the hypothalamic-pituitary-adrenal axis may contribute to the risk of suicide. “Witnessing the course of the disease in one’s parent” also may contribute the risk, the researchers wrote.
The analysis may have missed people with neurologic disorders diagnosed before 1977 if they did not have subsequent contact with a hospital, the investigators noted. In addition, diagnoses given in primary care were not included, suicide deaths may be underrecorded, and “adjusting for preexisting mental disorders could be viewed as overadjusting,” they wrote.
The study was supported by a grant from the Psychiatric Research Foundation in Denmark. The authors reported that they had no disclosures.
SOURCE: Erlangsen A et al. JAMA. 2020 Feb 4. doi: 10.1001/jama.2019.21834.
The absolute risk difference is small, but statistically significant. “These findings do not necessarily warrant changing the management of treatment for individual patients,” wrote Annette Erlangsen, PhD, a researcher at the Danish Research Institute for Suicide Prevention in Hellerup, and colleagues. “As with all patients, physicians should be aware of the potential for depression, demoralization, and suicide.”
In addition, dementia, Alzheimer’s disease, and intellectual disabilities may be associated with lower suicide rates, according to the study, which was published in JAMA.
“Plausible mechanisms” could underlie the association between neurologic disease and suicide, the authors wrote. A neurologic diagnosis “may constitute a distressing life event,” and the diseases may have psychological, physical, and psychiatric effects. Patients may see themselves as a burden or have less financial security. In addition, the diseases may entail “communication difficulties, poor sleep, and pain.” Neurologic diseases may alter brain circuitry and functioning and influence aggression and impulsivity. “People with neurologic disorders may also have easier access to toxic medication,” they added.
More than a dozen conditions examined
Prior studies have found associations between neurologic conditions and rates of suicide, but data have been inconclusive or inconsistent for some of the disorders. To examine whether people with neurologic disorders have higher suicide rates, relative to people without these disorders, the researchers conducted a retrospective study. They analyzed data from more than 7.3 million people aged 15 years or older who lived in Denmark between 1980 and 2016. The cohort included more than 1.2 million people with neurologic disorders. The investigators identified neurologic disorders using ICD codes for head injury, stroke, epilepsy, polyneuropathy, diseases of the myoneural junction, Parkinson’s disease, multiple sclerosis, CNS infections, meningitis, encephalitis, amyotrophic lateral sclerosis, Huntington’s disease, dementia, intellectual disability, and other brain disorders. They compared incidence rates using a Poisson regression model and adjusted for time period, sex, age, region, socioeconomic status, comorbidity, self-harm or psychiatric hospitalization prior to a neurologic diagnosis, and whether a person lived alone.
In all, 35,483 people in the cohort died by suicide at an average age of about 52 years; 77.4% were male. About 15% of those who died by suicide had a neurologic disorder. The suicide incidence rate among people with a neurologic disorder was 44.0 per 100,000 person-years, whereas the rate among people without a neurologic disorder was 20.1 per 100,000 person-years.
The adjusted incidence rate ratio for people with a neurologic disorder was 1.8. The rate ratio was highest during the 3 months after diagnosis, at 3.1. Huntington’s disease and amyotrophic lateral sclerosis were associated with “the largest excess adjusted [incidence rate ratios] of suicide mortality,” with a rate ratio of 4.9 for each condition, the researchers reported. The adjusted incidence rate ratio was 1.7 for head injury, 1.3 for stroke, 1.7 for epilepsy, 1.4 for intracerebral hemorrhage, 1.3 for cerebral infarction, 1.3 for subarachnoid hemorrhage, 1.7 for polyneuropathy and peripheral neuropathy, 2.2 for Guillain-Barré syndrome, 1.9 for diseases of myoneural junction and muscle, 1.8 for other brain disorders, 1.7 for Parkinson’s disease, 2.2 for multiple sclerosis, and 1.6 for CNS infection.
Compared with people without a neurologic condition, people with dementia, Alzheimer’s disease, and intellectual disabilities had lower suicide rates, with adjusted incidence rate ratios of 0.8, 0.2, and 0.6, respectively. “However, the adjusted [incidence rate ratio] for people with dementia during the first month after diagnosis was 3.0,” the researchers wrote.
In addition, the suicide rate increased with an increasing cumulative number of hospital contacts for neurologic conditions.
Overall incidence rates declined
“Over the study period, the suicide incidence rate for people with neurological disorders decreased from 78.6 per 100,000 person-years during the 1980-1999 years to 27.3 per 100,000 person-years during the 2000-2016 years,” wrote Dr. Erlangsen and colleagues. “The suicide incidence rate for those without a disorder decreased from 26.3 to 12.7 during the same time spans. ... The decline in the overall suicide rate over time did not affect the relative risk pattern.”
The decline in the general suicide rate in Denmark “has largely been attributed to means restriction, such as efforts to limit availability of firearms and particularly toxic medication,” the authors added.
In those time spans, the adjusted incidence rate ratio for suicide among those with dementia decreased from 2.4 to 1.0, and among those with multiple sclerosis from 2.0 to 1.0. “It is possible that the improvements observed for dementia and multiple sclerosis may be related to improvements in treatment and intensified community-based support,” Dr. Erlangsen and coauthors wrote.
When the researchers used people with rheumatoid arthritis as a reference group, those with a neurologic disorder had a higher suicide rate per 100,000 person-years, 30.2 versus 18.4. The adjusted incidence rate ratio for that comparison was 1.4.
In patients with Huntington’s disease, depression mediated by hyperactivity in the hypothalamic-pituitary-adrenal axis may contribute to the risk of suicide. “Witnessing the course of the disease in one’s parent” also may contribute the risk, the researchers wrote.
The analysis may have missed people with neurologic disorders diagnosed before 1977 if they did not have subsequent contact with a hospital, the investigators noted. In addition, diagnoses given in primary care were not included, suicide deaths may be underrecorded, and “adjusting for preexisting mental disorders could be viewed as overadjusting,” they wrote.
The study was supported by a grant from the Psychiatric Research Foundation in Denmark. The authors reported that they had no disclosures.
SOURCE: Erlangsen A et al. JAMA. 2020 Feb 4. doi: 10.1001/jama.2019.21834.
FROM JAMA
Presentation of a Rare Malignancy: Leiomyosarcoma of the Prostate (FULL)
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B). 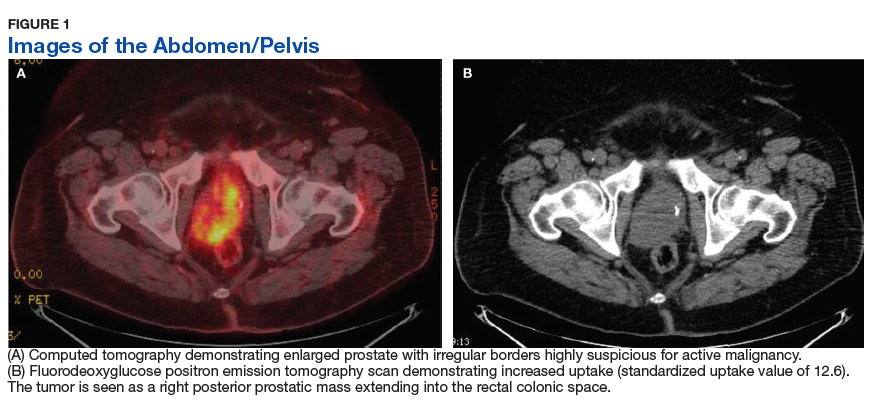
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues. 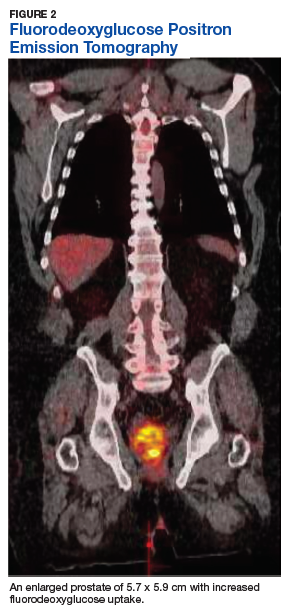
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B).
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues.
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B).
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues.
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
Primary Urethral Carcinoma With Nodal Metastasis (FULL)
The presentation of a fungating penile mass often indicates penile carcinoma, but providers should be aware of urethral carcinoma in the differential diagnosis.
Primary urethral carcinoma (PUC) is a rare but morbid disease, representing < 1% of all urologic malignancies.1 Up to one-third of male patients may present with nodal metastases.2-4 The overall survival (OS) for all male PUC is < 50% at 5 years and is lower still in patients with nodal involvement.4
Although surgical intervention, including radical resection, has been a mainstay in disease management, the presence of high-stage disease may warrant multimodal treatment with chemotherapy, radiation, and surgery. Recent series have described success with neoadjuvant and adjuvant chemoradiation, yet the optimal regimen remains unestablished.5,6 Although nodal disease is commonly encountered with proximal, high-stage tumors, this case exhibits a rare presentation of a distal fungating penile mass with low pathologic stage but rapid progression to nodal disease.
Case Presentation
A male veteran aged 77 years with a history of diabetes mellitus and stroke presented with obstructive urinary symptoms, gross hematuria, and 15-pound weight loss. Examination revealed a distal penile mass with purulent exudate at the meatus but no inguinal lymphadenopathy. Two fragments of this mass detached during office cystoscopy, and pathology revealed high-grade urothelial cell carcinoma (UCC). A magnetic resonance image of the pelvis with and without IV contrast revealed a 2.4-cm tumor in the glans penis with possible extension into the subcutaneous connective tissue of the penis and penile skin, without invasion of the corpora cavernosa/spongiosum or lymphadenopathy (Figure 1). 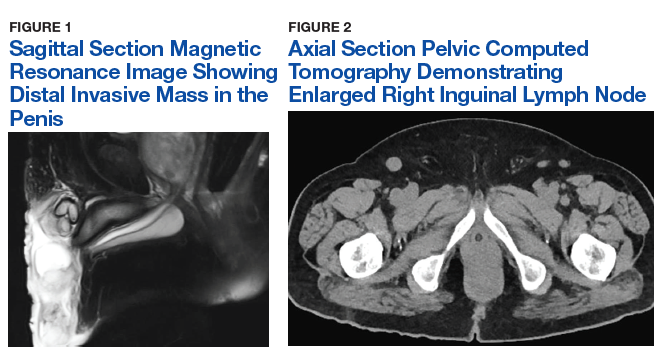
Prostatic urethral and random bladder biopsies, bilateral retrograde pyelograms, and selective ureteral washings revealed no abnormalities or signs of disease. Percutaneous biopsy of the inguinal node confirmed metastatic UCC. The patient underwent radical penectomy, creation of a perineal urethrostomy, and suprapubic cystostomy tube placement. Negative margins were confirmed on the urethral stump and corpus spongiosum. Final pathology revealed high-grade UCC with squamous differentiation on hematoxylin and eosin staining, arising from the penile urethra, invading the glans and corpus spongiosum, with no invasion of the corpus cavernosa (Figures 3 and 4).
Immunohistochemical stains were performed and strongly positive for cytokeratin 7 and p63. Final pathologic stage was described as pT2N1, with negative margins, indicating an American Joint Committee on Cancer classification of Stage III disease.7 The patient was referred postoperatively for adjuvant chemoradiation. 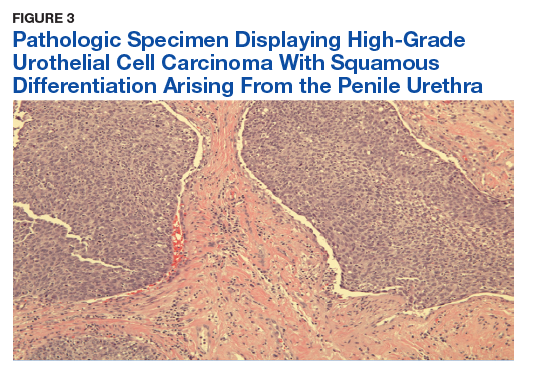

Discussion
The low incidence of PUC, coupled with a high morbidity/mortality rate, creates a difficult scenario in choosing the best oncologic management for this disease. National guidelines stratify treatment algorithms by stage and location of primary tumor, as these were found to be the 2 most important prognostic factors for men.1 The location of the primary tumor is most often in the bulbomembranous urethra, but up to one-third occur in the pendulous urethra.2
A recent review reported that UCC is the most common histologic subtype.4 When considering the differential diagnosis, a distal penile mass may represent a malignant penile lesion, such as squamous cell carcinoma, Buschke-Lowenstein tumor, Kaposi sarcoma, or precancerous lesions. Additional benign and infectious disorders include epidermoid and retention cysts, leukoplakia, balanitis xerotica obliterans, condyloma acuminatum, chancre/chancroid, lymphogranuloma venereum, granuloma inguinale, and tuberculosis. Clinical workup typically includes physical examination, cystourethroscopy and biopsy, chest X-ray, and pelvic/abdominal cross-sectional imaging.9,10 Magnetic resonance imaging of the abdomen and pelvis is ideal in identifying soft tissue structures and extension of tumor.
In male patients with PUC, nodal metastases are commonly seen at initial presentation in up to one-third of patients, while distant metastases may be present in up to 6% at presentation.2-4 When tumors arise from the anterior urethra, the primary lymphatic drainage is first to the inguinal lymph nodes, whereas posterior tumors drain to the pelvic lymph nodes. A multivariate analysis of men with PUC within the Surveillance, Epidemiology, and End Results database demonstrated an OS across all stages to be 46.2% and 29.3% at 5 and 10 years, respectively. Increased likelihood of death was predicted by advanced age, high grade/stage, systemic metastases, non-UCC histology, and the lack of surgery.4
Surgical intervention, including radical resection via penectomy, has been the mainstay in disease management and was first described by Marshall in 1957 for bulbar urethral cancer.11 In 1998, Gheiler and colleagues demonstrated that surgical resection alone yielded excellent outcomes in patients with low-stage disease with 89% of patients disease free at mean 42 months. This was in stark contrast to patients with advanced stage disease (T3 or N+) who exhibited a disease-free survival rate of 42% at the same follow-up interval and benefited from combined chemoradiation and surgical resection.3
In the presence of high-stage disease, multimodal therapy with chemotherapy, radiation, and/or surgery is warranted. A study in 2008 reviewed chemoradiation in which patients with PUC received a 5-week protocol of external beam radiotherapy to the genitals, inguinal/pelvic lymph nodes, plus an additional radiation bolus to the primary tumor.5 In the 18 patients reported, 15 had complete response to therapy, and only 4 patients required salvage surgical resection. The 7-year survival for the cohort was 72% with chemoradiation alone, with about half the population recurring or progressing at 7 years. However, all patients that avoided surgical resection went on to develop urethral strictures that required surgical therapy, 3 of which required complex reconstructive procedures.
To place this survival into context, the 1999 study by Dalbagni and colleagues reported a 5-year OS of 42% when surgical resection alone was performed in 40/46 men with PUC.2 Last, a large retrospective series of 44 patients reported mostly advanced-stage patients with PUC and analyzed patients treated with chemotherapy based on histologic pathology. The results demonstrated a 72% overall response rate to neoadjuvant chemotherapy, with a median OS of 32 months in patients undergoing chemotherapy vs 46 months in patients who underwent subsequent surgery. This study solidified that for patients with PUC involving the lymph nodes; optimal treatment includes neoadjuvant cisplatin-based chemotherapy followed by surgical resection.6
As medicine and oncologic therapies become more individualized, physicians are looking to new immunologic agents for systemic therapy. Immune checkpoint inhibitors were approved by the US Food and Drug Administration for UCC of the bladder in 2016.12 Unfortunately, due to the rarity of PUC and the recent development of immune checkpoint inhibitors, there have been no published reports of these or other immunotherapies in PUC. However, given the histologic similarity and pathogenesis, checkpoint inhibitors may have a future indication in the systemic management of this disease.
Conclusion
This patient’s PUC represents a rare presentation of a distal urethral carcinoma, T2-staged tumor, with rapid progression to nodal metastases. Additionally, the presentation of a fungating penile mass would usually indicate penile carcinoma, but providers should be aware of urethral carcinoma in the differential diagnosis. Notably, the patient was found to have progression to lymph node involvement during a mere 2-month period.
Recent case series have published encouraging results with neoadjuvant chemotherapy or chemoradiation.5,6 However, radical resection in men with T2 to T4 disease is associated with significantly higher cancer-specific survival. Given our concern of a loss to follow-up, we felt that radical resection of the primary tumor and adjuvant chemoradiation represented the patient’s best oncologic outcomes. Therefore, he underwent radical penectomy and creation of a perineal urethrostomy. As of his 6-month follow-up, he showed no evidence of disease, had returned to his preoperative functional status, and was referred for chemoradiation.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Swartz MA, Porter MP, Lin DW, Weiss NS. Incidence of primary urethral carcinoma in the United States. Urology. 2006;68(6):1164-1168.
2. Dalbagni G, Zhang ZF, Lacombe L, Herr HW. Male urethral carcinoma: analysis of treatment outcome. Urology. 1999;53(6):1126-1132.
3. Gheiler EL, Tefilli MV, Tiguert R, de Oliveira JG, Pontes JE, Wood DP Jr. Management of primary urethral cancer. Urology. 1998;52(3):487-493.
4. Rabbani F. Prognostic factors in male urethral cancer. Cancer. 2011;117(11):2426-2434.
5. Cohen MS, Triaca V, Billmeyer B, et al. Coordinated chemoradiation therapy with genital preservation for the treatment of primary invasive carcinoma of the male urethra. J Urol. 2008;179(2):536-541; discussion 541.
6. Dayyani F, Pettaway CA, Kamat AM, Munsell MF, Sircar K, Pagliaro LC. Retrospective analysis of survival outcomes and the role of cisplatin-based chemotherapy in patients with urethral carcinomas referred to medical oncologists. Urol Oncol. 2013;31(7):1171-1177.
7. American Joint Committee on Cancer. AJCC cancer staging manual. 8th ed. https://cancerstaging.org/references-tools/deskreferences/Documents/AJCC%20Cancer%20Staging%20Form%20Supplement.pdf. Updated June 5, 2018. Accessed January 22, 2019.
8. Gakis G, Witjes JA, Compérat E, et al. European Association of Urology guidelines on primary urethral carcinoma. https://uroweb.org/wp-content/uploads/EAU-Guidelines-Primary-Urethral-Carcinoma-2016-1.pdf. Updated March 2015. Accessed January 22, 2019
9. National Comprehensive Cancer Network. Bladder Cancer. Version 1.2019. https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Updated December 20, 2018. Accessed January 17, 2019.
10. Dayyani F, Hoffman K, Eifel P, et al. Management of advanced primary urethral carcinomas. BJU Int. 2014;114(1):25-31.
11. Marshall VF. Radical excision of locally extensive carcinoma of the deep male urethra. J Urol. 1957;78(3):252-264.
12. Hsu FS, Su CH, Huang KH. A comprehensive review of US FDA-approved immune checkpoint inhibitors in urothelial carcinoma. J Immunol Res. 2017;2017:6940546.
The presentation of a fungating penile mass often indicates penile carcinoma, but providers should be aware of urethral carcinoma in the differential diagnosis.
The presentation of a fungating penile mass often indicates penile carcinoma, but providers should be aware of urethral carcinoma in the differential diagnosis.
Primary urethral carcinoma (PUC) is a rare but morbid disease, representing < 1% of all urologic malignancies.1 Up to one-third of male patients may present with nodal metastases.2-4 The overall survival (OS) for all male PUC is < 50% at 5 years and is lower still in patients with nodal involvement.4
Although surgical intervention, including radical resection, has been a mainstay in disease management, the presence of high-stage disease may warrant multimodal treatment with chemotherapy, radiation, and surgery. Recent series have described success with neoadjuvant and adjuvant chemoradiation, yet the optimal regimen remains unestablished.5,6 Although nodal disease is commonly encountered with proximal, high-stage tumors, this case exhibits a rare presentation of a distal fungating penile mass with low pathologic stage but rapid progression to nodal disease.
Case Presentation
A male veteran aged 77 years with a history of diabetes mellitus and stroke presented with obstructive urinary symptoms, gross hematuria, and 15-pound weight loss. Examination revealed a distal penile mass with purulent exudate at the meatus but no inguinal lymphadenopathy. Two fragments of this mass detached during office cystoscopy, and pathology revealed high-grade urothelial cell carcinoma (UCC). A magnetic resonance image of the pelvis with and without IV contrast revealed a 2.4-cm tumor in the glans penis with possible extension into the subcutaneous connective tissue of the penis and penile skin, without invasion of the corpora cavernosa/spongiosum or lymphadenopathy (Figure 1).
Prostatic urethral and random bladder biopsies, bilateral retrograde pyelograms, and selective ureteral washings revealed no abnormalities or signs of disease. Percutaneous biopsy of the inguinal node confirmed metastatic UCC. The patient underwent radical penectomy, creation of a perineal urethrostomy, and suprapubic cystostomy tube placement. Negative margins were confirmed on the urethral stump and corpus spongiosum. Final pathology revealed high-grade UCC with squamous differentiation on hematoxylin and eosin staining, arising from the penile urethra, invading the glans and corpus spongiosum, with no invasion of the corpus cavernosa (Figures 3 and 4).
Immunohistochemical stains were performed and strongly positive for cytokeratin 7 and p63. Final pathologic stage was described as pT2N1, with negative margins, indicating an American Joint Committee on Cancer classification of Stage III disease.7 The patient was referred postoperatively for adjuvant chemoradiation.
Discussion
The low incidence of PUC, coupled with a high morbidity/mortality rate, creates a difficult scenario in choosing the best oncologic management for this disease. National guidelines stratify treatment algorithms by stage and location of primary tumor, as these were found to be the 2 most important prognostic factors for men.1 The location of the primary tumor is most often in the bulbomembranous urethra, but up to one-third occur in the pendulous urethra.2
A recent review reported that UCC is the most common histologic subtype.4 When considering the differential diagnosis, a distal penile mass may represent a malignant penile lesion, such as squamous cell carcinoma, Buschke-Lowenstein tumor, Kaposi sarcoma, or precancerous lesions. Additional benign and infectious disorders include epidermoid and retention cysts, leukoplakia, balanitis xerotica obliterans, condyloma acuminatum, chancre/chancroid, lymphogranuloma venereum, granuloma inguinale, and tuberculosis. Clinical workup typically includes physical examination, cystourethroscopy and biopsy, chest X-ray, and pelvic/abdominal cross-sectional imaging.9,10 Magnetic resonance imaging of the abdomen and pelvis is ideal in identifying soft tissue structures and extension of tumor.
In male patients with PUC, nodal metastases are commonly seen at initial presentation in up to one-third of patients, while distant metastases may be present in up to 6% at presentation.2-4 When tumors arise from the anterior urethra, the primary lymphatic drainage is first to the inguinal lymph nodes, whereas posterior tumors drain to the pelvic lymph nodes. A multivariate analysis of men with PUC within the Surveillance, Epidemiology, and End Results database demonstrated an OS across all stages to be 46.2% and 29.3% at 5 and 10 years, respectively. Increased likelihood of death was predicted by advanced age, high grade/stage, systemic metastases, non-UCC histology, and the lack of surgery.4
Surgical intervention, including radical resection via penectomy, has been the mainstay in disease management and was first described by Marshall in 1957 for bulbar urethral cancer.11 In 1998, Gheiler and colleagues demonstrated that surgical resection alone yielded excellent outcomes in patients with low-stage disease with 89% of patients disease free at mean 42 months. This was in stark contrast to patients with advanced stage disease (T3 or N+) who exhibited a disease-free survival rate of 42% at the same follow-up interval and benefited from combined chemoradiation and surgical resection.3
In the presence of high-stage disease, multimodal therapy with chemotherapy, radiation, and/or surgery is warranted. A study in 2008 reviewed chemoradiation in which patients with PUC received a 5-week protocol of external beam radiotherapy to the genitals, inguinal/pelvic lymph nodes, plus an additional radiation bolus to the primary tumor.5 In the 18 patients reported, 15 had complete response to therapy, and only 4 patients required salvage surgical resection. The 7-year survival for the cohort was 72% with chemoradiation alone, with about half the population recurring or progressing at 7 years. However, all patients that avoided surgical resection went on to develop urethral strictures that required surgical therapy, 3 of which required complex reconstructive procedures.
To place this survival into context, the 1999 study by Dalbagni and colleagues reported a 5-year OS of 42% when surgical resection alone was performed in 40/46 men with PUC.2 Last, a large retrospective series of 44 patients reported mostly advanced-stage patients with PUC and analyzed patients treated with chemotherapy based on histologic pathology. The results demonstrated a 72% overall response rate to neoadjuvant chemotherapy, with a median OS of 32 months in patients undergoing chemotherapy vs 46 months in patients who underwent subsequent surgery. This study solidified that for patients with PUC involving the lymph nodes; optimal treatment includes neoadjuvant cisplatin-based chemotherapy followed by surgical resection.6
As medicine and oncologic therapies become more individualized, physicians are looking to new immunologic agents for systemic therapy. Immune checkpoint inhibitors were approved by the US Food and Drug Administration for UCC of the bladder in 2016.12 Unfortunately, due to the rarity of PUC and the recent development of immune checkpoint inhibitors, there have been no published reports of these or other immunotherapies in PUC. However, given the histologic similarity and pathogenesis, checkpoint inhibitors may have a future indication in the systemic management of this disease.
Conclusion
This patient’s PUC represents a rare presentation of a distal urethral carcinoma, T2-staged tumor, with rapid progression to nodal metastases. Additionally, the presentation of a fungating penile mass would usually indicate penile carcinoma, but providers should be aware of urethral carcinoma in the differential diagnosis. Notably, the patient was found to have progression to lymph node involvement during a mere 2-month period.
Recent case series have published encouraging results with neoadjuvant chemotherapy or chemoradiation.5,6 However, radical resection in men with T2 to T4 disease is associated with significantly higher cancer-specific survival. Given our concern of a loss to follow-up, we felt that radical resection of the primary tumor and adjuvant chemoradiation represented the patient’s best oncologic outcomes. Therefore, he underwent radical penectomy and creation of a perineal urethrostomy. As of his 6-month follow-up, he showed no evidence of disease, had returned to his preoperative functional status, and was referred for chemoradiation.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Primary urethral carcinoma (PUC) is a rare but morbid disease, representing < 1% of all urologic malignancies.1 Up to one-third of male patients may present with nodal metastases.2-4 The overall survival (OS) for all male PUC is < 50% at 5 years and is lower still in patients with nodal involvement.4
Although surgical intervention, including radical resection, has been a mainstay in disease management, the presence of high-stage disease may warrant multimodal treatment with chemotherapy, radiation, and surgery. Recent series have described success with neoadjuvant and adjuvant chemoradiation, yet the optimal regimen remains unestablished.5,6 Although nodal disease is commonly encountered with proximal, high-stage tumors, this case exhibits a rare presentation of a distal fungating penile mass with low pathologic stage but rapid progression to nodal disease.
Case Presentation
A male veteran aged 77 years with a history of diabetes mellitus and stroke presented with obstructive urinary symptoms, gross hematuria, and 15-pound weight loss. Examination revealed a distal penile mass with purulent exudate at the meatus but no inguinal lymphadenopathy. Two fragments of this mass detached during office cystoscopy, and pathology revealed high-grade urothelial cell carcinoma (UCC). A magnetic resonance image of the pelvis with and without IV contrast revealed a 2.4-cm tumor in the glans penis with possible extension into the subcutaneous connective tissue of the penis and penile skin, without invasion of the corpora cavernosa/spongiosum or lymphadenopathy (Figure 1).
Prostatic urethral and random bladder biopsies, bilateral retrograde pyelograms, and selective ureteral washings revealed no abnormalities or signs of disease. Percutaneous biopsy of the inguinal node confirmed metastatic UCC. The patient underwent radical penectomy, creation of a perineal urethrostomy, and suprapubic cystostomy tube placement. Negative margins were confirmed on the urethral stump and corpus spongiosum. Final pathology revealed high-grade UCC with squamous differentiation on hematoxylin and eosin staining, arising from the penile urethra, invading the glans and corpus spongiosum, with no invasion of the corpus cavernosa (Figures 3 and 4).
Immunohistochemical stains were performed and strongly positive for cytokeratin 7 and p63. Final pathologic stage was described as pT2N1, with negative margins, indicating an American Joint Committee on Cancer classification of Stage III disease.7 The patient was referred postoperatively for adjuvant chemoradiation.
Discussion
The low incidence of PUC, coupled with a high morbidity/mortality rate, creates a difficult scenario in choosing the best oncologic management for this disease. National guidelines stratify treatment algorithms by stage and location of primary tumor, as these were found to be the 2 most important prognostic factors for men.1 The location of the primary tumor is most often in the bulbomembranous urethra, but up to one-third occur in the pendulous urethra.2
A recent review reported that UCC is the most common histologic subtype.4 When considering the differential diagnosis, a distal penile mass may represent a malignant penile lesion, such as squamous cell carcinoma, Buschke-Lowenstein tumor, Kaposi sarcoma, or precancerous lesions. Additional benign and infectious disorders include epidermoid and retention cysts, leukoplakia, balanitis xerotica obliterans, condyloma acuminatum, chancre/chancroid, lymphogranuloma venereum, granuloma inguinale, and tuberculosis. Clinical workup typically includes physical examination, cystourethroscopy and biopsy, chest X-ray, and pelvic/abdominal cross-sectional imaging.9,10 Magnetic resonance imaging of the abdomen and pelvis is ideal in identifying soft tissue structures and extension of tumor.
In male patients with PUC, nodal metastases are commonly seen at initial presentation in up to one-third of patients, while distant metastases may be present in up to 6% at presentation.2-4 When tumors arise from the anterior urethra, the primary lymphatic drainage is first to the inguinal lymph nodes, whereas posterior tumors drain to the pelvic lymph nodes. A multivariate analysis of men with PUC within the Surveillance, Epidemiology, and End Results database demonstrated an OS across all stages to be 46.2% and 29.3% at 5 and 10 years, respectively. Increased likelihood of death was predicted by advanced age, high grade/stage, systemic metastases, non-UCC histology, and the lack of surgery.4
Surgical intervention, including radical resection via penectomy, has been the mainstay in disease management and was first described by Marshall in 1957 for bulbar urethral cancer.11 In 1998, Gheiler and colleagues demonstrated that surgical resection alone yielded excellent outcomes in patients with low-stage disease with 89% of patients disease free at mean 42 months. This was in stark contrast to patients with advanced stage disease (T3 or N+) who exhibited a disease-free survival rate of 42% at the same follow-up interval and benefited from combined chemoradiation and surgical resection.3
In the presence of high-stage disease, multimodal therapy with chemotherapy, radiation, and/or surgery is warranted. A study in 2008 reviewed chemoradiation in which patients with PUC received a 5-week protocol of external beam radiotherapy to the genitals, inguinal/pelvic lymph nodes, plus an additional radiation bolus to the primary tumor.5 In the 18 patients reported, 15 had complete response to therapy, and only 4 patients required salvage surgical resection. The 7-year survival for the cohort was 72% with chemoradiation alone, with about half the population recurring or progressing at 7 years. However, all patients that avoided surgical resection went on to develop urethral strictures that required surgical therapy, 3 of which required complex reconstructive procedures.
To place this survival into context, the 1999 study by Dalbagni and colleagues reported a 5-year OS of 42% when surgical resection alone was performed in 40/46 men with PUC.2 Last, a large retrospective series of 44 patients reported mostly advanced-stage patients with PUC and analyzed patients treated with chemotherapy based on histologic pathology. The results demonstrated a 72% overall response rate to neoadjuvant chemotherapy, with a median OS of 32 months in patients undergoing chemotherapy vs 46 months in patients who underwent subsequent surgery. This study solidified that for patients with PUC involving the lymph nodes; optimal treatment includes neoadjuvant cisplatin-based chemotherapy followed by surgical resection.6
As medicine and oncologic therapies become more individualized, physicians are looking to new immunologic agents for systemic therapy. Immune checkpoint inhibitors were approved by the US Food and Drug Administration for UCC of the bladder in 2016.12 Unfortunately, due to the rarity of PUC and the recent development of immune checkpoint inhibitors, there have been no published reports of these or other immunotherapies in PUC. However, given the histologic similarity and pathogenesis, checkpoint inhibitors may have a future indication in the systemic management of this disease.
Conclusion
This patient’s PUC represents a rare presentation of a distal urethral carcinoma, T2-staged tumor, with rapid progression to nodal metastases. Additionally, the presentation of a fungating penile mass would usually indicate penile carcinoma, but providers should be aware of urethral carcinoma in the differential diagnosis. Notably, the patient was found to have progression to lymph node involvement during a mere 2-month period.
Recent case series have published encouraging results with neoadjuvant chemotherapy or chemoradiation.5,6 However, radical resection in men with T2 to T4 disease is associated with significantly higher cancer-specific survival. Given our concern of a loss to follow-up, we felt that radical resection of the primary tumor and adjuvant chemoradiation represented the patient’s best oncologic outcomes. Therefore, he underwent radical penectomy and creation of a perineal urethrostomy. As of his 6-month follow-up, he showed no evidence of disease, had returned to his preoperative functional status, and was referred for chemoradiation.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Swartz MA, Porter MP, Lin DW, Weiss NS. Incidence of primary urethral carcinoma in the United States. Urology. 2006;68(6):1164-1168.
2. Dalbagni G, Zhang ZF, Lacombe L, Herr HW. Male urethral carcinoma: analysis of treatment outcome. Urology. 1999;53(6):1126-1132.
3. Gheiler EL, Tefilli MV, Tiguert R, de Oliveira JG, Pontes JE, Wood DP Jr. Management of primary urethral cancer. Urology. 1998;52(3):487-493.
4. Rabbani F. Prognostic factors in male urethral cancer. Cancer. 2011;117(11):2426-2434.
5. Cohen MS, Triaca V, Billmeyer B, et al. Coordinated chemoradiation therapy with genital preservation for the treatment of primary invasive carcinoma of the male urethra. J Urol. 2008;179(2):536-541; discussion 541.
6. Dayyani F, Pettaway CA, Kamat AM, Munsell MF, Sircar K, Pagliaro LC. Retrospective analysis of survival outcomes and the role of cisplatin-based chemotherapy in patients with urethral carcinomas referred to medical oncologists. Urol Oncol. 2013;31(7):1171-1177.
7. American Joint Committee on Cancer. AJCC cancer staging manual. 8th ed. https://cancerstaging.org/references-tools/deskreferences/Documents/AJCC%20Cancer%20Staging%20Form%20Supplement.pdf. Updated June 5, 2018. Accessed January 22, 2019.
8. Gakis G, Witjes JA, Compérat E, et al. European Association of Urology guidelines on primary urethral carcinoma. https://uroweb.org/wp-content/uploads/EAU-Guidelines-Primary-Urethral-Carcinoma-2016-1.pdf. Updated March 2015. Accessed January 22, 2019
9. National Comprehensive Cancer Network. Bladder Cancer. Version 1.2019. https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Updated December 20, 2018. Accessed January 17, 2019.
10. Dayyani F, Hoffman K, Eifel P, et al. Management of advanced primary urethral carcinomas. BJU Int. 2014;114(1):25-31.
11. Marshall VF. Radical excision of locally extensive carcinoma of the deep male urethra. J Urol. 1957;78(3):252-264.
12. Hsu FS, Su CH, Huang KH. A comprehensive review of US FDA-approved immune checkpoint inhibitors in urothelial carcinoma. J Immunol Res. 2017;2017:6940546.
1. Swartz MA, Porter MP, Lin DW, Weiss NS. Incidence of primary urethral carcinoma in the United States. Urology. 2006;68(6):1164-1168.
2. Dalbagni G, Zhang ZF, Lacombe L, Herr HW. Male urethral carcinoma: analysis of treatment outcome. Urology. 1999;53(6):1126-1132.
3. Gheiler EL, Tefilli MV, Tiguert R, de Oliveira JG, Pontes JE, Wood DP Jr. Management of primary urethral cancer. Urology. 1998;52(3):487-493.
4. Rabbani F. Prognostic factors in male urethral cancer. Cancer. 2011;117(11):2426-2434.
5. Cohen MS, Triaca V, Billmeyer B, et al. Coordinated chemoradiation therapy with genital preservation for the treatment of primary invasive carcinoma of the male urethra. J Urol. 2008;179(2):536-541; discussion 541.
6. Dayyani F, Pettaway CA, Kamat AM, Munsell MF, Sircar K, Pagliaro LC. Retrospective analysis of survival outcomes and the role of cisplatin-based chemotherapy in patients with urethral carcinomas referred to medical oncologists. Urol Oncol. 2013;31(7):1171-1177.
7. American Joint Committee on Cancer. AJCC cancer staging manual. 8th ed. https://cancerstaging.org/references-tools/deskreferences/Documents/AJCC%20Cancer%20Staging%20Form%20Supplement.pdf. Updated June 5, 2018. Accessed January 22, 2019.
8. Gakis G, Witjes JA, Compérat E, et al. European Association of Urology guidelines on primary urethral carcinoma. https://uroweb.org/wp-content/uploads/EAU-Guidelines-Primary-Urethral-Carcinoma-2016-1.pdf. Updated March 2015. Accessed January 22, 2019
9. National Comprehensive Cancer Network. Bladder Cancer. Version 1.2019. https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Updated December 20, 2018. Accessed January 17, 2019.
10. Dayyani F, Hoffman K, Eifel P, et al. Management of advanced primary urethral carcinomas. BJU Int. 2014;114(1):25-31.
11. Marshall VF. Radical excision of locally extensive carcinoma of the deep male urethra. J Urol. 1957;78(3):252-264.
12. Hsu FS, Su CH, Huang KH. A comprehensive review of US FDA-approved immune checkpoint inhibitors in urothelial carcinoma. J Immunol Res. 2017;2017:6940546.