User login
Panel Endorses Surveillance for Low-Risk Prostate Cancer
An independent panel convened by the U.S. National Institutes of Health has endorsed the use of active surveillance and delay of treatment for men with localized, low-risk prostate cancer.
"Our panel found that many men with localized, low-risk prostate cancer should be closely monitored, permitting their treatment to be delayed until disease progression warrants it. Some of the men affected by prostate cancer will benefit from immediate treatment and others will benefit from observation," panel and conference chairperson Dr. Patricia A. Ganz said in a telebriefing.

However, monitoring or observational strategies, sometimes referred to as "watchful waiting" or "active surveillance," have not been uniformly studied, and available data do not yet point to clear follow-up protocols, said Dr. Ganz, director of the division of cancer prevention and control research at the Jonsson Comprehensive Cancer Center, University of California, Los Angeles.
The panel identified the combination of a prostate-specific antigen (PSA) level less than 10 ng/dL and a Gleason score of 6 or lower as the emerging consensus definition of "low-risk" disease. "Using this definition, we estimate that more than 100,000 men diagnosed with prostate cancer each year in the United States would be candidates for active monitoring rather than immediate treatment," she said.
Additional research will be needed to determine the point at which treatment might ultimately be needed for men who are being closely monitored. The panel recommended that such studies receiving federal funding should not be done at a single site, but rather should be multicenter studies with large patient populations.
The consensus about active surveillance came from increasing evidence that outcomes for men with low-risk disease are not better for those who undergo surgery or radiation therapy. Among the evidence considered was the newly reported findings of the Prostate Cancer Intervention Versus Observation Trial (PIVOT), the only randomized controlled trial conducted on men identified via PSA screening that compared watchful waiting with radical prostatectomy.
With a median follow-up of 10 years, there were no statistically significant differences in prostate cancer mortality or all-cause mortality. "Supporting data from additional cohort studies give us confidence that the risk of death is minimal in a low risk population," the panel wrote.
According to the document, only about 10% of men eligible for observational strategies choose this approach. The reasons for this are probably due both to physician communication that favors treatment and to patient expectations. "When men are given a diagnosis of cancer, it’s very difficult to decline the standard therapy for this disease, which is either surgery or radiation therapy," Dr. Ganz noted.
For this reason, the panel also endorsed consideration of a name change to remove the anxiety-provoking term cancer to describe this low-risk condition, much as was done with cervical intraepithelial neoplasia for early-stage cervical neoplasms and ductal carcinoma in situ for lower-risk breast lesions.
"Some of the men affected by prostate cancer will benefit from immediate treatment and others will benefit from observation."
They also discussed ways in which urologists and primary care physicians ordering PSA tests could be educated to counsel patients about the potential benefits of active surveillance and delaying treatment, Dr. Ganz said.
"Anything we can do to bring this into the consultation room so that the patient feels comfortable raising this issue with his physician ... [Stakeholders] now have an NIH-vetted document that describes this as a reasonable approach to the management of prostate cancer and for those reasons it can be very powerful," she said.
The 14-member state-of-the-science panel included experts in the fields of cancer prevention and control, urology, pathology, epidemiology, genetics, transplantation, bioethics, economics, health services research, shared decision-making, health communication, and community engagement.
Panel members were compensated for travel to the conference, but were not otherwise paid and have no additional conflicts of interest. The statement does not constitute federal policy.
The draft statement is posted online, and the final version will be posted by mid to late January 2012.
An independent panel convened by the U.S. National Institutes of Health has endorsed the use of active surveillance and delay of treatment for men with localized, low-risk prostate cancer.
"Our panel found that many men with localized, low-risk prostate cancer should be closely monitored, permitting their treatment to be delayed until disease progression warrants it. Some of the men affected by prostate cancer will benefit from immediate treatment and others will benefit from observation," panel and conference chairperson Dr. Patricia A. Ganz said in a telebriefing.
However, monitoring or observational strategies, sometimes referred to as "watchful waiting" or "active surveillance," have not been uniformly studied, and available data do not yet point to clear follow-up protocols, said Dr. Ganz, director of the division of cancer prevention and control research at the Jonsson Comprehensive Cancer Center, University of California, Los Angeles.
The panel identified the combination of a prostate-specific antigen (PSA) level less than 10 ng/dL and a Gleason score of 6 or lower as the emerging consensus definition of "low-risk" disease. "Using this definition, we estimate that more than 100,000 men diagnosed with prostate cancer each year in the United States would be candidates for active monitoring rather than immediate treatment," she said.
Additional research will be needed to determine the point at which treatment might ultimately be needed for men who are being closely monitored. The panel recommended that such studies receiving federal funding should not be done at a single site, but rather should be multicenter studies with large patient populations.
The consensus about active surveillance came from increasing evidence that outcomes for men with low-risk disease are not better for those who undergo surgery or radiation therapy. Among the evidence considered was the newly reported findings of the Prostate Cancer Intervention Versus Observation Trial (PIVOT), the only randomized controlled trial conducted on men identified via PSA screening that compared watchful waiting with radical prostatectomy.
With a median follow-up of 10 years, there were no statistically significant differences in prostate cancer mortality or all-cause mortality. "Supporting data from additional cohort studies give us confidence that the risk of death is minimal in a low risk population," the panel wrote.
According to the document, only about 10% of men eligible for observational strategies choose this approach. The reasons for this are probably due both to physician communication that favors treatment and to patient expectations. "When men are given a diagnosis of cancer, it’s very difficult to decline the standard therapy for this disease, which is either surgery or radiation therapy," Dr. Ganz noted.
For this reason, the panel also endorsed consideration of a name change to remove the anxiety-provoking term cancer to describe this low-risk condition, much as was done with cervical intraepithelial neoplasia for early-stage cervical neoplasms and ductal carcinoma in situ for lower-risk breast lesions.
"Some of the men affected by prostate cancer will benefit from immediate treatment and others will benefit from observation."
They also discussed ways in which urologists and primary care physicians ordering PSA tests could be educated to counsel patients about the potential benefits of active surveillance and delaying treatment, Dr. Ganz said.
"Anything we can do to bring this into the consultation room so that the patient feels comfortable raising this issue with his physician ... [Stakeholders] now have an NIH-vetted document that describes this as a reasonable approach to the management of prostate cancer and for those reasons it can be very powerful," she said.
The 14-member state-of-the-science panel included experts in the fields of cancer prevention and control, urology, pathology, epidemiology, genetics, transplantation, bioethics, economics, health services research, shared decision-making, health communication, and community engagement.
Panel members were compensated for travel to the conference, but were not otherwise paid and have no additional conflicts of interest. The statement does not constitute federal policy.
The draft statement is posted online, and the final version will be posted by mid to late January 2012.
An independent panel convened by the U.S. National Institutes of Health has endorsed the use of active surveillance and delay of treatment for men with localized, low-risk prostate cancer.
"Our panel found that many men with localized, low-risk prostate cancer should be closely monitored, permitting their treatment to be delayed until disease progression warrants it. Some of the men affected by prostate cancer will benefit from immediate treatment and others will benefit from observation," panel and conference chairperson Dr. Patricia A. Ganz said in a telebriefing.
However, monitoring or observational strategies, sometimes referred to as "watchful waiting" or "active surveillance," have not been uniformly studied, and available data do not yet point to clear follow-up protocols, said Dr. Ganz, director of the division of cancer prevention and control research at the Jonsson Comprehensive Cancer Center, University of California, Los Angeles.
The panel identified the combination of a prostate-specific antigen (PSA) level less than 10 ng/dL and a Gleason score of 6 or lower as the emerging consensus definition of "low-risk" disease. "Using this definition, we estimate that more than 100,000 men diagnosed with prostate cancer each year in the United States would be candidates for active monitoring rather than immediate treatment," she said.
Additional research will be needed to determine the point at which treatment might ultimately be needed for men who are being closely monitored. The panel recommended that such studies receiving federal funding should not be done at a single site, but rather should be multicenter studies with large patient populations.
The consensus about active surveillance came from increasing evidence that outcomes for men with low-risk disease are not better for those who undergo surgery or radiation therapy. Among the evidence considered was the newly reported findings of the Prostate Cancer Intervention Versus Observation Trial (PIVOT), the only randomized controlled trial conducted on men identified via PSA screening that compared watchful waiting with radical prostatectomy.
With a median follow-up of 10 years, there were no statistically significant differences in prostate cancer mortality or all-cause mortality. "Supporting data from additional cohort studies give us confidence that the risk of death is minimal in a low risk population," the panel wrote.
According to the document, only about 10% of men eligible for observational strategies choose this approach. The reasons for this are probably due both to physician communication that favors treatment and to patient expectations. "When men are given a diagnosis of cancer, it’s very difficult to decline the standard therapy for this disease, which is either surgery or radiation therapy," Dr. Ganz noted.
For this reason, the panel also endorsed consideration of a name change to remove the anxiety-provoking term cancer to describe this low-risk condition, much as was done with cervical intraepithelial neoplasia for early-stage cervical neoplasms and ductal carcinoma in situ for lower-risk breast lesions.
"Some of the men affected by prostate cancer will benefit from immediate treatment and others will benefit from observation."
They also discussed ways in which urologists and primary care physicians ordering PSA tests could be educated to counsel patients about the potential benefits of active surveillance and delaying treatment, Dr. Ganz said.
"Anything we can do to bring this into the consultation room so that the patient feels comfortable raising this issue with his physician ... [Stakeholders] now have an NIH-vetted document that describes this as a reasonable approach to the management of prostate cancer and for those reasons it can be very powerful," she said.
The 14-member state-of-the-science panel included experts in the fields of cancer prevention and control, urology, pathology, epidemiology, genetics, transplantation, bioethics, economics, health services research, shared decision-making, health communication, and community engagement.
Panel members were compensated for travel to the conference, but were not otherwise paid and have no additional conflicts of interest. The statement does not constitute federal policy.
The draft statement is posted online, and the final version will be posted by mid to late January 2012.
Major Finding: Many men with localized, low-risk prostate cancer should be closely monitored, permitting their treatment to be delayed until disease progression warrants it.
Data Source: Newly reported findings of the Prostate Cancer Intervention Versus Observation Trial (PIVOT).
Disclosures: Panel members were compensated for travel to the conference, but were not otherwise paid and have no additional conflicts of interest. The statement does not constitute federal policy.
Valproate Exposure Associated With Autism, Lower IQ
BALTIMORE – In utero exposure to valproate appears to increase the risk of significant adverse effects on fetal brain development that persist into childhood.
In two separate studies, children whose mothers took valproate during pregnancy had a higher risk for lower IQ and other cognitive deficiencies, as well as autism and other disorders along the autistic spectrum. "All women with epilepsy of childbearing potential should be informed of the risks. I feel that valproate should not be a first choice antiepileptic drug in women of childbearing potential," Dr. Kimford J. Meador, director of the Emory Epilepsy Center and professor of neurology at Emory University, Atlanta, said in an interview.

The cognition data come from the NEAD (Neurodevelopmental Effects of Antiepileptic Drugs) study, a prospective observational study that enrolled pregnant women who were using any of several antiepileptic-drug monotherapies from October 1999 through February 2004 in 25 epilepsy centers in the United States and the United Kingdom. Dr. Meador and his colleagues previously published interim NEAD data showing impaired cognitive function in 309 offspring at 3 years of age (N. Engl. J. Med. 2009;360:1597-1605).
In June 2011, the Food and Drug Administration issued a safety alert about the increased risk of impaired cognitive development in children exposed to valproate products in utero.
The investigators have now followed the children in the NEAD study up to 6 years of age. The primary study outcome is IQ at age 6 years as measured by the Differential Ability Scales (DAS), and a second analysis measures verbal and nonverbal cluster scores from the DAS.
In a multivariate analysis of the intent-to-treat sample of 310 children, IQ was lower among children exposed in utero to valproate, compared with children of mothers who took other antiepileptic drugs (AEDs) during pregnancy. Adjusted mean IQs were 105 for carbamazepine, 108 for lamotrigine, and 106 for phenytoin, compared with 99 in the children of mothers who took valproate. The difference between valproate and each of the other three medication groups was significant (P = .002, compared with all three groups combined).

Overall, maternal IQ was also strongly associated with their children’s IQ at 6 years (P less than .001), and both lower maternal age and lower gestational age were associated with lower IQ in the child. (P = .04 and .03, respectively). However, maternal IQ was not associated with child IQ among the offspring of mothers who took valproate, whereas it was strongly associated with child IQ for the other three AEDs, Dr. Meador reported.
The verbal cluster score was less than the nonverbal cluster score across all AEDs combined (P less than .0001) and for carbamazepine (P less than .028), lamotrigine (P less than .003), and valproate (P less than .005) individually, he said.
The autism findings were from a population-based cohort study of 655,691 children born between 1996 and 2006 to 428,431 mothers who were identified from the Danish Civil Registration System, a nationwide registry in Denmark. Information on AED prescriptions filled 30 days prior to and during pregnancy was obtained from the Danish National Prescription Registry, and children diagnosed with autism spectrum disorder and childhood autism were identified from the Danish Psychiatric Register, said Dr. Jakob Christensen of the department of neurology at Aarhus (Denmark) University Hospital.
The relative risk of autism spectrum disorder following valproate exposure during pregnancy was more than doubled (2.6), compared with children who were not exposed to antiepileptic medication during pregnancy. The relative risk of childhood autism was even higher, at 4.8. The relative risk of autism spectrum disorder was 2.6 following valproate monotherapy exposure and 2.5 following polytherapy that included valproate. The relative risk of childhood autism was 4.1 following valproate monotherapy and 6.8 following polytherapy that included valproate, Dr. Christensen reported at the meeting.
According to Dr. Meador, "There is a subgroup of women with primary generalized epilepsy who may only respond to valproate. I recommend trying the other AEDs first even in women with primary generalized epilepsy. If the woman is ultimately only controlled by valproate and decides to consider pregnancy, then the dose should be kept as low as possible."
The NEAD study was funded by the National Institute of Neurological Disorders and Stroke. Dr. Meador has worked as a consultant for the Epilepsy Study Consortium that receives money from NeuroPace, Upsher-Smith Laboratories, and Vivus Pharmaceuticals, but these funds are paid to Emory University and not to him directly. Fees for his consultant work for GlaxoSmithKline, Johnson & Johnson (Ortho McNeil), Medtronics, and UCB Pharma have gone to a charity of the company’s choice. In 2010, he received travel support from Sanofi-Aventis, a manufacturer of valproate. The Danish study was funded by the Danish Epilepsy Society. Dr. Christensen disclosed that he has received honoraria from UCB Pharma and Eisai.
BALTIMORE – In utero exposure to valproate appears to increase the risk of significant adverse effects on fetal brain development that persist into childhood.
In two separate studies, children whose mothers took valproate during pregnancy had a higher risk for lower IQ and other cognitive deficiencies, as well as autism and other disorders along the autistic spectrum. "All women with epilepsy of childbearing potential should be informed of the risks. I feel that valproate should not be a first choice antiepileptic drug in women of childbearing potential," Dr. Kimford J. Meador, director of the Emory Epilepsy Center and professor of neurology at Emory University, Atlanta, said in an interview.
The cognition data come from the NEAD (Neurodevelopmental Effects of Antiepileptic Drugs) study, a prospective observational study that enrolled pregnant women who were using any of several antiepileptic-drug monotherapies from October 1999 through February 2004 in 25 epilepsy centers in the United States and the United Kingdom. Dr. Meador and his colleagues previously published interim NEAD data showing impaired cognitive function in 309 offspring at 3 years of age (N. Engl. J. Med. 2009;360:1597-1605).
In June 2011, the Food and Drug Administration issued a safety alert about the increased risk of impaired cognitive development in children exposed to valproate products in utero.
The investigators have now followed the children in the NEAD study up to 6 years of age. The primary study outcome is IQ at age 6 years as measured by the Differential Ability Scales (DAS), and a second analysis measures verbal and nonverbal cluster scores from the DAS.
In a multivariate analysis of the intent-to-treat sample of 310 children, IQ was lower among children exposed in utero to valproate, compared with children of mothers who took other antiepileptic drugs (AEDs) during pregnancy. Adjusted mean IQs were 105 for carbamazepine, 108 for lamotrigine, and 106 for phenytoin, compared with 99 in the children of mothers who took valproate. The difference between valproate and each of the other three medication groups was significant (P = .002, compared with all three groups combined).
Overall, maternal IQ was also strongly associated with their children’s IQ at 6 years (P less than .001), and both lower maternal age and lower gestational age were associated with lower IQ in the child. (P = .04 and .03, respectively). However, maternal IQ was not associated with child IQ among the offspring of mothers who took valproate, whereas it was strongly associated with child IQ for the other three AEDs, Dr. Meador reported.
The verbal cluster score was less than the nonverbal cluster score across all AEDs combined (P less than .0001) and for carbamazepine (P less than .028), lamotrigine (P less than .003), and valproate (P less than .005) individually, he said.
The autism findings were from a population-based cohort study of 655,691 children born between 1996 and 2006 to 428,431 mothers who were identified from the Danish Civil Registration System, a nationwide registry in Denmark. Information on AED prescriptions filled 30 days prior to and during pregnancy was obtained from the Danish National Prescription Registry, and children diagnosed with autism spectrum disorder and childhood autism were identified from the Danish Psychiatric Register, said Dr. Jakob Christensen of the department of neurology at Aarhus (Denmark) University Hospital.
The relative risk of autism spectrum disorder following valproate exposure during pregnancy was more than doubled (2.6), compared with children who were not exposed to antiepileptic medication during pregnancy. The relative risk of childhood autism was even higher, at 4.8. The relative risk of autism spectrum disorder was 2.6 following valproate monotherapy exposure and 2.5 following polytherapy that included valproate. The relative risk of childhood autism was 4.1 following valproate monotherapy and 6.8 following polytherapy that included valproate, Dr. Christensen reported at the meeting.
According to Dr. Meador, "There is a subgroup of women with primary generalized epilepsy who may only respond to valproate. I recommend trying the other AEDs first even in women with primary generalized epilepsy. If the woman is ultimately only controlled by valproate and decides to consider pregnancy, then the dose should be kept as low as possible."
The NEAD study was funded by the National Institute of Neurological Disorders and Stroke. Dr. Meador has worked as a consultant for the Epilepsy Study Consortium that receives money from NeuroPace, Upsher-Smith Laboratories, and Vivus Pharmaceuticals, but these funds are paid to Emory University and not to him directly. Fees for his consultant work for GlaxoSmithKline, Johnson & Johnson (Ortho McNeil), Medtronics, and UCB Pharma have gone to a charity of the company’s choice. In 2010, he received travel support from Sanofi-Aventis, a manufacturer of valproate. The Danish study was funded by the Danish Epilepsy Society. Dr. Christensen disclosed that he has received honoraria from UCB Pharma and Eisai.
BALTIMORE – In utero exposure to valproate appears to increase the risk of significant adverse effects on fetal brain development that persist into childhood.
In two separate studies, children whose mothers took valproate during pregnancy had a higher risk for lower IQ and other cognitive deficiencies, as well as autism and other disorders along the autistic spectrum. "All women with epilepsy of childbearing potential should be informed of the risks. I feel that valproate should not be a first choice antiepileptic drug in women of childbearing potential," Dr. Kimford J. Meador, director of the Emory Epilepsy Center and professor of neurology at Emory University, Atlanta, said in an interview.
The cognition data come from the NEAD (Neurodevelopmental Effects of Antiepileptic Drugs) study, a prospective observational study that enrolled pregnant women who were using any of several antiepileptic-drug monotherapies from October 1999 through February 2004 in 25 epilepsy centers in the United States and the United Kingdom. Dr. Meador and his colleagues previously published interim NEAD data showing impaired cognitive function in 309 offspring at 3 years of age (N. Engl. J. Med. 2009;360:1597-1605).
In June 2011, the Food and Drug Administration issued a safety alert about the increased risk of impaired cognitive development in children exposed to valproate products in utero.
The investigators have now followed the children in the NEAD study up to 6 years of age. The primary study outcome is IQ at age 6 years as measured by the Differential Ability Scales (DAS), and a second analysis measures verbal and nonverbal cluster scores from the DAS.
In a multivariate analysis of the intent-to-treat sample of 310 children, IQ was lower among children exposed in utero to valproate, compared with children of mothers who took other antiepileptic drugs (AEDs) during pregnancy. Adjusted mean IQs were 105 for carbamazepine, 108 for lamotrigine, and 106 for phenytoin, compared with 99 in the children of mothers who took valproate. The difference between valproate and each of the other three medication groups was significant (P = .002, compared with all three groups combined).
Overall, maternal IQ was also strongly associated with their children’s IQ at 6 years (P less than .001), and both lower maternal age and lower gestational age were associated with lower IQ in the child. (P = .04 and .03, respectively). However, maternal IQ was not associated with child IQ among the offspring of mothers who took valproate, whereas it was strongly associated with child IQ for the other three AEDs, Dr. Meador reported.
The verbal cluster score was less than the nonverbal cluster score across all AEDs combined (P less than .0001) and for carbamazepine (P less than .028), lamotrigine (P less than .003), and valproate (P less than .005) individually, he said.
The autism findings were from a population-based cohort study of 655,691 children born between 1996 and 2006 to 428,431 mothers who were identified from the Danish Civil Registration System, a nationwide registry in Denmark. Information on AED prescriptions filled 30 days prior to and during pregnancy was obtained from the Danish National Prescription Registry, and children diagnosed with autism spectrum disorder and childhood autism were identified from the Danish Psychiatric Register, said Dr. Jakob Christensen of the department of neurology at Aarhus (Denmark) University Hospital.
The relative risk of autism spectrum disorder following valproate exposure during pregnancy was more than doubled (2.6), compared with children who were not exposed to antiepileptic medication during pregnancy. The relative risk of childhood autism was even higher, at 4.8. The relative risk of autism spectrum disorder was 2.6 following valproate monotherapy exposure and 2.5 following polytherapy that included valproate. The relative risk of childhood autism was 4.1 following valproate monotherapy and 6.8 following polytherapy that included valproate, Dr. Christensen reported at the meeting.
According to Dr. Meador, "There is a subgroup of women with primary generalized epilepsy who may only respond to valproate. I recommend trying the other AEDs first even in women with primary generalized epilepsy. If the woman is ultimately only controlled by valproate and decides to consider pregnancy, then the dose should be kept as low as possible."
The NEAD study was funded by the National Institute of Neurological Disorders and Stroke. Dr. Meador has worked as a consultant for the Epilepsy Study Consortium that receives money from NeuroPace, Upsher-Smith Laboratories, and Vivus Pharmaceuticals, but these funds are paid to Emory University and not to him directly. Fees for his consultant work for GlaxoSmithKline, Johnson & Johnson (Ortho McNeil), Medtronics, and UCB Pharma have gone to a charity of the company’s choice. In 2010, he received travel support from Sanofi-Aventis, a manufacturer of valproate. The Danish study was funded by the Danish Epilepsy Society. Dr. Christensen disclosed that he has received honoraria from UCB Pharma and Eisai.
FROM THE ANNUAL MEETING OF THE AMERICAN EPILEPSY SOCIETY
Implanted EEG Device Predicts Seizures in Early Study
BALTIMORE – A novel implantable device has demonstrated potential for predicting seizure onset in a preliminary analysis of 15 adult patients with medically refractory complex partial seizures.
The ambulatory intracranial EEG (iEEG) device, NeuroVista’s Seizure Advisory System (SAS), consists of electrodes that are implanted between the skull and the brain surface that continuously record electrical activity. The electrodes are connected by wires to a data storage device implanted in the chest. Signals are transmitted wirelessly to an external handheld device that processes the data and transmits visual and audible signals to the patient. A blue light signifies a low likelihood of seizures, white indicates medium susceptibility, and red alerts to a high likelihood of impending seizure.

"This is something we’ve never been able to do before, to predict when a seizure might happen, which potentially gives the opportunity for patients to make themselves safe, or possibly even take an acute-acting medication long-term. The uncertainty of when a seizure might occur is the most disabling part of seizures for most people. So to be able to have this sort of warning, to be able to structure day-to-day activities around [seizures] potentially, will mean a lot to people being able to control their lives," said Dr. Mark Cook, chair of medicine and director of neurosciences at St. Vincent’s Hospital, Melbourne. Dr. Cook reported the results of the study at the annual meeting of the American Epilepsy Society.
The 15 study subjects were all 18 years and older, had disabling partial and/or secondarily generalized partial seizures, and had failed therapeutic treatment with a minimum of two antiepileptic drugs. They were implanted at one of three clinical centers in Australia: Austin Health, The Royal Melbourne Hospital, and St. Vincent’s Hospital. At baseline, the patients reported experiencing 2-12 disabling partial onset seizures per month. Following intracranial implantation of the device, iEEG was collected to configure a patient-specific algorithm that identified periods of low, moderate, and high seizure likelihood.
In the data collection phase of the study, the estimated performance for the high and low likelihood advisories had to be statistically superior to a time-matched chance predictor with P value of .05 or less, and the high likelihood advisory sensitivity could not be statistically inferior to 65%. Patients who met those criteria entered an Advisory Phase where their system was configured to provide visual and audible advisories that indicate seizure likelihood.
In some instances, seizure incidence as recorded by the device was dramatically different from what the patient had reported. In one patient who initially reported having 7 seizures per month, the SAS recorded 104 per month, based on data covering 70 days. Another patient who reported 6 seizures per month actually had 80 per month, based on 126 days of data. Other patients overestimated their seizures, with one patient who reported having 3 seizures per month experiencing just 0.6 seizures per month, based on 209 days of data.
"Some patients overestimate the number of their seizures they’re having, but the amount by which [others] underestimate their seizures is very dramatic, sometimes by a factor of 10," Dr. Cook commented.
In the data collection (training) phase of the study, the sensitivity of the red advisory among 11 patients who completed the phase ranged from 0.65 to 1.00, with 10 of those patients meeting the performance criteria for the seizure advisories. Among 6 patients who have completed the subsequent 4-month advisory (prospective) study phase, red advisory sensitivity ranged from 0.56 to 1.00. In both study phases, there were no seizures during the low advisory (100% negative predictive value). The other four patients are still in the data collection phase.
The safety profile is consistent with published literature for strip electrodes and subclavicular implants, he said.
NeuroVista is continuing to study the clinical utility of the SAS, a company spokesman said.
Dr. Cook stated that he had no financial disclosures. The study was funded by NeuroVista, and four of the coinvestigators are company employees.
BALTIMORE – A novel implantable device has demonstrated potential for predicting seizure onset in a preliminary analysis of 15 adult patients with medically refractory complex partial seizures.
The ambulatory intracranial EEG (iEEG) device, NeuroVista’s Seizure Advisory System (SAS), consists of electrodes that are implanted between the skull and the brain surface that continuously record electrical activity. The electrodes are connected by wires to a data storage device implanted in the chest. Signals are transmitted wirelessly to an external handheld device that processes the data and transmits visual and audible signals to the patient. A blue light signifies a low likelihood of seizures, white indicates medium susceptibility, and red alerts to a high likelihood of impending seizure.
"This is something we’ve never been able to do before, to predict when a seizure might happen, which potentially gives the opportunity for patients to make themselves safe, or possibly even take an acute-acting medication long-term. The uncertainty of when a seizure might occur is the most disabling part of seizures for most people. So to be able to have this sort of warning, to be able to structure day-to-day activities around [seizures] potentially, will mean a lot to people being able to control their lives," said Dr. Mark Cook, chair of medicine and director of neurosciences at St. Vincent’s Hospital, Melbourne. Dr. Cook reported the results of the study at the annual meeting of the American Epilepsy Society.
The 15 study subjects were all 18 years and older, had disabling partial and/or secondarily generalized partial seizures, and had failed therapeutic treatment with a minimum of two antiepileptic drugs. They were implanted at one of three clinical centers in Australia: Austin Health, The Royal Melbourne Hospital, and St. Vincent’s Hospital. At baseline, the patients reported experiencing 2-12 disabling partial onset seizures per month. Following intracranial implantation of the device, iEEG was collected to configure a patient-specific algorithm that identified periods of low, moderate, and high seizure likelihood.
In the data collection phase of the study, the estimated performance for the high and low likelihood advisories had to be statistically superior to a time-matched chance predictor with P value of .05 or less, and the high likelihood advisory sensitivity could not be statistically inferior to 65%. Patients who met those criteria entered an Advisory Phase where their system was configured to provide visual and audible advisories that indicate seizure likelihood.
In some instances, seizure incidence as recorded by the device was dramatically different from what the patient had reported. In one patient who initially reported having 7 seizures per month, the SAS recorded 104 per month, based on data covering 70 days. Another patient who reported 6 seizures per month actually had 80 per month, based on 126 days of data. Other patients overestimated their seizures, with one patient who reported having 3 seizures per month experiencing just 0.6 seizures per month, based on 209 days of data.
"Some patients overestimate the number of their seizures they’re having, but the amount by which [others] underestimate their seizures is very dramatic, sometimes by a factor of 10," Dr. Cook commented.
In the data collection (training) phase of the study, the sensitivity of the red advisory among 11 patients who completed the phase ranged from 0.65 to 1.00, with 10 of those patients meeting the performance criteria for the seizure advisories. Among 6 patients who have completed the subsequent 4-month advisory (prospective) study phase, red advisory sensitivity ranged from 0.56 to 1.00. In both study phases, there were no seizures during the low advisory (100% negative predictive value). The other four patients are still in the data collection phase.
The safety profile is consistent with published literature for strip electrodes and subclavicular implants, he said.
NeuroVista is continuing to study the clinical utility of the SAS, a company spokesman said.
Dr. Cook stated that he had no financial disclosures. The study was funded by NeuroVista, and four of the coinvestigators are company employees.
BALTIMORE – A novel implantable device has demonstrated potential for predicting seizure onset in a preliminary analysis of 15 adult patients with medically refractory complex partial seizures.
The ambulatory intracranial EEG (iEEG) device, NeuroVista’s Seizure Advisory System (SAS), consists of electrodes that are implanted between the skull and the brain surface that continuously record electrical activity. The electrodes are connected by wires to a data storage device implanted in the chest. Signals are transmitted wirelessly to an external handheld device that processes the data and transmits visual and audible signals to the patient. A blue light signifies a low likelihood of seizures, white indicates medium susceptibility, and red alerts to a high likelihood of impending seizure.
"This is something we’ve never been able to do before, to predict when a seizure might happen, which potentially gives the opportunity for patients to make themselves safe, or possibly even take an acute-acting medication long-term. The uncertainty of when a seizure might occur is the most disabling part of seizures for most people. So to be able to have this sort of warning, to be able to structure day-to-day activities around [seizures] potentially, will mean a lot to people being able to control their lives," said Dr. Mark Cook, chair of medicine and director of neurosciences at St. Vincent’s Hospital, Melbourne. Dr. Cook reported the results of the study at the annual meeting of the American Epilepsy Society.
The 15 study subjects were all 18 years and older, had disabling partial and/or secondarily generalized partial seizures, and had failed therapeutic treatment with a minimum of two antiepileptic drugs. They were implanted at one of three clinical centers in Australia: Austin Health, The Royal Melbourne Hospital, and St. Vincent’s Hospital. At baseline, the patients reported experiencing 2-12 disabling partial onset seizures per month. Following intracranial implantation of the device, iEEG was collected to configure a patient-specific algorithm that identified periods of low, moderate, and high seizure likelihood.
In the data collection phase of the study, the estimated performance for the high and low likelihood advisories had to be statistically superior to a time-matched chance predictor with P value of .05 or less, and the high likelihood advisory sensitivity could not be statistically inferior to 65%. Patients who met those criteria entered an Advisory Phase where their system was configured to provide visual and audible advisories that indicate seizure likelihood.
In some instances, seizure incidence as recorded by the device was dramatically different from what the patient had reported. In one patient who initially reported having 7 seizures per month, the SAS recorded 104 per month, based on data covering 70 days. Another patient who reported 6 seizures per month actually had 80 per month, based on 126 days of data. Other patients overestimated their seizures, with one patient who reported having 3 seizures per month experiencing just 0.6 seizures per month, based on 209 days of data.
"Some patients overestimate the number of their seizures they’re having, but the amount by which [others] underestimate their seizures is very dramatic, sometimes by a factor of 10," Dr. Cook commented.
In the data collection (training) phase of the study, the sensitivity of the red advisory among 11 patients who completed the phase ranged from 0.65 to 1.00, with 10 of those patients meeting the performance criteria for the seizure advisories. Among 6 patients who have completed the subsequent 4-month advisory (prospective) study phase, red advisory sensitivity ranged from 0.56 to 1.00. In both study phases, there were no seizures during the low advisory (100% negative predictive value). The other four patients are still in the data collection phase.
The safety profile is consistent with published literature for strip electrodes and subclavicular implants, he said.
NeuroVista is continuing to study the clinical utility of the SAS, a company spokesman said.
Dr. Cook stated that he had no financial disclosures. The study was funded by NeuroVista, and four of the coinvestigators are company employees.
FROM THE ANNUAL MEETING OF THE AMERICAN EPILEPSY SOCIETY
Major Finding: The sensitivity of the device for time periods deemed to have a high likelihood of impending seizure ranged from 0.56 to 1.00 in six patients who completed a 4-month prospective phase of the study.
Data Source: A preliminary study of 15 patients implanted with NeuroVista’s Seizure Advisory System.
Disclosures: Dr. Cook stated that he had no financial disclosures. The study was funded by NeuroVista, and four of the coinvestigators are company employees.
FDA Issues Artificial Pancreas Guidance Document
The U.S. Food and Drug Administration has issued draft guidance to assist investigators and manufacturers in the development of artificial pancreas device systems to treat type 1 diabetes.
The 64-page document, issued Dec. 6, provides recommendations for design and testing that meet statutory requirements for safety and effectiveness for artificial pancreas devices, several of which are currently under development. These automated closed-loop systems combine a continuous glucose monitor (CGM), an insulin infusion pump, and a glucose meter for calibrating the monitor. The devices would work together, monitoring blood glucose levels and automatically infusing appropriate doses of insulin as determined by a computer algorithm.
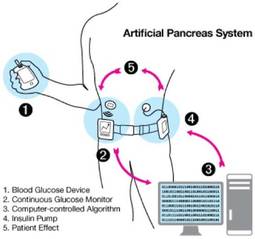
"While not a cure, an artificial pancreas could reduce dangerous high and low blood sugars, providing a better quality of life for those with diabetes and lowering the risk for future diabetes-related complications," said Charles "Chip" L. Zimliki, Ph.D., leader of the FDA’s Artificial Pancreas Working Group and Critical Path Initiative, in a media telebriefing.
One of the primary goals of diabetes researchers and of the diabetes community has been to develop "an automated system that could replace the endocrine portion of the pancreas to control blood glucose levels. People with type 1 diabetes must frequently monitor their blood sugar using a blood glucose meter throughout the day, and adjust their insulin dosing based upon these readings. It’s difficult, and we need devices to make it easier," Dr. Zimliki said.
The guidance addresses different types of future generation artificial pancreas devices, such as a treat-to-range system that would adjust insulin dosing if glucose levels approach a low or high threshold, and a fully automated treat-to-target system that would maintain target glucose levels at all times, without interaction from the user other than CGM calibration.
A three-phase clinical study progression is recommended to facilitate moving trials to outpatient settings as quickly as possible. Sponsors are permitted to use existing safety and effectiveness data for artificial pancreas device components, including data gathered from clinical studies conducted outside of the United States. They are also given the choice of either showing that the system provides glycemic control as well as standard therapies or that it provides better glycemic control when compared with other therapies, Dr. Zimliki said.
He noted that flexibility is a key feature of the artificial pancreas guidance, which is one of the few from FDA to be issued prior to the approval of an actual device. The document allows flexibility with regard to study size and duration, end points, and methodologies. "Because of the novelty of these systems, we really couldn’t be prescriptive in this guidance document. ... We have recommendations in there, and if people want to follow them, that’s great. But if they don’t, they just have to provide us justification for what they want to do."
Thus far, the FDA has approved 20 clinical trials evaluating various artificial pancreas device systems, and "the data are very encouraging," he said.
Regarding a possible timeline for device approval, Dr. Zimliki said that FDA’s main responsibility is to "develop a clear pathway forward" for investigators and manufacturers, and that "These systems aren’t going to be perfect right out of the gate, and there’s going to be an iterative step approach. ... We really don’t know the actual timeline, but we encourage this to happen as fast as possible."
The current guidance was informed by public comments on a previous one issued last June outlining FDA’s study expectations for a low-glucose suspend (LGS) device component that would respond to sensor readings of low or rapidly declining blood glucose levels by temporarily reducing or shutting down the delivery of insulin in order to avoid or mitigate hypoglycemia. The FDA "will be looking forward to" comments on the current document as well, Dr. Zimliki said. It will be published in the Federal Register in a few days.
The U.S. Food and Drug Administration has issued draft guidance to assist investigators and manufacturers in the development of artificial pancreas device systems to treat type 1 diabetes.
The 64-page document, issued Dec. 6, provides recommendations for design and testing that meet statutory requirements for safety and effectiveness for artificial pancreas devices, several of which are currently under development. These automated closed-loop systems combine a continuous glucose monitor (CGM), an insulin infusion pump, and a glucose meter for calibrating the monitor. The devices would work together, monitoring blood glucose levels and automatically infusing appropriate doses of insulin as determined by a computer algorithm.
"While not a cure, an artificial pancreas could reduce dangerous high and low blood sugars, providing a better quality of life for those with diabetes and lowering the risk for future diabetes-related complications," said Charles "Chip" L. Zimliki, Ph.D., leader of the FDA’s Artificial Pancreas Working Group and Critical Path Initiative, in a media telebriefing.
One of the primary goals of diabetes researchers and of the diabetes community has been to develop "an automated system that could replace the endocrine portion of the pancreas to control blood glucose levels. People with type 1 diabetes must frequently monitor their blood sugar using a blood glucose meter throughout the day, and adjust their insulin dosing based upon these readings. It’s difficult, and we need devices to make it easier," Dr. Zimliki said.
The guidance addresses different types of future generation artificial pancreas devices, such as a treat-to-range system that would adjust insulin dosing if glucose levels approach a low or high threshold, and a fully automated treat-to-target system that would maintain target glucose levels at all times, without interaction from the user other than CGM calibration.
A three-phase clinical study progression is recommended to facilitate moving trials to outpatient settings as quickly as possible. Sponsors are permitted to use existing safety and effectiveness data for artificial pancreas device components, including data gathered from clinical studies conducted outside of the United States. They are also given the choice of either showing that the system provides glycemic control as well as standard therapies or that it provides better glycemic control when compared with other therapies, Dr. Zimliki said.
He noted that flexibility is a key feature of the artificial pancreas guidance, which is one of the few from FDA to be issued prior to the approval of an actual device. The document allows flexibility with regard to study size and duration, end points, and methodologies. "Because of the novelty of these systems, we really couldn’t be prescriptive in this guidance document. ... We have recommendations in there, and if people want to follow them, that’s great. But if they don’t, they just have to provide us justification for what they want to do."
Thus far, the FDA has approved 20 clinical trials evaluating various artificial pancreas device systems, and "the data are very encouraging," he said.
Regarding a possible timeline for device approval, Dr. Zimliki said that FDA’s main responsibility is to "develop a clear pathway forward" for investigators and manufacturers, and that "These systems aren’t going to be perfect right out of the gate, and there’s going to be an iterative step approach. ... We really don’t know the actual timeline, but we encourage this to happen as fast as possible."
The current guidance was informed by public comments on a previous one issued last June outlining FDA’s study expectations for a low-glucose suspend (LGS) device component that would respond to sensor readings of low or rapidly declining blood glucose levels by temporarily reducing or shutting down the delivery of insulin in order to avoid or mitigate hypoglycemia. The FDA "will be looking forward to" comments on the current document as well, Dr. Zimliki said. It will be published in the Federal Register in a few days.
The U.S. Food and Drug Administration has issued draft guidance to assist investigators and manufacturers in the development of artificial pancreas device systems to treat type 1 diabetes.
The 64-page document, issued Dec. 6, provides recommendations for design and testing that meet statutory requirements for safety and effectiveness for artificial pancreas devices, several of which are currently under development. These automated closed-loop systems combine a continuous glucose monitor (CGM), an insulin infusion pump, and a glucose meter for calibrating the monitor. The devices would work together, monitoring blood glucose levels and automatically infusing appropriate doses of insulin as determined by a computer algorithm.
"While not a cure, an artificial pancreas could reduce dangerous high and low blood sugars, providing a better quality of life for those with diabetes and lowering the risk for future diabetes-related complications," said Charles "Chip" L. Zimliki, Ph.D., leader of the FDA’s Artificial Pancreas Working Group and Critical Path Initiative, in a media telebriefing.
One of the primary goals of diabetes researchers and of the diabetes community has been to develop "an automated system that could replace the endocrine portion of the pancreas to control blood glucose levels. People with type 1 diabetes must frequently monitor their blood sugar using a blood glucose meter throughout the day, and adjust their insulin dosing based upon these readings. It’s difficult, and we need devices to make it easier," Dr. Zimliki said.
The guidance addresses different types of future generation artificial pancreas devices, such as a treat-to-range system that would adjust insulin dosing if glucose levels approach a low or high threshold, and a fully automated treat-to-target system that would maintain target glucose levels at all times, without interaction from the user other than CGM calibration.
A three-phase clinical study progression is recommended to facilitate moving trials to outpatient settings as quickly as possible. Sponsors are permitted to use existing safety and effectiveness data for artificial pancreas device components, including data gathered from clinical studies conducted outside of the United States. They are also given the choice of either showing that the system provides glycemic control as well as standard therapies or that it provides better glycemic control when compared with other therapies, Dr. Zimliki said.
He noted that flexibility is a key feature of the artificial pancreas guidance, which is one of the few from FDA to be issued prior to the approval of an actual device. The document allows flexibility with regard to study size and duration, end points, and methodologies. "Because of the novelty of these systems, we really couldn’t be prescriptive in this guidance document. ... We have recommendations in there, and if people want to follow them, that’s great. But if they don’t, they just have to provide us justification for what they want to do."
Thus far, the FDA has approved 20 clinical trials evaluating various artificial pancreas device systems, and "the data are very encouraging," he said.
Regarding a possible timeline for device approval, Dr. Zimliki said that FDA’s main responsibility is to "develop a clear pathway forward" for investigators and manufacturers, and that "These systems aren’t going to be perfect right out of the gate, and there’s going to be an iterative step approach. ... We really don’t know the actual timeline, but we encourage this to happen as fast as possible."
The current guidance was informed by public comments on a previous one issued last June outlining FDA’s study expectations for a low-glucose suspend (LGS) device component that would respond to sensor readings of low or rapidly declining blood glucose levels by temporarily reducing or shutting down the delivery of insulin in order to avoid or mitigate hypoglycemia. The FDA "will be looking forward to" comments on the current document as well, Dr. Zimliki said. It will be published in the Federal Register in a few days.
Smallest Insulin Pump Yet Features Touch-Screen
The newest insulin pump system on the U.S. market is the smallest, and the first to feature a color touch screen.
The Food and Drug Administration granted clearance on Nov. 16 to Tandem Diabetes Care Inc. to market its t:slim Insulin Delivery System. It is the first to be cleared under the FDA’s Infusion Pump Improvement Initiative, which established requirements for infusion pump manufacturers and aimed to facilitate device improvements and increase user awareness.
According to a company statement, the t:slim was designed to make diabetes management easier to teach and to learn. Additional user-oriented features of the insulin pump include an eco-friendly rechargeable battery and USB connectivity to Web-based therapy management software.
Among the 1.5 million people in the United States with type 1 diabetes, about 20%-30% use an insulin pump, according to industry estimates. Clinical evidence supports the use of a pump to improve glycemic control and quality of life. "Tandem Diabetes Care believes that enhanced ease of use and attractive design will encourage more patients to consider the clinical benefits of insulin pump therapy," according to a company statement.
Tandem plans to launch its marketing of the device during the first half of 2012.
The newest insulin pump system on the U.S. market is the smallest, and the first to feature a color touch screen.
The Food and Drug Administration granted clearance on Nov. 16 to Tandem Diabetes Care Inc. to market its t:slim Insulin Delivery System. It is the first to be cleared under the FDA’s Infusion Pump Improvement Initiative, which established requirements for infusion pump manufacturers and aimed to facilitate device improvements and increase user awareness.
According to a company statement, the t:slim was designed to make diabetes management easier to teach and to learn. Additional user-oriented features of the insulin pump include an eco-friendly rechargeable battery and USB connectivity to Web-based therapy management software.
Among the 1.5 million people in the United States with type 1 diabetes, about 20%-30% use an insulin pump, according to industry estimates. Clinical evidence supports the use of a pump to improve glycemic control and quality of life. "Tandem Diabetes Care believes that enhanced ease of use and attractive design will encourage more patients to consider the clinical benefits of insulin pump therapy," according to a company statement.
Tandem plans to launch its marketing of the device during the first half of 2012.
The newest insulin pump system on the U.S. market is the smallest, and the first to feature a color touch screen.
The Food and Drug Administration granted clearance on Nov. 16 to Tandem Diabetes Care Inc. to market its t:slim Insulin Delivery System. It is the first to be cleared under the FDA’s Infusion Pump Improvement Initiative, which established requirements for infusion pump manufacturers and aimed to facilitate device improvements and increase user awareness.
According to a company statement, the t:slim was designed to make diabetes management easier to teach and to learn. Additional user-oriented features of the insulin pump include an eco-friendly rechargeable battery and USB connectivity to Web-based therapy management software.
Among the 1.5 million people in the United States with type 1 diabetes, about 20%-30% use an insulin pump, according to industry estimates. Clinical evidence supports the use of a pump to improve glycemic control and quality of life. "Tandem Diabetes Care believes that enhanced ease of use and attractive design will encourage more patients to consider the clinical benefits of insulin pump therapy," according to a company statement.
Tandem plans to launch its marketing of the device during the first half of 2012.
Safety Assurance Awaits Further Data for New Psoriasis Drugs
LAS VEGAS – New agents for treating psoriasis have shown great promise in terms of efficacy, but assurances of safety await further data from larger studies.
Thus far, phase II data show impressive efficacy and no worrisome safety signals for the anti-interleukin-17 agents AMG 827 and secukinumab and the small molecules apremilast and tofacitinib. However, the studies have been too small and of insufficient duration to definitively rule out cardiovascular, infectious, and cancer risks, said Dr. Kenneth B. Gordon at the Las Vegas Dermatology Seminar, sponsored by Skin Disease Education Foundation (SDEF).
"It is my hope that these drugs are going to be fantastic. We just have to maintain a level of concern and vigilance for both biologics and small molecules ... We just desperately need large trials," said Dr. Gordon, head of the division of dermatology at NorthShore University HealthSystem, Chicago.
The discovery of IL-17 as a key player in psoriatic plaque formation has led to a new understanding of psoriasis pathophysiology and has become a new target for drug development. However, experience with one agent that blocks IL-12/23 – which induces activated IL-17 – gives pause.
Briakinumab, an extremely effective anti-psoriatic agent that blocks IL 12/23, was withdrawn from development after phase III studies showed a signal – albeit statistically insignificant – for serious infections including cellulitis and pneumonia, MACE events (cardiac arrest, myocardial infarction, and stroke), and malignancies (nonmelanoma skin cancer and squamous cell carcinoma of the lung and nasopharynx).

The absolute MACE event numbers were small, but were counter to what would be expected from a systemic anti-inflammatory agent, which should reduce cardiovascular disease, Dr. Craig L. Leonardi, a clinical professor of dermatology at St. Louis University, said in a separate presentation.
According to Dr. Gordon, "It’s really an exciting time for new psoriatic therapies based on a better understanding of psoriasis pathophysiology. However, we are less excited about phase II data because of what happened to briakinumab in phase III."
It will be essential to determine the etiology of the adverse effects, he said. "If the effects are not related to how well the agent controls psoriasis but to mechanisms like IL-12 blockade that may not have relevance to IL-17, they may not show up [with the new agents in development]. That’s what the phase II studies suggest, but it’s too early to really make a final statement," Dr. Gordon said in an interview.
Phase II data on the anti-IL17 and small molecules were presented earlier this year at the European Academy of Dermatology and Venereology meeting in Lisbon and at the World Congress of Dermatology in Seoul. Among the findings:
• AMG 827: This fully-human monoclonal antibody binds to and blocks the IL-17 receptor. In a phase II, double-blind, randomized, placebo-controlled trial of 198 patients who were randomized to subcutaneous AMG 827 at 280 mg monthly; to 70, 140, or 210 mg every 2 weeks; or to placebo. The primary end point, PASI 75 response rate at week 12, was highest (83%) in patients who received 210 mg every 2 weeks. Moreover, at that dose, the proportion achieving a PASI 100 score, indicating no psoriasis activity, was 63%.
"This is an extraordinarily high-responding drug, similar only to briakinumab," Dr. Gordon commented.
Changes in neutrophil counts were seen, as to be expected from IL-17 and IL-12 blockade. "Some of the changes were significant. We need to keep an eye on this," he said.
• Secukinumab: This novel, fully human antibody to IL-17A was investigated in three separate phase II trials of patients with moderate to severe plaque psoriasis. An intravenous induction dose-ranging study of 100 patients yielded PASI 75 response rates at 12 weeks of 40%-83%. A subcutaneous dose-ranging study of 125 patients produced PASI 75 response rates of 19%-81%, and a subcutaneous regimen-finding trial involving 404 patients determined that a regimen of 150 mg at weeks 0, 1, 2, and 4 produced the best PASI 75 response at week 12, of 55%.
While the safety analysis showed no significant differences from placebo, there were two cases of cardiac disorders – one angina pectoris and one coronary artery disease – in the intravenous dose-ranging study and two cases listed as "cardiac disorders" in the regimen-finding study. "We don’t know if these are significant. We have to look at phase III trials," Dr. Gordon said.
• Apremilast: This small molecule, taken orally, works by inhibiting type 4 phosphodiesterase. In a randomized, placebo-controlled phase II trial of 352 patients with moderate to severe plaque psoriasis, the PASI 75 response rate at 16 weeks was 41% with a 30-mg twice-daily dose.
Adverse effects were dose-dependent. Adverse events that occurred in 5% or more of patients included headache, nausea, diarrhea, and upper respiratory tract infections. These typically occurred early in the course of treatment.
Serious adverse events included one myocardial infarction and one case of prostate cancer in the 30-mg BID group. But there was also a prostate cancer and a sudden death among the placebo recipients. "We really don’t have enough patients to look at adverse effects," Dr. Gordon said.
• Tofacitinib: This oral Janus kinase inhibitor demonstrated dose-dependent efficacy, with 67% of patients randomized at 15 mg twice a day achieving PASI 75 and "clear" or "almost clear." The phase II, 12-week, double-blind, placebo-controlled trial enrolled 197 patients with moderate to severe plaque psoriasis.
Concomitant overall decreases in hemoglobin and neutrophil counts from baseline were also dose-dependent. The finding did not specify the proportion of patients who had the decreases or the individual degree of change. "If everyone had a small change, it’s not going to bother me too much. But, if 10% of patients had a big change, it’s a really important finding. Those are the questions we need to ask," Dr. Gordon said.
Dr. Gordon said that so far the phase II safety data for the agents are encouraging, but enthusiasm should still be tempered. "When multiple drugs with the same mechanism have [similar] results, you start to feel more confident. But still, we need to see larger studies."
"It’s really an exciting time for new psoriatic therapies based on a better understanding of psoriasis pathophysiology."
Dr. Leonardi’s presentation focused on anti-IL 12/23 inhibitor treatment; he noted that the recently-approved monoclonal antibody ustekinumab binds to the same shared p40 subunit of IL-12 and IL-23 as briakinumab, the agent that was withdrawn from development in phase III. "There are more similarities than differences" between the two agents, he commented.
He and his colleagues recently conducted a meta-analysis of 22 randomized controlled clinical trials of biologic therapies comprising 10,183 patients with chronic plaque psoriasis, in which 10 of 3,179 patients receiving either ustekinumab or briakinumab experienced a MACE, compared with 0 events in 1,474 patients receiving placebo, for a MACE rate of 1.33 per 100 patient-years.
In contrast, no difference was seen among patients in the anti-TNF-alpha trials, with only 1 of 3,858 patients receiving anti-TNF-alpha agents experiencing a MACE, compared with 1 of 1,812 patients receiving placebo (JAMA 2011;306:864-71).
Although the difference for the anti-IL 12/23 agents was not statistically significant, the data set was not large enough to detect rare events. "This is a class effect in my mind," Dr. Leonardi said, adding that he uses ustekinumab as a second-line agent, after the TNF antagonists.
"It’s important to remember that all new drugs are ‘new’ ... We will learn more and more about these drugs as time goes on," he said.
Dr. Gordon disclosed that he has received research support or honoraria as a consultant from Abbott, Amgen, Centocor, Eli Lilly, Merck, Novartis, and Pfizer. Dr. Leonardi disclosed that he has had financial relationships with 23 companies that are involved in psoriasis treatment development, including Abbott and Centocor.
SDEF and this news organization are owned by Elsevier.
LAS VEGAS – New agents for treating psoriasis have shown great promise in terms of efficacy, but assurances of safety await further data from larger studies.
Thus far, phase II data show impressive efficacy and no worrisome safety signals for the anti-interleukin-17 agents AMG 827 and secukinumab and the small molecules apremilast and tofacitinib. However, the studies have been too small and of insufficient duration to definitively rule out cardiovascular, infectious, and cancer risks, said Dr. Kenneth B. Gordon at the Las Vegas Dermatology Seminar, sponsored by Skin Disease Education Foundation (SDEF).
"It is my hope that these drugs are going to be fantastic. We just have to maintain a level of concern and vigilance for both biologics and small molecules ... We just desperately need large trials," said Dr. Gordon, head of the division of dermatology at NorthShore University HealthSystem, Chicago.
The discovery of IL-17 as a key player in psoriatic plaque formation has led to a new understanding of psoriasis pathophysiology and has become a new target for drug development. However, experience with one agent that blocks IL-12/23 – which induces activated IL-17 – gives pause.
Briakinumab, an extremely effective anti-psoriatic agent that blocks IL 12/23, was withdrawn from development after phase III studies showed a signal – albeit statistically insignificant – for serious infections including cellulitis and pneumonia, MACE events (cardiac arrest, myocardial infarction, and stroke), and malignancies (nonmelanoma skin cancer and squamous cell carcinoma of the lung and nasopharynx).
The absolute MACE event numbers were small, but were counter to what would be expected from a systemic anti-inflammatory agent, which should reduce cardiovascular disease, Dr. Craig L. Leonardi, a clinical professor of dermatology at St. Louis University, said in a separate presentation.
According to Dr. Gordon, "It’s really an exciting time for new psoriatic therapies based on a better understanding of psoriasis pathophysiology. However, we are less excited about phase II data because of what happened to briakinumab in phase III."
It will be essential to determine the etiology of the adverse effects, he said. "If the effects are not related to how well the agent controls psoriasis but to mechanisms like IL-12 blockade that may not have relevance to IL-17, they may not show up [with the new agents in development]. That’s what the phase II studies suggest, but it’s too early to really make a final statement," Dr. Gordon said in an interview.
Phase II data on the anti-IL17 and small molecules were presented earlier this year at the European Academy of Dermatology and Venereology meeting in Lisbon and at the World Congress of Dermatology in Seoul. Among the findings:
• AMG 827: This fully-human monoclonal antibody binds to and blocks the IL-17 receptor. In a phase II, double-blind, randomized, placebo-controlled trial of 198 patients who were randomized to subcutaneous AMG 827 at 280 mg monthly; to 70, 140, or 210 mg every 2 weeks; or to placebo. The primary end point, PASI 75 response rate at week 12, was highest (83%) in patients who received 210 mg every 2 weeks. Moreover, at that dose, the proportion achieving a PASI 100 score, indicating no psoriasis activity, was 63%.
"This is an extraordinarily high-responding drug, similar only to briakinumab," Dr. Gordon commented.
Changes in neutrophil counts were seen, as to be expected from IL-17 and IL-12 blockade. "Some of the changes were significant. We need to keep an eye on this," he said.
• Secukinumab: This novel, fully human antibody to IL-17A was investigated in three separate phase II trials of patients with moderate to severe plaque psoriasis. An intravenous induction dose-ranging study of 100 patients yielded PASI 75 response rates at 12 weeks of 40%-83%. A subcutaneous dose-ranging study of 125 patients produced PASI 75 response rates of 19%-81%, and a subcutaneous regimen-finding trial involving 404 patients determined that a regimen of 150 mg at weeks 0, 1, 2, and 4 produced the best PASI 75 response at week 12, of 55%.
While the safety analysis showed no significant differences from placebo, there were two cases of cardiac disorders – one angina pectoris and one coronary artery disease – in the intravenous dose-ranging study and two cases listed as "cardiac disorders" in the regimen-finding study. "We don’t know if these are significant. We have to look at phase III trials," Dr. Gordon said.
• Apremilast: This small molecule, taken orally, works by inhibiting type 4 phosphodiesterase. In a randomized, placebo-controlled phase II trial of 352 patients with moderate to severe plaque psoriasis, the PASI 75 response rate at 16 weeks was 41% with a 30-mg twice-daily dose.
Adverse effects were dose-dependent. Adverse events that occurred in 5% or more of patients included headache, nausea, diarrhea, and upper respiratory tract infections. These typically occurred early in the course of treatment.
Serious adverse events included one myocardial infarction and one case of prostate cancer in the 30-mg BID group. But there was also a prostate cancer and a sudden death among the placebo recipients. "We really don’t have enough patients to look at adverse effects," Dr. Gordon said.
• Tofacitinib: This oral Janus kinase inhibitor demonstrated dose-dependent efficacy, with 67% of patients randomized at 15 mg twice a day achieving PASI 75 and "clear" or "almost clear." The phase II, 12-week, double-blind, placebo-controlled trial enrolled 197 patients with moderate to severe plaque psoriasis.
Concomitant overall decreases in hemoglobin and neutrophil counts from baseline were also dose-dependent. The finding did not specify the proportion of patients who had the decreases or the individual degree of change. "If everyone had a small change, it’s not going to bother me too much. But, if 10% of patients had a big change, it’s a really important finding. Those are the questions we need to ask," Dr. Gordon said.
Dr. Gordon said that so far the phase II safety data for the agents are encouraging, but enthusiasm should still be tempered. "When multiple drugs with the same mechanism have [similar] results, you start to feel more confident. But still, we need to see larger studies."
"It’s really an exciting time for new psoriatic therapies based on a better understanding of psoriasis pathophysiology."
Dr. Leonardi’s presentation focused on anti-IL 12/23 inhibitor treatment; he noted that the recently-approved monoclonal antibody ustekinumab binds to the same shared p40 subunit of IL-12 and IL-23 as briakinumab, the agent that was withdrawn from development in phase III. "There are more similarities than differences" between the two agents, he commented.
He and his colleagues recently conducted a meta-analysis of 22 randomized controlled clinical trials of biologic therapies comprising 10,183 patients with chronic plaque psoriasis, in which 10 of 3,179 patients receiving either ustekinumab or briakinumab experienced a MACE, compared with 0 events in 1,474 patients receiving placebo, for a MACE rate of 1.33 per 100 patient-years.
In contrast, no difference was seen among patients in the anti-TNF-alpha trials, with only 1 of 3,858 patients receiving anti-TNF-alpha agents experiencing a MACE, compared with 1 of 1,812 patients receiving placebo (JAMA 2011;306:864-71).
Although the difference for the anti-IL 12/23 agents was not statistically significant, the data set was not large enough to detect rare events. "This is a class effect in my mind," Dr. Leonardi said, adding that he uses ustekinumab as a second-line agent, after the TNF antagonists.
"It’s important to remember that all new drugs are ‘new’ ... We will learn more and more about these drugs as time goes on," he said.
Dr. Gordon disclosed that he has received research support or honoraria as a consultant from Abbott, Amgen, Centocor, Eli Lilly, Merck, Novartis, and Pfizer. Dr. Leonardi disclosed that he has had financial relationships with 23 companies that are involved in psoriasis treatment development, including Abbott and Centocor.
SDEF and this news organization are owned by Elsevier.
LAS VEGAS – New agents for treating psoriasis have shown great promise in terms of efficacy, but assurances of safety await further data from larger studies.
Thus far, phase II data show impressive efficacy and no worrisome safety signals for the anti-interleukin-17 agents AMG 827 and secukinumab and the small molecules apremilast and tofacitinib. However, the studies have been too small and of insufficient duration to definitively rule out cardiovascular, infectious, and cancer risks, said Dr. Kenneth B. Gordon at the Las Vegas Dermatology Seminar, sponsored by Skin Disease Education Foundation (SDEF).
"It is my hope that these drugs are going to be fantastic. We just have to maintain a level of concern and vigilance for both biologics and small molecules ... We just desperately need large trials," said Dr. Gordon, head of the division of dermatology at NorthShore University HealthSystem, Chicago.
The discovery of IL-17 as a key player in psoriatic plaque formation has led to a new understanding of psoriasis pathophysiology and has become a new target for drug development. However, experience with one agent that blocks IL-12/23 – which induces activated IL-17 – gives pause.
Briakinumab, an extremely effective anti-psoriatic agent that blocks IL 12/23, was withdrawn from development after phase III studies showed a signal – albeit statistically insignificant – for serious infections including cellulitis and pneumonia, MACE events (cardiac arrest, myocardial infarction, and stroke), and malignancies (nonmelanoma skin cancer and squamous cell carcinoma of the lung and nasopharynx).
The absolute MACE event numbers were small, but were counter to what would be expected from a systemic anti-inflammatory agent, which should reduce cardiovascular disease, Dr. Craig L. Leonardi, a clinical professor of dermatology at St. Louis University, said in a separate presentation.
According to Dr. Gordon, "It’s really an exciting time for new psoriatic therapies based on a better understanding of psoriasis pathophysiology. However, we are less excited about phase II data because of what happened to briakinumab in phase III."
It will be essential to determine the etiology of the adverse effects, he said. "If the effects are not related to how well the agent controls psoriasis but to mechanisms like IL-12 blockade that may not have relevance to IL-17, they may not show up [with the new agents in development]. That’s what the phase II studies suggest, but it’s too early to really make a final statement," Dr. Gordon said in an interview.
Phase II data on the anti-IL17 and small molecules were presented earlier this year at the European Academy of Dermatology and Venereology meeting in Lisbon and at the World Congress of Dermatology in Seoul. Among the findings:
• AMG 827: This fully-human monoclonal antibody binds to and blocks the IL-17 receptor. In a phase II, double-blind, randomized, placebo-controlled trial of 198 patients who were randomized to subcutaneous AMG 827 at 280 mg monthly; to 70, 140, or 210 mg every 2 weeks; or to placebo. The primary end point, PASI 75 response rate at week 12, was highest (83%) in patients who received 210 mg every 2 weeks. Moreover, at that dose, the proportion achieving a PASI 100 score, indicating no psoriasis activity, was 63%.
"This is an extraordinarily high-responding drug, similar only to briakinumab," Dr. Gordon commented.
Changes in neutrophil counts were seen, as to be expected from IL-17 and IL-12 blockade. "Some of the changes were significant. We need to keep an eye on this," he said.
• Secukinumab: This novel, fully human antibody to IL-17A was investigated in three separate phase II trials of patients with moderate to severe plaque psoriasis. An intravenous induction dose-ranging study of 100 patients yielded PASI 75 response rates at 12 weeks of 40%-83%. A subcutaneous dose-ranging study of 125 patients produced PASI 75 response rates of 19%-81%, and a subcutaneous regimen-finding trial involving 404 patients determined that a regimen of 150 mg at weeks 0, 1, 2, and 4 produced the best PASI 75 response at week 12, of 55%.
While the safety analysis showed no significant differences from placebo, there were two cases of cardiac disorders – one angina pectoris and one coronary artery disease – in the intravenous dose-ranging study and two cases listed as "cardiac disorders" in the regimen-finding study. "We don’t know if these are significant. We have to look at phase III trials," Dr. Gordon said.
• Apremilast: This small molecule, taken orally, works by inhibiting type 4 phosphodiesterase. In a randomized, placebo-controlled phase II trial of 352 patients with moderate to severe plaque psoriasis, the PASI 75 response rate at 16 weeks was 41% with a 30-mg twice-daily dose.
Adverse effects were dose-dependent. Adverse events that occurred in 5% or more of patients included headache, nausea, diarrhea, and upper respiratory tract infections. These typically occurred early in the course of treatment.
Serious adverse events included one myocardial infarction and one case of prostate cancer in the 30-mg BID group. But there was also a prostate cancer and a sudden death among the placebo recipients. "We really don’t have enough patients to look at adverse effects," Dr. Gordon said.
• Tofacitinib: This oral Janus kinase inhibitor demonstrated dose-dependent efficacy, with 67% of patients randomized at 15 mg twice a day achieving PASI 75 and "clear" or "almost clear." The phase II, 12-week, double-blind, placebo-controlled trial enrolled 197 patients with moderate to severe plaque psoriasis.
Concomitant overall decreases in hemoglobin and neutrophil counts from baseline were also dose-dependent. The finding did not specify the proportion of patients who had the decreases or the individual degree of change. "If everyone had a small change, it’s not going to bother me too much. But, if 10% of patients had a big change, it’s a really important finding. Those are the questions we need to ask," Dr. Gordon said.
Dr. Gordon said that so far the phase II safety data for the agents are encouraging, but enthusiasm should still be tempered. "When multiple drugs with the same mechanism have [similar] results, you start to feel more confident. But still, we need to see larger studies."
"It’s really an exciting time for new psoriatic therapies based on a better understanding of psoriasis pathophysiology."
Dr. Leonardi’s presentation focused on anti-IL 12/23 inhibitor treatment; he noted that the recently-approved monoclonal antibody ustekinumab binds to the same shared p40 subunit of IL-12 and IL-23 as briakinumab, the agent that was withdrawn from development in phase III. "There are more similarities than differences" between the two agents, he commented.
He and his colleagues recently conducted a meta-analysis of 22 randomized controlled clinical trials of biologic therapies comprising 10,183 patients with chronic plaque psoriasis, in which 10 of 3,179 patients receiving either ustekinumab or briakinumab experienced a MACE, compared with 0 events in 1,474 patients receiving placebo, for a MACE rate of 1.33 per 100 patient-years.
In contrast, no difference was seen among patients in the anti-TNF-alpha trials, with only 1 of 3,858 patients receiving anti-TNF-alpha agents experiencing a MACE, compared with 1 of 1,812 patients receiving placebo (JAMA 2011;306:864-71).
Although the difference for the anti-IL 12/23 agents was not statistically significant, the data set was not large enough to detect rare events. "This is a class effect in my mind," Dr. Leonardi said, adding that he uses ustekinumab as a second-line agent, after the TNF antagonists.
"It’s important to remember that all new drugs are ‘new’ ... We will learn more and more about these drugs as time goes on," he said.
Dr. Gordon disclosed that he has received research support or honoraria as a consultant from Abbott, Amgen, Centocor, Eli Lilly, Merck, Novartis, and Pfizer. Dr. Leonardi disclosed that he has had financial relationships with 23 companies that are involved in psoriasis treatment development, including Abbott and Centocor.
SDEF and this news organization are owned by Elsevier.
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
Reflux Surgery Redos Safe, Outcomes Not Always Optimal
Reoperation for failed antireflux surgery can be performed safely in experienced centers, but outcomes are not as good as with primary operations, according to the results of two new analyses.
Dr. Nicholas R.A. Symons of Imperial College, London, and his associates performed a systematic literature review of 20 studies comprising 930 operations in 922 patients. "We can conclude that laparoscopic revision antireflux surgery, when performed in units with an interest in this type of surgery, is feasible and safe but subject to somewhat greater risk of conversion, higher morbidity, longer hospital stay, and poorer outcomes than primary laparoscopic fundoplication," the researchers said (Am. J. Surg. 2011;202:336-43).
Similarly, based on their retrospective study of 275 patients, Dr. Omar Awais of the University of Pittsburgh and his associates said, "Redo antireflux surgery can be performed safely in experienced centers, and as expected, the outcomes after redo antireflux surgery are not as good as [those of] first-time procedures. The operative approach depends on the underlying cause of failure" (Ann. Thorac. Surg. 2011;92:1083-90).
Between 2.8% and 4.4% of patients who undergo laparoscopic fundoplication at a specialist center will require late reoperation for persistent or recurrent symptoms, and there may be an increased revision rate after primary laparoscopic antireflux surgery compared with an initial open approach. Revision of failed antireflux surgery increasingly is being performed laparoscopically, but data about this approach are limited, Dr. Symons and his associates noted.
Their analysis included 19 case-control series and one prospective case-control study. Patients ranged in age from 13 to 83 years, and 57% were female. Of the eight articles in which the mean time between initial fundoplication and revision was given, the mean interval was 45.5 months (range 2-360 months). Of 12 studies noting the number of previous fundoplications the patients had undergone, 47 procedures (6.9%) were second reoperations and 9 (1.3%) were third reoperations.
Of the 18 studies documenting the type of initial surgery, 62% were laparoscopic, 35% were performed via laparotomy, 3% via thoracotomy, and 0.2% using video-assisted thoracoscopic surgery. Nissen fundoplication and Toupet partial fundoplication were the most common initial and revision procedures. Reflux/heartburn, experienced by 61% of patients, and dysphagia in 31% were the most common indications for laparoscopic reoperation, the investigators reported. Surgical duration, reported in 13 of the case series had an overall mean of 166 minutes for 721 patients.
The overall rate of conversion from laparoscopic reoperation to open surgery – reported in all the studies – was 7.2%, while the rate for patients who had more than one previous fundoplication was 19%. The most common reason for conversion, reported in 16 studies, was adhesions from previous surgery. The overall 7.2% conversion rate is higher than that noted for primary surgery (3.7%), "but is not excessive given the increased complexity of revision surgery," they commented.
Postoperative complications, reported from 18 studies, occurred in 14%, ranging from 0% to 44%. There were two deaths, both occurring in the same study. Pneumothorax was the most common complication, in 2% (14/810). Mean length of stay, reported in 18 studies, ranged from 1.2 to 6 days. Satisfactory to excellent results were reported for 84% of the operations; 5% of patients had a further antireflux procedure.
While revision laparoscopic fundoplication does not confer excessive morbidity, evidence for the efficacy of this procedure "is far less convincing, mainly owing to the mixture of reporting time points, inconsistency of end point definitions, and methods of assessment between studies. There appears to be a larger proportion of patients undergoing a re-revision surgery than after primary fundoplication," Dr. Symons commented.
The University of Pittsburgh study included patients who underwent minimally invasive reoperative surgery after failed fundoplication from 1996 to 2008. The 275 patients had a median age of 52 years (range 17-88 years), and 11.3% had had more than one prior antireflux surgery. As with the systematic review, the most common presenting symptoms were heartburn (64%) and dysphagia (49.5%). The median time from the prior operation to the redo operation was 36 months.
Transmediastinal migration of the wrap or a recurrent hiatal hernia (64%) were the most common causes of failure of the prior antireflux operation. Esophageal shortening was noted in 43% of patients, and a defect in the crural repair was identified in 4.4%. The most common procedure during reoperation was a Nissen fundoplication with or without a Collis gastroplasty. Nearly all of the redo procedures (93%) were done with a minimally invasive approach. There were eight conversions to open surgery due to extensive adhesions or a recognized intraoperative perforation, Dr. Awais and his associates reported.
Major complications included postoperative leaks in 3.3%; atrial fibrillation in 2%; and bleeding, pulmonary embolism, and Clostridium difficile colitis, each in less than 1%. Reexploration was required in 1.4%, for complications related to leak or bleeding. There was no perioperative mortality. Length of stay ranged from 1 to 75 days (median 3 days).
During follow-up of up to 14.5 years (median 3.3 years), 11.3% had a failure of the redo operation, requiring surgical intervention. An esophagectomy was required in four patients. The estimated probability of freedom from failure was 95% at 1 year, 93% at 2 years, and 84% at 5 years. Age and partial fundoplication were significantly associated with failure of the redo operation. and there was a trend for multiple redo operations to be associated with failure, Dr. Awais and his associates said.
Dysphagia decreased significantly after the redo procedure in 135 patients, with dysphagia scores declining from 2.7 to 1.4. Scores on the GERD-Health Related Quality of Life questionnaire, available in 186 patients, were excellent in 52% and satisfactory in 33%.
Both Dr. Symons and Dr. Awais stated that they had no disclosures.
Reoperation for failed antireflux surgery can be performed safely in experienced centers, but outcomes are not as good as with primary operations, according to the results of two new analyses.
Dr. Nicholas R.A. Symons of Imperial College, London, and his associates performed a systematic literature review of 20 studies comprising 930 operations in 922 patients. "We can conclude that laparoscopic revision antireflux surgery, when performed in units with an interest in this type of surgery, is feasible and safe but subject to somewhat greater risk of conversion, higher morbidity, longer hospital stay, and poorer outcomes than primary laparoscopic fundoplication," the researchers said (Am. J. Surg. 2011;202:336-43).
Similarly, based on their retrospective study of 275 patients, Dr. Omar Awais of the University of Pittsburgh and his associates said, "Redo antireflux surgery can be performed safely in experienced centers, and as expected, the outcomes after redo antireflux surgery are not as good as [those of] first-time procedures. The operative approach depends on the underlying cause of failure" (Ann. Thorac. Surg. 2011;92:1083-90).
Between 2.8% and 4.4% of patients who undergo laparoscopic fundoplication at a specialist center will require late reoperation for persistent or recurrent symptoms, and there may be an increased revision rate after primary laparoscopic antireflux surgery compared with an initial open approach. Revision of failed antireflux surgery increasingly is being performed laparoscopically, but data about this approach are limited, Dr. Symons and his associates noted.
Their analysis included 19 case-control series and one prospective case-control study. Patients ranged in age from 13 to 83 years, and 57% were female. Of the eight articles in which the mean time between initial fundoplication and revision was given, the mean interval was 45.5 months (range 2-360 months). Of 12 studies noting the number of previous fundoplications the patients had undergone, 47 procedures (6.9%) were second reoperations and 9 (1.3%) were third reoperations.
Of the 18 studies documenting the type of initial surgery, 62% were laparoscopic, 35% were performed via laparotomy, 3% via thoracotomy, and 0.2% using video-assisted thoracoscopic surgery. Nissen fundoplication and Toupet partial fundoplication were the most common initial and revision procedures. Reflux/heartburn, experienced by 61% of patients, and dysphagia in 31% were the most common indications for laparoscopic reoperation, the investigators reported. Surgical duration, reported in 13 of the case series had an overall mean of 166 minutes for 721 patients.
The overall rate of conversion from laparoscopic reoperation to open surgery – reported in all the studies – was 7.2%, while the rate for patients who had more than one previous fundoplication was 19%. The most common reason for conversion, reported in 16 studies, was adhesions from previous surgery. The overall 7.2% conversion rate is higher than that noted for primary surgery (3.7%), "but is not excessive given the increased complexity of revision surgery," they commented.
Postoperative complications, reported from 18 studies, occurred in 14%, ranging from 0% to 44%. There were two deaths, both occurring in the same study. Pneumothorax was the most common complication, in 2% (14/810). Mean length of stay, reported in 18 studies, ranged from 1.2 to 6 days. Satisfactory to excellent results were reported for 84% of the operations; 5% of patients had a further antireflux procedure.
While revision laparoscopic fundoplication does not confer excessive morbidity, evidence for the efficacy of this procedure "is far less convincing, mainly owing to the mixture of reporting time points, inconsistency of end point definitions, and methods of assessment between studies. There appears to be a larger proportion of patients undergoing a re-revision surgery than after primary fundoplication," Dr. Symons commented.
The University of Pittsburgh study included patients who underwent minimally invasive reoperative surgery after failed fundoplication from 1996 to 2008. The 275 patients had a median age of 52 years (range 17-88 years), and 11.3% had had more than one prior antireflux surgery. As with the systematic review, the most common presenting symptoms were heartburn (64%) and dysphagia (49.5%). The median time from the prior operation to the redo operation was 36 months.
Transmediastinal migration of the wrap or a recurrent hiatal hernia (64%) were the most common causes of failure of the prior antireflux operation. Esophageal shortening was noted in 43% of patients, and a defect in the crural repair was identified in 4.4%. The most common procedure during reoperation was a Nissen fundoplication with or without a Collis gastroplasty. Nearly all of the redo procedures (93%) were done with a minimally invasive approach. There were eight conversions to open surgery due to extensive adhesions or a recognized intraoperative perforation, Dr. Awais and his associates reported.
Major complications included postoperative leaks in 3.3%; atrial fibrillation in 2%; and bleeding, pulmonary embolism, and Clostridium difficile colitis, each in less than 1%. Reexploration was required in 1.4%, for complications related to leak or bleeding. There was no perioperative mortality. Length of stay ranged from 1 to 75 days (median 3 days).
During follow-up of up to 14.5 years (median 3.3 years), 11.3% had a failure of the redo operation, requiring surgical intervention. An esophagectomy was required in four patients. The estimated probability of freedom from failure was 95% at 1 year, 93% at 2 years, and 84% at 5 years. Age and partial fundoplication were significantly associated with failure of the redo operation. and there was a trend for multiple redo operations to be associated with failure, Dr. Awais and his associates said.
Dysphagia decreased significantly after the redo procedure in 135 patients, with dysphagia scores declining from 2.7 to 1.4. Scores on the GERD-Health Related Quality of Life questionnaire, available in 186 patients, were excellent in 52% and satisfactory in 33%.
Both Dr. Symons and Dr. Awais stated that they had no disclosures.
Reoperation for failed antireflux surgery can be performed safely in experienced centers, but outcomes are not as good as with primary operations, according to the results of two new analyses.
Dr. Nicholas R.A. Symons of Imperial College, London, and his associates performed a systematic literature review of 20 studies comprising 930 operations in 922 patients. "We can conclude that laparoscopic revision antireflux surgery, when performed in units with an interest in this type of surgery, is feasible and safe but subject to somewhat greater risk of conversion, higher morbidity, longer hospital stay, and poorer outcomes than primary laparoscopic fundoplication," the researchers said (Am. J. Surg. 2011;202:336-43).
Similarly, based on their retrospective study of 275 patients, Dr. Omar Awais of the University of Pittsburgh and his associates said, "Redo antireflux surgery can be performed safely in experienced centers, and as expected, the outcomes after redo antireflux surgery are not as good as [those of] first-time procedures. The operative approach depends on the underlying cause of failure" (Ann. Thorac. Surg. 2011;92:1083-90).
Between 2.8% and 4.4% of patients who undergo laparoscopic fundoplication at a specialist center will require late reoperation for persistent or recurrent symptoms, and there may be an increased revision rate after primary laparoscopic antireflux surgery compared with an initial open approach. Revision of failed antireflux surgery increasingly is being performed laparoscopically, but data about this approach are limited, Dr. Symons and his associates noted.
Their analysis included 19 case-control series and one prospective case-control study. Patients ranged in age from 13 to 83 years, and 57% were female. Of the eight articles in which the mean time between initial fundoplication and revision was given, the mean interval was 45.5 months (range 2-360 months). Of 12 studies noting the number of previous fundoplications the patients had undergone, 47 procedures (6.9%) were second reoperations and 9 (1.3%) were third reoperations.
Of the 18 studies documenting the type of initial surgery, 62% were laparoscopic, 35% were performed via laparotomy, 3% via thoracotomy, and 0.2% using video-assisted thoracoscopic surgery. Nissen fundoplication and Toupet partial fundoplication were the most common initial and revision procedures. Reflux/heartburn, experienced by 61% of patients, and dysphagia in 31% were the most common indications for laparoscopic reoperation, the investigators reported. Surgical duration, reported in 13 of the case series had an overall mean of 166 minutes for 721 patients.
The overall rate of conversion from laparoscopic reoperation to open surgery – reported in all the studies – was 7.2%, while the rate for patients who had more than one previous fundoplication was 19%. The most common reason for conversion, reported in 16 studies, was adhesions from previous surgery. The overall 7.2% conversion rate is higher than that noted for primary surgery (3.7%), "but is not excessive given the increased complexity of revision surgery," they commented.
Postoperative complications, reported from 18 studies, occurred in 14%, ranging from 0% to 44%. There were two deaths, both occurring in the same study. Pneumothorax was the most common complication, in 2% (14/810). Mean length of stay, reported in 18 studies, ranged from 1.2 to 6 days. Satisfactory to excellent results were reported for 84% of the operations; 5% of patients had a further antireflux procedure.
While revision laparoscopic fundoplication does not confer excessive morbidity, evidence for the efficacy of this procedure "is far less convincing, mainly owing to the mixture of reporting time points, inconsistency of end point definitions, and methods of assessment between studies. There appears to be a larger proportion of patients undergoing a re-revision surgery than after primary fundoplication," Dr. Symons commented.
The University of Pittsburgh study included patients who underwent minimally invasive reoperative surgery after failed fundoplication from 1996 to 2008. The 275 patients had a median age of 52 years (range 17-88 years), and 11.3% had had more than one prior antireflux surgery. As with the systematic review, the most common presenting symptoms were heartburn (64%) and dysphagia (49.5%). The median time from the prior operation to the redo operation was 36 months.
Transmediastinal migration of the wrap or a recurrent hiatal hernia (64%) were the most common causes of failure of the prior antireflux operation. Esophageal shortening was noted in 43% of patients, and a defect in the crural repair was identified in 4.4%. The most common procedure during reoperation was a Nissen fundoplication with or without a Collis gastroplasty. Nearly all of the redo procedures (93%) were done with a minimally invasive approach. There were eight conversions to open surgery due to extensive adhesions or a recognized intraoperative perforation, Dr. Awais and his associates reported.
Major complications included postoperative leaks in 3.3%; atrial fibrillation in 2%; and bleeding, pulmonary embolism, and Clostridium difficile colitis, each in less than 1%. Reexploration was required in 1.4%, for complications related to leak or bleeding. There was no perioperative mortality. Length of stay ranged from 1 to 75 days (median 3 days).
During follow-up of up to 14.5 years (median 3.3 years), 11.3% had a failure of the redo operation, requiring surgical intervention. An esophagectomy was required in four patients. The estimated probability of freedom from failure was 95% at 1 year, 93% at 2 years, and 84% at 5 years. Age and partial fundoplication were significantly associated with failure of the redo operation. and there was a trend for multiple redo operations to be associated with failure, Dr. Awais and his associates said.
Dysphagia decreased significantly after the redo procedure in 135 patients, with dysphagia scores declining from 2.7 to 1.4. Scores on the GERD-Health Related Quality of Life questionnaire, available in 186 patients, were excellent in 52% and satisfactory in 33%.
Both Dr. Symons and Dr. Awais stated that they had no disclosures.
Meningococcal Vaccine Not Cost Effective in Infants, Toddlers
ATLANTA – Routine infant or toddler immunization against meningococcal disease does not appear to be a cost-effective measure, according to an assessment from a working group of the Advisory Committee on Immunization Practices.
That conclusion was presented to the full committee at its October meeting. If a conjugate meningococcal vaccine is licensed for use in infants before the next Centers for Disease Control and Prevention’s (CDC) Advisory Committee on Immunization Practices (ACIP) meeting Feb. 22-23, the committee will vote on it then, said Dr. Amanda Cohn of the CDC’s National Center for Immunization and Respiratory Disease (NCIRD).
The working group’s assessment was based primarily on the low proportion of preventable disease cases and the high cost of vaccinating infants or toddlers with meningococcal vaccine per case prevented. Moreover, although current meningococcal conjugate vaccines are safe and immunogenic, they show evidence of declining antibodies after about 3 years, suggesting that a booster dose at age 6 years would likely be needed to protect children until the routine 11-to 12-year-old (adolescent) immunization, Dr. Cohn noted.
Jessica MacNeil, MPH, also with NCIRD, presented epidemiologic data showing that meningococcal disease has been declining in all age groups. The data do not suggest this is due to the adolescent immunization program.** The first evidence of an impact of the adolescent immunization program was detected in 2008-2009, when the incidence was 0.14 per 100,000 population among those aged 11-19 years and 0.82 among children less than 1 year of age. In contrast, those rates in 2006-2007 were 0.27 and 1.07, respectively. Moreover, the largest proportion of cases in children aged less than 5 years is due to meningococcal serotype B, which is not included in the any of the vaccines, she noted.
Surveillance data from 1993-2009 show that while 86% of children under 5 years of age who contract meningococcal disease are hospitalized, the case-fatality ratio is low, ranging from 1% for serogroup Y to 10% for serogroup C. A large majority – 75%-80% of children under 5 years with meningococcal disease – survives and recover. Major complications – including skin necrosis, amputation, hearing loss, and death – are less frequent in infants than in adolescents, Ms. MacNeil noted.
In a cost-effectiveness analysis presented by Ismael Ortega-Sanchez, Ph.D., the cost per quality-adjusted life year (QALY) saved for either giving four doses of the vaccine to infants at 2, 4, 6, and 12 months or two doses to toddlers at 9 and 12 months varied with different assumptions of what the vaccine will cost and whether it was given during a season of high or low disease incidence. Assuming a cost of $60 per vaccine dose and an average incidence rate, the costs would be approximately $1,015,000* per QALY saved for infants and $1,036,000* per QALY saved for toddlers.
"Vaccinating infants or toddlers with meningococcal vaccine has a high cost per case prevented, even at a low vaccine price. Cost estimates are much higher than prior analyses because of declining incidence and shorter duration of protection. Infant vaccination prevents twice as many cases as toddler vaccination but at twice the cost," said Dr. Ortega-Sanchez, also with NCIRD.
No conjugate meningococcal vaccine is yet licensed for use in young infants. MenACWY-D (Sanofi Pasteur’s Menactra) is licensed for persons aged 2-55 years as a single dose. In April 2011, it was also licensed as a two-dose series for ages 9-23 months (on a 9- and 12-month schedule). MenACWY-CRM (Novartis’ Menveo), is also licensed as a single dose for ages 2-55 years, and an indication for a 4-dose infant series at 2, 4, 6, and 12 months is under review by the Food and Drug Administration. A combination product, HibMenCY-T (GlaxoSmithKline’s MenHibrix) is under FDA review for a 4 dose infant series, also at 2, 4, 6, and 12 months.
Sanofi Pasteur said in a statement, "With the current incidence of meningococcal disease at an all time low in the United States, a recommendation for children at highest risk is a prudent course of action. CDC has indicated that ACIP would consider making a routine infant recommendation in the future, should U.S. meningococcal disease incidence rates return to the historically higher levels observed prior to the introduction of meningococcal conjugate vaccines in adolescents.
"Sanofi Pasteur supports the ACIP’s recommendation calling for a 2-dose series of meningococcal vaccine for infants and children 9 through 23 months of age who are at high risk for getting meningococcal disease, including those traveling to countries where meningococcal infection is endemic, and those with human immunodeficiency virus (HIV) infection or certain complement component deficiencies. Given the rapid and potentially devastating nature of meningococcal disease and the incidence of infection in children under 1 year of age, we believe this recommendation will make significant strides in helping to protect those at greatest risk for the disease.
And, in a statement from Novartis, "When looking at the incidence of meningococcal disease, it is important to not only look at the number of cases but also the substantial life-long effects of the disease, which include significant psychosocial, economic, and emotional burden for victims’ families and their communities. Novartis believes that health care professionals and parents should have the choice to immunize infants against the potentially devastating consequences of this disease. We will continue our dialogue with the CDC and with ACIP prior to their vote on this matter in February, 2012.
"Infants are at highest risk for meningococcal disease due to the relative immaturity of their immune systems. Approximately 1 in 10 infants under 1 year of age die after contracting meningococcal disease, even with appropriate medical care, and as many as 1 in 5 will suffer serious life-long complications such as limb amputations, seizures, paralysis, hearing loss, and learning disabilities. The health care costs associated with meningococcal disease and the long-term management of its complications are substantial and present a significant public health burden.
"At Novartis, we are committed to eliminating meningococcal disease and we have made a significant effort to develop a safe and effective vaccine to help protect infants."
And, from GSK: "GlaxoSmithKline is committed to making MenHibrix available in the United States. We believe there is a clear public health benefit in recommending meningococcal vaccines for use in infants and toddlers in the United States. Rates of meningococcal disease are highest in infants and toddlers younger than 2 years of age. Approximately 300 cases of meningococcal disease from all serogroups were reported annually in the United States in this age group between 1998 and 2007."
As a CDC employee, Dr. Cohn has no conflicts of interest.
*Correction, 11/8/2011: An earlier version of this story gave the incorrect cost savings per QALY for infants and toddlers with certain doses of the vaccine.
**Correction, 11/9/2011: An earlier version of this story misstated the impact of the adolescent immunization program.
ATLANTA – Routine infant or toddler immunization against meningococcal disease does not appear to be a cost-effective measure, according to an assessment from a working group of the Advisory Committee on Immunization Practices.
That conclusion was presented to the full committee at its October meeting. If a conjugate meningococcal vaccine is licensed for use in infants before the next Centers for Disease Control and Prevention’s (CDC) Advisory Committee on Immunization Practices (ACIP) meeting Feb. 22-23, the committee will vote on it then, said Dr. Amanda Cohn of the CDC’s National Center for Immunization and Respiratory Disease (NCIRD).
The working group’s assessment was based primarily on the low proportion of preventable disease cases and the high cost of vaccinating infants or toddlers with meningococcal vaccine per case prevented. Moreover, although current meningococcal conjugate vaccines are safe and immunogenic, they show evidence of declining antibodies after about 3 years, suggesting that a booster dose at age 6 years would likely be needed to protect children until the routine 11-to 12-year-old (adolescent) immunization, Dr. Cohn noted.
Jessica MacNeil, MPH, also with NCIRD, presented epidemiologic data showing that meningococcal disease has been declining in all age groups. The data do not suggest this is due to the adolescent immunization program.** The first evidence of an impact of the adolescent immunization program was detected in 2008-2009, when the incidence was 0.14 per 100,000 population among those aged 11-19 years and 0.82 among children less than 1 year of age. In contrast, those rates in 2006-2007 were 0.27 and 1.07, respectively. Moreover, the largest proportion of cases in children aged less than 5 years is due to meningococcal serotype B, which is not included in the any of the vaccines, she noted.
Surveillance data from 1993-2009 show that while 86% of children under 5 years of age who contract meningococcal disease are hospitalized, the case-fatality ratio is low, ranging from 1% for serogroup Y to 10% for serogroup C. A large majority – 75%-80% of children under 5 years with meningococcal disease – survives and recover. Major complications – including skin necrosis, amputation, hearing loss, and death – are less frequent in infants than in adolescents, Ms. MacNeil noted.
In a cost-effectiveness analysis presented by Ismael Ortega-Sanchez, Ph.D., the cost per quality-adjusted life year (QALY) saved for either giving four doses of the vaccine to infants at 2, 4, 6, and 12 months or two doses to toddlers at 9 and 12 months varied with different assumptions of what the vaccine will cost and whether it was given during a season of high or low disease incidence. Assuming a cost of $60 per vaccine dose and an average incidence rate, the costs would be approximately $1,015,000* per QALY saved for infants and $1,036,000* per QALY saved for toddlers.
"Vaccinating infants or toddlers with meningococcal vaccine has a high cost per case prevented, even at a low vaccine price. Cost estimates are much higher than prior analyses because of declining incidence and shorter duration of protection. Infant vaccination prevents twice as many cases as toddler vaccination but at twice the cost," said Dr. Ortega-Sanchez, also with NCIRD.
No conjugate meningococcal vaccine is yet licensed for use in young infants. MenACWY-D (Sanofi Pasteur’s Menactra) is licensed for persons aged 2-55 years as a single dose. In April 2011, it was also licensed as a two-dose series for ages 9-23 months (on a 9- and 12-month schedule). MenACWY-CRM (Novartis’ Menveo), is also licensed as a single dose for ages 2-55 years, and an indication for a 4-dose infant series at 2, 4, 6, and 12 months is under review by the Food and Drug Administration. A combination product, HibMenCY-T (GlaxoSmithKline’s MenHibrix) is under FDA review for a 4 dose infant series, also at 2, 4, 6, and 12 months.
Sanofi Pasteur said in a statement, "With the current incidence of meningococcal disease at an all time low in the United States, a recommendation for children at highest risk is a prudent course of action. CDC has indicated that ACIP would consider making a routine infant recommendation in the future, should U.S. meningococcal disease incidence rates return to the historically higher levels observed prior to the introduction of meningococcal conjugate vaccines in adolescents.
"Sanofi Pasteur supports the ACIP’s recommendation calling for a 2-dose series of meningococcal vaccine for infants and children 9 through 23 months of age who are at high risk for getting meningococcal disease, including those traveling to countries where meningococcal infection is endemic, and those with human immunodeficiency virus (HIV) infection or certain complement component deficiencies. Given the rapid and potentially devastating nature of meningococcal disease and the incidence of infection in children under 1 year of age, we believe this recommendation will make significant strides in helping to protect those at greatest risk for the disease.
And, in a statement from Novartis, "When looking at the incidence of meningococcal disease, it is important to not only look at the number of cases but also the substantial life-long effects of the disease, which include significant psychosocial, economic, and emotional burden for victims’ families and their communities. Novartis believes that health care professionals and parents should have the choice to immunize infants against the potentially devastating consequences of this disease. We will continue our dialogue with the CDC and with ACIP prior to their vote on this matter in February, 2012.
"Infants are at highest risk for meningococcal disease due to the relative immaturity of their immune systems. Approximately 1 in 10 infants under 1 year of age die after contracting meningococcal disease, even with appropriate medical care, and as many as 1 in 5 will suffer serious life-long complications such as limb amputations, seizures, paralysis, hearing loss, and learning disabilities. The health care costs associated with meningococcal disease and the long-term management of its complications are substantial and present a significant public health burden.
"At Novartis, we are committed to eliminating meningococcal disease and we have made a significant effort to develop a safe and effective vaccine to help protect infants."
And, from GSK: "GlaxoSmithKline is committed to making MenHibrix available in the United States. We believe there is a clear public health benefit in recommending meningococcal vaccines for use in infants and toddlers in the United States. Rates of meningococcal disease are highest in infants and toddlers younger than 2 years of age. Approximately 300 cases of meningococcal disease from all serogroups were reported annually in the United States in this age group between 1998 and 2007."
As a CDC employee, Dr. Cohn has no conflicts of interest.
*Correction, 11/8/2011: An earlier version of this story gave the incorrect cost savings per QALY for infants and toddlers with certain doses of the vaccine.
**Correction, 11/9/2011: An earlier version of this story misstated the impact of the adolescent immunization program.
ATLANTA – Routine infant or toddler immunization against meningococcal disease does not appear to be a cost-effective measure, according to an assessment from a working group of the Advisory Committee on Immunization Practices.
That conclusion was presented to the full committee at its October meeting. If a conjugate meningococcal vaccine is licensed for use in infants before the next Centers for Disease Control and Prevention’s (CDC) Advisory Committee on Immunization Practices (ACIP) meeting Feb. 22-23, the committee will vote on it then, said Dr. Amanda Cohn of the CDC’s National Center for Immunization and Respiratory Disease (NCIRD).
The working group’s assessment was based primarily on the low proportion of preventable disease cases and the high cost of vaccinating infants or toddlers with meningococcal vaccine per case prevented. Moreover, although current meningococcal conjugate vaccines are safe and immunogenic, they show evidence of declining antibodies after about 3 years, suggesting that a booster dose at age 6 years would likely be needed to protect children until the routine 11-to 12-year-old (adolescent) immunization, Dr. Cohn noted.
Jessica MacNeil, MPH, also with NCIRD, presented epidemiologic data showing that meningococcal disease has been declining in all age groups. The data do not suggest this is due to the adolescent immunization program.** The first evidence of an impact of the adolescent immunization program was detected in 2008-2009, when the incidence was 0.14 per 100,000 population among those aged 11-19 years and 0.82 among children less than 1 year of age. In contrast, those rates in 2006-2007 were 0.27 and 1.07, respectively. Moreover, the largest proportion of cases in children aged less than 5 years is due to meningococcal serotype B, which is not included in the any of the vaccines, she noted.
Surveillance data from 1993-2009 show that while 86% of children under 5 years of age who contract meningococcal disease are hospitalized, the case-fatality ratio is low, ranging from 1% for serogroup Y to 10% for serogroup C. A large majority – 75%-80% of children under 5 years with meningococcal disease – survives and recover. Major complications – including skin necrosis, amputation, hearing loss, and death – are less frequent in infants than in adolescents, Ms. MacNeil noted.
In a cost-effectiveness analysis presented by Ismael Ortega-Sanchez, Ph.D., the cost per quality-adjusted life year (QALY) saved for either giving four doses of the vaccine to infants at 2, 4, 6, and 12 months or two doses to toddlers at 9 and 12 months varied with different assumptions of what the vaccine will cost and whether it was given during a season of high or low disease incidence. Assuming a cost of $60 per vaccine dose and an average incidence rate, the costs would be approximately $1,015,000* per QALY saved for infants and $1,036,000* per QALY saved for toddlers.
"Vaccinating infants or toddlers with meningococcal vaccine has a high cost per case prevented, even at a low vaccine price. Cost estimates are much higher than prior analyses because of declining incidence and shorter duration of protection. Infant vaccination prevents twice as many cases as toddler vaccination but at twice the cost," said Dr. Ortega-Sanchez, also with NCIRD.
No conjugate meningococcal vaccine is yet licensed for use in young infants. MenACWY-D (Sanofi Pasteur’s Menactra) is licensed for persons aged 2-55 years as a single dose. In April 2011, it was also licensed as a two-dose series for ages 9-23 months (on a 9- and 12-month schedule). MenACWY-CRM (Novartis’ Menveo), is also licensed as a single dose for ages 2-55 years, and an indication for a 4-dose infant series at 2, 4, 6, and 12 months is under review by the Food and Drug Administration. A combination product, HibMenCY-T (GlaxoSmithKline’s MenHibrix) is under FDA review for a 4 dose infant series, also at 2, 4, 6, and 12 months.
Sanofi Pasteur said in a statement, "With the current incidence of meningococcal disease at an all time low in the United States, a recommendation for children at highest risk is a prudent course of action. CDC has indicated that ACIP would consider making a routine infant recommendation in the future, should U.S. meningococcal disease incidence rates return to the historically higher levels observed prior to the introduction of meningococcal conjugate vaccines in adolescents.
"Sanofi Pasteur supports the ACIP’s recommendation calling for a 2-dose series of meningococcal vaccine for infants and children 9 through 23 months of age who are at high risk for getting meningococcal disease, including those traveling to countries where meningococcal infection is endemic, and those with human immunodeficiency virus (HIV) infection or certain complement component deficiencies. Given the rapid and potentially devastating nature of meningococcal disease and the incidence of infection in children under 1 year of age, we believe this recommendation will make significant strides in helping to protect those at greatest risk for the disease.
And, in a statement from Novartis, "When looking at the incidence of meningococcal disease, it is important to not only look at the number of cases but also the substantial life-long effects of the disease, which include significant psychosocial, economic, and emotional burden for victims’ families and their communities. Novartis believes that health care professionals and parents should have the choice to immunize infants against the potentially devastating consequences of this disease. We will continue our dialogue with the CDC and with ACIP prior to their vote on this matter in February, 2012.
"Infants are at highest risk for meningococcal disease due to the relative immaturity of their immune systems. Approximately 1 in 10 infants under 1 year of age die after contracting meningococcal disease, even with appropriate medical care, and as many as 1 in 5 will suffer serious life-long complications such as limb amputations, seizures, paralysis, hearing loss, and learning disabilities. The health care costs associated with meningococcal disease and the long-term management of its complications are substantial and present a significant public health burden.
"At Novartis, we are committed to eliminating meningococcal disease and we have made a significant effort to develop a safe and effective vaccine to help protect infants."
And, from GSK: "GlaxoSmithKline is committed to making MenHibrix available in the United States. We believe there is a clear public health benefit in recommending meningococcal vaccines for use in infants and toddlers in the United States. Rates of meningococcal disease are highest in infants and toddlers younger than 2 years of age. Approximately 300 cases of meningococcal disease from all serogroups were reported annually in the United States in this age group between 1998 and 2007."
As a CDC employee, Dr. Cohn has no conflicts of interest.
*Correction, 11/8/2011: An earlier version of this story gave the incorrect cost savings per QALY for infants and toddlers with certain doses of the vaccine.
**Correction, 11/9/2011: An earlier version of this story misstated the impact of the adolescent immunization program.
FROM A MEETING OF CENTERS FOR DISEASE CONTROL AND PREVENTION'S ADVISORY COMMITTEE ON IMMUNIZATION PRACTICES
Prescription Pain Pill Overdoses Quadrupled In Last Decade
Overdose deaths involving opioid pain relievers now exceed deaths from heroin and cocaine combined, according to a report from the Centers for Disease Control and Prevention.
Prescription drug overdoses have been increasing in the United States over the last decade, and by 2008 had reached 36,450 deaths – almost as many as from motor vehicle crashes (39,973). Opioid pain reliever (OPR) sales have also increased, "despite numerous warnings and recommendations over the past decade for voluntary education of providers about more cautious use of OPR[s]," said Dr. Leonard J. Paulozzi and his associates at the CDC’s division of unintentional injury prevention.
During 1999-2008, overdose death rates, sales, and substance abuse treatment admissions related to OPRs increased in parallel, with the overdose death rate in 2008 nearly four times the rate in 1999. Sales of OPRs in 2010 were four times those in 1999. The substance abuse treatment admission rate in 2009 was almost six times the rate in 1999. By 2010, enough OPRs were sold each year to medicate every American adult with 5 mg of hydrocodone every 4 hours for 1 month, the researchers said.
The report used death rates based on the National Vital Statistics System multiple cause of death files, age adjusted to the 2000 U.S. Census population. Deaths were attributed to drug overdose in 2008 at a rate of 11.9/100,000 population. Among those, a particular drug was specified for 74.5%. Of those 27,153 deaths, prescription drugs were a factor in 73.8%. And of those 20,044, OPRs were involved in 73.8% (14,800). Drug overdose death rates were 6.5/100,000 population for all prescription drugs and 4.8/100,000 for OPRs, compared with 2.8/100,000 for illicit drugs, including heroin, cocaine, hallucinogens, and stimulants (MMWR 2011;60:1-6).
Overdose resulted in 830,652 years of potential life lost, a number comparable to that of motor vehicle crashes. Overdose deaths varied fivefold by state, ranging from 5.5/100,000 population in Nebraska to 27.0/100,000 in New Mexico.
Middle-aged whites were more likely to die of an OPR overdose, compared with other races and age groups. Deaths rates involving OPRs among non-Hispanic whites and Native Americans/Alaska Natives were three times higher than the rates for blacks and Hispanic whites. Death rates from all categories of drug overdose were highest among people aged 35-54 years.
The investigators also looked at rates of nonmedical OPR use and annual drug sales, based on data from the 2008-2009 National Survey on Drug Use and Health and the Automation of Reports and Consolidated Orders System of the Drug Enforcement Administration. The prevalence of nonmedical use of OPRs during 2008-2009 ranged from 3.6% in Nebraska to 8.1% in Oklahoma. The rate of OPR sales ranged from 3.7 kg/10,000 population in Illinois to 12.6 kg in Florida. The highest sales rates were clustered in the Southeast and the Northwest.
Differences in OPR overdose mortality by race and ethnicity can’t explain the wide variation in death rates among states, nor can demographic differences fully explain the wide variations among states in the nonmedical use and sales of OPRs. Montana and Iowa, for example, both have largely non-Hispanic white populations. However, Montana’s rate of nonmedical OPR use was 5.3% vs. Iowa’s 3.6%, and Montana’s OPR sales were 8.4 kg/10,000 population, compared with Iowa’s 4.6 kg, Dr. Paulozzi and his associates noted.
In one study, 3% of physicians accounted for 62% of the OPRs prescribed, suggesting that the high-volume prescribers can have a major impact on the use of OPRs and overdose death rates. In Florida, the proliferation of illegitimate pain clinics – also called "pill mills" – appear to have contributed to increases in overdoses in that state.
"Public health interventions to reduce prescription drug overdose must strike a balance between reducing misuse and abuse and safeguarding legitimate access to treatment. To find this balance, health care providers should only use OPR[s] in carefully screened and monitored patients when non-OPR treatments have not been sufficient to treat pain, as recommended in evidence-based guidelines," the investigators wrote.
In addition, state professional licensing boards can take action against prescribers who misuse their licenses, and law enforcement agencies can take action against illegal activities. "Concerted attempts to address this problem, especially in states with high rates of OPR sales, nonmedical use, or overdose mortality, might help control the epidemic," Dr. Paulozzi and his associates concluded.
As CDC employees, the authors have no relevant financial disclosures.
Overdose deaths involving opioid pain relievers now exceed deaths from heroin and cocaine combined, according to a report from the Centers for Disease Control and Prevention.
Prescription drug overdoses have been increasing in the United States over the last decade, and by 2008 had reached 36,450 deaths – almost as many as from motor vehicle crashes (39,973). Opioid pain reliever (OPR) sales have also increased, "despite numerous warnings and recommendations over the past decade for voluntary education of providers about more cautious use of OPR[s]," said Dr. Leonard J. Paulozzi and his associates at the CDC’s division of unintentional injury prevention.
During 1999-2008, overdose death rates, sales, and substance abuse treatment admissions related to OPRs increased in parallel, with the overdose death rate in 2008 nearly four times the rate in 1999. Sales of OPRs in 2010 were four times those in 1999. The substance abuse treatment admission rate in 2009 was almost six times the rate in 1999. By 2010, enough OPRs were sold each year to medicate every American adult with 5 mg of hydrocodone every 4 hours for 1 month, the researchers said.
The report used death rates based on the National Vital Statistics System multiple cause of death files, age adjusted to the 2000 U.S. Census population. Deaths were attributed to drug overdose in 2008 at a rate of 11.9/100,000 population. Among those, a particular drug was specified for 74.5%. Of those 27,153 deaths, prescription drugs were a factor in 73.8%. And of those 20,044, OPRs were involved in 73.8% (14,800). Drug overdose death rates were 6.5/100,000 population for all prescription drugs and 4.8/100,000 for OPRs, compared with 2.8/100,000 for illicit drugs, including heroin, cocaine, hallucinogens, and stimulants (MMWR 2011;60:1-6).
Overdose resulted in 830,652 years of potential life lost, a number comparable to that of motor vehicle crashes. Overdose deaths varied fivefold by state, ranging from 5.5/100,000 population in Nebraska to 27.0/100,000 in New Mexico.
Middle-aged whites were more likely to die of an OPR overdose, compared with other races and age groups. Deaths rates involving OPRs among non-Hispanic whites and Native Americans/Alaska Natives were three times higher than the rates for blacks and Hispanic whites. Death rates from all categories of drug overdose were highest among people aged 35-54 years.
The investigators also looked at rates of nonmedical OPR use and annual drug sales, based on data from the 2008-2009 National Survey on Drug Use and Health and the Automation of Reports and Consolidated Orders System of the Drug Enforcement Administration. The prevalence of nonmedical use of OPRs during 2008-2009 ranged from 3.6% in Nebraska to 8.1% in Oklahoma. The rate of OPR sales ranged from 3.7 kg/10,000 population in Illinois to 12.6 kg in Florida. The highest sales rates were clustered in the Southeast and the Northwest.
Differences in OPR overdose mortality by race and ethnicity can’t explain the wide variation in death rates among states, nor can demographic differences fully explain the wide variations among states in the nonmedical use and sales of OPRs. Montana and Iowa, for example, both have largely non-Hispanic white populations. However, Montana’s rate of nonmedical OPR use was 5.3% vs. Iowa’s 3.6%, and Montana’s OPR sales were 8.4 kg/10,000 population, compared with Iowa’s 4.6 kg, Dr. Paulozzi and his associates noted.
In one study, 3% of physicians accounted for 62% of the OPRs prescribed, suggesting that the high-volume prescribers can have a major impact on the use of OPRs and overdose death rates. In Florida, the proliferation of illegitimate pain clinics – also called "pill mills" – appear to have contributed to increases in overdoses in that state.
"Public health interventions to reduce prescription drug overdose must strike a balance between reducing misuse and abuse and safeguarding legitimate access to treatment. To find this balance, health care providers should only use OPR[s] in carefully screened and monitored patients when non-OPR treatments have not been sufficient to treat pain, as recommended in evidence-based guidelines," the investigators wrote.
In addition, state professional licensing boards can take action against prescribers who misuse their licenses, and law enforcement agencies can take action against illegal activities. "Concerted attempts to address this problem, especially in states with high rates of OPR sales, nonmedical use, or overdose mortality, might help control the epidemic," Dr. Paulozzi and his associates concluded.
As CDC employees, the authors have no relevant financial disclosures.
Overdose deaths involving opioid pain relievers now exceed deaths from heroin and cocaine combined, according to a report from the Centers for Disease Control and Prevention.
Prescription drug overdoses have been increasing in the United States over the last decade, and by 2008 had reached 36,450 deaths – almost as many as from motor vehicle crashes (39,973). Opioid pain reliever (OPR) sales have also increased, "despite numerous warnings and recommendations over the past decade for voluntary education of providers about more cautious use of OPR[s]," said Dr. Leonard J. Paulozzi and his associates at the CDC’s division of unintentional injury prevention.
During 1999-2008, overdose death rates, sales, and substance abuse treatment admissions related to OPRs increased in parallel, with the overdose death rate in 2008 nearly four times the rate in 1999. Sales of OPRs in 2010 were four times those in 1999. The substance abuse treatment admission rate in 2009 was almost six times the rate in 1999. By 2010, enough OPRs were sold each year to medicate every American adult with 5 mg of hydrocodone every 4 hours for 1 month, the researchers said.
The report used death rates based on the National Vital Statistics System multiple cause of death files, age adjusted to the 2000 U.S. Census population. Deaths were attributed to drug overdose in 2008 at a rate of 11.9/100,000 population. Among those, a particular drug was specified for 74.5%. Of those 27,153 deaths, prescription drugs were a factor in 73.8%. And of those 20,044, OPRs were involved in 73.8% (14,800). Drug overdose death rates were 6.5/100,000 population for all prescription drugs and 4.8/100,000 for OPRs, compared with 2.8/100,000 for illicit drugs, including heroin, cocaine, hallucinogens, and stimulants (MMWR 2011;60:1-6).
Overdose resulted in 830,652 years of potential life lost, a number comparable to that of motor vehicle crashes. Overdose deaths varied fivefold by state, ranging from 5.5/100,000 population in Nebraska to 27.0/100,000 in New Mexico.
Middle-aged whites were more likely to die of an OPR overdose, compared with other races and age groups. Deaths rates involving OPRs among non-Hispanic whites and Native Americans/Alaska Natives were three times higher than the rates for blacks and Hispanic whites. Death rates from all categories of drug overdose were highest among people aged 35-54 years.
The investigators also looked at rates of nonmedical OPR use and annual drug sales, based on data from the 2008-2009 National Survey on Drug Use and Health and the Automation of Reports and Consolidated Orders System of the Drug Enforcement Administration. The prevalence of nonmedical use of OPRs during 2008-2009 ranged from 3.6% in Nebraska to 8.1% in Oklahoma. The rate of OPR sales ranged from 3.7 kg/10,000 population in Illinois to 12.6 kg in Florida. The highest sales rates were clustered in the Southeast and the Northwest.
Differences in OPR overdose mortality by race and ethnicity can’t explain the wide variation in death rates among states, nor can demographic differences fully explain the wide variations among states in the nonmedical use and sales of OPRs. Montana and Iowa, for example, both have largely non-Hispanic white populations. However, Montana’s rate of nonmedical OPR use was 5.3% vs. Iowa’s 3.6%, and Montana’s OPR sales were 8.4 kg/10,000 population, compared with Iowa’s 4.6 kg, Dr. Paulozzi and his associates noted.
In one study, 3% of physicians accounted for 62% of the OPRs prescribed, suggesting that the high-volume prescribers can have a major impact on the use of OPRs and overdose death rates. In Florida, the proliferation of illegitimate pain clinics – also called "pill mills" – appear to have contributed to increases in overdoses in that state.
"Public health interventions to reduce prescription drug overdose must strike a balance between reducing misuse and abuse and safeguarding legitimate access to treatment. To find this balance, health care providers should only use OPR[s] in carefully screened and monitored patients when non-OPR treatments have not been sufficient to treat pain, as recommended in evidence-based guidelines," the investigators wrote.
In addition, state professional licensing boards can take action against prescribers who misuse their licenses, and law enforcement agencies can take action against illegal activities. "Concerted attempts to address this problem, especially in states with high rates of OPR sales, nonmedical use, or overdose mortality, might help control the epidemic," Dr. Paulozzi and his associates concluded.
As CDC employees, the authors have no relevant financial disclosures.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
Major Finding: A total of 36,450 deaths were attributed to drug overdose in 2008, for a rate of 11.9/100,000 population.
Data Source: Age-adjusted drug overdose death rates were 6.5/100,000 population for all prescription drugs and 4.8/100,000 for opioid drugs, compared with just 2.8/100,000 for illicit drugs, including heroin, cocaine, hallucinogens, or stimulants.
Disclosures: As Centers for Disease Control and Prevention employees, the authors have no relevant financial disclosures.
Autologous Cells Improve Acne Scarring
WASHINGTON – The autologous cellular product azficel-T produced significant improvement in acne scarring compared with placebo, according to new study results reported at the annual meeting of the American Society for Dermatologic Surgery.
The product, Laviv (Fibrocell Science), was recently approved by the Food and Drug Administration for the treatment of moderate to severe nasolabial folds. It is the first FDA-approved personalized cell therapy for aesthetic use, according to Dr. Girish Munavalli of the dermatology department at Wake Forest University, Winston-Salem, N.C.
In the multicenter, randomized, double-blind, placebo-controlled study, skin biopsies were collected from 119 patients with moderate-to-severe depressed acne scarring of at least 9 cm2 for at least 3 years. The biopsies were used to produce individual fibroblasts for each patient. Up to three injections of 2 mL of autologous fibroblasts (10-20 million cells/mL) were given on one cheek and placebo on the other at 14-day intervals in 99 of the patients. Treatment was administered at a dose of 0.1 mL/cm2 into areas of acne scarring. Adverse events were recorded for each cheek.
At 4 months after completion of the treatment, a statistically significant higher percentage of patients had responded to treatment with azficel-T than to treatment with placebo, as rated by both the study investigators (58.7% vs. 42.2%, respectively) and patients (43.1% vs. 18.3%). Patient and evaluator assessments at earlier time points during the study also showed a significantly higher proportion of responses with azficel-T than with placebo at all but one assessment, reported Dr. Munavalli.
The study successfully met its two prospectively defined end points: a 2-point or greater improvement on a 5-point Subject Live Acne Scarring assessment scale, and a 1-point or greater reduction in cheek acne severity on a physician-assessed, validated 5-point Evaluator Live Acne Scar assessment scale.
All reported adverse events were mild or moderate in severity. No subjects experienced serious adverse events, discontinued treatment, or withdrew from the study. The incidence of adverse events occurring in the treatment areas was comparable between azficel-T and placebo. The most commonly reported adverse events were treatment-area erythema (occurring in 11.1%) and swelling (10.1%). In the azficel-T-treated area, erythema was moderate in 5 of the 12 subjects who reported it, and swelling was moderate in 5 of the 11 who reported it. In contrast, all treatment-related adverse events in the placebo-treated areas were mild.
Other adverse events with a possible relationship to study treatment included bruising, rash, irritation, nodules, pain, acne, induration, and headache, Dr. Munavalli reported.
"Our findings show that using a person's own collagen-producing cells in the form of Laviv may provide a promising new alternative to improve acne scarring with minimal downtime and an excellent safety profile," he noted.
Dr. Munavalli received research funding from Fibrocell Science.
WASHINGTON – The autologous cellular product azficel-T produced significant improvement in acne scarring compared with placebo, according to new study results reported at the annual meeting of the American Society for Dermatologic Surgery.
The product, Laviv (Fibrocell Science), was recently approved by the Food and Drug Administration for the treatment of moderate to severe nasolabial folds. It is the first FDA-approved personalized cell therapy for aesthetic use, according to Dr. Girish Munavalli of the dermatology department at Wake Forest University, Winston-Salem, N.C.
In the multicenter, randomized, double-blind, placebo-controlled study, skin biopsies were collected from 119 patients with moderate-to-severe depressed acne scarring of at least 9 cm2 for at least 3 years. The biopsies were used to produce individual fibroblasts for each patient. Up to three injections of 2 mL of autologous fibroblasts (10-20 million cells/mL) were given on one cheek and placebo on the other at 14-day intervals in 99 of the patients. Treatment was administered at a dose of 0.1 mL/cm2 into areas of acne scarring. Adverse events were recorded for each cheek.
At 4 months after completion of the treatment, a statistically significant higher percentage of patients had responded to treatment with azficel-T than to treatment with placebo, as rated by both the study investigators (58.7% vs. 42.2%, respectively) and patients (43.1% vs. 18.3%). Patient and evaluator assessments at earlier time points during the study also showed a significantly higher proportion of responses with azficel-T than with placebo at all but one assessment, reported Dr. Munavalli.
The study successfully met its two prospectively defined end points: a 2-point or greater improvement on a 5-point Subject Live Acne Scarring assessment scale, and a 1-point or greater reduction in cheek acne severity on a physician-assessed, validated 5-point Evaluator Live Acne Scar assessment scale.
All reported adverse events were mild or moderate in severity. No subjects experienced serious adverse events, discontinued treatment, or withdrew from the study. The incidence of adverse events occurring in the treatment areas was comparable between azficel-T and placebo. The most commonly reported adverse events were treatment-area erythema (occurring in 11.1%) and swelling (10.1%). In the azficel-T-treated area, erythema was moderate in 5 of the 12 subjects who reported it, and swelling was moderate in 5 of the 11 who reported it. In contrast, all treatment-related adverse events in the placebo-treated areas were mild.
Other adverse events with a possible relationship to study treatment included bruising, rash, irritation, nodules, pain, acne, induration, and headache, Dr. Munavalli reported.
"Our findings show that using a person's own collagen-producing cells in the form of Laviv may provide a promising new alternative to improve acne scarring with minimal downtime and an excellent safety profile," he noted.
Dr. Munavalli received research funding from Fibrocell Science.
WASHINGTON – The autologous cellular product azficel-T produced significant improvement in acne scarring compared with placebo, according to new study results reported at the annual meeting of the American Society for Dermatologic Surgery.
The product, Laviv (Fibrocell Science), was recently approved by the Food and Drug Administration for the treatment of moderate to severe nasolabial folds. It is the first FDA-approved personalized cell therapy for aesthetic use, according to Dr. Girish Munavalli of the dermatology department at Wake Forest University, Winston-Salem, N.C.
In the multicenter, randomized, double-blind, placebo-controlled study, skin biopsies were collected from 119 patients with moderate-to-severe depressed acne scarring of at least 9 cm2 for at least 3 years. The biopsies were used to produce individual fibroblasts for each patient. Up to three injections of 2 mL of autologous fibroblasts (10-20 million cells/mL) were given on one cheek and placebo on the other at 14-day intervals in 99 of the patients. Treatment was administered at a dose of 0.1 mL/cm2 into areas of acne scarring. Adverse events were recorded for each cheek.
At 4 months after completion of the treatment, a statistically significant higher percentage of patients had responded to treatment with azficel-T than to treatment with placebo, as rated by both the study investigators (58.7% vs. 42.2%, respectively) and patients (43.1% vs. 18.3%). Patient and evaluator assessments at earlier time points during the study also showed a significantly higher proportion of responses with azficel-T than with placebo at all but one assessment, reported Dr. Munavalli.
The study successfully met its two prospectively defined end points: a 2-point or greater improvement on a 5-point Subject Live Acne Scarring assessment scale, and a 1-point or greater reduction in cheek acne severity on a physician-assessed, validated 5-point Evaluator Live Acne Scar assessment scale.
All reported adverse events were mild or moderate in severity. No subjects experienced serious adverse events, discontinued treatment, or withdrew from the study. The incidence of adverse events occurring in the treatment areas was comparable between azficel-T and placebo. The most commonly reported adverse events were treatment-area erythema (occurring in 11.1%) and swelling (10.1%). In the azficel-T-treated area, erythema was moderate in 5 of the 12 subjects who reported it, and swelling was moderate in 5 of the 11 who reported it. In contrast, all treatment-related adverse events in the placebo-treated areas were mild.
Other adverse events with a possible relationship to study treatment included bruising, rash, irritation, nodules, pain, acne, induration, and headache, Dr. Munavalli reported.
"Our findings show that using a person's own collagen-producing cells in the form of Laviv may provide a promising new alternative to improve acne scarring with minimal downtime and an excellent safety profile," he noted.
Dr. Munavalli received research funding from Fibrocell Science.
FROM THE ANNUAL MEETING OF THE AMERICAN SOCIETY FOR DERMATOLOGIC SURGERY
Major Finding: At 4 months after completion of the treatment, a statistically significant higher percentage of patients had responded to treatment with Laviv than to treatment with placebo, as rated by both the study investigators (58.7% vs. 42.2%, respectively) and patients (43.1% vs. 18.3%).
Data Source: A multicenter, randomized, double-blind, placebo-controlled study of 119 patients who had moderate to severe depressed acne scarring of at least 9 cm2 for at least 3 years.
Disclosures: Dr. Munavalli received research funding from Fibrocell Science.