User login
Poor response to statins hikes risk of cardiovascular events
About half of patients taking statins for hyperlipidemia don’t adequately respond, leaving them at a 22% increased risk of cardiovascular disease, compared with optimal responders. 
Over 6 years, there were about 2,000 more cardiovascular events among those who failed to experience the national treatment target of at least a 40% reduction in LDL cholesterol, according to Stephen F. Weng, MD, and his colleagues. The report is in Heart.
Physicians’ choice of initial statin weighed heavily in the outcomes. Patients who ended up with an optimal response were more likely to get a more potent statin right off, while those with a poorer response were more likely to get a less-potent statin.
“This study provides ‘real world evidence’ that 50% of patients started on statins do not derive the intended therapeutic benefit from them, significantly increasing their risk of future cardiovascular disease,” wrote Dr. Weng of the University of Nottingham, England, and his colleagues. “These findings contribute to the debate on the effectiveness of statin therapy and highlight the need for personalized medicine in lipid management for patients.”
The study comprised 165,411 primary care patients who had hypercholesterolemia but were free of cardiovascular disease at baseline. Statins were prescribed with the goal of at least a 40% reduction in baseline LDL within 24 months of the start of therapy.
Patients had a mean age of 62 years, with a mean baseline LDL of 4.1 mmol/L (158 mg/dL). About 49% were women.
The primary endpoints were the number of patients who did not achieve the 40% or higher reduction in baseline LDL and the between-group risk differences in cardiovascular events (coronary heart disease, stroke or transient ischemic attack, peripheral vascular disease, cardiovascular death).
After 24 months, 51.2% of patients experienced a suboptimal LDL response, with a mean reduction of 2.1 mmol/L (81 mg/dL) compared with 3.1 mmol/L (120 mg/dL). Compared with optimal responders, these patients were significantly more likely to have received a low-potency statin (29% vs. 18%).
Incident cardiovascular events occurred in 14% of the overall group (coronary artery disease, 8%; stroke/TIA, 3%; peripheral vascular disease 1.9%; cardiovascular death, 1%). All of these outcomes were significantly more common among suboptimal responders than optimal responders.
During a mean of 6 years of follow-up, there were 22,798 cardiovascular disease events overall, with significantly more occurring in suboptimal than optimal responders (12,142 vs. 10,656). This translated to a cardiovascular disease rate of 22.6 and 19.7 per 1,000 person-years, respectively.
In a multivariate analysis controlling for age and baseline LDL level, suboptimal responders were 22% more likely to have a cardiovascular disease incident than were optimal responders.
Among suboptimal responders, every unit decrease of 1 mmol/L (39 mg/dL) conferred a significant 6% risk reduction in cardiovascular disease (odds ratio, 0.94).
The benefit was not universal, the authors pointed out. “In this group, the decreased risk remained significant for only stroke/TIA and was not significant for other constituent cardiovascular disease outcomes. However, in patients with an optimal response, an even greater protective effect of LDL reduction and future cardiovascular disease was seen [13%; OR, 0.87],” and this reduction was significant for all of the individual outcomes.
“The study also highlights the benefit of reducing LDL to optimal values, which would lead to better cardiovascular disease outcomes for patients currently on statins,” the authors concluded.
None of the authors had any relevant financial disclosures.
SOURCE: Weng S. et al. Heart 2019 Apr. doi: 10.1136/heartjnl-2018-314253.
Guidelines always look good on paper, but they’re only as good as their implementation, Márcio S. Bittencourt, MD, wrote in an accompanying editorial.
In the United Kingdom, the National Institute for Health and Care Excellence (NICE) guideline pinned effective statin therapy as a lowering of LDL cholesterol by at least 40%. This target aligns well with data accumulated in randomized controlled studies, but it doesn’t benefit patients unless it can be put into practice.
“An important step after a guideline publication is the assessment of its uptake among health practitioners and patients in the real world, as well as of the impact of its adherence on clinical outcomes. These analyses may not only verify its appropriateness, providing feedback for continuous improvement of recommendations, but also identify targets to optimize delivery of health to the society.”
To understand suboptimal statin response, we must understand the many possible reasons behind it – on the part of both physicians and patients.
Physicians may prefer to prescribe low-potency statins for several reasons, including unawareness of guideline recommendations, doubtfulness of better outcomes with higher potent statins or when a lower LDL is attained, and fear of adverse reactions or drug interactions, Dr. Bittencourt noted. “Moreover, doctors may be reluctant to up-titrate drugs when the treatment goals are not achieved, the so-called therapeutic inertia.”
In this study, for example, optimal responders were more likely to initially receive moderately potent statins. Suboptimal responders, on the other hand, were more likely to receive low-potency statins.
“This probably explains why baseline LDL was higher in optimal responders, indicating that higher LDL motivates the physician to be more aggressive upfront.”
Patients bring their own issues to the treatment table.
“Although an inter-individual response to statins may occur according to the genetic background, most cases where LDL response is less than expected are probably due to lack of adherence or persistence to the treatment. ... Of note, poor adherence to lipid-lowering therapy, together with low-intensity therapy, as opposed to high-intensity treatment, is associated with higher cardiovascular risk.”
Effective implementation of guidelines “has been a challenge for a long time. Both physicians and patients should be targets for approaches aiming at improving adherence to guidelines.”
For clinicians, these could include continuing medical education and simplified treatment algorithms. Patients, too, would benefit from some teaching.
“Patients and society should be educated on the scientific evidence documenting the benefits of lipid-lowering therapy, and antistatin propaganda based on pseudoscience should be strongly disavowed and demystified by health authorities.”
Dr. Bittencourt is an internist at the University Hospital San Paolo, Brazil.
Guidelines always look good on paper, but they’re only as good as their implementation, Márcio S. Bittencourt, MD, wrote in an accompanying editorial.
In the United Kingdom, the National Institute for Health and Care Excellence (NICE) guideline pinned effective statin therapy as a lowering of LDL cholesterol by at least 40%. This target aligns well with data accumulated in randomized controlled studies, but it doesn’t benefit patients unless it can be put into practice.
“An important step after a guideline publication is the assessment of its uptake among health practitioners and patients in the real world, as well as of the impact of its adherence on clinical outcomes. These analyses may not only verify its appropriateness, providing feedback for continuous improvement of recommendations, but also identify targets to optimize delivery of health to the society.”
To understand suboptimal statin response, we must understand the many possible reasons behind it – on the part of both physicians and patients.
Physicians may prefer to prescribe low-potency statins for several reasons, including unawareness of guideline recommendations, doubtfulness of better outcomes with higher potent statins or when a lower LDL is attained, and fear of adverse reactions or drug interactions, Dr. Bittencourt noted. “Moreover, doctors may be reluctant to up-titrate drugs when the treatment goals are not achieved, the so-called therapeutic inertia.”
In this study, for example, optimal responders were more likely to initially receive moderately potent statins. Suboptimal responders, on the other hand, were more likely to receive low-potency statins.
“This probably explains why baseline LDL was higher in optimal responders, indicating that higher LDL motivates the physician to be more aggressive upfront.”
Patients bring their own issues to the treatment table.
“Although an inter-individual response to statins may occur according to the genetic background, most cases where LDL response is less than expected are probably due to lack of adherence or persistence to the treatment. ... Of note, poor adherence to lipid-lowering therapy, together with low-intensity therapy, as opposed to high-intensity treatment, is associated with higher cardiovascular risk.”
Effective implementation of guidelines “has been a challenge for a long time. Both physicians and patients should be targets for approaches aiming at improving adherence to guidelines.”
For clinicians, these could include continuing medical education and simplified treatment algorithms. Patients, too, would benefit from some teaching.
“Patients and society should be educated on the scientific evidence documenting the benefits of lipid-lowering therapy, and antistatin propaganda based on pseudoscience should be strongly disavowed and demystified by health authorities.”
Dr. Bittencourt is an internist at the University Hospital San Paolo, Brazil.
Guidelines always look good on paper, but they’re only as good as their implementation, Márcio S. Bittencourt, MD, wrote in an accompanying editorial.
In the United Kingdom, the National Institute for Health and Care Excellence (NICE) guideline pinned effective statin therapy as a lowering of LDL cholesterol by at least 40%. This target aligns well with data accumulated in randomized controlled studies, but it doesn’t benefit patients unless it can be put into practice.
“An important step after a guideline publication is the assessment of its uptake among health practitioners and patients in the real world, as well as of the impact of its adherence on clinical outcomes. These analyses may not only verify its appropriateness, providing feedback for continuous improvement of recommendations, but also identify targets to optimize delivery of health to the society.”
To understand suboptimal statin response, we must understand the many possible reasons behind it – on the part of both physicians and patients.
Physicians may prefer to prescribe low-potency statins for several reasons, including unawareness of guideline recommendations, doubtfulness of better outcomes with higher potent statins or when a lower LDL is attained, and fear of adverse reactions or drug interactions, Dr. Bittencourt noted. “Moreover, doctors may be reluctant to up-titrate drugs when the treatment goals are not achieved, the so-called therapeutic inertia.”
In this study, for example, optimal responders were more likely to initially receive moderately potent statins. Suboptimal responders, on the other hand, were more likely to receive low-potency statins.
“This probably explains why baseline LDL was higher in optimal responders, indicating that higher LDL motivates the physician to be more aggressive upfront.”
Patients bring their own issues to the treatment table.
“Although an inter-individual response to statins may occur according to the genetic background, most cases where LDL response is less than expected are probably due to lack of adherence or persistence to the treatment. ... Of note, poor adherence to lipid-lowering therapy, together with low-intensity therapy, as opposed to high-intensity treatment, is associated with higher cardiovascular risk.”
Effective implementation of guidelines “has been a challenge for a long time. Both physicians and patients should be targets for approaches aiming at improving adherence to guidelines.”
For clinicians, these could include continuing medical education and simplified treatment algorithms. Patients, too, would benefit from some teaching.
“Patients and society should be educated on the scientific evidence documenting the benefits of lipid-lowering therapy, and antistatin propaganda based on pseudoscience should be strongly disavowed and demystified by health authorities.”
Dr. Bittencourt is an internist at the University Hospital San Paolo, Brazil.
About half of patients taking statins for hyperlipidemia don’t adequately respond, leaving them at a 22% increased risk of cardiovascular disease, compared with optimal responders.
Over 6 years, there were about 2,000 more cardiovascular events among those who failed to experience the national treatment target of at least a 40% reduction in LDL cholesterol, according to Stephen F. Weng, MD, and his colleagues. The report is in Heart.
Physicians’ choice of initial statin weighed heavily in the outcomes. Patients who ended up with an optimal response were more likely to get a more potent statin right off, while those with a poorer response were more likely to get a less-potent statin.
“This study provides ‘real world evidence’ that 50% of patients started on statins do not derive the intended therapeutic benefit from them, significantly increasing their risk of future cardiovascular disease,” wrote Dr. Weng of the University of Nottingham, England, and his colleagues. “These findings contribute to the debate on the effectiveness of statin therapy and highlight the need for personalized medicine in lipid management for patients.”
The study comprised 165,411 primary care patients who had hypercholesterolemia but were free of cardiovascular disease at baseline. Statins were prescribed with the goal of at least a 40% reduction in baseline LDL within 24 months of the start of therapy.
Patients had a mean age of 62 years, with a mean baseline LDL of 4.1 mmol/L (158 mg/dL). About 49% were women.
The primary endpoints were the number of patients who did not achieve the 40% or higher reduction in baseline LDL and the between-group risk differences in cardiovascular events (coronary heart disease, stroke or transient ischemic attack, peripheral vascular disease, cardiovascular death).
After 24 months, 51.2% of patients experienced a suboptimal LDL response, with a mean reduction of 2.1 mmol/L (81 mg/dL) compared with 3.1 mmol/L (120 mg/dL). Compared with optimal responders, these patients were significantly more likely to have received a low-potency statin (29% vs. 18%).
Incident cardiovascular events occurred in 14% of the overall group (coronary artery disease, 8%; stroke/TIA, 3%; peripheral vascular disease 1.9%; cardiovascular death, 1%). All of these outcomes were significantly more common among suboptimal responders than optimal responders.
During a mean of 6 years of follow-up, there were 22,798 cardiovascular disease events overall, with significantly more occurring in suboptimal than optimal responders (12,142 vs. 10,656). This translated to a cardiovascular disease rate of 22.6 and 19.7 per 1,000 person-years, respectively.
In a multivariate analysis controlling for age and baseline LDL level, suboptimal responders were 22% more likely to have a cardiovascular disease incident than were optimal responders.
Among suboptimal responders, every unit decrease of 1 mmol/L (39 mg/dL) conferred a significant 6% risk reduction in cardiovascular disease (odds ratio, 0.94).
The benefit was not universal, the authors pointed out. “In this group, the decreased risk remained significant for only stroke/TIA and was not significant for other constituent cardiovascular disease outcomes. However, in patients with an optimal response, an even greater protective effect of LDL reduction and future cardiovascular disease was seen [13%; OR, 0.87],” and this reduction was significant for all of the individual outcomes.
“The study also highlights the benefit of reducing LDL to optimal values, which would lead to better cardiovascular disease outcomes for patients currently on statins,” the authors concluded.
None of the authors had any relevant financial disclosures.
SOURCE: Weng S. et al. Heart 2019 Apr. doi: 10.1136/heartjnl-2018-314253.
About half of patients taking statins for hyperlipidemia don’t adequately respond, leaving them at a 22% increased risk of cardiovascular disease, compared with optimal responders.
Over 6 years, there were about 2,000 more cardiovascular events among those who failed to experience the national treatment target of at least a 40% reduction in LDL cholesterol, according to Stephen F. Weng, MD, and his colleagues. The report is in Heart.
Physicians’ choice of initial statin weighed heavily in the outcomes. Patients who ended up with an optimal response were more likely to get a more potent statin right off, while those with a poorer response were more likely to get a less-potent statin.
“This study provides ‘real world evidence’ that 50% of patients started on statins do not derive the intended therapeutic benefit from them, significantly increasing their risk of future cardiovascular disease,” wrote Dr. Weng of the University of Nottingham, England, and his colleagues. “These findings contribute to the debate on the effectiveness of statin therapy and highlight the need for personalized medicine in lipid management for patients.”
The study comprised 165,411 primary care patients who had hypercholesterolemia but were free of cardiovascular disease at baseline. Statins were prescribed with the goal of at least a 40% reduction in baseline LDL within 24 months of the start of therapy.
Patients had a mean age of 62 years, with a mean baseline LDL of 4.1 mmol/L (158 mg/dL). About 49% were women.
The primary endpoints were the number of patients who did not achieve the 40% or higher reduction in baseline LDL and the between-group risk differences in cardiovascular events (coronary heart disease, stroke or transient ischemic attack, peripheral vascular disease, cardiovascular death).
After 24 months, 51.2% of patients experienced a suboptimal LDL response, with a mean reduction of 2.1 mmol/L (81 mg/dL) compared with 3.1 mmol/L (120 mg/dL). Compared with optimal responders, these patients were significantly more likely to have received a low-potency statin (29% vs. 18%).
Incident cardiovascular events occurred in 14% of the overall group (coronary artery disease, 8%; stroke/TIA, 3%; peripheral vascular disease 1.9%; cardiovascular death, 1%). All of these outcomes were significantly more common among suboptimal responders than optimal responders.
During a mean of 6 years of follow-up, there were 22,798 cardiovascular disease events overall, with significantly more occurring in suboptimal than optimal responders (12,142 vs. 10,656). This translated to a cardiovascular disease rate of 22.6 and 19.7 per 1,000 person-years, respectively.
In a multivariate analysis controlling for age and baseline LDL level, suboptimal responders were 22% more likely to have a cardiovascular disease incident than were optimal responders.
Among suboptimal responders, every unit decrease of 1 mmol/L (39 mg/dL) conferred a significant 6% risk reduction in cardiovascular disease (odds ratio, 0.94).
The benefit was not universal, the authors pointed out. “In this group, the decreased risk remained significant for only stroke/TIA and was not significant for other constituent cardiovascular disease outcomes. However, in patients with an optimal response, an even greater protective effect of LDL reduction and future cardiovascular disease was seen [13%; OR, 0.87],” and this reduction was significant for all of the individual outcomes.
“The study also highlights the benefit of reducing LDL to optimal values, which would lead to better cardiovascular disease outcomes for patients currently on statins,” the authors concluded.
None of the authors had any relevant financial disclosures.
SOURCE: Weng S. et al. Heart 2019 Apr. doi: 10.1136/heartjnl-2018-314253.
FROM HEART
FDA orders companies to cease all sales of transvaginal mesh for POP repair

The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
BACE-1 inhibition worsens cognition in patients with prodromal Alzheimer’s disease
More bad news for Alzheimer’s research. Two more BACE inhibitors fall far short of the finish line.
Declining cognitive scores, falls, suicidal ideation, and liver enzyme abnormalities were all seen in clinical trials.
The news doesn’t bode well for the therapeutic target of BACE (beta-site APP cleaving enzyme) inhibition. BACE is one of the enzymes that trims the amyloid precursor protein (APP). Inhibiting it does reduce the amount of toxic amyloid-beta in cerebrospinal fluid, and amyloid plaque in the brain. But none of these molecules has shown clinical benefit in dementia patients, whether their disease is mild, or moderate or – now – prodromal. And it is now apparent that inhibiting BACE also produces serious off-target problems.
“BACE-1 inhibition certainly seemed to have a sound rationale assuming the basis for amyloid’s role in Alzheimer’s disease pathogenesis is a gain of toxicity,” Richard J. Caselli, MD, of the Mayo Clinic, Rochester, Minn., said in an interview. “That APP is important for Alzheimer’s pathogenesis still seems clear but whether amyloid-beta toxicity is the driving force is no longer clear. Further, interruption of the APP system disrupts more than amyloid-beta peptide, possibly explaining the adverse cognitive effects of BACE-1 inhibition shown exhibited now by three different BACE-1 inhibitors.”
Verubecestat
Researchers got their first dose of bad news regarding verubecestat at the 2017 Clinical Trials in Alzheimer’s Disease meeting. There, Michael F. Egan, MD, Merck’s associate vice president of clinical neuroscience, discussed the molecule’s failure to slow cognitive decline in patients with mild to moderate Alzheimer’s disease. There was plenty of biomarker evidence that the drug did block amyloid-beta production, but there also was a plethora of concerning adverse events, Dr. Egan said in an interview.
However, verubecestat still was being pursued in patients with prodromal Alzheimer’s disease. In February, Merck stopped the trial after a futility analysis and announced that the company was terminating studies of verubecestat in that population as well. In the April 11 issue of the New England Journal of Medicine, Dr. Egan and his colleagues report the full extent of verubecestat’s failure in prodromal patients, and the accompanying adverse events.
At the time of termination, 1,454 patients had been enrolled. Of these, 485 received 12 mg/day, 484 received 40 mg/day, and 485 received placebo. About half of each group completed 104 weeks of treatment in the study, which was designed to extend up to 5 years.
The primary outcome was change in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB). Seven secondary outcomes examined other cognitive and functional end points, along with changes in hippocampal volume on MRI and amyloid burden as determined in PET imaging.
Not only did verubecestat fail to slow cognitive decline, it appeared to exacerbate it. The mean change on the CDR-SB was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group, favoring placebo.
“In an exploratory analysis according to time point, scores on the CDR-SB were also higher [signifying more impairment of cognition and daily functioning] in the 40-mg group than in the placebo group at 13, 26, and 52 weeks ... suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group,” the investigators said.
Verubecestat also was associated with more conversions to Alzheimer’s disease. Per 100 patient-years, the Alzheimer’s disease event rates were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group. Compared with placebo, those taking 12-mg doses were 30% more likely to develop Alzheimer’s disease and those taking 40-mg doses were 38% more likely. The findings suggest that “verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease,” the investigators said.
The negative impact of verubecestat was apparent quite early in the study. “In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the [Composite Cognition Score-3 Domain] and the ADAS-Cog [Alzheimer’s Disease Assessment Scale–Cognitive Subscale] measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter.”
Results of the secondary end points, including the ADAS-Cog and the Mini-Mental State Exam, also indicated that verubecestat may have worsened cognitive performance.
Imaging outcomes were positive, however. Hippocampal volume was 6,448 mL in the 12-mg group, 6,469 mL in the 40-mg group, and 6,435 mL in the placebo group. Brain amyloid increased in the placebo group, as expected, and decreased in the active groups. The small group of patients who underwent cerebrospinal fluid sampling showed reductions of more than 60% in amyloid-beta and soluble APP-beta associated with verubecestat. These results show that the molecule was indeed hitting its intended target, but that doing so was not clinically beneficial.
Adverse events were more common in the verubecestat groups. These included rash, dermatitis, urticaria, sleep disturbance, weight loss, and cough. Hair coloring changed in 2.5% of patients in the 12-mg group and 5% of the 40-mg group, but in none of the subjects taking placebo.
Patients taking verubecestat were more likely to sustain falls and injuries and to express suicidal ideation.
The results of this trial differ from the study of verubecestat in mild to moderate Alzheimer’s disease, the investigators noted. Those patients did not decline cognitively as did those with prodromal disease.
“Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function. It is also possible that the effects of verubecestat are due to inhibition of BACE-2,” they said.
Atabecestat
In a research letter in the same issue of the New England Journal of Medicine (2019 Apr 11;380:1483-5), David Henley, MD, senior director of Janssen’s Alzheimer’s clinical development core, released similarly negative results from an interim analysis of EARLY (Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia) trial, a randomized study of the BACE-1 inhibitor candidate, atabecestat.
The phase 2 trial enrolled 557 patients with prodromal Alzheimer’s disease. The primary cognitive end point was change from baseline in the Preclinical Alzheimer’s Cognitive Composite (PACC) score.
This trial was discontinued in May 2018 because of liver-related adverse events, although safety follow-up continues. The research letter did not disclose details of the hepatic events, but a company press release from May 2018 referred to them in a general sense.
“Elevations of liver enzymes, which were serious in nature, have been observed in some study participants who received the Janssen BACE inhibitor, atabecestat. After a thorough evaluation of all available liver safety data from our studies, Janssen has concluded that the benefit-risk ratio is no longer favorable to continue development of atabecestat for people who have late-onset preclinical stage Alzheimer’s disease.”
Patients in EARLY were randomized to 5 mg, 25 mg, or placebo. As in the verubecestat trial, those randomized to placebo did better. The mean changes from baseline in the PACC score were −1.44 in the 25-mg group, −0.58 in the 5-mg group, and −0.32 in the placebo group.
“At month 6, the difference between the 25-mg group and the placebo group was −1.12 and the difference between the 5-mg group and the placebo group was −0.26, favoring placebo over the higher dose,” the authors said.
This theme reemerged in a secondary end point, the Repeatable Battery for the Assessment of Neuropsychological Status. The 25-mg group declined 3.58 points more than placebo, and the 5-mg group, 1.43 points more.
Adverse events were more common in the active groups and included depression, effects on sleep and dreams, and anxiety.
“The differences in cognitive performance between the groups are of uncertain clinical significance; however, given similar findings favoring placebo over BACE-1 inhibitors in other trial, we are communicating this potential signal of worsening cognitive function in the treated groups,” Dr. Henley said.
SOURCE: Egan MF et al. N Engl J Med. 2019 Apr 11;380:1408-20.
“Some trials fail because the experimental treatment proves to be no different than a control or standard intervention,” David Knopman, MD, wrote in an accompanying editorial (N Engl J Med. 2019 Apr 11;380:1476-8). “Others fail because of unacceptable side effects. In this issue of the Journal, an article by Egan et al. and a letter to the editor by Henley et al. (N Engl J Med. 2019 Apr 11;380:1483-5) describe a third reason for failure – a treatment worsens the target symptoms.
Certainly, beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibition makes sense when viewed in the light of the current understanding of Alzheimer’s disease neuropathology. The amyloid cascade hypothesis holds that toxic amyloid-beta fragments accumulate in the brain, form dense neuritic plaques, and lead to neuronal death and cognitive decline.
“The model is rooted in the inseparability of Alzheimer’s disease from abundant amyloid-beta pathologic features,” Dr. Knopman wrote. But, “Over the past 2 decades, the amyloid-beta–lowering strategy has been put to the test in trials of antiamyloid antibodies, none of which have been successful.”
Therefore, hitting amyloid at the source – the transmembrane cleavage domain – seemed important and, potentially, effective. But three BACE inhibitors (verubecestat, atabecestat, and lanabecestat) have shown similarly negative cognitive effects. “Together, these results suggest that preserved BACE-1 activity may be critical to normal synaptic functions. These observations place a limitation on how amyloid-beta lowering can be accomplished.”
It is possible that decreasing the level of BACE inhibition might ameliorate off-target effects and neuronal compromise but still be enough to reduce the generation of toxic amyloid-beta fragments, Dr. Knopman said. But, “Adjustments in the dose to a narrow window of BACE-1 inhibition would be difficult to accomplish in a clinical trial until there are peripheral biomarkers that reflect the activity of the agent in the brain.”
Thus far, most of the studied antiamyloid drugs have indeed reduced amyloid-beta levels, but none of those reductions affected cognition. A rethinking of amyloid-beta’s place in dementia progression may be in order.
“The dissociation between amyloid-beta lowering and cognitive benefits with both BACE-1 inhibition and antiamyloid antibody therapy is troubling. To be blunt, amyloid-beta lowering seems to be an ineffective approach, and it is time to focus on other targets to move therapeutics for Alzheimer’s disease forward.”
Dr. Knopman is a clinical neurologist and Alzheimer’s researcher at the Mayo Clinic, Rochester, Minn.
“Some trials fail because the experimental treatment proves to be no different than a control or standard intervention,” David Knopman, MD, wrote in an accompanying editorial (N Engl J Med. 2019 Apr 11;380:1476-8). “Others fail because of unacceptable side effects. In this issue of the Journal, an article by Egan et al. and a letter to the editor by Henley et al. (N Engl J Med. 2019 Apr 11;380:1483-5) describe a third reason for failure – a treatment worsens the target symptoms.
Certainly, beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibition makes sense when viewed in the light of the current understanding of Alzheimer’s disease neuropathology. The amyloid cascade hypothesis holds that toxic amyloid-beta fragments accumulate in the brain, form dense neuritic plaques, and lead to neuronal death and cognitive decline.
“The model is rooted in the inseparability of Alzheimer’s disease from abundant amyloid-beta pathologic features,” Dr. Knopman wrote. But, “Over the past 2 decades, the amyloid-beta–lowering strategy has been put to the test in trials of antiamyloid antibodies, none of which have been successful.”
Therefore, hitting amyloid at the source – the transmembrane cleavage domain – seemed important and, potentially, effective. But three BACE inhibitors (verubecestat, atabecestat, and lanabecestat) have shown similarly negative cognitive effects. “Together, these results suggest that preserved BACE-1 activity may be critical to normal synaptic functions. These observations place a limitation on how amyloid-beta lowering can be accomplished.”
It is possible that decreasing the level of BACE inhibition might ameliorate off-target effects and neuronal compromise but still be enough to reduce the generation of toxic amyloid-beta fragments, Dr. Knopman said. But, “Adjustments in the dose to a narrow window of BACE-1 inhibition would be difficult to accomplish in a clinical trial until there are peripheral biomarkers that reflect the activity of the agent in the brain.”
Thus far, most of the studied antiamyloid drugs have indeed reduced amyloid-beta levels, but none of those reductions affected cognition. A rethinking of amyloid-beta’s place in dementia progression may be in order.
“The dissociation between amyloid-beta lowering and cognitive benefits with both BACE-1 inhibition and antiamyloid antibody therapy is troubling. To be blunt, amyloid-beta lowering seems to be an ineffective approach, and it is time to focus on other targets to move therapeutics for Alzheimer’s disease forward.”
Dr. Knopman is a clinical neurologist and Alzheimer’s researcher at the Mayo Clinic, Rochester, Minn.
“Some trials fail because the experimental treatment proves to be no different than a control or standard intervention,” David Knopman, MD, wrote in an accompanying editorial (N Engl J Med. 2019 Apr 11;380:1476-8). “Others fail because of unacceptable side effects. In this issue of the Journal, an article by Egan et al. and a letter to the editor by Henley et al. (N Engl J Med. 2019 Apr 11;380:1483-5) describe a third reason for failure – a treatment worsens the target symptoms.
Certainly, beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibition makes sense when viewed in the light of the current understanding of Alzheimer’s disease neuropathology. The amyloid cascade hypothesis holds that toxic amyloid-beta fragments accumulate in the brain, form dense neuritic plaques, and lead to neuronal death and cognitive decline.
“The model is rooted in the inseparability of Alzheimer’s disease from abundant amyloid-beta pathologic features,” Dr. Knopman wrote. But, “Over the past 2 decades, the amyloid-beta–lowering strategy has been put to the test in trials of antiamyloid antibodies, none of which have been successful.”
Therefore, hitting amyloid at the source – the transmembrane cleavage domain – seemed important and, potentially, effective. But three BACE inhibitors (verubecestat, atabecestat, and lanabecestat) have shown similarly negative cognitive effects. “Together, these results suggest that preserved BACE-1 activity may be critical to normal synaptic functions. These observations place a limitation on how amyloid-beta lowering can be accomplished.”
It is possible that decreasing the level of BACE inhibition might ameliorate off-target effects and neuronal compromise but still be enough to reduce the generation of toxic amyloid-beta fragments, Dr. Knopman said. But, “Adjustments in the dose to a narrow window of BACE-1 inhibition would be difficult to accomplish in a clinical trial until there are peripheral biomarkers that reflect the activity of the agent in the brain.”
Thus far, most of the studied antiamyloid drugs have indeed reduced amyloid-beta levels, but none of those reductions affected cognition. A rethinking of amyloid-beta’s place in dementia progression may be in order.
“The dissociation between amyloid-beta lowering and cognitive benefits with both BACE-1 inhibition and antiamyloid antibody therapy is troubling. To be blunt, amyloid-beta lowering seems to be an ineffective approach, and it is time to focus on other targets to move therapeutics for Alzheimer’s disease forward.”
Dr. Knopman is a clinical neurologist and Alzheimer’s researcher at the Mayo Clinic, Rochester, Minn.
More bad news for Alzheimer’s research. Two more BACE inhibitors fall far short of the finish line.
Declining cognitive scores, falls, suicidal ideation, and liver enzyme abnormalities were all seen in clinical trials.
The news doesn’t bode well for the therapeutic target of BACE (beta-site APP cleaving enzyme) inhibition. BACE is one of the enzymes that trims the amyloid precursor protein (APP). Inhibiting it does reduce the amount of toxic amyloid-beta in cerebrospinal fluid, and amyloid plaque in the brain. But none of these molecules has shown clinical benefit in dementia patients, whether their disease is mild, or moderate or – now – prodromal. And it is now apparent that inhibiting BACE also produces serious off-target problems.
“BACE-1 inhibition certainly seemed to have a sound rationale assuming the basis for amyloid’s role in Alzheimer’s disease pathogenesis is a gain of toxicity,” Richard J. Caselli, MD, of the Mayo Clinic, Rochester, Minn., said in an interview. “That APP is important for Alzheimer’s pathogenesis still seems clear but whether amyloid-beta toxicity is the driving force is no longer clear. Further, interruption of the APP system disrupts more than amyloid-beta peptide, possibly explaining the adverse cognitive effects of BACE-1 inhibition shown exhibited now by three different BACE-1 inhibitors.”
Verubecestat
Researchers got their first dose of bad news regarding verubecestat at the 2017 Clinical Trials in Alzheimer’s Disease meeting. There, Michael F. Egan, MD, Merck’s associate vice president of clinical neuroscience, discussed the molecule’s failure to slow cognitive decline in patients with mild to moderate Alzheimer’s disease. There was plenty of biomarker evidence that the drug did block amyloid-beta production, but there also was a plethora of concerning adverse events, Dr. Egan said in an interview.
However, verubecestat still was being pursued in patients with prodromal Alzheimer’s disease. In February, Merck stopped the trial after a futility analysis and announced that the company was terminating studies of verubecestat in that population as well. In the April 11 issue of the New England Journal of Medicine, Dr. Egan and his colleagues report the full extent of verubecestat’s failure in prodromal patients, and the accompanying adverse events.
At the time of termination, 1,454 patients had been enrolled. Of these, 485 received 12 mg/day, 484 received 40 mg/day, and 485 received placebo. About half of each group completed 104 weeks of treatment in the study, which was designed to extend up to 5 years.
The primary outcome was change in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB). Seven secondary outcomes examined other cognitive and functional end points, along with changes in hippocampal volume on MRI and amyloid burden as determined in PET imaging.
Not only did verubecestat fail to slow cognitive decline, it appeared to exacerbate it. The mean change on the CDR-SB was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group, favoring placebo.
“In an exploratory analysis according to time point, scores on the CDR-SB were also higher [signifying more impairment of cognition and daily functioning] in the 40-mg group than in the placebo group at 13, 26, and 52 weeks ... suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group,” the investigators said.
Verubecestat also was associated with more conversions to Alzheimer’s disease. Per 100 patient-years, the Alzheimer’s disease event rates were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group. Compared with placebo, those taking 12-mg doses were 30% more likely to develop Alzheimer’s disease and those taking 40-mg doses were 38% more likely. The findings suggest that “verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease,” the investigators said.
The negative impact of verubecestat was apparent quite early in the study. “In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the [Composite Cognition Score-3 Domain] and the ADAS-Cog [Alzheimer’s Disease Assessment Scale–Cognitive Subscale] measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter.”
Results of the secondary end points, including the ADAS-Cog and the Mini-Mental State Exam, also indicated that verubecestat may have worsened cognitive performance.
Imaging outcomes were positive, however. Hippocampal volume was 6,448 mL in the 12-mg group, 6,469 mL in the 40-mg group, and 6,435 mL in the placebo group. Brain amyloid increased in the placebo group, as expected, and decreased in the active groups. The small group of patients who underwent cerebrospinal fluid sampling showed reductions of more than 60% in amyloid-beta and soluble APP-beta associated with verubecestat. These results show that the molecule was indeed hitting its intended target, but that doing so was not clinically beneficial.
Adverse events were more common in the verubecestat groups. These included rash, dermatitis, urticaria, sleep disturbance, weight loss, and cough. Hair coloring changed in 2.5% of patients in the 12-mg group and 5% of the 40-mg group, but in none of the subjects taking placebo.
Patients taking verubecestat were more likely to sustain falls and injuries and to express suicidal ideation.
The results of this trial differ from the study of verubecestat in mild to moderate Alzheimer’s disease, the investigators noted. Those patients did not decline cognitively as did those with prodromal disease.
“Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function. It is also possible that the effects of verubecestat are due to inhibition of BACE-2,” they said.
Atabecestat
In a research letter in the same issue of the New England Journal of Medicine (2019 Apr 11;380:1483-5), David Henley, MD, senior director of Janssen’s Alzheimer’s clinical development core, released similarly negative results from an interim analysis of EARLY (Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia) trial, a randomized study of the BACE-1 inhibitor candidate, atabecestat.
The phase 2 trial enrolled 557 patients with prodromal Alzheimer’s disease. The primary cognitive end point was change from baseline in the Preclinical Alzheimer’s Cognitive Composite (PACC) score.
This trial was discontinued in May 2018 because of liver-related adverse events, although safety follow-up continues. The research letter did not disclose details of the hepatic events, but a company press release from May 2018 referred to them in a general sense.
“Elevations of liver enzymes, which were serious in nature, have been observed in some study participants who received the Janssen BACE inhibitor, atabecestat. After a thorough evaluation of all available liver safety data from our studies, Janssen has concluded that the benefit-risk ratio is no longer favorable to continue development of atabecestat for people who have late-onset preclinical stage Alzheimer’s disease.”
Patients in EARLY were randomized to 5 mg, 25 mg, or placebo. As in the verubecestat trial, those randomized to placebo did better. The mean changes from baseline in the PACC score were −1.44 in the 25-mg group, −0.58 in the 5-mg group, and −0.32 in the placebo group.
“At month 6, the difference between the 25-mg group and the placebo group was −1.12 and the difference between the 5-mg group and the placebo group was −0.26, favoring placebo over the higher dose,” the authors said.
This theme reemerged in a secondary end point, the Repeatable Battery for the Assessment of Neuropsychological Status. The 25-mg group declined 3.58 points more than placebo, and the 5-mg group, 1.43 points more.
Adverse events were more common in the active groups and included depression, effects on sleep and dreams, and anxiety.
“The differences in cognitive performance between the groups are of uncertain clinical significance; however, given similar findings favoring placebo over BACE-1 inhibitors in other trial, we are communicating this potential signal of worsening cognitive function in the treated groups,” Dr. Henley said.
SOURCE: Egan MF et al. N Engl J Med. 2019 Apr 11;380:1408-20.
More bad news for Alzheimer’s research. Two more BACE inhibitors fall far short of the finish line.
Declining cognitive scores, falls, suicidal ideation, and liver enzyme abnormalities were all seen in clinical trials.
The news doesn’t bode well for the therapeutic target of BACE (beta-site APP cleaving enzyme) inhibition. BACE is one of the enzymes that trims the amyloid precursor protein (APP). Inhibiting it does reduce the amount of toxic amyloid-beta in cerebrospinal fluid, and amyloid plaque in the brain. But none of these molecules has shown clinical benefit in dementia patients, whether their disease is mild, or moderate or – now – prodromal. And it is now apparent that inhibiting BACE also produces serious off-target problems.
“BACE-1 inhibition certainly seemed to have a sound rationale assuming the basis for amyloid’s role in Alzheimer’s disease pathogenesis is a gain of toxicity,” Richard J. Caselli, MD, of the Mayo Clinic, Rochester, Minn., said in an interview. “That APP is important for Alzheimer’s pathogenesis still seems clear but whether amyloid-beta toxicity is the driving force is no longer clear. Further, interruption of the APP system disrupts more than amyloid-beta peptide, possibly explaining the adverse cognitive effects of BACE-1 inhibition shown exhibited now by three different BACE-1 inhibitors.”
Verubecestat
Researchers got their first dose of bad news regarding verubecestat at the 2017 Clinical Trials in Alzheimer’s Disease meeting. There, Michael F. Egan, MD, Merck’s associate vice president of clinical neuroscience, discussed the molecule’s failure to slow cognitive decline in patients with mild to moderate Alzheimer’s disease. There was plenty of biomarker evidence that the drug did block amyloid-beta production, but there also was a plethora of concerning adverse events, Dr. Egan said in an interview.
However, verubecestat still was being pursued in patients with prodromal Alzheimer’s disease. In February, Merck stopped the trial after a futility analysis and announced that the company was terminating studies of verubecestat in that population as well. In the April 11 issue of the New England Journal of Medicine, Dr. Egan and his colleagues report the full extent of verubecestat’s failure in prodromal patients, and the accompanying adverse events.
At the time of termination, 1,454 patients had been enrolled. Of these, 485 received 12 mg/day, 484 received 40 mg/day, and 485 received placebo. About half of each group completed 104 weeks of treatment in the study, which was designed to extend up to 5 years.
The primary outcome was change in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB). Seven secondary outcomes examined other cognitive and functional end points, along with changes in hippocampal volume on MRI and amyloid burden as determined in PET imaging.
Not only did verubecestat fail to slow cognitive decline, it appeared to exacerbate it. The mean change on the CDR-SB was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group, favoring placebo.
“In an exploratory analysis according to time point, scores on the CDR-SB were also higher [signifying more impairment of cognition and daily functioning] in the 40-mg group than in the placebo group at 13, 26, and 52 weeks ... suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group,” the investigators said.
Verubecestat also was associated with more conversions to Alzheimer’s disease. Per 100 patient-years, the Alzheimer’s disease event rates were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group. Compared with placebo, those taking 12-mg doses were 30% more likely to develop Alzheimer’s disease and those taking 40-mg doses were 38% more likely. The findings suggest that “verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease,” the investigators said.
The negative impact of verubecestat was apparent quite early in the study. “In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the [Composite Cognition Score-3 Domain] and the ADAS-Cog [Alzheimer’s Disease Assessment Scale–Cognitive Subscale] measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter.”
Results of the secondary end points, including the ADAS-Cog and the Mini-Mental State Exam, also indicated that verubecestat may have worsened cognitive performance.
Imaging outcomes were positive, however. Hippocampal volume was 6,448 mL in the 12-mg group, 6,469 mL in the 40-mg group, and 6,435 mL in the placebo group. Brain amyloid increased in the placebo group, as expected, and decreased in the active groups. The small group of patients who underwent cerebrospinal fluid sampling showed reductions of more than 60% in amyloid-beta and soluble APP-beta associated with verubecestat. These results show that the molecule was indeed hitting its intended target, but that doing so was not clinically beneficial.
Adverse events were more common in the verubecestat groups. These included rash, dermatitis, urticaria, sleep disturbance, weight loss, and cough. Hair coloring changed in 2.5% of patients in the 12-mg group and 5% of the 40-mg group, but in none of the subjects taking placebo.
Patients taking verubecestat were more likely to sustain falls and injuries and to express suicidal ideation.
The results of this trial differ from the study of verubecestat in mild to moderate Alzheimer’s disease, the investigators noted. Those patients did not decline cognitively as did those with prodromal disease.
“Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function. It is also possible that the effects of verubecestat are due to inhibition of BACE-2,” they said.
Atabecestat
In a research letter in the same issue of the New England Journal of Medicine (2019 Apr 11;380:1483-5), David Henley, MD, senior director of Janssen’s Alzheimer’s clinical development core, released similarly negative results from an interim analysis of EARLY (Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia) trial, a randomized study of the BACE-1 inhibitor candidate, atabecestat.
The phase 2 trial enrolled 557 patients with prodromal Alzheimer’s disease. The primary cognitive end point was change from baseline in the Preclinical Alzheimer’s Cognitive Composite (PACC) score.
This trial was discontinued in May 2018 because of liver-related adverse events, although safety follow-up continues. The research letter did not disclose details of the hepatic events, but a company press release from May 2018 referred to them in a general sense.
“Elevations of liver enzymes, which were serious in nature, have been observed in some study participants who received the Janssen BACE inhibitor, atabecestat. After a thorough evaluation of all available liver safety data from our studies, Janssen has concluded that the benefit-risk ratio is no longer favorable to continue development of atabecestat for people who have late-onset preclinical stage Alzheimer’s disease.”
Patients in EARLY were randomized to 5 mg, 25 mg, or placebo. As in the verubecestat trial, those randomized to placebo did better. The mean changes from baseline in the PACC score were −1.44 in the 25-mg group, −0.58 in the 5-mg group, and −0.32 in the placebo group.
“At month 6, the difference between the 25-mg group and the placebo group was −1.12 and the difference between the 5-mg group and the placebo group was −0.26, favoring placebo over the higher dose,” the authors said.
This theme reemerged in a secondary end point, the Repeatable Battery for the Assessment of Neuropsychological Status. The 25-mg group declined 3.58 points more than placebo, and the 5-mg group, 1.43 points more.
Adverse events were more common in the active groups and included depression, effects on sleep and dreams, and anxiety.
“The differences in cognitive performance between the groups are of uncertain clinical significance; however, given similar findings favoring placebo over BACE-1 inhibitors in other trial, we are communicating this potential signal of worsening cognitive function in the treated groups,” Dr. Henley said.
SOURCE: Egan MF et al. N Engl J Med. 2019 Apr 11;380:1408-20.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Ligelizumab maintains urticaria control for up to 1 year
WASHINGTON – in an open-label extension study, Diane Baker, MD, said at the annual meeting of the American Academy of Dermatology.
About 75% of the cohort experienced complete disease control at least once during the study. Novartis is developing ligelizumab (QGE031) as a treatment option for patients with spontaneous chronic urticaria (CSU) whose symptoms are inadequately controlled by H1-antihistamines. Like omalizumab (Xolair), which is approved in the United States and Europe for treatment of CSU, ligelizumab is a humanized anti-IgE monoclonal antibody. But the investigational agent binds to IgE with greater affinity than omalizumab, said Dr. Baker, a dermatologist who practices in Portland, Ore.
The extension study was a follow-up to a 12-week, phase 2, dose-finding trial of 382 CSU patients. In the study, which was not powered for efficacy endpoints, 51% of those who received 72 mg subcutaneously every 4 weeks had a Hives Severity Score of 0 by week 12, compared with 42% of those who received 240 mg every 4 weeks and 26% of those taking the omalizumab comparator. Additionally, 47% of those in the 72-mg group and 46% of the 240-mg group achieved a score of 0 on another indicator, the Urticaria Activity Score, which measures symptoms over 7 days (UAS7).
The extension study, which evaluated the 240-mg dose, showed the durability of that response, with 52% of those in the 240-mg group maintained a UAS7 of 0 at 1 year, according to Dr. Baker. By the end of the year, most patients (75.8%) had experienced at least one period of complete symptom control, and 84.0% experienced a UAS of 6 or lower at least once.
Adverse events were common in the cohort, with 84% experiencing at least one. But most (78%) were mild or moderate, and there was no clear side effect pattern, Dr. Baker said. Eight patients discontinued treatment because of an adverse event, and another eight dropped out because of lack of efficacy. Other reasons for discontinuation were pregnancy, protocol deviation, and physician or patient decision.
Novartis has launched two 1-year, phase 3 trials randomizing patients to 72 mg or 240 mg of ligelizumab or 300 mg of omalizumab every 4 weeks in a similar patient population, Dr. Baker said. PEARL 1 and PEARL 2, the largest pivotal trials to date in CSU, will enroll more than 2,000 patients, according to a company press release.
Dr. Baker is a clinical trials investigator for Novartis.
SOURCE: Baker D et al. AAD 2019, Session S034.
WASHINGTON – in an open-label extension study, Diane Baker, MD, said at the annual meeting of the American Academy of Dermatology.
About 75% of the cohort experienced complete disease control at least once during the study. Novartis is developing ligelizumab (QGE031) as a treatment option for patients with spontaneous chronic urticaria (CSU) whose symptoms are inadequately controlled by H1-antihistamines. Like omalizumab (Xolair), which is approved in the United States and Europe for treatment of CSU, ligelizumab is a humanized anti-IgE monoclonal antibody. But the investigational agent binds to IgE with greater affinity than omalizumab, said Dr. Baker, a dermatologist who practices in Portland, Ore.
The extension study was a follow-up to a 12-week, phase 2, dose-finding trial of 382 CSU patients. In the study, which was not powered for efficacy endpoints, 51% of those who received 72 mg subcutaneously every 4 weeks had a Hives Severity Score of 0 by week 12, compared with 42% of those who received 240 mg every 4 weeks and 26% of those taking the omalizumab comparator. Additionally, 47% of those in the 72-mg group and 46% of the 240-mg group achieved a score of 0 on another indicator, the Urticaria Activity Score, which measures symptoms over 7 days (UAS7).
The extension study, which evaluated the 240-mg dose, showed the durability of that response, with 52% of those in the 240-mg group maintained a UAS7 of 0 at 1 year, according to Dr. Baker. By the end of the year, most patients (75.8%) had experienced at least one period of complete symptom control, and 84.0% experienced a UAS of 6 or lower at least once.
Adverse events were common in the cohort, with 84% experiencing at least one. But most (78%) were mild or moderate, and there was no clear side effect pattern, Dr. Baker said. Eight patients discontinued treatment because of an adverse event, and another eight dropped out because of lack of efficacy. Other reasons for discontinuation were pregnancy, protocol deviation, and physician or patient decision.
Novartis has launched two 1-year, phase 3 trials randomizing patients to 72 mg or 240 mg of ligelizumab or 300 mg of omalizumab every 4 weeks in a similar patient population, Dr. Baker said. PEARL 1 and PEARL 2, the largest pivotal trials to date in CSU, will enroll more than 2,000 patients, according to a company press release.
Dr. Baker is a clinical trials investigator for Novartis.
SOURCE: Baker D et al. AAD 2019, Session S034.
WASHINGTON – in an open-label extension study, Diane Baker, MD, said at the annual meeting of the American Academy of Dermatology.
About 75% of the cohort experienced complete disease control at least once during the study. Novartis is developing ligelizumab (QGE031) as a treatment option for patients with spontaneous chronic urticaria (CSU) whose symptoms are inadequately controlled by H1-antihistamines. Like omalizumab (Xolair), which is approved in the United States and Europe for treatment of CSU, ligelizumab is a humanized anti-IgE monoclonal antibody. But the investigational agent binds to IgE with greater affinity than omalizumab, said Dr. Baker, a dermatologist who practices in Portland, Ore.
The extension study was a follow-up to a 12-week, phase 2, dose-finding trial of 382 CSU patients. In the study, which was not powered for efficacy endpoints, 51% of those who received 72 mg subcutaneously every 4 weeks had a Hives Severity Score of 0 by week 12, compared with 42% of those who received 240 mg every 4 weeks and 26% of those taking the omalizumab comparator. Additionally, 47% of those in the 72-mg group and 46% of the 240-mg group achieved a score of 0 on another indicator, the Urticaria Activity Score, which measures symptoms over 7 days (UAS7).
The extension study, which evaluated the 240-mg dose, showed the durability of that response, with 52% of those in the 240-mg group maintained a UAS7 of 0 at 1 year, according to Dr. Baker. By the end of the year, most patients (75.8%) had experienced at least one period of complete symptom control, and 84.0% experienced a UAS of 6 or lower at least once.
Adverse events were common in the cohort, with 84% experiencing at least one. But most (78%) were mild or moderate, and there was no clear side effect pattern, Dr. Baker said. Eight patients discontinued treatment because of an adverse event, and another eight dropped out because of lack of efficacy. Other reasons for discontinuation were pregnancy, protocol deviation, and physician or patient decision.
Novartis has launched two 1-year, phase 3 trials randomizing patients to 72 mg or 240 mg of ligelizumab or 300 mg of omalizumab every 4 weeks in a similar patient population, Dr. Baker said. PEARL 1 and PEARL 2, the largest pivotal trials to date in CSU, will enroll more than 2,000 patients, according to a company press release.
Dr. Baker is a clinical trials investigator for Novartis.
SOURCE: Baker D et al. AAD 2019, Session S034.
REPORTING FROM AAD 2019
Pembrolizumab benefits some patients with advanced cervical cancer
The checkpoint inhibitor pembrolizumab conferred a durable tumor response in patients with advanced cervical cancer, who were positive for programmed death ligand 1 (PD-L1).
Among 98 patients, all of whom had progressed despite prior treatment, overall response rate was 12.2% (95% confidence interval, 6.5%-20.4%), with three complete and nine partial responses. All 12 responses were in patients with PD-L1-positive tumors, wrote Hyun Cheol Chung, MD, and his colleagues. The report is in the Journal of Clinical Oncology.
The response curve had not peaked by the end of follow-up, suggesting further benefit of the antibody, said Dr. Chung of Yonsei Cancer Center, Seoul, South Korea. On the basis of these data, first reported at the 2018 meeting of the American Society of Clinical Oncology, the Food and Drug Administration approved the antibody for advanced cervical cancer last year.
“These results show that treatment with pembrolizumab offers a clinically meaningful therapeutic option for a subset of patients with previously treated advanced cervical cancer,” the researchers said. The approval makes pembrolizumab the first immunotherapy approved for the treatment of an advanced gynecologic malignancy.
The phase 2 KEYNOTE-158 study comprised 98 women (median age 46 years) with advanced cervical cancer with progression after chemotherapy. All received pembrolizumab 200 mg every 3 weeks for 2 years or until progression, intolerable toxicity, or physician or patient decision.
Most of the patients (83.7%) were positive for PD-L1. All had received at least one or more lines of chemotherapy before entering the study. The median follow-up was 10 months and the median treatment duration 2.9 months, with a range of 1 day-22 months.
By the study’s end, most patients (85.7%) had experienced disease progression or had died. Overall survival was poor even in the PD-L1-positive patients, with 82.9% experiencing progression or death. Overall mortality was 69.4% and 64.6%, respectively. However, PD-L1 patients did live longer, a median of 11 months, compared with 9.4 months in the entire cohort.
At 6 months, overall survival estimates were 75.2% in the total cohort and 80.2% in the PD-L1 patients; 12-month overall survival estimates were 41.4% and 47.3%, respectively.
However, among the 12 responders with PD-L1-postitive tumors, just one had died by the data cutoff, suggesting that longer treatment with pembrolizumab could be beneficial in this group.
“Responses typically occurred within 2.1 months and were durable, with a median duration of response that had not been reached after a median follow-up of 10.2 months and an estimated 90.9% of responses ongoing at 6 months” Dr. Chung and his colleagues wrote.
Adverse events were common (65.3%) with 12.2% graded as serious. But none were lethal, and only four patients discontinued pembrolizumab because of a treatment-related adverse event.
“Our results from an interim analysis of data from the phase II KEYNOTE-158 clinical trial show that pembrolizumab has promising antitumor activity in patients with previously treated advanced cervical cancer,” the researchers said. “These response rates are similar or superior to those observed with other treatment options in this setting.”
Pembrolizumab is now being evaluated in combination with concurrent chemoradiotherapy and with concurrent versus sequential chemoradiotherapy.
Merck & Co. sponsored the study.
SOURCE: Chung HC et al. J Clin Oncol. 2019 April 3. doi: 10.1200/JCO.18.01265.
The checkpoint inhibitor pembrolizumab conferred a durable tumor response in patients with advanced cervical cancer, who were positive for programmed death ligand 1 (PD-L1).
Among 98 patients, all of whom had progressed despite prior treatment, overall response rate was 12.2% (95% confidence interval, 6.5%-20.4%), with three complete and nine partial responses. All 12 responses were in patients with PD-L1-positive tumors, wrote Hyun Cheol Chung, MD, and his colleagues. The report is in the Journal of Clinical Oncology.
The response curve had not peaked by the end of follow-up, suggesting further benefit of the antibody, said Dr. Chung of Yonsei Cancer Center, Seoul, South Korea. On the basis of these data, first reported at the 2018 meeting of the American Society of Clinical Oncology, the Food and Drug Administration approved the antibody for advanced cervical cancer last year.
“These results show that treatment with pembrolizumab offers a clinically meaningful therapeutic option for a subset of patients with previously treated advanced cervical cancer,” the researchers said. The approval makes pembrolizumab the first immunotherapy approved for the treatment of an advanced gynecologic malignancy.
The phase 2 KEYNOTE-158 study comprised 98 women (median age 46 years) with advanced cervical cancer with progression after chemotherapy. All received pembrolizumab 200 mg every 3 weeks for 2 years or until progression, intolerable toxicity, or physician or patient decision.
Most of the patients (83.7%) were positive for PD-L1. All had received at least one or more lines of chemotherapy before entering the study. The median follow-up was 10 months and the median treatment duration 2.9 months, with a range of 1 day-22 months.
By the study’s end, most patients (85.7%) had experienced disease progression or had died. Overall survival was poor even in the PD-L1-positive patients, with 82.9% experiencing progression or death. Overall mortality was 69.4% and 64.6%, respectively. However, PD-L1 patients did live longer, a median of 11 months, compared with 9.4 months in the entire cohort.
At 6 months, overall survival estimates were 75.2% in the total cohort and 80.2% in the PD-L1 patients; 12-month overall survival estimates were 41.4% and 47.3%, respectively.
However, among the 12 responders with PD-L1-postitive tumors, just one had died by the data cutoff, suggesting that longer treatment with pembrolizumab could be beneficial in this group.
“Responses typically occurred within 2.1 months and were durable, with a median duration of response that had not been reached after a median follow-up of 10.2 months and an estimated 90.9% of responses ongoing at 6 months” Dr. Chung and his colleagues wrote.
Adverse events were common (65.3%) with 12.2% graded as serious. But none were lethal, and only four patients discontinued pembrolizumab because of a treatment-related adverse event.
“Our results from an interim analysis of data from the phase II KEYNOTE-158 clinical trial show that pembrolizumab has promising antitumor activity in patients with previously treated advanced cervical cancer,” the researchers said. “These response rates are similar or superior to those observed with other treatment options in this setting.”
Pembrolizumab is now being evaluated in combination with concurrent chemoradiotherapy and with concurrent versus sequential chemoradiotherapy.
Merck & Co. sponsored the study.
SOURCE: Chung HC et al. J Clin Oncol. 2019 April 3. doi: 10.1200/JCO.18.01265.
The checkpoint inhibitor pembrolizumab conferred a durable tumor response in patients with advanced cervical cancer, who were positive for programmed death ligand 1 (PD-L1).
Among 98 patients, all of whom had progressed despite prior treatment, overall response rate was 12.2% (95% confidence interval, 6.5%-20.4%), with three complete and nine partial responses. All 12 responses were in patients with PD-L1-positive tumors, wrote Hyun Cheol Chung, MD, and his colleagues. The report is in the Journal of Clinical Oncology.
The response curve had not peaked by the end of follow-up, suggesting further benefit of the antibody, said Dr. Chung of Yonsei Cancer Center, Seoul, South Korea. On the basis of these data, first reported at the 2018 meeting of the American Society of Clinical Oncology, the Food and Drug Administration approved the antibody for advanced cervical cancer last year.
“These results show that treatment with pembrolizumab offers a clinically meaningful therapeutic option for a subset of patients with previously treated advanced cervical cancer,” the researchers said. The approval makes pembrolizumab the first immunotherapy approved for the treatment of an advanced gynecologic malignancy.
The phase 2 KEYNOTE-158 study comprised 98 women (median age 46 years) with advanced cervical cancer with progression after chemotherapy. All received pembrolizumab 200 mg every 3 weeks for 2 years or until progression, intolerable toxicity, or physician or patient decision.
Most of the patients (83.7%) were positive for PD-L1. All had received at least one or more lines of chemotherapy before entering the study. The median follow-up was 10 months and the median treatment duration 2.9 months, with a range of 1 day-22 months.
By the study’s end, most patients (85.7%) had experienced disease progression or had died. Overall survival was poor even in the PD-L1-positive patients, with 82.9% experiencing progression or death. Overall mortality was 69.4% and 64.6%, respectively. However, PD-L1 patients did live longer, a median of 11 months, compared with 9.4 months in the entire cohort.
At 6 months, overall survival estimates were 75.2% in the total cohort and 80.2% in the PD-L1 patients; 12-month overall survival estimates were 41.4% and 47.3%, respectively.
However, among the 12 responders with PD-L1-postitive tumors, just one had died by the data cutoff, suggesting that longer treatment with pembrolizumab could be beneficial in this group.
“Responses typically occurred within 2.1 months and were durable, with a median duration of response that had not been reached after a median follow-up of 10.2 months and an estimated 90.9% of responses ongoing at 6 months” Dr. Chung and his colleagues wrote.
Adverse events were common (65.3%) with 12.2% graded as serious. But none were lethal, and only four patients discontinued pembrolizumab because of a treatment-related adverse event.
“Our results from an interim analysis of data from the phase II KEYNOTE-158 clinical trial show that pembrolizumab has promising antitumor activity in patients with previously treated advanced cervical cancer,” the researchers said. “These response rates are similar or superior to those observed with other treatment options in this setting.”
Pembrolizumab is now being evaluated in combination with concurrent chemoradiotherapy and with concurrent versus sequential chemoradiotherapy.
Merck & Co. sponsored the study.
SOURCE: Chung HC et al. J Clin Oncol. 2019 April 3. doi: 10.1200/JCO.18.01265.
FROM JOURNAL OF CLINICAL ONCOLOGY
Colchicine reduces inflammatory markers associated with metabolic syndrome
A small study offers a tantalizing hint that
The 3-month trial did not meet its primary endpoint – change in insulin sensitivity as measured by a glucose tolerance test – but it did hit several secondary goals, all of which were related to the inflammation that accompanies prediabetes, Jack A. Yanovski, MD, and colleagues wrote in Diabetes, Obesity, and Metabolism.
“Colchicine is well-known to have anti-inflammatory properties, although its effect on obesity-associated inflammation has not previously been investigated,” said Dr. Yanovski of the National institutes of Health and his coauthors. “Classically, it has been posited that colchicine blocks inflammation by impeding leukocyte locomotion, diapedesis, and, ultimately, recruitment to sites of inflammation. ... Recently, it has been shown that colchicine also inhibits the formation of the NLRP3 [NOD-like receptor family pyrin domain-containing 3] inflammasome, an important component of the obesity-associated inflammatory cascade.”
The NLRP3 inflammasome has been shown to play an important part in promoting the inflammatory state of obesity, the authors noted. When a cell senses danger, NLRP3 uses microtubules to create an inflammasome that then produces interleukin-1 beta gene and interleukin-18. One of colchicine’s known actions is to inhibit microtubule formation, suggesting that it could put the brakes on this process.
The study comprised 40 patients who had metabolic syndrome, significant insulin resistance, and elevated inflammatory markers. Among the exclusionary criteria were having a significant medical illness, a history of gout, and recent or current use of colchicine.
The patients were randomized to colchicine 0.6 mg or placebo twice daily for 3 months. No dietary advice was given during the study period. Of the 40 randomized patients, 37 completed the 3-month study, though none left because of adverse events.
Although there were no significant between-group differences in levels of fasting insulin, colchicine did significantly decrease inflammatory markers, compared with placebo. C-reactive protein dropped by 2.8 mg/L in the active group but increased slightly in the placebo group. The erythrocyte sedimentation rate also decreased in the colchicine group, compared with placebo (difference, –5.9 mm/hr; P = .07). The active group experienced an improvement in fasting insulin as measured by the homeostasis model assessment–estimated insulin resistance index and in glucose effectiveness, which suggests metabolic improvement.
“Larger trials are needed to investigate whether colchicine has efficacy in improving insulin resistance and/or preventing the onset of diabetes mellitus in at-risk individuals with obesity-associated inflammation,” the authors concluded.
The study was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development and by the National Institutes of Health. None of the authors reported any disclosures or conflicts of interest relating to this study.
SOURCE: Yanovski JA et al. Diabetes Obes Metab. 2019 Mar 14. doi: 10.1111/dom.13702.
A small study offers a tantalizing hint that
The 3-month trial did not meet its primary endpoint – change in insulin sensitivity as measured by a glucose tolerance test – but it did hit several secondary goals, all of which were related to the inflammation that accompanies prediabetes, Jack A. Yanovski, MD, and colleagues wrote in Diabetes, Obesity, and Metabolism.
“Colchicine is well-known to have anti-inflammatory properties, although its effect on obesity-associated inflammation has not previously been investigated,” said Dr. Yanovski of the National institutes of Health and his coauthors. “Classically, it has been posited that colchicine blocks inflammation by impeding leukocyte locomotion, diapedesis, and, ultimately, recruitment to sites of inflammation. ... Recently, it has been shown that colchicine also inhibits the formation of the NLRP3 [NOD-like receptor family pyrin domain-containing 3] inflammasome, an important component of the obesity-associated inflammatory cascade.”
The NLRP3 inflammasome has been shown to play an important part in promoting the inflammatory state of obesity, the authors noted. When a cell senses danger, NLRP3 uses microtubules to create an inflammasome that then produces interleukin-1 beta gene and interleukin-18. One of colchicine’s known actions is to inhibit microtubule formation, suggesting that it could put the brakes on this process.
The study comprised 40 patients who had metabolic syndrome, significant insulin resistance, and elevated inflammatory markers. Among the exclusionary criteria were having a significant medical illness, a history of gout, and recent or current use of colchicine.
The patients were randomized to colchicine 0.6 mg or placebo twice daily for 3 months. No dietary advice was given during the study period. Of the 40 randomized patients, 37 completed the 3-month study, though none left because of adverse events.
Although there were no significant between-group differences in levels of fasting insulin, colchicine did significantly decrease inflammatory markers, compared with placebo. C-reactive protein dropped by 2.8 mg/L in the active group but increased slightly in the placebo group. The erythrocyte sedimentation rate also decreased in the colchicine group, compared with placebo (difference, –5.9 mm/hr; P = .07). The active group experienced an improvement in fasting insulin as measured by the homeostasis model assessment–estimated insulin resistance index and in glucose effectiveness, which suggests metabolic improvement.
“Larger trials are needed to investigate whether colchicine has efficacy in improving insulin resistance and/or preventing the onset of diabetes mellitus in at-risk individuals with obesity-associated inflammation,” the authors concluded.
The study was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development and by the National Institutes of Health. None of the authors reported any disclosures or conflicts of interest relating to this study.
SOURCE: Yanovski JA et al. Diabetes Obes Metab. 2019 Mar 14. doi: 10.1111/dom.13702.
A small study offers a tantalizing hint that
The 3-month trial did not meet its primary endpoint – change in insulin sensitivity as measured by a glucose tolerance test – but it did hit several secondary goals, all of which were related to the inflammation that accompanies prediabetes, Jack A. Yanovski, MD, and colleagues wrote in Diabetes, Obesity, and Metabolism.
“Colchicine is well-known to have anti-inflammatory properties, although its effect on obesity-associated inflammation has not previously been investigated,” said Dr. Yanovski of the National institutes of Health and his coauthors. “Classically, it has been posited that colchicine blocks inflammation by impeding leukocyte locomotion, diapedesis, and, ultimately, recruitment to sites of inflammation. ... Recently, it has been shown that colchicine also inhibits the formation of the NLRP3 [NOD-like receptor family pyrin domain-containing 3] inflammasome, an important component of the obesity-associated inflammatory cascade.”
The NLRP3 inflammasome has been shown to play an important part in promoting the inflammatory state of obesity, the authors noted. When a cell senses danger, NLRP3 uses microtubules to create an inflammasome that then produces interleukin-1 beta gene and interleukin-18. One of colchicine’s known actions is to inhibit microtubule formation, suggesting that it could put the brakes on this process.
The study comprised 40 patients who had metabolic syndrome, significant insulin resistance, and elevated inflammatory markers. Among the exclusionary criteria were having a significant medical illness, a history of gout, and recent or current use of colchicine.
The patients were randomized to colchicine 0.6 mg or placebo twice daily for 3 months. No dietary advice was given during the study period. Of the 40 randomized patients, 37 completed the 3-month study, though none left because of adverse events.
Although there were no significant between-group differences in levels of fasting insulin, colchicine did significantly decrease inflammatory markers, compared with placebo. C-reactive protein dropped by 2.8 mg/L in the active group but increased slightly in the placebo group. The erythrocyte sedimentation rate also decreased in the colchicine group, compared with placebo (difference, –5.9 mm/hr; P = .07). The active group experienced an improvement in fasting insulin as measured by the homeostasis model assessment–estimated insulin resistance index and in glucose effectiveness, which suggests metabolic improvement.
“Larger trials are needed to investigate whether colchicine has efficacy in improving insulin resistance and/or preventing the onset of diabetes mellitus in at-risk individuals with obesity-associated inflammation,” the authors concluded.
The study was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development and by the National Institutes of Health. None of the authors reported any disclosures or conflicts of interest relating to this study.
SOURCE: Yanovski JA et al. Diabetes Obes Metab. 2019 Mar 14. doi: 10.1111/dom.13702.
FROM DIABETES, OBESITY, AND METABOLISM
Deuterium-altered ruxolitinib may be an effective treatment for alopecia areata
WASHINGTON – Yet another inhibitor of the Janus kinase enzyme has debuted with positive phase 2 results for patients with even longstanding alopecia areata.

About half of those who took 8 mg of CPT-543, a chemically altered form of ruxolitinib, twice a day for 24 weeks, regrew hair – including eyebrows and eyelashes – by at least 50%. The dose-ranging study also found that a 4 mg twice-daily dose promoted the same growth in 21%, James V. Cassella, PhD, said during a late breaking clinical trials session at the annual meeting of the American Academy of Dermatology.
Adverse events were mild, including headache, reported in 26% of the 8 mg group. However, investigators “are keeping an eye” on infections and blood chemistry, and in light of the confirmed increased risk of herpes zoster and suspected increased risk of thromboembolic events with ??ruxolitinib, said Dr. Cassella, chief development officer of Concert Pharmaceuticals, which is developing the molecule.
Ruxolitinib, an inhibitor of both JAK1 and JAK2, is available under the name Jakafi and is approved for the treatment of myelofibrosis and polycythemia vera. The addition of deuterium slows its metabolism, increasing half-life and bioavailability without affecting receptor selectivity or potency, according to the company.
The company’s (35). The primary endpoint was the proportion of patients with at least a 50% relative reduction in scalp hair loss as measured by the Severity of Alopecia Tool (SALT) at 24 weeks, from baseline. Secondarily, the trial examined response by alopecia subtype (patchy or complete) and individual SALT changes compared with baseline.
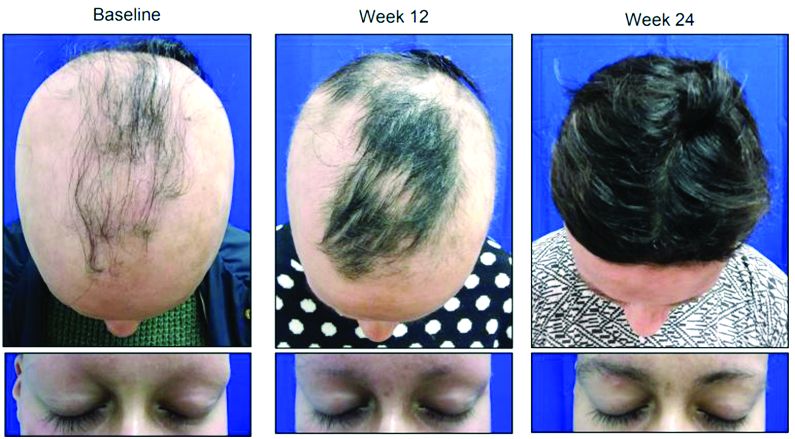
Patients were generally in their mid-30s, and about 75% were female. The mean current alopecia episode was about 5 years. The mean SALT score at baseline was about 89, with 100 being complete hair loss.
By week 24, 47% of those taking 8 mg twice daily and 21% of those taking 4 mg twice daily experienced the primary endpoint of at least 50% SALT reduction from baseline. Almost 9% of those taking placebo also hit the target. These patients all had patchy alopecia and may have been coming out of an alopecia episode during the trial, Dr. Cassella said.
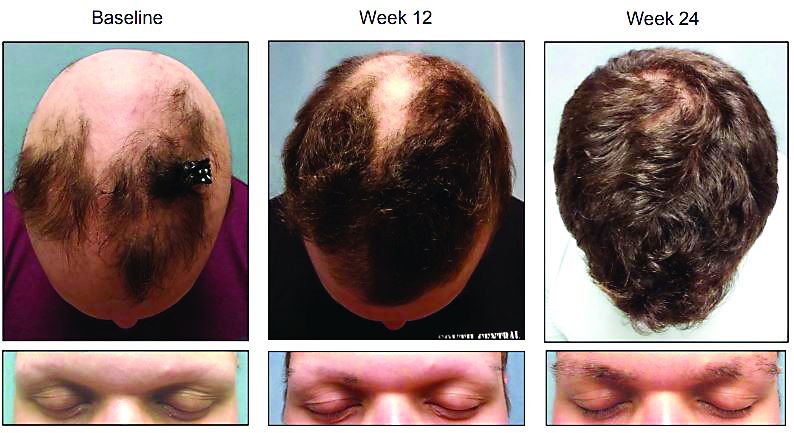
Week 12 was the inflection point for response division, with both active groups significantly outperforming the placebo group. By week 16, 30% of the 8 mg group and about 15% of the 4 mg group had already hit the primary endpoint. Response in the 4 mg group climbed slowly until the end of the trial, while in the 8 mg group, response ascended more quickly. Response was still trending upward when the study stopped.
“We think we have not hit the ceiling effect with this drug,” Dr. Cassella said. “There is some evidence that response would continue to increase after 24 weeks.”
Patchy alopecia and alopecia universalis appeared to respond best to treatment in both dosage groups. There was no response in either group for patients with alopecia ophiasis or totalis.
Headache was the most common adverse event, and appeared to be dose-dependent, occurring in 11% of placebo patients, 17% of the 4 mg group, and 26% of the 8 mg group. Six patients developed increased blood creatinine phosphokinase levels (one in the placebo group, three in the 4 mg group, and two in the 8 mg group). There were no thromboembolic events. Three patients in the placebo group and two in the 8 mg group discontinued the medication due to unspecified adverse events.
In early March, the company announced an open-label dose-finding study, which will randomize 60 patients with moderate-to-severe alopecia areata to either 8 mg twice daily or 16 mg once daily over a 24-week treatment period. Concert intends to conduct a food-effect trial to assess the relative bioavailability of oral doses of CTP-543 under fasted and fed conditions in 14 healthy volunteers in the first half of 2019.
SOURCE: Casella J. AAD 2019; S034, Abstract 11291.
WASHINGTON – Yet another inhibitor of the Janus kinase enzyme has debuted with positive phase 2 results for patients with even longstanding alopecia areata.
About half of those who took 8 mg of CPT-543, a chemically altered form of ruxolitinib, twice a day for 24 weeks, regrew hair – including eyebrows and eyelashes – by at least 50%. The dose-ranging study also found that a 4 mg twice-daily dose promoted the same growth in 21%, James V. Cassella, PhD, said during a late breaking clinical trials session at the annual meeting of the American Academy of Dermatology.
Adverse events were mild, including headache, reported in 26% of the 8 mg group. However, investigators “are keeping an eye” on infections and blood chemistry, and in light of the confirmed increased risk of herpes zoster and suspected increased risk of thromboembolic events with ??ruxolitinib, said Dr. Cassella, chief development officer of Concert Pharmaceuticals, which is developing the molecule.
Ruxolitinib, an inhibitor of both JAK1 and JAK2, is available under the name Jakafi and is approved for the treatment of myelofibrosis and polycythemia vera. The addition of deuterium slows its metabolism, increasing half-life and bioavailability without affecting receptor selectivity or potency, according to the company.
The company’s (35). The primary endpoint was the proportion of patients with at least a 50% relative reduction in scalp hair loss as measured by the Severity of Alopecia Tool (SALT) at 24 weeks, from baseline. Secondarily, the trial examined response by alopecia subtype (patchy or complete) and individual SALT changes compared with baseline.
Patients were generally in their mid-30s, and about 75% were female. The mean current alopecia episode was about 5 years. The mean SALT score at baseline was about 89, with 100 being complete hair loss.
By week 24, 47% of those taking 8 mg twice daily and 21% of those taking 4 mg twice daily experienced the primary endpoint of at least 50% SALT reduction from baseline. Almost 9% of those taking placebo also hit the target. These patients all had patchy alopecia and may have been coming out of an alopecia episode during the trial, Dr. Cassella said.
Week 12 was the inflection point for response division, with both active groups significantly outperforming the placebo group. By week 16, 30% of the 8 mg group and about 15% of the 4 mg group had already hit the primary endpoint. Response in the 4 mg group climbed slowly until the end of the trial, while in the 8 mg group, response ascended more quickly. Response was still trending upward when the study stopped.
“We think we have not hit the ceiling effect with this drug,” Dr. Cassella said. “There is some evidence that response would continue to increase after 24 weeks.”
Patchy alopecia and alopecia universalis appeared to respond best to treatment in both dosage groups. There was no response in either group for patients with alopecia ophiasis or totalis.
Headache was the most common adverse event, and appeared to be dose-dependent, occurring in 11% of placebo patients, 17% of the 4 mg group, and 26% of the 8 mg group. Six patients developed increased blood creatinine phosphokinase levels (one in the placebo group, three in the 4 mg group, and two in the 8 mg group). There were no thromboembolic events. Three patients in the placebo group and two in the 8 mg group discontinued the medication due to unspecified adverse events.
In early March, the company announced an open-label dose-finding study, which will randomize 60 patients with moderate-to-severe alopecia areata to either 8 mg twice daily or 16 mg once daily over a 24-week treatment period. Concert intends to conduct a food-effect trial to assess the relative bioavailability of oral doses of CTP-543 under fasted and fed conditions in 14 healthy volunteers in the first half of 2019.
SOURCE: Casella J. AAD 2019; S034, Abstract 11291.
WASHINGTON – Yet another inhibitor of the Janus kinase enzyme has debuted with positive phase 2 results for patients with even longstanding alopecia areata.
About half of those who took 8 mg of CPT-543, a chemically altered form of ruxolitinib, twice a day for 24 weeks, regrew hair – including eyebrows and eyelashes – by at least 50%. The dose-ranging study also found that a 4 mg twice-daily dose promoted the same growth in 21%, James V. Cassella, PhD, said during a late breaking clinical trials session at the annual meeting of the American Academy of Dermatology.
Adverse events were mild, including headache, reported in 26% of the 8 mg group. However, investigators “are keeping an eye” on infections and blood chemistry, and in light of the confirmed increased risk of herpes zoster and suspected increased risk of thromboembolic events with ??ruxolitinib, said Dr. Cassella, chief development officer of Concert Pharmaceuticals, which is developing the molecule.
Ruxolitinib, an inhibitor of both JAK1 and JAK2, is available under the name Jakafi and is approved for the treatment of myelofibrosis and polycythemia vera. The addition of deuterium slows its metabolism, increasing half-life and bioavailability without affecting receptor selectivity or potency, according to the company.
The company’s (35). The primary endpoint was the proportion of patients with at least a 50% relative reduction in scalp hair loss as measured by the Severity of Alopecia Tool (SALT) at 24 weeks, from baseline. Secondarily, the trial examined response by alopecia subtype (patchy or complete) and individual SALT changes compared with baseline.
Patients were generally in their mid-30s, and about 75% were female. The mean current alopecia episode was about 5 years. The mean SALT score at baseline was about 89, with 100 being complete hair loss.
By week 24, 47% of those taking 8 mg twice daily and 21% of those taking 4 mg twice daily experienced the primary endpoint of at least 50% SALT reduction from baseline. Almost 9% of those taking placebo also hit the target. These patients all had patchy alopecia and may have been coming out of an alopecia episode during the trial, Dr. Cassella said.
Week 12 was the inflection point for response division, with both active groups significantly outperforming the placebo group. By week 16, 30% of the 8 mg group and about 15% of the 4 mg group had already hit the primary endpoint. Response in the 4 mg group climbed slowly until the end of the trial, while in the 8 mg group, response ascended more quickly. Response was still trending upward when the study stopped.
“We think we have not hit the ceiling effect with this drug,” Dr. Cassella said. “There is some evidence that response would continue to increase after 24 weeks.”
Patchy alopecia and alopecia universalis appeared to respond best to treatment in both dosage groups. There was no response in either group for patients with alopecia ophiasis or totalis.
Headache was the most common adverse event, and appeared to be dose-dependent, occurring in 11% of placebo patients, 17% of the 4 mg group, and 26% of the 8 mg group. Six patients developed increased blood creatinine phosphokinase levels (one in the placebo group, three in the 4 mg group, and two in the 8 mg group). There were no thromboembolic events. Three patients in the placebo group and two in the 8 mg group discontinued the medication due to unspecified adverse events.
In early March, the company announced an open-label dose-finding study, which will randomize 60 patients with moderate-to-severe alopecia areata to either 8 mg twice daily or 16 mg once daily over a 24-week treatment period. Concert intends to conduct a food-effect trial to assess the relative bioavailability of oral doses of CTP-543 under fasted and fed conditions in 14 healthy volunteers in the first half of 2019.
SOURCE: Casella J. AAD 2019; S034, Abstract 11291.
REPORTING FROM AAD 19
Abatacept appears safe for RA patients with COPD
Concerns about abatacept (Orencia)-related lung disease in patients with rheumatoid arthritis appear to be unfounded, a large U.S. database review has determined.

Safety signals seen in a small subset of patients in the 2006 ASSURE study likely arose from chance, Samy Suissa, PhD, and his colleagues wrote in Seminars in Arthritis & Rheumatism. Patients in that study with preexisting chronic obstructive pulmonary disease (COPD) were 84% more likely to develop an exacerbation or other lung disorder than were those taking a placebo comparator.
The new database study contradicted that finding.
“Our study suggests that these numerical differences in ASSURE are expected results of random variation and thus compatible with chance,” Dr. Suissa of Jewish General Hospital and McGill University, both in Montreal, and his coauthors wrote. “Moreover, our findings are consistent with two studies of the safety of abatacept in the context of interstitial lung disease, albeit a different respiratory disease than COPD.”
The year-long ASSURE trial reported on the safety of abatacept in 959 patients versus 482 assigned to placebo. The safety signal arose in a subgroup of 54 patients with COPD, 37 of whom were assigned to abatacept and 17 to placebo. Among these were four serious respiratory adverse events (COPD exacerbation, worsening of COPD, bronchitis, and pneumonia) in four patients taking abatacept and none in the placebo arm.
“It is useful to note that this difference of 11% versus 0% rate is compatible with chance [exact P = .31], while the exact 95% confidence interval for the odds ratio is wide and includes unity ... The trial also reported more mild-moderate respiratory events with abatacept than with placebo [43.2% vs. 23.5%], including cough, rhonchi, COPD exacerbation, COPD, dyspnea, and nasal congestion. This difference resulted in an odds ration of 1.84,” with a wide confidence interval that included unity (0.48-8.63).
Nevertheless, these findings led to the addition of a warning in the prescribing insert: “Adult COPD patients treated with Orencia developed adverse events more frequently than those treated with placebo, including COPD exacerbations, cough, rhonchi, and dyspnea. Use of Orencia in patients with RA and COPD should be undertaken with caution and such patients should be monitored for worsening of their respiratory status.”
Dr. Suissa’s team used the U.S. MarketScan prescribing database to asses the risk of respiratory adverse events associated with abatacept, compared with other biologic disease-modifying antirheumatic drugs (DMARDs) in a real-world setting.
The cohort comprised 1,807 patients with rheumatoid arthritis and COPD who started a new prescription for abatacept, matched in time to 3,547 who initiated another biologic DMARD. The primary endpoint was the combined risk of hospitalization for COPD exacerbation, bronchitis, and hospitalized pneumonia or influenza.
The most common comparator biologic DMARDs were etanercept, adalimumab, rituximab, and infliximab.
For patients with COPD and comparator patients, the incidence rates for COPD exacerbation were 1.2 per 100 person-years with abatacept and 2.1 per 100 person-years with a different biologic DMARD; for bronchitis, the respective rates were 4.2 and 5.3; for hospitalized pneumonia or influenza, 3.6 and 2.6; and for outpatient pneumonia or flu, 14.7 and 14.4. For the combined endpoint, the incidence rate was 8.7 per 100 person-years with abatacept and 9.9 per 100 person-years with other biologic DMARDs.
The adjusted hazard ratio of the combined endpoint with abatacept versus that with other biologic DMARDs was a nonsignificant risk of 0.87. The hazard ratio with abatacept was 0.60 for hospitalized COPD; 0.80 for bronchitis; 1.39 for hospitalized pneumonia and flu; and 1.05 for outpatient pneumonia and flu. None of these associations was statistically significant.
“One exception was the risk of hospitalization for pneumonia or influenza, which was higher with abatacept among patients with more severe COPD,” at 6.99, than it was with other biologic DMARDs, the authors noted.
Dr. Suissa disclosed relationships with AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, and Novartis.
SOURCE: Suissa S et al. Semin Arthritis Rheum. 2019 Mar 16. doi: 10.1016j.semarthrit.2019.03.007.
Concerns about abatacept (Orencia)-related lung disease in patients with rheumatoid arthritis appear to be unfounded, a large U.S. database review has determined.
Safety signals seen in a small subset of patients in the 2006 ASSURE study likely arose from chance, Samy Suissa, PhD, and his colleagues wrote in Seminars in Arthritis & Rheumatism. Patients in that study with preexisting chronic obstructive pulmonary disease (COPD) were 84% more likely to develop an exacerbation or other lung disorder than were those taking a placebo comparator.
The new database study contradicted that finding.
“Our study suggests that these numerical differences in ASSURE are expected results of random variation and thus compatible with chance,” Dr. Suissa of Jewish General Hospital and McGill University, both in Montreal, and his coauthors wrote. “Moreover, our findings are consistent with two studies of the safety of abatacept in the context of interstitial lung disease, albeit a different respiratory disease than COPD.”
The year-long ASSURE trial reported on the safety of abatacept in 959 patients versus 482 assigned to placebo. The safety signal arose in a subgroup of 54 patients with COPD, 37 of whom were assigned to abatacept and 17 to placebo. Among these were four serious respiratory adverse events (COPD exacerbation, worsening of COPD, bronchitis, and pneumonia) in four patients taking abatacept and none in the placebo arm.
“It is useful to note that this difference of 11% versus 0% rate is compatible with chance [exact P = .31], while the exact 95% confidence interval for the odds ratio is wide and includes unity ... The trial also reported more mild-moderate respiratory events with abatacept than with placebo [43.2% vs. 23.5%], including cough, rhonchi, COPD exacerbation, COPD, dyspnea, and nasal congestion. This difference resulted in an odds ration of 1.84,” with a wide confidence interval that included unity (0.48-8.63).
Nevertheless, these findings led to the addition of a warning in the prescribing insert: “Adult COPD patients treated with Orencia developed adverse events more frequently than those treated with placebo, including COPD exacerbations, cough, rhonchi, and dyspnea. Use of Orencia in patients with RA and COPD should be undertaken with caution and such patients should be monitored for worsening of their respiratory status.”
Dr. Suissa’s team used the U.S. MarketScan prescribing database to asses the risk of respiratory adverse events associated with abatacept, compared with other biologic disease-modifying antirheumatic drugs (DMARDs) in a real-world setting.
The cohort comprised 1,807 patients with rheumatoid arthritis and COPD who started a new prescription for abatacept, matched in time to 3,547 who initiated another biologic DMARD. The primary endpoint was the combined risk of hospitalization for COPD exacerbation, bronchitis, and hospitalized pneumonia or influenza.
The most common comparator biologic DMARDs were etanercept, adalimumab, rituximab, and infliximab.
For patients with COPD and comparator patients, the incidence rates for COPD exacerbation were 1.2 per 100 person-years with abatacept and 2.1 per 100 person-years with a different biologic DMARD; for bronchitis, the respective rates were 4.2 and 5.3; for hospitalized pneumonia or influenza, 3.6 and 2.6; and for outpatient pneumonia or flu, 14.7 and 14.4. For the combined endpoint, the incidence rate was 8.7 per 100 person-years with abatacept and 9.9 per 100 person-years with other biologic DMARDs.
The adjusted hazard ratio of the combined endpoint with abatacept versus that with other biologic DMARDs was a nonsignificant risk of 0.87. The hazard ratio with abatacept was 0.60 for hospitalized COPD; 0.80 for bronchitis; 1.39 for hospitalized pneumonia and flu; and 1.05 for outpatient pneumonia and flu. None of these associations was statistically significant.
“One exception was the risk of hospitalization for pneumonia or influenza, which was higher with abatacept among patients with more severe COPD,” at 6.99, than it was with other biologic DMARDs, the authors noted.
Dr. Suissa disclosed relationships with AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, and Novartis.
SOURCE: Suissa S et al. Semin Arthritis Rheum. 2019 Mar 16. doi: 10.1016j.semarthrit.2019.03.007.
Concerns about abatacept (Orencia)-related lung disease in patients with rheumatoid arthritis appear to be unfounded, a large U.S. database review has determined.
Safety signals seen in a small subset of patients in the 2006 ASSURE study likely arose from chance, Samy Suissa, PhD, and his colleagues wrote in Seminars in Arthritis & Rheumatism. Patients in that study with preexisting chronic obstructive pulmonary disease (COPD) were 84% more likely to develop an exacerbation or other lung disorder than were those taking a placebo comparator.
The new database study contradicted that finding.
“Our study suggests that these numerical differences in ASSURE are expected results of random variation and thus compatible with chance,” Dr. Suissa of Jewish General Hospital and McGill University, both in Montreal, and his coauthors wrote. “Moreover, our findings are consistent with two studies of the safety of abatacept in the context of interstitial lung disease, albeit a different respiratory disease than COPD.”
The year-long ASSURE trial reported on the safety of abatacept in 959 patients versus 482 assigned to placebo. The safety signal arose in a subgroup of 54 patients with COPD, 37 of whom were assigned to abatacept and 17 to placebo. Among these were four serious respiratory adverse events (COPD exacerbation, worsening of COPD, bronchitis, and pneumonia) in four patients taking abatacept and none in the placebo arm.
“It is useful to note that this difference of 11% versus 0% rate is compatible with chance [exact P = .31], while the exact 95% confidence interval for the odds ratio is wide and includes unity ... The trial also reported more mild-moderate respiratory events with abatacept than with placebo [43.2% vs. 23.5%], including cough, rhonchi, COPD exacerbation, COPD, dyspnea, and nasal congestion. This difference resulted in an odds ration of 1.84,” with a wide confidence interval that included unity (0.48-8.63).
Nevertheless, these findings led to the addition of a warning in the prescribing insert: “Adult COPD patients treated with Orencia developed adverse events more frequently than those treated with placebo, including COPD exacerbations, cough, rhonchi, and dyspnea. Use of Orencia in patients with RA and COPD should be undertaken with caution and such patients should be monitored for worsening of their respiratory status.”
Dr. Suissa’s team used the U.S. MarketScan prescribing database to asses the risk of respiratory adverse events associated with abatacept, compared with other biologic disease-modifying antirheumatic drugs (DMARDs) in a real-world setting.
The cohort comprised 1,807 patients with rheumatoid arthritis and COPD who started a new prescription for abatacept, matched in time to 3,547 who initiated another biologic DMARD. The primary endpoint was the combined risk of hospitalization for COPD exacerbation, bronchitis, and hospitalized pneumonia or influenza.
The most common comparator biologic DMARDs were etanercept, adalimumab, rituximab, and infliximab.
For patients with COPD and comparator patients, the incidence rates for COPD exacerbation were 1.2 per 100 person-years with abatacept and 2.1 per 100 person-years with a different biologic DMARD; for bronchitis, the respective rates were 4.2 and 5.3; for hospitalized pneumonia or influenza, 3.6 and 2.6; and for outpatient pneumonia or flu, 14.7 and 14.4. For the combined endpoint, the incidence rate was 8.7 per 100 person-years with abatacept and 9.9 per 100 person-years with other biologic DMARDs.
The adjusted hazard ratio of the combined endpoint with abatacept versus that with other biologic DMARDs was a nonsignificant risk of 0.87. The hazard ratio with abatacept was 0.60 for hospitalized COPD; 0.80 for bronchitis; 1.39 for hospitalized pneumonia and flu; and 1.05 for outpatient pneumonia and flu. None of these associations was statistically significant.
“One exception was the risk of hospitalization for pneumonia or influenza, which was higher with abatacept among patients with more severe COPD,” at 6.99, than it was with other biologic DMARDs, the authors noted.
Dr. Suissa disclosed relationships with AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, and Novartis.
SOURCE: Suissa S et al. Semin Arthritis Rheum. 2019 Mar 16. doi: 10.1016j.semarthrit.2019.03.007.
FROM SEMINARS IN ARTHRITIS & RHEUMATISM
Amyloid brain imaging changed clinical management in 60% of MCI and dementia patients
.

Diagnoses changed from Alzheimer’s disease to non–Alzheimer’s disease in 25% of 11,409 patients and from non–Alzheimer’s disease to Alzheimer’s disease in 10.5%, reported Gil Rabinovici, MD, and his colleagues. The use of Alzheimer’s disease drugs doubled in amyloid-positive MCI patients, and increased by a third in amyloid-positive dementia patients. Physicians involved in the study said the scans provided key clinical information in 82% of cases with post-scan management changes.
Scans also benefited amyloid-negative patients. Before the scan, 71% of these carried an Alzheimer’s disease diagnosis; afterward, just 10% did, opening the way for an accurate diagnosis and more effective treatment.
The study was powered to detect a 30% or greater change in the MCI and dementia groups. The 60% change emphasize how useful amyloid PET scans could be in clinical practice, Dr. Rabinovici, the study’s lead author and principal investigator, said in a press statement.
“We are impressed by the magnitude of these results, which make it clear that amyloid PET imaging can have a major impact on how we diagnose and care for patients with Alzheimer’s disease and other forms of cognitive decline,” said Dr. Rabinovici of the University of California, San Francisco.
Alzheimer’s Association leaders were similarly pleased.
“These results present highly credible, large-scale evidence that amyloid PET imaging can be a powerful tool to improve the accuracy of Alzheimer’s diagnosis and lead to better medical management, especially in difficult-to-diagnose cases,” said Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association and a coauthor of the study. “It is important that amyloid PET imaging be more broadly accessible to those who need it.”

Next steps
Ultimately, investigators hope the nationwide-wide, open-label study will prove the clinical value of amyloid PET scanning and convince the Centers for Medicare & Medicaid Services to make the test a fully covered service for those who meet the appropriate use criteria set forth by the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging.
IDEAS’ second goal – showing that the scans improve health outcomes – is scheduled for 2020. These data are a key component of the CMS decision, but they might be a tough sell, Clifford R. Jack Jr., MD, and Ronald C. Petersen, MD, PhD, wrote in an accompanying editorial. Dr. Jack and Dr. Petersen are affiliated with the Mayo Clinic in Rochester, Minn.
“For CMS to cover the cost of amyloid PET, it must be demonstrated that the result of a scan has an effect on patient outcomes, not just patient care processes – and, without a disease-modifying therapy available, that might be a challenge,” they wrote.
IDEAS is a funding collaboration of the CMS, the Alzheimer’s Association, Avid Radiopharmaceuticals/Eli Lilly, General Electric Healthcare, Piramal Imaging, and the American College of Radiology. Dr. Rabinovici had no financial disclosures.
SOURCE: Rabinovici GD et al. JAMA. 2019 Apr 2. doi: 10.1001/jama.2019.2000.
Current clinical practice does not routinely include biomarkers, and if given a choice, most patients would prefer brain imaging to spinal fluid-based testing, so IDEAS may be making imaging-based biomarker characterization a real possibility in the future.

Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He made these comments in an interview.
Current clinical practice does not routinely include biomarkers, and if given a choice, most patients would prefer brain imaging to spinal fluid-based testing, so IDEAS may be making imaging-based biomarker characterization a real possibility in the future.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He made these comments in an interview.
Current clinical practice does not routinely include biomarkers, and if given a choice, most patients would prefer brain imaging to spinal fluid-based testing, so IDEAS may be making imaging-based biomarker characterization a real possibility in the future.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He made these comments in an interview.
.
Diagnoses changed from Alzheimer’s disease to non–Alzheimer’s disease in 25% of 11,409 patients and from non–Alzheimer’s disease to Alzheimer’s disease in 10.5%, reported Gil Rabinovici, MD, and his colleagues. The use of Alzheimer’s disease drugs doubled in amyloid-positive MCI patients, and increased by a third in amyloid-positive dementia patients. Physicians involved in the study said the scans provided key clinical information in 82% of cases with post-scan management changes.
Scans also benefited amyloid-negative patients. Before the scan, 71% of these carried an Alzheimer’s disease diagnosis; afterward, just 10% did, opening the way for an accurate diagnosis and more effective treatment.
The study was powered to detect a 30% or greater change in the MCI and dementia groups. The 60% change emphasize how useful amyloid PET scans could be in clinical practice, Dr. Rabinovici, the study’s lead author and principal investigator, said in a press statement.
“We are impressed by the magnitude of these results, which make it clear that amyloid PET imaging can have a major impact on how we diagnose and care for patients with Alzheimer’s disease and other forms of cognitive decline,” said Dr. Rabinovici of the University of California, San Francisco.
Alzheimer’s Association leaders were similarly pleased.
“These results present highly credible, large-scale evidence that amyloid PET imaging can be a powerful tool to improve the accuracy of Alzheimer’s diagnosis and lead to better medical management, especially in difficult-to-diagnose cases,” said Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association and a coauthor of the study. “It is important that amyloid PET imaging be more broadly accessible to those who need it.”
Next steps
Ultimately, investigators hope the nationwide-wide, open-label study will prove the clinical value of amyloid PET scanning and convince the Centers for Medicare & Medicaid Services to make the test a fully covered service for those who meet the appropriate use criteria set forth by the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging.
IDEAS’ second goal – showing that the scans improve health outcomes – is scheduled for 2020. These data are a key component of the CMS decision, but they might be a tough sell, Clifford R. Jack Jr., MD, and Ronald C. Petersen, MD, PhD, wrote in an accompanying editorial. Dr. Jack and Dr. Petersen are affiliated with the Mayo Clinic in Rochester, Minn.
“For CMS to cover the cost of amyloid PET, it must be demonstrated that the result of a scan has an effect on patient outcomes, not just patient care processes – and, without a disease-modifying therapy available, that might be a challenge,” they wrote.
IDEAS is a funding collaboration of the CMS, the Alzheimer’s Association, Avid Radiopharmaceuticals/Eli Lilly, General Electric Healthcare, Piramal Imaging, and the American College of Radiology. Dr. Rabinovici had no financial disclosures.
SOURCE: Rabinovici GD et al. JAMA. 2019 Apr 2. doi: 10.1001/jama.2019.2000.
.
Diagnoses changed from Alzheimer’s disease to non–Alzheimer’s disease in 25% of 11,409 patients and from non–Alzheimer’s disease to Alzheimer’s disease in 10.5%, reported Gil Rabinovici, MD, and his colleagues. The use of Alzheimer’s disease drugs doubled in amyloid-positive MCI patients, and increased by a third in amyloid-positive dementia patients. Physicians involved in the study said the scans provided key clinical information in 82% of cases with post-scan management changes.
Scans also benefited amyloid-negative patients. Before the scan, 71% of these carried an Alzheimer’s disease diagnosis; afterward, just 10% did, opening the way for an accurate diagnosis and more effective treatment.
The study was powered to detect a 30% or greater change in the MCI and dementia groups. The 60% change emphasize how useful amyloid PET scans could be in clinical practice, Dr. Rabinovici, the study’s lead author and principal investigator, said in a press statement.
“We are impressed by the magnitude of these results, which make it clear that amyloid PET imaging can have a major impact on how we diagnose and care for patients with Alzheimer’s disease and other forms of cognitive decline,” said Dr. Rabinovici of the University of California, San Francisco.
Alzheimer’s Association leaders were similarly pleased.
“These results present highly credible, large-scale evidence that amyloid PET imaging can be a powerful tool to improve the accuracy of Alzheimer’s diagnosis and lead to better medical management, especially in difficult-to-diagnose cases,” said Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association and a coauthor of the study. “It is important that amyloid PET imaging be more broadly accessible to those who need it.”
Next steps
Ultimately, investigators hope the nationwide-wide, open-label study will prove the clinical value of amyloid PET scanning and convince the Centers for Medicare & Medicaid Services to make the test a fully covered service for those who meet the appropriate use criteria set forth by the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging.
IDEAS’ second goal – showing that the scans improve health outcomes – is scheduled for 2020. These data are a key component of the CMS decision, but they might be a tough sell, Clifford R. Jack Jr., MD, and Ronald C. Petersen, MD, PhD, wrote in an accompanying editorial. Dr. Jack and Dr. Petersen are affiliated with the Mayo Clinic in Rochester, Minn.
“For CMS to cover the cost of amyloid PET, it must be demonstrated that the result of a scan has an effect on patient outcomes, not just patient care processes – and, without a disease-modifying therapy available, that might be a challenge,” they wrote.
IDEAS is a funding collaboration of the CMS, the Alzheimer’s Association, Avid Radiopharmaceuticals/Eli Lilly, General Electric Healthcare, Piramal Imaging, and the American College of Radiology. Dr. Rabinovici had no financial disclosures.
SOURCE: Rabinovici GD et al. JAMA. 2019 Apr 2. doi: 10.1001/jama.2019.2000.
FROM THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION
Corticosteroids: What is their place in pneumonia and sepsis?
Two experts drew on both personal experience and extant literature in the debate “Steroids for Pneumonia and Sepsis ... Do You Believe?” on Tuesday.

Daniel Dressler, MD, MSC, SFHM, of Emory University, Atlanta, and Daniel J. Brotman, MD, SFHM, director of the hospitalist program at Johns Hopkins Hospital, Baltimore, used a series of case studies to illustrate the conundrum. Despite their “pro” and “con” stances, though, both agreed in the end: First, do no harm.
There are no blanket recommendations for the use of steroids for pneumonia, because historically, studies have come to varied conclusions. However, Dr. Dressler, who advocated for the medications, said in an interview that recent publications paint a more complete picture.
“I think the newer studies in 2015 have made us more comfortable, because they look like there is more benefit” for steroids, especially among more severely ill patients, he noted. These international studies added more than 800 cases to the literature. A Spanish trial randomized 120 patients with high C-reactive protein to placebo or 0.5 mg/kg methylpred-nisolone every 12 hours for 5 days. There were fewer treatment failures in the prednisone group (13% vs. 31%), and fewer adverse clinical outcomes of intubation, shock, or death (3% vs. 14%). The number needed to treat to prevent one event was just six. (JAMA. 2015;313[7]:677-86).

In addition, a Cochrane meta-analysis analyzed 13 randomized trials, comprising more than 2,000 hospitalized patients. It found consistently lower rates of mortality, acute respiratory distress syndrome, early treatment failure, and hyperglycemia (Cochrane Database Syst Rev. 2017 Dec 13;12:CD007720).
“My recommendation is that inpatient clinicians should consider a brief course [5-7 days] of moderate-dose steroids (20-60 mg of prednisone equivalent) in patients admitted with CAP [community acquired pneumonia],” he said in an interview. “I personally give 40 mg prednisone for 5 days. I give even stronger consideration for severe CAP.”
Dr. Brotman countered with another set of articles from the literature, citing several studies with different conclusions. A separate meta-analysis of 12 trials appeared in the Annals of Internal Medicine (2015;163[7]:519-28). Half of the studies looked at outcomes in severe CAP, and the other half in less-severe cases. In the severe population, corticosteroids were associated with an overall decrease of about 40% in mortality, but that finding was driven largely by a single study; the others found nonsignificant decreases. The picture was less equivocal in the milder cases: Corticosteroids did not significantly reduce the risk of death.
In both groups, however, the drugs did significantly reduce the amount of hospital time. But this reduction came at a price, according to another review published in Clinical Infectious Diseases (2018 Jan 18;66[3]:346-54). Hospital stay was indeed reduced by a day, but there was no significant reduced risk of death (5.0% vs. 5.9% placebo). Similar rates of ICU admission and treatment failure, a doubling in the risk of hyperglycemia that required insulin, and a significantly higher risk of CAP-related rehospitalization (5.0% vs. 2.7%) rounded out the findings.
“Steroids may help patients feel better and have more reassuring vital signs and get out sooner, but at the expense of some toxicity, which might account for the readmissions,” Dr. Brotman said in an interview.
He then turned to the subject of sepsis. Before administering steroids for sepsis, physicians need to determine whether the powerful anti-inflammatory effect is worth the risks they carry. Adrenal failure is the biggest risk, Dr. Brotman said, citing last year’s ADRENAL study of 3,658 mechanically ventilated patients (N Engl J Med. 2018;378:797-808). They were randomized to a week of hydrocortisone 200 mg per day or placebo. The overall death rate was 28%, and steroids reduced the risk by only 5% (odds ratio, 0.95). The treatment group also had higher mean arterial pressure and lactate, a slower heart rate, and more serious diverse events, including hyperglycemia, hypernatremia, myopathy, and encephalopathy.
Initially, treated patients appeared to do better clinically, with a shorter period of ventilation, a shorter discharge from intensive care. But overall, there was no difference in ventilator-free days or hospital length of stay.“You may be improving clinical outcomes, but if you’re suppressing inflammation completely, you’re also suppressing a healthy response to an infectious process. There are some infections we need to be particularly cautious with, including tuberculosis,” Dr. Brotman added in the interview.
For his part, Dr. Dressler stated that the steroids-for-sepsis issue is “slightly murky.”
“A couple of new trials came out recently, and they lead us to reassess our thinking on this,” he said. Together, the studies comprised about 5,000 patients with septic shock – more than doubling the already studied cohort in 1 year. The reassessment came by means of a 2018 meta-analysis of all 9,000 patients. The findings actually led to new treatment guidelines, which were published in the British Medical Journal last year (2018;362:k3284).
The conclusion made a “weak recommendation” for corticosteroids in patients with sepsis. “Both steroids and no steroids are reasonable management options,” when also considering the overall clinical picture. For example, the recommendations advise against giving steroids to pregnant women, neonates, and patients with preexisting adrenal insufficiency.
However, the article noted, “Fully informed patients who value avoiding death over quality of life and function would likely choose corticosteroids.”
“I’m not sure these [studies] are changing what most people are doing,” Dr. Brotman countered in his interview. “I think the studies do help somewhat, because now we have enough numbers to suggest we can achieve a statistically significantly benefit. Septic shock is a life-threatening situation with a 40% risk of death. Now we can see that for every 50 people we treat with steroids, we can prevent 1 death. But that’s not the whole picture. Steroids won’t change the morality rate from 40% to 10%, but these studies do suggest that we can capture a small percent of people who may otherwise die.”
Dr. Dressler reported no financial disclosures. Dr. Brotman reported relationships with Bristol-Myers Squibb, Pfizer, and Portola.
Two experts drew on both personal experience and extant literature in the debate “Steroids for Pneumonia and Sepsis ... Do You Believe?” on Tuesday.
Daniel Dressler, MD, MSC, SFHM, of Emory University, Atlanta, and Daniel J. Brotman, MD, SFHM, director of the hospitalist program at Johns Hopkins Hospital, Baltimore, used a series of case studies to illustrate the conundrum. Despite their “pro” and “con” stances, though, both agreed in the end: First, do no harm.
There are no blanket recommendations for the use of steroids for pneumonia, because historically, studies have come to varied conclusions. However, Dr. Dressler, who advocated for the medications, said in an interview that recent publications paint a more complete picture.
“I think the newer studies in 2015 have made us more comfortable, because they look like there is more benefit” for steroids, especially among more severely ill patients, he noted. These international studies added more than 800 cases to the literature. A Spanish trial randomized 120 patients with high C-reactive protein to placebo or 0.5 mg/kg methylpred-nisolone every 12 hours for 5 days. There were fewer treatment failures in the prednisone group (13% vs. 31%), and fewer adverse clinical outcomes of intubation, shock, or death (3% vs. 14%). The number needed to treat to prevent one event was just six. (JAMA. 2015;313[7]:677-86).
In addition, a Cochrane meta-analysis analyzed 13 randomized trials, comprising more than 2,000 hospitalized patients. It found consistently lower rates of mortality, acute respiratory distress syndrome, early treatment failure, and hyperglycemia (Cochrane Database Syst Rev. 2017 Dec 13;12:CD007720).
“My recommendation is that inpatient clinicians should consider a brief course [5-7 days] of moderate-dose steroids (20-60 mg of prednisone equivalent) in patients admitted with CAP [community acquired pneumonia],” he said in an interview. “I personally give 40 mg prednisone for 5 days. I give even stronger consideration for severe CAP.”
Dr. Brotman countered with another set of articles from the literature, citing several studies with different conclusions. A separate meta-analysis of 12 trials appeared in the Annals of Internal Medicine (2015;163[7]:519-28). Half of the studies looked at outcomes in severe CAP, and the other half in less-severe cases. In the severe population, corticosteroids were associated with an overall decrease of about 40% in mortality, but that finding was driven largely by a single study; the others found nonsignificant decreases. The picture was less equivocal in the milder cases: Corticosteroids did not significantly reduce the risk of death.
In both groups, however, the drugs did significantly reduce the amount of hospital time. But this reduction came at a price, according to another review published in Clinical Infectious Diseases (2018 Jan 18;66[3]:346-54). Hospital stay was indeed reduced by a day, but there was no significant reduced risk of death (5.0% vs. 5.9% placebo). Similar rates of ICU admission and treatment failure, a doubling in the risk of hyperglycemia that required insulin, and a significantly higher risk of CAP-related rehospitalization (5.0% vs. 2.7%) rounded out the findings.
“Steroids may help patients feel better and have more reassuring vital signs and get out sooner, but at the expense of some toxicity, which might account for the readmissions,” Dr. Brotman said in an interview.
He then turned to the subject of sepsis. Before administering steroids for sepsis, physicians need to determine whether the powerful anti-inflammatory effect is worth the risks they carry. Adrenal failure is the biggest risk, Dr. Brotman said, citing last year’s ADRENAL study of 3,658 mechanically ventilated patients (N Engl J Med. 2018;378:797-808). They were randomized to a week of hydrocortisone 200 mg per day or placebo. The overall death rate was 28%, and steroids reduced the risk by only 5% (odds ratio, 0.95). The treatment group also had higher mean arterial pressure and lactate, a slower heart rate, and more serious diverse events, including hyperglycemia, hypernatremia, myopathy, and encephalopathy.
Initially, treated patients appeared to do better clinically, with a shorter period of ventilation, a shorter discharge from intensive care. But overall, there was no difference in ventilator-free days or hospital length of stay.“You may be improving clinical outcomes, but if you’re suppressing inflammation completely, you’re also suppressing a healthy response to an infectious process. There are some infections we need to be particularly cautious with, including tuberculosis,” Dr. Brotman added in the interview.
For his part, Dr. Dressler stated that the steroids-for-sepsis issue is “slightly murky.”
“A couple of new trials came out recently, and they lead us to reassess our thinking on this,” he said. Together, the studies comprised about 5,000 patients with septic shock – more than doubling the already studied cohort in 1 year. The reassessment came by means of a 2018 meta-analysis of all 9,000 patients. The findings actually led to new treatment guidelines, which were published in the British Medical Journal last year (2018;362:k3284).
The conclusion made a “weak recommendation” for corticosteroids in patients with sepsis. “Both steroids and no steroids are reasonable management options,” when also considering the overall clinical picture. For example, the recommendations advise against giving steroids to pregnant women, neonates, and patients with preexisting adrenal insufficiency.
However, the article noted, “Fully informed patients who value avoiding death over quality of life and function would likely choose corticosteroids.”
“I’m not sure these [studies] are changing what most people are doing,” Dr. Brotman countered in his interview. “I think the studies do help somewhat, because now we have enough numbers to suggest we can achieve a statistically significantly benefit. Septic shock is a life-threatening situation with a 40% risk of death. Now we can see that for every 50 people we treat with steroids, we can prevent 1 death. But that’s not the whole picture. Steroids won’t change the morality rate from 40% to 10%, but these studies do suggest that we can capture a small percent of people who may otherwise die.”
Dr. Dressler reported no financial disclosures. Dr. Brotman reported relationships with Bristol-Myers Squibb, Pfizer, and Portola.
Two experts drew on both personal experience and extant literature in the debate “Steroids for Pneumonia and Sepsis ... Do You Believe?” on Tuesday.
Daniel Dressler, MD, MSC, SFHM, of Emory University, Atlanta, and Daniel J. Brotman, MD, SFHM, director of the hospitalist program at Johns Hopkins Hospital, Baltimore, used a series of case studies to illustrate the conundrum. Despite their “pro” and “con” stances, though, both agreed in the end: First, do no harm.
There are no blanket recommendations for the use of steroids for pneumonia, because historically, studies have come to varied conclusions. However, Dr. Dressler, who advocated for the medications, said in an interview that recent publications paint a more complete picture.
“I think the newer studies in 2015 have made us more comfortable, because they look like there is more benefit” for steroids, especially among more severely ill patients, he noted. These international studies added more than 800 cases to the literature. A Spanish trial randomized 120 patients with high C-reactive protein to placebo or 0.5 mg/kg methylpred-nisolone every 12 hours for 5 days. There were fewer treatment failures in the prednisone group (13% vs. 31%), and fewer adverse clinical outcomes of intubation, shock, or death (3% vs. 14%). The number needed to treat to prevent one event was just six. (JAMA. 2015;313[7]:677-86).
In addition, a Cochrane meta-analysis analyzed 13 randomized trials, comprising more than 2,000 hospitalized patients. It found consistently lower rates of mortality, acute respiratory distress syndrome, early treatment failure, and hyperglycemia (Cochrane Database Syst Rev. 2017 Dec 13;12:CD007720).
“My recommendation is that inpatient clinicians should consider a brief course [5-7 days] of moderate-dose steroids (20-60 mg of prednisone equivalent) in patients admitted with CAP [community acquired pneumonia],” he said in an interview. “I personally give 40 mg prednisone for 5 days. I give even stronger consideration for severe CAP.”
Dr. Brotman countered with another set of articles from the literature, citing several studies with different conclusions. A separate meta-analysis of 12 trials appeared in the Annals of Internal Medicine (2015;163[7]:519-28). Half of the studies looked at outcomes in severe CAP, and the other half in less-severe cases. In the severe population, corticosteroids were associated with an overall decrease of about 40% in mortality, but that finding was driven largely by a single study; the others found nonsignificant decreases. The picture was less equivocal in the milder cases: Corticosteroids did not significantly reduce the risk of death.
In both groups, however, the drugs did significantly reduce the amount of hospital time. But this reduction came at a price, according to another review published in Clinical Infectious Diseases (2018 Jan 18;66[3]:346-54). Hospital stay was indeed reduced by a day, but there was no significant reduced risk of death (5.0% vs. 5.9% placebo). Similar rates of ICU admission and treatment failure, a doubling in the risk of hyperglycemia that required insulin, and a significantly higher risk of CAP-related rehospitalization (5.0% vs. 2.7%) rounded out the findings.
“Steroids may help patients feel better and have more reassuring vital signs and get out sooner, but at the expense of some toxicity, which might account for the readmissions,” Dr. Brotman said in an interview.
He then turned to the subject of sepsis. Before administering steroids for sepsis, physicians need to determine whether the powerful anti-inflammatory effect is worth the risks they carry. Adrenal failure is the biggest risk, Dr. Brotman said, citing last year’s ADRENAL study of 3,658 mechanically ventilated patients (N Engl J Med. 2018;378:797-808). They were randomized to a week of hydrocortisone 200 mg per day or placebo. The overall death rate was 28%, and steroids reduced the risk by only 5% (odds ratio, 0.95). The treatment group also had higher mean arterial pressure and lactate, a slower heart rate, and more serious diverse events, including hyperglycemia, hypernatremia, myopathy, and encephalopathy.
Initially, treated patients appeared to do better clinically, with a shorter period of ventilation, a shorter discharge from intensive care. But overall, there was no difference in ventilator-free days or hospital length of stay.“You may be improving clinical outcomes, but if you’re suppressing inflammation completely, you’re also suppressing a healthy response to an infectious process. There are some infections we need to be particularly cautious with, including tuberculosis,” Dr. Brotman added in the interview.
For his part, Dr. Dressler stated that the steroids-for-sepsis issue is “slightly murky.”
“A couple of new trials came out recently, and they lead us to reassess our thinking on this,” he said. Together, the studies comprised about 5,000 patients with septic shock – more than doubling the already studied cohort in 1 year. The reassessment came by means of a 2018 meta-analysis of all 9,000 patients. The findings actually led to new treatment guidelines, which were published in the British Medical Journal last year (2018;362:k3284).
The conclusion made a “weak recommendation” for corticosteroids in patients with sepsis. “Both steroids and no steroids are reasonable management options,” when also considering the overall clinical picture. For example, the recommendations advise against giving steroids to pregnant women, neonates, and patients with preexisting adrenal insufficiency.
However, the article noted, “Fully informed patients who value avoiding death over quality of life and function would likely choose corticosteroids.”
“I’m not sure these [studies] are changing what most people are doing,” Dr. Brotman countered in his interview. “I think the studies do help somewhat, because now we have enough numbers to suggest we can achieve a statistically significantly benefit. Septic shock is a life-threatening situation with a 40% risk of death. Now we can see that for every 50 people we treat with steroids, we can prevent 1 death. But that’s not the whole picture. Steroids won’t change the morality rate from 40% to 10%, but these studies do suggest that we can capture a small percent of people who may otherwise die.”
Dr. Dressler reported no financial disclosures. Dr. Brotman reported relationships with Bristol-Myers Squibb, Pfizer, and Portola.