User login
Alzheimer’s: Biomarkers, not cognition, will now define disorder
A new definition of Alzheimer’s disease based solely on biomarkers has the potential to strengthen clinical trials and change the way physicians talk to patients.
AB is the key to this classification paradigm – any patient with it (A+) is on the Alzheimer’s continuum. But only those with both amyloid and tau in the brain (A+T+) receive the “Alzheimer’s disease” classification. A third biomarker, neurodegeneration, may be either present or absent for an Alzheimer’s disease profile (N+ or N-). Cognitive staging adds important details, but remains secondary to the biomarker classification.

Jointly created by National Institute on Aging and the Alzheimer’s Association, the system – dubbed the NIA-AA Research Framework – represents a new, common language that researchers around the world may now use to generate and test Alzheimer’s hypotheses, and to optimize both epidemiologic studies and interventional trials. It will be especially important as Alzheimer’s prevention trials seek to target patients who are cognitively normal, yet harbor the neuropathological hallmarks of the disease.
This recasting adds Alzheimer’s to the list of biomarker-defined disorders, including hypertension, diabetes, and hyperlipidemia. It is a timely and necessary reframing, said Clifford Jack, MD, chair of the 20-member committee that created the paradigm. It appears in the April 10 issue of Alzheimer’s & Dementia.
“This is a fundamental change in the definition of Alzheimer’s disease,” Dr. Jack said in an interview. “We are advocating the disease be defined by its neuropathology [of plaques and tangles], which is specific to Alzheimer’s, and no longer by clinical symptoms which are not specific for any disease.”
One of the primary intents is to refine AD research cohorts, allowing pure stratification of patients who actually have the intended therapeutic targets of amyloid beta or tau. Without biomarker screening, up to 30% of subjects who enroll in AD drug trials don’t have the target pathologies – a situation researchers say contributes to the long string of failed Alzheimer’s drug studies.
For now, the system is intended only for research settings said Dr. Jack, an Alzheimer’s investigator at the Mayo Clinic, Rochester, Minn. But as biomarker testing comes of age and new less-expensive markers are discovered, the paradigm will likely be incorporated into clinical practice. The process can begin even now with a simple change in the way doctors talk to patients about Alzheimer’s, he said in an interview.
“We advocate people stop using the terms ‘probable or possible AD.’ A better term is ‘Alzheimer’s clinical syndrome.’ Without biomarkers, the clinical syndrome is the only thing you can know. What you can’t know is whether they do or don’t have Alzheimer’s disease. When I’m asked by physicians, ‘What do I tell my patients now?’ my very direct answer is ‘Tell them the truth.’ And the truth is that they have Alzheimer’s clinical syndrome and may or may not have Alzheimer’s disease.”
A reflection of evolving science
The research framework reflects advances in Alzheimer’s science that have occurred since the NIA last updated it AD diagnostic criteria in 2011. Those criteria divided the disease continuum into three phases largely based on cognitive symptoms, but were the first to recognize a presymptomatic AD phase.
- Preclinical: Brain changes, including amyloid buildup and other nerve cell changes already may be in progress but significant clinical symptoms are not yet evident.
- Mild cognitive impairment (MCI): A stage marked by symptoms of memory and/or other thinking problems that are greater than normal for a person’s age and education but that do not interfere with his or her independence. MCI may or may not progress to Alzheimer’s dementia.
- Alzheimer’s dementia: The final stage of the disease in which the symptoms of Alzheimer’s, such as memory loss, word-finding difficulties, and visual/spatial problems, are significant enough to impair a person’s ability to function independently.
The next 6 years brought striking advances in understanding the biology and pathology of AD, as well as technical advances in biomarker measurements. It became possible not only to measure AB and tau in cerebrospinal fluid but also to see these proteins in living brains with specialized PET ligands. It also became obvious that about a third of subjects in any given AD study didn’t have the disease-defining brain plaques and tangles – the therapeutic targets of all the largest drug studies to date. And while it’s clear that none of the interventions that have been through trials have exerted a significant benefit yet, “Treating people for a disease they don’t have can’t possibly help the results,” Dr. Jack said.
These research observations and revolutionary biomarker advances have reshaped the way researchers think about AD. To maximize research potential and to create a global classification standard that would unify studies as well, NIA and the Alzheimer’s Association convened several meetings to redefine Alzheimer’s disease biologically, by pathologic brain changes as measured by biomarkers. In this paradigm, cognitive dysfunction steps aside as the primary classification driver, becoming a symptom of AD rather than its definition.
“The way AD has historically been defined is by clinical symptoms: a progressive amnestic dementia was Alzheimer’s, and if there was no progressive amnestic dementia, it wasn’t,” Dr. Jack said. “Well, it turns out that both of those statements are wrong. About 30% of people with progressive amnestic dementia have other things causing it.”
It makes much more sense, he said, to define the disease based on its unique neuropathologic signature: amyloid beta plaques and tau neurofibrillary tangles in the brain.
The three-part key: A/T(N)
The NIA-AA research framework yields eight biomarker profiles with different combinations of amyloid (A), tau (T), and neuropathologic damage (N).
“Different measures have different roles,” Dr. Jack and his colleagues wrote in Alzheimer’s & Dementia. “Amyloid beta biomarkers determine whether or not an individual is in the Alzheimer’s continuum. Pathologic tau biomarkers determine if someone who is in the Alzheimer’s continuum has AD, because both amyloid beta and tau are required for a neuropathologic diagnosis of the disease. Neurodegenerative/neuronal injury biomarkers and cognitive symptoms, neither of which is specific for AD, are used only to stage severity not to define the presence of the Alzheimer’s continuum.”
The “N” category is not as cut and dried at the other biomarkers, the paper noted.
“Biomarkers in the (N) group are indicators of neurodegeneration or neuronal injury resulting from many causes; they are not specific for neurodegeneration due to AD. In any individual, the proportion of observed neurodegeneration/injury that can be attributed to AD versus other possible comorbid conditions (most of which have no extant biomarker) is unknown.”
The biomarker profiles are:
- A-T-(N): Normal AD biomarkers
- A+T-(N): Alzheimer’s pathologic change; Alzheimer’s continuum
- A+T+(N): Alzheimer’s disease; Alzheimer’s continuum
- A+T-(N)+: Alzheimer’s with suspected non Alzheimer’s pathologic change; Alzheimer’s continuum
- A-T+(N)-: Non-AD pathologic change
- A-T-(N)+: Non-AD pathologic change
- A-T+(N)+: Non-AD pathologic change
“This latter biomarker profile implies evidence of one or more neuropathologic processes other than AD and has been labeled ‘suspected non-Alzheimer’s pathophysiology, or SNAP,” according to the paper.
Cognitive staging further refines each person’s status. There are two clinical staging schemes in the framework. One is the familiar syndromal staging system of cognitively unimpaired, MCI, and dementia, which can be subdivided into mild, moderate, and severe. This can be applied to anyone with a biomarker profile.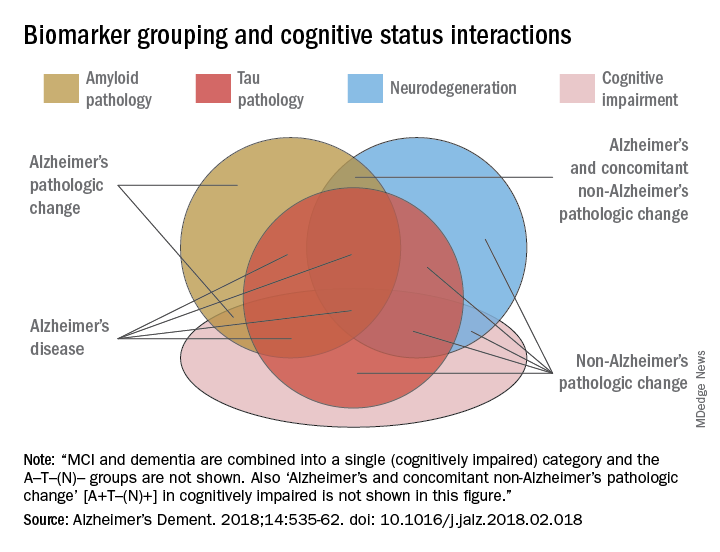
The second, a six-stage numerical clinical staging scheme, will apply only to those who are amyloid-positive and on the Alzheimer’s continuum. Stages run from 1 (unimpaired) to 6 (severe dementia). The numeric staging does not concentrate solely on cognition but also takes into account neurobehavioral and functional symptoms. It includes a transitional stage during which measures may be within population norms but have declined relative to the individual’s past performance.
The numeric staging scheme is intended to mesh with FDA guidance for clinical trials outcomes, the committee noted.
“A useful application envisioned for this numeric cognitive staging scheme is interventional trials. Indeed, the NIA-AA numeric staging scheme is intentionally very similar to the categorical system for staging AD outlined in recent FDA guidance for industry pertaining to developing drugs for treatment of early AD … it was our belief that harmonizing this aspect of the framework with FDA guidance would enhance cross fertilization between observational and interventional studies, which in turn would facilitate conduct of interventional clinical trials early in the disease process.”
The entire system yields a shorthand biomarker profile entirely unique to each subject. For example an A+T-(N)+ MCI profile suggests that both Alzheimer’s and non-Alzheimer’s pathologic change may be contributing to the cognitive impairment. A cognitive staging number could also be added.
This biomarker profile introduces the option of completely avoiding traditional AD nomenclature, the committee noted.
“Some investigators may prefer to not use the biomarker category terminology but instead simply report biomarker profile, i.e., A+T+(N)+ instead of ‘Alzheimer’s disease.’ An alternative is to combine the biomarker profile with a descriptive term – for example, ‘A+T+(N)+ with dementia’ instead of ‘Alzheimer’s disease with dementia’.”
Again, Dr. Jack cautioned, the paradigm is not intended for clinical use – at least not now. It relies entirely on biomarkers obtained by methods that are either invasive (lumbar puncture), unavailable outside research settings (tau scans), or very expensive when privately obtained (amyloid scans). Until this situation changes, the biomarker profile paradigm has little clinical impact.
IDEAS on the horizon
Change may be coming, however. The Alzheimer’s Association-sponsored Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study is assessing the clinical usefulness of amyloid PET scans and their impact on patient outcomes. The goal is to accumulate enough data to prove that amyloid scans are a cost-effective addition to the management of dementia patients. If federal payers agree and decide to cover amyloid scans, advocates hope that private insurers might follow suit.
An interim analysis of 4,000 scans, presented at the 2017 Alzheimer’s Association International Conference, was quite positive. Scan results changed patient management in 68% of cases, including refining dementia diagnoses, adding, stopping, or switching medications, and altering patient counseling.
IDEAS uses an FDA-approved amyloid imaging agent. But although several are under investigation, there are no approved tau PET ligands. However, other less-invasive and less-costly options may soon be developed, the committee noted. The search continues for a validated blood-based biomarker, including neurofilament light protein, plasma amyloid beta, and plasma tau.
“In the future, less-invasive/less-expensive blood-based biomarker tests - along with genetics, clinical, and demographic information - will likely play an important screening role in selecting individuals for more-expensive/more-invasive biomarker testing. This has been the history in other biologically defined diseases such as cardiovascular disease,” Dr. Jack and his colleagues noted in the paper.
In any case, however, without an effective treatment, much of the information conveyed by the biomarker profile paradigm remains, literally, academic, Dr. Jack said.
“If [the biomarker profile] were easy to determine and inexpensive, I imagine a lot of people would ask for it. Certainly many people would want to know, especially if they have a cognitive problem. People who have a family history, who may have Alzheimer’s pathology without the symptoms, might want to know. But the reality is that, until there’s a treatment that alters the course of this disease, finding out that you actually have Alzheimer’s is not going to enable you to change anything.”
The editors of Alzheimer’s & Dementia are seeking comment on the research framework. Letters and commentary can be submitted through June and will be considered for publication in an e-book, to be published sometime this summer, according to an accompanying editorial (https://doi.org/10/1016/j.jalz.2018.03.003).
Alzheimer’s & Dementia is the official journal of the Alzheimer’s Association. Dr. Jack has served on scientific advisory boards for Elan/Janssen AI, Bristol-Meyers Squibb, Eli Lilly, GE Healthcare, Siemens, and Eisai; received research support from Baxter International, Allon Therapeutics; and holds stock in Johnson & Johnson. Disclosures for other committee members can be found here.
SOURCE: Jack CR et al. Alzheimer’s Dement. 2018;14:535-62. doi: 10.1016/j.jalz.2018.02.018.
The biologically defined amyloid beta–tau–neuronal damage (ATN) framework is a logical and modern approach to Alzheimer’s disease (AD) diagnosis. It is hard to argue that more data are bad. Having such data on every patient would certainly be a luxury, but, with a few notable exceptions, the context in which this will most frequently occur is within the context of clinical trials.
While having this information does provide a biological basis for diagnosis, it does not account for non-AD contributions to the patient’s symptoms, which are found in more than half of all AD patients at autopsy; these non-AD pathologies also can influence clinical trial outcomes.

It also seems a bit ironic that the only meaningful manifestation of AD is now essentially left out of the diagnostic framework or relegated to nothing more than an adjective. Yet having a head full of amyloid means little if a person does not express symptoms (and vice versa), and we know that all people do not progress in the same way.
In the future, genomic and exposomic profiles may provide an even-more-nuanced picture, but further work is needed before that becomes a clinical reality. For now, the ATN biomarker framework represents the state of the art, though not an end.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale. He is also associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He has no relevant disclosures.
The biologically defined amyloid beta–tau–neuronal damage (ATN) framework is a logical and modern approach to Alzheimer’s disease (AD) diagnosis. It is hard to argue that more data are bad. Having such data on every patient would certainly be a luxury, but, with a few notable exceptions, the context in which this will most frequently occur is within the context of clinical trials.
While having this information does provide a biological basis for diagnosis, it does not account for non-AD contributions to the patient’s symptoms, which are found in more than half of all AD patients at autopsy; these non-AD pathologies also can influence clinical trial outcomes.
It also seems a bit ironic that the only meaningful manifestation of AD is now essentially left out of the diagnostic framework or relegated to nothing more than an adjective. Yet having a head full of amyloid means little if a person does not express symptoms (and vice versa), and we know that all people do not progress in the same way.
In the future, genomic and exposomic profiles may provide an even-more-nuanced picture, but further work is needed before that becomes a clinical reality. For now, the ATN biomarker framework represents the state of the art, though not an end.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale. He is also associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He has no relevant disclosures.
The biologically defined amyloid beta–tau–neuronal damage (ATN) framework is a logical and modern approach to Alzheimer’s disease (AD) diagnosis. It is hard to argue that more data are bad. Having such data on every patient would certainly be a luxury, but, with a few notable exceptions, the context in which this will most frequently occur is within the context of clinical trials.
While having this information does provide a biological basis for diagnosis, it does not account for non-AD contributions to the patient’s symptoms, which are found in more than half of all AD patients at autopsy; these non-AD pathologies also can influence clinical trial outcomes.
It also seems a bit ironic that the only meaningful manifestation of AD is now essentially left out of the diagnostic framework or relegated to nothing more than an adjective. Yet having a head full of amyloid means little if a person does not express symptoms (and vice versa), and we know that all people do not progress in the same way.
In the future, genomic and exposomic profiles may provide an even-more-nuanced picture, but further work is needed before that becomes a clinical reality. For now, the ATN biomarker framework represents the state of the art, though not an end.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale. He is also associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He has no relevant disclosures.
A new definition of Alzheimer’s disease based solely on biomarkers has the potential to strengthen clinical trials and change the way physicians talk to patients.
AB is the key to this classification paradigm – any patient with it (A+) is on the Alzheimer’s continuum. But only those with both amyloid and tau in the brain (A+T+) receive the “Alzheimer’s disease” classification. A third biomarker, neurodegeneration, may be either present or absent for an Alzheimer’s disease profile (N+ or N-). Cognitive staging adds important details, but remains secondary to the biomarker classification.
Jointly created by National Institute on Aging and the Alzheimer’s Association, the system – dubbed the NIA-AA Research Framework – represents a new, common language that researchers around the world may now use to generate and test Alzheimer’s hypotheses, and to optimize both epidemiologic studies and interventional trials. It will be especially important as Alzheimer’s prevention trials seek to target patients who are cognitively normal, yet harbor the neuropathological hallmarks of the disease.
This recasting adds Alzheimer’s to the list of biomarker-defined disorders, including hypertension, diabetes, and hyperlipidemia. It is a timely and necessary reframing, said Clifford Jack, MD, chair of the 20-member committee that created the paradigm. It appears in the April 10 issue of Alzheimer’s & Dementia.
“This is a fundamental change in the definition of Alzheimer’s disease,” Dr. Jack said in an interview. “We are advocating the disease be defined by its neuropathology [of plaques and tangles], which is specific to Alzheimer’s, and no longer by clinical symptoms which are not specific for any disease.”
One of the primary intents is to refine AD research cohorts, allowing pure stratification of patients who actually have the intended therapeutic targets of amyloid beta or tau. Without biomarker screening, up to 30% of subjects who enroll in AD drug trials don’t have the target pathologies – a situation researchers say contributes to the long string of failed Alzheimer’s drug studies.
For now, the system is intended only for research settings said Dr. Jack, an Alzheimer’s investigator at the Mayo Clinic, Rochester, Minn. But as biomarker testing comes of age and new less-expensive markers are discovered, the paradigm will likely be incorporated into clinical practice. The process can begin even now with a simple change in the way doctors talk to patients about Alzheimer’s, he said in an interview.
“We advocate people stop using the terms ‘probable or possible AD.’ A better term is ‘Alzheimer’s clinical syndrome.’ Without biomarkers, the clinical syndrome is the only thing you can know. What you can’t know is whether they do or don’t have Alzheimer’s disease. When I’m asked by physicians, ‘What do I tell my patients now?’ my very direct answer is ‘Tell them the truth.’ And the truth is that they have Alzheimer’s clinical syndrome and may or may not have Alzheimer’s disease.”
A reflection of evolving science
The research framework reflects advances in Alzheimer’s science that have occurred since the NIA last updated it AD diagnostic criteria in 2011. Those criteria divided the disease continuum into three phases largely based on cognitive symptoms, but were the first to recognize a presymptomatic AD phase.
- Preclinical: Brain changes, including amyloid buildup and other nerve cell changes already may be in progress but significant clinical symptoms are not yet evident.
- Mild cognitive impairment (MCI): A stage marked by symptoms of memory and/or other thinking problems that are greater than normal for a person’s age and education but that do not interfere with his or her independence. MCI may or may not progress to Alzheimer’s dementia.
- Alzheimer’s dementia: The final stage of the disease in which the symptoms of Alzheimer’s, such as memory loss, word-finding difficulties, and visual/spatial problems, are significant enough to impair a person’s ability to function independently.
The next 6 years brought striking advances in understanding the biology and pathology of AD, as well as technical advances in biomarker measurements. It became possible not only to measure AB and tau in cerebrospinal fluid but also to see these proteins in living brains with specialized PET ligands. It also became obvious that about a third of subjects in any given AD study didn’t have the disease-defining brain plaques and tangles – the therapeutic targets of all the largest drug studies to date. And while it’s clear that none of the interventions that have been through trials have exerted a significant benefit yet, “Treating people for a disease they don’t have can’t possibly help the results,” Dr. Jack said.
These research observations and revolutionary biomarker advances have reshaped the way researchers think about AD. To maximize research potential and to create a global classification standard that would unify studies as well, NIA and the Alzheimer’s Association convened several meetings to redefine Alzheimer’s disease biologically, by pathologic brain changes as measured by biomarkers. In this paradigm, cognitive dysfunction steps aside as the primary classification driver, becoming a symptom of AD rather than its definition.
“The way AD has historically been defined is by clinical symptoms: a progressive amnestic dementia was Alzheimer’s, and if there was no progressive amnestic dementia, it wasn’t,” Dr. Jack said. “Well, it turns out that both of those statements are wrong. About 30% of people with progressive amnestic dementia have other things causing it.”
It makes much more sense, he said, to define the disease based on its unique neuropathologic signature: amyloid beta plaques and tau neurofibrillary tangles in the brain.
The three-part key: A/T(N)
The NIA-AA research framework yields eight biomarker profiles with different combinations of amyloid (A), tau (T), and neuropathologic damage (N).
“Different measures have different roles,” Dr. Jack and his colleagues wrote in Alzheimer’s & Dementia. “Amyloid beta biomarkers determine whether or not an individual is in the Alzheimer’s continuum. Pathologic tau biomarkers determine if someone who is in the Alzheimer’s continuum has AD, because both amyloid beta and tau are required for a neuropathologic diagnosis of the disease. Neurodegenerative/neuronal injury biomarkers and cognitive symptoms, neither of which is specific for AD, are used only to stage severity not to define the presence of the Alzheimer’s continuum.”
The “N” category is not as cut and dried at the other biomarkers, the paper noted.
“Biomarkers in the (N) group are indicators of neurodegeneration or neuronal injury resulting from many causes; they are not specific for neurodegeneration due to AD. In any individual, the proportion of observed neurodegeneration/injury that can be attributed to AD versus other possible comorbid conditions (most of which have no extant biomarker) is unknown.”
The biomarker profiles are:
- A-T-(N): Normal AD biomarkers
- A+T-(N): Alzheimer’s pathologic change; Alzheimer’s continuum
- A+T+(N): Alzheimer’s disease; Alzheimer’s continuum
- A+T-(N)+: Alzheimer’s with suspected non Alzheimer’s pathologic change; Alzheimer’s continuum
- A-T+(N)-: Non-AD pathologic change
- A-T-(N)+: Non-AD pathologic change
- A-T+(N)+: Non-AD pathologic change
“This latter biomarker profile implies evidence of one or more neuropathologic processes other than AD and has been labeled ‘suspected non-Alzheimer’s pathophysiology, or SNAP,” according to the paper.
Cognitive staging further refines each person’s status. There are two clinical staging schemes in the framework. One is the familiar syndromal staging system of cognitively unimpaired, MCI, and dementia, which can be subdivided into mild, moderate, and severe. This can be applied to anyone with a biomarker profile.
The second, a six-stage numerical clinical staging scheme, will apply only to those who are amyloid-positive and on the Alzheimer’s continuum. Stages run from 1 (unimpaired) to 6 (severe dementia). The numeric staging does not concentrate solely on cognition but also takes into account neurobehavioral and functional symptoms. It includes a transitional stage during which measures may be within population norms but have declined relative to the individual’s past performance.
The numeric staging scheme is intended to mesh with FDA guidance for clinical trials outcomes, the committee noted.
“A useful application envisioned for this numeric cognitive staging scheme is interventional trials. Indeed, the NIA-AA numeric staging scheme is intentionally very similar to the categorical system for staging AD outlined in recent FDA guidance for industry pertaining to developing drugs for treatment of early AD … it was our belief that harmonizing this aspect of the framework with FDA guidance would enhance cross fertilization between observational and interventional studies, which in turn would facilitate conduct of interventional clinical trials early in the disease process.”
The entire system yields a shorthand biomarker profile entirely unique to each subject. For example an A+T-(N)+ MCI profile suggests that both Alzheimer’s and non-Alzheimer’s pathologic change may be contributing to the cognitive impairment. A cognitive staging number could also be added.
This biomarker profile introduces the option of completely avoiding traditional AD nomenclature, the committee noted.
“Some investigators may prefer to not use the biomarker category terminology but instead simply report biomarker profile, i.e., A+T+(N)+ instead of ‘Alzheimer’s disease.’ An alternative is to combine the biomarker profile with a descriptive term – for example, ‘A+T+(N)+ with dementia’ instead of ‘Alzheimer’s disease with dementia’.”
Again, Dr. Jack cautioned, the paradigm is not intended for clinical use – at least not now. It relies entirely on biomarkers obtained by methods that are either invasive (lumbar puncture), unavailable outside research settings (tau scans), or very expensive when privately obtained (amyloid scans). Until this situation changes, the biomarker profile paradigm has little clinical impact.
IDEAS on the horizon
Change may be coming, however. The Alzheimer’s Association-sponsored Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study is assessing the clinical usefulness of amyloid PET scans and their impact on patient outcomes. The goal is to accumulate enough data to prove that amyloid scans are a cost-effective addition to the management of dementia patients. If federal payers agree and decide to cover amyloid scans, advocates hope that private insurers might follow suit.
An interim analysis of 4,000 scans, presented at the 2017 Alzheimer’s Association International Conference, was quite positive. Scan results changed patient management in 68% of cases, including refining dementia diagnoses, adding, stopping, or switching medications, and altering patient counseling.
IDEAS uses an FDA-approved amyloid imaging agent. But although several are under investigation, there are no approved tau PET ligands. However, other less-invasive and less-costly options may soon be developed, the committee noted. The search continues for a validated blood-based biomarker, including neurofilament light protein, plasma amyloid beta, and plasma tau.
“In the future, less-invasive/less-expensive blood-based biomarker tests - along with genetics, clinical, and demographic information - will likely play an important screening role in selecting individuals for more-expensive/more-invasive biomarker testing. This has been the history in other biologically defined diseases such as cardiovascular disease,” Dr. Jack and his colleagues noted in the paper.
In any case, however, without an effective treatment, much of the information conveyed by the biomarker profile paradigm remains, literally, academic, Dr. Jack said.
“If [the biomarker profile] were easy to determine and inexpensive, I imagine a lot of people would ask for it. Certainly many people would want to know, especially if they have a cognitive problem. People who have a family history, who may have Alzheimer’s pathology without the symptoms, might want to know. But the reality is that, until there’s a treatment that alters the course of this disease, finding out that you actually have Alzheimer’s is not going to enable you to change anything.”
The editors of Alzheimer’s & Dementia are seeking comment on the research framework. Letters and commentary can be submitted through June and will be considered for publication in an e-book, to be published sometime this summer, according to an accompanying editorial (https://doi.org/10/1016/j.jalz.2018.03.003).
Alzheimer’s & Dementia is the official journal of the Alzheimer’s Association. Dr. Jack has served on scientific advisory boards for Elan/Janssen AI, Bristol-Meyers Squibb, Eli Lilly, GE Healthcare, Siemens, and Eisai; received research support from Baxter International, Allon Therapeutics; and holds stock in Johnson & Johnson. Disclosures for other committee members can be found here.
SOURCE: Jack CR et al. Alzheimer’s Dement. 2018;14:535-62. doi: 10.1016/j.jalz.2018.02.018.
A new definition of Alzheimer’s disease based solely on biomarkers has the potential to strengthen clinical trials and change the way physicians talk to patients.
AB is the key to this classification paradigm – any patient with it (A+) is on the Alzheimer’s continuum. But only those with both amyloid and tau in the brain (A+T+) receive the “Alzheimer’s disease” classification. A third biomarker, neurodegeneration, may be either present or absent for an Alzheimer’s disease profile (N+ or N-). Cognitive staging adds important details, but remains secondary to the biomarker classification.
Jointly created by National Institute on Aging and the Alzheimer’s Association, the system – dubbed the NIA-AA Research Framework – represents a new, common language that researchers around the world may now use to generate and test Alzheimer’s hypotheses, and to optimize both epidemiologic studies and interventional trials. It will be especially important as Alzheimer’s prevention trials seek to target patients who are cognitively normal, yet harbor the neuropathological hallmarks of the disease.
This recasting adds Alzheimer’s to the list of biomarker-defined disorders, including hypertension, diabetes, and hyperlipidemia. It is a timely and necessary reframing, said Clifford Jack, MD, chair of the 20-member committee that created the paradigm. It appears in the April 10 issue of Alzheimer’s & Dementia.
“This is a fundamental change in the definition of Alzheimer’s disease,” Dr. Jack said in an interview. “We are advocating the disease be defined by its neuropathology [of plaques and tangles], which is specific to Alzheimer’s, and no longer by clinical symptoms which are not specific for any disease.”
One of the primary intents is to refine AD research cohorts, allowing pure stratification of patients who actually have the intended therapeutic targets of amyloid beta or tau. Without biomarker screening, up to 30% of subjects who enroll in AD drug trials don’t have the target pathologies – a situation researchers say contributes to the long string of failed Alzheimer’s drug studies.
For now, the system is intended only for research settings said Dr. Jack, an Alzheimer’s investigator at the Mayo Clinic, Rochester, Minn. But as biomarker testing comes of age and new less-expensive markers are discovered, the paradigm will likely be incorporated into clinical practice. The process can begin even now with a simple change in the way doctors talk to patients about Alzheimer’s, he said in an interview.
“We advocate people stop using the terms ‘probable or possible AD.’ A better term is ‘Alzheimer’s clinical syndrome.’ Without biomarkers, the clinical syndrome is the only thing you can know. What you can’t know is whether they do or don’t have Alzheimer’s disease. When I’m asked by physicians, ‘What do I tell my patients now?’ my very direct answer is ‘Tell them the truth.’ And the truth is that they have Alzheimer’s clinical syndrome and may or may not have Alzheimer’s disease.”
A reflection of evolving science
The research framework reflects advances in Alzheimer’s science that have occurred since the NIA last updated it AD diagnostic criteria in 2011. Those criteria divided the disease continuum into three phases largely based on cognitive symptoms, but were the first to recognize a presymptomatic AD phase.
- Preclinical: Brain changes, including amyloid buildup and other nerve cell changes already may be in progress but significant clinical symptoms are not yet evident.
- Mild cognitive impairment (MCI): A stage marked by symptoms of memory and/or other thinking problems that are greater than normal for a person’s age and education but that do not interfere with his or her independence. MCI may or may not progress to Alzheimer’s dementia.
- Alzheimer’s dementia: The final stage of the disease in which the symptoms of Alzheimer’s, such as memory loss, word-finding difficulties, and visual/spatial problems, are significant enough to impair a person’s ability to function independently.
The next 6 years brought striking advances in understanding the biology and pathology of AD, as well as technical advances in biomarker measurements. It became possible not only to measure AB and tau in cerebrospinal fluid but also to see these proteins in living brains with specialized PET ligands. It also became obvious that about a third of subjects in any given AD study didn’t have the disease-defining brain plaques and tangles – the therapeutic targets of all the largest drug studies to date. And while it’s clear that none of the interventions that have been through trials have exerted a significant benefit yet, “Treating people for a disease they don’t have can’t possibly help the results,” Dr. Jack said.
These research observations and revolutionary biomarker advances have reshaped the way researchers think about AD. To maximize research potential and to create a global classification standard that would unify studies as well, NIA and the Alzheimer’s Association convened several meetings to redefine Alzheimer’s disease biologically, by pathologic brain changes as measured by biomarkers. In this paradigm, cognitive dysfunction steps aside as the primary classification driver, becoming a symptom of AD rather than its definition.
“The way AD has historically been defined is by clinical symptoms: a progressive amnestic dementia was Alzheimer’s, and if there was no progressive amnestic dementia, it wasn’t,” Dr. Jack said. “Well, it turns out that both of those statements are wrong. About 30% of people with progressive amnestic dementia have other things causing it.”
It makes much more sense, he said, to define the disease based on its unique neuropathologic signature: amyloid beta plaques and tau neurofibrillary tangles in the brain.
The three-part key: A/T(N)
The NIA-AA research framework yields eight biomarker profiles with different combinations of amyloid (A), tau (T), and neuropathologic damage (N).
“Different measures have different roles,” Dr. Jack and his colleagues wrote in Alzheimer’s & Dementia. “Amyloid beta biomarkers determine whether or not an individual is in the Alzheimer’s continuum. Pathologic tau biomarkers determine if someone who is in the Alzheimer’s continuum has AD, because both amyloid beta and tau are required for a neuropathologic diagnosis of the disease. Neurodegenerative/neuronal injury biomarkers and cognitive symptoms, neither of which is specific for AD, are used only to stage severity not to define the presence of the Alzheimer’s continuum.”
The “N” category is not as cut and dried at the other biomarkers, the paper noted.
“Biomarkers in the (N) group are indicators of neurodegeneration or neuronal injury resulting from many causes; they are not specific for neurodegeneration due to AD. In any individual, the proportion of observed neurodegeneration/injury that can be attributed to AD versus other possible comorbid conditions (most of which have no extant biomarker) is unknown.”
The biomarker profiles are:
- A-T-(N): Normal AD biomarkers
- A+T-(N): Alzheimer’s pathologic change; Alzheimer’s continuum
- A+T+(N): Alzheimer’s disease; Alzheimer’s continuum
- A+T-(N)+: Alzheimer’s with suspected non Alzheimer’s pathologic change; Alzheimer’s continuum
- A-T+(N)-: Non-AD pathologic change
- A-T-(N)+: Non-AD pathologic change
- A-T+(N)+: Non-AD pathologic change
“This latter biomarker profile implies evidence of one or more neuropathologic processes other than AD and has been labeled ‘suspected non-Alzheimer’s pathophysiology, or SNAP,” according to the paper.
Cognitive staging further refines each person’s status. There are two clinical staging schemes in the framework. One is the familiar syndromal staging system of cognitively unimpaired, MCI, and dementia, which can be subdivided into mild, moderate, and severe. This can be applied to anyone with a biomarker profile.
The second, a six-stage numerical clinical staging scheme, will apply only to those who are amyloid-positive and on the Alzheimer’s continuum. Stages run from 1 (unimpaired) to 6 (severe dementia). The numeric staging does not concentrate solely on cognition but also takes into account neurobehavioral and functional symptoms. It includes a transitional stage during which measures may be within population norms but have declined relative to the individual’s past performance.
The numeric staging scheme is intended to mesh with FDA guidance for clinical trials outcomes, the committee noted.
“A useful application envisioned for this numeric cognitive staging scheme is interventional trials. Indeed, the NIA-AA numeric staging scheme is intentionally very similar to the categorical system for staging AD outlined in recent FDA guidance for industry pertaining to developing drugs for treatment of early AD … it was our belief that harmonizing this aspect of the framework with FDA guidance would enhance cross fertilization between observational and interventional studies, which in turn would facilitate conduct of interventional clinical trials early in the disease process.”
The entire system yields a shorthand biomarker profile entirely unique to each subject. For example an A+T-(N)+ MCI profile suggests that both Alzheimer’s and non-Alzheimer’s pathologic change may be contributing to the cognitive impairment. A cognitive staging number could also be added.
This biomarker profile introduces the option of completely avoiding traditional AD nomenclature, the committee noted.
“Some investigators may prefer to not use the biomarker category terminology but instead simply report biomarker profile, i.e., A+T+(N)+ instead of ‘Alzheimer’s disease.’ An alternative is to combine the biomarker profile with a descriptive term – for example, ‘A+T+(N)+ with dementia’ instead of ‘Alzheimer’s disease with dementia’.”
Again, Dr. Jack cautioned, the paradigm is not intended for clinical use – at least not now. It relies entirely on biomarkers obtained by methods that are either invasive (lumbar puncture), unavailable outside research settings (tau scans), or very expensive when privately obtained (amyloid scans). Until this situation changes, the biomarker profile paradigm has little clinical impact.
IDEAS on the horizon
Change may be coming, however. The Alzheimer’s Association-sponsored Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study is assessing the clinical usefulness of amyloid PET scans and their impact on patient outcomes. The goal is to accumulate enough data to prove that amyloid scans are a cost-effective addition to the management of dementia patients. If federal payers agree and decide to cover amyloid scans, advocates hope that private insurers might follow suit.
An interim analysis of 4,000 scans, presented at the 2017 Alzheimer’s Association International Conference, was quite positive. Scan results changed patient management in 68% of cases, including refining dementia diagnoses, adding, stopping, or switching medications, and altering patient counseling.
IDEAS uses an FDA-approved amyloid imaging agent. But although several are under investigation, there are no approved tau PET ligands. However, other less-invasive and less-costly options may soon be developed, the committee noted. The search continues for a validated blood-based biomarker, including neurofilament light protein, plasma amyloid beta, and plasma tau.
“In the future, less-invasive/less-expensive blood-based biomarker tests - along with genetics, clinical, and demographic information - will likely play an important screening role in selecting individuals for more-expensive/more-invasive biomarker testing. This has been the history in other biologically defined diseases such as cardiovascular disease,” Dr. Jack and his colleagues noted in the paper.
In any case, however, without an effective treatment, much of the information conveyed by the biomarker profile paradigm remains, literally, academic, Dr. Jack said.
“If [the biomarker profile] were easy to determine and inexpensive, I imagine a lot of people would ask for it. Certainly many people would want to know, especially if they have a cognitive problem. People who have a family history, who may have Alzheimer’s pathology without the symptoms, might want to know. But the reality is that, until there’s a treatment that alters the course of this disease, finding out that you actually have Alzheimer’s is not going to enable you to change anything.”
The editors of Alzheimer’s & Dementia are seeking comment on the research framework. Letters and commentary can be submitted through June and will be considered for publication in an e-book, to be published sometime this summer, according to an accompanying editorial (https://doi.org/10/1016/j.jalz.2018.03.003).
Alzheimer’s & Dementia is the official journal of the Alzheimer’s Association. Dr. Jack has served on scientific advisory boards for Elan/Janssen AI, Bristol-Meyers Squibb, Eli Lilly, GE Healthcare, Siemens, and Eisai; received research support from Baxter International, Allon Therapeutics; and holds stock in Johnson & Johnson. Disclosures for other committee members can be found here.
SOURCE: Jack CR et al. Alzheimer’s Dement. 2018;14:535-62. doi: 10.1016/j.jalz.2018.02.018.
FROM ALZHEIMER’S & DEMENTIA
Simvastatin, atorvastatin cut mortality risk for sepsis patients
a large health care database review has determined.
Among almost 53,000 sepsis patients, those who had been taking simvastatin were 28% less likely to die within 30 days of a sepsis admission than were patients not taking a statin. Atorvastatin conferred a similar significant survival benefit, reducing the risk of death by 22%, Chien-Chang Lee, MD and his colleagues wrote in the April issue of the journal CHEST®.
The drugs also exert a direct antimicrobial effect, he asserted.
“Of note, simvastatin was shown by several reports to have the most potent antibacterial activity,” targeting both methicillin-resistant and -sensitive Staphylococcus aureus, as well as gram negative and positive bacteria.
Dr. Lee and his colleagues extracted mortality and statin prescription data from the Taiwan National Health Insurance Database from 2000-2011. They looked at 30- and 90-day mortality in 52,737 patients who developed sepsis; the statins of interest were atorvastatin, simvastatin, and rosuvastatin. Patients had to have been taking the medication for at least 30 days before sepsis onset to be included, and patients taking more than one statin were excluded from the analysis.
Patients were a mean of 69 years old. About half had a lower respiratory infection. The remainder had infections within the abdomen, the biliary or urinary tract, skin, or orthopedic infections. There were no significant differences in comorbidities or in other medications taken among the three statin groups or the nonusers.
Of the entire cohort, 17% died by 30 days and nearly 23% by 90 days. Compared with those who had never received a statin, the statin users were 12% less likely to die by 30 days (hazard ratio, 0.88). Mortality at 90 days was also decreased, when compared with nonusers (HR, 0.93).
Simvastatin demonstrated the greatest benefit, with a 28% decreased risk of 30-day mortality (HR, 0.72). Atorvastatin followed, with a 22% risk reduction (HR, 0.78). Rosuvastatin exerted a nonsignificant 13% benefit.
The authors then examined 90-day mortality risks for the patients with a propensity matching score using a subgroup comprising 536 simvastatin users, 536 atorvastatin users, and 536 rosuvastatin users. Simvastatin was associated with a 23% reduction in 30-day mortality risk (HR, 0.77) and atorvastatin with a 21% reduction (HR, 0.79), when compared with rosuvastatin.
Statins’ antimicrobial properties are probably partially caused by their inactivation of the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase pathway, Dr. Lee and his colleagues noted. In addition to being vital for cholesterol synthesis, this pathway “also contributes to the production of isoprenoids and lipid compounds that are essential for cell signaling and structure in the pathogen. Secondly, the chemical property of different types of statins may affect their targeting to bacteria. The lipophilic properties of simvastatin or atorvastatin may allow better binding to bacteria cell walls than the hydrophilic properties of rosuvastatin.”
The study was funded by the Taiwan National Science Foundation and Taiwan National Ministry of Science and Technology. Dr. Lee had no financial conflicts.
The statin-sepsis mortality link will probably never be definitively proven, but the study by Lee and colleagues gives us the best data so far on this intriguing connection, Steven Q. Simpson, MD and Joel D. Mermis, MD wrote in an accompanying editorial.
“It is unlikely that prospective randomized trials of statins for prevention of sepsis mortality will ever be undertaken, owing to the sheer number of patients that would require randomization in order to have adequate numbers who actually develop sepsis,” the colleagues wrote. “We believe that the next best thing to randomization and a prospective trial is exactly what the authors have done – identify a cohort, track them through time, even if nonconcurrently, and match cases to controls by propensity matching on important clinical characteristics.”
Nevertheless, the two said, “This brings us to one aspect of the study that leaves open a window for some doubt.”
Lee et al. extracted their data from a large national insurance claims database. These systems “are commonly believed to overestimate sepsis incidence,” Dr. Simpson and Dr. Mermis wrote. A 2009 U.S. study bore this out, they said. “That study showed that in the U.S in 2014, there were approximately 1.7 million cases of sepsis in a population of 330 million, for an annual incidence rate of five sepsis cases per 1,000 patient-years.”
However, a “quick calculation” of the Taiwan data suggests that the annual sepsis caseload is about 5,200 per year in a population of 23 million at risk – an annual incidence of only 0.2 cases per 1,000 patient-years.
“This represents an order of magnitude difference in sepsis incidence between the U.S. and Taiwan, providing some issues to ponder. Does Taiwan indeed have a lower incidence of sepsis by that much? If so, is the lower incidence related to genetics, environment, health care access, or other factors?
“Although Lee et al. have provided us with data of the highest quality that we can likely hope for, the book may not be quite closed, yet.”
Dr. Mermis and Dr. Simpson are pulmonologists at the University of Kansas, Kansas City. They made their comments in an editorial published in the April issue of CHEST® (Mermis JD and Simpson SQ. CHEST. 2018 April. doi: 10.1016/j.chest.2017.12.004.)
The statin-sepsis mortality link will probably never be definitively proven, but the study by Lee and colleagues gives us the best data so far on this intriguing connection, Steven Q. Simpson, MD and Joel D. Mermis, MD wrote in an accompanying editorial.
“It is unlikely that prospective randomized trials of statins for prevention of sepsis mortality will ever be undertaken, owing to the sheer number of patients that would require randomization in order to have adequate numbers who actually develop sepsis,” the colleagues wrote. “We believe that the next best thing to randomization and a prospective trial is exactly what the authors have done – identify a cohort, track them through time, even if nonconcurrently, and match cases to controls by propensity matching on important clinical characteristics.”
Nevertheless, the two said, “This brings us to one aspect of the study that leaves open a window for some doubt.”
Lee et al. extracted their data from a large national insurance claims database. These systems “are commonly believed to overestimate sepsis incidence,” Dr. Simpson and Dr. Mermis wrote. A 2009 U.S. study bore this out, they said. “That study showed that in the U.S in 2014, there were approximately 1.7 million cases of sepsis in a population of 330 million, for an annual incidence rate of five sepsis cases per 1,000 patient-years.”
However, a “quick calculation” of the Taiwan data suggests that the annual sepsis caseload is about 5,200 per year in a population of 23 million at risk – an annual incidence of only 0.2 cases per 1,000 patient-years.
“This represents an order of magnitude difference in sepsis incidence between the U.S. and Taiwan, providing some issues to ponder. Does Taiwan indeed have a lower incidence of sepsis by that much? If so, is the lower incidence related to genetics, environment, health care access, or other factors?
“Although Lee et al. have provided us with data of the highest quality that we can likely hope for, the book may not be quite closed, yet.”
Dr. Mermis and Dr. Simpson are pulmonologists at the University of Kansas, Kansas City. They made their comments in an editorial published in the April issue of CHEST® (Mermis JD and Simpson SQ. CHEST. 2018 April. doi: 10.1016/j.chest.2017.12.004.)
The statin-sepsis mortality link will probably never be definitively proven, but the study by Lee and colleagues gives us the best data so far on this intriguing connection, Steven Q. Simpson, MD and Joel D. Mermis, MD wrote in an accompanying editorial.
“It is unlikely that prospective randomized trials of statins for prevention of sepsis mortality will ever be undertaken, owing to the sheer number of patients that would require randomization in order to have adequate numbers who actually develop sepsis,” the colleagues wrote. “We believe that the next best thing to randomization and a prospective trial is exactly what the authors have done – identify a cohort, track them through time, even if nonconcurrently, and match cases to controls by propensity matching on important clinical characteristics.”
Nevertheless, the two said, “This brings us to one aspect of the study that leaves open a window for some doubt.”
Lee et al. extracted their data from a large national insurance claims database. These systems “are commonly believed to overestimate sepsis incidence,” Dr. Simpson and Dr. Mermis wrote. A 2009 U.S. study bore this out, they said. “That study showed that in the U.S in 2014, there were approximately 1.7 million cases of sepsis in a population of 330 million, for an annual incidence rate of five sepsis cases per 1,000 patient-years.”
However, a “quick calculation” of the Taiwan data suggests that the annual sepsis caseload is about 5,200 per year in a population of 23 million at risk – an annual incidence of only 0.2 cases per 1,000 patient-years.
“This represents an order of magnitude difference in sepsis incidence between the U.S. and Taiwan, providing some issues to ponder. Does Taiwan indeed have a lower incidence of sepsis by that much? If so, is the lower incidence related to genetics, environment, health care access, or other factors?
“Although Lee et al. have provided us with data of the highest quality that we can likely hope for, the book may not be quite closed, yet.”
Dr. Mermis and Dr. Simpson are pulmonologists at the University of Kansas, Kansas City. They made their comments in an editorial published in the April issue of CHEST® (Mermis JD and Simpson SQ. CHEST. 2018 April. doi: 10.1016/j.chest.2017.12.004.)
a large health care database review has determined.
Among almost 53,000 sepsis patients, those who had been taking simvastatin were 28% less likely to die within 30 days of a sepsis admission than were patients not taking a statin. Atorvastatin conferred a similar significant survival benefit, reducing the risk of death by 22%, Chien-Chang Lee, MD and his colleagues wrote in the April issue of the journal CHEST®.
The drugs also exert a direct antimicrobial effect, he asserted.
“Of note, simvastatin was shown by several reports to have the most potent antibacterial activity,” targeting both methicillin-resistant and -sensitive Staphylococcus aureus, as well as gram negative and positive bacteria.
Dr. Lee and his colleagues extracted mortality and statin prescription data from the Taiwan National Health Insurance Database from 2000-2011. They looked at 30- and 90-day mortality in 52,737 patients who developed sepsis; the statins of interest were atorvastatin, simvastatin, and rosuvastatin. Patients had to have been taking the medication for at least 30 days before sepsis onset to be included, and patients taking more than one statin were excluded from the analysis.
Patients were a mean of 69 years old. About half had a lower respiratory infection. The remainder had infections within the abdomen, the biliary or urinary tract, skin, or orthopedic infections. There were no significant differences in comorbidities or in other medications taken among the three statin groups or the nonusers.
Of the entire cohort, 17% died by 30 days and nearly 23% by 90 days. Compared with those who had never received a statin, the statin users were 12% less likely to die by 30 days (hazard ratio, 0.88). Mortality at 90 days was also decreased, when compared with nonusers (HR, 0.93).
Simvastatin demonstrated the greatest benefit, with a 28% decreased risk of 30-day mortality (HR, 0.72). Atorvastatin followed, with a 22% risk reduction (HR, 0.78). Rosuvastatin exerted a nonsignificant 13% benefit.
The authors then examined 90-day mortality risks for the patients with a propensity matching score using a subgroup comprising 536 simvastatin users, 536 atorvastatin users, and 536 rosuvastatin users. Simvastatin was associated with a 23% reduction in 30-day mortality risk (HR, 0.77) and atorvastatin with a 21% reduction (HR, 0.79), when compared with rosuvastatin.
Statins’ antimicrobial properties are probably partially caused by their inactivation of the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase pathway, Dr. Lee and his colleagues noted. In addition to being vital for cholesterol synthesis, this pathway “also contributes to the production of isoprenoids and lipid compounds that are essential for cell signaling and structure in the pathogen. Secondly, the chemical property of different types of statins may affect their targeting to bacteria. The lipophilic properties of simvastatin or atorvastatin may allow better binding to bacteria cell walls than the hydrophilic properties of rosuvastatin.”
The study was funded by the Taiwan National Science Foundation and Taiwan National Ministry of Science and Technology. Dr. Lee had no financial conflicts.
a large health care database review has determined.
Among almost 53,000 sepsis patients, those who had been taking simvastatin were 28% less likely to die within 30 days of a sepsis admission than were patients not taking a statin. Atorvastatin conferred a similar significant survival benefit, reducing the risk of death by 22%, Chien-Chang Lee, MD and his colleagues wrote in the April issue of the journal CHEST®.
The drugs also exert a direct antimicrobial effect, he asserted.
“Of note, simvastatin was shown by several reports to have the most potent antibacterial activity,” targeting both methicillin-resistant and -sensitive Staphylococcus aureus, as well as gram negative and positive bacteria.
Dr. Lee and his colleagues extracted mortality and statin prescription data from the Taiwan National Health Insurance Database from 2000-2011. They looked at 30- and 90-day mortality in 52,737 patients who developed sepsis; the statins of interest were atorvastatin, simvastatin, and rosuvastatin. Patients had to have been taking the medication for at least 30 days before sepsis onset to be included, and patients taking more than one statin were excluded from the analysis.
Patients were a mean of 69 years old. About half had a lower respiratory infection. The remainder had infections within the abdomen, the biliary or urinary tract, skin, or orthopedic infections. There were no significant differences in comorbidities or in other medications taken among the three statin groups or the nonusers.
Of the entire cohort, 17% died by 30 days and nearly 23% by 90 days. Compared with those who had never received a statin, the statin users were 12% less likely to die by 30 days (hazard ratio, 0.88). Mortality at 90 days was also decreased, when compared with nonusers (HR, 0.93).
Simvastatin demonstrated the greatest benefit, with a 28% decreased risk of 30-day mortality (HR, 0.72). Atorvastatin followed, with a 22% risk reduction (HR, 0.78). Rosuvastatin exerted a nonsignificant 13% benefit.
The authors then examined 90-day mortality risks for the patients with a propensity matching score using a subgroup comprising 536 simvastatin users, 536 atorvastatin users, and 536 rosuvastatin users. Simvastatin was associated with a 23% reduction in 30-day mortality risk (HR, 0.77) and atorvastatin with a 21% reduction (HR, 0.79), when compared with rosuvastatin.
Statins’ antimicrobial properties are probably partially caused by their inactivation of the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase pathway, Dr. Lee and his colleagues noted. In addition to being vital for cholesterol synthesis, this pathway “also contributes to the production of isoprenoids and lipid compounds that are essential for cell signaling and structure in the pathogen. Secondly, the chemical property of different types of statins may affect their targeting to bacteria. The lipophilic properties of simvastatin or atorvastatin may allow better binding to bacteria cell walls than the hydrophilic properties of rosuvastatin.”
The study was funded by the Taiwan National Science Foundation and Taiwan National Ministry of Science and Technology. Dr. Lee had no financial conflicts.
FROM CHEST
Key clinical point: Simvastatin and atorvastatin were associated with decreased mortality risk among sepsis patients.
Major finding: Compared with those not taking the drugs, those taking simvastatin were 28% less likely to die by 30 days, and those taking atorvastatin were 22% less likely.
Study details: The database study comprised almost 54,000 sepsis cases over 11 years.
Disclosures: The study was funded by the Taiwan National Science Foundation and Taiwan National Ministry of Science and Technology. Dr. Lee had no financial conflicts.
Source: Lee C-C et al. CHEST. 2018 April;153(4):769-70.
AbbVie, Samsung Bioepis settle suits with delayed U.S. entry for adalimumab biosimilar
A new adalimumab biosimilar will become available in the European Union later this year, but a court settlement will keep Samsung Bioepis’ competitor off U.S. shelves until 2023.
Under the settlement, AbbVie, which manufactures adalimumab (Humira), will grant Bioepis and its partner, Biogen, a nonexclusive license to the intellectual property relating to the antibody. Bioepis’ version, dubbed SB5 (Imraldi), will enter global markets in a staggered fashion, according to an AbbVie press statement. In most countries in the European Union, the license period will begin on Oct. 16, 2018. In the United States, Samsung Bioepis’ license period will begin on June 30, 2023, according to the Abbvie statement.
Biogen and Bioepis hailed the settlement as a victory, but Imraldi won’t be the first Humira biosimilar to break into the U.S. market. Last September, AbbVie settled a similar suit with Amgen, granting patent licenses for the global use and sale of its anti–tumor necrosis factor–alpha antibody, Amgevita/Amjevita. Amgen expects to launch Amgevita in Europe on Oct. 16, 2018, and Amjevita in the United States on Jan. 31, 2023. Samsung Bioepis’ U.S. license date will not be accelerated upon Amgen’s entry.
Ian Henshaw, Biogen’s global head of biosimilars, said the deal further strengthens the company’s European biosimilars reach.
“Biogen is a leader in the emerging field of biosimilars through Samsung Bioepis, our joint venture with Samsung BioLogics,” Mr. Henshaw said in a press statement. “Biogen already markets two biosimilars in Europe and the planned introduction of Imraldi on Oct. 16 could potentially expand patient choice by offering physicians more options to meet the needs of patients while delivering significant savings to healthcare systems.”
AbbVie focused on the settlement as a global recognition of its leadership role in developing the anti-TNF-alpha antibody.
“The Samsung Bioepis settlement reflects the strength and breadth of AbbVie’s intellectual property,” Laura Schumacher, the company’s general counsel, said in the Abbvie statement. “We continue to believe biosimilars will play an important role in our healthcare system, but we also believe it is important to protect our investment in innovation. This agreement accomplishes both objectives.”
Samsung Bioepis will pay royalties to AbbVie for licensing its adalimumab patents once its biosimilar product is launched. As is the case with the prior Amgen resolution, AbbVie will not make any payments to Samsung Bioepis. “All litigation pending between the parties, as well as all litigation with Samsung Bioepis’ European partner, Biogen, will be dismissed. The precise terms of the agreements are confidential,” the Abbvie statement said.
The settlement brings to a closing a flurry of lawsuits Samsung Bioepis filed against AbbVie in 2017.
A new adalimumab biosimilar will become available in the European Union later this year, but a court settlement will keep Samsung Bioepis’ competitor off U.S. shelves until 2023.
Under the settlement, AbbVie, which manufactures adalimumab (Humira), will grant Bioepis and its partner, Biogen, a nonexclusive license to the intellectual property relating to the antibody. Bioepis’ version, dubbed SB5 (Imraldi), will enter global markets in a staggered fashion, according to an AbbVie press statement. In most countries in the European Union, the license period will begin on Oct. 16, 2018. In the United States, Samsung Bioepis’ license period will begin on June 30, 2023, according to the Abbvie statement.
Biogen and Bioepis hailed the settlement as a victory, but Imraldi won’t be the first Humira biosimilar to break into the U.S. market. Last September, AbbVie settled a similar suit with Amgen, granting patent licenses for the global use and sale of its anti–tumor necrosis factor–alpha antibody, Amgevita/Amjevita. Amgen expects to launch Amgevita in Europe on Oct. 16, 2018, and Amjevita in the United States on Jan. 31, 2023. Samsung Bioepis’ U.S. license date will not be accelerated upon Amgen’s entry.
Ian Henshaw, Biogen’s global head of biosimilars, said the deal further strengthens the company’s European biosimilars reach.
“Biogen is a leader in the emerging field of biosimilars through Samsung Bioepis, our joint venture with Samsung BioLogics,” Mr. Henshaw said in a press statement. “Biogen already markets two biosimilars in Europe and the planned introduction of Imraldi on Oct. 16 could potentially expand patient choice by offering physicians more options to meet the needs of patients while delivering significant savings to healthcare systems.”
AbbVie focused on the settlement as a global recognition of its leadership role in developing the anti-TNF-alpha antibody.
“The Samsung Bioepis settlement reflects the strength and breadth of AbbVie’s intellectual property,” Laura Schumacher, the company’s general counsel, said in the Abbvie statement. “We continue to believe biosimilars will play an important role in our healthcare system, but we also believe it is important to protect our investment in innovation. This agreement accomplishes both objectives.”
Samsung Bioepis will pay royalties to AbbVie for licensing its adalimumab patents once its biosimilar product is launched. As is the case with the prior Amgen resolution, AbbVie will not make any payments to Samsung Bioepis. “All litigation pending between the parties, as well as all litigation with Samsung Bioepis’ European partner, Biogen, will be dismissed. The precise terms of the agreements are confidential,” the Abbvie statement said.
The settlement brings to a closing a flurry of lawsuits Samsung Bioepis filed against AbbVie in 2017.
A new adalimumab biosimilar will become available in the European Union later this year, but a court settlement will keep Samsung Bioepis’ competitor off U.S. shelves until 2023.
Under the settlement, AbbVie, which manufactures adalimumab (Humira), will grant Bioepis and its partner, Biogen, a nonexclusive license to the intellectual property relating to the antibody. Bioepis’ version, dubbed SB5 (Imraldi), will enter global markets in a staggered fashion, according to an AbbVie press statement. In most countries in the European Union, the license period will begin on Oct. 16, 2018. In the United States, Samsung Bioepis’ license period will begin on June 30, 2023, according to the Abbvie statement.
Biogen and Bioepis hailed the settlement as a victory, but Imraldi won’t be the first Humira biosimilar to break into the U.S. market. Last September, AbbVie settled a similar suit with Amgen, granting patent licenses for the global use and sale of its anti–tumor necrosis factor–alpha antibody, Amgevita/Amjevita. Amgen expects to launch Amgevita in Europe on Oct. 16, 2018, and Amjevita in the United States on Jan. 31, 2023. Samsung Bioepis’ U.S. license date will not be accelerated upon Amgen’s entry.
Ian Henshaw, Biogen’s global head of biosimilars, said the deal further strengthens the company’s European biosimilars reach.
“Biogen is a leader in the emerging field of biosimilars through Samsung Bioepis, our joint venture with Samsung BioLogics,” Mr. Henshaw said in a press statement. “Biogen already markets two biosimilars in Europe and the planned introduction of Imraldi on Oct. 16 could potentially expand patient choice by offering physicians more options to meet the needs of patients while delivering significant savings to healthcare systems.”
AbbVie focused on the settlement as a global recognition of its leadership role in developing the anti-TNF-alpha antibody.
“The Samsung Bioepis settlement reflects the strength and breadth of AbbVie’s intellectual property,” Laura Schumacher, the company’s general counsel, said in the Abbvie statement. “We continue to believe biosimilars will play an important role in our healthcare system, but we also believe it is important to protect our investment in innovation. This agreement accomplishes both objectives.”
Samsung Bioepis will pay royalties to AbbVie for licensing its adalimumab patents once its biosimilar product is launched. As is the case with the prior Amgen resolution, AbbVie will not make any payments to Samsung Bioepis. “All litigation pending between the parties, as well as all litigation with Samsung Bioepis’ European partner, Biogen, will be dismissed. The precise terms of the agreements are confidential,” the Abbvie statement said.
The settlement brings to a closing a flurry of lawsuits Samsung Bioepis filed against AbbVie in 2017.
Federal budget grants $1.8 billion to Alzheimer’s and dementia research
Congress has appropriated an additional $414 million for research into Alzheimer’s disease and other dementias – the full increase requested by the National Institutes of Health for fiscal year 2018. The boost brings the total AD funding available this year to $1.8 billion.
Bolstered by the mandate of the National Plan to Address Alzheimer’s Disease, to prevent or treat AD by 2025, the NIH is aiming higher still. Its draft FY2019 AD appropriation asks for another $597 million; if passed, nearly $2.4 billion could be available for AD research as soon as next October.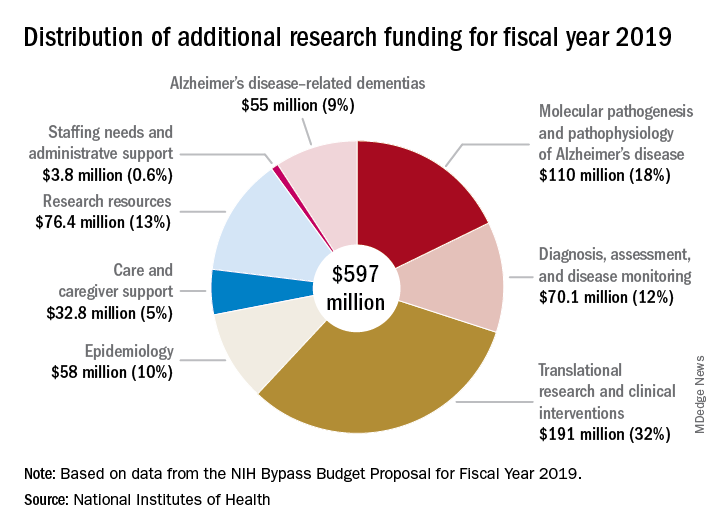
“For the third consecutive fiscal year, Congress has approved the Alzheimer’s Association’s appeal for a historic funding increase for Alzheimer’s and dementia research at the NIH,” Alzheimer’s Association president Harry Johns said in a press statement. “This decision demonstrates that Congress is deeply committed to providing the Alzheimer’s and dementia science community with the resources needed to move research forward.”
Several members of Congress championed the AD funding request, including Roy Blunt (R-Mo.), Patty Murray (D-Wash.), Nita Lowey (D-N.Y.), and Tom Cole (R-Okla.).
The forward momentum is on pace to continue into FY2019, according to Robert Egge, chief public policy officer and executive vice president of governmental affairs for the association. The NIH Bypass Budget, an estimate of how much additional funding is necessary to reach the 2025 goal, contains an additional $597 million appropriation for AD and other dementias.
“This is what we need to fund scientific projects that are meritorious and ready to go,” Mr. Egge said in an interview. “Congress has the assurance that this request is scientifically driven and that NIH is already thinking about how the money will be used.”
While a record-setting amount in the AD research world, this year’s $1.8 billion appropriation is a fraction of what other costly diseases receive. By comparison, the budget granted the National Cancer Institute $5.7 billion for its research programs.
“Compared to what the cost of the disease brings to Americans in terms of Medicare, Medicaid, and out of pocket expenses, it’s not that much,” Mr. Egge said. “But we have the opportunity to use this money to change these huge numbers that we’re facing.”

A 2013 report by the Rand Corporation found that, although Alzheimer’s affects fewer people than cancer or heart disease, the cost of treating and caring for those patients far outstrips spending in either of those other categories. The conclusions were perhaps even more startling, considering that it looked only at costs related solely to Alzheimer’s, not the cost of treating comorbid illnesses.
Long-term care was a key driver of the total cost in 2013, and remains the bulk of expenses today, Mr. Egge said. Transitions – going from home to nursing home to hospital – are terribly expensive, he noted. And although the Rand report didn’t include it, managing comorbid illnesses in Alzheimer’s is an enormous money drain. “Diabetes is just one example. It costs 80% more to manage diabetes in a patient with AD than in one without AD.”
The Facts and Figures report notes that the average 2017 per-person payout for Medicare beneficiaries was more than three times higher in AD patients than in those without the disease ($48,028 vs. $13,705). These are the kinds of numbers it takes to put partisan bickering on hold and grapple with tough decisions, Mr. Egge said.
“The fiscal argument is one thing that really impressed Congress. They do know how worried Americans are about this disease and how tough it is on families, but the growing fiscal impact has really focused them on addressing it.”
Congress has appropriated an additional $414 million for research into Alzheimer’s disease and other dementias – the full increase requested by the National Institutes of Health for fiscal year 2018. The boost brings the total AD funding available this year to $1.8 billion.
Bolstered by the mandate of the National Plan to Address Alzheimer’s Disease, to prevent or treat AD by 2025, the NIH is aiming higher still. Its draft FY2019 AD appropriation asks for another $597 million; if passed, nearly $2.4 billion could be available for AD research as soon as next October.
“For the third consecutive fiscal year, Congress has approved the Alzheimer’s Association’s appeal for a historic funding increase for Alzheimer’s and dementia research at the NIH,” Alzheimer’s Association president Harry Johns said in a press statement. “This decision demonstrates that Congress is deeply committed to providing the Alzheimer’s and dementia science community with the resources needed to move research forward.”
Several members of Congress championed the AD funding request, including Roy Blunt (R-Mo.), Patty Murray (D-Wash.), Nita Lowey (D-N.Y.), and Tom Cole (R-Okla.).
The forward momentum is on pace to continue into FY2019, according to Robert Egge, chief public policy officer and executive vice president of governmental affairs for the association. The NIH Bypass Budget, an estimate of how much additional funding is necessary to reach the 2025 goal, contains an additional $597 million appropriation for AD and other dementias.
“This is what we need to fund scientific projects that are meritorious and ready to go,” Mr. Egge said in an interview. “Congress has the assurance that this request is scientifically driven and that NIH is already thinking about how the money will be used.”
While a record-setting amount in the AD research world, this year’s $1.8 billion appropriation is a fraction of what other costly diseases receive. By comparison, the budget granted the National Cancer Institute $5.7 billion for its research programs.
“Compared to what the cost of the disease brings to Americans in terms of Medicare, Medicaid, and out of pocket expenses, it’s not that much,” Mr. Egge said. “But we have the opportunity to use this money to change these huge numbers that we’re facing.”
A 2013 report by the Rand Corporation found that, although Alzheimer’s affects fewer people than cancer or heart disease, the cost of treating and caring for those patients far outstrips spending in either of those other categories. The conclusions were perhaps even more startling, considering that it looked only at costs related solely to Alzheimer’s, not the cost of treating comorbid illnesses.
Long-term care was a key driver of the total cost in 2013, and remains the bulk of expenses today, Mr. Egge said. Transitions – going from home to nursing home to hospital – are terribly expensive, he noted. And although the Rand report didn’t include it, managing comorbid illnesses in Alzheimer’s is an enormous money drain. “Diabetes is just one example. It costs 80% more to manage diabetes in a patient with AD than in one without AD.”
The Facts and Figures report notes that the average 2017 per-person payout for Medicare beneficiaries was more than three times higher in AD patients than in those without the disease ($48,028 vs. $13,705). These are the kinds of numbers it takes to put partisan bickering on hold and grapple with tough decisions, Mr. Egge said.
“The fiscal argument is one thing that really impressed Congress. They do know how worried Americans are about this disease and how tough it is on families, but the growing fiscal impact has really focused them on addressing it.”
Congress has appropriated an additional $414 million for research into Alzheimer’s disease and other dementias – the full increase requested by the National Institutes of Health for fiscal year 2018. The boost brings the total AD funding available this year to $1.8 billion.
Bolstered by the mandate of the National Plan to Address Alzheimer’s Disease, to prevent or treat AD by 2025, the NIH is aiming higher still. Its draft FY2019 AD appropriation asks for another $597 million; if passed, nearly $2.4 billion could be available for AD research as soon as next October.
“For the third consecutive fiscal year, Congress has approved the Alzheimer’s Association’s appeal for a historic funding increase for Alzheimer’s and dementia research at the NIH,” Alzheimer’s Association president Harry Johns said in a press statement. “This decision demonstrates that Congress is deeply committed to providing the Alzheimer’s and dementia science community with the resources needed to move research forward.”
Several members of Congress championed the AD funding request, including Roy Blunt (R-Mo.), Patty Murray (D-Wash.), Nita Lowey (D-N.Y.), and Tom Cole (R-Okla.).
The forward momentum is on pace to continue into FY2019, according to Robert Egge, chief public policy officer and executive vice president of governmental affairs for the association. The NIH Bypass Budget, an estimate of how much additional funding is necessary to reach the 2025 goal, contains an additional $597 million appropriation for AD and other dementias.
“This is what we need to fund scientific projects that are meritorious and ready to go,” Mr. Egge said in an interview. “Congress has the assurance that this request is scientifically driven and that NIH is already thinking about how the money will be used.”
While a record-setting amount in the AD research world, this year’s $1.8 billion appropriation is a fraction of what other costly diseases receive. By comparison, the budget granted the National Cancer Institute $5.7 billion for its research programs.
“Compared to what the cost of the disease brings to Americans in terms of Medicare, Medicaid, and out of pocket expenses, it’s not that much,” Mr. Egge said. “But we have the opportunity to use this money to change these huge numbers that we’re facing.”
A 2013 report by the Rand Corporation found that, although Alzheimer’s affects fewer people than cancer or heart disease, the cost of treating and caring for those patients far outstrips spending in either of those other categories. The conclusions were perhaps even more startling, considering that it looked only at costs related solely to Alzheimer’s, not the cost of treating comorbid illnesses.
Long-term care was a key driver of the total cost in 2013, and remains the bulk of expenses today, Mr. Egge said. Transitions – going from home to nursing home to hospital – are terribly expensive, he noted. And although the Rand report didn’t include it, managing comorbid illnesses in Alzheimer’s is an enormous money drain. “Diabetes is just one example. It costs 80% more to manage diabetes in a patient with AD than in one without AD.”
The Facts and Figures report notes that the average 2017 per-person payout for Medicare beneficiaries was more than three times higher in AD patients than in those without the disease ($48,028 vs. $13,705). These are the kinds of numbers it takes to put partisan bickering on hold and grapple with tough decisions, Mr. Egge said.
“The fiscal argument is one thing that really impressed Congress. They do know how worried Americans are about this disease and how tough it is on families, but the growing fiscal impact has really focused them on addressing it.”
Preprint publishing challenges the status quo in medicine
Like an upstart quick-draw challenging a grizzled gunslinger, preprint servers are muscling in on the once-exclusive territory of scientific journals.
These online venues sidestep the time-honored but lengthy peer-review process in favor of instant data dissemination. By directly posting unreviewed papers, authors escape the months-long drudgery of peer review, stake an immediate claim on new ideas, and connect instantly with like-minded scientists whose feedback can mold this new idea into a sound scientific contribution.
“The caveat, of course, is that it may be crap.”
That’s the unvarnished truth of preprint publishing, said John Inglis, PhD – and he should know. As the cofounder of Cold Spring Harbor Laboratory’s bioRxiv, the largest-to-date preprint server for the biological sciences, he gives equal billing to both the lofty and the low, and lets them soar or sink by their own merit.
And many of them do soar, Dr. Inglis said. Of the more than 20,000 papers published since bioRxiv’s modest beginning in 2013, slightly more than 60% have gone on to peer-reviewed publication. The four most prolific sources of bioRxiv preprints are the research powerhouses of Stanford, Cambridge, Oxford, and Harvard. The twitterverse is virtually awash with #bioRxiv tags, which alert bioRxiv’s 18,000 followers to new papers in any of 27 subject areas. “We gave up counting 2 years ago, when we reached 100,000,” Dr. Inglis said.
BioRxiv, pronounced “bioarchive,” may be the largest preprint server for the biological sciences, but it’s not the only one. The Center for Open Science has created a preprint server search engine, which lists 25 such servers, a number of them in the life sciences.
PeerJ Preprints also offers a home for unreviewed papers, accepting “drafts of an article, abstract, or poster that has not yet been peer reviewed for formal publication.” Authors can submit a draft, incomplete, or final version, which can be online within 24 hours.
The bioRxiv model is coming to medicine, too. A new preprint server – to be called medRxiv – is expected to launch later in 2018 and will accept a wide range of papers on health and medicine, including clinical trial results.

Brand new or rebrand?
Preprint – or at least the concept of it – is nothing new, Dr. Inglis said. It’s simply the extension into the digital space of something that has been happening for many decades in the physical space.
Scientists have always written drafts of their papers and sent them out to friends and colleagues for feedback before unveiling them publicly. In the early 1990s, UC Berkeley astrophysicist Joanne Cohn began emailing unreviewed physics papers to colleagues. Within a couple of years, physicist Paul Ginsparg, PhD, of Cornell University, created a central repository for these papers at the Los Alamos National Laboratory. This repository became aRxiv, a central component of communication in the physical sciences, and the progenitor of the preprint servers now in existence.
The biological sciences were far behind this curve of open sharing, Dr. Inglis said. “I think some biologists were always aware of aRxiv and intrigued by it, but most were unconvinced that the habits and behaviors of research biologists would support a similar process.”
The competition inherent in research biology was likely a large driver of that lag. “Biological experiments are complicated, it takes a long time for ideas to evolve and results to arrive, and people are possessive of their data and ideas. They have always shared information through conferences, but there was a lot of hesitation about making this information available in an uncontrolled way, beyond the audiences at those meetings,” he said.
[polldaddy:9970002]
Nature Publishing Group first floated the preprint notion among biologists in 2006, with Nature Precedings. It published more than 2,000 papers before folding, rather suddenly, in 2012. A publisher’s statement simply said that the effort was “unsustainable as originally conceived.”
Commentators suspected the model was a financial bust, and indeed, preprint servers aren’t money machines. BioRxiv, proudly not for profit, was founded with financial support from Cold Spring Harbor Laboratory and survives largely on private grants. In April 2017, it received a grant for an undisclosed amount from the Chan Zuckerberg Initiative, established by Facebook founder Mark Zuckerberg and his wife, Priscilla Chan.
Who’s minding the data?
The screening process at bioRxiv is minimal, Dr. Inglis said. An in-house staff checks each paper for obvious flaws, like plagiarism, irrelevance, unacceptable article type, and offensive language. Then they’re sent out to a committee of affiliate scientists, which confirms that the manuscript is a research paper and that it contains science, without judging the quality of that science. Papers aren’t edited before being posted online.
Each bioRxiv paper gets a DOI link, and appears with the following disclaimer detailing the risks inherent in reading “unrefereed” science: “Because [peer review] can be lengthy, authors use the bioRxiv service to make their manuscripts available as ‘preprints’ before peer review, allowing other scientists to see, discuss, and comment on the findings immediately. Readers should therefore be aware that articles on bioRxiv have not been finalized by authors, might contain errors, and report information that has not yet been accepted or endorsed in any way by the scientific or medical community.”
From biology to medicine
The bioRxiv team is poised to jump into a different pool now – medical science. Although the launch date isn’t firm yet, medRxiv will go live sometime very soon, Dr. Inglis said. It’s a proposed partnership between Cold Spring Harbor Laboratory, the Yale-based YODA Project (Yale University Open Data Access Project), and BMJ. The medRxiv papers, like those posted to bioRxiv, will be screened but not peer reviewed or scrutinized for trial design, methodology, or interpretation of results.
The benefits of medRxiv will be more rapid communication of research results, increased opportunities for collaboration, the sharing of hard-to-publish outputs like quality innovations in health care, and greater transparency of clinical trials data, Dr. Inglis said. Despite this, he expects the same kind of push-back bioRxiv initially encountered, at least in the beginning.
“I expect we will be turning the clock back 5 years and find a lot of people who think this is potentially a bad thing, a risk that poor information or misinformation is going to be disseminated to a wider audience, which is exactly what we heard about bioRxiv,” he said. “But we hope that when medRxiv launches, it will demonstrate the same kind of gradual acceptance as people get more and more familiar with the preprint platform.”
The founders intend to build into the server policies to mitigate the risk from medically relevant information that hasn’t been peer reviewed, such as not accepting case studies or editorials and opinion pieces, he added.
While many find the preprint disclaimer acceptable on papers that have no immediate clinical impact, there is concern about applying it to papers that discuss patient treatment.
Howard Bauchner, MD, JAMA’s editor in chief, addressed it in an editorial published in September 2017. Although not explicitly directed at bioRxiv, Dr. Bauchner took a firm stance against shortcutting the evaluation of evidence that is often years in the making.
“New interest in preprint servers in clinical medicine increases the likelihood of premature dissemination and public consumption of clinical research findings prior to rigorous evaluation and peer review,” Dr. Bauchner wrote. “For most articles, public consumption of research findings prior to peer review will have little influence on health, but for some articles, the effect could be devastating for some patients if the results made public prior to peer review are wrong or incorrectly interpreted.”
Dr. Bauchner did not overstate the potential influence of unvetted science, as a January 2018 bioRxiv study on CRISPR gene editing clearly demonstrated. The paper by Carsten Charlesworth, a doctoral student at Stanford (Calif.) University, found that up to 79% of humans could already be immune to Crispr-Cas9, the gene-editing protein derived from Staphylococcus aureus and S. pyogenes. More than science geeks were reading: The report initially sent CRISPR stocks tumbling.
Aaron D. Viny, MD, is in general a hesitant fan of bioRxiv’s preprint platform. But he raised an eyebrow when he learned about medRxiv.
“The only pressure that I can see in regulating these reports is social media,” said Dr. Viny, a hematologic oncologist at Memorial Sloan Kettering, in New York. “The fear is that it will be misused in two different realms. The most dangerous and worrisome, of course, is for patients using the data to influence their care plan, when the data haven’t been vetted appropriately. But secondarily, how could it influence the economics of clinical trials? There is no shortage of hedge fund managers in biotech. These data could misinform a consultant who might know the area in a way that artificially exploits early research data. Could that permit someone to submit disingenuous data to manipulate the stock of a given pharmaceutical company? I don’t know how you police that kind of thing.”
Who’s loving it – and why?
There are plenty of reasons to support a thriving preprint community, said Jessica Polka, PhD, director of ASAPbio, (Accelerating Science and Publication in biology), a group that bills itself as a scientist-driven initiative to promote the productive use of preprints in the life sciences.
“Preprinting complements traditional journal publishing by allowing researchers to rapidly communicate their findings to the scientific community,” she said. “This, in turn, provides them with opportunities for earlier and broader feedback and a way to transparently demonstrate progress on a project. More importantly, the whole community benefits by having earlier access to research findings, which can accelerate the pace of discovery.”

The disclosures applied to every preprint paper are the publisher’s way of assuring this same awareness, she said. And preprints do need to be approached with some skepticism, as should peer-reviewed literature.
“The veracity of published papers is not always a given. An example is the 1998 vaccine paper [published in the Lancet] by Dr. Andrew Wakefield,” which launched the antivaccine movement. “But the answer to problems of reliability is to provide more information about the research and how it has been verified and evaluated, not less information. For example, confirmation bias can make it difficult to refute work that has been published. The current incentives for publishing negative results in a journal are not strong enough to reveal all of the information that could be useful to other researchers, but preprinting reduces the barrier to sharing negative results,” she said.
Swimming up the (main)stream
Universal peer-reviewed acceptance of preprints isn’t a done deal, Dr. Polka said. Journals are tussling with how to handle these papers. The Lancet clearly states that preprints don’t constitute prior publication and are welcome. The New England Journal of Medicine offers an uncontestable “no way.”
JAMA discourages submitting preprints, and will consider one only if the submitted version offers “meaningful new information” above what the preprint disseminated.
Cell Press has a slightly different take. They will consider papers previously posted on preprint services, but the policy applies only to the original submitted version of the paper. “We do not support posting of revisions that respond to editorial input and peer review or the final published version to preprint servers,” the policy notes.
In an interview, Deborah Sweet, PhD, the group’s vice president of editorial, elaborated on the policy. “In our view, one of the most important purposes of preprint posting is to gather feedback from the scientific community before a formal submission to a journal,” she said. “The ‘original submission’ term in our guidelines refers to the first version of the paper submitted to [Cell Press], which could include revisions made in response to community feedback on a preprint. After formal submission, we think it is most appropriate to incorporate and represent the value of the editorial and peer-review evaluation process in the final published journal article so that is clearly identifiable as the version of record.”
bioRxiv has made substantial inroads with dozens of other peer-reviewed journals. More than 100 – including a number of publications by EMBO Press and PLOS (Public Library of Science) – participate in bioRxiv’s B2J (BioRxiv-to-journal) direct-submission program.
With a few clicks, authors can transmit their bioRxiv manuscript files directly to these journals, without having to prepare separate submissions, Dr. Sweet said. Last year, Cell Press added two publications – Cell Reports and Structure – to the B2J program. “Once the paper is sent, it moves behind the scenes to the journal system and reappears as a formal submission,” she said. “In our process, before transferring the paper to the journal editors, authors have a chance to update the files (for example, to add a cover letter) and answer the standard questions that we ask, including ones about reviewer suggestions and exclusion requests. Once that step is done, the paper is handed over to the editorial team, and it’s ready to go for consideration in the same way as any other submission.”
Who’s reading?
Regardless of whether peer-review journals grant them legitimacy, preprints are getting a lot of views. A recent research letter, published in JAMA, looked at readership and online attention in 7,750 preprints posted from November 2013 to January 2017.
Primary author Stylianos Serghiou then selected 776 papers that had first appeared in bioRxiv, and matched them with 3,647 peer-reviewed articles lacking preprint exposure. He examined several publishing metrics for the papers, including views and downloads, citations in other sources, and Altmetric scores.
Altmetric tracks digital attention to scientific papers: Wikipedia citations, mentions in policy documents, blog discussions, and social media mentions including Facebook, Reddit, and Twitter. An Altmetric “attention score” of more than 20 corresponds to articles in the top 5% of readership, he said in an interview.
“Almost one in five of the bioRxiv preprints were getting these very high Almetric scores – much higher scores than articles that had no preprint posting,” Mr. Serghiou said in an interview.
Other findings include:
- The median number of preprint abstract views was 924, and the median number of PDF downloads was 321.
- In total, 18% of the preprints achieved an Altmetric score of more than 20.
- Of 7,750 preprints, 55% were accepted in a peer-reviewed publication within 24 months.
- Altmetric scores were significantly higher in articles in preprints (median 9.5 vs. 3.5).
The differences are probably related, at least in part, to the digital media savvy of preprint authors, Mr. Serghiou suggested. “We speculate that people who publish in bioRxiv may be more familiar with social media methods of making others aware of their work. They tend to be very good at using platforms like Twitter and Facebook to promote their results.”
Despite the high exposure scores, only 10% of bioRxiv articles get any posted comments or feedback – a key raison d’être for using a preprint service.
“Ten percent doesn’t sound like a very robust [feedback], but most journal articles get no comments whatsoever,” Dr. Inglis said. “And if they do, especially on the weekly magazines of science, comments may be from someone who has an ax to grind, or who doesn’t know much about the subject.”
What isn’t measured, in either volume or import, is the private communication a preprint engenders, Dr. Inglis said. “Feedback comes directly and privately to the author through email or at meetings or on the phone. We hear time and again that authors get hundreds of downloads after posting, and receive numerous contacts from colleagues who want to know more, to point out weaknesses, or request collaborations. These are the advantages we see from this potentially anxiety-provoking process of putting a manuscript out that has not been approved for publication. The entire purpose is to accelerate the speed of research by accelerating the speed of communication.”
Dr. Inglis, Dr. Sweet, and Dr. Polka are all employees of their respective companies. Dr. Viny and Mr. Serghiou both reported having no financial disclosures relevant to this article.
It’s another beautiful day on the upper east side of Manhattan. The sun shines through the window shades, my 2-year-old daughter sings to herself as she wakes up, my wife has just returned from an early-morning workout – all is right as rain.
My phone buzzes. My stomach clenches. It buzzes again. My Twitter alerts are here. I dread this part of my morning ritual – finding out if I’ve been scooped overnight by the massive inflow of scientific manuscripts reported to me by my army of scientific literature–searching Twitter bots.

But this massive data dump now has a #fakenews problem. It’s not Russian election meddling, it’s open source “preprint” publications. Nearly half of my morning list of Twitter alerts now are sourced from the latest uploads to bioRxiv. BioRxiv is an online site run by scientists at Cold Spring Harbor Laboratory and is composed of posting manuscripts without undergoing a peer-review process. Now, most commonly, these manuscripts are concurrently under review in the bona fide peer-review process elsewhere, but unrevised, they are uploaded directly for public consumption.
There was one recent tweet that highlighted some interesting logistical considerations for bioRxiv manuscripts in the peer-review process. The tweet from an unnamed laboratory complains that a peer reviewer is displeased with the authors citing their own bioRxiv paper, while the tweeter contends that all referenced information, online or otherwise, must be cited. Moreover, the reviewer brings up an accusation of self-plagiarism as the submitted manuscript is identical to the one on bioRxiv. While the latter just seems like a misunderstanding of the bioRxiv platform, the former is a really interesting question of whether bioRxiv represents data that can/should be referenced.
Proponents of the platform are excited that data is accessible sooner, that one’s latest and greatest scientific finding can be “scoop proof” by getting it online and marking one’s territory. Naysayers contend that, without peer review, the work cannot truly be part of the scientific literature and should be taken with great caution.
There is undoubtedly danger. Online media sources Gizmodo and the Motley Fool both reported that a January 2018 bioRxiv preprint resulted in a nearly 20% drop in stock prices of CRISPR biotechnology firms Editas Medicine and Intellia Therapeutics. The manuscript warned of the potential immunogenicity of CRISPR, suggesting that preexisting antibodies might limit its clinical application. Far more cynically, this highlights how a stock price could theoretically be artificially manipulated through preprint data.
The preprint is an open market response to the long, arduous process that peer review has become, but undoubtedly, peer review is an essential part of how we maintain transparency and accountability in science and medicine. It remains to be seen exactly how journal editors intend to use bioRxiv submissions in the appraisal of “novelty.”
How will the scientific community vet and referee the works, and will the title and conclusions of a scientifically flawed work permeate misleading information into the field and lay public? Would you let it influence your research or clinical practice? We will be finding out one tweet at a time.
Aaron D. Viny, MD, is with the Memorial Sloan Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. He reported having no relevant financial disclosures. Contact him on Twitter @TheDoctorIsVin.
It’s another beautiful day on the upper east side of Manhattan. The sun shines through the window shades, my 2-year-old daughter sings to herself as she wakes up, my wife has just returned from an early-morning workout – all is right as rain.
My phone buzzes. My stomach clenches. It buzzes again. My Twitter alerts are here. I dread this part of my morning ritual – finding out if I’ve been scooped overnight by the massive inflow of scientific manuscripts reported to me by my army of scientific literature–searching Twitter bots.
But this massive data dump now has a #fakenews problem. It’s not Russian election meddling, it’s open source “preprint” publications. Nearly half of my morning list of Twitter alerts now are sourced from the latest uploads to bioRxiv. BioRxiv is an online site run by scientists at Cold Spring Harbor Laboratory and is composed of posting manuscripts without undergoing a peer-review process. Now, most commonly, these manuscripts are concurrently under review in the bona fide peer-review process elsewhere, but unrevised, they are uploaded directly for public consumption.
There was one recent tweet that highlighted some interesting logistical considerations for bioRxiv manuscripts in the peer-review process. The tweet from an unnamed laboratory complains that a peer reviewer is displeased with the authors citing their own bioRxiv paper, while the tweeter contends that all referenced information, online or otherwise, must be cited. Moreover, the reviewer brings up an accusation of self-plagiarism as the submitted manuscript is identical to the one on bioRxiv. While the latter just seems like a misunderstanding of the bioRxiv platform, the former is a really interesting question of whether bioRxiv represents data that can/should be referenced.
Proponents of the platform are excited that data is accessible sooner, that one’s latest and greatest scientific finding can be “scoop proof” by getting it online and marking one’s territory. Naysayers contend that, without peer review, the work cannot truly be part of the scientific literature and should be taken with great caution.
There is undoubtedly danger. Online media sources Gizmodo and the Motley Fool both reported that a January 2018 bioRxiv preprint resulted in a nearly 20% drop in stock prices of CRISPR biotechnology firms Editas Medicine and Intellia Therapeutics. The manuscript warned of the potential immunogenicity of CRISPR, suggesting that preexisting antibodies might limit its clinical application. Far more cynically, this highlights how a stock price could theoretically be artificially manipulated through preprint data.
The preprint is an open market response to the long, arduous process that peer review has become, but undoubtedly, peer review is an essential part of how we maintain transparency and accountability in science and medicine. It remains to be seen exactly how journal editors intend to use bioRxiv submissions in the appraisal of “novelty.”
How will the scientific community vet and referee the works, and will the title and conclusions of a scientifically flawed work permeate misleading information into the field and lay public? Would you let it influence your research or clinical practice? We will be finding out one tweet at a time.
Aaron D. Viny, MD, is with the Memorial Sloan Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. He reported having no relevant financial disclosures. Contact him on Twitter @TheDoctorIsVin.
It’s another beautiful day on the upper east side of Manhattan. The sun shines through the window shades, my 2-year-old daughter sings to herself as she wakes up, my wife has just returned from an early-morning workout – all is right as rain.
My phone buzzes. My stomach clenches. It buzzes again. My Twitter alerts are here. I dread this part of my morning ritual – finding out if I’ve been scooped overnight by the massive inflow of scientific manuscripts reported to me by my army of scientific literature–searching Twitter bots.
But this massive data dump now has a #fakenews problem. It’s not Russian election meddling, it’s open source “preprint” publications. Nearly half of my morning list of Twitter alerts now are sourced from the latest uploads to bioRxiv. BioRxiv is an online site run by scientists at Cold Spring Harbor Laboratory and is composed of posting manuscripts without undergoing a peer-review process. Now, most commonly, these manuscripts are concurrently under review in the bona fide peer-review process elsewhere, but unrevised, they are uploaded directly for public consumption.
There was one recent tweet that highlighted some interesting logistical considerations for bioRxiv manuscripts in the peer-review process. The tweet from an unnamed laboratory complains that a peer reviewer is displeased with the authors citing their own bioRxiv paper, while the tweeter contends that all referenced information, online or otherwise, must be cited. Moreover, the reviewer brings up an accusation of self-plagiarism as the submitted manuscript is identical to the one on bioRxiv. While the latter just seems like a misunderstanding of the bioRxiv platform, the former is a really interesting question of whether bioRxiv represents data that can/should be referenced.
Proponents of the platform are excited that data is accessible sooner, that one’s latest and greatest scientific finding can be “scoop proof” by getting it online and marking one’s territory. Naysayers contend that, without peer review, the work cannot truly be part of the scientific literature and should be taken with great caution.
There is undoubtedly danger. Online media sources Gizmodo and the Motley Fool both reported that a January 2018 bioRxiv preprint resulted in a nearly 20% drop in stock prices of CRISPR biotechnology firms Editas Medicine and Intellia Therapeutics. The manuscript warned of the potential immunogenicity of CRISPR, suggesting that preexisting antibodies might limit its clinical application. Far more cynically, this highlights how a stock price could theoretically be artificially manipulated through preprint data.
The preprint is an open market response to the long, arduous process that peer review has become, but undoubtedly, peer review is an essential part of how we maintain transparency and accountability in science and medicine. It remains to be seen exactly how journal editors intend to use bioRxiv submissions in the appraisal of “novelty.”
How will the scientific community vet and referee the works, and will the title and conclusions of a scientifically flawed work permeate misleading information into the field and lay public? Would you let it influence your research or clinical practice? We will be finding out one tweet at a time.
Aaron D. Viny, MD, is with the Memorial Sloan Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. He reported having no relevant financial disclosures. Contact him on Twitter @TheDoctorIsVin.
Like an upstart quick-draw challenging a grizzled gunslinger, preprint servers are muscling in on the once-exclusive territory of scientific journals.
These online venues sidestep the time-honored but lengthy peer-review process in favor of instant data dissemination. By directly posting unreviewed papers, authors escape the months-long drudgery of peer review, stake an immediate claim on new ideas, and connect instantly with like-minded scientists whose feedback can mold this new idea into a sound scientific contribution.
“The caveat, of course, is that it may be crap.”
That’s the unvarnished truth of preprint publishing, said John Inglis, PhD – and he should know. As the cofounder of Cold Spring Harbor Laboratory’s bioRxiv, the largest-to-date preprint server for the biological sciences, he gives equal billing to both the lofty and the low, and lets them soar or sink by their own merit.
And many of them do soar, Dr. Inglis said. Of the more than 20,000 papers published since bioRxiv’s modest beginning in 2013, slightly more than 60% have gone on to peer-reviewed publication. The four most prolific sources of bioRxiv preprints are the research powerhouses of Stanford, Cambridge, Oxford, and Harvard. The twitterverse is virtually awash with #bioRxiv tags, which alert bioRxiv’s 18,000 followers to new papers in any of 27 subject areas. “We gave up counting 2 years ago, when we reached 100,000,” Dr. Inglis said.
BioRxiv, pronounced “bioarchive,” may be the largest preprint server for the biological sciences, but it’s not the only one. The Center for Open Science has created a preprint server search engine, which lists 25 such servers, a number of them in the life sciences.
PeerJ Preprints also offers a home for unreviewed papers, accepting “drafts of an article, abstract, or poster that has not yet been peer reviewed for formal publication.” Authors can submit a draft, incomplete, or final version, which can be online within 24 hours.
The bioRxiv model is coming to medicine, too. A new preprint server – to be called medRxiv – is expected to launch later in 2018 and will accept a wide range of papers on health and medicine, including clinical trial results.
Brand new or rebrand?
Preprint – or at least the concept of it – is nothing new, Dr. Inglis said. It’s simply the extension into the digital space of something that has been happening for many decades in the physical space.
Scientists have always written drafts of their papers and sent them out to friends and colleagues for feedback before unveiling them publicly. In the early 1990s, UC Berkeley astrophysicist Joanne Cohn began emailing unreviewed physics papers to colleagues. Within a couple of years, physicist Paul Ginsparg, PhD, of Cornell University, created a central repository for these papers at the Los Alamos National Laboratory. This repository became aRxiv, a central component of communication in the physical sciences, and the progenitor of the preprint servers now in existence.
The biological sciences were far behind this curve of open sharing, Dr. Inglis said. “I think some biologists were always aware of aRxiv and intrigued by it, but most were unconvinced that the habits and behaviors of research biologists would support a similar process.”
The competition inherent in research biology was likely a large driver of that lag. “Biological experiments are complicated, it takes a long time for ideas to evolve and results to arrive, and people are possessive of their data and ideas. They have always shared information through conferences, but there was a lot of hesitation about making this information available in an uncontrolled way, beyond the audiences at those meetings,” he said.
[polldaddy:9970002]
Nature Publishing Group first floated the preprint notion among biologists in 2006, with Nature Precedings. It published more than 2,000 papers before folding, rather suddenly, in 2012. A publisher’s statement simply said that the effort was “unsustainable as originally conceived.”
Commentators suspected the model was a financial bust, and indeed, preprint servers aren’t money machines. BioRxiv, proudly not for profit, was founded with financial support from Cold Spring Harbor Laboratory and survives largely on private grants. In April 2017, it received a grant for an undisclosed amount from the Chan Zuckerberg Initiative, established by Facebook founder Mark Zuckerberg and his wife, Priscilla Chan.
Who’s minding the data?
The screening process at bioRxiv is minimal, Dr. Inglis said. An in-house staff checks each paper for obvious flaws, like plagiarism, irrelevance, unacceptable article type, and offensive language. Then they’re sent out to a committee of affiliate scientists, which confirms that the manuscript is a research paper and that it contains science, without judging the quality of that science. Papers aren’t edited before being posted online.
Each bioRxiv paper gets a DOI link, and appears with the following disclaimer detailing the risks inherent in reading “unrefereed” science: “Because [peer review] can be lengthy, authors use the bioRxiv service to make their manuscripts available as ‘preprints’ before peer review, allowing other scientists to see, discuss, and comment on the findings immediately. Readers should therefore be aware that articles on bioRxiv have not been finalized by authors, might contain errors, and report information that has not yet been accepted or endorsed in any way by the scientific or medical community.”
From biology to medicine
The bioRxiv team is poised to jump into a different pool now – medical science. Although the launch date isn’t firm yet, medRxiv will go live sometime very soon, Dr. Inglis said. It’s a proposed partnership between Cold Spring Harbor Laboratory, the Yale-based YODA Project (Yale University Open Data Access Project), and BMJ. The medRxiv papers, like those posted to bioRxiv, will be screened but not peer reviewed or scrutinized for trial design, methodology, or interpretation of results.
The benefits of medRxiv will be more rapid communication of research results, increased opportunities for collaboration, the sharing of hard-to-publish outputs like quality innovations in health care, and greater transparency of clinical trials data, Dr. Inglis said. Despite this, he expects the same kind of push-back bioRxiv initially encountered, at least in the beginning.
“I expect we will be turning the clock back 5 years and find a lot of people who think this is potentially a bad thing, a risk that poor information or misinformation is going to be disseminated to a wider audience, which is exactly what we heard about bioRxiv,” he said. “But we hope that when medRxiv launches, it will demonstrate the same kind of gradual acceptance as people get more and more familiar with the preprint platform.”
The founders intend to build into the server policies to mitigate the risk from medically relevant information that hasn’t been peer reviewed, such as not accepting case studies or editorials and opinion pieces, he added.
While many find the preprint disclaimer acceptable on papers that have no immediate clinical impact, there is concern about applying it to papers that discuss patient treatment.
Howard Bauchner, MD, JAMA’s editor in chief, addressed it in an editorial published in September 2017. Although not explicitly directed at bioRxiv, Dr. Bauchner took a firm stance against shortcutting the evaluation of evidence that is often years in the making.
“New interest in preprint servers in clinical medicine increases the likelihood of premature dissemination and public consumption of clinical research findings prior to rigorous evaluation and peer review,” Dr. Bauchner wrote. “For most articles, public consumption of research findings prior to peer review will have little influence on health, but for some articles, the effect could be devastating for some patients if the results made public prior to peer review are wrong or incorrectly interpreted.”
Dr. Bauchner did not overstate the potential influence of unvetted science, as a January 2018 bioRxiv study on CRISPR gene editing clearly demonstrated. The paper by Carsten Charlesworth, a doctoral student at Stanford (Calif.) University, found that up to 79% of humans could already be immune to Crispr-Cas9, the gene-editing protein derived from Staphylococcus aureus and S. pyogenes. More than science geeks were reading: The report initially sent CRISPR stocks tumbling.
Aaron D. Viny, MD, is in general a hesitant fan of bioRxiv’s preprint platform. But he raised an eyebrow when he learned about medRxiv.
“The only pressure that I can see in regulating these reports is social media,” said Dr. Viny, a hematologic oncologist at Memorial Sloan Kettering, in New York. “The fear is that it will be misused in two different realms. The most dangerous and worrisome, of course, is for patients using the data to influence their care plan, when the data haven’t been vetted appropriately. But secondarily, how could it influence the economics of clinical trials? There is no shortage of hedge fund managers in biotech. These data could misinform a consultant who might know the area in a way that artificially exploits early research data. Could that permit someone to submit disingenuous data to manipulate the stock of a given pharmaceutical company? I don’t know how you police that kind of thing.”
Who’s loving it – and why?
There are plenty of reasons to support a thriving preprint community, said Jessica Polka, PhD, director of ASAPbio, (Accelerating Science and Publication in biology), a group that bills itself as a scientist-driven initiative to promote the productive use of preprints in the life sciences.
“Preprinting complements traditional journal publishing by allowing researchers to rapidly communicate their findings to the scientific community,” she said. “This, in turn, provides them with opportunities for earlier and broader feedback and a way to transparently demonstrate progress on a project. More importantly, the whole community benefits by having earlier access to research findings, which can accelerate the pace of discovery.”
The disclosures applied to every preprint paper are the publisher’s way of assuring this same awareness, she said. And preprints do need to be approached with some skepticism, as should peer-reviewed literature.
“The veracity of published papers is not always a given. An example is the 1998 vaccine paper [published in the Lancet] by Dr. Andrew Wakefield,” which launched the antivaccine movement. “But the answer to problems of reliability is to provide more information about the research and how it has been verified and evaluated, not less information. For example, confirmation bias can make it difficult to refute work that has been published. The current incentives for publishing negative results in a journal are not strong enough to reveal all of the information that could be useful to other researchers, but preprinting reduces the barrier to sharing negative results,” she said.
Swimming up the (main)stream
Universal peer-reviewed acceptance of preprints isn’t a done deal, Dr. Polka said. Journals are tussling with how to handle these papers. The Lancet clearly states that preprints don’t constitute prior publication and are welcome. The New England Journal of Medicine offers an uncontestable “no way.”
JAMA discourages submitting preprints, and will consider one only if the submitted version offers “meaningful new information” above what the preprint disseminated.
Cell Press has a slightly different take. They will consider papers previously posted on preprint services, but the policy applies only to the original submitted version of the paper. “We do not support posting of revisions that respond to editorial input and peer review or the final published version to preprint servers,” the policy notes.
In an interview, Deborah Sweet, PhD, the group’s vice president of editorial, elaborated on the policy. “In our view, one of the most important purposes of preprint posting is to gather feedback from the scientific community before a formal submission to a journal,” she said. “The ‘original submission’ term in our guidelines refers to the first version of the paper submitted to [Cell Press], which could include revisions made in response to community feedback on a preprint. After formal submission, we think it is most appropriate to incorporate and represent the value of the editorial and peer-review evaluation process in the final published journal article so that is clearly identifiable as the version of record.”
bioRxiv has made substantial inroads with dozens of other peer-reviewed journals. More than 100 – including a number of publications by EMBO Press and PLOS (Public Library of Science) – participate in bioRxiv’s B2J (BioRxiv-to-journal) direct-submission program.
With a few clicks, authors can transmit their bioRxiv manuscript files directly to these journals, without having to prepare separate submissions, Dr. Sweet said. Last year, Cell Press added two publications – Cell Reports and Structure – to the B2J program. “Once the paper is sent, it moves behind the scenes to the journal system and reappears as a formal submission,” she said. “In our process, before transferring the paper to the journal editors, authors have a chance to update the files (for example, to add a cover letter) and answer the standard questions that we ask, including ones about reviewer suggestions and exclusion requests. Once that step is done, the paper is handed over to the editorial team, and it’s ready to go for consideration in the same way as any other submission.”
Who’s reading?
Regardless of whether peer-review journals grant them legitimacy, preprints are getting a lot of views. A recent research letter, published in JAMA, looked at readership and online attention in 7,750 preprints posted from November 2013 to January 2017.
Primary author Stylianos Serghiou then selected 776 papers that had first appeared in bioRxiv, and matched them with 3,647 peer-reviewed articles lacking preprint exposure. He examined several publishing metrics for the papers, including views and downloads, citations in other sources, and Altmetric scores.
Altmetric tracks digital attention to scientific papers: Wikipedia citations, mentions in policy documents, blog discussions, and social media mentions including Facebook, Reddit, and Twitter. An Altmetric “attention score” of more than 20 corresponds to articles in the top 5% of readership, he said in an interview.
“Almost one in five of the bioRxiv preprints were getting these very high Almetric scores – much higher scores than articles that had no preprint posting,” Mr. Serghiou said in an interview.
Other findings include:
- The median number of preprint abstract views was 924, and the median number of PDF downloads was 321.
- In total, 18% of the preprints achieved an Altmetric score of more than 20.
- Of 7,750 preprints, 55% were accepted in a peer-reviewed publication within 24 months.
- Altmetric scores were significantly higher in articles in preprints (median 9.5 vs. 3.5).
The differences are probably related, at least in part, to the digital media savvy of preprint authors, Mr. Serghiou suggested. “We speculate that people who publish in bioRxiv may be more familiar with social media methods of making others aware of their work. They tend to be very good at using platforms like Twitter and Facebook to promote their results.”
Despite the high exposure scores, only 10% of bioRxiv articles get any posted comments or feedback – a key raison d’être for using a preprint service.
“Ten percent doesn’t sound like a very robust [feedback], but most journal articles get no comments whatsoever,” Dr. Inglis said. “And if they do, especially on the weekly magazines of science, comments may be from someone who has an ax to grind, or who doesn’t know much about the subject.”
What isn’t measured, in either volume or import, is the private communication a preprint engenders, Dr. Inglis said. “Feedback comes directly and privately to the author through email or at meetings or on the phone. We hear time and again that authors get hundreds of downloads after posting, and receive numerous contacts from colleagues who want to know more, to point out weaknesses, or request collaborations. These are the advantages we see from this potentially anxiety-provoking process of putting a manuscript out that has not been approved for publication. The entire purpose is to accelerate the speed of research by accelerating the speed of communication.”
Dr. Inglis, Dr. Sweet, and Dr. Polka are all employees of their respective companies. Dr. Viny and Mr. Serghiou both reported having no financial disclosures relevant to this article.
Like an upstart quick-draw challenging a grizzled gunslinger, preprint servers are muscling in on the once-exclusive territory of scientific journals.
These online venues sidestep the time-honored but lengthy peer-review process in favor of instant data dissemination. By directly posting unreviewed papers, authors escape the months-long drudgery of peer review, stake an immediate claim on new ideas, and connect instantly with like-minded scientists whose feedback can mold this new idea into a sound scientific contribution.
“The caveat, of course, is that it may be crap.”
That’s the unvarnished truth of preprint publishing, said John Inglis, PhD – and he should know. As the cofounder of Cold Spring Harbor Laboratory’s bioRxiv, the largest-to-date preprint server for the biological sciences, he gives equal billing to both the lofty and the low, and lets them soar or sink by their own merit.
And many of them do soar, Dr. Inglis said. Of the more than 20,000 papers published since bioRxiv’s modest beginning in 2013, slightly more than 60% have gone on to peer-reviewed publication. The four most prolific sources of bioRxiv preprints are the research powerhouses of Stanford, Cambridge, Oxford, and Harvard. The twitterverse is virtually awash with #bioRxiv tags, which alert bioRxiv’s 18,000 followers to new papers in any of 27 subject areas. “We gave up counting 2 years ago, when we reached 100,000,” Dr. Inglis said.
BioRxiv, pronounced “bioarchive,” may be the largest preprint server for the biological sciences, but it’s not the only one. The Center for Open Science has created a preprint server search engine, which lists 25 such servers, a number of them in the life sciences.
PeerJ Preprints also offers a home for unreviewed papers, accepting “drafts of an article, abstract, or poster that has not yet been peer reviewed for formal publication.” Authors can submit a draft, incomplete, or final version, which can be online within 24 hours.
The bioRxiv model is coming to medicine, too. A new preprint server – to be called medRxiv – is expected to launch later in 2018 and will accept a wide range of papers on health and medicine, including clinical trial results.
Brand new or rebrand?
Preprint – or at least the concept of it – is nothing new, Dr. Inglis said. It’s simply the extension into the digital space of something that has been happening for many decades in the physical space.
Scientists have always written drafts of their papers and sent them out to friends and colleagues for feedback before unveiling them publicly. In the early 1990s, UC Berkeley astrophysicist Joanne Cohn began emailing unreviewed physics papers to colleagues. Within a couple of years, physicist Paul Ginsparg, PhD, of Cornell University, created a central repository for these papers at the Los Alamos National Laboratory. This repository became aRxiv, a central component of communication in the physical sciences, and the progenitor of the preprint servers now in existence.
The biological sciences were far behind this curve of open sharing, Dr. Inglis said. “I think some biologists were always aware of aRxiv and intrigued by it, but most were unconvinced that the habits and behaviors of research biologists would support a similar process.”
The competition inherent in research biology was likely a large driver of that lag. “Biological experiments are complicated, it takes a long time for ideas to evolve and results to arrive, and people are possessive of their data and ideas. They have always shared information through conferences, but there was a lot of hesitation about making this information available in an uncontrolled way, beyond the audiences at those meetings,” he said.
[polldaddy:9970002]
Nature Publishing Group first floated the preprint notion among biologists in 2006, with Nature Precedings. It published more than 2,000 papers before folding, rather suddenly, in 2012. A publisher’s statement simply said that the effort was “unsustainable as originally conceived.”
Commentators suspected the model was a financial bust, and indeed, preprint servers aren’t money machines. BioRxiv, proudly not for profit, was founded with financial support from Cold Spring Harbor Laboratory and survives largely on private grants. In April 2017, it received a grant for an undisclosed amount from the Chan Zuckerberg Initiative, established by Facebook founder Mark Zuckerberg and his wife, Priscilla Chan.
Who’s minding the data?
The screening process at bioRxiv is minimal, Dr. Inglis said. An in-house staff checks each paper for obvious flaws, like plagiarism, irrelevance, unacceptable article type, and offensive language. Then they’re sent out to a committee of affiliate scientists, which confirms that the manuscript is a research paper and that it contains science, without judging the quality of that science. Papers aren’t edited before being posted online.
Each bioRxiv paper gets a DOI link, and appears with the following disclaimer detailing the risks inherent in reading “unrefereed” science: “Because [peer review] can be lengthy, authors use the bioRxiv service to make their manuscripts available as ‘preprints’ before peer review, allowing other scientists to see, discuss, and comment on the findings immediately. Readers should therefore be aware that articles on bioRxiv have not been finalized by authors, might contain errors, and report information that has not yet been accepted or endorsed in any way by the scientific or medical community.”
From biology to medicine
The bioRxiv team is poised to jump into a different pool now – medical science. Although the launch date isn’t firm yet, medRxiv will go live sometime very soon, Dr. Inglis said. It’s a proposed partnership between Cold Spring Harbor Laboratory, the Yale-based YODA Project (Yale University Open Data Access Project), and BMJ. The medRxiv papers, like those posted to bioRxiv, will be screened but not peer reviewed or scrutinized for trial design, methodology, or interpretation of results.
The benefits of medRxiv will be more rapid communication of research results, increased opportunities for collaboration, the sharing of hard-to-publish outputs like quality innovations in health care, and greater transparency of clinical trials data, Dr. Inglis said. Despite this, he expects the same kind of push-back bioRxiv initially encountered, at least in the beginning.
“I expect we will be turning the clock back 5 years and find a lot of people who think this is potentially a bad thing, a risk that poor information or misinformation is going to be disseminated to a wider audience, which is exactly what we heard about bioRxiv,” he said. “But we hope that when medRxiv launches, it will demonstrate the same kind of gradual acceptance as people get more and more familiar with the preprint platform.”
The founders intend to build into the server policies to mitigate the risk from medically relevant information that hasn’t been peer reviewed, such as not accepting case studies or editorials and opinion pieces, he added.
While many find the preprint disclaimer acceptable on papers that have no immediate clinical impact, there is concern about applying it to papers that discuss patient treatment.
Howard Bauchner, MD, JAMA’s editor in chief, addressed it in an editorial published in September 2017. Although not explicitly directed at bioRxiv, Dr. Bauchner took a firm stance against shortcutting the evaluation of evidence that is often years in the making.
“New interest in preprint servers in clinical medicine increases the likelihood of premature dissemination and public consumption of clinical research findings prior to rigorous evaluation and peer review,” Dr. Bauchner wrote. “For most articles, public consumption of research findings prior to peer review will have little influence on health, but for some articles, the effect could be devastating for some patients if the results made public prior to peer review are wrong or incorrectly interpreted.”
Dr. Bauchner did not overstate the potential influence of unvetted science, as a January 2018 bioRxiv study on CRISPR gene editing clearly demonstrated. The paper by Carsten Charlesworth, a doctoral student at Stanford (Calif.) University, found that up to 79% of humans could already be immune to Crispr-Cas9, the gene-editing protein derived from Staphylococcus aureus and S. pyogenes. More than science geeks were reading: The report initially sent CRISPR stocks tumbling.
Aaron D. Viny, MD, is in general a hesitant fan of bioRxiv’s preprint platform. But he raised an eyebrow when he learned about medRxiv.
“The only pressure that I can see in regulating these reports is social media,” said Dr. Viny, a hematologic oncologist at Memorial Sloan Kettering, in New York. “The fear is that it will be misused in two different realms. The most dangerous and worrisome, of course, is for patients using the data to influence their care plan, when the data haven’t been vetted appropriately. But secondarily, how could it influence the economics of clinical trials? There is no shortage of hedge fund managers in biotech. These data could misinform a consultant who might know the area in a way that artificially exploits early research data. Could that permit someone to submit disingenuous data to manipulate the stock of a given pharmaceutical company? I don’t know how you police that kind of thing.”
Who’s loving it – and why?
There are plenty of reasons to support a thriving preprint community, said Jessica Polka, PhD, director of ASAPbio, (Accelerating Science and Publication in biology), a group that bills itself as a scientist-driven initiative to promote the productive use of preprints in the life sciences.
“Preprinting complements traditional journal publishing by allowing researchers to rapidly communicate their findings to the scientific community,” she said. “This, in turn, provides them with opportunities for earlier and broader feedback and a way to transparently demonstrate progress on a project. More importantly, the whole community benefits by having earlier access to research findings, which can accelerate the pace of discovery.”
The disclosures applied to every preprint paper are the publisher’s way of assuring this same awareness, she said. And preprints do need to be approached with some skepticism, as should peer-reviewed literature.
“The veracity of published papers is not always a given. An example is the 1998 vaccine paper [published in the Lancet] by Dr. Andrew Wakefield,” which launched the antivaccine movement. “But the answer to problems of reliability is to provide more information about the research and how it has been verified and evaluated, not less information. For example, confirmation bias can make it difficult to refute work that has been published. The current incentives for publishing negative results in a journal are not strong enough to reveal all of the information that could be useful to other researchers, but preprinting reduces the barrier to sharing negative results,” she said.
Swimming up the (main)stream
Universal peer-reviewed acceptance of preprints isn’t a done deal, Dr. Polka said. Journals are tussling with how to handle these papers. The Lancet clearly states that preprints don’t constitute prior publication and are welcome. The New England Journal of Medicine offers an uncontestable “no way.”
JAMA discourages submitting preprints, and will consider one only if the submitted version offers “meaningful new information” above what the preprint disseminated.
Cell Press has a slightly different take. They will consider papers previously posted on preprint services, but the policy applies only to the original submitted version of the paper. “We do not support posting of revisions that respond to editorial input and peer review or the final published version to preprint servers,” the policy notes.
In an interview, Deborah Sweet, PhD, the group’s vice president of editorial, elaborated on the policy. “In our view, one of the most important purposes of preprint posting is to gather feedback from the scientific community before a formal submission to a journal,” she said. “The ‘original submission’ term in our guidelines refers to the first version of the paper submitted to [Cell Press], which could include revisions made in response to community feedback on a preprint. After formal submission, we think it is most appropriate to incorporate and represent the value of the editorial and peer-review evaluation process in the final published journal article so that is clearly identifiable as the version of record.”
bioRxiv has made substantial inroads with dozens of other peer-reviewed journals. More than 100 – including a number of publications by EMBO Press and PLOS (Public Library of Science) – participate in bioRxiv’s B2J (BioRxiv-to-journal) direct-submission program.
With a few clicks, authors can transmit their bioRxiv manuscript files directly to these journals, without having to prepare separate submissions, Dr. Sweet said. Last year, Cell Press added two publications – Cell Reports and Structure – to the B2J program. “Once the paper is sent, it moves behind the scenes to the journal system and reappears as a formal submission,” she said. “In our process, before transferring the paper to the journal editors, authors have a chance to update the files (for example, to add a cover letter) and answer the standard questions that we ask, including ones about reviewer suggestions and exclusion requests. Once that step is done, the paper is handed over to the editorial team, and it’s ready to go for consideration in the same way as any other submission.”
Who’s reading?
Regardless of whether peer-review journals grant them legitimacy, preprints are getting a lot of views. A recent research letter, published in JAMA, looked at readership and online attention in 7,750 preprints posted from November 2013 to January 2017.
Primary author Stylianos Serghiou then selected 776 papers that had first appeared in bioRxiv, and matched them with 3,647 peer-reviewed articles lacking preprint exposure. He examined several publishing metrics for the papers, including views and downloads, citations in other sources, and Altmetric scores.
Altmetric tracks digital attention to scientific papers: Wikipedia citations, mentions in policy documents, blog discussions, and social media mentions including Facebook, Reddit, and Twitter. An Altmetric “attention score” of more than 20 corresponds to articles in the top 5% of readership, he said in an interview.
“Almost one in five of the bioRxiv preprints were getting these very high Almetric scores – much higher scores than articles that had no preprint posting,” Mr. Serghiou said in an interview.
Other findings include:
- The median number of preprint abstract views was 924, and the median number of PDF downloads was 321.
- In total, 18% of the preprints achieved an Altmetric score of more than 20.
- Of 7,750 preprints, 55% were accepted in a peer-reviewed publication within 24 months.
- Altmetric scores were significantly higher in articles in preprints (median 9.5 vs. 3.5).
The differences are probably related, at least in part, to the digital media savvy of preprint authors, Mr. Serghiou suggested. “We speculate that people who publish in bioRxiv may be more familiar with social media methods of making others aware of their work. They tend to be very good at using platforms like Twitter and Facebook to promote their results.”
Despite the high exposure scores, only 10% of bioRxiv articles get any posted comments or feedback – a key raison d’être for using a preprint service.
“Ten percent doesn’t sound like a very robust [feedback], but most journal articles get no comments whatsoever,” Dr. Inglis said. “And if they do, especially on the weekly magazines of science, comments may be from someone who has an ax to grind, or who doesn’t know much about the subject.”
What isn’t measured, in either volume or import, is the private communication a preprint engenders, Dr. Inglis said. “Feedback comes directly and privately to the author through email or at meetings or on the phone. We hear time and again that authors get hundreds of downloads after posting, and receive numerous contacts from colleagues who want to know more, to point out weaknesses, or request collaborations. These are the advantages we see from this potentially anxiety-provoking process of putting a manuscript out that has not been approved for publication. The entire purpose is to accelerate the speed of research by accelerating the speed of communication.”
Dr. Inglis, Dr. Sweet, and Dr. Polka are all employees of their respective companies. Dr. Viny and Mr. Serghiou both reported having no financial disclosures relevant to this article.
Dexcom G6 gets FDA nod
from the Food and Drug Administration.
The Dexcom G6 is about 28% smaller than its predecessor, the G5, can be worn for up to 10 days – 43% longer than the G5 – and doesn’t require any finger-stick calibrations or treatment decisions. It’s the first FDA-approved integrated continuous glucose monitoring (iCGM) system that can link electronically to other compatible devices, including automated insulin dosing systems, insulin pumps, blood glucose meters, and other electronic devices used for diabetes management, the FDA said in a press statement. Its revamped sensor doesn’t interact with acetaminophen – another distinct advantage over the G5.
The device will be commercially available sometime this year, the Dexcom website noted.
The device also set a new premarketing review standard for CGM’s, which can now utilize the less-burdensome 510(k) clearance pathway. Until now, they have been treated as the highest-risk Class III medical devices.
According to the FDA statement, the agency “…recognized this as an opportunity to reduce the regulatory burden for this type of device by establishing criteria that would classify these as ‘moderate risk,’ class II medical devices with special controls.”
G6 was approved through this new pathway, dedicated to novel, low-to-moderate-risk devices that are not “substantially equivalent” to an already legally marketed device, the press statement said.

The FDA evaluated data from two clinical studies of the Dexcom G6, which included 324 adults and children aged 2 years and older with diabetes. Both studies included multiple clinical visits within a 10-day period where system readings were compared to a laboratory test method that measures blood glucose values. No serious adverse events were reported during the studies.
from the Food and Drug Administration.
The Dexcom G6 is about 28% smaller than its predecessor, the G5, can be worn for up to 10 days – 43% longer than the G5 – and doesn’t require any finger-stick calibrations or treatment decisions. It’s the first FDA-approved integrated continuous glucose monitoring (iCGM) system that can link electronically to other compatible devices, including automated insulin dosing systems, insulin pumps, blood glucose meters, and other electronic devices used for diabetes management, the FDA said in a press statement. Its revamped sensor doesn’t interact with acetaminophen – another distinct advantage over the G5.
The device will be commercially available sometime this year, the Dexcom website noted.
The device also set a new premarketing review standard for CGM’s, which can now utilize the less-burdensome 510(k) clearance pathway. Until now, they have been treated as the highest-risk Class III medical devices.
According to the FDA statement, the agency “…recognized this as an opportunity to reduce the regulatory burden for this type of device by establishing criteria that would classify these as ‘moderate risk,’ class II medical devices with special controls.”
G6 was approved through this new pathway, dedicated to novel, low-to-moderate-risk devices that are not “substantially equivalent” to an already legally marketed device, the press statement said.
The FDA evaluated data from two clinical studies of the Dexcom G6, which included 324 adults and children aged 2 years and older with diabetes. Both studies included multiple clinical visits within a 10-day period where system readings were compared to a laboratory test method that measures blood glucose values. No serious adverse events were reported during the studies.
from the Food and Drug Administration.
The Dexcom G6 is about 28% smaller than its predecessor, the G5, can be worn for up to 10 days – 43% longer than the G5 – and doesn’t require any finger-stick calibrations or treatment decisions. It’s the first FDA-approved integrated continuous glucose monitoring (iCGM) system that can link electronically to other compatible devices, including automated insulin dosing systems, insulin pumps, blood glucose meters, and other electronic devices used for diabetes management, the FDA said in a press statement. Its revamped sensor doesn’t interact with acetaminophen – another distinct advantage over the G5.
The device will be commercially available sometime this year, the Dexcom website noted.
The device also set a new premarketing review standard for CGM’s, which can now utilize the less-burdensome 510(k) clearance pathway. Until now, they have been treated as the highest-risk Class III medical devices.
According to the FDA statement, the agency “…recognized this as an opportunity to reduce the regulatory burden for this type of device by establishing criteria that would classify these as ‘moderate risk,’ class II medical devices with special controls.”
G6 was approved through this new pathway, dedicated to novel, low-to-moderate-risk devices that are not “substantially equivalent” to an already legally marketed device, the press statement said.
The FDA evaluated data from two clinical studies of the Dexcom G6, which included 324 adults and children aged 2 years and older with diabetes. Both studies included multiple clinical visits within a 10-day period where system readings were compared to a laboratory test method that measures blood glucose values. No serious adverse events were reported during the studies.
Early diagnosis of Alzheimer’s could save U.S. trillions over time
Alzheimer’s disease may cost the United States alone more than $1.3 trillion by 2050, but early diagnosis could be one way to mitigate at least some of that increase, a special report released by the Alzheimer’s Association says.
An improved clinical scenario, with 88% of patients diagnosed in the early stage of mild cognitive impairment (MCI), could save $231.4 billion in direct treatment and long-term care costs by that time, according to the report, contained in the 2018 Alzheimer’s Disease Facts and Figures. Extrapolated out to the full lifespan of everyone now alive in the United States, the 88% diagnostic scenario could reap $7 trillion in savings, the report noted. This benefit would comprise $3.3 trillion in Medicare savings, $2.3 trillion in Medicaid savings, and $1.4 trillion in other areas of spending, including out-of-pocket expenses and private insurance.
The improved clinical diagnosis picture could manifest if diagnoses were based solely on biomarkers rather than the current method, which relies largely on symptoms and performance on cognitive tests, without biomarker confirmation. The biomarker-based diagnostic algorithm has been proposed for research cohorts, but not for clinical care.
The diagnostic workup currently employed, which is most often not confirmed with biomarkers, “means that many people who are diagnosed with Alzheimer’s may in reality have MCI or dementia due to other causes,” the report noted. Studies consistently show that up to 30% of patients diagnosed with apparent Alzheimer’s actually have another source of cognitive dysfunction. The misdiagnosis gap haunts clinical trialists and makes a strong case for incorporating biomarkers, including amyloid imaging, into the diagnostic workup – something the Alzheimer’s Association is pushing for with its IDEAS study.
Diagnostic reliance on symptoms and cognitive test performance without the additional information provided by biomarkers can affect the confidence clinicians have in making a diagnosis and thereby delay a diagnosis, dementia specialist Marwan N. Sabbagh, MD, said when asked to comment on the report.
“The report by the Alzheimer’s Association underscores the fact that early diagnosis of dementia or MCI due to Alzheimer’s disease is important not only because it is good health care but because net savings can be realized. The simple fact is that physicians have been taught to approach a diagnosis of dementia as a diagnosis of exclusion and they have been told that a diagnosis can be absolutely attained only by biopsy or autopsy. The consequence of these messages is that there is a lack of confidence in the clinic diagnosis and a subsequent delay in making a diagnosis,” said Dr. Sabbagh, the Karsten Solheim Chair for Dementia, professor of neurology, and director of the Alzheimer’s and memory disorders division at the Barrow Neurological Institute, Phoenix. “The deployment of in vivo biomarkers will transform the diagnosis from one of exclusion to one of inclusion. The up front costs will be saved later in the course.”
Earlier diagnosis is also associated with greater per-person savings, the report noted. “Under the current status quo, an individual with Alzheimer’s has total projected health and long-term care costs of $424,000 (present value of future costs) from the year before MCI until death. Under the partial early diagnosis scenario, the average per-person cost for an individual with Alzheimer’s is projected to be $360,000, saving $64,000 per individual.”
The economic modeling study employed The Health Economics Medical Innovation Simulation (THEMIS), which uses data from the Health and Retirement Study (HRS), a nationally representative sample of adults aged 50 and older.
The simulated population included everyone alive in the United States in 2018 and assumed cognitive assessment beginning at age 50. The model did not assume that biomarkers would be used in the diagnostic process.
It included three scenarios:
• The current situation, in which many people never receive a diagnosis or receive it later in the disease.
• A partial early-diagnosis scenario, with 88% of Alzheimer’s patients diagnosed in the MCI stage.
• A full early diagnosis scenario, in which all Alzheimer’s patients receive an early MCI diagnosis.
The current situation of inaccurate or late diagnosis remains the most expensive scenario. The model projected a total expenditure of $47.1 trillion over the lifetime of everyone alive in the United States in 2018 ($23.1 trillion in Medicare costs, $11.8 trillion in Medicaid costs, and $12.1 trillion in other costs). The report also noted that this total doesn’t include the current expense of caring for everyone in the United States who has Alzheimer’s now.
The partial early diagnosis scenario assumes that everyone with Alzheimer’s has a 70% chance of being diagnosed with MCI every 2 years; this would yield a total diagnostic rate of 88%.
Under this scenario, the model projected a total care cost of $40.1 trillion – a $7 trillion benefit composed of $3.3 trillion in Medicare savings, $2.3 trillion in Medicaid savings, and $1.4 trillion in other savings.
“Thus, nearly all of the potential savings of early diagnosis can be realized under the partial early diagnosis scenario,” the report noted.
These savings would be realized over a long period, but there could be massive shorter-term benefits as well, the report said. Savings under the partial early-diagnosis scenario could be $31.8 billion in 2025 and $231.4 billion in 2050.
That would be good financial news, especially in light of the report’s current cost analysis. In 2018, the cost of caring for Alzheimer’s patients and those with other dementias is on track to exceed $277 billion, which is $18 billion more than the United States paid out last year. If the current diagnostic scenario and incidence rates continue unabated, the report projected an annual expense of $1.35 trillion for care in 2050.
Alzheimer’s disease may cost the United States alone more than $1.3 trillion by 2050, but early diagnosis could be one way to mitigate at least some of that increase, a special report released by the Alzheimer’s Association says.
An improved clinical scenario, with 88% of patients diagnosed in the early stage of mild cognitive impairment (MCI), could save $231.4 billion in direct treatment and long-term care costs by that time, according to the report, contained in the 2018 Alzheimer’s Disease Facts and Figures. Extrapolated out to the full lifespan of everyone now alive in the United States, the 88% diagnostic scenario could reap $7 trillion in savings, the report noted. This benefit would comprise $3.3 trillion in Medicare savings, $2.3 trillion in Medicaid savings, and $1.4 trillion in other areas of spending, including out-of-pocket expenses and private insurance.
The improved clinical diagnosis picture could manifest if diagnoses were based solely on biomarkers rather than the current method, which relies largely on symptoms and performance on cognitive tests, without biomarker confirmation. The biomarker-based diagnostic algorithm has been proposed for research cohorts, but not for clinical care.
The diagnostic workup currently employed, which is most often not confirmed with biomarkers, “means that many people who are diagnosed with Alzheimer’s may in reality have MCI or dementia due to other causes,” the report noted. Studies consistently show that up to 30% of patients diagnosed with apparent Alzheimer’s actually have another source of cognitive dysfunction. The misdiagnosis gap haunts clinical trialists and makes a strong case for incorporating biomarkers, including amyloid imaging, into the diagnostic workup – something the Alzheimer’s Association is pushing for with its IDEAS study.
Diagnostic reliance on symptoms and cognitive test performance without the additional information provided by biomarkers can affect the confidence clinicians have in making a diagnosis and thereby delay a diagnosis, dementia specialist Marwan N. Sabbagh, MD, said when asked to comment on the report.
“The report by the Alzheimer’s Association underscores the fact that early diagnosis of dementia or MCI due to Alzheimer’s disease is important not only because it is good health care but because net savings can be realized. The simple fact is that physicians have been taught to approach a diagnosis of dementia as a diagnosis of exclusion and they have been told that a diagnosis can be absolutely attained only by biopsy or autopsy. The consequence of these messages is that there is a lack of confidence in the clinic diagnosis and a subsequent delay in making a diagnosis,” said Dr. Sabbagh, the Karsten Solheim Chair for Dementia, professor of neurology, and director of the Alzheimer’s and memory disorders division at the Barrow Neurological Institute, Phoenix. “The deployment of in vivo biomarkers will transform the diagnosis from one of exclusion to one of inclusion. The up front costs will be saved later in the course.”
Earlier diagnosis is also associated with greater per-person savings, the report noted. “Under the current status quo, an individual with Alzheimer’s has total projected health and long-term care costs of $424,000 (present value of future costs) from the year before MCI until death. Under the partial early diagnosis scenario, the average per-person cost for an individual with Alzheimer’s is projected to be $360,000, saving $64,000 per individual.”
The economic modeling study employed The Health Economics Medical Innovation Simulation (THEMIS), which uses data from the Health and Retirement Study (HRS), a nationally representative sample of adults aged 50 and older.
The simulated population included everyone alive in the United States in 2018 and assumed cognitive assessment beginning at age 50. The model did not assume that biomarkers would be used in the diagnostic process.
It included three scenarios:
• The current situation, in which many people never receive a diagnosis or receive it later in the disease.
• A partial early-diagnosis scenario, with 88% of Alzheimer’s patients diagnosed in the MCI stage.
• A full early diagnosis scenario, in which all Alzheimer’s patients receive an early MCI diagnosis.
The current situation of inaccurate or late diagnosis remains the most expensive scenario. The model projected a total expenditure of $47.1 trillion over the lifetime of everyone alive in the United States in 2018 ($23.1 trillion in Medicare costs, $11.8 trillion in Medicaid costs, and $12.1 trillion in other costs). The report also noted that this total doesn’t include the current expense of caring for everyone in the United States who has Alzheimer’s now.
The partial early diagnosis scenario assumes that everyone with Alzheimer’s has a 70% chance of being diagnosed with MCI every 2 years; this would yield a total diagnostic rate of 88%.
Under this scenario, the model projected a total care cost of $40.1 trillion – a $7 trillion benefit composed of $3.3 trillion in Medicare savings, $2.3 trillion in Medicaid savings, and $1.4 trillion in other savings.
“Thus, nearly all of the potential savings of early diagnosis can be realized under the partial early diagnosis scenario,” the report noted.
These savings would be realized over a long period, but there could be massive shorter-term benefits as well, the report said. Savings under the partial early-diagnosis scenario could be $31.8 billion in 2025 and $231.4 billion in 2050.
That would be good financial news, especially in light of the report’s current cost analysis. In 2018, the cost of caring for Alzheimer’s patients and those with other dementias is on track to exceed $277 billion, which is $18 billion more than the United States paid out last year. If the current diagnostic scenario and incidence rates continue unabated, the report projected an annual expense of $1.35 trillion for care in 2050.
Alzheimer’s disease may cost the United States alone more than $1.3 trillion by 2050, but early diagnosis could be one way to mitigate at least some of that increase, a special report released by the Alzheimer’s Association says.
An improved clinical scenario, with 88% of patients diagnosed in the early stage of mild cognitive impairment (MCI), could save $231.4 billion in direct treatment and long-term care costs by that time, according to the report, contained in the 2018 Alzheimer’s Disease Facts and Figures. Extrapolated out to the full lifespan of everyone now alive in the United States, the 88% diagnostic scenario could reap $7 trillion in savings, the report noted. This benefit would comprise $3.3 trillion in Medicare savings, $2.3 trillion in Medicaid savings, and $1.4 trillion in other areas of spending, including out-of-pocket expenses and private insurance.
The improved clinical diagnosis picture could manifest if diagnoses were based solely on biomarkers rather than the current method, which relies largely on symptoms and performance on cognitive tests, without biomarker confirmation. The biomarker-based diagnostic algorithm has been proposed for research cohorts, but not for clinical care.
The diagnostic workup currently employed, which is most often not confirmed with biomarkers, “means that many people who are diagnosed with Alzheimer’s may in reality have MCI or dementia due to other causes,” the report noted. Studies consistently show that up to 30% of patients diagnosed with apparent Alzheimer’s actually have another source of cognitive dysfunction. The misdiagnosis gap haunts clinical trialists and makes a strong case for incorporating biomarkers, including amyloid imaging, into the diagnostic workup – something the Alzheimer’s Association is pushing for with its IDEAS study.
Diagnostic reliance on symptoms and cognitive test performance without the additional information provided by biomarkers can affect the confidence clinicians have in making a diagnosis and thereby delay a diagnosis, dementia specialist Marwan N. Sabbagh, MD, said when asked to comment on the report.
“The report by the Alzheimer’s Association underscores the fact that early diagnosis of dementia or MCI due to Alzheimer’s disease is important not only because it is good health care but because net savings can be realized. The simple fact is that physicians have been taught to approach a diagnosis of dementia as a diagnosis of exclusion and they have been told that a diagnosis can be absolutely attained only by biopsy or autopsy. The consequence of these messages is that there is a lack of confidence in the clinic diagnosis and a subsequent delay in making a diagnosis,” said Dr. Sabbagh, the Karsten Solheim Chair for Dementia, professor of neurology, and director of the Alzheimer’s and memory disorders division at the Barrow Neurological Institute, Phoenix. “The deployment of in vivo biomarkers will transform the diagnosis from one of exclusion to one of inclusion. The up front costs will be saved later in the course.”
Earlier diagnosis is also associated with greater per-person savings, the report noted. “Under the current status quo, an individual with Alzheimer’s has total projected health and long-term care costs of $424,000 (present value of future costs) from the year before MCI until death. Under the partial early diagnosis scenario, the average per-person cost for an individual with Alzheimer’s is projected to be $360,000, saving $64,000 per individual.”
The economic modeling study employed The Health Economics Medical Innovation Simulation (THEMIS), which uses data from the Health and Retirement Study (HRS), a nationally representative sample of adults aged 50 and older.
The simulated population included everyone alive in the United States in 2018 and assumed cognitive assessment beginning at age 50. The model did not assume that biomarkers would be used in the diagnostic process.
It included three scenarios:
• The current situation, in which many people never receive a diagnosis or receive it later in the disease.
• A partial early-diagnosis scenario, with 88% of Alzheimer’s patients diagnosed in the MCI stage.
• A full early diagnosis scenario, in which all Alzheimer’s patients receive an early MCI diagnosis.
The current situation of inaccurate or late diagnosis remains the most expensive scenario. The model projected a total expenditure of $47.1 trillion over the lifetime of everyone alive in the United States in 2018 ($23.1 trillion in Medicare costs, $11.8 trillion in Medicaid costs, and $12.1 trillion in other costs). The report also noted that this total doesn’t include the current expense of caring for everyone in the United States who has Alzheimer’s now.
The partial early diagnosis scenario assumes that everyone with Alzheimer’s has a 70% chance of being diagnosed with MCI every 2 years; this would yield a total diagnostic rate of 88%.
Under this scenario, the model projected a total care cost of $40.1 trillion – a $7 trillion benefit composed of $3.3 trillion in Medicare savings, $2.3 trillion in Medicaid savings, and $1.4 trillion in other savings.
“Thus, nearly all of the potential savings of early diagnosis can be realized under the partial early diagnosis scenario,” the report noted.
These savings would be realized over a long period, but there could be massive shorter-term benefits as well, the report said. Savings under the partial early-diagnosis scenario could be $31.8 billion in 2025 and $231.4 billion in 2050.
That would be good financial news, especially in light of the report’s current cost analysis. In 2018, the cost of caring for Alzheimer’s patients and those with other dementias is on track to exceed $277 billion, which is $18 billion more than the United States paid out last year. If the current diagnostic scenario and incidence rates continue unabated, the report projected an annual expense of $1.35 trillion for care in 2050.
Adapting consumer technology into GI practice
BOSTON – You can command Alexa to order pizza and spool up your favorite flick, but accessing digital health data remains a struggle. Michael Docktor, MD, wants to change that.
A pediatric gastroenterologist and the clinical director of innovation at Boston Children’s Hospital, Dr. Docktor believes that it’s just a matter of time before consumer-driven digital technology fundamentally changes the way physicians and patients interact.

“In medicine, we are often in the habit of trying to recreate the wheel,” he said during the “Digital Health in GI Disease” session at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology. “My hope and belief is that we can borrow from the best of the consumer technology world and apply that to our world in health care and GI specifically.”
Dr. Docktor shared some of the “tools and toys” that have come of his group’s program and also “exposed folks to things that they may not be thinking about traditionally in medicine.”
In particular, one area he focused on was how certain voice technologies are enhancing health care delivery, such as integration of Amazon’s Alexa. Consider Alexa a nurse on call.
he said. “We have placed a fairly large bet on voice in health care and built some skills for Alexa.”
He also highlighted a new virtual reality tool that was recently launched by Boston Children’s Hospital to help gastroenterologists better educate their patients.
HealthVoyager was developed in conjunction with Klick Health. The kid-friendly app lets children take a virtual ride through their GI tract. Clinicians draw in abnormal findings on a simplified template. The app then recreates those findings – lesions, polyps, or inflammatory changes – in the positions they actually occupy in the patient. It generates a QR code that’s given to the patient, allowing the child to access her imaging in a HIPAA-compliant manner.
It’s cool, sure, Dr. Docktor said. But does it bring any value to the physician-patient interaction?
“The challenge of digital health is to prove that there’s actual value, it’s not just a bunch of snazzy tech. Are patients really using it? Sharing it? Are they educating themselves and their family and their community? We want to study this clinically and validate whether or not it results in improved adherence and improved patient satisfaction.”
He covered other technologies, such as Chatbox and blockchain, and the roles they can play in health care.
In the not-too-distant future, Dr. Docktor envisions voice assistants integrated into daily medical practice. Amazon’s Alexa provides an aspirational goal, he said.
“We are seeing the rise of the voice assistant. By 2020, researchers predict that 50% of all Internet searches will happen just by voice. Voice interface, I believe, will be driving health care by interfacing with patients at home. I predict that over the next 5 years, most of us will have a medical encounter on a device like this. Technology is not a limiting factor in this scenario. It’s just red tape on the payer and provider side at this point.”

Dr. Docktor’s colleague, Carla E. Small, senior director of the Innovation & Digital Health Accelerator at Boston Children’s Hospital, provided another real-life example of his digital vision. The Innovation & Digital Health Accelerator is a division within the hospital devoted to identifying, nurturing, and implementing digital health care solutions.
“The world has moved to a technology-enabled health care environment, and we all have to be there along with it,” she said. “That also creates a great opportunity for those who have an interest in innovation. There is a lot of ground for changing the way we do things and really leveraging that creativity and innovation.”
One Accelerator product that’s up and running is Thermia. The online tool guides parents through the anxiety of managing a child’s fever.
Thermia quickly and easily allows concerned parents to interpret a child’s temperature and understand which steps they should consider taking. Parents enter their child’s age, temperature, weight, any associated symptoms like rash, sore throat, or GI upset, as well as comorbid medical conditions. An algorithm issues advice for treatment at home or, if the data suggest a risk or serious problem, suggests a visit to the pediatrician or the emergency room. Thermia also automatically calculates the dosage of over-the-counter antipyretic medications based on age and weight.
The Accelerator is investigating a host of other digital health products in different stages of concept, design, and execution. Health care simply has to embrace the digital trends that are changing the way people interact with their world.
The AGA Center for GI Innovation and Technology wants to hear the unique ways gastroenterologists are leveraging consumer technology in their practices. Send us an email at [email protected].
[email protected]
[email protected]
BOSTON – You can command Alexa to order pizza and spool up your favorite flick, but accessing digital health data remains a struggle. Michael Docktor, MD, wants to change that.
A pediatric gastroenterologist and the clinical director of innovation at Boston Children’s Hospital, Dr. Docktor believes that it’s just a matter of time before consumer-driven digital technology fundamentally changes the way physicians and patients interact.
“In medicine, we are often in the habit of trying to recreate the wheel,” he said during the “Digital Health in GI Disease” session at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology. “My hope and belief is that we can borrow from the best of the consumer technology world and apply that to our world in health care and GI specifically.”
Dr. Docktor shared some of the “tools and toys” that have come of his group’s program and also “exposed folks to things that they may not be thinking about traditionally in medicine.”
In particular, one area he focused on was how certain voice technologies are enhancing health care delivery, such as integration of Amazon’s Alexa. Consider Alexa a nurse on call.
he said. “We have placed a fairly large bet on voice in health care and built some skills for Alexa.”
He also highlighted a new virtual reality tool that was recently launched by Boston Children’s Hospital to help gastroenterologists better educate their patients.
HealthVoyager was developed in conjunction with Klick Health. The kid-friendly app lets children take a virtual ride through their GI tract. Clinicians draw in abnormal findings on a simplified template. The app then recreates those findings – lesions, polyps, or inflammatory changes – in the positions they actually occupy in the patient. It generates a QR code that’s given to the patient, allowing the child to access her imaging in a HIPAA-compliant manner.
It’s cool, sure, Dr. Docktor said. But does it bring any value to the physician-patient interaction?
“The challenge of digital health is to prove that there’s actual value, it’s not just a bunch of snazzy tech. Are patients really using it? Sharing it? Are they educating themselves and their family and their community? We want to study this clinically and validate whether or not it results in improved adherence and improved patient satisfaction.”
He covered other technologies, such as Chatbox and blockchain, and the roles they can play in health care.
In the not-too-distant future, Dr. Docktor envisions voice assistants integrated into daily medical practice. Amazon’s Alexa provides an aspirational goal, he said.
“We are seeing the rise of the voice assistant. By 2020, researchers predict that 50% of all Internet searches will happen just by voice. Voice interface, I believe, will be driving health care by interfacing with patients at home. I predict that over the next 5 years, most of us will have a medical encounter on a device like this. Technology is not a limiting factor in this scenario. It’s just red tape on the payer and provider side at this point.”
Dr. Docktor’s colleague, Carla E. Small, senior director of the Innovation & Digital Health Accelerator at Boston Children’s Hospital, provided another real-life example of his digital vision. The Innovation & Digital Health Accelerator is a division within the hospital devoted to identifying, nurturing, and implementing digital health care solutions.
“The world has moved to a technology-enabled health care environment, and we all have to be there along with it,” she said. “That also creates a great opportunity for those who have an interest in innovation. There is a lot of ground for changing the way we do things and really leveraging that creativity and innovation.”
One Accelerator product that’s up and running is Thermia. The online tool guides parents through the anxiety of managing a child’s fever.
Thermia quickly and easily allows concerned parents to interpret a child’s temperature and understand which steps they should consider taking. Parents enter their child’s age, temperature, weight, any associated symptoms like rash, sore throat, or GI upset, as well as comorbid medical conditions. An algorithm issues advice for treatment at home or, if the data suggest a risk or serious problem, suggests a visit to the pediatrician or the emergency room. Thermia also automatically calculates the dosage of over-the-counter antipyretic medications based on age and weight.
The Accelerator is investigating a host of other digital health products in different stages of concept, design, and execution. Health care simply has to embrace the digital trends that are changing the way people interact with their world.
The AGA Center for GI Innovation and Technology wants to hear the unique ways gastroenterologists are leveraging consumer technology in their practices. Send us an email at [email protected].
[email protected]
[email protected]
BOSTON – You can command Alexa to order pizza and spool up your favorite flick, but accessing digital health data remains a struggle. Michael Docktor, MD, wants to change that.
A pediatric gastroenterologist and the clinical director of innovation at Boston Children’s Hospital, Dr. Docktor believes that it’s just a matter of time before consumer-driven digital technology fundamentally changes the way physicians and patients interact.
“In medicine, we are often in the habit of trying to recreate the wheel,” he said during the “Digital Health in GI Disease” session at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology. “My hope and belief is that we can borrow from the best of the consumer technology world and apply that to our world in health care and GI specifically.”
Dr. Docktor shared some of the “tools and toys” that have come of his group’s program and also “exposed folks to things that they may not be thinking about traditionally in medicine.”
In particular, one area he focused on was how certain voice technologies are enhancing health care delivery, such as integration of Amazon’s Alexa. Consider Alexa a nurse on call.
he said. “We have placed a fairly large bet on voice in health care and built some skills for Alexa.”
He also highlighted a new virtual reality tool that was recently launched by Boston Children’s Hospital to help gastroenterologists better educate their patients.
HealthVoyager was developed in conjunction with Klick Health. The kid-friendly app lets children take a virtual ride through their GI tract. Clinicians draw in abnormal findings on a simplified template. The app then recreates those findings – lesions, polyps, or inflammatory changes – in the positions they actually occupy in the patient. It generates a QR code that’s given to the patient, allowing the child to access her imaging in a HIPAA-compliant manner.
It’s cool, sure, Dr. Docktor said. But does it bring any value to the physician-patient interaction?
“The challenge of digital health is to prove that there’s actual value, it’s not just a bunch of snazzy tech. Are patients really using it? Sharing it? Are they educating themselves and their family and their community? We want to study this clinically and validate whether or not it results in improved adherence and improved patient satisfaction.”
He covered other technologies, such as Chatbox and blockchain, and the roles they can play in health care.
In the not-too-distant future, Dr. Docktor envisions voice assistants integrated into daily medical practice. Amazon’s Alexa provides an aspirational goal, he said.
“We are seeing the rise of the voice assistant. By 2020, researchers predict that 50% of all Internet searches will happen just by voice. Voice interface, I believe, will be driving health care by interfacing with patients at home. I predict that over the next 5 years, most of us will have a medical encounter on a device like this. Technology is not a limiting factor in this scenario. It’s just red tape on the payer and provider side at this point.”
Dr. Docktor’s colleague, Carla E. Small, senior director of the Innovation & Digital Health Accelerator at Boston Children’s Hospital, provided another real-life example of his digital vision. The Innovation & Digital Health Accelerator is a division within the hospital devoted to identifying, nurturing, and implementing digital health care solutions.
“The world has moved to a technology-enabled health care environment, and we all have to be there along with it,” she said. “That also creates a great opportunity for those who have an interest in innovation. There is a lot of ground for changing the way we do things and really leveraging that creativity and innovation.”
One Accelerator product that’s up and running is Thermia. The online tool guides parents through the anxiety of managing a child’s fever.
Thermia quickly and easily allows concerned parents to interpret a child’s temperature and understand which steps they should consider taking. Parents enter their child’s age, temperature, weight, any associated symptoms like rash, sore throat, or GI upset, as well as comorbid medical conditions. An algorithm issues advice for treatment at home or, if the data suggest a risk or serious problem, suggests a visit to the pediatrician or the emergency room. Thermia also automatically calculates the dosage of over-the-counter antipyretic medications based on age and weight.
The Accelerator is investigating a host of other digital health products in different stages of concept, design, and execution. Health care simply has to embrace the digital trends that are changing the way people interact with their world.
The AGA Center for GI Innovation and Technology wants to hear the unique ways gastroenterologists are leveraging consumer technology in their practices. Send us an email at [email protected].
[email protected]
[email protected]
REPORTING FROM THE 2018 AGA TECH SUMMIT
Chemotherapy, metabolic pathway may affect CAR T-cell potential
Two critical factors – prior exposure to chemotherapy and a glycolytic metabolism – appear to degrade the potential of T cells to become chimeric antigen receptor–T cells.
Chemotherapy, especially with cyclophosphamide and doxorubicin, seems particularly toxic to T cells, damaging the mitochondria and decreasing the cells’ spare respiratory capacity – a measure of mitochondrial health, David Barrett, MD, said during a press briefing held in advance of the annual meeting of the American Association for Cancer Research.

These new findings may help explain why children with acute lymphoblastic leukemia (ALL) tend to respond so vigorously to CAR T treatment, and why T cells from patients with solid tumors simply don’t grow, or die soon after patient infusion, he said in an interview. They also suggest a benefit of harvesting T cells before any chemotherapy, a procedure Dr. Barrett and his colleagues have advocated.
“Based on these data we have altered our practice for T-cell therapy in high-risk leukemia patients. If we have a patient who may have a poor prognosis, we try to collect the cells early and store them before proceeding, because we know chemotherapy will progressively degrade them.”
There still is no successful CAR T-cell protocol for solid tumors, but Dr. Barrett said these findings eventually may help such patients, particularly if more advanced experiments in manipulating the cells’ metabolism prove successful.
He and his colleagues investigated why T cells from some patients result in a poor clinical product that either fails manufacture or does not proliferate in the patient. They examined T cells from 157 pediatric patients with a variety of cancers, including ALL, non-Hodgkin lymphoma, neuroblastoma, osteosarcoma, rhabdomyosarcoma, Wilms tumor, Hodgkin disease, chronic myelogenous leukemia, and Ewing sarcoma. The team obtained cells at diagnosis and after each cycle of chemotherapy.
They examined how well the cells grew in the transformation and expansion process. A “pass” was considered a fivefold expansion in response to CD3/CD28 exposure for 7 days. Normal donor cells typically expand 20- to 30-fold in this time.
Only T cells taken from ALL and Wilms tumor patients before chemotherapy achieved a pass, Dr. Barrett said. Most of the ALL expansions (80%) and half of the Wilms tumor expansions passed. “We noted very poor CAR T-cell potential in all the other tumor types – less than a 30% pass. We noted a decline in potential with cumulative chemotherapy in all cases, though this was particularly significant in children less than 3 years old.”
The team also used RNA profiling to look at the cells’ metabolic pathways. Dr. Barrett noted that T cells are highly metabolically adaptable, capable of using several different fuel types and switching from one to another. Glucose and fatty acids are frequent fuels. Most of the cells from patients with solid tumors exhibited a glycolytic metabolism, while cells from patients with ALL and Wilms tumor appeared to rely more on fatty acids.
“One is not inherently worse than the other,” he said. “But glycolysis appears to be a bad thing when we’re trying to turn them into CAR T cells. Those T cells were too exhausted to do anything.”
However, Dr. Barrett encouraged the cells to switch fuels by treating them in vitro with palmitic acid, the most common fatty acid in plants and animals.
“We were growing the cells in a media containing sugar, fatty acids, and amino acids,” he explained. “We just started overloading them with palmitic acid, which has a natural transporter on the T-cell surface, so it already had a good pathway to get into the cell. It helped restore some of the performance of these T cells in some assays, although it wasn’t a complete reversal. But it was encouraging that something as simple as providing an alternate fuel was enough to get some positive effect. Whether or not we would also have to block glucose use to get it to really work is something we continue to study.”
T cells that had been exposed to chemotherapy also did poorly. Cyclophosphamide and doxorubicin seemed particularly toxic. Cells with exposure to these two agents had severely depleted CAR T cell potential with very poor spare respiratory capacity. This is a marker of mitochondrial injury, Dr. Barrett said. “That wasn’t a huge surprise. We already knew that cyclophosphamide is very toxic to T cells.”
But the finding did suggest the simple intervention of harvesting T cells before chemotherapy, which is what Dr. Barrett and his colleagues now do in their high-risk ALL patients. Whether or not this would improve response in patients with solid tumors is still unknown.
He had no financial disclosures. This study was supported by the AACR, the Doris Duke Charitable Foundation Clinical Science Development Award, the Jeffrey Pride Foundation Research Award, and the St. Baldrick’s Foundation Scholar Award.
SOURCE: Barrett DM et al. AACR 2018, Abstract 1631.
Two critical factors – prior exposure to chemotherapy and a glycolytic metabolism – appear to degrade the potential of T cells to become chimeric antigen receptor–T cells.
Chemotherapy, especially with cyclophosphamide and doxorubicin, seems particularly toxic to T cells, damaging the mitochondria and decreasing the cells’ spare respiratory capacity – a measure of mitochondrial health, David Barrett, MD, said during a press briefing held in advance of the annual meeting of the American Association for Cancer Research.
These new findings may help explain why children with acute lymphoblastic leukemia (ALL) tend to respond so vigorously to CAR T treatment, and why T cells from patients with solid tumors simply don’t grow, or die soon after patient infusion, he said in an interview. They also suggest a benefit of harvesting T cells before any chemotherapy, a procedure Dr. Barrett and his colleagues have advocated.
“Based on these data we have altered our practice for T-cell therapy in high-risk leukemia patients. If we have a patient who may have a poor prognosis, we try to collect the cells early and store them before proceeding, because we know chemotherapy will progressively degrade them.”
There still is no successful CAR T-cell protocol for solid tumors, but Dr. Barrett said these findings eventually may help such patients, particularly if more advanced experiments in manipulating the cells’ metabolism prove successful.
He and his colleagues investigated why T cells from some patients result in a poor clinical product that either fails manufacture or does not proliferate in the patient. They examined T cells from 157 pediatric patients with a variety of cancers, including ALL, non-Hodgkin lymphoma, neuroblastoma, osteosarcoma, rhabdomyosarcoma, Wilms tumor, Hodgkin disease, chronic myelogenous leukemia, and Ewing sarcoma. The team obtained cells at diagnosis and after each cycle of chemotherapy.
They examined how well the cells grew in the transformation and expansion process. A “pass” was considered a fivefold expansion in response to CD3/CD28 exposure for 7 days. Normal donor cells typically expand 20- to 30-fold in this time.
Only T cells taken from ALL and Wilms tumor patients before chemotherapy achieved a pass, Dr. Barrett said. Most of the ALL expansions (80%) and half of the Wilms tumor expansions passed. “We noted very poor CAR T-cell potential in all the other tumor types – less than a 30% pass. We noted a decline in potential with cumulative chemotherapy in all cases, though this was particularly significant in children less than 3 years old.”
The team also used RNA profiling to look at the cells’ metabolic pathways. Dr. Barrett noted that T cells are highly metabolically adaptable, capable of using several different fuel types and switching from one to another. Glucose and fatty acids are frequent fuels. Most of the cells from patients with solid tumors exhibited a glycolytic metabolism, while cells from patients with ALL and Wilms tumor appeared to rely more on fatty acids.
“One is not inherently worse than the other,” he said. “But glycolysis appears to be a bad thing when we’re trying to turn them into CAR T cells. Those T cells were too exhausted to do anything.”
However, Dr. Barrett encouraged the cells to switch fuels by treating them in vitro with palmitic acid, the most common fatty acid in plants and animals.
“We were growing the cells in a media containing sugar, fatty acids, and amino acids,” he explained. “We just started overloading them with palmitic acid, which has a natural transporter on the T-cell surface, so it already had a good pathway to get into the cell. It helped restore some of the performance of these T cells in some assays, although it wasn’t a complete reversal. But it was encouraging that something as simple as providing an alternate fuel was enough to get some positive effect. Whether or not we would also have to block glucose use to get it to really work is something we continue to study.”
T cells that had been exposed to chemotherapy also did poorly. Cyclophosphamide and doxorubicin seemed particularly toxic. Cells with exposure to these two agents had severely depleted CAR T cell potential with very poor spare respiratory capacity. This is a marker of mitochondrial injury, Dr. Barrett said. “That wasn’t a huge surprise. We already knew that cyclophosphamide is very toxic to T cells.”
But the finding did suggest the simple intervention of harvesting T cells before chemotherapy, which is what Dr. Barrett and his colleagues now do in their high-risk ALL patients. Whether or not this would improve response in patients with solid tumors is still unknown.
He had no financial disclosures. This study was supported by the AACR, the Doris Duke Charitable Foundation Clinical Science Development Award, the Jeffrey Pride Foundation Research Award, and the St. Baldrick’s Foundation Scholar Award.
SOURCE: Barrett DM et al. AACR 2018, Abstract 1631.
Two critical factors – prior exposure to chemotherapy and a glycolytic metabolism – appear to degrade the potential of T cells to become chimeric antigen receptor–T cells.
Chemotherapy, especially with cyclophosphamide and doxorubicin, seems particularly toxic to T cells, damaging the mitochondria and decreasing the cells’ spare respiratory capacity – a measure of mitochondrial health, David Barrett, MD, said during a press briefing held in advance of the annual meeting of the American Association for Cancer Research.
These new findings may help explain why children with acute lymphoblastic leukemia (ALL) tend to respond so vigorously to CAR T treatment, and why T cells from patients with solid tumors simply don’t grow, or die soon after patient infusion, he said in an interview. They also suggest a benefit of harvesting T cells before any chemotherapy, a procedure Dr. Barrett and his colleagues have advocated.
“Based on these data we have altered our practice for T-cell therapy in high-risk leukemia patients. If we have a patient who may have a poor prognosis, we try to collect the cells early and store them before proceeding, because we know chemotherapy will progressively degrade them.”
There still is no successful CAR T-cell protocol for solid tumors, but Dr. Barrett said these findings eventually may help such patients, particularly if more advanced experiments in manipulating the cells’ metabolism prove successful.
He and his colleagues investigated why T cells from some patients result in a poor clinical product that either fails manufacture or does not proliferate in the patient. They examined T cells from 157 pediatric patients with a variety of cancers, including ALL, non-Hodgkin lymphoma, neuroblastoma, osteosarcoma, rhabdomyosarcoma, Wilms tumor, Hodgkin disease, chronic myelogenous leukemia, and Ewing sarcoma. The team obtained cells at diagnosis and after each cycle of chemotherapy.
They examined how well the cells grew in the transformation and expansion process. A “pass” was considered a fivefold expansion in response to CD3/CD28 exposure for 7 days. Normal donor cells typically expand 20- to 30-fold in this time.
Only T cells taken from ALL and Wilms tumor patients before chemotherapy achieved a pass, Dr. Barrett said. Most of the ALL expansions (80%) and half of the Wilms tumor expansions passed. “We noted very poor CAR T-cell potential in all the other tumor types – less than a 30% pass. We noted a decline in potential with cumulative chemotherapy in all cases, though this was particularly significant in children less than 3 years old.”
The team also used RNA profiling to look at the cells’ metabolic pathways. Dr. Barrett noted that T cells are highly metabolically adaptable, capable of using several different fuel types and switching from one to another. Glucose and fatty acids are frequent fuels. Most of the cells from patients with solid tumors exhibited a glycolytic metabolism, while cells from patients with ALL and Wilms tumor appeared to rely more on fatty acids.
“One is not inherently worse than the other,” he said. “But glycolysis appears to be a bad thing when we’re trying to turn them into CAR T cells. Those T cells were too exhausted to do anything.”
However, Dr. Barrett encouraged the cells to switch fuels by treating them in vitro with palmitic acid, the most common fatty acid in plants and animals.
“We were growing the cells in a media containing sugar, fatty acids, and amino acids,” he explained. “We just started overloading them with palmitic acid, which has a natural transporter on the T-cell surface, so it already had a good pathway to get into the cell. It helped restore some of the performance of these T cells in some assays, although it wasn’t a complete reversal. But it was encouraging that something as simple as providing an alternate fuel was enough to get some positive effect. Whether or not we would also have to block glucose use to get it to really work is something we continue to study.”
T cells that had been exposed to chemotherapy also did poorly. Cyclophosphamide and doxorubicin seemed particularly toxic. Cells with exposure to these two agents had severely depleted CAR T cell potential with very poor spare respiratory capacity. This is a marker of mitochondrial injury, Dr. Barrett said. “That wasn’t a huge surprise. We already knew that cyclophosphamide is very toxic to T cells.”
But the finding did suggest the simple intervention of harvesting T cells before chemotherapy, which is what Dr. Barrett and his colleagues now do in their high-risk ALL patients. Whether or not this would improve response in patients with solid tumors is still unknown.
He had no financial disclosures. This study was supported by the AACR, the Doris Duke Charitable Foundation Clinical Science Development Award, the Jeffrey Pride Foundation Research Award, and the St. Baldrick’s Foundation Scholar Award.
SOURCE: Barrett DM et al. AACR 2018, Abstract 1631.
FROM AACR 2018
Key clinical point: Prior exposure to chemotherapy may degrade the potential of T cells to become CAR T cells, suggesting a benefit of harvesting T cells before any chemotherapy.
Major finding: Only T cells taken from ALL and Wilm’s tumor patients before chemotherapy achieved a fivefold expansion in response to CD3/CD28 exposure for 7 days.
Study details: An examination of T cells from 157 pediatric patients with a variety of cancers at diagnosis and after each cycle of chemotherapy.
Disclosures: The study was supported by the American Association of Cancer Research, the Doris Duke Charitable Foundation Clinical Science Development Award, the Jeffrey Pride Foundation Research Award, and the St. Baldrick’s Foundation Scholar Award. Dr. Barrett and his coauthors had no financial disclosures.
Source: Barrett DM et al. AACR 2018, Abstract 1631.
Fulvestrant plus neratinib reversed treatment-acquired HER2 mutations in metastatic ER+ breast cancer
Dual therapy with fulvestrant and the irreversible HER2 kinase inhibitor neratinib reversed treatment-acquired hormone resistance in metastatic estrogen receptor (ER)–positive breast cancer cells.
Elaine Mardis, PhD, a spokesperson for the American Association of Cancer Research, hailed the research by Utthara Nayar, PhD, and colleagues as “groundbreaking and unexpected” during a briefing held in advance of the annual meeting of the American Association for Cancer Research. The lab experiments were part of a whole-exome sequencing study of metastatic ER-positive tumor biopsies from 168 patients, 12 of whom had acquired the HER2 mutations, said Dr. Nayar of the Dana-Farber Cancer Institute, Boston.
The findings have prompted a phase 2 trial of the combination, which is now recruiting patients, Dr. Nayar said. The 5-year study seeks 152 women with inoperable locally advanced or metastatic ER-positive breast cancer with a confirmed HER2-positive mutation. Patients will be randomized to the combination of neratinib and fulvestrant or to neratinib alone. The primary outcome is progression-free survival.
“We also hope to be able to develop upfront combinations to preempt the resistance and lead to more durable responses,” Dr. Nayar said.
All of the 168 patients who contributed metastatic tumor biopsy samples to the study had developed resistance to estrogen receptor treatments, including aromatase inhibitors, tamoxifen, and fulvestrant. Of these biopsies, 12 had HER2 mutations, 8 of which had been previously characterized as activating.
Dr. Nayar and colleagues examined the untreated primary tumors in five of these patients; there was no mutation in four, suggesting that the mutations were a response to treatment. “In these 80%, the mutations were acquired as tumors were exposed to treatment and not present in the original tumor,” Dr. Nayar said.
These acquired HER2 mutations were mutually exclusive with ER mutations, which suggested a different mechanism of resistance to ER-directed therapies, she noted in her abstract. The mutations conferred resistance to tamoxifen, fulvestrant, and palbociclib.
However, the combination of fulvestrant and neratinib, an irreversible HER2 kinase inhibitor, overcame resistance in these cells.
In addition to pioneering a potentially important therapy for treatment-resistant metastatic breast cancer, the study highlights the importance of gene sequencing metastatic tumors, said Nikhil Wagle, MD, Dr. Nayar’s colleague and deputy director of the Center for Cancer Precision Medicine at Dana-Farber.
“Our study highlights how important it is to profile resistant metastatic tumors since these tumors may harbor targetable mechanisms of resistance that were not present in the original tumor biopsy,” Dr. Wagle noted in a press statement. “Repeated sequencing of tumors can pinpoint new genetic changes that cause resistance to therapies. This in turn can enable physicians to personalize therapy depending on the specific genetic changes in a patient’s tumor over time.”
The study was supported by the Department of Defense, the National Cancer Institute, the Susan G. Komen Foundation, the Dana-Farber Cancer Center, and a number of other private funders. Dr. Wagle is a stockholder in Foundation Medicine. Dr. Nayar had no financial disclosure.
SOURCE: Nayer U et al. AACR 2018, Abstract 4952
Dual therapy with fulvestrant and the irreversible HER2 kinase inhibitor neratinib reversed treatment-acquired hormone resistance in metastatic estrogen receptor (ER)–positive breast cancer cells.
Elaine Mardis, PhD, a spokesperson for the American Association of Cancer Research, hailed the research by Utthara Nayar, PhD, and colleagues as “groundbreaking and unexpected” during a briefing held in advance of the annual meeting of the American Association for Cancer Research. The lab experiments were part of a whole-exome sequencing study of metastatic ER-positive tumor biopsies from 168 patients, 12 of whom had acquired the HER2 mutations, said Dr. Nayar of the Dana-Farber Cancer Institute, Boston.
The findings have prompted a phase 2 trial of the combination, which is now recruiting patients, Dr. Nayar said. The 5-year study seeks 152 women with inoperable locally advanced or metastatic ER-positive breast cancer with a confirmed HER2-positive mutation. Patients will be randomized to the combination of neratinib and fulvestrant or to neratinib alone. The primary outcome is progression-free survival.
“We also hope to be able to develop upfront combinations to preempt the resistance and lead to more durable responses,” Dr. Nayar said.
All of the 168 patients who contributed metastatic tumor biopsy samples to the study had developed resistance to estrogen receptor treatments, including aromatase inhibitors, tamoxifen, and fulvestrant. Of these biopsies, 12 had HER2 mutations, 8 of which had been previously characterized as activating.
Dr. Nayar and colleagues examined the untreated primary tumors in five of these patients; there was no mutation in four, suggesting that the mutations were a response to treatment. “In these 80%, the mutations were acquired as tumors were exposed to treatment and not present in the original tumor,” Dr. Nayar said.
These acquired HER2 mutations were mutually exclusive with ER mutations, which suggested a different mechanism of resistance to ER-directed therapies, she noted in her abstract. The mutations conferred resistance to tamoxifen, fulvestrant, and palbociclib.
However, the combination of fulvestrant and neratinib, an irreversible HER2 kinase inhibitor, overcame resistance in these cells.
In addition to pioneering a potentially important therapy for treatment-resistant metastatic breast cancer, the study highlights the importance of gene sequencing metastatic tumors, said Nikhil Wagle, MD, Dr. Nayar’s colleague and deputy director of the Center for Cancer Precision Medicine at Dana-Farber.
“Our study highlights how important it is to profile resistant metastatic tumors since these tumors may harbor targetable mechanisms of resistance that were not present in the original tumor biopsy,” Dr. Wagle noted in a press statement. “Repeated sequencing of tumors can pinpoint new genetic changes that cause resistance to therapies. This in turn can enable physicians to personalize therapy depending on the specific genetic changes in a patient’s tumor over time.”
The study was supported by the Department of Defense, the National Cancer Institute, the Susan G. Komen Foundation, the Dana-Farber Cancer Center, and a number of other private funders. Dr. Wagle is a stockholder in Foundation Medicine. Dr. Nayar had no financial disclosure.
SOURCE: Nayer U et al. AACR 2018, Abstract 4952
Dual therapy with fulvestrant and the irreversible HER2 kinase inhibitor neratinib reversed treatment-acquired hormone resistance in metastatic estrogen receptor (ER)–positive breast cancer cells.
Elaine Mardis, PhD, a spokesperson for the American Association of Cancer Research, hailed the research by Utthara Nayar, PhD, and colleagues as “groundbreaking and unexpected” during a briefing held in advance of the annual meeting of the American Association for Cancer Research. The lab experiments were part of a whole-exome sequencing study of metastatic ER-positive tumor biopsies from 168 patients, 12 of whom had acquired the HER2 mutations, said Dr. Nayar of the Dana-Farber Cancer Institute, Boston.
The findings have prompted a phase 2 trial of the combination, which is now recruiting patients, Dr. Nayar said. The 5-year study seeks 152 women with inoperable locally advanced or metastatic ER-positive breast cancer with a confirmed HER2-positive mutation. Patients will be randomized to the combination of neratinib and fulvestrant or to neratinib alone. The primary outcome is progression-free survival.
“We also hope to be able to develop upfront combinations to preempt the resistance and lead to more durable responses,” Dr. Nayar said.
All of the 168 patients who contributed metastatic tumor biopsy samples to the study had developed resistance to estrogen receptor treatments, including aromatase inhibitors, tamoxifen, and fulvestrant. Of these biopsies, 12 had HER2 mutations, 8 of which had been previously characterized as activating.
Dr. Nayar and colleagues examined the untreated primary tumors in five of these patients; there was no mutation in four, suggesting that the mutations were a response to treatment. “In these 80%, the mutations were acquired as tumors were exposed to treatment and not present in the original tumor,” Dr. Nayar said.
These acquired HER2 mutations were mutually exclusive with ER mutations, which suggested a different mechanism of resistance to ER-directed therapies, she noted in her abstract. The mutations conferred resistance to tamoxifen, fulvestrant, and palbociclib.
However, the combination of fulvestrant and neratinib, an irreversible HER2 kinase inhibitor, overcame resistance in these cells.
In addition to pioneering a potentially important therapy for treatment-resistant metastatic breast cancer, the study highlights the importance of gene sequencing metastatic tumors, said Nikhil Wagle, MD, Dr. Nayar’s colleague and deputy director of the Center for Cancer Precision Medicine at Dana-Farber.
“Our study highlights how important it is to profile resistant metastatic tumors since these tumors may harbor targetable mechanisms of resistance that were not present in the original tumor biopsy,” Dr. Wagle noted in a press statement. “Repeated sequencing of tumors can pinpoint new genetic changes that cause resistance to therapies. This in turn can enable physicians to personalize therapy depending on the specific genetic changes in a patient’s tumor over time.”
The study was supported by the Department of Defense, the National Cancer Institute, the Susan G. Komen Foundation, the Dana-Farber Cancer Center, and a number of other private funders. Dr. Wagle is a stockholder in Foundation Medicine. Dr. Nayar had no financial disclosure.
SOURCE: Nayer U et al. AACR 2018, Abstract 4952
FROM THE AACR 2018 ANNUAL MEETING
Key clinical point: The combination of fulvestrant and neratinib reversed acquired HER2 mutations in ER+ metastatic breast cancer cells.
Major finding: Of 168 biopsies, 12 had acquired HER2 mutations after hormone treatment; these mutations were reversed with the dual therapy.
Study details: The exome sequencing study comprised 168 biopsies, and the in vitro study comprised 12.
Disclosures: The study was supported by the Department of Defense, the National Cancer Institute, the Susan G. Komen Foundation, the Dana-Farber Cancer Institute, and other private funders. Dr. Wagle is a stockholder in Foundation Medicine. Dr. Nayar had no financial disclosure.
Source: Nayar U et al. AACR 2018, Abstract 4952