User login
Early mortality risk factors in adult sickle cell disease support ECG screening
Among adults with sickle cell disease, early mortality is associated with increasing tricuspid regurgitant jet velocity on ECG, reticulocyte count, brain natriuretic peptide levels, and patient age, and with decreasing fetal hemoglobin levels, according to a report published in Haematologica.
Although survival improved among children with sickle cell disease (SCD) following the Food and Drug Administration approval of hydroxyurea treatment in 1998, mortality remains high among adult patients. To examine the clinical and laboratory factors underlying early mortality in this patient population, researchers combined the findings from their single-center cohort study of 161 clinic patients and a meta-analysis of nine studies in the literature, for a total of 3,257 participants. This is “the largest number of SCD patients in whom risk factors for mortality have been evaluated in the hydroxyurea era,” said Poulami Maitra, PhD, of the department of biostatistics at the University of North Carolina, Chapel Hill, and her associates.
The clinic cohort had a median age of 36 years (range, 18-71 years), and there were 29 deaths during a median follow-up of 7.2 years. The median age at death was 48 years. Similarly, the median age at death ranged from 39.7 to 53 years in the meta-analysis.
In the combined cohort, patients who had a tricuspid regurgitant jet velocity of 2.5 m/s or more had three times greater risk of dying than did patients who had lower values, and the risk of dying was approximately doubled for every 1-U elevation in log(N-terminal pro-B type natriuretic peptide), which reflects increasing ventricular strain. Both links have been reported before. The findings confirm that recent clinical practice guidelines recommending periodic echocardiographic screening for these patients is warranted, the investigators said (Haematologica. 2017 Jan 19. doi: 10.3324/haematol.2016.153791).
The hazard of dying was 5% higher for every 1% increase in reticulocyte count, which suggests that hemolysis contributes to early mortality in these patients. The hazard of dying also was 3% lower for every 1% increase in fetal hemoglobin. This association with fetal hemoglobin is the basis for the development of drugs that raise that level, such as hydroxyurea.
Mortality was 30% higher for every 10-year increase in patient age, reflecting the fact that people with SCD show increasing end-organ damage over time that contributes to their mortality, Dr. Maitra and her associates said.
This work was supported by the National Institutes of Health and the North Carolina Sickle Cell Program. Dr. Maitra reported having no relevant financial disclosures; one of her associates reported serving as a consultant to Pfizer and Global Blood Therapeutics.
Among adults with sickle cell disease, early mortality is associated with increasing tricuspid regurgitant jet velocity on ECG, reticulocyte count, brain natriuretic peptide levels, and patient age, and with decreasing fetal hemoglobin levels, according to a report published in Haematologica.
Although survival improved among children with sickle cell disease (SCD) following the Food and Drug Administration approval of hydroxyurea treatment in 1998, mortality remains high among adult patients. To examine the clinical and laboratory factors underlying early mortality in this patient population, researchers combined the findings from their single-center cohort study of 161 clinic patients and a meta-analysis of nine studies in the literature, for a total of 3,257 participants. This is “the largest number of SCD patients in whom risk factors for mortality have been evaluated in the hydroxyurea era,” said Poulami Maitra, PhD, of the department of biostatistics at the University of North Carolina, Chapel Hill, and her associates.
The clinic cohort had a median age of 36 years (range, 18-71 years), and there were 29 deaths during a median follow-up of 7.2 years. The median age at death was 48 years. Similarly, the median age at death ranged from 39.7 to 53 years in the meta-analysis.
In the combined cohort, patients who had a tricuspid regurgitant jet velocity of 2.5 m/s or more had three times greater risk of dying than did patients who had lower values, and the risk of dying was approximately doubled for every 1-U elevation in log(N-terminal pro-B type natriuretic peptide), which reflects increasing ventricular strain. Both links have been reported before. The findings confirm that recent clinical practice guidelines recommending periodic echocardiographic screening for these patients is warranted, the investigators said (Haematologica. 2017 Jan 19. doi: 10.3324/haematol.2016.153791).
The hazard of dying was 5% higher for every 1% increase in reticulocyte count, which suggests that hemolysis contributes to early mortality in these patients. The hazard of dying also was 3% lower for every 1% increase in fetal hemoglobin. This association with fetal hemoglobin is the basis for the development of drugs that raise that level, such as hydroxyurea.
Mortality was 30% higher for every 10-year increase in patient age, reflecting the fact that people with SCD show increasing end-organ damage over time that contributes to their mortality, Dr. Maitra and her associates said.
This work was supported by the National Institutes of Health and the North Carolina Sickle Cell Program. Dr. Maitra reported having no relevant financial disclosures; one of her associates reported serving as a consultant to Pfizer and Global Blood Therapeutics.
Among adults with sickle cell disease, early mortality is associated with increasing tricuspid regurgitant jet velocity on ECG, reticulocyte count, brain natriuretic peptide levels, and patient age, and with decreasing fetal hemoglobin levels, according to a report published in Haematologica.
Although survival improved among children with sickle cell disease (SCD) following the Food and Drug Administration approval of hydroxyurea treatment in 1998, mortality remains high among adult patients. To examine the clinical and laboratory factors underlying early mortality in this patient population, researchers combined the findings from their single-center cohort study of 161 clinic patients and a meta-analysis of nine studies in the literature, for a total of 3,257 participants. This is “the largest number of SCD patients in whom risk factors for mortality have been evaluated in the hydroxyurea era,” said Poulami Maitra, PhD, of the department of biostatistics at the University of North Carolina, Chapel Hill, and her associates.
The clinic cohort had a median age of 36 years (range, 18-71 years), and there were 29 deaths during a median follow-up of 7.2 years. The median age at death was 48 years. Similarly, the median age at death ranged from 39.7 to 53 years in the meta-analysis.
In the combined cohort, patients who had a tricuspid regurgitant jet velocity of 2.5 m/s or more had three times greater risk of dying than did patients who had lower values, and the risk of dying was approximately doubled for every 1-U elevation in log(N-terminal pro-B type natriuretic peptide), which reflects increasing ventricular strain. Both links have been reported before. The findings confirm that recent clinical practice guidelines recommending periodic echocardiographic screening for these patients is warranted, the investigators said (Haematologica. 2017 Jan 19. doi: 10.3324/haematol.2016.153791).
The hazard of dying was 5% higher for every 1% increase in reticulocyte count, which suggests that hemolysis contributes to early mortality in these patients. The hazard of dying also was 3% lower for every 1% increase in fetal hemoglobin. This association with fetal hemoglobin is the basis for the development of drugs that raise that level, such as hydroxyurea.
Mortality was 30% higher for every 10-year increase in patient age, reflecting the fact that people with SCD show increasing end-organ damage over time that contributes to their mortality, Dr. Maitra and her associates said.
This work was supported by the National Institutes of Health and the North Carolina Sickle Cell Program. Dr. Maitra reported having no relevant financial disclosures; one of her associates reported serving as a consultant to Pfizer and Global Blood Therapeutics.
FROM HAEMATOLOGICA
Key clinical point:
Major finding: Patients who had a tricuspid regurgitant jet velocity of 2.5 m/s or more had three times greater risk of dying than did patients with lower values, and the risk of dying was approximately doubled for every 1-U elevation in log(N-terminal pro-B type natriuretic peptide).
Data source: A single-center cohort study combined with a meta-analysis of nine studies, involving a total of 3,257 adults who had sickle cell disease.
Disclosures: This work was supported by the National Institutes of Health and the North Carolina Sickle Cell Program. Dr. Maitra reported having no relevant financial disclosures; one of her associates reported serving as a consultant to Pfizer and Global Blood Therapeutics.
Annual nailfold videocapillaroscopy found prognostic in systemic sclerosis
Annual nailfold videocapillaroscopy can be used to predict disease progression in systemic sclerosis, according to a report in Seminars in Arthritis & Rheumatism.
Early and diffuse alterations in the microvasculature are a key feature of systemic sclerosis, and nailfold videocapillaroscopy can detect morphologic changes that reflect such alterations, including capillary loss, neoangiogenesis, giant capillaries, microhemorrhages, and the presence of avascular areas. The technique already has an established role in diagnosing systemic sclerosis, said Jérôme Avouac, MD, of the rheumatology department at Cochin Hospital, Paris, and his associates.

Two contiguous fields extending over 1 mm in the middle of each nailfold, corresponding to the distal row of capillaries, were assessed for four features: the number of capillaries, the presence of giant capillaries, microhemorrhages, and neoangiogenesis (defined as meandering, ramified, branching, bushy, bizarre capillaries and those with more than two crossings). A total of 72 patients (51%) showed significant progression of at least one of these features during follow-up.
A progressive loss of capillaries over time strongly predicted overall disease progression (hazard ratio, 4.35). Other significant predictors of overall disease progression included the development of new ischemic digital ulcers (HR, 5.33), progression of lung involvement (HR, 18.53), progression of skin fibrosis (HR, 4.22), and worsening of the Medsger severity score (HR, 5.26). In addition, the presence of neoangiogenesis at baseline, but not the progression of neoangiogenesis over time, also predicted overall disease progression (HR, 2.53), the development of new ischemic digital ulcers (HR, 2.60), progression of lung involvement (HR, 7.38), and worsening of the Medsger severity score (HR, 2.72), Dr. Avouac and his associates said (Semin Arthritis Rheum. 2017 Feb 10. doi: 10.1016/j.semarthrit.2017.02.006).
These findings demonstrate that serial videocapillaroscopy, which they described as a simple, safe, noninvasive, and inexpensive imaging technique, can be used in routine follow-up of systemic sclerosis to improve risk assessment, the investigators said.
The authors reported no financial support for this study and reported having no relevant financial disclosures.
Annual nailfold videocapillaroscopy can be used to predict disease progression in systemic sclerosis, according to a report in Seminars in Arthritis & Rheumatism.
Early and diffuse alterations in the microvasculature are a key feature of systemic sclerosis, and nailfold videocapillaroscopy can detect morphologic changes that reflect such alterations, including capillary loss, neoangiogenesis, giant capillaries, microhemorrhages, and the presence of avascular areas. The technique already has an established role in diagnosing systemic sclerosis, said Jérôme Avouac, MD, of the rheumatology department at Cochin Hospital, Paris, and his associates.
Two contiguous fields extending over 1 mm in the middle of each nailfold, corresponding to the distal row of capillaries, were assessed for four features: the number of capillaries, the presence of giant capillaries, microhemorrhages, and neoangiogenesis (defined as meandering, ramified, branching, bushy, bizarre capillaries and those with more than two crossings). A total of 72 patients (51%) showed significant progression of at least one of these features during follow-up.
A progressive loss of capillaries over time strongly predicted overall disease progression (hazard ratio, 4.35). Other significant predictors of overall disease progression included the development of new ischemic digital ulcers (HR, 5.33), progression of lung involvement (HR, 18.53), progression of skin fibrosis (HR, 4.22), and worsening of the Medsger severity score (HR, 5.26). In addition, the presence of neoangiogenesis at baseline, but not the progression of neoangiogenesis over time, also predicted overall disease progression (HR, 2.53), the development of new ischemic digital ulcers (HR, 2.60), progression of lung involvement (HR, 7.38), and worsening of the Medsger severity score (HR, 2.72), Dr. Avouac and his associates said (Semin Arthritis Rheum. 2017 Feb 10. doi: 10.1016/j.semarthrit.2017.02.006).
These findings demonstrate that serial videocapillaroscopy, which they described as a simple, safe, noninvasive, and inexpensive imaging technique, can be used in routine follow-up of systemic sclerosis to improve risk assessment, the investigators said.
The authors reported no financial support for this study and reported having no relevant financial disclosures.
Annual nailfold videocapillaroscopy can be used to predict disease progression in systemic sclerosis, according to a report in Seminars in Arthritis & Rheumatism.
Early and diffuse alterations in the microvasculature are a key feature of systemic sclerosis, and nailfold videocapillaroscopy can detect morphologic changes that reflect such alterations, including capillary loss, neoangiogenesis, giant capillaries, microhemorrhages, and the presence of avascular areas. The technique already has an established role in diagnosing systemic sclerosis, said Jérôme Avouac, MD, of the rheumatology department at Cochin Hospital, Paris, and his associates.
Two contiguous fields extending over 1 mm in the middle of each nailfold, corresponding to the distal row of capillaries, were assessed for four features: the number of capillaries, the presence of giant capillaries, microhemorrhages, and neoangiogenesis (defined as meandering, ramified, branching, bushy, bizarre capillaries and those with more than two crossings). A total of 72 patients (51%) showed significant progression of at least one of these features during follow-up.
A progressive loss of capillaries over time strongly predicted overall disease progression (hazard ratio, 4.35). Other significant predictors of overall disease progression included the development of new ischemic digital ulcers (HR, 5.33), progression of lung involvement (HR, 18.53), progression of skin fibrosis (HR, 4.22), and worsening of the Medsger severity score (HR, 5.26). In addition, the presence of neoangiogenesis at baseline, but not the progression of neoangiogenesis over time, also predicted overall disease progression (HR, 2.53), the development of new ischemic digital ulcers (HR, 2.60), progression of lung involvement (HR, 7.38), and worsening of the Medsger severity score (HR, 2.72), Dr. Avouac and his associates said (Semin Arthritis Rheum. 2017 Feb 10. doi: 10.1016/j.semarthrit.2017.02.006).
These findings demonstrate that serial videocapillaroscopy, which they described as a simple, safe, noninvasive, and inexpensive imaging technique, can be used in routine follow-up of systemic sclerosis to improve risk assessment, the investigators said.
The authors reported no financial support for this study and reported having no relevant financial disclosures.
Key clinical point: Annual nailfold videocapillaroscopy can be used to predict disease progression in systemic sclerosis.
Major finding: A progressive loss of capillaries over time strongly predicted overall disease progression (HR, 4.35).
Data source: A prospective single-center observational cohort study involving 140 patients followed for up to 5 years.
Disclosures: The authors reported no financial support for this study and reported having no relevant financial disclosures.
Lanadelumab reduced hereditary angioedema attacks by 88%-100%
Lanadelumab, a monoclonal antibody that inhibits kallikrein, reduced attacks of hereditary angioedema with C1 inhibitor deficiency by 88%-100% in a small, phase I trial.
Hereditary angioedema with C1 inhibitor deficiency is a rare disorder characterized by unpredictable, recurrent, and potentially life-threatening episodes of subcutaneous or submucosal swelling, typically affecting the hands and feet, abdomen, face, larynx, or genitourinary tract. It is caused by a deficiency or dysfunction of the C1 inhibitor, which regulates the complement, coagulation, and kallikrein-kinin cascades.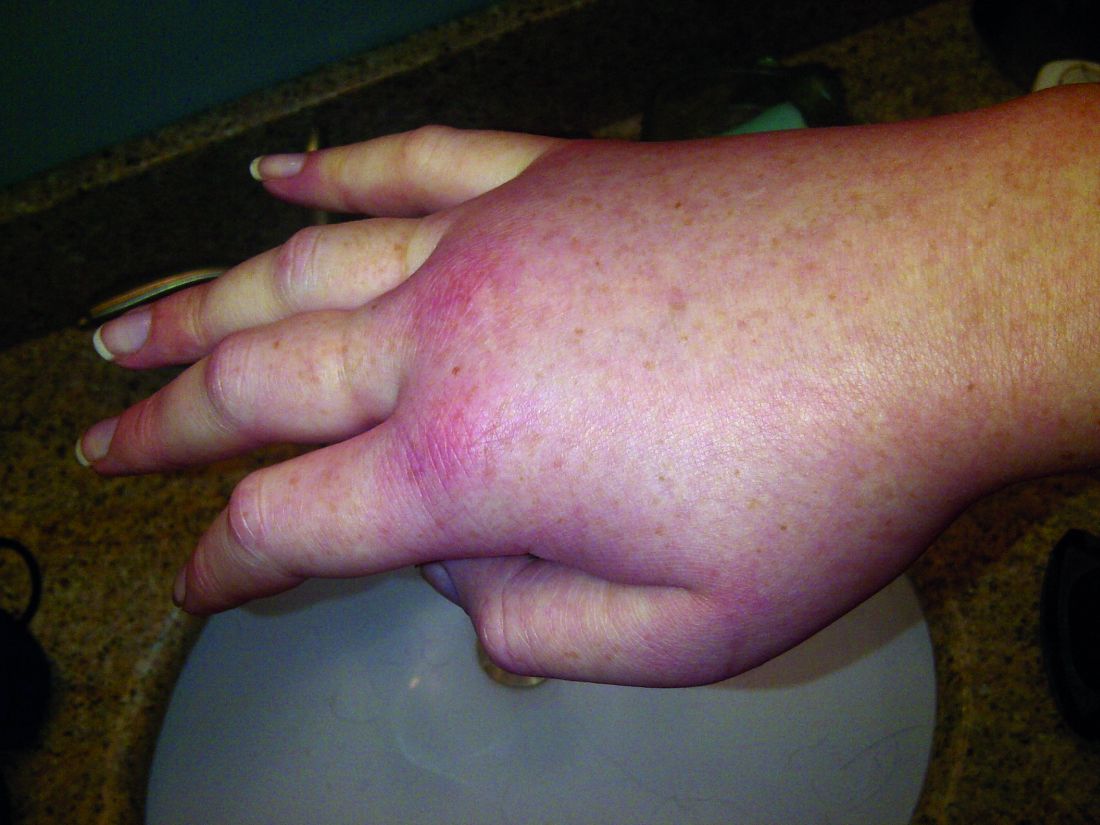
They performed a multicenter, double-blind, randomized study to assess the safety and adverse-effect profile of four doses of this new agent or placebo in 37 adults (aged 18-71 years, mean age, 39.9 years) who received two injections, 2 weeks apart, and were followed for 6 weeks. Given their histories, all the study participants had “a reasonable probability of having one or more attacks” during the study period, the researchers noted.
Four participants received a 30-mg dose, 4 received a 100-mg dose, 5 received a 300-mg dose, 11 received a 400-mg dose, and 13 received placebo.
There were no serious adverse events, no deaths, and no discontinuations of the study medication because of an adverse effect. One patient each developed severe adverse events: pain at the injection site that lasted for 1 minute and headache plus night sweats.
Pharmacodynamic assessments showed that lanadelumab inhibited kallikrein in a linear, dose-dependent manner, and the two higher doses reduced levels of cleaved high-molecular-weight kininogen to those reported in healthy control subjects. At the same time, the two higher doses decreased the number of attacks by 88% and 100%, respectively, compared with placebo.
All the patients in the 300-mg group and 9 of the 11 in the 400-mg group had no attacks during the study period, the investigators said.
These findings “provide proof of concept that lanadelumab has the potential to correct the pathophysiological abnormality underlying attacks of angioedema and may be a new therapeutic option for hereditary angioedema with C1 inhibitor deficiency,” Dr. Banerji and her associates said.
The HELP Study, a phase III trial assessing the safety and efficacy of 6 months of lanadelumab treatment, is now underway, they added.
The trial was sponsored by Dyax, which also participated in the study design, data collection and interpretation, and writing of the results. Dr. Banerji reported ties to Alnylam Pharmaceuticals, CSL Behring, Dyax, and Shire; her associates reported ties to numerous industry sources.
This preliminary study suggests that a new agent, in injections that would be convenient and widely accessible, could provide an unprecedented level of protection against angioedema.
If these findings are confirmed, and if lanadelumab is affordable, it could transform the way hereditary angioedema is managed and the life prospects for affected families.
Moreover, kallikrein is implicated in other forms of bradykinin-mediated angioedema, such as that associated with ACE inhibitors, and plays a key role in the generation of inflammation and pain. So, the sustained inhibition of kallikrein potentially could be beneficial for a much wider range of disorders.
Hilary J. Longhurst, MD, is at Barts Health National Health Service Trust, London. She reported having ties to BioCryst, CSL Behring, and Shire. Dr. Longhurst made these remarks in an editorial accompanying Dr. Banerji’s report (N Engl J Med. 2017 Feb 23;376[8]:788-9).
This preliminary study suggests that a new agent, in injections that would be convenient and widely accessible, could provide an unprecedented level of protection against angioedema.
If these findings are confirmed, and if lanadelumab is affordable, it could transform the way hereditary angioedema is managed and the life prospects for affected families.
Moreover, kallikrein is implicated in other forms of bradykinin-mediated angioedema, such as that associated with ACE inhibitors, and plays a key role in the generation of inflammation and pain. So, the sustained inhibition of kallikrein potentially could be beneficial for a much wider range of disorders.
Hilary J. Longhurst, MD, is at Barts Health National Health Service Trust, London. She reported having ties to BioCryst, CSL Behring, and Shire. Dr. Longhurst made these remarks in an editorial accompanying Dr. Banerji’s report (N Engl J Med. 2017 Feb 23;376[8]:788-9).
This preliminary study suggests that a new agent, in injections that would be convenient and widely accessible, could provide an unprecedented level of protection against angioedema.
If these findings are confirmed, and if lanadelumab is affordable, it could transform the way hereditary angioedema is managed and the life prospects for affected families.
Moreover, kallikrein is implicated in other forms of bradykinin-mediated angioedema, such as that associated with ACE inhibitors, and plays a key role in the generation of inflammation and pain. So, the sustained inhibition of kallikrein potentially could be beneficial for a much wider range of disorders.
Hilary J. Longhurst, MD, is at Barts Health National Health Service Trust, London. She reported having ties to BioCryst, CSL Behring, and Shire. Dr. Longhurst made these remarks in an editorial accompanying Dr. Banerji’s report (N Engl J Med. 2017 Feb 23;376[8]:788-9).
Lanadelumab, a monoclonal antibody that inhibits kallikrein, reduced attacks of hereditary angioedema with C1 inhibitor deficiency by 88%-100% in a small, phase I trial.
Hereditary angioedema with C1 inhibitor deficiency is a rare disorder characterized by unpredictable, recurrent, and potentially life-threatening episodes of subcutaneous or submucosal swelling, typically affecting the hands and feet, abdomen, face, larynx, or genitourinary tract. It is caused by a deficiency or dysfunction of the C1 inhibitor, which regulates the complement, coagulation, and kallikrein-kinin cascades.
They performed a multicenter, double-blind, randomized study to assess the safety and adverse-effect profile of four doses of this new agent or placebo in 37 adults (aged 18-71 years, mean age, 39.9 years) who received two injections, 2 weeks apart, and were followed for 6 weeks. Given their histories, all the study participants had “a reasonable probability of having one or more attacks” during the study period, the researchers noted.
Four participants received a 30-mg dose, 4 received a 100-mg dose, 5 received a 300-mg dose, 11 received a 400-mg dose, and 13 received placebo.
There were no serious adverse events, no deaths, and no discontinuations of the study medication because of an adverse effect. One patient each developed severe adverse events: pain at the injection site that lasted for 1 minute and headache plus night sweats.
Pharmacodynamic assessments showed that lanadelumab inhibited kallikrein in a linear, dose-dependent manner, and the two higher doses reduced levels of cleaved high-molecular-weight kininogen to those reported in healthy control subjects. At the same time, the two higher doses decreased the number of attacks by 88% and 100%, respectively, compared with placebo.
All the patients in the 300-mg group and 9 of the 11 in the 400-mg group had no attacks during the study period, the investigators said.
These findings “provide proof of concept that lanadelumab has the potential to correct the pathophysiological abnormality underlying attacks of angioedema and may be a new therapeutic option for hereditary angioedema with C1 inhibitor deficiency,” Dr. Banerji and her associates said.
The HELP Study, a phase III trial assessing the safety and efficacy of 6 months of lanadelumab treatment, is now underway, they added.
The trial was sponsored by Dyax, which also participated in the study design, data collection and interpretation, and writing of the results. Dr. Banerji reported ties to Alnylam Pharmaceuticals, CSL Behring, Dyax, and Shire; her associates reported ties to numerous industry sources.
Lanadelumab, a monoclonal antibody that inhibits kallikrein, reduced attacks of hereditary angioedema with C1 inhibitor deficiency by 88%-100% in a small, phase I trial.
Hereditary angioedema with C1 inhibitor deficiency is a rare disorder characterized by unpredictable, recurrent, and potentially life-threatening episodes of subcutaneous or submucosal swelling, typically affecting the hands and feet, abdomen, face, larynx, or genitourinary tract. It is caused by a deficiency or dysfunction of the C1 inhibitor, which regulates the complement, coagulation, and kallikrein-kinin cascades.
They performed a multicenter, double-blind, randomized study to assess the safety and adverse-effect profile of four doses of this new agent or placebo in 37 adults (aged 18-71 years, mean age, 39.9 years) who received two injections, 2 weeks apart, and were followed for 6 weeks. Given their histories, all the study participants had “a reasonable probability of having one or more attacks” during the study period, the researchers noted.
Four participants received a 30-mg dose, 4 received a 100-mg dose, 5 received a 300-mg dose, 11 received a 400-mg dose, and 13 received placebo.
There were no serious adverse events, no deaths, and no discontinuations of the study medication because of an adverse effect. One patient each developed severe adverse events: pain at the injection site that lasted for 1 minute and headache plus night sweats.
Pharmacodynamic assessments showed that lanadelumab inhibited kallikrein in a linear, dose-dependent manner, and the two higher doses reduced levels of cleaved high-molecular-weight kininogen to those reported in healthy control subjects. At the same time, the two higher doses decreased the number of attacks by 88% and 100%, respectively, compared with placebo.
All the patients in the 300-mg group and 9 of the 11 in the 400-mg group had no attacks during the study period, the investigators said.
These findings “provide proof of concept that lanadelumab has the potential to correct the pathophysiological abnormality underlying attacks of angioedema and may be a new therapeutic option for hereditary angioedema with C1 inhibitor deficiency,” Dr. Banerji and her associates said.
The HELP Study, a phase III trial assessing the safety and efficacy of 6 months of lanadelumab treatment, is now underway, they added.
The trial was sponsored by Dyax, which also participated in the study design, data collection and interpretation, and writing of the results. Dr. Banerji reported ties to Alnylam Pharmaceuticals, CSL Behring, Dyax, and Shire; her associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Lanadelumab, a monoclonal antibody that inhibits kallikrein, reduced attacks of hereditary angioedema with C1 inhibitor deficiency by 88%-100%.
Major finding: All the patients in the 300-mg group and 9 of the 11 in the 400-mg group had no angioedema attacks during the study period.
Data source: A multicenter, randomized, double-blind, placebo-controlled phase Ib trial involving 37 adults who had hereditary angioedema with C1 inhibitor deficiency.
Disclosures: The trial was sponsored by Dyax, which also participated in the study design, data collection and interpretation, and writing the results. Dr. Banerji reported ties to Alnylam Pharmaceuticals, CSL Behring, Dyax, and Shire; her associates reported ties to numerous industry sources.
Nonthoracic MRI safe in patients with cardiac devices
Nonthoracic MRI was safe in patients who had implanted non–MRI-conditional pacemakers or implantable cardioverter defibrillators, as long as they followed a specific safety protocol before and after the imaging procedure, according to a report published online Feb. 23 in the New England Journal of Medicine.
Patients with implanted cardiac devices have long been advised to avoid MRI because of the potential for the magnetic field to induce heating of the cardiac leads, which could in turn produce thermal injury to the myocardium and adversely affect the device’s function. Certain cardiac devices that have been proved to pose no such hazards have been designated by the Food and Drug Administration as “MRI conditional.” However, an estimated 2 million patients in the United States and another 6 million worldwide have devices that are not MRI conditional, and at least half of these patients are predicted to require an MRI during their lifetimes, said Robert J. Russo, MD, PhD, of Scripps Research Institute and the La Jolla (Calif.) Cardiovascular Research Institute, and his associates.
The MagnaSafe Registry was established to monitor device-related clinical events and device alterations among adults undergoing nonthoracic MRIs at 1.5 T. Dr. Russo and his associates analyzed data in this registry from 19 medical centers during a 5-year period. They assessed 1,000 MRIs in 818 patients with pacemakers and 500 MRIs in 428 patients with implantable cardioverter defibrillators who were followed for 6 months after the imaging procedures. Most of these scans involved the brain or spine, and the median duration of exposure to the magnetic field was 44 minutes.
According to the safety protocol, all devices were interrogated immediately before the MRI and, depending on those results, were programmed to no pacing or asynchronous pacing during the scan with all tachycardia and bradycardia therapies inactivated. Immediately after the scan, all devices were reprogrammed to baseline settings, a full device interrogation was repeated, and, if necessary, further reprogramming was performed to maintain adequate pacing and sensing. A physician, nurse practitioner, or physician’s assistant with cardiac expertise attended each scan.
There were no deaths, lead failures requiring immediate replacement, losses of capture, or full electrical resets associated with any of the 1,500 MRI scans.
Four patients developed atrial fibrillation, and two developed atrial flutter, during or after the MRI; three returned to sinus rhythm while still in the scanning room, and the other three did so within 49 hours. There were six cases requiring partial generator electrical resets. “Changes in device settings were common, but relatively few exceeded our prespecified threshold criteria for a clinically important change,” Dr. Russo and his associates wrote (N Engl J Med. 2017;376[8]:755-64).
Four patients reported feeling discomfort at the implant site during MRI, including one who felt a heating sensation and was removed from the scanner before completing the procedure. None of them had any further problems.
Some experts have suggested that to allow patients with cardiac devices to undergo MRI, the generators and leads could be removed before the procedure and replaced afterward. The findings of this study show that undergoing a nonthoracic MRI using this protocol would likely be a safer alternative, the investigators added.
This work was supported by St. Jude Medical, Biotronik, Boston Scientific, the Hewitt Foundation for Medical Research, and several philanthropic gifts. Dr. Russo reported ties to St. Jude Medical, Biotronik, Boston Scientific, and the Hewitt Foundation, and his associates reported ties to numerous industry sources.
Nonthoracic MRI was safe in patients who had implanted non–MRI-conditional pacemakers or implantable cardioverter defibrillators, as long as they followed a specific safety protocol before and after the imaging procedure, according to a report published online Feb. 23 in the New England Journal of Medicine.
Patients with implanted cardiac devices have long been advised to avoid MRI because of the potential for the magnetic field to induce heating of the cardiac leads, which could in turn produce thermal injury to the myocardium and adversely affect the device’s function. Certain cardiac devices that have been proved to pose no such hazards have been designated by the Food and Drug Administration as “MRI conditional.” However, an estimated 2 million patients in the United States and another 6 million worldwide have devices that are not MRI conditional, and at least half of these patients are predicted to require an MRI during their lifetimes, said Robert J. Russo, MD, PhD, of Scripps Research Institute and the La Jolla (Calif.) Cardiovascular Research Institute, and his associates.
The MagnaSafe Registry was established to monitor device-related clinical events and device alterations among adults undergoing nonthoracic MRIs at 1.5 T. Dr. Russo and his associates analyzed data in this registry from 19 medical centers during a 5-year period. They assessed 1,000 MRIs in 818 patients with pacemakers and 500 MRIs in 428 patients with implantable cardioverter defibrillators who were followed for 6 months after the imaging procedures. Most of these scans involved the brain or spine, and the median duration of exposure to the magnetic field was 44 minutes.
According to the safety protocol, all devices were interrogated immediately before the MRI and, depending on those results, were programmed to no pacing or asynchronous pacing during the scan with all tachycardia and bradycardia therapies inactivated. Immediately after the scan, all devices were reprogrammed to baseline settings, a full device interrogation was repeated, and, if necessary, further reprogramming was performed to maintain adequate pacing and sensing. A physician, nurse practitioner, or physician’s assistant with cardiac expertise attended each scan.
There were no deaths, lead failures requiring immediate replacement, losses of capture, or full electrical resets associated with any of the 1,500 MRI scans.
Four patients developed atrial fibrillation, and two developed atrial flutter, during or after the MRI; three returned to sinus rhythm while still in the scanning room, and the other three did so within 49 hours. There were six cases requiring partial generator electrical resets. “Changes in device settings were common, but relatively few exceeded our prespecified threshold criteria for a clinically important change,” Dr. Russo and his associates wrote (N Engl J Med. 2017;376[8]:755-64).
Four patients reported feeling discomfort at the implant site during MRI, including one who felt a heating sensation and was removed from the scanner before completing the procedure. None of them had any further problems.
Some experts have suggested that to allow patients with cardiac devices to undergo MRI, the generators and leads could be removed before the procedure and replaced afterward. The findings of this study show that undergoing a nonthoracic MRI using this protocol would likely be a safer alternative, the investigators added.
This work was supported by St. Jude Medical, Biotronik, Boston Scientific, the Hewitt Foundation for Medical Research, and several philanthropic gifts. Dr. Russo reported ties to St. Jude Medical, Biotronik, Boston Scientific, and the Hewitt Foundation, and his associates reported ties to numerous industry sources.
Nonthoracic MRI was safe in patients who had implanted non–MRI-conditional pacemakers or implantable cardioverter defibrillators, as long as they followed a specific safety protocol before and after the imaging procedure, according to a report published online Feb. 23 in the New England Journal of Medicine.
Patients with implanted cardiac devices have long been advised to avoid MRI because of the potential for the magnetic field to induce heating of the cardiac leads, which could in turn produce thermal injury to the myocardium and adversely affect the device’s function. Certain cardiac devices that have been proved to pose no such hazards have been designated by the Food and Drug Administration as “MRI conditional.” However, an estimated 2 million patients in the United States and another 6 million worldwide have devices that are not MRI conditional, and at least half of these patients are predicted to require an MRI during their lifetimes, said Robert J. Russo, MD, PhD, of Scripps Research Institute and the La Jolla (Calif.) Cardiovascular Research Institute, and his associates.
The MagnaSafe Registry was established to monitor device-related clinical events and device alterations among adults undergoing nonthoracic MRIs at 1.5 T. Dr. Russo and his associates analyzed data in this registry from 19 medical centers during a 5-year period. They assessed 1,000 MRIs in 818 patients with pacemakers and 500 MRIs in 428 patients with implantable cardioverter defibrillators who were followed for 6 months after the imaging procedures. Most of these scans involved the brain or spine, and the median duration of exposure to the magnetic field was 44 minutes.
According to the safety protocol, all devices were interrogated immediately before the MRI and, depending on those results, were programmed to no pacing or asynchronous pacing during the scan with all tachycardia and bradycardia therapies inactivated. Immediately after the scan, all devices were reprogrammed to baseline settings, a full device interrogation was repeated, and, if necessary, further reprogramming was performed to maintain adequate pacing and sensing. A physician, nurse practitioner, or physician’s assistant with cardiac expertise attended each scan.
There were no deaths, lead failures requiring immediate replacement, losses of capture, or full electrical resets associated with any of the 1,500 MRI scans.
Four patients developed atrial fibrillation, and two developed atrial flutter, during or after the MRI; three returned to sinus rhythm while still in the scanning room, and the other three did so within 49 hours. There were six cases requiring partial generator electrical resets. “Changes in device settings were common, but relatively few exceeded our prespecified threshold criteria for a clinically important change,” Dr. Russo and his associates wrote (N Engl J Med. 2017;376[8]:755-64).
Four patients reported feeling discomfort at the implant site during MRI, including one who felt a heating sensation and was removed from the scanner before completing the procedure. None of them had any further problems.
Some experts have suggested that to allow patients with cardiac devices to undergo MRI, the generators and leads could be removed before the procedure and replaced afterward. The findings of this study show that undergoing a nonthoracic MRI using this protocol would likely be a safer alternative, the investigators added.
This work was supported by St. Jude Medical, Biotronik, Boston Scientific, the Hewitt Foundation for Medical Research, and several philanthropic gifts. Dr. Russo reported ties to St. Jude Medical, Biotronik, Boston Scientific, and the Hewitt Foundation, and his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Nonthoracic MRI was safe in patients who had implanted non–MRI-conditional pacemakers or ICDs, as long as they followed a specific safety protocol before and after the imaging procedure.
Key numerical finding: No deaths, lead failures requiring immediate replacement, losses of capture, or full electrical resets were tied to any of the 1,500 MRI scans.
Data source: A U.S. registry–based cohort study of 1,000 MRIs involving patients with pacemakers and 500 MRIs involving patients with ICDs, performed during a 5-year period.
Disclosures: This work was supported by St. Jude Medical, Biotronik, Boston Scientific, the Hewitt Foundation for Medical Research, and several philanthropic gifts. Dr. Russo reported ties to St. Jude Medical, Biotronik, Boston Scientific, and the Hewitt Foundation, and his associates reported ties to numerous industry sources.
Osimertinib helps NSCLC that progresses despite EGFR-TKIs
Osimertinib, an epidermal growth factor receptor tyrosine kinase inhibitor selective for both EGFR and T790M mutations that render cancers resistant to EGFR-TKIs, yielded a high overall response rate, “encouraging” progression-free survival, and a durable treatment response in advanced non–small cell lung cancer (NSCLC) that had progressed despite EGFR-TKI therapy, according to a report published online Feb. 21 in the Journal of Clinical Oncology.
In a manufacturer-sponsored, open-label phase II trial, 198 patients in 10 countries took 80 mg of oral osimertinib once daily for a median duration of 13.2 months (range, 1-18 months). The overall response rate was 62%, and the median duration of response was 15.2 months. The disease control rate was 90%, said James Chih-Hsin Yang, MD, PhD, of National Taiwan Hospital, Taipei, and his associates.
The median progression-free survival was 12.3 months, and the treatment benefit was generally consistent across all subgroups of patients regardless of age, smoking status, previous therapies, and duration of treatment. Questionnaire responses showed that patients “had consistent and sustained improvements in key lung cancer symptoms including dyspnea, cough, chest pain, and pain in the arm or shoulder,” as well as in global health status and physical functioning. This is particularly noteworthy because some patients had received “many (up to 11) lines of cancer therapy before osimertinib,” the investigators reported (J Clin Oncol. 2017 Feb 21. doi: 10.1200/jco.2016.70.3223).
“We also report encouraging systemic progression-free survival with osimertinib in patients with CNS metastases, and a high CNS response rate (64%) in those with measurable CNS lesions,” they wrote. This finding is particularly important “because new pharmacologic strategies are needed to treat brain metastases, given the long-term complications of brain radiation,” they added.
Osimertinib was generally well tolerated, with 21% of patients having adverse effects leading to dose interruptions and 5% to dose reductions. Nine patients (3%) discontinued the agent because of adverse effects, which included interstitial lung disease (3 fatal cases), QT prolongation, a reduced neutrophil count, and severe vomiting and diarrhea.
This trial was sponsored by AstraZeneca. Dr. Yang and his associates reported ties to numerous industry sources.
Osimertinib, an epidermal growth factor receptor tyrosine kinase inhibitor selective for both EGFR and T790M mutations that render cancers resistant to EGFR-TKIs, yielded a high overall response rate, “encouraging” progression-free survival, and a durable treatment response in advanced non–small cell lung cancer (NSCLC) that had progressed despite EGFR-TKI therapy, according to a report published online Feb. 21 in the Journal of Clinical Oncology.
In a manufacturer-sponsored, open-label phase II trial, 198 patients in 10 countries took 80 mg of oral osimertinib once daily for a median duration of 13.2 months (range, 1-18 months). The overall response rate was 62%, and the median duration of response was 15.2 months. The disease control rate was 90%, said James Chih-Hsin Yang, MD, PhD, of National Taiwan Hospital, Taipei, and his associates.
The median progression-free survival was 12.3 months, and the treatment benefit was generally consistent across all subgroups of patients regardless of age, smoking status, previous therapies, and duration of treatment. Questionnaire responses showed that patients “had consistent and sustained improvements in key lung cancer symptoms including dyspnea, cough, chest pain, and pain in the arm or shoulder,” as well as in global health status and physical functioning. This is particularly noteworthy because some patients had received “many (up to 11) lines of cancer therapy before osimertinib,” the investigators reported (J Clin Oncol. 2017 Feb 21. doi: 10.1200/jco.2016.70.3223).
“We also report encouraging systemic progression-free survival with osimertinib in patients with CNS metastases, and a high CNS response rate (64%) in those with measurable CNS lesions,” they wrote. This finding is particularly important “because new pharmacologic strategies are needed to treat brain metastases, given the long-term complications of brain radiation,” they added.
Osimertinib was generally well tolerated, with 21% of patients having adverse effects leading to dose interruptions and 5% to dose reductions. Nine patients (3%) discontinued the agent because of adverse effects, which included interstitial lung disease (3 fatal cases), QT prolongation, a reduced neutrophil count, and severe vomiting and diarrhea.
This trial was sponsored by AstraZeneca. Dr. Yang and his associates reported ties to numerous industry sources.
Osimertinib, an epidermal growth factor receptor tyrosine kinase inhibitor selective for both EGFR and T790M mutations that render cancers resistant to EGFR-TKIs, yielded a high overall response rate, “encouraging” progression-free survival, and a durable treatment response in advanced non–small cell lung cancer (NSCLC) that had progressed despite EGFR-TKI therapy, according to a report published online Feb. 21 in the Journal of Clinical Oncology.
In a manufacturer-sponsored, open-label phase II trial, 198 patients in 10 countries took 80 mg of oral osimertinib once daily for a median duration of 13.2 months (range, 1-18 months). The overall response rate was 62%, and the median duration of response was 15.2 months. The disease control rate was 90%, said James Chih-Hsin Yang, MD, PhD, of National Taiwan Hospital, Taipei, and his associates.
The median progression-free survival was 12.3 months, and the treatment benefit was generally consistent across all subgroups of patients regardless of age, smoking status, previous therapies, and duration of treatment. Questionnaire responses showed that patients “had consistent and sustained improvements in key lung cancer symptoms including dyspnea, cough, chest pain, and pain in the arm or shoulder,” as well as in global health status and physical functioning. This is particularly noteworthy because some patients had received “many (up to 11) lines of cancer therapy before osimertinib,” the investigators reported (J Clin Oncol. 2017 Feb 21. doi: 10.1200/jco.2016.70.3223).
“We also report encouraging systemic progression-free survival with osimertinib in patients with CNS metastases, and a high CNS response rate (64%) in those with measurable CNS lesions,” they wrote. This finding is particularly important “because new pharmacologic strategies are needed to treat brain metastases, given the long-term complications of brain radiation,” they added.
Osimertinib was generally well tolerated, with 21% of patients having adverse effects leading to dose interruptions and 5% to dose reductions. Nine patients (3%) discontinued the agent because of adverse effects, which included interstitial lung disease (3 fatal cases), QT prolongation, a reduced neutrophil count, and severe vomiting and diarrhea.
This trial was sponsored by AstraZeneca. Dr. Yang and his associates reported ties to numerous industry sources.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Osimertinib yielded a high overall response rate, “encouraging” progression-free survival, and a durable treatment response in advanced non–small cell lung cancer that had progressed despite EGFR-TKI therapy.
Major finding: The overall response rate was 62%, and the median duration of response was 15.2 months.
Data source: An international manufacturer-sponsored, open-label phase II trial involving 198 patients treated for a mean of 13.2 months.
Disclosures: This trial was sponsored by AstraZeneca. Dr. Yang and his associates reported ties to numerous industry sources.
Hormone therapy prolongs PFS in rare low-grade serous cancer
Hormone maintenance therapy, when given after primary cytoreductive surgery and platinum-based chemotherapy, prolonged progression-free survival among women who had low-grade serous carcinoma of the ovary or peritoneum, a study showed.
Low-grade serous carcinoma (LGSC) is a rare histologic subtype that is somewhat resistant to conventional chemotherapy, so researchers have been searching for alternative or add-on treatments. To examine whether hormone maintenance therapy would be beneficial, the investigators analyzed information from a longitudinal database of patients with the malignancy who were treated at a single medical center.
They focused on 203 patients diagnosed as having stage II-IV disease of the ovary or peritoneum between 1981 and 2013, who underwent primary cytoreductive surgery followed by platinum-based chemotherapy. Seventy of these patients received hormone maintenance therapy for a median of 33 months (range, 1-223 months), taking letrozole, tamoxifen, leuprolide, anastrozole, medroxyprogesterone, or some combination of these agents. The remaining 133 patients took no hormone therapy and served as a control group, said David M. Gershenson, MD, and his associates at the University of Texas M.D. Anderson Cancer Center, Houston.
Women who took hormone maintenance therapy had a median progression-free survival of 64.9 months, compared with 26.4 months for the control group. This benefit was similar between women who had persistent disease after completing chemotherapy and those who were clinically disease free after completing chemotherapy, the investigators reported (J Clin Oncol. 2017 Feb 21. doi: 10.1200/jco.2016.71.0632).
“The findings of this hypothesis-generating study are potentially practice changing and warrant using a prospective trial design. A phase III randomized trial is currently under development” to compare hormone therapy against placebo in women with LGSC, Dr. Gersehnson and his associates noted.
They added that reports during the last decade showing that LGSC is resistant to platinum-based chemotherapy have led some clinicians to conclude that it is of no benefit at all and should be abandoned in this patient population. “In our view, that perspective is premature based on available data. Although LGSC is indolent and not as chemotherapy sensitive as high-grade serous carcinoma, it is not entirely chemotherapy resistant,” they wrote. Some women do respond, while “a high proportion … have stable disease for a period of time.”
Hormone maintenance therapy, when given after primary cytoreductive surgery and platinum-based chemotherapy, prolonged progression-free survival among women who had low-grade serous carcinoma of the ovary or peritoneum, a study showed.
Low-grade serous carcinoma (LGSC) is a rare histologic subtype that is somewhat resistant to conventional chemotherapy, so researchers have been searching for alternative or add-on treatments. To examine whether hormone maintenance therapy would be beneficial, the investigators analyzed information from a longitudinal database of patients with the malignancy who were treated at a single medical center.
They focused on 203 patients diagnosed as having stage II-IV disease of the ovary or peritoneum between 1981 and 2013, who underwent primary cytoreductive surgery followed by platinum-based chemotherapy. Seventy of these patients received hormone maintenance therapy for a median of 33 months (range, 1-223 months), taking letrozole, tamoxifen, leuprolide, anastrozole, medroxyprogesterone, or some combination of these agents. The remaining 133 patients took no hormone therapy and served as a control group, said David M. Gershenson, MD, and his associates at the University of Texas M.D. Anderson Cancer Center, Houston.
Women who took hormone maintenance therapy had a median progression-free survival of 64.9 months, compared with 26.4 months for the control group. This benefit was similar between women who had persistent disease after completing chemotherapy and those who were clinically disease free after completing chemotherapy, the investigators reported (J Clin Oncol. 2017 Feb 21. doi: 10.1200/jco.2016.71.0632).
“The findings of this hypothesis-generating study are potentially practice changing and warrant using a prospective trial design. A phase III randomized trial is currently under development” to compare hormone therapy against placebo in women with LGSC, Dr. Gersehnson and his associates noted.
They added that reports during the last decade showing that LGSC is resistant to platinum-based chemotherapy have led some clinicians to conclude that it is of no benefit at all and should be abandoned in this patient population. “In our view, that perspective is premature based on available data. Although LGSC is indolent and not as chemotherapy sensitive as high-grade serous carcinoma, it is not entirely chemotherapy resistant,” they wrote. Some women do respond, while “a high proportion … have stable disease for a period of time.”
Hormone maintenance therapy, when given after primary cytoreductive surgery and platinum-based chemotherapy, prolonged progression-free survival among women who had low-grade serous carcinoma of the ovary or peritoneum, a study showed.
Low-grade serous carcinoma (LGSC) is a rare histologic subtype that is somewhat resistant to conventional chemotherapy, so researchers have been searching for alternative or add-on treatments. To examine whether hormone maintenance therapy would be beneficial, the investigators analyzed information from a longitudinal database of patients with the malignancy who were treated at a single medical center.
They focused on 203 patients diagnosed as having stage II-IV disease of the ovary or peritoneum between 1981 and 2013, who underwent primary cytoreductive surgery followed by platinum-based chemotherapy. Seventy of these patients received hormone maintenance therapy for a median of 33 months (range, 1-223 months), taking letrozole, tamoxifen, leuprolide, anastrozole, medroxyprogesterone, or some combination of these agents. The remaining 133 patients took no hormone therapy and served as a control group, said David M. Gershenson, MD, and his associates at the University of Texas M.D. Anderson Cancer Center, Houston.
Women who took hormone maintenance therapy had a median progression-free survival of 64.9 months, compared with 26.4 months for the control group. This benefit was similar between women who had persistent disease after completing chemotherapy and those who were clinically disease free after completing chemotherapy, the investigators reported (J Clin Oncol. 2017 Feb 21. doi: 10.1200/jco.2016.71.0632).
“The findings of this hypothesis-generating study are potentially practice changing and warrant using a prospective trial design. A phase III randomized trial is currently under development” to compare hormone therapy against placebo in women with LGSC, Dr. Gersehnson and his associates noted.
They added that reports during the last decade showing that LGSC is resistant to platinum-based chemotherapy have led some clinicians to conclude that it is of no benefit at all and should be abandoned in this patient population. “In our view, that perspective is premature based on available data. Although LGSC is indolent and not as chemotherapy sensitive as high-grade serous carcinoma, it is not entirely chemotherapy resistant,” they wrote. Some women do respond, while “a high proportion … have stable disease for a period of time.”
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Hormone maintenance therapy given after primary cytoreductive surgery and platinum-based chemotherapy prolonged progression-free survival among women who had low-grade serous carcinoma of the ovary or peritoneum.
Major finding: Women who took hormone maintenance therapy had a median progression-free survival of 64.9 months, compared with 26.4 months for the control group.
Data source: A cohort study involving 203 patients enrolled in a longitudinal database of rare low-grade serous tumors who were followed for a median of 71 months.
Disclosures: This study was supported in part by the Sara Brown Musselman Fund for Serous Ovarian Cancer Research and the National Cancer Institute. Dr. Gershenson reported ties to Johnson & Johnson, Pfizer, Biogen Idec, Celgene, AbbVie, GlaxoSmithKline, Merck, and Clovis Oncology. His associates reported ties to numerous industry sources.
Light therapy eases Parkinson’s-related sleep disturbances
Light therapy significantly reduced excessive daytime sleepiness, improved sleep quality, decreased overnight awakenings, shortened sleep latency, enhanced daytime alertness and activity level, and improved motor symptoms in patients with Parkinson’s disease, according to a report published online Feb. 20 in JAMA Neurology.
The noninvasive, nonpharmacologic treatment was well tolerated, and patient adherence was excellent in a small, multicenter, randomized controlled trial. Light therapy is widely available as a treatment for several sleep and psychiatric disorders and is “relatively easy to prescribe and incorporate into a clinical practice,” said Aleksandar Videnovic, MD, of the department of neurology at Massachusetts General Hospital and the division of sleep medicine at Harvard Medical School, both in Boston, and his associates.
To assess the safety and efficacy of light therapy as a novel treatment for PD, they studied 31 adults (age range, 32-77 years) who had a mean disease duration of 6 years. These study participants were randomly assigned to use 1 hour of exposure to 10,000 lux of bright light (16 patients in the intervention group) or 1 hour of exposure to less than 300 lux of dim red light (15 control subjects) every morning and every afternoon for 2 weeks.
The study participants – 13 men and 18 women – also wore actigraphy monitors all day and all night, completed daily sleep diaries, and noted daytime sleepiness in a log every 2 hours, 3 days per week.
Bright light significantly improved excessive daytime sleepiness as measured by the Epworth Sleepiness Scale and self-reported alertness during wake time, as well as several sleep metrics such as overall sleep quality, overnight awakenings, and ease of falling asleep. All the patients in the intervention group reported being more refreshed in the mornings during the study period, as compared with baseline.
Light therapy also improved overall PD severity as measured by the Unified Parkinson’s Disease Rating Scale, particularly in scores related to activities of daily living and motor symptoms. Moreover, this effect persisted during the 2-week washout period after treatment was discontinued, Dr. Videnovic and his associates said (JAMA Neurol. 2017 Feb 20. doi: 10.1001/jamaneurol.2016.5192).
The treatment was well tolerated. In the intervention group, one patient reported headache and another sleepiness, and in the control group one patient reported itchy eyes. The effects resolved spontaneously, and neither lead to treatment withdrawal.
“Based on these results, the next logical step is to optimize various parameters of light therapy (e.g., intensity, duration, and wavelength) not only for impaired sleep and alertness but also for other motor and nonmotor manifestation of PD,” the investigators wrote.
A major limitation of this study was that exposure to ambient light throughout the day was not measured. Some people in the control group received as much or even more light exposure than those assigned to bright-light therapy. “Future studies may be more strict in controlling such exposures,” Dr. Videnovic and his associates said.
This study was supported by the National Parkinson Foundation and the National Institutes of Health. Dr. Videnovic reported having no relevant financial disclosures. One of his associates reported ties to Merck, Phillips, Eisai, and Teva.
The study by Dr. Videnovic and his associates is important because it introduces a new concept into the much-studied phenomenon of sleep disturbances in Parkinson’s disease.
The authors demonstrated that chronobiological interventions can be used therapeutically in PD. Accounting for circadian physiology also sets a new standard for future studies of sleep, nighttime wakefulness, and daytime function not only in PD but, it is hoped, in other diseases as well.
Birgit Högl, MD, is with the department of neurology at the Medical University of Innsbruck (Austria). She reported receiving honoraria as a speaker, advisory board member, or consultant from UCB, Otsuka, Lundbeck, Lilly, Axovant, AbbVie, Mundipharma, Benevolent Bio, and Janssen Cilag, and travel support from Habel Medizintechnik and Vivisol. Dr. Högl made these remarks in an editorial (JAMA Neurol. 2017 Feb 20. doi: 10.1001/jamaneurol.2016.5519) accompanying the report by Dr. Videnovic and his colleagues.
The study by Dr. Videnovic and his associates is important because it introduces a new concept into the much-studied phenomenon of sleep disturbances in Parkinson’s disease.
The authors demonstrated that chronobiological interventions can be used therapeutically in PD. Accounting for circadian physiology also sets a new standard for future studies of sleep, nighttime wakefulness, and daytime function not only in PD but, it is hoped, in other diseases as well.
Birgit Högl, MD, is with the department of neurology at the Medical University of Innsbruck (Austria). She reported receiving honoraria as a speaker, advisory board member, or consultant from UCB, Otsuka, Lundbeck, Lilly, Axovant, AbbVie, Mundipharma, Benevolent Bio, and Janssen Cilag, and travel support from Habel Medizintechnik and Vivisol. Dr. Högl made these remarks in an editorial (JAMA Neurol. 2017 Feb 20. doi: 10.1001/jamaneurol.2016.5519) accompanying the report by Dr. Videnovic and his colleagues.
The study by Dr. Videnovic and his associates is important because it introduces a new concept into the much-studied phenomenon of sleep disturbances in Parkinson’s disease.
The authors demonstrated that chronobiological interventions can be used therapeutically in PD. Accounting for circadian physiology also sets a new standard for future studies of sleep, nighttime wakefulness, and daytime function not only in PD but, it is hoped, in other diseases as well.
Birgit Högl, MD, is with the department of neurology at the Medical University of Innsbruck (Austria). She reported receiving honoraria as a speaker, advisory board member, or consultant from UCB, Otsuka, Lundbeck, Lilly, Axovant, AbbVie, Mundipharma, Benevolent Bio, and Janssen Cilag, and travel support from Habel Medizintechnik and Vivisol. Dr. Högl made these remarks in an editorial (JAMA Neurol. 2017 Feb 20. doi: 10.1001/jamaneurol.2016.5519) accompanying the report by Dr. Videnovic and his colleagues.
Light therapy significantly reduced excessive daytime sleepiness, improved sleep quality, decreased overnight awakenings, shortened sleep latency, enhanced daytime alertness and activity level, and improved motor symptoms in patients with Parkinson’s disease, according to a report published online Feb. 20 in JAMA Neurology.
The noninvasive, nonpharmacologic treatment was well tolerated, and patient adherence was excellent in a small, multicenter, randomized controlled trial. Light therapy is widely available as a treatment for several sleep and psychiatric disorders and is “relatively easy to prescribe and incorporate into a clinical practice,” said Aleksandar Videnovic, MD, of the department of neurology at Massachusetts General Hospital and the division of sleep medicine at Harvard Medical School, both in Boston, and his associates.
To assess the safety and efficacy of light therapy as a novel treatment for PD, they studied 31 adults (age range, 32-77 years) who had a mean disease duration of 6 years. These study participants were randomly assigned to use 1 hour of exposure to 10,000 lux of bright light (16 patients in the intervention group) or 1 hour of exposure to less than 300 lux of dim red light (15 control subjects) every morning and every afternoon for 2 weeks.
The study participants – 13 men and 18 women – also wore actigraphy monitors all day and all night, completed daily sleep diaries, and noted daytime sleepiness in a log every 2 hours, 3 days per week.
Bright light significantly improved excessive daytime sleepiness as measured by the Epworth Sleepiness Scale and self-reported alertness during wake time, as well as several sleep metrics such as overall sleep quality, overnight awakenings, and ease of falling asleep. All the patients in the intervention group reported being more refreshed in the mornings during the study period, as compared with baseline.
Light therapy also improved overall PD severity as measured by the Unified Parkinson’s Disease Rating Scale, particularly in scores related to activities of daily living and motor symptoms. Moreover, this effect persisted during the 2-week washout period after treatment was discontinued, Dr. Videnovic and his associates said (JAMA Neurol. 2017 Feb 20. doi: 10.1001/jamaneurol.2016.5192).
The treatment was well tolerated. In the intervention group, one patient reported headache and another sleepiness, and in the control group one patient reported itchy eyes. The effects resolved spontaneously, and neither lead to treatment withdrawal.
“Based on these results, the next logical step is to optimize various parameters of light therapy (e.g., intensity, duration, and wavelength) not only for impaired sleep and alertness but also for other motor and nonmotor manifestation of PD,” the investigators wrote.
A major limitation of this study was that exposure to ambient light throughout the day was not measured. Some people in the control group received as much or even more light exposure than those assigned to bright-light therapy. “Future studies may be more strict in controlling such exposures,” Dr. Videnovic and his associates said.
This study was supported by the National Parkinson Foundation and the National Institutes of Health. Dr. Videnovic reported having no relevant financial disclosures. One of his associates reported ties to Merck, Phillips, Eisai, and Teva.
Light therapy significantly reduced excessive daytime sleepiness, improved sleep quality, decreased overnight awakenings, shortened sleep latency, enhanced daytime alertness and activity level, and improved motor symptoms in patients with Parkinson’s disease, according to a report published online Feb. 20 in JAMA Neurology.
The noninvasive, nonpharmacologic treatment was well tolerated, and patient adherence was excellent in a small, multicenter, randomized controlled trial. Light therapy is widely available as a treatment for several sleep and psychiatric disorders and is “relatively easy to prescribe and incorporate into a clinical practice,” said Aleksandar Videnovic, MD, of the department of neurology at Massachusetts General Hospital and the division of sleep medicine at Harvard Medical School, both in Boston, and his associates.
To assess the safety and efficacy of light therapy as a novel treatment for PD, they studied 31 adults (age range, 32-77 years) who had a mean disease duration of 6 years. These study participants were randomly assigned to use 1 hour of exposure to 10,000 lux of bright light (16 patients in the intervention group) or 1 hour of exposure to less than 300 lux of dim red light (15 control subjects) every morning and every afternoon for 2 weeks.
The study participants – 13 men and 18 women – also wore actigraphy monitors all day and all night, completed daily sleep diaries, and noted daytime sleepiness in a log every 2 hours, 3 days per week.
Bright light significantly improved excessive daytime sleepiness as measured by the Epworth Sleepiness Scale and self-reported alertness during wake time, as well as several sleep metrics such as overall sleep quality, overnight awakenings, and ease of falling asleep. All the patients in the intervention group reported being more refreshed in the mornings during the study period, as compared with baseline.
Light therapy also improved overall PD severity as measured by the Unified Parkinson’s Disease Rating Scale, particularly in scores related to activities of daily living and motor symptoms. Moreover, this effect persisted during the 2-week washout period after treatment was discontinued, Dr. Videnovic and his associates said (JAMA Neurol. 2017 Feb 20. doi: 10.1001/jamaneurol.2016.5192).
The treatment was well tolerated. In the intervention group, one patient reported headache and another sleepiness, and in the control group one patient reported itchy eyes. The effects resolved spontaneously, and neither lead to treatment withdrawal.
“Based on these results, the next logical step is to optimize various parameters of light therapy (e.g., intensity, duration, and wavelength) not only for impaired sleep and alertness but also for other motor and nonmotor manifestation of PD,” the investigators wrote.
A major limitation of this study was that exposure to ambient light throughout the day was not measured. Some people in the control group received as much or even more light exposure than those assigned to bright-light therapy. “Future studies may be more strict in controlling such exposures,” Dr. Videnovic and his associates said.
This study was supported by the National Parkinson Foundation and the National Institutes of Health. Dr. Videnovic reported having no relevant financial disclosures. One of his associates reported ties to Merck, Phillips, Eisai, and Teva.
FROM JAMA NEUROLOGY
Key clinical point:
Major finding: Compared with a control condition, bright light significantly improved excessive daytime sleepiness as measured by the Epworth Sleepiness Scale and self-reported alertness during wake time.
Data source: A randomized controlled trial involving 31 adults with Parkinson’s disease–related sleep disturbances.
Disclosures: This study was supported by the National Parkinson Foundation and the National Institutes of Health. Dr. Videnovic reported having no relevant financial disclosures. One of his associates reported ties to Merck, Phillips, Eisai, and Teva.
RA-BEAM trial shows that baricitinib improves RA symptoms, slows joint damage
Oral baricitinib, when added to standard rheumatoid arthritis treatment, improved symptoms and slowed the progression of joint damage, compared with both placebo and adalimumab, according to a report published online Feb. 15 in the New England Journal of Medicine.
The JAK inhibitor’s beneficial effects were noted as early as the first week of treatment and persisted throughout 1 year of follow-up in an international manufacturer-sponsored phase III trial (RA-BEAM) involving 1,305 adults with moderate to severe active rheumatoid arthritis (RA). The study participants had not responded to methotrexate and had never been treated with biologics; the majority had previously received at least two conventional synthetic disease-modifying antirheumatic drugs (DMARDs), said Peter C. Taylor, MD, PhD, of the Nuffield Department of Orthopedics, Rheumatology, and Musculoskeletal Sciences and the Kennedy Institute of Rheumatology at the University of Oxford (England), and his associates.

The primary efficacy endpoint – the proportion of patients at week 12 who showed a response of 20% or more according to American College of Rheumatology criteria (ACR20) – was 70% for baricitinib, compared with 40% for placebo. Baricitinib also was superior to placebo regarding all the major secondary efficacy endpoints of the study, including scores on the Health Assessment Questionnaire–Disability Index; results on the Disease Activity Score for 28 joints using high-sensitivity C-reactive protein (DAS28-CRP); remission according to the Simplified Disease Activity Index criteria; and the patients’ daily diary reports on duration and severity of morning joint stiffness, worst level of tiredness, and worst level of pain.
In addition, baricitinib was “considered to be significantly superior to adalimumab” according to the ACR20 (61% for adalimumab) and the DAS28-CRP responses at week 12. Both baricitinib and adalimumab were associated with significant reductions in the radiographic progression of structural joint damage, compared with placebo, at week 24 and week 52, the investigators reported (N Engl J Med. 2017;376:652-62).
Regarding safety outcomes, adverse events including infections were more frequent with baricitinib and adalimumab than with placebo, as were decreases in neutrophil counts, increases in aminotransferase and creatinine levels, and increases in low-density lipoprotein cholesterol levels. At 1 year, three patients in the baricitinib group and two in the adalimumab group discontinued treatment because of liver abnormalities.
Baricitinib is currently under consideration for approval by the U.S. Food and Drug Administration and was approved for marketing in the European Union on Feb. 13.
The RA-BEAM trial was supported by Eli Lilly and Incyte. Dr. Taylor reported receiving personal fees from Eli Lilly, and he and his associates reported ties to numerous other industry sources.
Oral baricitinib, when added to standard rheumatoid arthritis treatment, improved symptoms and slowed the progression of joint damage, compared with both placebo and adalimumab, according to a report published online Feb. 15 in the New England Journal of Medicine.
The JAK inhibitor’s beneficial effects were noted as early as the first week of treatment and persisted throughout 1 year of follow-up in an international manufacturer-sponsored phase III trial (RA-BEAM) involving 1,305 adults with moderate to severe active rheumatoid arthritis (RA). The study participants had not responded to methotrexate and had never been treated with biologics; the majority had previously received at least two conventional synthetic disease-modifying antirheumatic drugs (DMARDs), said Peter C. Taylor, MD, PhD, of the Nuffield Department of Orthopedics, Rheumatology, and Musculoskeletal Sciences and the Kennedy Institute of Rheumatology at the University of Oxford (England), and his associates.
The primary efficacy endpoint – the proportion of patients at week 12 who showed a response of 20% or more according to American College of Rheumatology criteria (ACR20) – was 70% for baricitinib, compared with 40% for placebo. Baricitinib also was superior to placebo regarding all the major secondary efficacy endpoints of the study, including scores on the Health Assessment Questionnaire–Disability Index; results on the Disease Activity Score for 28 joints using high-sensitivity C-reactive protein (DAS28-CRP); remission according to the Simplified Disease Activity Index criteria; and the patients’ daily diary reports on duration and severity of morning joint stiffness, worst level of tiredness, and worst level of pain.
In addition, baricitinib was “considered to be significantly superior to adalimumab” according to the ACR20 (61% for adalimumab) and the DAS28-CRP responses at week 12. Both baricitinib and adalimumab were associated with significant reductions in the radiographic progression of structural joint damage, compared with placebo, at week 24 and week 52, the investigators reported (N Engl J Med. 2017;376:652-62).
Regarding safety outcomes, adverse events including infections were more frequent with baricitinib and adalimumab than with placebo, as were decreases in neutrophil counts, increases in aminotransferase and creatinine levels, and increases in low-density lipoprotein cholesterol levels. At 1 year, three patients in the baricitinib group and two in the adalimumab group discontinued treatment because of liver abnormalities.
Baricitinib is currently under consideration for approval by the U.S. Food and Drug Administration and was approved for marketing in the European Union on Feb. 13.
The RA-BEAM trial was supported by Eli Lilly and Incyte. Dr. Taylor reported receiving personal fees from Eli Lilly, and he and his associates reported ties to numerous other industry sources.
Oral baricitinib, when added to standard rheumatoid arthritis treatment, improved symptoms and slowed the progression of joint damage, compared with both placebo and adalimumab, according to a report published online Feb. 15 in the New England Journal of Medicine.
The JAK inhibitor’s beneficial effects were noted as early as the first week of treatment and persisted throughout 1 year of follow-up in an international manufacturer-sponsored phase III trial (RA-BEAM) involving 1,305 adults with moderate to severe active rheumatoid arthritis (RA). The study participants had not responded to methotrexate and had never been treated with biologics; the majority had previously received at least two conventional synthetic disease-modifying antirheumatic drugs (DMARDs), said Peter C. Taylor, MD, PhD, of the Nuffield Department of Orthopedics, Rheumatology, and Musculoskeletal Sciences and the Kennedy Institute of Rheumatology at the University of Oxford (England), and his associates.
The primary efficacy endpoint – the proportion of patients at week 12 who showed a response of 20% or more according to American College of Rheumatology criteria (ACR20) – was 70% for baricitinib, compared with 40% for placebo. Baricitinib also was superior to placebo regarding all the major secondary efficacy endpoints of the study, including scores on the Health Assessment Questionnaire–Disability Index; results on the Disease Activity Score for 28 joints using high-sensitivity C-reactive protein (DAS28-CRP); remission according to the Simplified Disease Activity Index criteria; and the patients’ daily diary reports on duration and severity of morning joint stiffness, worst level of tiredness, and worst level of pain.
In addition, baricitinib was “considered to be significantly superior to adalimumab” according to the ACR20 (61% for adalimumab) and the DAS28-CRP responses at week 12. Both baricitinib and adalimumab were associated with significant reductions in the radiographic progression of structural joint damage, compared with placebo, at week 24 and week 52, the investigators reported (N Engl J Med. 2017;376:652-62).
Regarding safety outcomes, adverse events including infections were more frequent with baricitinib and adalimumab than with placebo, as were decreases in neutrophil counts, increases in aminotransferase and creatinine levels, and increases in low-density lipoprotein cholesterol levels. At 1 year, three patients in the baricitinib group and two in the adalimumab group discontinued treatment because of liver abnormalities.
Baricitinib is currently under consideration for approval by the U.S. Food and Drug Administration and was approved for marketing in the European Union on Feb. 13.
The RA-BEAM trial was supported by Eli Lilly and Incyte. Dr. Taylor reported receiving personal fees from Eli Lilly, and he and his associates reported ties to numerous other industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: The primary efficacy end point – the proportion of patients at week 12 who showed an ACR20 response – was 70% for baricitinib, compared with 40% for placebo.
Data source: A manufacturer-sponsored, international, randomized, double-blind phase III clinical trial involving 1,305 adults with moderate to severe active RA.
Disclosures: This study was supported by Eli Lilly and Incyte. Dr. Taylor reported receiving personal fees from Eli Lilly, and he and his associates reported ties to numerous other industry sources.
More periviable infants survive without neurodevelopmental impairment
Among periviable infants born at 11 tertiary care centers in 2000 through 2011, the rate of survival without neurodevelopmental impairment increased a small but significant 4%, according to a report published online Feb. 16 in the New England Journal of Medicine.
The rate of survival with neurodevelopmental impairment also increased, although to a lesser extent (1%).
“These findings are important for guiding counseling and decision making with respect to periviable birth. Prognosis continues to be guarded; in the most recent epoch [time period in our study], mortality was 64%, and 43% of surviving infants had neurodevelopmental impairment,” they noted.
The investigators defined such impairment as moderate or severe cerebral palsy, Gross Motor Function Classification System level of at least 2 on a scale of 1-5, profound hearing loss requiring amplification in both ears, profound visual impairment in both eyes, or cognitive impairment such as a Mental Developmental Index score of less than 70 or a Cognitive Composite score of less than 85.
To examine time trends in the outcomes of periviable infants, Dr. Younge of Duke University, Durham, N.C., and her associates analyzed data from the network’s registry of births at 11 academic tertiary care centers nationwide. They focused on 4,274 infants who were born during 3 epochs – 2000-2003, 2004-2007, and 2008-2011 – and were evaluated for motor function, sensory impairment, and cognitive delay at a corrected age of 18-22 months.
The percentage of infants who survived without neurodevelopmental impairment increased over time, from 16% during the first epoch to 20% during the third epoch. However, the percentage who survived with neurodevelopmental impairment also increased, from 15% during the first epoch to 16% during the third epoch (New Engl. J. Med. 2017 Feb 16. doi: 10.1056/NEJMoa1605566).
The rates of active treatment of these periviable infants didn’t change significantly over time. Overall, 22% of infants born at 22 weeks, 71% of those born at 23 weeks, and 95% of those born at 24 weeks received active treatment at birth. Therefore, the overall decrease in mortality and the 4% improvement in neurodevelopmental outcomes wasn’t attributable to greater use of active treatment for periviable infants over time, said Dr. Younge and her associates.
Despite these small but significant improvements in outcomes, “the incidence of death, neurodevelopmental impairment, and other adverse outcomes remains high in this population,” they noted.
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institutes of Health, the National Center for Research Resources, and the National Center for Advancing Translational Sciences for the Neonatal Research Network’s Generic Database and Follow-up Studies. Dr. Younge reported having no relevant financial disclosures; two of her associates reported ties to Pediatrix Medical Group and rEVO Biologics.
The study by Younge et al. was limited in that it only included infants born in 11 academic tertiary care medical centers.
This study population represents only 4%-5% of periviable infants born in the United States, so the findings are not generalizable.
Prakesh S. Shah, MD, is in the department of pediatrics and the Institute of Health Policy, Management, and Evaluation at Mount Sinai Hospital, Toronto, and the University of Toronto. He reported having no relevant financial disclosures. Dr. Shah made these remarks in an editorial accompanying Dr. Younge’s report (N Engl J Med. 2017 Feb 16. doi: 10.1056/NEJMe1616539).
The study by Younge et al. was limited in that it only included infants born in 11 academic tertiary care medical centers.
This study population represents only 4%-5% of periviable infants born in the United States, so the findings are not generalizable.
Prakesh S. Shah, MD, is in the department of pediatrics and the Institute of Health Policy, Management, and Evaluation at Mount Sinai Hospital, Toronto, and the University of Toronto. He reported having no relevant financial disclosures. Dr. Shah made these remarks in an editorial accompanying Dr. Younge’s report (N Engl J Med. 2017 Feb 16. doi: 10.1056/NEJMe1616539).
The study by Younge et al. was limited in that it only included infants born in 11 academic tertiary care medical centers.
This study population represents only 4%-5% of periviable infants born in the United States, so the findings are not generalizable.
Prakesh S. Shah, MD, is in the department of pediatrics and the Institute of Health Policy, Management, and Evaluation at Mount Sinai Hospital, Toronto, and the University of Toronto. He reported having no relevant financial disclosures. Dr. Shah made these remarks in an editorial accompanying Dr. Younge’s report (N Engl J Med. 2017 Feb 16. doi: 10.1056/NEJMe1616539).
Among periviable infants born at 11 tertiary care centers in 2000 through 2011, the rate of survival without neurodevelopmental impairment increased a small but significant 4%, according to a report published online Feb. 16 in the New England Journal of Medicine.
The rate of survival with neurodevelopmental impairment also increased, although to a lesser extent (1%).
“These findings are important for guiding counseling and decision making with respect to periviable birth. Prognosis continues to be guarded; in the most recent epoch [time period in our study], mortality was 64%, and 43% of surviving infants had neurodevelopmental impairment,” they noted.
The investigators defined such impairment as moderate or severe cerebral palsy, Gross Motor Function Classification System level of at least 2 on a scale of 1-5, profound hearing loss requiring amplification in both ears, profound visual impairment in both eyes, or cognitive impairment such as a Mental Developmental Index score of less than 70 or a Cognitive Composite score of less than 85.
To examine time trends in the outcomes of periviable infants, Dr. Younge of Duke University, Durham, N.C., and her associates analyzed data from the network’s registry of births at 11 academic tertiary care centers nationwide. They focused on 4,274 infants who were born during 3 epochs – 2000-2003, 2004-2007, and 2008-2011 – and were evaluated for motor function, sensory impairment, and cognitive delay at a corrected age of 18-22 months.
The percentage of infants who survived without neurodevelopmental impairment increased over time, from 16% during the first epoch to 20% during the third epoch. However, the percentage who survived with neurodevelopmental impairment also increased, from 15% during the first epoch to 16% during the third epoch (New Engl. J. Med. 2017 Feb 16. doi: 10.1056/NEJMoa1605566).
The rates of active treatment of these periviable infants didn’t change significantly over time. Overall, 22% of infants born at 22 weeks, 71% of those born at 23 weeks, and 95% of those born at 24 weeks received active treatment at birth. Therefore, the overall decrease in mortality and the 4% improvement in neurodevelopmental outcomes wasn’t attributable to greater use of active treatment for periviable infants over time, said Dr. Younge and her associates.
Despite these small but significant improvements in outcomes, “the incidence of death, neurodevelopmental impairment, and other adverse outcomes remains high in this population,” they noted.
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institutes of Health, the National Center for Research Resources, and the National Center for Advancing Translational Sciences for the Neonatal Research Network’s Generic Database and Follow-up Studies. Dr. Younge reported having no relevant financial disclosures; two of her associates reported ties to Pediatrix Medical Group and rEVO Biologics.
Among periviable infants born at 11 tertiary care centers in 2000 through 2011, the rate of survival without neurodevelopmental impairment increased a small but significant 4%, according to a report published online Feb. 16 in the New England Journal of Medicine.
The rate of survival with neurodevelopmental impairment also increased, although to a lesser extent (1%).
“These findings are important for guiding counseling and decision making with respect to periviable birth. Prognosis continues to be guarded; in the most recent epoch [time period in our study], mortality was 64%, and 43% of surviving infants had neurodevelopmental impairment,” they noted.
The investigators defined such impairment as moderate or severe cerebral palsy, Gross Motor Function Classification System level of at least 2 on a scale of 1-5, profound hearing loss requiring amplification in both ears, profound visual impairment in both eyes, or cognitive impairment such as a Mental Developmental Index score of less than 70 or a Cognitive Composite score of less than 85.
To examine time trends in the outcomes of periviable infants, Dr. Younge of Duke University, Durham, N.C., and her associates analyzed data from the network’s registry of births at 11 academic tertiary care centers nationwide. They focused on 4,274 infants who were born during 3 epochs – 2000-2003, 2004-2007, and 2008-2011 – and were evaluated for motor function, sensory impairment, and cognitive delay at a corrected age of 18-22 months.
The percentage of infants who survived without neurodevelopmental impairment increased over time, from 16% during the first epoch to 20% during the third epoch. However, the percentage who survived with neurodevelopmental impairment also increased, from 15% during the first epoch to 16% during the third epoch (New Engl. J. Med. 2017 Feb 16. doi: 10.1056/NEJMoa1605566).
The rates of active treatment of these periviable infants didn’t change significantly over time. Overall, 22% of infants born at 22 weeks, 71% of those born at 23 weeks, and 95% of those born at 24 weeks received active treatment at birth. Therefore, the overall decrease in mortality and the 4% improvement in neurodevelopmental outcomes wasn’t attributable to greater use of active treatment for periviable infants over time, said Dr. Younge and her associates.
Despite these small but significant improvements in outcomes, “the incidence of death, neurodevelopmental impairment, and other adverse outcomes remains high in this population,” they noted.
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institutes of Health, the National Center for Research Resources, and the National Center for Advancing Translational Sciences for the Neonatal Research Network’s Generic Database and Follow-up Studies. Dr. Younge reported having no relevant financial disclosures; two of her associates reported ties to Pediatrix Medical Group and rEVO Biologics.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: The percentage of infants who survived without neurodevelopmental impairment increased over time, from 16% to 20%, as did the percentage who survived with neurodevelopmental impairment, from 15% to 16%.
Data source: A cohort study involving 4,274 infants in an NIH registry born at 22-24 weeks’ gestation and evaluated for neurodevelopmental impairment at a corrected age of 18-22 months.
Disclosures: This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institutes of Health, the National Center for Research Resources, and the National Center for Advancing Translational Sciences for the Neonatal Research Network’s Generic Database and Follow-up Studies. Dr. Younge reported having no relevant financial disclosures; two of her associates reported ties to Pediatrix Medical Group and rEVO Biologics.
Low-back pain: CBT, mindfulness benefits diminish over time
For patients with chronic low-back pain, the benefits of cognitive-behavioral therapy and mindfulness-based stress reduction that were reported at 6 months and 1 year largely disappeared by 2-year follow-up, according to a Research Letter to the Editor published Feb 14 in JAMA.
In a clinical trial involving 342 affected patients aged 20-70 years, participants were randomly assigned to receive CBT (113 patients), mindfulness-based stress reduction (MBSR, 116 patients), or usual care (113 patients) and followed up at 26 weeks. The two psychological interventions were delivered in 2-hour group sessions every week for 8 weeks. As previously reported, both CBT and MBSR reduced functional limitations and decreased the “bothersomeness” of the pain, as measured by scores on the modified Roland Disability Questionnaire, compared with usual care, said Daniel C. Cherkin, PhD of the Group Health Research Institute, Seattle, and his associates.
This study was limited in that few participants in the two intervention groups attended all eight of their weekly CBT or MBSR sessions, and only 60% attended six sessions.
Further study is needed to determine whether strategies to increase adherence to these therapies, or “booster sessions” added months after the eight scheduled sessions, would help maintain the short-term benefits, Dr. Cherkin and his associates said.
For patients with chronic low-back pain, the benefits of cognitive-behavioral therapy and mindfulness-based stress reduction that were reported at 6 months and 1 year largely disappeared by 2-year follow-up, according to a Research Letter to the Editor published Feb 14 in JAMA.
In a clinical trial involving 342 affected patients aged 20-70 years, participants were randomly assigned to receive CBT (113 patients), mindfulness-based stress reduction (MBSR, 116 patients), or usual care (113 patients) and followed up at 26 weeks. The two psychological interventions were delivered in 2-hour group sessions every week for 8 weeks. As previously reported, both CBT and MBSR reduced functional limitations and decreased the “bothersomeness” of the pain, as measured by scores on the modified Roland Disability Questionnaire, compared with usual care, said Daniel C. Cherkin, PhD of the Group Health Research Institute, Seattle, and his associates.
This study was limited in that few participants in the two intervention groups attended all eight of their weekly CBT or MBSR sessions, and only 60% attended six sessions.
Further study is needed to determine whether strategies to increase adherence to these therapies, or “booster sessions” added months after the eight scheduled sessions, would help maintain the short-term benefits, Dr. Cherkin and his associates said.
For patients with chronic low-back pain, the benefits of cognitive-behavioral therapy and mindfulness-based stress reduction that were reported at 6 months and 1 year largely disappeared by 2-year follow-up, according to a Research Letter to the Editor published Feb 14 in JAMA.
In a clinical trial involving 342 affected patients aged 20-70 years, participants were randomly assigned to receive CBT (113 patients), mindfulness-based stress reduction (MBSR, 116 patients), or usual care (113 patients) and followed up at 26 weeks. The two psychological interventions were delivered in 2-hour group sessions every week for 8 weeks. As previously reported, both CBT and MBSR reduced functional limitations and decreased the “bothersomeness” of the pain, as measured by scores on the modified Roland Disability Questionnaire, compared with usual care, said Daniel C. Cherkin, PhD of the Group Health Research Institute, Seattle, and his associates.
This study was limited in that few participants in the two intervention groups attended all eight of their weekly CBT or MBSR sessions, and only 60% attended six sessions.
Further study is needed to determine whether strategies to increase adherence to these therapies, or “booster sessions” added months after the eight scheduled sessions, would help maintain the short-term benefits, Dr. Cherkin and his associates said.
FROM JAMA
Key clinical point: For patients with chronic low-back pain, the benefits of cognitive-behavioral therapy and mindfulness-based stress reduction reported at 6 months and 1 year largely disappeared by 2 years.
Major finding: The differences among the three study groups in functional limitations and bothersomeness of back pain were no longer significant at 2 years, even though the proportion of participants who showed 30% or more improvement from baseline on the RDQ remained numerically higher for CBT (62%) and MBSR (55%) than for usual care (42%).
Data source: Extended follow-up of a randomized trial involving 342 adults with chronic low-back pain.
Disclosures: This work was supported by the National Center for Complementary and Integrative Health. Dr. Cherkin and his associates reported having no relevant financial disclosures.