User login
Why Are Prion Diseases on the Rise?
This transcript has been edited for clarity.
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here. 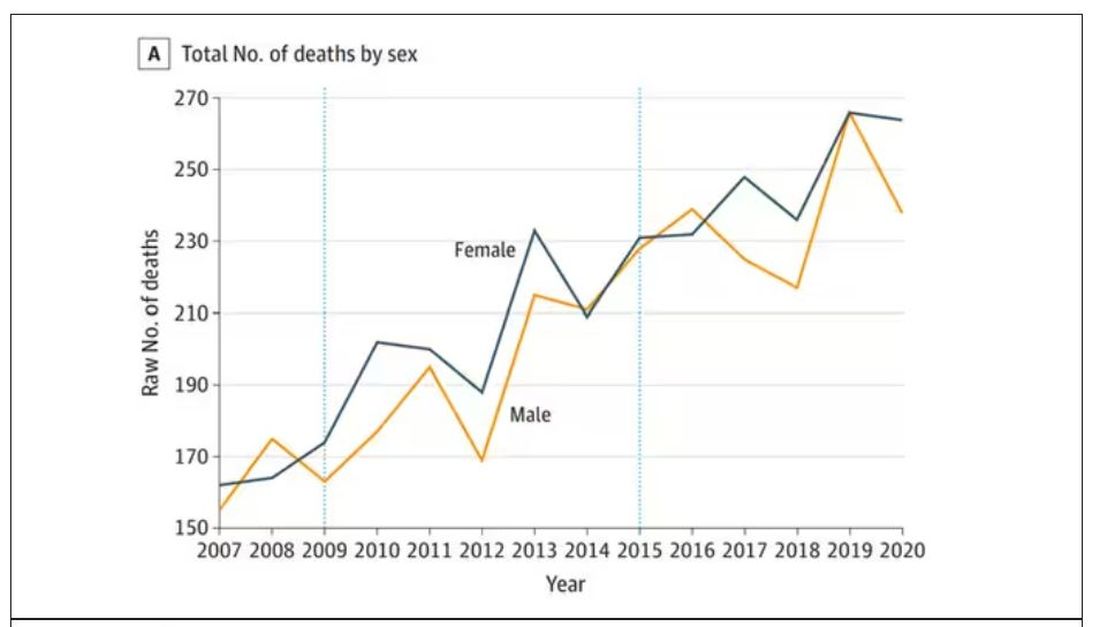
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here.
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here.
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Are you sure your patient is alive?
This transcript has been edited for clarity.
Much of my research focuses on what is known as clinical decision support — prompts and messages to providers to help them make good decisions for their patients. I know that these things can be annoying, which is exactly why I study them — to figure out which ones actually help.
When I got started on this about 10 years ago, we were learning a lot about how best to message providers about their patients. My team had developed a simple alert for acute kidney injury (AKI). We knew that providers often missed the diagnosis, so maybe letting them know would improve patient outcomes.
As we tested the alert, we got feedback, and I have kept an email from an ICU doctor from those early days. It read:
Dear Dr. Wilson: Thank you for the automated alert informing me that my patient had AKI. Regrettably, the alert fired about an hour after the patient had died. I feel that the information is less than actionable at this time.
Our early system had neglected to add a conditional flag ensuring that the patient was still alive at the time it sent the alert message. A small oversight, but one that had very large implications. Future studies would show that “false positive” alerts like this seriously degrade physician confidence in the system. And why wouldn’t they?
Not knowing the vital status of a patient can have major consequences.
Health systems send messages to their patients all the time: reminders of appointments, reminders for preventive care, reminders for vaccinations, and so on.
But what if the patient being reminded has died? It’s a waste of resources, of course, but more than that, it can be painful for their families and reflects poorly on the health care system. Of all the people who should know whether someone is alive or dead, shouldn’t their doctor be at the top of the list?
A new study in JAMA Internal Medicine quantifies this very phenomenon.
Researchers examined 11,658 primary care patients in their health system who met the criteria of being “seriously ill” and followed them for 2 years. During that period of time, 25% were recorded as deceased in the electronic health record. But 30.8% had died. That left 676 patients who had died, but were not known to have died, left in the system.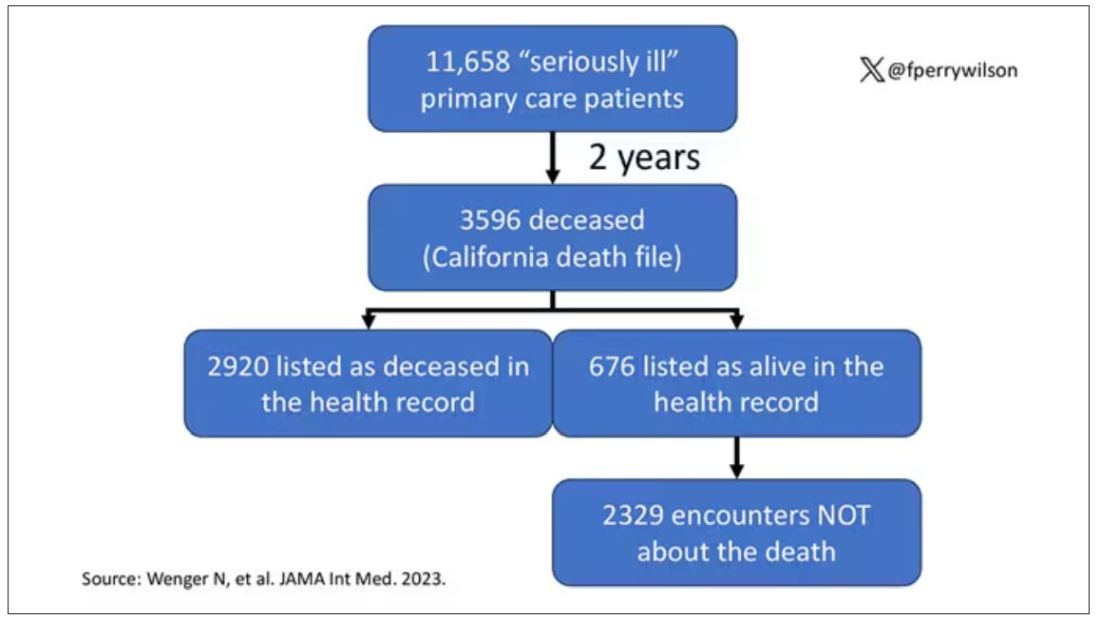
And those 676 were not left to rest in peace. They received 221 telephone and 338 health portal messages not related to death, and 920 letters reminding them about unmet primary care metrics like flu shots and cancer screening. Orders were entered into the health record for things like vaccines and routine screenings for 158 patients, and 310 future appointments — destined to be no-shows — were still on the books. One can only imagine the frustration of families checking their mail and finding yet another letter reminding their deceased loved one to get a mammogram.
How did the researchers figure out who had died? It turns out it’s not that hard. California keeps a record of all deaths in the state; they simply had to search it. Like all state death records, they tend to lag a bit so it’s not clinically terribly useful, but it works. California and most other states also have a very accurate and up-to-date death file which can only be used by law enforcement to investigate criminal activity and fraud; health care is left in the lurch.
Nationwide, there is the real-time fact of death service, supported by the National Association for Public Health Statistics and Information Systems. This allows employers to verify, in real time, whether the person applying for a job is alive. Healthcare systems are not allowed to use it.
Let’s also remember that very few people die in this country without some health care agency knowing about it and recording it. But sharing of medical information is so poor in the United States that your patient could die in a hospital one city away from you and you might not find out until you’re calling them to see why they missed a scheduled follow-up appointment.
These events — the embarrassing lack of knowledge about the very vital status of our patients — highlight a huge problem with health care in our country. The fragmented health care system is terrible at data sharing, in part because of poor protocols, in part because of unfounded concerns about patient privacy, and in part because of a tendency to hoard data that might be valuable in the future. It has to stop. We need to know how our patients are doing even when they are not sitting in front of us. When it comes to life and death, the knowledge is out there; we just can’t access it. Seems like a pretty easy fix.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com .
This transcript has been edited for clarity.
Much of my research focuses on what is known as clinical decision support — prompts and messages to providers to help them make good decisions for their patients. I know that these things can be annoying, which is exactly why I study them — to figure out which ones actually help.
When I got started on this about 10 years ago, we were learning a lot about how best to message providers about their patients. My team had developed a simple alert for acute kidney injury (AKI). We knew that providers often missed the diagnosis, so maybe letting them know would improve patient outcomes.
As we tested the alert, we got feedback, and I have kept an email from an ICU doctor from those early days. It read:
Dear Dr. Wilson: Thank you for the automated alert informing me that my patient had AKI. Regrettably, the alert fired about an hour after the patient had died. I feel that the information is less than actionable at this time.
Our early system had neglected to add a conditional flag ensuring that the patient was still alive at the time it sent the alert message. A small oversight, but one that had very large implications. Future studies would show that “false positive” alerts like this seriously degrade physician confidence in the system. And why wouldn’t they?
Not knowing the vital status of a patient can have major consequences.
Health systems send messages to their patients all the time: reminders of appointments, reminders for preventive care, reminders for vaccinations, and so on.
But what if the patient being reminded has died? It’s a waste of resources, of course, but more than that, it can be painful for their families and reflects poorly on the health care system. Of all the people who should know whether someone is alive or dead, shouldn’t their doctor be at the top of the list?
A new study in JAMA Internal Medicine quantifies this very phenomenon.
Researchers examined 11,658 primary care patients in their health system who met the criteria of being “seriously ill” and followed them for 2 years. During that period of time, 25% were recorded as deceased in the electronic health record. But 30.8% had died. That left 676 patients who had died, but were not known to have died, left in the system.
And those 676 were not left to rest in peace. They received 221 telephone and 338 health portal messages not related to death, and 920 letters reminding them about unmet primary care metrics like flu shots and cancer screening. Orders were entered into the health record for things like vaccines and routine screenings for 158 patients, and 310 future appointments — destined to be no-shows — were still on the books. One can only imagine the frustration of families checking their mail and finding yet another letter reminding their deceased loved one to get a mammogram.
How did the researchers figure out who had died? It turns out it’s not that hard. California keeps a record of all deaths in the state; they simply had to search it. Like all state death records, they tend to lag a bit so it’s not clinically terribly useful, but it works. California and most other states also have a very accurate and up-to-date death file which can only be used by law enforcement to investigate criminal activity and fraud; health care is left in the lurch.
Nationwide, there is the real-time fact of death service, supported by the National Association for Public Health Statistics and Information Systems. This allows employers to verify, in real time, whether the person applying for a job is alive. Healthcare systems are not allowed to use it.
Let’s also remember that very few people die in this country without some health care agency knowing about it and recording it. But sharing of medical information is so poor in the United States that your patient could die in a hospital one city away from you and you might not find out until you’re calling them to see why they missed a scheduled follow-up appointment.
These events — the embarrassing lack of knowledge about the very vital status of our patients — highlight a huge problem with health care in our country. The fragmented health care system is terrible at data sharing, in part because of poor protocols, in part because of unfounded concerns about patient privacy, and in part because of a tendency to hoard data that might be valuable in the future. It has to stop. We need to know how our patients are doing even when they are not sitting in front of us. When it comes to life and death, the knowledge is out there; we just can’t access it. Seems like a pretty easy fix.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com .
This transcript has been edited for clarity.
Much of my research focuses on what is known as clinical decision support — prompts and messages to providers to help them make good decisions for their patients. I know that these things can be annoying, which is exactly why I study them — to figure out which ones actually help.
When I got started on this about 10 years ago, we were learning a lot about how best to message providers about their patients. My team had developed a simple alert for acute kidney injury (AKI). We knew that providers often missed the diagnosis, so maybe letting them know would improve patient outcomes.
As we tested the alert, we got feedback, and I have kept an email from an ICU doctor from those early days. It read:
Dear Dr. Wilson: Thank you for the automated alert informing me that my patient had AKI. Regrettably, the alert fired about an hour after the patient had died. I feel that the information is less than actionable at this time.
Our early system had neglected to add a conditional flag ensuring that the patient was still alive at the time it sent the alert message. A small oversight, but one that had very large implications. Future studies would show that “false positive” alerts like this seriously degrade physician confidence in the system. And why wouldn’t they?
Not knowing the vital status of a patient can have major consequences.
Health systems send messages to their patients all the time: reminders of appointments, reminders for preventive care, reminders for vaccinations, and so on.
But what if the patient being reminded has died? It’s a waste of resources, of course, but more than that, it can be painful for their families and reflects poorly on the health care system. Of all the people who should know whether someone is alive or dead, shouldn’t their doctor be at the top of the list?
A new study in JAMA Internal Medicine quantifies this very phenomenon.
Researchers examined 11,658 primary care patients in their health system who met the criteria of being “seriously ill” and followed them for 2 years. During that period of time, 25% were recorded as deceased in the electronic health record. But 30.8% had died. That left 676 patients who had died, but were not known to have died, left in the system.
And those 676 were not left to rest in peace. They received 221 telephone and 338 health portal messages not related to death, and 920 letters reminding them about unmet primary care metrics like flu shots and cancer screening. Orders were entered into the health record for things like vaccines and routine screenings for 158 patients, and 310 future appointments — destined to be no-shows — were still on the books. One can only imagine the frustration of families checking their mail and finding yet another letter reminding their deceased loved one to get a mammogram.
How did the researchers figure out who had died? It turns out it’s not that hard. California keeps a record of all deaths in the state; they simply had to search it. Like all state death records, they tend to lag a bit so it’s not clinically terribly useful, but it works. California and most other states also have a very accurate and up-to-date death file which can only be used by law enforcement to investigate criminal activity and fraud; health care is left in the lurch.
Nationwide, there is the real-time fact of death service, supported by the National Association for Public Health Statistics and Information Systems. This allows employers to verify, in real time, whether the person applying for a job is alive. Healthcare systems are not allowed to use it.
Let’s also remember that very few people die in this country without some health care agency knowing about it and recording it. But sharing of medical information is so poor in the United States that your patient could die in a hospital one city away from you and you might not find out until you’re calling them to see why they missed a scheduled follow-up appointment.
These events — the embarrassing lack of knowledge about the very vital status of our patients — highlight a huge problem with health care in our country. The fragmented health care system is terrible at data sharing, in part because of poor protocols, in part because of unfounded concerns about patient privacy, and in part because of a tendency to hoard data that might be valuable in the future. It has to stop. We need to know how our patients are doing even when they are not sitting in front of us. When it comes to life and death, the knowledge is out there; we just can’t access it. Seems like a pretty easy fix.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com .
Is air filtration the best public health intervention against respiratory viruses?
This transcript has been edited for clarity.
When it comes to the public health fight against respiratory viruses – COVID, flu, RSV, and so on – it has always struck me as strange how staunchly basically any intervention is opposed. Masking was, of course, the prototypical entrenched warfare of opposing ideologies, with advocates pointing to studies suggesting the efficacy of masking to prevent transmission and advocating for broad masking recommendations, and detractors citing studies that suggested masks were ineffective and characterizing masking policies as fascist overreach. I’ll admit that I was always perplexed by this a bit, as that particular intervention seemed so benign – a bit annoying, I guess, but not crazy.
I have come to appreciate what I call status quo bias, which is the tendency to reject any policy, advice, or intervention that would force you, as an individual, to change your usual behavior. We just don’t like to do that. It has made me think that the most successful public health interventions might be the ones that take the individual out of the loop. And air quality control seems an ideal fit here. Here is a potential intervention where you, the individual, have to do precisely nothing. The status quo is preserved. We just, you know, have cleaner indoor air.
But even the suggestion of air treatment systems as a bulwark against respiratory virus transmission has been met with not just skepticism but cynicism, and perhaps even defeatism. It seems that there are those out there who think there really is nothing we can do. Sickness is interpreted in a Calvinistic framework: You become ill because it is your pre-destiny. But maybe air treatment could actually work. It seems like it might, if a new paper from PLOS One is to be believed.
What we’re talking about is a study titled “Bipolar Ionization Rapidly Inactivates Real-World, Airborne Concentrations of Infective Respiratory Viruses” – a highly controlled, laboratory-based analysis of a bipolar ionization system which seems to rapidly reduce viral counts in the air.
The proposed mechanism of action is pretty simple. The ionization system – which, don’t worry, has been shown not to produce ozone – spits out positively and negatively charged particles, which float around the test chamber, designed to look like a pretty standard room that you might find in an office or a school.
Virus is then injected into the chamber through an aerosolization machine, to achieve concentrations on the order of what you might get standing within 6 feet or so of someone actively infected with COVID while they are breathing and talking.
The idea is that those ions stick to the virus particles, similar to how a balloon sticks to the wall after you rub it on your hair, and that tends to cause them to clump together and settle on surfaces more rapidly, and thus get farther away from their ports of entry to the human system: nose, mouth, and eyes. But the ions may also interfere with viruses’ ability to bind to cellular receptors, even in the air.
To quantify viral infectivity, the researchers used a biological system. Basically, you take air samples and expose a petri dish of cells to them and see how many cells die. Fewer cells dying, less infective. Under control conditions, you can see that virus infectivity does decrease over time. Time zero here is the end of a SARS-CoV-2 aerosolization.
This may simply reflect the fact that virus particles settle out of the air. But As you can see, within about an hour, you have almost no infective virus detectable. That’s fairly impressive.
Now, I’m not saying that this is a panacea, but it is certainly worth considering the use of technologies like these if we are going to revamp the infrastructure of our offices and schools. And, of course, it would be nice to see this tested in a rigorous clinical trial with actual infected people, not cells, as the outcome. But I continue to be encouraged by interventions like this which, to be honest, ask very little of us as individuals. Maybe it’s time we accept the things, or people, that we cannot change.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When it comes to the public health fight against respiratory viruses – COVID, flu, RSV, and so on – it has always struck me as strange how staunchly basically any intervention is opposed. Masking was, of course, the prototypical entrenched warfare of opposing ideologies, with advocates pointing to studies suggesting the efficacy of masking to prevent transmission and advocating for broad masking recommendations, and detractors citing studies that suggested masks were ineffective and characterizing masking policies as fascist overreach. I’ll admit that I was always perplexed by this a bit, as that particular intervention seemed so benign – a bit annoying, I guess, but not crazy.
I have come to appreciate what I call status quo bias, which is the tendency to reject any policy, advice, or intervention that would force you, as an individual, to change your usual behavior. We just don’t like to do that. It has made me think that the most successful public health interventions might be the ones that take the individual out of the loop. And air quality control seems an ideal fit here. Here is a potential intervention where you, the individual, have to do precisely nothing. The status quo is preserved. We just, you know, have cleaner indoor air.
But even the suggestion of air treatment systems as a bulwark against respiratory virus transmission has been met with not just skepticism but cynicism, and perhaps even defeatism. It seems that there are those out there who think there really is nothing we can do. Sickness is interpreted in a Calvinistic framework: You become ill because it is your pre-destiny. But maybe air treatment could actually work. It seems like it might, if a new paper from PLOS One is to be believed.
What we’re talking about is a study titled “Bipolar Ionization Rapidly Inactivates Real-World, Airborne Concentrations of Infective Respiratory Viruses” – a highly controlled, laboratory-based analysis of a bipolar ionization system which seems to rapidly reduce viral counts in the air.
The proposed mechanism of action is pretty simple. The ionization system – which, don’t worry, has been shown not to produce ozone – spits out positively and negatively charged particles, which float around the test chamber, designed to look like a pretty standard room that you might find in an office or a school.
Virus is then injected into the chamber through an aerosolization machine, to achieve concentrations on the order of what you might get standing within 6 feet or so of someone actively infected with COVID while they are breathing and talking.
The idea is that those ions stick to the virus particles, similar to how a balloon sticks to the wall after you rub it on your hair, and that tends to cause them to clump together and settle on surfaces more rapidly, and thus get farther away from their ports of entry to the human system: nose, mouth, and eyes. But the ions may also interfere with viruses’ ability to bind to cellular receptors, even in the air.
To quantify viral infectivity, the researchers used a biological system. Basically, you take air samples and expose a petri dish of cells to them and see how many cells die. Fewer cells dying, less infective. Under control conditions, you can see that virus infectivity does decrease over time. Time zero here is the end of a SARS-CoV-2 aerosolization.
This may simply reflect the fact that virus particles settle out of the air. But As you can see, within about an hour, you have almost no infective virus detectable. That’s fairly impressive.
Now, I’m not saying that this is a panacea, but it is certainly worth considering the use of technologies like these if we are going to revamp the infrastructure of our offices and schools. And, of course, it would be nice to see this tested in a rigorous clinical trial with actual infected people, not cells, as the outcome. But I continue to be encouraged by interventions like this which, to be honest, ask very little of us as individuals. Maybe it’s time we accept the things, or people, that we cannot change.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When it comes to the public health fight against respiratory viruses – COVID, flu, RSV, and so on – it has always struck me as strange how staunchly basically any intervention is opposed. Masking was, of course, the prototypical entrenched warfare of opposing ideologies, with advocates pointing to studies suggesting the efficacy of masking to prevent transmission and advocating for broad masking recommendations, and detractors citing studies that suggested masks were ineffective and characterizing masking policies as fascist overreach. I’ll admit that I was always perplexed by this a bit, as that particular intervention seemed so benign – a bit annoying, I guess, but not crazy.
I have come to appreciate what I call status quo bias, which is the tendency to reject any policy, advice, or intervention that would force you, as an individual, to change your usual behavior. We just don’t like to do that. It has made me think that the most successful public health interventions might be the ones that take the individual out of the loop. And air quality control seems an ideal fit here. Here is a potential intervention where you, the individual, have to do precisely nothing. The status quo is preserved. We just, you know, have cleaner indoor air.
But even the suggestion of air treatment systems as a bulwark against respiratory virus transmission has been met with not just skepticism but cynicism, and perhaps even defeatism. It seems that there are those out there who think there really is nothing we can do. Sickness is interpreted in a Calvinistic framework: You become ill because it is your pre-destiny. But maybe air treatment could actually work. It seems like it might, if a new paper from PLOS One is to be believed.
What we’re talking about is a study titled “Bipolar Ionization Rapidly Inactivates Real-World, Airborne Concentrations of Infective Respiratory Viruses” – a highly controlled, laboratory-based analysis of a bipolar ionization system which seems to rapidly reduce viral counts in the air.
The proposed mechanism of action is pretty simple. The ionization system – which, don’t worry, has been shown not to produce ozone – spits out positively and negatively charged particles, which float around the test chamber, designed to look like a pretty standard room that you might find in an office or a school.
Virus is then injected into the chamber through an aerosolization machine, to achieve concentrations on the order of what you might get standing within 6 feet or so of someone actively infected with COVID while they are breathing and talking.
The idea is that those ions stick to the virus particles, similar to how a balloon sticks to the wall after you rub it on your hair, and that tends to cause them to clump together and settle on surfaces more rapidly, and thus get farther away from their ports of entry to the human system: nose, mouth, and eyes. But the ions may also interfere with viruses’ ability to bind to cellular receptors, even in the air.
To quantify viral infectivity, the researchers used a biological system. Basically, you take air samples and expose a petri dish of cells to them and see how many cells die. Fewer cells dying, less infective. Under control conditions, you can see that virus infectivity does decrease over time. Time zero here is the end of a SARS-CoV-2 aerosolization.
This may simply reflect the fact that virus particles settle out of the air. But As you can see, within about an hour, you have almost no infective virus detectable. That’s fairly impressive.
Now, I’m not saying that this is a panacea, but it is certainly worth considering the use of technologies like these if we are going to revamp the infrastructure of our offices and schools. And, of course, it would be nice to see this tested in a rigorous clinical trial with actual infected people, not cells, as the outcome. But I continue to be encouraged by interventions like this which, to be honest, ask very little of us as individuals. Maybe it’s time we accept the things, or people, that we cannot change.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Headache after drinking red wine? This could be why
This transcript has been edited for clarity.
Robert Louis Stevenson famously said, “Wine is bottled poetry.” And I think it works quite well. I’ve had wines that are simple, elegant, and unpretentious like Emily Dickinson, and passionate and mysterious like Pablo Neruda. And I’ve had wines that are more analogous to the limerick you might read scrawled on a rest-stop bathroom wall. Those ones give me headaches.
– and apparently it’s not just the alcohol.
Headaches are common, and headaches after drinking alcohol are particularly common. An interesting epidemiologic phenomenon, not yet adequately explained, is why red wine is associated with more headache than other forms of alcohol. There have been many studies fingering many suspects, from sulfites to tannins to various phenolic compounds, but none have really provided a concrete explanation for what might be going on.
A new hypothesis came to the fore on Nov. 20 in the journal Scientific Reports:
To understand the idea, first a reminder of what happens when you drink alcohol, physiologically.
Alcohol is metabolized by the enzyme alcohol dehydrogenase in the gut and then in the liver. That turns it into acetaldehyde, a toxic metabolite. In most of us, aldehyde dehydrogenase (ALDH) quickly metabolizes acetaldehyde to the inert acetate, which can be safely excreted.
I say “most of us” because some populations, particularly those with East Asian ancestry, have a mutation in the ALDH gene which can lead to accumulation of toxic acetaldehyde with alcohol consumption – leading to facial flushing, nausea, and headache.
We can also inhibit the enzyme medically. That’s what the drug disulfiram, also known as Antabuse, does. It doesn’t prevent you from wanting to drink; it makes the consequences of drinking incredibly aversive.
The researchers focused in on the aldehyde dehydrogenase enzyme and conducted a screening study. Are there any compounds in red wine that naturally inhibit ALDH?
The results pointed squarely at quercetin, and particularly its metabolite quercetin glucuronide, which, at 20 micromolar concentrations, inhibited about 80% of ALDH activity.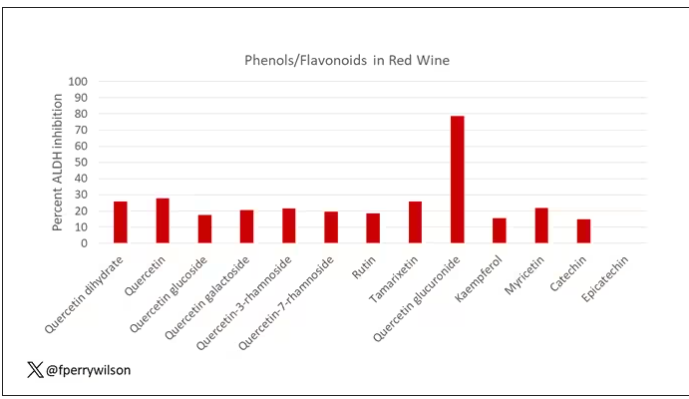
Quercetin is a flavonoid – a compound that gives color to a variety of vegetables and fruits, including grapes. In a test tube, it is an antioxidant, which is enough evidence to spawn a small quercetin-as-supplement industry, but there is no convincing evidence that it is medically useful. The authors then examined the concentration of quercetin glucuronide to achieve various inhibitions of ALDH, as you can see in this graph here.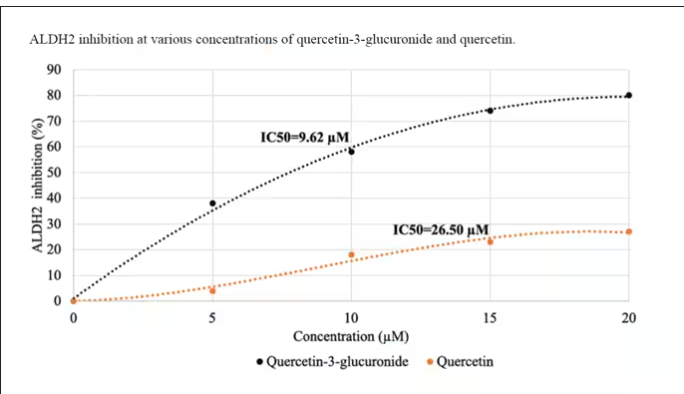
By about 10 micromolar, we see a decent amount of inhibition. Disulfiram is about 10 times more potent than that, but then again, you don’t drink three glasses of disulfiram with Thanksgiving dinner.
This is where this study stops. But it obviously tells us very little about what might be happening in the human body. For that, we need to ask the question: Can we get our quercetin levels to 10 micromolar? Is that remotely achievable?
Let’s start with how much quercetin there is in red wine. Like all things wine, it varies, but this study examining Australian wines found mean concentrations of 11 mg/L. The highest value I saw was close to 50 mg/L.
So let’s do some math. To make the numbers easy, let’s say you drank a liter of Australian wine, taking in 50 mg of quercetin glucuronide.
How much of that gets into your bloodstream? Some studies suggest a bioavailability of less than 1%, which basically means none and should probably put the quercetin hypothesis to bed. But there is some variation here too; it seems to depend on the form of quercetin you ingest.
Let’s say all 50 mg gets into your bloodstream. What blood concentration would that lead to? Well, I’ll keep the stoichiometry in the graphics and just say that if we assume that the volume of distribution of the compound is restricted to plasma alone, then you could achieve similar concentrations to what was done in petri dishes during this study.
Of course, if quercetin is really the culprit behind red wine headache, I have some questions: Why aren’t the Amazon reviews of quercetin supplements chock full of warnings not to take them with alcohol? And other foods have way higher quercetin concentration than wine, but you don’t hear people warning not to take your red onions with alcohol, or your capers, or lingonberries.
There’s some more work to be done here – most importantly, some human studies. Let’s give people wine with different amounts of quercetin and see what happens. Sign me up. Seriously.
As for Thanksgiving, it’s worth noting that cranberries have a lot of quercetin in them. So between the cranberry sauce, the Beaujolais, and your uncle ranting about the contrails again, the probability of headache is pretty darn high. Stay safe out there, and Happy Thanksgiving.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Robert Louis Stevenson famously said, “Wine is bottled poetry.” And I think it works quite well. I’ve had wines that are simple, elegant, and unpretentious like Emily Dickinson, and passionate and mysterious like Pablo Neruda. And I’ve had wines that are more analogous to the limerick you might read scrawled on a rest-stop bathroom wall. Those ones give me headaches.
– and apparently it’s not just the alcohol.
Headaches are common, and headaches after drinking alcohol are particularly common. An interesting epidemiologic phenomenon, not yet adequately explained, is why red wine is associated with more headache than other forms of alcohol. There have been many studies fingering many suspects, from sulfites to tannins to various phenolic compounds, but none have really provided a concrete explanation for what might be going on.
A new hypothesis came to the fore on Nov. 20 in the journal Scientific Reports:
To understand the idea, first a reminder of what happens when you drink alcohol, physiologically.
Alcohol is metabolized by the enzyme alcohol dehydrogenase in the gut and then in the liver. That turns it into acetaldehyde, a toxic metabolite. In most of us, aldehyde dehydrogenase (ALDH) quickly metabolizes acetaldehyde to the inert acetate, which can be safely excreted.
I say “most of us” because some populations, particularly those with East Asian ancestry, have a mutation in the ALDH gene which can lead to accumulation of toxic acetaldehyde with alcohol consumption – leading to facial flushing, nausea, and headache.
We can also inhibit the enzyme medically. That’s what the drug disulfiram, also known as Antabuse, does. It doesn’t prevent you from wanting to drink; it makes the consequences of drinking incredibly aversive.
The researchers focused in on the aldehyde dehydrogenase enzyme and conducted a screening study. Are there any compounds in red wine that naturally inhibit ALDH?
The results pointed squarely at quercetin, and particularly its metabolite quercetin glucuronide, which, at 20 micromolar concentrations, inhibited about 80% of ALDH activity.
Quercetin is a flavonoid – a compound that gives color to a variety of vegetables and fruits, including grapes. In a test tube, it is an antioxidant, which is enough evidence to spawn a small quercetin-as-supplement industry, but there is no convincing evidence that it is medically useful. The authors then examined the concentration of quercetin glucuronide to achieve various inhibitions of ALDH, as you can see in this graph here.
By about 10 micromolar, we see a decent amount of inhibition. Disulfiram is about 10 times more potent than that, but then again, you don’t drink three glasses of disulfiram with Thanksgiving dinner.
This is where this study stops. But it obviously tells us very little about what might be happening in the human body. For that, we need to ask the question: Can we get our quercetin levels to 10 micromolar? Is that remotely achievable?
Let’s start with how much quercetin there is in red wine. Like all things wine, it varies, but this study examining Australian wines found mean concentrations of 11 mg/L. The highest value I saw was close to 50 mg/L.
So let’s do some math. To make the numbers easy, let’s say you drank a liter of Australian wine, taking in 50 mg of quercetin glucuronide.
How much of that gets into your bloodstream? Some studies suggest a bioavailability of less than 1%, which basically means none and should probably put the quercetin hypothesis to bed. But there is some variation here too; it seems to depend on the form of quercetin you ingest.
Let’s say all 50 mg gets into your bloodstream. What blood concentration would that lead to? Well, I’ll keep the stoichiometry in the graphics and just say that if we assume that the volume of distribution of the compound is restricted to plasma alone, then you could achieve similar concentrations to what was done in petri dishes during this study.
Of course, if quercetin is really the culprit behind red wine headache, I have some questions: Why aren’t the Amazon reviews of quercetin supplements chock full of warnings not to take them with alcohol? And other foods have way higher quercetin concentration than wine, but you don’t hear people warning not to take your red onions with alcohol, or your capers, or lingonberries.
There’s some more work to be done here – most importantly, some human studies. Let’s give people wine with different amounts of quercetin and see what happens. Sign me up. Seriously.
As for Thanksgiving, it’s worth noting that cranberries have a lot of quercetin in them. So between the cranberry sauce, the Beaujolais, and your uncle ranting about the contrails again, the probability of headache is pretty darn high. Stay safe out there, and Happy Thanksgiving.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Robert Louis Stevenson famously said, “Wine is bottled poetry.” And I think it works quite well. I’ve had wines that are simple, elegant, and unpretentious like Emily Dickinson, and passionate and mysterious like Pablo Neruda. And I’ve had wines that are more analogous to the limerick you might read scrawled on a rest-stop bathroom wall. Those ones give me headaches.
– and apparently it’s not just the alcohol.
Headaches are common, and headaches after drinking alcohol are particularly common. An interesting epidemiologic phenomenon, not yet adequately explained, is why red wine is associated with more headache than other forms of alcohol. There have been many studies fingering many suspects, from sulfites to tannins to various phenolic compounds, but none have really provided a concrete explanation for what might be going on.
A new hypothesis came to the fore on Nov. 20 in the journal Scientific Reports:
To understand the idea, first a reminder of what happens when you drink alcohol, physiologically.
Alcohol is metabolized by the enzyme alcohol dehydrogenase in the gut and then in the liver. That turns it into acetaldehyde, a toxic metabolite. In most of us, aldehyde dehydrogenase (ALDH) quickly metabolizes acetaldehyde to the inert acetate, which can be safely excreted.
I say “most of us” because some populations, particularly those with East Asian ancestry, have a mutation in the ALDH gene which can lead to accumulation of toxic acetaldehyde with alcohol consumption – leading to facial flushing, nausea, and headache.
We can also inhibit the enzyme medically. That’s what the drug disulfiram, also known as Antabuse, does. It doesn’t prevent you from wanting to drink; it makes the consequences of drinking incredibly aversive.
The researchers focused in on the aldehyde dehydrogenase enzyme and conducted a screening study. Are there any compounds in red wine that naturally inhibit ALDH?
The results pointed squarely at quercetin, and particularly its metabolite quercetin glucuronide, which, at 20 micromolar concentrations, inhibited about 80% of ALDH activity.
Quercetin is a flavonoid – a compound that gives color to a variety of vegetables and fruits, including grapes. In a test tube, it is an antioxidant, which is enough evidence to spawn a small quercetin-as-supplement industry, but there is no convincing evidence that it is medically useful. The authors then examined the concentration of quercetin glucuronide to achieve various inhibitions of ALDH, as you can see in this graph here.
By about 10 micromolar, we see a decent amount of inhibition. Disulfiram is about 10 times more potent than that, but then again, you don’t drink three glasses of disulfiram with Thanksgiving dinner.
This is where this study stops. But it obviously tells us very little about what might be happening in the human body. For that, we need to ask the question: Can we get our quercetin levels to 10 micromolar? Is that remotely achievable?
Let’s start with how much quercetin there is in red wine. Like all things wine, it varies, but this study examining Australian wines found mean concentrations of 11 mg/L. The highest value I saw was close to 50 mg/L.
So let’s do some math. To make the numbers easy, let’s say you drank a liter of Australian wine, taking in 50 mg of quercetin glucuronide.
How much of that gets into your bloodstream? Some studies suggest a bioavailability of less than 1%, which basically means none and should probably put the quercetin hypothesis to bed. But there is some variation here too; it seems to depend on the form of quercetin you ingest.
Let’s say all 50 mg gets into your bloodstream. What blood concentration would that lead to? Well, I’ll keep the stoichiometry in the graphics and just say that if we assume that the volume of distribution of the compound is restricted to plasma alone, then you could achieve similar concentrations to what was done in petri dishes during this study.
Of course, if quercetin is really the culprit behind red wine headache, I have some questions: Why aren’t the Amazon reviews of quercetin supplements chock full of warnings not to take them with alcohol? And other foods have way higher quercetin concentration than wine, but you don’t hear people warning not to take your red onions with alcohol, or your capers, or lingonberries.
There’s some more work to be done here – most importantly, some human studies. Let’s give people wine with different amounts of quercetin and see what happens. Sign me up. Seriously.
As for Thanksgiving, it’s worth noting that cranberries have a lot of quercetin in them. So between the cranberry sauce, the Beaujolais, and your uncle ranting about the contrails again, the probability of headache is pretty darn high. Stay safe out there, and Happy Thanksgiving.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
The future of medicine is RNA
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Every once in a while, medicine changes in a fundamental way, and we may not realize it while it’s happening. I wasn’t around in 1928 when Fleming discovered penicillin; or in 1953 when Watson, Crick, and Franklin characterized the double-helical structure of DNA.
But looking at medicine today, there are essentially two places where I think we will see, in retrospect, that we were at a fundamental turning point. One is artificial intelligence, which gets so much attention and hype that I will simply say yes, this will change things, stay tuned.
The other is a bit more obscure, but I suspect it may be just as impactful. That other thing is

I want to start with the idea that many diseases are, fundamentally, a problem of proteins. In some cases, like hypercholesterolemia, the body produces too much protein; in others, like hemophilia, too little.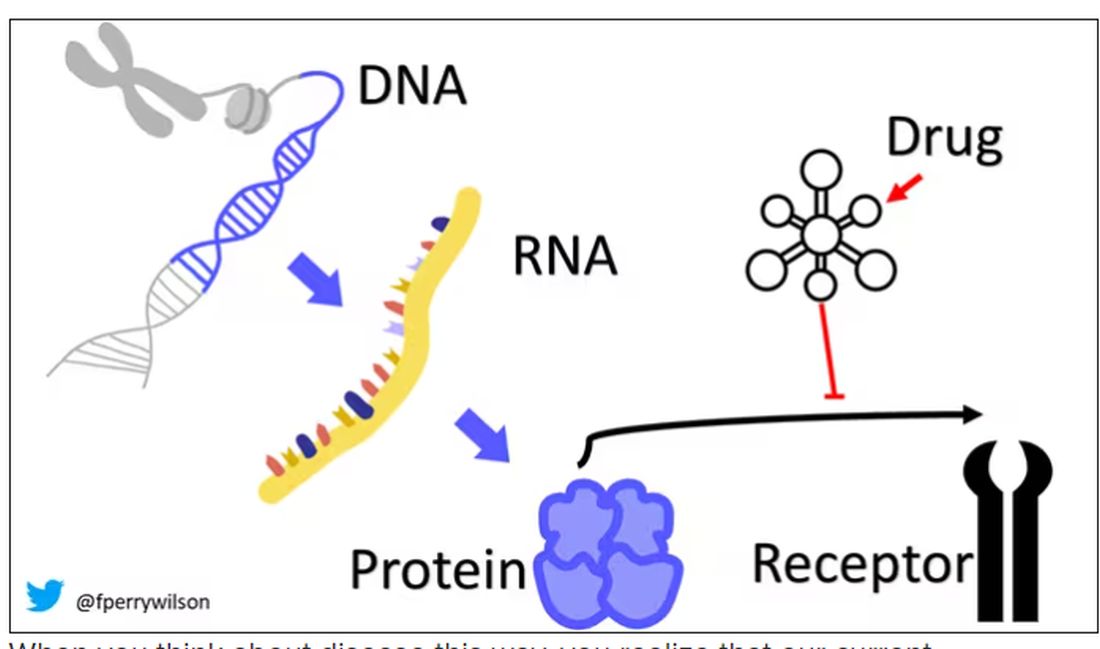
When you think about disease this way, you realize that our current medications take effect late in the disease game. We have these molecules that try to block a protein from its receptor, prevent a protein from cleaving another protein, or increase the rate that a protein is broken down. It’s all distal to the fundamental problem: the production of the bad protein in the first place.
Enter small inhibitory RNAs, or siRNAs for short, discovered in 1998 by Andrew Fire and Craig Mello at UMass Worcester. The two won the Nobel prize in medicine just 8 years later; that’s a really short time, highlighting just how important this discovery was. In contrast, Karikó and Weissman won the Nobel for mRNA vaccines this year, after inventing them 18 years ago.
siRNAs are the body’s way of targeting proteins for destruction before they are ever created. About 20 base pairs long, siRNAs seek out a complementary target mRNA, attach to it, and call in a group of proteins to destroy it. With the target mRNA gone, no protein can be created.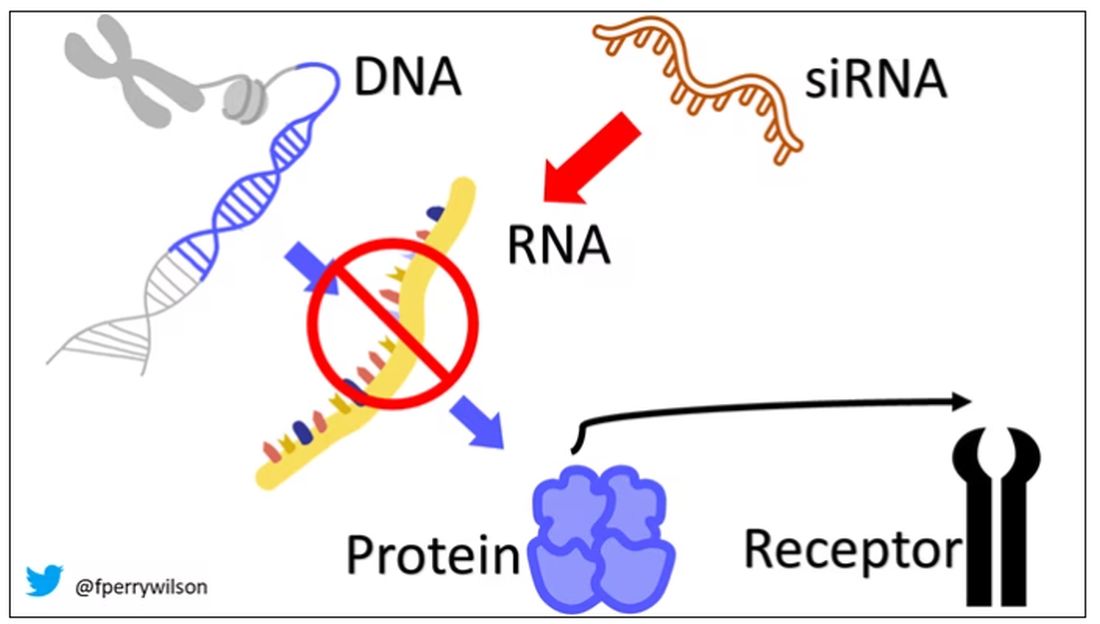
You see where this is going, right? How does high cholesterol kill you? Proteins. How does Staphylococcus aureus kill you? Proteins. Even viruses can’t replicate if their RNA is prevented from being turned into proteins.
So, how do we use siRNAs? A new paper appearing in JAMA describes a fairly impressive use case.
The background here is that higher levels of lipoprotein(a), an LDL-like protein, are associated with cardiovascular disease, heart attack, and stroke. But unfortunately, statins really don’t have any effect on lipoprotein(a) levels. Neither does diet. Your lipoprotein(a) level seems to be more or less hard-coded genetically.
So, what if we stop the genetic machinery from working? Enter lepodisiran, a drug from Eli Lilly. Unlike so many other medications, which are usually found in nature, purified, and synthesized, lepodisiran was created from scratch. It’s not hard. Thanks to the Human Genome Project, we know the genetic code for lipoprotein(a), so inventing an siRNA to target it specifically is trivial. That’s one of the key features of siRNA – you don’t have to find a chemical that binds strongly to some protein receptor, and worry about the off-target effects and all that nonsense. You just pick a protein you want to suppress and you suppress it.
Okay, it’s not that simple. siRNA is broken down very quickly by the body, so it needs to be targeted to the organ of interest – in this case, the liver, since that is where lipoprotein(a) is synthesized. Lepodisiran is targeted to the liver by this special targeting label here.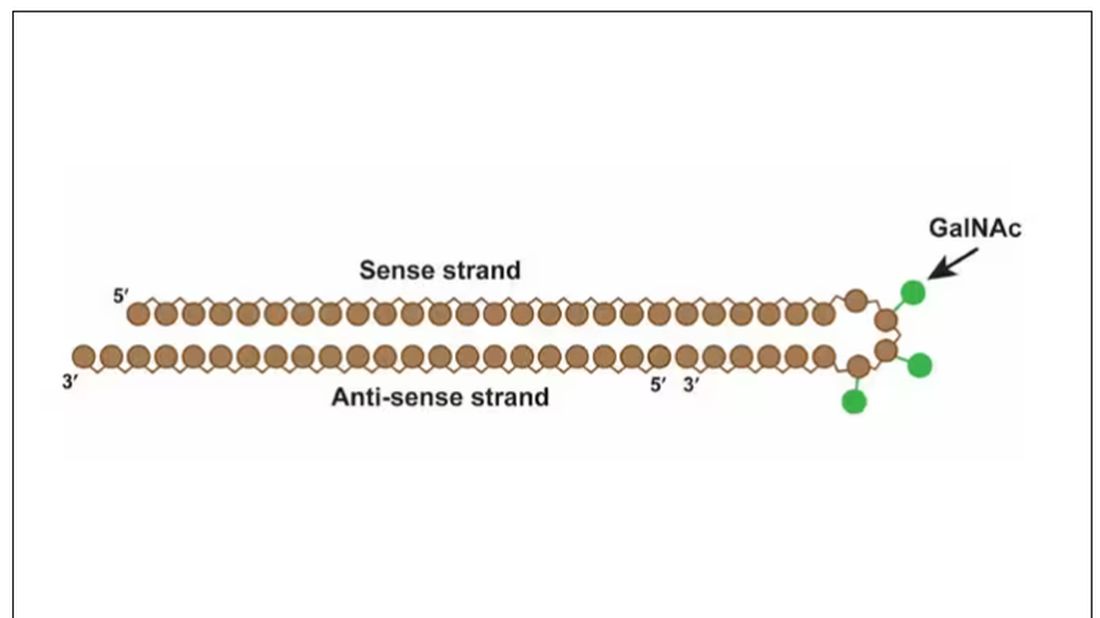
The report is a standard dose-escalation trial. Six patients, all with elevated lipoprotein(a) levels, were started with a 4-mg dose (two additional individuals got placebo). They were intensely monitored, spending 3 days in a research unit for multiple blood draws followed by weekly, and then biweekly outpatient visits. Once they had done well, the next group of six people received a higher dose (two more got placebo), and the process was repeated – six times total – until the highest dose, 608 mg, was reached.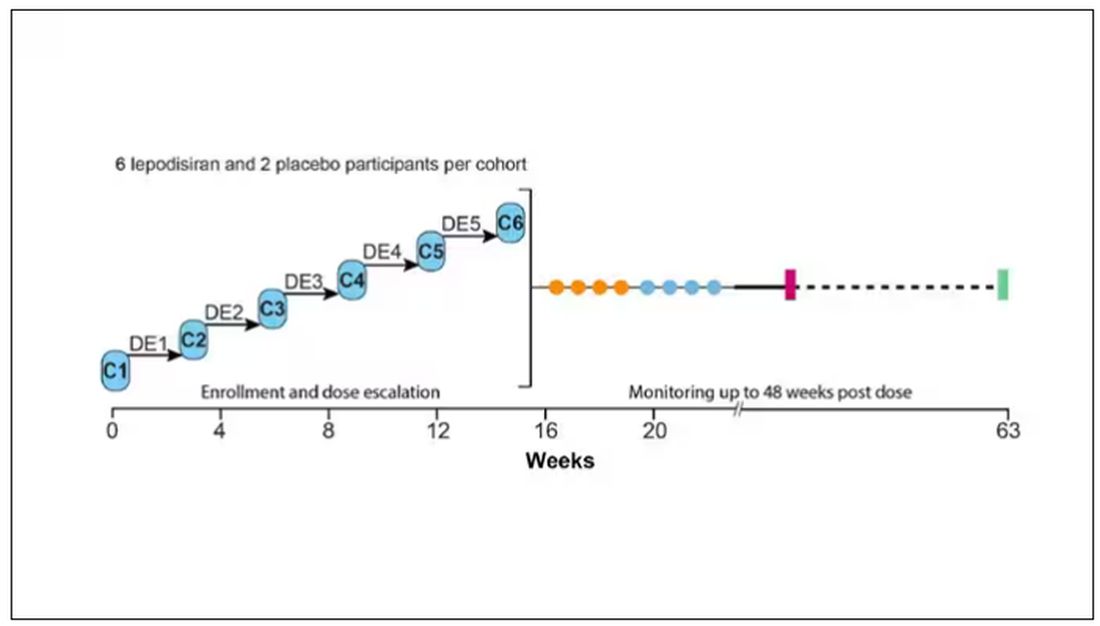
This is an injection, of course; siRNA wouldn’t withstand the harshness of the digestive system. And it’s only one injection. You can see from the blood concentration curves that within about 48 hours, circulating lepodisiran was not detectable.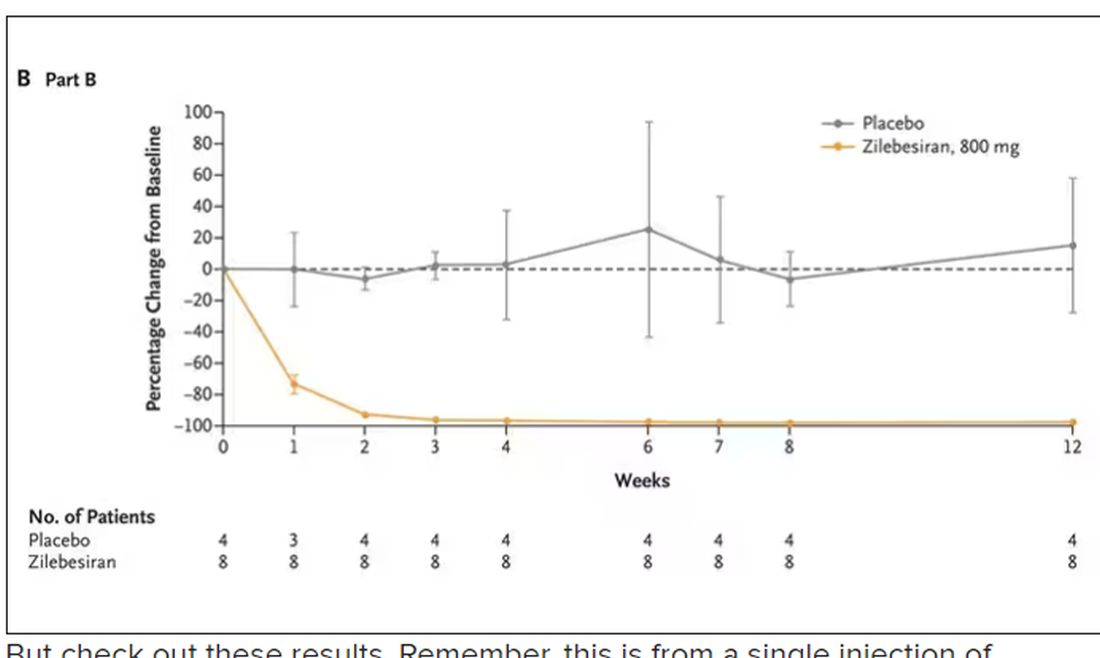
But check out these results. Remember, this is from a single injection of lepodisiran.
Lipoprotein(a) levels start to drop within a week of administration, and they stay down. In the higher-dose groups, levels are nearly undetectable a year after that injection.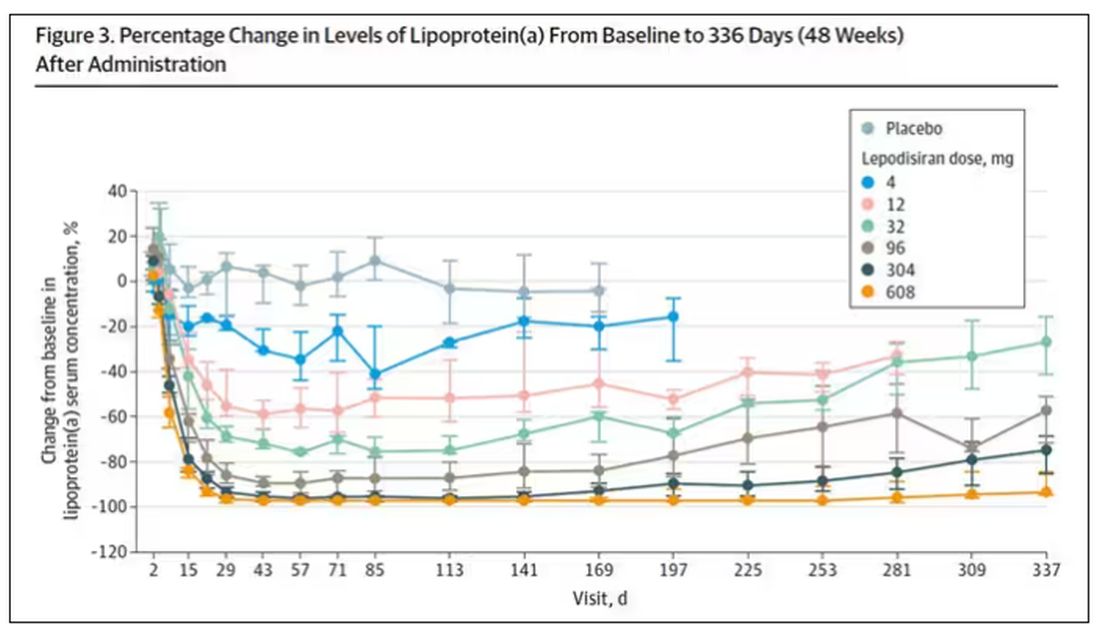
It was this graph that made me sit back and think that there might be something new under the sun. A single injection that can suppress protein synthesis for an entire year? If it really works, it changes the game.
Of course, this study wasn’t powered to look at important outcomes like heart attacks and strokes. It was primarily designed to assess safety, and the drug was pretty well tolerated, with similar rates of adverse events in the drug and placebo groups.
As crazy as it sounds, the real concern here might be that this drug is too good; is it safe to drop your lipoprotein(a) levels to zero for a year? I don’t know. But lower doses don’t have quite as strong an effect.
Trust me, these drugs are going to change things. They already are. In July, The New England Journal of Medicine published a study of zilebesiran, an siRNA that inhibits the production of angiotensinogen, to control blood pressure. Similar story: One injection led to a basically complete suppression of angiotensinogen and a sustained decrease in blood pressure.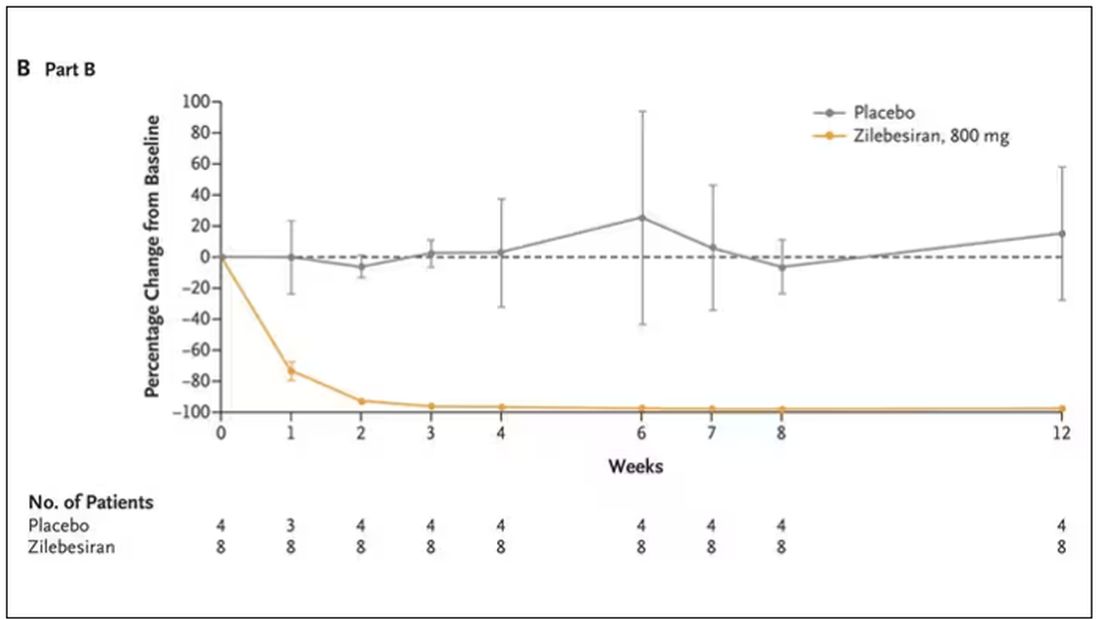
I’m not exaggerating when I say that there may come a time when you go to your doctor once a year, get your RNA shots, and don’t have to take any other medication from that point on. And that time may be, like, 5 years from now. It’s wild.
Seems to me that that rapid Nobel Prize was very well deserved.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships. This transcript has been edited for clarity.
A version of this article appeared on Medscape.com.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Every once in a while, medicine changes in a fundamental way, and we may not realize it while it’s happening. I wasn’t around in 1928 when Fleming discovered penicillin; or in 1953 when Watson, Crick, and Franklin characterized the double-helical structure of DNA.
But looking at medicine today, there are essentially two places where I think we will see, in retrospect, that we were at a fundamental turning point. One is artificial intelligence, which gets so much attention and hype that I will simply say yes, this will change things, stay tuned.
The other is a bit more obscure, but I suspect it may be just as impactful. That other thing is
I want to start with the idea that many diseases are, fundamentally, a problem of proteins. In some cases, like hypercholesterolemia, the body produces too much protein; in others, like hemophilia, too little.
When you think about disease this way, you realize that our current medications take effect late in the disease game. We have these molecules that try to block a protein from its receptor, prevent a protein from cleaving another protein, or increase the rate that a protein is broken down. It’s all distal to the fundamental problem: the production of the bad protein in the first place.
Enter small inhibitory RNAs, or siRNAs for short, discovered in 1998 by Andrew Fire and Craig Mello at UMass Worcester. The two won the Nobel prize in medicine just 8 years later; that’s a really short time, highlighting just how important this discovery was. In contrast, Karikó and Weissman won the Nobel for mRNA vaccines this year, after inventing them 18 years ago.
siRNAs are the body’s way of targeting proteins for destruction before they are ever created. About 20 base pairs long, siRNAs seek out a complementary target mRNA, attach to it, and call in a group of proteins to destroy it. With the target mRNA gone, no protein can be created.
You see where this is going, right? How does high cholesterol kill you? Proteins. How does Staphylococcus aureus kill you? Proteins. Even viruses can’t replicate if their RNA is prevented from being turned into proteins.
So, how do we use siRNAs? A new paper appearing in JAMA describes a fairly impressive use case.
The background here is that higher levels of lipoprotein(a), an LDL-like protein, are associated with cardiovascular disease, heart attack, and stroke. But unfortunately, statins really don’t have any effect on lipoprotein(a) levels. Neither does diet. Your lipoprotein(a) level seems to be more or less hard-coded genetically.
So, what if we stop the genetic machinery from working? Enter lepodisiran, a drug from Eli Lilly. Unlike so many other medications, which are usually found in nature, purified, and synthesized, lepodisiran was created from scratch. It’s not hard. Thanks to the Human Genome Project, we know the genetic code for lipoprotein(a), so inventing an siRNA to target it specifically is trivial. That’s one of the key features of siRNA – you don’t have to find a chemical that binds strongly to some protein receptor, and worry about the off-target effects and all that nonsense. You just pick a protein you want to suppress and you suppress it.
Okay, it’s not that simple. siRNA is broken down very quickly by the body, so it needs to be targeted to the organ of interest – in this case, the liver, since that is where lipoprotein(a) is synthesized. Lepodisiran is targeted to the liver by this special targeting label here.
The report is a standard dose-escalation trial. Six patients, all with elevated lipoprotein(a) levels, were started with a 4-mg dose (two additional individuals got placebo). They were intensely monitored, spending 3 days in a research unit for multiple blood draws followed by weekly, and then biweekly outpatient visits. Once they had done well, the next group of six people received a higher dose (two more got placebo), and the process was repeated – six times total – until the highest dose, 608 mg, was reached.
This is an injection, of course; siRNA wouldn’t withstand the harshness of the digestive system. And it’s only one injection. You can see from the blood concentration curves that within about 48 hours, circulating lepodisiran was not detectable.
But check out these results. Remember, this is from a single injection of lepodisiran.
Lipoprotein(a) levels start to drop within a week of administration, and they stay down. In the higher-dose groups, levels are nearly undetectable a year after that injection.
It was this graph that made me sit back and think that there might be something new under the sun. A single injection that can suppress protein synthesis for an entire year? If it really works, it changes the game.
Of course, this study wasn’t powered to look at important outcomes like heart attacks and strokes. It was primarily designed to assess safety, and the drug was pretty well tolerated, with similar rates of adverse events in the drug and placebo groups.
As crazy as it sounds, the real concern here might be that this drug is too good; is it safe to drop your lipoprotein(a) levels to zero for a year? I don’t know. But lower doses don’t have quite as strong an effect.
Trust me, these drugs are going to change things. They already are. In July, The New England Journal of Medicine published a study of zilebesiran, an siRNA that inhibits the production of angiotensinogen, to control blood pressure. Similar story: One injection led to a basically complete suppression of angiotensinogen and a sustained decrease in blood pressure.
I’m not exaggerating when I say that there may come a time when you go to your doctor once a year, get your RNA shots, and don’t have to take any other medication from that point on. And that time may be, like, 5 years from now. It’s wild.
Seems to me that that rapid Nobel Prize was very well deserved.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships. This transcript has been edited for clarity.
A version of this article appeared on Medscape.com.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Every once in a while, medicine changes in a fundamental way, and we may not realize it while it’s happening. I wasn’t around in 1928 when Fleming discovered penicillin; or in 1953 when Watson, Crick, and Franklin characterized the double-helical structure of DNA.
But looking at medicine today, there are essentially two places where I think we will see, in retrospect, that we were at a fundamental turning point. One is artificial intelligence, which gets so much attention and hype that I will simply say yes, this will change things, stay tuned.
The other is a bit more obscure, but I suspect it may be just as impactful. That other thing is
I want to start with the idea that many diseases are, fundamentally, a problem of proteins. In some cases, like hypercholesterolemia, the body produces too much protein; in others, like hemophilia, too little.
When you think about disease this way, you realize that our current medications take effect late in the disease game. We have these molecules that try to block a protein from its receptor, prevent a protein from cleaving another protein, or increase the rate that a protein is broken down. It’s all distal to the fundamental problem: the production of the bad protein in the first place.
Enter small inhibitory RNAs, or siRNAs for short, discovered in 1998 by Andrew Fire and Craig Mello at UMass Worcester. The two won the Nobel prize in medicine just 8 years later; that’s a really short time, highlighting just how important this discovery was. In contrast, Karikó and Weissman won the Nobel for mRNA vaccines this year, after inventing them 18 years ago.
siRNAs are the body’s way of targeting proteins for destruction before they are ever created. About 20 base pairs long, siRNAs seek out a complementary target mRNA, attach to it, and call in a group of proteins to destroy it. With the target mRNA gone, no protein can be created.
You see where this is going, right? How does high cholesterol kill you? Proteins. How does Staphylococcus aureus kill you? Proteins. Even viruses can’t replicate if their RNA is prevented from being turned into proteins.
So, how do we use siRNAs? A new paper appearing in JAMA describes a fairly impressive use case.
The background here is that higher levels of lipoprotein(a), an LDL-like protein, are associated with cardiovascular disease, heart attack, and stroke. But unfortunately, statins really don’t have any effect on lipoprotein(a) levels. Neither does diet. Your lipoprotein(a) level seems to be more or less hard-coded genetically.
So, what if we stop the genetic machinery from working? Enter lepodisiran, a drug from Eli Lilly. Unlike so many other medications, which are usually found in nature, purified, and synthesized, lepodisiran was created from scratch. It’s not hard. Thanks to the Human Genome Project, we know the genetic code for lipoprotein(a), so inventing an siRNA to target it specifically is trivial. That’s one of the key features of siRNA – you don’t have to find a chemical that binds strongly to some protein receptor, and worry about the off-target effects and all that nonsense. You just pick a protein you want to suppress and you suppress it.
Okay, it’s not that simple. siRNA is broken down very quickly by the body, so it needs to be targeted to the organ of interest – in this case, the liver, since that is where lipoprotein(a) is synthesized. Lepodisiran is targeted to the liver by this special targeting label here.
The report is a standard dose-escalation trial. Six patients, all with elevated lipoprotein(a) levels, were started with a 4-mg dose (two additional individuals got placebo). They were intensely monitored, spending 3 days in a research unit for multiple blood draws followed by weekly, and then biweekly outpatient visits. Once they had done well, the next group of six people received a higher dose (two more got placebo), and the process was repeated – six times total – until the highest dose, 608 mg, was reached.
This is an injection, of course; siRNA wouldn’t withstand the harshness of the digestive system. And it’s only one injection. You can see from the blood concentration curves that within about 48 hours, circulating lepodisiran was not detectable.
But check out these results. Remember, this is from a single injection of lepodisiran.
Lipoprotein(a) levels start to drop within a week of administration, and they stay down. In the higher-dose groups, levels are nearly undetectable a year after that injection.
It was this graph that made me sit back and think that there might be something new under the sun. A single injection that can suppress protein synthesis for an entire year? If it really works, it changes the game.
Of course, this study wasn’t powered to look at important outcomes like heart attacks and strokes. It was primarily designed to assess safety, and the drug was pretty well tolerated, with similar rates of adverse events in the drug and placebo groups.
As crazy as it sounds, the real concern here might be that this drug is too good; is it safe to drop your lipoprotein(a) levels to zero for a year? I don’t know. But lower doses don’t have quite as strong an effect.
Trust me, these drugs are going to change things. They already are. In July, The New England Journal of Medicine published a study of zilebesiran, an siRNA that inhibits the production of angiotensinogen, to control blood pressure. Similar story: One injection led to a basically complete suppression of angiotensinogen and a sustained decrease in blood pressure.
I’m not exaggerating when I say that there may come a time when you go to your doctor once a year, get your RNA shots, and don’t have to take any other medication from that point on. And that time may be, like, 5 years from now. It’s wild.
Seems to me that that rapid Nobel Prize was very well deserved.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships. This transcript has been edited for clarity.
A version of this article appeared on Medscape.com.
Even one night in the ED raises risk for death
This transcript has been edited for clarity.
As a consulting nephrologist, I go all over the hospital. Medicine floors, surgical floors, the ICU – I’ve even done consults in the operating room. And more and more, I do consults in the emergency department.
The reason I am doing more consults in the ED is not because the ED docs are getting gun shy with creatinine increases; it’s because patients are staying for extended periods in the ED despite being formally admitted to the hospital. It’s a phenomenon known as boarding, because there are simply not enough beds. You know the scene if you have ever been to a busy hospital: The ED is full to breaking, with patients on stretchers in hallways. It can often feel more like a warzone than a place for healing.
This is a huge problem.
The Joint Commission specifies that admitted patients should spend no more than 4 hours in the ED waiting for a bed in the hospital.
That is, based on what I’ve seen, hugely ambitious. But I should point out that I work in a hospital that runs near capacity all the time, and studies – from some of my Yale colleagues, actually – have shown that once hospital capacity exceeds 85%, boarding rates skyrocket.
I want to discuss some of the causes of extended boarding and some solutions. But before that, I should prove to you that this really matters, and for that we are going to dig in to a new study which suggests that ED boarding kills.
To put some hard numbers to the boarding problem, we turn to this paper out of France, appearing in JAMA Internal Medicine.
This is a unique study design. Basically, on a single day – Dec. 12, 2022 – researchers fanned out across France to 97 EDs and started counting patients. The study focused on those older than age 75 who were admitted to a hospital ward from the ED. The researchers then defined two groups: those who were sent up to the hospital floor before midnight, and those who spent at least from midnight until 8 AM in the ED (basically, people forced to sleep in the ED for a night). The middle-ground people who were sent up between midnight and 8 AM were excluded.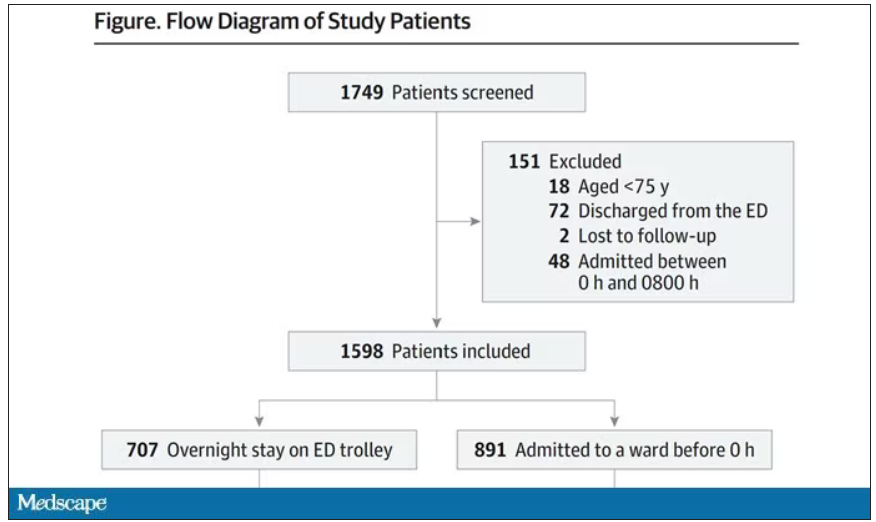
The baseline characteristics between the two groups of patients were pretty similar: median age around 86, 55% female. There were no significant differences in comorbidities. That said, comporting with previous studies, people in an urban ED, an academic ED, or a busy ED were much more likely to board overnight.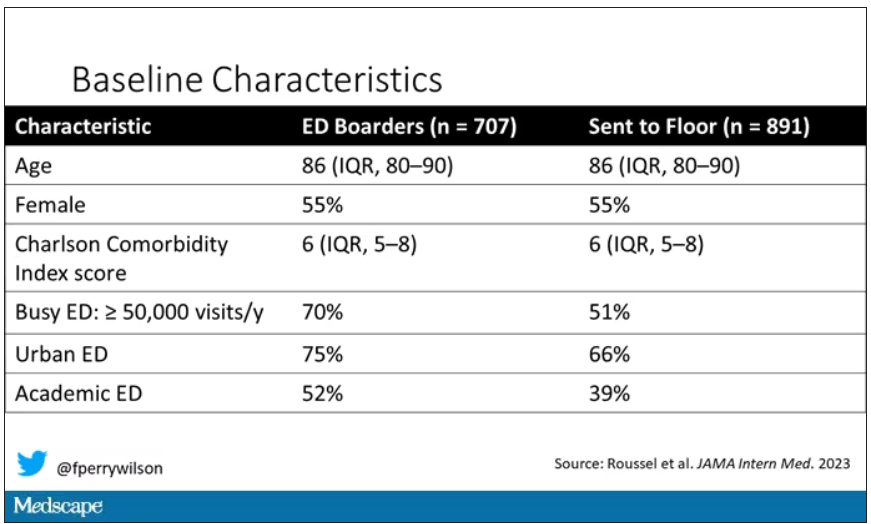
So, what we have are two similar groups of patients treated quite differently. Not quite a randomized trial, given the hospital differences, but not bad for purposes of analysis.
Here are the most important numbers from the trial:
This difference held up even after adjustment for patient and hospital characteristics. Put another way, you’d need to send 22 patients to the floor instead of boarding in the ED to save one life. Not a bad return on investment.
It’s not entirely clear what the mechanism for the excess mortality might be, but the researchers note that patients kept in the ED overnight were about twice as likely to have a fall during their hospital stay – not surprising, given the dangers of gurneys in hallways and the sleep deprivation that trying to rest in a busy ED engenders.
I should point out that this could be worse in the United States. French ED doctors continue to care for admitted patients boarding in the ED, whereas in many hospitals in the United States, admitted patients are the responsibility of the floor team, regardless of where they are, making it more likely that these individuals may be neglected.
So, if boarding in the ED is a life-threatening situation, why do we do it? What conditions predispose to this?
You’ll hear a lot of talk, mostly from hospital administrators, saying that this is simply a problem of supply and demand. There are not enough beds for the number of patients who need beds. And staffing shortages don’t help either.
However, they never want to talk about the reasons for the staffing shortages, like poor pay, poor support, and, of course, the moral injury of treating patients in hallways.
The issue of volume is real. We could do a lot to prevent ED visits and hospital admissions by providing better access to preventive and primary care and improving our outpatient mental health infrastructure. But I think this framing passes the buck a little.
Another reason ED boarding occurs is the way our health care system is paid for. If you are building a hospital, you have little incentive to build in excess capacity. The most efficient hospital, from a profit-and-loss standpoint, is one that is 100% full as often as possible. That may be fine at times, but throw in a respiratory virus or even a pandemic, and those systems fracture under the pressure.
Let us also remember that not all hospital beds are given to patients who acutely need hospital beds. Many beds, in many hospitals, are necessary to handle postoperative patients undergoing elective procedures. Those patients having a knee replacement or abdominoplasty don’t spend the night in the ED when they leave the OR; they go to a hospital bed. And those procedures are – let’s face it – more profitable than an ED admission for a medical issue. That’s why, even when hospitals expand the number of beds they have, they do it with an eye toward increasing the rate of those profitable procedures, not decreasing the burden faced by their ED.
For now, the band-aid to the solution might be to better triage individuals boarding in the ED for floor access, prioritizing those of older age, greater frailty, or more medical complexity. But it feels like a stop-gap measure as long as the incentives are aligned to view an empty hospital bed as a sign of failure in the health system instead of success.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
As a consulting nephrologist, I go all over the hospital. Medicine floors, surgical floors, the ICU – I’ve even done consults in the operating room. And more and more, I do consults in the emergency department.
The reason I am doing more consults in the ED is not because the ED docs are getting gun shy with creatinine increases; it’s because patients are staying for extended periods in the ED despite being formally admitted to the hospital. It’s a phenomenon known as boarding, because there are simply not enough beds. You know the scene if you have ever been to a busy hospital: The ED is full to breaking, with patients on stretchers in hallways. It can often feel more like a warzone than a place for healing.
This is a huge problem.
The Joint Commission specifies that admitted patients should spend no more than 4 hours in the ED waiting for a bed in the hospital.
That is, based on what I’ve seen, hugely ambitious. But I should point out that I work in a hospital that runs near capacity all the time, and studies – from some of my Yale colleagues, actually – have shown that once hospital capacity exceeds 85%, boarding rates skyrocket.
I want to discuss some of the causes of extended boarding and some solutions. But before that, I should prove to you that this really matters, and for that we are going to dig in to a new study which suggests that ED boarding kills.
To put some hard numbers to the boarding problem, we turn to this paper out of France, appearing in JAMA Internal Medicine.
This is a unique study design. Basically, on a single day – Dec. 12, 2022 – researchers fanned out across France to 97 EDs and started counting patients. The study focused on those older than age 75 who were admitted to a hospital ward from the ED. The researchers then defined two groups: those who were sent up to the hospital floor before midnight, and those who spent at least from midnight until 8 AM in the ED (basically, people forced to sleep in the ED for a night). The middle-ground people who were sent up between midnight and 8 AM were excluded.
The baseline characteristics between the two groups of patients were pretty similar: median age around 86, 55% female. There were no significant differences in comorbidities. That said, comporting with previous studies, people in an urban ED, an academic ED, or a busy ED were much more likely to board overnight.
So, what we have are two similar groups of patients treated quite differently. Not quite a randomized trial, given the hospital differences, but not bad for purposes of analysis.
Here are the most important numbers from the trial:
This difference held up even after adjustment for patient and hospital characteristics. Put another way, you’d need to send 22 patients to the floor instead of boarding in the ED to save one life. Not a bad return on investment.
It’s not entirely clear what the mechanism for the excess mortality might be, but the researchers note that patients kept in the ED overnight were about twice as likely to have a fall during their hospital stay – not surprising, given the dangers of gurneys in hallways and the sleep deprivation that trying to rest in a busy ED engenders.
I should point out that this could be worse in the United States. French ED doctors continue to care for admitted patients boarding in the ED, whereas in many hospitals in the United States, admitted patients are the responsibility of the floor team, regardless of where they are, making it more likely that these individuals may be neglected.
So, if boarding in the ED is a life-threatening situation, why do we do it? What conditions predispose to this?
You’ll hear a lot of talk, mostly from hospital administrators, saying that this is simply a problem of supply and demand. There are not enough beds for the number of patients who need beds. And staffing shortages don’t help either.
However, they never want to talk about the reasons for the staffing shortages, like poor pay, poor support, and, of course, the moral injury of treating patients in hallways.
The issue of volume is real. We could do a lot to prevent ED visits and hospital admissions by providing better access to preventive and primary care and improving our outpatient mental health infrastructure. But I think this framing passes the buck a little.
Another reason ED boarding occurs is the way our health care system is paid for. If you are building a hospital, you have little incentive to build in excess capacity. The most efficient hospital, from a profit-and-loss standpoint, is one that is 100% full as often as possible. That may be fine at times, but throw in a respiratory virus or even a pandemic, and those systems fracture under the pressure.
Let us also remember that not all hospital beds are given to patients who acutely need hospital beds. Many beds, in many hospitals, are necessary to handle postoperative patients undergoing elective procedures. Those patients having a knee replacement or abdominoplasty don’t spend the night in the ED when they leave the OR; they go to a hospital bed. And those procedures are – let’s face it – more profitable than an ED admission for a medical issue. That’s why, even when hospitals expand the number of beds they have, they do it with an eye toward increasing the rate of those profitable procedures, not decreasing the burden faced by their ED.
For now, the band-aid to the solution might be to better triage individuals boarding in the ED for floor access, prioritizing those of older age, greater frailty, or more medical complexity. But it feels like a stop-gap measure as long as the incentives are aligned to view an empty hospital bed as a sign of failure in the health system instead of success.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
As a consulting nephrologist, I go all over the hospital. Medicine floors, surgical floors, the ICU – I’ve even done consults in the operating room. And more and more, I do consults in the emergency department.
The reason I am doing more consults in the ED is not because the ED docs are getting gun shy with creatinine increases; it’s because patients are staying for extended periods in the ED despite being formally admitted to the hospital. It’s a phenomenon known as boarding, because there are simply not enough beds. You know the scene if you have ever been to a busy hospital: The ED is full to breaking, with patients on stretchers in hallways. It can often feel more like a warzone than a place for healing.
This is a huge problem.
The Joint Commission specifies that admitted patients should spend no more than 4 hours in the ED waiting for a bed in the hospital.
That is, based on what I’ve seen, hugely ambitious. But I should point out that I work in a hospital that runs near capacity all the time, and studies – from some of my Yale colleagues, actually – have shown that once hospital capacity exceeds 85%, boarding rates skyrocket.
I want to discuss some of the causes of extended boarding and some solutions. But before that, I should prove to you that this really matters, and for that we are going to dig in to a new study which suggests that ED boarding kills.
To put some hard numbers to the boarding problem, we turn to this paper out of France, appearing in JAMA Internal Medicine.
This is a unique study design. Basically, on a single day – Dec. 12, 2022 – researchers fanned out across France to 97 EDs and started counting patients. The study focused on those older than age 75 who were admitted to a hospital ward from the ED. The researchers then defined two groups: those who were sent up to the hospital floor before midnight, and those who spent at least from midnight until 8 AM in the ED (basically, people forced to sleep in the ED for a night). The middle-ground people who were sent up between midnight and 8 AM were excluded.
The baseline characteristics between the two groups of patients were pretty similar: median age around 86, 55% female. There were no significant differences in comorbidities. That said, comporting with previous studies, people in an urban ED, an academic ED, or a busy ED were much more likely to board overnight.
So, what we have are two similar groups of patients treated quite differently. Not quite a randomized trial, given the hospital differences, but not bad for purposes of analysis.
Here are the most important numbers from the trial:
This difference held up even after adjustment for patient and hospital characteristics. Put another way, you’d need to send 22 patients to the floor instead of boarding in the ED to save one life. Not a bad return on investment.
It’s not entirely clear what the mechanism for the excess mortality might be, but the researchers note that patients kept in the ED overnight were about twice as likely to have a fall during their hospital stay – not surprising, given the dangers of gurneys in hallways and the sleep deprivation that trying to rest in a busy ED engenders.
I should point out that this could be worse in the United States. French ED doctors continue to care for admitted patients boarding in the ED, whereas in many hospitals in the United States, admitted patients are the responsibility of the floor team, regardless of where they are, making it more likely that these individuals may be neglected.
So, if boarding in the ED is a life-threatening situation, why do we do it? What conditions predispose to this?
You’ll hear a lot of talk, mostly from hospital administrators, saying that this is simply a problem of supply and demand. There are not enough beds for the number of patients who need beds. And staffing shortages don’t help either.
However, they never want to talk about the reasons for the staffing shortages, like poor pay, poor support, and, of course, the moral injury of treating patients in hallways.
The issue of volume is real. We could do a lot to prevent ED visits and hospital admissions by providing better access to preventive and primary care and improving our outpatient mental health infrastructure. But I think this framing passes the buck a little.
Another reason ED boarding occurs is the way our health care system is paid for. If you are building a hospital, you have little incentive to build in excess capacity. The most efficient hospital, from a profit-and-loss standpoint, is one that is 100% full as often as possible. That may be fine at times, but throw in a respiratory virus or even a pandemic, and those systems fracture under the pressure.
Let us also remember that not all hospital beds are given to patients who acutely need hospital beds. Many beds, in many hospitals, are necessary to handle postoperative patients undergoing elective procedures. Those patients having a knee replacement or abdominoplasty don’t spend the night in the ED when they leave the OR; they go to a hospital bed. And those procedures are – let’s face it – more profitable than an ED admission for a medical issue. That’s why, even when hospitals expand the number of beds they have, they do it with an eye toward increasing the rate of those profitable procedures, not decreasing the burden faced by their ED.
For now, the band-aid to the solution might be to better triage individuals boarding in the ED for floor access, prioritizing those of older age, greater frailty, or more medical complexity. But it feels like a stop-gap measure as long as the incentives are aligned to view an empty hospital bed as a sign of failure in the health system instead of success.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This drug works, but wait till you hear what’s in it
This transcript has been edited for clarity.
As some of you may know, I do a fair amount of clinical research developing and evaluating artificial intelligence (AI) models, particularly machine learning algorithms that predict certain outcomes.
A thorny issue that comes up as algorithms have gotten more complicated is “explainability.” The problem is that AI can be a black box. Even if you have a model that is very accurate at predicting death, clinicians don’t trust it unless you can explain how it makes its predictions – how it works. “It just works” is not good enough to build trust.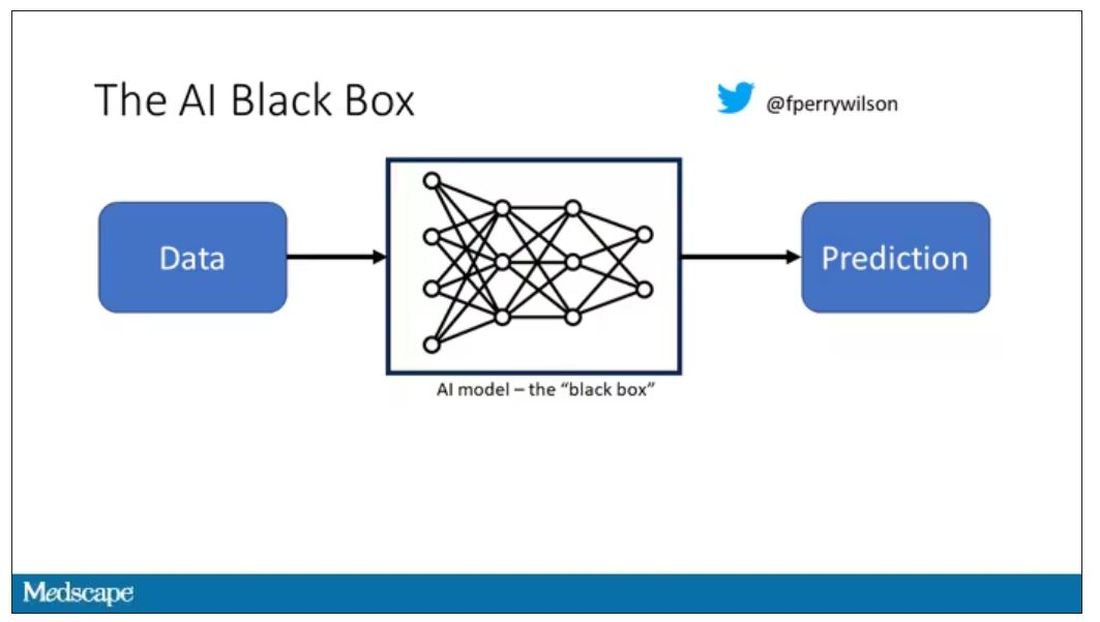
It’s easier to build trust when you’re talking about a medication rather than a computer program. When a new blood pressure drug comes out that lowers blood pressure, importantly, we know why it lowers blood pressure. Every drug has a mechanism of action and, for most of the drugs in our arsenal, we know what that mechanism is.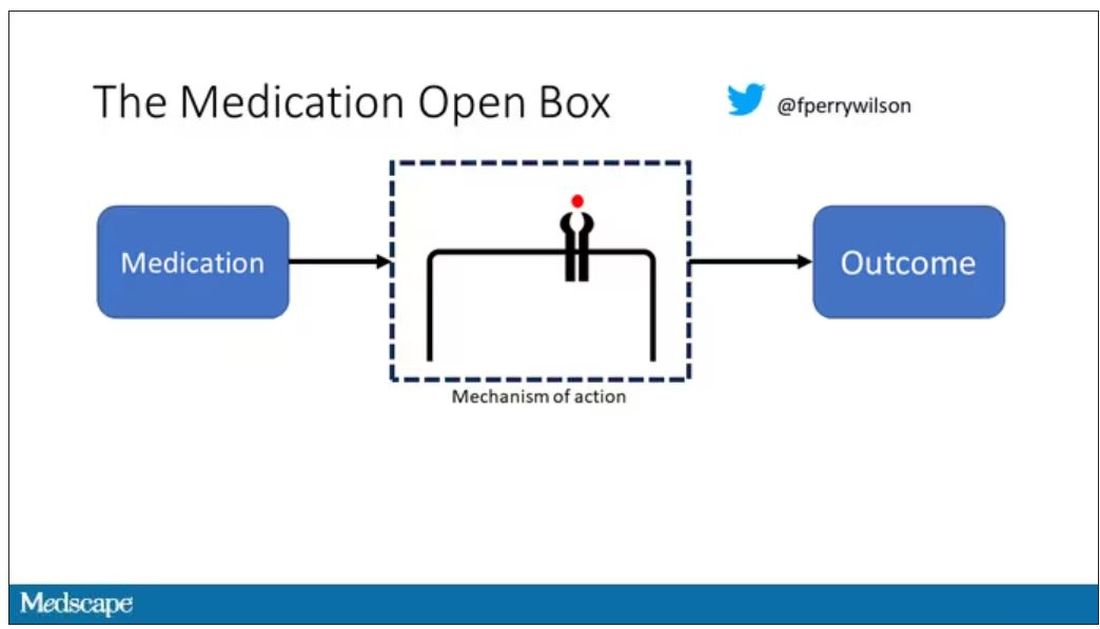
But what if there were a drug – or better yet, a treatment – that worked? And I can honestly say we have no idea how it works. That’s what came across my desk today in what I believe is the largest, most rigorous trial of a traditional Chinese medication in history.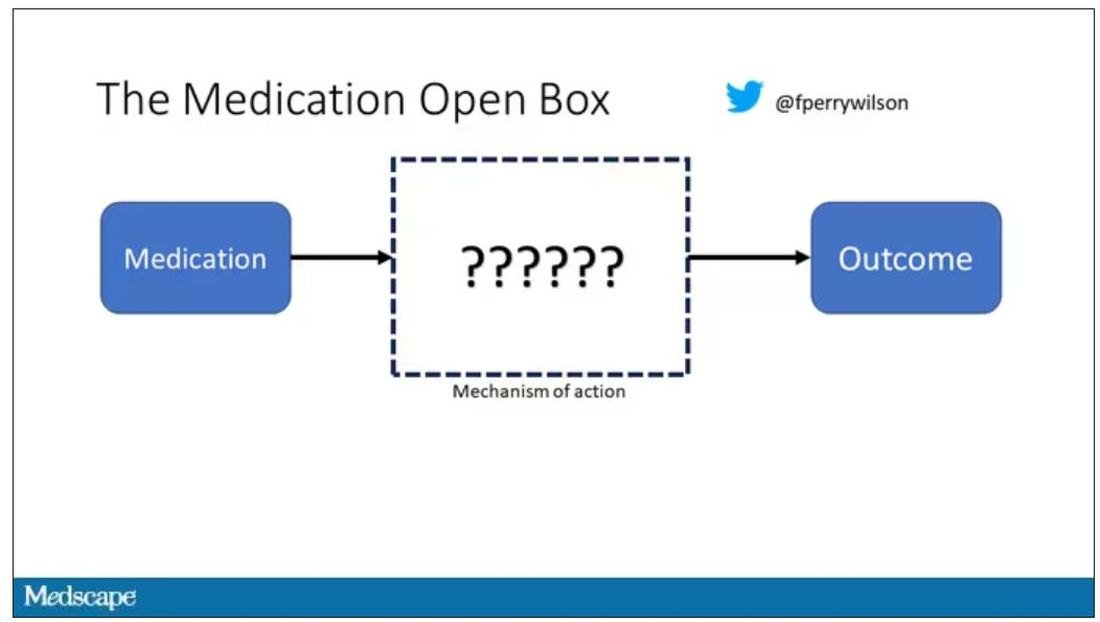
“Traditional Chinese medicine” is an omnibus term that refers to a class of therapies and health practices that are fundamentally different from how we practice medicine in the West.
It’s a highly personalized practice, with practitioners using often esoteric means to choose what substance to give what patient. That personalization makes traditional Chinese medicine nearly impossible to study in the typical randomized trial framework because treatments are not chosen solely on the basis of disease states.
The lack of scientific rigor in traditional Chinese medicine means that it is rife with practices and beliefs that can legitimately be called pseudoscience. As a nephrologist who has treated someone for “Chinese herb nephropathy,” I can tell you that some of the practices may be actively harmful.
But that doesn’t mean there is nothing there. I do not subscribe to the “argument from antiquity” – the idea that because something has been done for a long time it must be correct. But at the same time, traditional and non–science-based medicine practices could still identify therapies that work.
And with that, let me introduce you to Tongxinluo. Tongxinluo literally means “to open the network of the heart,” and it is a substance that has been used for centuries by traditional Chinese medicine practitioners to treat angina but was approved by the Chinese state medicine agency for use in 1996.
Like many traditional Chinese medicine preparations, Tongxinluo is not a single chemical – far from it. It is a powder made from a variety of plant and insect parts, as you can see here.
I can’t imagine running a trial of this concoction in the United States; I just don’t see an institutional review board signing off, given the ingredient list.
But let’s set that aside and talk about the study itself.
While I don’t have access to any primary data, the write-up of the study suggests that it was highly rigorous. Chinese researchers randomized 3,797 patients with ST-elevation MI to take Tongxinluo – four capsules, three times a day for 12 months – or matching placebo. The placebo was designed to look just like the Tongxinluo capsules and, if the capsules were opened, to smell like them as well.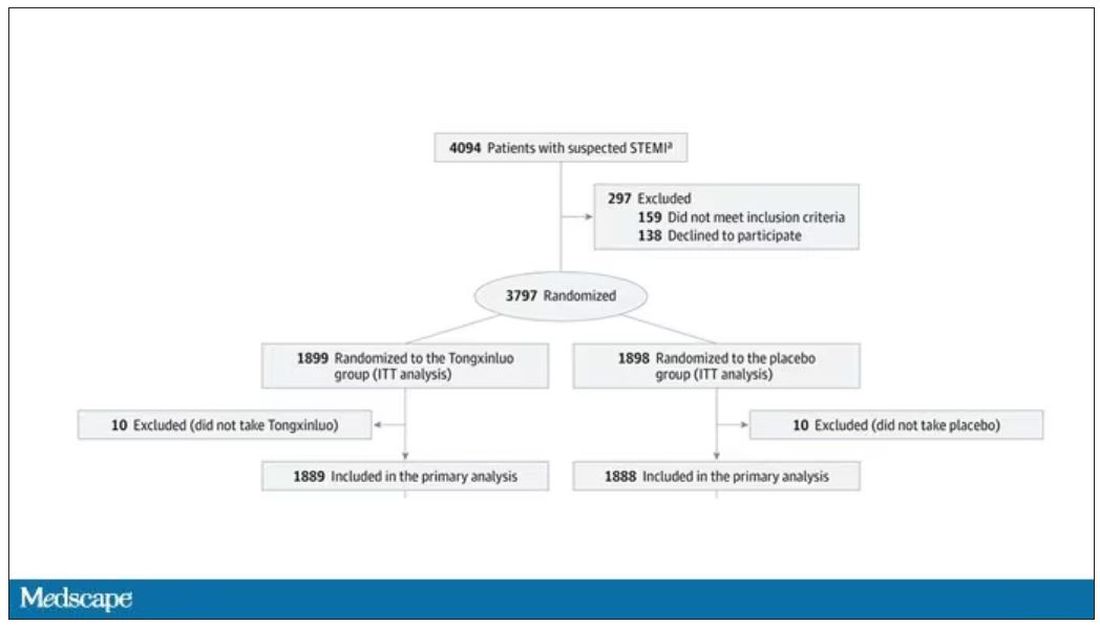
Researchers and participants were blinded, and the statistical analysis was done both by the primary team and an independent research agency, also in China.
And the results were pretty good. The primary outcome, 30-day major cardiovascular and cerebral events, were significantly lower in the intervention group than in the placebo group.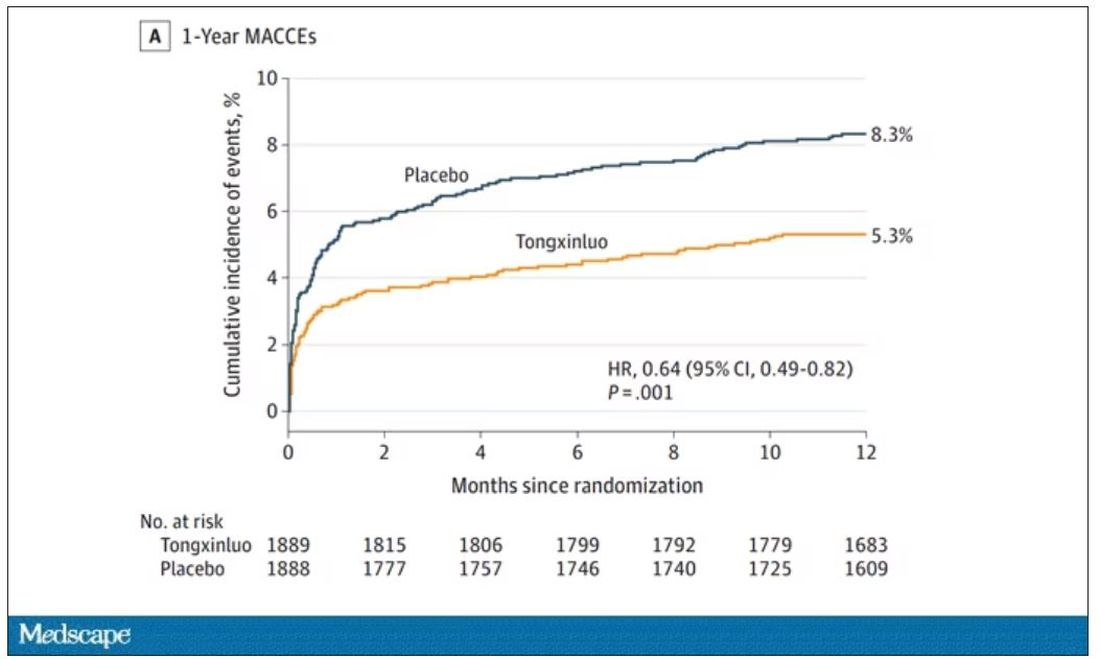
One-year outcomes were similarly good; 8.3% of the placebo group suffered a major cardiovascular or cerebral event in that time frame, compared with 5.3% of the Tongxinluo group. In short, if this were a pure chemical compound from a major pharmaceutical company, well, you might be seeing a new treatment for heart attack – and a boost in stock price.
But there are some issues here, generalizability being a big one. This study was done entirely in China, so its applicability to a more diverse population is unclear. Moreover, the quality of post-MI care in this study is quite a bit worse than what we’d see here in the United States, with just over 50% of patients being discharged on a beta-blocker, for example.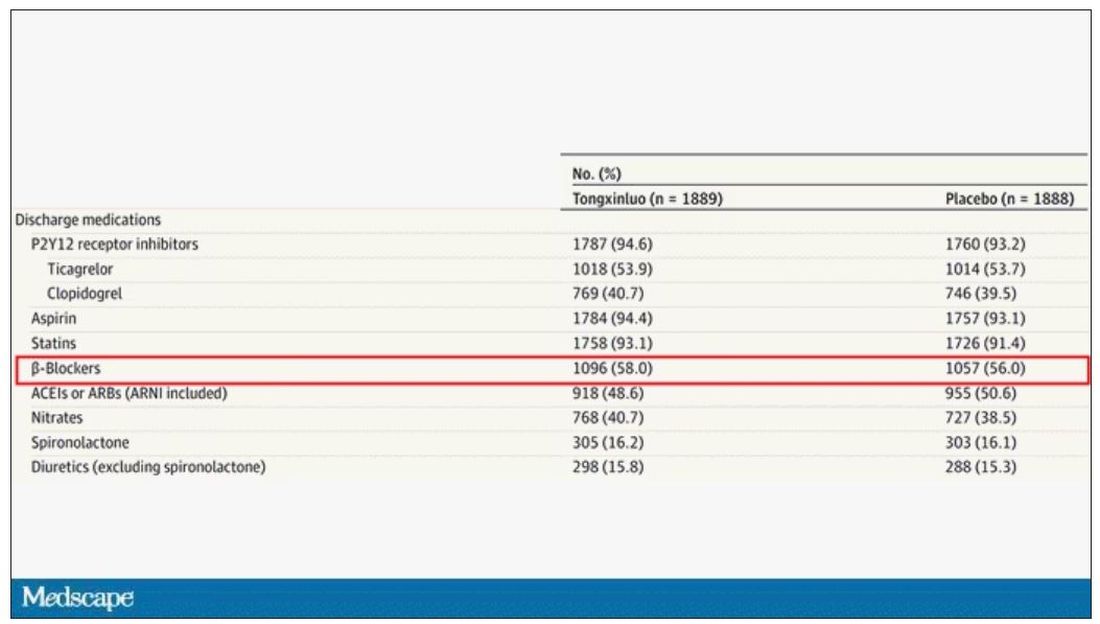
But issues of generalizability and potentially substandard supplementary treatments are the usual reasons we worry about new medication trials. And those concerns seem to pale before the big one I have here which is, you know – we don’t know why this works.
Is it the extract of leech in the preparation perhaps thinning the blood a bit? Or is it the antioxidants in the ginseng, or something from the Pacific centipede or the sandalwood?
This trial doesn’t read to me as a vindication of traditional Chinese medicine but rather as an example of missed opportunity. More rigorous scientific study over the centuries that Tongxinluo has been used could have identified one, or perhaps more, compounds with strong therapeutic potential.
Purity of medical substances is incredibly important. Pure substances have predictable effects and side effects. Pure substances interact with other treatments we give patients in predictable ways. Pure substances can be quantified for purity by third parties, they can be manufactured according to accepted standards, and they can be assessed for adulteration. In short, pure substances pose less risk.
Now, I know that may come off as particularly sterile. Some people will feel that a “natural” substance has some inherent benefit over pure compounds. And, of course, there is something soothing about imagining a traditional preparation handed down over centuries, being prepared with care by a single practitioner, in contrast to the sterile industrial processes of a for-profit pharmaceutical company. I get it. But natural is not the same as safe. I am glad I have access to purified aspirin and don’t have to chew willow bark. I like my pure penicillin and am glad I don’t have to make a mold slurry to treat a bacterial infection.
I applaud the researchers for subjecting Tongxinluo to the rigor of a well-designed trial. They have generated data that are incredibly exciting, but not because we have a new treatment for ST-elevation MI on our hands; it’s because we have a map to a new treatment. The next big thing in heart attack care is not the mixture that is Tongxinluo, but it might be in the mixture.
A version of this article first appeared on Medscape.com.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and on Medscape. He tweets @fperrywilson and his new book, “How Medicine Works and When It Doesn’t,” is available now.
This transcript has been edited for clarity.
As some of you may know, I do a fair amount of clinical research developing and evaluating artificial intelligence (AI) models, particularly machine learning algorithms that predict certain outcomes.
A thorny issue that comes up as algorithms have gotten more complicated is “explainability.” The problem is that AI can be a black box. Even if you have a model that is very accurate at predicting death, clinicians don’t trust it unless you can explain how it makes its predictions – how it works. “It just works” is not good enough to build trust.
It’s easier to build trust when you’re talking about a medication rather than a computer program. When a new blood pressure drug comes out that lowers blood pressure, importantly, we know why it lowers blood pressure. Every drug has a mechanism of action and, for most of the drugs in our arsenal, we know what that mechanism is.
But what if there were a drug – or better yet, a treatment – that worked? And I can honestly say we have no idea how it works. That’s what came across my desk today in what I believe is the largest, most rigorous trial of a traditional Chinese medication in history.
“Traditional Chinese medicine” is an omnibus term that refers to a class of therapies and health practices that are fundamentally different from how we practice medicine in the West.
It’s a highly personalized practice, with practitioners using often esoteric means to choose what substance to give what patient. That personalization makes traditional Chinese medicine nearly impossible to study in the typical randomized trial framework because treatments are not chosen solely on the basis of disease states.
The lack of scientific rigor in traditional Chinese medicine means that it is rife with practices and beliefs that can legitimately be called pseudoscience. As a nephrologist who has treated someone for “Chinese herb nephropathy,” I can tell you that some of the practices may be actively harmful.
But that doesn’t mean there is nothing there. I do not subscribe to the “argument from antiquity” – the idea that because something has been done for a long time it must be correct. But at the same time, traditional and non–science-based medicine practices could still identify therapies that work.
And with that, let me introduce you to Tongxinluo. Tongxinluo literally means “to open the network of the heart,” and it is a substance that has been used for centuries by traditional Chinese medicine practitioners to treat angina but was approved by the Chinese state medicine agency for use in 1996.
Like many traditional Chinese medicine preparations, Tongxinluo is not a single chemical – far from it. It is a powder made from a variety of plant and insect parts, as you can see here.
I can’t imagine running a trial of this concoction in the United States; I just don’t see an institutional review board signing off, given the ingredient list.
But let’s set that aside and talk about the study itself.
While I don’t have access to any primary data, the write-up of the study suggests that it was highly rigorous. Chinese researchers randomized 3,797 patients with ST-elevation MI to take Tongxinluo – four capsules, three times a day for 12 months – or matching placebo. The placebo was designed to look just like the Tongxinluo capsules and, if the capsules were opened, to smell like them as well.
Researchers and participants were blinded, and the statistical analysis was done both by the primary team and an independent research agency, also in China.
And the results were pretty good. The primary outcome, 30-day major cardiovascular and cerebral events, were significantly lower in the intervention group than in the placebo group.
One-year outcomes were similarly good; 8.3% of the placebo group suffered a major cardiovascular or cerebral event in that time frame, compared with 5.3% of the Tongxinluo group. In short, if this were a pure chemical compound from a major pharmaceutical company, well, you might be seeing a new treatment for heart attack – and a boost in stock price.
But there are some issues here, generalizability being a big one. This study was done entirely in China, so its applicability to a more diverse population is unclear. Moreover, the quality of post-MI care in this study is quite a bit worse than what we’d see here in the United States, with just over 50% of patients being discharged on a beta-blocker, for example.
But issues of generalizability and potentially substandard supplementary treatments are the usual reasons we worry about new medication trials. And those concerns seem to pale before the big one I have here which is, you know – we don’t know why this works.
Is it the extract of leech in the preparation perhaps thinning the blood a bit? Or is it the antioxidants in the ginseng, or something from the Pacific centipede or the sandalwood?
This trial doesn’t read to me as a vindication of traditional Chinese medicine but rather as an example of missed opportunity. More rigorous scientific study over the centuries that Tongxinluo has been used could have identified one, or perhaps more, compounds with strong therapeutic potential.
Purity of medical substances is incredibly important. Pure substances have predictable effects and side effects. Pure substances interact with other treatments we give patients in predictable ways. Pure substances can be quantified for purity by third parties, they can be manufactured according to accepted standards, and they can be assessed for adulteration. In short, pure substances pose less risk.
Now, I know that may come off as particularly sterile. Some people will feel that a “natural” substance has some inherent benefit over pure compounds. And, of course, there is something soothing about imagining a traditional preparation handed down over centuries, being prepared with care by a single practitioner, in contrast to the sterile industrial processes of a for-profit pharmaceutical company. I get it. But natural is not the same as safe. I am glad I have access to purified aspirin and don’t have to chew willow bark. I like my pure penicillin and am glad I don’t have to make a mold slurry to treat a bacterial infection.
I applaud the researchers for subjecting Tongxinluo to the rigor of a well-designed trial. They have generated data that are incredibly exciting, but not because we have a new treatment for ST-elevation MI on our hands; it’s because we have a map to a new treatment. The next big thing in heart attack care is not the mixture that is Tongxinluo, but it might be in the mixture.
A version of this article first appeared on Medscape.com.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and on Medscape. He tweets @fperrywilson and his new book, “How Medicine Works and When It Doesn’t,” is available now.
This transcript has been edited for clarity.
As some of you may know, I do a fair amount of clinical research developing and evaluating artificial intelligence (AI) models, particularly machine learning algorithms that predict certain outcomes.
A thorny issue that comes up as algorithms have gotten more complicated is “explainability.” The problem is that AI can be a black box. Even if you have a model that is very accurate at predicting death, clinicians don’t trust it unless you can explain how it makes its predictions – how it works. “It just works” is not good enough to build trust.
It’s easier to build trust when you’re talking about a medication rather than a computer program. When a new blood pressure drug comes out that lowers blood pressure, importantly, we know why it lowers blood pressure. Every drug has a mechanism of action and, for most of the drugs in our arsenal, we know what that mechanism is.
But what if there were a drug – or better yet, a treatment – that worked? And I can honestly say we have no idea how it works. That’s what came across my desk today in what I believe is the largest, most rigorous trial of a traditional Chinese medication in history.
“Traditional Chinese medicine” is an omnibus term that refers to a class of therapies and health practices that are fundamentally different from how we practice medicine in the West.
It’s a highly personalized practice, with practitioners using often esoteric means to choose what substance to give what patient. That personalization makes traditional Chinese medicine nearly impossible to study in the typical randomized trial framework because treatments are not chosen solely on the basis of disease states.
The lack of scientific rigor in traditional Chinese medicine means that it is rife with practices and beliefs that can legitimately be called pseudoscience. As a nephrologist who has treated someone for “Chinese herb nephropathy,” I can tell you that some of the practices may be actively harmful.
But that doesn’t mean there is nothing there. I do not subscribe to the “argument from antiquity” – the idea that because something has been done for a long time it must be correct. But at the same time, traditional and non–science-based medicine practices could still identify therapies that work.
And with that, let me introduce you to Tongxinluo. Tongxinluo literally means “to open the network of the heart,” and it is a substance that has been used for centuries by traditional Chinese medicine practitioners to treat angina but was approved by the Chinese state medicine agency for use in 1996.
Like many traditional Chinese medicine preparations, Tongxinluo is not a single chemical – far from it. It is a powder made from a variety of plant and insect parts, as you can see here.
I can’t imagine running a trial of this concoction in the United States; I just don’t see an institutional review board signing off, given the ingredient list.
But let’s set that aside and talk about the study itself.
While I don’t have access to any primary data, the write-up of the study suggests that it was highly rigorous. Chinese researchers randomized 3,797 patients with ST-elevation MI to take Tongxinluo – four capsules, three times a day for 12 months – or matching placebo. The placebo was designed to look just like the Tongxinluo capsules and, if the capsules were opened, to smell like them as well.
Researchers and participants were blinded, and the statistical analysis was done both by the primary team and an independent research agency, also in China.
And the results were pretty good. The primary outcome, 30-day major cardiovascular and cerebral events, were significantly lower in the intervention group than in the placebo group.
One-year outcomes were similarly good; 8.3% of the placebo group suffered a major cardiovascular or cerebral event in that time frame, compared with 5.3% of the Tongxinluo group. In short, if this were a pure chemical compound from a major pharmaceutical company, well, you might be seeing a new treatment for heart attack – and a boost in stock price.
But there are some issues here, generalizability being a big one. This study was done entirely in China, so its applicability to a more diverse population is unclear. Moreover, the quality of post-MI care in this study is quite a bit worse than what we’d see here in the United States, with just over 50% of patients being discharged on a beta-blocker, for example.
But issues of generalizability and potentially substandard supplementary treatments are the usual reasons we worry about new medication trials. And those concerns seem to pale before the big one I have here which is, you know – we don’t know why this works.
Is it the extract of leech in the preparation perhaps thinning the blood a bit? Or is it the antioxidants in the ginseng, or something from the Pacific centipede or the sandalwood?
This trial doesn’t read to me as a vindication of traditional Chinese medicine but rather as an example of missed opportunity. More rigorous scientific study over the centuries that Tongxinluo has been used could have identified one, or perhaps more, compounds with strong therapeutic potential.
Purity of medical substances is incredibly important. Pure substances have predictable effects and side effects. Pure substances interact with other treatments we give patients in predictable ways. Pure substances can be quantified for purity by third parties, they can be manufactured according to accepted standards, and they can be assessed for adulteration. In short, pure substances pose less risk.
Now, I know that may come off as particularly sterile. Some people will feel that a “natural” substance has some inherent benefit over pure compounds. And, of course, there is something soothing about imagining a traditional preparation handed down over centuries, being prepared with care by a single practitioner, in contrast to the sterile industrial processes of a for-profit pharmaceutical company. I get it. But natural is not the same as safe. I am glad I have access to purified aspirin and don’t have to chew willow bark. I like my pure penicillin and am glad I don’t have to make a mold slurry to treat a bacterial infection.
I applaud the researchers for subjecting Tongxinluo to the rigor of a well-designed trial. They have generated data that are incredibly exciting, but not because we have a new treatment for ST-elevation MI on our hands; it’s because we have a map to a new treatment. The next big thing in heart attack care is not the mixture that is Tongxinluo, but it might be in the mixture.
A version of this article first appeared on Medscape.com.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and on Medscape. He tweets @fperrywilson and his new book, “How Medicine Works and When It Doesn’t,” is available now.
AI in medicine has a major Cassandra problem
This transcript has been edited for clarity.
Today I’m going to talk to you about a study at the cutting edge of modern medicine, one that uses an artificial intelligence (AI) model to guide care. But before I do, I need to take you back to the late Bronze Age, to a city located on the coast of what is now Turkey.
Troy’s towering walls made it seem unassailable, but that would not stop the Achaeans and their fleet of black ships from making landfall, and, after a siege, destroying the city. The destruction of Troy, as told in the Iliad and the Aeneid, was foretold by Cassandra, the daughter of King Priam and Priestess of Troy.
Cassandra had been given the gift of prophecy by the god Apollo in exchange for her favors. But after the gift was bestowed, she rejected the bright god and, in his rage, he added a curse to her blessing: that no one would ever believe her prophecies.
Thus it was that when her brother Paris set off to Sparta to abduct Helen, she warned him that his actions would lead to the downfall of their great city. He, of course, ignored her.
And you know the rest of the story.
Why am I telling you the story of Cassandra of Troy when we’re supposed to be talking about AI in medicine? Because AI has a major Cassandra problem.
The recent history of AI, and particularly the subset of AI known as machine learning in medicine, has been characterized by an accuracy arms race.
The electronic health record allows for the collection of volumes of data orders of magnitude greater than what we have ever been able to collect before. And all that data can be crunched by various algorithms to make predictions about, well, anything – whether a patient will be transferred to the intensive care unit, whether a GI bleed will need an intervention, whether someone will die in the next year.
Studies in this area tend to rely on retrospective datasets, and as time has gone on, better algorithms and more data have led to better and better predictions. In some simpler cases, machine-learning models have achieved near-perfect accuracy – Cassandra-level accuracy – as in the reading of chest x-rays for pneumonia, for example.
But as Cassandra teaches us, even perfect prediction is useless if no one believes you, if they don’t change their behavior. And this is the central problem of AI in medicine today. Many people are focusing on accuracy of the prediction but have forgotten that high accuracy is just table stakes for an AI model to be useful. It has to not only be accurate, but its use also has to change outcomes for patients. We need to be able to save Troy.
The best way to determine whether an AI model will help patients is to treat a model like we treat a new medication and evaluate it through a randomized trial. That’s what researchers, led by Shannon Walker of Vanderbilt University, Nashville, Tenn., did in a paper appearing in JAMA Network Open.
The model in question was one that predicted venous thromboembolism – blood clots – in hospitalized children. The model took in a variety of data points from the health record: a history of blood clot, history of cancer, presence of a central line, a variety of lab values. And the predictive model was very good – maybe not Cassandra good, but it achieved an AUC of 0.90, which means it had very high accuracy.
But again, accuracy is just table stakes.
The authors deployed the model in the live health record and recorded the results. For half of the kids, that was all that happened; no one actually saw the predictions. For those randomized to the intervention, the hematology team would be notified when the risk for clot was calculated to be greater than 2.5%. The hematology team would then contact the primary team to discuss prophylactic anticoagulation.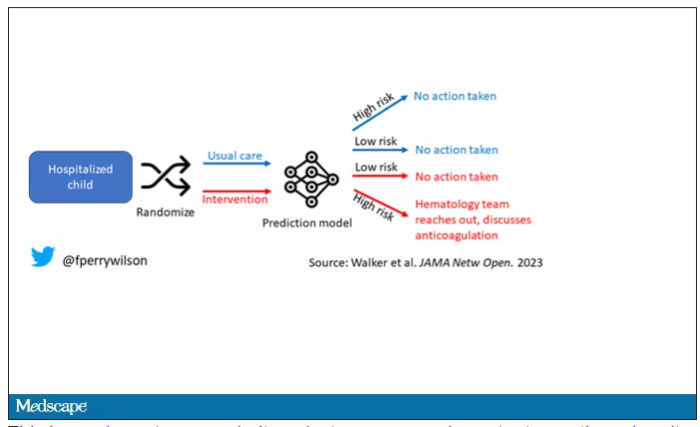
This is an elegant approach.
Let’s start with those table stakes – accuracy. The predictions were, by and large, pretty accurate in this trial. Of the 135 kids who developed blood clots, 121 had been flagged by the model in advance. That’s about 90%. The model flagged about 10% of kids who didn’t get a blood clot as well, but that’s not entirely surprising since the threshold for flagging was a 2.5% risk.
Given that the model preidentified almost every kid who would go on to develop a blood clot, it would make sense that kids randomized to the intervention would do better; after all, Cassandra was calling out her warnings.
But those kids didn’t do better. The rate of blood clot was no different between the group that used the accurate prediction model and the group that did not.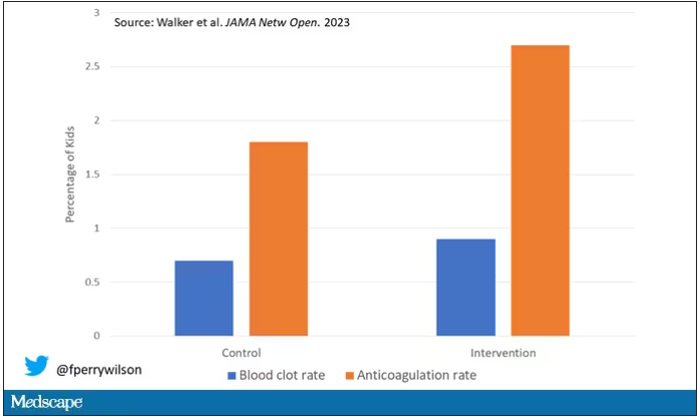
Why? Why does the use of an accurate model not necessarily improve outcomes?
First of all, a warning must lead to some change in management. Indeed, the kids in the intervention group were more likely to receive anticoagulation, but barely so. There were lots of reasons for this: physician preference, imminent discharge, active bleeding, and so on.
But let’s take a look at the 77 kids in the intervention arm who developed blood clots, because I think this is an instructive analysis.
Six of them did not meet the 2.5% threshold criteria, a case where the model missed its mark. Again, accuracy is table stakes.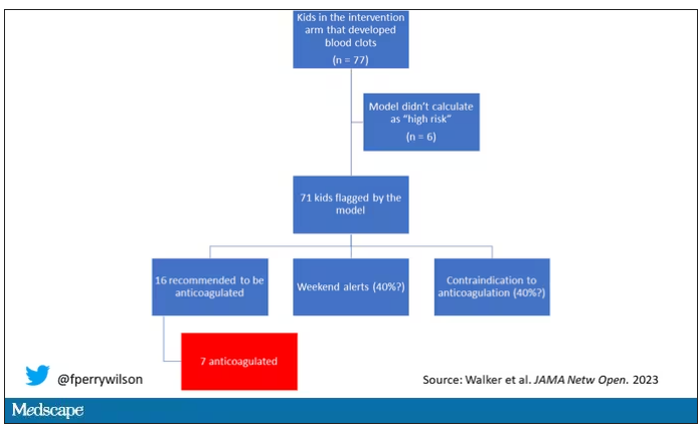
Of the remaining 71, only 16 got a recommendation from the hematologist to start anticoagulation. Why not more? Well, the model identified some of the high-risk kids on the weekend, and it seems that the study team did not contact treatment teams during that time. That may account for about 40% of these cases. The remainder had some contraindication to anticoagulation.
Most tellingly, of the 16 who did get a recommendation to start anticoagulation, the recommendation was followed in only seven patients.
This is the gap between accurate prediction and the ability to change outcomes for patients. A prediction is useless if it is wrong, for sure. But it’s also useless if you don’t tell anyone about it. It’s useless if you tell someone but they can’t do anything about it. And it’s useless if they could do something about it but choose not to.
That’s the gulf that these models need to cross at this point. So, the next time some slick company tells you how accurate their AI model is, ask them if accuracy is really the most important thing. If they say, “Well, yes, of course,” then tell them about Cassandra.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Today I’m going to talk to you about a study at the cutting edge of modern medicine, one that uses an artificial intelligence (AI) model to guide care. But before I do, I need to take you back to the late Bronze Age, to a city located on the coast of what is now Turkey.
Troy’s towering walls made it seem unassailable, but that would not stop the Achaeans and their fleet of black ships from making landfall, and, after a siege, destroying the city. The destruction of Troy, as told in the Iliad and the Aeneid, was foretold by Cassandra, the daughter of King Priam and Priestess of Troy.
Cassandra had been given the gift of prophecy by the god Apollo in exchange for her favors. But after the gift was bestowed, she rejected the bright god and, in his rage, he added a curse to her blessing: that no one would ever believe her prophecies.
Thus it was that when her brother Paris set off to Sparta to abduct Helen, she warned him that his actions would lead to the downfall of their great city. He, of course, ignored her.
And you know the rest of the story.
Why am I telling you the story of Cassandra of Troy when we’re supposed to be talking about AI in medicine? Because AI has a major Cassandra problem.
The recent history of AI, and particularly the subset of AI known as machine learning in medicine, has been characterized by an accuracy arms race.
The electronic health record allows for the collection of volumes of data orders of magnitude greater than what we have ever been able to collect before. And all that data can be crunched by various algorithms to make predictions about, well, anything – whether a patient will be transferred to the intensive care unit, whether a GI bleed will need an intervention, whether someone will die in the next year.
Studies in this area tend to rely on retrospective datasets, and as time has gone on, better algorithms and more data have led to better and better predictions. In some simpler cases, machine-learning models have achieved near-perfect accuracy – Cassandra-level accuracy – as in the reading of chest x-rays for pneumonia, for example.
But as Cassandra teaches us, even perfect prediction is useless if no one believes you, if they don’t change their behavior. And this is the central problem of AI in medicine today. Many people are focusing on accuracy of the prediction but have forgotten that high accuracy is just table stakes for an AI model to be useful. It has to not only be accurate, but its use also has to change outcomes for patients. We need to be able to save Troy.
The best way to determine whether an AI model will help patients is to treat a model like we treat a new medication and evaluate it through a randomized trial. That’s what researchers, led by Shannon Walker of Vanderbilt University, Nashville, Tenn., did in a paper appearing in JAMA Network Open.
The model in question was one that predicted venous thromboembolism – blood clots – in hospitalized children. The model took in a variety of data points from the health record: a history of blood clot, history of cancer, presence of a central line, a variety of lab values. And the predictive model was very good – maybe not Cassandra good, but it achieved an AUC of 0.90, which means it had very high accuracy.
But again, accuracy is just table stakes.
The authors deployed the model in the live health record and recorded the results. For half of the kids, that was all that happened; no one actually saw the predictions. For those randomized to the intervention, the hematology team would be notified when the risk for clot was calculated to be greater than 2.5%. The hematology team would then contact the primary team to discuss prophylactic anticoagulation.
This is an elegant approach.
Let’s start with those table stakes – accuracy. The predictions were, by and large, pretty accurate in this trial. Of the 135 kids who developed blood clots, 121 had been flagged by the model in advance. That’s about 90%. The model flagged about 10% of kids who didn’t get a blood clot as well, but that’s not entirely surprising since the threshold for flagging was a 2.5% risk.
Given that the model preidentified almost every kid who would go on to develop a blood clot, it would make sense that kids randomized to the intervention would do better; after all, Cassandra was calling out her warnings.
But those kids didn’t do better. The rate of blood clot was no different between the group that used the accurate prediction model and the group that did not.
Why? Why does the use of an accurate model not necessarily improve outcomes?
First of all, a warning must lead to some change in management. Indeed, the kids in the intervention group were more likely to receive anticoagulation, but barely so. There were lots of reasons for this: physician preference, imminent discharge, active bleeding, and so on.
But let’s take a look at the 77 kids in the intervention arm who developed blood clots, because I think this is an instructive analysis.
Six of them did not meet the 2.5% threshold criteria, a case where the model missed its mark. Again, accuracy is table stakes.
Of the remaining 71, only 16 got a recommendation from the hematologist to start anticoagulation. Why not more? Well, the model identified some of the high-risk kids on the weekend, and it seems that the study team did not contact treatment teams during that time. That may account for about 40% of these cases. The remainder had some contraindication to anticoagulation.
Most tellingly, of the 16 who did get a recommendation to start anticoagulation, the recommendation was followed in only seven patients.
This is the gap between accurate prediction and the ability to change outcomes for patients. A prediction is useless if it is wrong, for sure. But it’s also useless if you don’t tell anyone about it. It’s useless if you tell someone but they can’t do anything about it. And it’s useless if they could do something about it but choose not to.
That’s the gulf that these models need to cross at this point. So, the next time some slick company tells you how accurate their AI model is, ask them if accuracy is really the most important thing. If they say, “Well, yes, of course,” then tell them about Cassandra.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Today I’m going to talk to you about a study at the cutting edge of modern medicine, one that uses an artificial intelligence (AI) model to guide care. But before I do, I need to take you back to the late Bronze Age, to a city located on the coast of what is now Turkey.
Troy’s towering walls made it seem unassailable, but that would not stop the Achaeans and their fleet of black ships from making landfall, and, after a siege, destroying the city. The destruction of Troy, as told in the Iliad and the Aeneid, was foretold by Cassandra, the daughter of King Priam and Priestess of Troy.
Cassandra had been given the gift of prophecy by the god Apollo in exchange for her favors. But after the gift was bestowed, she rejected the bright god and, in his rage, he added a curse to her blessing: that no one would ever believe her prophecies.
Thus it was that when her brother Paris set off to Sparta to abduct Helen, she warned him that his actions would lead to the downfall of their great city. He, of course, ignored her.
And you know the rest of the story.
Why am I telling you the story of Cassandra of Troy when we’re supposed to be talking about AI in medicine? Because AI has a major Cassandra problem.
The recent history of AI, and particularly the subset of AI known as machine learning in medicine, has been characterized by an accuracy arms race.
The electronic health record allows for the collection of volumes of data orders of magnitude greater than what we have ever been able to collect before. And all that data can be crunched by various algorithms to make predictions about, well, anything – whether a patient will be transferred to the intensive care unit, whether a GI bleed will need an intervention, whether someone will die in the next year.
Studies in this area tend to rely on retrospective datasets, and as time has gone on, better algorithms and more data have led to better and better predictions. In some simpler cases, machine-learning models have achieved near-perfect accuracy – Cassandra-level accuracy – as in the reading of chest x-rays for pneumonia, for example.
But as Cassandra teaches us, even perfect prediction is useless if no one believes you, if they don’t change their behavior. And this is the central problem of AI in medicine today. Many people are focusing on accuracy of the prediction but have forgotten that high accuracy is just table stakes for an AI model to be useful. It has to not only be accurate, but its use also has to change outcomes for patients. We need to be able to save Troy.
The best way to determine whether an AI model will help patients is to treat a model like we treat a new medication and evaluate it through a randomized trial. That’s what researchers, led by Shannon Walker of Vanderbilt University, Nashville, Tenn., did in a paper appearing in JAMA Network Open.
The model in question was one that predicted venous thromboembolism – blood clots – in hospitalized children. The model took in a variety of data points from the health record: a history of blood clot, history of cancer, presence of a central line, a variety of lab values. And the predictive model was very good – maybe not Cassandra good, but it achieved an AUC of 0.90, which means it had very high accuracy.
But again, accuracy is just table stakes.
The authors deployed the model in the live health record and recorded the results. For half of the kids, that was all that happened; no one actually saw the predictions. For those randomized to the intervention, the hematology team would be notified when the risk for clot was calculated to be greater than 2.5%. The hematology team would then contact the primary team to discuss prophylactic anticoagulation.
This is an elegant approach.
Let’s start with those table stakes – accuracy. The predictions were, by and large, pretty accurate in this trial. Of the 135 kids who developed blood clots, 121 had been flagged by the model in advance. That’s about 90%. The model flagged about 10% of kids who didn’t get a blood clot as well, but that’s not entirely surprising since the threshold for flagging was a 2.5% risk.
Given that the model preidentified almost every kid who would go on to develop a blood clot, it would make sense that kids randomized to the intervention would do better; after all, Cassandra was calling out her warnings.
But those kids didn’t do better. The rate of blood clot was no different between the group that used the accurate prediction model and the group that did not.
Why? Why does the use of an accurate model not necessarily improve outcomes?
First of all, a warning must lead to some change in management. Indeed, the kids in the intervention group were more likely to receive anticoagulation, but barely so. There were lots of reasons for this: physician preference, imminent discharge, active bleeding, and so on.
But let’s take a look at the 77 kids in the intervention arm who developed blood clots, because I think this is an instructive analysis.
Six of them did not meet the 2.5% threshold criteria, a case where the model missed its mark. Again, accuracy is table stakes.
Of the remaining 71, only 16 got a recommendation from the hematologist to start anticoagulation. Why not more? Well, the model identified some of the high-risk kids on the weekend, and it seems that the study team did not contact treatment teams during that time. That may account for about 40% of these cases. The remainder had some contraindication to anticoagulation.
Most tellingly, of the 16 who did get a recommendation to start anticoagulation, the recommendation was followed in only seven patients.
This is the gap between accurate prediction and the ability to change outcomes for patients. A prediction is useless if it is wrong, for sure. But it’s also useless if you don’t tell anyone about it. It’s useless if you tell someone but they can’t do anything about it. And it’s useless if they could do something about it but choose not to.
That’s the gulf that these models need to cross at this point. So, the next time some slick company tells you how accurate their AI model is, ask them if accuracy is really the most important thing. If they say, “Well, yes, of course,” then tell them about Cassandra.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Every click you make, the EHR is watching you
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.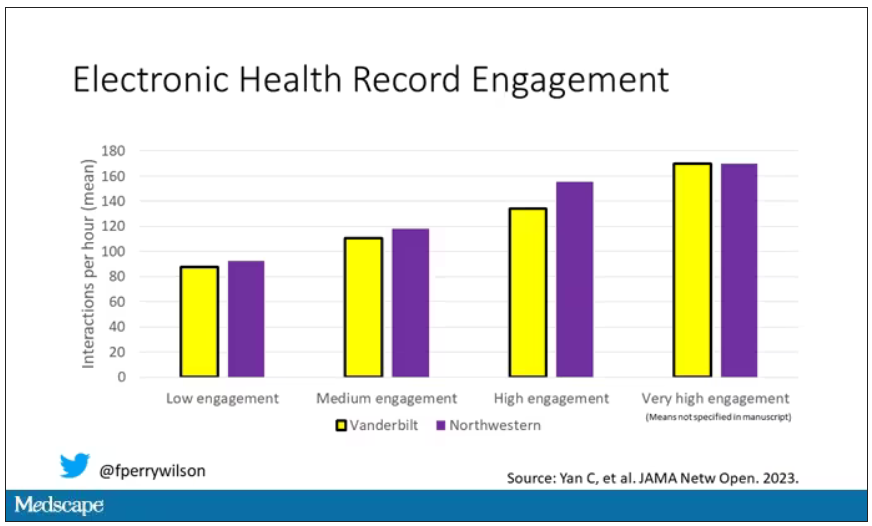
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.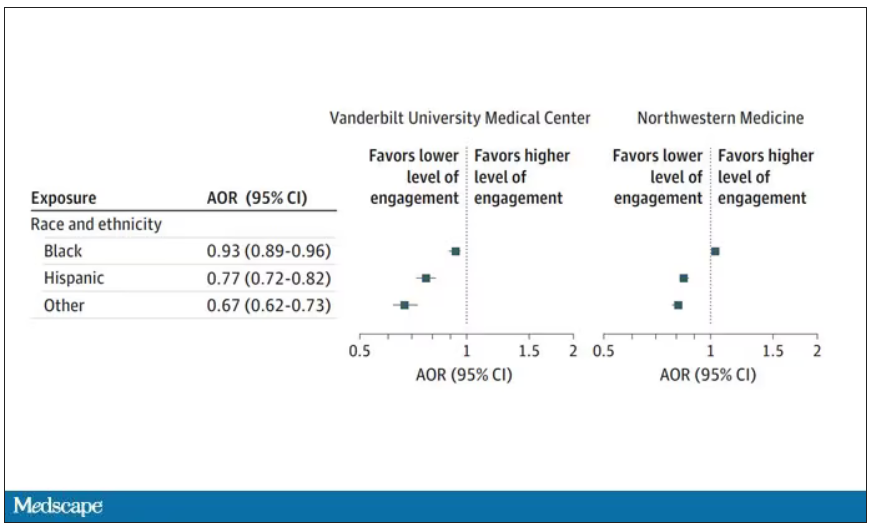
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.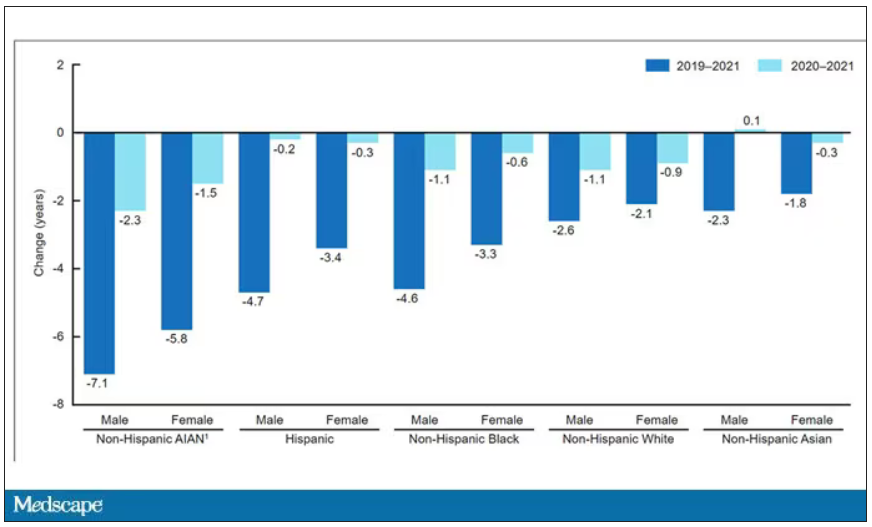
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
The surprising link between loneliness and Parkinson’s disease
This transcript has been edited for clarity.
On May 3, 2023, Surgeon General Vivek Murthy issued an advisory raising an alarm about what he called an “epidemic of loneliness” in the United States.
Now, I am not saying that Vivek Murthy read my book, “How Medicine Works and When It Doesn’t” – released in January and available in bookstores now – where, in chapter 11, I call attention to the problem of loneliness and its relationship to the exponential rise in deaths of despair. But Vivek, if you did, let me know. I could use the publicity.
No, of course the idea that loneliness is a public health issue is not new, but I’m glad to see it finally getting attention. At this point, studies have linked loneliness to heart disease, stroke, dementia, and premature death.
The UK Biobank is really a treasure trove of data for epidemiologists. I must see three to four studies a week coming out of this mega-dataset. This one, appearing in JAMA Neurology, caught my eye for its focus specifically on loneliness as a risk factor – something I’m hoping to see more of in the future.
The study examines data from just under 500,000 individuals in the United Kingdom who answered a survey including the question “Do you often feel lonely?” between 2006 and 2010; 18.4% of people answered yes. Individuals’ electronic health record data were then monitored over time to see who would get a new diagnosis code consistent with Parkinson’s disease. Through 2021, 2822 people did – that’s just over half a percent.
So, now we do the statistics thing. Of the nonlonely folks, 2,273 went on to develop Parkinson’s disease. Of those who said they often feel lonely, 549 people did. The raw numbers here, to be honest, aren’t that compelling. Lonely people had an absolute risk for Parkinson’s disease about 0.03% higher than that of nonlonely people. Put another way, you’d need to take over 3,000 lonely souls and make them not lonely to prevent 1 case of Parkinson’s disease.
Still, the costs of loneliness are not measured exclusively in Parkinson’s disease, and I would argue that the real risks here come from other sources: alcohol abuse, drug abuse, and suicide. Nevertheless, the weak but significant association with Parkinson’s disease reminds us that loneliness is a neurologic phenomenon. There is something about social connection that affects our brain in a way that is not just spiritual; it is actually biological.
Of course, people who say they are often lonely are different in other ways from people who report not being lonely. Lonely people, in this dataset, were younger, more likely to be female, less likely to have a college degree, in worse physical health, and engaged in more high-risk health behaviors like smoking.
The authors adjusted for all of these factors and found that, on the relative scale, lonely people were still about 20%-30% more likely to develop Parkinson’s disease.
So, what do we do about this? There is no pill for loneliness, and God help us if there ever is. Recognizing the problem is a good start. But there are some policy things we can do to reduce loneliness. We can invest in public spaces that bring people together – parks, museums, libraries – and public transportation. We can deal with tech companies that are so optimized at capturing our attention that we cease to engage with other humans. And, individually, we can just reach out a bit more. We’ve spent the past few pandemic years with our attention focused sharply inward. It’s time to look out again.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale University’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
On May 3, 2023, Surgeon General Vivek Murthy issued an advisory raising an alarm about what he called an “epidemic of loneliness” in the United States.
Now, I am not saying that Vivek Murthy read my book, “How Medicine Works and When It Doesn’t” – released in January and available in bookstores now – where, in chapter 11, I call attention to the problem of loneliness and its relationship to the exponential rise in deaths of despair. But Vivek, if you did, let me know. I could use the publicity.
No, of course the idea that loneliness is a public health issue is not new, but I’m glad to see it finally getting attention. At this point, studies have linked loneliness to heart disease, stroke, dementia, and premature death.
The UK Biobank is really a treasure trove of data for epidemiologists. I must see three to four studies a week coming out of this mega-dataset. This one, appearing in JAMA Neurology, caught my eye for its focus specifically on loneliness as a risk factor – something I’m hoping to see more of in the future.
The study examines data from just under 500,000 individuals in the United Kingdom who answered a survey including the question “Do you often feel lonely?” between 2006 and 2010; 18.4% of people answered yes. Individuals’ electronic health record data were then monitored over time to see who would get a new diagnosis code consistent with Parkinson’s disease. Through 2021, 2822 people did – that’s just over half a percent.
So, now we do the statistics thing. Of the nonlonely folks, 2,273 went on to develop Parkinson’s disease. Of those who said they often feel lonely, 549 people did. The raw numbers here, to be honest, aren’t that compelling. Lonely people had an absolute risk for Parkinson’s disease about 0.03% higher than that of nonlonely people. Put another way, you’d need to take over 3,000 lonely souls and make them not lonely to prevent 1 case of Parkinson’s disease.
Still, the costs of loneliness are not measured exclusively in Parkinson’s disease, and I would argue that the real risks here come from other sources: alcohol abuse, drug abuse, and suicide. Nevertheless, the weak but significant association with Parkinson’s disease reminds us that loneliness is a neurologic phenomenon. There is something about social connection that affects our brain in a way that is not just spiritual; it is actually biological.
Of course, people who say they are often lonely are different in other ways from people who report not being lonely. Lonely people, in this dataset, were younger, more likely to be female, less likely to have a college degree, in worse physical health, and engaged in more high-risk health behaviors like smoking.
The authors adjusted for all of these factors and found that, on the relative scale, lonely people were still about 20%-30% more likely to develop Parkinson’s disease.
So, what do we do about this? There is no pill for loneliness, and God help us if there ever is. Recognizing the problem is a good start. But there are some policy things we can do to reduce loneliness. We can invest in public spaces that bring people together – parks, museums, libraries – and public transportation. We can deal with tech companies that are so optimized at capturing our attention that we cease to engage with other humans. And, individually, we can just reach out a bit more. We’ve spent the past few pandemic years with our attention focused sharply inward. It’s time to look out again.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale University’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
On May 3, 2023, Surgeon General Vivek Murthy issued an advisory raising an alarm about what he called an “epidemic of loneliness” in the United States.
Now, I am not saying that Vivek Murthy read my book, “How Medicine Works and When It Doesn’t” – released in January and available in bookstores now – where, in chapter 11, I call attention to the problem of loneliness and its relationship to the exponential rise in deaths of despair. But Vivek, if you did, let me know. I could use the publicity.
No, of course the idea that loneliness is a public health issue is not new, but I’m glad to see it finally getting attention. At this point, studies have linked loneliness to heart disease, stroke, dementia, and premature death.
The UK Biobank is really a treasure trove of data for epidemiologists. I must see three to four studies a week coming out of this mega-dataset. This one, appearing in JAMA Neurology, caught my eye for its focus specifically on loneliness as a risk factor – something I’m hoping to see more of in the future.
The study examines data from just under 500,000 individuals in the United Kingdom who answered a survey including the question “Do you often feel lonely?” between 2006 and 2010; 18.4% of people answered yes. Individuals’ electronic health record data were then monitored over time to see who would get a new diagnosis code consistent with Parkinson’s disease. Through 2021, 2822 people did – that’s just over half a percent.
So, now we do the statistics thing. Of the nonlonely folks, 2,273 went on to develop Parkinson’s disease. Of those who said they often feel lonely, 549 people did. The raw numbers here, to be honest, aren’t that compelling. Lonely people had an absolute risk for Parkinson’s disease about 0.03% higher than that of nonlonely people. Put another way, you’d need to take over 3,000 lonely souls and make them not lonely to prevent 1 case of Parkinson’s disease.
Still, the costs of loneliness are not measured exclusively in Parkinson’s disease, and I would argue that the real risks here come from other sources: alcohol abuse, drug abuse, and suicide. Nevertheless, the weak but significant association with Parkinson’s disease reminds us that loneliness is a neurologic phenomenon. There is something about social connection that affects our brain in a way that is not just spiritual; it is actually biological.
Of course, people who say they are often lonely are different in other ways from people who report not being lonely. Lonely people, in this dataset, were younger, more likely to be female, less likely to have a college degree, in worse physical health, and engaged in more high-risk health behaviors like smoking.
The authors adjusted for all of these factors and found that, on the relative scale, lonely people were still about 20%-30% more likely to develop Parkinson’s disease.
So, what do we do about this? There is no pill for loneliness, and God help us if there ever is. Recognizing the problem is a good start. But there are some policy things we can do to reduce loneliness. We can invest in public spaces that bring people together – parks, museums, libraries – and public transportation. We can deal with tech companies that are so optimized at capturing our attention that we cease to engage with other humans. And, individually, we can just reach out a bit more. We’ve spent the past few pandemic years with our attention focused sharply inward. It’s time to look out again.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale University’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.