User login
What’s Eating You? Megalopyge opercularis
Lepidoptera is the second largest order of the class Insecta and comprises approximately 160,000 species of butterflies and moths classified among approximately 124 families and subfamilies. Venomous properties have been identified in 12 of these families, posing a serious threat to human health. 1
The clinical manifestations from Lepidoptera envenomation can range from general systemic symptoms such as fever and abdominal distress; to more complex focal affections including hemorrhage, ophthalmologic lesions, and irritation of the respiratory tracts; to less severe reactions of the skin, which are the most common presentation.1
Terminology
Lepidopterism is the term used to address a clinical spectrum of systemic manifestations from direct contact with venomous butterflies or moths and/or their products.2 Conversely, erucism is a term used to describe localized cutaneous reactions after direct contact with toxins from caterpillars.
Lepidopterism is derived from the Greek roots lepis, meaning scale, and pteron, meaning wing. The term erucism stems from the Latin word eruca, which means larva.2
Ideally, lepidopterism should refer solely to reactions from butterflies and moths—adult forms of insects with scaly wings—while erucism should refer to reactions from contact with caterpillars—the larval form of butterflies and moths.
In common use, lepidopterism can describe any reaction from caterpillars, moths, or adult butterflies, as well as any case of Lepidoptera exposure with only systemic manifestations, regardless of cutaneous findings. Concurrently, erucism has been defined as either any reaction from caterpillars or any skin reaction from contact with caterpillars or moths.2
Because caterpillars are the larval form of butterflies and moths, caterpillar-associated skin reactions also have been conveniently denominated caterpillar dermatitis.1 Henceforth in this article, both terms erucism and caterpillar dermatitis are used interchangeably.
Caterpillar Envenomation
Caterpillars cause the vast majority of adverse events from lepidopteran exposures.2 Envenomation by caterpillars might stand as the world’s most common envenomation given the larvae proximity to humans.3 Although involvement of internal organs (eg, renal failure), cerebral hemorrhage, and joint lesions can occur, skin manifestations are more predominant with the majority of species. Initial localized pain, edema, and erythema usually are present at the site of direct contact and subsequently progress toward maculopapular to bullous lesions, erosions, petechiae, necrosis, and ulceration depending on the offending species.1,4
Megalopyge opercularis
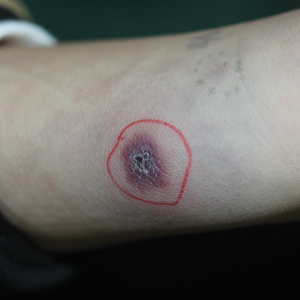
In the United States, more than 50 species of caterpillars have been identified as poisonous or venomous.5 Megalopyge opercularis (Figure 1), the larval form of the flannel moth, is an important cause of caterpillar-associated dermatitis in the southern United States.6,7 Megalopyge opercularis also is commonly known as the puss caterpillar, opossum bug, wooly slug, el perrito, tree asp, or Italian asp.6 This lepidopteran insect is mainly found in the southeastern and southcentral United States, with noted particular abundance in Texas, Louisiana, and Florida.6,8 The puss caterpillar has 2 generations per year; the first develops during the months of June to July, and the second develops from September to October, carrying seasonal health hazards.6,8

Megalopyge opercularis is tapered at the ends and can measure 2.5 to 3.5×1 cm at maturity. It is covered by silky, long-streaked, wavy hairs that may appear single colored or as a mix of colors—from white to gray to brown—forming a mid-dorsal crest.6 Beneath this furry coat, rows of short sharp spines are hidden. Upon contact with the human skin, these spines will break and discharge venom.1,6,8 Toxins contained within the hollow spines are thought to be produced by specialized basal cells, but there still is little knowledge about the dynamics and composition of the venom.1
Clinical Manifestations
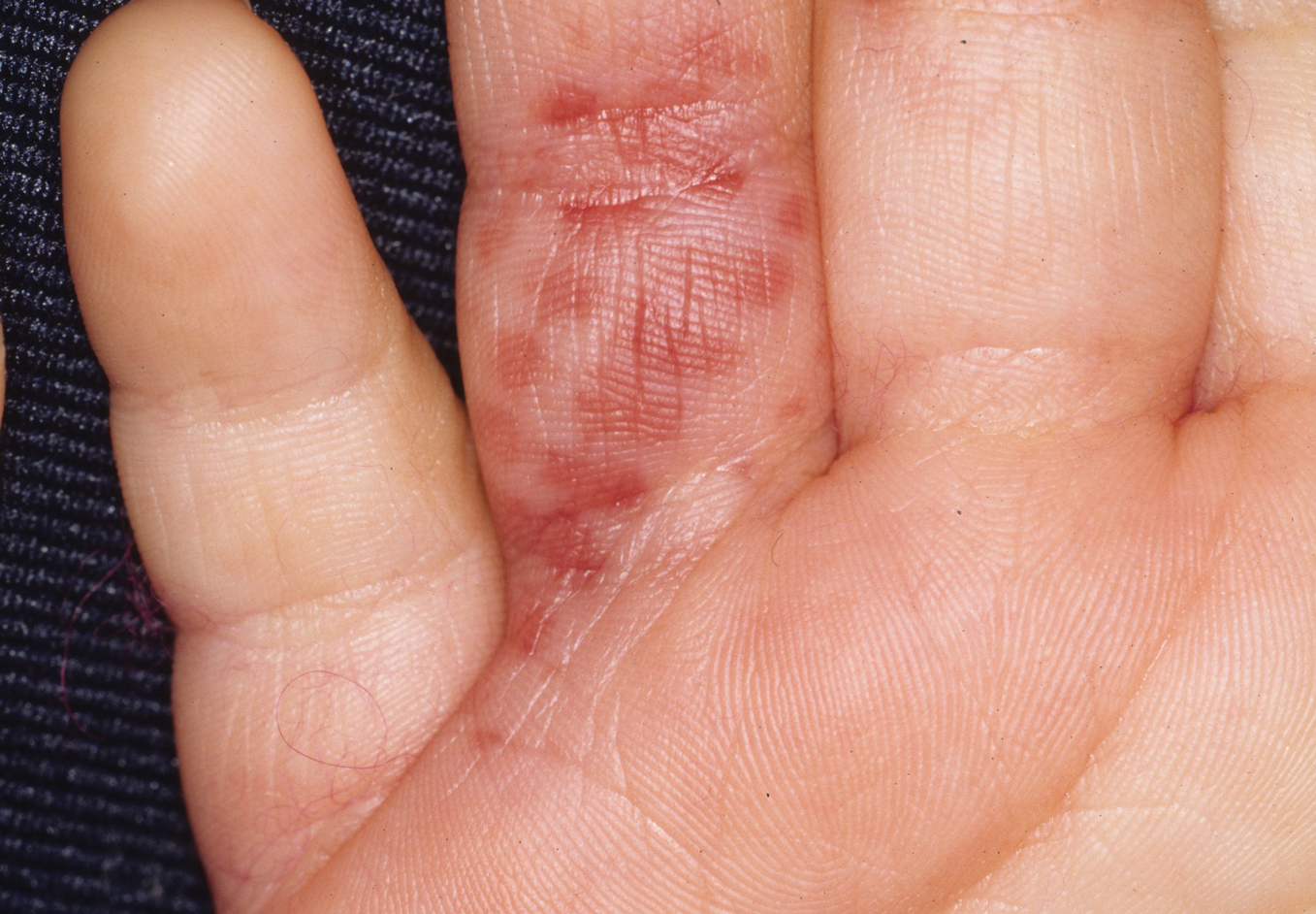
The severity of the reaction depends on the caterpillar’s size and the extent of contact.1,4 Contact with M opercularis instantly presents with a throbbing or burning pain that may be followed by localized erythema and rash.1,6 A characteristic gridlike pattern of erythematous macules develops, reflecting each site of puncture from the insect’s spines (Figure 2).8,9 Skin lesions can progress from erythematous macules to hemorrhagic vesicles or pustules, usually self-resolving after a few days. The reaction also can present with radiating pain to regional lymph nodes and numbness of the affected area.1,6,8 Moreover, some patients may report urticaria and pruritus.9
Envenomation by a puss caterpillar also can present with systemic manifestations including fever, headache, nausea, vomiting, shocklike symptoms, and seizures.1,6,7 Anaphylactic reaction is rare but also can present.7 Uncommon cases have been reported with severe abdominal pain and muscle spasm mimicking acute appendicitis and latrodectism, respectively.7,9
Diagnosis
The diagnosis of M opercularis envenomation is made clinically based on the morphology of the skin lesions and a history of probable exposure. Coexistent leukocytosis is likely, but laboratory testing is not warranted, as it is both nonspecific and insensitive.9
Management/Treatment
The most commonly reported immediate approaches to treatment involve attempts to remove the spines from the skin with tape (stripping), application of ice packs over the affected area, oral antihistamines, topical and intralesional anesthetics, regional nerve block, and oral analgesics.6,9 There have been several cases detailing the successful use of parenteral calcium gluconate,5,7 and diazepam has been used to treat severe muscle spasms. Anaphylactic reactions should be managed in a controlled monitored setting with subcutaneous epinephrine.7 Despite their common use, some data suggest that ice packs and mid- to high-potency topical steroids are ineffective.9
Incidence
From 2001 to 2005, a mean average of 94,552 annual cases of animal bites and stings were reported to poison control centers in the United States, of which 2094 were linked to caterpillars in this 5-year period.10 There were 3484 M opercularis caterpillar stings reported to the Texas Poison Center Network from 2000 to 2016.5,6 Given their ability to sting throughout their life cycle, thousands of M opercularis caterpillar stings can occur each year.1,6 Existing literature on M opercularis caterpillar stings mainly involves case reports with affections of the skin and oral mucosa, self-reported envenomation, and case studies.5,6,8
Although multiple health concerns associated with caterpillar envenomation have been reported worldwide, the lack of official epidemiologic reports highly suggests that this problem remains underestimated. There also may be many unreported cases because certain reactions are mild or self-limited and can even go unnoticed.11 Nonetheless, there is an evident rise of cases reported in the United States. According to the 2018 annual report of the American Association of Poison Control Centers, there were 2815 case mentions from caterpillar envenomation.12
In 1921 and 1952, some public schools in Texas were temporarily closed due to outbreaks of puss caterpillar–associated dermatitis.8 Similar outbreaks also have been reported in South Carolina, Virginia, and Oklahoma.9 Emerging data suggest that plant oil products and the pesticide cypermethrin may be helpful in controlling local infestations of the puss caterpillar.8
- Villas-Boas IM, Bonfa G, Tambourgi DV. Venomous caterpillars: from inoculation apparatus to venom composition and envenomation. Toxicon. 2018;153:39-52.
- Hossler EW. Caterpillars and moths: part I. dermatologic manifestations of encounters with Lepidoptera. J Am Acad Dermatol. 2010;62:1-10; quiz 11-12.
- Haddad Junior V, Amorim PC, Haddad Junior WT, et al. Venomous and poisonous arthropods: identification, clinical manifestations of envenomation, and treatments used in human injuries. Rev Soc Bras Med Trop. 2015;48:650-657.
- Haddad V Jr, Cardoso JL, Lupi O, et al. Tropical dermatology: venomous arthropods and human skin: part I. Insecta. J Am Acad Dermatol. 2012;67:331.e1-331.e14; quiz 345.
- Pappano DA, Trout Fryxell R, Warren M. Oral mucosal envenomation of an infant by a puss caterpillar. Pediatr Emerg Care. 2017;33:424-426.
- Forrester MB. Megalopyge opercularis caterpillar stings reported to Texas poison centers. Wilderness Environ Med. 2018;29:215-220.
- Hossler EW. Caterpillars and moths: part II. dermatologic manifestations of encounters with Lepidoptera. J Am Acad Dermatol. 2010;62:13-28; quiz 29-30.
- Eagleman DM. Envenomation by the asp caterpillar (Megalopyge opercularis). Clin Toxicol (Phila). 2008;46:201-205.
- Greene SC, Carey JM. Puss caterpillar envenomation: erucism mimicking appendicitis in a young child [published online May 23, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001514.
- Langley RL. Animal bites and stings reported by United States Poison Control Centers, 2001-2005. Wilderness Environ Med. 2008;19:7-14.
- Seldeslachts A, Peigneur S, Tytgat J. Caterpillar venom: a health hazard of the 21st century [published online May 30, 2020]. Biomedicines. doi:10.3390/biomedicines8060143.
- Gummin DD, Mowry JB, Spyker DA, et al. 2018 annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 36th annual report. Clin Toxicol (Phila). 2019;57:1220-1413.
Lepidoptera is the second largest order of the class Insecta and comprises approximately 160,000 species of butterflies and moths classified among approximately 124 families and subfamilies. Venomous properties have been identified in 12 of these families, posing a serious threat to human health. 1
The clinical manifestations from Lepidoptera envenomation can range from general systemic symptoms such as fever and abdominal distress; to more complex focal affections including hemorrhage, ophthalmologic lesions, and irritation of the respiratory tracts; to less severe reactions of the skin, which are the most common presentation.1
Terminology
Lepidopterism is the term used to address a clinical spectrum of systemic manifestations from direct contact with venomous butterflies or moths and/or their products.2 Conversely, erucism is a term used to describe localized cutaneous reactions after direct contact with toxins from caterpillars.
Lepidopterism is derived from the Greek roots lepis, meaning scale, and pteron, meaning wing. The term erucism stems from the Latin word eruca, which means larva.2
Ideally, lepidopterism should refer solely to reactions from butterflies and moths—adult forms of insects with scaly wings—while erucism should refer to reactions from contact with caterpillars—the larval form of butterflies and moths.
In common use, lepidopterism can describe any reaction from caterpillars, moths, or adult butterflies, as well as any case of Lepidoptera exposure with only systemic manifestations, regardless of cutaneous findings. Concurrently, erucism has been defined as either any reaction from caterpillars or any skin reaction from contact with caterpillars or moths.2
Because caterpillars are the larval form of butterflies and moths, caterpillar-associated skin reactions also have been conveniently denominated caterpillar dermatitis.1 Henceforth in this article, both terms erucism and caterpillar dermatitis are used interchangeably.
Caterpillar Envenomation
Caterpillars cause the vast majority of adverse events from lepidopteran exposures.2 Envenomation by caterpillars might stand as the world’s most common envenomation given the larvae proximity to humans.3 Although involvement of internal organs (eg, renal failure), cerebral hemorrhage, and joint lesions can occur, skin manifestations are more predominant with the majority of species. Initial localized pain, edema, and erythema usually are present at the site of direct contact and subsequently progress toward maculopapular to bullous lesions, erosions, petechiae, necrosis, and ulceration depending on the offending species.1,4
Megalopyge opercularis
In the United States, more than 50 species of caterpillars have been identified as poisonous or venomous.5 Megalopyge opercularis (Figure 1), the larval form of the flannel moth, is an important cause of caterpillar-associated dermatitis in the southern United States.6,7 Megalopyge opercularis also is commonly known as the puss caterpillar, opossum bug, wooly slug, el perrito, tree asp, or Italian asp.6 This lepidopteran insect is mainly found in the southeastern and southcentral United States, with noted particular abundance in Texas, Louisiana, and Florida.6,8 The puss caterpillar has 2 generations per year; the first develops during the months of June to July, and the second develops from September to October, carrying seasonal health hazards.6,8
Megalopyge opercularis is tapered at the ends and can measure 2.5 to 3.5×1 cm at maturity. It is covered by silky, long-streaked, wavy hairs that may appear single colored or as a mix of colors—from white to gray to brown—forming a mid-dorsal crest.6 Beneath this furry coat, rows of short sharp spines are hidden. Upon contact with the human skin, these spines will break and discharge venom.1,6,8 Toxins contained within the hollow spines are thought to be produced by specialized basal cells, but there still is little knowledge about the dynamics and composition of the venom.1
Clinical Manifestations
The severity of the reaction depends on the caterpillar’s size and the extent of contact.1,4 Contact with M opercularis instantly presents with a throbbing or burning pain that may be followed by localized erythema and rash.1,6 A characteristic gridlike pattern of erythematous macules develops, reflecting each site of puncture from the insect’s spines (Figure 2).8,9 Skin lesions can progress from erythematous macules to hemorrhagic vesicles or pustules, usually self-resolving after a few days. The reaction also can present with radiating pain to regional lymph nodes and numbness of the affected area.1,6,8 Moreover, some patients may report urticaria and pruritus.9
Envenomation by a puss caterpillar also can present with systemic manifestations including fever, headache, nausea, vomiting, shocklike symptoms, and seizures.1,6,7 Anaphylactic reaction is rare but also can present.7 Uncommon cases have been reported with severe abdominal pain and muscle spasm mimicking acute appendicitis and latrodectism, respectively.7,9
Diagnosis
The diagnosis of M opercularis envenomation is made clinically based on the morphology of the skin lesions and a history of probable exposure. Coexistent leukocytosis is likely, but laboratory testing is not warranted, as it is both nonspecific and insensitive.9
Management/Treatment
The most commonly reported immediate approaches to treatment involve attempts to remove the spines from the skin with tape (stripping), application of ice packs over the affected area, oral antihistamines, topical and intralesional anesthetics, regional nerve block, and oral analgesics.6,9 There have been several cases detailing the successful use of parenteral calcium gluconate,5,7 and diazepam has been used to treat severe muscle spasms. Anaphylactic reactions should be managed in a controlled monitored setting with subcutaneous epinephrine.7 Despite their common use, some data suggest that ice packs and mid- to high-potency topical steroids are ineffective.9
Incidence
From 2001 to 2005, a mean average of 94,552 annual cases of animal bites and stings were reported to poison control centers in the United States, of which 2094 were linked to caterpillars in this 5-year period.10 There were 3484 M opercularis caterpillar stings reported to the Texas Poison Center Network from 2000 to 2016.5,6 Given their ability to sting throughout their life cycle, thousands of M opercularis caterpillar stings can occur each year.1,6 Existing literature on M opercularis caterpillar stings mainly involves case reports with affections of the skin and oral mucosa, self-reported envenomation, and case studies.5,6,8
Although multiple health concerns associated with caterpillar envenomation have been reported worldwide, the lack of official epidemiologic reports highly suggests that this problem remains underestimated. There also may be many unreported cases because certain reactions are mild or self-limited and can even go unnoticed.11 Nonetheless, there is an evident rise of cases reported in the United States. According to the 2018 annual report of the American Association of Poison Control Centers, there were 2815 case mentions from caterpillar envenomation.12
In 1921 and 1952, some public schools in Texas were temporarily closed due to outbreaks of puss caterpillar–associated dermatitis.8 Similar outbreaks also have been reported in South Carolina, Virginia, and Oklahoma.9 Emerging data suggest that plant oil products and the pesticide cypermethrin may be helpful in controlling local infestations of the puss caterpillar.8
Lepidoptera is the second largest order of the class Insecta and comprises approximately 160,000 species of butterflies and moths classified among approximately 124 families and subfamilies. Venomous properties have been identified in 12 of these families, posing a serious threat to human health. 1
The clinical manifestations from Lepidoptera envenomation can range from general systemic symptoms such as fever and abdominal distress; to more complex focal affections including hemorrhage, ophthalmologic lesions, and irritation of the respiratory tracts; to less severe reactions of the skin, which are the most common presentation.1
Terminology
Lepidopterism is the term used to address a clinical spectrum of systemic manifestations from direct contact with venomous butterflies or moths and/or their products.2 Conversely, erucism is a term used to describe localized cutaneous reactions after direct contact with toxins from caterpillars.
Lepidopterism is derived from the Greek roots lepis, meaning scale, and pteron, meaning wing. The term erucism stems from the Latin word eruca, which means larva.2
Ideally, lepidopterism should refer solely to reactions from butterflies and moths—adult forms of insects with scaly wings—while erucism should refer to reactions from contact with caterpillars—the larval form of butterflies and moths.
In common use, lepidopterism can describe any reaction from caterpillars, moths, or adult butterflies, as well as any case of Lepidoptera exposure with only systemic manifestations, regardless of cutaneous findings. Concurrently, erucism has been defined as either any reaction from caterpillars or any skin reaction from contact with caterpillars or moths.2
Because caterpillars are the larval form of butterflies and moths, caterpillar-associated skin reactions also have been conveniently denominated caterpillar dermatitis.1 Henceforth in this article, both terms erucism and caterpillar dermatitis are used interchangeably.
Caterpillar Envenomation
Caterpillars cause the vast majority of adverse events from lepidopteran exposures.2 Envenomation by caterpillars might stand as the world’s most common envenomation given the larvae proximity to humans.3 Although involvement of internal organs (eg, renal failure), cerebral hemorrhage, and joint lesions can occur, skin manifestations are more predominant with the majority of species. Initial localized pain, edema, and erythema usually are present at the site of direct contact and subsequently progress toward maculopapular to bullous lesions, erosions, petechiae, necrosis, and ulceration depending on the offending species.1,4
Megalopyge opercularis
In the United States, more than 50 species of caterpillars have been identified as poisonous or venomous.5 Megalopyge opercularis (Figure 1), the larval form of the flannel moth, is an important cause of caterpillar-associated dermatitis in the southern United States.6,7 Megalopyge opercularis also is commonly known as the puss caterpillar, opossum bug, wooly slug, el perrito, tree asp, or Italian asp.6 This lepidopteran insect is mainly found in the southeastern and southcentral United States, with noted particular abundance in Texas, Louisiana, and Florida.6,8 The puss caterpillar has 2 generations per year; the first develops during the months of June to July, and the second develops from September to October, carrying seasonal health hazards.6,8
Megalopyge opercularis is tapered at the ends and can measure 2.5 to 3.5×1 cm at maturity. It is covered by silky, long-streaked, wavy hairs that may appear single colored or as a mix of colors—from white to gray to brown—forming a mid-dorsal crest.6 Beneath this furry coat, rows of short sharp spines are hidden. Upon contact with the human skin, these spines will break and discharge venom.1,6,8 Toxins contained within the hollow spines are thought to be produced by specialized basal cells, but there still is little knowledge about the dynamics and composition of the venom.1
Clinical Manifestations
The severity of the reaction depends on the caterpillar’s size and the extent of contact.1,4 Contact with M opercularis instantly presents with a throbbing or burning pain that may be followed by localized erythema and rash.1,6 A characteristic gridlike pattern of erythematous macules develops, reflecting each site of puncture from the insect’s spines (Figure 2).8,9 Skin lesions can progress from erythematous macules to hemorrhagic vesicles or pustules, usually self-resolving after a few days. The reaction also can present with radiating pain to regional lymph nodes and numbness of the affected area.1,6,8 Moreover, some patients may report urticaria and pruritus.9
Envenomation by a puss caterpillar also can present with systemic manifestations including fever, headache, nausea, vomiting, shocklike symptoms, and seizures.1,6,7 Anaphylactic reaction is rare but also can present.7 Uncommon cases have been reported with severe abdominal pain and muscle spasm mimicking acute appendicitis and latrodectism, respectively.7,9
Diagnosis
The diagnosis of M opercularis envenomation is made clinically based on the morphology of the skin lesions and a history of probable exposure. Coexistent leukocytosis is likely, but laboratory testing is not warranted, as it is both nonspecific and insensitive.9
Management/Treatment
The most commonly reported immediate approaches to treatment involve attempts to remove the spines from the skin with tape (stripping), application of ice packs over the affected area, oral antihistamines, topical and intralesional anesthetics, regional nerve block, and oral analgesics.6,9 There have been several cases detailing the successful use of parenteral calcium gluconate,5,7 and diazepam has been used to treat severe muscle spasms. Anaphylactic reactions should be managed in a controlled monitored setting with subcutaneous epinephrine.7 Despite their common use, some data suggest that ice packs and mid- to high-potency topical steroids are ineffective.9
Incidence
From 2001 to 2005, a mean average of 94,552 annual cases of animal bites and stings were reported to poison control centers in the United States, of which 2094 were linked to caterpillars in this 5-year period.10 There were 3484 M opercularis caterpillar stings reported to the Texas Poison Center Network from 2000 to 2016.5,6 Given their ability to sting throughout their life cycle, thousands of M opercularis caterpillar stings can occur each year.1,6 Existing literature on M opercularis caterpillar stings mainly involves case reports with affections of the skin and oral mucosa, self-reported envenomation, and case studies.5,6,8
Although multiple health concerns associated with caterpillar envenomation have been reported worldwide, the lack of official epidemiologic reports highly suggests that this problem remains underestimated. There also may be many unreported cases because certain reactions are mild or self-limited and can even go unnoticed.11 Nonetheless, there is an evident rise of cases reported in the United States. According to the 2018 annual report of the American Association of Poison Control Centers, there were 2815 case mentions from caterpillar envenomation.12
In 1921 and 1952, some public schools in Texas were temporarily closed due to outbreaks of puss caterpillar–associated dermatitis.8 Similar outbreaks also have been reported in South Carolina, Virginia, and Oklahoma.9 Emerging data suggest that plant oil products and the pesticide cypermethrin may be helpful in controlling local infestations of the puss caterpillar.8
- Villas-Boas IM, Bonfa G, Tambourgi DV. Venomous caterpillars: from inoculation apparatus to venom composition and envenomation. Toxicon. 2018;153:39-52.
- Hossler EW. Caterpillars and moths: part I. dermatologic manifestations of encounters with Lepidoptera. J Am Acad Dermatol. 2010;62:1-10; quiz 11-12.
- Haddad Junior V, Amorim PC, Haddad Junior WT, et al. Venomous and poisonous arthropods: identification, clinical manifestations of envenomation, and treatments used in human injuries. Rev Soc Bras Med Trop. 2015;48:650-657.
- Haddad V Jr, Cardoso JL, Lupi O, et al. Tropical dermatology: venomous arthropods and human skin: part I. Insecta. J Am Acad Dermatol. 2012;67:331.e1-331.e14; quiz 345.
- Pappano DA, Trout Fryxell R, Warren M. Oral mucosal envenomation of an infant by a puss caterpillar. Pediatr Emerg Care. 2017;33:424-426.
- Forrester MB. Megalopyge opercularis caterpillar stings reported to Texas poison centers. Wilderness Environ Med. 2018;29:215-220.
- Hossler EW. Caterpillars and moths: part II. dermatologic manifestations of encounters with Lepidoptera. J Am Acad Dermatol. 2010;62:13-28; quiz 29-30.
- Eagleman DM. Envenomation by the asp caterpillar (Megalopyge opercularis). Clin Toxicol (Phila). 2008;46:201-205.
- Greene SC, Carey JM. Puss caterpillar envenomation: erucism mimicking appendicitis in a young child [published online May 23, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001514.
- Langley RL. Animal bites and stings reported by United States Poison Control Centers, 2001-2005. Wilderness Environ Med. 2008;19:7-14.
- Seldeslachts A, Peigneur S, Tytgat J. Caterpillar venom: a health hazard of the 21st century [published online May 30, 2020]. Biomedicines. doi:10.3390/biomedicines8060143.
- Gummin DD, Mowry JB, Spyker DA, et al. 2018 annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 36th annual report. Clin Toxicol (Phila). 2019;57:1220-1413.
- Villas-Boas IM, Bonfa G, Tambourgi DV. Venomous caterpillars: from inoculation apparatus to venom composition and envenomation. Toxicon. 2018;153:39-52.
- Hossler EW. Caterpillars and moths: part I. dermatologic manifestations of encounters with Lepidoptera. J Am Acad Dermatol. 2010;62:1-10; quiz 11-12.
- Haddad Junior V, Amorim PC, Haddad Junior WT, et al. Venomous and poisonous arthropods: identification, clinical manifestations of envenomation, and treatments used in human injuries. Rev Soc Bras Med Trop. 2015;48:650-657.
- Haddad V Jr, Cardoso JL, Lupi O, et al. Tropical dermatology: venomous arthropods and human skin: part I. Insecta. J Am Acad Dermatol. 2012;67:331.e1-331.e14; quiz 345.
- Pappano DA, Trout Fryxell R, Warren M. Oral mucosal envenomation of an infant by a puss caterpillar. Pediatr Emerg Care. 2017;33:424-426.
- Forrester MB. Megalopyge opercularis caterpillar stings reported to Texas poison centers. Wilderness Environ Med. 2018;29:215-220.
- Hossler EW. Caterpillars and moths: part II. dermatologic manifestations of encounters with Lepidoptera. J Am Acad Dermatol. 2010;62:13-28; quiz 29-30.
- Eagleman DM. Envenomation by the asp caterpillar (Megalopyge opercularis). Clin Toxicol (Phila). 2008;46:201-205.
- Greene SC, Carey JM. Puss caterpillar envenomation: erucism mimicking appendicitis in a young child [published online May 23, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001514.
- Langley RL. Animal bites and stings reported by United States Poison Control Centers, 2001-2005. Wilderness Environ Med. 2008;19:7-14.
- Seldeslachts A, Peigneur S, Tytgat J. Caterpillar venom: a health hazard of the 21st century [published online May 30, 2020]. Biomedicines. doi:10.3390/biomedicines8060143.
- Gummin DD, Mowry JB, Spyker DA, et al. 2018 annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 36th annual report. Clin Toxicol (Phila). 2019;57:1220-1413.
Practice Points
- Megalopyge opercularis is the most widely distributed caterpillar species in the Americas, and envenomation by it can occur year-round.
- Skin reactions to M opercularis stings can present as maculopapular dermatitis, eczematous eruptions, or urticarial reactions.
- During the initial presentation, patients experience intense throbbing pain, yet the severity of symptoms depends on the caterpillar’s size and the extent of contact.
- A history of caterpillar exposure helps with diagnosis, and treatment remains empiric.
What’s Eating You? Bark Scorpions (Centruroides exilicauda and Centruroides sculpturatus)
Epidemiology and Identification
Centruroides is a common genus of bark scorpions in the United States with at least 21 species considered to be medically important, including the closely related Centruroides exilicauda and Centruroides sculpturatus.1 Scorpions can be recognized by a bulbous sac and pointed stinger at the end of a tail-like abdomen. They also have long lobsterlike pedipalps (pincers) for grasping their prey. Identifying characteristics for C exilicauda and C sculpturatus include a small, slender, yellow to light brown or tan body typically measuring 1.3 to 7.6 cm in length with a subaculear tooth or tubercle at the base of the stinger, a characteristic that is common to all Centruroides species (Figure).2 Some variability in size has been shown, with smaller scorpions found in increased elevations and cooler temperatures.1,3 Both C exilicauda and C sculpturatus are found in northern Mexico as well as the southwestern United States (eg, Arizona, New Mexico, Texas, California, Nevada).1 They have a preference for residing in or around trees and often are found on the underside of bark, stones, or tables as well as inside shoes or small cracks and crevices. Scorpions typically sting in self-defense, and stings commonly occur when humans attempt to move tables, put on shoes, or walk barefoot in scorpion-infested areas. Most stings occur from the end of spring through the end summer, but many may go unreported.1,4

The venom of the Centruroides genus includes peptides and proteins that play a fundamental role in toxic activity by impairing potassium, sodium, and calcium ion channels.1,3 Toxins have been shown to be species specific, functioning either in capturing prey or deterring predators. Intraspecies variability in toxins has been demonstrated, which may complicate the production of adequate antivenin.3 Many have thought that C exilicauda Wood and C sculpturatus Ewing are the same species, and the names have been used synonymously in the past; however, genetic and biochemical studies of their venom components have shown that they are distinct species and that C sculpturatus is the more dangerous of the two.5 The median lethal dose 50% of C sculpturatus was found to be 22.7 μg in CD1 mice.6
Envenomation and Clinical Manifestations
Stings from C exilicauda and C sculpturatus have been shown to cause fatality in children more often than in adults.7 In the United States, Arizona has the highest frequency of serious symptoms of envenomation as well as the highest hospital and intensive care unit admission rates.6 Envenomation results in an immediate sharp burning pain followed by numbness.4 Wounds can produce some regional lymph node swelling, ecchymosis, paresthesia, and lymphangitis. More often than not, however, wounds have little to no inflammation and are characterized only by pain.4 The puncture wound is too small to be seen, and C exilicauda and C sculpturatus venom do not cause local tissue destruction, an important factor in distinguishing it from other scorpion envenomations.
More severe complications that may follow are caused by the neurotoxin released by Centruroides stings. The toxin components can increase the duration and amplitude of the neuronal action potential and enhance the release of neurotransmitters such as acetylcholine and norepinephrine.8 Stings can lead to cranial nerve dysfunction and somatic skeletal neuromuscular dysfunction as well as autonomic dysfunction, specifically salivation, fever, tongue and muscle fasciculations, opsoclonus, vomiting, bronchoconstriction, diaphoresis, nystagmus, blurred vision, slurred speech, hypertension, rhabdomyolysis, stridor, wheezing, aspiration, anaphylaxis, and tachycardia, leading to cardiac and respiratory compromise.4,8 Some patients have experienced a decreased sense of smell or hearing and decreased fine motor movements.7 Although pancreatitis may occur with scorpion stings, it is not common for C exilicauda.9 Comorbidities such as cardiac disease and substance use disorders contribute to prolonged length of hospital stay and poor outcome.8
Treatment
Most Centruroides stings can be managed at home, but patients with more serious symptoms and children younger than 2 years should be taken to a hospital for treatment.7 If a patient reports only pain but shows no other signs of neurotoxicity, observation and pain relief with rest, ice, and elevation is appropriate management. Patients with severe manifestations have been treated with various combinations of lorazepam, glycopyrrolate, ipratropium bromide, and ondansetron, but the only treatment definitively shown to decrease time to symptom abatement is antivenin.7 It has been demonstrated that C exilicauda and C sculpturatus antivenin is relatively safe.7 Most patients, especially adults, do not die from C exilicauda and C sculpturatus stings; therefore, antivenin more commonly is symptom abating than it is lifesaving.10 In children, time to symptom resolution was decreased to fewer than 4 hours with antivenin, and there is a lower rate of inpatient admission when antivenin is administered.4,10,11 There is a low incidence of anaphylactic reaction after antivenin, but there have been reported cases of self-limited serum sickness after antivenin use that generally can be managed with antihistamines and corticosteroids.4,7
- Gonzalez-Santillan E, Possani LD. North American scorpion species of public health importance with reappraisal of historical epidemiology. Acta Tropica. 2018;187:264-274.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Carcamo-Noriega EN, Olamendi-Portugal T, Restano-Cassulini R, et al. Intraspecific variation of Centruroides sculpturatus scorpion venom from two regions of Arizona. Arch Biochem Biophys. 2018;638:52-57.
- Kang AM, Brooks DE. Nationwide scorpion exposures reported to US Poison Control centers from 2005 to 2015. J Med Toxicol. 2017;13:158-165.
- Valdez-Cruz N, Dávila S, Licea A, et al. Biochemical, genetic and physiological characterization of venom components from two species of scorpions: Centruroides exilicauda Wood and Centruroides sculpturatus Ewing. Biochimie. 2004;86:387-396.
- Jiménez-Vargas JM, Quintero-Hernández V, Gonzáles-Morales L, et al. Design and expression of recombinant toxins from Mexican scorpions of the genus Centruroides for production of antivenoms. Toxicon. 2017;128:5-14.
- Hurst NB, Lipe DN, Karpen SR, et al. Centruroides sculpturatus envenomation in three adult patients requiring treatment with antivenom. Clin Toxicol (Phila). 2018;56:294-296.
- O’Connor A, Padilla-Jones A, Ruha A. Severe bark scorpion envenomation in adults. Clin Toxicol. 2018;56:170-174.
- Berg R, Tarantino M. Envenomation by the scorpion Centruroides exilicauda (C sculpturatus): severe and unusual manifestations. Pediatrics. 1991;87:930-933.
- LoVecchio F, McBride C. Scorpion envenomations in young children in central Arizona. J Toxicol Clin Toxicol. 2003;41:937-940.
- Rodrigo C, Gnanathasan A. Management of scorpion envenoming: a systematic review and meta-analysis of controlled clinical trials. Syst Rev. 2017;6:74.
Epidemiology and Identification
Centruroides is a common genus of bark scorpions in the United States with at least 21 species considered to be medically important, including the closely related Centruroides exilicauda and Centruroides sculpturatus.1 Scorpions can be recognized by a bulbous sac and pointed stinger at the end of a tail-like abdomen. They also have long lobsterlike pedipalps (pincers) for grasping their prey. Identifying characteristics for C exilicauda and C sculpturatus include a small, slender, yellow to light brown or tan body typically measuring 1.3 to 7.6 cm in length with a subaculear tooth or tubercle at the base of the stinger, a characteristic that is common to all Centruroides species (Figure).2 Some variability in size has been shown, with smaller scorpions found in increased elevations and cooler temperatures.1,3 Both C exilicauda and C sculpturatus are found in northern Mexico as well as the southwestern United States (eg, Arizona, New Mexico, Texas, California, Nevada).1 They have a preference for residing in or around trees and often are found on the underside of bark, stones, or tables as well as inside shoes or small cracks and crevices. Scorpions typically sting in self-defense, and stings commonly occur when humans attempt to move tables, put on shoes, or walk barefoot in scorpion-infested areas. Most stings occur from the end of spring through the end summer, but many may go unreported.1,4
The venom of the Centruroides genus includes peptides and proteins that play a fundamental role in toxic activity by impairing potassium, sodium, and calcium ion channels.1,3 Toxins have been shown to be species specific, functioning either in capturing prey or deterring predators. Intraspecies variability in toxins has been demonstrated, which may complicate the production of adequate antivenin.3 Many have thought that C exilicauda Wood and C sculpturatus Ewing are the same species, and the names have been used synonymously in the past; however, genetic and biochemical studies of their venom components have shown that they are distinct species and that C sculpturatus is the more dangerous of the two.5 The median lethal dose 50% of C sculpturatus was found to be 22.7 μg in CD1 mice.6
Envenomation and Clinical Manifestations
Stings from C exilicauda and C sculpturatus have been shown to cause fatality in children more often than in adults.7 In the United States, Arizona has the highest frequency of serious symptoms of envenomation as well as the highest hospital and intensive care unit admission rates.6 Envenomation results in an immediate sharp burning pain followed by numbness.4 Wounds can produce some regional lymph node swelling, ecchymosis, paresthesia, and lymphangitis. More often than not, however, wounds have little to no inflammation and are characterized only by pain.4 The puncture wound is too small to be seen, and C exilicauda and C sculpturatus venom do not cause local tissue destruction, an important factor in distinguishing it from other scorpion envenomations.
More severe complications that may follow are caused by the neurotoxin released by Centruroides stings. The toxin components can increase the duration and amplitude of the neuronal action potential and enhance the release of neurotransmitters such as acetylcholine and norepinephrine.8 Stings can lead to cranial nerve dysfunction and somatic skeletal neuromuscular dysfunction as well as autonomic dysfunction, specifically salivation, fever, tongue and muscle fasciculations, opsoclonus, vomiting, bronchoconstriction, diaphoresis, nystagmus, blurred vision, slurred speech, hypertension, rhabdomyolysis, stridor, wheezing, aspiration, anaphylaxis, and tachycardia, leading to cardiac and respiratory compromise.4,8 Some patients have experienced a decreased sense of smell or hearing and decreased fine motor movements.7 Although pancreatitis may occur with scorpion stings, it is not common for C exilicauda.9 Comorbidities such as cardiac disease and substance use disorders contribute to prolonged length of hospital stay and poor outcome.8
Treatment
Most Centruroides stings can be managed at home, but patients with more serious symptoms and children younger than 2 years should be taken to a hospital for treatment.7 If a patient reports only pain but shows no other signs of neurotoxicity, observation and pain relief with rest, ice, and elevation is appropriate management. Patients with severe manifestations have been treated with various combinations of lorazepam, glycopyrrolate, ipratropium bromide, and ondansetron, but the only treatment definitively shown to decrease time to symptom abatement is antivenin.7 It has been demonstrated that C exilicauda and C sculpturatus antivenin is relatively safe.7 Most patients, especially adults, do not die from C exilicauda and C sculpturatus stings; therefore, antivenin more commonly is symptom abating than it is lifesaving.10 In children, time to symptom resolution was decreased to fewer than 4 hours with antivenin, and there is a lower rate of inpatient admission when antivenin is administered.4,10,11 There is a low incidence of anaphylactic reaction after antivenin, but there have been reported cases of self-limited serum sickness after antivenin use that generally can be managed with antihistamines and corticosteroids.4,7
Epidemiology and Identification
Centruroides is a common genus of bark scorpions in the United States with at least 21 species considered to be medically important, including the closely related Centruroides exilicauda and Centruroides sculpturatus.1 Scorpions can be recognized by a bulbous sac and pointed stinger at the end of a tail-like abdomen. They also have long lobsterlike pedipalps (pincers) for grasping their prey. Identifying characteristics for C exilicauda and C sculpturatus include a small, slender, yellow to light brown or tan body typically measuring 1.3 to 7.6 cm in length with a subaculear tooth or tubercle at the base of the stinger, a characteristic that is common to all Centruroides species (Figure).2 Some variability in size has been shown, with smaller scorpions found in increased elevations and cooler temperatures.1,3 Both C exilicauda and C sculpturatus are found in northern Mexico as well as the southwestern United States (eg, Arizona, New Mexico, Texas, California, Nevada).1 They have a preference for residing in or around trees and often are found on the underside of bark, stones, or tables as well as inside shoes or small cracks and crevices. Scorpions typically sting in self-defense, and stings commonly occur when humans attempt to move tables, put on shoes, or walk barefoot in scorpion-infested areas. Most stings occur from the end of spring through the end summer, but many may go unreported.1,4
The venom of the Centruroides genus includes peptides and proteins that play a fundamental role in toxic activity by impairing potassium, sodium, and calcium ion channels.1,3 Toxins have been shown to be species specific, functioning either in capturing prey or deterring predators. Intraspecies variability in toxins has been demonstrated, which may complicate the production of adequate antivenin.3 Many have thought that C exilicauda Wood and C sculpturatus Ewing are the same species, and the names have been used synonymously in the past; however, genetic and biochemical studies of their venom components have shown that they are distinct species and that C sculpturatus is the more dangerous of the two.5 The median lethal dose 50% of C sculpturatus was found to be 22.7 μg in CD1 mice.6
Envenomation and Clinical Manifestations
Stings from C exilicauda and C sculpturatus have been shown to cause fatality in children more often than in adults.7 In the United States, Arizona has the highest frequency of serious symptoms of envenomation as well as the highest hospital and intensive care unit admission rates.6 Envenomation results in an immediate sharp burning pain followed by numbness.4 Wounds can produce some regional lymph node swelling, ecchymosis, paresthesia, and lymphangitis. More often than not, however, wounds have little to no inflammation and are characterized only by pain.4 The puncture wound is too small to be seen, and C exilicauda and C sculpturatus venom do not cause local tissue destruction, an important factor in distinguishing it from other scorpion envenomations.
More severe complications that may follow are caused by the neurotoxin released by Centruroides stings. The toxin components can increase the duration and amplitude of the neuronal action potential and enhance the release of neurotransmitters such as acetylcholine and norepinephrine.8 Stings can lead to cranial nerve dysfunction and somatic skeletal neuromuscular dysfunction as well as autonomic dysfunction, specifically salivation, fever, tongue and muscle fasciculations, opsoclonus, vomiting, bronchoconstriction, diaphoresis, nystagmus, blurred vision, slurred speech, hypertension, rhabdomyolysis, stridor, wheezing, aspiration, anaphylaxis, and tachycardia, leading to cardiac and respiratory compromise.4,8 Some patients have experienced a decreased sense of smell or hearing and decreased fine motor movements.7 Although pancreatitis may occur with scorpion stings, it is not common for C exilicauda.9 Comorbidities such as cardiac disease and substance use disorders contribute to prolonged length of hospital stay and poor outcome.8
Treatment
Most Centruroides stings can be managed at home, but patients with more serious symptoms and children younger than 2 years should be taken to a hospital for treatment.7 If a patient reports only pain but shows no other signs of neurotoxicity, observation and pain relief with rest, ice, and elevation is appropriate management. Patients with severe manifestations have been treated with various combinations of lorazepam, glycopyrrolate, ipratropium bromide, and ondansetron, but the only treatment definitively shown to decrease time to symptom abatement is antivenin.7 It has been demonstrated that C exilicauda and C sculpturatus antivenin is relatively safe.7 Most patients, especially adults, do not die from C exilicauda and C sculpturatus stings; therefore, antivenin more commonly is symptom abating than it is lifesaving.10 In children, time to symptom resolution was decreased to fewer than 4 hours with antivenin, and there is a lower rate of inpatient admission when antivenin is administered.4,10,11 There is a low incidence of anaphylactic reaction after antivenin, but there have been reported cases of self-limited serum sickness after antivenin use that generally can be managed with antihistamines and corticosteroids.4,7
- Gonzalez-Santillan E, Possani LD. North American scorpion species of public health importance with reappraisal of historical epidemiology. Acta Tropica. 2018;187:264-274.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Carcamo-Noriega EN, Olamendi-Portugal T, Restano-Cassulini R, et al. Intraspecific variation of Centruroides sculpturatus scorpion venom from two regions of Arizona. Arch Biochem Biophys. 2018;638:52-57.
- Kang AM, Brooks DE. Nationwide scorpion exposures reported to US Poison Control centers from 2005 to 2015. J Med Toxicol. 2017;13:158-165.
- Valdez-Cruz N, Dávila S, Licea A, et al. Biochemical, genetic and physiological characterization of venom components from two species of scorpions: Centruroides exilicauda Wood and Centruroides sculpturatus Ewing. Biochimie. 2004;86:387-396.
- Jiménez-Vargas JM, Quintero-Hernández V, Gonzáles-Morales L, et al. Design and expression of recombinant toxins from Mexican scorpions of the genus Centruroides for production of antivenoms. Toxicon. 2017;128:5-14.
- Hurst NB, Lipe DN, Karpen SR, et al. Centruroides sculpturatus envenomation in three adult patients requiring treatment with antivenom. Clin Toxicol (Phila). 2018;56:294-296.
- O’Connor A, Padilla-Jones A, Ruha A. Severe bark scorpion envenomation in adults. Clin Toxicol. 2018;56:170-174.
- Berg R, Tarantino M. Envenomation by the scorpion Centruroides exilicauda (C sculpturatus): severe and unusual manifestations. Pediatrics. 1991;87:930-933.
- LoVecchio F, McBride C. Scorpion envenomations in young children in central Arizona. J Toxicol Clin Toxicol. 2003;41:937-940.
- Rodrigo C, Gnanathasan A. Management of scorpion envenoming: a systematic review and meta-analysis of controlled clinical trials. Syst Rev. 2017;6:74.
- Gonzalez-Santillan E, Possani LD. North American scorpion species of public health importance with reappraisal of historical epidemiology. Acta Tropica. 2018;187:264-274.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Carcamo-Noriega EN, Olamendi-Portugal T, Restano-Cassulini R, et al. Intraspecific variation of Centruroides sculpturatus scorpion venom from two regions of Arizona. Arch Biochem Biophys. 2018;638:52-57.
- Kang AM, Brooks DE. Nationwide scorpion exposures reported to US Poison Control centers from 2005 to 2015. J Med Toxicol. 2017;13:158-165.
- Valdez-Cruz N, Dávila S, Licea A, et al. Biochemical, genetic and physiological characterization of venom components from two species of scorpions: Centruroides exilicauda Wood and Centruroides sculpturatus Ewing. Biochimie. 2004;86:387-396.
- Jiménez-Vargas JM, Quintero-Hernández V, Gonzáles-Morales L, et al. Design and expression of recombinant toxins from Mexican scorpions of the genus Centruroides for production of antivenoms. Toxicon. 2017;128:5-14.
- Hurst NB, Lipe DN, Karpen SR, et al. Centruroides sculpturatus envenomation in three adult patients requiring treatment with antivenom. Clin Toxicol (Phila). 2018;56:294-296.
- O’Connor A, Padilla-Jones A, Ruha A. Severe bark scorpion envenomation in adults. Clin Toxicol. 2018;56:170-174.
- Berg R, Tarantino M. Envenomation by the scorpion Centruroides exilicauda (C sculpturatus): severe and unusual manifestations. Pediatrics. 1991;87:930-933.
- LoVecchio F, McBride C. Scorpion envenomations in young children in central Arizona. J Toxicol Clin Toxicol. 2003;41:937-940.
- Rodrigo C, Gnanathasan A. Management of scorpion envenoming: a systematic review and meta-analysis of controlled clinical trials. Syst Rev. 2017;6:74.
Practice Points
- Centruroides scorpions can inflict painful stings.
- Children are at greatest risk for systemic toxicity.

Tender White Lesions on the Groin
The Diagnosis: Candidal Intertrigo
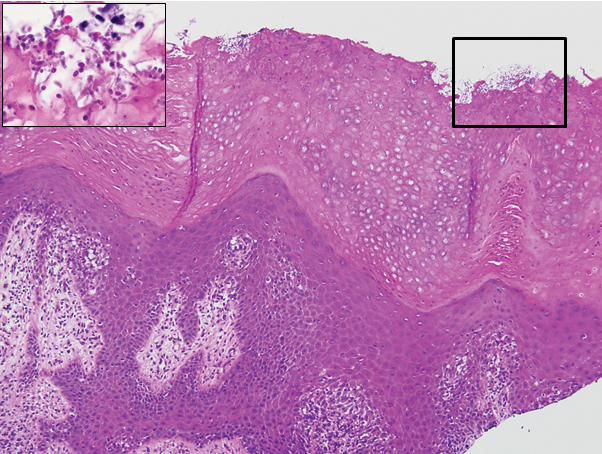
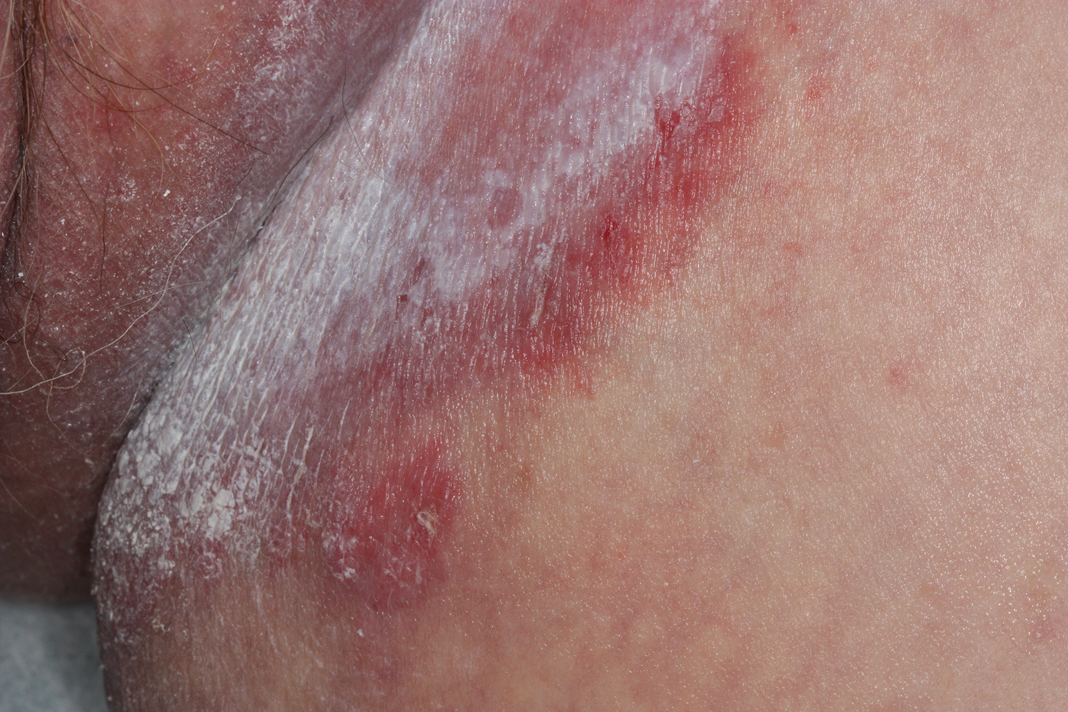
The biopsy confirmed a diagnosis of severe hyperkeratotic candidal intertrigo with no evidence of Hailey-Hailey disease. Hematoxylin and eosin- stained sections demonstrated irregular acanthosis and variable spongiosis. The stratum corneum was predominantly orthokeratotic with overlying psuedohyphae and yeast fungal elements (Figure 1).
Hyperimmunoglobulinemia E syndrome (HIES), also known as hyper-IgE syndrome or Job syndrome, is a rare immunodeficiency disorder characterized by an eczematous dermatitis-like rash, recurrent skin and lung abscesses, eosinophilia, and elevated serum IgE. Facial asymmetry, prominent forehead, deep-set eyes, broad nose, and roughened facial skin with large pores are characteristic of the sporadic and autosomal-recessive forms. Other common findings include retained primary teeth, hyperextensible joints, and recurrent mucocutaneous candidiasis.1
Although autosomal-dominant and autosomal-recessive inheritance patterns exist, sporadic mutations are the most common cause of HIES.2 Several genes have been implicated depending on the inheritance pattern. The majority of autosomal-dominant cases are associated with inactivating STAT3 (signal transducer and activator of transcription 3) mutations, whereas the majority of autosomal-recessive cases are associated with inactivating DOCK8 (dedicator of cytokinesis 8) mutations.1 Ultimately, all of these mutations lead to an impaired helper T cell (TH17) response, which is crucial for clearing fungal and extracellular bacterial infections.3
Skin eruptions typically are the first manifestation of HIES; they appear within the first week to month of life as papulopustular eruptions on the face and scalp and rapidly generalize to the rest of the body, favoring the shoulders, arms, chest, and buttocks. The pustules then coalesce into crusted plaques that resemble atopic dermatitis, frequently with superimposed Staphylococcus aureus infection. On microscopy, the pustules are folliculocentric and often contain eosinophils, whereas the plaques may contain intraepidermal collections of eosinophils.1
Mucocutaneous candidiasis is seen in approximately 60% of HIES cases and is closely linked to STAT3 inactivating mutations.3 Histologically, there is marked acanthosis with neutrophil exocytosis and abundant yeast and pseudohyphal forms within the stratum corneum (Figure 2).4 Cutaneous candidal infections typically require both oral and topical antifungal agents to clear the infection.3 Most cases of mucocutaneous candidiasis are caused by Candida albicans; however, other known culprits include Candida glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei.5,6 Of note, species identification and antifungal susceptibility studies may be useful in refractory cases, especially with C glabrata, which is known to acquire resistance to azoles, such as fluconazole, with emerging resistance to echinocandins.6

The differential diagnosis of this groin eruption included Hailey-Hailey disease; pemphigus vegetans, Hallopeau type; tinea cruris; and inverse psoriasis. Hailey-Hailey disease can be complicated by a superimposed candidal infection with similar clinical features, and biopsy may be required for definitive diagnosis. Hailey-Hailey disease typically presents with macerated fissured plaques that resemble macerated tissue paper with red fissures (Figure 3). Biopsy confirms full-thickness acantholysis resembling a dilapidated brick wall with minimal dyskeratosis.1 Pemphigus vegetans is a localized variant of pemphigus vulgaris with a predilection for flexural surfaces. The lesions progress to vegetating erosive plaques.4 The Hallopeau type often is studded with pustules and typically remains more localized than the Neumann type. Direct immunofluorescence demonstrates intercellular deposition of IgG and C3, and routine sections characteristically show pseudoepitheliomatous hyperplasia with intraepidermal eosinophilic microabscesses.1,4 Tinea cruris is characterized by erythematous annular lesions with raised scaly borders spreading down the inner thighs.7 The epidermis is variably spongiotic with parakeratosis, and neutrophils often present in a layered stratum corneum with basketweave keratin above a layer of more compact and eosinophilic keratin. Fungal stains, such as periodic acid-Schiff, will highlight the fungal hyphae within the stratum corneum. The inguinal folds are a typical location for inverse psoriasis, which generally appears as thin, sharply demarcated, shiny red plaques with less scale than plaque psoriasis.1 Psoriasiform hyperplasia with a diminished granular layer and tortuous papillary dermal vessels would be expected histologically.1
- James WD, Berger TG, Elston DM. Andrews' Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Schwartz RA, Tarlow MM. Dermatologic manifestations of Job syndrome. Medscape website. https://emedicine.medscape.com/article/1050852-overview. Updated April 22, 2019. Accessed March 28, 2020.
- Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int. 2012;61:191-196.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Pappas PG, Kauffman CA, Andes DR, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409-417.
- Center for Disease Control and Prevention. Antifungal resistance. https://www.cdc.gov/fungal/antifungal-resistance.html. Updated March 17, 2020. Accessed April 20, 2020.
- Tinea cruris. DermNet NZ website. https://www.dermnetnz.org/topics/tinea-cruris/. Published 2003. Accessed March 28, 2020.
The Diagnosis: Candidal Intertrigo
The biopsy confirmed a diagnosis of severe hyperkeratotic candidal intertrigo with no evidence of Hailey-Hailey disease. Hematoxylin and eosin- stained sections demonstrated irregular acanthosis and variable spongiosis. The stratum corneum was predominantly orthokeratotic with overlying psuedohyphae and yeast fungal elements (Figure 1).
Hyperimmunoglobulinemia E syndrome (HIES), also known as hyper-IgE syndrome or Job syndrome, is a rare immunodeficiency disorder characterized by an eczematous dermatitis-like rash, recurrent skin and lung abscesses, eosinophilia, and elevated serum IgE. Facial asymmetry, prominent forehead, deep-set eyes, broad nose, and roughened facial skin with large pores are characteristic of the sporadic and autosomal-recessive forms. Other common findings include retained primary teeth, hyperextensible joints, and recurrent mucocutaneous candidiasis.1
Although autosomal-dominant and autosomal-recessive inheritance patterns exist, sporadic mutations are the most common cause of HIES.2 Several genes have been implicated depending on the inheritance pattern. The majority of autosomal-dominant cases are associated with inactivating STAT3 (signal transducer and activator of transcription 3) mutations, whereas the majority of autosomal-recessive cases are associated with inactivating DOCK8 (dedicator of cytokinesis 8) mutations.1 Ultimately, all of these mutations lead to an impaired helper T cell (TH17) response, which is crucial for clearing fungal and extracellular bacterial infections.3
Skin eruptions typically are the first manifestation of HIES; they appear within the first week to month of life as papulopustular eruptions on the face and scalp and rapidly generalize to the rest of the body, favoring the shoulders, arms, chest, and buttocks. The pustules then coalesce into crusted plaques that resemble atopic dermatitis, frequently with superimposed Staphylococcus aureus infection. On microscopy, the pustules are folliculocentric and often contain eosinophils, whereas the plaques may contain intraepidermal collections of eosinophils.1
Mucocutaneous candidiasis is seen in approximately 60% of HIES cases and is closely linked to STAT3 inactivating mutations.3 Histologically, there is marked acanthosis with neutrophil exocytosis and abundant yeast and pseudohyphal forms within the stratum corneum (Figure 2).4 Cutaneous candidal infections typically require both oral and topical antifungal agents to clear the infection.3 Most cases of mucocutaneous candidiasis are caused by Candida albicans; however, other known culprits include Candida glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei.5,6 Of note, species identification and antifungal susceptibility studies may be useful in refractory cases, especially with C glabrata, which is known to acquire resistance to azoles, such as fluconazole, with emerging resistance to echinocandins.6
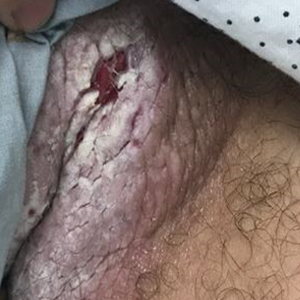
The differential diagnosis of this groin eruption included Hailey-Hailey disease; pemphigus vegetans, Hallopeau type; tinea cruris; and inverse psoriasis. Hailey-Hailey disease can be complicated by a superimposed candidal infection with similar clinical features, and biopsy may be required for definitive diagnosis. Hailey-Hailey disease typically presents with macerated fissured plaques that resemble macerated tissue paper with red fissures (Figure 3). Biopsy confirms full-thickness acantholysis resembling a dilapidated brick wall with minimal dyskeratosis.1 Pemphigus vegetans is a localized variant of pemphigus vulgaris with a predilection for flexural surfaces. The lesions progress to vegetating erosive plaques.4 The Hallopeau type often is studded with pustules and typically remains more localized than the Neumann type. Direct immunofluorescence demonstrates intercellular deposition of IgG and C3, and routine sections characteristically show pseudoepitheliomatous hyperplasia with intraepidermal eosinophilic microabscesses.1,4 Tinea cruris is characterized by erythematous annular lesions with raised scaly borders spreading down the inner thighs.7 The epidermis is variably spongiotic with parakeratosis, and neutrophils often present in a layered stratum corneum with basketweave keratin above a layer of more compact and eosinophilic keratin. Fungal stains, such as periodic acid-Schiff, will highlight the fungal hyphae within the stratum corneum. The inguinal folds are a typical location for inverse psoriasis, which generally appears as thin, sharply demarcated, shiny red plaques with less scale than plaque psoriasis.1 Psoriasiform hyperplasia with a diminished granular layer and tortuous papillary dermal vessels would be expected histologically.1
The Diagnosis: Candidal Intertrigo
The biopsy confirmed a diagnosis of severe hyperkeratotic candidal intertrigo with no evidence of Hailey-Hailey disease. Hematoxylin and eosin- stained sections demonstrated irregular acanthosis and variable spongiosis. The stratum corneum was predominantly orthokeratotic with overlying psuedohyphae and yeast fungal elements (Figure 1).
Hyperimmunoglobulinemia E syndrome (HIES), also known as hyper-IgE syndrome or Job syndrome, is a rare immunodeficiency disorder characterized by an eczematous dermatitis-like rash, recurrent skin and lung abscesses, eosinophilia, and elevated serum IgE. Facial asymmetry, prominent forehead, deep-set eyes, broad nose, and roughened facial skin with large pores are characteristic of the sporadic and autosomal-recessive forms. Other common findings include retained primary teeth, hyperextensible joints, and recurrent mucocutaneous candidiasis.1
Although autosomal-dominant and autosomal-recessive inheritance patterns exist, sporadic mutations are the most common cause of HIES.2 Several genes have been implicated depending on the inheritance pattern. The majority of autosomal-dominant cases are associated with inactivating STAT3 (signal transducer and activator of transcription 3) mutations, whereas the majority of autosomal-recessive cases are associated with inactivating DOCK8 (dedicator of cytokinesis 8) mutations.1 Ultimately, all of these mutations lead to an impaired helper T cell (TH17) response, which is crucial for clearing fungal and extracellular bacterial infections.3
Skin eruptions typically are the first manifestation of HIES; they appear within the first week to month of life as papulopustular eruptions on the face and scalp and rapidly generalize to the rest of the body, favoring the shoulders, arms, chest, and buttocks. The pustules then coalesce into crusted plaques that resemble atopic dermatitis, frequently with superimposed Staphylococcus aureus infection. On microscopy, the pustules are folliculocentric and often contain eosinophils, whereas the plaques may contain intraepidermal collections of eosinophils.1
Mucocutaneous candidiasis is seen in approximately 60% of HIES cases and is closely linked to STAT3 inactivating mutations.3 Histologically, there is marked acanthosis with neutrophil exocytosis and abundant yeast and pseudohyphal forms within the stratum corneum (Figure 2).4 Cutaneous candidal infections typically require both oral and topical antifungal agents to clear the infection.3 Most cases of mucocutaneous candidiasis are caused by Candida albicans; however, other known culprits include Candida glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei.5,6 Of note, species identification and antifungal susceptibility studies may be useful in refractory cases, especially with C glabrata, which is known to acquire resistance to azoles, such as fluconazole, with emerging resistance to echinocandins.6
The differential diagnosis of this groin eruption included Hailey-Hailey disease; pemphigus vegetans, Hallopeau type; tinea cruris; and inverse psoriasis. Hailey-Hailey disease can be complicated by a superimposed candidal infection with similar clinical features, and biopsy may be required for definitive diagnosis. Hailey-Hailey disease typically presents with macerated fissured plaques that resemble macerated tissue paper with red fissures (Figure 3). Biopsy confirms full-thickness acantholysis resembling a dilapidated brick wall with minimal dyskeratosis.1 Pemphigus vegetans is a localized variant of pemphigus vulgaris with a predilection for flexural surfaces. The lesions progress to vegetating erosive plaques.4 The Hallopeau type often is studded with pustules and typically remains more localized than the Neumann type. Direct immunofluorescence demonstrates intercellular deposition of IgG and C3, and routine sections characteristically show pseudoepitheliomatous hyperplasia with intraepidermal eosinophilic microabscesses.1,4 Tinea cruris is characterized by erythematous annular lesions with raised scaly borders spreading down the inner thighs.7 The epidermis is variably spongiotic with parakeratosis, and neutrophils often present in a layered stratum corneum with basketweave keratin above a layer of more compact and eosinophilic keratin. Fungal stains, such as periodic acid-Schiff, will highlight the fungal hyphae within the stratum corneum. The inguinal folds are a typical location for inverse psoriasis, which generally appears as thin, sharply demarcated, shiny red plaques with less scale than plaque psoriasis.1 Psoriasiform hyperplasia with a diminished granular layer and tortuous papillary dermal vessels would be expected histologically.1
- James WD, Berger TG, Elston DM. Andrews' Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Schwartz RA, Tarlow MM. Dermatologic manifestations of Job syndrome. Medscape website. https://emedicine.medscape.com/article/1050852-overview. Updated April 22, 2019. Accessed March 28, 2020.
- Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int. 2012;61:191-196.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Pappas PG, Kauffman CA, Andes DR, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409-417.
- Center for Disease Control and Prevention. Antifungal resistance. https://www.cdc.gov/fungal/antifungal-resistance.html. Updated March 17, 2020. Accessed April 20, 2020.
- Tinea cruris. DermNet NZ website. https://www.dermnetnz.org/topics/tinea-cruris/. Published 2003. Accessed March 28, 2020.
- James WD, Berger TG, Elston DM. Andrews' Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Schwartz RA, Tarlow MM. Dermatologic manifestations of Job syndrome. Medscape website. https://emedicine.medscape.com/article/1050852-overview. Updated April 22, 2019. Accessed March 28, 2020.
- Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int. 2012;61:191-196.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Pappas PG, Kauffman CA, Andes DR, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409-417.
- Center for Disease Control and Prevention. Antifungal resistance. https://www.cdc.gov/fungal/antifungal-resistance.html. Updated March 17, 2020. Accessed April 20, 2020.
- Tinea cruris. DermNet NZ website. https://www.dermnetnz.org/topics/tinea-cruris/. Published 2003. Accessed March 28, 2020.
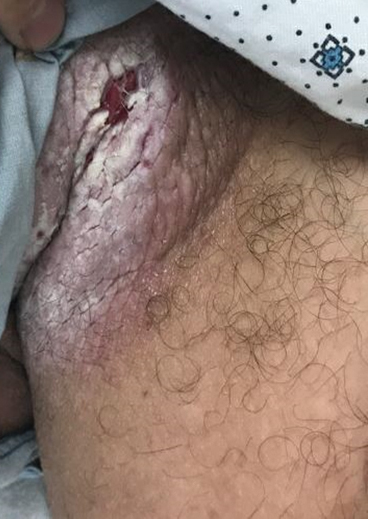
A 28-year-old man with a history of hyperimmunoglobulinemia E syndrome (previously known as Job syndrome), coarse facial features, and multiple skin and soft tissue infections presented to the university dermatology clinic with persistent white, macerated, fissured groin plaques that were present for months. The lesions were tender and pruritic with a burning sensation. Treatment with topical terbinafine and oral fluconazole was attempted without resolution of the eruption. A biopsy of the groin lesion was performed.
Climate Change and Expansion of Tick Geography
The expanding range of tick-borne diseases is a growing problem worldwide. Climate change plays a preeminent role in the expansion of tick species, especially for southern ticks in the United States such as Amblyomma species, which have introduced new pathogens to northern states.1-5 In addition to well-known tick-borne diseases, Amblyomma ticks have been implicated in the spread of emerging severe and potentially fatal viral illnesses, including Bourbon virus and Heartland virus.6 The increasing range of Amblyomma ticks also exposes new populations to tick-induced meat allergy (alpha-gal) syndrome, whereby development of specific IgE antibodies to the oligosaccharide galactose-alpha-1,3-galactose (alpha-gal) following tick bites results in severe allergic responses to consumption of
Amblyomma ticks have now been identified close to the Canadian border in Michigan and New York, and predictions of continued climate change raise the possibility of northward range expansion into all provinces of Canada from Alberta to Newfoundland and Labrador during the coming decades.8,9 Additional factors that contribute to the expanding range of many tick species include international travel, migratory patterns of birds, competition, and natural predators such as fire ants that feed on tick eggs and influence the feeding behavior of adults.10
Traditional methods of tick identification rely on gross morphology, including the presence of festoons, shape of the coxae where the legs attach, and markings on the hard overlying scutum. More recently, molecular identification has improved tick identification, leading to more accurate assessment of tick prevalence. These modern identification studies include analysis of 16S ribosomal DNA (rDNA), 12S rDNA, and ITS1 rDNA, and ITS2 rDNA genes.11
The spread of tick vectors has huge public health implications, and better methods to control tick populations are needed.12 New acaricides and growth regulators are being developed,13 and early spring applications of acaricides such as bifenthrin can suppress nymphs prior to the initiation of host-seeking activity.14 Controlled burns within tick habitats have proved helpful in reducing the risk for vector-borne disease.15,16 Personal protection is best accomplished with the use of a repellent together with clothing impregnated with an acaricide such as permethrin.17 Efforts to slow climate change and continued surveillance for the spread of tick vectors is urgently needed.
- Sanchez-Vicente S, Tagliafierro T, Coleman JL, et al. Polymicrobial nature of tick-borne diseases [published online September 10, 2019]. MBio. doi:10.1128/mBio.02055-19.
- Raghavan RK, Peterson AT, Cobos ME, et al. Current and future distribution of the Lone Star tick, Amblyomma americanum (L.) (Acari: Ixodidae) in North America. PLoS One. 2019;14:e0209082.
- Stafford KC 3rd, Molaei G, Little EAH, et al. Distribution and establishment of the Lone Star tick in Connecticut and implications for range expansion and public health. J Med Entomol. 2018;25:1561-1568.
- Gilliam ME, Rechkemmer WT, McCravy KW, et al. The influence of prescribed fire, habitat, and weather on Amblyomma americanum (Ixodida: Ixodidae) in West-Central Illinois, USA [published online March 22, 2018]. Insects. doi:10.3390/insects9020036.
- Sonenshine DE. Range expansion of tick disease vectors in North America: implications for spread of tick-borne disease [published online March 9, 2018]. Int J Environ Res Public Health. doi:10.3390/ijerph15030478.
- Savage HM, Godsey MS Jr, Panella NA, et al. Surveillance for tick-borne viruses near the location of a fatal human case of Bourbon virus (family Orthomyxoviridae: genus Thogotovirus) in eastern Kansas, 2015. J Med Entomol. 2018;55:701-705.
- Crispell G, Commins SP, Archer-Hartman SA, et al. Discovery of alpha-gal-containing antigens in North American tick species believed to induce red meat allergy. Front Immunol. 2019;10:1056.
- Gasmi S, Bouchard C, Ogden NH, et al. Evidence for increasing densities and geographic ranges of tick species of public health significance other than Ixodes scapularis in Québec, Canada. PLoS One. 2018;13:e0201924.
- Sagurova I, Ludwig A, Ogden NH, et al. Predicted northward expansion of the geographic range of the tick vector Amblyomma americanum in North America under future climate conditions. Environ Health Perspect. 2019;127:107014.
- Kjeldgaard MK, Takano OM, Bockoven AA, et al. Red imported fire ant (Solenopsis invicta) aggression influences the behavior of three hard tick species. Exp Appl Acarol. 2019;79:87-97.
- Abouelhassan EM, El-Gawady HM, Abdel-Aal AA, et al. Comparison of some molecular markers for tick species identification. J Arthropod Borne Dis. 2019;13:153-164.
- Jordan RA, Egizi A. The growing importance of lone star ticks in a Lyme disease endemic county: passive tick surveillance in Monmouth County, NJ, 2006–2016. PLoS One. 2019;14:e0211778.
- Showler AT, Donahue WA, Harlien JL, et al. Efficacy of novaluron + pyriproxyfen (Tekko Pro) insect growth regulators against Amblyomma americanum (Acari: Ixodidae), Rhipicephalus (Boophilus) annulatus, Rhipicephalus (Boophilus) microplus, and Rhipicephalus sanguineus. J Med Entomol. 2019;56:1338-1345.
- Schulze TL, Jordan RA. Early season applications of bifenthrin suppress host-seeking Ixodes scapularis and Amblyomma americanum (Acari: Ixodidae) nymphs [published online November 26, 2019]. J Med Entomol. doi:10.1093/jme/tjz202.
- Hodo CL, Forgacs D, Auckland LD, et al. Presence of diverse Rickettsia spp. and absence of Borrelia burgdorferi sensu lato in ticks in an East Texas forest with reduced tick density associated with controlled burns. Ticks Tick Borne Dis. 2020;11:101310.
- Gleim ER, Zemtsova GE, Berghaus RD, et al. Frequent prescribed fires can reduce risk of tick-borne diseases. Sci Rep. 2019;9:9974.
- Prose R, Breuner NE, Johnson TL, et al. Contact irritancy and toxicity of permethrin-treated clothing for Ixodes scapularis, Amblyomma americanum, and Dermacentor variabilis ticks (Acari: Ixodidae). J Med Entomol. 2018;55:1217-1224.
The expanding range of tick-borne diseases is a growing problem worldwide. Climate change plays a preeminent role in the expansion of tick species, especially for southern ticks in the United States such as Amblyomma species, which have introduced new pathogens to northern states.1-5 In addition to well-known tick-borne diseases, Amblyomma ticks have been implicated in the spread of emerging severe and potentially fatal viral illnesses, including Bourbon virus and Heartland virus.6 The increasing range of Amblyomma ticks also exposes new populations to tick-induced meat allergy (alpha-gal) syndrome, whereby development of specific IgE antibodies to the oligosaccharide galactose-alpha-1,3-galactose (alpha-gal) following tick bites results in severe allergic responses to consumption of
Amblyomma ticks have now been identified close to the Canadian border in Michigan and New York, and predictions of continued climate change raise the possibility of northward range expansion into all provinces of Canada from Alberta to Newfoundland and Labrador during the coming decades.8,9 Additional factors that contribute to the expanding range of many tick species include international travel, migratory patterns of birds, competition, and natural predators such as fire ants that feed on tick eggs and influence the feeding behavior of adults.10
Traditional methods of tick identification rely on gross morphology, including the presence of festoons, shape of the coxae where the legs attach, and markings on the hard overlying scutum. More recently, molecular identification has improved tick identification, leading to more accurate assessment of tick prevalence. These modern identification studies include analysis of 16S ribosomal DNA (rDNA), 12S rDNA, and ITS1 rDNA, and ITS2 rDNA genes.11
The spread of tick vectors has huge public health implications, and better methods to control tick populations are needed.12 New acaricides and growth regulators are being developed,13 and early spring applications of acaricides such as bifenthrin can suppress nymphs prior to the initiation of host-seeking activity.14 Controlled burns within tick habitats have proved helpful in reducing the risk for vector-borne disease.15,16 Personal protection is best accomplished with the use of a repellent together with clothing impregnated with an acaricide such as permethrin.17 Efforts to slow climate change and continued surveillance for the spread of tick vectors is urgently needed.
The expanding range of tick-borne diseases is a growing problem worldwide. Climate change plays a preeminent role in the expansion of tick species, especially for southern ticks in the United States such as Amblyomma species, which have introduced new pathogens to northern states.1-5 In addition to well-known tick-borne diseases, Amblyomma ticks have been implicated in the spread of emerging severe and potentially fatal viral illnesses, including Bourbon virus and Heartland virus.6 The increasing range of Amblyomma ticks also exposes new populations to tick-induced meat allergy (alpha-gal) syndrome, whereby development of specific IgE antibodies to the oligosaccharide galactose-alpha-1,3-galactose (alpha-gal) following tick bites results in severe allergic responses to consumption of
Amblyomma ticks have now been identified close to the Canadian border in Michigan and New York, and predictions of continued climate change raise the possibility of northward range expansion into all provinces of Canada from Alberta to Newfoundland and Labrador during the coming decades.8,9 Additional factors that contribute to the expanding range of many tick species include international travel, migratory patterns of birds, competition, and natural predators such as fire ants that feed on tick eggs and influence the feeding behavior of adults.10
Traditional methods of tick identification rely on gross morphology, including the presence of festoons, shape of the coxae where the legs attach, and markings on the hard overlying scutum. More recently, molecular identification has improved tick identification, leading to more accurate assessment of tick prevalence. These modern identification studies include analysis of 16S ribosomal DNA (rDNA), 12S rDNA, and ITS1 rDNA, and ITS2 rDNA genes.11
The spread of tick vectors has huge public health implications, and better methods to control tick populations are needed.12 New acaricides and growth regulators are being developed,13 and early spring applications of acaricides such as bifenthrin can suppress nymphs prior to the initiation of host-seeking activity.14 Controlled burns within tick habitats have proved helpful in reducing the risk for vector-borne disease.15,16 Personal protection is best accomplished with the use of a repellent together with clothing impregnated with an acaricide such as permethrin.17 Efforts to slow climate change and continued surveillance for the spread of tick vectors is urgently needed.
- Sanchez-Vicente S, Tagliafierro T, Coleman JL, et al. Polymicrobial nature of tick-borne diseases [published online September 10, 2019]. MBio. doi:10.1128/mBio.02055-19.
- Raghavan RK, Peterson AT, Cobos ME, et al. Current and future distribution of the Lone Star tick, Amblyomma americanum (L.) (Acari: Ixodidae) in North America. PLoS One. 2019;14:e0209082.
- Stafford KC 3rd, Molaei G, Little EAH, et al. Distribution and establishment of the Lone Star tick in Connecticut and implications for range expansion and public health. J Med Entomol. 2018;25:1561-1568.
- Gilliam ME, Rechkemmer WT, McCravy KW, et al. The influence of prescribed fire, habitat, and weather on Amblyomma americanum (Ixodida: Ixodidae) in West-Central Illinois, USA [published online March 22, 2018]. Insects. doi:10.3390/insects9020036.
- Sonenshine DE. Range expansion of tick disease vectors in North America: implications for spread of tick-borne disease [published online March 9, 2018]. Int J Environ Res Public Health. doi:10.3390/ijerph15030478.
- Savage HM, Godsey MS Jr, Panella NA, et al. Surveillance for tick-borne viruses near the location of a fatal human case of Bourbon virus (family Orthomyxoviridae: genus Thogotovirus) in eastern Kansas, 2015. J Med Entomol. 2018;55:701-705.
- Crispell G, Commins SP, Archer-Hartman SA, et al. Discovery of alpha-gal-containing antigens in North American tick species believed to induce red meat allergy. Front Immunol. 2019;10:1056.
- Gasmi S, Bouchard C, Ogden NH, et al. Evidence for increasing densities and geographic ranges of tick species of public health significance other than Ixodes scapularis in Québec, Canada. PLoS One. 2018;13:e0201924.
- Sagurova I, Ludwig A, Ogden NH, et al. Predicted northward expansion of the geographic range of the tick vector Amblyomma americanum in North America under future climate conditions. Environ Health Perspect. 2019;127:107014.
- Kjeldgaard MK, Takano OM, Bockoven AA, et al. Red imported fire ant (Solenopsis invicta) aggression influences the behavior of three hard tick species. Exp Appl Acarol. 2019;79:87-97.
- Abouelhassan EM, El-Gawady HM, Abdel-Aal AA, et al. Comparison of some molecular markers for tick species identification. J Arthropod Borne Dis. 2019;13:153-164.
- Jordan RA, Egizi A. The growing importance of lone star ticks in a Lyme disease endemic county: passive tick surveillance in Monmouth County, NJ, 2006–2016. PLoS One. 2019;14:e0211778.
- Showler AT, Donahue WA, Harlien JL, et al. Efficacy of novaluron + pyriproxyfen (Tekko Pro) insect growth regulators against Amblyomma americanum (Acari: Ixodidae), Rhipicephalus (Boophilus) annulatus, Rhipicephalus (Boophilus) microplus, and Rhipicephalus sanguineus. J Med Entomol. 2019;56:1338-1345.
- Schulze TL, Jordan RA. Early season applications of bifenthrin suppress host-seeking Ixodes scapularis and Amblyomma americanum (Acari: Ixodidae) nymphs [published online November 26, 2019]. J Med Entomol. doi:10.1093/jme/tjz202.
- Hodo CL, Forgacs D, Auckland LD, et al. Presence of diverse Rickettsia spp. and absence of Borrelia burgdorferi sensu lato in ticks in an East Texas forest with reduced tick density associated with controlled burns. Ticks Tick Borne Dis. 2020;11:101310.
- Gleim ER, Zemtsova GE, Berghaus RD, et al. Frequent prescribed fires can reduce risk of tick-borne diseases. Sci Rep. 2019;9:9974.
- Prose R, Breuner NE, Johnson TL, et al. Contact irritancy and toxicity of permethrin-treated clothing for Ixodes scapularis, Amblyomma americanum, and Dermacentor variabilis ticks (Acari: Ixodidae). J Med Entomol. 2018;55:1217-1224.
- Sanchez-Vicente S, Tagliafierro T, Coleman JL, et al. Polymicrobial nature of tick-borne diseases [published online September 10, 2019]. MBio. doi:10.1128/mBio.02055-19.
- Raghavan RK, Peterson AT, Cobos ME, et al. Current and future distribution of the Lone Star tick, Amblyomma americanum (L.) (Acari: Ixodidae) in North America. PLoS One. 2019;14:e0209082.
- Stafford KC 3rd, Molaei G, Little EAH, et al. Distribution and establishment of the Lone Star tick in Connecticut and implications for range expansion and public health. J Med Entomol. 2018;25:1561-1568.
- Gilliam ME, Rechkemmer WT, McCravy KW, et al. The influence of prescribed fire, habitat, and weather on Amblyomma americanum (Ixodida: Ixodidae) in West-Central Illinois, USA [published online March 22, 2018]. Insects. doi:10.3390/insects9020036.
- Sonenshine DE. Range expansion of tick disease vectors in North America: implications for spread of tick-borne disease [published online March 9, 2018]. Int J Environ Res Public Health. doi:10.3390/ijerph15030478.
- Savage HM, Godsey MS Jr, Panella NA, et al. Surveillance for tick-borne viruses near the location of a fatal human case of Bourbon virus (family Orthomyxoviridae: genus Thogotovirus) in eastern Kansas, 2015. J Med Entomol. 2018;55:701-705.
- Crispell G, Commins SP, Archer-Hartman SA, et al. Discovery of alpha-gal-containing antigens in North American tick species believed to induce red meat allergy. Front Immunol. 2019;10:1056.
- Gasmi S, Bouchard C, Ogden NH, et al. Evidence for increasing densities and geographic ranges of tick species of public health significance other than Ixodes scapularis in Québec, Canada. PLoS One. 2018;13:e0201924.
- Sagurova I, Ludwig A, Ogden NH, et al. Predicted northward expansion of the geographic range of the tick vector Amblyomma americanum in North America under future climate conditions. Environ Health Perspect. 2019;127:107014.
- Kjeldgaard MK, Takano OM, Bockoven AA, et al. Red imported fire ant (Solenopsis invicta) aggression influences the behavior of three hard tick species. Exp Appl Acarol. 2019;79:87-97.
- Abouelhassan EM, El-Gawady HM, Abdel-Aal AA, et al. Comparison of some molecular markers for tick species identification. J Arthropod Borne Dis. 2019;13:153-164.
- Jordan RA, Egizi A. The growing importance of lone star ticks in a Lyme disease endemic county: passive tick surveillance in Monmouth County, NJ, 2006–2016. PLoS One. 2019;14:e0211778.
- Showler AT, Donahue WA, Harlien JL, et al. Efficacy of novaluron + pyriproxyfen (Tekko Pro) insect growth regulators against Amblyomma americanum (Acari: Ixodidae), Rhipicephalus (Boophilus) annulatus, Rhipicephalus (Boophilus) microplus, and Rhipicephalus sanguineus. J Med Entomol. 2019;56:1338-1345.
- Schulze TL, Jordan RA. Early season applications of bifenthrin suppress host-seeking Ixodes scapularis and Amblyomma americanum (Acari: Ixodidae) nymphs [published online November 26, 2019]. J Med Entomol. doi:10.1093/jme/tjz202.
- Hodo CL, Forgacs D, Auckland LD, et al. Presence of diverse Rickettsia spp. and absence of Borrelia burgdorferi sensu lato in ticks in an East Texas forest with reduced tick density associated with controlled burns. Ticks Tick Borne Dis. 2020;11:101310.
- Gleim ER, Zemtsova GE, Berghaus RD, et al. Frequent prescribed fires can reduce risk of tick-borne diseases. Sci Rep. 2019;9:9974.
- Prose R, Breuner NE, Johnson TL, et al. Contact irritancy and toxicity of permethrin-treated clothing for Ixodes scapularis, Amblyomma americanum, and Dermacentor variabilis ticks (Acari: Ixodidae). J Med Entomol. 2018;55:1217-1224.

Multinodular Plaque on the Penis
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
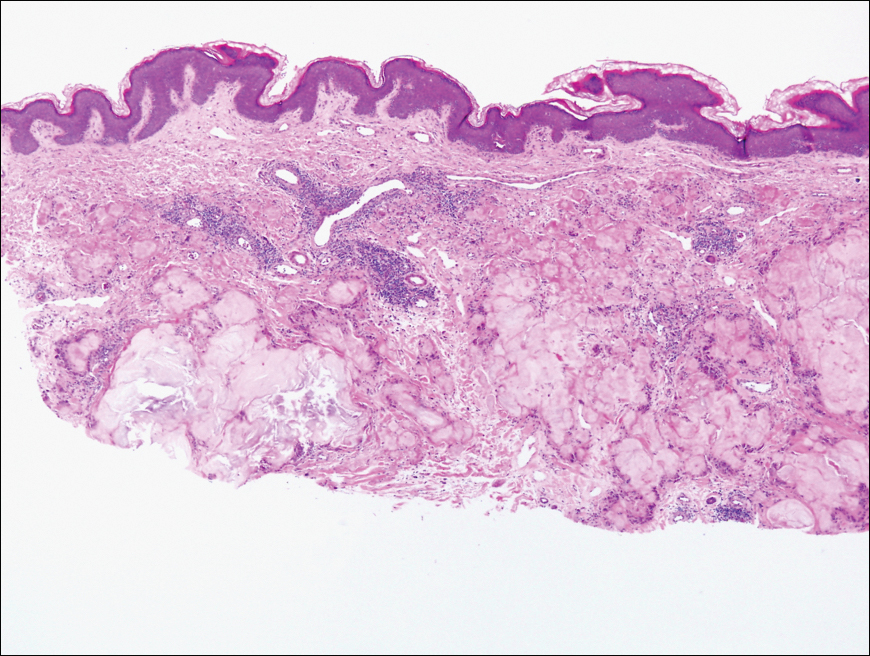
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
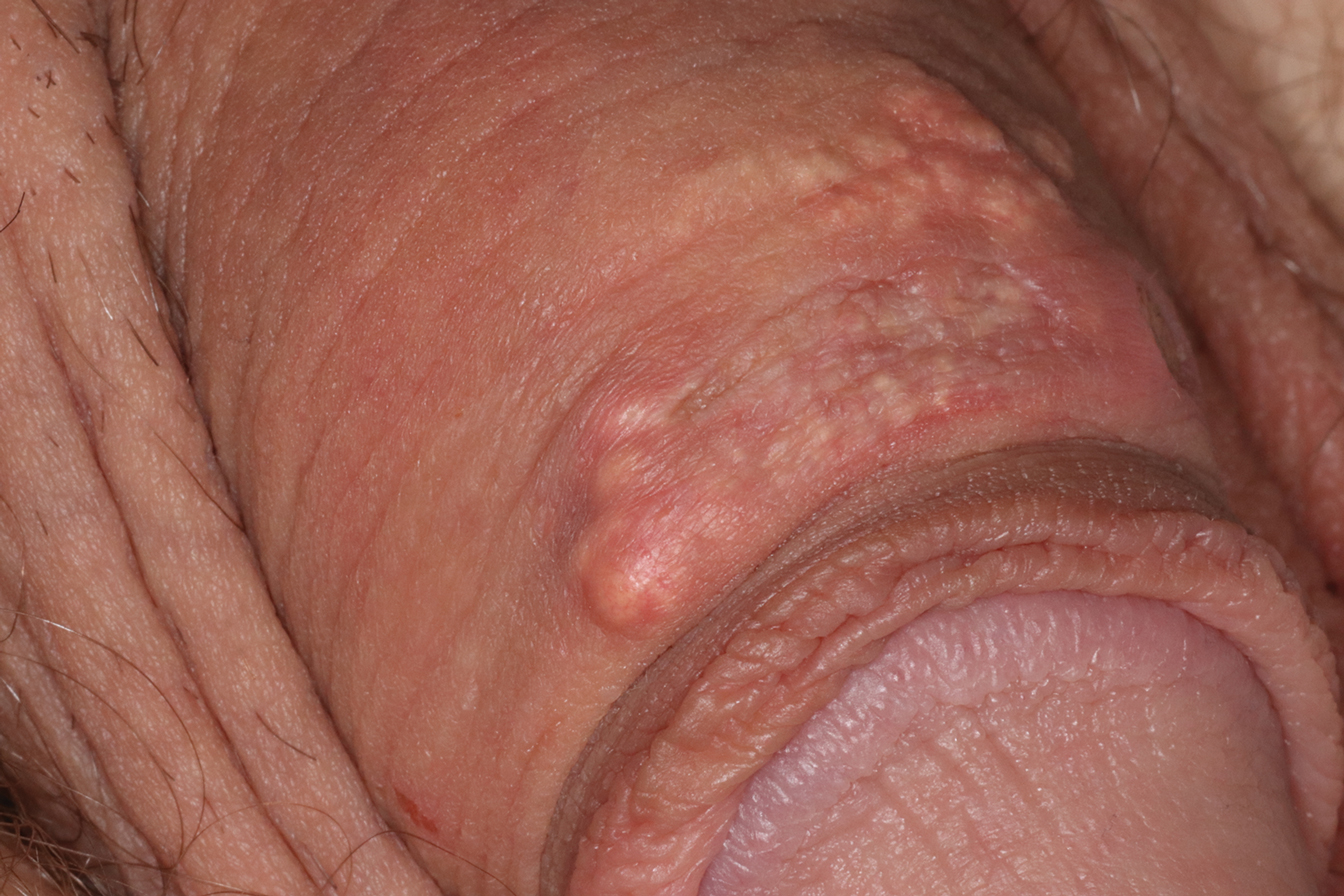
A 34-year-old man presented for evaluation of a slowly growing group of firm white bumps on the penis. The lesions were nontender and asymptomatic. Medical and family history was notable for gout, though he was not being treated. Physical examination revealed a 3-cm, firm, multinodular, chalky white plaque on the dorsal aspect of the penile shaft. A tangential biopsy was performed and sent for hematoxylin and eosin staining.
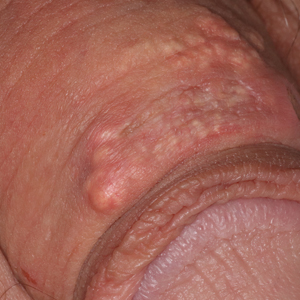
What’s Eating You? Human Body Lice (Pediculus humanus corporis)
Epidemiology and Transmission
Pediculus humanus corporis, commonly known as the human body louse, is one in a family of 3 ectoparasites of the same suborder that also encompasses pubic lice (Phthirus pubis) and head lice (Pediculus humanus capitis). Adults are approximately 2 mm in size, with the same life cycle as head lice (Figure 1). They require blood meals roughly 5 times per day and cannot survive longer than 2 days without feeding.1 Although similar in structure to head lice, body lice differ behaviorally in that they do not reside on their human host’s body; instead, they infest the host’s clothing, localizing to seams (Figure 2), and migrate to the host for blood meals. In fact, based on this behavior, genetic analysis of early human body lice has been used to postulate when clothing was first used by humans as well as to determine early human migration patterns.2,3
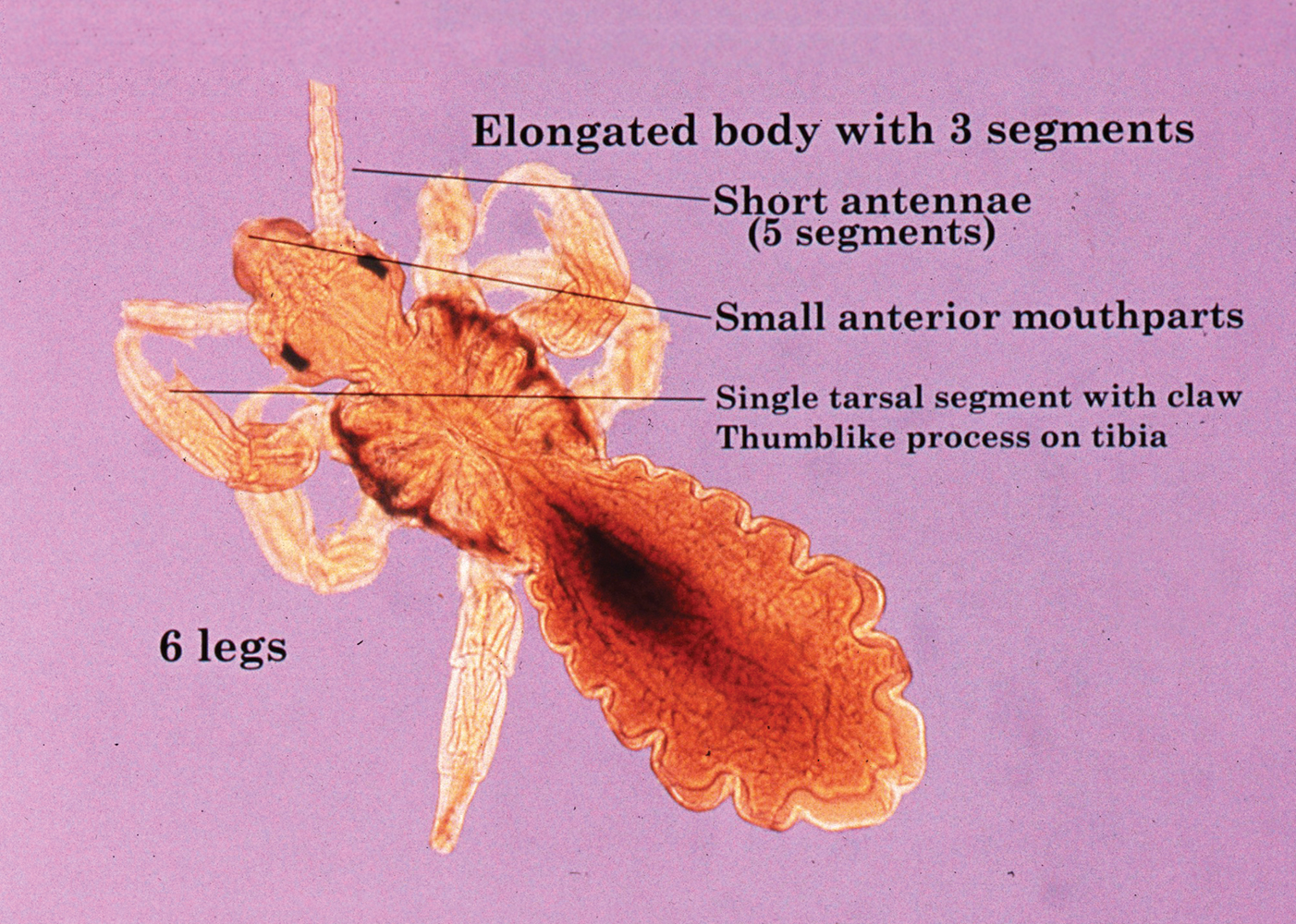

Although clinicians in developed countries may be less familiar with body lice compared to their counterparts, body lice nevertheless remain a global health concern in impoverished, densely populated areas, as well as in homeless populations due to poor hygiene. Transmission frequently occurs via physical contact with an affected individual and his/her personal items (eg, linens) via fomites.4,5 Body louse infestation is more prevalent in homeless individuals who sleep outside vs in shelters; a history of pubic lice and lack of regular bathing have been reported as additional risk factors.6 Outbreaks have been noted in the wake of natural disasters, in the setting of political upheavals, and in refugee camps, as well as in individuals seeking political asylum.7 Unlike head and pubic lice, body lice can serve as vectors for infectious diseases including Rickettsia prowazekii (epidemic typhus), Borrelia recurrentis (louse-borne relapsing fever), Bartonella quintana (trench fever), and Yersinia pestis (plague).5,8,9 Several Acinetobacter species were isolated from nearly one-third of collected body louse specimens in a French study.10 Additionally, serology for B quintana was found to be positive in up to 30% of cases in one United States urban homeless population.4
Clinical Manifestations
Patients often present with generalized pruritus, usually considerably more severe than with P humanus capitis, with lesions concentrated on the trunk.11 In addition to often impetiginized, self-inflicted excoriations, feeding sites may present as erythematous macules (Figure 3), papules, or papular urticaria with a central hemorrhagic punctum. Extensive infestation also can manifest as the colloquial vagabond disease, characterized by postinflammatory hyperpigmentation and thickening of the involved skin. Remarkably, patients also may present with considerable iron-deficiency anemia secondary to high parasite load and large volume blood feeding. Multiple case reports have demonstrated associated morbidity.12-14 The differential diagnosis for pediculosis may include scabies, lichen simplex chronicus, and eczematous dermatitis, though the clinician should prudently consider whether both scabies and pediculosis may be present, as coexistence is possible.4,15

Diagnosis
Diagnosis can be reached by visualizing adult lice, nymphs, or viable nits on the body or more commonly within inner clothing seams; nits also fluoresce under Wood light.15 Although dermoscopy has proven useful for increased sensitivity and differentiation between viable and hatched nits, the insects also can be viewed with the unaided eye.16
Treatment: New Concerns and Strategies
The mainstay of treatment for body lice has long consisted of thorough washing and drying of all clothing and linens in a hot dryer. Treatment can be augmented with the addition of pharmacotherapy, plus antibiotics as warranted for louse-borne disease. Pharmacologic intervention often is used in cases of mass infestation and is similar to head lice.
Options for head lice include topical permethrin, malathion, lindane, spinosad, benzyl alcohol, and ivermectin. Pyrethroids, derived from the chrysanthemum, generally are considered safe for human use with a side-effect profile limited to irritation and allergy17; however, neurotoxicity and leukemia are clinical concerns, with an association more recently shown between large-volume use of pyrethroids and acute lymphoblastic leukemia.18,19 Use of lindane is not recommended due to a greater potential for central nervous system neurotoxicity, manifested by seizures, with repeated large surface application. Malathion is problematic due to the risk for mucosal irritation, flammability of some formulations, and theoretical organophosphate poisoning, as its mechanism of action involves inhibition of acetylcholinesterase.15 However, in the context of head lice treatment, a randomized controlled trial reported no incidence of acetylcholinesterase inhibition.20 Spinosad, manufactured from the soil bacterium Saccharopolyspora spinosa, functions similarly by interfering with the nicotinic acetylcholine receptor and also carries a risk for skin irritation.21 Among all the treatment options, we prefer benzyl alcohol, particularly in the context of resistance, as it is effective via a physical mechanism of action and lacks notable neurotoxic effects to the host. Use of benzyl alcohol is approved for patients as young as 6 months; it functions by asphyxiating the lice via paralysis of the respiratory spiracle with occlusion by inert ingredients. Itching, episodic numbness, and scalp or mucosal irritation are possible complications of treatment.22
Treatment resistance of body lice has increased in recent years, warranting exploration of additional management strategies. Moreover, developing resistance to lindane and malathion has been reported.23 Resistance to pyrethroids has been attributed to mutations in a voltage-gated sodium channel, one of which was universally present in the sampling of a single population.24 A randomized controlled trial showed that off-label oral ivermectin 400 μg/kg was superior to malathion lotion 0.5% in difficult-to-treat cases of head lice25; utility of oral ivermectin also has been reported in body lice.26 In vitro studies also have shown promise for pursuing synergistic treatment of body lice with both ivermectin and antibiotics.27
A novel primary prophylaxis approach for at-risk homeless individuals recently utilized permethrin-impregnated underwear. Although the intervention provided short-term infestation improvement, longer-term use did not show improvement from placebo and also increased prevalence of permethrin-resistant haplotypes.2
- Veracx A, Raoult D. Biology and genetics of human head and body lice. Trends Parasitol. 2012;28:563-571.
- Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing. Curr Biol. 2003;13:1414-1417.
- Drali R, Mumcuoglu KY, Yesilyurt G, et al. Studies of ancient lice reveal unsuspected past migrations of vectors. Am J Trop Med Hyg. 2015;93:623-625.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Feldmeier H, Heukelbach J. Epidermal parasitic skin diseases: a neglected category of poverty-associated plagues. Bull World Health Organ. 2009;87:152-159.
- Arnaud A, Chosidow O, Detrez MA, et al. Prevalence of scabies and Pediculosis corporis among homeless people in the Paris region: results from two randomized cross-sectional surveys (HYTPEAC study). Br J Dermatol. 2016;174:104-112.
- Hytonen J, Khawaja T, Gronroos JO, et al. Louse-borne relapsing fever in Finland in two asylum seekers from Somalia. APMIS. 2017;125:59-62.
- Nordmann T, Feldt T, Bosselmann M, et al. Outbreak of louse-borne relapsing fever among urban dwellers in Arsi Zone, Central Ethiopia, from July to November 2016. Am J Trop Med Hyg. 2018;98:1599-1602.
- Louni M, Mana N, Bitam I, et al. Body lice of homeless people reveal the presence of several emerging bacterial pathogens in northern Algeria. PLoS Negl Trop Dis. 2018;12:E0006397.
- Candy K, Amanzougaghene N, Izri A, et al. Molecular survey of head and body lice, Pediculus humanus, in France. Vector Borne Zoonotic Dis. 2018;18:243-251.
Bolognia JL, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier Limited; 2018. - Nara A, Nagai H, Yamaguchi R, et al. An unusual autopsy case of lethal hypothermia exacerbated by body lice-induced severe anemia. Int J Legal Med. 2016;130:765-769.
- Althomali SA, Alzubaidi LM, Alkhaldi DM. Severe iron deficiency anaemia associated with heavy lice infestation in a young woman [published online November 5, 2015]. BMJ Case Rep. doi:10.1136/bcr-2015-212207.
- Hau V, Muhi-Iddin N. A ghost covered in lice: a case of severe blood loss with long-standing heavy pediculosis capitis infestation [published online December 19, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-206623.
- Diaz JH. Lice (Pediculosis). In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 9th ed. New York, NY: Elsevier; 2020:3482-3486.
- Martins LG, Bernardes Filho F, Quaresma MV, et al. Dermoscopy applied to pediculosis corporis diagnosis. An Bras Dermatol. 2014;89:513-514.
- Devore CD, Schutze GE; Council on School Health and Committee on Infectious Diseases, American Academy of Pediatrics. Head lice. Pediatrics. 2015;135:E1355-E1365.
- Shafer TJ, Meyer DA, Crofton KM. Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs. Environ Health Perspect. 2005;113:123-136.
- Ding G, Shi R, Gao Y, et al. Pyrethroid pesticide exposure and risk of childhood acute lymphocytic leukemia in Shanghai. Environ Sci Technol. 2012;46:13480-13487.
- Meinking TL, Vicaria M, Eyerdam DH, et al. A randomized, investigator-blinded, time-ranging study of the comparative efficacy of 0.5% malathion gel versus Ovide Lotion (0.5% malathion) or Nix Crème Rinse (1% permethrin) used as labeled, for the treatment of head lice. Pediatr Dermatol. 2007;24:405-411.
- McCormack PL. Spinosad: in pediculosis capitis. Am J Clin Dermatol. 2011;12:349-353.
- Meinking TL, Villar ME, Vicaria M, et al. The clinical trials supporting benzyl alcohol lotion 5% (Ulesfia): a safe and effective topical treatment for head lice (pediculosis humanus capitis). Pediatr Dermatol. 2010;27:19-24.
- Lebwohl M, Clark L, Levitt J. Therapy for head lice based on life cycle, resistance, and safety considerations. Pediatrics. 2007;119:965-974
- Drali R, Benkouiten S, Badiaga S, et al. Detection of a knockdown resistance mutation associated with permethrin resistance in the body louse Pediculus humanus corporis by use of melting curve analysis genotyping. J Clin Microbiol. 2012;50:2229-2233.
- Chosidow O, Giraudeau B, Cottrell J, et al. Oral ivermectin versus malathion lotion for difficult-to-treat head lice. N Engl J Med. 2010;362:896-905.
- Foucault C, Ranque S, Badiaga S, et al. Oral ivermectin in the treatment of body lice. J Infect Dis. 2006;193:474-476.
- Sangaré AK, Doumbo OK, Raoult D. Management and treatment of human lice [published online July 27, 2016]. Biomed Res Int. doi:10.1155/2016/8962685.
- Benkouiten S, Drali R, Badiaga S, et al. Effect of permethrin-impregnated underwear on body lice in sheltered homeless persons: a randomized controlled trial. JAMA Dermatol. 2014;150:273-279.
Epidemiology and Transmission
Pediculus humanus corporis, commonly known as the human body louse, is one in a family of 3 ectoparasites of the same suborder that also encompasses pubic lice (Phthirus pubis) and head lice (Pediculus humanus capitis). Adults are approximately 2 mm in size, with the same life cycle as head lice (Figure 1). They require blood meals roughly 5 times per day and cannot survive longer than 2 days without feeding.1 Although similar in structure to head lice, body lice differ behaviorally in that they do not reside on their human host’s body; instead, they infest the host’s clothing, localizing to seams (Figure 2), and migrate to the host for blood meals. In fact, based on this behavior, genetic analysis of early human body lice has been used to postulate when clothing was first used by humans as well as to determine early human migration patterns.2,3
Although clinicians in developed countries may be less familiar with body lice compared to their counterparts, body lice nevertheless remain a global health concern in impoverished, densely populated areas, as well as in homeless populations due to poor hygiene. Transmission frequently occurs via physical contact with an affected individual and his/her personal items (eg, linens) via fomites.4,5 Body louse infestation is more prevalent in homeless individuals who sleep outside vs in shelters; a history of pubic lice and lack of regular bathing have been reported as additional risk factors.6 Outbreaks have been noted in the wake of natural disasters, in the setting of political upheavals, and in refugee camps, as well as in individuals seeking political asylum.7 Unlike head and pubic lice, body lice can serve as vectors for infectious diseases including Rickettsia prowazekii (epidemic typhus), Borrelia recurrentis (louse-borne relapsing fever), Bartonella quintana (trench fever), and Yersinia pestis (plague).5,8,9 Several Acinetobacter species were isolated from nearly one-third of collected body louse specimens in a French study.10 Additionally, serology for B quintana was found to be positive in up to 30% of cases in one United States urban homeless population.4
Clinical Manifestations
Patients often present with generalized pruritus, usually considerably more severe than with P humanus capitis, with lesions concentrated on the trunk.11 In addition to often impetiginized, self-inflicted excoriations, feeding sites may present as erythematous macules (Figure 3), papules, or papular urticaria with a central hemorrhagic punctum. Extensive infestation also can manifest as the colloquial vagabond disease, characterized by postinflammatory hyperpigmentation and thickening of the involved skin. Remarkably, patients also may present with considerable iron-deficiency anemia secondary to high parasite load and large volume blood feeding. Multiple case reports have demonstrated associated morbidity.12-14 The differential diagnosis for pediculosis may include scabies, lichen simplex chronicus, and eczematous dermatitis, though the clinician should prudently consider whether both scabies and pediculosis may be present, as coexistence is possible.4,15
Diagnosis
Diagnosis can be reached by visualizing adult lice, nymphs, or viable nits on the body or more commonly within inner clothing seams; nits also fluoresce under Wood light.15 Although dermoscopy has proven useful for increased sensitivity and differentiation between viable and hatched nits, the insects also can be viewed with the unaided eye.16
Treatment: New Concerns and Strategies
The mainstay of treatment for body lice has long consisted of thorough washing and drying of all clothing and linens in a hot dryer. Treatment can be augmented with the addition of pharmacotherapy, plus antibiotics as warranted for louse-borne disease. Pharmacologic intervention often is used in cases of mass infestation and is similar to head lice.
Options for head lice include topical permethrin, malathion, lindane, spinosad, benzyl alcohol, and ivermectin. Pyrethroids, derived from the chrysanthemum, generally are considered safe for human use with a side-effect profile limited to irritation and allergy17; however, neurotoxicity and leukemia are clinical concerns, with an association more recently shown between large-volume use of pyrethroids and acute lymphoblastic leukemia.18,19 Use of lindane is not recommended due to a greater potential for central nervous system neurotoxicity, manifested by seizures, with repeated large surface application. Malathion is problematic due to the risk for mucosal irritation, flammability of some formulations, and theoretical organophosphate poisoning, as its mechanism of action involves inhibition of acetylcholinesterase.15 However, in the context of head lice treatment, a randomized controlled trial reported no incidence of acetylcholinesterase inhibition.20 Spinosad, manufactured from the soil bacterium Saccharopolyspora spinosa, functions similarly by interfering with the nicotinic acetylcholine receptor and also carries a risk for skin irritation.21 Among all the treatment options, we prefer benzyl alcohol, particularly in the context of resistance, as it is effective via a physical mechanism of action and lacks notable neurotoxic effects to the host. Use of benzyl alcohol is approved for patients as young as 6 months; it functions by asphyxiating the lice via paralysis of the respiratory spiracle with occlusion by inert ingredients. Itching, episodic numbness, and scalp or mucosal irritation are possible complications of treatment.22
Treatment resistance of body lice has increased in recent years, warranting exploration of additional management strategies. Moreover, developing resistance to lindane and malathion has been reported.23 Resistance to pyrethroids has been attributed to mutations in a voltage-gated sodium channel, one of which was universally present in the sampling of a single population.24 A randomized controlled trial showed that off-label oral ivermectin 400 μg/kg was superior to malathion lotion 0.5% in difficult-to-treat cases of head lice25; utility of oral ivermectin also has been reported in body lice.26 In vitro studies also have shown promise for pursuing synergistic treatment of body lice with both ivermectin and antibiotics.27
A novel primary prophylaxis approach for at-risk homeless individuals recently utilized permethrin-impregnated underwear. Although the intervention provided short-term infestation improvement, longer-term use did not show improvement from placebo and also increased prevalence of permethrin-resistant haplotypes.2
Epidemiology and Transmission
Pediculus humanus corporis, commonly known as the human body louse, is one in a family of 3 ectoparasites of the same suborder that also encompasses pubic lice (Phthirus pubis) and head lice (Pediculus humanus capitis). Adults are approximately 2 mm in size, with the same life cycle as head lice (Figure 1). They require blood meals roughly 5 times per day and cannot survive longer than 2 days without feeding.1 Although similar in structure to head lice, body lice differ behaviorally in that they do not reside on their human host’s body; instead, they infest the host’s clothing, localizing to seams (Figure 2), and migrate to the host for blood meals. In fact, based on this behavior, genetic analysis of early human body lice has been used to postulate when clothing was first used by humans as well as to determine early human migration patterns.2,3
Although clinicians in developed countries may be less familiar with body lice compared to their counterparts, body lice nevertheless remain a global health concern in impoverished, densely populated areas, as well as in homeless populations due to poor hygiene. Transmission frequently occurs via physical contact with an affected individual and his/her personal items (eg, linens) via fomites.4,5 Body louse infestation is more prevalent in homeless individuals who sleep outside vs in shelters; a history of pubic lice and lack of regular bathing have been reported as additional risk factors.6 Outbreaks have been noted in the wake of natural disasters, in the setting of political upheavals, and in refugee camps, as well as in individuals seeking political asylum.7 Unlike head and pubic lice, body lice can serve as vectors for infectious diseases including Rickettsia prowazekii (epidemic typhus), Borrelia recurrentis (louse-borne relapsing fever), Bartonella quintana (trench fever), and Yersinia pestis (plague).5,8,9 Several Acinetobacter species were isolated from nearly one-third of collected body louse specimens in a French study.10 Additionally, serology for B quintana was found to be positive in up to 30% of cases in one United States urban homeless population.4
Clinical Manifestations
Patients often present with generalized pruritus, usually considerably more severe than with P humanus capitis, with lesions concentrated on the trunk.11 In addition to often impetiginized, self-inflicted excoriations, feeding sites may present as erythematous macules (Figure 3), papules, or papular urticaria with a central hemorrhagic punctum. Extensive infestation also can manifest as the colloquial vagabond disease, characterized by postinflammatory hyperpigmentation and thickening of the involved skin. Remarkably, patients also may present with considerable iron-deficiency anemia secondary to high parasite load and large volume blood feeding. Multiple case reports have demonstrated associated morbidity.12-14 The differential diagnosis for pediculosis may include scabies, lichen simplex chronicus, and eczematous dermatitis, though the clinician should prudently consider whether both scabies and pediculosis may be present, as coexistence is possible.4,15
Diagnosis
Diagnosis can be reached by visualizing adult lice, nymphs, or viable nits on the body or more commonly within inner clothing seams; nits also fluoresce under Wood light.15 Although dermoscopy has proven useful for increased sensitivity and differentiation between viable and hatched nits, the insects also can be viewed with the unaided eye.16
Treatment: New Concerns and Strategies
The mainstay of treatment for body lice has long consisted of thorough washing and drying of all clothing and linens in a hot dryer. Treatment can be augmented with the addition of pharmacotherapy, plus antibiotics as warranted for louse-borne disease. Pharmacologic intervention often is used in cases of mass infestation and is similar to head lice.
Options for head lice include topical permethrin, malathion, lindane, spinosad, benzyl alcohol, and ivermectin. Pyrethroids, derived from the chrysanthemum, generally are considered safe for human use with a side-effect profile limited to irritation and allergy17; however, neurotoxicity and leukemia are clinical concerns, with an association more recently shown between large-volume use of pyrethroids and acute lymphoblastic leukemia.18,19 Use of lindane is not recommended due to a greater potential for central nervous system neurotoxicity, manifested by seizures, with repeated large surface application. Malathion is problematic due to the risk for mucosal irritation, flammability of some formulations, and theoretical organophosphate poisoning, as its mechanism of action involves inhibition of acetylcholinesterase.15 However, in the context of head lice treatment, a randomized controlled trial reported no incidence of acetylcholinesterase inhibition.20 Spinosad, manufactured from the soil bacterium Saccharopolyspora spinosa, functions similarly by interfering with the nicotinic acetylcholine receptor and also carries a risk for skin irritation.21 Among all the treatment options, we prefer benzyl alcohol, particularly in the context of resistance, as it is effective via a physical mechanism of action and lacks notable neurotoxic effects to the host. Use of benzyl alcohol is approved for patients as young as 6 months; it functions by asphyxiating the lice via paralysis of the respiratory spiracle with occlusion by inert ingredients. Itching, episodic numbness, and scalp or mucosal irritation are possible complications of treatment.22
Treatment resistance of body lice has increased in recent years, warranting exploration of additional management strategies. Moreover, developing resistance to lindane and malathion has been reported.23 Resistance to pyrethroids has been attributed to mutations in a voltage-gated sodium channel, one of which was universally present in the sampling of a single population.24 A randomized controlled trial showed that off-label oral ivermectin 400 μg/kg was superior to malathion lotion 0.5% in difficult-to-treat cases of head lice25; utility of oral ivermectin also has been reported in body lice.26 In vitro studies also have shown promise for pursuing synergistic treatment of body lice with both ivermectin and antibiotics.27
A novel primary prophylaxis approach for at-risk homeless individuals recently utilized permethrin-impregnated underwear. Although the intervention provided short-term infestation improvement, longer-term use did not show improvement from placebo and also increased prevalence of permethrin-resistant haplotypes.2
- Veracx A, Raoult D. Biology and genetics of human head and body lice. Trends Parasitol. 2012;28:563-571.
- Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing. Curr Biol. 2003;13:1414-1417.
- Drali R, Mumcuoglu KY, Yesilyurt G, et al. Studies of ancient lice reveal unsuspected past migrations of vectors. Am J Trop Med Hyg. 2015;93:623-625.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Feldmeier H, Heukelbach J. Epidermal parasitic skin diseases: a neglected category of poverty-associated plagues. Bull World Health Organ. 2009;87:152-159.
- Arnaud A, Chosidow O, Detrez MA, et al. Prevalence of scabies and Pediculosis corporis among homeless people in the Paris region: results from two randomized cross-sectional surveys (HYTPEAC study). Br J Dermatol. 2016;174:104-112.
- Hytonen J, Khawaja T, Gronroos JO, et al. Louse-borne relapsing fever in Finland in two asylum seekers from Somalia. APMIS. 2017;125:59-62.
- Nordmann T, Feldt T, Bosselmann M, et al. Outbreak of louse-borne relapsing fever among urban dwellers in Arsi Zone, Central Ethiopia, from July to November 2016. Am J Trop Med Hyg. 2018;98:1599-1602.
- Louni M, Mana N, Bitam I, et al. Body lice of homeless people reveal the presence of several emerging bacterial pathogens in northern Algeria. PLoS Negl Trop Dis. 2018;12:E0006397.
- Candy K, Amanzougaghene N, Izri A, et al. Molecular survey of head and body lice, Pediculus humanus, in France. Vector Borne Zoonotic Dis. 2018;18:243-251.
Bolognia JL, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier Limited; 2018. - Nara A, Nagai H, Yamaguchi R, et al. An unusual autopsy case of lethal hypothermia exacerbated by body lice-induced severe anemia. Int J Legal Med. 2016;130:765-769.
- Althomali SA, Alzubaidi LM, Alkhaldi DM. Severe iron deficiency anaemia associated with heavy lice infestation in a young woman [published online November 5, 2015]. BMJ Case Rep. doi:10.1136/bcr-2015-212207.
- Hau V, Muhi-Iddin N. A ghost covered in lice: a case of severe blood loss with long-standing heavy pediculosis capitis infestation [published online December 19, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-206623.
- Diaz JH. Lice (Pediculosis). In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 9th ed. New York, NY: Elsevier; 2020:3482-3486.
- Martins LG, Bernardes Filho F, Quaresma MV, et al. Dermoscopy applied to pediculosis corporis diagnosis. An Bras Dermatol. 2014;89:513-514.
- Devore CD, Schutze GE; Council on School Health and Committee on Infectious Diseases, American Academy of Pediatrics. Head lice. Pediatrics. 2015;135:E1355-E1365.
- Shafer TJ, Meyer DA, Crofton KM. Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs. Environ Health Perspect. 2005;113:123-136.
- Ding G, Shi R, Gao Y, et al. Pyrethroid pesticide exposure and risk of childhood acute lymphocytic leukemia in Shanghai. Environ Sci Technol. 2012;46:13480-13487.
- Meinking TL, Vicaria M, Eyerdam DH, et al. A randomized, investigator-blinded, time-ranging study of the comparative efficacy of 0.5% malathion gel versus Ovide Lotion (0.5% malathion) or Nix Crème Rinse (1% permethrin) used as labeled, for the treatment of head lice. Pediatr Dermatol. 2007;24:405-411.
- McCormack PL. Spinosad: in pediculosis capitis. Am J Clin Dermatol. 2011;12:349-353.
- Meinking TL, Villar ME, Vicaria M, et al. The clinical trials supporting benzyl alcohol lotion 5% (Ulesfia): a safe and effective topical treatment for head lice (pediculosis humanus capitis). Pediatr Dermatol. 2010;27:19-24.
- Lebwohl M, Clark L, Levitt J. Therapy for head lice based on life cycle, resistance, and safety considerations. Pediatrics. 2007;119:965-974
- Drali R, Benkouiten S, Badiaga S, et al. Detection of a knockdown resistance mutation associated with permethrin resistance in the body louse Pediculus humanus corporis by use of melting curve analysis genotyping. J Clin Microbiol. 2012;50:2229-2233.
- Chosidow O, Giraudeau B, Cottrell J, et al. Oral ivermectin versus malathion lotion for difficult-to-treat head lice. N Engl J Med. 2010;362:896-905.
- Foucault C, Ranque S, Badiaga S, et al. Oral ivermectin in the treatment of body lice. J Infect Dis. 2006;193:474-476.
- Sangaré AK, Doumbo OK, Raoult D. Management and treatment of human lice [published online July 27, 2016]. Biomed Res Int. doi:10.1155/2016/8962685.
- Benkouiten S, Drali R, Badiaga S, et al. Effect of permethrin-impregnated underwear on body lice in sheltered homeless persons: a randomized controlled trial. JAMA Dermatol. 2014;150:273-279.
- Veracx A, Raoult D. Biology and genetics of human head and body lice. Trends Parasitol. 2012;28:563-571.
- Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing. Curr Biol. 2003;13:1414-1417.
- Drali R, Mumcuoglu KY, Yesilyurt G, et al. Studies of ancient lice reveal unsuspected past migrations of vectors. Am J Trop Med Hyg. 2015;93:623-625.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Feldmeier H, Heukelbach J. Epidermal parasitic skin diseases: a neglected category of poverty-associated plagues. Bull World Health Organ. 2009;87:152-159.
- Arnaud A, Chosidow O, Detrez MA, et al. Prevalence of scabies and Pediculosis corporis among homeless people in the Paris region: results from two randomized cross-sectional surveys (HYTPEAC study). Br J Dermatol. 2016;174:104-112.
- Hytonen J, Khawaja T, Gronroos JO, et al. Louse-borne relapsing fever in Finland in two asylum seekers from Somalia. APMIS. 2017;125:59-62.
- Nordmann T, Feldt T, Bosselmann M, et al. Outbreak of louse-borne relapsing fever among urban dwellers in Arsi Zone, Central Ethiopia, from July to November 2016. Am J Trop Med Hyg. 2018;98:1599-1602.
- Louni M, Mana N, Bitam I, et al. Body lice of homeless people reveal the presence of several emerging bacterial pathogens in northern Algeria. PLoS Negl Trop Dis. 2018;12:E0006397.
- Candy K, Amanzougaghene N, Izri A, et al. Molecular survey of head and body lice, Pediculus humanus, in France. Vector Borne Zoonotic Dis. 2018;18:243-251.
Bolognia JL, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier Limited; 2018. - Nara A, Nagai H, Yamaguchi R, et al. An unusual autopsy case of lethal hypothermia exacerbated by body lice-induced severe anemia. Int J Legal Med. 2016;130:765-769.
- Althomali SA, Alzubaidi LM, Alkhaldi DM. Severe iron deficiency anaemia associated with heavy lice infestation in a young woman [published online November 5, 2015]. BMJ Case Rep. doi:10.1136/bcr-2015-212207.
- Hau V, Muhi-Iddin N. A ghost covered in lice: a case of severe blood loss with long-standing heavy pediculosis capitis infestation [published online December 19, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-206623.
- Diaz JH. Lice (Pediculosis). In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 9th ed. New York, NY: Elsevier; 2020:3482-3486.
- Martins LG, Bernardes Filho F, Quaresma MV, et al. Dermoscopy applied to pediculosis corporis diagnosis. An Bras Dermatol. 2014;89:513-514.
- Devore CD, Schutze GE; Council on School Health and Committee on Infectious Diseases, American Academy of Pediatrics. Head lice. Pediatrics. 2015;135:E1355-E1365.
- Shafer TJ, Meyer DA, Crofton KM. Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs. Environ Health Perspect. 2005;113:123-136.
- Ding G, Shi R, Gao Y, et al. Pyrethroid pesticide exposure and risk of childhood acute lymphocytic leukemia in Shanghai. Environ Sci Technol. 2012;46:13480-13487.
- Meinking TL, Vicaria M, Eyerdam DH, et al. A randomized, investigator-blinded, time-ranging study of the comparative efficacy of 0.5% malathion gel versus Ovide Lotion (0.5% malathion) or Nix Crème Rinse (1% permethrin) used as labeled, for the treatment of head lice. Pediatr Dermatol. 2007;24:405-411.
- McCormack PL. Spinosad: in pediculosis capitis. Am J Clin Dermatol. 2011;12:349-353.
- Meinking TL, Villar ME, Vicaria M, et al. The clinical trials supporting benzyl alcohol lotion 5% (Ulesfia): a safe and effective topical treatment for head lice (pediculosis humanus capitis). Pediatr Dermatol. 2010;27:19-24.
- Lebwohl M, Clark L, Levitt J. Therapy for head lice based on life cycle, resistance, and safety considerations. Pediatrics. 2007;119:965-974
- Drali R, Benkouiten S, Badiaga S, et al. Detection of a knockdown resistance mutation associated with permethrin resistance in the body louse Pediculus humanus corporis by use of melting curve analysis genotyping. J Clin Microbiol. 2012;50:2229-2233.
- Chosidow O, Giraudeau B, Cottrell J, et al. Oral ivermectin versus malathion lotion for difficult-to-treat head lice. N Engl J Med. 2010;362:896-905.
- Foucault C, Ranque S, Badiaga S, et al. Oral ivermectin in the treatment of body lice. J Infect Dis. 2006;193:474-476.
- Sangaré AK, Doumbo OK, Raoult D. Management and treatment of human lice [published online July 27, 2016]. Biomed Res Int. doi:10.1155/2016/8962685.
- Benkouiten S, Drali R, Badiaga S, et al. Effect of permethrin-impregnated underwear on body lice in sheltered homeless persons: a randomized controlled trial. JAMA Dermatol. 2014;150:273-279.
Practice Points
- Body lice reside in clothing, particularly folds and seams, and migrate to the host for blood meals. To evaluate for infestation, the clinician should not only look at the skin but also closely examine the patient’s clothing. Clothes also are a target for treatment via washing in hot water.
- Due to observed and theoretical adverse effects of other chemical treatments, benzyl alcohol is the authors’ choice for treatment of head lice.
- Oral ivermectin is a promising future treatment for body lice.
What’s Eating You? Vespids Revisited
Identification
The Hymenoptera order of insects includes Apidae (bees), Vespidae (wasps, yellow jackets, hornets), and Formicidae (fire ants). All 3 of these families of insects inject venom into their prey or as a defense mechanism via ovipositors in their abdomen. Vespids are the most aggressive and are found in each of the United States.1 They have membranous wings, broad antennae, and a nonbarbed stinger (Figure 1).2 The nonbarbed stinger of Vespidae differentiates them from Apidae and allows these insects to sting their prey multiple times. Vespids can build nests in the ground (yellow jackets), trees (hornets), or areas of cover such as window shutters (mud wasps). Because only the queens survive winter, larger populations do not develop until late summer when the most stings take place. Stings most often take place near the nest of the vespid or while the victim is eating outdoors.3

Envenomation
When vespids sting their prey they inject venom via their ovipositors.1 The venom is composed of a mixture of low-molecular-weight proteins, kinins, proteolytic enzymes, lipids, carbohydrates, and high-molecular-weight proteins that act as allergens.1,4,5 The proteolytic enzymes degrade the surrounding tissue, basophils become activated, and histamine is released secondary to mast cell degranulation, which results in vasodilation and an inflammatory response characterized by edema, erythema, warmth, and pain.1 The pain of the sting is immediate and can be intense; almost all victims are acutely aware of the discomforting sensation.4
Management of Reactions
Three types of reactions can be seen after a vespid sting: uncomplicated local reactions, large local reactions, and systemic reactions (SRs). The most common reaction is the self-limiting uncomplicated local reaction that includes a focal area of warmth, edema, erythema, induration, and tenderness.1 Treatment of this kind of reaction is supportive, with ice, nonsteroidal anti-inflammatory drugs, and H1 and H2 blockers being commonly used methods. Large local reactions (Figure 2) are similar to uncomplicated local reactions but are greater than 10 cm in diameter and last longer. The same symptomatic treatment may be used along with possible short (3–5 days) oral glucocorticoid (40–60 mg prednisone) or potent topical steroid administration if symptoms persist. Systemic reactions involve IgE-mediated generalized urticaria, angioedema, face swelling, stridor, bronchospasm, nausea, vomiting, flushing, and respiratory distress.1 Emergency management includes maintenance of airway, breathing, and circulation. Epinephrine injection commonly is employed and should be given via intramuscular injection into the anterolateral thigh; a dose of 0.3 to 0.5 mg can be repeatedly injected every 5 to 15 minutes, as needed.1
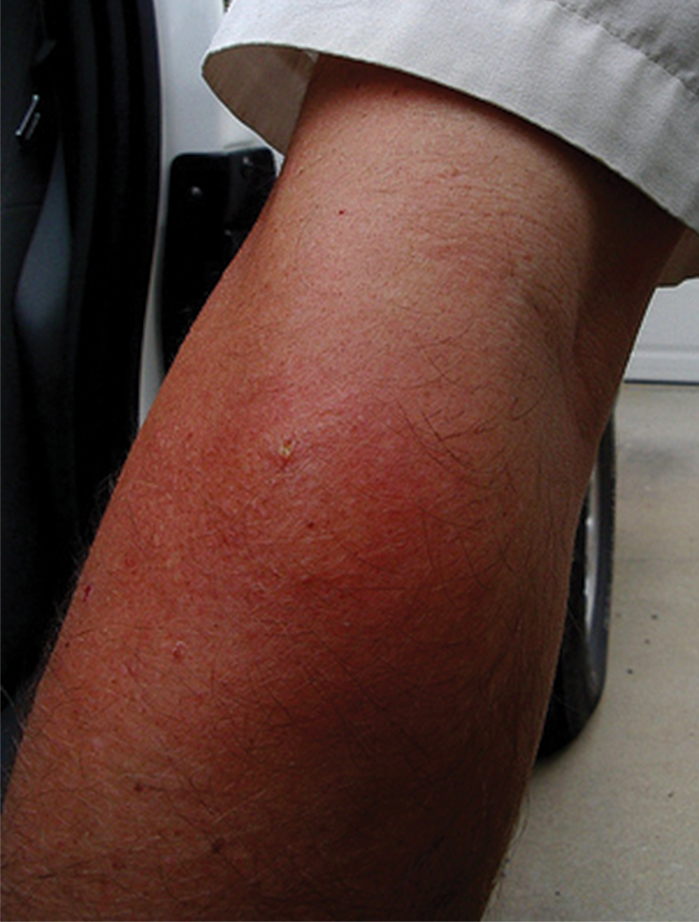
If an individual has an SR, it is recommended to go to an emergency department after stabilization for monitoring. Referral to an allergist for desensitization is appropriate. A radioallergosorbent test to measure allergen-specific IgE can be helpful to confirm an allergy.4 This test also should be done weeks after the incident because during the first few days IgE may be too low to measure. Once the allergy is confirmed, the desensitization with venom immunotherapy (VIT) can begin. Venom immunotherapy is effective and reduces a patient’s risk for recurrent SRs to less than 5% to 20%.6 A 2015 study recommended longer duration of VIT therapy due to risk for repeat SRs after discontinuing therapy. This study concluded that VIT is to be administered for 5 years, unless the patient is at high risk for SRs after VIT therapy—risk factors include older age, cardiopulmonary disease, SR during VIT treatment, mast cell disorders, and elevated serum tryptase—in which case VIT may have to be continued indefinitely. It is recommended that all patients with history of SR carry an epinephrine autoinjector in case of emergency.6
Epidemiologic data show a prevalence of 0.3% to 7.5% for self-reported SRs due to stings, with lower prevalence in children (0.15%–0.3%).4,7 An additional study looking at data from an allergy practice determined 24% of all cases of anaphylaxis were due to insect stings.5
Conclusion
Although many vespid stings can be managed symptomatically, it is imperative for patients and providers to be aware of the possible severe reactions that can take place. It is essential for providers to be aware of how to care for and treat large local reactions and SRs, as symptom recognition and timely treatment can improve patient safety and result in better outcomes.
- Arif F, Williams M. Hymenoptera Stings (Bee, Vespids and Ants). Treasure Island, FL: StatPearls Publishing LLC; 2019. https://www.ncbi.nlm.nih.gov/books/NBK518972/. Updated April 20, 2019. Accessed December 11, 2019.
- Elston, DM. Life-threatening stings, bites, infestations, and parasitic diseases. Clin Dermatol. 2005;23:164-170.
- Ulrich RM, Gabrielle H, Arthur H. Allergic reactions to stinging and biting insects. In: Rich RR, Fleisher T, Shearer W, et al, eds. Clinical Immunology: Principles and Practice. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2008:657-666.
- Biló BM, Rueff F, Mosbech H, et al. Diagnosis of hymenoptera venom allergy. Allergy. 2005;60:1339-1349.
- Schafer T, Przybilla B. IgE antibodies to hymenoptera venoms in the serum are common in the general population and are related to indication of atopy. Allergy. 1996;51:372-377.
- Ulrich MR, Johannes R. When can immunotherapy for insect sting allergy be stopped? J Allergy Clin Immunol. 2015;3:324-328.
- Abrishami MH, Boyd GK, Settipane GA. Prevalence of bee sting allergy in 2010 girl scouts. Acta Allergol. 1971;26:117-120.
Identification
The Hymenoptera order of insects includes Apidae (bees), Vespidae (wasps, yellow jackets, hornets), and Formicidae (fire ants). All 3 of these families of insects inject venom into their prey or as a defense mechanism via ovipositors in their abdomen. Vespids are the most aggressive and are found in each of the United States.1 They have membranous wings, broad antennae, and a nonbarbed stinger (Figure 1).2 The nonbarbed stinger of Vespidae differentiates them from Apidae and allows these insects to sting their prey multiple times. Vespids can build nests in the ground (yellow jackets), trees (hornets), or areas of cover such as window shutters (mud wasps). Because only the queens survive winter, larger populations do not develop until late summer when the most stings take place. Stings most often take place near the nest of the vespid or while the victim is eating outdoors.3
Envenomation
When vespids sting their prey they inject venom via their ovipositors.1 The venom is composed of a mixture of low-molecular-weight proteins, kinins, proteolytic enzymes, lipids, carbohydrates, and high-molecular-weight proteins that act as allergens.1,4,5 The proteolytic enzymes degrade the surrounding tissue, basophils become activated, and histamine is released secondary to mast cell degranulation, which results in vasodilation and an inflammatory response characterized by edema, erythema, warmth, and pain.1 The pain of the sting is immediate and can be intense; almost all victims are acutely aware of the discomforting sensation.4
Management of Reactions
Three types of reactions can be seen after a vespid sting: uncomplicated local reactions, large local reactions, and systemic reactions (SRs). The most common reaction is the self-limiting uncomplicated local reaction that includes a focal area of warmth, edema, erythema, induration, and tenderness.1 Treatment of this kind of reaction is supportive, with ice, nonsteroidal anti-inflammatory drugs, and H1 and H2 blockers being commonly used methods. Large local reactions (Figure 2) are similar to uncomplicated local reactions but are greater than 10 cm in diameter and last longer. The same symptomatic treatment may be used along with possible short (3–5 days) oral glucocorticoid (40–60 mg prednisone) or potent topical steroid administration if symptoms persist. Systemic reactions involve IgE-mediated generalized urticaria, angioedema, face swelling, stridor, bronchospasm, nausea, vomiting, flushing, and respiratory distress.1 Emergency management includes maintenance of airway, breathing, and circulation. Epinephrine injection commonly is employed and should be given via intramuscular injection into the anterolateral thigh; a dose of 0.3 to 0.5 mg can be repeatedly injected every 5 to 15 minutes, as needed.1
If an individual has an SR, it is recommended to go to an emergency department after stabilization for monitoring. Referral to an allergist for desensitization is appropriate. A radioallergosorbent test to measure allergen-specific IgE can be helpful to confirm an allergy.4 This test also should be done weeks after the incident because during the first few days IgE may be too low to measure. Once the allergy is confirmed, the desensitization with venom immunotherapy (VIT) can begin. Venom immunotherapy is effective and reduces a patient’s risk for recurrent SRs to less than 5% to 20%.6 A 2015 study recommended longer duration of VIT therapy due to risk for repeat SRs after discontinuing therapy. This study concluded that VIT is to be administered for 5 years, unless the patient is at high risk for SRs after VIT therapy—risk factors include older age, cardiopulmonary disease, SR during VIT treatment, mast cell disorders, and elevated serum tryptase—in which case VIT may have to be continued indefinitely. It is recommended that all patients with history of SR carry an epinephrine autoinjector in case of emergency.6
Epidemiologic data show a prevalence of 0.3% to 7.5% for self-reported SRs due to stings, with lower prevalence in children (0.15%–0.3%).4,7 An additional study looking at data from an allergy practice determined 24% of all cases of anaphylaxis were due to insect stings.5
Conclusion
Although many vespid stings can be managed symptomatically, it is imperative for patients and providers to be aware of the possible severe reactions that can take place. It is essential for providers to be aware of how to care for and treat large local reactions and SRs, as symptom recognition and timely treatment can improve patient safety and result in better outcomes.
Identification
The Hymenoptera order of insects includes Apidae (bees), Vespidae (wasps, yellow jackets, hornets), and Formicidae (fire ants). All 3 of these families of insects inject venom into their prey or as a defense mechanism via ovipositors in their abdomen. Vespids are the most aggressive and are found in each of the United States.1 They have membranous wings, broad antennae, and a nonbarbed stinger (Figure 1).2 The nonbarbed stinger of Vespidae differentiates them from Apidae and allows these insects to sting their prey multiple times. Vespids can build nests in the ground (yellow jackets), trees (hornets), or areas of cover such as window shutters (mud wasps). Because only the queens survive winter, larger populations do not develop until late summer when the most stings take place. Stings most often take place near the nest of the vespid or while the victim is eating outdoors.3
Envenomation
When vespids sting their prey they inject venom via their ovipositors.1 The venom is composed of a mixture of low-molecular-weight proteins, kinins, proteolytic enzymes, lipids, carbohydrates, and high-molecular-weight proteins that act as allergens.1,4,5 The proteolytic enzymes degrade the surrounding tissue, basophils become activated, and histamine is released secondary to mast cell degranulation, which results in vasodilation and an inflammatory response characterized by edema, erythema, warmth, and pain.1 The pain of the sting is immediate and can be intense; almost all victims are acutely aware of the discomforting sensation.4
Management of Reactions
Three types of reactions can be seen after a vespid sting: uncomplicated local reactions, large local reactions, and systemic reactions (SRs). The most common reaction is the self-limiting uncomplicated local reaction that includes a focal area of warmth, edema, erythema, induration, and tenderness.1 Treatment of this kind of reaction is supportive, with ice, nonsteroidal anti-inflammatory drugs, and H1 and H2 blockers being commonly used methods. Large local reactions (Figure 2) are similar to uncomplicated local reactions but are greater than 10 cm in diameter and last longer. The same symptomatic treatment may be used along with possible short (3–5 days) oral glucocorticoid (40–60 mg prednisone) or potent topical steroid administration if symptoms persist. Systemic reactions involve IgE-mediated generalized urticaria, angioedema, face swelling, stridor, bronchospasm, nausea, vomiting, flushing, and respiratory distress.1 Emergency management includes maintenance of airway, breathing, and circulation. Epinephrine injection commonly is employed and should be given via intramuscular injection into the anterolateral thigh; a dose of 0.3 to 0.5 mg can be repeatedly injected every 5 to 15 minutes, as needed.1
If an individual has an SR, it is recommended to go to an emergency department after stabilization for monitoring. Referral to an allergist for desensitization is appropriate. A radioallergosorbent test to measure allergen-specific IgE can be helpful to confirm an allergy.4 This test also should be done weeks after the incident because during the first few days IgE may be too low to measure. Once the allergy is confirmed, the desensitization with venom immunotherapy (VIT) can begin. Venom immunotherapy is effective and reduces a patient’s risk for recurrent SRs to less than 5% to 20%.6 A 2015 study recommended longer duration of VIT therapy due to risk for repeat SRs after discontinuing therapy. This study concluded that VIT is to be administered for 5 years, unless the patient is at high risk for SRs after VIT therapy—risk factors include older age, cardiopulmonary disease, SR during VIT treatment, mast cell disorders, and elevated serum tryptase—in which case VIT may have to be continued indefinitely. It is recommended that all patients with history of SR carry an epinephrine autoinjector in case of emergency.6
Epidemiologic data show a prevalence of 0.3% to 7.5% for self-reported SRs due to stings, with lower prevalence in children (0.15%–0.3%).4,7 An additional study looking at data from an allergy practice determined 24% of all cases of anaphylaxis were due to insect stings.5
Conclusion
Although many vespid stings can be managed symptomatically, it is imperative for patients and providers to be aware of the possible severe reactions that can take place. It is essential for providers to be aware of how to care for and treat large local reactions and SRs, as symptom recognition and timely treatment can improve patient safety and result in better outcomes.
- Arif F, Williams M. Hymenoptera Stings (Bee, Vespids and Ants). Treasure Island, FL: StatPearls Publishing LLC; 2019. https://www.ncbi.nlm.nih.gov/books/NBK518972/. Updated April 20, 2019. Accessed December 11, 2019.
- Elston, DM. Life-threatening stings, bites, infestations, and parasitic diseases. Clin Dermatol. 2005;23:164-170.
- Ulrich RM, Gabrielle H, Arthur H. Allergic reactions to stinging and biting insects. In: Rich RR, Fleisher T, Shearer W, et al, eds. Clinical Immunology: Principles and Practice. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2008:657-666.
- Biló BM, Rueff F, Mosbech H, et al. Diagnosis of hymenoptera venom allergy. Allergy. 2005;60:1339-1349.
- Schafer T, Przybilla B. IgE antibodies to hymenoptera venoms in the serum are common in the general population and are related to indication of atopy. Allergy. 1996;51:372-377.
- Ulrich MR, Johannes R. When can immunotherapy for insect sting allergy be stopped? J Allergy Clin Immunol. 2015;3:324-328.
- Abrishami MH, Boyd GK, Settipane GA. Prevalence of bee sting allergy in 2010 girl scouts. Acta Allergol. 1971;26:117-120.
- Arif F, Williams M. Hymenoptera Stings (Bee, Vespids and Ants). Treasure Island, FL: StatPearls Publishing LLC; 2019. https://www.ncbi.nlm.nih.gov/books/NBK518972/. Updated April 20, 2019. Accessed December 11, 2019.
- Elston, DM. Life-threatening stings, bites, infestations, and parasitic diseases. Clin Dermatol. 2005;23:164-170.
- Ulrich RM, Gabrielle H, Arthur H. Allergic reactions to stinging and biting insects. In: Rich RR, Fleisher T, Shearer W, et al, eds. Clinical Immunology: Principles and Practice. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2008:657-666.
- Biló BM, Rueff F, Mosbech H, et al. Diagnosis of hymenoptera venom allergy. Allergy. 2005;60:1339-1349.
- Schafer T, Przybilla B. IgE antibodies to hymenoptera venoms in the serum are common in the general population and are related to indication of atopy. Allergy. 1996;51:372-377.
- Ulrich MR, Johannes R. When can immunotherapy for insect sting allergy be stopped? J Allergy Clin Immunol. 2015;3:324-328.
- Abrishami MH, Boyd GK, Settipane GA. Prevalence of bee sting allergy in 2010 girl scouts. Acta Allergol. 1971;26:117-120.
Practice Points
- Most vespid stings can be managed with nonsteroidal anti-inflammatory drugs, ice, and antihistamines.
- For systemic reactions, prompt recognition and initiation of intramuscular epinephrine is recommended.
- In patients with confirmed allergy, recent data now suggest at least 5 years of venom immunotherapy and potentially lifelong for specific patients.

What’s Eating You? Blister Beetles Revisited
Classification
Blister beetles are both a scourge and the source of medical cantharidin (Figure 1). The term blister beetle refers to 3 families of the order Coleoptera: Meloidae, Oedemeridae, and Staphylinidae (Figure 2).

Meloidae is the most well-known family of blister beetles, with more than 200 species worldwide identified as a cause of blistering dermatitis.1 The most notorious is Lytta vesicatoria, also known as the Spanish fly. Although some blister beetles are inconspicuous in appearance, most are brightly colored and easily spotted, making them attractive to small children.2 They may be attracted to fluorescent lighting and commonly enter through open windows.3 Blister beetles do not bite or sting; rather, they release cantharidin via hemolymph, an oily yellow substance that is copiously expressed from the leg joints when disturbed by rubbing or pressing.1,3-5 Blistering also is associated with exposure to the contents of crushed beetles, which is the source of pharmacologic cantharidin.2,4,6
Oedemeridae are the smallest and least known beetles within the Coleoptera family. They often are called false blister beetles, which is a misnomer. Oedemeridae beetles also produce cantharidin, similar to Meloidae beetles; however, because Oedemeridae beetles are smaller, the vesiculobullous eruptions also are less pronounced.1
A third well-known family of blister beetles is the Staphylinidae family. Rove beetles (genus Paederus) differ from the Meloidae and Oedemeridae families in that they produce the vesicant pederin rather than cantharidin. Pederin causes a more violent eruption called dermatitis linearis, which often is associated with intense urticaria prior to blistering.1 Rove beetles are found in moist environments and tend to favor tropical and subtropical climates. They may emerge in large numbers after heavy rainfall.
Clinical Presentation
Blistering dermatitis—caused by exposure to cantharidin (families Meloidae and Oedemeridae) or pederin (genus Paederus)—begins as burning and tingling within minutes of exposure. Bullae later develop, often in a linear fashion, with subsequent bursting and crust formation.1,3 Secondary infection can occur.5 Exposure occurring on the elbows, knees, or mirroring skin folds results in lesions on opposing surfaces that come into contact, otherwise known as kissing lesions.1,3 Acantholysis of suprabasal keratinocytes can be seen on histologic sections of blisters (Figure 3)1,7 due to cantharidin activation of the serine/threonine protein phosphatases that cause detachment of tonofilaments from desmosomes.1,8 Washing of exposed sites with soap or alcohol can potentially prevent development of blistering dermatitis; however, lesions usually heal without complication when treated with topical antibiotics.3
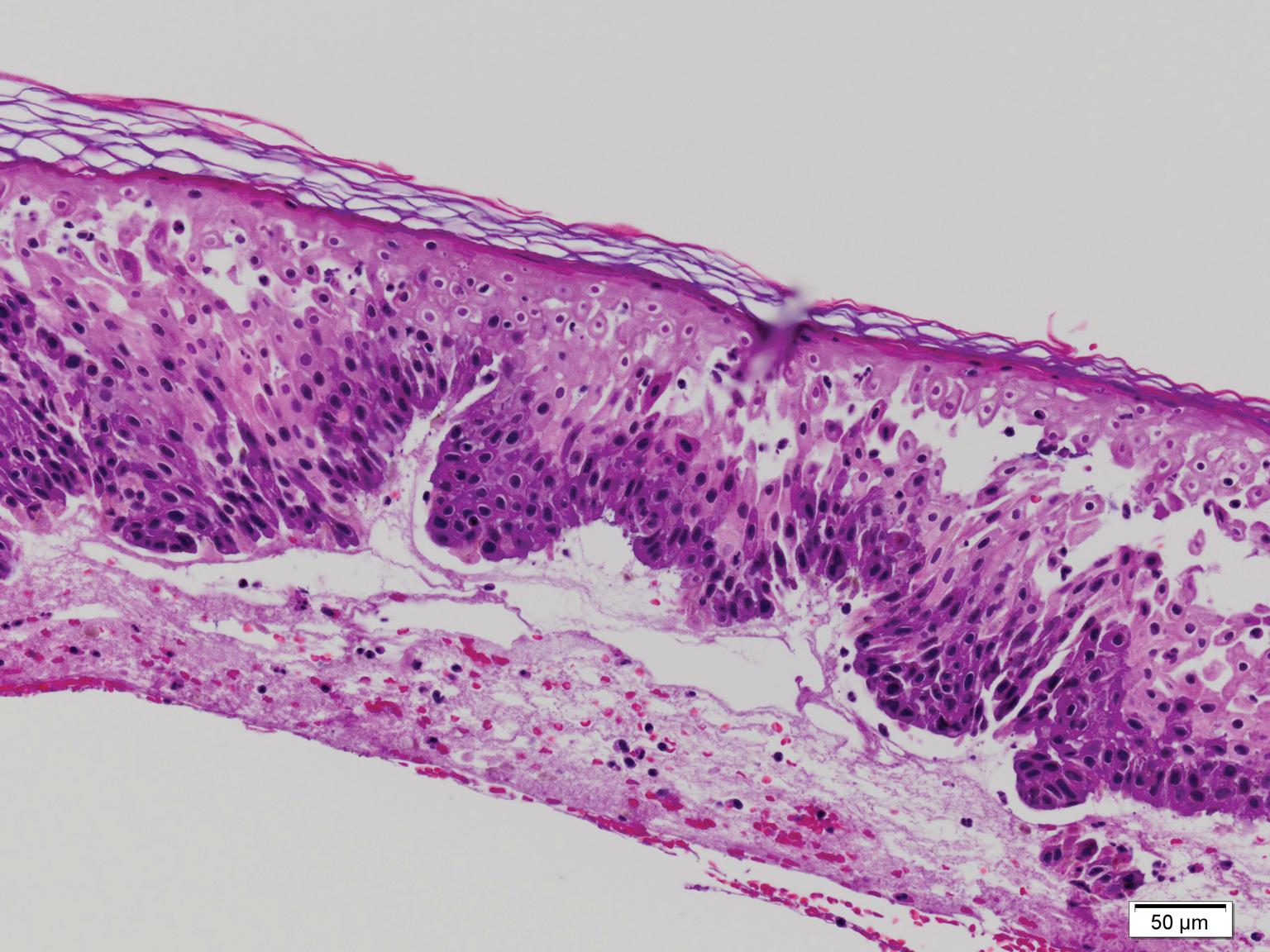
If blister beetles are ingested, they can cause poisoning characterized by abdominal pain and hematuria.1,3,9,10 In fact, blister beetle ingestion by animals consuming baled hay is an important agricultural and economical implication. Several cases of fatal farm animal ingestion resulting in gastrointestinal erosion have been reported.4,6
Medicinal Properties
Medicinal use of cantharidin has been recorded as early as the 19th century and is believed to have been part of ancient medicinal practices in China, Spain, South Africa, and pre-Columbian America for its vesicant, abortifacient, and supposed aphrodisiac properties.2,4,8
Toxicity from internal ingestion has been well documented.1,3,9,10 The Mylabris and Epicauta genera of the family Meloidae, most commonly the Spanish fly, are used for modern extraction of cantharidin. Up to 5% of the dry weight of a blister beetle can be constituted by cantharidin.4
Although its use has diminished due to limited availability, cantharidin is still used for treatment of common warts, periungual warts, and molluscum contagiosum. It is a favorable choice for treating pediatric patients because of the tolerability and painlessness of application. Due to its acantholytic properties, warts generally slough with the cantharidin-induced blister.11
In recent years, cantharidin has been studied for its anticancer properties. It has been shown to weaken cancer cell antioxidant properties by interfering with glutathione-related enzymes, thus inducing oxidative damage. Additionally, cantharidin is associated with decreased mitochondrial cytochrome C and increased cytosolic cytochrome C, an important signal for apoptosis.12 Furthermore, cantharidin recently was shown to increase expression of proapoptotic proteins and decrease expression of antiapoptotic proteins causing cell death in nasopharyngeal carcinoma, making it a potential anticancer treatment.13
Conclusion
Blister beetles, long known for their production of vesicant agents, are both a cause of as well as a potential treatment of dermatologic disease. The blistering associated with exposure to a disturbed beetle generally is mild and heals without scarring if vital areas such as the eyelids are not affected.
- Nicholls DS, Christmas TI, Greig DE. Oedemerid blister beetle dermatosis: a review. J Am Acad Dermatol. 1990;22:815-819.
- Percino-Daniel N, Buckley D, García-París M. Pharmacological properties of blister beetles (Coleoptera: Meloidae) promoted their integration into the cultural heritage of native rural Spain as inferred by vernacular names diversity, traditions, and mitochondrial DNA. J Ethnopharmacol. 2013;147:570-583.
- James WD, Berger TG, Elston DM. Parasitic infestations, stings, and bites. In: James WD, Berger TG, Elston DM, eds. Andrew’s Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016:418-450.
- Selander RB, Fasulo TR. Featured creatures: blister beetles. University of Florida/IFAS Entomology and Nematology website. http://entnemdept.ufl.edu/creatures/urban/medical/blister_beetles.htm. Published October 2000. Revised September 2010. Accessed November 12, 2019.
- Wijerathne BTB. Blister mystery. Wilderness Environ Med. 2017;28:271-272.
- Penrith ML, Naude TW. Mortality in chickens associated with blister beetle consumption. J S Afr Vet Assoc. 1996;67:97-99.
- Yell JA, Burge SM, Dean D. Cantharidin-induced acantholysis: adhesion molecules, proteases, and related proteins. Br J Dermatol. 1994;130:148-157.
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine-threonine protein phosphatases types 1 and 2a. FEBS Letters. 1993;330:283-286.
- Al-Binali AM, Shabana M, Al-Fifi S, et al. Cantharidin poisoning due to blister beetle ingestion in children: two case reports and a review of clinical presentations. Sultan Qaboos Univ Med J. 2010;10:258-261.
- Tagwireyi D, Ball DE, Loga PJ, et al. Cantharidin poisoning due to “blister beetle” ingestion. Toxicon. 2000;38:1865-1869.
- Al-Dawsari NA, Masterpol KS. Cantharidin in dermatology. Skinmed. 2016;14:111-114.
- Verma AK, Prasad SB. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, epicauta hirticornis. Anticancer Agents Med Chem. 2013;13:1096-1114.
- Chen AW, Tseng YS, Lin CC, et al. Norcantharidin induce apoptosis in human nasopharyngeal carcinoma through caspase and mitochondrial pathway. Environ Toxicol. 2018;33:343-350.
Classification
Blister beetles are both a scourge and the source of medical cantharidin (Figure 1). The term blister beetle refers to 3 families of the order Coleoptera: Meloidae, Oedemeridae, and Staphylinidae (Figure 2).
Meloidae is the most well-known family of blister beetles, with more than 200 species worldwide identified as a cause of blistering dermatitis.1 The most notorious is Lytta vesicatoria, also known as the Spanish fly. Although some blister beetles are inconspicuous in appearance, most are brightly colored and easily spotted, making them attractive to small children.2 They may be attracted to fluorescent lighting and commonly enter through open windows.3 Blister beetles do not bite or sting; rather, they release cantharidin via hemolymph, an oily yellow substance that is copiously expressed from the leg joints when disturbed by rubbing or pressing.1,3-5 Blistering also is associated with exposure to the contents of crushed beetles, which is the source of pharmacologic cantharidin.2,4,6
Oedemeridae are the smallest and least known beetles within the Coleoptera family. They often are called false blister beetles, which is a misnomer. Oedemeridae beetles also produce cantharidin, similar to Meloidae beetles; however, because Oedemeridae beetles are smaller, the vesiculobullous eruptions also are less pronounced.1
A third well-known family of blister beetles is the Staphylinidae family. Rove beetles (genus Paederus) differ from the Meloidae and Oedemeridae families in that they produce the vesicant pederin rather than cantharidin. Pederin causes a more violent eruption called dermatitis linearis, which often is associated with intense urticaria prior to blistering.1 Rove beetles are found in moist environments and tend to favor tropical and subtropical climates. They may emerge in large numbers after heavy rainfall.
Clinical Presentation
Blistering dermatitis—caused by exposure to cantharidin (families Meloidae and Oedemeridae) or pederin (genus Paederus)—begins as burning and tingling within minutes of exposure. Bullae later develop, often in a linear fashion, with subsequent bursting and crust formation.1,3 Secondary infection can occur.5 Exposure occurring on the elbows, knees, or mirroring skin folds results in lesions on opposing surfaces that come into contact, otherwise known as kissing lesions.1,3 Acantholysis of suprabasal keratinocytes can be seen on histologic sections of blisters (Figure 3)1,7 due to cantharidin activation of the serine/threonine protein phosphatases that cause detachment of tonofilaments from desmosomes.1,8 Washing of exposed sites with soap or alcohol can potentially prevent development of blistering dermatitis; however, lesions usually heal without complication when treated with topical antibiotics.3
If blister beetles are ingested, they can cause poisoning characterized by abdominal pain and hematuria.1,3,9,10 In fact, blister beetle ingestion by animals consuming baled hay is an important agricultural and economical implication. Several cases of fatal farm animal ingestion resulting in gastrointestinal erosion have been reported.4,6
Medicinal Properties
Medicinal use of cantharidin has been recorded as early as the 19th century and is believed to have been part of ancient medicinal practices in China, Spain, South Africa, and pre-Columbian America for its vesicant, abortifacient, and supposed aphrodisiac properties.2,4,8
Toxicity from internal ingestion has been well documented.1,3,9,10 The Mylabris and Epicauta genera of the family Meloidae, most commonly the Spanish fly, are used for modern extraction of cantharidin. Up to 5% of the dry weight of a blister beetle can be constituted by cantharidin.4
Although its use has diminished due to limited availability, cantharidin is still used for treatment of common warts, periungual warts, and molluscum contagiosum. It is a favorable choice for treating pediatric patients because of the tolerability and painlessness of application. Due to its acantholytic properties, warts generally slough with the cantharidin-induced blister.11
In recent years, cantharidin has been studied for its anticancer properties. It has been shown to weaken cancer cell antioxidant properties by interfering with glutathione-related enzymes, thus inducing oxidative damage. Additionally, cantharidin is associated with decreased mitochondrial cytochrome C and increased cytosolic cytochrome C, an important signal for apoptosis.12 Furthermore, cantharidin recently was shown to increase expression of proapoptotic proteins and decrease expression of antiapoptotic proteins causing cell death in nasopharyngeal carcinoma, making it a potential anticancer treatment.13
Conclusion
Blister beetles, long known for their production of vesicant agents, are both a cause of as well as a potential treatment of dermatologic disease. The blistering associated with exposure to a disturbed beetle generally is mild and heals without scarring if vital areas such as the eyelids are not affected.
Classification
Blister beetles are both a scourge and the source of medical cantharidin (Figure 1). The term blister beetle refers to 3 families of the order Coleoptera: Meloidae, Oedemeridae, and Staphylinidae (Figure 2).
Meloidae is the most well-known family of blister beetles, with more than 200 species worldwide identified as a cause of blistering dermatitis.1 The most notorious is Lytta vesicatoria, also known as the Spanish fly. Although some blister beetles are inconspicuous in appearance, most are brightly colored and easily spotted, making them attractive to small children.2 They may be attracted to fluorescent lighting and commonly enter through open windows.3 Blister beetles do not bite or sting; rather, they release cantharidin via hemolymph, an oily yellow substance that is copiously expressed from the leg joints when disturbed by rubbing or pressing.1,3-5 Blistering also is associated with exposure to the contents of crushed beetles, which is the source of pharmacologic cantharidin.2,4,6
Oedemeridae are the smallest and least known beetles within the Coleoptera family. They often are called false blister beetles, which is a misnomer. Oedemeridae beetles also produce cantharidin, similar to Meloidae beetles; however, because Oedemeridae beetles are smaller, the vesiculobullous eruptions also are less pronounced.1
A third well-known family of blister beetles is the Staphylinidae family. Rove beetles (genus Paederus) differ from the Meloidae and Oedemeridae families in that they produce the vesicant pederin rather than cantharidin. Pederin causes a more violent eruption called dermatitis linearis, which often is associated with intense urticaria prior to blistering.1 Rove beetles are found in moist environments and tend to favor tropical and subtropical climates. They may emerge in large numbers after heavy rainfall.
Clinical Presentation
Blistering dermatitis—caused by exposure to cantharidin (families Meloidae and Oedemeridae) or pederin (genus Paederus)—begins as burning and tingling within minutes of exposure. Bullae later develop, often in a linear fashion, with subsequent bursting and crust formation.1,3 Secondary infection can occur.5 Exposure occurring on the elbows, knees, or mirroring skin folds results in lesions on opposing surfaces that come into contact, otherwise known as kissing lesions.1,3 Acantholysis of suprabasal keratinocytes can be seen on histologic sections of blisters (Figure 3)1,7 due to cantharidin activation of the serine/threonine protein phosphatases that cause detachment of tonofilaments from desmosomes.1,8 Washing of exposed sites with soap or alcohol can potentially prevent development of blistering dermatitis; however, lesions usually heal without complication when treated with topical antibiotics.3
If blister beetles are ingested, they can cause poisoning characterized by abdominal pain and hematuria.1,3,9,10 In fact, blister beetle ingestion by animals consuming baled hay is an important agricultural and economical implication. Several cases of fatal farm animal ingestion resulting in gastrointestinal erosion have been reported.4,6
Medicinal Properties
Medicinal use of cantharidin has been recorded as early as the 19th century and is believed to have been part of ancient medicinal practices in China, Spain, South Africa, and pre-Columbian America for its vesicant, abortifacient, and supposed aphrodisiac properties.2,4,8
Toxicity from internal ingestion has been well documented.1,3,9,10 The Mylabris and Epicauta genera of the family Meloidae, most commonly the Spanish fly, are used for modern extraction of cantharidin. Up to 5% of the dry weight of a blister beetle can be constituted by cantharidin.4
Although its use has diminished due to limited availability, cantharidin is still used for treatment of common warts, periungual warts, and molluscum contagiosum. It is a favorable choice for treating pediatric patients because of the tolerability and painlessness of application. Due to its acantholytic properties, warts generally slough with the cantharidin-induced blister.11
In recent years, cantharidin has been studied for its anticancer properties. It has been shown to weaken cancer cell antioxidant properties by interfering with glutathione-related enzymes, thus inducing oxidative damage. Additionally, cantharidin is associated with decreased mitochondrial cytochrome C and increased cytosolic cytochrome C, an important signal for apoptosis.12 Furthermore, cantharidin recently was shown to increase expression of proapoptotic proteins and decrease expression of antiapoptotic proteins causing cell death in nasopharyngeal carcinoma, making it a potential anticancer treatment.13
Conclusion
Blister beetles, long known for their production of vesicant agents, are both a cause of as well as a potential treatment of dermatologic disease. The blistering associated with exposure to a disturbed beetle generally is mild and heals without scarring if vital areas such as the eyelids are not affected.
- Nicholls DS, Christmas TI, Greig DE. Oedemerid blister beetle dermatosis: a review. J Am Acad Dermatol. 1990;22:815-819.
- Percino-Daniel N, Buckley D, García-París M. Pharmacological properties of blister beetles (Coleoptera: Meloidae) promoted their integration into the cultural heritage of native rural Spain as inferred by vernacular names diversity, traditions, and mitochondrial DNA. J Ethnopharmacol. 2013;147:570-583.
- James WD, Berger TG, Elston DM. Parasitic infestations, stings, and bites. In: James WD, Berger TG, Elston DM, eds. Andrew’s Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016:418-450.
- Selander RB, Fasulo TR. Featured creatures: blister beetles. University of Florida/IFAS Entomology and Nematology website. http://entnemdept.ufl.edu/creatures/urban/medical/blister_beetles.htm. Published October 2000. Revised September 2010. Accessed November 12, 2019.
- Wijerathne BTB. Blister mystery. Wilderness Environ Med. 2017;28:271-272.
- Penrith ML, Naude TW. Mortality in chickens associated with blister beetle consumption. J S Afr Vet Assoc. 1996;67:97-99.
- Yell JA, Burge SM, Dean D. Cantharidin-induced acantholysis: adhesion molecules, proteases, and related proteins. Br J Dermatol. 1994;130:148-157.
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine-threonine protein phosphatases types 1 and 2a. FEBS Letters. 1993;330:283-286.
- Al-Binali AM, Shabana M, Al-Fifi S, et al. Cantharidin poisoning due to blister beetle ingestion in children: two case reports and a review of clinical presentations. Sultan Qaboos Univ Med J. 2010;10:258-261.
- Tagwireyi D, Ball DE, Loga PJ, et al. Cantharidin poisoning due to “blister beetle” ingestion. Toxicon. 2000;38:1865-1869.
- Al-Dawsari NA, Masterpol KS. Cantharidin in dermatology. Skinmed. 2016;14:111-114.
- Verma AK, Prasad SB. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, epicauta hirticornis. Anticancer Agents Med Chem. 2013;13:1096-1114.
- Chen AW, Tseng YS, Lin CC, et al. Norcantharidin induce apoptosis in human nasopharyngeal carcinoma through caspase and mitochondrial pathway. Environ Toxicol. 2018;33:343-350.
- Nicholls DS, Christmas TI, Greig DE. Oedemerid blister beetle dermatosis: a review. J Am Acad Dermatol. 1990;22:815-819.
- Percino-Daniel N, Buckley D, García-París M. Pharmacological properties of blister beetles (Coleoptera: Meloidae) promoted their integration into the cultural heritage of native rural Spain as inferred by vernacular names diversity, traditions, and mitochondrial DNA. J Ethnopharmacol. 2013;147:570-583.
- James WD, Berger TG, Elston DM. Parasitic infestations, stings, and bites. In: James WD, Berger TG, Elston DM, eds. Andrew’s Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016:418-450.
- Selander RB, Fasulo TR. Featured creatures: blister beetles. University of Florida/IFAS Entomology and Nematology website. http://entnemdept.ufl.edu/creatures/urban/medical/blister_beetles.htm. Published October 2000. Revised September 2010. Accessed November 12, 2019.
- Wijerathne BTB. Blister mystery. Wilderness Environ Med. 2017;28:271-272.
- Penrith ML, Naude TW. Mortality in chickens associated with blister beetle consumption. J S Afr Vet Assoc. 1996;67:97-99.
- Yell JA, Burge SM, Dean D. Cantharidin-induced acantholysis: adhesion molecules, proteases, and related proteins. Br J Dermatol. 1994;130:148-157.
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine-threonine protein phosphatases types 1 and 2a. FEBS Letters. 1993;330:283-286.
- Al-Binali AM, Shabana M, Al-Fifi S, et al. Cantharidin poisoning due to blister beetle ingestion in children: two case reports and a review of clinical presentations. Sultan Qaboos Univ Med J. 2010;10:258-261.
- Tagwireyi D, Ball DE, Loga PJ, et al. Cantharidin poisoning due to “blister beetle” ingestion. Toxicon. 2000;38:1865-1869.
- Al-Dawsari NA, Masterpol KS. Cantharidin in dermatology. Skinmed. 2016;14:111-114.
- Verma AK, Prasad SB. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, epicauta hirticornis. Anticancer Agents Med Chem. 2013;13:1096-1114.
- Chen AW, Tseng YS, Lin CC, et al. Norcantharidin induce apoptosis in human nasopharyngeal carcinoma through caspase and mitochondrial pathway. Environ Toxicol. 2018;33:343-350.
Practice Points
- Exposure to cantharidin presents initially with burning and tingling before progressing to bullae formation, sometimes in a linear fashion. Rupture of bullae and crust formation can occur.
- Washing of the exposed skin with soap and water may prevent development of blistering dermatitis.
- Clinical use of cantharidin is favorable in treating pediatric patients with common warts and molluscum contagiosum due to tolerability and painlessness of application.

Solitary Papule on the Nose
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
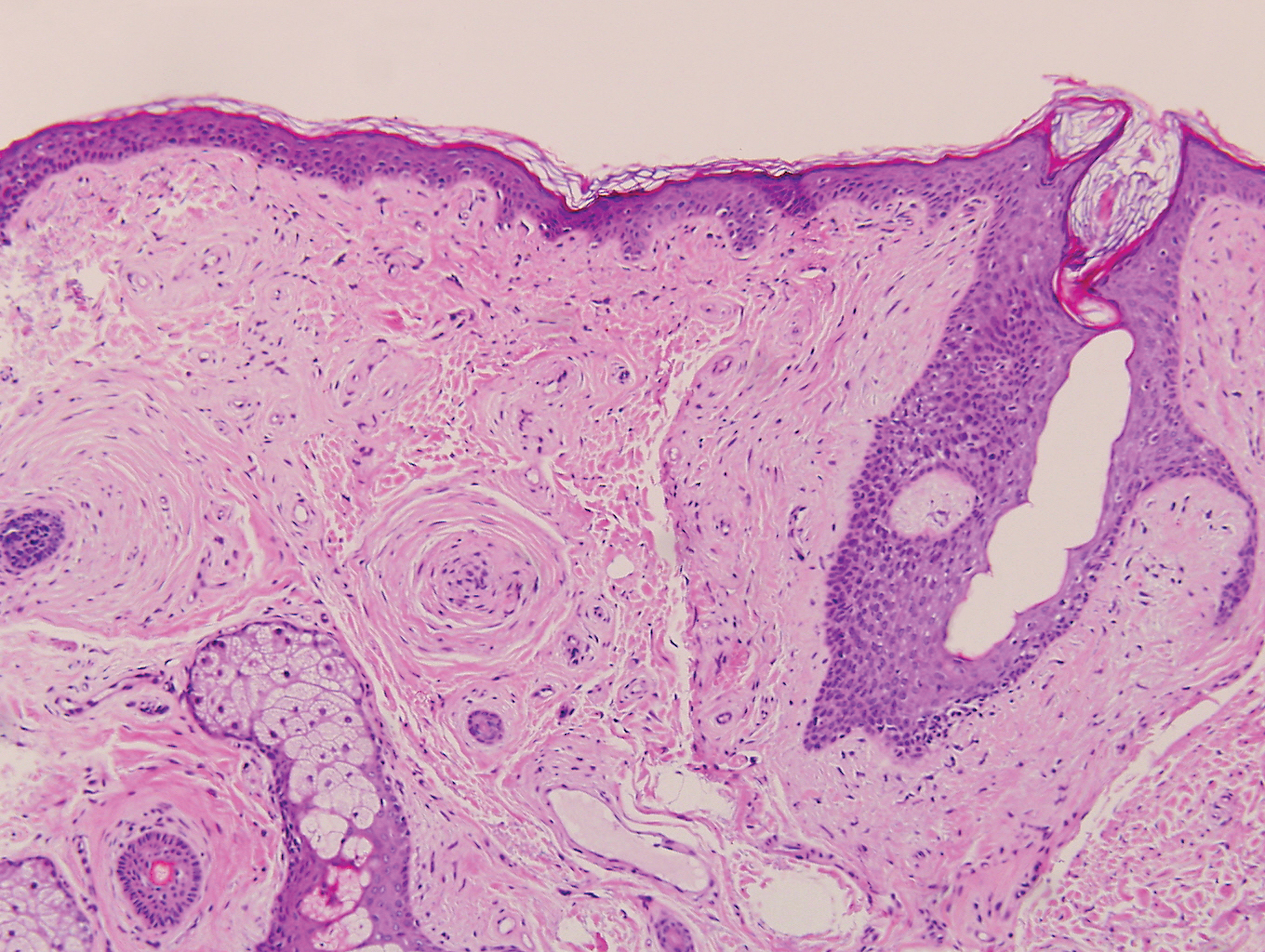
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
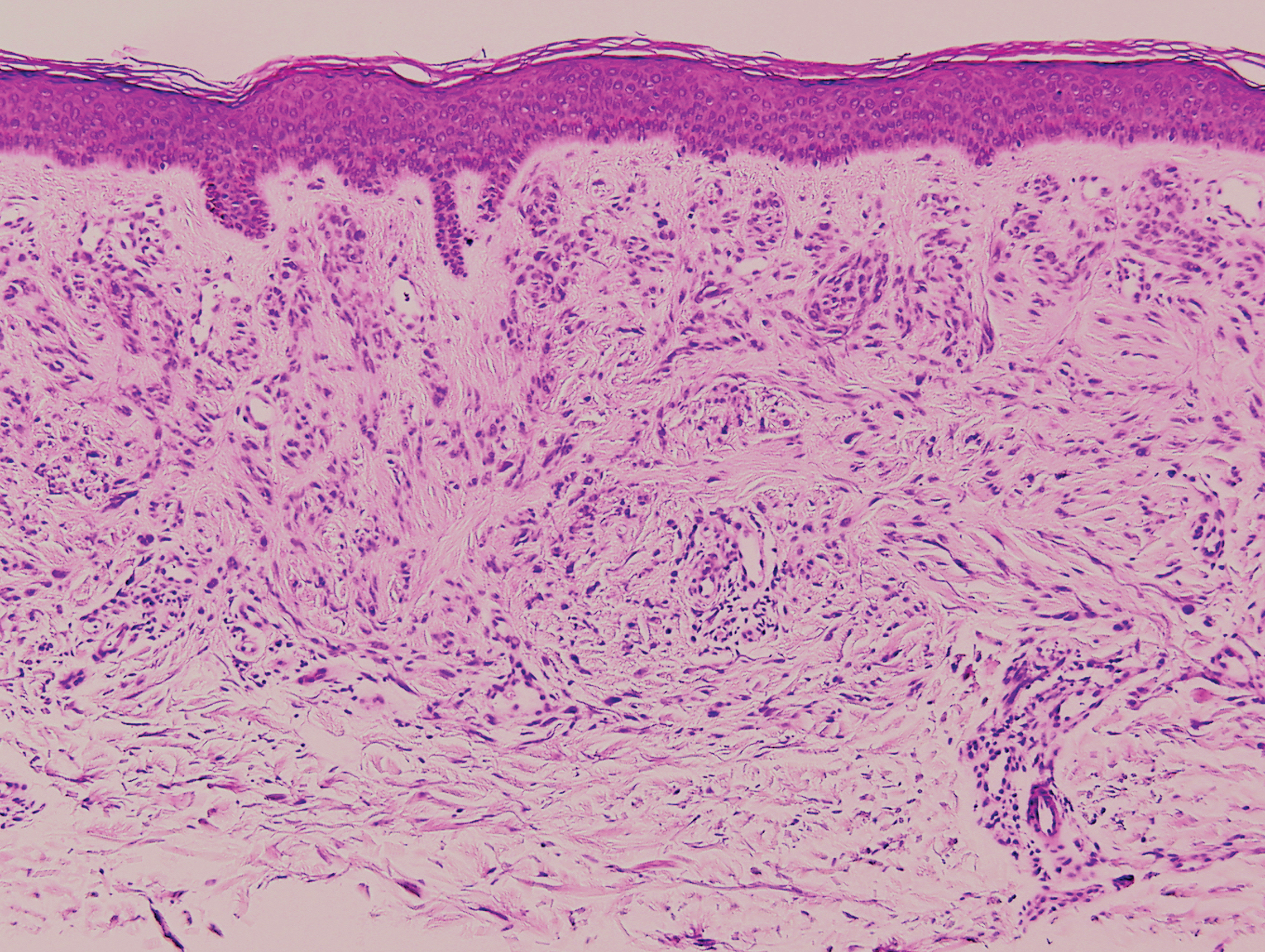
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
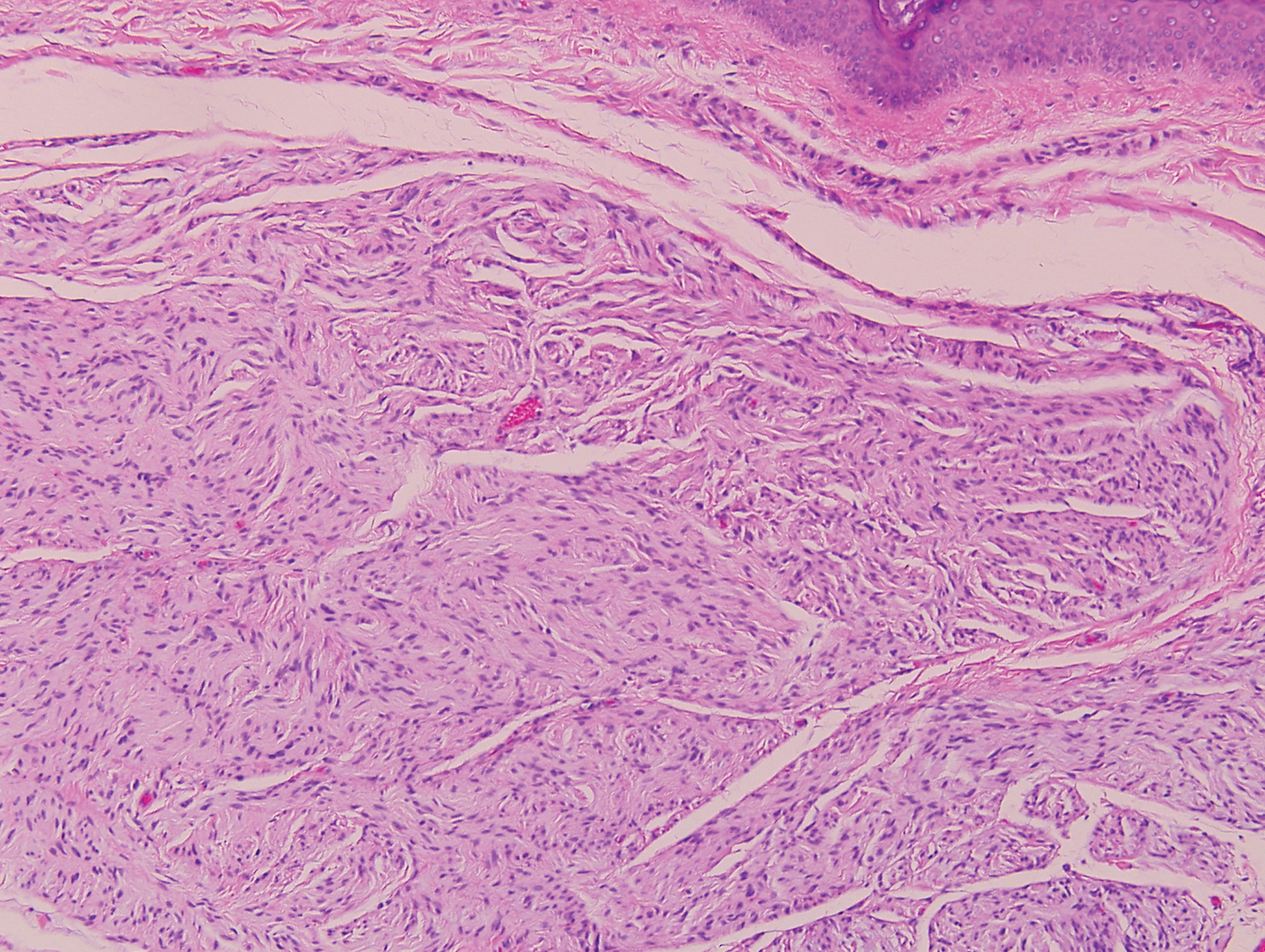
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
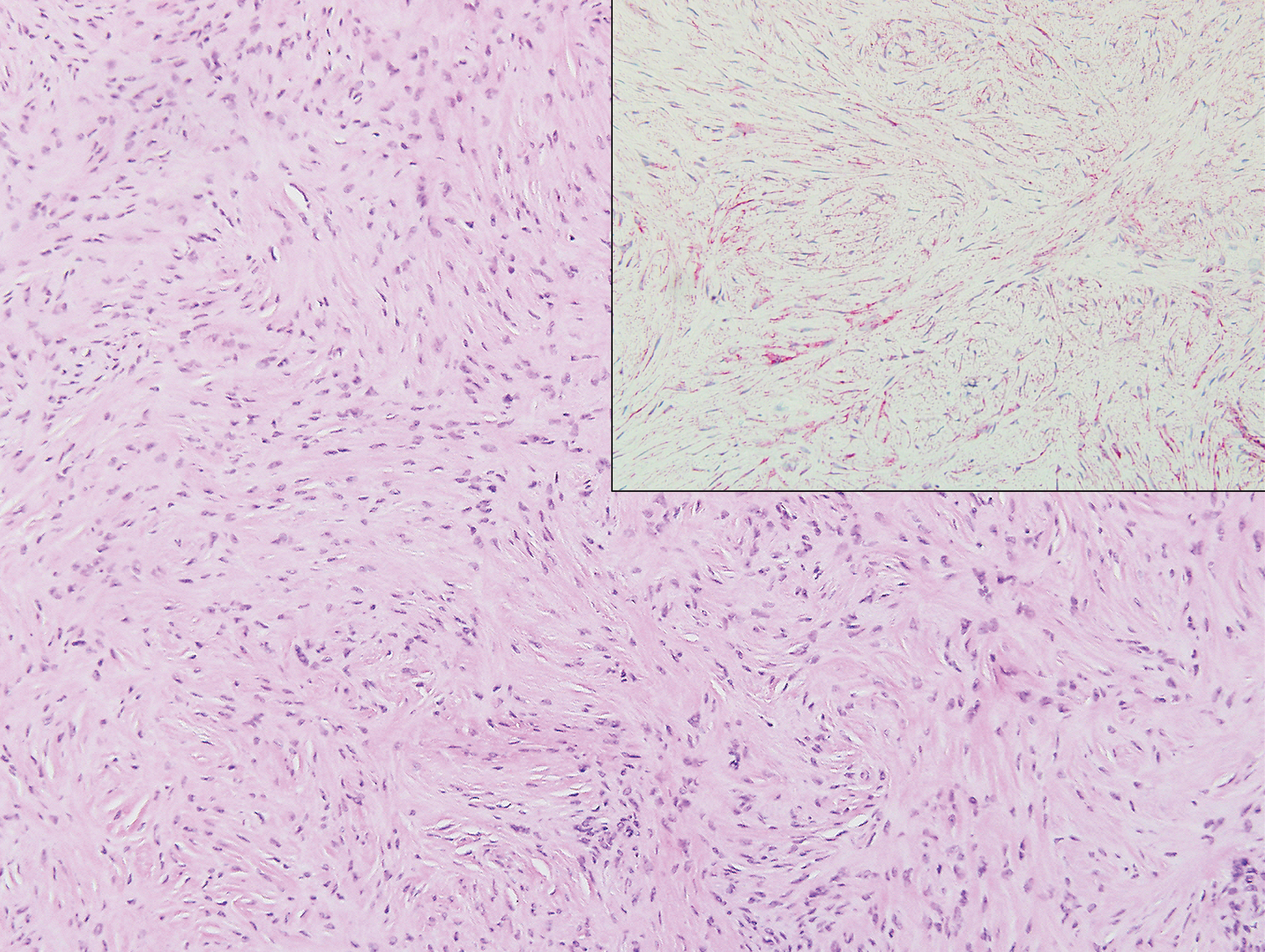
A 25-year-old man presented with a flesh-colored papule on the left side of the nose of 2 years' duration.
Systemic Epstein-Barr Virus–Positive T-cell Lymphoma of Childhood
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
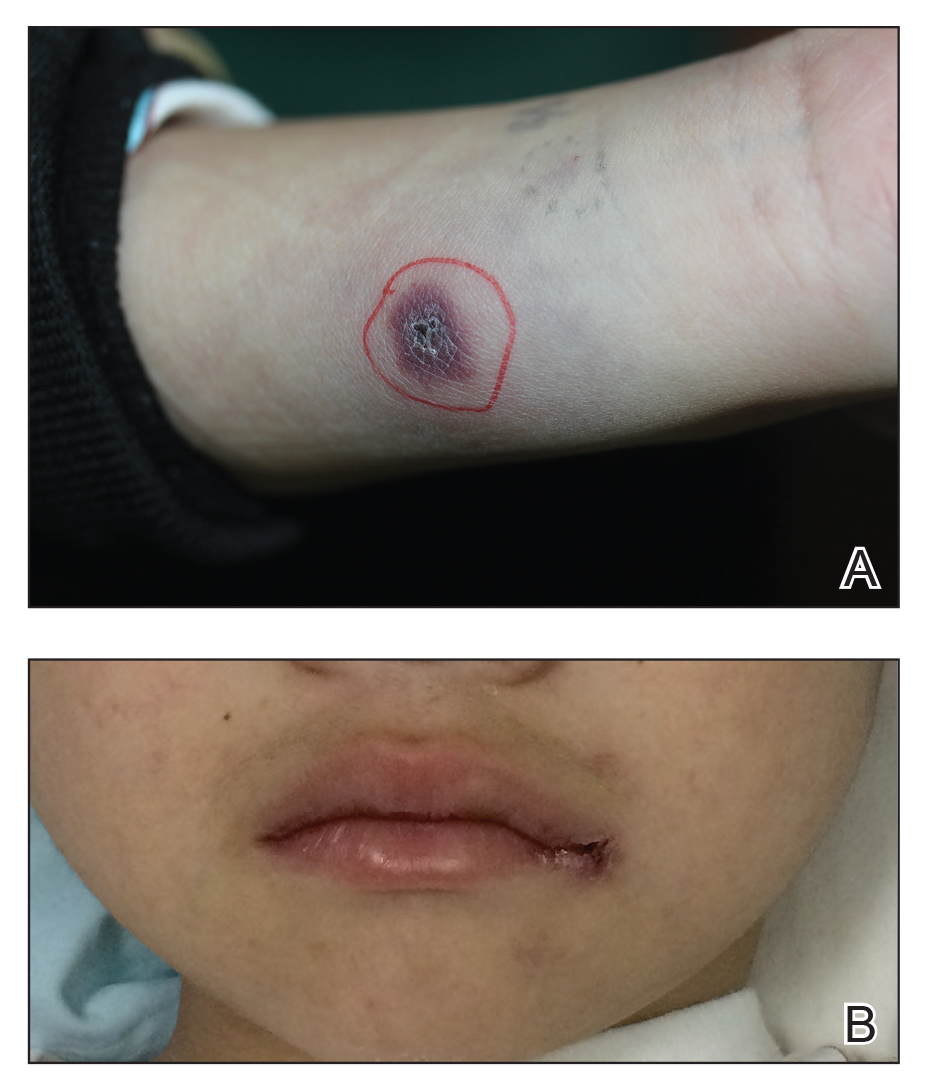
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
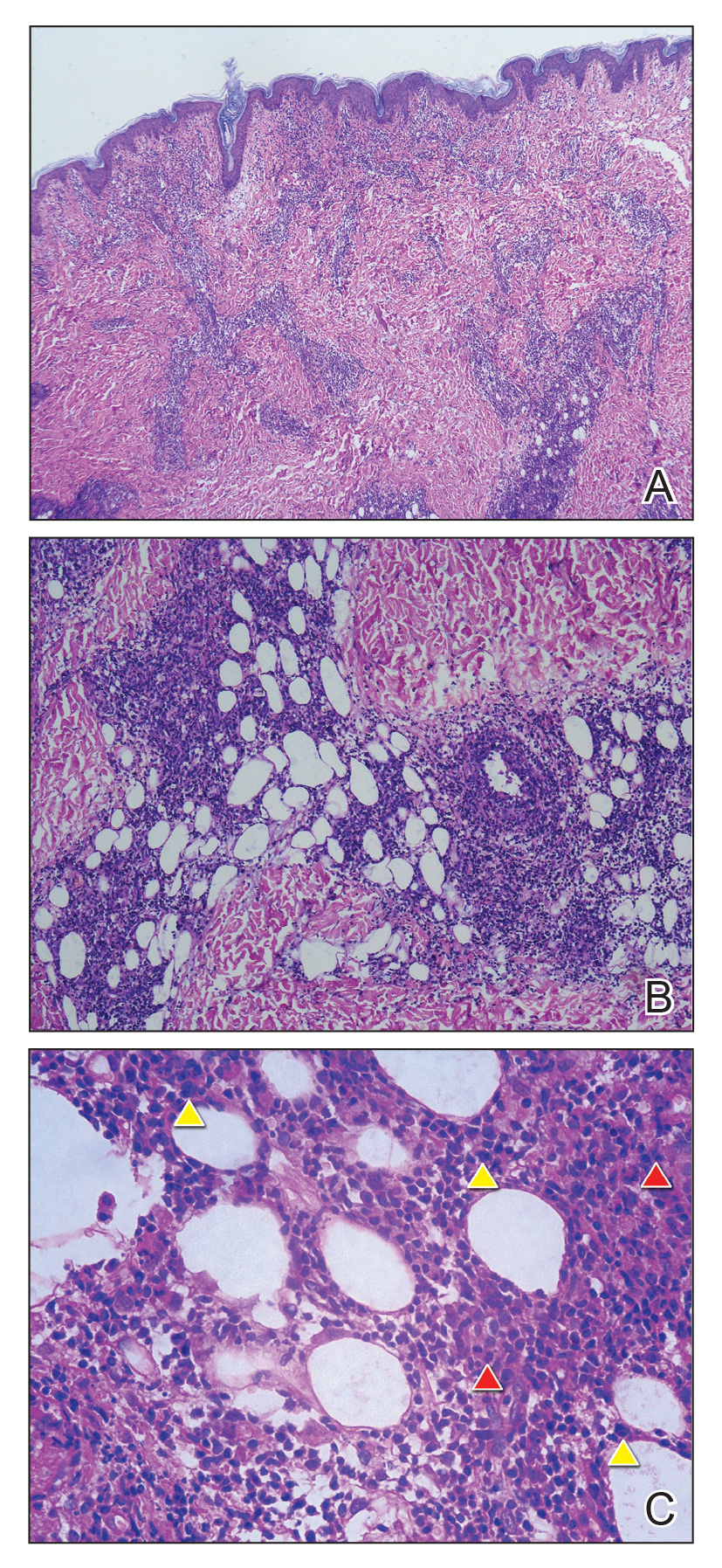
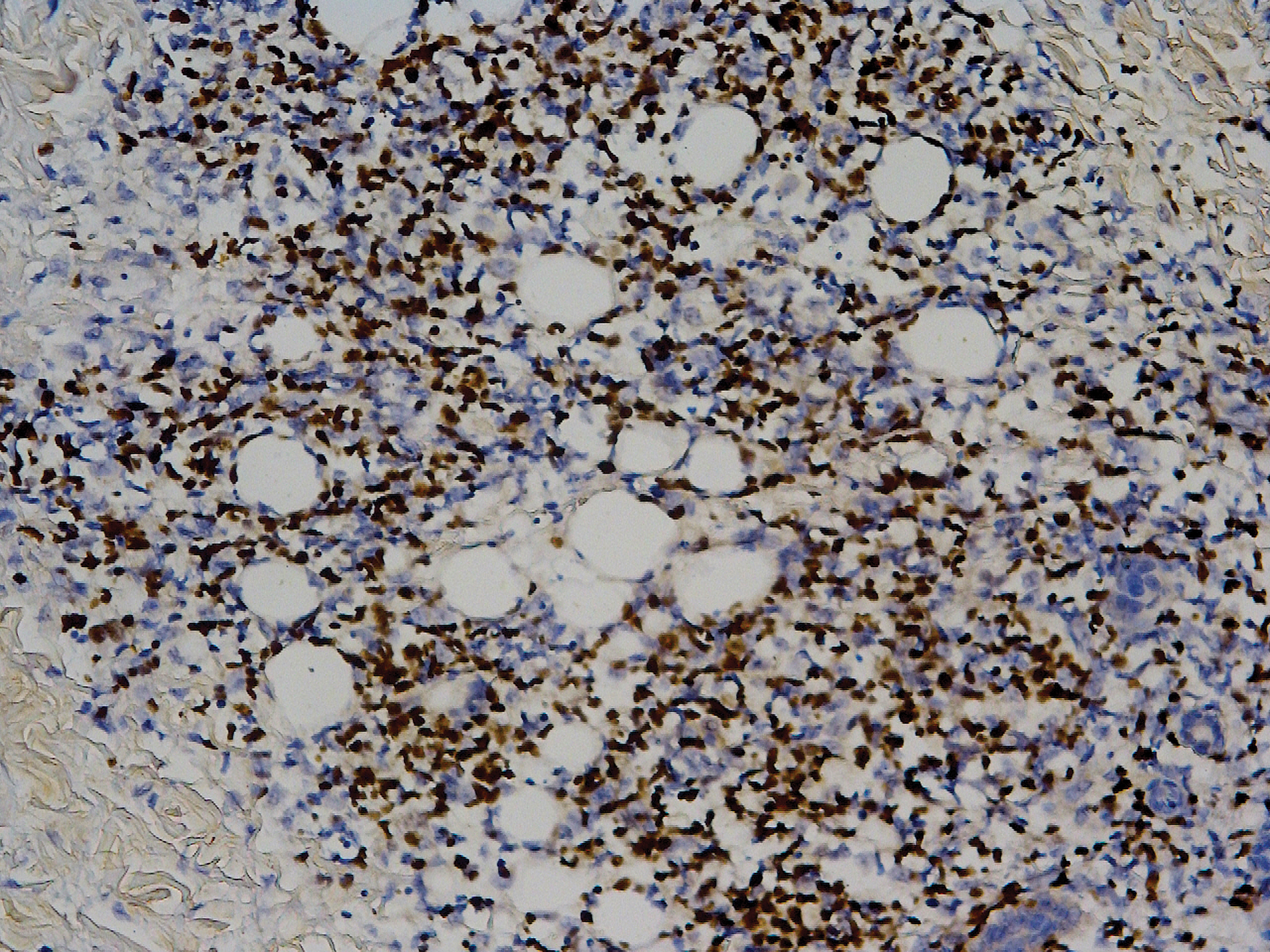
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Practice Points
- Systemic Epstein-Barr virus (EBV)–positive T-cell lymphoma of childhood is a fulminant illness with a predilection for Asians and indigenous populations from Mexico and Central and South America. In most patients, the disease has an aggressive clinical course with high mortality.
- The disease often is complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. When these severe complications are absent, the prognosis might be better.
- In situ hybridization for EBV-encoded RNA and for T-cell receptor gene rearrangements is an important tool to establish the diagnosis as well as for treatment options and predicting the prognosis.