User login
Imaging guides BCC laser ablation
KISSIMMEE, FLA. – Reflective confocal microscopic imaging successfully guided carbon dioxide laser ablation of basal cell carcinomas, and imaging results fully matched those from Mohs histology, a small study found.
“Our results suggest that reflective confocal microscopy can accurately guide carbon dioxide laser ablation of superficial and early nodular basal cell carcinomas,” said Dr. Brian Hibler of Memorial Sloan Kettering Cancer Center in New York. The technique provides a real-time, noninvasive way to delineate the tumor area before ablation and to check for residual tumor between passes with the laser, he said at the annual meeting of the American Society for Laser Medicine and surgery.
While conventional and Mohs microscopic surgeries remain the gold standard for removing basal carcinomas (BCC), surgery is not an option for some patients because of tumor location, comorbidities, or personal preferences, Dr. Hibler noted. Past studies have reported good clinical and cosmetic outcomes with laser ablation of BCCs, but use of the modality has been limited because there was no way to assess response without excision or biopsies. Reflective confocal microscopy (RCM) uses a low-powered laser system that provides cellular-level imaging and can distinguish BCCs, he said.
Dr. Hibler and his colleagues performed baseline RCM of eight BCCs (three on the trunk, three on the extremities, and two on the head and neck) from seven patients aged 29-83 years. Two patients were men and five were women. The patients then underwent carbon dioxide laser ablation with a wavelength of 10,600 nm, pulse duration of 750 microseconds, and fluence of 7.5 J/cm2, using a square pattern and density of 30%. If RCM revealed residual BCC, the researchers repeated the process up to two more times, for a maximum of three passes. They then removed the entire lesion using Mohs micrographic surgery, performing vertical histologic sectioning of the tissue.
Reflective confocal microscopy generated reliable cellular-level images in real time on the tumor surface and up to 150 mcm deep, Dr. Hibler reported. Tissue from BCCs appears as dense nodular areas with adjacent spaces and red blood cell trafficking, he noted. Microscopy results were consistent with Mohs histology findings in all eight cases, including six in which the tumor was completely removed and two with residual tumor. One of these two cases was the only infiltrative BCC in the series, while the other might have been tissue artifact, Dr. Hibler said.
Patients experienced no adverse effects from the interventions, Dr. Hibler reported. “Future studies are planned are planned without Mohs, so we can use reflective confocal microscopy to longitudinally monitor for recurrence,” he added.
The study won an award at the meeting.
Dr. Hibler reported no funding sources and made no disclosures.
KISSIMMEE, FLA. – Reflective confocal microscopic imaging successfully guided carbon dioxide laser ablation of basal cell carcinomas, and imaging results fully matched those from Mohs histology, a small study found.
“Our results suggest that reflective confocal microscopy can accurately guide carbon dioxide laser ablation of superficial and early nodular basal cell carcinomas,” said Dr. Brian Hibler of Memorial Sloan Kettering Cancer Center in New York. The technique provides a real-time, noninvasive way to delineate the tumor area before ablation and to check for residual tumor between passes with the laser, he said at the annual meeting of the American Society for Laser Medicine and surgery.
While conventional and Mohs microscopic surgeries remain the gold standard for removing basal carcinomas (BCC), surgery is not an option for some patients because of tumor location, comorbidities, or personal preferences, Dr. Hibler noted. Past studies have reported good clinical and cosmetic outcomes with laser ablation of BCCs, but use of the modality has been limited because there was no way to assess response without excision or biopsies. Reflective confocal microscopy (RCM) uses a low-powered laser system that provides cellular-level imaging and can distinguish BCCs, he said.
Dr. Hibler and his colleagues performed baseline RCM of eight BCCs (three on the trunk, three on the extremities, and two on the head and neck) from seven patients aged 29-83 years. Two patients were men and five were women. The patients then underwent carbon dioxide laser ablation with a wavelength of 10,600 nm, pulse duration of 750 microseconds, and fluence of 7.5 J/cm2, using a square pattern and density of 30%. If RCM revealed residual BCC, the researchers repeated the process up to two more times, for a maximum of three passes. They then removed the entire lesion using Mohs micrographic surgery, performing vertical histologic sectioning of the tissue.
Reflective confocal microscopy generated reliable cellular-level images in real time on the tumor surface and up to 150 mcm deep, Dr. Hibler reported. Tissue from BCCs appears as dense nodular areas with adjacent spaces and red blood cell trafficking, he noted. Microscopy results were consistent with Mohs histology findings in all eight cases, including six in which the tumor was completely removed and two with residual tumor. One of these two cases was the only infiltrative BCC in the series, while the other might have been tissue artifact, Dr. Hibler said.
Patients experienced no adverse effects from the interventions, Dr. Hibler reported. “Future studies are planned are planned without Mohs, so we can use reflective confocal microscopy to longitudinally monitor for recurrence,” he added.
The study won an award at the meeting.
Dr. Hibler reported no funding sources and made no disclosures.
KISSIMMEE, FLA. – Reflective confocal microscopic imaging successfully guided carbon dioxide laser ablation of basal cell carcinomas, and imaging results fully matched those from Mohs histology, a small study found.
“Our results suggest that reflective confocal microscopy can accurately guide carbon dioxide laser ablation of superficial and early nodular basal cell carcinomas,” said Dr. Brian Hibler of Memorial Sloan Kettering Cancer Center in New York. The technique provides a real-time, noninvasive way to delineate the tumor area before ablation and to check for residual tumor between passes with the laser, he said at the annual meeting of the American Society for Laser Medicine and surgery.
While conventional and Mohs microscopic surgeries remain the gold standard for removing basal carcinomas (BCC), surgery is not an option for some patients because of tumor location, comorbidities, or personal preferences, Dr. Hibler noted. Past studies have reported good clinical and cosmetic outcomes with laser ablation of BCCs, but use of the modality has been limited because there was no way to assess response without excision or biopsies. Reflective confocal microscopy (RCM) uses a low-powered laser system that provides cellular-level imaging and can distinguish BCCs, he said.
Dr. Hibler and his colleagues performed baseline RCM of eight BCCs (three on the trunk, three on the extremities, and two on the head and neck) from seven patients aged 29-83 years. Two patients were men and five were women. The patients then underwent carbon dioxide laser ablation with a wavelength of 10,600 nm, pulse duration of 750 microseconds, and fluence of 7.5 J/cm2, using a square pattern and density of 30%. If RCM revealed residual BCC, the researchers repeated the process up to two more times, for a maximum of three passes. They then removed the entire lesion using Mohs micrographic surgery, performing vertical histologic sectioning of the tissue.
Reflective confocal microscopy generated reliable cellular-level images in real time on the tumor surface and up to 150 mcm deep, Dr. Hibler reported. Tissue from BCCs appears as dense nodular areas with adjacent spaces and red blood cell trafficking, he noted. Microscopy results were consistent with Mohs histology findings in all eight cases, including six in which the tumor was completely removed and two with residual tumor. One of these two cases was the only infiltrative BCC in the series, while the other might have been tissue artifact, Dr. Hibler said.
Patients experienced no adverse effects from the interventions, Dr. Hibler reported. “Future studies are planned are planned without Mohs, so we can use reflective confocal microscopy to longitudinally monitor for recurrence,” he added.
The study won an award at the meeting.
Dr. Hibler reported no funding sources and made no disclosures.
Key clinical point: Reflective confocal microscopy offers noninvasive, real-time imaging to guide laser ablation of basal cell carcinomas.
Major finding: Results from RCM matched those from Mohs histology in all patients.
Data source: Prospective study of eight BCCs in seven patients.
Disclosures: Dr. Hibler reported no funding sources and made no disclosures.
Radiofrequency improves lower face laxity
KISSIMMEE, FLA. – Three treatments with a noninvasive radiofrequency device significantly improved mild to moderate laxity of the lower face and neck, a prospective study of 30 patients showed.
Downtime after treatment ranged from 2 to 4 days, and adverse effects included mild stippling and purpura at the treatment sites, Dr. Girish Munavalli reported at the annual meeting of the American Society for Laser Medicine and Surgery.

Aging causes progressive loosening of the tissues of the lower face and neck, which manifests as narrowing of the cervicomental angle (between the neck and the lowest point under the chin) and the gnathion angle (between the lowest point under the chin and the most anterior or prominent point on the front of the chin). Historically, patients resorted to invasive surgical lifting procedures to restore these angles, said Dr. Munavalli of Wake Forest University, Charlotte, N.C.
To explore noninvasive alternatives, he and his associates conducted a pilot study of a bipolar fractionated microneedle radiofrequency device that consisted of a handpiece and a 1 cm2 square disposable microneedle tip with 49 proximally insulated 34-G microneedle electrodes. The study comprised 7 men and 23 women aged 37-71 years. One physician treated all patients, and each patient underwent three treatment sessions lasting about 45 minutes and spaced a month apart.
Each session included three passes across the lower face and submental area at a maximum tissue depth of 1-3 mm, Dr. Munavalli said. Thicker skin requires deeper needle penetration, which in turn requires more treatment energy and longer pulse duration, he noted. Therefore, the physician performed the first pass at a depth of about 2.5 mm with an energy setting of 9-11 and a pulse duration of 280-320 msec, the second pass at a depth of 1.5 mm with a setting of 8-9 and a pulse duration of 230-250 msec, and the third pass at a depth of 1 mm with a setting of 6 and a pulse duration of 160 msec.
Six months after the end of treatment, patients underwent computerized measurements of the cervicomental and gnathion angles, and a panel of blinded investigators compared pre- and posttreatment photographs, Dr. Munavalli said. On average, the cervicomental angle increased by 27 degrees (range, 18 to 36 degrees; P < .01), and the gnathion angle increased by 16 degrees (range, 12 to 20 degrees; P < .01). The blinded assessors correctly chose the posttreatment photographs 90% of the time. Taken together, the results suggest that the device is a safe and effective option for improving laxity of the lower face and neck, Dr. Munavalli concluded.
Lutronic Corp is the maker of the Infini device, which the FDA approved for dermatologic use in July 2013. Dr. Munavalli reported no funding sources or financial disclosures.
KISSIMMEE, FLA. – Three treatments with a noninvasive radiofrequency device significantly improved mild to moderate laxity of the lower face and neck, a prospective study of 30 patients showed.
Downtime after treatment ranged from 2 to 4 days, and adverse effects included mild stippling and purpura at the treatment sites, Dr. Girish Munavalli reported at the annual meeting of the American Society for Laser Medicine and Surgery.
Aging causes progressive loosening of the tissues of the lower face and neck, which manifests as narrowing of the cervicomental angle (between the neck and the lowest point under the chin) and the gnathion angle (between the lowest point under the chin and the most anterior or prominent point on the front of the chin). Historically, patients resorted to invasive surgical lifting procedures to restore these angles, said Dr. Munavalli of Wake Forest University, Charlotte, N.C.
To explore noninvasive alternatives, he and his associates conducted a pilot study of a bipolar fractionated microneedle radiofrequency device that consisted of a handpiece and a 1 cm2 square disposable microneedle tip with 49 proximally insulated 34-G microneedle electrodes. The study comprised 7 men and 23 women aged 37-71 years. One physician treated all patients, and each patient underwent three treatment sessions lasting about 45 minutes and spaced a month apart.
Each session included three passes across the lower face and submental area at a maximum tissue depth of 1-3 mm, Dr. Munavalli said. Thicker skin requires deeper needle penetration, which in turn requires more treatment energy and longer pulse duration, he noted. Therefore, the physician performed the first pass at a depth of about 2.5 mm with an energy setting of 9-11 and a pulse duration of 280-320 msec, the second pass at a depth of 1.5 mm with a setting of 8-9 and a pulse duration of 230-250 msec, and the third pass at a depth of 1 mm with a setting of 6 and a pulse duration of 160 msec.
Six months after the end of treatment, patients underwent computerized measurements of the cervicomental and gnathion angles, and a panel of blinded investigators compared pre- and posttreatment photographs, Dr. Munavalli said. On average, the cervicomental angle increased by 27 degrees (range, 18 to 36 degrees; P < .01), and the gnathion angle increased by 16 degrees (range, 12 to 20 degrees; P < .01). The blinded assessors correctly chose the posttreatment photographs 90% of the time. Taken together, the results suggest that the device is a safe and effective option for improving laxity of the lower face and neck, Dr. Munavalli concluded.
Lutronic Corp is the maker of the Infini device, which the FDA approved for dermatologic use in July 2013. Dr. Munavalli reported no funding sources or financial disclosures.
KISSIMMEE, FLA. – Three treatments with a noninvasive radiofrequency device significantly improved mild to moderate laxity of the lower face and neck, a prospective study of 30 patients showed.
Downtime after treatment ranged from 2 to 4 days, and adverse effects included mild stippling and purpura at the treatment sites, Dr. Girish Munavalli reported at the annual meeting of the American Society for Laser Medicine and Surgery.
Aging causes progressive loosening of the tissues of the lower face and neck, which manifests as narrowing of the cervicomental angle (between the neck and the lowest point under the chin) and the gnathion angle (between the lowest point under the chin and the most anterior or prominent point on the front of the chin). Historically, patients resorted to invasive surgical lifting procedures to restore these angles, said Dr. Munavalli of Wake Forest University, Charlotte, N.C.
To explore noninvasive alternatives, he and his associates conducted a pilot study of a bipolar fractionated microneedle radiofrequency device that consisted of a handpiece and a 1 cm2 square disposable microneedle tip with 49 proximally insulated 34-G microneedle electrodes. The study comprised 7 men and 23 women aged 37-71 years. One physician treated all patients, and each patient underwent three treatment sessions lasting about 45 minutes and spaced a month apart.
Each session included three passes across the lower face and submental area at a maximum tissue depth of 1-3 mm, Dr. Munavalli said. Thicker skin requires deeper needle penetration, which in turn requires more treatment energy and longer pulse duration, he noted. Therefore, the physician performed the first pass at a depth of about 2.5 mm with an energy setting of 9-11 and a pulse duration of 280-320 msec, the second pass at a depth of 1.5 mm with a setting of 8-9 and a pulse duration of 230-250 msec, and the third pass at a depth of 1 mm with a setting of 6 and a pulse duration of 160 msec.
Six months after the end of treatment, patients underwent computerized measurements of the cervicomental and gnathion angles, and a panel of blinded investigators compared pre- and posttreatment photographs, Dr. Munavalli said. On average, the cervicomental angle increased by 27 degrees (range, 18 to 36 degrees; P < .01), and the gnathion angle increased by 16 degrees (range, 12 to 20 degrees; P < .01). The blinded assessors correctly chose the posttreatment photographs 90% of the time. Taken together, the results suggest that the device is a safe and effective option for improving laxity of the lower face and neck, Dr. Munavalli concluded.
Lutronic Corp is the maker of the Infini device, which the FDA approved for dermatologic use in July 2013. Dr. Munavalli reported no funding sources or financial disclosures.
Key clinical point: A noninvasive bipolar fractionated microneedle radiofrequency device significantly improved laxity of the lower face and neck.
Major finding: After three treatment sessions, the cervicomental and gnathion angles increased by an average of 27 degrees and 16 degrees, respectively.
Data source: Prospective study of 30 patients with mild to moderate laxity of the lower face and neck.
Disclosures: Dr. Munavalli reported no funding sources or financial disclosures.

Fractional laser resurfacing plus ALA-PDT upped AK clearance
KISSIMMEE, FLA. – Fractional carbon dioxide laser resurfacing followed by 30 minutes of aminolevulinic acid plus blue light photodynamic therapy cleared 94% of actinic keratoses, significantly more than ALA-PDT alone, according to the findings of a randomized, single-blinded, split-face study of 20 patients.
Laser resurfacing was associated with worse short-term erythema, but erythema resolved in about 7 days and was not associated with other adverse events, Dr. Macrene Alexiades-Armenakas said at the annual meeting of the American Society for Laser Medicine and Surgery.
Historically, two sessions of 20% topical ALA and blue light PDT have yielded actinic keratosis cure rates of 78% to 89%, but only with ALA incubation times of 14-18 hours, said Dr. Alexiades-Armenakas, associate clinical professor at Yale University, New Haven, Conn.
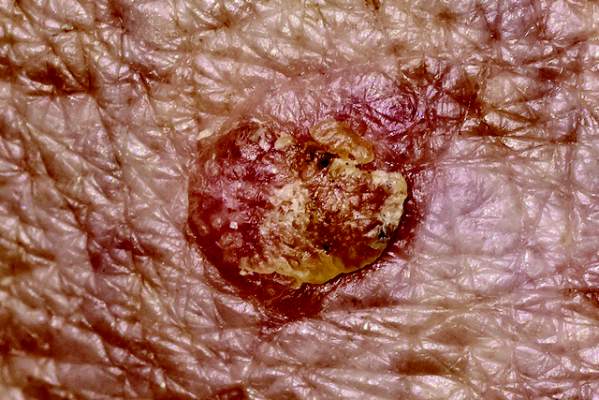
“Increasing drug penetration may serve to enhance PDT efficacy and shorten incubation time,” she said.
To test that hypothesis, she compared the safety and efficacy of 15- and 30-minute incubations of ALA and blue light PDT, with or without CO2 laser resurfacing. After cleaning patients’ faces with acetone wipes and applying a topical anesthetic for 1 hour, she randomly selected one half of each patient’s face for pretreatment with fractional CO2 laser, using settings of 15-28 W, 500 mcm dot spacing, and 600-800 microsecond dwell time.
Next, she applied 5-ALA to the entire face, then performed blue light illumination for 1,000 seconds. Half of the 20 patients were randomly assigned ALA incubation times of 15 minutes, while the other half underwent 30-minute incubations. She rechecked patients at 1 week, 4 weeks, and 8 weeks, and took digital photographs at baseline and at each recheck using identical lighting conditions. A blinded evaluator scored each side of each face, defining clearance as complete regression of actinic keratosis.
At 8 weeks, the rate of complete clearance for the 10 patients who underwent 15-minute ALA incubations was 88% for laser resurfacing followed by ALA-PDT, compared with 74% for ALA-PDT alone (P < .05), Dr. Alexiades-Armenakas reported. Clearance rates for the 30-minute incubation group were 94% for laser followed by ALA-PDT and 82% for ALA-PDT alone (P < .05).
Skin treated only with ALA-PDT developed minimal to moderate erythema that resolved within 5-7 days for all patients, but the laser-resurfaced skin developed “moderate to significant” erythema that resolved within 5-7 days with home care, she said.
Taken together, the results indicate that fractional CO2 laser treatment yields safe and effective clearance of actinic keratoses with “ultra-short” incubation times, Dr. Alexiades-Armenakas said.
Deka manufactures the fractional CO2 laser tested in the study, and DUSA Pharmaceuticals manufactures the blue light PDT device and the ALA product. Dr. Alexiades-Armenakas reported receiving clinical research grants from Deka, DUSA Pharmaceuticals, Alma, and Syneron.
KISSIMMEE, FLA. – Fractional carbon dioxide laser resurfacing followed by 30 minutes of aminolevulinic acid plus blue light photodynamic therapy cleared 94% of actinic keratoses, significantly more than ALA-PDT alone, according to the findings of a randomized, single-blinded, split-face study of 20 patients.
Laser resurfacing was associated with worse short-term erythema, but erythema resolved in about 7 days and was not associated with other adverse events, Dr. Macrene Alexiades-Armenakas said at the annual meeting of the American Society for Laser Medicine and Surgery.
Historically, two sessions of 20% topical ALA and blue light PDT have yielded actinic keratosis cure rates of 78% to 89%, but only with ALA incubation times of 14-18 hours, said Dr. Alexiades-Armenakas, associate clinical professor at Yale University, New Haven, Conn.
“Increasing drug penetration may serve to enhance PDT efficacy and shorten incubation time,” she said.
To test that hypothesis, she compared the safety and efficacy of 15- and 30-minute incubations of ALA and blue light PDT, with or without CO2 laser resurfacing. After cleaning patients’ faces with acetone wipes and applying a topical anesthetic for 1 hour, she randomly selected one half of each patient’s face for pretreatment with fractional CO2 laser, using settings of 15-28 W, 500 mcm dot spacing, and 600-800 microsecond dwell time.
Next, she applied 5-ALA to the entire face, then performed blue light illumination for 1,000 seconds. Half of the 20 patients were randomly assigned ALA incubation times of 15 minutes, while the other half underwent 30-minute incubations. She rechecked patients at 1 week, 4 weeks, and 8 weeks, and took digital photographs at baseline and at each recheck using identical lighting conditions. A blinded evaluator scored each side of each face, defining clearance as complete regression of actinic keratosis.
At 8 weeks, the rate of complete clearance for the 10 patients who underwent 15-minute ALA incubations was 88% for laser resurfacing followed by ALA-PDT, compared with 74% for ALA-PDT alone (P < .05), Dr. Alexiades-Armenakas reported. Clearance rates for the 30-minute incubation group were 94% for laser followed by ALA-PDT and 82% for ALA-PDT alone (P < .05).
Skin treated only with ALA-PDT developed minimal to moderate erythema that resolved within 5-7 days for all patients, but the laser-resurfaced skin developed “moderate to significant” erythema that resolved within 5-7 days with home care, she said.
Taken together, the results indicate that fractional CO2 laser treatment yields safe and effective clearance of actinic keratoses with “ultra-short” incubation times, Dr. Alexiades-Armenakas said.
Deka manufactures the fractional CO2 laser tested in the study, and DUSA Pharmaceuticals manufactures the blue light PDT device and the ALA product. Dr. Alexiades-Armenakas reported receiving clinical research grants from Deka, DUSA Pharmaceuticals, Alma, and Syneron.
KISSIMMEE, FLA. – Fractional carbon dioxide laser resurfacing followed by 30 minutes of aminolevulinic acid plus blue light photodynamic therapy cleared 94% of actinic keratoses, significantly more than ALA-PDT alone, according to the findings of a randomized, single-blinded, split-face study of 20 patients.
Laser resurfacing was associated with worse short-term erythema, but erythema resolved in about 7 days and was not associated with other adverse events, Dr. Macrene Alexiades-Armenakas said at the annual meeting of the American Society for Laser Medicine and Surgery.
Historically, two sessions of 20% topical ALA and blue light PDT have yielded actinic keratosis cure rates of 78% to 89%, but only with ALA incubation times of 14-18 hours, said Dr. Alexiades-Armenakas, associate clinical professor at Yale University, New Haven, Conn.
“Increasing drug penetration may serve to enhance PDT efficacy and shorten incubation time,” she said.
To test that hypothesis, she compared the safety and efficacy of 15- and 30-minute incubations of ALA and blue light PDT, with or without CO2 laser resurfacing. After cleaning patients’ faces with acetone wipes and applying a topical anesthetic for 1 hour, she randomly selected one half of each patient’s face for pretreatment with fractional CO2 laser, using settings of 15-28 W, 500 mcm dot spacing, and 600-800 microsecond dwell time.
Next, she applied 5-ALA to the entire face, then performed blue light illumination for 1,000 seconds. Half of the 20 patients were randomly assigned ALA incubation times of 15 minutes, while the other half underwent 30-minute incubations. She rechecked patients at 1 week, 4 weeks, and 8 weeks, and took digital photographs at baseline and at each recheck using identical lighting conditions. A blinded evaluator scored each side of each face, defining clearance as complete regression of actinic keratosis.
At 8 weeks, the rate of complete clearance for the 10 patients who underwent 15-minute ALA incubations was 88% for laser resurfacing followed by ALA-PDT, compared with 74% for ALA-PDT alone (P < .05), Dr. Alexiades-Armenakas reported. Clearance rates for the 30-minute incubation group were 94% for laser followed by ALA-PDT and 82% for ALA-PDT alone (P < .05).
Skin treated only with ALA-PDT developed minimal to moderate erythema that resolved within 5-7 days for all patients, but the laser-resurfaced skin developed “moderate to significant” erythema that resolved within 5-7 days with home care, she said.
Taken together, the results indicate that fractional CO2 laser treatment yields safe and effective clearance of actinic keratoses with “ultra-short” incubation times, Dr. Alexiades-Armenakas said.
Deka manufactures the fractional CO2 laser tested in the study, and DUSA Pharmaceuticals manufactures the blue light PDT device and the ALA product. Dr. Alexiades-Armenakas reported receiving clinical research grants from Deka, DUSA Pharmaceuticals, Alma, and Syneron.
AT LASER 2015
Key clinical point: Performing fractional CO2 laser resurfacing before aminolevulinic acid blue light photodynamic therapy cleared significantly more actinic keratoses than ALA-PDT alone.
Major finding: For the 30-minute incubation group, laser plus ALA-PDT cleared 94% of actinic keratoses, compared with 82% for ALA-PDT alone.
Data source: A randomized, single-blinded, split-face study of 20 patients.
Disclosures: Deka made the fractional CO2 laser tested in the study and DUSA Pharmaceuticals made the blue light PDT device and the ALA product. Dr. Alexiades-Armenakas reported receiving clinical research grants from Deka, DUSA Pharmaceuticals, Alma, and Syneron.
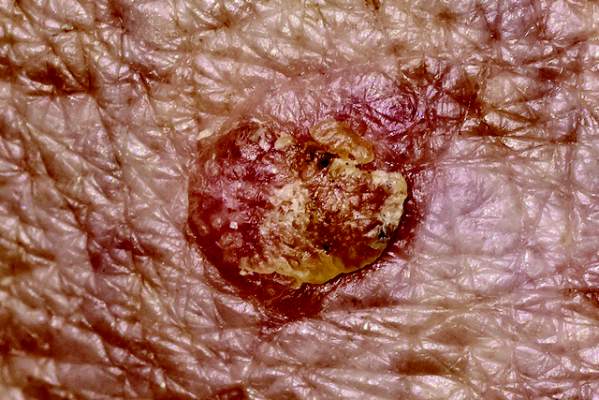
Intradermal ALA-PDT linked to long-term remission in BCC
KISSIMMEE, FLA. – Using a needle-free device to inject nodular basal cell carcinomas with intralesional 5-aminolevulinic acid before photodynamic therapy led to complete, years-long remissions and few side effects in a small case series.
“This approach represents an interesting alternative to Mohs, for sure,” Dr. Daniel Barolet said at the annual meeting of the American Society for Laser Medicine and Surgery. “The secret is in the injector nozzle, which lets you inject with multiple openings to get the best uniformity around the tumor.”
Mohs micrographic surgery remains the standard for basal cell carcinoma (BCC) in high-risk sites, and the number of Mohs surgeries has approximately doubled since 2001, said Dr. Barolet, adjunct professor of dermatology at McGill University in Montreal.
Mohs, however, can cause scarring, and BCCs recur in about 4% of patients. In contrast, photodynamic therapy (PDT) is associated with less scarring and pain, fewer complications, shorter recovery times, and lower costs, although the recurrence rate is about 14%, he noted.
Since PDT alone does not efficiently penetrate thick tumor volumes, it works best with pretreatment using agents such as aminolevulinic acid (ALA).
Using needles to inject the tumor, however, can cause pain, vascular damage, vasoconstriction, deep purpura, necrosis, and infection. “Because of this, no-needle injection is an interesting avenue for PDT,” he noted. Needle-free devices currently are used to inject insulin and to administer some vaccines. They are “virtually painless,” noninvasive, and tissue sparing, he said.
To explore the potential role for needle-free injection in ALA-PDT, Dr. Barolot used a prototype high-speed jet to deliver intralesional 5-ALA in the nodular facial BCCs of four patients. He then performed photoactivation with a red light–emitting diode, with continuous wave at 630 nm, irradiance at 50 mW/cm2, and total fluence 50-100 J/cm2.
Patients had no evidence of clinical or histopathologic recurrence for up to 7 years after treatment, Dr. Barolet reported. They experienced mild crusting at treated sites for up to a week after treatment, but no other adverse effects. Two patients needed a second treatment 2 months after the initial treatment to achieve complete remission. “Excellent cosmesis was obtained,” he added, pointing to before and after photos that showed no evidence of lesions several months after treatment.
Multicenter clinical trials are needed to further evaluate the modality, but the preliminary data suggest that intralesional PDT is a reasonable alternative to Mohs for BCCs in high-risk body sites, as long as lesions are few in number and do not affect large areas of the body, Dr. Barolet said.
The modality is especially well suited to “tricky” areas of the body that are difficult to treat with Mohs, he said.
“Developing a user-friendly, disposable no-needle injector will make it much easier for users,” he added. For low-risk BCCs in low-risk sites, conventional treatments such as surgical excision remain the best option, he said.
Dr. Barolet reported no funding sources for the study and said he had no relevant financial disclosures.
KISSIMMEE, FLA. – Using a needle-free device to inject nodular basal cell carcinomas with intralesional 5-aminolevulinic acid before photodynamic therapy led to complete, years-long remissions and few side effects in a small case series.
“This approach represents an interesting alternative to Mohs, for sure,” Dr. Daniel Barolet said at the annual meeting of the American Society for Laser Medicine and Surgery. “The secret is in the injector nozzle, which lets you inject with multiple openings to get the best uniformity around the tumor.”
Mohs micrographic surgery remains the standard for basal cell carcinoma (BCC) in high-risk sites, and the number of Mohs surgeries has approximately doubled since 2001, said Dr. Barolet, adjunct professor of dermatology at McGill University in Montreal.
Mohs, however, can cause scarring, and BCCs recur in about 4% of patients. In contrast, photodynamic therapy (PDT) is associated with less scarring and pain, fewer complications, shorter recovery times, and lower costs, although the recurrence rate is about 14%, he noted.
Since PDT alone does not efficiently penetrate thick tumor volumes, it works best with pretreatment using agents such as aminolevulinic acid (ALA).
Using needles to inject the tumor, however, can cause pain, vascular damage, vasoconstriction, deep purpura, necrosis, and infection. “Because of this, no-needle injection is an interesting avenue for PDT,” he noted. Needle-free devices currently are used to inject insulin and to administer some vaccines. They are “virtually painless,” noninvasive, and tissue sparing, he said.
To explore the potential role for needle-free injection in ALA-PDT, Dr. Barolot used a prototype high-speed jet to deliver intralesional 5-ALA in the nodular facial BCCs of four patients. He then performed photoactivation with a red light–emitting diode, with continuous wave at 630 nm, irradiance at 50 mW/cm2, and total fluence 50-100 J/cm2.
Patients had no evidence of clinical or histopathologic recurrence for up to 7 years after treatment, Dr. Barolet reported. They experienced mild crusting at treated sites for up to a week after treatment, but no other adverse effects. Two patients needed a second treatment 2 months after the initial treatment to achieve complete remission. “Excellent cosmesis was obtained,” he added, pointing to before and after photos that showed no evidence of lesions several months after treatment.
Multicenter clinical trials are needed to further evaluate the modality, but the preliminary data suggest that intralesional PDT is a reasonable alternative to Mohs for BCCs in high-risk body sites, as long as lesions are few in number and do not affect large areas of the body, Dr. Barolet said.
The modality is especially well suited to “tricky” areas of the body that are difficult to treat with Mohs, he said.
“Developing a user-friendly, disposable no-needle injector will make it much easier for users,” he added. For low-risk BCCs in low-risk sites, conventional treatments such as surgical excision remain the best option, he said.
Dr. Barolet reported no funding sources for the study and said he had no relevant financial disclosures.
KISSIMMEE, FLA. – Using a needle-free device to inject nodular basal cell carcinomas with intralesional 5-aminolevulinic acid before photodynamic therapy led to complete, years-long remissions and few side effects in a small case series.
“This approach represents an interesting alternative to Mohs, for sure,” Dr. Daniel Barolet said at the annual meeting of the American Society for Laser Medicine and Surgery. “The secret is in the injector nozzle, which lets you inject with multiple openings to get the best uniformity around the tumor.”
Mohs micrographic surgery remains the standard for basal cell carcinoma (BCC) in high-risk sites, and the number of Mohs surgeries has approximately doubled since 2001, said Dr. Barolet, adjunct professor of dermatology at McGill University in Montreal.
Mohs, however, can cause scarring, and BCCs recur in about 4% of patients. In contrast, photodynamic therapy (PDT) is associated with less scarring and pain, fewer complications, shorter recovery times, and lower costs, although the recurrence rate is about 14%, he noted.
Since PDT alone does not efficiently penetrate thick tumor volumes, it works best with pretreatment using agents such as aminolevulinic acid (ALA).
Using needles to inject the tumor, however, can cause pain, vascular damage, vasoconstriction, deep purpura, necrosis, and infection. “Because of this, no-needle injection is an interesting avenue for PDT,” he noted. Needle-free devices currently are used to inject insulin and to administer some vaccines. They are “virtually painless,” noninvasive, and tissue sparing, he said.
To explore the potential role for needle-free injection in ALA-PDT, Dr. Barolot used a prototype high-speed jet to deliver intralesional 5-ALA in the nodular facial BCCs of four patients. He then performed photoactivation with a red light–emitting diode, with continuous wave at 630 nm, irradiance at 50 mW/cm2, and total fluence 50-100 J/cm2.
Patients had no evidence of clinical or histopathologic recurrence for up to 7 years after treatment, Dr. Barolet reported. They experienced mild crusting at treated sites for up to a week after treatment, but no other adverse effects. Two patients needed a second treatment 2 months after the initial treatment to achieve complete remission. “Excellent cosmesis was obtained,” he added, pointing to before and after photos that showed no evidence of lesions several months after treatment.
Multicenter clinical trials are needed to further evaluate the modality, but the preliminary data suggest that intralesional PDT is a reasonable alternative to Mohs for BCCs in high-risk body sites, as long as lesions are few in number and do not affect large areas of the body, Dr. Barolet said.
The modality is especially well suited to “tricky” areas of the body that are difficult to treat with Mohs, he said.
“Developing a user-friendly, disposable no-needle injector will make it much easier for users,” he added. For low-risk BCCs in low-risk sites, conventional treatments such as surgical excision remain the best option, he said.
Dr. Barolet reported no funding sources for the study and said he had no relevant financial disclosures.
AT LASER 2015
Key clinical point: Intralesional 5-ALA-PDT is a potential alternative to Mohs micrographic surgery for treating basal cell carcinomas in high-risk sites.
Major finding: Four treated patients experienced resolution of recurrent basal cell carcinomas for up to 7 years.
Data source: Series of four cases of recurrent nodular facial basal cell carcinomas.
Disclosures: Dr. Barolet reported no funding sources and declared no relevant financial disclosures.
Skin patch hastened Q-switched laser tattoo removal
KISSIMMEE, FLA. – A perfluorodecalin-infused patch lessened reactive whitening and accelerated clearing in 65% of tattoos treated with 755-nm Q-switched lasers in a single-center, split-tattoo study.
“Most subjects showed obvious accelerated clearing on the patch side of the tattoo, and preferred the patch-treated side compared with the control side,” Dr. Brian Biesman said at the annual meeting of the American Society for Laser Medicine and Surgery. “Despite the small number of patients, the study was powered sufficiently to address our qualitative question: Was there an enhanced rate of clearance relative to control when the PFD [perfluorodecalin] patch is used?”

Passing a laser across a tattoo triggers an immediate whitening reaction that blocks light and takes about 20 minutes to resolve, said Dr. Biesman, who is clinical assistant professor at Vanderbilt University and director of the Nashville Centre for Laser and Facial Surgery. Removing tattoos with Q-switched lasers can take months to years because it involves making a single pass each month. A past study showed that topical PFD resolved whitening in seconds, allowing operators to make multiple passes in 5 minutes. but the patch seems to have additional dermoprotective properties that enable patients to tolerate higher fluence as well as multiple passes, Dr. Biesman said.
The study included 17 patients with Fitzpatrick skin types I through III who had previously untreated, dark blue or black ink tattoos measuring less than 100 cm2 in area. Patients with suntans, blood-borne diseases, oral retinoid exposure in the past 12 months, or lidocaine allergies were excluded, he said. All patients underwent monthly treatments with a conventional Q-switched alexandrite laser after pretreatment with topical lidocaine. The control (uncovered) half of each tattoo received a single laser pass, while the half covered by the patch received the maximum tolerable fluence and number of passes in 5 minutes.
The PFD patch was associated with substantially faster clearance, compared with the control, in 11 of 17 patients (65%). Patients tolerated 1.5 to 1.8 times greater fluence and about four passes per session on the patch side. They also developed no serious or unexpected side effects, said Dr. Biesman.
Responses to the patch varied widely, however, ranging from no visible improvement after many sessions to more than 80% greater clearance after two sessions, compared with the control side. Larger studies would be needed to examine predictors of success, he added, noting that the composition of tattoo ink affects response, and that clinicians rarely, if ever, know which inks were used.
It also remains unclear whether faster clearing was the result of multiple passes or higher fluence. The PFD patch reduces scatter in the dermis and epidermis, which could facilitate use of higher fluence, he said.
On April 20, the Food and Drug Administration cleared the PFD patch for use as an accessory to tattoo removal with a 755-nm Q-switched alexandrite laser in Fitzpatrick skin types I through III under the agency’s 510(k) medical device regulatory process, according to Dr. Biesman.
ON Light Sciences manufactures the patch. Dr. Biesman reported receiving grant support and holding ownership interest in ON Light Sciences and also reported financial relationships with a number of other pharmaceutical and device companies.
KISSIMMEE, FLA. – A perfluorodecalin-infused patch lessened reactive whitening and accelerated clearing in 65% of tattoos treated with 755-nm Q-switched lasers in a single-center, split-tattoo study.
“Most subjects showed obvious accelerated clearing on the patch side of the tattoo, and preferred the patch-treated side compared with the control side,” Dr. Brian Biesman said at the annual meeting of the American Society for Laser Medicine and Surgery. “Despite the small number of patients, the study was powered sufficiently to address our qualitative question: Was there an enhanced rate of clearance relative to control when the PFD [perfluorodecalin] patch is used?”
Passing a laser across a tattoo triggers an immediate whitening reaction that blocks light and takes about 20 minutes to resolve, said Dr. Biesman, who is clinical assistant professor at Vanderbilt University and director of the Nashville Centre for Laser and Facial Surgery. Removing tattoos with Q-switched lasers can take months to years because it involves making a single pass each month. A past study showed that topical PFD resolved whitening in seconds, allowing operators to make multiple passes in 5 minutes. but the patch seems to have additional dermoprotective properties that enable patients to tolerate higher fluence as well as multiple passes, Dr. Biesman said.
The study included 17 patients with Fitzpatrick skin types I through III who had previously untreated, dark blue or black ink tattoos measuring less than 100 cm2 in area. Patients with suntans, blood-borne diseases, oral retinoid exposure in the past 12 months, or lidocaine allergies were excluded, he said. All patients underwent monthly treatments with a conventional Q-switched alexandrite laser after pretreatment with topical lidocaine. The control (uncovered) half of each tattoo received a single laser pass, while the half covered by the patch received the maximum tolerable fluence and number of passes in 5 minutes.
The PFD patch was associated with substantially faster clearance, compared with the control, in 11 of 17 patients (65%). Patients tolerated 1.5 to 1.8 times greater fluence and about four passes per session on the patch side. They also developed no serious or unexpected side effects, said Dr. Biesman.
Responses to the patch varied widely, however, ranging from no visible improvement after many sessions to more than 80% greater clearance after two sessions, compared with the control side. Larger studies would be needed to examine predictors of success, he added, noting that the composition of tattoo ink affects response, and that clinicians rarely, if ever, know which inks were used.
It also remains unclear whether faster clearing was the result of multiple passes or higher fluence. The PFD patch reduces scatter in the dermis and epidermis, which could facilitate use of higher fluence, he said.
On April 20, the Food and Drug Administration cleared the PFD patch for use as an accessory to tattoo removal with a 755-nm Q-switched alexandrite laser in Fitzpatrick skin types I through III under the agency’s 510(k) medical device regulatory process, according to Dr. Biesman.
ON Light Sciences manufactures the patch. Dr. Biesman reported receiving grant support and holding ownership interest in ON Light Sciences and also reported financial relationships with a number of other pharmaceutical and device companies.
KISSIMMEE, FLA. – A perfluorodecalin-infused patch lessened reactive whitening and accelerated clearing in 65% of tattoos treated with 755-nm Q-switched lasers in a single-center, split-tattoo study.
“Most subjects showed obvious accelerated clearing on the patch side of the tattoo, and preferred the patch-treated side compared with the control side,” Dr. Brian Biesman said at the annual meeting of the American Society for Laser Medicine and Surgery. “Despite the small number of patients, the study was powered sufficiently to address our qualitative question: Was there an enhanced rate of clearance relative to control when the PFD [perfluorodecalin] patch is used?”
Passing a laser across a tattoo triggers an immediate whitening reaction that blocks light and takes about 20 minutes to resolve, said Dr. Biesman, who is clinical assistant professor at Vanderbilt University and director of the Nashville Centre for Laser and Facial Surgery. Removing tattoos with Q-switched lasers can take months to years because it involves making a single pass each month. A past study showed that topical PFD resolved whitening in seconds, allowing operators to make multiple passes in 5 minutes. but the patch seems to have additional dermoprotective properties that enable patients to tolerate higher fluence as well as multiple passes, Dr. Biesman said.
The study included 17 patients with Fitzpatrick skin types I through III who had previously untreated, dark blue or black ink tattoos measuring less than 100 cm2 in area. Patients with suntans, blood-borne diseases, oral retinoid exposure in the past 12 months, or lidocaine allergies were excluded, he said. All patients underwent monthly treatments with a conventional Q-switched alexandrite laser after pretreatment with topical lidocaine. The control (uncovered) half of each tattoo received a single laser pass, while the half covered by the patch received the maximum tolerable fluence and number of passes in 5 minutes.
The PFD patch was associated with substantially faster clearance, compared with the control, in 11 of 17 patients (65%). Patients tolerated 1.5 to 1.8 times greater fluence and about four passes per session on the patch side. They also developed no serious or unexpected side effects, said Dr. Biesman.
Responses to the patch varied widely, however, ranging from no visible improvement after many sessions to more than 80% greater clearance after two sessions, compared with the control side. Larger studies would be needed to examine predictors of success, he added, noting that the composition of tattoo ink affects response, and that clinicians rarely, if ever, know which inks were used.
It also remains unclear whether faster clearing was the result of multiple passes or higher fluence. The PFD patch reduces scatter in the dermis and epidermis, which could facilitate use of higher fluence, he said.
On April 20, the Food and Drug Administration cleared the PFD patch for use as an accessory to tattoo removal with a 755-nm Q-switched alexandrite laser in Fitzpatrick skin types I through III under the agency’s 510(k) medical device regulatory process, according to Dr. Biesman.
ON Light Sciences manufactures the patch. Dr. Biesman reported receiving grant support and holding ownership interest in ON Light Sciences and also reported financial relationships with a number of other pharmaceutical and device companies.
AT LASER 2015
Key clinical point: A perfluorodecalin-infused skin patch reduced whitening and facilitated clearance of tattoos treated with 755-nm Q-switched lasers.
Major finding: Nearly two-thirds (11 of 17) of tattoos cleared more rapidly with the PFD patch than without.
Data source: Split-tattoo study of 17 patients with black or blue ink tattoos.
Disclosures: ON Light Sciences makes the patch tested in the study. Dr. Biesman disclosed grant support and ownership interest in ON Light Sciences and financial relationships with a number of other pharmaceutical and device companies.

Simplified PESI identified low-risk pulmonary embolism
A simplified version of the Pulmonary Embolism Severity Index identified patients with acute pulmonary embolism who were at low risk of adverse events and might be suitable for outpatient care, investigators reported in Academic Emergency Medicine.
“Although guidelines, such as those from the American College of Chest Physicians, recommend outpatient treatment for selected PE patients at low risk of recurrence, existing evidence for the outpatient management of patients with PE is derived from small cohorts of patients from outside the United States,” said Dr. Gregory J. Fermann of the University of Cincinnati department of emergency medicine and his associates.
“The results of this analysis provide further support that risk stratification of PE patients may allow a cohort of low-risk patients to be treated in a clinical decision unit or by a closely monitored outpatient strategy. Such an approach might relieve some of the burden placed on the emergency department (Acad. Emerg. Med. 2015;22:299-307).”
The PESI has been shown to identify patients at increased risk of death and adverse outcome events after acute PE. The simplified PESI has 6 of the PESI’s 11 variables, but remains accurate in assessing PE severity, the researchers said. They carried out a post hoc analysis of simplified PESI scores and outcomes among 4,831 acute PE patients from the phase III Einstein PE study, in which rivaroxaban was found noninferior to an enoxaparin–vitamin K antagonist combination in terms of the risk of recurrent venous thromboembolism and clinically important bleeding events (N. Engl. J. Med. 2012;366:1287-97).
Roughly half (53.6%) of the patients had a score of 0, one-third (36.7%) had a score of 1, and 9.7% had a score of 2 or 3, the researchers reported. Higher simplified PESI scores were associated with increased risk of almost all adverse outcomes measured, including recurrent VTE, fatal PE, all-cause mortality, and major bleeding. Patients with scores of 0 or 1 had low rates of major adverse events during the first 30 days of treatment, regardless of which protocol they received.
However, the incidence of major bleeds up to 30 days was lower in the rivaroxaban group than in the standard treatment group, especially if patients’ simplified PESI scores were greater than 0. Scores of 2 or 3 were associated with greater risk of recurrent VTE, fatal PE, all-cause mortality, and major bleeding at all time points and in both treatment groups.
Bayer HealthCare Pharmaceuticals and Janssen Research & Development funded the study. Dr. Fermann reported an advisory relationship with Janssen and research funding from Cardiorentis, Trevena, Novartis, Siemens, and Pfizer. Two coauthors reported employment with Bayer, and two other coauthors reported financial and advisory relationships with several other pharmaceutical companies.
A simplified version of the Pulmonary Embolism Severity Index identified patients with acute pulmonary embolism who were at low risk of adverse events and might be suitable for outpatient care, investigators reported in Academic Emergency Medicine.
“Although guidelines, such as those from the American College of Chest Physicians, recommend outpatient treatment for selected PE patients at low risk of recurrence, existing evidence for the outpatient management of patients with PE is derived from small cohorts of patients from outside the United States,” said Dr. Gregory J. Fermann of the University of Cincinnati department of emergency medicine and his associates.
“The results of this analysis provide further support that risk stratification of PE patients may allow a cohort of low-risk patients to be treated in a clinical decision unit or by a closely monitored outpatient strategy. Such an approach might relieve some of the burden placed on the emergency department (Acad. Emerg. Med. 2015;22:299-307).”
The PESI has been shown to identify patients at increased risk of death and adverse outcome events after acute PE. The simplified PESI has 6 of the PESI’s 11 variables, but remains accurate in assessing PE severity, the researchers said. They carried out a post hoc analysis of simplified PESI scores and outcomes among 4,831 acute PE patients from the phase III Einstein PE study, in which rivaroxaban was found noninferior to an enoxaparin–vitamin K antagonist combination in terms of the risk of recurrent venous thromboembolism and clinically important bleeding events (N. Engl. J. Med. 2012;366:1287-97).
Roughly half (53.6%) of the patients had a score of 0, one-third (36.7%) had a score of 1, and 9.7% had a score of 2 or 3, the researchers reported. Higher simplified PESI scores were associated with increased risk of almost all adverse outcomes measured, including recurrent VTE, fatal PE, all-cause mortality, and major bleeding. Patients with scores of 0 or 1 had low rates of major adverse events during the first 30 days of treatment, regardless of which protocol they received.
However, the incidence of major bleeds up to 30 days was lower in the rivaroxaban group than in the standard treatment group, especially if patients’ simplified PESI scores were greater than 0. Scores of 2 or 3 were associated with greater risk of recurrent VTE, fatal PE, all-cause mortality, and major bleeding at all time points and in both treatment groups.
Bayer HealthCare Pharmaceuticals and Janssen Research & Development funded the study. Dr. Fermann reported an advisory relationship with Janssen and research funding from Cardiorentis, Trevena, Novartis, Siemens, and Pfizer. Two coauthors reported employment with Bayer, and two other coauthors reported financial and advisory relationships with several other pharmaceutical companies.
A simplified version of the Pulmonary Embolism Severity Index identified patients with acute pulmonary embolism who were at low risk of adverse events and might be suitable for outpatient care, investigators reported in Academic Emergency Medicine.
“Although guidelines, such as those from the American College of Chest Physicians, recommend outpatient treatment for selected PE patients at low risk of recurrence, existing evidence for the outpatient management of patients with PE is derived from small cohorts of patients from outside the United States,” said Dr. Gregory J. Fermann of the University of Cincinnati department of emergency medicine and his associates.
“The results of this analysis provide further support that risk stratification of PE patients may allow a cohort of low-risk patients to be treated in a clinical decision unit or by a closely monitored outpatient strategy. Such an approach might relieve some of the burden placed on the emergency department (Acad. Emerg. Med. 2015;22:299-307).”
The PESI has been shown to identify patients at increased risk of death and adverse outcome events after acute PE. The simplified PESI has 6 of the PESI’s 11 variables, but remains accurate in assessing PE severity, the researchers said. They carried out a post hoc analysis of simplified PESI scores and outcomes among 4,831 acute PE patients from the phase III Einstein PE study, in which rivaroxaban was found noninferior to an enoxaparin–vitamin K antagonist combination in terms of the risk of recurrent venous thromboembolism and clinically important bleeding events (N. Engl. J. Med. 2012;366:1287-97).
Roughly half (53.6%) of the patients had a score of 0, one-third (36.7%) had a score of 1, and 9.7% had a score of 2 or 3, the researchers reported. Higher simplified PESI scores were associated with increased risk of almost all adverse outcomes measured, including recurrent VTE, fatal PE, all-cause mortality, and major bleeding. Patients with scores of 0 or 1 had low rates of major adverse events during the first 30 days of treatment, regardless of which protocol they received.
However, the incidence of major bleeds up to 30 days was lower in the rivaroxaban group than in the standard treatment group, especially if patients’ simplified PESI scores were greater than 0. Scores of 2 or 3 were associated with greater risk of recurrent VTE, fatal PE, all-cause mortality, and major bleeding at all time points and in both treatment groups.
Bayer HealthCare Pharmaceuticals and Janssen Research & Development funded the study. Dr. Fermann reported an advisory relationship with Janssen and research funding from Cardiorentis, Trevena, Novartis, Siemens, and Pfizer. Two coauthors reported employment with Bayer, and two other coauthors reported financial and advisory relationships with several other pharmaceutical companies.
Key clinical point: The simplified version of the PESI identified low-risk pulmonary embolism patients.
Major finding: Patients with scores of 0 or 1 had low rates of major adverse events during the first 30 days, regardless of which treatment they received.
Data source: Post hoc analysis of simplified PESI scores and outcomes among 4,831 patients with acute pulmonary embolism who received either rivaroxaban or an enoxaparin–vitamin K antagonist combination.
Disclosures: Bayer HealthCare Pharmaceuticals and Janssen Research & Development funded the study. Dr. Fermann reported an advisory relationship with Janssen and research funding from Cardiorentis, Trevena, Novartis, Siemens, and Pfizer. Two coauthors reported employment with Bayer, and two other coauthors reported relationships with several other pharmaceutical companies.
Angiojet system found safe, effective in lower-extremity deep venous thrombosis
More than 80% of patients with lower-extremity deep venous thrombosis who underwent endovascular treatment with the Angiojet rheolytic thrombectomy system were free of rethrombosis a year later, based on final results from the PEARL registry study.
Almost 4% of patients had bleeding events after treatment, but none of these events was tied to use of the Angiojet system, reported Dr. Mark Garcia of Mount Sinai Medical Center in New York and his associates.

“PEARL registry data demonstrate that rheolytic pharmacomechanical catheter-directed thrombolysis treatment of deep venous thrombosis is safe and effective, and can potentially reduce the need for concomitant catheter-directed thrombolysis and intensive care,” the researchers wrote.
The rates of venous thromboembolism are rising, and the number of affected adults is expected to double in the next 40 years as the population ages and experiences recurrent episodes. Lower-extremity deep venous thrombosis (DVT) is especially likely to recur or to develop complications such as pulmonary embolism and post-thrombotic syndrome. For this reason, practice guidelines now advocate early removal of iliofemoral clots if patients are functional, have a good life expectancy, are within 14 days of symptom onset, and are unlikely to develop bleeding complications. Options for clot removal include catheter-directed thrombolysis (CDT) or pharmacomechanical CDT, which combines catheterization with intervention to break up or aspirate the clot while infusing it with a thrombolytic drug, said the investigators (J. Vasc. Interv. Radiol. 2015 Mar. 27 [doi:10.1016/j.jvir.2015.01.036]).
The PEARL registry study prospectively followed patients who underwent PCDT for arterial or venous thrombosis with the AngioJet thrombectomy catheter system. Researchers analyzed data from 329 patients with severe lower-extremity DVT who were treated at 32 sites in the United States and Europe between 2007 and 2013. Two-thirds of the patients underwent Angiojet thrombectomy within 2 weeks of symptom onset, while 19% were treated within 15 to 30 days and 14% were treated for chronic lesions. A total of 57% of patients were men, and the average age at onset was 52 years. The cohort’s most prevalent risk factors for thrombosis included a history of DVT, preexisting caval filters, past or current tobacco use, and prior pulmonary embolism, the investigators reported.
Grade III (100%) clot removal was possible without needing to use CDT in 39% of patients. Most of these patients underwent PCDT alone (lasting a median of 2 hours), while the rest underwent rheolytic thrombectomy without a lytic agent (median, 1.4 hours). However, just over half of patients underwent PCDT and catheter-directed thrombolysis, lasting a median of 22 hours, and 9% underwent rheolytic thrombectomy with CDT (median, 41 hours). About three-quarters of patients had procedures lasting under 24 hours, and about one in three were done within 6 hours. Also, 86% of procedures required no more than two catheter laboratory sessions.
Three months after treatment, 94% of patients were free from rethrombosis, and 87% and 83% of the cohort remained so at 6 and 12 months, respectively, the researchers added. Even patients with chronic thrombi improved so much on the 12-item Short-Form Health Survey that their scores approximated population norms with a year of treatment, they said.
A total of nine patients (2.7%) had adverse events possibly related to treatment, including one case of acute renal failure, said the investigators. Clinicians should follow recommendations for hydration and limit run time to four minutes in a free-flowing vessel to prevent that outcome, they added.
Dr. Garcia and his associates reported being paid consultants for Boston Scientific, which makes the Angiojet thrombectomy catheter system and funded the study. Dr. Garcia also reported grant funds and consulting fees from BTG/EKOS and Cook. Four coauthors reported receiving consulting fees from Cordis, Cook, Medtronic, AstraZeneca, and Covidien.
More than 80% of patients with lower-extremity deep venous thrombosis who underwent endovascular treatment with the Angiojet rheolytic thrombectomy system were free of rethrombosis a year later, based on final results from the PEARL registry study.
Almost 4% of patients had bleeding events after treatment, but none of these events was tied to use of the Angiojet system, reported Dr. Mark Garcia of Mount Sinai Medical Center in New York and his associates.
“PEARL registry data demonstrate that rheolytic pharmacomechanical catheter-directed thrombolysis treatment of deep venous thrombosis is safe and effective, and can potentially reduce the need for concomitant catheter-directed thrombolysis and intensive care,” the researchers wrote.
The rates of venous thromboembolism are rising, and the number of affected adults is expected to double in the next 40 years as the population ages and experiences recurrent episodes. Lower-extremity deep venous thrombosis (DVT) is especially likely to recur or to develop complications such as pulmonary embolism and post-thrombotic syndrome. For this reason, practice guidelines now advocate early removal of iliofemoral clots if patients are functional, have a good life expectancy, are within 14 days of symptom onset, and are unlikely to develop bleeding complications. Options for clot removal include catheter-directed thrombolysis (CDT) or pharmacomechanical CDT, which combines catheterization with intervention to break up or aspirate the clot while infusing it with a thrombolytic drug, said the investigators (J. Vasc. Interv. Radiol. 2015 Mar. 27 [doi:10.1016/j.jvir.2015.01.036]).
The PEARL registry study prospectively followed patients who underwent PCDT for arterial or venous thrombosis with the AngioJet thrombectomy catheter system. Researchers analyzed data from 329 patients with severe lower-extremity DVT who were treated at 32 sites in the United States and Europe between 2007 and 2013. Two-thirds of the patients underwent Angiojet thrombectomy within 2 weeks of symptom onset, while 19% were treated within 15 to 30 days and 14% were treated for chronic lesions. A total of 57% of patients were men, and the average age at onset was 52 years. The cohort’s most prevalent risk factors for thrombosis included a history of DVT, preexisting caval filters, past or current tobacco use, and prior pulmonary embolism, the investigators reported.
Grade III (100%) clot removal was possible without needing to use CDT in 39% of patients. Most of these patients underwent PCDT alone (lasting a median of 2 hours), while the rest underwent rheolytic thrombectomy without a lytic agent (median, 1.4 hours). However, just over half of patients underwent PCDT and catheter-directed thrombolysis, lasting a median of 22 hours, and 9% underwent rheolytic thrombectomy with CDT (median, 41 hours). About three-quarters of patients had procedures lasting under 24 hours, and about one in three were done within 6 hours. Also, 86% of procedures required no more than two catheter laboratory sessions.
Three months after treatment, 94% of patients were free from rethrombosis, and 87% and 83% of the cohort remained so at 6 and 12 months, respectively, the researchers added. Even patients with chronic thrombi improved so much on the 12-item Short-Form Health Survey that their scores approximated population norms with a year of treatment, they said.
A total of nine patients (2.7%) had adverse events possibly related to treatment, including one case of acute renal failure, said the investigators. Clinicians should follow recommendations for hydration and limit run time to four minutes in a free-flowing vessel to prevent that outcome, they added.
Dr. Garcia and his associates reported being paid consultants for Boston Scientific, which makes the Angiojet thrombectomy catheter system and funded the study. Dr. Garcia also reported grant funds and consulting fees from BTG/EKOS and Cook. Four coauthors reported receiving consulting fees from Cordis, Cook, Medtronic, AstraZeneca, and Covidien.
More than 80% of patients with lower-extremity deep venous thrombosis who underwent endovascular treatment with the Angiojet rheolytic thrombectomy system were free of rethrombosis a year later, based on final results from the PEARL registry study.
Almost 4% of patients had bleeding events after treatment, but none of these events was tied to use of the Angiojet system, reported Dr. Mark Garcia of Mount Sinai Medical Center in New York and his associates.
“PEARL registry data demonstrate that rheolytic pharmacomechanical catheter-directed thrombolysis treatment of deep venous thrombosis is safe and effective, and can potentially reduce the need for concomitant catheter-directed thrombolysis and intensive care,” the researchers wrote.
The rates of venous thromboembolism are rising, and the number of affected adults is expected to double in the next 40 years as the population ages and experiences recurrent episodes. Lower-extremity deep venous thrombosis (DVT) is especially likely to recur or to develop complications such as pulmonary embolism and post-thrombotic syndrome. For this reason, practice guidelines now advocate early removal of iliofemoral clots if patients are functional, have a good life expectancy, are within 14 days of symptom onset, and are unlikely to develop bleeding complications. Options for clot removal include catheter-directed thrombolysis (CDT) or pharmacomechanical CDT, which combines catheterization with intervention to break up or aspirate the clot while infusing it with a thrombolytic drug, said the investigators (J. Vasc. Interv. Radiol. 2015 Mar. 27 [doi:10.1016/j.jvir.2015.01.036]).
The PEARL registry study prospectively followed patients who underwent PCDT for arterial or venous thrombosis with the AngioJet thrombectomy catheter system. Researchers analyzed data from 329 patients with severe lower-extremity DVT who were treated at 32 sites in the United States and Europe between 2007 and 2013. Two-thirds of the patients underwent Angiojet thrombectomy within 2 weeks of symptom onset, while 19% were treated within 15 to 30 days and 14% were treated for chronic lesions. A total of 57% of patients were men, and the average age at onset was 52 years. The cohort’s most prevalent risk factors for thrombosis included a history of DVT, preexisting caval filters, past or current tobacco use, and prior pulmonary embolism, the investigators reported.
Grade III (100%) clot removal was possible without needing to use CDT in 39% of patients. Most of these patients underwent PCDT alone (lasting a median of 2 hours), while the rest underwent rheolytic thrombectomy without a lytic agent (median, 1.4 hours). However, just over half of patients underwent PCDT and catheter-directed thrombolysis, lasting a median of 22 hours, and 9% underwent rheolytic thrombectomy with CDT (median, 41 hours). About three-quarters of patients had procedures lasting under 24 hours, and about one in three were done within 6 hours. Also, 86% of procedures required no more than two catheter laboratory sessions.
Three months after treatment, 94% of patients were free from rethrombosis, and 87% and 83% of the cohort remained so at 6 and 12 months, respectively, the researchers added. Even patients with chronic thrombi improved so much on the 12-item Short-Form Health Survey that their scores approximated population norms with a year of treatment, they said.
A total of nine patients (2.7%) had adverse events possibly related to treatment, including one case of acute renal failure, said the investigators. Clinicians should follow recommendations for hydration and limit run time to four minutes in a free-flowing vessel to prevent that outcome, they added.
Dr. Garcia and his associates reported being paid consultants for Boston Scientific, which makes the Angiojet thrombectomy catheter system and funded the study. Dr. Garcia also reported grant funds and consulting fees from BTG/EKOS and Cook. Four coauthors reported receiving consulting fees from Cordis, Cook, Medtronic, AstraZeneca, and Covidien.
FROM THE JOURNAL OF VASCULAR INTERVENTIONAL RADIOLOGY
Key clinical point: Endovascular treatment with the Angiojet rheolytic thrombectomy systemwas found safe and effective for lower-extremity deep venous thrombosis.
Major finding: 83% of patients were free of rethrombosis a year later, and only one suffered a serious adverse event (acute renal failure).
Data source: A multicenter prospective registry study of 329 patients who underwent Angiojet thrombectomy with or without catheter-directed thrombolysis.
Disclosures: Dr. Garcia and his associates reported being paid consultants for Boston Scientific, which makes the Angiojet thrombectomy catheter system and funded the study. Dr. Garcia also reported grant funds and consulting fees from BTG/EKOS and Cook. Four coauthors reported receiving consulting fees from Cordis, Cook, Medtronic, AstraZeneca, and Covidien.
Early goal-directed therapy did not improve septic shock outcomes
For patients in early septic shock, goal-directed therapy based on continuously monitoring central venous oxygen saturation did not improve outcomes compared with standard care, according to a multicenter randomized trial reported in the New England Journal of Medicine and at the annual meeting of the International Society on Intensive Care and Emergency Medicine.
All-cause mortality at 90 days was 29% for both study arms, and costs of care were similar, reported Paul R. Mouncey of the Intensive Care National Audit and Research Centre, London, and his associates. The trial is the last of three planned studies of early, goal-directed therapy (EGDT), “all of which showed that EGDT was not superior to usual care,” the investigators wrote (N. Engl. J. Med. 2015 Mar. 17 [doi:10.1056/NEJMoa1500896]).
Support for EGDT originated mainly from a single-center, proof-of-concept study in 2001 by Dr. Emanuel Rivers and associates, who reported lower hospital mortality and length of stay when patients in early septic shock received 6 hours of treatment aimed at optimizing oxygen transport (N. Engl. J. Med. 2001;345:1368-77).
The protocol called for continuously monitoring central venous pressure, mean arterial pressure, and central venous oxygen saturation to guide use of intravenous fluids, vasoactive drugs, and red-cell transfusions. But uptake of EGDT has been limited, and clinicians have been concerned about the validity of the results and the complexity, risks, and costs of implementing EGDT, Mr. Mouncey and his associates said.
For the study, they randomized 1,260 patients with signs of early septic shock to either EGDT or standard care. All patients received intravenous antibiotics and adequate fluid resuscitation. The EGDT group also underwent a 6-hour resuscitation protocol that included monitoring central venous oxygen saturation, with additional treatment decisions left up to the treating clinician.
In all, 29.5% of patients who received EGDT died within 90 days, as did 29.2% of patients who received standard care (P = .90). The EGDT group received more intravenous fluids, vasoactive drugs, and red-cell transfusions and had significantly worse organ-failure scores, more days of advanced cardiovascular support, and longer ICU stays, but had no improvement in health-related quality of life or other secondary outcomes, the researchers reported.
“On average, EGDT increased costs, and the probability that it was cost-effective was below 20%,” they wrote. “Our results suggest that techniques used in usual resuscitation have evolved over the 15 years since the landmark study by Rivers et al.”
The United Kingdom National Institute for Health Research health technology assessment program funded the study. Nine coauthors reported receiving grants from the NIHR. One coauthor reported grants from ImaCor and Cheetah Medical. The other authors declared no relevant conflicts of interest.
For patients in early septic shock, goal-directed therapy based on continuously monitoring central venous oxygen saturation did not improve outcomes compared with standard care, according to a multicenter randomized trial reported in the New England Journal of Medicine and at the annual meeting of the International Society on Intensive Care and Emergency Medicine.
All-cause mortality at 90 days was 29% for both study arms, and costs of care were similar, reported Paul R. Mouncey of the Intensive Care National Audit and Research Centre, London, and his associates. The trial is the last of three planned studies of early, goal-directed therapy (EGDT), “all of which showed that EGDT was not superior to usual care,” the investigators wrote (N. Engl. J. Med. 2015 Mar. 17 [doi:10.1056/NEJMoa1500896]).
Support for EGDT originated mainly from a single-center, proof-of-concept study in 2001 by Dr. Emanuel Rivers and associates, who reported lower hospital mortality and length of stay when patients in early septic shock received 6 hours of treatment aimed at optimizing oxygen transport (N. Engl. J. Med. 2001;345:1368-77).
The protocol called for continuously monitoring central venous pressure, mean arterial pressure, and central venous oxygen saturation to guide use of intravenous fluids, vasoactive drugs, and red-cell transfusions. But uptake of EGDT has been limited, and clinicians have been concerned about the validity of the results and the complexity, risks, and costs of implementing EGDT, Mr. Mouncey and his associates said.
For the study, they randomized 1,260 patients with signs of early septic shock to either EGDT or standard care. All patients received intravenous antibiotics and adequate fluid resuscitation. The EGDT group also underwent a 6-hour resuscitation protocol that included monitoring central venous oxygen saturation, with additional treatment decisions left up to the treating clinician.
In all, 29.5% of patients who received EGDT died within 90 days, as did 29.2% of patients who received standard care (P = .90). The EGDT group received more intravenous fluids, vasoactive drugs, and red-cell transfusions and had significantly worse organ-failure scores, more days of advanced cardiovascular support, and longer ICU stays, but had no improvement in health-related quality of life or other secondary outcomes, the researchers reported.
“On average, EGDT increased costs, and the probability that it was cost-effective was below 20%,” they wrote. “Our results suggest that techniques used in usual resuscitation have evolved over the 15 years since the landmark study by Rivers et al.”
The United Kingdom National Institute for Health Research health technology assessment program funded the study. Nine coauthors reported receiving grants from the NIHR. One coauthor reported grants from ImaCor and Cheetah Medical. The other authors declared no relevant conflicts of interest.
For patients in early septic shock, goal-directed therapy based on continuously monitoring central venous oxygen saturation did not improve outcomes compared with standard care, according to a multicenter randomized trial reported in the New England Journal of Medicine and at the annual meeting of the International Society on Intensive Care and Emergency Medicine.
All-cause mortality at 90 days was 29% for both study arms, and costs of care were similar, reported Paul R. Mouncey of the Intensive Care National Audit and Research Centre, London, and his associates. The trial is the last of three planned studies of early, goal-directed therapy (EGDT), “all of which showed that EGDT was not superior to usual care,” the investigators wrote (N. Engl. J. Med. 2015 Mar. 17 [doi:10.1056/NEJMoa1500896]).
Support for EGDT originated mainly from a single-center, proof-of-concept study in 2001 by Dr. Emanuel Rivers and associates, who reported lower hospital mortality and length of stay when patients in early septic shock received 6 hours of treatment aimed at optimizing oxygen transport (N. Engl. J. Med. 2001;345:1368-77).
The protocol called for continuously monitoring central venous pressure, mean arterial pressure, and central venous oxygen saturation to guide use of intravenous fluids, vasoactive drugs, and red-cell transfusions. But uptake of EGDT has been limited, and clinicians have been concerned about the validity of the results and the complexity, risks, and costs of implementing EGDT, Mr. Mouncey and his associates said.
For the study, they randomized 1,260 patients with signs of early septic shock to either EGDT or standard care. All patients received intravenous antibiotics and adequate fluid resuscitation. The EGDT group also underwent a 6-hour resuscitation protocol that included monitoring central venous oxygen saturation, with additional treatment decisions left up to the treating clinician.
In all, 29.5% of patients who received EGDT died within 90 days, as did 29.2% of patients who received standard care (P = .90). The EGDT group received more intravenous fluids, vasoactive drugs, and red-cell transfusions and had significantly worse organ-failure scores, more days of advanced cardiovascular support, and longer ICU stays, but had no improvement in health-related quality of life or other secondary outcomes, the researchers reported.
“On average, EGDT increased costs, and the probability that it was cost-effective was below 20%,” they wrote. “Our results suggest that techniques used in usual resuscitation have evolved over the 15 years since the landmark study by Rivers et al.”
The United Kingdom National Institute for Health Research health technology assessment program funded the study. Nine coauthors reported receiving grants from the NIHR. One coauthor reported grants from ImaCor and Cheetah Medical. The other authors declared no relevant conflicts of interest.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: A strict program of early goal-directed therapy did not improve outcomes in patients with early septic shock.
Major finding: In all, 29.5% of patients who received EGDT died, as did 29.2% of patients who received standard care (P = .90).
Data source: Open, multicenter, randomized controlled parallel-group trial of 1,260 patients with early septic shock.
Disclosures: The U.K. National Institute for Health Research health technology assessment program funded the study. Nine coauthors reported receiving grants from the NIHR. One coauthor reported grants from ImaCor and Cheetah Medical. The other authors declared no relevant conflicts of interest.
Apremilast meets psoriasis endpoints at week 32
SAN FRANCISCO – Apremilast met its primary endpoint at week 32 with no new or serious safety signals among patients with moderate to severe plaque psoriasis, according to phase III data from the ongoing LIBERATE study.
At week 16, 40% of patients who received oral apremilast achieved PASI-75, compared with 12% of the placebo group, Dr. Kristian Reich reported at the annual meeting of the American Academy of Dermatology.
By week 32, PASI-75 response for apremilast had further risen to 53%, he added. “This drug may not be a quick fix, but the longer you give it, the higher the response,” said Dr. Reich of SCIderm Research Institute and Dermatologikum Hamburg in Germany.
The randomized, double-blind LIBERATE study compared the safety and efficacy of apremilast (30 mg twice daily) and injectable etanercept (50 mg weekly) with placebo among 250 patients with plaque psoriasis who had not previously received biologic therapy. Patients received one of the three treatments through week 16, and then all were switched to or continued apremilast.
At week 16, PASI-75 response rates were 40% for apremilast, 48% for etanercept, and 12% for placebo (P < .0001 for apremilast versus placebo), reported Dr. Reich. The high rate of response to placebo might have occurred because patients in the study were treatment naive, he said. At week 32, PASI-75 response rates were 53% for patients who received apremilast from baseline, and 61% for patients who switched from etanercept to apremilast at week 16.
Based on the results, “I would probably use apremilast more in the moderate [psoriasis] space than in the severe space, but its efficacy does not correlate with baseline disease severity, as far as I know,” said Dr. Reich. Apremilast also beat placebo in analyses of secondary endpoints, including static physician global assessment of clear or almost clear and the Dermatology Quality of Life Index, he added.
Safety analyses showed that the drug was generally well tolerated. Fewer than 4% of patients discontinued treatment because of adverse events, the most common of which were diarrhea, nausea, vomiting, and headache (including tension headache). No new side effects emerged after patients switched from etanercept to apremilast at week 16, he said.
A post hoc analysis found that apremilast was noninferior to etanercept (P > .05) in terms of PASI-75, although the study was not powered to directly compare the two biologics, Dr. Reich noted.
Celgene Corporation makes apremilast and sponsored the study. Dr. Reich reported serving as a speaker for and receiving honoraria from Celgene.
SAN FRANCISCO – Apremilast met its primary endpoint at week 32 with no new or serious safety signals among patients with moderate to severe plaque psoriasis, according to phase III data from the ongoing LIBERATE study.
At week 16, 40% of patients who received oral apremilast achieved PASI-75, compared with 12% of the placebo group, Dr. Kristian Reich reported at the annual meeting of the American Academy of Dermatology.
By week 32, PASI-75 response for apremilast had further risen to 53%, he added. “This drug may not be a quick fix, but the longer you give it, the higher the response,” said Dr. Reich of SCIderm Research Institute and Dermatologikum Hamburg in Germany.
The randomized, double-blind LIBERATE study compared the safety and efficacy of apremilast (30 mg twice daily) and injectable etanercept (50 mg weekly) with placebo among 250 patients with plaque psoriasis who had not previously received biologic therapy. Patients received one of the three treatments through week 16, and then all were switched to or continued apremilast.
At week 16, PASI-75 response rates were 40% for apremilast, 48% for etanercept, and 12% for placebo (P < .0001 for apremilast versus placebo), reported Dr. Reich. The high rate of response to placebo might have occurred because patients in the study were treatment naive, he said. At week 32, PASI-75 response rates were 53% for patients who received apremilast from baseline, and 61% for patients who switched from etanercept to apremilast at week 16.
Based on the results, “I would probably use apremilast more in the moderate [psoriasis] space than in the severe space, but its efficacy does not correlate with baseline disease severity, as far as I know,” said Dr. Reich. Apremilast also beat placebo in analyses of secondary endpoints, including static physician global assessment of clear or almost clear and the Dermatology Quality of Life Index, he added.
Safety analyses showed that the drug was generally well tolerated. Fewer than 4% of patients discontinued treatment because of adverse events, the most common of which were diarrhea, nausea, vomiting, and headache (including tension headache). No new side effects emerged after patients switched from etanercept to apremilast at week 16, he said.
A post hoc analysis found that apremilast was noninferior to etanercept (P > .05) in terms of PASI-75, although the study was not powered to directly compare the two biologics, Dr. Reich noted.
Celgene Corporation makes apremilast and sponsored the study. Dr. Reich reported serving as a speaker for and receiving honoraria from Celgene.
SAN FRANCISCO – Apremilast met its primary endpoint at week 32 with no new or serious safety signals among patients with moderate to severe plaque psoriasis, according to phase III data from the ongoing LIBERATE study.
At week 16, 40% of patients who received oral apremilast achieved PASI-75, compared with 12% of the placebo group, Dr. Kristian Reich reported at the annual meeting of the American Academy of Dermatology.
By week 32, PASI-75 response for apremilast had further risen to 53%, he added. “This drug may not be a quick fix, but the longer you give it, the higher the response,” said Dr. Reich of SCIderm Research Institute and Dermatologikum Hamburg in Germany.
The randomized, double-blind LIBERATE study compared the safety and efficacy of apremilast (30 mg twice daily) and injectable etanercept (50 mg weekly) with placebo among 250 patients with plaque psoriasis who had not previously received biologic therapy. Patients received one of the three treatments through week 16, and then all were switched to or continued apremilast.
At week 16, PASI-75 response rates were 40% for apremilast, 48% for etanercept, and 12% for placebo (P < .0001 for apremilast versus placebo), reported Dr. Reich. The high rate of response to placebo might have occurred because patients in the study were treatment naive, he said. At week 32, PASI-75 response rates were 53% for patients who received apremilast from baseline, and 61% for patients who switched from etanercept to apremilast at week 16.
Based on the results, “I would probably use apremilast more in the moderate [psoriasis] space than in the severe space, but its efficacy does not correlate with baseline disease severity, as far as I know,” said Dr. Reich. Apremilast also beat placebo in analyses of secondary endpoints, including static physician global assessment of clear or almost clear and the Dermatology Quality of Life Index, he added.
Safety analyses showed that the drug was generally well tolerated. Fewer than 4% of patients discontinued treatment because of adverse events, the most common of which were diarrhea, nausea, vomiting, and headache (including tension headache). No new side effects emerged after patients switched from etanercept to apremilast at week 16, he said.
A post hoc analysis found that apremilast was noninferior to etanercept (P > .05) in terms of PASI-75, although the study was not powered to directly compare the two biologics, Dr. Reich noted.
Celgene Corporation makes apremilast and sponsored the study. Dr. Reich reported serving as a speaker for and receiving honoraria from Celgene.
AT THE AAD ANNUAL MEETING
Key clinical point: Oral apremilast beat placebo and showed durable efficacy in patients with moderate to severe plaque psoriasis.
Major finding: PASI-75 response rates were 40% at week 16 (compared with 12% for placebo) and 53% at week 32.
Data source: Ongoing phase IIIb study of apremilast, etanercept, and placebo among 250 patients with moderate to severe plaque psoriasis.
Disclosures: Celgene Corporation makes apremilast and sponsored the study. Dr. Reich reported serving as a speaker for and receiving honoraria from Celgene.
Ixekizumab met psoriasis endpoints by week 12, with durable response at 60 weeks
SAN FRANCISCO – More than 80% of psoriasis patients who received ixekizumab achieved a 75% reduction in the Psoriasis Area and Severity Index score and static Physician Global Assessment scores of clear or almost clear skin at 12 weeks, based on the results of a phase III trial.
In addition, 35% of patients who received 80 mg ixekizumab twice monthly for 12 weeks achieved complete resolution of their psoriasis plaques (PASI 100), reported Dr. Kenneth Gordon of Northwestern University in Chicago. “The overall safety profile for ixekizumab was acceptable during both the induction and maintenance phases,” Dr. Gordon said at the annual meeting of the American Academy of Dermatology.
Ixekizumab is a monoclonal antibody that targets interleukin (IL)-17A, a major cytokine in the pathogenesis of psoriasis. Dr. Gordon and his associates conducted a randomized induction and withdrawal trial that compared twice-monthly or monthly treatment of the biologic with placebo among 1,296 patients. During the 12-week induction phase, 431 patients received placebo, 433 received 80 mg ixekizumab every 2 weeks, and 432 received 80 mg ixekizumab every 4 weeks, said Dr. Gordon. He and his associates then rerandomized the responders (based on PASI 75 and static Patient Global Assessment [sPGA] scores) to one of the three protocols, and followed these patients until week 60.
About 80% of patients who received ixekizumab every 2 or 4 weeks achieved sPGA scores of 0 or 1 (clear or almost clear) at 12 weeks, compared with 3% of the placebo group, said Dr. Gordon. Rates of PASI 75 response at 12 weeks were 89% for patients treated twice monthly, 83% for patients treated monthly, and 4% for the placebo group. Rates of PASI 100 were 35%, 33%, and 0%, respectively.
Among initial responders who were rerandomized to monthly ixekizumab, 73% maintained sPGA scores of clear or almost clear at week 60, while 78% maintained or achieved PASI 75, and 52% maintained or achieved PASI 100, said Dr. Gordon. The investigators also found a significant positive linear correlation between PASI response and scores on the Dermatology Quality of Life Index (DLQI), with P values ranging from less than .001 to less than .002, Dr. Gordon reported. “We are seeing that clearance is very important in quality of life,” he added. “More patients reported no itching or other negative impact on quality of life with higher levels of response.”
Serious adverse events at week 12 affected 1.4% of the twice-monthly ixekizumab group, 2.8% of the monthly ixekizumab group, and 1.2% of the placebo group, Dr. Gordon reported. Rates of candidiasis were similar among all three arms. Between weeks 12 and 60, the monthly treatment group had three major adverse cardiac events, but exposure-adjusted rates of adverse events for this group were similar to those during the induction period, he added.
Eli Lilly sponsored the trial. Dr. Gordon reported receiving research funding from Eli Lilly and several other pharmaceutical companies.
SAN FRANCISCO – More than 80% of psoriasis patients who received ixekizumab achieved a 75% reduction in the Psoriasis Area and Severity Index score and static Physician Global Assessment scores of clear or almost clear skin at 12 weeks, based on the results of a phase III trial.
In addition, 35% of patients who received 80 mg ixekizumab twice monthly for 12 weeks achieved complete resolution of their psoriasis plaques (PASI 100), reported Dr. Kenneth Gordon of Northwestern University in Chicago. “The overall safety profile for ixekizumab was acceptable during both the induction and maintenance phases,” Dr. Gordon said at the annual meeting of the American Academy of Dermatology.
Ixekizumab is a monoclonal antibody that targets interleukin (IL)-17A, a major cytokine in the pathogenesis of psoriasis. Dr. Gordon and his associates conducted a randomized induction and withdrawal trial that compared twice-monthly or monthly treatment of the biologic with placebo among 1,296 patients. During the 12-week induction phase, 431 patients received placebo, 433 received 80 mg ixekizumab every 2 weeks, and 432 received 80 mg ixekizumab every 4 weeks, said Dr. Gordon. He and his associates then rerandomized the responders (based on PASI 75 and static Patient Global Assessment [sPGA] scores) to one of the three protocols, and followed these patients until week 60.
About 80% of patients who received ixekizumab every 2 or 4 weeks achieved sPGA scores of 0 or 1 (clear or almost clear) at 12 weeks, compared with 3% of the placebo group, said Dr. Gordon. Rates of PASI 75 response at 12 weeks were 89% for patients treated twice monthly, 83% for patients treated monthly, and 4% for the placebo group. Rates of PASI 100 were 35%, 33%, and 0%, respectively.
Among initial responders who were rerandomized to monthly ixekizumab, 73% maintained sPGA scores of clear or almost clear at week 60, while 78% maintained or achieved PASI 75, and 52% maintained or achieved PASI 100, said Dr. Gordon. The investigators also found a significant positive linear correlation between PASI response and scores on the Dermatology Quality of Life Index (DLQI), with P values ranging from less than .001 to less than .002, Dr. Gordon reported. “We are seeing that clearance is very important in quality of life,” he added. “More patients reported no itching or other negative impact on quality of life with higher levels of response.”
Serious adverse events at week 12 affected 1.4% of the twice-monthly ixekizumab group, 2.8% of the monthly ixekizumab group, and 1.2% of the placebo group, Dr. Gordon reported. Rates of candidiasis were similar among all three arms. Between weeks 12 and 60, the monthly treatment group had three major adverse cardiac events, but exposure-adjusted rates of adverse events for this group were similar to those during the induction period, he added.
Eli Lilly sponsored the trial. Dr. Gordon reported receiving research funding from Eli Lilly and several other pharmaceutical companies.
SAN FRANCISCO – More than 80% of psoriasis patients who received ixekizumab achieved a 75% reduction in the Psoriasis Area and Severity Index score and static Physician Global Assessment scores of clear or almost clear skin at 12 weeks, based on the results of a phase III trial.
In addition, 35% of patients who received 80 mg ixekizumab twice monthly for 12 weeks achieved complete resolution of their psoriasis plaques (PASI 100), reported Dr. Kenneth Gordon of Northwestern University in Chicago. “The overall safety profile for ixekizumab was acceptable during both the induction and maintenance phases,” Dr. Gordon said at the annual meeting of the American Academy of Dermatology.
Ixekizumab is a monoclonal antibody that targets interleukin (IL)-17A, a major cytokine in the pathogenesis of psoriasis. Dr. Gordon and his associates conducted a randomized induction and withdrawal trial that compared twice-monthly or monthly treatment of the biologic with placebo among 1,296 patients. During the 12-week induction phase, 431 patients received placebo, 433 received 80 mg ixekizumab every 2 weeks, and 432 received 80 mg ixekizumab every 4 weeks, said Dr. Gordon. He and his associates then rerandomized the responders (based on PASI 75 and static Patient Global Assessment [sPGA] scores) to one of the three protocols, and followed these patients until week 60.
About 80% of patients who received ixekizumab every 2 or 4 weeks achieved sPGA scores of 0 or 1 (clear or almost clear) at 12 weeks, compared with 3% of the placebo group, said Dr. Gordon. Rates of PASI 75 response at 12 weeks were 89% for patients treated twice monthly, 83% for patients treated monthly, and 4% for the placebo group. Rates of PASI 100 were 35%, 33%, and 0%, respectively.
Among initial responders who were rerandomized to monthly ixekizumab, 73% maintained sPGA scores of clear or almost clear at week 60, while 78% maintained or achieved PASI 75, and 52% maintained or achieved PASI 100, said Dr. Gordon. The investigators also found a significant positive linear correlation between PASI response and scores on the Dermatology Quality of Life Index (DLQI), with P values ranging from less than .001 to less than .002, Dr. Gordon reported. “We are seeing that clearance is very important in quality of life,” he added. “More patients reported no itching or other negative impact on quality of life with higher levels of response.”
Serious adverse events at week 12 affected 1.4% of the twice-monthly ixekizumab group, 2.8% of the monthly ixekizumab group, and 1.2% of the placebo group, Dr. Gordon reported. Rates of candidiasis were similar among all three arms. Between weeks 12 and 60, the monthly treatment group had three major adverse cardiac events, but exposure-adjusted rates of adverse events for this group were similar to those during the induction period, he added.
Eli Lilly sponsored the trial. Dr. Gordon reported receiving research funding from Eli Lilly and several other pharmaceutical companies.
AT THE AAD ANNUAL MEETING
Key clinical point: The investigational anti-IL-17A antibody ixekizumab met its endpoints at week 12 in patients with plaque psoriasis, and showed durability of response at 60 weeks.
Major finding: More than 80% of treated patients achieved PASI 75 and static Physician Global Assessment scores of clear or almost clear skin at 12 weeks.
Data source: A phase III trial of 1,296 patients with plaque psoriasis.
Disclosures: Eli Lilly sponsored the trial. Dr. Gordon reported receiving research funding from Eli Lilly and several other pharmaceutical companies.