User login
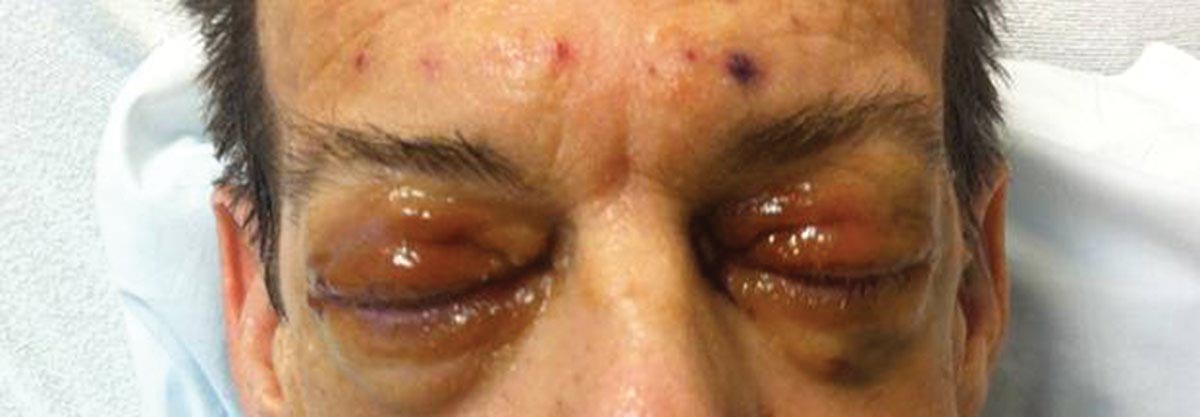
Postoperative Patient Suddenly Worsens
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
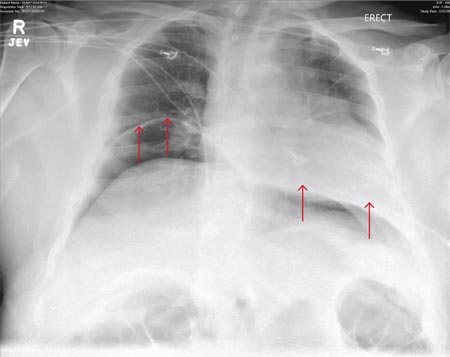
A 55-year-old man undergoes an elective craniotomy for tumor resection, with uneventful preoperative and intraoperative stages. Immediately postoperative, however, he experiences seizures. Noncontrast CT of the head is negative except for postoperative changes. The patient is placed in the ICU for close monitoring. He is slowly improving when, on the fifth postoperative day, tachypnea and dyspnea are observed. The patient is afebrile. His blood pressure is 116/70 mm Hg; pulse, 90 beats/min; respiratory rate, 30 breaths/min; and O2 saturation, 98%. A stat portable chest radiograph is obtained. What is your impression?
Leg Swelling Accompanied by Weight Gain
In the past two weeks, a 59-year-old postmenopausal woman has noticed swelling in her legs and a 10-lb weight gain. For the past three days, she has also had a vague, aching pain in the right upper abdominal quadrant, which surprises her, since her gall bladder was removed long ago. There is no prior history of chest pain, dyspnea, or systemic hypertension.
The patient does have a history of paroxysmal atrial fibrillation, palpitations, and pulmonary hypertension. She is chronically obese and has hypothyroidism. Surgical history is significant for cholecystectomy, hysterectomy, and left breast lumpectomy with axillary node dissection.
Her job at a local factory, assembling components for pressure washer pumps, requires her to sit for extended periods. She smokes 1.5 packs of cigarettes per day, a habit that began when she was 16. She drinks one or two beers daily and admits she has “many more” on the weekends. She has used marijuana in the recent past but not in the past month. She denies use of any other illicit drugs or homeopathic medications.
Her medication list includes levothyroxine sodium and ibuprofen. She says she’s “supposed to be taking some kind of heart medication” but hasn’t taken it for several months (and cannot remember the name). It was prescribed for her when she was on vacation in the Florida Keys and experienced similar symptoms. She sheepishly admits to trying her husband’s sildenafil, as she’s been told it works for pulmonary hypertension. She is allergic to sulfa and tetracycline.
Review of systems is remarkable for bilateral hip and ankle pain, which she attributes to her weight. She has had no change in bowel or bladder function. The remainder of the review is unremarkable.
Physical exam reveals a weight of 297 lb and height of 5’6”. Vital signs include a blood pressure of 128/88 mm Hg; pulse, 90 beats/min; respiratory rate, 14 breaths/min-1; temperature, 98.2°F; and O2 saturation, 98.2%.
She is morbidly obese and in no distress. Pertinent physical findings include elevated jugular venous return, bilateral rales in both lung bases, a soft, early diastolic murmur best heard at the left lower sternal border, and a regular rate and rhythm. She also has mild tenderness to deep palpation in the right upper abdominal quadrant. Her lower extremities demonstrate 3+ pitting edema to the level of the knees bilaterally. There are no skin lesions, and the neurologic exam is grossly intact.
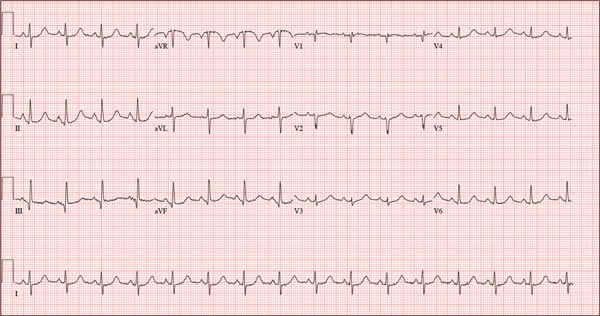
As part of her workup, you order an ECG, which reveals a ventricular rate of 94 beats/min; PR interval, 130 ms; QRS duration, 76 ms; QT/QTc interval, 394/492 ms; P axis, 50°; R axis, 80°; and T axis, 47°. What is your interpretation?
ANSWER
Pertinent findings on this ECG include normal sinus rhythm, right atrial enlargement, and a prolonged QT interval. Criteria for right atrial enlargement include P waves > 2.5 mm in leads II, III, and aVF and > 1.5 mm in leads V1 and V2. A prolonged QT interval is evidenced by a QTc > 470 ms using Bazett’s formula (QTc = QT divided by the square root of the RR interval).
The patient’s symptoms and ECG finding of right atrial enlargement coincide with pulmonary hypertension and right-sided heart failure. The prolonged QT interval may be due to her history of hypothyroidism; however, this has not been confirmed.
In the past two weeks, a 59-year-old postmenopausal woman has noticed swelling in her legs and a 10-lb weight gain. For the past three days, she has also had a vague, aching pain in the right upper abdominal quadrant, which surprises her, since her gall bladder was removed long ago. There is no prior history of chest pain, dyspnea, or systemic hypertension.
The patient does have a history of paroxysmal atrial fibrillation, palpitations, and pulmonary hypertension. She is chronically obese and has hypothyroidism. Surgical history is significant for cholecystectomy, hysterectomy, and left breast lumpectomy with axillary node dissection.
Her job at a local factory, assembling components for pressure washer pumps, requires her to sit for extended periods. She smokes 1.5 packs of cigarettes per day, a habit that began when she was 16. She drinks one or two beers daily and admits she has “many more” on the weekends. She has used marijuana in the recent past but not in the past month. She denies use of any other illicit drugs or homeopathic medications.
Her medication list includes levothyroxine sodium and ibuprofen. She says she’s “supposed to be taking some kind of heart medication” but hasn’t taken it for several months (and cannot remember the name). It was prescribed for her when she was on vacation in the Florida Keys and experienced similar symptoms. She sheepishly admits to trying her husband’s sildenafil, as she’s been told it works for pulmonary hypertension. She is allergic to sulfa and tetracycline.
Review of systems is remarkable for bilateral hip and ankle pain, which she attributes to her weight. She has had no change in bowel or bladder function. The remainder of the review is unremarkable.
Physical exam reveals a weight of 297 lb and height of 5’6”. Vital signs include a blood pressure of 128/88 mm Hg; pulse, 90 beats/min; respiratory rate, 14 breaths/min-1; temperature, 98.2°F; and O2 saturation, 98.2%.
She is morbidly obese and in no distress. Pertinent physical findings include elevated jugular venous return, bilateral rales in both lung bases, a soft, early diastolic murmur best heard at the left lower sternal border, and a regular rate and rhythm. She also has mild tenderness to deep palpation in the right upper abdominal quadrant. Her lower extremities demonstrate 3+ pitting edema to the level of the knees bilaterally. There are no skin lesions, and the neurologic exam is grossly intact.
As part of her workup, you order an ECG, which reveals a ventricular rate of 94 beats/min; PR interval, 130 ms; QRS duration, 76 ms; QT/QTc interval, 394/492 ms; P axis, 50°; R axis, 80°; and T axis, 47°. What is your interpretation?
ANSWER
Pertinent findings on this ECG include normal sinus rhythm, right atrial enlargement, and a prolonged QT interval. Criteria for right atrial enlargement include P waves > 2.5 mm in leads II, III, and aVF and > 1.5 mm in leads V1 and V2. A prolonged QT interval is evidenced by a QTc > 470 ms using Bazett’s formula (QTc = QT divided by the square root of the RR interval).
The patient’s symptoms and ECG finding of right atrial enlargement coincide with pulmonary hypertension and right-sided heart failure. The prolonged QT interval may be due to her history of hypothyroidism; however, this has not been confirmed.
In the past two weeks, a 59-year-old postmenopausal woman has noticed swelling in her legs and a 10-lb weight gain. For the past three days, she has also had a vague, aching pain in the right upper abdominal quadrant, which surprises her, since her gall bladder was removed long ago. There is no prior history of chest pain, dyspnea, or systemic hypertension.
The patient does have a history of paroxysmal atrial fibrillation, palpitations, and pulmonary hypertension. She is chronically obese and has hypothyroidism. Surgical history is significant for cholecystectomy, hysterectomy, and left breast lumpectomy with axillary node dissection.
Her job at a local factory, assembling components for pressure washer pumps, requires her to sit for extended periods. She smokes 1.5 packs of cigarettes per day, a habit that began when she was 16. She drinks one or two beers daily and admits she has “many more” on the weekends. She has used marijuana in the recent past but not in the past month. She denies use of any other illicit drugs or homeopathic medications.
Her medication list includes levothyroxine sodium and ibuprofen. She says she’s “supposed to be taking some kind of heart medication” but hasn’t taken it for several months (and cannot remember the name). It was prescribed for her when she was on vacation in the Florida Keys and experienced similar symptoms. She sheepishly admits to trying her husband’s sildenafil, as she’s been told it works for pulmonary hypertension. She is allergic to sulfa and tetracycline.
Review of systems is remarkable for bilateral hip and ankle pain, which she attributes to her weight. She has had no change in bowel or bladder function. The remainder of the review is unremarkable.
Physical exam reveals a weight of 297 lb and height of 5’6”. Vital signs include a blood pressure of 128/88 mm Hg; pulse, 90 beats/min; respiratory rate, 14 breaths/min-1; temperature, 98.2°F; and O2 saturation, 98.2%.
She is morbidly obese and in no distress. Pertinent physical findings include elevated jugular venous return, bilateral rales in both lung bases, a soft, early diastolic murmur best heard at the left lower sternal border, and a regular rate and rhythm. She also has mild tenderness to deep palpation in the right upper abdominal quadrant. Her lower extremities demonstrate 3+ pitting edema to the level of the knees bilaterally. There are no skin lesions, and the neurologic exam is grossly intact.
As part of her workup, you order an ECG, which reveals a ventricular rate of 94 beats/min; PR interval, 130 ms; QRS duration, 76 ms; QT/QTc interval, 394/492 ms; P axis, 50°; R axis, 80°; and T axis, 47°. What is your interpretation?
ANSWER
Pertinent findings on this ECG include normal sinus rhythm, right atrial enlargement, and a prolonged QT interval. Criteria for right atrial enlargement include P waves > 2.5 mm in leads II, III, and aVF and > 1.5 mm in leads V1 and V2. A prolonged QT interval is evidenced by a QTc > 470 ms using Bazett’s formula (QTc = QT divided by the square root of the RR interval).
The patient’s symptoms and ECG finding of right atrial enlargement coincide with pulmonary hypertension and right-sided heart failure. The prolonged QT interval may be due to her history of hypothyroidism; however, this has not been confirmed.
Those symptoms first appeared two weeks ago. Now, this woman also has abdominal pain. What does an ECG add to the clinical picture?
As Problem Spreads, Man Seeks Help
ANSWER
The correct answer is Schamberg disease (choice “b”), a benign form of capillaritis; see Discussion for more information.
Scurvy patients can present with ecchymosis (among other findings that were missing in this case). But scurvy (choice “a”) is rare, and by the time the disease is evident, the patient is typically quite ill.
Cutaneous T-cell lymphoma (choice “c”) can manifest as purpuric annular lesions. However, these would be unlikely to take the distributive pattern seen in this case, and they usually have an atrophic surface.
Thrombocytopenia (choice “d”) and other coagulopathies, although rightly considered, would probably manifest in other ways as well (ie, not just cutaneously).
Continue for the discussion >>
DISCUSSION
Schamberg disease is typical of a whole class of conditions in which red blood cells (RBCs) are extravasated from slightly damaged capillaries. This results in nonblanchable purpura and subsequent hemosiderin staining caused by phagocytosis of the RBCs by macrophages. Clinically, this family of diseases present as cayenne pepper–colored macules, most of which are annular in configuration.
Schamberg is, by far, the most common of these conditions. This presentation was typical: manifestation on the knees and ankles followed by upward spread (hence the condition’s other name, progressive pigmentary purpura). Usually resolving on their own within months, these lesions are almost always asymptomatic—but nonetheless alarming to the patient.
Other equally benign, self-limited forms of capillaritis include those in which lesions are pruritic (eg, purpura of Doucas and Kapetanakis) or lichenoid (purpura of Gougerot-Blum). Another example is lichen aureus, in which only one or two lesions, more gold than brown, appear on the legs of younger patients.
There are many theories as to these conditions’ cause, the most common of which is increased intravascular pressure secondary to dependence. However, if this were so, we’d likely see a great deal more cases, since many patients have problems related to venous insufficiency.
In some cases, skin biopsy (usually 4-mm punch) is necessary to rule out more serious diseases, such as an early form of cutaneous T-cell lymphoma. When a coagulopathy is suspected, blood work is necessary to confirm or rule out the diagnosis. In this case, there was no reason to suspect coagulopathy (or scurvy), since no other signs were seen.
This patient was educated about his diagnosis and provided Web-based resources he could consult. Various treatments—topical steroids, increased vitamin C intake, and increased exposure to UV light—have been tried but with disappointing results.
ANSWER
The correct answer is Schamberg disease (choice “b”), a benign form of capillaritis; see Discussion for more information.
Scurvy patients can present with ecchymosis (among other findings that were missing in this case). But scurvy (choice “a”) is rare, and by the time the disease is evident, the patient is typically quite ill.
Cutaneous T-cell lymphoma (choice “c”) can manifest as purpuric annular lesions. However, these would be unlikely to take the distributive pattern seen in this case, and they usually have an atrophic surface.
Thrombocytopenia (choice “d”) and other coagulopathies, although rightly considered, would probably manifest in other ways as well (ie, not just cutaneously).
Continue for the discussion >>
DISCUSSION
Schamberg disease is typical of a whole class of conditions in which red blood cells (RBCs) are extravasated from slightly damaged capillaries. This results in nonblanchable purpura and subsequent hemosiderin staining caused by phagocytosis of the RBCs by macrophages. Clinically, this family of diseases present as cayenne pepper–colored macules, most of which are annular in configuration.
Schamberg is, by far, the most common of these conditions. This presentation was typical: manifestation on the knees and ankles followed by upward spread (hence the condition’s other name, progressive pigmentary purpura). Usually resolving on their own within months, these lesions are almost always asymptomatic—but nonetheless alarming to the patient.
Other equally benign, self-limited forms of capillaritis include those in which lesions are pruritic (eg, purpura of Doucas and Kapetanakis) or lichenoid (purpura of Gougerot-Blum). Another example is lichen aureus, in which only one or two lesions, more gold than brown, appear on the legs of younger patients.
There are many theories as to these conditions’ cause, the most common of which is increased intravascular pressure secondary to dependence. However, if this were so, we’d likely see a great deal more cases, since many patients have problems related to venous insufficiency.
In some cases, skin biopsy (usually 4-mm punch) is necessary to rule out more serious diseases, such as an early form of cutaneous T-cell lymphoma. When a coagulopathy is suspected, blood work is necessary to confirm or rule out the diagnosis. In this case, there was no reason to suspect coagulopathy (or scurvy), since no other signs were seen.
This patient was educated about his diagnosis and provided Web-based resources he could consult. Various treatments—topical steroids, increased vitamin C intake, and increased exposure to UV light—have been tried but with disappointing results.
ANSWER
The correct answer is Schamberg disease (choice “b”), a benign form of capillaritis; see Discussion for more information.
Scurvy patients can present with ecchymosis (among other findings that were missing in this case). But scurvy (choice “a”) is rare, and by the time the disease is evident, the patient is typically quite ill.
Cutaneous T-cell lymphoma (choice “c”) can manifest as purpuric annular lesions. However, these would be unlikely to take the distributive pattern seen in this case, and they usually have an atrophic surface.
Thrombocytopenia (choice “d”) and other coagulopathies, although rightly considered, would probably manifest in other ways as well (ie, not just cutaneously).
Continue for the discussion >>
DISCUSSION
Schamberg disease is typical of a whole class of conditions in which red blood cells (RBCs) are extravasated from slightly damaged capillaries. This results in nonblanchable purpura and subsequent hemosiderin staining caused by phagocytosis of the RBCs by macrophages. Clinically, this family of diseases present as cayenne pepper–colored macules, most of which are annular in configuration.
Schamberg is, by far, the most common of these conditions. This presentation was typical: manifestation on the knees and ankles followed by upward spread (hence the condition’s other name, progressive pigmentary purpura). Usually resolving on their own within months, these lesions are almost always asymptomatic—but nonetheless alarming to the patient.
Other equally benign, self-limited forms of capillaritis include those in which lesions are pruritic (eg, purpura of Doucas and Kapetanakis) or lichenoid (purpura of Gougerot-Blum). Another example is lichen aureus, in which only one or two lesions, more gold than brown, appear on the legs of younger patients.
There are many theories as to these conditions’ cause, the most common of which is increased intravascular pressure secondary to dependence. However, if this were so, we’d likely see a great deal more cases, since many patients have problems related to venous insufficiency.
In some cases, skin biopsy (usually 4-mm punch) is necessary to rule out more serious diseases, such as an early form of cutaneous T-cell lymphoma. When a coagulopathy is suspected, blood work is necessary to confirm or rule out the diagnosis. In this case, there was no reason to suspect coagulopathy (or scurvy), since no other signs were seen.
This patient was educated about his diagnosis and provided Web-based resources he could consult. Various treatments—topical steroids, increased vitamin C intake, and increased exposure to UV light—have been tried but with disappointing results.
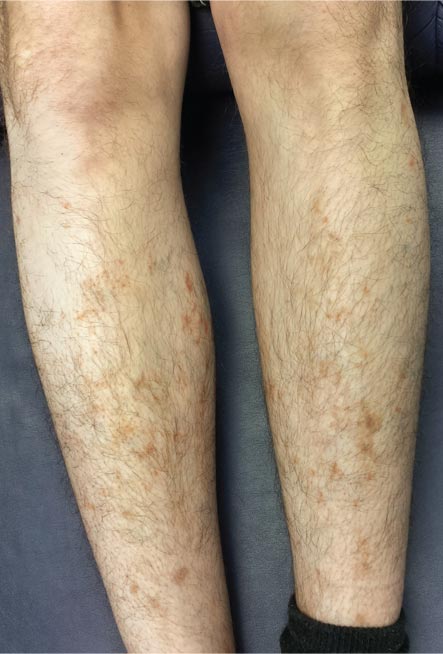
For several months, a 30-year-old man has had an asymptomatic rash on his legs. The lesions first appeared on his lower legs and ankles; over the subsequent months, they have spread upward. Now, the rash reaches to just below his knees. During this time, he has had two bouts of strep throat, both adequately treated. He denies any other skin problems and has no relevant family history. The patient denies alcohol or drug abuse and is not taking any prescription medications. Prior to referral to dermatology, he was seen in two urgent care clinics; at one, he received a diagnosis of fungal infection and at the other, of “vitamin deficiency.” He was given a month-long course of terbinafine (250 mg/d) that produced no change in his rash. He achieved the same (non)result from an increased intake of vitamins. Examination reveals annular reddish brown macules, measuring 1 to 3 cm, sparsely distributed from the knees to just above the ankles on both legs. The lesions are a bit more densely arrayed on the anterior legs. There is no palpable component to any of them and no discernable surface scale. Digital pressure fails to blanch the lesions. The hairs and follicles on the patient’s legs appear normal. There are no notable skin changes elsewhere, and the patient is alert, oriented, and in no distress.

Friable Nodule on the Back
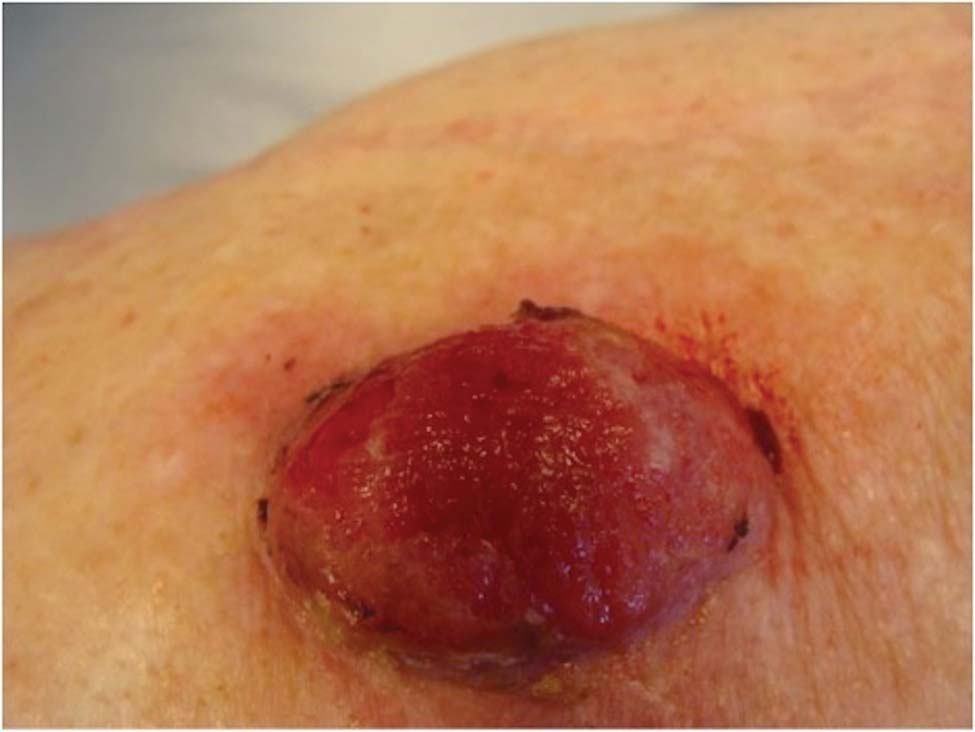
The Diagnosis: Spindle Cell Malignant Melanoma With Perineural Invasion
The incidence of melanoma has steadily increased in the United States since the 1930s when the incidence was reported at 1.0 per 100,000.1 In 1973 melanoma incidence was 6.8 per 100,000, and by 2007 the rate increased to 20.1 per 100,000.2 The American Cancer Society projects 73,870 new cases of melanoma in 2015, with melanoma as the fifth most common cancer in males and the seventh most common in females.3 Melanoma-related deaths are projected to be 9940. The lifetime probability of developing melanoma is 1 in 34 for males and 1 in 53 for females. Twice as many males are estimated to have melanoma-related deaths compared to females. The 5-year relative survival rate is 93% for white individuals and 75% for black individuals.3
Spindle cell melanoma is a rare variant of melanoma that was originally described by Conley et al4 in 1971. The lesion represents 2% to 4% of all melanomas and presents in older patients on sun-exposed skin as a pink or variably pigmented nodule measuring an average of 2 cm.5 Males are affected more than females, and prominent neural invasion is present in 30% to 100% of cases.6-10 Neural invasion can result in nerve palsies and/or dysesthesia. Because half of these lesions are amelanotic, they are often clinically misdiagnosed prior to biopsy.11
Histopathologically, these lesions can be quite challenging. Spindle cell melanoma histologically is an intradermal lesion composed of spindle cells distributed in bundles, fascicles, or nests, or singly between collagen fibers of the dermis. Other spindle tumors such as spindle cell squamous cell carcinoma, atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiosarcoma, and leiomyosarcoma have a similar presentation with hematoxylin and eosin stain. The diagnostic process for spindle cell tumors is greatly aided by immunohistologic analysis. Spindle cell melanoma usually shows immunoreactivity with S-100. Human melanoma black 45, CD57, and neuron-specific enolase usually do not stain, and CD68 has been demonstrated in a minority of cases.
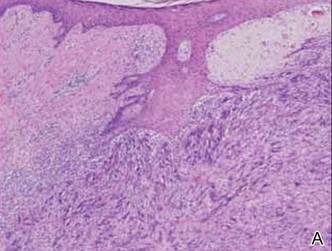
The initial biopsy specimen in our case displayed a dense dermal atypical spindle cell proliferation with hematoxylin and eosin stain. The differential diagnosis of the proliferation included desmoplastic melanoma, spindle cell squamous cell carcinoma, leiomyosarcoma, angiosarcoma, and atypical fibroxanthoma. Immunostains were used to further study the lesional biopsy. Cytokeratin 34bE12, CK5/6, cerium ammonium molybdate 5.2, CK7, epithelial membrane antigen, CK18, high-molecular-weight cytokeratin, S-100, Melan-A, human melanoma black 45, smooth muscle actin, desmin, CD68, CD34, CD10, and p63 were studied. The atypical dermal spindle cells were positive for S-100. S-100 and Melan-A highlighted an increased number of single melanocytes at the dermoepidermal junction. Other markers were negative.
A 1.0-cm wide excision to fascia was performed. Routine hematoxylin and eosin stain showed atypical dermal spindle cells with perineural invasion (Figure 1). The malignant spindle cells were S-100 positive (Figure 2). The spindle cells were negative for high-molecular-weight cytokeratin, CK5/6, p63, Melan-A, A103, microphthalmia, and tyrosinase. These findings confirm melanoma of the spindle cell type.
|
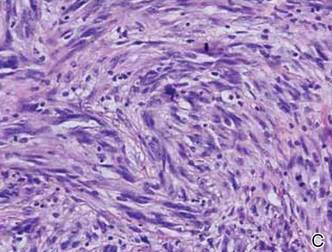
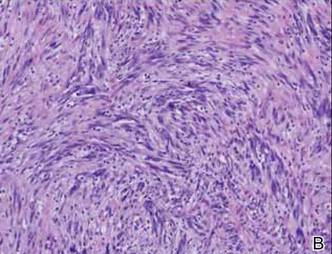
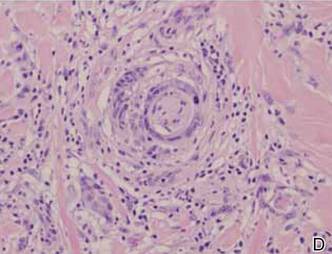
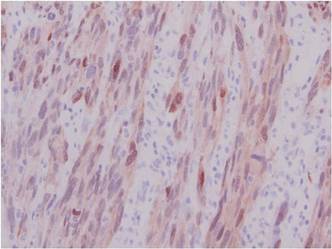
The lesion was a Clark level V melanoma with a Breslow thickness of at least 12 mm. Perineural invasion was noted and the mitotic index was 7 cells/mm2. Vascular and lymphatic invasion was negative and ulceration was present. Tumor-infiltrating lymphocytes were brisk, resulting in a pathologic staging of pT4NXMX.
On initial histologic study with hematoxylin and eosin stain, spindle cell melanoma lesions tend to be generally quite thick. Manganoni et al12 reported in their series a Breslow thickness ranging from 2.1 to 12 mm with a mean of 5.8 mm.
Treatment in our case involved a second wide incision to fascia with an additional 2-cm margin. The role of sentinel lymph node biopsy (SLNB) remains undefined. Patients with spindle cell melanoma have a lower frequency of positive sentinel lymph nodes than nondesmoplastic melanomas.13 As such, the need to perform SLNB has not been determined.13-15 Our patient had a history of breast cancer. The lesion appeared on the left side of the back and she pre-viously had a complete axillary lymphadenectomy on the left axillae. She declined SLNB. A systematic workup by the oncology service was negative. Continued follow-up has revealed no recurrent disease and recent workup was negative for metastatic disease.
Many studies report the increased incidence of local recurrence after excision for spindle cell melanoma as compared to non–spindle cell melanoma,16-18 which is likely related to perineural invasion as in our patient.
Spindle cell melanoma is a rare tumor that is often amelanotic and difficult to diagnose clinically. Routine hematoxylin and eosin staining shows a dermal spindle cell tumor. Immunohistochemical study is of great aid in defining the tumor. The clinician and pathologist must work together to correctly diagnose and treat this lesion.
1. Mikkilineni R, Weinstock MA. Epidemiology. In: Sober AJ, Haluska FG, eds. Atlas of Clinical Oncology: Skin Cancer. London, England: BC Decker; 2001:1-15.
2. Rigel D. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204-209.
3. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed February 10, 2015.
4. Conley J, Lattes R, Orr W. Desmoplastic malignant melanoma (a rare variant of malignant melanoma. Cancer. 1971;28:914-936.
5. Repertinger SK, Teruya B, Sarma DP. Common spindle cell malignant neoplasms of the skin: differential diagnosis and review of the literature. Internet J Dermatol. 2009;7. https://ispub.com/IJD/7/2/11747. Accessed February 10, 2015.
6. Chang PC, Fischbein NJ, McCalmont TH, et al. Perineural spread of malignant melanoma of the head and neck: clinical and imaging features. AJNR Am J Neuroradial. 2004;25:5-11.
7. Cruz J, Reis-Filho JS, Lopes JM. Malignant peripheral nerve sheath tumor-like primary cutaneous melanoma. J Clin Pathol. 2004;57:218-220.
8. Tsao H, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma. Semin Cutan Med Surg. 1997;16:45-47.
9. Kossard S, Doherty E, Murray E. Neurotropic melanoma. a variant of desmoplastic melanoma. Arch Dermatol. 1997;7:907-912.
10. Bruijn JA, Salasche S, Sober AJ, et al. Desmoplastic melanoma: clinicopathologic aspects of six cases. Dermatology. 1992;185:3-8.
11. Jesitus J. Desmoplastic melanoma. Dermatology Times. March 2009:1-2.
12. Manganoni AM, Farisoglio C, Bassissi S, et al. Desmoplastic melanoma: report of 5 cases. Dermatol Res Pract. 2009;2009:679010.
13. Gyorki DE, Busam K, Panageas K, et al. Sentinel lymph node biopsy for patients with desmoplastic melanoma. Ann Surg Oncol. 2003;10:403-407.
14. Livestro DP, Muzikansky A, Kaine EM, et al. of desmoplastic melanoma: a case-control comparison with other melanomas. J Clin Oncol. 2005;23:6739-6746.
15. Pawlik TM, Ross MI, Prieto VG, et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer. 2006;106:900-906.
16. Smithers HM, McLeod GR, Little JH. Desmoplastic melanoma: patterns of recurrence. World J Surg. 1992;16:186-190.
17. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology. 2004;36:445-451.
18. Bruijn JA, Mihm MC Jr, Barnhill RL. Desmoplastic melanoma. Histopathology. 1992;20:197-205.
The Diagnosis: Spindle Cell Malignant Melanoma With Perineural Invasion
The incidence of melanoma has steadily increased in the United States since the 1930s when the incidence was reported at 1.0 per 100,000.1 In 1973 melanoma incidence was 6.8 per 100,000, and by 2007 the rate increased to 20.1 per 100,000.2 The American Cancer Society projects 73,870 new cases of melanoma in 2015, with melanoma as the fifth most common cancer in males and the seventh most common in females.3 Melanoma-related deaths are projected to be 9940. The lifetime probability of developing melanoma is 1 in 34 for males and 1 in 53 for females. Twice as many males are estimated to have melanoma-related deaths compared to females. The 5-year relative survival rate is 93% for white individuals and 75% for black individuals.3
Spindle cell melanoma is a rare variant of melanoma that was originally described by Conley et al4 in 1971. The lesion represents 2% to 4% of all melanomas and presents in older patients on sun-exposed skin as a pink or variably pigmented nodule measuring an average of 2 cm.5 Males are affected more than females, and prominent neural invasion is present in 30% to 100% of cases.6-10 Neural invasion can result in nerve palsies and/or dysesthesia. Because half of these lesions are amelanotic, they are often clinically misdiagnosed prior to biopsy.11
Histopathologically, these lesions can be quite challenging. Spindle cell melanoma histologically is an intradermal lesion composed of spindle cells distributed in bundles, fascicles, or nests, or singly between collagen fibers of the dermis. Other spindle tumors such as spindle cell squamous cell carcinoma, atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiosarcoma, and leiomyosarcoma have a similar presentation with hematoxylin and eosin stain. The diagnostic process for spindle cell tumors is greatly aided by immunohistologic analysis. Spindle cell melanoma usually shows immunoreactivity with S-100. Human melanoma black 45, CD57, and neuron-specific enolase usually do not stain, and CD68 has been demonstrated in a minority of cases.
The initial biopsy specimen in our case displayed a dense dermal atypical spindle cell proliferation with hematoxylin and eosin stain. The differential diagnosis of the proliferation included desmoplastic melanoma, spindle cell squamous cell carcinoma, leiomyosarcoma, angiosarcoma, and atypical fibroxanthoma. Immunostains were used to further study the lesional biopsy. Cytokeratin 34bE12, CK5/6, cerium ammonium molybdate 5.2, CK7, epithelial membrane antigen, CK18, high-molecular-weight cytokeratin, S-100, Melan-A, human melanoma black 45, smooth muscle actin, desmin, CD68, CD34, CD10, and p63 were studied. The atypical dermal spindle cells were positive for S-100. S-100 and Melan-A highlighted an increased number of single melanocytes at the dermoepidermal junction. Other markers were negative.
A 1.0-cm wide excision to fascia was performed. Routine hematoxylin and eosin stain showed atypical dermal spindle cells with perineural invasion (Figure 1). The malignant spindle cells were S-100 positive (Figure 2). The spindle cells were negative for high-molecular-weight cytokeratin, CK5/6, p63, Melan-A, A103, microphthalmia, and tyrosinase. These findings confirm melanoma of the spindle cell type.
|
The lesion was a Clark level V melanoma with a Breslow thickness of at least 12 mm. Perineural invasion was noted and the mitotic index was 7 cells/mm2. Vascular and lymphatic invasion was negative and ulceration was present. Tumor-infiltrating lymphocytes were brisk, resulting in a pathologic staging of pT4NXMX.
On initial histologic study with hematoxylin and eosin stain, spindle cell melanoma lesions tend to be generally quite thick. Manganoni et al12 reported in their series a Breslow thickness ranging from 2.1 to 12 mm with a mean of 5.8 mm.
Treatment in our case involved a second wide incision to fascia with an additional 2-cm margin. The role of sentinel lymph node biopsy (SLNB) remains undefined. Patients with spindle cell melanoma have a lower frequency of positive sentinel lymph nodes than nondesmoplastic melanomas.13 As such, the need to perform SLNB has not been determined.13-15 Our patient had a history of breast cancer. The lesion appeared on the left side of the back and she pre-viously had a complete axillary lymphadenectomy on the left axillae. She declined SLNB. A systematic workup by the oncology service was negative. Continued follow-up has revealed no recurrent disease and recent workup was negative for metastatic disease.
Many studies report the increased incidence of local recurrence after excision for spindle cell melanoma as compared to non–spindle cell melanoma,16-18 which is likely related to perineural invasion as in our patient.
Spindle cell melanoma is a rare tumor that is often amelanotic and difficult to diagnose clinically. Routine hematoxylin and eosin staining shows a dermal spindle cell tumor. Immunohistochemical study is of great aid in defining the tumor. The clinician and pathologist must work together to correctly diagnose and treat this lesion.
The Diagnosis: Spindle Cell Malignant Melanoma With Perineural Invasion
The incidence of melanoma has steadily increased in the United States since the 1930s when the incidence was reported at 1.0 per 100,000.1 In 1973 melanoma incidence was 6.8 per 100,000, and by 2007 the rate increased to 20.1 per 100,000.2 The American Cancer Society projects 73,870 new cases of melanoma in 2015, with melanoma as the fifth most common cancer in males and the seventh most common in females.3 Melanoma-related deaths are projected to be 9940. The lifetime probability of developing melanoma is 1 in 34 for males and 1 in 53 for females. Twice as many males are estimated to have melanoma-related deaths compared to females. The 5-year relative survival rate is 93% for white individuals and 75% for black individuals.3
Spindle cell melanoma is a rare variant of melanoma that was originally described by Conley et al4 in 1971. The lesion represents 2% to 4% of all melanomas and presents in older patients on sun-exposed skin as a pink or variably pigmented nodule measuring an average of 2 cm.5 Males are affected more than females, and prominent neural invasion is present in 30% to 100% of cases.6-10 Neural invasion can result in nerve palsies and/or dysesthesia. Because half of these lesions are amelanotic, they are often clinically misdiagnosed prior to biopsy.11
Histopathologically, these lesions can be quite challenging. Spindle cell melanoma histologically is an intradermal lesion composed of spindle cells distributed in bundles, fascicles, or nests, or singly between collagen fibers of the dermis. Other spindle tumors such as spindle cell squamous cell carcinoma, atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiosarcoma, and leiomyosarcoma have a similar presentation with hematoxylin and eosin stain. The diagnostic process for spindle cell tumors is greatly aided by immunohistologic analysis. Spindle cell melanoma usually shows immunoreactivity with S-100. Human melanoma black 45, CD57, and neuron-specific enolase usually do not stain, and CD68 has been demonstrated in a minority of cases.
The initial biopsy specimen in our case displayed a dense dermal atypical spindle cell proliferation with hematoxylin and eosin stain. The differential diagnosis of the proliferation included desmoplastic melanoma, spindle cell squamous cell carcinoma, leiomyosarcoma, angiosarcoma, and atypical fibroxanthoma. Immunostains were used to further study the lesional biopsy. Cytokeratin 34bE12, CK5/6, cerium ammonium molybdate 5.2, CK7, epithelial membrane antigen, CK18, high-molecular-weight cytokeratin, S-100, Melan-A, human melanoma black 45, smooth muscle actin, desmin, CD68, CD34, CD10, and p63 were studied. The atypical dermal spindle cells were positive for S-100. S-100 and Melan-A highlighted an increased number of single melanocytes at the dermoepidermal junction. Other markers were negative.
A 1.0-cm wide excision to fascia was performed. Routine hematoxylin and eosin stain showed atypical dermal spindle cells with perineural invasion (Figure 1). The malignant spindle cells were S-100 positive (Figure 2). The spindle cells were negative for high-molecular-weight cytokeratin, CK5/6, p63, Melan-A, A103, microphthalmia, and tyrosinase. These findings confirm melanoma of the spindle cell type.
|
The lesion was a Clark level V melanoma with a Breslow thickness of at least 12 mm. Perineural invasion was noted and the mitotic index was 7 cells/mm2. Vascular and lymphatic invasion was negative and ulceration was present. Tumor-infiltrating lymphocytes were brisk, resulting in a pathologic staging of pT4NXMX.
On initial histologic study with hematoxylin and eosin stain, spindle cell melanoma lesions tend to be generally quite thick. Manganoni et al12 reported in their series a Breslow thickness ranging from 2.1 to 12 mm with a mean of 5.8 mm.
Treatment in our case involved a second wide incision to fascia with an additional 2-cm margin. The role of sentinel lymph node biopsy (SLNB) remains undefined. Patients with spindle cell melanoma have a lower frequency of positive sentinel lymph nodes than nondesmoplastic melanomas.13 As such, the need to perform SLNB has not been determined.13-15 Our patient had a history of breast cancer. The lesion appeared on the left side of the back and she pre-viously had a complete axillary lymphadenectomy on the left axillae. She declined SLNB. A systematic workup by the oncology service was negative. Continued follow-up has revealed no recurrent disease and recent workup was negative for metastatic disease.
Many studies report the increased incidence of local recurrence after excision for spindle cell melanoma as compared to non–spindle cell melanoma,16-18 which is likely related to perineural invasion as in our patient.
Spindle cell melanoma is a rare tumor that is often amelanotic and difficult to diagnose clinically. Routine hematoxylin and eosin staining shows a dermal spindle cell tumor. Immunohistochemical study is of great aid in defining the tumor. The clinician and pathologist must work together to correctly diagnose and treat this lesion.
1. Mikkilineni R, Weinstock MA. Epidemiology. In: Sober AJ, Haluska FG, eds. Atlas of Clinical Oncology: Skin Cancer. London, England: BC Decker; 2001:1-15.
2. Rigel D. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204-209.
3. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed February 10, 2015.
4. Conley J, Lattes R, Orr W. Desmoplastic malignant melanoma (a rare variant of malignant melanoma. Cancer. 1971;28:914-936.
5. Repertinger SK, Teruya B, Sarma DP. Common spindle cell malignant neoplasms of the skin: differential diagnosis and review of the literature. Internet J Dermatol. 2009;7. https://ispub.com/IJD/7/2/11747. Accessed February 10, 2015.
6. Chang PC, Fischbein NJ, McCalmont TH, et al. Perineural spread of malignant melanoma of the head and neck: clinical and imaging features. AJNR Am J Neuroradial. 2004;25:5-11.
7. Cruz J, Reis-Filho JS, Lopes JM. Malignant peripheral nerve sheath tumor-like primary cutaneous melanoma. J Clin Pathol. 2004;57:218-220.
8. Tsao H, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma. Semin Cutan Med Surg. 1997;16:45-47.
9. Kossard S, Doherty E, Murray E. Neurotropic melanoma. a variant of desmoplastic melanoma. Arch Dermatol. 1997;7:907-912.
10. Bruijn JA, Salasche S, Sober AJ, et al. Desmoplastic melanoma: clinicopathologic aspects of six cases. Dermatology. 1992;185:3-8.
11. Jesitus J. Desmoplastic melanoma. Dermatology Times. March 2009:1-2.
12. Manganoni AM, Farisoglio C, Bassissi S, et al. Desmoplastic melanoma: report of 5 cases. Dermatol Res Pract. 2009;2009:679010.
13. Gyorki DE, Busam K, Panageas K, et al. Sentinel lymph node biopsy for patients with desmoplastic melanoma. Ann Surg Oncol. 2003;10:403-407.
14. Livestro DP, Muzikansky A, Kaine EM, et al. of desmoplastic melanoma: a case-control comparison with other melanomas. J Clin Oncol. 2005;23:6739-6746.
15. Pawlik TM, Ross MI, Prieto VG, et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer. 2006;106:900-906.
16. Smithers HM, McLeod GR, Little JH. Desmoplastic melanoma: patterns of recurrence. World J Surg. 1992;16:186-190.
17. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology. 2004;36:445-451.
18. Bruijn JA, Mihm MC Jr, Barnhill RL. Desmoplastic melanoma. Histopathology. 1992;20:197-205.
1. Mikkilineni R, Weinstock MA. Epidemiology. In: Sober AJ, Haluska FG, eds. Atlas of Clinical Oncology: Skin Cancer. London, England: BC Decker; 2001:1-15.
2. Rigel D. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204-209.
3. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed February 10, 2015.
4. Conley J, Lattes R, Orr W. Desmoplastic malignant melanoma (a rare variant of malignant melanoma. Cancer. 1971;28:914-936.
5. Repertinger SK, Teruya B, Sarma DP. Common spindle cell malignant neoplasms of the skin: differential diagnosis and review of the literature. Internet J Dermatol. 2009;7. https://ispub.com/IJD/7/2/11747. Accessed February 10, 2015.
6. Chang PC, Fischbein NJ, McCalmont TH, et al. Perineural spread of malignant melanoma of the head and neck: clinical and imaging features. AJNR Am J Neuroradial. 2004;25:5-11.
7. Cruz J, Reis-Filho JS, Lopes JM. Malignant peripheral nerve sheath tumor-like primary cutaneous melanoma. J Clin Pathol. 2004;57:218-220.
8. Tsao H, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma. Semin Cutan Med Surg. 1997;16:45-47.
9. Kossard S, Doherty E, Murray E. Neurotropic melanoma. a variant of desmoplastic melanoma. Arch Dermatol. 1997;7:907-912.
10. Bruijn JA, Salasche S, Sober AJ, et al. Desmoplastic melanoma: clinicopathologic aspects of six cases. Dermatology. 1992;185:3-8.
11. Jesitus J. Desmoplastic melanoma. Dermatology Times. March 2009:1-2.
12. Manganoni AM, Farisoglio C, Bassissi S, et al. Desmoplastic melanoma: report of 5 cases. Dermatol Res Pract. 2009;2009:679010.
13. Gyorki DE, Busam K, Panageas K, et al. Sentinel lymph node biopsy for patients with desmoplastic melanoma. Ann Surg Oncol. 2003;10:403-407.
14. Livestro DP, Muzikansky A, Kaine EM, et al. of desmoplastic melanoma: a case-control comparison with other melanomas. J Clin Oncol. 2005;23:6739-6746.
15. Pawlik TM, Ross MI, Prieto VG, et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer. 2006;106:900-906.
16. Smithers HM, McLeod GR, Little JH. Desmoplastic melanoma: patterns of recurrence. World J Surg. 1992;16:186-190.
17. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology. 2004;36:445-451.
18. Bruijn JA, Mihm MC Jr, Barnhill RL. Desmoplastic melanoma. Histopathology. 1992;20:197-205.
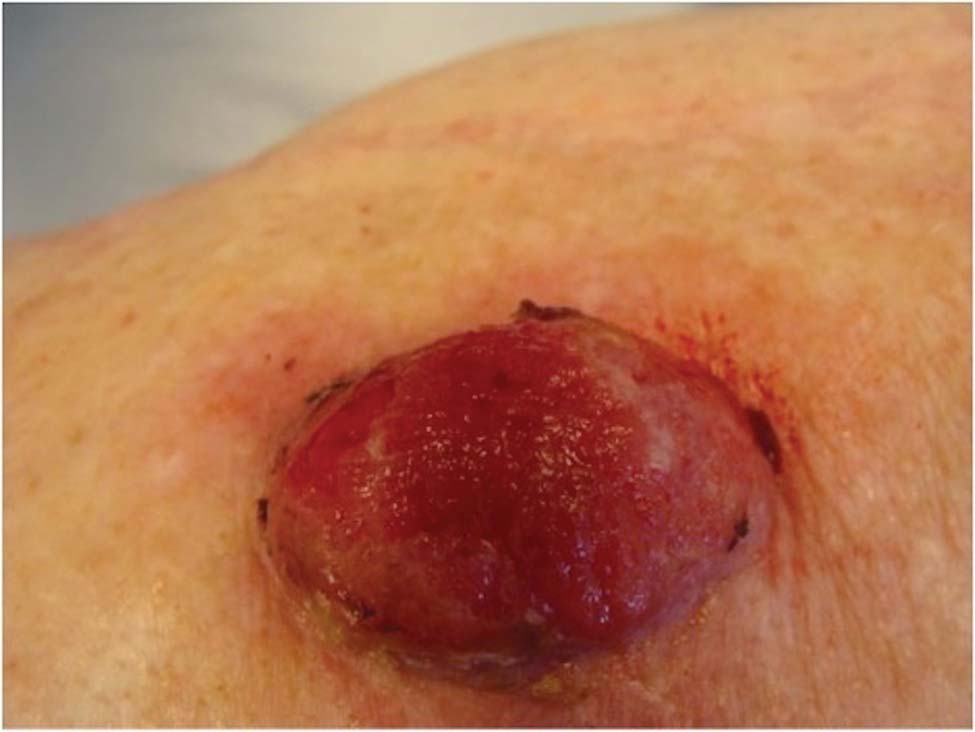
A 78-year-old woman presented with a large friable, sharply demarcated nodule of 3 months’ duration on the left side of the back. The lesion occasionally bled but was otherwise asymptomatic. There was no perilesional paresthesia. The patient’s medical history included hypertension, depression, chronic obstructive pulmonary disease, breast cancer, osteoporosis, and aortic valve disease.
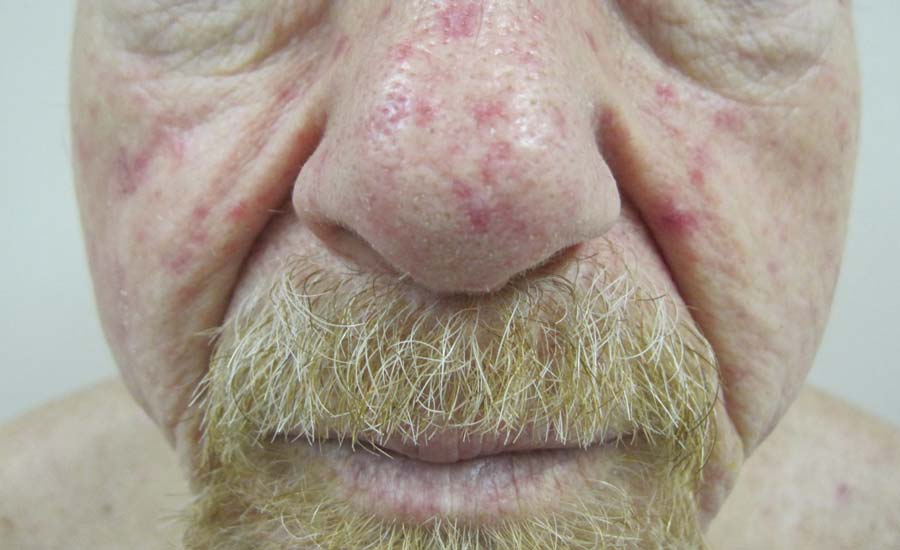
Telangiectases on the Cheeks and Nose
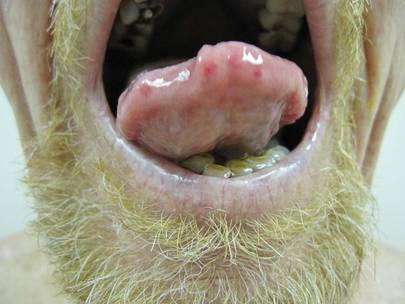
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
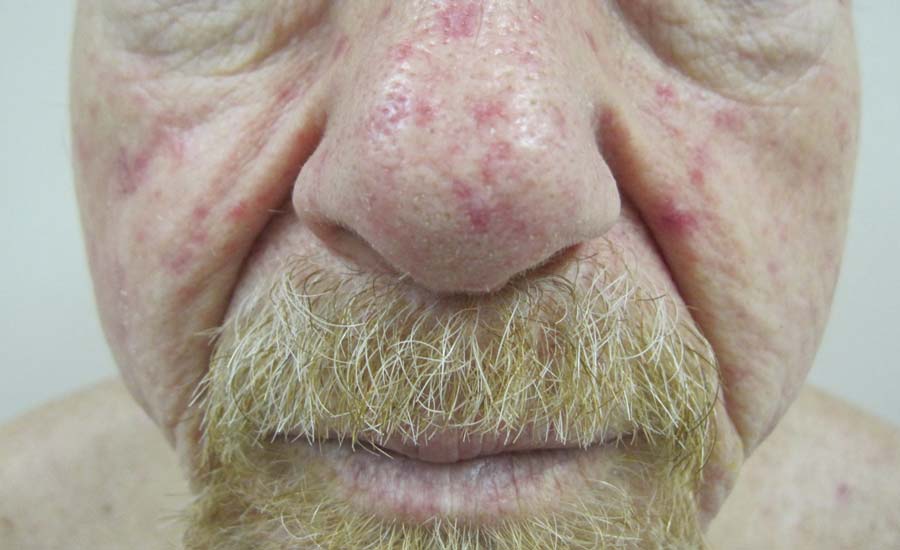
A 68-year-old man presented for evaluation of numerous telangiectases that developed on the cheeks, nose, lips, tongue, and fingers. He denied any history of gastrointestinal tract bleeding but admitted to numerous nosebleeds in the recent past. Family history indicated that his maternal grandmother died of an aneurysm, his mother died of a cerebral hemorrhage, 2 paternal cousins had hemochromatosis, and 2 brothers were healthy.
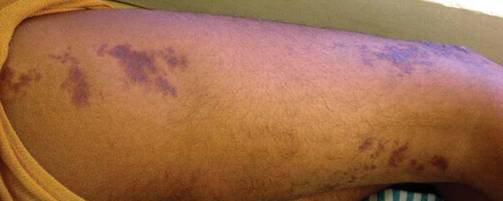
Bluish Red Verrucous Lesions on the Leg
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
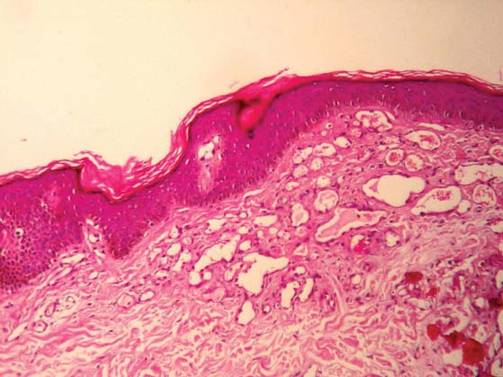
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
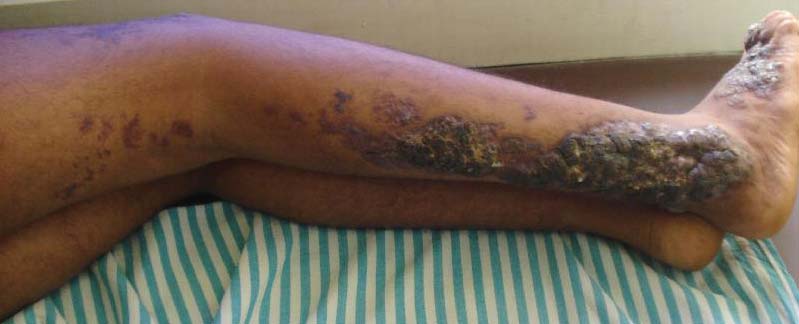
An 18-year-old man presented with a history of multiple bluish verrucous lesions over the right leg. A small nodule on the lateral aspect of the right ankle was present at birth; it increased in size and number and gradually extended to the right buttock. He had recurrent bleeding and infection over the lesions. No other remarkable comorbidities were noted. Dermatologic examination revealed multiple well-circumscribed, bluish red, verrucous lesions distributed linearly along the lateral aspect of the leg. The surface of the lesions was verruciform and showed crusting at places. The second and third toes on the right foot were involved. On the buttock, multiple well-defined, bluish red plaques were present. Both limbs were of equal length.
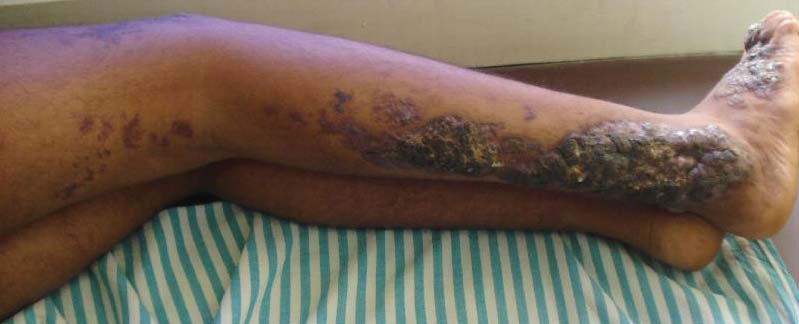
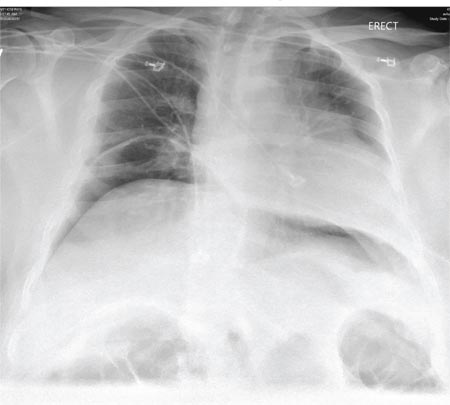
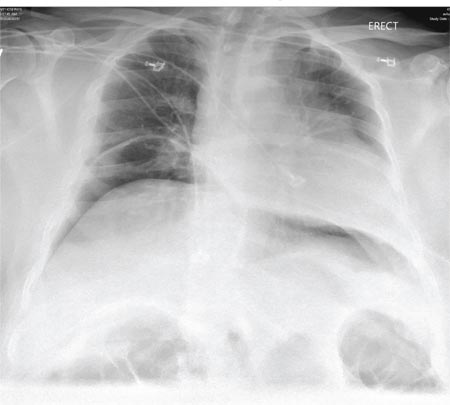
An Incidental Finding During Neuro Evaluation
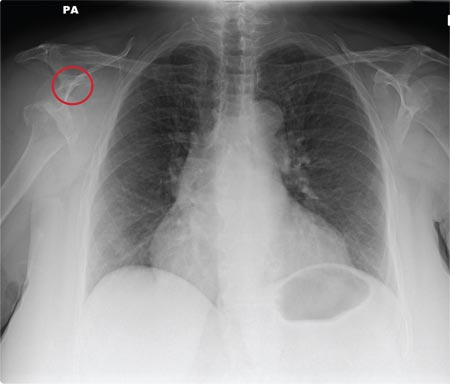
ANSWER
The radiograph shows a normal-appearing chest. Of note, though, is an anterior dislocation of the right shoulder. In addition, there is a fracture within the greater tuberosity of the right humerus.
Prompt orthopedic evaluation is obtained. In further discussion with the family, it was revealed that the patient had been experiencing falls recently; this injury was most likely the result of one.
ANSWER
The radiograph shows a normal-appearing chest. Of note, though, is an anterior dislocation of the right shoulder. In addition, there is a fracture within the greater tuberosity of the right humerus.
Prompt orthopedic evaluation is obtained. In further discussion with the family, it was revealed that the patient had been experiencing falls recently; this injury was most likely the result of one.
ANSWER
The radiograph shows a normal-appearing chest. Of note, though, is an anterior dislocation of the right shoulder. In addition, there is a fracture within the greater tuberosity of the right humerus.
Prompt orthopedic evaluation is obtained. In further discussion with the family, it was revealed that the patient had been experiencing falls recently; this injury was most likely the result of one.
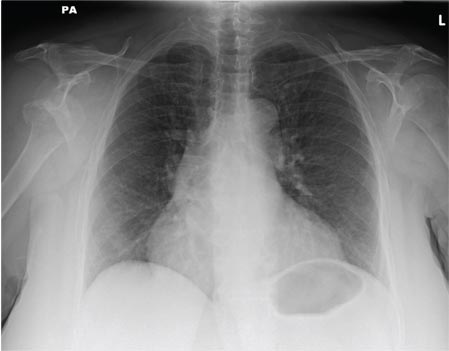
A 65-year-old woman is transferred to your facility from an outlying hospital for evaluation of a brain tumor. Family members found the patient sitting on the sofa, with a decreased level of consciousness. There was reported “seizure-type activity.” When she arrived at the outlying hospital, the patient was noted to have right-side weakness. Stat CT of the head demonstrated a fairly large parasagittal mass, and the patient was urgently transferred to your facility for neurosurgical evaluation. Primary survey on arrival shows an older female who is awake, alert, and in no obvious distress. Vital signs are normal. She has fairly pronounced right upper extremity weakness, more proximally than distally. Otherwise, the exam grossly appears normal. The patient’s initial imaging studies were sent with her on a CD. As you are trying to view the images of the brain, a chest radiograph pops up on your screen. What is your impression?
What Does This Man Need (Besides Milk & Cookies)?
ANSWER
This ECG is representative of sinus rhythm with second-degree atrioventricular block with 2:1 conduction; possible left atrial enlargement; and ST-T wave abnormalities suspicious for lateral ischemia.
Sinus rhythm is evidenced by the P waves that march through at a rate that is consistently double that of the QRS rate (82 beats/min). The PR interval in the conducted beats remains constant, with every other P wave blocked from conducting into the ventricles.
The biphasic P wave seen in lead V1 does not meet criteria for left atrial enlargement (P wave in lead I ≥ 110 ms, terminal negative P wave in lead V1 ≥ 1 mm2) but is suspicious. Finally, ST-T wave elevations in leads V2-V4 are suspicious for ventricular septal ischemia.
The patient underwent placement of a dual-chamber permanent pacemaker. He has done well since.
ANSWER
This ECG is representative of sinus rhythm with second-degree atrioventricular block with 2:1 conduction; possible left atrial enlargement; and ST-T wave abnormalities suspicious for lateral ischemia.
Sinus rhythm is evidenced by the P waves that march through at a rate that is consistently double that of the QRS rate (82 beats/min). The PR interval in the conducted beats remains constant, with every other P wave blocked from conducting into the ventricles.
The biphasic P wave seen in lead V1 does not meet criteria for left atrial enlargement (P wave in lead I ≥ 110 ms, terminal negative P wave in lead V1 ≥ 1 mm2) but is suspicious. Finally, ST-T wave elevations in leads V2-V4 are suspicious for ventricular septal ischemia.
The patient underwent placement of a dual-chamber permanent pacemaker. He has done well since.
ANSWER
This ECG is representative of sinus rhythm with second-degree atrioventricular block with 2:1 conduction; possible left atrial enlargement; and ST-T wave abnormalities suspicious for lateral ischemia.
Sinus rhythm is evidenced by the P waves that march through at a rate that is consistently double that of the QRS rate (82 beats/min). The PR interval in the conducted beats remains constant, with every other P wave blocked from conducting into the ventricles.
The biphasic P wave seen in lead V1 does not meet criteria for left atrial enlargement (P wave in lead I ≥ 110 ms, terminal negative P wave in lead V1 ≥ 1 mm2) but is suspicious. Finally, ST-T wave elevations in leads V2-V4 are suspicious for ventricular septal ischemia.
The patient underwent placement of a dual-chamber permanent pacemaker. He has done well since.

A 74-year-old man has been a resident of a skilled nursing facility for seven years and is well known to the staff. This morning, when the medical assistant performed a routine vital sign check, she noticed the patient’s heart rate was in the 40s. This newly discovered bradycardia, coupled with a four-day history of lethargy, prompts the facility to transfer the patient to your emergency department for evaluation. The patient has a history of hypertension, hypothyroidism, chronic obstructive pulmonary disease, GERD, osteoarthritis, and dementia. Surgical history includes appendectomy, cholecystectomy, and left hip replacement. The patient’s multiple chronic conditions are well managed with medications, including a b-blocker, hydrochlorothiazide, levothyroxine, and an inhaler. He receives protein and vitamin supplements daily and is allergic to penicillin. There is a remote history of smoking (from his youth and tour of duty in the Korean War), although the patient says he hasn’t smoked in 30 years. He has “never touched” alcohol, because his father died of complications from alcoholism at age 45. The patient’s wife died of a stroke 11 years ago. His son (and family) visit him twice weekly, bringing chocolate milk and cookies that the patient anxiously awaits. The review of systems is remarkable for a recent cold (resolved), urinary retention, and loose stools. The patient’s appetite is intact. He also exhibits evidence of short-term memory loss; however, this is sporadic. Vital signs on arrival include a blood pressure of 158/88 mm Hg; pulse, 48 beats/min and regular; respiratory rate, 14 breaths/min; and temperature, 97.6°F. His weight is 174 lb and his height, 69 in. Pertinent findings on the physical exam include mild cataracts bilaterally, a right carotid bruit, no evidence of elevated neck veins, and late expiratory wheezes in both bases. The cardiac exam is remarkable for a regular rhythm with a heart rate of 42 beats/min. There is a grade II/VI early systolic murmur at the left upper sternal border but no radiation, extra heart sounds, or rubs. The abdomen is soft and nontender, with old surgical scars, and the abdominal aorta is easily palpable. The extremities exhibit full range of motion without peripheral edema, and osteoarthritic changes are evident in both hands. An ECG shows a ventricular rate of 43 beats/min; PR interval, 198 ms; QRS duration, 96 ms; QT/QTc interval, 464/392 ms; P axis, 60°; R axis, 4°; and T axis, 107°. What is your interpretation of this ECG?
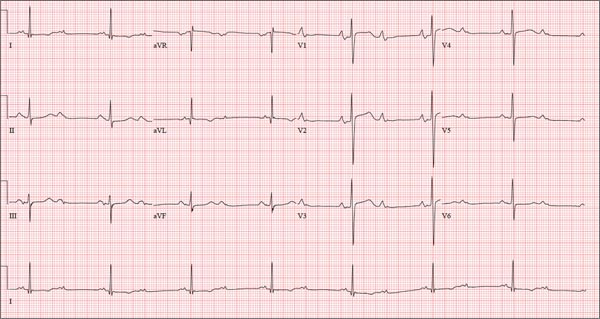
Boy Has Had “Bald Spot” Since Birth
ANSWER
The answer is temporal triangular alopecia (choice “d”), an unusual form of permanent hair loss preferentially affecting the exact area depicted in this case.
Alopecia areata (choice “a”) involves localized hair loss. By contrast, this patient never had hair in this area to lose.
Nevus sebaceous (choice “b”) is a congenital hamartoma that is typically hairless; there are no follicles, and the bumpy, rough surface is composed of sebaceous globules.
Cutis aplasia (choice “c”) manifests with hairless lesions, but there is marked aplasia of the skin as well and no surface adnexae, let alone hairs or follicles.
DISCUSSION
Temporal triangular alopecia (TTA) is an unusual type of alopecia. Of unknown origin, it usually affects this area of the scalp—and usually unilaterally. Approximately one-third of TTA patients are born with the condition; the rest develop it in the first two to three years of life. As in this case, it is often wrongly attributed to the use of forceps but has nothing to do with trauma. One school of thought holds that TTA is probably an inherited condition—but others disagree.
TTA was originally known as congenital triangular alopecia. However, when enough cases had been accumulated to accurately determine the nature of the condition, it was realized that TTA is not always congenital or triangular. Thus, a new name was bestowed.
The hallmark of TTA is the normal number of hair follicles that only grow vellus hairs. The solitary peripheral tuft of terminal dark hairs is typical of TTA and thus a confirmatory finding.
TREATMENT/PROGNOSIS
TTA is by definition permanent. Since there’s no inflammation (a key difference from alopecia areata), steroids are useless. The only successful treatment for TTA, if any is attempted, is hair transplantation. As of this writing, the family is mulling this treatment option.
ANSWER
The answer is temporal triangular alopecia (choice “d”), an unusual form of permanent hair loss preferentially affecting the exact area depicted in this case.
Alopecia areata (choice “a”) involves localized hair loss. By contrast, this patient never had hair in this area to lose.
Nevus sebaceous (choice “b”) is a congenital hamartoma that is typically hairless; there are no follicles, and the bumpy, rough surface is composed of sebaceous globules.
Cutis aplasia (choice “c”) manifests with hairless lesions, but there is marked aplasia of the skin as well and no surface adnexae, let alone hairs or follicles.
DISCUSSION
Temporal triangular alopecia (TTA) is an unusual type of alopecia. Of unknown origin, it usually affects this area of the scalp—and usually unilaterally. Approximately one-third of TTA patients are born with the condition; the rest develop it in the first two to three years of life. As in this case, it is often wrongly attributed to the use of forceps but has nothing to do with trauma. One school of thought holds that TTA is probably an inherited condition—but others disagree.
TTA was originally known as congenital triangular alopecia. However, when enough cases had been accumulated to accurately determine the nature of the condition, it was realized that TTA is not always congenital or triangular. Thus, a new name was bestowed.
The hallmark of TTA is the normal number of hair follicles that only grow vellus hairs. The solitary peripheral tuft of terminal dark hairs is typical of TTA and thus a confirmatory finding.
TREATMENT/PROGNOSIS
TTA is by definition permanent. Since there’s no inflammation (a key difference from alopecia areata), steroids are useless. The only successful treatment for TTA, if any is attempted, is hair transplantation. As of this writing, the family is mulling this treatment option.
ANSWER
The answer is temporal triangular alopecia (choice “d”), an unusual form of permanent hair loss preferentially affecting the exact area depicted in this case.
Alopecia areata (choice “a”) involves localized hair loss. By contrast, this patient never had hair in this area to lose.
Nevus sebaceous (choice “b”) is a congenital hamartoma that is typically hairless; there are no follicles, and the bumpy, rough surface is composed of sebaceous globules.
Cutis aplasia (choice “c”) manifests with hairless lesions, but there is marked aplasia of the skin as well and no surface adnexae, let alone hairs or follicles.
DISCUSSION
Temporal triangular alopecia (TTA) is an unusual type of alopecia. Of unknown origin, it usually affects this area of the scalp—and usually unilaterally. Approximately one-third of TTA patients are born with the condition; the rest develop it in the first two to three years of life. As in this case, it is often wrongly attributed to the use of forceps but has nothing to do with trauma. One school of thought holds that TTA is probably an inherited condition—but others disagree.
TTA was originally known as congenital triangular alopecia. However, when enough cases had been accumulated to accurately determine the nature of the condition, it was realized that TTA is not always congenital or triangular. Thus, a new name was bestowed.
The hallmark of TTA is the normal number of hair follicles that only grow vellus hairs. The solitary peripheral tuft of terminal dark hairs is typical of TTA and thus a confirmatory finding.
TREATMENT/PROGNOSIS
TTA is by definition permanent. Since there’s no inflammation (a key difference from alopecia areata), steroids are useless. The only successful treatment for TTA, if any is attempted, is hair transplantation. As of this writing, the family is mulling this treatment option.
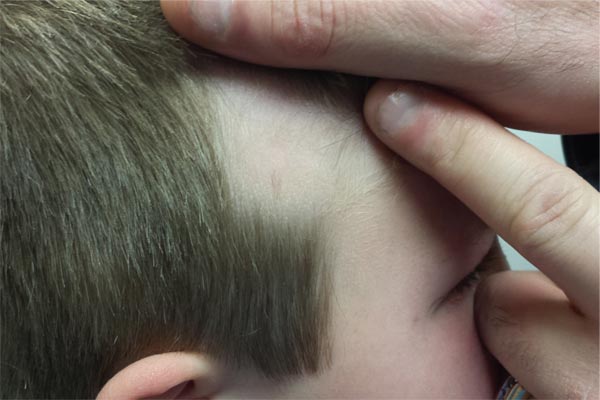
Since birth, this 8-year-old boy has had a “bald spot” on his scalp. The pediatrician who attended the birth suggested trauma as the cause, since forceps were used to facilitate delivery. But the problem has failed to resolve, leaving the boy an object of ridicule among his classmates. According to the patient’s parents, there has never been any broken skin or hair growth in the area. There is no family history of similar problems, and the child’s health history is unremarkable. The child’s current pediatrician, who made the referral to dermatology, suggested the lesion might be a form of nevus sebaceous. The affected site is roughly triangular, measures about 3 cm on each side, and is located just inside the temporal scalp. The hair loss in this sharply circumscribed area is almost complete, with a lone tuft of darker terminal hairs on the inferior aspect of the site. No redness or epidermal disturbance (eg, scaling) is noted. Dermatoscopic examination (with 10x magnification) reveals a normal number of follicles and hairs. The latter are vellus hairs, except for the aforementioned solitary tuft. The rest of the scalp, including the same location on the opposite side, is free of any significant changes.

Waxy Indurated Plaques on the Eyelids
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
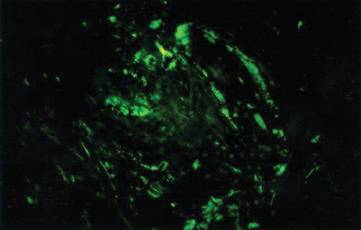
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
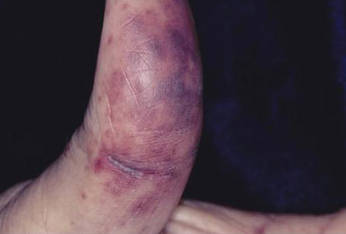
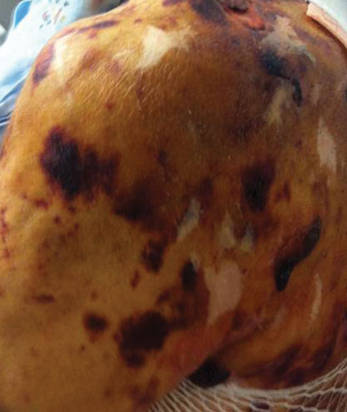
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
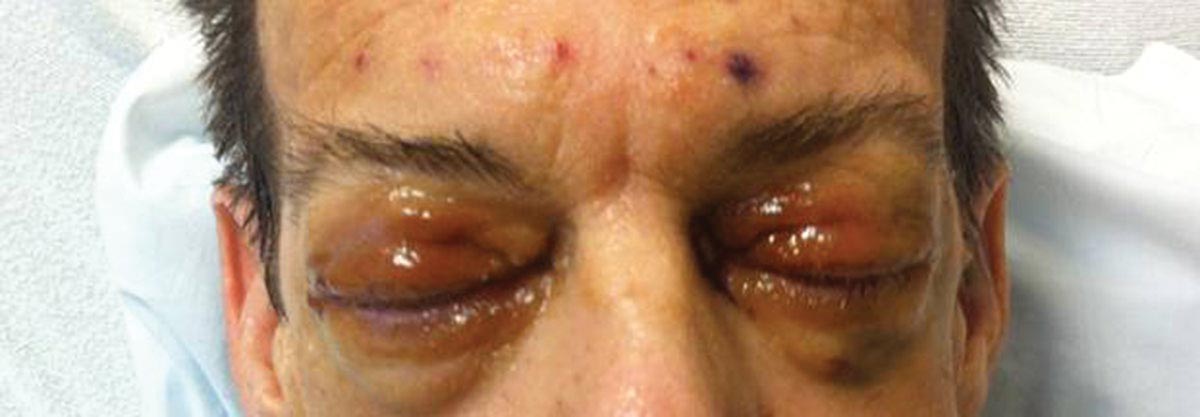
A 57-year-old man who underwent an orthotopic liver transplant 10 months prior for autoimmune hepatitis developed a rash on the trunk, arms, and legs. A dermatology consultation was requested. On evaluation, the patient was noted to have purpuric patches on the trunk and extremities, as well as prominent, bilateral, waxy and indurated periorbital plaques. The patient’s medical history revealed anemia, osteopenia, multiple bone fractures, and renal impairment.