User login
Study supports palliative care in HSCT recipients
![]()
Photo by Chad McNeeley
Palliative care can be beneficial for patients undergoing hematopoietic stem cell transplant (HSCT) to treat hematologic malignancies, according to research published in JAMA.
The single-center study suggested that palliative care can improve HSCT recipients’ quality of life, relieve symptoms associated with the procedure, and reduce depression and anxiety.
Researchers observed such benefits during hospitalization for HSCT and a few months later.
In addition, caregivers of patients receiving palliative care experienced less depression and were better at coping with the stress associated with the illness of their loved one.
“Palliative care clinicians are increasingly asked to help care for patients with solid tumors but are rarely consulted for patients with hematologic malignancies, especially those receiving therapy designed to cure their disease,” said study author Areej El-Jawahri, MD, of Massachusette General Hospital in Boston.
“The physical and psychological symptoms associated with HSCT are sometimes regarded as expected and unavoidable, which, combined with the persistent misperception that equates palliative care with end-of-life care, has contributed to a lack of involvement of palliative care clinicians in the care of these patients.”
Intervention
Dr El-Jawahri and her colleagues studied 160 patients who underwent autologous or allogeneic HSCT to treat a variety of hematologic malignancies from August 2014 into January 2016.
Participants were randomized to receive either standard care (n=79) or the palliative care intervention (n=81).
Within 3 days of their admission to the hospital, patients in the intervention group had an initial meeting with a palliative care clinician—a physician or advance practice nurse—who continued to meet with them at least twice a week during their hospitalization.
At the meetings, which could be attended by a family member or friend of the patient, clinicians first focused on establishing a rapport with patients and their caregivers.
Clinicians addressed ways of managing the physical and psychological symptoms patients were experiencing and provided support and strategies for coping with distress. Patients received an average of 8 palliative care visits during their hospitalizations, which lasted on average 20 days.
At the outset of the study and 2 weeks into the process, a time when symptoms tend to be at their worst, patients in both groups and participating caregivers completed questionnaires assessing their mood and quality of life.
Patients also completed questionnaires asking about symptoms of their illness and those associated with the procedure. Patients completed additional assessments 3 months after HSCT as well.
Results
The study’s primary endpoint was change in quality of life from baseline to week 2. Patients receiving the palliative care intervention had significantly better quality of life scores at week 2 than patients in the control group.
Also at the 2-week mark, patients receiving the palliative care intervention reported lower levels of depression, anxiety, and symptoms than the control group, but there was no significant difference between the groups with regard to fatigue.
At 3 months, patients receiving the palliative care intervention still had higher quality of life scores and less depression than controls, but there were no significant between-group differences in anxiety, fatigue, or symptom burden.
Caregivers attended 42% of the palliative care sessions. At the 2-week assessment, caregivers in the intervention group were found to have fewer depressive symptoms and improved coping skills, compared with caregivers in the control group.
“Caregivers play a crucial role in supporting patients during the transplant process, and they are substantially impacted as they watch their loved ones struggle with side effects that can be emotionally challenging,” Dr El-Jawahri said.
She and her colleagues noted that additional, larger studies are needed to assess caregiver impacts more completely, to replicate patient results at centers with more diverse patient populations, to assess the inclusion of more complete palliative care teams, to collect cost data, and to adapt the palliative care intervention to assist patients receiving other potentially curative treatment for hematologic or other cancers. ![]()
![]()
Photo by Chad McNeeley
Palliative care can be beneficial for patients undergoing hematopoietic stem cell transplant (HSCT) to treat hematologic malignancies, according to research published in JAMA.
The single-center study suggested that palliative care can improve HSCT recipients’ quality of life, relieve symptoms associated with the procedure, and reduce depression and anxiety.
Researchers observed such benefits during hospitalization for HSCT and a few months later.
In addition, caregivers of patients receiving palliative care experienced less depression and were better at coping with the stress associated with the illness of their loved one.
“Palliative care clinicians are increasingly asked to help care for patients with solid tumors but are rarely consulted for patients with hematologic malignancies, especially those receiving therapy designed to cure their disease,” said study author Areej El-Jawahri, MD, of Massachusette General Hospital in Boston.
“The physical and psychological symptoms associated with HSCT are sometimes regarded as expected and unavoidable, which, combined with the persistent misperception that equates palliative care with end-of-life care, has contributed to a lack of involvement of palliative care clinicians in the care of these patients.”
Intervention
Dr El-Jawahri and her colleagues studied 160 patients who underwent autologous or allogeneic HSCT to treat a variety of hematologic malignancies from August 2014 into January 2016.
Participants were randomized to receive either standard care (n=79) or the palliative care intervention (n=81).
Within 3 days of their admission to the hospital, patients in the intervention group had an initial meeting with a palliative care clinician—a physician or advance practice nurse—who continued to meet with them at least twice a week during their hospitalization.
At the meetings, which could be attended by a family member or friend of the patient, clinicians first focused on establishing a rapport with patients and their caregivers.
Clinicians addressed ways of managing the physical and psychological symptoms patients were experiencing and provided support and strategies for coping with distress. Patients received an average of 8 palliative care visits during their hospitalizations, which lasted on average 20 days.
At the outset of the study and 2 weeks into the process, a time when symptoms tend to be at their worst, patients in both groups and participating caregivers completed questionnaires assessing their mood and quality of life.
Patients also completed questionnaires asking about symptoms of their illness and those associated with the procedure. Patients completed additional assessments 3 months after HSCT as well.
Results
The study’s primary endpoint was change in quality of life from baseline to week 2. Patients receiving the palliative care intervention had significantly better quality of life scores at week 2 than patients in the control group.
Also at the 2-week mark, patients receiving the palliative care intervention reported lower levels of depression, anxiety, and symptoms than the control group, but there was no significant difference between the groups with regard to fatigue.
At 3 months, patients receiving the palliative care intervention still had higher quality of life scores and less depression than controls, but there were no significant between-group differences in anxiety, fatigue, or symptom burden.
Caregivers attended 42% of the palliative care sessions. At the 2-week assessment, caregivers in the intervention group were found to have fewer depressive symptoms and improved coping skills, compared with caregivers in the control group.
“Caregivers play a crucial role in supporting patients during the transplant process, and they are substantially impacted as they watch their loved ones struggle with side effects that can be emotionally challenging,” Dr El-Jawahri said.
She and her colleagues noted that additional, larger studies are needed to assess caregiver impacts more completely, to replicate patient results at centers with more diverse patient populations, to assess the inclusion of more complete palliative care teams, to collect cost data, and to adapt the palliative care intervention to assist patients receiving other potentially curative treatment for hematologic or other cancers. ![]()
![]()
Photo by Chad McNeeley
Palliative care can be beneficial for patients undergoing hematopoietic stem cell transplant (HSCT) to treat hematologic malignancies, according to research published in JAMA.
The single-center study suggested that palliative care can improve HSCT recipients’ quality of life, relieve symptoms associated with the procedure, and reduce depression and anxiety.
Researchers observed such benefits during hospitalization for HSCT and a few months later.
In addition, caregivers of patients receiving palliative care experienced less depression and were better at coping with the stress associated with the illness of their loved one.
“Palliative care clinicians are increasingly asked to help care for patients with solid tumors but are rarely consulted for patients with hematologic malignancies, especially those receiving therapy designed to cure their disease,” said study author Areej El-Jawahri, MD, of Massachusette General Hospital in Boston.
“The physical and psychological symptoms associated with HSCT are sometimes regarded as expected and unavoidable, which, combined with the persistent misperception that equates palliative care with end-of-life care, has contributed to a lack of involvement of palliative care clinicians in the care of these patients.”
Intervention
Dr El-Jawahri and her colleagues studied 160 patients who underwent autologous or allogeneic HSCT to treat a variety of hematologic malignancies from August 2014 into January 2016.
Participants were randomized to receive either standard care (n=79) or the palliative care intervention (n=81).
Within 3 days of their admission to the hospital, patients in the intervention group had an initial meeting with a palliative care clinician—a physician or advance practice nurse—who continued to meet with them at least twice a week during their hospitalization.
At the meetings, which could be attended by a family member or friend of the patient, clinicians first focused on establishing a rapport with patients and their caregivers.
Clinicians addressed ways of managing the physical and psychological symptoms patients were experiencing and provided support and strategies for coping with distress. Patients received an average of 8 palliative care visits during their hospitalizations, which lasted on average 20 days.
At the outset of the study and 2 weeks into the process, a time when symptoms tend to be at their worst, patients in both groups and participating caregivers completed questionnaires assessing their mood and quality of life.
Patients also completed questionnaires asking about symptoms of their illness and those associated with the procedure. Patients completed additional assessments 3 months after HSCT as well.
Results
The study’s primary endpoint was change in quality of life from baseline to week 2. Patients receiving the palliative care intervention had significantly better quality of life scores at week 2 than patients in the control group.
Also at the 2-week mark, patients receiving the palliative care intervention reported lower levels of depression, anxiety, and symptoms than the control group, but there was no significant difference between the groups with regard to fatigue.
At 3 months, patients receiving the palliative care intervention still had higher quality of life scores and less depression than controls, but there were no significant between-group differences in anxiety, fatigue, or symptom burden.
Caregivers attended 42% of the palliative care sessions. At the 2-week assessment, caregivers in the intervention group were found to have fewer depressive symptoms and improved coping skills, compared with caregivers in the control group.
“Caregivers play a crucial role in supporting patients during the transplant process, and they are substantially impacted as they watch their loved ones struggle with side effects that can be emotionally challenging,” Dr El-Jawahri said.
She and her colleagues noted that additional, larger studies are needed to assess caregiver impacts more completely, to replicate patient results at centers with more diverse patient populations, to assess the inclusion of more complete palliative care teams, to collect cost data, and to adapt the palliative care intervention to assist patients receiving other potentially curative treatment for hematologic or other cancers. ![]()
Antiviral appears active against tough-to-treat CMV
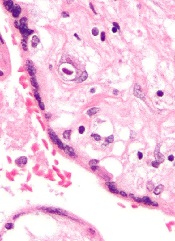
NEW ORLEANS—The antiviral agent maribavir has shown activity against cytomegalovirus (CMV) in a phase 2 trial.
The trial included transplant recipients with a CMV infection that was resistant or refractory to prior treatment with (val)ganciclovir or foscarnet.
Sixty-seven percent of patients treated with varying doses of maribavir for up to 24 weeks had no detectable levels of CMV in their plasma within 6 weeks of starting treatment.
However, CMV infection did recur in some of these patients.
Dysgeusia was the most commonly reported treatment-emergent adverse event (AE), and one patient died of an AE that was considered possibly related to maribavir.
These results were presented at IDWeek 2016 (abstract 78). The study was sponsored by Shire.
“Cytomegalovirus infection that is resistant or refractory to standard therapy in transplant patients is associated with significant complications and high mortality,” said study investigator Genovefa Papanicolaou, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“The phase 2 findings support further research to confirm these results among patients who have limited options to combat the infection.”
Maribavir is a member of a class of drugs called benzimidazole ribosides. Maribavir is thought to inhibit viral DNA assembly and egress of viral capsids from the nucleus of infected cells. The drug has not been shown to affect the maturation of viral DNA or affect the viral DNA polymerase.
This phase 2 study of maribavir included 120 patients ages 12 and older with CMV infection (≥1000 DNA copies/mL of blood plasma) that was resistant or refractory to (val)ganciclovir or foscarnet.
Forty-seven of the patients had received a hematopoietic stem cell transplant, and 73 had a solid organ transplant. The patients’ median age was 55 (range, 18 to 74).
The patients were randomized to 1 of 3 twice-daily oral doses of maribavir—400 mg, 800 mg, or 1200 mg—for up to 24 weeks of treatment.
Efficacy
The study’s primary efficacy endpoint was the proportion of patients with confirmed undetectable plasma CMV DNA within 6 weeks of treatment.
Overall, 67% (80/120) of patients met the primary efficacy endpoint. This included 70% (n=28) of patients in the 400 mg group, 63% (n=25) in the 800 mg group, and 67% (n=27) in the 1200 mg group.
CMV infection recurred in 30 patients, including 7 in the 400 mg group, 11 in the 800 mg group, and 12 in the 1200 mg group.
Safety
The primary safety analysis was focused on the incidence of treatment-emergent AEs. The incidence was 78% (n=93) overall, including 78% (n=31) in the 400 mg group, 80% (n=32) in the 800 mg group, and 75% (n=30) in the 1200 mg group.
Twenty-seven percent of patients died due to any AE, 1 of which (multi-organ failure) was considered possibly related to maribavir.
Forty-one patients (34%) discontinued treatment with maribavir due to an AE, including 17 patients who discontinued due to CMV infection.
Dysgeusia was the most common treatment-emergent AE and led to treatment discontinuation in 1 patient. Dysgeusia occurred in 65% (n=78) of all patients, including 60% (n=24) in the 400 mg group, 63% (n=25) in the 800 mg group, and 73% (n=29) in the 1200 mg group.
Other treatment-emergent AEs (occurring in at least 20% of patients) for all doses included nausea, vomiting, CMV infection, diarrhea, fatigue, and anemia. Immunosuppressant drug level increases were reported as an AE in 10% of all patients. ![]()
NEW ORLEANS—The antiviral agent maribavir has shown activity against cytomegalovirus (CMV) in a phase 2 trial.
The trial included transplant recipients with a CMV infection that was resistant or refractory to prior treatment with (val)ganciclovir or foscarnet.
Sixty-seven percent of patients treated with varying doses of maribavir for up to 24 weeks had no detectable levels of CMV in their plasma within 6 weeks of starting treatment.
However, CMV infection did recur in some of these patients.
Dysgeusia was the most commonly reported treatment-emergent adverse event (AE), and one patient died of an AE that was considered possibly related to maribavir.
These results were presented at IDWeek 2016 (abstract 78). The study was sponsored by Shire.
“Cytomegalovirus infection that is resistant or refractory to standard therapy in transplant patients is associated with significant complications and high mortality,” said study investigator Genovefa Papanicolaou, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“The phase 2 findings support further research to confirm these results among patients who have limited options to combat the infection.”
Maribavir is a member of a class of drugs called benzimidazole ribosides. Maribavir is thought to inhibit viral DNA assembly and egress of viral capsids from the nucleus of infected cells. The drug has not been shown to affect the maturation of viral DNA or affect the viral DNA polymerase.
This phase 2 study of maribavir included 120 patients ages 12 and older with CMV infection (≥1000 DNA copies/mL of blood plasma) that was resistant or refractory to (val)ganciclovir or foscarnet.
Forty-seven of the patients had received a hematopoietic stem cell transplant, and 73 had a solid organ transplant. The patients’ median age was 55 (range, 18 to 74).
The patients were randomized to 1 of 3 twice-daily oral doses of maribavir—400 mg, 800 mg, or 1200 mg—for up to 24 weeks of treatment.
Efficacy
The study’s primary efficacy endpoint was the proportion of patients with confirmed undetectable plasma CMV DNA within 6 weeks of treatment.
Overall, 67% (80/120) of patients met the primary efficacy endpoint. This included 70% (n=28) of patients in the 400 mg group, 63% (n=25) in the 800 mg group, and 67% (n=27) in the 1200 mg group.
CMV infection recurred in 30 patients, including 7 in the 400 mg group, 11 in the 800 mg group, and 12 in the 1200 mg group.
Safety
The primary safety analysis was focused on the incidence of treatment-emergent AEs. The incidence was 78% (n=93) overall, including 78% (n=31) in the 400 mg group, 80% (n=32) in the 800 mg group, and 75% (n=30) in the 1200 mg group.
Twenty-seven percent of patients died due to any AE, 1 of which (multi-organ failure) was considered possibly related to maribavir.
Forty-one patients (34%) discontinued treatment with maribavir due to an AE, including 17 patients who discontinued due to CMV infection.
Dysgeusia was the most common treatment-emergent AE and led to treatment discontinuation in 1 patient. Dysgeusia occurred in 65% (n=78) of all patients, including 60% (n=24) in the 400 mg group, 63% (n=25) in the 800 mg group, and 73% (n=29) in the 1200 mg group.
Other treatment-emergent AEs (occurring in at least 20% of patients) for all doses included nausea, vomiting, CMV infection, diarrhea, fatigue, and anemia. Immunosuppressant drug level increases were reported as an AE in 10% of all patients. ![]()
NEW ORLEANS—The antiviral agent maribavir has shown activity against cytomegalovirus (CMV) in a phase 2 trial.
The trial included transplant recipients with a CMV infection that was resistant or refractory to prior treatment with (val)ganciclovir or foscarnet.
Sixty-seven percent of patients treated with varying doses of maribavir for up to 24 weeks had no detectable levels of CMV in their plasma within 6 weeks of starting treatment.
However, CMV infection did recur in some of these patients.
Dysgeusia was the most commonly reported treatment-emergent adverse event (AE), and one patient died of an AE that was considered possibly related to maribavir.
These results were presented at IDWeek 2016 (abstract 78). The study was sponsored by Shire.
“Cytomegalovirus infection that is resistant or refractory to standard therapy in transplant patients is associated with significant complications and high mortality,” said study investigator Genovefa Papanicolaou, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“The phase 2 findings support further research to confirm these results among patients who have limited options to combat the infection.”
Maribavir is a member of a class of drugs called benzimidazole ribosides. Maribavir is thought to inhibit viral DNA assembly and egress of viral capsids from the nucleus of infected cells. The drug has not been shown to affect the maturation of viral DNA or affect the viral DNA polymerase.
This phase 2 study of maribavir included 120 patients ages 12 and older with CMV infection (≥1000 DNA copies/mL of blood plasma) that was resistant or refractory to (val)ganciclovir or foscarnet.
Forty-seven of the patients had received a hematopoietic stem cell transplant, and 73 had a solid organ transplant. The patients’ median age was 55 (range, 18 to 74).
The patients were randomized to 1 of 3 twice-daily oral doses of maribavir—400 mg, 800 mg, or 1200 mg—for up to 24 weeks of treatment.
Efficacy
The study’s primary efficacy endpoint was the proportion of patients with confirmed undetectable plasma CMV DNA within 6 weeks of treatment.
Overall, 67% (80/120) of patients met the primary efficacy endpoint. This included 70% (n=28) of patients in the 400 mg group, 63% (n=25) in the 800 mg group, and 67% (n=27) in the 1200 mg group.
CMV infection recurred in 30 patients, including 7 in the 400 mg group, 11 in the 800 mg group, and 12 in the 1200 mg group.
Safety
The primary safety analysis was focused on the incidence of treatment-emergent AEs. The incidence was 78% (n=93) overall, including 78% (n=31) in the 400 mg group, 80% (n=32) in the 800 mg group, and 75% (n=30) in the 1200 mg group.
Twenty-seven percent of patients died due to any AE, 1 of which (multi-organ failure) was considered possibly related to maribavir.
Forty-one patients (34%) discontinued treatment with maribavir due to an AE, including 17 patients who discontinued due to CMV infection.
Dysgeusia was the most common treatment-emergent AE and led to treatment discontinuation in 1 patient. Dysgeusia occurred in 65% (n=78) of all patients, including 60% (n=24) in the 400 mg group, 63% (n=25) in the 800 mg group, and 73% (n=29) in the 1200 mg group.
Other treatment-emergent AEs (occurring in at least 20% of patients) for all doses included nausea, vomiting, CMV infection, diarrhea, fatigue, and anemia. Immunosuppressant drug level increases were reported as an AE in 10% of all patients. ![]()
Drug can fight adenovirus in HSCT recipients
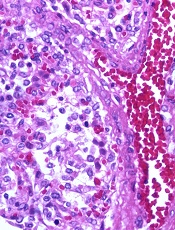
Image by Yale Rosen
NEW ORLEANS—Interim results of a phase 3 trial suggest brincidofovir can treat adenovirus (AdV) infection in recipients of allogeneic hematopoietic stem cell transplant (HSCT).
Both pediatric and adult patients experienced a decline in AdV viral load after brincidofovir treatment, but pediatric patients were more likely to respond.
Overall survival rates were better for patients who had a rapid response and were therefore better among pediatric patients than adults.
Investigators said the adverse events (AEs) in this study were consistent with the known safety profile of brincidofovir.
Michael Grimley, MD, of Cincinnati Children’s Hospital in Ohio, and his colleagues presented these results at IDWeek 2016 (abstract 2339). The research was supported by Chimerix, the company developing brincidofovir.
This trial, known as AdVise, was designed to evaluate brincidofovir for the treatment of AdV infection in pediatric and adult patients divided into 3 cohorts:
- Cohort A consists of allogeneic HSCT recipients with asymptomatic or limited AdV infection
- Cohort B consists of allogeneic HSCT recipients with disseminated AdV disease
- Cohort C consists of autologous HSCT recipients, solid organ transplant recipients, and other immunocompromised patients.
All patients were assigned to 12 weeks of oral brincidofovir, administered twice weekly. An additional 12 weeks of treatment was allowed in patients with ongoing or recurrent infection. After completing treatment, all patients were followed until week 36.
Interim analysis
The investigators examined outcomes at 24 weeks after the first brincidofovir dose (12 weeks after prescribed dosing duration) in 158 patients, including:
- Cohort A—23 adults and 43 pediatric patients
- Cohort B—35 adults and 57 pediatric patients.
The investigators noted that many of the patients did not complete the study. The team said this is a reflection of the significant mortality risk of AdV because most of these patients died before they could finish.
Sixty-five percent of adults and 33% of children in Cohort A did not complete the study. The same was true for 71% of adults and 49% of children in Cohort B.
Mortality
The study’s primary efficacy endpoint is all-cause mortality at day 60 after the first brincidofovir dose in allogeneic HSCT recipients with disseminated AdV disease (Cohort B). All-cause mortality at day 60 in this cohort was 19% in pediatric patients and 43% in adults.
In Cohorts A and B, all-cause mortality at 24 weeks was lower in children than adults.
At 24 weeks, pediatric all-cause mortality was 33% in Cohort A and 42% in Cohort B. Adult all-cause mortality was 48% in Cohort A and 71% in Cohort B.
AdV-related mortality at 24 weeks in pediatric patients was 9% in Cohort A and 14% in Cohort B. AdV-related mortality in adults was 4% in Cohort A and 46% in Cohort B.
Declines in viremia
In Cohort A, 61% of patients achieved undetectable viremia at the end of treatment—43% of adults and 70% of children.
In Cohort B, 49% of patients achieved undetectable viremia at the end of treatment—29% of adults and 63% of children.
The median time to undetectable AdV viremia was 43 days (range, 8 to non-estimable) for adults in Cohort A, 14 days (range, 5 to 23) for children in Cohort A, non-estimable (range, 29 days to non-estimable) for adults in Cohort B, and 22 days (range, 15 to 36) for children in Cohort B.
Link between response and survival
The investigators conducted post-hoc analyses to assess the correlation between rapid virologic response to brincidofovir and time to subsequent mortality.
The team compared patients who responded to treatment—defined as achieving a ≥ 2-log10 copies/mL decline, undetectable AdV viremia at week 4, or undetectable AdV viremia at week 6—with non-responders.
Fifty percent of adults and 84% of children who were still alive at week 4 had achieved a ≥ 2 log decline or undetectable AdV viremia at that time.
This type of response was associated with improved survival at week 24. In adults, the mortality rate was 46% in responders and 85% in non-responders (P=0.03). In pediatric patients, the mortality rate was 25% in responders and 71% in non-responders (P=0.01).
In patients who were alive at week 6, 42% of adults and 68% of children achieved undetectable AdV viremia by that time.
This response was associated with improved survival at week 24. In adults, the mortality rate was 30% in responders and 86% in non-responders (P=0.001). In pediatric patients, the mortality rate was 18% in responders and 54% in non-responders (P=0.01).
Safety
All adults had treatment-emergent AEs, as did all pediatric patients in Cohort B and 95% of pediatric patients in Cohort A.
The most common treatment-emergent AEs were gastrointestinal (GI) events, which occurred in 70% of adults and 81% of children in Cohort A, as well as 83% of adults and 74% of children in Cohort B.
Acute graft-versus-host disease (GVHD) was also common, occurring in 22% of adults and 37% of children in Cohort A and 43% of adults and 40% of children in Cohort B. Some patients did have acute GVHD at baseline, however—22%, 26%, 34%, and 19%, respectively.
The percentage of patients with AEs leading to treatment discontinuation was 26% for adults and 28% for children in Cohort A and 31% for adults and 14% for children in Cohort B.
Overall, 20% of pediatric patients and 29% of adults discontinued brincidofovir due to AEs. GI events were cited as the most common reason—5% and 14%, respectively.
The investigators said there were no events reported that were suggestive of drug-related nephrotoxicity or myelosuppression. ![]()
Image by Yale Rosen
NEW ORLEANS—Interim results of a phase 3 trial suggest brincidofovir can treat adenovirus (AdV) infection in recipients of allogeneic hematopoietic stem cell transplant (HSCT).
Both pediatric and adult patients experienced a decline in AdV viral load after brincidofovir treatment, but pediatric patients were more likely to respond.
Overall survival rates were better for patients who had a rapid response and were therefore better among pediatric patients than adults.
Investigators said the adverse events (AEs) in this study were consistent with the known safety profile of brincidofovir.
Michael Grimley, MD, of Cincinnati Children’s Hospital in Ohio, and his colleagues presented these results at IDWeek 2016 (abstract 2339). The research was supported by Chimerix, the company developing brincidofovir.
This trial, known as AdVise, was designed to evaluate brincidofovir for the treatment of AdV infection in pediatric and adult patients divided into 3 cohorts:
- Cohort A consists of allogeneic HSCT recipients with asymptomatic or limited AdV infection
- Cohort B consists of allogeneic HSCT recipients with disseminated AdV disease
- Cohort C consists of autologous HSCT recipients, solid organ transplant recipients, and other immunocompromised patients.
All patients were assigned to 12 weeks of oral brincidofovir, administered twice weekly. An additional 12 weeks of treatment was allowed in patients with ongoing or recurrent infection. After completing treatment, all patients were followed until week 36.
Interim analysis
The investigators examined outcomes at 24 weeks after the first brincidofovir dose (12 weeks after prescribed dosing duration) in 158 patients, including:
- Cohort A—23 adults and 43 pediatric patients
- Cohort B—35 adults and 57 pediatric patients.
The investigators noted that many of the patients did not complete the study. The team said this is a reflection of the significant mortality risk of AdV because most of these patients died before they could finish.
Sixty-five percent of adults and 33% of children in Cohort A did not complete the study. The same was true for 71% of adults and 49% of children in Cohort B.
Mortality
The study’s primary efficacy endpoint is all-cause mortality at day 60 after the first brincidofovir dose in allogeneic HSCT recipients with disseminated AdV disease (Cohort B). All-cause mortality at day 60 in this cohort was 19% in pediatric patients and 43% in adults.
In Cohorts A and B, all-cause mortality at 24 weeks was lower in children than adults.
At 24 weeks, pediatric all-cause mortality was 33% in Cohort A and 42% in Cohort B. Adult all-cause mortality was 48% in Cohort A and 71% in Cohort B.
AdV-related mortality at 24 weeks in pediatric patients was 9% in Cohort A and 14% in Cohort B. AdV-related mortality in adults was 4% in Cohort A and 46% in Cohort B.
Declines in viremia
In Cohort A, 61% of patients achieved undetectable viremia at the end of treatment—43% of adults and 70% of children.
In Cohort B, 49% of patients achieved undetectable viremia at the end of treatment—29% of adults and 63% of children.
The median time to undetectable AdV viremia was 43 days (range, 8 to non-estimable) for adults in Cohort A, 14 days (range, 5 to 23) for children in Cohort A, non-estimable (range, 29 days to non-estimable) for adults in Cohort B, and 22 days (range, 15 to 36) for children in Cohort B.
Link between response and survival
The investigators conducted post-hoc analyses to assess the correlation between rapid virologic response to brincidofovir and time to subsequent mortality.
The team compared patients who responded to treatment—defined as achieving a ≥ 2-log10 copies/mL decline, undetectable AdV viremia at week 4, or undetectable AdV viremia at week 6—with non-responders.
Fifty percent of adults and 84% of children who were still alive at week 4 had achieved a ≥ 2 log decline or undetectable AdV viremia at that time.
This type of response was associated with improved survival at week 24. In adults, the mortality rate was 46% in responders and 85% in non-responders (P=0.03). In pediatric patients, the mortality rate was 25% in responders and 71% in non-responders (P=0.01).
In patients who were alive at week 6, 42% of adults and 68% of children achieved undetectable AdV viremia by that time.
This response was associated with improved survival at week 24. In adults, the mortality rate was 30% in responders and 86% in non-responders (P=0.001). In pediatric patients, the mortality rate was 18% in responders and 54% in non-responders (P=0.01).
Safety
All adults had treatment-emergent AEs, as did all pediatric patients in Cohort B and 95% of pediatric patients in Cohort A.
The most common treatment-emergent AEs were gastrointestinal (GI) events, which occurred in 70% of adults and 81% of children in Cohort A, as well as 83% of adults and 74% of children in Cohort B.
Acute graft-versus-host disease (GVHD) was also common, occurring in 22% of adults and 37% of children in Cohort A and 43% of adults and 40% of children in Cohort B. Some patients did have acute GVHD at baseline, however—22%, 26%, 34%, and 19%, respectively.
The percentage of patients with AEs leading to treatment discontinuation was 26% for adults and 28% for children in Cohort A and 31% for adults and 14% for children in Cohort B.
Overall, 20% of pediatric patients and 29% of adults discontinued brincidofovir due to AEs. GI events were cited as the most common reason—5% and 14%, respectively.
The investigators said there were no events reported that were suggestive of drug-related nephrotoxicity or myelosuppression. ![]()
Image by Yale Rosen
NEW ORLEANS—Interim results of a phase 3 trial suggest brincidofovir can treat adenovirus (AdV) infection in recipients of allogeneic hematopoietic stem cell transplant (HSCT).
Both pediatric and adult patients experienced a decline in AdV viral load after brincidofovir treatment, but pediatric patients were more likely to respond.
Overall survival rates were better for patients who had a rapid response and were therefore better among pediatric patients than adults.
Investigators said the adverse events (AEs) in this study were consistent with the known safety profile of brincidofovir.
Michael Grimley, MD, of Cincinnati Children’s Hospital in Ohio, and his colleagues presented these results at IDWeek 2016 (abstract 2339). The research was supported by Chimerix, the company developing brincidofovir.
This trial, known as AdVise, was designed to evaluate brincidofovir for the treatment of AdV infection in pediatric and adult patients divided into 3 cohorts:
- Cohort A consists of allogeneic HSCT recipients with asymptomatic or limited AdV infection
- Cohort B consists of allogeneic HSCT recipients with disseminated AdV disease
- Cohort C consists of autologous HSCT recipients, solid organ transplant recipients, and other immunocompromised patients.
All patients were assigned to 12 weeks of oral brincidofovir, administered twice weekly. An additional 12 weeks of treatment was allowed in patients with ongoing or recurrent infection. After completing treatment, all patients were followed until week 36.
Interim analysis
The investigators examined outcomes at 24 weeks after the first brincidofovir dose (12 weeks after prescribed dosing duration) in 158 patients, including:
- Cohort A—23 adults and 43 pediatric patients
- Cohort B—35 adults and 57 pediatric patients.
The investigators noted that many of the patients did not complete the study. The team said this is a reflection of the significant mortality risk of AdV because most of these patients died before they could finish.
Sixty-five percent of adults and 33% of children in Cohort A did not complete the study. The same was true for 71% of adults and 49% of children in Cohort B.
Mortality
The study’s primary efficacy endpoint is all-cause mortality at day 60 after the first brincidofovir dose in allogeneic HSCT recipients with disseminated AdV disease (Cohort B). All-cause mortality at day 60 in this cohort was 19% in pediatric patients and 43% in adults.
In Cohorts A and B, all-cause mortality at 24 weeks was lower in children than adults.
At 24 weeks, pediatric all-cause mortality was 33% in Cohort A and 42% in Cohort B. Adult all-cause mortality was 48% in Cohort A and 71% in Cohort B.
AdV-related mortality at 24 weeks in pediatric patients was 9% in Cohort A and 14% in Cohort B. AdV-related mortality in adults was 4% in Cohort A and 46% in Cohort B.
Declines in viremia
In Cohort A, 61% of patients achieved undetectable viremia at the end of treatment—43% of adults and 70% of children.
In Cohort B, 49% of patients achieved undetectable viremia at the end of treatment—29% of adults and 63% of children.
The median time to undetectable AdV viremia was 43 days (range, 8 to non-estimable) for adults in Cohort A, 14 days (range, 5 to 23) for children in Cohort A, non-estimable (range, 29 days to non-estimable) for adults in Cohort B, and 22 days (range, 15 to 36) for children in Cohort B.
Link between response and survival
The investigators conducted post-hoc analyses to assess the correlation between rapid virologic response to brincidofovir and time to subsequent mortality.
The team compared patients who responded to treatment—defined as achieving a ≥ 2-log10 copies/mL decline, undetectable AdV viremia at week 4, or undetectable AdV viremia at week 6—with non-responders.
Fifty percent of adults and 84% of children who were still alive at week 4 had achieved a ≥ 2 log decline or undetectable AdV viremia at that time.
This type of response was associated with improved survival at week 24. In adults, the mortality rate was 46% in responders and 85% in non-responders (P=0.03). In pediatric patients, the mortality rate was 25% in responders and 71% in non-responders (P=0.01).
In patients who were alive at week 6, 42% of adults and 68% of children achieved undetectable AdV viremia by that time.
This response was associated with improved survival at week 24. In adults, the mortality rate was 30% in responders and 86% in non-responders (P=0.001). In pediatric patients, the mortality rate was 18% in responders and 54% in non-responders (P=0.01).
Safety
All adults had treatment-emergent AEs, as did all pediatric patients in Cohort B and 95% of pediatric patients in Cohort A.
The most common treatment-emergent AEs were gastrointestinal (GI) events, which occurred in 70% of adults and 81% of children in Cohort A, as well as 83% of adults and 74% of children in Cohort B.
Acute graft-versus-host disease (GVHD) was also common, occurring in 22% of adults and 37% of children in Cohort A and 43% of adults and 40% of children in Cohort B. Some patients did have acute GVHD at baseline, however—22%, 26%, 34%, and 19%, respectively.
The percentage of patients with AEs leading to treatment discontinuation was 26% for adults and 28% for children in Cohort A and 31% for adults and 14% for children in Cohort B.
Overall, 20% of pediatric patients and 29% of adults discontinued brincidofovir due to AEs. GI events were cited as the most common reason—5% and 14%, respectively.
The investigators said there were no events reported that were suggestive of drug-related nephrotoxicity or myelosuppression. ![]()
Results support continued study of CMV vaccine

CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
FDA grants drug orphan designation for GVHD

Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted orphan drug designation to ALXN1007 for the treatment of acute graft-versus-host disease (GVHD).
ALXN1007 is an anti-inflammatory monoclonal antibody targeting complement protein C5a.
The drug is being developed
by Alexion Pharmaceuticals, Inc.
It is currently under investigation in a phase 2 trial of patients with acute GVHD of the lower gastrointestinal tract (GI-GVHD).
Results from this trial were presented at the 21st Congress of the European Hematology Association (abstract LB2269).
The presentation included 15 patients with newly diagnosed, biopsy-confirmed acute GI-GVHD. The patients had a median age of 60 (range, 25-69), and 60% were male.
Patients had acute myeloid leukemia/myelodysplastic syndrome (n=8), acute lymphoblastic leukemia (n=2), acute lymphocytic leukemia (n=1), acute myeloblastic leukemia (n=1), aplastic anemia (n=1), cutaneous T-cell lymphoma (n=1), and mantle cell lymphoma (n=1).
Most patients received transplants from matched, unrelated donors (n=11); 3 had matched, related donors; and 1 had a mismatched donor. Ten patients received peripheral blood grafts, 4 received cord blood, and 1 received a bone marrow transplant.
Patients had grade 1 (n=7), grade 2 (n=2), and grade 3 (n=6) acute GI-GVHD.
The patients received weekly doses of ALXN1007 at 10 mg/kg, in combination with methylprednisolone at an initial dose of 2 mg/kg, through day 56.
Thirteen patients were evaluable for efficacy. One patient experienced leukemia relapse at day 18, and 1 withdrew from the study early.
The overall acute GVHD response rate was 77% (10/13), both at day 28 and day 56. The complete GI-GVHD response rate was 69% at day 28 and 77% at day 56.
At day 180, the nonrelapse mortality rate was 12.5%, and the overall survival rate was 69.2%.
All of the patients had treatment-emergent adverse events (AEs), and 11 patients (69%) had serious treatment-emergent AEs.
Five patients experienced a total of 12 treatment-related AEs (1 case each)—adenovirus infection, bronchopulmonary aspergillosis, chills, corona virus infection, viral cystitis, Epstein-Barr virus infection, hypersensitivity, influenza, influenza-like illness, infusion-related reaction, respiratory syncytial virus infection, and tremor.
There were 6 deaths, but none were considered treatment-related.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
ALXN1007 has orphan designation from the European Commission as well. ![]()
Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted orphan drug designation to ALXN1007 for the treatment of acute graft-versus-host disease (GVHD).
ALXN1007 is an anti-inflammatory monoclonal antibody targeting complement protein C5a.
The drug is being developed
by Alexion Pharmaceuticals, Inc.
It is currently under investigation in a phase 2 trial of patients with acute GVHD of the lower gastrointestinal tract (GI-GVHD).
Results from this trial were presented at the 21st Congress of the European Hematology Association (abstract LB2269).
The presentation included 15 patients with newly diagnosed, biopsy-confirmed acute GI-GVHD. The patients had a median age of 60 (range, 25-69), and 60% were male.
Patients had acute myeloid leukemia/myelodysplastic syndrome (n=8), acute lymphoblastic leukemia (n=2), acute lymphocytic leukemia (n=1), acute myeloblastic leukemia (n=1), aplastic anemia (n=1), cutaneous T-cell lymphoma (n=1), and mantle cell lymphoma (n=1).
Most patients received transplants from matched, unrelated donors (n=11); 3 had matched, related donors; and 1 had a mismatched donor. Ten patients received peripheral blood grafts, 4 received cord blood, and 1 received a bone marrow transplant.
Patients had grade 1 (n=7), grade 2 (n=2), and grade 3 (n=6) acute GI-GVHD.
The patients received weekly doses of ALXN1007 at 10 mg/kg, in combination with methylprednisolone at an initial dose of 2 mg/kg, through day 56.
Thirteen patients were evaluable for efficacy. One patient experienced leukemia relapse at day 18, and 1 withdrew from the study early.
The overall acute GVHD response rate was 77% (10/13), both at day 28 and day 56. The complete GI-GVHD response rate was 69% at day 28 and 77% at day 56.
At day 180, the nonrelapse mortality rate was 12.5%, and the overall survival rate was 69.2%.
All of the patients had treatment-emergent adverse events (AEs), and 11 patients (69%) had serious treatment-emergent AEs.
Five patients experienced a total of 12 treatment-related AEs (1 case each)—adenovirus infection, bronchopulmonary aspergillosis, chills, corona virus infection, viral cystitis, Epstein-Barr virus infection, hypersensitivity, influenza, influenza-like illness, infusion-related reaction, respiratory syncytial virus infection, and tremor.
There were 6 deaths, but none were considered treatment-related.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
ALXN1007 has orphan designation from the European Commission as well. ![]()
Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted orphan drug designation to ALXN1007 for the treatment of acute graft-versus-host disease (GVHD).
ALXN1007 is an anti-inflammatory monoclonal antibody targeting complement protein C5a.
The drug is being developed
by Alexion Pharmaceuticals, Inc.
It is currently under investigation in a phase 2 trial of patients with acute GVHD of the lower gastrointestinal tract (GI-GVHD).
Results from this trial were presented at the 21st Congress of the European Hematology Association (abstract LB2269).
The presentation included 15 patients with newly diagnosed, biopsy-confirmed acute GI-GVHD. The patients had a median age of 60 (range, 25-69), and 60% were male.
Patients had acute myeloid leukemia/myelodysplastic syndrome (n=8), acute lymphoblastic leukemia (n=2), acute lymphocytic leukemia (n=1), acute myeloblastic leukemia (n=1), aplastic anemia (n=1), cutaneous T-cell lymphoma (n=1), and mantle cell lymphoma (n=1).
Most patients received transplants from matched, unrelated donors (n=11); 3 had matched, related donors; and 1 had a mismatched donor. Ten patients received peripheral blood grafts, 4 received cord blood, and 1 received a bone marrow transplant.
Patients had grade 1 (n=7), grade 2 (n=2), and grade 3 (n=6) acute GI-GVHD.
The patients received weekly doses of ALXN1007 at 10 mg/kg, in combination with methylprednisolone at an initial dose of 2 mg/kg, through day 56.
Thirteen patients were evaluable for efficacy. One patient experienced leukemia relapse at day 18, and 1 withdrew from the study early.
The overall acute GVHD response rate was 77% (10/13), both at day 28 and day 56. The complete GI-GVHD response rate was 69% at day 28 and 77% at day 56.
At day 180, the nonrelapse mortality rate was 12.5%, and the overall survival rate was 69.2%.
All of the patients had treatment-emergent adverse events (AEs), and 11 patients (69%) had serious treatment-emergent AEs.
Five patients experienced a total of 12 treatment-related AEs (1 case each)—adenovirus infection, bronchopulmonary aspergillosis, chills, corona virus infection, viral cystitis, Epstein-Barr virus infection, hypersensitivity, influenza, influenza-like illness, infusion-related reaction, respiratory syncytial virus infection, and tremor.
There were 6 deaths, but none were considered treatment-related.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
ALXN1007 has orphan designation from the European Commission as well. ![]()
EBV-CTLs accepted into EMA’s PRIME program
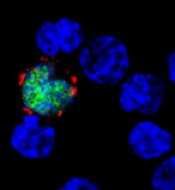
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The European Medicines Agency (EMA) has accepted into its Priority Medicines (PRIME) program an allogeneic cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs).
EBV-CTLs have been accepted as a treatment for patients with rituximab-refractory EBV-associated post-transplant lymphoproliferative disorder (EBV-PTLD).
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs.
The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate the potential to benefit patients with unmet medical need through early clinical or nonclinical data.
About EBV-CTLs
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc.
To create EBV-CTLs, T cells are collected from the blood of third-party donors and exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
The EBV-CTLs are designed to find cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTLs are currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results from 2 studies of EBV-CTLs in rituximab-refractory PTLD were presented at the 2015 AACR Annual Meeting.
Atara Bio’s EBV-CTLs have been classified as an advanced therapy medicinal product by the EMA and have orphan designation in the European Union. ![]()
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The European Medicines Agency (EMA) has accepted into its Priority Medicines (PRIME) program an allogeneic cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs).
EBV-CTLs have been accepted as a treatment for patients with rituximab-refractory EBV-associated post-transplant lymphoproliferative disorder (EBV-PTLD).
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs.
The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate the potential to benefit patients with unmet medical need through early clinical or nonclinical data.
About EBV-CTLs
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc.
To create EBV-CTLs, T cells are collected from the blood of third-party donors and exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
The EBV-CTLs are designed to find cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTLs are currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results from 2 studies of EBV-CTLs in rituximab-refractory PTLD were presented at the 2015 AACR Annual Meeting.
Atara Bio’s EBV-CTLs have been classified as an advanced therapy medicinal product by the EMA and have orphan designation in the European Union. ![]()
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The European Medicines Agency (EMA) has accepted into its Priority Medicines (PRIME) program an allogeneic cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs).
EBV-CTLs have been accepted as a treatment for patients with rituximab-refractory EBV-associated post-transplant lymphoproliferative disorder (EBV-PTLD).
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs.
The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate the potential to benefit patients with unmet medical need through early clinical or nonclinical data.
About EBV-CTLs
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc.
To create EBV-CTLs, T cells are collected from the blood of third-party donors and exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
The EBV-CTLs are designed to find cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTLs are currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results from 2 studies of EBV-CTLs in rituximab-refractory PTLD were presented at the 2015 AACR Annual Meeting.
Atara Bio’s EBV-CTLs have been classified as an advanced therapy medicinal product by the EMA and have orphan designation in the European Union. ![]()
COMP recommends orphan designation for CMV-CTLs
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for a cytomegalovirus-specific cytotoxic T-lymphocyte product (CMV-CTLs) intended to treat CMV infection in patients with impaired cell-mediated immunity.
The CMV-CTLs are designed to find and kill cells expressing CMV.
To create CMV-CTLs, T cells are collected from the blood of third-party donors and then exposed to CMV antigens.
The resulting activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
The CMV-CTLs are being developed by Atara Biotherapeutics, Inc.
The cells are currently under investigation in a pair of phase 2 trials (NCT01646645 and NCT02136797).
Results of a phase 1 trial (published in Biology of Blood and Marrow Transplantation in 2015) suggested CMV-CTLs are safe and can clear CMV infection in patients who have undergone allogeneic hematopoietic stem cell transplant.
The trial included 17 transplant recipients with CMV viremia or clinical infection that persisted despite prolonged treatment with antiviral drugs. Fourteen of the patients had received T-cell-depleted transplants without graft-versus-host disease (GVHD) prophylaxis.
Sixteen of the patients received CMV-CTLs created using cells derived from their transplant donor, and 1 patient received cells from a third-party donor.
Fifteen patients achieved clearance of CMV viremia, including 3 of the 5 patients with overt disease and the patient who received cells from a third-party donor.
In addition, the researchers said CMV-CTLs were well-tolerated. None of the patients experienced fever, alterations in vital signs, or other toxicities during the first 48 hours of observation.
None of the patients developed manifestations of de novo acute GVHD, and GHVD did not worsen in either of the 2 patients who had GVHD prior to infusion.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for a cytomegalovirus-specific cytotoxic T-lymphocyte product (CMV-CTLs) intended to treat CMV infection in patients with impaired cell-mediated immunity.
The CMV-CTLs are designed to find and kill cells expressing CMV.
To create CMV-CTLs, T cells are collected from the blood of third-party donors and then exposed to CMV antigens.
The resulting activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
The CMV-CTLs are being developed by Atara Biotherapeutics, Inc.
The cells are currently under investigation in a pair of phase 2 trials (NCT01646645 and NCT02136797).
Results of a phase 1 trial (published in Biology of Blood and Marrow Transplantation in 2015) suggested CMV-CTLs are safe and can clear CMV infection in patients who have undergone allogeneic hematopoietic stem cell transplant.
The trial included 17 transplant recipients with CMV viremia or clinical infection that persisted despite prolonged treatment with antiviral drugs. Fourteen of the patients had received T-cell-depleted transplants without graft-versus-host disease (GVHD) prophylaxis.
Sixteen of the patients received CMV-CTLs created using cells derived from their transplant donor, and 1 patient received cells from a third-party donor.
Fifteen patients achieved clearance of CMV viremia, including 3 of the 5 patients with overt disease and the patient who received cells from a third-party donor.
In addition, the researchers said CMV-CTLs were well-tolerated. None of the patients experienced fever, alterations in vital signs, or other toxicities during the first 48 hours of observation.
None of the patients developed manifestations of de novo acute GVHD, and GHVD did not worsen in either of the 2 patients who had GVHD prior to infusion.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for a cytomegalovirus-specific cytotoxic T-lymphocyte product (CMV-CTLs) intended to treat CMV infection in patients with impaired cell-mediated immunity.
The CMV-CTLs are designed to find and kill cells expressing CMV.
To create CMV-CTLs, T cells are collected from the blood of third-party donors and then exposed to CMV antigens.
The resulting activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
The CMV-CTLs are being developed by Atara Biotherapeutics, Inc.
The cells are currently under investigation in a pair of phase 2 trials (NCT01646645 and NCT02136797).
Results of a phase 1 trial (published in Biology of Blood and Marrow Transplantation in 2015) suggested CMV-CTLs are safe and can clear CMV infection in patients who have undergone allogeneic hematopoietic stem cell transplant.
The trial included 17 transplant recipients with CMV viremia or clinical infection that persisted despite prolonged treatment with antiviral drugs. Fourteen of the patients had received T-cell-depleted transplants without graft-versus-host disease (GVHD) prophylaxis.
Sixteen of the patients received CMV-CTLs created using cells derived from their transplant donor, and 1 patient received cells from a third-party donor.
Fifteen patients achieved clearance of CMV viremia, including 3 of the 5 patients with overt disease and the patient who received cells from a third-party donor.
In addition, the researchers said CMV-CTLs were well-tolerated. None of the patients experienced fever, alterations in vital signs, or other toxicities during the first 48 hours of observation.
None of the patients developed manifestations of de novo acute GVHD, and GHVD did not worsen in either of the 2 patients who had GVHD prior to infusion.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
Cord blood product granted breakthrough designation

Photo courtesy of NHS
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for NiCord®, a product in development as a graft modality for patients undergoing hematopoietic stem cell transplant to treat high-risk hematologic malignancies.
NiCord consists of cells from a single unit of umbilical cord blood cultured in nicotinamide (a vitamin B derivative) and cytokines that are typically used for expansion (thrombopoietin, interleukin 6, FLT3 ligand, and stem cell factor).
Data from the pilot and phase 1/2 studies of NiCord have suggested the therapy can provide a clinically meaningful improvement in time to neutrophil engraftment over traditional cord blood transplant.
Additional research presented at EBMT 2016 showed that patients who received NiCord had fewer infections, shorter hospital stays, quicker platelet engraftment, and improved non-relapse mortality when compared to patients who received a traditional cord blood transplant.
The phase 3 registration study of NiCord is scheduled to begin before the end of this year.
In addition to breakthrough designation, NiCord also has orphan designation from the FDA as a treatment for patients with acute lymphoblastic leukemia, acute myeloid leukemia, Hodgkin lymphoma, or myelodysplastic syndromes.
NiCord is being developed by Gamida Cell.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Photo courtesy of NHS
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for NiCord®, a product in development as a graft modality for patients undergoing hematopoietic stem cell transplant to treat high-risk hematologic malignancies.
NiCord consists of cells from a single unit of umbilical cord blood cultured in nicotinamide (a vitamin B derivative) and cytokines that are typically used for expansion (thrombopoietin, interleukin 6, FLT3 ligand, and stem cell factor).
Data from the pilot and phase 1/2 studies of NiCord have suggested the therapy can provide a clinically meaningful improvement in time to neutrophil engraftment over traditional cord blood transplant.
Additional research presented at EBMT 2016 showed that patients who received NiCord had fewer infections, shorter hospital stays, quicker platelet engraftment, and improved non-relapse mortality when compared to patients who received a traditional cord blood transplant.
The phase 3 registration study of NiCord is scheduled to begin before the end of this year.
In addition to breakthrough designation, NiCord also has orphan designation from the FDA as a treatment for patients with acute lymphoblastic leukemia, acute myeloid leukemia, Hodgkin lymphoma, or myelodysplastic syndromes.
NiCord is being developed by Gamida Cell.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Photo courtesy of NHS
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for NiCord®, a product in development as a graft modality for patients undergoing hematopoietic stem cell transplant to treat high-risk hematologic malignancies.
NiCord consists of cells from a single unit of umbilical cord blood cultured in nicotinamide (a vitamin B derivative) and cytokines that are typically used for expansion (thrombopoietin, interleukin 6, FLT3 ligand, and stem cell factor).
Data from the pilot and phase 1/2 studies of NiCord have suggested the therapy can provide a clinically meaningful improvement in time to neutrophil engraftment over traditional cord blood transplant.
Additional research presented at EBMT 2016 showed that patients who received NiCord had fewer infections, shorter hospital stays, quicker platelet engraftment, and improved non-relapse mortality when compared to patients who received a traditional cord blood transplant.
The phase 3 registration study of NiCord is scheduled to begin before the end of this year.
In addition to breakthrough designation, NiCord also has orphan designation from the FDA as a treatment for patients with acute lymphoblastic leukemia, acute myeloid leukemia, Hodgkin lymphoma, or myelodysplastic syndromes.
NiCord is being developed by Gamida Cell.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Vitamin D affects HSPC production, team says
Photo by Ian Johnston
The availability of vitamin D during embryonic development can affect hematopoietic stem and progenitor cells (HSPCs), according to research published in Cell Reports.
Experiments with zebrafish embryos suggested that vitamin D acts directly on HSPCs to increase proliferation.
Similarly, in HSPCs from human umbilical cords, treatment with vitamin D enhanced hematopoietic colony numbers.
Researchers therefore theorized that vitamin D supplementation might be useful for HSPC expansion prior to transplant.
“We clearly showed that not getting enough vitamin D can alter how blood stem cells are formed,” said study author Trista North, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Vitamin D was having a direct response on the blood stem cells, and it changed what those cells did in terms of multiplying and staying alive.”
The researchers found, in both human and zebrafish tissue, that 1,25(OH)D3 (active vitamin D3) had an impact on HSPC production and function.
Investigation into the mechanism revealed that CXCL8-CXCR1/2 signaling functions downstream of 1,25(OH)D3-mediated vitamin D receptor stimulation to directly regulate HSPC production and expansion.
“What was surprising was that vitamin D is having an impact so early,” Dr North said. “We really only thought about vitamin D in terms of bone development and maintenance, but we clearly show that, whether they were zebrafish or human blood stem cells, they can respond directly to the nutrient.”
One caveat is the researchers did face difficulty testing the response in mice, as the animals don’t have the same vitamin D inflammatory targets observed in zebrafish and humans.
Additionally, the team didn’t know the vitamin D levels in the umbilical cord blood samples they tested, which may have influenced the outcome of their analysis.
As a next step, Dr North and her colleagues hope to test cord blood samples for which they know the vitamin D status to see if umbilical cords with healthy levels respond better or worse to stimulation than cords from vitamin-D-deficient donors.
Photo by Ian Johnston
The availability of vitamin D during embryonic development can affect hematopoietic stem and progenitor cells (HSPCs), according to research published in Cell Reports.
Experiments with zebrafish embryos suggested that vitamin D acts directly on HSPCs to increase proliferation.
Similarly, in HSPCs from human umbilical cords, treatment with vitamin D enhanced hematopoietic colony numbers.
Researchers therefore theorized that vitamin D supplementation might be useful for HSPC expansion prior to transplant.
“We clearly showed that not getting enough vitamin D can alter how blood stem cells are formed,” said study author Trista North, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Vitamin D was having a direct response on the blood stem cells, and it changed what those cells did in terms of multiplying and staying alive.”
The researchers found, in both human and zebrafish tissue, that 1,25(OH)D3 (active vitamin D3) had an impact on HSPC production and function.
Investigation into the mechanism revealed that CXCL8-CXCR1/2 signaling functions downstream of 1,25(OH)D3-mediated vitamin D receptor stimulation to directly regulate HSPC production and expansion.
“What was surprising was that vitamin D is having an impact so early,” Dr North said. “We really only thought about vitamin D in terms of bone development and maintenance, but we clearly show that, whether they were zebrafish or human blood stem cells, they can respond directly to the nutrient.”
One caveat is the researchers did face difficulty testing the response in mice, as the animals don’t have the same vitamin D inflammatory targets observed in zebrafish and humans.
Additionally, the team didn’t know the vitamin D levels in the umbilical cord blood samples they tested, which may have influenced the outcome of their analysis.
As a next step, Dr North and her colleagues hope to test cord blood samples for which they know the vitamin D status to see if umbilical cords with healthy levels respond better or worse to stimulation than cords from vitamin-D-deficient donors.
Photo by Ian Johnston
The availability of vitamin D during embryonic development can affect hematopoietic stem and progenitor cells (HSPCs), according to research published in Cell Reports.
Experiments with zebrafish embryos suggested that vitamin D acts directly on HSPCs to increase proliferation.
Similarly, in HSPCs from human umbilical cords, treatment with vitamin D enhanced hematopoietic colony numbers.
Researchers therefore theorized that vitamin D supplementation might be useful for HSPC expansion prior to transplant.
“We clearly showed that not getting enough vitamin D can alter how blood stem cells are formed,” said study author Trista North, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Vitamin D was having a direct response on the blood stem cells, and it changed what those cells did in terms of multiplying and staying alive.”
The researchers found, in both human and zebrafish tissue, that 1,25(OH)D3 (active vitamin D3) had an impact on HSPC production and function.
Investigation into the mechanism revealed that CXCL8-CXCR1/2 signaling functions downstream of 1,25(OH)D3-mediated vitamin D receptor stimulation to directly regulate HSPC production and expansion.
“What was surprising was that vitamin D is having an impact so early,” Dr North said. “We really only thought about vitamin D in terms of bone development and maintenance, but we clearly show that, whether they were zebrafish or human blood stem cells, they can respond directly to the nutrient.”
One caveat is the researchers did face difficulty testing the response in mice, as the animals don’t have the same vitamin D inflammatory targets observed in zebrafish and humans.
Additionally, the team didn’t know the vitamin D levels in the umbilical cord blood samples they tested, which may have influenced the outcome of their analysis.
As a next step, Dr North and her colleagues hope to test cord blood samples for which they know the vitamin D status to see if umbilical cords with healthy levels respond better or worse to stimulation than cords from vitamin-D-deficient donors.
Therapy granted orphan designation to prevent GVHD

Image from PLOS ONE
The US Food and Drug Administration (FDA) has granted orphan drug designation to a programmed cellular immunotherapy known as ProTmune™.
The designation is for ProTmune to be used as graft-versus-host disease (GVHD) prophylaxis in patients undergoing allogeneic hematopoietic stem cell transplant (HSCT).
This indication covers a range of diseases, including hematologic malignancies and genetic disorders.
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
About ProTmune
ProTmune is produced by modulating a donor-sourced, mobilized peripheral blood graft ex vivo with 2 small molecules—FT1050 and FT4145—to enhance the biological properties and therapeutic function of the graft’s immune cells.
The programmed mobilized peripheral blood graft is administered to a patient as a one-time intravenous infusion.
ProTmune is being developed by Fate Therapeutics, Inc.
The company is conducting a phase 1/2 trial testing ProTmune for the prevention of acute GVHD and cytomegalovirus infection in adults with hematologic malignancies who are undergoing allogeneic HSCT.
ProTmune was previously granted fast track designation from the FDA.
“The granting of both orphan drug and fast track designations for ProTmune validates the product candidate’s unique therapeutic potential to address life-threatening complications and improve the curative potential of allogeneic [HSCT],” said Scott Wolchko, president and chief executive officer of Fate Therapeutics.
“Graft-versus-host disease is a significant cause of morbidity and mortality in patients undergoing allogeneic [HSCT], and there are no FDA-approved therapies to prevent its occurrence. Through our development of ProTmune, we seek to transform the allogeneic [HSCT] paradigm by providing immunocompromised patients a therapeutically optimized donor graft containing immune cells with reduced alloreactivity and enhanced infection-fighting and anti-tumor properties.”
Image from PLOS ONE
The US Food and Drug Administration (FDA) has granted orphan drug designation to a programmed cellular immunotherapy known as ProTmune™.
The designation is for ProTmune to be used as graft-versus-host disease (GVHD) prophylaxis in patients undergoing allogeneic hematopoietic stem cell transplant (HSCT).
This indication covers a range of diseases, including hematologic malignancies and genetic disorders.
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
About ProTmune
ProTmune is produced by modulating a donor-sourced, mobilized peripheral blood graft ex vivo with 2 small molecules—FT1050 and FT4145—to enhance the biological properties and therapeutic function of the graft’s immune cells.
The programmed mobilized peripheral blood graft is administered to a patient as a one-time intravenous infusion.
ProTmune is being developed by Fate Therapeutics, Inc.
The company is conducting a phase 1/2 trial testing ProTmune for the prevention of acute GVHD and cytomegalovirus infection in adults with hematologic malignancies who are undergoing allogeneic HSCT.
ProTmune was previously granted fast track designation from the FDA.
“The granting of both orphan drug and fast track designations for ProTmune validates the product candidate’s unique therapeutic potential to address life-threatening complications and improve the curative potential of allogeneic [HSCT],” said Scott Wolchko, president and chief executive officer of Fate Therapeutics.
“Graft-versus-host disease is a significant cause of morbidity and mortality in patients undergoing allogeneic [HSCT], and there are no FDA-approved therapies to prevent its occurrence. Through our development of ProTmune, we seek to transform the allogeneic [HSCT] paradigm by providing immunocompromised patients a therapeutically optimized donor graft containing immune cells with reduced alloreactivity and enhanced infection-fighting and anti-tumor properties.”
Image from PLOS ONE
The US Food and Drug Administration (FDA) has granted orphan drug designation to a programmed cellular immunotherapy known as ProTmune™.
The designation is for ProTmune to be used as graft-versus-host disease (GVHD) prophylaxis in patients undergoing allogeneic hematopoietic stem cell transplant (HSCT).
This indication covers a range of diseases, including hematologic malignancies and genetic disorders.
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
About ProTmune
ProTmune is produced by modulating a donor-sourced, mobilized peripheral blood graft ex vivo with 2 small molecules—FT1050 and FT4145—to enhance the biological properties and therapeutic function of the graft’s immune cells.
The programmed mobilized peripheral blood graft is administered to a patient as a one-time intravenous infusion.
ProTmune is being developed by Fate Therapeutics, Inc.
The company is conducting a phase 1/2 trial testing ProTmune for the prevention of acute GVHD and cytomegalovirus infection in adults with hematologic malignancies who are undergoing allogeneic HSCT.
ProTmune was previously granted fast track designation from the FDA.
“The granting of both orphan drug and fast track designations for ProTmune validates the product candidate’s unique therapeutic potential to address life-threatening complications and improve the curative potential of allogeneic [HSCT],” said Scott Wolchko, president and chief executive officer of Fate Therapeutics.
“Graft-versus-host disease is a significant cause of morbidity and mortality in patients undergoing allogeneic [HSCT], and there are no FDA-approved therapies to prevent its occurrence. Through our development of ProTmune, we seek to transform the allogeneic [HSCT] paradigm by providing immunocompromised patients a therapeutically optimized donor graft containing immune cells with reduced alloreactivity and enhanced infection-fighting and anti-tumor properties.”