User login
Bites and Stings

Hymenoptera
The order Hymenoptera of the phylum Arthropoda can be divided into three subgroups that are medically relevant: (1) Apidae (Apids), which include the honeybee and bumblebee; (2) Vespidae, (Vespids) which include yellow jackets, hornets and wasps; and (3) Formicidae (ants).6
Bees and Wasps
The main allergens in Apid venom are phospholipase A2, hyaluronidase, and melittin. Melittin, the main component, is a membrane active polypeptide that causes degranulation of basophils and mast cells. The allergens in Vespid venom are phospholipase, hyaluronidase, and antigen 5. As all Hymenoptera share some of these components, cross-sensitization may occur and individuals may be allergic to more than one species.7

Occasionally after multiple stings, patients present with symptoms of a systemic toxic reaction. This is often seen in an Africanized bee attack. These so-called “killer bees” are hybrids of African bees that escaped from laboratories in Brazil in the 1950s and spread northward; they are found in most of the warmer US states. Their venom is not more toxic than that of any other bee, but Africanized honeybees are more aggressive and respond to a perceived threat in far greater numbers. The reaction that results from multiple stings is systemic and may resemble anaphylaxis. Common symptoms include nausea, vomiting, and diarrhea, as well as lightheadedness and syncope. Interestingly, urticaria and bronchospasm are not universally present, even though respiratory failure and cardiac arrest may occur. Other symptoms include renal failure with acute tubular necrosis, myoglobinuria or hemoglobinuria, hepatic failure, and disseminated intravascular coagulation (DIC).10,11 In addition, there have been reports of unusual reactions such as vasculitis, nephrosis, neuritis, encephalitis, and serum sickness. Late-appearing symptoms usually start several days to weeks after a sting and tend last for a prolonged period of time. Serum sickness tends to appear 5 to 14 days after exposure and consists of fever, malaise, headache, urticaria, lymphadenopathy, and polyarthritis.12 Of note, patients who have venom-induced serum sickness may be at risk for anaphylaxis after subsequent stings and may therefore be suitable candidates for venom immunotherapy.13
Anaphylaxis
The definition of anaphylaxis is not universally agreed upon. The American Academy of Allergy, Asthma and Immunology defines anaphylaxis as a serious allergic response that often involves swelling, hives, hypotension and, in severe cases, shock. A major difference between anaphylaxis and other allergic reactions is that anaphylaxis typically involves more than one body system.14 The clinical features of anaphylaxis from insect stings are the same as those from other causes, typically generalized urticaria, facial flushing, and angioedema. Abdominal cramping, nausea, vomiting, and diarrhea are also seen. Life-threatening symptoms include stridor, circulatory collapse with shock, and bronchospasm. Symptoms usually begin 10 to 20 minutes after a sting, and almost all will develop within 6 hours. Interestingly, symptoms may recur 8 to 12 hours after the initial reaction.15-18
Management
Immediate removal of the bee stinger is the most important principle as it precludes any further venom transfer. Traditional teaching recommended scraping the stinger out to avoid squeezing remaining venom into the tissues; however, involuntary muscle contractions of the gland continue after the stinger detaches, and the venom is quickly exhausted. Thus, immediate removal of the stinger is crucial, though the method of removal is now thought irrelevant.19
The mainstay of therapy for serious reactions is intramuscular (IM) epinephrine. The initial dosing is 0.3 to 0.5 mg (0.3 to 0.5 mL of 1:1000 concentration) in adults, and 0.01 mg/kg in children (maximum 0.3 mg). The injection should be IM and not subcutaneous, as IM dosing provides higher and more consistent and rapid peak blood epinephrine levels.20 Concomitant intravenous (IV) administration of standard antihistamines, often diphenhydramine 1 mg/kg (generally 25-50 mg) and histamine-2 receptor antagonists (typically ranitidine 50 mg) are also recommended. The administration of steroids (methylprednisolone 125 mg IV or prednisone 60 mg orally) is traditionally recommended and thought to help potentiate the effect of other interventional measures.20 Bronchospasm, if present, is treated with nebulized β-agonists (albuterol). Hypotension may develop and requires significant crystalloid infusion—often several liters. If hypotension persists despite adequate fluid replacement, vasopressor therapy is recommended.
If a patient does not respond to initial treatment and cardiovascular (CV) collapse is evident, IV infusion of epinephrine should be initiated. Epinephrine, 100 mcg (0.1 mg) IV, should be given as a 1:100,000 dilution. This can be done by placing epinephrine, 0.1 mg (0.1 mL of the 1:1000 dilution), in 10 mL of normal saline solution and infusing it over 5 to 10 minutes (a rate of 1 to 2 mL/min). If the patient is refractory to the initial bolus, then an epinephrine infusion can be started by placing epinephrine, 1 mg (1.0 mL of the 1:1000 dilution), in 500 mL of 5% dextrose in water or NS and administering at a rate of 1 to 4 mcg per minute (0.5 to 2 mL/min), titrating to effect.20 Antivenins have been studied for treatment, but none are commercially available at this time.21 Patients with anaphylaxis associated with severe signs and symptoms, including any evidence of CV collapse, should be admitted to the hospital for aggressive therapy and monitoring. Persons with mild-to-moderate reactions should be observed for 4 to 6 hours to monitor for late occurring symptoms. Outpatient therapy with antihistamines, oral steroids, and a prescription for an epinephrine auto-injector—including training on proper administration prior to discharge—are strongly recommended.22 Follow-up with an allergist is also indicated in patients with significant reactions, as skin testing and immunotherapy may be beneficial to prevent anaphylaxis during future exposures.
Ants

Fire ant venom is composed of an insoluble alkaloid, and crossreactivity with the venom of other Hymenopteras species does exist. Stings generally result in a papule, which evolves into a sterile pustule. Localized necrosis, scarring, and secondary infection can occur. Systemic reactions with angioedema and urticaria have been documented, which can sometimes lead to fatalities.24
Treatment
Treatment of fire ant stings begin and end with local wound care. If the reaction is systemic, a treatment plan similar to that outlined in the treatment section for bees and wasps is indicated.
Araneae
The order Araneae of the phylum Arthropoda includes over 34,000 species of spiders divided into 105 families. Of those, only half a dozen are medically relevant and only three are commonly encountered in the United States. These include Loxosceles (most notably, the brown recluse spider), Tegeneria (mainly the hobo brown spider) and Latrodectus (includes the black widow spider). True spiders have a worldwide distribution and tend to thrive in heavily populated areas, resulting in many biting episodes per year. Data from the AAPPC’s most recent annual report listed 9,255 single spider-bite exposures in 2012, with one associated death.3
Spiders are carnivores and use venom to paralyze their prey. They are generally not a threat to humans as their fangs are too small to penetrate human skin, and the amount of venom injected is too small to produce toxicity. Thus, reactions resulting from a spider bite are typically limited to a localized reaction. Fortunately, most bites only require supportive medical therapy.
Loxosceles
Loxosceles are present worldwide, but L reclusa (the “brown recluse spider”) accounts for a significant number of envenomations in the United States. The AAPCC’s 2012 data notes 1,365 cases of exposure to the brown recluse spider with 510 of those victims seeking medical care.3 In many instances, clinicians attribute necrotic bites to the brown recluse spider, however, confirmation is often lacking. Loxosceles are nocturnal, and they are found both indoors and outdoors—mostly in dark and dry areas such as basements, closets, and woodpiles. These spiders are shy, but may bite when threatened. Their venom contains enzymes, including hyaluronidase and sphingomyelinase. Though rare, wounds can become necrotic due to neutrophil activation, platelet aggregation, and thrombosis.25 The most common reaction to a Loxosceles bite is a mild painless erythematous lesion that becomes firm and generally heals over several days to weeks. In severe reactions, erythema, edema, and pruritus initially develop, followed within 24 to 72 hours by a hemorrhagic bulla surrounded by blanched skin. This leads to the “red, white, and blue sign” (ie, erythema, blanching, and ecchymosis). Infrequently, the ecchymotic area becomes necrotic and ulcerates in 3 to 5 days. The differential diagnoses should include necrotizing fasciitis, erythema chronicum migrans (from Borrelia-infected tick bites), and anthrax. Ulcerated lesions may result in significant cosmetic defect. Healing may take up to 2 weeks, and skin grafting is occasionally required.26

Treatment begins with the usual supportive measures, including analgesia, ice, elevation, and a light compression dressing. Antibiotics are not indicated, unless there are signs of secondary infection. Serial evaluation for wound checks should be arranged. If ulceration develops, surgical debridement may be required. The vast majority of bites heal with supportive care alone, and aggressive medical therapy is usually not warranted.27Patients with systemic manifestations should be admitted to the hospital for further care. There is no evidence-based literature to guide therapy. Many therapies have been tried with variable results and there remains no definitive standard of care.
Treatment regimens include antihistamines, antivenin, colchicine, dapsone, hyperbaric oxygen, cyproheptadine, surgical excision, and steroids.28 Dapsone continues to be widely advocated worldwide despite its known adverse effects—most notably hemolysis and methemoglobinemia. Antivenin administration has shown some promise in animal models, but its efficacy in humans is still unclear.29
Tegenaria
The Tegenaria agrestis or hobo spider is a native of Europe and central Asia and is only found in the northwest part of the United States. It is considered aggressive and tends to bite even with only mild provocation. The clinical presentation, inclusive of systemic reactions, is similar to that of the brown recluse spider. Similarly, there is no proven treatment. Surgical wound resection and skin grafting should be considered and is at times required.
Latrodectus.
Latrodectus, also known as widow spiders, are found worldwide. Five species are commonly found in the United States, but the black widow is the most well known. Only three of the species are actually black. Other varieties are typically brown or red. However, almost all Latrodectus spiders have a characteristic orange-red hourglass-shaped marking (Figure 3). Widow spiders aggressively defend their webs, and are most often found in woodpiles, basements, garages, and sheds. Most bites occur in the warmer months, between April and October.

Latrodectus antivenin is very effective, often resolving symptoms rapidly and reducing the duration of illness—even when administered up to 90 hours postenvenomation.32 This antivenin is derived from horse serum, and hypersensitivity reactions are possible. One death from anaphylaxis has been reported in the United States after antivenin was given undiluted via IV push; however, slow administration of diluted antivenin is considered safe.33
Diptera
The order Diptera of the phylum Arthropoda includes over 240,000 species. Among those, the mosquitoes and flies are the most medically relevant.
Mosquitoes
An actual mosquito bite itself causes minimal trauma and is not usually felt by the victim. However, the local anesthetic that is injected into the wound at the time of the attack causes local tissue damage. Mosquito bites can lead to both immediate and delayed reactions. Typical immediate reactions are of short duration and include edema, erythema, and pruritus. More severe reactions are extremely rare and consist of skin necrosis. Delayed skin reactions are fairly common, but tend to last longer, persisting for days or even weeks. Treatment is symptomatic, usually with antihistamines and NSAIDS.
Patients can acquire an allergy to mosquito saliva over time and develop increasingly pronounced edematous and pruritic lesions with subsequent bites. They can also experience fever, malaise, generalized edema, as well as severe nausea and vomiting.
Systemic or anaphylactic reactions are not known to occur. Instead, the greatest danger occurs with the transmission of life-threatening diseases. Malaria, yellow fever, dengue hemorrhagic fever, and different types of equine encephalitis are all transmitted by mosquito bites. One interesting newcomer to the United States is the West Nile virus (WNV), which has spread rapidly since its introduction in 2003. Over 1,850 cases were reported in 22 different states over the initial 8 months. Acute symptoms are mild in the majority of patients, but a small minority can experience fatal disease. Neurological symptoms include tumor, myoclonus, and Parkinsonism. An irreversible poliomyelitis-like syndrome may also develop. In addition to WNV, St Louis encephalitis and equine encephalitis also remain important pathogens in the United States.34 Prevention of bites is crucial and includes the use of pyrethroid-impregnated mosquito netting, repellents, and oral malaria prophylaxis. N,N-diethyl-3-methylbenzamide (DEET) remains the most effective mosquito repellent.35 Although toxic reactions are rare, they do occur and anaphylaxis has been reported. 36,37
Flies
Flies are blood-sucking insects that feed by stabbing and piercing their victim’s skin. Their bites always cause some degree of pain and pruritus. Allergic reactions are possible, though not as severe as those produced by Hymenoptera venom. Treatment is largely symptomatic with ice, oral antihistamine, analgesics. and topical or oral steroids as needed. Secondary bite infection is a concern and antibiotics are sometimes necessary.
Shiponaptera
The order Shinoptera includes fleas and lice. All produce very similar lesions, making diagnosis difficult. One concern with these bites is the development of secondary infections, especially in children. The skin should be washed with soap and water. Calamine, cool soaks, and oral or topical antihistamine may all be helpful to reduce symptoms.
Fleas
With fleas, as with mosquitoes, there is additionally a concern for transmission of life-threatening diseases. Fleas transmit bubonic plague, endemic typhus, brucellosis, melioidosis, and erysipeloid. Fortunately, effective oral and injectable formulations for both dogs and cats are now available to control fleas on most family pets.
Lice
Hemiptera

Lepidoptera
The order Lepidoptera includes butterflies and moths and their caterpillars. Symptoms that result from contact with this class of insects are referred to as lepidopterism. Caterpillars have hair or spines for protection, which are also sometimes connected to a venom gland. Contact with these spines usually causes localized skin irritation and pruritus. Megalopyge opercularis, also known as the “puss caterpillar,” is mainly found in the southeastern United States and accounts for the majority of envenomation cases in this country. Intense local burning pain is typical at the site of contact and is followed by a grid-like pattern of hemorrhagic papules, which appear 2 to 3 hours after exposure and may last for several days. Regional lymphadenopathy is common. Other symptoms include headache, fever, hypotension, and convulsions. No deaths have ever been reported.
As there is no antivenin available for lepidopterism, treatment is mostly supportive. If spines are visible following contact, they should be removed with adhesive tape. Antihistamines and steroids may be used for symptom control. In patients with hypotension, IV fluids and IV epinephrine may be required.43
Coleoptera
The order Coleoptera includes a large number of beetles, though clinically significant envenomation occurs only with blister beetles. There are over 1,500 species of blister beetles worldwide, approximately 2,002 of which are in the United States. The blister beetle responsible for most of the medically significant envenomations is Cantharis vesicatoria—also known as “Spanish fly.” Of note, the Spanish fly is not naturally found in the United States.

Cantharidin-containing substances are sometimes used medicinally in wart removal preparations and are also sold for their purported aphrodisiac effects (the associated vascular congestion and urethral inflammation are interpreted as enhanced sexuality). Transdermal absorption or ingestion may lead to systemic toxicity with severe vomiting, hematemesis, abdominal pain, diarrhea, hematuria, renal failure, etc. Death has been reported after large ingestions.
Treatment is largely supportive. The skin should be irrigated thoroughly after exposure, followed by local wound care. Patients who present after ingestion should be admitted to the hospital for further treatment and care.47
Conclusion
Knowledge of a vast array of stinging insects and spiders is important for any clinician, but the appropriate evaluation and treatment of bites and envenomations are crucial for EPs. Most exposures can be treated with supportive care, while others require in-depth knowledge and clinical expertise.
Dr Deljoui is a former resident, department of emergency medicine, Eastern Virginia Medical School, Norfolk; and current critical care fellow, University of Maryland, Baltimore.
Dr Knapp is an associate professor and residency program director, department of emergency medicine, Eastern Virginia Medical School, Norfolk.
- White J. Bites and stings from venomous animals: a global overview. The Drug Monit. 2000;22(1):65-68.
- Oten EJ. Venomous animal injuries. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 1. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:794-807.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229. doi:10.3109/15563650.2013.863906. https://aapcc.s3.amazonaws.com/pdfs/annual_reports/2012_NPDS_Annual_Report.pdf. Accessed April 2, 2014.
- Liberman P, Camargo CA, Bohike K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol. 2006;97(5):596-602.
- Barnard JH. Studies of 400 Hymenoptera sting deaths in the United States. J Allergy Clin Immunol. 1973;52(5):259-264.
- Frazier CA. Insect Allergy: Allergic and Toxic Reactions to Insects and Other Arthropods. 2nd Ed. St Louis, MO: WH Green; 1987:421.
- King TP, Spangfort MD. Structure and biology of stinging insect venom allergens. Int Arch Allergy Immunol. 2000;123(2):99-106.
- Antonicelli L, Bilo MB, Bonifazi F. Epidemiology of Hymenoptera allergy. Curr Opin Allergy Clin Immunol.2002;2(4);341-346.
- Mauriello PM, Barde SH. Natural history of large local reactions from stinging insects. J Allergy Clin Immunol. 1984;74(4 Pt 1):494-498.
- Díaz-Sánchez CL, Lifshitz-Guinzberg A, Ignacio-Ibarra G, Halabe-Cherem J, Quinones-Galvan A. Survival after massive (>2,000) Africanized honey bee stings. Arch Intern Med. 1998;158(8):925-927.
- Elston DM. Life-threatening stings, bites, infestations and parasitic diseases. Clin Dermatol. 2005;23(2):164-170.
- Lazoglu AH1, Boglioli LR, Taff ML, Rosenbluth M, Macris NT. Serum sickness reaction following multiple insect stings. Ann Allergy Asthma Immunol. 1995;75(6 Pt 1):522-524.
- Reisman RE, Livingston A. Late-onset allergic reactions, including serum sickness, after insect stings. J Allergy Clin Immunol. 1989;84(3);331-337.
- Anaphylaxis. American Academy of Allergy, Asthma & Immunology Web site. http://www.aaaai.org/conditions-and-treatments/conditions-a-to-zsearch/anaphylaxis.aspx. Accessed April 2, 2014.
- Brown H, Benton HS. Allergy to the Hymenoptera. V. Clinical study of 400 patients. Arch Intern Med. 1970;125(4):665-669.
- Frazier CA. Allergic reactions to insect stings: a review of 180 cases. South Med J. 1964;57;1023-1034.
- Mueller HL. Further experiences with severe allergic reactions to insect stings. N Engl J Med. 1959;161:374-377.
- Lockey RF, Turkeltaub PC, Baird-Warren IA, et al. The Hymenoptera venom study I, 1979-1982: demographics and history-sting data. J Allergy Clin Immunol. 1988;82(3 Pt 1):370-381.
- Schneir AB, Clark RF. Bites and stings. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011:chap120;585-596.
- Rowe BH, Gaeta T. Anaphylaxis, acute allergic reactions, and angioedema. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011:chap 6;52-54.
- Jones RG1, Corteling RL, Bhogal G, Landon J. A novel Fab-based antivenom for the treatment of mass bee attacks. Am J Trop Med Hyg. 1999;61(3):361-366.
- National Institutes of Health, US Department of Health and Human Services, National Insitute of Allergy and Infectious Diseases. Guidelines for the Diagnosis and Management of Food Allergy in the United States. Summary of the NIAID-Sponsored Expert Panel Report. Bethesda, MD: National Institutes of Health; 2010. NIH Publication No. 11-7700. http://www.niaid.nih.gov/topics/foodAllergy/clinical/Documents/FAGuidelinesExecSummary.pdf. Accessed April 2, 2014.
- Kemp SF, deShazo RD, Moffitt JE, Williams DF, Buhner WA 2nd. Expanding habitat of the imported fire ant (Solenopsis invicta): a public health concern. J Allergy Clin Immunol. 2000;105(4):683-691.
- Fernández-Meléndez S, Miranda A, García-González JJ, Barber D, Lombardero M. Anaphylaxis caused by imported red fire ant stings in Málaga, Spain. J Investig Allergol Immunol. 2007;17(1):48,49.
- Swanson DL. Bites of brown recluse spiders and suspected necrotic arachnidism. N Engl J Med. 2005;352(7):700-707.
- Saucier JR. Arachnid envenomation. Emerg Med Clin North Am. 2004;22(2):405-422.
- Wright SW, Wrenn KD, Murray L, Seger D. Clinical presentation and outcome of brown recluse spider bite. Ann Emerg Med. 1997;30(1):28-32.
- Phillips S, Kohn M, Baker D, et al. Therapy of brown spider envenomation: a controlled trial of hyperbaric oxygen, dapsone, and cyproheptadine. Ann Emerg Med. 1995;25(3):363-368.
- Pauli I, Puka J, Gubert IC, Minozzo JC. The efficacy of antivenom in loxoscelism treatment. Toxicon. 2006;48(2):123-127.
- Ushkaryov YA, Volynski KE, Ashton AC. The multiple actions of black widow spider toxins and their selective use in neurosecretion studies. Toxicon. 2004;43(5):527-542.
- Clark RF, Wethern-Kestner S, Vance MV, Gerkin R. Clinical presentation and treatment of black widow spider envenomation: a review of 163 cases. Ann Emerg Med. 1992;21(7):782-787.
- O’Malley GF, Dart RC, Kuffner EF. Successful treatment of latrodectism with antivenom after 90 hours. N Engl J Med. 1999;340(8):657.
- Clark RF. The safety and efficacy of antivenin Latrodectus mactans. J Toxicol Clin Toxicol. 2001;39(2):125-127.
- Sejvar JJ, Haddad MB, Tierney BC. Neurologic manifestations and outcome of West Nile virus infection [published correction appears in JAMA. 2003;290(10):1318]. JAMA. 2003;290(4):511-515.
- Brown M, Herbert AA. Insect repellents: an overview. J Am Acad Dermatol. 1997;36(2 Pt 1):243-249.
- Fradin MS. Mosquitoes and mosquito repellents: a clinician’s quide. Ann Intern Med. 1998;128(11):931-940.
- Miller JD. Anaphylaxis associated with insect repellent. N Engl J Med. 1982;307(21):1341,1342.
- Spach DH, Kanter AS, Dougherty MJ, et al. Bartonella (Rochalimaea) quintana bacteremia in inner-city patients with chronic alcoholism. N Engl J Med. 1995;332(7): 424-428.
- Jackson LA, Spach DH, Kippen DA, et al. Seroprevalence to Bartonella quintana among patients at a community clinic in downtown Seattle. J Infect Dis. 1996;173(4):1023-1026.
- Sundnes KO. Epidemic of louse-borne relapsing fever in Ethiopia. Lancet. 1993;342(8881):1213-1215.
- Vetter R. Kissing bugs (Triatoma) and the skin. Dermatol Online J. 2001;7(1):6. http://escholarship.org/uc/item/59k2m8wt. Accessed April 2, 2014.
- Stucki A, Ludwig R. Images in clinical medicine. Bedbug bites. N Engl J Med. 2008; 359:10)1047.
- Kuspis DA, Rawlins JE, Krenzelok EP. Human exposures to stinging caterpillars: Lophocampa caryae exposures. Am J Emerg Med. 2001;19(5):396-398.
- Moed L, Shwayder TA, 0.Chang MW. Cantharidin revisited: a blistering defense of an ancient medicine. Arch Dermatol. 2001;137(10):1357-1360.
Hymenoptera
The order Hymenoptera of the phylum Arthropoda can be divided into three subgroups that are medically relevant: (1) Apidae (Apids), which include the honeybee and bumblebee; (2) Vespidae, (Vespids) which include yellow jackets, hornets and wasps; and (3) Formicidae (ants).6
Bees and Wasps
The main allergens in Apid venom are phospholipase A2, hyaluronidase, and melittin. Melittin, the main component, is a membrane active polypeptide that causes degranulation of basophils and mast cells. The allergens in Vespid venom are phospholipase, hyaluronidase, and antigen 5. As all Hymenoptera share some of these components, cross-sensitization may occur and individuals may be allergic to more than one species.7
Occasionally after multiple stings, patients present with symptoms of a systemic toxic reaction. This is often seen in an Africanized bee attack. These so-called “killer bees” are hybrids of African bees that escaped from laboratories in Brazil in the 1950s and spread northward; they are found in most of the warmer US states. Their venom is not more toxic than that of any other bee, but Africanized honeybees are more aggressive and respond to a perceived threat in far greater numbers. The reaction that results from multiple stings is systemic and may resemble anaphylaxis. Common symptoms include nausea, vomiting, and diarrhea, as well as lightheadedness and syncope. Interestingly, urticaria and bronchospasm are not universally present, even though respiratory failure and cardiac arrest may occur. Other symptoms include renal failure with acute tubular necrosis, myoglobinuria or hemoglobinuria, hepatic failure, and disseminated intravascular coagulation (DIC).10,11 In addition, there have been reports of unusual reactions such as vasculitis, nephrosis, neuritis, encephalitis, and serum sickness. Late-appearing symptoms usually start several days to weeks after a sting and tend last for a prolonged period of time. Serum sickness tends to appear 5 to 14 days after exposure and consists of fever, malaise, headache, urticaria, lymphadenopathy, and polyarthritis.12 Of note, patients who have venom-induced serum sickness may be at risk for anaphylaxis after subsequent stings and may therefore be suitable candidates for venom immunotherapy.13
Anaphylaxis
The definition of anaphylaxis is not universally agreed upon. The American Academy of Allergy, Asthma and Immunology defines anaphylaxis as a serious allergic response that often involves swelling, hives, hypotension and, in severe cases, shock. A major difference between anaphylaxis and other allergic reactions is that anaphylaxis typically involves more than one body system.14 The clinical features of anaphylaxis from insect stings are the same as those from other causes, typically generalized urticaria, facial flushing, and angioedema. Abdominal cramping, nausea, vomiting, and diarrhea are also seen. Life-threatening symptoms include stridor, circulatory collapse with shock, and bronchospasm. Symptoms usually begin 10 to 20 minutes after a sting, and almost all will develop within 6 hours. Interestingly, symptoms may recur 8 to 12 hours after the initial reaction.15-18
Management
Immediate removal of the bee stinger is the most important principle as it precludes any further venom transfer. Traditional teaching recommended scraping the stinger out to avoid squeezing remaining venom into the tissues; however, involuntary muscle contractions of the gland continue after the stinger detaches, and the venom is quickly exhausted. Thus, immediate removal of the stinger is crucial, though the method of removal is now thought irrelevant.19
The mainstay of therapy for serious reactions is intramuscular (IM) epinephrine. The initial dosing is 0.3 to 0.5 mg (0.3 to 0.5 mL of 1:1000 concentration) in adults, and 0.01 mg/kg in children (maximum 0.3 mg). The injection should be IM and not subcutaneous, as IM dosing provides higher and more consistent and rapid peak blood epinephrine levels.20 Concomitant intravenous (IV) administration of standard antihistamines, often diphenhydramine 1 mg/kg (generally 25-50 mg) and histamine-2 receptor antagonists (typically ranitidine 50 mg) are also recommended. The administration of steroids (methylprednisolone 125 mg IV or prednisone 60 mg orally) is traditionally recommended and thought to help potentiate the effect of other interventional measures.20 Bronchospasm, if present, is treated with nebulized β-agonists (albuterol). Hypotension may develop and requires significant crystalloid infusion—often several liters. If hypotension persists despite adequate fluid replacement, vasopressor therapy is recommended.
If a patient does not respond to initial treatment and cardiovascular (CV) collapse is evident, IV infusion of epinephrine should be initiated. Epinephrine, 100 mcg (0.1 mg) IV, should be given as a 1:100,000 dilution. This can be done by placing epinephrine, 0.1 mg (0.1 mL of the 1:1000 dilution), in 10 mL of normal saline solution and infusing it over 5 to 10 minutes (a rate of 1 to 2 mL/min). If the patient is refractory to the initial bolus, then an epinephrine infusion can be started by placing epinephrine, 1 mg (1.0 mL of the 1:1000 dilution), in 500 mL of 5% dextrose in water or NS and administering at a rate of 1 to 4 mcg per minute (0.5 to 2 mL/min), titrating to effect.20 Antivenins have been studied for treatment, but none are commercially available at this time.21 Patients with anaphylaxis associated with severe signs and symptoms, including any evidence of CV collapse, should be admitted to the hospital for aggressive therapy and monitoring. Persons with mild-to-moderate reactions should be observed for 4 to 6 hours to monitor for late occurring symptoms. Outpatient therapy with antihistamines, oral steroids, and a prescription for an epinephrine auto-injector—including training on proper administration prior to discharge—are strongly recommended.22 Follow-up with an allergist is also indicated in patients with significant reactions, as skin testing and immunotherapy may be beneficial to prevent anaphylaxis during future exposures.
Ants
Fire ant venom is composed of an insoluble alkaloid, and crossreactivity with the venom of other Hymenopteras species does exist. Stings generally result in a papule, which evolves into a sterile pustule. Localized necrosis, scarring, and secondary infection can occur. Systemic reactions with angioedema and urticaria have been documented, which can sometimes lead to fatalities.24
Treatment
Treatment of fire ant stings begin and end with local wound care. If the reaction is systemic, a treatment plan similar to that outlined in the treatment section for bees and wasps is indicated.
Araneae
The order Araneae of the phylum Arthropoda includes over 34,000 species of spiders divided into 105 families. Of those, only half a dozen are medically relevant and only three are commonly encountered in the United States. These include Loxosceles (most notably, the brown recluse spider), Tegeneria (mainly the hobo brown spider) and Latrodectus (includes the black widow spider). True spiders have a worldwide distribution and tend to thrive in heavily populated areas, resulting in many biting episodes per year. Data from the AAPPC’s most recent annual report listed 9,255 single spider-bite exposures in 2012, with one associated death.3
Spiders are carnivores and use venom to paralyze their prey. They are generally not a threat to humans as their fangs are too small to penetrate human skin, and the amount of venom injected is too small to produce toxicity. Thus, reactions resulting from a spider bite are typically limited to a localized reaction. Fortunately, most bites only require supportive medical therapy.
Loxosceles
Loxosceles are present worldwide, but L reclusa (the “brown recluse spider”) accounts for a significant number of envenomations in the United States. The AAPCC’s 2012 data notes 1,365 cases of exposure to the brown recluse spider with 510 of those victims seeking medical care.3 In many instances, clinicians attribute necrotic bites to the brown recluse spider, however, confirmation is often lacking. Loxosceles are nocturnal, and they are found both indoors and outdoors—mostly in dark and dry areas such as basements, closets, and woodpiles. These spiders are shy, but may bite when threatened. Their venom contains enzymes, including hyaluronidase and sphingomyelinase. Though rare, wounds can become necrotic due to neutrophil activation, platelet aggregation, and thrombosis.25 The most common reaction to a Loxosceles bite is a mild painless erythematous lesion that becomes firm and generally heals over several days to weeks. In severe reactions, erythema, edema, and pruritus initially develop, followed within 24 to 72 hours by a hemorrhagic bulla surrounded by blanched skin. This leads to the “red, white, and blue sign” (ie, erythema, blanching, and ecchymosis). Infrequently, the ecchymotic area becomes necrotic and ulcerates in 3 to 5 days. The differential diagnoses should include necrotizing fasciitis, erythema chronicum migrans (from Borrelia-infected tick bites), and anthrax. Ulcerated lesions may result in significant cosmetic defect. Healing may take up to 2 weeks, and skin grafting is occasionally required.26
Treatment begins with the usual supportive measures, including analgesia, ice, elevation, and a light compression dressing. Antibiotics are not indicated, unless there are signs of secondary infection. Serial evaluation for wound checks should be arranged. If ulceration develops, surgical debridement may be required. The vast majority of bites heal with supportive care alone, and aggressive medical therapy is usually not warranted.27Patients with systemic manifestations should be admitted to the hospital for further care. There is no evidence-based literature to guide therapy. Many therapies have been tried with variable results and there remains no definitive standard of care.
Treatment regimens include antihistamines, antivenin, colchicine, dapsone, hyperbaric oxygen, cyproheptadine, surgical excision, and steroids.28 Dapsone continues to be widely advocated worldwide despite its known adverse effects—most notably hemolysis and methemoglobinemia. Antivenin administration has shown some promise in animal models, but its efficacy in humans is still unclear.29
Tegenaria
The Tegenaria agrestis or hobo spider is a native of Europe and central Asia and is only found in the northwest part of the United States. It is considered aggressive and tends to bite even with only mild provocation. The clinical presentation, inclusive of systemic reactions, is similar to that of the brown recluse spider. Similarly, there is no proven treatment. Surgical wound resection and skin grafting should be considered and is at times required.
Latrodectus.
Latrodectus, also known as widow spiders, are found worldwide. Five species are commonly found in the United States, but the black widow is the most well known. Only three of the species are actually black. Other varieties are typically brown or red. However, almost all Latrodectus spiders have a characteristic orange-red hourglass-shaped marking (Figure 3). Widow spiders aggressively defend their webs, and are most often found in woodpiles, basements, garages, and sheds. Most bites occur in the warmer months, between April and October.
Latrodectus antivenin is very effective, often resolving symptoms rapidly and reducing the duration of illness—even when administered up to 90 hours postenvenomation.32 This antivenin is derived from horse serum, and hypersensitivity reactions are possible. One death from anaphylaxis has been reported in the United States after antivenin was given undiluted via IV push; however, slow administration of diluted antivenin is considered safe.33
Diptera
The order Diptera of the phylum Arthropoda includes over 240,000 species. Among those, the mosquitoes and flies are the most medically relevant.
Mosquitoes
An actual mosquito bite itself causes minimal trauma and is not usually felt by the victim. However, the local anesthetic that is injected into the wound at the time of the attack causes local tissue damage. Mosquito bites can lead to both immediate and delayed reactions. Typical immediate reactions are of short duration and include edema, erythema, and pruritus. More severe reactions are extremely rare and consist of skin necrosis. Delayed skin reactions are fairly common, but tend to last longer, persisting for days or even weeks. Treatment is symptomatic, usually with antihistamines and NSAIDS.
Patients can acquire an allergy to mosquito saliva over time and develop increasingly pronounced edematous and pruritic lesions with subsequent bites. They can also experience fever, malaise, generalized edema, as well as severe nausea and vomiting.
Systemic or anaphylactic reactions are not known to occur. Instead, the greatest danger occurs with the transmission of life-threatening diseases. Malaria, yellow fever, dengue hemorrhagic fever, and different types of equine encephalitis are all transmitted by mosquito bites. One interesting newcomer to the United States is the West Nile virus (WNV), which has spread rapidly since its introduction in 2003. Over 1,850 cases were reported in 22 different states over the initial 8 months. Acute symptoms are mild in the majority of patients, but a small minority can experience fatal disease. Neurological symptoms include tumor, myoclonus, and Parkinsonism. An irreversible poliomyelitis-like syndrome may also develop. In addition to WNV, St Louis encephalitis and equine encephalitis also remain important pathogens in the United States.34 Prevention of bites is crucial and includes the use of pyrethroid-impregnated mosquito netting, repellents, and oral malaria prophylaxis. N,N-diethyl-3-methylbenzamide (DEET) remains the most effective mosquito repellent.35 Although toxic reactions are rare, they do occur and anaphylaxis has been reported. 36,37
Flies
Flies are blood-sucking insects that feed by stabbing and piercing their victim’s skin. Their bites always cause some degree of pain and pruritus. Allergic reactions are possible, though not as severe as those produced by Hymenoptera venom. Treatment is largely symptomatic with ice, oral antihistamine, analgesics. and topical or oral steroids as needed. Secondary bite infection is a concern and antibiotics are sometimes necessary.
Shiponaptera
The order Shinoptera includes fleas and lice. All produce very similar lesions, making diagnosis difficult. One concern with these bites is the development of secondary infections, especially in children. The skin should be washed with soap and water. Calamine, cool soaks, and oral or topical antihistamine may all be helpful to reduce symptoms.
Fleas
With fleas, as with mosquitoes, there is additionally a concern for transmission of life-threatening diseases. Fleas transmit bubonic plague, endemic typhus, brucellosis, melioidosis, and erysipeloid. Fortunately, effective oral and injectable formulations for both dogs and cats are now available to control fleas on most family pets.
Lice
Hemiptera
Lepidoptera
The order Lepidoptera includes butterflies and moths and their caterpillars. Symptoms that result from contact with this class of insects are referred to as lepidopterism. Caterpillars have hair or spines for protection, which are also sometimes connected to a venom gland. Contact with these spines usually causes localized skin irritation and pruritus. Megalopyge opercularis, also known as the “puss caterpillar,” is mainly found in the southeastern United States and accounts for the majority of envenomation cases in this country. Intense local burning pain is typical at the site of contact and is followed by a grid-like pattern of hemorrhagic papules, which appear 2 to 3 hours after exposure and may last for several days. Regional lymphadenopathy is common. Other symptoms include headache, fever, hypotension, and convulsions. No deaths have ever been reported.
As there is no antivenin available for lepidopterism, treatment is mostly supportive. If spines are visible following contact, they should be removed with adhesive tape. Antihistamines and steroids may be used for symptom control. In patients with hypotension, IV fluids and IV epinephrine may be required.43
Coleoptera
The order Coleoptera includes a large number of beetles, though clinically significant envenomation occurs only with blister beetles. There are over 1,500 species of blister beetles worldwide, approximately 2,002 of which are in the United States. The blister beetle responsible for most of the medically significant envenomations is Cantharis vesicatoria—also known as “Spanish fly.” Of note, the Spanish fly is not naturally found in the United States.
Cantharidin-containing substances are sometimes used medicinally in wart removal preparations and are also sold for their purported aphrodisiac effects (the associated vascular congestion and urethral inflammation are interpreted as enhanced sexuality). Transdermal absorption or ingestion may lead to systemic toxicity with severe vomiting, hematemesis, abdominal pain, diarrhea, hematuria, renal failure, etc. Death has been reported after large ingestions.
Treatment is largely supportive. The skin should be irrigated thoroughly after exposure, followed by local wound care. Patients who present after ingestion should be admitted to the hospital for further treatment and care.47
Conclusion
Knowledge of a vast array of stinging insects and spiders is important for any clinician, but the appropriate evaluation and treatment of bites and envenomations are crucial for EPs. Most exposures can be treated with supportive care, while others require in-depth knowledge and clinical expertise.
Dr Deljoui is a former resident, department of emergency medicine, Eastern Virginia Medical School, Norfolk; and current critical care fellow, University of Maryland, Baltimore.
Dr Knapp is an associate professor and residency program director, department of emergency medicine, Eastern Virginia Medical School, Norfolk.
Hymenoptera
The order Hymenoptera of the phylum Arthropoda can be divided into three subgroups that are medically relevant: (1) Apidae (Apids), which include the honeybee and bumblebee; (2) Vespidae, (Vespids) which include yellow jackets, hornets and wasps; and (3) Formicidae (ants).6
Bees and Wasps
The main allergens in Apid venom are phospholipase A2, hyaluronidase, and melittin. Melittin, the main component, is a membrane active polypeptide that causes degranulation of basophils and mast cells. The allergens in Vespid venom are phospholipase, hyaluronidase, and antigen 5. As all Hymenoptera share some of these components, cross-sensitization may occur and individuals may be allergic to more than one species.7
Occasionally after multiple stings, patients present with symptoms of a systemic toxic reaction. This is often seen in an Africanized bee attack. These so-called “killer bees” are hybrids of African bees that escaped from laboratories in Brazil in the 1950s and spread northward; they are found in most of the warmer US states. Their venom is not more toxic than that of any other bee, but Africanized honeybees are more aggressive and respond to a perceived threat in far greater numbers. The reaction that results from multiple stings is systemic and may resemble anaphylaxis. Common symptoms include nausea, vomiting, and diarrhea, as well as lightheadedness and syncope. Interestingly, urticaria and bronchospasm are not universally present, even though respiratory failure and cardiac arrest may occur. Other symptoms include renal failure with acute tubular necrosis, myoglobinuria or hemoglobinuria, hepatic failure, and disseminated intravascular coagulation (DIC).10,11 In addition, there have been reports of unusual reactions such as vasculitis, nephrosis, neuritis, encephalitis, and serum sickness. Late-appearing symptoms usually start several days to weeks after a sting and tend last for a prolonged period of time. Serum sickness tends to appear 5 to 14 days after exposure and consists of fever, malaise, headache, urticaria, lymphadenopathy, and polyarthritis.12 Of note, patients who have venom-induced serum sickness may be at risk for anaphylaxis after subsequent stings and may therefore be suitable candidates for venom immunotherapy.13
Anaphylaxis
The definition of anaphylaxis is not universally agreed upon. The American Academy of Allergy, Asthma and Immunology defines anaphylaxis as a serious allergic response that often involves swelling, hives, hypotension and, in severe cases, shock. A major difference between anaphylaxis and other allergic reactions is that anaphylaxis typically involves more than one body system.14 The clinical features of anaphylaxis from insect stings are the same as those from other causes, typically generalized urticaria, facial flushing, and angioedema. Abdominal cramping, nausea, vomiting, and diarrhea are also seen. Life-threatening symptoms include stridor, circulatory collapse with shock, and bronchospasm. Symptoms usually begin 10 to 20 minutes after a sting, and almost all will develop within 6 hours. Interestingly, symptoms may recur 8 to 12 hours after the initial reaction.15-18
Management
Immediate removal of the bee stinger is the most important principle as it precludes any further venom transfer. Traditional teaching recommended scraping the stinger out to avoid squeezing remaining venom into the tissues; however, involuntary muscle contractions of the gland continue after the stinger detaches, and the venom is quickly exhausted. Thus, immediate removal of the stinger is crucial, though the method of removal is now thought irrelevant.19
The mainstay of therapy for serious reactions is intramuscular (IM) epinephrine. The initial dosing is 0.3 to 0.5 mg (0.3 to 0.5 mL of 1:1000 concentration) in adults, and 0.01 mg/kg in children (maximum 0.3 mg). The injection should be IM and not subcutaneous, as IM dosing provides higher and more consistent and rapid peak blood epinephrine levels.20 Concomitant intravenous (IV) administration of standard antihistamines, often diphenhydramine 1 mg/kg (generally 25-50 mg) and histamine-2 receptor antagonists (typically ranitidine 50 mg) are also recommended. The administration of steroids (methylprednisolone 125 mg IV or prednisone 60 mg orally) is traditionally recommended and thought to help potentiate the effect of other interventional measures.20 Bronchospasm, if present, is treated with nebulized β-agonists (albuterol). Hypotension may develop and requires significant crystalloid infusion—often several liters. If hypotension persists despite adequate fluid replacement, vasopressor therapy is recommended.
If a patient does not respond to initial treatment and cardiovascular (CV) collapse is evident, IV infusion of epinephrine should be initiated. Epinephrine, 100 mcg (0.1 mg) IV, should be given as a 1:100,000 dilution. This can be done by placing epinephrine, 0.1 mg (0.1 mL of the 1:1000 dilution), in 10 mL of normal saline solution and infusing it over 5 to 10 minutes (a rate of 1 to 2 mL/min). If the patient is refractory to the initial bolus, then an epinephrine infusion can be started by placing epinephrine, 1 mg (1.0 mL of the 1:1000 dilution), in 500 mL of 5% dextrose in water or NS and administering at a rate of 1 to 4 mcg per minute (0.5 to 2 mL/min), titrating to effect.20 Antivenins have been studied for treatment, but none are commercially available at this time.21 Patients with anaphylaxis associated with severe signs and symptoms, including any evidence of CV collapse, should be admitted to the hospital for aggressive therapy and monitoring. Persons with mild-to-moderate reactions should be observed for 4 to 6 hours to monitor for late occurring symptoms. Outpatient therapy with antihistamines, oral steroids, and a prescription for an epinephrine auto-injector—including training on proper administration prior to discharge—are strongly recommended.22 Follow-up with an allergist is also indicated in patients with significant reactions, as skin testing and immunotherapy may be beneficial to prevent anaphylaxis during future exposures.
Ants
Fire ant venom is composed of an insoluble alkaloid, and crossreactivity with the venom of other Hymenopteras species does exist. Stings generally result in a papule, which evolves into a sterile pustule. Localized necrosis, scarring, and secondary infection can occur. Systemic reactions with angioedema and urticaria have been documented, which can sometimes lead to fatalities.24
Treatment
Treatment of fire ant stings begin and end with local wound care. If the reaction is systemic, a treatment plan similar to that outlined in the treatment section for bees and wasps is indicated.
Araneae
The order Araneae of the phylum Arthropoda includes over 34,000 species of spiders divided into 105 families. Of those, only half a dozen are medically relevant and only three are commonly encountered in the United States. These include Loxosceles (most notably, the brown recluse spider), Tegeneria (mainly the hobo brown spider) and Latrodectus (includes the black widow spider). True spiders have a worldwide distribution and tend to thrive in heavily populated areas, resulting in many biting episodes per year. Data from the AAPPC’s most recent annual report listed 9,255 single spider-bite exposures in 2012, with one associated death.3
Spiders are carnivores and use venom to paralyze their prey. They are generally not a threat to humans as their fangs are too small to penetrate human skin, and the amount of venom injected is too small to produce toxicity. Thus, reactions resulting from a spider bite are typically limited to a localized reaction. Fortunately, most bites only require supportive medical therapy.
Loxosceles
Loxosceles are present worldwide, but L reclusa (the “brown recluse spider”) accounts for a significant number of envenomations in the United States. The AAPCC’s 2012 data notes 1,365 cases of exposure to the brown recluse spider with 510 of those victims seeking medical care.3 In many instances, clinicians attribute necrotic bites to the brown recluse spider, however, confirmation is often lacking. Loxosceles are nocturnal, and they are found both indoors and outdoors—mostly in dark and dry areas such as basements, closets, and woodpiles. These spiders are shy, but may bite when threatened. Their venom contains enzymes, including hyaluronidase and sphingomyelinase. Though rare, wounds can become necrotic due to neutrophil activation, platelet aggregation, and thrombosis.25 The most common reaction to a Loxosceles bite is a mild painless erythematous lesion that becomes firm and generally heals over several days to weeks. In severe reactions, erythema, edema, and pruritus initially develop, followed within 24 to 72 hours by a hemorrhagic bulla surrounded by blanched skin. This leads to the “red, white, and blue sign” (ie, erythema, blanching, and ecchymosis). Infrequently, the ecchymotic area becomes necrotic and ulcerates in 3 to 5 days. The differential diagnoses should include necrotizing fasciitis, erythema chronicum migrans (from Borrelia-infected tick bites), and anthrax. Ulcerated lesions may result in significant cosmetic defect. Healing may take up to 2 weeks, and skin grafting is occasionally required.26
Treatment begins with the usual supportive measures, including analgesia, ice, elevation, and a light compression dressing. Antibiotics are not indicated, unless there are signs of secondary infection. Serial evaluation for wound checks should be arranged. If ulceration develops, surgical debridement may be required. The vast majority of bites heal with supportive care alone, and aggressive medical therapy is usually not warranted.27Patients with systemic manifestations should be admitted to the hospital for further care. There is no evidence-based literature to guide therapy. Many therapies have been tried with variable results and there remains no definitive standard of care.
Treatment regimens include antihistamines, antivenin, colchicine, dapsone, hyperbaric oxygen, cyproheptadine, surgical excision, and steroids.28 Dapsone continues to be widely advocated worldwide despite its known adverse effects—most notably hemolysis and methemoglobinemia. Antivenin administration has shown some promise in animal models, but its efficacy in humans is still unclear.29
Tegenaria
The Tegenaria agrestis or hobo spider is a native of Europe and central Asia and is only found in the northwest part of the United States. It is considered aggressive and tends to bite even with only mild provocation. The clinical presentation, inclusive of systemic reactions, is similar to that of the brown recluse spider. Similarly, there is no proven treatment. Surgical wound resection and skin grafting should be considered and is at times required.
Latrodectus.
Latrodectus, also known as widow spiders, are found worldwide. Five species are commonly found in the United States, but the black widow is the most well known. Only three of the species are actually black. Other varieties are typically brown or red. However, almost all Latrodectus spiders have a characteristic orange-red hourglass-shaped marking (Figure 3). Widow spiders aggressively defend their webs, and are most often found in woodpiles, basements, garages, and sheds. Most bites occur in the warmer months, between April and October.
Latrodectus antivenin is very effective, often resolving symptoms rapidly and reducing the duration of illness—even when administered up to 90 hours postenvenomation.32 This antivenin is derived from horse serum, and hypersensitivity reactions are possible. One death from anaphylaxis has been reported in the United States after antivenin was given undiluted via IV push; however, slow administration of diluted antivenin is considered safe.33
Diptera
The order Diptera of the phylum Arthropoda includes over 240,000 species. Among those, the mosquitoes and flies are the most medically relevant.
Mosquitoes
An actual mosquito bite itself causes minimal trauma and is not usually felt by the victim. However, the local anesthetic that is injected into the wound at the time of the attack causes local tissue damage. Mosquito bites can lead to both immediate and delayed reactions. Typical immediate reactions are of short duration and include edema, erythema, and pruritus. More severe reactions are extremely rare and consist of skin necrosis. Delayed skin reactions are fairly common, but tend to last longer, persisting for days or even weeks. Treatment is symptomatic, usually with antihistamines and NSAIDS.
Patients can acquire an allergy to mosquito saliva over time and develop increasingly pronounced edematous and pruritic lesions with subsequent bites. They can also experience fever, malaise, generalized edema, as well as severe nausea and vomiting.
Systemic or anaphylactic reactions are not known to occur. Instead, the greatest danger occurs with the transmission of life-threatening diseases. Malaria, yellow fever, dengue hemorrhagic fever, and different types of equine encephalitis are all transmitted by mosquito bites. One interesting newcomer to the United States is the West Nile virus (WNV), which has spread rapidly since its introduction in 2003. Over 1,850 cases were reported in 22 different states over the initial 8 months. Acute symptoms are mild in the majority of patients, but a small minority can experience fatal disease. Neurological symptoms include tumor, myoclonus, and Parkinsonism. An irreversible poliomyelitis-like syndrome may also develop. In addition to WNV, St Louis encephalitis and equine encephalitis also remain important pathogens in the United States.34 Prevention of bites is crucial and includes the use of pyrethroid-impregnated mosquito netting, repellents, and oral malaria prophylaxis. N,N-diethyl-3-methylbenzamide (DEET) remains the most effective mosquito repellent.35 Although toxic reactions are rare, they do occur and anaphylaxis has been reported. 36,37
Flies
Flies are blood-sucking insects that feed by stabbing and piercing their victim’s skin. Their bites always cause some degree of pain and pruritus. Allergic reactions are possible, though not as severe as those produced by Hymenoptera venom. Treatment is largely symptomatic with ice, oral antihistamine, analgesics. and topical or oral steroids as needed. Secondary bite infection is a concern and antibiotics are sometimes necessary.
Shiponaptera
The order Shinoptera includes fleas and lice. All produce very similar lesions, making diagnosis difficult. One concern with these bites is the development of secondary infections, especially in children. The skin should be washed with soap and water. Calamine, cool soaks, and oral or topical antihistamine may all be helpful to reduce symptoms.
Fleas
With fleas, as with mosquitoes, there is additionally a concern for transmission of life-threatening diseases. Fleas transmit bubonic plague, endemic typhus, brucellosis, melioidosis, and erysipeloid. Fortunately, effective oral and injectable formulations for both dogs and cats are now available to control fleas on most family pets.
Lice
Hemiptera
Lepidoptera
The order Lepidoptera includes butterflies and moths and their caterpillars. Symptoms that result from contact with this class of insects are referred to as lepidopterism. Caterpillars have hair or spines for protection, which are also sometimes connected to a venom gland. Contact with these spines usually causes localized skin irritation and pruritus. Megalopyge opercularis, also known as the “puss caterpillar,” is mainly found in the southeastern United States and accounts for the majority of envenomation cases in this country. Intense local burning pain is typical at the site of contact and is followed by a grid-like pattern of hemorrhagic papules, which appear 2 to 3 hours after exposure and may last for several days. Regional lymphadenopathy is common. Other symptoms include headache, fever, hypotension, and convulsions. No deaths have ever been reported.
As there is no antivenin available for lepidopterism, treatment is mostly supportive. If spines are visible following contact, they should be removed with adhesive tape. Antihistamines and steroids may be used for symptom control. In patients with hypotension, IV fluids and IV epinephrine may be required.43
Coleoptera
The order Coleoptera includes a large number of beetles, though clinically significant envenomation occurs only with blister beetles. There are over 1,500 species of blister beetles worldwide, approximately 2,002 of which are in the United States. The blister beetle responsible for most of the medically significant envenomations is Cantharis vesicatoria—also known as “Spanish fly.” Of note, the Spanish fly is not naturally found in the United States.
Cantharidin-containing substances are sometimes used medicinally in wart removal preparations and are also sold for their purported aphrodisiac effects (the associated vascular congestion and urethral inflammation are interpreted as enhanced sexuality). Transdermal absorption or ingestion may lead to systemic toxicity with severe vomiting, hematemesis, abdominal pain, diarrhea, hematuria, renal failure, etc. Death has been reported after large ingestions.
Treatment is largely supportive. The skin should be irrigated thoroughly after exposure, followed by local wound care. Patients who present after ingestion should be admitted to the hospital for further treatment and care.47
Conclusion
Knowledge of a vast array of stinging insects and spiders is important for any clinician, but the appropriate evaluation and treatment of bites and envenomations are crucial for EPs. Most exposures can be treated with supportive care, while others require in-depth knowledge and clinical expertise.
Dr Deljoui is a former resident, department of emergency medicine, Eastern Virginia Medical School, Norfolk; and current critical care fellow, University of Maryland, Baltimore.
Dr Knapp is an associate professor and residency program director, department of emergency medicine, Eastern Virginia Medical School, Norfolk.
- White J. Bites and stings from venomous animals: a global overview. The Drug Monit. 2000;22(1):65-68.
- Oten EJ. Venomous animal injuries. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 1. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:794-807.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229. doi:10.3109/15563650.2013.863906. https://aapcc.s3.amazonaws.com/pdfs/annual_reports/2012_NPDS_Annual_Report.pdf. Accessed April 2, 2014.
- Liberman P, Camargo CA, Bohike K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol. 2006;97(5):596-602.
- Barnard JH. Studies of 400 Hymenoptera sting deaths in the United States. J Allergy Clin Immunol. 1973;52(5):259-264.
- Frazier CA. Insect Allergy: Allergic and Toxic Reactions to Insects and Other Arthropods. 2nd Ed. St Louis, MO: WH Green; 1987:421.
- King TP, Spangfort MD. Structure and biology of stinging insect venom allergens. Int Arch Allergy Immunol. 2000;123(2):99-106.
- Antonicelli L, Bilo MB, Bonifazi F. Epidemiology of Hymenoptera allergy. Curr Opin Allergy Clin Immunol.2002;2(4);341-346.
- Mauriello PM, Barde SH. Natural history of large local reactions from stinging insects. J Allergy Clin Immunol. 1984;74(4 Pt 1):494-498.
- Díaz-Sánchez CL, Lifshitz-Guinzberg A, Ignacio-Ibarra G, Halabe-Cherem J, Quinones-Galvan A. Survival after massive (>2,000) Africanized honey bee stings. Arch Intern Med. 1998;158(8):925-927.
- Elston DM. Life-threatening stings, bites, infestations and parasitic diseases. Clin Dermatol. 2005;23(2):164-170.
- Lazoglu AH1, Boglioli LR, Taff ML, Rosenbluth M, Macris NT. Serum sickness reaction following multiple insect stings. Ann Allergy Asthma Immunol. 1995;75(6 Pt 1):522-524.
- Reisman RE, Livingston A. Late-onset allergic reactions, including serum sickness, after insect stings. J Allergy Clin Immunol. 1989;84(3);331-337.
- Anaphylaxis. American Academy of Allergy, Asthma & Immunology Web site. http://www.aaaai.org/conditions-and-treatments/conditions-a-to-zsearch/anaphylaxis.aspx. Accessed April 2, 2014.
- Brown H, Benton HS. Allergy to the Hymenoptera. V. Clinical study of 400 patients. Arch Intern Med. 1970;125(4):665-669.
- Frazier CA. Allergic reactions to insect stings: a review of 180 cases. South Med J. 1964;57;1023-1034.
- Mueller HL. Further experiences with severe allergic reactions to insect stings. N Engl J Med. 1959;161:374-377.
- Lockey RF, Turkeltaub PC, Baird-Warren IA, et al. The Hymenoptera venom study I, 1979-1982: demographics and history-sting data. J Allergy Clin Immunol. 1988;82(3 Pt 1):370-381.
- Schneir AB, Clark RF. Bites and stings. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011:chap120;585-596.
- Rowe BH, Gaeta T. Anaphylaxis, acute allergic reactions, and angioedema. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011:chap 6;52-54.
- Jones RG1, Corteling RL, Bhogal G, Landon J. A novel Fab-based antivenom for the treatment of mass bee attacks. Am J Trop Med Hyg. 1999;61(3):361-366.
- National Institutes of Health, US Department of Health and Human Services, National Insitute of Allergy and Infectious Diseases. Guidelines for the Diagnosis and Management of Food Allergy in the United States. Summary of the NIAID-Sponsored Expert Panel Report. Bethesda, MD: National Institutes of Health; 2010. NIH Publication No. 11-7700. http://www.niaid.nih.gov/topics/foodAllergy/clinical/Documents/FAGuidelinesExecSummary.pdf. Accessed April 2, 2014.
- Kemp SF, deShazo RD, Moffitt JE, Williams DF, Buhner WA 2nd. Expanding habitat of the imported fire ant (Solenopsis invicta): a public health concern. J Allergy Clin Immunol. 2000;105(4):683-691.
- Fernández-Meléndez S, Miranda A, García-González JJ, Barber D, Lombardero M. Anaphylaxis caused by imported red fire ant stings in Málaga, Spain. J Investig Allergol Immunol. 2007;17(1):48,49.
- Swanson DL. Bites of brown recluse spiders and suspected necrotic arachnidism. N Engl J Med. 2005;352(7):700-707.
- Saucier JR. Arachnid envenomation. Emerg Med Clin North Am. 2004;22(2):405-422.
- Wright SW, Wrenn KD, Murray L, Seger D. Clinical presentation and outcome of brown recluse spider bite. Ann Emerg Med. 1997;30(1):28-32.
- Phillips S, Kohn M, Baker D, et al. Therapy of brown spider envenomation: a controlled trial of hyperbaric oxygen, dapsone, and cyproheptadine. Ann Emerg Med. 1995;25(3):363-368.
- Pauli I, Puka J, Gubert IC, Minozzo JC. The efficacy of antivenom in loxoscelism treatment. Toxicon. 2006;48(2):123-127.
- Ushkaryov YA, Volynski KE, Ashton AC. The multiple actions of black widow spider toxins and their selective use in neurosecretion studies. Toxicon. 2004;43(5):527-542.
- Clark RF, Wethern-Kestner S, Vance MV, Gerkin R. Clinical presentation and treatment of black widow spider envenomation: a review of 163 cases. Ann Emerg Med. 1992;21(7):782-787.
- O’Malley GF, Dart RC, Kuffner EF. Successful treatment of latrodectism with antivenom after 90 hours. N Engl J Med. 1999;340(8):657.
- Clark RF. The safety and efficacy of antivenin Latrodectus mactans. J Toxicol Clin Toxicol. 2001;39(2):125-127.
- Sejvar JJ, Haddad MB, Tierney BC. Neurologic manifestations and outcome of West Nile virus infection [published correction appears in JAMA. 2003;290(10):1318]. JAMA. 2003;290(4):511-515.
- Brown M, Herbert AA. Insect repellents: an overview. J Am Acad Dermatol. 1997;36(2 Pt 1):243-249.
- Fradin MS. Mosquitoes and mosquito repellents: a clinician’s quide. Ann Intern Med. 1998;128(11):931-940.
- Miller JD. Anaphylaxis associated with insect repellent. N Engl J Med. 1982;307(21):1341,1342.
- Spach DH, Kanter AS, Dougherty MJ, et al. Bartonella (Rochalimaea) quintana bacteremia in inner-city patients with chronic alcoholism. N Engl J Med. 1995;332(7): 424-428.
- Jackson LA, Spach DH, Kippen DA, et al. Seroprevalence to Bartonella quintana among patients at a community clinic in downtown Seattle. J Infect Dis. 1996;173(4):1023-1026.
- Sundnes KO. Epidemic of louse-borne relapsing fever in Ethiopia. Lancet. 1993;342(8881):1213-1215.
- Vetter R. Kissing bugs (Triatoma) and the skin. Dermatol Online J. 2001;7(1):6. http://escholarship.org/uc/item/59k2m8wt. Accessed April 2, 2014.
- Stucki A, Ludwig R. Images in clinical medicine. Bedbug bites. N Engl J Med. 2008; 359:10)1047.
- Kuspis DA, Rawlins JE, Krenzelok EP. Human exposures to stinging caterpillars: Lophocampa caryae exposures. Am J Emerg Med. 2001;19(5):396-398.
- Moed L, Shwayder TA, 0.Chang MW. Cantharidin revisited: a blistering defense of an ancient medicine. Arch Dermatol. 2001;137(10):1357-1360.
- White J. Bites and stings from venomous animals: a global overview. The Drug Monit. 2000;22(1):65-68.
- Oten EJ. Venomous animal injuries. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 1. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:794-807.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229. doi:10.3109/15563650.2013.863906. https://aapcc.s3.amazonaws.com/pdfs/annual_reports/2012_NPDS_Annual_Report.pdf. Accessed April 2, 2014.
- Liberman P, Camargo CA, Bohike K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol. 2006;97(5):596-602.
- Barnard JH. Studies of 400 Hymenoptera sting deaths in the United States. J Allergy Clin Immunol. 1973;52(5):259-264.
- Frazier CA. Insect Allergy: Allergic and Toxic Reactions to Insects and Other Arthropods. 2nd Ed. St Louis, MO: WH Green; 1987:421.
- King TP, Spangfort MD. Structure and biology of stinging insect venom allergens. Int Arch Allergy Immunol. 2000;123(2):99-106.
- Antonicelli L, Bilo MB, Bonifazi F. Epidemiology of Hymenoptera allergy. Curr Opin Allergy Clin Immunol.2002;2(4);341-346.
- Mauriello PM, Barde SH. Natural history of large local reactions from stinging insects. J Allergy Clin Immunol. 1984;74(4 Pt 1):494-498.
- Díaz-Sánchez CL, Lifshitz-Guinzberg A, Ignacio-Ibarra G, Halabe-Cherem J, Quinones-Galvan A. Survival after massive (>2,000) Africanized honey bee stings. Arch Intern Med. 1998;158(8):925-927.
- Elston DM. Life-threatening stings, bites, infestations and parasitic diseases. Clin Dermatol. 2005;23(2):164-170.
- Lazoglu AH1, Boglioli LR, Taff ML, Rosenbluth M, Macris NT. Serum sickness reaction following multiple insect stings. Ann Allergy Asthma Immunol. 1995;75(6 Pt 1):522-524.
- Reisman RE, Livingston A. Late-onset allergic reactions, including serum sickness, after insect stings. J Allergy Clin Immunol. 1989;84(3);331-337.
- Anaphylaxis. American Academy of Allergy, Asthma & Immunology Web site. http://www.aaaai.org/conditions-and-treatments/conditions-a-to-zsearch/anaphylaxis.aspx. Accessed April 2, 2014.
- Brown H, Benton HS. Allergy to the Hymenoptera. V. Clinical study of 400 patients. Arch Intern Med. 1970;125(4):665-669.
- Frazier CA. Allergic reactions to insect stings: a review of 180 cases. South Med J. 1964;57;1023-1034.
- Mueller HL. Further experiences with severe allergic reactions to insect stings. N Engl J Med. 1959;161:374-377.
- Lockey RF, Turkeltaub PC, Baird-Warren IA, et al. The Hymenoptera venom study I, 1979-1982: demographics and history-sting data. J Allergy Clin Immunol. 1988;82(3 Pt 1):370-381.
- Schneir AB, Clark RF. Bites and stings. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011:chap120;585-596.
- Rowe BH, Gaeta T. Anaphylaxis, acute allergic reactions, and angioedema. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011:chap 6;52-54.
- Jones RG1, Corteling RL, Bhogal G, Landon J. A novel Fab-based antivenom for the treatment of mass bee attacks. Am J Trop Med Hyg. 1999;61(3):361-366.
- National Institutes of Health, US Department of Health and Human Services, National Insitute of Allergy and Infectious Diseases. Guidelines for the Diagnosis and Management of Food Allergy in the United States. Summary of the NIAID-Sponsored Expert Panel Report. Bethesda, MD: National Institutes of Health; 2010. NIH Publication No. 11-7700. http://www.niaid.nih.gov/topics/foodAllergy/clinical/Documents/FAGuidelinesExecSummary.pdf. Accessed April 2, 2014.
- Kemp SF, deShazo RD, Moffitt JE, Williams DF, Buhner WA 2nd. Expanding habitat of the imported fire ant (Solenopsis invicta): a public health concern. J Allergy Clin Immunol. 2000;105(4):683-691.
- Fernández-Meléndez S, Miranda A, García-González JJ, Barber D, Lombardero M. Anaphylaxis caused by imported red fire ant stings in Málaga, Spain. J Investig Allergol Immunol. 2007;17(1):48,49.
- Swanson DL. Bites of brown recluse spiders and suspected necrotic arachnidism. N Engl J Med. 2005;352(7):700-707.
- Saucier JR. Arachnid envenomation. Emerg Med Clin North Am. 2004;22(2):405-422.
- Wright SW, Wrenn KD, Murray L, Seger D. Clinical presentation and outcome of brown recluse spider bite. Ann Emerg Med. 1997;30(1):28-32.
- Phillips S, Kohn M, Baker D, et al. Therapy of brown spider envenomation: a controlled trial of hyperbaric oxygen, dapsone, and cyproheptadine. Ann Emerg Med. 1995;25(3):363-368.
- Pauli I, Puka J, Gubert IC, Minozzo JC. The efficacy of antivenom in loxoscelism treatment. Toxicon. 2006;48(2):123-127.
- Ushkaryov YA, Volynski KE, Ashton AC. The multiple actions of black widow spider toxins and their selective use in neurosecretion studies. Toxicon. 2004;43(5):527-542.
- Clark RF, Wethern-Kestner S, Vance MV, Gerkin R. Clinical presentation and treatment of black widow spider envenomation: a review of 163 cases. Ann Emerg Med. 1992;21(7):782-787.
- O’Malley GF, Dart RC, Kuffner EF. Successful treatment of latrodectism with antivenom after 90 hours. N Engl J Med. 1999;340(8):657.
- Clark RF. The safety and efficacy of antivenin Latrodectus mactans. J Toxicol Clin Toxicol. 2001;39(2):125-127.
- Sejvar JJ, Haddad MB, Tierney BC. Neurologic manifestations and outcome of West Nile virus infection [published correction appears in JAMA. 2003;290(10):1318]. JAMA. 2003;290(4):511-515.
- Brown M, Herbert AA. Insect repellents: an overview. J Am Acad Dermatol. 1997;36(2 Pt 1):243-249.
- Fradin MS. Mosquitoes and mosquito repellents: a clinician’s quide. Ann Intern Med. 1998;128(11):931-940.
- Miller JD. Anaphylaxis associated with insect repellent. N Engl J Med. 1982;307(21):1341,1342.
- Spach DH, Kanter AS, Dougherty MJ, et al. Bartonella (Rochalimaea) quintana bacteremia in inner-city patients with chronic alcoholism. N Engl J Med. 1995;332(7): 424-428.
- Jackson LA, Spach DH, Kippen DA, et al. Seroprevalence to Bartonella quintana among patients at a community clinic in downtown Seattle. J Infect Dis. 1996;173(4):1023-1026.
- Sundnes KO. Epidemic of louse-borne relapsing fever in Ethiopia. Lancet. 1993;342(8881):1213-1215.
- Vetter R. Kissing bugs (Triatoma) and the skin. Dermatol Online J. 2001;7(1):6. http://escholarship.org/uc/item/59k2m8wt. Accessed April 2, 2014.
- Stucki A, Ludwig R. Images in clinical medicine. Bedbug bites. N Engl J Med. 2008; 359:10)1047.
- Kuspis DA, Rawlins JE, Krenzelok EP. Human exposures to stinging caterpillars: Lophocampa caryae exposures. Am J Emerg Med. 2001;19(5):396-398.
- Moed L, Shwayder TA, 0.Chang MW. Cantharidin revisited: a blistering defense of an ancient medicine. Arch Dermatol. 2001;137(10):1357-1360.

Case Studies in Toxicology: A Patchwork of Problems in Parkinson Patients
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?

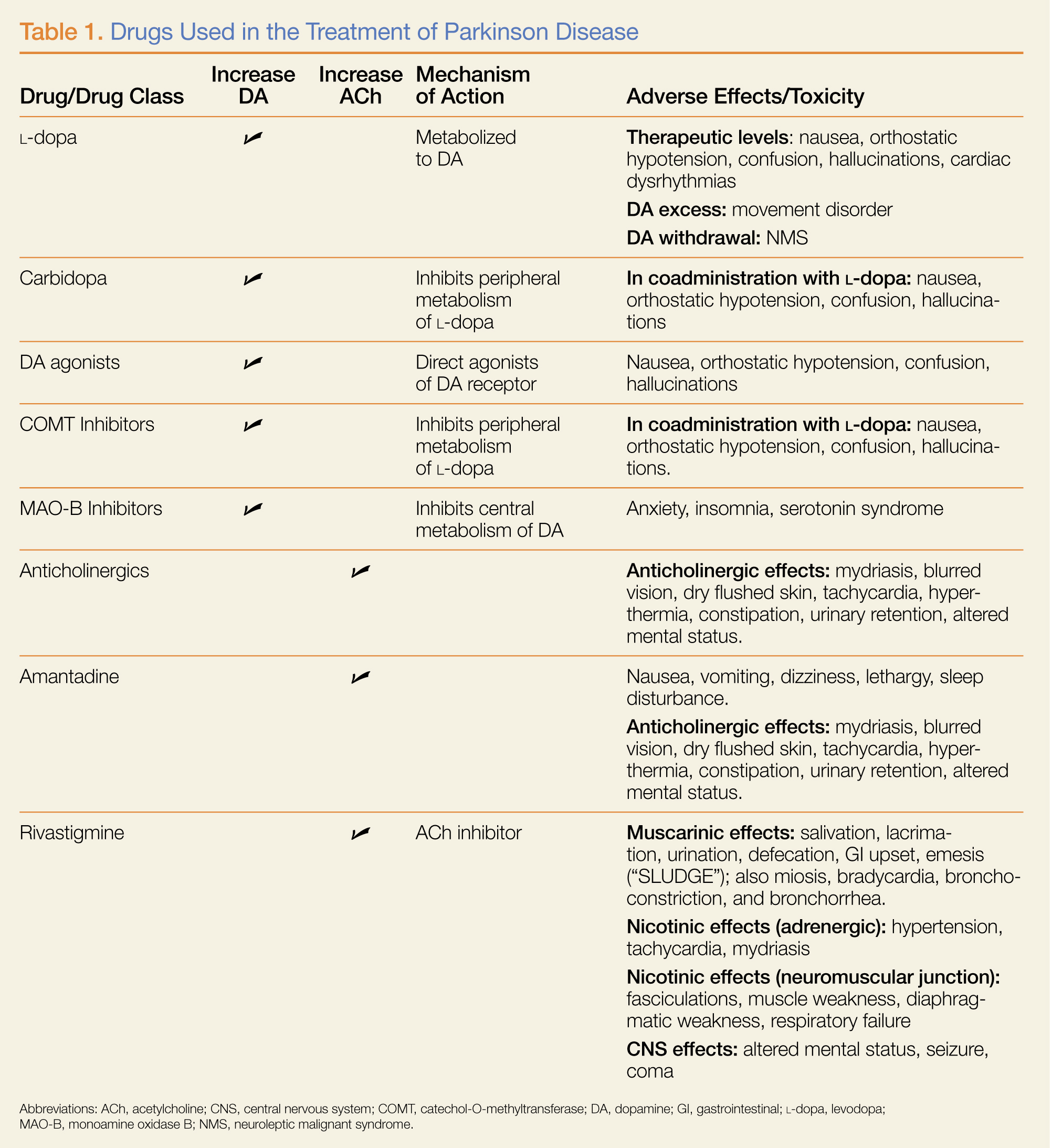
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.

Case Report: Nasal Septal Abscess
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
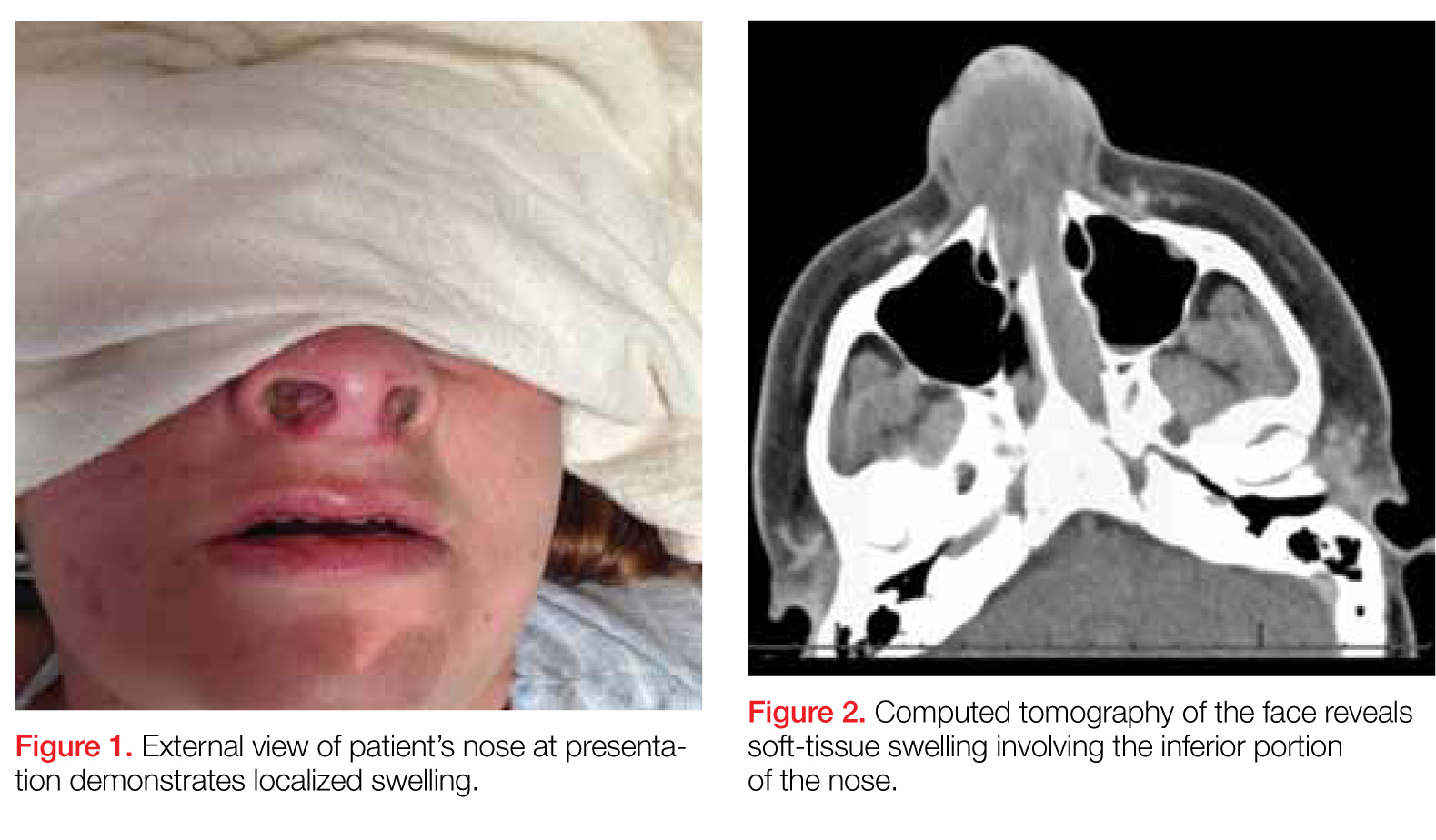
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
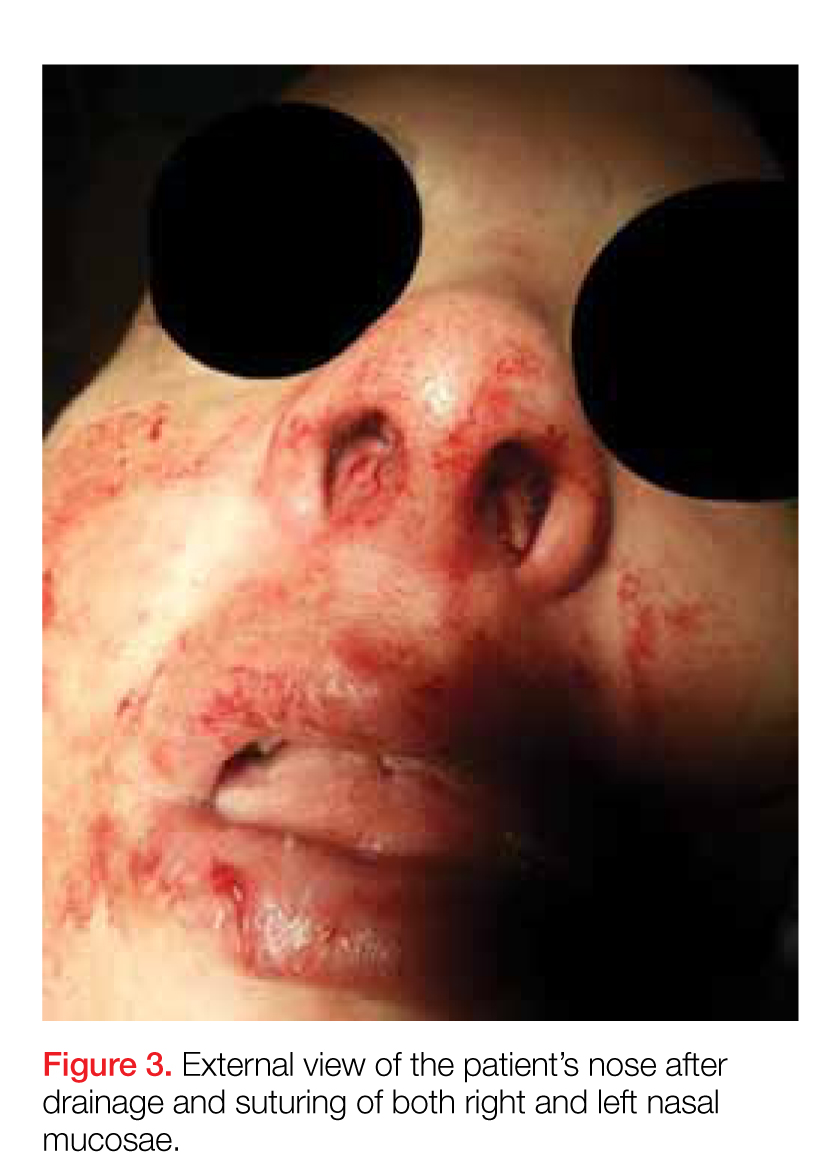
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.

Poland Syndrome: A Congenital Abnormality Mimicking a Traumatic Injury
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.

Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.
Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.
Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
Physicians are major source for frequent opioid misusers
Most people who misuse opioid pain relievers cite friends and relatives as their sources for the drugs, but more of the people who misuse these agents most often – those who take them from 200 to 365 days of the year – obtain their opioids from physicians’ prescriptions than from any other single source, according to a report published online March 3 in JAMA Internal Medicine.
"These results underscore the need for interventions targeting prescribing behaviors, in addition to those targeting medication sharing, selling, and diversion," the report’s authors warned.
It is a commonly cited statistic that most people who misuse opioid pain relievers obtain the drugs from family and friends for free, so many interventions to stop such misuse focus on patients. But few studies have examined whether the source of these drugs, and thus an appropriate target for interventions, might differ according to the frequency of misuse.
To study this issue, researchers analyzed data from the National Survey on Drug Use and Health, an annual survey that provides information on drug use among U.S. residents aged 12 years and older.
Survey data from 2008 through 2011 identified 11,018,735 respondents who said they misused an opioid pain reliever either by obtaining the drug without a prescription or by getting a prescription but taking the drug strictly because of the feeling or experience it provided. The source of the drug differed according to the frequency of use: As the days of use increased, the likelihood that the user obtained the drug from a friend or family member decreased, and the likelihood that he or she obtained the drug from a physician rose, said Christopher M. Jones, Pharm.D., and his associates at the National Center for Injury Prevention and Control, Centers for Disease Control and Prevention, Atlanta.
Among people who misused opioids only 1-29 days of the year, 54.4% said they got them from a friend or relative for free – the most popular source. In contrast, only 18% of this patient group said the opioids were prescribed by one or more physicians.
But the percentage of patients who obtained misused opioids through physician prescriptions steadily rose with increasing use.
Among those who misused the drugs 200-365 days a year, the top source was physician prescriptions, 27.3% of users, followed by opioids obtained for free from friends or relatives, 26.4%. A total of 23.2% of users said they bought their opioids from friends or relatives, while another 15% of frequent users bought their drugs from dealers or strangers.
"This pattern is similar to that of patients in opioid treatment programs, who cite dealers and physicians as frequent sources," Dr. Jones and his associates said in a Research Letter to the Editor (JAMA Intern. Med. 2014 March 3 [doi:10.1001/jamainternmed.2013.12809]).
"Many abusers of opioid pain relievers are going directly to doctors for their drugs," CDC Director Tom Frieden commented in a statement. "Health care providers need to screen for abuse risk and prescribe judiciously by checking past records in state prescription drug monitoring programs. It’s time we stop the source and treat the troubled."
"The essential steps health care providers can take to curb this serious health problem include more judicious prescribing, use of prescription-drug–monitoring programs, and screening patients for abuse before prescribing opioids," the study authors noted.
The federal government is encouraging the development of abuse-deterrent opioid formulations, the CDC noted, and requiring companies that make extended-release and long-acting opioids to offer prescribers educational programs about the understanding the risks of opioid therapy; choosing, managing, and monitoring patients; and counseling patients on safe use of opioids.
The Centers for Disease Control and Prevention supported the study. No financial conflicts of interest were reported.
Most people who misuse opioid pain relievers cite friends and relatives as their sources for the drugs, but more of the people who misuse these agents most often – those who take them from 200 to 365 days of the year – obtain their opioids from physicians’ prescriptions than from any other single source, according to a report published online March 3 in JAMA Internal Medicine.
"These results underscore the need for interventions targeting prescribing behaviors, in addition to those targeting medication sharing, selling, and diversion," the report’s authors warned.
It is a commonly cited statistic that most people who misuse opioid pain relievers obtain the drugs from family and friends for free, so many interventions to stop such misuse focus on patients. But few studies have examined whether the source of these drugs, and thus an appropriate target for interventions, might differ according to the frequency of misuse.
To study this issue, researchers analyzed data from the National Survey on Drug Use and Health, an annual survey that provides information on drug use among U.S. residents aged 12 years and older.
Survey data from 2008 through 2011 identified 11,018,735 respondents who said they misused an opioid pain reliever either by obtaining the drug without a prescription or by getting a prescription but taking the drug strictly because of the feeling or experience it provided. The source of the drug differed according to the frequency of use: As the days of use increased, the likelihood that the user obtained the drug from a friend or family member decreased, and the likelihood that he or she obtained the drug from a physician rose, said Christopher M. Jones, Pharm.D., and his associates at the National Center for Injury Prevention and Control, Centers for Disease Control and Prevention, Atlanta.
Among people who misused opioids only 1-29 days of the year, 54.4% said they got them from a friend or relative for free – the most popular source. In contrast, only 18% of this patient group said the opioids were prescribed by one or more physicians.
But the percentage of patients who obtained misused opioids through physician prescriptions steadily rose with increasing use.
Among those who misused the drugs 200-365 days a year, the top source was physician prescriptions, 27.3% of users, followed by opioids obtained for free from friends or relatives, 26.4%. A total of 23.2% of users said they bought their opioids from friends or relatives, while another 15% of frequent users bought their drugs from dealers or strangers.
"This pattern is similar to that of patients in opioid treatment programs, who cite dealers and physicians as frequent sources," Dr. Jones and his associates said in a Research Letter to the Editor (JAMA Intern. Med. 2014 March 3 [doi:10.1001/jamainternmed.2013.12809]).
"Many abusers of opioid pain relievers are going directly to doctors for their drugs," CDC Director Tom Frieden commented in a statement. "Health care providers need to screen for abuse risk and prescribe judiciously by checking past records in state prescription drug monitoring programs. It’s time we stop the source and treat the troubled."
"The essential steps health care providers can take to curb this serious health problem include more judicious prescribing, use of prescription-drug–monitoring programs, and screening patients for abuse before prescribing opioids," the study authors noted.
The federal government is encouraging the development of abuse-deterrent opioid formulations, the CDC noted, and requiring companies that make extended-release and long-acting opioids to offer prescribers educational programs about the understanding the risks of opioid therapy; choosing, managing, and monitoring patients; and counseling patients on safe use of opioids.
The Centers for Disease Control and Prevention supported the study. No financial conflicts of interest were reported.
Most people who misuse opioid pain relievers cite friends and relatives as their sources for the drugs, but more of the people who misuse these agents most often – those who take them from 200 to 365 days of the year – obtain their opioids from physicians’ prescriptions than from any other single source, according to a report published online March 3 in JAMA Internal Medicine.
"These results underscore the need for interventions targeting prescribing behaviors, in addition to those targeting medication sharing, selling, and diversion," the report’s authors warned.
It is a commonly cited statistic that most people who misuse opioid pain relievers obtain the drugs from family and friends for free, so many interventions to stop such misuse focus on patients. But few studies have examined whether the source of these drugs, and thus an appropriate target for interventions, might differ according to the frequency of misuse.
To study this issue, researchers analyzed data from the National Survey on Drug Use and Health, an annual survey that provides information on drug use among U.S. residents aged 12 years and older.
Survey data from 2008 through 2011 identified 11,018,735 respondents who said they misused an opioid pain reliever either by obtaining the drug without a prescription or by getting a prescription but taking the drug strictly because of the feeling or experience it provided. The source of the drug differed according to the frequency of use: As the days of use increased, the likelihood that the user obtained the drug from a friend or family member decreased, and the likelihood that he or she obtained the drug from a physician rose, said Christopher M. Jones, Pharm.D., and his associates at the National Center for Injury Prevention and Control, Centers for Disease Control and Prevention, Atlanta.
Among people who misused opioids only 1-29 days of the year, 54.4% said they got them from a friend or relative for free – the most popular source. In contrast, only 18% of this patient group said the opioids were prescribed by one or more physicians.
But the percentage of patients who obtained misused opioids through physician prescriptions steadily rose with increasing use.
Among those who misused the drugs 200-365 days a year, the top source was physician prescriptions, 27.3% of users, followed by opioids obtained for free from friends or relatives, 26.4%. A total of 23.2% of users said they bought their opioids from friends or relatives, while another 15% of frequent users bought their drugs from dealers or strangers.
"This pattern is similar to that of patients in opioid treatment programs, who cite dealers and physicians as frequent sources," Dr. Jones and his associates said in a Research Letter to the Editor (JAMA Intern. Med. 2014 March 3 [doi:10.1001/jamainternmed.2013.12809]).
"Many abusers of opioid pain relievers are going directly to doctors for their drugs," CDC Director Tom Frieden commented in a statement. "Health care providers need to screen for abuse risk and prescribe judiciously by checking past records in state prescription drug monitoring programs. It’s time we stop the source and treat the troubled."
"The essential steps health care providers can take to curb this serious health problem include more judicious prescribing, use of prescription-drug–monitoring programs, and screening patients for abuse before prescribing opioids," the study authors noted.
The federal government is encouraging the development of abuse-deterrent opioid formulations, the CDC noted, and requiring companies that make extended-release and long-acting opioids to offer prescribers educational programs about the understanding the risks of opioid therapy; choosing, managing, and monitoring patients; and counseling patients on safe use of opioids.
The Centers for Disease Control and Prevention supported the study. No financial conflicts of interest were reported.
FROM JAMA INTERNAL MEDICINE
Major Finding: Opioid pain relievers were prescribed by a physician for 18% of people who used them only 1-29 days of the year, but that percentage steadily rose with increasing use, so that 27.3% of people who used the drugs 200-365 days/year obtained them via physician prescription, more than any other single source.
Data Source: An analysis of data on 11,018,735 survey respondents aged 12 years and older who reported misusing opioid pain relievers during a 4-year period.
Disclosures: The Centers for Disease Control and Prevention supported the study. No financial conflicts of interest were reported.

There’s No Place Like Home… for Carbon Monoxide Poisoning
Case
An 84-year-old woman with a history of hypertension and dyslipidemia and her husband, an 88-year-old man with a history of dementia and coronary artery disease, presented to the ED via EMS after neighbors discovered the woman lying on her living room floor, responding only to painful stimuli. Earlier in the evening, the same neighbors had helped the husband to bed after noticing that he had become lethargic. The EMS report indicated that a car had been left running in a closed garage of the patients’ home. The fire department identified an ambient carbon monoxide (CO) concentration of 88 ppm.
Upon arrival to the ED, the woman’s vital signs were: blood pressure (BP), 130/74 mm Hg; heart rate (HR), 63 beats/minute; respiratory rate (RR), 16 breaths/minute; temperature, 99°F. Oxygen saturation was 99% on room air. Her husband’s vital signs were: BP, 150/66 mm Hg; HR, 59 beats/minute; RR, 19 breaths/minute; temperature, 98°F; oxygen saturation was 98% on room air.
What is carbon monoxide poisoning?
Carbon monoxide is a colorless and odorless toxic gas produced by incomplete combustion of carbon-based fuel. Common sources in the United States include portable generators, gas-powered furnaces, cooking appliances, poorly ventilated home-heating systems, and motor vehicles (Box 1).1
Carbon monoxide is the leading cause of unintentional poisoning deaths in the United States,1 resulting in more than 20,000 ED visits and 2,000 hospital admissions. Nearly three-fourths of these deaths are due to exposures in the home, with more than half occurring during the months of November through February.2,3 The average cost of a hospital admission for confirmed CO poisoning is over $11,000, with a cumulative nationwide total cost of over $26 million per year. While the hospitalization rate for persons aged 18 to 44 years is only 6.7%, the admittance rates for persons aged 65 to 84 years and older than 85 years are 33% and 43%, respectively.3 Although there has been a slight decline in the incidence of CO poisoning over the past 10 years, it is still a public health concern (Figure 1).2
Who is most susceptible to motor vehicle-related carbon monoxide poisoning?
The US Centers for Disease Control and Prevention (CDC) reports that motor vehicles are the second most common source of CO exposure.4 A study of US news media reports covering a 2.5-year period revealed that 8% of such poisonings were the result of a motor vehicle left running in a garage—the overall mortality rate of which is suggested to be significantly higher than that of other sources of CO exposure.5
Approximately 430 deaths per year are caused by unintentional, nonfire-related CO poisoning,6 and the CDC reports the death rate is highest in persons older than age 65 years.1 The death rate from these exposures is more than three times higher in men than women (Figure 2).6 In addition, older patients are disproportionately affected: In US news media-reported cases of CO poisoning that included patient age, 29% occurred in persons older than age 80 years.5 Moreover, in approximately one-third of motor vehicle-related deaths due to CO poisoning, nearly all of patients older than age 80 years were found dead at the scene of exposure. These reports suggest that the elderly are at greater risk for CO exposure due to age-related cognitive changes, physical inability to escape a toxic environment once becoming symptomatic, and a greater susceptibility to poisoning due to comorbid conditions.5
Case Continued
The husband and wife’s initial carboxyhemoglobin concentrations in this case were 35% and 13%, respectively. Both were treated with hyperbaric oxygen (HBO) without complication. During their inpatient stay, the woman noted that their home did not have a CO detector.
What is the role of hyperbaric oxygen therapy as a treatment option for CO poisoning?
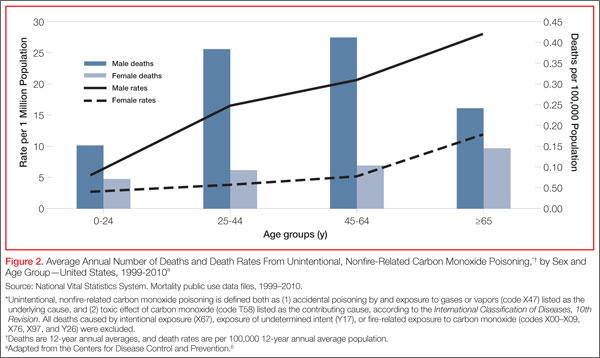
Hyperbaric oxygen therapy greatly accelerates the dissociation of hemoglobin from CO, reduces free radical-related cellular damage, and may have a role in preventing adverse neurological sequelae in the setting of CO poisoning. Although controversy exists, HBO therapy is generally indicated in select patients with elevated CO levels and abnormal neurological findings, cardiovascular findings, or persistent metabolic acidosis. While few ED patients with CO exposure receive HBO therapy, over 20% of patients requiring inpatient hospitalization receive treatment.3
What preventive measures can be taken to reduce motor vehicle-related CO poisoning?
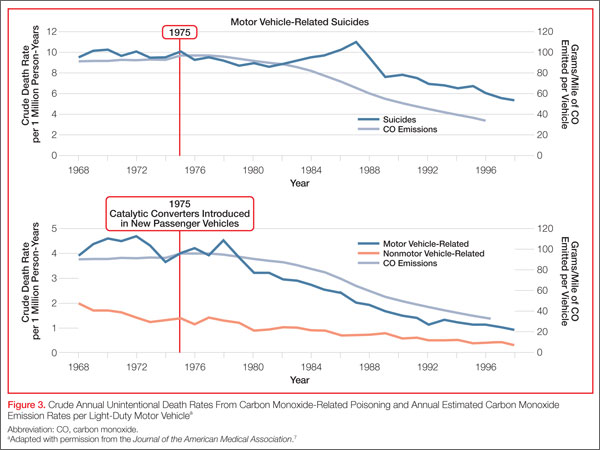
The literature supports the enforcement of motor vehicle emissions standards and the proper use of home CO detectors as primary preventive strategies. Computerized data from the CDC, US Census Bureau, and US Environmental Protection Agency from 1968 to 1998 were used to evaluate the influence of national vehicle emissions policies on CO-related mortality. The Clean Air Act of 1970 set environmental limits on CO emissions from automobiles at 15.0 g/mile in 1975; the EPA further reduced this standard to 3.4 g/mile for automobiles manufactured after 1981. After the enforcement of standards set forth by the Clean Air Act and the introduction of the catalytic converter in 1975, CO emissions from automobiles decreased by an estimated 76.3%, and unintentional motor vehicle-related CO deaths declined by 81.3% (Figure 3).7 (Catalytic converters contain elements [eg, platinum] that catalyze the oxidation of CO to carbon dioxide.)
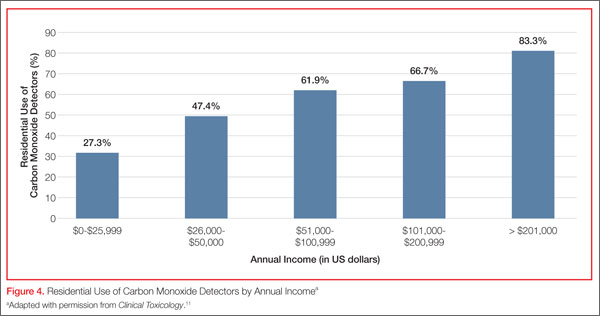
Since CO exposure occurs primarily in the home, the installation of battery-powered or battery-backed CO alarms—both in the home and garage—can prevent poisoning. These detectors are inexpensive and available at common retail stores. Unfortunately, despite the easy availability and access to CO detectors, only 39 states currently have legislation mandating their use, and approximately two-thirds of the states with existing legislation only require CO detectors in newly built structures.8
In 2010, the state of New York enacted legislation known as “Amanda’s Law,” (named after a teenaged girl whose death was caused by CO poisoning from a defective boiler) mandating CO detectors in all one- and two-family homes with heating sources that may emit CO or have attached garages. However, an industry survey in 2011 found that nearly half of New York families were not aware of this law.9 The two largest surveys on home CO detector use—those conducted by the US Census Bureau and CDC—estimate the national rate of having a working CO detector in a home is 32% to 40%, with a lower prevalence among those living in manufactured housing, renting a home, or living below the poverty level.10
What is the utilization of CO detectors by ED patients?


Case conclusion
After hospital admission and treatment, both patients were discharged on hospital day 2 with a return to a baseline mental status. Neither patient reported neurological sequelae or new cognitive changes when a follow up call was placed more than 6 months after HBO treatment. The couple furthermore reported that they installed a CO detector upon their return home.
Dr West is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr McGregor is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr Touger is an associate professor of clinical emergency medicine, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York. He is also medical director of the Jacobi Medical Center hyperbaric chamber.
Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Centers for Disease Control and Prevention. Carbon monoxide-related deaths—United States, 1999-2004. MMWR Morb Mortal Wkly Rep. 2007;56(50):1309-1312.
- Centers for Disease Control and Prevention. Carbon mononoxide exposures—United States, 2000-2009. MMWR Morb Mortal Wkly Rep. 2011;60(30):1014-1017.
- Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664.
- Centers for Disease Control and Prevention. Nonfatal, unintentional, non-fire-related carbon monoxide exposures—United States, 2004-2006. MMWR Morb Mortal Wkly Rep. 2008;57(33):896-899.
- Hampson NB. Residential carbon monoxide poisoning from motor vehicles. Am J Emerg Med. 2011;29(1):75-77.
- Centers for Disease Control and Prevention. Average annual number of deaths and death rates from unintentional, non-fire-related carbon monoxide poisoning, by sex and age group—United States, 1999–2010. MMWR Morb Mortal Wkly Rep. 2014;63(3):65.
- Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
- Carbon monoxide detectors: state statutes. National Conference of State Legislatures Web site. http://www.ncsl.org/research/environment-and-natural-resources/carbon-monoxide-detectors-state-statutes.aspx. Accessed March 11, 2014.
- Survey results: New York homeowners and the risk of carbon monoxide poisoning. Kidde Web site. http://www.kidde.com/PressRoom/Pages/SurveyResultsNYHomeownersCORisks.aspx. Accessed March 11, 2014.
- Iqbal S, Clower JH, King M, Bell J, Yip YF. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012;127(5):486-496.
- Johnson-Arbor K, Liebman DL, Carter EM. A survey of residential carbon monoxide detector utilization among Connecticut Emergency Department patients. Clin Toxicol (Phila). 2012;50(5):384-389.
Case
An 84-year-old woman with a history of hypertension and dyslipidemia and her husband, an 88-year-old man with a history of dementia and coronary artery disease, presented to the ED via EMS after neighbors discovered the woman lying on her living room floor, responding only to painful stimuli. Earlier in the evening, the same neighbors had helped the husband to bed after noticing that he had become lethargic. The EMS report indicated that a car had been left running in a closed garage of the patients’ home. The fire department identified an ambient carbon monoxide (CO) concentration of 88 ppm.
Upon arrival to the ED, the woman’s vital signs were: blood pressure (BP), 130/74 mm Hg; heart rate (HR), 63 beats/minute; respiratory rate (RR), 16 breaths/minute; temperature, 99°F. Oxygen saturation was 99% on room air. Her husband’s vital signs were: BP, 150/66 mm Hg; HR, 59 beats/minute; RR, 19 breaths/minute; temperature, 98°F; oxygen saturation was 98% on room air.
What is carbon monoxide poisoning?
Carbon monoxide is a colorless and odorless toxic gas produced by incomplete combustion of carbon-based fuel. Common sources in the United States include portable generators, gas-powered furnaces, cooking appliances, poorly ventilated home-heating systems, and motor vehicles (Box 1).1
Carbon monoxide is the leading cause of unintentional poisoning deaths in the United States,1 resulting in more than 20,000 ED visits and 2,000 hospital admissions. Nearly three-fourths of these deaths are due to exposures in the home, with more than half occurring during the months of November through February.2,3 The average cost of a hospital admission for confirmed CO poisoning is over $11,000, with a cumulative nationwide total cost of over $26 million per year. While the hospitalization rate for persons aged 18 to 44 years is only 6.7%, the admittance rates for persons aged 65 to 84 years and older than 85 years are 33% and 43%, respectively.3 Although there has been a slight decline in the incidence of CO poisoning over the past 10 years, it is still a public health concern (Figure 1).2
Who is most susceptible to motor vehicle-related carbon monoxide poisoning?
The US Centers for Disease Control and Prevention (CDC) reports that motor vehicles are the second most common source of CO exposure.4 A study of US news media reports covering a 2.5-year period revealed that 8% of such poisonings were the result of a motor vehicle left running in a garage—the overall mortality rate of which is suggested to be significantly higher than that of other sources of CO exposure.5
Approximately 430 deaths per year are caused by unintentional, nonfire-related CO poisoning,6 and the CDC reports the death rate is highest in persons older than age 65 years.1 The death rate from these exposures is more than three times higher in men than women (Figure 2).6 In addition, older patients are disproportionately affected: In US news media-reported cases of CO poisoning that included patient age, 29% occurred in persons older than age 80 years.5 Moreover, in approximately one-third of motor vehicle-related deaths due to CO poisoning, nearly all of patients older than age 80 years were found dead at the scene of exposure. These reports suggest that the elderly are at greater risk for CO exposure due to age-related cognitive changes, physical inability to escape a toxic environment once becoming symptomatic, and a greater susceptibility to poisoning due to comorbid conditions.5
Case Continued
The husband and wife’s initial carboxyhemoglobin concentrations in this case were 35% and 13%, respectively. Both were treated with hyperbaric oxygen (HBO) without complication. During their inpatient stay, the woman noted that their home did not have a CO detector.
What is the role of hyperbaric oxygen therapy as a treatment option for CO poisoning?
Hyperbaric oxygen therapy greatly accelerates the dissociation of hemoglobin from CO, reduces free radical-related cellular damage, and may have a role in preventing adverse neurological sequelae in the setting of CO poisoning. Although controversy exists, HBO therapy is generally indicated in select patients with elevated CO levels and abnormal neurological findings, cardiovascular findings, or persistent metabolic acidosis. While few ED patients with CO exposure receive HBO therapy, over 20% of patients requiring inpatient hospitalization receive treatment.3
What preventive measures can be taken to reduce motor vehicle-related CO poisoning?
The literature supports the enforcement of motor vehicle emissions standards and the proper use of home CO detectors as primary preventive strategies. Computerized data from the CDC, US Census Bureau, and US Environmental Protection Agency from 1968 to 1998 were used to evaluate the influence of national vehicle emissions policies on CO-related mortality. The Clean Air Act of 1970 set environmental limits on CO emissions from automobiles at 15.0 g/mile in 1975; the EPA further reduced this standard to 3.4 g/mile for automobiles manufactured after 1981. After the enforcement of standards set forth by the Clean Air Act and the introduction of the catalytic converter in 1975, CO emissions from automobiles decreased by an estimated 76.3%, and unintentional motor vehicle-related CO deaths declined by 81.3% (Figure 3).7 (Catalytic converters contain elements [eg, platinum] that catalyze the oxidation of CO to carbon dioxide.)
Since CO exposure occurs primarily in the home, the installation of battery-powered or battery-backed CO alarms—both in the home and garage—can prevent poisoning. These detectors are inexpensive and available at common retail stores. Unfortunately, despite the easy availability and access to CO detectors, only 39 states currently have legislation mandating their use, and approximately two-thirds of the states with existing legislation only require CO detectors in newly built structures.8
In 2010, the state of New York enacted legislation known as “Amanda’s Law,” (named after a teenaged girl whose death was caused by CO poisoning from a defective boiler) mandating CO detectors in all one- and two-family homes with heating sources that may emit CO or have attached garages. However, an industry survey in 2011 found that nearly half of New York families were not aware of this law.9 The two largest surveys on home CO detector use—those conducted by the US Census Bureau and CDC—estimate the national rate of having a working CO detector in a home is 32% to 40%, with a lower prevalence among those living in manufactured housing, renting a home, or living below the poverty level.10
What is the utilization of CO detectors by ED patients?
Case conclusion
After hospital admission and treatment, both patients were discharged on hospital day 2 with a return to a baseline mental status. Neither patient reported neurological sequelae or new cognitive changes when a follow up call was placed more than 6 months after HBO treatment. The couple furthermore reported that they installed a CO detector upon their return home.
Dr West is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr McGregor is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr Touger is an associate professor of clinical emergency medicine, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York. He is also medical director of the Jacobi Medical Center hyperbaric chamber.
Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
An 84-year-old woman with a history of hypertension and dyslipidemia and her husband, an 88-year-old man with a history of dementia and coronary artery disease, presented to the ED via EMS after neighbors discovered the woman lying on her living room floor, responding only to painful stimuli. Earlier in the evening, the same neighbors had helped the husband to bed after noticing that he had become lethargic. The EMS report indicated that a car had been left running in a closed garage of the patients’ home. The fire department identified an ambient carbon monoxide (CO) concentration of 88 ppm.
Upon arrival to the ED, the woman’s vital signs were: blood pressure (BP), 130/74 mm Hg; heart rate (HR), 63 beats/minute; respiratory rate (RR), 16 breaths/minute; temperature, 99°F. Oxygen saturation was 99% on room air. Her husband’s vital signs were: BP, 150/66 mm Hg; HR, 59 beats/minute; RR, 19 breaths/minute; temperature, 98°F; oxygen saturation was 98% on room air.
What is carbon monoxide poisoning?
Carbon monoxide is a colorless and odorless toxic gas produced by incomplete combustion of carbon-based fuel. Common sources in the United States include portable generators, gas-powered furnaces, cooking appliances, poorly ventilated home-heating systems, and motor vehicles (Box 1).1
Carbon monoxide is the leading cause of unintentional poisoning deaths in the United States,1 resulting in more than 20,000 ED visits and 2,000 hospital admissions. Nearly three-fourths of these deaths are due to exposures in the home, with more than half occurring during the months of November through February.2,3 The average cost of a hospital admission for confirmed CO poisoning is over $11,000, with a cumulative nationwide total cost of over $26 million per year. While the hospitalization rate for persons aged 18 to 44 years is only 6.7%, the admittance rates for persons aged 65 to 84 years and older than 85 years are 33% and 43%, respectively.3 Although there has been a slight decline in the incidence of CO poisoning over the past 10 years, it is still a public health concern (Figure 1).2
Who is most susceptible to motor vehicle-related carbon monoxide poisoning?
The US Centers for Disease Control and Prevention (CDC) reports that motor vehicles are the second most common source of CO exposure.4 A study of US news media reports covering a 2.5-year period revealed that 8% of such poisonings were the result of a motor vehicle left running in a garage—the overall mortality rate of which is suggested to be significantly higher than that of other sources of CO exposure.5
Approximately 430 deaths per year are caused by unintentional, nonfire-related CO poisoning,6 and the CDC reports the death rate is highest in persons older than age 65 years.1 The death rate from these exposures is more than three times higher in men than women (Figure 2).6 In addition, older patients are disproportionately affected: In US news media-reported cases of CO poisoning that included patient age, 29% occurred in persons older than age 80 years.5 Moreover, in approximately one-third of motor vehicle-related deaths due to CO poisoning, nearly all of patients older than age 80 years were found dead at the scene of exposure. These reports suggest that the elderly are at greater risk for CO exposure due to age-related cognitive changes, physical inability to escape a toxic environment once becoming symptomatic, and a greater susceptibility to poisoning due to comorbid conditions.5
Case Continued
The husband and wife’s initial carboxyhemoglobin concentrations in this case were 35% and 13%, respectively. Both were treated with hyperbaric oxygen (HBO) without complication. During their inpatient stay, the woman noted that their home did not have a CO detector.
What is the role of hyperbaric oxygen therapy as a treatment option for CO poisoning?
Hyperbaric oxygen therapy greatly accelerates the dissociation of hemoglobin from CO, reduces free radical-related cellular damage, and may have a role in preventing adverse neurological sequelae in the setting of CO poisoning. Although controversy exists, HBO therapy is generally indicated in select patients with elevated CO levels and abnormal neurological findings, cardiovascular findings, or persistent metabolic acidosis. While few ED patients with CO exposure receive HBO therapy, over 20% of patients requiring inpatient hospitalization receive treatment.3
What preventive measures can be taken to reduce motor vehicle-related CO poisoning?
The literature supports the enforcement of motor vehicle emissions standards and the proper use of home CO detectors as primary preventive strategies. Computerized data from the CDC, US Census Bureau, and US Environmental Protection Agency from 1968 to 1998 were used to evaluate the influence of national vehicle emissions policies on CO-related mortality. The Clean Air Act of 1970 set environmental limits on CO emissions from automobiles at 15.0 g/mile in 1975; the EPA further reduced this standard to 3.4 g/mile for automobiles manufactured after 1981. After the enforcement of standards set forth by the Clean Air Act and the introduction of the catalytic converter in 1975, CO emissions from automobiles decreased by an estimated 76.3%, and unintentional motor vehicle-related CO deaths declined by 81.3% (Figure 3).7 (Catalytic converters contain elements [eg, platinum] that catalyze the oxidation of CO to carbon dioxide.)
Since CO exposure occurs primarily in the home, the installation of battery-powered or battery-backed CO alarms—both in the home and garage—can prevent poisoning. These detectors are inexpensive and available at common retail stores. Unfortunately, despite the easy availability and access to CO detectors, only 39 states currently have legislation mandating their use, and approximately two-thirds of the states with existing legislation only require CO detectors in newly built structures.8
In 2010, the state of New York enacted legislation known as “Amanda’s Law,” (named after a teenaged girl whose death was caused by CO poisoning from a defective boiler) mandating CO detectors in all one- and two-family homes with heating sources that may emit CO or have attached garages. However, an industry survey in 2011 found that nearly half of New York families were not aware of this law.9 The two largest surveys on home CO detector use—those conducted by the US Census Bureau and CDC—estimate the national rate of having a working CO detector in a home is 32% to 40%, with a lower prevalence among those living in manufactured housing, renting a home, or living below the poverty level.10
What is the utilization of CO detectors by ED patients?
Case conclusion
After hospital admission and treatment, both patients were discharged on hospital day 2 with a return to a baseline mental status. Neither patient reported neurological sequelae or new cognitive changes when a follow up call was placed more than 6 months after HBO treatment. The couple furthermore reported that they installed a CO detector upon their return home.
Dr West is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr McGregor is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr Touger is an associate professor of clinical emergency medicine, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York. He is also medical director of the Jacobi Medical Center hyperbaric chamber.
Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Centers for Disease Control and Prevention. Carbon monoxide-related deaths—United States, 1999-2004. MMWR Morb Mortal Wkly Rep. 2007;56(50):1309-1312.
- Centers for Disease Control and Prevention. Carbon mononoxide exposures—United States, 2000-2009. MMWR Morb Mortal Wkly Rep. 2011;60(30):1014-1017.
- Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664.
- Centers for Disease Control and Prevention. Nonfatal, unintentional, non-fire-related carbon monoxide exposures—United States, 2004-2006. MMWR Morb Mortal Wkly Rep. 2008;57(33):896-899.
- Hampson NB. Residential carbon monoxide poisoning from motor vehicles. Am J Emerg Med. 2011;29(1):75-77.
- Centers for Disease Control and Prevention. Average annual number of deaths and death rates from unintentional, non-fire-related carbon monoxide poisoning, by sex and age group—United States, 1999–2010. MMWR Morb Mortal Wkly Rep. 2014;63(3):65.
- Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
- Carbon monoxide detectors: state statutes. National Conference of State Legislatures Web site. http://www.ncsl.org/research/environment-and-natural-resources/carbon-monoxide-detectors-state-statutes.aspx. Accessed March 11, 2014.
- Survey results: New York homeowners and the risk of carbon monoxide poisoning. Kidde Web site. http://www.kidde.com/PressRoom/Pages/SurveyResultsNYHomeownersCORisks.aspx. Accessed March 11, 2014.
- Iqbal S, Clower JH, King M, Bell J, Yip YF. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012;127(5):486-496.
- Johnson-Arbor K, Liebman DL, Carter EM. A survey of residential carbon monoxide detector utilization among Connecticut Emergency Department patients. Clin Toxicol (Phila). 2012;50(5):384-389.
- Centers for Disease Control and Prevention. Carbon monoxide-related deaths—United States, 1999-2004. MMWR Morb Mortal Wkly Rep. 2007;56(50):1309-1312.
- Centers for Disease Control and Prevention. Carbon mononoxide exposures—United States, 2000-2009. MMWR Morb Mortal Wkly Rep. 2011;60(30):1014-1017.
- Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664.
- Centers for Disease Control and Prevention. Nonfatal, unintentional, non-fire-related carbon monoxide exposures—United States, 2004-2006. MMWR Morb Mortal Wkly Rep. 2008;57(33):896-899.
- Hampson NB. Residential carbon monoxide poisoning from motor vehicles. Am J Emerg Med. 2011;29(1):75-77.
- Centers for Disease Control and Prevention. Average annual number of deaths and death rates from unintentional, non-fire-related carbon monoxide poisoning, by sex and age group—United States, 1999–2010. MMWR Morb Mortal Wkly Rep. 2014;63(3):65.
- Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
- Carbon monoxide detectors: state statutes. National Conference of State Legislatures Web site. http://www.ncsl.org/research/environment-and-natural-resources/carbon-monoxide-detectors-state-statutes.aspx. Accessed March 11, 2014.
- Survey results: New York homeowners and the risk of carbon monoxide poisoning. Kidde Web site. http://www.kidde.com/PressRoom/Pages/SurveyResultsNYHomeownersCORisks.aspx. Accessed March 11, 2014.
- Iqbal S, Clower JH, King M, Bell J, Yip YF. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012;127(5):486-496.
- Johnson-Arbor K, Liebman DL, Carter EM. A survey of residential carbon monoxide detector utilization among Connecticut Emergency Department patients. Clin Toxicol (Phila). 2012;50(5):384-389.

Pancytopenia warning added to label of hepatitis C drug boceprevir
A warning about cases of pancytopenia linked to the use of the oral hepatitis C antiviral drug boceprevir has been added to the drug’s label, the Food and Drug Administration has announced.
There have been postmarketing reports of "serious cases" of pancytopenia in people treated with boceprevir in combination with peginterferon alfa and ribavirin, according to the FDA. This information has been added to the "Warnings and Precautions" section of the label, with the recommendation that "complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at treatment weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate."
Approved in 2011, boceprevir is an HCV Ns3/4A protease inhibitor approved for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adults with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders. Boceprevir, in a 200-mg capsule formulation, is marketed as Victrelis by Merck Sharp & Dohme; the recommended dose is 800 mg, three times a day.
The boceprevir Medication Guide will also be updated with this information, according to the FDA.
Serious adverse events associated with boceprevir should be reported to the FDA at 800-332-1088 or at MedWatch.
A warning about cases of pancytopenia linked to the use of the oral hepatitis C antiviral drug boceprevir has been added to the drug’s label, the Food and Drug Administration has announced.
There have been postmarketing reports of "serious cases" of pancytopenia in people treated with boceprevir in combination with peginterferon alfa and ribavirin, according to the FDA. This information has been added to the "Warnings and Precautions" section of the label, with the recommendation that "complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at treatment weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate."
Approved in 2011, boceprevir is an HCV Ns3/4A protease inhibitor approved for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adults with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders. Boceprevir, in a 200-mg capsule formulation, is marketed as Victrelis by Merck Sharp & Dohme; the recommended dose is 800 mg, three times a day.
The boceprevir Medication Guide will also be updated with this information, according to the FDA.
Serious adverse events associated with boceprevir should be reported to the FDA at 800-332-1088 or at MedWatch.
A warning about cases of pancytopenia linked to the use of the oral hepatitis C antiviral drug boceprevir has been added to the drug’s label, the Food and Drug Administration has announced.
There have been postmarketing reports of "serious cases" of pancytopenia in people treated with boceprevir in combination with peginterferon alfa and ribavirin, according to the FDA. This information has been added to the "Warnings and Precautions" section of the label, with the recommendation that "complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at treatment weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate."
Approved in 2011, boceprevir is an HCV Ns3/4A protease inhibitor approved for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adults with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders. Boceprevir, in a 200-mg capsule formulation, is marketed as Victrelis by Merck Sharp & Dohme; the recommended dose is 800 mg, three times a day.
The boceprevir Medication Guide will also be updated with this information, according to the FDA.
Serious adverse events associated with boceprevir should be reported to the FDA at 800-332-1088 or at MedWatch.
Case Studies in Toxicology: The Acclaimed Zombie-Apocalypse Drug—Is it Just an Illusion?
Dr Takematsu is a senior fellow of medical toxicology, department of emergency medicine, New York University School of Medicine and New York City Poison Control Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
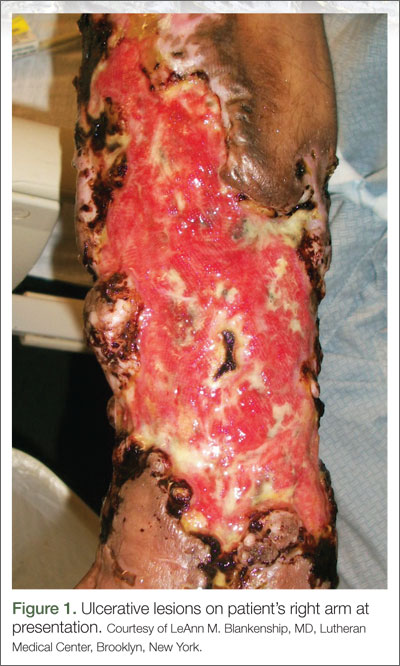
Case
A 50-year-old man with a 16-year history of injection heroin abuse presented to the ED complaining of ulcerative lesions on his right arm, which he stated had become worse over the past 3 months. He claimed the skin lesions, which involved his entire right arm, had grown “wider and deeper” since he started using the drug “Krokodil.” He further noted that he obtained the product from four different drug suppliers but did not know how it was prepared.
On presentation, his vital signs were: blood pressure, 135/78 mm Hg; heart rate, 92 beats/minute; respiratory rate, 14 breaths/minute; temperature, 98.2˚ F. Oxygen saturation was 100% on room air. Physical examination of the right arm was notable for broad ulcers that exposed fat tissue and muscle and involved the entire forearm circumferentially (Figure 1). There were no signs of acute infection such as erythema, warmth, or abscess formation. The remainder of the physical examination was unremarkable.
What is Krokodil?
The name Krokodil is derived from the Russian word for crocodile, and stems from the greenish, severely damaged “crocodile-like” skin lesions purportedly caused by subcutaneous injection of this opioid derivative. Krokodil is not a specific drug but rather it describes the product derived from an attempt to synthesize desomorphine, the core ingredient. Desomorphine is a short-acting opioid analogue that has a reported potency eight to 10 times greater than morphine (Figure 2).
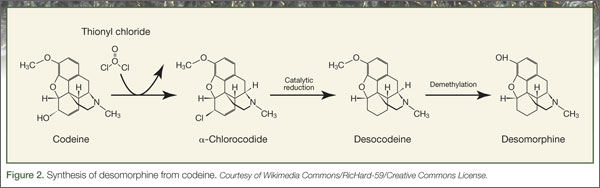
Also called “Russian Magic,” Krokodil has been used in Russia since 2003 following the country’s major restrictions on the importation of heroin. Since desomorphine can be synthesized from codeine, which was available in over-the-counter medications in Russia until 2012, drug users have turned to it as an inexpensive heroin substitute. (The average cost of a codeine-containing product is about 120 Rubles [$4.00] per 10-pack.1)
The chemical process of synthesizing desomorphine typically involves mixing one to five packs of codeine-based analgesics1 with a solvent (eg, paint thinner, gasoline), along with iodine and red phosphorous, which can be obtained from the striking pads of matchboxes.2 This produces a yield equivalent to 500 Rubles of heroin, making it an attractive alternative to low-income drug users.1 However, the yields are poor, and the starting products vary based on the codeine source. No systemic analysis of Krokodil has been performed to assess the purity and concentration of desomorphine in the resulting product.
What are the risks of using Krokodil?
Krokodil is typically self-administered by either intravenous (IV) or subcutaneous (“skin-popping”) injection. Solvents and contaminants in the product can damage tissue with which they come into contact. Thus, IV use of this product causes venous scarring and collapse3; when venous access is exhausted, or following infiltration, the subcutaneous route may lead to necrosis and ulceration of the skin. These lesions are said to be used by illicit drug users as a “shooter’s patch” to inject drugs,4 leading to further skin damage. In addition to the local dermal effects, systemic inflammation results from the dissemination of the components of the product, causing neurological, solid-organ, and other effects.
Has Krokodil reached the United States?
Because of the horrifying appearance of the skin lesions, Krokodil has been called by many descriptive names such as the “flesh-eating drug” and “zombie-apocalypse drug,” which in turn has led to somewhat sensationalistic media reporting. There is, however, no clear evidence of entry of desomorphine or of Krokodil use in the United States, and most authorities feel either is unlikely to occur. This speculation is supported by the low cost and easy availability of heroin in the United States compared to Russia. Of note, although Russia banned the nonprescription sale of products containing codeine in November 2012, the ban has had minimal impact on the rate of necrotic skin lesions.
To date, none of the so-called cases of Krokodil-associated lesions reported in the United States have confirmatory analytical testing, and diagnoses have been based primarily on history or morphological features of the wound. Furthermore, no reference laboratories in the United States have identified desomorphine in any of their tested samples. Since the skin lesions are not pathognomonic of Krokodil use and share morphologic features with all forms of subcutaneous drug use, analytical confirmation is critical to identifying this as an emerging drug trend. Therefore, users of product sold or brewed as Krokodil may develop skin lesions regardless of desomorphine content due to contaminants in the injected product.
What causes the skin lesions?
Desomorphine itself is not likely the cause of the skin lesions. In its pure form, there is no reason to expect this compound would induce any specific changes in the skin that would lead to necrosis. In fact, similar chemical syntheses of desomorphine produced in other countries, such as in the Czech Republic and New Zealand, have not led to analogous skin lesions. Ulcerative skin damage is most likely caused by the inflammatory contaminants in Krokodil. Injection of solvents and other chemicals into the subcutaneous space lead to skin damage and provide a bed for the development of indolent infections. This mechanism is similar to that leading to epidemics, in which similar lesions have been associated with black-tar heroin injection and drug contamination with Bacillus anthracis.5,6
Some people, however, point out the severity of the skin lesions associated with the use of Krokodil compared to other injection drugs. This may be related to the need for frequent administration of Krokodil due to its short duration of action, in turn leading to repeated exposure to the impure solvents.
Treatment of associated lesions involves wound care, including debridement, topical care, and antibiotics; amputation may be required in severe cases.
Case conclusion
The patient in this case was taken to the operating room for debridement of the right arm. The pathology results of the tissue showed ulceration, abscess, acute and chronic inflamed granulation tissue, fibrosis, and necrosis. A magnetic resonance image of the arm showed no signs of osteomyelitis. A blood sample sent for analysis was negative for desomorphine. The patient was stable after the surgery, and was discharged with follow-up instructions and referral for drug abuse counseling.
- Grund JP, Latypov A, Harris M. Breaking worse: The emergence of krokodil and excessive injuries among people who inject drugs in Eurasia. Int J Drug Policy. 2013;24(4):265-274.
- Gahr M, Freudenmann RW, Hiemke C, Gunst IM, Connemann BJ, Schönfeldt-Lecuona C. Desomorphine goes “crocodile.” J Addict Dis. 2012;31(4):407-412.
- Pieper B, Kirsner RS, Templin TN, Birk TJ. Injection drug use: an understudied cause of venous disease. Arch Dermatol. 2007;143(10):1305-1309.
- Iyer S, Subramanian P, Pabari A. A devastating complication of “skin popping.” Surgeon. 2011;9(5):295-297.
- Dunbar NM, Harruff RC. Necrotizing fasciitis: manifestations, microbiology and connection with black tar heroin. J Forensic Sci. 2007;52(4):920-923.
- Grunow R, Klee SR, Beyer W, et al. Anthrax among heroin users in Europe possibly caused by same Bacillus anthracis strain since 2000. E Euro Surveill. 2013;18(13):pii=20437.
Dr Takematsu is a senior fellow of medical toxicology, department of emergency medicine, New York University School of Medicine and New York City Poison Control Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 50-year-old man with a 16-year history of injection heroin abuse presented to the ED complaining of ulcerative lesions on his right arm, which he stated had become worse over the past 3 months. He claimed the skin lesions, which involved his entire right arm, had grown “wider and deeper” since he started using the drug “Krokodil.” He further noted that he obtained the product from four different drug suppliers but did not know how it was prepared.
On presentation, his vital signs were: blood pressure, 135/78 mm Hg; heart rate, 92 beats/minute; respiratory rate, 14 breaths/minute; temperature, 98.2˚ F. Oxygen saturation was 100% on room air. Physical examination of the right arm was notable for broad ulcers that exposed fat tissue and muscle and involved the entire forearm circumferentially (Figure 1). There were no signs of acute infection such as erythema, warmth, or abscess formation. The remainder of the physical examination was unremarkable.
What is Krokodil?
The name Krokodil is derived from the Russian word for crocodile, and stems from the greenish, severely damaged “crocodile-like” skin lesions purportedly caused by subcutaneous injection of this opioid derivative. Krokodil is not a specific drug but rather it describes the product derived from an attempt to synthesize desomorphine, the core ingredient. Desomorphine is a short-acting opioid analogue that has a reported potency eight to 10 times greater than morphine (Figure 2).
Also called “Russian Magic,” Krokodil has been used in Russia since 2003 following the country’s major restrictions on the importation of heroin. Since desomorphine can be synthesized from codeine, which was available in over-the-counter medications in Russia until 2012, drug users have turned to it as an inexpensive heroin substitute. (The average cost of a codeine-containing product is about 120 Rubles [$4.00] per 10-pack.1)
The chemical process of synthesizing desomorphine typically involves mixing one to five packs of codeine-based analgesics1 with a solvent (eg, paint thinner, gasoline), along with iodine and red phosphorous, which can be obtained from the striking pads of matchboxes.2 This produces a yield equivalent to 500 Rubles of heroin, making it an attractive alternative to low-income drug users.1 However, the yields are poor, and the starting products vary based on the codeine source. No systemic analysis of Krokodil has been performed to assess the purity and concentration of desomorphine in the resulting product.
What are the risks of using Krokodil?
Krokodil is typically self-administered by either intravenous (IV) or subcutaneous (“skin-popping”) injection. Solvents and contaminants in the product can damage tissue with which they come into contact. Thus, IV use of this product causes venous scarring and collapse3; when venous access is exhausted, or following infiltration, the subcutaneous route may lead to necrosis and ulceration of the skin. These lesions are said to be used by illicit drug users as a “shooter’s patch” to inject drugs,4 leading to further skin damage. In addition to the local dermal effects, systemic inflammation results from the dissemination of the components of the product, causing neurological, solid-organ, and other effects.
Has Krokodil reached the United States?
Because of the horrifying appearance of the skin lesions, Krokodil has been called by many descriptive names such as the “flesh-eating drug” and “zombie-apocalypse drug,” which in turn has led to somewhat sensationalistic media reporting. There is, however, no clear evidence of entry of desomorphine or of Krokodil use in the United States, and most authorities feel either is unlikely to occur. This speculation is supported by the low cost and easy availability of heroin in the United States compared to Russia. Of note, although Russia banned the nonprescription sale of products containing codeine in November 2012, the ban has had minimal impact on the rate of necrotic skin lesions.
To date, none of the so-called cases of Krokodil-associated lesions reported in the United States have confirmatory analytical testing, and diagnoses have been based primarily on history or morphological features of the wound. Furthermore, no reference laboratories in the United States have identified desomorphine in any of their tested samples. Since the skin lesions are not pathognomonic of Krokodil use and share morphologic features with all forms of subcutaneous drug use, analytical confirmation is critical to identifying this as an emerging drug trend. Therefore, users of product sold or brewed as Krokodil may develop skin lesions regardless of desomorphine content due to contaminants in the injected product.
What causes the skin lesions?
Desomorphine itself is not likely the cause of the skin lesions. In its pure form, there is no reason to expect this compound would induce any specific changes in the skin that would lead to necrosis. In fact, similar chemical syntheses of desomorphine produced in other countries, such as in the Czech Republic and New Zealand, have not led to analogous skin lesions. Ulcerative skin damage is most likely caused by the inflammatory contaminants in Krokodil. Injection of solvents and other chemicals into the subcutaneous space lead to skin damage and provide a bed for the development of indolent infections. This mechanism is similar to that leading to epidemics, in which similar lesions have been associated with black-tar heroin injection and drug contamination with Bacillus anthracis.5,6
Some people, however, point out the severity of the skin lesions associated with the use of Krokodil compared to other injection drugs. This may be related to the need for frequent administration of Krokodil due to its short duration of action, in turn leading to repeated exposure to the impure solvents.
Treatment of associated lesions involves wound care, including debridement, topical care, and antibiotics; amputation may be required in severe cases.
Case conclusion
The patient in this case was taken to the operating room for debridement of the right arm. The pathology results of the tissue showed ulceration, abscess, acute and chronic inflamed granulation tissue, fibrosis, and necrosis. A magnetic resonance image of the arm showed no signs of osteomyelitis. A blood sample sent for analysis was negative for desomorphine. The patient was stable after the surgery, and was discharged with follow-up instructions and referral for drug abuse counseling.
Dr Takematsu is a senior fellow of medical toxicology, department of emergency medicine, New York University School of Medicine and New York City Poison Control Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 50-year-old man with a 16-year history of injection heroin abuse presented to the ED complaining of ulcerative lesions on his right arm, which he stated had become worse over the past 3 months. He claimed the skin lesions, which involved his entire right arm, had grown “wider and deeper” since he started using the drug “Krokodil.” He further noted that he obtained the product from four different drug suppliers but did not know how it was prepared.
On presentation, his vital signs were: blood pressure, 135/78 mm Hg; heart rate, 92 beats/minute; respiratory rate, 14 breaths/minute; temperature, 98.2˚ F. Oxygen saturation was 100% on room air. Physical examination of the right arm was notable for broad ulcers that exposed fat tissue and muscle and involved the entire forearm circumferentially (Figure 1). There were no signs of acute infection such as erythema, warmth, or abscess formation. The remainder of the physical examination was unremarkable.
What is Krokodil?
The name Krokodil is derived from the Russian word for crocodile, and stems from the greenish, severely damaged “crocodile-like” skin lesions purportedly caused by subcutaneous injection of this opioid derivative. Krokodil is not a specific drug but rather it describes the product derived from an attempt to synthesize desomorphine, the core ingredient. Desomorphine is a short-acting opioid analogue that has a reported potency eight to 10 times greater than morphine (Figure 2).
Also called “Russian Magic,” Krokodil has been used in Russia since 2003 following the country’s major restrictions on the importation of heroin. Since desomorphine can be synthesized from codeine, which was available in over-the-counter medications in Russia until 2012, drug users have turned to it as an inexpensive heroin substitute. (The average cost of a codeine-containing product is about 120 Rubles [$4.00] per 10-pack.1)
The chemical process of synthesizing desomorphine typically involves mixing one to five packs of codeine-based analgesics1 with a solvent (eg, paint thinner, gasoline), along with iodine and red phosphorous, which can be obtained from the striking pads of matchboxes.2 This produces a yield equivalent to 500 Rubles of heroin, making it an attractive alternative to low-income drug users.1 However, the yields are poor, and the starting products vary based on the codeine source. No systemic analysis of Krokodil has been performed to assess the purity and concentration of desomorphine in the resulting product.
What are the risks of using Krokodil?
Krokodil is typically self-administered by either intravenous (IV) or subcutaneous (“skin-popping”) injection. Solvents and contaminants in the product can damage tissue with which they come into contact. Thus, IV use of this product causes venous scarring and collapse3; when venous access is exhausted, or following infiltration, the subcutaneous route may lead to necrosis and ulceration of the skin. These lesions are said to be used by illicit drug users as a “shooter’s patch” to inject drugs,4 leading to further skin damage. In addition to the local dermal effects, systemic inflammation results from the dissemination of the components of the product, causing neurological, solid-organ, and other effects.
Has Krokodil reached the United States?
Because of the horrifying appearance of the skin lesions, Krokodil has been called by many descriptive names such as the “flesh-eating drug” and “zombie-apocalypse drug,” which in turn has led to somewhat sensationalistic media reporting. There is, however, no clear evidence of entry of desomorphine or of Krokodil use in the United States, and most authorities feel either is unlikely to occur. This speculation is supported by the low cost and easy availability of heroin in the United States compared to Russia. Of note, although Russia banned the nonprescription sale of products containing codeine in November 2012, the ban has had minimal impact on the rate of necrotic skin lesions.
To date, none of the so-called cases of Krokodil-associated lesions reported in the United States have confirmatory analytical testing, and diagnoses have been based primarily on history or morphological features of the wound. Furthermore, no reference laboratories in the United States have identified desomorphine in any of their tested samples. Since the skin lesions are not pathognomonic of Krokodil use and share morphologic features with all forms of subcutaneous drug use, analytical confirmation is critical to identifying this as an emerging drug trend. Therefore, users of product sold or brewed as Krokodil may develop skin lesions regardless of desomorphine content due to contaminants in the injected product.
What causes the skin lesions?
Desomorphine itself is not likely the cause of the skin lesions. In its pure form, there is no reason to expect this compound would induce any specific changes in the skin that would lead to necrosis. In fact, similar chemical syntheses of desomorphine produced in other countries, such as in the Czech Republic and New Zealand, have not led to analogous skin lesions. Ulcerative skin damage is most likely caused by the inflammatory contaminants in Krokodil. Injection of solvents and other chemicals into the subcutaneous space lead to skin damage and provide a bed for the development of indolent infections. This mechanism is similar to that leading to epidemics, in which similar lesions have been associated with black-tar heroin injection and drug contamination with Bacillus anthracis.5,6
Some people, however, point out the severity of the skin lesions associated with the use of Krokodil compared to other injection drugs. This may be related to the need for frequent administration of Krokodil due to its short duration of action, in turn leading to repeated exposure to the impure solvents.
Treatment of associated lesions involves wound care, including debridement, topical care, and antibiotics; amputation may be required in severe cases.
Case conclusion
The patient in this case was taken to the operating room for debridement of the right arm. The pathology results of the tissue showed ulceration, abscess, acute and chronic inflamed granulation tissue, fibrosis, and necrosis. A magnetic resonance image of the arm showed no signs of osteomyelitis. A blood sample sent for analysis was negative for desomorphine. The patient was stable after the surgery, and was discharged with follow-up instructions and referral for drug abuse counseling.
- Grund JP, Latypov A, Harris M. Breaking worse: The emergence of krokodil and excessive injuries among people who inject drugs in Eurasia. Int J Drug Policy. 2013;24(4):265-274.
- Gahr M, Freudenmann RW, Hiemke C, Gunst IM, Connemann BJ, Schönfeldt-Lecuona C. Desomorphine goes “crocodile.” J Addict Dis. 2012;31(4):407-412.
- Pieper B, Kirsner RS, Templin TN, Birk TJ. Injection drug use: an understudied cause of venous disease. Arch Dermatol. 2007;143(10):1305-1309.
- Iyer S, Subramanian P, Pabari A. A devastating complication of “skin popping.” Surgeon. 2011;9(5):295-297.
- Dunbar NM, Harruff RC. Necrotizing fasciitis: manifestations, microbiology and connection with black tar heroin. J Forensic Sci. 2007;52(4):920-923.
- Grunow R, Klee SR, Beyer W, et al. Anthrax among heroin users in Europe possibly caused by same Bacillus anthracis strain since 2000. E Euro Surveill. 2013;18(13):pii=20437.
- Grund JP, Latypov A, Harris M. Breaking worse: The emergence of krokodil and excessive injuries among people who inject drugs in Eurasia. Int J Drug Policy. 2013;24(4):265-274.
- Gahr M, Freudenmann RW, Hiemke C, Gunst IM, Connemann BJ, Schönfeldt-Lecuona C. Desomorphine goes “crocodile.” J Addict Dis. 2012;31(4):407-412.
- Pieper B, Kirsner RS, Templin TN, Birk TJ. Injection drug use: an understudied cause of venous disease. Arch Dermatol. 2007;143(10):1305-1309.
- Iyer S, Subramanian P, Pabari A. A devastating complication of “skin popping.” Surgeon. 2011;9(5):295-297.
- Dunbar NM, Harruff RC. Necrotizing fasciitis: manifestations, microbiology and connection with black tar heroin. J Forensic Sci. 2007;52(4):920-923.
- Grunow R, Klee SR, Beyer W, et al. Anthrax among heroin users in Europe possibly caused by same Bacillus anthracis strain since 2000. E Euro Surveill. 2013;18(13):pii=20437.

Case Studies in Toxicology: Tiny Bubbles (Or, the Dangers of Cleaning Your Fruit)
Case
A previously healthy 32-year-old man presented to the ED after unintentionally ingesting a mouthful of concentrated (35%) hydrogen peroxide (H2O2) from an unmarked bottle he kept in his refrigerator. Upon realizing his error, he immediately drank a liter of water, which promptly induced vomiting. In the ED, the patient complained of mild throat and chest discomfort as well as “abdominal fullness.”
His initial vital signs were: blood pressure, 140/92 mm Hg; heart rate, 93 beats/minute; respiratory rate, 18 breaths/minute; temperature, 96.4° F. Oxygen saturation was 98% on room air. Physical examination revealed tenderness in the epigastric region with no peritoneal findings. Oropharynx and chest examination were normal, and standard laboratory investigations were all within normal limits.
What are the potential exposures to hydrogen peroxide?
Hydrogen peroxide is a colorless and odorless liquid. Solutions with concentrations ranging from 3% to 5% have many household applications, including use as a wound disinfectant and dentifrice; dilute solutions are also utilized for similar purposes in the hospital setting. Industrial-strength H2O2 (concentrations of 10% to 35%) is employed to bleach textiles and paper, and higher concentrations (70% to 90%) are used as an oxygen source for rocket engines.
Consumer application of concentrated H2O2 solutions has become increasingly common. Some, like this patient, clean the surfaces of fruits and vegetables with H2O2 to decrease transmission of bacteria during cutting.1 More concerning, however, is the purported medicinal benefits of ingesting “food-grade” (35%) H2O2 mixed with water—touted on many Internet sites as a treatment for illnesses such as emphysema, cancer, anemia, and HIV.2 Sometimes referred to as “hyperoxygenation therapy,” this so-called treatment has not been approved by the US Food and Drug Administration for any such purpose.3 When diluted sufficiently, this concoction is not harmful but unlikely to provide any health benefits.
Dr Lucyk is a fellow of medical toxicology in the department of emergency medicine at the New York University School of Medicine and the New York City Poison Control Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
What are the toxic effects of concentrated hydrogen peroxide?
Injury from concentrated H2O2 consumption is primarily from either direct caustic injury or the embolic obstruction of blood flow. Following ingestion, the enzyme catalase metabolizes the breakdown of H2O2 in accordance with the following equation: 2H2O2(aq) → 2H2O(l) + O2(g) + heat. A single milliliter of 35% H2O2 results in the liberation of 100 mL of O2. (The more common 3% household solution generates 10 mL of oxygen per 1 ml of H2O2.) The creation of a large intragastric pressure gradient from the liberation of gas, coupled with the caustic and exothermic injury of the bowel mucosa, may contribute to the movement of oxygen through epithelial interstices into the circulation.In addition, and perhaps more importantly, absorption of intact H2O2 with subsequent metabolism by catalase in the blood liberates oxygen directly within the vasculature. Oxygen bubbles may coalesce in blood circulation and occlude vascular flow. In canine studies, elevated oxygen tension in the portal venous system led to cessation of mesenteric flow in arteries and veins, though the mechanism of action is unclear.4 Furthermore, coalescence of bubbles can lead to disruption of bowel-cell architecture, fibrin plugging of capillaries, venous thrombosis, and infarction of tissues.4
Cases of cardiac and cerebral gas embolism have been reported, and present similarly to patients with diving-related decompression injuries (eg, stroke-like syndromes).5,6 The proposed mechanism for these latter effects involves the metabolism of H2O2 in the systemic circulation with production of oxygen bubbles. In the presence of an atrial septal defect, bubbles may move from the right atrium to the arterial circulation.7
Toxicity and death from H2O2 exposure associated with the historical treatment of inspissated meconium,4 as well as the irrigation of wounds,8 has been reported in the medical literature. Ingestion of a 3% solution is generally benign, resulting at worst in gastrointestinal symptoms or throat irritation.9 Rarely does significant toxicity occur at this low concentration,5 with the vast majority of such cases involving concentrated solutions of 35%.
Case continuation
Case 2

Based on this patient’s continued symptoms, an abdominal radiograph was obtained to assess the presence of portal venous air. Although radiographic findings were normal, continued abdominal examination findings warranted a subsequent abdominal computed tomography (CT) scan, which revealed the presence of extensive air throughout the portal venous system (Figure.).
Do all patients presenting with H2O2 ingestion require imaging to assess for the presence of portal venous air?
Reportedly, ingestion of as little as a “sip” or “mouthful” of 35% H2O2 has resulted in venous and arterial gas embolism,6 occasionally with severe consequences, but no current consensus guidelines exist regarding imaging requirements. Some toxicologists and hyperbaric physicians believe that the presence of portal venous air does not adversely impact a patient’s prognosis or necessitate treatment, and therefore a workup is unnecessary. Others, however, suggest that the presence of portal venous air indicates oversaturation of oxygen in the blood, placing the patient at increased risk for cardiac and cerebral air embolism. Neither one of these theories is well supported in the literature. Although practice patterns vary by institution, it is reasonable that all patients presenting with abdominal complaints after ingestion of H2O2 undergo CT imaging to assess for portal venous air.
If portal venous air is detected, do patients require hyperbaric oxygen therapy?
The management of patients with portal venous gas following H2O2
Hyperbaric therapy increases the amount of oxygen that can be dissolved in the blood, thereby decreasing bubble formation and allowing transport of dissolved oxygen to the lungs where it can be exhaled. Some patients with portal venous air experience significant pain and portal venous hypertension, which may respond rapidly to this therapy.10 Based on available literature, hyperbaric therapy is reasonable for patients with significant abdominal pain and portal venous air following H2O2 ingestion; less controversial is the role of hyperbaric therapy in those with cerebral air embolism. Multiple case reports of patients with significant neurologic findings demonstrate resolution of symptoms following hyperbaric therapy.6
Case conclusion
Hyperbaric oxygen therapy was recommended for the patient in this case, but transfer to a hyperbaric facility was not possible. He was instead admitted to the hospital for continuous monitoring. Over the next 12 hours, his symptoms gradually resolved, and a repeat CT scan the following day showed complete resolution of the portal venous gas. The patient was subsequently discharged without any sequelae.
- Ukuku DO, Bari ML, Kawamoto S, Isshiki K. Use of hydrogen peroxide in combination with nisin, sodium lactate and citric acid for reducing transfer of bacterial pathogens from whole melon surfaces to fresh-cut pieces. Int J Food Microbiol. 2005;104(2):225-233.
- 35% H2O2 hydrogen peroxide food grade certified benefits. The One Minute Miracle Web site. http:// www.theoneminutemiracleinc.com/pages/h2o2- benefits/. Accessed November 20, 2013.
- FDA warns consumers against drinking high-strength hydrogen peroxide for medicinal use: ingestion can lead to serious health risk and death [news release]. Silver Spring, MD: US Food and Drug Administration; July 27, 2006. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ 2006/ucm108701.htm. Accessed November 20, 2013.
- Shaw A, Cooperman A, Fusco J. Gas embolism produced by hydrogen peroxide. N Engl J Med. 1967;277(5):238-241.
- Cina SJ, Downs JC, Conradi SE. Hydrogen peroxide: a source of lethal oxygen embolism. Case report and review of the literature. Am J Forensic Med Pathol. 1994;15(1):44-50.
- Rider SP, Jackson SB, Rusyniak DE. Cerebral air gas embolism from concentrated hydrogen peroxide ingestion. Clin Toxicol (Phila). 2008;46(9):815-818.
- French LK, Horowitz BZ, McKeown NJ. Hydrogen peroxide ingestion associated with portal venous gas and treatment with hyperbaric oxygen: a case series and review of the literature. Clin Toxicol (Phila). 2010;48(6):533-538.
- Bassan MM, Dudai M, Shalev O. Near-fatal systemic oxygen embolism due to wound irrigation with hydrogen peroxide. Postgrad Med J. 1982;58(681):448-450.
- Henry MC, Wheeler J, Mofenson HC, et al. Hydrogen peroxide 3% exposures. J Toxicol Clin Toxicol. 1996;34(3):323-327.
- Papafragkou S, Gasparyan A, Batista R, Scott P. Treatment of portal venous gas embolism with hyperbaric oxygen after accidental ingestion of hydrogen peroxide: a case report and review of the literature. J Emerg Med. 2012;43(1):e21-e23.
Case
A previously healthy 32-year-old man presented to the ED after unintentionally ingesting a mouthful of concentrated (35%) hydrogen peroxide (H2O2) from an unmarked bottle he kept in his refrigerator. Upon realizing his error, he immediately drank a liter of water, which promptly induced vomiting. In the ED, the patient complained of mild throat and chest discomfort as well as “abdominal fullness.”
His initial vital signs were: blood pressure, 140/92 mm Hg; heart rate, 93 beats/minute; respiratory rate, 18 breaths/minute; temperature, 96.4° F. Oxygen saturation was 98% on room air. Physical examination revealed tenderness in the epigastric region with no peritoneal findings. Oropharynx and chest examination were normal, and standard laboratory investigations were all within normal limits.
What are the potential exposures to hydrogen peroxide?
Hydrogen peroxide is a colorless and odorless liquid. Solutions with concentrations ranging from 3% to 5% have many household applications, including use as a wound disinfectant and dentifrice; dilute solutions are also utilized for similar purposes in the hospital setting. Industrial-strength H2O2 (concentrations of 10% to 35%) is employed to bleach textiles and paper, and higher concentrations (70% to 90%) are used as an oxygen source for rocket engines.
Consumer application of concentrated H2O2 solutions has become increasingly common. Some, like this patient, clean the surfaces of fruits and vegetables with H2O2 to decrease transmission of bacteria during cutting.1 More concerning, however, is the purported medicinal benefits of ingesting “food-grade” (35%) H2O2 mixed with water—touted on many Internet sites as a treatment for illnesses such as emphysema, cancer, anemia, and HIV.2 Sometimes referred to as “hyperoxygenation therapy,” this so-called treatment has not been approved by the US Food and Drug Administration for any such purpose.3 When diluted sufficiently, this concoction is not harmful but unlikely to provide any health benefits.
Dr Lucyk is a fellow of medical toxicology in the department of emergency medicine at the New York University School of Medicine and the New York City Poison Control Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
What are the toxic effects of concentrated hydrogen peroxide?
Injury from concentrated H2O2 consumption is primarily from either direct caustic injury or the embolic obstruction of blood flow. Following ingestion, the enzyme catalase metabolizes the breakdown of H2O2 in accordance with the following equation: 2H2O2(aq) → 2H2O(l) + O2(g) + heat. A single milliliter of 35% H2O2 results in the liberation of 100 mL of O2. (The more common 3% household solution generates 10 mL of oxygen per 1 ml of H2O2.) The creation of a large intragastric pressure gradient from the liberation of gas, coupled with the caustic and exothermic injury of the bowel mucosa, may contribute to the movement of oxygen through epithelial interstices into the circulation.In addition, and perhaps more importantly, absorption of intact H2O2 with subsequent metabolism by catalase in the blood liberates oxygen directly within the vasculature. Oxygen bubbles may coalesce in blood circulation and occlude vascular flow. In canine studies, elevated oxygen tension in the portal venous system led to cessation of mesenteric flow in arteries and veins, though the mechanism of action is unclear.4 Furthermore, coalescence of bubbles can lead to disruption of bowel-cell architecture, fibrin plugging of capillaries, venous thrombosis, and infarction of tissues.4
Cases of cardiac and cerebral gas embolism have been reported, and present similarly to patients with diving-related decompression injuries (eg, stroke-like syndromes).5,6 The proposed mechanism for these latter effects involves the metabolism of H2O2 in the systemic circulation with production of oxygen bubbles. In the presence of an atrial septal defect, bubbles may move from the right atrium to the arterial circulation.7
Toxicity and death from H2O2 exposure associated with the historical treatment of inspissated meconium,4 as well as the irrigation of wounds,8 has been reported in the medical literature. Ingestion of a 3% solution is generally benign, resulting at worst in gastrointestinal symptoms or throat irritation.9 Rarely does significant toxicity occur at this low concentration,5 with the vast majority of such cases involving concentrated solutions of 35%.
Case continuation
Case 2
Based on this patient’s continued symptoms, an abdominal radiograph was obtained to assess the presence of portal venous air. Although radiographic findings were normal, continued abdominal examination findings warranted a subsequent abdominal computed tomography (CT) scan, which revealed the presence of extensive air throughout the portal venous system (Figure.).
Do all patients presenting with H2O2 ingestion require imaging to assess for the presence of portal venous air?
Reportedly, ingestion of as little as a “sip” or “mouthful” of 35% H2O2 has resulted in venous and arterial gas embolism,6 occasionally with severe consequences, but no current consensus guidelines exist regarding imaging requirements. Some toxicologists and hyperbaric physicians believe that the presence of portal venous air does not adversely impact a patient’s prognosis or necessitate treatment, and therefore a workup is unnecessary. Others, however, suggest that the presence of portal venous air indicates oversaturation of oxygen in the blood, placing the patient at increased risk for cardiac and cerebral air embolism. Neither one of these theories is well supported in the literature. Although practice patterns vary by institution, it is reasonable that all patients presenting with abdominal complaints after ingestion of H2O2 undergo CT imaging to assess for portal venous air.
If portal venous air is detected, do patients require hyperbaric oxygen therapy?
The management of patients with portal venous gas following H2O2
Hyperbaric therapy increases the amount of oxygen that can be dissolved in the blood, thereby decreasing bubble formation and allowing transport of dissolved oxygen to the lungs where it can be exhaled. Some patients with portal venous air experience significant pain and portal venous hypertension, which may respond rapidly to this therapy.10 Based on available literature, hyperbaric therapy is reasonable for patients with significant abdominal pain and portal venous air following H2O2 ingestion; less controversial is the role of hyperbaric therapy in those with cerebral air embolism. Multiple case reports of patients with significant neurologic findings demonstrate resolution of symptoms following hyperbaric therapy.6
Case conclusion
Hyperbaric oxygen therapy was recommended for the patient in this case, but transfer to a hyperbaric facility was not possible. He was instead admitted to the hospital for continuous monitoring. Over the next 12 hours, his symptoms gradually resolved, and a repeat CT scan the following day showed complete resolution of the portal venous gas. The patient was subsequently discharged without any sequelae.
Case
A previously healthy 32-year-old man presented to the ED after unintentionally ingesting a mouthful of concentrated (35%) hydrogen peroxide (H2O2) from an unmarked bottle he kept in his refrigerator. Upon realizing his error, he immediately drank a liter of water, which promptly induced vomiting. In the ED, the patient complained of mild throat and chest discomfort as well as “abdominal fullness.”
His initial vital signs were: blood pressure, 140/92 mm Hg; heart rate, 93 beats/minute; respiratory rate, 18 breaths/minute; temperature, 96.4° F. Oxygen saturation was 98% on room air. Physical examination revealed tenderness in the epigastric region with no peritoneal findings. Oropharynx and chest examination were normal, and standard laboratory investigations were all within normal limits.
What are the potential exposures to hydrogen peroxide?
Hydrogen peroxide is a colorless and odorless liquid. Solutions with concentrations ranging from 3% to 5% have many household applications, including use as a wound disinfectant and dentifrice; dilute solutions are also utilized for similar purposes in the hospital setting. Industrial-strength H2O2 (concentrations of 10% to 35%) is employed to bleach textiles and paper, and higher concentrations (70% to 90%) are used as an oxygen source for rocket engines.
Consumer application of concentrated H2O2 solutions has become increasingly common. Some, like this patient, clean the surfaces of fruits and vegetables with H2O2 to decrease transmission of bacteria during cutting.1 More concerning, however, is the purported medicinal benefits of ingesting “food-grade” (35%) H2O2 mixed with water—touted on many Internet sites as a treatment for illnesses such as emphysema, cancer, anemia, and HIV.2 Sometimes referred to as “hyperoxygenation therapy,” this so-called treatment has not been approved by the US Food and Drug Administration for any such purpose.3 When diluted sufficiently, this concoction is not harmful but unlikely to provide any health benefits.
Dr Lucyk is a fellow of medical toxicology in the department of emergency medicine at the New York University School of Medicine and the New York City Poison Control Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
What are the toxic effects of concentrated hydrogen peroxide?
Injury from concentrated H2O2 consumption is primarily from either direct caustic injury or the embolic obstruction of blood flow. Following ingestion, the enzyme catalase metabolizes the breakdown of H2O2 in accordance with the following equation: 2H2O2(aq) → 2H2O(l) + O2(g) + heat. A single milliliter of 35% H2O2 results in the liberation of 100 mL of O2. (The more common 3% household solution generates 10 mL of oxygen per 1 ml of H2O2.) The creation of a large intragastric pressure gradient from the liberation of gas, coupled with the caustic and exothermic injury of the bowel mucosa, may contribute to the movement of oxygen through epithelial interstices into the circulation.In addition, and perhaps more importantly, absorption of intact H2O2 with subsequent metabolism by catalase in the blood liberates oxygen directly within the vasculature. Oxygen bubbles may coalesce in blood circulation and occlude vascular flow. In canine studies, elevated oxygen tension in the portal venous system led to cessation of mesenteric flow in arteries and veins, though the mechanism of action is unclear.4 Furthermore, coalescence of bubbles can lead to disruption of bowel-cell architecture, fibrin plugging of capillaries, venous thrombosis, and infarction of tissues.4
Cases of cardiac and cerebral gas embolism have been reported, and present similarly to patients with diving-related decompression injuries (eg, stroke-like syndromes).5,6 The proposed mechanism for these latter effects involves the metabolism of H2O2 in the systemic circulation with production of oxygen bubbles. In the presence of an atrial septal defect, bubbles may move from the right atrium to the arterial circulation.7
Toxicity and death from H2O2 exposure associated with the historical treatment of inspissated meconium,4 as well as the irrigation of wounds,8 has been reported in the medical literature. Ingestion of a 3% solution is generally benign, resulting at worst in gastrointestinal symptoms or throat irritation.9 Rarely does significant toxicity occur at this low concentration,5 with the vast majority of such cases involving concentrated solutions of 35%.
Case continuation
Case 2
Based on this patient’s continued symptoms, an abdominal radiograph was obtained to assess the presence of portal venous air. Although radiographic findings were normal, continued abdominal examination findings warranted a subsequent abdominal computed tomography (CT) scan, which revealed the presence of extensive air throughout the portal venous system (Figure.).
Do all patients presenting with H2O2 ingestion require imaging to assess for the presence of portal venous air?
Reportedly, ingestion of as little as a “sip” or “mouthful” of 35% H2O2 has resulted in venous and arterial gas embolism,6 occasionally with severe consequences, but no current consensus guidelines exist regarding imaging requirements. Some toxicologists and hyperbaric physicians believe that the presence of portal venous air does not adversely impact a patient’s prognosis or necessitate treatment, and therefore a workup is unnecessary. Others, however, suggest that the presence of portal venous air indicates oversaturation of oxygen in the blood, placing the patient at increased risk for cardiac and cerebral air embolism. Neither one of these theories is well supported in the literature. Although practice patterns vary by institution, it is reasonable that all patients presenting with abdominal complaints after ingestion of H2O2 undergo CT imaging to assess for portal venous air.
If portal venous air is detected, do patients require hyperbaric oxygen therapy?
The management of patients with portal venous gas following H2O2
Hyperbaric therapy increases the amount of oxygen that can be dissolved in the blood, thereby decreasing bubble formation and allowing transport of dissolved oxygen to the lungs where it can be exhaled. Some patients with portal venous air experience significant pain and portal venous hypertension, which may respond rapidly to this therapy.10 Based on available literature, hyperbaric therapy is reasonable for patients with significant abdominal pain and portal venous air following H2O2 ingestion; less controversial is the role of hyperbaric therapy in those with cerebral air embolism. Multiple case reports of patients with significant neurologic findings demonstrate resolution of symptoms following hyperbaric therapy.6
Case conclusion
Hyperbaric oxygen therapy was recommended for the patient in this case, but transfer to a hyperbaric facility was not possible. He was instead admitted to the hospital for continuous monitoring. Over the next 12 hours, his symptoms gradually resolved, and a repeat CT scan the following day showed complete resolution of the portal venous gas. The patient was subsequently discharged without any sequelae.
- Ukuku DO, Bari ML, Kawamoto S, Isshiki K. Use of hydrogen peroxide in combination with nisin, sodium lactate and citric acid for reducing transfer of bacterial pathogens from whole melon surfaces to fresh-cut pieces. Int J Food Microbiol. 2005;104(2):225-233.
- 35% H2O2 hydrogen peroxide food grade certified benefits. The One Minute Miracle Web site. http:// www.theoneminutemiracleinc.com/pages/h2o2- benefits/. Accessed November 20, 2013.
- FDA warns consumers against drinking high-strength hydrogen peroxide for medicinal use: ingestion can lead to serious health risk and death [news release]. Silver Spring, MD: US Food and Drug Administration; July 27, 2006. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ 2006/ucm108701.htm. Accessed November 20, 2013.
- Shaw A, Cooperman A, Fusco J. Gas embolism produced by hydrogen peroxide. N Engl J Med. 1967;277(5):238-241.
- Cina SJ, Downs JC, Conradi SE. Hydrogen peroxide: a source of lethal oxygen embolism. Case report and review of the literature. Am J Forensic Med Pathol. 1994;15(1):44-50.
- Rider SP, Jackson SB, Rusyniak DE. Cerebral air gas embolism from concentrated hydrogen peroxide ingestion. Clin Toxicol (Phila). 2008;46(9):815-818.
- French LK, Horowitz BZ, McKeown NJ. Hydrogen peroxide ingestion associated with portal venous gas and treatment with hyperbaric oxygen: a case series and review of the literature. Clin Toxicol (Phila). 2010;48(6):533-538.
- Bassan MM, Dudai M, Shalev O. Near-fatal systemic oxygen embolism due to wound irrigation with hydrogen peroxide. Postgrad Med J. 1982;58(681):448-450.
- Henry MC, Wheeler J, Mofenson HC, et al. Hydrogen peroxide 3% exposures. J Toxicol Clin Toxicol. 1996;34(3):323-327.
- Papafragkou S, Gasparyan A, Batista R, Scott P. Treatment of portal venous gas embolism with hyperbaric oxygen after accidental ingestion of hydrogen peroxide: a case report and review of the literature. J Emerg Med. 2012;43(1):e21-e23.
- Ukuku DO, Bari ML, Kawamoto S, Isshiki K. Use of hydrogen peroxide in combination with nisin, sodium lactate and citric acid for reducing transfer of bacterial pathogens from whole melon surfaces to fresh-cut pieces. Int J Food Microbiol. 2005;104(2):225-233.
- 35% H2O2 hydrogen peroxide food grade certified benefits. The One Minute Miracle Web site. http:// www.theoneminutemiracleinc.com/pages/h2o2- benefits/. Accessed November 20, 2013.
- FDA warns consumers against drinking high-strength hydrogen peroxide for medicinal use: ingestion can lead to serious health risk and death [news release]. Silver Spring, MD: US Food and Drug Administration; July 27, 2006. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ 2006/ucm108701.htm. Accessed November 20, 2013.
- Shaw A, Cooperman A, Fusco J. Gas embolism produced by hydrogen peroxide. N Engl J Med. 1967;277(5):238-241.
- Cina SJ, Downs JC, Conradi SE. Hydrogen peroxide: a source of lethal oxygen embolism. Case report and review of the literature. Am J Forensic Med Pathol. 1994;15(1):44-50.
- Rider SP, Jackson SB, Rusyniak DE. Cerebral air gas embolism from concentrated hydrogen peroxide ingestion. Clin Toxicol (Phila). 2008;46(9):815-818.
- French LK, Horowitz BZ, McKeown NJ. Hydrogen peroxide ingestion associated with portal venous gas and treatment with hyperbaric oxygen: a case series and review of the literature. Clin Toxicol (Phila). 2010;48(6):533-538.
- Bassan MM, Dudai M, Shalev O. Near-fatal systemic oxygen embolism due to wound irrigation with hydrogen peroxide. Postgrad Med J. 1982;58(681):448-450.
- Henry MC, Wheeler J, Mofenson HC, et al. Hydrogen peroxide 3% exposures. J Toxicol Clin Toxicol. 1996;34(3):323-327.
- Papafragkou S, Gasparyan A, Batista R, Scott P. Treatment of portal venous gas embolism with hyperbaric oxygen after accidental ingestion of hydrogen peroxide: a case report and review of the literature. J Emerg Med. 2012;43(1):e21-e23.

New definition of kidney injury is more predictive of mortality
The newly proposed consensus definition of acute kidney injury in patients with cirrhosis accurately predicts 30-day mortality and other adverse outcomes in this patient population much better than the current, more rigid definition would, according to a report in the December issue of Gastroenterology (doi:10.1053/j.gastro.2013.08.051).
In what they described as the largest prospective study of this topic to date, researchers found that the recently proposed, broader redefinition of acute kidney injury (AKI) correctly identified which patients were likely to die, develop severe complications such as organ failure, or require longer hospitalization, even when the AKI was transient and resolved completely after treatment.
Courtesy American Gastroenterological Association
More than half of the patients in this study who had episodes of AKI according to the new definition did not meet the criteria of the old definition. So using the new definition will help identify these high-risk patients at an earlier stage of renal dysfunction, "well before the stringent diagnostic criteria of [the old definition] are reached," when they will have a better treatment response, said Dr. Florence Wong of the division of gastroenterology, University of Toronto, and her associates.
The old definition of AKI required the presence of hepatorenal syndrome, with a serum creatinine level of greater than 2.5 mg/dL. This meant that patients with less severe renal dysfunction didn’t qualify and weren’t treated. But emerging evidence indicates that even mild degrees of renal dysfunction signal a poor prognosis, and that serum creatinine alone doesn’t accurately reflect renal dysfunction in advanced cirrhosis.
So the International Ascites Club and the Acute Dialysis Quality Initiative (ADQI) group proposed that acute kidney injury in cirrhosis should be redefined as an increase in serum creatinine level of 0.3 mg/dL or greater within 48 hours, or a 50% increase in serum creatinine level from a stable baseline reading within the previous 6 months, regardless of final serum creatinine level.
Dr. Wong and her colleagues assessed the new definition in a cohort of 337 cirrhotic patients treated during a 2-year period at 12 North American medical centers who were admitted with a bacterial infection (287 subjects) or who developed a bacterial infection during hospitalization (50 subjects). The most common infections were urinary tract infection (27% of patients), spontaneous bacterial peritonitis (21%), skin infection (14%), pneumonia (10%), and spontaneous bacteremia with no clear source of infection (9%).
Approximately half of these patients (49%) developed at least one episode of AKI during hospitalization. The 30-day mortality was significantly higher for those who developed AKI according to the new definition (34% mortality) than in those who did not (7% mortality), the investigators said.
Most patients who developed AKI had only a transient case, and their renal function completely recovered. Yet their subsequent mortality within 30 days was twice as high as that for patients who didn’t have any AKI.
The negative predictive value of the new definition of AKI was 93%, and the positive predictive value was 34%.
This study was supported in part by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Center for Research Resources. No financial conflicts of interest were reported.
Renal dysfunction in patients with cirrhosis is often associated with sepsis. This combination involves a very high probability of death. Recently, the concept of acute kidney injury has been proposed to be extended also to renal failure occurring in patients with cirrhosis. AKI should overcome limitations associated with a fixed creatinine threshold, ensure rapid identification of renal dysfunction, and allow timely treatment in patients with hepatorenal syndrome. However, AKI should also overcome the skepticism of those who wish not to abandon previous definitions.
The recent paper of Dr. Wong and her colleagues explored the impact of AKI in 337 hospitalized patients with cirrhosis. Two-hundred eighty-seven patients had bacterial infection at admission, and 93 developed it during hospitalization. Overall, 68 patients died from multiorgan failure, whereas only 7% of patients without AKI died. Mortality ranged from 15% in patients who recovered from AKI to 80% in those who did not. Moreover, 76 patients (23%) developed a second infection, often associated with invasive procedures! An elevated Model for End-Stage Liver Disease score and a second infection were factors independently associated with AKI. Accordingly, the development of AKI in cirrhosis, even if reversible, was shown to be a strong predictor of short survival.
These findings show that, in cirrhosis, even small creatinine changes (0.3 mg) are clinically relevant, and that AKI is probably a hallmark of hemodynamic instability with a risk of multiorgan failure and death. The altered hemodynamics in patients with cirrhosis cause central hypovolemia. Aiming at protecting our patients from infection and AKI, we should also pay more attention to clinical procedures that raise serum creatinine level.
Dr. Francesco Salerno is in the department of internal medicine, at the Policlinico IRCCS San Donato, University of Milan (Italy); Dr. Vincenzo La Mura is with the Fondazione IRCCS Ca'Granda, in the department of gastroenterologia-1 of the Hospital Maggiore Policlinico, Milan. They reported no relevant financial conflicts.
Renal dysfunction in patients with cirrhosis is often associated with sepsis. This combination involves a very high probability of death. Recently, the concept of acute kidney injury has been proposed to be extended also to renal failure occurring in patients with cirrhosis. AKI should overcome limitations associated with a fixed creatinine threshold, ensure rapid identification of renal dysfunction, and allow timely treatment in patients with hepatorenal syndrome. However, AKI should also overcome the skepticism of those who wish not to abandon previous definitions.
The recent paper of Dr. Wong and her colleagues explored the impact of AKI in 337 hospitalized patients with cirrhosis. Two-hundred eighty-seven patients had bacterial infection at admission, and 93 developed it during hospitalization. Overall, 68 patients died from multiorgan failure, whereas only 7% of patients without AKI died. Mortality ranged from 15% in patients who recovered from AKI to 80% in those who did not. Moreover, 76 patients (23%) developed a second infection, often associated with invasive procedures! An elevated Model for End-Stage Liver Disease score and a second infection were factors independently associated with AKI. Accordingly, the development of AKI in cirrhosis, even if reversible, was shown to be a strong predictor of short survival.
These findings show that, in cirrhosis, even small creatinine changes (0.3 mg) are clinically relevant, and that AKI is probably a hallmark of hemodynamic instability with a risk of multiorgan failure and death. The altered hemodynamics in patients with cirrhosis cause central hypovolemia. Aiming at protecting our patients from infection and AKI, we should also pay more attention to clinical procedures that raise serum creatinine level.
Dr. Francesco Salerno is in the department of internal medicine, at the Policlinico IRCCS San Donato, University of Milan (Italy); Dr. Vincenzo La Mura is with the Fondazione IRCCS Ca'Granda, in the department of gastroenterologia-1 of the Hospital Maggiore Policlinico, Milan. They reported no relevant financial conflicts.
Renal dysfunction in patients with cirrhosis is often associated with sepsis. This combination involves a very high probability of death. Recently, the concept of acute kidney injury has been proposed to be extended also to renal failure occurring in patients with cirrhosis. AKI should overcome limitations associated with a fixed creatinine threshold, ensure rapid identification of renal dysfunction, and allow timely treatment in patients with hepatorenal syndrome. However, AKI should also overcome the skepticism of those who wish not to abandon previous definitions.
The recent paper of Dr. Wong and her colleagues explored the impact of AKI in 337 hospitalized patients with cirrhosis. Two-hundred eighty-seven patients had bacterial infection at admission, and 93 developed it during hospitalization. Overall, 68 patients died from multiorgan failure, whereas only 7% of patients without AKI died. Mortality ranged from 15% in patients who recovered from AKI to 80% in those who did not. Moreover, 76 patients (23%) developed a second infection, often associated with invasive procedures! An elevated Model for End-Stage Liver Disease score and a second infection were factors independently associated with AKI. Accordingly, the development of AKI in cirrhosis, even if reversible, was shown to be a strong predictor of short survival.
These findings show that, in cirrhosis, even small creatinine changes (0.3 mg) are clinically relevant, and that AKI is probably a hallmark of hemodynamic instability with a risk of multiorgan failure and death. The altered hemodynamics in patients with cirrhosis cause central hypovolemia. Aiming at protecting our patients from infection and AKI, we should also pay more attention to clinical procedures that raise serum creatinine level.
Dr. Francesco Salerno is in the department of internal medicine, at the Policlinico IRCCS San Donato, University of Milan (Italy); Dr. Vincenzo La Mura is with the Fondazione IRCCS Ca'Granda, in the department of gastroenterologia-1 of the Hospital Maggiore Policlinico, Milan. They reported no relevant financial conflicts.
The newly proposed consensus definition of acute kidney injury in patients with cirrhosis accurately predicts 30-day mortality and other adverse outcomes in this patient population much better than the current, more rigid definition would, according to a report in the December issue of Gastroenterology (doi:10.1053/j.gastro.2013.08.051).
In what they described as the largest prospective study of this topic to date, researchers found that the recently proposed, broader redefinition of acute kidney injury (AKI) correctly identified which patients were likely to die, develop severe complications such as organ failure, or require longer hospitalization, even when the AKI was transient and resolved completely after treatment.
Courtesy American Gastroenterological Association
More than half of the patients in this study who had episodes of AKI according to the new definition did not meet the criteria of the old definition. So using the new definition will help identify these high-risk patients at an earlier stage of renal dysfunction, "well before the stringent diagnostic criteria of [the old definition] are reached," when they will have a better treatment response, said Dr. Florence Wong of the division of gastroenterology, University of Toronto, and her associates.
The old definition of AKI required the presence of hepatorenal syndrome, with a serum creatinine level of greater than 2.5 mg/dL. This meant that patients with less severe renal dysfunction didn’t qualify and weren’t treated. But emerging evidence indicates that even mild degrees of renal dysfunction signal a poor prognosis, and that serum creatinine alone doesn’t accurately reflect renal dysfunction in advanced cirrhosis.
So the International Ascites Club and the Acute Dialysis Quality Initiative (ADQI) group proposed that acute kidney injury in cirrhosis should be redefined as an increase in serum creatinine level of 0.3 mg/dL or greater within 48 hours, or a 50% increase in serum creatinine level from a stable baseline reading within the previous 6 months, regardless of final serum creatinine level.
Dr. Wong and her colleagues assessed the new definition in a cohort of 337 cirrhotic patients treated during a 2-year period at 12 North American medical centers who were admitted with a bacterial infection (287 subjects) or who developed a bacterial infection during hospitalization (50 subjects). The most common infections were urinary tract infection (27% of patients), spontaneous bacterial peritonitis (21%), skin infection (14%), pneumonia (10%), and spontaneous bacteremia with no clear source of infection (9%).
Approximately half of these patients (49%) developed at least one episode of AKI during hospitalization. The 30-day mortality was significantly higher for those who developed AKI according to the new definition (34% mortality) than in those who did not (7% mortality), the investigators said.
Most patients who developed AKI had only a transient case, and their renal function completely recovered. Yet their subsequent mortality within 30 days was twice as high as that for patients who didn’t have any AKI.
The negative predictive value of the new definition of AKI was 93%, and the positive predictive value was 34%.
This study was supported in part by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Center for Research Resources. No financial conflicts of interest were reported.
The newly proposed consensus definition of acute kidney injury in patients with cirrhosis accurately predicts 30-day mortality and other adverse outcomes in this patient population much better than the current, more rigid definition would, according to a report in the December issue of Gastroenterology (doi:10.1053/j.gastro.2013.08.051).
In what they described as the largest prospective study of this topic to date, researchers found that the recently proposed, broader redefinition of acute kidney injury (AKI) correctly identified which patients were likely to die, develop severe complications such as organ failure, or require longer hospitalization, even when the AKI was transient and resolved completely after treatment.
Courtesy American Gastroenterological Association
More than half of the patients in this study who had episodes of AKI according to the new definition did not meet the criteria of the old definition. So using the new definition will help identify these high-risk patients at an earlier stage of renal dysfunction, "well before the stringent diagnostic criteria of [the old definition] are reached," when they will have a better treatment response, said Dr. Florence Wong of the division of gastroenterology, University of Toronto, and her associates.
The old definition of AKI required the presence of hepatorenal syndrome, with a serum creatinine level of greater than 2.5 mg/dL. This meant that patients with less severe renal dysfunction didn’t qualify and weren’t treated. But emerging evidence indicates that even mild degrees of renal dysfunction signal a poor prognosis, and that serum creatinine alone doesn’t accurately reflect renal dysfunction in advanced cirrhosis.
So the International Ascites Club and the Acute Dialysis Quality Initiative (ADQI) group proposed that acute kidney injury in cirrhosis should be redefined as an increase in serum creatinine level of 0.3 mg/dL or greater within 48 hours, or a 50% increase in serum creatinine level from a stable baseline reading within the previous 6 months, regardless of final serum creatinine level.
Dr. Wong and her colleagues assessed the new definition in a cohort of 337 cirrhotic patients treated during a 2-year period at 12 North American medical centers who were admitted with a bacterial infection (287 subjects) or who developed a bacterial infection during hospitalization (50 subjects). The most common infections were urinary tract infection (27% of patients), spontaneous bacterial peritonitis (21%), skin infection (14%), pneumonia (10%), and spontaneous bacteremia with no clear source of infection (9%).
Approximately half of these patients (49%) developed at least one episode of AKI during hospitalization. The 30-day mortality was significantly higher for those who developed AKI according to the new definition (34% mortality) than in those who did not (7% mortality), the investigators said.
Most patients who developed AKI had only a transient case, and their renal function completely recovered. Yet their subsequent mortality within 30 days was twice as high as that for patients who didn’t have any AKI.
The negative predictive value of the new definition of AKI was 93%, and the positive predictive value was 34%.
This study was supported in part by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Center for Research Resources. No financial conflicts of interest were reported.
FROM GASTROENTEROLOGY
Major finding: 30-day mortality was significantly higher for those who developed acute kidney injury according to a new definition (34% mortality) than in those who did not (7% mortality).
Data source: A cohort study of 337 inpatients at 12 North American medical centers who had cirrhosis and a bacterial infection, half of whom developed AKI.
Disclosures: This study was supported in part by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Center for Research Resources. No financial conflicts of interest were reported.