User login
Trichodysplasia Spinulosa in the Setting of Colon Cancer
Case Report
An 82-year-old woman presented to the clinic with a rash on the face that had been present for a few months. She denied any treatment or prior occurrence. Her medical history was remarkable for non-Hodgkin lymphoma that had been successfully treated with chemotherapy 4 years prior. Additionally, she recently had been diagnosed with stage IV colon cancer. She reported that surgery had been scheduled and she would start adjuvant chemotherapy soon after.
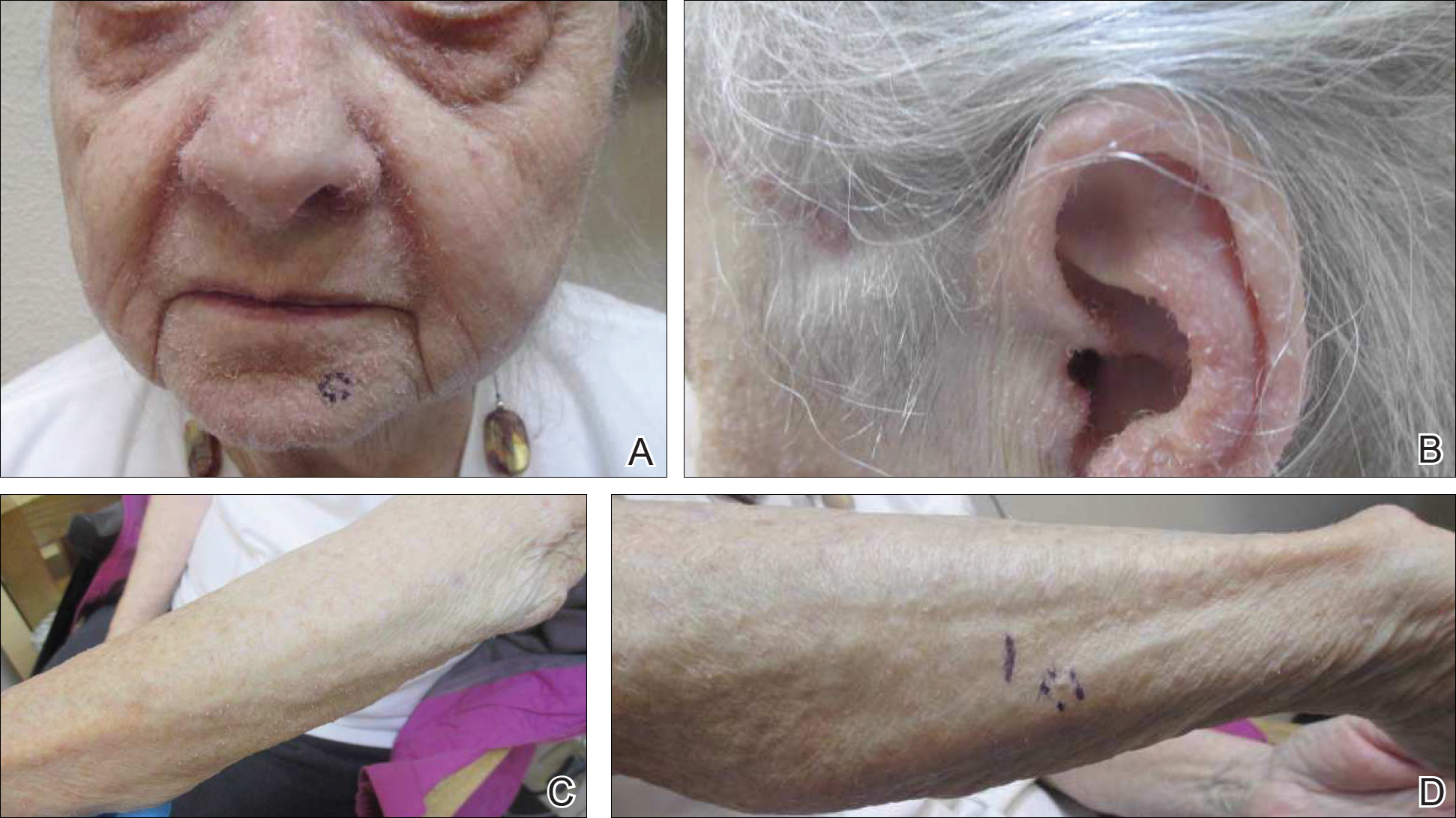
On physical examination she exhibited perioral and perinasal erythematous papules with sparing of the vermilion border. A diagnosis of perioral dermatitis was made, and she was started on topical metronidazole. At 1-month follow-up, her condition had slightly worsened and she was subsequently started on doxycycline. When she returned to the clinic again the following month, physical examination revealed agminated folliculocentric papules with central spicules on the face, nose, ears, upper extremities (Figure 1), and trunk. The differential diagnosis included multiple minute digitate hyperkeratosis, spiculosis of multiple myeloma, and trichodysplasia spinulosa (TS).
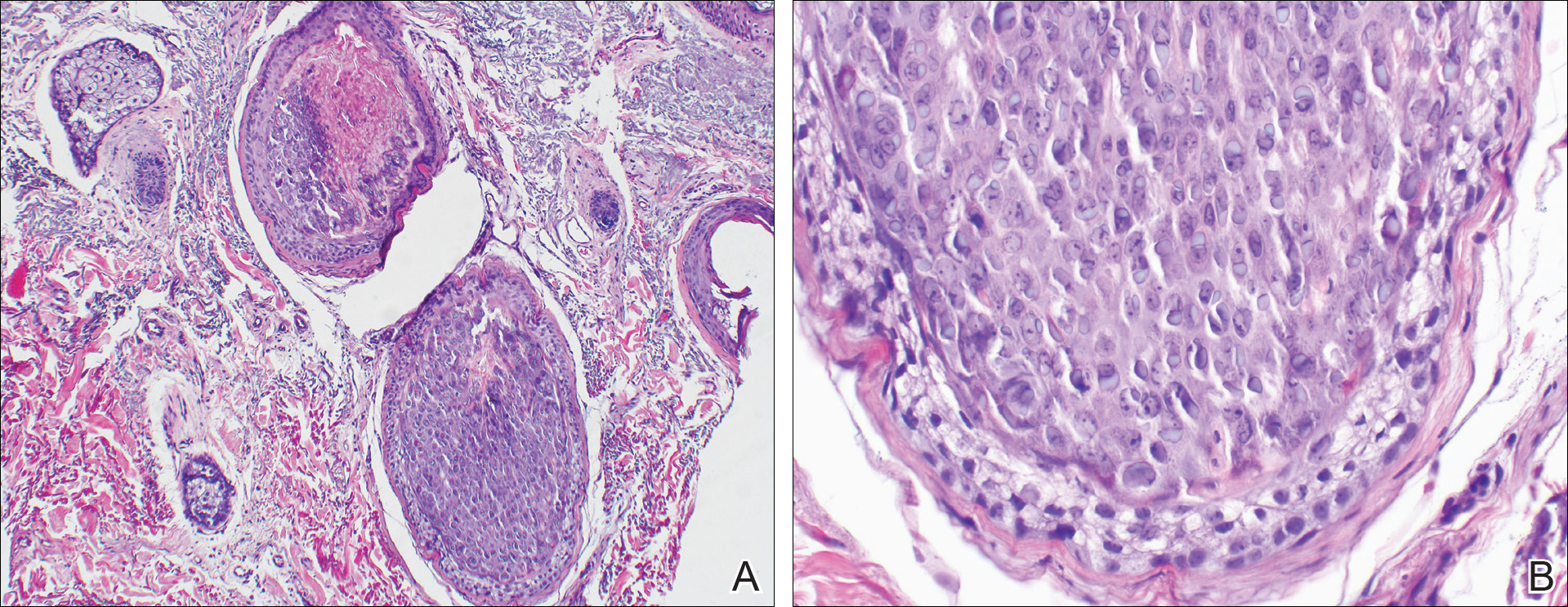
A punch biopsy of 2 separate papules on the face and upper extremity revealed dilated follicles with enlarged trichohyalin granules and dyskeratosis (Figure 2), consistent with TS. Additional testing such as electron microscopy or polymerase chain reaction was not performed to keep the patient’s medical costs down; also, the strong clinical and histopathologic evidence did not warrant further testing.
The plan was to start split-face treatment with topical acyclovir and a topical retinoid to see which agent was more effective, but the patient declined until her chemotherapy regimen had concluded. Unfortunately, the patient died 3 months later due to colon cancer.
Comment
History and Presentation
Trichodysplasia spinulosa was first recognized as hairlike hyperkeratosis.1 The name by which it is currently known was later championed by Haycox et al.2 They reported a case of a 44-year-old man who underwent a combined renal-pancreas transplant and while taking immunosuppressive medication developed erythematous papules with follicular spinous processes and progressive alopecia.2 Other synonymous terms used for this condition include pilomatrix dysplasia, cyclosporine-induced folliculodystrophy, virus-associated trichodysplasia,3 and follicular dystrophy of immunosuppression.4 Trichodysplasia spinulosa can affect both adult and pediatric immunocompromised patients, including organ transplant recipients on immunosuppressants and cancer patients on chemotherapy.3 The condition also has been reported to precede the recurrence of lymphoma.5
Etiology
The connection of TS with a viral etiology was first demonstrated in 1999, and subsequently it was confirmed to be a polyomavirus.2 The family name of Polyomaviridae possesses a Greek derivation with poly- meaning many and -oma meaning cancer.3 This name was given after the polyomavirus induced multiple tumors in mice.3,6 This viral family consists of multiple naked viruses with a surrounding icosahedral capsid containing 3 structural proteins known as VP1, VP2, and VP3. Their life cycle is characterized by early and late phases with respective early and late protein formation.3
Polyomavirus infections maintain an asymptomatic and latent course in immunocompetent patients.7 The prevalence and manifestation of these viruses change when the host’s immune system is altered. The first identified JC virus and BK virus of the same family have been found at increased frequencies in blood and lymphoid tissue during host immunosuppression.6 Moreover, the Merkel cell polyomavirus detected in Merkel cell carcinoma is well documented in the dermatologic literature.6,8
A specific polyomavirus has been implicated in the majority of TS cases and has subsequently received the name of TS polyomavirus.9 As a polyomavirus, it similarly produces capsid antigens and large/small T antigens. Among the viral protein antigens produced, the large tumor or LT antigen represents one of the most potent viral proteins. It has been postulated to inhibit the retinoblastoma family of proteins, leading to increased inner root sheath cells that allow for further viral replication.9,10
The disease presents with folliculocentric papules localized mainly on the central face and ears, which grow central keratin spines or spicules that can become 1 to 3 mm in length. Coinciding alopecia and madarosis also may be present.9
Diagnosis
Histologic examination reveals abnormal follicular maturation and distension. Additionally, increased proliferation and amount of trichohyalin is seen within the inner root sheath cells. Further testing via viral culture, polymerase chain reaction, electron microscopy, or immunohistochemical stains can confirm the diagnosis. Such testing may not be warranted in all cases given that classic clinical findings coupled with routine histopathology staining can provide enough evidence.10,11
Management
Currently, a universal successful treatment for TS does not exist. There have been anecdotal successes reported with topical medications such as cidofovir ointment 1%, acyclovir combined with 2-deoxy-D-glucose and epigallocatechin, corticosteroids, topical tacrolimus, topical retinoids, and imiquimod. Additionally, success has been seen with oral minocycline, oral retinoids, valacyclovir, and valganciclovir, with the latter showing the best results. Patients also have shown improvement after modifying their immunosuppressive treatment regimen.10,12
Conclusion
Given the previously published case of TS preceding the recurrence of lymphoma,5 we notified our patient’s oncologist of this potential risk. Her history of lymphoma and immunosuppressive treatment 4 years prior may represent the etiology of the cutaneous presentation; however, the TS with concurrent colon cancer presented prior to starting immunosuppressive therapy, suggesting that it also may have been a paraneoplastic process and not just a sign of immunosuppression. Therefore, we recommend that patients who present with TS should be evaluated for underlying malignancy if not already diagnosed.
- Linke M, Geraud C, Sauer C, et al. Follicular erythematous papules with keratotic spicules. Acta Derm Venereol . 2014;94:493-494.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases [published online September 12, 2011]. Patholog Res Int. 2011;2011:123491.
- Matthews MR, Wang RC, Reddick RL, et al. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa–associated human polyomavirus. J Cutan Pathol. 2011;38:420-431.
- Osswald SS, Kulick KB, Tomaszewski MM, et al. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol. 2007;34:721-725.
- Dalianis T, Hirsch HH. Human polyomavirus in disease and cancer. Virology. 2013;437:63-72.
- Tsuzuki S, Fukumoto H, Mine S, et al. Detection of trichodysplasia spinulosa–associated polyomavirus in a fatal case of myocarditis in a seven-month-old girl. Int J Clin Exp Pathol. 2014;7:5308-5312.
- Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa–associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infect Dis. 2012;12:383.
- Van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- Krichhof MG, Shojania K, Hull MW, et al. Trichodysplasia spinulosa: rare presentation of polyomavirus infection in immunocompromised patients. J Cutan Med Surg. 2014;18:430-435.
- Rianthavorn P, Posuwan N, Payungporn S, et al. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J Exp Med. 2012;228:197-204.
- Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Arch Dermatol. 2012;148:219-223.
Case Report
An 82-year-old woman presented to the clinic with a rash on the face that had been present for a few months. She denied any treatment or prior occurrence. Her medical history was remarkable for non-Hodgkin lymphoma that had been successfully treated with chemotherapy 4 years prior. Additionally, she recently had been diagnosed with stage IV colon cancer. She reported that surgery had been scheduled and she would start adjuvant chemotherapy soon after.
On physical examination she exhibited perioral and perinasal erythematous papules with sparing of the vermilion border. A diagnosis of perioral dermatitis was made, and she was started on topical metronidazole. At 1-month follow-up, her condition had slightly worsened and she was subsequently started on doxycycline. When she returned to the clinic again the following month, physical examination revealed agminated folliculocentric papules with central spicules on the face, nose, ears, upper extremities (Figure 1), and trunk. The differential diagnosis included multiple minute digitate hyperkeratosis, spiculosis of multiple myeloma, and trichodysplasia spinulosa (TS).
A punch biopsy of 2 separate papules on the face and upper extremity revealed dilated follicles with enlarged trichohyalin granules and dyskeratosis (Figure 2), consistent with TS. Additional testing such as electron microscopy or polymerase chain reaction was not performed to keep the patient’s medical costs down; also, the strong clinical and histopathologic evidence did not warrant further testing.
The plan was to start split-face treatment with topical acyclovir and a topical retinoid to see which agent was more effective, but the patient declined until her chemotherapy regimen had concluded. Unfortunately, the patient died 3 months later due to colon cancer.
Comment
History and Presentation
Trichodysplasia spinulosa was first recognized as hairlike hyperkeratosis.1 The name by which it is currently known was later championed by Haycox et al.2 They reported a case of a 44-year-old man who underwent a combined renal-pancreas transplant and while taking immunosuppressive medication developed erythematous papules with follicular spinous processes and progressive alopecia.2 Other synonymous terms used for this condition include pilomatrix dysplasia, cyclosporine-induced folliculodystrophy, virus-associated trichodysplasia,3 and follicular dystrophy of immunosuppression.4 Trichodysplasia spinulosa can affect both adult and pediatric immunocompromised patients, including organ transplant recipients on immunosuppressants and cancer patients on chemotherapy.3 The condition also has been reported to precede the recurrence of lymphoma.5
Etiology
The connection of TS with a viral etiology was first demonstrated in 1999, and subsequently it was confirmed to be a polyomavirus.2 The family name of Polyomaviridae possesses a Greek derivation with poly- meaning many and -oma meaning cancer.3 This name was given after the polyomavirus induced multiple tumors in mice.3,6 This viral family consists of multiple naked viruses with a surrounding icosahedral capsid containing 3 structural proteins known as VP1, VP2, and VP3. Their life cycle is characterized by early and late phases with respective early and late protein formation.3
Polyomavirus infections maintain an asymptomatic and latent course in immunocompetent patients.7 The prevalence and manifestation of these viruses change when the host’s immune system is altered. The first identified JC virus and BK virus of the same family have been found at increased frequencies in blood and lymphoid tissue during host immunosuppression.6 Moreover, the Merkel cell polyomavirus detected in Merkel cell carcinoma is well documented in the dermatologic literature.6,8
A specific polyomavirus has been implicated in the majority of TS cases and has subsequently received the name of TS polyomavirus.9 As a polyomavirus, it similarly produces capsid antigens and large/small T antigens. Among the viral protein antigens produced, the large tumor or LT antigen represents one of the most potent viral proteins. It has been postulated to inhibit the retinoblastoma family of proteins, leading to increased inner root sheath cells that allow for further viral replication.9,10
The disease presents with folliculocentric papules localized mainly on the central face and ears, which grow central keratin spines or spicules that can become 1 to 3 mm in length. Coinciding alopecia and madarosis also may be present.9
Diagnosis
Histologic examination reveals abnormal follicular maturation and distension. Additionally, increased proliferation and amount of trichohyalin is seen within the inner root sheath cells. Further testing via viral culture, polymerase chain reaction, electron microscopy, or immunohistochemical stains can confirm the diagnosis. Such testing may not be warranted in all cases given that classic clinical findings coupled with routine histopathology staining can provide enough evidence.10,11
Management
Currently, a universal successful treatment for TS does not exist. There have been anecdotal successes reported with topical medications such as cidofovir ointment 1%, acyclovir combined with 2-deoxy-D-glucose and epigallocatechin, corticosteroids, topical tacrolimus, topical retinoids, and imiquimod. Additionally, success has been seen with oral minocycline, oral retinoids, valacyclovir, and valganciclovir, with the latter showing the best results. Patients also have shown improvement after modifying their immunosuppressive treatment regimen.10,12
Conclusion
Given the previously published case of TS preceding the recurrence of lymphoma,5 we notified our patient’s oncologist of this potential risk. Her history of lymphoma and immunosuppressive treatment 4 years prior may represent the etiology of the cutaneous presentation; however, the TS with concurrent colon cancer presented prior to starting immunosuppressive therapy, suggesting that it also may have been a paraneoplastic process and not just a sign of immunosuppression. Therefore, we recommend that patients who present with TS should be evaluated for underlying malignancy if not already diagnosed.
Case Report
An 82-year-old woman presented to the clinic with a rash on the face that had been present for a few months. She denied any treatment or prior occurrence. Her medical history was remarkable for non-Hodgkin lymphoma that had been successfully treated with chemotherapy 4 years prior. Additionally, she recently had been diagnosed with stage IV colon cancer. She reported that surgery had been scheduled and she would start adjuvant chemotherapy soon after.
On physical examination she exhibited perioral and perinasal erythematous papules with sparing of the vermilion border. A diagnosis of perioral dermatitis was made, and she was started on topical metronidazole. At 1-month follow-up, her condition had slightly worsened and she was subsequently started on doxycycline. When she returned to the clinic again the following month, physical examination revealed agminated folliculocentric papules with central spicules on the face, nose, ears, upper extremities (Figure 1), and trunk. The differential diagnosis included multiple minute digitate hyperkeratosis, spiculosis of multiple myeloma, and trichodysplasia spinulosa (TS).
A punch biopsy of 2 separate papules on the face and upper extremity revealed dilated follicles with enlarged trichohyalin granules and dyskeratosis (Figure 2), consistent with TS. Additional testing such as electron microscopy or polymerase chain reaction was not performed to keep the patient’s medical costs down; also, the strong clinical and histopathologic evidence did not warrant further testing.
The plan was to start split-face treatment with topical acyclovir and a topical retinoid to see which agent was more effective, but the patient declined until her chemotherapy regimen had concluded. Unfortunately, the patient died 3 months later due to colon cancer.
Comment
History and Presentation
Trichodysplasia spinulosa was first recognized as hairlike hyperkeratosis.1 The name by which it is currently known was later championed by Haycox et al.2 They reported a case of a 44-year-old man who underwent a combined renal-pancreas transplant and while taking immunosuppressive medication developed erythematous papules with follicular spinous processes and progressive alopecia.2 Other synonymous terms used for this condition include pilomatrix dysplasia, cyclosporine-induced folliculodystrophy, virus-associated trichodysplasia,3 and follicular dystrophy of immunosuppression.4 Trichodysplasia spinulosa can affect both adult and pediatric immunocompromised patients, including organ transplant recipients on immunosuppressants and cancer patients on chemotherapy.3 The condition also has been reported to precede the recurrence of lymphoma.5
Etiology
The connection of TS with a viral etiology was first demonstrated in 1999, and subsequently it was confirmed to be a polyomavirus.2 The family name of Polyomaviridae possesses a Greek derivation with poly- meaning many and -oma meaning cancer.3 This name was given after the polyomavirus induced multiple tumors in mice.3,6 This viral family consists of multiple naked viruses with a surrounding icosahedral capsid containing 3 structural proteins known as VP1, VP2, and VP3. Their life cycle is characterized by early and late phases with respective early and late protein formation.3
Polyomavirus infections maintain an asymptomatic and latent course in immunocompetent patients.7 The prevalence and manifestation of these viruses change when the host’s immune system is altered. The first identified JC virus and BK virus of the same family have been found at increased frequencies in blood and lymphoid tissue during host immunosuppression.6 Moreover, the Merkel cell polyomavirus detected in Merkel cell carcinoma is well documented in the dermatologic literature.6,8
A specific polyomavirus has been implicated in the majority of TS cases and has subsequently received the name of TS polyomavirus.9 As a polyomavirus, it similarly produces capsid antigens and large/small T antigens. Among the viral protein antigens produced, the large tumor or LT antigen represents one of the most potent viral proteins. It has been postulated to inhibit the retinoblastoma family of proteins, leading to increased inner root sheath cells that allow for further viral replication.9,10
The disease presents with folliculocentric papules localized mainly on the central face and ears, which grow central keratin spines or spicules that can become 1 to 3 mm in length. Coinciding alopecia and madarosis also may be present.9
Diagnosis
Histologic examination reveals abnormal follicular maturation and distension. Additionally, increased proliferation and amount of trichohyalin is seen within the inner root sheath cells. Further testing via viral culture, polymerase chain reaction, electron microscopy, or immunohistochemical stains can confirm the diagnosis. Such testing may not be warranted in all cases given that classic clinical findings coupled with routine histopathology staining can provide enough evidence.10,11
Management
Currently, a universal successful treatment for TS does not exist. There have been anecdotal successes reported with topical medications such as cidofovir ointment 1%, acyclovir combined with 2-deoxy-D-glucose and epigallocatechin, corticosteroids, topical tacrolimus, topical retinoids, and imiquimod. Additionally, success has been seen with oral minocycline, oral retinoids, valacyclovir, and valganciclovir, with the latter showing the best results. Patients also have shown improvement after modifying their immunosuppressive treatment regimen.10,12
Conclusion
Given the previously published case of TS preceding the recurrence of lymphoma,5 we notified our patient’s oncologist of this potential risk. Her history of lymphoma and immunosuppressive treatment 4 years prior may represent the etiology of the cutaneous presentation; however, the TS with concurrent colon cancer presented prior to starting immunosuppressive therapy, suggesting that it also may have been a paraneoplastic process and not just a sign of immunosuppression. Therefore, we recommend that patients who present with TS should be evaluated for underlying malignancy if not already diagnosed.
- Linke M, Geraud C, Sauer C, et al. Follicular erythematous papules with keratotic spicules. Acta Derm Venereol . 2014;94:493-494.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases [published online September 12, 2011]. Patholog Res Int. 2011;2011:123491.
- Matthews MR, Wang RC, Reddick RL, et al. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa–associated human polyomavirus. J Cutan Pathol. 2011;38:420-431.
- Osswald SS, Kulick KB, Tomaszewski MM, et al. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol. 2007;34:721-725.
- Dalianis T, Hirsch HH. Human polyomavirus in disease and cancer. Virology. 2013;437:63-72.
- Tsuzuki S, Fukumoto H, Mine S, et al. Detection of trichodysplasia spinulosa–associated polyomavirus in a fatal case of myocarditis in a seven-month-old girl. Int J Clin Exp Pathol. 2014;7:5308-5312.
- Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa–associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infect Dis. 2012;12:383.
- Van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- Krichhof MG, Shojania K, Hull MW, et al. Trichodysplasia spinulosa: rare presentation of polyomavirus infection in immunocompromised patients. J Cutan Med Surg. 2014;18:430-435.
- Rianthavorn P, Posuwan N, Payungporn S, et al. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J Exp Med. 2012;228:197-204.
- Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Arch Dermatol. 2012;148:219-223.
- Linke M, Geraud C, Sauer C, et al. Follicular erythematous papules with keratotic spicules. Acta Derm Venereol . 2014;94:493-494.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases [published online September 12, 2011]. Patholog Res Int. 2011;2011:123491.
- Matthews MR, Wang RC, Reddick RL, et al. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa–associated human polyomavirus. J Cutan Pathol. 2011;38:420-431.
- Osswald SS, Kulick KB, Tomaszewski MM, et al. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol. 2007;34:721-725.
- Dalianis T, Hirsch HH. Human polyomavirus in disease and cancer. Virology. 2013;437:63-72.
- Tsuzuki S, Fukumoto H, Mine S, et al. Detection of trichodysplasia spinulosa–associated polyomavirus in a fatal case of myocarditis in a seven-month-old girl. Int J Clin Exp Pathol. 2014;7:5308-5312.
- Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa–associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infect Dis. 2012;12:383.
- Van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- Krichhof MG, Shojania K, Hull MW, et al. Trichodysplasia spinulosa: rare presentation of polyomavirus infection in immunocompromised patients. J Cutan Med Surg. 2014;18:430-435.
- Rianthavorn P, Posuwan N, Payungporn S, et al. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J Exp Med. 2012;228:197-204.
- Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Arch Dermatol. 2012;148:219-223.
Practice Points
- Rashes have a life span and can evolve with time.
- If apparent straightforward conditions do not appear to respond to standard therapy, start to think outside the box for underlying potential causes.

Identifying and Treating CNS Vasculitis
Often difficult to diagnose, CNS vasculitis is a serious condition that can be treated effectively if addressed early.
HILTON HEAD, SC—Vasculitis is a general term for a group of uncommon diseases involving inflammation of blood vessels, which can lead to the occlusion or destruction of the vessels and to ischemia of the tissues supplied by those vessels. CNS vasculitis can be a primary disease or occur secondary to infections or as part of a systemic vasculitis or systemic inflammatory (eg, rheumatologic) disease. Without prompt diagnosis and treatment, patients are at high risk of permanent neurologic disability or death, according to a presentation at the 41st Annual Contemporary Clinical Neurology Symposium.

If diagnosed and treated early, CNS vasculitis has a good prognosis, but delayed intervention can result in severe morbidity or mortality, said Siddharama Pawate, MD, Associate Professor of Neurology at the Vanderbilt University Medical Center in Nashville. “You have to do extensive workups sometimes before you can make a diagnosis,” he said, citing the need for neurology, rheumatology, and infectious disease input. “Investigations include serologic testing, CSF analysis, MRI, and brain biopsy.”
Rare But With a High Risk of Morbidity
The 2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitis adopted an extensive list of names for the numerous manifestations of vasculitis. These include the following:
• Large-vessel vasculitis (eg, giant cell arteritis [GCA])
• Medium-vessel vasculitis (eg, polyarteritis nodosa)
• Small-vessel vasculitis (eg, microscopic polyangiitis and granulomatosis with polyangiitis [Wegener’s granulomatosis])
• Variable-vessel vasculitis (eg, Behçet’s disease)
• Single-organ vasculitis (eg, primary CNS vasculitis)
• Vasculitis associated with systemic disease (eg, rheumatoid vasculitis)
• Vasculitis associated with probable etiology (eg, hepatitis B virus-associated or cancer-associated vasculitis).
CNS vasculitis may be grouped into two larger categories—infectious CNS vasculitis and immune-mediated CNS vasculitis. Dr. Pawate provided a brief overview of infectious causes of vasculitis including bacteria, mycobacteria, varicella-zoster, fungi, and neurocysticercosis, but devoted the main part of his presentation to the latter of the two categories, including a focus on primary CNS vasculitis (PCNSV), which is also known as primary angiitis of the CNS (PACNS). Dr. Pawate offered an analysis of what is entailed in the recognition, diagnosis, and treatment of CNS vasculitis in general and its immune-mediated variants more specifically.
CNS vasculitis may occur as part of a broader systemic vasculitis. GCA is a medical emergency that can cause permanent visual loss if not diagnosed and treated early. Vision loss occurs most often due to anterior ischemic optic neuropathy but also central retinal artery occlusion and posterior ischemic optic neuropathy. The American College of Rheumatology criteria for GCA diagnosis include three of the five following core features: age 50 or older at onset, new-onset headaches, temporal artery abnormality, elevated erythrocyte sedimentation rate of at least 50 mm/h, and abnormal temporal artery biopsy. High-dose steroids should be started if there is suspicion, without waiting for biopsy results. Recently, the anti-IL6 monoclonal antibody tocilizumab was approved by the FDA as a treatment for GCA. Granulomatosis with polyangiitis most often causes peripheral neuropathy, but can cause a small or medium vessel CNS vasculitis. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) is characterized by the triad of asthma, hypereosinophilia, and necrotizing vasculitis, usually in that order. CNS involvement is common as part of the vasculitis. CNS vasculitis may also be seen in Behçet’s disease manifesting as dural sinus thrombosis and arterial occlusion or aneurysm.
In systemic lupus erythematosus, vasculitis has a prevalence ranging between 11% and 36%, but CNS involvement is much less common. Case reports of CNS vasculitis in patients with rheumatoid arthritis are rare.
Primary CNS Vasculitis
Primary CNS vasculitis causes inflammation of mostly small- to medium-sized leptomeningeal and parenchymal arterial vessels. It is rare—a 2007 retrospective analysis by Salvarani et al of 101 cases estimated the average annual incidence to be 2.4 cases per one million person-years. Onset tends to occur in middle age, with a median age at diagnosis of 50 and a similar frequency in males and females.
Clinical presentation of PCNSV can be acute, subacute, chronic, or recurrent. Depending on the area of the brain or spinal cord that is affected, patients can present with a wide variety of neurologic complaints. Headache is the most common symptom—found in 50% to 78% of patients—followed by altered cognition and persistent neurologic deficits. Stroke and transient ischemic attack involving multiple vascular areas occur in 30% to 50% of patients.
The diagnostic gold standard for PCNSV is brain parenchymal/leptomeningeal biopsy. The most common diagnoses are granulomatous angiitis of the CNS (58%), lymphocytic PACNS (28%), and necrotizing vasculitis (14%). More than 90% of patients have abnormalities on MRI, and the most common imaging findings are cortical and subcortical infarcts, leptomeningeal enhancement, intracranial hemorrhage, and areas of increased signal on FLAIR and T2-weighted sequences. It has been increasingly recognized that differential diagnosis for PCNSV should include reversible cerebral vasoconstriction syndrome (RCVS), which is characterized by reversible multifocal narrowing of the cerebral arteries—typically preceded by sudden, severe thunderclap headaches with or without associated neurologic deficits. RCVS typically resolves in approximately 12 weeks.
Regarding treatment for PCNSV, Dr. Pawate recommended starting with high-dose steroids—eg, IV methylprednisolone, 1,000 mg daily for five days, with a prolonged oral prednisone taper starting at 1 mg/kg/day—and six to nine months of IV cyclophosphamide pulse therapy, 500–750 mg/m2 every two to four weeks for more severe cases. Case studies have shown rituximab and mycophenolate to be effective. “I had one patient initially on cyclophosphamide for six months who we [then] maintained on mycophenolate for five years and then stopped immunosuppression,” Dr. Pawate said. “She did quite well.”
—Fred Balzac
Suggested Reading
Abdel Razek AA, Alvarez H, Bagg S, et al. Imaging spectrum of CNS vasculitis. Radiographics. 2014;34(4):873-894.
Chow FC, Marra CM, Cho TA. Cerebrovascular disease in central nervous system infections. Semin Neurol. 2011;31(3):286-306.
Hajj-Ali RA, Calabrese LH. Diagnosis and classification of central nervous system vasculitis. J Autoimmun. 2014;48-49:149-152.
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
Salvarani C, Brown RD Jr, Calamia KT, et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol. 2007;62(5):442-451.
Scolding NJ. Central nervous system vasculitis. Semin Immunopathol. 2009;31(4):527-536.
Often difficult to diagnose, CNS vasculitis is a serious condition that can be treated effectively if addressed early.
Often difficult to diagnose, CNS vasculitis is a serious condition that can be treated effectively if addressed early.
HILTON HEAD, SC—Vasculitis is a general term for a group of uncommon diseases involving inflammation of blood vessels, which can lead to the occlusion or destruction of the vessels and to ischemia of the tissues supplied by those vessels. CNS vasculitis can be a primary disease or occur secondary to infections or as part of a systemic vasculitis or systemic inflammatory (eg, rheumatologic) disease. Without prompt diagnosis and treatment, patients are at high risk of permanent neurologic disability or death, according to a presentation at the 41st Annual Contemporary Clinical Neurology Symposium.
If diagnosed and treated early, CNS vasculitis has a good prognosis, but delayed intervention can result in severe morbidity or mortality, said Siddharama Pawate, MD, Associate Professor of Neurology at the Vanderbilt University Medical Center in Nashville. “You have to do extensive workups sometimes before you can make a diagnosis,” he said, citing the need for neurology, rheumatology, and infectious disease input. “Investigations include serologic testing, CSF analysis, MRI, and brain biopsy.”
Rare But With a High Risk of Morbidity
The 2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitis adopted an extensive list of names for the numerous manifestations of vasculitis. These include the following:
• Large-vessel vasculitis (eg, giant cell arteritis [GCA])
• Medium-vessel vasculitis (eg, polyarteritis nodosa)
• Small-vessel vasculitis (eg, microscopic polyangiitis and granulomatosis with polyangiitis [Wegener’s granulomatosis])
• Variable-vessel vasculitis (eg, Behçet’s disease)
• Single-organ vasculitis (eg, primary CNS vasculitis)
• Vasculitis associated with systemic disease (eg, rheumatoid vasculitis)
• Vasculitis associated with probable etiology (eg, hepatitis B virus-associated or cancer-associated vasculitis).
CNS vasculitis may be grouped into two larger categories—infectious CNS vasculitis and immune-mediated CNS vasculitis. Dr. Pawate provided a brief overview of infectious causes of vasculitis including bacteria, mycobacteria, varicella-zoster, fungi, and neurocysticercosis, but devoted the main part of his presentation to the latter of the two categories, including a focus on primary CNS vasculitis (PCNSV), which is also known as primary angiitis of the CNS (PACNS). Dr. Pawate offered an analysis of what is entailed in the recognition, diagnosis, and treatment of CNS vasculitis in general and its immune-mediated variants more specifically.
CNS vasculitis may occur as part of a broader systemic vasculitis. GCA is a medical emergency that can cause permanent visual loss if not diagnosed and treated early. Vision loss occurs most often due to anterior ischemic optic neuropathy but also central retinal artery occlusion and posterior ischemic optic neuropathy. The American College of Rheumatology criteria for GCA diagnosis include three of the five following core features: age 50 or older at onset, new-onset headaches, temporal artery abnormality, elevated erythrocyte sedimentation rate of at least 50 mm/h, and abnormal temporal artery biopsy. High-dose steroids should be started if there is suspicion, without waiting for biopsy results. Recently, the anti-IL6 monoclonal antibody tocilizumab was approved by the FDA as a treatment for GCA. Granulomatosis with polyangiitis most often causes peripheral neuropathy, but can cause a small or medium vessel CNS vasculitis. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) is characterized by the triad of asthma, hypereosinophilia, and necrotizing vasculitis, usually in that order. CNS involvement is common as part of the vasculitis. CNS vasculitis may also be seen in Behçet’s disease manifesting as dural sinus thrombosis and arterial occlusion or aneurysm.
In systemic lupus erythematosus, vasculitis has a prevalence ranging between 11% and 36%, but CNS involvement is much less common. Case reports of CNS vasculitis in patients with rheumatoid arthritis are rare.
Primary CNS Vasculitis
Primary CNS vasculitis causes inflammation of mostly small- to medium-sized leptomeningeal and parenchymal arterial vessels. It is rare—a 2007 retrospective analysis by Salvarani et al of 101 cases estimated the average annual incidence to be 2.4 cases per one million person-years. Onset tends to occur in middle age, with a median age at diagnosis of 50 and a similar frequency in males and females.
Clinical presentation of PCNSV can be acute, subacute, chronic, or recurrent. Depending on the area of the brain or spinal cord that is affected, patients can present with a wide variety of neurologic complaints. Headache is the most common symptom—found in 50% to 78% of patients—followed by altered cognition and persistent neurologic deficits. Stroke and transient ischemic attack involving multiple vascular areas occur in 30% to 50% of patients.
The diagnostic gold standard for PCNSV is brain parenchymal/leptomeningeal biopsy. The most common diagnoses are granulomatous angiitis of the CNS (58%), lymphocytic PACNS (28%), and necrotizing vasculitis (14%). More than 90% of patients have abnormalities on MRI, and the most common imaging findings are cortical and subcortical infarcts, leptomeningeal enhancement, intracranial hemorrhage, and areas of increased signal on FLAIR and T2-weighted sequences. It has been increasingly recognized that differential diagnosis for PCNSV should include reversible cerebral vasoconstriction syndrome (RCVS), which is characterized by reversible multifocal narrowing of the cerebral arteries—typically preceded by sudden, severe thunderclap headaches with or without associated neurologic deficits. RCVS typically resolves in approximately 12 weeks.
Regarding treatment for PCNSV, Dr. Pawate recommended starting with high-dose steroids—eg, IV methylprednisolone, 1,000 mg daily for five days, with a prolonged oral prednisone taper starting at 1 mg/kg/day—and six to nine months of IV cyclophosphamide pulse therapy, 500–750 mg/m2 every two to four weeks for more severe cases. Case studies have shown rituximab and mycophenolate to be effective. “I had one patient initially on cyclophosphamide for six months who we [then] maintained on mycophenolate for five years and then stopped immunosuppression,” Dr. Pawate said. “She did quite well.”
—Fred Balzac
Suggested Reading
Abdel Razek AA, Alvarez H, Bagg S, et al. Imaging spectrum of CNS vasculitis. Radiographics. 2014;34(4):873-894.
Chow FC, Marra CM, Cho TA. Cerebrovascular disease in central nervous system infections. Semin Neurol. 2011;31(3):286-306.
Hajj-Ali RA, Calabrese LH. Diagnosis and classification of central nervous system vasculitis. J Autoimmun. 2014;48-49:149-152.
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
Salvarani C, Brown RD Jr, Calamia KT, et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol. 2007;62(5):442-451.
Scolding NJ. Central nervous system vasculitis. Semin Immunopathol. 2009;31(4):527-536.
HILTON HEAD, SC—Vasculitis is a general term for a group of uncommon diseases involving inflammation of blood vessels, which can lead to the occlusion or destruction of the vessels and to ischemia of the tissues supplied by those vessels. CNS vasculitis can be a primary disease or occur secondary to infections or as part of a systemic vasculitis or systemic inflammatory (eg, rheumatologic) disease. Without prompt diagnosis and treatment, patients are at high risk of permanent neurologic disability or death, according to a presentation at the 41st Annual Contemporary Clinical Neurology Symposium.
If diagnosed and treated early, CNS vasculitis has a good prognosis, but delayed intervention can result in severe morbidity or mortality, said Siddharama Pawate, MD, Associate Professor of Neurology at the Vanderbilt University Medical Center in Nashville. “You have to do extensive workups sometimes before you can make a diagnosis,” he said, citing the need for neurology, rheumatology, and infectious disease input. “Investigations include serologic testing, CSF analysis, MRI, and brain biopsy.”
Rare But With a High Risk of Morbidity
The 2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitis adopted an extensive list of names for the numerous manifestations of vasculitis. These include the following:
• Large-vessel vasculitis (eg, giant cell arteritis [GCA])
• Medium-vessel vasculitis (eg, polyarteritis nodosa)
• Small-vessel vasculitis (eg, microscopic polyangiitis and granulomatosis with polyangiitis [Wegener’s granulomatosis])
• Variable-vessel vasculitis (eg, Behçet’s disease)
• Single-organ vasculitis (eg, primary CNS vasculitis)
• Vasculitis associated with systemic disease (eg, rheumatoid vasculitis)
• Vasculitis associated with probable etiology (eg, hepatitis B virus-associated or cancer-associated vasculitis).
CNS vasculitis may be grouped into two larger categories—infectious CNS vasculitis and immune-mediated CNS vasculitis. Dr. Pawate provided a brief overview of infectious causes of vasculitis including bacteria, mycobacteria, varicella-zoster, fungi, and neurocysticercosis, but devoted the main part of his presentation to the latter of the two categories, including a focus on primary CNS vasculitis (PCNSV), which is also known as primary angiitis of the CNS (PACNS). Dr. Pawate offered an analysis of what is entailed in the recognition, diagnosis, and treatment of CNS vasculitis in general and its immune-mediated variants more specifically.
CNS vasculitis may occur as part of a broader systemic vasculitis. GCA is a medical emergency that can cause permanent visual loss if not diagnosed and treated early. Vision loss occurs most often due to anterior ischemic optic neuropathy but also central retinal artery occlusion and posterior ischemic optic neuropathy. The American College of Rheumatology criteria for GCA diagnosis include three of the five following core features: age 50 or older at onset, new-onset headaches, temporal artery abnormality, elevated erythrocyte sedimentation rate of at least 50 mm/h, and abnormal temporal artery biopsy. High-dose steroids should be started if there is suspicion, without waiting for biopsy results. Recently, the anti-IL6 monoclonal antibody tocilizumab was approved by the FDA as a treatment for GCA. Granulomatosis with polyangiitis most often causes peripheral neuropathy, but can cause a small or medium vessel CNS vasculitis. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) is characterized by the triad of asthma, hypereosinophilia, and necrotizing vasculitis, usually in that order. CNS involvement is common as part of the vasculitis. CNS vasculitis may also be seen in Behçet’s disease manifesting as dural sinus thrombosis and arterial occlusion or aneurysm.
In systemic lupus erythematosus, vasculitis has a prevalence ranging between 11% and 36%, but CNS involvement is much less common. Case reports of CNS vasculitis in patients with rheumatoid arthritis are rare.
Primary CNS Vasculitis
Primary CNS vasculitis causes inflammation of mostly small- to medium-sized leptomeningeal and parenchymal arterial vessels. It is rare—a 2007 retrospective analysis by Salvarani et al of 101 cases estimated the average annual incidence to be 2.4 cases per one million person-years. Onset tends to occur in middle age, with a median age at diagnosis of 50 and a similar frequency in males and females.
Clinical presentation of PCNSV can be acute, subacute, chronic, or recurrent. Depending on the area of the brain or spinal cord that is affected, patients can present with a wide variety of neurologic complaints. Headache is the most common symptom—found in 50% to 78% of patients—followed by altered cognition and persistent neurologic deficits. Stroke and transient ischemic attack involving multiple vascular areas occur in 30% to 50% of patients.
The diagnostic gold standard for PCNSV is brain parenchymal/leptomeningeal biopsy. The most common diagnoses are granulomatous angiitis of the CNS (58%), lymphocytic PACNS (28%), and necrotizing vasculitis (14%). More than 90% of patients have abnormalities on MRI, and the most common imaging findings are cortical and subcortical infarcts, leptomeningeal enhancement, intracranial hemorrhage, and areas of increased signal on FLAIR and T2-weighted sequences. It has been increasingly recognized that differential diagnosis for PCNSV should include reversible cerebral vasoconstriction syndrome (RCVS), which is characterized by reversible multifocal narrowing of the cerebral arteries—typically preceded by sudden, severe thunderclap headaches with or without associated neurologic deficits. RCVS typically resolves in approximately 12 weeks.
Regarding treatment for PCNSV, Dr. Pawate recommended starting with high-dose steroids—eg, IV methylprednisolone, 1,000 mg daily for five days, with a prolonged oral prednisone taper starting at 1 mg/kg/day—and six to nine months of IV cyclophosphamide pulse therapy, 500–750 mg/m2 every two to four weeks for more severe cases. Case studies have shown rituximab and mycophenolate to be effective. “I had one patient initially on cyclophosphamide for six months who we [then] maintained on mycophenolate for five years and then stopped immunosuppression,” Dr. Pawate said. “She did quite well.”
—Fred Balzac
Suggested Reading
Abdel Razek AA, Alvarez H, Bagg S, et al. Imaging spectrum of CNS vasculitis. Radiographics. 2014;34(4):873-894.
Chow FC, Marra CM, Cho TA. Cerebrovascular disease in central nervous system infections. Semin Neurol. 2011;31(3):286-306.
Hajj-Ali RA, Calabrese LH. Diagnosis and classification of central nervous system vasculitis. J Autoimmun. 2014;48-49:149-152.
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
Salvarani C, Brown RD Jr, Calamia KT, et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol. 2007;62(5):442-451.
Scolding NJ. Central nervous system vasculitis. Semin Immunopathol. 2009;31(4):527-536.
Diagnosis, Pathology, and Treatment of bvFTD Pose Challenges
Certain clinical features may indicate bvFTD, and off-label treatments may provide benefits.
HILTON HEAD, SC—Behavioral variant frontotemporal dementia (bvFTD), a clinically and pathologically heterogenous condition, can be difficult to distinguish from other forms of dementia or frontotemporal disease. “Although clinical symptoms vary based on which part of the brain is affected, there is a significant amount of overlap, with different pathologies causing the same type of syndrome,” said Richard Ryan Darby, MD, Assistant Professor of Neurology at Vanderbilt University Medical School in Nashville. “In a large proportion of patients, behavioral changes can lead to criminal behavior.” Future research examining brain lesion networks may shed light on this condition, said Dr. Darby at the 41st Annual Contemporary Clinical Neurology Symposium.

Clinical Features and Diagnosis
The incidence of bvFTD is equal to that of Alzheimer’s disease. However, patients with bvFTD tend to be younger: between the ages of 45 and 65. “Social and behavioral features predominate,” said Dr. Darby. “If a patient in this age range with no previous psychiatric history presents to you with a new psychiatric diagnosis such as schizophrenia or bipolar disease, this should raise your suspicion of bvFTD.” These psychiatric problems often cause considerable disruptions in patients’ lives. They often have problems at work and lose their source of income. “Patients often have no insight about their symptoms,” said Dr. Darby.
A differential diagnosis of bvFTD is possible in patients with three or more of the following six clinical features: socially inappropriate behavior (eg, eating from the trash or walking around naked at inappropriate times); lack of empathy; apathy; stereotyped or repetitive behavior (eg, saying things repeatedly, pacing); hyperorality (eg, eating uncontrollably, particularly sweet foods); and executive dysfunction, especially when memory is preserved.
“When you talk to caregivers, they often say, ‘This is not the person I married,’ or, ‘This is not my father. He seems like a different person,’” said Dr. Darby. An MRI or a PET scan showing changes in the frontotemporal lobes is a firm basis for a diagnosis of bvFTD, said Dr. Darby. Genetic testing for autosomal dominant mutation or pathology or an autopsy provides a definite diagnosis.
Pathology
Between 40% and 50% of patients with bvFTD have tau pathology, said Dr. Darby. This pathology includes the classic Pick body form of tau, tufted astrocytes (which are associated with clinical symptoms of progressive supranuclear palsy), and astrocytic plaques (which are associated with symptoms of corticobasal degeneration). Similarly, between 40% and 50% of patients with bvFTD have a TAR DNA-binding protein 43 (TDP-43) pathology. TDP-43 Type A pathology is associated with perirolandic seizures, Type B is associated with
Mutations in C9orf72 occur in 13% to 50% of patients with bvFTD who have genetic mutations. ALS and parkinsonism are common clinical presentations in patients with this genetic mutation. MAPT and GRN mutations are present in 5% to 20% of cases, and each can present clinically as parkinsonism.
Treatment
SSRIs, antipsychotics, anticonvulsants, and stimulants have been used to treat apathy, disinhibition, compulsive behaviors, agitation, and inappropriate behavior in patients with bvFTD. “There are no FDA-approved treatments for bvFTD, and the evidence for [these agents] has been mostly reported in case studies and small clinical trials,” said Dr. Darby. Among the SSRIs, paroxetine improved repetitive behavior in open-label trials, but not in a randomized controlled trial. Sertraline and citalopram have been studied in open-label trials, and trazodone was examined in a randomized controlled crossover trial involving 26 patients.
The FDA issued a black box warning against the use of atypical antipsychotics in patients with dementia because they entail a risk of cardiac- and infection-related mortality. “For patients with bvFTD tau pathology in particular, there is a risk of extrapyramidal adverse effects,” said Dr. Darby. A series of case reports of risperidone and aripiprazole provided evidence of symptom improvement, as did an open label study of olanzapine. Quetiapine improved agitation in a case series but failed to show benefit in a double-blind crossover trial of eight patients with FTD.
Case series have shown evidence that antiepileptic drugs (eg, valproic acid, topiramate, and carbamazepine) have a stabilizing effect. “Stimulants are tried in some patients, but should be used with caution,” said Dr. Darby.
Criminality
Between 37% and 57% of patients with bvFTD engage in criminal behavior. “Approximately 10% to 15% of the time, a patient’s getting in trouble with the law is the reason for the initial presentation,” said Dr. Darby. The types of crimes described in case reports include pedophilia, public masturbation, hit and run, traffic violations, and theft.
“Murder and violent crimes occur but are rare. Crimes committed by patients with bvFTD are usually reactive,” said Dr. Darby. “When asked, they can tell you whether a specific act is right or wrong; however, they don’t show remorse for criminal behavior.” It is not clear whether executive dysfunction and the inability to reason, social perception and the inability to empathize, or differences in moral decision making are the reasons for changes in patients’ behavior, he said.
“One idea is that a network of brain regions, not just one part of the brain, is responsible for formulating the complex concept of morality.” In 2017, Dr. Darby and colleagues systematically mapped brain lesions with a documented temporal association with criminal behavior in 17 patients who were identified through a literature search. Criminal behavior included white collar crimes, and 12 of 17 patients had committed violent crimes. Fifteen cases had no history of criminal behavior before the lesion, and the behavior resolved following treatment of the lesion in two cases.
No single brain region had been damaged in all cases. Because lesion-induced symptoms can arise from sites connected to the lesion location, the investigators identified these sites in the cases. The network of these sites included regions involved in morality, value-based decision making, and theory of mind, but not regions involved in cognitive control or empathy. Darby and colleagues replicated these results in a separate cohort of 23 cases in which a temporal relationship between brain lesions and criminal behavior was plausible, but not definite.
Prior research suggests that the areas associated with criminal behavior in patients with brain lesions closely resemble the areas typically affected in patients with bvFTD. Prospective studies are needed to further elucidate these results, Dr. Darby concluded.
—Adriene Marshall
Suggested Reading
Darby RR, Horn A, Cushman F, Fox MD. Lesion network localization of criminal behavior. Proc Natl Acad Sci U S A. 2018;115(3):601-606.
Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329-3345.
Certain clinical features may indicate bvFTD, and off-label treatments may provide benefits.
Certain clinical features may indicate bvFTD, and off-label treatments may provide benefits.
HILTON HEAD, SC—Behavioral variant frontotemporal dementia (bvFTD), a clinically and pathologically heterogenous condition, can be difficult to distinguish from other forms of dementia or frontotemporal disease. “Although clinical symptoms vary based on which part of the brain is affected, there is a significant amount of overlap, with different pathologies causing the same type of syndrome,” said Richard Ryan Darby, MD, Assistant Professor of Neurology at Vanderbilt University Medical School in Nashville. “In a large proportion of patients, behavioral changes can lead to criminal behavior.” Future research examining brain lesion networks may shed light on this condition, said Dr. Darby at the 41st Annual Contemporary Clinical Neurology Symposium.
Clinical Features and Diagnosis
The incidence of bvFTD is equal to that of Alzheimer’s disease. However, patients with bvFTD tend to be younger: between the ages of 45 and 65. “Social and behavioral features predominate,” said Dr. Darby. “If a patient in this age range with no previous psychiatric history presents to you with a new psychiatric diagnosis such as schizophrenia or bipolar disease, this should raise your suspicion of bvFTD.” These psychiatric problems often cause considerable disruptions in patients’ lives. They often have problems at work and lose their source of income. “Patients often have no insight about their symptoms,” said Dr. Darby.
A differential diagnosis of bvFTD is possible in patients with three or more of the following six clinical features: socially inappropriate behavior (eg, eating from the trash or walking around naked at inappropriate times); lack of empathy; apathy; stereotyped or repetitive behavior (eg, saying things repeatedly, pacing); hyperorality (eg, eating uncontrollably, particularly sweet foods); and executive dysfunction, especially when memory is preserved.
“When you talk to caregivers, they often say, ‘This is not the person I married,’ or, ‘This is not my father. He seems like a different person,’” said Dr. Darby. An MRI or a PET scan showing changes in the frontotemporal lobes is a firm basis for a diagnosis of bvFTD, said Dr. Darby. Genetic testing for autosomal dominant mutation or pathology or an autopsy provides a definite diagnosis.
Pathology
Between 40% and 50% of patients with bvFTD have tau pathology, said Dr. Darby. This pathology includes the classic Pick body form of tau, tufted astrocytes (which are associated with clinical symptoms of progressive supranuclear palsy), and astrocytic plaques (which are associated with symptoms of corticobasal degeneration). Similarly, between 40% and 50% of patients with bvFTD have a TAR DNA-binding protein 43 (TDP-43) pathology. TDP-43 Type A pathology is associated with perirolandic seizures, Type B is associated with
Mutations in C9orf72 occur in 13% to 50% of patients with bvFTD who have genetic mutations. ALS and parkinsonism are common clinical presentations in patients with this genetic mutation. MAPT and GRN mutations are present in 5% to 20% of cases, and each can present clinically as parkinsonism.
Treatment
SSRIs, antipsychotics, anticonvulsants, and stimulants have been used to treat apathy, disinhibition, compulsive behaviors, agitation, and inappropriate behavior in patients with bvFTD. “There are no FDA-approved treatments for bvFTD, and the evidence for [these agents] has been mostly reported in case studies and small clinical trials,” said Dr. Darby. Among the SSRIs, paroxetine improved repetitive behavior in open-label trials, but not in a randomized controlled trial. Sertraline and citalopram have been studied in open-label trials, and trazodone was examined in a randomized controlled crossover trial involving 26 patients.
The FDA issued a black box warning against the use of atypical antipsychotics in patients with dementia because they entail a risk of cardiac- and infection-related mortality. “For patients with bvFTD tau pathology in particular, there is a risk of extrapyramidal adverse effects,” said Dr. Darby. A series of case reports of risperidone and aripiprazole provided evidence of symptom improvement, as did an open label study of olanzapine. Quetiapine improved agitation in a case series but failed to show benefit in a double-blind crossover trial of eight patients with FTD.
Case series have shown evidence that antiepileptic drugs (eg, valproic acid, topiramate, and carbamazepine) have a stabilizing effect. “Stimulants are tried in some patients, but should be used with caution,” said Dr. Darby.
Criminality
Between 37% and 57% of patients with bvFTD engage in criminal behavior. “Approximately 10% to 15% of the time, a patient’s getting in trouble with the law is the reason for the initial presentation,” said Dr. Darby. The types of crimes described in case reports include pedophilia, public masturbation, hit and run, traffic violations, and theft.
“Murder and violent crimes occur but are rare. Crimes committed by patients with bvFTD are usually reactive,” said Dr. Darby. “When asked, they can tell you whether a specific act is right or wrong; however, they don’t show remorse for criminal behavior.” It is not clear whether executive dysfunction and the inability to reason, social perception and the inability to empathize, or differences in moral decision making are the reasons for changes in patients’ behavior, he said.
“One idea is that a network of brain regions, not just one part of the brain, is responsible for formulating the complex concept of morality.” In 2017, Dr. Darby and colleagues systematically mapped brain lesions with a documented temporal association with criminal behavior in 17 patients who were identified through a literature search. Criminal behavior included white collar crimes, and 12 of 17 patients had committed violent crimes. Fifteen cases had no history of criminal behavior before the lesion, and the behavior resolved following treatment of the lesion in two cases.
No single brain region had been damaged in all cases. Because lesion-induced symptoms can arise from sites connected to the lesion location, the investigators identified these sites in the cases. The network of these sites included regions involved in morality, value-based decision making, and theory of mind, but not regions involved in cognitive control or empathy. Darby and colleagues replicated these results in a separate cohort of 23 cases in which a temporal relationship between brain lesions and criminal behavior was plausible, but not definite.
Prior research suggests that the areas associated with criminal behavior in patients with brain lesions closely resemble the areas typically affected in patients with bvFTD. Prospective studies are needed to further elucidate these results, Dr. Darby concluded.
—Adriene Marshall
Suggested Reading
Darby RR, Horn A, Cushman F, Fox MD. Lesion network localization of criminal behavior. Proc Natl Acad Sci U S A. 2018;115(3):601-606.
Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329-3345.
HILTON HEAD, SC—Behavioral variant frontotemporal dementia (bvFTD), a clinically and pathologically heterogenous condition, can be difficult to distinguish from other forms of dementia or frontotemporal disease. “Although clinical symptoms vary based on which part of the brain is affected, there is a significant amount of overlap, with different pathologies causing the same type of syndrome,” said Richard Ryan Darby, MD, Assistant Professor of Neurology at Vanderbilt University Medical School in Nashville. “In a large proportion of patients, behavioral changes can lead to criminal behavior.” Future research examining brain lesion networks may shed light on this condition, said Dr. Darby at the 41st Annual Contemporary Clinical Neurology Symposium.
Clinical Features and Diagnosis
The incidence of bvFTD is equal to that of Alzheimer’s disease. However, patients with bvFTD tend to be younger: between the ages of 45 and 65. “Social and behavioral features predominate,” said Dr. Darby. “If a patient in this age range with no previous psychiatric history presents to you with a new psychiatric diagnosis such as schizophrenia or bipolar disease, this should raise your suspicion of bvFTD.” These psychiatric problems often cause considerable disruptions in patients’ lives. They often have problems at work and lose their source of income. “Patients often have no insight about their symptoms,” said Dr. Darby.
A differential diagnosis of bvFTD is possible in patients with three or more of the following six clinical features: socially inappropriate behavior (eg, eating from the trash or walking around naked at inappropriate times); lack of empathy; apathy; stereotyped or repetitive behavior (eg, saying things repeatedly, pacing); hyperorality (eg, eating uncontrollably, particularly sweet foods); and executive dysfunction, especially when memory is preserved.
“When you talk to caregivers, they often say, ‘This is not the person I married,’ or, ‘This is not my father. He seems like a different person,’” said Dr. Darby. An MRI or a PET scan showing changes in the frontotemporal lobes is a firm basis for a diagnosis of bvFTD, said Dr. Darby. Genetic testing for autosomal dominant mutation or pathology or an autopsy provides a definite diagnosis.
Pathology
Between 40% and 50% of patients with bvFTD have tau pathology, said Dr. Darby. This pathology includes the classic Pick body form of tau, tufted astrocytes (which are associated with clinical symptoms of progressive supranuclear palsy), and astrocytic plaques (which are associated with symptoms of corticobasal degeneration). Similarly, between 40% and 50% of patients with bvFTD have a TAR DNA-binding protein 43 (TDP-43) pathology. TDP-43 Type A pathology is associated with perirolandic seizures, Type B is associated with
Mutations in C9orf72 occur in 13% to 50% of patients with bvFTD who have genetic mutations. ALS and parkinsonism are common clinical presentations in patients with this genetic mutation. MAPT and GRN mutations are present in 5% to 20% of cases, and each can present clinically as parkinsonism.
Treatment
SSRIs, antipsychotics, anticonvulsants, and stimulants have been used to treat apathy, disinhibition, compulsive behaviors, agitation, and inappropriate behavior in patients with bvFTD. “There are no FDA-approved treatments for bvFTD, and the evidence for [these agents] has been mostly reported in case studies and small clinical trials,” said Dr. Darby. Among the SSRIs, paroxetine improved repetitive behavior in open-label trials, but not in a randomized controlled trial. Sertraline and citalopram have been studied in open-label trials, and trazodone was examined in a randomized controlled crossover trial involving 26 patients.
The FDA issued a black box warning against the use of atypical antipsychotics in patients with dementia because they entail a risk of cardiac- and infection-related mortality. “For patients with bvFTD tau pathology in particular, there is a risk of extrapyramidal adverse effects,” said Dr. Darby. A series of case reports of risperidone and aripiprazole provided evidence of symptom improvement, as did an open label study of olanzapine. Quetiapine improved agitation in a case series but failed to show benefit in a double-blind crossover trial of eight patients with FTD.
Case series have shown evidence that antiepileptic drugs (eg, valproic acid, topiramate, and carbamazepine) have a stabilizing effect. “Stimulants are tried in some patients, but should be used with caution,” said Dr. Darby.
Criminality
Between 37% and 57% of patients with bvFTD engage in criminal behavior. “Approximately 10% to 15% of the time, a patient’s getting in trouble with the law is the reason for the initial presentation,” said Dr. Darby. The types of crimes described in case reports include pedophilia, public masturbation, hit and run, traffic violations, and theft.
“Murder and violent crimes occur but are rare. Crimes committed by patients with bvFTD are usually reactive,” said Dr. Darby. “When asked, they can tell you whether a specific act is right or wrong; however, they don’t show remorse for criminal behavior.” It is not clear whether executive dysfunction and the inability to reason, social perception and the inability to empathize, or differences in moral decision making are the reasons for changes in patients’ behavior, he said.
“One idea is that a network of brain regions, not just one part of the brain, is responsible for formulating the complex concept of morality.” In 2017, Dr. Darby and colleagues systematically mapped brain lesions with a documented temporal association with criminal behavior in 17 patients who were identified through a literature search. Criminal behavior included white collar crimes, and 12 of 17 patients had committed violent crimes. Fifteen cases had no history of criminal behavior before the lesion, and the behavior resolved following treatment of the lesion in two cases.
No single brain region had been damaged in all cases. Because lesion-induced symptoms can arise from sites connected to the lesion location, the investigators identified these sites in the cases. The network of these sites included regions involved in morality, value-based decision making, and theory of mind, but not regions involved in cognitive control or empathy. Darby and colleagues replicated these results in a separate cohort of 23 cases in which a temporal relationship between brain lesions and criminal behavior was plausible, but not definite.
Prior research suggests that the areas associated with criminal behavior in patients with brain lesions closely resemble the areas typically affected in patients with bvFTD. Prospective studies are needed to further elucidate these results, Dr. Darby concluded.
—Adriene Marshall
Suggested Reading
Darby RR, Horn A, Cushman F, Fox MD. Lesion network localization of criminal behavior. Proc Natl Acad Sci U S A. 2018;115(3):601-606.
Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329-3345.
FDA Grants Fund Rare Disease Research
Twelve FDA grants fund new clinical trials to advance the development of medical products for the treatment of rare diseases.
On September 24, 2018, the FDA announced that it awarded 12 new clinical trial research grants totaling more than $18 million over the next four years to enhance the development of medical products for patients with rare diseases. These new grants were awarded to principal investigators from academia and industry across the country.
“Developing a treatment for a rare disease can be especially challenging. Given the often small number of patients affected by certain very rare diseases, there can be limited markets for new treatments, and as a result fewer resources devoted to researching these opportunities,” said FDA Commissioner Scott Gottlieb, MD. “The FDA is committed to doing its part to facilitate continued progress toward more treatments, and even potential cures, for patients with rare diseases. New scientific advances offer more opportunities to develop these potential cures. With efficient regulation, proper incentives for product development, and the continued support of patients, providers, and researchers, we have more opportunities to pursue these advances than ever before. For 35 years, the FDA has provided much-needed financial support for clinical trials of potentially life-changing treatments for patients with rare diseases. This funding helps support early-stage development activities targeting rare diseases that do not have effective treatments. By providing seed capital, these FDA-administered grants enable researchers to prove out important concepts. The FDA grants also provide some important recognition to promising development programs that ultimately can help researchers attract additional funding.”

The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. This program is funded by Congressional appropriations and encourages clinical development of drugs, biologics, medical devices, or medical foods for use in rare diseases. The grants are intended for clinical studies evaluating the safety and effectiveness of products that could either result in, or substantially contribute to, the FDA approval of products targeted to the treatment of rare diseases. Grant applications were reviewed and evaluated for scientific and technical merit by more than 100 rare disease experts, which included representatives from academia, the NIH, and the FDA.
The grant recipients, principal investigators, and approximate funding amounts, listed alphabetically, are:
Alkeus Pharmaceuticals, Inc (Cambridge, Massachusetts), Leonide Saad, phase 2 study of ALK-001 for the treatment of Stargardt disease—$1.75 million over four years
Arizona State University–Tempe Campus (Tempe, Arizona), Keith Lindor, phase 2 study of oral vancomycin for the treatment of primary sclerosing cholangitis—$2 million over four years
Cedars-Sinai Medical Center (Los Angeles), Shlomo Melmed, phase 2 study of seliciclib for the treatment of Cushing disease—$2 million over four years
Columbia University (New York), Yvonne Saenger, phase 1 study of talimogene laherparepvec for the treatment for advanced pancreatic cancer—$750,000 over three years
Emory University (Atlanta), Eric Sorscher, phase 1/ 2 study of Ad/PNP fludarabine for the treatment of head and neck squamous cell carcinoma—$1.5 million over three years
Fibrocell Technologies, Inc (Exton, Pennsylvania), John Maslowski, phase 1/2 study of gene-modified ex-vivo autologous fibroblasts for the treatment of dystrophic epidermolysis bullosa—$1.5 million over four years
Johns Hopkins University (Baltimore), Amy Dezern, phase 1/2 study of CD8-reduced T cells for the treatment of myelodysplastic syndrome or acute myeloid leukemia—$750,000 over three years
Oncolmmune, Inc (Rockville, Maryland) Yang Liu, phase 2b study of CD24Fc for the prevention of graft versus host disease—$2 million over four years
Patagonia Pharmaceuticals, LLC (Woodcliff Lake, New Jersey), Zachary Rome, phase 2 study of PAT-001 (isotretinoin) for the treatment of congenital ichthyosis—$1.5 million over three years
The General Hospital Corporation (Boston), Stephanie Seminara, phase 2 study of kisspeptin for the treatment of dopamine agonist intolerant hyperprolactinemia—$1.4 million over four years
University of Minnesota (Minneapolis), Kyriakie Sarafoglou, phase 2a study of subcutaneous hydrocortisone infusion pump for the treatment of congenital adrenal hyperplasia—$1.4 million over three years
University of North Carolina at Chapel Hill (Chapel Hill, North Carolina), Matthew Laughon, phase 2 study of sildenafil for the prevention of bronchopulmonary dysplasia—$2 million over four years.
“Since its creation in 1983, the Orphan Products Grants Program has provided more than $400 million to fund more than 600 new clinical studies,” said Debra Lewis, OD, Acting Director of the FDA’s Office of Orphan Products Development. “We are encouraged to see so much interest in our grants program and are pleased to support research for a variety of rare diseases that have little, or no, treatment options for patients.”
One-third of the new awards aim to accelerate cancer research by enrolling patients with rare forms of cancer, including advanced pancreatic cancer, head and neck squamous cell carcinoma, myelodysplastic syndrome, and acute myeloid leukemia. Another 25% of the new awards fund studies evaluating drug products for rare endocrine disorders, including Cushing disease, dopamine agonist intolerant hyperprolactinemia, and congenital adrenal hyperplasia. Another study addresses an unmet need in primary sclerosing cholangitis, a rare, chronic, and potentially serious bile duct disease.
About 42% of the grants fund studies that enroll children and adolescents, targeting a variety of rare diseases in children such as Stargardt disease, a juvenile genetic eye disorder that causes progressive vision loss; dystrophic epidermolysis bullosa, a genetic condition that causes the skin to be fragile resulting in painful blisters; and bronchopulmonary dysplasia, a serious lung condition that affects infants.
To date, the program’s grants have supported research that led to the marketing approval of more than 60 orphan products. Among the recent product approvals which were supported by studies funded by this grants program are a marketing approval for a much-needed treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults with multidrug resistant HIV-1 infection and another approval to reduce the acute complications of sickle cell disease in adult and pediatric patients.
The FDA is also currently supporting six natural history studies for rare diseases to further advance the mission of bringing new therapies to market.
Twelve FDA grants fund new clinical trials to advance the development of medical products for the treatment of rare diseases.
Twelve FDA grants fund new clinical trials to advance the development of medical products for the treatment of rare diseases.
On September 24, 2018, the FDA announced that it awarded 12 new clinical trial research grants totaling more than $18 million over the next four years to enhance the development of medical products for patients with rare diseases. These new grants were awarded to principal investigators from academia and industry across the country.
“Developing a treatment for a rare disease can be especially challenging. Given the often small number of patients affected by certain very rare diseases, there can be limited markets for new treatments, and as a result fewer resources devoted to researching these opportunities,” said FDA Commissioner Scott Gottlieb, MD. “The FDA is committed to doing its part to facilitate continued progress toward more treatments, and even potential cures, for patients with rare diseases. New scientific advances offer more opportunities to develop these potential cures. With efficient regulation, proper incentives for product development, and the continued support of patients, providers, and researchers, we have more opportunities to pursue these advances than ever before. For 35 years, the FDA has provided much-needed financial support for clinical trials of potentially life-changing treatments for patients with rare diseases. This funding helps support early-stage development activities targeting rare diseases that do not have effective treatments. By providing seed capital, these FDA-administered grants enable researchers to prove out important concepts. The FDA grants also provide some important recognition to promising development programs that ultimately can help researchers attract additional funding.”
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. This program is funded by Congressional appropriations and encourages clinical development of drugs, biologics, medical devices, or medical foods for use in rare diseases. The grants are intended for clinical studies evaluating the safety and effectiveness of products that could either result in, or substantially contribute to, the FDA approval of products targeted to the treatment of rare diseases. Grant applications were reviewed and evaluated for scientific and technical merit by more than 100 rare disease experts, which included representatives from academia, the NIH, and the FDA.
The grant recipients, principal investigators, and approximate funding amounts, listed alphabetically, are:
Alkeus Pharmaceuticals, Inc (Cambridge, Massachusetts), Leonide Saad, phase 2 study of ALK-001 for the treatment of Stargardt disease—$1.75 million over four years
Arizona State University–Tempe Campus (Tempe, Arizona), Keith Lindor, phase 2 study of oral vancomycin for the treatment of primary sclerosing cholangitis—$2 million over four years
Cedars-Sinai Medical Center (Los Angeles), Shlomo Melmed, phase 2 study of seliciclib for the treatment of Cushing disease—$2 million over four years
Columbia University (New York), Yvonne Saenger, phase 1 study of talimogene laherparepvec for the treatment for advanced pancreatic cancer—$750,000 over three years
Emory University (Atlanta), Eric Sorscher, phase 1/ 2 study of Ad/PNP fludarabine for the treatment of head and neck squamous cell carcinoma—$1.5 million over three years
Fibrocell Technologies, Inc (Exton, Pennsylvania), John Maslowski, phase 1/2 study of gene-modified ex-vivo autologous fibroblasts for the treatment of dystrophic epidermolysis bullosa—$1.5 million over four years
Johns Hopkins University (Baltimore), Amy Dezern, phase 1/2 study of CD8-reduced T cells for the treatment of myelodysplastic syndrome or acute myeloid leukemia—$750,000 over three years
Oncolmmune, Inc (Rockville, Maryland) Yang Liu, phase 2b study of CD24Fc for the prevention of graft versus host disease—$2 million over four years
Patagonia Pharmaceuticals, LLC (Woodcliff Lake, New Jersey), Zachary Rome, phase 2 study of PAT-001 (isotretinoin) for the treatment of congenital ichthyosis—$1.5 million over three years
The General Hospital Corporation (Boston), Stephanie Seminara, phase 2 study of kisspeptin for the treatment of dopamine agonist intolerant hyperprolactinemia—$1.4 million over four years
University of Minnesota (Minneapolis), Kyriakie Sarafoglou, phase 2a study of subcutaneous hydrocortisone infusion pump for the treatment of congenital adrenal hyperplasia—$1.4 million over three years
University of North Carolina at Chapel Hill (Chapel Hill, North Carolina), Matthew Laughon, phase 2 study of sildenafil for the prevention of bronchopulmonary dysplasia—$2 million over four years.
“Since its creation in 1983, the Orphan Products Grants Program has provided more than $400 million to fund more than 600 new clinical studies,” said Debra Lewis, OD, Acting Director of the FDA’s Office of Orphan Products Development. “We are encouraged to see so much interest in our grants program and are pleased to support research for a variety of rare diseases that have little, or no, treatment options for patients.”
One-third of the new awards aim to accelerate cancer research by enrolling patients with rare forms of cancer, including advanced pancreatic cancer, head and neck squamous cell carcinoma, myelodysplastic syndrome, and acute myeloid leukemia. Another 25% of the new awards fund studies evaluating drug products for rare endocrine disorders, including Cushing disease, dopamine agonist intolerant hyperprolactinemia, and congenital adrenal hyperplasia. Another study addresses an unmet need in primary sclerosing cholangitis, a rare, chronic, and potentially serious bile duct disease.
About 42% of the grants fund studies that enroll children and adolescents, targeting a variety of rare diseases in children such as Stargardt disease, a juvenile genetic eye disorder that causes progressive vision loss; dystrophic epidermolysis bullosa, a genetic condition that causes the skin to be fragile resulting in painful blisters; and bronchopulmonary dysplasia, a serious lung condition that affects infants.
To date, the program’s grants have supported research that led to the marketing approval of more than 60 orphan products. Among the recent product approvals which were supported by studies funded by this grants program are a marketing approval for a much-needed treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults with multidrug resistant HIV-1 infection and another approval to reduce the acute complications of sickle cell disease in adult and pediatric patients.
The FDA is also currently supporting six natural history studies for rare diseases to further advance the mission of bringing new therapies to market.
On September 24, 2018, the FDA announced that it awarded 12 new clinical trial research grants totaling more than $18 million over the next four years to enhance the development of medical products for patients with rare diseases. These new grants were awarded to principal investigators from academia and industry across the country.
“Developing a treatment for a rare disease can be especially challenging. Given the often small number of patients affected by certain very rare diseases, there can be limited markets for new treatments, and as a result fewer resources devoted to researching these opportunities,” said FDA Commissioner Scott Gottlieb, MD. “The FDA is committed to doing its part to facilitate continued progress toward more treatments, and even potential cures, for patients with rare diseases. New scientific advances offer more opportunities to develop these potential cures. With efficient regulation, proper incentives for product development, and the continued support of patients, providers, and researchers, we have more opportunities to pursue these advances than ever before. For 35 years, the FDA has provided much-needed financial support for clinical trials of potentially life-changing treatments for patients with rare diseases. This funding helps support early-stage development activities targeting rare diseases that do not have effective treatments. By providing seed capital, these FDA-administered grants enable researchers to prove out important concepts. The FDA grants also provide some important recognition to promising development programs that ultimately can help researchers attract additional funding.”
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. This program is funded by Congressional appropriations and encourages clinical development of drugs, biologics, medical devices, or medical foods for use in rare diseases. The grants are intended for clinical studies evaluating the safety and effectiveness of products that could either result in, or substantially contribute to, the FDA approval of products targeted to the treatment of rare diseases. Grant applications were reviewed and evaluated for scientific and technical merit by more than 100 rare disease experts, which included representatives from academia, the NIH, and the FDA.
The grant recipients, principal investigators, and approximate funding amounts, listed alphabetically, are:
Alkeus Pharmaceuticals, Inc (Cambridge, Massachusetts), Leonide Saad, phase 2 study of ALK-001 for the treatment of Stargardt disease—$1.75 million over four years
Arizona State University–Tempe Campus (Tempe, Arizona), Keith Lindor, phase 2 study of oral vancomycin for the treatment of primary sclerosing cholangitis—$2 million over four years
Cedars-Sinai Medical Center (Los Angeles), Shlomo Melmed, phase 2 study of seliciclib for the treatment of Cushing disease—$2 million over four years
Columbia University (New York), Yvonne Saenger, phase 1 study of talimogene laherparepvec for the treatment for advanced pancreatic cancer—$750,000 over three years
Emory University (Atlanta), Eric Sorscher, phase 1/ 2 study of Ad/PNP fludarabine for the treatment of head and neck squamous cell carcinoma—$1.5 million over three years
Fibrocell Technologies, Inc (Exton, Pennsylvania), John Maslowski, phase 1/2 study of gene-modified ex-vivo autologous fibroblasts for the treatment of dystrophic epidermolysis bullosa—$1.5 million over four years
Johns Hopkins University (Baltimore), Amy Dezern, phase 1/2 study of CD8-reduced T cells for the treatment of myelodysplastic syndrome or acute myeloid leukemia—$750,000 over three years
Oncolmmune, Inc (Rockville, Maryland) Yang Liu, phase 2b study of CD24Fc for the prevention of graft versus host disease—$2 million over four years
Patagonia Pharmaceuticals, LLC (Woodcliff Lake, New Jersey), Zachary Rome, phase 2 study of PAT-001 (isotretinoin) for the treatment of congenital ichthyosis—$1.5 million over three years
The General Hospital Corporation (Boston), Stephanie Seminara, phase 2 study of kisspeptin for the treatment of dopamine agonist intolerant hyperprolactinemia—$1.4 million over four years
University of Minnesota (Minneapolis), Kyriakie Sarafoglou, phase 2a study of subcutaneous hydrocortisone infusion pump for the treatment of congenital adrenal hyperplasia—$1.4 million over three years
University of North Carolina at Chapel Hill (Chapel Hill, North Carolina), Matthew Laughon, phase 2 study of sildenafil for the prevention of bronchopulmonary dysplasia—$2 million over four years.
“Since its creation in 1983, the Orphan Products Grants Program has provided more than $400 million to fund more than 600 new clinical studies,” said Debra Lewis, OD, Acting Director of the FDA’s Office of Orphan Products Development. “We are encouraged to see so much interest in our grants program and are pleased to support research for a variety of rare diseases that have little, or no, treatment options for patients.”
One-third of the new awards aim to accelerate cancer research by enrolling patients with rare forms of cancer, including advanced pancreatic cancer, head and neck squamous cell carcinoma, myelodysplastic syndrome, and acute myeloid leukemia. Another 25% of the new awards fund studies evaluating drug products for rare endocrine disorders, including Cushing disease, dopamine agonist intolerant hyperprolactinemia, and congenital adrenal hyperplasia. Another study addresses an unmet need in primary sclerosing cholangitis, a rare, chronic, and potentially serious bile duct disease.
About 42% of the grants fund studies that enroll children and adolescents, targeting a variety of rare diseases in children such as Stargardt disease, a juvenile genetic eye disorder that causes progressive vision loss; dystrophic epidermolysis bullosa, a genetic condition that causes the skin to be fragile resulting in painful blisters; and bronchopulmonary dysplasia, a serious lung condition that affects infants.
To date, the program’s grants have supported research that led to the marketing approval of more than 60 orphan products. Among the recent product approvals which were supported by studies funded by this grants program are a marketing approval for a much-needed treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults with multidrug resistant HIV-1 infection and another approval to reduce the acute complications of sickle cell disease in adult and pediatric patients.
The FDA is also currently supporting six natural history studies for rare diseases to further advance the mission of bringing new therapies to market.
Adult-Onset Still Disease: Persistent Pruritic Papular Rash With Unique Histopathologic Findings
Adult-onset Still disease (AOSD) is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, evanescent skin rash, and lymphadenopathy. 1 The most commonly used criteria for diagnosing AOSD are the Yamaguchi criteria. 2 The major criteria include high fever for more than 1 week, arthralgia for more than 2 weeks, leukocytosis, and an evanescent skin rash. The minor criteria consist of sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and negative rheumatoid factor and antinuclear antibodies. Classically, the skin rash is described as an evanescent, salmon-colored erythema involving the extremities. Nevertheless, unusual cutaneous eruptions have been reported in AOSD, including persistent pruritic papules and plaques. 3 Importantly, this atypical rash demonstrates specific histologic findings that are not found on routine histopathology of a typical evanescent rash. We describe 2 patients with this atypical cutaneous eruption along with the unique histopathologic findings of AOSD.
Case Reports
Patient 1
A 23-year-old Chinese woman presented with periodic fevers, persistent rash, and joint pain of 2 years’ duration. Her medical history included splenectomy for hepatosplenomegaly as well as evaluation by hematology for lymphadenopathy; a cervical lymph node biopsy showed lymphoid and follicular hyperplasia.
Twenty days later, the patient was referred to the dermatology department for evaluation of the persistent rash. The patient described a history of flushing of the face, severe joint pain in both arms and legs, aching muscles, and persistent sore throat. The patient did not report any history of drug ingestion. Physical examination revealed a fever (temperature, 39.2°C); swollen nontender lymph nodes in the neck, axillae, and groin; and salmon-colored and hyperpigmented patches and thin plaques over the neck, chest, abdomen, and arms (Figure 1). A splenectomy scar also was noted. Peripheral blood was collected for laboratory analyses, which revealed transaminitis and moderate hyperferritinemia (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. The patient was admitted to the hospital, and a skin biopsy was performed. Histology showed superficial dyskeratotic keratinocytes and sparse perivascular infiltration of neutrophils in the upper dermis (Figure 2).
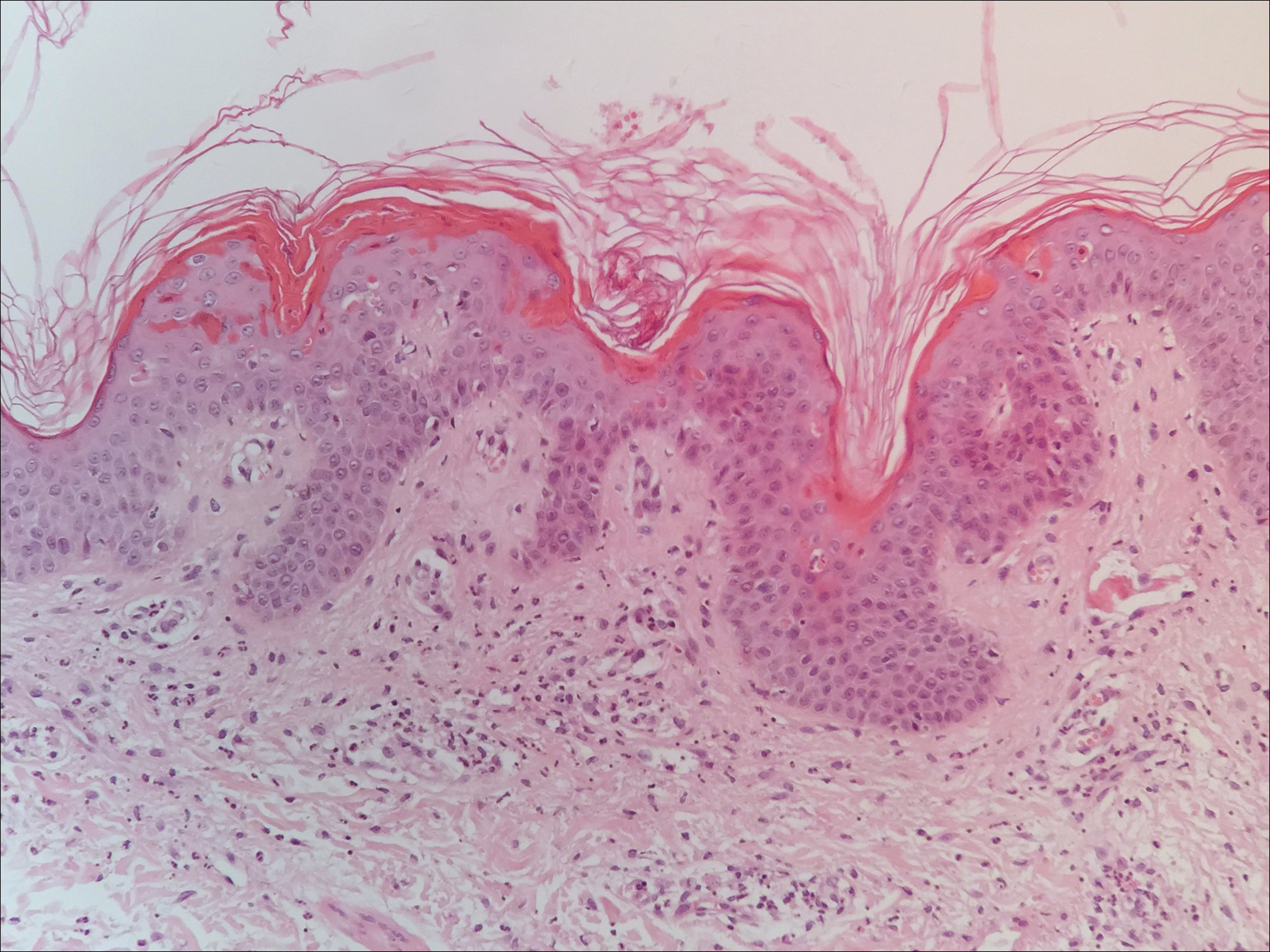
The patient was diagnosed with AOSD based on fulfillment of the Yamaguchi criteria.2 She was treated with methylprednisolone 60 mg daily and was discharged 14 days later. At 16-month follow-up, the patient demonstrated complete resolution of symptoms with a maintenance dose of prednisolone (7.5 mg daily).
Patient 2
A 23-year-old black woman presented to the emergency department 3 months postpartum with recurrent high fevers, worsening joint pain, and persistent itchy rash of 2 months’ duration. The patient had no history of travel, autoimmune disease, or sick contacts. She occasionally took aspirin for joint pain. Physical examination revealed a fever (temperature, 39.1°C) along with hyperpigmented patches and thin scaly hyperpigmented papules coalescing into a poorly demarcated V-shaped plaque on the upper back and posterior neck, extending to the chest in a shawl-like distribution (Figure 3). Submental lymphadenopathy was present. The spleen was not palpable.
Peripheral blood was collected for laboratory analysis and demonstrated transaminitis and a markedly high ferritin level (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. Skin biopsy was performed and demonstrated many necrotic keratinocytes, singly and in aggregates, distributed from the spinous layer to the stratum corneum. A neutrophilic infiltrate was present in the papillary dermis (Figure 4).
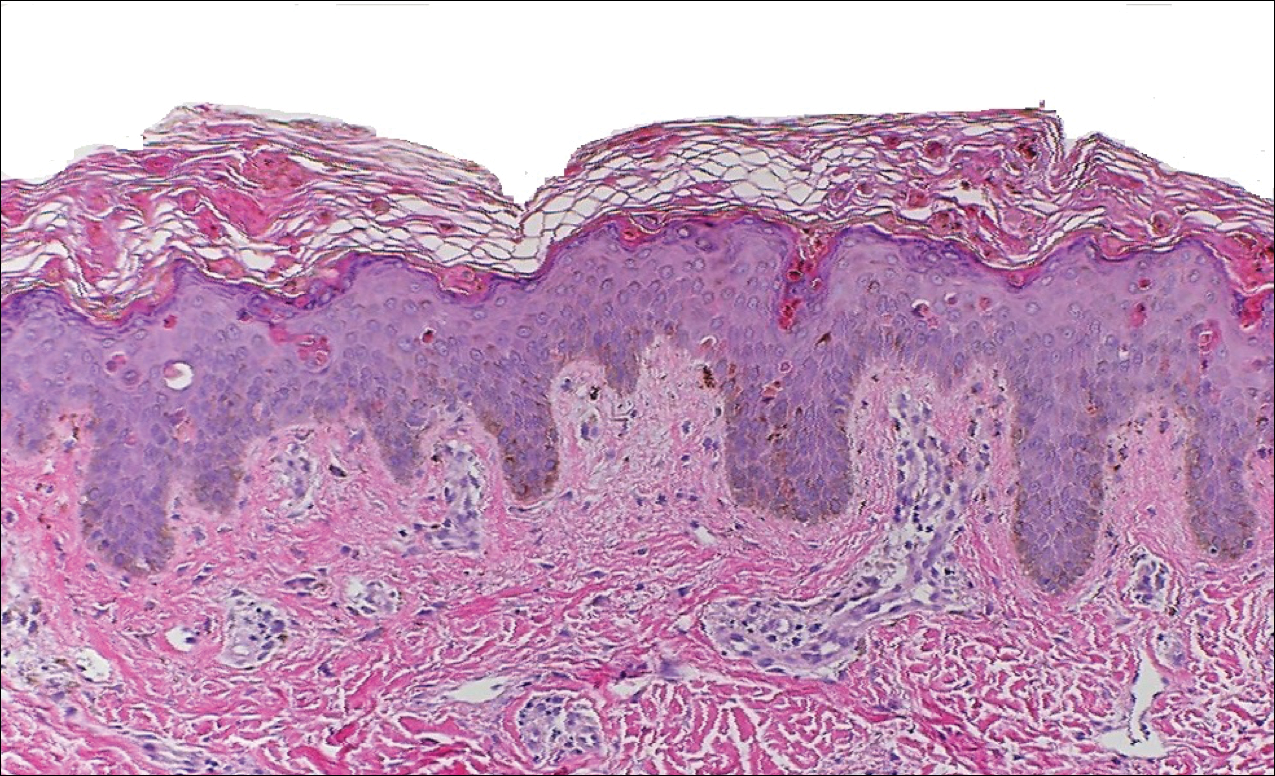
The patient met the Yamaguchi criteria and was subsequently diagnosed with AOSD. She was treated with intravenous methylprednisolone 20 mg every 8 hours and was discharged 1 week later on oral prednisone 60 mg daily to be tapered over a period of months. At 2-week follow-up, the patient continued to experience rash and joint pain; oral methotrexate 10 mg weekly was added to her regimen, as well as vitamin D, calcium, and folic acid supplementation. At the next 2-week follow-up the patient noted improvement in the rash as well as the joint pain, but both still persisted. Prednisone was decreased to 50 mg daily and methotrexate was increased to 15 mg weekly. The patient continued to show improvement over the subsequent 3 months, during which prednisone was tapered to 10 mg daily and methotrexate was increased to 20 mg weekly. The patient showed resolution of symptoms at 3-month follow-up on this regimen, with plans to continue the prednisone taper and maintain methotrexate dosing.
Comment
Adult-onset Still disease is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, salmon-pink evanescent erythema, and lymphadenopathy.2 The condition also can cause liver dysfunction, splenomegaly, pericarditis, pleuritis, renal dysfunction, and a reactive hemophagocytic syndrome.1 Furthermore, one review of the literature described an association with delayed-onset malignancy.4 Early diagnosis is important yet challenging, as AOSD is a diagnosis of exclusion. The Yamaguchi criteria are the most widely used method of diagnosis and demonstrate more than 90% sensitivity.In addition to the Yamaguchi criteria, marked hyperferritinemia is characteristic of AOSD and can act as an indicator of disease activity.5 Interestingly, both of our patients had elevated ferritin levels, with patient 2 showing marked elevation (Table). In both patients, all major criteria were fulfilled, except the typical skin rash.
The skin rash in AOSD, classically consisting of an evanescent, salmon-pink erythema predominantly involving the extremities, has been observed in up to 87% of AOSD patients.5 The histology of the typical evanescent rash is nonspecific, characterized by a relatively sparse, perivascular, mixed inflammatory infiltrate. Notably, other skin manifestations may be found in patients with AOSD.1,2,5-16 Persistent pruritic papules and plaques are the most commonly reported nonclassical rash, presenting as erythematous, slightly scaly papules and plaques with a linear configuration typically on the trunk.2 Both of our patients presented with this atypical eruption. Importantly, the histopathology of this unique rash displays distinctive features, which can aid in early diagnosis. Findings include dyskeratotic keratinocytes in the cornified layers as well as in the epidermis, and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis. These findings were evident in both histopathologic studies of our patients (Figures 2 and 4). Although not present in our patients, dermal mucin deposition has been demonstrated in some reports.1,13,15
A 2015 review of the literature yielded 30 cases of AOSD with pruritic persistent papules and plaques.4 The study confirmed a linear, erythematous or brown rash on the back and neck in the majority of cases. Histologic findings were congruent with those reported in our 2 cases: necrotic keratinocytes in the upper epidermis with a neutrophilic infiltrate in the upper dermis without vasculitis. Most patients showed rapid resolution of the rash and symptoms with the use of prednisone, prednisolone, or intravenous pulsed methylprednisolone. Interestingly, a range of presentations were noted, including prurigo pigmentosalike urticarial papules; lichenoid papules; and dermatographismlike, dermatomyositislike, and lichen amyloidosis–like rashes.4 In our report, patient 2 presented with a rash in a dermat-omyositislike shawl distribution. It has been suggested that patients with dermatomyositislike rashes require more potent immunotherapy as compared to patients with other rash morphologies.4 The need for methotrexate in addition to a prednisone taper in the clinical course of patient 2 lends further support to this observation.
Conclusion
A clinically and pathologically distinct form of cutaneous disease—AOSD with persistent pruritic papules and plaques—was observed in our 2 patients. These histopathologic findings facilitated timely diagnosis in both patients. A range of clinical morphologies may exist in AOSD, an awareness of which is paramount. Adult-onset Still disease should be included in the differential diagnosis of a dermatomyositislike presentation in a shawl distribution. Prompt diagnosis is essential to ensure adequate therapy.
- Yamamoto T. Cutaneous manifestations associated with adult-onset Still’s disease: important diagnostic values. Rheumatol Int. 2012;32:2233-2237.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-431.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associates with delayed malignancy [published online June 6, 2015]. J Am Acad Dermatol. 2015;73:294-303.
- Schwarz-Eywill M, Heilig B, Bauer H, et al. Evaluation of serum ferritin as a marker for adult Still’s disease activity. Ann Rheum Dis. 1992;51:683-685.
- Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17:1058-1063.
- Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241-242.
- Lübbe J, Hofer M, Chavaz P, et al. Adult onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710-713.
- Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still’s disease. Dermatology. 2001;202:333-335.
- Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still’s disease. Int J Dermatol. 2003;42:824-825.
- Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still’s disease. Ann Dermatol Venereol. 2005;132:693-696.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still’s disease. Am J Dermatopathol. 2007;49:194-196.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still’s disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Azeck AG, Littlewood SM. Adult-onset Still’s disease with atypical cutaneous features. J Eur Acad Dermatol Venereol. 2005;19:360-363.
Adult-onset Still disease (AOSD) is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, evanescent skin rash, and lymphadenopathy. 1 The most commonly used criteria for diagnosing AOSD are the Yamaguchi criteria. 2 The major criteria include high fever for more than 1 week, arthralgia for more than 2 weeks, leukocytosis, and an evanescent skin rash. The minor criteria consist of sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and negative rheumatoid factor and antinuclear antibodies. Classically, the skin rash is described as an evanescent, salmon-colored erythema involving the extremities. Nevertheless, unusual cutaneous eruptions have been reported in AOSD, including persistent pruritic papules and plaques. 3 Importantly, this atypical rash demonstrates specific histologic findings that are not found on routine histopathology of a typical evanescent rash. We describe 2 patients with this atypical cutaneous eruption along with the unique histopathologic findings of AOSD.
Case Reports
Patient 1
A 23-year-old Chinese woman presented with periodic fevers, persistent rash, and joint pain of 2 years’ duration. Her medical history included splenectomy for hepatosplenomegaly as well as evaluation by hematology for lymphadenopathy; a cervical lymph node biopsy showed lymphoid and follicular hyperplasia.
Twenty days later, the patient was referred to the dermatology department for evaluation of the persistent rash. The patient described a history of flushing of the face, severe joint pain in both arms and legs, aching muscles, and persistent sore throat. The patient did not report any history of drug ingestion. Physical examination revealed a fever (temperature, 39.2°C); swollen nontender lymph nodes in the neck, axillae, and groin; and salmon-colored and hyperpigmented patches and thin plaques over the neck, chest, abdomen, and arms (Figure 1). A splenectomy scar also was noted. Peripheral blood was collected for laboratory analyses, which revealed transaminitis and moderate hyperferritinemia (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. The patient was admitted to the hospital, and a skin biopsy was performed. Histology showed superficial dyskeratotic keratinocytes and sparse perivascular infiltration of neutrophils in the upper dermis (Figure 2).
The patient was diagnosed with AOSD based on fulfillment of the Yamaguchi criteria.2 She was treated with methylprednisolone 60 mg daily and was discharged 14 days later. At 16-month follow-up, the patient demonstrated complete resolution of symptoms with a maintenance dose of prednisolone (7.5 mg daily).
Patient 2
A 23-year-old black woman presented to the emergency department 3 months postpartum with recurrent high fevers, worsening joint pain, and persistent itchy rash of 2 months’ duration. The patient had no history of travel, autoimmune disease, or sick contacts. She occasionally took aspirin for joint pain. Physical examination revealed a fever (temperature, 39.1°C) along with hyperpigmented patches and thin scaly hyperpigmented papules coalescing into a poorly demarcated V-shaped plaque on the upper back and posterior neck, extending to the chest in a shawl-like distribution (Figure 3). Submental lymphadenopathy was present. The spleen was not palpable.
Peripheral blood was collected for laboratory analysis and demonstrated transaminitis and a markedly high ferritin level (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. Skin biopsy was performed and demonstrated many necrotic keratinocytes, singly and in aggregates, distributed from the spinous layer to the stratum corneum. A neutrophilic infiltrate was present in the papillary dermis (Figure 4).
The patient met the Yamaguchi criteria and was subsequently diagnosed with AOSD. She was treated with intravenous methylprednisolone 20 mg every 8 hours and was discharged 1 week later on oral prednisone 60 mg daily to be tapered over a period of months. At 2-week follow-up, the patient continued to experience rash and joint pain; oral methotrexate 10 mg weekly was added to her regimen, as well as vitamin D, calcium, and folic acid supplementation. At the next 2-week follow-up the patient noted improvement in the rash as well as the joint pain, but both still persisted. Prednisone was decreased to 50 mg daily and methotrexate was increased to 15 mg weekly. The patient continued to show improvement over the subsequent 3 months, during which prednisone was tapered to 10 mg daily and methotrexate was increased to 20 mg weekly. The patient showed resolution of symptoms at 3-month follow-up on this regimen, with plans to continue the prednisone taper and maintain methotrexate dosing.
Comment
Adult-onset Still disease is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, salmon-pink evanescent erythema, and lymphadenopathy.2 The condition also can cause liver dysfunction, splenomegaly, pericarditis, pleuritis, renal dysfunction, and a reactive hemophagocytic syndrome.1 Furthermore, one review of the literature described an association with delayed-onset malignancy.4 Early diagnosis is important yet challenging, as AOSD is a diagnosis of exclusion. The Yamaguchi criteria are the most widely used method of diagnosis and demonstrate more than 90% sensitivity.In addition to the Yamaguchi criteria, marked hyperferritinemia is characteristic of AOSD and can act as an indicator of disease activity.5 Interestingly, both of our patients had elevated ferritin levels, with patient 2 showing marked elevation (Table). In both patients, all major criteria were fulfilled, except the typical skin rash.
The skin rash in AOSD, classically consisting of an evanescent, salmon-pink erythema predominantly involving the extremities, has been observed in up to 87% of AOSD patients.5 The histology of the typical evanescent rash is nonspecific, characterized by a relatively sparse, perivascular, mixed inflammatory infiltrate. Notably, other skin manifestations may be found in patients with AOSD.1,2,5-16 Persistent pruritic papules and plaques are the most commonly reported nonclassical rash, presenting as erythematous, slightly scaly papules and plaques with a linear configuration typically on the trunk.2 Both of our patients presented with this atypical eruption. Importantly, the histopathology of this unique rash displays distinctive features, which can aid in early diagnosis. Findings include dyskeratotic keratinocytes in the cornified layers as well as in the epidermis, and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis. These findings were evident in both histopathologic studies of our patients (Figures 2 and 4). Although not present in our patients, dermal mucin deposition has been demonstrated in some reports.1,13,15
A 2015 review of the literature yielded 30 cases of AOSD with pruritic persistent papules and plaques.4 The study confirmed a linear, erythematous or brown rash on the back and neck in the majority of cases. Histologic findings were congruent with those reported in our 2 cases: necrotic keratinocytes in the upper epidermis with a neutrophilic infiltrate in the upper dermis without vasculitis. Most patients showed rapid resolution of the rash and symptoms with the use of prednisone, prednisolone, or intravenous pulsed methylprednisolone. Interestingly, a range of presentations were noted, including prurigo pigmentosalike urticarial papules; lichenoid papules; and dermatographismlike, dermatomyositislike, and lichen amyloidosis–like rashes.4 In our report, patient 2 presented with a rash in a dermat-omyositislike shawl distribution. It has been suggested that patients with dermatomyositislike rashes require more potent immunotherapy as compared to patients with other rash morphologies.4 The need for methotrexate in addition to a prednisone taper in the clinical course of patient 2 lends further support to this observation.
Conclusion
A clinically and pathologically distinct form of cutaneous disease—AOSD with persistent pruritic papules and plaques—was observed in our 2 patients. These histopathologic findings facilitated timely diagnosis in both patients. A range of clinical morphologies may exist in AOSD, an awareness of which is paramount. Adult-onset Still disease should be included in the differential diagnosis of a dermatomyositislike presentation in a shawl distribution. Prompt diagnosis is essential to ensure adequate therapy.
Adult-onset Still disease (AOSD) is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, evanescent skin rash, and lymphadenopathy. 1 The most commonly used criteria for diagnosing AOSD are the Yamaguchi criteria. 2 The major criteria include high fever for more than 1 week, arthralgia for more than 2 weeks, leukocytosis, and an evanescent skin rash. The minor criteria consist of sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and negative rheumatoid factor and antinuclear antibodies. Classically, the skin rash is described as an evanescent, salmon-colored erythema involving the extremities. Nevertheless, unusual cutaneous eruptions have been reported in AOSD, including persistent pruritic papules and plaques. 3 Importantly, this atypical rash demonstrates specific histologic findings that are not found on routine histopathology of a typical evanescent rash. We describe 2 patients with this atypical cutaneous eruption along with the unique histopathologic findings of AOSD.
Case Reports
Patient 1
A 23-year-old Chinese woman presented with periodic fevers, persistent rash, and joint pain of 2 years’ duration. Her medical history included splenectomy for hepatosplenomegaly as well as evaluation by hematology for lymphadenopathy; a cervical lymph node biopsy showed lymphoid and follicular hyperplasia.
Twenty days later, the patient was referred to the dermatology department for evaluation of the persistent rash. The patient described a history of flushing of the face, severe joint pain in both arms and legs, aching muscles, and persistent sore throat. The patient did not report any history of drug ingestion. Physical examination revealed a fever (temperature, 39.2°C); swollen nontender lymph nodes in the neck, axillae, and groin; and salmon-colored and hyperpigmented patches and thin plaques over the neck, chest, abdomen, and arms (Figure 1). A splenectomy scar also was noted. Peripheral blood was collected for laboratory analyses, which revealed transaminitis and moderate hyperferritinemia (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. The patient was admitted to the hospital, and a skin biopsy was performed. Histology showed superficial dyskeratotic keratinocytes and sparse perivascular infiltration of neutrophils in the upper dermis (Figure 2).
The patient was diagnosed with AOSD based on fulfillment of the Yamaguchi criteria.2 She was treated with methylprednisolone 60 mg daily and was discharged 14 days later. At 16-month follow-up, the patient demonstrated complete resolution of symptoms with a maintenance dose of prednisolone (7.5 mg daily).
Patient 2
A 23-year-old black woman presented to the emergency department 3 months postpartum with recurrent high fevers, worsening joint pain, and persistent itchy rash of 2 months’ duration. The patient had no history of travel, autoimmune disease, or sick contacts. She occasionally took aspirin for joint pain. Physical examination revealed a fever (temperature, 39.1°C) along with hyperpigmented patches and thin scaly hyperpigmented papules coalescing into a poorly demarcated V-shaped plaque on the upper back and posterior neck, extending to the chest in a shawl-like distribution (Figure 3). Submental lymphadenopathy was present. The spleen was not palpable.
Peripheral blood was collected for laboratory analysis and demonstrated transaminitis and a markedly high ferritin level (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. Skin biopsy was performed and demonstrated many necrotic keratinocytes, singly and in aggregates, distributed from the spinous layer to the stratum corneum. A neutrophilic infiltrate was present in the papillary dermis (Figure 4).
The patient met the Yamaguchi criteria and was subsequently diagnosed with AOSD. She was treated with intravenous methylprednisolone 20 mg every 8 hours and was discharged 1 week later on oral prednisone 60 mg daily to be tapered over a period of months. At 2-week follow-up, the patient continued to experience rash and joint pain; oral methotrexate 10 mg weekly was added to her regimen, as well as vitamin D, calcium, and folic acid supplementation. At the next 2-week follow-up the patient noted improvement in the rash as well as the joint pain, but both still persisted. Prednisone was decreased to 50 mg daily and methotrexate was increased to 15 mg weekly. The patient continued to show improvement over the subsequent 3 months, during which prednisone was tapered to 10 mg daily and methotrexate was increased to 20 mg weekly. The patient showed resolution of symptoms at 3-month follow-up on this regimen, with plans to continue the prednisone taper and maintain methotrexate dosing.
Comment
Adult-onset Still disease is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, salmon-pink evanescent erythema, and lymphadenopathy.2 The condition also can cause liver dysfunction, splenomegaly, pericarditis, pleuritis, renal dysfunction, and a reactive hemophagocytic syndrome.1 Furthermore, one review of the literature described an association with delayed-onset malignancy.4 Early diagnosis is important yet challenging, as AOSD is a diagnosis of exclusion. The Yamaguchi criteria are the most widely used method of diagnosis and demonstrate more than 90% sensitivity.In addition to the Yamaguchi criteria, marked hyperferritinemia is characteristic of AOSD and can act as an indicator of disease activity.5 Interestingly, both of our patients had elevated ferritin levels, with patient 2 showing marked elevation (Table). In both patients, all major criteria were fulfilled, except the typical skin rash.
The skin rash in AOSD, classically consisting of an evanescent, salmon-pink erythema predominantly involving the extremities, has been observed in up to 87% of AOSD patients.5 The histology of the typical evanescent rash is nonspecific, characterized by a relatively sparse, perivascular, mixed inflammatory infiltrate. Notably, other skin manifestations may be found in patients with AOSD.1,2,5-16 Persistent pruritic papules and plaques are the most commonly reported nonclassical rash, presenting as erythematous, slightly scaly papules and plaques with a linear configuration typically on the trunk.2 Both of our patients presented with this atypical eruption. Importantly, the histopathology of this unique rash displays distinctive features, which can aid in early diagnosis. Findings include dyskeratotic keratinocytes in the cornified layers as well as in the epidermis, and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis. These findings were evident in both histopathologic studies of our patients (Figures 2 and 4). Although not present in our patients, dermal mucin deposition has been demonstrated in some reports.1,13,15
A 2015 review of the literature yielded 30 cases of AOSD with pruritic persistent papules and plaques.4 The study confirmed a linear, erythematous or brown rash on the back and neck in the majority of cases. Histologic findings were congruent with those reported in our 2 cases: necrotic keratinocytes in the upper epidermis with a neutrophilic infiltrate in the upper dermis without vasculitis. Most patients showed rapid resolution of the rash and symptoms with the use of prednisone, prednisolone, or intravenous pulsed methylprednisolone. Interestingly, a range of presentations were noted, including prurigo pigmentosalike urticarial papules; lichenoid papules; and dermatographismlike, dermatomyositislike, and lichen amyloidosis–like rashes.4 In our report, patient 2 presented with a rash in a dermat-omyositislike shawl distribution. It has been suggested that patients with dermatomyositislike rashes require more potent immunotherapy as compared to patients with other rash morphologies.4 The need for methotrexate in addition to a prednisone taper in the clinical course of patient 2 lends further support to this observation.
Conclusion
A clinically and pathologically distinct form of cutaneous disease—AOSD with persistent pruritic papules and plaques—was observed in our 2 patients. These histopathologic findings facilitated timely diagnosis in both patients. A range of clinical morphologies may exist in AOSD, an awareness of which is paramount. Adult-onset Still disease should be included in the differential diagnosis of a dermatomyositislike presentation in a shawl distribution. Prompt diagnosis is essential to ensure adequate therapy.
- Yamamoto T. Cutaneous manifestations associated with adult-onset Still’s disease: important diagnostic values. Rheumatol Int. 2012;32:2233-2237.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-431.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associates with delayed malignancy [published online June 6, 2015]. J Am Acad Dermatol. 2015;73:294-303.
- Schwarz-Eywill M, Heilig B, Bauer H, et al. Evaluation of serum ferritin as a marker for adult Still’s disease activity. Ann Rheum Dis. 1992;51:683-685.
- Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17:1058-1063.
- Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241-242.
- Lübbe J, Hofer M, Chavaz P, et al. Adult onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710-713.
- Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still’s disease. Dermatology. 2001;202:333-335.
- Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still’s disease. Int J Dermatol. 2003;42:824-825.
- Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still’s disease. Ann Dermatol Venereol. 2005;132:693-696.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still’s disease. Am J Dermatopathol. 2007;49:194-196.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still’s disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Azeck AG, Littlewood SM. Adult-onset Still’s disease with atypical cutaneous features. J Eur Acad Dermatol Venereol. 2005;19:360-363.
- Yamamoto T. Cutaneous manifestations associated with adult-onset Still’s disease: important diagnostic values. Rheumatol Int. 2012;32:2233-2237.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-431.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associates with delayed malignancy [published online June 6, 2015]. J Am Acad Dermatol. 2015;73:294-303.
- Schwarz-Eywill M, Heilig B, Bauer H, et al. Evaluation of serum ferritin as a marker for adult Still’s disease activity. Ann Rheum Dis. 1992;51:683-685.
- Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17:1058-1063.
- Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241-242.
- Lübbe J, Hofer M, Chavaz P, et al. Adult onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710-713.
- Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still’s disease. Dermatology. 2001;202:333-335.
- Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still’s disease. Int J Dermatol. 2003;42:824-825.
- Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still’s disease. Ann Dermatol Venereol. 2005;132:693-696.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still’s disease. Am J Dermatopathol. 2007;49:194-196.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still’s disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Azeck AG, Littlewood SM. Adult-onset Still’s disease with atypical cutaneous features. J Eur Acad Dermatol Venereol. 2005;19:360-363.
Practice Points
- Serologic testing and skin biopsy are necessary in the timely and appropriate diagnosis of adult-onset Still disease (AOSD).
- In patients with a persistent pruritic papular rash, consider AOSD if there is a supporting history.
- Skin biopsy is diagnostic of AOSD with the unique histopathologic findings of dyskeratotic keratinocytes in the cornified layers as well as in the epidermis and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis.
New System Classifies Idiopathic Inflammatory Myopathies
A clinical and serologic approach to identifying these disorders may eliminate the need for muscle biopsy.
A new system that incorporates clinical and serologic data may help classify idiopathic inflammatory myopathies, according to an analysis published online ahead of print September 10 in JAMA Neurology.
By analyzing the patterns of relationships between 47 variables in this observational, retrospective cohort study, investigators identified four clusters of patients that corresponded to known subtypes of idiopathic inflammatory myopathy. Myositis-specific autoantibodies played a key role in predicting whether a patient belonged in a given cluster, according to the investigators. Myositis-specific antibodies known to be associated with certain subgroups were observed in the corresponding clusters that the researchers identified.
“This [finding] emphasizes that muscle biopsy may no longer be necessary for diagnosis of idiopathic inflammatory myopathies in patients with myositis-specific antibodies and corresponding phenotypes,” said Kubéraka Mariampillai, PhD, of the Université Pierre et Marie Curie, Institut National de la Santé et de la Recherche Médicale (INSERM) in Paris, and colleagues.
The study was based on data for 260 patients in the database of the French Myositis Network. Patients’ mean age was 60, and 63% were women.
Investigators conducted a multiple correspondence analysis based on 47 selected variables, including age, ethnicity, historical and recent diagnoses, dermatologic changes, creatine kinase levels, myositis-specific antibodies, and pathologic characteristics. They identified four subgroups of patients corresponding to dermatomyositis, inclusion body myositis, immune-mediated necrotizing myopathy, and antisynthetase syndrome.
Using decisional algorithm trees, investigators found that the pathologic data were “dispensable,” said the authors. The best tree omitted variables related to muscle biopsy and had a 78% correct estimation based on antisynthetase syndrome antibodies, dermatomyositis rash, and finger flexor scores of 3 or less, said the investigators. “The classification quality of the tree was appreciated on the basis of all classification criteria, with an overall sensitivity of 77.0% and a specificity of 92.0%.”
Patients with polymyositis were included in the study, but were grouped mainly in the clusters corresponding to immune-mediated necrotizing myopathy and antisynthetase syndrome. “This finding indicates that patients with polymyositis do not represent a subgroup of patients, and use of this term should probably be discontinued,” Dr. Mariampillai and colleagues concluded.
—Andrew D. Bowser
Mariampillai K, Granger B, Amelin D, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol. 2018 Sep 10 [Epub ahead of print].
A clinical and serologic approach to identifying these disorders may eliminate the need for muscle biopsy.
A clinical and serologic approach to identifying these disorders may eliminate the need for muscle biopsy.
A new system that incorporates clinical and serologic data may help classify idiopathic inflammatory myopathies, according to an analysis published online ahead of print September 10 in JAMA Neurology.
By analyzing the patterns of relationships between 47 variables in this observational, retrospective cohort study, investigators identified four clusters of patients that corresponded to known subtypes of idiopathic inflammatory myopathy. Myositis-specific autoantibodies played a key role in predicting whether a patient belonged in a given cluster, according to the investigators. Myositis-specific antibodies known to be associated with certain subgroups were observed in the corresponding clusters that the researchers identified.
“This [finding] emphasizes that muscle biopsy may no longer be necessary for diagnosis of idiopathic inflammatory myopathies in patients with myositis-specific antibodies and corresponding phenotypes,” said Kubéraka Mariampillai, PhD, of the Université Pierre et Marie Curie, Institut National de la Santé et de la Recherche Médicale (INSERM) in Paris, and colleagues.
The study was based on data for 260 patients in the database of the French Myositis Network. Patients’ mean age was 60, and 63% were women.
Investigators conducted a multiple correspondence analysis based on 47 selected variables, including age, ethnicity, historical and recent diagnoses, dermatologic changes, creatine kinase levels, myositis-specific antibodies, and pathologic characteristics. They identified four subgroups of patients corresponding to dermatomyositis, inclusion body myositis, immune-mediated necrotizing myopathy, and antisynthetase syndrome.
Using decisional algorithm trees, investigators found that the pathologic data were “dispensable,” said the authors. The best tree omitted variables related to muscle biopsy and had a 78% correct estimation based on antisynthetase syndrome antibodies, dermatomyositis rash, and finger flexor scores of 3 or less, said the investigators. “The classification quality of the tree was appreciated on the basis of all classification criteria, with an overall sensitivity of 77.0% and a specificity of 92.0%.”
Patients with polymyositis were included in the study, but were grouped mainly in the clusters corresponding to immune-mediated necrotizing myopathy and antisynthetase syndrome. “This finding indicates that patients with polymyositis do not represent a subgroup of patients, and use of this term should probably be discontinued,” Dr. Mariampillai and colleagues concluded.
—Andrew D. Bowser
Mariampillai K, Granger B, Amelin D, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol. 2018 Sep 10 [Epub ahead of print].
A new system that incorporates clinical and serologic data may help classify idiopathic inflammatory myopathies, according to an analysis published online ahead of print September 10 in JAMA Neurology.
By analyzing the patterns of relationships between 47 variables in this observational, retrospective cohort study, investigators identified four clusters of patients that corresponded to known subtypes of idiopathic inflammatory myopathy. Myositis-specific autoantibodies played a key role in predicting whether a patient belonged in a given cluster, according to the investigators. Myositis-specific antibodies known to be associated with certain subgroups were observed in the corresponding clusters that the researchers identified.
“This [finding] emphasizes that muscle biopsy may no longer be necessary for diagnosis of idiopathic inflammatory myopathies in patients with myositis-specific antibodies and corresponding phenotypes,” said Kubéraka Mariampillai, PhD, of the Université Pierre et Marie Curie, Institut National de la Santé et de la Recherche Médicale (INSERM) in Paris, and colleagues.
The study was based on data for 260 patients in the database of the French Myositis Network. Patients’ mean age was 60, and 63% were women.
Investigators conducted a multiple correspondence analysis based on 47 selected variables, including age, ethnicity, historical and recent diagnoses, dermatologic changes, creatine kinase levels, myositis-specific antibodies, and pathologic characteristics. They identified four subgroups of patients corresponding to dermatomyositis, inclusion body myositis, immune-mediated necrotizing myopathy, and antisynthetase syndrome.
Using decisional algorithm trees, investigators found that the pathologic data were “dispensable,” said the authors. The best tree omitted variables related to muscle biopsy and had a 78% correct estimation based on antisynthetase syndrome antibodies, dermatomyositis rash, and finger flexor scores of 3 or less, said the investigators. “The classification quality of the tree was appreciated on the basis of all classification criteria, with an overall sensitivity of 77.0% and a specificity of 92.0%.”
Patients with polymyositis were included in the study, but were grouped mainly in the clusters corresponding to immune-mediated necrotizing myopathy and antisynthetase syndrome. “This finding indicates that patients with polymyositis do not represent a subgroup of patients, and use of this term should probably be discontinued,” Dr. Mariampillai and colleagues concluded.
—Andrew D. Bowser
Mariampillai K, Granger B, Amelin D, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol. 2018 Sep 10 [Epub ahead of print].
Early Treatment Improves Outcomes in Neuromyelitis Optica Spectrum Disorder
Research has improved understanding of the disorder’s pathology and indicated which treatments are most beneficial.
HILTON HEAD, SC—Neuromyelitis optica spectrum disorder (NMOSD) can result in severe disability, but early diagnosis and treatment increase the likelihood that a patient will regain his or her baseline function, according to an overview provided at the 41st Annual Contemporary Clinical Neurology Symposium. Increased understanding of NMOSD has led to new diagnostic criteria, and emerging data are clarifying the question of effective treatments.
NMOSD As a Distinct Disorder
NMOSD originally was recognized as an inflammatory disorder of the CNS that causes transverse myelitis and optic neuritis, said Siddharama Pawate, MD, Associate Professor of Neurology at Vanderbilt University Medical Center in Nashville. Although neurologists first considered NMOSD a variant of multiple sclerosis (MS), the former has several features that distinguish it from the latter. These features include exceptionally severe relapses, spinal cord lesions that span more than three vertebral segments, and CSF that reveals pleocytosis and high protein levels. In addition, some MS treatments such as interferons, fingolimod, and natalizumab usually exacerbate, rather than mitigate, NMOSD.

In 2004, researchers found that antibodies against aquaporin-4 (AQP4) were almost 100% specific for NMOSD. Astrocytes and ependymal cells, but not oligodendrocytes or neurons, express AQP4. When anti-AQP4 antibodies bind to the membrane of an astrocyte, they disrupt the blood–brain barrier and eventually cause the astrocyte to die. The death of astrocytes promotes secondary damage of oligodendrocytes and neurons. Because of these processes, swelling in the spinal cord and the optic nerve are prominent features of NMOSD on MRI, said Dr. Pawate. The swelling, in turn, can lead to vascular compromise and necrosis, thus
Clinical Presentations of NMOSD
Approximately 75% of patients with NMOSD present with optic neuritis. The next most common clinical presentation is transverse myelitis, which may include paraparesis or quadriparesis, loss of sensation, and bladder or bowel dysfunction. About 35% of patients present with transverse myelitis. Optic neuritis and transverse myelitis occur simultaneously in about 10% of patients who present with NMOSD. “Unlike the MS lesions that are mostly in the white matter, NMOSD lesions in the spinal cord involve gray matter and white matter,” said Dr. Pawate. Other clinical features specific to NMOSD include severe neuropathic pain, tonic spasms that last for as long as 90 seconds, and pruritus. The latter symptom responds well to gabapentin, said Dr. Pawate.
NMOSD entails more severe optic neuritis than that associated with MS. It can be bilateral and lead to complete loss of vision. Optic neuritis usually is longitudinally extensive in NMOSD. A lesion length of 17.6 mm suffices to distinguish NMOSD from MS, and a length of more than 35 mm is approximately 100% specific for the former disorder. Swelling can cause necrosis in the optic nerve and result in poor recovery of vision. Furthermore, homonymous hemianopsia can happen in NMOSD due to damage to the optic tracts, but is rare in MS.
The clinical presentation of NMOSD also may include area postrema syndrome, which entails intractable nausea and vomiting. Patients may have cerebral or cerebellar lesions, symptomatic narcolepsy, or endocrine dysfunction (eg, syndrome of inappropriate antidiuretic hormone secretion).
The 2015 Diagnostic Criteria
Deepening understanding of NMOSD led to the development of new diagnostic criteria in 2015. The criteria identify optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome with MRI lesions typical of NMOSD, and symptomatic cerebral syndrome with MRI lesions typical of NMOSD as the six core clinical characteristics. If the patient tests positive for AQP4 antibodies and has one core clinical characteristic, a diagnosis of NMOSD is appropriate. If the patient tests negative for AQP4 antibodies, he or she must have two or more core clinical characteristics (at least one of which should be optic neuritis, acute myelitis, or area postrema syndrome) that are disseminated in space for a diagnosis of NMOSD to be appropriate. In both cases, alternative diagnoses also must be excluded.
Some patients who test negative for AQP4 antibodies have myelin oligodendrocyte glycoprotein (MOG) antibodies. Although AQP4-mediated NMOSD and MOG-mediated NMOSD are clinically similar, they are distinct diseases, said Dr. Pawate. Pathology primarily affects myelin, not astrocytes, in MOG-positive NMOSD. Patients with MOG-positive NMOSD also tend not to have relapses, and the disorder has a better prognosis, compared with AQP4-positive NMOSD.
Between 15% and 30% of patients with NMOSD have oligoclonal bands, and 20% have an elevated IgG index. Unlike in MS, however, these findings tend to be transient. In addition, as much as 30% of patients with NMOSD may have other comorbid autoimmune disorders. A review of the literature indicated that 22 autoimmune conditions, including myasthenia gravis, ulcerative colitis, hypothyroidism, and thrombocytopenia, have been observed in patients with NMOSD. “We think that this indicates a heightened autoimmune response in general in those patients,” said Dr. Pawate.
Treatment of NMOSD
Treatment of NMOSD is based on the principle that relapses, which can be severe, result in disability. The disease does not progress between relapses, unlike MS. Therefore, the consensus is that relapses should be treated promptly and aggressively. Maintenance immunosuppression may prevent future relapses, and other symptoms can be managed as needed. “I have had a patient for 10 years now who, after the first attack, has not had any more attacks and is living fairly normally,” said Dr. Pawate.
Evidence supports plasma exchange as a standard treatment for relapses in NMOSD. It requires five to seven sessions and 1.5 volumes. This treatment removes antibodies and other soluble disease mediators, such as complement. Bonnan and Cabre found that administering plasma exchange early in the relapse can mitigate astrocyte dysfunction and prevent neuronal death.
In a 2012 study, patients with optic neuritis were treated with IV corticosteroids or IV corticosteroids plus plasma exchange. Approximately 75% of patients treated with plasma exchange had a final visual acuity better than 20/40, compared with 39% of patients who received steroids alone. About 13% of patients treated with plasma exchange had a final visual acuity worse than 20/200, compared with 56% of patients who received steroids alone.
In 2017, Bonnan et al found that short delay to plasma exchange is the strongest predictor of outcome in severe attacks of NMOSD. The rate of good recovery was approximately 80% when plasma exchange was performed within a day or so of relapse onset. Plasma exchange also was effective when administered at a week after onset. The therapeutic window closes at approximately three weeks after onset, said Dr. Pawate.
Neurologists should begin maintenance immunosuppression immediately, said Dr. Pawate. Rituximab has the best evidential support for this indication, but the drug can be expensive, and insurance reimbursement is not easy to obtain. Only one formal publication has examined mycophenolate, but neurologists have a lot of clinical experience with this treatment. It takes three to four months before mycophenolate achieves its full efficacy, so bridge therapy is required. Mealy et al found that with optimal dosing, rituximab reduces patients’ relapse rate by 94%, mycophenolate reduces it by 90%, and azathioprine reduces it by 72%.
In one case series, tocilizumab, a monoclonal antibody targeting IL-6, was effective in patients who had not responded to rituximab. Eculizumab, a monoclonal antibody targeting the complement factor C5a, may be another option, based on recent reports. Maintenance immunosuppression should continue for at least five years, and indefinitely for patients with AQP4 antibodies, said Dr. Pawate.
If it is not clear whether the diagnosis is NMOSD or MS, a neurologist should treat the patient for NMOSD, said Dr. Pawate. Mycophenolate and rituximab, the two most commonly used NMOSD treatments, are effective against MS as well, but several treatments for MS, such as natalizumab, fingolimod, and interferon beta, may exacerbate NMOSD.
A clinical evaluation is the best way to monitor the treatment’s effect, said Dr. Pawate. “Make sure they are not having any new symptoms, new vision complaints, new motor weakness, or sensory complaints. MRI is of limited value in treatment monitoring…. Basically, nothing substitutes for talking to the patient and performing an examination.”
—Erik Greb
Suggested Reading
Bonnan M, Cabre P. Plasma exchange in severe attacks of neuromyelitis optica. Mult Scler Int. 2012; 2012:787630.
Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89(4):346-351.
Hyun JW, Jeong IH, Joung A, et al. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016;86(19):1772-1779.
Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity. 2014;47(3):154-161.
Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum-autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106-2112.
Mealy MA, Wingerchuk DM, Palace J, et al. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. 2014;71(3):324-330.
Merle H, Olindo S, Jeannin S, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. 2012;130(7):858-862.
Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(4):663-679.
Research has improved understanding of the disorder’s pathology and indicated which treatments are most beneficial.
Research has improved understanding of the disorder’s pathology and indicated which treatments are most beneficial.
HILTON HEAD, SC—Neuromyelitis optica spectrum disorder (NMOSD) can result in severe disability, but early diagnosis and treatment increase the likelihood that a patient will regain his or her baseline function, according to an overview provided at the 41st Annual Contemporary Clinical Neurology Symposium. Increased understanding of NMOSD has led to new diagnostic criteria, and emerging data are clarifying the question of effective treatments.
NMOSD As a Distinct Disorder
NMOSD originally was recognized as an inflammatory disorder of the CNS that causes transverse myelitis and optic neuritis, said Siddharama Pawate, MD, Associate Professor of Neurology at Vanderbilt University Medical Center in Nashville. Although neurologists first considered NMOSD a variant of multiple sclerosis (MS), the former has several features that distinguish it from the latter. These features include exceptionally severe relapses, spinal cord lesions that span more than three vertebral segments, and CSF that reveals pleocytosis and high protein levels. In addition, some MS treatments such as interferons, fingolimod, and natalizumab usually exacerbate, rather than mitigate, NMOSD.
In 2004, researchers found that antibodies against aquaporin-4 (AQP4) were almost 100% specific for NMOSD. Astrocytes and ependymal cells, but not oligodendrocytes or neurons, express AQP4. When anti-AQP4 antibodies bind to the membrane of an astrocyte, they disrupt the blood–brain barrier and eventually cause the astrocyte to die. The death of astrocytes promotes secondary damage of oligodendrocytes and neurons. Because of these processes, swelling in the spinal cord and the optic nerve are prominent features of NMOSD on MRI, said Dr. Pawate. The swelling, in turn, can lead to vascular compromise and necrosis, thus
Clinical Presentations of NMOSD
Approximately 75% of patients with NMOSD present with optic neuritis. The next most common clinical presentation is transverse myelitis, which may include paraparesis or quadriparesis, loss of sensation, and bladder or bowel dysfunction. About 35% of patients present with transverse myelitis. Optic neuritis and transverse myelitis occur simultaneously in about 10% of patients who present with NMOSD. “Unlike the MS lesions that are mostly in the white matter, NMOSD lesions in the spinal cord involve gray matter and white matter,” said Dr. Pawate. Other clinical features specific to NMOSD include severe neuropathic pain, tonic spasms that last for as long as 90 seconds, and pruritus. The latter symptom responds well to gabapentin, said Dr. Pawate.
NMOSD entails more severe optic neuritis than that associated with MS. It can be bilateral and lead to complete loss of vision. Optic neuritis usually is longitudinally extensive in NMOSD. A lesion length of 17.6 mm suffices to distinguish NMOSD from MS, and a length of more than 35 mm is approximately 100% specific for the former disorder. Swelling can cause necrosis in the optic nerve and result in poor recovery of vision. Furthermore, homonymous hemianopsia can happen in NMOSD due to damage to the optic tracts, but is rare in MS.
The clinical presentation of NMOSD also may include area postrema syndrome, which entails intractable nausea and vomiting. Patients may have cerebral or cerebellar lesions, symptomatic narcolepsy, or endocrine dysfunction (eg, syndrome of inappropriate antidiuretic hormone secretion).
The 2015 Diagnostic Criteria
Deepening understanding of NMOSD led to the development of new diagnostic criteria in 2015. The criteria identify optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome with MRI lesions typical of NMOSD, and symptomatic cerebral syndrome with MRI lesions typical of NMOSD as the six core clinical characteristics. If the patient tests positive for AQP4 antibodies and has one core clinical characteristic, a diagnosis of NMOSD is appropriate. If the patient tests negative for AQP4 antibodies, he or she must have two or more core clinical characteristics (at least one of which should be optic neuritis, acute myelitis, or area postrema syndrome) that are disseminated in space for a diagnosis of NMOSD to be appropriate. In both cases, alternative diagnoses also must be excluded.
Some patients who test negative for AQP4 antibodies have myelin oligodendrocyte glycoprotein (MOG) antibodies. Although AQP4-mediated NMOSD and MOG-mediated NMOSD are clinically similar, they are distinct diseases, said Dr. Pawate. Pathology primarily affects myelin, not astrocytes, in MOG-positive NMOSD. Patients with MOG-positive NMOSD also tend not to have relapses, and the disorder has a better prognosis, compared with AQP4-positive NMOSD.
Between 15% and 30% of patients with NMOSD have oligoclonal bands, and 20% have an elevated IgG index. Unlike in MS, however, these findings tend to be transient. In addition, as much as 30% of patients with NMOSD may have other comorbid autoimmune disorders. A review of the literature indicated that 22 autoimmune conditions, including myasthenia gravis, ulcerative colitis, hypothyroidism, and thrombocytopenia, have been observed in patients with NMOSD. “We think that this indicates a heightened autoimmune response in general in those patients,” said Dr. Pawate.
Treatment of NMOSD
Treatment of NMOSD is based on the principle that relapses, which can be severe, result in disability. The disease does not progress between relapses, unlike MS. Therefore, the consensus is that relapses should be treated promptly and aggressively. Maintenance immunosuppression may prevent future relapses, and other symptoms can be managed as needed. “I have had a patient for 10 years now who, after the first attack, has not had any more attacks and is living fairly normally,” said Dr. Pawate.
Evidence supports plasma exchange as a standard treatment for relapses in NMOSD. It requires five to seven sessions and 1.5 volumes. This treatment removes antibodies and other soluble disease mediators, such as complement. Bonnan and Cabre found that administering plasma exchange early in the relapse can mitigate astrocyte dysfunction and prevent neuronal death.
In a 2012 study, patients with optic neuritis were treated with IV corticosteroids or IV corticosteroids plus plasma exchange. Approximately 75% of patients treated with plasma exchange had a final visual acuity better than 20/40, compared with 39% of patients who received steroids alone. About 13% of patients treated with plasma exchange had a final visual acuity worse than 20/200, compared with 56% of patients who received steroids alone.
In 2017, Bonnan et al found that short delay to plasma exchange is the strongest predictor of outcome in severe attacks of NMOSD. The rate of good recovery was approximately 80% when plasma exchange was performed within a day or so of relapse onset. Plasma exchange also was effective when administered at a week after onset. The therapeutic window closes at approximately three weeks after onset, said Dr. Pawate.
Neurologists should begin maintenance immunosuppression immediately, said Dr. Pawate. Rituximab has the best evidential support for this indication, but the drug can be expensive, and insurance reimbursement is not easy to obtain. Only one formal publication has examined mycophenolate, but neurologists have a lot of clinical experience with this treatment. It takes three to four months before mycophenolate achieves its full efficacy, so bridge therapy is required. Mealy et al found that with optimal dosing, rituximab reduces patients’ relapse rate by 94%, mycophenolate reduces it by 90%, and azathioprine reduces it by 72%.
In one case series, tocilizumab, a monoclonal antibody targeting IL-6, was effective in patients who had not responded to rituximab. Eculizumab, a monoclonal antibody targeting the complement factor C5a, may be another option, based on recent reports. Maintenance immunosuppression should continue for at least five years, and indefinitely for patients with AQP4 antibodies, said Dr. Pawate.
If it is not clear whether the diagnosis is NMOSD or MS, a neurologist should treat the patient for NMOSD, said Dr. Pawate. Mycophenolate and rituximab, the two most commonly used NMOSD treatments, are effective against MS as well, but several treatments for MS, such as natalizumab, fingolimod, and interferon beta, may exacerbate NMOSD.
A clinical evaluation is the best way to monitor the treatment’s effect, said Dr. Pawate. “Make sure they are not having any new symptoms, new vision complaints, new motor weakness, or sensory complaints. MRI is of limited value in treatment monitoring…. Basically, nothing substitutes for talking to the patient and performing an examination.”
—Erik Greb
Suggested Reading
Bonnan M, Cabre P. Plasma exchange in severe attacks of neuromyelitis optica. Mult Scler Int. 2012; 2012:787630.
Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89(4):346-351.
Hyun JW, Jeong IH, Joung A, et al. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016;86(19):1772-1779.
Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity. 2014;47(3):154-161.
Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum-autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106-2112.
Mealy MA, Wingerchuk DM, Palace J, et al. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. 2014;71(3):324-330.
Merle H, Olindo S, Jeannin S, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. 2012;130(7):858-862.
Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(4):663-679.
HILTON HEAD, SC—Neuromyelitis optica spectrum disorder (NMOSD) can result in severe disability, but early diagnosis and treatment increase the likelihood that a patient will regain his or her baseline function, according to an overview provided at the 41st Annual Contemporary Clinical Neurology Symposium. Increased understanding of NMOSD has led to new diagnostic criteria, and emerging data are clarifying the question of effective treatments.
NMOSD As a Distinct Disorder
NMOSD originally was recognized as an inflammatory disorder of the CNS that causes transverse myelitis and optic neuritis, said Siddharama Pawate, MD, Associate Professor of Neurology at Vanderbilt University Medical Center in Nashville. Although neurologists first considered NMOSD a variant of multiple sclerosis (MS), the former has several features that distinguish it from the latter. These features include exceptionally severe relapses, spinal cord lesions that span more than three vertebral segments, and CSF that reveals pleocytosis and high protein levels. In addition, some MS treatments such as interferons, fingolimod, and natalizumab usually exacerbate, rather than mitigate, NMOSD.
In 2004, researchers found that antibodies against aquaporin-4 (AQP4) were almost 100% specific for NMOSD. Astrocytes and ependymal cells, but not oligodendrocytes or neurons, express AQP4. When anti-AQP4 antibodies bind to the membrane of an astrocyte, they disrupt the blood–brain barrier and eventually cause the astrocyte to die. The death of astrocytes promotes secondary damage of oligodendrocytes and neurons. Because of these processes, swelling in the spinal cord and the optic nerve are prominent features of NMOSD on MRI, said Dr. Pawate. The swelling, in turn, can lead to vascular compromise and necrosis, thus
Clinical Presentations of NMOSD
Approximately 75% of patients with NMOSD present with optic neuritis. The next most common clinical presentation is transverse myelitis, which may include paraparesis or quadriparesis, loss of sensation, and bladder or bowel dysfunction. About 35% of patients present with transverse myelitis. Optic neuritis and transverse myelitis occur simultaneously in about 10% of patients who present with NMOSD. “Unlike the MS lesions that are mostly in the white matter, NMOSD lesions in the spinal cord involve gray matter and white matter,” said Dr. Pawate. Other clinical features specific to NMOSD include severe neuropathic pain, tonic spasms that last for as long as 90 seconds, and pruritus. The latter symptom responds well to gabapentin, said Dr. Pawate.
NMOSD entails more severe optic neuritis than that associated with MS. It can be bilateral and lead to complete loss of vision. Optic neuritis usually is longitudinally extensive in NMOSD. A lesion length of 17.6 mm suffices to distinguish NMOSD from MS, and a length of more than 35 mm is approximately 100% specific for the former disorder. Swelling can cause necrosis in the optic nerve and result in poor recovery of vision. Furthermore, homonymous hemianopsia can happen in NMOSD due to damage to the optic tracts, but is rare in MS.
The clinical presentation of NMOSD also may include area postrema syndrome, which entails intractable nausea and vomiting. Patients may have cerebral or cerebellar lesions, symptomatic narcolepsy, or endocrine dysfunction (eg, syndrome of inappropriate antidiuretic hormone secretion).
The 2015 Diagnostic Criteria
Deepening understanding of NMOSD led to the development of new diagnostic criteria in 2015. The criteria identify optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome with MRI lesions typical of NMOSD, and symptomatic cerebral syndrome with MRI lesions typical of NMOSD as the six core clinical characteristics. If the patient tests positive for AQP4 antibodies and has one core clinical characteristic, a diagnosis of NMOSD is appropriate. If the patient tests negative for AQP4 antibodies, he or she must have two or more core clinical characteristics (at least one of which should be optic neuritis, acute myelitis, or area postrema syndrome) that are disseminated in space for a diagnosis of NMOSD to be appropriate. In both cases, alternative diagnoses also must be excluded.
Some patients who test negative for AQP4 antibodies have myelin oligodendrocyte glycoprotein (MOG) antibodies. Although AQP4-mediated NMOSD and MOG-mediated NMOSD are clinically similar, they are distinct diseases, said Dr. Pawate. Pathology primarily affects myelin, not astrocytes, in MOG-positive NMOSD. Patients with MOG-positive NMOSD also tend not to have relapses, and the disorder has a better prognosis, compared with AQP4-positive NMOSD.
Between 15% and 30% of patients with NMOSD have oligoclonal bands, and 20% have an elevated IgG index. Unlike in MS, however, these findings tend to be transient. In addition, as much as 30% of patients with NMOSD may have other comorbid autoimmune disorders. A review of the literature indicated that 22 autoimmune conditions, including myasthenia gravis, ulcerative colitis, hypothyroidism, and thrombocytopenia, have been observed in patients with NMOSD. “We think that this indicates a heightened autoimmune response in general in those patients,” said Dr. Pawate.
Treatment of NMOSD
Treatment of NMOSD is based on the principle that relapses, which can be severe, result in disability. The disease does not progress between relapses, unlike MS. Therefore, the consensus is that relapses should be treated promptly and aggressively. Maintenance immunosuppression may prevent future relapses, and other symptoms can be managed as needed. “I have had a patient for 10 years now who, after the first attack, has not had any more attacks and is living fairly normally,” said Dr. Pawate.
Evidence supports plasma exchange as a standard treatment for relapses in NMOSD. It requires five to seven sessions and 1.5 volumes. This treatment removes antibodies and other soluble disease mediators, such as complement. Bonnan and Cabre found that administering plasma exchange early in the relapse can mitigate astrocyte dysfunction and prevent neuronal death.
In a 2012 study, patients with optic neuritis were treated with IV corticosteroids or IV corticosteroids plus plasma exchange. Approximately 75% of patients treated with plasma exchange had a final visual acuity better than 20/40, compared with 39% of patients who received steroids alone. About 13% of patients treated with plasma exchange had a final visual acuity worse than 20/200, compared with 56% of patients who received steroids alone.
In 2017, Bonnan et al found that short delay to plasma exchange is the strongest predictor of outcome in severe attacks of NMOSD. The rate of good recovery was approximately 80% when plasma exchange was performed within a day or so of relapse onset. Plasma exchange also was effective when administered at a week after onset. The therapeutic window closes at approximately three weeks after onset, said Dr. Pawate.
Neurologists should begin maintenance immunosuppression immediately, said Dr. Pawate. Rituximab has the best evidential support for this indication, but the drug can be expensive, and insurance reimbursement is not easy to obtain. Only one formal publication has examined mycophenolate, but neurologists have a lot of clinical experience with this treatment. It takes three to four months before mycophenolate achieves its full efficacy, so bridge therapy is required. Mealy et al found that with optimal dosing, rituximab reduces patients’ relapse rate by 94%, mycophenolate reduces it by 90%, and azathioprine reduces it by 72%.
In one case series, tocilizumab, a monoclonal antibody targeting IL-6, was effective in patients who had not responded to rituximab. Eculizumab, a monoclonal antibody targeting the complement factor C5a, may be another option, based on recent reports. Maintenance immunosuppression should continue for at least five years, and indefinitely for patients with AQP4 antibodies, said Dr. Pawate.
If it is not clear whether the diagnosis is NMOSD or MS, a neurologist should treat the patient for NMOSD, said Dr. Pawate. Mycophenolate and rituximab, the two most commonly used NMOSD treatments, are effective against MS as well, but several treatments for MS, such as natalizumab, fingolimod, and interferon beta, may exacerbate NMOSD.
A clinical evaluation is the best way to monitor the treatment’s effect, said Dr. Pawate. “Make sure they are not having any new symptoms, new vision complaints, new motor weakness, or sensory complaints. MRI is of limited value in treatment monitoring…. Basically, nothing substitutes for talking to the patient and performing an examination.”
—Erik Greb
Suggested Reading
Bonnan M, Cabre P. Plasma exchange in severe attacks of neuromyelitis optica. Mult Scler Int. 2012; 2012:787630.
Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89(4):346-351.
Hyun JW, Jeong IH, Joung A, et al. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016;86(19):1772-1779.
Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity. 2014;47(3):154-161.
Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum-autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106-2112.
Mealy MA, Wingerchuk DM, Palace J, et al. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. 2014;71(3):324-330.
Merle H, Olindo S, Jeannin S, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. 2012;130(7):858-862.
Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(4):663-679.
IgA vasculitis may be more common in adults than assumed
LAS VEGAS – IgA vasculitis has a reputation as an illness of childhood, but rheumatologist Alexandra Villa-Forte, MD, MPH, cautioned colleagues that it can strike adults, too, often in a much more severe form. And, she warned, it’s likely not as rare as physicians assume.
“I believe it’s more common in adults than reported. There’s a huge problem with establishing the right way to make the diagnosis in adults, which is why it is missed,” said Dr. Villa-Forte of the Cleveland Clinic, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
But even if IgA vasculitis (IV) is diagnosed correctly in adults, there are many questions about how to move forward, she said. “Treatment in adults remains a problem since we don’t have data.”
IV is also known as Henoch-Schönlein purpura, or HSP, and “spring fever” because it often appears in children in the spring following an upper respiratory infection.
The condition causes vasculitis, the swelling of small blood vessels in organs such as the skin, joints, kidneys, and intestines. Leaking blood vessels can cause skin rashes known as purpura.
The estimated ranges of disease are high, with the annual incidence in children estimated at 3-26 per 100,000 and in adults at 0.1-1.8 per 100,000. Dr. Villa-Forte noted that the male-to-female ratio is 1.5, and she said the condition is less common in African Americans.
“In children, this is a disease that is frequently self-limited. Most of them don’t need treatment, and most of them do not relapse,” Dr. Villa-Forte said. “In adults, it’s more resistant to treatment, frequently chronic, and frequently relapsing over the years.”
There are other differences in IV between children and adults. “In children, there is a clear seasonal pattern of disease that is not seen in adults,” she said, and it’s linked to preceding infections.
“In adults, there are multiple causes, and most of the time they’re not identified,” she said.
As for diagnosis, she suggests looking at clinical presentation and whether tissue biopsy shows cutaneous leukocytoclastic vasculitis with IgA deposits. She cautioned that increased serum IgA is seen in about 50% of adult patients, making it an unreliable indicator.
Prognosis is much better for children than adults. According to a 2014 study, 80% of children completely recover, compared with 40% of adults. Persistent hematuria or proteinuria occurs in 30% of children and 60% of adults, respectively, while chronic renal failure occurs in 2% of children and 10% of adults (J Korean Med Sci. 2014 Feb;29[2]:198-203).
It’s possible that the latter number may be higher, with as many as 30% of adults developing chronic kidney disease (CKD), Dr. Villa-Forte said.
An estimated 97% of nephritis develops within the first 6 months of disease onset in adults, she said, “and in adults, the active renal disease can persist for over 20 years.”
Guidelines suggest that patients be monitored for CKD for 6 months after disease onset, but Dr. Villa-Forte said it’s better to monitor them for 12 months.
What about treatment? “The major challenge in adults is the real absence between correlation between initial presentation and long-term renal outcome,” she said. “That makes for a difficult choice in terms of treatment selection.”
Fever and cutaneous lesions above the waist may predict renal involvement, she said, although that isn’t confirmed, and increasing proteinuria is a probable factor predicting progression/complications.
According to Dr. Villa-Forte, the value of early treatment hasn’t been proved. Due to the risk of kidney problems, she said, “we don’t feel like we can just watch patients – that we need to do something. But there’s not good data supporting that.”
Various protocols involving steroids, early plasmapheresis, rituximab (Rituxan), and other drug regimens lack evidence, she said, although use of steroids in early nephritis may be beneficial in adults.
Dr. Villa-Forte had no relevant disclosures.
Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – IgA vasculitis has a reputation as an illness of childhood, but rheumatologist Alexandra Villa-Forte, MD, MPH, cautioned colleagues that it can strike adults, too, often in a much more severe form. And, she warned, it’s likely not as rare as physicians assume.
“I believe it’s more common in adults than reported. There’s a huge problem with establishing the right way to make the diagnosis in adults, which is why it is missed,” said Dr. Villa-Forte of the Cleveland Clinic, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
But even if IgA vasculitis (IV) is diagnosed correctly in adults, there are many questions about how to move forward, she said. “Treatment in adults remains a problem since we don’t have data.”
IV is also known as Henoch-Schönlein purpura, or HSP, and “spring fever” because it often appears in children in the spring following an upper respiratory infection.
The condition causes vasculitis, the swelling of small blood vessels in organs such as the skin, joints, kidneys, and intestines. Leaking blood vessels can cause skin rashes known as purpura.
The estimated ranges of disease are high, with the annual incidence in children estimated at 3-26 per 100,000 and in adults at 0.1-1.8 per 100,000. Dr. Villa-Forte noted that the male-to-female ratio is 1.5, and she said the condition is less common in African Americans.
“In children, this is a disease that is frequently self-limited. Most of them don’t need treatment, and most of them do not relapse,” Dr. Villa-Forte said. “In adults, it’s more resistant to treatment, frequently chronic, and frequently relapsing over the years.”
There are other differences in IV between children and adults. “In children, there is a clear seasonal pattern of disease that is not seen in adults,” she said, and it’s linked to preceding infections.
“In adults, there are multiple causes, and most of the time they’re not identified,” she said.
As for diagnosis, she suggests looking at clinical presentation and whether tissue biopsy shows cutaneous leukocytoclastic vasculitis with IgA deposits. She cautioned that increased serum IgA is seen in about 50% of adult patients, making it an unreliable indicator.
Prognosis is much better for children than adults. According to a 2014 study, 80% of children completely recover, compared with 40% of adults. Persistent hematuria or proteinuria occurs in 30% of children and 60% of adults, respectively, while chronic renal failure occurs in 2% of children and 10% of adults (J Korean Med Sci. 2014 Feb;29[2]:198-203).
It’s possible that the latter number may be higher, with as many as 30% of adults developing chronic kidney disease (CKD), Dr. Villa-Forte said.
An estimated 97% of nephritis develops within the first 6 months of disease onset in adults, she said, “and in adults, the active renal disease can persist for over 20 years.”
Guidelines suggest that patients be monitored for CKD for 6 months after disease onset, but Dr. Villa-Forte said it’s better to monitor them for 12 months.
What about treatment? “The major challenge in adults is the real absence between correlation between initial presentation and long-term renal outcome,” she said. “That makes for a difficult choice in terms of treatment selection.”
Fever and cutaneous lesions above the waist may predict renal involvement, she said, although that isn’t confirmed, and increasing proteinuria is a probable factor predicting progression/complications.
According to Dr. Villa-Forte, the value of early treatment hasn’t been proved. Due to the risk of kidney problems, she said, “we don’t feel like we can just watch patients – that we need to do something. But there’s not good data supporting that.”
Various protocols involving steroids, early plasmapheresis, rituximab (Rituxan), and other drug regimens lack evidence, she said, although use of steroids in early nephritis may be beneficial in adults.
Dr. Villa-Forte had no relevant disclosures.
Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – IgA vasculitis has a reputation as an illness of childhood, but rheumatologist Alexandra Villa-Forte, MD, MPH, cautioned colleagues that it can strike adults, too, often in a much more severe form. And, she warned, it’s likely not as rare as physicians assume.
“I believe it’s more common in adults than reported. There’s a huge problem with establishing the right way to make the diagnosis in adults, which is why it is missed,” said Dr. Villa-Forte of the Cleveland Clinic, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
But even if IgA vasculitis (IV) is diagnosed correctly in adults, there are many questions about how to move forward, she said. “Treatment in adults remains a problem since we don’t have data.”
IV is also known as Henoch-Schönlein purpura, or HSP, and “spring fever” because it often appears in children in the spring following an upper respiratory infection.
The condition causes vasculitis, the swelling of small blood vessels in organs such as the skin, joints, kidneys, and intestines. Leaking blood vessels can cause skin rashes known as purpura.
The estimated ranges of disease are high, with the annual incidence in children estimated at 3-26 per 100,000 and in adults at 0.1-1.8 per 100,000. Dr. Villa-Forte noted that the male-to-female ratio is 1.5, and she said the condition is less common in African Americans.
“In children, this is a disease that is frequently self-limited. Most of them don’t need treatment, and most of them do not relapse,” Dr. Villa-Forte said. “In adults, it’s more resistant to treatment, frequently chronic, and frequently relapsing over the years.”
There are other differences in IV between children and adults. “In children, there is a clear seasonal pattern of disease that is not seen in adults,” she said, and it’s linked to preceding infections.
“In adults, there are multiple causes, and most of the time they’re not identified,” she said.
As for diagnosis, she suggests looking at clinical presentation and whether tissue biopsy shows cutaneous leukocytoclastic vasculitis with IgA deposits. She cautioned that increased serum IgA is seen in about 50% of adult patients, making it an unreliable indicator.
Prognosis is much better for children than adults. According to a 2014 study, 80% of children completely recover, compared with 40% of adults. Persistent hematuria or proteinuria occurs in 30% of children and 60% of adults, respectively, while chronic renal failure occurs in 2% of children and 10% of adults (J Korean Med Sci. 2014 Feb;29[2]:198-203).
It’s possible that the latter number may be higher, with as many as 30% of adults developing chronic kidney disease (CKD), Dr. Villa-Forte said.
An estimated 97% of nephritis develops within the first 6 months of disease onset in adults, she said, “and in adults, the active renal disease can persist for over 20 years.”
Guidelines suggest that patients be monitored for CKD for 6 months after disease onset, but Dr. Villa-Forte said it’s better to monitor them for 12 months.
What about treatment? “The major challenge in adults is the real absence between correlation between initial presentation and long-term renal outcome,” she said. “That makes for a difficult choice in terms of treatment selection.”
Fever and cutaneous lesions above the waist may predict renal involvement, she said, although that isn’t confirmed, and increasing proteinuria is a probable factor predicting progression/complications.
According to Dr. Villa-Forte, the value of early treatment hasn’t been proved. Due to the risk of kidney problems, she said, “we don’t feel like we can just watch patients – that we need to do something. But there’s not good data supporting that.”
Various protocols involving steroids, early plasmapheresis, rituximab (Rituxan), and other drug regimens lack evidence, she said, although use of steroids in early nephritis may be beneficial in adults.
Dr. Villa-Forte had no relevant disclosures.
Global Academy for Medical Education and this news organization are owned by the same parent company.
REPORTING FROM THE ANNUAL PERSPECTIVES IN RHEUMATIC DISEASES
Identifying and Managing MS Relapse
Multiple sclerosis (MS) is a chronic, autoimmune-mediated disorder of the central nervous system that affects more than 40,000 people in the United States. About 85% of cases are categorized as relapsing remitting multiple sclerosis (RRMS), based on the clinical and radiographic pattern of focal demyelination in different regions of the brain and spinal cord over time. Though not fully understood, the pathophysiology of RRMS involves axonal degeneration and inflammatory demyelination; the latter is considered a relapse in patients with an established MS diagnosis.
OVERVIEW
MS relapse can have a significant impact on patients’ short- and long-term function, quality of life, and finances. Relapse may be identified via
- New neurologic symptoms reported by the patient
- New neurologic findings on physical examination
- New radiographic findings on contrast-enhanced MRI of the central nervous system, or
- Abnormal results of cerebrospinal fluid analysis.
Patient-reported symptoms and abnormal signs identified on physical exam should correspond with the area of the central nervous system affected. In some cases, patients may have radiographic evidence of relapse without symptoms or signs.
It is essential for health care providers to identify relapse, as it is an important marker of disease activity that may warrant treatment—particularly if symptoms are impacting function or if there is optic neuritis. MS relapse is also an indicator of suboptimal response to disease-modifying therapies.
Treatment of relapse is one component of RRMS management, which also includes symptom management and use of disease-modifying therapy to reduce risk for disease activity and decline in function.
DIAGNOSING RELAPSE
Because risk for MS relapse cannot be predicted, both patients and providers need to have a high index of suspicion in the setting of new neurologic symptoms or decline in function. Relapse should be considered when these symptoms last longer than 24 hours in the absence of fever or infection. The clinical features of relapse should have corresponding radiographic evidence of active demyelination on contrast-enhanced MRI.
A pseudo-relapse is characterized by new or worsening neurologic symptoms lasting longer than 24 hours with concurrent fever, infection, or other metabolic derangement. Pseudo-relapse does not show radiographic evidence of active demyelination on contrast-enhanced MRI.
Continue to: Aggravation of longstanding neurologic symptoms...
Aggravation of longstanding neurologic symptoms is not considered a relapse, as no new radiographic evidence of disease progression will be seen on MRI. Factors that may contribute to aggravation of established symptoms include an increase in core body temperature, sleep deprivation, and psychosocial stress.
When a patient with suspected or diagnosed RRMS presents with new neurologic symptoms of more than 24 hours’ duration, the first step is to conduct a physical exam to assess for objective evidence of neurologic deficits and signs of infection, including fever. The provider should also order select laboratory testing—including a complete blood cell count and urinalysis with culture—to exclude infection. In certain cases, contrast-enhanced MRI of the brain and/or spine may be ordered; however, this may delay treatment initiation if the study cannot be promptly scheduled.
Evaluation and management should involve communication, if not face-to-face consultation, with the patient’s neurology provider who is responsible for MS management.
IMMEDIATE MANAGEMENT
Acute relapses are managed with anti-inflammatory agents. For some patients, treatment may provide symptomatic relief, shorten the recovery phase, and improve motor function. Long-term benefits have not been demonstrated, except in patients with optic neuritis.
Firstline therapy for MS relapse is high-dose corticosteroids, which can be administered at home, at an ambulatory infusion center, or (in some cases) in a hospital setting. The preferred regimen is methylprednisolone (1 g IV for 3-5 d), with or without prednisone taper. Another option is dexamethasone (80 mg bid for 3-5 d), with or without prednisone taper.1-3
Continue to: Common adverse effects of corticosteroids include...
Common adverse effects of corticosteroids include headache, emotional lability, insomnia, glucose intolerance, hypertension, dyspepsia, and exacerbation of psychiatric conditions; drug interactions should also be considered. Patients with diabetes may need to be admitted to the hospital for glycemic monitoring and control.

High-dose corticosteroids are associated with a rare, non–dose-dependent risk for aseptic femoral necrosis. For patients who are refractory to or not candidates for corticosteroids, adrenocorticotropic hormone (ACTH) gel (80 U/d IM or subQ daily for 10 d) is an option. This medication may be better tolerated, although it is much more expensive than corticosteroids. Plasmapheresis and IV immunoglobulin are also options for patients with refractory symptoms or contraindications to recommended therapies.1-3
ONGOING MANAGEMENT
Once a treatment plan is initiated, providers should carefully follow the patient’s response in terms of adverse effects, symptom improvement, and functional recovery. Those with refractory symptoms may need additional doses of the initial therapy or an alternative therapy.
The relapse recovery period may last several months and be complete or incomplete, so providers may also need to manage neurologic symptoms and functional deficits (with pharmacologic and/or nonpharmacologic options). Patients who have had a relapse should also meet with their neurology provider to discuss their disease-modifying therapy plan, since relapse indicates a suboptimal response to current therapy.
1. Bevan C, Gelfand JM. Therapeutic management of severe relapses in multiple sclerosis. Curr Treat Options Neurol. 2015;17(4):17.
2. Frohman TC, O’Donoghue DL, Northrop D, eds. Multiple Sclerosis for the Physician Assistant. National Multiple Sclerosis Society. 2011.
3. Giesser B, ed. Primer on Multiple Sclerosis . 2nd ed. Oxford, UK: Oxford University Press ; 2015.
Clinician Reviews in partnership with

MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's response was authored by Nicholas M. Hudak is a PA in the Department of Neurology, Clinical Coordinator of the PA Program, and an Associate Professor in the Department of Community & Family Medicine, at the Duke University School of Medicine in Durham, North Carolina.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's response was authored by Nicholas M. Hudak is a PA in the Department of Neurology, Clinical Coordinator of the PA Program, and an Associate Professor in the Department of Community & Family Medicine, at the Duke University School of Medicine in Durham, North Carolina.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's response was authored by Nicholas M. Hudak is a PA in the Department of Neurology, Clinical Coordinator of the PA Program, and an Associate Professor in the Department of Community & Family Medicine, at the Duke University School of Medicine in Durham, North Carolina.
Multiple sclerosis (MS) is a chronic, autoimmune-mediated disorder of the central nervous system that affects more than 40,000 people in the United States. About 85% of cases are categorized as relapsing remitting multiple sclerosis (RRMS), based on the clinical and radiographic pattern of focal demyelination in different regions of the brain and spinal cord over time. Though not fully understood, the pathophysiology of RRMS involves axonal degeneration and inflammatory demyelination; the latter is considered a relapse in patients with an established MS diagnosis.
OVERVIEW
MS relapse can have a significant impact on patients’ short- and long-term function, quality of life, and finances. Relapse may be identified via
- New neurologic symptoms reported by the patient
- New neurologic findings on physical examination
- New radiographic findings on contrast-enhanced MRI of the central nervous system, or
- Abnormal results of cerebrospinal fluid analysis.
Patient-reported symptoms and abnormal signs identified on physical exam should correspond with the area of the central nervous system affected. In some cases, patients may have radiographic evidence of relapse without symptoms or signs.
It is essential for health care providers to identify relapse, as it is an important marker of disease activity that may warrant treatment—particularly if symptoms are impacting function or if there is optic neuritis. MS relapse is also an indicator of suboptimal response to disease-modifying therapies.
Treatment of relapse is one component of RRMS management, which also includes symptom management and use of disease-modifying therapy to reduce risk for disease activity and decline in function.
DIAGNOSING RELAPSE
Because risk for MS relapse cannot be predicted, both patients and providers need to have a high index of suspicion in the setting of new neurologic symptoms or decline in function. Relapse should be considered when these symptoms last longer than 24 hours in the absence of fever or infection. The clinical features of relapse should have corresponding radiographic evidence of active demyelination on contrast-enhanced MRI.
A pseudo-relapse is characterized by new or worsening neurologic symptoms lasting longer than 24 hours with concurrent fever, infection, or other metabolic derangement. Pseudo-relapse does not show radiographic evidence of active demyelination on contrast-enhanced MRI.
Continue to: Aggravation of longstanding neurologic symptoms...
Aggravation of longstanding neurologic symptoms is not considered a relapse, as no new radiographic evidence of disease progression will be seen on MRI. Factors that may contribute to aggravation of established symptoms include an increase in core body temperature, sleep deprivation, and psychosocial stress.
When a patient with suspected or diagnosed RRMS presents with new neurologic symptoms of more than 24 hours’ duration, the first step is to conduct a physical exam to assess for objective evidence of neurologic deficits and signs of infection, including fever. The provider should also order select laboratory testing—including a complete blood cell count and urinalysis with culture—to exclude infection. In certain cases, contrast-enhanced MRI of the brain and/or spine may be ordered; however, this may delay treatment initiation if the study cannot be promptly scheduled.
Evaluation and management should involve communication, if not face-to-face consultation, with the patient’s neurology provider who is responsible for MS management.
IMMEDIATE MANAGEMENT
Acute relapses are managed with anti-inflammatory agents. For some patients, treatment may provide symptomatic relief, shorten the recovery phase, and improve motor function. Long-term benefits have not been demonstrated, except in patients with optic neuritis.
Firstline therapy for MS relapse is high-dose corticosteroids, which can be administered at home, at an ambulatory infusion center, or (in some cases) in a hospital setting. The preferred regimen is methylprednisolone (1 g IV for 3-5 d), with or without prednisone taper. Another option is dexamethasone (80 mg bid for 3-5 d), with or without prednisone taper.1-3
Continue to: Common adverse effects of corticosteroids include...
Common adverse effects of corticosteroids include headache, emotional lability, insomnia, glucose intolerance, hypertension, dyspepsia, and exacerbation of psychiatric conditions; drug interactions should also be considered. Patients with diabetes may need to be admitted to the hospital for glycemic monitoring and control.
High-dose corticosteroids are associated with a rare, non–dose-dependent risk for aseptic femoral necrosis. For patients who are refractory to or not candidates for corticosteroids, adrenocorticotropic hormone (ACTH) gel (80 U/d IM or subQ daily for 10 d) is an option. This medication may be better tolerated, although it is much more expensive than corticosteroids. Plasmapheresis and IV immunoglobulin are also options for patients with refractory symptoms or contraindications to recommended therapies.1-3
ONGOING MANAGEMENT
Once a treatment plan is initiated, providers should carefully follow the patient’s response in terms of adverse effects, symptom improvement, and functional recovery. Those with refractory symptoms may need additional doses of the initial therapy or an alternative therapy.
The relapse recovery period may last several months and be complete or incomplete, so providers may also need to manage neurologic symptoms and functional deficits (with pharmacologic and/or nonpharmacologic options). Patients who have had a relapse should also meet with their neurology provider to discuss their disease-modifying therapy plan, since relapse indicates a suboptimal response to current therapy.
Multiple sclerosis (MS) is a chronic, autoimmune-mediated disorder of the central nervous system that affects more than 40,000 people in the United States. About 85% of cases are categorized as relapsing remitting multiple sclerosis (RRMS), based on the clinical and radiographic pattern of focal demyelination in different regions of the brain and spinal cord over time. Though not fully understood, the pathophysiology of RRMS involves axonal degeneration and inflammatory demyelination; the latter is considered a relapse in patients with an established MS diagnosis.
OVERVIEW
MS relapse can have a significant impact on patients’ short- and long-term function, quality of life, and finances. Relapse may be identified via
- New neurologic symptoms reported by the patient
- New neurologic findings on physical examination
- New radiographic findings on contrast-enhanced MRI of the central nervous system, or
- Abnormal results of cerebrospinal fluid analysis.
Patient-reported symptoms and abnormal signs identified on physical exam should correspond with the area of the central nervous system affected. In some cases, patients may have radiographic evidence of relapse without symptoms or signs.
It is essential for health care providers to identify relapse, as it is an important marker of disease activity that may warrant treatment—particularly if symptoms are impacting function or if there is optic neuritis. MS relapse is also an indicator of suboptimal response to disease-modifying therapies.
Treatment of relapse is one component of RRMS management, which also includes symptom management and use of disease-modifying therapy to reduce risk for disease activity and decline in function.
DIAGNOSING RELAPSE
Because risk for MS relapse cannot be predicted, both patients and providers need to have a high index of suspicion in the setting of new neurologic symptoms or decline in function. Relapse should be considered when these symptoms last longer than 24 hours in the absence of fever or infection. The clinical features of relapse should have corresponding radiographic evidence of active demyelination on contrast-enhanced MRI.
A pseudo-relapse is characterized by new or worsening neurologic symptoms lasting longer than 24 hours with concurrent fever, infection, or other metabolic derangement. Pseudo-relapse does not show radiographic evidence of active demyelination on contrast-enhanced MRI.
Continue to: Aggravation of longstanding neurologic symptoms...
Aggravation of longstanding neurologic symptoms is not considered a relapse, as no new radiographic evidence of disease progression will be seen on MRI. Factors that may contribute to aggravation of established symptoms include an increase in core body temperature, sleep deprivation, and psychosocial stress.
When a patient with suspected or diagnosed RRMS presents with new neurologic symptoms of more than 24 hours’ duration, the first step is to conduct a physical exam to assess for objective evidence of neurologic deficits and signs of infection, including fever. The provider should also order select laboratory testing—including a complete blood cell count and urinalysis with culture—to exclude infection. In certain cases, contrast-enhanced MRI of the brain and/or spine may be ordered; however, this may delay treatment initiation if the study cannot be promptly scheduled.
Evaluation and management should involve communication, if not face-to-face consultation, with the patient’s neurology provider who is responsible for MS management.
IMMEDIATE MANAGEMENT
Acute relapses are managed with anti-inflammatory agents. For some patients, treatment may provide symptomatic relief, shorten the recovery phase, and improve motor function. Long-term benefits have not been demonstrated, except in patients with optic neuritis.
Firstline therapy for MS relapse is high-dose corticosteroids, which can be administered at home, at an ambulatory infusion center, or (in some cases) in a hospital setting. The preferred regimen is methylprednisolone (1 g IV for 3-5 d), with or without prednisone taper. Another option is dexamethasone (80 mg bid for 3-5 d), with or without prednisone taper.1-3
Continue to: Common adverse effects of corticosteroids include...
Common adverse effects of corticosteroids include headache, emotional lability, insomnia, glucose intolerance, hypertension, dyspepsia, and exacerbation of psychiatric conditions; drug interactions should also be considered. Patients with diabetes may need to be admitted to the hospital for glycemic monitoring and control.
High-dose corticosteroids are associated with a rare, non–dose-dependent risk for aseptic femoral necrosis. For patients who are refractory to or not candidates for corticosteroids, adrenocorticotropic hormone (ACTH) gel (80 U/d IM or subQ daily for 10 d) is an option. This medication may be better tolerated, although it is much more expensive than corticosteroids. Plasmapheresis and IV immunoglobulin are also options for patients with refractory symptoms or contraindications to recommended therapies.1-3
ONGOING MANAGEMENT
Once a treatment plan is initiated, providers should carefully follow the patient’s response in terms of adverse effects, symptom improvement, and functional recovery. Those with refractory symptoms may need additional doses of the initial therapy or an alternative therapy.
The relapse recovery period may last several months and be complete or incomplete, so providers may also need to manage neurologic symptoms and functional deficits (with pharmacologic and/or nonpharmacologic options). Patients who have had a relapse should also meet with their neurology provider to discuss their disease-modifying therapy plan, since relapse indicates a suboptimal response to current therapy.
1. Bevan C, Gelfand JM. Therapeutic management of severe relapses in multiple sclerosis. Curr Treat Options Neurol. 2015;17(4):17.
2. Frohman TC, O’Donoghue DL, Northrop D, eds. Multiple Sclerosis for the Physician Assistant. National Multiple Sclerosis Society. 2011.
3. Giesser B, ed. Primer on Multiple Sclerosis . 2nd ed. Oxford, UK: Oxford University Press ; 2015.
1. Bevan C, Gelfand JM. Therapeutic management of severe relapses in multiple sclerosis. Curr Treat Options Neurol. 2015;17(4):17.
2. Frohman TC, O’Donoghue DL, Northrop D, eds. Multiple Sclerosis for the Physician Assistant. National Multiple Sclerosis Society. 2011.
3. Giesser B, ed. Primer on Multiple Sclerosis . 2nd ed. Oxford, UK: Oxford University Press ; 2015.

Subcutaneous Actemra approved for systemic JIA
The Food and Drug Administration has approved the subcutaneous formulation of Actemra (tocilizumab) for systemic juvenile idiopathic arthritis (SJIA) for patients aged 2 years and older, according to a press release from its developer, Genentech. The intravenous formulation was approved in 2011 for this indication.

The approval is based on data the JIGSAW-118 study. This 52-week, open-label, multicenter, phase 1b pharmacokinetic/pharmacodynamic bridging study was designed to determine the appropriate dosing regimen by treating 51 patients with SJIA according to body weight.
The safety profile of subcutaneous tocilizumab was similar to that seen with intravenous tocilizumab, although there were more injection-site reactions seen with the subcutaneous formulation. Its efficacy was extrapolated based on the drug’s pharmacokinetic profile seen with IV tocilizumab in SJIA patients and with subcutaneous tocilizumab in patients with rheumatoid arthritis.
SJIA is a rare disease with limited treatment options, according to the press release. In general, JIA affects almost 300,000 children in the United States, and about 10% of those cases are SJIA.
The Food and Drug Administration has approved the subcutaneous formulation of Actemra (tocilizumab) for systemic juvenile idiopathic arthritis (SJIA) for patients aged 2 years and older, according to a press release from its developer, Genentech. The intravenous formulation was approved in 2011 for this indication.
The approval is based on data the JIGSAW-118 study. This 52-week, open-label, multicenter, phase 1b pharmacokinetic/pharmacodynamic bridging study was designed to determine the appropriate dosing regimen by treating 51 patients with SJIA according to body weight.
The safety profile of subcutaneous tocilizumab was similar to that seen with intravenous tocilizumab, although there were more injection-site reactions seen with the subcutaneous formulation. Its efficacy was extrapolated based on the drug’s pharmacokinetic profile seen with IV tocilizumab in SJIA patients and with subcutaneous tocilizumab in patients with rheumatoid arthritis.
SJIA is a rare disease with limited treatment options, according to the press release. In general, JIA affects almost 300,000 children in the United States, and about 10% of those cases are SJIA.
The Food and Drug Administration has approved the subcutaneous formulation of Actemra (tocilizumab) for systemic juvenile idiopathic arthritis (SJIA) for patients aged 2 years and older, according to a press release from its developer, Genentech. The intravenous formulation was approved in 2011 for this indication.
The approval is based on data the JIGSAW-118 study. This 52-week, open-label, multicenter, phase 1b pharmacokinetic/pharmacodynamic bridging study was designed to determine the appropriate dosing regimen by treating 51 patients with SJIA according to body weight.
The safety profile of subcutaneous tocilizumab was similar to that seen with intravenous tocilizumab, although there were more injection-site reactions seen with the subcutaneous formulation. Its efficacy was extrapolated based on the drug’s pharmacokinetic profile seen with IV tocilizumab in SJIA patients and with subcutaneous tocilizumab in patients with rheumatoid arthritis.
SJIA is a rare disease with limited treatment options, according to the press release. In general, JIA affects almost 300,000 children in the United States, and about 10% of those cases are SJIA.