User login
Clinical Scoring System May Help Diagnose Acute Flaccid Myelitis
Proposed diagnostic criteria require validation.
CHICAGO—Use of the CDC case definition for acute flaccid myelitis may lead to misdiagnosis in patients with other conditions, according to research presented at the 47th Annual Meeting of the Child Neurology Society. A review of 45 reported cases of acute flaccid myelitis found that 12 of the patients had other diseases, such as polyradiculoneuropathy, transverse myelitis, spinal cord ischemia, clinically isolated syndrome, meningitis, and myelopathy due to severe Chiari I malformation, said Matthew Elrick, MD, PhD, Clinical Fellow in Neurology at Johns Hopkins Hospital in Baltimore, and colleagues.
Acute flaccid myelitis is a polio-like illness of acute spinal motor neuron injury following viral infection. Recent outbreaks have been reported, but clinical diagnostic criteria are lacking. Clinical characteristics may help differentiate acute flaccid myelitis from other causes of myelopathy, Dr. Elrick said.
Dr. Elrick and colleagues analyzed cases from two patient cohorts. One cohort included patients who were recruited nationwide based on the CDC case definition of acute flaccid myelitis. The other cohort included patients who were referred to the Johns Hopkins Transverse Myelitis Center and received a diagnosis of acute flaccid myelitis. The researchers reviewed patients’ records and imaging data. An independent neurologist reviewed a subset of cases to confirm inter-rater reliability. Characteristics that differed significantly between patients with and without acute flaccid myelitis were used to build a clinical scoring system.
The CDC case definition includes clinical criteria (ie, an illness with onset of acute flaccid limb weakness), confirmatory laboratory evidence (ie, MRI showing spinal cord lesion largely restricted to gray matter and spanning one or more vertebral segments), and supportive laboratory evidence (ie, CSF with pleocytosis). Clinically compatible cases with confirmatory laboratory evidence are considered confirmed, and clinically compatible cases with supportive laboratory evidence are considered probable.
The investigators based their proposed criteria on the physiologic understanding of acute flaccid myelitis as a disease of the motor neuron with features similar to those of poliomyelitis. They refined the criteria based on features commonly seen in apparent cases (eg, weakness involving the limbs, neck, face, or bulbar muscles and prodromal illness with fever, respiratory symptoms, or gastrointestinal symptoms). In addition, they incorporated rule-out criteria to exclude mimics.
Based on the results of their analysis, they developed a clinical scoring system to aid in the bedside diagnosis of acute flaccid myelitis by considering features such as asymmetric onset, decreased tone, and absence of increased tone.
The proposed criteria and clinical scoring system require validation and may be updated in light of data from recent cases of acute flaccid myelitis, Dr. Elrick said
Proposed diagnostic criteria require validation.
Proposed diagnostic criteria require validation.
CHICAGO—Use of the CDC case definition for acute flaccid myelitis may lead to misdiagnosis in patients with other conditions, according to research presented at the 47th Annual Meeting of the Child Neurology Society. A review of 45 reported cases of acute flaccid myelitis found that 12 of the patients had other diseases, such as polyradiculoneuropathy, transverse myelitis, spinal cord ischemia, clinically isolated syndrome, meningitis, and myelopathy due to severe Chiari I malformation, said Matthew Elrick, MD, PhD, Clinical Fellow in Neurology at Johns Hopkins Hospital in Baltimore, and colleagues.
Acute flaccid myelitis is a polio-like illness of acute spinal motor neuron injury following viral infection. Recent outbreaks have been reported, but clinical diagnostic criteria are lacking. Clinical characteristics may help differentiate acute flaccid myelitis from other causes of myelopathy, Dr. Elrick said.
Dr. Elrick and colleagues analyzed cases from two patient cohorts. One cohort included patients who were recruited nationwide based on the CDC case definition of acute flaccid myelitis. The other cohort included patients who were referred to the Johns Hopkins Transverse Myelitis Center and received a diagnosis of acute flaccid myelitis. The researchers reviewed patients’ records and imaging data. An independent neurologist reviewed a subset of cases to confirm inter-rater reliability. Characteristics that differed significantly between patients with and without acute flaccid myelitis were used to build a clinical scoring system.
The CDC case definition includes clinical criteria (ie, an illness with onset of acute flaccid limb weakness), confirmatory laboratory evidence (ie, MRI showing spinal cord lesion largely restricted to gray matter and spanning one or more vertebral segments), and supportive laboratory evidence (ie, CSF with pleocytosis). Clinically compatible cases with confirmatory laboratory evidence are considered confirmed, and clinically compatible cases with supportive laboratory evidence are considered probable.
The investigators based their proposed criteria on the physiologic understanding of acute flaccid myelitis as a disease of the motor neuron with features similar to those of poliomyelitis. They refined the criteria based on features commonly seen in apparent cases (eg, weakness involving the limbs, neck, face, or bulbar muscles and prodromal illness with fever, respiratory symptoms, or gastrointestinal symptoms). In addition, they incorporated rule-out criteria to exclude mimics.
Based on the results of their analysis, they developed a clinical scoring system to aid in the bedside diagnosis of acute flaccid myelitis by considering features such as asymmetric onset, decreased tone, and absence of increased tone.
The proposed criteria and clinical scoring system require validation and may be updated in light of data from recent cases of acute flaccid myelitis, Dr. Elrick said
CHICAGO—Use of the CDC case definition for acute flaccid myelitis may lead to misdiagnosis in patients with other conditions, according to research presented at the 47th Annual Meeting of the Child Neurology Society. A review of 45 reported cases of acute flaccid myelitis found that 12 of the patients had other diseases, such as polyradiculoneuropathy, transverse myelitis, spinal cord ischemia, clinically isolated syndrome, meningitis, and myelopathy due to severe Chiari I malformation, said Matthew Elrick, MD, PhD, Clinical Fellow in Neurology at Johns Hopkins Hospital in Baltimore, and colleagues.
Acute flaccid myelitis is a polio-like illness of acute spinal motor neuron injury following viral infection. Recent outbreaks have been reported, but clinical diagnostic criteria are lacking. Clinical characteristics may help differentiate acute flaccid myelitis from other causes of myelopathy, Dr. Elrick said.
Dr. Elrick and colleagues analyzed cases from two patient cohorts. One cohort included patients who were recruited nationwide based on the CDC case definition of acute flaccid myelitis. The other cohort included patients who were referred to the Johns Hopkins Transverse Myelitis Center and received a diagnosis of acute flaccid myelitis. The researchers reviewed patients’ records and imaging data. An independent neurologist reviewed a subset of cases to confirm inter-rater reliability. Characteristics that differed significantly between patients with and without acute flaccid myelitis were used to build a clinical scoring system.
The CDC case definition includes clinical criteria (ie, an illness with onset of acute flaccid limb weakness), confirmatory laboratory evidence (ie, MRI showing spinal cord lesion largely restricted to gray matter and spanning one or more vertebral segments), and supportive laboratory evidence (ie, CSF with pleocytosis). Clinically compatible cases with confirmatory laboratory evidence are considered confirmed, and clinically compatible cases with supportive laboratory evidence are considered probable.
The investigators based their proposed criteria on the physiologic understanding of acute flaccid myelitis as a disease of the motor neuron with features similar to those of poliomyelitis. They refined the criteria based on features commonly seen in apparent cases (eg, weakness involving the limbs, neck, face, or bulbar muscles and prodromal illness with fever, respiratory symptoms, or gastrointestinal symptoms). In addition, they incorporated rule-out criteria to exclude mimics.
Based on the results of their analysis, they developed a clinical scoring system to aid in the bedside diagnosis of acute flaccid myelitis by considering features such as asymmetric onset, decreased tone, and absence of increased tone.
The proposed criteria and clinical scoring system require validation and may be updated in light of data from recent cases of acute flaccid myelitis, Dr. Elrick said
OV-101 shows promise for Angelman syndrome
SEATTLE – A novel extrasynaptic gamma-aminobutyric acid (GABA)–receptor agonist called OV-101 was safe and well-tolerated in adult and adolescent Angelman syndrome patients in a 12-week phase 2 trial. In a secondary analysis, the treatment appeared to improve sleep.

Angelman syndrome is associated with a microdeletion on chromosome 15 encompassing the ubiquitin protein ligase E3a (UBE3A) gene. The resulting loss of expression of the UBE3A protein leads to increases in the uptake of GABA and reduces levels of extrasynaptic GABA. Patients with Angelman syndrome typically have motor dysfunction, often extreme: “These kids are very excitable, very active, and they have lots of trouble with sleep,” said Alex Kolevzon, MD, professor of psychiatry and pediatrics at the Icahn School of Medicine at Mount Sinai, in an interview.
Dr. Kolevzon presented the results at a poster session at the annual meeting of the American Academy of Child and Adolescent Psychiatry.
The study was conducted at 12 sites in the United States and 1 in Israel. Ovid Pharmaceuticals plans to apply to the Food and Drug Administration later this year for approval. There is no existing drug for Angelman syndrome, and the study provided good safety reassurance. “There were some side effects, but for the most part we considered them mild, and only four (out of 88 subjects) discontinued because of side effects,” said Dr. Kolevzon.
The researchers used actigraphy to gain a more objective measure of sleep in the study participants. They randomized 88 patients with Angelman syndrome (aged 13-49 years) to receive placebo in the morning and 15 mg of OV-101 at night, 10 mg OVID-101 in the morning and 15 mg OVID-101 at night, or placebo both in the morning and at night.
Pyrexia occurred in 24% of the group who received the active drug only at night, 3% of the group given the twice-daily dose, and 7% of the placebo group. Seizures occurred in 7% of the once-daily group and 10% of the twice-daily group; seizures were not noted in the placebo group.
The main efficacy outcome measure was the Clinical Global Impressions-9 (CGI-9) scale. The once-daily group had a significant benefit in the sleep domain at 12 weeks, compared with placebo (difference, –0.77; P = .0141), but the twice-daily group had only a trend toward improvement in sleep (difference, –0.45; P = .1407).
Both active therapy groups had significant improvement in CGI-9 measures after 12 weeks of treatment compared to placebo – the twice-daily group (P = .0206, Fisher’s Exact Test) and the once-daily group (P = .0006, mixed model repeated measures analysis).
The actigraphy analysis, conducted in the 45% of patients who could tolerate its use, found that, compared to placebo, the once-daily dosing group experienced an 25.7 minute improvement in latency to sleep onset (P = .0147), as well an approximately 50 minute reduction in sleep time during the day, and a 3.65% improvement in sleep efficiency.
OV-101 has the potential to treat other conditions as well. “Obviously there are a lot of neurodevelopmental disorders where you see dysregulation between the GABAergic and glutamergic systems. This is a drug that has a unique effect on the GABAergic system. It’s already being studied in Fragile X syndrome, where we see this same kind of dysregulation and excess excitation,” said Dr. Kolevzon.
Dr. Kolevzon is a consultant for several drug companies including Ovid Therapeutics.
SOURCE: AACAP 2018. New Research Poster 3.1.
SEATTLE – A novel extrasynaptic gamma-aminobutyric acid (GABA)–receptor agonist called OV-101 was safe and well-tolerated in adult and adolescent Angelman syndrome patients in a 12-week phase 2 trial. In a secondary analysis, the treatment appeared to improve sleep.
Angelman syndrome is associated with a microdeletion on chromosome 15 encompassing the ubiquitin protein ligase E3a (UBE3A) gene. The resulting loss of expression of the UBE3A protein leads to increases in the uptake of GABA and reduces levels of extrasynaptic GABA. Patients with Angelman syndrome typically have motor dysfunction, often extreme: “These kids are very excitable, very active, and they have lots of trouble with sleep,” said Alex Kolevzon, MD, professor of psychiatry and pediatrics at the Icahn School of Medicine at Mount Sinai, in an interview.
Dr. Kolevzon presented the results at a poster session at the annual meeting of the American Academy of Child and Adolescent Psychiatry.
The study was conducted at 12 sites in the United States and 1 in Israel. Ovid Pharmaceuticals plans to apply to the Food and Drug Administration later this year for approval. There is no existing drug for Angelman syndrome, and the study provided good safety reassurance. “There were some side effects, but for the most part we considered them mild, and only four (out of 88 subjects) discontinued because of side effects,” said Dr. Kolevzon.
The researchers used actigraphy to gain a more objective measure of sleep in the study participants. They randomized 88 patients with Angelman syndrome (aged 13-49 years) to receive placebo in the morning and 15 mg of OV-101 at night, 10 mg OVID-101 in the morning and 15 mg OVID-101 at night, or placebo both in the morning and at night.
Pyrexia occurred in 24% of the group who received the active drug only at night, 3% of the group given the twice-daily dose, and 7% of the placebo group. Seizures occurred in 7% of the once-daily group and 10% of the twice-daily group; seizures were not noted in the placebo group.
The main efficacy outcome measure was the Clinical Global Impressions-9 (CGI-9) scale. The once-daily group had a significant benefit in the sleep domain at 12 weeks, compared with placebo (difference, –0.77; P = .0141), but the twice-daily group had only a trend toward improvement in sleep (difference, –0.45; P = .1407).
Both active therapy groups had significant improvement in CGI-9 measures after 12 weeks of treatment compared to placebo – the twice-daily group (P = .0206, Fisher’s Exact Test) and the once-daily group (P = .0006, mixed model repeated measures analysis).
The actigraphy analysis, conducted in the 45% of patients who could tolerate its use, found that, compared to placebo, the once-daily dosing group experienced an 25.7 minute improvement in latency to sleep onset (P = .0147), as well an approximately 50 minute reduction in sleep time during the day, and a 3.65% improvement in sleep efficiency.
OV-101 has the potential to treat other conditions as well. “Obviously there are a lot of neurodevelopmental disorders where you see dysregulation between the GABAergic and glutamergic systems. This is a drug that has a unique effect on the GABAergic system. It’s already being studied in Fragile X syndrome, where we see this same kind of dysregulation and excess excitation,” said Dr. Kolevzon.
Dr. Kolevzon is a consultant for several drug companies including Ovid Therapeutics.
SOURCE: AACAP 2018. New Research Poster 3.1.
SEATTLE – A novel extrasynaptic gamma-aminobutyric acid (GABA)–receptor agonist called OV-101 was safe and well-tolerated in adult and adolescent Angelman syndrome patients in a 12-week phase 2 trial. In a secondary analysis, the treatment appeared to improve sleep.
Angelman syndrome is associated with a microdeletion on chromosome 15 encompassing the ubiquitin protein ligase E3a (UBE3A) gene. The resulting loss of expression of the UBE3A protein leads to increases in the uptake of GABA and reduces levels of extrasynaptic GABA. Patients with Angelman syndrome typically have motor dysfunction, often extreme: “These kids are very excitable, very active, and they have lots of trouble with sleep,” said Alex Kolevzon, MD, professor of psychiatry and pediatrics at the Icahn School of Medicine at Mount Sinai, in an interview.
Dr. Kolevzon presented the results at a poster session at the annual meeting of the American Academy of Child and Adolescent Psychiatry.
The study was conducted at 12 sites in the United States and 1 in Israel. Ovid Pharmaceuticals plans to apply to the Food and Drug Administration later this year for approval. There is no existing drug for Angelman syndrome, and the study provided good safety reassurance. “There were some side effects, but for the most part we considered them mild, and only four (out of 88 subjects) discontinued because of side effects,” said Dr. Kolevzon.
The researchers used actigraphy to gain a more objective measure of sleep in the study participants. They randomized 88 patients with Angelman syndrome (aged 13-49 years) to receive placebo in the morning and 15 mg of OV-101 at night, 10 mg OVID-101 in the morning and 15 mg OVID-101 at night, or placebo both in the morning and at night.
Pyrexia occurred in 24% of the group who received the active drug only at night, 3% of the group given the twice-daily dose, and 7% of the placebo group. Seizures occurred in 7% of the once-daily group and 10% of the twice-daily group; seizures were not noted in the placebo group.
The main efficacy outcome measure was the Clinical Global Impressions-9 (CGI-9) scale. The once-daily group had a significant benefit in the sleep domain at 12 weeks, compared with placebo (difference, –0.77; P = .0141), but the twice-daily group had only a trend toward improvement in sleep (difference, –0.45; P = .1407).
Both active therapy groups had significant improvement in CGI-9 measures after 12 weeks of treatment compared to placebo – the twice-daily group (P = .0206, Fisher’s Exact Test) and the once-daily group (P = .0006, mixed model repeated measures analysis).
The actigraphy analysis, conducted in the 45% of patients who could tolerate its use, found that, compared to placebo, the once-daily dosing group experienced an 25.7 minute improvement in latency to sleep onset (P = .0147), as well an approximately 50 minute reduction in sleep time during the day, and a 3.65% improvement in sleep efficiency.
OV-101 has the potential to treat other conditions as well. “Obviously there are a lot of neurodevelopmental disorders where you see dysregulation between the GABAergic and glutamergic systems. This is a drug that has a unique effect on the GABAergic system. It’s already being studied in Fragile X syndrome, where we see this same kind of dysregulation and excess excitation,” said Dr. Kolevzon.
Dr. Kolevzon is a consultant for several drug companies including Ovid Therapeutics.
SOURCE: AACAP 2018. New Research Poster 3.1.
REPORTING FROM AACAP 2018
Key clinical point: A new drug may improve sleep outcomes in Angelman Syndrome.
Major finding: Patients who received a single daily dose of OV-101 scored better than study participants given placebo on the Clinical Global Impressions-Improvement scale.
Study details: Randomized, controlled phase 2 trial (n = 88).
Disclosures: The study was funded by Ovid Therapeutics. Dr. Kolevzon is a consultant for Ovid Therapeutics and several other drug companies.
Source: AACAP 2018 New Research Poster 3.1. .
Cognitive and Behavioral Problems Increase With ALS Disease Stage
Data raise the question of whether cognitive and behavioral change should be included in ALS diagnostic criteria.
Cognitive deficits and behavioral symptoms that are specific to amyotrophic lateral sclerosis (ALS) occur more frequently as the disease progresses, according to research published online ahead of print September 12 in Neurology. Few patients are free of neuropsychologic impairment when they reach the final stage of the disease. It might be appropriate to include cognitive and behavioral change in the diagnostic criteria and future staging systems for ALS, said the authors.
In 2013, Elamin et al suggested that cognitive change in ALS may be associated with indirect measures of disease progression, such as total score on the ALS Functional Rating Scale-Revised. Christopher Crockford, PhD, a researcher at the University of Edinburgh, and colleagues sought to determine whether the cognitive and behavioral symptoms in ALS were more prevalent at more advanced stages of disease.

The Role of Letter Fluency Impairment
They conducted a multicenter, cross-sectional, observational study that included 161 patients from Edinburgh, Dublin, and London with possible, probable, or definite ALS, according to revised El Escorial diagnostic criteria. The researchers also recruited 80 healthy, matched controls. Through interviews, Dr. Crockford and colleagues elicited demographic and clinical data. They measured participants’ clinical staging with the King’s Clinical Staging System and neuropsychologic status with the Edinburgh Cognitive and Behavioral ALS Screen (ECAS).
The investigators did not observe significant differences between the patient and control groups in background variables. Approximately 67% of patients with ALS were male, compared with 60% of controls. Mean age at testing was approximately 61 in both groups. Among patients with ALS, 40 were in Stage 1, 45 were in Stage 2, 22 were in Stage 3, and 54 were in Stage 4.
Compared with controls, patients with ALS had significantly worse cognitive performance on all domains of the ECAS except visuospatial functioning. Dr. Crockford’s group found a significant cross-sectional effect across disease stages for ALS-specific functions (eg, executive, language, and letter fluency) and ECAS total score. They did not find a similar effect for ALS-nonspecific functions (eg, memory and visuospatial). In addition, rates of ALS-specific impairment and behavioral change increased with increasing disease stage.
Letter fluency impairment may explain the relationship between cognitive function and disease stage, said the authors. They observed higher rates of all behavioral problems in later King’s stages. Bulbar signs were significantly related to ALS-specific scores, ECAS total score, and behavioral scores. Site of onset was not related to these scores, however.
Intervention programs to alleviate the effect of patients’ neuropsychologic impairment on caregivers may be appropriate, said the authors. “Furthermore, clinicians should be cognizant of current neuropsychologic status when prescribing life-prolonging interventions to patients and implement support structures for those with a neuropsychologic impairment,” they added.
Informing Patients and Caregivers
Although Dr. Crockford and colleagues focused on the behavioral and cognitive effects of ALS, the disease may affect mental health as well, said Paul Wicks, PhD, Vice President of Innovation at PatientsLikeMe in Cambridge, Massachusetts, and Steven M. Albert, PhD, Professor and Chair of Behavioral and Community Health Sciences at the University of Pittsburgh, in an accompanying editorial. The data show that the rates of major depression and depressed mood increase with increasing disease stage.
Drs. Wicks and Albert cited a survey in which 90% of patients and caregivers reported that their doctors had not told them that cognitive or psychologic symptoms can arise in ALS. “In our experience, colleagues report keeping the information from patients in order to spare them further distress,” they said. Yet most respondents to this survey reported that they would have liked to have been informed about these symptoms.
“Educating patients and caregivers that cognitive change is a part of ALS should be no different from similar discussions to be had in multiple sclerosis, Parkinson disease, and a range of other conditions,” said Drs. Wicks and Albert. “Keeping the truth from patients and caregivers is not protective; it is paternalistic, and it is time to stop. Only by facing up to the hard truth that one of the most dreaded conditions in medicine is even worse than we previously acknowledged can we take stock, marshal our resources, and make renewed plans to defeat our common enemy.”
Suggested Reading
Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018 Sep 12 [Epub ahead of print].
Wicks P, Albert SM. It’s time to stop saying “the mind is unaffected” in ALS. Neurology. 2018 Sep 12 [Epub ahead of print].
Data raise the question of whether cognitive and behavioral change should be included in ALS diagnostic criteria.
Data raise the question of whether cognitive and behavioral change should be included in ALS diagnostic criteria.
Cognitive deficits and behavioral symptoms that are specific to amyotrophic lateral sclerosis (ALS) occur more frequently as the disease progresses, according to research published online ahead of print September 12 in Neurology. Few patients are free of neuropsychologic impairment when they reach the final stage of the disease. It might be appropriate to include cognitive and behavioral change in the diagnostic criteria and future staging systems for ALS, said the authors.
In 2013, Elamin et al suggested that cognitive change in ALS may be associated with indirect measures of disease progression, such as total score on the ALS Functional Rating Scale-Revised. Christopher Crockford, PhD, a researcher at the University of Edinburgh, and colleagues sought to determine whether the cognitive and behavioral symptoms in ALS were more prevalent at more advanced stages of disease.
The Role of Letter Fluency Impairment
They conducted a multicenter, cross-sectional, observational study that included 161 patients from Edinburgh, Dublin, and London with possible, probable, or definite ALS, according to revised El Escorial diagnostic criteria. The researchers also recruited 80 healthy, matched controls. Through interviews, Dr. Crockford and colleagues elicited demographic and clinical data. They measured participants’ clinical staging with the King’s Clinical Staging System and neuropsychologic status with the Edinburgh Cognitive and Behavioral ALS Screen (ECAS).
The investigators did not observe significant differences between the patient and control groups in background variables. Approximately 67% of patients with ALS were male, compared with 60% of controls. Mean age at testing was approximately 61 in both groups. Among patients with ALS, 40 were in Stage 1, 45 were in Stage 2, 22 were in Stage 3, and 54 were in Stage 4.
Compared with controls, patients with ALS had significantly worse cognitive performance on all domains of the ECAS except visuospatial functioning. Dr. Crockford’s group found a significant cross-sectional effect across disease stages for ALS-specific functions (eg, executive, language, and letter fluency) and ECAS total score. They did not find a similar effect for ALS-nonspecific functions (eg, memory and visuospatial). In addition, rates of ALS-specific impairment and behavioral change increased with increasing disease stage.
Letter fluency impairment may explain the relationship between cognitive function and disease stage, said the authors. They observed higher rates of all behavioral problems in later King’s stages. Bulbar signs were significantly related to ALS-specific scores, ECAS total score, and behavioral scores. Site of onset was not related to these scores, however.
Intervention programs to alleviate the effect of patients’ neuropsychologic impairment on caregivers may be appropriate, said the authors. “Furthermore, clinicians should be cognizant of current neuropsychologic status when prescribing life-prolonging interventions to patients and implement support structures for those with a neuropsychologic impairment,” they added.
Informing Patients and Caregivers
Although Dr. Crockford and colleagues focused on the behavioral and cognitive effects of ALS, the disease may affect mental health as well, said Paul Wicks, PhD, Vice President of Innovation at PatientsLikeMe in Cambridge, Massachusetts, and Steven M. Albert, PhD, Professor and Chair of Behavioral and Community Health Sciences at the University of Pittsburgh, in an accompanying editorial. The data show that the rates of major depression and depressed mood increase with increasing disease stage.
Drs. Wicks and Albert cited a survey in which 90% of patients and caregivers reported that their doctors had not told them that cognitive or psychologic symptoms can arise in ALS. “In our experience, colleagues report keeping the information from patients in order to spare them further distress,” they said. Yet most respondents to this survey reported that they would have liked to have been informed about these symptoms.
“Educating patients and caregivers that cognitive change is a part of ALS should be no different from similar discussions to be had in multiple sclerosis, Parkinson disease, and a range of other conditions,” said Drs. Wicks and Albert. “Keeping the truth from patients and caregivers is not protective; it is paternalistic, and it is time to stop. Only by facing up to the hard truth that one of the most dreaded conditions in medicine is even worse than we previously acknowledged can we take stock, marshal our resources, and make renewed plans to defeat our common enemy.”
Suggested Reading
Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018 Sep 12 [Epub ahead of print].
Wicks P, Albert SM. It’s time to stop saying “the mind is unaffected” in ALS. Neurology. 2018 Sep 12 [Epub ahead of print].
Cognitive deficits and behavioral symptoms that are specific to amyotrophic lateral sclerosis (ALS) occur more frequently as the disease progresses, according to research published online ahead of print September 12 in Neurology. Few patients are free of neuropsychologic impairment when they reach the final stage of the disease. It might be appropriate to include cognitive and behavioral change in the diagnostic criteria and future staging systems for ALS, said the authors.
In 2013, Elamin et al suggested that cognitive change in ALS may be associated with indirect measures of disease progression, such as total score on the ALS Functional Rating Scale-Revised. Christopher Crockford, PhD, a researcher at the University of Edinburgh, and colleagues sought to determine whether the cognitive and behavioral symptoms in ALS were more prevalent at more advanced stages of disease.
The Role of Letter Fluency Impairment
They conducted a multicenter, cross-sectional, observational study that included 161 patients from Edinburgh, Dublin, and London with possible, probable, or definite ALS, according to revised El Escorial diagnostic criteria. The researchers also recruited 80 healthy, matched controls. Through interviews, Dr. Crockford and colleagues elicited demographic and clinical data. They measured participants’ clinical staging with the King’s Clinical Staging System and neuropsychologic status with the Edinburgh Cognitive and Behavioral ALS Screen (ECAS).
The investigators did not observe significant differences between the patient and control groups in background variables. Approximately 67% of patients with ALS were male, compared with 60% of controls. Mean age at testing was approximately 61 in both groups. Among patients with ALS, 40 were in Stage 1, 45 were in Stage 2, 22 were in Stage 3, and 54 were in Stage 4.
Compared with controls, patients with ALS had significantly worse cognitive performance on all domains of the ECAS except visuospatial functioning. Dr. Crockford’s group found a significant cross-sectional effect across disease stages for ALS-specific functions (eg, executive, language, and letter fluency) and ECAS total score. They did not find a similar effect for ALS-nonspecific functions (eg, memory and visuospatial). In addition, rates of ALS-specific impairment and behavioral change increased with increasing disease stage.
Letter fluency impairment may explain the relationship between cognitive function and disease stage, said the authors. They observed higher rates of all behavioral problems in later King’s stages. Bulbar signs were significantly related to ALS-specific scores, ECAS total score, and behavioral scores. Site of onset was not related to these scores, however.
Intervention programs to alleviate the effect of patients’ neuropsychologic impairment on caregivers may be appropriate, said the authors. “Furthermore, clinicians should be cognizant of current neuropsychologic status when prescribing life-prolonging interventions to patients and implement support structures for those with a neuropsychologic impairment,” they added.
Informing Patients and Caregivers
Although Dr. Crockford and colleagues focused on the behavioral and cognitive effects of ALS, the disease may affect mental health as well, said Paul Wicks, PhD, Vice President of Innovation at PatientsLikeMe in Cambridge, Massachusetts, and Steven M. Albert, PhD, Professor and Chair of Behavioral and Community Health Sciences at the University of Pittsburgh, in an accompanying editorial. The data show that the rates of major depression and depressed mood increase with increasing disease stage.
Drs. Wicks and Albert cited a survey in which 90% of patients and caregivers reported that their doctors had not told them that cognitive or psychologic symptoms can arise in ALS. “In our experience, colleagues report keeping the information from patients in order to spare them further distress,” they said. Yet most respondents to this survey reported that they would have liked to have been informed about these symptoms.
“Educating patients and caregivers that cognitive change is a part of ALS should be no different from similar discussions to be had in multiple sclerosis, Parkinson disease, and a range of other conditions,” said Drs. Wicks and Albert. “Keeping the truth from patients and caregivers is not protective; it is paternalistic, and it is time to stop. Only by facing up to the hard truth that one of the most dreaded conditions in medicine is even worse than we previously acknowledged can we take stock, marshal our resources, and make renewed plans to defeat our common enemy.”
Suggested Reading
Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018 Sep 12 [Epub ahead of print].
Wicks P, Albert SM. It’s time to stop saying “the mind is unaffected” in ALS. Neurology. 2018 Sep 12 [Epub ahead of print].
FDA approves Sympazan for Lennox-Gastaut syndrome
, according to a release from its developer. Final approval came after the orphan drug designation period for the previously marketed formulation, Onfi, came to an end in October.

LGS is a severe form of epilepsy; it can present with multiple types of seizures, as well as intellectual disabilities. Patients with LGS can have difficulty swallowing tablets or large volumes of oral suspension – which was previously the only way clobazam was delivered – because of physical limitations or behavioral or compliance issues. According to the press release from Aquestive Therapeutics, the Sympazan oral film might be able to get around those difficulties and reduce care burdens, especially with patients who are resistant to or even combative about treatment.
The approval is based on multiple pharmacokinetic studies that altogether showed that the oral film is bioequivalent to clobazam tablets and has a similar safety profile.
In a phase 3 study of 238 patients with LGS, clobazam tablets were shown to reduce drop seizures (those that involved falls) by 41% at low doses and by 68% at high doses versus a reduction of 12% seen with placebo (P less than .05 for all doses vs. placebo).
There is a risk of profound sedation when clobazam is used alongside benzodiazepines; there is also a risk of sedation and somnolence if it is used concomitantly with alcohol or other CNS depressants. Other risks associated with clobazam include suicidal ideation and behavior, serious dermatologic reactions, and physical and psychological dependence. The most common adverse reactions included constipation, pyrexia, lethargy, and drooling.
Full prescribing information can be found on the FDA website.
, according to a release from its developer. Final approval came after the orphan drug designation period for the previously marketed formulation, Onfi, came to an end in October.
LGS is a severe form of epilepsy; it can present with multiple types of seizures, as well as intellectual disabilities. Patients with LGS can have difficulty swallowing tablets or large volumes of oral suspension – which was previously the only way clobazam was delivered – because of physical limitations or behavioral or compliance issues. According to the press release from Aquestive Therapeutics, the Sympazan oral film might be able to get around those difficulties and reduce care burdens, especially with patients who are resistant to or even combative about treatment.
The approval is based on multiple pharmacokinetic studies that altogether showed that the oral film is bioequivalent to clobazam tablets and has a similar safety profile.
In a phase 3 study of 238 patients with LGS, clobazam tablets were shown to reduce drop seizures (those that involved falls) by 41% at low doses and by 68% at high doses versus a reduction of 12% seen with placebo (P less than .05 for all doses vs. placebo).
There is a risk of profound sedation when clobazam is used alongside benzodiazepines; there is also a risk of sedation and somnolence if it is used concomitantly with alcohol or other CNS depressants. Other risks associated with clobazam include suicidal ideation and behavior, serious dermatologic reactions, and physical and psychological dependence. The most common adverse reactions included constipation, pyrexia, lethargy, and drooling.
Full prescribing information can be found on the FDA website.
, according to a release from its developer. Final approval came after the orphan drug designation period for the previously marketed formulation, Onfi, came to an end in October.
LGS is a severe form of epilepsy; it can present with multiple types of seizures, as well as intellectual disabilities. Patients with LGS can have difficulty swallowing tablets or large volumes of oral suspension – which was previously the only way clobazam was delivered – because of physical limitations or behavioral or compliance issues. According to the press release from Aquestive Therapeutics, the Sympazan oral film might be able to get around those difficulties and reduce care burdens, especially with patients who are resistant to or even combative about treatment.
The approval is based on multiple pharmacokinetic studies that altogether showed that the oral film is bioequivalent to clobazam tablets and has a similar safety profile.
In a phase 3 study of 238 patients with LGS, clobazam tablets were shown to reduce drop seizures (those that involved falls) by 41% at low doses and by 68% at high doses versus a reduction of 12% seen with placebo (P less than .05 for all doses vs. placebo).
There is a risk of profound sedation when clobazam is used alongside benzodiazepines; there is also a risk of sedation and somnolence if it is used concomitantly with alcohol or other CNS depressants. Other risks associated with clobazam include suicidal ideation and behavior, serious dermatologic reactions, and physical and psychological dependence. The most common adverse reactions included constipation, pyrexia, lethargy, and drooling.
Full prescribing information can be found on the FDA website.
Triggers May Guide Treatment of New Daily Persistent Headache
Aggressive initial therapy is appropriate because the disorder becomes increasingly refractory with time.
ASHEVILLE, NC—New daily persistent headache is rare and difficult to treat. Although neurologists may be tempted to try a series of treatments until the patient improves, therapeutic success is more likely if the neurologist can identify a triggering event, said Todd Rozen, MD, a neurologist at Mayo Clinic in Jacksonville, Florida. Elements of the patient’s history or clinical examination also can guide treatment, he added at the Eighth Annual Scientific Meeting of the Southern Headache Society.
Patients Remember the Onset
New daily persistent headache was first described in 1986, and few researchers have studied it. It is a persistent headache with a clearly remembered onset. “Most [patients] can name the date it began or at least the month,” said Dr. Rozen. The headache becomes unremitting within 24 hours and must be present for longer than three months, according to the current diagnostic criteria. Patients may have a remitting form, a relapsing-remitting form, or a refractory form of the headache. The age of onset “can be as early as in the mid to late teens or early 20s, especially in the female population,” said Dr. Rozen. Age of onset also depends on the triggering event.
The pain typically is bilateral and moderate to severe. Although many patients may present with a tension-type headache, more than 60% have migrainous symptoms such as nausea, photophobia, or phonophobia, said Dr. Rozen.
The disorder is more common among women than among men. Between 10% and 13% of patients who present to headache clinics have new daily persistent headache, said Dr. Rozen. “It is either becoming more prevalent in the office, or we are better at recognizing it.”
Comparing Effects on the Genders
For a study published in 2016, Dr. Rozen examined 97 patients (65 women) with new daily persistent headache. Approximately 53% of patients could not identify a triggering event for their headache, which makes treatment “much more difficult,” said Dr. Rozen. Although the mean age of onset was younger in women (32.4) than men (35.8), the age of onset was the same between genders when Dr. Rozen examined for individual triggers.
The frequency of individual triggering events also was the same between genders, and these results suggest that each trigger may be associated with a discrete pathogenesis. Triggers included infection or flulike illness (22%), stressful life event (9%), surgery (9%), and other (7%). All patients who had identified surgery as a trigger had been intubated, and Dr. Rozen hypothesized that their headaches were cervicogenic. The younger patients who had undergone surgery were hypermobile, and the older patients had neck arthritis as predisposing risk factors for neck irritation with intubation.
A Somatoform Disorder?
The stubbornly refractory nature of this disorder has aroused the suspicion that it may be somatoform. In 2017, Uniyal and colleagues found that somatization, generalized anxiety disorder, depression, and catastrophization were more common in patients with new daily persistent headache, compared with patients with chronic low back pain and healthy controls.
Interpreting these data is difficult, said Dr. Rozen. They may indicate that these psychiatric comorbidities are risk factors for new daily persistent headache. An equally plausible interpretation is that these patients have a different disorder (eg, Ehlers-Danlos syndrome) that encompasses these common traits. Finally, symptoms such as depression and catastrophization may be sequelae of, rather than risk factors for, new daily persistent headache.
Researchers have found imaging abnormalities to be rare in patients with new daily persistent headache. About two-thirds of patients in a 2002 study had normal MRI or CT results, and the rest had nonspecific findings unrelated to the headache. Dr. Rozen found that white matter lesions were uncommon in patients with this disorder, except for those with a history of migraine or cardiovascular or cerebrovascular risk factors. CSF likewise generally is normal in patients with new daily persistent headache.
Triggers Suggest Treatments
Goadsby proposed in 2011 that new daily persistent headache is a syndrome rather than a single disorder. “I’m completely in agreement,” said Dr. Rozen. “However, I do believe that individuals who have the same triggering event have the same pathogenesis.” Identifying the triggering event and understanding the temporal profile of the first headache can enable the choice of appropriate therapy, he added.
A patient whose persistent headache begins with a thunderclap onset likely has a prolonged cerebral artery vasospasm. Dr. Rozen treated a patient whose initial headache was a thunderclap; imaging ultimately revealed that she had a vasospasm. Her headache responded to nimodipine within days. Nimodipine generally provides relief within three to five days, said Dr. Rozen. If it worsens the headache, then the patient does not have vasospasm, he added.
Many patients with new daily persistent headache have a physical presentation that suggests Marfan syndrome. This observation led Dr. Rozen to hypothesize that cervical hypermobility is a risk factor for new daily persistent headache. Hypermobile patients may put significant stress on the C1, C2, and C3 joints, which are “where the trigeminal–cervical complex comes together,” said Dr. Rozen. A long plane ride or appointment with the dentist could trigger new daily persistent headache. Treatment with onabotulinumtoxinA often helps these patients. High cervical blocks also can bring relief, said Dr. Rozen.
He and his colleagues recently identified a new subset of patients with new daily persistent headache. They were older female patients with a mean age of 57 who suddenly developed the disorder. Most of them reported that the pain was worst before they got out of bed in the morning. Within seconds of assuming the Trendelenburg position, these patients had intensified pain and nausea, suggesting CSF hypertension. The patients all responded to acetazolamide or spironolactone, which lowered CSF pressure. “I think these individuals developed cerebral vein insufficiency because of estrogen withdrawal based on their age. Plus, the majority were overweight, which can also raise baseline CSF pressure.”
Examination Should Incorporate Imaging
All patients with new daily persistent headache should undergo imaging, including a brain MRI with and without gadolinium, plus an MR venogram, which can identify CSF leaks and a cerebral vein thrombosis, which are leading secondary causes of the disorder. Neurologists could examine patients’ viral titers in addition if the history suggests a post infectious trigger. A lumbar puncture and measurement of opening CSF pressure are appropriate for patients who have not responded to medication.
Evidence From the Literature
The literature possibly supports the efficacy of several treatments in new daily persistent headache, but includes no placebo-controlled trials for them. Dr. Rozen found doxycycline to be helpful for several patients with elevated CSF tumor necrosis factor alpha.
Marmura and colleagues found that mexiletine reduced the severity of pain in patients with refractory new daily persistent headache. The treatment did not reduce headache frequency, however, and side effects were common.
In a retrospective study, Prakash et al followed 63 patients with new daily persistent headache for five years. They found that patients who received IV methyl prednisolone and sodium valproate had a better response than patients who received other therapies. They called for prospective and controlled studies to confirm this observation.
In general, aggressive initial therapy is warranted, “especially if you meet an individual within one year of headache onset,” said Dr. Rozen. The likelihood of response to therapy appears to decline with the duration of the headache. “Infusion therapy or inpatient therapy with IV medications, even with standard migraine protocols, may help break the cycle,” Dr. Rozen concluded.
—Erik Greb
Suggested Reading
Goadsby PJ. New daily persistent headache: a syndrome, not a discrete disorder. Headache. 2011;51(4):650-653.
Marmura MJ, Passero FC Jr, Young WB. Mexiletine for refractory chronic daily headache: a report of nine cases. Headache. 2008;48(10):1506-1510.
Prakash S, Saini S, Rana KR, Mahato P. Refining clinical features and therapeutic options of new daily persistent headache: a retrospective study of 63 patients in India. J Headache Pain. 2012;13(6):477-485.
Rozen TD. A new subtype of chronic daily headache presenting in older women. J Womens Health (Larchmt). 2018;27(2):203-208.
Rozen TD. Triggering events and new daily persistent headache: age and gender differences and insights on pathogenesis-a clinic-based study. Headache. 2016;56(1):164-173.
Uniyal R, Paliwal VK, Tripathi A. Psychiatric comorbidity in new daily persistent headache: a cross-sectional study. Eur J Pain. 2017;21(6):1031-1038.
Aggressive initial therapy is appropriate because the disorder becomes increasingly refractory with time.
Aggressive initial therapy is appropriate because the disorder becomes increasingly refractory with time.
ASHEVILLE, NC—New daily persistent headache is rare and difficult to treat. Although neurologists may be tempted to try a series of treatments until the patient improves, therapeutic success is more likely if the neurologist can identify a triggering event, said Todd Rozen, MD, a neurologist at Mayo Clinic in Jacksonville, Florida. Elements of the patient’s history or clinical examination also can guide treatment, he added at the Eighth Annual Scientific Meeting of the Southern Headache Society.
Patients Remember the Onset
New daily persistent headache was first described in 1986, and few researchers have studied it. It is a persistent headache with a clearly remembered onset. “Most [patients] can name the date it began or at least the month,” said Dr. Rozen. The headache becomes unremitting within 24 hours and must be present for longer than three months, according to the current diagnostic criteria. Patients may have a remitting form, a relapsing-remitting form, or a refractory form of the headache. The age of onset “can be as early as in the mid to late teens or early 20s, especially in the female population,” said Dr. Rozen. Age of onset also depends on the triggering event.
The pain typically is bilateral and moderate to severe. Although many patients may present with a tension-type headache, more than 60% have migrainous symptoms such as nausea, photophobia, or phonophobia, said Dr. Rozen.
The disorder is more common among women than among men. Between 10% and 13% of patients who present to headache clinics have new daily persistent headache, said Dr. Rozen. “It is either becoming more prevalent in the office, or we are better at recognizing it.”
Comparing Effects on the Genders
For a study published in 2016, Dr. Rozen examined 97 patients (65 women) with new daily persistent headache. Approximately 53% of patients could not identify a triggering event for their headache, which makes treatment “much more difficult,” said Dr. Rozen. Although the mean age of onset was younger in women (32.4) than men (35.8), the age of onset was the same between genders when Dr. Rozen examined for individual triggers.
The frequency of individual triggering events also was the same between genders, and these results suggest that each trigger may be associated with a discrete pathogenesis. Triggers included infection or flulike illness (22%), stressful life event (9%), surgery (9%), and other (7%). All patients who had identified surgery as a trigger had been intubated, and Dr. Rozen hypothesized that their headaches were cervicogenic. The younger patients who had undergone surgery were hypermobile, and the older patients had neck arthritis as predisposing risk factors for neck irritation with intubation.
A Somatoform Disorder?
The stubbornly refractory nature of this disorder has aroused the suspicion that it may be somatoform. In 2017, Uniyal and colleagues found that somatization, generalized anxiety disorder, depression, and catastrophization were more common in patients with new daily persistent headache, compared with patients with chronic low back pain and healthy controls.
Interpreting these data is difficult, said Dr. Rozen. They may indicate that these psychiatric comorbidities are risk factors for new daily persistent headache. An equally plausible interpretation is that these patients have a different disorder (eg, Ehlers-Danlos syndrome) that encompasses these common traits. Finally, symptoms such as depression and catastrophization may be sequelae of, rather than risk factors for, new daily persistent headache.
Researchers have found imaging abnormalities to be rare in patients with new daily persistent headache. About two-thirds of patients in a 2002 study had normal MRI or CT results, and the rest had nonspecific findings unrelated to the headache. Dr. Rozen found that white matter lesions were uncommon in patients with this disorder, except for those with a history of migraine or cardiovascular or cerebrovascular risk factors. CSF likewise generally is normal in patients with new daily persistent headache.
Triggers Suggest Treatments
Goadsby proposed in 2011 that new daily persistent headache is a syndrome rather than a single disorder. “I’m completely in agreement,” said Dr. Rozen. “However, I do believe that individuals who have the same triggering event have the same pathogenesis.” Identifying the triggering event and understanding the temporal profile of the first headache can enable the choice of appropriate therapy, he added.
A patient whose persistent headache begins with a thunderclap onset likely has a prolonged cerebral artery vasospasm. Dr. Rozen treated a patient whose initial headache was a thunderclap; imaging ultimately revealed that she had a vasospasm. Her headache responded to nimodipine within days. Nimodipine generally provides relief within three to five days, said Dr. Rozen. If it worsens the headache, then the patient does not have vasospasm, he added.
Many patients with new daily persistent headache have a physical presentation that suggests Marfan syndrome. This observation led Dr. Rozen to hypothesize that cervical hypermobility is a risk factor for new daily persistent headache. Hypermobile patients may put significant stress on the C1, C2, and C3 joints, which are “where the trigeminal–cervical complex comes together,” said Dr. Rozen. A long plane ride or appointment with the dentist could trigger new daily persistent headache. Treatment with onabotulinumtoxinA often helps these patients. High cervical blocks also can bring relief, said Dr. Rozen.
He and his colleagues recently identified a new subset of patients with new daily persistent headache. They were older female patients with a mean age of 57 who suddenly developed the disorder. Most of them reported that the pain was worst before they got out of bed in the morning. Within seconds of assuming the Trendelenburg position, these patients had intensified pain and nausea, suggesting CSF hypertension. The patients all responded to acetazolamide or spironolactone, which lowered CSF pressure. “I think these individuals developed cerebral vein insufficiency because of estrogen withdrawal based on their age. Plus, the majority were overweight, which can also raise baseline CSF pressure.”
Examination Should Incorporate Imaging
All patients with new daily persistent headache should undergo imaging, including a brain MRI with and without gadolinium, plus an MR venogram, which can identify CSF leaks and a cerebral vein thrombosis, which are leading secondary causes of the disorder. Neurologists could examine patients’ viral titers in addition if the history suggests a post infectious trigger. A lumbar puncture and measurement of opening CSF pressure are appropriate for patients who have not responded to medication.
Evidence From the Literature
The literature possibly supports the efficacy of several treatments in new daily persistent headache, but includes no placebo-controlled trials for them. Dr. Rozen found doxycycline to be helpful for several patients with elevated CSF tumor necrosis factor alpha.
Marmura and colleagues found that mexiletine reduced the severity of pain in patients with refractory new daily persistent headache. The treatment did not reduce headache frequency, however, and side effects were common.
In a retrospective study, Prakash et al followed 63 patients with new daily persistent headache for five years. They found that patients who received IV methyl prednisolone and sodium valproate had a better response than patients who received other therapies. They called for prospective and controlled studies to confirm this observation.
In general, aggressive initial therapy is warranted, “especially if you meet an individual within one year of headache onset,” said Dr. Rozen. The likelihood of response to therapy appears to decline with the duration of the headache. “Infusion therapy or inpatient therapy with IV medications, even with standard migraine protocols, may help break the cycle,” Dr. Rozen concluded.
—Erik Greb
Suggested Reading
Goadsby PJ. New daily persistent headache: a syndrome, not a discrete disorder. Headache. 2011;51(4):650-653.
Marmura MJ, Passero FC Jr, Young WB. Mexiletine for refractory chronic daily headache: a report of nine cases. Headache. 2008;48(10):1506-1510.
Prakash S, Saini S, Rana KR, Mahato P. Refining clinical features and therapeutic options of new daily persistent headache: a retrospective study of 63 patients in India. J Headache Pain. 2012;13(6):477-485.
Rozen TD. A new subtype of chronic daily headache presenting in older women. J Womens Health (Larchmt). 2018;27(2):203-208.
Rozen TD. Triggering events and new daily persistent headache: age and gender differences and insights on pathogenesis-a clinic-based study. Headache. 2016;56(1):164-173.
Uniyal R, Paliwal VK, Tripathi A. Psychiatric comorbidity in new daily persistent headache: a cross-sectional study. Eur J Pain. 2017;21(6):1031-1038.
ASHEVILLE, NC—New daily persistent headache is rare and difficult to treat. Although neurologists may be tempted to try a series of treatments until the patient improves, therapeutic success is more likely if the neurologist can identify a triggering event, said Todd Rozen, MD, a neurologist at Mayo Clinic in Jacksonville, Florida. Elements of the patient’s history or clinical examination also can guide treatment, he added at the Eighth Annual Scientific Meeting of the Southern Headache Society.
Patients Remember the Onset
New daily persistent headache was first described in 1986, and few researchers have studied it. It is a persistent headache with a clearly remembered onset. “Most [patients] can name the date it began or at least the month,” said Dr. Rozen. The headache becomes unremitting within 24 hours and must be present for longer than three months, according to the current diagnostic criteria. Patients may have a remitting form, a relapsing-remitting form, or a refractory form of the headache. The age of onset “can be as early as in the mid to late teens or early 20s, especially in the female population,” said Dr. Rozen. Age of onset also depends on the triggering event.
The pain typically is bilateral and moderate to severe. Although many patients may present with a tension-type headache, more than 60% have migrainous symptoms such as nausea, photophobia, or phonophobia, said Dr. Rozen.
The disorder is more common among women than among men. Between 10% and 13% of patients who present to headache clinics have new daily persistent headache, said Dr. Rozen. “It is either becoming more prevalent in the office, or we are better at recognizing it.”
Comparing Effects on the Genders
For a study published in 2016, Dr. Rozen examined 97 patients (65 women) with new daily persistent headache. Approximately 53% of patients could not identify a triggering event for their headache, which makes treatment “much more difficult,” said Dr. Rozen. Although the mean age of onset was younger in women (32.4) than men (35.8), the age of onset was the same between genders when Dr. Rozen examined for individual triggers.
The frequency of individual triggering events also was the same between genders, and these results suggest that each trigger may be associated with a discrete pathogenesis. Triggers included infection or flulike illness (22%), stressful life event (9%), surgery (9%), and other (7%). All patients who had identified surgery as a trigger had been intubated, and Dr. Rozen hypothesized that their headaches were cervicogenic. The younger patients who had undergone surgery were hypermobile, and the older patients had neck arthritis as predisposing risk factors for neck irritation with intubation.
A Somatoform Disorder?
The stubbornly refractory nature of this disorder has aroused the suspicion that it may be somatoform. In 2017, Uniyal and colleagues found that somatization, generalized anxiety disorder, depression, and catastrophization were more common in patients with new daily persistent headache, compared with patients with chronic low back pain and healthy controls.
Interpreting these data is difficult, said Dr. Rozen. They may indicate that these psychiatric comorbidities are risk factors for new daily persistent headache. An equally plausible interpretation is that these patients have a different disorder (eg, Ehlers-Danlos syndrome) that encompasses these common traits. Finally, symptoms such as depression and catastrophization may be sequelae of, rather than risk factors for, new daily persistent headache.
Researchers have found imaging abnormalities to be rare in patients with new daily persistent headache. About two-thirds of patients in a 2002 study had normal MRI or CT results, and the rest had nonspecific findings unrelated to the headache. Dr. Rozen found that white matter lesions were uncommon in patients with this disorder, except for those with a history of migraine or cardiovascular or cerebrovascular risk factors. CSF likewise generally is normal in patients with new daily persistent headache.
Triggers Suggest Treatments
Goadsby proposed in 2011 that new daily persistent headache is a syndrome rather than a single disorder. “I’m completely in agreement,” said Dr. Rozen. “However, I do believe that individuals who have the same triggering event have the same pathogenesis.” Identifying the triggering event and understanding the temporal profile of the first headache can enable the choice of appropriate therapy, he added.
A patient whose persistent headache begins with a thunderclap onset likely has a prolonged cerebral artery vasospasm. Dr. Rozen treated a patient whose initial headache was a thunderclap; imaging ultimately revealed that she had a vasospasm. Her headache responded to nimodipine within days. Nimodipine generally provides relief within three to five days, said Dr. Rozen. If it worsens the headache, then the patient does not have vasospasm, he added.
Many patients with new daily persistent headache have a physical presentation that suggests Marfan syndrome. This observation led Dr. Rozen to hypothesize that cervical hypermobility is a risk factor for new daily persistent headache. Hypermobile patients may put significant stress on the C1, C2, and C3 joints, which are “where the trigeminal–cervical complex comes together,” said Dr. Rozen. A long plane ride or appointment with the dentist could trigger new daily persistent headache. Treatment with onabotulinumtoxinA often helps these patients. High cervical blocks also can bring relief, said Dr. Rozen.
He and his colleagues recently identified a new subset of patients with new daily persistent headache. They were older female patients with a mean age of 57 who suddenly developed the disorder. Most of them reported that the pain was worst before they got out of bed in the morning. Within seconds of assuming the Trendelenburg position, these patients had intensified pain and nausea, suggesting CSF hypertension. The patients all responded to acetazolamide or spironolactone, which lowered CSF pressure. “I think these individuals developed cerebral vein insufficiency because of estrogen withdrawal based on their age. Plus, the majority were overweight, which can also raise baseline CSF pressure.”
Examination Should Incorporate Imaging
All patients with new daily persistent headache should undergo imaging, including a brain MRI with and without gadolinium, plus an MR venogram, which can identify CSF leaks and a cerebral vein thrombosis, which are leading secondary causes of the disorder. Neurologists could examine patients’ viral titers in addition if the history suggests a post infectious trigger. A lumbar puncture and measurement of opening CSF pressure are appropriate for patients who have not responded to medication.
Evidence From the Literature
The literature possibly supports the efficacy of several treatments in new daily persistent headache, but includes no placebo-controlled trials for them. Dr. Rozen found doxycycline to be helpful for several patients with elevated CSF tumor necrosis factor alpha.
Marmura and colleagues found that mexiletine reduced the severity of pain in patients with refractory new daily persistent headache. The treatment did not reduce headache frequency, however, and side effects were common.
In a retrospective study, Prakash et al followed 63 patients with new daily persistent headache for five years. They found that patients who received IV methyl prednisolone and sodium valproate had a better response than patients who received other therapies. They called for prospective and controlled studies to confirm this observation.
In general, aggressive initial therapy is warranted, “especially if you meet an individual within one year of headache onset,” said Dr. Rozen. The likelihood of response to therapy appears to decline with the duration of the headache. “Infusion therapy or inpatient therapy with IV medications, even with standard migraine protocols, may help break the cycle,” Dr. Rozen concluded.
—Erik Greb
Suggested Reading
Goadsby PJ. New daily persistent headache: a syndrome, not a discrete disorder. Headache. 2011;51(4):650-653.
Marmura MJ, Passero FC Jr, Young WB. Mexiletine for refractory chronic daily headache: a report of nine cases. Headache. 2008;48(10):1506-1510.
Prakash S, Saini S, Rana KR, Mahato P. Refining clinical features and therapeutic options of new daily persistent headache: a retrospective study of 63 patients in India. J Headache Pain. 2012;13(6):477-485.
Rozen TD. A new subtype of chronic daily headache presenting in older women. J Womens Health (Larchmt). 2018;27(2):203-208.
Rozen TD. Triggering events and new daily persistent headache: age and gender differences and insights on pathogenesis-a clinic-based study. Headache. 2016;56(1):164-173.
Uniyal R, Paliwal VK, Tripathi A. Psychiatric comorbidity in new daily persistent headache: a cross-sectional study. Eur J Pain. 2017;21(6):1031-1038.
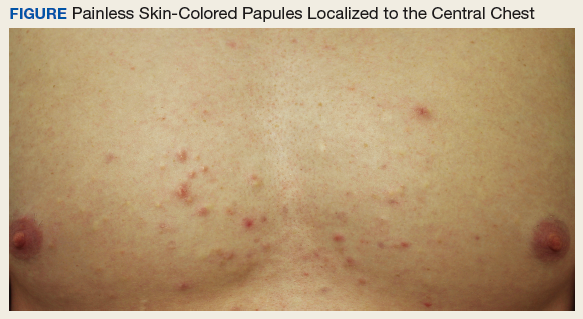
Skin-Colored Papules on the Chest
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
Concomitant Fibrofolliculoma and Trichodiscoma on the Abdomen
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
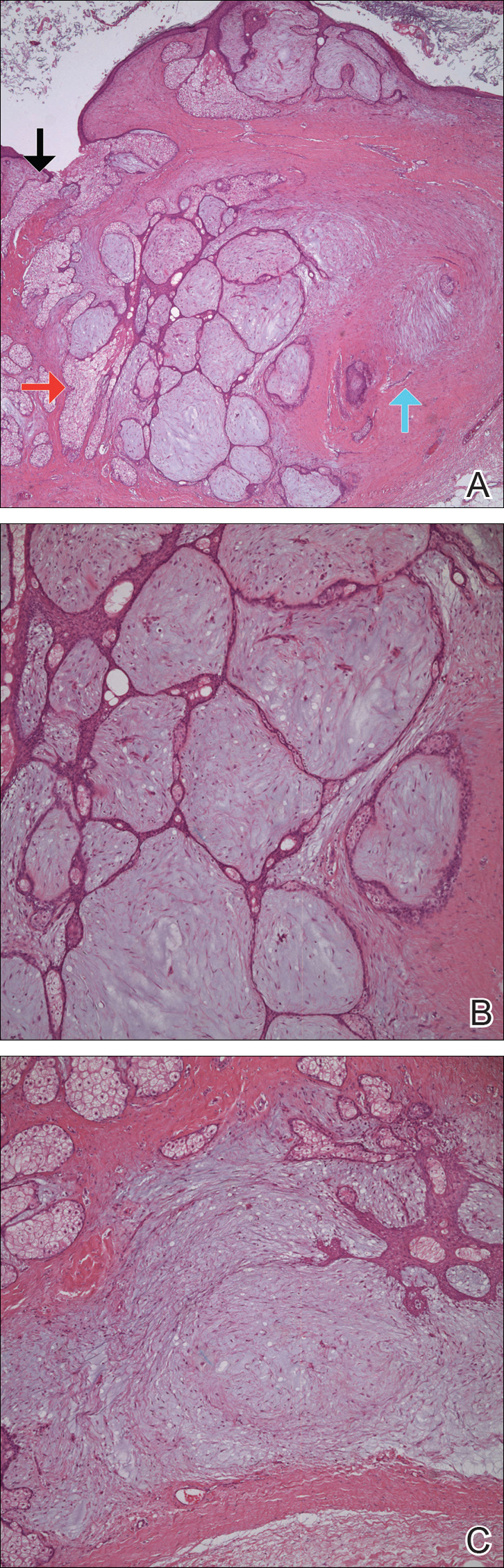
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.

Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Practice Points
- Fibrofolliculoma and trichodiscoma are flesh-colored adnexal tumors that arise from or around hair follicles.
- It is important to recognize these entities, as they can be related to Birt-Hogg-Dubé syndrome.
Pediatric indications appear in one-third of orphan drug approvals
WASHINGTON – Pediatric indications appeared in approximately 36% of orphan drug approvals between 2000 and 2017, according to data from the Food and Drug Administration.
“Given the impact of rare disease on children, it is of particular interest to better understand where new drug approvals and advances are occurring, through the lens of pediatrics,” said Kathleen L. Miller, Ph.D., and Michael Lanthier of the Food and Drug Administration in Silver Spring, Md. They presented their findings in a poster at the NORD Rare Summit, held by the National Organization for Rare Disorders.
Overall, 31% of 314 orphan drug approvals by the FDA between 2000 and 2017 had a pediatric and adult indication, 5% had pediatric-only indications, and 64% had adult-only indications.
For the 112 pediatric indication approvals, 14% were for inherited blood disorders, 14% for inherited metabolic disorders, 13% for rare cancers, 10% for antidotes and medical countermeasures, 9% for infectious diseases, 8% for auto-inflammatory diseases, 7% for neurologic disorders, and 25% for other conditions.
Approximately 63% of the total orphan drug approvals during the study period were for rare cancers or genetic disorders (138 approvals and 59 approvals, respectively). Although 90% of the rare cancer approvals were for adults only, 84% of genetic disorder drug approvals had pediatric indications, Dr. Miller and Mr. Lanthier noted.
When analyzed by submission type, pediatric indications were included in 52% of new orphan formulations, 35% of orphan secondary indication approvals, and 32% of new molecular entity approvals.
Although more research is needed, the results suggest that “pediatric indications represent a sizable proportion of orphan drug approvals in both a range of therapeutic areas and in a range of approval types,” the investigators concluded.
The researchers are employed by the FDA, which sponsored the study. They had no financial conflicts to disclose.
WASHINGTON – Pediatric indications appeared in approximately 36% of orphan drug approvals between 2000 and 2017, according to data from the Food and Drug Administration.
“Given the impact of rare disease on children, it is of particular interest to better understand where new drug approvals and advances are occurring, through the lens of pediatrics,” said Kathleen L. Miller, Ph.D., and Michael Lanthier of the Food and Drug Administration in Silver Spring, Md. They presented their findings in a poster at the NORD Rare Summit, held by the National Organization for Rare Disorders.
Overall, 31% of 314 orphan drug approvals by the FDA between 2000 and 2017 had a pediatric and adult indication, 5% had pediatric-only indications, and 64% had adult-only indications.
For the 112 pediatric indication approvals, 14% were for inherited blood disorders, 14% for inherited metabolic disorders, 13% for rare cancers, 10% for antidotes and medical countermeasures, 9% for infectious diseases, 8% for auto-inflammatory diseases, 7% for neurologic disorders, and 25% for other conditions.
Approximately 63% of the total orphan drug approvals during the study period were for rare cancers or genetic disorders (138 approvals and 59 approvals, respectively). Although 90% of the rare cancer approvals were for adults only, 84% of genetic disorder drug approvals had pediatric indications, Dr. Miller and Mr. Lanthier noted.
When analyzed by submission type, pediatric indications were included in 52% of new orphan formulations, 35% of orphan secondary indication approvals, and 32% of new molecular entity approvals.
Although more research is needed, the results suggest that “pediatric indications represent a sizable proportion of orphan drug approvals in both a range of therapeutic areas and in a range of approval types,” the investigators concluded.
The researchers are employed by the FDA, which sponsored the study. They had no financial conflicts to disclose.
WASHINGTON – Pediatric indications appeared in approximately 36% of orphan drug approvals between 2000 and 2017, according to data from the Food and Drug Administration.
“Given the impact of rare disease on children, it is of particular interest to better understand where new drug approvals and advances are occurring, through the lens of pediatrics,” said Kathleen L. Miller, Ph.D., and Michael Lanthier of the Food and Drug Administration in Silver Spring, Md. They presented their findings in a poster at the NORD Rare Summit, held by the National Organization for Rare Disorders.
Overall, 31% of 314 orphan drug approvals by the FDA between 2000 and 2017 had a pediatric and adult indication, 5% had pediatric-only indications, and 64% had adult-only indications.
For the 112 pediatric indication approvals, 14% were for inherited blood disorders, 14% for inherited metabolic disorders, 13% for rare cancers, 10% for antidotes and medical countermeasures, 9% for infectious diseases, 8% for auto-inflammatory diseases, 7% for neurologic disorders, and 25% for other conditions.
Approximately 63% of the total orphan drug approvals during the study period were for rare cancers or genetic disorders (138 approvals and 59 approvals, respectively). Although 90% of the rare cancer approvals were for adults only, 84% of genetic disorder drug approvals had pediatric indications, Dr. Miller and Mr. Lanthier noted.
When analyzed by submission type, pediatric indications were included in 52% of new orphan formulations, 35% of orphan secondary indication approvals, and 32% of new molecular entity approvals.
Although more research is needed, the results suggest that “pediatric indications represent a sizable proportion of orphan drug approvals in both a range of therapeutic areas and in a range of approval types,” the investigators concluded.
The researchers are employed by the FDA, which sponsored the study. They had no financial conflicts to disclose.
REPORTING FROM NORD SUMMIT 2018
Key clinical point:
Major finding: A total of 112 orphan drug approvals between 2000 and 2017 included a pediatric indication.
Study details: The data come from a review of 314 orphan drug approvals by the FDA between 2000 and 2017.
Disclosures: The researchers are employed by the Food and Drug Administration, which sponsored the study. They had no financial conflicts to disclose.
What Are the Clinical, Laboratory, and Electrodiagnostic Features of Zinc Deficiency-Induced Peripheral Neuropathy?
Reduced tendon reflexes and an abnormal Romberg test may be common in patients with this disorder.
WASHINGTON, DC—Patients with zinc deficiency-induced peripheral neuropathy may present with paresthesia, gait abnormalities, sensory deficits, reduced tendon reflexes, an abnormal Romberg test, and increased CSF protein, according to a study presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine.
Recognition of the features of zinc deficiency-induced peripheral neuropathy may help neurologists diagnose the disorder and manage patients, researchers said.
“Zinc, an essential trace element, plays a critical role in maintaining normal structural and functional conditions in the body,” said lead author Favio C. Bumanlag, Chief Technologist in the Department of Neurology at the Lewis Katz School of Medicine at Temple University in Philadelphia. “Peripheral nerves are susceptible to damage when zinc deficiency occurs.... Recognition of [zinc deficiency-induced peripheral neuropathy] will help physicians and technologists effectively manage patients.”
To study the clinical and electrophysiologic features of zinc deficiency-induced peripheral neuropathy, Mr. Bumanlag and Jin Luo, MD, PhD, Professor of Neurology and Pharmacology at Temple University, retrospectively reviewed charts in their neuromuscular clinic and EMG laboratory database to identify patients with peripheral neuropathy and zinc deficiency. They included charts from between January 1, 2015, and December 31, 2017, in their review. They excluded patients with abnormal copper levels.
Mr. Bumanlag and Dr. Luo obtained information about patients’ clinical presentations, past medical histories, BMI, neurologic examinations, and laboratory results. They also examined patients’ needle electromyograms and nerve conduction studies.
In all, they identified 12 patients with peripheral neuropathy and zinc deficiency. Patients had a mean age of 55.1. Six were female. Patients’ mean zinc level was 52.5 μg/dL, with a range of 37 μg/dL to 58 μg/dL (reference, 56–134 μg/dL). Mean copper level was 107.6 μg/dL, with a range of 84 μg/dL to 173 μg/dL (reference, 72–166 μg/dL). Eleven of the 12 patients had received an electrophysiologic evaluation.
Notable findings in presentation included paresthesia in 75 and gait abnormalities in 42%. One patient was obese (8%), and three patients had diarrhea (25%). Neurologic examination showed sensory deficits in 83%, reduced tendon reflexes in 67%, and an abnormal Romberg test in 67%. Four of five patients had increased CSF protein. Electrophysiologic evaluations showed features of demyelinating peripheral neuropathy (28%) and distally active denervation in the lower extremities.
“Zinc participates in more than 200 enzymatic reactions,” said the researchers. “Unfortunately, zinc deficiency-induced peripheral neuropathy is often misdiagnosed or delayed in diagnosis. Literature on zinc deficiency-induced peripheral neuropathy is sparse.”
Reduced tendon reflexes and an abnormal Romberg test may be common in patients with this disorder.
Reduced tendon reflexes and an abnormal Romberg test may be common in patients with this disorder.
WASHINGTON, DC—Patients with zinc deficiency-induced peripheral neuropathy may present with paresthesia, gait abnormalities, sensory deficits, reduced tendon reflexes, an abnormal Romberg test, and increased CSF protein, according to a study presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine.
Recognition of the features of zinc deficiency-induced peripheral neuropathy may help neurologists diagnose the disorder and manage patients, researchers said.
“Zinc, an essential trace element, plays a critical role in maintaining normal structural and functional conditions in the body,” said lead author Favio C. Bumanlag, Chief Technologist in the Department of Neurology at the Lewis Katz School of Medicine at Temple University in Philadelphia. “Peripheral nerves are susceptible to damage when zinc deficiency occurs.... Recognition of [zinc deficiency-induced peripheral neuropathy] will help physicians and technologists effectively manage patients.”
To study the clinical and electrophysiologic features of zinc deficiency-induced peripheral neuropathy, Mr. Bumanlag and Jin Luo, MD, PhD, Professor of Neurology and Pharmacology at Temple University, retrospectively reviewed charts in their neuromuscular clinic and EMG laboratory database to identify patients with peripheral neuropathy and zinc deficiency. They included charts from between January 1, 2015, and December 31, 2017, in their review. They excluded patients with abnormal copper levels.
Mr. Bumanlag and Dr. Luo obtained information about patients’ clinical presentations, past medical histories, BMI, neurologic examinations, and laboratory results. They also examined patients’ needle electromyograms and nerve conduction studies.
In all, they identified 12 patients with peripheral neuropathy and zinc deficiency. Patients had a mean age of 55.1. Six were female. Patients’ mean zinc level was 52.5 μg/dL, with a range of 37 μg/dL to 58 μg/dL (reference, 56–134 μg/dL). Mean copper level was 107.6 μg/dL, with a range of 84 μg/dL to 173 μg/dL (reference, 72–166 μg/dL). Eleven of the 12 patients had received an electrophysiologic evaluation.
Notable findings in presentation included paresthesia in 75 and gait abnormalities in 42%. One patient was obese (8%), and three patients had diarrhea (25%). Neurologic examination showed sensory deficits in 83%, reduced tendon reflexes in 67%, and an abnormal Romberg test in 67%. Four of five patients had increased CSF protein. Electrophysiologic evaluations showed features of demyelinating peripheral neuropathy (28%) and distally active denervation in the lower extremities.
“Zinc participates in more than 200 enzymatic reactions,” said the researchers. “Unfortunately, zinc deficiency-induced peripheral neuropathy is often misdiagnosed or delayed in diagnosis. Literature on zinc deficiency-induced peripheral neuropathy is sparse.”
WASHINGTON, DC—Patients with zinc deficiency-induced peripheral neuropathy may present with paresthesia, gait abnormalities, sensory deficits, reduced tendon reflexes, an abnormal Romberg test, and increased CSF protein, according to a study presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine.
Recognition of the features of zinc deficiency-induced peripheral neuropathy may help neurologists diagnose the disorder and manage patients, researchers said.
“Zinc, an essential trace element, plays a critical role in maintaining normal structural and functional conditions in the body,” said lead author Favio C. Bumanlag, Chief Technologist in the Department of Neurology at the Lewis Katz School of Medicine at Temple University in Philadelphia. “Peripheral nerves are susceptible to damage when zinc deficiency occurs.... Recognition of [zinc deficiency-induced peripheral neuropathy] will help physicians and technologists effectively manage patients.”
To study the clinical and electrophysiologic features of zinc deficiency-induced peripheral neuropathy, Mr. Bumanlag and Jin Luo, MD, PhD, Professor of Neurology and Pharmacology at Temple University, retrospectively reviewed charts in their neuromuscular clinic and EMG laboratory database to identify patients with peripheral neuropathy and zinc deficiency. They included charts from between January 1, 2015, and December 31, 2017, in their review. They excluded patients with abnormal copper levels.
Mr. Bumanlag and Dr. Luo obtained information about patients’ clinical presentations, past medical histories, BMI, neurologic examinations, and laboratory results. They also examined patients’ needle electromyograms and nerve conduction studies.
In all, they identified 12 patients with peripheral neuropathy and zinc deficiency. Patients had a mean age of 55.1. Six were female. Patients’ mean zinc level was 52.5 μg/dL, with a range of 37 μg/dL to 58 μg/dL (reference, 56–134 μg/dL). Mean copper level was 107.6 μg/dL, with a range of 84 μg/dL to 173 μg/dL (reference, 72–166 μg/dL). Eleven of the 12 patients had received an electrophysiologic evaluation.
Notable findings in presentation included paresthesia in 75 and gait abnormalities in 42%. One patient was obese (8%), and three patients had diarrhea (25%). Neurologic examination showed sensory deficits in 83%, reduced tendon reflexes in 67%, and an abnormal Romberg test in 67%. Four of five patients had increased CSF protein. Electrophysiologic evaluations showed features of demyelinating peripheral neuropathy (28%) and distally active denervation in the lower extremities.
“Zinc participates in more than 200 enzymatic reactions,” said the researchers. “Unfortunately, zinc deficiency-induced peripheral neuropathy is often misdiagnosed or delayed in diagnosis. Literature on zinc deficiency-induced peripheral neuropathy is sparse.”
Does Thymectomy Benefit Patients With Anti-MuSK Myasthenia Gravis?
Favorable clinical outcomes are not more likely in patients with anti-MuSK myasthenia gravis who undergo thymectomy versus patients who do not.
WASHINGTON, DC—Among patients with anti-muscle-specific kinase (MuSK) myasthenia gravis, thymectomy is not associated with greater likelihood of clinical improvement, according to an analysis of data from a multicenter cohort study. The results were presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM).
Although a randomized trial has demonstrated benefit from thymectomy in nonthymomatous antiacetylcholine receptor (AChR) antibody positive generalized myasthenia gravis, observational studies suggest that thymectomy may not be efficacious in anti-MuSK myasthenia gravis. Histologic studies have found that patients with anti-MuSK myasthenia gravis have less hyperplastic thymic tissue, compared with patients with anti-AChR myasthenia gravis.
To evaluate the therapeutic impact of thymectomy in patients with anti-MuSK myasthenia gravis, Katherine Clifford, a medical student at the University of Vermont Larner College of Medicine in Burlington, and colleagues analyzed data from a multicenter, retrospective, blinded review of rituximab treatment in patients with anti-MuSK myasthenia gravis. The primary outcome was favorable outcome on the Myasthenia Gravis Foundation of America (MGFA) Post-Intervention Status (PIS). The researchers defined a favorable outcome as an MGFA PIS score of minimal manifestations or better.
Secondary outcomes included prednisone dose; use of other immunosuppressant medications, IV immunoglobulin (IVIG), or plasma exchange (PLEX) treatment; and Myasthenia Gravis Status and Treatment Intensity (MGSTI).
Baseline characteristics were similar between patients with anti-MuSK myasthenia gravis who underwent thymectomy (n = 26) and those who did not (n = 29), including treatment with rituximab (42% vs 45%). Median follow-up was more than three years.
At last visit, 35% (nine of 26) of patients who underwent thymectomy had a favorable outcome, compared with 55% (16 of 29) of patients who did not undergo thymectomy. In addition, 69% of patients who underwent thymectomy were taking prednisone, compared with 41% of patients who did not undergo thymectomy (median dose, 10 mg/day vs 0 mg/day).
“After controlling for rituximab, baseline prednisone, and final IVIG/PLEX treatment, thymectomy was not associated with greater likelihood of favorable clinical outcome, but broad confidence intervals cannot exclude therapeutic effect (odds ratio, 0.43),” the investigators reported.

“The recent MGTX trial clearly demonstrated the benefit of thymectomy for patients with AChR antibody positive myasthenia gravis,” said A. Gordon Smith, MD, Cochair of the AANEM Annual Meeting Program Committee. “Ms. Clifford and her colleagues now provide compelling data suggesting thymectomy may not be effective in MuSK-positive myasthenia gravis.”
The study’s follow-up is long enough for the findings to be clinically “relevant to all physicians treating myasthenia gravis,” said Robert W. Irwin, MD, Cochair of the AANEM Annual Meeting Program Committee.
Favorable clinical outcomes are not more likely in patients with anti-MuSK myasthenia gravis who undergo thymectomy versus patients who do not.
Favorable clinical outcomes are not more likely in patients with anti-MuSK myasthenia gravis who undergo thymectomy versus patients who do not.
WASHINGTON, DC—Among patients with anti-muscle-specific kinase (MuSK) myasthenia gravis, thymectomy is not associated with greater likelihood of clinical improvement, according to an analysis of data from a multicenter cohort study. The results were presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM).
Although a randomized trial has demonstrated benefit from thymectomy in nonthymomatous antiacetylcholine receptor (AChR) antibody positive generalized myasthenia gravis, observational studies suggest that thymectomy may not be efficacious in anti-MuSK myasthenia gravis. Histologic studies have found that patients with anti-MuSK myasthenia gravis have less hyperplastic thymic tissue, compared with patients with anti-AChR myasthenia gravis.
To evaluate the therapeutic impact of thymectomy in patients with anti-MuSK myasthenia gravis, Katherine Clifford, a medical student at the University of Vermont Larner College of Medicine in Burlington, and colleagues analyzed data from a multicenter, retrospective, blinded review of rituximab treatment in patients with anti-MuSK myasthenia gravis. The primary outcome was favorable outcome on the Myasthenia Gravis Foundation of America (MGFA) Post-Intervention Status (PIS). The researchers defined a favorable outcome as an MGFA PIS score of minimal manifestations or better.
Secondary outcomes included prednisone dose; use of other immunosuppressant medications, IV immunoglobulin (IVIG), or plasma exchange (PLEX) treatment; and Myasthenia Gravis Status and Treatment Intensity (MGSTI).
Baseline characteristics were similar between patients with anti-MuSK myasthenia gravis who underwent thymectomy (n = 26) and those who did not (n = 29), including treatment with rituximab (42% vs 45%). Median follow-up was more than three years.
At last visit, 35% (nine of 26) of patients who underwent thymectomy had a favorable outcome, compared with 55% (16 of 29) of patients who did not undergo thymectomy. In addition, 69% of patients who underwent thymectomy were taking prednisone, compared with 41% of patients who did not undergo thymectomy (median dose, 10 mg/day vs 0 mg/day).
“After controlling for rituximab, baseline prednisone, and final IVIG/PLEX treatment, thymectomy was not associated with greater likelihood of favorable clinical outcome, but broad confidence intervals cannot exclude therapeutic effect (odds ratio, 0.43),” the investigators reported.
“The recent MGTX trial clearly demonstrated the benefit of thymectomy for patients with AChR antibody positive myasthenia gravis,” said A. Gordon Smith, MD, Cochair of the AANEM Annual Meeting Program Committee. “Ms. Clifford and her colleagues now provide compelling data suggesting thymectomy may not be effective in MuSK-positive myasthenia gravis.”
The study’s follow-up is long enough for the findings to be clinically “relevant to all physicians treating myasthenia gravis,” said Robert W. Irwin, MD, Cochair of the AANEM Annual Meeting Program Committee.
WASHINGTON, DC—Among patients with anti-muscle-specific kinase (MuSK) myasthenia gravis, thymectomy is not associated with greater likelihood of clinical improvement, according to an analysis of data from a multicenter cohort study. The results were presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM).
Although a randomized trial has demonstrated benefit from thymectomy in nonthymomatous antiacetylcholine receptor (AChR) antibody positive generalized myasthenia gravis, observational studies suggest that thymectomy may not be efficacious in anti-MuSK myasthenia gravis. Histologic studies have found that patients with anti-MuSK myasthenia gravis have less hyperplastic thymic tissue, compared with patients with anti-AChR myasthenia gravis.
To evaluate the therapeutic impact of thymectomy in patients with anti-MuSK myasthenia gravis, Katherine Clifford, a medical student at the University of Vermont Larner College of Medicine in Burlington, and colleagues analyzed data from a multicenter, retrospective, blinded review of rituximab treatment in patients with anti-MuSK myasthenia gravis. The primary outcome was favorable outcome on the Myasthenia Gravis Foundation of America (MGFA) Post-Intervention Status (PIS). The researchers defined a favorable outcome as an MGFA PIS score of minimal manifestations or better.
Secondary outcomes included prednisone dose; use of other immunosuppressant medications, IV immunoglobulin (IVIG), or plasma exchange (PLEX) treatment; and Myasthenia Gravis Status and Treatment Intensity (MGSTI).
Baseline characteristics were similar between patients with anti-MuSK myasthenia gravis who underwent thymectomy (n = 26) and those who did not (n = 29), including treatment with rituximab (42% vs 45%). Median follow-up was more than three years.
At last visit, 35% (nine of 26) of patients who underwent thymectomy had a favorable outcome, compared with 55% (16 of 29) of patients who did not undergo thymectomy. In addition, 69% of patients who underwent thymectomy were taking prednisone, compared with 41% of patients who did not undergo thymectomy (median dose, 10 mg/day vs 0 mg/day).
“After controlling for rituximab, baseline prednisone, and final IVIG/PLEX treatment, thymectomy was not associated with greater likelihood of favorable clinical outcome, but broad confidence intervals cannot exclude therapeutic effect (odds ratio, 0.43),” the investigators reported.
“The recent MGTX trial clearly demonstrated the benefit of thymectomy for patients with AChR antibody positive myasthenia gravis,” said A. Gordon Smith, MD, Cochair of the AANEM Annual Meeting Program Committee. “Ms. Clifford and her colleagues now provide compelling data suggesting thymectomy may not be effective in MuSK-positive myasthenia gravis.”
The study’s follow-up is long enough for the findings to be clinically “relevant to all physicians treating myasthenia gravis,” said Robert W. Irwin, MD, Cochair of the AANEM Annual Meeting Program Committee.