User login
Adaptive therapy borrows from nature to keep rhabdomyosarcoma in check
In 1859, Charles Darwin published “On the Origin of Species,” which outlined his world-shaking theory of evolution and its core principle of natural selection caused by environmental pressures that may determine whether an organism adapts and survives, or remains static, languishes, and eventually dies out.
The same forces that have influenced the size and shape of the beaks of finches in the Galapagos Islands, the length of giraffe necks in Africa, and the intestinal microbiomes of the nearly 8 billion human inhabitants of this planet also control whether malignant cells thrive and multiply, wither and die when assaulted by chemotherapy, or go into hiding, mutating and waiting for their next opportunity to erupt again and metastasize.
The ability of malignant cells to adapt to environmental pressures is “cancer’s most lethal and sophisticated property,” said Damon R. Reed, MD, program leader of the adolescent/young adult program at Moffitt Cancer Center in Tampa, Fla.
Dr. Reed and colleagues are developing methods to meet cancer on its own terms, applying evolutionary principles to the treatment of childhood fusion-positive rhabdomyosarcoma in an innovative, and some would say audacious, clinical trial.
Adaptive versus conventional therapy
The trial, now recruiting, is designed to evaluate each of four different strategies for chemotherapy schedules in patients with newly diagnosed metastatic fusion-positive rhabdomyosarcoma.
The trial contains four arms, three of which consist of either conventional chemotherapy based on published clinical trials, moving a second-line therapy to the first line, or adding maintenance therapy, all of which have the goal of inducing as many complete remissions as possible.
The remaining adaptive therapy arm, however, is entirely novel in approach, with therapy using a combination of chemotherapy drugs that will be started and interrupted based on tumor responses, with resumption of therapy on an adaptive schedule unique to each patient. The goal of treatment for patients enrolled in this arm will be prolongation of the time to disease progression, rather than complete remission.
Although some people might consider the adaptive therapy approach to be sacrificing the hope for a cure in exchange for palliation, the hard truth is that patients with fusion-positive rhabdomyosarcoma (in contrast to those with fusion-negative disease) have a dismal prognosis following relapse after up-front intensified therapy.
Instead, because a cure is exceedingly unlikely in patients with metastatic disease, the conventional idea of delivering the maximum tolerated dose of chemotherapy until disease progression could be replaced by an approach based on understanding of the evolution of cancer cells under selective pressures, Dr. Reed and colleagues contend.
“Although adaptive therapy would represent a major paradigm shift in pediatric oncology, this approach would exploit the chemotherapy-sensitive population to prevent the emergence of resistant populations, optimizing tumor control with less toxicity,” they wrote in a commentary published online in the journal Cancer.1
Poor survival with advanced disease
Childhood rhabdomyosarcoma (RMS) is a form of soft tissue sarcoma of mesenchymal origin. Approximately 25% of cases are parameningeal, arising from sites adjacent to the meninges of the nasopharynx, middle ear, paranasal sinuses, orbit, and other regions of the head and neck. Approximately 31% of cases arise in the genitourinary tract and 13% in the extremities, and other tumors occur less commonly in the trunk, chest wall, perineal/anal region, and abdomen.
The overall 5-year survival rate is approximately 71%.1
However, for patients with high-risk disease, a group that includes children 10 years of age or older with widespread disease with or without an activating PAX/FOX01 gene fusion, 5-year survival ranges from just 20% to 30% (Cancer Facts & Figures 2020).
“Among patients with metastatic disease, there is a clear difference in overall survival between those who have fusion-positive disease, where the 5-year overall survival is about 19%, and patients with fusion-negative disease,” said Douglas S. Hawkins, MD, chair of the children’s oncology group and professor of pediatrics at the University of Washington, Seattle, and associate chief in the division of hematology/oncology at Seattle Children’s Hospital.
Patients with fusion-negative disease can be further classified into those with multiple metastatic sites, with a 5-year overall survival rate of approximately 45%, and those with a single metastatic site, with a 5-year overall survival rate of 70%, he said in an interview.
“So when we talk about metastatic rhabdomyosarcoma, there actually is a diversity of outcomes, between really bad – those with fusion-positive disease – and not terrible – not great, but not terrible – for a selected group of patients with fusion-negative disease,” Dr. Hawkins said.
The poor prognosis for patients with metastatic fusion-positive disease prompted Dr. Reed and colleagues to rethink the entire approach to advanced cancers.
“If someone has a sarcoma, we know that we need to do surgery and radiation to the area, we know that localized disease does better than metastatic disease, and we generally hit it with some kind of chemotherapy that we call ‘standard of care,’ ” he said in an interview.
This approach is largely effective in some forms of cancer of bone and soft tissues, such as Ewing sarcoma, he notes, which has 5-year survival rates below 20% when treated with surgery and radiation only, but with the addition of chemotherapy has 5-year overall survival rates as high as 80%.
“At other times, with other sarcomas, the cure rate is abysmal, but we still call it standard of care,” Dr. Reed said.
For example, patients with metastatic fusion-positive RMS may have an initial response to chemotherapy, but most will eventually experience relapse and die of the disease.
“With some of the most common treatments, 70% of patients will have their cancers shrink by more than 50%, which is a major response, but the vast majority of them will have a recurrence later on,” Dr. Hawkins said.
He noted that the standard of care for patients with metastatic rhabdomyosarcoma, both with and without the PAX/FOX01 fusion, is chemotherapy, generally with the VAC regimen (vincristine, actinomycin D, and cyclophosphamide), although other agents such as doxorubicin, ifosfamide, etoposide, or irinotecan have also been tried, with little effect on event-free survival or overall survival rates.
A life too brief
Ricky Huff and his family know the course that the disease can take only too well. In 2015, his 5-month-old son, Theo, was diagnosed with metastatic rhabdomyosarcoma and put under the care of Damon Reed at Moffitt.
“During the whole course of treatment – I’m sure like many other parents – apart from relying on Damon and his treatment expertise to try to determine the best treatment options, I was reading everything under the sun to try to get a working knowledge of what Theo was up against, what his treatment and clinical trial options were, and what was the state of the science,” Mr. Huff says.
Unfortunately, the characteristics of Theo’s disease, including his very young age at onset and diagnosis of stage 4 metastatic disease, conspired against him, and despite undergoing 14 months of chemotherapy, Theo died of the disease in October 2016, 5 months shy of what would have been his second birthday.
In their grief, Mr. Huff, a real estate lawyer with a practice in Clearwater, Fla., and his wife, Leah, were determined to help other families of children with cancer and settled on the National Pediatric Cancer Foundation. Mr. Huff joined the board of directors of the foundation, which is collaborating with Moffitt Cancer Center on the adaptive therapy trial.
An evolutionary primer (cancer edition)
To get a better idea of just how adaptive therapy works, it is helpful to view cancer cells through the lens of species development, adaptation, extinction, and evolution.
“Cancer cells compete against each other in a dynamic environment. Their tumor ecosystems exhibit spatial and temporal fluctuations in blood-borne nutrients, oxygen, growth factors, immune cells, and hormones,” Dr. Reed and colleagues wrote.
These influences can affect genetically identical cancer cells, which may begin to diverge from one another depending on their location in a tumor and the availability of nutrients, which in turn can result in two once-identical cells exhibiting different transcription rates for growth factors.
“Ultimately, this may affect the rate of progression through the cell cycle, leading to distinct rates of proliferation and mutational acquisition,” they wrote.
The diverging subpopulations will begin to develop different methods for adapting to the tumor microenvironment, with unique strategies for both accelerating growth and avoiding hazards such as chemotherapy drugs or radiation, the investigators explained.
“By the time a cancer becomes clinically apparent, cancer cells have transformed from a single clone into a diverse community of cell types evolving in response to a spatially and temporally heterogeneous tumor environment. Theoretically, a 10-gram cancer may contain the same order of magnitude of cancer cells as there are humans on earth, with tremendous diversity of phenotypes and environments,” they wrote.
Survival of the fittest
The competition of individuals within and between species described by Darwin also applies to cancer cells, in their interactions both with each other and with stromal cells and immune cells resulting in “the progressive replacement of less fit phenotypes by those that are more fit,” Dr. Reed and colleagues explained.
And just like the old joke about two hikers trying to escape from a charging grizzly bear (one says, “This is futile – we can’t outrun a grizzly,” and the other says, “I only have to outrun you!”), cancer cells only need to be more resistant to therapeutic attack than normal cells that are critical to function.
“This may explain why initial responses in certain solid tumors (notably rhabdomyosarcoma) do not predict eventual survival. The sensitivities of the dominant cancer cell populations dictate the initial response, but it is the ecology and evolution of the rare and more resistant populations that determine cure or relapse,” they wrote.
The endangered species list
As with many types of cancer, the current approach to treating pediatric sarcomas with curative intent is with a “first strike” approach, treating patients with surgery, radiation, and cytotoxic chemotherapy at the maximum tolerated dose for as long as needed or until unacceptable toxicities occur, with the intention of wiping out all cancer cells without permanently injuring normal cells.
The evolutionary analogy to this approach is a mass extinction event such as the meteor strike that is believed to have wiped out the dinosaurs roughly 66 million years ago. Fossil evidence suggests that the cataclysmic event resulted in the atmosphere being blanketed with dust particles that blocked sunlight and caused massive die-off of plants that dinosaurs needed to survive and were ill-adapted to do without.
In contrast, populations of smaller, more adaptable species of microbes, insects, and animals, including our mammalian ancestors, were able to survive and eventually flourish.
Many patients with localized cancers may be cured with up-front therapy, but others will have residual disease from populations of cells that are intrinsically resistant to therapy or have developed new evasion strategies.
Strike two and the MVP
Dr. Reed and colleagues liken the approach of second-line therapy for treatment of relapsed or refractory disease to the concept of “background extinctions,” using the fate of the passenger pigeon as an example of how a second-strike therapeutic strategy works.
Although the popular conception is that the passenger pigeon was hunted to extinction by humans, the species in fact died out because of many different factors, including loss of habitat, isolation of populations leading to a loss of genetic diversity, and disruption of breeding habits.
“Once first strikes of deforestation and hunting reduced the birds to small, fragmented populations, a series of what would otherwise have been minor second strikes pushed the passenger pigeon below its extinction threshold, or minimum viable population,” they said.
The analogy, as it applies to cancer therapy, is the use of second-line or follow-on therapy with one or more agents that the residual cells are at least in theory not resistant to. In the case of fusion-positive rhabdomyosarcoma, the drug most commonly added in the second-strike approach is vinorelbine.2
“Second strikes should be timed to occur around the time when the first strike has achieved its greatest effect, presumably at the point when the disease becomes clinically undetectable or at a measurable nadir,” Dr. Reed and colleagues wrote. “Ideally, second-strike therapies should have modes of action that require different resistance strategies by the cancer cells than those needed for resistance to the first strike.”
Adaptive therapy
As Dr. Reed and colleagues note, despite optimal therapy, 94% of patients with metastatic fusion-positive rhabdomyosarcoma will experience a relapse within 3 years of diagnosis.1 Clearly the scorched earth or “throw everything you have it” approach no longer works, and that’s where adaptive therapy comes in.
Here again, the authors rely on nature, or rather human interaction with nature, to devise a strategy for keeping the disease at bay when extinction of all cancerous cells cannot be achieved.
They cite the example of agricultural integrated pest management, which seeks to keep harmful insects in check by treating them to suppress but not completely destroy a population, then stopping the use of pesticides, and resuming only when the insect population spikes and again becomes a threat to crops.
“The goal is to limit crop damage while retaining the sensitivity of the insects to the pesticides. Resistance most often comes at a cost. In the absence of the pesticide, sensitive individuals will outcompete resistant individuals,” they wrote.
Adaptive therapy uses the same approach to reduce selection pressures that foster resistance, with patients treated only until a specific, predetermined response is achieved in the dominant population of chemosensitive cells. The treatment is then interrupted and reintroduced only when the tumor rebounds to a certain predetermined size.
In this scenario, cells that retain sensitivity to chemotherapy will be able to reproduce and proliferate more rapidly than drug-resistant cells, and the therapy can then be reintroduced. This strategy is less likely to cause the development and proliferation of resistant cells than conventional intensified chemotherapy, Dr. Reed and colleagues contend.
Putting it to the test
The clinical trial that Dr. Reed and colleagues have initiated, officially titled “Evolutionary Inspired Therapy for Newly Diagnosed, Metastatic, Fusion Positive Rhabdomyosarcoma,” (NCT04388839) contains four arms: three experimental and one active comparator arm.
“We won’t randomize; we don’t feel that it would be fair to randomize patients, because these arms are so different from each other,” Dr. Reed said.
Arm A is the experimental first-strike arm, a 42-week course containing cyclophosphamide delivered intravenously over 60 minutes at a dose ranging from 220 mg to 1200 mg, vinorelbine delivered in an IV push over 6-10 minutes with a dose ranging from 4 mg to 25 mg, and actinomycin D administered via IV over 3-5 minutes at a dose ranging from 0.025 mg to 0.04 mg.
“The idea is that we take the standard of care, and we add a drug – vinorelbine – to make it stronger,” Dr. Reed said. “The idea is that the resistant cell, the cell that escapes, if we start hitting it on day 1 with vinorelbine, we might be able to drive it to extinction.”
Arm B, the second experimental arm, is the second-strike and maintenance arm, in which patients will receive conventional doses of vincristine, actinomycin D, and cyclophosphamide (VAC) until complete response (CR) for 12-42 weeks, and will then be switched to up to 2 years of maintenance with vinorelbine and oral cyclophosphamide.
“Vinorelbine will be added when the cancer is declining or first goes into remission. We try not to wait 42 weeks, which is too long we think, by which time the cancer may be fully adapted and resistant,” he explained.
Arm C is the adaptive therapy arm, in which patients will receive VAC that starts and stops based on response, with the goal of prolonging time to disease progression rather than achieving CR.
Arm D is the active comparator arm, consisting of conventional chemotherapy based on published clinical trials, such as VAC for 42 weeks, or other standard-of-care regimens that may include irinotecan, doxorubicin, ifosfamide, and/or etoposide.
A change in thinking
Dr. Reed acknowledges that Arm C, the adaptive therapy arm, “definitely represents a change in thinking for pediatric oncology.”
“The idea is that if you could do this perfectly well, you would be able to take a patient who is diagnosed today and essentially ‘pause’ their disease for a while. Then 5 years from now, if there is a better medicine, you would have gotten that patient to that medicine.”
The optimal approach to treating metastatic fusion-positive rhabdomyosarcoma may be similar to that used for treatment of acute lymphoblastic leukemia, with induction, consolidation, and maintenance and the option of delayed intensification, he said.
“But we’re so far away from knowing which series to do that we just need to show that any series – any changing it up – is helpful.”
Dr. Reed said that when he started presenting the concept of adaptive therapy in clinical meetings in 2017, “I was told to come up with a better idea. There were several people who instantly got it, but most people would instantly get angry.”
The common refrain was that adaptive therapy was “giving up.”
But minds began to change in 2018, following presentation at the annual meeting of the American Society of Clinical Oncology of a European study showing that adding 6 months of low-dose chemotherapy maintenance to standard therapy improved the 5-year overall survival rate of pediatric rhabdomyosarcoma from 73.7% to 86.6%.2
Before presenting the idea of adaptive therapy to his colleagues, he ran it by the parents of children with advanced sarcomas, and many were on board with it, he said.
Ricky Huff said that had the option of adaptive therapy been available for Theo, he and his wife would have been willing to try it.
“Of course, everyone has the ability in hindsight to apply critical thinking to decisions that you made or could have made,” he said. “I think is true for many parents, who if they’re presented with information about options will say ‘well if there’s a 1 percent chance, I want that chance for my child, especially for a 5-month-old.”
The decision to choose adaptive therapy is a difficult decision to make, whether for oneself or for one’s son, because it isn’t curative.
“My wife and I have since had a conversation about this, and I do think we would have considered it, although through a lot of difficult conversations,” he said.
“After we got the pathology, knowing that it was metastatic, fusion-positive, and given his age, just doing a brief literature review on my own, I knew what we were up against using 20-year-old treatments, and that the chance of a cure was very, very small.”
If parents of children with metastatic, poor-prognosis rhabdomyosarcoma could be made to understand that adaptive therapy would entail shorter and fewer hospital stays, and cumulatively less toxic chemotherapy, and could prolong the lives of their children, the option might be more acceptable, he said.
And as Dr. Reed mentioned, prolonging time to progression offers hope of additional therapies to come.
“The whole time that my son was being treated, I hoped that there was going to be something else that came out, that a new trial would be launched because they found a way to drug a mutation, or treat it with immunotherapy – something that was going to give us a better option.”
Asked whether he would be willing to share his experiences in this article, Mr. Huff said that “I am willing to, in whatever small way I can, make an impact, and hopefully save another family from what we experienced.”
References
1. Reed DR et al. Cancer. 2020 Jun 1;126(11):2577-87 2. Bisogno G et al. J Clin Oncol. 2018;36:18_suppl,LBA-2
In 1859, Charles Darwin published “On the Origin of Species,” which outlined his world-shaking theory of evolution and its core principle of natural selection caused by environmental pressures that may determine whether an organism adapts and survives, or remains static, languishes, and eventually dies out.
The same forces that have influenced the size and shape of the beaks of finches in the Galapagos Islands, the length of giraffe necks in Africa, and the intestinal microbiomes of the nearly 8 billion human inhabitants of this planet also control whether malignant cells thrive and multiply, wither and die when assaulted by chemotherapy, or go into hiding, mutating and waiting for their next opportunity to erupt again and metastasize.
The ability of malignant cells to adapt to environmental pressures is “cancer’s most lethal and sophisticated property,” said Damon R. Reed, MD, program leader of the adolescent/young adult program at Moffitt Cancer Center in Tampa, Fla.
Dr. Reed and colleagues are developing methods to meet cancer on its own terms, applying evolutionary principles to the treatment of childhood fusion-positive rhabdomyosarcoma in an innovative, and some would say audacious, clinical trial.
Adaptive versus conventional therapy
The trial, now recruiting, is designed to evaluate each of four different strategies for chemotherapy schedules in patients with newly diagnosed metastatic fusion-positive rhabdomyosarcoma.
The trial contains four arms, three of which consist of either conventional chemotherapy based on published clinical trials, moving a second-line therapy to the first line, or adding maintenance therapy, all of which have the goal of inducing as many complete remissions as possible.
The remaining adaptive therapy arm, however, is entirely novel in approach, with therapy using a combination of chemotherapy drugs that will be started and interrupted based on tumor responses, with resumption of therapy on an adaptive schedule unique to each patient. The goal of treatment for patients enrolled in this arm will be prolongation of the time to disease progression, rather than complete remission.
Although some people might consider the adaptive therapy approach to be sacrificing the hope for a cure in exchange for palliation, the hard truth is that patients with fusion-positive rhabdomyosarcoma (in contrast to those with fusion-negative disease) have a dismal prognosis following relapse after up-front intensified therapy.
Instead, because a cure is exceedingly unlikely in patients with metastatic disease, the conventional idea of delivering the maximum tolerated dose of chemotherapy until disease progression could be replaced by an approach based on understanding of the evolution of cancer cells under selective pressures, Dr. Reed and colleagues contend.
“Although adaptive therapy would represent a major paradigm shift in pediatric oncology, this approach would exploit the chemotherapy-sensitive population to prevent the emergence of resistant populations, optimizing tumor control with less toxicity,” they wrote in a commentary published online in the journal Cancer.1
Poor survival with advanced disease
Childhood rhabdomyosarcoma (RMS) is a form of soft tissue sarcoma of mesenchymal origin. Approximately 25% of cases are parameningeal, arising from sites adjacent to the meninges of the nasopharynx, middle ear, paranasal sinuses, orbit, and other regions of the head and neck. Approximately 31% of cases arise in the genitourinary tract and 13% in the extremities, and other tumors occur less commonly in the trunk, chest wall, perineal/anal region, and abdomen.
The overall 5-year survival rate is approximately 71%.1
However, for patients with high-risk disease, a group that includes children 10 years of age or older with widespread disease with or without an activating PAX/FOX01 gene fusion, 5-year survival ranges from just 20% to 30% (Cancer Facts & Figures 2020).
“Among patients with metastatic disease, there is a clear difference in overall survival between those who have fusion-positive disease, where the 5-year overall survival is about 19%, and patients with fusion-negative disease,” said Douglas S. Hawkins, MD, chair of the children’s oncology group and professor of pediatrics at the University of Washington, Seattle, and associate chief in the division of hematology/oncology at Seattle Children’s Hospital.
Patients with fusion-negative disease can be further classified into those with multiple metastatic sites, with a 5-year overall survival rate of approximately 45%, and those with a single metastatic site, with a 5-year overall survival rate of 70%, he said in an interview.
“So when we talk about metastatic rhabdomyosarcoma, there actually is a diversity of outcomes, between really bad – those with fusion-positive disease – and not terrible – not great, but not terrible – for a selected group of patients with fusion-negative disease,” Dr. Hawkins said.
The poor prognosis for patients with metastatic fusion-positive disease prompted Dr. Reed and colleagues to rethink the entire approach to advanced cancers.
“If someone has a sarcoma, we know that we need to do surgery and radiation to the area, we know that localized disease does better than metastatic disease, and we generally hit it with some kind of chemotherapy that we call ‘standard of care,’ ” he said in an interview.
This approach is largely effective in some forms of cancer of bone and soft tissues, such as Ewing sarcoma, he notes, which has 5-year survival rates below 20% when treated with surgery and radiation only, but with the addition of chemotherapy has 5-year overall survival rates as high as 80%.
“At other times, with other sarcomas, the cure rate is abysmal, but we still call it standard of care,” Dr. Reed said.
For example, patients with metastatic fusion-positive RMS may have an initial response to chemotherapy, but most will eventually experience relapse and die of the disease.
“With some of the most common treatments, 70% of patients will have their cancers shrink by more than 50%, which is a major response, but the vast majority of them will have a recurrence later on,” Dr. Hawkins said.
He noted that the standard of care for patients with metastatic rhabdomyosarcoma, both with and without the PAX/FOX01 fusion, is chemotherapy, generally with the VAC regimen (vincristine, actinomycin D, and cyclophosphamide), although other agents such as doxorubicin, ifosfamide, etoposide, or irinotecan have also been tried, with little effect on event-free survival or overall survival rates.
A life too brief
Ricky Huff and his family know the course that the disease can take only too well. In 2015, his 5-month-old son, Theo, was diagnosed with metastatic rhabdomyosarcoma and put under the care of Damon Reed at Moffitt.
“During the whole course of treatment – I’m sure like many other parents – apart from relying on Damon and his treatment expertise to try to determine the best treatment options, I was reading everything under the sun to try to get a working knowledge of what Theo was up against, what his treatment and clinical trial options were, and what was the state of the science,” Mr. Huff says.
Unfortunately, the characteristics of Theo’s disease, including his very young age at onset and diagnosis of stage 4 metastatic disease, conspired against him, and despite undergoing 14 months of chemotherapy, Theo died of the disease in October 2016, 5 months shy of what would have been his second birthday.
In their grief, Mr. Huff, a real estate lawyer with a practice in Clearwater, Fla., and his wife, Leah, were determined to help other families of children with cancer and settled on the National Pediatric Cancer Foundation. Mr. Huff joined the board of directors of the foundation, which is collaborating with Moffitt Cancer Center on the adaptive therapy trial.
An evolutionary primer (cancer edition)
To get a better idea of just how adaptive therapy works, it is helpful to view cancer cells through the lens of species development, adaptation, extinction, and evolution.
“Cancer cells compete against each other in a dynamic environment. Their tumor ecosystems exhibit spatial and temporal fluctuations in blood-borne nutrients, oxygen, growth factors, immune cells, and hormones,” Dr. Reed and colleagues wrote.
These influences can affect genetically identical cancer cells, which may begin to diverge from one another depending on their location in a tumor and the availability of nutrients, which in turn can result in two once-identical cells exhibiting different transcription rates for growth factors.
“Ultimately, this may affect the rate of progression through the cell cycle, leading to distinct rates of proliferation and mutational acquisition,” they wrote.
The diverging subpopulations will begin to develop different methods for adapting to the tumor microenvironment, with unique strategies for both accelerating growth and avoiding hazards such as chemotherapy drugs or radiation, the investigators explained.
“By the time a cancer becomes clinically apparent, cancer cells have transformed from a single clone into a diverse community of cell types evolving in response to a spatially and temporally heterogeneous tumor environment. Theoretically, a 10-gram cancer may contain the same order of magnitude of cancer cells as there are humans on earth, with tremendous diversity of phenotypes and environments,” they wrote.
Survival of the fittest
The competition of individuals within and between species described by Darwin also applies to cancer cells, in their interactions both with each other and with stromal cells and immune cells resulting in “the progressive replacement of less fit phenotypes by those that are more fit,” Dr. Reed and colleagues explained.
And just like the old joke about two hikers trying to escape from a charging grizzly bear (one says, “This is futile – we can’t outrun a grizzly,” and the other says, “I only have to outrun you!”), cancer cells only need to be more resistant to therapeutic attack than normal cells that are critical to function.
“This may explain why initial responses in certain solid tumors (notably rhabdomyosarcoma) do not predict eventual survival. The sensitivities of the dominant cancer cell populations dictate the initial response, but it is the ecology and evolution of the rare and more resistant populations that determine cure or relapse,” they wrote.
The endangered species list
As with many types of cancer, the current approach to treating pediatric sarcomas with curative intent is with a “first strike” approach, treating patients with surgery, radiation, and cytotoxic chemotherapy at the maximum tolerated dose for as long as needed or until unacceptable toxicities occur, with the intention of wiping out all cancer cells without permanently injuring normal cells.
The evolutionary analogy to this approach is a mass extinction event such as the meteor strike that is believed to have wiped out the dinosaurs roughly 66 million years ago. Fossil evidence suggests that the cataclysmic event resulted in the atmosphere being blanketed with dust particles that blocked sunlight and caused massive die-off of plants that dinosaurs needed to survive and were ill-adapted to do without.
In contrast, populations of smaller, more adaptable species of microbes, insects, and animals, including our mammalian ancestors, were able to survive and eventually flourish.
Many patients with localized cancers may be cured with up-front therapy, but others will have residual disease from populations of cells that are intrinsically resistant to therapy or have developed new evasion strategies.
Strike two and the MVP
Dr. Reed and colleagues liken the approach of second-line therapy for treatment of relapsed or refractory disease to the concept of “background extinctions,” using the fate of the passenger pigeon as an example of how a second-strike therapeutic strategy works.
Although the popular conception is that the passenger pigeon was hunted to extinction by humans, the species in fact died out because of many different factors, including loss of habitat, isolation of populations leading to a loss of genetic diversity, and disruption of breeding habits.
“Once first strikes of deforestation and hunting reduced the birds to small, fragmented populations, a series of what would otherwise have been minor second strikes pushed the passenger pigeon below its extinction threshold, or minimum viable population,” they said.
The analogy, as it applies to cancer therapy, is the use of second-line or follow-on therapy with one or more agents that the residual cells are at least in theory not resistant to. In the case of fusion-positive rhabdomyosarcoma, the drug most commonly added in the second-strike approach is vinorelbine.2
“Second strikes should be timed to occur around the time when the first strike has achieved its greatest effect, presumably at the point when the disease becomes clinically undetectable or at a measurable nadir,” Dr. Reed and colleagues wrote. “Ideally, second-strike therapies should have modes of action that require different resistance strategies by the cancer cells than those needed for resistance to the first strike.”
Adaptive therapy
As Dr. Reed and colleagues note, despite optimal therapy, 94% of patients with metastatic fusion-positive rhabdomyosarcoma will experience a relapse within 3 years of diagnosis.1 Clearly the scorched earth or “throw everything you have it” approach no longer works, and that’s where adaptive therapy comes in.
Here again, the authors rely on nature, or rather human interaction with nature, to devise a strategy for keeping the disease at bay when extinction of all cancerous cells cannot be achieved.
They cite the example of agricultural integrated pest management, which seeks to keep harmful insects in check by treating them to suppress but not completely destroy a population, then stopping the use of pesticides, and resuming only when the insect population spikes and again becomes a threat to crops.
“The goal is to limit crop damage while retaining the sensitivity of the insects to the pesticides. Resistance most often comes at a cost. In the absence of the pesticide, sensitive individuals will outcompete resistant individuals,” they wrote.
Adaptive therapy uses the same approach to reduce selection pressures that foster resistance, with patients treated only until a specific, predetermined response is achieved in the dominant population of chemosensitive cells. The treatment is then interrupted and reintroduced only when the tumor rebounds to a certain predetermined size.
In this scenario, cells that retain sensitivity to chemotherapy will be able to reproduce and proliferate more rapidly than drug-resistant cells, and the therapy can then be reintroduced. This strategy is less likely to cause the development and proliferation of resistant cells than conventional intensified chemotherapy, Dr. Reed and colleagues contend.
Putting it to the test
The clinical trial that Dr. Reed and colleagues have initiated, officially titled “Evolutionary Inspired Therapy for Newly Diagnosed, Metastatic, Fusion Positive Rhabdomyosarcoma,” (NCT04388839) contains four arms: three experimental and one active comparator arm.
“We won’t randomize; we don’t feel that it would be fair to randomize patients, because these arms are so different from each other,” Dr. Reed said.
Arm A is the experimental first-strike arm, a 42-week course containing cyclophosphamide delivered intravenously over 60 minutes at a dose ranging from 220 mg to 1200 mg, vinorelbine delivered in an IV push over 6-10 minutes with a dose ranging from 4 mg to 25 mg, and actinomycin D administered via IV over 3-5 minutes at a dose ranging from 0.025 mg to 0.04 mg.
“The idea is that we take the standard of care, and we add a drug – vinorelbine – to make it stronger,” Dr. Reed said. “The idea is that the resistant cell, the cell that escapes, if we start hitting it on day 1 with vinorelbine, we might be able to drive it to extinction.”
Arm B, the second experimental arm, is the second-strike and maintenance arm, in which patients will receive conventional doses of vincristine, actinomycin D, and cyclophosphamide (VAC) until complete response (CR) for 12-42 weeks, and will then be switched to up to 2 years of maintenance with vinorelbine and oral cyclophosphamide.
“Vinorelbine will be added when the cancer is declining or first goes into remission. We try not to wait 42 weeks, which is too long we think, by which time the cancer may be fully adapted and resistant,” he explained.
Arm C is the adaptive therapy arm, in which patients will receive VAC that starts and stops based on response, with the goal of prolonging time to disease progression rather than achieving CR.
Arm D is the active comparator arm, consisting of conventional chemotherapy based on published clinical trials, such as VAC for 42 weeks, or other standard-of-care regimens that may include irinotecan, doxorubicin, ifosfamide, and/or etoposide.
A change in thinking
Dr. Reed acknowledges that Arm C, the adaptive therapy arm, “definitely represents a change in thinking for pediatric oncology.”
“The idea is that if you could do this perfectly well, you would be able to take a patient who is diagnosed today and essentially ‘pause’ their disease for a while. Then 5 years from now, if there is a better medicine, you would have gotten that patient to that medicine.”
The optimal approach to treating metastatic fusion-positive rhabdomyosarcoma may be similar to that used for treatment of acute lymphoblastic leukemia, with induction, consolidation, and maintenance and the option of delayed intensification, he said.
“But we’re so far away from knowing which series to do that we just need to show that any series – any changing it up – is helpful.”
Dr. Reed said that when he started presenting the concept of adaptive therapy in clinical meetings in 2017, “I was told to come up with a better idea. There were several people who instantly got it, but most people would instantly get angry.”
The common refrain was that adaptive therapy was “giving up.”
But minds began to change in 2018, following presentation at the annual meeting of the American Society of Clinical Oncology of a European study showing that adding 6 months of low-dose chemotherapy maintenance to standard therapy improved the 5-year overall survival rate of pediatric rhabdomyosarcoma from 73.7% to 86.6%.2
Before presenting the idea of adaptive therapy to his colleagues, he ran it by the parents of children with advanced sarcomas, and many were on board with it, he said.
Ricky Huff said that had the option of adaptive therapy been available for Theo, he and his wife would have been willing to try it.
“Of course, everyone has the ability in hindsight to apply critical thinking to decisions that you made or could have made,” he said. “I think is true for many parents, who if they’re presented with information about options will say ‘well if there’s a 1 percent chance, I want that chance for my child, especially for a 5-month-old.”
The decision to choose adaptive therapy is a difficult decision to make, whether for oneself or for one’s son, because it isn’t curative.
“My wife and I have since had a conversation about this, and I do think we would have considered it, although through a lot of difficult conversations,” he said.
“After we got the pathology, knowing that it was metastatic, fusion-positive, and given his age, just doing a brief literature review on my own, I knew what we were up against using 20-year-old treatments, and that the chance of a cure was very, very small.”
If parents of children with metastatic, poor-prognosis rhabdomyosarcoma could be made to understand that adaptive therapy would entail shorter and fewer hospital stays, and cumulatively less toxic chemotherapy, and could prolong the lives of their children, the option might be more acceptable, he said.
And as Dr. Reed mentioned, prolonging time to progression offers hope of additional therapies to come.
“The whole time that my son was being treated, I hoped that there was going to be something else that came out, that a new trial would be launched because they found a way to drug a mutation, or treat it with immunotherapy – something that was going to give us a better option.”
Asked whether he would be willing to share his experiences in this article, Mr. Huff said that “I am willing to, in whatever small way I can, make an impact, and hopefully save another family from what we experienced.”
References
1. Reed DR et al. Cancer. 2020 Jun 1;126(11):2577-87 2. Bisogno G et al. J Clin Oncol. 2018;36:18_suppl,LBA-2
In 1859, Charles Darwin published “On the Origin of Species,” which outlined his world-shaking theory of evolution and its core principle of natural selection caused by environmental pressures that may determine whether an organism adapts and survives, or remains static, languishes, and eventually dies out.
The same forces that have influenced the size and shape of the beaks of finches in the Galapagos Islands, the length of giraffe necks in Africa, and the intestinal microbiomes of the nearly 8 billion human inhabitants of this planet also control whether malignant cells thrive and multiply, wither and die when assaulted by chemotherapy, or go into hiding, mutating and waiting for their next opportunity to erupt again and metastasize.
The ability of malignant cells to adapt to environmental pressures is “cancer’s most lethal and sophisticated property,” said Damon R. Reed, MD, program leader of the adolescent/young adult program at Moffitt Cancer Center in Tampa, Fla.
Dr. Reed and colleagues are developing methods to meet cancer on its own terms, applying evolutionary principles to the treatment of childhood fusion-positive rhabdomyosarcoma in an innovative, and some would say audacious, clinical trial.
Adaptive versus conventional therapy
The trial, now recruiting, is designed to evaluate each of four different strategies for chemotherapy schedules in patients with newly diagnosed metastatic fusion-positive rhabdomyosarcoma.
The trial contains four arms, three of which consist of either conventional chemotherapy based on published clinical trials, moving a second-line therapy to the first line, or adding maintenance therapy, all of which have the goal of inducing as many complete remissions as possible.
The remaining adaptive therapy arm, however, is entirely novel in approach, with therapy using a combination of chemotherapy drugs that will be started and interrupted based on tumor responses, with resumption of therapy on an adaptive schedule unique to each patient. The goal of treatment for patients enrolled in this arm will be prolongation of the time to disease progression, rather than complete remission.
Although some people might consider the adaptive therapy approach to be sacrificing the hope for a cure in exchange for palliation, the hard truth is that patients with fusion-positive rhabdomyosarcoma (in contrast to those with fusion-negative disease) have a dismal prognosis following relapse after up-front intensified therapy.
Instead, because a cure is exceedingly unlikely in patients with metastatic disease, the conventional idea of delivering the maximum tolerated dose of chemotherapy until disease progression could be replaced by an approach based on understanding of the evolution of cancer cells under selective pressures, Dr. Reed and colleagues contend.
“Although adaptive therapy would represent a major paradigm shift in pediatric oncology, this approach would exploit the chemotherapy-sensitive population to prevent the emergence of resistant populations, optimizing tumor control with less toxicity,” they wrote in a commentary published online in the journal Cancer.1
Poor survival with advanced disease
Childhood rhabdomyosarcoma (RMS) is a form of soft tissue sarcoma of mesenchymal origin. Approximately 25% of cases are parameningeal, arising from sites adjacent to the meninges of the nasopharynx, middle ear, paranasal sinuses, orbit, and other regions of the head and neck. Approximately 31% of cases arise in the genitourinary tract and 13% in the extremities, and other tumors occur less commonly in the trunk, chest wall, perineal/anal region, and abdomen.
The overall 5-year survival rate is approximately 71%.1
However, for patients with high-risk disease, a group that includes children 10 years of age or older with widespread disease with or without an activating PAX/FOX01 gene fusion, 5-year survival ranges from just 20% to 30% (Cancer Facts & Figures 2020).
“Among patients with metastatic disease, there is a clear difference in overall survival between those who have fusion-positive disease, where the 5-year overall survival is about 19%, and patients with fusion-negative disease,” said Douglas S. Hawkins, MD, chair of the children’s oncology group and professor of pediatrics at the University of Washington, Seattle, and associate chief in the division of hematology/oncology at Seattle Children’s Hospital.
Patients with fusion-negative disease can be further classified into those with multiple metastatic sites, with a 5-year overall survival rate of approximately 45%, and those with a single metastatic site, with a 5-year overall survival rate of 70%, he said in an interview.
“So when we talk about metastatic rhabdomyosarcoma, there actually is a diversity of outcomes, between really bad – those with fusion-positive disease – and not terrible – not great, but not terrible – for a selected group of patients with fusion-negative disease,” Dr. Hawkins said.
The poor prognosis for patients with metastatic fusion-positive disease prompted Dr. Reed and colleagues to rethink the entire approach to advanced cancers.
“If someone has a sarcoma, we know that we need to do surgery and radiation to the area, we know that localized disease does better than metastatic disease, and we generally hit it with some kind of chemotherapy that we call ‘standard of care,’ ” he said in an interview.
This approach is largely effective in some forms of cancer of bone and soft tissues, such as Ewing sarcoma, he notes, which has 5-year survival rates below 20% when treated with surgery and radiation only, but with the addition of chemotherapy has 5-year overall survival rates as high as 80%.
“At other times, with other sarcomas, the cure rate is abysmal, but we still call it standard of care,” Dr. Reed said.
For example, patients with metastatic fusion-positive RMS may have an initial response to chemotherapy, but most will eventually experience relapse and die of the disease.
“With some of the most common treatments, 70% of patients will have their cancers shrink by more than 50%, which is a major response, but the vast majority of them will have a recurrence later on,” Dr. Hawkins said.
He noted that the standard of care for patients with metastatic rhabdomyosarcoma, both with and without the PAX/FOX01 fusion, is chemotherapy, generally with the VAC regimen (vincristine, actinomycin D, and cyclophosphamide), although other agents such as doxorubicin, ifosfamide, etoposide, or irinotecan have also been tried, with little effect on event-free survival or overall survival rates.
A life too brief
Ricky Huff and his family know the course that the disease can take only too well. In 2015, his 5-month-old son, Theo, was diagnosed with metastatic rhabdomyosarcoma and put under the care of Damon Reed at Moffitt.
“During the whole course of treatment – I’m sure like many other parents – apart from relying on Damon and his treatment expertise to try to determine the best treatment options, I was reading everything under the sun to try to get a working knowledge of what Theo was up against, what his treatment and clinical trial options were, and what was the state of the science,” Mr. Huff says.
Unfortunately, the characteristics of Theo’s disease, including his very young age at onset and diagnosis of stage 4 metastatic disease, conspired against him, and despite undergoing 14 months of chemotherapy, Theo died of the disease in October 2016, 5 months shy of what would have been his second birthday.
In their grief, Mr. Huff, a real estate lawyer with a practice in Clearwater, Fla., and his wife, Leah, were determined to help other families of children with cancer and settled on the National Pediatric Cancer Foundation. Mr. Huff joined the board of directors of the foundation, which is collaborating with Moffitt Cancer Center on the adaptive therapy trial.
An evolutionary primer (cancer edition)
To get a better idea of just how adaptive therapy works, it is helpful to view cancer cells through the lens of species development, adaptation, extinction, and evolution.
“Cancer cells compete against each other in a dynamic environment. Their tumor ecosystems exhibit spatial and temporal fluctuations in blood-borne nutrients, oxygen, growth factors, immune cells, and hormones,” Dr. Reed and colleagues wrote.
These influences can affect genetically identical cancer cells, which may begin to diverge from one another depending on their location in a tumor and the availability of nutrients, which in turn can result in two once-identical cells exhibiting different transcription rates for growth factors.
“Ultimately, this may affect the rate of progression through the cell cycle, leading to distinct rates of proliferation and mutational acquisition,” they wrote.
The diverging subpopulations will begin to develop different methods for adapting to the tumor microenvironment, with unique strategies for both accelerating growth and avoiding hazards such as chemotherapy drugs or radiation, the investigators explained.
“By the time a cancer becomes clinically apparent, cancer cells have transformed from a single clone into a diverse community of cell types evolving in response to a spatially and temporally heterogeneous tumor environment. Theoretically, a 10-gram cancer may contain the same order of magnitude of cancer cells as there are humans on earth, with tremendous diversity of phenotypes and environments,” they wrote.
Survival of the fittest
The competition of individuals within and between species described by Darwin also applies to cancer cells, in their interactions both with each other and with stromal cells and immune cells resulting in “the progressive replacement of less fit phenotypes by those that are more fit,” Dr. Reed and colleagues explained.
And just like the old joke about two hikers trying to escape from a charging grizzly bear (one says, “This is futile – we can’t outrun a grizzly,” and the other says, “I only have to outrun you!”), cancer cells only need to be more resistant to therapeutic attack than normal cells that are critical to function.
“This may explain why initial responses in certain solid tumors (notably rhabdomyosarcoma) do not predict eventual survival. The sensitivities of the dominant cancer cell populations dictate the initial response, but it is the ecology and evolution of the rare and more resistant populations that determine cure or relapse,” they wrote.
The endangered species list
As with many types of cancer, the current approach to treating pediatric sarcomas with curative intent is with a “first strike” approach, treating patients with surgery, radiation, and cytotoxic chemotherapy at the maximum tolerated dose for as long as needed or until unacceptable toxicities occur, with the intention of wiping out all cancer cells without permanently injuring normal cells.
The evolutionary analogy to this approach is a mass extinction event such as the meteor strike that is believed to have wiped out the dinosaurs roughly 66 million years ago. Fossil evidence suggests that the cataclysmic event resulted in the atmosphere being blanketed with dust particles that blocked sunlight and caused massive die-off of plants that dinosaurs needed to survive and were ill-adapted to do without.
In contrast, populations of smaller, more adaptable species of microbes, insects, and animals, including our mammalian ancestors, were able to survive and eventually flourish.
Many patients with localized cancers may be cured with up-front therapy, but others will have residual disease from populations of cells that are intrinsically resistant to therapy or have developed new evasion strategies.
Strike two and the MVP
Dr. Reed and colleagues liken the approach of second-line therapy for treatment of relapsed or refractory disease to the concept of “background extinctions,” using the fate of the passenger pigeon as an example of how a second-strike therapeutic strategy works.
Although the popular conception is that the passenger pigeon was hunted to extinction by humans, the species in fact died out because of many different factors, including loss of habitat, isolation of populations leading to a loss of genetic diversity, and disruption of breeding habits.
“Once first strikes of deforestation and hunting reduced the birds to small, fragmented populations, a series of what would otherwise have been minor second strikes pushed the passenger pigeon below its extinction threshold, or minimum viable population,” they said.
The analogy, as it applies to cancer therapy, is the use of second-line or follow-on therapy with one or more agents that the residual cells are at least in theory not resistant to. In the case of fusion-positive rhabdomyosarcoma, the drug most commonly added in the second-strike approach is vinorelbine.2
“Second strikes should be timed to occur around the time when the first strike has achieved its greatest effect, presumably at the point when the disease becomes clinically undetectable or at a measurable nadir,” Dr. Reed and colleagues wrote. “Ideally, second-strike therapies should have modes of action that require different resistance strategies by the cancer cells than those needed for resistance to the first strike.”
Adaptive therapy
As Dr. Reed and colleagues note, despite optimal therapy, 94% of patients with metastatic fusion-positive rhabdomyosarcoma will experience a relapse within 3 years of diagnosis.1 Clearly the scorched earth or “throw everything you have it” approach no longer works, and that’s where adaptive therapy comes in.
Here again, the authors rely on nature, or rather human interaction with nature, to devise a strategy for keeping the disease at bay when extinction of all cancerous cells cannot be achieved.
They cite the example of agricultural integrated pest management, which seeks to keep harmful insects in check by treating them to suppress but not completely destroy a population, then stopping the use of pesticides, and resuming only when the insect population spikes and again becomes a threat to crops.
“The goal is to limit crop damage while retaining the sensitivity of the insects to the pesticides. Resistance most often comes at a cost. In the absence of the pesticide, sensitive individuals will outcompete resistant individuals,” they wrote.
Adaptive therapy uses the same approach to reduce selection pressures that foster resistance, with patients treated only until a specific, predetermined response is achieved in the dominant population of chemosensitive cells. The treatment is then interrupted and reintroduced only when the tumor rebounds to a certain predetermined size.
In this scenario, cells that retain sensitivity to chemotherapy will be able to reproduce and proliferate more rapidly than drug-resistant cells, and the therapy can then be reintroduced. This strategy is less likely to cause the development and proliferation of resistant cells than conventional intensified chemotherapy, Dr. Reed and colleagues contend.
Putting it to the test
The clinical trial that Dr. Reed and colleagues have initiated, officially titled “Evolutionary Inspired Therapy for Newly Diagnosed, Metastatic, Fusion Positive Rhabdomyosarcoma,” (NCT04388839) contains four arms: three experimental and one active comparator arm.
“We won’t randomize; we don’t feel that it would be fair to randomize patients, because these arms are so different from each other,” Dr. Reed said.
Arm A is the experimental first-strike arm, a 42-week course containing cyclophosphamide delivered intravenously over 60 minutes at a dose ranging from 220 mg to 1200 mg, vinorelbine delivered in an IV push over 6-10 minutes with a dose ranging from 4 mg to 25 mg, and actinomycin D administered via IV over 3-5 minutes at a dose ranging from 0.025 mg to 0.04 mg.
“The idea is that we take the standard of care, and we add a drug – vinorelbine – to make it stronger,” Dr. Reed said. “The idea is that the resistant cell, the cell that escapes, if we start hitting it on day 1 with vinorelbine, we might be able to drive it to extinction.”
Arm B, the second experimental arm, is the second-strike and maintenance arm, in which patients will receive conventional doses of vincristine, actinomycin D, and cyclophosphamide (VAC) until complete response (CR) for 12-42 weeks, and will then be switched to up to 2 years of maintenance with vinorelbine and oral cyclophosphamide.
“Vinorelbine will be added when the cancer is declining or first goes into remission. We try not to wait 42 weeks, which is too long we think, by which time the cancer may be fully adapted and resistant,” he explained.
Arm C is the adaptive therapy arm, in which patients will receive VAC that starts and stops based on response, with the goal of prolonging time to disease progression rather than achieving CR.
Arm D is the active comparator arm, consisting of conventional chemotherapy based on published clinical trials, such as VAC for 42 weeks, or other standard-of-care regimens that may include irinotecan, doxorubicin, ifosfamide, and/or etoposide.
A change in thinking
Dr. Reed acknowledges that Arm C, the adaptive therapy arm, “definitely represents a change in thinking for pediatric oncology.”
“The idea is that if you could do this perfectly well, you would be able to take a patient who is diagnosed today and essentially ‘pause’ their disease for a while. Then 5 years from now, if there is a better medicine, you would have gotten that patient to that medicine.”
The optimal approach to treating metastatic fusion-positive rhabdomyosarcoma may be similar to that used for treatment of acute lymphoblastic leukemia, with induction, consolidation, and maintenance and the option of delayed intensification, he said.
“But we’re so far away from knowing which series to do that we just need to show that any series – any changing it up – is helpful.”
Dr. Reed said that when he started presenting the concept of adaptive therapy in clinical meetings in 2017, “I was told to come up with a better idea. There were several people who instantly got it, but most people would instantly get angry.”
The common refrain was that adaptive therapy was “giving up.”
But minds began to change in 2018, following presentation at the annual meeting of the American Society of Clinical Oncology of a European study showing that adding 6 months of low-dose chemotherapy maintenance to standard therapy improved the 5-year overall survival rate of pediatric rhabdomyosarcoma from 73.7% to 86.6%.2
Before presenting the idea of adaptive therapy to his colleagues, he ran it by the parents of children with advanced sarcomas, and many were on board with it, he said.
Ricky Huff said that had the option of adaptive therapy been available for Theo, he and his wife would have been willing to try it.
“Of course, everyone has the ability in hindsight to apply critical thinking to decisions that you made or could have made,” he said. “I think is true for many parents, who if they’re presented with information about options will say ‘well if there’s a 1 percent chance, I want that chance for my child, especially for a 5-month-old.”
The decision to choose adaptive therapy is a difficult decision to make, whether for oneself or for one’s son, because it isn’t curative.
“My wife and I have since had a conversation about this, and I do think we would have considered it, although through a lot of difficult conversations,” he said.
“After we got the pathology, knowing that it was metastatic, fusion-positive, and given his age, just doing a brief literature review on my own, I knew what we were up against using 20-year-old treatments, and that the chance of a cure was very, very small.”
If parents of children with metastatic, poor-prognosis rhabdomyosarcoma could be made to understand that adaptive therapy would entail shorter and fewer hospital stays, and cumulatively less toxic chemotherapy, and could prolong the lives of their children, the option might be more acceptable, he said.
And as Dr. Reed mentioned, prolonging time to progression offers hope of additional therapies to come.
“The whole time that my son was being treated, I hoped that there was going to be something else that came out, that a new trial would be launched because they found a way to drug a mutation, or treat it with immunotherapy – something that was going to give us a better option.”
Asked whether he would be willing to share his experiences in this article, Mr. Huff said that “I am willing to, in whatever small way I can, make an impact, and hopefully save another family from what we experienced.”
References
1. Reed DR et al. Cancer. 2020 Jun 1;126(11):2577-87 2. Bisogno G et al. J Clin Oncol. 2018;36:18_suppl,LBA-2
Mesothelioma trials: Moving toward improved survival
Although mesothelioma continues to be a very difficult disease to treat and one with a poor prognosis, new and emerging therapeutic developments hold the promise of extending survival for appropriately selected patients.
Following years of little to no movement, encouraging advances in treatment have been seen on the immunotherapy front. Immune checkpoint inhibitors have demonstrated acceptable safety and promising efficacy in the treatment of unresectable malignant pleural mesothelioma (MPM), including an overall survival advantage over standard-of-care first-line chemotherapy. Beyond systemic therapy, the development of new radiation techniques to complement current, more conservative surgical approaches is likewise encouraging, though further randomized clinical trial data is awaited to determine the potential impact on survival.
Longer survival would be good news for the estimated 3,000 individuals diagnosed with MPM each year in the United States. Overall, the outlook for patients with this rare cancer remains unfavorable, with a 5-year survival rate of about 11%, according to data from the U.S. Surveillance, Epidemiology and End Results (SEER) Program.
One factor underlying that grim survival statistic is a relative lack of investment in the development of drugs specific to rare cancers, as compared to more common malignancies, said Anne S. Tsao, MD, professor and director of the mesothelioma program at the University of Texas MD Anderson Cancer Center in Houston.
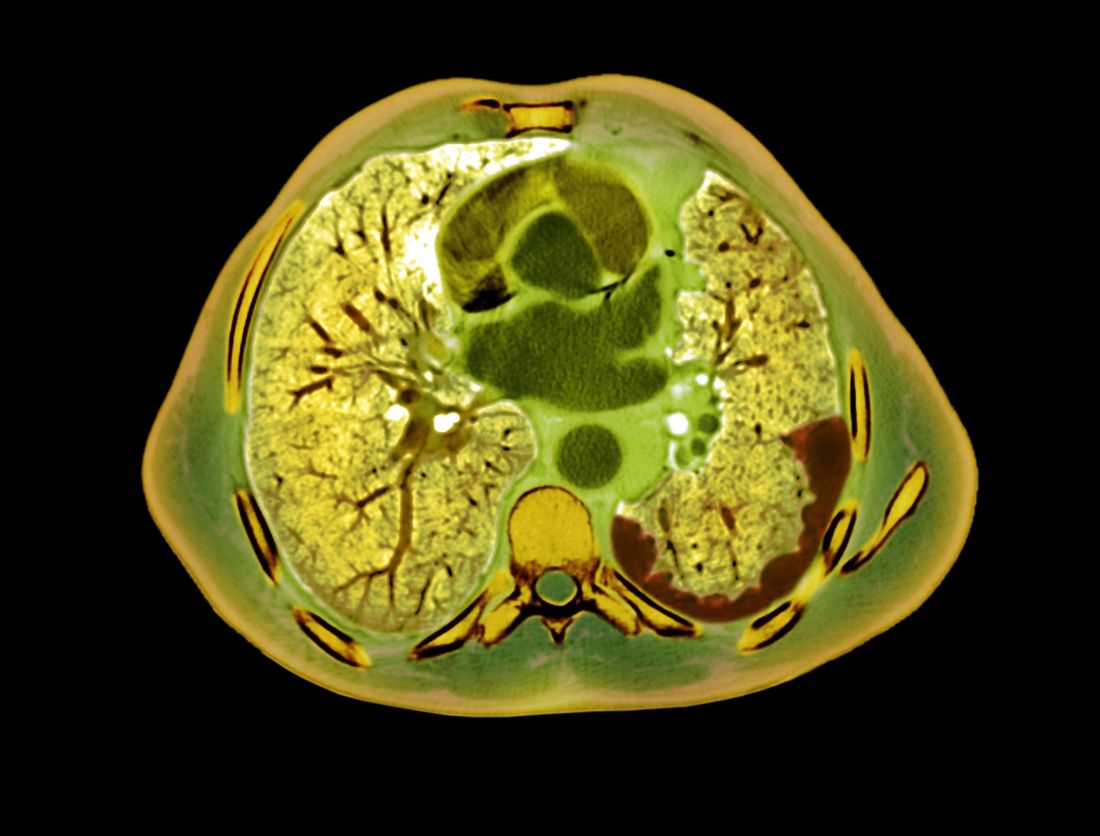
On the plus side, the wave of research for more common cancers has yielded a number of agents, including the immune checkpoint inhibitors such as nivolumab, ipilimumab, pembrolizumab, and durvalumab, that hold promise in rare tumor types as well.
“I think that mesothelioma has benefited from that, because these all are agents that have been developed for other solid tumors that are then brought into mesothelioma,” Dr. Tsao said in an interview. “So there’s always a lag time, but nevertheless, of course we are thrilled that we have additional treatment options for these patients.”
Checkpoint inhibitors
Multiple checkpoint inhibitors have received Food and Drug Administration approval for the treatment of non–small cell lung cancer (NSCLC) over the past few years. Because many mesothelioma doctors also treat NSCLC, bringing those agents into the mesothelioma sphere was not a very difficult jump, Dr. Tsao said.
Checkpoint inhibitors got a foothold in mesothelioma, much like in NSCLC, by demonstrating clear benefit in the salvage setting, according to Dr. Tsao.
Pembrolizumab, nivolumab, and avelumab were evaluated in phase 1b/2 clinical trials and real-world cohorts that demonstrated response rates of around 20%, median progression-free survival of 4 months, and median overall survival (OS) around 12 months in patients with previously treated MPM.
Although results of those early-stage studies had to be interpreted with caution, they nonetheless suggested a slight edge for these checkpoint inhibitors over historical data, according to the authors of a recent article in Cancer Treatment Reviews.1 On the basis of phase 1 and 2 data, current clinical practice guidelines from the National Comprehensive Cancer Network2 list pembrolizumab and the combination of nivolumab and ipilimumab as options for MPM patients who have received previous therapy. Phase 3 trials have also been launched, including PROMISE-meso, which is comparing pembrolizumab to single-agent chemotherapy in advanced, pretreated MPM3, and CONFIRM, which pits nivolumab against placebo in relapsed MPM.4
On the front lines
Encouraging results in previously treated MPM led to the evaluation of checkpoint inhibitors as first-line therapy. Notably, the FDA approved nivolumab given with ipilimumab for the treatment of patients with unresectable MPM in October 2020, making that combination the first immunotherapy regimen to receive an indication in this disease.
The FDA approval was based on prespecified interim analysis of CheckMate 743, a phase 3 study that included 605 patients randomly allocated to nivolumab plus ipilimumab or to placebo.
At the interim analysis, median OS was 18.1 months for nivolumab plus ipilimumab, versus just 14.1 months for placebo (hazard ratio, 0.74; 96.6% confidence interval, 0.60-0.91; P = 0.0020), according to results of the study published in the Lancet.5 The 2-year OS rate was 41% for the immunotherapy combination and 27% for placebo. Grade 3-4 treatment-related adverse events were seen in 30% of the immunotherapy-treated patients and 32% of the chemotherapy-treated patients.
The magnitude of nivolumab-ipilimumab benefit appeared to be largest among patients with non-epithelioid MPM subtypes (sarcomatoid and biphasic), owing to the inferior impact of chemotherapy in these patients, with a median OS of just 8.8 months, according to investigators.
That’s not to say that immunotherapy didn’t work for patients with epithelioid histology. The benefit of nivolumab-ipilimumab was consistent for non-epithelioid and epithelioid patient subsets, with median OS of 18.1 and 18.7 months, respectively, results of subgroup analysis showed.
According to Dr. Tsao, those results reflect the extremely poor prognosis and pressing need for effective therapy early in the course of treatment for patients with non-epithelioid histology.
“You have to get the most effective therapy into these patients as quickly as you can,” she explained. “If you can get the more effective treatment and early, then you’ll see a longer-term benefit for them.”
Role of the PD-L1 biomarker
Despite this progress, one key hurdle has been determining the role of the PD-L1 biomarker in mesothelioma. In NSCLC, PD-L1 is often used to determine which patients will benefit from immune checkpoint inhibitors. In mesothelioma, the correlations have been more elusive.
Among patients in the CheckMate 743 study treated with nivolumab plus ipilimumab, OS was not significantly different for those with PD-L1 expression levels of less than 1% and those with 1% or greater, investigators said. Moreover, PD-L1 expression wasn’t a stratification factor in the study.
“When looking at all of the studies, it appears that the checkpoint inhibitors can truly benefit a certain percentage of mesothelioma patients, but we can’t pick them out just yet,” Dr. Tsao said.
“So our recommendation is to offer [checkpoint inhibitor therapy] at some point in their treatment, whether it’s first, second, or third line,” she continued. “They can get some benefit, and even in those if you don’t get a great response, you can still get disease stabilization, which in and of itself can be highly beneficial.”
Future directions
Immune checkpoint inhibitor–based combination regimens and cellular therapy represent promising directions forward in MPM research. There are several notable phase 3 trials of checkpoint inhibitors plus chemotherapy and targeted therapy going forward, plus intriguing data emerging on the potential role of chimeric antigen receptor (CAR) T-cell therapy in this setting.
One phase 3 trial to watch is IND277, which is comparing pembrolizumab plus cisplatin/pemetrexed chemotherapy to cisplatin/pemetrexed alone; that trial has enrolled 520 participants and has an estimated primary completion date in July 2022, according to the ClinicalTrials.gov website. Another is BEAT-Meso, a comparison of atezolizumab plus bevacizumab and chemotherapy against bevacizumab and chemotherapy, which has an estimated enrollment of 400 participants and primary completion date of January 2024. A third trial of interest is DREAM3R, which compares durvalumab plus chemotherapy followed by durvalumab maintenance to standard chemotherapy followed by observation. That study should enroll 480 participants and has an estimated primary completion date of April 2025.
CAR T-cell therapy, while best known for its emerging role in the treatment of hematologic malignancies, may also have a place in mesothelioma therapy one day. In a recently published report, investigators described a first-in-human phase I study of a mesothelin-targeted CAR T-cell therapy given in combination with pembrolizumab. Among 18 MPM patients who received pembrolizumab safely, median OS from time of CAR T-cell infusion was 23.9 months and 1-year OS was 83%, according to investigators.6An OS of nearly 24 months is “very encouraging” and compares favorably with historical results with systemic therapy in this difficult-to-treat disease, said Jacques P. Fontaine, MD, a thoracic surgeon and section head of mesothelioma research and treatment center at Moffitt Cancer Center in Tampa, Fla.
“It’s huge, but you have to take into account that this [OS] is still less than 2 years,” Dr. Fontaine said in an interview. “There’s still a lot of work to be done.”
Radiotherapy making an IMPRINT
Meanwhile, new developments in the multimodality treatment of resectable MPM are progressing and have the potential to extend survival among patients who undergo lung-sparing surgery.
Less aggressive intervention is increasingly the preferred approach to surgery in this patient population. That shift is supported by studies showing that lung-sparing pleurectomy-decortication (P/D) resulted in less morbidity and potentially better survival outcomes than extrapleural pneumonectomy (EPP), according to Andreas Rimner, MD, associate attending physician and director of thoracic radiation oncology research at Memorial Sloan Kettering Cancer Center in New York.
However, it is more challenging to deliver radiotherapy safely in patients who have undergone P/D as compared with patients who have undergone EPP, according to Dr. Rimner.
“When there’s no lung in place [as in EPP], it’s pretty simple – you just treat the entire empty chest to kill any microscopic cells that may still be left behind,” he said in an interview. “But now we have a situation where both lungs are still in place, and they are very radiation sensitive, so that’s not an easy feat.”
Driven by the limitations of conventional radiation, Dr. Rimner and colleagues developed a novel technique known as hemithoracic intensity-modulated pleural radiation therapy (IMPRINT) that allows more precise application of radiotherapy.
In a phase 2 study published in 2016, IMPRINT was found to be safe, with an acceptable rate of radiation pneumonitis (30% grade 2 or 3), according to investigators.7
Subsequent studies have demonstrated encouraging clinical outcomes, including a 20.2-month median OS for IMPRINT versus 12.3 months for conventional adjuvant radiotherapy in a retrospective study of 209 patients who underwent P/D between 1975 and 2015.8 Those findings led to the development of a phase 3 trial known as NRG-LU006 that is evaluating P/D plus chemotherapy with or without adjuvant IMPRINT in an estimated 150 patients. The study has a primary endpoint of OS, and an estimated primary completion date in July 2025, according to ClinicalTrials.gov.
Dr. Rimner said he’s optimistic about the prospects of this study, particularly with recently published results of a phase 3 study in which Italian investigators demonstrated an OS benefit of IMPRINT over palliative radiation in patients with nonmetastatic MPM.9
“That’s more data and rationale that shows there is good reason to believe that we are adding something here with this radiation technique,” said Dr. Rimner.
Dr. Fontaine, the thoracic surgeon and mesothelioma research head at Moffitt Cancer Center, said he’s hoping to see a substantial impact of IMPRINT on disease-free survival (DFS) once results of NRG-LU006 are available.
“I think DFS plays a role that we’ve underestimated over the last few years for sure,” he said.
For a patient with MPM, a short DFS can be anxiety provoking and may have negative impacts on quality of life, even despite a long OS, he explained.
“In terms of your outlook on life, how many times you have to go see a doctor, and how you enjoy life, there’s a big difference between the two,” he said.
Dr. Tsao provided disclosures related to Ariad, AstraZeneca, BMS, Boehringer Ingelheim, Eli Lilly, EMD Serono, Epizyme, Genentech, Huron, Merck, Millennium, Novartis, Polaris, Roche, Seattle Genetics, SELLAS Life Sciences Group, and Takeda. Dr. Fontaine reported no relevant disclosures. Dr. Rimner reported disclosures related to Bristol-Myers Squibb, GE Healthcare, Varian Medical Systems, and Boehringer Ingelheim.
References
1. Parikh K et al. Cancer Treat Rev. 2021 Sept 1;99:102250.
2. National Comprehensive Cancer Network (NCCN) Guidelines. Malignant Pleural Mesothelioma. Version 2.2021, published 2021 Feb 16. Accessed 2021 Aug 30. https://www.nccn.org/professionals/physician_gls/pdf/mpm.pdf
3. Popat S et al. Ann Oncol. 2020;31(12):1734-45.
4. Fennell D et al. Journal of Thoracic Oncology. 2021 Mar 1;16(3):S62.
5. Baas P et al. [published correction appears in Lancet. 2021 Feb 20;397(10275):670]. Lancet. 2021 Jan 30;397(10272):375-86.
6. Adusumilli PS et al. Cancer Discov. 2021 Jul 15;candisc.0407.2021.
7. Rimner A et al. J Clin Oncol. 2016;34(23):2761-8.
8. Shaikh F et al. J Thorac Oncol. 2017;12(6):993-1000.
9. Trovo M et al. Int J Radiat Oncol Biol Phys. 2021;109(5):1368-76.
Although mesothelioma continues to be a very difficult disease to treat and one with a poor prognosis, new and emerging therapeutic developments hold the promise of extending survival for appropriately selected patients.
Following years of little to no movement, encouraging advances in treatment have been seen on the immunotherapy front. Immune checkpoint inhibitors have demonstrated acceptable safety and promising efficacy in the treatment of unresectable malignant pleural mesothelioma (MPM), including an overall survival advantage over standard-of-care first-line chemotherapy. Beyond systemic therapy, the development of new radiation techniques to complement current, more conservative surgical approaches is likewise encouraging, though further randomized clinical trial data is awaited to determine the potential impact on survival.
Longer survival would be good news for the estimated 3,000 individuals diagnosed with MPM each year in the United States. Overall, the outlook for patients with this rare cancer remains unfavorable, with a 5-year survival rate of about 11%, according to data from the U.S. Surveillance, Epidemiology and End Results (SEER) Program.
One factor underlying that grim survival statistic is a relative lack of investment in the development of drugs specific to rare cancers, as compared to more common malignancies, said Anne S. Tsao, MD, professor and director of the mesothelioma program at the University of Texas MD Anderson Cancer Center in Houston.
On the plus side, the wave of research for more common cancers has yielded a number of agents, including the immune checkpoint inhibitors such as nivolumab, ipilimumab, pembrolizumab, and durvalumab, that hold promise in rare tumor types as well.
“I think that mesothelioma has benefited from that, because these all are agents that have been developed for other solid tumors that are then brought into mesothelioma,” Dr. Tsao said in an interview. “So there’s always a lag time, but nevertheless, of course we are thrilled that we have additional treatment options for these patients.”
Checkpoint inhibitors
Multiple checkpoint inhibitors have received Food and Drug Administration approval for the treatment of non–small cell lung cancer (NSCLC) over the past few years. Because many mesothelioma doctors also treat NSCLC, bringing those agents into the mesothelioma sphere was not a very difficult jump, Dr. Tsao said.
Checkpoint inhibitors got a foothold in mesothelioma, much like in NSCLC, by demonstrating clear benefit in the salvage setting, according to Dr. Tsao.
Pembrolizumab, nivolumab, and avelumab were evaluated in phase 1b/2 clinical trials and real-world cohorts that demonstrated response rates of around 20%, median progression-free survival of 4 months, and median overall survival (OS) around 12 months in patients with previously treated MPM.
Although results of those early-stage studies had to be interpreted with caution, they nonetheless suggested a slight edge for these checkpoint inhibitors over historical data, according to the authors of a recent article in Cancer Treatment Reviews.1 On the basis of phase 1 and 2 data, current clinical practice guidelines from the National Comprehensive Cancer Network2 list pembrolizumab and the combination of nivolumab and ipilimumab as options for MPM patients who have received previous therapy. Phase 3 trials have also been launched, including PROMISE-meso, which is comparing pembrolizumab to single-agent chemotherapy in advanced, pretreated MPM3, and CONFIRM, which pits nivolumab against placebo in relapsed MPM.4
On the front lines
Encouraging results in previously treated MPM led to the evaluation of checkpoint inhibitors as first-line therapy. Notably, the FDA approved nivolumab given with ipilimumab for the treatment of patients with unresectable MPM in October 2020, making that combination the first immunotherapy regimen to receive an indication in this disease.
The FDA approval was based on prespecified interim analysis of CheckMate 743, a phase 3 study that included 605 patients randomly allocated to nivolumab plus ipilimumab or to placebo.
At the interim analysis, median OS was 18.1 months for nivolumab plus ipilimumab, versus just 14.1 months for placebo (hazard ratio, 0.74; 96.6% confidence interval, 0.60-0.91; P = 0.0020), according to results of the study published in the Lancet.5 The 2-year OS rate was 41% for the immunotherapy combination and 27% for placebo. Grade 3-4 treatment-related adverse events were seen in 30% of the immunotherapy-treated patients and 32% of the chemotherapy-treated patients.
The magnitude of nivolumab-ipilimumab benefit appeared to be largest among patients with non-epithelioid MPM subtypes (sarcomatoid and biphasic), owing to the inferior impact of chemotherapy in these patients, with a median OS of just 8.8 months, according to investigators.
That’s not to say that immunotherapy didn’t work for patients with epithelioid histology. The benefit of nivolumab-ipilimumab was consistent for non-epithelioid and epithelioid patient subsets, with median OS of 18.1 and 18.7 months, respectively, results of subgroup analysis showed.
According to Dr. Tsao, those results reflect the extremely poor prognosis and pressing need for effective therapy early in the course of treatment for patients with non-epithelioid histology.
“You have to get the most effective therapy into these patients as quickly as you can,” she explained. “If you can get the more effective treatment and early, then you’ll see a longer-term benefit for them.”
Role of the PD-L1 biomarker
Despite this progress, one key hurdle has been determining the role of the PD-L1 biomarker in mesothelioma. In NSCLC, PD-L1 is often used to determine which patients will benefit from immune checkpoint inhibitors. In mesothelioma, the correlations have been more elusive.
Among patients in the CheckMate 743 study treated with nivolumab plus ipilimumab, OS was not significantly different for those with PD-L1 expression levels of less than 1% and those with 1% or greater, investigators said. Moreover, PD-L1 expression wasn’t a stratification factor in the study.
“When looking at all of the studies, it appears that the checkpoint inhibitors can truly benefit a certain percentage of mesothelioma patients, but we can’t pick them out just yet,” Dr. Tsao said.
“So our recommendation is to offer [checkpoint inhibitor therapy] at some point in their treatment, whether it’s first, second, or third line,” she continued. “They can get some benefit, and even in those if you don’t get a great response, you can still get disease stabilization, which in and of itself can be highly beneficial.”
Future directions
Immune checkpoint inhibitor–based combination regimens and cellular therapy represent promising directions forward in MPM research. There are several notable phase 3 trials of checkpoint inhibitors plus chemotherapy and targeted therapy going forward, plus intriguing data emerging on the potential role of chimeric antigen receptor (CAR) T-cell therapy in this setting.
One phase 3 trial to watch is IND277, which is comparing pembrolizumab plus cisplatin/pemetrexed chemotherapy to cisplatin/pemetrexed alone; that trial has enrolled 520 participants and has an estimated primary completion date in July 2022, according to the ClinicalTrials.gov website. Another is BEAT-Meso, a comparison of atezolizumab plus bevacizumab and chemotherapy against bevacizumab and chemotherapy, which has an estimated enrollment of 400 participants and primary completion date of January 2024. A third trial of interest is DREAM3R, which compares durvalumab plus chemotherapy followed by durvalumab maintenance to standard chemotherapy followed by observation. That study should enroll 480 participants and has an estimated primary completion date of April 2025.
CAR T-cell therapy, while best known for its emerging role in the treatment of hematologic malignancies, may also have a place in mesothelioma therapy one day. In a recently published report, investigators described a first-in-human phase I study of a mesothelin-targeted CAR T-cell therapy given in combination with pembrolizumab. Among 18 MPM patients who received pembrolizumab safely, median OS from time of CAR T-cell infusion was 23.9 months and 1-year OS was 83%, according to investigators.6An OS of nearly 24 months is “very encouraging” and compares favorably with historical results with systemic therapy in this difficult-to-treat disease, said Jacques P. Fontaine, MD, a thoracic surgeon and section head of mesothelioma research and treatment center at Moffitt Cancer Center in Tampa, Fla.
“It’s huge, but you have to take into account that this [OS] is still less than 2 years,” Dr. Fontaine said in an interview. “There’s still a lot of work to be done.”
Radiotherapy making an IMPRINT
Meanwhile, new developments in the multimodality treatment of resectable MPM are progressing and have the potential to extend survival among patients who undergo lung-sparing surgery.
Less aggressive intervention is increasingly the preferred approach to surgery in this patient population. That shift is supported by studies showing that lung-sparing pleurectomy-decortication (P/D) resulted in less morbidity and potentially better survival outcomes than extrapleural pneumonectomy (EPP), according to Andreas Rimner, MD, associate attending physician and director of thoracic radiation oncology research at Memorial Sloan Kettering Cancer Center in New York.
However, it is more challenging to deliver radiotherapy safely in patients who have undergone P/D as compared with patients who have undergone EPP, according to Dr. Rimner.
“When there’s no lung in place [as in EPP], it’s pretty simple – you just treat the entire empty chest to kill any microscopic cells that may still be left behind,” he said in an interview. “But now we have a situation where both lungs are still in place, and they are very radiation sensitive, so that’s not an easy feat.”
Driven by the limitations of conventional radiation, Dr. Rimner and colleagues developed a novel technique known as hemithoracic intensity-modulated pleural radiation therapy (IMPRINT) that allows more precise application of radiotherapy.
In a phase 2 study published in 2016, IMPRINT was found to be safe, with an acceptable rate of radiation pneumonitis (30% grade 2 or 3), according to investigators.7
Subsequent studies have demonstrated encouraging clinical outcomes, including a 20.2-month median OS for IMPRINT versus 12.3 months for conventional adjuvant radiotherapy in a retrospective study of 209 patients who underwent P/D between 1975 and 2015.8 Those findings led to the development of a phase 3 trial known as NRG-LU006 that is evaluating P/D plus chemotherapy with or without adjuvant IMPRINT in an estimated 150 patients. The study has a primary endpoint of OS, and an estimated primary completion date in July 2025, according to ClinicalTrials.gov.
Dr. Rimner said he’s optimistic about the prospects of this study, particularly with recently published results of a phase 3 study in which Italian investigators demonstrated an OS benefit of IMPRINT over palliative radiation in patients with nonmetastatic MPM.9
“That’s more data and rationale that shows there is good reason to believe that we are adding something here with this radiation technique,” said Dr. Rimner.
Dr. Fontaine, the thoracic surgeon and mesothelioma research head at Moffitt Cancer Center, said he’s hoping to see a substantial impact of IMPRINT on disease-free survival (DFS) once results of NRG-LU006 are available.
“I think DFS plays a role that we’ve underestimated over the last few years for sure,” he said.
For a patient with MPM, a short DFS can be anxiety provoking and may have negative impacts on quality of life, even despite a long OS, he explained.
“In terms of your outlook on life, how many times you have to go see a doctor, and how you enjoy life, there’s a big difference between the two,” he said.
Dr. Tsao provided disclosures related to Ariad, AstraZeneca, BMS, Boehringer Ingelheim, Eli Lilly, EMD Serono, Epizyme, Genentech, Huron, Merck, Millennium, Novartis, Polaris, Roche, Seattle Genetics, SELLAS Life Sciences Group, and Takeda. Dr. Fontaine reported no relevant disclosures. Dr. Rimner reported disclosures related to Bristol-Myers Squibb, GE Healthcare, Varian Medical Systems, and Boehringer Ingelheim.
References
1. Parikh K et al. Cancer Treat Rev. 2021 Sept 1;99:102250.
2. National Comprehensive Cancer Network (NCCN) Guidelines. Malignant Pleural Mesothelioma. Version 2.2021, published 2021 Feb 16. Accessed 2021 Aug 30. https://www.nccn.org/professionals/physician_gls/pdf/mpm.pdf
3. Popat S et al. Ann Oncol. 2020;31(12):1734-45.
4. Fennell D et al. Journal of Thoracic Oncology. 2021 Mar 1;16(3):S62.
5. Baas P et al. [published correction appears in Lancet. 2021 Feb 20;397(10275):670]. Lancet. 2021 Jan 30;397(10272):375-86.
6. Adusumilli PS et al. Cancer Discov. 2021 Jul 15;candisc.0407.2021.
7. Rimner A et al. J Clin Oncol. 2016;34(23):2761-8.
8. Shaikh F et al. J Thorac Oncol. 2017;12(6):993-1000.
9. Trovo M et al. Int J Radiat Oncol Biol Phys. 2021;109(5):1368-76.
Although mesothelioma continues to be a very difficult disease to treat and one with a poor prognosis, new and emerging therapeutic developments hold the promise of extending survival for appropriately selected patients.
Following years of little to no movement, encouraging advances in treatment have been seen on the immunotherapy front. Immune checkpoint inhibitors have demonstrated acceptable safety and promising efficacy in the treatment of unresectable malignant pleural mesothelioma (MPM), including an overall survival advantage over standard-of-care first-line chemotherapy. Beyond systemic therapy, the development of new radiation techniques to complement current, more conservative surgical approaches is likewise encouraging, though further randomized clinical trial data is awaited to determine the potential impact on survival.
Longer survival would be good news for the estimated 3,000 individuals diagnosed with MPM each year in the United States. Overall, the outlook for patients with this rare cancer remains unfavorable, with a 5-year survival rate of about 11%, according to data from the U.S. Surveillance, Epidemiology and End Results (SEER) Program.
One factor underlying that grim survival statistic is a relative lack of investment in the development of drugs specific to rare cancers, as compared to more common malignancies, said Anne S. Tsao, MD, professor and director of the mesothelioma program at the University of Texas MD Anderson Cancer Center in Houston.
On the plus side, the wave of research for more common cancers has yielded a number of agents, including the immune checkpoint inhibitors such as nivolumab, ipilimumab, pembrolizumab, and durvalumab, that hold promise in rare tumor types as well.
“I think that mesothelioma has benefited from that, because these all are agents that have been developed for other solid tumors that are then brought into mesothelioma,” Dr. Tsao said in an interview. “So there’s always a lag time, but nevertheless, of course we are thrilled that we have additional treatment options for these patients.”
Checkpoint inhibitors
Multiple checkpoint inhibitors have received Food and Drug Administration approval for the treatment of non–small cell lung cancer (NSCLC) over the past few years. Because many mesothelioma doctors also treat NSCLC, bringing those agents into the mesothelioma sphere was not a very difficult jump, Dr. Tsao said.
Checkpoint inhibitors got a foothold in mesothelioma, much like in NSCLC, by demonstrating clear benefit in the salvage setting, according to Dr. Tsao.
Pembrolizumab, nivolumab, and avelumab were evaluated in phase 1b/2 clinical trials and real-world cohorts that demonstrated response rates of around 20%, median progression-free survival of 4 months, and median overall survival (OS) around 12 months in patients with previously treated MPM.
Although results of those early-stage studies had to be interpreted with caution, they nonetheless suggested a slight edge for these checkpoint inhibitors over historical data, according to the authors of a recent article in Cancer Treatment Reviews.1 On the basis of phase 1 and 2 data, current clinical practice guidelines from the National Comprehensive Cancer Network2 list pembrolizumab and the combination of nivolumab and ipilimumab as options for MPM patients who have received previous therapy. Phase 3 trials have also been launched, including PROMISE-meso, which is comparing pembrolizumab to single-agent chemotherapy in advanced, pretreated MPM3, and CONFIRM, which pits nivolumab against placebo in relapsed MPM.4
On the front lines
Encouraging results in previously treated MPM led to the evaluation of checkpoint inhibitors as first-line therapy. Notably, the FDA approved nivolumab given with ipilimumab for the treatment of patients with unresectable MPM in October 2020, making that combination the first immunotherapy regimen to receive an indication in this disease.
The FDA approval was based on prespecified interim analysis of CheckMate 743, a phase 3 study that included 605 patients randomly allocated to nivolumab plus ipilimumab or to placebo.
At the interim analysis, median OS was 18.1 months for nivolumab plus ipilimumab, versus just 14.1 months for placebo (hazard ratio, 0.74; 96.6% confidence interval, 0.60-0.91; P = 0.0020), according to results of the study published in the Lancet.5 The 2-year OS rate was 41% for the immunotherapy combination and 27% for placebo. Grade 3-4 treatment-related adverse events were seen in 30% of the immunotherapy-treated patients and 32% of the chemotherapy-treated patients.
The magnitude of nivolumab-ipilimumab benefit appeared to be largest among patients with non-epithelioid MPM subtypes (sarcomatoid and biphasic), owing to the inferior impact of chemotherapy in these patients, with a median OS of just 8.8 months, according to investigators.
That’s not to say that immunotherapy didn’t work for patients with epithelioid histology. The benefit of nivolumab-ipilimumab was consistent for non-epithelioid and epithelioid patient subsets, with median OS of 18.1 and 18.7 months, respectively, results of subgroup analysis showed.
According to Dr. Tsao, those results reflect the extremely poor prognosis and pressing need for effective therapy early in the course of treatment for patients with non-epithelioid histology.
“You have to get the most effective therapy into these patients as quickly as you can,” she explained. “If you can get the more effective treatment and early, then you’ll see a longer-term benefit for them.”
Role of the PD-L1 biomarker
Despite this progress, one key hurdle has been determining the role of the PD-L1 biomarker in mesothelioma. In NSCLC, PD-L1 is often used to determine which patients will benefit from immune checkpoint inhibitors. In mesothelioma, the correlations have been more elusive.
Among patients in the CheckMate 743 study treated with nivolumab plus ipilimumab, OS was not significantly different for those with PD-L1 expression levels of less than 1% and those with 1% or greater, investigators said. Moreover, PD-L1 expression wasn’t a stratification factor in the study.
“When looking at all of the studies, it appears that the checkpoint inhibitors can truly benefit a certain percentage of mesothelioma patients, but we can’t pick them out just yet,” Dr. Tsao said.
“So our recommendation is to offer [checkpoint inhibitor therapy] at some point in their treatment, whether it’s first, second, or third line,” she continued. “They can get some benefit, and even in those if you don’t get a great response, you can still get disease stabilization, which in and of itself can be highly beneficial.”
Future directions
Immune checkpoint inhibitor–based combination regimens and cellular therapy represent promising directions forward in MPM research. There are several notable phase 3 trials of checkpoint inhibitors plus chemotherapy and targeted therapy going forward, plus intriguing data emerging on the potential role of chimeric antigen receptor (CAR) T-cell therapy in this setting.
One phase 3 trial to watch is IND277, which is comparing pembrolizumab plus cisplatin/pemetrexed chemotherapy to cisplatin/pemetrexed alone; that trial has enrolled 520 participants and has an estimated primary completion date in July 2022, according to the ClinicalTrials.gov website. Another is BEAT-Meso, a comparison of atezolizumab plus bevacizumab and chemotherapy against bevacizumab and chemotherapy, which has an estimated enrollment of 400 participants and primary completion date of January 2024. A third trial of interest is DREAM3R, which compares durvalumab plus chemotherapy followed by durvalumab maintenance to standard chemotherapy followed by observation. That study should enroll 480 participants and has an estimated primary completion date of April 2025.
CAR T-cell therapy, while best known for its emerging role in the treatment of hematologic malignancies, may also have a place in mesothelioma therapy one day. In a recently published report, investigators described a first-in-human phase I study of a mesothelin-targeted CAR T-cell therapy given in combination with pembrolizumab. Among 18 MPM patients who received pembrolizumab safely, median OS from time of CAR T-cell infusion was 23.9 months and 1-year OS was 83%, according to investigators.6An OS of nearly 24 months is “very encouraging” and compares favorably with historical results with systemic therapy in this difficult-to-treat disease, said Jacques P. Fontaine, MD, a thoracic surgeon and section head of mesothelioma research and treatment center at Moffitt Cancer Center in Tampa, Fla.
“It’s huge, but you have to take into account that this [OS] is still less than 2 years,” Dr. Fontaine said in an interview. “There’s still a lot of work to be done.”
Radiotherapy making an IMPRINT
Meanwhile, new developments in the multimodality treatment of resectable MPM are progressing and have the potential to extend survival among patients who undergo lung-sparing surgery.
Less aggressive intervention is increasingly the preferred approach to surgery in this patient population. That shift is supported by studies showing that lung-sparing pleurectomy-decortication (P/D) resulted in less morbidity and potentially better survival outcomes than extrapleural pneumonectomy (EPP), according to Andreas Rimner, MD, associate attending physician and director of thoracic radiation oncology research at Memorial Sloan Kettering Cancer Center in New York.
However, it is more challenging to deliver radiotherapy safely in patients who have undergone P/D as compared with patients who have undergone EPP, according to Dr. Rimner.
“When there’s no lung in place [as in EPP], it’s pretty simple – you just treat the entire empty chest to kill any microscopic cells that may still be left behind,” he said in an interview. “But now we have a situation where both lungs are still in place, and they are very radiation sensitive, so that’s not an easy feat.”
Driven by the limitations of conventional radiation, Dr. Rimner and colleagues developed a novel technique known as hemithoracic intensity-modulated pleural radiation therapy (IMPRINT) that allows more precise application of radiotherapy.
In a phase 2 study published in 2016, IMPRINT was found to be safe, with an acceptable rate of radiation pneumonitis (30% grade 2 or 3), according to investigators.7
Subsequent studies have demonstrated encouraging clinical outcomes, including a 20.2-month median OS for IMPRINT versus 12.3 months for conventional adjuvant radiotherapy in a retrospective study of 209 patients who underwent P/D between 1975 and 2015.8 Those findings led to the development of a phase 3 trial known as NRG-LU006 that is evaluating P/D plus chemotherapy with or without adjuvant IMPRINT in an estimated 150 patients. The study has a primary endpoint of OS, and an estimated primary completion date in July 2025, according to ClinicalTrials.gov.
Dr. Rimner said he’s optimistic about the prospects of this study, particularly with recently published results of a phase 3 study in which Italian investigators demonstrated an OS benefit of IMPRINT over palliative radiation in patients with nonmetastatic MPM.9
“That’s more data and rationale that shows there is good reason to believe that we are adding something here with this radiation technique,” said Dr. Rimner.
Dr. Fontaine, the thoracic surgeon and mesothelioma research head at Moffitt Cancer Center, said he’s hoping to see a substantial impact of IMPRINT on disease-free survival (DFS) once results of NRG-LU006 are available.
“I think DFS plays a role that we’ve underestimated over the last few years for sure,” he said.
For a patient with MPM, a short DFS can be anxiety provoking and may have negative impacts on quality of life, even despite a long OS, he explained.
“In terms of your outlook on life, how many times you have to go see a doctor, and how you enjoy life, there’s a big difference between the two,” he said.
Dr. Tsao provided disclosures related to Ariad, AstraZeneca, BMS, Boehringer Ingelheim, Eli Lilly, EMD Serono, Epizyme, Genentech, Huron, Merck, Millennium, Novartis, Polaris, Roche, Seattle Genetics, SELLAS Life Sciences Group, and Takeda. Dr. Fontaine reported no relevant disclosures. Dr. Rimner reported disclosures related to Bristol-Myers Squibb, GE Healthcare, Varian Medical Systems, and Boehringer Ingelheim.
References
1. Parikh K et al. Cancer Treat Rev. 2021 Sept 1;99:102250.
2. National Comprehensive Cancer Network (NCCN) Guidelines. Malignant Pleural Mesothelioma. Version 2.2021, published 2021 Feb 16. Accessed 2021 Aug 30. https://www.nccn.org/professionals/physician_gls/pdf/mpm.pdf
3. Popat S et al. Ann Oncol. 2020;31(12):1734-45.
4. Fennell D et al. Journal of Thoracic Oncology. 2021 Mar 1;16(3):S62.
5. Baas P et al. [published correction appears in Lancet. 2021 Feb 20;397(10275):670]. Lancet. 2021 Jan 30;397(10272):375-86.
6. Adusumilli PS et al. Cancer Discov. 2021 Jul 15;candisc.0407.2021.
7. Rimner A et al. J Clin Oncol. 2016;34(23):2761-8.
8. Shaikh F et al. J Thorac Oncol. 2017;12(6):993-1000.
9. Trovo M et al. Int J Radiat Oncol Biol Phys. 2021;109(5):1368-76.
A Fatal Case of Hemophagocytic Lymphohistiocytosis Secondary to Anti-MDA5–Positive Dermatomyositis
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
PRACTICE POINTS
- Anti-MDA5 (melanoma differentiation–associated gene 5 antibody)–positive dermatomyositis associated with hemophagocytic lymphohistiocytosis is a rare and aggressive condition associated with a poor prognosis, and there is no standard treatment.
- Dermatomyositis-associated liver injury is not well defined.
Giant cell arteritis fast-track clinics pave way for greater ultrasound use in U.S.
Temporal artery biopsy has been the standard for diagnosing giant cell arteritis (GCA), but vascular ultrasound, a procedure that’s less invasive, less time-intensive, less expensive, and more convenient, has gained widespread use in Europe, and now clinics in the United States are adopting this approach and moving toward having rheumatologists take on the role of ultrasonographer.
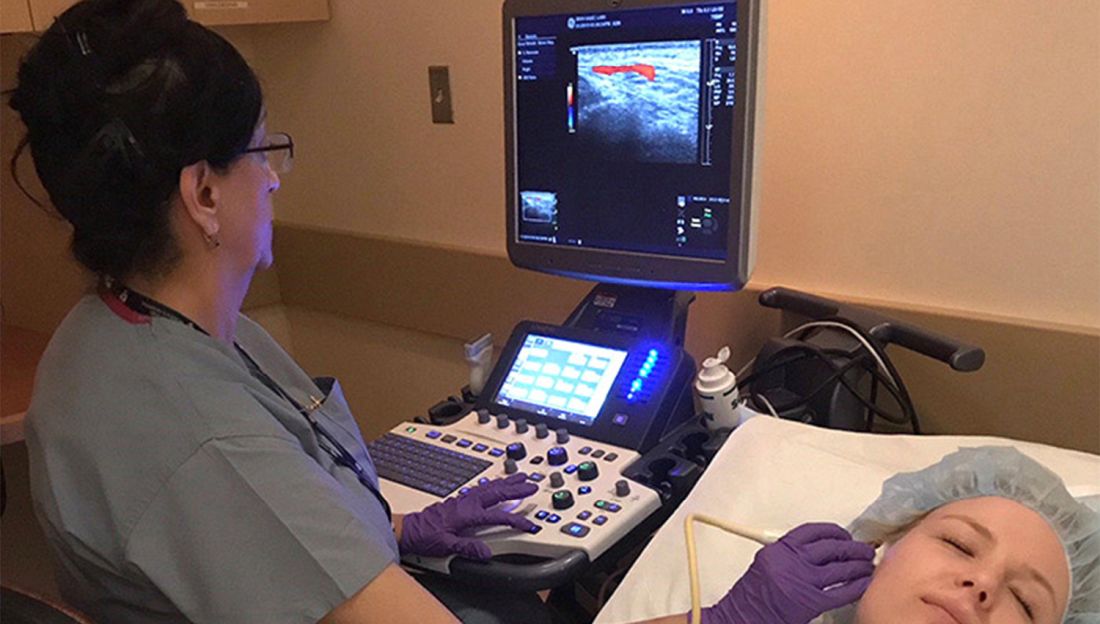
However, directors at these clinics – known as GCA fast-track clinics – caution that the bar can be high for adopting vascular ultrasound (VUS) as a tool to diagnose GCA. Ultrasonographers need specialized training to perform the task and an adequate caseload to maintain their skills. Clinics also need to be outfitted with high-definition ultrasound machines.
“It definitely takes adequate training and learning of how to adjust the settings on the ultrasound machine to be able to visualize the findings appropriately,” said Minna Kohler, MD, director of the rheumatology and musculoskeletal ultrasound program at Massachusetts General Hospital in Boston, which has its own GCA fast-track clinic.

“And the clinical context is very important,” she said. “If you have a high suspicion for someone with temporal arteritis, or GCA, and the patient has been on steroids for weeks before you see them, the ultrasound findings may not show signs of the disease. In those cases in which the imaging is equivocal, we would still pursue a biopsy.”
The idea of the fast-track clinic is as the name implies: to quickly confirm the presence of GCA in a matter of hours, or days at the most, in an outpatient setting without the hassles of a biopsy. Temporal artery biopsy (TAB), by comparison, “is more costly because it requires operating room time, a surgical consultation, and surgery time, whereas ultrasound is a very inexpensive exam since it’s done in the clinic by the rheumatologist,” Dr. Kohler said.
European experience
Use of VUS to diagnose GCA is supplanting TAB in Europe and other countries. In Denmark alone – with a population of 6 million – three outpatient fast-track clinics are operating. The United States, with a population more than 50 times larger than Denmark’s, has six.
Stavros Chrysidis, MD, PhD, chief of rheumatology at the Hospital South West Jutland, one of the fast-track clinic sites in Denmark, led a recent multicenter study, known as EUREKA, of VUS in patients with suspected GCA. He and his colleagues reported in The Lancet Rheumatology that the sensitivity and specificity of VUS was superior to TAB in confirming a diagnosis of GCA. Dr. Chrysidis has instructed U.S. rheumatologists and ultrasonographers in performing and interpreting VUS for GCA.
The study emphasizes the importance of training for ultrasonographers, said Dr. Chrysidis, who regularly performs VUS at his institution. “The most important finding is that, when we apply VUS by systematically trained ultrasonographers using appropriate equipment in appropriate settings, it has excellent diagnostic accuracy on GCA,” he told this news organization.
He noted that The Lancet Rheumatology report is the first multicenter study of VUS for diagnosing GCA in which all the ultrasonographers participated in a standardized training program, which his group developed. “Ultrasound is very operator dependent,” he said. “That’s why the training is very important.”
The training occurred over a year and included two workshops consisting of 5 days of theoretical training on VUS; supervised hands-on evaluation of healthy individuals and patients with known GCA; and evaluation of ultrasound images. Over the course of the year, trainees performed at least 50 VUS evaluations, half of which were in patients with confirmed GCA. During the training period, an external rheumatologist with broad experience in VUS made the final diagnosis.
“The equipment and settings are very important because ultrasound can be very time consuming if you are not educated well and if your equipment is not adjusted well,” Dr. Chrysidis said. The equipment must be calibrated beforehand “so you don’t spend time on adjustments.”
For diagnosing temporal artery anomalies, the ultrasound equipment must have a resolution of 0.3-0.4 mm, he said. “When you have a transducer of 10 MHz, you cannot visualize changes smaller than 5 mm.”
The EUREKA study stated that VUS could replace TAB as a first-line diagnostic tool for GCA – provided the ultrasonographers are systematically trained and the equipment and settings are appropriate. In the Jutland clinic, VUS already has replaced TAB, Dr. Chrysidis said.
“In my department since 2017, when we started the fast-track clinic after the EUREKA study was terminated, we have performed three temporal artery biopsies in the last 4 years, and we screen 60-70 patients per year because we use ultrasound as the primary imaging,” he said. In cases when the results are inconclusive, they order a PET scan. “We don’t perform biopsies anymore,” he said.
U.S. fast-track clinic models
The fast-track clinic models in the United States vary. Results of a survey of the U.S. clinics were presented as an abstract at the 2021 American College of Rheumatology annual meeting. Some centers have a vasculitis specialist obtain and interpret the imaging. At others, a vasculitis specialist refers patients to a VUS-trained rheumatologist to perform and interpret the test. Another approach is to have vasculitis specialists refer patients to ultrasound technicians trained in VUS, with a vascular surgeon interpreting the images and either a VUS-trained rheumatologist or vascular medicine specialist verifying the images.
The take-home message of that survey is that “ultrasound evaluation should be considered in the hands of experts, realizing that not everyone has that skill set, but if it is available, it’s a way to expedite diagnosis and it can be helpful in managing the GCA patient in an appropriate way, quicker than trying to schedule cross-sectional imaging,” said Massachusetts General’s Dr. Kohler, who is a coauthor of the abstract. “Certainly, cross-sectional imaging also plays an important role, but when it comes to confirming whether to continue with treatment or not for a very serious condition, ultrasound is a quick way to get the answer.”
In addition to the fast-track clinic at Massachusetts General, the survey included fast-track clinics at the University of Washington, Seattle; Brigham and Women’s Hospital, Boston; Loma Linda (Calif.) University; University of California, Los Angeles; and at a private practice, Arthritis and Rheumatism Associates in the Washington area.
Advantages of VUS vs. TAB
At Massachusetts General, some of the rheumatologists are trained to perform VUS. The rheumatologists also perform the clinical evaluation of suspected cases of GCA. The advantage of VUS, Dr. Kohler said, is that the answer is “right there”; that is, the imaging yields a diagnosis almost instantaneously whereas a biopsy must be sent to a lab for analysis.
“Since a lot of patients with suspected vasculitis may already come to us on steroid therapy, and if there’s a low probability or low suspicion for vasculitis, [VUS] actually confirms that, and we’re able to taper prednisone or steroids quickly rather than keep them on a prolonged course.”
Alison Bays, MD, MPH, of the department of rheumatology at the University of Washington, said that the advantages of avoiding biopsy aren’t to be overlooked. “Temporal artery biopsies are invasive and carry surgical risks, especially as many of our patients are elderly,” she told this news organization.

“These patients occasionally refuse biopsy, but the acceptance of ultrasound is high,” Dr. Bays said. “Scheduling surgery can be more complicated, resulting in delays to biopsy and potentially higher rates of false negatives. Additionally, ultrasound has resulted in a higher rate of diagnosis with GCA as TAB misses large-vessel involvement.” The fast-track clinic at the university has evaluated 250 patients since it opened in 2017.
Dr. Bays and colleagues published the first United States study of a fast-track protocol using vascular sonographers. “Our group has demonstrated that fast-track clinics can rapidly and effectively evaluate patients, and we demonstrated a different method of evaluation using vascular sonographers rather than rheumatologists to do the vascular ultrasound,” she said. “It utilizes the familiarity vascular sonographers already have with imaging blood vessels.”
She added that the TABUL study in the United Kingdom in 2016 demonstrated that VUS yielded a savings of $686 (£484 as reported in the study), compared with TAB. “Further studies need to be done in the United States,” she said. “Beyond direct comparison of costs, reduction in steroid burden due to quick evaluation and diagnosis may carry additional benefits.”
At Brigham and Women’s Hospital, the division of rheumatology and the vascular section of the cardiology division collaborate on the GCA evaluation, said Sara Tedeschi, MD, MPH, codirector of the fast-track clinic there. Trained vascular technologists credentialed in the procedure and specifically trained in using VUS for evaluating arteritis perform the VUS. Cardiologists with a subspecialty in vascular medicine interpret the studies.

VUS patients have a rheumatology evaluation just before they have the ultrasound. “The rheumatology evaluation is then able to incorporate information from the VUS together with laboratory data, the patient’s history, and physical examination,” Dr. Tedeschi said.
“If the rheumatologist recommends a temporal artery biopsy as a next step, we arrange this with vascular surgery,” she said. “If the rheumatologist recommends other imaging such as MRA [magnetic resonance angiography] or PET-CT, we frequently review the images together with our colleagues in cardiovascular radiology and/or nuclear medicine.”
But in the United States, it will take some time for GCA fast-track clinics to become the standard, Dr. Tedeschi said. “Temporal artery biopsy may be faster to arrange in certain practice settings if VUS is not already being employed for giant cell arteritis evaluation,” she said.
Dr. Bays recognized this limitation, saying, “We are hoping that in the future, the American College of Rheumatology will consider vascular ultrasound as a first-line diagnostic test in diagnosis as rheumatologists and vascular sonographers gain familiarity over time.”
But that would mean that centers performing VUS for GCA would have to meet rigorous standards for the procedure. “With that, standardization of a protocol, high-quality equipment, and adequate training are necessary to ensure quality and reduce the chance of false-positive or false-negative results,” she said.
Dr. Chrysidis, Dr. Bays, and Dr. Tedeschi have disclosed no relevant financial relationships. Dr. Kohler is a board member of Ultrasound School of North American Rheumatologists.
A version of this article first appeared on Medscape.com.
Temporal artery biopsy has been the standard for diagnosing giant cell arteritis (GCA), but vascular ultrasound, a procedure that’s less invasive, less time-intensive, less expensive, and more convenient, has gained widespread use in Europe, and now clinics in the United States are adopting this approach and moving toward having rheumatologists take on the role of ultrasonographer.
However, directors at these clinics – known as GCA fast-track clinics – caution that the bar can be high for adopting vascular ultrasound (VUS) as a tool to diagnose GCA. Ultrasonographers need specialized training to perform the task and an adequate caseload to maintain their skills. Clinics also need to be outfitted with high-definition ultrasound machines.
“It definitely takes adequate training and learning of how to adjust the settings on the ultrasound machine to be able to visualize the findings appropriately,” said Minna Kohler, MD, director of the rheumatology and musculoskeletal ultrasound program at Massachusetts General Hospital in Boston, which has its own GCA fast-track clinic.
“And the clinical context is very important,” she said. “If you have a high suspicion for someone with temporal arteritis, or GCA, and the patient has been on steroids for weeks before you see them, the ultrasound findings may not show signs of the disease. In those cases in which the imaging is equivocal, we would still pursue a biopsy.”
The idea of the fast-track clinic is as the name implies: to quickly confirm the presence of GCA in a matter of hours, or days at the most, in an outpatient setting without the hassles of a biopsy. Temporal artery biopsy (TAB), by comparison, “is more costly because it requires operating room time, a surgical consultation, and surgery time, whereas ultrasound is a very inexpensive exam since it’s done in the clinic by the rheumatologist,” Dr. Kohler said.
European experience
Use of VUS to diagnose GCA is supplanting TAB in Europe and other countries. In Denmark alone – with a population of 6 million – three outpatient fast-track clinics are operating. The United States, with a population more than 50 times larger than Denmark’s, has six.
Stavros Chrysidis, MD, PhD, chief of rheumatology at the Hospital South West Jutland, one of the fast-track clinic sites in Denmark, led a recent multicenter study, known as EUREKA, of VUS in patients with suspected GCA. He and his colleagues reported in The Lancet Rheumatology that the sensitivity and specificity of VUS was superior to TAB in confirming a diagnosis of GCA. Dr. Chrysidis has instructed U.S. rheumatologists and ultrasonographers in performing and interpreting VUS for GCA.
The study emphasizes the importance of training for ultrasonographers, said Dr. Chrysidis, who regularly performs VUS at his institution. “The most important finding is that, when we apply VUS by systematically trained ultrasonographers using appropriate equipment in appropriate settings, it has excellent diagnostic accuracy on GCA,” he told this news organization.
He noted that The Lancet Rheumatology report is the first multicenter study of VUS for diagnosing GCA in which all the ultrasonographers participated in a standardized training program, which his group developed. “Ultrasound is very operator dependent,” he said. “That’s why the training is very important.”
The training occurred over a year and included two workshops consisting of 5 days of theoretical training on VUS; supervised hands-on evaluation of healthy individuals and patients with known GCA; and evaluation of ultrasound images. Over the course of the year, trainees performed at least 50 VUS evaluations, half of which were in patients with confirmed GCA. During the training period, an external rheumatologist with broad experience in VUS made the final diagnosis.
“The equipment and settings are very important because ultrasound can be very time consuming if you are not educated well and if your equipment is not adjusted well,” Dr. Chrysidis said. The equipment must be calibrated beforehand “so you don’t spend time on adjustments.”
For diagnosing temporal artery anomalies, the ultrasound equipment must have a resolution of 0.3-0.4 mm, he said. “When you have a transducer of 10 MHz, you cannot visualize changes smaller than 5 mm.”
The EUREKA study stated that VUS could replace TAB as a first-line diagnostic tool for GCA – provided the ultrasonographers are systematically trained and the equipment and settings are appropriate. In the Jutland clinic, VUS already has replaced TAB, Dr. Chrysidis said.
“In my department since 2017, when we started the fast-track clinic after the EUREKA study was terminated, we have performed three temporal artery biopsies in the last 4 years, and we screen 60-70 patients per year because we use ultrasound as the primary imaging,” he said. In cases when the results are inconclusive, they order a PET scan. “We don’t perform biopsies anymore,” he said.
U.S. fast-track clinic models
The fast-track clinic models in the United States vary. Results of a survey of the U.S. clinics were presented as an abstract at the 2021 American College of Rheumatology annual meeting. Some centers have a vasculitis specialist obtain and interpret the imaging. At others, a vasculitis specialist refers patients to a VUS-trained rheumatologist to perform and interpret the test. Another approach is to have vasculitis specialists refer patients to ultrasound technicians trained in VUS, with a vascular surgeon interpreting the images and either a VUS-trained rheumatologist or vascular medicine specialist verifying the images.
The take-home message of that survey is that “ultrasound evaluation should be considered in the hands of experts, realizing that not everyone has that skill set, but if it is available, it’s a way to expedite diagnosis and it can be helpful in managing the GCA patient in an appropriate way, quicker than trying to schedule cross-sectional imaging,” said Massachusetts General’s Dr. Kohler, who is a coauthor of the abstract. “Certainly, cross-sectional imaging also plays an important role, but when it comes to confirming whether to continue with treatment or not for a very serious condition, ultrasound is a quick way to get the answer.”
In addition to the fast-track clinic at Massachusetts General, the survey included fast-track clinics at the University of Washington, Seattle; Brigham and Women’s Hospital, Boston; Loma Linda (Calif.) University; University of California, Los Angeles; and at a private practice, Arthritis and Rheumatism Associates in the Washington area.
Advantages of VUS vs. TAB
At Massachusetts General, some of the rheumatologists are trained to perform VUS. The rheumatologists also perform the clinical evaluation of suspected cases of GCA. The advantage of VUS, Dr. Kohler said, is that the answer is “right there”; that is, the imaging yields a diagnosis almost instantaneously whereas a biopsy must be sent to a lab for analysis.
“Since a lot of patients with suspected vasculitis may already come to us on steroid therapy, and if there’s a low probability or low suspicion for vasculitis, [VUS] actually confirms that, and we’re able to taper prednisone or steroids quickly rather than keep them on a prolonged course.”
Alison Bays, MD, MPH, of the department of rheumatology at the University of Washington, said that the advantages of avoiding biopsy aren’t to be overlooked. “Temporal artery biopsies are invasive and carry surgical risks, especially as many of our patients are elderly,” she told this news organization.
“These patients occasionally refuse biopsy, but the acceptance of ultrasound is high,” Dr. Bays said. “Scheduling surgery can be more complicated, resulting in delays to biopsy and potentially higher rates of false negatives. Additionally, ultrasound has resulted in a higher rate of diagnosis with GCA as TAB misses large-vessel involvement.” The fast-track clinic at the university has evaluated 250 patients since it opened in 2017.
Dr. Bays and colleagues published the first United States study of a fast-track protocol using vascular sonographers. “Our group has demonstrated that fast-track clinics can rapidly and effectively evaluate patients, and we demonstrated a different method of evaluation using vascular sonographers rather than rheumatologists to do the vascular ultrasound,” she said. “It utilizes the familiarity vascular sonographers already have with imaging blood vessels.”
She added that the TABUL study in the United Kingdom in 2016 demonstrated that VUS yielded a savings of $686 (£484 as reported in the study), compared with TAB. “Further studies need to be done in the United States,” she said. “Beyond direct comparison of costs, reduction in steroid burden due to quick evaluation and diagnosis may carry additional benefits.”
At Brigham and Women’s Hospital, the division of rheumatology and the vascular section of the cardiology division collaborate on the GCA evaluation, said Sara Tedeschi, MD, MPH, codirector of the fast-track clinic there. Trained vascular technologists credentialed in the procedure and specifically trained in using VUS for evaluating arteritis perform the VUS. Cardiologists with a subspecialty in vascular medicine interpret the studies.
VUS patients have a rheumatology evaluation just before they have the ultrasound. “The rheumatology evaluation is then able to incorporate information from the VUS together with laboratory data, the patient’s history, and physical examination,” Dr. Tedeschi said.
“If the rheumatologist recommends a temporal artery biopsy as a next step, we arrange this with vascular surgery,” she said. “If the rheumatologist recommends other imaging such as MRA [magnetic resonance angiography] or PET-CT, we frequently review the images together with our colleagues in cardiovascular radiology and/or nuclear medicine.”
But in the United States, it will take some time for GCA fast-track clinics to become the standard, Dr. Tedeschi said. “Temporal artery biopsy may be faster to arrange in certain practice settings if VUS is not already being employed for giant cell arteritis evaluation,” she said.
Dr. Bays recognized this limitation, saying, “We are hoping that in the future, the American College of Rheumatology will consider vascular ultrasound as a first-line diagnostic test in diagnosis as rheumatologists and vascular sonographers gain familiarity over time.”
But that would mean that centers performing VUS for GCA would have to meet rigorous standards for the procedure. “With that, standardization of a protocol, high-quality equipment, and adequate training are necessary to ensure quality and reduce the chance of false-positive or false-negative results,” she said.
Dr. Chrysidis, Dr. Bays, and Dr. Tedeschi have disclosed no relevant financial relationships. Dr. Kohler is a board member of Ultrasound School of North American Rheumatologists.
A version of this article first appeared on Medscape.com.
Temporal artery biopsy has been the standard for diagnosing giant cell arteritis (GCA), but vascular ultrasound, a procedure that’s less invasive, less time-intensive, less expensive, and more convenient, has gained widespread use in Europe, and now clinics in the United States are adopting this approach and moving toward having rheumatologists take on the role of ultrasonographer.
However, directors at these clinics – known as GCA fast-track clinics – caution that the bar can be high for adopting vascular ultrasound (VUS) as a tool to diagnose GCA. Ultrasonographers need specialized training to perform the task and an adequate caseload to maintain their skills. Clinics also need to be outfitted with high-definition ultrasound machines.
“It definitely takes adequate training and learning of how to adjust the settings on the ultrasound machine to be able to visualize the findings appropriately,” said Minna Kohler, MD, director of the rheumatology and musculoskeletal ultrasound program at Massachusetts General Hospital in Boston, which has its own GCA fast-track clinic.
“And the clinical context is very important,” she said. “If you have a high suspicion for someone with temporal arteritis, or GCA, and the patient has been on steroids for weeks before you see them, the ultrasound findings may not show signs of the disease. In those cases in which the imaging is equivocal, we would still pursue a biopsy.”
The idea of the fast-track clinic is as the name implies: to quickly confirm the presence of GCA in a matter of hours, or days at the most, in an outpatient setting without the hassles of a biopsy. Temporal artery biopsy (TAB), by comparison, “is more costly because it requires operating room time, a surgical consultation, and surgery time, whereas ultrasound is a very inexpensive exam since it’s done in the clinic by the rheumatologist,” Dr. Kohler said.
European experience
Use of VUS to diagnose GCA is supplanting TAB in Europe and other countries. In Denmark alone – with a population of 6 million – three outpatient fast-track clinics are operating. The United States, with a population more than 50 times larger than Denmark’s, has six.
Stavros Chrysidis, MD, PhD, chief of rheumatology at the Hospital South West Jutland, one of the fast-track clinic sites in Denmark, led a recent multicenter study, known as EUREKA, of VUS in patients with suspected GCA. He and his colleagues reported in The Lancet Rheumatology that the sensitivity and specificity of VUS was superior to TAB in confirming a diagnosis of GCA. Dr. Chrysidis has instructed U.S. rheumatologists and ultrasonographers in performing and interpreting VUS for GCA.
The study emphasizes the importance of training for ultrasonographers, said Dr. Chrysidis, who regularly performs VUS at his institution. “The most important finding is that, when we apply VUS by systematically trained ultrasonographers using appropriate equipment in appropriate settings, it has excellent diagnostic accuracy on GCA,” he told this news organization.
He noted that The Lancet Rheumatology report is the first multicenter study of VUS for diagnosing GCA in which all the ultrasonographers participated in a standardized training program, which his group developed. “Ultrasound is very operator dependent,” he said. “That’s why the training is very important.”
The training occurred over a year and included two workshops consisting of 5 days of theoretical training on VUS; supervised hands-on evaluation of healthy individuals and patients with known GCA; and evaluation of ultrasound images. Over the course of the year, trainees performed at least 50 VUS evaluations, half of which were in patients with confirmed GCA. During the training period, an external rheumatologist with broad experience in VUS made the final diagnosis.
“The equipment and settings are very important because ultrasound can be very time consuming if you are not educated well and if your equipment is not adjusted well,” Dr. Chrysidis said. The equipment must be calibrated beforehand “so you don’t spend time on adjustments.”
For diagnosing temporal artery anomalies, the ultrasound equipment must have a resolution of 0.3-0.4 mm, he said. “When you have a transducer of 10 MHz, you cannot visualize changes smaller than 5 mm.”
The EUREKA study stated that VUS could replace TAB as a first-line diagnostic tool for GCA – provided the ultrasonographers are systematically trained and the equipment and settings are appropriate. In the Jutland clinic, VUS already has replaced TAB, Dr. Chrysidis said.
“In my department since 2017, when we started the fast-track clinic after the EUREKA study was terminated, we have performed three temporal artery biopsies in the last 4 years, and we screen 60-70 patients per year because we use ultrasound as the primary imaging,” he said. In cases when the results are inconclusive, they order a PET scan. “We don’t perform biopsies anymore,” he said.
U.S. fast-track clinic models
The fast-track clinic models in the United States vary. Results of a survey of the U.S. clinics were presented as an abstract at the 2021 American College of Rheumatology annual meeting. Some centers have a vasculitis specialist obtain and interpret the imaging. At others, a vasculitis specialist refers patients to a VUS-trained rheumatologist to perform and interpret the test. Another approach is to have vasculitis specialists refer patients to ultrasound technicians trained in VUS, with a vascular surgeon interpreting the images and either a VUS-trained rheumatologist or vascular medicine specialist verifying the images.
The take-home message of that survey is that “ultrasound evaluation should be considered in the hands of experts, realizing that not everyone has that skill set, but if it is available, it’s a way to expedite diagnosis and it can be helpful in managing the GCA patient in an appropriate way, quicker than trying to schedule cross-sectional imaging,” said Massachusetts General’s Dr. Kohler, who is a coauthor of the abstract. “Certainly, cross-sectional imaging also plays an important role, but when it comes to confirming whether to continue with treatment or not for a very serious condition, ultrasound is a quick way to get the answer.”
In addition to the fast-track clinic at Massachusetts General, the survey included fast-track clinics at the University of Washington, Seattle; Brigham and Women’s Hospital, Boston; Loma Linda (Calif.) University; University of California, Los Angeles; and at a private practice, Arthritis and Rheumatism Associates in the Washington area.
Advantages of VUS vs. TAB
At Massachusetts General, some of the rheumatologists are trained to perform VUS. The rheumatologists also perform the clinical evaluation of suspected cases of GCA. The advantage of VUS, Dr. Kohler said, is that the answer is “right there”; that is, the imaging yields a diagnosis almost instantaneously whereas a biopsy must be sent to a lab for analysis.
“Since a lot of patients with suspected vasculitis may already come to us on steroid therapy, and if there’s a low probability or low suspicion for vasculitis, [VUS] actually confirms that, and we’re able to taper prednisone or steroids quickly rather than keep them on a prolonged course.”
Alison Bays, MD, MPH, of the department of rheumatology at the University of Washington, said that the advantages of avoiding biopsy aren’t to be overlooked. “Temporal artery biopsies are invasive and carry surgical risks, especially as many of our patients are elderly,” she told this news organization.
“These patients occasionally refuse biopsy, but the acceptance of ultrasound is high,” Dr. Bays said. “Scheduling surgery can be more complicated, resulting in delays to biopsy and potentially higher rates of false negatives. Additionally, ultrasound has resulted in a higher rate of diagnosis with GCA as TAB misses large-vessel involvement.” The fast-track clinic at the university has evaluated 250 patients since it opened in 2017.
Dr. Bays and colleagues published the first United States study of a fast-track protocol using vascular sonographers. “Our group has demonstrated that fast-track clinics can rapidly and effectively evaluate patients, and we demonstrated a different method of evaluation using vascular sonographers rather than rheumatologists to do the vascular ultrasound,” she said. “It utilizes the familiarity vascular sonographers already have with imaging blood vessels.”
She added that the TABUL study in the United Kingdom in 2016 demonstrated that VUS yielded a savings of $686 (£484 as reported in the study), compared with TAB. “Further studies need to be done in the United States,” she said. “Beyond direct comparison of costs, reduction in steroid burden due to quick evaluation and diagnosis may carry additional benefits.”
At Brigham and Women’s Hospital, the division of rheumatology and the vascular section of the cardiology division collaborate on the GCA evaluation, said Sara Tedeschi, MD, MPH, codirector of the fast-track clinic there. Trained vascular technologists credentialed in the procedure and specifically trained in using VUS for evaluating arteritis perform the VUS. Cardiologists with a subspecialty in vascular medicine interpret the studies.
VUS patients have a rheumatology evaluation just before they have the ultrasound. “The rheumatology evaluation is then able to incorporate information from the VUS together with laboratory data, the patient’s history, and physical examination,” Dr. Tedeschi said.
“If the rheumatologist recommends a temporal artery biopsy as a next step, we arrange this with vascular surgery,” she said. “If the rheumatologist recommends other imaging such as MRA [magnetic resonance angiography] or PET-CT, we frequently review the images together with our colleagues in cardiovascular radiology and/or nuclear medicine.”
But in the United States, it will take some time for GCA fast-track clinics to become the standard, Dr. Tedeschi said. “Temporal artery biopsy may be faster to arrange in certain practice settings if VUS is not already being employed for giant cell arteritis evaluation,” she said.
Dr. Bays recognized this limitation, saying, “We are hoping that in the future, the American College of Rheumatology will consider vascular ultrasound as a first-line diagnostic test in diagnosis as rheumatologists and vascular sonographers gain familiarity over time.”
But that would mean that centers performing VUS for GCA would have to meet rigorous standards for the procedure. “With that, standardization of a protocol, high-quality equipment, and adequate training are necessary to ensure quality and reduce the chance of false-positive or false-negative results,” she said.
Dr. Chrysidis, Dr. Bays, and Dr. Tedeschi have disclosed no relevant financial relationships. Dr. Kohler is a board member of Ultrasound School of North American Rheumatologists.
A version of this article first appeared on Medscape.com.
Genomic screening of healthy newborns gets more popular
Even before their baby is born, parents face some tough questions: Home birth or hospital? Cloth or disposable diapers? Breast, bottle, or both? But advances in genetic sequencing technology mean that parents will soon face yet another choice: whether to sequence their newborn’s DNA for an overview of the baby’s entire genome.
Genetic testing has been used for decades to diagnose conditions even before birth. But DNA sequencing technologies, once expensive and tough to access, are now rapid and cheap enough that doctors could order genomic screening for any infant, regardless of health status.
The possibility has raised many questions about the ethical, legal, and social repercussions of doing so. One of the biggest sticking points of sequencing newborns is the potential psychosocial fallout for families of such wide-scale use of genetic screening.
“There’s a narrative of catastrophic distress,” says Robert Green, MD, a geneticist at Harvard Medical School and lead investigator on the BabySeq study, which is evaluating the medical, social, and economic consequences of newborn genetic screening. The concern is that parents learning that their child carries a gene variant related to cancer or heart disease will become “incredibly anxious and distressed,” he says. “And it’s not an unreasonable speculation.”
But Dr. Green’s team found no evidence of such anxiety in the results from a randomized trial it conducted, published in JAMA Pediatrics. In the meantime, Genomics England announced it would begin a pilot study involving whole-genome sequencing of up to 200,000 babies. The first goal is to identify severe disease that starts in childhood, but the information would also be stored and used to detect drug sensitivities and conditions that come up later in life.
The large U.K. project is a bold move, according to David Amor, PhD, a pediatric geneticist at Murdoch Children’s Research Institute in Australia, who says its time has come. Geneticists have been accused of thinking their field involves unique pitfalls, compared with the rest of medicine, he points out, and that doctors need to protect patients and families from the potential harm genetic testing poses.
“But it is becoming apparent that that’s not really the case,” he says, and “maybe there’s not a whole lot special about genetics – it’s just medicine.”
When a first-draft copy of the human genome was published in 2001, scientists and doctors hailed the start of a new era of precision medicine. Knowing our genome sequence was expected to lead to a better grasp on our individual disease risks. Yet even as technologies advanced, clinical genetics remained focused on diagnosis rather than screening, according to Lilian Downie, a clinical genetics PhD candidate at the University of Melbourne. She calls the difference subtle but important.
Diagnostic genetic testing confirms whether a person has a specific condition, whereas genetic screening tests evaluate someone’s risk of getting an illness. Both approaches use sequencing, but they answer different questions, explains Ms. Downie.
Diagnosing disease versus predicting future illness
Genetic testing is on the upswing for both purposes, whether clinically for diagnosis or through direct-to-consumer screening-oriented services like 23andMe. Scientists began to note that many people carried disease-related genetic variants without having signs of disease. In some cases, a variant that is mathematically linked to a disease simply doesn’t cause it. In other cases, though, even if the gene variant contributes to a disease, not everyone who carries the genetic change will get the condition.
This potential disconnect between having a variant and developing the condition is a big problem, says Katie Stoll, a genetic counselor and executive director of the Genetic Support Foundation in Olympia, WA.
“It’s more complicated than just looking at one gene variant and one outcome,” she says. Without a sure link between the two, this information could unnecessarily entail “some pretty big emotional and financial costs.”
Ms. Stoll and others in the genetics field who share similar concerns are one reason the BabySeq project was first funded back in 2015. Although the overall aim of the initiative is to answer questions about the value of genomic sequencing in newborn screening, the media and scientific attention has focused on the psychosocial impact of healthy newborn sequencing, says Dr. Green. In the study published in JAMA Pediatrics, his group focused on these issues, too.
For that randomized trial, they enrolled 325 families, 257 with healthy babies and 68 whose babies had spent time in neonatal intensive care. Enrolled infants were randomly given standard care alone or standard care with genomic sequencing added on. The genomic sequencing report contained information about the presence of genetic variants associated with disease that start in childhood. Parents also could choose whether to learn about genetic risks for conditions that start in adulthood, such as cancer.
Boston-based Tina Moniz was one of those parents. When her first daughter was born in Jan. 2016, someone from the BabySeq study asked her and her husband if they would like to take part. The decision was simple for the couple.
“I didn’t hesitate,” she says. “To me, knowledge is power.”
Using screening tools for parental and marital distress and parent-child bonding, the research evaluated BabySeq families at 3 and 10 months after parents received the sequencing results. The investigators found no significant differences in any of these measures between screened and unscreened families. Ms. Moniz learned that her daughter’s only concerning result was being a carrier for cystic fibrosis. Rather than finding this information anxiety-provoking, Ms. Moniz considered it to be reassuring.
“My mom brain worries about so many things, but at least I know I don’t have to add genetic disease to the list,” she says.
But Ms. Stoll, who wasn’t involved in the BabySeq study, isn’t as convinced. She says that less than 10% of the families approached about the trial ultimately agreed to take part, suggesting potential bias in the selection process. Most participants were white, well-educated, and well-off, making it hard to generalize the study’s results.
What’s more, the standard care involved meeting with a genetic counselor and giving a detailed family history, neither of which is routinely offered to new parents, Ms. Stoll says. These study features leave her unconvinced that healthy newborn genetic screening is beneficial.
“We can’t assume these psychosocial consequences will be true for everyone,” she says.
Follow-up and treatment needed
Traditional newborn screening relies on blood biochemical tests to detect and diagnose metabolic diseases. This approach still outperforms DNA sequencing in trials, says Cynthia Powell, MD, a pediatric geneticist at the University of North Carolina at Chapel Hill, who wasn’t involved with the BabySeq study. Despite the enthusiasm for genomics, this kind of screening won’t replace newborn biochemical screening anytime soon, she says.
“There are some states that have only one geneticist available, so should we really be doing this if we can’t provide the necessary follow-up and treatment for these babies?” she asks.
Still, Dr. Powell says, the BabySeq study helps advance understanding of what the infrastructure needs are for widespread use of DNA sequencing in newborns. She says those needs include appropriate consent processes, access to genetic counselors to discuss testing, and referrals for further testing and treatment in those babies with concerning results.
The BabySeq program will also guide new initiatives, like the pilot program that Genomics England launched in Sept. 2021. As part of that project, the U.K. group intends to look into how practical whole-genome sequencing for newborn screening would be and look at the risks, benefits, and limits of its widespread use.
“For the first time, we’re putting real data into these questions that people have basically just speculated and hypothesized and created narratives about,” Dr. Green says.
But for now, the findings on the psychosocial effects of general newborn genomic screening show that “we should consider genetics to be just one more arrow in our medical quiver.”
A version of this article first appeared on WebMD.com.
Even before their baby is born, parents face some tough questions: Home birth or hospital? Cloth or disposable diapers? Breast, bottle, or both? But advances in genetic sequencing technology mean that parents will soon face yet another choice: whether to sequence their newborn’s DNA for an overview of the baby’s entire genome.
Genetic testing has been used for decades to diagnose conditions even before birth. But DNA sequencing technologies, once expensive and tough to access, are now rapid and cheap enough that doctors could order genomic screening for any infant, regardless of health status.
The possibility has raised many questions about the ethical, legal, and social repercussions of doing so. One of the biggest sticking points of sequencing newborns is the potential psychosocial fallout for families of such wide-scale use of genetic screening.
“There’s a narrative of catastrophic distress,” says Robert Green, MD, a geneticist at Harvard Medical School and lead investigator on the BabySeq study, which is evaluating the medical, social, and economic consequences of newborn genetic screening. The concern is that parents learning that their child carries a gene variant related to cancer or heart disease will become “incredibly anxious and distressed,” he says. “And it’s not an unreasonable speculation.”
But Dr. Green’s team found no evidence of such anxiety in the results from a randomized trial it conducted, published in JAMA Pediatrics. In the meantime, Genomics England announced it would begin a pilot study involving whole-genome sequencing of up to 200,000 babies. The first goal is to identify severe disease that starts in childhood, but the information would also be stored and used to detect drug sensitivities and conditions that come up later in life.
The large U.K. project is a bold move, according to David Amor, PhD, a pediatric geneticist at Murdoch Children’s Research Institute in Australia, who says its time has come. Geneticists have been accused of thinking their field involves unique pitfalls, compared with the rest of medicine, he points out, and that doctors need to protect patients and families from the potential harm genetic testing poses.
“But it is becoming apparent that that’s not really the case,” he says, and “maybe there’s not a whole lot special about genetics – it’s just medicine.”
When a first-draft copy of the human genome was published in 2001, scientists and doctors hailed the start of a new era of precision medicine. Knowing our genome sequence was expected to lead to a better grasp on our individual disease risks. Yet even as technologies advanced, clinical genetics remained focused on diagnosis rather than screening, according to Lilian Downie, a clinical genetics PhD candidate at the University of Melbourne. She calls the difference subtle but important.
Diagnostic genetic testing confirms whether a person has a specific condition, whereas genetic screening tests evaluate someone’s risk of getting an illness. Both approaches use sequencing, but they answer different questions, explains Ms. Downie.
Diagnosing disease versus predicting future illness
Genetic testing is on the upswing for both purposes, whether clinically for diagnosis or through direct-to-consumer screening-oriented services like 23andMe. Scientists began to note that many people carried disease-related genetic variants without having signs of disease. In some cases, a variant that is mathematically linked to a disease simply doesn’t cause it. In other cases, though, even if the gene variant contributes to a disease, not everyone who carries the genetic change will get the condition.
This potential disconnect between having a variant and developing the condition is a big problem, says Katie Stoll, a genetic counselor and executive director of the Genetic Support Foundation in Olympia, WA.
“It’s more complicated than just looking at one gene variant and one outcome,” she says. Without a sure link between the two, this information could unnecessarily entail “some pretty big emotional and financial costs.”
Ms. Stoll and others in the genetics field who share similar concerns are one reason the BabySeq project was first funded back in 2015. Although the overall aim of the initiative is to answer questions about the value of genomic sequencing in newborn screening, the media and scientific attention has focused on the psychosocial impact of healthy newborn sequencing, says Dr. Green. In the study published in JAMA Pediatrics, his group focused on these issues, too.
For that randomized trial, they enrolled 325 families, 257 with healthy babies and 68 whose babies had spent time in neonatal intensive care. Enrolled infants were randomly given standard care alone or standard care with genomic sequencing added on. The genomic sequencing report contained information about the presence of genetic variants associated with disease that start in childhood. Parents also could choose whether to learn about genetic risks for conditions that start in adulthood, such as cancer.
Boston-based Tina Moniz was one of those parents. When her first daughter was born in Jan. 2016, someone from the BabySeq study asked her and her husband if they would like to take part. The decision was simple for the couple.
“I didn’t hesitate,” she says. “To me, knowledge is power.”
Using screening tools for parental and marital distress and parent-child bonding, the research evaluated BabySeq families at 3 and 10 months after parents received the sequencing results. The investigators found no significant differences in any of these measures between screened and unscreened families. Ms. Moniz learned that her daughter’s only concerning result was being a carrier for cystic fibrosis. Rather than finding this information anxiety-provoking, Ms. Moniz considered it to be reassuring.
“My mom brain worries about so many things, but at least I know I don’t have to add genetic disease to the list,” she says.
But Ms. Stoll, who wasn’t involved in the BabySeq study, isn’t as convinced. She says that less than 10% of the families approached about the trial ultimately agreed to take part, suggesting potential bias in the selection process. Most participants were white, well-educated, and well-off, making it hard to generalize the study’s results.
What’s more, the standard care involved meeting with a genetic counselor and giving a detailed family history, neither of which is routinely offered to new parents, Ms. Stoll says. These study features leave her unconvinced that healthy newborn genetic screening is beneficial.
“We can’t assume these psychosocial consequences will be true for everyone,” she says.
Follow-up and treatment needed
Traditional newborn screening relies on blood biochemical tests to detect and diagnose metabolic diseases. This approach still outperforms DNA sequencing in trials, says Cynthia Powell, MD, a pediatric geneticist at the University of North Carolina at Chapel Hill, who wasn’t involved with the BabySeq study. Despite the enthusiasm for genomics, this kind of screening won’t replace newborn biochemical screening anytime soon, she says.
“There are some states that have only one geneticist available, so should we really be doing this if we can’t provide the necessary follow-up and treatment for these babies?” she asks.
Still, Dr. Powell says, the BabySeq study helps advance understanding of what the infrastructure needs are for widespread use of DNA sequencing in newborns. She says those needs include appropriate consent processes, access to genetic counselors to discuss testing, and referrals for further testing and treatment in those babies with concerning results.
The BabySeq program will also guide new initiatives, like the pilot program that Genomics England launched in Sept. 2021. As part of that project, the U.K. group intends to look into how practical whole-genome sequencing for newborn screening would be and look at the risks, benefits, and limits of its widespread use.
“For the first time, we’re putting real data into these questions that people have basically just speculated and hypothesized and created narratives about,” Dr. Green says.
But for now, the findings on the psychosocial effects of general newborn genomic screening show that “we should consider genetics to be just one more arrow in our medical quiver.”
A version of this article first appeared on WebMD.com.
Even before their baby is born, parents face some tough questions: Home birth or hospital? Cloth or disposable diapers? Breast, bottle, or both? But advances in genetic sequencing technology mean that parents will soon face yet another choice: whether to sequence their newborn’s DNA for an overview of the baby’s entire genome.
Genetic testing has been used for decades to diagnose conditions even before birth. But DNA sequencing technologies, once expensive and tough to access, are now rapid and cheap enough that doctors could order genomic screening for any infant, regardless of health status.
The possibility has raised many questions about the ethical, legal, and social repercussions of doing so. One of the biggest sticking points of sequencing newborns is the potential psychosocial fallout for families of such wide-scale use of genetic screening.
“There’s a narrative of catastrophic distress,” says Robert Green, MD, a geneticist at Harvard Medical School and lead investigator on the BabySeq study, which is evaluating the medical, social, and economic consequences of newborn genetic screening. The concern is that parents learning that their child carries a gene variant related to cancer or heart disease will become “incredibly anxious and distressed,” he says. “And it’s not an unreasonable speculation.”
But Dr. Green’s team found no evidence of such anxiety in the results from a randomized trial it conducted, published in JAMA Pediatrics. In the meantime, Genomics England announced it would begin a pilot study involving whole-genome sequencing of up to 200,000 babies. The first goal is to identify severe disease that starts in childhood, but the information would also be stored and used to detect drug sensitivities and conditions that come up later in life.
The large U.K. project is a bold move, according to David Amor, PhD, a pediatric geneticist at Murdoch Children’s Research Institute in Australia, who says its time has come. Geneticists have been accused of thinking their field involves unique pitfalls, compared with the rest of medicine, he points out, and that doctors need to protect patients and families from the potential harm genetic testing poses.
“But it is becoming apparent that that’s not really the case,” he says, and “maybe there’s not a whole lot special about genetics – it’s just medicine.”
When a first-draft copy of the human genome was published in 2001, scientists and doctors hailed the start of a new era of precision medicine. Knowing our genome sequence was expected to lead to a better grasp on our individual disease risks. Yet even as technologies advanced, clinical genetics remained focused on diagnosis rather than screening, according to Lilian Downie, a clinical genetics PhD candidate at the University of Melbourne. She calls the difference subtle but important.
Diagnostic genetic testing confirms whether a person has a specific condition, whereas genetic screening tests evaluate someone’s risk of getting an illness. Both approaches use sequencing, but they answer different questions, explains Ms. Downie.
Diagnosing disease versus predicting future illness
Genetic testing is on the upswing for both purposes, whether clinically for diagnosis or through direct-to-consumer screening-oriented services like 23andMe. Scientists began to note that many people carried disease-related genetic variants without having signs of disease. In some cases, a variant that is mathematically linked to a disease simply doesn’t cause it. In other cases, though, even if the gene variant contributes to a disease, not everyone who carries the genetic change will get the condition.
This potential disconnect between having a variant and developing the condition is a big problem, says Katie Stoll, a genetic counselor and executive director of the Genetic Support Foundation in Olympia, WA.
“It’s more complicated than just looking at one gene variant and one outcome,” she says. Without a sure link between the two, this information could unnecessarily entail “some pretty big emotional and financial costs.”
Ms. Stoll and others in the genetics field who share similar concerns are one reason the BabySeq project was first funded back in 2015. Although the overall aim of the initiative is to answer questions about the value of genomic sequencing in newborn screening, the media and scientific attention has focused on the psychosocial impact of healthy newborn sequencing, says Dr. Green. In the study published in JAMA Pediatrics, his group focused on these issues, too.
For that randomized trial, they enrolled 325 families, 257 with healthy babies and 68 whose babies had spent time in neonatal intensive care. Enrolled infants were randomly given standard care alone or standard care with genomic sequencing added on. The genomic sequencing report contained information about the presence of genetic variants associated with disease that start in childhood. Parents also could choose whether to learn about genetic risks for conditions that start in adulthood, such as cancer.
Boston-based Tina Moniz was one of those parents. When her first daughter was born in Jan. 2016, someone from the BabySeq study asked her and her husband if they would like to take part. The decision was simple for the couple.
“I didn’t hesitate,” she says. “To me, knowledge is power.”
Using screening tools for parental and marital distress and parent-child bonding, the research evaluated BabySeq families at 3 and 10 months after parents received the sequencing results. The investigators found no significant differences in any of these measures between screened and unscreened families. Ms. Moniz learned that her daughter’s only concerning result was being a carrier for cystic fibrosis. Rather than finding this information anxiety-provoking, Ms. Moniz considered it to be reassuring.
“My mom brain worries about so many things, but at least I know I don’t have to add genetic disease to the list,” she says.
But Ms. Stoll, who wasn’t involved in the BabySeq study, isn’t as convinced. She says that less than 10% of the families approached about the trial ultimately agreed to take part, suggesting potential bias in the selection process. Most participants were white, well-educated, and well-off, making it hard to generalize the study’s results.
What’s more, the standard care involved meeting with a genetic counselor and giving a detailed family history, neither of which is routinely offered to new parents, Ms. Stoll says. These study features leave her unconvinced that healthy newborn genetic screening is beneficial.
“We can’t assume these psychosocial consequences will be true for everyone,” she says.
Follow-up and treatment needed
Traditional newborn screening relies on blood biochemical tests to detect and diagnose metabolic diseases. This approach still outperforms DNA sequencing in trials, says Cynthia Powell, MD, a pediatric geneticist at the University of North Carolina at Chapel Hill, who wasn’t involved with the BabySeq study. Despite the enthusiasm for genomics, this kind of screening won’t replace newborn biochemical screening anytime soon, she says.
“There are some states that have only one geneticist available, so should we really be doing this if we can’t provide the necessary follow-up and treatment for these babies?” she asks.
Still, Dr. Powell says, the BabySeq study helps advance understanding of what the infrastructure needs are for widespread use of DNA sequencing in newborns. She says those needs include appropriate consent processes, access to genetic counselors to discuss testing, and referrals for further testing and treatment in those babies with concerning results.
The BabySeq program will also guide new initiatives, like the pilot program that Genomics England launched in Sept. 2021. As part of that project, the U.K. group intends to look into how practical whole-genome sequencing for newborn screening would be and look at the risks, benefits, and limits of its widespread use.
“For the first time, we’re putting real data into these questions that people have basically just speculated and hypothesized and created narratives about,” Dr. Green says.
But for now, the findings on the psychosocial effects of general newborn genomic screening show that “we should consider genetics to be just one more arrow in our medical quiver.”
A version of this article first appeared on WebMD.com.
Multiple Lesions With Recurrent Bleeding
The Diagnosis: Nevoid Basal Cell Carcinoma Syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare autosomal-dominant disorder that increases the risk for developing various carcinomas and affects multiple organ systems. Nevoid basal cell carcinoma syndrome is estimated at 1 per 40,000 to 60,000 individuals with no sexual predilection.1,2 Pathogenesis of NBCCS occurs through molecular alterations in the dormant hedgehog signaling pathway, causing constitutive signaling activity and a loss of function in the tumor suppressor patched 1 gene, PTCH1. As a result, the inhibition of smoothened oncogenes is released, Gli proteins are activated, and the hedgehog signaling pathway is no longer quiescent.2 Additional loss of function in the suppressor of fused homolog protein, a negative regulator of the hedgehog pathway, allows for further tumor proliferation. The crucial role these genes play in the hedgehog signaling pathway and their mutation association with NBCCS allows for molecular confirmation in the diagnosis of NBCCS. Allelic losses at the PTCH1 gene site are thought to occur in approximately 70% of NBCCS patients.2
Diagnosis of NBCCS is based on genetic testing to examine pathogenic gene variants, notably in the PTCH1 gene, and identification of characteristic clinical findings.2 Diagnosis of NBCCS requires either 2 minor suggestive criteria and 1 major suggestive criterion, 2 major suggestive criteria and 1 minor suggestive criterion, or 1 major suggestive criterion with molecular confirmation. The presence of basal cell carcinomas (BCCs) before 20 years of age or an excessive numbers of BCCs, keratocystic odontogenic tumors (KOTs), palmar or plantar pitting, and first-degree relatives with NBCCS are classified as major suggestive criteria.2 Nevoid basal cell carcinoma syndrome patients typically have BCCs that crust, ulcerate, or bleed. Minor suggestive criteria for NBCCS are rib abnormalities, skeletal malformations, macrocephaly, cleft lip or palate, and desmoplastic medulloblastoma.2-4 Suppressor of fused homolog protein mutations may increase the risk for desmoplastic medulloblastoma in NBCCS patients. Our patient had 4 of the major suggestive criteria, including a history of KOTs, multiple BCCs, first-degree relatives with NBCCS, and palmar or plantar pitting (bottom quiz image), while having 1 minor suggestive criterion of frontal bossing.
Patients with NBCCS have high phenotypic variability, as their skin carcinomas do not have the classic features of pearly surfaces or corkscrew telangiectasia that typically are associated with BCCs.1 Basal cell carcinomas in NBCCS-affected individuals usually are indistinguishable from sporadic lesions that arise in sun-exposed areas, making NBCCS difficult to diagnose. These sporadic lesions often are misdiagnosed as psoriatic or eczematous lesions, and additional subsequent examination is required. The findings of multiple papules and plaques spanning the body as well as lesions with rolled borders and ulcerated bases, indicative of BCCs, aid dermatologists in distinguishing benign lesions from those of NBCCS.1
Additional differential diagnoses are required to distinguish NBCCS from other similar inherited skin disorders that are characterized by BCCs. The presence of multiple incidental BCCs early in life remains a histopathologic clue for NBCCS diagnosis, as opposed to Rombo syndrome, in which BCCs often develop in adulthood.2,4 In addition, although both Bazex syndrome and Muir-Torre syndrome are characterized by the early onset of BCCs, the lack of skeletal abnormalities and palmar and plantar pitting distinguish these entities from NBCCS.2,4 Furthermore, though psoriasis also can present on the scalp, clinical presentation often includes well-demarcated and symmetric plaques that are erythematous and silvery, all of which were not present in our patient and typically are not seen in NBCCS.5
The recommended treatment of NBCCS is vismodegib, a specific oncogene inhibitor. This medication suppresses the hedgehog signaling pathway by inhibiting smoothened oncogenes and downstream target molecules, thereby decreasing tumor proliferation.6 In doing so, vismodegib inhibits the development of new BCCs while reducing the burden of present ones. Additionally, vismodegib appears to effectively treat KOTs. If successful, this medication may be able to suppress KOTs in patients with NBCCS and thus facilitate surgery.6 Additional hedgehog inhibitors include patidegib, sonidegib, and itraconazole. Patidegib gel 2% currently is in phase 3 clinical trials for evaluation of efficacy and safety in treatment of NBCCS. Sonidegib is approved for the treatment of locally advanced BCCs in the United States and the European Union and for both locally advanced BCCs and metastatic BCCs in Switzerland and Australia.7 Further research is needed before recommending antifungal itraconazole for NBCCS clinical use.8 Other medications for localized areas include topical application of 5-fluorouracil and imiquimod.2
- Sangehra R, Grewal P. Gorlin syndrome presentation and the importance of differential diagnosis of skin cancer: a case report. J Pharm Pharm Sci. 2018;21:222-224.
- Bresler S, Padwa B, Granter S. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Head Neck Pathol. 2016;10:119-124.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- Evans G, Farndon P. Nevoid basal cell carcinoma syndrome. GeneReviews [Internet]. University of Washington; 1993-2020.
- Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63:278-285.
- Booms P, Harth M, Sader R, et al. Vismodegib hedgehog-signaling inhibition and treatment of basal cell carcinomas as well as keratocystic odontogenic tumors in Gorlin syndrome. Ann Maxillofac Surg. 2015;5:14-19.
- Gutzmer R, Soloon J. Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target Oncol. 2019;14:253-267.
- Leavitt E, Lask G, Martin S. Sonic hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr Treat Options Oncol. 2019;20:84.
The Diagnosis: Nevoid Basal Cell Carcinoma Syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare autosomal-dominant disorder that increases the risk for developing various carcinomas and affects multiple organ systems. Nevoid basal cell carcinoma syndrome is estimated at 1 per 40,000 to 60,000 individuals with no sexual predilection.1,2 Pathogenesis of NBCCS occurs through molecular alterations in the dormant hedgehog signaling pathway, causing constitutive signaling activity and a loss of function in the tumor suppressor patched 1 gene, PTCH1. As a result, the inhibition of smoothened oncogenes is released, Gli proteins are activated, and the hedgehog signaling pathway is no longer quiescent.2 Additional loss of function in the suppressor of fused homolog protein, a negative regulator of the hedgehog pathway, allows for further tumor proliferation. The crucial role these genes play in the hedgehog signaling pathway and their mutation association with NBCCS allows for molecular confirmation in the diagnosis of NBCCS. Allelic losses at the PTCH1 gene site are thought to occur in approximately 70% of NBCCS patients.2
Diagnosis of NBCCS is based on genetic testing to examine pathogenic gene variants, notably in the PTCH1 gene, and identification of characteristic clinical findings.2 Diagnosis of NBCCS requires either 2 minor suggestive criteria and 1 major suggestive criterion, 2 major suggestive criteria and 1 minor suggestive criterion, or 1 major suggestive criterion with molecular confirmation. The presence of basal cell carcinomas (BCCs) before 20 years of age or an excessive numbers of BCCs, keratocystic odontogenic tumors (KOTs), palmar or plantar pitting, and first-degree relatives with NBCCS are classified as major suggestive criteria.2 Nevoid basal cell carcinoma syndrome patients typically have BCCs that crust, ulcerate, or bleed. Minor suggestive criteria for NBCCS are rib abnormalities, skeletal malformations, macrocephaly, cleft lip or palate, and desmoplastic medulloblastoma.2-4 Suppressor of fused homolog protein mutations may increase the risk for desmoplastic medulloblastoma in NBCCS patients. Our patient had 4 of the major suggestive criteria, including a history of KOTs, multiple BCCs, first-degree relatives with NBCCS, and palmar or plantar pitting (bottom quiz image), while having 1 minor suggestive criterion of frontal bossing.
Patients with NBCCS have high phenotypic variability, as their skin carcinomas do not have the classic features of pearly surfaces or corkscrew telangiectasia that typically are associated with BCCs.1 Basal cell carcinomas in NBCCS-affected individuals usually are indistinguishable from sporadic lesions that arise in sun-exposed areas, making NBCCS difficult to diagnose. These sporadic lesions often are misdiagnosed as psoriatic or eczematous lesions, and additional subsequent examination is required. The findings of multiple papules and plaques spanning the body as well as lesions with rolled borders and ulcerated bases, indicative of BCCs, aid dermatologists in distinguishing benign lesions from those of NBCCS.1
Additional differential diagnoses are required to distinguish NBCCS from other similar inherited skin disorders that are characterized by BCCs. The presence of multiple incidental BCCs early in life remains a histopathologic clue for NBCCS diagnosis, as opposed to Rombo syndrome, in which BCCs often develop in adulthood.2,4 In addition, although both Bazex syndrome and Muir-Torre syndrome are characterized by the early onset of BCCs, the lack of skeletal abnormalities and palmar and plantar pitting distinguish these entities from NBCCS.2,4 Furthermore, though psoriasis also can present on the scalp, clinical presentation often includes well-demarcated and symmetric plaques that are erythematous and silvery, all of which were not present in our patient and typically are not seen in NBCCS.5
The recommended treatment of NBCCS is vismodegib, a specific oncogene inhibitor. This medication suppresses the hedgehog signaling pathway by inhibiting smoothened oncogenes and downstream target molecules, thereby decreasing tumor proliferation.6 In doing so, vismodegib inhibits the development of new BCCs while reducing the burden of present ones. Additionally, vismodegib appears to effectively treat KOTs. If successful, this medication may be able to suppress KOTs in patients with NBCCS and thus facilitate surgery.6 Additional hedgehog inhibitors include patidegib, sonidegib, and itraconazole. Patidegib gel 2% currently is in phase 3 clinical trials for evaluation of efficacy and safety in treatment of NBCCS. Sonidegib is approved for the treatment of locally advanced BCCs in the United States and the European Union and for both locally advanced BCCs and metastatic BCCs in Switzerland and Australia.7 Further research is needed before recommending antifungal itraconazole for NBCCS clinical use.8 Other medications for localized areas include topical application of 5-fluorouracil and imiquimod.2
The Diagnosis: Nevoid Basal Cell Carcinoma Syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare autosomal-dominant disorder that increases the risk for developing various carcinomas and affects multiple organ systems. Nevoid basal cell carcinoma syndrome is estimated at 1 per 40,000 to 60,000 individuals with no sexual predilection.1,2 Pathogenesis of NBCCS occurs through molecular alterations in the dormant hedgehog signaling pathway, causing constitutive signaling activity and a loss of function in the tumor suppressor patched 1 gene, PTCH1. As a result, the inhibition of smoothened oncogenes is released, Gli proteins are activated, and the hedgehog signaling pathway is no longer quiescent.2 Additional loss of function in the suppressor of fused homolog protein, a negative regulator of the hedgehog pathway, allows for further tumor proliferation. The crucial role these genes play in the hedgehog signaling pathway and their mutation association with NBCCS allows for molecular confirmation in the diagnosis of NBCCS. Allelic losses at the PTCH1 gene site are thought to occur in approximately 70% of NBCCS patients.2
Diagnosis of NBCCS is based on genetic testing to examine pathogenic gene variants, notably in the PTCH1 gene, and identification of characteristic clinical findings.2 Diagnosis of NBCCS requires either 2 minor suggestive criteria and 1 major suggestive criterion, 2 major suggestive criteria and 1 minor suggestive criterion, or 1 major suggestive criterion with molecular confirmation. The presence of basal cell carcinomas (BCCs) before 20 years of age or an excessive numbers of BCCs, keratocystic odontogenic tumors (KOTs), palmar or plantar pitting, and first-degree relatives with NBCCS are classified as major suggestive criteria.2 Nevoid basal cell carcinoma syndrome patients typically have BCCs that crust, ulcerate, or bleed. Minor suggestive criteria for NBCCS are rib abnormalities, skeletal malformations, macrocephaly, cleft lip or palate, and desmoplastic medulloblastoma.2-4 Suppressor of fused homolog protein mutations may increase the risk for desmoplastic medulloblastoma in NBCCS patients. Our patient had 4 of the major suggestive criteria, including a history of KOTs, multiple BCCs, first-degree relatives with NBCCS, and palmar or plantar pitting (bottom quiz image), while having 1 minor suggestive criterion of frontal bossing.
Patients with NBCCS have high phenotypic variability, as their skin carcinomas do not have the classic features of pearly surfaces or corkscrew telangiectasia that typically are associated with BCCs.1 Basal cell carcinomas in NBCCS-affected individuals usually are indistinguishable from sporadic lesions that arise in sun-exposed areas, making NBCCS difficult to diagnose. These sporadic lesions often are misdiagnosed as psoriatic or eczematous lesions, and additional subsequent examination is required. The findings of multiple papules and plaques spanning the body as well as lesions with rolled borders and ulcerated bases, indicative of BCCs, aid dermatologists in distinguishing benign lesions from those of NBCCS.1
Additional differential diagnoses are required to distinguish NBCCS from other similar inherited skin disorders that are characterized by BCCs. The presence of multiple incidental BCCs early in life remains a histopathologic clue for NBCCS diagnosis, as opposed to Rombo syndrome, in which BCCs often develop in adulthood.2,4 In addition, although both Bazex syndrome and Muir-Torre syndrome are characterized by the early onset of BCCs, the lack of skeletal abnormalities and palmar and plantar pitting distinguish these entities from NBCCS.2,4 Furthermore, though psoriasis also can present on the scalp, clinical presentation often includes well-demarcated and symmetric plaques that are erythematous and silvery, all of which were not present in our patient and typically are not seen in NBCCS.5
The recommended treatment of NBCCS is vismodegib, a specific oncogene inhibitor. This medication suppresses the hedgehog signaling pathway by inhibiting smoothened oncogenes and downstream target molecules, thereby decreasing tumor proliferation.6 In doing so, vismodegib inhibits the development of new BCCs while reducing the burden of present ones. Additionally, vismodegib appears to effectively treat KOTs. If successful, this medication may be able to suppress KOTs in patients with NBCCS and thus facilitate surgery.6 Additional hedgehog inhibitors include patidegib, sonidegib, and itraconazole. Patidegib gel 2% currently is in phase 3 clinical trials for evaluation of efficacy and safety in treatment of NBCCS. Sonidegib is approved for the treatment of locally advanced BCCs in the United States and the European Union and for both locally advanced BCCs and metastatic BCCs in Switzerland and Australia.7 Further research is needed before recommending antifungal itraconazole for NBCCS clinical use.8 Other medications for localized areas include topical application of 5-fluorouracil and imiquimod.2
- Sangehra R, Grewal P. Gorlin syndrome presentation and the importance of differential diagnosis of skin cancer: a case report. J Pharm Pharm Sci. 2018;21:222-224.
- Bresler S, Padwa B, Granter S. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Head Neck Pathol. 2016;10:119-124.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- Evans G, Farndon P. Nevoid basal cell carcinoma syndrome. GeneReviews [Internet]. University of Washington; 1993-2020.
- Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63:278-285.
- Booms P, Harth M, Sader R, et al. Vismodegib hedgehog-signaling inhibition and treatment of basal cell carcinomas as well as keratocystic odontogenic tumors in Gorlin syndrome. Ann Maxillofac Surg. 2015;5:14-19.
- Gutzmer R, Soloon J. Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target Oncol. 2019;14:253-267.
- Leavitt E, Lask G, Martin S. Sonic hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr Treat Options Oncol. 2019;20:84.
- Sangehra R, Grewal P. Gorlin syndrome presentation and the importance of differential diagnosis of skin cancer: a case report. J Pharm Pharm Sci. 2018;21:222-224.
- Bresler S, Padwa B, Granter S. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Head Neck Pathol. 2016;10:119-124.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- Evans G, Farndon P. Nevoid basal cell carcinoma syndrome. GeneReviews [Internet]. University of Washington; 1993-2020.
- Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63:278-285.
- Booms P, Harth M, Sader R, et al. Vismodegib hedgehog-signaling inhibition and treatment of basal cell carcinomas as well as keratocystic odontogenic tumors in Gorlin syndrome. Ann Maxillofac Surg. 2015;5:14-19.
- Gutzmer R, Soloon J. Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target Oncol. 2019;14:253-267.
- Leavitt E, Lask G, Martin S. Sonic hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr Treat Options Oncol. 2019;20:84.

A 63-year-old man with frontal bossing and a history of jaw cysts presented with numerous lesions on the scalp, trunk, and legs with recurrent bleeding. Both of his siblings had similar findings. Many lesions had been present for at least 40 years. Physical examination revealed a large, irregular, atrophic, hyperpigmented plaque on the central scalp with multiple translucent hyperpigmented papules at the periphery (top). Similar papules and plaques were present at the temples, around the waist, and on the distal lower extremities, leading to surgical excision of the largest leg lesions. In addition, there were many pinpoint pits on both palms (bottom). A biopsy was submitted for review, which confirmed basal cell carcinomas on the scalp.
Validity of commercial serologic tests for dermatomyositis still questionable
, according to Jeffrey P. Callen, MD.

That’s because the validity and reproducibility of testing in commercial laboratories remain questionable, Dr. Callen, professor of medicine and chief of the division of dermatology at the University of Louisville, Ky., said during MedscapeLive’s annual Las Vegas Dermatology Seminar. “The testing in research laboratories is not widely available and the results are often delayed by weeks to months,” he said.
In addition, while the associations between antibody results and risks of malignancy or pulmonary disease are “statistically valid,” he said, “there are patients with disease in whom antibodies are not present and those without associated disease in whom the testing was positive.” For example, there are patients positive for anti–transition initiation factor (TIF)-1gamma but don’t have a malignancy, “and the ones with anti-MDA-5 tend to have pulmonary disease, but there are patients with anti-MDA-5 who don’t have pulmonary disease.”
Compared with patients with systemic lupus erythematosus, patients with dermatomyositis tend to have more itching and they tend of have fewer serologic abnormalities, such as anti-Ro/SS-A antibody, “but there is overlap,” Dr. Callen said. “The reason to differentiate cutaneous lupus erythematosus from dermatomyositis is because we think that patients who have amyopathic dermatomyositis still have an increased risk of having or developing an internal malignancy,” he added. Another differentiating point that is substantive is the presence of Gottron papules.
In a recent development related to antibody testing, researchers demonstrated that the IgG2 isotype of anti-TIF-1gamma antibodies is a biomarker of cancer and mortality in adult dermatomyositis.
According to population-based studies, about 20%-25% of dermatomyositis patients have had, have, or will develop a cancer (Lancet 2001;357: 96-100). Amyopathic dermatomyositis patients may also have cancer. Polymyositis patients generally have lower rates and their risk of subsequent malignancy is much closer to that of the general population, suggesting that the presence of the association is due to a “diagnostic suspicion bias,” Dr. Callen said.
A large-scale multicenter cohort study that set out to identify the risk factors and prognosis of patients with cancer-associated myositis found that ovarian cancer seems to be overrepresented. The only serologic abnormality that was statistically significant was anti-TIF-1gamma antibody (P less than .001). Patients with cancer-associated myositis also have less overall survival compared with those with non–cancer-associated myositis (P = .004), with malignancy being the primary cause of death (P less than .001).
In what is believed to be the largest study of its kind, Dr. Callen and colleagues retrospectively examined the prevalence of malignancy and screening practices in 400 dermatomyositis patients. Of the 400 patients, 48 (12%) had malignancies, and 21 cancers (40%) were diagnosed within 1 year of the dermatomyositis diagnosis. Both classic dermatomyositis and amyopathic dermatomyositis were associated with cancer, and 27 patients (6.8%) had a cancer at the time of diagnosis. Of those, 59% were asymptomatic; their cancers were discovered with CT scans, suggesting that “blind” screening is effective in identifying cancers in DM patients.
Dr. Callen’s malignancy evaluation includes chest x-ray, CT of the chest and abdomen, stool Hematest in all dermatomyositis patients; a mammogram, pelvic ultrasound and/or CT of the pelvis in women; and age, race or ethnicity-related testing. “I generally reevaluate patients annually for 3 years, because data from epidemiologic studies suggest that after 3 years [from the initial diagnosis], the rates of malignancy return toward normal,” he said. “I also evaluate any new symptom that might be suggestive of malignancy. The remaining issue is how to handle a patient in remission for several years, but who develops a relapse. What I do is perform another malignancy assessment.”
According to results from a meta-analysis of risk factors and systematic review of screening approaches, factors that increase malignancy risk include dermatomyositis subtype (risk ratio, 2.21), older age (weighted mean difference 11.19), male gender (RR, 1.53), dysphagia (RR, 2.09), cutaneous necrosis (RR, 2.73), and positive anti-TIF-1gamma (RR, 4.41).
Factors associated with a decreased risk of malignancy include polymyositis (RR, 0.49), clinically amyopathic dermatomyositis subtypes (RR, 0.44), Raynaud’s phenomenon (RR, 0.61), interstitial lung disease (RR, 0.49), very high serum creatine kinase (WMD –1189.96) or lactate dehydrogenase levels (WMD –336.53), and anti-Jo1 (RR, 0.45) or anti-EJ (RR, 0.17) positivity.
The analysis also found that CT scanning of the thorax, abdomen and pelvis appeared to yield a high proportion of underlying asymptomatic cancers. Limited evidence relating to the utility of tumor markers and 18F-FDG PET/CT was available.
As for treatment, the use of tofacitinib for cutaneous lesions of dermatomyositis has been suggested in various studies. In a recent open-label study of 10 patients with dermatomyositis who took extended release the JAK inhibitor tofacitinib 11 mg daily for 12 weeks, half experienced moderate improvement in disease activity, and the other half experienced minimal improvement. JAK inhibitors have been used in patients with juvenile dermatomyositis.
Dr. Callen’s treatment approach with dermatomyositis patients includes recommendations for sunscreens and protective clothing, plus assessment of vitamin D levels. “I will use topical emollients, corticosteroids, and calcineurin inhibitors,” he said. “Antimalarials might be used. I generally reach for methotrexate or mycophenolate mofetil relatively early. IVIG has also been studied.” Off-label therapies that have been used include dapsone, thalidomide, leflunomide, sirolimus, chlorambucil, etanercept, infliximab, rituximab, apremilast, tofacitinib, lenabasum, and low-dose naltrexone.
Dr. Callen disclosed that he is a consultant to Genentech and is a member of the safety monitoring committee for Principia Biopharma. He holds equity in Celgene, Pfizer, 3M, Johnson & Johnson, Merck, Abbott Laboratories, AbbVie, Procter & Gamble, Gilead, Allergen, and Amgen.
MedscapeLive and this news organization are owned by the same parent company.
, according to Jeffrey P. Callen, MD.
That’s because the validity and reproducibility of testing in commercial laboratories remain questionable, Dr. Callen, professor of medicine and chief of the division of dermatology at the University of Louisville, Ky., said during MedscapeLive’s annual Las Vegas Dermatology Seminar. “The testing in research laboratories is not widely available and the results are often delayed by weeks to months,” he said.
In addition, while the associations between antibody results and risks of malignancy or pulmonary disease are “statistically valid,” he said, “there are patients with disease in whom antibodies are not present and those without associated disease in whom the testing was positive.” For example, there are patients positive for anti–transition initiation factor (TIF)-1gamma but don’t have a malignancy, “and the ones with anti-MDA-5 tend to have pulmonary disease, but there are patients with anti-MDA-5 who don’t have pulmonary disease.”
Compared with patients with systemic lupus erythematosus, patients with dermatomyositis tend to have more itching and they tend of have fewer serologic abnormalities, such as anti-Ro/SS-A antibody, “but there is overlap,” Dr. Callen said. “The reason to differentiate cutaneous lupus erythematosus from dermatomyositis is because we think that patients who have amyopathic dermatomyositis still have an increased risk of having or developing an internal malignancy,” he added. Another differentiating point that is substantive is the presence of Gottron papules.
In a recent development related to antibody testing, researchers demonstrated that the IgG2 isotype of anti-TIF-1gamma antibodies is a biomarker of cancer and mortality in adult dermatomyositis.
According to population-based studies, about 20%-25% of dermatomyositis patients have had, have, or will develop a cancer (Lancet 2001;357: 96-100). Amyopathic dermatomyositis patients may also have cancer. Polymyositis patients generally have lower rates and their risk of subsequent malignancy is much closer to that of the general population, suggesting that the presence of the association is due to a “diagnostic suspicion bias,” Dr. Callen said.
A large-scale multicenter cohort study that set out to identify the risk factors and prognosis of patients with cancer-associated myositis found that ovarian cancer seems to be overrepresented. The only serologic abnormality that was statistically significant was anti-TIF-1gamma antibody (P less than .001). Patients with cancer-associated myositis also have less overall survival compared with those with non–cancer-associated myositis (P = .004), with malignancy being the primary cause of death (P less than .001).
In what is believed to be the largest study of its kind, Dr. Callen and colleagues retrospectively examined the prevalence of malignancy and screening practices in 400 dermatomyositis patients. Of the 400 patients, 48 (12%) had malignancies, and 21 cancers (40%) were diagnosed within 1 year of the dermatomyositis diagnosis. Both classic dermatomyositis and amyopathic dermatomyositis were associated with cancer, and 27 patients (6.8%) had a cancer at the time of diagnosis. Of those, 59% were asymptomatic; their cancers were discovered with CT scans, suggesting that “blind” screening is effective in identifying cancers in DM patients.
Dr. Callen’s malignancy evaluation includes chest x-ray, CT of the chest and abdomen, stool Hematest in all dermatomyositis patients; a mammogram, pelvic ultrasound and/or CT of the pelvis in women; and age, race or ethnicity-related testing. “I generally reevaluate patients annually for 3 years, because data from epidemiologic studies suggest that after 3 years [from the initial diagnosis], the rates of malignancy return toward normal,” he said. “I also evaluate any new symptom that might be suggestive of malignancy. The remaining issue is how to handle a patient in remission for several years, but who develops a relapse. What I do is perform another malignancy assessment.”
According to results from a meta-analysis of risk factors and systematic review of screening approaches, factors that increase malignancy risk include dermatomyositis subtype (risk ratio, 2.21), older age (weighted mean difference 11.19), male gender (RR, 1.53), dysphagia (RR, 2.09), cutaneous necrosis (RR, 2.73), and positive anti-TIF-1gamma (RR, 4.41).
Factors associated with a decreased risk of malignancy include polymyositis (RR, 0.49), clinically amyopathic dermatomyositis subtypes (RR, 0.44), Raynaud’s phenomenon (RR, 0.61), interstitial lung disease (RR, 0.49), very high serum creatine kinase (WMD –1189.96) or lactate dehydrogenase levels (WMD –336.53), and anti-Jo1 (RR, 0.45) or anti-EJ (RR, 0.17) positivity.
The analysis also found that CT scanning of the thorax, abdomen and pelvis appeared to yield a high proportion of underlying asymptomatic cancers. Limited evidence relating to the utility of tumor markers and 18F-FDG PET/CT was available.
As for treatment, the use of tofacitinib for cutaneous lesions of dermatomyositis has been suggested in various studies. In a recent open-label study of 10 patients with dermatomyositis who took extended release the JAK inhibitor tofacitinib 11 mg daily for 12 weeks, half experienced moderate improvement in disease activity, and the other half experienced minimal improvement. JAK inhibitors have been used in patients with juvenile dermatomyositis.
Dr. Callen’s treatment approach with dermatomyositis patients includes recommendations for sunscreens and protective clothing, plus assessment of vitamin D levels. “I will use topical emollients, corticosteroids, and calcineurin inhibitors,” he said. “Antimalarials might be used. I generally reach for methotrexate or mycophenolate mofetil relatively early. IVIG has also been studied.” Off-label therapies that have been used include dapsone, thalidomide, leflunomide, sirolimus, chlorambucil, etanercept, infliximab, rituximab, apremilast, tofacitinib, lenabasum, and low-dose naltrexone.
Dr. Callen disclosed that he is a consultant to Genentech and is a member of the safety monitoring committee for Principia Biopharma. He holds equity in Celgene, Pfizer, 3M, Johnson & Johnson, Merck, Abbott Laboratories, AbbVie, Procter & Gamble, Gilead, Allergen, and Amgen.
MedscapeLive and this news organization are owned by the same parent company.
, according to Jeffrey P. Callen, MD.
That’s because the validity and reproducibility of testing in commercial laboratories remain questionable, Dr. Callen, professor of medicine and chief of the division of dermatology at the University of Louisville, Ky., said during MedscapeLive’s annual Las Vegas Dermatology Seminar. “The testing in research laboratories is not widely available and the results are often delayed by weeks to months,” he said.
In addition, while the associations between antibody results and risks of malignancy or pulmonary disease are “statistically valid,” he said, “there are patients with disease in whom antibodies are not present and those without associated disease in whom the testing was positive.” For example, there are patients positive for anti–transition initiation factor (TIF)-1gamma but don’t have a malignancy, “and the ones with anti-MDA-5 tend to have pulmonary disease, but there are patients with anti-MDA-5 who don’t have pulmonary disease.”
Compared with patients with systemic lupus erythematosus, patients with dermatomyositis tend to have more itching and they tend of have fewer serologic abnormalities, such as anti-Ro/SS-A antibody, “but there is overlap,” Dr. Callen said. “The reason to differentiate cutaneous lupus erythematosus from dermatomyositis is because we think that patients who have amyopathic dermatomyositis still have an increased risk of having or developing an internal malignancy,” he added. Another differentiating point that is substantive is the presence of Gottron papules.
In a recent development related to antibody testing, researchers demonstrated that the IgG2 isotype of anti-TIF-1gamma antibodies is a biomarker of cancer and mortality in adult dermatomyositis.
According to population-based studies, about 20%-25% of dermatomyositis patients have had, have, or will develop a cancer (Lancet 2001;357: 96-100). Amyopathic dermatomyositis patients may also have cancer. Polymyositis patients generally have lower rates and their risk of subsequent malignancy is much closer to that of the general population, suggesting that the presence of the association is due to a “diagnostic suspicion bias,” Dr. Callen said.
A large-scale multicenter cohort study that set out to identify the risk factors and prognosis of patients with cancer-associated myositis found that ovarian cancer seems to be overrepresented. The only serologic abnormality that was statistically significant was anti-TIF-1gamma antibody (P less than .001). Patients with cancer-associated myositis also have less overall survival compared with those with non–cancer-associated myositis (P = .004), with malignancy being the primary cause of death (P less than .001).
In what is believed to be the largest study of its kind, Dr. Callen and colleagues retrospectively examined the prevalence of malignancy and screening practices in 400 dermatomyositis patients. Of the 400 patients, 48 (12%) had malignancies, and 21 cancers (40%) were diagnosed within 1 year of the dermatomyositis diagnosis. Both classic dermatomyositis and amyopathic dermatomyositis were associated with cancer, and 27 patients (6.8%) had a cancer at the time of diagnosis. Of those, 59% were asymptomatic; their cancers were discovered with CT scans, suggesting that “blind” screening is effective in identifying cancers in DM patients.
Dr. Callen’s malignancy evaluation includes chest x-ray, CT of the chest and abdomen, stool Hematest in all dermatomyositis patients; a mammogram, pelvic ultrasound and/or CT of the pelvis in women; and age, race or ethnicity-related testing. “I generally reevaluate patients annually for 3 years, because data from epidemiologic studies suggest that after 3 years [from the initial diagnosis], the rates of malignancy return toward normal,” he said. “I also evaluate any new symptom that might be suggestive of malignancy. The remaining issue is how to handle a patient in remission for several years, but who develops a relapse. What I do is perform another malignancy assessment.”
According to results from a meta-analysis of risk factors and systematic review of screening approaches, factors that increase malignancy risk include dermatomyositis subtype (risk ratio, 2.21), older age (weighted mean difference 11.19), male gender (RR, 1.53), dysphagia (RR, 2.09), cutaneous necrosis (RR, 2.73), and positive anti-TIF-1gamma (RR, 4.41).
Factors associated with a decreased risk of malignancy include polymyositis (RR, 0.49), clinically amyopathic dermatomyositis subtypes (RR, 0.44), Raynaud’s phenomenon (RR, 0.61), interstitial lung disease (RR, 0.49), very high serum creatine kinase (WMD –1189.96) or lactate dehydrogenase levels (WMD –336.53), and anti-Jo1 (RR, 0.45) or anti-EJ (RR, 0.17) positivity.
The analysis also found that CT scanning of the thorax, abdomen and pelvis appeared to yield a high proportion of underlying asymptomatic cancers. Limited evidence relating to the utility of tumor markers and 18F-FDG PET/CT was available.
As for treatment, the use of tofacitinib for cutaneous lesions of dermatomyositis has been suggested in various studies. In a recent open-label study of 10 patients with dermatomyositis who took extended release the JAK inhibitor tofacitinib 11 mg daily for 12 weeks, half experienced moderate improvement in disease activity, and the other half experienced minimal improvement. JAK inhibitors have been used in patients with juvenile dermatomyositis.
Dr. Callen’s treatment approach with dermatomyositis patients includes recommendations for sunscreens and protective clothing, plus assessment of vitamin D levels. “I will use topical emollients, corticosteroids, and calcineurin inhibitors,” he said. “Antimalarials might be used. I generally reach for methotrexate or mycophenolate mofetil relatively early. IVIG has also been studied.” Off-label therapies that have been used include dapsone, thalidomide, leflunomide, sirolimus, chlorambucil, etanercept, infliximab, rituximab, apremilast, tofacitinib, lenabasum, and low-dose naltrexone.
Dr. Callen disclosed that he is a consultant to Genentech and is a member of the safety monitoring committee for Principia Biopharma. He holds equity in Celgene, Pfizer, 3M, Johnson & Johnson, Merck, Abbott Laboratories, AbbVie, Procter & Gamble, Gilead, Allergen, and Amgen.
MedscapeLive and this news organization are owned by the same parent company.
FROM THE MEDSCAPELIVE LAS VEGAS DERMATOLOGY SEMINAR
FDA approves new interferon for polycythemia vera
, according to an agency press release.

Besremi has a longer half-life than do other pegylated interferon-alfas, allowing for dosing every 2 weeks instead of weekly. If red blood cell counts remain normal for a year, patients have the option of switching to once-monthly dosing. As with similar products, Besremi is self-administered as a subcutaneous injection.
It’s the first interferon approved in the United States specifically for polycythemia vera. Besremi is also approved for upfront therapy, unlike FDA’s first approval for the condition, the oral JAK inhibitor ruxolitinib (Jakafi), which is indicated only after hydroxyurea failure.
Taiwan-based maker PharmaEssentia said in another press release that it will roll Besremi out to the U.S. market in the coming weeks.
“As we begin working closely with the community to integrate this important treatment into clinical practice, we also continue to expand our scientific efforts to unlock the full potential of our pioneering molecule,” said Ko-Chung Lin, PhD, the company’s CEO.
As for unlocking the full potential, Besremi is under investigation for other interferon indications, including myelofibrosis, leukemia, and chronic hepatitis.
The FDA’s approval was based on results in 51 adults treated for an average of 5 years; 31 (61%) had a complete hematologic response, defined as a hematocrit below 45% with no phlebotomy for at least 2 months, plus normal platelet and white cell counts, normal spleen size, and no blood clots.
“Noninferiority to hydroxyurea regarding haematological response and normal spleen size was not shown at 12 months. However, response to ropeginterferon alfa-2b continued to increase over time with improved responses compared with hydroxyurea at 36 months,” investigators noted in an earlier report (Lancet Haematol. 2020 Mar;7[3]:e196-e208).
Besremi carries the same boxed warning as those of peginterferon alfa-2b (Pegintron) and peginterferon alfa-2a (Pegasys), which notes the risk of life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Related contraindications include severe depression and other psychiatric problems; liver impairment; serious or untreated autoimmune disease, and immunosuppression following organ transplant.
Influenza-like illness, arthralgia, fatigue, pruritis, nasopharyngitis, and musculoskeletal pain were the most common adverse events in studies, occurring in over 40% of subjects. Urinary tract infections, transient ischemic attacks, and depression were the most frequent serious complications, occurring in over 4%.
Labeling also notes the risk for fetal harm and the need for effective contraception.
Besremi was approved in Europe in 2019 and is approved in Taiwan and South Korea.
Polycythemia vera is a rare condition thought to be caused by acquired bone marrow stem cell mutations that trigger an overproduction of red blood cells. Patients are at increased risk of blood clots and emboli, and subsequent heart attacks, strokes, and other problems. There’s also the risk of transformation to secondary myelofibrosis or leukemia.
, according to an agency press release.
Besremi has a longer half-life than do other pegylated interferon-alfas, allowing for dosing every 2 weeks instead of weekly. If red blood cell counts remain normal for a year, patients have the option of switching to once-monthly dosing. As with similar products, Besremi is self-administered as a subcutaneous injection.
It’s the first interferon approved in the United States specifically for polycythemia vera. Besremi is also approved for upfront therapy, unlike FDA’s first approval for the condition, the oral JAK inhibitor ruxolitinib (Jakafi), which is indicated only after hydroxyurea failure.
Taiwan-based maker PharmaEssentia said in another press release that it will roll Besremi out to the U.S. market in the coming weeks.
“As we begin working closely with the community to integrate this important treatment into clinical practice, we also continue to expand our scientific efforts to unlock the full potential of our pioneering molecule,” said Ko-Chung Lin, PhD, the company’s CEO.
As for unlocking the full potential, Besremi is under investigation for other interferon indications, including myelofibrosis, leukemia, and chronic hepatitis.
The FDA’s approval was based on results in 51 adults treated for an average of 5 years; 31 (61%) had a complete hematologic response, defined as a hematocrit below 45% with no phlebotomy for at least 2 months, plus normal platelet and white cell counts, normal spleen size, and no blood clots.
“Noninferiority to hydroxyurea regarding haematological response and normal spleen size was not shown at 12 months. However, response to ropeginterferon alfa-2b continued to increase over time with improved responses compared with hydroxyurea at 36 months,” investigators noted in an earlier report (Lancet Haematol. 2020 Mar;7[3]:e196-e208).
Besremi carries the same boxed warning as those of peginterferon alfa-2b (Pegintron) and peginterferon alfa-2a (Pegasys), which notes the risk of life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Related contraindications include severe depression and other psychiatric problems; liver impairment; serious or untreated autoimmune disease, and immunosuppression following organ transplant.
Influenza-like illness, arthralgia, fatigue, pruritis, nasopharyngitis, and musculoskeletal pain were the most common adverse events in studies, occurring in over 40% of subjects. Urinary tract infections, transient ischemic attacks, and depression were the most frequent serious complications, occurring in over 4%.
Labeling also notes the risk for fetal harm and the need for effective contraception.
Besremi was approved in Europe in 2019 and is approved in Taiwan and South Korea.
Polycythemia vera is a rare condition thought to be caused by acquired bone marrow stem cell mutations that trigger an overproduction of red blood cells. Patients are at increased risk of blood clots and emboli, and subsequent heart attacks, strokes, and other problems. There’s also the risk of transformation to secondary myelofibrosis or leukemia.
, according to an agency press release.
Besremi has a longer half-life than do other pegylated interferon-alfas, allowing for dosing every 2 weeks instead of weekly. If red blood cell counts remain normal for a year, patients have the option of switching to once-monthly dosing. As with similar products, Besremi is self-administered as a subcutaneous injection.
It’s the first interferon approved in the United States specifically for polycythemia vera. Besremi is also approved for upfront therapy, unlike FDA’s first approval for the condition, the oral JAK inhibitor ruxolitinib (Jakafi), which is indicated only after hydroxyurea failure.
Taiwan-based maker PharmaEssentia said in another press release that it will roll Besremi out to the U.S. market in the coming weeks.
“As we begin working closely with the community to integrate this important treatment into clinical practice, we also continue to expand our scientific efforts to unlock the full potential of our pioneering molecule,” said Ko-Chung Lin, PhD, the company’s CEO.
As for unlocking the full potential, Besremi is under investigation for other interferon indications, including myelofibrosis, leukemia, and chronic hepatitis.
The FDA’s approval was based on results in 51 adults treated for an average of 5 years; 31 (61%) had a complete hematologic response, defined as a hematocrit below 45% with no phlebotomy for at least 2 months, plus normal platelet and white cell counts, normal spleen size, and no blood clots.
“Noninferiority to hydroxyurea regarding haematological response and normal spleen size was not shown at 12 months. However, response to ropeginterferon alfa-2b continued to increase over time with improved responses compared with hydroxyurea at 36 months,” investigators noted in an earlier report (Lancet Haematol. 2020 Mar;7[3]:e196-e208).
Besremi carries the same boxed warning as those of peginterferon alfa-2b (Pegintron) and peginterferon alfa-2a (Pegasys), which notes the risk of life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Related contraindications include severe depression and other psychiatric problems; liver impairment; serious or untreated autoimmune disease, and immunosuppression following organ transplant.
Influenza-like illness, arthralgia, fatigue, pruritis, nasopharyngitis, and musculoskeletal pain were the most common adverse events in studies, occurring in over 40% of subjects. Urinary tract infections, transient ischemic attacks, and depression were the most frequent serious complications, occurring in over 4%.
Labeling also notes the risk for fetal harm and the need for effective contraception.
Besremi was approved in Europe in 2019 and is approved in Taiwan and South Korea.
Polycythemia vera is a rare condition thought to be caused by acquired bone marrow stem cell mutations that trigger an overproduction of red blood cells. Patients are at increased risk of blood clots and emboli, and subsequent heart attacks, strokes, and other problems. There’s also the risk of transformation to secondary myelofibrosis or leukemia.
ADVOCATE: Avacopan shows renal benefits in ANCA vasculitis
Treatment of antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis and renal disease with the oral C5a receptor inhibitor avacopan (Tavneos, ChemoCentryx) provides significant recovery of kidney function, compared with prednisone, particularly in patients with severe kidney disease, novel research indicates.

The new analysis underscores that “the real value of avacopan is that we can now expect to get our patients steroid free,” said first author David R.W. Jayne, MD, a professor of clinical autoimmunity at the University of Cambridge (England), when presenting the findings at the American Society of Nephrology’s Kidney Week 2021.
“Whether or not we’re brave enough to initiate treatment without steroids, I think that will perhaps come with some patient experience,” he added.
The findings are from a subanalysis of renal effects in the phase 3 ADVOCATE trial, which was published in February 2021 in the New England Journal of Medicine and included 330 patients with ANCA-associated vasculitis.
The trial in large part led to the U.S. approval of avacopan by the Food and Drug Administration in October as an adjunctive treatment for adults with severe active ANCA-associated vasculitis in combination with standard therapy including glucocorticoids.
The approval was greeted with enthusiasm as suggesting a much-needed option to help reduce, or even potentially eliminate, the need for glucocorticoids and their side effects. Other agents included in treatment regimens for ANCA-associated vasculitis include cyclophosphamide and rituximab.
Dr. Jayne emphasized that, before avacopan, treatment options had been limited.
“There is nothing else new in the clinic apart from rituximab, which we have now been using for almost 20 years,” he said in an interview. “Avacopan is new, the mode of action is different from any drugs in use at the moment, and the speed of action is very quick.”
The need to more closely investigate the trial’s renal outcomes in this new analysis was important because the high mortality rates in ANCA-associated vasculitis – a rare systemic autoimmune disease causing overactivation of complement resulting in inflammation of small blood vessels – is largely driven by those with MPO and PR3 autoantibody renal vasculitis, Dr. Jayne explained.

Commenting on the study, J. Charles Jennette, MD, a professor of pathology and laboratory medicine and professor of medicine at the University of North Carolina at Chapel Hill, said the new findings on renal outcomes, such as proteinuria, may offer key insights on avacopan’s efficacy.
“To me, the most impressive outcome of the ADVOCATE Phase 3 trial was the more rapid reduction in hematuria and proteinuria with avacopan compared to conventional prednisone therapy,” he said in an interview.
Recovery of eGFR with avacopan best in those with severe renal disease
In the trial, patients with ANCA-associated vasculitis were randomized 1:1 to treatment with oral avacopan 30 mg twice daily or oral prednisone on a tapering schedule.
All patients also received background immunosuppression – about two-thirds received rituximab and a third received cyclophosphamide – followed by azathioprine.
The main study results showed similar rates of remission in both groups at week 26 and a superior remission rate with avacopan, in terms of sustained remission, at week 52 (65.7% vs. 54.9%; P < .001).
Approximately 80% of patients in the trial had renal involvement of ANCA vasculitis, the focus of the new analysis, and they had a baseline mean estimated glomerular filtration rate (eGFR) of 45 mL/min per 1.73 m2.
Among those with renal involvement, patients treated with avacopan had a significantly greater eGFR recovery, compared with the prednisone group at week 26 (P = .046) and week 52 (P < .029).
The strongest improvements were observed among patients with moderate to severe kidney damage, who had a mean eGFR of 21 mL/min per 1.73 m2 at baseline. Among those patients, the mean increase in eGFR was 13.7 mL/min per 1.73 m2 in the avacopan-treated group (n = 52) versus 8.2 mL/min per 1.73 m2 in the prednisone group (n = 48; P < .01) by week 52.
Improvements in urinary albumin:creatinine ratios (UACR) of as much as 40% were also observed in the avacopan group within the first 4 weeks of treatment, while no changes were observed in the same period in the prednisone group.
In other findings, the study also showed more rapid declines in proteinuria within 4 weeks in the avacopan group, and fewer patients had hematuria and there were greater reductions in MCP-1 in avacopan-treated patients at week 52, Dr. Jayne reported.
In terms of safety, there were no differences between the groups, with trends of fewer deaths and severe adverse events in the avacopan group.
“We found that the improved recovery of eGFR with avacopan was accentuated among those with more severe renal disease,” Dr. Jayne said.
He noted that, while the study’s aim was for the avacopan group to be steroid free, the patients received brief, reduced doses of about a third of the normal oral steroid dose early in the trial. However, using a Glucocorticoid Toxicity Index, the authors found those in the avacopan group did have fewer glucocorticoid-related adverse events.
Future issues to be examined include what happens when avacopan is discontinued and whether there will be a high relapse rate, Dr. Jayne noted.
Overall, however, “we anticipate that with longer-term follow-up, this better eGFR recovery will have a [favorable] effect on kidney failure and potentially mortality risk in these patients,” he concluded.
Targeted therapy is good for patients and doctors
Expanding upon his comments regarding the new drug, Dr. Jennette said it implies “that the C5a receptor inhibitor was targeting an event that blocks injury more quickly and effectively than prednisone.”
“This may be because prednisone has more complex pharmacodynamics and less targeted effects than a C5a receptor inhibitor,” he said.
Overall, the findings bode well for a potentially beneficial therapy, he added. “We have entered a new era of more targeted therapies, for example, targeted B-cell therapy using an anti-CD20 antibody, and targeted complement-mediated injury therapy using C5a receptor inhibitor.”
“The validation of this targeted therapy to block complement-mediated autoimmune inflammatory injury is another advance toward targeted precision therapy versus empirical therapy. This will be good for the doctors and good for the patients,” Dr. Jennette concluded.
The study was funded by ChemoCentryx. Dr. Jayne has reported receiving grants and/or consulting for AstraZeneca, ChemoCentryx, GlaxoSmithKline, MiroBio, Vifor, and Roche/Genentech. Dr. Jennette has received funding from ChemoCentryx for preclinical validation studies of avacopan in a mouse model of ANCA glomerulonephritis.
A version of this article first appeared on Medscape.com.
Treatment of antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis and renal disease with the oral C5a receptor inhibitor avacopan (Tavneos, ChemoCentryx) provides significant recovery of kidney function, compared with prednisone, particularly in patients with severe kidney disease, novel research indicates.
The new analysis underscores that “the real value of avacopan is that we can now expect to get our patients steroid free,” said first author David R.W. Jayne, MD, a professor of clinical autoimmunity at the University of Cambridge (England), when presenting the findings at the American Society of Nephrology’s Kidney Week 2021.
“Whether or not we’re brave enough to initiate treatment without steroids, I think that will perhaps come with some patient experience,” he added.
The findings are from a subanalysis of renal effects in the phase 3 ADVOCATE trial, which was published in February 2021 in the New England Journal of Medicine and included 330 patients with ANCA-associated vasculitis.
The trial in large part led to the U.S. approval of avacopan by the Food and Drug Administration in October as an adjunctive treatment for adults with severe active ANCA-associated vasculitis in combination with standard therapy including glucocorticoids.
The approval was greeted with enthusiasm as suggesting a much-needed option to help reduce, or even potentially eliminate, the need for glucocorticoids and their side effects. Other agents included in treatment regimens for ANCA-associated vasculitis include cyclophosphamide and rituximab.
Dr. Jayne emphasized that, before avacopan, treatment options had been limited.
“There is nothing else new in the clinic apart from rituximab, which we have now been using for almost 20 years,” he said in an interview. “Avacopan is new, the mode of action is different from any drugs in use at the moment, and the speed of action is very quick.”
The need to more closely investigate the trial’s renal outcomes in this new analysis was important because the high mortality rates in ANCA-associated vasculitis – a rare systemic autoimmune disease causing overactivation of complement resulting in inflammation of small blood vessels – is largely driven by those with MPO and PR3 autoantibody renal vasculitis, Dr. Jayne explained.
Commenting on the study, J. Charles Jennette, MD, a professor of pathology and laboratory medicine and professor of medicine at the University of North Carolina at Chapel Hill, said the new findings on renal outcomes, such as proteinuria, may offer key insights on avacopan’s efficacy.
“To me, the most impressive outcome of the ADVOCATE Phase 3 trial was the more rapid reduction in hematuria and proteinuria with avacopan compared to conventional prednisone therapy,” he said in an interview.
Recovery of eGFR with avacopan best in those with severe renal disease
In the trial, patients with ANCA-associated vasculitis were randomized 1:1 to treatment with oral avacopan 30 mg twice daily or oral prednisone on a tapering schedule.
All patients also received background immunosuppression – about two-thirds received rituximab and a third received cyclophosphamide – followed by azathioprine.
The main study results showed similar rates of remission in both groups at week 26 and a superior remission rate with avacopan, in terms of sustained remission, at week 52 (65.7% vs. 54.9%; P < .001).
Approximately 80% of patients in the trial had renal involvement of ANCA vasculitis, the focus of the new analysis, and they had a baseline mean estimated glomerular filtration rate (eGFR) of 45 mL/min per 1.73 m2.
Among those with renal involvement, patients treated with avacopan had a significantly greater eGFR recovery, compared with the prednisone group at week 26 (P = .046) and week 52 (P < .029).
The strongest improvements were observed among patients with moderate to severe kidney damage, who had a mean eGFR of 21 mL/min per 1.73 m2 at baseline. Among those patients, the mean increase in eGFR was 13.7 mL/min per 1.73 m2 in the avacopan-treated group (n = 52) versus 8.2 mL/min per 1.73 m2 in the prednisone group (n = 48; P < .01) by week 52.
Improvements in urinary albumin:creatinine ratios (UACR) of as much as 40% were also observed in the avacopan group within the first 4 weeks of treatment, while no changes were observed in the same period in the prednisone group.
In other findings, the study also showed more rapid declines in proteinuria within 4 weeks in the avacopan group, and fewer patients had hematuria and there were greater reductions in MCP-1 in avacopan-treated patients at week 52, Dr. Jayne reported.
In terms of safety, there were no differences between the groups, with trends of fewer deaths and severe adverse events in the avacopan group.
“We found that the improved recovery of eGFR with avacopan was accentuated among those with more severe renal disease,” Dr. Jayne said.
He noted that, while the study’s aim was for the avacopan group to be steroid free, the patients received brief, reduced doses of about a third of the normal oral steroid dose early in the trial. However, using a Glucocorticoid Toxicity Index, the authors found those in the avacopan group did have fewer glucocorticoid-related adverse events.
Future issues to be examined include what happens when avacopan is discontinued and whether there will be a high relapse rate, Dr. Jayne noted.
Overall, however, “we anticipate that with longer-term follow-up, this better eGFR recovery will have a [favorable] effect on kidney failure and potentially mortality risk in these patients,” he concluded.
Targeted therapy is good for patients and doctors
Expanding upon his comments regarding the new drug, Dr. Jennette said it implies “that the C5a receptor inhibitor was targeting an event that blocks injury more quickly and effectively than prednisone.”
“This may be because prednisone has more complex pharmacodynamics and less targeted effects than a C5a receptor inhibitor,” he said.
Overall, the findings bode well for a potentially beneficial therapy, he added. “We have entered a new era of more targeted therapies, for example, targeted B-cell therapy using an anti-CD20 antibody, and targeted complement-mediated injury therapy using C5a receptor inhibitor.”
“The validation of this targeted therapy to block complement-mediated autoimmune inflammatory injury is another advance toward targeted precision therapy versus empirical therapy. This will be good for the doctors and good for the patients,” Dr. Jennette concluded.
The study was funded by ChemoCentryx. Dr. Jayne has reported receiving grants and/or consulting for AstraZeneca, ChemoCentryx, GlaxoSmithKline, MiroBio, Vifor, and Roche/Genentech. Dr. Jennette has received funding from ChemoCentryx for preclinical validation studies of avacopan in a mouse model of ANCA glomerulonephritis.
A version of this article first appeared on Medscape.com.
Treatment of antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis and renal disease with the oral C5a receptor inhibitor avacopan (Tavneos, ChemoCentryx) provides significant recovery of kidney function, compared with prednisone, particularly in patients with severe kidney disease, novel research indicates.
The new analysis underscores that “the real value of avacopan is that we can now expect to get our patients steroid free,” said first author David R.W. Jayne, MD, a professor of clinical autoimmunity at the University of Cambridge (England), when presenting the findings at the American Society of Nephrology’s Kidney Week 2021.
“Whether or not we’re brave enough to initiate treatment without steroids, I think that will perhaps come with some patient experience,” he added.
The findings are from a subanalysis of renal effects in the phase 3 ADVOCATE trial, which was published in February 2021 in the New England Journal of Medicine and included 330 patients with ANCA-associated vasculitis.
The trial in large part led to the U.S. approval of avacopan by the Food and Drug Administration in October as an adjunctive treatment for adults with severe active ANCA-associated vasculitis in combination with standard therapy including glucocorticoids.
The approval was greeted with enthusiasm as suggesting a much-needed option to help reduce, or even potentially eliminate, the need for glucocorticoids and their side effects. Other agents included in treatment regimens for ANCA-associated vasculitis include cyclophosphamide and rituximab.
Dr. Jayne emphasized that, before avacopan, treatment options had been limited.
“There is nothing else new in the clinic apart from rituximab, which we have now been using for almost 20 years,” he said in an interview. “Avacopan is new, the mode of action is different from any drugs in use at the moment, and the speed of action is very quick.”
The need to more closely investigate the trial’s renal outcomes in this new analysis was important because the high mortality rates in ANCA-associated vasculitis – a rare systemic autoimmune disease causing overactivation of complement resulting in inflammation of small blood vessels – is largely driven by those with MPO and PR3 autoantibody renal vasculitis, Dr. Jayne explained.
Commenting on the study, J. Charles Jennette, MD, a professor of pathology and laboratory medicine and professor of medicine at the University of North Carolina at Chapel Hill, said the new findings on renal outcomes, such as proteinuria, may offer key insights on avacopan’s efficacy.
“To me, the most impressive outcome of the ADVOCATE Phase 3 trial was the more rapid reduction in hematuria and proteinuria with avacopan compared to conventional prednisone therapy,” he said in an interview.
Recovery of eGFR with avacopan best in those with severe renal disease
In the trial, patients with ANCA-associated vasculitis were randomized 1:1 to treatment with oral avacopan 30 mg twice daily or oral prednisone on a tapering schedule.
All patients also received background immunosuppression – about two-thirds received rituximab and a third received cyclophosphamide – followed by azathioprine.
The main study results showed similar rates of remission in both groups at week 26 and a superior remission rate with avacopan, in terms of sustained remission, at week 52 (65.7% vs. 54.9%; P < .001).
Approximately 80% of patients in the trial had renal involvement of ANCA vasculitis, the focus of the new analysis, and they had a baseline mean estimated glomerular filtration rate (eGFR) of 45 mL/min per 1.73 m2.
Among those with renal involvement, patients treated with avacopan had a significantly greater eGFR recovery, compared with the prednisone group at week 26 (P = .046) and week 52 (P < .029).
The strongest improvements were observed among patients with moderate to severe kidney damage, who had a mean eGFR of 21 mL/min per 1.73 m2 at baseline. Among those patients, the mean increase in eGFR was 13.7 mL/min per 1.73 m2 in the avacopan-treated group (n = 52) versus 8.2 mL/min per 1.73 m2 in the prednisone group (n = 48; P < .01) by week 52.
Improvements in urinary albumin:creatinine ratios (UACR) of as much as 40% were also observed in the avacopan group within the first 4 weeks of treatment, while no changes were observed in the same period in the prednisone group.
In other findings, the study also showed more rapid declines in proteinuria within 4 weeks in the avacopan group, and fewer patients had hematuria and there were greater reductions in MCP-1 in avacopan-treated patients at week 52, Dr. Jayne reported.
In terms of safety, there were no differences between the groups, with trends of fewer deaths and severe adverse events in the avacopan group.
“We found that the improved recovery of eGFR with avacopan was accentuated among those with more severe renal disease,” Dr. Jayne said.
He noted that, while the study’s aim was for the avacopan group to be steroid free, the patients received brief, reduced doses of about a third of the normal oral steroid dose early in the trial. However, using a Glucocorticoid Toxicity Index, the authors found those in the avacopan group did have fewer glucocorticoid-related adverse events.
Future issues to be examined include what happens when avacopan is discontinued and whether there will be a high relapse rate, Dr. Jayne noted.
Overall, however, “we anticipate that with longer-term follow-up, this better eGFR recovery will have a [favorable] effect on kidney failure and potentially mortality risk in these patients,” he concluded.
Targeted therapy is good for patients and doctors
Expanding upon his comments regarding the new drug, Dr. Jennette said it implies “that the C5a receptor inhibitor was targeting an event that blocks injury more quickly and effectively than prednisone.”
“This may be because prednisone has more complex pharmacodynamics and less targeted effects than a C5a receptor inhibitor,” he said.
Overall, the findings bode well for a potentially beneficial therapy, he added. “We have entered a new era of more targeted therapies, for example, targeted B-cell therapy using an anti-CD20 antibody, and targeted complement-mediated injury therapy using C5a receptor inhibitor.”
“The validation of this targeted therapy to block complement-mediated autoimmune inflammatory injury is another advance toward targeted precision therapy versus empirical therapy. This will be good for the doctors and good for the patients,” Dr. Jennette concluded.
The study was funded by ChemoCentryx. Dr. Jayne has reported receiving grants and/or consulting for AstraZeneca, ChemoCentryx, GlaxoSmithKline, MiroBio, Vifor, and Roche/Genentech. Dr. Jennette has received funding from ChemoCentryx for preclinical validation studies of avacopan in a mouse model of ANCA glomerulonephritis.
A version of this article first appeared on Medscape.com.
FROM KIDNEY WEEK 2021
Drug combo at outset of polyarticular JIA benefits patients most
Initiating treatment of polyarticular juvenile idiopathic arthritis (polyJIA) with both a conventional synthetic disease-modifying antirheumatic drug and a biologic DMARD resulted in more patients achieving clinical inactive disease 2 years later than did starting with only a csDMARD and stepping up to a biologic, according to data presented at the virtual annual meeting of the American College of Rheumatology.

“The 24-month results support the 12-month primary results that suggested that the early-combination group was superior and that, at 24 months, more early combination CTP [consensus treatment plan] patients achieve CID [clinical inactive disease], compared to step up,” Yukiko Kimura, MD, division chief of pediatric rheumatology at HMH Hackensack (N.J.) University Medical Center, told attendees. “This suggests that starting biologics early in polyJIA may lead to better long-term outcomes in many patients.”
Dr. Kimura noted that polyarticular JIA patients are already at risk for poor outcomes, and initial therapy can especially impact outcomes. Further, little evidence exists to suggest when the best time is to start biologics, a gap this study aimed to address.
Diane Brown, MD, PhD, a pediatric rheumatologist at Children’s Hospital Los Angeles who was not involved in the study, was pleased to see the results, which she said support her own preferences and practice patterns.
“Starting sooner with combination therapy, taking advantage of the advances with biologics and our long history with methotrexate at the same time, gives better outcomes for the long run,” Dr. Brown said in an interview. “Having studies like this to back up my own recommendations can be very powerful when talking to families, and it is absolutely invaluable when battling with insurance companies who always want you to take the cheapest road.”
Study details
The findings were an update of 12-month results in the CARRA STOP-JIA study that enrolled 400 untreated patients with polyJIA and compared three Childhood Arthritis and Rheumatology Research Alliance (CARRA) CTPs. Overall, 49.5% of participants received biologics within 3 months of starting the study. For these updated results, 275 participants had complete data at 24 months for the three CTPs:
- A step-up group of 177 patients who started therapy with a csDMARD and added a biologic if needed at least 3 months later
- An early-combination group of 73 patients who started therapy with a csDMARD and biologic together
- A biologic-first group of 25 patients who started with biologic monotherapy, adding a csDMARD only if needed at least 3 months later.
The primary outcome was the percentage of participants who reached CID without taking glucocorticoids at 24 months. Since the participants were not randomized, the researchers made adjustments to account for baseline differences between the groups, including differences in JIA categories, number of active joints, physician global assessment of disease activity, and the clinical Juvenile Arthritis Disease Activity Score based on 10 joints (cJADAS10).
At 24 months in an intention to treat analysis, 59.4% of the early-combination group had achieved CID, compared with 48% of the biologic-first group and 40.1% of the step-up group (P = .009 for early combination vs. step up). All three groups had improved since the 12-month time point, when 37% of the early-combination group, 24% of the biologic-first group, and 32% of the step-up group had reached CID.
There were no significant differences between the groups in secondary outcomes of achieving cJADAS10 inactive disease of 2.5 or less or 70% improvement in pediatric ACR response criteria at 24 months. All groups improved in PROMIS pain interference or mobility measures from baseline. Most of the 17 severe adverse events were infections.
Moving from step-up therapy to early-combination treatment
Dr. Brown said that she spent many years in her practice using the step-up therapy because it was difficult to get insurance companies to pay for biologics without first showing that methotrexate was insufficient.
”But methotrexate takes so long to control the disease that you need a lot of steroids, with all of their side effects, at least temporarily, or you must simply accept a longer period of active and symptomatic disease before you get to that desired state of clinically inactive disease,” Dr. Brown said. “And during that time, you can be accumulating what may be permanent damage to joints, as well as increase in risk of contractures and deconditioning for that child who is too uncomfortable to move and exercise and play normally.”
Dr. Brown is also wary of using a biologic as an initial therapy by itself because the actions of biologics are so specific. ”I like to back up the powerful, rapid, and specific actions of a biologic with the broader, if slower, action of methotrexate to minimize chances that the immune system is going to find a way around blockade of a single cytokine by your biologic,” she said.
While patient preference will also play a role in what CTP patients with polyJIA start with, Dr. Brown said that she believes more medication upfront can result in less medication and better outcomes in the long run, as the findings of this study suggest. The results here are helpful when speaking with families who are anxious about “so much medicine” or “such powerful medicines,” she said. ”I hope it will also help ease the fears of other providers who share the same concerns about ‘so much medicine.’ ”
The study’s biggest limitation is not being a randomized, controlled trial, but Dr. Brown said the researchers demonstrated effectively that the disease burden remains similar across the groups at baseline.
”It would also be useful to have a clear breakdown of adverse events and opportunistic infections because an excess of opportunistic infections would be a key concern with early combination therapy,” she said, although she added that the study overall was a ”beautiful example of the value of registry data.”
Dr. Kimura emphasized that polyJIA remains a challenging disease to treat, with 40%-60% of participants not reaching CID at 24 months. The registry follow-up will continue for up to 10 years to hopefully provide more information about longer-term outcomes from different treatments.
The research was funded by a grant from Genentech to CARRA. Dr. Kimura reported royalties from UpToDate and salary support from CARRA. Dr. Brown had no disclosures.
Initiating treatment of polyarticular juvenile idiopathic arthritis (polyJIA) with both a conventional synthetic disease-modifying antirheumatic drug and a biologic DMARD resulted in more patients achieving clinical inactive disease 2 years later than did starting with only a csDMARD and stepping up to a biologic, according to data presented at the virtual annual meeting of the American College of Rheumatology.
“The 24-month results support the 12-month primary results that suggested that the early-combination group was superior and that, at 24 months, more early combination CTP [consensus treatment plan] patients achieve CID [clinical inactive disease], compared to step up,” Yukiko Kimura, MD, division chief of pediatric rheumatology at HMH Hackensack (N.J.) University Medical Center, told attendees. “This suggests that starting biologics early in polyJIA may lead to better long-term outcomes in many patients.”
Dr. Kimura noted that polyarticular JIA patients are already at risk for poor outcomes, and initial therapy can especially impact outcomes. Further, little evidence exists to suggest when the best time is to start biologics, a gap this study aimed to address.
Diane Brown, MD, PhD, a pediatric rheumatologist at Children’s Hospital Los Angeles who was not involved in the study, was pleased to see the results, which she said support her own preferences and practice patterns.
“Starting sooner with combination therapy, taking advantage of the advances with biologics and our long history with methotrexate at the same time, gives better outcomes for the long run,” Dr. Brown said in an interview. “Having studies like this to back up my own recommendations can be very powerful when talking to families, and it is absolutely invaluable when battling with insurance companies who always want you to take the cheapest road.”
Study details
The findings were an update of 12-month results in the CARRA STOP-JIA study that enrolled 400 untreated patients with polyJIA and compared three Childhood Arthritis and Rheumatology Research Alliance (CARRA) CTPs. Overall, 49.5% of participants received biologics within 3 months of starting the study. For these updated results, 275 participants had complete data at 24 months for the three CTPs:
- A step-up group of 177 patients who started therapy with a csDMARD and added a biologic if needed at least 3 months later
- An early-combination group of 73 patients who started therapy with a csDMARD and biologic together
- A biologic-first group of 25 patients who started with biologic monotherapy, adding a csDMARD only if needed at least 3 months later.
The primary outcome was the percentage of participants who reached CID without taking glucocorticoids at 24 months. Since the participants were not randomized, the researchers made adjustments to account for baseline differences between the groups, including differences in JIA categories, number of active joints, physician global assessment of disease activity, and the clinical Juvenile Arthritis Disease Activity Score based on 10 joints (cJADAS10).
At 24 months in an intention to treat analysis, 59.4% of the early-combination group had achieved CID, compared with 48% of the biologic-first group and 40.1% of the step-up group (P = .009 for early combination vs. step up). All three groups had improved since the 12-month time point, when 37% of the early-combination group, 24% of the biologic-first group, and 32% of the step-up group had reached CID.
There were no significant differences between the groups in secondary outcomes of achieving cJADAS10 inactive disease of 2.5 or less or 70% improvement in pediatric ACR response criteria at 24 months. All groups improved in PROMIS pain interference or mobility measures from baseline. Most of the 17 severe adverse events were infections.
Moving from step-up therapy to early-combination treatment
Dr. Brown said that she spent many years in her practice using the step-up therapy because it was difficult to get insurance companies to pay for biologics without first showing that methotrexate was insufficient.
”But methotrexate takes so long to control the disease that you need a lot of steroids, with all of their side effects, at least temporarily, or you must simply accept a longer period of active and symptomatic disease before you get to that desired state of clinically inactive disease,” Dr. Brown said. “And during that time, you can be accumulating what may be permanent damage to joints, as well as increase in risk of contractures and deconditioning for that child who is too uncomfortable to move and exercise and play normally.”
Dr. Brown is also wary of using a biologic as an initial therapy by itself because the actions of biologics are so specific. ”I like to back up the powerful, rapid, and specific actions of a biologic with the broader, if slower, action of methotrexate to minimize chances that the immune system is going to find a way around blockade of a single cytokine by your biologic,” she said.
While patient preference will also play a role in what CTP patients with polyJIA start with, Dr. Brown said that she believes more medication upfront can result in less medication and better outcomes in the long run, as the findings of this study suggest. The results here are helpful when speaking with families who are anxious about “so much medicine” or “such powerful medicines,” she said. ”I hope it will also help ease the fears of other providers who share the same concerns about ‘so much medicine.’ ”
The study’s biggest limitation is not being a randomized, controlled trial, but Dr. Brown said the researchers demonstrated effectively that the disease burden remains similar across the groups at baseline.
”It would also be useful to have a clear breakdown of adverse events and opportunistic infections because an excess of opportunistic infections would be a key concern with early combination therapy,” she said, although she added that the study overall was a ”beautiful example of the value of registry data.”
Dr. Kimura emphasized that polyJIA remains a challenging disease to treat, with 40%-60% of participants not reaching CID at 24 months. The registry follow-up will continue for up to 10 years to hopefully provide more information about longer-term outcomes from different treatments.
The research was funded by a grant from Genentech to CARRA. Dr. Kimura reported royalties from UpToDate and salary support from CARRA. Dr. Brown had no disclosures.
Initiating treatment of polyarticular juvenile idiopathic arthritis (polyJIA) with both a conventional synthetic disease-modifying antirheumatic drug and a biologic DMARD resulted in more patients achieving clinical inactive disease 2 years later than did starting with only a csDMARD and stepping up to a biologic, according to data presented at the virtual annual meeting of the American College of Rheumatology.
“The 24-month results support the 12-month primary results that suggested that the early-combination group was superior and that, at 24 months, more early combination CTP [consensus treatment plan] patients achieve CID [clinical inactive disease], compared to step up,” Yukiko Kimura, MD, division chief of pediatric rheumatology at HMH Hackensack (N.J.) University Medical Center, told attendees. “This suggests that starting biologics early in polyJIA may lead to better long-term outcomes in many patients.”
Dr. Kimura noted that polyarticular JIA patients are already at risk for poor outcomes, and initial therapy can especially impact outcomes. Further, little evidence exists to suggest when the best time is to start biologics, a gap this study aimed to address.
Diane Brown, MD, PhD, a pediatric rheumatologist at Children’s Hospital Los Angeles who was not involved in the study, was pleased to see the results, which she said support her own preferences and practice patterns.
“Starting sooner with combination therapy, taking advantage of the advances with biologics and our long history with methotrexate at the same time, gives better outcomes for the long run,” Dr. Brown said in an interview. “Having studies like this to back up my own recommendations can be very powerful when talking to families, and it is absolutely invaluable when battling with insurance companies who always want you to take the cheapest road.”
Study details
The findings were an update of 12-month results in the CARRA STOP-JIA study that enrolled 400 untreated patients with polyJIA and compared three Childhood Arthritis and Rheumatology Research Alliance (CARRA) CTPs. Overall, 49.5% of participants received biologics within 3 months of starting the study. For these updated results, 275 participants had complete data at 24 months for the three CTPs:
- A step-up group of 177 patients who started therapy with a csDMARD and added a biologic if needed at least 3 months later
- An early-combination group of 73 patients who started therapy with a csDMARD and biologic together
- A biologic-first group of 25 patients who started with biologic monotherapy, adding a csDMARD only if needed at least 3 months later.
The primary outcome was the percentage of participants who reached CID without taking glucocorticoids at 24 months. Since the participants were not randomized, the researchers made adjustments to account for baseline differences between the groups, including differences in JIA categories, number of active joints, physician global assessment of disease activity, and the clinical Juvenile Arthritis Disease Activity Score based on 10 joints (cJADAS10).
At 24 months in an intention to treat analysis, 59.4% of the early-combination group had achieved CID, compared with 48% of the biologic-first group and 40.1% of the step-up group (P = .009 for early combination vs. step up). All three groups had improved since the 12-month time point, when 37% of the early-combination group, 24% of the biologic-first group, and 32% of the step-up group had reached CID.
There were no significant differences between the groups in secondary outcomes of achieving cJADAS10 inactive disease of 2.5 or less or 70% improvement in pediatric ACR response criteria at 24 months. All groups improved in PROMIS pain interference or mobility measures from baseline. Most of the 17 severe adverse events were infections.
Moving from step-up therapy to early-combination treatment
Dr. Brown said that she spent many years in her practice using the step-up therapy because it was difficult to get insurance companies to pay for biologics without first showing that methotrexate was insufficient.
”But methotrexate takes so long to control the disease that you need a lot of steroids, with all of their side effects, at least temporarily, or you must simply accept a longer period of active and symptomatic disease before you get to that desired state of clinically inactive disease,” Dr. Brown said. “And during that time, you can be accumulating what may be permanent damage to joints, as well as increase in risk of contractures and deconditioning for that child who is too uncomfortable to move and exercise and play normally.”
Dr. Brown is also wary of using a biologic as an initial therapy by itself because the actions of biologics are so specific. ”I like to back up the powerful, rapid, and specific actions of a biologic with the broader, if slower, action of methotrexate to minimize chances that the immune system is going to find a way around blockade of a single cytokine by your biologic,” she said.
While patient preference will also play a role in what CTP patients with polyJIA start with, Dr. Brown said that she believes more medication upfront can result in less medication and better outcomes in the long run, as the findings of this study suggest. The results here are helpful when speaking with families who are anxious about “so much medicine” or “such powerful medicines,” she said. ”I hope it will also help ease the fears of other providers who share the same concerns about ‘so much medicine.’ ”
The study’s biggest limitation is not being a randomized, controlled trial, but Dr. Brown said the researchers demonstrated effectively that the disease burden remains similar across the groups at baseline.
”It would also be useful to have a clear breakdown of adverse events and opportunistic infections because an excess of opportunistic infections would be a key concern with early combination therapy,” she said, although she added that the study overall was a ”beautiful example of the value of registry data.”
Dr. Kimura emphasized that polyJIA remains a challenging disease to treat, with 40%-60% of participants not reaching CID at 24 months. The registry follow-up will continue for up to 10 years to hopefully provide more information about longer-term outcomes from different treatments.
The research was funded by a grant from Genentech to CARRA. Dr. Kimura reported royalties from UpToDate and salary support from CARRA. Dr. Brown had no disclosures.
FROM ACR 2021