User login
More biosimilars reach the market in efforts to improve access and cut costs
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Finding your practice home base
As summer winds down and we begin to gear up to return to school or work, I was thinking about new and returning hem-onc residents, fellows, and young attendings and a question I routinely get from them: what should I do next in my career? I always answer by holding up 3 fingers and telling them that they can practice 1, at a university hospital; 2, at a university teaching affiliate; or 3, at a community hospital or practice with a little or no university affiliation. These days, trainees in hematology-oncology are often advised to be highly specialty-specific when they plan their long-term careers and to focus on a particular cancer or hematologic disorder. That is fine if you want to remain in an academic or university-based practice, but not if community practice is your preference. So, what are the differences among these 3 options?
Option 1, to remain in a university setting where you can be highly focused and specialized in a single narrowly defined area, could be satisfying, but keep in mind that the institution expects results! You will be carefully monitored for research output and teaching and administration commitments, and your interaction with patients could add up to less than 50% of your time. Publication and grant renewal will also play a role and therefore take up your time.
If you are considering option 2 – to work at a university teaching affiliate hospital – you need to bear in mind that you likely will see a patient population with a much broader range of diagnoses than would be the case with the first option. Patient care for option 2 will take up more than 50% of your time, so it might be a little more challenging to stay current, but perhaps more refreshing if you enjoy contact with patients. Teaching, research, and administration will surely be available, and publication and grant renewal will play as big or small a role as you want.
Option 3 would be to join a community hospital or practice where the primary focus is on patient care and the diagnoses will span the hematology and oncology spectrum. This type of practice can be very demanding of one’s time, but as rewarding as the other options, especially if you value contact with patients. With this option, one is more likely to practice as a generalist, perhaps with an emphasis in one of the hem-onc specialties, but able to treat a cluster of different types of cancer as well.
I always advise trainees to be sure they ask physicians practicing in each of these options to give examples of what their best and worst days are like so that they can get some idea of what the daily humdrum and challenges would encompass. What did I choose? I have always gone with option 2 and have been very happy in that setting.
In this issue…
More biosimilars head our way. Turning to the current issue of the journal, on page e181, Dr Jane de Lartigue discusses 2 new biosimilars recently approved by the United States Food and Drug Administration (FDA) – epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for chemotherapy-induced anemia (CIA), and pegfilgrastim-jmdb (Fulphila; Mylan and Biocon) for prevention of febrile neutropenia. As Dr de Lartigue notes, biosimilars are copies of FDA-approved biologic drugs that cannot be identical to the reference drug but demonstrate a high similarity to it. In this case, the reference drug for epoetin alfa-epbx is epoetin alfa (Epogen/Procrit, Amgen) and for pegfilgrastim-jmdb, it is pegfilgrastim (Neulasta, Amgen). As the reference drugs’ patents expire, biosimilars are being developed to increase competition in the marketplace in an effort to reduce costs and improve patient access to these therapies. Indeed, the FDA is working to streamline the biosimilar approval process to facilitate that access.
Reading this article got me thinking about something I often have to consider in the course of my work: transfusion versus erythropoiesis-stimulating agents (ESAs)? Recombinant erythropoietin drugs such as the biosimilar, epoetin alfa-epbx, and its reference drug are grouped together as ESAs, and have been used to treat CIA since the late 1980s. However, there were a few trials that used higher-dose ESA or set high hemoglobin targets, and their findings suggested that ESAs may shorten survival in patients with cancer or increase tumor growth, or both. The use of ESAs took a nosedive after the 2007 decision by the FDA’s Oncologic Drugs Advisory Committee to rein in their use for a hard start of ESA treatment at less than 10 g/dL hemoglobin, and not higher. Subsequent trials addressed the concerns about survival and tumor growth. A meta-analysis of 60 randomized, placebo-controlled trials of ESAs in CIA found that there was no difference in overall survival between the study and control groups.1 Likewise, findings from an FDA-mandated trial with epoetin alfa (Procrit) in patients with metastatic breast cancer have reported that there was no significant difference in overall survival between the study and control groups.2 The results of a second FDA-mandated trial with darbepoetin alfa (Aranesp, Amgen) in patients with metastatic lung cancer are expected to be released soon. The FDA lifted the ESA Risk Evaluation and Mitigation Strategy based on those findings. However, many practitioners, both young and old, continue to shy away from using ESAs because of the FDA black box warning that remains in place despite the latest data.3The use of transfusion ticked up reciprocally with the decline in ESA use, but perhaps we should re-evaluate the use of these agents in our practice, especially now that the less costly, equally safe and effective biosimilars are becoming available and we have the new survival data. Transfusions are time consuming and have side effects, including allergic reaction and infection risk, whereas ESAs are easily administered by injection, which patients might find preferable.
Malignancies in patients with HIV-AIDS. On page e188, Koppaka and colleagues report on a study in India of the patterns of malignancies in patients with HIV-AIDS. I began my career just as the first reports of what became known as HIV-AIDS emerged, and we were all mystified by what was killing these patients and the curious hematologic and oncologic problems they developed. Back then, the patients were profoundly immunosuppressed, and the immunosuppression cancers of non-Hodgkin lymphoma, usually higher grade, and Kaposi sarcoma were most prevalent and today are collectively labeled AIDS-defining malignancies (ADMs).
Fast forward to present day, and we have extremely effective antiretroviral therapies that have resulted in a significant reduction in mortality among HIV-infected individuals who are now living long enough to get what we call non–AIDS-defining malignancies (NADMs) such as anal or cervical cancers, hepatoma (hepatocellular carcinoma), Hodgkin lymphoma, and lung cancer. Of note is that these NADMs are all highly viral associated, with anal and cervical cancers linked to infection with the human papillomavirus; hepatoma linked to the hepatitis B/C viruses; Hodgkin lymphoma to the Epstein-Barr virus; and lung cancer, possibly also HPV. Fortunately, these days we can use standard-dose chemoradiation therapy for all HIV-related cancers because the patients’ immune systems are much better reconstituted and the modern-day antiretroviral therapies have much less drug–drug interaction thanks to the advent of the integrase inhibitors. The researchers give an excellent breakdown of the occurrence of these malignancies, as well as an analysis of the correlation between CD4 counts and the different malignancies.
Immunotherapy-related side effects in the ED. What happens when our patients who are on immunotherapy end up in the emergency department (ED) with therapy-related symptoms? And what can the treating oncologist do to help the ED physician achieve the best possible outcome for the patient? I spoke to Dr Maura Sammon, an ED physician, about some of the more common of these side effects – lung, gastrointestinal, rash, and endocrine-related problems – and she describes in detail how physicians in the ED would triage and treat the patient. Dr Sammon also emphasizes the importance of communication: first, between the treating oncologist and patient, about the differences between chemotherapy and immunotherapy; and second, between the ED physician and the treating oncologist as soon as possible after the patient has presented to ensure a good outcome. The interview is part of The JCSO Interview series. It is jam-packed with useful, how-to information, and you can read a transcript of it on page e216 of this issue, or you can listen to it online.4
We round off the issue with a selection of Case Reports (pp. e200-e209), an original report on the characteristics of urgent palliative cancer care consultations encountered by radiation oncologists (p. e193), and a New Therapies feature, also by Dr de Lartigue, focusing on the rarity and complexities of sarcomas (p. e210).
Those are my dog-day-of-summer thoughts as we head toward another Labor Day and a new academic year. Since we are all online now, we encourage you to listen to my bimonthly podcast of each issue on our website at www.jcso-online.com, and of course, follow us on Twitter (@jcs_onc) and Instagram (@jcsoncology) and like us on Facebook.
1. Glaspy J, Crawford J, Vansteenkiste J, et al. Erythropoiesis-stimulating agents in oncology: a study-level meta-analysis of survival and other safety outcomes. Br J Cancer. 2010;102(2):301-315.
2. Leyland-Jones B, Bondarenko I, Nemsadze G, et al. A randomized, open-label, multicenter, phase III study of epoetin alfa versus best standard of care in anemic patients with metastatic breast cancer receiving standard chemotherapy. J Clin Oncol. 2016;34:1197-1207.
3. US Food and Drug Administration release. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Last updated April 13, 2017. Accessed August 20, 2018.
4. Henry D, Sammon M. Treating immunotherapy-related AEs in the emergency department [Audio]. https://www.mdedge.com/jcso/article/171966/patient-survivor-care/treating-immunotherapy-related-aes-emergency-department. Published August 6, 2018.
As summer winds down and we begin to gear up to return to school or work, I was thinking about new and returning hem-onc residents, fellows, and young attendings and a question I routinely get from them: what should I do next in my career? I always answer by holding up 3 fingers and telling them that they can practice 1, at a university hospital; 2, at a university teaching affiliate; or 3, at a community hospital or practice with a little or no university affiliation. These days, trainees in hematology-oncology are often advised to be highly specialty-specific when they plan their long-term careers and to focus on a particular cancer or hematologic disorder. That is fine if you want to remain in an academic or university-based practice, but not if community practice is your preference. So, what are the differences among these 3 options?
Option 1, to remain in a university setting where you can be highly focused and specialized in a single narrowly defined area, could be satisfying, but keep in mind that the institution expects results! You will be carefully monitored for research output and teaching and administration commitments, and your interaction with patients could add up to less than 50% of your time. Publication and grant renewal will also play a role and therefore take up your time.
If you are considering option 2 – to work at a university teaching affiliate hospital – you need to bear in mind that you likely will see a patient population with a much broader range of diagnoses than would be the case with the first option. Patient care for option 2 will take up more than 50% of your time, so it might be a little more challenging to stay current, but perhaps more refreshing if you enjoy contact with patients. Teaching, research, and administration will surely be available, and publication and grant renewal will play as big or small a role as you want.
Option 3 would be to join a community hospital or practice where the primary focus is on patient care and the diagnoses will span the hematology and oncology spectrum. This type of practice can be very demanding of one’s time, but as rewarding as the other options, especially if you value contact with patients. With this option, one is more likely to practice as a generalist, perhaps with an emphasis in one of the hem-onc specialties, but able to treat a cluster of different types of cancer as well.
I always advise trainees to be sure they ask physicians practicing in each of these options to give examples of what their best and worst days are like so that they can get some idea of what the daily humdrum and challenges would encompass. What did I choose? I have always gone with option 2 and have been very happy in that setting.
In this issue…
More biosimilars head our way. Turning to the current issue of the journal, on page e181, Dr Jane de Lartigue discusses 2 new biosimilars recently approved by the United States Food and Drug Administration (FDA) – epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for chemotherapy-induced anemia (CIA), and pegfilgrastim-jmdb (Fulphila; Mylan and Biocon) for prevention of febrile neutropenia. As Dr de Lartigue notes, biosimilars are copies of FDA-approved biologic drugs that cannot be identical to the reference drug but demonstrate a high similarity to it. In this case, the reference drug for epoetin alfa-epbx is epoetin alfa (Epogen/Procrit, Amgen) and for pegfilgrastim-jmdb, it is pegfilgrastim (Neulasta, Amgen). As the reference drugs’ patents expire, biosimilars are being developed to increase competition in the marketplace in an effort to reduce costs and improve patient access to these therapies. Indeed, the FDA is working to streamline the biosimilar approval process to facilitate that access.
Reading this article got me thinking about something I often have to consider in the course of my work: transfusion versus erythropoiesis-stimulating agents (ESAs)? Recombinant erythropoietin drugs such as the biosimilar, epoetin alfa-epbx, and its reference drug are grouped together as ESAs, and have been used to treat CIA since the late 1980s. However, there were a few trials that used higher-dose ESA or set high hemoglobin targets, and their findings suggested that ESAs may shorten survival in patients with cancer or increase tumor growth, or both. The use of ESAs took a nosedive after the 2007 decision by the FDA’s Oncologic Drugs Advisory Committee to rein in their use for a hard start of ESA treatment at less than 10 g/dL hemoglobin, and not higher. Subsequent trials addressed the concerns about survival and tumor growth. A meta-analysis of 60 randomized, placebo-controlled trials of ESAs in CIA found that there was no difference in overall survival between the study and control groups.1 Likewise, findings from an FDA-mandated trial with epoetin alfa (Procrit) in patients with metastatic breast cancer have reported that there was no significant difference in overall survival between the study and control groups.2 The results of a second FDA-mandated trial with darbepoetin alfa (Aranesp, Amgen) in patients with metastatic lung cancer are expected to be released soon. The FDA lifted the ESA Risk Evaluation and Mitigation Strategy based on those findings. However, many practitioners, both young and old, continue to shy away from using ESAs because of the FDA black box warning that remains in place despite the latest data.3The use of transfusion ticked up reciprocally with the decline in ESA use, but perhaps we should re-evaluate the use of these agents in our practice, especially now that the less costly, equally safe and effective biosimilars are becoming available and we have the new survival data. Transfusions are time consuming and have side effects, including allergic reaction and infection risk, whereas ESAs are easily administered by injection, which patients might find preferable.
Malignancies in patients with HIV-AIDS. On page e188, Koppaka and colleagues report on a study in India of the patterns of malignancies in patients with HIV-AIDS. I began my career just as the first reports of what became known as HIV-AIDS emerged, and we were all mystified by what was killing these patients and the curious hematologic and oncologic problems they developed. Back then, the patients were profoundly immunosuppressed, and the immunosuppression cancers of non-Hodgkin lymphoma, usually higher grade, and Kaposi sarcoma were most prevalent and today are collectively labeled AIDS-defining malignancies (ADMs).
Fast forward to present day, and we have extremely effective antiretroviral therapies that have resulted in a significant reduction in mortality among HIV-infected individuals who are now living long enough to get what we call non–AIDS-defining malignancies (NADMs) such as anal or cervical cancers, hepatoma (hepatocellular carcinoma), Hodgkin lymphoma, and lung cancer. Of note is that these NADMs are all highly viral associated, with anal and cervical cancers linked to infection with the human papillomavirus; hepatoma linked to the hepatitis B/C viruses; Hodgkin lymphoma to the Epstein-Barr virus; and lung cancer, possibly also HPV. Fortunately, these days we can use standard-dose chemoradiation therapy for all HIV-related cancers because the patients’ immune systems are much better reconstituted and the modern-day antiretroviral therapies have much less drug–drug interaction thanks to the advent of the integrase inhibitors. The researchers give an excellent breakdown of the occurrence of these malignancies, as well as an analysis of the correlation between CD4 counts and the different malignancies.
Immunotherapy-related side effects in the ED. What happens when our patients who are on immunotherapy end up in the emergency department (ED) with therapy-related symptoms? And what can the treating oncologist do to help the ED physician achieve the best possible outcome for the patient? I spoke to Dr Maura Sammon, an ED physician, about some of the more common of these side effects – lung, gastrointestinal, rash, and endocrine-related problems – and she describes in detail how physicians in the ED would triage and treat the patient. Dr Sammon also emphasizes the importance of communication: first, between the treating oncologist and patient, about the differences between chemotherapy and immunotherapy; and second, between the ED physician and the treating oncologist as soon as possible after the patient has presented to ensure a good outcome. The interview is part of The JCSO Interview series. It is jam-packed with useful, how-to information, and you can read a transcript of it on page e216 of this issue, or you can listen to it online.4
We round off the issue with a selection of Case Reports (pp. e200-e209), an original report on the characteristics of urgent palliative cancer care consultations encountered by radiation oncologists (p. e193), and a New Therapies feature, also by Dr de Lartigue, focusing on the rarity and complexities of sarcomas (p. e210).
Those are my dog-day-of-summer thoughts as we head toward another Labor Day and a new academic year. Since we are all online now, we encourage you to listen to my bimonthly podcast of each issue on our website at www.jcso-online.com, and of course, follow us on Twitter (@jcs_onc) and Instagram (@jcsoncology) and like us on Facebook.
As summer winds down and we begin to gear up to return to school or work, I was thinking about new and returning hem-onc residents, fellows, and young attendings and a question I routinely get from them: what should I do next in my career? I always answer by holding up 3 fingers and telling them that they can practice 1, at a university hospital; 2, at a university teaching affiliate; or 3, at a community hospital or practice with a little or no university affiliation. These days, trainees in hematology-oncology are often advised to be highly specialty-specific when they plan their long-term careers and to focus on a particular cancer or hematologic disorder. That is fine if you want to remain in an academic or university-based practice, but not if community practice is your preference. So, what are the differences among these 3 options?
Option 1, to remain in a university setting where you can be highly focused and specialized in a single narrowly defined area, could be satisfying, but keep in mind that the institution expects results! You will be carefully monitored for research output and teaching and administration commitments, and your interaction with patients could add up to less than 50% of your time. Publication and grant renewal will also play a role and therefore take up your time.
If you are considering option 2 – to work at a university teaching affiliate hospital – you need to bear in mind that you likely will see a patient population with a much broader range of diagnoses than would be the case with the first option. Patient care for option 2 will take up more than 50% of your time, so it might be a little more challenging to stay current, but perhaps more refreshing if you enjoy contact with patients. Teaching, research, and administration will surely be available, and publication and grant renewal will play as big or small a role as you want.
Option 3 would be to join a community hospital or practice where the primary focus is on patient care and the diagnoses will span the hematology and oncology spectrum. This type of practice can be very demanding of one’s time, but as rewarding as the other options, especially if you value contact with patients. With this option, one is more likely to practice as a generalist, perhaps with an emphasis in one of the hem-onc specialties, but able to treat a cluster of different types of cancer as well.
I always advise trainees to be sure they ask physicians practicing in each of these options to give examples of what their best and worst days are like so that they can get some idea of what the daily humdrum and challenges would encompass. What did I choose? I have always gone with option 2 and have been very happy in that setting.
In this issue…
More biosimilars head our way. Turning to the current issue of the journal, on page e181, Dr Jane de Lartigue discusses 2 new biosimilars recently approved by the United States Food and Drug Administration (FDA) – epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for chemotherapy-induced anemia (CIA), and pegfilgrastim-jmdb (Fulphila; Mylan and Biocon) for prevention of febrile neutropenia. As Dr de Lartigue notes, biosimilars are copies of FDA-approved biologic drugs that cannot be identical to the reference drug but demonstrate a high similarity to it. In this case, the reference drug for epoetin alfa-epbx is epoetin alfa (Epogen/Procrit, Amgen) and for pegfilgrastim-jmdb, it is pegfilgrastim (Neulasta, Amgen). As the reference drugs’ patents expire, biosimilars are being developed to increase competition in the marketplace in an effort to reduce costs and improve patient access to these therapies. Indeed, the FDA is working to streamline the biosimilar approval process to facilitate that access.
Reading this article got me thinking about something I often have to consider in the course of my work: transfusion versus erythropoiesis-stimulating agents (ESAs)? Recombinant erythropoietin drugs such as the biosimilar, epoetin alfa-epbx, and its reference drug are grouped together as ESAs, and have been used to treat CIA since the late 1980s. However, there were a few trials that used higher-dose ESA or set high hemoglobin targets, and their findings suggested that ESAs may shorten survival in patients with cancer or increase tumor growth, or both. The use of ESAs took a nosedive after the 2007 decision by the FDA’s Oncologic Drugs Advisory Committee to rein in their use for a hard start of ESA treatment at less than 10 g/dL hemoglobin, and not higher. Subsequent trials addressed the concerns about survival and tumor growth. A meta-analysis of 60 randomized, placebo-controlled trials of ESAs in CIA found that there was no difference in overall survival between the study and control groups.1 Likewise, findings from an FDA-mandated trial with epoetin alfa (Procrit) in patients with metastatic breast cancer have reported that there was no significant difference in overall survival between the study and control groups.2 The results of a second FDA-mandated trial with darbepoetin alfa (Aranesp, Amgen) in patients with metastatic lung cancer are expected to be released soon. The FDA lifted the ESA Risk Evaluation and Mitigation Strategy based on those findings. However, many practitioners, both young and old, continue to shy away from using ESAs because of the FDA black box warning that remains in place despite the latest data.3The use of transfusion ticked up reciprocally with the decline in ESA use, but perhaps we should re-evaluate the use of these agents in our practice, especially now that the less costly, equally safe and effective biosimilars are becoming available and we have the new survival data. Transfusions are time consuming and have side effects, including allergic reaction and infection risk, whereas ESAs are easily administered by injection, which patients might find preferable.
Malignancies in patients with HIV-AIDS. On page e188, Koppaka and colleagues report on a study in India of the patterns of malignancies in patients with HIV-AIDS. I began my career just as the first reports of what became known as HIV-AIDS emerged, and we were all mystified by what was killing these patients and the curious hematologic and oncologic problems they developed. Back then, the patients were profoundly immunosuppressed, and the immunosuppression cancers of non-Hodgkin lymphoma, usually higher grade, and Kaposi sarcoma were most prevalent and today are collectively labeled AIDS-defining malignancies (ADMs).
Fast forward to present day, and we have extremely effective antiretroviral therapies that have resulted in a significant reduction in mortality among HIV-infected individuals who are now living long enough to get what we call non–AIDS-defining malignancies (NADMs) such as anal or cervical cancers, hepatoma (hepatocellular carcinoma), Hodgkin lymphoma, and lung cancer. Of note is that these NADMs are all highly viral associated, with anal and cervical cancers linked to infection with the human papillomavirus; hepatoma linked to the hepatitis B/C viruses; Hodgkin lymphoma to the Epstein-Barr virus; and lung cancer, possibly also HPV. Fortunately, these days we can use standard-dose chemoradiation therapy for all HIV-related cancers because the patients’ immune systems are much better reconstituted and the modern-day antiretroviral therapies have much less drug–drug interaction thanks to the advent of the integrase inhibitors. The researchers give an excellent breakdown of the occurrence of these malignancies, as well as an analysis of the correlation between CD4 counts and the different malignancies.
Immunotherapy-related side effects in the ED. What happens when our patients who are on immunotherapy end up in the emergency department (ED) with therapy-related symptoms? And what can the treating oncologist do to help the ED physician achieve the best possible outcome for the patient? I spoke to Dr Maura Sammon, an ED physician, about some of the more common of these side effects – lung, gastrointestinal, rash, and endocrine-related problems – and she describes in detail how physicians in the ED would triage and treat the patient. Dr Sammon also emphasizes the importance of communication: first, between the treating oncologist and patient, about the differences between chemotherapy and immunotherapy; and second, between the ED physician and the treating oncologist as soon as possible after the patient has presented to ensure a good outcome. The interview is part of The JCSO Interview series. It is jam-packed with useful, how-to information, and you can read a transcript of it on page e216 of this issue, or you can listen to it online.4
We round off the issue with a selection of Case Reports (pp. e200-e209), an original report on the characteristics of urgent palliative cancer care consultations encountered by radiation oncologists (p. e193), and a New Therapies feature, also by Dr de Lartigue, focusing on the rarity and complexities of sarcomas (p. e210).
Those are my dog-day-of-summer thoughts as we head toward another Labor Day and a new academic year. Since we are all online now, we encourage you to listen to my bimonthly podcast of each issue on our website at www.jcso-online.com, and of course, follow us on Twitter (@jcs_onc) and Instagram (@jcsoncology) and like us on Facebook.
1. Glaspy J, Crawford J, Vansteenkiste J, et al. Erythropoiesis-stimulating agents in oncology: a study-level meta-analysis of survival and other safety outcomes. Br J Cancer. 2010;102(2):301-315.
2. Leyland-Jones B, Bondarenko I, Nemsadze G, et al. A randomized, open-label, multicenter, phase III study of epoetin alfa versus best standard of care in anemic patients with metastatic breast cancer receiving standard chemotherapy. J Clin Oncol. 2016;34:1197-1207.
3. US Food and Drug Administration release. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Last updated April 13, 2017. Accessed August 20, 2018.
4. Henry D, Sammon M. Treating immunotherapy-related AEs in the emergency department [Audio]. https://www.mdedge.com/jcso/article/171966/patient-survivor-care/treating-immunotherapy-related-aes-emergency-department. Published August 6, 2018.
1. Glaspy J, Crawford J, Vansteenkiste J, et al. Erythropoiesis-stimulating agents in oncology: a study-level meta-analysis of survival and other safety outcomes. Br J Cancer. 2010;102(2):301-315.
2. Leyland-Jones B, Bondarenko I, Nemsadze G, et al. A randomized, open-label, multicenter, phase III study of epoetin alfa versus best standard of care in anemic patients with metastatic breast cancer receiving standard chemotherapy. J Clin Oncol. 2016;34:1197-1207.
3. US Food and Drug Administration release. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Last updated April 13, 2017. Accessed August 20, 2018.
4. Henry D, Sammon M. Treating immunotherapy-related AEs in the emergency department [Audio]. https://www.mdedge.com/jcso/article/171966/patient-survivor-care/treating-immunotherapy-related-aes-emergency-department. Published August 6, 2018.
Carcinoma of the colon in a child
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).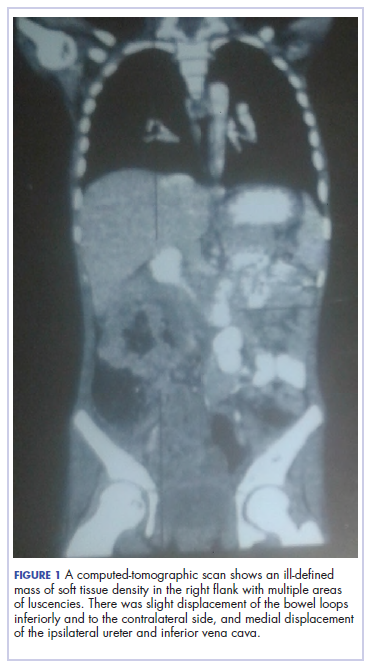
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).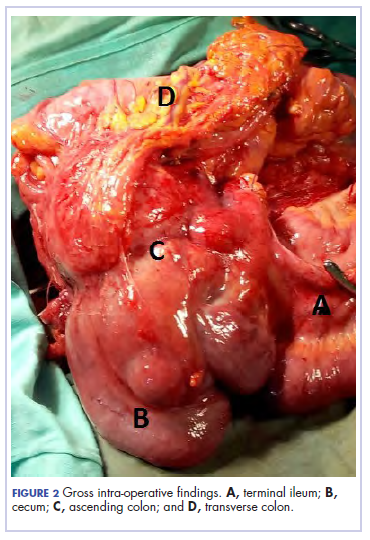
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).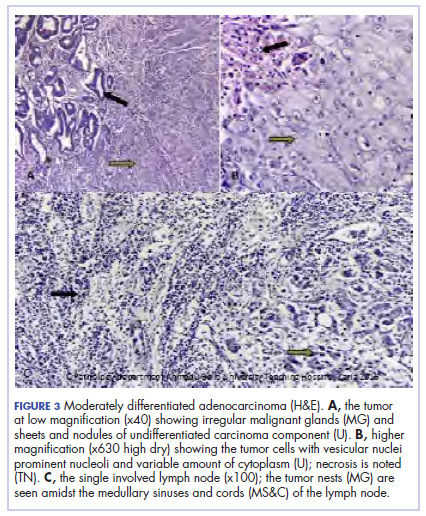
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.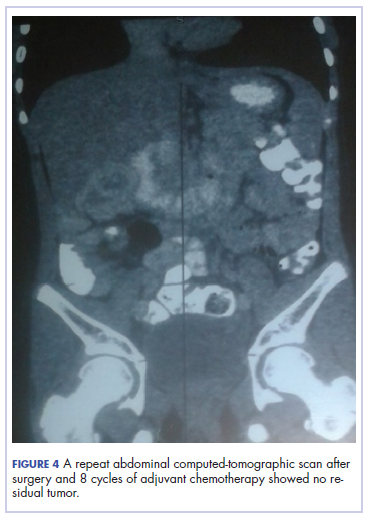
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
An unusual case of primary cardiac prosthetic valve-associated lymphoma
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
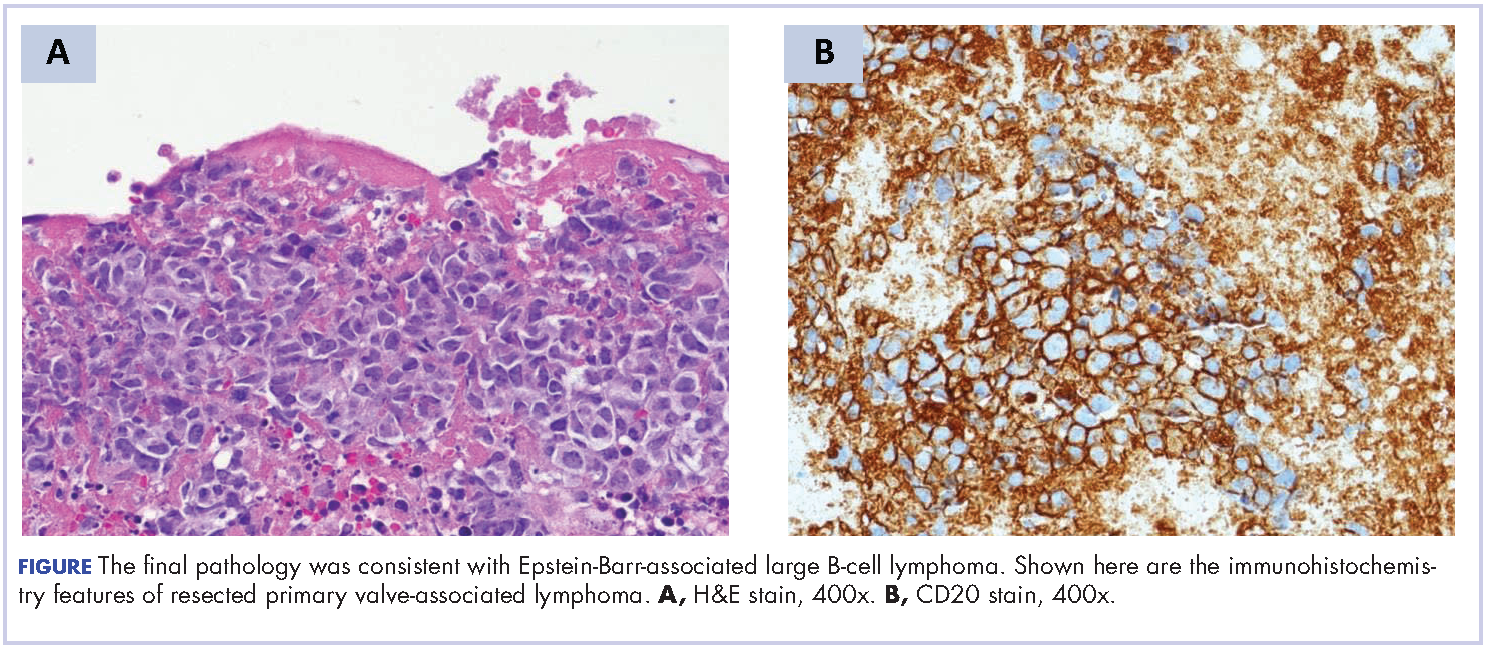
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
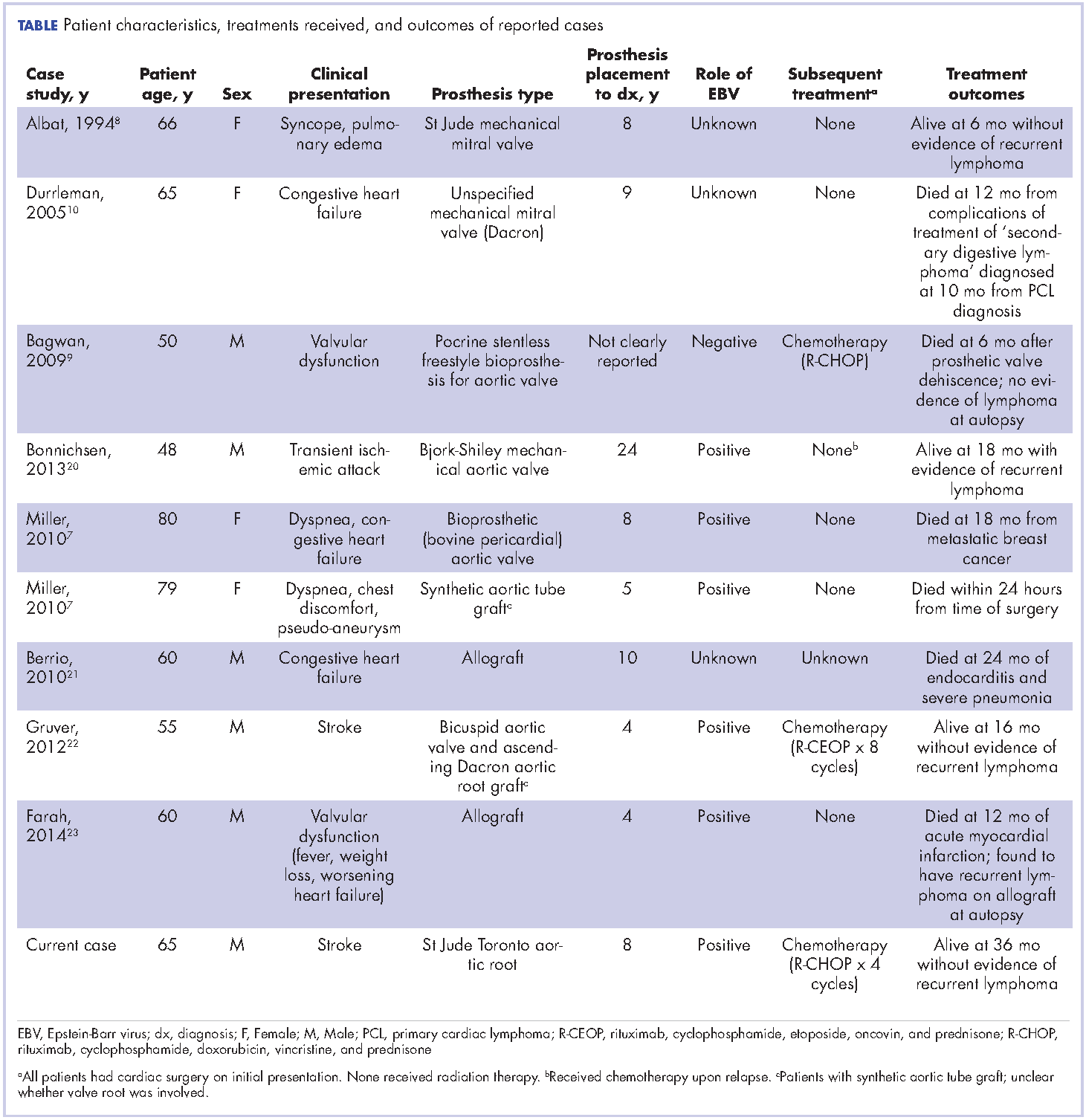
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Durable response to pralatrexate for aggressive PTCL subtypes
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.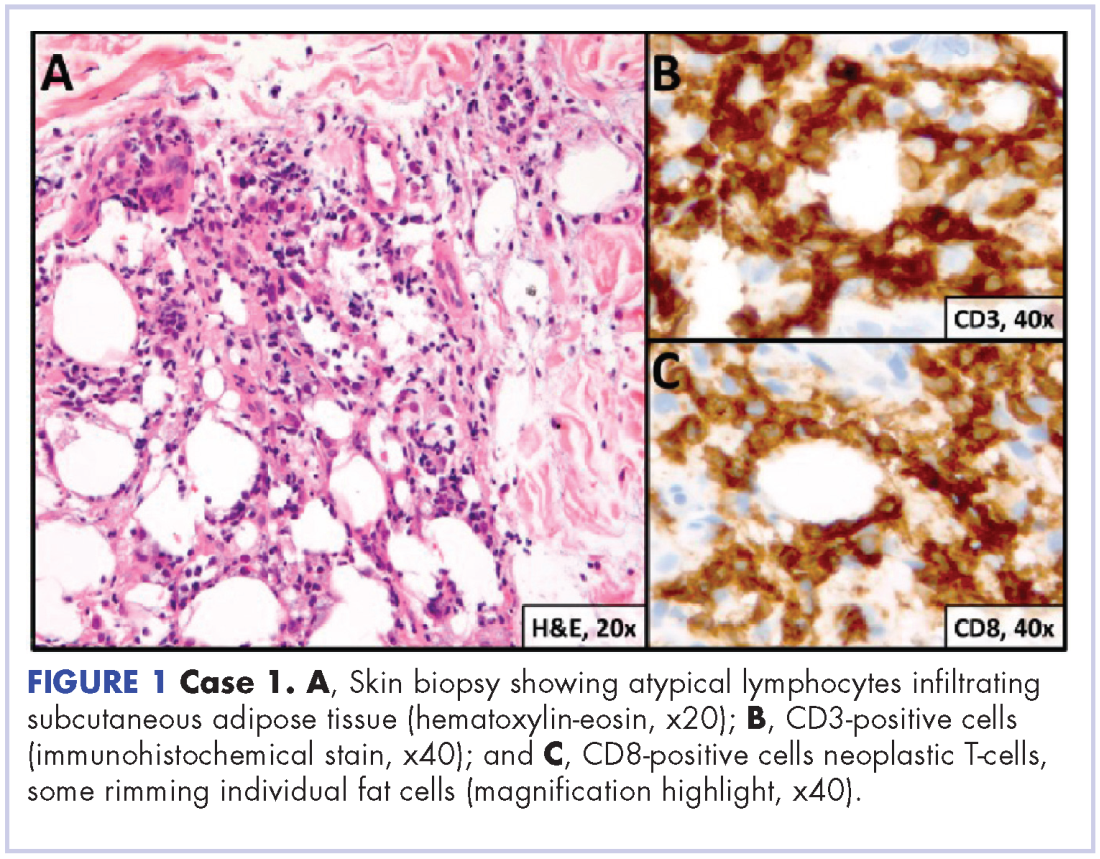
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.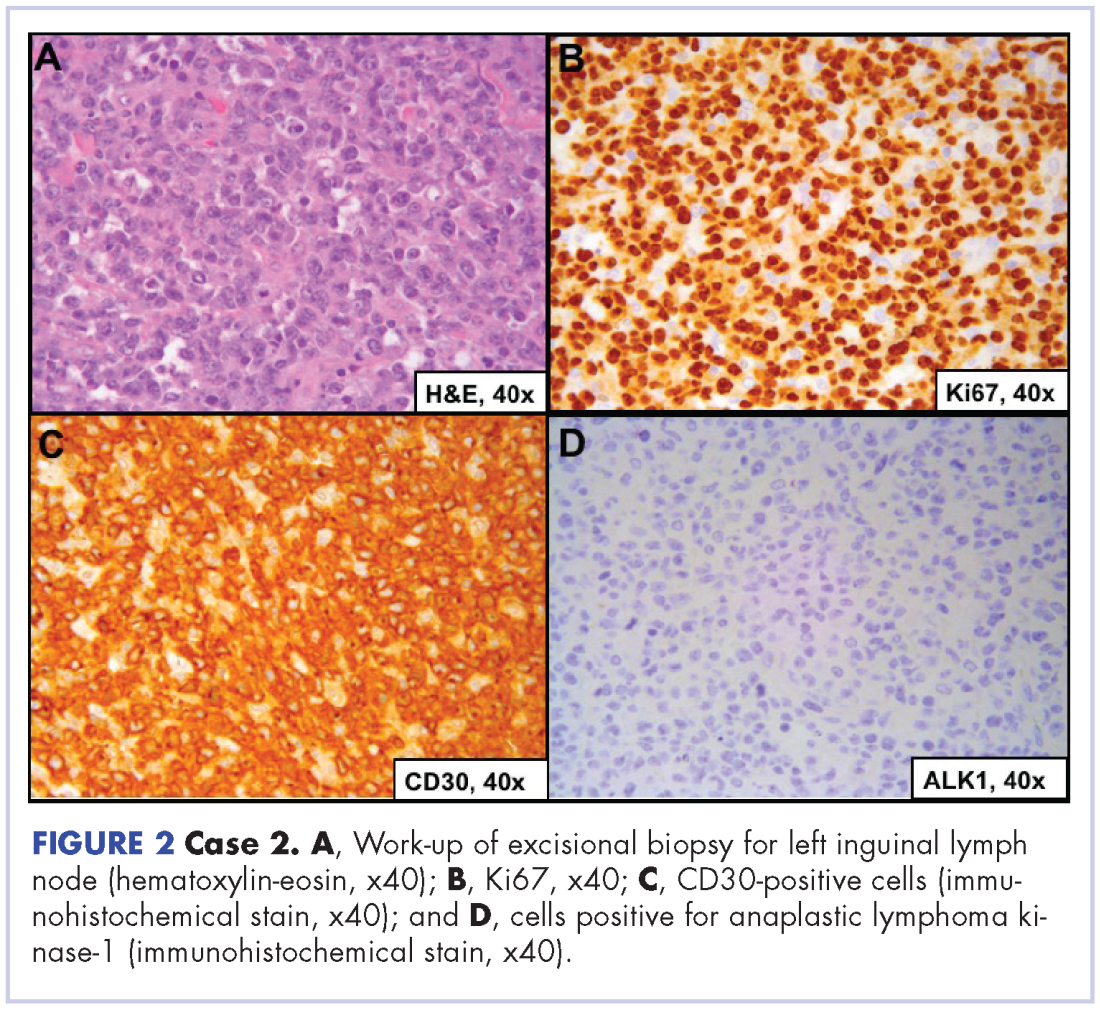
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).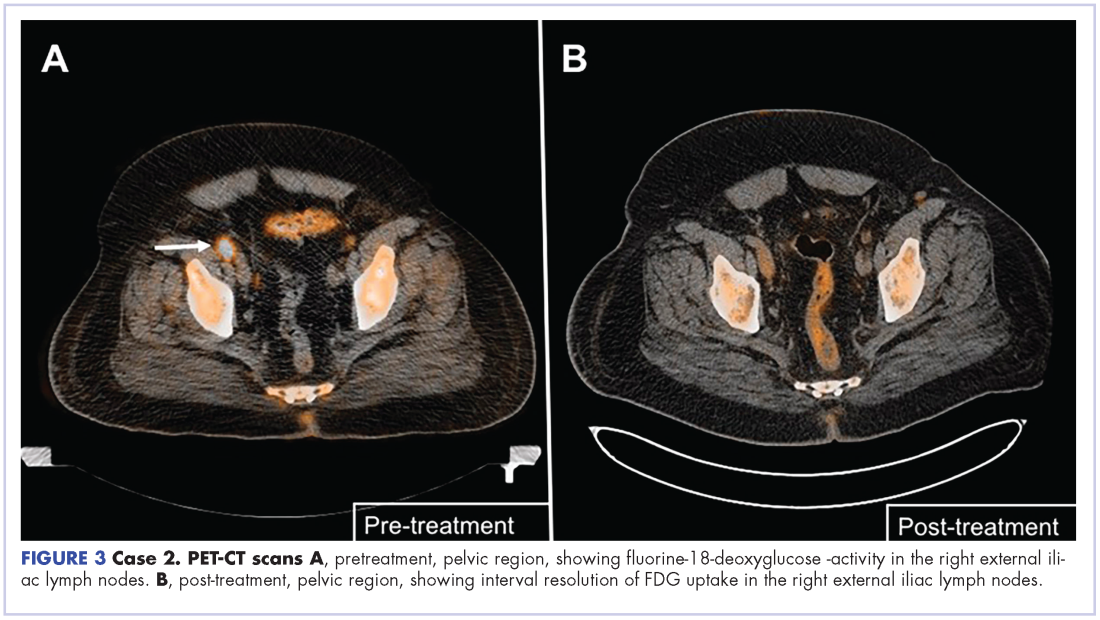
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
New myeloma drugs improve response and extend survival
DR HENRY I thought we might discuss some cases of patients with myeloma, starting with a relatively simple case and ending with one that is a little more complicated. For the first case, we have a 56-year-old healthy man with IgG kappa myeloma whose work-up shows he has multiple lytic bone lesions. He has normal renal function, normal calcium, and he’s transplant-eligible by other health issues. I’ll leave the cytogenetics up to you if that changes your approach. How would you develop or pose some options for this man’s treatment to begin with?
DR ANDERSON It’s important to start out by saying that we, in myeloma, have many new classes of drugs and many new opportunities to choose from to treat this patient.1 As you know, we have proteasome inhibitors, the first-generation bortezomib, then carfilzomib and ixazomib. We have the immunomodulatory drugs (IMiDs), thalidomide, and now lenalidomide and pomalidomide. We have a histone deacetylase (HDAC) inhibitor approved called panobinostat, and we have 2 monoclonal antibodies approved, elotuzumab and daratumumab. These classes of medicine have made it possible for 20 different Food and Drug Administration (FDA) approvals in the last 10-15 years. These agents, having been tested in advanced myeloma, have moved toward initial management.
This person is 50 years old. He has adequate liver, heart, lung, and kidney function, so he would be eligible for high-dose therapy and stem-cell transplantation. In terms of initial management, there are many options (Figure 1). We strongly recommend that triplet therapy be used initially. The most common triplets would be lenalidomide, bortezomib, and dexamethasone (RVD)2,3 or cyclophosphamide, bortezomib, and dexamethasone (CyBorD).4 If this man had neuropathy, perhaps carfilzomib, the second-generation proteasome inhibitor, with lenalidomide and dexamethasone could have been used. Why do we use these? The extent and frequency of response with these triplets is nearly universal overall response rate, with three-quarters very good partial and half-complete responses, including minimal residual disease negative responses. In this patient, we would therefore recommend treatment with either RVD or CyBorD for several cycles to maximal response.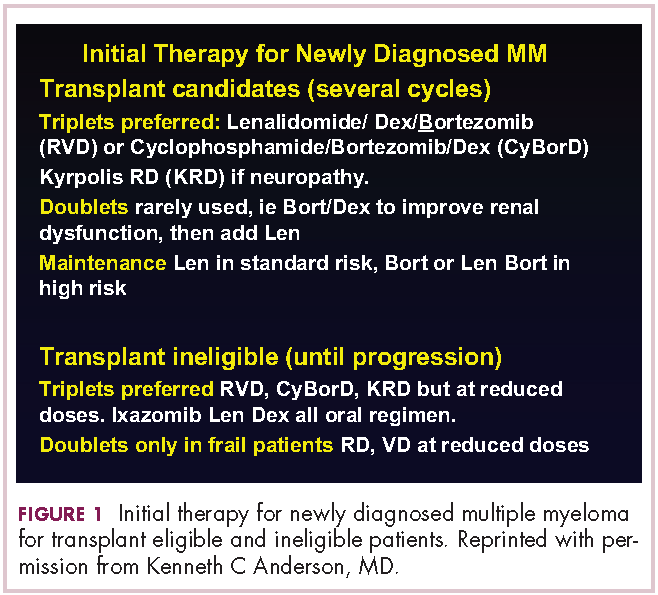
He would then have autologous stem cells collected, and it is still the standard of care to proceed to high-dose melphalan and a single high-dose therapy and stem-cell transplantation. The cytogenetics are important: if this patient has standard-risk multiple myeloma, then lenalidomide maintenance would be given after transplant. It is now FDA-approved for this purpose because it can prolong both progression-free and – most importantly – overall survival.5 Standard-risk cytogenetics might, for example, include hyperdiploidy or translocation 11;14. On the other hand, if his myeloma were high-risk and characterized, for example, by 17p deletion, we would carry out the same induction and transplantation, but we would alter the maintenance to incorporate a proteasome inhibitor. Lenalidomide and bortezomib, for example, could be combined. Early data show that using combined maintenance therapy with lenalidomide and bortezomib, can overcome the early relapses that are characteristic of high-risk disease.6
Because of the extent and frequency of response to combination novel therapies, we have undertaken with our French colleagues a clinical trial of RVD in newly diagnosed patients – such as this patient – followed by stem-cell collection in all patients (Figure 2). Then there is a randomization to either early high-dose therapy, melphalan, and autologous stem-cell transplantation, followed by lenalidomide maintenance; or in the other cohort, harvesting of stem cells, additional RVD, and then maintenance with lenalidomide, saving the stem-cell transplant for later.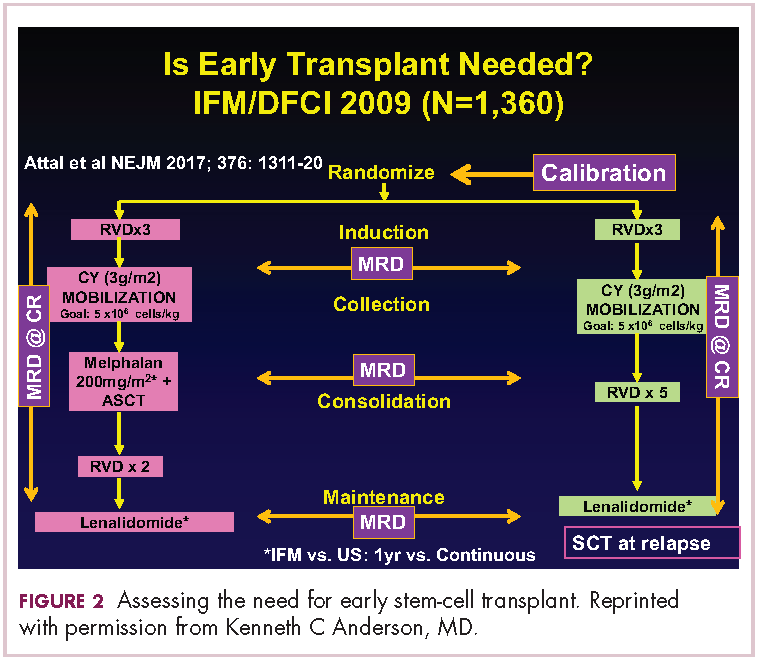
The French portion of this trial was reported in the New England Journal of Medicine earlier in 2017.7 It showed that patients who received RVD, high-dose melphalan, stem-cell transplant, and had 1 year of lenalidomide maintenance, had a progression-free survival advantage of about 1 year, without an overall survival advantage; compared with those patients who received RVD and lenalidomide maintenance, saving the transplant for later. I would hasten to add that lenalidomide maintenance was given for only 1 year in this trial, and patients in the RVD-only or RVD-and-transplant arms of this trial relapsed after the lenalidomide maintenance was discontinued.
The American portion of this trial is identical. That is, RVD induction is being given and all patients have a stem-cell collection. Half of the patients then go to high-dose melphalan and stem-cell transplant early, and half of them have the transplant only later at the time of relapse. A major difference, however, is that in both the RVD-only and RVD-and-transplant cohorts, patients receive lenalidomide maintenance until progression. This trial has been ongoing since 2009 and is still ongoing, which tells us that patients in both arms – the RVD-only as well as the RVD-and-transplant arms – are doing well.
In the recent STAMINA trial, all patients underwent a single high-dose therapy and transplant. Then there was a randomization to lenalidomide maintenance only in 1 cohort; a randomization to consolidation with RVD posttransplant followed by lenalidomide maintenance in the second cohort; or a randomization to a second high-dose melphalan and stem-cell transplant followed by lenalidomide maintenance in the third cohort.8 I mention this because the outcomes in all 3 cohorts was similar.
I believe this tells us strongly that high-dose therapy and stem-cell transplantation twice – so-called tandem transplant – is no longer a major option in multiple myeloma. For now, however, in this patient, the standard of care would be to undergo induction therapy with triplet, novel combination treatment. Then, stem cells would be collected and high-dose therapy stem-cell transplant would be done, followed either by lenalidomide maintenance for standard disease or lenalidomide and bortezomib maintenance for high-risk disease. We won’t really know if we can delay transplant until the trials I’ve mentioned totally read out. In my clinical practice, if patients have had a major response to their induction therapy and have stem cells harvested, we can then offer them the opportunity to use maintenance therapy and save the transplant as a potential option for later, when myeloma relapses.
DR HENRY In summary then, this would be, in 2017, off-protocol while the data is pending: it’s reasonable to get a deep induction response, collect stem cells, have a discussion with the patient, and then consider high-dose therapy or not.
DR ANDERSON Yes. I think it’s reasonable to discuss it. We need to be open and honest with patients that the standard of care remains transplant, that you incorporate novel treatments before the transplant and novel treatments as maintenance after the transplant. The happy news is that the outcome, especially for patients who have standard-risk myeloma, is at least a decade or longer progression-free survival. It’s an optimistic picture. The data in terms of transplant being needed or not, will come within the next several years.
For now, it is a standard of care to use 1 high-dose melphalan and stem-cell transplant in this setting. I will add into our discussion with patients – besides the opportunity to harvest stem cells and think about whether one needs to do a transplant early on or not – is the issue of toxicity. High-dose melphalan by itself has a small but real secondary incidence of cancer, myelodysplasia, or leukemia. If one uses lenalidomide maintenance after melphalan transplantation treatment, that risk of secondary cancer is slightly increased.
In my experience, if patients have achieved a complete response with induction therapy only, it’s not unreasonable to offer early transplant and be clear that’s the standard of care. The alternative is maintenance with lenalidomide, knowing once the stem cells have been harvested, that transplantation can be an option to treat relapsed myeloma. We have many other options available as well. Time will tell in terms of the ongoing randomized trials as to whether transplant remains central to our treatment paradigm.
DR HENRY This leads us to our second patient. Here we have an older man of 74 years. He’s a professional piano player, so we want to try to avoid peripheral neuropathy in him. He has some mild renal insufficiency and some coronary artery disease, so he’s deemed transplant-ineligible. He has IgG kappa myeloma, and he’s brand new. What would you consider to be options for him for treatment?
DR ANDERSON This brings up the issue of a transplant-ineligible patient. He has significant comorbidity that would make transplantation an increased risk. What we would recommend in such a patient is still triplet induction therapy incorporating novel agents (Figure 1). Lenalidomide, the immunomodulatory drug, can safely be given in the context of neuropathy because it does not cause significant neuropathy. It would need to be dose modified, depending on the degree of renal insufficiency. We would recommend also including proteasome inhibitors. Bortezomib, the first-generation proteasome inhibitor, would be contraindicated because it does have a small but real attendant neuropathy. If, however, it is given weekly and subcutaneously, the risk of attendant neuropathy is quite low. In this patient, therefore, one could start with lenalidomide and bortezomib weekly and subcutaneously,1,2 with a very early and vigilant follow-up for the earliest signs of neuropathy, so as not to allow it to develop and compromise his career.
Alternatively, one could use a proteasome inhibitor that does not have attendant neuropathy. Carfilzomib, the second-generation proteasome inhibitor, does not have neuropathy.9 But we would need to have caution here, because this patient has a history of coronary artery disease, and carfilzomib has a very small, but real, incidence of cardiac toxicity so would need to be used judiciously in this setting. The third proteasome inhibitor, ixazomib, is the next-generation bortezomib-class proteasome inhibitor, and it’s oral.10 It has less neuropathy than does bortezomib, so in my view is a very realistic option for him together with lenalidomide. It does have a small incidence of neuropathy, so close monitoring for neuropathy would be indicated. We could use lenalidomide–dexamethasone as a doublet and avoid neuropathy,11 but usually doublets are reserved only for frail patients.
My recommendation, therefore, would be RVD with the bortezomib weekly or subcutaneously, or alternatively, lenalidomide, ixazomib, dexamethasone as an all-oral regimen as induction therapy. In my view, this 74-year-old patient with comorbidity is not a transplant candidate. However, one can be very optimistic with this patient. The likelihood that he could have myeloma as a chronic illness and die from something else is quite high. Initial induction triplet therapy would achieve a very high response extent and frequency. The durability would be long, especially with lenalidomide maintenance if it’s standard-risk myeloma or lenalidomide and a proteasome inhibitor, probably ixazomib in this setting, if he were to have high-risk myeloma.
If myeloma relapses, then there are many options that could be used in this patient and achieve years of progression-free and overall survival. Indeed, he is 74 years old and will respond very well to induction triplet therapy, with many years’ duration of response due to continuous lenalidomide or lenalidomide and proteasome inhibitor maintenance. Then there are many effective options to treat relapsed therapy using triplet novel agents. Therefore, his lifespan is unlikely to be shortened by multiple myeloma.
DR HENRY It’s so incredible compared with what it was when I trained. The next patient, a 45-year-old woman with IgG lambda myeloma, has had RVD induction and responded. She had lenalidomide maintenance, but then she progressed, and she got her stem-cell transplant, and she’s progressing after that. I guess we’re looking here to fold in some of the newer agents. How you would you do that in this patient?DR ANDERSON Yes. I think one of the most remarkable and exciting developments with myeloma is the rapid approval of the novel classes of agents that I mentioned earlier – the proteasome inhibitors, the immunomodulatory drugs, the HDAC inhibitor, and the monoclonal antibodies.1 They’re particularly relevant in a patient such as this one, whose myeloma has relapsed after what would be considered standard therapy for a young person with standard-risk myeloma. This patient had RVD and maintenance therapy, and then progressed. The transplant was given for relapsed myeloma. The opportunity to use stem-cell transplant in patients when myeloma becomes active after maintenance should not be forgotten as it can be very effective. In all the trials done to date in which early versus late transplant are compared, there have been similar outcomes. Therefore, if the transplant isn’t done early, don’t forget that it’s an option at the time the myeloma progresses. I do want to mention, that there are lots of options for relapsed myeloma (Figures 3 and 4). I mentioned RVD or CyBorD as initial triplet therapies.2-4 In North America, those are the 2 most common regimens. If myeloma then relapses and is resistant to RVD or to CyBorD, then we need to identify alternatives.

We also need to think about the comorbidities in the patient – issues such as age, neuropathy, presence of renal dysfunction, and other clinical factors. And we need to think about what treatment they’ve had in the past. This patient has had RVD, maintenance with lenalidomide, and a stem-cell transplant. We can offer patients a variety of therapies, but in the context of resistance to the first-generation proteasome inhibitor bortezomib and the first-generation immunomodulatory drug lenalidomide, we would strongly recommend the second-generation immunomodulatory drug pomalidomide12 together with a second-generation proteasome inhibitor, be that carfilzomib13 or ixazomib.14 When one uses the second-generation IMiDs and proteasome inhibitors together, there’s a very high frequency of response in the order of 70%-80%, which lasts years.
Besides carfilzomib and ixazomib proteasome inhibitors, we also have elotuzumab and daratumumab, the monoclonal antibodies.15-17 These agents have been FDA approved to treat patients such as this one who has had 1-3 previous therapies for their myeloma. All of them have been approved in randomized phase 3 trials compared with lenalidomide-dexamethasone in the control arm.13-15,17 They’ve all been found to be superior. Although lenalidomide-dexamethasone combined with daratumumab, ixazomib, elotuzumab, or carfilzomib is superior to lenalidomide in relapsed myeloma, the situation in North America, as in this patient, is usually that patients have had lenalidomide-dexamethasone as part of their initial treatment and their myeloma is refractory to lenalidomide.
Hence, we recommend, that we go to the second-generation pomalidomide and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Having said that, the treatment paradigm is evolving. For example, the monoclonal antibody daratumumab was initially approved by the FDA in multiply relapsed disease as a single agent because it achieves a 30% response rate.16 It now has been moved earlier into the first relapse of multiple myeloma, where it achieves much higher response rates when combined with lenalidomide–dexamethasone or combined with bortezomib–dexamethasone.17,18 Response rates of 70%-80% can be achieved, including minimal residual disease negative complete responses.
Today, in a patient who has had RVD transplant and myeloma has returned, we would recommend second-generation IMiDs, pomalidomide, and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Data for daratumumab combined with lenalidomide-dexamethasone or with bortezomib-dexamethasone, look very promising. We need, however, to see more experience of daratumumab together with lenalidomide-dexamethasone or daratumumab together with bortezomib-dexamethasone in patients whose myeloma is refractory to RVD, that is, patients whose myeloma has returned after RVD induction treatment. Of note, pomalidomide, dexamethasone, and daratumumab have just been approved by the FDA and may also be active even in myeloma recurring after RVD treatment.19
Daratumumab in combination will be moving earlier and earlier and may be appropriate to treat the first relapse. I do want to stress, however, that at present I save daratumumab for the second or greater relapse. Daratumumab is active even when relapse occurs after treatment with second-generation IMiDs and proteasome inhibitors.
DR HENRY Before we close, I have a couple practical questions with these antibodies. Daratumumab has the track record of first-treatment severe reactions and long infusion times. How long are you anticipating the first daratumumab treatment takes? There has been some talk that maybe splitting it in half and going over 2 days is easier on the patient and the infusion center. Have you done that?
DR ANDERSON Yes, I think that’s a very important point. We need to be thinking – first and foremost – about efficacy of our therapy. Equally important, however, are the safety profile and the user-friendliness for the patient. Daratumumab infusions are quite long – on the order of 8 hours or longer on day 1 of infusion. And to date, all the clinical trials have used daratumumab infusions weekly for 8 treatments, followed by 8 treatments given every 2 weeks. Then monthly daratumumab is given as a maintenance therapy. Thus, there is a requirement for multiple outpatient clinic visits that can be prolonged.
One of the opportunities that’s being tested is to give daratumumab subcutaneously. While this is being evaluated in protocols now, the results that have been reported at our national meetings look to be quite promising in terms of efficacy, similar to results with the intravenous administration. Obviously, this would allow for a much more convenient clinic visit and shorter time for the patients being treated.
I should mention that the other antibody, elotuzumab, has been approved in combination with lenalidomide and dexamethasone.15 The infusions with lenalidomide, dexamethasone, and the antibody elotuzumab are much shorter, on the order of 2- or 3-hour visits. The place for elotuzumab in the management of relapsed myeloma is yet to be totally defined. We tend to use it now in the setting of more indolent relapses, where patients might have a slowly rising monoclonal protein. Elotuzumab-lenalidomide-dexamethasone has maintained an overall survival advantage at 4 years compared with lenalidomide-dexamethasone when used in relapsed myeloma.
We are quite excited about both antibodies. Daratumumab tends to get most of the activity, as it achieves responses as a single agent,16 and the depth of the responses are markedly increased when it’s combined with lenalidomide-dexamethasone or bortezomib-dexamethasone.17,18 However, one shouldn’t forget elotuzumab15 based on its tolerability and the survival advantage I mentioned at 4 years.
The final point is that we think about myeloma genetically at the time of diagnosis and relapse in terms of standard or high-risk disease. One of the hallmarks of high-risk disease has been 17P deletion or P53 dysfunction. One of the most exciting outcomes of the development of monoclonal antibodies has been the responses observed even in the context of P53 deletion. Clearly, antibody-mediated cellular cytotoxicity, complement-mediated cytotoxicity, and other mechanisms of action of these antibodies do not require normal P53 function. The important point, therefore, is that what has previously been thought of as high-risk disease can nowadays be effectively treated with these new immune treatments, correlating with the marked improvement in survival and overall outcome.
DR HENRY We have outlined 3 kinds of myeloma patients we see, and especially interesting is the last patient, who has relapsed and then progressed, and in whom newer drugs have a role. Thank you for such a complete and thorough discussion, Dr Anderson.
1. Kumar SK, Callander NS, Alsina M, et al. Multiple Myeloma, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:230-269.
2. Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly-diagnosed multiple myeloma. Blood. 2010;116:679-686.
3. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777: a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
4. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127:2569-2574.
5. McCarthy PL, Owzar K, Hofmeister CC, et al. A phase III study of lenalidomide after transplant for multiple myeloma. N Engl J Med. 2012;366:1770-1781.
6. Nooka AK, Kaufman JL, Muppidi S, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib, and dexamethasone (RVD) in high risk myeloma patients. Leukemia. 2014;28:690-693.
7. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib and dexamethasone with transplantation in myeloma. N Engl J Med. 2017;376:1311-1320.
8. Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (len) and dexamethasone (RVD) consolidation with len maintenance (ACM), tandem autohct with len maintenance (TAM) and autohct with len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702 – StaMINA Trial). Abstract LBA-1. Presented at the 2016 ASH Annual Meeting, December 6, 2016; San Diego, CA.
9. Dytfeld D, Jasielec J, Griffith KA, et al: Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162-164.
10. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503-1512
11. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. for the FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371:906-917.
12. Richardson P, Siegel D, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826-1832.
13. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. for the ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142-152
14. Moreau P, Masszi T, Grzasko N, et al. for the TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;28;374:1621-1634.
15. Lonial S, Dimopoulos M, Palumbo A, et al. for the ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621-631.
16. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD 38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207-1219.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. for the CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
19. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974-981.
DR HENRY I thought we might discuss some cases of patients with myeloma, starting with a relatively simple case and ending with one that is a little more complicated. For the first case, we have a 56-year-old healthy man with IgG kappa myeloma whose work-up shows he has multiple lytic bone lesions. He has normal renal function, normal calcium, and he’s transplant-eligible by other health issues. I’ll leave the cytogenetics up to you if that changes your approach. How would you develop or pose some options for this man’s treatment to begin with?
DR ANDERSON It’s important to start out by saying that we, in myeloma, have many new classes of drugs and many new opportunities to choose from to treat this patient.1 As you know, we have proteasome inhibitors, the first-generation bortezomib, then carfilzomib and ixazomib. We have the immunomodulatory drugs (IMiDs), thalidomide, and now lenalidomide and pomalidomide. We have a histone deacetylase (HDAC) inhibitor approved called panobinostat, and we have 2 monoclonal antibodies approved, elotuzumab and daratumumab. These classes of medicine have made it possible for 20 different Food and Drug Administration (FDA) approvals in the last 10-15 years. These agents, having been tested in advanced myeloma, have moved toward initial management.
This person is 50 years old. He has adequate liver, heart, lung, and kidney function, so he would be eligible for high-dose therapy and stem-cell transplantation. In terms of initial management, there are many options (Figure 1). We strongly recommend that triplet therapy be used initially. The most common triplets would be lenalidomide, bortezomib, and dexamethasone (RVD)2,3 or cyclophosphamide, bortezomib, and dexamethasone (CyBorD).4 If this man had neuropathy, perhaps carfilzomib, the second-generation proteasome inhibitor, with lenalidomide and dexamethasone could have been used. Why do we use these? The extent and frequency of response with these triplets is nearly universal overall response rate, with three-quarters very good partial and half-complete responses, including minimal residual disease negative responses. In this patient, we would therefore recommend treatment with either RVD or CyBorD for several cycles to maximal response.
He would then have autologous stem cells collected, and it is still the standard of care to proceed to high-dose melphalan and a single high-dose therapy and stem-cell transplantation. The cytogenetics are important: if this patient has standard-risk multiple myeloma, then lenalidomide maintenance would be given after transplant. It is now FDA-approved for this purpose because it can prolong both progression-free and – most importantly – overall survival.5 Standard-risk cytogenetics might, for example, include hyperdiploidy or translocation 11;14. On the other hand, if his myeloma were high-risk and characterized, for example, by 17p deletion, we would carry out the same induction and transplantation, but we would alter the maintenance to incorporate a proteasome inhibitor. Lenalidomide and bortezomib, for example, could be combined. Early data show that using combined maintenance therapy with lenalidomide and bortezomib, can overcome the early relapses that are characteristic of high-risk disease.6
Because of the extent and frequency of response to combination novel therapies, we have undertaken with our French colleagues a clinical trial of RVD in newly diagnosed patients – such as this patient – followed by stem-cell collection in all patients (Figure 2). Then there is a randomization to either early high-dose therapy, melphalan, and autologous stem-cell transplantation, followed by lenalidomide maintenance; or in the other cohort, harvesting of stem cells, additional RVD, and then maintenance with lenalidomide, saving the stem-cell transplant for later.
The French portion of this trial was reported in the New England Journal of Medicine earlier in 2017.7 It showed that patients who received RVD, high-dose melphalan, stem-cell transplant, and had 1 year of lenalidomide maintenance, had a progression-free survival advantage of about 1 year, without an overall survival advantage; compared with those patients who received RVD and lenalidomide maintenance, saving the transplant for later. I would hasten to add that lenalidomide maintenance was given for only 1 year in this trial, and patients in the RVD-only or RVD-and-transplant arms of this trial relapsed after the lenalidomide maintenance was discontinued.
The American portion of this trial is identical. That is, RVD induction is being given and all patients have a stem-cell collection. Half of the patients then go to high-dose melphalan and stem-cell transplant early, and half of them have the transplant only later at the time of relapse. A major difference, however, is that in both the RVD-only and RVD-and-transplant cohorts, patients receive lenalidomide maintenance until progression. This trial has been ongoing since 2009 and is still ongoing, which tells us that patients in both arms – the RVD-only as well as the RVD-and-transplant arms – are doing well.
In the recent STAMINA trial, all patients underwent a single high-dose therapy and transplant. Then there was a randomization to lenalidomide maintenance only in 1 cohort; a randomization to consolidation with RVD posttransplant followed by lenalidomide maintenance in the second cohort; or a randomization to a second high-dose melphalan and stem-cell transplant followed by lenalidomide maintenance in the third cohort.8 I mention this because the outcomes in all 3 cohorts was similar.
I believe this tells us strongly that high-dose therapy and stem-cell transplantation twice – so-called tandem transplant – is no longer a major option in multiple myeloma. For now, however, in this patient, the standard of care would be to undergo induction therapy with triplet, novel combination treatment. Then, stem cells would be collected and high-dose therapy stem-cell transplant would be done, followed either by lenalidomide maintenance for standard disease or lenalidomide and bortezomib maintenance for high-risk disease. We won’t really know if we can delay transplant until the trials I’ve mentioned totally read out. In my clinical practice, if patients have had a major response to their induction therapy and have stem cells harvested, we can then offer them the opportunity to use maintenance therapy and save the transplant as a potential option for later, when myeloma relapses.
DR HENRY In summary then, this would be, in 2017, off-protocol while the data is pending: it’s reasonable to get a deep induction response, collect stem cells, have a discussion with the patient, and then consider high-dose therapy or not.
DR ANDERSON Yes. I think it’s reasonable to discuss it. We need to be open and honest with patients that the standard of care remains transplant, that you incorporate novel treatments before the transplant and novel treatments as maintenance after the transplant. The happy news is that the outcome, especially for patients who have standard-risk myeloma, is at least a decade or longer progression-free survival. It’s an optimistic picture. The data in terms of transplant being needed or not, will come within the next several years.
For now, it is a standard of care to use 1 high-dose melphalan and stem-cell transplant in this setting. I will add into our discussion with patients – besides the opportunity to harvest stem cells and think about whether one needs to do a transplant early on or not – is the issue of toxicity. High-dose melphalan by itself has a small but real secondary incidence of cancer, myelodysplasia, or leukemia. If one uses lenalidomide maintenance after melphalan transplantation treatment, that risk of secondary cancer is slightly increased.
In my experience, if patients have achieved a complete response with induction therapy only, it’s not unreasonable to offer early transplant and be clear that’s the standard of care. The alternative is maintenance with lenalidomide, knowing once the stem cells have been harvested, that transplantation can be an option to treat relapsed myeloma. We have many other options available as well. Time will tell in terms of the ongoing randomized trials as to whether transplant remains central to our treatment paradigm.
DR HENRY This leads us to our second patient. Here we have an older man of 74 years. He’s a professional piano player, so we want to try to avoid peripheral neuropathy in him. He has some mild renal insufficiency and some coronary artery disease, so he’s deemed transplant-ineligible. He has IgG kappa myeloma, and he’s brand new. What would you consider to be options for him for treatment?
DR ANDERSON This brings up the issue of a transplant-ineligible patient. He has significant comorbidity that would make transplantation an increased risk. What we would recommend in such a patient is still triplet induction therapy incorporating novel agents (Figure 1). Lenalidomide, the immunomodulatory drug, can safely be given in the context of neuropathy because it does not cause significant neuropathy. It would need to be dose modified, depending on the degree of renal insufficiency. We would recommend also including proteasome inhibitors. Bortezomib, the first-generation proteasome inhibitor, would be contraindicated because it does have a small but real attendant neuropathy. If, however, it is given weekly and subcutaneously, the risk of attendant neuropathy is quite low. In this patient, therefore, one could start with lenalidomide and bortezomib weekly and subcutaneously,1,2 with a very early and vigilant follow-up for the earliest signs of neuropathy, so as not to allow it to develop and compromise his career.
Alternatively, one could use a proteasome inhibitor that does not have attendant neuropathy. Carfilzomib, the second-generation proteasome inhibitor, does not have neuropathy.9 But we would need to have caution here, because this patient has a history of coronary artery disease, and carfilzomib has a very small, but real, incidence of cardiac toxicity so would need to be used judiciously in this setting. The third proteasome inhibitor, ixazomib, is the next-generation bortezomib-class proteasome inhibitor, and it’s oral.10 It has less neuropathy than does bortezomib, so in my view is a very realistic option for him together with lenalidomide. It does have a small incidence of neuropathy, so close monitoring for neuropathy would be indicated. We could use lenalidomide–dexamethasone as a doublet and avoid neuropathy,11 but usually doublets are reserved only for frail patients.
My recommendation, therefore, would be RVD with the bortezomib weekly or subcutaneously, or alternatively, lenalidomide, ixazomib, dexamethasone as an all-oral regimen as induction therapy. In my view, this 74-year-old patient with comorbidity is not a transplant candidate. However, one can be very optimistic with this patient. The likelihood that he could have myeloma as a chronic illness and die from something else is quite high. Initial induction triplet therapy would achieve a very high response extent and frequency. The durability would be long, especially with lenalidomide maintenance if it’s standard-risk myeloma or lenalidomide and a proteasome inhibitor, probably ixazomib in this setting, if he were to have high-risk myeloma.
If myeloma relapses, then there are many options that could be used in this patient and achieve years of progression-free and overall survival. Indeed, he is 74 years old and will respond very well to induction triplet therapy, with many years’ duration of response due to continuous lenalidomide or lenalidomide and proteasome inhibitor maintenance. Then there are many effective options to treat relapsed therapy using triplet novel agents. Therefore, his lifespan is unlikely to be shortened by multiple myeloma.
DR HENRY It’s so incredible compared with what it was when I trained. The next patient, a 45-year-old woman with IgG lambda myeloma, has had RVD induction and responded. She had lenalidomide maintenance, but then she progressed, and she got her stem-cell transplant, and she’s progressing after that. I guess we’re looking here to fold in some of the newer agents. How you would you do that in this patient?DR ANDERSON Yes. I think one of the most remarkable and exciting developments with myeloma is the rapid approval of the novel classes of agents that I mentioned earlier – the proteasome inhibitors, the immunomodulatory drugs, the HDAC inhibitor, and the monoclonal antibodies.1 They’re particularly relevant in a patient such as this one, whose myeloma has relapsed after what would be considered standard therapy for a young person with standard-risk myeloma. This patient had RVD and maintenance therapy, and then progressed. The transplant was given for relapsed myeloma. The opportunity to use stem-cell transplant in patients when myeloma becomes active after maintenance should not be forgotten as it can be very effective. In all the trials done to date in which early versus late transplant are compared, there have been similar outcomes. Therefore, if the transplant isn’t done early, don’t forget that it’s an option at the time the myeloma progresses. I do want to mention, that there are lots of options for relapsed myeloma (Figures 3 and 4). I mentioned RVD or CyBorD as initial triplet therapies.2-4 In North America, those are the 2 most common regimens. If myeloma then relapses and is resistant to RVD or to CyBorD, then we need to identify alternatives.
We also need to think about the comorbidities in the patient – issues such as age, neuropathy, presence of renal dysfunction, and other clinical factors. And we need to think about what treatment they’ve had in the past. This patient has had RVD, maintenance with lenalidomide, and a stem-cell transplant. We can offer patients a variety of therapies, but in the context of resistance to the first-generation proteasome inhibitor bortezomib and the first-generation immunomodulatory drug lenalidomide, we would strongly recommend the second-generation immunomodulatory drug pomalidomide12 together with a second-generation proteasome inhibitor, be that carfilzomib13 or ixazomib.14 When one uses the second-generation IMiDs and proteasome inhibitors together, there’s a very high frequency of response in the order of 70%-80%, which lasts years.
Besides carfilzomib and ixazomib proteasome inhibitors, we also have elotuzumab and daratumumab, the monoclonal antibodies.15-17 These agents have been FDA approved to treat patients such as this one who has had 1-3 previous therapies for their myeloma. All of them have been approved in randomized phase 3 trials compared with lenalidomide-dexamethasone in the control arm.13-15,17 They’ve all been found to be superior. Although lenalidomide-dexamethasone combined with daratumumab, ixazomib, elotuzumab, or carfilzomib is superior to lenalidomide in relapsed myeloma, the situation in North America, as in this patient, is usually that patients have had lenalidomide-dexamethasone as part of their initial treatment and their myeloma is refractory to lenalidomide.
Hence, we recommend, that we go to the second-generation pomalidomide and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Having said that, the treatment paradigm is evolving. For example, the monoclonal antibody daratumumab was initially approved by the FDA in multiply relapsed disease as a single agent because it achieves a 30% response rate.16 It now has been moved earlier into the first relapse of multiple myeloma, where it achieves much higher response rates when combined with lenalidomide–dexamethasone or combined with bortezomib–dexamethasone.17,18 Response rates of 70%-80% can be achieved, including minimal residual disease negative complete responses.
Today, in a patient who has had RVD transplant and myeloma has returned, we would recommend second-generation IMiDs, pomalidomide, and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Data for daratumumab combined with lenalidomide-dexamethasone or with bortezomib-dexamethasone, look very promising. We need, however, to see more experience of daratumumab together with lenalidomide-dexamethasone or daratumumab together with bortezomib-dexamethasone in patients whose myeloma is refractory to RVD, that is, patients whose myeloma has returned after RVD induction treatment. Of note, pomalidomide, dexamethasone, and daratumumab have just been approved by the FDA and may also be active even in myeloma recurring after RVD treatment.19
Daratumumab in combination will be moving earlier and earlier and may be appropriate to treat the first relapse. I do want to stress, however, that at present I save daratumumab for the second or greater relapse. Daratumumab is active even when relapse occurs after treatment with second-generation IMiDs and proteasome inhibitors.
DR HENRY Before we close, I have a couple practical questions with these antibodies. Daratumumab has the track record of first-treatment severe reactions and long infusion times. How long are you anticipating the first daratumumab treatment takes? There has been some talk that maybe splitting it in half and going over 2 days is easier on the patient and the infusion center. Have you done that?
DR ANDERSON Yes, I think that’s a very important point. We need to be thinking – first and foremost – about efficacy of our therapy. Equally important, however, are the safety profile and the user-friendliness for the patient. Daratumumab infusions are quite long – on the order of 8 hours or longer on day 1 of infusion. And to date, all the clinical trials have used daratumumab infusions weekly for 8 treatments, followed by 8 treatments given every 2 weeks. Then monthly daratumumab is given as a maintenance therapy. Thus, there is a requirement for multiple outpatient clinic visits that can be prolonged.
One of the opportunities that’s being tested is to give daratumumab subcutaneously. While this is being evaluated in protocols now, the results that have been reported at our national meetings look to be quite promising in terms of efficacy, similar to results with the intravenous administration. Obviously, this would allow for a much more convenient clinic visit and shorter time for the patients being treated.
I should mention that the other antibody, elotuzumab, has been approved in combination with lenalidomide and dexamethasone.15 The infusions with lenalidomide, dexamethasone, and the antibody elotuzumab are much shorter, on the order of 2- or 3-hour visits. The place for elotuzumab in the management of relapsed myeloma is yet to be totally defined. We tend to use it now in the setting of more indolent relapses, where patients might have a slowly rising monoclonal protein. Elotuzumab-lenalidomide-dexamethasone has maintained an overall survival advantage at 4 years compared with lenalidomide-dexamethasone when used in relapsed myeloma.
We are quite excited about both antibodies. Daratumumab tends to get most of the activity, as it achieves responses as a single agent,16 and the depth of the responses are markedly increased when it’s combined with lenalidomide-dexamethasone or bortezomib-dexamethasone.17,18 However, one shouldn’t forget elotuzumab15 based on its tolerability and the survival advantage I mentioned at 4 years.
The final point is that we think about myeloma genetically at the time of diagnosis and relapse in terms of standard or high-risk disease. One of the hallmarks of high-risk disease has been 17P deletion or P53 dysfunction. One of the most exciting outcomes of the development of monoclonal antibodies has been the responses observed even in the context of P53 deletion. Clearly, antibody-mediated cellular cytotoxicity, complement-mediated cytotoxicity, and other mechanisms of action of these antibodies do not require normal P53 function. The important point, therefore, is that what has previously been thought of as high-risk disease can nowadays be effectively treated with these new immune treatments, correlating with the marked improvement in survival and overall outcome.
DR HENRY We have outlined 3 kinds of myeloma patients we see, and especially interesting is the last patient, who has relapsed and then progressed, and in whom newer drugs have a role. Thank you for such a complete and thorough discussion, Dr Anderson.
DR HENRY I thought we might discuss some cases of patients with myeloma, starting with a relatively simple case and ending with one that is a little more complicated. For the first case, we have a 56-year-old healthy man with IgG kappa myeloma whose work-up shows he has multiple lytic bone lesions. He has normal renal function, normal calcium, and he’s transplant-eligible by other health issues. I’ll leave the cytogenetics up to you if that changes your approach. How would you develop or pose some options for this man’s treatment to begin with?
DR ANDERSON It’s important to start out by saying that we, in myeloma, have many new classes of drugs and many new opportunities to choose from to treat this patient.1 As you know, we have proteasome inhibitors, the first-generation bortezomib, then carfilzomib and ixazomib. We have the immunomodulatory drugs (IMiDs), thalidomide, and now lenalidomide and pomalidomide. We have a histone deacetylase (HDAC) inhibitor approved called panobinostat, and we have 2 monoclonal antibodies approved, elotuzumab and daratumumab. These classes of medicine have made it possible for 20 different Food and Drug Administration (FDA) approvals in the last 10-15 years. These agents, having been tested in advanced myeloma, have moved toward initial management.
This person is 50 years old. He has adequate liver, heart, lung, and kidney function, so he would be eligible for high-dose therapy and stem-cell transplantation. In terms of initial management, there are many options (Figure 1). We strongly recommend that triplet therapy be used initially. The most common triplets would be lenalidomide, bortezomib, and dexamethasone (RVD)2,3 or cyclophosphamide, bortezomib, and dexamethasone (CyBorD).4 If this man had neuropathy, perhaps carfilzomib, the second-generation proteasome inhibitor, with lenalidomide and dexamethasone could have been used. Why do we use these? The extent and frequency of response with these triplets is nearly universal overall response rate, with three-quarters very good partial and half-complete responses, including minimal residual disease negative responses. In this patient, we would therefore recommend treatment with either RVD or CyBorD for several cycles to maximal response.
He would then have autologous stem cells collected, and it is still the standard of care to proceed to high-dose melphalan and a single high-dose therapy and stem-cell transplantation. The cytogenetics are important: if this patient has standard-risk multiple myeloma, then lenalidomide maintenance would be given after transplant. It is now FDA-approved for this purpose because it can prolong both progression-free and – most importantly – overall survival.5 Standard-risk cytogenetics might, for example, include hyperdiploidy or translocation 11;14. On the other hand, if his myeloma were high-risk and characterized, for example, by 17p deletion, we would carry out the same induction and transplantation, but we would alter the maintenance to incorporate a proteasome inhibitor. Lenalidomide and bortezomib, for example, could be combined. Early data show that using combined maintenance therapy with lenalidomide and bortezomib, can overcome the early relapses that are characteristic of high-risk disease.6
Because of the extent and frequency of response to combination novel therapies, we have undertaken with our French colleagues a clinical trial of RVD in newly diagnosed patients – such as this patient – followed by stem-cell collection in all patients (Figure 2). Then there is a randomization to either early high-dose therapy, melphalan, and autologous stem-cell transplantation, followed by lenalidomide maintenance; or in the other cohort, harvesting of stem cells, additional RVD, and then maintenance with lenalidomide, saving the stem-cell transplant for later.
The French portion of this trial was reported in the New England Journal of Medicine earlier in 2017.7 It showed that patients who received RVD, high-dose melphalan, stem-cell transplant, and had 1 year of lenalidomide maintenance, had a progression-free survival advantage of about 1 year, without an overall survival advantage; compared with those patients who received RVD and lenalidomide maintenance, saving the transplant for later. I would hasten to add that lenalidomide maintenance was given for only 1 year in this trial, and patients in the RVD-only or RVD-and-transplant arms of this trial relapsed after the lenalidomide maintenance was discontinued.
The American portion of this trial is identical. That is, RVD induction is being given and all patients have a stem-cell collection. Half of the patients then go to high-dose melphalan and stem-cell transplant early, and half of them have the transplant only later at the time of relapse. A major difference, however, is that in both the RVD-only and RVD-and-transplant cohorts, patients receive lenalidomide maintenance until progression. This trial has been ongoing since 2009 and is still ongoing, which tells us that patients in both arms – the RVD-only as well as the RVD-and-transplant arms – are doing well.
In the recent STAMINA trial, all patients underwent a single high-dose therapy and transplant. Then there was a randomization to lenalidomide maintenance only in 1 cohort; a randomization to consolidation with RVD posttransplant followed by lenalidomide maintenance in the second cohort; or a randomization to a second high-dose melphalan and stem-cell transplant followed by lenalidomide maintenance in the third cohort.8 I mention this because the outcomes in all 3 cohorts was similar.
I believe this tells us strongly that high-dose therapy and stem-cell transplantation twice – so-called tandem transplant – is no longer a major option in multiple myeloma. For now, however, in this patient, the standard of care would be to undergo induction therapy with triplet, novel combination treatment. Then, stem cells would be collected and high-dose therapy stem-cell transplant would be done, followed either by lenalidomide maintenance for standard disease or lenalidomide and bortezomib maintenance for high-risk disease. We won’t really know if we can delay transplant until the trials I’ve mentioned totally read out. In my clinical practice, if patients have had a major response to their induction therapy and have stem cells harvested, we can then offer them the opportunity to use maintenance therapy and save the transplant as a potential option for later, when myeloma relapses.
DR HENRY In summary then, this would be, in 2017, off-protocol while the data is pending: it’s reasonable to get a deep induction response, collect stem cells, have a discussion with the patient, and then consider high-dose therapy or not.
DR ANDERSON Yes. I think it’s reasonable to discuss it. We need to be open and honest with patients that the standard of care remains transplant, that you incorporate novel treatments before the transplant and novel treatments as maintenance after the transplant. The happy news is that the outcome, especially for patients who have standard-risk myeloma, is at least a decade or longer progression-free survival. It’s an optimistic picture. The data in terms of transplant being needed or not, will come within the next several years.
For now, it is a standard of care to use 1 high-dose melphalan and stem-cell transplant in this setting. I will add into our discussion with patients – besides the opportunity to harvest stem cells and think about whether one needs to do a transplant early on or not – is the issue of toxicity. High-dose melphalan by itself has a small but real secondary incidence of cancer, myelodysplasia, or leukemia. If one uses lenalidomide maintenance after melphalan transplantation treatment, that risk of secondary cancer is slightly increased.
In my experience, if patients have achieved a complete response with induction therapy only, it’s not unreasonable to offer early transplant and be clear that’s the standard of care. The alternative is maintenance with lenalidomide, knowing once the stem cells have been harvested, that transplantation can be an option to treat relapsed myeloma. We have many other options available as well. Time will tell in terms of the ongoing randomized trials as to whether transplant remains central to our treatment paradigm.
DR HENRY This leads us to our second patient. Here we have an older man of 74 years. He’s a professional piano player, so we want to try to avoid peripheral neuropathy in him. He has some mild renal insufficiency and some coronary artery disease, so he’s deemed transplant-ineligible. He has IgG kappa myeloma, and he’s brand new. What would you consider to be options for him for treatment?
DR ANDERSON This brings up the issue of a transplant-ineligible patient. He has significant comorbidity that would make transplantation an increased risk. What we would recommend in such a patient is still triplet induction therapy incorporating novel agents (Figure 1). Lenalidomide, the immunomodulatory drug, can safely be given in the context of neuropathy because it does not cause significant neuropathy. It would need to be dose modified, depending on the degree of renal insufficiency. We would recommend also including proteasome inhibitors. Bortezomib, the first-generation proteasome inhibitor, would be contraindicated because it does have a small but real attendant neuropathy. If, however, it is given weekly and subcutaneously, the risk of attendant neuropathy is quite low. In this patient, therefore, one could start with lenalidomide and bortezomib weekly and subcutaneously,1,2 with a very early and vigilant follow-up for the earliest signs of neuropathy, so as not to allow it to develop and compromise his career.
Alternatively, one could use a proteasome inhibitor that does not have attendant neuropathy. Carfilzomib, the second-generation proteasome inhibitor, does not have neuropathy.9 But we would need to have caution here, because this patient has a history of coronary artery disease, and carfilzomib has a very small, but real, incidence of cardiac toxicity so would need to be used judiciously in this setting. The third proteasome inhibitor, ixazomib, is the next-generation bortezomib-class proteasome inhibitor, and it’s oral.10 It has less neuropathy than does bortezomib, so in my view is a very realistic option for him together with lenalidomide. It does have a small incidence of neuropathy, so close monitoring for neuropathy would be indicated. We could use lenalidomide–dexamethasone as a doublet and avoid neuropathy,11 but usually doublets are reserved only for frail patients.
My recommendation, therefore, would be RVD with the bortezomib weekly or subcutaneously, or alternatively, lenalidomide, ixazomib, dexamethasone as an all-oral regimen as induction therapy. In my view, this 74-year-old patient with comorbidity is not a transplant candidate. However, one can be very optimistic with this patient. The likelihood that he could have myeloma as a chronic illness and die from something else is quite high. Initial induction triplet therapy would achieve a very high response extent and frequency. The durability would be long, especially with lenalidomide maintenance if it’s standard-risk myeloma or lenalidomide and a proteasome inhibitor, probably ixazomib in this setting, if he were to have high-risk myeloma.
If myeloma relapses, then there are many options that could be used in this patient and achieve years of progression-free and overall survival. Indeed, he is 74 years old and will respond very well to induction triplet therapy, with many years’ duration of response due to continuous lenalidomide or lenalidomide and proteasome inhibitor maintenance. Then there are many effective options to treat relapsed therapy using triplet novel agents. Therefore, his lifespan is unlikely to be shortened by multiple myeloma.
DR HENRY It’s so incredible compared with what it was when I trained. The next patient, a 45-year-old woman with IgG lambda myeloma, has had RVD induction and responded. She had lenalidomide maintenance, but then she progressed, and she got her stem-cell transplant, and she’s progressing after that. I guess we’re looking here to fold in some of the newer agents. How you would you do that in this patient?DR ANDERSON Yes. I think one of the most remarkable and exciting developments with myeloma is the rapid approval of the novel classes of agents that I mentioned earlier – the proteasome inhibitors, the immunomodulatory drugs, the HDAC inhibitor, and the monoclonal antibodies.1 They’re particularly relevant in a patient such as this one, whose myeloma has relapsed after what would be considered standard therapy for a young person with standard-risk myeloma. This patient had RVD and maintenance therapy, and then progressed. The transplant was given for relapsed myeloma. The opportunity to use stem-cell transplant in patients when myeloma becomes active after maintenance should not be forgotten as it can be very effective. In all the trials done to date in which early versus late transplant are compared, there have been similar outcomes. Therefore, if the transplant isn’t done early, don’t forget that it’s an option at the time the myeloma progresses. I do want to mention, that there are lots of options for relapsed myeloma (Figures 3 and 4). I mentioned RVD or CyBorD as initial triplet therapies.2-4 In North America, those are the 2 most common regimens. If myeloma then relapses and is resistant to RVD or to CyBorD, then we need to identify alternatives.
We also need to think about the comorbidities in the patient – issues such as age, neuropathy, presence of renal dysfunction, and other clinical factors. And we need to think about what treatment they’ve had in the past. This patient has had RVD, maintenance with lenalidomide, and a stem-cell transplant. We can offer patients a variety of therapies, but in the context of resistance to the first-generation proteasome inhibitor bortezomib and the first-generation immunomodulatory drug lenalidomide, we would strongly recommend the second-generation immunomodulatory drug pomalidomide12 together with a second-generation proteasome inhibitor, be that carfilzomib13 or ixazomib.14 When one uses the second-generation IMiDs and proteasome inhibitors together, there’s a very high frequency of response in the order of 70%-80%, which lasts years.
Besides carfilzomib and ixazomib proteasome inhibitors, we also have elotuzumab and daratumumab, the monoclonal antibodies.15-17 These agents have been FDA approved to treat patients such as this one who has had 1-3 previous therapies for their myeloma. All of them have been approved in randomized phase 3 trials compared with lenalidomide-dexamethasone in the control arm.13-15,17 They’ve all been found to be superior. Although lenalidomide-dexamethasone combined with daratumumab, ixazomib, elotuzumab, or carfilzomib is superior to lenalidomide in relapsed myeloma, the situation in North America, as in this patient, is usually that patients have had lenalidomide-dexamethasone as part of their initial treatment and their myeloma is refractory to lenalidomide.
Hence, we recommend, that we go to the second-generation pomalidomide and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Having said that, the treatment paradigm is evolving. For example, the monoclonal antibody daratumumab was initially approved by the FDA in multiply relapsed disease as a single agent because it achieves a 30% response rate.16 It now has been moved earlier into the first relapse of multiple myeloma, where it achieves much higher response rates when combined with lenalidomide–dexamethasone or combined with bortezomib–dexamethasone.17,18 Response rates of 70%-80% can be achieved, including minimal residual disease negative complete responses.
Today, in a patient who has had RVD transplant and myeloma has returned, we would recommend second-generation IMiDs, pomalidomide, and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Data for daratumumab combined with lenalidomide-dexamethasone or with bortezomib-dexamethasone, look very promising. We need, however, to see more experience of daratumumab together with lenalidomide-dexamethasone or daratumumab together with bortezomib-dexamethasone in patients whose myeloma is refractory to RVD, that is, patients whose myeloma has returned after RVD induction treatment. Of note, pomalidomide, dexamethasone, and daratumumab have just been approved by the FDA and may also be active even in myeloma recurring after RVD treatment.19
Daratumumab in combination will be moving earlier and earlier and may be appropriate to treat the first relapse. I do want to stress, however, that at present I save daratumumab for the second or greater relapse. Daratumumab is active even when relapse occurs after treatment with second-generation IMiDs and proteasome inhibitors.
DR HENRY Before we close, I have a couple practical questions with these antibodies. Daratumumab has the track record of first-treatment severe reactions and long infusion times. How long are you anticipating the first daratumumab treatment takes? There has been some talk that maybe splitting it in half and going over 2 days is easier on the patient and the infusion center. Have you done that?
DR ANDERSON Yes, I think that’s a very important point. We need to be thinking – first and foremost – about efficacy of our therapy. Equally important, however, are the safety profile and the user-friendliness for the patient. Daratumumab infusions are quite long – on the order of 8 hours or longer on day 1 of infusion. And to date, all the clinical trials have used daratumumab infusions weekly for 8 treatments, followed by 8 treatments given every 2 weeks. Then monthly daratumumab is given as a maintenance therapy. Thus, there is a requirement for multiple outpatient clinic visits that can be prolonged.
One of the opportunities that’s being tested is to give daratumumab subcutaneously. While this is being evaluated in protocols now, the results that have been reported at our national meetings look to be quite promising in terms of efficacy, similar to results with the intravenous administration. Obviously, this would allow for a much more convenient clinic visit and shorter time for the patients being treated.
I should mention that the other antibody, elotuzumab, has been approved in combination with lenalidomide and dexamethasone.15 The infusions with lenalidomide, dexamethasone, and the antibody elotuzumab are much shorter, on the order of 2- or 3-hour visits. The place for elotuzumab in the management of relapsed myeloma is yet to be totally defined. We tend to use it now in the setting of more indolent relapses, where patients might have a slowly rising monoclonal protein. Elotuzumab-lenalidomide-dexamethasone has maintained an overall survival advantage at 4 years compared with lenalidomide-dexamethasone when used in relapsed myeloma.
We are quite excited about both antibodies. Daratumumab tends to get most of the activity, as it achieves responses as a single agent,16 and the depth of the responses are markedly increased when it’s combined with lenalidomide-dexamethasone or bortezomib-dexamethasone.17,18 However, one shouldn’t forget elotuzumab15 based on its tolerability and the survival advantage I mentioned at 4 years.
The final point is that we think about myeloma genetically at the time of diagnosis and relapse in terms of standard or high-risk disease. One of the hallmarks of high-risk disease has been 17P deletion or P53 dysfunction. One of the most exciting outcomes of the development of monoclonal antibodies has been the responses observed even in the context of P53 deletion. Clearly, antibody-mediated cellular cytotoxicity, complement-mediated cytotoxicity, and other mechanisms of action of these antibodies do not require normal P53 function. The important point, therefore, is that what has previously been thought of as high-risk disease can nowadays be effectively treated with these new immune treatments, correlating with the marked improvement in survival and overall outcome.
DR HENRY We have outlined 3 kinds of myeloma patients we see, and especially interesting is the last patient, who has relapsed and then progressed, and in whom newer drugs have a role. Thank you for such a complete and thorough discussion, Dr Anderson.
1. Kumar SK, Callander NS, Alsina M, et al. Multiple Myeloma, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:230-269.
2. Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly-diagnosed multiple myeloma. Blood. 2010;116:679-686.
3. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777: a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
4. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127:2569-2574.
5. McCarthy PL, Owzar K, Hofmeister CC, et al. A phase III study of lenalidomide after transplant for multiple myeloma. N Engl J Med. 2012;366:1770-1781.
6. Nooka AK, Kaufman JL, Muppidi S, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib, and dexamethasone (RVD) in high risk myeloma patients. Leukemia. 2014;28:690-693.
7. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib and dexamethasone with transplantation in myeloma. N Engl J Med. 2017;376:1311-1320.
8. Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (len) and dexamethasone (RVD) consolidation with len maintenance (ACM), tandem autohct with len maintenance (TAM) and autohct with len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702 – StaMINA Trial). Abstract LBA-1. Presented at the 2016 ASH Annual Meeting, December 6, 2016; San Diego, CA.
9. Dytfeld D, Jasielec J, Griffith KA, et al: Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162-164.
10. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503-1512
11. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. for the FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371:906-917.
12. Richardson P, Siegel D, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826-1832.
13. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. for the ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142-152
14. Moreau P, Masszi T, Grzasko N, et al. for the TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;28;374:1621-1634.
15. Lonial S, Dimopoulos M, Palumbo A, et al. for the ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621-631.
16. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD 38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207-1219.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. for the CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
19. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974-981.
1. Kumar SK, Callander NS, Alsina M, et al. Multiple Myeloma, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:230-269.
2. Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly-diagnosed multiple myeloma. Blood. 2010;116:679-686.
3. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777: a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
4. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127:2569-2574.
5. McCarthy PL, Owzar K, Hofmeister CC, et al. A phase III study of lenalidomide after transplant for multiple myeloma. N Engl J Med. 2012;366:1770-1781.
6. Nooka AK, Kaufman JL, Muppidi S, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib, and dexamethasone (RVD) in high risk myeloma patients. Leukemia. 2014;28:690-693.
7. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib and dexamethasone with transplantation in myeloma. N Engl J Med. 2017;376:1311-1320.
8. Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (len) and dexamethasone (RVD) consolidation with len maintenance (ACM), tandem autohct with len maintenance (TAM) and autohct with len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702 – StaMINA Trial). Abstract LBA-1. Presented at the 2016 ASH Annual Meeting, December 6, 2016; San Diego, CA.
9. Dytfeld D, Jasielec J, Griffith KA, et al: Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162-164.
10. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503-1512
11. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. for the FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371:906-917.
12. Richardson P, Siegel D, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826-1832.
13. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. for the ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142-152
14. Moreau P, Masszi T, Grzasko N, et al. for the TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;28;374:1621-1634.
15. Lonial S, Dimopoulos M, Palumbo A, et al. for the ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621-631.
16. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD 38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207-1219.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. for the CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
19. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974-981.
Caution urged over real-world bleeding risk with ibrutinib
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to an almost 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Paul R. Kunk, MD, of University of Virginia, Charlottesville, and his colleagues. Their report is in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting but the authors suggested that this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria. These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice,” the researchers wrote.
They conducted a review of patients attending their center and associated regional clinics between January 2012 and May 2016. They identified 70 patients, average age 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%) and mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström macroglobulinemia (1%).
The analysis showed that bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis. However, major bleeding, defined as grade 3, occurred in 13 patients (19%), a figure that the authors noted was greater than the rate of around 7% reported by clinical trials.
Of these patients, seven were taking combined antiplatelet and anticoagulant therapy, four were taking antiplatelets alone, one was taking an anticoagulant agent alone, and one was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding included antiplatelet or anticoagulant medication, the combination of the two medications or interacting medications, anemia (hemoglobin less than 12 g/dL) and an elevated international normalized ratio (greater than 1.5).
However, in a multivariate analysis, only combined antiplatelet and anticoagulant use (hazard ratio, 20.0; 95% confidence interval, 2.1-200.0; P less than .01) and an elevated INR (HR, 4.6; 95% CI, 1.1-19.6; P less than .01) remained statistically significant.
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, they said their findings confirmed “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.
“As ibrutinib increases in use, it is paramount to increase awareness of the known adverse events. This is especially important given the association of ibrutinib use with atrial fibrillation,” they wrote.
They noted that their trial was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive,” they noted.
SOURCE: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to an almost 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Paul R. Kunk, MD, of University of Virginia, Charlottesville, and his colleagues. Their report is in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting but the authors suggested that this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria. These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice,” the researchers wrote.
They conducted a review of patients attending their center and associated regional clinics between January 2012 and May 2016. They identified 70 patients, average age 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%) and mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström macroglobulinemia (1%).
The analysis showed that bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis. However, major bleeding, defined as grade 3, occurred in 13 patients (19%), a figure that the authors noted was greater than the rate of around 7% reported by clinical trials.
Of these patients, seven were taking combined antiplatelet and anticoagulant therapy, four were taking antiplatelets alone, one was taking an anticoagulant agent alone, and one was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding included antiplatelet or anticoagulant medication, the combination of the two medications or interacting medications, anemia (hemoglobin less than 12 g/dL) and an elevated international normalized ratio (greater than 1.5).
However, in a multivariate analysis, only combined antiplatelet and anticoagulant use (hazard ratio, 20.0; 95% confidence interval, 2.1-200.0; P less than .01) and an elevated INR (HR, 4.6; 95% CI, 1.1-19.6; P less than .01) remained statistically significant.
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, they said their findings confirmed “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.
“As ibrutinib increases in use, it is paramount to increase awareness of the known adverse events. This is especially important given the association of ibrutinib use with atrial fibrillation,” they wrote.
They noted that their trial was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive,” they noted.
SOURCE: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to an almost 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Paul R. Kunk, MD, of University of Virginia, Charlottesville, and his colleagues. Their report is in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting but the authors suggested that this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria. These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice,” the researchers wrote.
They conducted a review of patients attending their center and associated regional clinics between January 2012 and May 2016. They identified 70 patients, average age 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%) and mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström macroglobulinemia (1%).
The analysis showed that bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis. However, major bleeding, defined as grade 3, occurred in 13 patients (19%), a figure that the authors noted was greater than the rate of around 7% reported by clinical trials.
Of these patients, seven were taking combined antiplatelet and anticoagulant therapy, four were taking antiplatelets alone, one was taking an anticoagulant agent alone, and one was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding included antiplatelet or anticoagulant medication, the combination of the two medications or interacting medications, anemia (hemoglobin less than 12 g/dL) and an elevated international normalized ratio (greater than 1.5).
However, in a multivariate analysis, only combined antiplatelet and anticoagulant use (hazard ratio, 20.0; 95% confidence interval, 2.1-200.0; P less than .01) and an elevated INR (HR, 4.6; 95% CI, 1.1-19.6; P less than .01) remained statistically significant.
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, they said their findings confirmed “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.
“As ibrutinib increases in use, it is paramount to increase awareness of the known adverse events. This is especially important given the association of ibrutinib use with atrial fibrillation,” they wrote.
They noted that their trial was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive,” they noted.
SOURCE: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point: Clinicians should exercise caution when prescribing antiplatelet and anticoagulant medications in people taking the Bruton tyrosine kinase inhibitor ibrutinib.
Major finding: The use of both antiplatelet and anticoagulant therapy significantly increased the risk of a major bleed event (HR, 19.2; 95% CI, 2.3-166.7; P less than .01) in patients also taking ibrutinib.
Study details: A retrospective analysis of prescription data from 70 patients seen at a single U.S. cancer center and its regional clinics between January 2012 and May 2016.
Disclosures: Two of the authors reported receiving clinical trial support from Acerta and Abbvie.
Source: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
Variants in five genes signal TNBC risk
Women with germline pathogenic variants in five genes are at high risk for developing triple-negative breast cancer, and have a greater than 20% lifetime risk for breast cancer in general, results of a large study suggest.
Multigene testing of nearly 11,000 women with triple-negative breast cancer (TNBC; lacking estrogen, progesterone, and human epidermal growth factor receptors) showed that germline pathogenic variants in BARD1, BRCA1, BRCA2, PALB2, and RAD51D were associated with risk for clinical TNBC ranging from nearly sixfold to more than 16-fold higher than that of women without the genetic variants, reported Fergus J. Couch, PhD, of the Mayo Clinic, Rochester, Minn., and his colleagues.
“The results suggest that all TNBC patients should undergo multigene panel testing, regardless of age at diagnosis or family history of cancer, for improved cancer risk assessment and because of the ongoing development of targeted therapeutic approaches for TNBC patients with mutations in predisposition genes,” they wrote. Their report is in the Journal of the National Cancer Institute.
Although National Comprehensive Cancer Network guidelines recommend testing for the cancer predisposition genes BRCA1 and BRCA2 in women with a TNBC diagnosis at age 60 or younger or those with a family history of breast and/or ovarian cancer, the picture is less clear regarding genetic predisposition to TNBC, the authors noted.
“[R]ecommendations for testing of other genes are not fully established because the risks of TNBC associated with mutations in cancer predisposition genes have not been established. Thus, a better understanding of gene-specific risks for TNBC is needed to identify the genes that should be tested in the setting of TNBC,” they wrote.
The investigators looked for associations between deleterious mutations in cancer predisposition genes and TNBC among 8,753 patients with TNBC testing with a 21-gene assay, and among 2,148 women tested for 17 genes in studies conducted by the Triple Negative Breast Cancer Consortium.
They found that among white women, germline pathogenic variants were associated with the following odds ratios (OR) for TNBC (all P values less than .0001):
- BRCA2 = 5.42
- BARD1 = 5.92
- RAD51D = 6.97
- PALB2 = 14.41
- BRCA1 = 16.27
Although there were insufficient data on the risks for African American women, an exploratory analysis showed that risks for TNBC associated with specific pathogenic variants were similar to those for white women, the authors said.
The pathogenic variants were detected in 12% of all patients in the study.
“Continued study of gene-specific risks for breast cancer subtypes may lead to tailored medical management recommendations for PV [pathogenic variant] carriers. Consistent with this hypothesis, initial studies evaluating intensified screening in high-risk women have suggested that a decrease in mortality from TNBC can be achieved,” they wrote.
The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
SOURCE: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
Women with germline pathogenic variants in five genes are at high risk for developing triple-negative breast cancer, and have a greater than 20% lifetime risk for breast cancer in general, results of a large study suggest.
Multigene testing of nearly 11,000 women with triple-negative breast cancer (TNBC; lacking estrogen, progesterone, and human epidermal growth factor receptors) showed that germline pathogenic variants in BARD1, BRCA1, BRCA2, PALB2, and RAD51D were associated with risk for clinical TNBC ranging from nearly sixfold to more than 16-fold higher than that of women without the genetic variants, reported Fergus J. Couch, PhD, of the Mayo Clinic, Rochester, Minn., and his colleagues.
“The results suggest that all TNBC patients should undergo multigene panel testing, regardless of age at diagnosis or family history of cancer, for improved cancer risk assessment and because of the ongoing development of targeted therapeutic approaches for TNBC patients with mutations in predisposition genes,” they wrote. Their report is in the Journal of the National Cancer Institute.
Although National Comprehensive Cancer Network guidelines recommend testing for the cancer predisposition genes BRCA1 and BRCA2 in women with a TNBC diagnosis at age 60 or younger or those with a family history of breast and/or ovarian cancer, the picture is less clear regarding genetic predisposition to TNBC, the authors noted.
“[R]ecommendations for testing of other genes are not fully established because the risks of TNBC associated with mutations in cancer predisposition genes have not been established. Thus, a better understanding of gene-specific risks for TNBC is needed to identify the genes that should be tested in the setting of TNBC,” they wrote.
The investigators looked for associations between deleterious mutations in cancer predisposition genes and TNBC among 8,753 patients with TNBC testing with a 21-gene assay, and among 2,148 women tested for 17 genes in studies conducted by the Triple Negative Breast Cancer Consortium.
They found that among white women, germline pathogenic variants were associated with the following odds ratios (OR) for TNBC (all P values less than .0001):
- BRCA2 = 5.42
- BARD1 = 5.92
- RAD51D = 6.97
- PALB2 = 14.41
- BRCA1 = 16.27
Although there were insufficient data on the risks for African American women, an exploratory analysis showed that risks for TNBC associated with specific pathogenic variants were similar to those for white women, the authors said.
The pathogenic variants were detected in 12% of all patients in the study.
“Continued study of gene-specific risks for breast cancer subtypes may lead to tailored medical management recommendations for PV [pathogenic variant] carriers. Consistent with this hypothesis, initial studies evaluating intensified screening in high-risk women have suggested that a decrease in mortality from TNBC can be achieved,” they wrote.
The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
SOURCE: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
Women with germline pathogenic variants in five genes are at high risk for developing triple-negative breast cancer, and have a greater than 20% lifetime risk for breast cancer in general, results of a large study suggest.
Multigene testing of nearly 11,000 women with triple-negative breast cancer (TNBC; lacking estrogen, progesterone, and human epidermal growth factor receptors) showed that germline pathogenic variants in BARD1, BRCA1, BRCA2, PALB2, and RAD51D were associated with risk for clinical TNBC ranging from nearly sixfold to more than 16-fold higher than that of women without the genetic variants, reported Fergus J. Couch, PhD, of the Mayo Clinic, Rochester, Minn., and his colleagues.
“The results suggest that all TNBC patients should undergo multigene panel testing, regardless of age at diagnosis or family history of cancer, for improved cancer risk assessment and because of the ongoing development of targeted therapeutic approaches for TNBC patients with mutations in predisposition genes,” they wrote. Their report is in the Journal of the National Cancer Institute.
Although National Comprehensive Cancer Network guidelines recommend testing for the cancer predisposition genes BRCA1 and BRCA2 in women with a TNBC diagnosis at age 60 or younger or those with a family history of breast and/or ovarian cancer, the picture is less clear regarding genetic predisposition to TNBC, the authors noted.
“[R]ecommendations for testing of other genes are not fully established because the risks of TNBC associated with mutations in cancer predisposition genes have not been established. Thus, a better understanding of gene-specific risks for TNBC is needed to identify the genes that should be tested in the setting of TNBC,” they wrote.
The investigators looked for associations between deleterious mutations in cancer predisposition genes and TNBC among 8,753 patients with TNBC testing with a 21-gene assay, and among 2,148 women tested for 17 genes in studies conducted by the Triple Negative Breast Cancer Consortium.
They found that among white women, germline pathogenic variants were associated with the following odds ratios (OR) for TNBC (all P values less than .0001):
- BRCA2 = 5.42
- BARD1 = 5.92
- RAD51D = 6.97
- PALB2 = 14.41
- BRCA1 = 16.27
Although there were insufficient data on the risks for African American women, an exploratory analysis showed that risks for TNBC associated with specific pathogenic variants were similar to those for white women, the authors said.
The pathogenic variants were detected in 12% of all patients in the study.
“Continued study of gene-specific risks for breast cancer subtypes may lead to tailored medical management recommendations for PV [pathogenic variant] carriers. Consistent with this hypothesis, initial studies evaluating intensified screening in high-risk women have suggested that a decrease in mortality from TNBC can be achieved,” they wrote.
The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
SOURCE: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
FROM JOURNAL OF THE NATIONAL CANCER INSTITUTE
Key clinical point: Pathogenic variants in five cancer predisposition genes are associated with significantly increased risk for triple negative breast cancer (TNBC).
Major finding: Pathogenic variants in BRCA1 were associated with a more than 16-fold risk for TNBC.
Study details: Retrospective review of multigene assay testing in 10,901 women with TNBC.
Disclosures: The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
Source: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
Broad genomic testing of NSCLC in community oncology disappoints
results of a retrospective cohort study reported in JAMA suggest.
Investigators led by Carolyn J. Presley, MD, a thoracic and geriatric medical oncologist at the Ohio State University Comprehensive Cancer Center, Columbus, assessed outcomes among more than 5,500 patients with advanced nonsquamous NSCLC treated mainly in U.S. community practices. Overall, 15% had broad-based genomic testing (next-generation sequencing evaluating more than 30 cancer genes).
Main results showed that, among the patients having broad testing, less than 5% received a targeted treatment based on results that were not attainable with routine testing for common alterations in EGFR and ALK genes. Moreover, survival after broad testing was not better than that after routine testing.
“This study highlights how broad-based genomic sequencing has disseminated beyond traditional research settings ahead of a demonstrated association with better survival,” Dr. Presley and her coinvestigators write. They speculate that community uptake is being driven by the ease and cost of ordering a single comprehensive test, perceived benefit, attempts to conserve tissue, and hopes of improved survival if a targeted treatment is available.
“The lack of an association between broad-based genomic sequencing and survival is likely multifactorial,” the investigators maintain. “First, there were few genetic alterations identified with available targeted treatments. Second, even among those patients for whom targeted treatments were available, the treatments may not yield a substantial survival benefit or patients may not have had access to targeted agents due to financial barriers. Decision support for clinicians once they receive broad-based genomic sequencing results may also be needed.”
Study details
Dr. Presley and colleagues used the Flatiron Health Database to identify patients with advanced NSCLC who received care at 191 oncology practices across the United States during 2011-2016. The 5,688 patients studied had stage IIIB, stage IV, or unresectable nonsquamous NSCLC and received at least one line of treatment.
Overall, 15.4% received broad-based genomic sequencing of their tumor, while the rest received routine testing for EGFR and/or ALK alterations only, according to the results reported.
In the broadly tested group, merely 4.5% were given targeted treatment based on testing results. Another 9.8% received routine EGFR/ALK-targeted treatment, and 85.1% did not receive any targeted treatment.
The 12-month unadjusted mortality rate was 49.2% for patients undergoing broad testing, compared with 35.9% for patients undergoing routine testing.
In an instrumental variable analysis done to account for confounding, the 12-month predicted probability of death was 41.1% after broad testing and 44.4% after routine testing (P = .63).
Findings were similar in a propensity score–matched survival analysis (42.0% vs. 45.1%; hazard ratio, 0.92; P = .40), although there was some suggestion of a benefit of broad testing over routine testing in a Kaplan-Meier analysis among the entire unmatched cohort (HR, 0.69; P less than .001).
“Improved access to research clinical trials in the community setting may improve use of mutational data,” the investigators speculate. “Given the paucity of targeted agents, efforts to increase access to broad-based genomic sequencing should be paired with efforts to facilitate clinical trial enrollment.”
Dr. Presley disclosed that she receives grants from the Yale Lung SPORE Career Development Award, the Robert Wood Johnson/Veterans Affairs Clinical Scholars Program, and The Ohio State University K12 Training Grant for clinical faculty investigators. The study was funded by the Veterans Affairs Robert Wood Johnson Clinical Scholar Program and the Yale Lung SPORE Career Development Award.
SOURCE: Presley CJ et al. JAMA. 2018 Aug 7. doi: 10.1001/jama.2018.9824.
There still may be a role for broad-based genomic testing in patients with NSCLC treated in community oncology practices, according to editorialists Paul A. Bunn Jr., MD, and Dara L. Aisner, MD, PhD. They discussed several study limitations that leave the matter unsettled.
Importantly, the majority of patients in whom this testing identified a potentially treatable alteration did not receive the treatment. “This gap between finding and treating molecular alterations in the community-based clinical setting highlights the reality that obtaining more tumor genomic information must be complemented with clinician education and decision support to understand the importance of matched therapy, and demonstrates a strength of harnessing EMR data to identify potential gaps in practice,” they maintain.
The study did not assess important outcomes other than survival, such as progression-free survival and response rate, Dr. Bunn and Dr. Aisner further note. Previous research has shown that tyrosine kinase inhibitors, for example, improve some of these outcomes without altering survival.
Another limitation was that the study period predated regulatory approval of some relevant targeted therapies and came shortly on the heels of approval of a targeted therapy for ALK rearrangements. And additional therapies are in the pipeline.
“[T]he incremental value of a cutoff of 30 genes analyzed may place the bar too high to appreciate a survival advantage and the tissue, time, and cost savings due to next-generation sequencing were not considered,” the editorialists point out. The optimal number of genes is unclear and likely to change over time.
Finally, the reports oncologists receive from broad-based genomic sequencing may be long and complex, which may deter them from pursuing appropriate therapy, Dr. Bunn and Dr. Aisner propose.
“The study… provides important insights into how broad-based genomic sequencing is used in the community oncology setting, where the majority of patients with advanced NSCLC in the United States receive care,” they conclude. “However, the limitations of this investigation suggest that the authors’ conclusion that broad testing is not warranted should be tempered to ensure that patients receive the right therapy for the right alteration at the right time.”
Paul A. Bunn Jr., MD, is with the University of Colorado Cancer Center and department of medical oncology, University of Colorado, Denver and Dara L. Aisner, MD, PhD, is with the University of Colorado Cancer Center and department of pathology, University of Colorado, Aurora. These comments were excerpted from an accompanying editorial .
There still may be a role for broad-based genomic testing in patients with NSCLC treated in community oncology practices, according to editorialists Paul A. Bunn Jr., MD, and Dara L. Aisner, MD, PhD. They discussed several study limitations that leave the matter unsettled.
Importantly, the majority of patients in whom this testing identified a potentially treatable alteration did not receive the treatment. “This gap between finding and treating molecular alterations in the community-based clinical setting highlights the reality that obtaining more tumor genomic information must be complemented with clinician education and decision support to understand the importance of matched therapy, and demonstrates a strength of harnessing EMR data to identify potential gaps in practice,” they maintain.
The study did not assess important outcomes other than survival, such as progression-free survival and response rate, Dr. Bunn and Dr. Aisner further note. Previous research has shown that tyrosine kinase inhibitors, for example, improve some of these outcomes without altering survival.
Another limitation was that the study period predated regulatory approval of some relevant targeted therapies and came shortly on the heels of approval of a targeted therapy for ALK rearrangements. And additional therapies are in the pipeline.
“[T]he incremental value of a cutoff of 30 genes analyzed may place the bar too high to appreciate a survival advantage and the tissue, time, and cost savings due to next-generation sequencing were not considered,” the editorialists point out. The optimal number of genes is unclear and likely to change over time.
Finally, the reports oncologists receive from broad-based genomic sequencing may be long and complex, which may deter them from pursuing appropriate therapy, Dr. Bunn and Dr. Aisner propose.
“The study… provides important insights into how broad-based genomic sequencing is used in the community oncology setting, where the majority of patients with advanced NSCLC in the United States receive care,” they conclude. “However, the limitations of this investigation suggest that the authors’ conclusion that broad testing is not warranted should be tempered to ensure that patients receive the right therapy for the right alteration at the right time.”
Paul A. Bunn Jr., MD, is with the University of Colorado Cancer Center and department of medical oncology, University of Colorado, Denver and Dara L. Aisner, MD, PhD, is with the University of Colorado Cancer Center and department of pathology, University of Colorado, Aurora. These comments were excerpted from an accompanying editorial .
There still may be a role for broad-based genomic testing in patients with NSCLC treated in community oncology practices, according to editorialists Paul A. Bunn Jr., MD, and Dara L. Aisner, MD, PhD. They discussed several study limitations that leave the matter unsettled.
Importantly, the majority of patients in whom this testing identified a potentially treatable alteration did not receive the treatment. “This gap between finding and treating molecular alterations in the community-based clinical setting highlights the reality that obtaining more tumor genomic information must be complemented with clinician education and decision support to understand the importance of matched therapy, and demonstrates a strength of harnessing EMR data to identify potential gaps in practice,” they maintain.
The study did not assess important outcomes other than survival, such as progression-free survival and response rate, Dr. Bunn and Dr. Aisner further note. Previous research has shown that tyrosine kinase inhibitors, for example, improve some of these outcomes without altering survival.
Another limitation was that the study period predated regulatory approval of some relevant targeted therapies and came shortly on the heels of approval of a targeted therapy for ALK rearrangements. And additional therapies are in the pipeline.
“[T]he incremental value of a cutoff of 30 genes analyzed may place the bar too high to appreciate a survival advantage and the tissue, time, and cost savings due to next-generation sequencing were not considered,” the editorialists point out. The optimal number of genes is unclear and likely to change over time.
Finally, the reports oncologists receive from broad-based genomic sequencing may be long and complex, which may deter them from pursuing appropriate therapy, Dr. Bunn and Dr. Aisner propose.
“The study… provides important insights into how broad-based genomic sequencing is used in the community oncology setting, where the majority of patients with advanced NSCLC in the United States receive care,” they conclude. “However, the limitations of this investigation suggest that the authors’ conclusion that broad testing is not warranted should be tempered to ensure that patients receive the right therapy for the right alteration at the right time.”
Paul A. Bunn Jr., MD, is with the University of Colorado Cancer Center and department of medical oncology, University of Colorado, Denver and Dara L. Aisner, MD, PhD, is with the University of Colorado Cancer Center and department of pathology, University of Colorado, Aurora. These comments were excerpted from an accompanying editorial .
results of a retrospective cohort study reported in JAMA suggest.
Investigators led by Carolyn J. Presley, MD, a thoracic and geriatric medical oncologist at the Ohio State University Comprehensive Cancer Center, Columbus, assessed outcomes among more than 5,500 patients with advanced nonsquamous NSCLC treated mainly in U.S. community practices. Overall, 15% had broad-based genomic testing (next-generation sequencing evaluating more than 30 cancer genes).
Main results showed that, among the patients having broad testing, less than 5% received a targeted treatment based on results that were not attainable with routine testing for common alterations in EGFR and ALK genes. Moreover, survival after broad testing was not better than that after routine testing.
“This study highlights how broad-based genomic sequencing has disseminated beyond traditional research settings ahead of a demonstrated association with better survival,” Dr. Presley and her coinvestigators write. They speculate that community uptake is being driven by the ease and cost of ordering a single comprehensive test, perceived benefit, attempts to conserve tissue, and hopes of improved survival if a targeted treatment is available.
“The lack of an association between broad-based genomic sequencing and survival is likely multifactorial,” the investigators maintain. “First, there were few genetic alterations identified with available targeted treatments. Second, even among those patients for whom targeted treatments were available, the treatments may not yield a substantial survival benefit or patients may not have had access to targeted agents due to financial barriers. Decision support for clinicians once they receive broad-based genomic sequencing results may also be needed.”
Study details
Dr. Presley and colleagues used the Flatiron Health Database to identify patients with advanced NSCLC who received care at 191 oncology practices across the United States during 2011-2016. The 5,688 patients studied had stage IIIB, stage IV, or unresectable nonsquamous NSCLC and received at least one line of treatment.
Overall, 15.4% received broad-based genomic sequencing of their tumor, while the rest received routine testing for EGFR and/or ALK alterations only, according to the results reported.
In the broadly tested group, merely 4.5% were given targeted treatment based on testing results. Another 9.8% received routine EGFR/ALK-targeted treatment, and 85.1% did not receive any targeted treatment.
The 12-month unadjusted mortality rate was 49.2% for patients undergoing broad testing, compared with 35.9% for patients undergoing routine testing.
In an instrumental variable analysis done to account for confounding, the 12-month predicted probability of death was 41.1% after broad testing and 44.4% after routine testing (P = .63).
Findings were similar in a propensity score–matched survival analysis (42.0% vs. 45.1%; hazard ratio, 0.92; P = .40), although there was some suggestion of a benefit of broad testing over routine testing in a Kaplan-Meier analysis among the entire unmatched cohort (HR, 0.69; P less than .001).
“Improved access to research clinical trials in the community setting may improve use of mutational data,” the investigators speculate. “Given the paucity of targeted agents, efforts to increase access to broad-based genomic sequencing should be paired with efforts to facilitate clinical trial enrollment.”
Dr. Presley disclosed that she receives grants from the Yale Lung SPORE Career Development Award, the Robert Wood Johnson/Veterans Affairs Clinical Scholars Program, and The Ohio State University K12 Training Grant for clinical faculty investigators. The study was funded by the Veterans Affairs Robert Wood Johnson Clinical Scholar Program and the Yale Lung SPORE Career Development Award.
SOURCE: Presley CJ et al. JAMA. 2018 Aug 7. doi: 10.1001/jama.2018.9824.
results of a retrospective cohort study reported in JAMA suggest.
Investigators led by Carolyn J. Presley, MD, a thoracic and geriatric medical oncologist at the Ohio State University Comprehensive Cancer Center, Columbus, assessed outcomes among more than 5,500 patients with advanced nonsquamous NSCLC treated mainly in U.S. community practices. Overall, 15% had broad-based genomic testing (next-generation sequencing evaluating more than 30 cancer genes).
Main results showed that, among the patients having broad testing, less than 5% received a targeted treatment based on results that were not attainable with routine testing for common alterations in EGFR and ALK genes. Moreover, survival after broad testing was not better than that after routine testing.
“This study highlights how broad-based genomic sequencing has disseminated beyond traditional research settings ahead of a demonstrated association with better survival,” Dr. Presley and her coinvestigators write. They speculate that community uptake is being driven by the ease and cost of ordering a single comprehensive test, perceived benefit, attempts to conserve tissue, and hopes of improved survival if a targeted treatment is available.
“The lack of an association between broad-based genomic sequencing and survival is likely multifactorial,” the investigators maintain. “First, there were few genetic alterations identified with available targeted treatments. Second, even among those patients for whom targeted treatments were available, the treatments may not yield a substantial survival benefit or patients may not have had access to targeted agents due to financial barriers. Decision support for clinicians once they receive broad-based genomic sequencing results may also be needed.”
Study details
Dr. Presley and colleagues used the Flatiron Health Database to identify patients with advanced NSCLC who received care at 191 oncology practices across the United States during 2011-2016. The 5,688 patients studied had stage IIIB, stage IV, or unresectable nonsquamous NSCLC and received at least one line of treatment.
Overall, 15.4% received broad-based genomic sequencing of their tumor, while the rest received routine testing for EGFR and/or ALK alterations only, according to the results reported.
In the broadly tested group, merely 4.5% were given targeted treatment based on testing results. Another 9.8% received routine EGFR/ALK-targeted treatment, and 85.1% did not receive any targeted treatment.
The 12-month unadjusted mortality rate was 49.2% for patients undergoing broad testing, compared with 35.9% for patients undergoing routine testing.
In an instrumental variable analysis done to account for confounding, the 12-month predicted probability of death was 41.1% after broad testing and 44.4% after routine testing (P = .63).
Findings were similar in a propensity score–matched survival analysis (42.0% vs. 45.1%; hazard ratio, 0.92; P = .40), although there was some suggestion of a benefit of broad testing over routine testing in a Kaplan-Meier analysis among the entire unmatched cohort (HR, 0.69; P less than .001).
“Improved access to research clinical trials in the community setting may improve use of mutational data,” the investigators speculate. “Given the paucity of targeted agents, efforts to increase access to broad-based genomic sequencing should be paired with efforts to facilitate clinical trial enrollment.”
Dr. Presley disclosed that she receives grants from the Yale Lung SPORE Career Development Award, the Robert Wood Johnson/Veterans Affairs Clinical Scholars Program, and The Ohio State University K12 Training Grant for clinical faculty investigators. The study was funded by the Veterans Affairs Robert Wood Johnson Clinical Scholar Program and the Yale Lung SPORE Career Development Award.
SOURCE: Presley CJ et al. JAMA. 2018 Aug 7. doi: 10.1001/jama.2018.9824.
FROM JAMA
Key clinical point: In community oncology, broad-based genomic sequencing of NSCLC does not improve survival when compared with routine testing.
Major finding: The 12-month mortality rate was 49.2% for patients undergoing broad-based genomic sequencing and 35.9% for patients undergoing routine testing solely for EGFR and/or ALK alterations.
Study details: A retrospective cohort study of 5,688 patients with advanced nonsquamous NSCLC treated in 191 U.S. community practices.
Disclosures: Dr. Presley disclosed that she receives grants from the Yale Lung SPORE Career Development Award, the Robert Wood Johnson/Veterans Affairs Clinical Scholars Program, and The Ohio State University K12 Training Grant for clinical faculty investigators. The study was funded by the Veterans Affairs Robert Wood Johnson Clinical Scholar Program and the Yale Lung SPORE Career Development Award.
Source: Presley CJ et al. JAMA. 2018 Aug 7. doi: 10.1001/jama.2018.9824.
ASCO calls for expanding clinical trial eligibility
The American Society of Clinical Oncology and Friends of Cancer Research have submitted recommended language to the Food and Drug Administration for ways to expand eligibility criteria for cancer clinical trials.
The recommendations address five specific areas that were identified as most likely to restrict participation, but least likely to affect the safety of participants, and include minimum age requirements for trial enrollment, HIV/AIDS status, brain metastases, organ dysfunction, and prior and concurrent malignancies.
“Eligibility criteria ensure patient safety, but if they are overly strict, they can jeopardize accrual for clinical trials and reduce the ability to apply trial results to treating patients with cancer in clinical practice,” ASCO President Monica M. Bertagnolli, MD, said in a statement. “These guidance documents help trial sponsors understand how to modernize eligibility criteria and ensure that trial participants more accurately reflect the patients who will receive a drug after approval.”
The two organizations launched an effort to update clinical trial eligibility criteria in 2016 and published a joint statement in 2017. The letter to the FDA and the rationale and instructions for expanding eligibility criteria in each of the five areas can be found here on the Friends of Cancer Research website.
SOURCE: Friends of Cancer and ASCO letter to the FDA.
The American Society of Clinical Oncology and Friends of Cancer Research have submitted recommended language to the Food and Drug Administration for ways to expand eligibility criteria for cancer clinical trials.
The recommendations address five specific areas that were identified as most likely to restrict participation, but least likely to affect the safety of participants, and include minimum age requirements for trial enrollment, HIV/AIDS status, brain metastases, organ dysfunction, and prior and concurrent malignancies.
“Eligibility criteria ensure patient safety, but if they are overly strict, they can jeopardize accrual for clinical trials and reduce the ability to apply trial results to treating patients with cancer in clinical practice,” ASCO President Monica M. Bertagnolli, MD, said in a statement. “These guidance documents help trial sponsors understand how to modernize eligibility criteria and ensure that trial participants more accurately reflect the patients who will receive a drug after approval.”
The two organizations launched an effort to update clinical trial eligibility criteria in 2016 and published a joint statement in 2017. The letter to the FDA and the rationale and instructions for expanding eligibility criteria in each of the five areas can be found here on the Friends of Cancer Research website.
SOURCE: Friends of Cancer and ASCO letter to the FDA.
The American Society of Clinical Oncology and Friends of Cancer Research have submitted recommended language to the Food and Drug Administration for ways to expand eligibility criteria for cancer clinical trials.
The recommendations address five specific areas that were identified as most likely to restrict participation, but least likely to affect the safety of participants, and include minimum age requirements for trial enrollment, HIV/AIDS status, brain metastases, organ dysfunction, and prior and concurrent malignancies.
“Eligibility criteria ensure patient safety, but if they are overly strict, they can jeopardize accrual for clinical trials and reduce the ability to apply trial results to treating patients with cancer in clinical practice,” ASCO President Monica M. Bertagnolli, MD, said in a statement. “These guidance documents help trial sponsors understand how to modernize eligibility criteria and ensure that trial participants more accurately reflect the patients who will receive a drug after approval.”
The two organizations launched an effort to update clinical trial eligibility criteria in 2016 and published a joint statement in 2017. The letter to the FDA and the rationale and instructions for expanding eligibility criteria in each of the five areas can be found here on the Friends of Cancer Research website.
SOURCE: Friends of Cancer and ASCO letter to the FDA.
Key clinical point: ASCO and Friends of Cancer have submitted draft recommendations to the FDA for expanding cancer clinical trial participation.
Major finding: The organizations recommend addressing minimum age requirements, HIV/AIDS status, brain metastases, organ dysfunction, and prior and concurrent malignancies.
Data source: Draft guidance produced by ASCO and Friends of Cancer Research and submitted to the FDA.
Disclosures: Individual members of the working groups were not listed and conflicts of interest were not disclosed.
Source: Friends of Cancer and ASCO letter to the FDA.