User login
Opioids, benzodiazepines carry greater risk of COPD-related hospitalization
according to recent research from Annals of the American Thoracic Society.
In addition, the risk of hospitalization because of respiratory events for patients with chronic obstructive pulmonary disease (COPD) was greater when opioid and benzodiazepine medications were combined, compared with patients who did not take either medication, Jacques G. Baillargeon, PhD, of the department of preventive medicine and community health at the University of Texas, Galveston, and colleagues wrote.
“Patients with COPD and their physicians should judiciously assess the risks and benefits of opioids and benzodiazepines, alone and in combination, and preferentially recommend nonopioid and nonbenzodiazepine approaches for pain, sleep, and anxiety management in patients with COPD,” the investigators wrote.
The researchers performed a case-control study of 3,232 Medicare beneficiary cases of COPD patients who were aged at least 66 years. Patients were included if they experienced a hospitalization related to a COPD-related adverse event with a respiratory diagnosis in 2014 and then matched to one or two control patients (total, 6,247 patients) based on age at hospitalization, gender, COPD medication, COPD complexity, obstructive sleep apnea, and socioeconomic status. COPD complexity was assigned to three levels (low, moderate, high) and calculated using the patient’s comorbid respiratory conditions and associated medical procedures in the 12 months prior to their hospitalization.
They found that, in the 30 days before COPD-related hospitalization, use of opioids was associated with greater likelihood of hospitalization (adjusted odds ratio, 1.73; 95% confidence interval, 1.52-1.97), as was use of benzodiazepines (aOR, 1.42; 95% CI, 1.21-1.66). When patients used both opioids and benzodiazepines, they had a significantly higher risk of hospitalization, compared with patients who did not use opioids or benzodiazepines (aOR, 2.32; 95% CI, 1.94-2.77).
In the 60 days prior to hospitalization, there was also a greater likelihood of hospitalization among COPD patients who used opioids (aOR, 1.66; 95% CI, 1.47-1.88), benzodiazepines (aOR, 1.44; 95% CI, 1.24-1.67), and both opioids and benzodiazepines (aOR, 2.27; 95% CI, 1.93-2.67); at 90 days, this higher risk of hospitalization persisted among COPD patients taking opioids (aOR, 1.58; 95% CI, 1.40-1.78), benzodiazepines (aOR, 1.40; 95% CI, 1.20-1.63), and both opioids and benzodiazepines (aOR, 2.21; 95% CI, 1.88-2.59).
The researchers acknowledged that one potential limitation in the study was how COPD diagnoses were obtained through coding performed by clinicians instead of from laboratory testing. Confounding by COPD indication and severity; use of over-the-counter medication or opioids and benzodiazepines received illegally; and lack of analyses of potential confounders such as diet, alcohol use, smoking status and herbal supplement use were other limitations.
This study was supported by an award from the National Center for Advancing Translational Sciences and National Institutes of Health. Dr. Baillargeon had no disclosures.
SOURCE: Baillargeon JG et al. Ann Am Thorac Soc. 2019 Oct 1. doi: 10.1513/AnnalsATS.201901-024OC.
according to recent research from Annals of the American Thoracic Society.
In addition, the risk of hospitalization because of respiratory events for patients with chronic obstructive pulmonary disease (COPD) was greater when opioid and benzodiazepine medications were combined, compared with patients who did not take either medication, Jacques G. Baillargeon, PhD, of the department of preventive medicine and community health at the University of Texas, Galveston, and colleagues wrote.
“Patients with COPD and their physicians should judiciously assess the risks and benefits of opioids and benzodiazepines, alone and in combination, and preferentially recommend nonopioid and nonbenzodiazepine approaches for pain, sleep, and anxiety management in patients with COPD,” the investigators wrote.
The researchers performed a case-control study of 3,232 Medicare beneficiary cases of COPD patients who were aged at least 66 years. Patients were included if they experienced a hospitalization related to a COPD-related adverse event with a respiratory diagnosis in 2014 and then matched to one or two control patients (total, 6,247 patients) based on age at hospitalization, gender, COPD medication, COPD complexity, obstructive sleep apnea, and socioeconomic status. COPD complexity was assigned to three levels (low, moderate, high) and calculated using the patient’s comorbid respiratory conditions and associated medical procedures in the 12 months prior to their hospitalization.
They found that, in the 30 days before COPD-related hospitalization, use of opioids was associated with greater likelihood of hospitalization (adjusted odds ratio, 1.73; 95% confidence interval, 1.52-1.97), as was use of benzodiazepines (aOR, 1.42; 95% CI, 1.21-1.66). When patients used both opioids and benzodiazepines, they had a significantly higher risk of hospitalization, compared with patients who did not use opioids or benzodiazepines (aOR, 2.32; 95% CI, 1.94-2.77).
In the 60 days prior to hospitalization, there was also a greater likelihood of hospitalization among COPD patients who used opioids (aOR, 1.66; 95% CI, 1.47-1.88), benzodiazepines (aOR, 1.44; 95% CI, 1.24-1.67), and both opioids and benzodiazepines (aOR, 2.27; 95% CI, 1.93-2.67); at 90 days, this higher risk of hospitalization persisted among COPD patients taking opioids (aOR, 1.58; 95% CI, 1.40-1.78), benzodiazepines (aOR, 1.40; 95% CI, 1.20-1.63), and both opioids and benzodiazepines (aOR, 2.21; 95% CI, 1.88-2.59).
The researchers acknowledged that one potential limitation in the study was how COPD diagnoses were obtained through coding performed by clinicians instead of from laboratory testing. Confounding by COPD indication and severity; use of over-the-counter medication or opioids and benzodiazepines received illegally; and lack of analyses of potential confounders such as diet, alcohol use, smoking status and herbal supplement use were other limitations.
This study was supported by an award from the National Center for Advancing Translational Sciences and National Institutes of Health. Dr. Baillargeon had no disclosures.
SOURCE: Baillargeon JG et al. Ann Am Thorac Soc. 2019 Oct 1. doi: 10.1513/AnnalsATS.201901-024OC.
according to recent research from Annals of the American Thoracic Society.
In addition, the risk of hospitalization because of respiratory events for patients with chronic obstructive pulmonary disease (COPD) was greater when opioid and benzodiazepine medications were combined, compared with patients who did not take either medication, Jacques G. Baillargeon, PhD, of the department of preventive medicine and community health at the University of Texas, Galveston, and colleagues wrote.
“Patients with COPD and their physicians should judiciously assess the risks and benefits of opioids and benzodiazepines, alone and in combination, and preferentially recommend nonopioid and nonbenzodiazepine approaches for pain, sleep, and anxiety management in patients with COPD,” the investigators wrote.
The researchers performed a case-control study of 3,232 Medicare beneficiary cases of COPD patients who were aged at least 66 years. Patients were included if they experienced a hospitalization related to a COPD-related adverse event with a respiratory diagnosis in 2014 and then matched to one or two control patients (total, 6,247 patients) based on age at hospitalization, gender, COPD medication, COPD complexity, obstructive sleep apnea, and socioeconomic status. COPD complexity was assigned to three levels (low, moderate, high) and calculated using the patient’s comorbid respiratory conditions and associated medical procedures in the 12 months prior to their hospitalization.
They found that, in the 30 days before COPD-related hospitalization, use of opioids was associated with greater likelihood of hospitalization (adjusted odds ratio, 1.73; 95% confidence interval, 1.52-1.97), as was use of benzodiazepines (aOR, 1.42; 95% CI, 1.21-1.66). When patients used both opioids and benzodiazepines, they had a significantly higher risk of hospitalization, compared with patients who did not use opioids or benzodiazepines (aOR, 2.32; 95% CI, 1.94-2.77).
In the 60 days prior to hospitalization, there was also a greater likelihood of hospitalization among COPD patients who used opioids (aOR, 1.66; 95% CI, 1.47-1.88), benzodiazepines (aOR, 1.44; 95% CI, 1.24-1.67), and both opioids and benzodiazepines (aOR, 2.27; 95% CI, 1.93-2.67); at 90 days, this higher risk of hospitalization persisted among COPD patients taking opioids (aOR, 1.58; 95% CI, 1.40-1.78), benzodiazepines (aOR, 1.40; 95% CI, 1.20-1.63), and both opioids and benzodiazepines (aOR, 2.21; 95% CI, 1.88-2.59).
The researchers acknowledged that one potential limitation in the study was how COPD diagnoses were obtained through coding performed by clinicians instead of from laboratory testing. Confounding by COPD indication and severity; use of over-the-counter medication or opioids and benzodiazepines received illegally; and lack of analyses of potential confounders such as diet, alcohol use, smoking status and herbal supplement use were other limitations.
This study was supported by an award from the National Center for Advancing Translational Sciences and National Institutes of Health. Dr. Baillargeon had no disclosures.
SOURCE: Baillargeon JG et al. Ann Am Thorac Soc. 2019 Oct 1. doi: 10.1513/AnnalsATS.201901-024OC.
FROM ANNALS OF THE AMERICAN THORACIC SOCIETY
Clinical interventions for global drug use need updating
Public health approach requires greater emphasis on harms, benefits of substance use.
Strategies aimed at reducing drug-related harm should be informed by evidence, and recognize the contribution of social and economic factors to drug use, report the authors of a series of four papers published in The Lancet.
Louisa Degenhardt, PhD, and coauthors wrote in the first paper that, although the availability and use of drugs have been transformed over recent decades – including the emergence of hundreds of new psychoactive substances – professional and public policy has not yet adapted to those new realities (Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9).
, in a way that you don’t see in other areas of public health,” Dr. Degenhardt, of the National Drug and Alcohol Research Centre at the University of New South Wales in Sydney, said in an interview. “There has been an increasing level of awareness of issues but also level of recognition that we need to have hard evidence to work out the best ways to respond.”
The paper by Dr. Degenhardt and coauthors addressed the issue of opioid use and dependence around the world, citing evidence that in 2017, 40.5 million people were dependent on opioids and 109,500 deaths were attributable to opioid overdose. An effective treatment exists in the form of opioid agonists methadone and buprenorphine, both of which are recognized as World Health Organization essential medicines.
While the best evidence for positive outcomes from opioid agonist treatment is in people using illicit opioids such as heroin, there is also evidence for their effectiveness in people with pharmaceutical opioid dependence. A study in Kentucky suggested that scaling up the use and retention of opioid agonist treatment, including in prison, could prevent 57% of overdose deaths among injecting drug users.
“Despite strong evidence for the effectiveness of a range of interventions to improve the health and well-being of people who are dependent on opioids, coverage is low, even in high-income countries,” the authors wrote. They also called for international efforts to eliminate marketing strategies that have contributed to the increase in opioid prescription and harms in North America.
The second paper examined the public health implications of legalizing cannabis for medicinal and recreational use (Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1). Cannabis has been considered an illicit drug for more than 50 years but recently has been decriminalized or legalized in many parts of the world in recognition of the lower levels of harm, compared with other illicit substances.
Cannabis is used to treat a range of medical conditions, including muscle spasticity in multiple sclerosis. It also is used to treat pain, nausea, and vomiting in palliative care, and to reduce seizures in epilepsy. However, the authors noted that the evidence for many medical applications was absent, and that weakly regulated medical cannabis programs in some U.S. states were blurring the boundaries between medicinal and nonmedicinal use.
They also wrote that the public health effects of legalization could not be assessed, because legalization had happened only in the last 5 years.
“A major determinant of the public health effect of cannabis legalization will be the effect that it has on alcohol use,” they wrote. “The substitution of cannabis for alcohol would produce substantial public health gains, but any increase in the combined use of alcohol and cannabis could increase harm.”
The authors also looked at the effect of use of stimulants such as cocaine and amphetamines. While their use is associated with higher mortality, increased incidence of HIV and hepatitis C infection, poor mental health, and increased risk of cardiovascular events, no effective pharmacotherapies are available, and psychosocial interventions such as cognitive-behavioral therapy have only a weak effect.
“Many governments rely on punitive responses, such as involuntary detention in drug centers, despite the absence of evidence for their effectiveness and their potential to increase harm,” the authors wrote. “Substantial research investment is needed to develop more effective, innovative, and impactful prevention and treatment” (Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5).
They focused on interventions to prevent the transmission of blood-borne and sexually transmitted infections – such as the provision of safe injecting equipment, condoms or pre-exposure prophylaxis against HIV – and improve treatment of these, and interventions to prevent and treat overdose, injury, and other harms.
The final paper in the series explored new psychoactive substances, such as synthetic cannabinoids, stimulants, hallucinogens, and dissociative and depressant substances (Peacock A et al. Lancet 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7).
There really needs to be massive change in systems in terms of the way monitoring occurs and the speed with which new drugs are identified, Dr. Degenhardt said in the interview. She also said the risks that are identified need to be communicated more effectively.
“At the moment, the way that drug surveillance works in most countries, things come and then particular drugs may spread in use, cause massive harm, and all of our systems of detecting and responding are not fit to detect those things in a timely way and disseminate information to reduce those risks.”
The papers were supported by European Monitoring Centre on Drugs and Drug Addiction, and the Australian National Drug and Alcohol Research Centre. The authors declared support from a range of institutions and funding bodies, and several also declared unrelated grants, funding, and other support from the pharmaceutical sector.
SOURCES: Degenhardt L et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9; Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1; Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5; and Peacock A et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7.
Public health approach requires greater emphasis on harms, benefits of substance use.
Public health approach requires greater emphasis on harms, benefits of substance use.
Strategies aimed at reducing drug-related harm should be informed by evidence, and recognize the contribution of social and economic factors to drug use, report the authors of a series of four papers published in The Lancet.
Louisa Degenhardt, PhD, and coauthors wrote in the first paper that, although the availability and use of drugs have been transformed over recent decades – including the emergence of hundreds of new psychoactive substances – professional and public policy has not yet adapted to those new realities (Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9).
, in a way that you don’t see in other areas of public health,” Dr. Degenhardt, of the National Drug and Alcohol Research Centre at the University of New South Wales in Sydney, said in an interview. “There has been an increasing level of awareness of issues but also level of recognition that we need to have hard evidence to work out the best ways to respond.”
The paper by Dr. Degenhardt and coauthors addressed the issue of opioid use and dependence around the world, citing evidence that in 2017, 40.5 million people were dependent on opioids and 109,500 deaths were attributable to opioid overdose. An effective treatment exists in the form of opioid agonists methadone and buprenorphine, both of which are recognized as World Health Organization essential medicines.
While the best evidence for positive outcomes from opioid agonist treatment is in people using illicit opioids such as heroin, there is also evidence for their effectiveness in people with pharmaceutical opioid dependence. A study in Kentucky suggested that scaling up the use and retention of opioid agonist treatment, including in prison, could prevent 57% of overdose deaths among injecting drug users.
“Despite strong evidence for the effectiveness of a range of interventions to improve the health and well-being of people who are dependent on opioids, coverage is low, even in high-income countries,” the authors wrote. They also called for international efforts to eliminate marketing strategies that have contributed to the increase in opioid prescription and harms in North America.
The second paper examined the public health implications of legalizing cannabis for medicinal and recreational use (Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1). Cannabis has been considered an illicit drug for more than 50 years but recently has been decriminalized or legalized in many parts of the world in recognition of the lower levels of harm, compared with other illicit substances.
Cannabis is used to treat a range of medical conditions, including muscle spasticity in multiple sclerosis. It also is used to treat pain, nausea, and vomiting in palliative care, and to reduce seizures in epilepsy. However, the authors noted that the evidence for many medical applications was absent, and that weakly regulated medical cannabis programs in some U.S. states were blurring the boundaries between medicinal and nonmedicinal use.
They also wrote that the public health effects of legalization could not be assessed, because legalization had happened only in the last 5 years.
“A major determinant of the public health effect of cannabis legalization will be the effect that it has on alcohol use,” they wrote. “The substitution of cannabis for alcohol would produce substantial public health gains, but any increase in the combined use of alcohol and cannabis could increase harm.”
The authors also looked at the effect of use of stimulants such as cocaine and amphetamines. While their use is associated with higher mortality, increased incidence of HIV and hepatitis C infection, poor mental health, and increased risk of cardiovascular events, no effective pharmacotherapies are available, and psychosocial interventions such as cognitive-behavioral therapy have only a weak effect.
“Many governments rely on punitive responses, such as involuntary detention in drug centers, despite the absence of evidence for their effectiveness and their potential to increase harm,” the authors wrote. “Substantial research investment is needed to develop more effective, innovative, and impactful prevention and treatment” (Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5).
They focused on interventions to prevent the transmission of blood-borne and sexually transmitted infections – such as the provision of safe injecting equipment, condoms or pre-exposure prophylaxis against HIV – and improve treatment of these, and interventions to prevent and treat overdose, injury, and other harms.
The final paper in the series explored new psychoactive substances, such as synthetic cannabinoids, stimulants, hallucinogens, and dissociative and depressant substances (Peacock A et al. Lancet 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7).
There really needs to be massive change in systems in terms of the way monitoring occurs and the speed with which new drugs are identified, Dr. Degenhardt said in the interview. She also said the risks that are identified need to be communicated more effectively.
“At the moment, the way that drug surveillance works in most countries, things come and then particular drugs may spread in use, cause massive harm, and all of our systems of detecting and responding are not fit to detect those things in a timely way and disseminate information to reduce those risks.”
The papers were supported by European Monitoring Centre on Drugs and Drug Addiction, and the Australian National Drug and Alcohol Research Centre. The authors declared support from a range of institutions and funding bodies, and several also declared unrelated grants, funding, and other support from the pharmaceutical sector.
SOURCES: Degenhardt L et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9; Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1; Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5; and Peacock A et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7.
Strategies aimed at reducing drug-related harm should be informed by evidence, and recognize the contribution of social and economic factors to drug use, report the authors of a series of four papers published in The Lancet.
Louisa Degenhardt, PhD, and coauthors wrote in the first paper that, although the availability and use of drugs have been transformed over recent decades – including the emergence of hundreds of new psychoactive substances – professional and public policy has not yet adapted to those new realities (Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9).
, in a way that you don’t see in other areas of public health,” Dr. Degenhardt, of the National Drug and Alcohol Research Centre at the University of New South Wales in Sydney, said in an interview. “There has been an increasing level of awareness of issues but also level of recognition that we need to have hard evidence to work out the best ways to respond.”
The paper by Dr. Degenhardt and coauthors addressed the issue of opioid use and dependence around the world, citing evidence that in 2017, 40.5 million people were dependent on opioids and 109,500 deaths were attributable to opioid overdose. An effective treatment exists in the form of opioid agonists methadone and buprenorphine, both of which are recognized as World Health Organization essential medicines.
While the best evidence for positive outcomes from opioid agonist treatment is in people using illicit opioids such as heroin, there is also evidence for their effectiveness in people with pharmaceutical opioid dependence. A study in Kentucky suggested that scaling up the use and retention of opioid agonist treatment, including in prison, could prevent 57% of overdose deaths among injecting drug users.
“Despite strong evidence for the effectiveness of a range of interventions to improve the health and well-being of people who are dependent on opioids, coverage is low, even in high-income countries,” the authors wrote. They also called for international efforts to eliminate marketing strategies that have contributed to the increase in opioid prescription and harms in North America.
The second paper examined the public health implications of legalizing cannabis for medicinal and recreational use (Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1). Cannabis has been considered an illicit drug for more than 50 years but recently has been decriminalized or legalized in many parts of the world in recognition of the lower levels of harm, compared with other illicit substances.
Cannabis is used to treat a range of medical conditions, including muscle spasticity in multiple sclerosis. It also is used to treat pain, nausea, and vomiting in palliative care, and to reduce seizures in epilepsy. However, the authors noted that the evidence for many medical applications was absent, and that weakly regulated medical cannabis programs in some U.S. states were blurring the boundaries between medicinal and nonmedicinal use.
They also wrote that the public health effects of legalization could not be assessed, because legalization had happened only in the last 5 years.
“A major determinant of the public health effect of cannabis legalization will be the effect that it has on alcohol use,” they wrote. “The substitution of cannabis for alcohol would produce substantial public health gains, but any increase in the combined use of alcohol and cannabis could increase harm.”
The authors also looked at the effect of use of stimulants such as cocaine and amphetamines. While their use is associated with higher mortality, increased incidence of HIV and hepatitis C infection, poor mental health, and increased risk of cardiovascular events, no effective pharmacotherapies are available, and psychosocial interventions such as cognitive-behavioral therapy have only a weak effect.
“Many governments rely on punitive responses, such as involuntary detention in drug centers, despite the absence of evidence for their effectiveness and their potential to increase harm,” the authors wrote. “Substantial research investment is needed to develop more effective, innovative, and impactful prevention and treatment” (Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5).
They focused on interventions to prevent the transmission of blood-borne and sexually transmitted infections – such as the provision of safe injecting equipment, condoms or pre-exposure prophylaxis against HIV – and improve treatment of these, and interventions to prevent and treat overdose, injury, and other harms.
The final paper in the series explored new psychoactive substances, such as synthetic cannabinoids, stimulants, hallucinogens, and dissociative and depressant substances (Peacock A et al. Lancet 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7).
There really needs to be massive change in systems in terms of the way monitoring occurs and the speed with which new drugs are identified, Dr. Degenhardt said in the interview. She also said the risks that are identified need to be communicated more effectively.
“At the moment, the way that drug surveillance works in most countries, things come and then particular drugs may spread in use, cause massive harm, and all of our systems of detecting and responding are not fit to detect those things in a timely way and disseminate information to reduce those risks.”
The papers were supported by European Monitoring Centre on Drugs and Drug Addiction, and the Australian National Drug and Alcohol Research Centre. The authors declared support from a range of institutions and funding bodies, and several also declared unrelated grants, funding, and other support from the pharmaceutical sector.
SOURCES: Degenhardt L et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9; Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1; Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5; and Peacock A et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7.
FROM THE LANCET
Key clinical point: People with drug use disorders around the world need evidence-based and nonjudgmental clinical care.
Major finding: Many interventions aimed at reducing the harm of illicit drug use are not informed by evidence.
Study details: Series of four papers reviewing the evidence on cannabinoids, opioids, new psychoactive substances, and stimulants.
Disclosures: The papers were supported by European Monitoring Centre on Drugs and Drug Addiction, and the Australian National Drug and Alcohol Research Centre. The authors declared support from a range of institutions and funding bodies, and several also declared unrelated grants, funding, and other support from the pharmaceutical sector.
Sources: Degenhardt L et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32229-9; Hall W et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)31789-1; Farrell M et al. Lancet. 2019 Oct 23. doi: 10.1016/S0140-6736(19)32230-5; and Peacock A et al. Lancet 2019 Oct 23. doi: 10.1016/S0140-6736(19)32231-7.
Is carpal tunnel syndrome the tip of the iceberg?
He takes the following medications: felodipine and atorvastatin. On exam, his blood pressure is 110/60 mm Hg, and his pulse is 90 beats per minute.

A cardiac examination found normal heart sounds with no murmurs.
A chest examination found dullness to percussion at both bases and rales.
A chest x-ray showed bilateral effusions and mild pulmonary edema.
The brain natriuretic peptide test found a level of 1,300 picograms/mL.
An ECG found increased ventricular wall thickness, an ejection fraction of 32%, and normal aortic and mitral valves.
What history would be the most helpful in making a diagnosis?
A. History of prostate cancer
B. History of carpal tunnel syndrome
C. History of playing professional football
D. History of hyperlipidemia
E. History of ulcerative colitis
The correct answer here would be B. history of carpal tunnel syndrome (CTS). This patient has clinical heart failure, without a history of clinical ischemic disease. The differential diagnosis for causes of heart failure is long, with the most common causes being chronic hypertension and ischemic heart disease. Other common causes include chronic untreated sleep apnea and valvular heart disease.
This patient really does not have clear reasons for having clinical heart failure. His cardiovascular risk factors have been well controlled, and no valvular disease was found on ECG.
Several recent reports have raised the importance of a history of CTS significantly increasing the likelihood of amyloidosis being the cause of underlying heart failure.
CTS is such a common clinical entity that it is easy to not appreciate its presence as a clue to possible amyloid cardiomyopathy. Fosbøl et al. reported that a diagnosis of CTS was associated with a higher incidence of heart failure (hazard ratio, 1.54; CI, 1.45-1.64).1 They found a highly increased risk of amyloid (HR, 12.2) in patients who had surgery for CTS.
Sperry et al. found that over 10% of patients who underwent carpal tunnel release stained for amyloid on biopsy specimens, and that concomitant cardiac evaluation identified patients with cardiac involvement.2
Pinney et al. found that 48% of patients with transthyretin amyloidosis had a history of CTS.3
In a retrospective study of patients with wild-type transthyretin amyloid (253), patients with hereditary transthyretin amyloid (136), and asymptomatic gene carriers (77), participants were screened for a history of spinal stenosis and CTS.4 Almost 60% of the patients with amyloid had a history of CTS, and 11% had a history of spinal stenosis. Patients with CTS and hereditary amyloid had thicker interventricular septums, higher left ventricular mass, and lower Karnovsky index than those without CTS.
The diagnosis of CTS, especially in those who need surgery for treatment or have bilateral disease, should make us consider the possibility of underlying amyloidosis.
Pearl: In patients who have heart failure and a history of CTS, amyloidosis should be considered as a cause.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and serves as third-year medical student clerkship director at that university. Contact Dr. Paauw at [email protected].
References
1. Fosbøl EL et al. J Am Coll Cardiol. 2019;74:15-23.
2. Sperry BW et al. J Am Coll Cardiol. 2018 Oct 23;72(17):2040-50.
3. Pinney JH et al. J Am Heart Assoc. 2013 Apr 22;2(2):e000098.
4. Aus dem Siepen F et al. Clin Res Cardiol. 2019 Apr 5. doi: 10.1007/s00392-019-01467-1.
He takes the following medications: felodipine and atorvastatin. On exam, his blood pressure is 110/60 mm Hg, and his pulse is 90 beats per minute.
A cardiac examination found normal heart sounds with no murmurs.
A chest examination found dullness to percussion at both bases and rales.
A chest x-ray showed bilateral effusions and mild pulmonary edema.
The brain natriuretic peptide test found a level of 1,300 picograms/mL.
An ECG found increased ventricular wall thickness, an ejection fraction of 32%, and normal aortic and mitral valves.
What history would be the most helpful in making a diagnosis?
A. History of prostate cancer
B. History of carpal tunnel syndrome
C. History of playing professional football
D. History of hyperlipidemia
E. History of ulcerative colitis
The correct answer here would be B. history of carpal tunnel syndrome (CTS). This patient has clinical heart failure, without a history of clinical ischemic disease. The differential diagnosis for causes of heart failure is long, with the most common causes being chronic hypertension and ischemic heart disease. Other common causes include chronic untreated sleep apnea and valvular heart disease.
This patient really does not have clear reasons for having clinical heart failure. His cardiovascular risk factors have been well controlled, and no valvular disease was found on ECG.
Several recent reports have raised the importance of a history of CTS significantly increasing the likelihood of amyloidosis being the cause of underlying heart failure.
CTS is such a common clinical entity that it is easy to not appreciate its presence as a clue to possible amyloid cardiomyopathy. Fosbøl et al. reported that a diagnosis of CTS was associated with a higher incidence of heart failure (hazard ratio, 1.54; CI, 1.45-1.64).1 They found a highly increased risk of amyloid (HR, 12.2) in patients who had surgery for CTS.
Sperry et al. found that over 10% of patients who underwent carpal tunnel release stained for amyloid on biopsy specimens, and that concomitant cardiac evaluation identified patients with cardiac involvement.2
Pinney et al. found that 48% of patients with transthyretin amyloidosis had a history of CTS.3
In a retrospective study of patients with wild-type transthyretin amyloid (253), patients with hereditary transthyretin amyloid (136), and asymptomatic gene carriers (77), participants were screened for a history of spinal stenosis and CTS.4 Almost 60% of the patients with amyloid had a history of CTS, and 11% had a history of spinal stenosis. Patients with CTS and hereditary amyloid had thicker interventricular septums, higher left ventricular mass, and lower Karnovsky index than those without CTS.
The diagnosis of CTS, especially in those who need surgery for treatment or have bilateral disease, should make us consider the possibility of underlying amyloidosis.
Pearl: In patients who have heart failure and a history of CTS, amyloidosis should be considered as a cause.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and serves as third-year medical student clerkship director at that university. Contact Dr. Paauw at [email protected].
References
1. Fosbøl EL et al. J Am Coll Cardiol. 2019;74:15-23.
2. Sperry BW et al. J Am Coll Cardiol. 2018 Oct 23;72(17):2040-50.
3. Pinney JH et al. J Am Heart Assoc. 2013 Apr 22;2(2):e000098.
4. Aus dem Siepen F et al. Clin Res Cardiol. 2019 Apr 5. doi: 10.1007/s00392-019-01467-1.
He takes the following medications: felodipine and atorvastatin. On exam, his blood pressure is 110/60 mm Hg, and his pulse is 90 beats per minute.
A cardiac examination found normal heart sounds with no murmurs.
A chest examination found dullness to percussion at both bases and rales.
A chest x-ray showed bilateral effusions and mild pulmonary edema.
The brain natriuretic peptide test found a level of 1,300 picograms/mL.
An ECG found increased ventricular wall thickness, an ejection fraction of 32%, and normal aortic and mitral valves.
What history would be the most helpful in making a diagnosis?
A. History of prostate cancer
B. History of carpal tunnel syndrome
C. History of playing professional football
D. History of hyperlipidemia
E. History of ulcerative colitis
The correct answer here would be B. history of carpal tunnel syndrome (CTS). This patient has clinical heart failure, without a history of clinical ischemic disease. The differential diagnosis for causes of heart failure is long, with the most common causes being chronic hypertension and ischemic heart disease. Other common causes include chronic untreated sleep apnea and valvular heart disease.
This patient really does not have clear reasons for having clinical heart failure. His cardiovascular risk factors have been well controlled, and no valvular disease was found on ECG.
Several recent reports have raised the importance of a history of CTS significantly increasing the likelihood of amyloidosis being the cause of underlying heart failure.
CTS is such a common clinical entity that it is easy to not appreciate its presence as a clue to possible amyloid cardiomyopathy. Fosbøl et al. reported that a diagnosis of CTS was associated with a higher incidence of heart failure (hazard ratio, 1.54; CI, 1.45-1.64).1 They found a highly increased risk of amyloid (HR, 12.2) in patients who had surgery for CTS.
Sperry et al. found that over 10% of patients who underwent carpal tunnel release stained for amyloid on biopsy specimens, and that concomitant cardiac evaluation identified patients with cardiac involvement.2
Pinney et al. found that 48% of patients with transthyretin amyloidosis had a history of CTS.3
In a retrospective study of patients with wild-type transthyretin amyloid (253), patients with hereditary transthyretin amyloid (136), and asymptomatic gene carriers (77), participants were screened for a history of spinal stenosis and CTS.4 Almost 60% of the patients with amyloid had a history of CTS, and 11% had a history of spinal stenosis. Patients with CTS and hereditary amyloid had thicker interventricular septums, higher left ventricular mass, and lower Karnovsky index than those without CTS.
The diagnosis of CTS, especially in those who need surgery for treatment or have bilateral disease, should make us consider the possibility of underlying amyloidosis.
Pearl: In patients who have heart failure and a history of CTS, amyloidosis should be considered as a cause.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and serves as third-year medical student clerkship director at that university. Contact Dr. Paauw at [email protected].
References
1. Fosbøl EL et al. J Am Coll Cardiol. 2019;74:15-23.
2. Sperry BW et al. J Am Coll Cardiol. 2018 Oct 23;72(17):2040-50.
3. Pinney JH et al. J Am Heart Assoc. 2013 Apr 22;2(2):e000098.
4. Aus dem Siepen F et al. Clin Res Cardiol. 2019 Apr 5. doi: 10.1007/s00392-019-01467-1.
Drug crisis continues to evolve beyond opioids
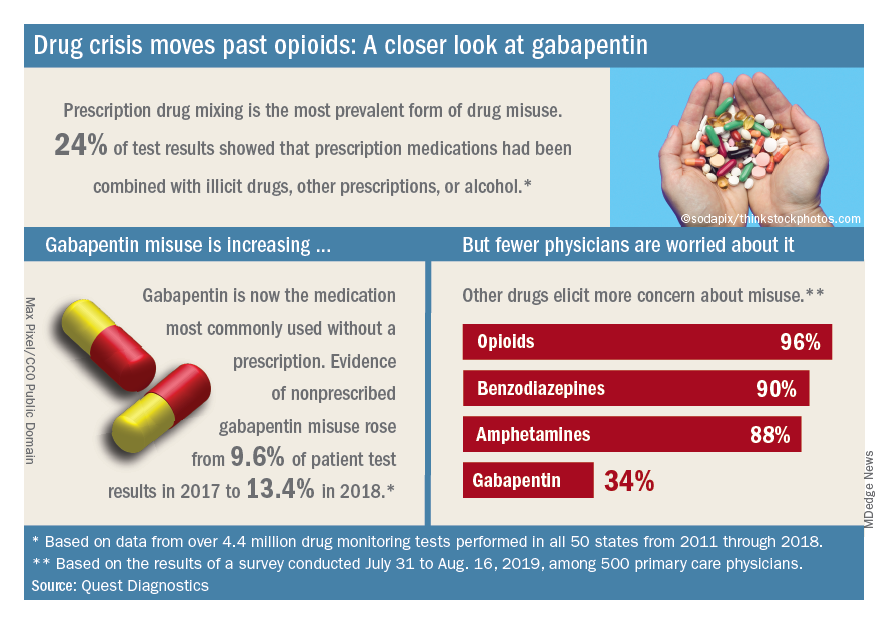
Almost three-quarters of primary care physicians believe that their patients will take their controlled medications as prescribed, but more than half of drug-monitoring lab tests show signs of misuse, according to a new report from Quest Diagnostics.
“ and may miss some of the drug misuse risks affecting their patients,” report coauthor Harvey W. Kaufman, MD, Quest’s senior medical director, said in a written statement.
Analysis of more than 4.4 million drug-monitoring tests showed that 51% involved an inconsistent result, such as detection of a nonprescribed drug or nondetection of a drug that was prescribed. The report also included a survey of 500 primary care physicians, of whom 72% said they trusted their patents to properly use opioids and other controlled substances.
“The intersection of these two data sets reveals, for the first time, the contrast between physician expectations about patient drug use and the evolution of the drug epidemic and actual patient behavior, as revealed by objective lab data, amid a national drug crisis that claimed an estimated 68,500 lives last year,” the report said.
A majority (62%) of the physicians surveyed also said that the opioid crisis will evolve into a new prescription drug crisis, and even more (72%) think that patients with chronic pain will use illicit drugs if they cannot get prescription opioids. Evidence from the drug test dataset suggests that “misuse of nonprescribed fentanyl and nonprescribed gabapentin warrant[s] a closer look,” the report said. In the survey, 78% of respondents reported prescribing gabapentin as an alternative to opioids for patients with chronic pain.
Those two drugs, along with alcohol, are the only three drug groups for which misuse increased from 2017 to 2018, and both are frequently involved in drug mixing, which is the most common form of misuse. Gabapentin went from 9.6% of all nonprescribed misuse in 2017 to 13.4% in 2018, an increase of 40%. Nonprescribed fentanyl was found in 64% of test results that were positive for heroin and 24% that were positive for cocaine, the Quest data showed.
The survey results, however, suggest that gabapentin is not on physicians’ radar, as only 34% said that they were concerned about its misuse, compared with 96% for opioids and 90% for benzodiazepines, according to the report.
“While gabapentin may not have opioids’ addictive potential, it can exaggerate euphoric effects when combined with opioids or anxiety medications. This drug mixing is dangerous,” said report coauthor Jeffrey Gudin, MD, senior medical advisor, prescription drug monitoring, for Quest Diagnostics.
The survey was conducted online among family physicians, general practitioners, and internists from July 31 to Aug. 16, 2019, by the Harris Poll on behalf of Quest and Center for Addiction. The test result data were collected in all 50 states and Washington, D.C., from 2011 to 2018, and results from drug rehabilitation clinics and addiction specialists were excluded from the analysis, so actual misuse rates are probably higher than reported.
Almost three-quarters of primary care physicians believe that their patients will take their controlled medications as prescribed, but more than half of drug-monitoring lab tests show signs of misuse, according to a new report from Quest Diagnostics.
“ and may miss some of the drug misuse risks affecting their patients,” report coauthor Harvey W. Kaufman, MD, Quest’s senior medical director, said in a written statement.
Analysis of more than 4.4 million drug-monitoring tests showed that 51% involved an inconsistent result, such as detection of a nonprescribed drug or nondetection of a drug that was prescribed. The report also included a survey of 500 primary care physicians, of whom 72% said they trusted their patents to properly use opioids and other controlled substances.
“The intersection of these two data sets reveals, for the first time, the contrast between physician expectations about patient drug use and the evolution of the drug epidemic and actual patient behavior, as revealed by objective lab data, amid a national drug crisis that claimed an estimated 68,500 lives last year,” the report said.
A majority (62%) of the physicians surveyed also said that the opioid crisis will evolve into a new prescription drug crisis, and even more (72%) think that patients with chronic pain will use illicit drugs if they cannot get prescription opioids. Evidence from the drug test dataset suggests that “misuse of nonprescribed fentanyl and nonprescribed gabapentin warrant[s] a closer look,” the report said. In the survey, 78% of respondents reported prescribing gabapentin as an alternative to opioids for patients with chronic pain.
Those two drugs, along with alcohol, are the only three drug groups for which misuse increased from 2017 to 2018, and both are frequently involved in drug mixing, which is the most common form of misuse. Gabapentin went from 9.6% of all nonprescribed misuse in 2017 to 13.4% in 2018, an increase of 40%. Nonprescribed fentanyl was found in 64% of test results that were positive for heroin and 24% that were positive for cocaine, the Quest data showed.
The survey results, however, suggest that gabapentin is not on physicians’ radar, as only 34% said that they were concerned about its misuse, compared with 96% for opioids and 90% for benzodiazepines, according to the report.
“While gabapentin may not have opioids’ addictive potential, it can exaggerate euphoric effects when combined with opioids or anxiety medications. This drug mixing is dangerous,” said report coauthor Jeffrey Gudin, MD, senior medical advisor, prescription drug monitoring, for Quest Diagnostics.
The survey was conducted online among family physicians, general practitioners, and internists from July 31 to Aug. 16, 2019, by the Harris Poll on behalf of Quest and Center for Addiction. The test result data were collected in all 50 states and Washington, D.C., from 2011 to 2018, and results from drug rehabilitation clinics and addiction specialists were excluded from the analysis, so actual misuse rates are probably higher than reported.
Almost three-quarters of primary care physicians believe that their patients will take their controlled medications as prescribed, but more than half of drug-monitoring lab tests show signs of misuse, according to a new report from Quest Diagnostics.
“ and may miss some of the drug misuse risks affecting their patients,” report coauthor Harvey W. Kaufman, MD, Quest’s senior medical director, said in a written statement.
Analysis of more than 4.4 million drug-monitoring tests showed that 51% involved an inconsistent result, such as detection of a nonprescribed drug or nondetection of a drug that was prescribed. The report also included a survey of 500 primary care physicians, of whom 72% said they trusted their patents to properly use opioids and other controlled substances.
“The intersection of these two data sets reveals, for the first time, the contrast between physician expectations about patient drug use and the evolution of the drug epidemic and actual patient behavior, as revealed by objective lab data, amid a national drug crisis that claimed an estimated 68,500 lives last year,” the report said.
A majority (62%) of the physicians surveyed also said that the opioid crisis will evolve into a new prescription drug crisis, and even more (72%) think that patients with chronic pain will use illicit drugs if they cannot get prescription opioids. Evidence from the drug test dataset suggests that “misuse of nonprescribed fentanyl and nonprescribed gabapentin warrant[s] a closer look,” the report said. In the survey, 78% of respondents reported prescribing gabapentin as an alternative to opioids for patients with chronic pain.
Those two drugs, along with alcohol, are the only three drug groups for which misuse increased from 2017 to 2018, and both are frequently involved in drug mixing, which is the most common form of misuse. Gabapentin went from 9.6% of all nonprescribed misuse in 2017 to 13.4% in 2018, an increase of 40%. Nonprescribed fentanyl was found in 64% of test results that were positive for heroin and 24% that were positive for cocaine, the Quest data showed.
The survey results, however, suggest that gabapentin is not on physicians’ radar, as only 34% said that they were concerned about its misuse, compared with 96% for opioids and 90% for benzodiazepines, according to the report.
“While gabapentin may not have opioids’ addictive potential, it can exaggerate euphoric effects when combined with opioids or anxiety medications. This drug mixing is dangerous,” said report coauthor Jeffrey Gudin, MD, senior medical advisor, prescription drug monitoring, for Quest Diagnostics.
The survey was conducted online among family physicians, general practitioners, and internists from July 31 to Aug. 16, 2019, by the Harris Poll on behalf of Quest and Center for Addiction. The test result data were collected in all 50 states and Washington, D.C., from 2011 to 2018, and results from drug rehabilitation clinics and addiction specialists were excluded from the analysis, so actual misuse rates are probably higher than reported.
FDA approves Reyvow for acute migraine treatment
The Food and Drug Administration has approved lasmiditan (Reyvow) for acute treatment of migraines with and without auras in adults.

The agency’s Oct. 11 announcement said the approval is based on results from a pair of randomized, double-blind, placebo-controlled trials that included 3,177 adult patients with a history of migraine with and without aura. The percentage of patients whose pain and most bothersome migraine symptom (nausea, light sensitivity, or sound sensitivity) resolved after 2 hours was higher in patients receiving lasmiditan than in patients receiving placebo.
Lasmiditan is a serotonin 5-hydroxytryptamine1F–receptor agonist, giving it a unique mechanism of action as compared with other migraine treatments.
The most common adverse events associated with lasmiditan include dizziness, fatigue, paresthesia, and sedation. There is a risk of driving impairment while taking the medication, and patients are advised not to operate or drive machinery for 8 hours after taking lasmiditan.
“Reyvow is a new option for the acute treatment of migraine, a painful condition that affects one in seven Americans. We know that the migraine community is keenly interested in additional treatment options, and we remain committed to continuing to work with stakeholders to promote the development of new therapies for the acute and preventive treatment of migraine,” said Nick Kozauer, MD, acting deputy director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research.
Eli Lilly, the drug’s manufacturer, said in a news release that “the recommended controlled substance classification for Reyvow is currently under review by the Drug Enforcement Administration and is expected within 90 days of today’s FDA approval, after which Reyvow will be available to patients in retail pharmacies” in oral doses of 50 mg, 100 mg, and 200 mg.
The Food and Drug Administration has approved lasmiditan (Reyvow) for acute treatment of migraines with and without auras in adults.
The agency’s Oct. 11 announcement said the approval is based on results from a pair of randomized, double-blind, placebo-controlled trials that included 3,177 adult patients with a history of migraine with and without aura. The percentage of patients whose pain and most bothersome migraine symptom (nausea, light sensitivity, or sound sensitivity) resolved after 2 hours was higher in patients receiving lasmiditan than in patients receiving placebo.
Lasmiditan is a serotonin 5-hydroxytryptamine1F–receptor agonist, giving it a unique mechanism of action as compared with other migraine treatments.
The most common adverse events associated with lasmiditan include dizziness, fatigue, paresthesia, and sedation. There is a risk of driving impairment while taking the medication, and patients are advised not to operate or drive machinery for 8 hours after taking lasmiditan.
“Reyvow is a new option for the acute treatment of migraine, a painful condition that affects one in seven Americans. We know that the migraine community is keenly interested in additional treatment options, and we remain committed to continuing to work with stakeholders to promote the development of new therapies for the acute and preventive treatment of migraine,” said Nick Kozauer, MD, acting deputy director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research.
Eli Lilly, the drug’s manufacturer, said in a news release that “the recommended controlled substance classification for Reyvow is currently under review by the Drug Enforcement Administration and is expected within 90 days of today’s FDA approval, after which Reyvow will be available to patients in retail pharmacies” in oral doses of 50 mg, 100 mg, and 200 mg.
The Food and Drug Administration has approved lasmiditan (Reyvow) for acute treatment of migraines with and without auras in adults.
The agency’s Oct. 11 announcement said the approval is based on results from a pair of randomized, double-blind, placebo-controlled trials that included 3,177 adult patients with a history of migraine with and without aura. The percentage of patients whose pain and most bothersome migraine symptom (nausea, light sensitivity, or sound sensitivity) resolved after 2 hours was higher in patients receiving lasmiditan than in patients receiving placebo.
Lasmiditan is a serotonin 5-hydroxytryptamine1F–receptor agonist, giving it a unique mechanism of action as compared with other migraine treatments.
The most common adverse events associated with lasmiditan include dizziness, fatigue, paresthesia, and sedation. There is a risk of driving impairment while taking the medication, and patients are advised not to operate or drive machinery for 8 hours after taking lasmiditan.
“Reyvow is a new option for the acute treatment of migraine, a painful condition that affects one in seven Americans. We know that the migraine community is keenly interested in additional treatment options, and we remain committed to continuing to work with stakeholders to promote the development of new therapies for the acute and preventive treatment of migraine,” said Nick Kozauer, MD, acting deputy director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research.
Eli Lilly, the drug’s manufacturer, said in a news release that “the recommended controlled substance classification for Reyvow is currently under review by the Drug Enforcement Administration and is expected within 90 days of today’s FDA approval, after which Reyvow will be available to patients in retail pharmacies” in oral doses of 50 mg, 100 mg, and 200 mg.
Preop IV dexamethasone conveys relief after total knee surgery
Patients given a single preoperative dose of intravenous dexamethasone had significantly less pain after total knee arthroplasty than did those given a placebo in a randomized controlled study of 100 adults.
“Corticosteroids were introduced several years ago for relieving postoperative pain in total joint replacement but, unfortunately, are not widely used due to surgeons’ concerns and the limited supporting evidence,” wrote Nattapol Tammachote, MD, of Thammasat University, Khlong Luang, Pathumthani, Thailand, and colleagues.
In a study published in the Journal of Arthroplasty, the researchers randomized 50 adults undergoing unilateral total knee surgery to a preoperative IV dexamethasone dose of 0.15 mg/kg diluted with normal saline or saline placebo. Patients, who were aged 50-85 years, were assessed every 3 hours after surgery, up to 48 hours; the primary outcomes were pain level, using the visual analog pain scale (VAS), and morphine use.
Overall, patients in the treatment group reported significant reductions on the VAS in mean pain scores of 11 points at rest and 15 points with knee movement. No significant differences in morphine use were noted between groups overall or at 12-hour intervals post-surgery.
In the first 24-48 hours after surgery dexamethasone was associated with a significantly lower rate of nausea and vomiting vs. placebo (58% vs. 84%), and a lower average C-reactive protein level (89 mg/L vs. 167 mg/L) at 48 hours after surgery. Hospital stays averaged 3 days for both groups, and no wound infections were reported.
Scores on tests of knee function using the modified Western Ontario and McMaster University Osteoarthritis Index scores and range of motion of the knee at three months were similar between the groups.
The study findings were limited by several factors, including the small sample size and use of multimodal pain control that may have impacted morphine use, a lack of data on hyperglycemia, and variation in doses of ketorolac given to patients in both groups, the researchers noted.
The results nevertheless support the potential of preoperative dexamethasone as “a promising approach in postoperative pain management and may be suitable for patients with contraindication to multimodal pain regimens,” they concluded.
The researchers reported no financial conflicts.
SOURCE: Tammachote N et al. J Arthroplasty. 2019. doi: https://doi.org/10.1016/ j.arth.2019.09.002.
Patients given a single preoperative dose of intravenous dexamethasone had significantly less pain after total knee arthroplasty than did those given a placebo in a randomized controlled study of 100 adults.
“Corticosteroids were introduced several years ago for relieving postoperative pain in total joint replacement but, unfortunately, are not widely used due to surgeons’ concerns and the limited supporting evidence,” wrote Nattapol Tammachote, MD, of Thammasat University, Khlong Luang, Pathumthani, Thailand, and colleagues.
In a study published in the Journal of Arthroplasty, the researchers randomized 50 adults undergoing unilateral total knee surgery to a preoperative IV dexamethasone dose of 0.15 mg/kg diluted with normal saline or saline placebo. Patients, who were aged 50-85 years, were assessed every 3 hours after surgery, up to 48 hours; the primary outcomes were pain level, using the visual analog pain scale (VAS), and morphine use.
Overall, patients in the treatment group reported significant reductions on the VAS in mean pain scores of 11 points at rest and 15 points with knee movement. No significant differences in morphine use were noted between groups overall or at 12-hour intervals post-surgery.
In the first 24-48 hours after surgery dexamethasone was associated with a significantly lower rate of nausea and vomiting vs. placebo (58% vs. 84%), and a lower average C-reactive protein level (89 mg/L vs. 167 mg/L) at 48 hours after surgery. Hospital stays averaged 3 days for both groups, and no wound infections were reported.
Scores on tests of knee function using the modified Western Ontario and McMaster University Osteoarthritis Index scores and range of motion of the knee at three months were similar between the groups.
The study findings were limited by several factors, including the small sample size and use of multimodal pain control that may have impacted morphine use, a lack of data on hyperglycemia, and variation in doses of ketorolac given to patients in both groups, the researchers noted.
The results nevertheless support the potential of preoperative dexamethasone as “a promising approach in postoperative pain management and may be suitable for patients with contraindication to multimodal pain regimens,” they concluded.
The researchers reported no financial conflicts.
SOURCE: Tammachote N et al. J Arthroplasty. 2019. doi: https://doi.org/10.1016/ j.arth.2019.09.002.
Patients given a single preoperative dose of intravenous dexamethasone had significantly less pain after total knee arthroplasty than did those given a placebo in a randomized controlled study of 100 adults.
“Corticosteroids were introduced several years ago for relieving postoperative pain in total joint replacement but, unfortunately, are not widely used due to surgeons’ concerns and the limited supporting evidence,” wrote Nattapol Tammachote, MD, of Thammasat University, Khlong Luang, Pathumthani, Thailand, and colleagues.
In a study published in the Journal of Arthroplasty, the researchers randomized 50 adults undergoing unilateral total knee surgery to a preoperative IV dexamethasone dose of 0.15 mg/kg diluted with normal saline or saline placebo. Patients, who were aged 50-85 years, were assessed every 3 hours after surgery, up to 48 hours; the primary outcomes were pain level, using the visual analog pain scale (VAS), and morphine use.
Overall, patients in the treatment group reported significant reductions on the VAS in mean pain scores of 11 points at rest and 15 points with knee movement. No significant differences in morphine use were noted between groups overall or at 12-hour intervals post-surgery.
In the first 24-48 hours after surgery dexamethasone was associated with a significantly lower rate of nausea and vomiting vs. placebo (58% vs. 84%), and a lower average C-reactive protein level (89 mg/L vs. 167 mg/L) at 48 hours after surgery. Hospital stays averaged 3 days for both groups, and no wound infections were reported.
Scores on tests of knee function using the modified Western Ontario and McMaster University Osteoarthritis Index scores and range of motion of the knee at three months were similar between the groups.
The study findings were limited by several factors, including the small sample size and use of multimodal pain control that may have impacted morphine use, a lack of data on hyperglycemia, and variation in doses of ketorolac given to patients in both groups, the researchers noted.
The results nevertheless support the potential of preoperative dexamethasone as “a promising approach in postoperative pain management and may be suitable for patients with contraindication to multimodal pain regimens,” they concluded.
The researchers reported no financial conflicts.
SOURCE: Tammachote N et al. J Arthroplasty. 2019. doi: https://doi.org/10.1016/ j.arth.2019.09.002.
FROM THE JOURNAL OF ARTHROPLASTY
Preop pain perceptions drive outcomes after knee surgery
Adult athletes who underwent knee surgery and had higher levels of preoperative pain catastrophizing were significantly less likely to return to preinjury activity, based on data from 101 individuals.

Pain is highly subjective, and pain perception can play a role in postsurgical outcomes, but the relationships among preoperative pain perception and short-term outcomes including returning to sports have not been well-studied, wrote Joshua S. Everhart, MD, of The Ohio State University Wexner Medical Center, Columbus, and colleagues.
In a study published in the Journal of Science and Medicine in Sport, the researchers assessed 101 adult athletes who underwent knee surgery at a single center. The average age of the patients was 33 years, and 49 were women.
Pain perception and coping were assessed via the McGill Pain questionnaire (SF-MPQ), Pain Catastrophizing Scale (PCS), Pain Coping Measure (PCM), and the brief COPE subscales of acceptance, denial, positive reframing, and use of instrumental support.
Patients who were severe pain catastrophizers (defined as scores greater than 36 on the Pain Catastrophizing Scale) had increased odds of not returning to a similar level of sport (OR 11.3).
Higher scores on the brief COPE subscale of “use of instrumental support” (instruments designed to help patients cope with pain) had a protective effect on returning to preinjury activity (OR 0.72 per point increase). However, higher COPE-denial scores were significantly associated with lower odds of improvement in kinesiophobia (OR 0.43).
Patients with greater levels of problem-focused coping had significantly greater improvement in International Knee Documentation Committee (IKDC) scores, as did patients who were older and more active.
“Specific coping strategies appear to moderate the effect of pain perceptions on postoperative outcomes, with some coping strategies being protective and others being harmful,” the researchers said.
The findings were limited by several factors including the use of multiple comparisons, the inability to assess the impact of pain perception after knee rehabilitation independent of surgery, and the small number of some uncommon procedures, the researchers noted.
However, the results suggest that “recognition of pain perception and coping styles early on in treatment may help sports medicine providers identify patients at risk for an unsatisfactory subjective outcome,” they concluded.
The researchers had no financial conflicts to disclose.
SOURCE: Everhart JS et al. J Sci Med Sport. 2019. doi: 10.1016/j.jsams.2019.09.011.
Adult athletes who underwent knee surgery and had higher levels of preoperative pain catastrophizing were significantly less likely to return to preinjury activity, based on data from 101 individuals.
Pain is highly subjective, and pain perception can play a role in postsurgical outcomes, but the relationships among preoperative pain perception and short-term outcomes including returning to sports have not been well-studied, wrote Joshua S. Everhart, MD, of The Ohio State University Wexner Medical Center, Columbus, and colleagues.
In a study published in the Journal of Science and Medicine in Sport, the researchers assessed 101 adult athletes who underwent knee surgery at a single center. The average age of the patients was 33 years, and 49 were women.
Pain perception and coping were assessed via the McGill Pain questionnaire (SF-MPQ), Pain Catastrophizing Scale (PCS), Pain Coping Measure (PCM), and the brief COPE subscales of acceptance, denial, positive reframing, and use of instrumental support.
Patients who were severe pain catastrophizers (defined as scores greater than 36 on the Pain Catastrophizing Scale) had increased odds of not returning to a similar level of sport (OR 11.3).
Higher scores on the brief COPE subscale of “use of instrumental support” (instruments designed to help patients cope with pain) had a protective effect on returning to preinjury activity (OR 0.72 per point increase). However, higher COPE-denial scores were significantly associated with lower odds of improvement in kinesiophobia (OR 0.43).
Patients with greater levels of problem-focused coping had significantly greater improvement in International Knee Documentation Committee (IKDC) scores, as did patients who were older and more active.
“Specific coping strategies appear to moderate the effect of pain perceptions on postoperative outcomes, with some coping strategies being protective and others being harmful,” the researchers said.
The findings were limited by several factors including the use of multiple comparisons, the inability to assess the impact of pain perception after knee rehabilitation independent of surgery, and the small number of some uncommon procedures, the researchers noted.
However, the results suggest that “recognition of pain perception and coping styles early on in treatment may help sports medicine providers identify patients at risk for an unsatisfactory subjective outcome,” they concluded.
The researchers had no financial conflicts to disclose.
SOURCE: Everhart JS et al. J Sci Med Sport. 2019. doi: 10.1016/j.jsams.2019.09.011.
Adult athletes who underwent knee surgery and had higher levels of preoperative pain catastrophizing were significantly less likely to return to preinjury activity, based on data from 101 individuals.
Pain is highly subjective, and pain perception can play a role in postsurgical outcomes, but the relationships among preoperative pain perception and short-term outcomes including returning to sports have not been well-studied, wrote Joshua S. Everhart, MD, of The Ohio State University Wexner Medical Center, Columbus, and colleagues.
In a study published in the Journal of Science and Medicine in Sport, the researchers assessed 101 adult athletes who underwent knee surgery at a single center. The average age of the patients was 33 years, and 49 were women.
Pain perception and coping were assessed via the McGill Pain questionnaire (SF-MPQ), Pain Catastrophizing Scale (PCS), Pain Coping Measure (PCM), and the brief COPE subscales of acceptance, denial, positive reframing, and use of instrumental support.
Patients who were severe pain catastrophizers (defined as scores greater than 36 on the Pain Catastrophizing Scale) had increased odds of not returning to a similar level of sport (OR 11.3).
Higher scores on the brief COPE subscale of “use of instrumental support” (instruments designed to help patients cope with pain) had a protective effect on returning to preinjury activity (OR 0.72 per point increase). However, higher COPE-denial scores were significantly associated with lower odds of improvement in kinesiophobia (OR 0.43).
Patients with greater levels of problem-focused coping had significantly greater improvement in International Knee Documentation Committee (IKDC) scores, as did patients who were older and more active.
“Specific coping strategies appear to moderate the effect of pain perceptions on postoperative outcomes, with some coping strategies being protective and others being harmful,” the researchers said.
The findings were limited by several factors including the use of multiple comparisons, the inability to assess the impact of pain perception after knee rehabilitation independent of surgery, and the small number of some uncommon procedures, the researchers noted.
However, the results suggest that “recognition of pain perception and coping styles early on in treatment may help sports medicine providers identify patients at risk for an unsatisfactory subjective outcome,” they concluded.
The researchers had no financial conflicts to disclose.
SOURCE: Everhart JS et al. J Sci Med Sport. 2019. doi: 10.1016/j.jsams.2019.09.011.
FROM THE JOURNAL OF SCIENCE AND MEDICINE IN SPORT
FDA approves afamelanotide for treatment of rare condition with light-induced pain
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.

This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
Consider centralized pain in patients with rheumatic disease
Las Vegas – A fibromyalgia survey may provide important information about the degree to which patients with rheumatic disease experience centralized pain. This information may guide treatment decisions, said Daniel J. Clauw, MD, professor of anesthesiology, rheumatology, and psychiatry and director of the Chronic Pain and Fatigue Research Center at the University of Michigan in Ann Arbor.

The questionnaire that Dr. Clauw uses is a patient self-report survey for the assessment of fibromyalgia based on criteria in the 2011 modification of the American College of Rheumatology preliminary diagnostic criteria for fibromyalgia. In it, he asks patients to report where they experience pain throughout the body and symptoms such as fatigue, sleep problems, and memory problems. The survey predicts outcomes of surgery for osteoarthritis better than x-rays, MRI scans, or psychological factors do, he said.
Physicians should ask every patient with chronic pain, including patients with OA, rheumatoid arthritis, or lupus, to complete the survey, Dr. Clauw said at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “This score will tell you the degree to which their central nervous system is augmenting or amplifying what is going on in their body,” he said. “And the higher their score is, the more you should treat them like you would someone with fibromyalgia, even if their underlying disease might be an autoimmune disease.”
Physicians should not use a cutoff of 13 points on the fibromyalgia measure to define whether a patient has the disease, as has been done in the past, he said. The threshold is arbitrary, he said. “We should not think about fibromyalgia as ‘yes’ or ‘no.’ We should think of the degree of fibromyalgia that people have.”
A poor relationship between pain and imaging
Some patients who have severe knee OA on imaging walk without pain. Other patients have normal x-rays, but severe pain. “There is a terrible relationship between what you see on a knee x-ray or an MRI and whether someone has pain,” Dr. Clauw said. Furthermore, the poor relationship between imaging and pain is common across chronic pain conditions, he said.
This phenomenon may occur because pain manifests in different ways, similar to there being multiple ways to adjust the volume of an electric guitar, he said. How hard the strings are strummed affects the volume. But so does the amplifier setting. “In these centralized pain conditions, the problem is an amplifier problem, not a guitar problem,” he said. “The amplifier, i.e., the central nervous system, is set too high.”
Researchers have found that people who have severe OA of the knee on x-ray but do not experience pain “have a very low amplifier setting,” he said. That is, they are nontender and less sensitive to pain. Most of these patients are men. “On average, men have a much lower amplifier setting than women,” he said. “This is also why ... women have 1.5 to 2 times the rate of any type of chronic pain than men, because on average women have a higher amplifier setting. ... In OA, at any given age, men and women have the exact same percentage of radiographic OA. But if you look at the clinical condition of OA, it is always two-thirds women, one-third men.”
Opioid responsiveness
To examine whether fibromyalgia survey results correlate with outcomes after knee and hip arthroplasty, Dr. Clauw and colleagues conducted a prospective, observational cohort study that included approximately 500 people. Patients completed the questionnaire on the day of surgery.
Patients with higher levels of fibromyalgia were less responsive to opioids. “For each 1-point increase in the fibromyalgia score, people needed about one more hydrocodone tablet in the first 24-48 hours to control their pain,” he said (Anesthesiology. 2013 Dec;119[6]:1434-43). In addition, each 1-point increase in the fibromyalgia score made people about 25% less likely to have a 50% improvement in knee pain level after 6 months (Arthritis Rheumatol. 2015 May;67[5]:1386-94). The correlations were independent of psychological factors. In addition, the associations were linear. “There was nothing magical about a fibromyalgia score of 13,” Dr. Clauw said.
Dr. Clauw is a coauthor of a study to be presented at the 2019 American College of Rheumatology/Association of Rheumatology Professionals annual meeting that found pain centralization in patients with RA is associated with poor response to disease-modifying antirheumatic drugs (DMARDs).
Prior studies in patients with RA have found that the degree of fibromyalgia is a better predictor of pain and disability than erythrocyte sedimentation rate or the number of swollen joints.
Diagnosed cases are the “tip of the iceberg”
Researchers at Dr. Clauw’s institution have identified dozens of patients undergoing knee surgery who met criteria for fibromyalgia but had not received the diagnosis. “This is at the University of Michigan, which is the epicenter for fibromyalgia research. If we are not seeing fibromyalgia superimposed on OA in our patients, no one is seeing it,” he said.
Patients with diagnosed fibromyalgia are “the tip of the iceberg,” he said. “There are far greater numbers of individuals whose primary diagnosis is OA, RA, lupus, ankylosing spondylitis, cancer pain, or sickle cell disease that have the same fundamental problem as fibromyalgia patients. But you do not see it because you label them as having an autoimmune disease or osteoarthritis. And that is at your peril and at their peril. Because treating that individual as if all of their pain and other symptoms are due to a problem out on the periphery will not make that person better.”
Patients with high levels of centralized pain may be less responsive to peripherally directed therapies such as surgery or injections, Dr. Clauw said. Pharmacologic options for patients with centralized pain include gabapentinoids (e.g., pregabalin and gabapentin), serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and milnacipran), and tricyclic compounds (e.g., amitriptyline and cyclobenzaprine), he said. “Opioids are going to be quite unlikely to help these individuals,” he said. “In fact, it is likely that opioids will make this kind of pain worse.”
Dr. Clauw is a consultant for Aptinyx, Daiichi Sankyo, Eli Lilly, Intec Pharma, Pfizer, Samumed, Theravance, Tonix, and Zynerba Pharma. He has received grant or research support from Aptinyx and Pfizer and is an expert witness.
Global Academy for Medical Education and this news organization are owned by the same parent company.
Las Vegas – A fibromyalgia survey may provide important information about the degree to which patients with rheumatic disease experience centralized pain. This information may guide treatment decisions, said Daniel J. Clauw, MD, professor of anesthesiology, rheumatology, and psychiatry and director of the Chronic Pain and Fatigue Research Center at the University of Michigan in Ann Arbor.
The questionnaire that Dr. Clauw uses is a patient self-report survey for the assessment of fibromyalgia based on criteria in the 2011 modification of the American College of Rheumatology preliminary diagnostic criteria for fibromyalgia. In it, he asks patients to report where they experience pain throughout the body and symptoms such as fatigue, sleep problems, and memory problems. The survey predicts outcomes of surgery for osteoarthritis better than x-rays, MRI scans, or psychological factors do, he said.
Physicians should ask every patient with chronic pain, including patients with OA, rheumatoid arthritis, or lupus, to complete the survey, Dr. Clauw said at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “This score will tell you the degree to which their central nervous system is augmenting or amplifying what is going on in their body,” he said. “And the higher their score is, the more you should treat them like you would someone with fibromyalgia, even if their underlying disease might be an autoimmune disease.”
Physicians should not use a cutoff of 13 points on the fibromyalgia measure to define whether a patient has the disease, as has been done in the past, he said. The threshold is arbitrary, he said. “We should not think about fibromyalgia as ‘yes’ or ‘no.’ We should think of the degree of fibromyalgia that people have.”
A poor relationship between pain and imaging
Some patients who have severe knee OA on imaging walk without pain. Other patients have normal x-rays, but severe pain. “There is a terrible relationship between what you see on a knee x-ray or an MRI and whether someone has pain,” Dr. Clauw said. Furthermore, the poor relationship between imaging and pain is common across chronic pain conditions, he said.
This phenomenon may occur because pain manifests in different ways, similar to there being multiple ways to adjust the volume of an electric guitar, he said. How hard the strings are strummed affects the volume. But so does the amplifier setting. “In these centralized pain conditions, the problem is an amplifier problem, not a guitar problem,” he said. “The amplifier, i.e., the central nervous system, is set too high.”
Researchers have found that people who have severe OA of the knee on x-ray but do not experience pain “have a very low amplifier setting,” he said. That is, they are nontender and less sensitive to pain. Most of these patients are men. “On average, men have a much lower amplifier setting than women,” he said. “This is also why ... women have 1.5 to 2 times the rate of any type of chronic pain than men, because on average women have a higher amplifier setting. ... In OA, at any given age, men and women have the exact same percentage of radiographic OA. But if you look at the clinical condition of OA, it is always two-thirds women, one-third men.”
Opioid responsiveness
To examine whether fibromyalgia survey results correlate with outcomes after knee and hip arthroplasty, Dr. Clauw and colleagues conducted a prospective, observational cohort study that included approximately 500 people. Patients completed the questionnaire on the day of surgery.
Patients with higher levels of fibromyalgia were less responsive to opioids. “For each 1-point increase in the fibromyalgia score, people needed about one more hydrocodone tablet in the first 24-48 hours to control their pain,” he said (Anesthesiology. 2013 Dec;119[6]:1434-43). In addition, each 1-point increase in the fibromyalgia score made people about 25% less likely to have a 50% improvement in knee pain level after 6 months (Arthritis Rheumatol. 2015 May;67[5]:1386-94). The correlations were independent of psychological factors. In addition, the associations were linear. “There was nothing magical about a fibromyalgia score of 13,” Dr. Clauw said.
Dr. Clauw is a coauthor of a study to be presented at the 2019 American College of Rheumatology/Association of Rheumatology Professionals annual meeting that found pain centralization in patients with RA is associated with poor response to disease-modifying antirheumatic drugs (DMARDs).
Prior studies in patients with RA have found that the degree of fibromyalgia is a better predictor of pain and disability than erythrocyte sedimentation rate or the number of swollen joints.
Diagnosed cases are the “tip of the iceberg”
Researchers at Dr. Clauw’s institution have identified dozens of patients undergoing knee surgery who met criteria for fibromyalgia but had not received the diagnosis. “This is at the University of Michigan, which is the epicenter for fibromyalgia research. If we are not seeing fibromyalgia superimposed on OA in our patients, no one is seeing it,” he said.
Patients with diagnosed fibromyalgia are “the tip of the iceberg,” he said. “There are far greater numbers of individuals whose primary diagnosis is OA, RA, lupus, ankylosing spondylitis, cancer pain, or sickle cell disease that have the same fundamental problem as fibromyalgia patients. But you do not see it because you label them as having an autoimmune disease or osteoarthritis. And that is at your peril and at their peril. Because treating that individual as if all of their pain and other symptoms are due to a problem out on the periphery will not make that person better.”
Patients with high levels of centralized pain may be less responsive to peripherally directed therapies such as surgery or injections, Dr. Clauw said. Pharmacologic options for patients with centralized pain include gabapentinoids (e.g., pregabalin and gabapentin), serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and milnacipran), and tricyclic compounds (e.g., amitriptyline and cyclobenzaprine), he said. “Opioids are going to be quite unlikely to help these individuals,” he said. “In fact, it is likely that opioids will make this kind of pain worse.”
Dr. Clauw is a consultant for Aptinyx, Daiichi Sankyo, Eli Lilly, Intec Pharma, Pfizer, Samumed, Theravance, Tonix, and Zynerba Pharma. He has received grant or research support from Aptinyx and Pfizer and is an expert witness.
Global Academy for Medical Education and this news organization are owned by the same parent company.
Las Vegas – A fibromyalgia survey may provide important information about the degree to which patients with rheumatic disease experience centralized pain. This information may guide treatment decisions, said Daniel J. Clauw, MD, professor of anesthesiology, rheumatology, and psychiatry and director of the Chronic Pain and Fatigue Research Center at the University of Michigan in Ann Arbor.
The questionnaire that Dr. Clauw uses is a patient self-report survey for the assessment of fibromyalgia based on criteria in the 2011 modification of the American College of Rheumatology preliminary diagnostic criteria for fibromyalgia. In it, he asks patients to report where they experience pain throughout the body and symptoms such as fatigue, sleep problems, and memory problems. The survey predicts outcomes of surgery for osteoarthritis better than x-rays, MRI scans, or psychological factors do, he said.
Physicians should ask every patient with chronic pain, including patients with OA, rheumatoid arthritis, or lupus, to complete the survey, Dr. Clauw said at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “This score will tell you the degree to which their central nervous system is augmenting or amplifying what is going on in their body,” he said. “And the higher their score is, the more you should treat them like you would someone with fibromyalgia, even if their underlying disease might be an autoimmune disease.”
Physicians should not use a cutoff of 13 points on the fibromyalgia measure to define whether a patient has the disease, as has been done in the past, he said. The threshold is arbitrary, he said. “We should not think about fibromyalgia as ‘yes’ or ‘no.’ We should think of the degree of fibromyalgia that people have.”
A poor relationship between pain and imaging
Some patients who have severe knee OA on imaging walk without pain. Other patients have normal x-rays, but severe pain. “There is a terrible relationship between what you see on a knee x-ray or an MRI and whether someone has pain,” Dr. Clauw said. Furthermore, the poor relationship between imaging and pain is common across chronic pain conditions, he said.
This phenomenon may occur because pain manifests in different ways, similar to there being multiple ways to adjust the volume of an electric guitar, he said. How hard the strings are strummed affects the volume. But so does the amplifier setting. “In these centralized pain conditions, the problem is an amplifier problem, not a guitar problem,” he said. “The amplifier, i.e., the central nervous system, is set too high.”
Researchers have found that people who have severe OA of the knee on x-ray but do not experience pain “have a very low amplifier setting,” he said. That is, they are nontender and less sensitive to pain. Most of these patients are men. “On average, men have a much lower amplifier setting than women,” he said. “This is also why ... women have 1.5 to 2 times the rate of any type of chronic pain than men, because on average women have a higher amplifier setting. ... In OA, at any given age, men and women have the exact same percentage of radiographic OA. But if you look at the clinical condition of OA, it is always two-thirds women, one-third men.”
Opioid responsiveness
To examine whether fibromyalgia survey results correlate with outcomes after knee and hip arthroplasty, Dr. Clauw and colleagues conducted a prospective, observational cohort study that included approximately 500 people. Patients completed the questionnaire on the day of surgery.
Patients with higher levels of fibromyalgia were less responsive to opioids. “For each 1-point increase in the fibromyalgia score, people needed about one more hydrocodone tablet in the first 24-48 hours to control their pain,” he said (Anesthesiology. 2013 Dec;119[6]:1434-43). In addition, each 1-point increase in the fibromyalgia score made people about 25% less likely to have a 50% improvement in knee pain level after 6 months (Arthritis Rheumatol. 2015 May;67[5]:1386-94). The correlations were independent of psychological factors. In addition, the associations were linear. “There was nothing magical about a fibromyalgia score of 13,” Dr. Clauw said.
Dr. Clauw is a coauthor of a study to be presented at the 2019 American College of Rheumatology/Association of Rheumatology Professionals annual meeting that found pain centralization in patients with RA is associated with poor response to disease-modifying antirheumatic drugs (DMARDs).
Prior studies in patients with RA have found that the degree of fibromyalgia is a better predictor of pain and disability than erythrocyte sedimentation rate or the number of swollen joints.
Diagnosed cases are the “tip of the iceberg”
Researchers at Dr. Clauw’s institution have identified dozens of patients undergoing knee surgery who met criteria for fibromyalgia but had not received the diagnosis. “This is at the University of Michigan, which is the epicenter for fibromyalgia research. If we are not seeing fibromyalgia superimposed on OA in our patients, no one is seeing it,” he said.
Patients with diagnosed fibromyalgia are “the tip of the iceberg,” he said. “There are far greater numbers of individuals whose primary diagnosis is OA, RA, lupus, ankylosing spondylitis, cancer pain, or sickle cell disease that have the same fundamental problem as fibromyalgia patients. But you do not see it because you label them as having an autoimmune disease or osteoarthritis. And that is at your peril and at their peril. Because treating that individual as if all of their pain and other symptoms are due to a problem out on the periphery will not make that person better.”
Patients with high levels of centralized pain may be less responsive to peripherally directed therapies such as surgery or injections, Dr. Clauw said. Pharmacologic options for patients with centralized pain include gabapentinoids (e.g., pregabalin and gabapentin), serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and milnacipran), and tricyclic compounds (e.g., amitriptyline and cyclobenzaprine), he said. “Opioids are going to be quite unlikely to help these individuals,” he said. “In fact, it is likely that opioids will make this kind of pain worse.”
Dr. Clauw is a consultant for Aptinyx, Daiichi Sankyo, Eli Lilly, Intec Pharma, Pfizer, Samumed, Theravance, Tonix, and Zynerba Pharma. He has received grant or research support from Aptinyx and Pfizer and is an expert witness.
Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM PRD 2019
Persistent rash on the sole
A 52-year-old Chinese woman presented to a tertiary hospital in Singapore with a 3-month history of persistent and intermittently painful rashes over her right calf and foot (FIGURE). The patient had pancytopenia due to ongoing chemotherapy for metastatic nasopharyngeal carcinoma. She was systemically well and denied other dermatoses. Examination demonstrated scattered crops of tense hemorrhagic vesicles, each surrounded by a livid purpuric base, over the right plantar aspect of the foot, with areas of eschar over the right medial hallux. No allodynia, hyperaesthesia, or lymphadenopathy was noted.
A punch biopsy of an intact vesicle was performed.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis:
Herpes zoster
Histopathologic examination showed full-thickness epidermal necrosis with ballooning degeneration resulting in an intra-epidermal blister. Multinucleated keratinocytes with nuclear moulding were seen within the blister cavity. Grocott-Gomori methenamine-silver (GMS), acid-fast, and Gram stains were negative. Granular immunoglobulin (Ig) G, IgM, and C3 were seen intramurally. DNA analysis of vesicular fluid was positive for varicella zoster virus (VZV). A diagnosis of herpes zoster (HZ) of the right S1 dermatome with primary obliterative vasculitis was established.
Immunocompromised people—those who have impaired T-cell immunity (eg, recipients of organ or hematopoietic stem-cell transplants), take immunosuppressive therapy, or have lymphoma, leukemia, or human immunodeficiency virus (HIV) infection—have an increased risk for HZ. For example, in patients with acquired immunodeficiency syndrome (AIDS), HZ uniquely manifests as recurrent shingles. An estimated 20% to 30% of HIV-infected patients will have more than 1 episode of HZ, which may involve the same or different dermatomes.1,2 Furthermore, HZ in this population is more commonly associated with atypical presentations.3
What an atypical presentation may look like
In immunocompromised patients, HZ may present with atypical cutaneous manifestations or with atypical generalized symptoms.
Atypical cutaneous manifestations, as in disseminated zoster, manifest with multiple hyperkeratotic papules (3-20 mm in diameter) that follow no dermatomal pattern. These lesions may be chronic, persisting for months or years, and may be associated with acyclovir-resistant strains of VZV.2,3 Another dermatologic variant is ecthymatous VZV, which manifests with multiple large (10-30 mm) punched-out ulcerations with a central black eschar and a peripheral rim of vesicles.4 Viral folliculitis—in which infection is limited to the hair follicle, with no associated blisters—has also been reported in atypical HZ.5
Our patient presented with hemorrhagic vesicles mimicking vasculitic lesions, which had persisted over a 3-month period with intermittent localized pain. It has been proposed that in atypical presentations, the reactivated VZV spreads transaxonally from adjacent nerves to the outermost adventitial layer of the arterial wall, leading to a vasculitic appearance of the vesicles.6 Viral-induced vasculitis may also result either directly from infection of the blood vessels or secondary to vascular damage from an inflammatory immune complex–mediated reaction, cell-mediated hypersensitivity, or inflammation due to immune dysregulation.7,8
Continue to: Differential includes vesiculobullous conditions
Differential includes vesiculobullous conditions
There are several important items to consider in the differential.
Cutaneous vasculitis, in severe cases, may manifest with vesicles or bullae that resemble the lesions seen in HZ. However, its unilateral nature and distribution distinguish it.
Angioinvasive fungal infections in immunocompromised patients may manifest with scattered ulceronecrotic lesions to purpuric vesiculobullous dermatoses.9 However, no fungal organisms were seen on GMS staining of the biopsied tissue.
Atypical hand-foot-and-mouth disease tends to affect adults and is associated with Coxsackievirus A6 infection.10 It may manifest as generalized vesiculobullous exanthem resembling varicella. The chronic nature and restricted extent of the patient’s rash made this diagnosis unlikely.
Successful management depends on timely identification
Although most cases of HZ can be diagnosed clinically, atypical rashes may require a biopsy and direct immunofluorescence assay for VZV antigen or a polymerase-chain-reaction (PCR) assay for VZV DNA in cells from the base of blisters. Therefore, it is important to consider the diagnosis of HZ in immunocompromised patients presenting with an atypical rash to avoid misdiagnosis and costly testing.
Continue to: Our patient was treated...
Our patient was treated with oral acyclovir 800 mg 5 times/day for 10 days, with prompt resolution of her rash.
CORRESPONDENCE
Joel Hua-Liang Lim, MBBS, MRCP, MMed, 1 Mandalay Road, Singapore 308205; [email protected]
1. LeBoit PE, Limova M, Yen TS, et al. Chronic verrucous varicella-zoster virus infection in patients with the acquired immunodeficiency syndrome (AIDS): histologic and molecular biologic findings. Am J Dermatopathol. 1992;14:1-7.
2. Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
3. Weinberg JM, Mysliwiec A, Turiansky GW, et al. Viral folliculitis: atypical presentations of herpes simplex, herpes zoster, and molluscum contagiosum. Arch Dermatol. 1997;133:983-986.
4. Gilson IH, Barnett JH, Conant MA, et al. Disseminated ecthymatous herpes varicella zoster virus infection in patients with acquired immunodeficiency syndrome. J Am Acad Dermatol. 1989;20:637-642.
5. Løkke BJ, Weismann K, Mathiesen L, et al. Atypical varicella-zoster infection in AIDS. Acta Derm Venereol. 1993;73:123-125.
6. Uhoda I, Piérard-Franchimont C, Piérard GE. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement. Dermatology. 2000;200:173-175.
7. Teng GG, Chatham WW. Vasculitis related to viral and other microbial agents. Best Pract Res Clin Rheumatol. 2015;29:226-243.
8. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16:12.
9. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
10. Lott JP, Liu K, Landry M-L, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
A 52-year-old Chinese woman presented to a tertiary hospital in Singapore with a 3-month history of persistent and intermittently painful rashes over her right calf and foot (FIGURE). The patient had pancytopenia due to ongoing chemotherapy for metastatic nasopharyngeal carcinoma. She was systemically well and denied other dermatoses. Examination demonstrated scattered crops of tense hemorrhagic vesicles, each surrounded by a livid purpuric base, over the right plantar aspect of the foot, with areas of eschar over the right medial hallux. No allodynia, hyperaesthesia, or lymphadenopathy was noted.
A punch biopsy of an intact vesicle was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis:
Herpes zoster
Histopathologic examination showed full-thickness epidermal necrosis with ballooning degeneration resulting in an intra-epidermal blister. Multinucleated keratinocytes with nuclear moulding were seen within the blister cavity. Grocott-Gomori methenamine-silver (GMS), acid-fast, and Gram stains were negative. Granular immunoglobulin (Ig) G, IgM, and C3 were seen intramurally. DNA analysis of vesicular fluid was positive for varicella zoster virus (VZV). A diagnosis of herpes zoster (HZ) of the right S1 dermatome with primary obliterative vasculitis was established.
Immunocompromised people—those who have impaired T-cell immunity (eg, recipients of organ or hematopoietic stem-cell transplants), take immunosuppressive therapy, or have lymphoma, leukemia, or human immunodeficiency virus (HIV) infection—have an increased risk for HZ. For example, in patients with acquired immunodeficiency syndrome (AIDS), HZ uniquely manifests as recurrent shingles. An estimated 20% to 30% of HIV-infected patients will have more than 1 episode of HZ, which may involve the same or different dermatomes.1,2 Furthermore, HZ in this population is more commonly associated with atypical presentations.3
What an atypical presentation may look like
In immunocompromised patients, HZ may present with atypical cutaneous manifestations or with atypical generalized symptoms.
Atypical cutaneous manifestations, as in disseminated zoster, manifest with multiple hyperkeratotic papules (3-20 mm in diameter) that follow no dermatomal pattern. These lesions may be chronic, persisting for months or years, and may be associated with acyclovir-resistant strains of VZV.2,3 Another dermatologic variant is ecthymatous VZV, which manifests with multiple large (10-30 mm) punched-out ulcerations with a central black eschar and a peripheral rim of vesicles.4 Viral folliculitis—in which infection is limited to the hair follicle, with no associated blisters—has also been reported in atypical HZ.5
Our patient presented with hemorrhagic vesicles mimicking vasculitic lesions, which had persisted over a 3-month period with intermittent localized pain. It has been proposed that in atypical presentations, the reactivated VZV spreads transaxonally from adjacent nerves to the outermost adventitial layer of the arterial wall, leading to a vasculitic appearance of the vesicles.6 Viral-induced vasculitis may also result either directly from infection of the blood vessels or secondary to vascular damage from an inflammatory immune complex–mediated reaction, cell-mediated hypersensitivity, or inflammation due to immune dysregulation.7,8
Continue to: Differential includes vesiculobullous conditions
Differential includes vesiculobullous conditions
There are several important items to consider in the differential.
Cutaneous vasculitis, in severe cases, may manifest with vesicles or bullae that resemble the lesions seen in HZ. However, its unilateral nature and distribution distinguish it.
Angioinvasive fungal infections in immunocompromised patients may manifest with scattered ulceronecrotic lesions to purpuric vesiculobullous dermatoses.9 However, no fungal organisms were seen on GMS staining of the biopsied tissue.
Atypical hand-foot-and-mouth disease tends to affect adults and is associated with Coxsackievirus A6 infection.10 It may manifest as generalized vesiculobullous exanthem resembling varicella. The chronic nature and restricted extent of the patient’s rash made this diagnosis unlikely.
Successful management depends on timely identification
Although most cases of HZ can be diagnosed clinically, atypical rashes may require a biopsy and direct immunofluorescence assay for VZV antigen or a polymerase-chain-reaction (PCR) assay for VZV DNA in cells from the base of blisters. Therefore, it is important to consider the diagnosis of HZ in immunocompromised patients presenting with an atypical rash to avoid misdiagnosis and costly testing.
Continue to: Our patient was treated...
Our patient was treated with oral acyclovir 800 mg 5 times/day for 10 days, with prompt resolution of her rash.
CORRESPONDENCE
Joel Hua-Liang Lim, MBBS, MRCP, MMed, 1 Mandalay Road, Singapore 308205; [email protected]
A 52-year-old Chinese woman presented to a tertiary hospital in Singapore with a 3-month history of persistent and intermittently painful rashes over her right calf and foot (FIGURE). The patient had pancytopenia due to ongoing chemotherapy for metastatic nasopharyngeal carcinoma. She was systemically well and denied other dermatoses. Examination demonstrated scattered crops of tense hemorrhagic vesicles, each surrounded by a livid purpuric base, over the right plantar aspect of the foot, with areas of eschar over the right medial hallux. No allodynia, hyperaesthesia, or lymphadenopathy was noted.
A punch biopsy of an intact vesicle was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis:
Herpes zoster
Histopathologic examination showed full-thickness epidermal necrosis with ballooning degeneration resulting in an intra-epidermal blister. Multinucleated keratinocytes with nuclear moulding were seen within the blister cavity. Grocott-Gomori methenamine-silver (GMS), acid-fast, and Gram stains were negative. Granular immunoglobulin (Ig) G, IgM, and C3 were seen intramurally. DNA analysis of vesicular fluid was positive for varicella zoster virus (VZV). A diagnosis of herpes zoster (HZ) of the right S1 dermatome with primary obliterative vasculitis was established.
Immunocompromised people—those who have impaired T-cell immunity (eg, recipients of organ or hematopoietic stem-cell transplants), take immunosuppressive therapy, or have lymphoma, leukemia, or human immunodeficiency virus (HIV) infection—have an increased risk for HZ. For example, in patients with acquired immunodeficiency syndrome (AIDS), HZ uniquely manifests as recurrent shingles. An estimated 20% to 30% of HIV-infected patients will have more than 1 episode of HZ, which may involve the same or different dermatomes.1,2 Furthermore, HZ in this population is more commonly associated with atypical presentations.3
What an atypical presentation may look like
In immunocompromised patients, HZ may present with atypical cutaneous manifestations or with atypical generalized symptoms.
Atypical cutaneous manifestations, as in disseminated zoster, manifest with multiple hyperkeratotic papules (3-20 mm in diameter) that follow no dermatomal pattern. These lesions may be chronic, persisting for months or years, and may be associated with acyclovir-resistant strains of VZV.2,3 Another dermatologic variant is ecthymatous VZV, which manifests with multiple large (10-30 mm) punched-out ulcerations with a central black eschar and a peripheral rim of vesicles.4 Viral folliculitis—in which infection is limited to the hair follicle, with no associated blisters—has also been reported in atypical HZ.5
Our patient presented with hemorrhagic vesicles mimicking vasculitic lesions, which had persisted over a 3-month period with intermittent localized pain. It has been proposed that in atypical presentations, the reactivated VZV spreads transaxonally from adjacent nerves to the outermost adventitial layer of the arterial wall, leading to a vasculitic appearance of the vesicles.6 Viral-induced vasculitis may also result either directly from infection of the blood vessels or secondary to vascular damage from an inflammatory immune complex–mediated reaction, cell-mediated hypersensitivity, or inflammation due to immune dysregulation.7,8
Continue to: Differential includes vesiculobullous conditions
Differential includes vesiculobullous conditions
There are several important items to consider in the differential.
Cutaneous vasculitis, in severe cases, may manifest with vesicles or bullae that resemble the lesions seen in HZ. However, its unilateral nature and distribution distinguish it.
Angioinvasive fungal infections in immunocompromised patients may manifest with scattered ulceronecrotic lesions to purpuric vesiculobullous dermatoses.9 However, no fungal organisms were seen on GMS staining of the biopsied tissue.
Atypical hand-foot-and-mouth disease tends to affect adults and is associated with Coxsackievirus A6 infection.10 It may manifest as generalized vesiculobullous exanthem resembling varicella. The chronic nature and restricted extent of the patient’s rash made this diagnosis unlikely.
Successful management depends on timely identification
Although most cases of HZ can be diagnosed clinically, atypical rashes may require a biopsy and direct immunofluorescence assay for VZV antigen or a polymerase-chain-reaction (PCR) assay for VZV DNA in cells from the base of blisters. Therefore, it is important to consider the diagnosis of HZ in immunocompromised patients presenting with an atypical rash to avoid misdiagnosis and costly testing.
Continue to: Our patient was treated...
Our patient was treated with oral acyclovir 800 mg 5 times/day for 10 days, with prompt resolution of her rash.
CORRESPONDENCE
Joel Hua-Liang Lim, MBBS, MRCP, MMed, 1 Mandalay Road, Singapore 308205; [email protected]
1. LeBoit PE, Limova M, Yen TS, et al. Chronic verrucous varicella-zoster virus infection in patients with the acquired immunodeficiency syndrome (AIDS): histologic and molecular biologic findings. Am J Dermatopathol. 1992;14:1-7.
2. Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
3. Weinberg JM, Mysliwiec A, Turiansky GW, et al. Viral folliculitis: atypical presentations of herpes simplex, herpes zoster, and molluscum contagiosum. Arch Dermatol. 1997;133:983-986.
4. Gilson IH, Barnett JH, Conant MA, et al. Disseminated ecthymatous herpes varicella zoster virus infection in patients with acquired immunodeficiency syndrome. J Am Acad Dermatol. 1989;20:637-642.
5. Løkke BJ, Weismann K, Mathiesen L, et al. Atypical varicella-zoster infection in AIDS. Acta Derm Venereol. 1993;73:123-125.
6. Uhoda I, Piérard-Franchimont C, Piérard GE. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement. Dermatology. 2000;200:173-175.
7. Teng GG, Chatham WW. Vasculitis related to viral and other microbial agents. Best Pract Res Clin Rheumatol. 2015;29:226-243.
8. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16:12.
9. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
10. Lott JP, Liu K, Landry M-L, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
1. LeBoit PE, Limova M, Yen TS, et al. Chronic verrucous varicella-zoster virus infection in patients with the acquired immunodeficiency syndrome (AIDS): histologic and molecular biologic findings. Am J Dermatopathol. 1992;14:1-7.
2. Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
3. Weinberg JM, Mysliwiec A, Turiansky GW, et al. Viral folliculitis: atypical presentations of herpes simplex, herpes zoster, and molluscum contagiosum. Arch Dermatol. 1997;133:983-986.
4. Gilson IH, Barnett JH, Conant MA, et al. Disseminated ecthymatous herpes varicella zoster virus infection in patients with acquired immunodeficiency syndrome. J Am Acad Dermatol. 1989;20:637-642.
5. Løkke BJ, Weismann K, Mathiesen L, et al. Atypical varicella-zoster infection in AIDS. Acta Derm Venereol. 1993;73:123-125.
6. Uhoda I, Piérard-Franchimont C, Piérard GE. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement. Dermatology. 2000;200:173-175.
7. Teng GG, Chatham WW. Vasculitis related to viral and other microbial agents. Best Pract Res Clin Rheumatol. 2015;29:226-243.
8. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16:12.
9. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
10. Lott JP, Liu K, Landry M-L, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
