User login
Tips for Living With Tardive Dyskinesia
Click here to download the PDF.

Click here to download the PDF.
Click here to download the PDF.

Tips for Living With Suspected CTE
Click here to download the PDF. 
Click here to download the PDF.
Click here to download the PDF.

New histopathologic marker may aid dermatomyositis diagnosis
The detection of sarcoplasmic myxovirus resistance A expression in immunohistochemical analysis of muscle biopsy in patients suspected of having dermatomyositis may add greater sensitivity for the diagnosis when compared with conventional pathologic hallmarks of the disease, according to findings from a retrospective cohort study.
Myxovirus resistance A (MxA) is one of the type 1 interferon–inducible proteins whose overexpression is believed to play a role in the pathogenesis of dermatomyositis, and MxA expression has rarely been observed in other idiopathic inflammatory myopathies, said first author Akinori Uruha, MD, PhD, of the National Center of Neurology and Psychiatry, Tokyo, and his colleagues. They compared MxA expression in muscle biopsy samples from definite, probable, and possible dermatomyositis cases as well as other idiopathic inflammatory myopathies and other control conditions to assess its value against other muscle pathologic markers of dermatomyositis, such as the presence of perifascicular atrophy (PFA) and capillary membrane attack complex (MAC) deposition (Neurology. 2016 Dec 30. doi: 10.1212/WNL.0000000000003568).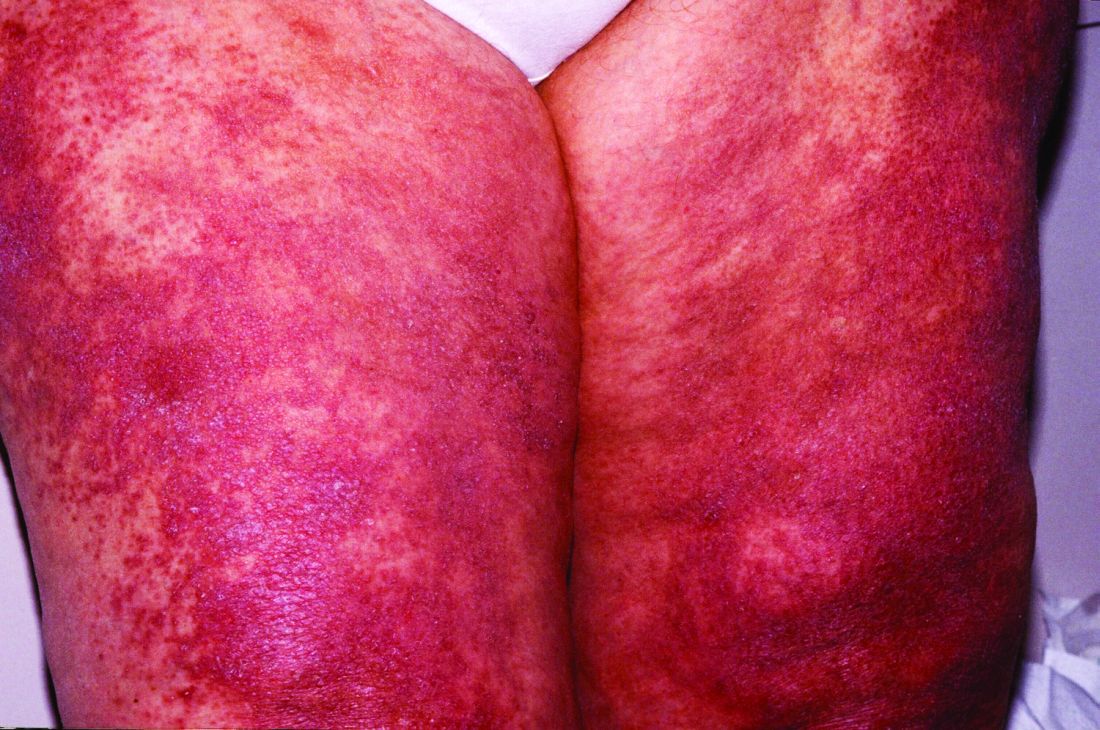
The investigators studied muscle biopsy samples collected from 154 consecutive patients with idiopathic inflammatory myopathies seen from all over Japan, including 34 with dermatomyositis (10 juvenile cases), 8 with polymyositis (1 juvenile), 16 with anti–tRNA-synthetase antibody–associated myopathy (ASM); 46 with immune-mediated necrotizing myopathy (IMNM), and 50 with inclusion body myositis. The IMNM cases involved included 24 with anti–signal recognition particle (SRP) antibodies, 6 with anti–3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibodies, and 16 without anti-SRP, anti-HMGCR, or anti–tRNA-synthetase antibodies (3 juvenile patients). They used 51 patients with muscular dystrophy and 26 with neuropathies as controls.
Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Of 18 cases with probable dermatomyositis, defined as typical skin rash but a lack of PFA, 8 (44%) showed sarcoplasmic MxA expression, and its sensitivity was 90% in juvenile cases overall and 63% in adult patients. Only 3 (17%) of the 18 showed capillary MAC deposition. Sarcoplasmic MxA expression occurred in all 12 patients with definite dermatomyositis, defined by the typical skin rash plus presence of PFA, whereas only 7 (58%) showed capillary MAC deposition. Among the four patients with possible dermatomyositis (PFA present but lacking typical skin rash), all showed sarcoplasmic MxA expression, compared with just two showing capillary MAC deposition.
In all other patients without definite, probable, or possible dermatomyositis, only two were positive for sarcoplasmic MxA expression (one with ASM and one with IMNM).
Dr. Uruha and his associates said that the results are “clearly demonstrating that sarcoplasmic MxA expression should be an excellent diagnostic marker of [dermatomyositis].”
The authors noted that the study was limited by the fact that they could not obtain full information about dermatomyositis-associated antibodies, and because other proteins of type 1 interferon signature are known to be upregulated in dermatomyositis, additional studies will need to determine which of the proteins is a better diagnostic marker.
The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
The detection of sarcoplasmic myxovirus resistance A expression in immunohistochemical analysis of muscle biopsy in patients suspected of having dermatomyositis may add greater sensitivity for the diagnosis when compared with conventional pathologic hallmarks of the disease, according to findings from a retrospective cohort study.
Myxovirus resistance A (MxA) is one of the type 1 interferon–inducible proteins whose overexpression is believed to play a role in the pathogenesis of dermatomyositis, and MxA expression has rarely been observed in other idiopathic inflammatory myopathies, said first author Akinori Uruha, MD, PhD, of the National Center of Neurology and Psychiatry, Tokyo, and his colleagues. They compared MxA expression in muscle biopsy samples from definite, probable, and possible dermatomyositis cases as well as other idiopathic inflammatory myopathies and other control conditions to assess its value against other muscle pathologic markers of dermatomyositis, such as the presence of perifascicular atrophy (PFA) and capillary membrane attack complex (MAC) deposition (Neurology. 2016 Dec 30. doi: 10.1212/WNL.0000000000003568).
The investigators studied muscle biopsy samples collected from 154 consecutive patients with idiopathic inflammatory myopathies seen from all over Japan, including 34 with dermatomyositis (10 juvenile cases), 8 with polymyositis (1 juvenile), 16 with anti–tRNA-synthetase antibody–associated myopathy (ASM); 46 with immune-mediated necrotizing myopathy (IMNM), and 50 with inclusion body myositis. The IMNM cases involved included 24 with anti–signal recognition particle (SRP) antibodies, 6 with anti–3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibodies, and 16 without anti-SRP, anti-HMGCR, or anti–tRNA-synthetase antibodies (3 juvenile patients). They used 51 patients with muscular dystrophy and 26 with neuropathies as controls.
Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Of 18 cases with probable dermatomyositis, defined as typical skin rash but a lack of PFA, 8 (44%) showed sarcoplasmic MxA expression, and its sensitivity was 90% in juvenile cases overall and 63% in adult patients. Only 3 (17%) of the 18 showed capillary MAC deposition. Sarcoplasmic MxA expression occurred in all 12 patients with definite dermatomyositis, defined by the typical skin rash plus presence of PFA, whereas only 7 (58%) showed capillary MAC deposition. Among the four patients with possible dermatomyositis (PFA present but lacking typical skin rash), all showed sarcoplasmic MxA expression, compared with just two showing capillary MAC deposition.
In all other patients without definite, probable, or possible dermatomyositis, only two were positive for sarcoplasmic MxA expression (one with ASM and one with IMNM).
Dr. Uruha and his associates said that the results are “clearly demonstrating that sarcoplasmic MxA expression should be an excellent diagnostic marker of [dermatomyositis].”
The authors noted that the study was limited by the fact that they could not obtain full information about dermatomyositis-associated antibodies, and because other proteins of type 1 interferon signature are known to be upregulated in dermatomyositis, additional studies will need to determine which of the proteins is a better diagnostic marker.
The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
The detection of sarcoplasmic myxovirus resistance A expression in immunohistochemical analysis of muscle biopsy in patients suspected of having dermatomyositis may add greater sensitivity for the diagnosis when compared with conventional pathologic hallmarks of the disease, according to findings from a retrospective cohort study.
Myxovirus resistance A (MxA) is one of the type 1 interferon–inducible proteins whose overexpression is believed to play a role in the pathogenesis of dermatomyositis, and MxA expression has rarely been observed in other idiopathic inflammatory myopathies, said first author Akinori Uruha, MD, PhD, of the National Center of Neurology and Psychiatry, Tokyo, and his colleagues. They compared MxA expression in muscle biopsy samples from definite, probable, and possible dermatomyositis cases as well as other idiopathic inflammatory myopathies and other control conditions to assess its value against other muscle pathologic markers of dermatomyositis, such as the presence of perifascicular atrophy (PFA) and capillary membrane attack complex (MAC) deposition (Neurology. 2016 Dec 30. doi: 10.1212/WNL.0000000000003568).
The investigators studied muscle biopsy samples collected from 154 consecutive patients with idiopathic inflammatory myopathies seen from all over Japan, including 34 with dermatomyositis (10 juvenile cases), 8 with polymyositis (1 juvenile), 16 with anti–tRNA-synthetase antibody–associated myopathy (ASM); 46 with immune-mediated necrotizing myopathy (IMNM), and 50 with inclusion body myositis. The IMNM cases involved included 24 with anti–signal recognition particle (SRP) antibodies, 6 with anti–3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibodies, and 16 without anti-SRP, anti-HMGCR, or anti–tRNA-synthetase antibodies (3 juvenile patients). They used 51 patients with muscular dystrophy and 26 with neuropathies as controls.
Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Of 18 cases with probable dermatomyositis, defined as typical skin rash but a lack of PFA, 8 (44%) showed sarcoplasmic MxA expression, and its sensitivity was 90% in juvenile cases overall and 63% in adult patients. Only 3 (17%) of the 18 showed capillary MAC deposition. Sarcoplasmic MxA expression occurred in all 12 patients with definite dermatomyositis, defined by the typical skin rash plus presence of PFA, whereas only 7 (58%) showed capillary MAC deposition. Among the four patients with possible dermatomyositis (PFA present but lacking typical skin rash), all showed sarcoplasmic MxA expression, compared with just two showing capillary MAC deposition.
In all other patients without definite, probable, or possible dermatomyositis, only two were positive for sarcoplasmic MxA expression (one with ASM and one with IMNM).
Dr. Uruha and his associates said that the results are “clearly demonstrating that sarcoplasmic MxA expression should be an excellent diagnostic marker of [dermatomyositis].”
The authors noted that the study was limited by the fact that they could not obtain full information about dermatomyositis-associated antibodies, and because other proteins of type 1 interferon signature are known to be upregulated in dermatomyositis, additional studies will need to determine which of the proteins is a better diagnostic marker.
The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
FROM NEUROLOGY
Key clinical point:
Major finding: Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Data source: A retrospective cohort study of 154 patients with idiopathic inflammatory myopathies, 51 with muscular dystrophy, and 26 with neuropathies.
Disclosures: The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
Meningococcal conjugate vaccination may be associated with increased risk of Bell’s palsy
A postlicensure safety study of a meningococcal conjugate vaccine in Southern California has shown that the vaccine may be associated with an increase in the risk of Bell’s palsy, but only if the vaccine is taken concomitantly with another vaccine.
Researchers set out to evaluate the safety of one quadrivalent meningococcal conjugate vaccine, MenACWY-CRM. Two MenACWY vaccines are currently licensed in the United States; MenACWY-D is the other. The vaccines underwent studies on the road to approval, but researchers saw an absence of data about how the vaccine was faring in routine clinical use.
Researchers looked through the electronic health records of the study population for “26 prespecified events of interest (EOIs) under investigation, including neurologic, rheumatologic, hematologic, endocrine, renal, pediatric, and pediatric infectious disease EOIs. Occurrence of incident episodes of these EOIs was identified during a 1-year observation period after the index vaccination for each individual.”
They identified 4,240 EOIs, but dismissed 3,000 of them as probable preexisting conditions. With what was left, some of the EOIs did not occur at all (such as Guillain-Barré syndrome, myasthenia gravis, or systemic lupus erythematosus). Of transverse myelitis and autoimmune hemolytic anemia, among others, there was only 1 case.
Seizure, iridocyclitis, Hashimoto’s disease, and anaphylaxis initially showed statistically significant risk incidence, but were all ruled out (of the hypothesis of possible cause by vaccination) by further review from a physician investigator.
But in the case of Bell’s palsy, the independent case review committee did not rule out the possibility that the MenACWY-CRM vaccine increased the risk incidence of the condition.
However, the increased risk was present only for subjects who received a concomitant vaccine along with the MenACWY-CRM, such as Tdap, influenza, or human papillomavirus vaccine. “Stratified analyses demonstrated an increased risk for Bell’s palsy in subjects receiving concomitant vaccines (risk incidence, 5.0; 95% confidence interval, 1.4-17.8), and no increased risk for those without concomitant vaccine (RI, 1.1; 95% CI, 0.2-5.5),” Dr. Tseng and his coauthors wrote. All eight cases of Bell’s palsy resolved completely.
They concluded, “we observed a temporal association between occurrence of Bell’s palsy and receipt of MenACWY-CRM concomitantly with other vaccines. The association needs further investigation because it could be due to chance, concomitant vaccination, or underlying medical history predisposing to Bell’s palsy.”
Dr. Tseng and numerous coauthors reported receiving research support from Novartis Vaccines, the sponsor of the study. Three coauthors were employees of Novartis at the time of the study.
A postlicensure safety study of a meningococcal conjugate vaccine in Southern California has shown that the vaccine may be associated with an increase in the risk of Bell’s palsy, but only if the vaccine is taken concomitantly with another vaccine.
Researchers set out to evaluate the safety of one quadrivalent meningococcal conjugate vaccine, MenACWY-CRM. Two MenACWY vaccines are currently licensed in the United States; MenACWY-D is the other. The vaccines underwent studies on the road to approval, but researchers saw an absence of data about how the vaccine was faring in routine clinical use.
Researchers looked through the electronic health records of the study population for “26 prespecified events of interest (EOIs) under investigation, including neurologic, rheumatologic, hematologic, endocrine, renal, pediatric, and pediatric infectious disease EOIs. Occurrence of incident episodes of these EOIs was identified during a 1-year observation period after the index vaccination for each individual.”
They identified 4,240 EOIs, but dismissed 3,000 of them as probable preexisting conditions. With what was left, some of the EOIs did not occur at all (such as Guillain-Barré syndrome, myasthenia gravis, or systemic lupus erythematosus). Of transverse myelitis and autoimmune hemolytic anemia, among others, there was only 1 case.
Seizure, iridocyclitis, Hashimoto’s disease, and anaphylaxis initially showed statistically significant risk incidence, but were all ruled out (of the hypothesis of possible cause by vaccination) by further review from a physician investigator.
But in the case of Bell’s palsy, the independent case review committee did not rule out the possibility that the MenACWY-CRM vaccine increased the risk incidence of the condition.
However, the increased risk was present only for subjects who received a concomitant vaccine along with the MenACWY-CRM, such as Tdap, influenza, or human papillomavirus vaccine. “Stratified analyses demonstrated an increased risk for Bell’s palsy in subjects receiving concomitant vaccines (risk incidence, 5.0; 95% confidence interval, 1.4-17.8), and no increased risk for those without concomitant vaccine (RI, 1.1; 95% CI, 0.2-5.5),” Dr. Tseng and his coauthors wrote. All eight cases of Bell’s palsy resolved completely.
They concluded, “we observed a temporal association between occurrence of Bell’s palsy and receipt of MenACWY-CRM concomitantly with other vaccines. The association needs further investigation because it could be due to chance, concomitant vaccination, or underlying medical history predisposing to Bell’s palsy.”
Dr. Tseng and numerous coauthors reported receiving research support from Novartis Vaccines, the sponsor of the study. Three coauthors were employees of Novartis at the time of the study.
A postlicensure safety study of a meningococcal conjugate vaccine in Southern California has shown that the vaccine may be associated with an increase in the risk of Bell’s palsy, but only if the vaccine is taken concomitantly with another vaccine.
Researchers set out to evaluate the safety of one quadrivalent meningococcal conjugate vaccine, MenACWY-CRM. Two MenACWY vaccines are currently licensed in the United States; MenACWY-D is the other. The vaccines underwent studies on the road to approval, but researchers saw an absence of data about how the vaccine was faring in routine clinical use.
Researchers looked through the electronic health records of the study population for “26 prespecified events of interest (EOIs) under investigation, including neurologic, rheumatologic, hematologic, endocrine, renal, pediatric, and pediatric infectious disease EOIs. Occurrence of incident episodes of these EOIs was identified during a 1-year observation period after the index vaccination for each individual.”
They identified 4,240 EOIs, but dismissed 3,000 of them as probable preexisting conditions. With what was left, some of the EOIs did not occur at all (such as Guillain-Barré syndrome, myasthenia gravis, or systemic lupus erythematosus). Of transverse myelitis and autoimmune hemolytic anemia, among others, there was only 1 case.
Seizure, iridocyclitis, Hashimoto’s disease, and anaphylaxis initially showed statistically significant risk incidence, but were all ruled out (of the hypothesis of possible cause by vaccination) by further review from a physician investigator.
But in the case of Bell’s palsy, the independent case review committee did not rule out the possibility that the MenACWY-CRM vaccine increased the risk incidence of the condition.
However, the increased risk was present only for subjects who received a concomitant vaccine along with the MenACWY-CRM, such as Tdap, influenza, or human papillomavirus vaccine. “Stratified analyses demonstrated an increased risk for Bell’s palsy in subjects receiving concomitant vaccines (risk incidence, 5.0; 95% confidence interval, 1.4-17.8), and no increased risk for those without concomitant vaccine (RI, 1.1; 95% CI, 0.2-5.5),” Dr. Tseng and his coauthors wrote. All eight cases of Bell’s palsy resolved completely.
They concluded, “we observed a temporal association between occurrence of Bell’s palsy and receipt of MenACWY-CRM concomitantly with other vaccines. The association needs further investigation because it could be due to chance, concomitant vaccination, or underlying medical history predisposing to Bell’s palsy.”
Dr. Tseng and numerous coauthors reported receiving research support from Novartis Vaccines, the sponsor of the study. Three coauthors were employees of Novartis at the time of the study.
What’s New in Rare Neuromuscular Disorders?
LAS VEGAS—At the American Academy of Neurology’s Fall 2016 Conference, Robert M. Pascuzzi, MD, presented several case videos to illustrate recent advances in caring for patients with rare neuromuscular disorders, including myasthenia gravis, Lambert-Eaton syndrome, Duchenne muscular dystrophy, and amyotrophic lateral sclerosis (ALS). Dr. Pascuzzi is Chair of the Department of Neurology at Indiana University School of Medicine in Indianapolis.

Myasthenia Gravis
The first case, a 55-year-old woman with a six-month history of double vision and swallowing trouble, also expressed the Cogan lid twitch, which is “pathognomonic for myasthenia gravis,” Dr. Pascuzzi said.
“It’s nice to have a test to confirm your clinical suspicions,” he said, noting that antibodies to the acetylcholine receptor (AChR) yield “almost perfect specificity” for myasthenia gravis. Other antibodies in use or under study to confirm this diagnosis include muscle-specific kinase (MuSK), low-density lipoprotein receptor-related protein 4 (LRP4), agrin, and cortactin.
The recently completed International Thymectomy Trial, which followed 126 patients for three years, found that those randomly assigned to transsternal thymectomy plus prednisone versus prednisone alone had significantly improved clinical outcomes, as measured on the Quantitative Myasthenia Gravis scale. Patients also required less prednisone, 44 mg versus 60 mg (alternate days), and fewer patients in the thymectomy group required azathioprine (17% versus 48%) or were hospitalized for exacerbations (9% versus 37%).
“Up until this study, nobody was completely sure this treatment worked,” Dr. Pascuzzi said.
Lambert-Eaton Syndrome
In the second case, a woman presented with a six-month history of falling down after her legs gave out on her. She also complained of dry mouth and, on examination, had proximal weakness and no muscle stretch reflexes.
Serology and EMG testing confirmed Lambert-Eaton myasthenic syndrome, a paraneoplastic syndrome. Half the patients with this syndrome have small cell carcinoma, Dr. Pascuzzi said, usually lung, due to smoking. In 64% of cases, the SOX1 antibody is positive.
Treatment approaches include treating the cancer; and 3,4 diaminopyridine, pyridostigmine, and immunotherapy.
Duchenne Muscular Dystrophy
A six-year-old boy with progressive clumsiness was found to have Duchenne muscular dystrophy, a diagnosis with “huge” genetic implications for his three younger brothers, Dr. Pascuzzi said. By age 20, patients are usually on ventilators.
Recently, the FDA granted accelerated approval to the first drug to treat Duchenne muscular dystrophy—Exondys 51 (eteplirsen). The drug “is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13% of the population with Duchenne muscular dystrophy,” the FDA noted.
However, eteplirsen is not without controversy, Dr. Pascuzzi said. “There’s no documented clinical benefit. If you show there is a little more dystrophin being deposited and built in muscle, what does that mean? As of today, nobody knows. So that is an issue I think the FDA Advisory Panel and other people are worried about. And then, at $300,000 a year, it is not a financially insignificant impact.”
As the drug is used, further studies and data “will eventually show that it does or does not have clinical benefit,” Dr. Pascuzzi said. Patients must undergo testing for the mutation. Reported adverse events include balance issues and vomiting.
Amyotrophic Lateral Sclerosis
The third and fourth cases focused on a 53-year-old man with 12 months of progressive decline in speech and swallowing, and a woman with similar symptoms who also would laugh for no apparent reason. Both cases were displaying the pseudobulbar affect of ALS.
About 5% to 10% of all ALS cases are familial and, in 40% to 50%, the most common gene is C9orf72. Less common is the SOD1 gene (20%), with those carrying its A4V mutation generally dying within one year. Other mutations include TDP-43 (5%), and FUS (5%).
Dr. Pascuzzi cautioned that cognitive impairment among patients with ALS can be as high as 40% and cannot usually be detected on a bedside exam. Up to 15% of patients “get a frank, overt, clinically significant dementia, usually a frontotemporal syndrome.”
Treatment approaches include use of multidisciplinary clinics, ankle-foot orthosis, tools for activities of daily living, bilevel positive airway pressure, chin tuck, and power chairs; drugs include riluzole, dextromethorphan/quinidine for pseudobulbar affect, and drugs for spasticity.
He said it is important to address fear, depression, and anxiety in patients with ALS, and not to overlook the caregiver: “We are slowly but surely melting down our caregivers.”
A recent study in Lancet Neurology helped Dr. Pascuzzi and colleagues decide against diaphragm pacing for patients with respiratory insufficiency due to ALS, due to the potential for harm. “I would be cautious about pursuing this,” he said.
Despite the many failed controlled trials over the past 20 years, much investigational work is being done on ALS therapeutics, from injection of antisense molecules to tirasemtiv to stem cells, immune therapy, neuro-protection, and high fat/calorie diet, “so there is reason to be hopeful.”
Stem cell transplantation, in particular, is something he said his patients always ask him about. Two forms, “both important,” have undergone fairly extensive clinical investigation.
In the first, spinal cord-derived neural stem cells are transplanted into the spinal cord.
“The people that drive this work are convinced this is not stem cells that turn into motor neurons and replace lost or failing motor neurons, but instead these are stem cells that provide an environment that is more favorable for sick motor neurons,” Dr. Pascuzzi said. “They secrete nerve growth factors and transport proteins to improve the environment.”
The second approach, autologous stem cell treatment, is a “culture-based method for inducing mesenchymal stem cells to secrete neurotrophic factors,” he said, which has been “shown to be protective in several animal models of neurodegenerative diseases.”
—Debra Hughes
Suggested Reading
DiPALS Writing Committee on behalf of the DiPALS Study Group Collaborators. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): a multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015;14(9);883-892.
Glass JD, Hertzberg VS, Boulis NM, et al. Transplantation of spinal cord-derived neural stem cells for ALS: analysis of phase 1 and 2 trials. Neurology. 2016;87(4):392-400.
Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol. 2016;73(3):337-344.
U.S. Food and Drug Administration. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA News Release. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm. Accessed November 4, 2016.
Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375(6):511-522.
LAS VEGAS—At the American Academy of Neurology’s Fall 2016 Conference, Robert M. Pascuzzi, MD, presented several case videos to illustrate recent advances in caring for patients with rare neuromuscular disorders, including myasthenia gravis, Lambert-Eaton syndrome, Duchenne muscular dystrophy, and amyotrophic lateral sclerosis (ALS). Dr. Pascuzzi is Chair of the Department of Neurology at Indiana University School of Medicine in Indianapolis.
Myasthenia Gravis
The first case, a 55-year-old woman with a six-month history of double vision and swallowing trouble, also expressed the Cogan lid twitch, which is “pathognomonic for myasthenia gravis,” Dr. Pascuzzi said.
“It’s nice to have a test to confirm your clinical suspicions,” he said, noting that antibodies to the acetylcholine receptor (AChR) yield “almost perfect specificity” for myasthenia gravis. Other antibodies in use or under study to confirm this diagnosis include muscle-specific kinase (MuSK), low-density lipoprotein receptor-related protein 4 (LRP4), agrin, and cortactin.
The recently completed International Thymectomy Trial, which followed 126 patients for three years, found that those randomly assigned to transsternal thymectomy plus prednisone versus prednisone alone had significantly improved clinical outcomes, as measured on the Quantitative Myasthenia Gravis scale. Patients also required less prednisone, 44 mg versus 60 mg (alternate days), and fewer patients in the thymectomy group required azathioprine (17% versus 48%) or were hospitalized for exacerbations (9% versus 37%).
“Up until this study, nobody was completely sure this treatment worked,” Dr. Pascuzzi said.
Lambert-Eaton Syndrome
In the second case, a woman presented with a six-month history of falling down after her legs gave out on her. She also complained of dry mouth and, on examination, had proximal weakness and no muscle stretch reflexes.
Serology and EMG testing confirmed Lambert-Eaton myasthenic syndrome, a paraneoplastic syndrome. Half the patients with this syndrome have small cell carcinoma, Dr. Pascuzzi said, usually lung, due to smoking. In 64% of cases, the SOX1 antibody is positive.
Treatment approaches include treating the cancer; and 3,4 diaminopyridine, pyridostigmine, and immunotherapy.
Duchenne Muscular Dystrophy
A six-year-old boy with progressive clumsiness was found to have Duchenne muscular dystrophy, a diagnosis with “huge” genetic implications for his three younger brothers, Dr. Pascuzzi said. By age 20, patients are usually on ventilators.
Recently, the FDA granted accelerated approval to the first drug to treat Duchenne muscular dystrophy—Exondys 51 (eteplirsen). The drug “is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13% of the population with Duchenne muscular dystrophy,” the FDA noted.
However, eteplirsen is not without controversy, Dr. Pascuzzi said. “There’s no documented clinical benefit. If you show there is a little more dystrophin being deposited and built in muscle, what does that mean? As of today, nobody knows. So that is an issue I think the FDA Advisory Panel and other people are worried about. And then, at $300,000 a year, it is not a financially insignificant impact.”
As the drug is used, further studies and data “will eventually show that it does or does not have clinical benefit,” Dr. Pascuzzi said. Patients must undergo testing for the mutation. Reported adverse events include balance issues and vomiting.
Amyotrophic Lateral Sclerosis
The third and fourth cases focused on a 53-year-old man with 12 months of progressive decline in speech and swallowing, and a woman with similar symptoms who also would laugh for no apparent reason. Both cases were displaying the pseudobulbar affect of ALS.
About 5% to 10% of all ALS cases are familial and, in 40% to 50%, the most common gene is C9orf72. Less common is the SOD1 gene (20%), with those carrying its A4V mutation generally dying within one year. Other mutations include TDP-43 (5%), and FUS (5%).
Dr. Pascuzzi cautioned that cognitive impairment among patients with ALS can be as high as 40% and cannot usually be detected on a bedside exam. Up to 15% of patients “get a frank, overt, clinically significant dementia, usually a frontotemporal syndrome.”
Treatment approaches include use of multidisciplinary clinics, ankle-foot orthosis, tools for activities of daily living, bilevel positive airway pressure, chin tuck, and power chairs; drugs include riluzole, dextromethorphan/quinidine for pseudobulbar affect, and drugs for spasticity.
He said it is important to address fear, depression, and anxiety in patients with ALS, and not to overlook the caregiver: “We are slowly but surely melting down our caregivers.”
A recent study in Lancet Neurology helped Dr. Pascuzzi and colleagues decide against diaphragm pacing for patients with respiratory insufficiency due to ALS, due to the potential for harm. “I would be cautious about pursuing this,” he said.
Despite the many failed controlled trials over the past 20 years, much investigational work is being done on ALS therapeutics, from injection of antisense molecules to tirasemtiv to stem cells, immune therapy, neuro-protection, and high fat/calorie diet, “so there is reason to be hopeful.”
Stem cell transplantation, in particular, is something he said his patients always ask him about. Two forms, “both important,” have undergone fairly extensive clinical investigation.
In the first, spinal cord-derived neural stem cells are transplanted into the spinal cord.
“The people that drive this work are convinced this is not stem cells that turn into motor neurons and replace lost or failing motor neurons, but instead these are stem cells that provide an environment that is more favorable for sick motor neurons,” Dr. Pascuzzi said. “They secrete nerve growth factors and transport proteins to improve the environment.”
The second approach, autologous stem cell treatment, is a “culture-based method for inducing mesenchymal stem cells to secrete neurotrophic factors,” he said, which has been “shown to be protective in several animal models of neurodegenerative diseases.”
—Debra Hughes
Suggested Reading
DiPALS Writing Committee on behalf of the DiPALS Study Group Collaborators. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): a multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015;14(9);883-892.
Glass JD, Hertzberg VS, Boulis NM, et al. Transplantation of spinal cord-derived neural stem cells for ALS: analysis of phase 1 and 2 trials. Neurology. 2016;87(4):392-400.
Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol. 2016;73(3):337-344.
U.S. Food and Drug Administration. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA News Release. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm. Accessed November 4, 2016.
Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375(6):511-522.
LAS VEGAS—At the American Academy of Neurology’s Fall 2016 Conference, Robert M. Pascuzzi, MD, presented several case videos to illustrate recent advances in caring for patients with rare neuromuscular disorders, including myasthenia gravis, Lambert-Eaton syndrome, Duchenne muscular dystrophy, and amyotrophic lateral sclerosis (ALS). Dr. Pascuzzi is Chair of the Department of Neurology at Indiana University School of Medicine in Indianapolis.
Myasthenia Gravis
The first case, a 55-year-old woman with a six-month history of double vision and swallowing trouble, also expressed the Cogan lid twitch, which is “pathognomonic for myasthenia gravis,” Dr. Pascuzzi said.
“It’s nice to have a test to confirm your clinical suspicions,” he said, noting that antibodies to the acetylcholine receptor (AChR) yield “almost perfect specificity” for myasthenia gravis. Other antibodies in use or under study to confirm this diagnosis include muscle-specific kinase (MuSK), low-density lipoprotein receptor-related protein 4 (LRP4), agrin, and cortactin.
The recently completed International Thymectomy Trial, which followed 126 patients for three years, found that those randomly assigned to transsternal thymectomy plus prednisone versus prednisone alone had significantly improved clinical outcomes, as measured on the Quantitative Myasthenia Gravis scale. Patients also required less prednisone, 44 mg versus 60 mg (alternate days), and fewer patients in the thymectomy group required azathioprine (17% versus 48%) or were hospitalized for exacerbations (9% versus 37%).
“Up until this study, nobody was completely sure this treatment worked,” Dr. Pascuzzi said.
Lambert-Eaton Syndrome
In the second case, a woman presented with a six-month history of falling down after her legs gave out on her. She also complained of dry mouth and, on examination, had proximal weakness and no muscle stretch reflexes.
Serology and EMG testing confirmed Lambert-Eaton myasthenic syndrome, a paraneoplastic syndrome. Half the patients with this syndrome have small cell carcinoma, Dr. Pascuzzi said, usually lung, due to smoking. In 64% of cases, the SOX1 antibody is positive.
Treatment approaches include treating the cancer; and 3,4 diaminopyridine, pyridostigmine, and immunotherapy.
Duchenne Muscular Dystrophy
A six-year-old boy with progressive clumsiness was found to have Duchenne muscular dystrophy, a diagnosis with “huge” genetic implications for his three younger brothers, Dr. Pascuzzi said. By age 20, patients are usually on ventilators.
Recently, the FDA granted accelerated approval to the first drug to treat Duchenne muscular dystrophy—Exondys 51 (eteplirsen). The drug “is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13% of the population with Duchenne muscular dystrophy,” the FDA noted.
However, eteplirsen is not without controversy, Dr. Pascuzzi said. “There’s no documented clinical benefit. If you show there is a little more dystrophin being deposited and built in muscle, what does that mean? As of today, nobody knows. So that is an issue I think the FDA Advisory Panel and other people are worried about. And then, at $300,000 a year, it is not a financially insignificant impact.”
As the drug is used, further studies and data “will eventually show that it does or does not have clinical benefit,” Dr. Pascuzzi said. Patients must undergo testing for the mutation. Reported adverse events include balance issues and vomiting.
Amyotrophic Lateral Sclerosis
The third and fourth cases focused on a 53-year-old man with 12 months of progressive decline in speech and swallowing, and a woman with similar symptoms who also would laugh for no apparent reason. Both cases were displaying the pseudobulbar affect of ALS.
About 5% to 10% of all ALS cases are familial and, in 40% to 50%, the most common gene is C9orf72. Less common is the SOD1 gene (20%), with those carrying its A4V mutation generally dying within one year. Other mutations include TDP-43 (5%), and FUS (5%).
Dr. Pascuzzi cautioned that cognitive impairment among patients with ALS can be as high as 40% and cannot usually be detected on a bedside exam. Up to 15% of patients “get a frank, overt, clinically significant dementia, usually a frontotemporal syndrome.”
Treatment approaches include use of multidisciplinary clinics, ankle-foot orthosis, tools for activities of daily living, bilevel positive airway pressure, chin tuck, and power chairs; drugs include riluzole, dextromethorphan/quinidine for pseudobulbar affect, and drugs for spasticity.
He said it is important to address fear, depression, and anxiety in patients with ALS, and not to overlook the caregiver: “We are slowly but surely melting down our caregivers.”
A recent study in Lancet Neurology helped Dr. Pascuzzi and colleagues decide against diaphragm pacing for patients with respiratory insufficiency due to ALS, due to the potential for harm. “I would be cautious about pursuing this,” he said.
Despite the many failed controlled trials over the past 20 years, much investigational work is being done on ALS therapeutics, from injection of antisense molecules to tirasemtiv to stem cells, immune therapy, neuro-protection, and high fat/calorie diet, “so there is reason to be hopeful.”
Stem cell transplantation, in particular, is something he said his patients always ask him about. Two forms, “both important,” have undergone fairly extensive clinical investigation.
In the first, spinal cord-derived neural stem cells are transplanted into the spinal cord.
“The people that drive this work are convinced this is not stem cells that turn into motor neurons and replace lost or failing motor neurons, but instead these are stem cells that provide an environment that is more favorable for sick motor neurons,” Dr. Pascuzzi said. “They secrete nerve growth factors and transport proteins to improve the environment.”
The second approach, autologous stem cell treatment, is a “culture-based method for inducing mesenchymal stem cells to secrete neurotrophic factors,” he said, which has been “shown to be protective in several animal models of neurodegenerative diseases.”
—Debra Hughes
Suggested Reading
DiPALS Writing Committee on behalf of the DiPALS Study Group Collaborators. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): a multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015;14(9);883-892.
Glass JD, Hertzberg VS, Boulis NM, et al. Transplantation of spinal cord-derived neural stem cells for ALS: analysis of phase 1 and 2 trials. Neurology. 2016;87(4):392-400.
Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol. 2016;73(3):337-344.
U.S. Food and Drug Administration. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA News Release. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm. Accessed November 4, 2016.
Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375(6):511-522.

Is physician-assisted suicide compatible with the Hippocratic Oath?
Do we stand by our Hippocratic Oath when providing physician-assisted suicide?
Physician-assisted suicide (PAS) is a form of euthanasia in which a physician provides the patient with the pertinent information, and, in certain cases, a prescription for the necessary lethal drugs, so that the individual can willingly and successfully terminate his or her own life. The justification for PAS is the compassionate relief of intractable human suffering. Euthanasia and PAS are accepted practices in European countries such as the Netherlands, Belgium, and Luxembourg.

Such patients are mentally incapable of making the difficult decision to end their life and therefore should require a psychiatric evaluation to include counseling prior to making a decision to engage in PAS. For patients with mental illness, PAS is even more problematic than for terminally ill patients, because patients may lack the capacity to make rational and responsible decisions. Indeed, there are sizable loopholes where our medical system lacks safeguards and similarly lacks the requirements for a thorough, pre-PAS mental health examination, family notifications, and consultations, and for the minimally necessary legal pressure required to ensure patient cooperation.
Critical role of mental health workers
Although PAS has been legalized in those five U.S. states, its support and cases have stalled in recent years, indicating serious ethical concerns, mostly because of multileveled challenges of combating and delineating cultural stereotypes, quantifying mental capacity, gauging quality of life, and deciding where to situate psychiatrists in the PAS decision.
The psychiatrist’s role is being debated. In the United States, opponents take issue with current PAS-legal state’s legislation regarding psychiatric evaluations. For example, Oregon stipulates a psychiatrist referral only in cases where a physician other than a psychiatrist believes the patient’s judgment is impaired. It’s agreed that psychiatrists have the best skill set to assess a patient’s perceptions. Other PAS-legal states require a psychiatrist or psychologist assessment before making the decision. Unfortunately, though, physicians have rarely referred these patients to psychiatrists before offering PAS as an option.
PAS opponents target standards by which concepts like “quality of life” or “contributing member of society” are judged – specifically, that “unbearable suffering” and its ramifications are ill defined – people whose lives are deemed “not worth living” (including the terminally ill) would be susceptible to “sympathetic death” via PAS that might result from PAS legalization. Opponents also argue that recognizing a suicide “right” contradicts that a significant number of suicide attempters have mental illness and need help. They say that legalizing PAS would enable mentally incapacitated people to commit the irreversible act based on their distorted perceptions without providing them the expected assistance from their profession.1
The use of euthanasia or PAS gradually is trending from physically terminally ill patients toward psychiatrically complex patients. There are cases in which euthanasia or PAS was requested by psychiatric patients who had chronic psychiatric, medical, and psychosocial histories rather than purely physical ailment histories. In one study, Scott Y. H. Kim, MD, PhD, and his associates reviewed cases in the Netherlands in which either euthanasia or PAS for psychiatric disorders was deployed. The granted PAS requests appeared to involve physician judgment without psychiatric input.
The study reviewed 66 cases: 55% of patients had chronic severe conditions with extensive histories of attempted suicides and psychiatric hospitalizations, demonstrating that the granted euthanasia and PAS requests had involved extensive evaluations. However, 11% of cases had no independent psychiatric input, and 24% involved disagreement among the physicians.2 PAS proponents and opponents support the involvement and expertise of psychiatrists in all of this.
Psychiatrists have long contended that suicide attempts are often a “cry for help,” not an earnest act to end one’s life. Legalizing PAS tells suicidal individuals that society does not care whether they live or die – a truly un-Hippocratic stance. The stereotypes tossed around in the PAS debate, which could mean life or death, need to be unpacked with specific criteria attached, rather than preconceptions.
Autonomy of patients and implications of PAS
In the PAS debate, there are serious life concerns exhibited by terminally ill/psychiatrically ill patients over losing their autonomy and becoming a burden to their families and caregivers. Stereotypes must be deconstructed, yes, but such patients generally have been considered to be rendered, by the sum effect of their illnesses, mentally incapable of making the decision to end their lives. Addressing this order of patient life concerns generally has been the realm of social workers, psychologists, and, in the most desirable cases, psychiatrists. Therefore, even in keeping with established practice, some form of competent psychiatric evaluation should be required before considering PAS as a viable option.
The call for this requirement is backed up by a study, which reported that 47% of patients who had considered or committed suicide previously were diagnosed with psychiatric disorders, and 15% had undiagnosed psychiatric disorders.3 Also, inventories of thwarted suicide attempters reveal that most lack the conviction to end their lives, and this obverse phenomenon is backed by a study that revealed that 75% of 96 suicidal patients were found to be ambivalent in their intent to end their lives. Suicide attempters simply may be trying to communicate with people in their lives in order to test their love and care.4 This indicates that those who attempt suicide are predominantly psychiatrically ill and prone to distorted perceptions, impaired judgment, frustration, escapism, and manipulative guilt.
Suicide is abhorrent to most human beings’ sensibilities, because humanity has an innate will to live.5 It is wrong to offer patients life-ending options when they might rejuvenate or ease into death naturally. With the debilitated or elderly, PAS could violate their human rights. If the attitude that they are a burden to society persists, then a certain segment of society might be tempted to avoid their intergenerational obligations to elders, particularly those elders presenting with concurrent mental illnesses. PAS would additionally fuel that problem. This would represent a profound injustice and gross violation of human dignity, while also serving as a denial of basic human rights, particularly the right to life and care.6
Conclusion
The complex physical, psychological, and social challenges associated with PAS and the difficulty in enforcing its laws necessitate more adept alternatives. Instead of conditionally legalizing suicide, we should ease patient suffering with compassion and calibrated treatment.6,7
Terminally ill patients often suffer from depression, affecting their rationality. Adequate counseling, medical care, and psychological care should be the response to terminally ill patients considering suicide. If society accepts that ending a life is a reasonable response to human suffering that could otherwise be treated or reversed, then those with the most serious psychiatric conditions are destined to become more vulnerable. Therefore, physicians and psychiatrists should instead work harder to help patients recover, rather than participating in their quest for death.
Simplistic legal and regulatory oversight is insufficient, because the questions evoked by PAS are complicated by life, death, and ethics. Further research is needed to look into the mechanisms and morality of how psychiatrists and other physicians will make judgments and recommendations that are vital elements in developing regulatory oversight on PAS. Finally, future studies should look at which practices have been successful and which might be deemed unethical.
References
1. Clin Med (Lond). 2010 Aug;109(4):323-5.
2. JAMA Psychiatry. 2016;73(4):362-8.
3. “The Final Months: A Study of the Lives of 134 Persons Who Committed Suicide,” New York: Oxford University Press, 1981.
4. “Why We Shouldn’t Legalize Assisting Suicide, Part I.” Department of Medical Ethics, National Right to Life Committee.
5. “Working with Suicidal Individuals: A Guide to Providing Understanding, Assessment and Support,” Philadelphia: Jessica Kingsley Publishers, 2010.
6. “Always Care, Never Kill: How Physician-Assisted Suicide Endangers the Weak, Corrupts Medicine, Compromises the Family, and Violates Human Dignity and Equality,” Washington: The Heritage Foundation, 2015.
7. Ann Intern Med. 2012 Jan 3;156(1 Pt 2):73-104.
Dr. Ahmed is a 2nd-year resident in the psychiatry & behavioral sciences department at the Nassau University Medical Center, New York. Over the last 3 years, he has published and presented papers at national and international forums. His interests include public social psychiatry, health care policy, health disparities, mental health stigma, and undiagnosed and overdiagnosed psychiatric illnesses in children. Dr. Ahmed is a member of the American Psychiatric Association, the American Society of Clinical Psychopharmacology, and the American Association for Social Psychiatry.
Do we stand by our Hippocratic Oath when providing physician-assisted suicide?
Physician-assisted suicide (PAS) is a form of euthanasia in which a physician provides the patient with the pertinent information, and, in certain cases, a prescription for the necessary lethal drugs, so that the individual can willingly and successfully terminate his or her own life. The justification for PAS is the compassionate relief of intractable human suffering. Euthanasia and PAS are accepted practices in European countries such as the Netherlands, Belgium, and Luxembourg.
Such patients are mentally incapable of making the difficult decision to end their life and therefore should require a psychiatric evaluation to include counseling prior to making a decision to engage in PAS. For patients with mental illness, PAS is even more problematic than for terminally ill patients, because patients may lack the capacity to make rational and responsible decisions. Indeed, there are sizable loopholes where our medical system lacks safeguards and similarly lacks the requirements for a thorough, pre-PAS mental health examination, family notifications, and consultations, and for the minimally necessary legal pressure required to ensure patient cooperation.
Critical role of mental health workers
Although PAS has been legalized in those five U.S. states, its support and cases have stalled in recent years, indicating serious ethical concerns, mostly because of multileveled challenges of combating and delineating cultural stereotypes, quantifying mental capacity, gauging quality of life, and deciding where to situate psychiatrists in the PAS decision.
The psychiatrist’s role is being debated. In the United States, opponents take issue with current PAS-legal state’s legislation regarding psychiatric evaluations. For example, Oregon stipulates a psychiatrist referral only in cases where a physician other than a psychiatrist believes the patient’s judgment is impaired. It’s agreed that psychiatrists have the best skill set to assess a patient’s perceptions. Other PAS-legal states require a psychiatrist or psychologist assessment before making the decision. Unfortunately, though, physicians have rarely referred these patients to psychiatrists before offering PAS as an option.
PAS opponents target standards by which concepts like “quality of life” or “contributing member of society” are judged – specifically, that “unbearable suffering” and its ramifications are ill defined – people whose lives are deemed “not worth living” (including the terminally ill) would be susceptible to “sympathetic death” via PAS that might result from PAS legalization. Opponents also argue that recognizing a suicide “right” contradicts that a significant number of suicide attempters have mental illness and need help. They say that legalizing PAS would enable mentally incapacitated people to commit the irreversible act based on their distorted perceptions without providing them the expected assistance from their profession.1
The use of euthanasia or PAS gradually is trending from physically terminally ill patients toward psychiatrically complex patients. There are cases in which euthanasia or PAS was requested by psychiatric patients who had chronic psychiatric, medical, and psychosocial histories rather than purely physical ailment histories. In one study, Scott Y. H. Kim, MD, PhD, and his associates reviewed cases in the Netherlands in which either euthanasia or PAS for psychiatric disorders was deployed. The granted PAS requests appeared to involve physician judgment without psychiatric input.
The study reviewed 66 cases: 55% of patients had chronic severe conditions with extensive histories of attempted suicides and psychiatric hospitalizations, demonstrating that the granted euthanasia and PAS requests had involved extensive evaluations. However, 11% of cases had no independent psychiatric input, and 24% involved disagreement among the physicians.2 PAS proponents and opponents support the involvement and expertise of psychiatrists in all of this.
Psychiatrists have long contended that suicide attempts are often a “cry for help,” not an earnest act to end one’s life. Legalizing PAS tells suicidal individuals that society does not care whether they live or die – a truly un-Hippocratic stance. The stereotypes tossed around in the PAS debate, which could mean life or death, need to be unpacked with specific criteria attached, rather than preconceptions.
Autonomy of patients and implications of PAS
In the PAS debate, there are serious life concerns exhibited by terminally ill/psychiatrically ill patients over losing their autonomy and becoming a burden to their families and caregivers. Stereotypes must be deconstructed, yes, but such patients generally have been considered to be rendered, by the sum effect of their illnesses, mentally incapable of making the decision to end their lives. Addressing this order of patient life concerns generally has been the realm of social workers, psychologists, and, in the most desirable cases, psychiatrists. Therefore, even in keeping with established practice, some form of competent psychiatric evaluation should be required before considering PAS as a viable option.
The call for this requirement is backed up by a study, which reported that 47% of patients who had considered or committed suicide previously were diagnosed with psychiatric disorders, and 15% had undiagnosed psychiatric disorders.3 Also, inventories of thwarted suicide attempters reveal that most lack the conviction to end their lives, and this obverse phenomenon is backed by a study that revealed that 75% of 96 suicidal patients were found to be ambivalent in their intent to end their lives. Suicide attempters simply may be trying to communicate with people in their lives in order to test their love and care.4 This indicates that those who attempt suicide are predominantly psychiatrically ill and prone to distorted perceptions, impaired judgment, frustration, escapism, and manipulative guilt.
Suicide is abhorrent to most human beings’ sensibilities, because humanity has an innate will to live.5 It is wrong to offer patients life-ending options when they might rejuvenate or ease into death naturally. With the debilitated or elderly, PAS could violate their human rights. If the attitude that they are a burden to society persists, then a certain segment of society might be tempted to avoid their intergenerational obligations to elders, particularly those elders presenting with concurrent mental illnesses. PAS would additionally fuel that problem. This would represent a profound injustice and gross violation of human dignity, while also serving as a denial of basic human rights, particularly the right to life and care.6
Conclusion
The complex physical, psychological, and social challenges associated with PAS and the difficulty in enforcing its laws necessitate more adept alternatives. Instead of conditionally legalizing suicide, we should ease patient suffering with compassion and calibrated treatment.6,7
Terminally ill patients often suffer from depression, affecting their rationality. Adequate counseling, medical care, and psychological care should be the response to terminally ill patients considering suicide. If society accepts that ending a life is a reasonable response to human suffering that could otherwise be treated or reversed, then those with the most serious psychiatric conditions are destined to become more vulnerable. Therefore, physicians and psychiatrists should instead work harder to help patients recover, rather than participating in their quest for death.
Simplistic legal and regulatory oversight is insufficient, because the questions evoked by PAS are complicated by life, death, and ethics. Further research is needed to look into the mechanisms and morality of how psychiatrists and other physicians will make judgments and recommendations that are vital elements in developing regulatory oversight on PAS. Finally, future studies should look at which practices have been successful and which might be deemed unethical.
References
1. Clin Med (Lond). 2010 Aug;109(4):323-5.
2. JAMA Psychiatry. 2016;73(4):362-8.
3. “The Final Months: A Study of the Lives of 134 Persons Who Committed Suicide,” New York: Oxford University Press, 1981.
4. “Why We Shouldn’t Legalize Assisting Suicide, Part I.” Department of Medical Ethics, National Right to Life Committee.
5. “Working with Suicidal Individuals: A Guide to Providing Understanding, Assessment and Support,” Philadelphia: Jessica Kingsley Publishers, 2010.
6. “Always Care, Never Kill: How Physician-Assisted Suicide Endangers the Weak, Corrupts Medicine, Compromises the Family, and Violates Human Dignity and Equality,” Washington: The Heritage Foundation, 2015.
7. Ann Intern Med. 2012 Jan 3;156(1 Pt 2):73-104.
Dr. Ahmed is a 2nd-year resident in the psychiatry & behavioral sciences department at the Nassau University Medical Center, New York. Over the last 3 years, he has published and presented papers at national and international forums. His interests include public social psychiatry, health care policy, health disparities, mental health stigma, and undiagnosed and overdiagnosed psychiatric illnesses in children. Dr. Ahmed is a member of the American Psychiatric Association, the American Society of Clinical Psychopharmacology, and the American Association for Social Psychiatry.
Do we stand by our Hippocratic Oath when providing physician-assisted suicide?
Physician-assisted suicide (PAS) is a form of euthanasia in which a physician provides the patient with the pertinent information, and, in certain cases, a prescription for the necessary lethal drugs, so that the individual can willingly and successfully terminate his or her own life. The justification for PAS is the compassionate relief of intractable human suffering. Euthanasia and PAS are accepted practices in European countries such as the Netherlands, Belgium, and Luxembourg.
Such patients are mentally incapable of making the difficult decision to end their life and therefore should require a psychiatric evaluation to include counseling prior to making a decision to engage in PAS. For patients with mental illness, PAS is even more problematic than for terminally ill patients, because patients may lack the capacity to make rational and responsible decisions. Indeed, there are sizable loopholes where our medical system lacks safeguards and similarly lacks the requirements for a thorough, pre-PAS mental health examination, family notifications, and consultations, and for the minimally necessary legal pressure required to ensure patient cooperation.
Critical role of mental health workers
Although PAS has been legalized in those five U.S. states, its support and cases have stalled in recent years, indicating serious ethical concerns, mostly because of multileveled challenges of combating and delineating cultural stereotypes, quantifying mental capacity, gauging quality of life, and deciding where to situate psychiatrists in the PAS decision.
The psychiatrist’s role is being debated. In the United States, opponents take issue with current PAS-legal state’s legislation regarding psychiatric evaluations. For example, Oregon stipulates a psychiatrist referral only in cases where a physician other than a psychiatrist believes the patient’s judgment is impaired. It’s agreed that psychiatrists have the best skill set to assess a patient’s perceptions. Other PAS-legal states require a psychiatrist or psychologist assessment before making the decision. Unfortunately, though, physicians have rarely referred these patients to psychiatrists before offering PAS as an option.
PAS opponents target standards by which concepts like “quality of life” or “contributing member of society” are judged – specifically, that “unbearable suffering” and its ramifications are ill defined – people whose lives are deemed “not worth living” (including the terminally ill) would be susceptible to “sympathetic death” via PAS that might result from PAS legalization. Opponents also argue that recognizing a suicide “right” contradicts that a significant number of suicide attempters have mental illness and need help. They say that legalizing PAS would enable mentally incapacitated people to commit the irreversible act based on their distorted perceptions without providing them the expected assistance from their profession.1
The use of euthanasia or PAS gradually is trending from physically terminally ill patients toward psychiatrically complex patients. There are cases in which euthanasia or PAS was requested by psychiatric patients who had chronic psychiatric, medical, and psychosocial histories rather than purely physical ailment histories. In one study, Scott Y. H. Kim, MD, PhD, and his associates reviewed cases in the Netherlands in which either euthanasia or PAS for psychiatric disorders was deployed. The granted PAS requests appeared to involve physician judgment without psychiatric input.
The study reviewed 66 cases: 55% of patients had chronic severe conditions with extensive histories of attempted suicides and psychiatric hospitalizations, demonstrating that the granted euthanasia and PAS requests had involved extensive evaluations. However, 11% of cases had no independent psychiatric input, and 24% involved disagreement among the physicians.2 PAS proponents and opponents support the involvement and expertise of psychiatrists in all of this.
Psychiatrists have long contended that suicide attempts are often a “cry for help,” not an earnest act to end one’s life. Legalizing PAS tells suicidal individuals that society does not care whether they live or die – a truly un-Hippocratic stance. The stereotypes tossed around in the PAS debate, which could mean life or death, need to be unpacked with specific criteria attached, rather than preconceptions.
Autonomy of patients and implications of PAS
In the PAS debate, there are serious life concerns exhibited by terminally ill/psychiatrically ill patients over losing their autonomy and becoming a burden to their families and caregivers. Stereotypes must be deconstructed, yes, but such patients generally have been considered to be rendered, by the sum effect of their illnesses, mentally incapable of making the decision to end their lives. Addressing this order of patient life concerns generally has been the realm of social workers, psychologists, and, in the most desirable cases, psychiatrists. Therefore, even in keeping with established practice, some form of competent psychiatric evaluation should be required before considering PAS as a viable option.
The call for this requirement is backed up by a study, which reported that 47% of patients who had considered or committed suicide previously were diagnosed with psychiatric disorders, and 15% had undiagnosed psychiatric disorders.3 Also, inventories of thwarted suicide attempters reveal that most lack the conviction to end their lives, and this obverse phenomenon is backed by a study that revealed that 75% of 96 suicidal patients were found to be ambivalent in their intent to end their lives. Suicide attempters simply may be trying to communicate with people in their lives in order to test their love and care.4 This indicates that those who attempt suicide are predominantly psychiatrically ill and prone to distorted perceptions, impaired judgment, frustration, escapism, and manipulative guilt.
Suicide is abhorrent to most human beings’ sensibilities, because humanity has an innate will to live.5 It is wrong to offer patients life-ending options when they might rejuvenate or ease into death naturally. With the debilitated or elderly, PAS could violate their human rights. If the attitude that they are a burden to society persists, then a certain segment of society might be tempted to avoid their intergenerational obligations to elders, particularly those elders presenting with concurrent mental illnesses. PAS would additionally fuel that problem. This would represent a profound injustice and gross violation of human dignity, while also serving as a denial of basic human rights, particularly the right to life and care.6
Conclusion
The complex physical, psychological, and social challenges associated with PAS and the difficulty in enforcing its laws necessitate more adept alternatives. Instead of conditionally legalizing suicide, we should ease patient suffering with compassion and calibrated treatment.6,7
Terminally ill patients often suffer from depression, affecting their rationality. Adequate counseling, medical care, and psychological care should be the response to terminally ill patients considering suicide. If society accepts that ending a life is a reasonable response to human suffering that could otherwise be treated or reversed, then those with the most serious psychiatric conditions are destined to become more vulnerable. Therefore, physicians and psychiatrists should instead work harder to help patients recover, rather than participating in their quest for death.
Simplistic legal and regulatory oversight is insufficient, because the questions evoked by PAS are complicated by life, death, and ethics. Further research is needed to look into the mechanisms and morality of how psychiatrists and other physicians will make judgments and recommendations that are vital elements in developing regulatory oversight on PAS. Finally, future studies should look at which practices have been successful and which might be deemed unethical.
References
1. Clin Med (Lond). 2010 Aug;109(4):323-5.
2. JAMA Psychiatry. 2016;73(4):362-8.
3. “The Final Months: A Study of the Lives of 134 Persons Who Committed Suicide,” New York: Oxford University Press, 1981.
4. “Why We Shouldn’t Legalize Assisting Suicide, Part I.” Department of Medical Ethics, National Right to Life Committee.
5. “Working with Suicidal Individuals: A Guide to Providing Understanding, Assessment and Support,” Philadelphia: Jessica Kingsley Publishers, 2010.
6. “Always Care, Never Kill: How Physician-Assisted Suicide Endangers the Weak, Corrupts Medicine, Compromises the Family, and Violates Human Dignity and Equality,” Washington: The Heritage Foundation, 2015.
7. Ann Intern Med. 2012 Jan 3;156(1 Pt 2):73-104.
Dr. Ahmed is a 2nd-year resident in the psychiatry & behavioral sciences department at the Nassau University Medical Center, New York. Over the last 3 years, he has published and presented papers at national and international forums. His interests include public social psychiatry, health care policy, health disparities, mental health stigma, and undiagnosed and overdiagnosed psychiatric illnesses in children. Dr. Ahmed is a member of the American Psychiatric Association, the American Society of Clinical Psychopharmacology, and the American Association for Social Psychiatry.
What Are New and Potential Therapies for Neuromuscular Disorders?
HILTON HEAD, SC—Biologics, stem cells, and gene therapy may hold promise as treatments for neuromuscular disorders. Ongoing clinical trials are poised to inform treatment strategies and may identify new therapies for amyotrophic lateral sclerosis (ALS), myasthenia gravis, muscular dystrophy, and neuropathy, according to a lecture delivered at the 39th Annual Contemporary Clinical Neurology Symposium.
Recent developments in neuromuscular disorders include the FDA’s approval of a new drug for Duchenne muscular dystrophy and the publication of a randomized trial that found that thymectomy improves clinical outcomes in patients with myasthenia gravis.

ALS
In ALS, many drugs have been studied with little success, said Amanda C. Peltier, MD, Associate Professor of Neurology at Vanderbilt University in Nashville. Riluzole, an approved drug for ALS that may result in a three-month increase in life expectancy or time to mechanical ventilation, targets excitotoxicity. Other drugs targeting excitotoxicity (eg, ceftriaxone and talampanel) have not been found to be effective, however. Agents that target oxidative stress, inflammatory response, protein degradation pathways, mitochondrial dysfunction, and apoptotic pathways have not provided benefit, Dr. Peltier said.
Most trials in ALS use time to mechanical ventilation, time to death, or rate of decline on the ALS Functional Rating Scale as end points. These end points are “very gross measures of how people do,” and researchers are seeking better ways to measure progression in ALS, she said.
Two phase II stem cell studies in ALS are ongoing—the BrainStorm trial in Hassadah, Israel, and the Neuralstem Trial at Emory University and the University of Michigan. Safety data for the Neuralstem Trial that were presented at the 68th Annual Meeting of the American Academy of Neurology (AAN) showed that patients experienced no significant worsening after injections of stem cells in the cervical and lumbar cord. Dose escalation therapy currently is being tested.
Several phase III studies in ALS are under way, including a study of tirasemtiv, an activator of the skeletal muscle troponin complex. “The goal of this [therapy] is to increase muscle force in ALS patients. It is not actually acting at the motor neuron itself,” Dr. Peltier said. In addition, investigators are re-studying creatine, an organic acid involved in ATP formation, at doses of 5 g to 10 g per day. Researchers also are studying masitinib, a biologic targeting inflammation.
Muscular Dystrophies
“There is more gene therapy going on in muscular dystrophy than has ever happened in my lifetime,” Dr. Peltier said. Neuromuscular diseases are caused by genetic mutations in structural proteins that connect the muscle cell membrane to the actin–myosin complex. Dystrophin, one of the largest proteins in the body, is mutated in Duchenne muscular dystrophy and Becker muscular dystrophy.
Some patients with Duchenne muscular dystrophy have a frameshift mutation that results in nonfunctional dystrophin. One treatment strategy entails using an antisense oligonucleotide fragment that binds to messenger RNA to create a more functional protein. “The biggest problem is that those mutations are not the vast majority of mutations in Duchenne,” said Dr. Peltier. “A large proportion of our Duchenne patients are not going to be able to benefit from this technology.”
The FDA on September 19 approved eteplirsen (Exondys 51) injection to treat patients with Duchenne muscular dystrophy who have a confirmed mutation of the dystrophin gene that is amenable to exon 51 skipping. About 13% of people with Duchenne muscular dystrophy have this mutation. The FDA approved eteplirsen under an accelerated pathway that provides patients with access to the drug while the drug developer conducts trials to verify clinical benefit. The FDA said that the approval was based on an increase of dystrophin in skeletal muscle that was observed in some treated patients. Clinical benefit of eteplirsen has not been established.
“The approval of eteplirsen is a positive first step in gene therapy,” Dr. Peltier said. “The FDA is requiring further clinical trials to establish whether clinical improvement will occur, and hopefully these [trials] will be positive.” Gene therapy may have applications for treating other dystrophies and genetic disorders, and the FDA is considering gene therapy for spinal muscular atrophy that is based on increasing messenger RNA transcripts. “Hopefully this is a signal that the FDA is open to gene therapy for these devastating neuromuscular disorders,” she said.
Idebenone, an analog of coenzyme Q10, may reduce loss of respiratory function in patients with Duchenne muscular dystrophy, according to a post hoc analysis of the phase III Duchenne Muscular Dystrophy Long-term Idebenone Study (DELOS). Patients who received idebenone had a lower risk of bronchopulmonary adverse events and a reduced need for systemic antibiotics, compared with patients who received placebo.
Ataluren, a drug intended to make red blood cells less sensitive to premature stop codons, resulted in faster walk times in the Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy (ACT DMD) study. “So far, it seems to be promising,” Dr. Peltier said.
Many patients with Becker or Duchenne muscular dystrophy develop cardiomyopathy. Drugs that typically are used for heart failure and ischemic cardiomyopathy have not been studied in this patient population, however. An ongoing study comparing ACE inhibitors and beta blockers aims to determine the best strategy for treating cardiomyopathy in this population, she said.
In addition, investigators are studying whether follistatin, delivered by an adeno-associated virus, inhibits the degradation of muscle cells in muscular dystrophy and sporadic inclusion body myositis. A recombinant version of histidyl-tRNA synthetase, aTyr1940, is being studied in facioscapulohumeral muscular dystrophy and limb girdle dystrophies.
Among patients with myotonic dystrophy, a small study found that methylphenidate significantly reduces daytime sleepiness, Dr. Peltier said.
Biologics
Several biologics are being studied as treatments for various neuromuscular disorders. In a phase III study in patients with refractory generalized myasthenia gravis, eculizumab, which inhibits complements and the membrane attack complex, did not result in a significant improvement in Myasthenia Gravis–Activities of Daily Living Profile score. Eculizumab did, however, significantly improve a secondary end point, Quantitative Myasthenia Gravis total score.
A phase II trial to evaluate rituximab in myasthenia gravis has completed enrollment. Results may be available in approximately a year. Rituximab also is being studied in juvenile and adult dermatomyositis and chronic inflammatory demyelinating polyneuropathy (CIDP), Dr. Peltier said.
Etanercept and infliximab, treatments for rheumatoid arthritis that act on tumor necrosis factor alpha, are being studied in dermatomyositis and polymyositis. Tocilizumab, a treatment for rheumatoid arthritis that inhibits IL-6 receptors, is being studied in those disorders as well.
Alemtuzumab has been shown to stabilize quantitative muscle testing in sporadic inclusion body myositis and may be helpful in refractory CIDP, Dr. Peltier said.
Myasthenia Gravis
Researchers in August published the results of a randomized trial that found that thymectomy improves clinical outcomes over three years in patients with nonthymomatous myasthenia gravis. Patients who underwent thymectomy and received alternate-day prednisone had a lower time-weighted average Quantitative Myasthenia Gravis score, compared with patients who received alternate-day prednisone alone. Thymectomy had been a mainstay in the treatment of myasthenia gravis, although prior nonrandomized studies had not provided conclusive evidence of its benefit. “The MGTX trial was based on the traditional method of thymectomy requiring sternotomy,” she said. “Further trials comparing the effectiveness of full sternotomy versus newer methods utilizing transcervical approaches or partial sternotomy will most likely be coming.”
A trial of subcutaneous IV immunoglobulin (IVIg) in myasthenia gravis is enrolling patients. Additional data to support the use of IVIg in myasthenia gravis would improve patient outcomes due to the ease of delivering IVIg versus plasma exchange, Dr. Peltier said.
Neuropathy
Investigators have pooled data for patients with neuropathy who have received IVIg. The INSIGHTS Quality Improvement Registry includes data from 585 patients. Researchers have found that patients who fulfill European Federation of Neurological Societies or AAN definite or probable criteria for CIDP or multifocal motor neuropathy respond better to IVIg. This finding “makes sense,” Dr. Peltier said. “If we are more confident of the diagnosis, then it is very likely that IVIg will perform better.”
Among patients with type 2 diabetic neuropathy, exercise significantly improves epidermal nerve fiber density. “Exercise has been interesting, in that it may actually reverse some of the effects” of neuropathy in type 2 diabetes, Dr. Peltier said. “It may be that you will see more exercise studies in the future.”
—Jake Remaly
HILTON HEAD, SC—Biologics, stem cells, and gene therapy may hold promise as treatments for neuromuscular disorders. Ongoing clinical trials are poised to inform treatment strategies and may identify new therapies for amyotrophic lateral sclerosis (ALS), myasthenia gravis, muscular dystrophy, and neuropathy, according to a lecture delivered at the 39th Annual Contemporary Clinical Neurology Symposium.
Recent developments in neuromuscular disorders include the FDA’s approval of a new drug for Duchenne muscular dystrophy and the publication of a randomized trial that found that thymectomy improves clinical outcomes in patients with myasthenia gravis.
ALS
In ALS, many drugs have been studied with little success, said Amanda C. Peltier, MD, Associate Professor of Neurology at Vanderbilt University in Nashville. Riluzole, an approved drug for ALS that may result in a three-month increase in life expectancy or time to mechanical ventilation, targets excitotoxicity. Other drugs targeting excitotoxicity (eg, ceftriaxone and talampanel) have not been found to be effective, however. Agents that target oxidative stress, inflammatory response, protein degradation pathways, mitochondrial dysfunction, and apoptotic pathways have not provided benefit, Dr. Peltier said.
Most trials in ALS use time to mechanical ventilation, time to death, or rate of decline on the ALS Functional Rating Scale as end points. These end points are “very gross measures of how people do,” and researchers are seeking better ways to measure progression in ALS, she said.
Two phase II stem cell studies in ALS are ongoing—the BrainStorm trial in Hassadah, Israel, and the Neuralstem Trial at Emory University and the University of Michigan. Safety data for the Neuralstem Trial that were presented at the 68th Annual Meeting of the American Academy of Neurology (AAN) showed that patients experienced no significant worsening after injections of stem cells in the cervical and lumbar cord. Dose escalation therapy currently is being tested.
Several phase III studies in ALS are under way, including a study of tirasemtiv, an activator of the skeletal muscle troponin complex. “The goal of this [therapy] is to increase muscle force in ALS patients. It is not actually acting at the motor neuron itself,” Dr. Peltier said. In addition, investigators are re-studying creatine, an organic acid involved in ATP formation, at doses of 5 g to 10 g per day. Researchers also are studying masitinib, a biologic targeting inflammation.
Muscular Dystrophies
“There is more gene therapy going on in muscular dystrophy than has ever happened in my lifetime,” Dr. Peltier said. Neuromuscular diseases are caused by genetic mutations in structural proteins that connect the muscle cell membrane to the actin–myosin complex. Dystrophin, one of the largest proteins in the body, is mutated in Duchenne muscular dystrophy and Becker muscular dystrophy.
Some patients with Duchenne muscular dystrophy have a frameshift mutation that results in nonfunctional dystrophin. One treatment strategy entails using an antisense oligonucleotide fragment that binds to messenger RNA to create a more functional protein. “The biggest problem is that those mutations are not the vast majority of mutations in Duchenne,” said Dr. Peltier. “A large proportion of our Duchenne patients are not going to be able to benefit from this technology.”
The FDA on September 19 approved eteplirsen (Exondys 51) injection to treat patients with Duchenne muscular dystrophy who have a confirmed mutation of the dystrophin gene that is amenable to exon 51 skipping. About 13% of people with Duchenne muscular dystrophy have this mutation. The FDA approved eteplirsen under an accelerated pathway that provides patients with access to the drug while the drug developer conducts trials to verify clinical benefit. The FDA said that the approval was based on an increase of dystrophin in skeletal muscle that was observed in some treated patients. Clinical benefit of eteplirsen has not been established.
“The approval of eteplirsen is a positive first step in gene therapy,” Dr. Peltier said. “The FDA is requiring further clinical trials to establish whether clinical improvement will occur, and hopefully these [trials] will be positive.” Gene therapy may have applications for treating other dystrophies and genetic disorders, and the FDA is considering gene therapy for spinal muscular atrophy that is based on increasing messenger RNA transcripts. “Hopefully this is a signal that the FDA is open to gene therapy for these devastating neuromuscular disorders,” she said.
Idebenone, an analog of coenzyme Q10, may reduce loss of respiratory function in patients with Duchenne muscular dystrophy, according to a post hoc analysis of the phase III Duchenne Muscular Dystrophy Long-term Idebenone Study (DELOS). Patients who received idebenone had a lower risk of bronchopulmonary adverse events and a reduced need for systemic antibiotics, compared with patients who received placebo.
Ataluren, a drug intended to make red blood cells less sensitive to premature stop codons, resulted in faster walk times in the Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy (ACT DMD) study. “So far, it seems to be promising,” Dr. Peltier said.
Many patients with Becker or Duchenne muscular dystrophy develop cardiomyopathy. Drugs that typically are used for heart failure and ischemic cardiomyopathy have not been studied in this patient population, however. An ongoing study comparing ACE inhibitors and beta blockers aims to determine the best strategy for treating cardiomyopathy in this population, she said.
In addition, investigators are studying whether follistatin, delivered by an adeno-associated virus, inhibits the degradation of muscle cells in muscular dystrophy and sporadic inclusion body myositis. A recombinant version of histidyl-tRNA synthetase, aTyr1940, is being studied in facioscapulohumeral muscular dystrophy and limb girdle dystrophies.
Among patients with myotonic dystrophy, a small study found that methylphenidate significantly reduces daytime sleepiness, Dr. Peltier said.
Biologics
Several biologics are being studied as treatments for various neuromuscular disorders. In a phase III study in patients with refractory generalized myasthenia gravis, eculizumab, which inhibits complements and the membrane attack complex, did not result in a significant improvement in Myasthenia Gravis–Activities of Daily Living Profile score. Eculizumab did, however, significantly improve a secondary end point, Quantitative Myasthenia Gravis total score.
A phase II trial to evaluate rituximab in myasthenia gravis has completed enrollment. Results may be available in approximately a year. Rituximab also is being studied in juvenile and adult dermatomyositis and chronic inflammatory demyelinating polyneuropathy (CIDP), Dr. Peltier said.
Etanercept and infliximab, treatments for rheumatoid arthritis that act on tumor necrosis factor alpha, are being studied in dermatomyositis and polymyositis. Tocilizumab, a treatment for rheumatoid arthritis that inhibits IL-6 receptors, is being studied in those disorders as well.
Alemtuzumab has been shown to stabilize quantitative muscle testing in sporadic inclusion body myositis and may be helpful in refractory CIDP, Dr. Peltier said.
Myasthenia Gravis
Researchers in August published the results of a randomized trial that found that thymectomy improves clinical outcomes over three years in patients with nonthymomatous myasthenia gravis. Patients who underwent thymectomy and received alternate-day prednisone had a lower time-weighted average Quantitative Myasthenia Gravis score, compared with patients who received alternate-day prednisone alone. Thymectomy had been a mainstay in the treatment of myasthenia gravis, although prior nonrandomized studies had not provided conclusive evidence of its benefit. “The MGTX trial was based on the traditional method of thymectomy requiring sternotomy,” she said. “Further trials comparing the effectiveness of full sternotomy versus newer methods utilizing transcervical approaches or partial sternotomy will most likely be coming.”
A trial of subcutaneous IV immunoglobulin (IVIg) in myasthenia gravis is enrolling patients. Additional data to support the use of IVIg in myasthenia gravis would improve patient outcomes due to the ease of delivering IVIg versus plasma exchange, Dr. Peltier said.
Neuropathy
Investigators have pooled data for patients with neuropathy who have received IVIg. The INSIGHTS Quality Improvement Registry includes data from 585 patients. Researchers have found that patients who fulfill European Federation of Neurological Societies or AAN definite or probable criteria for CIDP or multifocal motor neuropathy respond better to IVIg. This finding “makes sense,” Dr. Peltier said. “If we are more confident of the diagnosis, then it is very likely that IVIg will perform better.”
Among patients with type 2 diabetic neuropathy, exercise significantly improves epidermal nerve fiber density. “Exercise has been interesting, in that it may actually reverse some of the effects” of neuropathy in type 2 diabetes, Dr. Peltier said. “It may be that you will see more exercise studies in the future.”
—Jake Remaly
HILTON HEAD, SC—Biologics, stem cells, and gene therapy may hold promise as treatments for neuromuscular disorders. Ongoing clinical trials are poised to inform treatment strategies and may identify new therapies for amyotrophic lateral sclerosis (ALS), myasthenia gravis, muscular dystrophy, and neuropathy, according to a lecture delivered at the 39th Annual Contemporary Clinical Neurology Symposium.
Recent developments in neuromuscular disorders include the FDA’s approval of a new drug for Duchenne muscular dystrophy and the publication of a randomized trial that found that thymectomy improves clinical outcomes in patients with myasthenia gravis.
ALS
In ALS, many drugs have been studied with little success, said Amanda C. Peltier, MD, Associate Professor of Neurology at Vanderbilt University in Nashville. Riluzole, an approved drug for ALS that may result in a three-month increase in life expectancy or time to mechanical ventilation, targets excitotoxicity. Other drugs targeting excitotoxicity (eg, ceftriaxone and talampanel) have not been found to be effective, however. Agents that target oxidative stress, inflammatory response, protein degradation pathways, mitochondrial dysfunction, and apoptotic pathways have not provided benefit, Dr. Peltier said.
Most trials in ALS use time to mechanical ventilation, time to death, or rate of decline on the ALS Functional Rating Scale as end points. These end points are “very gross measures of how people do,” and researchers are seeking better ways to measure progression in ALS, she said.
Two phase II stem cell studies in ALS are ongoing—the BrainStorm trial in Hassadah, Israel, and the Neuralstem Trial at Emory University and the University of Michigan. Safety data for the Neuralstem Trial that were presented at the 68th Annual Meeting of the American Academy of Neurology (AAN) showed that patients experienced no significant worsening after injections of stem cells in the cervical and lumbar cord. Dose escalation therapy currently is being tested.
Several phase III studies in ALS are under way, including a study of tirasemtiv, an activator of the skeletal muscle troponin complex. “The goal of this [therapy] is to increase muscle force in ALS patients. It is not actually acting at the motor neuron itself,” Dr. Peltier said. In addition, investigators are re-studying creatine, an organic acid involved in ATP formation, at doses of 5 g to 10 g per day. Researchers also are studying masitinib, a biologic targeting inflammation.
Muscular Dystrophies
“There is more gene therapy going on in muscular dystrophy than has ever happened in my lifetime,” Dr. Peltier said. Neuromuscular diseases are caused by genetic mutations in structural proteins that connect the muscle cell membrane to the actin–myosin complex. Dystrophin, one of the largest proteins in the body, is mutated in Duchenne muscular dystrophy and Becker muscular dystrophy.
Some patients with Duchenne muscular dystrophy have a frameshift mutation that results in nonfunctional dystrophin. One treatment strategy entails using an antisense oligonucleotide fragment that binds to messenger RNA to create a more functional protein. “The biggest problem is that those mutations are not the vast majority of mutations in Duchenne,” said Dr. Peltier. “A large proportion of our Duchenne patients are not going to be able to benefit from this technology.”
The FDA on September 19 approved eteplirsen (Exondys 51) injection to treat patients with Duchenne muscular dystrophy who have a confirmed mutation of the dystrophin gene that is amenable to exon 51 skipping. About 13% of people with Duchenne muscular dystrophy have this mutation. The FDA approved eteplirsen under an accelerated pathway that provides patients with access to the drug while the drug developer conducts trials to verify clinical benefit. The FDA said that the approval was based on an increase of dystrophin in skeletal muscle that was observed in some treated patients. Clinical benefit of eteplirsen has not been established.
“The approval of eteplirsen is a positive first step in gene therapy,” Dr. Peltier said. “The FDA is requiring further clinical trials to establish whether clinical improvement will occur, and hopefully these [trials] will be positive.” Gene therapy may have applications for treating other dystrophies and genetic disorders, and the FDA is considering gene therapy for spinal muscular atrophy that is based on increasing messenger RNA transcripts. “Hopefully this is a signal that the FDA is open to gene therapy for these devastating neuromuscular disorders,” she said.
Idebenone, an analog of coenzyme Q10, may reduce loss of respiratory function in patients with Duchenne muscular dystrophy, according to a post hoc analysis of the phase III Duchenne Muscular Dystrophy Long-term Idebenone Study (DELOS). Patients who received idebenone had a lower risk of bronchopulmonary adverse events and a reduced need for systemic antibiotics, compared with patients who received placebo.
Ataluren, a drug intended to make red blood cells less sensitive to premature stop codons, resulted in faster walk times in the Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy (ACT DMD) study. “So far, it seems to be promising,” Dr. Peltier said.
Many patients with Becker or Duchenne muscular dystrophy develop cardiomyopathy. Drugs that typically are used for heart failure and ischemic cardiomyopathy have not been studied in this patient population, however. An ongoing study comparing ACE inhibitors and beta blockers aims to determine the best strategy for treating cardiomyopathy in this population, she said.
In addition, investigators are studying whether follistatin, delivered by an adeno-associated virus, inhibits the degradation of muscle cells in muscular dystrophy and sporadic inclusion body myositis. A recombinant version of histidyl-tRNA synthetase, aTyr1940, is being studied in facioscapulohumeral muscular dystrophy and limb girdle dystrophies.
Among patients with myotonic dystrophy, a small study found that methylphenidate significantly reduces daytime sleepiness, Dr. Peltier said.
Biologics
Several biologics are being studied as treatments for various neuromuscular disorders. In a phase III study in patients with refractory generalized myasthenia gravis, eculizumab, which inhibits complements and the membrane attack complex, did not result in a significant improvement in Myasthenia Gravis–Activities of Daily Living Profile score. Eculizumab did, however, significantly improve a secondary end point, Quantitative Myasthenia Gravis total score.
A phase II trial to evaluate rituximab in myasthenia gravis has completed enrollment. Results may be available in approximately a year. Rituximab also is being studied in juvenile and adult dermatomyositis and chronic inflammatory demyelinating polyneuropathy (CIDP), Dr. Peltier said.
Etanercept and infliximab, treatments for rheumatoid arthritis that act on tumor necrosis factor alpha, are being studied in dermatomyositis and polymyositis. Tocilizumab, a treatment for rheumatoid arthritis that inhibits IL-6 receptors, is being studied in those disorders as well.
Alemtuzumab has been shown to stabilize quantitative muscle testing in sporadic inclusion body myositis and may be helpful in refractory CIDP, Dr. Peltier said.
Myasthenia Gravis
Researchers in August published the results of a randomized trial that found that thymectomy improves clinical outcomes over three years in patients with nonthymomatous myasthenia gravis. Patients who underwent thymectomy and received alternate-day prednisone had a lower time-weighted average Quantitative Myasthenia Gravis score, compared with patients who received alternate-day prednisone alone. Thymectomy had been a mainstay in the treatment of myasthenia gravis, although prior nonrandomized studies had not provided conclusive evidence of its benefit. “The MGTX trial was based on the traditional method of thymectomy requiring sternotomy,” she said. “Further trials comparing the effectiveness of full sternotomy versus newer methods utilizing transcervical approaches or partial sternotomy will most likely be coming.”
A trial of subcutaneous IV immunoglobulin (IVIg) in myasthenia gravis is enrolling patients. Additional data to support the use of IVIg in myasthenia gravis would improve patient outcomes due to the ease of delivering IVIg versus plasma exchange, Dr. Peltier said.
Neuropathy
Investigators have pooled data for patients with neuropathy who have received IVIg. The INSIGHTS Quality Improvement Registry includes data from 585 patients. Researchers have found that patients who fulfill European Federation of Neurological Societies or AAN definite or probable criteria for CIDP or multifocal motor neuropathy respond better to IVIg. This finding “makes sense,” Dr. Peltier said. “If we are more confident of the diagnosis, then it is very likely that IVIg will perform better.”
Among patients with type 2 diabetic neuropathy, exercise significantly improves epidermal nerve fiber density. “Exercise has been interesting, in that it may actually reverse some of the effects” of neuropathy in type 2 diabetes, Dr. Peltier said. “It may be that you will see more exercise studies in the future.”
—Jake Remaly

Further evidence links Zika, Guillain-Barré syndrome
Evidence of Zika virus found in Colombian patients with Guillain-Barré syndrome supports the theory that Zika virus infection and Guillain-Barré syndrome are related and could occur parainfectiously, according to a study published in the New England Journal of Medicine.
“Our study provides virologic evidence of [Zika virus] infection in patients with Guillain-Barré syndrome,” wrote Beatriz Parra, PhD, of the Hospital Universitario del Valle in Valle del Cauca, Colombia, and her coauthors (N Engl J Med. 2016 Oct 5. doi: 10.1056/NEJMoa1605564).
Results indicated that 66 (97%) of subjects had symptoms consistent with a Zika virus infection prior to the onset of Guillain-Barré syndrome. The median number of days between onset of Zika-like symptoms and the onset of Guillain-Barré syndrome was found to be 7 days (interquartile range, 3-10 days). Seventeen (40%) of the 42 patients who underwent laboratory testing tested positive for Zika virus RNA in their sample, with 16 of those 17 positive tests coming from urine samples. Additionally, 18 of the 42 laboratory-tested subjects had “clinical and immunologic findings [that] supported” a Zika virus infection.
“The onset of the Guillain-Barré syndrome can parallel the onset of systemic manifestations of [Zika virus] infection, indicating a so-called parainfectious onset, which suggests that factors different from the known postinfectious mechanisms may be present in [Zika virus]–related Guillain-Barré syndrome,” the authors explained, adding that 20 (48%) of the 42 laboratory-tested subjects had a parainfectious onset.
“RT-PCR testing of urine is a valuable diagnostic tool for the identification of [Zika virus] infection in patients with Guillain-Barré syndrome,” Dr. Parra and her coauthors concluded.
The study was funded by the Bart McLean Fund for Neuroimmunology Research, Johns Hopkins Project Restore, and the Universidad del Valle. Dr. Parra reported no relevant financial disclosures.
Dr. Parra and her colleagues report in the Journal the results of a prospective study of 68 Colombian patients who had a syndrome consistent with the Guillain-Barré syndrome, 66 of whom had previously had symptoms of Zika virus (ZIKV) infection. Major strengths of this study include the documentation of a temporal relationship between the Guillain-Barré syndrome and ZIKV infection (marked by a substantial increase in the incidence of the Guillain-Barré syndrome after the introduction of ZIKV, from 20 to 90 cases per month throughout Colombia), the criteria applied for the diagnosis of the Guillain-Barré syndrome, and the molecular and serologic flavivirus data from analyses of serum, cerebrospinal fluid, and urine.
The difficulties in diagnosing ZIKV infection are borne out in this study, as only 17 patients had definitive laboratory evidence of recent ZIKV infection. Of these 17 patients, only 14 had electrophysiologic data consistent with the Guillain-Barré syndrome and therefore could have met Brighton level 1 diagnostic criteria for the syndrome. Among the 25 ZIKV PCR–negative patients, dengue virus (DENV) IgG antibodies were present in the cerebrospinal fluid of 12 patients and in the serum of 10 patients, and serum DENV IgM test results were positive in 1. These data raise the possibility of primary DENV infection and false-positive ZIKV serologic test results from cross reactivity.
Overall, the study by Dr. Parra and her colleagues supports the association between ZIKV and the Guillain-Barré syndrome, although confirmation in another cohort would strengthen this assertion. Although high rates of seropositivity may prove protective against further waves of ZIKV-related Guillain-Barré syndrome in Central and South America, the ZIKV pandemic is just beginning in North America and Africa, and an increase in the incidence of the Guillain-Barré syndrome may follow.
Jennifer A. Frontera, MD, is with the Cerebrovascular Center at the Cleveland Clinic in Cleveland. Ivan R.F. da Silva. MD, is with the Federal Fluminense University in Niterói, Brazil. These comments were adapted from their editorial accompanying the study ( N Engl J Med. 2016 Oct 5. doi: 10.1056/NEJMe1611840 ).
Dr. Parra and her colleagues report in the Journal the results of a prospective study of 68 Colombian patients who had a syndrome consistent with the Guillain-Barré syndrome, 66 of whom had previously had symptoms of Zika virus (ZIKV) infection. Major strengths of this study include the documentation of a temporal relationship between the Guillain-Barré syndrome and ZIKV infection (marked by a substantial increase in the incidence of the Guillain-Barré syndrome after the introduction of ZIKV, from 20 to 90 cases per month throughout Colombia), the criteria applied for the diagnosis of the Guillain-Barré syndrome, and the molecular and serologic flavivirus data from analyses of serum, cerebrospinal fluid, and urine.
The difficulties in diagnosing ZIKV infection are borne out in this study, as only 17 patients had definitive laboratory evidence of recent ZIKV infection. Of these 17 patients, only 14 had electrophysiologic data consistent with the Guillain-Barré syndrome and therefore could have met Brighton level 1 diagnostic criteria for the syndrome. Among the 25 ZIKV PCR–negative patients, dengue virus (DENV) IgG antibodies were present in the cerebrospinal fluid of 12 patients and in the serum of 10 patients, and serum DENV IgM test results were positive in 1. These data raise the possibility of primary DENV infection and false-positive ZIKV serologic test results from cross reactivity.
Overall, the study by Dr. Parra and her colleagues supports the association between ZIKV and the Guillain-Barré syndrome, although confirmation in another cohort would strengthen this assertion. Although high rates of seropositivity may prove protective against further waves of ZIKV-related Guillain-Barré syndrome in Central and South America, the ZIKV pandemic is just beginning in North America and Africa, and an increase in the incidence of the Guillain-Barré syndrome may follow.
Jennifer A. Frontera, MD, is with the Cerebrovascular Center at the Cleveland Clinic in Cleveland. Ivan R.F. da Silva. MD, is with the Federal Fluminense University in Niterói, Brazil. These comments were adapted from their editorial accompanying the study ( N Engl J Med. 2016 Oct 5. doi: 10.1056/NEJMe1611840 ).
Dr. Parra and her colleagues report in the Journal the results of a prospective study of 68 Colombian patients who had a syndrome consistent with the Guillain-Barré syndrome, 66 of whom had previously had symptoms of Zika virus (ZIKV) infection. Major strengths of this study include the documentation of a temporal relationship between the Guillain-Barré syndrome and ZIKV infection (marked by a substantial increase in the incidence of the Guillain-Barré syndrome after the introduction of ZIKV, from 20 to 90 cases per month throughout Colombia), the criteria applied for the diagnosis of the Guillain-Barré syndrome, and the molecular and serologic flavivirus data from analyses of serum, cerebrospinal fluid, and urine.
The difficulties in diagnosing ZIKV infection are borne out in this study, as only 17 patients had definitive laboratory evidence of recent ZIKV infection. Of these 17 patients, only 14 had electrophysiologic data consistent with the Guillain-Barré syndrome and therefore could have met Brighton level 1 diagnostic criteria for the syndrome. Among the 25 ZIKV PCR–negative patients, dengue virus (DENV) IgG antibodies were present in the cerebrospinal fluid of 12 patients and in the serum of 10 patients, and serum DENV IgM test results were positive in 1. These data raise the possibility of primary DENV infection and false-positive ZIKV serologic test results from cross reactivity.
Overall, the study by Dr. Parra and her colleagues supports the association between ZIKV and the Guillain-Barré syndrome, although confirmation in another cohort would strengthen this assertion. Although high rates of seropositivity may prove protective against further waves of ZIKV-related Guillain-Barré syndrome in Central and South America, the ZIKV pandemic is just beginning in North America and Africa, and an increase in the incidence of the Guillain-Barré syndrome may follow.
Jennifer A. Frontera, MD, is with the Cerebrovascular Center at the Cleveland Clinic in Cleveland. Ivan R.F. da Silva. MD, is with the Federal Fluminense University in Niterói, Brazil. These comments were adapted from their editorial accompanying the study ( N Engl J Med. 2016 Oct 5. doi: 10.1056/NEJMe1611840 ).
Evidence of Zika virus found in Colombian patients with Guillain-Barré syndrome supports the theory that Zika virus infection and Guillain-Barré syndrome are related and could occur parainfectiously, according to a study published in the New England Journal of Medicine.
“Our study provides virologic evidence of [Zika virus] infection in patients with Guillain-Barré syndrome,” wrote Beatriz Parra, PhD, of the Hospital Universitario del Valle in Valle del Cauca, Colombia, and her coauthors (N Engl J Med. 2016 Oct 5. doi: 10.1056/NEJMoa1605564).
Results indicated that 66 (97%) of subjects had symptoms consistent with a Zika virus infection prior to the onset of Guillain-Barré syndrome. The median number of days between onset of Zika-like symptoms and the onset of Guillain-Barré syndrome was found to be 7 days (interquartile range, 3-10 days). Seventeen (40%) of the 42 patients who underwent laboratory testing tested positive for Zika virus RNA in their sample, with 16 of those 17 positive tests coming from urine samples. Additionally, 18 of the 42 laboratory-tested subjects had “clinical and immunologic findings [that] supported” a Zika virus infection.
“The onset of the Guillain-Barré syndrome can parallel the onset of systemic manifestations of [Zika virus] infection, indicating a so-called parainfectious onset, which suggests that factors different from the known postinfectious mechanisms may be present in [Zika virus]–related Guillain-Barré syndrome,” the authors explained, adding that 20 (48%) of the 42 laboratory-tested subjects had a parainfectious onset.
“RT-PCR testing of urine is a valuable diagnostic tool for the identification of [Zika virus] infection in patients with Guillain-Barré syndrome,” Dr. Parra and her coauthors concluded.
The study was funded by the Bart McLean Fund for Neuroimmunology Research, Johns Hopkins Project Restore, and the Universidad del Valle. Dr. Parra reported no relevant financial disclosures.
Evidence of Zika virus found in Colombian patients with Guillain-Barré syndrome supports the theory that Zika virus infection and Guillain-Barré syndrome are related and could occur parainfectiously, according to a study published in the New England Journal of Medicine.
“Our study provides virologic evidence of [Zika virus] infection in patients with Guillain-Barré syndrome,” wrote Beatriz Parra, PhD, of the Hospital Universitario del Valle in Valle del Cauca, Colombia, and her coauthors (N Engl J Med. 2016 Oct 5. doi: 10.1056/NEJMoa1605564).
Results indicated that 66 (97%) of subjects had symptoms consistent with a Zika virus infection prior to the onset of Guillain-Barré syndrome. The median number of days between onset of Zika-like symptoms and the onset of Guillain-Barré syndrome was found to be 7 days (interquartile range, 3-10 days). Seventeen (40%) of the 42 patients who underwent laboratory testing tested positive for Zika virus RNA in their sample, with 16 of those 17 positive tests coming from urine samples. Additionally, 18 of the 42 laboratory-tested subjects had “clinical and immunologic findings [that] supported” a Zika virus infection.
“The onset of the Guillain-Barré syndrome can parallel the onset of systemic manifestations of [Zika virus] infection, indicating a so-called parainfectious onset, which suggests that factors different from the known postinfectious mechanisms may be present in [Zika virus]–related Guillain-Barré syndrome,” the authors explained, adding that 20 (48%) of the 42 laboratory-tested subjects had a parainfectious onset.
“RT-PCR testing of urine is a valuable diagnostic tool for the identification of [Zika virus] infection in patients with Guillain-Barré syndrome,” Dr. Parra and her coauthors concluded.
The study was funded by the Bart McLean Fund for Neuroimmunology Research, Johns Hopkins Project Restore, and the Universidad del Valle. Dr. Parra reported no relevant financial disclosures.
Key clinical point:
Major finding: 97% of subjects had Zika-like symptoms prior to onset of GBS, and 40% of GBS patients who underwent RT-PCR testing were positive for Zika virus.
Data source: Study of 68 GBS patients from six hospitals in Colombia, with 42 patients undergoing RT-PCR analysis.
Disclosures: Funding provided by the Bart McLean Fund for Neuroimmunology Research, Johns Hopkins Project Restore, and the Universidad del Valle. Dr. Parra reported no relevant financial disclosures.
Incontinence trial finds small advantage for Botox over sacral neuromodulation
OnabotulinumtoxinA decreased daily episodes of urinary incontinence by a small amount, compared with sacral neuromodulation, but did not appear to impact several quality of life measures and raised the rates of urinary tract infection and self-catheterization, according to findings from a comparative effectiveness study.
In an open-label randomized trial directly comparing the two approaches for refractory urgency incontinence, onabotulinumtoxinA showed a statistically significant advantage over sacral neuromodulation, but whether this translates into a clinically significant difference is unclear.
“Overall, these findings make it uncertain whether onabotulinumtoxinA provides a clinically important net benefit, compared with sacral neuromodulation,” said Cindy L. Amundsen, MD, of Duke University, Durham N.C., and her associates.
Noting that a recent systematic review of the literature found insufficient evidence to recommend one of these treatments over the other, the investigators performed their study at nine medical centers participating in the National Institutes of Health’s Pelvic Floor Disorder Network. Study participants included 386 women who had a minimum of six urgency incontinence episodes per day and whose symptoms persisted despite treatment with at least one behavioral or physical therapy intervention and at least two medical therapies. They were followed up at 6 months.
In the intention-to-treat analysis, the 190 women who received a single injection of onabotulinumtoxinA showed a mean reduction of 3.9 daily episodes of urinary incontinence, compared with a reduction of 3.3 episodes for the 174 women who underwent sacral neuromodulation. The onabotulinumtoxinA group also showed slightly greater improvement on the Overactive Bladder Short Form score for symptom bother and on the Overactive Bladder Satisfaction of Treatment questionnaire, Dr. Amundsen and her associates reported (JAMA. 2016;316[13]:1366-1374).
However, there were no significant differences between the two study groups in measures of convenience, adverse effects, treatment preference, or other quality of life factors. And onabotulinumtoxinA was associated with a higher rate of urinary tract infection (35% vs. 11%) and of intermittent self-catheterization (8% vs. 0% at 1 month).
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the NIH Office of Research on Women’s Health. Dr. Amundsen reported having no relevant financial disclosures; two of her associates reported ties to Pfizer, Medtronic (maker of the InterStim sacral neuromodulation device), Allergan (maker of Botox), and Axonics.
OnabotulinumtoxinA decreased daily episodes of urinary incontinence by a small amount, compared with sacral neuromodulation, but did not appear to impact several quality of life measures and raised the rates of urinary tract infection and self-catheterization, according to findings from a comparative effectiveness study.
In an open-label randomized trial directly comparing the two approaches for refractory urgency incontinence, onabotulinumtoxinA showed a statistically significant advantage over sacral neuromodulation, but whether this translates into a clinically significant difference is unclear.
“Overall, these findings make it uncertain whether onabotulinumtoxinA provides a clinically important net benefit, compared with sacral neuromodulation,” said Cindy L. Amundsen, MD, of Duke University, Durham N.C., and her associates.
Noting that a recent systematic review of the literature found insufficient evidence to recommend one of these treatments over the other, the investigators performed their study at nine medical centers participating in the National Institutes of Health’s Pelvic Floor Disorder Network. Study participants included 386 women who had a minimum of six urgency incontinence episodes per day and whose symptoms persisted despite treatment with at least one behavioral or physical therapy intervention and at least two medical therapies. They were followed up at 6 months.
In the intention-to-treat analysis, the 190 women who received a single injection of onabotulinumtoxinA showed a mean reduction of 3.9 daily episodes of urinary incontinence, compared with a reduction of 3.3 episodes for the 174 women who underwent sacral neuromodulation. The onabotulinumtoxinA group also showed slightly greater improvement on the Overactive Bladder Short Form score for symptom bother and on the Overactive Bladder Satisfaction of Treatment questionnaire, Dr. Amundsen and her associates reported (JAMA. 2016;316[13]:1366-1374).
However, there were no significant differences between the two study groups in measures of convenience, adverse effects, treatment preference, or other quality of life factors. And onabotulinumtoxinA was associated with a higher rate of urinary tract infection (35% vs. 11%) and of intermittent self-catheterization (8% vs. 0% at 1 month).
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the NIH Office of Research on Women’s Health. Dr. Amundsen reported having no relevant financial disclosures; two of her associates reported ties to Pfizer, Medtronic (maker of the InterStim sacral neuromodulation device), Allergan (maker of Botox), and Axonics.
OnabotulinumtoxinA decreased daily episodes of urinary incontinence by a small amount, compared with sacral neuromodulation, but did not appear to impact several quality of life measures and raised the rates of urinary tract infection and self-catheterization, according to findings from a comparative effectiveness study.
In an open-label randomized trial directly comparing the two approaches for refractory urgency incontinence, onabotulinumtoxinA showed a statistically significant advantage over sacral neuromodulation, but whether this translates into a clinically significant difference is unclear.
“Overall, these findings make it uncertain whether onabotulinumtoxinA provides a clinically important net benefit, compared with sacral neuromodulation,” said Cindy L. Amundsen, MD, of Duke University, Durham N.C., and her associates.
Noting that a recent systematic review of the literature found insufficient evidence to recommend one of these treatments over the other, the investigators performed their study at nine medical centers participating in the National Institutes of Health’s Pelvic Floor Disorder Network. Study participants included 386 women who had a minimum of six urgency incontinence episodes per day and whose symptoms persisted despite treatment with at least one behavioral or physical therapy intervention and at least two medical therapies. They were followed up at 6 months.
In the intention-to-treat analysis, the 190 women who received a single injection of onabotulinumtoxinA showed a mean reduction of 3.9 daily episodes of urinary incontinence, compared with a reduction of 3.3 episodes for the 174 women who underwent sacral neuromodulation. The onabotulinumtoxinA group also showed slightly greater improvement on the Overactive Bladder Short Form score for symptom bother and on the Overactive Bladder Satisfaction of Treatment questionnaire, Dr. Amundsen and her associates reported (JAMA. 2016;316[13]:1366-1374).
However, there were no significant differences between the two study groups in measures of convenience, adverse effects, treatment preference, or other quality of life factors. And onabotulinumtoxinA was associated with a higher rate of urinary tract infection (35% vs. 11%) and of intermittent self-catheterization (8% vs. 0% at 1 month).
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the NIH Office of Research on Women’s Health. Dr. Amundsen reported having no relevant financial disclosures; two of her associates reported ties to Pfizer, Medtronic (maker of the InterStim sacral neuromodulation device), Allergan (maker of Botox), and Axonics.
Key clinical point:
Major finding: The 190 women who received a single injection of onabotulinumtoxinA showed a mean reduction of 3.9 daily episodes of urinary incontinence, compared with a reduction of 3.3 episodes for the 174 women who underwent sacral neuromodulation.
Data source: A multicenter open-label randomized trial involving 386 women followed for 6 months.
Disclosures: This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the NIH Office of Research on Women’s Health. Dr. Amundsen reported having no relevant financial disclosures; two of her associates reported ties to Pfizer, Medtronic (maker of the InterStim sacral neuromodulation device), Allergan (maker of Botox), and Axonics.
FDA approves Duchenne muscular dystrophy treatment under ‘accelerated pathway’
The first treatment for Duchenne muscular dystrophy has been greenlighted by the Food and Drug Administration.
The injectable eteplirsen (Exondys 51) was approved under the accelerated approval pathway, designed to fast-track medicines thought to exceed the benefits of existing treatments for life-threatening diseases, and was also granted priority review and an orphan drug designation. Eteplirsen is specifically indicated for patients who have a confirmed mutation of the dystrophin gene predisposed to exon 51 skipping. This includes about 13% of the population with Duchenne muscular dystrophy, which occurs in about 1 of every 3,600 male infants worldwide.

“In rare diseases, new drug development is especially challenging due to the small numbers of people affected by each disease and the lack of medical understanding of many disorders,” Janet Woodcock, MD, the FDA’s director of the Center for Drug Evaluation and Research (CDER), said in a statement.
The FDA found that data submitted by Sarepta Therapeutics sufficiently demonstrated an increase in dystrophin production, raising the possibility that there may be clinical benefit in this patient cohort; however, because eteplirsen’s actual clinical benefit has not been established, the FDA is requiring Sarepta to conduct a clinical trial. The study will assess whether eteplirsen improves motor function of this patient population. If the trial fails, the FDA is likely to withdraw approval.
“Accelerated approval makes this drug available to patients based on initial data, but we eagerly await learning more about the efficacy of this drug through a confirmatory clinical trial,” Dr. Woodcock said.
The accelerated approval of eteplirsen is based on the surrogate endpoint of dystrophin increase in skeletal muscle observed in some patients given a trial of the drug. The drug’s tentative labeling shows that in a small, randomized trial, three of eight boys who received either 30 mg/kg or 50 mg/kg per week of eteplirsen experienced balance disorder and vomiting. Contact dermatitis also was reported in two of the boys treated with eteplirsen. None of these adverse reactions were reported in four boys who received placebo.
In subsequent studies of 88 boys given either 30 mg/kg or 50 mg/kg per week of eteplirsen for up to 208 weeks, adverse reactions were reported at rates of 10% or higher, including vomiting, contusion, excoriation, arthralgia, rash, catheter site pain, and upper respiratory tract infection.
Priority review status is granted to an investigational drug based on its potential to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan drug designation provides incentives such as clinical trial tax credits, user fee waiver, and eligibility for orphan drug exclusivity to assist and encourage the development of drugs for rare diseases.
In a letter to CDER staff, Dr. Woodcock said that “The approval of Exondys 51 reflects FDA’s ability to apply flexibility to address challenges we often see with rare, life-threatening diseases – while remaining within our statutory framework. In this case, flexibility is warranted because of the life-threatening nature of the disease; the lack of available therapy; the fact that the intended population is a small subset of an already rare disease; and the fact that this is a life-limiting disease of children. These factors, combined with the dystrophin production data – and the drug’s low risk profile – led the Agency to approve the drug under the accelerated approval pathway.”
Dr. Woodcock noted that in April 2016, members of an advisory committee recommended that there was not substantial evidence that the drug is effective in providing clinical benefit and also voted 7-6 against accelerated approval because of uncertainties about the dystrophin data presented by the sponsor. But Sarepta later submitted additional data showing substantial evidence of dystrophin production, although the amount of dystrophin produced was only a small fraction of the normal level, she said.
On Twitter @whitneymcknight
The first treatment for Duchenne muscular dystrophy has been greenlighted by the Food and Drug Administration.
The injectable eteplirsen (Exondys 51) was approved under the accelerated approval pathway, designed to fast-track medicines thought to exceed the benefits of existing treatments for life-threatening diseases, and was also granted priority review and an orphan drug designation. Eteplirsen is specifically indicated for patients who have a confirmed mutation of the dystrophin gene predisposed to exon 51 skipping. This includes about 13% of the population with Duchenne muscular dystrophy, which occurs in about 1 of every 3,600 male infants worldwide.
“In rare diseases, new drug development is especially challenging due to the small numbers of people affected by each disease and the lack of medical understanding of many disorders,” Janet Woodcock, MD, the FDA’s director of the Center for Drug Evaluation and Research (CDER), said in a statement.
The FDA found that data submitted by Sarepta Therapeutics sufficiently demonstrated an increase in dystrophin production, raising the possibility that there may be clinical benefit in this patient cohort; however, because eteplirsen’s actual clinical benefit has not been established, the FDA is requiring Sarepta to conduct a clinical trial. The study will assess whether eteplirsen improves motor function of this patient population. If the trial fails, the FDA is likely to withdraw approval.
“Accelerated approval makes this drug available to patients based on initial data, but we eagerly await learning more about the efficacy of this drug through a confirmatory clinical trial,” Dr. Woodcock said.
The accelerated approval of eteplirsen is based on the surrogate endpoint of dystrophin increase in skeletal muscle observed in some patients given a trial of the drug. The drug’s tentative labeling shows that in a small, randomized trial, three of eight boys who received either 30 mg/kg or 50 mg/kg per week of eteplirsen experienced balance disorder and vomiting. Contact dermatitis also was reported in two of the boys treated with eteplirsen. None of these adverse reactions were reported in four boys who received placebo.
In subsequent studies of 88 boys given either 30 mg/kg or 50 mg/kg per week of eteplirsen for up to 208 weeks, adverse reactions were reported at rates of 10% or higher, including vomiting, contusion, excoriation, arthralgia, rash, catheter site pain, and upper respiratory tract infection.
Priority review status is granted to an investigational drug based on its potential to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan drug designation provides incentives such as clinical trial tax credits, user fee waiver, and eligibility for orphan drug exclusivity to assist and encourage the development of drugs for rare diseases.
In a letter to CDER staff, Dr. Woodcock said that “The approval of Exondys 51 reflects FDA’s ability to apply flexibility to address challenges we often see with rare, life-threatening diseases – while remaining within our statutory framework. In this case, flexibility is warranted because of the life-threatening nature of the disease; the lack of available therapy; the fact that the intended population is a small subset of an already rare disease; and the fact that this is a life-limiting disease of children. These factors, combined with the dystrophin production data – and the drug’s low risk profile – led the Agency to approve the drug under the accelerated approval pathway.”
Dr. Woodcock noted that in April 2016, members of an advisory committee recommended that there was not substantial evidence that the drug is effective in providing clinical benefit and also voted 7-6 against accelerated approval because of uncertainties about the dystrophin data presented by the sponsor. But Sarepta later submitted additional data showing substantial evidence of dystrophin production, although the amount of dystrophin produced was only a small fraction of the normal level, she said.
On Twitter @whitneymcknight
The first treatment for Duchenne muscular dystrophy has been greenlighted by the Food and Drug Administration.
The injectable eteplirsen (Exondys 51) was approved under the accelerated approval pathway, designed to fast-track medicines thought to exceed the benefits of existing treatments for life-threatening diseases, and was also granted priority review and an orphan drug designation. Eteplirsen is specifically indicated for patients who have a confirmed mutation of the dystrophin gene predisposed to exon 51 skipping. This includes about 13% of the population with Duchenne muscular dystrophy, which occurs in about 1 of every 3,600 male infants worldwide.
“In rare diseases, new drug development is especially challenging due to the small numbers of people affected by each disease and the lack of medical understanding of many disorders,” Janet Woodcock, MD, the FDA’s director of the Center for Drug Evaluation and Research (CDER), said in a statement.
The FDA found that data submitted by Sarepta Therapeutics sufficiently demonstrated an increase in dystrophin production, raising the possibility that there may be clinical benefit in this patient cohort; however, because eteplirsen’s actual clinical benefit has not been established, the FDA is requiring Sarepta to conduct a clinical trial. The study will assess whether eteplirsen improves motor function of this patient population. If the trial fails, the FDA is likely to withdraw approval.
“Accelerated approval makes this drug available to patients based on initial data, but we eagerly await learning more about the efficacy of this drug through a confirmatory clinical trial,” Dr. Woodcock said.
The accelerated approval of eteplirsen is based on the surrogate endpoint of dystrophin increase in skeletal muscle observed in some patients given a trial of the drug. The drug’s tentative labeling shows that in a small, randomized trial, three of eight boys who received either 30 mg/kg or 50 mg/kg per week of eteplirsen experienced balance disorder and vomiting. Contact dermatitis also was reported in two of the boys treated with eteplirsen. None of these adverse reactions were reported in four boys who received placebo.
In subsequent studies of 88 boys given either 30 mg/kg or 50 mg/kg per week of eteplirsen for up to 208 weeks, adverse reactions were reported at rates of 10% or higher, including vomiting, contusion, excoriation, arthralgia, rash, catheter site pain, and upper respiratory tract infection.
Priority review status is granted to an investigational drug based on its potential to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan drug designation provides incentives such as clinical trial tax credits, user fee waiver, and eligibility for orphan drug exclusivity to assist and encourage the development of drugs for rare diseases.
In a letter to CDER staff, Dr. Woodcock said that “The approval of Exondys 51 reflects FDA’s ability to apply flexibility to address challenges we often see with rare, life-threatening diseases – while remaining within our statutory framework. In this case, flexibility is warranted because of the life-threatening nature of the disease; the lack of available therapy; the fact that the intended population is a small subset of an already rare disease; and the fact that this is a life-limiting disease of children. These factors, combined with the dystrophin production data – and the drug’s low risk profile – led the Agency to approve the drug under the accelerated approval pathway.”
Dr. Woodcock noted that in April 2016, members of an advisory committee recommended that there was not substantial evidence that the drug is effective in providing clinical benefit and also voted 7-6 against accelerated approval because of uncertainties about the dystrophin data presented by the sponsor. But Sarepta later submitted additional data showing substantial evidence of dystrophin production, although the amount of dystrophin produced was only a small fraction of the normal level, she said.
On Twitter @whitneymcknight
