User login
FDA approves new formulation of deferasirox for iron chelation
The Food and Drug Administration approved a new formulation of deferasirox as a once-daily oral tablet for iron chelation, Novartis announced.
The product, to be marketed as Jadenu, is indicated for chronic iron overload due to blood transfusions in patients 2 years of age and older, and chronic iron overload in non–transfusion-dependent thalassemia in patients 10 years of age or older. Novartis said it is the only once-daily oral iron chelator that can be swallowed whole, with or without food.
Jadenu is a reformulation of Exjade, a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
The new product received its green light under the FDA’s accelerated approval process based on a reduction of liver iron concentrations and serum ferritin levels. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials, Novartis said.
Nausea, vomiting, diarrhea, stomach pain, increases in kidney laboratory values, and skin rash were the most common side effects reported in deferasirox clinical trials. Novartis warned that the drug may cause serious kidney problems, liver problems, and bleeding in the stomach or intestines, and in some cases, death from these complications. The company added that it is not known if Jadenu is safe or effective when taken with other iron chelation therapy, and controlled clinical trials of deferasirox for patients with myelodysplastic syndromes and chronic iron overload due to blood transfusions have not been performed.
Full prescribing information is available at http://tinyurl.com/nspjlek.
The Food and Drug Administration approved a new formulation of deferasirox as a once-daily oral tablet for iron chelation, Novartis announced.
The product, to be marketed as Jadenu, is indicated for chronic iron overload due to blood transfusions in patients 2 years of age and older, and chronic iron overload in non–transfusion-dependent thalassemia in patients 10 years of age or older. Novartis said it is the only once-daily oral iron chelator that can be swallowed whole, with or without food.
Jadenu is a reformulation of Exjade, a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
The new product received its green light under the FDA’s accelerated approval process based on a reduction of liver iron concentrations and serum ferritin levels. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials, Novartis said.
Nausea, vomiting, diarrhea, stomach pain, increases in kidney laboratory values, and skin rash were the most common side effects reported in deferasirox clinical trials. Novartis warned that the drug may cause serious kidney problems, liver problems, and bleeding in the stomach or intestines, and in some cases, death from these complications. The company added that it is not known if Jadenu is safe or effective when taken with other iron chelation therapy, and controlled clinical trials of deferasirox for patients with myelodysplastic syndromes and chronic iron overload due to blood transfusions have not been performed.
Full prescribing information is available at http://tinyurl.com/nspjlek.
The Food and Drug Administration approved a new formulation of deferasirox as a once-daily oral tablet for iron chelation, Novartis announced.
The product, to be marketed as Jadenu, is indicated for chronic iron overload due to blood transfusions in patients 2 years of age and older, and chronic iron overload in non–transfusion-dependent thalassemia in patients 10 years of age or older. Novartis said it is the only once-daily oral iron chelator that can be swallowed whole, with or without food.
Jadenu is a reformulation of Exjade, a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
The new product received its green light under the FDA’s accelerated approval process based on a reduction of liver iron concentrations and serum ferritin levels. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials, Novartis said.
Nausea, vomiting, diarrhea, stomach pain, increases in kidney laboratory values, and skin rash were the most common side effects reported in deferasirox clinical trials. Novartis warned that the drug may cause serious kidney problems, liver problems, and bleeding in the stomach or intestines, and in some cases, death from these complications. The company added that it is not known if Jadenu is safe or effective when taken with other iron chelation therapy, and controlled clinical trials of deferasirox for patients with myelodysplastic syndromes and chronic iron overload due to blood transfusions have not been performed.
Full prescribing information is available at http://tinyurl.com/nspjlek.
Understanding the role of del(7q) in MDS
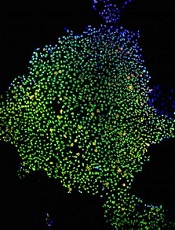
Image by James Thomson
A new study has improved researchers’ understanding of a genetic defect associated with myelodysplastic syndromes (MDS), and the team hopes this will ultimately help us correct the defect in patients.
The researchers used induced pluripotent stem cells (iPSCs) to study del(7q), a chromosomal abnormality found in patients with MDS.
This provided the team with new insight into how the deletion contributes to MDS development.
Steven D. Nimer, MD, of the Sylvester Comprehensive Cancer Center at the University of Miami in Florida, and his colleagues described this work in Nature Biotechnology.
To determine how del(7q) contributes to MDS development, the researchers isolated hematopoietic cells from a patient with del(7q) and reprogrammed them into iPSCs. These iPSCs recapitulated MDS-associated phenotypes, including impaired hematopoietic differentiation.
The researchers also showed that, by engineering heterozygous chromosome 7q loss, they could recapitulate in normal iPSCs the characteristics they observed in the del(7q) iPSCs.
So the team was not surprised to find that disease phenotypes were rescued when del(7q) iPSC lines acquired a duplicate copy of chromosome 7q material.
The researchers also found that hemizygosity of chromosome 7q reduced the expression of many genes in the chromosome7q-deleted region. But gene expression was restored upon chromosome 7q dosage correction.
“[W]e were able to pinpoint a region on chromosome 7 that is critical and were able to identify candidate genes residing there that may cause this disease,” said study author Eirini Papapetrou, MD, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
Focusing on hits residing in this region, 7q32.3–7q36.1, the researchers found 10 genes that were enriched (by >1.5-fold) recurrently.
Further investigation confirmed that 4 of these genes—HIPK2, ATP6V0E2, LUC7L2, and EZH2—could partially rescue the emergence of CD45+ hematopoietic progenitors when overexpressed in del(7q) iPSCs.
The researchers conducted additional experiments focusing on EZH2 alone and found that haploinsufficiency of EZH2 decreases cells’ hematopoietic potential.
But it seems the hematopoietic defects caused by chromosome 7q hemizygosity are mediated through the haploinsufficiency of EZH2 in combination with 1 or more additional genes, which might include LUC7L2, HIPK2, ATP6V0E2, and/or other genes. ![]()
Image by James Thomson
A new study has improved researchers’ understanding of a genetic defect associated with myelodysplastic syndromes (MDS), and the team hopes this will ultimately help us correct the defect in patients.
The researchers used induced pluripotent stem cells (iPSCs) to study del(7q), a chromosomal abnormality found in patients with MDS.
This provided the team with new insight into how the deletion contributes to MDS development.
Steven D. Nimer, MD, of the Sylvester Comprehensive Cancer Center at the University of Miami in Florida, and his colleagues described this work in Nature Biotechnology.
To determine how del(7q) contributes to MDS development, the researchers isolated hematopoietic cells from a patient with del(7q) and reprogrammed them into iPSCs. These iPSCs recapitulated MDS-associated phenotypes, including impaired hematopoietic differentiation.
The researchers also showed that, by engineering heterozygous chromosome 7q loss, they could recapitulate in normal iPSCs the characteristics they observed in the del(7q) iPSCs.
So the team was not surprised to find that disease phenotypes were rescued when del(7q) iPSC lines acquired a duplicate copy of chromosome 7q material.
The researchers also found that hemizygosity of chromosome 7q reduced the expression of many genes in the chromosome7q-deleted region. But gene expression was restored upon chromosome 7q dosage correction.
“[W]e were able to pinpoint a region on chromosome 7 that is critical and were able to identify candidate genes residing there that may cause this disease,” said study author Eirini Papapetrou, MD, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
Focusing on hits residing in this region, 7q32.3–7q36.1, the researchers found 10 genes that were enriched (by >1.5-fold) recurrently.
Further investigation confirmed that 4 of these genes—HIPK2, ATP6V0E2, LUC7L2, and EZH2—could partially rescue the emergence of CD45+ hematopoietic progenitors when overexpressed in del(7q) iPSCs.
The researchers conducted additional experiments focusing on EZH2 alone and found that haploinsufficiency of EZH2 decreases cells’ hematopoietic potential.
But it seems the hematopoietic defects caused by chromosome 7q hemizygosity are mediated through the haploinsufficiency of EZH2 in combination with 1 or more additional genes, which might include LUC7L2, HIPK2, ATP6V0E2, and/or other genes. ![]()
Image by James Thomson
A new study has improved researchers’ understanding of a genetic defect associated with myelodysplastic syndromes (MDS), and the team hopes this will ultimately help us correct the defect in patients.
The researchers used induced pluripotent stem cells (iPSCs) to study del(7q), a chromosomal abnormality found in patients with MDS.
This provided the team with new insight into how the deletion contributes to MDS development.
Steven D. Nimer, MD, of the Sylvester Comprehensive Cancer Center at the University of Miami in Florida, and his colleagues described this work in Nature Biotechnology.
To determine how del(7q) contributes to MDS development, the researchers isolated hematopoietic cells from a patient with del(7q) and reprogrammed them into iPSCs. These iPSCs recapitulated MDS-associated phenotypes, including impaired hematopoietic differentiation.
The researchers also showed that, by engineering heterozygous chromosome 7q loss, they could recapitulate in normal iPSCs the characteristics they observed in the del(7q) iPSCs.
So the team was not surprised to find that disease phenotypes were rescued when del(7q) iPSC lines acquired a duplicate copy of chromosome 7q material.
The researchers also found that hemizygosity of chromosome 7q reduced the expression of many genes in the chromosome7q-deleted region. But gene expression was restored upon chromosome 7q dosage correction.
“[W]e were able to pinpoint a region on chromosome 7 that is critical and were able to identify candidate genes residing there that may cause this disease,” said study author Eirini Papapetrou, MD, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
Focusing on hits residing in this region, 7q32.3–7q36.1, the researchers found 10 genes that were enriched (by >1.5-fold) recurrently.
Further investigation confirmed that 4 of these genes—HIPK2, ATP6V0E2, LUC7L2, and EZH2—could partially rescue the emergence of CD45+ hematopoietic progenitors when overexpressed in del(7q) iPSCs.
The researchers conducted additional experiments focusing on EZH2 alone and found that haploinsufficiency of EZH2 decreases cells’ hematopoietic potential.
But it seems the hematopoietic defects caused by chromosome 7q hemizygosity are mediated through the haploinsufficiency of EZH2 in combination with 1 or more additional genes, which might include LUC7L2, HIPK2, ATP6V0E2, and/or other genes. ![]()
Iron chelation improves survival in lower-risk MDS

Photo courtesy of ASH
SAN FRANCISCO—Iron chelation therapy significantly improves survival for patients with lower-risk myelodysplastic syndrome (MDS) and delays the progression to acute myeloid leukemia (AML), a new study suggests.
“There is a signal for survival with an impressive difference with chelation therapy,” said study investigator Roger Lyons, MD, of Cancer Care Centers of South Texas in San Antonio.
“If this is real, then everyone with lower-risk MDS will go on chelation therapy upfront.”
Dr Lyons presented the results of this research at the 2014 ASH Annual Meeting (abstract 1350).
He and his colleagues initiated a US registry to collect prospective data on clinical outcomes of patients with lower-risk MDS who received chelation or non-chelation therapy.
The registry enrolled 599 adult patients, with a median age of 76 years, from 107 US centers. The patients had transfusional iron overload with serum ferritin ≥ 1000 µg/L and/or ≥ 20 packed red blood cell units and/or ≥ 6 units every 12 weeks.
Patients were divided into 2 groups: those who had never been chelated and those who had used iron chelation. The researchers also looked at a subgroup of patients: those who received chelation therapy for 6 months or more.
The team evaluated patients every 5 months for 5 years or until death, assessing patient characteristics, survival, disease status, comorbidities, cause of death, and MDS therapy.
At enrollment, the 271 chelated patients had a greater median number of lifetime units transfused compared to the 328 non-chelated patients. Additionally, fewer patients receiving chelation therapy had cardiac, vascular, or endocrine concomitant conditions.
Of the chelated patients, 187 (69%) were chelated with deferasirox, 40 (14.8%) with deferasirox and deferoxamine, and 32 (11.8%) with deferoxamine. For 12 patients (4.5%), the researchers did not know which chelator was used.
The cumulative duration of chelation was 18.9 months in patients who had ever used iron chelation and 27 months in patients with at least 6 months of iron chelation.
Patient outcomes
“From the date of diagnosis, the overall survival for patients receiving chelation therapy was significantly longer than for patients receiving non-chelation therapy, including those with cardiovascular or endocrine concomitant conditions,” Dr Lyons noted.
However, there was a potential clinical bias in patient selection, since patients with longer predicted survival may have been chosen for chelation therapy.
At 5 years of follow-up, the mortality rate was 72.9% for non-chelated patients and 59.4% for patients who received chelation therapy (P=0.0005).
Among patients chelated for 6 months or more, the mortality rate was 56.9% (P=0.0002, compared to non-chelated patients). The most common causes of death were MDS/AML and cardiac conditions.
The time from MDS diagnosis to AML progression was significantly longer for chelated patients than for non-chelated patients—72.1 months and 46.4 months, respectively (P<0.0001).
Among patients chelated for 6 months or more, the time to AML transformation was 78.8 months (P<0.0001, compared to non-chelated patients).
Twice as many patients developed AML in the non-chelation group (n=34, 10.4%) than in the chelation group (n=17, 6.3%). However, this difference was not statistically significant.
Taken together, these results suggest chelation can benefit patients with lower-risk MDS, according to Dr Lyons and his colleagues.
“If you think a lower-risk MDS patient will live 1 or 2 years or is a candidate for transplant, get the patient’s iron levels down by chelation, if possible,” Dr Lyons advised.
Three researchers involved in this study are employed by Novartis, and 1 reported research funding from the company, which manufactures deferasirox (Exjade). ![]()
Photo courtesy of ASH
SAN FRANCISCO—Iron chelation therapy significantly improves survival for patients with lower-risk myelodysplastic syndrome (MDS) and delays the progression to acute myeloid leukemia (AML), a new study suggests.
“There is a signal for survival with an impressive difference with chelation therapy,” said study investigator Roger Lyons, MD, of Cancer Care Centers of South Texas in San Antonio.
“If this is real, then everyone with lower-risk MDS will go on chelation therapy upfront.”
Dr Lyons presented the results of this research at the 2014 ASH Annual Meeting (abstract 1350).
He and his colleagues initiated a US registry to collect prospective data on clinical outcomes of patients with lower-risk MDS who received chelation or non-chelation therapy.
The registry enrolled 599 adult patients, with a median age of 76 years, from 107 US centers. The patients had transfusional iron overload with serum ferritin ≥ 1000 µg/L and/or ≥ 20 packed red blood cell units and/or ≥ 6 units every 12 weeks.
Patients were divided into 2 groups: those who had never been chelated and those who had used iron chelation. The researchers also looked at a subgroup of patients: those who received chelation therapy for 6 months or more.
The team evaluated patients every 5 months for 5 years or until death, assessing patient characteristics, survival, disease status, comorbidities, cause of death, and MDS therapy.
At enrollment, the 271 chelated patients had a greater median number of lifetime units transfused compared to the 328 non-chelated patients. Additionally, fewer patients receiving chelation therapy had cardiac, vascular, or endocrine concomitant conditions.
Of the chelated patients, 187 (69%) were chelated with deferasirox, 40 (14.8%) with deferasirox and deferoxamine, and 32 (11.8%) with deferoxamine. For 12 patients (4.5%), the researchers did not know which chelator was used.
The cumulative duration of chelation was 18.9 months in patients who had ever used iron chelation and 27 months in patients with at least 6 months of iron chelation.
Patient outcomes
“From the date of diagnosis, the overall survival for patients receiving chelation therapy was significantly longer than for patients receiving non-chelation therapy, including those with cardiovascular or endocrine concomitant conditions,” Dr Lyons noted.
However, there was a potential clinical bias in patient selection, since patients with longer predicted survival may have been chosen for chelation therapy.
At 5 years of follow-up, the mortality rate was 72.9% for non-chelated patients and 59.4% for patients who received chelation therapy (P=0.0005).
Among patients chelated for 6 months or more, the mortality rate was 56.9% (P=0.0002, compared to non-chelated patients). The most common causes of death were MDS/AML and cardiac conditions.
The time from MDS diagnosis to AML progression was significantly longer for chelated patients than for non-chelated patients—72.1 months and 46.4 months, respectively (P<0.0001).
Among patients chelated for 6 months or more, the time to AML transformation was 78.8 months (P<0.0001, compared to non-chelated patients).
Twice as many patients developed AML in the non-chelation group (n=34, 10.4%) than in the chelation group (n=17, 6.3%). However, this difference was not statistically significant.
Taken together, these results suggest chelation can benefit patients with lower-risk MDS, according to Dr Lyons and his colleagues.
“If you think a lower-risk MDS patient will live 1 or 2 years or is a candidate for transplant, get the patient’s iron levels down by chelation, if possible,” Dr Lyons advised.
Three researchers involved in this study are employed by Novartis, and 1 reported research funding from the company, which manufactures deferasirox (Exjade). ![]()
Photo courtesy of ASH
SAN FRANCISCO—Iron chelation therapy significantly improves survival for patients with lower-risk myelodysplastic syndrome (MDS) and delays the progression to acute myeloid leukemia (AML), a new study suggests.
“There is a signal for survival with an impressive difference with chelation therapy,” said study investigator Roger Lyons, MD, of Cancer Care Centers of South Texas in San Antonio.
“If this is real, then everyone with lower-risk MDS will go on chelation therapy upfront.”
Dr Lyons presented the results of this research at the 2014 ASH Annual Meeting (abstract 1350).
He and his colleagues initiated a US registry to collect prospective data on clinical outcomes of patients with lower-risk MDS who received chelation or non-chelation therapy.
The registry enrolled 599 adult patients, with a median age of 76 years, from 107 US centers. The patients had transfusional iron overload with serum ferritin ≥ 1000 µg/L and/or ≥ 20 packed red blood cell units and/or ≥ 6 units every 12 weeks.
Patients were divided into 2 groups: those who had never been chelated and those who had used iron chelation. The researchers also looked at a subgroup of patients: those who received chelation therapy for 6 months or more.
The team evaluated patients every 5 months for 5 years or until death, assessing patient characteristics, survival, disease status, comorbidities, cause of death, and MDS therapy.
At enrollment, the 271 chelated patients had a greater median number of lifetime units transfused compared to the 328 non-chelated patients. Additionally, fewer patients receiving chelation therapy had cardiac, vascular, or endocrine concomitant conditions.
Of the chelated patients, 187 (69%) were chelated with deferasirox, 40 (14.8%) with deferasirox and deferoxamine, and 32 (11.8%) with deferoxamine. For 12 patients (4.5%), the researchers did not know which chelator was used.
The cumulative duration of chelation was 18.9 months in patients who had ever used iron chelation and 27 months in patients with at least 6 months of iron chelation.
Patient outcomes
“From the date of diagnosis, the overall survival for patients receiving chelation therapy was significantly longer than for patients receiving non-chelation therapy, including those with cardiovascular or endocrine concomitant conditions,” Dr Lyons noted.
However, there was a potential clinical bias in patient selection, since patients with longer predicted survival may have been chosen for chelation therapy.
At 5 years of follow-up, the mortality rate was 72.9% for non-chelated patients and 59.4% for patients who received chelation therapy (P=0.0005).
Among patients chelated for 6 months or more, the mortality rate was 56.9% (P=0.0002, compared to non-chelated patients). The most common causes of death were MDS/AML and cardiac conditions.
The time from MDS diagnosis to AML progression was significantly longer for chelated patients than for non-chelated patients—72.1 months and 46.4 months, respectively (P<0.0001).
Among patients chelated for 6 months or more, the time to AML transformation was 78.8 months (P<0.0001, compared to non-chelated patients).
Twice as many patients developed AML in the non-chelation group (n=34, 10.4%) than in the chelation group (n=17, 6.3%). However, this difference was not statistically significant.
Taken together, these results suggest chelation can benefit patients with lower-risk MDS, according to Dr Lyons and his colleagues.
“If you think a lower-risk MDS patient will live 1 or 2 years or is a candidate for transplant, get the patient’s iron levels down by chelation, if possible,” Dr Lyons advised.
Three researchers involved in this study are employed by Novartis, and 1 reported research funding from the company, which manufactures deferasirox (Exjade). ![]()
Two activin receptor fusion proteins show promise in anemia

site of the ASH Annual Meeting
Photo courtesy of ASH
SAN FRANCISCO—Two activin receptor fusion proteins, luspatercept and sotatercept, increased hemoglobin levels and transfusion independence in patients with β-thalassemia and myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML), respectively, in phase 2 trials.
Luspatercept is a type IIB activin receptor, while sotatercept is type IIA. Both impact late-stage erythropoiesis and improve anemia.
Investigators reported the trial results at the 2014 ASH Annual Meeting.
Luspatercept in β-thalassemia
Antonio G. Piga, MD, of Turin University in Italy, explained that luspatercept binds to GDF11 and other ligands in the TGF-β superfamily and promotes late-stage erythroid maturation.
The study was designed in the US and conducted abroad, he said, because while β-thalassemia is rare in the US, it is not so in Europe.
Investigators evaluated whether luspatercept could increase hemoglobin levels 1.5 g/dL or more for at least 2 weeks in non-transfusion-dependent (NTD) patients.
And in transfusion-dependent (TD) patients, luspatercept was expected to decrease the transfusion burden by 20% or more over 12 weeks.
Thirty patients, 7 TD and 23 NTD, received an injection of luspatercept every 3 weeks for 3 months at doses ranging from 0.2 to 1.0 mg/kg.
The median age was 35, and 53% of patients were male. Eighty-three percent had had a splenectomy.
Luspatercept efficacy
Three-quarters of patients treated with 0.8 to 1.0 mg/kg increased their hemoglobin levels or reduced their transfusion burden.
Of the NTD patients, 8 of 12 with iron overload at baseline experienced a reduction in liver iron concentration of 1 mg or more at 16 weeks.
And in the TD group, “All patients had clinically improved reduction of transfusion dependence,” Dr Piga said.
They had a more than 60% reduction in transfusion burden over 12 weeks. This included 2 patients with β0 β0 genotype, who experienced a 79% and 75% reduction.
“There was a trend to lower liver iron concentration in TD patients,” Dr Piga noted, “except in 1 patient.”
And 5 of 5 TD patients experienced decreases in serum ferritin ranging from 12% to 60%.
Luspatercept safety
Luspatercept did not cause any treatment-related serious or severe adverse events. The most common adverse events were bone pain (20%), headache (17%), myalgia (13%), and asthenia (10%).
There was 1 grade 3 dose-limiting toxicity of worsening lumbar spine bone pain, and 3 patients discontinued early, 1 each with occipital headache, ankle pain, and back pain.
Luspatercept had beneficial effects on other complications of the disease, Dr Piga noted, such as the healing of leg ulcers in the 3 patients with this complication, 1 who is just ending the trial.
With these promising results, Dr Piga said the investigators are “anxious to start phase 3.”
Dr Piga reported the data as abstract 53. The study was supported by Acceleron Pharma and Celgene Corporation.
Sotatercept in MDS and CMML with anemia
Rami Komrokji, MD, of the Moffit Cancer Center in Tampa, Florida, explained that sotaterept increases the release of mature erythrocytes into circulation by a mechanism distinct from erythropoietin.
Sotatercept was shown to stimulate erythropoiesis and increase hemoglobin levels in healthy volunteers, so investigators undertook to study its potential to treat anemia.
They conducted a phase 2 dose-finding study to determine the best effective dose in patients with anemia and lower-risk MDS or nonproliferative CMML who were refractory to erythropoiesis-stimulating agents (ESAs).
Investigators evaluated 53 patients who had anemia of 9 g/dL or less requiring 2 or more units of red blood cells (RBCs) in the 12 weeks prior to enrollment.
Their white blood cell counts had to be under 13,000/μL, and they had to have no response, loss of response, or low chance of response to ESAs, reflected by serum erythropoietin of more than 500 mIU/mL.
Patients were a median age of 71, and 70% were male.
They received subcutaneous sotatercept at dose levels of 0.1, 0.3, 0.5, or 1.0 mg/kg once every 3 weeks for up to 24 months following the first treatment.
Sotatercept efficacy
The investigators evaluated efficacy for the entire cohort as well as in subgroups of patients with high transfusion burden (HTB) and low transfusion burden (LTB). Patients were defined as HTB if they required RBC transfusions of 4 or more units every 8 weeks and LTB as less than 4 units per 8 weeks.
Overall, 45% (24/53) of the evaluable patients achieved hematologic improvement as defined by IWG 2006 criteria.
Forty-two percent of HTB patients had a reduction in their transfusion burden of 4 or more RBC units per 8 weeks, with a median duration of longest response of 106 days (range, 62 to 345+). Eleven percent (5/44) achieved RBC transfusion independence of 56 days or more.
Sixty-three percent (5/8) of LTB patients achieved both RBC transfusion independence of 56 days or more and a mean hemoglobin increase of 1.5 mg/dL or more for at least 8 weeks.
Their maximum mean hemoglobin increase ranged from 1.9 to 4.4 g/dL, and the mean duration of RBC transfusion independence ranged from 76 to 233+ days. Of these 8 patients, 67% were in the 1.0 mg/kg cohort.
Sotatercept safety
“Most of the adverse events were not necessarily related to the treatment,” Dr Komrokji said, “and they were grade 1 or grade 2 toxicity.”
Twenty of 54* patients (37%) experienced 1 or more treatment-related adverse events, the most common of which were fatigue/asthenia (13%), headache (9%), decreased appetite (7%), nausea (7%), and dyspnea (6%).
Three patients discontinued the study due to treatment-emergent adverse events that were possibly related to sotatercept. One was for grade 2 hemolytic anemia, 1 for grade 3 hypertension, and 1 for grade 2 muscle weakness.
Dr Komrokji concluded saying the results showed “promising evidence of clinical activity” in these ESA-refractory, anemic, lower-risk MDS and CMML patients who have a “challenging and unmet need for treatment.”
He indicated that further exploration of sotatercept at higher dose levels and for longer treatment periods is planned and ongoing.
He presented the data as abstract 3251. The study was supported by Celgene Corporation. ![]()
*One patient was excluded from the efficacy analysis due to a protocol violation.
site of the ASH Annual Meeting
Photo courtesy of ASH
SAN FRANCISCO—Two activin receptor fusion proteins, luspatercept and sotatercept, increased hemoglobin levels and transfusion independence in patients with β-thalassemia and myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML), respectively, in phase 2 trials.
Luspatercept is a type IIB activin receptor, while sotatercept is type IIA. Both impact late-stage erythropoiesis and improve anemia.
Investigators reported the trial results at the 2014 ASH Annual Meeting.
Luspatercept in β-thalassemia
Antonio G. Piga, MD, of Turin University in Italy, explained that luspatercept binds to GDF11 and other ligands in the TGF-β superfamily and promotes late-stage erythroid maturation.
The study was designed in the US and conducted abroad, he said, because while β-thalassemia is rare in the US, it is not so in Europe.
Investigators evaluated whether luspatercept could increase hemoglobin levels 1.5 g/dL or more for at least 2 weeks in non-transfusion-dependent (NTD) patients.
And in transfusion-dependent (TD) patients, luspatercept was expected to decrease the transfusion burden by 20% or more over 12 weeks.
Thirty patients, 7 TD and 23 NTD, received an injection of luspatercept every 3 weeks for 3 months at doses ranging from 0.2 to 1.0 mg/kg.
The median age was 35, and 53% of patients were male. Eighty-three percent had had a splenectomy.
Luspatercept efficacy
Three-quarters of patients treated with 0.8 to 1.0 mg/kg increased their hemoglobin levels or reduced their transfusion burden.
Of the NTD patients, 8 of 12 with iron overload at baseline experienced a reduction in liver iron concentration of 1 mg or more at 16 weeks.
And in the TD group, “All patients had clinically improved reduction of transfusion dependence,” Dr Piga said.
They had a more than 60% reduction in transfusion burden over 12 weeks. This included 2 patients with β0 β0 genotype, who experienced a 79% and 75% reduction.
“There was a trend to lower liver iron concentration in TD patients,” Dr Piga noted, “except in 1 patient.”
And 5 of 5 TD patients experienced decreases in serum ferritin ranging from 12% to 60%.
Luspatercept safety
Luspatercept did not cause any treatment-related serious or severe adverse events. The most common adverse events were bone pain (20%), headache (17%), myalgia (13%), and asthenia (10%).
There was 1 grade 3 dose-limiting toxicity of worsening lumbar spine bone pain, and 3 patients discontinued early, 1 each with occipital headache, ankle pain, and back pain.
Luspatercept had beneficial effects on other complications of the disease, Dr Piga noted, such as the healing of leg ulcers in the 3 patients with this complication, 1 who is just ending the trial.
With these promising results, Dr Piga said the investigators are “anxious to start phase 3.”
Dr Piga reported the data as abstract 53. The study was supported by Acceleron Pharma and Celgene Corporation.
Sotatercept in MDS and CMML with anemia
Rami Komrokji, MD, of the Moffit Cancer Center in Tampa, Florida, explained that sotaterept increases the release of mature erythrocytes into circulation by a mechanism distinct from erythropoietin.
Sotatercept was shown to stimulate erythropoiesis and increase hemoglobin levels in healthy volunteers, so investigators undertook to study its potential to treat anemia.
They conducted a phase 2 dose-finding study to determine the best effective dose in patients with anemia and lower-risk MDS or nonproliferative CMML who were refractory to erythropoiesis-stimulating agents (ESAs).
Investigators evaluated 53 patients who had anemia of 9 g/dL or less requiring 2 or more units of red blood cells (RBCs) in the 12 weeks prior to enrollment.
Their white blood cell counts had to be under 13,000/μL, and they had to have no response, loss of response, or low chance of response to ESAs, reflected by serum erythropoietin of more than 500 mIU/mL.
Patients were a median age of 71, and 70% were male.
They received subcutaneous sotatercept at dose levels of 0.1, 0.3, 0.5, or 1.0 mg/kg once every 3 weeks for up to 24 months following the first treatment.
Sotatercept efficacy
The investigators evaluated efficacy for the entire cohort as well as in subgroups of patients with high transfusion burden (HTB) and low transfusion burden (LTB). Patients were defined as HTB if they required RBC transfusions of 4 or more units every 8 weeks and LTB as less than 4 units per 8 weeks.
Overall, 45% (24/53) of the evaluable patients achieved hematologic improvement as defined by IWG 2006 criteria.
Forty-two percent of HTB patients had a reduction in their transfusion burden of 4 or more RBC units per 8 weeks, with a median duration of longest response of 106 days (range, 62 to 345+). Eleven percent (5/44) achieved RBC transfusion independence of 56 days or more.
Sixty-three percent (5/8) of LTB patients achieved both RBC transfusion independence of 56 days or more and a mean hemoglobin increase of 1.5 mg/dL or more for at least 8 weeks.
Their maximum mean hemoglobin increase ranged from 1.9 to 4.4 g/dL, and the mean duration of RBC transfusion independence ranged from 76 to 233+ days. Of these 8 patients, 67% were in the 1.0 mg/kg cohort.
Sotatercept safety
“Most of the adverse events were not necessarily related to the treatment,” Dr Komrokji said, “and they were grade 1 or grade 2 toxicity.”
Twenty of 54* patients (37%) experienced 1 or more treatment-related adverse events, the most common of which were fatigue/asthenia (13%), headache (9%), decreased appetite (7%), nausea (7%), and dyspnea (6%).
Three patients discontinued the study due to treatment-emergent adverse events that were possibly related to sotatercept. One was for grade 2 hemolytic anemia, 1 for grade 3 hypertension, and 1 for grade 2 muscle weakness.
Dr Komrokji concluded saying the results showed “promising evidence of clinical activity” in these ESA-refractory, anemic, lower-risk MDS and CMML patients who have a “challenging and unmet need for treatment.”
He indicated that further exploration of sotatercept at higher dose levels and for longer treatment periods is planned and ongoing.
He presented the data as abstract 3251. The study was supported by Celgene Corporation. ![]()
*One patient was excluded from the efficacy analysis due to a protocol violation.
site of the ASH Annual Meeting
Photo courtesy of ASH
SAN FRANCISCO—Two activin receptor fusion proteins, luspatercept and sotatercept, increased hemoglobin levels and transfusion independence in patients with β-thalassemia and myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML), respectively, in phase 2 trials.
Luspatercept is a type IIB activin receptor, while sotatercept is type IIA. Both impact late-stage erythropoiesis and improve anemia.
Investigators reported the trial results at the 2014 ASH Annual Meeting.
Luspatercept in β-thalassemia
Antonio G. Piga, MD, of Turin University in Italy, explained that luspatercept binds to GDF11 and other ligands in the TGF-β superfamily and promotes late-stage erythroid maturation.
The study was designed in the US and conducted abroad, he said, because while β-thalassemia is rare in the US, it is not so in Europe.
Investigators evaluated whether luspatercept could increase hemoglobin levels 1.5 g/dL or more for at least 2 weeks in non-transfusion-dependent (NTD) patients.
And in transfusion-dependent (TD) patients, luspatercept was expected to decrease the transfusion burden by 20% or more over 12 weeks.
Thirty patients, 7 TD and 23 NTD, received an injection of luspatercept every 3 weeks for 3 months at doses ranging from 0.2 to 1.0 mg/kg.
The median age was 35, and 53% of patients were male. Eighty-three percent had had a splenectomy.
Luspatercept efficacy
Three-quarters of patients treated with 0.8 to 1.0 mg/kg increased their hemoglobin levels or reduced their transfusion burden.
Of the NTD patients, 8 of 12 with iron overload at baseline experienced a reduction in liver iron concentration of 1 mg or more at 16 weeks.
And in the TD group, “All patients had clinically improved reduction of transfusion dependence,” Dr Piga said.
They had a more than 60% reduction in transfusion burden over 12 weeks. This included 2 patients with β0 β0 genotype, who experienced a 79% and 75% reduction.
“There was a trend to lower liver iron concentration in TD patients,” Dr Piga noted, “except in 1 patient.”
And 5 of 5 TD patients experienced decreases in serum ferritin ranging from 12% to 60%.
Luspatercept safety
Luspatercept did not cause any treatment-related serious or severe adverse events. The most common adverse events were bone pain (20%), headache (17%), myalgia (13%), and asthenia (10%).
There was 1 grade 3 dose-limiting toxicity of worsening lumbar spine bone pain, and 3 patients discontinued early, 1 each with occipital headache, ankle pain, and back pain.
Luspatercept had beneficial effects on other complications of the disease, Dr Piga noted, such as the healing of leg ulcers in the 3 patients with this complication, 1 who is just ending the trial.
With these promising results, Dr Piga said the investigators are “anxious to start phase 3.”
Dr Piga reported the data as abstract 53. The study was supported by Acceleron Pharma and Celgene Corporation.
Sotatercept in MDS and CMML with anemia
Rami Komrokji, MD, of the Moffit Cancer Center in Tampa, Florida, explained that sotaterept increases the release of mature erythrocytes into circulation by a mechanism distinct from erythropoietin.
Sotatercept was shown to stimulate erythropoiesis and increase hemoglobin levels in healthy volunteers, so investigators undertook to study its potential to treat anemia.
They conducted a phase 2 dose-finding study to determine the best effective dose in patients with anemia and lower-risk MDS or nonproliferative CMML who were refractory to erythropoiesis-stimulating agents (ESAs).
Investigators evaluated 53 patients who had anemia of 9 g/dL or less requiring 2 or more units of red blood cells (RBCs) in the 12 weeks prior to enrollment.
Their white blood cell counts had to be under 13,000/μL, and they had to have no response, loss of response, or low chance of response to ESAs, reflected by serum erythropoietin of more than 500 mIU/mL.
Patients were a median age of 71, and 70% were male.
They received subcutaneous sotatercept at dose levels of 0.1, 0.3, 0.5, or 1.0 mg/kg once every 3 weeks for up to 24 months following the first treatment.
Sotatercept efficacy
The investigators evaluated efficacy for the entire cohort as well as in subgroups of patients with high transfusion burden (HTB) and low transfusion burden (LTB). Patients were defined as HTB if they required RBC transfusions of 4 or more units every 8 weeks and LTB as less than 4 units per 8 weeks.
Overall, 45% (24/53) of the evaluable patients achieved hematologic improvement as defined by IWG 2006 criteria.
Forty-two percent of HTB patients had a reduction in their transfusion burden of 4 or more RBC units per 8 weeks, with a median duration of longest response of 106 days (range, 62 to 345+). Eleven percent (5/44) achieved RBC transfusion independence of 56 days or more.
Sixty-three percent (5/8) of LTB patients achieved both RBC transfusion independence of 56 days or more and a mean hemoglobin increase of 1.5 mg/dL or more for at least 8 weeks.
Their maximum mean hemoglobin increase ranged from 1.9 to 4.4 g/dL, and the mean duration of RBC transfusion independence ranged from 76 to 233+ days. Of these 8 patients, 67% were in the 1.0 mg/kg cohort.
Sotatercept safety
“Most of the adverse events were not necessarily related to the treatment,” Dr Komrokji said, “and they were grade 1 or grade 2 toxicity.”
Twenty of 54* patients (37%) experienced 1 or more treatment-related adverse events, the most common of which were fatigue/asthenia (13%), headache (9%), decreased appetite (7%), nausea (7%), and dyspnea (6%).
Three patients discontinued the study due to treatment-emergent adverse events that were possibly related to sotatercept. One was for grade 2 hemolytic anemia, 1 for grade 3 hypertension, and 1 for grade 2 muscle weakness.
Dr Komrokji concluded saying the results showed “promising evidence of clinical activity” in these ESA-refractory, anemic, lower-risk MDS and CMML patients who have a “challenging and unmet need for treatment.”
He indicated that further exploration of sotatercept at higher dose levels and for longer treatment periods is planned and ongoing.
He presented the data as abstract 3251. The study was supported by Celgene Corporation. ![]()
*One patient was excluded from the efficacy analysis due to a protocol violation.
Investigational sotatercept improves heme parameters in MDS
SAN FRANCISCO – A first-in-class investigational agent called sotatercept appears to be safe and to improve hematologic parameters in patients with lower-risk myelodysplastic syndrome or nonproliferative chronic myelomonocytic leukemia and anemia requiring transfusion, a study showed.
In the open-label phase II dose-finding study of sotatercept in patients with myelodysplastic syndrome (MDS) or nonproliferative chronic myelomonocytic leukemia (CMML), hematologic improvement according to International Working Group (IWG) 2006 criteria was seen in 24 of 53 evaluable patients, said Dr. Rami Komrokji of the Moffitt Cancer Center,Tampa.
The patients were all refractory to, or were deemed to have a low chance of responding to, an erythropoiesis-stimulating agent (ESA), Dr. Komrokji said at the annual meeting of the American Society of Hematology.

“A medication like sotatercept would probably have a role in the management of anemia in lower-risk MDS patients. The treatment is administered every 3 weeks, which makes it also logistically easier for the patients to get the treatment. I don’t think we have seen any safety concern, at least at this point, about the chronic use of this medication,” he said in an interview.
Sotatercept (ACE-011) is an activin type IIA receptor fusion protein that acts on late-stage erythropoiesis to increase the release of mature erythrocytes into circulation. The mechanism of action is distinct from that of erythropoietins such as epoetin alfa (Procrit, Epogen) or darbapoietin alfa (Aranesp).
In clinical trials with healthy volunteers, sotatercept has been shown to increase hemoglobin levels, suggesting that it could help to reduce anemia and perhaps lessen dependence on transfusions among patients with lower-risk MDS, Dr. Komrokji said.
He and his colleagues at centers in the United States and France enrolled patients with low-risk or intermediate-1–risk MDS as defined by the International Prognostic Scoring System (IPSS), or nonproliferative CMML (fewer than 13,000 white blood cells per microliter). The patients had to have anemia requiring at least 2 red blood cell (RBC) transfusions in the 12 weeks before enrollment for hemoglobin levels below 9.0 g/dL, and no response, loss of response, or a low chance of response to an ESA. Those patients with serum erythropoietin levels greater than 500 mIU/mL were considered to have a low chance of responding to an ESA.
The patients received subcutaneous injections of sotatercept at doses of 0.1, 0.3, 0.5, or 1.0 mg/kg once every 3 weeks.
As noted, the rate of overall hematologic improvement by IWG 2006 criteria was 45%, occurring in 24 of 53 patients available for evaluation. Five of 44 patients with a high transfusion burden (4 or more RBC units required within 8 weeks) were able to be free of RBC transfusions for at least 8 weeks, as were 5 of 9 with a low transfusion burden (fewer than 4 RBC units over a period of 8 weeks).
Looking at the efficacy in patients with a high transfusion burden, the investigators found that 4 of 6 assigned to the 0.3-mg/kg dose group and 8 of 14 assigned to the 1-mg/kg dose group had a reduction in transfusion burden. The median duration of effect was 106 days, with the longest response lasting for 150 days.
There were no major adverse events in the study, and no apparent increase in risk for thrombosis, as had been seen in some studies of ESAs. Another theoretical risk with this type of agent is hypertension, but there was only one grade 3 case and no grade 4 cases of hypertension in the study, Dr. Komrokji said.
Sotatercept is currently in phase II trials for anemia related to hematologic malignancies and other diseases.
SAN FRANCISCO – A first-in-class investigational agent called sotatercept appears to be safe and to improve hematologic parameters in patients with lower-risk myelodysplastic syndrome or nonproliferative chronic myelomonocytic leukemia and anemia requiring transfusion, a study showed.
In the open-label phase II dose-finding study of sotatercept in patients with myelodysplastic syndrome (MDS) or nonproliferative chronic myelomonocytic leukemia (CMML), hematologic improvement according to International Working Group (IWG) 2006 criteria was seen in 24 of 53 evaluable patients, said Dr. Rami Komrokji of the Moffitt Cancer Center,Tampa.
The patients were all refractory to, or were deemed to have a low chance of responding to, an erythropoiesis-stimulating agent (ESA), Dr. Komrokji said at the annual meeting of the American Society of Hematology.
“A medication like sotatercept would probably have a role in the management of anemia in lower-risk MDS patients. The treatment is administered every 3 weeks, which makes it also logistically easier for the patients to get the treatment. I don’t think we have seen any safety concern, at least at this point, about the chronic use of this medication,” he said in an interview.
Sotatercept (ACE-011) is an activin type IIA receptor fusion protein that acts on late-stage erythropoiesis to increase the release of mature erythrocytes into circulation. The mechanism of action is distinct from that of erythropoietins such as epoetin alfa (Procrit, Epogen) or darbapoietin alfa (Aranesp).
In clinical trials with healthy volunteers, sotatercept has been shown to increase hemoglobin levels, suggesting that it could help to reduce anemia and perhaps lessen dependence on transfusions among patients with lower-risk MDS, Dr. Komrokji said.
He and his colleagues at centers in the United States and France enrolled patients with low-risk or intermediate-1–risk MDS as defined by the International Prognostic Scoring System (IPSS), or nonproliferative CMML (fewer than 13,000 white blood cells per microliter). The patients had to have anemia requiring at least 2 red blood cell (RBC) transfusions in the 12 weeks before enrollment for hemoglobin levels below 9.0 g/dL, and no response, loss of response, or a low chance of response to an ESA. Those patients with serum erythropoietin levels greater than 500 mIU/mL were considered to have a low chance of responding to an ESA.
The patients received subcutaneous injections of sotatercept at doses of 0.1, 0.3, 0.5, or 1.0 mg/kg once every 3 weeks.
As noted, the rate of overall hematologic improvement by IWG 2006 criteria was 45%, occurring in 24 of 53 patients available for evaluation. Five of 44 patients with a high transfusion burden (4 or more RBC units required within 8 weeks) were able to be free of RBC transfusions for at least 8 weeks, as were 5 of 9 with a low transfusion burden (fewer than 4 RBC units over a period of 8 weeks).
Looking at the efficacy in patients with a high transfusion burden, the investigators found that 4 of 6 assigned to the 0.3-mg/kg dose group and 8 of 14 assigned to the 1-mg/kg dose group had a reduction in transfusion burden. The median duration of effect was 106 days, with the longest response lasting for 150 days.
There were no major adverse events in the study, and no apparent increase in risk for thrombosis, as had been seen in some studies of ESAs. Another theoretical risk with this type of agent is hypertension, but there was only one grade 3 case and no grade 4 cases of hypertension in the study, Dr. Komrokji said.
Sotatercept is currently in phase II trials for anemia related to hematologic malignancies and other diseases.
SAN FRANCISCO – A first-in-class investigational agent called sotatercept appears to be safe and to improve hematologic parameters in patients with lower-risk myelodysplastic syndrome or nonproliferative chronic myelomonocytic leukemia and anemia requiring transfusion, a study showed.
In the open-label phase II dose-finding study of sotatercept in patients with myelodysplastic syndrome (MDS) or nonproliferative chronic myelomonocytic leukemia (CMML), hematologic improvement according to International Working Group (IWG) 2006 criteria was seen in 24 of 53 evaluable patients, said Dr. Rami Komrokji of the Moffitt Cancer Center,Tampa.
The patients were all refractory to, or were deemed to have a low chance of responding to, an erythropoiesis-stimulating agent (ESA), Dr. Komrokji said at the annual meeting of the American Society of Hematology.
“A medication like sotatercept would probably have a role in the management of anemia in lower-risk MDS patients. The treatment is administered every 3 weeks, which makes it also logistically easier for the patients to get the treatment. I don’t think we have seen any safety concern, at least at this point, about the chronic use of this medication,” he said in an interview.
Sotatercept (ACE-011) is an activin type IIA receptor fusion protein that acts on late-stage erythropoiesis to increase the release of mature erythrocytes into circulation. The mechanism of action is distinct from that of erythropoietins such as epoetin alfa (Procrit, Epogen) or darbapoietin alfa (Aranesp).
In clinical trials with healthy volunteers, sotatercept has been shown to increase hemoglobin levels, suggesting that it could help to reduce anemia and perhaps lessen dependence on transfusions among patients with lower-risk MDS, Dr. Komrokji said.
He and his colleagues at centers in the United States and France enrolled patients with low-risk or intermediate-1–risk MDS as defined by the International Prognostic Scoring System (IPSS), or nonproliferative CMML (fewer than 13,000 white blood cells per microliter). The patients had to have anemia requiring at least 2 red blood cell (RBC) transfusions in the 12 weeks before enrollment for hemoglobin levels below 9.0 g/dL, and no response, loss of response, or a low chance of response to an ESA. Those patients with serum erythropoietin levels greater than 500 mIU/mL were considered to have a low chance of responding to an ESA.
The patients received subcutaneous injections of sotatercept at doses of 0.1, 0.3, 0.5, or 1.0 mg/kg once every 3 weeks.
As noted, the rate of overall hematologic improvement by IWG 2006 criteria was 45%, occurring in 24 of 53 patients available for evaluation. Five of 44 patients with a high transfusion burden (4 or more RBC units required within 8 weeks) were able to be free of RBC transfusions for at least 8 weeks, as were 5 of 9 with a low transfusion burden (fewer than 4 RBC units over a period of 8 weeks).
Looking at the efficacy in patients with a high transfusion burden, the investigators found that 4 of 6 assigned to the 0.3-mg/kg dose group and 8 of 14 assigned to the 1-mg/kg dose group had a reduction in transfusion burden. The median duration of effect was 106 days, with the longest response lasting for 150 days.
There were no major adverse events in the study, and no apparent increase in risk for thrombosis, as had been seen in some studies of ESAs. Another theoretical risk with this type of agent is hypertension, but there was only one grade 3 case and no grade 4 cases of hypertension in the study, Dr. Komrokji said.
Sotatercept is currently in phase II trials for anemia related to hematologic malignancies and other diseases.
Key clinical point: Sotatercept is a first-in-its-class agent that stimulates erythropoiesis through a mechanism different from that of erythropoietins.
Major finding: The rate of overall hematologic improvement by IWG 2006 criteria was 45%, occurring in 24 of 53 patients available for evaluation.
Data source: An ongoing phase II study with data available on 53 patients with MDS or nonproliferative CMML.
Disclosures: The study is sponsored by Celgene. Dr. Komrokji reported consulting for and receiving research funding from the company.

Vitiligo, alopecia more likely in GVHD when donor is female and recipient is male
The development of vitiligo or alopecia areata is not common in patients with chronic graft-versus-host disease, but it is significantly more likely to occur when the donor was female, especially when the recipient was male, according to a report published online Sept. 10 in JAMA Dermatology.
Fifteen case reports and small series in the literature have reported the development of vitiligo or alopecia areata after allogeneic hematopoietic stem cell transplantation, most often among patients who first developed chronic graft-versus-host disease (GVHD) after the procedure. To further explore the frequency of these two skin autoimmune manifestations in GVHD and to identify associated risk factors, researchers performed a retrospective cross-sectional analysis involving 282 adult and pediatric patients referred to the National Institutes of Health Clinical Center for GVHD in 2004-2013.
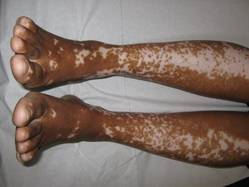
They identified 15 GVHD patients with vitiligo (4.9%) and 2 with alopecia areata (0.7%); one of these patients had both skin disorders. Most of the 15 patients had undergone stem-cell transplantation to treat chronic myelogenous leukemia (CML) (5 patients) or acute leukemia or myelodysplastic syndrome (5 patients), and most had had a human leukocyte antigen–identical donor. Twelve of the 15 developed the skin disorder after having GVHD for more than 1 year, said Rena C. Zuo of the National Cancer Institute’s dermatology branch and her associates.
"Notably, CML accounts for only about 300 of 7,892 (3.8%) allogeneic hematopoietic stem-cell transplantations per year in the United States, a relative minority among indicated diseases," they said.
In what they described as the first study to identify an association between donor/recipient sex mismatch and the development of concomitant autoimmunity in patients with chronic GVHD, the investigators found that 14 of the 15 patients who developed vitiligo or alopecia areata had female donors, 2 of whom had previously given birth; the gender of the donor in the 15th case was unknown. Nine of these 14 recipients were male, "resulting in a female-to-male sex mismatch in [at least] 64% of cases," Ms. Zuo and her colleagues said (JAMA Dermatol. 2014 Sept. 10 [doi:10.1001/jamadermatol.2014.1550]).
Both parous female donors and donor-recipient sex mismatch are known risk factors for GVHD. The risk of autoimmunity in female-to-male transplants "may reflect the activity of skin-homing donor T cells specific for recipient minor histocompatibility antigens encoded by Y-chromosome genes, a mechanism previously implicated in both GVHD and graft-versus-tumor responses," the investigators added.
This study was supported by the National Institutes of Health and the National Cancer Institute. Ms. Zuo and her associates reported no financial conflicts of interest.
The development of vitiligo or alopecia areata is not common in patients with chronic graft-versus-host disease, but it is significantly more likely to occur when the donor was female, especially when the recipient was male, according to a report published online Sept. 10 in JAMA Dermatology.
Fifteen case reports and small series in the literature have reported the development of vitiligo or alopecia areata after allogeneic hematopoietic stem cell transplantation, most often among patients who first developed chronic graft-versus-host disease (GVHD) after the procedure. To further explore the frequency of these two skin autoimmune manifestations in GVHD and to identify associated risk factors, researchers performed a retrospective cross-sectional analysis involving 282 adult and pediatric patients referred to the National Institutes of Health Clinical Center for GVHD in 2004-2013.
They identified 15 GVHD patients with vitiligo (4.9%) and 2 with alopecia areata (0.7%); one of these patients had both skin disorders. Most of the 15 patients had undergone stem-cell transplantation to treat chronic myelogenous leukemia (CML) (5 patients) or acute leukemia or myelodysplastic syndrome (5 patients), and most had had a human leukocyte antigen–identical donor. Twelve of the 15 developed the skin disorder after having GVHD for more than 1 year, said Rena C. Zuo of the National Cancer Institute’s dermatology branch and her associates.
"Notably, CML accounts for only about 300 of 7,892 (3.8%) allogeneic hematopoietic stem-cell transplantations per year in the United States, a relative minority among indicated diseases," they said.
In what they described as the first study to identify an association between donor/recipient sex mismatch and the development of concomitant autoimmunity in patients with chronic GVHD, the investigators found that 14 of the 15 patients who developed vitiligo or alopecia areata had female donors, 2 of whom had previously given birth; the gender of the donor in the 15th case was unknown. Nine of these 14 recipients were male, "resulting in a female-to-male sex mismatch in [at least] 64% of cases," Ms. Zuo and her colleagues said (JAMA Dermatol. 2014 Sept. 10 [doi:10.1001/jamadermatol.2014.1550]).
Both parous female donors and donor-recipient sex mismatch are known risk factors for GVHD. The risk of autoimmunity in female-to-male transplants "may reflect the activity of skin-homing donor T cells specific for recipient minor histocompatibility antigens encoded by Y-chromosome genes, a mechanism previously implicated in both GVHD and graft-versus-tumor responses," the investigators added.
This study was supported by the National Institutes of Health and the National Cancer Institute. Ms. Zuo and her associates reported no financial conflicts of interest.
The development of vitiligo or alopecia areata is not common in patients with chronic graft-versus-host disease, but it is significantly more likely to occur when the donor was female, especially when the recipient was male, according to a report published online Sept. 10 in JAMA Dermatology.
Fifteen case reports and small series in the literature have reported the development of vitiligo or alopecia areata after allogeneic hematopoietic stem cell transplantation, most often among patients who first developed chronic graft-versus-host disease (GVHD) after the procedure. To further explore the frequency of these two skin autoimmune manifestations in GVHD and to identify associated risk factors, researchers performed a retrospective cross-sectional analysis involving 282 adult and pediatric patients referred to the National Institutes of Health Clinical Center for GVHD in 2004-2013.
They identified 15 GVHD patients with vitiligo (4.9%) and 2 with alopecia areata (0.7%); one of these patients had both skin disorders. Most of the 15 patients had undergone stem-cell transplantation to treat chronic myelogenous leukemia (CML) (5 patients) or acute leukemia or myelodysplastic syndrome (5 patients), and most had had a human leukocyte antigen–identical donor. Twelve of the 15 developed the skin disorder after having GVHD for more than 1 year, said Rena C. Zuo of the National Cancer Institute’s dermatology branch and her associates.
"Notably, CML accounts for only about 300 of 7,892 (3.8%) allogeneic hematopoietic stem-cell transplantations per year in the United States, a relative minority among indicated diseases," they said.
In what they described as the first study to identify an association between donor/recipient sex mismatch and the development of concomitant autoimmunity in patients with chronic GVHD, the investigators found that 14 of the 15 patients who developed vitiligo or alopecia areata had female donors, 2 of whom had previously given birth; the gender of the donor in the 15th case was unknown. Nine of these 14 recipients were male, "resulting in a female-to-male sex mismatch in [at least] 64% of cases," Ms. Zuo and her colleagues said (JAMA Dermatol. 2014 Sept. 10 [doi:10.1001/jamadermatol.2014.1550]).
Both parous female donors and donor-recipient sex mismatch are known risk factors for GVHD. The risk of autoimmunity in female-to-male transplants "may reflect the activity of skin-homing donor T cells specific for recipient minor histocompatibility antigens encoded by Y-chromosome genes, a mechanism previously implicated in both GVHD and graft-versus-tumor responses," the investigators added.
This study was supported by the National Institutes of Health and the National Cancer Institute. Ms. Zuo and her associates reported no financial conflicts of interest.
FROM JAMA DERMATOLOGY
Key clinical point: Female-to-male stem cell donation ups the risk for vitiligo in GVHD.
Major finding: 14 of the 15 patients who developed vitiligo or alopecia areata had female donors (the gender of the 15th donor was unknown), and 9 of these 14 recipients were male.
Data source: A retrospective cross-sectional analysis involving 282 adults and children with chronic GVHD, 15 of whom developed vitiligo or alopecia areata.
Disclosures: This study was supported by the National Institutes of Health and the National Cancer Institute. Dr. Zuo and her associates reported no financial conflicts of interest.
FDA approves generic decitabine for MDS

Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved decitabine for injection, a generic version of Dacogen, to treat patients with myelodysplastic syndromes (MDS).
Decitabine is indicated for previously treated and untreated patients with de novo and secondary MDS of all French-American-British subtypes—refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia—as well as intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Decitabine will be marketed in 20 mL single-dose glass vials containing 50 mg decitabine—the same size and strength as the brand name drug. The dosing regimen is identical as well.
InnoPharma developed the generic formulation of decitabine and entered into an agreement with Sandoz Inc. Sandoz will sell, market, and distribute decitabine in the US. InnoPharma is set to be acquired by Pfizer Inc., but the transaction is subject to US regulatory approval.
The FDA approved another generic form of decitabine for the treatment of MDS in July 2013. That drug is a product of Dr Reddy’s Laboratories Limited.
Dacogen has been FDA-approved to treat MDS since May 2006. Dacogen is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals Inc. ![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved decitabine for injection, a generic version of Dacogen, to treat patients with myelodysplastic syndromes (MDS).
Decitabine is indicated for previously treated and untreated patients with de novo and secondary MDS of all French-American-British subtypes—refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia—as well as intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Decitabine will be marketed in 20 mL single-dose glass vials containing 50 mg decitabine—the same size and strength as the brand name drug. The dosing regimen is identical as well.
InnoPharma developed the generic formulation of decitabine and entered into an agreement with Sandoz Inc. Sandoz will sell, market, and distribute decitabine in the US. InnoPharma is set to be acquired by Pfizer Inc., but the transaction is subject to US regulatory approval.
The FDA approved another generic form of decitabine for the treatment of MDS in July 2013. That drug is a product of Dr Reddy’s Laboratories Limited.
Dacogen has been FDA-approved to treat MDS since May 2006. Dacogen is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals Inc. ![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved decitabine for injection, a generic version of Dacogen, to treat patients with myelodysplastic syndromes (MDS).
Decitabine is indicated for previously treated and untreated patients with de novo and secondary MDS of all French-American-British subtypes—refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia—as well as intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Decitabine will be marketed in 20 mL single-dose glass vials containing 50 mg decitabine—the same size and strength as the brand name drug. The dosing regimen is identical as well.
InnoPharma developed the generic formulation of decitabine and entered into an agreement with Sandoz Inc. Sandoz will sell, market, and distribute decitabine in the US. InnoPharma is set to be acquired by Pfizer Inc., but the transaction is subject to US regulatory approval.
The FDA approved another generic form of decitabine for the treatment of MDS in July 2013. That drug is a product of Dr Reddy’s Laboratories Limited.
Dacogen has been FDA-approved to treat MDS since May 2006. Dacogen is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals Inc. ![]()
Drugs can increase risk of MDS and AML
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
AG-221 sparked durable responses in hematologic cancers
MILAN – The investigational drug AG-221 induced responses in more than half of patients with advanced IDH2 mutation–positive hematologic cancers, updated phase I data showed.
Among 25 evaluable patients, 14 responded to treatment with AG-221: 6 had complete responses, 2 had complete responses with incomplete platelet count recovery, 1 had a complete response with incomplete hematologic recovery, and 5 had partial responses.
Twelve of 14 responses are ongoing, and five patients with stable disease remain on study.
Responses are seen in acute myelogenous leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia, Dr. Stéphane de Botton said during a late-breaking abstract session at the annual congress of the European Hematology Association.
"Very interestingly, at least in five patients, the duration of the responses has reached greater than 2.5 months," he said.
AG-221 is a first-in-class inhibitor of the isocitrate dehydrogenase–2 (IDH2) protein and was just granted orphan drug status by the Food and Drug Administration for the treatment of acute myelogenous leukemia (AML).
About 15% of patients with AML carry an IDH2 mutation, as do 5% with myelodysplastic syndromes.
An initial report from this phase I, multicenter study showed five of seven patients evaluable at that time had a complete response or platelet count recovery after treatment with AG-221.
Of the 35 patients now enrolled, 27 had relapsed or refractory AML, 4 had myelodysplastic syndromes, 2 had untreated AML, 1 had chronic myelomonocytic leukemia, and 1 had granulocytic sarcoma. Patients’ mean age was 66 years, 31 had IDH2 R140Q mutations, and 4 had IDH2 R172K mutations.
Up to 100% plasma 2-hydroxyglutarate inhibition was seen after multiple doses in R140Q patients and up to 60% plasma 2-HG inhibition, in R172K patients, said Dr. de Botton, head of hematology, Institut Gustave Roussy, Villejuif, France.
Dose escalation has continued with single-agent oral dosing ranging from 30 mg twice daily to 150 mg once daily in 28-day cycles. The results were very similar with 50 mg b.i.d. and 100 mg every day.
AG-221 was also "remarkably well tolerated," with the maximum tolerated dose yet to be reached, he said.
The majority of adverse events are grade 1 and 2, notably edema, leukocytosis, nausea, sepsis, and thrombocytopenia.
Grade 3 or higher events in 18 evaluable patients included 3 cases each of thrombocytopenia and febrile neutropenia, 2 of leukocytosis, and 1 case each of diarrhea and rash, he said.
There have been four possible treatment-related serious adverse events: grade 3 confusion and grade 5 respiratory failure in a patient with severe sepsis, one case of grade 3 leukocytosis along with grade 3 anorexia and grade 1 nausea, one case of grade 3 diarrhea, and one case of grade 3 leukocytosis.
There have been seven deaths within 30 days of study drug termination: five stemming from complications of disease-related sepsis, one from complications of a humeral fracture not related to the study drug, and one from stroke, also unrelated to treatment, Dr. de Botton said.
"AG-221 is safe and well tolerated to date with durable clinical activity," he concluded.
Dr. de Botton reported research funding from Agios Pharmaceuticals, the study sponsor.
MILAN – The investigational drug AG-221 induced responses in more than half of patients with advanced IDH2 mutation–positive hematologic cancers, updated phase I data showed.
Among 25 evaluable patients, 14 responded to treatment with AG-221: 6 had complete responses, 2 had complete responses with incomplete platelet count recovery, 1 had a complete response with incomplete hematologic recovery, and 5 had partial responses.
Twelve of 14 responses are ongoing, and five patients with stable disease remain on study.
Responses are seen in acute myelogenous leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia, Dr. Stéphane de Botton said during a late-breaking abstract session at the annual congress of the European Hematology Association.
"Very interestingly, at least in five patients, the duration of the responses has reached greater than 2.5 months," he said.
AG-221 is a first-in-class inhibitor of the isocitrate dehydrogenase–2 (IDH2) protein and was just granted orphan drug status by the Food and Drug Administration for the treatment of acute myelogenous leukemia (AML).
About 15% of patients with AML carry an IDH2 mutation, as do 5% with myelodysplastic syndromes.
An initial report from this phase I, multicenter study showed five of seven patients evaluable at that time had a complete response or platelet count recovery after treatment with AG-221.
Of the 35 patients now enrolled, 27 had relapsed or refractory AML, 4 had myelodysplastic syndromes, 2 had untreated AML, 1 had chronic myelomonocytic leukemia, and 1 had granulocytic sarcoma. Patients’ mean age was 66 years, 31 had IDH2 R140Q mutations, and 4 had IDH2 R172K mutations.
Up to 100% plasma 2-hydroxyglutarate inhibition was seen after multiple doses in R140Q patients and up to 60% plasma 2-HG inhibition, in R172K patients, said Dr. de Botton, head of hematology, Institut Gustave Roussy, Villejuif, France.
Dose escalation has continued with single-agent oral dosing ranging from 30 mg twice daily to 150 mg once daily in 28-day cycles. The results were very similar with 50 mg b.i.d. and 100 mg every day.
AG-221 was also "remarkably well tolerated," with the maximum tolerated dose yet to be reached, he said.
The majority of adverse events are grade 1 and 2, notably edema, leukocytosis, nausea, sepsis, and thrombocytopenia.
Grade 3 or higher events in 18 evaluable patients included 3 cases each of thrombocytopenia and febrile neutropenia, 2 of leukocytosis, and 1 case each of diarrhea and rash, he said.
There have been four possible treatment-related serious adverse events: grade 3 confusion and grade 5 respiratory failure in a patient with severe sepsis, one case of grade 3 leukocytosis along with grade 3 anorexia and grade 1 nausea, one case of grade 3 diarrhea, and one case of grade 3 leukocytosis.
There have been seven deaths within 30 days of study drug termination: five stemming from complications of disease-related sepsis, one from complications of a humeral fracture not related to the study drug, and one from stroke, also unrelated to treatment, Dr. de Botton said.
"AG-221 is safe and well tolerated to date with durable clinical activity," he concluded.
Dr. de Botton reported research funding from Agios Pharmaceuticals, the study sponsor.
MILAN – The investigational drug AG-221 induced responses in more than half of patients with advanced IDH2 mutation–positive hematologic cancers, updated phase I data showed.
Among 25 evaluable patients, 14 responded to treatment with AG-221: 6 had complete responses, 2 had complete responses with incomplete platelet count recovery, 1 had a complete response with incomplete hematologic recovery, and 5 had partial responses.
Twelve of 14 responses are ongoing, and five patients with stable disease remain on study.
Responses are seen in acute myelogenous leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia, Dr. Stéphane de Botton said during a late-breaking abstract session at the annual congress of the European Hematology Association.
"Very interestingly, at least in five patients, the duration of the responses has reached greater than 2.5 months," he said.
AG-221 is a first-in-class inhibitor of the isocitrate dehydrogenase–2 (IDH2) protein and was just granted orphan drug status by the Food and Drug Administration for the treatment of acute myelogenous leukemia (AML).
About 15% of patients with AML carry an IDH2 mutation, as do 5% with myelodysplastic syndromes.
An initial report from this phase I, multicenter study showed five of seven patients evaluable at that time had a complete response or platelet count recovery after treatment with AG-221.
Of the 35 patients now enrolled, 27 had relapsed or refractory AML, 4 had myelodysplastic syndromes, 2 had untreated AML, 1 had chronic myelomonocytic leukemia, and 1 had granulocytic sarcoma. Patients’ mean age was 66 years, 31 had IDH2 R140Q mutations, and 4 had IDH2 R172K mutations.
Up to 100% plasma 2-hydroxyglutarate inhibition was seen after multiple doses in R140Q patients and up to 60% plasma 2-HG inhibition, in R172K patients, said Dr. de Botton, head of hematology, Institut Gustave Roussy, Villejuif, France.
Dose escalation has continued with single-agent oral dosing ranging from 30 mg twice daily to 150 mg once daily in 28-day cycles. The results were very similar with 50 mg b.i.d. and 100 mg every day.
AG-221 was also "remarkably well tolerated," with the maximum tolerated dose yet to be reached, he said.
The majority of adverse events are grade 1 and 2, notably edema, leukocytosis, nausea, sepsis, and thrombocytopenia.
Grade 3 or higher events in 18 evaluable patients included 3 cases each of thrombocytopenia and febrile neutropenia, 2 of leukocytosis, and 1 case each of diarrhea and rash, he said.
There have been four possible treatment-related serious adverse events: grade 3 confusion and grade 5 respiratory failure in a patient with severe sepsis, one case of grade 3 leukocytosis along with grade 3 anorexia and grade 1 nausea, one case of grade 3 diarrhea, and one case of grade 3 leukocytosis.
There have been seven deaths within 30 days of study drug termination: five stemming from complications of disease-related sepsis, one from complications of a humeral fracture not related to the study drug, and one from stroke, also unrelated to treatment, Dr. de Botton said.
"AG-221 is safe and well tolerated to date with durable clinical activity," he concluded.
Dr. de Botton reported research funding from Agios Pharmaceuticals, the study sponsor.
AT THE EHA CONGRESS
Key clinical point: AG-221 could shift the treatment for IDH2 mutation-positive hematologic cancers.
Major finding: Fourteen of 25 patients responded to treatment with AG-221.
Data source: A prospective phase I dose-escalation study in 35 patients with hematologic cancers.
Disclosures: Dr. de Botton reported research funding from Agios Pharmaceuticals, the study sponsor.
Inhibitor shows promise for hematologic disorders

Photo courtesy of EHA
MILAN—The IDH2 inhibitor AG-221 is well-tolerated and exhibits durable clinical activity in patients with hematologic disorders, results of a phase 1 study suggest.
The drug prompted responses in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML).
Fourteen of 25 patients achieved a response, and 12 of those responses are ongoing.
Most adverse events (AEs) were grade 1 or 2 in nature. However, 4 patients did have serious AEs that were possibly related to treatment.
Stéphane de Botton, MD, PhD, of Institut Gustave Roussy in Villejuif, France, presented these results at the 19th Annual Congress of the European Hematology Association (EHA) as abstract LB2434.
Dr de Botton and his colleagues enrolled 35 patients who had a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at 30 mg BID (n=7), 50 mg BID (n=7), 75 mg BID (n=6), 100 mg QD (n=5), 100 mg BID (n=5), or 150 mg QD (n=5). Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
Safety data
“AG-221 was remarkable well-tolerated, and the [maximum tolerated dose] has not been reached,” Dr de Botton said. “The majority of adverse events were grade 1 and 2.”
Eighteen patients were evaluable for safety. AEs of all grades included nausea (n=4), pyrexia (n=4), thrombocytopenia (n=4), anemia (n=3), dizziness (n=3), febrile neutropenia (n=3), peripheral edema (n=3), sepsis (n=3), cough (n=2), diarrhea (n=2), fatigue (n=2), leukocytosis (n=2), neutropenia (n=2), petechiae (n=2), and rash (n=2).
Grade 3 or higher AEs included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1). Dr de Botton noted that diarrhea and rash were not expected events.
Four patients had serious AEs possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Seven patients died within 30 days of study drug termination: 4 in the 30-mg cohort, 2 in the 50-mg cohort, and 1 in the 100-mg-BID cohort.
Five deaths were due to complications of disease-related sepsis (all in cycle 1), 1 complication of a humeral fracture, and 1 complication of a stroke.
Activity and response data
The researchers observed high AG-221 accumulation after multiple doses. And results were “really very similar” between the 30-mg-BID cohort and the 100-mg-QD cohort, Dr de Botton noted.
He also said AG-221 was “very efficient” at inhibiting 2-HG in the plasma. 2-HG was inhibited up to 100% in subjects with R140Q mutations and up to 60% in subjects with R172K mutations.
Twenty-five patients were evaluable for response. The remaining 10 patients did not have day-28 marrow assessments, either due to early termination (n=7) or receiving less than 28 days of treatment although they were still on the study (n=3).
In all, there were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery (CRps), 1 CR with incomplete hematologic recovery (CRi), and 5 partial responses (PRs). Five patients had stable disease (SD), and 6 had progressive disease (PD).
The most responses occurred in the 50-mg group, which had 3 CRs, 1 CRi, and 1 PR. This was followed by the 30-mg group, which had 2 CRs, 1 CRp, and 1 PR.
“The majority of responses occurred in cycle 1,” Dr de Botton noted, “except in the first cohort [30 mg], where responses occurred late, at the end of cycle 3 and cycle 4.”
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CRp, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with SD remain on study.
This study is sponsored by Celgene Corporation and Agios Pharmaceuticals Inc., the companies developing AG-221. ![]()
Photo courtesy of EHA
MILAN—The IDH2 inhibitor AG-221 is well-tolerated and exhibits durable clinical activity in patients with hematologic disorders, results of a phase 1 study suggest.
The drug prompted responses in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML).
Fourteen of 25 patients achieved a response, and 12 of those responses are ongoing.
Most adverse events (AEs) were grade 1 or 2 in nature. However, 4 patients did have serious AEs that were possibly related to treatment.
Stéphane de Botton, MD, PhD, of Institut Gustave Roussy in Villejuif, France, presented these results at the 19th Annual Congress of the European Hematology Association (EHA) as abstract LB2434.
Dr de Botton and his colleagues enrolled 35 patients who had a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at 30 mg BID (n=7), 50 mg BID (n=7), 75 mg BID (n=6), 100 mg QD (n=5), 100 mg BID (n=5), or 150 mg QD (n=5). Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
Safety data
“AG-221 was remarkable well-tolerated, and the [maximum tolerated dose] has not been reached,” Dr de Botton said. “The majority of adverse events were grade 1 and 2.”
Eighteen patients were evaluable for safety. AEs of all grades included nausea (n=4), pyrexia (n=4), thrombocytopenia (n=4), anemia (n=3), dizziness (n=3), febrile neutropenia (n=3), peripheral edema (n=3), sepsis (n=3), cough (n=2), diarrhea (n=2), fatigue (n=2), leukocytosis (n=2), neutropenia (n=2), petechiae (n=2), and rash (n=2).
Grade 3 or higher AEs included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1). Dr de Botton noted that diarrhea and rash were not expected events.
Four patients had serious AEs possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Seven patients died within 30 days of study drug termination: 4 in the 30-mg cohort, 2 in the 50-mg cohort, and 1 in the 100-mg-BID cohort.
Five deaths were due to complications of disease-related sepsis (all in cycle 1), 1 complication of a humeral fracture, and 1 complication of a stroke.
Activity and response data
The researchers observed high AG-221 accumulation after multiple doses. And results were “really very similar” between the 30-mg-BID cohort and the 100-mg-QD cohort, Dr de Botton noted.
He also said AG-221 was “very efficient” at inhibiting 2-HG in the plasma. 2-HG was inhibited up to 100% in subjects with R140Q mutations and up to 60% in subjects with R172K mutations.
Twenty-five patients were evaluable for response. The remaining 10 patients did not have day-28 marrow assessments, either due to early termination (n=7) or receiving less than 28 days of treatment although they were still on the study (n=3).
In all, there were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery (CRps), 1 CR with incomplete hematologic recovery (CRi), and 5 partial responses (PRs). Five patients had stable disease (SD), and 6 had progressive disease (PD).
The most responses occurred in the 50-mg group, which had 3 CRs, 1 CRi, and 1 PR. This was followed by the 30-mg group, which had 2 CRs, 1 CRp, and 1 PR.
“The majority of responses occurred in cycle 1,” Dr de Botton noted, “except in the first cohort [30 mg], where responses occurred late, at the end of cycle 3 and cycle 4.”
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CRp, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with SD remain on study.
This study is sponsored by Celgene Corporation and Agios Pharmaceuticals Inc., the companies developing AG-221. ![]()
Photo courtesy of EHA
MILAN—The IDH2 inhibitor AG-221 is well-tolerated and exhibits durable clinical activity in patients with hematologic disorders, results of a phase 1 study suggest.
The drug prompted responses in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML).
Fourteen of 25 patients achieved a response, and 12 of those responses are ongoing.
Most adverse events (AEs) were grade 1 or 2 in nature. However, 4 patients did have serious AEs that were possibly related to treatment.
Stéphane de Botton, MD, PhD, of Institut Gustave Roussy in Villejuif, France, presented these results at the 19th Annual Congress of the European Hematology Association (EHA) as abstract LB2434.
Dr de Botton and his colleagues enrolled 35 patients who had a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at 30 mg BID (n=7), 50 mg BID (n=7), 75 mg BID (n=6), 100 mg QD (n=5), 100 mg BID (n=5), or 150 mg QD (n=5). Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
Safety data
“AG-221 was remarkable well-tolerated, and the [maximum tolerated dose] has not been reached,” Dr de Botton said. “The majority of adverse events were grade 1 and 2.”
Eighteen patients were evaluable for safety. AEs of all grades included nausea (n=4), pyrexia (n=4), thrombocytopenia (n=4), anemia (n=3), dizziness (n=3), febrile neutropenia (n=3), peripheral edema (n=3), sepsis (n=3), cough (n=2), diarrhea (n=2), fatigue (n=2), leukocytosis (n=2), neutropenia (n=2), petechiae (n=2), and rash (n=2).
Grade 3 or higher AEs included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1). Dr de Botton noted that diarrhea and rash were not expected events.
Four patients had serious AEs possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Seven patients died within 30 days of study drug termination: 4 in the 30-mg cohort, 2 in the 50-mg cohort, and 1 in the 100-mg-BID cohort.
Five deaths were due to complications of disease-related sepsis (all in cycle 1), 1 complication of a humeral fracture, and 1 complication of a stroke.
Activity and response data
The researchers observed high AG-221 accumulation after multiple doses. And results were “really very similar” between the 30-mg-BID cohort and the 100-mg-QD cohort, Dr de Botton noted.
He also said AG-221 was “very efficient” at inhibiting 2-HG in the plasma. 2-HG was inhibited up to 100% in subjects with R140Q mutations and up to 60% in subjects with R172K mutations.
Twenty-five patients were evaluable for response. The remaining 10 patients did not have day-28 marrow assessments, either due to early termination (n=7) or receiving less than 28 days of treatment although they were still on the study (n=3).
In all, there were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery (CRps), 1 CR with incomplete hematologic recovery (CRi), and 5 partial responses (PRs). Five patients had stable disease (SD), and 6 had progressive disease (PD).
The most responses occurred in the 50-mg group, which had 3 CRs, 1 CRi, and 1 PR. This was followed by the 30-mg group, which had 2 CRs, 1 CRp, and 1 PR.
“The majority of responses occurred in cycle 1,” Dr de Botton noted, “except in the first cohort [30 mg], where responses occurred late, at the end of cycle 3 and cycle 4.”
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CRp, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with SD remain on study.
This study is sponsored by Celgene Corporation and Agios Pharmaceuticals Inc., the companies developing AG-221. ![]()