User login
Inhibitor could treat range of hematologic disorders

A small molecule that targets the sonic Hedgehog signaling pathway has advanced to phase 2 trials in a range of hematologic disorders.
In a phase 1 study, the inhibitor, PF-04449913, exhibited activity in adults with leukemias, myelodysplastic syndromes (MDS), and myelofibrosis (MF).
Sixty percent of the patients studied experienced treatment-related adverse events (AEs), but there were no treatment-related deaths. Most deaths were disease-related.
Researchers detailed the results of this trial in The Lancet Haematology. The study was funded by Pfizer, the company developing PF-04449913, as well as the California Institute for Regenerative Medicine and European Leukemia Net.
Preclinical research showed that PF-04449913 forces dormant cancer stem cells in the bone marrow to begin differentiating and exit into the blood stream where they can be destroyed by chemotherapy agents targeting dividing cells.
“This drug gets that unwanted house guests to leave and never come back,” said Catriona Jamieson, MD, PhD, of University of California, San Diego School of Medicine.
“It’s a significant step forward in treating people with refractory or resistant myeloid leukemia, myelodysplastic syndrome, and myelofibrosis. It’s a bonus that the drug can be administered as easily as an aspirin, in a single, daily, oral tablet.”
For the first-in-human study, Dr Jamieson and her colleagues evaluated PF-04449913 in 47 adult patients. Twenty-eight of them had acute myeloid leukemia (AML), 6 had MDS, 5 had chronic myeloid leukemia (CML), 1 had chronic myelomonocytic leukemia (CMML), and 7 had MF.
Eighty-five percent of patients (n=40) had an ECOG performance status of 0-1. Eighty-one percent (n=38) had received previous systemic treatment, and 47% (n=22) had received 3 or more previous treatment regimens.
Patients received escalating daily doses of PF-04449913 in 28-day cycles. Treatment cycles were repeated until a patient experienced unacceptable AEs without evidence of clinical improvement. Patients who showed clinical activity without experiencing serious AEs received additional treatment cycles.
Dosing and AEs
Patients received PF-04449913 once daily at 5 mg (n=3), 10 mg (n=3), 20 mg (n=4), 40 mg (n=4), 80 mg (n=8), 120 mg (n=3), 180 mg (n=3), 270 mg (n=5), 400 mg (n=9), or 600 mg (n=5).
The researchers found the maximum-tolerated dose to be 400 mg once daily. The mean half-life was 23.9 hours in this dose group, and pharmacokinetics seemed to be dose-proportional.
Two patients experienced dose-limiting toxicities, 1 in the 80 mg group (grade 3 hypoxia and grade 3 pleural effusion), and 1 in the 600 mg group (grade 3 peripheral edema).
In all, 60% of patients (n=28) experienced treatment-related AEs. The most common were dysgeusia (28%), decreased appetite (19%), and alopecia (15%). There were 3 grade 4 AEs—1 case of neutropenia and 2 cases of thrombocytopenia.
There were 15 deaths, none of which were treatment-related. Eleven deaths were disease-related, and the remaining 4 were related to infection.
Clinical activity
The researchers said there was “some suggestion of clinical activity” in 23 patients (49%).
Of the 5 patients with CML (2 chronic phase and 3 blast phase), 1 patient with blast phase CML had a partial cytogenetic response to PF-04449913.
Of the 6 patients with MDS and 1 with CMML, 4 had stable disease after treatment. Two of these patients had hematologic improvement.
Two of the 7 patients with MF had clinical improvement.
Of the 28 patients with AML, 16 showed evidence of possible biological activity. One patient had a complete response and 4 had a partial response with incomplete hematologic recovery. Four AML patients had minor responses, and 7 had stable disease.
Given these results, PF-04449913 is now being investigated in 5 phase 2 trials of hematologic disorders, 4 of which are recruiting participants.
“Our hope is that this drug will enable more effective treatment to begin earlier and that, with earlier intervention, we can alter the course of disease and remove the need for, or improve the chances of success with, bone marrow transplantation,” Dr Jamieson said. “It’s all about reducing the burden of disease by intervening early.” ![]()
A small molecule that targets the sonic Hedgehog signaling pathway has advanced to phase 2 trials in a range of hematologic disorders.
In a phase 1 study, the inhibitor, PF-04449913, exhibited activity in adults with leukemias, myelodysplastic syndromes (MDS), and myelofibrosis (MF).
Sixty percent of the patients studied experienced treatment-related adverse events (AEs), but there were no treatment-related deaths. Most deaths were disease-related.
Researchers detailed the results of this trial in The Lancet Haematology. The study was funded by Pfizer, the company developing PF-04449913, as well as the California Institute for Regenerative Medicine and European Leukemia Net.
Preclinical research showed that PF-04449913 forces dormant cancer stem cells in the bone marrow to begin differentiating and exit into the blood stream where they can be destroyed by chemotherapy agents targeting dividing cells.
“This drug gets that unwanted house guests to leave and never come back,” said Catriona Jamieson, MD, PhD, of University of California, San Diego School of Medicine.
“It’s a significant step forward in treating people with refractory or resistant myeloid leukemia, myelodysplastic syndrome, and myelofibrosis. It’s a bonus that the drug can be administered as easily as an aspirin, in a single, daily, oral tablet.”
For the first-in-human study, Dr Jamieson and her colleagues evaluated PF-04449913 in 47 adult patients. Twenty-eight of them had acute myeloid leukemia (AML), 6 had MDS, 5 had chronic myeloid leukemia (CML), 1 had chronic myelomonocytic leukemia (CMML), and 7 had MF.
Eighty-five percent of patients (n=40) had an ECOG performance status of 0-1. Eighty-one percent (n=38) had received previous systemic treatment, and 47% (n=22) had received 3 or more previous treatment regimens.
Patients received escalating daily doses of PF-04449913 in 28-day cycles. Treatment cycles were repeated until a patient experienced unacceptable AEs without evidence of clinical improvement. Patients who showed clinical activity without experiencing serious AEs received additional treatment cycles.
Dosing and AEs
Patients received PF-04449913 once daily at 5 mg (n=3), 10 mg (n=3), 20 mg (n=4), 40 mg (n=4), 80 mg (n=8), 120 mg (n=3), 180 mg (n=3), 270 mg (n=5), 400 mg (n=9), or 600 mg (n=5).
The researchers found the maximum-tolerated dose to be 400 mg once daily. The mean half-life was 23.9 hours in this dose group, and pharmacokinetics seemed to be dose-proportional.
Two patients experienced dose-limiting toxicities, 1 in the 80 mg group (grade 3 hypoxia and grade 3 pleural effusion), and 1 in the 600 mg group (grade 3 peripheral edema).
In all, 60% of patients (n=28) experienced treatment-related AEs. The most common were dysgeusia (28%), decreased appetite (19%), and alopecia (15%). There were 3 grade 4 AEs—1 case of neutropenia and 2 cases of thrombocytopenia.
There were 15 deaths, none of which were treatment-related. Eleven deaths were disease-related, and the remaining 4 were related to infection.
Clinical activity
The researchers said there was “some suggestion of clinical activity” in 23 patients (49%).
Of the 5 patients with CML (2 chronic phase and 3 blast phase), 1 patient with blast phase CML had a partial cytogenetic response to PF-04449913.
Of the 6 patients with MDS and 1 with CMML, 4 had stable disease after treatment. Two of these patients had hematologic improvement.
Two of the 7 patients with MF had clinical improvement.
Of the 28 patients with AML, 16 showed evidence of possible biological activity. One patient had a complete response and 4 had a partial response with incomplete hematologic recovery. Four AML patients had minor responses, and 7 had stable disease.
Given these results, PF-04449913 is now being investigated in 5 phase 2 trials of hematologic disorders, 4 of which are recruiting participants.
“Our hope is that this drug will enable more effective treatment to begin earlier and that, with earlier intervention, we can alter the course of disease and remove the need for, or improve the chances of success with, bone marrow transplantation,” Dr Jamieson said. “It’s all about reducing the burden of disease by intervening early.” ![]()
A small molecule that targets the sonic Hedgehog signaling pathway has advanced to phase 2 trials in a range of hematologic disorders.
In a phase 1 study, the inhibitor, PF-04449913, exhibited activity in adults with leukemias, myelodysplastic syndromes (MDS), and myelofibrosis (MF).
Sixty percent of the patients studied experienced treatment-related adverse events (AEs), but there were no treatment-related deaths. Most deaths were disease-related.
Researchers detailed the results of this trial in The Lancet Haematology. The study was funded by Pfizer, the company developing PF-04449913, as well as the California Institute for Regenerative Medicine and European Leukemia Net.
Preclinical research showed that PF-04449913 forces dormant cancer stem cells in the bone marrow to begin differentiating and exit into the blood stream where they can be destroyed by chemotherapy agents targeting dividing cells.
“This drug gets that unwanted house guests to leave and never come back,” said Catriona Jamieson, MD, PhD, of University of California, San Diego School of Medicine.
“It’s a significant step forward in treating people with refractory or resistant myeloid leukemia, myelodysplastic syndrome, and myelofibrosis. It’s a bonus that the drug can be administered as easily as an aspirin, in a single, daily, oral tablet.”
For the first-in-human study, Dr Jamieson and her colleagues evaluated PF-04449913 in 47 adult patients. Twenty-eight of them had acute myeloid leukemia (AML), 6 had MDS, 5 had chronic myeloid leukemia (CML), 1 had chronic myelomonocytic leukemia (CMML), and 7 had MF.
Eighty-five percent of patients (n=40) had an ECOG performance status of 0-1. Eighty-one percent (n=38) had received previous systemic treatment, and 47% (n=22) had received 3 or more previous treatment regimens.
Patients received escalating daily doses of PF-04449913 in 28-day cycles. Treatment cycles were repeated until a patient experienced unacceptable AEs without evidence of clinical improvement. Patients who showed clinical activity without experiencing serious AEs received additional treatment cycles.
Dosing and AEs
Patients received PF-04449913 once daily at 5 mg (n=3), 10 mg (n=3), 20 mg (n=4), 40 mg (n=4), 80 mg (n=8), 120 mg (n=3), 180 mg (n=3), 270 mg (n=5), 400 mg (n=9), or 600 mg (n=5).
The researchers found the maximum-tolerated dose to be 400 mg once daily. The mean half-life was 23.9 hours in this dose group, and pharmacokinetics seemed to be dose-proportional.
Two patients experienced dose-limiting toxicities, 1 in the 80 mg group (grade 3 hypoxia and grade 3 pleural effusion), and 1 in the 600 mg group (grade 3 peripheral edema).
In all, 60% of patients (n=28) experienced treatment-related AEs. The most common were dysgeusia (28%), decreased appetite (19%), and alopecia (15%). There were 3 grade 4 AEs—1 case of neutropenia and 2 cases of thrombocytopenia.
There were 15 deaths, none of which were treatment-related. Eleven deaths were disease-related, and the remaining 4 were related to infection.
Clinical activity
The researchers said there was “some suggestion of clinical activity” in 23 patients (49%).
Of the 5 patients with CML (2 chronic phase and 3 blast phase), 1 patient with blast phase CML had a partial cytogenetic response to PF-04449913.
Of the 6 patients with MDS and 1 with CMML, 4 had stable disease after treatment. Two of these patients had hematologic improvement.
Two of the 7 patients with MF had clinical improvement.
Of the 28 patients with AML, 16 showed evidence of possible biological activity. One patient had a complete response and 4 had a partial response with incomplete hematologic recovery. Four AML patients had minor responses, and 7 had stable disease.
Given these results, PF-04449913 is now being investigated in 5 phase 2 trials of hematologic disorders, 4 of which are recruiting participants.
“Our hope is that this drug will enable more effective treatment to begin earlier and that, with earlier intervention, we can alter the course of disease and remove the need for, or improve the chances of success with, bone marrow transplantation,” Dr Jamieson said. “It’s all about reducing the burden of disease by intervening early.” ![]()
Genetic mutation identifies favorable prognosis MDS
Among patients with refractory anemia with ring sideroblasts, the presence of a common mutation in SF3B1 appears to be a marker for an indolent clinical course and favorable outcome compared to patients with wild-type SF3B1, European investigators reported.
The gene SF3B1, which encodes for a splicing factor subunit, is frequently mutated in cases of chronic lymphocytic leukemia and myelodysplastic syndromes.
“SF3B1 mutation is a major determinant of disease phenotype and clinical outcome in MDS [myelodysplastic syndrome] with ring sideroblasts. SF3B1-mutated MDS is characterized by homogeneous hematologic features, favorable prognosis, and restricted patterns of co-mutated genes and clonal evolution. Overall, these results strongly support the recognition of MDS associated with SF3B1 mutation as a distinct MDS subtype. Conversely, SF3B1-negative MDS with ring sideroblasts represents a subset with a high prevalence of TP53 mutations and worse outcome that should be taken into consideration in clinical decision-making,” the study authors conclude.
Dr. Luca Malcovati and his colleagues from the University of Pavia, Italy, and other European centers, conducted a mutational analysis of 293 patients with myeloid neoplasms and 1% or more ring sideroblasts. They found somatic mutations in SF3B1 in 129 of 159 patients with refractory anemia with ring sideroblasts (RARS) or refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS). In contrast, there was a significantly lower prevalence of SF3B1 mutations among 50 patients with myelodysplastic/myeloproliferative neoplasm (MDS/MPN), and among 84 additional patients with other myeloid diseases under the World Health Organization classification of disorders of hematopoietic and lymphoid tissues (P < .001).
In multivariable analyses controlling for demographic and disease-related factors, patients with SF3B1 mutations had significantly better overall survival (hazard ratio, 0.37; P = .003), as well as a lower cumulative incidence of disease progression (HR, 0.31; P = .018), compared with patients with wild-type SF3B1 (Blood 2015;126[2]:233-41).
Mutations in SF3B1 were predictive of better outcomes among patients with RARS, RCMD-RS, and in patients with MDS without excess blasts.
When they looked at other mutations, the investigators found that in patients with SF3B1 mutations, the mutations in DNA methylation genes were associated with the presence of multilineage dysplasia, but this association had no significant effect on clinical outcomes.
Among patients with wild-type SB3B1, mutations in TP53 were frequently seen, and these mutations were associated with poor outcomes.
Gene sequencing efforts in myeloid malignancies have largely charted the mutational “landscape.” This map allows us to (1) have some idea of the fundamental biology underlying the disease, (2) define potential drug targets, and (3) refine outcome expectations, especially when there are no “knockout” therapies (as in chronic myeloid leukemia). The consequence is also the further subclassification of myeloid malignancies, thus making relatively rare diseases into extremely rare ones. One obvious challenge is to cleverly design clinical studies given the myriad subcategories of disease. The higher bar is understanding the biology of how the various mutations and pathways merge to cause disease. The work Malcovati et al., along with the other fine studies noted above, gets us one step farther down the road to cures.
Dr. Jerald Radich of Fred Hutchinson Cancer Research Center, Seattle, made his comment in an accompanying editorial.
Gene sequencing efforts in myeloid malignancies have largely charted the mutational “landscape.” This map allows us to (1) have some idea of the fundamental biology underlying the disease, (2) define potential drug targets, and (3) refine outcome expectations, especially when there are no “knockout” therapies (as in chronic myeloid leukemia). The consequence is also the further subclassification of myeloid malignancies, thus making relatively rare diseases into extremely rare ones. One obvious challenge is to cleverly design clinical studies given the myriad subcategories of disease. The higher bar is understanding the biology of how the various mutations and pathways merge to cause disease. The work Malcovati et al., along with the other fine studies noted above, gets us one step farther down the road to cures.
Dr. Jerald Radich of Fred Hutchinson Cancer Research Center, Seattle, made his comment in an accompanying editorial.
Gene sequencing efforts in myeloid malignancies have largely charted the mutational “landscape.” This map allows us to (1) have some idea of the fundamental biology underlying the disease, (2) define potential drug targets, and (3) refine outcome expectations, especially when there are no “knockout” therapies (as in chronic myeloid leukemia). The consequence is also the further subclassification of myeloid malignancies, thus making relatively rare diseases into extremely rare ones. One obvious challenge is to cleverly design clinical studies given the myriad subcategories of disease. The higher bar is understanding the biology of how the various mutations and pathways merge to cause disease. The work Malcovati et al., along with the other fine studies noted above, gets us one step farther down the road to cures.
Dr. Jerald Radich of Fred Hutchinson Cancer Research Center, Seattle, made his comment in an accompanying editorial.
Among patients with refractory anemia with ring sideroblasts, the presence of a common mutation in SF3B1 appears to be a marker for an indolent clinical course and favorable outcome compared to patients with wild-type SF3B1, European investigators reported.
The gene SF3B1, which encodes for a splicing factor subunit, is frequently mutated in cases of chronic lymphocytic leukemia and myelodysplastic syndromes.
“SF3B1 mutation is a major determinant of disease phenotype and clinical outcome in MDS [myelodysplastic syndrome] with ring sideroblasts. SF3B1-mutated MDS is characterized by homogeneous hematologic features, favorable prognosis, and restricted patterns of co-mutated genes and clonal evolution. Overall, these results strongly support the recognition of MDS associated with SF3B1 mutation as a distinct MDS subtype. Conversely, SF3B1-negative MDS with ring sideroblasts represents a subset with a high prevalence of TP53 mutations and worse outcome that should be taken into consideration in clinical decision-making,” the study authors conclude.
Dr. Luca Malcovati and his colleagues from the University of Pavia, Italy, and other European centers, conducted a mutational analysis of 293 patients with myeloid neoplasms and 1% or more ring sideroblasts. They found somatic mutations in SF3B1 in 129 of 159 patients with refractory anemia with ring sideroblasts (RARS) or refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS). In contrast, there was a significantly lower prevalence of SF3B1 mutations among 50 patients with myelodysplastic/myeloproliferative neoplasm (MDS/MPN), and among 84 additional patients with other myeloid diseases under the World Health Organization classification of disorders of hematopoietic and lymphoid tissues (P < .001).
In multivariable analyses controlling for demographic and disease-related factors, patients with SF3B1 mutations had significantly better overall survival (hazard ratio, 0.37; P = .003), as well as a lower cumulative incidence of disease progression (HR, 0.31; P = .018), compared with patients with wild-type SF3B1 (Blood 2015;126[2]:233-41).
Mutations in SF3B1 were predictive of better outcomes among patients with RARS, RCMD-RS, and in patients with MDS without excess blasts.
When they looked at other mutations, the investigators found that in patients with SF3B1 mutations, the mutations in DNA methylation genes were associated with the presence of multilineage dysplasia, but this association had no significant effect on clinical outcomes.
Among patients with wild-type SB3B1, mutations in TP53 were frequently seen, and these mutations were associated with poor outcomes.
Among patients with refractory anemia with ring sideroblasts, the presence of a common mutation in SF3B1 appears to be a marker for an indolent clinical course and favorable outcome compared to patients with wild-type SF3B1, European investigators reported.
The gene SF3B1, which encodes for a splicing factor subunit, is frequently mutated in cases of chronic lymphocytic leukemia and myelodysplastic syndromes.
“SF3B1 mutation is a major determinant of disease phenotype and clinical outcome in MDS [myelodysplastic syndrome] with ring sideroblasts. SF3B1-mutated MDS is characterized by homogeneous hematologic features, favorable prognosis, and restricted patterns of co-mutated genes and clonal evolution. Overall, these results strongly support the recognition of MDS associated with SF3B1 mutation as a distinct MDS subtype. Conversely, SF3B1-negative MDS with ring sideroblasts represents a subset with a high prevalence of TP53 mutations and worse outcome that should be taken into consideration in clinical decision-making,” the study authors conclude.
Dr. Luca Malcovati and his colleagues from the University of Pavia, Italy, and other European centers, conducted a mutational analysis of 293 patients with myeloid neoplasms and 1% or more ring sideroblasts. They found somatic mutations in SF3B1 in 129 of 159 patients with refractory anemia with ring sideroblasts (RARS) or refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS). In contrast, there was a significantly lower prevalence of SF3B1 mutations among 50 patients with myelodysplastic/myeloproliferative neoplasm (MDS/MPN), and among 84 additional patients with other myeloid diseases under the World Health Organization classification of disorders of hematopoietic and lymphoid tissues (P < .001).
In multivariable analyses controlling for demographic and disease-related factors, patients with SF3B1 mutations had significantly better overall survival (hazard ratio, 0.37; P = .003), as well as a lower cumulative incidence of disease progression (HR, 0.31; P = .018), compared with patients with wild-type SF3B1 (Blood 2015;126[2]:233-41).
Mutations in SF3B1 were predictive of better outcomes among patients with RARS, RCMD-RS, and in patients with MDS without excess blasts.
When they looked at other mutations, the investigators found that in patients with SF3B1 mutations, the mutations in DNA methylation genes were associated with the presence of multilineage dysplasia, but this association had no significant effect on clinical outcomes.
Among patients with wild-type SB3B1, mutations in TP53 were frequently seen, and these mutations were associated with poor outcomes.
FROM BLOOD
Key clinical point: Mutations in SF3B1 identify a subset of patients with MDS with favorable prognosis.
Major finding: Patients with SF3B1 had a hazard ratio for death of 0.37, compared with patients with unmutated (wild-type) SF3B1.
Data source: Mutational analysis of 293 patients with myeloid neoplasms with 1% of more ring sideroblasts followed in centers in Italy, Sweden, and Denmark.
Disclosures: The study was supported by grants from Associazione Italiana per la Ricerca sul Cancro, Fondo per gli Investimenti della Ricerca di Base, and Ministero dell’Istruzione, dell’Università e della Ricerca PRIN 2010-2011, Fondazione Veronesi and Regione Lombardia/Fondazione Cariplo, and Associazione Italiana per la Ricerca sul Cancro IG. The authors and Dr. Radich reported no conflicts of interest.
Clonal hematopoiesis explored in aplastic anemia
Clonal hematopoiesis was detected in DNA samples from approximately half of 439 patients with aplastic anemia, and a third of the study population carried mutations in candidate genes that correlated with clinical outcomes, according to a report published online July 2 in the New England Journal of Medicine.
Most patients with aplastic anemia respond to immunosuppressive therapy or bone marrow transplantation, but about 15% later develop myelodysplastic syndromes, acute myeloid leukemia (AML), or both. Historically, this has been attributed to “clonal evolution,” but a more accurate term is clonal hematopoiesis. However, not all patients with clonal hematopoiesis go on to develop late myelodysplastic syndromes or AML, said Dr. Tetsuichi Yoshizato of the department of pathology and tumor biology at Kyoto (Japan) University and associates.
To clarify the role of clonal hematopoiesis in aplastic anemia, the investigators analyzed DNA in blood, bone marrow, and buccal samples from 439 patients with bone marrow failure who were treated at three specialized centers in the United States and Japan.
Targeted sequencing of a panel of genes that are recurrently mutated in myeloid cancers was performed; 249 mutations were detected in candidate genes for myelodysplastic syndromes/AML in 36% of the study population. And about one-third of patients whose DNA harbored mutations had multiple (as many as 7) mutations. The most frequently mutated genes were BCOR and BCORL1 (in 9.3% of patients), PIGA (7.5%), DNMT3A (8.4%), and ASXL1 (6.2%), which together accounted for 77% of all mutation-positive patients, the investigators reported.
In addition, 47% of patients had expanded hematopoietic cell clones. Clones carrying certain mutations were associated with a better response to immunosuppressive treatment, while clones carrying several other mutations were associated with a poor treatment response, lower survival, and progression to myelodysplastic syndromes/AML. Mutations in PIGA and BCOR and BCORL1 correlated with a better response to immunosuppressive therapy and better overall and progression-free survival; mutations in a subgroup of genes that included DNMT3A and ASXL1 were associated with worse outcomes.
The pattern of mutations in individual patients, however, varied markedly over time and was often unpredictable. “It should be underscored that the complex dynamics of clonal hematopoiesis are highly variable and not necessarily determinative,” Dr. Yoshizato and associates said (N. Engl. J. Med. 2015 July 2 [doi:10.1056/NEJMoa1414799]).
Although further genetic research is needed before these findings can be applied clinically to guide prognosis and treatment, they already “have implications for bone marrow failure, for early events in leukemogenesis, and for normal aging,” the investigators added.
Clonal hematopoiesis was detected in DNA samples from approximately half of 439 patients with aplastic anemia, and a third of the study population carried mutations in candidate genes that correlated with clinical outcomes, according to a report published online July 2 in the New England Journal of Medicine.
Most patients with aplastic anemia respond to immunosuppressive therapy or bone marrow transplantation, but about 15% later develop myelodysplastic syndromes, acute myeloid leukemia (AML), or both. Historically, this has been attributed to “clonal evolution,” but a more accurate term is clonal hematopoiesis. However, not all patients with clonal hematopoiesis go on to develop late myelodysplastic syndromes or AML, said Dr. Tetsuichi Yoshizato of the department of pathology and tumor biology at Kyoto (Japan) University and associates.
To clarify the role of clonal hematopoiesis in aplastic anemia, the investigators analyzed DNA in blood, bone marrow, and buccal samples from 439 patients with bone marrow failure who were treated at three specialized centers in the United States and Japan.
Targeted sequencing of a panel of genes that are recurrently mutated in myeloid cancers was performed; 249 mutations were detected in candidate genes for myelodysplastic syndromes/AML in 36% of the study population. And about one-third of patients whose DNA harbored mutations had multiple (as many as 7) mutations. The most frequently mutated genes were BCOR and BCORL1 (in 9.3% of patients), PIGA (7.5%), DNMT3A (8.4%), and ASXL1 (6.2%), which together accounted for 77% of all mutation-positive patients, the investigators reported.
In addition, 47% of patients had expanded hematopoietic cell clones. Clones carrying certain mutations were associated with a better response to immunosuppressive treatment, while clones carrying several other mutations were associated with a poor treatment response, lower survival, and progression to myelodysplastic syndromes/AML. Mutations in PIGA and BCOR and BCORL1 correlated with a better response to immunosuppressive therapy and better overall and progression-free survival; mutations in a subgroup of genes that included DNMT3A and ASXL1 were associated with worse outcomes.
The pattern of mutations in individual patients, however, varied markedly over time and was often unpredictable. “It should be underscored that the complex dynamics of clonal hematopoiesis are highly variable and not necessarily determinative,” Dr. Yoshizato and associates said (N. Engl. J. Med. 2015 July 2 [doi:10.1056/NEJMoa1414799]).
Although further genetic research is needed before these findings can be applied clinically to guide prognosis and treatment, they already “have implications for bone marrow failure, for early events in leukemogenesis, and for normal aging,” the investigators added.
Clonal hematopoiesis was detected in DNA samples from approximately half of 439 patients with aplastic anemia, and a third of the study population carried mutations in candidate genes that correlated with clinical outcomes, according to a report published online July 2 in the New England Journal of Medicine.
Most patients with aplastic anemia respond to immunosuppressive therapy or bone marrow transplantation, but about 15% later develop myelodysplastic syndromes, acute myeloid leukemia (AML), or both. Historically, this has been attributed to “clonal evolution,” but a more accurate term is clonal hematopoiesis. However, not all patients with clonal hematopoiesis go on to develop late myelodysplastic syndromes or AML, said Dr. Tetsuichi Yoshizato of the department of pathology and tumor biology at Kyoto (Japan) University and associates.
To clarify the role of clonal hematopoiesis in aplastic anemia, the investigators analyzed DNA in blood, bone marrow, and buccal samples from 439 patients with bone marrow failure who were treated at three specialized centers in the United States and Japan.
Targeted sequencing of a panel of genes that are recurrently mutated in myeloid cancers was performed; 249 mutations were detected in candidate genes for myelodysplastic syndromes/AML in 36% of the study population. And about one-third of patients whose DNA harbored mutations had multiple (as many as 7) mutations. The most frequently mutated genes were BCOR and BCORL1 (in 9.3% of patients), PIGA (7.5%), DNMT3A (8.4%), and ASXL1 (6.2%), which together accounted for 77% of all mutation-positive patients, the investigators reported.
In addition, 47% of patients had expanded hematopoietic cell clones. Clones carrying certain mutations were associated with a better response to immunosuppressive treatment, while clones carrying several other mutations were associated with a poor treatment response, lower survival, and progression to myelodysplastic syndromes/AML. Mutations in PIGA and BCOR and BCORL1 correlated with a better response to immunosuppressive therapy and better overall and progression-free survival; mutations in a subgroup of genes that included DNMT3A and ASXL1 were associated with worse outcomes.
The pattern of mutations in individual patients, however, varied markedly over time and was often unpredictable. “It should be underscored that the complex dynamics of clonal hematopoiesis are highly variable and not necessarily determinative,” Dr. Yoshizato and associates said (N. Engl. J. Med. 2015 July 2 [doi:10.1056/NEJMoa1414799]).
Although further genetic research is needed before these findings can be applied clinically to guide prognosis and treatment, they already “have implications for bone marrow failure, for early events in leukemogenesis, and for normal aging,” the investigators added.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Clonal hematopoiesis was detected in 47% of 439 patients with aplastic anemia, and some of the mutations were related to clinical outcomes.
Major finding: The most frequently mutated genes were BCOR and BCORL1 (in 9.3% of patients), PIGA (7.5%), DNMT3A (8.4%), and ASXL1 (6.2%), which together accounted for 77% of all mutation-positive patients.
Data source: DNA analysis of blood, bone marrow, and buccal samples from 439 patients with aplastic anemia treated at three medical centers in the United States and Japan.
Disclosures: This work was supported by the Ministry of Health, Labor, and Welfare of Japan; the Japan Society for the Promotion of Science; the National Heart, Lung, and Blood Institute; the Aplastic Anemia and MDS International Foundation; and the Scott Hamilton Cancer Alliance for Research, Education, and Survivorship Foundation. Dr. Yoshizato reported having no relevant financial disclosures; an associate reported receiving a grant from Daiichi-Sankyo unrelated to this work.
CD8 cell dose predicts outcomes in allogeneic stem cell transplantation with reduced-intensity conditioning
Peripheral blood stem cell (PBSC) grafts with high doses of CD8 cells were associated with significantly lower relapse risk and improved survival in patients who were treated for hematologic malignancies with reduced-intensity conditioning (RIC) hematopoietic allogeneic stem cell transplantation (allo-HSCT), according to a report online in the Journal of Clinical Oncology.
A multivariate analysis showed that CD8 cell dose was an independent predictor of relapse (adjusted hazard ratio [aHR], 0.43; P = .009), relapse-free survival (aHR, 0.50; P = .006), and overall survival (aHR, 0.57; P = .04). The data showed a linear association between CD8 cell dose and outcomes, and further analysis identified an optimum cutoff of CD8 cell dose (0.72 x 108 CD8 cells per kg) to segregate survival outcomes. Patients who received grafts with CD8 cell doses above the cutoff had significantly improved regression-free and overall survival (P = .005 and P = .007, respectively).
“These findings indicate that improved survival after RIC transplantations could be achieved by optimizing donor selection and PBSC collection to increase the likelihood of mobilizing grafts containing high CD8 cell doses,” wrote Dr. Ran Reshef of the department of medicine at the Hospital of the University of Pennsylvania, Philadelphia, and colleagues (Journ. Clin. Onc. 2015 June 8 [doi:10.1200/JCO.2014.60.1203]).
Younger donors were more likely to have CD8 cell doses above the cutoff (CD8hi), however, only 53% of donors younger than 30 years had CD8hi grafts. To find methods to predict graft composition during donor screening, the investigators studied 21 randomly selected allo-HSCT donors. They found no correlations between CD8 graft content and clinical variables such as weight, sex, viral serologies, or apheresis parameters. Donors with a higher proportion of CD8 cells donated grafts with higher CD8 cell dose, but the presence of higher CD4 counts negated this. Screening for the relative proportions of CD8 and CD4 cells identifies donors most likely to mobilize CD8hi grafts.
“This is also a practical consideration because the assay is rapid, is routinely performed in clinical laboratories, and can easily be done at the time of confirmatory HLA [human leukocyte antigen] typing,” the authors noted. Since the relationship between CD8 dose and survival is linear, the higher the dose the better, even if it is below the cutoff.
Previous studies showed conflicting results regarding the outcome of RIC transplantation with younger unrelated donors versus older sibling donors. Donor age inversely correlates with CD8 cell dose, and the results of this study showed that overall survival was significantly better with younger unrelated donors with a CD8hi graft, compared with older sibling donors (P = .03). No such benefit was observed with younger unrelated donors with CD8lo grafts (P = .28), indicating the benefit may rely on CD8 cell dose.
The study evaluated 200 patients with hematologic malignancy who underwent allo-HSCT with fludarabine plus busulfan conditioning from 2007 to 2014 at the Abramson Cancer Center, University of Pennsylvania in Philadelphia. The cumulative relapse incidence was 42% at 1 year and 47% at 5 years. The most common diseases in the cohort were acute myeloid leukemia, myelodysplastic syndrome, and non-Hodgkin lymphoma.
High CD8 dose was associated with an increased, but nonsignificant risk of chronic graft-versus-host disease (GVHD); the risk for nonrelapse mortality was not associated with cell doses.
Peripheral blood stem cell (PBSC) grafts with high doses of CD8 cells were associated with significantly lower relapse risk and improved survival in patients who were treated for hematologic malignancies with reduced-intensity conditioning (RIC) hematopoietic allogeneic stem cell transplantation (allo-HSCT), according to a report online in the Journal of Clinical Oncology.
A multivariate analysis showed that CD8 cell dose was an independent predictor of relapse (adjusted hazard ratio [aHR], 0.43; P = .009), relapse-free survival (aHR, 0.50; P = .006), and overall survival (aHR, 0.57; P = .04). The data showed a linear association between CD8 cell dose and outcomes, and further analysis identified an optimum cutoff of CD8 cell dose (0.72 x 108 CD8 cells per kg) to segregate survival outcomes. Patients who received grafts with CD8 cell doses above the cutoff had significantly improved regression-free and overall survival (P = .005 and P = .007, respectively).
“These findings indicate that improved survival after RIC transplantations could be achieved by optimizing donor selection and PBSC collection to increase the likelihood of mobilizing grafts containing high CD8 cell doses,” wrote Dr. Ran Reshef of the department of medicine at the Hospital of the University of Pennsylvania, Philadelphia, and colleagues (Journ. Clin. Onc. 2015 June 8 [doi:10.1200/JCO.2014.60.1203]).
Younger donors were more likely to have CD8 cell doses above the cutoff (CD8hi), however, only 53% of donors younger than 30 years had CD8hi grafts. To find methods to predict graft composition during donor screening, the investigators studied 21 randomly selected allo-HSCT donors. They found no correlations between CD8 graft content and clinical variables such as weight, sex, viral serologies, or apheresis parameters. Donors with a higher proportion of CD8 cells donated grafts with higher CD8 cell dose, but the presence of higher CD4 counts negated this. Screening for the relative proportions of CD8 and CD4 cells identifies donors most likely to mobilize CD8hi grafts.
“This is also a practical consideration because the assay is rapid, is routinely performed in clinical laboratories, and can easily be done at the time of confirmatory HLA [human leukocyte antigen] typing,” the authors noted. Since the relationship between CD8 dose and survival is linear, the higher the dose the better, even if it is below the cutoff.
Previous studies showed conflicting results regarding the outcome of RIC transplantation with younger unrelated donors versus older sibling donors. Donor age inversely correlates with CD8 cell dose, and the results of this study showed that overall survival was significantly better with younger unrelated donors with a CD8hi graft, compared with older sibling donors (P = .03). No such benefit was observed with younger unrelated donors with CD8lo grafts (P = .28), indicating the benefit may rely on CD8 cell dose.
The study evaluated 200 patients with hematologic malignancy who underwent allo-HSCT with fludarabine plus busulfan conditioning from 2007 to 2014 at the Abramson Cancer Center, University of Pennsylvania in Philadelphia. The cumulative relapse incidence was 42% at 1 year and 47% at 5 years. The most common diseases in the cohort were acute myeloid leukemia, myelodysplastic syndrome, and non-Hodgkin lymphoma.
High CD8 dose was associated with an increased, but nonsignificant risk of chronic graft-versus-host disease (GVHD); the risk for nonrelapse mortality was not associated with cell doses.
Peripheral blood stem cell (PBSC) grafts with high doses of CD8 cells were associated with significantly lower relapse risk and improved survival in patients who were treated for hematologic malignancies with reduced-intensity conditioning (RIC) hematopoietic allogeneic stem cell transplantation (allo-HSCT), according to a report online in the Journal of Clinical Oncology.
A multivariate analysis showed that CD8 cell dose was an independent predictor of relapse (adjusted hazard ratio [aHR], 0.43; P = .009), relapse-free survival (aHR, 0.50; P = .006), and overall survival (aHR, 0.57; P = .04). The data showed a linear association between CD8 cell dose and outcomes, and further analysis identified an optimum cutoff of CD8 cell dose (0.72 x 108 CD8 cells per kg) to segregate survival outcomes. Patients who received grafts with CD8 cell doses above the cutoff had significantly improved regression-free and overall survival (P = .005 and P = .007, respectively).
“These findings indicate that improved survival after RIC transplantations could be achieved by optimizing donor selection and PBSC collection to increase the likelihood of mobilizing grafts containing high CD8 cell doses,” wrote Dr. Ran Reshef of the department of medicine at the Hospital of the University of Pennsylvania, Philadelphia, and colleagues (Journ. Clin. Onc. 2015 June 8 [doi:10.1200/JCO.2014.60.1203]).
Younger donors were more likely to have CD8 cell doses above the cutoff (CD8hi), however, only 53% of donors younger than 30 years had CD8hi grafts. To find methods to predict graft composition during donor screening, the investigators studied 21 randomly selected allo-HSCT donors. They found no correlations between CD8 graft content and clinical variables such as weight, sex, viral serologies, or apheresis parameters. Donors with a higher proportion of CD8 cells donated grafts with higher CD8 cell dose, but the presence of higher CD4 counts negated this. Screening for the relative proportions of CD8 and CD4 cells identifies donors most likely to mobilize CD8hi grafts.
“This is also a practical consideration because the assay is rapid, is routinely performed in clinical laboratories, and can easily be done at the time of confirmatory HLA [human leukocyte antigen] typing,” the authors noted. Since the relationship between CD8 dose and survival is linear, the higher the dose the better, even if it is below the cutoff.
Previous studies showed conflicting results regarding the outcome of RIC transplantation with younger unrelated donors versus older sibling donors. Donor age inversely correlates with CD8 cell dose, and the results of this study showed that overall survival was significantly better with younger unrelated donors with a CD8hi graft, compared with older sibling donors (P = .03). No such benefit was observed with younger unrelated donors with CD8lo grafts (P = .28), indicating the benefit may rely on CD8 cell dose.
The study evaluated 200 patients with hematologic malignancy who underwent allo-HSCT with fludarabine plus busulfan conditioning from 2007 to 2014 at the Abramson Cancer Center, University of Pennsylvania in Philadelphia. The cumulative relapse incidence was 42% at 1 year and 47% at 5 years. The most common diseases in the cohort were acute myeloid leukemia, myelodysplastic syndrome, and non-Hodgkin lymphoma.
High CD8 dose was associated with an increased, but nonsignificant risk of chronic graft-versus-host disease (GVHD); the risk for nonrelapse mortality was not associated with cell doses.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: High CD8 cell dose in peripheral blood stem cell grafts was associated with reduced risk of relapse and increased survival in allogeneic stem cell transplantation with reduced-intensity conditioning.
Major finding: With a cumulative relapse incidence of 42% at 1 year, CD8 cell dose was an independent predictor of relapse (aHR, 0.43; P = .009), relapse-free survival (0.50; P = .006), and overall survival (0.57; P = .04).
Data source: The retrospective, single-institution study evaluated 200 patients who underwent peripheral blood alloHSCT with fludarabine plus busulfan conditioning from 2007 to 2014. Analysis of graft T-cell content used 21 randomly selected donors.
Disclosures: Dr. Reshef reported having consulting or advisory roles with Celgene, Spectrum Pharmaceuticals, Tobira Therapeutics, and Teva Pharmaceutical Industries. Many of his coauthors reported having financial relationships with industry.
ASCO: PERSIST-1 – pacritinib tops best available therapy in myelofibrosis
CHICAGO – Pacritinib, an investigational oral inhibitor of Janus kinase 2 (JAK2), reduced splenomegaly and alleviated other symptoms in patients with myelofibrosis, in a randomized phase III trial reported at the annual meeting of the American Society of Clinical Oncology.
After 24 weeks of treatment, patients in the pacritinib arm were about four times more likely to have a sizable reduction in spleen volume than peers in the best available therapy arm, reported lead study author Dr. Ruben A. Mesa, deputy director of the Mayo Clinic Cancer Center in Scottsdale, Arizona.
Of special note, the drug was not associated with increased anemia or thrombocytopenia. In fact, it was safe in the subset of patients who had thrombocytopenia at baseline, a group currently having an unmet need for treatments because they cannot receive ruxolitinib (Jakafi), a dual JAK1 and JAK2 inhibitor that is associated with thrombocytopenia.

“Based on these preliminary results, pacritinib may represent a very important agent for individuals with advanced disease and may have impact on the disease course,” Dr. Mesa commented. Additionally, the findings warrant studies of combination therapy with other potentially disease-modifying agents in myeloproliferative neoplasms.
“I think pacritinib for myelofibrosis represents an advance in our field,” commented invited discussant Dr. Lloyd E. Damon, a professor of medicine and director of hematologic malignancies and bone marrow transplant at the University of California, San Francisco.
The trial’s findings have a number of implications going forward, he said. “There are several avenues yet to explore with these types of agents; for instance, what is the role of JAK inhibitors in those who are actually JAK2 mutated, and for that matter, those who are actually calreticulin mutated, and for that matter, those for whom there is no known mutation? The JAK inhibitors so far are directed against wild type,” he noted. “And should we be seeking to develop agents which are very specific against JAK gene products that are mutated or calreticulin gene products that are mutated vis-a-vis what we are seeing in the FLT3 inhibitors?”
Session attendee Dr. Harry Erba, a professor of medicine and director of the hematologic malignancy program, University of Alabama at Birmingham, commented, “With ruxolitinib, I’d always assumed that the improvement in quality of life was due to the JAK1 inhibition and decreasing of inflammatory cytokine signaling. In this study, the benefit in terms of the total symptom score was maybe a little bit less robust, comparing two very different studies – only 20% or 25%.”
“I think when it comes to the mechanism of symptom improvement, JAK2 probably still remains a key piece. As we look at the entire portfolio of JAK inhibitors that have been tested, we see improvement in symptoms whether they hit JAK1 or not,” Dr. Mesa replied.
“Was there a difference in the responses between the spleen-related and the more inflammatory-related symptoms?” Dr. Erba further asked. “Also, the other thing I was struck by, with ruxolitinib, it seems to be such a quick response in terms of quality of life and symptoms, and here there just seemed to be a more gradual improvement in time.”
“We did not see a strong difference between spleen- and non–spleen-related improvements,” Dr. Mesa replied, and the majority of responses seen with pacritinib were still “fairly rapid,” occurring within 4-8 weeks.
The CTI-funded trial – known as PERSIST-1 (A Randomized Controlled Phase 3 Study of Oral Pacritinib Versus Best Available Therapy in Patients with Primary Myelofibrosis, Post-Polycythemia Vera Myelofibrosis, or Post–Essential Thrombocythemia Myelofibrosis) – was unique in allowing patients to enroll regardless of platelet count, Dr. Mesa noted.
The patients were randomized in 2:1 ratio to receive pacritinib or best available therapy. The latter typically consisted of off-label agents such as erythropoietin-stimulating agents, immunomodulatory drugs, and hydroxyurea; ruxolitinib was not permitted. Crossover was allowed, and 79% of patients in the best available therapy arm eventually did go on to receive pacritinib.
Median follow-up was 8.4 months. In intention-to-treat analyses, at 24 weeks, 19.1% of patients in the pacritinib arm had a reduction of at least 35% in spleen volume, compared with only 4.7% in the best available therapy arm (P = .0003). The findings were similar in the subsets with a platelet count of less than 50,000 per microliter (22.9% vs. 0%) and less than 100,000 per microliter (16.7% vs. 0%).
Patients in the pacritinib arm were less likely to die if they had a spleen volume reduction of at least 20%, but longer follow-up is needed to determine if the drug improves survival, according to Dr. Mesa, who disclosed that he receives honoraria from and has a consulting or advisory role with Novartis, and receives research funding from Celgene, CTI, Incyte, and Gilead Sciences.
The proportion of patients having at least a one-half reduction in Total Symptom Score was 24.5% with pacritinib and 6.5% with best available therapy (P < .0001).
“We did not see any significant drug-emergent thrombocytopenia,” Dr. Mesa reported. In fact, among patients who entered the trial with a platelet count of less than 50,000 per microliter, those in the pacritinib arm had a significant, steady improvement in platelet count. “This could be multifactorial, from reduced splenic sequestration amongst other beneficial features,” he proposed
Among patients who were red cell transfusion dependent at baseline, 25.7% in the pacritinib arm achieved transfusion independence, compared with none in the control arm (P = .04).
The most common nonhematologic grade 3 or 4 adverse event with pacritinib was diarrhea (5% vs. 0%), while the most common hematologic grade 3 or 4 adverse event was anemia (16.8% vs. 15.1%).
Dr. Mesa noted that an ongoing sister trial, PERSIST-2, is still open to accrual. “This is a trial exclusively for patients with thrombocytopenia and allows individuals who have previously received JAK inhibitor therapy, with patients being randomized to the dose tested in the PERSIST-1 study or a b.i.d. dosing with similar goals and endpoints,” he elaborated.
CHICAGO – Pacritinib, an investigational oral inhibitor of Janus kinase 2 (JAK2), reduced splenomegaly and alleviated other symptoms in patients with myelofibrosis, in a randomized phase III trial reported at the annual meeting of the American Society of Clinical Oncology.
After 24 weeks of treatment, patients in the pacritinib arm were about four times more likely to have a sizable reduction in spleen volume than peers in the best available therapy arm, reported lead study author Dr. Ruben A. Mesa, deputy director of the Mayo Clinic Cancer Center in Scottsdale, Arizona.
Of special note, the drug was not associated with increased anemia or thrombocytopenia. In fact, it was safe in the subset of patients who had thrombocytopenia at baseline, a group currently having an unmet need for treatments because they cannot receive ruxolitinib (Jakafi), a dual JAK1 and JAK2 inhibitor that is associated with thrombocytopenia.
“Based on these preliminary results, pacritinib may represent a very important agent for individuals with advanced disease and may have impact on the disease course,” Dr. Mesa commented. Additionally, the findings warrant studies of combination therapy with other potentially disease-modifying agents in myeloproliferative neoplasms.
“I think pacritinib for myelofibrosis represents an advance in our field,” commented invited discussant Dr. Lloyd E. Damon, a professor of medicine and director of hematologic malignancies and bone marrow transplant at the University of California, San Francisco.
The trial’s findings have a number of implications going forward, he said. “There are several avenues yet to explore with these types of agents; for instance, what is the role of JAK inhibitors in those who are actually JAK2 mutated, and for that matter, those who are actually calreticulin mutated, and for that matter, those for whom there is no known mutation? The JAK inhibitors so far are directed against wild type,” he noted. “And should we be seeking to develop agents which are very specific against JAK gene products that are mutated or calreticulin gene products that are mutated vis-a-vis what we are seeing in the FLT3 inhibitors?”
Session attendee Dr. Harry Erba, a professor of medicine and director of the hematologic malignancy program, University of Alabama at Birmingham, commented, “With ruxolitinib, I’d always assumed that the improvement in quality of life was due to the JAK1 inhibition and decreasing of inflammatory cytokine signaling. In this study, the benefit in terms of the total symptom score was maybe a little bit less robust, comparing two very different studies – only 20% or 25%.”
“I think when it comes to the mechanism of symptom improvement, JAK2 probably still remains a key piece. As we look at the entire portfolio of JAK inhibitors that have been tested, we see improvement in symptoms whether they hit JAK1 or not,” Dr. Mesa replied.
“Was there a difference in the responses between the spleen-related and the more inflammatory-related symptoms?” Dr. Erba further asked. “Also, the other thing I was struck by, with ruxolitinib, it seems to be such a quick response in terms of quality of life and symptoms, and here there just seemed to be a more gradual improvement in time.”
“We did not see a strong difference between spleen- and non–spleen-related improvements,” Dr. Mesa replied, and the majority of responses seen with pacritinib were still “fairly rapid,” occurring within 4-8 weeks.
The CTI-funded trial – known as PERSIST-1 (A Randomized Controlled Phase 3 Study of Oral Pacritinib Versus Best Available Therapy in Patients with Primary Myelofibrosis, Post-Polycythemia Vera Myelofibrosis, or Post–Essential Thrombocythemia Myelofibrosis) – was unique in allowing patients to enroll regardless of platelet count, Dr. Mesa noted.
The patients were randomized in 2:1 ratio to receive pacritinib or best available therapy. The latter typically consisted of off-label agents such as erythropoietin-stimulating agents, immunomodulatory drugs, and hydroxyurea; ruxolitinib was not permitted. Crossover was allowed, and 79% of patients in the best available therapy arm eventually did go on to receive pacritinib.
Median follow-up was 8.4 months. In intention-to-treat analyses, at 24 weeks, 19.1% of patients in the pacritinib arm had a reduction of at least 35% in spleen volume, compared with only 4.7% in the best available therapy arm (P = .0003). The findings were similar in the subsets with a platelet count of less than 50,000 per microliter (22.9% vs. 0%) and less than 100,000 per microliter (16.7% vs. 0%).
Patients in the pacritinib arm were less likely to die if they had a spleen volume reduction of at least 20%, but longer follow-up is needed to determine if the drug improves survival, according to Dr. Mesa, who disclosed that he receives honoraria from and has a consulting or advisory role with Novartis, and receives research funding from Celgene, CTI, Incyte, and Gilead Sciences.
The proportion of patients having at least a one-half reduction in Total Symptom Score was 24.5% with pacritinib and 6.5% with best available therapy (P < .0001).
“We did not see any significant drug-emergent thrombocytopenia,” Dr. Mesa reported. In fact, among patients who entered the trial with a platelet count of less than 50,000 per microliter, those in the pacritinib arm had a significant, steady improvement in platelet count. “This could be multifactorial, from reduced splenic sequestration amongst other beneficial features,” he proposed
Among patients who were red cell transfusion dependent at baseline, 25.7% in the pacritinib arm achieved transfusion independence, compared with none in the control arm (P = .04).
The most common nonhematologic grade 3 or 4 adverse event with pacritinib was diarrhea (5% vs. 0%), while the most common hematologic grade 3 or 4 adverse event was anemia (16.8% vs. 15.1%).
Dr. Mesa noted that an ongoing sister trial, PERSIST-2, is still open to accrual. “This is a trial exclusively for patients with thrombocytopenia and allows individuals who have previously received JAK inhibitor therapy, with patients being randomized to the dose tested in the PERSIST-1 study or a b.i.d. dosing with similar goals and endpoints,” he elaborated.
CHICAGO – Pacritinib, an investigational oral inhibitor of Janus kinase 2 (JAK2), reduced splenomegaly and alleviated other symptoms in patients with myelofibrosis, in a randomized phase III trial reported at the annual meeting of the American Society of Clinical Oncology.
After 24 weeks of treatment, patients in the pacritinib arm were about four times more likely to have a sizable reduction in spleen volume than peers in the best available therapy arm, reported lead study author Dr. Ruben A. Mesa, deputy director of the Mayo Clinic Cancer Center in Scottsdale, Arizona.
Of special note, the drug was not associated with increased anemia or thrombocytopenia. In fact, it was safe in the subset of patients who had thrombocytopenia at baseline, a group currently having an unmet need for treatments because they cannot receive ruxolitinib (Jakafi), a dual JAK1 and JAK2 inhibitor that is associated with thrombocytopenia.
“Based on these preliminary results, pacritinib may represent a very important agent for individuals with advanced disease and may have impact on the disease course,” Dr. Mesa commented. Additionally, the findings warrant studies of combination therapy with other potentially disease-modifying agents in myeloproliferative neoplasms.
“I think pacritinib for myelofibrosis represents an advance in our field,” commented invited discussant Dr. Lloyd E. Damon, a professor of medicine and director of hematologic malignancies and bone marrow transplant at the University of California, San Francisco.
The trial’s findings have a number of implications going forward, he said. “There are several avenues yet to explore with these types of agents; for instance, what is the role of JAK inhibitors in those who are actually JAK2 mutated, and for that matter, those who are actually calreticulin mutated, and for that matter, those for whom there is no known mutation? The JAK inhibitors so far are directed against wild type,” he noted. “And should we be seeking to develop agents which are very specific against JAK gene products that are mutated or calreticulin gene products that are mutated vis-a-vis what we are seeing in the FLT3 inhibitors?”
Session attendee Dr. Harry Erba, a professor of medicine and director of the hematologic malignancy program, University of Alabama at Birmingham, commented, “With ruxolitinib, I’d always assumed that the improvement in quality of life was due to the JAK1 inhibition and decreasing of inflammatory cytokine signaling. In this study, the benefit in terms of the total symptom score was maybe a little bit less robust, comparing two very different studies – only 20% or 25%.”
“I think when it comes to the mechanism of symptom improvement, JAK2 probably still remains a key piece. As we look at the entire portfolio of JAK inhibitors that have been tested, we see improvement in symptoms whether they hit JAK1 or not,” Dr. Mesa replied.
“Was there a difference in the responses between the spleen-related and the more inflammatory-related symptoms?” Dr. Erba further asked. “Also, the other thing I was struck by, with ruxolitinib, it seems to be such a quick response in terms of quality of life and symptoms, and here there just seemed to be a more gradual improvement in time.”
“We did not see a strong difference between spleen- and non–spleen-related improvements,” Dr. Mesa replied, and the majority of responses seen with pacritinib were still “fairly rapid,” occurring within 4-8 weeks.
The CTI-funded trial – known as PERSIST-1 (A Randomized Controlled Phase 3 Study of Oral Pacritinib Versus Best Available Therapy in Patients with Primary Myelofibrosis, Post-Polycythemia Vera Myelofibrosis, or Post–Essential Thrombocythemia Myelofibrosis) – was unique in allowing patients to enroll regardless of platelet count, Dr. Mesa noted.
The patients were randomized in 2:1 ratio to receive pacritinib or best available therapy. The latter typically consisted of off-label agents such as erythropoietin-stimulating agents, immunomodulatory drugs, and hydroxyurea; ruxolitinib was not permitted. Crossover was allowed, and 79% of patients in the best available therapy arm eventually did go on to receive pacritinib.
Median follow-up was 8.4 months. In intention-to-treat analyses, at 24 weeks, 19.1% of patients in the pacritinib arm had a reduction of at least 35% in spleen volume, compared with only 4.7% in the best available therapy arm (P = .0003). The findings were similar in the subsets with a platelet count of less than 50,000 per microliter (22.9% vs. 0%) and less than 100,000 per microliter (16.7% vs. 0%).
Patients in the pacritinib arm were less likely to die if they had a spleen volume reduction of at least 20%, but longer follow-up is needed to determine if the drug improves survival, according to Dr. Mesa, who disclosed that he receives honoraria from and has a consulting or advisory role with Novartis, and receives research funding from Celgene, CTI, Incyte, and Gilead Sciences.
The proportion of patients having at least a one-half reduction in Total Symptom Score was 24.5% with pacritinib and 6.5% with best available therapy (P < .0001).
“We did not see any significant drug-emergent thrombocytopenia,” Dr. Mesa reported. In fact, among patients who entered the trial with a platelet count of less than 50,000 per microliter, those in the pacritinib arm had a significant, steady improvement in platelet count. “This could be multifactorial, from reduced splenic sequestration amongst other beneficial features,” he proposed
Among patients who were red cell transfusion dependent at baseline, 25.7% in the pacritinib arm achieved transfusion independence, compared with none in the control arm (P = .04).
The most common nonhematologic grade 3 or 4 adverse event with pacritinib was diarrhea (5% vs. 0%), while the most common hematologic grade 3 or 4 adverse event was anemia (16.8% vs. 15.1%).
Dr. Mesa noted that an ongoing sister trial, PERSIST-2, is still open to accrual. “This is a trial exclusively for patients with thrombocytopenia and allows individuals who have previously received JAK inhibitor therapy, with patients being randomized to the dose tested in the PERSIST-1 study or a b.i.d. dosing with similar goals and endpoints,” he elaborated.
AT THE 2015 ASCO ANNUAL MEETING
Key clinical point: Pacritinib is superior to best available therapy for alleviating splenomegaly and other symptoms of myelofibrosis.
Major finding: Patients were more likely to have a 35% or greater reduction in spleen volume with pacritinib (19.1% vs. 4.7%).
Data source: A randomized phase III trial in 327 patients with myelofibrosis or similar neoplasias.
Disclosures: Dr. Mesa disclosed that he receives honoraria from and has a consulting or advisory role with Novartis, and receives research funding from Celgene, CTI, Incyte, and Gilead Sciences. The trial was funded by CTI.

Dexrazoxane Tx did not affect overall survival in pediatric leukemia and lymphoma
Exposure to dexrazoxane among pediatric patients with leukemia or lymphoma did not affect overall mortality during a median follow-up period of 12.6 years, according to a report published online in the Journal of Clinical Oncology.
Aggregated data from three Children’s Oncology Group trials showed that among 1,008 pediatric patients who received treatment with doxorubicin with or without dexrazoxane (DRZ) from 1996 to 2001, exposure to DRZ was not associated with an increased risk of relapse (HR, 0.81; 95% CI, 0.60-1.08) or death (HR, 1.03; 0.73-1.45). Comparing DRZ with non-DRZ treatment groups at 10 years, the cumulative incidence of relapse was 16.1% vs. 19.1% (difference, – 3.0%; 95% CI, – 7.9% to 0.2%) and overall mortality was 12.8% vs. 12.2% (difference, – 0.6%; 95% CI, – 3.5% to 4.7%). The three trials (P9404, P9425, and P9426) evaluated individually likewise did not show significant differences in relapse or mortality rates.
Although studies in adults show a positive effect of DRZ on heart failure rates after anthracycline therapy, concern over DRZ interference with cancer therapies and a possible link to second cancers have limited its use in children and prompted Dr. Eric Chow of the Fred Hutchinson Cancer Research Center, Seattle, and his colleagues to assess the effect of DRZ on mortality.
The investigators wrote that DRZ “does not appear to interfere with cancer treatment efficacy, in terms of original cancer mortality or overall risk of relapse. Although the risk for secondary cancer mortality (mainly as a result of AML/MDS [acute myeloid leukemia/myelodysplastic syndrome]) was greater among those exposed to DRZ, the overall number of events was small, and the differences were not statistically significant,” the investigators said. (J. Clin. Oncol. 2015 May 26 [doi:10.1200/JCO.2014.59.4473])
Aggregated data from the three trials shows that the 10-year mortality rate of AML/MDS was 1.4% for those treated with DRZ (seven patients), compared with 0.8% for those treated without DRZ (five patients).
The beneficial effects of DRZ in decreasing the risk of heart failure have been observed in trials of adult patients, but the results for survivors of childhood cancers have been inconclusive because heart failure may develop over a longer time period in children. With the median age of survivors in this study of 24 years, significant differences in cardiac mortality due to DRZ use are not detectable. To evaluate DRZ as a cardioprotectant, a new Children’s Oncology Group study (Effects of Dexrazoxane Hydrochloride on Biomarkers Associated With Cardiomyopathy and Heart Failure After Cancer Treatment [HEART]) will determine the cardiovascular health of individuals in the three trials P9404, P9425, and P9426.
“Given that second cancers and symptomatic cardiac disease appear to be by far the two most common categories of serious late effects (in terms of both absolute and relative risks) among long-term childhood cancer survivors as a group … with cumulative incidences of each approaching 20% by age 50 years, any strategy that offers the promise of reduced cardiotoxicity without being offset by second cancers is highly attractive,” Dr. Chow and his associates wrote.
The study was supported by the National Institutes of Health, St. Baldrick’s Foundation, and the Leukemia and Lymphoma Society. Dr. Chow reported having no relevant financial conflicts. Three of his coauthors reported having financial relationships with industry.
Exposure to dexrazoxane among pediatric patients with leukemia or lymphoma did not affect overall mortality during a median follow-up period of 12.6 years, according to a report published online in the Journal of Clinical Oncology.
Aggregated data from three Children’s Oncology Group trials showed that among 1,008 pediatric patients who received treatment with doxorubicin with or without dexrazoxane (DRZ) from 1996 to 2001, exposure to DRZ was not associated with an increased risk of relapse (HR, 0.81; 95% CI, 0.60-1.08) or death (HR, 1.03; 0.73-1.45). Comparing DRZ with non-DRZ treatment groups at 10 years, the cumulative incidence of relapse was 16.1% vs. 19.1% (difference, – 3.0%; 95% CI, – 7.9% to 0.2%) and overall mortality was 12.8% vs. 12.2% (difference, – 0.6%; 95% CI, – 3.5% to 4.7%). The three trials (P9404, P9425, and P9426) evaluated individually likewise did not show significant differences in relapse or mortality rates.
Although studies in adults show a positive effect of DRZ on heart failure rates after anthracycline therapy, concern over DRZ interference with cancer therapies and a possible link to second cancers have limited its use in children and prompted Dr. Eric Chow of the Fred Hutchinson Cancer Research Center, Seattle, and his colleagues to assess the effect of DRZ on mortality.
The investigators wrote that DRZ “does not appear to interfere with cancer treatment efficacy, in terms of original cancer mortality or overall risk of relapse. Although the risk for secondary cancer mortality (mainly as a result of AML/MDS [acute myeloid leukemia/myelodysplastic syndrome]) was greater among those exposed to DRZ, the overall number of events was small, and the differences were not statistically significant,” the investigators said. (J. Clin. Oncol. 2015 May 26 [doi:10.1200/JCO.2014.59.4473])
Aggregated data from the three trials shows that the 10-year mortality rate of AML/MDS was 1.4% for those treated with DRZ (seven patients), compared with 0.8% for those treated without DRZ (five patients).
The beneficial effects of DRZ in decreasing the risk of heart failure have been observed in trials of adult patients, but the results for survivors of childhood cancers have been inconclusive because heart failure may develop over a longer time period in children. With the median age of survivors in this study of 24 years, significant differences in cardiac mortality due to DRZ use are not detectable. To evaluate DRZ as a cardioprotectant, a new Children’s Oncology Group study (Effects of Dexrazoxane Hydrochloride on Biomarkers Associated With Cardiomyopathy and Heart Failure After Cancer Treatment [HEART]) will determine the cardiovascular health of individuals in the three trials P9404, P9425, and P9426.
“Given that second cancers and symptomatic cardiac disease appear to be by far the two most common categories of serious late effects (in terms of both absolute and relative risks) among long-term childhood cancer survivors as a group … with cumulative incidences of each approaching 20% by age 50 years, any strategy that offers the promise of reduced cardiotoxicity without being offset by second cancers is highly attractive,” Dr. Chow and his associates wrote.
The study was supported by the National Institutes of Health, St. Baldrick’s Foundation, and the Leukemia and Lymphoma Society. Dr. Chow reported having no relevant financial conflicts. Three of his coauthors reported having financial relationships with industry.
Exposure to dexrazoxane among pediatric patients with leukemia or lymphoma did not affect overall mortality during a median follow-up period of 12.6 years, according to a report published online in the Journal of Clinical Oncology.
Aggregated data from three Children’s Oncology Group trials showed that among 1,008 pediatric patients who received treatment with doxorubicin with or without dexrazoxane (DRZ) from 1996 to 2001, exposure to DRZ was not associated with an increased risk of relapse (HR, 0.81; 95% CI, 0.60-1.08) or death (HR, 1.03; 0.73-1.45). Comparing DRZ with non-DRZ treatment groups at 10 years, the cumulative incidence of relapse was 16.1% vs. 19.1% (difference, – 3.0%; 95% CI, – 7.9% to 0.2%) and overall mortality was 12.8% vs. 12.2% (difference, – 0.6%; 95% CI, – 3.5% to 4.7%). The three trials (P9404, P9425, and P9426) evaluated individually likewise did not show significant differences in relapse or mortality rates.
Although studies in adults show a positive effect of DRZ on heart failure rates after anthracycline therapy, concern over DRZ interference with cancer therapies and a possible link to second cancers have limited its use in children and prompted Dr. Eric Chow of the Fred Hutchinson Cancer Research Center, Seattle, and his colleagues to assess the effect of DRZ on mortality.
The investigators wrote that DRZ “does not appear to interfere with cancer treatment efficacy, in terms of original cancer mortality or overall risk of relapse. Although the risk for secondary cancer mortality (mainly as a result of AML/MDS [acute myeloid leukemia/myelodysplastic syndrome]) was greater among those exposed to DRZ, the overall number of events was small, and the differences were not statistically significant,” the investigators said. (J. Clin. Oncol. 2015 May 26 [doi:10.1200/JCO.2014.59.4473])
Aggregated data from the three trials shows that the 10-year mortality rate of AML/MDS was 1.4% for those treated with DRZ (seven patients), compared with 0.8% for those treated without DRZ (five patients).
The beneficial effects of DRZ in decreasing the risk of heart failure have been observed in trials of adult patients, but the results for survivors of childhood cancers have been inconclusive because heart failure may develop over a longer time period in children. With the median age of survivors in this study of 24 years, significant differences in cardiac mortality due to DRZ use are not detectable. To evaluate DRZ as a cardioprotectant, a new Children’s Oncology Group study (Effects of Dexrazoxane Hydrochloride on Biomarkers Associated With Cardiomyopathy and Heart Failure After Cancer Treatment [HEART]) will determine the cardiovascular health of individuals in the three trials P9404, P9425, and P9426.
“Given that second cancers and symptomatic cardiac disease appear to be by far the two most common categories of serious late effects (in terms of both absolute and relative risks) among long-term childhood cancer survivors as a group … with cumulative incidences of each approaching 20% by age 50 years, any strategy that offers the promise of reduced cardiotoxicity without being offset by second cancers is highly attractive,” Dr. Chow and his associates wrote.
The study was supported by the National Institutes of Health, St. Baldrick’s Foundation, and the Leukemia and Lymphoma Society. Dr. Chow reported having no relevant financial conflicts. Three of his coauthors reported having financial relationships with industry.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Treatment with dexrazoxane was not associated with an increased risk for cancer relapse or death.
Major finding: For pediatric patients with leukemia and lymphoma, the cumulative incidence of relapse at 10 years was 16.1% with DRZ, compared with 19.1% without DRZ (difference, – 3.0%; 95% CI, – 7.9% to 0.2%); overall mortality was 12.8% with DRZ vs. 12.2% without DRZ (difference, – 0.6%; 95% CI, – 3.5% to 4.7%).
Data source: Aggregated Children’s Oncology Group trials enrolling 1,008 pediatric patients with leukemia or lymphoma who were randomized to receive doxorubicin with or without DRZ from 1996 to 2001.
Disclosures: The study was supported by the National Institutes of Health, St. Baldrick’s Foundation, and the Leukemia and Lymphoma Society. Dr. Chow reported having no relevant financial conflicts. Three of his coauthors reported having financial relationships with industry.
Team links telomere degeneration and MDS
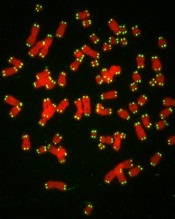
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
Drug can alleviate transfusion dependence in non-del-5q MDS

Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Inhibitor improves OS in poor-prognosis MDS

WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
Drug shows promise for lower-risk MDS

WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()