User login
Novel AKT inhibitor active against MM cells
A novel inhibitor of AKT pathway signaling showed significant cytotoxic activity in mouse models and in human cells isolated from patients with primary or relapsed multiple myeloma (MM), investigators reported.
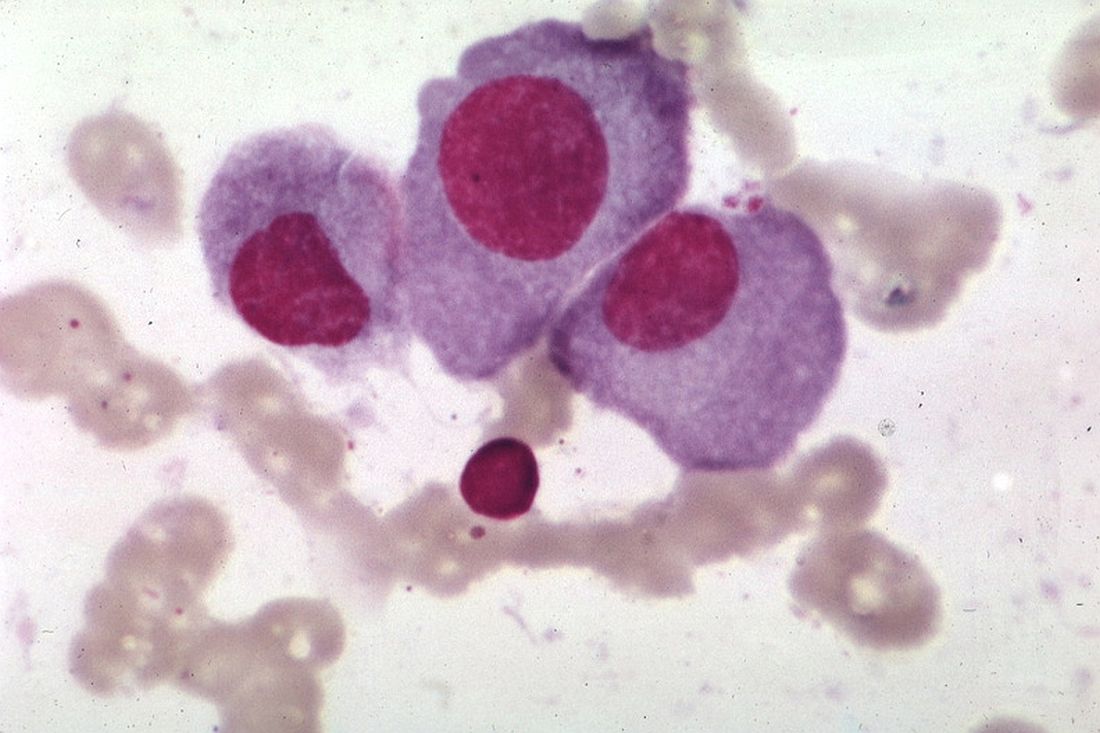
The experimental agent, labeled HS1793, is a derivative of the naturally occurring antioxidant compound resveratrol. In preclinical studies, HS1793 was shown to offer “great promise in eliminating MM cells and improving therapeutic responses in primary and relapsed/refractory MM patients,” according to Jin Han, MD, PhD, of Inje University in Busan, South Korea, and colleagues.
In a series of experiments, described in the journal Cancer Letters, the investigators demonstrated that HS1793 decreased AKT signaling to induce mitochondria-mediated cell death in multiple myeloma cells, and was cytotoxic and specific for myeloma cells in a mouse model of human metastatic myeloma, and in samples of human multiple myeloma cells.
When activated, AKT promotes oncogenesis by in turn activating other downstream pathways involved in proliferation or survival of malignant cells.
“AKT is frequently activated in MM cells and the incidence of AKT activation correlates positively with disease activity,” the authors noted.
They first screened 400 compounds, and narrowed in on resveratrol analogs, eventually choosing HS1793 as the most promising candidate.
This first experiment found evidence that suggested that the compound inhibits AKT activation by interfering with the interaction between AKT and its promoter HSP90.
They then showed in human MM cell lines that the antimyeloma action of HS1793 appeared to be from a dose-dependent effect that allowed for mitochondria-mediated programmed cell death.
In a separate series of experiments, they found that the inhibition by HS1793 of AKT/HSP90 interaction results in cell death by suppressing nuclear factor kappa–B (NF-KB) pathway signaling. The investigators had previously reported that a different compound, an inhibitor of spindle protein kinesin, induced MM cell death via inhibition of NF-KB signaling.
Next, the investigators showed that HS1793-induced cell death was caused by the direct inhibition of AKT that in turn suppressed NF-KB activation.
Finally, they showed in a mouse model of multiple myeloma metastatic to bone that HS1793 “dramatically decreased” lytic skull and femur lesions in treated mice, compared with mice treated with a vehicle placebo, and increased survival of the mice that received the AKT inhibitor.
They also showed that HS1793 was cytotoxic to multiple myeloma cells but not to normal plasma cells isolated from patients with MM.
“Given that HS1793 treatment specifically induced the death of primary and relapsed MM cells, HS1793 offers excellent translational potential as a novel MM therapy,” they wrote.
The study was supported by grants from the Korean government. The researchers reported having no potential conflicts of interest.
SOURCE: Song IS et al. Cancer Lett. 2018;432:205-15.
A novel inhibitor of AKT pathway signaling showed significant cytotoxic activity in mouse models and in human cells isolated from patients with primary or relapsed multiple myeloma (MM), investigators reported.
The experimental agent, labeled HS1793, is a derivative of the naturally occurring antioxidant compound resveratrol. In preclinical studies, HS1793 was shown to offer “great promise in eliminating MM cells and improving therapeutic responses in primary and relapsed/refractory MM patients,” according to Jin Han, MD, PhD, of Inje University in Busan, South Korea, and colleagues.
In a series of experiments, described in the journal Cancer Letters, the investigators demonstrated that HS1793 decreased AKT signaling to induce mitochondria-mediated cell death in multiple myeloma cells, and was cytotoxic and specific for myeloma cells in a mouse model of human metastatic myeloma, and in samples of human multiple myeloma cells.
When activated, AKT promotes oncogenesis by in turn activating other downstream pathways involved in proliferation or survival of malignant cells.
“AKT is frequently activated in MM cells and the incidence of AKT activation correlates positively with disease activity,” the authors noted.
They first screened 400 compounds, and narrowed in on resveratrol analogs, eventually choosing HS1793 as the most promising candidate.
This first experiment found evidence that suggested that the compound inhibits AKT activation by interfering with the interaction between AKT and its promoter HSP90.
They then showed in human MM cell lines that the antimyeloma action of HS1793 appeared to be from a dose-dependent effect that allowed for mitochondria-mediated programmed cell death.
In a separate series of experiments, they found that the inhibition by HS1793 of AKT/HSP90 interaction results in cell death by suppressing nuclear factor kappa–B (NF-KB) pathway signaling. The investigators had previously reported that a different compound, an inhibitor of spindle protein kinesin, induced MM cell death via inhibition of NF-KB signaling.
Next, the investigators showed that HS1793-induced cell death was caused by the direct inhibition of AKT that in turn suppressed NF-KB activation.
Finally, they showed in a mouse model of multiple myeloma metastatic to bone that HS1793 “dramatically decreased” lytic skull and femur lesions in treated mice, compared with mice treated with a vehicle placebo, and increased survival of the mice that received the AKT inhibitor.
They also showed that HS1793 was cytotoxic to multiple myeloma cells but not to normal plasma cells isolated from patients with MM.
“Given that HS1793 treatment specifically induced the death of primary and relapsed MM cells, HS1793 offers excellent translational potential as a novel MM therapy,” they wrote.
The study was supported by grants from the Korean government. The researchers reported having no potential conflicts of interest.
SOURCE: Song IS et al. Cancer Lett. 2018;432:205-15.
A novel inhibitor of AKT pathway signaling showed significant cytotoxic activity in mouse models and in human cells isolated from patients with primary or relapsed multiple myeloma (MM), investigators reported.
The experimental agent, labeled HS1793, is a derivative of the naturally occurring antioxidant compound resveratrol. In preclinical studies, HS1793 was shown to offer “great promise in eliminating MM cells and improving therapeutic responses in primary and relapsed/refractory MM patients,” according to Jin Han, MD, PhD, of Inje University in Busan, South Korea, and colleagues.
In a series of experiments, described in the journal Cancer Letters, the investigators demonstrated that HS1793 decreased AKT signaling to induce mitochondria-mediated cell death in multiple myeloma cells, and was cytotoxic and specific for myeloma cells in a mouse model of human metastatic myeloma, and in samples of human multiple myeloma cells.
When activated, AKT promotes oncogenesis by in turn activating other downstream pathways involved in proliferation or survival of malignant cells.
“AKT is frequently activated in MM cells and the incidence of AKT activation correlates positively with disease activity,” the authors noted.
They first screened 400 compounds, and narrowed in on resveratrol analogs, eventually choosing HS1793 as the most promising candidate.
This first experiment found evidence that suggested that the compound inhibits AKT activation by interfering with the interaction between AKT and its promoter HSP90.
They then showed in human MM cell lines that the antimyeloma action of HS1793 appeared to be from a dose-dependent effect that allowed for mitochondria-mediated programmed cell death.
In a separate series of experiments, they found that the inhibition by HS1793 of AKT/HSP90 interaction results in cell death by suppressing nuclear factor kappa–B (NF-KB) pathway signaling. The investigators had previously reported that a different compound, an inhibitor of spindle protein kinesin, induced MM cell death via inhibition of NF-KB signaling.
Next, the investigators showed that HS1793-induced cell death was caused by the direct inhibition of AKT that in turn suppressed NF-KB activation.
Finally, they showed in a mouse model of multiple myeloma metastatic to bone that HS1793 “dramatically decreased” lytic skull and femur lesions in treated mice, compared with mice treated with a vehicle placebo, and increased survival of the mice that received the AKT inhibitor.
They also showed that HS1793 was cytotoxic to multiple myeloma cells but not to normal plasma cells isolated from patients with MM.
“Given that HS1793 treatment specifically induced the death of primary and relapsed MM cells, HS1793 offers excellent translational potential as a novel MM therapy,” they wrote.
The study was supported by grants from the Korean government. The researchers reported having no potential conflicts of interest.
SOURCE: Song IS et al. Cancer Lett. 2018;432:205-15.
FROM CANCER LETTERS
Key clinical point:
Major finding: HS1793 showed significant multiple myeloma cytotoxicity in mouse models and in human cells isolated from patients with primary or relapsed/refractory myeloma.
Study details: Preclinical investigations in cell lines, murine models, and isolated human multiple myeloma cells.
Disclosures: The study was supported by grants from the Korean government. The researchers reported having no potential conflicts of interest.
Source: Song IS et al. Cancer Lett. 2018:432:205-15.
Occurrence of Skeletal-Related Events in Multiple Myeloma and Prostate Cancer Patients Receiving Standard Versus Extended-Interval Zoledronic Acid
Background/Purpose: Dysregulation of osteoclast activity and uncontrolled bone resorption are hallmarks of multiple myeloma and metastatic prostate cancer, predisposing patients to net bone loss and pathologic fractures. Zoledronic acid, a bisphosphonate, induces osteoclast apoptosis and reduces bone resorption, reducing fracture risk, but the optimal dosing interval is the subject of current clinical debate. Historically, the standard dosing interval has been every 4 weeks, but recent research demonstrated no difference in the rate of skeletal-related events (SREs)—fracture, spinal compression, bone irradiation, or surgery—when zoledronic acid was dosed every 12 weeks. The primary objective of this study was to determine if extending the zoledronic acid dosing interval would increase the incidence of SREs in a Veteran population.
Methods: Retrospective observational analysis of multiple myeloma and prostate cancer patients who received zoledronic acid. Patients were stratified by zoledronic acid dosing interval (standard or extended). Baseline data, duration of treatment, type and incidence of SREs, and incidence of osteonecrosis of the jaw (ONJ) were determined for each group. Pearson’s chi-square test was used to determine statistical significance.
Results: One hundred twenty-three patients were eligible for inclusion based on prespecified criteria. No difference in the rate of SREs was found between the standard- and extended-interval dosing groups (30.6% vs 22.9%, P = 0.374). All instances of ONJ occurred in the standard-interval dosing group, but the difference in incidence between groups was not statistically significant (2.5% vs 0%, P = .347). Subgroup analysis did not reveal a difference between multiple myeloma and metastatic prostate cancer in the incidence of SREs (42.9% vs 14.3%, P = .172; and 28% vs 25%, P = .753, respectively) or ONJ (4.8% vs 0%, P = .451; and 2% vs 0%, P = .577, respectively).
Conclusions/Impliacations: Based on our results, extending the zoledronic acid dosing interval does not increase the incidence of SREs. Dosing zoledronic acid every three months offers a potential avenue to increase Veteran compliance and decrease the chance for adverse drug reactions without compromising therapeutic benefit.
Background/Purpose: Dysregulation of osteoclast activity and uncontrolled bone resorption are hallmarks of multiple myeloma and metastatic prostate cancer, predisposing patients to net bone loss and pathologic fractures. Zoledronic acid, a bisphosphonate, induces osteoclast apoptosis and reduces bone resorption, reducing fracture risk, but the optimal dosing interval is the subject of current clinical debate. Historically, the standard dosing interval has been every 4 weeks, but recent research demonstrated no difference in the rate of skeletal-related events (SREs)—fracture, spinal compression, bone irradiation, or surgery—when zoledronic acid was dosed every 12 weeks. The primary objective of this study was to determine if extending the zoledronic acid dosing interval would increase the incidence of SREs in a Veteran population.
Methods: Retrospective observational analysis of multiple myeloma and prostate cancer patients who received zoledronic acid. Patients were stratified by zoledronic acid dosing interval (standard or extended). Baseline data, duration of treatment, type and incidence of SREs, and incidence of osteonecrosis of the jaw (ONJ) were determined for each group. Pearson’s chi-square test was used to determine statistical significance.
Results: One hundred twenty-three patients were eligible for inclusion based on prespecified criteria. No difference in the rate of SREs was found between the standard- and extended-interval dosing groups (30.6% vs 22.9%, P = 0.374). All instances of ONJ occurred in the standard-interval dosing group, but the difference in incidence between groups was not statistically significant (2.5% vs 0%, P = .347). Subgroup analysis did not reveal a difference between multiple myeloma and metastatic prostate cancer in the incidence of SREs (42.9% vs 14.3%, P = .172; and 28% vs 25%, P = .753, respectively) or ONJ (4.8% vs 0%, P = .451; and 2% vs 0%, P = .577, respectively).
Conclusions/Impliacations: Based on our results, extending the zoledronic acid dosing interval does not increase the incidence of SREs. Dosing zoledronic acid every three months offers a potential avenue to increase Veteran compliance and decrease the chance for adverse drug reactions without compromising therapeutic benefit.
Background/Purpose: Dysregulation of osteoclast activity and uncontrolled bone resorption are hallmarks of multiple myeloma and metastatic prostate cancer, predisposing patients to net bone loss and pathologic fractures. Zoledronic acid, a bisphosphonate, induces osteoclast apoptosis and reduces bone resorption, reducing fracture risk, but the optimal dosing interval is the subject of current clinical debate. Historically, the standard dosing interval has been every 4 weeks, but recent research demonstrated no difference in the rate of skeletal-related events (SREs)—fracture, spinal compression, bone irradiation, or surgery—when zoledronic acid was dosed every 12 weeks. The primary objective of this study was to determine if extending the zoledronic acid dosing interval would increase the incidence of SREs in a Veteran population.
Methods: Retrospective observational analysis of multiple myeloma and prostate cancer patients who received zoledronic acid. Patients were stratified by zoledronic acid dosing interval (standard or extended). Baseline data, duration of treatment, type and incidence of SREs, and incidence of osteonecrosis of the jaw (ONJ) were determined for each group. Pearson’s chi-square test was used to determine statistical significance.
Results: One hundred twenty-three patients were eligible for inclusion based on prespecified criteria. No difference in the rate of SREs was found between the standard- and extended-interval dosing groups (30.6% vs 22.9%, P = 0.374). All instances of ONJ occurred in the standard-interval dosing group, but the difference in incidence between groups was not statistically significant (2.5% vs 0%, P = .347). Subgroup analysis did not reveal a difference between multiple myeloma and metastatic prostate cancer in the incidence of SREs (42.9% vs 14.3%, P = .172; and 28% vs 25%, P = .753, respectively) or ONJ (4.8% vs 0%, P = .451; and 2% vs 0%, P = .577, respectively).
Conclusions/Impliacations: Based on our results, extending the zoledronic acid dosing interval does not increase the incidence of SREs. Dosing zoledronic acid every three months offers a potential avenue to increase Veteran compliance and decrease the chance for adverse drug reactions without compromising therapeutic benefit.
New guidelines on antimicrobial prophylaxis

Experts have published updated guidelines on antimicrobial prophylaxis for adults with cancer-related immunosuppression.
The guidelines include antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions, such as hand hygiene, that may reduce infection risk.
The guidelines were developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA) and published in the Journal of Clinical Oncology.
For the most part, the expert panel that created these guidelines endorsed the previous ASCO recommendations, published in 2013.
However, the panel considered six new studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Recommendations
The ASCO/IDSA guidelines say health care providers should systematically assess the risk of febrile neutropenia, taking into account patient-, cancer-, and treatment-related factors.
Fluoroquinolone prophylaxis is recommended for patients at high risk of febrile neutropenia or profound, protracted neutropenia. This includes most patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) and patients undergoing hematopoietic stem cell transplant (HSCT) who are treated with myeloablative conditioning regimens.
Antifungal prophylaxis with an oral triazole or parenteral echinocandin is recommended for patients at risk of profound, protracted neutropenia, which includes HSCT recipients and most patients with AML/MDS.
However, neither antifungal nor antibiotic prophylaxis are routinely recommended for patients with solid tumors.
Prophylaxis with a nucleoside analog, such as acyclovir, is recommended in patients who are herpes simplex virus–seropositive and are undergoing allogeneic HSCT or leukemia induction.
Pneumocystis jirovecii prophylaxis, such as trimethoprim-sulfamethoxazole, is recommended for patients receiving chemotherapy regimens associated with a greater than 3.5% risk for pneumonia from P jirovecii.
Treatment with a nucleoside reverse transcription inhibitor, such as entecavir or tenofovir, is recommended for patients at high risk of hepatitis B virus reactivation.
Yearly influenza vaccination with an inactivated quadrivalent vaccine is recommended for all patients undergoing chemotherapy for malignancy as well as their family members, household contacts, and health care providers.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce the risk of pathogen transmission, according to the guidelines.
The guidelines also note that outpatients who develop neutropenia following cancer therapy should avoid prolonged contact with environments that have high concentrations of airborne fungal spores.
The guidelines do not recommend interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks. “Evidence of clinical benefit is lacking” for those interventions, the expert panel said.
Members of the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
Experts have published updated guidelines on antimicrobial prophylaxis for adults with cancer-related immunosuppression.
The guidelines include antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions, such as hand hygiene, that may reduce infection risk.
The guidelines were developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA) and published in the Journal of Clinical Oncology.
For the most part, the expert panel that created these guidelines endorsed the previous ASCO recommendations, published in 2013.
However, the panel considered six new studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Recommendations
The ASCO/IDSA guidelines say health care providers should systematically assess the risk of febrile neutropenia, taking into account patient-, cancer-, and treatment-related factors.
Fluoroquinolone prophylaxis is recommended for patients at high risk of febrile neutropenia or profound, protracted neutropenia. This includes most patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) and patients undergoing hematopoietic stem cell transplant (HSCT) who are treated with myeloablative conditioning regimens.
Antifungal prophylaxis with an oral triazole or parenteral echinocandin is recommended for patients at risk of profound, protracted neutropenia, which includes HSCT recipients and most patients with AML/MDS.
However, neither antifungal nor antibiotic prophylaxis are routinely recommended for patients with solid tumors.
Prophylaxis with a nucleoside analog, such as acyclovir, is recommended in patients who are herpes simplex virus–seropositive and are undergoing allogeneic HSCT or leukemia induction.
Pneumocystis jirovecii prophylaxis, such as trimethoprim-sulfamethoxazole, is recommended for patients receiving chemotherapy regimens associated with a greater than 3.5% risk for pneumonia from P jirovecii.
Treatment with a nucleoside reverse transcription inhibitor, such as entecavir or tenofovir, is recommended for patients at high risk of hepatitis B virus reactivation.
Yearly influenza vaccination with an inactivated quadrivalent vaccine is recommended for all patients undergoing chemotherapy for malignancy as well as their family members, household contacts, and health care providers.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce the risk of pathogen transmission, according to the guidelines.
The guidelines also note that outpatients who develop neutropenia following cancer therapy should avoid prolonged contact with environments that have high concentrations of airborne fungal spores.
The guidelines do not recommend interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks. “Evidence of clinical benefit is lacking” for those interventions, the expert panel said.
Members of the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
Experts have published updated guidelines on antimicrobial prophylaxis for adults with cancer-related immunosuppression.
The guidelines include antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions, such as hand hygiene, that may reduce infection risk.
The guidelines were developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA) and published in the Journal of Clinical Oncology.
For the most part, the expert panel that created these guidelines endorsed the previous ASCO recommendations, published in 2013.
However, the panel considered six new studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Recommendations
The ASCO/IDSA guidelines say health care providers should systematically assess the risk of febrile neutropenia, taking into account patient-, cancer-, and treatment-related factors.
Fluoroquinolone prophylaxis is recommended for patients at high risk of febrile neutropenia or profound, protracted neutropenia. This includes most patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) and patients undergoing hematopoietic stem cell transplant (HSCT) who are treated with myeloablative conditioning regimens.
Antifungal prophylaxis with an oral triazole or parenteral echinocandin is recommended for patients at risk of profound, protracted neutropenia, which includes HSCT recipients and most patients with AML/MDS.
However, neither antifungal nor antibiotic prophylaxis are routinely recommended for patients with solid tumors.
Prophylaxis with a nucleoside analog, such as acyclovir, is recommended in patients who are herpes simplex virus–seropositive and are undergoing allogeneic HSCT or leukemia induction.
Pneumocystis jirovecii prophylaxis, such as trimethoprim-sulfamethoxazole, is recommended for patients receiving chemotherapy regimens associated with a greater than 3.5% risk for pneumonia from P jirovecii.
Treatment with a nucleoside reverse transcription inhibitor, such as entecavir or tenofovir, is recommended for patients at high risk of hepatitis B virus reactivation.
Yearly influenza vaccination with an inactivated quadrivalent vaccine is recommended for all patients undergoing chemotherapy for malignancy as well as their family members, household contacts, and health care providers.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce the risk of pathogen transmission, according to the guidelines.
The guidelines also note that outpatients who develop neutropenia following cancer therapy should avoid prolonged contact with environments that have high concentrations of airborne fungal spores.
The guidelines do not recommend interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks. “Evidence of clinical benefit is lacking” for those interventions, the expert panel said.
Members of the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
Daratumumab approved in Europe for new myeloma indication
The drug is now authorized for use in combination with bortezomib, melphalan, and prednisone (VMP) to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant, according to a press release published on the Genmab website.
Daratumumab was previously approved by the European Commission (EC) for use in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat adults with MM who have received at least one prior therapy.
In addition, daratumumab is approved by the EC as monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who had disease progression on their last therapy.
The EC’s latest approval for daratumumab is based on results from the phase 3 ALCYONE trial. Results from this study were presented at the 2017 annual meeting of the American Society of Hematology and simultaneously published in the New England Journal of Medicine.
ALCYONE enrolled 706 patients with newly diagnosed MM who were not eligible for high-dose chemotherapy with autologous stem cell transplant. Patients were randomized to receive VMP or daratumumab plus VMP (D-VMP).
The overall response rates were 91% in the D-VMP arm and 74% in the VMP arm (P less than.0001), and rates of complete response were 43% and 24%, respectively. Rates of minimal residual disease negativity were 22% and 6%, respectively.
The median progression-free survival (PFS) was not reached in the D-VMP arm and was 18.1 months in the VMP arm. The 12-month PFS was 87% and 76%, respectively, and the 18-month PFS was 72% and 50%, respectively.
The most common treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (50% and 53%), thrombocytopenia (49% and 54%), anemia (28% and 38%), peripheral sensory neuropathy (28% and 34%), upper respiratory tract infection (26% and 14%), diarrhea (24% and 25%), pyrexia (23% and 21%), and nausea (21% and 22%).
Infusion-related reactions occurred in 28% of patients in the D-VMP arm and in none of those in the VMP arm.
The rate of grade 3/4 infections was higher in the D-VMP arm than the VMP arm – 23% and 15%, respectively. In both arms, most infections resolved.
The most common grade 3/4 treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (40% and 39%), thrombocytopenia (34% and 38%), and anemia (16% and 20%).
The rate of discontinuation caused by adverse events was 5% in the D-VMP arm and 9% in the VMP arm.
The drug is now authorized for use in combination with bortezomib, melphalan, and prednisone (VMP) to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant, according to a press release published on the Genmab website.
Daratumumab was previously approved by the European Commission (EC) for use in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat adults with MM who have received at least one prior therapy.
In addition, daratumumab is approved by the EC as monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who had disease progression on their last therapy.
The EC’s latest approval for daratumumab is based on results from the phase 3 ALCYONE trial. Results from this study were presented at the 2017 annual meeting of the American Society of Hematology and simultaneously published in the New England Journal of Medicine.
ALCYONE enrolled 706 patients with newly diagnosed MM who were not eligible for high-dose chemotherapy with autologous stem cell transplant. Patients were randomized to receive VMP or daratumumab plus VMP (D-VMP).
The overall response rates were 91% in the D-VMP arm and 74% in the VMP arm (P less than.0001), and rates of complete response were 43% and 24%, respectively. Rates of minimal residual disease negativity were 22% and 6%, respectively.
The median progression-free survival (PFS) was not reached in the D-VMP arm and was 18.1 months in the VMP arm. The 12-month PFS was 87% and 76%, respectively, and the 18-month PFS was 72% and 50%, respectively.
The most common treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (50% and 53%), thrombocytopenia (49% and 54%), anemia (28% and 38%), peripheral sensory neuropathy (28% and 34%), upper respiratory tract infection (26% and 14%), diarrhea (24% and 25%), pyrexia (23% and 21%), and nausea (21% and 22%).
Infusion-related reactions occurred in 28% of patients in the D-VMP arm and in none of those in the VMP arm.
The rate of grade 3/4 infections was higher in the D-VMP arm than the VMP arm – 23% and 15%, respectively. In both arms, most infections resolved.
The most common grade 3/4 treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (40% and 39%), thrombocytopenia (34% and 38%), and anemia (16% and 20%).
The rate of discontinuation caused by adverse events was 5% in the D-VMP arm and 9% in the VMP arm.
The drug is now authorized for use in combination with bortezomib, melphalan, and prednisone (VMP) to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant, according to a press release published on the Genmab website.
Daratumumab was previously approved by the European Commission (EC) for use in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat adults with MM who have received at least one prior therapy.
In addition, daratumumab is approved by the EC as monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who had disease progression on their last therapy.
The EC’s latest approval for daratumumab is based on results from the phase 3 ALCYONE trial. Results from this study were presented at the 2017 annual meeting of the American Society of Hematology and simultaneously published in the New England Journal of Medicine.
ALCYONE enrolled 706 patients with newly diagnosed MM who were not eligible for high-dose chemotherapy with autologous stem cell transplant. Patients were randomized to receive VMP or daratumumab plus VMP (D-VMP).
The overall response rates were 91% in the D-VMP arm and 74% in the VMP arm (P less than.0001), and rates of complete response were 43% and 24%, respectively. Rates of minimal residual disease negativity were 22% and 6%, respectively.
The median progression-free survival (PFS) was not reached in the D-VMP arm and was 18.1 months in the VMP arm. The 12-month PFS was 87% and 76%, respectively, and the 18-month PFS was 72% and 50%, respectively.
The most common treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (50% and 53%), thrombocytopenia (49% and 54%), anemia (28% and 38%), peripheral sensory neuropathy (28% and 34%), upper respiratory tract infection (26% and 14%), diarrhea (24% and 25%), pyrexia (23% and 21%), and nausea (21% and 22%).
Infusion-related reactions occurred in 28% of patients in the D-VMP arm and in none of those in the VMP arm.
The rate of grade 3/4 infections was higher in the D-VMP arm than the VMP arm – 23% and 15%, respectively. In both arms, most infections resolved.
The most common grade 3/4 treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (40% and 39%), thrombocytopenia (34% and 38%), and anemia (16% and 20%).
The rate of discontinuation caused by adverse events was 5% in the D-VMP arm and 9% in the VMP arm.
Daratumumab approved for new indication in MM
The European Commission (EC) has approved a new indication for daratumumab (Darzalex®).
The drug is now authorized for use in combination with bortezomib, melphalan, and prednisone (VMP) to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant.
Daratumumab was previously approved by the EC for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, to treat adults with MM who have received at least one prior therapy.
In addition, daratumumab is EC-approved as monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on their last therapy.
The EC’s latest approval for daratumumab is based on results from the phase 3 ALCYONE (MMY3007) study.
Results from this study were presented at the 2017 ASH Annual Meeting and simultaneously published in The New England Journal of Medicine.
ALCYONE enrolled 706 patients with newly diagnosed MM who were not eligible for high-dose chemotherapy with autologous stem cell transplant. Patients were randomized to receive VMP or daratumumab plus VMP (D-VMP).
The overall response rates were 91% in the D-VMP arm and 74% in the VMP arm (P<0.0001). Rates of complete response were 43% and 24%, respectively. Rates of minimal residual disease negativity were 22% and 6%, respectively.
The median progression-free survival (PFS) was not reached in the D-VMP arm and was 18.1 months in the VMP arm. The 12-month PFS was 87% and 76%, respectively. The 18-month PFS was 72% and 50%, respectively.
The most common treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (50% and 53%), thrombocytopenia (49% and 54%), anemia (28% and 38%), peripheral sensory neuropathy (28% and 34%), upper respiratory tract infection (26% and 14%), diarrhea (24% and 25%), pyrexia (23% and 21%), and nausea (21% and 22%).
Infusion-related reactions occurred in 28% of patients in the D-VMP arm and 0% of those in the VMP arm.
The rate of grade 3/4 infections was higher in the D-VMP arm than the VMP arm—23% and 15%, respectively. In both arms, most infections resolved.
The most common grade 3/4 treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (40% and 39%), thrombocytopenia (34% and 38%), and anemia (16% and 20%).
The rate of discontinuation due to adverse events was 5% in the D-VMP arm and 9% in the VMP arm.
The European Commission (EC) has approved a new indication for daratumumab (Darzalex®).
The drug is now authorized for use in combination with bortezomib, melphalan, and prednisone (VMP) to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant.
Daratumumab was previously approved by the EC for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, to treat adults with MM who have received at least one prior therapy.
In addition, daratumumab is EC-approved as monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on their last therapy.
The EC’s latest approval for daratumumab is based on results from the phase 3 ALCYONE (MMY3007) study.
Results from this study were presented at the 2017 ASH Annual Meeting and simultaneously published in The New England Journal of Medicine.
ALCYONE enrolled 706 patients with newly diagnosed MM who were not eligible for high-dose chemotherapy with autologous stem cell transplant. Patients were randomized to receive VMP or daratumumab plus VMP (D-VMP).
The overall response rates were 91% in the D-VMP arm and 74% in the VMP arm (P<0.0001). Rates of complete response were 43% and 24%, respectively. Rates of minimal residual disease negativity were 22% and 6%, respectively.
The median progression-free survival (PFS) was not reached in the D-VMP arm and was 18.1 months in the VMP arm. The 12-month PFS was 87% and 76%, respectively. The 18-month PFS was 72% and 50%, respectively.
The most common treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (50% and 53%), thrombocytopenia (49% and 54%), anemia (28% and 38%), peripheral sensory neuropathy (28% and 34%), upper respiratory tract infection (26% and 14%), diarrhea (24% and 25%), pyrexia (23% and 21%), and nausea (21% and 22%).
Infusion-related reactions occurred in 28% of patients in the D-VMP arm and 0% of those in the VMP arm.
The rate of grade 3/4 infections was higher in the D-VMP arm than the VMP arm—23% and 15%, respectively. In both arms, most infections resolved.
The most common grade 3/4 treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (40% and 39%), thrombocytopenia (34% and 38%), and anemia (16% and 20%).
The rate of discontinuation due to adverse events was 5% in the D-VMP arm and 9% in the VMP arm.
The European Commission (EC) has approved a new indication for daratumumab (Darzalex®).
The drug is now authorized for use in combination with bortezomib, melphalan, and prednisone (VMP) to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant.
Daratumumab was previously approved by the EC for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, to treat adults with MM who have received at least one prior therapy.
In addition, daratumumab is EC-approved as monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on their last therapy.
The EC’s latest approval for daratumumab is based on results from the phase 3 ALCYONE (MMY3007) study.
Results from this study were presented at the 2017 ASH Annual Meeting and simultaneously published in The New England Journal of Medicine.
ALCYONE enrolled 706 patients with newly diagnosed MM who were not eligible for high-dose chemotherapy with autologous stem cell transplant. Patients were randomized to receive VMP or daratumumab plus VMP (D-VMP).
The overall response rates were 91% in the D-VMP arm and 74% in the VMP arm (P<0.0001). Rates of complete response were 43% and 24%, respectively. Rates of minimal residual disease negativity were 22% and 6%, respectively.
The median progression-free survival (PFS) was not reached in the D-VMP arm and was 18.1 months in the VMP arm. The 12-month PFS was 87% and 76%, respectively. The 18-month PFS was 72% and 50%, respectively.
The most common treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (50% and 53%), thrombocytopenia (49% and 54%), anemia (28% and 38%), peripheral sensory neuropathy (28% and 34%), upper respiratory tract infection (26% and 14%), diarrhea (24% and 25%), pyrexia (23% and 21%), and nausea (21% and 22%).
Infusion-related reactions occurred in 28% of patients in the D-VMP arm and 0% of those in the VMP arm.
The rate of grade 3/4 infections was higher in the D-VMP arm than the VMP arm—23% and 15%, respectively. In both arms, most infections resolved.
The most common grade 3/4 treatment-emergent adverse events (in the D-VMP and VMP arms, respectively) were neutropenia (40% and 39%), thrombocytopenia (34% and 38%), and anemia (16% and 20%).
The rate of discontinuation due to adverse events was 5% in the D-VMP arm and 9% in the VMP arm.
Fracture risk tied to death in women with MM
Preexisting osteoporosis is an important risk factor for mortality in postmenopausal women who develop multiple myeloma (MM), according to researchers.
They found that high fracture risk was associated with an increased risk of death in postmenopausal females with MM, independent of other clinical risk factors.
Ashley E. Rosko, MD, of Ohio State University in Columbus, and her colleagues reported these findings in Clinical Lymphoma, Myeloma and Leukemia.
The researchers studied 362 subjects in the Women’s Health Initiative data set who developed MM but had no history of any cancer at baseline. The women were between 50 and 79 years of age and postmenopausal when they were originally recruited at 40 US centers between 1993 and 1998.
Dr. Rosko and her colleagues calculated bone health for the women using the Fracture Risk Assessment Tool (FRAX), a web-based tool that calculates 10-year probability of hip and other major osteoporotic fractures.
Ninety-eight of the subjects were classified as having high FRAX scores, defined as a 10-year probability of 3% or greater for hip fracture or 20% or greater for other major osteoporosis-related fractures.
With a median follow-up of 10.5 years, the adjusted risk of death was elevated in women with high FRAX scores, with a covariate-adjusted hazard ratio of 1.51 (95% confidence interval, 1.01-2.25; P=0.044) versus women with low FRAX scores.
Of the 362 patients, 226 died during the follow-up period. That included 72% (n=71) of women with high FRAX scores and 59% (n=155) of women with low FRAX scores.
These findings suggest osteoporosis is an “important comorbidity” in women who develop MM, according to Dr. Rosko and her coauthors.
“Recognizing osteoporosis as a risk factor associated with multiple myeloma mortality is an important prognostic factor in postmenopausal women,” the researchers wrote.
This work was supported, in part, by the National Cancer Institute. The researchers reported no relevant financial disclosures.
Preexisting osteoporosis is an important risk factor for mortality in postmenopausal women who develop multiple myeloma (MM), according to researchers.
They found that high fracture risk was associated with an increased risk of death in postmenopausal females with MM, independent of other clinical risk factors.
Ashley E. Rosko, MD, of Ohio State University in Columbus, and her colleagues reported these findings in Clinical Lymphoma, Myeloma and Leukemia.
The researchers studied 362 subjects in the Women’s Health Initiative data set who developed MM but had no history of any cancer at baseline. The women were between 50 and 79 years of age and postmenopausal when they were originally recruited at 40 US centers between 1993 and 1998.
Dr. Rosko and her colleagues calculated bone health for the women using the Fracture Risk Assessment Tool (FRAX), a web-based tool that calculates 10-year probability of hip and other major osteoporotic fractures.
Ninety-eight of the subjects were classified as having high FRAX scores, defined as a 10-year probability of 3% or greater for hip fracture or 20% or greater for other major osteoporosis-related fractures.
With a median follow-up of 10.5 years, the adjusted risk of death was elevated in women with high FRAX scores, with a covariate-adjusted hazard ratio of 1.51 (95% confidence interval, 1.01-2.25; P=0.044) versus women with low FRAX scores.
Of the 362 patients, 226 died during the follow-up period. That included 72% (n=71) of women with high FRAX scores and 59% (n=155) of women with low FRAX scores.
These findings suggest osteoporosis is an “important comorbidity” in women who develop MM, according to Dr. Rosko and her coauthors.
“Recognizing osteoporosis as a risk factor associated with multiple myeloma mortality is an important prognostic factor in postmenopausal women,” the researchers wrote.
This work was supported, in part, by the National Cancer Institute. The researchers reported no relevant financial disclosures.
Preexisting osteoporosis is an important risk factor for mortality in postmenopausal women who develop multiple myeloma (MM), according to researchers.
They found that high fracture risk was associated with an increased risk of death in postmenopausal females with MM, independent of other clinical risk factors.
Ashley E. Rosko, MD, of Ohio State University in Columbus, and her colleagues reported these findings in Clinical Lymphoma, Myeloma and Leukemia.
The researchers studied 362 subjects in the Women’s Health Initiative data set who developed MM but had no history of any cancer at baseline. The women were between 50 and 79 years of age and postmenopausal when they were originally recruited at 40 US centers between 1993 and 1998.
Dr. Rosko and her colleagues calculated bone health for the women using the Fracture Risk Assessment Tool (FRAX), a web-based tool that calculates 10-year probability of hip and other major osteoporotic fractures.
Ninety-eight of the subjects were classified as having high FRAX scores, defined as a 10-year probability of 3% or greater for hip fracture or 20% or greater for other major osteoporosis-related fractures.
With a median follow-up of 10.5 years, the adjusted risk of death was elevated in women with high FRAX scores, with a covariate-adjusted hazard ratio of 1.51 (95% confidence interval, 1.01-2.25; P=0.044) versus women with low FRAX scores.
Of the 362 patients, 226 died during the follow-up period. That included 72% (n=71) of women with high FRAX scores and 59% (n=155) of women with low FRAX scores.
These findings suggest osteoporosis is an “important comorbidity” in women who develop MM, according to Dr. Rosko and her coauthors.
“Recognizing osteoporosis as a risk factor associated with multiple myeloma mortality is an important prognostic factor in postmenopausal women,” the researchers wrote.
This work was supported, in part, by the National Cancer Institute. The researchers reported no relevant financial disclosures.
Fracture risk linked to mortality in women with myeloma
Preexisting osteoporosis is an important risk factor for mortality risk in cancer-free postmenopausal women who go on to develop multiple myeloma, results of a recent analysis suggest.
High fracture risk was associated with an increased risk of death, independent of other clinical risk factors, in this analysis of postmenopausal women in the Women’s Health Initiative (WHI) data set.
The findings help define osteoporosis as an important prognostic factor associated with mortality in postmenopausal women who develop myeloma, according to study author Ashley E. Rosko, MD, of Ohio State University, Columbus, and her colleagues.
“Osteoporosis is highly prevalent in aging adults, and very little is known on how this comorbid condition contributes to outcomes in individuals who develop myeloma,” wrote Dr. Rosko and her coauthors. Their report is in the journal Clinical Lymphoma, Myeloma and Leukemia.
The analysis involved 362 women in the WHI data set who developed myeloma and had no history of any cancer at baseline. Women in the WHI were between 50 and 79 years of age and postmenopausal at baseline when originally recruited at 40 U.S. centers between 1993 and 1998.
Dr. Rosko and her colleagues calculated bone health for women in the data set using the Fracture Risk Assessment Tool (FRAX), a web-based tool that calculates 10-year probability of hip and other major osteoporotic fractures.
Of the 362 women who developed myeloma, 98 were classified as having high FRAX scores, defined as a 10-year probability of 3% or greater for hip fracture, or 20% or greater for other major osteoporosis-related fractures.
With a median follow-up of 10.5 years, the adjusted risk of death was elevated in women with high FRAX scores, according to investigators, with a covariate-adjusted hazard ratio of 1.51 (95% confidence interval, 1.01-2.25; P = .044) versus women with low FRAX scores.
Of the 362 patients who developed myeloma, 226 died during the follow-up period. That included 71 women with high FRAX scores, or 72% of that subset; and 155 women with low FRAX scores, or 59% of that subset, investigators reported.
These findings suggest osteoporosis is an “important comorbidity” in women who develop multiple myeloma, Dr. Rosko and her coauthors said in a discussion of the study results.
“Recognizing osteoporosis as a risk factor associated with multiple myeloma mortality is an important prognostic factor in postmenopausal women,” they said.
This investigation was supported in part by the National Cancer Institute. The researchers reported having no relevant financial disclosures.
SOURCE: Rosko AE et al. Clin Lymphoma Myeloma Leuk. 2018 Sep;18(9):597-602.e1.
Preexisting osteoporosis is an important risk factor for mortality risk in cancer-free postmenopausal women who go on to develop multiple myeloma, results of a recent analysis suggest.
High fracture risk was associated with an increased risk of death, independent of other clinical risk factors, in this analysis of postmenopausal women in the Women’s Health Initiative (WHI) data set.
The findings help define osteoporosis as an important prognostic factor associated with mortality in postmenopausal women who develop myeloma, according to study author Ashley E. Rosko, MD, of Ohio State University, Columbus, and her colleagues.
“Osteoporosis is highly prevalent in aging adults, and very little is known on how this comorbid condition contributes to outcomes in individuals who develop myeloma,” wrote Dr. Rosko and her coauthors. Their report is in the journal Clinical Lymphoma, Myeloma and Leukemia.
The analysis involved 362 women in the WHI data set who developed myeloma and had no history of any cancer at baseline. Women in the WHI were between 50 and 79 years of age and postmenopausal at baseline when originally recruited at 40 U.S. centers between 1993 and 1998.
Dr. Rosko and her colleagues calculated bone health for women in the data set using the Fracture Risk Assessment Tool (FRAX), a web-based tool that calculates 10-year probability of hip and other major osteoporotic fractures.
Of the 362 women who developed myeloma, 98 were classified as having high FRAX scores, defined as a 10-year probability of 3% or greater for hip fracture, or 20% or greater for other major osteoporosis-related fractures.
With a median follow-up of 10.5 years, the adjusted risk of death was elevated in women with high FRAX scores, according to investigators, with a covariate-adjusted hazard ratio of 1.51 (95% confidence interval, 1.01-2.25; P = .044) versus women with low FRAX scores.
Of the 362 patients who developed myeloma, 226 died during the follow-up period. That included 71 women with high FRAX scores, or 72% of that subset; and 155 women with low FRAX scores, or 59% of that subset, investigators reported.
These findings suggest osteoporosis is an “important comorbidity” in women who develop multiple myeloma, Dr. Rosko and her coauthors said in a discussion of the study results.
“Recognizing osteoporosis as a risk factor associated with multiple myeloma mortality is an important prognostic factor in postmenopausal women,” they said.
This investigation was supported in part by the National Cancer Institute. The researchers reported having no relevant financial disclosures.
SOURCE: Rosko AE et al. Clin Lymphoma Myeloma Leuk. 2018 Sep;18(9):597-602.e1.
Preexisting osteoporosis is an important risk factor for mortality risk in cancer-free postmenopausal women who go on to develop multiple myeloma, results of a recent analysis suggest.
High fracture risk was associated with an increased risk of death, independent of other clinical risk factors, in this analysis of postmenopausal women in the Women’s Health Initiative (WHI) data set.
The findings help define osteoporosis as an important prognostic factor associated with mortality in postmenopausal women who develop myeloma, according to study author Ashley E. Rosko, MD, of Ohio State University, Columbus, and her colleagues.
“Osteoporosis is highly prevalent in aging adults, and very little is known on how this comorbid condition contributes to outcomes in individuals who develop myeloma,” wrote Dr. Rosko and her coauthors. Their report is in the journal Clinical Lymphoma, Myeloma and Leukemia.
The analysis involved 362 women in the WHI data set who developed myeloma and had no history of any cancer at baseline. Women in the WHI were between 50 and 79 years of age and postmenopausal at baseline when originally recruited at 40 U.S. centers between 1993 and 1998.
Dr. Rosko and her colleagues calculated bone health for women in the data set using the Fracture Risk Assessment Tool (FRAX), a web-based tool that calculates 10-year probability of hip and other major osteoporotic fractures.
Of the 362 women who developed myeloma, 98 were classified as having high FRAX scores, defined as a 10-year probability of 3% or greater for hip fracture, or 20% or greater for other major osteoporosis-related fractures.
With a median follow-up of 10.5 years, the adjusted risk of death was elevated in women with high FRAX scores, according to investigators, with a covariate-adjusted hazard ratio of 1.51 (95% confidence interval, 1.01-2.25; P = .044) versus women with low FRAX scores.
Of the 362 patients who developed myeloma, 226 died during the follow-up period. That included 71 women with high FRAX scores, or 72% of that subset; and 155 women with low FRAX scores, or 59% of that subset, investigators reported.
These findings suggest osteoporosis is an “important comorbidity” in women who develop multiple myeloma, Dr. Rosko and her coauthors said in a discussion of the study results.
“Recognizing osteoporosis as a risk factor associated with multiple myeloma mortality is an important prognostic factor in postmenopausal women,” they said.
This investigation was supported in part by the National Cancer Institute. The researchers reported having no relevant financial disclosures.
SOURCE: Rosko AE et al. Clin Lymphoma Myeloma Leuk. 2018 Sep;18(9):597-602.e1.
FROM CLINICAL LYMPHOMA, MYELOMA AND LEUKEMIA
Key clinical point:
Major finding: Risk of death was elevated in women at high risk of fracture (covariate-adjusted hazard ratio, 1.51; 95% confidence interval, 1.01-2.25; P = .044) versus women with low fracture risk.
Study details: Retrospective analysis of the Women’s Health Initiative data set including 362 postmenopausal women who were cancer free at baseline and developed myeloma over the course of study follow-up.
Disclosures: The analysis was supported in part by the National Cancer Institute. The researchers reported having no relevant financial disclosures.
Source: Rosko AE et al. Clin Lymphoma Myeloma Leuk. 2018 Sep;18(9):597-602.e1.
Elotuzumab under review for relapsed/refractory myeloma
The Food and Drug Administration has granted .
![]()
Bristol-Myers Squibb is seeking approval for elotuzumab in combination with pomalidomide and low-dose dexamethasone to treat patients with relapsed/refractory multiple myeloma who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved for use in combination with lenalidomide and dexamethasone to treat multiple myeloma patients who have received between one and three prior therapies.
The elotuzumab application is supported by data from ELOQUENT-3, a randomized, phase 2 study that evaluated the addition of elotuzumab to pomalidomide and low-dose dexamethasone in patients with relapsed/refractory multiple myeloma.
Researchers presented findings from this study at the annual congress of the European Hematology Association in June 2018.
The overall response rate was 53% in the elotuzumab, pomalidomide, and low-dose dexamethasone (EPd) arm and 26% in the pomalidomide and low-dose dexamethasone (Pd) arm. The median progression-free survival was 10.3 months in the EPd arm and 4.7 months in the Pd arm (hazard ratio, 0.54; P = .0078), the researchers reported.
The researchers also reported that adverse events in the EPd arm were consistent with expectations based on previous results with elotuzumab and pomalidomide regimens.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The priority review typically shortens the time to an approval decision by a few months. The agency is expected to make a decision on elotuzumab by Dec. 27, 2018.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The Food and Drug Administration has granted .
![]()
Bristol-Myers Squibb is seeking approval for elotuzumab in combination with pomalidomide and low-dose dexamethasone to treat patients with relapsed/refractory multiple myeloma who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved for use in combination with lenalidomide and dexamethasone to treat multiple myeloma patients who have received between one and three prior therapies.
The elotuzumab application is supported by data from ELOQUENT-3, a randomized, phase 2 study that evaluated the addition of elotuzumab to pomalidomide and low-dose dexamethasone in patients with relapsed/refractory multiple myeloma.
Researchers presented findings from this study at the annual congress of the European Hematology Association in June 2018.
The overall response rate was 53% in the elotuzumab, pomalidomide, and low-dose dexamethasone (EPd) arm and 26% in the pomalidomide and low-dose dexamethasone (Pd) arm. The median progression-free survival was 10.3 months in the EPd arm and 4.7 months in the Pd arm (hazard ratio, 0.54; P = .0078), the researchers reported.
The researchers also reported that adverse events in the EPd arm were consistent with expectations based on previous results with elotuzumab and pomalidomide regimens.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The priority review typically shortens the time to an approval decision by a few months. The agency is expected to make a decision on elotuzumab by Dec. 27, 2018.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The Food and Drug Administration has granted .
![]()
Bristol-Myers Squibb is seeking approval for elotuzumab in combination with pomalidomide and low-dose dexamethasone to treat patients with relapsed/refractory multiple myeloma who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved for use in combination with lenalidomide and dexamethasone to treat multiple myeloma patients who have received between one and three prior therapies.
The elotuzumab application is supported by data from ELOQUENT-3, a randomized, phase 2 study that evaluated the addition of elotuzumab to pomalidomide and low-dose dexamethasone in patients with relapsed/refractory multiple myeloma.
Researchers presented findings from this study at the annual congress of the European Hematology Association in June 2018.
The overall response rate was 53% in the elotuzumab, pomalidomide, and low-dose dexamethasone (EPd) arm and 26% in the pomalidomide and low-dose dexamethasone (Pd) arm. The median progression-free survival was 10.3 months in the EPd arm and 4.7 months in the Pd arm (hazard ratio, 0.54; P = .0078), the researchers reported.
The researchers also reported that adverse events in the EPd arm were consistent with expectations based on previous results with elotuzumab and pomalidomide regimens.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The priority review typically shortens the time to an approval decision by a few months. The agency is expected to make a decision on elotuzumab by Dec. 27, 2018.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Study reveals ‘complete mental health’ among cancer survivors

New research suggests cancer survivors are just as likely as people without a history of cancer to have complete mental health (CMH), which is defined as “optimal functioning” and the “absence of psychopathology.”
In a study of nearly 11,000 Canadians, 77.5% of cancer survivors and 76.8% of people with no cancer history had CMH.
As for patients who were battling cancer at the time of the study, 66.1% had CMH.
Esme Fuller-Thomson, PhD, and Keri West, both of the University of Toronto in Canada, conducted this research and reported the findings in Aging & Mental Health.
“Cancer patients were doing much better than we had expected,” Dr. Fuller-Thomson said. “Two-thirds met our very stringent criteria for complete mental health . . . . The news for cancer survivors was even better, with three-quarters living in complete mental health, which is a prevalence comparable to that of individuals with no cancer history.”
This study included a nationally representative sample of Canadian community dwellers age 50 and older. Subjects had current cancer (n=438), previous cancer (n=1174), or no cancer history (n=9279).
Data were obtained from Statistics Canada’s 2012 Canadian Community Health Survey-Mental Health.
To meet criteria for CMH, subjects had to have all of the following:
- Absence of mental illness, addictions, and suicidal thoughts in the past year
- Almost daily happiness or life satisfaction in the past month
- Psychosocial well-being.
The prevalence of CMH was 77.5% in cancer survivors and 76.8% in subjects who had never had cancer. Both were significantly higher than the 66.1% prevalence of CMH in current cancer patients (P<0.001).
In a multivariable model adjusted for demographics, current cancer patients had 45% lower odds of CMH compared to subjects with no cancer history (odds ratio [OR]=0.55). The odds of CMH were comparable for cancer survivors and those without a history of cancer (OR=0.98).
The researchers also conducted a multivariable analysis in which they adjusted for “all relevant factors,” which included demographics as well as adverse childhood events, socioeconomic status, health variables, lifetime mental illness, etc.
In this analysis, current cancer patients had 37% lower odds of CMH than subjects with no cancer history (OR=0.63). And cancer survivors had comparable odds of CMH as those with no cancer history (OR=1.06).
The researchers identified several factors that were associated with CMH in the population affected by cancer.
“Among those with former or current cancer, the odds of complete mental health were higher for women, white, married, and older respondents, as well as those with higher income and those who did not have disabling pain nor functional limitations,” West said.
“We found that earlier difficulties cast a long shadow. Those who had been physically abused during their childhood and those who had ever had depression or anxiety disorders were less likely to be in complete mental health.”
West and Dr Fuller-Thomson emphasized that these results are only correlational, and it is impossible to determine causality due to the cross-sectional and observational nature of the study.
The pair also said future longitudinal research is needed to improve understanding of what pathways improve resilience and recovery among cancer patients.
New research suggests cancer survivors are just as likely as people without a history of cancer to have complete mental health (CMH), which is defined as “optimal functioning” and the “absence of psychopathology.”
In a study of nearly 11,000 Canadians, 77.5% of cancer survivors and 76.8% of people with no cancer history had CMH.
As for patients who were battling cancer at the time of the study, 66.1% had CMH.
Esme Fuller-Thomson, PhD, and Keri West, both of the University of Toronto in Canada, conducted this research and reported the findings in Aging & Mental Health.
“Cancer patients were doing much better than we had expected,” Dr. Fuller-Thomson said. “Two-thirds met our very stringent criteria for complete mental health . . . . The news for cancer survivors was even better, with three-quarters living in complete mental health, which is a prevalence comparable to that of individuals with no cancer history.”
This study included a nationally representative sample of Canadian community dwellers age 50 and older. Subjects had current cancer (n=438), previous cancer (n=1174), or no cancer history (n=9279).
Data were obtained from Statistics Canada’s 2012 Canadian Community Health Survey-Mental Health.
To meet criteria for CMH, subjects had to have all of the following:
- Absence of mental illness, addictions, and suicidal thoughts in the past year
- Almost daily happiness or life satisfaction in the past month
- Psychosocial well-being.
The prevalence of CMH was 77.5% in cancer survivors and 76.8% in subjects who had never had cancer. Both were significantly higher than the 66.1% prevalence of CMH in current cancer patients (P<0.001).
In a multivariable model adjusted for demographics, current cancer patients had 45% lower odds of CMH compared to subjects with no cancer history (odds ratio [OR]=0.55). The odds of CMH were comparable for cancer survivors and those without a history of cancer (OR=0.98).
The researchers also conducted a multivariable analysis in which they adjusted for “all relevant factors,” which included demographics as well as adverse childhood events, socioeconomic status, health variables, lifetime mental illness, etc.
In this analysis, current cancer patients had 37% lower odds of CMH than subjects with no cancer history (OR=0.63). And cancer survivors had comparable odds of CMH as those with no cancer history (OR=1.06).
The researchers identified several factors that were associated with CMH in the population affected by cancer.
“Among those with former or current cancer, the odds of complete mental health were higher for women, white, married, and older respondents, as well as those with higher income and those who did not have disabling pain nor functional limitations,” West said.
“We found that earlier difficulties cast a long shadow. Those who had been physically abused during their childhood and those who had ever had depression or anxiety disorders were less likely to be in complete mental health.”
West and Dr Fuller-Thomson emphasized that these results are only correlational, and it is impossible to determine causality due to the cross-sectional and observational nature of the study.
The pair also said future longitudinal research is needed to improve understanding of what pathways improve resilience and recovery among cancer patients.
New research suggests cancer survivors are just as likely as people without a history of cancer to have complete mental health (CMH), which is defined as “optimal functioning” and the “absence of psychopathology.”
In a study of nearly 11,000 Canadians, 77.5% of cancer survivors and 76.8% of people with no cancer history had CMH.
As for patients who were battling cancer at the time of the study, 66.1% had CMH.
Esme Fuller-Thomson, PhD, and Keri West, both of the University of Toronto in Canada, conducted this research and reported the findings in Aging & Mental Health.
“Cancer patients were doing much better than we had expected,” Dr. Fuller-Thomson said. “Two-thirds met our very stringent criteria for complete mental health . . . . The news for cancer survivors was even better, with three-quarters living in complete mental health, which is a prevalence comparable to that of individuals with no cancer history.”
This study included a nationally representative sample of Canadian community dwellers age 50 and older. Subjects had current cancer (n=438), previous cancer (n=1174), or no cancer history (n=9279).
Data were obtained from Statistics Canada’s 2012 Canadian Community Health Survey-Mental Health.
To meet criteria for CMH, subjects had to have all of the following:
- Absence of mental illness, addictions, and suicidal thoughts in the past year
- Almost daily happiness or life satisfaction in the past month
- Psychosocial well-being.
The prevalence of CMH was 77.5% in cancer survivors and 76.8% in subjects who had never had cancer. Both were significantly higher than the 66.1% prevalence of CMH in current cancer patients (P<0.001).
In a multivariable model adjusted for demographics, current cancer patients had 45% lower odds of CMH compared to subjects with no cancer history (odds ratio [OR]=0.55). The odds of CMH were comparable for cancer survivors and those without a history of cancer (OR=0.98).
The researchers also conducted a multivariable analysis in which they adjusted for “all relevant factors,” which included demographics as well as adverse childhood events, socioeconomic status, health variables, lifetime mental illness, etc.
In this analysis, current cancer patients had 37% lower odds of CMH than subjects with no cancer history (OR=0.63). And cancer survivors had comparable odds of CMH as those with no cancer history (OR=1.06).
The researchers identified several factors that were associated with CMH in the population affected by cancer.
“Among those with former or current cancer, the odds of complete mental health were higher for women, white, married, and older respondents, as well as those with higher income and those who did not have disabling pain nor functional limitations,” West said.
“We found that earlier difficulties cast a long shadow. Those who had been physically abused during their childhood and those who had ever had depression or anxiety disorders were less likely to be in complete mental health.”
West and Dr Fuller-Thomson emphasized that these results are only correlational, and it is impossible to determine causality due to the cross-sectional and observational nature of the study.
The pair also said future longitudinal research is needed to improve understanding of what pathways improve resilience and recovery among cancer patients.
Drug receives priority review for second MM indication
The US Food and Drug Administration (FDA) has accepted for priority review a supplemental biologics license application (sBLA) for elotuzumab (Empliciti).
With this sBLA, Bristol-Myers Squibb Company is seeking approval for elotuzumab in combination with pomalidomide and low-dose dexamethasone to treat patients with relapsed/refractory multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
The FDA expects to make a decision on the sBLA for elotuzumab by December 27, 2018.
Elotuzumab is already FDA-approved for use in combination with lenalidomide and dexamethasone to treat MM patients who have received one to three prior therapies.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Supporting trial
The sBLA for elotuzumab is supported by data from ELOQUENT-3, a randomized, phase 2 study in which researchers evaluated the addition of elotuzumab to pomalidomide and low-dose dexamethasone in patients with relapsed/refractory MM.
Data from this study were presented at the 23rd Congress of the European Hematology Association in June.
The trial randomized 117 MM patients who received two or more prior therapies and were either refractory or relapsed and refractory to lenalidomide and a proteasome inhibitor.
The patients were randomized to receive either elotuzumab, pomalidomide, and low-dose dexamethasone (EPd; n=60) or pomalidomide and low-dose dexamethasone (Pd; n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53% in the EPd arm and 26% in the Pd arm (odds ratio=3.25; P=0.0029).
The median progression-free survival was 10.3 months in the EPd arm and 4.7 months in the Pd arm (hazard ratio=0.54, P=0.0078).
Overall survival data were not mature at last follow-up, but there was a trend favoring EPd over Pd (hazard ratio=0.62).
The researchers said adverse events (AEs) in the EPd arm were consistent with expectations based on previous results with elotuzumab and pomalidomide regimens.
Grade 3-4 nonhematologic AEs (in the EPd and Pd arms, respectively) included constipation (2% and 0%), hyperglycemia (8% and 7%), bone pain (3% and 0%), dyspnea (3% and 2%), fatigue (0% and 4%), respiratory tract infection (0% and 2%), and upper respiratory tract infection (0% and 2%).
Grade 3-4 hematologic AEs (in the EPd and Pd arms, respectively) included anemia (10% and 20%), neutropenia (13% and 27%), thrombocytopenia (8% and 5%), and lymphopenia (8% and 2%).
Grade 3-4 AEs of special interest (in the EPd and Pd arms, respectively) included infections (13% and 22%), vascular disorders (3% and 0%), cardiac disorders (7% and 4%), and neoplasms (2% and 11%).
In the EPd arm, grade 5 AEs included infection (n=3), cardiac failure (n=1), and general physical health deterioration (n=1).
In the Pd arm, grade 5 AEs included malignant neoplasm progression (n=4), infection (n=1), multiple organ failure and infection (n=1), myocardial infarction (n=1), and plasma cell myeloma (n=1).
The US Food and Drug Administration (FDA) has accepted for priority review a supplemental biologics license application (sBLA) for elotuzumab (Empliciti).
With this sBLA, Bristol-Myers Squibb Company is seeking approval for elotuzumab in combination with pomalidomide and low-dose dexamethasone to treat patients with relapsed/refractory multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
The FDA expects to make a decision on the sBLA for elotuzumab by December 27, 2018.
Elotuzumab is already FDA-approved for use in combination with lenalidomide and dexamethasone to treat MM patients who have received one to three prior therapies.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Supporting trial
The sBLA for elotuzumab is supported by data from ELOQUENT-3, a randomized, phase 2 study in which researchers evaluated the addition of elotuzumab to pomalidomide and low-dose dexamethasone in patients with relapsed/refractory MM.
Data from this study were presented at the 23rd Congress of the European Hematology Association in June.
The trial randomized 117 MM patients who received two or more prior therapies and were either refractory or relapsed and refractory to lenalidomide and a proteasome inhibitor.
The patients were randomized to receive either elotuzumab, pomalidomide, and low-dose dexamethasone (EPd; n=60) or pomalidomide and low-dose dexamethasone (Pd; n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53% in the EPd arm and 26% in the Pd arm (odds ratio=3.25; P=0.0029).
The median progression-free survival was 10.3 months in the EPd arm and 4.7 months in the Pd arm (hazard ratio=0.54, P=0.0078).
Overall survival data were not mature at last follow-up, but there was a trend favoring EPd over Pd (hazard ratio=0.62).
The researchers said adverse events (AEs) in the EPd arm were consistent with expectations based on previous results with elotuzumab and pomalidomide regimens.
Grade 3-4 nonhematologic AEs (in the EPd and Pd arms, respectively) included constipation (2% and 0%), hyperglycemia (8% and 7%), bone pain (3% and 0%), dyspnea (3% and 2%), fatigue (0% and 4%), respiratory tract infection (0% and 2%), and upper respiratory tract infection (0% and 2%).
Grade 3-4 hematologic AEs (in the EPd and Pd arms, respectively) included anemia (10% and 20%), neutropenia (13% and 27%), thrombocytopenia (8% and 5%), and lymphopenia (8% and 2%).
Grade 3-4 AEs of special interest (in the EPd and Pd arms, respectively) included infections (13% and 22%), vascular disorders (3% and 0%), cardiac disorders (7% and 4%), and neoplasms (2% and 11%).
In the EPd arm, grade 5 AEs included infection (n=3), cardiac failure (n=1), and general physical health deterioration (n=1).
In the Pd arm, grade 5 AEs included malignant neoplasm progression (n=4), infection (n=1), multiple organ failure and infection (n=1), myocardial infarction (n=1), and plasma cell myeloma (n=1).
The US Food and Drug Administration (FDA) has accepted for priority review a supplemental biologics license application (sBLA) for elotuzumab (Empliciti).
With this sBLA, Bristol-Myers Squibb Company is seeking approval for elotuzumab in combination with pomalidomide and low-dose dexamethasone to treat patients with relapsed/refractory multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
The FDA expects to make a decision on the sBLA for elotuzumab by December 27, 2018.
Elotuzumab is already FDA-approved for use in combination with lenalidomide and dexamethasone to treat MM patients who have received one to three prior therapies.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Supporting trial
The sBLA for elotuzumab is supported by data from ELOQUENT-3, a randomized, phase 2 study in which researchers evaluated the addition of elotuzumab to pomalidomide and low-dose dexamethasone in patients with relapsed/refractory MM.
Data from this study were presented at the 23rd Congress of the European Hematology Association in June.
The trial randomized 117 MM patients who received two or more prior therapies and were either refractory or relapsed and refractory to lenalidomide and a proteasome inhibitor.
The patients were randomized to receive either elotuzumab, pomalidomide, and low-dose dexamethasone (EPd; n=60) or pomalidomide and low-dose dexamethasone (Pd; n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53% in the EPd arm and 26% in the Pd arm (odds ratio=3.25; P=0.0029).
The median progression-free survival was 10.3 months in the EPd arm and 4.7 months in the Pd arm (hazard ratio=0.54, P=0.0078).
Overall survival data were not mature at last follow-up, but there was a trend favoring EPd over Pd (hazard ratio=0.62).
The researchers said adverse events (AEs) in the EPd arm were consistent with expectations based on previous results with elotuzumab and pomalidomide regimens.
Grade 3-4 nonhematologic AEs (in the EPd and Pd arms, respectively) included constipation (2% and 0%), hyperglycemia (8% and 7%), bone pain (3% and 0%), dyspnea (3% and 2%), fatigue (0% and 4%), respiratory tract infection (0% and 2%), and upper respiratory tract infection (0% and 2%).
Grade 3-4 hematologic AEs (in the EPd and Pd arms, respectively) included anemia (10% and 20%), neutropenia (13% and 27%), thrombocytopenia (8% and 5%), and lymphopenia (8% and 2%).
Grade 3-4 AEs of special interest (in the EPd and Pd arms, respectively) included infections (13% and 22%), vascular disorders (3% and 0%), cardiac disorders (7% and 4%), and neoplasms (2% and 11%).
In the EPd arm, grade 5 AEs included infection (n=3), cardiac failure (n=1), and general physical health deterioration (n=1).
In the Pd arm, grade 5 AEs included malignant neoplasm progression (n=4), infection (n=1), multiple organ failure and infection (n=1), myocardial infarction (n=1), and plasma cell myeloma (n=1).