User login
EXPECT: Omalizumab pregnancy safety data called ‘reassuring’
HOUSTON – Babies born to mothers who took omalizumab just before or during pregnancy exhibited birth defects that are in line with the general population, according to data from the EXPECT registry.
EXPECT (the Xolair Pregnancy Registry) is an ongoing, prospective observational study of pregnant women who received at least 1 dose of omalizumab within 2 months of conception or during their pregnancies.

Dr. Jennifer Namazy of the Scripps Clinic in La Jolla, Calif., presented data from women enrolled between September 2006 and November 2013 at the annual meeting of the American Academy of Allergy, Asthma, and Immunology. The registry aims to enroll 250 women.
Dr. Namazy reported on 186 of the 207 expectant mothers for whom outcomes are recorded. Asthma severity was available for 164. Most (105 or 64%) had severe asthma, while 55 (34%) had moderate asthma, and 4 (2.4%) had mild disease. Each received omalizumab during the first trimester of pregnancy.
A total of 178 births were recorded, 174 of which were live. Of the 170 singletons, 24 (14%) were born at less than 37 weeks’ gestation and of those 3 (13%) were considered small for gestational age (weight less than 10th percentile for gestational age).
Of 140 single-born infants who were carried to full term and also had relevant weight data, 4 (3%) had low birth weight (less than 2.5 kg) and 16 (11%) were considered small for gestational age. Overall, 27 (15%) infants had confirmed congenital anomalies and 11 (6.2%) had a major birth defect.
“This is all very reassuring data,” Dr. Namazy said in an interview. “In terms of the outcomes – major defects and conditional defects – these results are not too unusual, [compared with] the general population. But when we’re looking at infant outcomes, it’s not too different from the severe asthma population.”
She noted, however, that “it’s hard to make large-scale conclusions from this sample size. When you’re talking about congenital malformations, they’re so rare and uncommon that you need a very large population in order to reach any kind of conclusion. But looking at these [data], we’re reassured because there isn’t any consistent information that’s disconcerting; it’s all over the spectrum, and so there’s no significant increase in the rate of birth rate defects that we’re seeing.”
Omalizumab is marketed by Genentech as Xolair. The EXPECT study is funded by Genentech and Novartis Pharma AG. Dr. Namazy is affiliated with Genentech.
HOUSTON – Babies born to mothers who took omalizumab just before or during pregnancy exhibited birth defects that are in line with the general population, according to data from the EXPECT registry.
EXPECT (the Xolair Pregnancy Registry) is an ongoing, prospective observational study of pregnant women who received at least 1 dose of omalizumab within 2 months of conception or during their pregnancies.
Dr. Jennifer Namazy of the Scripps Clinic in La Jolla, Calif., presented data from women enrolled between September 2006 and November 2013 at the annual meeting of the American Academy of Allergy, Asthma, and Immunology. The registry aims to enroll 250 women.
Dr. Namazy reported on 186 of the 207 expectant mothers for whom outcomes are recorded. Asthma severity was available for 164. Most (105 or 64%) had severe asthma, while 55 (34%) had moderate asthma, and 4 (2.4%) had mild disease. Each received omalizumab during the first trimester of pregnancy.
A total of 178 births were recorded, 174 of which were live. Of the 170 singletons, 24 (14%) were born at less than 37 weeks’ gestation and of those 3 (13%) were considered small for gestational age (weight less than 10th percentile for gestational age).
Of 140 single-born infants who were carried to full term and also had relevant weight data, 4 (3%) had low birth weight (less than 2.5 kg) and 16 (11%) were considered small for gestational age. Overall, 27 (15%) infants had confirmed congenital anomalies and 11 (6.2%) had a major birth defect.
“This is all very reassuring data,” Dr. Namazy said in an interview. “In terms of the outcomes – major defects and conditional defects – these results are not too unusual, [compared with] the general population. But when we’re looking at infant outcomes, it’s not too different from the severe asthma population.”
She noted, however, that “it’s hard to make large-scale conclusions from this sample size. When you’re talking about congenital malformations, they’re so rare and uncommon that you need a very large population in order to reach any kind of conclusion. But looking at these [data], we’re reassured because there isn’t any consistent information that’s disconcerting; it’s all over the spectrum, and so there’s no significant increase in the rate of birth rate defects that we’re seeing.”
Omalizumab is marketed by Genentech as Xolair. The EXPECT study is funded by Genentech and Novartis Pharma AG. Dr. Namazy is affiliated with Genentech.
HOUSTON – Babies born to mothers who took omalizumab just before or during pregnancy exhibited birth defects that are in line with the general population, according to data from the EXPECT registry.
EXPECT (the Xolair Pregnancy Registry) is an ongoing, prospective observational study of pregnant women who received at least 1 dose of omalizumab within 2 months of conception or during their pregnancies.
Dr. Jennifer Namazy of the Scripps Clinic in La Jolla, Calif., presented data from women enrolled between September 2006 and November 2013 at the annual meeting of the American Academy of Allergy, Asthma, and Immunology. The registry aims to enroll 250 women.
Dr. Namazy reported on 186 of the 207 expectant mothers for whom outcomes are recorded. Asthma severity was available for 164. Most (105 or 64%) had severe asthma, while 55 (34%) had moderate asthma, and 4 (2.4%) had mild disease. Each received omalizumab during the first trimester of pregnancy.
A total of 178 births were recorded, 174 of which were live. Of the 170 singletons, 24 (14%) were born at less than 37 weeks’ gestation and of those 3 (13%) were considered small for gestational age (weight less than 10th percentile for gestational age).
Of 140 single-born infants who were carried to full term and also had relevant weight data, 4 (3%) had low birth weight (less than 2.5 kg) and 16 (11%) were considered small for gestational age. Overall, 27 (15%) infants had confirmed congenital anomalies and 11 (6.2%) had a major birth defect.
“This is all very reassuring data,” Dr. Namazy said in an interview. “In terms of the outcomes – major defects and conditional defects – these results are not too unusual, [compared with] the general population. But when we’re looking at infant outcomes, it’s not too different from the severe asthma population.”
She noted, however, that “it’s hard to make large-scale conclusions from this sample size. When you’re talking about congenital malformations, they’re so rare and uncommon that you need a very large population in order to reach any kind of conclusion. But looking at these [data], we’re reassured because there isn’t any consistent information that’s disconcerting; it’s all over the spectrum, and so there’s no significant increase in the rate of birth rate defects that we’re seeing.”
Omalizumab is marketed by Genentech as Xolair. The EXPECT study is funded by Genentech and Novartis Pharma AG. Dr. Namazy is affiliated with Genentech.
AT 2015 AAAAI ANNUAL MEETING
Key clinical point: Omalizumab does not appear to increase the risk of birth defects or adverse outcomes in pregnant women and their babies.
Major finding: Of 140 singleton infants carried to full term and for whom relevant weight data was available, 4 (2.9%) had low birth weight and 16 (11.4%) were considered small for gestational age, 27 (15.2%) had congenital anomalies, and 11 (6.2%) had a major birth defect.
Data source: EXPECT trial – an ongoing observational study of 207 prospectively enrolled women.
Disclosures: EXPECT is funded by Genentech and Novartis Pharma AG. Dr. Namazy is affiliated with Genentech.

Guideline clarifies first-line treatment for allergic rhinitis
First-line treatment for allergic rhinitis should include intranasal steroids, as well as less-sedating second-generation oral antihistamines for patients whose primary complaints are sneezing and itching, according to a new clinical practice guideline published online Feb. 2 in Otolaryngology–Head and Neck Surgery.
In contrast, sinonasal imaging should not be routine when patients first present with symptoms consistent with allergic rhinitis, and oral leukotriene receptor antagonists are not recommended as first-line therapy, said Dr. Michael D. Seidman of Henry Ford West Bloomfield (Mich.) Hospital and chair of the guideline working group, and his associates.
Dr. Seidman and a panel of 20 experts in otolaryngology, allergy and immunology, internal medicine, family medicine, pediatrics, sleep medicine, advanced practice nursing, complementary and alternative medicine, and consumer advocacy developed the new practice guideline to enable clinicians in all settings to improve patient care and reduce harmful or unnecessary variations in care for allergic rhinitis.
“The guideline is intended to focus on a limited number of quality improvement opportunities deemed most important by the working group and is not intended to be a comprehensive reference for diagnosing and managing allergic rhinitis,” the authors noted.
During the course of 1 year, the working group reviewed 1,605 randomized, controlled trials, 31 existing clinical practice guidelines, and 390 systematic reviews of the literature regarding allergic rhinitis in adults and children older than age 2 years. They then compiled 14 key recommendations that underwent extensive peer review, which have been published online and as a supplement to the February issue (Otolaryngol. Head Neck Surg. 2015;152:S1-S43).
In addition to the recommendations noted above, the guideline advises:
* Clinicians should diagnose allergic rhinitis when patients present with a history and physical exam consistent with the disorder (including clear rhinorrhea, nasal congestion, pale discoloration of the nasal mucosa, and red, watery eyes) plus symptoms of nasal congestion, runny nose, itchy nose, or sneezing.
* Clinicians should perform and interpret (or refer patients for) specific IgE allergy testing for allergic rhinitis that doesn’t respond to empiric treatment, or when the diagnosis is uncertain, or when identifying the specific causative allergen would allow targeted therapy.
* Clinicians should assess diagnosed patients for associated conditions such as asthma, atopic dermatitis, sleep-disordered breathing, conjunctivitis, rhinosinusitis, and otitis media, and should document that in the medical record.
* Clinicians should offer (or refer patients for) sublingual or subcutaneous immunotherapy when allergic rhinitis doesn’t respond adequately to pharmacologic therapy.
* Clinicians may advise avoidance of known allergens or controlling the patient’s environment by such measures as removing pets, using air filtration systems, using dust-mite–reducing covers for bedding, and using acaricides.
* Clinicians may offer intranasal antihistamines for patients with seasonal, perennial or episodic allergic rhinitis.*
* Clinicians may offer (or refer patients for) reduction of the inferior turbinates for patients who have nasal airway obstruction or enlarged turbinates.
* Clinicians may offer (or refer patient for) acupuncture if they are interested in nonpharmacologic therapy.
*Clinicians may offer combination pharmacologic therapy in patients with allergic rhinitis who have inadequate response to pharmacologic monotherapy.
The working group offered no recommendations concerning herbal therapy for allergic rhinitis, because of the limited literature on those substances and concern about their safety.
The full text of the guideline and its supporting data are available free of charge at www.entnet.org. In addition, an algorithm of the guideline’s action statements and a table of common allergic rhinitis clinical scenarios are available as quick reference guides for clinicians.
The American Academy of Otolaryngology–Head and Neck Surgery Foundation funded the guideline. Dr. Seidman reported being medical director of the Scientific Advisory Board of Visalus, founder of the Body Language Vitamin, and holder of six patents related to dietary supplements, aircraft, and middle ear and brain implants. His associates reported ties to Acclarent/Johnson/Johnson, FirstLine Medical, GlaxoSmithKline, Intersect, MEDA, Medtronic, Merck, Mylan, Novartis, TEVA, Transit of Venus, Sanofi, Sunovion Pharmaceuticals, and WellPoint.
*Correction, 2/18/2015: An earlier version of this story misstated the guideline for the use of intranasal antihistamines.
First-line treatment for allergic rhinitis should include intranasal steroids, as well as less-sedating second-generation oral antihistamines for patients whose primary complaints are sneezing and itching, according to a new clinical practice guideline published online Feb. 2 in Otolaryngology–Head and Neck Surgery.
In contrast, sinonasal imaging should not be routine when patients first present with symptoms consistent with allergic rhinitis, and oral leukotriene receptor antagonists are not recommended as first-line therapy, said Dr. Michael D. Seidman of Henry Ford West Bloomfield (Mich.) Hospital and chair of the guideline working group, and his associates.
Dr. Seidman and a panel of 20 experts in otolaryngology, allergy and immunology, internal medicine, family medicine, pediatrics, sleep medicine, advanced practice nursing, complementary and alternative medicine, and consumer advocacy developed the new practice guideline to enable clinicians in all settings to improve patient care and reduce harmful or unnecessary variations in care for allergic rhinitis.
“The guideline is intended to focus on a limited number of quality improvement opportunities deemed most important by the working group and is not intended to be a comprehensive reference for diagnosing and managing allergic rhinitis,” the authors noted.
During the course of 1 year, the working group reviewed 1,605 randomized, controlled trials, 31 existing clinical practice guidelines, and 390 systematic reviews of the literature regarding allergic rhinitis in adults and children older than age 2 years. They then compiled 14 key recommendations that underwent extensive peer review, which have been published online and as a supplement to the February issue (Otolaryngol. Head Neck Surg. 2015;152:S1-S43).
In addition to the recommendations noted above, the guideline advises:
* Clinicians should diagnose allergic rhinitis when patients present with a history and physical exam consistent with the disorder (including clear rhinorrhea, nasal congestion, pale discoloration of the nasal mucosa, and red, watery eyes) plus symptoms of nasal congestion, runny nose, itchy nose, or sneezing.
* Clinicians should perform and interpret (or refer patients for) specific IgE allergy testing for allergic rhinitis that doesn’t respond to empiric treatment, or when the diagnosis is uncertain, or when identifying the specific causative allergen would allow targeted therapy.
* Clinicians should assess diagnosed patients for associated conditions such as asthma, atopic dermatitis, sleep-disordered breathing, conjunctivitis, rhinosinusitis, and otitis media, and should document that in the medical record.
* Clinicians should offer (or refer patients for) sublingual or subcutaneous immunotherapy when allergic rhinitis doesn’t respond adequately to pharmacologic therapy.
* Clinicians may advise avoidance of known allergens or controlling the patient’s environment by such measures as removing pets, using air filtration systems, using dust-mite–reducing covers for bedding, and using acaricides.
* Clinicians may offer intranasal antihistamines for patients with seasonal, perennial or episodic allergic rhinitis.*
* Clinicians may offer (or refer patients for) reduction of the inferior turbinates for patients who have nasal airway obstruction or enlarged turbinates.
* Clinicians may offer (or refer patient for) acupuncture if they are interested in nonpharmacologic therapy.
*Clinicians may offer combination pharmacologic therapy in patients with allergic rhinitis who have inadequate response to pharmacologic monotherapy.
The working group offered no recommendations concerning herbal therapy for allergic rhinitis, because of the limited literature on those substances and concern about their safety.
The full text of the guideline and its supporting data are available free of charge at www.entnet.org. In addition, an algorithm of the guideline’s action statements and a table of common allergic rhinitis clinical scenarios are available as quick reference guides for clinicians.
The American Academy of Otolaryngology–Head and Neck Surgery Foundation funded the guideline. Dr. Seidman reported being medical director of the Scientific Advisory Board of Visalus, founder of the Body Language Vitamin, and holder of six patents related to dietary supplements, aircraft, and middle ear and brain implants. His associates reported ties to Acclarent/Johnson/Johnson, FirstLine Medical, GlaxoSmithKline, Intersect, MEDA, Medtronic, Merck, Mylan, Novartis, TEVA, Transit of Venus, Sanofi, Sunovion Pharmaceuticals, and WellPoint.
*Correction, 2/18/2015: An earlier version of this story misstated the guideline for the use of intranasal antihistamines.
First-line treatment for allergic rhinitis should include intranasal steroids, as well as less-sedating second-generation oral antihistamines for patients whose primary complaints are sneezing and itching, according to a new clinical practice guideline published online Feb. 2 in Otolaryngology–Head and Neck Surgery.
In contrast, sinonasal imaging should not be routine when patients first present with symptoms consistent with allergic rhinitis, and oral leukotriene receptor antagonists are not recommended as first-line therapy, said Dr. Michael D. Seidman of Henry Ford West Bloomfield (Mich.) Hospital and chair of the guideline working group, and his associates.
Dr. Seidman and a panel of 20 experts in otolaryngology, allergy and immunology, internal medicine, family medicine, pediatrics, sleep medicine, advanced practice nursing, complementary and alternative medicine, and consumer advocacy developed the new practice guideline to enable clinicians in all settings to improve patient care and reduce harmful or unnecessary variations in care for allergic rhinitis.
“The guideline is intended to focus on a limited number of quality improvement opportunities deemed most important by the working group and is not intended to be a comprehensive reference for diagnosing and managing allergic rhinitis,” the authors noted.
During the course of 1 year, the working group reviewed 1,605 randomized, controlled trials, 31 existing clinical practice guidelines, and 390 systematic reviews of the literature regarding allergic rhinitis in adults and children older than age 2 years. They then compiled 14 key recommendations that underwent extensive peer review, which have been published online and as a supplement to the February issue (Otolaryngol. Head Neck Surg. 2015;152:S1-S43).
In addition to the recommendations noted above, the guideline advises:
* Clinicians should diagnose allergic rhinitis when patients present with a history and physical exam consistent with the disorder (including clear rhinorrhea, nasal congestion, pale discoloration of the nasal mucosa, and red, watery eyes) plus symptoms of nasal congestion, runny nose, itchy nose, or sneezing.
* Clinicians should perform and interpret (or refer patients for) specific IgE allergy testing for allergic rhinitis that doesn’t respond to empiric treatment, or when the diagnosis is uncertain, or when identifying the specific causative allergen would allow targeted therapy.
* Clinicians should assess diagnosed patients for associated conditions such as asthma, atopic dermatitis, sleep-disordered breathing, conjunctivitis, rhinosinusitis, and otitis media, and should document that in the medical record.
* Clinicians should offer (or refer patients for) sublingual or subcutaneous immunotherapy when allergic rhinitis doesn’t respond adequately to pharmacologic therapy.
* Clinicians may advise avoidance of known allergens or controlling the patient’s environment by such measures as removing pets, using air filtration systems, using dust-mite–reducing covers for bedding, and using acaricides.
* Clinicians may offer intranasal antihistamines for patients with seasonal, perennial or episodic allergic rhinitis.*
* Clinicians may offer (or refer patients for) reduction of the inferior turbinates for patients who have nasal airway obstruction or enlarged turbinates.
* Clinicians may offer (or refer patient for) acupuncture if they are interested in nonpharmacologic therapy.
*Clinicians may offer combination pharmacologic therapy in patients with allergic rhinitis who have inadequate response to pharmacologic monotherapy.
The working group offered no recommendations concerning herbal therapy for allergic rhinitis, because of the limited literature on those substances and concern about their safety.
The full text of the guideline and its supporting data are available free of charge at www.entnet.org. In addition, an algorithm of the guideline’s action statements and a table of common allergic rhinitis clinical scenarios are available as quick reference guides for clinicians.
The American Academy of Otolaryngology–Head and Neck Surgery Foundation funded the guideline. Dr. Seidman reported being medical director of the Scientific Advisory Board of Visalus, founder of the Body Language Vitamin, and holder of six patents related to dietary supplements, aircraft, and middle ear and brain implants. His associates reported ties to Acclarent/Johnson/Johnson, FirstLine Medical, GlaxoSmithKline, Intersect, MEDA, Medtronic, Merck, Mylan, Novartis, TEVA, Transit of Venus, Sanofi, Sunovion Pharmaceuticals, and WellPoint.
*Correction, 2/18/2015: An earlier version of this story misstated the guideline for the use of intranasal antihistamines.
FROM OTOLARYNGOLOGY–HEAD AND NECK SURGERY
Key clinical point: First-line treatment for allergic rhinitis should include intranasal steroids and second-generation oral antihistamines, and should not include leukotriene receptor antagonists or sinonasal imaging studies.
Major finding: A panel of 20 experts took 1 year to review the literature and develop action items focusing on a limited number of quality improvement opportunities they deemed most important to improve patient care.
Data source: A review of 1,605 randomized, controlled trials, 31 sets of practice guidelines, and 390 systematic reviews regarding allergic rhinitis, and a compilation of 14 recommendations for managing the disorder.
Disclosures: The American Academy of Otolaryngology–Head and Neck Surgery Foundation funded the guideline. Dr. Seidman reported being medical director of the Scientific Advisory Board of Visalus, founder of the Body Language Vitamin, and holder of six patents related to dietary supplements, aircraft, and middle ear and brain implants. His associates reported ties to Acclarent/Johnson/Johnson, FirstLine Medical, GlaxoSmithKline, Intersect, MEDA, Medtronic, Merck, Mylan, Novartis, TEVA, Transit of Venus, Sanofi, Sunovion Pharmaceuticals, and WellPoint.
Icatibant rapidly resolved ACE inhibitor–induced angioedema
Angioedema caused by ACE inhibitors resolved 70% more rapidly with icatibant than did standard therapy in a multicenter phase II study in Germany, which was reported online Jan. 29 in the New England Journal of Medicine.
Because of the increasing use of ACE inhibitors, approximately one-third of all cases of angioedema treated in emergency departments now are attributed to these agents. The current standard ED treatment of ACE inhibitor–induced angioedema is glucocorticoids plus antihistamines. However, patients generally don’t respond to this therapy, likely because this form of angioedema isn’t a histamine-mediated reaction. Instead, it is thought by some to be a bradykinin-mediated reaction, said Dr. Murat Bas of the department of otorhinolaryngology, Technische Universität München (Germany), and his associates.
Icatibant injections (Firazyr) are approved by the Food and Drug Administration for the treatment of acute attacks of hereditary angioedema in adults 18 years of age and older. The drug also is being studied in the United States for the treatment of ACE-inhibitor–induced angioedema.
Since ACE inhibitors interfere with the breakdown of bradykinin, and bradykinin-mediated hereditary angioedema is usually treated with bradykinin-receptor antagonists such as icatibant, the investigators performed a double-blind randomized trial comparing subcutaneous icatibant against standard treatment in 27 adults who presented to four German EDs during a 1.5-year period.
The primary endpoint – the time to complete resolution of ACE inhibitor–induced angioedema – was 8 hours with icatibant and 27 hours with standard therapy. Angioedema resolved within 4 hours in five patients (38%) given icatibant; none of the patients given standard therapy responded that quickly. The onset of symptom relief was 2 hours with icatibant and 12 hours with standard glucocorticoids plus antihistamines, a significant difference as judged by the study participants and the researchers. Also, the physician-assessed severity of angioedema began to abate within 1 hour of icatibant administration and within 8 hours for standard treatment (N. Engl. J. Med. 2015 Jan. 29 [doi:10.1056/NEJMoa1312524]).
“Although the sample size in this trial was too small to allow for a robust evaluation of safety, no patient discontinued participation in the study owing to adverse events,” Dr. Bas and his associates added.
Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Angioedema caused by ACE inhibitors resolved 70% more rapidly with icatibant than did standard therapy in a multicenter phase II study in Germany, which was reported online Jan. 29 in the New England Journal of Medicine.
Because of the increasing use of ACE inhibitors, approximately one-third of all cases of angioedema treated in emergency departments now are attributed to these agents. The current standard ED treatment of ACE inhibitor–induced angioedema is glucocorticoids plus antihistamines. However, patients generally don’t respond to this therapy, likely because this form of angioedema isn’t a histamine-mediated reaction. Instead, it is thought by some to be a bradykinin-mediated reaction, said Dr. Murat Bas of the department of otorhinolaryngology, Technische Universität München (Germany), and his associates.
Icatibant injections (Firazyr) are approved by the Food and Drug Administration for the treatment of acute attacks of hereditary angioedema in adults 18 years of age and older. The drug also is being studied in the United States for the treatment of ACE-inhibitor–induced angioedema.
Since ACE inhibitors interfere with the breakdown of bradykinin, and bradykinin-mediated hereditary angioedema is usually treated with bradykinin-receptor antagonists such as icatibant, the investigators performed a double-blind randomized trial comparing subcutaneous icatibant against standard treatment in 27 adults who presented to four German EDs during a 1.5-year period.
The primary endpoint – the time to complete resolution of ACE inhibitor–induced angioedema – was 8 hours with icatibant and 27 hours with standard therapy. Angioedema resolved within 4 hours in five patients (38%) given icatibant; none of the patients given standard therapy responded that quickly. The onset of symptom relief was 2 hours with icatibant and 12 hours with standard glucocorticoids plus antihistamines, a significant difference as judged by the study participants and the researchers. Also, the physician-assessed severity of angioedema began to abate within 1 hour of icatibant administration and within 8 hours for standard treatment (N. Engl. J. Med. 2015 Jan. 29 [doi:10.1056/NEJMoa1312524]).
“Although the sample size in this trial was too small to allow for a robust evaluation of safety, no patient discontinued participation in the study owing to adverse events,” Dr. Bas and his associates added.
Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Angioedema caused by ACE inhibitors resolved 70% more rapidly with icatibant than did standard therapy in a multicenter phase II study in Germany, which was reported online Jan. 29 in the New England Journal of Medicine.
Because of the increasing use of ACE inhibitors, approximately one-third of all cases of angioedema treated in emergency departments now are attributed to these agents. The current standard ED treatment of ACE inhibitor–induced angioedema is glucocorticoids plus antihistamines. However, patients generally don’t respond to this therapy, likely because this form of angioedema isn’t a histamine-mediated reaction. Instead, it is thought by some to be a bradykinin-mediated reaction, said Dr. Murat Bas of the department of otorhinolaryngology, Technische Universität München (Germany), and his associates.
Icatibant injections (Firazyr) are approved by the Food and Drug Administration for the treatment of acute attacks of hereditary angioedema in adults 18 years of age and older. The drug also is being studied in the United States for the treatment of ACE-inhibitor–induced angioedema.
Since ACE inhibitors interfere with the breakdown of bradykinin, and bradykinin-mediated hereditary angioedema is usually treated with bradykinin-receptor antagonists such as icatibant, the investigators performed a double-blind randomized trial comparing subcutaneous icatibant against standard treatment in 27 adults who presented to four German EDs during a 1.5-year period.
The primary endpoint – the time to complete resolution of ACE inhibitor–induced angioedema – was 8 hours with icatibant and 27 hours with standard therapy. Angioedema resolved within 4 hours in five patients (38%) given icatibant; none of the patients given standard therapy responded that quickly. The onset of symptom relief was 2 hours with icatibant and 12 hours with standard glucocorticoids plus antihistamines, a significant difference as judged by the study participants and the researchers. Also, the physician-assessed severity of angioedema began to abate within 1 hour of icatibant administration and within 8 hours for standard treatment (N. Engl. J. Med. 2015 Jan. 29 [doi:10.1056/NEJMoa1312524]).
“Although the sample size in this trial was too small to allow for a robust evaluation of safety, no patient discontinued participation in the study owing to adverse events,” Dr. Bas and his associates added.
Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Key clinical point: Icatibant may prove to be a more effective treatment than glucocorticoids and antihistamines for ACE inhibitor–induced angioedema.
Major finding: The time to complete resolution of ACE inhibitor–induced angioedema was 8 hours with icatibant and 27 hours with standard therapy.
Data source: A multicenter double-blind randomized phase II clinical trial involving 27 adults hospitalized in Germany for ACE inhibitor–induced angioedema during a 1.5-year period.
Disclosures: This study was supported by an educational grant from Shire and by the Federal Ministry of Education and Research of Germany. Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Link Between Early Exposure to Acetaminophen and Childhood Asthma Found Weak, Overstated
The reported link between early life exposure to acetaminophen and the development of asthma in children is “weak” and “overstated” based on currently available evidence, according to a report published by the Archives of Disease in Childhood.
In a review of currently available data culled from Embase and PubMed databases, 1,192 relevant studies conducted between 1967 and 2013 were analyzed, of which 11 were included for analysis. Of these 11 studies, 5 found “increased odds” that exposure to acetaminophen during the first trimester of pregnancy could lead to development of asthma (pooled odds ratio, 1.39); however, there was a high degree of between-study heterogeneity among the trials (I2 = 64.2%, P = .03), reported Dr. M. Cheelo of the University of Melbourne, and associates.
Of those five, only two studies examined the effects of acetaminophen exposure during the second trimester, but attained widely disparate results: Study one reported an OR of 1.06, while the other reported an OR of 2.15, with I2 = 80%. Two studies also tested acetaminophen exposure during the third trimester and found a “weak association,” with a pooled OR of 1.17. Three studies look at acetaminophen exposure through an entire pregnancy, but all had “significant heterogeneity” in their findings (OR = 1.65, 1.22, and 0.74; I2 = 89%). Only one study that was examined adjusted for respiratory tract infections during pregnancy, but according to the authors, “all studies that adjusted for early life respiratory tract infections found a reduction in the association between [acetaminophen] exposure and subsequent childhood asthma” (Arch. Dis. Child. 2014 [doi:10.1136/archdischild-2012-303043]).
The other 6 of the 11 total studies examined acetaminophen exposure over the first 2 years of life. Three of these studies found a “weak positive association,” as did four studies directly comparing children with and without acetaminophen exposure. All but one study adjusted results for respiratory tract infections during pregnancy, which caused a “moderate attenuation of the association between frequency of [acetaminophen] intake and childhood asthma.” Consequently, investigators concluded that “evidence of an association between early life [acetaminophen] and asthma is often overstated, and there is currently insufficient evidence to support changing guidelines in the use of this medicine.”
The authors reported no relevant financial conflicts of interest.
The reported link between early life exposure to acetaminophen and the development of asthma in children is “weak” and “overstated” based on currently available evidence, according to a report published by the Archives of Disease in Childhood.
In a review of currently available data culled from Embase and PubMed databases, 1,192 relevant studies conducted between 1967 and 2013 were analyzed, of which 11 were included for analysis. Of these 11 studies, 5 found “increased odds” that exposure to acetaminophen during the first trimester of pregnancy could lead to development of asthma (pooled odds ratio, 1.39); however, there was a high degree of between-study heterogeneity among the trials (I2 = 64.2%, P = .03), reported Dr. M. Cheelo of the University of Melbourne, and associates.
Of those five, only two studies examined the effects of acetaminophen exposure during the second trimester, but attained widely disparate results: Study one reported an OR of 1.06, while the other reported an OR of 2.15, with I2 = 80%. Two studies also tested acetaminophen exposure during the third trimester and found a “weak association,” with a pooled OR of 1.17. Three studies look at acetaminophen exposure through an entire pregnancy, but all had “significant heterogeneity” in their findings (OR = 1.65, 1.22, and 0.74; I2 = 89%). Only one study that was examined adjusted for respiratory tract infections during pregnancy, but according to the authors, “all studies that adjusted for early life respiratory tract infections found a reduction in the association between [acetaminophen] exposure and subsequent childhood asthma” (Arch. Dis. Child. 2014 [doi:10.1136/archdischild-2012-303043]).
The other 6 of the 11 total studies examined acetaminophen exposure over the first 2 years of life. Three of these studies found a “weak positive association,” as did four studies directly comparing children with and without acetaminophen exposure. All but one study adjusted results for respiratory tract infections during pregnancy, which caused a “moderate attenuation of the association between frequency of [acetaminophen] intake and childhood asthma.” Consequently, investigators concluded that “evidence of an association between early life [acetaminophen] and asthma is often overstated, and there is currently insufficient evidence to support changing guidelines in the use of this medicine.”
The authors reported no relevant financial conflicts of interest.
The reported link between early life exposure to acetaminophen and the development of asthma in children is “weak” and “overstated” based on currently available evidence, according to a report published by the Archives of Disease in Childhood.
In a review of currently available data culled from Embase and PubMed databases, 1,192 relevant studies conducted between 1967 and 2013 were analyzed, of which 11 were included for analysis. Of these 11 studies, 5 found “increased odds” that exposure to acetaminophen during the first trimester of pregnancy could lead to development of asthma (pooled odds ratio, 1.39); however, there was a high degree of between-study heterogeneity among the trials (I2 = 64.2%, P = .03), reported Dr. M. Cheelo of the University of Melbourne, and associates.
Of those five, only two studies examined the effects of acetaminophen exposure during the second trimester, but attained widely disparate results: Study one reported an OR of 1.06, while the other reported an OR of 2.15, with I2 = 80%. Two studies also tested acetaminophen exposure during the third trimester and found a “weak association,” with a pooled OR of 1.17. Three studies look at acetaminophen exposure through an entire pregnancy, but all had “significant heterogeneity” in their findings (OR = 1.65, 1.22, and 0.74; I2 = 89%). Only one study that was examined adjusted for respiratory tract infections during pregnancy, but according to the authors, “all studies that adjusted for early life respiratory tract infections found a reduction in the association between [acetaminophen] exposure and subsequent childhood asthma” (Arch. Dis. Child. 2014 [doi:10.1136/archdischild-2012-303043]).
The other 6 of the 11 total studies examined acetaminophen exposure over the first 2 years of life. Three of these studies found a “weak positive association,” as did four studies directly comparing children with and without acetaminophen exposure. All but one study adjusted results for respiratory tract infections during pregnancy, which caused a “moderate attenuation of the association between frequency of [acetaminophen] intake and childhood asthma.” Consequently, investigators concluded that “evidence of an association between early life [acetaminophen] and asthma is often overstated, and there is currently insufficient evidence to support changing guidelines in the use of this medicine.”
The authors reported no relevant financial conflicts of interest.

FDA Approves Pediatric Dosage of QNASL
The Food and Drug Administration has approved the 40-mcg dose of QNASL for use in the treatment of nasal symptoms associated with allergic rhinitis in children aged 4-11 years, making it the first waterless nasal allergy spray approved for use in children as young as 4 years old.
“Through the availability of QNASL [beclomethasone dipropionate] 40 mcg, we are aiming to aid children and their caregivers in better managing the burdensome symptoms associated with nasal allergies,” said Dr. Tushar Shah, senior vice president of Teva Global Respiratory Research and Development.
The FDA approved beclomethasone dipropionate 40 mcg based on data from three double-blind, placebo-controlled studies of children aged 4-11 years. In those studies, once-daily beclomethasone dipropionate 40 mcg alleviated allergy symptoms in children with both seasonal and perennial allergic rhinitis with a minimum of adverse effects. The most common side effects were nosebleed and ulcers, which was “consistent with those seen in previous clinical studies of QNASL Nasal Aerosol,” according to a statement from the manufacturer.
QNASL40 mcg is expected to become available in February 2015.
The Food and Drug Administration has approved the 40-mcg dose of QNASL for use in the treatment of nasal symptoms associated with allergic rhinitis in children aged 4-11 years, making it the first waterless nasal allergy spray approved for use in children as young as 4 years old.
“Through the availability of QNASL [beclomethasone dipropionate] 40 mcg, we are aiming to aid children and their caregivers in better managing the burdensome symptoms associated with nasal allergies,” said Dr. Tushar Shah, senior vice president of Teva Global Respiratory Research and Development.
The FDA approved beclomethasone dipropionate 40 mcg based on data from three double-blind, placebo-controlled studies of children aged 4-11 years. In those studies, once-daily beclomethasone dipropionate 40 mcg alleviated allergy symptoms in children with both seasonal and perennial allergic rhinitis with a minimum of adverse effects. The most common side effects were nosebleed and ulcers, which was “consistent with those seen in previous clinical studies of QNASL Nasal Aerosol,” according to a statement from the manufacturer.
QNASL40 mcg is expected to become available in February 2015.
The Food and Drug Administration has approved the 40-mcg dose of QNASL for use in the treatment of nasal symptoms associated with allergic rhinitis in children aged 4-11 years, making it the first waterless nasal allergy spray approved for use in children as young as 4 years old.
“Through the availability of QNASL [beclomethasone dipropionate] 40 mcg, we are aiming to aid children and their caregivers in better managing the burdensome symptoms associated with nasal allergies,” said Dr. Tushar Shah, senior vice president of Teva Global Respiratory Research and Development.
The FDA approved beclomethasone dipropionate 40 mcg based on data from three double-blind, placebo-controlled studies of children aged 4-11 years. In those studies, once-daily beclomethasone dipropionate 40 mcg alleviated allergy symptoms in children with both seasonal and perennial allergic rhinitis with a minimum of adverse effects. The most common side effects were nosebleed and ulcers, which was “consistent with those seen in previous clinical studies of QNASL Nasal Aerosol,” according to a statement from the manufacturer.
QNASL40 mcg is expected to become available in February 2015.
FDA approves pediatric dosage of QNASL
The Food and Drug Administration has approved the 40-mcg dose of QNASL for use in the treatment of nasal symptoms associated with allergic rhinitis in children aged 4-11 years, making it the first waterless nasal allergy spray approved for use in children as young as 4 years old.
“Through the availability of QNASL [beclomethasone dipropionate] 40 mcg, we are aiming to aid children and their caregivers in better managing the burdensome symptoms associated with nasal allergies,” said Dr. Tushar Shah, senior vice president of Teva Global Respiratory Research and Development.
The FDA approved beclomethasone dipropionate 40 mcg based on data from three double-blind, placebo-controlled studies of children aged 4-11 years. In those studies, once-daily beclomethasone dipropionate 40 mcg alleviated allergy symptoms in children with both seasonal and perennial allergic rhinitis with a minimum of adverse effects. The most common side effects were nosebleed and ulcers, which was “consistent with those seen in previous clinical studies of QNASL Nasal Aerosol,” according to a statement from the manufacturer.
QNASL40 mcg is expected to become available in February 2015.
The Food and Drug Administration has approved the 40-mcg dose of QNASL for use in the treatment of nasal symptoms associated with allergic rhinitis in children aged 4-11 years, making it the first waterless nasal allergy spray approved for use in children as young as 4 years old.
“Through the availability of QNASL [beclomethasone dipropionate] 40 mcg, we are aiming to aid children and their caregivers in better managing the burdensome symptoms associated with nasal allergies,” said Dr. Tushar Shah, senior vice president of Teva Global Respiratory Research and Development.
The FDA approved beclomethasone dipropionate 40 mcg based on data from three double-blind, placebo-controlled studies of children aged 4-11 years. In those studies, once-daily beclomethasone dipropionate 40 mcg alleviated allergy symptoms in children with both seasonal and perennial allergic rhinitis with a minimum of adverse effects. The most common side effects were nosebleed and ulcers, which was “consistent with those seen in previous clinical studies of QNASL Nasal Aerosol,” according to a statement from the manufacturer.
QNASL40 mcg is expected to become available in February 2015.
The Food and Drug Administration has approved the 40-mcg dose of QNASL for use in the treatment of nasal symptoms associated with allergic rhinitis in children aged 4-11 years, making it the first waterless nasal allergy spray approved for use in children as young as 4 years old.
“Through the availability of QNASL [beclomethasone dipropionate] 40 mcg, we are aiming to aid children and their caregivers in better managing the burdensome symptoms associated with nasal allergies,” said Dr. Tushar Shah, senior vice president of Teva Global Respiratory Research and Development.
The FDA approved beclomethasone dipropionate 40 mcg based on data from three double-blind, placebo-controlled studies of children aged 4-11 years. In those studies, once-daily beclomethasone dipropionate 40 mcg alleviated allergy symptoms in children with both seasonal and perennial allergic rhinitis with a minimum of adverse effects. The most common side effects were nosebleed and ulcers, which was “consistent with those seen in previous clinical studies of QNASL Nasal Aerosol,” according to a statement from the manufacturer.
QNASL40 mcg is expected to become available in February 2015.
Don’t be scared of red eye, expert says
LAS VEGAS – Few conditions worry parents or school nurses more than when a child develops red eye, but how do you as the treating clinician know when to worry?
“Our challenge is to make the right diagnosis, not to worsen the problem, to figure when to refer, and to make that mother who had to take off from work to bring her child into the office – somehow we have to make her happy,” Dr. David B. Granet said at a pediatric update sponsored by the American Academy of Pediatrics California District 9.

Concomitant pain or photophobia typically means that something other than bacterial conjunctivitis is at play, said Dr. Granet, professor of ophthalmology and pediatrics at the University of California, San Diego. “Is there contact lens use?” he asked. “Is there proptosis or a history of trauma or injury? How long has it been going on? Most bacterial and viral infections will eventually go away. Is there a corneal opacity? Is there cellulitis, loss of vision, or herpes simplex virus?”
If parents call in suspecting that their child’s eye has been contaminated with a chemical, instruct them to irrigate the eye before they head to the emergency department, he advised. “Whoever’s answering the phone in your office ought to be able to separate out what’s worrisome and what’s not,” he said. “Like everything else we do, the history matters.”
For children who present to your office, consider “anything that can go wrong to make the eye red,” he continued, including nasolacrimal duct obstruction, adnexal disease, foreign body/trauma, uveitis, neoplasm, structural change, or conjunctivitis. “Has the vision changed? If so, that’s your vital sign for referral,” he said. “It’s generally a better sign to have both eyes involved with redness than just one. One eye involved means herpes simplex virus, uveitis, or trauma. Both eyes involved usually means infective or allergic conjunctivitis.”
The three most common conditions that cause a red or pink eye are allergic, bacterial, and viral conjunctivitis. Allergic conjunctivitis “is not just itching; that’s the symptom,” Dr. Granet said. “You get redness, swelling of the conjunctiva, lid edema, mucous discharge, and tearing. All of these occur when the patient rubs their eye. The best treatment for allergic conjunctivitis is avoidance of the allergen.”
He also recommended that affected children wash their hair before they go to sleep. “If their hair has been catching allergen all day long and they lie down on their pillow and start to roll [their head around in] it, that can cause a reaction,” he said.
Ketotifen fumarate (Zaditor) is an available over-the-counter treatment option, but olopatadine HCl (Pataday) is the most popular prescription written by pediatricians. “If you give any antihistamine, in low doses you start to prevent the release of histamine,” Dr. Granet said. “As the dose increases, you have a catastrophic event and you start to destruct the mast cell.”
Viral conjunctivitis usually affects older children and presents as a unilateral condition, then affects the fellow eye. It may be associated with pharyngitis and preauricular or submandibular adenopathy. Bacterial conjunctivitis, on the other hand, typically affects preschool-aged children, is often bilateral but can be unilateral, and yields mucopurulent discharge with matting. It is not associated with adenopathy, but it may be associated with otitis media, and it’s highly contagious. Topical antibiotic ointment therapy is indicated for bacterial conjunctivitis “not because this is a deadly disease, but because we want to reduce the chance for spread,” Dr. Granet said. “We know that communicable diseases are responsible for loss of 164 million school days each year. Additionally, there is a significant cost to a family when a parent misses work. Finally, if the diagnosis is in doubt, treatment with an antibiotic geared to work within a few days will help identify masquerade diseases early.”
Because of concerns about antibiotic resistance, fluoroquinolones are often the first choice for treating bacterial conjunctivitis. Dr. Granet led a multicenter comparison of moxifloxacin versus polymyxin B sulfate–trimethoprim ophthalmic solution in the speed of clinical efficacy for the treatment of bacterial conjunctivitis (J. Pediatr. Ophthalmol. Strabismus 2008;45:340-9). The investigators found that after day 2 of treatment, clinical cure was achieved by 81% of kids in the moxifloxacin group, compared with 44% of those in the polymyxin B sulfate–trimethoprim group. In addition, only 2.3% of kids in the moxifloxacin group were nonresponders, compared with 19.5% of those in the polymyxin B sulfate–trimethoprim group.
Common treatments for viral conjunctivitis include hygiene-related approaches like hand washing and not sharing towels and glasses. But these only prevent spread and don’t make the disease go away faster. The infection usually resolves in about 2 weeks.
Dr. Granet disclosed that he is a member of the speakers bureau for Alcon Labs and is a consultant for Diopsys.
On Twitter @dougbrunk
LAS VEGAS – Few conditions worry parents or school nurses more than when a child develops red eye, but how do you as the treating clinician know when to worry?
“Our challenge is to make the right diagnosis, not to worsen the problem, to figure when to refer, and to make that mother who had to take off from work to bring her child into the office – somehow we have to make her happy,” Dr. David B. Granet said at a pediatric update sponsored by the American Academy of Pediatrics California District 9.
Concomitant pain or photophobia typically means that something other than bacterial conjunctivitis is at play, said Dr. Granet, professor of ophthalmology and pediatrics at the University of California, San Diego. “Is there contact lens use?” he asked. “Is there proptosis or a history of trauma or injury? How long has it been going on? Most bacterial and viral infections will eventually go away. Is there a corneal opacity? Is there cellulitis, loss of vision, or herpes simplex virus?”
If parents call in suspecting that their child’s eye has been contaminated with a chemical, instruct them to irrigate the eye before they head to the emergency department, he advised. “Whoever’s answering the phone in your office ought to be able to separate out what’s worrisome and what’s not,” he said. “Like everything else we do, the history matters.”
For children who present to your office, consider “anything that can go wrong to make the eye red,” he continued, including nasolacrimal duct obstruction, adnexal disease, foreign body/trauma, uveitis, neoplasm, structural change, or conjunctivitis. “Has the vision changed? If so, that’s your vital sign for referral,” he said. “It’s generally a better sign to have both eyes involved with redness than just one. One eye involved means herpes simplex virus, uveitis, or trauma. Both eyes involved usually means infective or allergic conjunctivitis.”
The three most common conditions that cause a red or pink eye are allergic, bacterial, and viral conjunctivitis. Allergic conjunctivitis “is not just itching; that’s the symptom,” Dr. Granet said. “You get redness, swelling of the conjunctiva, lid edema, mucous discharge, and tearing. All of these occur when the patient rubs their eye. The best treatment for allergic conjunctivitis is avoidance of the allergen.”
He also recommended that affected children wash their hair before they go to sleep. “If their hair has been catching allergen all day long and they lie down on their pillow and start to roll [their head around in] it, that can cause a reaction,” he said.
Ketotifen fumarate (Zaditor) is an available over-the-counter treatment option, but olopatadine HCl (Pataday) is the most popular prescription written by pediatricians. “If you give any antihistamine, in low doses you start to prevent the release of histamine,” Dr. Granet said. “As the dose increases, you have a catastrophic event and you start to destruct the mast cell.”
Viral conjunctivitis usually affects older children and presents as a unilateral condition, then affects the fellow eye. It may be associated with pharyngitis and preauricular or submandibular adenopathy. Bacterial conjunctivitis, on the other hand, typically affects preschool-aged children, is often bilateral but can be unilateral, and yields mucopurulent discharge with matting. It is not associated with adenopathy, but it may be associated with otitis media, and it’s highly contagious. Topical antibiotic ointment therapy is indicated for bacterial conjunctivitis “not because this is a deadly disease, but because we want to reduce the chance for spread,” Dr. Granet said. “We know that communicable diseases are responsible for loss of 164 million school days each year. Additionally, there is a significant cost to a family when a parent misses work. Finally, if the diagnosis is in doubt, treatment with an antibiotic geared to work within a few days will help identify masquerade diseases early.”
Because of concerns about antibiotic resistance, fluoroquinolones are often the first choice for treating bacterial conjunctivitis. Dr. Granet led a multicenter comparison of moxifloxacin versus polymyxin B sulfate–trimethoprim ophthalmic solution in the speed of clinical efficacy for the treatment of bacterial conjunctivitis (J. Pediatr. Ophthalmol. Strabismus 2008;45:340-9). The investigators found that after day 2 of treatment, clinical cure was achieved by 81% of kids in the moxifloxacin group, compared with 44% of those in the polymyxin B sulfate–trimethoprim group. In addition, only 2.3% of kids in the moxifloxacin group were nonresponders, compared with 19.5% of those in the polymyxin B sulfate–trimethoprim group.
Common treatments for viral conjunctivitis include hygiene-related approaches like hand washing and not sharing towels and glasses. But these only prevent spread and don’t make the disease go away faster. The infection usually resolves in about 2 weeks.
Dr. Granet disclosed that he is a member of the speakers bureau for Alcon Labs and is a consultant for Diopsys.
On Twitter @dougbrunk
LAS VEGAS – Few conditions worry parents or school nurses more than when a child develops red eye, but how do you as the treating clinician know when to worry?
“Our challenge is to make the right diagnosis, not to worsen the problem, to figure when to refer, and to make that mother who had to take off from work to bring her child into the office – somehow we have to make her happy,” Dr. David B. Granet said at a pediatric update sponsored by the American Academy of Pediatrics California District 9.
Concomitant pain or photophobia typically means that something other than bacterial conjunctivitis is at play, said Dr. Granet, professor of ophthalmology and pediatrics at the University of California, San Diego. “Is there contact lens use?” he asked. “Is there proptosis or a history of trauma or injury? How long has it been going on? Most bacterial and viral infections will eventually go away. Is there a corneal opacity? Is there cellulitis, loss of vision, or herpes simplex virus?”
If parents call in suspecting that their child’s eye has been contaminated with a chemical, instruct them to irrigate the eye before they head to the emergency department, he advised. “Whoever’s answering the phone in your office ought to be able to separate out what’s worrisome and what’s not,” he said. “Like everything else we do, the history matters.”
For children who present to your office, consider “anything that can go wrong to make the eye red,” he continued, including nasolacrimal duct obstruction, adnexal disease, foreign body/trauma, uveitis, neoplasm, structural change, or conjunctivitis. “Has the vision changed? If so, that’s your vital sign for referral,” he said. “It’s generally a better sign to have both eyes involved with redness than just one. One eye involved means herpes simplex virus, uveitis, or trauma. Both eyes involved usually means infective or allergic conjunctivitis.”
The three most common conditions that cause a red or pink eye are allergic, bacterial, and viral conjunctivitis. Allergic conjunctivitis “is not just itching; that’s the symptom,” Dr. Granet said. “You get redness, swelling of the conjunctiva, lid edema, mucous discharge, and tearing. All of these occur when the patient rubs their eye. The best treatment for allergic conjunctivitis is avoidance of the allergen.”
He also recommended that affected children wash their hair before they go to sleep. “If their hair has been catching allergen all day long and they lie down on their pillow and start to roll [their head around in] it, that can cause a reaction,” he said.
Ketotifen fumarate (Zaditor) is an available over-the-counter treatment option, but olopatadine HCl (Pataday) is the most popular prescription written by pediatricians. “If you give any antihistamine, in low doses you start to prevent the release of histamine,” Dr. Granet said. “As the dose increases, you have a catastrophic event and you start to destruct the mast cell.”
Viral conjunctivitis usually affects older children and presents as a unilateral condition, then affects the fellow eye. It may be associated with pharyngitis and preauricular or submandibular adenopathy. Bacterial conjunctivitis, on the other hand, typically affects preschool-aged children, is often bilateral but can be unilateral, and yields mucopurulent discharge with matting. It is not associated with adenopathy, but it may be associated with otitis media, and it’s highly contagious. Topical antibiotic ointment therapy is indicated for bacterial conjunctivitis “not because this is a deadly disease, but because we want to reduce the chance for spread,” Dr. Granet said. “We know that communicable diseases are responsible for loss of 164 million school days each year. Additionally, there is a significant cost to a family when a parent misses work. Finally, if the diagnosis is in doubt, treatment with an antibiotic geared to work within a few days will help identify masquerade diseases early.”
Because of concerns about antibiotic resistance, fluoroquinolones are often the first choice for treating bacterial conjunctivitis. Dr. Granet led a multicenter comparison of moxifloxacin versus polymyxin B sulfate–trimethoprim ophthalmic solution in the speed of clinical efficacy for the treatment of bacterial conjunctivitis (J. Pediatr. Ophthalmol. Strabismus 2008;45:340-9). The investigators found that after day 2 of treatment, clinical cure was achieved by 81% of kids in the moxifloxacin group, compared with 44% of those in the polymyxin B sulfate–trimethoprim group. In addition, only 2.3% of kids in the moxifloxacin group were nonresponders, compared with 19.5% of those in the polymyxin B sulfate–trimethoprim group.
Common treatments for viral conjunctivitis include hygiene-related approaches like hand washing and not sharing towels and glasses. But these only prevent spread and don’t make the disease go away faster. The infection usually resolves in about 2 weeks.
Dr. Granet disclosed that he is a member of the speakers bureau for Alcon Labs and is a consultant for Diopsys.
On Twitter @dougbrunk
EXPERT ANALYSIS AT PEDIATRIC UPDATE

Identifying statin-associated autoimmune necrotizing myopathy
Statins are among the most widely prescribed drugs, as they reduce cardiovascular risk very effectively. They work by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a key enzyme in cholesterol biosynthesis. Although most patients tolerate statins well, muscle-related toxicity can limit the use of these drugs.
Recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Proper recognition of this ominous complication is important to prevent further damage from statin use.
STATIN-ASSOCIATED MUSCLE EFFECTS: A SPECTRUM
Muscle symptoms are among the best known and most important side effects of statins, ranging from asymptomatic, mild elevation of creatine kinase and benign myalgias to life-threatening rhabdomyolysis. As the various terms are often used inconsistently in the literature, we will briefly review each entity to put immune-necrotizing myopathy in its proper context on the spectrum of statin-associated muscle symptoms.
Myalgia
Myalgia, ie, muscle pain without elevation of muscle enzymes, is the most common side effect of statins. The incidence is about 10% in observational studies1,2; however, the incidence in the real world of clinical practice seems much higher. Myalgias can present as widespread pain or as localized pain, usually in the lower extremities. Muscle cramps and tendonitis-related pain are commonly reported.
Several clinical predictors of increased risk of statin-associated muscle pain were noted in the Prediction of Muscular Risk in Observational Conditions (PRIMO) study,2 assessing mild to moderate muscular symptoms in patients on high-dose statins. These included a history of muscle pain with another lipid-lowering agent, a history of cramps, a history of elevated creatine kinase levels, a personal or family history of muscle symptoms, untreated hypothyroidism, and a background of fibromyalgia-like symptoms. The incidence of muscle symptoms increased with the level of physical activity, and the median time of symptom onset was 1 month after starting statin therapy or titrating to a high dose.
In patients with myalgia alone, symptoms often improve when the statin is stopped.
Myopathy, myositis
The general terms myopathy and myositis have been used to refer to elevated muscle enzymes together with the muscle symptoms of pain, cramps, soreness, or weakness. An analysis of 21 clinical trials of statin therapy found that myopathy (creatine kinase level more than 10 times the upper limit of normal) occurred in 5 patients per 100,000 person-years.3 The incidence of myopathy increases when statins are used in high doses.
In another cohort of patients seen for statin intolerance,1 conventional risk factors for overt myositis included renal disease, diabetes, and thyroid disease. Electrolyte abnormalities did not differ between statin-tolerant and statin-intolerant patients.1
The search for genetic indicators of risk
In 2008, the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group conducted a genome-wide association study to identify genetic variants associated with myopathy with high-dose statin therapy.4 Eighty-five patients who had developed definite myopathy (muscle symptoms with enzyme levels more than 10 times the upper limit of normal) or incipient myopathy (asymptomatic or symptomatic, but enzyme levels at least three times the upper limit of normal) while taking simvastatin 80 mg were compared with 90 controls taking the same daily dose. The study reported variants in the SLCO1B1 gene as “strongly associated with an increased risk of statin-associated myopathy.”4
SLCO1B1 encodes the peptide responsible for hepatic uptake of statins, thus affecting the blood level of these drugs. Patients in the study who had the C variant, which predisposes to higher blood levels of statins, were at higher risk of myopathy than those with the T variant. The odds ratio for myopathy increased from 4.4 in heterozygotes for the C allele to 17.4 for homozygotes. This effect was similar even with lower doses of statins.4
We believe that these findings provide a strong basis for genetic testing of patients who may be at risk of statin-associated myopathy.
Rechallenging with a different statin
Often, patients with myopathy can be rechallenged with a different statin agent. In a study done in a lipid clinic, patients identified as having simvastatin-associated myopathy were given another statin.5 Between 15% and 42% tolerated the second statin, with no statistically significant difference between the tolerability rates with the different agents used (atorvastatin, rosuvastatin, pravastatin, and fluvastatin).
Rhabdomyolysis
Rhabdomyolysis, the most devastating complication of statin use, is marked by a creatine kinase level more than 10 times the upper limit of normal or greater than 10,000 IU/L, resulting from acute and massive destruction of muscle fibers and release of their contents into the bloodstream.
But rhabdomyolysis is a clinical syndrome, not solely an alarming increase in muscle enzyme levels. It can include renal failure and death. The rate of occurrence is low (1/100,000), but it can occur at any point in treatment.6 An analysis of Canadian and US case reports of statin-associated rhabdomyolysis showed an average of 824 cases each year.7 A dose-response relationship was observed with higher statin doses.
But statin-associated myopathy may not stop when the drug is stopped
The types of muscle toxicity discussed above stem from direct myotoxic effects of statins and are thought to be related to the blood concentration of the statin.6,8 They may be limited by genetic susceptibility. Mechanisms may include a change in muscle membrane excitability caused by modulation in membrane cholesterol levels, impaired mitochondrial function and calcium signaling, induction of apoptosis, and increased lipid peroxidation. Stopping the offending drug can often halt these downstream effects—hence the dictum that statin myopathy is self-limiting and should resolve with cessation of the drug.
But as we discuss in the following sections, some patients on statin therapy develop an immune-mediated myopathy that does not resolve with discontinuation of the statin and that may only resolve with immunosuppressive therapy.
STATINS AND AUTOIMMUNE NECROTIZING MYOPATHY
At the Johns Hopkins Myositis Center, a group of patients was identified who had necrotizing myopathy on biopsy but no known underlying condition or associated autoantibodies. In an attempt to establish an autoimmune basis for their disease, the sera from 26 patients were screened for novel antibodies.9 Sera from 16 of the patients immunoprecipitated a pair of proteins (sizes 100 kd and 200 kd), indicating these patients had an antibody to these proteins. This finding was highly specific for necrotizing myopathy when compared with controls, ie, other patients with myositis. Patients who had this finding displayed proximal muscle weakness, elevated muscle enzyme levels, and myopathic findings on electromyography; 63% had been exposed to statins before the onset of weakness, and when only patients over age 50 were included, the number rose to 83%.
This association of statins with necrotizing myopathy had been previously noted by two other groups. Needham et al10 described eight patients who, while on statins, developed myopathy that continued to worsen despite cessation of the drugs. An analysis of their muscle pathology revealed myofiber necrosis with little inflammatory infiltrate, as well as widespread up-regulation of expression of major histocompatibility complex class 1. These patients required immunosuppressive treatment (prednisone and methotrexate) to control their disease.
Grable-Esposito et al11 corroborated this finding by identifying 25 additional patients who developed a similar necrotizing myopathy while on statins.11 They also noted a significantly higher frequency of statin use in patients with necrotizing myopathy than in age-matched controls with polymyositis or inclusion-body myositis.
The researchers at the Johns Hopkins Myositis Center noted the similarity between these two patient groups and their own group of patients with necrotizing myopathy. Thus, a follow-up study was done to identify the 100-kg and 200-kd autoantigens observed in their earlier study. Exposure to a statin was found to up-regulate the expression of the two molecules.12 HMGCR was hypothesized as being the 100-kd antigen, because of its 97-kd molecular weight, and also because statin treatment had already been shown to up-regulate the expression of HMGCR.13 The researchers concluded that HMGCR was indeed the 100-kd antigen, with no distinctive antibodies recognizing the 200-kd protein. Although the 200-kd protein was once postulated to be a dimer of the 100-kd protein, its identity remains unknown.
The anti-HMGCR antibody was then screened for in a cohort of 750 myositis patients. The 16 patients previously found to have anti-200/100 were all positive for anti-HMGCR antibody. An additional 45 patients from the cohort (6%) were anti-HMGCR-positive by enzyme-linked immunosorbent assay, and all had necrotizing myopathy. Patients with other types of myopathy, including inflammatory myopathy, do not possess this antibody.12
The HMGCR antibody was quite specific for immune-mediated necrotizing myopathy, and this suggested that statins were capable of triggering an immune-mediated myopathy that is then perpetuated even if the drug is discontinued. As it was also demonstrated that statins increase the expression of HMGCR in muscle as well as in regenerating cells, the process may be sustained through persistently increased HMGCR expression associated with muscle repair.12
The C allele of the SLCO1B1 gene, which has been associated with statin-associated myopathy, was not increased in this population of patients positive for anti-HMGCR. Follow-up studies of the prevalence of anti-HMGCR in statin users in the Atherosclerosis Risk in Communities (ARIC) cohort, including those with self-limited statin myotoxicity, have also shown the absence of this antibody.14 This shows that anti-HMGCR is not found in the majority of statin-exposed patients and is highly specific for autoimmune myopathy. This also suggests that statin-associated autoimmune myopathy represents a pathologic process that is distinct from self-limited statin intolerance.
HOW THE CONDITION PRESENTS

Immune-mediated statin myopathy presents similarly to other idiopathic inflammatory myopathies such as polymyositis (Table 1). Symptoms often develop in a subacute to chronic course and can occur at any time with statin treatment. In one study,10 the average duration of statin use before the onset of weakness was 3 years (range 2 months to 10 years). In some patients whose statin had been stopped because of abnormal creatine kinase levels, weakness developed later, at a range of 0.5 to 20 months. Even low doses of statins (such as 10 mg of simvastatin) have been found to trigger this condition.10
Patients uniformly develop symmetric proximal arm and leg weakness, and distal weakness can also occur.11 Other features have included dysphagia, arthralgias, myalgias, and Raynaud phenomenon.9 Men and women are represented in roughly equal numbers.
The muscle enzymes are strikingly elevated in this disease, with a mean creatine kinase value of 10,333 IU/L at initial presentation.9 Although the creatine kinase level may be very elevated, patients often do not present with weakness until a certain threshold value is reached, in contrast with patients with anti-signal recognition particle necrotizing myopathy, who can present with profound weakness at a lower level. Hence, by the time patients are clinically symptomatic, the process may have been going on for some time. Despite the seemingly massive leak in muscle enzymes, the patients do not develop rhabdomyolysis. Inflammatory markers need not be elevated, and an association with other antibodies such as antinuclear antibody is not often seen.
Magnetic resonance imaging of the thigh has shown muscle edema in all patients.9 In decreasing order of frequency, other findings are atrophy, fatty replacement, and fascial edema.
Electromyography of involved muscle has shown irritable myopathy in most patients (88%) and nonirritable myopathy in a few.
Muscle biopsy studies have shown prominent necrotic and regenerating fibers without significant inflammatory infiltrate.11 There is also myophagocytosis of necrotic fibers and diffuse or focal up-regulation of major histocompatability complex class I expression.10
PATIENTS WITH ANTI-HMGCR WHO HAVE AND WHO HAVE NEVER TAKEN STATINS
When the anti-HMGCR antibody was tested for in the Johns Hopkins cohort,12 33% of patients with a necrotizing myopathy associated with this antibody had never taken a statin. The two groups were clinically indistinguishable, save for a few aspects. Compared with patients who had taken a statin, those who had never taken a statin were younger (mean age 37 vs 59), had higher levels of creatine kinase (13,392 vs 7,881 IU/L), and had a different race distribution (46.7% vs 86.7% white). Initial HMGCR antibody levels were also noted to correlate with creatine kinase levels and strength in statin-exposed patients but not in those who had never taken a statin.15
We hypothesize that in patients who have never taken a statin, other genetic or environmental factors may be the cause of the increased HMGCR expression, which then triggers the autoimmune response. Until further data are gathered, we should probably treat these patients as we treat those who develop this disease after taking a statin, and avoid giving them statins altogether.
MANAGEMENT OF STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Treatment of statin-associated autoimmune necrotizing myopathy can be challenging and requires immunosuppressive drugs.
Statin therapy should be stopped once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy. Electromyography detects myopathy and shows chronicity, distribution, and degree of severity. Although not necessary for diagnosis, magnetic resonance imaging helps to evaluate the extent of muscle involvement and damage and provides guidance when choosing a site for muscle biopsy. Muscle biopsy is necessary to determine the actual pathology and to exclude mimics such as dystrophy or metabolic myopathies.
When an immune-mediated myopathy is confirmed, prompt referral to a rheumatologist or a neuromuscular specialist is recommended.
Steroids are usually the first-line treatment for this disease. Other immunosuppressives, such as methotrexate, azathioprine, mycophenolate mofetil, and rituximab have been used with varying levels of success. In our experience, intravenous immunoglobulin has been particularly beneficial for refractory cases. With treatment, muscle enzyme levels and weakness improve, but relapses can occur. The ideal choice of immunosuppressive therapy and the duration of therapy are currently under investigation.
Rechallenge with another statin
At this time, the issue of rechallenging the patient with another statin has not been clarified. Given the autoimmune nature of the disease, we would avoid exposing the patient to a known trigger. However, this may be a difficult decision in patients with cardiovascular risk factors who require statins for primary or secondary prevention. We suggest using alternative cholesterol-lowering agents first and using them in combination if needed.
We have had some success in maintaining a handful of patients on a statin while treating them concurrently with immunosuppression. This is not ideal because they are constantly being exposed to the likely trigger for their disease, but it may be unavoidable if statins are deemed absolutely necessary. We have also had a patient with known statin-associated immune-mediated necrotizing myopathy who later became profoundly weak after another physician started her on a newer-generation agent, pitavastatin. This suggests to us that rechallenging patients, even with a different statin, can have deleterious effects.
IMPLICATIONS FOR CLINICAL PRACTICE
The true prevalence of statin-associated autoimmune myopathy in practice is unknown. In the Johns Hopkins Myositis Center cohort of patients with suspected myopathy, anti-HMGCR was found in 6% of the patients and was the second most frequent antibody found after anti-Jo1.12
Given the frequency of muscle-related complaints in patients on statins, we recommend obtaining baseline muscle enzyme measurements before starting statin therapy. As recommended by the National Lipid Association Statin Safety Assessment Task Force, the creatine kinase level should be measured when a patient develops muscular complaints, to help gauge the severity of the disease and to help decide whether to continue therapy.8 Random testing of the creatine kinase level in asymptomatic patients is not recommended.
At present, the diagnosis of statin-associated autoimmune necrotizing myopathy is based on a combination of findings—elevated muscle enzyme levels, muscle weakness, irritable findings on electromyography, and necrotizing myopathy on biopsy in a patient on a statin. The finding of the HMGCR antibody confirms the diagnosis. A test for this antibody is now commercially available in the United States. We suggest testing for the antibody in the following scenarios:
- A persistently elevated or rising creatine kinase, aspartate aminotransferase, alanine aminotransferase, or aldolase level after the statin is stopped; although no fixed creatine kinase level has been determined, a level above 1,000 U/L would be a reasonable cutoff at which to test
- Muscle symptoms (proximal or distal weakness) that persist 12 weeks after statin cessation regardless of the creatine kinase level, especially if the patient has dysphagia
- The finding of muscle irritability on electromyography or diffuse muscle edema on magnetic resonance imaging when testing for other myositis-specific antibodies is negative
- Muscle biopsy showing necrotizing myopathy with little or no inflammation.
In addition, since necrotizing myopathy is known to be associated with malignancy and since necrotizing myopathy is more common in older people, who are also more likely to be taking a statin, an age-appropriate malignancy evaluation is warranted as well.
- Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol 2011; 5:299–307.
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C–60C.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Fung EC, Crook MA. Statin myopathy: a lipid clinic experience on the tolerability of statin rechallenge. Cardiovasc Ther 2012; 30:e212–e218.
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008; 8:333–338.
- Holbrook A, Wright M, Sung M, Ribic C, Baker S. Statin-associated rhabdomyolysis: is there a dose-response relationship? Can J Cardiol 2011; 27:146–151.
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–94C.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766.
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007; 17:194–200.
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41:185–190.
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425–430.
- Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
Statins are among the most widely prescribed drugs, as they reduce cardiovascular risk very effectively. They work by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a key enzyme in cholesterol biosynthesis. Although most patients tolerate statins well, muscle-related toxicity can limit the use of these drugs.
Recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Proper recognition of this ominous complication is important to prevent further damage from statin use.
STATIN-ASSOCIATED MUSCLE EFFECTS: A SPECTRUM
Muscle symptoms are among the best known and most important side effects of statins, ranging from asymptomatic, mild elevation of creatine kinase and benign myalgias to life-threatening rhabdomyolysis. As the various terms are often used inconsistently in the literature, we will briefly review each entity to put immune-necrotizing myopathy in its proper context on the spectrum of statin-associated muscle symptoms.
Myalgia
Myalgia, ie, muscle pain without elevation of muscle enzymes, is the most common side effect of statins. The incidence is about 10% in observational studies1,2; however, the incidence in the real world of clinical practice seems much higher. Myalgias can present as widespread pain or as localized pain, usually in the lower extremities. Muscle cramps and tendonitis-related pain are commonly reported.
Several clinical predictors of increased risk of statin-associated muscle pain were noted in the Prediction of Muscular Risk in Observational Conditions (PRIMO) study,2 assessing mild to moderate muscular symptoms in patients on high-dose statins. These included a history of muscle pain with another lipid-lowering agent, a history of cramps, a history of elevated creatine kinase levels, a personal or family history of muscle symptoms, untreated hypothyroidism, and a background of fibromyalgia-like symptoms. The incidence of muscle symptoms increased with the level of physical activity, and the median time of symptom onset was 1 month after starting statin therapy or titrating to a high dose.
In patients with myalgia alone, symptoms often improve when the statin is stopped.
Myopathy, myositis
The general terms myopathy and myositis have been used to refer to elevated muscle enzymes together with the muscle symptoms of pain, cramps, soreness, or weakness. An analysis of 21 clinical trials of statin therapy found that myopathy (creatine kinase level more than 10 times the upper limit of normal) occurred in 5 patients per 100,000 person-years.3 The incidence of myopathy increases when statins are used in high doses.
In another cohort of patients seen for statin intolerance,1 conventional risk factors for overt myositis included renal disease, diabetes, and thyroid disease. Electrolyte abnormalities did not differ between statin-tolerant and statin-intolerant patients.1
The search for genetic indicators of risk
In 2008, the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group conducted a genome-wide association study to identify genetic variants associated with myopathy with high-dose statin therapy.4 Eighty-five patients who had developed definite myopathy (muscle symptoms with enzyme levels more than 10 times the upper limit of normal) or incipient myopathy (asymptomatic or symptomatic, but enzyme levels at least three times the upper limit of normal) while taking simvastatin 80 mg were compared with 90 controls taking the same daily dose. The study reported variants in the SLCO1B1 gene as “strongly associated with an increased risk of statin-associated myopathy.”4
SLCO1B1 encodes the peptide responsible for hepatic uptake of statins, thus affecting the blood level of these drugs. Patients in the study who had the C variant, which predisposes to higher blood levels of statins, were at higher risk of myopathy than those with the T variant. The odds ratio for myopathy increased from 4.4 in heterozygotes for the C allele to 17.4 for homozygotes. This effect was similar even with lower doses of statins.4
We believe that these findings provide a strong basis for genetic testing of patients who may be at risk of statin-associated myopathy.
Rechallenging with a different statin
Often, patients with myopathy can be rechallenged with a different statin agent. In a study done in a lipid clinic, patients identified as having simvastatin-associated myopathy were given another statin.5 Between 15% and 42% tolerated the second statin, with no statistically significant difference between the tolerability rates with the different agents used (atorvastatin, rosuvastatin, pravastatin, and fluvastatin).
Rhabdomyolysis
Rhabdomyolysis, the most devastating complication of statin use, is marked by a creatine kinase level more than 10 times the upper limit of normal or greater than 10,000 IU/L, resulting from acute and massive destruction of muscle fibers and release of their contents into the bloodstream.
But rhabdomyolysis is a clinical syndrome, not solely an alarming increase in muscle enzyme levels. It can include renal failure and death. The rate of occurrence is low (1/100,000), but it can occur at any point in treatment.6 An analysis of Canadian and US case reports of statin-associated rhabdomyolysis showed an average of 824 cases each year.7 A dose-response relationship was observed with higher statin doses.
But statin-associated myopathy may not stop when the drug is stopped
The types of muscle toxicity discussed above stem from direct myotoxic effects of statins and are thought to be related to the blood concentration of the statin.6,8 They may be limited by genetic susceptibility. Mechanisms may include a change in muscle membrane excitability caused by modulation in membrane cholesterol levels, impaired mitochondrial function and calcium signaling, induction of apoptosis, and increased lipid peroxidation. Stopping the offending drug can often halt these downstream effects—hence the dictum that statin myopathy is self-limiting and should resolve with cessation of the drug.
But as we discuss in the following sections, some patients on statin therapy develop an immune-mediated myopathy that does not resolve with discontinuation of the statin and that may only resolve with immunosuppressive therapy.
STATINS AND AUTOIMMUNE NECROTIZING MYOPATHY
At the Johns Hopkins Myositis Center, a group of patients was identified who had necrotizing myopathy on biopsy but no known underlying condition or associated autoantibodies. In an attempt to establish an autoimmune basis for their disease, the sera from 26 patients were screened for novel antibodies.9 Sera from 16 of the patients immunoprecipitated a pair of proteins (sizes 100 kd and 200 kd), indicating these patients had an antibody to these proteins. This finding was highly specific for necrotizing myopathy when compared with controls, ie, other patients with myositis. Patients who had this finding displayed proximal muscle weakness, elevated muscle enzyme levels, and myopathic findings on electromyography; 63% had been exposed to statins before the onset of weakness, and when only patients over age 50 were included, the number rose to 83%.
This association of statins with necrotizing myopathy had been previously noted by two other groups. Needham et al10 described eight patients who, while on statins, developed myopathy that continued to worsen despite cessation of the drugs. An analysis of their muscle pathology revealed myofiber necrosis with little inflammatory infiltrate, as well as widespread up-regulation of expression of major histocompatibility complex class 1. These patients required immunosuppressive treatment (prednisone and methotrexate) to control their disease.
Grable-Esposito et al11 corroborated this finding by identifying 25 additional patients who developed a similar necrotizing myopathy while on statins.11 They also noted a significantly higher frequency of statin use in patients with necrotizing myopathy than in age-matched controls with polymyositis or inclusion-body myositis.
The researchers at the Johns Hopkins Myositis Center noted the similarity between these two patient groups and their own group of patients with necrotizing myopathy. Thus, a follow-up study was done to identify the 100-kg and 200-kd autoantigens observed in their earlier study. Exposure to a statin was found to up-regulate the expression of the two molecules.12 HMGCR was hypothesized as being the 100-kd antigen, because of its 97-kd molecular weight, and also because statin treatment had already been shown to up-regulate the expression of HMGCR.13 The researchers concluded that HMGCR was indeed the 100-kd antigen, with no distinctive antibodies recognizing the 200-kd protein. Although the 200-kd protein was once postulated to be a dimer of the 100-kd protein, its identity remains unknown.
The anti-HMGCR antibody was then screened for in a cohort of 750 myositis patients. The 16 patients previously found to have anti-200/100 were all positive for anti-HMGCR antibody. An additional 45 patients from the cohort (6%) were anti-HMGCR-positive by enzyme-linked immunosorbent assay, and all had necrotizing myopathy. Patients with other types of myopathy, including inflammatory myopathy, do not possess this antibody.12
The HMGCR antibody was quite specific for immune-mediated necrotizing myopathy, and this suggested that statins were capable of triggering an immune-mediated myopathy that is then perpetuated even if the drug is discontinued. As it was also demonstrated that statins increase the expression of HMGCR in muscle as well as in regenerating cells, the process may be sustained through persistently increased HMGCR expression associated with muscle repair.12
The C allele of the SLCO1B1 gene, which has been associated with statin-associated myopathy, was not increased in this population of patients positive for anti-HMGCR. Follow-up studies of the prevalence of anti-HMGCR in statin users in the Atherosclerosis Risk in Communities (ARIC) cohort, including those with self-limited statin myotoxicity, have also shown the absence of this antibody.14 This shows that anti-HMGCR is not found in the majority of statin-exposed patients and is highly specific for autoimmune myopathy. This also suggests that statin-associated autoimmune myopathy represents a pathologic process that is distinct from self-limited statin intolerance.
HOW THE CONDITION PRESENTS
Immune-mediated statin myopathy presents similarly to other idiopathic inflammatory myopathies such as polymyositis (Table 1). Symptoms often develop in a subacute to chronic course and can occur at any time with statin treatment. In one study,10 the average duration of statin use before the onset of weakness was 3 years (range 2 months to 10 years). In some patients whose statin had been stopped because of abnormal creatine kinase levels, weakness developed later, at a range of 0.5 to 20 months. Even low doses of statins (such as 10 mg of simvastatin) have been found to trigger this condition.10
Patients uniformly develop symmetric proximal arm and leg weakness, and distal weakness can also occur.11 Other features have included dysphagia, arthralgias, myalgias, and Raynaud phenomenon.9 Men and women are represented in roughly equal numbers.
The muscle enzymes are strikingly elevated in this disease, with a mean creatine kinase value of 10,333 IU/L at initial presentation.9 Although the creatine kinase level may be very elevated, patients often do not present with weakness until a certain threshold value is reached, in contrast with patients with anti-signal recognition particle necrotizing myopathy, who can present with profound weakness at a lower level. Hence, by the time patients are clinically symptomatic, the process may have been going on for some time. Despite the seemingly massive leak in muscle enzymes, the patients do not develop rhabdomyolysis. Inflammatory markers need not be elevated, and an association with other antibodies such as antinuclear antibody is not often seen.
Magnetic resonance imaging of the thigh has shown muscle edema in all patients.9 In decreasing order of frequency, other findings are atrophy, fatty replacement, and fascial edema.
Electromyography of involved muscle has shown irritable myopathy in most patients (88%) and nonirritable myopathy in a few.
Muscle biopsy studies have shown prominent necrotic and regenerating fibers without significant inflammatory infiltrate.11 There is also myophagocytosis of necrotic fibers and diffuse or focal up-regulation of major histocompatability complex class I expression.10
PATIENTS WITH ANTI-HMGCR WHO HAVE AND WHO HAVE NEVER TAKEN STATINS
When the anti-HMGCR antibody was tested for in the Johns Hopkins cohort,12 33% of patients with a necrotizing myopathy associated with this antibody had never taken a statin. The two groups were clinically indistinguishable, save for a few aspects. Compared with patients who had taken a statin, those who had never taken a statin were younger (mean age 37 vs 59), had higher levels of creatine kinase (13,392 vs 7,881 IU/L), and had a different race distribution (46.7% vs 86.7% white). Initial HMGCR antibody levels were also noted to correlate with creatine kinase levels and strength in statin-exposed patients but not in those who had never taken a statin.15
We hypothesize that in patients who have never taken a statin, other genetic or environmental factors may be the cause of the increased HMGCR expression, which then triggers the autoimmune response. Until further data are gathered, we should probably treat these patients as we treat those who develop this disease after taking a statin, and avoid giving them statins altogether.
MANAGEMENT OF STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Treatment of statin-associated autoimmune necrotizing myopathy can be challenging and requires immunosuppressive drugs.
Statin therapy should be stopped once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy. Electromyography detects myopathy and shows chronicity, distribution, and degree of severity. Although not necessary for diagnosis, magnetic resonance imaging helps to evaluate the extent of muscle involvement and damage and provides guidance when choosing a site for muscle biopsy. Muscle biopsy is necessary to determine the actual pathology and to exclude mimics such as dystrophy or metabolic myopathies.
When an immune-mediated myopathy is confirmed, prompt referral to a rheumatologist or a neuromuscular specialist is recommended.
Steroids are usually the first-line treatment for this disease. Other immunosuppressives, such as methotrexate, azathioprine, mycophenolate mofetil, and rituximab have been used with varying levels of success. In our experience, intravenous immunoglobulin has been particularly beneficial for refractory cases. With treatment, muscle enzyme levels and weakness improve, but relapses can occur. The ideal choice of immunosuppressive therapy and the duration of therapy are currently under investigation.
Rechallenge with another statin
At this time, the issue of rechallenging the patient with another statin has not been clarified. Given the autoimmune nature of the disease, we would avoid exposing the patient to a known trigger. However, this may be a difficult decision in patients with cardiovascular risk factors who require statins for primary or secondary prevention. We suggest using alternative cholesterol-lowering agents first and using them in combination if needed.
We have had some success in maintaining a handful of patients on a statin while treating them concurrently with immunosuppression. This is not ideal because they are constantly being exposed to the likely trigger for their disease, but it may be unavoidable if statins are deemed absolutely necessary. We have also had a patient with known statin-associated immune-mediated necrotizing myopathy who later became profoundly weak after another physician started her on a newer-generation agent, pitavastatin. This suggests to us that rechallenging patients, even with a different statin, can have deleterious effects.
IMPLICATIONS FOR CLINICAL PRACTICE
The true prevalence of statin-associated autoimmune myopathy in practice is unknown. In the Johns Hopkins Myositis Center cohort of patients with suspected myopathy, anti-HMGCR was found in 6% of the patients and was the second most frequent antibody found after anti-Jo1.12
Given the frequency of muscle-related complaints in patients on statins, we recommend obtaining baseline muscle enzyme measurements before starting statin therapy. As recommended by the National Lipid Association Statin Safety Assessment Task Force, the creatine kinase level should be measured when a patient develops muscular complaints, to help gauge the severity of the disease and to help decide whether to continue therapy.8 Random testing of the creatine kinase level in asymptomatic patients is not recommended.
At present, the diagnosis of statin-associated autoimmune necrotizing myopathy is based on a combination of findings—elevated muscle enzyme levels, muscle weakness, irritable findings on electromyography, and necrotizing myopathy on biopsy in a patient on a statin. The finding of the HMGCR antibody confirms the diagnosis. A test for this antibody is now commercially available in the United States. We suggest testing for the antibody in the following scenarios:
- A persistently elevated or rising creatine kinase, aspartate aminotransferase, alanine aminotransferase, or aldolase level after the statin is stopped; although no fixed creatine kinase level has been determined, a level above 1,000 U/L would be a reasonable cutoff at which to test
- Muscle symptoms (proximal or distal weakness) that persist 12 weeks after statin cessation regardless of the creatine kinase level, especially if the patient has dysphagia
- The finding of muscle irritability on electromyography or diffuse muscle edema on magnetic resonance imaging when testing for other myositis-specific antibodies is negative
- Muscle biopsy showing necrotizing myopathy with little or no inflammation.
In addition, since necrotizing myopathy is known to be associated with malignancy and since necrotizing myopathy is more common in older people, who are also more likely to be taking a statin, an age-appropriate malignancy evaluation is warranted as well.
Statins are among the most widely prescribed drugs, as they reduce cardiovascular risk very effectively. They work by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a key enzyme in cholesterol biosynthesis. Although most patients tolerate statins well, muscle-related toxicity can limit the use of these drugs.
Recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Proper recognition of this ominous complication is important to prevent further damage from statin use.
STATIN-ASSOCIATED MUSCLE EFFECTS: A SPECTRUM
Muscle symptoms are among the best known and most important side effects of statins, ranging from asymptomatic, mild elevation of creatine kinase and benign myalgias to life-threatening rhabdomyolysis. As the various terms are often used inconsistently in the literature, we will briefly review each entity to put immune-necrotizing myopathy in its proper context on the spectrum of statin-associated muscle symptoms.
Myalgia
Myalgia, ie, muscle pain without elevation of muscle enzymes, is the most common side effect of statins. The incidence is about 10% in observational studies1,2; however, the incidence in the real world of clinical practice seems much higher. Myalgias can present as widespread pain or as localized pain, usually in the lower extremities. Muscle cramps and tendonitis-related pain are commonly reported.
Several clinical predictors of increased risk of statin-associated muscle pain were noted in the Prediction of Muscular Risk in Observational Conditions (PRIMO) study,2 assessing mild to moderate muscular symptoms in patients on high-dose statins. These included a history of muscle pain with another lipid-lowering agent, a history of cramps, a history of elevated creatine kinase levels, a personal or family history of muscle symptoms, untreated hypothyroidism, and a background of fibromyalgia-like symptoms. The incidence of muscle symptoms increased with the level of physical activity, and the median time of symptom onset was 1 month after starting statin therapy or titrating to a high dose.
In patients with myalgia alone, symptoms often improve when the statin is stopped.
Myopathy, myositis
The general terms myopathy and myositis have been used to refer to elevated muscle enzymes together with the muscle symptoms of pain, cramps, soreness, or weakness. An analysis of 21 clinical trials of statin therapy found that myopathy (creatine kinase level more than 10 times the upper limit of normal) occurred in 5 patients per 100,000 person-years.3 The incidence of myopathy increases when statins are used in high doses.
In another cohort of patients seen for statin intolerance,1 conventional risk factors for overt myositis included renal disease, diabetes, and thyroid disease. Electrolyte abnormalities did not differ between statin-tolerant and statin-intolerant patients.1
The search for genetic indicators of risk
In 2008, the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group conducted a genome-wide association study to identify genetic variants associated with myopathy with high-dose statin therapy.4 Eighty-five patients who had developed definite myopathy (muscle symptoms with enzyme levels more than 10 times the upper limit of normal) or incipient myopathy (asymptomatic or symptomatic, but enzyme levels at least three times the upper limit of normal) while taking simvastatin 80 mg were compared with 90 controls taking the same daily dose. The study reported variants in the SLCO1B1 gene as “strongly associated with an increased risk of statin-associated myopathy.”4
SLCO1B1 encodes the peptide responsible for hepatic uptake of statins, thus affecting the blood level of these drugs. Patients in the study who had the C variant, which predisposes to higher blood levels of statins, were at higher risk of myopathy than those with the T variant. The odds ratio for myopathy increased from 4.4 in heterozygotes for the C allele to 17.4 for homozygotes. This effect was similar even with lower doses of statins.4
We believe that these findings provide a strong basis for genetic testing of patients who may be at risk of statin-associated myopathy.
Rechallenging with a different statin
Often, patients with myopathy can be rechallenged with a different statin agent. In a study done in a lipid clinic, patients identified as having simvastatin-associated myopathy were given another statin.5 Between 15% and 42% tolerated the second statin, with no statistically significant difference between the tolerability rates with the different agents used (atorvastatin, rosuvastatin, pravastatin, and fluvastatin).
Rhabdomyolysis
Rhabdomyolysis, the most devastating complication of statin use, is marked by a creatine kinase level more than 10 times the upper limit of normal or greater than 10,000 IU/L, resulting from acute and massive destruction of muscle fibers and release of their contents into the bloodstream.
But rhabdomyolysis is a clinical syndrome, not solely an alarming increase in muscle enzyme levels. It can include renal failure and death. The rate of occurrence is low (1/100,000), but it can occur at any point in treatment.6 An analysis of Canadian and US case reports of statin-associated rhabdomyolysis showed an average of 824 cases each year.7 A dose-response relationship was observed with higher statin doses.
But statin-associated myopathy may not stop when the drug is stopped
The types of muscle toxicity discussed above stem from direct myotoxic effects of statins and are thought to be related to the blood concentration of the statin.6,8 They may be limited by genetic susceptibility. Mechanisms may include a change in muscle membrane excitability caused by modulation in membrane cholesterol levels, impaired mitochondrial function and calcium signaling, induction of apoptosis, and increased lipid peroxidation. Stopping the offending drug can often halt these downstream effects—hence the dictum that statin myopathy is self-limiting and should resolve with cessation of the drug.
But as we discuss in the following sections, some patients on statin therapy develop an immune-mediated myopathy that does not resolve with discontinuation of the statin and that may only resolve with immunosuppressive therapy.
STATINS AND AUTOIMMUNE NECROTIZING MYOPATHY
At the Johns Hopkins Myositis Center, a group of patients was identified who had necrotizing myopathy on biopsy but no known underlying condition or associated autoantibodies. In an attempt to establish an autoimmune basis for their disease, the sera from 26 patients were screened for novel antibodies.9 Sera from 16 of the patients immunoprecipitated a pair of proteins (sizes 100 kd and 200 kd), indicating these patients had an antibody to these proteins. This finding was highly specific for necrotizing myopathy when compared with controls, ie, other patients with myositis. Patients who had this finding displayed proximal muscle weakness, elevated muscle enzyme levels, and myopathic findings on electromyography; 63% had been exposed to statins before the onset of weakness, and when only patients over age 50 were included, the number rose to 83%.
This association of statins with necrotizing myopathy had been previously noted by two other groups. Needham et al10 described eight patients who, while on statins, developed myopathy that continued to worsen despite cessation of the drugs. An analysis of their muscle pathology revealed myofiber necrosis with little inflammatory infiltrate, as well as widespread up-regulation of expression of major histocompatibility complex class 1. These patients required immunosuppressive treatment (prednisone and methotrexate) to control their disease.
Grable-Esposito et al11 corroborated this finding by identifying 25 additional patients who developed a similar necrotizing myopathy while on statins.11 They also noted a significantly higher frequency of statin use in patients with necrotizing myopathy than in age-matched controls with polymyositis or inclusion-body myositis.
The researchers at the Johns Hopkins Myositis Center noted the similarity between these two patient groups and their own group of patients with necrotizing myopathy. Thus, a follow-up study was done to identify the 100-kg and 200-kd autoantigens observed in their earlier study. Exposure to a statin was found to up-regulate the expression of the two molecules.12 HMGCR was hypothesized as being the 100-kd antigen, because of its 97-kd molecular weight, and also because statin treatment had already been shown to up-regulate the expression of HMGCR.13 The researchers concluded that HMGCR was indeed the 100-kd antigen, with no distinctive antibodies recognizing the 200-kd protein. Although the 200-kd protein was once postulated to be a dimer of the 100-kd protein, its identity remains unknown.
The anti-HMGCR antibody was then screened for in a cohort of 750 myositis patients. The 16 patients previously found to have anti-200/100 were all positive for anti-HMGCR antibody. An additional 45 patients from the cohort (6%) were anti-HMGCR-positive by enzyme-linked immunosorbent assay, and all had necrotizing myopathy. Patients with other types of myopathy, including inflammatory myopathy, do not possess this antibody.12
The HMGCR antibody was quite specific for immune-mediated necrotizing myopathy, and this suggested that statins were capable of triggering an immune-mediated myopathy that is then perpetuated even if the drug is discontinued. As it was also demonstrated that statins increase the expression of HMGCR in muscle as well as in regenerating cells, the process may be sustained through persistently increased HMGCR expression associated with muscle repair.12
The C allele of the SLCO1B1 gene, which has been associated with statin-associated myopathy, was not increased in this population of patients positive for anti-HMGCR. Follow-up studies of the prevalence of anti-HMGCR in statin users in the Atherosclerosis Risk in Communities (ARIC) cohort, including those with self-limited statin myotoxicity, have also shown the absence of this antibody.14 This shows that anti-HMGCR is not found in the majority of statin-exposed patients and is highly specific for autoimmune myopathy. This also suggests that statin-associated autoimmune myopathy represents a pathologic process that is distinct from self-limited statin intolerance.
HOW THE CONDITION PRESENTS
Immune-mediated statin myopathy presents similarly to other idiopathic inflammatory myopathies such as polymyositis (Table 1). Symptoms often develop in a subacute to chronic course and can occur at any time with statin treatment. In one study,10 the average duration of statin use before the onset of weakness was 3 years (range 2 months to 10 years). In some patients whose statin had been stopped because of abnormal creatine kinase levels, weakness developed later, at a range of 0.5 to 20 months. Even low doses of statins (such as 10 mg of simvastatin) have been found to trigger this condition.10
Patients uniformly develop symmetric proximal arm and leg weakness, and distal weakness can also occur.11 Other features have included dysphagia, arthralgias, myalgias, and Raynaud phenomenon.9 Men and women are represented in roughly equal numbers.
The muscle enzymes are strikingly elevated in this disease, with a mean creatine kinase value of 10,333 IU/L at initial presentation.9 Although the creatine kinase level may be very elevated, patients often do not present with weakness until a certain threshold value is reached, in contrast with patients with anti-signal recognition particle necrotizing myopathy, who can present with profound weakness at a lower level. Hence, by the time patients are clinically symptomatic, the process may have been going on for some time. Despite the seemingly massive leak in muscle enzymes, the patients do not develop rhabdomyolysis. Inflammatory markers need not be elevated, and an association with other antibodies such as antinuclear antibody is not often seen.
Magnetic resonance imaging of the thigh has shown muscle edema in all patients.9 In decreasing order of frequency, other findings are atrophy, fatty replacement, and fascial edema.
Electromyography of involved muscle has shown irritable myopathy in most patients (88%) and nonirritable myopathy in a few.
Muscle biopsy studies have shown prominent necrotic and regenerating fibers without significant inflammatory infiltrate.11 There is also myophagocytosis of necrotic fibers and diffuse or focal up-regulation of major histocompatability complex class I expression.10
PATIENTS WITH ANTI-HMGCR WHO HAVE AND WHO HAVE NEVER TAKEN STATINS
When the anti-HMGCR antibody was tested for in the Johns Hopkins cohort,12 33% of patients with a necrotizing myopathy associated with this antibody had never taken a statin. The two groups were clinically indistinguishable, save for a few aspects. Compared with patients who had taken a statin, those who had never taken a statin were younger (mean age 37 vs 59), had higher levels of creatine kinase (13,392 vs 7,881 IU/L), and had a different race distribution (46.7% vs 86.7% white). Initial HMGCR antibody levels were also noted to correlate with creatine kinase levels and strength in statin-exposed patients but not in those who had never taken a statin.15
We hypothesize that in patients who have never taken a statin, other genetic or environmental factors may be the cause of the increased HMGCR expression, which then triggers the autoimmune response. Until further data are gathered, we should probably treat these patients as we treat those who develop this disease after taking a statin, and avoid giving them statins altogether.
MANAGEMENT OF STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Treatment of statin-associated autoimmune necrotizing myopathy can be challenging and requires immunosuppressive drugs.
Statin therapy should be stopped once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy. Electromyography detects myopathy and shows chronicity, distribution, and degree of severity. Although not necessary for diagnosis, magnetic resonance imaging helps to evaluate the extent of muscle involvement and damage and provides guidance when choosing a site for muscle biopsy. Muscle biopsy is necessary to determine the actual pathology and to exclude mimics such as dystrophy or metabolic myopathies.
When an immune-mediated myopathy is confirmed, prompt referral to a rheumatologist or a neuromuscular specialist is recommended.
Steroids are usually the first-line treatment for this disease. Other immunosuppressives, such as methotrexate, azathioprine, mycophenolate mofetil, and rituximab have been used with varying levels of success. In our experience, intravenous immunoglobulin has been particularly beneficial for refractory cases. With treatment, muscle enzyme levels and weakness improve, but relapses can occur. The ideal choice of immunosuppressive therapy and the duration of therapy are currently under investigation.
Rechallenge with another statin
At this time, the issue of rechallenging the patient with another statin has not been clarified. Given the autoimmune nature of the disease, we would avoid exposing the patient to a known trigger. However, this may be a difficult decision in patients with cardiovascular risk factors who require statins for primary or secondary prevention. We suggest using alternative cholesterol-lowering agents first and using them in combination if needed.
We have had some success in maintaining a handful of patients on a statin while treating them concurrently with immunosuppression. This is not ideal because they are constantly being exposed to the likely trigger for their disease, but it may be unavoidable if statins are deemed absolutely necessary. We have also had a patient with known statin-associated immune-mediated necrotizing myopathy who later became profoundly weak after another physician started her on a newer-generation agent, pitavastatin. This suggests to us that rechallenging patients, even with a different statin, can have deleterious effects.
IMPLICATIONS FOR CLINICAL PRACTICE
The true prevalence of statin-associated autoimmune myopathy in practice is unknown. In the Johns Hopkins Myositis Center cohort of patients with suspected myopathy, anti-HMGCR was found in 6% of the patients and was the second most frequent antibody found after anti-Jo1.12
Given the frequency of muscle-related complaints in patients on statins, we recommend obtaining baseline muscle enzyme measurements before starting statin therapy. As recommended by the National Lipid Association Statin Safety Assessment Task Force, the creatine kinase level should be measured when a patient develops muscular complaints, to help gauge the severity of the disease and to help decide whether to continue therapy.8 Random testing of the creatine kinase level in asymptomatic patients is not recommended.
At present, the diagnosis of statin-associated autoimmune necrotizing myopathy is based on a combination of findings—elevated muscle enzyme levels, muscle weakness, irritable findings on electromyography, and necrotizing myopathy on biopsy in a patient on a statin. The finding of the HMGCR antibody confirms the diagnosis. A test for this antibody is now commercially available in the United States. We suggest testing for the antibody in the following scenarios:
- A persistently elevated or rising creatine kinase, aspartate aminotransferase, alanine aminotransferase, or aldolase level after the statin is stopped; although no fixed creatine kinase level has been determined, a level above 1,000 U/L would be a reasonable cutoff at which to test
- Muscle symptoms (proximal or distal weakness) that persist 12 weeks after statin cessation regardless of the creatine kinase level, especially if the patient has dysphagia
- The finding of muscle irritability on electromyography or diffuse muscle edema on magnetic resonance imaging when testing for other myositis-specific antibodies is negative
- Muscle biopsy showing necrotizing myopathy with little or no inflammation.
In addition, since necrotizing myopathy is known to be associated with malignancy and since necrotizing myopathy is more common in older people, who are also more likely to be taking a statin, an age-appropriate malignancy evaluation is warranted as well.
- Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol 2011; 5:299–307.
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C–60C.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Fung EC, Crook MA. Statin myopathy: a lipid clinic experience on the tolerability of statin rechallenge. Cardiovasc Ther 2012; 30:e212–e218.
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008; 8:333–338.
- Holbrook A, Wright M, Sung M, Ribic C, Baker S. Statin-associated rhabdomyolysis: is there a dose-response relationship? Can J Cardiol 2011; 27:146–151.
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–94C.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766.
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007; 17:194–200.
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41:185–190.
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425–430.
- Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
- Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol 2011; 5:299–307.
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C–60C.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Fung EC, Crook MA. Statin myopathy: a lipid clinic experience on the tolerability of statin rechallenge. Cardiovasc Ther 2012; 30:e212–e218.
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008; 8:333–338.
- Holbrook A, Wright M, Sung M, Ribic C, Baker S. Statin-associated rhabdomyolysis: is there a dose-response relationship? Can J Cardiol 2011; 27:146–151.
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–94C.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766.
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007; 17:194–200.
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41:185–190.
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425–430.
- Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
KEY POINTS
- Most cases of muscle symptoms associated with statin use are a direct effect of the statin on the muscle and resolve after the statin is discontinued.
- In contrast to simple myalgia or myositis, statin-associated autoimmune necrotizing myopathy can persist or even arise de novo after the statin is stopped.
- This condition presents with symmetric proximal arm and leg weakness and striking elevations of muscle enzymes such as creatine kinase.
- Treatment can be challenging and requires immunosuppressive drugs; referral to a specialist is recommended.
- Statin therapy should be discontinued once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy.
Sarcoidal infiltration of tattoos
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
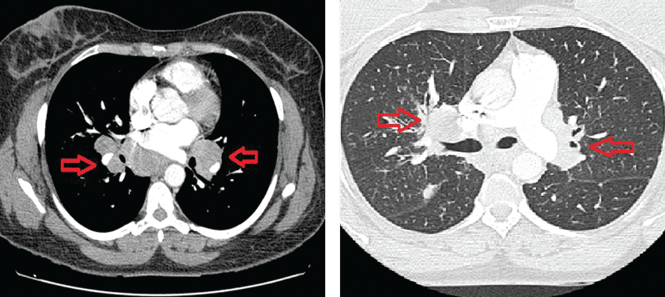
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
Link between early exposure to acetaminophen and childhood asthma found weak, overstated
The reported link between early life exposure to acetaminophen and the development of asthma in children is “weak” and “overstated” based on currently available evidence, according to a report published by the Archives of Disease in Childhood.
In a review of currently available data culled from Embase and PubMed databases, 1,192 relevant studies conducted between 1967 and 2013 were analyzed, of which 11 were included for analysis. Of these 11 studies, 5 found “increased odds” that exposure to acetaminophen during the first trimester of pregnancy could lead to development of asthma (pooled odds ratio, 1.39); however, there was a high degree of between-study heterogeneity among the trials (I2 = 64.2%, P = .03), reported Dr. M. Cheelo of the University of Melbourne, and associates.

Of those five, only two studies examined the effects of acetaminophen exposure during the second trimester, but attained widely disparate results: Study one reported an OR of 1.06, while the other reported an OR of 2.15, with I2 = 80%. Two studies also tested acetaminophen exposure during the third trimester and found a “weak association,” with a pooled OR of 1.17. Three studies look at acetaminophen exposure through an entire pregnancy, but all had “significant heterogeneity” in their findings (OR = 1.65, 1.22, and 0.74; I2 = 89%). Only one study that was examined adjusted for respiratory tract infections during pregnancy, but according to the authors, “all studies that adjusted for early life respiratory tract infections found a reduction in the association between [acetaminophen] exposure and subsequent childhood asthma” (Arch. Dis. Child. 2014 [doi:10.1136/archdischild-2012-303043]).
The other 6 of the 11 total studies examined acetaminophen exposure over the first 2 years of life. Three of these studies found a “weak positive association,” as did four studies directly comparing children with and without acetaminophen exposure. All but one study adjusted results for respiratory tract infections during pregnancy, which caused a “moderate attenuation of the association between frequency of [acetaminophen] intake and childhood asthma.” Consequently, investigators concluded that “evidence of an association between early life [acetaminophen] and asthma is often overstated, and there is currently insufficient evidence to support changing guidelines in the use of this medicine.”
The authors reported no relevant financial conflicts of interest.
The reported link between early life exposure to acetaminophen and the development of asthma in children is “weak” and “overstated” based on currently available evidence, according to a report published by the Archives of Disease in Childhood.
In a review of currently available data culled from Embase and PubMed databases, 1,192 relevant studies conducted between 1967 and 2013 were analyzed, of which 11 were included for analysis. Of these 11 studies, 5 found “increased odds” that exposure to acetaminophen during the first trimester of pregnancy could lead to development of asthma (pooled odds ratio, 1.39); however, there was a high degree of between-study heterogeneity among the trials (I2 = 64.2%, P = .03), reported Dr. M. Cheelo of the University of Melbourne, and associates.
Of those five, only two studies examined the effects of acetaminophen exposure during the second trimester, but attained widely disparate results: Study one reported an OR of 1.06, while the other reported an OR of 2.15, with I2 = 80%. Two studies also tested acetaminophen exposure during the third trimester and found a “weak association,” with a pooled OR of 1.17. Three studies look at acetaminophen exposure through an entire pregnancy, but all had “significant heterogeneity” in their findings (OR = 1.65, 1.22, and 0.74; I2 = 89%). Only one study that was examined adjusted for respiratory tract infections during pregnancy, but according to the authors, “all studies that adjusted for early life respiratory tract infections found a reduction in the association between [acetaminophen] exposure and subsequent childhood asthma” (Arch. Dis. Child. 2014 [doi:10.1136/archdischild-2012-303043]).
The other 6 of the 11 total studies examined acetaminophen exposure over the first 2 years of life. Three of these studies found a “weak positive association,” as did four studies directly comparing children with and without acetaminophen exposure. All but one study adjusted results for respiratory tract infections during pregnancy, which caused a “moderate attenuation of the association between frequency of [acetaminophen] intake and childhood asthma.” Consequently, investigators concluded that “evidence of an association between early life [acetaminophen] and asthma is often overstated, and there is currently insufficient evidence to support changing guidelines in the use of this medicine.”
The authors reported no relevant financial conflicts of interest.
The reported link between early life exposure to acetaminophen and the development of asthma in children is “weak” and “overstated” based on currently available evidence, according to a report published by the Archives of Disease in Childhood.
In a review of currently available data culled from Embase and PubMed databases, 1,192 relevant studies conducted between 1967 and 2013 were analyzed, of which 11 were included for analysis. Of these 11 studies, 5 found “increased odds” that exposure to acetaminophen during the first trimester of pregnancy could lead to development of asthma (pooled odds ratio, 1.39); however, there was a high degree of between-study heterogeneity among the trials (I2 = 64.2%, P = .03), reported Dr. M. Cheelo of the University of Melbourne, and associates.
Of those five, only two studies examined the effects of acetaminophen exposure during the second trimester, but attained widely disparate results: Study one reported an OR of 1.06, while the other reported an OR of 2.15, with I2 = 80%. Two studies also tested acetaminophen exposure during the third trimester and found a “weak association,” with a pooled OR of 1.17. Three studies look at acetaminophen exposure through an entire pregnancy, but all had “significant heterogeneity” in their findings (OR = 1.65, 1.22, and 0.74; I2 = 89%). Only one study that was examined adjusted for respiratory tract infections during pregnancy, but according to the authors, “all studies that adjusted for early life respiratory tract infections found a reduction in the association between [acetaminophen] exposure and subsequent childhood asthma” (Arch. Dis. Child. 2014 [doi:10.1136/archdischild-2012-303043]).
The other 6 of the 11 total studies examined acetaminophen exposure over the first 2 years of life. Three of these studies found a “weak positive association,” as did four studies directly comparing children with and without acetaminophen exposure. All but one study adjusted results for respiratory tract infections during pregnancy, which caused a “moderate attenuation of the association between frequency of [acetaminophen] intake and childhood asthma.” Consequently, investigators concluded that “evidence of an association between early life [acetaminophen] and asthma is often overstated, and there is currently insufficient evidence to support changing guidelines in the use of this medicine.”
The authors reported no relevant financial conflicts of interest.
FROM THE ARCHIVES OF DISEASE IN CHILDHOOD
Key clinical point: Current evidence regarding purported link between early life exposure to acetaminophen and development of childhood asthma is weak and often overstated.
Major finding: Meta-analysis of 11 studies found disparate results linking acetaminophen exposure and childhood asthma, and these results generally were attenuated when adjusted for data related to respiratory tract infections during pregnancy and subsequent childhood asthma.
Data source: Meta-analysis of 11 observational cohort studies.
Disclosures: The authors reported no relevant financial conflicts of interest.
