User login
The Importance of Sex of Patient in the Management of Femoroacetabular Impingement
Femoroacetabular impingement (FAI), a recently described hip condition in adolescents and young adults, results from abnormal physical contact between the proximal femur and the acetabulum.1 FAI is usually characterized by the site of the predominant morphologic abnormality—proximal femur (cam-type FAI), acetabulum (pincer-type FAI), or both (mixed impingement). Cam-type FAI is typified by the aspherical extension of the articular surface at the anterosuperior head–neck junction of the proximal femur with loss of the normal offset. With hip motion, especially in the maximal ranges of flexion and internal rotation, the aspherical proximal femur repeatedly contacts the anterosuperior acetabulum, damaging the chondrolabral junction and ultimately the labrum itself. In pincer-type impingement, femoral head overcoverage caused by acetabular retroversion and/or coxa profunda directly damages the anterior labrum when the acetabular rim contacts the proximal femur during physiologic motion. “Contrecoup” injury of the posterior-inferior acetabular cartilage may also occur. Over time, recurrent microtrauma to the acetabular cartilage and/or labrum may lead to degenerative changes of the hip and ultimately to premature osteoarthritis.1,2
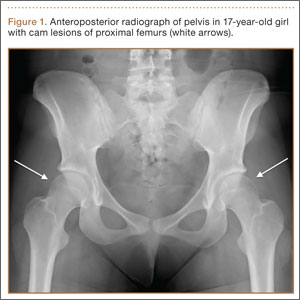
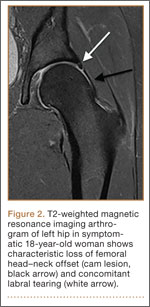
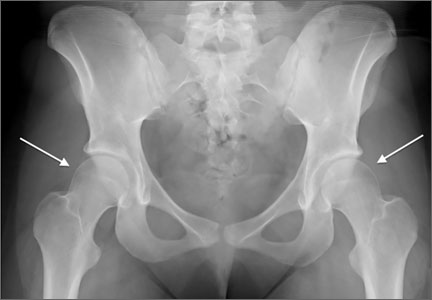
Patients with FAI typically present with groin pain that may be activity-related or that may occur with prolonged sitting with the hip in a flexed position. Physical examination findings suggestive of FAI include decreased passive internal hip rotation and reproducible pain with adduction and internal rotation of the flexed hip—the impingement sign, or the flexion, adduction, and internal rotation (FADIR) test.3 Diagnostic imaging evaluation initially includes radiographs of the pelvis and hips. These radiographs may show a “pistol-grip” deformity and/or decreased head–neck offset (as determined by increased alpha angle) in the setting of cam-type impingement (Figure 1).4 Pincer-type impingement may be associated with a crossover sign, coxa profunda, and an increased center-edge angle (CEA). Advanced imaging studies, such as computed tomography (CT), magnetic resonance imaging (MRI) arthrogram, and delayed gadolinium-enhanced MRI of cartilage (dGEMRIC), are commonly used to better delineate bony deformity and concomitant injuries of the labrum and cartilage (Figure 2).
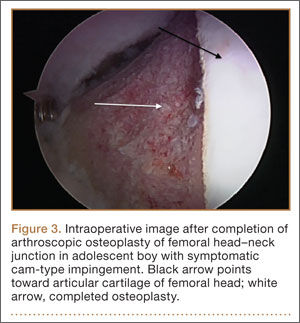
Treatment for FAI often consists initially of activity modification, use of anti-inflammatory medications, and physical therapy. Intra-articular corticosteroid injections may be used both diagnostically and therapeutically. When nonsurgical measures fail to adequately relieve symptoms, surgery may be warranted. Whether performed open or arthroscopically, surgery is directed first at correcting the underlying osseous abnormality—performing an osteoplasty of the proximal femur to remove the cam lesion, performing an acetabular osteoplasty (“rim-trimming”) to address a focal pincer lesion, and/or performing a periacetabular osteotomy to decrease global acetabular overcoverage (Figure 3).5
Sex-Based Differences in FAI Incidence
Traditionally, it was thought that cam-type impingement occurred predominantly in young, athletic males, whereas pincer-type impingement resulting from acetabular overcoverage occurred primarily in females during their fourth decade. However, our understanding of the sex-based differences in the incidence and presentation of FAI has evolved, and it is now clear that the interplay of sex, radiographic signs of impingement, and development of symptoms requiring treatment is more complex.
In recent large population-based studies, investigators have attempted to better characterize the sex-based differences in the incidence of osseous FAI deformity. Gosvig and colleagues2 examined radiographic and questionnaire outcomes of 3620 patients (age range, 21-90 years) and found that males were more likely than females to have a pistol-grip deformity of the hip (19.6% vs 5.2%); that deep acetabular sockets were common in both sexes (15.2% vs 19.4%); and that the presence of pistol-grip deformity or deep socket was significantly associated with development of osteoarthritis, independent of sex.
In a study of 2081 asymptomatic patients (mean age, 18.6 years), Laborie and colleagues4 reported similar radiographic findings. Males were significantly more likely than females to have a cam-type deformity, as evidenced by pistol-grip deformity, focal prominence of the femoral neck, and/or flattening of the lateral aspect of the femoral head. Males were also more likely than females to have a pincer deformity, though radiographic signs of pincer deformity—a crossover sign, excessive acetabular coverage (defined by increased CEA), and a posterior wall sign—were common in both sexes, occurring in 16.6% of females and 34.3% of males. Bilateral findings of FAI-associated deformity were also more common in males than in females, both for cam-type deformity (24.7% vs 6.3%) and pincer-type deformity (21.7% vs 9.7%).
Sex-Based Differences in FAI Presentation
In males and females, the clinical presentation of FAI is similar—insidious onset of deep groin pain, often exacerbated with activity, and physical examination findings of decreased hip motion (particularly internal rotation) and a positive impingement test.3 Nevertheless, the sexes’ clinical presentation differs in several ways. Specifically, in a study using 3-dimensional CT to assess bony deformity in both symptomatic and asymptomatic patients, Beaulé and colleagues6 reported that alpha angles were significantly higher in symptomatic males than in symptomatic females (73.3° vs 58.7°). Hetsroni and colleagues7 recently reported similar results in a study of 217 symptomatic young adults treated arthroscopically for hip pain. Preoperative CT showed that alpha angles were significantly larger in males than in females (63.6° vs 47.8°). The authors postulated that females may be more likely to be symptomatic in the setting of smaller cam lesions because of the increased peak hip flexion and frontal plane motion commonly demonstrated by females during drop landings in sport. The authors further hypothesized that sex differences in muscle mass (which contributes to dynamic hip stability) and ligamentous laxity (a component of static hip stability) may result in larger physiologic ranges of motion for many females. As a result, bony impingement may occur in the setting of smaller anatomical lesions in females. The authors further noted that, compared with their male counterparts, females being treated for symptomatic FAI had significantly more femoral and acetabular anteversion.
Another male–female presentation difference involves symptom bilaterality. Specifically, males are significantly more likely than females to have symptomatic FAI involving both hips. In a recent study of 646 patients who underwent hip arthroscopy for symptomatic FAI during a 2-year period, Klingenstein and colleagues8 found that females constituted 48.2% of unilateral arthroscopy patients but only 34.8% of bilateral arthroscopy patients. The odds ratio of males treated for both hips, compared with females, was 1.7 (95% confidence interval, 1.16–2.54).
Last, it has been reported that, on clinical presentation, hip function scores are significantly lower in females than in males. In a recent study of 612 cases of symptomatic FAI treated with hip arthroscopy, Malviya and colleagues9 found that females had significantly lower quality-of-life scores both before and after surgery. Hetsroni and colleagues7 reported similar findings, with females having significantly lower preoperative modified Harris Hip Scores and lower Hip Outcome Scores in the domains of Activities of Daily Living and Sports.
Sex-Based Differences in FAI Treatment
and Outcomes
Surgical treatment of FAI is focused on identifying the source of hip pain and dysfunction—be it osseous lesion, labral tearing, chondral injury, or iliopsoas tendonitis—and treating it accordingly, regardless of sex. Most studies of this approach find consistent improvement in the short-term and midterm outcome scores for a majority of patients. However, relatively few studies have focused specifically on sex in determining the percentage of patients who require surgical treatment, in deciding the type of surgery that should be performed, or in measuring surgical outcomes in patients with symptomatic FAI.
In their review of 23 studies of FAI surgery, Ng and colleagues10 found that, of 970 patients, 608 (62.7%) were male and 362 (37.3%) were female. Similarly higher rates for males were previously published.5,11 More recently, Clohisy and colleagues12 reported on the descriptive epidemiology of patients having surgery for FAI at 8 different medical centers in North America. Fifty-five percent of the hips surgically treated for symptomatic FAI were females’. The authors speculated that this unexpectedly high rate could have resulted from US and Canadian female athletes’ increasingly higher level of sports participation. The results of this study, one of the largest examining the rate of surgery for males and females with FAI, suggest that females are more likely to have surgery for symptomatic FAI despite being less likely to have radiographic evidence of impingement. Our understanding of this phenomenon continues to advance.
In a recent prospective study, Krych and colleagues13 evaluated the clinical outcomes of FAI surgeries (labral débridement, labral repair) in an all-female patient cohort. Female patients with symptomatic FAI were randomized to undergo either labral débridement or labral repair. There were clinical improvements in both groups, but, compared with labral débridement patients, labral repair patients had more significantly improved Hip Outcome Scores in the domains of Activities of Daily Living and Sports, as well as better subjective outcomes. Although the study did not compare female patients with male patients, it does provide evidence that female patients specifically may benefit more from labral repair than from labral débridement alone.
With respect to different surgical treatments for male and female patients, Hetsroni and colleagues7 introduced the idea of sex-specific treatment when they noted more hip anteversion in their study’s female patients than in its male patients. They suggested that, because the anterosuperior acetabulum is subjected to a high amount of stress during weight-bearing and gait, this area in females with suspected pincer lesions should be rim-trimmed judiciously to avoid increasing the stress and perhaps even hastening the development of degenerative disease. Last, though several authors have noted that hip function scores are lower in females than in males on presentation, it has also been reported that females demonstrate more improvement in functional scores after surgery.9 This may be important information to discuss during preoperative counseling about expected goals and outcomes.
Conclusion
Femoroacetabular impingement is a common clinical entity that affects both males and females. However, sexual dimorphism in FAI incidence, presentation, treatment, and outcomes has recently been described in the literature (Table). Being aware of these sex-based differences and tailoring patient evaluation and management accordingly will likely result in optimal outcomes for each person who presents with symptomatic FAI.

1. Ganz R, Parvizi J, Beck M, Leunig M, Notzli H, Siebenrock KA. Femoroacetabular impingement: a cause for osteoarthritis of the hip. Clin Orthop. 2003;(417):112-120.
2. Gosvig KK, Jacobsen S, Sonne-Holm S, Palm H, Troelsen A. Prevalence of malformations of the hip joint and their relationship to sex, groin pain, and risk of osteoarthritis: a population-based survey. J Bone Joint Surg Am. 2010;92(5):1162-1169.
3. Philippon MJ, Maxwell RB, Johnston TL, Schenker M, Briggs KK. Clinical presentation of femoroacetabular impingement. Knee Surg Sports Traumatol Arthrosc. 2007;15(8):1041-1047.
4. Laborie LB, Lehmann TG, Engesaeter IO, Eastwood DM, Engesaeter LB, Rosendahl K. Prevalence of radiographic findings thought to be associated with femoroacetabular impingement in a population-based cohort of 2081 healthy young adults. Radiology. 2011;260(2):494-502.
5. Clohisy JC, St John LC, Schutz AL. Surgical treatment of femoroacetabular impingement: a systematic review of the literature. Clin Orthop. 2010;468(2):555-564.
6. Beaulé PE, Zaragoza E, Motamedi K, Copelan N, Dorey FJ. Three-dimensional computed tomography of the hip in the assessment of femoroacetabular impingement. J Orthop Res. 2005;23(6):1286-1292.
7. Hetsroni I, Dela Torre K, Duke G, Lyman S, Kelly BT. Sex differences of hip morphology in young adults with hip pain and labral tears. Arthroscopy. 2013;29(1):54-63.
8. Klingenstein GG, Zbeda RM, Bedi A, Magennis E, Kelly BT. Prevalence and preoperative demographic and radiographic predictors of bilateral femoroacetabular impingement. Am J Sports Med. 2013;41(4):762-768.
9. Malviya A, Stafford GH, Villar RN. Impact of arthroscopy of the hip for femoroacetabular impingement on quality of life at a mean follow-up of 3.2 years. J Bone Joint Surg Br. 2012;94(4):466-470.
10. Ng VY, Arora N, Best TM, Pan X, Ellis TJ. Efficacy of surgery for femoroacetabular impingement: a systematic review. Am J Sports Med. 2010;38(11):2337-2345.
11. Matsuda DK, Carlisle JC, Arthurs SC, Wierks CH, Philippon MJ. Comparative systematic review of the open dislocation, mini-open, and arthroscopic surgeries for femoroacetabular impingement. Arthroscopy. 2011;27(2):252-269.
12. Clohisy JC, Baca G, Beaule PE, et al. Descriptive epidemiology of femoroacetabular impingement: a North American cohort of patients undergoing surgery. Am J Sports Med. 2013;41(6):1348-1356.
13. Krych AJ, Thompson M, Knutson Z, Scoon J, Coleman SH. Arthroscopic labral repair versus selective labral debridement in female patients with femoroacetabular impingement: a prospective randomized study. Arthroscopy. 2013;29(1):46-53.
Femoroacetabular impingement (FAI), a recently described hip condition in adolescents and young adults, results from abnormal physical contact between the proximal femur and the acetabulum.1 FAI is usually characterized by the site of the predominant morphologic abnormality—proximal femur (cam-type FAI), acetabulum (pincer-type FAI), or both (mixed impingement). Cam-type FAI is typified by the aspherical extension of the articular surface at the anterosuperior head–neck junction of the proximal femur with loss of the normal offset. With hip motion, especially in the maximal ranges of flexion and internal rotation, the aspherical proximal femur repeatedly contacts the anterosuperior acetabulum, damaging the chondrolabral junction and ultimately the labrum itself. In pincer-type impingement, femoral head overcoverage caused by acetabular retroversion and/or coxa profunda directly damages the anterior labrum when the acetabular rim contacts the proximal femur during physiologic motion. “Contrecoup” injury of the posterior-inferior acetabular cartilage may also occur. Over time, recurrent microtrauma to the acetabular cartilage and/or labrum may lead to degenerative changes of the hip and ultimately to premature osteoarthritis.1,2
Patients with FAI typically present with groin pain that may be activity-related or that may occur with prolonged sitting with the hip in a flexed position. Physical examination findings suggestive of FAI include decreased passive internal hip rotation and reproducible pain with adduction and internal rotation of the flexed hip—the impingement sign, or the flexion, adduction, and internal rotation (FADIR) test.3 Diagnostic imaging evaluation initially includes radiographs of the pelvis and hips. These radiographs may show a “pistol-grip” deformity and/or decreased head–neck offset (as determined by increased alpha angle) in the setting of cam-type impingement (Figure 1).4 Pincer-type impingement may be associated with a crossover sign, coxa profunda, and an increased center-edge angle (CEA). Advanced imaging studies, such as computed tomography (CT), magnetic resonance imaging (MRI) arthrogram, and delayed gadolinium-enhanced MRI of cartilage (dGEMRIC), are commonly used to better delineate bony deformity and concomitant injuries of the labrum and cartilage (Figure 2).
Treatment for FAI often consists initially of activity modification, use of anti-inflammatory medications, and physical therapy. Intra-articular corticosteroid injections may be used both diagnostically and therapeutically. When nonsurgical measures fail to adequately relieve symptoms, surgery may be warranted. Whether performed open or arthroscopically, surgery is directed first at correcting the underlying osseous abnormality—performing an osteoplasty of the proximal femur to remove the cam lesion, performing an acetabular osteoplasty (“rim-trimming”) to address a focal pincer lesion, and/or performing a periacetabular osteotomy to decrease global acetabular overcoverage (Figure 3).5
Sex-Based Differences in FAI Incidence
Traditionally, it was thought that cam-type impingement occurred predominantly in young, athletic males, whereas pincer-type impingement resulting from acetabular overcoverage occurred primarily in females during their fourth decade. However, our understanding of the sex-based differences in the incidence and presentation of FAI has evolved, and it is now clear that the interplay of sex, radiographic signs of impingement, and development of symptoms requiring treatment is more complex.
In recent large population-based studies, investigators have attempted to better characterize the sex-based differences in the incidence of osseous FAI deformity. Gosvig and colleagues2 examined radiographic and questionnaire outcomes of 3620 patients (age range, 21-90 years) and found that males were more likely than females to have a pistol-grip deformity of the hip (19.6% vs 5.2%); that deep acetabular sockets were common in both sexes (15.2% vs 19.4%); and that the presence of pistol-grip deformity or deep socket was significantly associated with development of osteoarthritis, independent of sex.
In a study of 2081 asymptomatic patients (mean age, 18.6 years), Laborie and colleagues4 reported similar radiographic findings. Males were significantly more likely than females to have a cam-type deformity, as evidenced by pistol-grip deformity, focal prominence of the femoral neck, and/or flattening of the lateral aspect of the femoral head. Males were also more likely than females to have a pincer deformity, though radiographic signs of pincer deformity—a crossover sign, excessive acetabular coverage (defined by increased CEA), and a posterior wall sign—were common in both sexes, occurring in 16.6% of females and 34.3% of males. Bilateral findings of FAI-associated deformity were also more common in males than in females, both for cam-type deformity (24.7% vs 6.3%) and pincer-type deformity (21.7% vs 9.7%).
Sex-Based Differences in FAI Presentation
In males and females, the clinical presentation of FAI is similar—insidious onset of deep groin pain, often exacerbated with activity, and physical examination findings of decreased hip motion (particularly internal rotation) and a positive impingement test.3 Nevertheless, the sexes’ clinical presentation differs in several ways. Specifically, in a study using 3-dimensional CT to assess bony deformity in both symptomatic and asymptomatic patients, Beaulé and colleagues6 reported that alpha angles were significantly higher in symptomatic males than in symptomatic females (73.3° vs 58.7°). Hetsroni and colleagues7 recently reported similar results in a study of 217 symptomatic young adults treated arthroscopically for hip pain. Preoperative CT showed that alpha angles were significantly larger in males than in females (63.6° vs 47.8°). The authors postulated that females may be more likely to be symptomatic in the setting of smaller cam lesions because of the increased peak hip flexion and frontal plane motion commonly demonstrated by females during drop landings in sport. The authors further hypothesized that sex differences in muscle mass (which contributes to dynamic hip stability) and ligamentous laxity (a component of static hip stability) may result in larger physiologic ranges of motion for many females. As a result, bony impingement may occur in the setting of smaller anatomical lesions in females. The authors further noted that, compared with their male counterparts, females being treated for symptomatic FAI had significantly more femoral and acetabular anteversion.
Another male–female presentation difference involves symptom bilaterality. Specifically, males are significantly more likely than females to have symptomatic FAI involving both hips. In a recent study of 646 patients who underwent hip arthroscopy for symptomatic FAI during a 2-year period, Klingenstein and colleagues8 found that females constituted 48.2% of unilateral arthroscopy patients but only 34.8% of bilateral arthroscopy patients. The odds ratio of males treated for both hips, compared with females, was 1.7 (95% confidence interval, 1.16–2.54).
Last, it has been reported that, on clinical presentation, hip function scores are significantly lower in females than in males. In a recent study of 612 cases of symptomatic FAI treated with hip arthroscopy, Malviya and colleagues9 found that females had significantly lower quality-of-life scores both before and after surgery. Hetsroni and colleagues7 reported similar findings, with females having significantly lower preoperative modified Harris Hip Scores and lower Hip Outcome Scores in the domains of Activities of Daily Living and Sports.
Sex-Based Differences in FAI Treatment
and Outcomes
Surgical treatment of FAI is focused on identifying the source of hip pain and dysfunction—be it osseous lesion, labral tearing, chondral injury, or iliopsoas tendonitis—and treating it accordingly, regardless of sex. Most studies of this approach find consistent improvement in the short-term and midterm outcome scores for a majority of patients. However, relatively few studies have focused specifically on sex in determining the percentage of patients who require surgical treatment, in deciding the type of surgery that should be performed, or in measuring surgical outcomes in patients with symptomatic FAI.
In their review of 23 studies of FAI surgery, Ng and colleagues10 found that, of 970 patients, 608 (62.7%) were male and 362 (37.3%) were female. Similarly higher rates for males were previously published.5,11 More recently, Clohisy and colleagues12 reported on the descriptive epidemiology of patients having surgery for FAI at 8 different medical centers in North America. Fifty-five percent of the hips surgically treated for symptomatic FAI were females’. The authors speculated that this unexpectedly high rate could have resulted from US and Canadian female athletes’ increasingly higher level of sports participation. The results of this study, one of the largest examining the rate of surgery for males and females with FAI, suggest that females are more likely to have surgery for symptomatic FAI despite being less likely to have radiographic evidence of impingement. Our understanding of this phenomenon continues to advance.
In a recent prospective study, Krych and colleagues13 evaluated the clinical outcomes of FAI surgeries (labral débridement, labral repair) in an all-female patient cohort. Female patients with symptomatic FAI were randomized to undergo either labral débridement or labral repair. There were clinical improvements in both groups, but, compared with labral débridement patients, labral repair patients had more significantly improved Hip Outcome Scores in the domains of Activities of Daily Living and Sports, as well as better subjective outcomes. Although the study did not compare female patients with male patients, it does provide evidence that female patients specifically may benefit more from labral repair than from labral débridement alone.
With respect to different surgical treatments for male and female patients, Hetsroni and colleagues7 introduced the idea of sex-specific treatment when they noted more hip anteversion in their study’s female patients than in its male patients. They suggested that, because the anterosuperior acetabulum is subjected to a high amount of stress during weight-bearing and gait, this area in females with suspected pincer lesions should be rim-trimmed judiciously to avoid increasing the stress and perhaps even hastening the development of degenerative disease. Last, though several authors have noted that hip function scores are lower in females than in males on presentation, it has also been reported that females demonstrate more improvement in functional scores after surgery.9 This may be important information to discuss during preoperative counseling about expected goals and outcomes.
Conclusion
Femoroacetabular impingement is a common clinical entity that affects both males and females. However, sexual dimorphism in FAI incidence, presentation, treatment, and outcomes has recently been described in the literature (Table). Being aware of these sex-based differences and tailoring patient evaluation and management accordingly will likely result in optimal outcomes for each person who presents with symptomatic FAI.
Femoroacetabular impingement (FAI), a recently described hip condition in adolescents and young adults, results from abnormal physical contact between the proximal femur and the acetabulum.1 FAI is usually characterized by the site of the predominant morphologic abnormality—proximal femur (cam-type FAI), acetabulum (pincer-type FAI), or both (mixed impingement). Cam-type FAI is typified by the aspherical extension of the articular surface at the anterosuperior head–neck junction of the proximal femur with loss of the normal offset. With hip motion, especially in the maximal ranges of flexion and internal rotation, the aspherical proximal femur repeatedly contacts the anterosuperior acetabulum, damaging the chondrolabral junction and ultimately the labrum itself. In pincer-type impingement, femoral head overcoverage caused by acetabular retroversion and/or coxa profunda directly damages the anterior labrum when the acetabular rim contacts the proximal femur during physiologic motion. “Contrecoup” injury of the posterior-inferior acetabular cartilage may also occur. Over time, recurrent microtrauma to the acetabular cartilage and/or labrum may lead to degenerative changes of the hip and ultimately to premature osteoarthritis.1,2
Patients with FAI typically present with groin pain that may be activity-related or that may occur with prolonged sitting with the hip in a flexed position. Physical examination findings suggestive of FAI include decreased passive internal hip rotation and reproducible pain with adduction and internal rotation of the flexed hip—the impingement sign, or the flexion, adduction, and internal rotation (FADIR) test.3 Diagnostic imaging evaluation initially includes radiographs of the pelvis and hips. These radiographs may show a “pistol-grip” deformity and/or decreased head–neck offset (as determined by increased alpha angle) in the setting of cam-type impingement (Figure 1).4 Pincer-type impingement may be associated with a crossover sign, coxa profunda, and an increased center-edge angle (CEA). Advanced imaging studies, such as computed tomography (CT), magnetic resonance imaging (MRI) arthrogram, and delayed gadolinium-enhanced MRI of cartilage (dGEMRIC), are commonly used to better delineate bony deformity and concomitant injuries of the labrum and cartilage (Figure 2).
Treatment for FAI often consists initially of activity modification, use of anti-inflammatory medications, and physical therapy. Intra-articular corticosteroid injections may be used both diagnostically and therapeutically. When nonsurgical measures fail to adequately relieve symptoms, surgery may be warranted. Whether performed open or arthroscopically, surgery is directed first at correcting the underlying osseous abnormality—performing an osteoplasty of the proximal femur to remove the cam lesion, performing an acetabular osteoplasty (“rim-trimming”) to address a focal pincer lesion, and/or performing a periacetabular osteotomy to decrease global acetabular overcoverage (Figure 3).5
Sex-Based Differences in FAI Incidence
Traditionally, it was thought that cam-type impingement occurred predominantly in young, athletic males, whereas pincer-type impingement resulting from acetabular overcoverage occurred primarily in females during their fourth decade. However, our understanding of the sex-based differences in the incidence and presentation of FAI has evolved, and it is now clear that the interplay of sex, radiographic signs of impingement, and development of symptoms requiring treatment is more complex.
In recent large population-based studies, investigators have attempted to better characterize the sex-based differences in the incidence of osseous FAI deformity. Gosvig and colleagues2 examined radiographic and questionnaire outcomes of 3620 patients (age range, 21-90 years) and found that males were more likely than females to have a pistol-grip deformity of the hip (19.6% vs 5.2%); that deep acetabular sockets were common in both sexes (15.2% vs 19.4%); and that the presence of pistol-grip deformity or deep socket was significantly associated with development of osteoarthritis, independent of sex.
In a study of 2081 asymptomatic patients (mean age, 18.6 years), Laborie and colleagues4 reported similar radiographic findings. Males were significantly more likely than females to have a cam-type deformity, as evidenced by pistol-grip deformity, focal prominence of the femoral neck, and/or flattening of the lateral aspect of the femoral head. Males were also more likely than females to have a pincer deformity, though radiographic signs of pincer deformity—a crossover sign, excessive acetabular coverage (defined by increased CEA), and a posterior wall sign—were common in both sexes, occurring in 16.6% of females and 34.3% of males. Bilateral findings of FAI-associated deformity were also more common in males than in females, both for cam-type deformity (24.7% vs 6.3%) and pincer-type deformity (21.7% vs 9.7%).
Sex-Based Differences in FAI Presentation
In males and females, the clinical presentation of FAI is similar—insidious onset of deep groin pain, often exacerbated with activity, and physical examination findings of decreased hip motion (particularly internal rotation) and a positive impingement test.3 Nevertheless, the sexes’ clinical presentation differs in several ways. Specifically, in a study using 3-dimensional CT to assess bony deformity in both symptomatic and asymptomatic patients, Beaulé and colleagues6 reported that alpha angles were significantly higher in symptomatic males than in symptomatic females (73.3° vs 58.7°). Hetsroni and colleagues7 recently reported similar results in a study of 217 symptomatic young adults treated arthroscopically for hip pain. Preoperative CT showed that alpha angles were significantly larger in males than in females (63.6° vs 47.8°). The authors postulated that females may be more likely to be symptomatic in the setting of smaller cam lesions because of the increased peak hip flexion and frontal plane motion commonly demonstrated by females during drop landings in sport. The authors further hypothesized that sex differences in muscle mass (which contributes to dynamic hip stability) and ligamentous laxity (a component of static hip stability) may result in larger physiologic ranges of motion for many females. As a result, bony impingement may occur in the setting of smaller anatomical lesions in females. The authors further noted that, compared with their male counterparts, females being treated for symptomatic FAI had significantly more femoral and acetabular anteversion.
Another male–female presentation difference involves symptom bilaterality. Specifically, males are significantly more likely than females to have symptomatic FAI involving both hips. In a recent study of 646 patients who underwent hip arthroscopy for symptomatic FAI during a 2-year period, Klingenstein and colleagues8 found that females constituted 48.2% of unilateral arthroscopy patients but only 34.8% of bilateral arthroscopy patients. The odds ratio of males treated for both hips, compared with females, was 1.7 (95% confidence interval, 1.16–2.54).
Last, it has been reported that, on clinical presentation, hip function scores are significantly lower in females than in males. In a recent study of 612 cases of symptomatic FAI treated with hip arthroscopy, Malviya and colleagues9 found that females had significantly lower quality-of-life scores both before and after surgery. Hetsroni and colleagues7 reported similar findings, with females having significantly lower preoperative modified Harris Hip Scores and lower Hip Outcome Scores in the domains of Activities of Daily Living and Sports.
Sex-Based Differences in FAI Treatment
and Outcomes
Surgical treatment of FAI is focused on identifying the source of hip pain and dysfunction—be it osseous lesion, labral tearing, chondral injury, or iliopsoas tendonitis—and treating it accordingly, regardless of sex. Most studies of this approach find consistent improvement in the short-term and midterm outcome scores for a majority of patients. However, relatively few studies have focused specifically on sex in determining the percentage of patients who require surgical treatment, in deciding the type of surgery that should be performed, or in measuring surgical outcomes in patients with symptomatic FAI.
In their review of 23 studies of FAI surgery, Ng and colleagues10 found that, of 970 patients, 608 (62.7%) were male and 362 (37.3%) were female. Similarly higher rates for males were previously published.5,11 More recently, Clohisy and colleagues12 reported on the descriptive epidemiology of patients having surgery for FAI at 8 different medical centers in North America. Fifty-five percent of the hips surgically treated for symptomatic FAI were females’. The authors speculated that this unexpectedly high rate could have resulted from US and Canadian female athletes’ increasingly higher level of sports participation. The results of this study, one of the largest examining the rate of surgery for males and females with FAI, suggest that females are more likely to have surgery for symptomatic FAI despite being less likely to have radiographic evidence of impingement. Our understanding of this phenomenon continues to advance.
In a recent prospective study, Krych and colleagues13 evaluated the clinical outcomes of FAI surgeries (labral débridement, labral repair) in an all-female patient cohort. Female patients with symptomatic FAI were randomized to undergo either labral débridement or labral repair. There were clinical improvements in both groups, but, compared with labral débridement patients, labral repair patients had more significantly improved Hip Outcome Scores in the domains of Activities of Daily Living and Sports, as well as better subjective outcomes. Although the study did not compare female patients with male patients, it does provide evidence that female patients specifically may benefit more from labral repair than from labral débridement alone.
With respect to different surgical treatments for male and female patients, Hetsroni and colleagues7 introduced the idea of sex-specific treatment when they noted more hip anteversion in their study’s female patients than in its male patients. They suggested that, because the anterosuperior acetabulum is subjected to a high amount of stress during weight-bearing and gait, this area in females with suspected pincer lesions should be rim-trimmed judiciously to avoid increasing the stress and perhaps even hastening the development of degenerative disease. Last, though several authors have noted that hip function scores are lower in females than in males on presentation, it has also been reported that females demonstrate more improvement in functional scores after surgery.9 This may be important information to discuss during preoperative counseling about expected goals and outcomes.
Conclusion
Femoroacetabular impingement is a common clinical entity that affects both males and females. However, sexual dimorphism in FAI incidence, presentation, treatment, and outcomes has recently been described in the literature (Table). Being aware of these sex-based differences and tailoring patient evaluation and management accordingly will likely result in optimal outcomes for each person who presents with symptomatic FAI.
1. Ganz R, Parvizi J, Beck M, Leunig M, Notzli H, Siebenrock KA. Femoroacetabular impingement: a cause for osteoarthritis of the hip. Clin Orthop. 2003;(417):112-120.
2. Gosvig KK, Jacobsen S, Sonne-Holm S, Palm H, Troelsen A. Prevalence of malformations of the hip joint and their relationship to sex, groin pain, and risk of osteoarthritis: a population-based survey. J Bone Joint Surg Am. 2010;92(5):1162-1169.
3. Philippon MJ, Maxwell RB, Johnston TL, Schenker M, Briggs KK. Clinical presentation of femoroacetabular impingement. Knee Surg Sports Traumatol Arthrosc. 2007;15(8):1041-1047.
4. Laborie LB, Lehmann TG, Engesaeter IO, Eastwood DM, Engesaeter LB, Rosendahl K. Prevalence of radiographic findings thought to be associated with femoroacetabular impingement in a population-based cohort of 2081 healthy young adults. Radiology. 2011;260(2):494-502.
5. Clohisy JC, St John LC, Schutz AL. Surgical treatment of femoroacetabular impingement: a systematic review of the literature. Clin Orthop. 2010;468(2):555-564.
6. Beaulé PE, Zaragoza E, Motamedi K, Copelan N, Dorey FJ. Three-dimensional computed tomography of the hip in the assessment of femoroacetabular impingement. J Orthop Res. 2005;23(6):1286-1292.
7. Hetsroni I, Dela Torre K, Duke G, Lyman S, Kelly BT. Sex differences of hip morphology in young adults with hip pain and labral tears. Arthroscopy. 2013;29(1):54-63.
8. Klingenstein GG, Zbeda RM, Bedi A, Magennis E, Kelly BT. Prevalence and preoperative demographic and radiographic predictors of bilateral femoroacetabular impingement. Am J Sports Med. 2013;41(4):762-768.
9. Malviya A, Stafford GH, Villar RN. Impact of arthroscopy of the hip for femoroacetabular impingement on quality of life at a mean follow-up of 3.2 years. J Bone Joint Surg Br. 2012;94(4):466-470.
10. Ng VY, Arora N, Best TM, Pan X, Ellis TJ. Efficacy of surgery for femoroacetabular impingement: a systematic review. Am J Sports Med. 2010;38(11):2337-2345.
11. Matsuda DK, Carlisle JC, Arthurs SC, Wierks CH, Philippon MJ. Comparative systematic review of the open dislocation, mini-open, and arthroscopic surgeries for femoroacetabular impingement. Arthroscopy. 2011;27(2):252-269.
12. Clohisy JC, Baca G, Beaule PE, et al. Descriptive epidemiology of femoroacetabular impingement: a North American cohort of patients undergoing surgery. Am J Sports Med. 2013;41(6):1348-1356.
13. Krych AJ, Thompson M, Knutson Z, Scoon J, Coleman SH. Arthroscopic labral repair versus selective labral debridement in female patients with femoroacetabular impingement: a prospective randomized study. Arthroscopy. 2013;29(1):46-53.
1. Ganz R, Parvizi J, Beck M, Leunig M, Notzli H, Siebenrock KA. Femoroacetabular impingement: a cause for osteoarthritis of the hip. Clin Orthop. 2003;(417):112-120.
2. Gosvig KK, Jacobsen S, Sonne-Holm S, Palm H, Troelsen A. Prevalence of malformations of the hip joint and their relationship to sex, groin pain, and risk of osteoarthritis: a population-based survey. J Bone Joint Surg Am. 2010;92(5):1162-1169.
3. Philippon MJ, Maxwell RB, Johnston TL, Schenker M, Briggs KK. Clinical presentation of femoroacetabular impingement. Knee Surg Sports Traumatol Arthrosc. 2007;15(8):1041-1047.
4. Laborie LB, Lehmann TG, Engesaeter IO, Eastwood DM, Engesaeter LB, Rosendahl K. Prevalence of radiographic findings thought to be associated with femoroacetabular impingement in a population-based cohort of 2081 healthy young adults. Radiology. 2011;260(2):494-502.
5. Clohisy JC, St John LC, Schutz AL. Surgical treatment of femoroacetabular impingement: a systematic review of the literature. Clin Orthop. 2010;468(2):555-564.
6. Beaulé PE, Zaragoza E, Motamedi K, Copelan N, Dorey FJ. Three-dimensional computed tomography of the hip in the assessment of femoroacetabular impingement. J Orthop Res. 2005;23(6):1286-1292.
7. Hetsroni I, Dela Torre K, Duke G, Lyman S, Kelly BT. Sex differences of hip morphology in young adults with hip pain and labral tears. Arthroscopy. 2013;29(1):54-63.
8. Klingenstein GG, Zbeda RM, Bedi A, Magennis E, Kelly BT. Prevalence and preoperative demographic and radiographic predictors of bilateral femoroacetabular impingement. Am J Sports Med. 2013;41(4):762-768.
9. Malviya A, Stafford GH, Villar RN. Impact of arthroscopy of the hip for femoroacetabular impingement on quality of life at a mean follow-up of 3.2 years. J Bone Joint Surg Br. 2012;94(4):466-470.
10. Ng VY, Arora N, Best TM, Pan X, Ellis TJ. Efficacy of surgery for femoroacetabular impingement: a systematic review. Am J Sports Med. 2010;38(11):2337-2345.
11. Matsuda DK, Carlisle JC, Arthurs SC, Wierks CH, Philippon MJ. Comparative systematic review of the open dislocation, mini-open, and arthroscopic surgeries for femoroacetabular impingement. Arthroscopy. 2011;27(2):252-269.
12. Clohisy JC, Baca G, Beaule PE, et al. Descriptive epidemiology of femoroacetabular impingement: a North American cohort of patients undergoing surgery. Am J Sports Med. 2013;41(6):1348-1356.
13. Krych AJ, Thompson M, Knutson Z, Scoon J, Coleman SH. Arthroscopic labral repair versus selective labral debridement in female patients with femoroacetabular impingement: a prospective randomized study. Arthroscopy. 2013;29(1):46-53.
ACP: Avoid ECG, MPI cardiac screening in low-risk patients
Clinicians should not screen for cardiac disease in asymptomatic, low-risk adults using resting or stress electrocardiography, stress echocardiography, or stress myocardial perfusion imaging , according to new guidelines from the American College of Physicians.
“There is no evidence that cardiac screening of low-risk adults with resting or stress ECG, stress echocardiography, or stress MPI improves outcomes, but it is associated with increased costs and potential harms,” wrote the guideline’s author, Dr. Roger Chou, associate professor of medicine at Oregon Health & Science University, Portland.
The recommendation is based on a systematic literature review, recommendations from the U.S. Preventive Services Task Force, and American College of Cardiology guidelines. The new ACP clinical guideline was published March 17 in Annals of Internal Medicine (doi: 10.7326/M14-1225).
“What we are saying here is that, as physicians, we have responsibility to understand what the pretest probability is, and what the likelihood is that someone actually has disease – and if it’s low enough, then doing the screening test is going to cause a lot more false positives than true positives,” Dr. Robert Centor, regional dean of the Huntsville Medical Campus of the University of Alabama at Birmingham, explained in an interview.
“Even if it is a true positive, there is no evidence that we can find that finding that heart disease will do anything other than lead someone to do a procedure that we have no evidence will improve their outcomes,” added Dr. Centor, chair of the ACP Board of Regents.
Despite existing recommendations to the contrary, physicians are increasingly performing these tests on low-risk patients, the ACP cautioned.
For example, a Consumer Reports survey found that “39% of asymptomatic adults without high blood pressure or a high cholesterol level reported having ECG within the past 5 years, and 12% reported undergoing exercise ECG,” Dr. Chou wrote in his report on behalf of the ACP High Value Care Task Force. More than half of those patients said their physicians recommended the tests as part of their routine health care.

The rise in the use of such tests is likely the result of a combination of factors, Dr. Centor said. Those factors include money (patients see no out-of-pocket cost and thus don’t consider the cost of tests in their decision making), direct-to-consumer advertising, fear on behalf of physicians that they might miss a diagnosis, and a lack of understanding by patients on the adverse effects of screening if they are at low risk for heart disease.
Dr. Chou identified a number of potential harms related to unnecessary screenings, including sudden death or hospitalization during stress tests; adverse events from pharmacologics used to induce stress; radiation exposure from myocardial perfusion imaging; false positive results that, in turn, lead to anxiety by the patient and additional unnecessary tests and treatments; disease labeling; and downstream harms from follow-up testing and interventions.
“To be most effective, efforts to reduce the use of imaging should be multifocal and should address clinician behavior, patient expectations, direct-to-consumer screening programs, and financial incentives,” Dr. Chou explained.
In low-risk patients, physicians instead should “focus on treating modifiable risk factors (such as smoking, diabetes, hypertension, hyperlipidemia, and overweight) and encouraging healthy levels of exercise,” according to the guideline.
Clinicians should not screen for cardiac disease in asymptomatic, low-risk adults using resting or stress electrocardiography, stress echocardiography, or stress myocardial perfusion imaging , according to new guidelines from the American College of Physicians.
“There is no evidence that cardiac screening of low-risk adults with resting or stress ECG, stress echocardiography, or stress MPI improves outcomes, but it is associated with increased costs and potential harms,” wrote the guideline’s author, Dr. Roger Chou, associate professor of medicine at Oregon Health & Science University, Portland.
The recommendation is based on a systematic literature review, recommendations from the U.S. Preventive Services Task Force, and American College of Cardiology guidelines. The new ACP clinical guideline was published March 17 in Annals of Internal Medicine (doi: 10.7326/M14-1225).
“What we are saying here is that, as physicians, we have responsibility to understand what the pretest probability is, and what the likelihood is that someone actually has disease – and if it’s low enough, then doing the screening test is going to cause a lot more false positives than true positives,” Dr. Robert Centor, regional dean of the Huntsville Medical Campus of the University of Alabama at Birmingham, explained in an interview.
“Even if it is a true positive, there is no evidence that we can find that finding that heart disease will do anything other than lead someone to do a procedure that we have no evidence will improve their outcomes,” added Dr. Centor, chair of the ACP Board of Regents.
Despite existing recommendations to the contrary, physicians are increasingly performing these tests on low-risk patients, the ACP cautioned.
For example, a Consumer Reports survey found that “39% of asymptomatic adults without high blood pressure or a high cholesterol level reported having ECG within the past 5 years, and 12% reported undergoing exercise ECG,” Dr. Chou wrote in his report on behalf of the ACP High Value Care Task Force. More than half of those patients said their physicians recommended the tests as part of their routine health care.
The rise in the use of such tests is likely the result of a combination of factors, Dr. Centor said. Those factors include money (patients see no out-of-pocket cost and thus don’t consider the cost of tests in their decision making), direct-to-consumer advertising, fear on behalf of physicians that they might miss a diagnosis, and a lack of understanding by patients on the adverse effects of screening if they are at low risk for heart disease.
Dr. Chou identified a number of potential harms related to unnecessary screenings, including sudden death or hospitalization during stress tests; adverse events from pharmacologics used to induce stress; radiation exposure from myocardial perfusion imaging; false positive results that, in turn, lead to anxiety by the patient and additional unnecessary tests and treatments; disease labeling; and downstream harms from follow-up testing and interventions.
“To be most effective, efforts to reduce the use of imaging should be multifocal and should address clinician behavior, patient expectations, direct-to-consumer screening programs, and financial incentives,” Dr. Chou explained.
In low-risk patients, physicians instead should “focus on treating modifiable risk factors (such as smoking, diabetes, hypertension, hyperlipidemia, and overweight) and encouraging healthy levels of exercise,” according to the guideline.
Clinicians should not screen for cardiac disease in asymptomatic, low-risk adults using resting or stress electrocardiography, stress echocardiography, or stress myocardial perfusion imaging , according to new guidelines from the American College of Physicians.
“There is no evidence that cardiac screening of low-risk adults with resting or stress ECG, stress echocardiography, or stress MPI improves outcomes, but it is associated with increased costs and potential harms,” wrote the guideline’s author, Dr. Roger Chou, associate professor of medicine at Oregon Health & Science University, Portland.
The recommendation is based on a systematic literature review, recommendations from the U.S. Preventive Services Task Force, and American College of Cardiology guidelines. The new ACP clinical guideline was published March 17 in Annals of Internal Medicine (doi: 10.7326/M14-1225).
“What we are saying here is that, as physicians, we have responsibility to understand what the pretest probability is, and what the likelihood is that someone actually has disease – and if it’s low enough, then doing the screening test is going to cause a lot more false positives than true positives,” Dr. Robert Centor, regional dean of the Huntsville Medical Campus of the University of Alabama at Birmingham, explained in an interview.
“Even if it is a true positive, there is no evidence that we can find that finding that heart disease will do anything other than lead someone to do a procedure that we have no evidence will improve their outcomes,” added Dr. Centor, chair of the ACP Board of Regents.
Despite existing recommendations to the contrary, physicians are increasingly performing these tests on low-risk patients, the ACP cautioned.
For example, a Consumer Reports survey found that “39% of asymptomatic adults without high blood pressure or a high cholesterol level reported having ECG within the past 5 years, and 12% reported undergoing exercise ECG,” Dr. Chou wrote in his report on behalf of the ACP High Value Care Task Force. More than half of those patients said their physicians recommended the tests as part of their routine health care.
The rise in the use of such tests is likely the result of a combination of factors, Dr. Centor said. Those factors include money (patients see no out-of-pocket cost and thus don’t consider the cost of tests in their decision making), direct-to-consumer advertising, fear on behalf of physicians that they might miss a diagnosis, and a lack of understanding by patients on the adverse effects of screening if they are at low risk for heart disease.
Dr. Chou identified a number of potential harms related to unnecessary screenings, including sudden death or hospitalization during stress tests; adverse events from pharmacologics used to induce stress; radiation exposure from myocardial perfusion imaging; false positive results that, in turn, lead to anxiety by the patient and additional unnecessary tests and treatments; disease labeling; and downstream harms from follow-up testing and interventions.
“To be most effective, efforts to reduce the use of imaging should be multifocal and should address clinician behavior, patient expectations, direct-to-consumer screening programs, and financial incentives,” Dr. Chou explained.
In low-risk patients, physicians instead should “focus on treating modifiable risk factors (such as smoking, diabetes, hypertension, hyperlipidemia, and overweight) and encouraging healthy levels of exercise,” according to the guideline.
FROM ANNALS OF INTERNAL MEDICINE
VIDEO: Did the PROMISE trial keep its promise?
SAN DIEGO – Patients with new-onset, stable chest pain account for millions of stress tests annually in the United States, but randomized data are limited on which test is best and the impact of testing on clinical outcomes.
Results from the prospective PROMISE trial, presented at the annual meeting of the American College of Cardiology, show there is no Holy Grail testing strategy. First-line testing with CT angiography did not reduce hard clinical events compared with functional testing, but did cut the number of patients undergoing an invasive catheterization showing no obstructive coronary artery disease.
Listen here for our interview with ACC president Dr. Patrick O’Gara on how these results will impact patient care and potentially influence current guideline recommendations.
Dr. O’Gara reported no relevant financial conflicts.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Patients with new-onset, stable chest pain account for millions of stress tests annually in the United States, but randomized data are limited on which test is best and the impact of testing on clinical outcomes.
Results from the prospective PROMISE trial, presented at the annual meeting of the American College of Cardiology, show there is no Holy Grail testing strategy. First-line testing with CT angiography did not reduce hard clinical events compared with functional testing, but did cut the number of patients undergoing an invasive catheterization showing no obstructive coronary artery disease.
Listen here for our interview with ACC president Dr. Patrick O’Gara on how these results will impact patient care and potentially influence current guideline recommendations.
Dr. O’Gara reported no relevant financial conflicts.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Patients with new-onset, stable chest pain account for millions of stress tests annually in the United States, but randomized data are limited on which test is best and the impact of testing on clinical outcomes.
Results from the prospective PROMISE trial, presented at the annual meeting of the American College of Cardiology, show there is no Holy Grail testing strategy. First-line testing with CT angiography did not reduce hard clinical events compared with functional testing, but did cut the number of patients undergoing an invasive catheterization showing no obstructive coronary artery disease.
Listen here for our interview with ACC president Dr. Patrick O’Gara on how these results will impact patient care and potentially influence current guideline recommendations.
Dr. O’Gara reported no relevant financial conflicts.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ACC 15

SCOT-HEART: CT angiography scores big in stable chest pain
SAN DIEGO – The addition of CT angiography to standard care changed the diagnosis and treatment of one in four patients with stable chest pain in the prospective, randomized SCOT-HEART trial.
CT angiography (CTA) also reduced coronary heart disease deaths or myocardial infarctions by 38% after a median follow-up of 1.7 years, although the finding was of borderline significance (hazard ratio, 0.62; P = .053).
A post-hoc, landmark analysis, however, that accounted for the roughly 6-week delay between the clinic visit and implementation or alteration of therapy based on CTA findings, showed a halving of these outcomes (HR, 0.50; P = .015), chief investigator Dr. David Newby reported at the annual meeting of the American College of Cardiology.
SCOT-HEART (Scottish COmputed Tomography of the Heart trial) involved 4,146 patients referred from chest pain clinics across Scotland for assessment of suspected angina due to coronary artery disease, of whom 47% were diagnosed in the clinic with coronary heart disease and 36% with angina due to coronary heart disease. Patients were then evenly randomized to standard care involving cardiovascular risk assessment with the ASSIGN Score alone or with CTA.
When attending clinicians reviewed the cases at 6 weeks, the diagnosis of coronary heart disease (CHD) changed in 25% of patients assigned CTA vs. only 1% assigned standard care alone and the diagnosis of angina due to CHD changed in 23% vs. 1% (P value < .001 for both), Dr. Newby, from the University of Edinburgh, said.
Clinicians reported that CTA significantly increased the certainty (Relative risk, 2.56; P < .0001) and frequency (RR, 1.09; P = .017) of the diagnosis of CHD and increased the certainty of a diagnosis of angina due to CHD (RR, 1.79; P < .0001), but had no effect on its frequency (RR, 0.93; P = .12).
Overall, 63% of patients had evidence of CHD on CTA, with 25% having obstructive disease.
CTA altered subsequent testing in 15% of patients vs. only 1% with standard care (P < .001). CTA use was associated with the cancellation of 121 functional stress tests and 29 invasive coronary angiography exams. CTA also prompted 94 new angiograms vs. just 8 with standard care, but this was mainly the result of the exclusion or discovery of obstructive coronary heart disease, including triple vessel disease, Dr. Newby observed. CT was associated with a nonsignificant increase in coronary revascularizations (11.2% vs. 9.7%; HR, 1.19; P .06).
The changes in diagnosis and testing were associated with changes in subsequent treatment in 23% of CTA patients vs. 5% of standard care patients overall (P < .001), including recommendations and cancellations for preventive and anti-anginal therapies. The results were also simultaneously published online (Lancet 2015 [doi:10.19016/S0140-6736(15)060291-4]).
The most impressive aspect of SCOT-HEART was the strong trend for improved outcomes in patients for which therapeutic alterations were made, Dr. Eric Peterson, with the Duke University in Durham, N.C., commented.
“This sets the standard for how we perform and evaluate whether CT can improve outcomes for patients,” he said.
In an editorial accompanying the report,, Dr. Pamela Douglas, from the Duke Clinical Research Institute in Durham, N.C., called the finding of reduced death and MI “intriguing,” but urged caution in its interpretation because it was one of 22 secondary end points and the absolute difference between groups was only 16 events (Lancet 2015; [doi: 10.1016/S0140-6736(15)60463-9]). Earlier in the meeting, she reported that the PROMISE trial found no difference in its primary composite end point of all-cause death, nonfatal myocardial infarction, unstable angina hospitalization, and major cardiovascular procedural complications among chest pain patients evaluated with CTA or functional testing.
Finally, it was noted that radiation exposure in SCOT-HEART (median 4.1 mSv) was substantially lower than that reported in PROMISE, a finding Dr. Newby said he could not explain.
SAN DIEGO – The addition of CT angiography to standard care changed the diagnosis and treatment of one in four patients with stable chest pain in the prospective, randomized SCOT-HEART trial.
CT angiography (CTA) also reduced coronary heart disease deaths or myocardial infarctions by 38% after a median follow-up of 1.7 years, although the finding was of borderline significance (hazard ratio, 0.62; P = .053).
A post-hoc, landmark analysis, however, that accounted for the roughly 6-week delay between the clinic visit and implementation or alteration of therapy based on CTA findings, showed a halving of these outcomes (HR, 0.50; P = .015), chief investigator Dr. David Newby reported at the annual meeting of the American College of Cardiology.
SCOT-HEART (Scottish COmputed Tomography of the Heart trial) involved 4,146 patients referred from chest pain clinics across Scotland for assessment of suspected angina due to coronary artery disease, of whom 47% were diagnosed in the clinic with coronary heart disease and 36% with angina due to coronary heart disease. Patients were then evenly randomized to standard care involving cardiovascular risk assessment with the ASSIGN Score alone or with CTA.
When attending clinicians reviewed the cases at 6 weeks, the diagnosis of coronary heart disease (CHD) changed in 25% of patients assigned CTA vs. only 1% assigned standard care alone and the diagnosis of angina due to CHD changed in 23% vs. 1% (P value < .001 for both), Dr. Newby, from the University of Edinburgh, said.
Clinicians reported that CTA significantly increased the certainty (Relative risk, 2.56; P < .0001) and frequency (RR, 1.09; P = .017) of the diagnosis of CHD and increased the certainty of a diagnosis of angina due to CHD (RR, 1.79; P < .0001), but had no effect on its frequency (RR, 0.93; P = .12).
Overall, 63% of patients had evidence of CHD on CTA, with 25% having obstructive disease.
CTA altered subsequent testing in 15% of patients vs. only 1% with standard care (P < .001). CTA use was associated with the cancellation of 121 functional stress tests and 29 invasive coronary angiography exams. CTA also prompted 94 new angiograms vs. just 8 with standard care, but this was mainly the result of the exclusion or discovery of obstructive coronary heart disease, including triple vessel disease, Dr. Newby observed. CT was associated with a nonsignificant increase in coronary revascularizations (11.2% vs. 9.7%; HR, 1.19; P .06).
The changes in diagnosis and testing were associated with changes in subsequent treatment in 23% of CTA patients vs. 5% of standard care patients overall (P < .001), including recommendations and cancellations for preventive and anti-anginal therapies. The results were also simultaneously published online (Lancet 2015 [doi:10.19016/S0140-6736(15)060291-4]).
The most impressive aspect of SCOT-HEART was the strong trend for improved outcomes in patients for which therapeutic alterations were made, Dr. Eric Peterson, with the Duke University in Durham, N.C., commented.
“This sets the standard for how we perform and evaluate whether CT can improve outcomes for patients,” he said.
In an editorial accompanying the report,, Dr. Pamela Douglas, from the Duke Clinical Research Institute in Durham, N.C., called the finding of reduced death and MI “intriguing,” but urged caution in its interpretation because it was one of 22 secondary end points and the absolute difference between groups was only 16 events (Lancet 2015; [doi: 10.1016/S0140-6736(15)60463-9]). Earlier in the meeting, she reported that the PROMISE trial found no difference in its primary composite end point of all-cause death, nonfatal myocardial infarction, unstable angina hospitalization, and major cardiovascular procedural complications among chest pain patients evaluated with CTA or functional testing.
Finally, it was noted that radiation exposure in SCOT-HEART (median 4.1 mSv) was substantially lower than that reported in PROMISE, a finding Dr. Newby said he could not explain.
SAN DIEGO – The addition of CT angiography to standard care changed the diagnosis and treatment of one in four patients with stable chest pain in the prospective, randomized SCOT-HEART trial.
CT angiography (CTA) also reduced coronary heart disease deaths or myocardial infarctions by 38% after a median follow-up of 1.7 years, although the finding was of borderline significance (hazard ratio, 0.62; P = .053).
A post-hoc, landmark analysis, however, that accounted for the roughly 6-week delay between the clinic visit and implementation or alteration of therapy based on CTA findings, showed a halving of these outcomes (HR, 0.50; P = .015), chief investigator Dr. David Newby reported at the annual meeting of the American College of Cardiology.
SCOT-HEART (Scottish COmputed Tomography of the Heart trial) involved 4,146 patients referred from chest pain clinics across Scotland for assessment of suspected angina due to coronary artery disease, of whom 47% were diagnosed in the clinic with coronary heart disease and 36% with angina due to coronary heart disease. Patients were then evenly randomized to standard care involving cardiovascular risk assessment with the ASSIGN Score alone or with CTA.
When attending clinicians reviewed the cases at 6 weeks, the diagnosis of coronary heart disease (CHD) changed in 25% of patients assigned CTA vs. only 1% assigned standard care alone and the diagnosis of angina due to CHD changed in 23% vs. 1% (P value < .001 for both), Dr. Newby, from the University of Edinburgh, said.
Clinicians reported that CTA significantly increased the certainty (Relative risk, 2.56; P < .0001) and frequency (RR, 1.09; P = .017) of the diagnosis of CHD and increased the certainty of a diagnosis of angina due to CHD (RR, 1.79; P < .0001), but had no effect on its frequency (RR, 0.93; P = .12).
Overall, 63% of patients had evidence of CHD on CTA, with 25% having obstructive disease.
CTA altered subsequent testing in 15% of patients vs. only 1% with standard care (P < .001). CTA use was associated with the cancellation of 121 functional stress tests and 29 invasive coronary angiography exams. CTA also prompted 94 new angiograms vs. just 8 with standard care, but this was mainly the result of the exclusion or discovery of obstructive coronary heart disease, including triple vessel disease, Dr. Newby observed. CT was associated with a nonsignificant increase in coronary revascularizations (11.2% vs. 9.7%; HR, 1.19; P .06).
The changes in diagnosis and testing were associated with changes in subsequent treatment in 23% of CTA patients vs. 5% of standard care patients overall (P < .001), including recommendations and cancellations for preventive and anti-anginal therapies. The results were also simultaneously published online (Lancet 2015 [doi:10.19016/S0140-6736(15)060291-4]).
The most impressive aspect of SCOT-HEART was the strong trend for improved outcomes in patients for which therapeutic alterations were made, Dr. Eric Peterson, with the Duke University in Durham, N.C., commented.
“This sets the standard for how we perform and evaluate whether CT can improve outcomes for patients,” he said.
In an editorial accompanying the report,, Dr. Pamela Douglas, from the Duke Clinical Research Institute in Durham, N.C., called the finding of reduced death and MI “intriguing,” but urged caution in its interpretation because it was one of 22 secondary end points and the absolute difference between groups was only 16 events (Lancet 2015; [doi: 10.1016/S0140-6736(15)60463-9]). Earlier in the meeting, she reported that the PROMISE trial found no difference in its primary composite end point of all-cause death, nonfatal myocardial infarction, unstable angina hospitalization, and major cardiovascular procedural complications among chest pain patients evaluated with CTA or functional testing.
Finally, it was noted that radiation exposure in SCOT-HEART (median 4.1 mSv) was substantially lower than that reported in PROMISE, a finding Dr. Newby said he could not explain.
AT ACC 2015
Key clinical point:CTA clarifies the diagnosis and leads to major alterations in testing and treatments in patients with suspected angina due to coronary heart disease.
Major finding:The addition of CT angiography changed the diagnosis and treatment of one in four patients with stable chest pain.
Data source: SCOT-HEART, a prospective, randomized study in 4,146 patients with new-onset, stable chest pain.
Disclosures: The study was funded by the Chief Scientist Office of the Scottish Government Health and Social Care Directorates, with supplementary awards from Edinburgh and Lothian’s Health Foundation Trust and the Heart Diseases Research Fund. Dr. Newby reported consultant fees and honoraria from Eli-Lilly, Roche, Toshiba, Pfizer, AstraZeneca, MSD, BMS, Boeringer Ingelheim, GlaxoSmithKline.
CT scans comparable to functional testing for CAD
SAN DIEGO – Initial anatomic testing with CT angiography yielded similar clinical outcomes to functional testing in chest pain patients evaluated for coronary artery disease in the PROMISE study.
After an average of 25 months, the primary composite end point of all-cause death, nonfatal myocardial infarction, unstable angina hospitalization, and major cardiovascular procedural complications occurred in 3.3% of the CTA patients and 3.0% of the functional-testing patients (adjusted hazard ratio, 1.04; P = .75).
The CTA strategy may offer a slight advantage, however, in terms of fewer invasive catheterizations without evidence of obstructive coronary artery disease (3.4% vs. 4.3%; P = .022) and a higher proportion of catheterizations with obstructive CAD (72.1% vs. 47.5%) getting revascularization (6.2% vs. 3.2%) or coronary artery bypass grafting (72 events vs. 38 events).
“Our results suggest that CTA is a viable alternative to functional testing. These real-world results should inform noninvasive testing choices in clinical care as well as provide guidance to future studies of diagnostic strategies in suspected heart disease,” lead study author Dr. Pamela Douglas reported at the annual meeting of the American College of Cardiology and simultaneously published online (N. Engl. J. Med. 2015 [DOI:10.1056/NEJoa1415516].

PROMISE (Prospective Multicenter Imaging Study for the Evaluation of Chest Pain) enrolled 10,003 symptomatic outpatients requiring nonemergent, noninvasive testing for suspected CAD who were older than 54 years for men or older than 64 years for women with no risk factors, or aged 45-54 years for men and 50-64 years for women with at least one cardiac risk factor.
Patients were randomly assigned to anatomical testing with CTA or functional testing including a nuclear stress test (67%), stress echocardiography (23%), or exercise electrocardiogram (10%). The patients had an average of 2.5 risk cardiovascular risk factors and half were women.
Radiation exposure was higher overall in the CTA group than the functional-testing group (mean 12.0 mSv vs. 10.1 mSv; P < .001), largely because 33% of the functional group had no exposure. Exposure was lower, however, in CTA patients compared with those for whom a nuclear test was specified at randomization as their first intended functional test (12.0 mSv vs. 14.1 mSv; P < .001), Dr. Douglas, from Duke Clinical Research Institute in Durham, N.C., said.
During a discussion of the results, Dr. Elliott Antman, associate dean for clinical and translational research at Harvard University, Boston, said CT angiography can’t officially be described as noninferior to functional testing because PROMISE was designed as a superiority trial with a noninferiority margin that was exceeded by the confidence intervals for the primary end point and questioned how clinicians should use the results.
“What I can say to the next patient is that they can be incredibly assured about their overall prognosis, which is a nontrivial thing to say that they have a very, very low likelihood of a bad event in the next 2 years no matter what we do,” Dr. Douglas responded. “I can offer a test choice that will have no difference in major health events like death or nonfatal MI, but I can offer a test that potentially has lower radiation and has a better triage function to the cath lab where you have less likelihood of ending up in the cath lab not needing to be there because you do not have obstructive disease.”
Though PROMISE may influence practice, it is unclear whether the noninferiority issue will impact its ability to change U.S. guidelines, which currently include a IIb recommendation that CTA be considered in the evaluation of patients with chest pain.
“Technically two randomized controlled trials are needed before you get evidence level A, but we have 10,000 patients who were well studied here, so I would anticipate a big change actually in the guidelines from a use criteria, but we shall see,” Dr. Douglas said.
SAN DIEGO – Initial anatomic testing with CT angiography yielded similar clinical outcomes to functional testing in chest pain patients evaluated for coronary artery disease in the PROMISE study.
After an average of 25 months, the primary composite end point of all-cause death, nonfatal myocardial infarction, unstable angina hospitalization, and major cardiovascular procedural complications occurred in 3.3% of the CTA patients and 3.0% of the functional-testing patients (adjusted hazard ratio, 1.04; P = .75).
The CTA strategy may offer a slight advantage, however, in terms of fewer invasive catheterizations without evidence of obstructive coronary artery disease (3.4% vs. 4.3%; P = .022) and a higher proportion of catheterizations with obstructive CAD (72.1% vs. 47.5%) getting revascularization (6.2% vs. 3.2%) or coronary artery bypass grafting (72 events vs. 38 events).
“Our results suggest that CTA is a viable alternative to functional testing. These real-world results should inform noninvasive testing choices in clinical care as well as provide guidance to future studies of diagnostic strategies in suspected heart disease,” lead study author Dr. Pamela Douglas reported at the annual meeting of the American College of Cardiology and simultaneously published online (N. Engl. J. Med. 2015 [DOI:10.1056/NEJoa1415516].
PROMISE (Prospective Multicenter Imaging Study for the Evaluation of Chest Pain) enrolled 10,003 symptomatic outpatients requiring nonemergent, noninvasive testing for suspected CAD who were older than 54 years for men or older than 64 years for women with no risk factors, or aged 45-54 years for men and 50-64 years for women with at least one cardiac risk factor.
Patients were randomly assigned to anatomical testing with CTA or functional testing including a nuclear stress test (67%), stress echocardiography (23%), or exercise electrocardiogram (10%). The patients had an average of 2.5 risk cardiovascular risk factors and half were women.
Radiation exposure was higher overall in the CTA group than the functional-testing group (mean 12.0 mSv vs. 10.1 mSv; P < .001), largely because 33% of the functional group had no exposure. Exposure was lower, however, in CTA patients compared with those for whom a nuclear test was specified at randomization as their first intended functional test (12.0 mSv vs. 14.1 mSv; P < .001), Dr. Douglas, from Duke Clinical Research Institute in Durham, N.C., said.
During a discussion of the results, Dr. Elliott Antman, associate dean for clinical and translational research at Harvard University, Boston, said CT angiography can’t officially be described as noninferior to functional testing because PROMISE was designed as a superiority trial with a noninferiority margin that was exceeded by the confidence intervals for the primary end point and questioned how clinicians should use the results.
“What I can say to the next patient is that they can be incredibly assured about their overall prognosis, which is a nontrivial thing to say that they have a very, very low likelihood of a bad event in the next 2 years no matter what we do,” Dr. Douglas responded. “I can offer a test choice that will have no difference in major health events like death or nonfatal MI, but I can offer a test that potentially has lower radiation and has a better triage function to the cath lab where you have less likelihood of ending up in the cath lab not needing to be there because you do not have obstructive disease.”
Though PROMISE may influence practice, it is unclear whether the noninferiority issue will impact its ability to change U.S. guidelines, which currently include a IIb recommendation that CTA be considered in the evaluation of patients with chest pain.
“Technically two randomized controlled trials are needed before you get evidence level A, but we have 10,000 patients who were well studied here, so I would anticipate a big change actually in the guidelines from a use criteria, but we shall see,” Dr. Douglas said.
SAN DIEGO – Initial anatomic testing with CT angiography yielded similar clinical outcomes to functional testing in chest pain patients evaluated for coronary artery disease in the PROMISE study.
After an average of 25 months, the primary composite end point of all-cause death, nonfatal myocardial infarction, unstable angina hospitalization, and major cardiovascular procedural complications occurred in 3.3% of the CTA patients and 3.0% of the functional-testing patients (adjusted hazard ratio, 1.04; P = .75).
The CTA strategy may offer a slight advantage, however, in terms of fewer invasive catheterizations without evidence of obstructive coronary artery disease (3.4% vs. 4.3%; P = .022) and a higher proportion of catheterizations with obstructive CAD (72.1% vs. 47.5%) getting revascularization (6.2% vs. 3.2%) or coronary artery bypass grafting (72 events vs. 38 events).
“Our results suggest that CTA is a viable alternative to functional testing. These real-world results should inform noninvasive testing choices in clinical care as well as provide guidance to future studies of diagnostic strategies in suspected heart disease,” lead study author Dr. Pamela Douglas reported at the annual meeting of the American College of Cardiology and simultaneously published online (N. Engl. J. Med. 2015 [DOI:10.1056/NEJoa1415516].
PROMISE (Prospective Multicenter Imaging Study for the Evaluation of Chest Pain) enrolled 10,003 symptomatic outpatients requiring nonemergent, noninvasive testing for suspected CAD who were older than 54 years for men or older than 64 years for women with no risk factors, or aged 45-54 years for men and 50-64 years for women with at least one cardiac risk factor.
Patients were randomly assigned to anatomical testing with CTA or functional testing including a nuclear stress test (67%), stress echocardiography (23%), or exercise electrocardiogram (10%). The patients had an average of 2.5 risk cardiovascular risk factors and half were women.
Radiation exposure was higher overall in the CTA group than the functional-testing group (mean 12.0 mSv vs. 10.1 mSv; P < .001), largely because 33% of the functional group had no exposure. Exposure was lower, however, in CTA patients compared with those for whom a nuclear test was specified at randomization as their first intended functional test (12.0 mSv vs. 14.1 mSv; P < .001), Dr. Douglas, from Duke Clinical Research Institute in Durham, N.C., said.
During a discussion of the results, Dr. Elliott Antman, associate dean for clinical and translational research at Harvard University, Boston, said CT angiography can’t officially be described as noninferior to functional testing because PROMISE was designed as a superiority trial with a noninferiority margin that was exceeded by the confidence intervals for the primary end point and questioned how clinicians should use the results.
“What I can say to the next patient is that they can be incredibly assured about their overall prognosis, which is a nontrivial thing to say that they have a very, very low likelihood of a bad event in the next 2 years no matter what we do,” Dr. Douglas responded. “I can offer a test choice that will have no difference in major health events like death or nonfatal MI, but I can offer a test that potentially has lower radiation and has a better triage function to the cath lab where you have less likelihood of ending up in the cath lab not needing to be there because you do not have obstructive disease.”
Though PROMISE may influence practice, it is unclear whether the noninferiority issue will impact its ability to change U.S. guidelines, which currently include a IIb recommendation that CTA be considered in the evaluation of patients with chest pain.
“Technically two randomized controlled trials are needed before you get evidence level A, but we have 10,000 patients who were well studied here, so I would anticipate a big change actually in the guidelines from a use criteria, but we shall see,” Dr. Douglas said.
AT ACC 2015
Key clinical point: Clinical outcomes are comparable with CT angiography and functional testing for suspected coronary artery disease in symptomatic patients.
Major finding: The primary end point occurred in 3.3% of the CTA group and 3.0% of the functional testing group (HR, 1.04; P = .75).
Data source: Prospective, randomized study in 10,003 symptomatic patients with suspected coronary artery disease.
Disclosures: The study was funded by the National Heart, Lung, and Blood Institute. Dr. Douglas reported numerous conflicts. Dr. Antman reported no financial disclosures.

More coronary artery calcification seen with sedentary behavior
Sedentary individuals have a significant increase in a marker of subclinical heart disease, according to a recent study. Each additional hour spent seated per day was associated with a 14% increase in the amount of coronary artery calcification seen on CT imaging.
Even for those who exercise vigorously but also spend many hours sitting, prolonged sedentary activity was independently associated with greater CAC. The effect held true even after adjustment for other known cardiac risk factors, according to Dr. Jacquelyn Kulinski of the Medical College of Wisconsin, Milwaukee, and colleagues at Dallas’ University of Texas Southwestern Medical Center. The results were released in advance of their presentation on March 15 at the annual meeting of the American College of Cardiology in San Diego.

Using data from wrist accelerometers in the Dallas Heart Study, a multi-ethnic, population-based sample of adults residing in Dallas, Tex., Dr. Kulinski and colleagues assessed the activity of 2,031 participants without known cardiovascular disease. The median age was 50, about 62% were female, and about half of the participants were black.
The accelerometer, worn on the nondominant hand, captured 7 consecutive days of minute-by-minute activity, and could differentiate between sedentary behavior (sitting or reclining during waking hours), nonexercise activity (such as light movement around the house or a workplace), and more vigorous, purposeful exercise. CAC was assessed via two cardiac CT scans and a standardized scoring system.
Overall, participants had an average 5 hours of sedentary time per day, with a range of 2-12 hours. Higher amounts of sedentary time were associated with being older, having a higher body mass index, and having diabetes or hypertension.
The multivariate analysis included adjustments for patient demographics, clinical characteristics, and the amount to which participants participated in moderate to vigorous physical activity. The researchers found that each additional hour of sedentary time during the day was associated with a statistically significant, 14% increase in CAC. Somewhat surprisingly, Dr. Kulinski noted, there was no association between the amount of moderate to vigorous physical activity and the amount of CAC detected.
Sedentary behavior, sometimes called “sitting disease,” has previously been identified as an independent risk factor for heart disease. These findings, she said, further bolster the concept that “lack of exercise and too much sitting are independent risk factors for CAD and death.”
Dr. Kulinski noted that although fitness is one of the strongest predictors of cardiac health and longevity, there has not been a clear demonstration that increased amounts of exercise are associated with less CAC. This has been true although CAC is an established marker of early formation of the plaque that can lead to coronary artery blockage. The present study suggests that sedentary behaviors, rather than lack of vigorous exercise, may contribute more to the development of CAC than had previously been known.
ACC Vice President Richard Chazal of Lee Memorial Health System, Fort Myers, Fla., remarked in a press briefing before the meeting that this information should be reassuring to patients. “Sometimes just moving around some, even short of a vigorous exercise program, is important,” said Dr. Chazal. “Regular exercise further reduces risk, and it may do so by a mechanism distinct from coronary artery calcification.”
“On a positive note,” Dr. Kulinski said, “reducing daily sitting time by even 1-2 hours could have a significant and positive impact on future cardiovascular health, and this should really be investigated in future studies.”
Dr. James de Lemos disclosed ties with Amgen, DiaDexus, Novo Nordisk, St. Jude Medical, Abbott Diagnostics, and Siemen’s Diagnostics. Dr. Jarrett Berry reports ties with Nihon and Merck. The remaining authors have no disclosures.
Sedentary individuals have a significant increase in a marker of subclinical heart disease, according to a recent study. Each additional hour spent seated per day was associated with a 14% increase in the amount of coronary artery calcification seen on CT imaging.
Even for those who exercise vigorously but also spend many hours sitting, prolonged sedentary activity was independently associated with greater CAC. The effect held true even after adjustment for other known cardiac risk factors, according to Dr. Jacquelyn Kulinski of the Medical College of Wisconsin, Milwaukee, and colleagues at Dallas’ University of Texas Southwestern Medical Center. The results were released in advance of their presentation on March 15 at the annual meeting of the American College of Cardiology in San Diego.
Using data from wrist accelerometers in the Dallas Heart Study, a multi-ethnic, population-based sample of adults residing in Dallas, Tex., Dr. Kulinski and colleagues assessed the activity of 2,031 participants without known cardiovascular disease. The median age was 50, about 62% were female, and about half of the participants were black.
The accelerometer, worn on the nondominant hand, captured 7 consecutive days of minute-by-minute activity, and could differentiate between sedentary behavior (sitting or reclining during waking hours), nonexercise activity (such as light movement around the house or a workplace), and more vigorous, purposeful exercise. CAC was assessed via two cardiac CT scans and a standardized scoring system.
Overall, participants had an average 5 hours of sedentary time per day, with a range of 2-12 hours. Higher amounts of sedentary time were associated with being older, having a higher body mass index, and having diabetes or hypertension.
The multivariate analysis included adjustments for patient demographics, clinical characteristics, and the amount to which participants participated in moderate to vigorous physical activity. The researchers found that each additional hour of sedentary time during the day was associated with a statistically significant, 14% increase in CAC. Somewhat surprisingly, Dr. Kulinski noted, there was no association between the amount of moderate to vigorous physical activity and the amount of CAC detected.
Sedentary behavior, sometimes called “sitting disease,” has previously been identified as an independent risk factor for heart disease. These findings, she said, further bolster the concept that “lack of exercise and too much sitting are independent risk factors for CAD and death.”
Dr. Kulinski noted that although fitness is one of the strongest predictors of cardiac health and longevity, there has not been a clear demonstration that increased amounts of exercise are associated with less CAC. This has been true although CAC is an established marker of early formation of the plaque that can lead to coronary artery blockage. The present study suggests that sedentary behaviors, rather than lack of vigorous exercise, may contribute more to the development of CAC than had previously been known.
ACC Vice President Richard Chazal of Lee Memorial Health System, Fort Myers, Fla., remarked in a press briefing before the meeting that this information should be reassuring to patients. “Sometimes just moving around some, even short of a vigorous exercise program, is important,” said Dr. Chazal. “Regular exercise further reduces risk, and it may do so by a mechanism distinct from coronary artery calcification.”
“On a positive note,” Dr. Kulinski said, “reducing daily sitting time by even 1-2 hours could have a significant and positive impact on future cardiovascular health, and this should really be investigated in future studies.”
Dr. James de Lemos disclosed ties with Amgen, DiaDexus, Novo Nordisk, St. Jude Medical, Abbott Diagnostics, and Siemen’s Diagnostics. Dr. Jarrett Berry reports ties with Nihon and Merck. The remaining authors have no disclosures.
Sedentary individuals have a significant increase in a marker of subclinical heart disease, according to a recent study. Each additional hour spent seated per day was associated with a 14% increase in the amount of coronary artery calcification seen on CT imaging.
Even for those who exercise vigorously but also spend many hours sitting, prolonged sedentary activity was independently associated with greater CAC. The effect held true even after adjustment for other known cardiac risk factors, according to Dr. Jacquelyn Kulinski of the Medical College of Wisconsin, Milwaukee, and colleagues at Dallas’ University of Texas Southwestern Medical Center. The results were released in advance of their presentation on March 15 at the annual meeting of the American College of Cardiology in San Diego.
Using data from wrist accelerometers in the Dallas Heart Study, a multi-ethnic, population-based sample of adults residing in Dallas, Tex., Dr. Kulinski and colleagues assessed the activity of 2,031 participants without known cardiovascular disease. The median age was 50, about 62% were female, and about half of the participants were black.
The accelerometer, worn on the nondominant hand, captured 7 consecutive days of minute-by-minute activity, and could differentiate between sedentary behavior (sitting or reclining during waking hours), nonexercise activity (such as light movement around the house or a workplace), and more vigorous, purposeful exercise. CAC was assessed via two cardiac CT scans and a standardized scoring system.
Overall, participants had an average 5 hours of sedentary time per day, with a range of 2-12 hours. Higher amounts of sedentary time were associated with being older, having a higher body mass index, and having diabetes or hypertension.
The multivariate analysis included adjustments for patient demographics, clinical characteristics, and the amount to which participants participated in moderate to vigorous physical activity. The researchers found that each additional hour of sedentary time during the day was associated with a statistically significant, 14% increase in CAC. Somewhat surprisingly, Dr. Kulinski noted, there was no association between the amount of moderate to vigorous physical activity and the amount of CAC detected.
Sedentary behavior, sometimes called “sitting disease,” has previously been identified as an independent risk factor for heart disease. These findings, she said, further bolster the concept that “lack of exercise and too much sitting are independent risk factors for CAD and death.”
Dr. Kulinski noted that although fitness is one of the strongest predictors of cardiac health and longevity, there has not been a clear demonstration that increased amounts of exercise are associated with less CAC. This has been true although CAC is an established marker of early formation of the plaque that can lead to coronary artery blockage. The present study suggests that sedentary behaviors, rather than lack of vigorous exercise, may contribute more to the development of CAC than had previously been known.
ACC Vice President Richard Chazal of Lee Memorial Health System, Fort Myers, Fla., remarked in a press briefing before the meeting that this information should be reassuring to patients. “Sometimes just moving around some, even short of a vigorous exercise program, is important,” said Dr. Chazal. “Regular exercise further reduces risk, and it may do so by a mechanism distinct from coronary artery calcification.”
“On a positive note,” Dr. Kulinski said, “reducing daily sitting time by even 1-2 hours could have a significant and positive impact on future cardiovascular health, and this should really be investigated in future studies.”
Dr. James de Lemos disclosed ties with Amgen, DiaDexus, Novo Nordisk, St. Jude Medical, Abbott Diagnostics, and Siemen’s Diagnostics. Dr. Jarrett Berry reports ties with Nihon and Merck. The remaining authors have no disclosures.
FROM ACC 15
Key clinical point: Increased sedentary time was independently associated with increased coronary artery calcification.
Major findings: In a multivariate analysis, each increased hour of sitting time was associated with a 14% increase in CAC.
Data source: Examination via logistic regression and multivariate analysis of activity data from 2,031 participants in the multi-ethnic, population-based Dallas Heart Study.
Disclosures: Dr. James de Lemos disclosed ties with Amgen, DiaDexus, Novo Nordisk, St. Jude Medical, Abbott Diagnostics, and Siemen’s Diagnostics. Dr. Jarrett Berry reports ties with Nihon and Merck. The remaining authors have no disclosures.

Coffee drinking linked to lower subclinical atherosclerosis
Light to moderate coffee drinking – less than 5 cups per day – is associated with decreased coronary artery calcium, and thus decreased risk of cardiovascular disease, according to an analysis of a large cohort published online March 2 in Heart.
Coffee consumption’s effect on cardiovascular health has been controversial, even though the bulk of the substantial evidence collected to date suggests that it is cardioprotective. To clarify the association between coffee drinking and CVD, researchers performed a cross-sectional analysis of data from a large cohort study of asymptomatic young and middle-age South Korean adults attending a comprehensive health screening during a 3-year period. Members of the cohort (mean age 41 years) completed a detailed food-frequency questionnaire and underwent cardiac CT to measure coronary artery calcium, a marker of subclinical coronary atherosclerosis that predicts future heart disease, said Dr. Yuni Choi of Sungkyunkwan University, Seoul, South Korea, and her associates.

For this analysis, results for 25,138 participants who had no clinical evidence of CVD were assessed. The large sample size allowed for the data to be adjusted to account for numerous confounding factors such as medication use, personal and family medical history, physical activity level, alcohol consumption, smoking status, and sociodemographic factors.
Coffee consumption correlated with coronary artery calcium in a U-shaped pattern: Adults who drank up to 3 cups of coffee per day (light intake) had a decreased prevalence of subclinical coronary athersclerosis, those who drank 3-4 cups per day (moderate intake) had the lowest prevalence, and those who drank 5 or more cups per day had an increased prevalence, compared with people who didn’t drink coffee. Coronary artery calcium ratios that compared coffee drinkers with nondrinkers were 0.86 at less than 1 cup per day, 0.82 at 1-2 cups per day, 0.78 at 3-4 cups per day, and 1.77 at 5 or more cups per day. The association between coffee intake and coronary artery calcium scores remained consistent across all subgroup analyses and in sensitivity analyses, the investigators said (Heart 2015 March 2 [doi:10.1136/heartjnl-2014-306663]).
The cross-sectional design of this study means that it can establish only an association, not causality, between coffee intake and CVD risk. “Further research is needed to confirm our findings and establish the biological basis of coffee’s potential preventive effects on coronary artery disease,” Dr. Choi and her associates wrote.
But the evidence already is strong enough that for the first time this year, dietary guidelines will likely state that moderate coffee consumption appears to be cardioprotective and “can be incorporated into a healthy dietary pattern.” Coffee drinkers need only minimize the sugars and fats they add to their coffee, in the form of sweeteners and creamers, to benefit from the beverage, the 2015 Dietary Guidelines Advisory Committee recommended in a report submitted for review to the secretaries of the U.S. Department of Health & Human Services and the U.S. Department of Agriculture in February.
Light to moderate coffee drinking – less than 5 cups per day – is associated with decreased coronary artery calcium, and thus decreased risk of cardiovascular disease, according to an analysis of a large cohort published online March 2 in Heart.
Coffee consumption’s effect on cardiovascular health has been controversial, even though the bulk of the substantial evidence collected to date suggests that it is cardioprotective. To clarify the association between coffee drinking and CVD, researchers performed a cross-sectional analysis of data from a large cohort study of asymptomatic young and middle-age South Korean adults attending a comprehensive health screening during a 3-year period. Members of the cohort (mean age 41 years) completed a detailed food-frequency questionnaire and underwent cardiac CT to measure coronary artery calcium, a marker of subclinical coronary atherosclerosis that predicts future heart disease, said Dr. Yuni Choi of Sungkyunkwan University, Seoul, South Korea, and her associates.
For this analysis, results for 25,138 participants who had no clinical evidence of CVD were assessed. The large sample size allowed for the data to be adjusted to account for numerous confounding factors such as medication use, personal and family medical history, physical activity level, alcohol consumption, smoking status, and sociodemographic factors.
Coffee consumption correlated with coronary artery calcium in a U-shaped pattern: Adults who drank up to 3 cups of coffee per day (light intake) had a decreased prevalence of subclinical coronary athersclerosis, those who drank 3-4 cups per day (moderate intake) had the lowest prevalence, and those who drank 5 or more cups per day had an increased prevalence, compared with people who didn’t drink coffee. Coronary artery calcium ratios that compared coffee drinkers with nondrinkers were 0.86 at less than 1 cup per day, 0.82 at 1-2 cups per day, 0.78 at 3-4 cups per day, and 1.77 at 5 or more cups per day. The association between coffee intake and coronary artery calcium scores remained consistent across all subgroup analyses and in sensitivity analyses, the investigators said (Heart 2015 March 2 [doi:10.1136/heartjnl-2014-306663]).
The cross-sectional design of this study means that it can establish only an association, not causality, between coffee intake and CVD risk. “Further research is needed to confirm our findings and establish the biological basis of coffee’s potential preventive effects on coronary artery disease,” Dr. Choi and her associates wrote.
But the evidence already is strong enough that for the first time this year, dietary guidelines will likely state that moderate coffee consumption appears to be cardioprotective and “can be incorporated into a healthy dietary pattern.” Coffee drinkers need only minimize the sugars and fats they add to their coffee, in the form of sweeteners and creamers, to benefit from the beverage, the 2015 Dietary Guidelines Advisory Committee recommended in a report submitted for review to the secretaries of the U.S. Department of Health & Human Services and the U.S. Department of Agriculture in February.
Light to moderate coffee drinking – less than 5 cups per day – is associated with decreased coronary artery calcium, and thus decreased risk of cardiovascular disease, according to an analysis of a large cohort published online March 2 in Heart.
Coffee consumption’s effect on cardiovascular health has been controversial, even though the bulk of the substantial evidence collected to date suggests that it is cardioprotective. To clarify the association between coffee drinking and CVD, researchers performed a cross-sectional analysis of data from a large cohort study of asymptomatic young and middle-age South Korean adults attending a comprehensive health screening during a 3-year period. Members of the cohort (mean age 41 years) completed a detailed food-frequency questionnaire and underwent cardiac CT to measure coronary artery calcium, a marker of subclinical coronary atherosclerosis that predicts future heart disease, said Dr. Yuni Choi of Sungkyunkwan University, Seoul, South Korea, and her associates.
For this analysis, results for 25,138 participants who had no clinical evidence of CVD were assessed. The large sample size allowed for the data to be adjusted to account for numerous confounding factors such as medication use, personal and family medical history, physical activity level, alcohol consumption, smoking status, and sociodemographic factors.
Coffee consumption correlated with coronary artery calcium in a U-shaped pattern: Adults who drank up to 3 cups of coffee per day (light intake) had a decreased prevalence of subclinical coronary athersclerosis, those who drank 3-4 cups per day (moderate intake) had the lowest prevalence, and those who drank 5 or more cups per day had an increased prevalence, compared with people who didn’t drink coffee. Coronary artery calcium ratios that compared coffee drinkers with nondrinkers were 0.86 at less than 1 cup per day, 0.82 at 1-2 cups per day, 0.78 at 3-4 cups per day, and 1.77 at 5 or more cups per day. The association between coffee intake and coronary artery calcium scores remained consistent across all subgroup analyses and in sensitivity analyses, the investigators said (Heart 2015 March 2 [doi:10.1136/heartjnl-2014-306663]).
The cross-sectional design of this study means that it can establish only an association, not causality, between coffee intake and CVD risk. “Further research is needed to confirm our findings and establish the biological basis of coffee’s potential preventive effects on coronary artery disease,” Dr. Choi and her associates wrote.
But the evidence already is strong enough that for the first time this year, dietary guidelines will likely state that moderate coffee consumption appears to be cardioprotective and “can be incorporated into a healthy dietary pattern.” Coffee drinkers need only minimize the sugars and fats they add to their coffee, in the form of sweeteners and creamers, to benefit from the beverage, the 2015 Dietary Guidelines Advisory Committee recommended in a report submitted for review to the secretaries of the U.S. Department of Health & Human Services and the U.S. Department of Agriculture in February.
Key clinical point: Light to moderate coffee drinking is associated with decreased coronary artery calcium, an indicator of subclinical atherosclerosis.
Major finding: Coronary artery calcium ratios that compared coffee drinkers with nondrinkers were 0.86 at less than 1 cup per day, 0.82 at 1-2 cups per day, 0.78 at 3-4 cups per day, and 1.77 at 5 or more cups per day.
Data source: A cross-sectional analysis of data from a cohort study involving 25,138 asymptomatic Korean adults who underwent a comprehensive health examination including cardiac CT for coronary artery calcium scoring.
Disclosures: Dr. Choi and her associates reported having no financial disclosures.

Emergency Imaging
Case
A 49-year-old man presented to the ED with low-back pain. Radiographs of the lumbosacral spine were obtained; a coned-down frontal representative radiograph of the lower lumbar spine and sacrum is presented (Figure 1a).
What is the diagnosis? Is additional imaging necessary? If so, why?
Answer
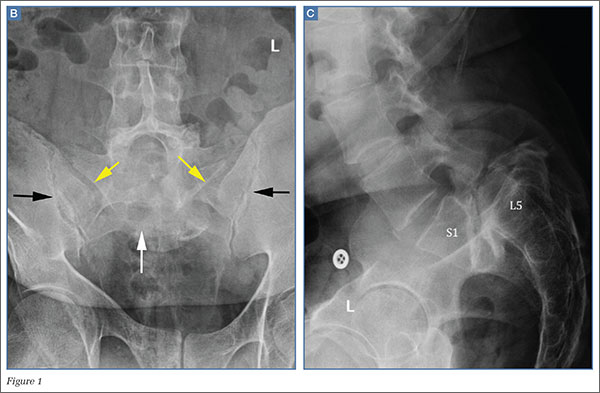
The frontal radiograph (Figure 1b) shows an “inverted Napoleon hat” sign,1 indicating high-grade anterolisthesis of L5 on S1 (less frequently, this sign also may be indicative of severe lumbar lordosis at the lumbosacral junction).
Anterolisthesis refers to anterior displacement of a vertebral body with respect to the vertebral body immediately below it. The dome of the “inverted hat” is formed by the anteroinferior endplate of the L5 vertebral body (white arrow, Figure 1b), and the tapered edges of the inverted hat are formed by the anteroinferiorly displaced/rotated L5 transverse processes (yellow arrows, Figure 1b). The contours of the L5 transverse processes project medial to the normal sacroiliac joints (black arrows, Figure 1b).
If the inverted Napoleon hat sign is identified on a single frontal view (eg, anteroposterior abdomen or pelvis radiograph), a lateral radiograph of the lumbosacral spine should be obtained to further evaluate the degree of L5-S1 anterolisthesis.
There are five grades of anterolisthesis, each based on quartiles of anterior displacement. Grade 1 anterolisthesis refers to less than 25% anterior displacement; grade 2 refers to 25% to 50% anterior displacement; grade 3 refers to 50% to 75% anterior displacement; and grade 4 refers to greater than 75% anterior displacement. In grade 5 anterolisthesis (also referred to as spondyloptosis), there is 100% anterior displacement with resultant anteroinferior slippage of the displaced vertebral body, which consequently lies anterior to the vertebral body below it. In this patient, the lateral radiograph of the lumbosacral spine (Figure 1c) demonstrates 100% anterior displacement of the L5 vertebral body with respect to S1, compatible with grade 5 anterolisthesis (spondyloptosis).
Spondylolisthesis is the more generalized term that includes all sagittal misalignments of the spine—more specifically, anterolisthesis for anterior displacement or retrolisthesis for posterior displacement. By convention, the displacement is named by the directional displacement of the more superior vertebral body in relation to the vertebral body immediately below it. Spondylolisthesis can be further delineated into one of six etiologic categories according to the modified Newman classification: congenital/dysplastic, spondylolytic, degenerative, traumatic, pathologic, or postsurgical.1,2 Of these six etiologies, spondylolysis (ie, defect of the pars interarticularis) and degenerative change (ie, disc degeneration and facet arthrosis) are the two most common causes of spondylolisthesis. High-grade spondylolisthesis (grade 3 or 4) typically requires bilateral spondylolysis (pars defects), while lower grade spondylolisthesis (particularly grade 1) can occur with any of the above etiologies. As seen in this case, anterolisthesis of L5 on S1 has the greatest effect on the exiting L5 nerve roots, and patients may present with low-back pain and/or hamstring tightness.1
With respect to imaging modalities, noncontrast computed tomography (CT) is of low utility in the workup of high-grade anterolisthesis, since it only serves to demonstrate the bilateral pars defects that are already presumed to be present—even if not seen radiographically. Conversely, magnetic resonance imaging (MRI), or CT myelography in patients in whom MRI is contraindicated, allows evaluation of the exiting nerve roots at the level of the spondylolisthesis. In addition to the spondylolisthesis, MRI can also reveal nerve impingement at other levels that might not be radiographically apparent (eg, neural foraminal stenosis that occurs due to disc herniation, facet arthrosis, and/or ligamentum flavum hypertrophy).
Management of high-grade spondylolisthesis remains controversial.3,4 Surgical treatment options include instrumented fusion, noninstrumented fusion, and L5 corpectomy with L4-S1 fusion. Fusion is generally recommended for patients with radicular symptoms, chronic incapacitating low-back pain, or risk of progression to spondyloptosis.4
Dr Bartolotta is an assistant professor of radiology at Weill Cornell Medical College in New York City, and an assistant attending radiologist at New York-Presbyterian Hospital/Weill Cornell Medical Center. Dr Hentel is an associate professor of clinical radiology at Weill Cornell Medical College, New York. He is also chief of emergency/musculoskeletal imaging and executive vice-chairman for the department of radiology at New York-Presbyterian Hospital/Weill Cornell Medical Center; and associate editor, imaging, of the EMERGENCY MEDICINE editorial board.
- Talangbayan LE. The inverted Napoleon’s hat sign. Radiology. 2007;243(2): 603-604.
- Wiltse LL, Newman PH, Macnab I. Classification of spondylolisis and spondylolisthesis. Clin Orthop Relat Res. 1976;117:23–29.
- Hart RA, Domes CM, Goodwin B, et al. High-grade spondylolisthesis treated using a modified Bohlman technique: results among multiple surgeons. J Neurosurg Spine. 2014;20(5):523-530.
- Lengert R, Charles YP, Walter A, Schuller S, Godet J, Steib JP. Posterior surgery in high-grade spondylolisthesis. Orthop Traumatol Surg Res. 2014;100(5):481-484.
Case
A 49-year-old man presented to the ED with low-back pain. Radiographs of the lumbosacral spine were obtained; a coned-down frontal representative radiograph of the lower lumbar spine and sacrum is presented (Figure 1a).
What is the diagnosis? Is additional imaging necessary? If so, why?
Answer
The frontal radiograph (Figure 1b) shows an “inverted Napoleon hat” sign,1 indicating high-grade anterolisthesis of L5 on S1 (less frequently, this sign also may be indicative of severe lumbar lordosis at the lumbosacral junction).
Anterolisthesis refers to anterior displacement of a vertebral body with respect to the vertebral body immediately below it. The dome of the “inverted hat” is formed by the anteroinferior endplate of the L5 vertebral body (white arrow, Figure 1b), and the tapered edges of the inverted hat are formed by the anteroinferiorly displaced/rotated L5 transverse processes (yellow arrows, Figure 1b). The contours of the L5 transverse processes project medial to the normal sacroiliac joints (black arrows, Figure 1b).
If the inverted Napoleon hat sign is identified on a single frontal view (eg, anteroposterior abdomen or pelvis radiograph), a lateral radiograph of the lumbosacral spine should be obtained to further evaluate the degree of L5-S1 anterolisthesis.
There are five grades of anterolisthesis, each based on quartiles of anterior displacement. Grade 1 anterolisthesis refers to less than 25% anterior displacement; grade 2 refers to 25% to 50% anterior displacement; grade 3 refers to 50% to 75% anterior displacement; and grade 4 refers to greater than 75% anterior displacement. In grade 5 anterolisthesis (also referred to as spondyloptosis), there is 100% anterior displacement with resultant anteroinferior slippage of the displaced vertebral body, which consequently lies anterior to the vertebral body below it. In this patient, the lateral radiograph of the lumbosacral spine (Figure 1c) demonstrates 100% anterior displacement of the L5 vertebral body with respect to S1, compatible with grade 5 anterolisthesis (spondyloptosis).
Spondylolisthesis is the more generalized term that includes all sagittal misalignments of the spine—more specifically, anterolisthesis for anterior displacement or retrolisthesis for posterior displacement. By convention, the displacement is named by the directional displacement of the more superior vertebral body in relation to the vertebral body immediately below it. Spondylolisthesis can be further delineated into one of six etiologic categories according to the modified Newman classification: congenital/dysplastic, spondylolytic, degenerative, traumatic, pathologic, or postsurgical.1,2 Of these six etiologies, spondylolysis (ie, defect of the pars interarticularis) and degenerative change (ie, disc degeneration and facet arthrosis) are the two most common causes of spondylolisthesis. High-grade spondylolisthesis (grade 3 or 4) typically requires bilateral spondylolysis (pars defects), while lower grade spondylolisthesis (particularly grade 1) can occur with any of the above etiologies. As seen in this case, anterolisthesis of L5 on S1 has the greatest effect on the exiting L5 nerve roots, and patients may present with low-back pain and/or hamstring tightness.1
With respect to imaging modalities, noncontrast computed tomography (CT) is of low utility in the workup of high-grade anterolisthesis, since it only serves to demonstrate the bilateral pars defects that are already presumed to be present—even if not seen radiographically. Conversely, magnetic resonance imaging (MRI), or CT myelography in patients in whom MRI is contraindicated, allows evaluation of the exiting nerve roots at the level of the spondylolisthesis. In addition to the spondylolisthesis, MRI can also reveal nerve impingement at other levels that might not be radiographically apparent (eg, neural foraminal stenosis that occurs due to disc herniation, facet arthrosis, and/or ligamentum flavum hypertrophy).
Management of high-grade spondylolisthesis remains controversial.3,4 Surgical treatment options include instrumented fusion, noninstrumented fusion, and L5 corpectomy with L4-S1 fusion. Fusion is generally recommended for patients with radicular symptoms, chronic incapacitating low-back pain, or risk of progression to spondyloptosis.4
Dr Bartolotta is an assistant professor of radiology at Weill Cornell Medical College in New York City, and an assistant attending radiologist at New York-Presbyterian Hospital/Weill Cornell Medical Center. Dr Hentel is an associate professor of clinical radiology at Weill Cornell Medical College, New York. He is also chief of emergency/musculoskeletal imaging and executive vice-chairman for the department of radiology at New York-Presbyterian Hospital/Weill Cornell Medical Center; and associate editor, imaging, of the EMERGENCY MEDICINE editorial board.
Case
A 49-year-old man presented to the ED with low-back pain. Radiographs of the lumbosacral spine were obtained; a coned-down frontal representative radiograph of the lower lumbar spine and sacrum is presented (Figure 1a).
What is the diagnosis? Is additional imaging necessary? If so, why?
Answer
The frontal radiograph (Figure 1b) shows an “inverted Napoleon hat” sign,1 indicating high-grade anterolisthesis of L5 on S1 (less frequently, this sign also may be indicative of severe lumbar lordosis at the lumbosacral junction).
Anterolisthesis refers to anterior displacement of a vertebral body with respect to the vertebral body immediately below it. The dome of the “inverted hat” is formed by the anteroinferior endplate of the L5 vertebral body (white arrow, Figure 1b), and the tapered edges of the inverted hat are formed by the anteroinferiorly displaced/rotated L5 transverse processes (yellow arrows, Figure 1b). The contours of the L5 transverse processes project medial to the normal sacroiliac joints (black arrows, Figure 1b).
If the inverted Napoleon hat sign is identified on a single frontal view (eg, anteroposterior abdomen or pelvis radiograph), a lateral radiograph of the lumbosacral spine should be obtained to further evaluate the degree of L5-S1 anterolisthesis.
There are five grades of anterolisthesis, each based on quartiles of anterior displacement. Grade 1 anterolisthesis refers to less than 25% anterior displacement; grade 2 refers to 25% to 50% anterior displacement; grade 3 refers to 50% to 75% anterior displacement; and grade 4 refers to greater than 75% anterior displacement. In grade 5 anterolisthesis (also referred to as spondyloptosis), there is 100% anterior displacement with resultant anteroinferior slippage of the displaced vertebral body, which consequently lies anterior to the vertebral body below it. In this patient, the lateral radiograph of the lumbosacral spine (Figure 1c) demonstrates 100% anterior displacement of the L5 vertebral body with respect to S1, compatible with grade 5 anterolisthesis (spondyloptosis).
Spondylolisthesis is the more generalized term that includes all sagittal misalignments of the spine—more specifically, anterolisthesis for anterior displacement or retrolisthesis for posterior displacement. By convention, the displacement is named by the directional displacement of the more superior vertebral body in relation to the vertebral body immediately below it. Spondylolisthesis can be further delineated into one of six etiologic categories according to the modified Newman classification: congenital/dysplastic, spondylolytic, degenerative, traumatic, pathologic, or postsurgical.1,2 Of these six etiologies, spondylolysis (ie, defect of the pars interarticularis) and degenerative change (ie, disc degeneration and facet arthrosis) are the two most common causes of spondylolisthesis. High-grade spondylolisthesis (grade 3 or 4) typically requires bilateral spondylolysis (pars defects), while lower grade spondylolisthesis (particularly grade 1) can occur with any of the above etiologies. As seen in this case, anterolisthesis of L5 on S1 has the greatest effect on the exiting L5 nerve roots, and patients may present with low-back pain and/or hamstring tightness.1
With respect to imaging modalities, noncontrast computed tomography (CT) is of low utility in the workup of high-grade anterolisthesis, since it only serves to demonstrate the bilateral pars defects that are already presumed to be present—even if not seen radiographically. Conversely, magnetic resonance imaging (MRI), or CT myelography in patients in whom MRI is contraindicated, allows evaluation of the exiting nerve roots at the level of the spondylolisthesis. In addition to the spondylolisthesis, MRI can also reveal nerve impingement at other levels that might not be radiographically apparent (eg, neural foraminal stenosis that occurs due to disc herniation, facet arthrosis, and/or ligamentum flavum hypertrophy).
Management of high-grade spondylolisthesis remains controversial.3,4 Surgical treatment options include instrumented fusion, noninstrumented fusion, and L5 corpectomy with L4-S1 fusion. Fusion is generally recommended for patients with radicular symptoms, chronic incapacitating low-back pain, or risk of progression to spondyloptosis.4
Dr Bartolotta is an assistant professor of radiology at Weill Cornell Medical College in New York City, and an assistant attending radiologist at New York-Presbyterian Hospital/Weill Cornell Medical Center. Dr Hentel is an associate professor of clinical radiology at Weill Cornell Medical College, New York. He is also chief of emergency/musculoskeletal imaging and executive vice-chairman for the department of radiology at New York-Presbyterian Hospital/Weill Cornell Medical Center; and associate editor, imaging, of the EMERGENCY MEDICINE editorial board.
- Talangbayan LE. The inverted Napoleon’s hat sign. Radiology. 2007;243(2): 603-604.
- Wiltse LL, Newman PH, Macnab I. Classification of spondylolisis and spondylolisthesis. Clin Orthop Relat Res. 1976;117:23–29.
- Hart RA, Domes CM, Goodwin B, et al. High-grade spondylolisthesis treated using a modified Bohlman technique: results among multiple surgeons. J Neurosurg Spine. 2014;20(5):523-530.
- Lengert R, Charles YP, Walter A, Schuller S, Godet J, Steib JP. Posterior surgery in high-grade spondylolisthesis. Orthop Traumatol Surg Res. 2014;100(5):481-484.
- Talangbayan LE. The inverted Napoleon’s hat sign. Radiology. 2007;243(2): 603-604.
- Wiltse LL, Newman PH, Macnab I. Classification of spondylolisis and spondylolisthesis. Clin Orthop Relat Res. 1976;117:23–29.
- Hart RA, Domes CM, Goodwin B, et al. High-grade spondylolisthesis treated using a modified Bohlman technique: results among multiple surgeons. J Neurosurg Spine. 2014;20(5):523-530.
- Lengert R, Charles YP, Walter A, Schuller S, Godet J, Steib JP. Posterior surgery in high-grade spondylolisthesis. Orthop Traumatol Surg Res. 2014;100(5):481-484.
Managing aneurysmal subarachnoid hemorrhage: It takes a team
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.


The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
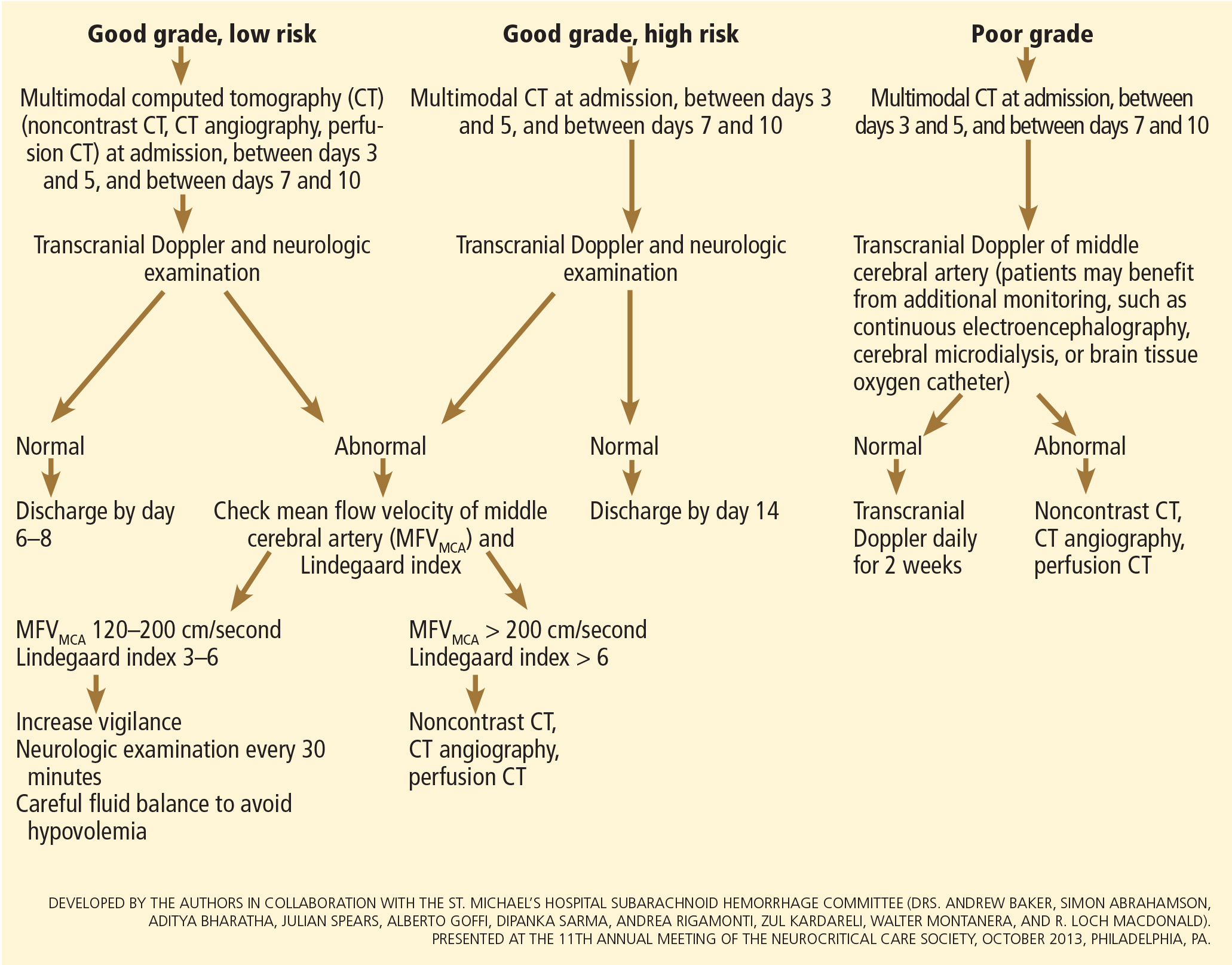
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
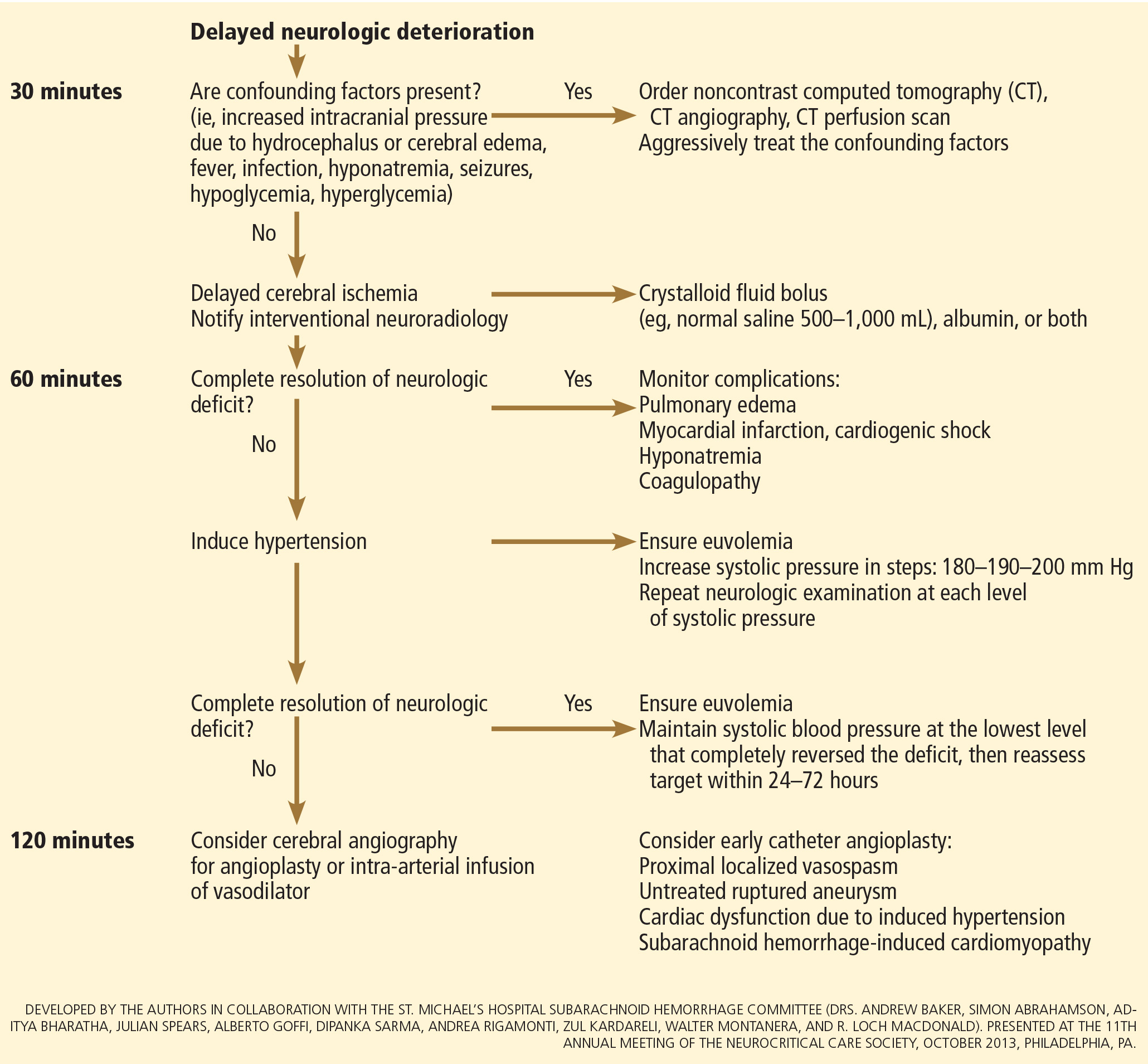
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
KEY POINTS
- The key symptom is the abrupt onset of severe headache, commonly described as “the worst headache of my life.
- Computed tomography without contrast should be done promptly when this condition is suspected.
- Outcomes are improved when patients are managed in a high-volume center with a specialized neurointensive care unit and access to an interdisciplinary team.
- Early aneurysm repair by surgical clipping or endovascular coiling is considered the standard of care and is the best strategy to reduce the risk of rebleeding.
- Medical and neurologic complications are extremely common and include hydrocephalus, increased intracranial pressure, seizures, delayed cerebral ischemia, hyponatremia, hypovolemia, and cardiac and pulmonary abnormalities.
Does this patient need ultrasonography of the leg to evaluate for deep vein thrombosis?
A 38-year-old woman presents to the emergency department after experiencing several days of swelling and mild discomfort in her left calf. She denies chest pain or shortness of breath. She does not recall antecedent trauma, is a nonsmoker, is healthy, and takes no medications apart from a multivitamin. She has not undergone any surgical procedure, has not been hospitalized recently, and has no history of venous thromboembolic disease. She says she started an aerobics program 1 week ago.
On examination, her left lower leg is mildly swollen, but the difference in calf circumference between the right and left legs is less than 1 cm. There is no erythema, no pitting edema, and only mild and rather diffuse tenderness of the calf. A urine pregnancy test is negative and her D-dimer level is 350 ng/mL (reference range < 500 ng/mL). Does she require ultrasonography of the left leg to evaluate for deep vein thrombosis (DVT)?
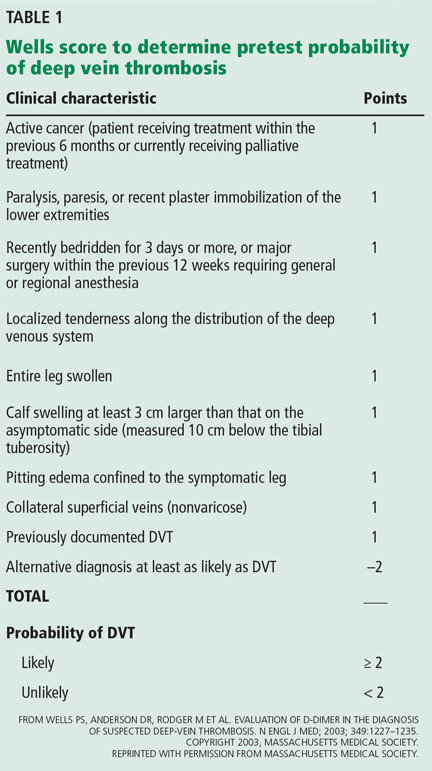
This patient does not need confirmatory ultrasonography, as her normal D-dimer level of 350 ng/mL is enough to rule out DVT. Her low probability of having DVT is further supported by her Wells score (Table 1), a tool that can help rule out DVT and reduce the need for further testing. DVT is unlikely if a patient’s Wells score is less than 2, and this patient’s score is –1. She receives 1 point for swelling of her left lower leg, but injury from her recent aerobic exercise is at least as likely as DVT to account for her symptoms (–2 points).
GUIDELINES AND CHOOSING WISELY
Compression ultrasonography is the study most commonly used to evaluate for DVT. The diagnosis is made if either the femoral or popliteal vein is noncompressible.1 In a patient with no history of DVT, the sensitivity of compression ultrasonography is 94%, and its specificity is 98%.
Several guidelines recommend using a clinical decision rule to establish the probability of venous thromboembolic disease before any additional diagnostic testing such as D-dimer measurement or ultrasonography.2–4 A number of clinical decision rules exist for DVT, but the Wells score is the most studied and validated.1 It incorporates the patient’s risk factors, symptoms, and signs to categorize the probability of DVT as low, moderate, or high and has been further modified to classify the risk as either likely or unlikely (Table 1).5
Guidelines from the American College of Chest Physicians (2012), Scottish Intercollegiate Guidelines Network (2010), and American Academy of Family Physicians and American College of Physicians (2007) recommend against performing imaging if a high-sensitivity D-dimer test is negative in a patient in whom the pretest probability of DVT is unlikely.2–4 Enzyme-linked immunofluorescence assays, microplate enzyme-linked immunosorbent assays, and latex quantitative assays are considered high-sensitivity D-dimer tests, having 96%, 94%, and 93% sensitivity, respectively, in ruling out DVT.1 Other D-dimer tests have lower sensitivity and cannot comfortably rule out DVT even if the results are negative.
Since D-dimer measurement is a sensitive but not specific test, it should be used only to rule out DVT—not to rule it in. Moreover, compression ultrasonography may be indicated to rule out other causes of the patient’s symptoms.

The guidelines caution against D-dimer testing if the patient has a comorbid condition that can by itself raise or lower the D-dimer level, leading one to falsely conclude the patient has or does not have DVT (Table 2).1–4 In these instances, the pretest probability of DVT may be higher than calculated by a clinical prediction rule, and compression ultrasonography may be an appropriate initial test.4 Compression ultrasonography is also recommended as a confirmatory test in low-risk patients who have a positive D-dimer test or as an initial test in patients at higher risk for DVT.2–4
If a patient has a low pretest probability of DVT as defined by the Wells score and a normal high-sensitivity D-dimer measurement, then ordering imaging studies is a questionable practice according to statements by the American College of Physicians, American College of Emergency Physicians, European Society of Cardiology, American Academy of Family Physicians, and Scottish Intercollegiate Guidelines Network.
HARMS OF ULTRASONOGRAPHY
Although ultrasonography is generally well tolerated, it may be unnecessary. Combining a prediction rule (to assess the probability) with D-dimer testing (to rule out DVT) can significantly reduce the use of ultrasonography and the associated cost.
Wells et al5 calculated that clinicians could cut back on ultrasonographic testing by 39% by not doing it in those who had a low pretest probability and a negative D-dimer test result.5 In that patient population, fewer than 1% of patients were later found to have DVT.
Ordering compression ultrasonography as additional testing may lead to a false-positive result and to additional unnecessary testing and treatments that would inconvenience the patient, increase the risk of serious complications such as bleeding, and incur increased costs. Cost considerations should include not only the cost of the test and its interpretation, but also the workup and treatment of false-positive results, patient time missed from work while being tested, and potential associated costs for patients who need to be evaluated in the emergency department to obtain same-day testing.
THE CLINICAL BOTTOM LINE
Our patient’s Wells score indicates that DVT is unlikely. A negative D-dimer test is sufficient to rule out DVT, and further testing is unnecessary.
- Huisman MV, Klok FA. Diagnostic management of acute deep vein thrombosis and pulmonary embolism. J Thromb Haemost 2013; 11:412–422.
- Bates SM, Jaeschke R, Stevens EM, et al. Antithrombotic therapy and prevention of thrombosis, 9th edition: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(2 suppl):e351S–e418S.
- Scottish Intercollegiate Guidelines Network (SIGN). Prevention and management of venous thromboembolism. A national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2010: http://sign.ac.uk/guidelines/fulltext/122/index.html. Accessed February 6, 2015.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Intern Med 2007; 146:454–458.
- Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 2003; 349:1227–1235.
A 38-year-old woman presents to the emergency department after experiencing several days of swelling and mild discomfort in her left calf. She denies chest pain or shortness of breath. She does not recall antecedent trauma, is a nonsmoker, is healthy, and takes no medications apart from a multivitamin. She has not undergone any surgical procedure, has not been hospitalized recently, and has no history of venous thromboembolic disease. She says she started an aerobics program 1 week ago.
On examination, her left lower leg is mildly swollen, but the difference in calf circumference between the right and left legs is less than 1 cm. There is no erythema, no pitting edema, and only mild and rather diffuse tenderness of the calf. A urine pregnancy test is negative and her D-dimer level is 350 ng/mL (reference range < 500 ng/mL). Does she require ultrasonography of the left leg to evaluate for deep vein thrombosis (DVT)?
This patient does not need confirmatory ultrasonography, as her normal D-dimer level of 350 ng/mL is enough to rule out DVT. Her low probability of having DVT is further supported by her Wells score (Table 1), a tool that can help rule out DVT and reduce the need for further testing. DVT is unlikely if a patient’s Wells score is less than 2, and this patient’s score is –1. She receives 1 point for swelling of her left lower leg, but injury from her recent aerobic exercise is at least as likely as DVT to account for her symptoms (–2 points).
GUIDELINES AND CHOOSING WISELY
Compression ultrasonography is the study most commonly used to evaluate for DVT. The diagnosis is made if either the femoral or popliteal vein is noncompressible.1 In a patient with no history of DVT, the sensitivity of compression ultrasonography is 94%, and its specificity is 98%.
Several guidelines recommend using a clinical decision rule to establish the probability of venous thromboembolic disease before any additional diagnostic testing such as D-dimer measurement or ultrasonography.2–4 A number of clinical decision rules exist for DVT, but the Wells score is the most studied and validated.1 It incorporates the patient’s risk factors, symptoms, and signs to categorize the probability of DVT as low, moderate, or high and has been further modified to classify the risk as either likely or unlikely (Table 1).5
Guidelines from the American College of Chest Physicians (2012), Scottish Intercollegiate Guidelines Network (2010), and American Academy of Family Physicians and American College of Physicians (2007) recommend against performing imaging if a high-sensitivity D-dimer test is negative in a patient in whom the pretest probability of DVT is unlikely.2–4 Enzyme-linked immunofluorescence assays, microplate enzyme-linked immunosorbent assays, and latex quantitative assays are considered high-sensitivity D-dimer tests, having 96%, 94%, and 93% sensitivity, respectively, in ruling out DVT.1 Other D-dimer tests have lower sensitivity and cannot comfortably rule out DVT even if the results are negative.
Since D-dimer measurement is a sensitive but not specific test, it should be used only to rule out DVT—not to rule it in. Moreover, compression ultrasonography may be indicated to rule out other causes of the patient’s symptoms.
The guidelines caution against D-dimer testing if the patient has a comorbid condition that can by itself raise or lower the D-dimer level, leading one to falsely conclude the patient has or does not have DVT (Table 2).1–4 In these instances, the pretest probability of DVT may be higher than calculated by a clinical prediction rule, and compression ultrasonography may be an appropriate initial test.4 Compression ultrasonography is also recommended as a confirmatory test in low-risk patients who have a positive D-dimer test or as an initial test in patients at higher risk for DVT.2–4
If a patient has a low pretest probability of DVT as defined by the Wells score and a normal high-sensitivity D-dimer measurement, then ordering imaging studies is a questionable practice according to statements by the American College of Physicians, American College of Emergency Physicians, European Society of Cardiology, American Academy of Family Physicians, and Scottish Intercollegiate Guidelines Network.
HARMS OF ULTRASONOGRAPHY
Although ultrasonography is generally well tolerated, it may be unnecessary. Combining a prediction rule (to assess the probability) with D-dimer testing (to rule out DVT) can significantly reduce the use of ultrasonography and the associated cost.
Wells et al5 calculated that clinicians could cut back on ultrasonographic testing by 39% by not doing it in those who had a low pretest probability and a negative D-dimer test result.5 In that patient population, fewer than 1% of patients were later found to have DVT.
Ordering compression ultrasonography as additional testing may lead to a false-positive result and to additional unnecessary testing and treatments that would inconvenience the patient, increase the risk of serious complications such as bleeding, and incur increased costs. Cost considerations should include not only the cost of the test and its interpretation, but also the workup and treatment of false-positive results, patient time missed from work while being tested, and potential associated costs for patients who need to be evaluated in the emergency department to obtain same-day testing.
THE CLINICAL BOTTOM LINE
Our patient’s Wells score indicates that DVT is unlikely. A negative D-dimer test is sufficient to rule out DVT, and further testing is unnecessary.
A 38-year-old woman presents to the emergency department after experiencing several days of swelling and mild discomfort in her left calf. She denies chest pain or shortness of breath. She does not recall antecedent trauma, is a nonsmoker, is healthy, and takes no medications apart from a multivitamin. She has not undergone any surgical procedure, has not been hospitalized recently, and has no history of venous thromboembolic disease. She says she started an aerobics program 1 week ago.
On examination, her left lower leg is mildly swollen, but the difference in calf circumference between the right and left legs is less than 1 cm. There is no erythema, no pitting edema, and only mild and rather diffuse tenderness of the calf. A urine pregnancy test is negative and her D-dimer level is 350 ng/mL (reference range < 500 ng/mL). Does she require ultrasonography of the left leg to evaluate for deep vein thrombosis (DVT)?
This patient does not need confirmatory ultrasonography, as her normal D-dimer level of 350 ng/mL is enough to rule out DVT. Her low probability of having DVT is further supported by her Wells score (Table 1), a tool that can help rule out DVT and reduce the need for further testing. DVT is unlikely if a patient’s Wells score is less than 2, and this patient’s score is –1. She receives 1 point for swelling of her left lower leg, but injury from her recent aerobic exercise is at least as likely as DVT to account for her symptoms (–2 points).
GUIDELINES AND CHOOSING WISELY
Compression ultrasonography is the study most commonly used to evaluate for DVT. The diagnosis is made if either the femoral or popliteal vein is noncompressible.1 In a patient with no history of DVT, the sensitivity of compression ultrasonography is 94%, and its specificity is 98%.
Several guidelines recommend using a clinical decision rule to establish the probability of venous thromboembolic disease before any additional diagnostic testing such as D-dimer measurement or ultrasonography.2–4 A number of clinical decision rules exist for DVT, but the Wells score is the most studied and validated.1 It incorporates the patient’s risk factors, symptoms, and signs to categorize the probability of DVT as low, moderate, or high and has been further modified to classify the risk as either likely or unlikely (Table 1).5
Guidelines from the American College of Chest Physicians (2012), Scottish Intercollegiate Guidelines Network (2010), and American Academy of Family Physicians and American College of Physicians (2007) recommend against performing imaging if a high-sensitivity D-dimer test is negative in a patient in whom the pretest probability of DVT is unlikely.2–4 Enzyme-linked immunofluorescence assays, microplate enzyme-linked immunosorbent assays, and latex quantitative assays are considered high-sensitivity D-dimer tests, having 96%, 94%, and 93% sensitivity, respectively, in ruling out DVT.1 Other D-dimer tests have lower sensitivity and cannot comfortably rule out DVT even if the results are negative.
Since D-dimer measurement is a sensitive but not specific test, it should be used only to rule out DVT—not to rule it in. Moreover, compression ultrasonography may be indicated to rule out other causes of the patient’s symptoms.
The guidelines caution against D-dimer testing if the patient has a comorbid condition that can by itself raise or lower the D-dimer level, leading one to falsely conclude the patient has or does not have DVT (Table 2).1–4 In these instances, the pretest probability of DVT may be higher than calculated by a clinical prediction rule, and compression ultrasonography may be an appropriate initial test.4 Compression ultrasonography is also recommended as a confirmatory test in low-risk patients who have a positive D-dimer test or as an initial test in patients at higher risk for DVT.2–4
If a patient has a low pretest probability of DVT as defined by the Wells score and a normal high-sensitivity D-dimer measurement, then ordering imaging studies is a questionable practice according to statements by the American College of Physicians, American College of Emergency Physicians, European Society of Cardiology, American Academy of Family Physicians, and Scottish Intercollegiate Guidelines Network.
HARMS OF ULTRASONOGRAPHY
Although ultrasonography is generally well tolerated, it may be unnecessary. Combining a prediction rule (to assess the probability) with D-dimer testing (to rule out DVT) can significantly reduce the use of ultrasonography and the associated cost.
Wells et al5 calculated that clinicians could cut back on ultrasonographic testing by 39% by not doing it in those who had a low pretest probability and a negative D-dimer test result.5 In that patient population, fewer than 1% of patients were later found to have DVT.
Ordering compression ultrasonography as additional testing may lead to a false-positive result and to additional unnecessary testing and treatments that would inconvenience the patient, increase the risk of serious complications such as bleeding, and incur increased costs. Cost considerations should include not only the cost of the test and its interpretation, but also the workup and treatment of false-positive results, patient time missed from work while being tested, and potential associated costs for patients who need to be evaluated in the emergency department to obtain same-day testing.
THE CLINICAL BOTTOM LINE
Our patient’s Wells score indicates that DVT is unlikely. A negative D-dimer test is sufficient to rule out DVT, and further testing is unnecessary.
- Huisman MV, Klok FA. Diagnostic management of acute deep vein thrombosis and pulmonary embolism. J Thromb Haemost 2013; 11:412–422.
- Bates SM, Jaeschke R, Stevens EM, et al. Antithrombotic therapy and prevention of thrombosis, 9th edition: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(2 suppl):e351S–e418S.
- Scottish Intercollegiate Guidelines Network (SIGN). Prevention and management of venous thromboembolism. A national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2010: http://sign.ac.uk/guidelines/fulltext/122/index.html. Accessed February 6, 2015.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Intern Med 2007; 146:454–458.
- Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 2003; 349:1227–1235.
- Huisman MV, Klok FA. Diagnostic management of acute deep vein thrombosis and pulmonary embolism. J Thromb Haemost 2013; 11:412–422.
- Bates SM, Jaeschke R, Stevens EM, et al. Antithrombotic therapy and prevention of thrombosis, 9th edition: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(2 suppl):e351S–e418S.
- Scottish Intercollegiate Guidelines Network (SIGN). Prevention and management of venous thromboembolism. A national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2010: http://sign.ac.uk/guidelines/fulltext/122/index.html. Accessed February 6, 2015.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Intern Med 2007; 146:454–458.
- Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 2003; 349:1227–1235.