User login
Insurance is a matter of life or death for lymphoma patients
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.

A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
FROM BLOOD
Key clinical point:
Major finding: The risk for death among patients under age 65 with no insurance, Medicaid, or Medicare was nearly twice that of similar patients with private health insurance.
Study details: Review of data on 43,648 patients with follicular lymphoma in the National Cancer Database.
Disclosures: The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
Source: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
FDA warns against azithromycin in blood or lymph node cancers
The Food and Drug Administration has issued a in patients with blood or lymph node cancers who have received donor stem cell transplants.

This use of azithromycin can lead to increased risk of cancer relapse and death in this population. The FDA is continuing to review data and is expected to issue further recommendations.
Patients with blood or lymph node cancers are at an increased risk of bronchiolitis obliterans syndrome after donor stem cell transplant; although azithromycin is not approved for prevention of this condition, the antibiotic is sometimes prescribed for that purpose.
A French study of 480 patients was undertaken to assess the effectiveness of this prophylaxis but revealed the increased risk of relapse and death and was halted 13 months after completing enrollment. The rate of cancer relapse was 32.9% in the azithromycin group and just 20.8% in the placebo group; the 2-year survival rate was 56.6% in the azithromycin group and 70.1% in the placebo group (JAMA 2017;318[6]:557-66).
Bronchiolitis obliterans syndrome is marked by inflammation and scarring of the airways that leads to severe shortness of breath and dry cough. There are no known effective antibiotic treatments for prophylaxis of the condition, according to the FDA.
FDA officials are advising physicians not to prescribe long-term azithromycin in this population. Patients who have had a stem cell transplant and are already taking the antibiotic, should consult a doctor before discontinuing.
The manufacturer of brand name azithromycin (Zithromax) has issued a Dear Healthcare Provider letter about the safety issue, and more information can be found in the FDA’s safety announcement.
The Food and Drug Administration has issued a in patients with blood or lymph node cancers who have received donor stem cell transplants.
This use of azithromycin can lead to increased risk of cancer relapse and death in this population. The FDA is continuing to review data and is expected to issue further recommendations.
Patients with blood or lymph node cancers are at an increased risk of bronchiolitis obliterans syndrome after donor stem cell transplant; although azithromycin is not approved for prevention of this condition, the antibiotic is sometimes prescribed for that purpose.
A French study of 480 patients was undertaken to assess the effectiveness of this prophylaxis but revealed the increased risk of relapse and death and was halted 13 months after completing enrollment. The rate of cancer relapse was 32.9% in the azithromycin group and just 20.8% in the placebo group; the 2-year survival rate was 56.6% in the azithromycin group and 70.1% in the placebo group (JAMA 2017;318[6]:557-66).
Bronchiolitis obliterans syndrome is marked by inflammation and scarring of the airways that leads to severe shortness of breath and dry cough. There are no known effective antibiotic treatments for prophylaxis of the condition, according to the FDA.
FDA officials are advising physicians not to prescribe long-term azithromycin in this population. Patients who have had a stem cell transplant and are already taking the antibiotic, should consult a doctor before discontinuing.
The manufacturer of brand name azithromycin (Zithromax) has issued a Dear Healthcare Provider letter about the safety issue, and more information can be found in the FDA’s safety announcement.
The Food and Drug Administration has issued a in patients with blood or lymph node cancers who have received donor stem cell transplants.
This use of azithromycin can lead to increased risk of cancer relapse and death in this population. The FDA is continuing to review data and is expected to issue further recommendations.
Patients with blood or lymph node cancers are at an increased risk of bronchiolitis obliterans syndrome after donor stem cell transplant; although azithromycin is not approved for prevention of this condition, the antibiotic is sometimes prescribed for that purpose.
A French study of 480 patients was undertaken to assess the effectiveness of this prophylaxis but revealed the increased risk of relapse and death and was halted 13 months after completing enrollment. The rate of cancer relapse was 32.9% in the azithromycin group and just 20.8% in the placebo group; the 2-year survival rate was 56.6% in the azithromycin group and 70.1% in the placebo group (JAMA 2017;318[6]:557-66).
Bronchiolitis obliterans syndrome is marked by inflammation and scarring of the airways that leads to severe shortness of breath and dry cough. There are no known effective antibiotic treatments for prophylaxis of the condition, according to the FDA.
FDA officials are advising physicians not to prescribe long-term azithromycin in this population. Patients who have had a stem cell transplant and are already taking the antibiotic, should consult a doctor before discontinuing.
The manufacturer of brand name azithromycin (Zithromax) has issued a Dear Healthcare Provider letter about the safety issue, and more information can be found in the FDA’s safety announcement.
Advances in Hematology and Oncology (August 2018)
Click here to access August 2018 Advances In Hematology and Oncology Digital Edition.
Table of Contents
- VHA Practice Guideline Recommendations for Diffuse Gliomas
- Positivity Rates in Head and Neck Cancer in the VA
- Prevalence of Cancer in Thyroid Nodules in a Veteran Population
- Fatal Drug-Resistant Invasive Infection in a 56-Year-Old Immunosuppressed Man
- Immune Checkpoint Inhibitors for Urothelial Cancer: An Update on New Therapies
- Research News: RCC. Adrenal Tumors, Myeloma Therapy, Cancer Clusters, Hodgkin Lymphoma
- Coordination of Prostate Cance Care Between Primary Care and Oncology
- Open Clinical Trials for Patients With Lung Cancers

Click here to access August 2018 Advances In Hematology and Oncology Digital Edition.
Table of Contents
- VHA Practice Guideline Recommendations for Diffuse Gliomas
- Positivity Rates in Head and Neck Cancer in the VA
- Prevalence of Cancer in Thyroid Nodules in a Veteran Population
- Fatal Drug-Resistant Invasive Infection in a 56-Year-Old Immunosuppressed Man
- Immune Checkpoint Inhibitors for Urothelial Cancer: An Update on New Therapies
- Research News: RCC. Adrenal Tumors, Myeloma Therapy, Cancer Clusters, Hodgkin Lymphoma
- Coordination of Prostate Cance Care Between Primary Care and Oncology
- Open Clinical Trials for Patients With Lung Cancers
Click here to access August 2018 Advances In Hematology and Oncology Digital Edition.
Table of Contents
- VHA Practice Guideline Recommendations for Diffuse Gliomas
- Positivity Rates in Head and Neck Cancer in the VA
- Prevalence of Cancer in Thyroid Nodules in a Veteran Population
- Fatal Drug-Resistant Invasive Infection in a 56-Year-Old Immunosuppressed Man
- Immune Checkpoint Inhibitors for Urothelial Cancer: An Update on New Therapies
- Research News: RCC. Adrenal Tumors, Myeloma Therapy, Cancer Clusters, Hodgkin Lymphoma
- Coordination of Prostate Cance Care Between Primary Care and Oncology
- Open Clinical Trials for Patients With Lung Cancers
Do Erythropoiesis-Stimulating Agents Have a Risk Evaluation and Mitigation Strategy? (FULL)
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
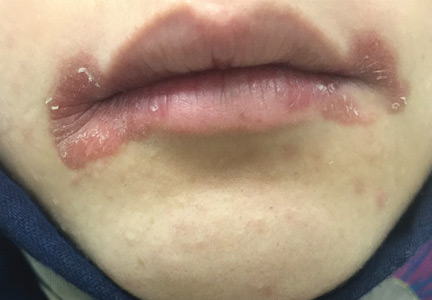
Angular cheilitis induced by iron deficiency anemia
A 20-year-old woman had a 4-month history of painful red erosions around the mouth. She had no dysphagia or fatigue and no history of diarrhea, gluten intolerance, or diabetes mellitus. An antifungal-antibacterial ointment prescribed by her dentist had provided no relief.

- Hemoglobin 8.0 g/dL (reference range for females 12.3–15.3)
- Mean corpuscular volume 62 fL (80–100)
- Serum ferritin 1.3 ng/mL (15–200)
- Reticulocyte count 0.86% (0.5–1.5)
- White blood cell count 9.8 × 109/L (4.5–11.0)
- Platelet count 450 × 109/L (150–400).
Vitamin B12 and folate levels were normal, and tests for antitissue transglutaminase and antinuclear antibodies were negative.
Based on these results, the diagnosis was angular cheilitis from iron deficiency anemia. Treatment with oral ferrous gluconate 300 mg twice daily cleared the cheilitis, and after 4 weeks of this treatment, the hemoglobin level increased to 9.8 g/dL, the serum ferritin increased to 7 ng/mL, and the reticulocyte count increased to 2.6%. She was advised to continue taking oral iron tablets for another 3 months until the hemoglobin level reached 12.0 g/dL.
During 2 years of follow-up, she had no recurrence of angular cheilitis, and her hemoglobin and serum ferritin levels remained normal. Ferrous gluconate was her only medication from the time of her diagnosis.
A BROAD DIFFERENTIAL DIAGNOSIS
Angular cheilitis (perlèche) is an inflammatory condition characterized by erosive inflammation at one or both angles of the mouth. It typically presents as erythema, scaling, fissuring, and ulceration. A wide variety of factors, including nutritional deficiencies, local and systemic factors, and drug side effects, may produce cheilitis.1,2
Nutritional deficiencies account for 25% of all cases of angular cheilitis3 and include iron deficiency and deficiencies of the B vitamins riboflavin (B2), niacin (B3), pyridoxine (B6), and cyanocobalamin (B12).1
Local causes include infection with Candida albicans or Staphylococcus aureus and allergic contact dermatitis. Common causes of allergic contact dermatitis include lipstick, toothpaste, mouthwash, cosmetics, sunscreen, fragrance, metals such as nickel, and dental appliances.1
Systemic diseases associated with angular cheilitis include xerostomia, inflammatory bowel disease, Sjögren syndrome, glucagonoma, and human immunodeficiency virus.1
Drugs that cause angular cheilitis include isotretinoin, sorafenib (antineoplastic kinase inhibitor), and ointments or creams such as neomycin sulfate–polymyxin B sulfate, bacitracin, idoxuridine, and steroids.1,4
Conditions that mimic angular cheilitis include herpes simplex type 1 (herpes labialis) and actinic cheilitis. Herpes labialis, characterized by burning sensation, itching, or paresthesia, usually precedes a recurrence of vesicles that eventually ulcerate or form a crust and heal without a crust. Herpes labialis often recurs, affecting the vermilion border and lasting approximately 1 week.
Actinic cheilitis, a premalignant condition that commonly involves the lower lip with sparing of the corners of the mouth, is caused by excessive sun exposure. Patients often have persistent dryness and cracking of the lips.
In our patient, angular cheilitis was the main clinical manifestation of iron deficiency anemia, highlighting the importance of looking for iron deficiency in affected patients without a more obvious cause.
- Park KK, Brodell RT, Helms SE. Angular cheilitis, part 2: nutritional, systemic, and drug-related causes and treatment. Cutis 2011; 88(1):27–32. pmid:21877503
- Park KK, Brodell RT, Helms SE. Angular cheilitis, part 1: local etiologies. Cutis 2011; 87(6):289–295. pmid:21838086
- Konstantinidis AB, Hatziotis JH. Angular cheilosis: an analysis of 156 cases. J Oral Med 1984; 39(4):199–206. pmid:6594458
- Yang CH, Lin WC, Chuang CK, et al. Hand-foot skin reaction in patients treated with sorafenib: a clinicopathological study of cutaneous manifestations due to multitargeted kinase inhibitor therapy. Br J Dermatol 2008; 158(3):592–596. doi:10.1111/j.1365-2133.2007.08357.x
A 20-year-old woman had a 4-month history of painful red erosions around the mouth. She had no dysphagia or fatigue and no history of diarrhea, gluten intolerance, or diabetes mellitus. An antifungal-antibacterial ointment prescribed by her dentist had provided no relief.
- Hemoglobin 8.0 g/dL (reference range for females 12.3–15.3)
- Mean corpuscular volume 62 fL (80–100)
- Serum ferritin 1.3 ng/mL (15–200)
- Reticulocyte count 0.86% (0.5–1.5)
- White blood cell count 9.8 × 109/L (4.5–11.0)
- Platelet count 450 × 109/L (150–400).
Vitamin B12 and folate levels were normal, and tests for antitissue transglutaminase and antinuclear antibodies were negative.
Based on these results, the diagnosis was angular cheilitis from iron deficiency anemia. Treatment with oral ferrous gluconate 300 mg twice daily cleared the cheilitis, and after 4 weeks of this treatment, the hemoglobin level increased to 9.8 g/dL, the serum ferritin increased to 7 ng/mL, and the reticulocyte count increased to 2.6%. She was advised to continue taking oral iron tablets for another 3 months until the hemoglobin level reached 12.0 g/dL.
During 2 years of follow-up, she had no recurrence of angular cheilitis, and her hemoglobin and serum ferritin levels remained normal. Ferrous gluconate was her only medication from the time of her diagnosis.
A BROAD DIFFERENTIAL DIAGNOSIS
Angular cheilitis (perlèche) is an inflammatory condition characterized by erosive inflammation at one or both angles of the mouth. It typically presents as erythema, scaling, fissuring, and ulceration. A wide variety of factors, including nutritional deficiencies, local and systemic factors, and drug side effects, may produce cheilitis.1,2
Nutritional deficiencies account for 25% of all cases of angular cheilitis3 and include iron deficiency and deficiencies of the B vitamins riboflavin (B2), niacin (B3), pyridoxine (B6), and cyanocobalamin (B12).1
Local causes include infection with Candida albicans or Staphylococcus aureus and allergic contact dermatitis. Common causes of allergic contact dermatitis include lipstick, toothpaste, mouthwash, cosmetics, sunscreen, fragrance, metals such as nickel, and dental appliances.1
Systemic diseases associated with angular cheilitis include xerostomia, inflammatory bowel disease, Sjögren syndrome, glucagonoma, and human immunodeficiency virus.1
Drugs that cause angular cheilitis include isotretinoin, sorafenib (antineoplastic kinase inhibitor), and ointments or creams such as neomycin sulfate–polymyxin B sulfate, bacitracin, idoxuridine, and steroids.1,4
Conditions that mimic angular cheilitis include herpes simplex type 1 (herpes labialis) and actinic cheilitis. Herpes labialis, characterized by burning sensation, itching, or paresthesia, usually precedes a recurrence of vesicles that eventually ulcerate or form a crust and heal without a crust. Herpes labialis often recurs, affecting the vermilion border and lasting approximately 1 week.
Actinic cheilitis, a premalignant condition that commonly involves the lower lip with sparing of the corners of the mouth, is caused by excessive sun exposure. Patients often have persistent dryness and cracking of the lips.
In our patient, angular cheilitis was the main clinical manifestation of iron deficiency anemia, highlighting the importance of looking for iron deficiency in affected patients without a more obvious cause.
A 20-year-old woman had a 4-month history of painful red erosions around the mouth. She had no dysphagia or fatigue and no history of diarrhea, gluten intolerance, or diabetes mellitus. An antifungal-antibacterial ointment prescribed by her dentist had provided no relief.
- Hemoglobin 8.0 g/dL (reference range for females 12.3–15.3)
- Mean corpuscular volume 62 fL (80–100)
- Serum ferritin 1.3 ng/mL (15–200)
- Reticulocyte count 0.86% (0.5–1.5)
- White blood cell count 9.8 × 109/L (4.5–11.0)
- Platelet count 450 × 109/L (150–400).
Vitamin B12 and folate levels were normal, and tests for antitissue transglutaminase and antinuclear antibodies were negative.
Based on these results, the diagnosis was angular cheilitis from iron deficiency anemia. Treatment with oral ferrous gluconate 300 mg twice daily cleared the cheilitis, and after 4 weeks of this treatment, the hemoglobin level increased to 9.8 g/dL, the serum ferritin increased to 7 ng/mL, and the reticulocyte count increased to 2.6%. She was advised to continue taking oral iron tablets for another 3 months until the hemoglobin level reached 12.0 g/dL.
During 2 years of follow-up, she had no recurrence of angular cheilitis, and her hemoglobin and serum ferritin levels remained normal. Ferrous gluconate was her only medication from the time of her diagnosis.
A BROAD DIFFERENTIAL DIAGNOSIS
Angular cheilitis (perlèche) is an inflammatory condition characterized by erosive inflammation at one or both angles of the mouth. It typically presents as erythema, scaling, fissuring, and ulceration. A wide variety of factors, including nutritional deficiencies, local and systemic factors, and drug side effects, may produce cheilitis.1,2
Nutritional deficiencies account for 25% of all cases of angular cheilitis3 and include iron deficiency and deficiencies of the B vitamins riboflavin (B2), niacin (B3), pyridoxine (B6), and cyanocobalamin (B12).1
Local causes include infection with Candida albicans or Staphylococcus aureus and allergic contact dermatitis. Common causes of allergic contact dermatitis include lipstick, toothpaste, mouthwash, cosmetics, sunscreen, fragrance, metals such as nickel, and dental appliances.1
Systemic diseases associated with angular cheilitis include xerostomia, inflammatory bowel disease, Sjögren syndrome, glucagonoma, and human immunodeficiency virus.1
Drugs that cause angular cheilitis include isotretinoin, sorafenib (antineoplastic kinase inhibitor), and ointments or creams such as neomycin sulfate–polymyxin B sulfate, bacitracin, idoxuridine, and steroids.1,4
Conditions that mimic angular cheilitis include herpes simplex type 1 (herpes labialis) and actinic cheilitis. Herpes labialis, characterized by burning sensation, itching, or paresthesia, usually precedes a recurrence of vesicles that eventually ulcerate or form a crust and heal without a crust. Herpes labialis often recurs, affecting the vermilion border and lasting approximately 1 week.
Actinic cheilitis, a premalignant condition that commonly involves the lower lip with sparing of the corners of the mouth, is caused by excessive sun exposure. Patients often have persistent dryness and cracking of the lips.
In our patient, angular cheilitis was the main clinical manifestation of iron deficiency anemia, highlighting the importance of looking for iron deficiency in affected patients without a more obvious cause.
- Park KK, Brodell RT, Helms SE. Angular cheilitis, part 2: nutritional, systemic, and drug-related causes and treatment. Cutis 2011; 88(1):27–32. pmid:21877503
- Park KK, Brodell RT, Helms SE. Angular cheilitis, part 1: local etiologies. Cutis 2011; 87(6):289–295. pmid:21838086
- Konstantinidis AB, Hatziotis JH. Angular cheilosis: an analysis of 156 cases. J Oral Med 1984; 39(4):199–206. pmid:6594458
- Yang CH, Lin WC, Chuang CK, et al. Hand-foot skin reaction in patients treated with sorafenib: a clinicopathological study of cutaneous manifestations due to multitargeted kinase inhibitor therapy. Br J Dermatol 2008; 158(3):592–596. doi:10.1111/j.1365-2133.2007.08357.x
- Park KK, Brodell RT, Helms SE. Angular cheilitis, part 2: nutritional, systemic, and drug-related causes and treatment. Cutis 2011; 88(1):27–32. pmid:21877503
- Park KK, Brodell RT, Helms SE. Angular cheilitis, part 1: local etiologies. Cutis 2011; 87(6):289–295. pmid:21838086
- Konstantinidis AB, Hatziotis JH. Angular cheilosis: an analysis of 156 cases. J Oral Med 1984; 39(4):199–206. pmid:6594458
- Yang CH, Lin WC, Chuang CK, et al. Hand-foot skin reaction in patients treated with sorafenib: a clinicopathological study of cutaneous manifestations due to multitargeted kinase inhibitor therapy. Br J Dermatol 2008; 158(3):592–596. doi:10.1111/j.1365-2133.2007.08357.x
Late mortality risk after childhood BMT is substantial, persistent
Children who undergo allogeneic blood or marrow transplantation (BMT) remain at an elevated risk of premature death even 25 years after the procedure, results of large, retrospective cohort study suggest.
Despite a significant decrease over several decades, the risk of all-cause mortality remained elevated, compared with the general population, according to this study of individuals who had BMT performed in childhood between 1974 and 2010.
“These findings emphasize the need for lifelong follow-up care after allogeneic BMT performed in childhood,” reported Anna Sällfors Holmqvist, MD, PhD, of the department of clinical sciences at Skåne University Hospital, Lund University, Sweden, and her associates.
, Dr. Holmqvist and her colleagues reported in JAMA Oncology.
Their retrospective analysis included 1,388 individuals who lived at least 2 years after allogeneic BMT performed in childhood at one of three centers: the University of Alabama at Birmingham; the University of Minnesota, Minneapolis; and City of Hope, Duarte, Calif.
There were 295 deaths over a median of 14.9 years of follow-up, for an overall survival rate of 79.3% at 20 years after BMT, reported Dr. Holmqvist and her associates. The three leading causes of death were infection or chronic graft-versus-host disease in 49.6% of cases, primary disease in 24.6%, and later malignancies in 18.4%.
Relative to the general population, the cohort had a 14.4-fold increased risk of premature death (95% confidence interval, 12.8-16.1), compared with the general population. Relative mortality was highest 2-5 years after BMT and dropped substantially after that but remained elevated – even 25 years or more after the procedure, the investigators noted.
Mortality decreased significantly over the 3 decades evaluated in this study. The rate of all-cause, 10-year cumulative mortality was 18.9% before 1990, 12.9% from 1990 to 1999, and 11.0% from 2000 to 2010 (P = .002).
That decrease in cumulative mortality over time could not be explained by changes in transplant practice over those three time periods, according to results of a mediation analysis performed by Dr. Holmqvist and her associates.
That finding suggests that unmeasured variables might underlie the decrease in late mortality, the investigators said.
Those unmeasured variables might include supportive care strategies, management of chronic graft-versus-host disease, or improved patient selection, they noted.
Dr. Holmqvist and her associates cited as one limitation their reliance on death certificates for causes of death. In addition, the causes of death for 51 of the 295 deceased patients were lacking.
The study was supported in part by grants from the National Cancer Institute, the Leukemia Lymphoma Society, and the Swedish Childhood Cancer Foundation. Dr. Holmqvist and her associates reported no conflicts of interest.
SOURCE: Holmqvist AS et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2453.
Children who undergo allogeneic blood or marrow transplantation (BMT) remain at an elevated risk of premature death even 25 years after the procedure, results of large, retrospective cohort study suggest.
Despite a significant decrease over several decades, the risk of all-cause mortality remained elevated, compared with the general population, according to this study of individuals who had BMT performed in childhood between 1974 and 2010.
“These findings emphasize the need for lifelong follow-up care after allogeneic BMT performed in childhood,” reported Anna Sällfors Holmqvist, MD, PhD, of the department of clinical sciences at Skåne University Hospital, Lund University, Sweden, and her associates.
, Dr. Holmqvist and her colleagues reported in JAMA Oncology.
Their retrospective analysis included 1,388 individuals who lived at least 2 years after allogeneic BMT performed in childhood at one of three centers: the University of Alabama at Birmingham; the University of Minnesota, Minneapolis; and City of Hope, Duarte, Calif.
There were 295 deaths over a median of 14.9 years of follow-up, for an overall survival rate of 79.3% at 20 years after BMT, reported Dr. Holmqvist and her associates. The three leading causes of death were infection or chronic graft-versus-host disease in 49.6% of cases, primary disease in 24.6%, and later malignancies in 18.4%.
Relative to the general population, the cohort had a 14.4-fold increased risk of premature death (95% confidence interval, 12.8-16.1), compared with the general population. Relative mortality was highest 2-5 years after BMT and dropped substantially after that but remained elevated – even 25 years or more after the procedure, the investigators noted.
Mortality decreased significantly over the 3 decades evaluated in this study. The rate of all-cause, 10-year cumulative mortality was 18.9% before 1990, 12.9% from 1990 to 1999, and 11.0% from 2000 to 2010 (P = .002).
That decrease in cumulative mortality over time could not be explained by changes in transplant practice over those three time periods, according to results of a mediation analysis performed by Dr. Holmqvist and her associates.
That finding suggests that unmeasured variables might underlie the decrease in late mortality, the investigators said.
Those unmeasured variables might include supportive care strategies, management of chronic graft-versus-host disease, or improved patient selection, they noted.
Dr. Holmqvist and her associates cited as one limitation their reliance on death certificates for causes of death. In addition, the causes of death for 51 of the 295 deceased patients were lacking.
The study was supported in part by grants from the National Cancer Institute, the Leukemia Lymphoma Society, and the Swedish Childhood Cancer Foundation. Dr. Holmqvist and her associates reported no conflicts of interest.
SOURCE: Holmqvist AS et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2453.
Children who undergo allogeneic blood or marrow transplantation (BMT) remain at an elevated risk of premature death even 25 years after the procedure, results of large, retrospective cohort study suggest.
Despite a significant decrease over several decades, the risk of all-cause mortality remained elevated, compared with the general population, according to this study of individuals who had BMT performed in childhood between 1974 and 2010.
“These findings emphasize the need for lifelong follow-up care after allogeneic BMT performed in childhood,” reported Anna Sällfors Holmqvist, MD, PhD, of the department of clinical sciences at Skåne University Hospital, Lund University, Sweden, and her associates.
, Dr. Holmqvist and her colleagues reported in JAMA Oncology.
Their retrospective analysis included 1,388 individuals who lived at least 2 years after allogeneic BMT performed in childhood at one of three centers: the University of Alabama at Birmingham; the University of Minnesota, Minneapolis; and City of Hope, Duarte, Calif.
There were 295 deaths over a median of 14.9 years of follow-up, for an overall survival rate of 79.3% at 20 years after BMT, reported Dr. Holmqvist and her associates. The three leading causes of death were infection or chronic graft-versus-host disease in 49.6% of cases, primary disease in 24.6%, and later malignancies in 18.4%.
Relative to the general population, the cohort had a 14.4-fold increased risk of premature death (95% confidence interval, 12.8-16.1), compared with the general population. Relative mortality was highest 2-5 years after BMT and dropped substantially after that but remained elevated – even 25 years or more after the procedure, the investigators noted.
Mortality decreased significantly over the 3 decades evaluated in this study. The rate of all-cause, 10-year cumulative mortality was 18.9% before 1990, 12.9% from 1990 to 1999, and 11.0% from 2000 to 2010 (P = .002).
That decrease in cumulative mortality over time could not be explained by changes in transplant practice over those three time periods, according to results of a mediation analysis performed by Dr. Holmqvist and her associates.
That finding suggests that unmeasured variables might underlie the decrease in late mortality, the investigators said.
Those unmeasured variables might include supportive care strategies, management of chronic graft-versus-host disease, or improved patient selection, they noted.
Dr. Holmqvist and her associates cited as one limitation their reliance on death certificates for causes of death. In addition, the causes of death for 51 of the 295 deceased patients were lacking.
The study was supported in part by grants from the National Cancer Institute, the Leukemia Lymphoma Society, and the Swedish Childhood Cancer Foundation. Dr. Holmqvist and her associates reported no conflicts of interest.
SOURCE: Holmqvist AS et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2453.
FROM JAMA ONCOLOGY
Key clinical point: Individuals undergoing allogeneic blood or marrow transplantation (BMT) in childhood require careful follow-up for many years because of a persistent elevated risk of premature death.
Major finding: Risk of premature death was increased 14.4-fold, compared with the general population (95% confidence interval, 12.8-16.1).
Study details: A retrospective cohort study including 1,388 individuals living 2 years or more after allogeneic BMT performed in childhood.
Disclosures: The study was supported in part by grants from the National Cancer Institute, the Leukemia Lymphoma Society, and the Swedish Childhood Cancer Foundation. Dr. Holmqvist and her coauthors reported no conflicts of interest.
Source: Holmqvist AS et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2453.
Colorectal cancer: New observations, new implications
The incidence and mortality of colorectal cancer (CRC) have declined by 3% per year over the past 10-15 years – a remarkable achievement. The decline in incidence has been dramatic for individuals over age 50 years, who are targeted by screening. However, the reduction in CRC risk does not apply to all populations in the United States. New epidemiologic trends and observations point to patient demographics and regional variation as potential risk factors. While such observations provide what I call “blurry snapshots,” they may well have important implications for our approach to screening and prevention.

What are the reasons? There are several personal and environmental factors that could be contributing. Obesity and metabolic syndrome are risk factors for CRC and have been more commonly developing in childhood over the past 40 years. Alteration of the microbiome could also potentially predispose one to developing CRC. The use of antibiotics in childhood was more common for some of these younger generations than it was for the preceding generations, and antibiotics have been introduced into the food industry to fatten animals. The introduction of food chemicals could also either alter the microbiome and/or promote inflammation, which could lead to neoplasia. Exposure to more ambient radiation may be another risk factor.
These hypotheses are biologically plausible – but untested. Nevertheless, this observational trend does have implications for clinicians. First, studies have shown that up to 20% of CRCs before age 50 years are associated with germline mutations, while others are associated with a family history of CRC. Therefore, it is important to capture and update family history. In addition, there is evidence that individuals aged 40-49 years with rectal bleeding have a higher risk of advanced adenomas, so our threshold for performing diagnostic colonoscopy should be lowered. African Americans also have a higher risk of CRC at a younger age than do other racial groups and might benefit from early screening at age 45 years. Notably, recent recommendations from the American Cancer Society call for consideration of screening everyone at age 45 years.
There is substantial state-to-state and county-to-county variation in the incidence and mortality of CRC. While some of this variation can be explained by racial variation, smoking, obesity, and social determinants of health, there are “hot-spots” that may defy easy explanation. There has been very little research about environmental factors (air, water, and ambient radiation). Two regions at particularly high risk are the Mississippi Delta region and Appalachia – areas where water pollution could be a factor. The substantial county-to-county variation within these high-risk areas points to a potential environmental culprit, but further research is needed.
For the GI community, there are several implications to be found in these changing demographics and risks. For one, we may need to consider expanding our risk concepts to include not only genetic and personal risk factors but also environmental factors. To mitigate risk, providers and public health officials may need to then target these high-risk areas for more intensive screening efforts.
Dr. Lieberman is a professor of medicine and chief of gastroenterology and hepatology at Oregon Health and Science University in Portland. He has no conflicts of interest. Dr. Lieberman made his comments during the AGA Institute Presidential Plenary at the Annual Digestive Disease Week.
The incidence and mortality of colorectal cancer (CRC) have declined by 3% per year over the past 10-15 years – a remarkable achievement. The decline in incidence has been dramatic for individuals over age 50 years, who are targeted by screening. However, the reduction in CRC risk does not apply to all populations in the United States. New epidemiologic trends and observations point to patient demographics and regional variation as potential risk factors. While such observations provide what I call “blurry snapshots,” they may well have important implications for our approach to screening and prevention.
What are the reasons? There are several personal and environmental factors that could be contributing. Obesity and metabolic syndrome are risk factors for CRC and have been more commonly developing in childhood over the past 40 years. Alteration of the microbiome could also potentially predispose one to developing CRC. The use of antibiotics in childhood was more common for some of these younger generations than it was for the preceding generations, and antibiotics have been introduced into the food industry to fatten animals. The introduction of food chemicals could also either alter the microbiome and/or promote inflammation, which could lead to neoplasia. Exposure to more ambient radiation may be another risk factor.
These hypotheses are biologically plausible – but untested. Nevertheless, this observational trend does have implications for clinicians. First, studies have shown that up to 20% of CRCs before age 50 years are associated with germline mutations, while others are associated with a family history of CRC. Therefore, it is important to capture and update family history. In addition, there is evidence that individuals aged 40-49 years with rectal bleeding have a higher risk of advanced adenomas, so our threshold for performing diagnostic colonoscopy should be lowered. African Americans also have a higher risk of CRC at a younger age than do other racial groups and might benefit from early screening at age 45 years. Notably, recent recommendations from the American Cancer Society call for consideration of screening everyone at age 45 years.
There is substantial state-to-state and county-to-county variation in the incidence and mortality of CRC. While some of this variation can be explained by racial variation, smoking, obesity, and social determinants of health, there are “hot-spots” that may defy easy explanation. There has been very little research about environmental factors (air, water, and ambient radiation). Two regions at particularly high risk are the Mississippi Delta region and Appalachia – areas where water pollution could be a factor. The substantial county-to-county variation within these high-risk areas points to a potential environmental culprit, but further research is needed.
For the GI community, there are several implications to be found in these changing demographics and risks. For one, we may need to consider expanding our risk concepts to include not only genetic and personal risk factors but also environmental factors. To mitigate risk, providers and public health officials may need to then target these high-risk areas for more intensive screening efforts.
Dr. Lieberman is a professor of medicine and chief of gastroenterology and hepatology at Oregon Health and Science University in Portland. He has no conflicts of interest. Dr. Lieberman made his comments during the AGA Institute Presidential Plenary at the Annual Digestive Disease Week.
The incidence and mortality of colorectal cancer (CRC) have declined by 3% per year over the past 10-15 years – a remarkable achievement. The decline in incidence has been dramatic for individuals over age 50 years, who are targeted by screening. However, the reduction in CRC risk does not apply to all populations in the United States. New epidemiologic trends and observations point to patient demographics and regional variation as potential risk factors. While such observations provide what I call “blurry snapshots,” they may well have important implications for our approach to screening and prevention.
What are the reasons? There are several personal and environmental factors that could be contributing. Obesity and metabolic syndrome are risk factors for CRC and have been more commonly developing in childhood over the past 40 years. Alteration of the microbiome could also potentially predispose one to developing CRC. The use of antibiotics in childhood was more common for some of these younger generations than it was for the preceding generations, and antibiotics have been introduced into the food industry to fatten animals. The introduction of food chemicals could also either alter the microbiome and/or promote inflammation, which could lead to neoplasia. Exposure to more ambient radiation may be another risk factor.
These hypotheses are biologically plausible – but untested. Nevertheless, this observational trend does have implications for clinicians. First, studies have shown that up to 20% of CRCs before age 50 years are associated with germline mutations, while others are associated with a family history of CRC. Therefore, it is important to capture and update family history. In addition, there is evidence that individuals aged 40-49 years with rectal bleeding have a higher risk of advanced adenomas, so our threshold for performing diagnostic colonoscopy should be lowered. African Americans also have a higher risk of CRC at a younger age than do other racial groups and might benefit from early screening at age 45 years. Notably, recent recommendations from the American Cancer Society call for consideration of screening everyone at age 45 years.
There is substantial state-to-state and county-to-county variation in the incidence and mortality of CRC. While some of this variation can be explained by racial variation, smoking, obesity, and social determinants of health, there are “hot-spots” that may defy easy explanation. There has been very little research about environmental factors (air, water, and ambient radiation). Two regions at particularly high risk are the Mississippi Delta region and Appalachia – areas where water pollution could be a factor. The substantial county-to-county variation within these high-risk areas points to a potential environmental culprit, but further research is needed.
For the GI community, there are several implications to be found in these changing demographics and risks. For one, we may need to consider expanding our risk concepts to include not only genetic and personal risk factors but also environmental factors. To mitigate risk, providers and public health officials may need to then target these high-risk areas for more intensive screening efforts.
Dr. Lieberman is a professor of medicine and chief of gastroenterology and hepatology at Oregon Health and Science University in Portland. He has no conflicts of interest. Dr. Lieberman made his comments during the AGA Institute Presidential Plenary at the Annual Digestive Disease Week.
In Ghana, SCD research is meeting patients on home turf
WASHINGTON – Sometimes, the hardest part of solving a problem is figuring out how to work around misaligned resources, and so it has been with sickle cell disease research.

“From my point of view, what I call the geographical disparity in sickle cell disease research can be explained by the fact that the majority of affected individuals are living in the East, and the overwhelming majority of the research takes place in the West,” said Solomon Ofori-Acquah, PhD. He and three physician collaborators from Ghana shared their roadmap to conducting clinical trials in West Africa during an “East Meets West” session of the annual symposium of the Foundation for Sickle Cell Disease Research.
In Ghana, not far from where scientists now believe the hemoglobin sickling mutation originated, fully 2% of newborns have SCD; this translates into 16,000 new cases per year in a population of just 28 million, compared with the 2,000 new SCD cases seen annually in the entire United States. And access even to proven therapies can be limited; historically, little to no clinical drug development work has been conducted in this part of the world.
In the United States, half of the SCD trials that were withdrawn or terminated listed recruitment and retention of study participants as a factor in the study’s discontinuation, said Amma Owusu-Ansah, MD. “I see what we are doing as a very feasible solution to the problem of inadequate accrual to studies in the U.S.,” said Dr. Owusu-Ansah, a hematologist at the University of Pittsburgh’s Center for Translational and International Hematology (CTIH), where she serves as clinical director.
From the African perspective, hosting clinical trials – and building a robust infrastructure to do so – may help alleviate the delay in translation of disease-modifying therapies for SCD to Africa, where most people with the disease live, she said.
An existing example of resource sharing is the Human Heredity & Health in Africa (H3Africa) initiative, said Dr. Ofori-Acquah, who directs the CTIH and also holds an appointment at the University of Ghana. The project, funded by the National Institutes of Health and the Wellcome Trust, “aims to facilitate a contemporary research approach to the study of genomics and environmental determinants of common diseases with the goal of improving the health of African populations,” according to the H3Africa website. Within this framework of 40 research centers conducting genomics research and biobanking, several discrete projects aim to expand knowledge of sickle cell disease.
“All of these networks are going to study thousands of patients,” said Dr. Ofori-Acquah. “I see the H3 as a mechanism to accelerate genomics research in sickle cell disease.”

“We created a research team and built capacity for future work…. Ghana, and Africa, are capable of conducting clinical trials to global standards and producing quality data,” she said.
The story of one clinical trial is illustrative of the challenges and strengths of the multinational approach.
The phase 1b trial of a novel treatment for sickle cell disease, NVX-508, began with an initial hurdle of lack of access to emergency care at the study site, said Dr. Owusu-Ansah, a study investigator. Her first reaction, she said, was, “Well, we can’t do this, because we don’t have access to a big staff and emergency facilities.”
But after consulting with colleagues, she realized a shift in mindset was needed: “Rather than focus on what we don’t have, what do we actually have available? We have relationships we have built with institutions,” including the oldest SCD clinic in Ghana, the Ghana Institute of Clinical Genetics (GICG). This facility sits next door to a hospital with 24-hour care, Korle Bu Teaching Hospital (KBTH), a major tertiary care and referral center.
Open since 1974, the KBTH-allied GICG provides comprehensive outpatient health care to teens and adult with SCD. Currently, more than 25,000 SCD patients are registered at GICG; about half have the HbSS genotype, and another 40% have the HbSC genotype, said Yvonne Dei-Adomakoh, MD. Dr. Dei-Adomakoh of the University of Ghana is an investigator for an upcoming phase 3 trial to test voxelotor against placebo in SCD.
The GICG is working hard to become a site where clinical trials, as well as research and development, are embedded into clinic functions. In this way, not only will research be advanced for all those with SCD, but advances will be more easily incorporated into clinical care, said Dr. Dei-Adomakoh.
Dr. Owusu-Ansah noted that the facility offers a pharmacy, a laboratory, exam rooms, and information technology and medical record resources. Importantly, GICG is already staffed with physicians and allied health personnel with SCD expertise.
The University of Ghana campus is home to one of Africa’s leading biomedical research facilities, a sophisticated 11,000-square-foot laboratory that can perform testing ranging from polymerase chain reactions to DNA sequencing to genotyping and flow cytometry; it also houses a laboratory animal facility. This laboratory, the Noguchi Memorial Institute for Medical Research, also offers administrative, scientific, and research support, and houses an institutional review board.
The problem of the Noguchi laboratory site’s distance from the 24-hour support of KBTH has been solved by arranging to have an ambulance with paramedics available on site during the clinical trials.
Some other challenges the investigators discovered highlighted less-obvious infrastructure deficits; keeping a refrigerated chain of custody for biological samples, for example, can be difficult. In preparation for the trials, much basic laboratory and clinical equipment has been updated.
Conducting a U.S.-registered clinical trial in Ghana doesn’t obviate the need to meet that country’s considerable regulatory hurdles, said Dr. Owusu-Ansah. Requirements include a full regulatory submission to, and physical inspection by, Ghana’s FDA. Ghana also requires that the principal investigator must live in Ghana for the duration of the trial and that key study personnel complete Ghanaian good clinical practices training, she said.
The University of Pittsburgh is a U.S. partner in the NVX-508 study, and it was non-negotiable for that institution that a clinical trial monitor visit the African study sites. The institution’s institutional review board was sensitive to the importance of protecting vulnerable populations, and needed to hear complete plans for risk assessment, data protection, and compensation for Ghanaian study participants, Dr. Owusu-Ansah said.
But, in a turn of events typical of the ups and downs of drug development, the phase 1 trial had passed most of the administrative hurdles when in July the drug’s sponsor, NuvOx Pharma, suspended the NVX-508 trial to focus on other areas. For now, the trial registration has been withdrawn on clinicaltrials.gov and the new drug application is inactive. But Dr. Owusu-Ansah said study preparations could resume in the future, if the drug is made available to investigators.
Dr. Owusu-Ansah reported that she has received salary support from NuvOx Pharma. Dr. Segbefia reported that she has received support from Daiichi-Sankyo and Eli Lilly and Company.
WASHINGTON – Sometimes, the hardest part of solving a problem is figuring out how to work around misaligned resources, and so it has been with sickle cell disease research.
“From my point of view, what I call the geographical disparity in sickle cell disease research can be explained by the fact that the majority of affected individuals are living in the East, and the overwhelming majority of the research takes place in the West,” said Solomon Ofori-Acquah, PhD. He and three physician collaborators from Ghana shared their roadmap to conducting clinical trials in West Africa during an “East Meets West” session of the annual symposium of the Foundation for Sickle Cell Disease Research.
In Ghana, not far from where scientists now believe the hemoglobin sickling mutation originated, fully 2% of newborns have SCD; this translates into 16,000 new cases per year in a population of just 28 million, compared with the 2,000 new SCD cases seen annually in the entire United States. And access even to proven therapies can be limited; historically, little to no clinical drug development work has been conducted in this part of the world.
In the United States, half of the SCD trials that were withdrawn or terminated listed recruitment and retention of study participants as a factor in the study’s discontinuation, said Amma Owusu-Ansah, MD. “I see what we are doing as a very feasible solution to the problem of inadequate accrual to studies in the U.S.,” said Dr. Owusu-Ansah, a hematologist at the University of Pittsburgh’s Center for Translational and International Hematology (CTIH), where she serves as clinical director.
From the African perspective, hosting clinical trials – and building a robust infrastructure to do so – may help alleviate the delay in translation of disease-modifying therapies for SCD to Africa, where most people with the disease live, she said.
An existing example of resource sharing is the Human Heredity & Health in Africa (H3Africa) initiative, said Dr. Ofori-Acquah, who directs the CTIH and also holds an appointment at the University of Ghana. The project, funded by the National Institutes of Health and the Wellcome Trust, “aims to facilitate a contemporary research approach to the study of genomics and environmental determinants of common diseases with the goal of improving the health of African populations,” according to the H3Africa website. Within this framework of 40 research centers conducting genomics research and biobanking, several discrete projects aim to expand knowledge of sickle cell disease.
“All of these networks are going to study thousands of patients,” said Dr. Ofori-Acquah. “I see the H3 as a mechanism to accelerate genomics research in sickle cell disease.”
“We created a research team and built capacity for future work…. Ghana, and Africa, are capable of conducting clinical trials to global standards and producing quality data,” she said.
The story of one clinical trial is illustrative of the challenges and strengths of the multinational approach.
The phase 1b trial of a novel treatment for sickle cell disease, NVX-508, began with an initial hurdle of lack of access to emergency care at the study site, said Dr. Owusu-Ansah, a study investigator. Her first reaction, she said, was, “Well, we can’t do this, because we don’t have access to a big staff and emergency facilities.”
But after consulting with colleagues, she realized a shift in mindset was needed: “Rather than focus on what we don’t have, what do we actually have available? We have relationships we have built with institutions,” including the oldest SCD clinic in Ghana, the Ghana Institute of Clinical Genetics (GICG). This facility sits next door to a hospital with 24-hour care, Korle Bu Teaching Hospital (KBTH), a major tertiary care and referral center.
Open since 1974, the KBTH-allied GICG provides comprehensive outpatient health care to teens and adult with SCD. Currently, more than 25,000 SCD patients are registered at GICG; about half have the HbSS genotype, and another 40% have the HbSC genotype, said Yvonne Dei-Adomakoh, MD. Dr. Dei-Adomakoh of the University of Ghana is an investigator for an upcoming phase 3 trial to test voxelotor against placebo in SCD.
The GICG is working hard to become a site where clinical trials, as well as research and development, are embedded into clinic functions. In this way, not only will research be advanced for all those with SCD, but advances will be more easily incorporated into clinical care, said Dr. Dei-Adomakoh.
Dr. Owusu-Ansah noted that the facility offers a pharmacy, a laboratory, exam rooms, and information technology and medical record resources. Importantly, GICG is already staffed with physicians and allied health personnel with SCD expertise.
The University of Ghana campus is home to one of Africa’s leading biomedical research facilities, a sophisticated 11,000-square-foot laboratory that can perform testing ranging from polymerase chain reactions to DNA sequencing to genotyping and flow cytometry; it also houses a laboratory animal facility. This laboratory, the Noguchi Memorial Institute for Medical Research, also offers administrative, scientific, and research support, and houses an institutional review board.
The problem of the Noguchi laboratory site’s distance from the 24-hour support of KBTH has been solved by arranging to have an ambulance with paramedics available on site during the clinical trials.
Some other challenges the investigators discovered highlighted less-obvious infrastructure deficits; keeping a refrigerated chain of custody for biological samples, for example, can be difficult. In preparation for the trials, much basic laboratory and clinical equipment has been updated.
Conducting a U.S.-registered clinical trial in Ghana doesn’t obviate the need to meet that country’s considerable regulatory hurdles, said Dr. Owusu-Ansah. Requirements include a full regulatory submission to, and physical inspection by, Ghana’s FDA. Ghana also requires that the principal investigator must live in Ghana for the duration of the trial and that key study personnel complete Ghanaian good clinical practices training, she said.
The University of Pittsburgh is a U.S. partner in the NVX-508 study, and it was non-negotiable for that institution that a clinical trial monitor visit the African study sites. The institution’s institutional review board was sensitive to the importance of protecting vulnerable populations, and needed to hear complete plans for risk assessment, data protection, and compensation for Ghanaian study participants, Dr. Owusu-Ansah said.
But, in a turn of events typical of the ups and downs of drug development, the phase 1 trial had passed most of the administrative hurdles when in July the drug’s sponsor, NuvOx Pharma, suspended the NVX-508 trial to focus on other areas. For now, the trial registration has been withdrawn on clinicaltrials.gov and the new drug application is inactive. But Dr. Owusu-Ansah said study preparations could resume in the future, if the drug is made available to investigators.
Dr. Owusu-Ansah reported that she has received salary support from NuvOx Pharma. Dr. Segbefia reported that she has received support from Daiichi-Sankyo and Eli Lilly and Company.
WASHINGTON – Sometimes, the hardest part of solving a problem is figuring out how to work around misaligned resources, and so it has been with sickle cell disease research.
“From my point of view, what I call the geographical disparity in sickle cell disease research can be explained by the fact that the majority of affected individuals are living in the East, and the overwhelming majority of the research takes place in the West,” said Solomon Ofori-Acquah, PhD. He and three physician collaborators from Ghana shared their roadmap to conducting clinical trials in West Africa during an “East Meets West” session of the annual symposium of the Foundation for Sickle Cell Disease Research.
In Ghana, not far from where scientists now believe the hemoglobin sickling mutation originated, fully 2% of newborns have SCD; this translates into 16,000 new cases per year in a population of just 28 million, compared with the 2,000 new SCD cases seen annually in the entire United States. And access even to proven therapies can be limited; historically, little to no clinical drug development work has been conducted in this part of the world.
In the United States, half of the SCD trials that were withdrawn or terminated listed recruitment and retention of study participants as a factor in the study’s discontinuation, said Amma Owusu-Ansah, MD. “I see what we are doing as a very feasible solution to the problem of inadequate accrual to studies in the U.S.,” said Dr. Owusu-Ansah, a hematologist at the University of Pittsburgh’s Center for Translational and International Hematology (CTIH), where she serves as clinical director.
From the African perspective, hosting clinical trials – and building a robust infrastructure to do so – may help alleviate the delay in translation of disease-modifying therapies for SCD to Africa, where most people with the disease live, she said.
An existing example of resource sharing is the Human Heredity & Health in Africa (H3Africa) initiative, said Dr. Ofori-Acquah, who directs the CTIH and also holds an appointment at the University of Ghana. The project, funded by the National Institutes of Health and the Wellcome Trust, “aims to facilitate a contemporary research approach to the study of genomics and environmental determinants of common diseases with the goal of improving the health of African populations,” according to the H3Africa website. Within this framework of 40 research centers conducting genomics research and biobanking, several discrete projects aim to expand knowledge of sickle cell disease.
“All of these networks are going to study thousands of patients,” said Dr. Ofori-Acquah. “I see the H3 as a mechanism to accelerate genomics research in sickle cell disease.”
“We created a research team and built capacity for future work…. Ghana, and Africa, are capable of conducting clinical trials to global standards and producing quality data,” she said.
The story of one clinical trial is illustrative of the challenges and strengths of the multinational approach.
The phase 1b trial of a novel treatment for sickle cell disease, NVX-508, began with an initial hurdle of lack of access to emergency care at the study site, said Dr. Owusu-Ansah, a study investigator. Her first reaction, she said, was, “Well, we can’t do this, because we don’t have access to a big staff and emergency facilities.”
But after consulting with colleagues, she realized a shift in mindset was needed: “Rather than focus on what we don’t have, what do we actually have available? We have relationships we have built with institutions,” including the oldest SCD clinic in Ghana, the Ghana Institute of Clinical Genetics (GICG). This facility sits next door to a hospital with 24-hour care, Korle Bu Teaching Hospital (KBTH), a major tertiary care and referral center.
Open since 1974, the KBTH-allied GICG provides comprehensive outpatient health care to teens and adult with SCD. Currently, more than 25,000 SCD patients are registered at GICG; about half have the HbSS genotype, and another 40% have the HbSC genotype, said Yvonne Dei-Adomakoh, MD. Dr. Dei-Adomakoh of the University of Ghana is an investigator for an upcoming phase 3 trial to test voxelotor against placebo in SCD.
The GICG is working hard to become a site where clinical trials, as well as research and development, are embedded into clinic functions. In this way, not only will research be advanced for all those with SCD, but advances will be more easily incorporated into clinical care, said Dr. Dei-Adomakoh.
Dr. Owusu-Ansah noted that the facility offers a pharmacy, a laboratory, exam rooms, and information technology and medical record resources. Importantly, GICG is already staffed with physicians and allied health personnel with SCD expertise.
The University of Ghana campus is home to one of Africa’s leading biomedical research facilities, a sophisticated 11,000-square-foot laboratory that can perform testing ranging from polymerase chain reactions to DNA sequencing to genotyping and flow cytometry; it also houses a laboratory animal facility. This laboratory, the Noguchi Memorial Institute for Medical Research, also offers administrative, scientific, and research support, and houses an institutional review board.
The problem of the Noguchi laboratory site’s distance from the 24-hour support of KBTH has been solved by arranging to have an ambulance with paramedics available on site during the clinical trials.
Some other challenges the investigators discovered highlighted less-obvious infrastructure deficits; keeping a refrigerated chain of custody for biological samples, for example, can be difficult. In preparation for the trials, much basic laboratory and clinical equipment has been updated.
Conducting a U.S.-registered clinical trial in Ghana doesn’t obviate the need to meet that country’s considerable regulatory hurdles, said Dr. Owusu-Ansah. Requirements include a full regulatory submission to, and physical inspection by, Ghana’s FDA. Ghana also requires that the principal investigator must live in Ghana for the duration of the trial and that key study personnel complete Ghanaian good clinical practices training, she said.
The University of Pittsburgh is a U.S. partner in the NVX-508 study, and it was non-negotiable for that institution that a clinical trial monitor visit the African study sites. The institution’s institutional review board was sensitive to the importance of protecting vulnerable populations, and needed to hear complete plans for risk assessment, data protection, and compensation for Ghanaian study participants, Dr. Owusu-Ansah said.
But, in a turn of events typical of the ups and downs of drug development, the phase 1 trial had passed most of the administrative hurdles when in July the drug’s sponsor, NuvOx Pharma, suspended the NVX-508 trial to focus on other areas. For now, the trial registration has been withdrawn on clinicaltrials.gov and the new drug application is inactive. But Dr. Owusu-Ansah said study preparations could resume in the future, if the drug is made available to investigators.
Dr. Owusu-Ansah reported that she has received salary support from NuvOx Pharma. Dr. Segbefia reported that she has received support from Daiichi-Sankyo and Eli Lilly and Company.
EXPERT ANALYSIS FROM FSCDR 2018
Former Smokers Motivate Quitters
In 2012, the CDC launched the “Tips from Former Smokers” campaign. It was memorable and emotionally forceful—one woman who had oral and throat cancer delivered her ad through an artificial voicebox—but did it have an impact on actual quitting rates?
No study had been done to assess the campaign’s combined, multiyear impact until CDC researchers looked at sustained (6 month) cigarette abstinence during the first 4 years of the campaign (2012-2015).
They found that the Tips campaign led to about 9.15 million total quit attempts. Based on an assumed 5.7% abstinence rate for people attempting to quit, this amounts to approximately 522,000 sustained quits.
The researchers say their findings indicate that the comprehensive approach combining evidence-based messages with the promotion of cessation resources was highly successful. Their finding of more than half-million sustained quits underscores the critical role of national tobacco education campaigns as a “counterpoint” to the substantial pro-tobacco advertising and promotion.
In 2012, the CDC launched the “Tips from Former Smokers” campaign. It was memorable and emotionally forceful—one woman who had oral and throat cancer delivered her ad through an artificial voicebox—but did it have an impact on actual quitting rates?
No study had been done to assess the campaign’s combined, multiyear impact until CDC researchers looked at sustained (6 month) cigarette abstinence during the first 4 years of the campaign (2012-2015).
They found that the Tips campaign led to about 9.15 million total quit attempts. Based on an assumed 5.7% abstinence rate for people attempting to quit, this amounts to approximately 522,000 sustained quits.
The researchers say their findings indicate that the comprehensive approach combining evidence-based messages with the promotion of cessation resources was highly successful. Their finding of more than half-million sustained quits underscores the critical role of national tobacco education campaigns as a “counterpoint” to the substantial pro-tobacco advertising and promotion.
In 2012, the CDC launched the “Tips from Former Smokers” campaign. It was memorable and emotionally forceful—one woman who had oral and throat cancer delivered her ad through an artificial voicebox—but did it have an impact on actual quitting rates?
No study had been done to assess the campaign’s combined, multiyear impact until CDC researchers looked at sustained (6 month) cigarette abstinence during the first 4 years of the campaign (2012-2015).
They found that the Tips campaign led to about 9.15 million total quit attempts. Based on an assumed 5.7% abstinence rate for people attempting to quit, this amounts to approximately 522,000 sustained quits.
The researchers say their findings indicate that the comprehensive approach combining evidence-based messages with the promotion of cessation resources was highly successful. Their finding of more than half-million sustained quits underscores the critical role of national tobacco education campaigns as a “counterpoint” to the substantial pro-tobacco advertising and promotion.
Hematology and Oncology Federal Health Data Trends (FULL)
Cancer research is a high priority for the DoD and especially for the VA. Researchers in both agencies played an important role in the early stages of the Cancer Moonshot. As part of this initiative, the VA, DoD, and National Cancer Institute joined forces in the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) project to develop a system to quickly identify unique targets and pathways of cancer for better interventions.
The VA also will provide access to the Million Veteran Program database, and > 20 years of electronic health records data for analysis using the U.S. Department of Energy’s advanced computer systems. The enhanced computational infrastructure provided by the departments will facilitate new studies of cancer genomics. The research will begin with prostate cancer, and it is hoped that the project will help researchers distinguish between those prostate cancers that require aggressive management and the more benign cancers that are less likely to progress.
According to the latest VA budget, its researchers are conducting a broad array of research on cancers common in the veteran population, including prostate, lung, colorectal, bladder, kidney, pancreatic, skin, esophageal, and femalespecific cancers (such as breast and cervical cancer), as well as lymphomas and melanomas. For example, one study is focused on improving palliative care for patients with advanced cancer, and another will enroll 50,000 veterans to compare colorectal cancer screening strategies.
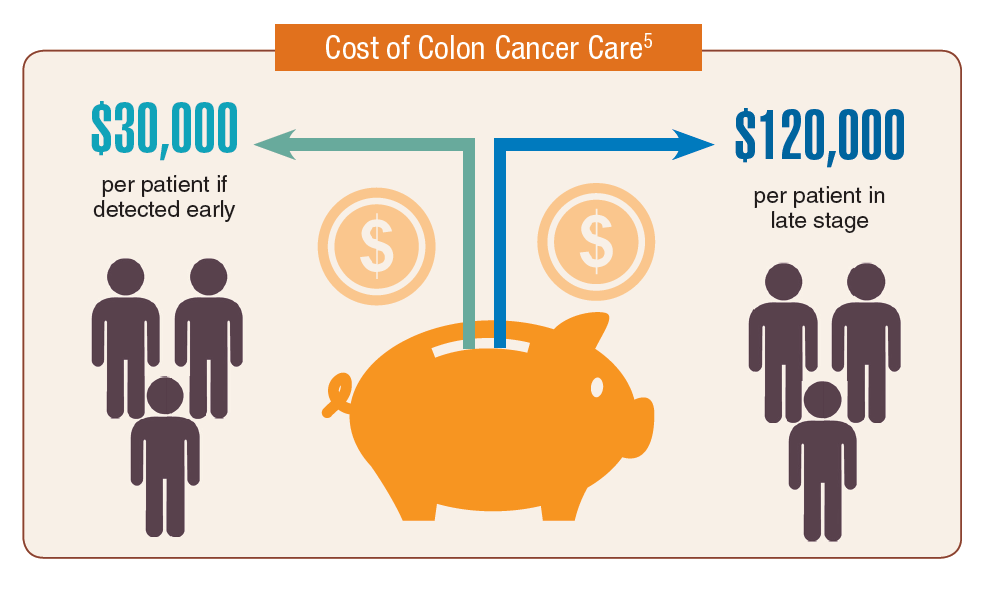
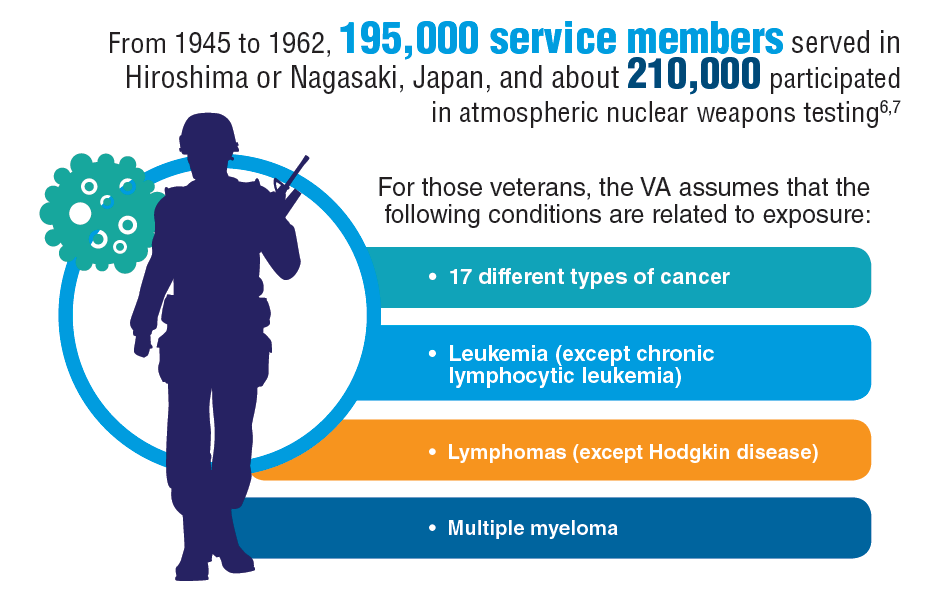
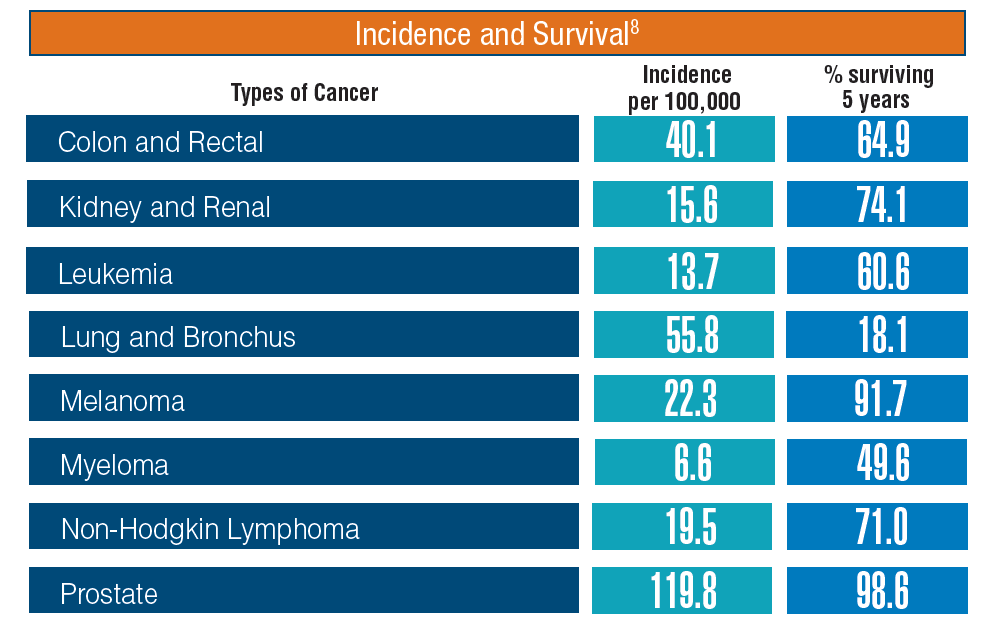
Click here to read the digital edition.
Cancer research is a high priority for the DoD and especially for the VA. Researchers in both agencies played an important role in the early stages of the Cancer Moonshot. As part of this initiative, the VA, DoD, and National Cancer Institute joined forces in the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) project to develop a system to quickly identify unique targets and pathways of cancer for better interventions.
The VA also will provide access to the Million Veteran Program database, and > 20 years of electronic health records data for analysis using the U.S. Department of Energy’s advanced computer systems. The enhanced computational infrastructure provided by the departments will facilitate new studies of cancer genomics. The research will begin with prostate cancer, and it is hoped that the project will help researchers distinguish between those prostate cancers that require aggressive management and the more benign cancers that are less likely to progress.
According to the latest VA budget, its researchers are conducting a broad array of research on cancers common in the veteran population, including prostate, lung, colorectal, bladder, kidney, pancreatic, skin, esophageal, and femalespecific cancers (such as breast and cervical cancer), as well as lymphomas and melanomas. For example, one study is focused on improving palliative care for patients with advanced cancer, and another will enroll 50,000 veterans to compare colorectal cancer screening strategies.
Click here to read the digital edition.
Cancer research is a high priority for the DoD and especially for the VA. Researchers in both agencies played an important role in the early stages of the Cancer Moonshot. As part of this initiative, the VA, DoD, and National Cancer Institute joined forces in the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) project to develop a system to quickly identify unique targets and pathways of cancer for better interventions.
The VA also will provide access to the Million Veteran Program database, and > 20 years of electronic health records data for analysis using the U.S. Department of Energy’s advanced computer systems. The enhanced computational infrastructure provided by the departments will facilitate new studies of cancer genomics. The research will begin with prostate cancer, and it is hoped that the project will help researchers distinguish between those prostate cancers that require aggressive management and the more benign cancers that are less likely to progress.
According to the latest VA budget, its researchers are conducting a broad array of research on cancers common in the veteran population, including prostate, lung, colorectal, bladder, kidney, pancreatic, skin, esophageal, and femalespecific cancers (such as breast and cervical cancer), as well as lymphomas and melanomas. For example, one study is focused on improving palliative care for patients with advanced cancer, and another will enroll 50,000 veterans to compare colorectal cancer screening strategies.