User login
FDA approves Nivestym, second biosimilar to Neupogen
Nivestym (filgrastim-aafi), a biosimilar to Neupogen (filgrastim) was approved July 20 by the Food and Drug Administration, according to a statement provided by the agency. Nivestym is the second biosimilar to Neupogen to be approved in the United States.

- Patients with cancer receiving myelosuppressive chemotherapy.
- Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy.
- Patients with cancer undergoing bone marrow transplantation.
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy.
- Patients with severe chronic neutropenia.
According to a press release from Pfizer, the manufacturer of the biosimilar, Nivestym is expected to be available in the United States at a significant discount to the current wholesale acquisition cost of Neupogen, which is not inclusive of discounts to payers, providers, distributors, and other purchasing organizations.
The FDA statement notes that a biosimilar is approved based on a showing that it is highly similar to an already approved biologic product, known as a reference product. The biosimilar also must be shown to have no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Prescribing information is available here.
Nivestym (filgrastim-aafi), a biosimilar to Neupogen (filgrastim) was approved July 20 by the Food and Drug Administration, according to a statement provided by the agency. Nivestym is the second biosimilar to Neupogen to be approved in the United States.
- Patients with cancer receiving myelosuppressive chemotherapy.
- Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy.
- Patients with cancer undergoing bone marrow transplantation.
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy.
- Patients with severe chronic neutropenia.
According to a press release from Pfizer, the manufacturer of the biosimilar, Nivestym is expected to be available in the United States at a significant discount to the current wholesale acquisition cost of Neupogen, which is not inclusive of discounts to payers, providers, distributors, and other purchasing organizations.
The FDA statement notes that a biosimilar is approved based on a showing that it is highly similar to an already approved biologic product, known as a reference product. The biosimilar also must be shown to have no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Prescribing information is available here.
Nivestym (filgrastim-aafi), a biosimilar to Neupogen (filgrastim) was approved July 20 by the Food and Drug Administration, according to a statement provided by the agency. Nivestym is the second biosimilar to Neupogen to be approved in the United States.
- Patients with cancer receiving myelosuppressive chemotherapy.
- Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy.
- Patients with cancer undergoing bone marrow transplantation.
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy.
- Patients with severe chronic neutropenia.
According to a press release from Pfizer, the manufacturer of the biosimilar, Nivestym is expected to be available in the United States at a significant discount to the current wholesale acquisition cost of Neupogen, which is not inclusive of discounts to payers, providers, distributors, and other purchasing organizations.
The FDA statement notes that a biosimilar is approved based on a showing that it is highly similar to an already approved biologic product, known as a reference product. The biosimilar also must be shown to have no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Prescribing information is available here.
FDA approves IDH1 inhibitor for relapsed/refractory AML
The Food and Drug Administration has approved ivosidenib (Tibsovo) as the first treatment of adult patients with relapsed/refractory acute myeloid leukemia (AML) and an isocitrate dehydrogenase-1 (IDH1) mutation.
More specifically, the oral treatment has been approved for patients whose mutations have been identified by the Abbott RealTime IDH1 assay, a companion diagnostic test.
The approval was based on results from a phase 1, open-label, single-arm, multicenter, dose-escalation and expansion trial of adult patients in this AML population. The primary end point was combined complete remission and complete remission with partial hematologic improvement; this combined rate was 32.8%, and the median duration of this remission was 8.2 months.
The most serious adverse events included differentiation syndrome, QTc prolongation, and Guillain-Barré syndrome. Other adverse reactions included fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, and constipation.
Ivosidenib is marketed as Tibsovo by Agios Pharmaceuticals. The RealTime IDH1 Assay is marketed by Abbott Laboratories.
The Food and Drug Administration has approved ivosidenib (Tibsovo) as the first treatment of adult patients with relapsed/refractory acute myeloid leukemia (AML) and an isocitrate dehydrogenase-1 (IDH1) mutation.
More specifically, the oral treatment has been approved for patients whose mutations have been identified by the Abbott RealTime IDH1 assay, a companion diagnostic test.
The approval was based on results from a phase 1, open-label, single-arm, multicenter, dose-escalation and expansion trial of adult patients in this AML population. The primary end point was combined complete remission and complete remission with partial hematologic improvement; this combined rate was 32.8%, and the median duration of this remission was 8.2 months.
The most serious adverse events included differentiation syndrome, QTc prolongation, and Guillain-Barré syndrome. Other adverse reactions included fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, and constipation.
Ivosidenib is marketed as Tibsovo by Agios Pharmaceuticals. The RealTime IDH1 Assay is marketed by Abbott Laboratories.
The Food and Drug Administration has approved ivosidenib (Tibsovo) as the first treatment of adult patients with relapsed/refractory acute myeloid leukemia (AML) and an isocitrate dehydrogenase-1 (IDH1) mutation.
More specifically, the oral treatment has been approved for patients whose mutations have been identified by the Abbott RealTime IDH1 assay, a companion diagnostic test.
The approval was based on results from a phase 1, open-label, single-arm, multicenter, dose-escalation and expansion trial of adult patients in this AML population. The primary end point was combined complete remission and complete remission with partial hematologic improvement; this combined rate was 32.8%, and the median duration of this remission was 8.2 months.
The most serious adverse events included differentiation syndrome, QTc prolongation, and Guillain-Barré syndrome. Other adverse reactions included fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, and constipation.
Ivosidenib is marketed as Tibsovo by Agios Pharmaceuticals. The RealTime IDH1 Assay is marketed by Abbott Laboratories.
Phase 3 results shed light on L-glutamine use in SCD
Children and adults with sickle cell disease who received L-glutamine alone or with hydroxyurea had a median number of pain episodes that was 25% lower than those who received placebo, according to newly published results from the phase 3 trial that led to the agent’s approval in 2017.
The median number of hospitalizations was 33% lower among individuals receiving L-glutamine than it was among those receiving placebo, in results reported by investigators led by Yutaka Niihara, MD, of the University of California, Los Angeles, and Emmaus Medical.
Blood test results showed persistent elevation of mean corpuscular volume, indicating adherence to hydroxyurea therapy and suggesting that the effect of L-glutamine might be additive, Dr. Niihara and his coauthors wrote in the New England Journal of Medicine.
“L-glutamine thus provides an alternative therapy for those who decline treatment with hydroxyurea or who may have unacceptable side effects from hydroxyurea, as well as an additive therapy to lower the incidence of pain crises for those who may have suboptimal response to hydroxyurea,” they wrote.
The multicenter, randomized, placebo-controlled, double-blind, phase 3 trial by Dr. Niihara and his colleagues included 230 children and adults with sickle cell anemia or sickle-beta0-thalassemia and two or more pain crises in the previous year.
Participants at 31 sites across the United States were randomized to receive L-glutamine powder (n = 152) or placebo (n = 78) orally twice weekly for 48 weeks, followed by a 3-week tapering period. Two-thirds received concomitant hydroxyurea during the trial.
Participants were contacted by telephone weekly during the study to encourage adherence.
A total of 156 individuals completed the study, including 97 of 152 (63.8%) in the L-glutamine arm and 59 of 78 (75.6%) in the placebo arm. The most common reasons for discontinuation were withdrawal of consent, nonadherence, or reasons classified as “other,” according to investigators.
The primary end point was the number of pain crises through week 48 of the trial. A median of 3.0 pain crises occurred in the L-glutamine group, compared with 4.0 in the placebo group (P = .005). Additionally, the median number of hospitalizations was 2.0 for the L-glutamine group versus 3.0 for the placebo group (P = .005).
Nausea, arm or leg pain, and back pain all had an incidence in the L-glutamine group that was 5% higher than in the placebo group, investigators reported.
Based on these results, the Food and Drug administration approved oral L-glutamine powder to reduce the acute complications of sickle cell disease in patients 5 years of age and older in July 2017.
The reasons for study withdrawal were similar in the L-glutamine and placebo groups, despite the higher withdrawal rate in the L-glutamine group, investigators said in a discussion of their results. “Recruitment and retention in a year-long study is difficult in an already burdened population,” they wrote.
The overall noncompletion rate was 32%, similar to the 35% rate seen in a recent multicenter trial of crizanlizumab in patients with sickle cell disease, they added.
Dr. Niihara is the founder and CEO of Emmaus Medical, which sponsored the trial. Other coauthors also reported disclosures related to Emmaus Medical and other companies.
SOURCE: Niihara Y et al. N Engl J Med. 2018 Jul 19;379(3):226-35.
Results of this phase 3 trial were “much awaited” and illustrate the efficacy of L-glutamine in reducing the number of acute vasoocclusive episodes in patients with sickle cell disease.
However, as with any new breakthrough in medicine, there are now compelling questions that need to be answered, Caterina P. Minniti, MD, said in an accompanying editorial.
How to handle cost is one such question. One year of treatment with pharmaceutical-grade L-glutamine carries an estimated cost of $40,515 versus $1,700 for a year of hydroxyurea, but whether the price tag will hinder prescribing of the newer agent has yet to be seen, according to Dr. Minniti.
“This agent certainly has been slow to enter the market because prescribing L-glutamine for patients requires many steps, which may dissuade busy practitioners from actively prescribing it,” she said.
Another question is whether it should be used alongside hydroxyurea, as was done in two-thirds of patients in the present trial. Concomitant use is possible and “most likely advantageous” given that L-glutamine has a different toxicity profile and putatively different mechanism of action from hydroxyurea, Dr. Minniti said.
Who should receive L-glutamine is another important question. Dr. Minniti said that, based on previous trial data, caution may be warranted in giving L-glutamine to patients with significant renal and hepatic dysfunction, but she added that its role could be broad.
“In the absence of specific guidelines, I believe that L-glutamine may be prescribed to persons older than 5 years of age who have any sickle genotype and continue to have episodes of acute disease exacerbations despite appropriate use of hydroxyurea or to those who cannot or do not use hydroxyurea,” she said in the editorial.
Caterina P. Minniti, MD, is with the division of hematology at Montefiore Medical Center at Einstein College of Medicine, New York. These comments are excerpted from her accompanying editorial ( N Engl J Med. 2018;379:292-4 ). Dr. Minniti reported disclosures related to Global Blood Therapeutics and Bayer, along with a patent pending for a topical sodium nitrite formulation.
Results of this phase 3 trial were “much awaited” and illustrate the efficacy of L-glutamine in reducing the number of acute vasoocclusive episodes in patients with sickle cell disease.
However, as with any new breakthrough in medicine, there are now compelling questions that need to be answered, Caterina P. Minniti, MD, said in an accompanying editorial.
How to handle cost is one such question. One year of treatment with pharmaceutical-grade L-glutamine carries an estimated cost of $40,515 versus $1,700 for a year of hydroxyurea, but whether the price tag will hinder prescribing of the newer agent has yet to be seen, according to Dr. Minniti.
“This agent certainly has been slow to enter the market because prescribing L-glutamine for patients requires many steps, which may dissuade busy practitioners from actively prescribing it,” she said.
Another question is whether it should be used alongside hydroxyurea, as was done in two-thirds of patients in the present trial. Concomitant use is possible and “most likely advantageous” given that L-glutamine has a different toxicity profile and putatively different mechanism of action from hydroxyurea, Dr. Minniti said.
Who should receive L-glutamine is another important question. Dr. Minniti said that, based on previous trial data, caution may be warranted in giving L-glutamine to patients with significant renal and hepatic dysfunction, but she added that its role could be broad.
“In the absence of specific guidelines, I believe that L-glutamine may be prescribed to persons older than 5 years of age who have any sickle genotype and continue to have episodes of acute disease exacerbations despite appropriate use of hydroxyurea or to those who cannot or do not use hydroxyurea,” she said in the editorial.
Caterina P. Minniti, MD, is with the division of hematology at Montefiore Medical Center at Einstein College of Medicine, New York. These comments are excerpted from her accompanying editorial ( N Engl J Med. 2018;379:292-4 ). Dr. Minniti reported disclosures related to Global Blood Therapeutics and Bayer, along with a patent pending for a topical sodium nitrite formulation.
Results of this phase 3 trial were “much awaited” and illustrate the efficacy of L-glutamine in reducing the number of acute vasoocclusive episodes in patients with sickle cell disease.
However, as with any new breakthrough in medicine, there are now compelling questions that need to be answered, Caterina P. Minniti, MD, said in an accompanying editorial.
How to handle cost is one such question. One year of treatment with pharmaceutical-grade L-glutamine carries an estimated cost of $40,515 versus $1,700 for a year of hydroxyurea, but whether the price tag will hinder prescribing of the newer agent has yet to be seen, according to Dr. Minniti.
“This agent certainly has been slow to enter the market because prescribing L-glutamine for patients requires many steps, which may dissuade busy practitioners from actively prescribing it,” she said.
Another question is whether it should be used alongside hydroxyurea, as was done in two-thirds of patients in the present trial. Concomitant use is possible and “most likely advantageous” given that L-glutamine has a different toxicity profile and putatively different mechanism of action from hydroxyurea, Dr. Minniti said.
Who should receive L-glutamine is another important question. Dr. Minniti said that, based on previous trial data, caution may be warranted in giving L-glutamine to patients with significant renal and hepatic dysfunction, but she added that its role could be broad.
“In the absence of specific guidelines, I believe that L-glutamine may be prescribed to persons older than 5 years of age who have any sickle genotype and continue to have episodes of acute disease exacerbations despite appropriate use of hydroxyurea or to those who cannot or do not use hydroxyurea,” she said in the editorial.
Caterina P. Minniti, MD, is with the division of hematology at Montefiore Medical Center at Einstein College of Medicine, New York. These comments are excerpted from her accompanying editorial ( N Engl J Med. 2018;379:292-4 ). Dr. Minniti reported disclosures related to Global Blood Therapeutics and Bayer, along with a patent pending for a topical sodium nitrite formulation.
Children and adults with sickle cell disease who received L-glutamine alone or with hydroxyurea had a median number of pain episodes that was 25% lower than those who received placebo, according to newly published results from the phase 3 trial that led to the agent’s approval in 2017.
The median number of hospitalizations was 33% lower among individuals receiving L-glutamine than it was among those receiving placebo, in results reported by investigators led by Yutaka Niihara, MD, of the University of California, Los Angeles, and Emmaus Medical.
Blood test results showed persistent elevation of mean corpuscular volume, indicating adherence to hydroxyurea therapy and suggesting that the effect of L-glutamine might be additive, Dr. Niihara and his coauthors wrote in the New England Journal of Medicine.
“L-glutamine thus provides an alternative therapy for those who decline treatment with hydroxyurea or who may have unacceptable side effects from hydroxyurea, as well as an additive therapy to lower the incidence of pain crises for those who may have suboptimal response to hydroxyurea,” they wrote.
The multicenter, randomized, placebo-controlled, double-blind, phase 3 trial by Dr. Niihara and his colleagues included 230 children and adults with sickle cell anemia or sickle-beta0-thalassemia and two or more pain crises in the previous year.
Participants at 31 sites across the United States were randomized to receive L-glutamine powder (n = 152) or placebo (n = 78) orally twice weekly for 48 weeks, followed by a 3-week tapering period. Two-thirds received concomitant hydroxyurea during the trial.
Participants were contacted by telephone weekly during the study to encourage adherence.
A total of 156 individuals completed the study, including 97 of 152 (63.8%) in the L-glutamine arm and 59 of 78 (75.6%) in the placebo arm. The most common reasons for discontinuation were withdrawal of consent, nonadherence, or reasons classified as “other,” according to investigators.
The primary end point was the number of pain crises through week 48 of the trial. A median of 3.0 pain crises occurred in the L-glutamine group, compared with 4.0 in the placebo group (P = .005). Additionally, the median number of hospitalizations was 2.0 for the L-glutamine group versus 3.0 for the placebo group (P = .005).
Nausea, arm or leg pain, and back pain all had an incidence in the L-glutamine group that was 5% higher than in the placebo group, investigators reported.
Based on these results, the Food and Drug administration approved oral L-glutamine powder to reduce the acute complications of sickle cell disease in patients 5 years of age and older in July 2017.
The reasons for study withdrawal were similar in the L-glutamine and placebo groups, despite the higher withdrawal rate in the L-glutamine group, investigators said in a discussion of their results. “Recruitment and retention in a year-long study is difficult in an already burdened population,” they wrote.
The overall noncompletion rate was 32%, similar to the 35% rate seen in a recent multicenter trial of crizanlizumab in patients with sickle cell disease, they added.
Dr. Niihara is the founder and CEO of Emmaus Medical, which sponsored the trial. Other coauthors also reported disclosures related to Emmaus Medical and other companies.
SOURCE: Niihara Y et al. N Engl J Med. 2018 Jul 19;379(3):226-35.
Children and adults with sickle cell disease who received L-glutamine alone or with hydroxyurea had a median number of pain episodes that was 25% lower than those who received placebo, according to newly published results from the phase 3 trial that led to the agent’s approval in 2017.
The median number of hospitalizations was 33% lower among individuals receiving L-glutamine than it was among those receiving placebo, in results reported by investigators led by Yutaka Niihara, MD, of the University of California, Los Angeles, and Emmaus Medical.
Blood test results showed persistent elevation of mean corpuscular volume, indicating adherence to hydroxyurea therapy and suggesting that the effect of L-glutamine might be additive, Dr. Niihara and his coauthors wrote in the New England Journal of Medicine.
“L-glutamine thus provides an alternative therapy for those who decline treatment with hydroxyurea or who may have unacceptable side effects from hydroxyurea, as well as an additive therapy to lower the incidence of pain crises for those who may have suboptimal response to hydroxyurea,” they wrote.
The multicenter, randomized, placebo-controlled, double-blind, phase 3 trial by Dr. Niihara and his colleagues included 230 children and adults with sickle cell anemia or sickle-beta0-thalassemia and two or more pain crises in the previous year.
Participants at 31 sites across the United States were randomized to receive L-glutamine powder (n = 152) or placebo (n = 78) orally twice weekly for 48 weeks, followed by a 3-week tapering period. Two-thirds received concomitant hydroxyurea during the trial.
Participants were contacted by telephone weekly during the study to encourage adherence.
A total of 156 individuals completed the study, including 97 of 152 (63.8%) in the L-glutamine arm and 59 of 78 (75.6%) in the placebo arm. The most common reasons for discontinuation were withdrawal of consent, nonadherence, or reasons classified as “other,” according to investigators.
The primary end point was the number of pain crises through week 48 of the trial. A median of 3.0 pain crises occurred in the L-glutamine group, compared with 4.0 in the placebo group (P = .005). Additionally, the median number of hospitalizations was 2.0 for the L-glutamine group versus 3.0 for the placebo group (P = .005).
Nausea, arm or leg pain, and back pain all had an incidence in the L-glutamine group that was 5% higher than in the placebo group, investigators reported.
Based on these results, the Food and Drug administration approved oral L-glutamine powder to reduce the acute complications of sickle cell disease in patients 5 years of age and older in July 2017.
The reasons for study withdrawal were similar in the L-glutamine and placebo groups, despite the higher withdrawal rate in the L-glutamine group, investigators said in a discussion of their results. “Recruitment and retention in a year-long study is difficult in an already burdened population,” they wrote.
The overall noncompletion rate was 32%, similar to the 35% rate seen in a recent multicenter trial of crizanlizumab in patients with sickle cell disease, they added.
Dr. Niihara is the founder and CEO of Emmaus Medical, which sponsored the trial. Other coauthors also reported disclosures related to Emmaus Medical and other companies.
SOURCE: Niihara Y et al. N Engl J Med. 2018 Jul 19;379(3):226-35.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: The median number of pain crises was 3.0 in the L-glutamine group, compared with 4.0 in the placebo group (P = .005).
Study details: A multicenter, randomized, placebo-controlled, double-blind, phase 3 trial including 230 chidren and adults with sickle cell anemia or sickle-beta0-thalassemia and two or more pain crises in the previous year.
Disclosures: Dr. Niihara is the founder and CEO of Emmaus Medical, which sponsored the trial. Other coauthors also reported disclosures related to Emmaus Medical and other companies. Source: Niihara Y et al. N Engl J Med. 2018 Jul 19;379(3):226-35.
For men with SCD and priapism, hypoxia may prompt RBC adhesion
WASHINGTON – For male patients with sickle cell disease, priapism can be more than just painful and embarrassing. The prolonged erections prompted by vasoocclusive events in the penis may lead to irreversible impotence, but little is known about risk factors for priapism, which remains a difficult-to-treat complication of the disease.
In males with HbSS sickle cell disease (SCD) and priapism, RBC adhesion is increased in hypoxic conditions, according to preliminary findings from work using a newly developed biochip that mimics microvascular conditions in SCD. This significant level of adhesion prompted by hypoxia was not seen in men who did not have priapism, according to study coauthor Erina Quinn, a research assistant in hematology and oncology at Case Western Reserve University, Cleveland, who presented the results at the annual meeting of the Foundation for Sickle Cell Disease Research.
When hemoglobin desaturation occurs, polymerization can be increased, leading to increased end-organ damage, Ms. Quinn said. The biochip is “an effort to measure cellular adhesion in a clinically meaningful way.” The tool can detect hemoglobin phenotype, differentiating among HbSS, HbSbeta+, and HbSC. It can also measure the degree of hemolysis and RBC deformability.
The biochip “mimics postcapillary flow conditions in microchannels,” Ms. Quinn said. The device forces blood samples through microchannels that are at the diameter of smaller venules, approximately 50 mcm, and at a physiological flow rate ranging from 1-13 mm/sec. The microfluidic channels are coated with laminin, a subendothelial matrix protein implicated in RBC adhesion. A second microfluidic biochip mimics hypoxic conditions.
The study enrolled 26 men with the HbSS genotype, 14 of whom reported priapism, and assessed RBC adhesion in blood samples run though both the SCD-modeled biochip and the hypoxia biochip. Investigators also assessed contemporaneous in vivo hemoglobin desaturation, and looked for associations with the in vitro biochip findings.
Of the 26 participants, 16 also had either nocturnal or exertional hemoglobin desaturation. In addition, 10 participants had both priapism and desaturations. These data were collected by retrospective chart review and patient survey.
Patients with priapism were a mean age of 34 years, compared with a mean age of 29 years for the other participants, a nonsignificant difference. There were no significant differences in mean hemoglobin or bilirubin levels, or in reticulocyte counts, between the two groups.
However, white blood count, absolute neutrophil count, and lactate dehydrogenase levels were significantly higher for men with priapism (P = .022, .037, and .008, respectively). Ferritin levels were higher as well, at a mean 2,433 (plus or minus 2,234) mcg/L for those with priapism, compared with a mean 269 (plus or minus 3,015) mcg/L for those without priapism (P = .031).
When absolute reticulocyte count was mapped against lactate dehydrogenase levels to create a measure of degree of hemolysis, “individuals with priapism had a more hemolytic lab profile,” said Ms. Quinn (P = .0186).
Though 10 of 14 men with priapism had hemoglobin desaturation, compared with 5 of 12 who did not have priapism, the difference was not statistically significant.
When the researchers compared microchip analysis of RBC adhesion, though, they found marked differences in RBC adhesion in hypoxic versus nonhypoxic conditions. Significantly more RBCs were adherent under hypoxic conditions – in the hypoxic biochip – for the patients with priapism than for patients without priapism (mean, 529 vs. 3,268 adherent cells; P = .016).
Though numbers were small, RBCs from patients with reported priapism and hemoglobin desaturation in vivo showed increased hypoxia enhanced adhesion in vitro (P = .013), Ms. Quinn said. These was no significant difference between adhesion in normoxic and hypoxic conditions for the patients without priapism.
Future directions of work with the biochip include prospective identification of desaturation events and better characterization of nocturnal symptoms, Ms. Quinn said. The investigators also plan to see whether treatment with supplemental oxygen affects RBC adhesion.
The research was supported by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
WASHINGTON – For male patients with sickle cell disease, priapism can be more than just painful and embarrassing. The prolonged erections prompted by vasoocclusive events in the penis may lead to irreversible impotence, but little is known about risk factors for priapism, which remains a difficult-to-treat complication of the disease.
In males with HbSS sickle cell disease (SCD) and priapism, RBC adhesion is increased in hypoxic conditions, according to preliminary findings from work using a newly developed biochip that mimics microvascular conditions in SCD. This significant level of adhesion prompted by hypoxia was not seen in men who did not have priapism, according to study coauthor Erina Quinn, a research assistant in hematology and oncology at Case Western Reserve University, Cleveland, who presented the results at the annual meeting of the Foundation for Sickle Cell Disease Research.
When hemoglobin desaturation occurs, polymerization can be increased, leading to increased end-organ damage, Ms. Quinn said. The biochip is “an effort to measure cellular adhesion in a clinically meaningful way.” The tool can detect hemoglobin phenotype, differentiating among HbSS, HbSbeta+, and HbSC. It can also measure the degree of hemolysis and RBC deformability.
The biochip “mimics postcapillary flow conditions in microchannels,” Ms. Quinn said. The device forces blood samples through microchannels that are at the diameter of smaller venules, approximately 50 mcm, and at a physiological flow rate ranging from 1-13 mm/sec. The microfluidic channels are coated with laminin, a subendothelial matrix protein implicated in RBC adhesion. A second microfluidic biochip mimics hypoxic conditions.
The study enrolled 26 men with the HbSS genotype, 14 of whom reported priapism, and assessed RBC adhesion in blood samples run though both the SCD-modeled biochip and the hypoxia biochip. Investigators also assessed contemporaneous in vivo hemoglobin desaturation, and looked for associations with the in vitro biochip findings.
Of the 26 participants, 16 also had either nocturnal or exertional hemoglobin desaturation. In addition, 10 participants had both priapism and desaturations. These data were collected by retrospective chart review and patient survey.
Patients with priapism were a mean age of 34 years, compared with a mean age of 29 years for the other participants, a nonsignificant difference. There were no significant differences in mean hemoglobin or bilirubin levels, or in reticulocyte counts, between the two groups.
However, white blood count, absolute neutrophil count, and lactate dehydrogenase levels were significantly higher for men with priapism (P = .022, .037, and .008, respectively). Ferritin levels were higher as well, at a mean 2,433 (plus or minus 2,234) mcg/L for those with priapism, compared with a mean 269 (plus or minus 3,015) mcg/L for those without priapism (P = .031).
When absolute reticulocyte count was mapped against lactate dehydrogenase levels to create a measure of degree of hemolysis, “individuals with priapism had a more hemolytic lab profile,” said Ms. Quinn (P = .0186).
Though 10 of 14 men with priapism had hemoglobin desaturation, compared with 5 of 12 who did not have priapism, the difference was not statistically significant.
When the researchers compared microchip analysis of RBC adhesion, though, they found marked differences in RBC adhesion in hypoxic versus nonhypoxic conditions. Significantly more RBCs were adherent under hypoxic conditions – in the hypoxic biochip – for the patients with priapism than for patients without priapism (mean, 529 vs. 3,268 adherent cells; P = .016).
Though numbers were small, RBCs from patients with reported priapism and hemoglobin desaturation in vivo showed increased hypoxia enhanced adhesion in vitro (P = .013), Ms. Quinn said. These was no significant difference between adhesion in normoxic and hypoxic conditions for the patients without priapism.
Future directions of work with the biochip include prospective identification of desaturation events and better characterization of nocturnal symptoms, Ms. Quinn said. The investigators also plan to see whether treatment with supplemental oxygen affects RBC adhesion.
The research was supported by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
WASHINGTON – For male patients with sickle cell disease, priapism can be more than just painful and embarrassing. The prolonged erections prompted by vasoocclusive events in the penis may lead to irreversible impotence, but little is known about risk factors for priapism, which remains a difficult-to-treat complication of the disease.
In males with HbSS sickle cell disease (SCD) and priapism, RBC adhesion is increased in hypoxic conditions, according to preliminary findings from work using a newly developed biochip that mimics microvascular conditions in SCD. This significant level of adhesion prompted by hypoxia was not seen in men who did not have priapism, according to study coauthor Erina Quinn, a research assistant in hematology and oncology at Case Western Reserve University, Cleveland, who presented the results at the annual meeting of the Foundation for Sickle Cell Disease Research.
When hemoglobin desaturation occurs, polymerization can be increased, leading to increased end-organ damage, Ms. Quinn said. The biochip is “an effort to measure cellular adhesion in a clinically meaningful way.” The tool can detect hemoglobin phenotype, differentiating among HbSS, HbSbeta+, and HbSC. It can also measure the degree of hemolysis and RBC deformability.
The biochip “mimics postcapillary flow conditions in microchannels,” Ms. Quinn said. The device forces blood samples through microchannels that are at the diameter of smaller venules, approximately 50 mcm, and at a physiological flow rate ranging from 1-13 mm/sec. The microfluidic channels are coated with laminin, a subendothelial matrix protein implicated in RBC adhesion. A second microfluidic biochip mimics hypoxic conditions.
The study enrolled 26 men with the HbSS genotype, 14 of whom reported priapism, and assessed RBC adhesion in blood samples run though both the SCD-modeled biochip and the hypoxia biochip. Investigators also assessed contemporaneous in vivo hemoglobin desaturation, and looked for associations with the in vitro biochip findings.
Of the 26 participants, 16 also had either nocturnal or exertional hemoglobin desaturation. In addition, 10 participants had both priapism and desaturations. These data were collected by retrospective chart review and patient survey.
Patients with priapism were a mean age of 34 years, compared with a mean age of 29 years for the other participants, a nonsignificant difference. There were no significant differences in mean hemoglobin or bilirubin levels, or in reticulocyte counts, between the two groups.
However, white blood count, absolute neutrophil count, and lactate dehydrogenase levels were significantly higher for men with priapism (P = .022, .037, and .008, respectively). Ferritin levels were higher as well, at a mean 2,433 (plus or minus 2,234) mcg/L for those with priapism, compared with a mean 269 (plus or minus 3,015) mcg/L for those without priapism (P = .031).
When absolute reticulocyte count was mapped against lactate dehydrogenase levels to create a measure of degree of hemolysis, “individuals with priapism had a more hemolytic lab profile,” said Ms. Quinn (P = .0186).
Though 10 of 14 men with priapism had hemoglobin desaturation, compared with 5 of 12 who did not have priapism, the difference was not statistically significant.
When the researchers compared microchip analysis of RBC adhesion, though, they found marked differences in RBC adhesion in hypoxic versus nonhypoxic conditions. Significantly more RBCs were adherent under hypoxic conditions – in the hypoxic biochip – for the patients with priapism than for patients without priapism (mean, 529 vs. 3,268 adherent cells; P = .016).
Though numbers were small, RBCs from patients with reported priapism and hemoglobin desaturation in vivo showed increased hypoxia enhanced adhesion in vitro (P = .013), Ms. Quinn said. These was no significant difference between adhesion in normoxic and hypoxic conditions for the patients without priapism.
Future directions of work with the biochip include prospective identification of desaturation events and better characterization of nocturnal symptoms, Ms. Quinn said. The investigators also plan to see whether treatment with supplemental oxygen affects RBC adhesion.
The research was supported by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
REPORTING FROM FSCDR 2018
Key clinical point: RBC adhesion was increased, but only in hypoxia, for men with sickle cell disease and priapism.
Major finding: Men who had desaturations and priapism had significantly higher RBC adhesion than those without priapism (P = .013).
Study details: An in vitro and in vivo study of 26 men with HbSS sickle cell disease, with and without priapism.
Disclosures: The study was funded by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
Pfizer launches phase 3 gene therapy study in hemophilia B
Pfizer has begun early work on a phase 3 study of an investigational gene therapy to treat hemophilia B.
Pfizer, along with Spark Therapeutics, launched a phase 3 lead-in study at multiple centers to evaluate the efficacy and safety of current factor IX prophylaxis replacement therapy in the usual care setting. The efficacy data from the lead-in study will become the within-subject control group for patients who enroll in the next part of the
Fidanacogene elaparvovec is a vector containing a bioengineered adeno-associated virus capsid and a high-activity human coagulation factor IX gene. In an ongoing phase 1/2 trial of fidanacogene elaparvovec, all 15 participants with hemophilia B were able to discontinue routine infusion of factor IX concentrates without serious adverse events, according to data released by Pfizer and Spark Therapeutics in May 2018.
Pfizer has begun early work on a phase 3 study of an investigational gene therapy to treat hemophilia B.
Pfizer, along with Spark Therapeutics, launched a phase 3 lead-in study at multiple centers to evaluate the efficacy and safety of current factor IX prophylaxis replacement therapy in the usual care setting. The efficacy data from the lead-in study will become the within-subject control group for patients who enroll in the next part of the
Fidanacogene elaparvovec is a vector containing a bioengineered adeno-associated virus capsid and a high-activity human coagulation factor IX gene. In an ongoing phase 1/2 trial of fidanacogene elaparvovec, all 15 participants with hemophilia B were able to discontinue routine infusion of factor IX concentrates without serious adverse events, according to data released by Pfizer and Spark Therapeutics in May 2018.
Pfizer has begun early work on a phase 3 study of an investigational gene therapy to treat hemophilia B.
Pfizer, along with Spark Therapeutics, launched a phase 3 lead-in study at multiple centers to evaluate the efficacy and safety of current factor IX prophylaxis replacement therapy in the usual care setting. The efficacy data from the lead-in study will become the within-subject control group for patients who enroll in the next part of the
Fidanacogene elaparvovec is a vector containing a bioengineered adeno-associated virus capsid and a high-activity human coagulation factor IX gene. In an ongoing phase 1/2 trial of fidanacogene elaparvovec, all 15 participants with hemophilia B were able to discontinue routine infusion of factor IX concentrates without serious adverse events, according to data released by Pfizer and Spark Therapeutics in May 2018.
Are We Beating Cancer—Finally?
Cancer death rates continue to decline in the US in all major racial and ethnic groups, according to the National Cancer Institute’s (NCI) latest Annual Report to the Nation on the Status of Cancer. The data are an “encouraging indicator of progress” in cancer research, says NCI Director Ned Sharpless, MD. “It’s clear that interventions are having an impact.”
Overall incidence, or rates of new cancers, dropped by 1.8% in men and 1.4% in women from 1999 to 2015. Between 2011 and 2015, death rates dropped for 11 of the 18 most common cancer types in men and 14 of the 20 most common types in women. The researchers say the “significant declines” also hold “significant differences” in rate by sex, race, and ethnicity. For example, black men and white women had the highest incidence rates, and black men and black women had the highest death rates.
However, over the same period, death rates for cancers of the liver, pancreas, and brain and nervous system rose in both men and women. Death rates for cancer of the uterus rose (the researchers say obesity is thought to be a contributing factor) and death rates for cancers of the oral cavity and pharynx and soft tissue increased in men, perhaps associated with human papillomavirus infection.
In a companion study, when researchers explored prostate cancer trends in more detail they found overall prostate cancer incidence rates declined an average of 6.5% each year between 2007 and 2014, from 163 new cases per 100,000 men to 104 new cases. Still, after a 2-decade steady decline, rates leveled off. Incidence of distant disease rose from 7.8 new cases per 100,000 to 9.2, but there was no increase in the rates of cases with aggressive histologic grade.
Interestingly, the researchers also report a decline in recent prostate-specific antigen screening between 2010 and 2013 national surveys. “The increase in late-stage disease and the flattening of the mortality trended occurred contemporaneously with the observed decrease in PSA screening,” said Serban Negoita, MD, DrPH, of NCI’s Surveillance Research Program. However, while “suggestive,” Negoita adds, their observation does not demonstrate causality: many factors contribute to incidence and mortality, such as improvements in staging and treating cancer.
Cancer death rates continue to decline in the US in all major racial and ethnic groups, according to the National Cancer Institute’s (NCI) latest Annual Report to the Nation on the Status of Cancer. The data are an “encouraging indicator of progress” in cancer research, says NCI Director Ned Sharpless, MD. “It’s clear that interventions are having an impact.”
Overall incidence, or rates of new cancers, dropped by 1.8% in men and 1.4% in women from 1999 to 2015. Between 2011 and 2015, death rates dropped for 11 of the 18 most common cancer types in men and 14 of the 20 most common types in women. The researchers say the “significant declines” also hold “significant differences” in rate by sex, race, and ethnicity. For example, black men and white women had the highest incidence rates, and black men and black women had the highest death rates.
However, over the same period, death rates for cancers of the liver, pancreas, and brain and nervous system rose in both men and women. Death rates for cancer of the uterus rose (the researchers say obesity is thought to be a contributing factor) and death rates for cancers of the oral cavity and pharynx and soft tissue increased in men, perhaps associated with human papillomavirus infection.
In a companion study, when researchers explored prostate cancer trends in more detail they found overall prostate cancer incidence rates declined an average of 6.5% each year between 2007 and 2014, from 163 new cases per 100,000 men to 104 new cases. Still, after a 2-decade steady decline, rates leveled off. Incidence of distant disease rose from 7.8 new cases per 100,000 to 9.2, but there was no increase in the rates of cases with aggressive histologic grade.
Interestingly, the researchers also report a decline in recent prostate-specific antigen screening between 2010 and 2013 national surveys. “The increase in late-stage disease and the flattening of the mortality trended occurred contemporaneously with the observed decrease in PSA screening,” said Serban Negoita, MD, DrPH, of NCI’s Surveillance Research Program. However, while “suggestive,” Negoita adds, their observation does not demonstrate causality: many factors contribute to incidence and mortality, such as improvements in staging and treating cancer.
Cancer death rates continue to decline in the US in all major racial and ethnic groups, according to the National Cancer Institute’s (NCI) latest Annual Report to the Nation on the Status of Cancer. The data are an “encouraging indicator of progress” in cancer research, says NCI Director Ned Sharpless, MD. “It’s clear that interventions are having an impact.”
Overall incidence, or rates of new cancers, dropped by 1.8% in men and 1.4% in women from 1999 to 2015. Between 2011 and 2015, death rates dropped for 11 of the 18 most common cancer types in men and 14 of the 20 most common types in women. The researchers say the “significant declines” also hold “significant differences” in rate by sex, race, and ethnicity. For example, black men and white women had the highest incidence rates, and black men and black women had the highest death rates.
However, over the same period, death rates for cancers of the liver, pancreas, and brain and nervous system rose in both men and women. Death rates for cancer of the uterus rose (the researchers say obesity is thought to be a contributing factor) and death rates for cancers of the oral cavity and pharynx and soft tissue increased in men, perhaps associated with human papillomavirus infection.
In a companion study, when researchers explored prostate cancer trends in more detail they found overall prostate cancer incidence rates declined an average of 6.5% each year between 2007 and 2014, from 163 new cases per 100,000 men to 104 new cases. Still, after a 2-decade steady decline, rates leveled off. Incidence of distant disease rose from 7.8 new cases per 100,000 to 9.2, but there was no increase in the rates of cases with aggressive histologic grade.
Interestingly, the researchers also report a decline in recent prostate-specific antigen screening between 2010 and 2013 national surveys. “The increase in late-stage disease and the flattening of the mortality trended occurred contemporaneously with the observed decrease in PSA screening,” said Serban Negoita, MD, DrPH, of NCI’s Surveillance Research Program. However, while “suggestive,” Negoita adds, their observation does not demonstrate causality: many factors contribute to incidence and mortality, such as improvements in staging and treating cancer.
FDA gives green light to freeze-dried plasma in combat
The Department of Defense has received emergency use authorization from the Food and Drug Administration to use pathogen-reduced, leukocyte-depleted, freeze-dried plasma for the emergency treatment of hemorrhage and coagulopathy in combat situations.
Hemorrhage and coagulopathy are the leading causes of preventable deaths among combat trauma casualties. While plasma contains proteins that help clot blood and thus can treat these conditions, it isn’t feasible to keep it on hand for combat emergencies in the field because of logistical and operational requirements, such as refrigeration or thawing periods. This freeze-dried plasma product, on the other hand, can be easily reconstituted in situations in which refrigeration isn’t possible.
The FDA authorization allows for the use of a French-made, powdered, freeze-dried product. This emergency use authorization came about in part because of a joint program established between the FDA and the Department of Defense in January 2018.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country, by expediting the development and availability of safe and effective, priority medical products that are essential to the health of our military service members,” said FDA commissioner Scott Gottlieb, MD. “This is especially true when it comes to products used to treat injuries in a potential battlefield setting.”
More information about this emergency use authorization can be found in the FDA’s full press announcement.
The Department of Defense has received emergency use authorization from the Food and Drug Administration to use pathogen-reduced, leukocyte-depleted, freeze-dried plasma for the emergency treatment of hemorrhage and coagulopathy in combat situations.
Hemorrhage and coagulopathy are the leading causes of preventable deaths among combat trauma casualties. While plasma contains proteins that help clot blood and thus can treat these conditions, it isn’t feasible to keep it on hand for combat emergencies in the field because of logistical and operational requirements, such as refrigeration or thawing periods. This freeze-dried plasma product, on the other hand, can be easily reconstituted in situations in which refrigeration isn’t possible.
The FDA authorization allows for the use of a French-made, powdered, freeze-dried product. This emergency use authorization came about in part because of a joint program established between the FDA and the Department of Defense in January 2018.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country, by expediting the development and availability of safe and effective, priority medical products that are essential to the health of our military service members,” said FDA commissioner Scott Gottlieb, MD. “This is especially true when it comes to products used to treat injuries in a potential battlefield setting.”
More information about this emergency use authorization can be found in the FDA’s full press announcement.
The Department of Defense has received emergency use authorization from the Food and Drug Administration to use pathogen-reduced, leukocyte-depleted, freeze-dried plasma for the emergency treatment of hemorrhage and coagulopathy in combat situations.
Hemorrhage and coagulopathy are the leading causes of preventable deaths among combat trauma casualties. While plasma contains proteins that help clot blood and thus can treat these conditions, it isn’t feasible to keep it on hand for combat emergencies in the field because of logistical and operational requirements, such as refrigeration or thawing periods. This freeze-dried plasma product, on the other hand, can be easily reconstituted in situations in which refrigeration isn’t possible.
The FDA authorization allows for the use of a French-made, powdered, freeze-dried product. This emergency use authorization came about in part because of a joint program established between the FDA and the Department of Defense in January 2018.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country, by expediting the development and availability of safe and effective, priority medical products that are essential to the health of our military service members,” said FDA commissioner Scott Gottlieb, MD. “This is especially true when it comes to products used to treat injuries in a potential battlefield setting.”
More information about this emergency use authorization can be found in the FDA’s full press announcement.
Diagnosis and Management of Aggressive B-Cell Non-Hodgkin Lymphoma
Abstract
- Objective: To review the diagnosis and management of aggressive B-cell non-Hodgkin lymphoma (NHL).
- Methods: Review of the literature.
- Results: NHL comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years.
- Conclusion: Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features [1]. While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women [2]. NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides [3], immunosuppression associated with HIV infection [4], autoimmune disorders [5], iatrogenically induced immune suppression in the post-transplant and other settings [6], family history of NHL [7], and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL [8]. In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma [9]. Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas [10]. Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes [11–13], including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Workup
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. 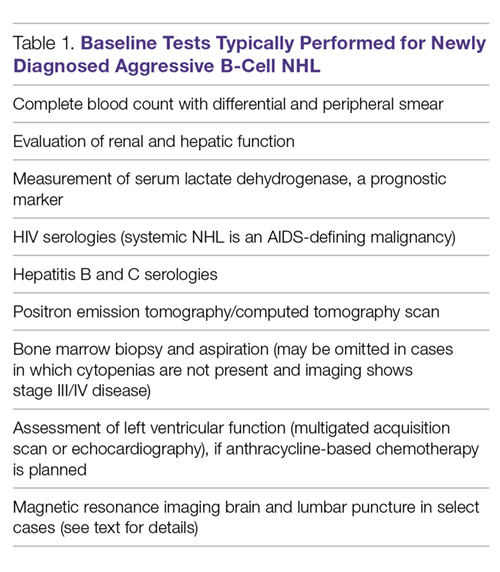
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL [14]. This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases [2]. It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes [15].
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races [16]. There is a slight male predominance (55%), median age at diagnosis is 65 years [16,17] and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue [18]. Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis [19].
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease [20]. The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement [21], this feature does not carry the same poor prognosis associated with transformed disease [22] or DLBCL involvement of the bone marrow [23].
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein [24], and about 70% express the BCL-6 protein [25]. C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL [26].
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). 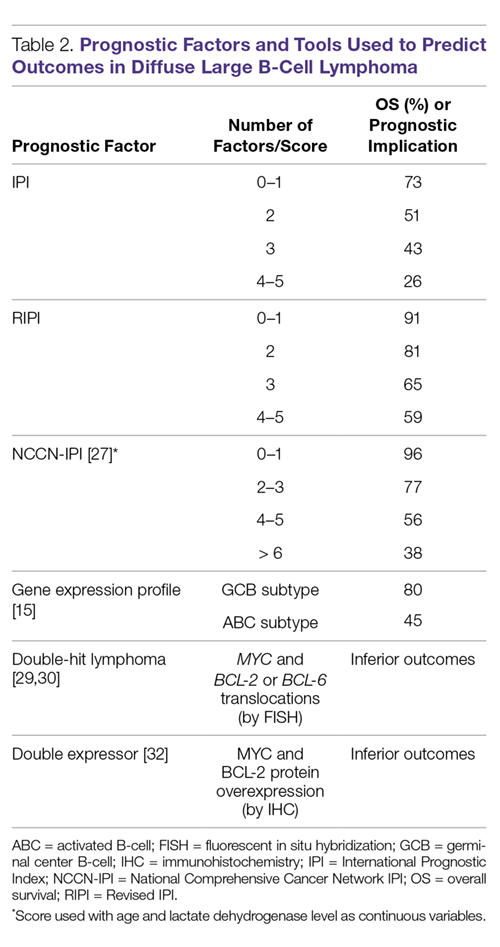
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP [28]. The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis [29,30]. Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma [1]. In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL [31,32]. Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy. DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years [33]. The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS [34,35].
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues [36] studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms [37], with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT [36].
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed [38]. Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]) [39]; dose-dense therapy (R-CHOP-14); replacement of rituximab with obinutuzuimab [40]; addition of novel agents such as bortezomib [41], lenalidomide[42], or ibrutinib [43,44] to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide [45], enzastaurin [46], and everolimus [47]. Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit [48]. More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however [49]. As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%) [50,51]. Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes [52], with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission [53].
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively [54]. A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF) [55]. In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months [56]. For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series [57].
Relapsed/Refractory Disease. Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype [58]. However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone [59]. The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort [60].
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT [61]. Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained [62]. However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL [64]. PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL [20]. It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious [64]. Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease [65], with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL [66]. PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis [67]. This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT [68–70]. The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78% [71] when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases [72,73]. For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80% [74–76] In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease [77]. This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe [78] MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease [79]. Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases [80]. In rare cases, CD5 or cyclin D1 may be negative [80]. Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression [81]. Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome [82]. In cyclin D1–negative cases, SOX11 expression may help with diagnosis [83]. A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range [84]. In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range [85–87]. Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004 [88], and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010 [89]. The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients [90]. The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement [91].
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course [92], but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy. For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission [87,93,94]. Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial [93]. At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy [95]. However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach [96]. Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP [97,98].
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years [99]. In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib [100,101], the BTK inhibitors ibrutinib [102,103] and acalabrutinib [104], and the immunomodulatory agent lenalidomide [105].
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission [95]. Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease [95,106]. A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma [107]. The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90% [107]. Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients [108]. In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT [109].
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70% [110,111]. High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse [112]. However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease [113].
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients [114]. For the minority of LBL patients with a B-cell phenotype, emerging options for relapsed/refractory LBL/ALL such as inotuzumab, blinatumomab, or anti-CD19 CAR T-cell therapy should be considered. These are not options for the majority who have a T-cell phenotype, and treatment options for these patients are limited to conventional relapsed/refractory ALL and aggressive NHL regimens.
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Corresponding author: Timothy S. Fenske, MD, Division of Hematology & Oncology, Medical College of Wisconsin, 9200 W. Wisconsin Ave., Milwaukee, WI 53226.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
Abstract
- Objective: To review the diagnosis and management of aggressive B-cell non-Hodgkin lymphoma (NHL).
- Methods: Review of the literature.
- Results: NHL comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years.
- Conclusion: Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features [1]. While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women [2]. NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides [3], immunosuppression associated with HIV infection [4], autoimmune disorders [5], iatrogenically induced immune suppression in the post-transplant and other settings [6], family history of NHL [7], and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL [8]. In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma [9]. Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas [10]. Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes [11–13], including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Workup
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL [14]. This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases [2]. It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes [15].
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races [16]. There is a slight male predominance (55%), median age at diagnosis is 65 years [16,17] and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue [18]. Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis [19].
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease [20]. The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement [21], this feature does not carry the same poor prognosis associated with transformed disease [22] or DLBCL involvement of the bone marrow [23].
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein [24], and about 70% express the BCL-6 protein [25]. C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL [26].
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2).
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP [28]. The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis [29,30]. Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma [1]. In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL [31,32]. Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy. DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years [33]. The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS [34,35].
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues [36] studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms [37], with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT [36].
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed [38]. Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]) [39]; dose-dense therapy (R-CHOP-14); replacement of rituximab with obinutuzuimab [40]; addition of novel agents such as bortezomib [41], lenalidomide[42], or ibrutinib [43,44] to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide [45], enzastaurin [46], and everolimus [47]. Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit [48]. More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however [49]. As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%) [50,51]. Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes [52], with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission [53].
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively [54]. A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF) [55]. In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months [56]. For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series [57].
Relapsed/Refractory Disease. Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype [58]. However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone [59]. The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort [60].
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT [61]. Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained [62]. However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL [64]. PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL [20]. It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious [64]. Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease [65], with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL [66]. PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis [67]. This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT [68–70]. The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78% [71] when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases [72,73]. For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80% [74–76] In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease [77]. This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe [78] MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease [79]. Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases [80]. In rare cases, CD5 or cyclin D1 may be negative [80]. Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression [81]. Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome [82]. In cyclin D1–negative cases, SOX11 expression may help with diagnosis [83]. A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range [84]. In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range [85–87]. Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004 [88], and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010 [89]. The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients [90]. The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement [91].
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course [92], but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy. For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission [87,93,94]. Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial [93]. At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy [95]. However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach [96]. Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP [97,98].
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years [99]. In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib [100,101], the BTK inhibitors ibrutinib [102,103] and acalabrutinib [104], and the immunomodulatory agent lenalidomide [105].
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission [95]. Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease [95,106]. A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma [107]. The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90% [107]. Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients [108]. In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT [109].
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70% [110,111]. High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse [112]. However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease [113].
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients [114]. For the minority of LBL patients with a B-cell phenotype, emerging options for relapsed/refractory LBL/ALL such as inotuzumab, blinatumomab, or anti-CD19 CAR T-cell therapy should be considered. These are not options for the majority who have a T-cell phenotype, and treatment options for these patients are limited to conventional relapsed/refractory ALL and aggressive NHL regimens.
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Corresponding author: Timothy S. Fenske, MD, Division of Hematology & Oncology, Medical College of Wisconsin, 9200 W. Wisconsin Ave., Milwaukee, WI 53226.
Abstract
- Objective: To review the diagnosis and management of aggressive B-cell non-Hodgkin lymphoma (NHL).
- Methods: Review of the literature.
- Results: NHL comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years.
- Conclusion: Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features [1]. While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women [2]. NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides [3], immunosuppression associated with HIV infection [4], autoimmune disorders [5], iatrogenically induced immune suppression in the post-transplant and other settings [6], family history of NHL [7], and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL [8]. In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma [9]. Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas [10]. Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes [11–13], including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Workup
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL [14]. This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases [2]. It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes [15].
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races [16]. There is a slight male predominance (55%), median age at diagnosis is 65 years [16,17] and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue [18]. Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis [19].
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease [20]. The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement [21], this feature does not carry the same poor prognosis associated with transformed disease [22] or DLBCL involvement of the bone marrow [23].
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein [24], and about 70% express the BCL-6 protein [25]. C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL [26].
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2).
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP [28]. The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis [29,30]. Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma [1]. In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL [31,32]. Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy. DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years [33]. The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS [34,35].
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues [36] studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms [37], with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT [36].
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed [38]. Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]) [39]; dose-dense therapy (R-CHOP-14); replacement of rituximab with obinutuzuimab [40]; addition of novel agents such as bortezomib [41], lenalidomide[42], or ibrutinib [43,44] to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide [45], enzastaurin [46], and everolimus [47]. Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit [48]. More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however [49]. As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%) [50,51]. Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes [52], with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission [53].
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively [54]. A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF) [55]. In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months [56]. For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series [57].
Relapsed/Refractory Disease. Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype [58]. However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone [59]. The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort [60].
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT [61]. Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained [62]. However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL [64]. PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL [20]. It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious [64]. Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease [65], with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL [66]. PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis [67]. This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT [68–70]. The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78% [71] when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases [72,73]. For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80% [74–76] In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease [77]. This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe [78] MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease [79]. Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases [80]. In rare cases, CD5 or cyclin D1 may be negative [80]. Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression [81]. Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome [82]. In cyclin D1–negative cases, SOX11 expression may help with diagnosis [83]. A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range [84]. In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range [85–87]. Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004 [88], and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010 [89]. The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients [90]. The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement [91].
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course [92], but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy. For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission [87,93,94]. Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial [93]. At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy [95]. However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach [96]. Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP [97,98].
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years [99]. In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib [100,101], the BTK inhibitors ibrutinib [102,103] and acalabrutinib [104], and the immunomodulatory agent lenalidomide [105].
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission [95]. Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease [95,106]. A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma [107]. The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90% [107]. Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients [108]. In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT [109].
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70% [110,111]. High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse [112]. However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease [113].
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients [114]. For the minority of LBL patients with a B-cell phenotype, emerging options for relapsed/refractory LBL/ALL such as inotuzumab, blinatumomab, or anti-CD19 CAR T-cell therapy should be considered. These are not options for the majority who have a T-cell phenotype, and treatment options for these patients are limited to conventional relapsed/refractory ALL and aggressive NHL regimens.
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Corresponding author: Timothy S. Fenske, MD, Division of Hematology & Oncology, Medical College of Wisconsin, 9200 W. Wisconsin Ave., Milwaukee, WI 53226.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
Long-term follow-up of monoclonal gammopathy of undetermined significance
Clinical question: What is the expected clinical progression of patients with monoclonal gammopathy of undetermined significance (MGUS)?

Study design: Prospective, observational cohort study.
Setting: Single institution in Minnesota.
Synopsis: Investigators identified 1,395 patients with MGUS during 1960-1994, with a median follow-up of 34 years. Progression to multiple myeloma, plasma cell disorders, or lymphoid disorders was noted in 147 patients (11%), which represents a 6.5-times higher risk for these disorders, compared with the age/sex–adjusted control population.
Two risk factors were associated with progression of disease: elevated serum M protein (greater than 1.5 g/dL) and an abnormal serum free light chain ratio. Risk of progression at 20 years in patients with both of these risk factors was 55% in patients with IgM subtypes and 30% in patients with non-IgM subtypes. With a single risk factor, risk of progression at 20 years was 41% and 20%, respectively. With no risk factors the risk of progression at 20 years was 19% and 7%. Overall expected survival was shorter in patients with MGUS versus that in the age/sex–matched control population.
Bottom line: Patients with MGUS have a shorter life expectancy than the general population, and the IgM subtype is associated with a greater risk of progression at 20 years, compared with the non-IgM subtype.
Citation: Kyle RA et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Eng J Med. 2018 Jan 18;378(3):241-9.
Dr. Thota is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Clinical question: What is the expected clinical progression of patients with monoclonal gammopathy of undetermined significance (MGUS)?
Study design: Prospective, observational cohort study.
Setting: Single institution in Minnesota.
Synopsis: Investigators identified 1,395 patients with MGUS during 1960-1994, with a median follow-up of 34 years. Progression to multiple myeloma, plasma cell disorders, or lymphoid disorders was noted in 147 patients (11%), which represents a 6.5-times higher risk for these disorders, compared with the age/sex–adjusted control population.
Two risk factors were associated with progression of disease: elevated serum M protein (greater than 1.5 g/dL) and an abnormal serum free light chain ratio. Risk of progression at 20 years in patients with both of these risk factors was 55% in patients with IgM subtypes and 30% in patients with non-IgM subtypes. With a single risk factor, risk of progression at 20 years was 41% and 20%, respectively. With no risk factors the risk of progression at 20 years was 19% and 7%. Overall expected survival was shorter in patients with MGUS versus that in the age/sex–matched control population.
Bottom line: Patients with MGUS have a shorter life expectancy than the general population, and the IgM subtype is associated with a greater risk of progression at 20 years, compared with the non-IgM subtype.
Citation: Kyle RA et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Eng J Med. 2018 Jan 18;378(3):241-9.
Dr. Thota is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Clinical question: What is the expected clinical progression of patients with monoclonal gammopathy of undetermined significance (MGUS)?
Study design: Prospective, observational cohort study.
Setting: Single institution in Minnesota.
Synopsis: Investigators identified 1,395 patients with MGUS during 1960-1994, with a median follow-up of 34 years. Progression to multiple myeloma, plasma cell disorders, or lymphoid disorders was noted in 147 patients (11%), which represents a 6.5-times higher risk for these disorders, compared with the age/sex–adjusted control population.
Two risk factors were associated with progression of disease: elevated serum M protein (greater than 1.5 g/dL) and an abnormal serum free light chain ratio. Risk of progression at 20 years in patients with both of these risk factors was 55% in patients with IgM subtypes and 30% in patients with non-IgM subtypes. With a single risk factor, risk of progression at 20 years was 41% and 20%, respectively. With no risk factors the risk of progression at 20 years was 19% and 7%. Overall expected survival was shorter in patients with MGUS versus that in the age/sex–matched control population.
Bottom line: Patients with MGUS have a shorter life expectancy than the general population, and the IgM subtype is associated with a greater risk of progression at 20 years, compared with the non-IgM subtype.
Citation: Kyle RA et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Eng J Med. 2018 Jan 18;378(3):241-9.
Dr. Thota is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Low platelets linked to pregnancy complications
A study that characterized the occurrence and frequency of thrombocytopenia throughout the course of pregnancy found a significant decline in platelet counts during the course of pregnancy, and significant differences between pregnant and nonpregnant women. However, the study – published in the New England Journal of Medicine – also found that women with pregnancy-related complications were more likely to have platelet counts less than 150,000/mm3, even in the absence of known causes of thrombocytopenia.
Jessica Reese, PhD, and her coinvestigators at the University of Oklahoma, Oklahoma City, used data from pregnant women who delivered at a single site from 2011 to 2014. In all, 4,568 women from the study group had uncomplicated pregnancies, and 2,586 had pregnancy-related complications. To be included in the complicated pregnancy group, women needed a diagnosis of hypertension, diabetes, eclampsia or preeclampsia, or abnormal placentation. Another 197 women had preexisting disorders known to be associated with thrombocytopenia.
For the women with uncomplicated pregnancies, Dr. Reese and her colleagues compared platelet counts with those of nonpregnant women who participated in the National Health and Nutrition Examination Survey (NHANES) from 1999 to 2012, using a stratified analysis that accounted for age and racial or ethnic background and excluding NHANES participants with cancer, diabetes, or hypertension.
To look at platelet levels across types of pregnancies and in comparison with nonpregnant women, the investigators established three cutpoints, grouping women into those who had a platelet count of at least 150,000/mm3, those with platelet counts less than 100,000/mm3 but at least 80,000/mm3, and those with platelet counts less than 80,000/mm3.
Only 1% of women with uncomplicated pregnancies had platelet counts less than 100,000/mm3 during pregnancy or at delivery, and just 5 women (0.1%) had unexplained platelet counts below 80,000/mm3. Seven more women with platelet counts less than 80,000/mm3 had an identified cause for their thrombocytopenia.
Overall, mean platelet counts were lower for the women with uncomplicated pregnancies during the first trimester than for nonpregnant women (251,000 vs. 273,000/mm3). These values fell throughout pregnancy to a mean of 217,000/mm3 by the time of delivery at a mean gestation of 39.0 weeks (P less than .001 for all time points). However, mean platelet counts rebounded by the time a postpartum value was obtained at a mean 7.1 weeks after delivery, to 264,000/mm3, a value that wasn’t significantly different from the nonpregnant cohort’s platelet counts.
When the investigators looked at mean platelet counts by trimester, they saw no difference between those with uncomplicated and complicated pregnancies until the third trimester. Then, “mean platelet counts decreased at a greater rate among women with pregnancy-related complications,” wrote Dr. Reese and her colleagues; 11.9% of women with complicated pregnancies had platelet counts below 150,000/mm3, while this level was seen in 9.9% of women without complications of pregnancy (P = .01).
At delivery, 2.3% (n = 59) of women with complicated pregnancies had platelet counts below 100,000/mm3, and 31 of these women had counts below 80,000/mm3, representing a significantly higher rate of thrombocytopenia at delivery than seen in the uncomplicated group (P less than .001).
In discussion, Dr. Reese and her coauthors examined the possible mechanisms for decreased levels of circulating platelets during pregnancy. Volume dilution from increased plasma volume is one well-accepted reason. Others include accumulation of platelets within the spleen, which increases in size by about 50% during pregnancy; similarly, the placenta’s circulation is similar to that of the spleen, so platelets may also accumulate there, the authors said. Further support for the placental mechanism comes from the lower average platelet counts for women with twin pregnancies.
The study’s relatively broad definition of pregnancy-related complications may have had the effect of lessening the difference in mean platelet counts between the complicated and uncomplicated pregnancy groups, the investigators acknowledged. Still, their study population had rates of these complications similar to those of the United States population, they said. “Therefore, our data may accurately reflect the platelet counts in women with these pregnancy-related complications,” they noted.
“Severe thrombocytopenia is rare, even in women with pregnancy-related complications,” concluded Dr. Reese and her colleagues. “Our data suggest that, for women with an uncomplicated pregnancy who have a platelet count of less than 100,000/mm3, a cause of thrombocytopenia other than the pregnancy itself should be considered.”
SOURCE: Reese J et al. N Engl J Med. 2018;379:32-43.
A study that characterized the occurrence and frequency of thrombocytopenia throughout the course of pregnancy found a significant decline in platelet counts during the course of pregnancy, and significant differences between pregnant and nonpregnant women. However, the study – published in the New England Journal of Medicine – also found that women with pregnancy-related complications were more likely to have platelet counts less than 150,000/mm3, even in the absence of known causes of thrombocytopenia.
Jessica Reese, PhD, and her coinvestigators at the University of Oklahoma, Oklahoma City, used data from pregnant women who delivered at a single site from 2011 to 2014. In all, 4,568 women from the study group had uncomplicated pregnancies, and 2,586 had pregnancy-related complications. To be included in the complicated pregnancy group, women needed a diagnosis of hypertension, diabetes, eclampsia or preeclampsia, or abnormal placentation. Another 197 women had preexisting disorders known to be associated with thrombocytopenia.
For the women with uncomplicated pregnancies, Dr. Reese and her colleagues compared platelet counts with those of nonpregnant women who participated in the National Health and Nutrition Examination Survey (NHANES) from 1999 to 2012, using a stratified analysis that accounted for age and racial or ethnic background and excluding NHANES participants with cancer, diabetes, or hypertension.
To look at platelet levels across types of pregnancies and in comparison with nonpregnant women, the investigators established three cutpoints, grouping women into those who had a platelet count of at least 150,000/mm3, those with platelet counts less than 100,000/mm3 but at least 80,000/mm3, and those with platelet counts less than 80,000/mm3.
Only 1% of women with uncomplicated pregnancies had platelet counts less than 100,000/mm3 during pregnancy or at delivery, and just 5 women (0.1%) had unexplained platelet counts below 80,000/mm3. Seven more women with platelet counts less than 80,000/mm3 had an identified cause for their thrombocytopenia.
Overall, mean platelet counts were lower for the women with uncomplicated pregnancies during the first trimester than for nonpregnant women (251,000 vs. 273,000/mm3). These values fell throughout pregnancy to a mean of 217,000/mm3 by the time of delivery at a mean gestation of 39.0 weeks (P less than .001 for all time points). However, mean platelet counts rebounded by the time a postpartum value was obtained at a mean 7.1 weeks after delivery, to 264,000/mm3, a value that wasn’t significantly different from the nonpregnant cohort’s platelet counts.
When the investigators looked at mean platelet counts by trimester, they saw no difference between those with uncomplicated and complicated pregnancies until the third trimester. Then, “mean platelet counts decreased at a greater rate among women with pregnancy-related complications,” wrote Dr. Reese and her colleagues; 11.9% of women with complicated pregnancies had platelet counts below 150,000/mm3, while this level was seen in 9.9% of women without complications of pregnancy (P = .01).
At delivery, 2.3% (n = 59) of women with complicated pregnancies had platelet counts below 100,000/mm3, and 31 of these women had counts below 80,000/mm3, representing a significantly higher rate of thrombocytopenia at delivery than seen in the uncomplicated group (P less than .001).
In discussion, Dr. Reese and her coauthors examined the possible mechanisms for decreased levels of circulating platelets during pregnancy. Volume dilution from increased plasma volume is one well-accepted reason. Others include accumulation of platelets within the spleen, which increases in size by about 50% during pregnancy; similarly, the placenta’s circulation is similar to that of the spleen, so platelets may also accumulate there, the authors said. Further support for the placental mechanism comes from the lower average platelet counts for women with twin pregnancies.
The study’s relatively broad definition of pregnancy-related complications may have had the effect of lessening the difference in mean platelet counts between the complicated and uncomplicated pregnancy groups, the investigators acknowledged. Still, their study population had rates of these complications similar to those of the United States population, they said. “Therefore, our data may accurately reflect the platelet counts in women with these pregnancy-related complications,” they noted.
“Severe thrombocytopenia is rare, even in women with pregnancy-related complications,” concluded Dr. Reese and her colleagues. “Our data suggest that, for women with an uncomplicated pregnancy who have a platelet count of less than 100,000/mm3, a cause of thrombocytopenia other than the pregnancy itself should be considered.”
SOURCE: Reese J et al. N Engl J Med. 2018;379:32-43.
A study that characterized the occurrence and frequency of thrombocytopenia throughout the course of pregnancy found a significant decline in platelet counts during the course of pregnancy, and significant differences between pregnant and nonpregnant women. However, the study – published in the New England Journal of Medicine – also found that women with pregnancy-related complications were more likely to have platelet counts less than 150,000/mm3, even in the absence of known causes of thrombocytopenia.
Jessica Reese, PhD, and her coinvestigators at the University of Oklahoma, Oklahoma City, used data from pregnant women who delivered at a single site from 2011 to 2014. In all, 4,568 women from the study group had uncomplicated pregnancies, and 2,586 had pregnancy-related complications. To be included in the complicated pregnancy group, women needed a diagnosis of hypertension, diabetes, eclampsia or preeclampsia, or abnormal placentation. Another 197 women had preexisting disorders known to be associated with thrombocytopenia.
For the women with uncomplicated pregnancies, Dr. Reese and her colleagues compared platelet counts with those of nonpregnant women who participated in the National Health and Nutrition Examination Survey (NHANES) from 1999 to 2012, using a stratified analysis that accounted for age and racial or ethnic background and excluding NHANES participants with cancer, diabetes, or hypertension.
To look at platelet levels across types of pregnancies and in comparison with nonpregnant women, the investigators established three cutpoints, grouping women into those who had a platelet count of at least 150,000/mm3, those with platelet counts less than 100,000/mm3 but at least 80,000/mm3, and those with platelet counts less than 80,000/mm3.
Only 1% of women with uncomplicated pregnancies had platelet counts less than 100,000/mm3 during pregnancy or at delivery, and just 5 women (0.1%) had unexplained platelet counts below 80,000/mm3. Seven more women with platelet counts less than 80,000/mm3 had an identified cause for their thrombocytopenia.
Overall, mean platelet counts were lower for the women with uncomplicated pregnancies during the first trimester than for nonpregnant women (251,000 vs. 273,000/mm3). These values fell throughout pregnancy to a mean of 217,000/mm3 by the time of delivery at a mean gestation of 39.0 weeks (P less than .001 for all time points). However, mean platelet counts rebounded by the time a postpartum value was obtained at a mean 7.1 weeks after delivery, to 264,000/mm3, a value that wasn’t significantly different from the nonpregnant cohort’s platelet counts.
When the investigators looked at mean platelet counts by trimester, they saw no difference between those with uncomplicated and complicated pregnancies until the third trimester. Then, “mean platelet counts decreased at a greater rate among women with pregnancy-related complications,” wrote Dr. Reese and her colleagues; 11.9% of women with complicated pregnancies had platelet counts below 150,000/mm3, while this level was seen in 9.9% of women without complications of pregnancy (P = .01).
At delivery, 2.3% (n = 59) of women with complicated pregnancies had platelet counts below 100,000/mm3, and 31 of these women had counts below 80,000/mm3, representing a significantly higher rate of thrombocytopenia at delivery than seen in the uncomplicated group (P less than .001).
In discussion, Dr. Reese and her coauthors examined the possible mechanisms for decreased levels of circulating platelets during pregnancy. Volume dilution from increased plasma volume is one well-accepted reason. Others include accumulation of platelets within the spleen, which increases in size by about 50% during pregnancy; similarly, the placenta’s circulation is similar to that of the spleen, so platelets may also accumulate there, the authors said. Further support for the placental mechanism comes from the lower average platelet counts for women with twin pregnancies.
The study’s relatively broad definition of pregnancy-related complications may have had the effect of lessening the difference in mean platelet counts between the complicated and uncomplicated pregnancy groups, the investigators acknowledged. Still, their study population had rates of these complications similar to those of the United States population, they said. “Therefore, our data may accurately reflect the platelet counts in women with these pregnancy-related complications,” they noted.
“Severe thrombocytopenia is rare, even in women with pregnancy-related complications,” concluded Dr. Reese and her colleagues. “Our data suggest that, for women with an uncomplicated pregnancy who have a platelet count of less than 100,000/mm3, a cause of thrombocytopenia other than the pregnancy itself should be considered.”
SOURCE: Reese J et al. N Engl J Med. 2018;379:32-43.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Platelet counts less than 150,000/mm3 were more common in complicated pregnancies.
Major finding: Platelet counts were below 150,000/mm3 in 11.9% of complicated versus 9.9% of uncomplicated pregnancies at the time of delivery (P = .01).
Study details: Review of records of 7,351 pregnant women delivering at a single site, compared with NHANES data for 8,885 nonpregnant women.
Disclosures: The National Institutes of Health funded the study. The authors reported having no conflicts of interest.
Source: Reese J et al. N Engl J Med. 2018;379:32-43.