User login
The role of aldosterone receptor antagonists in the management of heart failure: An update
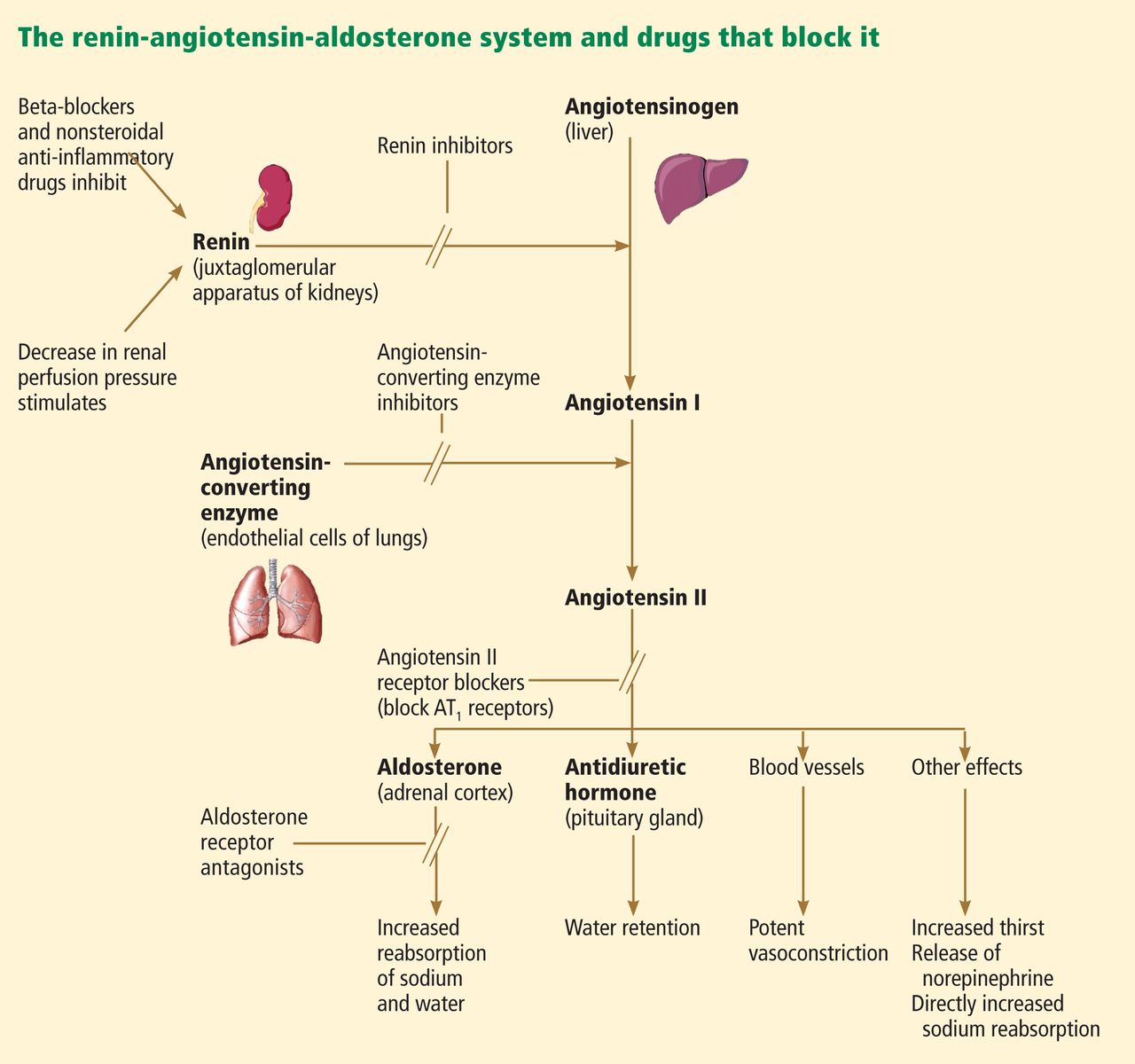
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23

Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
KEY POINTS
- Although caution is advised in starting ARAs, these drugs are commonly underused in heart failure.
- Aldosterone “escape” can blunt the effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. This is the rationale for also using ARAs.
- The major trials of ARAs in heart failure to date have been the Randomized Aldactone Evaluation Study (RALES), the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), and the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF).
- Close monitoring is essential when starting an ARA, as severe hyperkalemia and renal insufficiency can occur.
A woman with a swollen uvula
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
Immune thrombocytopenia in adults: An update
Immune thrombocytopenia (ITP), formerly known as idiopathic thrombocytopenic purpura, is an autoimmune disorder characterized by a low platelet count and increased risk of mucocutaneous bleeding. During the last decade its management has changed, with the advent of new medications and with increased awareness of treatment side effects. This article will focus on the pathophysiology, diagnosis, and management of ITP in adults.
A SLIGHT FEMALE PREDOMINANCE UNTIL AGE 65
The estimated age-adjusted prevalence of ITP in the United States is 9.5 to 23.6 cases per 100,000.1 In a recent study in the United Kingdom, the incidence was 4.4 per 100,000 patient-years among women and 3.4 among men.2 A slight female predominance was seen until age 65; thereafter, the incidence rates in men and women were about equal.
INCREASED PLATELET DESTRUCTION AND DECREASED PRODUCTION
ITP is a complex immune process in which cellular and humoral immunity are involved in the destruction of platelets3 as well as impaired platelet production. Several theories have emerged in the last decade to explain this autoimmune process.
Autoantibodies form against platelets
The triggering event for antibody initiation in ITP is unknown.3 Autoantibodies (mostly immunoglobulin G [IgG] but sometimes IgM and IgA) are produced against the platelet membrane glycoprotein GPIIb-IIIa. The antibody-coated platelets are rapidly cleared by the reticuloendothelial system in the spleen and liver, in a process mediated by Fc-receptor expression on macrophages and dendritic cells. Autoantibodies may also affect platelet production by inhibiting megakaryocyte maturation and inducing apoptosis.4,5
Patients with ITP also have CD4+ T cells that are autoreactive to GPIIb-IIIa and that stimulate B-cell clones to produce antiplatelet antibodies. Although autoreactive T cells are present in healthy individuals, they appear to be activated in patients with ITP by exposure to fragments of GPIIb-IIIa rather than native GPIIb-IIIa proteins.6 Activated macrophages internalize antibody-coated platelets and degrade GPIIb-IIIa and other glycoproteins to form “cryptic” epitopes that are expressed on the macrophage surface as novel peptides that induce further proliferation of CD4+ T-cell clones. Epitope spread thereby sustains a continuous loop that amplifies the production of GPIIb-IIIa antibodies.7
Defective T-regulatory cells appear to be critical to the pathogenesis of ITP by breaking self-tolerance, allowing the autoimmune process to progress.8 This, together with several other immune mechanisms such as molecular mimicry, abnormal cytokine profile, and B-cell abnormalities, may lead to enhanced platelet clearance.9
In addition to destroying platelets, antibodies may impair platelet production.10 Good evidence for platelets being underproduced in patients with ITP is that treating with thrombopoietin agonists results in increased platelet counts.
A DIAGNOSIS OF EXCLUSION
ITP is defined as isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.11 No diagnostic criteria currently exist, and the diagnosis is established only after excluding other causes of thrombocytopenia.
A recent report12 from an international working group established a platelet count threshold of less than 100 × 109/L for diagnosing ITP, down from the previous threshold of 150 × 109/L. The panel also recommended using the term “immune” rather than “idiopathic” thrombocytopenia, emphasizing the role of underlying immune mechanisms. The term “purpura” was removed, because many patients have no or minimal signs of bleeding at the time of diagnosis.12
The 2011 American Society of Hematology’s evidenced-based guidelines for the treatment of ITP present the most recent authoritative diagnostic and therapeutic recommendations.13
ITP is considered to be primary if it occurs in isolation, and secondary if it is associated with an underlying disorder. It is further classified according to its duration since diagnosis: newly diagnosed (< 3 months), persistent (3−12 months), and chronic (> 12 months).
In adults, ITP tends to be chronic, presenting with a more indolent course than in childhood, and unlike childhood ITP, infrequently following a viral infection.
Clinical features associated with ITP are related to thrombocytopenia: petechiae (pinpoint microvascular hemorrhages that do not blanch with pressure), purpura (appearing like large bruises), epistaxis (nosebleeds), menorrhagia, gum bleeding, and other types of mucocutaneous bleeding. Other common clinical features include fatigue, impaired quality of life, and treatment-related side effects (eg, infection).14

A low platelet count may be the sole initial manifestation. The patient’s history, physical examination, blood counts, and findings on blood smear are essential to rule out other diagnoses. Few diagnostic tests are useful in the initial evaluation (Table 1). Abnormalities in the blood count or blood smear may be further investigated with bone marrow biopsy but is not required if the patient has typical features of ITP, regardless of age.
Because there are no specific criteria for diagnosing ITP, other causes of thrombocytopenia must be excluded. The differential diagnosis can be further classified as ITP due to other underlying disease (ie, secondary ITP) vs nonautoimmune causes that are frequently encountered in clinical practice.
SECONDARY ITP
The differential diagnosis of thrombocytopenia due to known underlying immune disease includes the following:
Drug-induced ITP
Recurrent episodes of acute thrombocytopenia not explained by other causes should trigger consideration of drug-induced thrombocytopenia. 11 Patients should be questioned about drug use, especially of sulfonamides, antiepileptics, and quinine. Thrombocytopenia usually occurs 5 to 7 days after beginning the inciting drug for the first time and more quickly when the drug is given intermittently. Heparin is the most common cause of drug-related thrombocytopenia among hospitalized patients; the mechanism is unique and involves formation of a heparin-PF4 immune complex.
Human immunodeficiency virus infection
Approximately 40% of patients with human immunodeficiency virus (HIV) infection develop thrombocytopenia at some time.15 HIV infection can initially manifest as isolated thrombocytopenia and is sometimes clinically indistinguishable from chronic ITP, making it an important consideration in a newly diagnosed case of thrombocytopenia.
The mechanism of thrombocytopenia in early HIV is similar to that in primary ITP: as the disease progresses, low platelet counts can result from ineffective hematopoiesis due to megakaryocyte infection and marrow infiltration.16
Hepatitis C virus infection
Hepatitis C virus (HCV) infection can also cause immune thrombocytopenia. A recent study demonstrated the potential of the HCV core envelope protein 1 to induce antiplatelet antibodies (to platelet surface integrin GPIIIa49-66) by molecular mimicry.17 Other causes of thrombocytopenia in HCV infection may be related to chronic liver disease, such as portal hypertension-related hypersplenism, as well as decreased thrombopoietin production.18 Antiviral treatment with pegylated interferon may also cause mild thrombocytopenia.19
Helicobacter pylori
The association between H pylori infection and ITP remains uncertain. Eradication of infection appears to completely correct ITP in some places where the prevalence of H pylori is high (eg, Italy and Japan) but not in the United States and Canada, where the prevalence is low.20 The different response may be due not only to the differences in prevalence, but to different H pylori genotypes: most H pylori strains in Japan express CagA, whereas the frequency of CagA-positive strains is much lower in western countries.20
In areas where eradication therapy may be useful, the presence of H pylori infection should be determined by either a urea breath test or stool antigen testing.
Lymphoproliferative disorders
Secondary forms of ITP can occur in association with chronic lymphocytic leukemia, non-Hodgkin lymphoma, and Hodgkin lymphoma. These diagnoses should especially be considered in patients presenting with thrombocytopenia accompanied by systemic illness. ITP occurs in at least 2% of patients with chronic lymphocytic leukemia and is usually difficult to distinguish from thrombocytopenia secondary to marrow infiltration or from fludarabine (Fludora) therapy.21
It is especially important to determine if a lymphoproliferative disorder is present because it changes the treatment of ITP. Treatment of ITP complicating chronic lymphocytic leukemia is challenging and includes corticosteroids and steroid-sparing agents such as cyclosporine (Gengraf, Neoral, Sandimmune), rituximab (Rituxan), and intravenous immunoglobulin.22
Systemic lupus erythematosus and other autoimmune diseases
Thrombocytopenia is a frequent clinical manifestation of systemic lupus erythematosus, occurring in 7% to 30% of patients,23 and is an independent risk factor for death.24 Lupus should be suspected in patients with ITP who have multiorgan involvement and other clinical and laboratory abnormalities. A small percentage of patients with ITP (about 2%−5%) develop lupus after several years.21
Thrombocytopenia can also result from other autoimmune disorders such as antiphospholipid antibody syndrome25 and autoimmune thyroid diseases as well as immunodeficient states such as IgA deficiency and common variable immunodeficiency with low IgG levels.
NONAUTOIMMUNE THROMBOCYTOPENIA
Thrombocytopenia can also be caused by a number of nonautoimmune conditions.
Pseudothrombocytopenia
Pseudothrombocytopenia can occur if ex-vivo agglutination of platelets is induced by antiplatelet antibodies to EDTA, a standard blood anticoagulant. Automated counters cannot differentiate the agglutinated platelet clumps from individual cells such as red cells. This can frequently be overcome by running the counts in a citrate or ACD reagent tube. A peripheral blood smear can demonstrate whether platelet clumps are present.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura presents with thrombocytopenia, purpura, and anemia. Associated clinical abnormalities (fever, neurologic symptoms, and renal failure) and the presence of fragmented red cells on blood smear help to distinguish it from ITP. Plasma exchange is the treatment of choice.
Gestational thrombocytopenia
Five percent of pregnant women develop mild thrombocytopenia (platelet counts typically > 70 × 109/L) near the end of gestation.26 It requires no treatment and resolves after delivery. The fetus’ platelet count remains unaffected.
Gestational thrombocytopenia should be differentiated from the severe thrombocytopenia of preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count), which requires immediate attention.
Myelodysplastic syndrome
Myelodysplastic syndrome is common among elderly patients and should be considered in cases of unexplained cytopenia and abnormalities in the peripheral blood smear suggestive of dysplastic cytologic features. It can be diagnosed by bone marrow biopsy. Thrombocytopenia occurs in about 40% to 65% of cases of myelodysplastic syndrome.27
MANAGE ITP TO KEEP PLATELET COUNT ABOVE 30 × 109/L
ITP does not necessarily require treatment, and the initial challenge is to determine whether treatment or observation is indicated. Treatment is based on two major factors: the platelet count and degree of bleeding. The goals of management are to achieve a safe platelet count to prevent serious bleeding while minimizing treatment-related toxicity.7

Adults with platelet counts of less than 30 × 109/L are usually treated. In multiple large cohort studies, patients with platelet counts above that level have been safely observed without treatment.11,28
Table 2 outlines a comprehensive approach to therapy.
INITIAL TREATMENT: STEROIDS AND IMMUNOGLOBULINS
Oral corticosteroids are the initial agents of choice
Oral prednisone 1 mg/kg/day in tapering doses for 4 to 6 weeks is the most common initial regimen. Other regimens, such as high-dose dexamethasone (Decadron) (40 mg daily for 4 days per month) for several cycles, have been reported to be more effective29 but have not been studied in head-to-head trials with oral prednisone.
Due to their effectiveness, low cost, and convenience of use, corticosteroids have been the backbone of initial treatment in ITP. However, in most patients the platelet count decreases once the dose is tapered or stopped; remission is sustained in only 10% to 30% of cases.30 Continuation of corticosteroids is limited by long-term complications such as opportunistic infections, osteoporosis, and emotional lability.31
Intravenous immunoglobulin and anti-D immunoglobulin are alternatives
Intravenous immunoglobulin is recommended for patients who have not responded to corticosteroids and is often used in pregnancy. It is thought to act by blocking Fc receptors in the reticuloendothelial system. Intravenous immunoglobulin rapidly increases platelet counts in 65% to 80% of patients,32 but the effect is transient and the drug requires frequent administration. It is usually well tolerated, although about 5% of patients experience headache, chills, myalgias, arthralgias, and back pain. Rare, serious complications include thrombotic events, anaphylaxis (in IgA-deficient patients), and renal failure.
Anti-D immunoglobulin, a pooled IgG product, is derived from the plasma of Rh(D)-negative donors and can be given only to patients who are Rh(D)-positive. Response rates as high as 70% have been reported, with platelet effects lasting for more than 21 days.33 Studies have shown better results at a high dose (75 μg/kg) than with the approved dose of 50 μg/kg.34
Anti-D immunoglobulin can also be given intermittently whenever the platelet count falls below a specific level (ie, 30 × 109/L). This allows some patients to avoid splenectomy and may even trigger long-term remission.32
Common side effects of anti-D immunoglobulin include fever and chills; these can be prevented by premedication with acetaminophen or corticosteroids. Rare but fatal cases of intravascular hemolysis, renal failure, and disseminated intravascular coagulation have been reported, precluding its use for ITP in some countries, including those of the European Union.
Emergency treatment: Combination therapy
Evidence-based guidelines are limited for treating patients with active bleeding or who are at high risk of bleeding. For uncontrolled bleeding, a combination of first-line therapies is recommended, using prednisone and intravenous immunoglobulin.35 Other options include high-dose methylprednisolone and platelet transfusions, alone or in combination with intravenous immunoglobulin.36
SECOND-LINE TREATMENTS
Splenectomy produces complete remission in most patients
Patients who relapse and have a platelet count of less than 20 × 109/L are traditionally considered for splenectomy. More than two-thirds of patients respond with no need for further treatment.37
Although splenectomy has the highest rate of durable platelet response, the risks associated with surgery are an important concern. Even with a laparoscopic splenectomy, complications occur in 10% of patients and death in 0.2%. Long-term risks include the rare occurrence of sepsis with an estimated mortality rate of 0.73 per 1,000 patient-years, and possible increased risk of thrombosis.38,39
Adherence to recommended vaccination protocols and early administration of antibiotics for systemic febrile illness reduce the risk of sepsis.40 Patients are advised to receive immunization against encapsulated bacteria with pneumococcal, Haemophilus influenzae type b, and meningococcal vaccines. These vaccines should be given at least 2 weeks before elective splenectomy.41
Treatment of patients refractory to splenectomy is challenging and requires further immunosuppressive therapy, which is associated with an increased risk of infections and infection-related deaths.42
Rituximab in addition to or possibly instead of splenectomy
Rituximab (Rituxan) is a chimeric anti-CD20 monoclonal antibody that targets B cells. Although initially approved for treatment of lymphomas, rituximab has gained popularity in treating ITP due to its safety profile and ability to deplete CD20+ B cells responsible for antiplatelet antibody production by Fc-mediated cell lysis.
In the largest systematic review of published reports of rituximab use in ITP (19 studies, 313 patients), Arnold and colleagues43 reported an overall platelet response (defined as platelet count > 50 × 109/L) in 62.5% (95% confidence interval [CI] 52.6%−72.5%) of patients. The median duration of response was 10.5 months (range 3–20), and median follow-up was 9.5 months (range 2–25). Nearly all patients had received corticosteroid treatment and half of them had undergone splenectomy.
Rituximab has also been investigated as an alternative to splenectomy. In a prospective, single-arm, phase 2 trial, 60 patients with chronic ITP (platelet counts < 30 × 109/L) for whom one or more previous treatments had failed received rituximab infusions and were followed for up to 2 years. A good response (defined as a platelet count ≥ 50 × 109/L, with at least a doubling from baseline) was obtained in 24 (40%) of 60 patients (95% CI 28%–52%) at 1 year and 33.3% at 2 years. The authors concluded that rituximab could be used as a presplenectomy therapeutic option, particularly in patients with chronic ITP who are at increased surgical risk or who are reluctant to undergo surgery.44 Based on these results, rituximab may spare some patients from splenectomy, or at least delay it. However, it has never been tested in randomized controlled trials to establish its role as a splenectomy-sparing agent in ITP.
Side effects include infusion reactions, which are usually mild but in rare cases can be severe. Recently, progressive multifocal leukoencephalopathy has been recognized as a complication of rituximab treatment in patients with lymphoproliferative and autoimmune disorders.45 Although this complication is rare in patients with ITP, careful monitoring is required until additional long-term safety data are available.
Thrombopoietic receptor agonists require continuous treatment
In the early 1990s, recombinant thrombopoietin was tested in clinical studies. These were halted when antibodies developed to recombinant thrombopoietin that cross-reacted with endogenous thrombopoietin, resulting in severe thrombocytopenia.46
This led to the development of nonimmunogenic thrombopoietin receptor agonists that mimic the effect of thrombopoietin and stimulate the production of platelets. In 2008, the US Food and Drug Administration approved two drugs of this class for treating ITP: romiplostim (Nplate) and eltrombopag (Promacta). They are mainly used to treat patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Although well tolerated and effective in increasing platelet counts, these agents share common drawbacks. They do not modify the course of the disease, they are used only to sustain the platelet count, they require repeated administration, and they must be given for about 7 days to achieve an adequate platelet response, so they cannot be used in emergencies. Long-term adverse effects include bone marrow fibrosis and thrombosis.
Romiplostim is a synthetic peptide capable of binding to the thrombopoietin receptor c-Mpl. It has no sequence homology with endogenous thrombopoietin,47 so does not induce cross-reacting antibodies. It has a half-life of 120 to 160 hours and is usually given subcutaneously 1 to 10 μg/kg weekly.
Phase III clinical trials have shown the effectiveness of romiplostim in attaining a durable platelet response (platelet count > 50 × 109/L) in splenectomized and nonsplenectomized populations. It is well tolerated, and only two uncommon serious adverse effects have been reported: bone marrow reticulin formation and thromboembolism.48
A long-term open-label extension study of 142 patients treated with romiplostim for up to 156 weeks showed that 124 (87%) achieved a platelet count of more than 50 × 109/L at some point, and 84% of patients were able to reduce or discontinue concurrent medications for ITP.49
Kuter et al,50 in a randomized controlled trial, confirmed the efficacy of romiplostim in attaining durable increased platelet counts. Patients treated with romiplostim at a mean weekly dose of 3.9 μg/kg ± 2.1 μg/kg demonstrated a higher rate of platelet response, lower incidence of treatment failure, and improved quality of life vs patients treated with standard care.
Eltrombopag is a nonpeptide thrombopoietin agonist that binds to the transmembrane domain of the thrombopoietin receptor and stimulates the proliferation and differentiation of megakaryocytes in bone marrow. It is given orally in doses of 25 to 75 mg daily.
Eltrombopag has been shown to be effective in increasing platelet counts in chronic ITP.51 In a phase III trial conducted by Cheng and colleagues, 197 patients were randomized to eltrombopag or placebo.52 Patients treated with eltrombopag were eight times more likely to achieve platelet counts of more than 50 × 109/L during the 6-month treatment period (odds ratio 8.2, 95% CI 4.32–15.38, P < .001) vs placebo. Patients treated with eltrombopag had fewer bleeding episodes and were more likely to reduce or discontinue the dose of concurrent ITP medications. The only significant side effect seen was a rise in aminotransferases (seen in 7% of eltrombopag recipients vs 2% with placebo).52
Additional thrombopoietin agonists under investigation include ARK-501, totrombopag, and LGD-4665. MDX-33, a monoclonal antibody against the Fc-receptor, is also being studied; it acts by preventing opsonization of autoantibody-coated platelets.53
THIRD-LINE TREATMENTS FOR REFRACTORY CASES
Patients with ITP that is resistant to standard therapies have an increased risk of death, disease, and treatment-related complications.28,42
Combination chemotherapy
Immunosuppressants such as azathioprine (Imuran), cyclosporine (Neoral, Sandimmune), cyclophosphamide (Cytoxan), and mycophenolate (CellCept) were used in the past in single-agent regimens with some efficacy, but their use was limited due to drug-related toxicity and a low safety profile.3 However, there is increasing evidence for a role of combination chemotherapy to treat chronic refractory ITP to achieve greater efficacy and fewer adverse effects.54
Arnold and colleagues55 reported that combined azathioprine, mycophenolate, and cyclosporine achieved an overall response (platelet count > 30 × 109/L and doubling of the baseline) in 14 (73.7%) of 19 patients with chronic refractory ITP, lasting a median of 24 months.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation has provided remission in a limited number of patients. However, it is associated with fatal toxicities such as graft-vs-host disease and septicemia, and therefore it is reserved for severe refractory ITP with bleeding complications unresponsive to other therapies.56,57
THERAPY FOR SECONDARY ITP DEPENDS ON THE CAUSE
Treatments for secondary ITP vary depending on the cause of thrombocytopenia and are often more complex than therapy for primary disease. Optimal management involves treating the underlying condition (eg, chronic lymphocytic leukemia or systemic lupus erythematosus).
Drug-induced thrombocytopenia requires prompt recognition and withdrawal of the inciting agent.
Treating ITP due to HCV infection primarily involves antiviral agents to suppress viral replication. If treating ITP is required, then intravenous immunoglobulin is preferable to glucocorticoids because of the risk of increasing viral load with the latter.58 Eltrombopag may effectively increase platelet counts, allowing patients to receive interferon therapy for HCV.59 However, a recent study was halted due to increased incidence of portal vein thrombosis, raising concerns about the safety of eltrombopag for patients with chronic liver disease.60
Secondary ITP due to HIV infection should always be treated first with antivirals targeting HIV unless thrombocytopenia-related bleeding complications warrant treatment. If treatment for ITP is necessary, it should include corticosteroids, intravenous immunoglobulin, or anti-D immunoglobulin as first-line therapy.
Eradication therapy for H pylori is recommended for patients who are positive for the organism based on urea breath testing, stool antigen testing, or endoscopic biopsies.
- Feudjo-Tepie MA, Robinson NJ, Bennett D. Prevalence of diagnosed chronic immune thrombocytopenic purpura in the US: analysis of a large US claim database: a rebuttal. J Thromb Haemost 2008; 6:711–712.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol 2009; 83:83–89.
- Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist 2009; 14:12–21.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet auto-antibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Houwerzijl EJ, Blom NR, van der Want JJ, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004; 103:500–506.
- Kuwana M, Kaburaki J, Kitasato H, et al. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by autoreactive T cells in patients with immune thrombocytopenic purpura. Blood 2001; 98:130–139.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–858.
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol 2010; 17:590–595.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94:759–762.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207.
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011; 86:420–429.
- Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1275–1297.
- Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998; 91:1479–1495.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Peck-Radosavljevic M. Thrombocytopenia in liver disease. Can J Gastroenterol 2000; 14(suppl D):60D–66D.
- Roomer R, Hansen BE, Janssen HL, de Knegt RJ. Thrombocytopenia and the risk of bleeding during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. J Hepatol 2010; 53:455–459.
- Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113:1231–1240.
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113:6511–6521.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010; 23:47–59.
- Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:2243–2254.
- Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000; 39:399–406.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46:1019–1027.
- Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 1993; 329:1463–1466.
- Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007; 109:1705–1714.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Cheng Y, Wong RS, Soo YO, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 2003; 349:831–836.
- Bromberg ME. Immune thrombocytopenic purpura—the changing therapeutic landscape. N Engl J Med 2006; 355:1643–1645.
- Guidry JA, George JN, Vesely SK, Kennison SM, Terrell DR. Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83:175–182.
- Cooper N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1317–1327.
- Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 1997; 89:2689–2700.
- Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001; 112:1076–1078.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83:122–125.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Schilling RF. Estimating the risk for sepsis after splenectomy in hereditary spherocytosis. Ann Intern Med 1995; 122:187–188.
- Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood 2009; 114:2861–2868.
- Davies JM, Barnes R, Milligan D; British Committee for Standards in Haematology. Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med 2002; 2:440–443.
- Centers for Disease Control and Prevention (CDC). Recommended adult immunization schedule—United States, 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1–4.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104:956–960.
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146:25–33.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 2008; 112:999–1004.
- Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113:4834–4840.
- Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001; 98:3241–3248.
- Kuter DJ. New thrombopoietic growth factors. Blood 2007; 109:4607–4616.
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009; 113:2161–2171.
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363:1889–1899.
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:641–648.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011; 377:393–402.
- Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert Opin Investig Drugs 2009; 18:805–819.
- Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526–3531.
- Arnold DM, Nazi I, Santos A, et al. Combination immunosuppressant therapy for patients with chronic refractory immune thrombocytopenic purpura. Blood 2010; 115:29–31.
- Passweg JR, Rabusin M. Hematopoetic stem cell transplantation for immune thrombocytopenia and other refractory autoimmune cytopenias. Autoimmunity 2008; 41:660–665.
- Huhn RD, Fogarty PF, Nakamura R, et al. High-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopenia. Blood 2003; 101:71–77.
- Magrin S, Craxi A, Fabiano C, et al. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology 1994; 19:273–279.
- McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med 2007; 357:2227–2236.
- US Department of Health & Human Services. Promacta (eltrombopag): Portal Venous System Thromboses in Study of Patients With Chronic Liver Disease http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm211796.htm. Accessed June 27, 2012.
Immune thrombocytopenia (ITP), formerly known as idiopathic thrombocytopenic purpura, is an autoimmune disorder characterized by a low platelet count and increased risk of mucocutaneous bleeding. During the last decade its management has changed, with the advent of new medications and with increased awareness of treatment side effects. This article will focus on the pathophysiology, diagnosis, and management of ITP in adults.
A SLIGHT FEMALE PREDOMINANCE UNTIL AGE 65
The estimated age-adjusted prevalence of ITP in the United States is 9.5 to 23.6 cases per 100,000.1 In a recent study in the United Kingdom, the incidence was 4.4 per 100,000 patient-years among women and 3.4 among men.2 A slight female predominance was seen until age 65; thereafter, the incidence rates in men and women were about equal.
INCREASED PLATELET DESTRUCTION AND DECREASED PRODUCTION
ITP is a complex immune process in which cellular and humoral immunity are involved in the destruction of platelets3 as well as impaired platelet production. Several theories have emerged in the last decade to explain this autoimmune process.
Autoantibodies form against platelets
The triggering event for antibody initiation in ITP is unknown.3 Autoantibodies (mostly immunoglobulin G [IgG] but sometimes IgM and IgA) are produced against the platelet membrane glycoprotein GPIIb-IIIa. The antibody-coated platelets are rapidly cleared by the reticuloendothelial system in the spleen and liver, in a process mediated by Fc-receptor expression on macrophages and dendritic cells. Autoantibodies may also affect platelet production by inhibiting megakaryocyte maturation and inducing apoptosis.4,5
Patients with ITP also have CD4+ T cells that are autoreactive to GPIIb-IIIa and that stimulate B-cell clones to produce antiplatelet antibodies. Although autoreactive T cells are present in healthy individuals, they appear to be activated in patients with ITP by exposure to fragments of GPIIb-IIIa rather than native GPIIb-IIIa proteins.6 Activated macrophages internalize antibody-coated platelets and degrade GPIIb-IIIa and other glycoproteins to form “cryptic” epitopes that are expressed on the macrophage surface as novel peptides that induce further proliferation of CD4+ T-cell clones. Epitope spread thereby sustains a continuous loop that amplifies the production of GPIIb-IIIa antibodies.7
Defective T-regulatory cells appear to be critical to the pathogenesis of ITP by breaking self-tolerance, allowing the autoimmune process to progress.8 This, together with several other immune mechanisms such as molecular mimicry, abnormal cytokine profile, and B-cell abnormalities, may lead to enhanced platelet clearance.9
In addition to destroying platelets, antibodies may impair platelet production.10 Good evidence for platelets being underproduced in patients with ITP is that treating with thrombopoietin agonists results in increased platelet counts.
A DIAGNOSIS OF EXCLUSION
ITP is defined as isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.11 No diagnostic criteria currently exist, and the diagnosis is established only after excluding other causes of thrombocytopenia.
A recent report12 from an international working group established a platelet count threshold of less than 100 × 109/L for diagnosing ITP, down from the previous threshold of 150 × 109/L. The panel also recommended using the term “immune” rather than “idiopathic” thrombocytopenia, emphasizing the role of underlying immune mechanisms. The term “purpura” was removed, because many patients have no or minimal signs of bleeding at the time of diagnosis.12
The 2011 American Society of Hematology’s evidenced-based guidelines for the treatment of ITP present the most recent authoritative diagnostic and therapeutic recommendations.13
ITP is considered to be primary if it occurs in isolation, and secondary if it is associated with an underlying disorder. It is further classified according to its duration since diagnosis: newly diagnosed (< 3 months), persistent (3−12 months), and chronic (> 12 months).
In adults, ITP tends to be chronic, presenting with a more indolent course than in childhood, and unlike childhood ITP, infrequently following a viral infection.
Clinical features associated with ITP are related to thrombocytopenia: petechiae (pinpoint microvascular hemorrhages that do not blanch with pressure), purpura (appearing like large bruises), epistaxis (nosebleeds), menorrhagia, gum bleeding, and other types of mucocutaneous bleeding. Other common clinical features include fatigue, impaired quality of life, and treatment-related side effects (eg, infection).14
A low platelet count may be the sole initial manifestation. The patient’s history, physical examination, blood counts, and findings on blood smear are essential to rule out other diagnoses. Few diagnostic tests are useful in the initial evaluation (Table 1). Abnormalities in the blood count or blood smear may be further investigated with bone marrow biopsy but is not required if the patient has typical features of ITP, regardless of age.
Because there are no specific criteria for diagnosing ITP, other causes of thrombocytopenia must be excluded. The differential diagnosis can be further classified as ITP due to other underlying disease (ie, secondary ITP) vs nonautoimmune causes that are frequently encountered in clinical practice.
SECONDARY ITP
The differential diagnosis of thrombocytopenia due to known underlying immune disease includes the following:
Drug-induced ITP
Recurrent episodes of acute thrombocytopenia not explained by other causes should trigger consideration of drug-induced thrombocytopenia. 11 Patients should be questioned about drug use, especially of sulfonamides, antiepileptics, and quinine. Thrombocytopenia usually occurs 5 to 7 days after beginning the inciting drug for the first time and more quickly when the drug is given intermittently. Heparin is the most common cause of drug-related thrombocytopenia among hospitalized patients; the mechanism is unique and involves formation of a heparin-PF4 immune complex.
Human immunodeficiency virus infection
Approximately 40% of patients with human immunodeficiency virus (HIV) infection develop thrombocytopenia at some time.15 HIV infection can initially manifest as isolated thrombocytopenia and is sometimes clinically indistinguishable from chronic ITP, making it an important consideration in a newly diagnosed case of thrombocytopenia.
The mechanism of thrombocytopenia in early HIV is similar to that in primary ITP: as the disease progresses, low platelet counts can result from ineffective hematopoiesis due to megakaryocyte infection and marrow infiltration.16
Hepatitis C virus infection
Hepatitis C virus (HCV) infection can also cause immune thrombocytopenia. A recent study demonstrated the potential of the HCV core envelope protein 1 to induce antiplatelet antibodies (to platelet surface integrin GPIIIa49-66) by molecular mimicry.17 Other causes of thrombocytopenia in HCV infection may be related to chronic liver disease, such as portal hypertension-related hypersplenism, as well as decreased thrombopoietin production.18 Antiviral treatment with pegylated interferon may also cause mild thrombocytopenia.19
Helicobacter pylori
The association between H pylori infection and ITP remains uncertain. Eradication of infection appears to completely correct ITP in some places where the prevalence of H pylori is high (eg, Italy and Japan) but not in the United States and Canada, where the prevalence is low.20 The different response may be due not only to the differences in prevalence, but to different H pylori genotypes: most H pylori strains in Japan express CagA, whereas the frequency of CagA-positive strains is much lower in western countries.20
In areas where eradication therapy may be useful, the presence of H pylori infection should be determined by either a urea breath test or stool antigen testing.
Lymphoproliferative disorders
Secondary forms of ITP can occur in association with chronic lymphocytic leukemia, non-Hodgkin lymphoma, and Hodgkin lymphoma. These diagnoses should especially be considered in patients presenting with thrombocytopenia accompanied by systemic illness. ITP occurs in at least 2% of patients with chronic lymphocytic leukemia and is usually difficult to distinguish from thrombocytopenia secondary to marrow infiltration or from fludarabine (Fludora) therapy.21
It is especially important to determine if a lymphoproliferative disorder is present because it changes the treatment of ITP. Treatment of ITP complicating chronic lymphocytic leukemia is challenging and includes corticosteroids and steroid-sparing agents such as cyclosporine (Gengraf, Neoral, Sandimmune), rituximab (Rituxan), and intravenous immunoglobulin.22
Systemic lupus erythematosus and other autoimmune diseases
Thrombocytopenia is a frequent clinical manifestation of systemic lupus erythematosus, occurring in 7% to 30% of patients,23 and is an independent risk factor for death.24 Lupus should be suspected in patients with ITP who have multiorgan involvement and other clinical and laboratory abnormalities. A small percentage of patients with ITP (about 2%−5%) develop lupus after several years.21
Thrombocytopenia can also result from other autoimmune disorders such as antiphospholipid antibody syndrome25 and autoimmune thyroid diseases as well as immunodeficient states such as IgA deficiency and common variable immunodeficiency with low IgG levels.
NONAUTOIMMUNE THROMBOCYTOPENIA
Thrombocytopenia can also be caused by a number of nonautoimmune conditions.
Pseudothrombocytopenia
Pseudothrombocytopenia can occur if ex-vivo agglutination of platelets is induced by antiplatelet antibodies to EDTA, a standard blood anticoagulant. Automated counters cannot differentiate the agglutinated platelet clumps from individual cells such as red cells. This can frequently be overcome by running the counts in a citrate or ACD reagent tube. A peripheral blood smear can demonstrate whether platelet clumps are present.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura presents with thrombocytopenia, purpura, and anemia. Associated clinical abnormalities (fever, neurologic symptoms, and renal failure) and the presence of fragmented red cells on blood smear help to distinguish it from ITP. Plasma exchange is the treatment of choice.
Gestational thrombocytopenia
Five percent of pregnant women develop mild thrombocytopenia (platelet counts typically > 70 × 109/L) near the end of gestation.26 It requires no treatment and resolves after delivery. The fetus’ platelet count remains unaffected.
Gestational thrombocytopenia should be differentiated from the severe thrombocytopenia of preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count), which requires immediate attention.
Myelodysplastic syndrome
Myelodysplastic syndrome is common among elderly patients and should be considered in cases of unexplained cytopenia and abnormalities in the peripheral blood smear suggestive of dysplastic cytologic features. It can be diagnosed by bone marrow biopsy. Thrombocytopenia occurs in about 40% to 65% of cases of myelodysplastic syndrome.27
MANAGE ITP TO KEEP PLATELET COUNT ABOVE 30 × 109/L
ITP does not necessarily require treatment, and the initial challenge is to determine whether treatment or observation is indicated. Treatment is based on two major factors: the platelet count and degree of bleeding. The goals of management are to achieve a safe platelet count to prevent serious bleeding while minimizing treatment-related toxicity.7
Adults with platelet counts of less than 30 × 109/L are usually treated. In multiple large cohort studies, patients with platelet counts above that level have been safely observed without treatment.11,28
Table 2 outlines a comprehensive approach to therapy.
INITIAL TREATMENT: STEROIDS AND IMMUNOGLOBULINS
Oral corticosteroids are the initial agents of choice
Oral prednisone 1 mg/kg/day in tapering doses for 4 to 6 weeks is the most common initial regimen. Other regimens, such as high-dose dexamethasone (Decadron) (40 mg daily for 4 days per month) for several cycles, have been reported to be more effective29 but have not been studied in head-to-head trials with oral prednisone.
Due to their effectiveness, low cost, and convenience of use, corticosteroids have been the backbone of initial treatment in ITP. However, in most patients the platelet count decreases once the dose is tapered or stopped; remission is sustained in only 10% to 30% of cases.30 Continuation of corticosteroids is limited by long-term complications such as opportunistic infections, osteoporosis, and emotional lability.31
Intravenous immunoglobulin and anti-D immunoglobulin are alternatives
Intravenous immunoglobulin is recommended for patients who have not responded to corticosteroids and is often used in pregnancy. It is thought to act by blocking Fc receptors in the reticuloendothelial system. Intravenous immunoglobulin rapidly increases platelet counts in 65% to 80% of patients,32 but the effect is transient and the drug requires frequent administration. It is usually well tolerated, although about 5% of patients experience headache, chills, myalgias, arthralgias, and back pain. Rare, serious complications include thrombotic events, anaphylaxis (in IgA-deficient patients), and renal failure.
Anti-D immunoglobulin, a pooled IgG product, is derived from the plasma of Rh(D)-negative donors and can be given only to patients who are Rh(D)-positive. Response rates as high as 70% have been reported, with platelet effects lasting for more than 21 days.33 Studies have shown better results at a high dose (75 μg/kg) than with the approved dose of 50 μg/kg.34
Anti-D immunoglobulin can also be given intermittently whenever the platelet count falls below a specific level (ie, 30 × 109/L). This allows some patients to avoid splenectomy and may even trigger long-term remission.32
Common side effects of anti-D immunoglobulin include fever and chills; these can be prevented by premedication with acetaminophen or corticosteroids. Rare but fatal cases of intravascular hemolysis, renal failure, and disseminated intravascular coagulation have been reported, precluding its use for ITP in some countries, including those of the European Union.
Emergency treatment: Combination therapy
Evidence-based guidelines are limited for treating patients with active bleeding or who are at high risk of bleeding. For uncontrolled bleeding, a combination of first-line therapies is recommended, using prednisone and intravenous immunoglobulin.35 Other options include high-dose methylprednisolone and platelet transfusions, alone or in combination with intravenous immunoglobulin.36
SECOND-LINE TREATMENTS
Splenectomy produces complete remission in most patients
Patients who relapse and have a platelet count of less than 20 × 109/L are traditionally considered for splenectomy. More than two-thirds of patients respond with no need for further treatment.37
Although splenectomy has the highest rate of durable platelet response, the risks associated with surgery are an important concern. Even with a laparoscopic splenectomy, complications occur in 10% of patients and death in 0.2%. Long-term risks include the rare occurrence of sepsis with an estimated mortality rate of 0.73 per 1,000 patient-years, and possible increased risk of thrombosis.38,39
Adherence to recommended vaccination protocols and early administration of antibiotics for systemic febrile illness reduce the risk of sepsis.40 Patients are advised to receive immunization against encapsulated bacteria with pneumococcal, Haemophilus influenzae type b, and meningococcal vaccines. These vaccines should be given at least 2 weeks before elective splenectomy.41
Treatment of patients refractory to splenectomy is challenging and requires further immunosuppressive therapy, which is associated with an increased risk of infections and infection-related deaths.42
Rituximab in addition to or possibly instead of splenectomy
Rituximab (Rituxan) is a chimeric anti-CD20 monoclonal antibody that targets B cells. Although initially approved for treatment of lymphomas, rituximab has gained popularity in treating ITP due to its safety profile and ability to deplete CD20+ B cells responsible for antiplatelet antibody production by Fc-mediated cell lysis.
In the largest systematic review of published reports of rituximab use in ITP (19 studies, 313 patients), Arnold and colleagues43 reported an overall platelet response (defined as platelet count > 50 × 109/L) in 62.5% (95% confidence interval [CI] 52.6%−72.5%) of patients. The median duration of response was 10.5 months (range 3–20), and median follow-up was 9.5 months (range 2–25). Nearly all patients had received corticosteroid treatment and half of them had undergone splenectomy.
Rituximab has also been investigated as an alternative to splenectomy. In a prospective, single-arm, phase 2 trial, 60 patients with chronic ITP (platelet counts < 30 × 109/L) for whom one or more previous treatments had failed received rituximab infusions and were followed for up to 2 years. A good response (defined as a platelet count ≥ 50 × 109/L, with at least a doubling from baseline) was obtained in 24 (40%) of 60 patients (95% CI 28%–52%) at 1 year and 33.3% at 2 years. The authors concluded that rituximab could be used as a presplenectomy therapeutic option, particularly in patients with chronic ITP who are at increased surgical risk or who are reluctant to undergo surgery.44 Based on these results, rituximab may spare some patients from splenectomy, or at least delay it. However, it has never been tested in randomized controlled trials to establish its role as a splenectomy-sparing agent in ITP.
Side effects include infusion reactions, which are usually mild but in rare cases can be severe. Recently, progressive multifocal leukoencephalopathy has been recognized as a complication of rituximab treatment in patients with lymphoproliferative and autoimmune disorders.45 Although this complication is rare in patients with ITP, careful monitoring is required until additional long-term safety data are available.
Thrombopoietic receptor agonists require continuous treatment
In the early 1990s, recombinant thrombopoietin was tested in clinical studies. These were halted when antibodies developed to recombinant thrombopoietin that cross-reacted with endogenous thrombopoietin, resulting in severe thrombocytopenia.46
This led to the development of nonimmunogenic thrombopoietin receptor agonists that mimic the effect of thrombopoietin and stimulate the production of platelets. In 2008, the US Food and Drug Administration approved two drugs of this class for treating ITP: romiplostim (Nplate) and eltrombopag (Promacta). They are mainly used to treat patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Although well tolerated and effective in increasing platelet counts, these agents share common drawbacks. They do not modify the course of the disease, they are used only to sustain the platelet count, they require repeated administration, and they must be given for about 7 days to achieve an adequate platelet response, so they cannot be used in emergencies. Long-term adverse effects include bone marrow fibrosis and thrombosis.
Romiplostim is a synthetic peptide capable of binding to the thrombopoietin receptor c-Mpl. It has no sequence homology with endogenous thrombopoietin,47 so does not induce cross-reacting antibodies. It has a half-life of 120 to 160 hours and is usually given subcutaneously 1 to 10 μg/kg weekly.
Phase III clinical trials have shown the effectiveness of romiplostim in attaining a durable platelet response (platelet count > 50 × 109/L) in splenectomized and nonsplenectomized populations. It is well tolerated, and only two uncommon serious adverse effects have been reported: bone marrow reticulin formation and thromboembolism.48
A long-term open-label extension study of 142 patients treated with romiplostim for up to 156 weeks showed that 124 (87%) achieved a platelet count of more than 50 × 109/L at some point, and 84% of patients were able to reduce or discontinue concurrent medications for ITP.49
Kuter et al,50 in a randomized controlled trial, confirmed the efficacy of romiplostim in attaining durable increased platelet counts. Patients treated with romiplostim at a mean weekly dose of 3.9 μg/kg ± 2.1 μg/kg demonstrated a higher rate of platelet response, lower incidence of treatment failure, and improved quality of life vs patients treated with standard care.
Eltrombopag is a nonpeptide thrombopoietin agonist that binds to the transmembrane domain of the thrombopoietin receptor and stimulates the proliferation and differentiation of megakaryocytes in bone marrow. It is given orally in doses of 25 to 75 mg daily.
Eltrombopag has been shown to be effective in increasing platelet counts in chronic ITP.51 In a phase III trial conducted by Cheng and colleagues, 197 patients were randomized to eltrombopag or placebo.52 Patients treated with eltrombopag were eight times more likely to achieve platelet counts of more than 50 × 109/L during the 6-month treatment period (odds ratio 8.2, 95% CI 4.32–15.38, P < .001) vs placebo. Patients treated with eltrombopag had fewer bleeding episodes and were more likely to reduce or discontinue the dose of concurrent ITP medications. The only significant side effect seen was a rise in aminotransferases (seen in 7% of eltrombopag recipients vs 2% with placebo).52
Additional thrombopoietin agonists under investigation include ARK-501, totrombopag, and LGD-4665. MDX-33, a monoclonal antibody against the Fc-receptor, is also being studied; it acts by preventing opsonization of autoantibody-coated platelets.53
THIRD-LINE TREATMENTS FOR REFRACTORY CASES
Patients with ITP that is resistant to standard therapies have an increased risk of death, disease, and treatment-related complications.28,42
Combination chemotherapy
Immunosuppressants such as azathioprine (Imuran), cyclosporine (Neoral, Sandimmune), cyclophosphamide (Cytoxan), and mycophenolate (CellCept) were used in the past in single-agent regimens with some efficacy, but their use was limited due to drug-related toxicity and a low safety profile.3 However, there is increasing evidence for a role of combination chemotherapy to treat chronic refractory ITP to achieve greater efficacy and fewer adverse effects.54
Arnold and colleagues55 reported that combined azathioprine, mycophenolate, and cyclosporine achieved an overall response (platelet count > 30 × 109/L and doubling of the baseline) in 14 (73.7%) of 19 patients with chronic refractory ITP, lasting a median of 24 months.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation has provided remission in a limited number of patients. However, it is associated with fatal toxicities such as graft-vs-host disease and septicemia, and therefore it is reserved for severe refractory ITP with bleeding complications unresponsive to other therapies.56,57
THERAPY FOR SECONDARY ITP DEPENDS ON THE CAUSE
Treatments for secondary ITP vary depending on the cause of thrombocytopenia and are often more complex than therapy for primary disease. Optimal management involves treating the underlying condition (eg, chronic lymphocytic leukemia or systemic lupus erythematosus).
Drug-induced thrombocytopenia requires prompt recognition and withdrawal of the inciting agent.
Treating ITP due to HCV infection primarily involves antiviral agents to suppress viral replication. If treating ITP is required, then intravenous immunoglobulin is preferable to glucocorticoids because of the risk of increasing viral load with the latter.58 Eltrombopag may effectively increase platelet counts, allowing patients to receive interferon therapy for HCV.59 However, a recent study was halted due to increased incidence of portal vein thrombosis, raising concerns about the safety of eltrombopag for patients with chronic liver disease.60
Secondary ITP due to HIV infection should always be treated first with antivirals targeting HIV unless thrombocytopenia-related bleeding complications warrant treatment. If treatment for ITP is necessary, it should include corticosteroids, intravenous immunoglobulin, or anti-D immunoglobulin as first-line therapy.
Eradication therapy for H pylori is recommended for patients who are positive for the organism based on urea breath testing, stool antigen testing, or endoscopic biopsies.
Immune thrombocytopenia (ITP), formerly known as idiopathic thrombocytopenic purpura, is an autoimmune disorder characterized by a low platelet count and increased risk of mucocutaneous bleeding. During the last decade its management has changed, with the advent of new medications and with increased awareness of treatment side effects. This article will focus on the pathophysiology, diagnosis, and management of ITP in adults.
A SLIGHT FEMALE PREDOMINANCE UNTIL AGE 65
The estimated age-adjusted prevalence of ITP in the United States is 9.5 to 23.6 cases per 100,000.1 In a recent study in the United Kingdom, the incidence was 4.4 per 100,000 patient-years among women and 3.4 among men.2 A slight female predominance was seen until age 65; thereafter, the incidence rates in men and women were about equal.
INCREASED PLATELET DESTRUCTION AND DECREASED PRODUCTION
ITP is a complex immune process in which cellular and humoral immunity are involved in the destruction of platelets3 as well as impaired platelet production. Several theories have emerged in the last decade to explain this autoimmune process.
Autoantibodies form against platelets
The triggering event for antibody initiation in ITP is unknown.3 Autoantibodies (mostly immunoglobulin G [IgG] but sometimes IgM and IgA) are produced against the platelet membrane glycoprotein GPIIb-IIIa. The antibody-coated platelets are rapidly cleared by the reticuloendothelial system in the spleen and liver, in a process mediated by Fc-receptor expression on macrophages and dendritic cells. Autoantibodies may also affect platelet production by inhibiting megakaryocyte maturation and inducing apoptosis.4,5
Patients with ITP also have CD4+ T cells that are autoreactive to GPIIb-IIIa and that stimulate B-cell clones to produce antiplatelet antibodies. Although autoreactive T cells are present in healthy individuals, they appear to be activated in patients with ITP by exposure to fragments of GPIIb-IIIa rather than native GPIIb-IIIa proteins.6 Activated macrophages internalize antibody-coated platelets and degrade GPIIb-IIIa and other glycoproteins to form “cryptic” epitopes that are expressed on the macrophage surface as novel peptides that induce further proliferation of CD4+ T-cell clones. Epitope spread thereby sustains a continuous loop that amplifies the production of GPIIb-IIIa antibodies.7
Defective T-regulatory cells appear to be critical to the pathogenesis of ITP by breaking self-tolerance, allowing the autoimmune process to progress.8 This, together with several other immune mechanisms such as molecular mimicry, abnormal cytokine profile, and B-cell abnormalities, may lead to enhanced platelet clearance.9
In addition to destroying platelets, antibodies may impair platelet production.10 Good evidence for platelets being underproduced in patients with ITP is that treating with thrombopoietin agonists results in increased platelet counts.
A DIAGNOSIS OF EXCLUSION
ITP is defined as isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.11 No diagnostic criteria currently exist, and the diagnosis is established only after excluding other causes of thrombocytopenia.
A recent report12 from an international working group established a platelet count threshold of less than 100 × 109/L for diagnosing ITP, down from the previous threshold of 150 × 109/L. The panel also recommended using the term “immune” rather than “idiopathic” thrombocytopenia, emphasizing the role of underlying immune mechanisms. The term “purpura” was removed, because many patients have no or minimal signs of bleeding at the time of diagnosis.12
The 2011 American Society of Hematology’s evidenced-based guidelines for the treatment of ITP present the most recent authoritative diagnostic and therapeutic recommendations.13
ITP is considered to be primary if it occurs in isolation, and secondary if it is associated with an underlying disorder. It is further classified according to its duration since diagnosis: newly diagnosed (< 3 months), persistent (3−12 months), and chronic (> 12 months).
In adults, ITP tends to be chronic, presenting with a more indolent course than in childhood, and unlike childhood ITP, infrequently following a viral infection.
Clinical features associated with ITP are related to thrombocytopenia: petechiae (pinpoint microvascular hemorrhages that do not blanch with pressure), purpura (appearing like large bruises), epistaxis (nosebleeds), menorrhagia, gum bleeding, and other types of mucocutaneous bleeding. Other common clinical features include fatigue, impaired quality of life, and treatment-related side effects (eg, infection).14
A low platelet count may be the sole initial manifestation. The patient’s history, physical examination, blood counts, and findings on blood smear are essential to rule out other diagnoses. Few diagnostic tests are useful in the initial evaluation (Table 1). Abnormalities in the blood count or blood smear may be further investigated with bone marrow biopsy but is not required if the patient has typical features of ITP, regardless of age.
Because there are no specific criteria for diagnosing ITP, other causes of thrombocytopenia must be excluded. The differential diagnosis can be further classified as ITP due to other underlying disease (ie, secondary ITP) vs nonautoimmune causes that are frequently encountered in clinical practice.
SECONDARY ITP
The differential diagnosis of thrombocytopenia due to known underlying immune disease includes the following:
Drug-induced ITP
Recurrent episodes of acute thrombocytopenia not explained by other causes should trigger consideration of drug-induced thrombocytopenia. 11 Patients should be questioned about drug use, especially of sulfonamides, antiepileptics, and quinine. Thrombocytopenia usually occurs 5 to 7 days after beginning the inciting drug for the first time and more quickly when the drug is given intermittently. Heparin is the most common cause of drug-related thrombocytopenia among hospitalized patients; the mechanism is unique and involves formation of a heparin-PF4 immune complex.
Human immunodeficiency virus infection
Approximately 40% of patients with human immunodeficiency virus (HIV) infection develop thrombocytopenia at some time.15 HIV infection can initially manifest as isolated thrombocytopenia and is sometimes clinically indistinguishable from chronic ITP, making it an important consideration in a newly diagnosed case of thrombocytopenia.
The mechanism of thrombocytopenia in early HIV is similar to that in primary ITP: as the disease progresses, low platelet counts can result from ineffective hematopoiesis due to megakaryocyte infection and marrow infiltration.16
Hepatitis C virus infection
Hepatitis C virus (HCV) infection can also cause immune thrombocytopenia. A recent study demonstrated the potential of the HCV core envelope protein 1 to induce antiplatelet antibodies (to platelet surface integrin GPIIIa49-66) by molecular mimicry.17 Other causes of thrombocytopenia in HCV infection may be related to chronic liver disease, such as portal hypertension-related hypersplenism, as well as decreased thrombopoietin production.18 Antiviral treatment with pegylated interferon may also cause mild thrombocytopenia.19
Helicobacter pylori
The association between H pylori infection and ITP remains uncertain. Eradication of infection appears to completely correct ITP in some places where the prevalence of H pylori is high (eg, Italy and Japan) but not in the United States and Canada, where the prevalence is low.20 The different response may be due not only to the differences in prevalence, but to different H pylori genotypes: most H pylori strains in Japan express CagA, whereas the frequency of CagA-positive strains is much lower in western countries.20
In areas where eradication therapy may be useful, the presence of H pylori infection should be determined by either a urea breath test or stool antigen testing.
Lymphoproliferative disorders
Secondary forms of ITP can occur in association with chronic lymphocytic leukemia, non-Hodgkin lymphoma, and Hodgkin lymphoma. These diagnoses should especially be considered in patients presenting with thrombocytopenia accompanied by systemic illness. ITP occurs in at least 2% of patients with chronic lymphocytic leukemia and is usually difficult to distinguish from thrombocytopenia secondary to marrow infiltration or from fludarabine (Fludora) therapy.21
It is especially important to determine if a lymphoproliferative disorder is present because it changes the treatment of ITP. Treatment of ITP complicating chronic lymphocytic leukemia is challenging and includes corticosteroids and steroid-sparing agents such as cyclosporine (Gengraf, Neoral, Sandimmune), rituximab (Rituxan), and intravenous immunoglobulin.22
Systemic lupus erythematosus and other autoimmune diseases
Thrombocytopenia is a frequent clinical manifestation of systemic lupus erythematosus, occurring in 7% to 30% of patients,23 and is an independent risk factor for death.24 Lupus should be suspected in patients with ITP who have multiorgan involvement and other clinical and laboratory abnormalities. A small percentage of patients with ITP (about 2%−5%) develop lupus after several years.21
Thrombocytopenia can also result from other autoimmune disorders such as antiphospholipid antibody syndrome25 and autoimmune thyroid diseases as well as immunodeficient states such as IgA deficiency and common variable immunodeficiency with low IgG levels.
NONAUTOIMMUNE THROMBOCYTOPENIA
Thrombocytopenia can also be caused by a number of nonautoimmune conditions.
Pseudothrombocytopenia
Pseudothrombocytopenia can occur if ex-vivo agglutination of platelets is induced by antiplatelet antibodies to EDTA, a standard blood anticoagulant. Automated counters cannot differentiate the agglutinated platelet clumps from individual cells such as red cells. This can frequently be overcome by running the counts in a citrate or ACD reagent tube. A peripheral blood smear can demonstrate whether platelet clumps are present.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura presents with thrombocytopenia, purpura, and anemia. Associated clinical abnormalities (fever, neurologic symptoms, and renal failure) and the presence of fragmented red cells on blood smear help to distinguish it from ITP. Plasma exchange is the treatment of choice.
Gestational thrombocytopenia
Five percent of pregnant women develop mild thrombocytopenia (platelet counts typically > 70 × 109/L) near the end of gestation.26 It requires no treatment and resolves after delivery. The fetus’ platelet count remains unaffected.
Gestational thrombocytopenia should be differentiated from the severe thrombocytopenia of preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count), which requires immediate attention.
Myelodysplastic syndrome
Myelodysplastic syndrome is common among elderly patients and should be considered in cases of unexplained cytopenia and abnormalities in the peripheral blood smear suggestive of dysplastic cytologic features. It can be diagnosed by bone marrow biopsy. Thrombocytopenia occurs in about 40% to 65% of cases of myelodysplastic syndrome.27
MANAGE ITP TO KEEP PLATELET COUNT ABOVE 30 × 109/L
ITP does not necessarily require treatment, and the initial challenge is to determine whether treatment or observation is indicated. Treatment is based on two major factors: the platelet count and degree of bleeding. The goals of management are to achieve a safe platelet count to prevent serious bleeding while minimizing treatment-related toxicity.7
Adults with platelet counts of less than 30 × 109/L are usually treated. In multiple large cohort studies, patients with platelet counts above that level have been safely observed without treatment.11,28
Table 2 outlines a comprehensive approach to therapy.
INITIAL TREATMENT: STEROIDS AND IMMUNOGLOBULINS
Oral corticosteroids are the initial agents of choice
Oral prednisone 1 mg/kg/day in tapering doses for 4 to 6 weeks is the most common initial regimen. Other regimens, such as high-dose dexamethasone (Decadron) (40 mg daily for 4 days per month) for several cycles, have been reported to be more effective29 but have not been studied in head-to-head trials with oral prednisone.
Due to their effectiveness, low cost, and convenience of use, corticosteroids have been the backbone of initial treatment in ITP. However, in most patients the platelet count decreases once the dose is tapered or stopped; remission is sustained in only 10% to 30% of cases.30 Continuation of corticosteroids is limited by long-term complications such as opportunistic infections, osteoporosis, and emotional lability.31
Intravenous immunoglobulin and anti-D immunoglobulin are alternatives
Intravenous immunoglobulin is recommended for patients who have not responded to corticosteroids and is often used in pregnancy. It is thought to act by blocking Fc receptors in the reticuloendothelial system. Intravenous immunoglobulin rapidly increases platelet counts in 65% to 80% of patients,32 but the effect is transient and the drug requires frequent administration. It is usually well tolerated, although about 5% of patients experience headache, chills, myalgias, arthralgias, and back pain. Rare, serious complications include thrombotic events, anaphylaxis (in IgA-deficient patients), and renal failure.
Anti-D immunoglobulin, a pooled IgG product, is derived from the plasma of Rh(D)-negative donors and can be given only to patients who are Rh(D)-positive. Response rates as high as 70% have been reported, with platelet effects lasting for more than 21 days.33 Studies have shown better results at a high dose (75 μg/kg) than with the approved dose of 50 μg/kg.34
Anti-D immunoglobulin can also be given intermittently whenever the platelet count falls below a specific level (ie, 30 × 109/L). This allows some patients to avoid splenectomy and may even trigger long-term remission.32
Common side effects of anti-D immunoglobulin include fever and chills; these can be prevented by premedication with acetaminophen or corticosteroids. Rare but fatal cases of intravascular hemolysis, renal failure, and disseminated intravascular coagulation have been reported, precluding its use for ITP in some countries, including those of the European Union.
Emergency treatment: Combination therapy
Evidence-based guidelines are limited for treating patients with active bleeding or who are at high risk of bleeding. For uncontrolled bleeding, a combination of first-line therapies is recommended, using prednisone and intravenous immunoglobulin.35 Other options include high-dose methylprednisolone and platelet transfusions, alone or in combination with intravenous immunoglobulin.36
SECOND-LINE TREATMENTS
Splenectomy produces complete remission in most patients
Patients who relapse and have a platelet count of less than 20 × 109/L are traditionally considered for splenectomy. More than two-thirds of patients respond with no need for further treatment.37
Although splenectomy has the highest rate of durable platelet response, the risks associated with surgery are an important concern. Even with a laparoscopic splenectomy, complications occur in 10% of patients and death in 0.2%. Long-term risks include the rare occurrence of sepsis with an estimated mortality rate of 0.73 per 1,000 patient-years, and possible increased risk of thrombosis.38,39
Adherence to recommended vaccination protocols and early administration of antibiotics for systemic febrile illness reduce the risk of sepsis.40 Patients are advised to receive immunization against encapsulated bacteria with pneumococcal, Haemophilus influenzae type b, and meningococcal vaccines. These vaccines should be given at least 2 weeks before elective splenectomy.41
Treatment of patients refractory to splenectomy is challenging and requires further immunosuppressive therapy, which is associated with an increased risk of infections and infection-related deaths.42
Rituximab in addition to or possibly instead of splenectomy
Rituximab (Rituxan) is a chimeric anti-CD20 monoclonal antibody that targets B cells. Although initially approved for treatment of lymphomas, rituximab has gained popularity in treating ITP due to its safety profile and ability to deplete CD20+ B cells responsible for antiplatelet antibody production by Fc-mediated cell lysis.
In the largest systematic review of published reports of rituximab use in ITP (19 studies, 313 patients), Arnold and colleagues43 reported an overall platelet response (defined as platelet count > 50 × 109/L) in 62.5% (95% confidence interval [CI] 52.6%−72.5%) of patients. The median duration of response was 10.5 months (range 3–20), and median follow-up was 9.5 months (range 2–25). Nearly all patients had received corticosteroid treatment and half of them had undergone splenectomy.
Rituximab has also been investigated as an alternative to splenectomy. In a prospective, single-arm, phase 2 trial, 60 patients with chronic ITP (platelet counts < 30 × 109/L) for whom one or more previous treatments had failed received rituximab infusions and were followed for up to 2 years. A good response (defined as a platelet count ≥ 50 × 109/L, with at least a doubling from baseline) was obtained in 24 (40%) of 60 patients (95% CI 28%–52%) at 1 year and 33.3% at 2 years. The authors concluded that rituximab could be used as a presplenectomy therapeutic option, particularly in patients with chronic ITP who are at increased surgical risk or who are reluctant to undergo surgery.44 Based on these results, rituximab may spare some patients from splenectomy, or at least delay it. However, it has never been tested in randomized controlled trials to establish its role as a splenectomy-sparing agent in ITP.
Side effects include infusion reactions, which are usually mild but in rare cases can be severe. Recently, progressive multifocal leukoencephalopathy has been recognized as a complication of rituximab treatment in patients with lymphoproliferative and autoimmune disorders.45 Although this complication is rare in patients with ITP, careful monitoring is required until additional long-term safety data are available.
Thrombopoietic receptor agonists require continuous treatment
In the early 1990s, recombinant thrombopoietin was tested in clinical studies. These were halted when antibodies developed to recombinant thrombopoietin that cross-reacted with endogenous thrombopoietin, resulting in severe thrombocytopenia.46
This led to the development of nonimmunogenic thrombopoietin receptor agonists that mimic the effect of thrombopoietin and stimulate the production of platelets. In 2008, the US Food and Drug Administration approved two drugs of this class for treating ITP: romiplostim (Nplate) and eltrombopag (Promacta). They are mainly used to treat patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Although well tolerated and effective in increasing platelet counts, these agents share common drawbacks. They do not modify the course of the disease, they are used only to sustain the platelet count, they require repeated administration, and they must be given for about 7 days to achieve an adequate platelet response, so they cannot be used in emergencies. Long-term adverse effects include bone marrow fibrosis and thrombosis.
Romiplostim is a synthetic peptide capable of binding to the thrombopoietin receptor c-Mpl. It has no sequence homology with endogenous thrombopoietin,47 so does not induce cross-reacting antibodies. It has a half-life of 120 to 160 hours and is usually given subcutaneously 1 to 10 μg/kg weekly.
Phase III clinical trials have shown the effectiveness of romiplostim in attaining a durable platelet response (platelet count > 50 × 109/L) in splenectomized and nonsplenectomized populations. It is well tolerated, and only two uncommon serious adverse effects have been reported: bone marrow reticulin formation and thromboembolism.48
A long-term open-label extension study of 142 patients treated with romiplostim for up to 156 weeks showed that 124 (87%) achieved a platelet count of more than 50 × 109/L at some point, and 84% of patients were able to reduce or discontinue concurrent medications for ITP.49
Kuter et al,50 in a randomized controlled trial, confirmed the efficacy of romiplostim in attaining durable increased platelet counts. Patients treated with romiplostim at a mean weekly dose of 3.9 μg/kg ± 2.1 μg/kg demonstrated a higher rate of platelet response, lower incidence of treatment failure, and improved quality of life vs patients treated with standard care.
Eltrombopag is a nonpeptide thrombopoietin agonist that binds to the transmembrane domain of the thrombopoietin receptor and stimulates the proliferation and differentiation of megakaryocytes in bone marrow. It is given orally in doses of 25 to 75 mg daily.
Eltrombopag has been shown to be effective in increasing platelet counts in chronic ITP.51 In a phase III trial conducted by Cheng and colleagues, 197 patients were randomized to eltrombopag or placebo.52 Patients treated with eltrombopag were eight times more likely to achieve platelet counts of more than 50 × 109/L during the 6-month treatment period (odds ratio 8.2, 95% CI 4.32–15.38, P < .001) vs placebo. Patients treated with eltrombopag had fewer bleeding episodes and were more likely to reduce or discontinue the dose of concurrent ITP medications. The only significant side effect seen was a rise in aminotransferases (seen in 7% of eltrombopag recipients vs 2% with placebo).52
Additional thrombopoietin agonists under investigation include ARK-501, totrombopag, and LGD-4665. MDX-33, a monoclonal antibody against the Fc-receptor, is also being studied; it acts by preventing opsonization of autoantibody-coated platelets.53
THIRD-LINE TREATMENTS FOR REFRACTORY CASES
Patients with ITP that is resistant to standard therapies have an increased risk of death, disease, and treatment-related complications.28,42
Combination chemotherapy
Immunosuppressants such as azathioprine (Imuran), cyclosporine (Neoral, Sandimmune), cyclophosphamide (Cytoxan), and mycophenolate (CellCept) were used in the past in single-agent regimens with some efficacy, but their use was limited due to drug-related toxicity and a low safety profile.3 However, there is increasing evidence for a role of combination chemotherapy to treat chronic refractory ITP to achieve greater efficacy and fewer adverse effects.54
Arnold and colleagues55 reported that combined azathioprine, mycophenolate, and cyclosporine achieved an overall response (platelet count > 30 × 109/L and doubling of the baseline) in 14 (73.7%) of 19 patients with chronic refractory ITP, lasting a median of 24 months.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation has provided remission in a limited number of patients. However, it is associated with fatal toxicities such as graft-vs-host disease and septicemia, and therefore it is reserved for severe refractory ITP with bleeding complications unresponsive to other therapies.56,57
THERAPY FOR SECONDARY ITP DEPENDS ON THE CAUSE
Treatments for secondary ITP vary depending on the cause of thrombocytopenia and are often more complex than therapy for primary disease. Optimal management involves treating the underlying condition (eg, chronic lymphocytic leukemia or systemic lupus erythematosus).
Drug-induced thrombocytopenia requires prompt recognition and withdrawal of the inciting agent.
Treating ITP due to HCV infection primarily involves antiviral agents to suppress viral replication. If treating ITP is required, then intravenous immunoglobulin is preferable to glucocorticoids because of the risk of increasing viral load with the latter.58 Eltrombopag may effectively increase platelet counts, allowing patients to receive interferon therapy for HCV.59 However, a recent study was halted due to increased incidence of portal vein thrombosis, raising concerns about the safety of eltrombopag for patients with chronic liver disease.60
Secondary ITP due to HIV infection should always be treated first with antivirals targeting HIV unless thrombocytopenia-related bleeding complications warrant treatment. If treatment for ITP is necessary, it should include corticosteroids, intravenous immunoglobulin, or anti-D immunoglobulin as first-line therapy.
Eradication therapy for H pylori is recommended for patients who are positive for the organism based on urea breath testing, stool antigen testing, or endoscopic biopsies.
- Feudjo-Tepie MA, Robinson NJ, Bennett D. Prevalence of diagnosed chronic immune thrombocytopenic purpura in the US: analysis of a large US claim database: a rebuttal. J Thromb Haemost 2008; 6:711–712.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol 2009; 83:83–89.
- Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist 2009; 14:12–21.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet auto-antibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Houwerzijl EJ, Blom NR, van der Want JJ, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004; 103:500–506.
- Kuwana M, Kaburaki J, Kitasato H, et al. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by autoreactive T cells in patients with immune thrombocytopenic purpura. Blood 2001; 98:130–139.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–858.
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol 2010; 17:590–595.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94:759–762.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207.
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011; 86:420–429.
- Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1275–1297.
- Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998; 91:1479–1495.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Peck-Radosavljevic M. Thrombocytopenia in liver disease. Can J Gastroenterol 2000; 14(suppl D):60D–66D.
- Roomer R, Hansen BE, Janssen HL, de Knegt RJ. Thrombocytopenia and the risk of bleeding during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. J Hepatol 2010; 53:455–459.
- Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113:1231–1240.
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113:6511–6521.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010; 23:47–59.
- Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:2243–2254.
- Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000; 39:399–406.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46:1019–1027.
- Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 1993; 329:1463–1466.
- Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007; 109:1705–1714.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Cheng Y, Wong RS, Soo YO, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 2003; 349:831–836.
- Bromberg ME. Immune thrombocytopenic purpura—the changing therapeutic landscape. N Engl J Med 2006; 355:1643–1645.
- Guidry JA, George JN, Vesely SK, Kennison SM, Terrell DR. Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83:175–182.
- Cooper N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1317–1327.
- Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 1997; 89:2689–2700.
- Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001; 112:1076–1078.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83:122–125.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Schilling RF. Estimating the risk for sepsis after splenectomy in hereditary spherocytosis. Ann Intern Med 1995; 122:187–188.
- Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood 2009; 114:2861–2868.
- Davies JM, Barnes R, Milligan D; British Committee for Standards in Haematology. Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med 2002; 2:440–443.
- Centers for Disease Control and Prevention (CDC). Recommended adult immunization schedule—United States, 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1–4.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104:956–960.
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146:25–33.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 2008; 112:999–1004.
- Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113:4834–4840.
- Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001; 98:3241–3248.
- Kuter DJ. New thrombopoietic growth factors. Blood 2007; 109:4607–4616.
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009; 113:2161–2171.
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363:1889–1899.
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:641–648.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011; 377:393–402.
- Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert Opin Investig Drugs 2009; 18:805–819.
- Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526–3531.
- Arnold DM, Nazi I, Santos A, et al. Combination immunosuppressant therapy for patients with chronic refractory immune thrombocytopenic purpura. Blood 2010; 115:29–31.
- Passweg JR, Rabusin M. Hematopoetic stem cell transplantation for immune thrombocytopenia and other refractory autoimmune cytopenias. Autoimmunity 2008; 41:660–665.
- Huhn RD, Fogarty PF, Nakamura R, et al. High-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopenia. Blood 2003; 101:71–77.
- Magrin S, Craxi A, Fabiano C, et al. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology 1994; 19:273–279.
- McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med 2007; 357:2227–2236.
- US Department of Health & Human Services. Promacta (eltrombopag): Portal Venous System Thromboses in Study of Patients With Chronic Liver Disease http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm211796.htm. Accessed June 27, 2012.
- Feudjo-Tepie MA, Robinson NJ, Bennett D. Prevalence of diagnosed chronic immune thrombocytopenic purpura in the US: analysis of a large US claim database: a rebuttal. J Thromb Haemost 2008; 6:711–712.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol 2009; 83:83–89.
- Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist 2009; 14:12–21.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet auto-antibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Houwerzijl EJ, Blom NR, van der Want JJ, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004; 103:500–506.
- Kuwana M, Kaburaki J, Kitasato H, et al. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by autoreactive T cells in patients with immune thrombocytopenic purpura. Blood 2001; 98:130–139.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–858.
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol 2010; 17:590–595.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94:759–762.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207.
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011; 86:420–429.
- Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1275–1297.
- Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998; 91:1479–1495.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Peck-Radosavljevic M. Thrombocytopenia in liver disease. Can J Gastroenterol 2000; 14(suppl D):60D–66D.
- Roomer R, Hansen BE, Janssen HL, de Knegt RJ. Thrombocytopenia and the risk of bleeding during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. J Hepatol 2010; 53:455–459.
- Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113:1231–1240.
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113:6511–6521.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010; 23:47–59.
- Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:2243–2254.
- Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000; 39:399–406.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46:1019–1027.
- Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 1993; 329:1463–1466.
- Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007; 109:1705–1714.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Cheng Y, Wong RS, Soo YO, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 2003; 349:831–836.
- Bromberg ME. Immune thrombocytopenic purpura—the changing therapeutic landscape. N Engl J Med 2006; 355:1643–1645.
- Guidry JA, George JN, Vesely SK, Kennison SM, Terrell DR. Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83:175–182.
- Cooper N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1317–1327.
- Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 1997; 89:2689–2700.
- Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001; 112:1076–1078.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83:122–125.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Schilling RF. Estimating the risk for sepsis after splenectomy in hereditary spherocytosis. Ann Intern Med 1995; 122:187–188.
- Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood 2009; 114:2861–2868.
- Davies JM, Barnes R, Milligan D; British Committee for Standards in Haematology. Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med 2002; 2:440–443.
- Centers for Disease Control and Prevention (CDC). Recommended adult immunization schedule—United States, 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1–4.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104:956–960.
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146:25–33.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 2008; 112:999–1004.
- Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113:4834–4840.
- Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001; 98:3241–3248.
- Kuter DJ. New thrombopoietic growth factors. Blood 2007; 109:4607–4616.
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009; 113:2161–2171.
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363:1889–1899.
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:641–648.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011; 377:393–402.
- Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert Opin Investig Drugs 2009; 18:805–819.
- Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526–3531.
- Arnold DM, Nazi I, Santos A, et al. Combination immunosuppressant therapy for patients with chronic refractory immune thrombocytopenic purpura. Blood 2010; 115:29–31.
- Passweg JR, Rabusin M. Hematopoetic stem cell transplantation for immune thrombocytopenia and other refractory autoimmune cytopenias. Autoimmunity 2008; 41:660–665.
- Huhn RD, Fogarty PF, Nakamura R, et al. High-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopenia. Blood 2003; 101:71–77.
- Magrin S, Craxi A, Fabiano C, et al. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology 1994; 19:273–279.
- McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med 2007; 357:2227–2236.
- US Department of Health & Human Services. Promacta (eltrombopag): Portal Venous System Thromboses in Study of Patients With Chronic Liver Disease http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm211796.htm. Accessed June 27, 2012.
KEY POINTS
- Secondary ITP can be drug-induced or be a manifestation of human immunodeficiency virus (HIV), hepatitis C virus (HCV), a lymphoproliferative disorder, or systemic lupus erythematosus.
- Nonautoimmune conditions should also be considered, including pseudothrombocytopenia (a laboratory artifact induced by EDTA), thrombotic thrombocytopenic purpura, thrombocytopenia in pregnancy, and myelodysplastic syndrome (common in the elderly).
- Treatment is indicated to keep the platelet count above 30 × 109/L or to control bleeding.
- Initial treatment usually begins with glucocorticoids, with the duration limited by side effects.
- Patients for whom glucocorticoids fail generally require splenectomy, rituximab, or thrombopoietin receptor agonists.
Atrial fibrillation: New drugs, devices, and procedures
Although many developments have occurred in the last decade for managing atrial fibrillation, challenges remain. New and emerging alternatives to warfarin (Coumadin) for anticoagulation therapy prevent stroke marginally better and pose slightly less risk of hemorrhage, but they have important drawbacks.
The antiarrhythmic drug dronedarone (Multaq) has been found to offer only temporary benefit for persistent atrial fibrillation, and significant risks have emerged.
Radiofrequency ablation is gaining prominence, but repeat procedures are sometimes necessary.
An investigational device can be implanted via percutaneous catheter in the left atrial appendage to prevent embolization. It is too soon to know its eventual role in clinical practice.
This article reviews the results of clinical trials of these new treatments and discusses their role in clinical practice.
CHALLENGES OF ANTICOAGULATION
The main focus of managing atrial fibrillation is on alleviating symptoms, by either rate control or rhythm control. The other focus is on preventing stroke—a devastating outcome—with anticoagulation therapy.
For deciding whether to give warfarin to patients with atrial fibrillation, the six-point CHADS2 score is a crude but effective way of assessing the risk of stroke based on the following risk factors: congestive heart failure, hypertension, age 75 years or older, and diabetes (1 point each); or a history of stroke or transient ischemic attack (2 points).1 Warfarin is given if patients have a score of at least 2 points.
Warfarin has a narrow therapeutic window, with a higher risk of ischemic stroke if the international normalized ratio (INR) is less than 2.0,2 and a higher risk of intracranial hemorrhage if the INR is more than 3.0.3 Keeping the INR in the therapeutic range is difficult because of variations in diet, concurrent medications, and other factors.
The percent of time that the INR is within the therapeutic range predicts the risk of adverse events. Connolly et al4 showed that the cumulative risk of stroke, myocardial infarction, systemic embolism, or vascular death was no better with warfarin than with clopidogrel (Plavix) plus aspirin if the INR was in the therapeutic range less than 65% of the time, but the risk was significantly less if the INR was in the therapeutic range more than 65% of the time.
Also, comparing warfarin with the combination of aspirin and clopidogrel, Verheugt5 found that the rates of stroke of any kind, of disabling and fatal stroke, and of stroke per major bleed were lower in patients taking warfarin. Although many physicians prefer aspirin plus clopidogrel because of concerns about bleeding with warfarin, the rates of major bleeding were about the same in the two groups.
In a trial in patients for whom warfarin was “unsuitable,”6 the combination of aspirin plus clopidogrel was associated with a lower rate of stroke than aspirin alone (2.4% per year vs 3.3% per year, relative risk 0.762) but a higher rate of major bleeding events (2.0% per year vs 1.3% per year, relative risk 1.57).
NEW ALTERNATIVES TO WARFARIN

Because of the problems with warfarin, alternatives have been sought for many years. Several new oral anticoagulants are available or are being developed,7 including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis) and the direct factor II (thrombin) inhibitor dabigatran (Pradaxa) (Table 1).
Dabigatran’s advantages and drawbacks
Dabigatran has been on the market for more than a year and has gained rapid acceptance. The dosage is 150 mg twice a day, or 75 mg twice a day if renal function is impaired. Cleared by the kidneys, it has a half-life of 12 to 17 hours; 75% is cleared within 24 hours. For a patient who needs surgery that poses a low risk of bleeding, the general recommendation is to stop dabigatran the night before the surgical procedure. For operations with a greater risk of bleeding, many surgeons recommend stopping the drug 3 or 4 days before.
Advantages of dabigatran include that it is not influenced by diet and that the onset of therapeutic benefit is within 1 hour. Although some drugs affect dabigatran, drug interactions are more troublesome with warfarin.
A serious concern about dabigatran and the other new agents is that if a bleeding problem arises, the effects of these drugs are not reversible by administration of fresh frozen plasma. Dabigatran is reversible by dialysis; however, if a patient is also hypotensive, dialysis is not an option, and simply waiting for the drug to clear is the only choice.
Another drawback is that therapeutic levels cannot be monitored. If a patient taking warfarin requires cardioversion, the INR is carefully monitored for several weeks beforehand to reduce the risk of stroke. With dabigatran, there is no way to know if a patient is actually taking the drug as prescribed.
Clinical trials show that alternatives are marginally better than warfarin
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,8 dabigatran was associated with a significantly lower incidence of intracranial hemorrhage, combined strokes, and systemic embolization than warfarin. The incidence of major bleeds was slightly lower with dabigatran. Although dabigatran performed better, the differences were small and would not require patients to change from warfarin if they are already doing well.
Apixaban and rivaroxaban are other alternatives to warfarin, with different mechanisms of action and metabolism. Although rivaroxaban’s half-life is similar to that of apixaban and dabigatran, it is being marketed as allowing once-daily dosing instead of twice-daily.
Recent randomized controlled clinical trials of the new drugs include:
- The Apixaban Versus Acetylsalicylic Acid (ASA) to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment (AVERROES) trial,9 which compared apixaban and aspirin
- The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial comparing apixaban and warfarin10
- The Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF),11 comparing rivaroxaban and warfarin
- RE-LY,8 comparing dabigatran and warfarin.
In the ARISTOTLE,10 ROCKET AF,11 and RE-LY trials,8 the time that the warfarin patients’ INRs were in the therapeutic range varied from 55% to 68%. This seems low and is a problem when trying to compare therapies, but is probably about as high as one can expect in the real world.
In AVERROES,9 the combined rate of stroke and embolism was higher with aspirin than with apixaban. In the other trials, the rates were slightly better with the new drugs than with warfarin, and the rates of major hemorrhage and hemorrhagic stroke were only slightly higher with warfarin than with the new drugs. Because the differences in benefits and risks are so small, the main advantage of the newer drugs will probably be for patients who have difficulty staying in the therapeutic INR range on warfarin.
RATE CONTROL VS RESTORATION OF SINUS RHYTHM
Evidence is insufficient to determine the risk of very-long-term asymptomatic atrial fibrillation in patients on appropriate anticoagulation. Rate control is an option for asymptomatic patients but provides no change in quality of life and no definitive reduction in the risk of stroke. The main argument for restoring normal sinus rhythm in patients with mild to moderate symptoms is that it improves exercise capacity. The need for anticoagulation persists when patients are converted to sinus rhythm because the risk of recurrent atrial fibrillation remains high.
For patients with symptomatic atrial fibrillation, rate control is sometimes achieved with beta-blockers or calcium channel blockers. Rate control may be augmented with the addition of digoxin, but when used alone digoxin generally does not control the rate of atrial fibrillation. However, in many cases of atrial fibrillation, symptoms are not rate-related, and cardioversion to normal sinus rhythm should be attempted. In such cases, the symptoms may be attributable to a loss of atrial transport function.
Patients with the following risk factors should be admitted to the hospital to start antiarrhythmic drugs:
- Borderline or a long QTc interval at baseline (> 450 msec)
- Treatment with dofetilide (Tikosyn) because of its effects on the QT interval
- Heart failure or poor left-ventricular function
- Sinus node dysfunction
- Significant atrioventricular conduction disease.
Selecting an antiarrhythmic drug
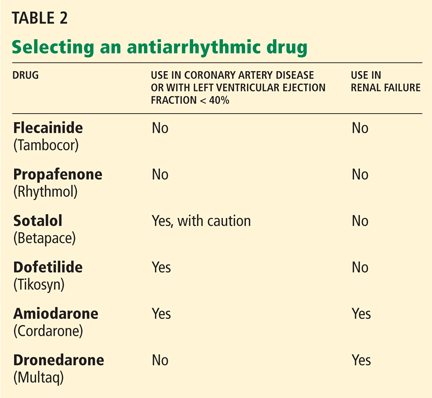
Any of the antiarrhythmic drugs listed in Table 2 can be used for a patient with lone atrial fibrillation (ie, not caused by underlying heart disease). The choice of drug should be determined by whether coronary artery disease or renal failure is present as well. Liver disease or chronic obstructive pulmonary disease also may affect this decision.
Benefits of dronedarone are mixed
In a randomized trial of dronedarone vs placebo in patients with atrial fibrillation, the rate of death and the rate of first hospitalization due to a cardiovascular event at 21 months were significantly lower with dronedarone.12 No difference was found between the two groups in the rate of death from all causes, but fewer people died of cardiovascular causes in the dronedarone group. More patients taking dronedarone developed bradycardia, QT-interval prolongation, nausea, diarrhea, rash, or a higher serum creatinine level. Gastrointestinal side effects are often a problem with dronedarone: 20% to 30% of patients cannot tolerate the drug.
Dronedarone may cause a small rise in creatinine, and although this effect should be monitored, by itself it should not be interpreted as impairment of renal function. In a study in healthy people,13 dronedarone caused a 10% to 15% increase in serum creatinine, but the glomerular filtration rate was unchanged, as were renal plasma flow and anion secretion.
Another trial, in patients with severe heart failure, found that patients taking dronedarone had higher rates of hospitalization and overall mortality, raising serious concern about the safety of this drug in patients with advanced heart failure.14
Singh et al15 pooled the data from two multicenter, randomized trials that compared dronedarone with placebo for maintaining sinus rhythm in patients with atrial fibrillation or flutter. The mean time to the recurrence of atrial fibrillation was 116 days with dronedarone and 53 days with placebo. Other trials also showed longer times to recurrence and lower recurrence rates with dronedarone. Although the differences were statistically significant, they may not be clinically meaningful for patients.
Dronedarone is structurally similar to amiodarone (Cordarone), but the two drugs work differently. A meta-analysis of clinical trials16 found that amiodarone recipients had a lower rate of recurrence of atrial fibrillation than did those receiving dronedarone.
Two safety warnings for dronedarone
In January 2011, the US Food and Drug Administration (FDA) issued an alert about cases of rare but severe liver injury in patients treated with dronedarone, including two cases of acute liver failure leading to liver transplantation.17
The Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS)18 compared dronedarone and placebo in patients with permanent atrial fibrillation. More people died or had serious cardiovascular adverse events in the dronedarone group. The study was stopped early after data monitoring showed that rates of death, stroke, and hospitalization for heart failure were two times higher in patients receiving dronedarone. This prompted the FDA to issue another safety alert in July 2011.
Interestingly, the PALLAS study did not set out to determine whether dronedarone controls atrial fibrillation, as the study patients had long-standing, persistent atrial fibrillation. The study was designed only to determine if the drug reduces the rate of adverse events; it clearly does not, and the study shows that dronedarone should not be used to control the heart rate in patients with persistent atrial fibrillation. Instead, its use is best restricted to patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
ABLATION OF ATRIAL FIBRILLATION
Another way to try to restore sinus rhythm is to destroy or isolate the area that is generating the abnormal beats via a catheter-based procedure.
Radiofrequency ablation is generally tried in patients in whom one or two drugs have failed to control atrial fibrillation. Direct comparisons show that ablation is superior to drug therapy and is effective in about 75% of patients with paroxysmal atrial fibrillation vs 20% to 40% of patients on drug therapy. Ablation plus drug therapy is often more effective than either treatment alone.
Mechanisms of atrial fibrillation and ablation
In many cases, atrial fibrillation is stimulated by vagal and sympathetic inputs to the atrium that enter around the pulmonary veins and trigger electrical activations in the area, generating spiraling, reentering circuits. Focal atrial fibrillation also originates predominantly in the pulmonary veins. Ablation of tissue widely circumscribing the mouth of the pulmonary veins prevents the electrical signal from exiting into the atrium.
In about 11% to 37% of cases, atrial fibrillation originates elsewhere, eg, in the left atrium, in the superior vena cava, or in the vein of Marshall. Techniques have evolved to also ablate these regions.
Anticoagulation therapy is recommended before the procedure, and patients at low risk should continue it for a minimum of 2 months afterward. Patients with a higher CHADS2 score should receive anticoagulation therapy for at least 1 year. The consensus statement by the Heart Rhythm Society19 recommends that patients remain on warfarin or one of the newer anticoagulants if their CHADS2 score is 2 or higher. This is because patients have a significant risk of recurrence of atrial fibrillation after radiofrequency ablation, so if their stroke risk is high they should remain on anticoagulant therapy.
Ablation is usually effective, but it carries rare but serious risks
The efficacy of a single radiofrequency ablation procedure is in the range of 60% to 80% for paroxysmal atrial fibrillation and 40% to 60% for persistent atrial fibrillation. The Second International Ablation Registry20 shows a success rate of about 75% in patients with paroxysmal atrial fibrillation and about 65% in patients with persistent and permanent atrial fibrillation. Registry data are often more favorable because reporting is optional, but these results are consistent with those from experienced medical centers. Rates of suppression of atrial fibrillation are higher in patients who also take antiarrhythmic drugs, making a “hybrid” approach useful when ablation alone fails.
According to a worldwide survey, the risk of serious complications is 4.5%. These include stroke (0.23%), tamponade (1.3%), and pulmonary vein stenosis (< 0.29%). The esophagus lies just behind the right atrium, and burning through and creating a fistula between them occurs in about 0.04% of cases and is almost uniformly fatal.20
A second ablation procedure is sometimes indicated for the recurrence of atrial fibrillation, which is almost always caused by recovery of the pulmonary veins. Bhargava et al21 found that the success rate at Cleveland Clinic for a single procedure for paroxysmal atrial fibrillation was 77%, and that it was 92% after a repeat procedure. For persistent atrial fibrillation, success rates were 76% after the first procedure and 90% after the second. Even for long-standing persistent atrial fibrillation (ie, lasting more than 1 year), 80% success was achieved after two procedures. Patients who are less likely to have a successful ablation procedure are those with long-standing atrial fibrillation and coexisting heart disease, including severe valvular disease, although mitral regurgitation sometimes improves if sinus rhythm can be maintained.
The need for a second procedure
After ablation, patients should be closely monitored for a recurrence of atrial fibrillation. Continuous monitoring with implantable cardiac monitor loop recorders can detect unrecognized episodes of arrhythmia. Long-term follow-up is also required to track outcomes and quality of life.
According to the Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation,19 atrial fibrillation recurs after ablation in about 35% to 60% of patients in the first 3 months, with recurrence rates after 1 year ranging from 5% to 16%. The rate of success is determined by the skill of the surgeon, underlying heart disease, attention to follow-up, and how success is defined.
Freedom from recurrence early on is a good predictor that late recurrence is unlikely. Patients who only have a very early recurrence (within the first 4 weeks) are more likely to have long-term freedom from atrial fibrillation tha those who have recurrences after that time.22
In a study of 831 patients, Hussein et al23 found recurrence rates of 24% between months 3 to 13 following ablation and 9% after 12 months. At 55 months, 79% were free from atrial fibrillation without drugs, 11% were free of atrial fibrillation with medications, and 5% had refractory atrial fibrillation.
Recurrence—whether early or late—was more likely to occur in people with persistent vs paroxysmal atrial fibrillation. Other risk factors for late recurrence included older age and larger left atrial size (which is also a risk factor for recurrence on drug therapy). Although recurrent arrhythmia was most often atrial fibrillation, atrial flutter also occurred frequently (in 27% of patients with late recurrence). Three patients (4% of patients with late recurrence) developed atrial tachycardia.23
In patients with early recurrence, 81% underwent repeat ablation, all of whom had recovery of one or more pulmonary veins. After the second ablation, 21% had recurrence, 65% of whom were controlled by medications.23
Whether a patient should undergo subsequent ablation procedures depends on the severity of symptoms, the likelihood of success (based on an educated guess), and the patient’s willingness to undergo another procedure.
ATRIAL APPENDAGE OCCLUSION DEVICE UNDER INVESTIGATION
New devices are being investigated that occlude the left atrial appendage to try to prevent embolization.
The Watchman device, resembling an umbrella, is implanted via a percutaneous catheter in the left atrial appendage, closing it off to preclude a thrombus from forming in the appendage and embolizing to the body. Clinical trials showed that patients who received a device had a slightly lower risk of stroke than otherwise seen in clinical practice.24 Safety and efficacy are still being determined.
The device cannot be deployed in a patient with an existing thrombus because of the danger of dislodging the thrombus, allowing it to embolize.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the national Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335:540–546.
- Hylek EM, Singer DE. Risk factors for intracranial hemorrhage in outpatients taking warfarin. Ann Intern Med 1994; 120:897–902.
- Connolly SJ, Pogue J, Eikelboom J, et al; ACTIVE W Investigators. Benefit of oral anticoagulant over antiplatelet therapy in atrial fibrillation depends on the quality of international normalized ratio control achieved by centers and countries as measured by time in therapeutic range. Circulation 2008; 118:2029–2037.
- Verheugt FWA. Who is ineligible for warfarin in atrial fibrillation? Lancet 2009; 374:510–511.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Harenberg J. New anticoagulants in atrial fibrillation. Semin Thromb Hemost 2009; 35:574–585.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Connolly SJ, Eikelboom J, Joyner C, et al; AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364:806–817.
- Granger CB, Alexander JH, McMurray JJV, et al; for the ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med August 28, 2011; 10.1056/nejmoa1107039.
- Patel MR, Mahaffey KW, Garg J, et al; the ROCKET AF Steering Committee. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Hohnloser SH, Crijns HJ, van Eickels M, et al; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:688–678.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Kóber L, Torp-Pederson C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 2009; 54:1089–1095.
- US Food and Drug Administration. FDA drug safety communication: severe liver injury associated with the use of dronedarone (marketed as Multaq). http://www.fda.gov/drugs/drugsafety/ucm240011.htm. Accessed July 5, 2012.
- Connolly SJ, Camm AJ, Halperin JL, et al; for the PALLAS Investigators. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011; 365:2268–2276.
- HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. Heart Rhythm 2007; 4:1–46.
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3:32–38.
- Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm 2009; 6:1403–1412.
- Themistoclakis S, Schweikert RA, Sliba WI, et al. Clinical predictors and relationship between early and late atrial tachyarrhythmias after pulmonary vein antrum isolation. Heart Rhythm 2008; 5:679–685.
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4:271–278.
- Holmes DR, Reddy VY, Turi ZG, et al; for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
Although many developments have occurred in the last decade for managing atrial fibrillation, challenges remain. New and emerging alternatives to warfarin (Coumadin) for anticoagulation therapy prevent stroke marginally better and pose slightly less risk of hemorrhage, but they have important drawbacks.
The antiarrhythmic drug dronedarone (Multaq) has been found to offer only temporary benefit for persistent atrial fibrillation, and significant risks have emerged.
Radiofrequency ablation is gaining prominence, but repeat procedures are sometimes necessary.
An investigational device can be implanted via percutaneous catheter in the left atrial appendage to prevent embolization. It is too soon to know its eventual role in clinical practice.
This article reviews the results of clinical trials of these new treatments and discusses their role in clinical practice.
CHALLENGES OF ANTICOAGULATION
The main focus of managing atrial fibrillation is on alleviating symptoms, by either rate control or rhythm control. The other focus is on preventing stroke—a devastating outcome—with anticoagulation therapy.
For deciding whether to give warfarin to patients with atrial fibrillation, the six-point CHADS2 score is a crude but effective way of assessing the risk of stroke based on the following risk factors: congestive heart failure, hypertension, age 75 years or older, and diabetes (1 point each); or a history of stroke or transient ischemic attack (2 points).1 Warfarin is given if patients have a score of at least 2 points.
Warfarin has a narrow therapeutic window, with a higher risk of ischemic stroke if the international normalized ratio (INR) is less than 2.0,2 and a higher risk of intracranial hemorrhage if the INR is more than 3.0.3 Keeping the INR in the therapeutic range is difficult because of variations in diet, concurrent medications, and other factors.
The percent of time that the INR is within the therapeutic range predicts the risk of adverse events. Connolly et al4 showed that the cumulative risk of stroke, myocardial infarction, systemic embolism, or vascular death was no better with warfarin than with clopidogrel (Plavix) plus aspirin if the INR was in the therapeutic range less than 65% of the time, but the risk was significantly less if the INR was in the therapeutic range more than 65% of the time.
Also, comparing warfarin with the combination of aspirin and clopidogrel, Verheugt5 found that the rates of stroke of any kind, of disabling and fatal stroke, and of stroke per major bleed were lower in patients taking warfarin. Although many physicians prefer aspirin plus clopidogrel because of concerns about bleeding with warfarin, the rates of major bleeding were about the same in the two groups.
In a trial in patients for whom warfarin was “unsuitable,”6 the combination of aspirin plus clopidogrel was associated with a lower rate of stroke than aspirin alone (2.4% per year vs 3.3% per year, relative risk 0.762) but a higher rate of major bleeding events (2.0% per year vs 1.3% per year, relative risk 1.57).
NEW ALTERNATIVES TO WARFARIN
Because of the problems with warfarin, alternatives have been sought for many years. Several new oral anticoagulants are available or are being developed,7 including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis) and the direct factor II (thrombin) inhibitor dabigatran (Pradaxa) (Table 1).
Dabigatran’s advantages and drawbacks
Dabigatran has been on the market for more than a year and has gained rapid acceptance. The dosage is 150 mg twice a day, or 75 mg twice a day if renal function is impaired. Cleared by the kidneys, it has a half-life of 12 to 17 hours; 75% is cleared within 24 hours. For a patient who needs surgery that poses a low risk of bleeding, the general recommendation is to stop dabigatran the night before the surgical procedure. For operations with a greater risk of bleeding, many surgeons recommend stopping the drug 3 or 4 days before.
Advantages of dabigatran include that it is not influenced by diet and that the onset of therapeutic benefit is within 1 hour. Although some drugs affect dabigatran, drug interactions are more troublesome with warfarin.
A serious concern about dabigatran and the other new agents is that if a bleeding problem arises, the effects of these drugs are not reversible by administration of fresh frozen plasma. Dabigatran is reversible by dialysis; however, if a patient is also hypotensive, dialysis is not an option, and simply waiting for the drug to clear is the only choice.
Another drawback is that therapeutic levels cannot be monitored. If a patient taking warfarin requires cardioversion, the INR is carefully monitored for several weeks beforehand to reduce the risk of stroke. With dabigatran, there is no way to know if a patient is actually taking the drug as prescribed.
Clinical trials show that alternatives are marginally better than warfarin
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,8 dabigatran was associated with a significantly lower incidence of intracranial hemorrhage, combined strokes, and systemic embolization than warfarin. The incidence of major bleeds was slightly lower with dabigatran. Although dabigatran performed better, the differences were small and would not require patients to change from warfarin if they are already doing well.
Apixaban and rivaroxaban are other alternatives to warfarin, with different mechanisms of action and metabolism. Although rivaroxaban’s half-life is similar to that of apixaban and dabigatran, it is being marketed as allowing once-daily dosing instead of twice-daily.
Recent randomized controlled clinical trials of the new drugs include:
- The Apixaban Versus Acetylsalicylic Acid (ASA) to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment (AVERROES) trial,9 which compared apixaban and aspirin
- The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial comparing apixaban and warfarin10
- The Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF),11 comparing rivaroxaban and warfarin
- RE-LY,8 comparing dabigatran and warfarin.
In the ARISTOTLE,10 ROCKET AF,11 and RE-LY trials,8 the time that the warfarin patients’ INRs were in the therapeutic range varied from 55% to 68%. This seems low and is a problem when trying to compare therapies, but is probably about as high as one can expect in the real world.
In AVERROES,9 the combined rate of stroke and embolism was higher with aspirin than with apixaban. In the other trials, the rates were slightly better with the new drugs than with warfarin, and the rates of major hemorrhage and hemorrhagic stroke were only slightly higher with warfarin than with the new drugs. Because the differences in benefits and risks are so small, the main advantage of the newer drugs will probably be for patients who have difficulty staying in the therapeutic INR range on warfarin.
RATE CONTROL VS RESTORATION OF SINUS RHYTHM
Evidence is insufficient to determine the risk of very-long-term asymptomatic atrial fibrillation in patients on appropriate anticoagulation. Rate control is an option for asymptomatic patients but provides no change in quality of life and no definitive reduction in the risk of stroke. The main argument for restoring normal sinus rhythm in patients with mild to moderate symptoms is that it improves exercise capacity. The need for anticoagulation persists when patients are converted to sinus rhythm because the risk of recurrent atrial fibrillation remains high.
For patients with symptomatic atrial fibrillation, rate control is sometimes achieved with beta-blockers or calcium channel blockers. Rate control may be augmented with the addition of digoxin, but when used alone digoxin generally does not control the rate of atrial fibrillation. However, in many cases of atrial fibrillation, symptoms are not rate-related, and cardioversion to normal sinus rhythm should be attempted. In such cases, the symptoms may be attributable to a loss of atrial transport function.
Patients with the following risk factors should be admitted to the hospital to start antiarrhythmic drugs:
- Borderline or a long QTc interval at baseline (> 450 msec)
- Treatment with dofetilide (Tikosyn) because of its effects on the QT interval
- Heart failure or poor left-ventricular function
- Sinus node dysfunction
- Significant atrioventricular conduction disease.
Selecting an antiarrhythmic drug
Any of the antiarrhythmic drugs listed in Table 2 can be used for a patient with lone atrial fibrillation (ie, not caused by underlying heart disease). The choice of drug should be determined by whether coronary artery disease or renal failure is present as well. Liver disease or chronic obstructive pulmonary disease also may affect this decision.
Benefits of dronedarone are mixed
In a randomized trial of dronedarone vs placebo in patients with atrial fibrillation, the rate of death and the rate of first hospitalization due to a cardiovascular event at 21 months were significantly lower with dronedarone.12 No difference was found between the two groups in the rate of death from all causes, but fewer people died of cardiovascular causes in the dronedarone group. More patients taking dronedarone developed bradycardia, QT-interval prolongation, nausea, diarrhea, rash, or a higher serum creatinine level. Gastrointestinal side effects are often a problem with dronedarone: 20% to 30% of patients cannot tolerate the drug.
Dronedarone may cause a small rise in creatinine, and although this effect should be monitored, by itself it should not be interpreted as impairment of renal function. In a study in healthy people,13 dronedarone caused a 10% to 15% increase in serum creatinine, but the glomerular filtration rate was unchanged, as were renal plasma flow and anion secretion.
Another trial, in patients with severe heart failure, found that patients taking dronedarone had higher rates of hospitalization and overall mortality, raising serious concern about the safety of this drug in patients with advanced heart failure.14
Singh et al15 pooled the data from two multicenter, randomized trials that compared dronedarone with placebo for maintaining sinus rhythm in patients with atrial fibrillation or flutter. The mean time to the recurrence of atrial fibrillation was 116 days with dronedarone and 53 days with placebo. Other trials also showed longer times to recurrence and lower recurrence rates with dronedarone. Although the differences were statistically significant, they may not be clinically meaningful for patients.
Dronedarone is structurally similar to amiodarone (Cordarone), but the two drugs work differently. A meta-analysis of clinical trials16 found that amiodarone recipients had a lower rate of recurrence of atrial fibrillation than did those receiving dronedarone.
Two safety warnings for dronedarone
In January 2011, the US Food and Drug Administration (FDA) issued an alert about cases of rare but severe liver injury in patients treated with dronedarone, including two cases of acute liver failure leading to liver transplantation.17
The Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS)18 compared dronedarone and placebo in patients with permanent atrial fibrillation. More people died or had serious cardiovascular adverse events in the dronedarone group. The study was stopped early after data monitoring showed that rates of death, stroke, and hospitalization for heart failure were two times higher in patients receiving dronedarone. This prompted the FDA to issue another safety alert in July 2011.
Interestingly, the PALLAS study did not set out to determine whether dronedarone controls atrial fibrillation, as the study patients had long-standing, persistent atrial fibrillation. The study was designed only to determine if the drug reduces the rate of adverse events; it clearly does not, and the study shows that dronedarone should not be used to control the heart rate in patients with persistent atrial fibrillation. Instead, its use is best restricted to patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
ABLATION OF ATRIAL FIBRILLATION
Another way to try to restore sinus rhythm is to destroy or isolate the area that is generating the abnormal beats via a catheter-based procedure.
Radiofrequency ablation is generally tried in patients in whom one or two drugs have failed to control atrial fibrillation. Direct comparisons show that ablation is superior to drug therapy and is effective in about 75% of patients with paroxysmal atrial fibrillation vs 20% to 40% of patients on drug therapy. Ablation plus drug therapy is often more effective than either treatment alone.
Mechanisms of atrial fibrillation and ablation
In many cases, atrial fibrillation is stimulated by vagal and sympathetic inputs to the atrium that enter around the pulmonary veins and trigger electrical activations in the area, generating spiraling, reentering circuits. Focal atrial fibrillation also originates predominantly in the pulmonary veins. Ablation of tissue widely circumscribing the mouth of the pulmonary veins prevents the electrical signal from exiting into the atrium.
In about 11% to 37% of cases, atrial fibrillation originates elsewhere, eg, in the left atrium, in the superior vena cava, or in the vein of Marshall. Techniques have evolved to also ablate these regions.
Anticoagulation therapy is recommended before the procedure, and patients at low risk should continue it for a minimum of 2 months afterward. Patients with a higher CHADS2 score should receive anticoagulation therapy for at least 1 year. The consensus statement by the Heart Rhythm Society19 recommends that patients remain on warfarin or one of the newer anticoagulants if their CHADS2 score is 2 or higher. This is because patients have a significant risk of recurrence of atrial fibrillation after radiofrequency ablation, so if their stroke risk is high they should remain on anticoagulant therapy.
Ablation is usually effective, but it carries rare but serious risks
The efficacy of a single radiofrequency ablation procedure is in the range of 60% to 80% for paroxysmal atrial fibrillation and 40% to 60% for persistent atrial fibrillation. The Second International Ablation Registry20 shows a success rate of about 75% in patients with paroxysmal atrial fibrillation and about 65% in patients with persistent and permanent atrial fibrillation. Registry data are often more favorable because reporting is optional, but these results are consistent with those from experienced medical centers. Rates of suppression of atrial fibrillation are higher in patients who also take antiarrhythmic drugs, making a “hybrid” approach useful when ablation alone fails.
According to a worldwide survey, the risk of serious complications is 4.5%. These include stroke (0.23%), tamponade (1.3%), and pulmonary vein stenosis (< 0.29%). The esophagus lies just behind the right atrium, and burning through and creating a fistula between them occurs in about 0.04% of cases and is almost uniformly fatal.20
A second ablation procedure is sometimes indicated for the recurrence of atrial fibrillation, which is almost always caused by recovery of the pulmonary veins. Bhargava et al21 found that the success rate at Cleveland Clinic for a single procedure for paroxysmal atrial fibrillation was 77%, and that it was 92% after a repeat procedure. For persistent atrial fibrillation, success rates were 76% after the first procedure and 90% after the second. Even for long-standing persistent atrial fibrillation (ie, lasting more than 1 year), 80% success was achieved after two procedures. Patients who are less likely to have a successful ablation procedure are those with long-standing atrial fibrillation and coexisting heart disease, including severe valvular disease, although mitral regurgitation sometimes improves if sinus rhythm can be maintained.
The need for a second procedure
After ablation, patients should be closely monitored for a recurrence of atrial fibrillation. Continuous monitoring with implantable cardiac monitor loop recorders can detect unrecognized episodes of arrhythmia. Long-term follow-up is also required to track outcomes and quality of life.
According to the Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation,19 atrial fibrillation recurs after ablation in about 35% to 60% of patients in the first 3 months, with recurrence rates after 1 year ranging from 5% to 16%. The rate of success is determined by the skill of the surgeon, underlying heart disease, attention to follow-up, and how success is defined.
Freedom from recurrence early on is a good predictor that late recurrence is unlikely. Patients who only have a very early recurrence (within the first 4 weeks) are more likely to have long-term freedom from atrial fibrillation tha those who have recurrences after that time.22
In a study of 831 patients, Hussein et al23 found recurrence rates of 24% between months 3 to 13 following ablation and 9% after 12 months. At 55 months, 79% were free from atrial fibrillation without drugs, 11% were free of atrial fibrillation with medications, and 5% had refractory atrial fibrillation.
Recurrence—whether early or late—was more likely to occur in people with persistent vs paroxysmal atrial fibrillation. Other risk factors for late recurrence included older age and larger left atrial size (which is also a risk factor for recurrence on drug therapy). Although recurrent arrhythmia was most often atrial fibrillation, atrial flutter also occurred frequently (in 27% of patients with late recurrence). Three patients (4% of patients with late recurrence) developed atrial tachycardia.23
In patients with early recurrence, 81% underwent repeat ablation, all of whom had recovery of one or more pulmonary veins. After the second ablation, 21% had recurrence, 65% of whom were controlled by medications.23
Whether a patient should undergo subsequent ablation procedures depends on the severity of symptoms, the likelihood of success (based on an educated guess), and the patient’s willingness to undergo another procedure.
ATRIAL APPENDAGE OCCLUSION DEVICE UNDER INVESTIGATION
New devices are being investigated that occlude the left atrial appendage to try to prevent embolization.
The Watchman device, resembling an umbrella, is implanted via a percutaneous catheter in the left atrial appendage, closing it off to preclude a thrombus from forming in the appendage and embolizing to the body. Clinical trials showed that patients who received a device had a slightly lower risk of stroke than otherwise seen in clinical practice.24 Safety and efficacy are still being determined.
The device cannot be deployed in a patient with an existing thrombus because of the danger of dislodging the thrombus, allowing it to embolize.
Although many developments have occurred in the last decade for managing atrial fibrillation, challenges remain. New and emerging alternatives to warfarin (Coumadin) for anticoagulation therapy prevent stroke marginally better and pose slightly less risk of hemorrhage, but they have important drawbacks.
The antiarrhythmic drug dronedarone (Multaq) has been found to offer only temporary benefit for persistent atrial fibrillation, and significant risks have emerged.
Radiofrequency ablation is gaining prominence, but repeat procedures are sometimes necessary.
An investigational device can be implanted via percutaneous catheter in the left atrial appendage to prevent embolization. It is too soon to know its eventual role in clinical practice.
This article reviews the results of clinical trials of these new treatments and discusses their role in clinical practice.
CHALLENGES OF ANTICOAGULATION
The main focus of managing atrial fibrillation is on alleviating symptoms, by either rate control or rhythm control. The other focus is on preventing stroke—a devastating outcome—with anticoagulation therapy.
For deciding whether to give warfarin to patients with atrial fibrillation, the six-point CHADS2 score is a crude but effective way of assessing the risk of stroke based on the following risk factors: congestive heart failure, hypertension, age 75 years or older, and diabetes (1 point each); or a history of stroke or transient ischemic attack (2 points).1 Warfarin is given if patients have a score of at least 2 points.
Warfarin has a narrow therapeutic window, with a higher risk of ischemic stroke if the international normalized ratio (INR) is less than 2.0,2 and a higher risk of intracranial hemorrhage if the INR is more than 3.0.3 Keeping the INR in the therapeutic range is difficult because of variations in diet, concurrent medications, and other factors.
The percent of time that the INR is within the therapeutic range predicts the risk of adverse events. Connolly et al4 showed that the cumulative risk of stroke, myocardial infarction, systemic embolism, or vascular death was no better with warfarin than with clopidogrel (Plavix) plus aspirin if the INR was in the therapeutic range less than 65% of the time, but the risk was significantly less if the INR was in the therapeutic range more than 65% of the time.
Also, comparing warfarin with the combination of aspirin and clopidogrel, Verheugt5 found that the rates of stroke of any kind, of disabling and fatal stroke, and of stroke per major bleed were lower in patients taking warfarin. Although many physicians prefer aspirin plus clopidogrel because of concerns about bleeding with warfarin, the rates of major bleeding were about the same in the two groups.
In a trial in patients for whom warfarin was “unsuitable,”6 the combination of aspirin plus clopidogrel was associated with a lower rate of stroke than aspirin alone (2.4% per year vs 3.3% per year, relative risk 0.762) but a higher rate of major bleeding events (2.0% per year vs 1.3% per year, relative risk 1.57).
NEW ALTERNATIVES TO WARFARIN
Because of the problems with warfarin, alternatives have been sought for many years. Several new oral anticoagulants are available or are being developed,7 including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis) and the direct factor II (thrombin) inhibitor dabigatran (Pradaxa) (Table 1).
Dabigatran’s advantages and drawbacks
Dabigatran has been on the market for more than a year and has gained rapid acceptance. The dosage is 150 mg twice a day, or 75 mg twice a day if renal function is impaired. Cleared by the kidneys, it has a half-life of 12 to 17 hours; 75% is cleared within 24 hours. For a patient who needs surgery that poses a low risk of bleeding, the general recommendation is to stop dabigatran the night before the surgical procedure. For operations with a greater risk of bleeding, many surgeons recommend stopping the drug 3 or 4 days before.
Advantages of dabigatran include that it is not influenced by diet and that the onset of therapeutic benefit is within 1 hour. Although some drugs affect dabigatran, drug interactions are more troublesome with warfarin.
A serious concern about dabigatran and the other new agents is that if a bleeding problem arises, the effects of these drugs are not reversible by administration of fresh frozen plasma. Dabigatran is reversible by dialysis; however, if a patient is also hypotensive, dialysis is not an option, and simply waiting for the drug to clear is the only choice.
Another drawback is that therapeutic levels cannot be monitored. If a patient taking warfarin requires cardioversion, the INR is carefully monitored for several weeks beforehand to reduce the risk of stroke. With dabigatran, there is no way to know if a patient is actually taking the drug as prescribed.
Clinical trials show that alternatives are marginally better than warfarin
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,8 dabigatran was associated with a significantly lower incidence of intracranial hemorrhage, combined strokes, and systemic embolization than warfarin. The incidence of major bleeds was slightly lower with dabigatran. Although dabigatran performed better, the differences were small and would not require patients to change from warfarin if they are already doing well.
Apixaban and rivaroxaban are other alternatives to warfarin, with different mechanisms of action and metabolism. Although rivaroxaban’s half-life is similar to that of apixaban and dabigatran, it is being marketed as allowing once-daily dosing instead of twice-daily.
Recent randomized controlled clinical trials of the new drugs include:
- The Apixaban Versus Acetylsalicylic Acid (ASA) to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment (AVERROES) trial,9 which compared apixaban and aspirin
- The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial comparing apixaban and warfarin10
- The Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF),11 comparing rivaroxaban and warfarin
- RE-LY,8 comparing dabigatran and warfarin.
In the ARISTOTLE,10 ROCKET AF,11 and RE-LY trials,8 the time that the warfarin patients’ INRs were in the therapeutic range varied from 55% to 68%. This seems low and is a problem when trying to compare therapies, but is probably about as high as one can expect in the real world.
In AVERROES,9 the combined rate of stroke and embolism was higher with aspirin than with apixaban. In the other trials, the rates were slightly better with the new drugs than with warfarin, and the rates of major hemorrhage and hemorrhagic stroke were only slightly higher with warfarin than with the new drugs. Because the differences in benefits and risks are so small, the main advantage of the newer drugs will probably be for patients who have difficulty staying in the therapeutic INR range on warfarin.
RATE CONTROL VS RESTORATION OF SINUS RHYTHM
Evidence is insufficient to determine the risk of very-long-term asymptomatic atrial fibrillation in patients on appropriate anticoagulation. Rate control is an option for asymptomatic patients but provides no change in quality of life and no definitive reduction in the risk of stroke. The main argument for restoring normal sinus rhythm in patients with mild to moderate symptoms is that it improves exercise capacity. The need for anticoagulation persists when patients are converted to sinus rhythm because the risk of recurrent atrial fibrillation remains high.
For patients with symptomatic atrial fibrillation, rate control is sometimes achieved with beta-blockers or calcium channel blockers. Rate control may be augmented with the addition of digoxin, but when used alone digoxin generally does not control the rate of atrial fibrillation. However, in many cases of atrial fibrillation, symptoms are not rate-related, and cardioversion to normal sinus rhythm should be attempted. In such cases, the symptoms may be attributable to a loss of atrial transport function.
Patients with the following risk factors should be admitted to the hospital to start antiarrhythmic drugs:
- Borderline or a long QTc interval at baseline (> 450 msec)
- Treatment with dofetilide (Tikosyn) because of its effects on the QT interval
- Heart failure or poor left-ventricular function
- Sinus node dysfunction
- Significant atrioventricular conduction disease.
Selecting an antiarrhythmic drug
Any of the antiarrhythmic drugs listed in Table 2 can be used for a patient with lone atrial fibrillation (ie, not caused by underlying heart disease). The choice of drug should be determined by whether coronary artery disease or renal failure is present as well. Liver disease or chronic obstructive pulmonary disease also may affect this decision.
Benefits of dronedarone are mixed
In a randomized trial of dronedarone vs placebo in patients with atrial fibrillation, the rate of death and the rate of first hospitalization due to a cardiovascular event at 21 months were significantly lower with dronedarone.12 No difference was found between the two groups in the rate of death from all causes, but fewer people died of cardiovascular causes in the dronedarone group. More patients taking dronedarone developed bradycardia, QT-interval prolongation, nausea, diarrhea, rash, or a higher serum creatinine level. Gastrointestinal side effects are often a problem with dronedarone: 20% to 30% of patients cannot tolerate the drug.
Dronedarone may cause a small rise in creatinine, and although this effect should be monitored, by itself it should not be interpreted as impairment of renal function. In a study in healthy people,13 dronedarone caused a 10% to 15% increase in serum creatinine, but the glomerular filtration rate was unchanged, as were renal plasma flow and anion secretion.
Another trial, in patients with severe heart failure, found that patients taking dronedarone had higher rates of hospitalization and overall mortality, raising serious concern about the safety of this drug in patients with advanced heart failure.14
Singh et al15 pooled the data from two multicenter, randomized trials that compared dronedarone with placebo for maintaining sinus rhythm in patients with atrial fibrillation or flutter. The mean time to the recurrence of atrial fibrillation was 116 days with dronedarone and 53 days with placebo. Other trials also showed longer times to recurrence and lower recurrence rates with dronedarone. Although the differences were statistically significant, they may not be clinically meaningful for patients.
Dronedarone is structurally similar to amiodarone (Cordarone), but the two drugs work differently. A meta-analysis of clinical trials16 found that amiodarone recipients had a lower rate of recurrence of atrial fibrillation than did those receiving dronedarone.
Two safety warnings for dronedarone
In January 2011, the US Food and Drug Administration (FDA) issued an alert about cases of rare but severe liver injury in patients treated with dronedarone, including two cases of acute liver failure leading to liver transplantation.17
The Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS)18 compared dronedarone and placebo in patients with permanent atrial fibrillation. More people died or had serious cardiovascular adverse events in the dronedarone group. The study was stopped early after data monitoring showed that rates of death, stroke, and hospitalization for heart failure were two times higher in patients receiving dronedarone. This prompted the FDA to issue another safety alert in July 2011.
Interestingly, the PALLAS study did not set out to determine whether dronedarone controls atrial fibrillation, as the study patients had long-standing, persistent atrial fibrillation. The study was designed only to determine if the drug reduces the rate of adverse events; it clearly does not, and the study shows that dronedarone should not be used to control the heart rate in patients with persistent atrial fibrillation. Instead, its use is best restricted to patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
ABLATION OF ATRIAL FIBRILLATION
Another way to try to restore sinus rhythm is to destroy or isolate the area that is generating the abnormal beats via a catheter-based procedure.
Radiofrequency ablation is generally tried in patients in whom one or two drugs have failed to control atrial fibrillation. Direct comparisons show that ablation is superior to drug therapy and is effective in about 75% of patients with paroxysmal atrial fibrillation vs 20% to 40% of patients on drug therapy. Ablation plus drug therapy is often more effective than either treatment alone.
Mechanisms of atrial fibrillation and ablation
In many cases, atrial fibrillation is stimulated by vagal and sympathetic inputs to the atrium that enter around the pulmonary veins and trigger electrical activations in the area, generating spiraling, reentering circuits. Focal atrial fibrillation also originates predominantly in the pulmonary veins. Ablation of tissue widely circumscribing the mouth of the pulmonary veins prevents the electrical signal from exiting into the atrium.
In about 11% to 37% of cases, atrial fibrillation originates elsewhere, eg, in the left atrium, in the superior vena cava, or in the vein of Marshall. Techniques have evolved to also ablate these regions.
Anticoagulation therapy is recommended before the procedure, and patients at low risk should continue it for a minimum of 2 months afterward. Patients with a higher CHADS2 score should receive anticoagulation therapy for at least 1 year. The consensus statement by the Heart Rhythm Society19 recommends that patients remain on warfarin or one of the newer anticoagulants if their CHADS2 score is 2 or higher. This is because patients have a significant risk of recurrence of atrial fibrillation after radiofrequency ablation, so if their stroke risk is high they should remain on anticoagulant therapy.
Ablation is usually effective, but it carries rare but serious risks
The efficacy of a single radiofrequency ablation procedure is in the range of 60% to 80% for paroxysmal atrial fibrillation and 40% to 60% for persistent atrial fibrillation. The Second International Ablation Registry20 shows a success rate of about 75% in patients with paroxysmal atrial fibrillation and about 65% in patients with persistent and permanent atrial fibrillation. Registry data are often more favorable because reporting is optional, but these results are consistent with those from experienced medical centers. Rates of suppression of atrial fibrillation are higher in patients who also take antiarrhythmic drugs, making a “hybrid” approach useful when ablation alone fails.
According to a worldwide survey, the risk of serious complications is 4.5%. These include stroke (0.23%), tamponade (1.3%), and pulmonary vein stenosis (< 0.29%). The esophagus lies just behind the right atrium, and burning through and creating a fistula between them occurs in about 0.04% of cases and is almost uniformly fatal.20
A second ablation procedure is sometimes indicated for the recurrence of atrial fibrillation, which is almost always caused by recovery of the pulmonary veins. Bhargava et al21 found that the success rate at Cleveland Clinic for a single procedure for paroxysmal atrial fibrillation was 77%, and that it was 92% after a repeat procedure. For persistent atrial fibrillation, success rates were 76% after the first procedure and 90% after the second. Even for long-standing persistent atrial fibrillation (ie, lasting more than 1 year), 80% success was achieved after two procedures. Patients who are less likely to have a successful ablation procedure are those with long-standing atrial fibrillation and coexisting heart disease, including severe valvular disease, although mitral regurgitation sometimes improves if sinus rhythm can be maintained.
The need for a second procedure
After ablation, patients should be closely monitored for a recurrence of atrial fibrillation. Continuous monitoring with implantable cardiac monitor loop recorders can detect unrecognized episodes of arrhythmia. Long-term follow-up is also required to track outcomes and quality of life.
According to the Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation,19 atrial fibrillation recurs after ablation in about 35% to 60% of patients in the first 3 months, with recurrence rates after 1 year ranging from 5% to 16%. The rate of success is determined by the skill of the surgeon, underlying heart disease, attention to follow-up, and how success is defined.
Freedom from recurrence early on is a good predictor that late recurrence is unlikely. Patients who only have a very early recurrence (within the first 4 weeks) are more likely to have long-term freedom from atrial fibrillation tha those who have recurrences after that time.22
In a study of 831 patients, Hussein et al23 found recurrence rates of 24% between months 3 to 13 following ablation and 9% after 12 months. At 55 months, 79% were free from atrial fibrillation without drugs, 11% were free of atrial fibrillation with medications, and 5% had refractory atrial fibrillation.
Recurrence—whether early or late—was more likely to occur in people with persistent vs paroxysmal atrial fibrillation. Other risk factors for late recurrence included older age and larger left atrial size (which is also a risk factor for recurrence on drug therapy). Although recurrent arrhythmia was most often atrial fibrillation, atrial flutter also occurred frequently (in 27% of patients with late recurrence). Three patients (4% of patients with late recurrence) developed atrial tachycardia.23
In patients with early recurrence, 81% underwent repeat ablation, all of whom had recovery of one or more pulmonary veins. After the second ablation, 21% had recurrence, 65% of whom were controlled by medications.23
Whether a patient should undergo subsequent ablation procedures depends on the severity of symptoms, the likelihood of success (based on an educated guess), and the patient’s willingness to undergo another procedure.
ATRIAL APPENDAGE OCCLUSION DEVICE UNDER INVESTIGATION
New devices are being investigated that occlude the left atrial appendage to try to prevent embolization.
The Watchman device, resembling an umbrella, is implanted via a percutaneous catheter in the left atrial appendage, closing it off to preclude a thrombus from forming in the appendage and embolizing to the body. Clinical trials showed that patients who received a device had a slightly lower risk of stroke than otherwise seen in clinical practice.24 Safety and efficacy are still being determined.
The device cannot be deployed in a patient with an existing thrombus because of the danger of dislodging the thrombus, allowing it to embolize.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the national Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335:540–546.
- Hylek EM, Singer DE. Risk factors for intracranial hemorrhage in outpatients taking warfarin. Ann Intern Med 1994; 120:897–902.
- Connolly SJ, Pogue J, Eikelboom J, et al; ACTIVE W Investigators. Benefit of oral anticoagulant over antiplatelet therapy in atrial fibrillation depends on the quality of international normalized ratio control achieved by centers and countries as measured by time in therapeutic range. Circulation 2008; 118:2029–2037.
- Verheugt FWA. Who is ineligible for warfarin in atrial fibrillation? Lancet 2009; 374:510–511.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Harenberg J. New anticoagulants in atrial fibrillation. Semin Thromb Hemost 2009; 35:574–585.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Connolly SJ, Eikelboom J, Joyner C, et al; AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364:806–817.
- Granger CB, Alexander JH, McMurray JJV, et al; for the ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med August 28, 2011; 10.1056/nejmoa1107039.
- Patel MR, Mahaffey KW, Garg J, et al; the ROCKET AF Steering Committee. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Hohnloser SH, Crijns HJ, van Eickels M, et al; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:688–678.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Kóber L, Torp-Pederson C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 2009; 54:1089–1095.
- US Food and Drug Administration. FDA drug safety communication: severe liver injury associated with the use of dronedarone (marketed as Multaq). http://www.fda.gov/drugs/drugsafety/ucm240011.htm. Accessed July 5, 2012.
- Connolly SJ, Camm AJ, Halperin JL, et al; for the PALLAS Investigators. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011; 365:2268–2276.
- HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. Heart Rhythm 2007; 4:1–46.
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3:32–38.
- Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm 2009; 6:1403–1412.
- Themistoclakis S, Schweikert RA, Sliba WI, et al. Clinical predictors and relationship between early and late atrial tachyarrhythmias after pulmonary vein antrum isolation. Heart Rhythm 2008; 5:679–685.
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4:271–278.
- Holmes DR, Reddy VY, Turi ZG, et al; for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the national Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335:540–546.
- Hylek EM, Singer DE. Risk factors for intracranial hemorrhage in outpatients taking warfarin. Ann Intern Med 1994; 120:897–902.
- Connolly SJ, Pogue J, Eikelboom J, et al; ACTIVE W Investigators. Benefit of oral anticoagulant over antiplatelet therapy in atrial fibrillation depends on the quality of international normalized ratio control achieved by centers and countries as measured by time in therapeutic range. Circulation 2008; 118:2029–2037.
- Verheugt FWA. Who is ineligible for warfarin in atrial fibrillation? Lancet 2009; 374:510–511.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Harenberg J. New anticoagulants in atrial fibrillation. Semin Thromb Hemost 2009; 35:574–585.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Connolly SJ, Eikelboom J, Joyner C, et al; AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364:806–817.
- Granger CB, Alexander JH, McMurray JJV, et al; for the ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med August 28, 2011; 10.1056/nejmoa1107039.
- Patel MR, Mahaffey KW, Garg J, et al; the ROCKET AF Steering Committee. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Hohnloser SH, Crijns HJ, van Eickels M, et al; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:688–678.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Kóber L, Torp-Pederson C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 2009; 54:1089–1095.
- US Food and Drug Administration. FDA drug safety communication: severe liver injury associated with the use of dronedarone (marketed as Multaq). http://www.fda.gov/drugs/drugsafety/ucm240011.htm. Accessed July 5, 2012.
- Connolly SJ, Camm AJ, Halperin JL, et al; for the PALLAS Investigators. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011; 365:2268–2276.
- HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. Heart Rhythm 2007; 4:1–46.
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3:32–38.
- Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm 2009; 6:1403–1412.
- Themistoclakis S, Schweikert RA, Sliba WI, et al. Clinical predictors and relationship between early and late atrial tachyarrhythmias after pulmonary vein antrum isolation. Heart Rhythm 2008; 5:679–685.
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4:271–278.
- Holmes DR, Reddy VY, Turi ZG, et al; for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
KEY POINTS
- Warfarin is as safe as—and more effective than—the combination of aspirin and clopidogrel (Plavix) if the international normalized ratio is in the therapeutic range 65% of the time or more.
- New anticoagulants are promising alternatives to warfarin, but they also pose risks. Patients who are doing well on warfarin need not change.
- Several antiarrhythmic drugs are available to control symptomatic atrial fibrillation. Dronedarone (Multaq) should only be considered for patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
- Ablation is often effective in controlling atrial fibrillation, but recurrence is common. Early recurrence often subsides, but late recurrence often requires a repeat procedure.
Psoriasis: Evolving treatment for a complex disease
Much has changed in our understanding of psoriasis over the past decade, which is having a major effect on its treatment.
Although topical corticosteroids and phototherapy remain mainstays of treatment, a variety of biologic agents have given new hope to those with the most severe forms of the disease. We are also beginning to understand that patients with psoriasis are at greater risk of cardiovascular disease, though the exact nature of that risk and how we should respond remains unclear. Finally, genome-wide association studies are just beginning to unravel the genetic basis of psoriasis.
In this paper, we review the epidemiology and impact of psoriasis, current views of its pathogenesis, its varied clinical forms, and its treatment.
PSORIASIS IMPOSES A GREAT BURDEN
Psoriasis is common, with a reported prevalence ranging from approximately 2%1 to 4.7%.2 It can manifest at any age, but it is most common in two age groups, ie, 20 to 30 years and 50 to 60 years.
For the patient, the burden is great, affecting physical, psychological, and occupational well-being. In fact, patients with psoriasis report quality-of-life impairment equal to or worse than that in patients with cancer or heart disease.3,4 Notably, functional disability secondary to psoriatic arthritis has been reported in up to 19% of psoriatic arthritis patients, and this negatively affects quality of life.5
In 2004, the annual direct medical costs of psoriasis in the United States were estimated to exceed $1 billion. Its indirect costs, measured as missed days and loss of productivity at work, are estimated to exceed the direct costs by $15 billion annually.6,7
Linked to cardiovascular and other diseases
Studies in the past 10 years have uncovered a link between psoriasis, metabolic syndrome, and cardiovascular disease.8–13 Specifically, patients with severe psoriasis are at higher risk of myocardial infarction and cardiovascular death than control patients. Interestingly, the risk decreases with age; patients at greatest risk are young men with severe psoriasis.8–10
In a large cohort study in the United Kingdom7 comparing patients with and without psoriasis, the hazard ratio for cardiovascular death in patients with severe psoriasis was 1.57 (95% confidence interval 1.26–1.96). This translated to 3.5 excess deaths per 1,000 patient-years. These patients were also at higher risk of death from malignancies, chronic lower respiratory disease, diabetes, dementia, infection, kidney disease, and unknown causes.
How much of the risk is due to psoriasis itself, its treatments, associated behaviors, or other factors requires more study. However, some evidence points to the dysregulation of the immune system, notably chronic elevation of pro-inflammatory cytokines.
Psoriasis and its comorbid conditions are thought to arise from chronically elevated levels of cytokines such as tumor necrosis factor alpha (TNF-alpha), interleukin 1 beta (IL-1 beta), and IL-17. These cytokines impair insulin signaling, deregulate lipid metabolism, and increase atherosclerotic changes in the coronary, cerebral, and peripheral arteries. In addition, several other diseases that involve the immune system occur more frequently with psoriasis, including Crohn disease, ulcerative colitis, lymphoma, obesity, and type 2 diabetes.1,8,14–18
In view of the prevalence of these comorbid conditions and the risks they pose, primary care physicians should consider screening patients with severe psoriasis for metabolic disorders and cardiovascular risk factors and promptly begin preventive therapies.19 Unfortunately, to date, there are no consensus guidelines as to the appropriate screening tests or secondary cardiovascular preventive measures for patients with severe psoriasis.
A VICIOUS CIRCLE OF INFLAMMATION AND KERATINOCYTE PROLIFERATION
The hallmark of plaque psoriasis is chronic inflammation in the skin, leading to keratinocyte proliferation.
External and internal triggers that have been identified include cutaneous injury (eg, sunburn, drug rash, viral exanthems), infections (eg, streptococcal), hypocalcemia, pregnancy, psychogenic stress, drugs (eg, lithium, interferon, beta-blockers, and antimalarials), alcohol, smoking, and obesity.20–23
As reviewed by Nestle et al,24 the initiation of lesion formation is still poorly understood but is thought to occur when a trigger (physical trauma, bacterial product, cellular stress) causes DNA to be released from keratinocytes. DNA forms a complex with the antimicrobial protein LL-37 and activates plasmacytoid dendritic cells (PDCs) via toll-like receptor 9. Activated PDCs release type I interferons, which in turn activate myeloid dendritic cells. Myeloid dendritic cells release IL-20 locally, which speeds keratinocyte proliferation.
A subset of myeloid dendritic cells leaves the dermis and migrates to local lymph nodes, where they release IL-23 and activate naive T cells. T helper 1 (Th1) and Th17 cells are recruited to the lesions and begin producing numerous cytokines, including interferon gamma, IL-17, and IL-22. This cytokine milieu increases keratinocyte proliferation and causes the keratinocytes to secrete antimicrobial proteins (LL-37, beta defensins), chemokines, and S100 proteins. These soluble factors have three main functions: stimulation of dendritic cells to release more IL-23, recruitment of neutrophils to the epidermis, and activation of dermal fibroblasts.
This cycle of keratinocytes activating dendritic cells, dendritic cells activating T cells, and T cells activating keratinocytes appears to be the main force maintaining the disease.24 It is unclear, however, whether this applies to all forms of psoriasis or only to plaque psoriasis.
Genetic factors discovered
In recent years, genome-wide association studies have identified at least 10 psoriasis-susceptibility loci that involve functioning of the immune system.25 These genes include those of the major histocompatibility complex, cytokines, receptors, and beta-defensins.
Of specific interest, polymorphisms in the IL-12/IL-13 receptor, the p40 subunit of IL-12 and IL-23, and the p19 subunit of IL-23 strongly associate with psoriasis, supporting their critical role in the disease process and providing targets for medical therapy.26
PSORIASIS HAS SEVERAL CLINICAL PHENOTYPES
Psoriasis has several clinical variants, each with a distinct clinical course and response to treatment.27 Moreover, many patients present with more than one variant.
Plaque psoriasis
Plaques can persist for several months to years, even in the same location, and only about 5% of patients report complete remission for up to 5 years.
Inverse psoriasis
Guttate psoriasis
Erythrodermic psoriasis
Approximately 1% to 2.25% of all patients with psoriasis develop this severe form, affecting more than 75% of the body surface area. It presents as generalized erythema, which is the most prominent feature, and it is often associated with superficial desquamation, hair loss, nail dystrophy, and systemic symptoms such as fever, chills, malaise, or high-output cardiac failure. There may be a history of preceding characteristic psoriatic plaques, recent withdrawal of treatment (usually corticosteroids), phototoxicity, or infection.
Conversely, approximately 25% of all patients with erythroderma have underlying psoriasis.28
Pustular psoriasis
There are several forms of pustular psoriasis, including generalized pustular psoriasis, annular pustular psoriasis, impetigo herpetiformis (pustular psoriasis of pregnancy), and palmoplantar pustulosis. However, there is some evidence to suggest that palmoplantar pustulosis may be distinct from psoriasis.29
Several triggers have been identified, including pregnancy, rapid tapering of medications, hypocalcemia, infection, or topical irritants.
Generalized pustular psoriasis, annular pustular psoriasis, and impetigo herpetiformis often present in association with fever and other systemic symptoms and, if left untreated, can result in life-threatening complications including bacterial superinfection, sepsis, dehydration, and, in rare cases, acute respiratory distress secondary to aseptic pneumonitis.30
Placental insufficiency resulting in stillbirth or neonatal death and other fetal abnormalities can occur in severe pustular psoriasis of pregnancy.31
Psoriatic arthritis
Psoriatic arthritis is a seronegative inflammatory spondyloarthropathy that can result in erosive arthritis in up to 57% of cases and functional disability in up to 19%.32 Although rare in the general population, it affects approximately 6% to 10% of psoriasis patients and up to 40% of patients with severe psoriasis.33 In 70% of cases, psoriasis precedes the onset of arthritis by about 10 years, and approximately 10% to 15% of patients simultaneously present with psoriasis and arthritis or develop arthritis before skin involvement.5,34
Patients complain of joint discomfort that is most prominent after periods of prolonged rest. Patterns of involvement are extremely variable but have been reported as an asymmetric oligoarthritis (involving four or fewer joints) or polyarthritis (involving more than four joints) in most patients. A rheumatoid arthritis-like presentation with a symmetric polyarthropathy involving the small and medium-sized joints has also been reported, making it difficult to clinically distinguish this from rheumatoid arthritis.
A distal interphalangeal-predominant pattern is reported in 5% to 10% of patients. Axial disease resembling ankylosing spondylitis occurs only in 5% of patients. Arthritis mutilans, characterized by severe, rapidly progressive joint inflammation, joint destruction, and deformity, occurs rarely. Enthesitis, ie, inflammation at the point of attachment of tendons or ligaments to bone, is present in up to 42% of patients.5,35
Nail disease
Disease severity also varies
Disease severity also differs among patients. An estimated 80% of patients have mild to moderate disease and 20% have moderate to severe disease, which includes disease involving more than 5% of the body surface or involvement of the face, hands, feet, or genitalia.1
The Psoriasis Area and Severity Index (PASI) is an objective measure used in clinical trials. It incorporates the amount of redness, scaling, and induration of each psoriatic lesion over the body surface involved. A 75% improvement in the PASI score (PASI-75) is regarded as clinically significant.37
PSORIASIS IS DIAGNOSED CLINICALLY
In most cases, the diagnosis of psoriasis is made clinically and is straightforward. However, in more difficult cases, biopsy may be needed. In particular:
- The plaques of psoriasis may be confused with squamous cell carcinoma in situ, dermatophyte infection, or cutaneous T-cell lymphoma, especially if they are treatment-resistant.
- Guttate psoriasis may be difficult to distinguish from pityriasis rosea.
- Erythrodermic psoriasis must be distinguished from other causes of erythroderma, including Sézary syndrome, pityriasis rubra pilaris, and drug reactions.
- Intertrigo, candidiasis, extramammary Paget disease, squamous cell carcinoma, and contact dermatitis all may mimic inverse psoriasis.
- Palmoplantar pustulosis may be difficult to differentiate from dyshidrotic eczema.
- Generalized pustular psoriasis should be distinguished from a pustular drug eruption (acute generalized exanthematous pustular drug eruption or acute generalized exanthematous pustulosis), impetigo, candidiasis, or an autoimmune blistering disorder such as pemphigus.
TREATMENT OF LIMITED DISEASE
Topical corticosteroids
A topical corticosteroid, either by itself or combined with a steroid-sparing agent, is the first-line therapy for patients with limited disease. The potency required for treatment should be based on the extent of disease and on the location, the choice of vehicle, and the patient’s preference and age.
Several double-blind studies have assessed the efficacy of various topical corticosteroids in treating psoriasis. In general, super-potent (class I) and potent (class II) topical corticosteroids are more efficacious than mild or moderate corticosteroids.38 Class I and class II steroids include agents such as clobetasol propionate 0.05% (Temovate), betamethasone dipropionate 0.05% (Diprolene), fluocinonide 0.05% (Lidex), and desoximetasone 0.25% (Topicort).
Use of class I steroids should be limited to an initial treatment course of twice-daily application for 2 to 4 weeks in an effort to avoid some of the local toxicities such as skin atrophy, telangiectasia, and striae. Decreasing class I topical steroid use to 1 to 2 times per week with the gradual introduction of a steroid-sparing agent following the initial 2 to 4 weeks of treatment is advised.
Steroid-sparing agents
Steroid-sparing agents include vitamin D analogues, retinoids, and tacrolimus ointment (Protopic).
Vitamin D analogues and retinoids are thought to decrease keratinocyte proliferation and enhance keratinocyte differentiation.39 The vitamin D analogues are also considered first-line topical agents and include calcipotriol (Dovonex), calcipotriene (Dovonex), and calcitriol (Vectical). To prevent hypercalcemia, use of more than 100 g of vitamin D analogues per week should be avoided.39
Treatment of inverse psoriasis and scalp psoriasis may be challenging
The areas affected in inverse psoriasis, such as the genitalia and axillae, are more prone to side effects when potent topical steroids are used because of increased absorption and occlusion in these areas. Agents that minimize irritation and toxicity in sensitive areas, such as topical tacrolimus, less-potent topical steroids, or calcitriol, can be used.39
For scalp psoriasis, alternative vehicles such as shampoos, gels, solutions, oils, sprays, and foams have improved patient compliance and efficacy of treatment.40
PHOTOTHERAPY FOR SEVERE DISEASE
Narrow-band ultraviolet B
Narrow-band ultraviolet B, ie, light confined to wavelengths of 311 to 313 nm, is a first-line treatment for moderate to severe psoriasis, either as monotherapy or in combination with other treatments. It is an especially attractive option in patients who are on medications or who have comorbidities that may preclude treatment with other systemic agents.
The mechanism of action may be via immunosuppressive effects on Langerhans cells, alteration of cytokines and adhesion molecules that lead to an increase in Th2 cells, and induction of apoptosis of T lymphocytes. Additionally, ultraviolet light affects the proliferation and differentiation of keratinocytes.41
Dosing is based on skin type, and treatment usually involves two or three visits per week for a total of 15 to 20 treatments, with additional therapy for maintenance. Adding acitretin (Soriatane), with close monitoring of aspartate aminotransferase and alanine aminotransferase levels and the patient’s lipid panel, can be considered in treatment-resistant cases.42
Psoralen combined with ultraviolet A
Psoralen combined with ultraviolet A is another option. It can be considered if narrow-band ultraviolet B treatment fails. It is also useful for dark-skinned patients and those with thicker plaques because ultraviolet A penetrates deeper than ultraviolet B. Oral or topical treatment with psoralen is followed by ultraviolet A treatment.
The duration of remission is much longer with psoralen plus ultraviolet A than with narrow-band ultraviolet B. However, this treatment caries a significant risk of cutaneous squamous cell carcinoma and melanoma, especially in light-skinned people and those who receive high doses of ultraviolet A (200 or more treatments) or cyclosporine.40,41,43–46 Long-term effects include photoaging, lentigines, and telangiectasias. As a consequence of these well-established side effects, this treatment is used less frequently.
Cautions with phototherapy
Careful screening and caution should be used in patients who have:
- Fair skin that tends to burn easily
- A history of arsenic intake or treatment with ionizing radiation
- A history of use of photosensitizing medications (fluoroquinolone antibiotics, doxycycline, hydrochlorothiazide)
- A history of melanoma or atypical nevi
- Multiple risk factors for melanoma
- A history of nonmelanoma skin cancer
- Immunosuppression due to organ transplantation.
ORAL THERAPIES FOR SEVERE PSORIASIS
Patients who have severe psoriasis—ie, affecting more than 5% of the body surface or debilitating disease affecting the palms, soles, or genitalia—are best managed with systemic medications, especially if they do not have access to phototherapy.20
Methotrexate
In 1972, the US Food and Drug Administration (FDA) approved methotrexate for treating severe psoriasis.42 In studies of methotrexate at doses of 15 to 20 mg weekly, 36% to 68% of patients with severe plaque psoriasis achieved a PASI-75 score.40,42,47
Dosages of methotrexate for treating severe psoriasis range from 7.5 to 25 mg once a week. Patients should also receive a folate supplement of 1 to 5 mg every day except the day they take methotrexate. The folate is to protect against gastrointestinal side effects, bone marrow suppression, and hepatic toxicity associated with methotrexate.
Other side effects of methotrexate include pulmonary fibrosis and stomatitis. Pregnancy, nursing, alcoholism, chronic liver disease, immunodeficiency syndromes, bone-marrow hypoplasia, leukopenia, thrombocytopenia, anemia, and hypersensitivity to methotrexate are all contraindications to methotrexate use.
The National Psoriasis Foundation, in its 2009 guidelines for the use of methotrexate in treating psoriasis,48 recommends obtaining a complete blood cell count with platelets, blood urea nitrogen, creatinine, and liver function tests at baseline and at 1- to 3-month intervals thereafter.
Liver biopsies were previously recommended for patients receiving methotrexate long-term when the cumulative dose of therapy reached 1.5 g. However, given the invasive nature of the liver biopsy procedure and the low incidence of methotrexate-induced hepatotoxicity, this recommendation has been revised.
For patients with no significant risk factors for hepatic toxicity (eg, obesity, diabetes, hyperlipidemia, hepatitis, or history of or current alcohol consumption) and normal liver function tests, liver biopsy should be considered when a cumulative methotrexate dose of 3.5 to 4.0 g is reached. Alternatively, one may choose to continue to monitor the patient without liver biopsy or to switch to another medication, if possible.42,48
Patients at high risk should be monitored more carefully, and liver biopsy should be considered soon after starting methotrexate and repeated after every 1.0 to 1.5 g.48
No reliable noninvasive measures to evaluate for liver fibrosis are routinely available in the United States. Serial measurements of serum type III procollagen aminopeptide have been reported to correlate with the risk of developing liver fibrosis; however, this test is readily available only in Europe.49
Cyclosporine
Cyclosporine (Gengraf, Neoral, Sandimmune) is very effective for treating psoriasis, especially erythrodermic psoriasis. It is often used only short-term or as a bridge to other maintenance therapies because it has a rapid onset and because long-term therapy (3 to 5 years) is associated with a risk of glomerulosclerosis.50
Cyclosporine works by decreasing T-cell activation by binding cyclophilin, which leads to inhibition of transcription of calcineurin and nuclear factor of activated T cells.51 Given at doses of 2.5 to 5 mg/kg/day, cyclosporine has been shown to result in rapid improvement in up to 80% to 90% of psoriatic patients.52,53
The initial recommended dose of cyclosporine is usually 2.5 to 3 mg/kg/day in two divided doses, which is maintained for 4 weeks and then increased by 0.5 mg/kg/day until the disease is stable.42
Nephrotoxicity and hypertension are cyclosporine’s most serious side effects. Blood urea nitrogen, creatinine, and blood pressure should be monitored at baseline and then twice a month for the first 3 months and once monthly thereafter. Liver function tests, complete blood cell count, lipid profile, magnesium, uric acid, and potassium should also be checked every month.
Cyclosporine also increases the risk of cutaneous squamous cell carcinoma, especially in patients who have received psoralen plus ultraviolet A treatment.42
Patients with hypersensitivity to cyclosporine, a history of chronic infection (eg, tuberculosis, hepatitis B, hepatitis C), renal insufficiency, or a history of systemic malignancy should not receive cyclosporine.
Acitretin
Acitretin, an oral retinoid, has been used for several years to treat psoriasis. Its onset is slow, typically ranging from 3 to 6 months, and its effects are dose-dependent. It is most effective as a maintenance therapy, usually after the disease has been stabilized by agents such as cyclosporine, or in combination with other treatments such as phototherapy.42 Acitretin has been shown to be effective in patients with pustular psoriasis.54
Acitretin does not alter the immune system and has not been shown to have significant cumulative toxicities. Serum triglycerides are monitored closely, since acitretin can lead to hypertriglyceridemia.
All retinoids, including acitretin, are in pregnancy category X and should therefore be avoided during pregnancy. Although its half-life is only 49 hours, acitretin may be transformed to etretinate either spontaneously or as a result of alcohol ingestion. Etretinate has a half-life of 168 days and can take up to 3 years to be eliminated from the body. Therefore, acitretin is contraindicated in women who plan to become pregnant or who do not agree to use adequate contraception for 3 years after the drug is discontinued.42
Biologic agents
Advances in our understanding of the pathogenesis of psoriasis have resulted in more specific, targeted therapy.
Alefacept (Amevive) is a human Fc IgG1 receptor fused to the alpha subunit of LFA3. It binds to CD2, blocks costimulatory signaling, and induces apoptosis in activated memory T cells.
Alefacept was the first biologic agent approved by the FDA for the treatment of psoriasis and one of the few biologic agents to induce long-term remission.55 However, its use has declined because few patients achieved significant clearance of their psoriasis and its onset of action was much slower than that of other medications.56
The currently approved biologic therapies commonly used for moderate to severe psoriasis include the TNF-alpha inhibitors and ustekinumab (Stelara).
The TNF-alpha inhibitors include infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira). They are generally well tolerated and highly effective. However, TNF-alpha inhibitors and other biologic agents are contraindicated in patients with serious infection, a personal history or a family history in a first-degree relative of demyelinating disease, or class III or IV congestive heart failure. Patients should be screened for active infection, including tuberculosis and hepatitis B, since reactivation has been reported following initiation of TNF-alpha inhibitors.1
Adalimumab is a human monoclonal antibody against TNF-alpha. It binds to soluble and membrane-bound TNF-alpha and prevents it from binding to p55 and p75 cell-surface TNF receptors.
The dosing schedule for adalimumab is 80 mg subcutaneously for the first week, followed by 40 mg subcutaneously the next week, and then 40 mg subcutaneously every 2 weeks thereafter.1
Etanercept is a recombinant human TNF-alpha receptor (p75) protein fused with the Fc portion of IgG1, which binds to soluble TNF-alpha.57 Dosing for etanercept is 50 mg subcutaneously twice weekly for the first 12 weeks, followed by 50 mg weekly thereafter.
Infliximab is a chimeric antibody composed of a human IgG1 constant region fused to a mouse variable region that binds to both soluble and membrane-bound TNF-alpha.58 Infliximab is given as an infusion at a dose of 5 mg/kg over 2 to 3 hours at weeks 0, 2, and 6, and then every 8 weeks thereafter.
Efficacy of TNF inhibitors. There are no specific guidelines for the sequence of initiation of TNF inhibitors because no studies have directly compared the efficacy of these medications. However, response to infliximab is relatively rapid compared with adalimumab and etanercept.
In a phase III clinical trial,59 as many as 80% of patients achieved PASI-75 clearance of their psoriasis after three doses of infliximab. Interestingly, only 61% of patients maintained PASI-75 clearance by week 50. This loss of efficacy of infliximab is also reported with other TNF-alpha inhibitors and is thought to be secondary to the development of antibodies to the drugs. For infliximab, this loss of efficacy is less when infliximab is given continuously rather than on an as-needed basis. Simultaneous treatment with methotrexate is also thought to decrease the development of antibodies to infliximab.60
Ustekinumab is an monoclonal antibody directed against the common p40 subunit of IL-12 and IL-23, which have been shown to be at increased levels in psoriatic lesions and important for the pathogenesis of psoriasis.
Between 66% and 76% of patients treated with ustekinumab achieved significant clearance of their disease after 12 weeks of treatment in two large phase III multicenter, randomized, double-blind, placebo-controlled trials.61,62
Dosing of ustekinumab is weight-based. For those weighing less than 100 kg, ustekinumab is given at 45 mg subcutaneously at baseline, at 4 weeks, and every 12 weeks thereafter. The same dosing schedule is used for those weighing more than 100 kg, but the dose is increased to 90 mg.
Guidelines for monitoring patients while on ustekinumab are similar to those for other biologic agents. Information on long-term toxicities is still being collected. However, injection-site reactions, serious infections, malignancies, and a single case of reversible posterior leukoencephalopathy have been reported.20
While biologic agents are significantly more expensive than the conventional therapies discussed above and insurance coverage for these agents varies, they have demonstrated superior efficacy and may be indicated for patients with recalcitrant moderate to severe psoriasis for whom multiple types of treatment have failed.
FOR PSORIATIC ARTHRITIS: SYSTEMIC MEDICATIONS
For patients with known or questionable psoriatic arthritis, evaluation by a rheumatologist is highly recommended.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are usually first-line in the treatment of mild psoriatic arthritis. If after 2 to 3 months of therapy with NSAIDs no benefit is achieved, treatment with methotrexate as monotherapy is a practical consideration because of its low cost. However, methotrexate as a monotherapy has not been shown to prevent radiologic progression of disease.5,32
The TNF-alpha inhibitors have been shown to have similar efficacy when compared among each other in the treatment of psoriatic arthritis.32,63 Based on radiologic evidence, ustekinumab has not shown to be as efficacious as the TNF-alpha inhibitors for treating psoriatic arthritis. Therefore, TNF inhibitors should be considered first-line in the treatment of psoriatic arthritis.21,64
Few studies have been done on the efficacy or sequence of therapies that should be used in the treatment of psoriatic arthritis. The American Academy of Dermatology’s Psoriasis Guidelines of Care recommend adding a TNF-alpha inhibitor or switching to a TNF-alpha inhibitor if no significant improvement is achieved after 12 to 16 weeks of treatment with oral methotrexate.20
FOR ERYTHRODERMIC PSORIASIS: MEDICATIONS THAT ACT PROMPTLY
The care of erythrodermic psoriatic patients is distinct from that of other psoriatic patients because of their associated systemic symptoms. Care should be taken to rule out sepsis, as this is a reported trigger of erythrodermic psoriasis.28
Systemic medications with a quick onset, such as oral cyclosporine, are recommended. Infliximab has also been reported to be beneficial because of its rapid onset.28
TREATMENT BASED ON THE TYPE AND THE SEVERITY OF PSORIASIS
The treatment of psoriasis can be as complex as the disease it itself and should be based on the type and the severity of psoriasis. Recognition of the various manifestations of psoriasis is important for effective treatment. However, in patients with moderate to severe psoriasis, atypical presentations, or recalcitrant disease, referral to a specialist is recommended.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008; 58:826–850.
- Christophers E. Psoriasis—epidemiology and clinical spectrum. Clin Exp Dermatol 2001; 26:314–320.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Weiss SC, Kimball AB, Liewehr DJ, Blauvelt A, Turner ML, Emanuel EJ. Quantifying the harmful effect of psoriasis on health-related quality of life. J Am Acad Dermatol 2002; 47:512–518.
- Garg A, Gladman D. Recognizing psoriatic arthritis in the dermatology clinic. J Am Acad Dermatol 2010; 63:733–748.
- Kimball AB, Yu AP, Signorovitch J, et al. The effects of adalimumab treatment and psoriasis severity on self-reported work productivity and activity impairment for patients with moderate to severe psoriasis. J Am Acad Dermatol 2012; 66:e67–e76.
- Schmitt JM, Ford DE. Work limitations and productivity loss are associated with health-related quality of life but not with clinical severity in patients with psoriasis. Dermatology 2006; 213:102–110.
- Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Abuabara K, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the U.K. Br J Dermatol 2010; 163:586–592.
- Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med 2011; 270:147–157.
- Lin HW, Wang KH, Lin HC, Lin HC. Increased risk of acute myocardial infarction in patients with psoriasis: a 5-year population-based study in Taiwan. J Am Acad Dermatol 2011; 64:495–501.
- Bremmer S, Van Voorhees AS, Hsu S, et al; National Psoriasis Foundation. Obesity and psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol 2010; 63:1058–1069.
- Tobin AM, Veale DJ, Fitzgerald O, et al. Cardiovascular disease and risk factors in patients with psoriasis and psoriatic arthritis. J Rheumatol 2010; 37:1386–1394.
- Najarian DJ, Gottlieb AB. Connections between psoriasis and Crohn’s disease. J Am Acad Dermatol 2003; 48:805–821.
- Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol 2006; 55:829–835.
- Shapiro J, Cohen AD, Weitzman D, Tal R, David M. Psoriasis and cardiovascular risk factors: a case-control study on inpatients comparing psoriasis to dermatitis. J Am Acad Dermatol 2012; 66:252–258.
- Gelfand JM, Shin DB, Neimann AL, Wang X, Margolis DJ, Troxel AB. The risk of lymphoma in patients with psoriasis. J Invest Dermatol 2006; 126:2194–2201.
- Chen YJ, Wu CY, Chen TJ, et al. The risk of cancer in patients with psoriasis: a population-based cohort study in Taiwan. J Am Acad Dermatol 2011; 65:84–91.
- Friedewald VE, Cather JC, Gelfand JM, et al. AJC editor’s consensus: psoriasis and coronary artery disease. Am J Cardiol 2008; 102:1631–1643.
- American Academy of Dermatology Work Group; Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. J Am Acad Dermatol 2011; 65:137–174.
- Mallbris L, Larsson P, Bergqvist S, Vingård E, Granath F, Ståhle M. Psoriasis phenotype at disease onset: clinical characterization of 400 adult cases. J Invest Dermatol 2005; 124:499–504.
- Armstrong AW, Armstrong EJ, Fuller EN, Sockolov ME, Voyles SV. Smoking and pathogenesis of psoriasis: a review of oxidative, inflammatory and genetic mechanisms. Br J Dermatol 2011; 165:1162–1168.
- Qureshi AA, Dominguez PL, Choi HK, Han J, Curhan G. Alcohol intake and risk of incident psoriasis in US women: a prospective study. Arch Dermatol 2010; 146:1364–1369.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2; Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 2010; 42:985–990.
- Nair RP, Duffin KC, Helms C, et al; Collaborative Association Study of Psoriasis. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 2009; 41:199–204.
- Griffiths CE, Christophers E, Barker JN, et al. A classification of psoriasis vulgaris according to phenotype. Br J Dermatol 2007; 156:258–262.
- Rosenbach M, Hsu S, Korman NJ, et al; National Psoriasis Foundation Medical Board. Treatment of erythrodermic psoriasis: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol 2010; 62:655–662.
- Mrowietz U, van de Kerkhof PC. Management of palmoplantar pustulosis: do we need to change? Br J Dermatol 2011; 164:942–946.
- Kluger N, Bessis D, Guillot B, Girard C. Acute respiratory distress syndrome complicating generalized pustular psoriasis (psoriasis-associated aseptic pneumonitis). J Am Acad Dermatol 2011; 64:1154–1158.
- Roth MM. Pregnancy dermatoses: diagnosis, management, and controversies. Am J Clin Dermatol 2011; 12:25–41.
- Gottlieb A, Korman NJ, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol 2008; 58:851–864.
- Ogdie A, Gelfand JM. Identification of risk factors for psoriatic arthritis: scientific opportunity meets clinical need. Arch Dermatol 2010; 146:785–788.
- Gelfand JM, Gladman DD, Mease PJ, et al. Epidemiology of psoriatic arthritis in the population of the United States. J Am Acad Dermatol 2005; 53:573.
- Moll JM, Wright V. Psoriatic arthritis. Semin Arthritis Rheum 1973; 3:55–78.
- McGonagle D. Enthesitis: an autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J Eur Acad Dermatol Venereol 2009; 23(suppl 1):9–13.
- Feldman SR, Krueger GG. Psoriasis assessment tools in clinical trials. Ann Rheum Dis 2005; 64(suppl 2):ii65–ii68.
- Mason J, Mason AR, Cork MJ. Topical preparations for the treatment of psoriasis: a systematic review. Br J Dermatol 2002; 146:351–364.
- Menter A, Korman NJ, Elmets CA, et al; American Academy of Dermatology. Guidelines of care for the management of psoriasis and psoriatic arthritis. Section 3. Guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol 2009; 60:643–659.
- Zivkovich AH, Feldman SR. Are ointments better than other vehicles for corticosteroid treatment of psoriasis? J Drugs Dermatol 2009; 8:570–572.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 5. Guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J Am Acad Dermatol 2010; 62:114–135.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. Guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol 2009; 61:451–485.
- Murase JE, Lee EE, Koo J. Effect of ethnicity on the risk of developing nonmelanoma skin cancer following long-term PUVA therapy. Int J Dermatol 2005; 44:1016–1021.
- Stern RS, Lunder EJ. Risk of squamous cell carcinoma and methoxsalen (psoralen) and UV-A radiation (PUVA). A meta-analysis. Arch Dermatol 1998; 134:1582–1585.
- Stern RS, Väkevä LH. Noncutaneous malignant tumors in the PUVA follow-up study: 1975–1996. J Invest Dermatol 1997; 108:897–900.
- Patel RV, Clark LN, Lebwohl M, Weinberg JM. Treatments for psoriasis and the risk of malignancy. J Am Acad Dermatol 2009; 60:1001–1017.
- Flytström I, Stenberg B, Svensson A, Bergbrant IM. Methotrexate vs. ciclosporin in psoriasis: effectiveness, quality of life and safety. A randomized controlled trial. Br J Dermatol 2008; 158:116–121.
- Kalb RE, Strober B, Weinstein G, Lebwohl M. Methotrexate and psoriasis: 2009 National Psoriasis Foundation Consensus Conference. J Am Acad Dermatol 2009; 60:824–837.
- Zachariae H, Heickendorff L, Søgaard H. The value of aminoterminal propeptide of type III procollagen in routine screening for methotrexate-induced liver fibrosis: a 10-year follow-up. Br J Dermatol 2001; 144:100–103.
- Lowe NJ, Wieder JM, Rosenbach A, et al. Long-term low-dose cyclosporine therapy for severe psoriasis: effects on renal function and structure. J Am Acad Dermatol 1996; 35:710–719.
- Gottlieb AB, Grossman RM, Khandke L, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol 1992; 98:302–309.
- Ellis CN, Fradin MS, Messana JM, et al. Cyclosporine for plaque-type psoriasis. Results of a multidose, double-blind trial. N Engl J Med 1991; 324:277–284.
- Faerber L, Braeutigam M, Weidinger G, et al. Cyclosporine in severe psoriasis. Results of a meta-analysis in 579 patients. Am J Clin Dermatol 2001; 2:41–47.
- Ozawa A, Ohkido M, Haruki Y, et al. Treatments of generalized pustular psoriasis: a multicenter study in Japan. J Dermatol 1999; 26:141–149.
- Krueger GG, Ellis CN. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol 2003; 148:784–788.
- Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE; Alefacept Clinical Study Group. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol 2003; 139:719–727.
- Gottlieb AB, Matheson RT, Lowe N, et al. A randomized trial of etanercept as monotherapy for psoriasis. Arch Dermatol 2003; 139:1627–1632.
- Gottlieb AB, Masud S, Ramamurthi R, et al. Pharmacodynamic and pharmacokinetic response to anti-tumor necrosis factor-alpha monoclonal antibody (infliximab) treatment of moderate to severe psoriasis vulgaris. J Am Acad Dermatol 2003; 48:68–75.
- Reich K, Nestle FO, Papp K, et al; EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Menter A, Feldman SR, Weinstein GD, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol 2007; 56:31.e1–31.e15.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Kimball AB, Papp KA, et al; PHOENIX 1 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Griffiths CE, Strober BE, van de Kerkhof P, et al; ACCEPT Study Group. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 2010; 362:118–128.
- Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009; 373:633–640.
Much has changed in our understanding of psoriasis over the past decade, which is having a major effect on its treatment.
Although topical corticosteroids and phototherapy remain mainstays of treatment, a variety of biologic agents have given new hope to those with the most severe forms of the disease. We are also beginning to understand that patients with psoriasis are at greater risk of cardiovascular disease, though the exact nature of that risk and how we should respond remains unclear. Finally, genome-wide association studies are just beginning to unravel the genetic basis of psoriasis.
In this paper, we review the epidemiology and impact of psoriasis, current views of its pathogenesis, its varied clinical forms, and its treatment.
PSORIASIS IMPOSES A GREAT BURDEN
Psoriasis is common, with a reported prevalence ranging from approximately 2%1 to 4.7%.2 It can manifest at any age, but it is most common in two age groups, ie, 20 to 30 years and 50 to 60 years.
For the patient, the burden is great, affecting physical, psychological, and occupational well-being. In fact, patients with psoriasis report quality-of-life impairment equal to or worse than that in patients with cancer or heart disease.3,4 Notably, functional disability secondary to psoriatic arthritis has been reported in up to 19% of psoriatic arthritis patients, and this negatively affects quality of life.5
In 2004, the annual direct medical costs of psoriasis in the United States were estimated to exceed $1 billion. Its indirect costs, measured as missed days and loss of productivity at work, are estimated to exceed the direct costs by $15 billion annually.6,7
Linked to cardiovascular and other diseases
Studies in the past 10 years have uncovered a link between psoriasis, metabolic syndrome, and cardiovascular disease.8–13 Specifically, patients with severe psoriasis are at higher risk of myocardial infarction and cardiovascular death than control patients. Interestingly, the risk decreases with age; patients at greatest risk are young men with severe psoriasis.8–10
In a large cohort study in the United Kingdom7 comparing patients with and without psoriasis, the hazard ratio for cardiovascular death in patients with severe psoriasis was 1.57 (95% confidence interval 1.26–1.96). This translated to 3.5 excess deaths per 1,000 patient-years. These patients were also at higher risk of death from malignancies, chronic lower respiratory disease, diabetes, dementia, infection, kidney disease, and unknown causes.
How much of the risk is due to psoriasis itself, its treatments, associated behaviors, or other factors requires more study. However, some evidence points to the dysregulation of the immune system, notably chronic elevation of pro-inflammatory cytokines.
Psoriasis and its comorbid conditions are thought to arise from chronically elevated levels of cytokines such as tumor necrosis factor alpha (TNF-alpha), interleukin 1 beta (IL-1 beta), and IL-17. These cytokines impair insulin signaling, deregulate lipid metabolism, and increase atherosclerotic changes in the coronary, cerebral, and peripheral arteries. In addition, several other diseases that involve the immune system occur more frequently with psoriasis, including Crohn disease, ulcerative colitis, lymphoma, obesity, and type 2 diabetes.1,8,14–18
In view of the prevalence of these comorbid conditions and the risks they pose, primary care physicians should consider screening patients with severe psoriasis for metabolic disorders and cardiovascular risk factors and promptly begin preventive therapies.19 Unfortunately, to date, there are no consensus guidelines as to the appropriate screening tests or secondary cardiovascular preventive measures for patients with severe psoriasis.
A VICIOUS CIRCLE OF INFLAMMATION AND KERATINOCYTE PROLIFERATION
The hallmark of plaque psoriasis is chronic inflammation in the skin, leading to keratinocyte proliferation.
External and internal triggers that have been identified include cutaneous injury (eg, sunburn, drug rash, viral exanthems), infections (eg, streptococcal), hypocalcemia, pregnancy, psychogenic stress, drugs (eg, lithium, interferon, beta-blockers, and antimalarials), alcohol, smoking, and obesity.20–23
As reviewed by Nestle et al,24 the initiation of lesion formation is still poorly understood but is thought to occur when a trigger (physical trauma, bacterial product, cellular stress) causes DNA to be released from keratinocytes. DNA forms a complex with the antimicrobial protein LL-37 and activates plasmacytoid dendritic cells (PDCs) via toll-like receptor 9. Activated PDCs release type I interferons, which in turn activate myeloid dendritic cells. Myeloid dendritic cells release IL-20 locally, which speeds keratinocyte proliferation.
A subset of myeloid dendritic cells leaves the dermis and migrates to local lymph nodes, where they release IL-23 and activate naive T cells. T helper 1 (Th1) and Th17 cells are recruited to the lesions and begin producing numerous cytokines, including interferon gamma, IL-17, and IL-22. This cytokine milieu increases keratinocyte proliferation and causes the keratinocytes to secrete antimicrobial proteins (LL-37, beta defensins), chemokines, and S100 proteins. These soluble factors have three main functions: stimulation of dendritic cells to release more IL-23, recruitment of neutrophils to the epidermis, and activation of dermal fibroblasts.
This cycle of keratinocytes activating dendritic cells, dendritic cells activating T cells, and T cells activating keratinocytes appears to be the main force maintaining the disease.24 It is unclear, however, whether this applies to all forms of psoriasis or only to plaque psoriasis.
Genetic factors discovered
In recent years, genome-wide association studies have identified at least 10 psoriasis-susceptibility loci that involve functioning of the immune system.25 These genes include those of the major histocompatibility complex, cytokines, receptors, and beta-defensins.
Of specific interest, polymorphisms in the IL-12/IL-13 receptor, the p40 subunit of IL-12 and IL-23, and the p19 subunit of IL-23 strongly associate with psoriasis, supporting their critical role in the disease process and providing targets for medical therapy.26
PSORIASIS HAS SEVERAL CLINICAL PHENOTYPES
Psoriasis has several clinical variants, each with a distinct clinical course and response to treatment.27 Moreover, many patients present with more than one variant.
Plaque psoriasis
Plaques can persist for several months to years, even in the same location, and only about 5% of patients report complete remission for up to 5 years.
Inverse psoriasis
Guttate psoriasis
Erythrodermic psoriasis
Approximately 1% to 2.25% of all patients with psoriasis develop this severe form, affecting more than 75% of the body surface area. It presents as generalized erythema, which is the most prominent feature, and it is often associated with superficial desquamation, hair loss, nail dystrophy, and systemic symptoms such as fever, chills, malaise, or high-output cardiac failure. There may be a history of preceding characteristic psoriatic plaques, recent withdrawal of treatment (usually corticosteroids), phototoxicity, or infection.
Conversely, approximately 25% of all patients with erythroderma have underlying psoriasis.28
Pustular psoriasis
There are several forms of pustular psoriasis, including generalized pustular psoriasis, annular pustular psoriasis, impetigo herpetiformis (pustular psoriasis of pregnancy), and palmoplantar pustulosis. However, there is some evidence to suggest that palmoplantar pustulosis may be distinct from psoriasis.29
Several triggers have been identified, including pregnancy, rapid tapering of medications, hypocalcemia, infection, or topical irritants.
Generalized pustular psoriasis, annular pustular psoriasis, and impetigo herpetiformis often present in association with fever and other systemic symptoms and, if left untreated, can result in life-threatening complications including bacterial superinfection, sepsis, dehydration, and, in rare cases, acute respiratory distress secondary to aseptic pneumonitis.30
Placental insufficiency resulting in stillbirth or neonatal death and other fetal abnormalities can occur in severe pustular psoriasis of pregnancy.31
Psoriatic arthritis
Psoriatic arthritis is a seronegative inflammatory spondyloarthropathy that can result in erosive arthritis in up to 57% of cases and functional disability in up to 19%.32 Although rare in the general population, it affects approximately 6% to 10% of psoriasis patients and up to 40% of patients with severe psoriasis.33 In 70% of cases, psoriasis precedes the onset of arthritis by about 10 years, and approximately 10% to 15% of patients simultaneously present with psoriasis and arthritis or develop arthritis before skin involvement.5,34
Patients complain of joint discomfort that is most prominent after periods of prolonged rest. Patterns of involvement are extremely variable but have been reported as an asymmetric oligoarthritis (involving four or fewer joints) or polyarthritis (involving more than four joints) in most patients. A rheumatoid arthritis-like presentation with a symmetric polyarthropathy involving the small and medium-sized joints has also been reported, making it difficult to clinically distinguish this from rheumatoid arthritis.
A distal interphalangeal-predominant pattern is reported in 5% to 10% of patients. Axial disease resembling ankylosing spondylitis occurs only in 5% of patients. Arthritis mutilans, characterized by severe, rapidly progressive joint inflammation, joint destruction, and deformity, occurs rarely. Enthesitis, ie, inflammation at the point of attachment of tendons or ligaments to bone, is present in up to 42% of patients.5,35
Nail disease
Disease severity also varies
Disease severity also differs among patients. An estimated 80% of patients have mild to moderate disease and 20% have moderate to severe disease, which includes disease involving more than 5% of the body surface or involvement of the face, hands, feet, or genitalia.1
The Psoriasis Area and Severity Index (PASI) is an objective measure used in clinical trials. It incorporates the amount of redness, scaling, and induration of each psoriatic lesion over the body surface involved. A 75% improvement in the PASI score (PASI-75) is regarded as clinically significant.37
PSORIASIS IS DIAGNOSED CLINICALLY
In most cases, the diagnosis of psoriasis is made clinically and is straightforward. However, in more difficult cases, biopsy may be needed. In particular:
- The plaques of psoriasis may be confused with squamous cell carcinoma in situ, dermatophyte infection, or cutaneous T-cell lymphoma, especially if they are treatment-resistant.
- Guttate psoriasis may be difficult to distinguish from pityriasis rosea.
- Erythrodermic psoriasis must be distinguished from other causes of erythroderma, including Sézary syndrome, pityriasis rubra pilaris, and drug reactions.
- Intertrigo, candidiasis, extramammary Paget disease, squamous cell carcinoma, and contact dermatitis all may mimic inverse psoriasis.
- Palmoplantar pustulosis may be difficult to differentiate from dyshidrotic eczema.
- Generalized pustular psoriasis should be distinguished from a pustular drug eruption (acute generalized exanthematous pustular drug eruption or acute generalized exanthematous pustulosis), impetigo, candidiasis, or an autoimmune blistering disorder such as pemphigus.
TREATMENT OF LIMITED DISEASE
Topical corticosteroids
A topical corticosteroid, either by itself or combined with a steroid-sparing agent, is the first-line therapy for patients with limited disease. The potency required for treatment should be based on the extent of disease and on the location, the choice of vehicle, and the patient’s preference and age.
Several double-blind studies have assessed the efficacy of various topical corticosteroids in treating psoriasis. In general, super-potent (class I) and potent (class II) topical corticosteroids are more efficacious than mild or moderate corticosteroids.38 Class I and class II steroids include agents such as clobetasol propionate 0.05% (Temovate), betamethasone dipropionate 0.05% (Diprolene), fluocinonide 0.05% (Lidex), and desoximetasone 0.25% (Topicort).
Use of class I steroids should be limited to an initial treatment course of twice-daily application for 2 to 4 weeks in an effort to avoid some of the local toxicities such as skin atrophy, telangiectasia, and striae. Decreasing class I topical steroid use to 1 to 2 times per week with the gradual introduction of a steroid-sparing agent following the initial 2 to 4 weeks of treatment is advised.
Steroid-sparing agents
Steroid-sparing agents include vitamin D analogues, retinoids, and tacrolimus ointment (Protopic).
Vitamin D analogues and retinoids are thought to decrease keratinocyte proliferation and enhance keratinocyte differentiation.39 The vitamin D analogues are also considered first-line topical agents and include calcipotriol (Dovonex), calcipotriene (Dovonex), and calcitriol (Vectical). To prevent hypercalcemia, use of more than 100 g of vitamin D analogues per week should be avoided.39
Treatment of inverse psoriasis and scalp psoriasis may be challenging
The areas affected in inverse psoriasis, such as the genitalia and axillae, are more prone to side effects when potent topical steroids are used because of increased absorption and occlusion in these areas. Agents that minimize irritation and toxicity in sensitive areas, such as topical tacrolimus, less-potent topical steroids, or calcitriol, can be used.39
For scalp psoriasis, alternative vehicles such as shampoos, gels, solutions, oils, sprays, and foams have improved patient compliance and efficacy of treatment.40
PHOTOTHERAPY FOR SEVERE DISEASE
Narrow-band ultraviolet B
Narrow-band ultraviolet B, ie, light confined to wavelengths of 311 to 313 nm, is a first-line treatment for moderate to severe psoriasis, either as monotherapy or in combination with other treatments. It is an especially attractive option in patients who are on medications or who have comorbidities that may preclude treatment with other systemic agents.
The mechanism of action may be via immunosuppressive effects on Langerhans cells, alteration of cytokines and adhesion molecules that lead to an increase in Th2 cells, and induction of apoptosis of T lymphocytes. Additionally, ultraviolet light affects the proliferation and differentiation of keratinocytes.41
Dosing is based on skin type, and treatment usually involves two or three visits per week for a total of 15 to 20 treatments, with additional therapy for maintenance. Adding acitretin (Soriatane), with close monitoring of aspartate aminotransferase and alanine aminotransferase levels and the patient’s lipid panel, can be considered in treatment-resistant cases.42
Psoralen combined with ultraviolet A
Psoralen combined with ultraviolet A is another option. It can be considered if narrow-band ultraviolet B treatment fails. It is also useful for dark-skinned patients and those with thicker plaques because ultraviolet A penetrates deeper than ultraviolet B. Oral or topical treatment with psoralen is followed by ultraviolet A treatment.
The duration of remission is much longer with psoralen plus ultraviolet A than with narrow-band ultraviolet B. However, this treatment caries a significant risk of cutaneous squamous cell carcinoma and melanoma, especially in light-skinned people and those who receive high doses of ultraviolet A (200 or more treatments) or cyclosporine.40,41,43–46 Long-term effects include photoaging, lentigines, and telangiectasias. As a consequence of these well-established side effects, this treatment is used less frequently.
Cautions with phototherapy
Careful screening and caution should be used in patients who have:
- Fair skin that tends to burn easily
- A history of arsenic intake or treatment with ionizing radiation
- A history of use of photosensitizing medications (fluoroquinolone antibiotics, doxycycline, hydrochlorothiazide)
- A history of melanoma or atypical nevi
- Multiple risk factors for melanoma
- A history of nonmelanoma skin cancer
- Immunosuppression due to organ transplantation.
ORAL THERAPIES FOR SEVERE PSORIASIS
Patients who have severe psoriasis—ie, affecting more than 5% of the body surface or debilitating disease affecting the palms, soles, or genitalia—are best managed with systemic medications, especially if they do not have access to phototherapy.20
Methotrexate
In 1972, the US Food and Drug Administration (FDA) approved methotrexate for treating severe psoriasis.42 In studies of methotrexate at doses of 15 to 20 mg weekly, 36% to 68% of patients with severe plaque psoriasis achieved a PASI-75 score.40,42,47
Dosages of methotrexate for treating severe psoriasis range from 7.5 to 25 mg once a week. Patients should also receive a folate supplement of 1 to 5 mg every day except the day they take methotrexate. The folate is to protect against gastrointestinal side effects, bone marrow suppression, and hepatic toxicity associated with methotrexate.
Other side effects of methotrexate include pulmonary fibrosis and stomatitis. Pregnancy, nursing, alcoholism, chronic liver disease, immunodeficiency syndromes, bone-marrow hypoplasia, leukopenia, thrombocytopenia, anemia, and hypersensitivity to methotrexate are all contraindications to methotrexate use.
The National Psoriasis Foundation, in its 2009 guidelines for the use of methotrexate in treating psoriasis,48 recommends obtaining a complete blood cell count with platelets, blood urea nitrogen, creatinine, and liver function tests at baseline and at 1- to 3-month intervals thereafter.
Liver biopsies were previously recommended for patients receiving methotrexate long-term when the cumulative dose of therapy reached 1.5 g. However, given the invasive nature of the liver biopsy procedure and the low incidence of methotrexate-induced hepatotoxicity, this recommendation has been revised.
For patients with no significant risk factors for hepatic toxicity (eg, obesity, diabetes, hyperlipidemia, hepatitis, or history of or current alcohol consumption) and normal liver function tests, liver biopsy should be considered when a cumulative methotrexate dose of 3.5 to 4.0 g is reached. Alternatively, one may choose to continue to monitor the patient without liver biopsy or to switch to another medication, if possible.42,48
Patients at high risk should be monitored more carefully, and liver biopsy should be considered soon after starting methotrexate and repeated after every 1.0 to 1.5 g.48
No reliable noninvasive measures to evaluate for liver fibrosis are routinely available in the United States. Serial measurements of serum type III procollagen aminopeptide have been reported to correlate with the risk of developing liver fibrosis; however, this test is readily available only in Europe.49
Cyclosporine
Cyclosporine (Gengraf, Neoral, Sandimmune) is very effective for treating psoriasis, especially erythrodermic psoriasis. It is often used only short-term or as a bridge to other maintenance therapies because it has a rapid onset and because long-term therapy (3 to 5 years) is associated with a risk of glomerulosclerosis.50
Cyclosporine works by decreasing T-cell activation by binding cyclophilin, which leads to inhibition of transcription of calcineurin and nuclear factor of activated T cells.51 Given at doses of 2.5 to 5 mg/kg/day, cyclosporine has been shown to result in rapid improvement in up to 80% to 90% of psoriatic patients.52,53
The initial recommended dose of cyclosporine is usually 2.5 to 3 mg/kg/day in two divided doses, which is maintained for 4 weeks and then increased by 0.5 mg/kg/day until the disease is stable.42
Nephrotoxicity and hypertension are cyclosporine’s most serious side effects. Blood urea nitrogen, creatinine, and blood pressure should be monitored at baseline and then twice a month for the first 3 months and once monthly thereafter. Liver function tests, complete blood cell count, lipid profile, magnesium, uric acid, and potassium should also be checked every month.
Cyclosporine also increases the risk of cutaneous squamous cell carcinoma, especially in patients who have received psoralen plus ultraviolet A treatment.42
Patients with hypersensitivity to cyclosporine, a history of chronic infection (eg, tuberculosis, hepatitis B, hepatitis C), renal insufficiency, or a history of systemic malignancy should not receive cyclosporine.
Acitretin
Acitretin, an oral retinoid, has been used for several years to treat psoriasis. Its onset is slow, typically ranging from 3 to 6 months, and its effects are dose-dependent. It is most effective as a maintenance therapy, usually after the disease has been stabilized by agents such as cyclosporine, or in combination with other treatments such as phototherapy.42 Acitretin has been shown to be effective in patients with pustular psoriasis.54
Acitretin does not alter the immune system and has not been shown to have significant cumulative toxicities. Serum triglycerides are monitored closely, since acitretin can lead to hypertriglyceridemia.
All retinoids, including acitretin, are in pregnancy category X and should therefore be avoided during pregnancy. Although its half-life is only 49 hours, acitretin may be transformed to etretinate either spontaneously or as a result of alcohol ingestion. Etretinate has a half-life of 168 days and can take up to 3 years to be eliminated from the body. Therefore, acitretin is contraindicated in women who plan to become pregnant or who do not agree to use adequate contraception for 3 years after the drug is discontinued.42
Biologic agents
Advances in our understanding of the pathogenesis of psoriasis have resulted in more specific, targeted therapy.
Alefacept (Amevive) is a human Fc IgG1 receptor fused to the alpha subunit of LFA3. It binds to CD2, blocks costimulatory signaling, and induces apoptosis in activated memory T cells.
Alefacept was the first biologic agent approved by the FDA for the treatment of psoriasis and one of the few biologic agents to induce long-term remission.55 However, its use has declined because few patients achieved significant clearance of their psoriasis and its onset of action was much slower than that of other medications.56
The currently approved biologic therapies commonly used for moderate to severe psoriasis include the TNF-alpha inhibitors and ustekinumab (Stelara).
The TNF-alpha inhibitors include infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira). They are generally well tolerated and highly effective. However, TNF-alpha inhibitors and other biologic agents are contraindicated in patients with serious infection, a personal history or a family history in a first-degree relative of demyelinating disease, or class III or IV congestive heart failure. Patients should be screened for active infection, including tuberculosis and hepatitis B, since reactivation has been reported following initiation of TNF-alpha inhibitors.1
Adalimumab is a human monoclonal antibody against TNF-alpha. It binds to soluble and membrane-bound TNF-alpha and prevents it from binding to p55 and p75 cell-surface TNF receptors.
The dosing schedule for adalimumab is 80 mg subcutaneously for the first week, followed by 40 mg subcutaneously the next week, and then 40 mg subcutaneously every 2 weeks thereafter.1
Etanercept is a recombinant human TNF-alpha receptor (p75) protein fused with the Fc portion of IgG1, which binds to soluble TNF-alpha.57 Dosing for etanercept is 50 mg subcutaneously twice weekly for the first 12 weeks, followed by 50 mg weekly thereafter.
Infliximab is a chimeric antibody composed of a human IgG1 constant region fused to a mouse variable region that binds to both soluble and membrane-bound TNF-alpha.58 Infliximab is given as an infusion at a dose of 5 mg/kg over 2 to 3 hours at weeks 0, 2, and 6, and then every 8 weeks thereafter.
Efficacy of TNF inhibitors. There are no specific guidelines for the sequence of initiation of TNF inhibitors because no studies have directly compared the efficacy of these medications. However, response to infliximab is relatively rapid compared with adalimumab and etanercept.
In a phase III clinical trial,59 as many as 80% of patients achieved PASI-75 clearance of their psoriasis after three doses of infliximab. Interestingly, only 61% of patients maintained PASI-75 clearance by week 50. This loss of efficacy of infliximab is also reported with other TNF-alpha inhibitors and is thought to be secondary to the development of antibodies to the drugs. For infliximab, this loss of efficacy is less when infliximab is given continuously rather than on an as-needed basis. Simultaneous treatment with methotrexate is also thought to decrease the development of antibodies to infliximab.60
Ustekinumab is an monoclonal antibody directed against the common p40 subunit of IL-12 and IL-23, which have been shown to be at increased levels in psoriatic lesions and important for the pathogenesis of psoriasis.
Between 66% and 76% of patients treated with ustekinumab achieved significant clearance of their disease after 12 weeks of treatment in two large phase III multicenter, randomized, double-blind, placebo-controlled trials.61,62
Dosing of ustekinumab is weight-based. For those weighing less than 100 kg, ustekinumab is given at 45 mg subcutaneously at baseline, at 4 weeks, and every 12 weeks thereafter. The same dosing schedule is used for those weighing more than 100 kg, but the dose is increased to 90 mg.
Guidelines for monitoring patients while on ustekinumab are similar to those for other biologic agents. Information on long-term toxicities is still being collected. However, injection-site reactions, serious infections, malignancies, and a single case of reversible posterior leukoencephalopathy have been reported.20
While biologic agents are significantly more expensive than the conventional therapies discussed above and insurance coverage for these agents varies, they have demonstrated superior efficacy and may be indicated for patients with recalcitrant moderate to severe psoriasis for whom multiple types of treatment have failed.
FOR PSORIATIC ARTHRITIS: SYSTEMIC MEDICATIONS
For patients with known or questionable psoriatic arthritis, evaluation by a rheumatologist is highly recommended.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are usually first-line in the treatment of mild psoriatic arthritis. If after 2 to 3 months of therapy with NSAIDs no benefit is achieved, treatment with methotrexate as monotherapy is a practical consideration because of its low cost. However, methotrexate as a monotherapy has not been shown to prevent radiologic progression of disease.5,32
The TNF-alpha inhibitors have been shown to have similar efficacy when compared among each other in the treatment of psoriatic arthritis.32,63 Based on radiologic evidence, ustekinumab has not shown to be as efficacious as the TNF-alpha inhibitors for treating psoriatic arthritis. Therefore, TNF inhibitors should be considered first-line in the treatment of psoriatic arthritis.21,64
Few studies have been done on the efficacy or sequence of therapies that should be used in the treatment of psoriatic arthritis. The American Academy of Dermatology’s Psoriasis Guidelines of Care recommend adding a TNF-alpha inhibitor or switching to a TNF-alpha inhibitor if no significant improvement is achieved after 12 to 16 weeks of treatment with oral methotrexate.20
FOR ERYTHRODERMIC PSORIASIS: MEDICATIONS THAT ACT PROMPTLY
The care of erythrodermic psoriatic patients is distinct from that of other psoriatic patients because of their associated systemic symptoms. Care should be taken to rule out sepsis, as this is a reported trigger of erythrodermic psoriasis.28
Systemic medications with a quick onset, such as oral cyclosporine, are recommended. Infliximab has also been reported to be beneficial because of its rapid onset.28
TREATMENT BASED ON THE TYPE AND THE SEVERITY OF PSORIASIS
The treatment of psoriasis can be as complex as the disease it itself and should be based on the type and the severity of psoriasis. Recognition of the various manifestations of psoriasis is important for effective treatment. However, in patients with moderate to severe psoriasis, atypical presentations, or recalcitrant disease, referral to a specialist is recommended.
Much has changed in our understanding of psoriasis over the past decade, which is having a major effect on its treatment.
Although topical corticosteroids and phototherapy remain mainstays of treatment, a variety of biologic agents have given new hope to those with the most severe forms of the disease. We are also beginning to understand that patients with psoriasis are at greater risk of cardiovascular disease, though the exact nature of that risk and how we should respond remains unclear. Finally, genome-wide association studies are just beginning to unravel the genetic basis of psoriasis.
In this paper, we review the epidemiology and impact of psoriasis, current views of its pathogenesis, its varied clinical forms, and its treatment.
PSORIASIS IMPOSES A GREAT BURDEN
Psoriasis is common, with a reported prevalence ranging from approximately 2%1 to 4.7%.2 It can manifest at any age, but it is most common in two age groups, ie, 20 to 30 years and 50 to 60 years.
For the patient, the burden is great, affecting physical, psychological, and occupational well-being. In fact, patients with psoriasis report quality-of-life impairment equal to or worse than that in patients with cancer or heart disease.3,4 Notably, functional disability secondary to psoriatic arthritis has been reported in up to 19% of psoriatic arthritis patients, and this negatively affects quality of life.5
In 2004, the annual direct medical costs of psoriasis in the United States were estimated to exceed $1 billion. Its indirect costs, measured as missed days and loss of productivity at work, are estimated to exceed the direct costs by $15 billion annually.6,7
Linked to cardiovascular and other diseases
Studies in the past 10 years have uncovered a link between psoriasis, metabolic syndrome, and cardiovascular disease.8–13 Specifically, patients with severe psoriasis are at higher risk of myocardial infarction and cardiovascular death than control patients. Interestingly, the risk decreases with age; patients at greatest risk are young men with severe psoriasis.8–10
In a large cohort study in the United Kingdom7 comparing patients with and without psoriasis, the hazard ratio for cardiovascular death in patients with severe psoriasis was 1.57 (95% confidence interval 1.26–1.96). This translated to 3.5 excess deaths per 1,000 patient-years. These patients were also at higher risk of death from malignancies, chronic lower respiratory disease, diabetes, dementia, infection, kidney disease, and unknown causes.
How much of the risk is due to psoriasis itself, its treatments, associated behaviors, or other factors requires more study. However, some evidence points to the dysregulation of the immune system, notably chronic elevation of pro-inflammatory cytokines.
Psoriasis and its comorbid conditions are thought to arise from chronically elevated levels of cytokines such as tumor necrosis factor alpha (TNF-alpha), interleukin 1 beta (IL-1 beta), and IL-17. These cytokines impair insulin signaling, deregulate lipid metabolism, and increase atherosclerotic changes in the coronary, cerebral, and peripheral arteries. In addition, several other diseases that involve the immune system occur more frequently with psoriasis, including Crohn disease, ulcerative colitis, lymphoma, obesity, and type 2 diabetes.1,8,14–18
In view of the prevalence of these comorbid conditions and the risks they pose, primary care physicians should consider screening patients with severe psoriasis for metabolic disorders and cardiovascular risk factors and promptly begin preventive therapies.19 Unfortunately, to date, there are no consensus guidelines as to the appropriate screening tests or secondary cardiovascular preventive measures for patients with severe psoriasis.
A VICIOUS CIRCLE OF INFLAMMATION AND KERATINOCYTE PROLIFERATION
The hallmark of plaque psoriasis is chronic inflammation in the skin, leading to keratinocyte proliferation.
External and internal triggers that have been identified include cutaneous injury (eg, sunburn, drug rash, viral exanthems), infections (eg, streptococcal), hypocalcemia, pregnancy, psychogenic stress, drugs (eg, lithium, interferon, beta-blockers, and antimalarials), alcohol, smoking, and obesity.20–23
As reviewed by Nestle et al,24 the initiation of lesion formation is still poorly understood but is thought to occur when a trigger (physical trauma, bacterial product, cellular stress) causes DNA to be released from keratinocytes. DNA forms a complex with the antimicrobial protein LL-37 and activates plasmacytoid dendritic cells (PDCs) via toll-like receptor 9. Activated PDCs release type I interferons, which in turn activate myeloid dendritic cells. Myeloid dendritic cells release IL-20 locally, which speeds keratinocyte proliferation.
A subset of myeloid dendritic cells leaves the dermis and migrates to local lymph nodes, where they release IL-23 and activate naive T cells. T helper 1 (Th1) and Th17 cells are recruited to the lesions and begin producing numerous cytokines, including interferon gamma, IL-17, and IL-22. This cytokine milieu increases keratinocyte proliferation and causes the keratinocytes to secrete antimicrobial proteins (LL-37, beta defensins), chemokines, and S100 proteins. These soluble factors have three main functions: stimulation of dendritic cells to release more IL-23, recruitment of neutrophils to the epidermis, and activation of dermal fibroblasts.
This cycle of keratinocytes activating dendritic cells, dendritic cells activating T cells, and T cells activating keratinocytes appears to be the main force maintaining the disease.24 It is unclear, however, whether this applies to all forms of psoriasis or only to plaque psoriasis.
Genetic factors discovered
In recent years, genome-wide association studies have identified at least 10 psoriasis-susceptibility loci that involve functioning of the immune system.25 These genes include those of the major histocompatibility complex, cytokines, receptors, and beta-defensins.
Of specific interest, polymorphisms in the IL-12/IL-13 receptor, the p40 subunit of IL-12 and IL-23, and the p19 subunit of IL-23 strongly associate with psoriasis, supporting their critical role in the disease process and providing targets for medical therapy.26
PSORIASIS HAS SEVERAL CLINICAL PHENOTYPES
Psoriasis has several clinical variants, each with a distinct clinical course and response to treatment.27 Moreover, many patients present with more than one variant.
Plaque psoriasis
Plaques can persist for several months to years, even in the same location, and only about 5% of patients report complete remission for up to 5 years.
Inverse psoriasis
Guttate psoriasis
Erythrodermic psoriasis
Approximately 1% to 2.25% of all patients with psoriasis develop this severe form, affecting more than 75% of the body surface area. It presents as generalized erythema, which is the most prominent feature, and it is often associated with superficial desquamation, hair loss, nail dystrophy, and systemic symptoms such as fever, chills, malaise, or high-output cardiac failure. There may be a history of preceding characteristic psoriatic plaques, recent withdrawal of treatment (usually corticosteroids), phototoxicity, or infection.
Conversely, approximately 25% of all patients with erythroderma have underlying psoriasis.28
Pustular psoriasis
There are several forms of pustular psoriasis, including generalized pustular psoriasis, annular pustular psoriasis, impetigo herpetiformis (pustular psoriasis of pregnancy), and palmoplantar pustulosis. However, there is some evidence to suggest that palmoplantar pustulosis may be distinct from psoriasis.29
Several triggers have been identified, including pregnancy, rapid tapering of medications, hypocalcemia, infection, or topical irritants.
Generalized pustular psoriasis, annular pustular psoriasis, and impetigo herpetiformis often present in association with fever and other systemic symptoms and, if left untreated, can result in life-threatening complications including bacterial superinfection, sepsis, dehydration, and, in rare cases, acute respiratory distress secondary to aseptic pneumonitis.30
Placental insufficiency resulting in stillbirth or neonatal death and other fetal abnormalities can occur in severe pustular psoriasis of pregnancy.31
Psoriatic arthritis
Psoriatic arthritis is a seronegative inflammatory spondyloarthropathy that can result in erosive arthritis in up to 57% of cases and functional disability in up to 19%.32 Although rare in the general population, it affects approximately 6% to 10% of psoriasis patients and up to 40% of patients with severe psoriasis.33 In 70% of cases, psoriasis precedes the onset of arthritis by about 10 years, and approximately 10% to 15% of patients simultaneously present with psoriasis and arthritis or develop arthritis before skin involvement.5,34
Patients complain of joint discomfort that is most prominent after periods of prolonged rest. Patterns of involvement are extremely variable but have been reported as an asymmetric oligoarthritis (involving four or fewer joints) or polyarthritis (involving more than four joints) in most patients. A rheumatoid arthritis-like presentation with a symmetric polyarthropathy involving the small and medium-sized joints has also been reported, making it difficult to clinically distinguish this from rheumatoid arthritis.
A distal interphalangeal-predominant pattern is reported in 5% to 10% of patients. Axial disease resembling ankylosing spondylitis occurs only in 5% of patients. Arthritis mutilans, characterized by severe, rapidly progressive joint inflammation, joint destruction, and deformity, occurs rarely. Enthesitis, ie, inflammation at the point of attachment of tendons or ligaments to bone, is present in up to 42% of patients.5,35
Nail disease
Disease severity also varies
Disease severity also differs among patients. An estimated 80% of patients have mild to moderate disease and 20% have moderate to severe disease, which includes disease involving more than 5% of the body surface or involvement of the face, hands, feet, or genitalia.1
The Psoriasis Area and Severity Index (PASI) is an objective measure used in clinical trials. It incorporates the amount of redness, scaling, and induration of each psoriatic lesion over the body surface involved. A 75% improvement in the PASI score (PASI-75) is regarded as clinically significant.37
PSORIASIS IS DIAGNOSED CLINICALLY
In most cases, the diagnosis of psoriasis is made clinically and is straightforward. However, in more difficult cases, biopsy may be needed. In particular:
- The plaques of psoriasis may be confused with squamous cell carcinoma in situ, dermatophyte infection, or cutaneous T-cell lymphoma, especially if they are treatment-resistant.
- Guttate psoriasis may be difficult to distinguish from pityriasis rosea.
- Erythrodermic psoriasis must be distinguished from other causes of erythroderma, including Sézary syndrome, pityriasis rubra pilaris, and drug reactions.
- Intertrigo, candidiasis, extramammary Paget disease, squamous cell carcinoma, and contact dermatitis all may mimic inverse psoriasis.
- Palmoplantar pustulosis may be difficult to differentiate from dyshidrotic eczema.
- Generalized pustular psoriasis should be distinguished from a pustular drug eruption (acute generalized exanthematous pustular drug eruption or acute generalized exanthematous pustulosis), impetigo, candidiasis, or an autoimmune blistering disorder such as pemphigus.
TREATMENT OF LIMITED DISEASE
Topical corticosteroids
A topical corticosteroid, either by itself or combined with a steroid-sparing agent, is the first-line therapy for patients with limited disease. The potency required for treatment should be based on the extent of disease and on the location, the choice of vehicle, and the patient’s preference and age.
Several double-blind studies have assessed the efficacy of various topical corticosteroids in treating psoriasis. In general, super-potent (class I) and potent (class II) topical corticosteroids are more efficacious than mild or moderate corticosteroids.38 Class I and class II steroids include agents such as clobetasol propionate 0.05% (Temovate), betamethasone dipropionate 0.05% (Diprolene), fluocinonide 0.05% (Lidex), and desoximetasone 0.25% (Topicort).
Use of class I steroids should be limited to an initial treatment course of twice-daily application for 2 to 4 weeks in an effort to avoid some of the local toxicities such as skin atrophy, telangiectasia, and striae. Decreasing class I topical steroid use to 1 to 2 times per week with the gradual introduction of a steroid-sparing agent following the initial 2 to 4 weeks of treatment is advised.
Steroid-sparing agents
Steroid-sparing agents include vitamin D analogues, retinoids, and tacrolimus ointment (Protopic).
Vitamin D analogues and retinoids are thought to decrease keratinocyte proliferation and enhance keratinocyte differentiation.39 The vitamin D analogues are also considered first-line topical agents and include calcipotriol (Dovonex), calcipotriene (Dovonex), and calcitriol (Vectical). To prevent hypercalcemia, use of more than 100 g of vitamin D analogues per week should be avoided.39
Treatment of inverse psoriasis and scalp psoriasis may be challenging
The areas affected in inverse psoriasis, such as the genitalia and axillae, are more prone to side effects when potent topical steroids are used because of increased absorption and occlusion in these areas. Agents that minimize irritation and toxicity in sensitive areas, such as topical tacrolimus, less-potent topical steroids, or calcitriol, can be used.39
For scalp psoriasis, alternative vehicles such as shampoos, gels, solutions, oils, sprays, and foams have improved patient compliance and efficacy of treatment.40
PHOTOTHERAPY FOR SEVERE DISEASE
Narrow-band ultraviolet B
Narrow-band ultraviolet B, ie, light confined to wavelengths of 311 to 313 nm, is a first-line treatment for moderate to severe psoriasis, either as monotherapy or in combination with other treatments. It is an especially attractive option in patients who are on medications or who have comorbidities that may preclude treatment with other systemic agents.
The mechanism of action may be via immunosuppressive effects on Langerhans cells, alteration of cytokines and adhesion molecules that lead to an increase in Th2 cells, and induction of apoptosis of T lymphocytes. Additionally, ultraviolet light affects the proliferation and differentiation of keratinocytes.41
Dosing is based on skin type, and treatment usually involves two or three visits per week for a total of 15 to 20 treatments, with additional therapy for maintenance. Adding acitretin (Soriatane), with close monitoring of aspartate aminotransferase and alanine aminotransferase levels and the patient’s lipid panel, can be considered in treatment-resistant cases.42
Psoralen combined with ultraviolet A
Psoralen combined with ultraviolet A is another option. It can be considered if narrow-band ultraviolet B treatment fails. It is also useful for dark-skinned patients and those with thicker plaques because ultraviolet A penetrates deeper than ultraviolet B. Oral or topical treatment with psoralen is followed by ultraviolet A treatment.
The duration of remission is much longer with psoralen plus ultraviolet A than with narrow-band ultraviolet B. However, this treatment caries a significant risk of cutaneous squamous cell carcinoma and melanoma, especially in light-skinned people and those who receive high doses of ultraviolet A (200 or more treatments) or cyclosporine.40,41,43–46 Long-term effects include photoaging, lentigines, and telangiectasias. As a consequence of these well-established side effects, this treatment is used less frequently.
Cautions with phototherapy
Careful screening and caution should be used in patients who have:
- Fair skin that tends to burn easily
- A history of arsenic intake or treatment with ionizing radiation
- A history of use of photosensitizing medications (fluoroquinolone antibiotics, doxycycline, hydrochlorothiazide)
- A history of melanoma or atypical nevi
- Multiple risk factors for melanoma
- A history of nonmelanoma skin cancer
- Immunosuppression due to organ transplantation.
ORAL THERAPIES FOR SEVERE PSORIASIS
Patients who have severe psoriasis—ie, affecting more than 5% of the body surface or debilitating disease affecting the palms, soles, or genitalia—are best managed with systemic medications, especially if they do not have access to phototherapy.20
Methotrexate
In 1972, the US Food and Drug Administration (FDA) approved methotrexate for treating severe psoriasis.42 In studies of methotrexate at doses of 15 to 20 mg weekly, 36% to 68% of patients with severe plaque psoriasis achieved a PASI-75 score.40,42,47
Dosages of methotrexate for treating severe psoriasis range from 7.5 to 25 mg once a week. Patients should also receive a folate supplement of 1 to 5 mg every day except the day they take methotrexate. The folate is to protect against gastrointestinal side effects, bone marrow suppression, and hepatic toxicity associated with methotrexate.
Other side effects of methotrexate include pulmonary fibrosis and stomatitis. Pregnancy, nursing, alcoholism, chronic liver disease, immunodeficiency syndromes, bone-marrow hypoplasia, leukopenia, thrombocytopenia, anemia, and hypersensitivity to methotrexate are all contraindications to methotrexate use.
The National Psoriasis Foundation, in its 2009 guidelines for the use of methotrexate in treating psoriasis,48 recommends obtaining a complete blood cell count with platelets, blood urea nitrogen, creatinine, and liver function tests at baseline and at 1- to 3-month intervals thereafter.
Liver biopsies were previously recommended for patients receiving methotrexate long-term when the cumulative dose of therapy reached 1.5 g. However, given the invasive nature of the liver biopsy procedure and the low incidence of methotrexate-induced hepatotoxicity, this recommendation has been revised.
For patients with no significant risk factors for hepatic toxicity (eg, obesity, diabetes, hyperlipidemia, hepatitis, or history of or current alcohol consumption) and normal liver function tests, liver biopsy should be considered when a cumulative methotrexate dose of 3.5 to 4.0 g is reached. Alternatively, one may choose to continue to monitor the patient without liver biopsy or to switch to another medication, if possible.42,48
Patients at high risk should be monitored more carefully, and liver biopsy should be considered soon after starting methotrexate and repeated after every 1.0 to 1.5 g.48
No reliable noninvasive measures to evaluate for liver fibrosis are routinely available in the United States. Serial measurements of serum type III procollagen aminopeptide have been reported to correlate with the risk of developing liver fibrosis; however, this test is readily available only in Europe.49
Cyclosporine
Cyclosporine (Gengraf, Neoral, Sandimmune) is very effective for treating psoriasis, especially erythrodermic psoriasis. It is often used only short-term or as a bridge to other maintenance therapies because it has a rapid onset and because long-term therapy (3 to 5 years) is associated with a risk of glomerulosclerosis.50
Cyclosporine works by decreasing T-cell activation by binding cyclophilin, which leads to inhibition of transcription of calcineurin and nuclear factor of activated T cells.51 Given at doses of 2.5 to 5 mg/kg/day, cyclosporine has been shown to result in rapid improvement in up to 80% to 90% of psoriatic patients.52,53
The initial recommended dose of cyclosporine is usually 2.5 to 3 mg/kg/day in two divided doses, which is maintained for 4 weeks and then increased by 0.5 mg/kg/day until the disease is stable.42
Nephrotoxicity and hypertension are cyclosporine’s most serious side effects. Blood urea nitrogen, creatinine, and blood pressure should be monitored at baseline and then twice a month for the first 3 months and once monthly thereafter. Liver function tests, complete blood cell count, lipid profile, magnesium, uric acid, and potassium should also be checked every month.
Cyclosporine also increases the risk of cutaneous squamous cell carcinoma, especially in patients who have received psoralen plus ultraviolet A treatment.42
Patients with hypersensitivity to cyclosporine, a history of chronic infection (eg, tuberculosis, hepatitis B, hepatitis C), renal insufficiency, or a history of systemic malignancy should not receive cyclosporine.
Acitretin
Acitretin, an oral retinoid, has been used for several years to treat psoriasis. Its onset is slow, typically ranging from 3 to 6 months, and its effects are dose-dependent. It is most effective as a maintenance therapy, usually after the disease has been stabilized by agents such as cyclosporine, or in combination with other treatments such as phototherapy.42 Acitretin has been shown to be effective in patients with pustular psoriasis.54
Acitretin does not alter the immune system and has not been shown to have significant cumulative toxicities. Serum triglycerides are monitored closely, since acitretin can lead to hypertriglyceridemia.
All retinoids, including acitretin, are in pregnancy category X and should therefore be avoided during pregnancy. Although its half-life is only 49 hours, acitretin may be transformed to etretinate either spontaneously or as a result of alcohol ingestion. Etretinate has a half-life of 168 days and can take up to 3 years to be eliminated from the body. Therefore, acitretin is contraindicated in women who plan to become pregnant or who do not agree to use adequate contraception for 3 years after the drug is discontinued.42
Biologic agents
Advances in our understanding of the pathogenesis of psoriasis have resulted in more specific, targeted therapy.
Alefacept (Amevive) is a human Fc IgG1 receptor fused to the alpha subunit of LFA3. It binds to CD2, blocks costimulatory signaling, and induces apoptosis in activated memory T cells.
Alefacept was the first biologic agent approved by the FDA for the treatment of psoriasis and one of the few biologic agents to induce long-term remission.55 However, its use has declined because few patients achieved significant clearance of their psoriasis and its onset of action was much slower than that of other medications.56
The currently approved biologic therapies commonly used for moderate to severe psoriasis include the TNF-alpha inhibitors and ustekinumab (Stelara).
The TNF-alpha inhibitors include infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira). They are generally well tolerated and highly effective. However, TNF-alpha inhibitors and other biologic agents are contraindicated in patients with serious infection, a personal history or a family history in a first-degree relative of demyelinating disease, or class III or IV congestive heart failure. Patients should be screened for active infection, including tuberculosis and hepatitis B, since reactivation has been reported following initiation of TNF-alpha inhibitors.1
Adalimumab is a human monoclonal antibody against TNF-alpha. It binds to soluble and membrane-bound TNF-alpha and prevents it from binding to p55 and p75 cell-surface TNF receptors.
The dosing schedule for adalimumab is 80 mg subcutaneously for the first week, followed by 40 mg subcutaneously the next week, and then 40 mg subcutaneously every 2 weeks thereafter.1
Etanercept is a recombinant human TNF-alpha receptor (p75) protein fused with the Fc portion of IgG1, which binds to soluble TNF-alpha.57 Dosing for etanercept is 50 mg subcutaneously twice weekly for the first 12 weeks, followed by 50 mg weekly thereafter.
Infliximab is a chimeric antibody composed of a human IgG1 constant region fused to a mouse variable region that binds to both soluble and membrane-bound TNF-alpha.58 Infliximab is given as an infusion at a dose of 5 mg/kg over 2 to 3 hours at weeks 0, 2, and 6, and then every 8 weeks thereafter.
Efficacy of TNF inhibitors. There are no specific guidelines for the sequence of initiation of TNF inhibitors because no studies have directly compared the efficacy of these medications. However, response to infliximab is relatively rapid compared with adalimumab and etanercept.
In a phase III clinical trial,59 as many as 80% of patients achieved PASI-75 clearance of their psoriasis after three doses of infliximab. Interestingly, only 61% of patients maintained PASI-75 clearance by week 50. This loss of efficacy of infliximab is also reported with other TNF-alpha inhibitors and is thought to be secondary to the development of antibodies to the drugs. For infliximab, this loss of efficacy is less when infliximab is given continuously rather than on an as-needed basis. Simultaneous treatment with methotrexate is also thought to decrease the development of antibodies to infliximab.60
Ustekinumab is an monoclonal antibody directed against the common p40 subunit of IL-12 and IL-23, which have been shown to be at increased levels in psoriatic lesions and important for the pathogenesis of psoriasis.
Between 66% and 76% of patients treated with ustekinumab achieved significant clearance of their disease after 12 weeks of treatment in two large phase III multicenter, randomized, double-blind, placebo-controlled trials.61,62
Dosing of ustekinumab is weight-based. For those weighing less than 100 kg, ustekinumab is given at 45 mg subcutaneously at baseline, at 4 weeks, and every 12 weeks thereafter. The same dosing schedule is used for those weighing more than 100 kg, but the dose is increased to 90 mg.
Guidelines for monitoring patients while on ustekinumab are similar to those for other biologic agents. Information on long-term toxicities is still being collected. However, injection-site reactions, serious infections, malignancies, and a single case of reversible posterior leukoencephalopathy have been reported.20
While biologic agents are significantly more expensive than the conventional therapies discussed above and insurance coverage for these agents varies, they have demonstrated superior efficacy and may be indicated for patients with recalcitrant moderate to severe psoriasis for whom multiple types of treatment have failed.
FOR PSORIATIC ARTHRITIS: SYSTEMIC MEDICATIONS
For patients with known or questionable psoriatic arthritis, evaluation by a rheumatologist is highly recommended.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are usually first-line in the treatment of mild psoriatic arthritis. If after 2 to 3 months of therapy with NSAIDs no benefit is achieved, treatment with methotrexate as monotherapy is a practical consideration because of its low cost. However, methotrexate as a monotherapy has not been shown to prevent radiologic progression of disease.5,32
The TNF-alpha inhibitors have been shown to have similar efficacy when compared among each other in the treatment of psoriatic arthritis.32,63 Based on radiologic evidence, ustekinumab has not shown to be as efficacious as the TNF-alpha inhibitors for treating psoriatic arthritis. Therefore, TNF inhibitors should be considered first-line in the treatment of psoriatic arthritis.21,64
Few studies have been done on the efficacy or sequence of therapies that should be used in the treatment of psoriatic arthritis. The American Academy of Dermatology’s Psoriasis Guidelines of Care recommend adding a TNF-alpha inhibitor or switching to a TNF-alpha inhibitor if no significant improvement is achieved after 12 to 16 weeks of treatment with oral methotrexate.20
FOR ERYTHRODERMIC PSORIASIS: MEDICATIONS THAT ACT PROMPTLY
The care of erythrodermic psoriatic patients is distinct from that of other psoriatic patients because of their associated systemic symptoms. Care should be taken to rule out sepsis, as this is a reported trigger of erythrodermic psoriasis.28
Systemic medications with a quick onset, such as oral cyclosporine, are recommended. Infliximab has also been reported to be beneficial because of its rapid onset.28
TREATMENT BASED ON THE TYPE AND THE SEVERITY OF PSORIASIS
The treatment of psoriasis can be as complex as the disease it itself and should be based on the type and the severity of psoriasis. Recognition of the various manifestations of psoriasis is important for effective treatment. However, in patients with moderate to severe psoriasis, atypical presentations, or recalcitrant disease, referral to a specialist is recommended.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008; 58:826–850.
- Christophers E. Psoriasis—epidemiology and clinical spectrum. Clin Exp Dermatol 2001; 26:314–320.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Weiss SC, Kimball AB, Liewehr DJ, Blauvelt A, Turner ML, Emanuel EJ. Quantifying the harmful effect of psoriasis on health-related quality of life. J Am Acad Dermatol 2002; 47:512–518.
- Garg A, Gladman D. Recognizing psoriatic arthritis in the dermatology clinic. J Am Acad Dermatol 2010; 63:733–748.
- Kimball AB, Yu AP, Signorovitch J, et al. The effects of adalimumab treatment and psoriasis severity on self-reported work productivity and activity impairment for patients with moderate to severe psoriasis. J Am Acad Dermatol 2012; 66:e67–e76.
- Schmitt JM, Ford DE. Work limitations and productivity loss are associated with health-related quality of life but not with clinical severity in patients with psoriasis. Dermatology 2006; 213:102–110.
- Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Abuabara K, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the U.K. Br J Dermatol 2010; 163:586–592.
- Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med 2011; 270:147–157.
- Lin HW, Wang KH, Lin HC, Lin HC. Increased risk of acute myocardial infarction in patients with psoriasis: a 5-year population-based study in Taiwan. J Am Acad Dermatol 2011; 64:495–501.
- Bremmer S, Van Voorhees AS, Hsu S, et al; National Psoriasis Foundation. Obesity and psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol 2010; 63:1058–1069.
- Tobin AM, Veale DJ, Fitzgerald O, et al. Cardiovascular disease and risk factors in patients with psoriasis and psoriatic arthritis. J Rheumatol 2010; 37:1386–1394.
- Najarian DJ, Gottlieb AB. Connections between psoriasis and Crohn’s disease. J Am Acad Dermatol 2003; 48:805–821.
- Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol 2006; 55:829–835.
- Shapiro J, Cohen AD, Weitzman D, Tal R, David M. Psoriasis and cardiovascular risk factors: a case-control study on inpatients comparing psoriasis to dermatitis. J Am Acad Dermatol 2012; 66:252–258.
- Gelfand JM, Shin DB, Neimann AL, Wang X, Margolis DJ, Troxel AB. The risk of lymphoma in patients with psoriasis. J Invest Dermatol 2006; 126:2194–2201.
- Chen YJ, Wu CY, Chen TJ, et al. The risk of cancer in patients with psoriasis: a population-based cohort study in Taiwan. J Am Acad Dermatol 2011; 65:84–91.
- Friedewald VE, Cather JC, Gelfand JM, et al. AJC editor’s consensus: psoriasis and coronary artery disease. Am J Cardiol 2008; 102:1631–1643.
- American Academy of Dermatology Work Group; Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. J Am Acad Dermatol 2011; 65:137–174.
- Mallbris L, Larsson P, Bergqvist S, Vingård E, Granath F, Ståhle M. Psoriasis phenotype at disease onset: clinical characterization of 400 adult cases. J Invest Dermatol 2005; 124:499–504.
- Armstrong AW, Armstrong EJ, Fuller EN, Sockolov ME, Voyles SV. Smoking and pathogenesis of psoriasis: a review of oxidative, inflammatory and genetic mechanisms. Br J Dermatol 2011; 165:1162–1168.
- Qureshi AA, Dominguez PL, Choi HK, Han J, Curhan G. Alcohol intake and risk of incident psoriasis in US women: a prospective study. Arch Dermatol 2010; 146:1364–1369.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2; Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 2010; 42:985–990.
- Nair RP, Duffin KC, Helms C, et al; Collaborative Association Study of Psoriasis. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 2009; 41:199–204.
- Griffiths CE, Christophers E, Barker JN, et al. A classification of psoriasis vulgaris according to phenotype. Br J Dermatol 2007; 156:258–262.
- Rosenbach M, Hsu S, Korman NJ, et al; National Psoriasis Foundation Medical Board. Treatment of erythrodermic psoriasis: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol 2010; 62:655–662.
- Mrowietz U, van de Kerkhof PC. Management of palmoplantar pustulosis: do we need to change? Br J Dermatol 2011; 164:942–946.
- Kluger N, Bessis D, Guillot B, Girard C. Acute respiratory distress syndrome complicating generalized pustular psoriasis (psoriasis-associated aseptic pneumonitis). J Am Acad Dermatol 2011; 64:1154–1158.
- Roth MM. Pregnancy dermatoses: diagnosis, management, and controversies. Am J Clin Dermatol 2011; 12:25–41.
- Gottlieb A, Korman NJ, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol 2008; 58:851–864.
- Ogdie A, Gelfand JM. Identification of risk factors for psoriatic arthritis: scientific opportunity meets clinical need. Arch Dermatol 2010; 146:785–788.
- Gelfand JM, Gladman DD, Mease PJ, et al. Epidemiology of psoriatic arthritis in the population of the United States. J Am Acad Dermatol 2005; 53:573.
- Moll JM, Wright V. Psoriatic arthritis. Semin Arthritis Rheum 1973; 3:55–78.
- McGonagle D. Enthesitis: an autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J Eur Acad Dermatol Venereol 2009; 23(suppl 1):9–13.
- Feldman SR, Krueger GG. Psoriasis assessment tools in clinical trials. Ann Rheum Dis 2005; 64(suppl 2):ii65–ii68.
- Mason J, Mason AR, Cork MJ. Topical preparations for the treatment of psoriasis: a systematic review. Br J Dermatol 2002; 146:351–364.
- Menter A, Korman NJ, Elmets CA, et al; American Academy of Dermatology. Guidelines of care for the management of psoriasis and psoriatic arthritis. Section 3. Guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol 2009; 60:643–659.
- Zivkovich AH, Feldman SR. Are ointments better than other vehicles for corticosteroid treatment of psoriasis? J Drugs Dermatol 2009; 8:570–572.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 5. Guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J Am Acad Dermatol 2010; 62:114–135.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. Guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol 2009; 61:451–485.
- Murase JE, Lee EE, Koo J. Effect of ethnicity on the risk of developing nonmelanoma skin cancer following long-term PUVA therapy. Int J Dermatol 2005; 44:1016–1021.
- Stern RS, Lunder EJ. Risk of squamous cell carcinoma and methoxsalen (psoralen) and UV-A radiation (PUVA). A meta-analysis. Arch Dermatol 1998; 134:1582–1585.
- Stern RS, Väkevä LH. Noncutaneous malignant tumors in the PUVA follow-up study: 1975–1996. J Invest Dermatol 1997; 108:897–900.
- Patel RV, Clark LN, Lebwohl M, Weinberg JM. Treatments for psoriasis and the risk of malignancy. J Am Acad Dermatol 2009; 60:1001–1017.
- Flytström I, Stenberg B, Svensson A, Bergbrant IM. Methotrexate vs. ciclosporin in psoriasis: effectiveness, quality of life and safety. A randomized controlled trial. Br J Dermatol 2008; 158:116–121.
- Kalb RE, Strober B, Weinstein G, Lebwohl M. Methotrexate and psoriasis: 2009 National Psoriasis Foundation Consensus Conference. J Am Acad Dermatol 2009; 60:824–837.
- Zachariae H, Heickendorff L, Søgaard H. The value of aminoterminal propeptide of type III procollagen in routine screening for methotrexate-induced liver fibrosis: a 10-year follow-up. Br J Dermatol 2001; 144:100–103.
- Lowe NJ, Wieder JM, Rosenbach A, et al. Long-term low-dose cyclosporine therapy for severe psoriasis: effects on renal function and structure. J Am Acad Dermatol 1996; 35:710–719.
- Gottlieb AB, Grossman RM, Khandke L, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol 1992; 98:302–309.
- Ellis CN, Fradin MS, Messana JM, et al. Cyclosporine for plaque-type psoriasis. Results of a multidose, double-blind trial. N Engl J Med 1991; 324:277–284.
- Faerber L, Braeutigam M, Weidinger G, et al. Cyclosporine in severe psoriasis. Results of a meta-analysis in 579 patients. Am J Clin Dermatol 2001; 2:41–47.
- Ozawa A, Ohkido M, Haruki Y, et al. Treatments of generalized pustular psoriasis: a multicenter study in Japan. J Dermatol 1999; 26:141–149.
- Krueger GG, Ellis CN. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol 2003; 148:784–788.
- Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE; Alefacept Clinical Study Group. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol 2003; 139:719–727.
- Gottlieb AB, Matheson RT, Lowe N, et al. A randomized trial of etanercept as monotherapy for psoriasis. Arch Dermatol 2003; 139:1627–1632.
- Gottlieb AB, Masud S, Ramamurthi R, et al. Pharmacodynamic and pharmacokinetic response to anti-tumor necrosis factor-alpha monoclonal antibody (infliximab) treatment of moderate to severe psoriasis vulgaris. J Am Acad Dermatol 2003; 48:68–75.
- Reich K, Nestle FO, Papp K, et al; EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Menter A, Feldman SR, Weinstein GD, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol 2007; 56:31.e1–31.e15.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Kimball AB, Papp KA, et al; PHOENIX 1 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Griffiths CE, Strober BE, van de Kerkhof P, et al; ACCEPT Study Group. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 2010; 362:118–128.
- Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009; 373:633–640.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008; 58:826–850.
- Christophers E. Psoriasis—epidemiology and clinical spectrum. Clin Exp Dermatol 2001; 26:314–320.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Weiss SC, Kimball AB, Liewehr DJ, Blauvelt A, Turner ML, Emanuel EJ. Quantifying the harmful effect of psoriasis on health-related quality of life. J Am Acad Dermatol 2002; 47:512–518.
- Garg A, Gladman D. Recognizing psoriatic arthritis in the dermatology clinic. J Am Acad Dermatol 2010; 63:733–748.
- Kimball AB, Yu AP, Signorovitch J, et al. The effects of adalimumab treatment and psoriasis severity on self-reported work productivity and activity impairment for patients with moderate to severe psoriasis. J Am Acad Dermatol 2012; 66:e67–e76.
- Schmitt JM, Ford DE. Work limitations and productivity loss are associated with health-related quality of life but not with clinical severity in patients with psoriasis. Dermatology 2006; 213:102–110.
- Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Abuabara K, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the U.K. Br J Dermatol 2010; 163:586–592.
- Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med 2011; 270:147–157.
- Lin HW, Wang KH, Lin HC, Lin HC. Increased risk of acute myocardial infarction in patients with psoriasis: a 5-year population-based study in Taiwan. J Am Acad Dermatol 2011; 64:495–501.
- Bremmer S, Van Voorhees AS, Hsu S, et al; National Psoriasis Foundation. Obesity and psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol 2010; 63:1058–1069.
- Tobin AM, Veale DJ, Fitzgerald O, et al. Cardiovascular disease and risk factors in patients with psoriasis and psoriatic arthritis. J Rheumatol 2010; 37:1386–1394.
- Najarian DJ, Gottlieb AB. Connections between psoriasis and Crohn’s disease. J Am Acad Dermatol 2003; 48:805–821.
- Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol 2006; 55:829–835.
- Shapiro J, Cohen AD, Weitzman D, Tal R, David M. Psoriasis and cardiovascular risk factors: a case-control study on inpatients comparing psoriasis to dermatitis. J Am Acad Dermatol 2012; 66:252–258.
- Gelfand JM, Shin DB, Neimann AL, Wang X, Margolis DJ, Troxel AB. The risk of lymphoma in patients with psoriasis. J Invest Dermatol 2006; 126:2194–2201.
- Chen YJ, Wu CY, Chen TJ, et al. The risk of cancer in patients with psoriasis: a population-based cohort study in Taiwan. J Am Acad Dermatol 2011; 65:84–91.
- Friedewald VE, Cather JC, Gelfand JM, et al. AJC editor’s consensus: psoriasis and coronary artery disease. Am J Cardiol 2008; 102:1631–1643.
- American Academy of Dermatology Work Group; Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. J Am Acad Dermatol 2011; 65:137–174.
- Mallbris L, Larsson P, Bergqvist S, Vingård E, Granath F, Ståhle M. Psoriasis phenotype at disease onset: clinical characterization of 400 adult cases. J Invest Dermatol 2005; 124:499–504.
- Armstrong AW, Armstrong EJ, Fuller EN, Sockolov ME, Voyles SV. Smoking and pathogenesis of psoriasis: a review of oxidative, inflammatory and genetic mechanisms. Br J Dermatol 2011; 165:1162–1168.
- Qureshi AA, Dominguez PL, Choi HK, Han J, Curhan G. Alcohol intake and risk of incident psoriasis in US women: a prospective study. Arch Dermatol 2010; 146:1364–1369.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2; Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 2010; 42:985–990.
- Nair RP, Duffin KC, Helms C, et al; Collaborative Association Study of Psoriasis. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 2009; 41:199–204.
- Griffiths CE, Christophers E, Barker JN, et al. A classification of psoriasis vulgaris according to phenotype. Br J Dermatol 2007; 156:258–262.
- Rosenbach M, Hsu S, Korman NJ, et al; National Psoriasis Foundation Medical Board. Treatment of erythrodermic psoriasis: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol 2010; 62:655–662.
- Mrowietz U, van de Kerkhof PC. Management of palmoplantar pustulosis: do we need to change? Br J Dermatol 2011; 164:942–946.
- Kluger N, Bessis D, Guillot B, Girard C. Acute respiratory distress syndrome complicating generalized pustular psoriasis (psoriasis-associated aseptic pneumonitis). J Am Acad Dermatol 2011; 64:1154–1158.
- Roth MM. Pregnancy dermatoses: diagnosis, management, and controversies. Am J Clin Dermatol 2011; 12:25–41.
- Gottlieb A, Korman NJ, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol 2008; 58:851–864.
- Ogdie A, Gelfand JM. Identification of risk factors for psoriatic arthritis: scientific opportunity meets clinical need. Arch Dermatol 2010; 146:785–788.
- Gelfand JM, Gladman DD, Mease PJ, et al. Epidemiology of psoriatic arthritis in the population of the United States. J Am Acad Dermatol 2005; 53:573.
- Moll JM, Wright V. Psoriatic arthritis. Semin Arthritis Rheum 1973; 3:55–78.
- McGonagle D. Enthesitis: an autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J Eur Acad Dermatol Venereol 2009; 23(suppl 1):9–13.
- Feldman SR, Krueger GG. Psoriasis assessment tools in clinical trials. Ann Rheum Dis 2005; 64(suppl 2):ii65–ii68.
- Mason J, Mason AR, Cork MJ. Topical preparations for the treatment of psoriasis: a systematic review. Br J Dermatol 2002; 146:351–364.
- Menter A, Korman NJ, Elmets CA, et al; American Academy of Dermatology. Guidelines of care for the management of psoriasis and psoriatic arthritis. Section 3. Guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol 2009; 60:643–659.
- Zivkovich AH, Feldman SR. Are ointments better than other vehicles for corticosteroid treatment of psoriasis? J Drugs Dermatol 2009; 8:570–572.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 5. Guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J Am Acad Dermatol 2010; 62:114–135.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. Guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol 2009; 61:451–485.
- Murase JE, Lee EE, Koo J. Effect of ethnicity on the risk of developing nonmelanoma skin cancer following long-term PUVA therapy. Int J Dermatol 2005; 44:1016–1021.
- Stern RS, Lunder EJ. Risk of squamous cell carcinoma and methoxsalen (psoralen) and UV-A radiation (PUVA). A meta-analysis. Arch Dermatol 1998; 134:1582–1585.
- Stern RS, Väkevä LH. Noncutaneous malignant tumors in the PUVA follow-up study: 1975–1996. J Invest Dermatol 1997; 108:897–900.
- Patel RV, Clark LN, Lebwohl M, Weinberg JM. Treatments for psoriasis and the risk of malignancy. J Am Acad Dermatol 2009; 60:1001–1017.
- Flytström I, Stenberg B, Svensson A, Bergbrant IM. Methotrexate vs. ciclosporin in psoriasis: effectiveness, quality of life and safety. A randomized controlled trial. Br J Dermatol 2008; 158:116–121.
- Kalb RE, Strober B, Weinstein G, Lebwohl M. Methotrexate and psoriasis: 2009 National Psoriasis Foundation Consensus Conference. J Am Acad Dermatol 2009; 60:824–837.
- Zachariae H, Heickendorff L, Søgaard H. The value of aminoterminal propeptide of type III procollagen in routine screening for methotrexate-induced liver fibrosis: a 10-year follow-up. Br J Dermatol 2001; 144:100–103.
- Lowe NJ, Wieder JM, Rosenbach A, et al. Long-term low-dose cyclosporine therapy for severe psoriasis: effects on renal function and structure. J Am Acad Dermatol 1996; 35:710–719.
- Gottlieb AB, Grossman RM, Khandke L, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol 1992; 98:302–309.
- Ellis CN, Fradin MS, Messana JM, et al. Cyclosporine for plaque-type psoriasis. Results of a multidose, double-blind trial. N Engl J Med 1991; 324:277–284.
- Faerber L, Braeutigam M, Weidinger G, et al. Cyclosporine in severe psoriasis. Results of a meta-analysis in 579 patients. Am J Clin Dermatol 2001; 2:41–47.
- Ozawa A, Ohkido M, Haruki Y, et al. Treatments of generalized pustular psoriasis: a multicenter study in Japan. J Dermatol 1999; 26:141–149.
- Krueger GG, Ellis CN. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol 2003; 148:784–788.
- Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE; Alefacept Clinical Study Group. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol 2003; 139:719–727.
- Gottlieb AB, Matheson RT, Lowe N, et al. A randomized trial of etanercept as monotherapy for psoriasis. Arch Dermatol 2003; 139:1627–1632.
- Gottlieb AB, Masud S, Ramamurthi R, et al. Pharmacodynamic and pharmacokinetic response to anti-tumor necrosis factor-alpha monoclonal antibody (infliximab) treatment of moderate to severe psoriasis vulgaris. J Am Acad Dermatol 2003; 48:68–75.
- Reich K, Nestle FO, Papp K, et al; EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Menter A, Feldman SR, Weinstein GD, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol 2007; 56:31.e1–31.e15.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Kimball AB, Papp KA, et al; PHOENIX 1 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Griffiths CE, Strober BE, van de Kerkhof P, et al; ACCEPT Study Group. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 2010; 362:118–128.
- Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009; 373:633–640.
KEY POINTS
- Studies in the past 10 years have uncovered a link between psoriasis, metabolic syndrome, and cardiovascular disease. Interestingly, the risk grows less with age; patients at greatest risk are young men with severe psoriasis.
- The most common presentation of psoriasis is plaque psoriasis. However, there are several other clinical variations of psoriasis, each of which has a distinct response to treatment and may be associated with significant systemic symptoms.
- Tumor necrosis factor inhibitors should be considered first-line in the treatment of psoriatic arthritis.
- Phototherapy and systemic medications including methotrexate, acitretin (Soriatane), cyclosporine (Gengraf, Neoral, Sandimmune), and biologic agents are the most effective treatments for moderate-to-severe psoriasis.
Advances in Lung Cancer Evaluation and Management

Supplement Editors:
Nathan Pennell, MD, PhD, and Peter Mazzone, MD
Contents
Lung cancer screening: Examining the issues
Peter Mazzone
Treatment implication of the new lung cancer staging system
Cristina P. Rodriguez
Bronchoscopy and endobronchial ultrasound for diagnosis and staging of lung cancer
Francisco Aécio Almeida
Preoperative evaluation of the lung resection candidate
Peter Mazzone
Video-assisted thoracoscopic surgery for the treatment of lung cancer
Sudish Murthy
Stereotactic body radiotherapy for stage I non–small cell lung cancer
Kevin Stephans
Locally advanced non–small cell lung cancer: What is the optimal concurrent chemoradiation regimen?
Gregory M.M. Videtic
The role of surgery for locally advanced non–small cell lung cancer
David P. Mason
The role of adjuvant chemotherapy in early-stage and locally advanced non–small cell lung cancer
Marc Shapiro
Selection of chemotherapy for patients with advanced non–small cell lung cancer
Nathan A. Pennell
The emerging role of palliative medicine in the treatment of lung cancer patients
Mellar P. Davis
Personalized targeted therapy in advanced non–small cell lung cancer
Patrick C. Ma
Supplement Editors:
Nathan Pennell, MD, PhD, and Peter Mazzone, MD
Contents
Lung cancer screening: Examining the issues
Peter Mazzone
Treatment implication of the new lung cancer staging system
Cristina P. Rodriguez
Bronchoscopy and endobronchial ultrasound for diagnosis and staging of lung cancer
Francisco Aécio Almeida
Preoperative evaluation of the lung resection candidate
Peter Mazzone
Video-assisted thoracoscopic surgery for the treatment of lung cancer
Sudish Murthy
Stereotactic body radiotherapy for stage I non–small cell lung cancer
Kevin Stephans
Locally advanced non–small cell lung cancer: What is the optimal concurrent chemoradiation regimen?
Gregory M.M. Videtic
The role of surgery for locally advanced non–small cell lung cancer
David P. Mason
The role of adjuvant chemotherapy in early-stage and locally advanced non–small cell lung cancer
Marc Shapiro
Selection of chemotherapy for patients with advanced non–small cell lung cancer
Nathan A. Pennell
The emerging role of palliative medicine in the treatment of lung cancer patients
Mellar P. Davis
Personalized targeted therapy in advanced non–small cell lung cancer
Patrick C. Ma
Supplement Editors:
Nathan Pennell, MD, PhD, and Peter Mazzone, MD
Contents
Lung cancer screening: Examining the issues
Peter Mazzone
Treatment implication of the new lung cancer staging system
Cristina P. Rodriguez
Bronchoscopy and endobronchial ultrasound for diagnosis and staging of lung cancer
Francisco Aécio Almeida
Preoperative evaluation of the lung resection candidate
Peter Mazzone
Video-assisted thoracoscopic surgery for the treatment of lung cancer
Sudish Murthy
Stereotactic body radiotherapy for stage I non–small cell lung cancer
Kevin Stephans
Locally advanced non–small cell lung cancer: What is the optimal concurrent chemoradiation regimen?
Gregory M.M. Videtic
The role of surgery for locally advanced non–small cell lung cancer
David P. Mason
The role of adjuvant chemotherapy in early-stage and locally advanced non–small cell lung cancer
Marc Shapiro
Selection of chemotherapy for patients with advanced non–small cell lung cancer
Nathan A. Pennell
The emerging role of palliative medicine in the treatment of lung cancer patients
Mellar P. Davis
Personalized targeted therapy in advanced non–small cell lung cancer
Patrick C. Ma

Antireflux surgery in the proton pump inhibitor era
For most patients with gastroesophageal reflux disease (GERD), a proton pump inhibitor (PPI) is the first choice for treatment.1 But some patients have symptoms that persist despite PPI therapy, some desire surgery despite successful PPI therapy, and some have persistent extraesophageal symptoms or other complications of reflux. For these patients, surgery is an option.2
In this article, we review the management of GERD and clarify the indications for antireflux surgery based on evidence of safety and efficacy.
GERD DEFINED: SYMPTOMS OR COMPLICATIONS
Defining the role of antireflux surgery is difficult, given the variety of presentations and the absence of a gold standard for diagnosing GERD. Most adults experience several episodes of physiologic reflux daily without symptoms.3 But a broad array of symptoms have been attributed to GERD, including chest pain, cough, and sore throat, and some conditions caused by acid reflux (eg, Barrett esophagus) can be asymptomatic.4,5

HEARTBURN ISN’T ALWAYS GERD
Typical GERD presents with the classic symptoms of pyrosis (heartburn) or acid regurgitation, or both.
Although these symptoms are often thought to be specific for GERD, other causes of esophageal injury— eg, eosinophilic esophagitis, infection (Candida, cytomegalovirus, herpes simplex virus), pill-induced esophagitis, or radiation therapy—can produce similar symptoms. Other causes, including coronary artery disease, biliary colic, foregut malignancy, or peptic ulcer disease, should also be considered in patients with supposedly typical GERD. Life-threatening mimics of GERD, such as unstable angina, should be excluded if they are likely, before proceeding with evaluating for possible GERD. Therefore, the initial history and examination should focus on appropriate diagnosis, with careful delineation of symptom quality.
Alarm features for advanced pathology6–8 include involuntary weight loss, dysphagia, vomiting, evidence of gastrointestinal blood loss, anemia, chest pain, and an epigastric mass.7 Admittedly, these features are only mediocre for detecting or excluding gastric or esophageal cancer, with a sensitivity of 67% and a specificity 66%.9 Nevertheless, they should prompt an endoscopic examination. In patients who have alarm features but have not yet been treated for GERD, upper endoscopy can identify an abnormality in about 60% of patients.10–12
PPIs HAVE REPLACED ANTACIDS AND HISTAMINE-2 RECEPTOR ANTAGONISTS
When the symptoms suggest GERD and no alarm features are present, an initial trial of the following lifestyle changes is reasonable:
- Avoiding acidic or refluxogenic foods (coffee, alcohol, chocolate, peppermint, fatty foods, citrus foods)
- Avoiding certain medications (anticholinergics, estrogens, calcium-channel blockers, nitroglycerine, benzodiazepines)
- Losing weight
- Quitting smoking
- Raising the head of the bed
- Staying upright for 2 to 3 hours after meals.
For someone with mild symptoms, these changes pose minimal risk. Unfortunately, they are unlikely to provide adequate symptom control for most patients.13–17
Before PPIs were invented, drug therapy for GERD symptoms that did not resolve with lifestyle changes consisted of antacids and, later, histamine-2 receptor antagonists. When maximal therapy failed to control symptoms, fundoplication surgery was considered an appropriate next step.
PPIs substantially changed the management of GERD, suppressing acid secretion much better than histamine-2 receptor antagonists. Taken 30 minutes before breakfast, a single daily dose of a PPI normalizes esophageal acid exposure in 67% of patients.18 Adding a second dose 30 minutes before dinner raises the number to more than 90%.19
PPIs have consistently outperformed histamine-2 blockers in the healing of esophagitis and in improving heartburn symptoms and are now the first-line medical therapy for uncomplicated GERD.6,8,20–25
WHEN PPIs WORK, SURGERY OFFERS NO ADVANTAGE
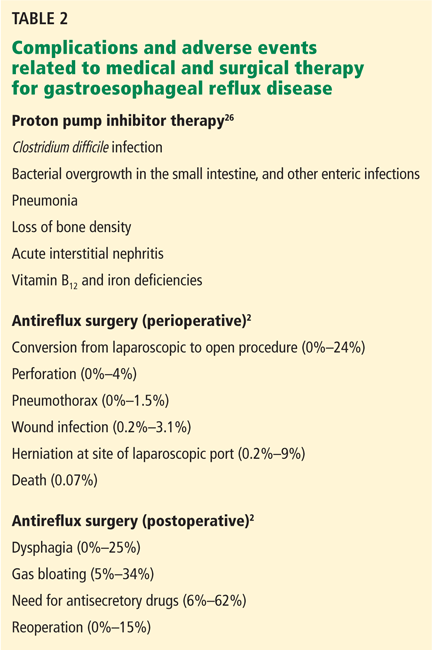
The LOTUS trial (Long-Term Usage of Esomeprazole vs Surgery for Treatment of Chronic GERD) compared long-term drug therapy with surgery to maintain remission of symptoms in GERD.27 In this trial, 554 patients whose symptoms initially responded to the PPI esomeprazole (Nexium) were randomized to continue to receive esomeprazole (n = 266) or to undergo laparoscopic antireflux surgery (288 were randomly assigned, and 248 had the operation). Dose adjustment of the esomeprazole was allowed (20–40 mg/day). A total of 372 patients completed 5 years of follow-up (192 esomeprazole, 180 surgery).
Symptoms stayed in remission in 92% of the esomeprazole group and 85% of the surgery group (P = .048). However, the difference was no longer statistically significant after modeling the effects of study dropout. The rate of severe adverse events was similar in both groups: 24.1% with esomeprazole and 28.6% with surgery.
These findings indicate that if symptoms fully abate with medical therapy, surgery offers no advantage. In addition, patients who desire surgery in the hope of avoiding lifelong drug therapy should be made aware that drug therapy and reoperation are often necessary after surgery.28 In most cases, antireflux surgery is unnecessary for patients whose GERD fully responds to PPI therapy.
IF PPIs FAIL, FURTHER TESTING NEEDED
But many patients who take PPIs still have symptoms, even though these drugs suppress acid secretion and heal esophagitis. In fact, symptoms completely resolve in only about one-half of patients with erosive disease and one-third of those without erosive disease.21
Reasons for an incomplete symptomatic response to PPIs are various. Acid reflux can persist, but this accounts for only 10% of cases.29 About one-third of patients have persistent reflux that is weakly acidic, with a pH higher than 4.29. However, most patients with persistent typical GERD symptoms do not have significant, persistent reflux, or their symptoms are not related to reflux events. In these cases, an alternative cause of the refractory symptoms should be sought.
Further diagnostic testing is indicated when symptoms persist despite PPI therapy. Upper endoscopy will reveal an abnormality such as persistent erosive esophagitis, eosinophilic esophagitis, esophageal stricture, Barrett esophagus, or esophageal cancer in roughly 10% of patients in whom empiric therapy fails.10
Although patients with persistent symptoms have not been enrolled in many randomized controlled trials, a multivariate analysis showed that failure of medical therapy heralds a poor response to surgery.30 Data such as these have led most experts to discourage fundoplication for such patients now, unlike in the pre-PPI era.
pH and intraluminal impedance testing
However, this recommendation against surgery is not a hard-and-fast rule.
In patients with esophageal regurgitation, most will not achieve adequate relief of symptoms with PPI therapy alone.34 The therapeutic gain of PPI therapy vs placebo averaged just 17% in seven randomized, controlled trials, more than 20% less than the response rate for heartburn.34 This is likely because of structural abnormalities such as reduced lower esophageal sphincter pressure, hiatal hernia, or delayed gastric emptying. Antireflux surgery can correct these structural abnormalities or prevent them from causing so much trouble; however, the presence of true regurgitation should first be confirmed by MII testing. If regurgitation is confirmed, antireflux surgery is warranted, particularly in patients with nocturnal symptoms who may be at high risk of aspiration. With careful patient selection, regurgitation symptoms improve in about 90% after surgery.2
In patients with heartburn, if esophageal acid exposure continues to be abnormal on MII-pH testing, then an escalation of therapy may improve symptoms, particularly if symptoms occur during reflux or if they partially responded to PPI therapy. Options in this scenario include alteration or intensification of acid-suppressive therapy, treatment with baclofen (Lioresal), and antireflux surgery.18,35,36 In randomized controlled trials of patients whose symptoms partially responded to PPIs, antireflux surgery has performed similarly to PPIs in terms of improving typical GERD symptoms, particularly regurgitation.27,37–41 Although this scenario is a reasonable indication for antireflux surgery, recommendations should be made with appropriate restraint since it is not easily reversible, some patients experience complications, and up to one-third will have no therapeutic benefit.30
Nonacid reflux. In some cases, MII-pH testing during PPI therapy will reveal reflux of weakly acidic (pH > 4) or alkaline stomach contents, often called “nonacid reflux.”29 Nonacid reflux is often present in patients with esophagitis that persists despite PPI therapy, indicating that even weakly acidic stomach contents can injure the mucosa.42 Since intensifying the acid-suppressive therapy is unlikely to improve these symptoms, antireflux surgery may have a role.
In one study,43 nonacid reflux was well controlled by laparoscopic Nissen fundoplication, although 15 (48%) of 31 patients had persistent symptoms of GERD after surgery. No patient had a strong symptom correlation with postoperative reflux events, suggesting an alternative cause of the persistent symptoms. Therefore, antireflux surgery for nonacid reflux should be limited exclusively to patients with strong symptom correlation, and even then it should be considered with restraint, given the limited evidence for benefit and the potential for harm.
If testing is negative. In studies investigating the diagnostic yield of MII-pH testing, more than 50% of patients who had refractory symptoms had a negative MII-pH test.29 In such situations, when the symptoms are strongly correlated with reflux events, the patient is said to have “esophageal hypersensitivity.” A few small studies have suggested that such patients may benefit from surgery, but these data have not been replicated in randomized controlled trials.32
When the testing is negative and there is no correlation between the patient’s symptoms and reflux events, the patient is unlikely to benefit from antireflux surgery. Care of these patients is beyond the scope of this review.
SURGERY RARELY IMPROVES COUGH, ASTHMA, OR LARYNGITIS
GERD has been implicated as a cause of chronic cough, asthma, and laryngitis, although each of these has many potential causes.44–46 Despite these associations, the evidence for therapeutic benefit from antireflux therapy is weak.
PPI therapy shows no benefit over placebo for chronic cough of uncertain etiology, but some benefit if GERD is objectively demonstrated.47 Laryngitis resolved in just 15% of patients on esomeprazole vs 16% of patients on placebo after excluding patients with moderate to severe heartburn.48
In a large randomized controlled trial in patients with asthma, there was no overall improvement in peak flow for the PPI group vs the placebo group, although significant improvement occurred in patients with heartburn and nocturnal respiratory symptoms.46
Potent antisecretory therapy seems to improve extraesophageal symptoms when typical GERD symptoms are also present, but it otherwise has shown little evidence of benefit.
The evidence for a benefit from antireflux surgery in patients with extraesophageal GERD syndromes is even more limited. Although one systematic review49 found that cough and other laryngeal symptoms improved in 60% to 100% of patients with objective evidence of GERD who underwent fundoplication, virtually all of the studies were uncontrolled case series.49
The lone randomized controlled trial in the systematic review compared Nissen fundoplication with ranitidine (Zantac) or antacids only for patients with asthma and GERD, and found no significant difference in peak expiratory flow among the three groups after 2 years. However, asthma symptom scores improved in 75% of the surgical group, 9% of the medical group, and 4% of the control group.50
In a study that was not included in the prior systematic review, patients with laryngopharyngeal reflux unresponsive to aggressive acid suppression who subsequently underwent fundoplication fared no better than those who did not.51
Thus, based on the available data, antireflux surgery is only rarely indicated for extraesophageal symptoms, especially in patients who have no typical GERD symptoms or in patients whose symptoms are refractory to medical therapy.
SURGERY FOR EROSIVE ESOPHAGITIS OR BARRETT ESOPHAGUS IF PPI FAILS
Lifelong antireflux therapy is indicated for patients with severe erosive esophagitis or Barrett esophagus. Erosive esophagitis recurs in more than 80% within 12 months of discontinuing antisecretory therapy.52 Both Barrett esophagus and esophageal adenocarcinoma are strongly associated with GERD, and nearly 10% of patients with chronic reflux have Barrett esophagus.53,54 It is suspected that suppressing reflux reduces the rate of progression of Barrett esophagus to esophageal adenocarcinoma, but this remains to be proven.
Perhaps the strongest indication for surgery in the PPI era is for patients who have persistent symptoms and severe erosive esophagitis (Los Angeles grade C or D) despite high-dose PPI therapy. If other causes of persistent esophagitis have been ruled out, fundoplication can induce healing and improve symptoms.55,56 In these cases, surgery is done to induce remission of the disease when maximal medical therapy has been truly unsuccessful.
Randomized controlled trials suggest that medical and surgical therapies are equally effective for preventing the recurrence of erosive esophagitis or the progression of Barrett esophagus. In a study of 225 patients, at 7 years of follow-up, esophagitis had recurred in 10.4% of patients on omeprazole vs 11.8% of those who had undergone antireflux surgery.40 Similarly, open Nissen fundoplication was no different from drug therapy (histamine-2 receptor antagonist or PPI) for progression of Barrett esophagus over a median of 5 years.57 A meta-analysis with nearly 5,000 person-years each in the medical and surgical groups also found no significant difference in rates of cancer progression.58
Notably, symptoms such as dysphagia, flatulence, and the inability to burp occurred significantly more often in the surgical groups in these studies.
In view of these data, antireflux surgery has no significant advantage over medical therapy for maintaining healing of erosive esophagitis or preventing progression of Barrett esophagus. Thus, it should be reserved for patients who do not desire lifelong drug therapy, provided they understand that there is no therapeutic advantage for their esophagitis or for Barrett esophagus.
SPECIFIC INDICATIONS FOR ANTIREFLUX SURGERY
Now that we have PPIs, several situations remain in which surgery for GERD is either indicated or worth considering.
Antireflux surgery is clearly indicated for:
- Patients with erosive esophagitis that does not heal with maximal drug therapy
- Patients with volume regurgitation, particularly if it occurs at night or if there is evidence of aspiration
- Patients who require lifelong treatment for reflux but who have had a serious adverse event related to PPI therapy, such as refractory Clostridium difficile infection.
Antireflux surgery is also worth considering in patients who for personal reasons wish to avoid long-term or lifelong drug therapy.
Patients should be informed, however, that antireflux surgery has not been shown to be better than medical therapy for maintaining remission of symptoms, for preventing progression of Barrett esophagus, or for maintaining healing of erosive esophagitis. Medical therapy is still the first option for these patients.
Surgery may also be considered in patients with persistent symptoms who have a partial response to medical therapy, who show persistent acidic or weakly acidic reflux on MII-pH testing, and whose symptoms have been correlated with reflux events. Although surgery is not sure to improve their symptoms, benefit is more likely in this patient population compared with those without these characteristics.
Extraesophageal GERD
In patients suspected of having extraesophageal GERD, surgery should be considered if typical GERD symptoms are present and improve with PPI therapy, if the extraesophageal syndrome partially responds to PPI therapy, and if MII-pH testing demonstrates a correlation between symptoms and reflux. Surgery may have a stronger indication in this setting if the patient has nocturnal reflux or extraesophageal symptoms.
When is surgery not an option?
In general, surgery should not be considered in patients who do not have a partial response to PPI therapy or who do not have a strong symptom-reflux correlation on MII-pH testing. In all cases of failed medical therapy without persistent severe erosive disease, the threshold for opting for surgery should be high, given the uncertain response of these patients to surgery.
Peristaltic dysfunction is a relative but not an absolute contraindication to surgery.59
RISKS, BENEFITS OF SURGERY FOR GERD
The patient’s preference for surgery over drug therapy should always be balanced against the risks of surgery, including both short-term and long-term adverse events, to allow the patient to make an adequately informed decision (Table 2).2,26
Adverse events associated with PPI therapy are rare and in many cases the association is debatable.26 Nonetheless, long-term PPI therapy has been most strongly associated with an increased risk of C difficile infection and other enteric infections, although the absolute risk of these events remains low.
Complication rates after antireflux surgery depend on the surgeon’s experience and technique. Death is exceedingly rare. In most high-volume centers, the need to convert from laparoscopic to open fundoplication occurs in fewer than 2.4% of patients.2
Potential perioperative complications include perforation (4%), wound infection (3%), and pneumothorax (2%).2
Antireflux surgery is also associated with a significant risk of dysphagia, bloating, an inability to burp, and excessive flatulence, all of which can markedly impair the quality of life.
A major consideration is that fundoplication is generally irreversible. Reoperation rates have been reported to range from 0% to 15%.2 Furthermore, up to 50% of patients still need medical therapy after surgery.60,61 Of note, only about 25% of patients on medical therapy after surgery will actually have an abnormal pH study.61
MORE STUDY NEEDED
Future studies directly comparing medical and surgical therapy for carefully selected patients with extraesophageal manifestations of GERD and refractory symptoms should help further delineate outcome in this difficult group of patients.
Under development are new drugs that may inhibit transient relaxation of the lower esophageal sphincter, as well as minimally invasive procedures, which may alter the indications for surgery in coming years.36
Acknowledgment: The research for this article was supported in part by a grant from the National Institutes of Health (T32 DK07634).
- Finks JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc 2006; 20:1698–1701.
- Stefanidis D, Hope WW, Kohn GP, Reardon PR, Richardson WS, Fanelli RD; SAGES Guidelines Committee. Guidelines for surgical treatment of gastroesophageal reflux disease. Surg Endosc 2010; 24:2647–2669.
- Richter JE. Typical and atypical presentations of gastroesophageal reflux disease. The role of esophageal testing in diagnosis and management. Gastroenterol Clin North Am 1996; 25:75–102.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Dickman R, Kim JL, Camargo L, et al. Correlation of gastroesophageal reflux disease symptoms characteristics with long-segment Barrett’s esophagus. Dis Esophagus 2006; 19:360–365.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Armstrong D, Marshall JK, Chiba N, et al; Canadian Association of Gastroenterology GERD Consensus Group. Canadian Consensus Conference on the management of gastroesophageal reflux disease in adults - update 2004. Can J Gastroenterol 2005; 19:15–35.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.
- Vakil N, Moayyedi P, Fennerty MB, Talley NJ. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: systematic review and meta-analysis. Gastroenterology 2006; 131:390–401.
- Poh CH, Gasiorowska A, Navarro-Rodriguez T, et al. Upper GI tract findings in patients with heartburn in whom proton pump inhibitor treatment failed versus those not receiving antireflux treatment. Gastrointest Endosc 2010; 71:28–34.
- Dickman R, Mattek N, Holub J, Peters D, Fass R. Prevalence of upper gastrointestinal tract findings in patients with noncardiac chest pain versus those with gastroesophageal reflux disease (GERD)-related symptoms: results from a national endoscopic database. Am J Gastroenterol 2007; 102:1173–1179.
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6–13.
- Fraser-Moodie CA, Norton B, Gornall C, Magnago S, Weale AR, Holmes GK. Weight loss has an independent beneficial effect on symptoms of gastro-oesophageal reflux in patients who are overweight. Scand J Gastroenterol 1999; 34:337–340.
- Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Bodymass index and symptoms of gastroesophageal reflux in women. N Engl J Med 2006; 354:2340–2348.
- Kjellin A, Ramel S, Rössner S, Thor K. Gastroesophageal reflux in obese patients is not reduced by weight reduction. Scand J Gastroenterol 1996; 31:1047–1051.
- Waring JP, Eastwood TF, Austin JM, Sanowski RA. The immediate effects of cessation of cigarette smoking on gastroesophageal reflux. Am J Gastroenterol 1989; 84:1076–1078.
- Pehl C, Waizenhoefer A, Wendl B, Schmidt T, Schepp W, Pfeiffer A. Effect of low and high fat meals on lower esophageal sphincter motility and gastroesophageal reflux in healthy subjects. Am J Gastroenterol 1999; 94:1192–1196.
- Bajbouj M, Becker V, Phillip V, Wilhelm D, Schmid RM, Meining A. High-dose esomeprazole for treatment of symptomatic refractory gastroesophageal reflux disease—a prospective pH-metry/impedance-controlled study. Digestion 2009; 80:112–118.
- Charbel S, Khandwala F, Vaezi MF. The role of esophageal pH monitoring in symptomatic patients on PPI therapy. Am J Gastroenterol 2005; 100:283–289.
- Khan M, Santana J, Donnellan C, Preston C, Moayyedi P. Medical treatments in the short term management of reflux oesophagitis. Cochrane Database Syst Rev 2007;CD003244.
- Dean BB, Gano AD, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2:656–664.
- Sabesin SM, Berlin RG, Humphries TJ, Bradstreet DC, Walton-Bowen KL, Zaidi S. Famotidine relieves symptoms of gastroesophageal reflux disease and heals erosions and ulcerations. Results of a multicenter, placebo-controlled, dose-ranging study. USA Merck Gastroesophageal Reflux Disease Study Group. Arch Intern Med 1991; 151:2394–2400.
- van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004;CD002095.
- Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology 1997; 112:1798–1810.
- Venables TL, Newland RD, Patel AC, Hole J, Wilcock C, Turbitt ML. Omeprazole 10 milligrams once daily, omeprazole 20 milligrams once daily, or ranitidine 150 milligrams twice daily, evaluated as initial therapy for the relief of symptoms of gastro-oesophageal reflux disease in general practice. Scand J Gastroenterol 1997; 32:965–973.
- Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med 2011; 78:39–49.
- Galmiche JP, Hatlebakk J, Attwood S, et al; LOTUS Trial Collaborators. Laparoscopic antireflux surgery vs esomeprazole treatment for chronic GERD: the LOTUS randomized clinical trial. JAMA 2011; 305:1969–1977.
- Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: followup of a randomized controlled trial. JAMA 2001; 285:2331–2338.
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55:1398–1402.
- Campos GM, Peters JH, DeMeester TR, et al. Multivariate analysis of factors predicting outcome after laparoscopic Nissen fundoplication. J Gastrointest Surg 1999; 3:292–300.
- Becker V, Bajbouj M, Waller K, Schmid RM, Meining A. Clinical trial: persistent gastro-oesophageal reflux symptoms despite standard therapy with proton pump inhibitors - a follow-up study of intraluminal-impedance guided therapy. Aliment Pharmacol Ther 2007; 26:1355–1360.
- Mainie I, Tutuian R, Agrawal A, Adams D, Castell DO. Combined multichannel intraluminal impedance-pH monitoring to select patients with persistent gastro-oesophageal reflux for laparoscopic Nissen fundoplication. Br J Surg 2006; 93:1483–1487.
- del Genio G, Tolone S, del Genio F, et al. Prospective assessment of patient selection for antireflux surgery by combined multichannel intraluminal impedance pH monitoring. J Gastrointest Surg 2008; 12:1491–1496.
- Kahrilas PJ, Howden CW, Hughes N. Response of regurgitation to proton pump inhibitor therapy in clinical trials of gastroesophageal reflux disease. Am J Gastroenterol 2011; 106:1419–1425.
- Koek GH, Sifrim D, Lerut T, Janssens J, Tack J. Effect of the GABA(B) agonist baclofen in patients with symptoms and duodeno-gastro-oesophageal reflux refractory to proton pump inhibitors. Gut 2003; 52:1397–1402.
- Boeckxstaens GE. Reflux inhibitors: a new approach for GERD? Curr Opin Pharmacol 2008; 8:685–689.
- Anvari M, Allen C, Marshall J, et al. A randomized controlled trial of laparoscopic nissen fundoplication versus proton pump inhibitors for treatment of patients with chronic gastroesophageal reflux disease: One-year follow-up. Surg Innov 2006; 13:238–249.
- Mahon D, Rhodes M, Decadt B, et al. Randomized clinical trial of laparoscopic Nissen fundoplication compared with proton-pump inhibitors for treatment of chronic gastro-oesophageal reflux. Br J Surg 2005; 92:695–699.
- Mehta S, Bennett J, Mahon D, Rhodes M. Prospective trial of laparoscopic nissen fundoplication versus proton pump inhibitor therapy for gastroesophageal reflux disease: Seven-year follow-up. J Gastrointest Surg 2006; 10:1312–1316.
- Lundell L, Miettinen P, Myrvold HE, et al; Nordic GORD Study Group. Seven-year follow-up of a randomized clinical trial comparing proton-pump inhibition with surgical therapy for reflux oesophagitis. Br J Surg 2007; 94:198–203.
- Lundell L, Attwood S, Ell C, et al; LOTUS trial collaborators. Comparing laparoscopic antireflux surgery with esomeprazole in the management of patients with chronic gastro-oesophageal reflux disease: a 3-year interim analysis of the LOTUS trial. Gut 2008; 57:1207–1213.
- Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 2011; 33:601–606.
- Broeders JA, Bredenoord AJ, Hazebroek EJ, Broeders IA, Gooszen HG, Smout AJ. Effects of anti-reflux surgery on weakly acidic reflux and belching. Gut 2011; 60:435–441.
- American Gastroenterological Association medical position statement: guidelines on the use of esophageal pH recording. Gastroenterology 1996; 110:1981.
- el-Serag HB, Sonnenberg A. Comorbid occurrence of laryngeal or pulmonary disease with esophagitis in United States military veterans. Gastroenterology 1997; 113:755–760.
- Kiljander TO, Laitinen JO. The prevalence of gastroesophageal reflux disease in adult asthmatics. Chest 2004; 126:1490–1494.
- Chang AB, Lasserson TJ, Kiljander TO, Connor FL, Gaffney JT, Garske LA. Systematic review and meta-analysis of randomised controlled trials of gastro-oesophageal reflux interventions for chronic cough associated with gastro-oesophageal reflux. BMJ 2006; 332:11–17.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Iqbal M, Batch AJ, Spychal RT, Cooper BT. Outcome of surgical fundoplication for extraesophageal (atypical) manifestations of gastroesophageal reflux disease in adults: a systematic review. J Laparoendosc Adv Surg Tech A 2008; 18:789–796.
- Sontag SJ, O’Connell S, Khandelwal S, et al. Asthmatics with gastroesophageal reflux: long term results of a randomized trial of medical and surgical antireflux therapies. Am J Gastroenterol 2003; 98:987–999.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- Johnson DA, Benjamin SB, Vakil NB, et al. Esomeprazole once daily for 6 months is effective therapy for maintaining healed erosive esophagitis and for controlling gastroesophageal reflux disease symptoms: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Gastroenterol 2001; 96:27–34.
- Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987; 92:118–124.
- Westhoff B, Brotze S, Weston A, et al. The frequency of Barrett’s esophagus in high-risk patients with chronic GERD. Gastrointest Endosc 2005; 61:226–231.
- Rosenthal R, Peterli R, Guenin MO, von Flüe M, Ackermann C. Laparoscopic antireflux surgery: long-term outcomes and quality of life. J Laparoendosc Adv Surg Tech A 2006; 16:557–561.
- Broeders JA, Draaisma WA, Bredenoord AJ, Smout AJ, Broeders IA, Gooszen HG. Long-term outcome of Nissen fundoplication in non-erosive and erosive gastro-oesophageal reflux disease. Br J Surg 2010; 97:845–352.
- Parrilla P, Martínez de Haro LF, Ortiz A, et al. Long-term results of a randomized prospective study comparing medical and surgical treatment of Barrett’s esophagus. Ann Surg 2003; 237:291–298.
- Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol 2003; 98:2390–2394.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and antireflux medication use after antireflux surgery. Clin Gastroenterol Hepatol 2006; 4:299–305.
- Lord RV, Kaminski A, Oberg S, et al. Absence of gastroesophageal reflux disease in a majority of patients taking acid suppression medications after Nissen fundoplication. J Gastrointest Surg 2002; 6:3–9.
For most patients with gastroesophageal reflux disease (GERD), a proton pump inhibitor (PPI) is the first choice for treatment.1 But some patients have symptoms that persist despite PPI therapy, some desire surgery despite successful PPI therapy, and some have persistent extraesophageal symptoms or other complications of reflux. For these patients, surgery is an option.2
In this article, we review the management of GERD and clarify the indications for antireflux surgery based on evidence of safety and efficacy.
GERD DEFINED: SYMPTOMS OR COMPLICATIONS
Defining the role of antireflux surgery is difficult, given the variety of presentations and the absence of a gold standard for diagnosing GERD. Most adults experience several episodes of physiologic reflux daily without symptoms.3 But a broad array of symptoms have been attributed to GERD, including chest pain, cough, and sore throat, and some conditions caused by acid reflux (eg, Barrett esophagus) can be asymptomatic.4,5
HEARTBURN ISN’T ALWAYS GERD
Typical GERD presents with the classic symptoms of pyrosis (heartburn) or acid regurgitation, or both.
Although these symptoms are often thought to be specific for GERD, other causes of esophageal injury— eg, eosinophilic esophagitis, infection (Candida, cytomegalovirus, herpes simplex virus), pill-induced esophagitis, or radiation therapy—can produce similar symptoms. Other causes, including coronary artery disease, biliary colic, foregut malignancy, or peptic ulcer disease, should also be considered in patients with supposedly typical GERD. Life-threatening mimics of GERD, such as unstable angina, should be excluded if they are likely, before proceeding with evaluating for possible GERD. Therefore, the initial history and examination should focus on appropriate diagnosis, with careful delineation of symptom quality.
Alarm features for advanced pathology6–8 include involuntary weight loss, dysphagia, vomiting, evidence of gastrointestinal blood loss, anemia, chest pain, and an epigastric mass.7 Admittedly, these features are only mediocre for detecting or excluding gastric or esophageal cancer, with a sensitivity of 67% and a specificity 66%.9 Nevertheless, they should prompt an endoscopic examination. In patients who have alarm features but have not yet been treated for GERD, upper endoscopy can identify an abnormality in about 60% of patients.10–12
PPIs HAVE REPLACED ANTACIDS AND HISTAMINE-2 RECEPTOR ANTAGONISTS
When the symptoms suggest GERD and no alarm features are present, an initial trial of the following lifestyle changes is reasonable:
- Avoiding acidic or refluxogenic foods (coffee, alcohol, chocolate, peppermint, fatty foods, citrus foods)
- Avoiding certain medications (anticholinergics, estrogens, calcium-channel blockers, nitroglycerine, benzodiazepines)
- Losing weight
- Quitting smoking
- Raising the head of the bed
- Staying upright for 2 to 3 hours after meals.
For someone with mild symptoms, these changes pose minimal risk. Unfortunately, they are unlikely to provide adequate symptom control for most patients.13–17
Before PPIs were invented, drug therapy for GERD symptoms that did not resolve with lifestyle changes consisted of antacids and, later, histamine-2 receptor antagonists. When maximal therapy failed to control symptoms, fundoplication surgery was considered an appropriate next step.
PPIs substantially changed the management of GERD, suppressing acid secretion much better than histamine-2 receptor antagonists. Taken 30 minutes before breakfast, a single daily dose of a PPI normalizes esophageal acid exposure in 67% of patients.18 Adding a second dose 30 minutes before dinner raises the number to more than 90%.19
PPIs have consistently outperformed histamine-2 blockers in the healing of esophagitis and in improving heartburn symptoms and are now the first-line medical therapy for uncomplicated GERD.6,8,20–25
WHEN PPIs WORK, SURGERY OFFERS NO ADVANTAGE
The LOTUS trial (Long-Term Usage of Esomeprazole vs Surgery for Treatment of Chronic GERD) compared long-term drug therapy with surgery to maintain remission of symptoms in GERD.27 In this trial, 554 patients whose symptoms initially responded to the PPI esomeprazole (Nexium) were randomized to continue to receive esomeprazole (n = 266) or to undergo laparoscopic antireflux surgery (288 were randomly assigned, and 248 had the operation). Dose adjustment of the esomeprazole was allowed (20–40 mg/day). A total of 372 patients completed 5 years of follow-up (192 esomeprazole, 180 surgery).
Symptoms stayed in remission in 92% of the esomeprazole group and 85% of the surgery group (P = .048). However, the difference was no longer statistically significant after modeling the effects of study dropout. The rate of severe adverse events was similar in both groups: 24.1% with esomeprazole and 28.6% with surgery.
These findings indicate that if symptoms fully abate with medical therapy, surgery offers no advantage. In addition, patients who desire surgery in the hope of avoiding lifelong drug therapy should be made aware that drug therapy and reoperation are often necessary after surgery.28 In most cases, antireflux surgery is unnecessary for patients whose GERD fully responds to PPI therapy.
IF PPIs FAIL, FURTHER TESTING NEEDED
But many patients who take PPIs still have symptoms, even though these drugs suppress acid secretion and heal esophagitis. In fact, symptoms completely resolve in only about one-half of patients with erosive disease and one-third of those without erosive disease.21
Reasons for an incomplete symptomatic response to PPIs are various. Acid reflux can persist, but this accounts for only 10% of cases.29 About one-third of patients have persistent reflux that is weakly acidic, with a pH higher than 4.29. However, most patients with persistent typical GERD symptoms do not have significant, persistent reflux, or their symptoms are not related to reflux events. In these cases, an alternative cause of the refractory symptoms should be sought.
Further diagnostic testing is indicated when symptoms persist despite PPI therapy. Upper endoscopy will reveal an abnormality such as persistent erosive esophagitis, eosinophilic esophagitis, esophageal stricture, Barrett esophagus, or esophageal cancer in roughly 10% of patients in whom empiric therapy fails.10
Although patients with persistent symptoms have not been enrolled in many randomized controlled trials, a multivariate analysis showed that failure of medical therapy heralds a poor response to surgery.30 Data such as these have led most experts to discourage fundoplication for such patients now, unlike in the pre-PPI era.
pH and intraluminal impedance testing
However, this recommendation against surgery is not a hard-and-fast rule.
In patients with esophageal regurgitation, most will not achieve adequate relief of symptoms with PPI therapy alone.34 The therapeutic gain of PPI therapy vs placebo averaged just 17% in seven randomized, controlled trials, more than 20% less than the response rate for heartburn.34 This is likely because of structural abnormalities such as reduced lower esophageal sphincter pressure, hiatal hernia, or delayed gastric emptying. Antireflux surgery can correct these structural abnormalities or prevent them from causing so much trouble; however, the presence of true regurgitation should first be confirmed by MII testing. If regurgitation is confirmed, antireflux surgery is warranted, particularly in patients with nocturnal symptoms who may be at high risk of aspiration. With careful patient selection, regurgitation symptoms improve in about 90% after surgery.2
In patients with heartburn, if esophageal acid exposure continues to be abnormal on MII-pH testing, then an escalation of therapy may improve symptoms, particularly if symptoms occur during reflux or if they partially responded to PPI therapy. Options in this scenario include alteration or intensification of acid-suppressive therapy, treatment with baclofen (Lioresal), and antireflux surgery.18,35,36 In randomized controlled trials of patients whose symptoms partially responded to PPIs, antireflux surgery has performed similarly to PPIs in terms of improving typical GERD symptoms, particularly regurgitation.27,37–41 Although this scenario is a reasonable indication for antireflux surgery, recommendations should be made with appropriate restraint since it is not easily reversible, some patients experience complications, and up to one-third will have no therapeutic benefit.30
Nonacid reflux. In some cases, MII-pH testing during PPI therapy will reveal reflux of weakly acidic (pH > 4) or alkaline stomach contents, often called “nonacid reflux.”29 Nonacid reflux is often present in patients with esophagitis that persists despite PPI therapy, indicating that even weakly acidic stomach contents can injure the mucosa.42 Since intensifying the acid-suppressive therapy is unlikely to improve these symptoms, antireflux surgery may have a role.
In one study,43 nonacid reflux was well controlled by laparoscopic Nissen fundoplication, although 15 (48%) of 31 patients had persistent symptoms of GERD after surgery. No patient had a strong symptom correlation with postoperative reflux events, suggesting an alternative cause of the persistent symptoms. Therefore, antireflux surgery for nonacid reflux should be limited exclusively to patients with strong symptom correlation, and even then it should be considered with restraint, given the limited evidence for benefit and the potential for harm.
If testing is negative. In studies investigating the diagnostic yield of MII-pH testing, more than 50% of patients who had refractory symptoms had a negative MII-pH test.29 In such situations, when the symptoms are strongly correlated with reflux events, the patient is said to have “esophageal hypersensitivity.” A few small studies have suggested that such patients may benefit from surgery, but these data have not been replicated in randomized controlled trials.32
When the testing is negative and there is no correlation between the patient’s symptoms and reflux events, the patient is unlikely to benefit from antireflux surgery. Care of these patients is beyond the scope of this review.
SURGERY RARELY IMPROVES COUGH, ASTHMA, OR LARYNGITIS
GERD has been implicated as a cause of chronic cough, asthma, and laryngitis, although each of these has many potential causes.44–46 Despite these associations, the evidence for therapeutic benefit from antireflux therapy is weak.
PPI therapy shows no benefit over placebo for chronic cough of uncertain etiology, but some benefit if GERD is objectively demonstrated.47 Laryngitis resolved in just 15% of patients on esomeprazole vs 16% of patients on placebo after excluding patients with moderate to severe heartburn.48
In a large randomized controlled trial in patients with asthma, there was no overall improvement in peak flow for the PPI group vs the placebo group, although significant improvement occurred in patients with heartburn and nocturnal respiratory symptoms.46
Potent antisecretory therapy seems to improve extraesophageal symptoms when typical GERD symptoms are also present, but it otherwise has shown little evidence of benefit.
The evidence for a benefit from antireflux surgery in patients with extraesophageal GERD syndromes is even more limited. Although one systematic review49 found that cough and other laryngeal symptoms improved in 60% to 100% of patients with objective evidence of GERD who underwent fundoplication, virtually all of the studies were uncontrolled case series.49
The lone randomized controlled trial in the systematic review compared Nissen fundoplication with ranitidine (Zantac) or antacids only for patients with asthma and GERD, and found no significant difference in peak expiratory flow among the three groups after 2 years. However, asthma symptom scores improved in 75% of the surgical group, 9% of the medical group, and 4% of the control group.50
In a study that was not included in the prior systematic review, patients with laryngopharyngeal reflux unresponsive to aggressive acid suppression who subsequently underwent fundoplication fared no better than those who did not.51
Thus, based on the available data, antireflux surgery is only rarely indicated for extraesophageal symptoms, especially in patients who have no typical GERD symptoms or in patients whose symptoms are refractory to medical therapy.
SURGERY FOR EROSIVE ESOPHAGITIS OR BARRETT ESOPHAGUS IF PPI FAILS
Lifelong antireflux therapy is indicated for patients with severe erosive esophagitis or Barrett esophagus. Erosive esophagitis recurs in more than 80% within 12 months of discontinuing antisecretory therapy.52 Both Barrett esophagus and esophageal adenocarcinoma are strongly associated with GERD, and nearly 10% of patients with chronic reflux have Barrett esophagus.53,54 It is suspected that suppressing reflux reduces the rate of progression of Barrett esophagus to esophageal adenocarcinoma, but this remains to be proven.
Perhaps the strongest indication for surgery in the PPI era is for patients who have persistent symptoms and severe erosive esophagitis (Los Angeles grade C or D) despite high-dose PPI therapy. If other causes of persistent esophagitis have been ruled out, fundoplication can induce healing and improve symptoms.55,56 In these cases, surgery is done to induce remission of the disease when maximal medical therapy has been truly unsuccessful.
Randomized controlled trials suggest that medical and surgical therapies are equally effective for preventing the recurrence of erosive esophagitis or the progression of Barrett esophagus. In a study of 225 patients, at 7 years of follow-up, esophagitis had recurred in 10.4% of patients on omeprazole vs 11.8% of those who had undergone antireflux surgery.40 Similarly, open Nissen fundoplication was no different from drug therapy (histamine-2 receptor antagonist or PPI) for progression of Barrett esophagus over a median of 5 years.57 A meta-analysis with nearly 5,000 person-years each in the medical and surgical groups also found no significant difference in rates of cancer progression.58
Notably, symptoms such as dysphagia, flatulence, and the inability to burp occurred significantly more often in the surgical groups in these studies.
In view of these data, antireflux surgery has no significant advantage over medical therapy for maintaining healing of erosive esophagitis or preventing progression of Barrett esophagus. Thus, it should be reserved for patients who do not desire lifelong drug therapy, provided they understand that there is no therapeutic advantage for their esophagitis or for Barrett esophagus.
SPECIFIC INDICATIONS FOR ANTIREFLUX SURGERY
Now that we have PPIs, several situations remain in which surgery for GERD is either indicated or worth considering.
Antireflux surgery is clearly indicated for:
- Patients with erosive esophagitis that does not heal with maximal drug therapy
- Patients with volume regurgitation, particularly if it occurs at night or if there is evidence of aspiration
- Patients who require lifelong treatment for reflux but who have had a serious adverse event related to PPI therapy, such as refractory Clostridium difficile infection.
Antireflux surgery is also worth considering in patients who for personal reasons wish to avoid long-term or lifelong drug therapy.
Patients should be informed, however, that antireflux surgery has not been shown to be better than medical therapy for maintaining remission of symptoms, for preventing progression of Barrett esophagus, or for maintaining healing of erosive esophagitis. Medical therapy is still the first option for these patients.
Surgery may also be considered in patients with persistent symptoms who have a partial response to medical therapy, who show persistent acidic or weakly acidic reflux on MII-pH testing, and whose symptoms have been correlated with reflux events. Although surgery is not sure to improve their symptoms, benefit is more likely in this patient population compared with those without these characteristics.
Extraesophageal GERD
In patients suspected of having extraesophageal GERD, surgery should be considered if typical GERD symptoms are present and improve with PPI therapy, if the extraesophageal syndrome partially responds to PPI therapy, and if MII-pH testing demonstrates a correlation between symptoms and reflux. Surgery may have a stronger indication in this setting if the patient has nocturnal reflux or extraesophageal symptoms.
When is surgery not an option?
In general, surgery should not be considered in patients who do not have a partial response to PPI therapy or who do not have a strong symptom-reflux correlation on MII-pH testing. In all cases of failed medical therapy without persistent severe erosive disease, the threshold for opting for surgery should be high, given the uncertain response of these patients to surgery.
Peristaltic dysfunction is a relative but not an absolute contraindication to surgery.59
RISKS, BENEFITS OF SURGERY FOR GERD
The patient’s preference for surgery over drug therapy should always be balanced against the risks of surgery, including both short-term and long-term adverse events, to allow the patient to make an adequately informed decision (Table 2).2,26
Adverse events associated with PPI therapy are rare and in many cases the association is debatable.26 Nonetheless, long-term PPI therapy has been most strongly associated with an increased risk of C difficile infection and other enteric infections, although the absolute risk of these events remains low.
Complication rates after antireflux surgery depend on the surgeon’s experience and technique. Death is exceedingly rare. In most high-volume centers, the need to convert from laparoscopic to open fundoplication occurs in fewer than 2.4% of patients.2
Potential perioperative complications include perforation (4%), wound infection (3%), and pneumothorax (2%).2
Antireflux surgery is also associated with a significant risk of dysphagia, bloating, an inability to burp, and excessive flatulence, all of which can markedly impair the quality of life.
A major consideration is that fundoplication is generally irreversible. Reoperation rates have been reported to range from 0% to 15%.2 Furthermore, up to 50% of patients still need medical therapy after surgery.60,61 Of note, only about 25% of patients on medical therapy after surgery will actually have an abnormal pH study.61
MORE STUDY NEEDED
Future studies directly comparing medical and surgical therapy for carefully selected patients with extraesophageal manifestations of GERD and refractory symptoms should help further delineate outcome in this difficult group of patients.
Under development are new drugs that may inhibit transient relaxation of the lower esophageal sphincter, as well as minimally invasive procedures, which may alter the indications for surgery in coming years.36
Acknowledgment: The research for this article was supported in part by a grant from the National Institutes of Health (T32 DK07634).
For most patients with gastroesophageal reflux disease (GERD), a proton pump inhibitor (PPI) is the first choice for treatment.1 But some patients have symptoms that persist despite PPI therapy, some desire surgery despite successful PPI therapy, and some have persistent extraesophageal symptoms or other complications of reflux. For these patients, surgery is an option.2
In this article, we review the management of GERD and clarify the indications for antireflux surgery based on evidence of safety and efficacy.
GERD DEFINED: SYMPTOMS OR COMPLICATIONS
Defining the role of antireflux surgery is difficult, given the variety of presentations and the absence of a gold standard for diagnosing GERD. Most adults experience several episodes of physiologic reflux daily without symptoms.3 But a broad array of symptoms have been attributed to GERD, including chest pain, cough, and sore throat, and some conditions caused by acid reflux (eg, Barrett esophagus) can be asymptomatic.4,5
HEARTBURN ISN’T ALWAYS GERD
Typical GERD presents with the classic symptoms of pyrosis (heartburn) or acid regurgitation, or both.
Although these symptoms are often thought to be specific for GERD, other causes of esophageal injury— eg, eosinophilic esophagitis, infection (Candida, cytomegalovirus, herpes simplex virus), pill-induced esophagitis, or radiation therapy—can produce similar symptoms. Other causes, including coronary artery disease, biliary colic, foregut malignancy, or peptic ulcer disease, should also be considered in patients with supposedly typical GERD. Life-threatening mimics of GERD, such as unstable angina, should be excluded if they are likely, before proceeding with evaluating for possible GERD. Therefore, the initial history and examination should focus on appropriate diagnosis, with careful delineation of symptom quality.
Alarm features for advanced pathology6–8 include involuntary weight loss, dysphagia, vomiting, evidence of gastrointestinal blood loss, anemia, chest pain, and an epigastric mass.7 Admittedly, these features are only mediocre for detecting or excluding gastric or esophageal cancer, with a sensitivity of 67% and a specificity 66%.9 Nevertheless, they should prompt an endoscopic examination. In patients who have alarm features but have not yet been treated for GERD, upper endoscopy can identify an abnormality in about 60% of patients.10–12
PPIs HAVE REPLACED ANTACIDS AND HISTAMINE-2 RECEPTOR ANTAGONISTS
When the symptoms suggest GERD and no alarm features are present, an initial trial of the following lifestyle changes is reasonable:
- Avoiding acidic or refluxogenic foods (coffee, alcohol, chocolate, peppermint, fatty foods, citrus foods)
- Avoiding certain medications (anticholinergics, estrogens, calcium-channel blockers, nitroglycerine, benzodiazepines)
- Losing weight
- Quitting smoking
- Raising the head of the bed
- Staying upright for 2 to 3 hours after meals.
For someone with mild symptoms, these changes pose minimal risk. Unfortunately, they are unlikely to provide adequate symptom control for most patients.13–17
Before PPIs were invented, drug therapy for GERD symptoms that did not resolve with lifestyle changes consisted of antacids and, later, histamine-2 receptor antagonists. When maximal therapy failed to control symptoms, fundoplication surgery was considered an appropriate next step.
PPIs substantially changed the management of GERD, suppressing acid secretion much better than histamine-2 receptor antagonists. Taken 30 minutes before breakfast, a single daily dose of a PPI normalizes esophageal acid exposure in 67% of patients.18 Adding a second dose 30 minutes before dinner raises the number to more than 90%.19
PPIs have consistently outperformed histamine-2 blockers in the healing of esophagitis and in improving heartburn symptoms and are now the first-line medical therapy for uncomplicated GERD.6,8,20–25
WHEN PPIs WORK, SURGERY OFFERS NO ADVANTAGE
The LOTUS trial (Long-Term Usage of Esomeprazole vs Surgery for Treatment of Chronic GERD) compared long-term drug therapy with surgery to maintain remission of symptoms in GERD.27 In this trial, 554 patients whose symptoms initially responded to the PPI esomeprazole (Nexium) were randomized to continue to receive esomeprazole (n = 266) or to undergo laparoscopic antireflux surgery (288 were randomly assigned, and 248 had the operation). Dose adjustment of the esomeprazole was allowed (20–40 mg/day). A total of 372 patients completed 5 years of follow-up (192 esomeprazole, 180 surgery).
Symptoms stayed in remission in 92% of the esomeprazole group and 85% of the surgery group (P = .048). However, the difference was no longer statistically significant after modeling the effects of study dropout. The rate of severe adverse events was similar in both groups: 24.1% with esomeprazole and 28.6% with surgery.
These findings indicate that if symptoms fully abate with medical therapy, surgery offers no advantage. In addition, patients who desire surgery in the hope of avoiding lifelong drug therapy should be made aware that drug therapy and reoperation are often necessary after surgery.28 In most cases, antireflux surgery is unnecessary for patients whose GERD fully responds to PPI therapy.
IF PPIs FAIL, FURTHER TESTING NEEDED
But many patients who take PPIs still have symptoms, even though these drugs suppress acid secretion and heal esophagitis. In fact, symptoms completely resolve in only about one-half of patients with erosive disease and one-third of those without erosive disease.21
Reasons for an incomplete symptomatic response to PPIs are various. Acid reflux can persist, but this accounts for only 10% of cases.29 About one-third of patients have persistent reflux that is weakly acidic, with a pH higher than 4.29. However, most patients with persistent typical GERD symptoms do not have significant, persistent reflux, or their symptoms are not related to reflux events. In these cases, an alternative cause of the refractory symptoms should be sought.
Further diagnostic testing is indicated when symptoms persist despite PPI therapy. Upper endoscopy will reveal an abnormality such as persistent erosive esophagitis, eosinophilic esophagitis, esophageal stricture, Barrett esophagus, or esophageal cancer in roughly 10% of patients in whom empiric therapy fails.10
Although patients with persistent symptoms have not been enrolled in many randomized controlled trials, a multivariate analysis showed that failure of medical therapy heralds a poor response to surgery.30 Data such as these have led most experts to discourage fundoplication for such patients now, unlike in the pre-PPI era.
pH and intraluminal impedance testing
However, this recommendation against surgery is not a hard-and-fast rule.
In patients with esophageal regurgitation, most will not achieve adequate relief of symptoms with PPI therapy alone.34 The therapeutic gain of PPI therapy vs placebo averaged just 17% in seven randomized, controlled trials, more than 20% less than the response rate for heartburn.34 This is likely because of structural abnormalities such as reduced lower esophageal sphincter pressure, hiatal hernia, or delayed gastric emptying. Antireflux surgery can correct these structural abnormalities or prevent them from causing so much trouble; however, the presence of true regurgitation should first be confirmed by MII testing. If regurgitation is confirmed, antireflux surgery is warranted, particularly in patients with nocturnal symptoms who may be at high risk of aspiration. With careful patient selection, regurgitation symptoms improve in about 90% after surgery.2
In patients with heartburn, if esophageal acid exposure continues to be abnormal on MII-pH testing, then an escalation of therapy may improve symptoms, particularly if symptoms occur during reflux or if they partially responded to PPI therapy. Options in this scenario include alteration or intensification of acid-suppressive therapy, treatment with baclofen (Lioresal), and antireflux surgery.18,35,36 In randomized controlled trials of patients whose symptoms partially responded to PPIs, antireflux surgery has performed similarly to PPIs in terms of improving typical GERD symptoms, particularly regurgitation.27,37–41 Although this scenario is a reasonable indication for antireflux surgery, recommendations should be made with appropriate restraint since it is not easily reversible, some patients experience complications, and up to one-third will have no therapeutic benefit.30
Nonacid reflux. In some cases, MII-pH testing during PPI therapy will reveal reflux of weakly acidic (pH > 4) or alkaline stomach contents, often called “nonacid reflux.”29 Nonacid reflux is often present in patients with esophagitis that persists despite PPI therapy, indicating that even weakly acidic stomach contents can injure the mucosa.42 Since intensifying the acid-suppressive therapy is unlikely to improve these symptoms, antireflux surgery may have a role.
In one study,43 nonacid reflux was well controlled by laparoscopic Nissen fundoplication, although 15 (48%) of 31 patients had persistent symptoms of GERD after surgery. No patient had a strong symptom correlation with postoperative reflux events, suggesting an alternative cause of the persistent symptoms. Therefore, antireflux surgery for nonacid reflux should be limited exclusively to patients with strong symptom correlation, and even then it should be considered with restraint, given the limited evidence for benefit and the potential for harm.
If testing is negative. In studies investigating the diagnostic yield of MII-pH testing, more than 50% of patients who had refractory symptoms had a negative MII-pH test.29 In such situations, when the symptoms are strongly correlated with reflux events, the patient is said to have “esophageal hypersensitivity.” A few small studies have suggested that such patients may benefit from surgery, but these data have not been replicated in randomized controlled trials.32
When the testing is negative and there is no correlation between the patient’s symptoms and reflux events, the patient is unlikely to benefit from antireflux surgery. Care of these patients is beyond the scope of this review.
SURGERY RARELY IMPROVES COUGH, ASTHMA, OR LARYNGITIS
GERD has been implicated as a cause of chronic cough, asthma, and laryngitis, although each of these has many potential causes.44–46 Despite these associations, the evidence for therapeutic benefit from antireflux therapy is weak.
PPI therapy shows no benefit over placebo for chronic cough of uncertain etiology, but some benefit if GERD is objectively demonstrated.47 Laryngitis resolved in just 15% of patients on esomeprazole vs 16% of patients on placebo after excluding patients with moderate to severe heartburn.48
In a large randomized controlled trial in patients with asthma, there was no overall improvement in peak flow for the PPI group vs the placebo group, although significant improvement occurred in patients with heartburn and nocturnal respiratory symptoms.46
Potent antisecretory therapy seems to improve extraesophageal symptoms when typical GERD symptoms are also present, but it otherwise has shown little evidence of benefit.
The evidence for a benefit from antireflux surgery in patients with extraesophageal GERD syndromes is even more limited. Although one systematic review49 found that cough and other laryngeal symptoms improved in 60% to 100% of patients with objective evidence of GERD who underwent fundoplication, virtually all of the studies were uncontrolled case series.49
The lone randomized controlled trial in the systematic review compared Nissen fundoplication with ranitidine (Zantac) or antacids only for patients with asthma and GERD, and found no significant difference in peak expiratory flow among the three groups after 2 years. However, asthma symptom scores improved in 75% of the surgical group, 9% of the medical group, and 4% of the control group.50
In a study that was not included in the prior systematic review, patients with laryngopharyngeal reflux unresponsive to aggressive acid suppression who subsequently underwent fundoplication fared no better than those who did not.51
Thus, based on the available data, antireflux surgery is only rarely indicated for extraesophageal symptoms, especially in patients who have no typical GERD symptoms or in patients whose symptoms are refractory to medical therapy.
SURGERY FOR EROSIVE ESOPHAGITIS OR BARRETT ESOPHAGUS IF PPI FAILS
Lifelong antireflux therapy is indicated for patients with severe erosive esophagitis or Barrett esophagus. Erosive esophagitis recurs in more than 80% within 12 months of discontinuing antisecretory therapy.52 Both Barrett esophagus and esophageal adenocarcinoma are strongly associated with GERD, and nearly 10% of patients with chronic reflux have Barrett esophagus.53,54 It is suspected that suppressing reflux reduces the rate of progression of Barrett esophagus to esophageal adenocarcinoma, but this remains to be proven.
Perhaps the strongest indication for surgery in the PPI era is for patients who have persistent symptoms and severe erosive esophagitis (Los Angeles grade C or D) despite high-dose PPI therapy. If other causes of persistent esophagitis have been ruled out, fundoplication can induce healing and improve symptoms.55,56 In these cases, surgery is done to induce remission of the disease when maximal medical therapy has been truly unsuccessful.
Randomized controlled trials suggest that medical and surgical therapies are equally effective for preventing the recurrence of erosive esophagitis or the progression of Barrett esophagus. In a study of 225 patients, at 7 years of follow-up, esophagitis had recurred in 10.4% of patients on omeprazole vs 11.8% of those who had undergone antireflux surgery.40 Similarly, open Nissen fundoplication was no different from drug therapy (histamine-2 receptor antagonist or PPI) for progression of Barrett esophagus over a median of 5 years.57 A meta-analysis with nearly 5,000 person-years each in the medical and surgical groups also found no significant difference in rates of cancer progression.58
Notably, symptoms such as dysphagia, flatulence, and the inability to burp occurred significantly more often in the surgical groups in these studies.
In view of these data, antireflux surgery has no significant advantage over medical therapy for maintaining healing of erosive esophagitis or preventing progression of Barrett esophagus. Thus, it should be reserved for patients who do not desire lifelong drug therapy, provided they understand that there is no therapeutic advantage for their esophagitis or for Barrett esophagus.
SPECIFIC INDICATIONS FOR ANTIREFLUX SURGERY
Now that we have PPIs, several situations remain in which surgery for GERD is either indicated or worth considering.
Antireflux surgery is clearly indicated for:
- Patients with erosive esophagitis that does not heal with maximal drug therapy
- Patients with volume regurgitation, particularly if it occurs at night or if there is evidence of aspiration
- Patients who require lifelong treatment for reflux but who have had a serious adverse event related to PPI therapy, such as refractory Clostridium difficile infection.
Antireflux surgery is also worth considering in patients who for personal reasons wish to avoid long-term or lifelong drug therapy.
Patients should be informed, however, that antireflux surgery has not been shown to be better than medical therapy for maintaining remission of symptoms, for preventing progression of Barrett esophagus, or for maintaining healing of erosive esophagitis. Medical therapy is still the first option for these patients.
Surgery may also be considered in patients with persistent symptoms who have a partial response to medical therapy, who show persistent acidic or weakly acidic reflux on MII-pH testing, and whose symptoms have been correlated with reflux events. Although surgery is not sure to improve their symptoms, benefit is more likely in this patient population compared with those without these characteristics.
Extraesophageal GERD
In patients suspected of having extraesophageal GERD, surgery should be considered if typical GERD symptoms are present and improve with PPI therapy, if the extraesophageal syndrome partially responds to PPI therapy, and if MII-pH testing demonstrates a correlation between symptoms and reflux. Surgery may have a stronger indication in this setting if the patient has nocturnal reflux or extraesophageal symptoms.
When is surgery not an option?
In general, surgery should not be considered in patients who do not have a partial response to PPI therapy or who do not have a strong symptom-reflux correlation on MII-pH testing. In all cases of failed medical therapy without persistent severe erosive disease, the threshold for opting for surgery should be high, given the uncertain response of these patients to surgery.
Peristaltic dysfunction is a relative but not an absolute contraindication to surgery.59
RISKS, BENEFITS OF SURGERY FOR GERD
The patient’s preference for surgery over drug therapy should always be balanced against the risks of surgery, including both short-term and long-term adverse events, to allow the patient to make an adequately informed decision (Table 2).2,26
Adverse events associated with PPI therapy are rare and in many cases the association is debatable.26 Nonetheless, long-term PPI therapy has been most strongly associated with an increased risk of C difficile infection and other enteric infections, although the absolute risk of these events remains low.
Complication rates after antireflux surgery depend on the surgeon’s experience and technique. Death is exceedingly rare. In most high-volume centers, the need to convert from laparoscopic to open fundoplication occurs in fewer than 2.4% of patients.2
Potential perioperative complications include perforation (4%), wound infection (3%), and pneumothorax (2%).2
Antireflux surgery is also associated with a significant risk of dysphagia, bloating, an inability to burp, and excessive flatulence, all of which can markedly impair the quality of life.
A major consideration is that fundoplication is generally irreversible. Reoperation rates have been reported to range from 0% to 15%.2 Furthermore, up to 50% of patients still need medical therapy after surgery.60,61 Of note, only about 25% of patients on medical therapy after surgery will actually have an abnormal pH study.61
MORE STUDY NEEDED
Future studies directly comparing medical and surgical therapy for carefully selected patients with extraesophageal manifestations of GERD and refractory symptoms should help further delineate outcome in this difficult group of patients.
Under development are new drugs that may inhibit transient relaxation of the lower esophageal sphincter, as well as minimally invasive procedures, which may alter the indications for surgery in coming years.36
Acknowledgment: The research for this article was supported in part by a grant from the National Institutes of Health (T32 DK07634).
- Finks JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc 2006; 20:1698–1701.
- Stefanidis D, Hope WW, Kohn GP, Reardon PR, Richardson WS, Fanelli RD; SAGES Guidelines Committee. Guidelines for surgical treatment of gastroesophageal reflux disease. Surg Endosc 2010; 24:2647–2669.
- Richter JE. Typical and atypical presentations of gastroesophageal reflux disease. The role of esophageal testing in diagnosis and management. Gastroenterol Clin North Am 1996; 25:75–102.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Dickman R, Kim JL, Camargo L, et al. Correlation of gastroesophageal reflux disease symptoms characteristics with long-segment Barrett’s esophagus. Dis Esophagus 2006; 19:360–365.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Armstrong D, Marshall JK, Chiba N, et al; Canadian Association of Gastroenterology GERD Consensus Group. Canadian Consensus Conference on the management of gastroesophageal reflux disease in adults - update 2004. Can J Gastroenterol 2005; 19:15–35.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.
- Vakil N, Moayyedi P, Fennerty MB, Talley NJ. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: systematic review and meta-analysis. Gastroenterology 2006; 131:390–401.
- Poh CH, Gasiorowska A, Navarro-Rodriguez T, et al. Upper GI tract findings in patients with heartburn in whom proton pump inhibitor treatment failed versus those not receiving antireflux treatment. Gastrointest Endosc 2010; 71:28–34.
- Dickman R, Mattek N, Holub J, Peters D, Fass R. Prevalence of upper gastrointestinal tract findings in patients with noncardiac chest pain versus those with gastroesophageal reflux disease (GERD)-related symptoms: results from a national endoscopic database. Am J Gastroenterol 2007; 102:1173–1179.
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6–13.
- Fraser-Moodie CA, Norton B, Gornall C, Magnago S, Weale AR, Holmes GK. Weight loss has an independent beneficial effect on symptoms of gastro-oesophageal reflux in patients who are overweight. Scand J Gastroenterol 1999; 34:337–340.
- Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Bodymass index and symptoms of gastroesophageal reflux in women. N Engl J Med 2006; 354:2340–2348.
- Kjellin A, Ramel S, Rössner S, Thor K. Gastroesophageal reflux in obese patients is not reduced by weight reduction. Scand J Gastroenterol 1996; 31:1047–1051.
- Waring JP, Eastwood TF, Austin JM, Sanowski RA. The immediate effects of cessation of cigarette smoking on gastroesophageal reflux. Am J Gastroenterol 1989; 84:1076–1078.
- Pehl C, Waizenhoefer A, Wendl B, Schmidt T, Schepp W, Pfeiffer A. Effect of low and high fat meals on lower esophageal sphincter motility and gastroesophageal reflux in healthy subjects. Am J Gastroenterol 1999; 94:1192–1196.
- Bajbouj M, Becker V, Phillip V, Wilhelm D, Schmid RM, Meining A. High-dose esomeprazole for treatment of symptomatic refractory gastroesophageal reflux disease—a prospective pH-metry/impedance-controlled study. Digestion 2009; 80:112–118.
- Charbel S, Khandwala F, Vaezi MF. The role of esophageal pH monitoring in symptomatic patients on PPI therapy. Am J Gastroenterol 2005; 100:283–289.
- Khan M, Santana J, Donnellan C, Preston C, Moayyedi P. Medical treatments in the short term management of reflux oesophagitis. Cochrane Database Syst Rev 2007;CD003244.
- Dean BB, Gano AD, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2:656–664.
- Sabesin SM, Berlin RG, Humphries TJ, Bradstreet DC, Walton-Bowen KL, Zaidi S. Famotidine relieves symptoms of gastroesophageal reflux disease and heals erosions and ulcerations. Results of a multicenter, placebo-controlled, dose-ranging study. USA Merck Gastroesophageal Reflux Disease Study Group. Arch Intern Med 1991; 151:2394–2400.
- van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004;CD002095.
- Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology 1997; 112:1798–1810.
- Venables TL, Newland RD, Patel AC, Hole J, Wilcock C, Turbitt ML. Omeprazole 10 milligrams once daily, omeprazole 20 milligrams once daily, or ranitidine 150 milligrams twice daily, evaluated as initial therapy for the relief of symptoms of gastro-oesophageal reflux disease in general practice. Scand J Gastroenterol 1997; 32:965–973.
- Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med 2011; 78:39–49.
- Galmiche JP, Hatlebakk J, Attwood S, et al; LOTUS Trial Collaborators. Laparoscopic antireflux surgery vs esomeprazole treatment for chronic GERD: the LOTUS randomized clinical trial. JAMA 2011; 305:1969–1977.
- Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: followup of a randomized controlled trial. JAMA 2001; 285:2331–2338.
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55:1398–1402.
- Campos GM, Peters JH, DeMeester TR, et al. Multivariate analysis of factors predicting outcome after laparoscopic Nissen fundoplication. J Gastrointest Surg 1999; 3:292–300.
- Becker V, Bajbouj M, Waller K, Schmid RM, Meining A. Clinical trial: persistent gastro-oesophageal reflux symptoms despite standard therapy with proton pump inhibitors - a follow-up study of intraluminal-impedance guided therapy. Aliment Pharmacol Ther 2007; 26:1355–1360.
- Mainie I, Tutuian R, Agrawal A, Adams D, Castell DO. Combined multichannel intraluminal impedance-pH monitoring to select patients with persistent gastro-oesophageal reflux for laparoscopic Nissen fundoplication. Br J Surg 2006; 93:1483–1487.
- del Genio G, Tolone S, del Genio F, et al. Prospective assessment of patient selection for antireflux surgery by combined multichannel intraluminal impedance pH monitoring. J Gastrointest Surg 2008; 12:1491–1496.
- Kahrilas PJ, Howden CW, Hughes N. Response of regurgitation to proton pump inhibitor therapy in clinical trials of gastroesophageal reflux disease. Am J Gastroenterol 2011; 106:1419–1425.
- Koek GH, Sifrim D, Lerut T, Janssens J, Tack J. Effect of the GABA(B) agonist baclofen in patients with symptoms and duodeno-gastro-oesophageal reflux refractory to proton pump inhibitors. Gut 2003; 52:1397–1402.
- Boeckxstaens GE. Reflux inhibitors: a new approach for GERD? Curr Opin Pharmacol 2008; 8:685–689.
- Anvari M, Allen C, Marshall J, et al. A randomized controlled trial of laparoscopic nissen fundoplication versus proton pump inhibitors for treatment of patients with chronic gastroesophageal reflux disease: One-year follow-up. Surg Innov 2006; 13:238–249.
- Mahon D, Rhodes M, Decadt B, et al. Randomized clinical trial of laparoscopic Nissen fundoplication compared with proton-pump inhibitors for treatment of chronic gastro-oesophageal reflux. Br J Surg 2005; 92:695–699.
- Mehta S, Bennett J, Mahon D, Rhodes M. Prospective trial of laparoscopic nissen fundoplication versus proton pump inhibitor therapy for gastroesophageal reflux disease: Seven-year follow-up. J Gastrointest Surg 2006; 10:1312–1316.
- Lundell L, Miettinen P, Myrvold HE, et al; Nordic GORD Study Group. Seven-year follow-up of a randomized clinical trial comparing proton-pump inhibition with surgical therapy for reflux oesophagitis. Br J Surg 2007; 94:198–203.
- Lundell L, Attwood S, Ell C, et al; LOTUS trial collaborators. Comparing laparoscopic antireflux surgery with esomeprazole in the management of patients with chronic gastro-oesophageal reflux disease: a 3-year interim analysis of the LOTUS trial. Gut 2008; 57:1207–1213.
- Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 2011; 33:601–606.
- Broeders JA, Bredenoord AJ, Hazebroek EJ, Broeders IA, Gooszen HG, Smout AJ. Effects of anti-reflux surgery on weakly acidic reflux and belching. Gut 2011; 60:435–441.
- American Gastroenterological Association medical position statement: guidelines on the use of esophageal pH recording. Gastroenterology 1996; 110:1981.
- el-Serag HB, Sonnenberg A. Comorbid occurrence of laryngeal or pulmonary disease with esophagitis in United States military veterans. Gastroenterology 1997; 113:755–760.
- Kiljander TO, Laitinen JO. The prevalence of gastroesophageal reflux disease in adult asthmatics. Chest 2004; 126:1490–1494.
- Chang AB, Lasserson TJ, Kiljander TO, Connor FL, Gaffney JT, Garske LA. Systematic review and meta-analysis of randomised controlled trials of gastro-oesophageal reflux interventions for chronic cough associated with gastro-oesophageal reflux. BMJ 2006; 332:11–17.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Iqbal M, Batch AJ, Spychal RT, Cooper BT. Outcome of surgical fundoplication for extraesophageal (atypical) manifestations of gastroesophageal reflux disease in adults: a systematic review. J Laparoendosc Adv Surg Tech A 2008; 18:789–796.
- Sontag SJ, O’Connell S, Khandelwal S, et al. Asthmatics with gastroesophageal reflux: long term results of a randomized trial of medical and surgical antireflux therapies. Am J Gastroenterol 2003; 98:987–999.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- Johnson DA, Benjamin SB, Vakil NB, et al. Esomeprazole once daily for 6 months is effective therapy for maintaining healed erosive esophagitis and for controlling gastroesophageal reflux disease symptoms: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Gastroenterol 2001; 96:27–34.
- Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987; 92:118–124.
- Westhoff B, Brotze S, Weston A, et al. The frequency of Barrett’s esophagus in high-risk patients with chronic GERD. Gastrointest Endosc 2005; 61:226–231.
- Rosenthal R, Peterli R, Guenin MO, von Flüe M, Ackermann C. Laparoscopic antireflux surgery: long-term outcomes and quality of life. J Laparoendosc Adv Surg Tech A 2006; 16:557–561.
- Broeders JA, Draaisma WA, Bredenoord AJ, Smout AJ, Broeders IA, Gooszen HG. Long-term outcome of Nissen fundoplication in non-erosive and erosive gastro-oesophageal reflux disease. Br J Surg 2010; 97:845–352.
- Parrilla P, Martínez de Haro LF, Ortiz A, et al. Long-term results of a randomized prospective study comparing medical and surgical treatment of Barrett’s esophagus. Ann Surg 2003; 237:291–298.
- Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol 2003; 98:2390–2394.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and antireflux medication use after antireflux surgery. Clin Gastroenterol Hepatol 2006; 4:299–305.
- Lord RV, Kaminski A, Oberg S, et al. Absence of gastroesophageal reflux disease in a majority of patients taking acid suppression medications after Nissen fundoplication. J Gastrointest Surg 2002; 6:3–9.
- Finks JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc 2006; 20:1698–1701.
- Stefanidis D, Hope WW, Kohn GP, Reardon PR, Richardson WS, Fanelli RD; SAGES Guidelines Committee. Guidelines for surgical treatment of gastroesophageal reflux disease. Surg Endosc 2010; 24:2647–2669.
- Richter JE. Typical and atypical presentations of gastroesophageal reflux disease. The role of esophageal testing in diagnosis and management. Gastroenterol Clin North Am 1996; 25:75–102.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Dickman R, Kim JL, Camargo L, et al. Correlation of gastroesophageal reflux disease symptoms characteristics with long-segment Barrett’s esophagus. Dis Esophagus 2006; 19:360–365.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Armstrong D, Marshall JK, Chiba N, et al; Canadian Association of Gastroenterology GERD Consensus Group. Canadian Consensus Conference on the management of gastroesophageal reflux disease in adults - update 2004. Can J Gastroenterol 2005; 19:15–35.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.
- Vakil N, Moayyedi P, Fennerty MB, Talley NJ. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: systematic review and meta-analysis. Gastroenterology 2006; 131:390–401.
- Poh CH, Gasiorowska A, Navarro-Rodriguez T, et al. Upper GI tract findings in patients with heartburn in whom proton pump inhibitor treatment failed versus those not receiving antireflux treatment. Gastrointest Endosc 2010; 71:28–34.
- Dickman R, Mattek N, Holub J, Peters D, Fass R. Prevalence of upper gastrointestinal tract findings in patients with noncardiac chest pain versus those with gastroesophageal reflux disease (GERD)-related symptoms: results from a national endoscopic database. Am J Gastroenterol 2007; 102:1173–1179.
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6–13.
- Fraser-Moodie CA, Norton B, Gornall C, Magnago S, Weale AR, Holmes GK. Weight loss has an independent beneficial effect on symptoms of gastro-oesophageal reflux in patients who are overweight. Scand J Gastroenterol 1999; 34:337–340.
- Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Bodymass index and symptoms of gastroesophageal reflux in women. N Engl J Med 2006; 354:2340–2348.
- Kjellin A, Ramel S, Rössner S, Thor K. Gastroesophageal reflux in obese patients is not reduced by weight reduction. Scand J Gastroenterol 1996; 31:1047–1051.
- Waring JP, Eastwood TF, Austin JM, Sanowski RA. The immediate effects of cessation of cigarette smoking on gastroesophageal reflux. Am J Gastroenterol 1989; 84:1076–1078.
- Pehl C, Waizenhoefer A, Wendl B, Schmidt T, Schepp W, Pfeiffer A. Effect of low and high fat meals on lower esophageal sphincter motility and gastroesophageal reflux in healthy subjects. Am J Gastroenterol 1999; 94:1192–1196.
- Bajbouj M, Becker V, Phillip V, Wilhelm D, Schmid RM, Meining A. High-dose esomeprazole for treatment of symptomatic refractory gastroesophageal reflux disease—a prospective pH-metry/impedance-controlled study. Digestion 2009; 80:112–118.
- Charbel S, Khandwala F, Vaezi MF. The role of esophageal pH monitoring in symptomatic patients on PPI therapy. Am J Gastroenterol 2005; 100:283–289.
- Khan M, Santana J, Donnellan C, Preston C, Moayyedi P. Medical treatments in the short term management of reflux oesophagitis. Cochrane Database Syst Rev 2007;CD003244.
- Dean BB, Gano AD, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2:656–664.
- Sabesin SM, Berlin RG, Humphries TJ, Bradstreet DC, Walton-Bowen KL, Zaidi S. Famotidine relieves symptoms of gastroesophageal reflux disease and heals erosions and ulcerations. Results of a multicenter, placebo-controlled, dose-ranging study. USA Merck Gastroesophageal Reflux Disease Study Group. Arch Intern Med 1991; 151:2394–2400.
- van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004;CD002095.
- Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology 1997; 112:1798–1810.
- Venables TL, Newland RD, Patel AC, Hole J, Wilcock C, Turbitt ML. Omeprazole 10 milligrams once daily, omeprazole 20 milligrams once daily, or ranitidine 150 milligrams twice daily, evaluated as initial therapy for the relief of symptoms of gastro-oesophageal reflux disease in general practice. Scand J Gastroenterol 1997; 32:965–973.
- Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med 2011; 78:39–49.
- Galmiche JP, Hatlebakk J, Attwood S, et al; LOTUS Trial Collaborators. Laparoscopic antireflux surgery vs esomeprazole treatment for chronic GERD: the LOTUS randomized clinical trial. JAMA 2011; 305:1969–1977.
- Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: followup of a randomized controlled trial. JAMA 2001; 285:2331–2338.
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55:1398–1402.
- Campos GM, Peters JH, DeMeester TR, et al. Multivariate analysis of factors predicting outcome after laparoscopic Nissen fundoplication. J Gastrointest Surg 1999; 3:292–300.
- Becker V, Bajbouj M, Waller K, Schmid RM, Meining A. Clinical trial: persistent gastro-oesophageal reflux symptoms despite standard therapy with proton pump inhibitors - a follow-up study of intraluminal-impedance guided therapy. Aliment Pharmacol Ther 2007; 26:1355–1360.
- Mainie I, Tutuian R, Agrawal A, Adams D, Castell DO. Combined multichannel intraluminal impedance-pH monitoring to select patients with persistent gastro-oesophageal reflux for laparoscopic Nissen fundoplication. Br J Surg 2006; 93:1483–1487.
- del Genio G, Tolone S, del Genio F, et al. Prospective assessment of patient selection for antireflux surgery by combined multichannel intraluminal impedance pH monitoring. J Gastrointest Surg 2008; 12:1491–1496.
- Kahrilas PJ, Howden CW, Hughes N. Response of regurgitation to proton pump inhibitor therapy in clinical trials of gastroesophageal reflux disease. Am J Gastroenterol 2011; 106:1419–1425.
- Koek GH, Sifrim D, Lerut T, Janssens J, Tack J. Effect of the GABA(B) agonist baclofen in patients with symptoms and duodeno-gastro-oesophageal reflux refractory to proton pump inhibitors. Gut 2003; 52:1397–1402.
- Boeckxstaens GE. Reflux inhibitors: a new approach for GERD? Curr Opin Pharmacol 2008; 8:685–689.
- Anvari M, Allen C, Marshall J, et al. A randomized controlled trial of laparoscopic nissen fundoplication versus proton pump inhibitors for treatment of patients with chronic gastroesophageal reflux disease: One-year follow-up. Surg Innov 2006; 13:238–249.
- Mahon D, Rhodes M, Decadt B, et al. Randomized clinical trial of laparoscopic Nissen fundoplication compared with proton-pump inhibitors for treatment of chronic gastro-oesophageal reflux. Br J Surg 2005; 92:695–699.
- Mehta S, Bennett J, Mahon D, Rhodes M. Prospective trial of laparoscopic nissen fundoplication versus proton pump inhibitor therapy for gastroesophageal reflux disease: Seven-year follow-up. J Gastrointest Surg 2006; 10:1312–1316.
- Lundell L, Miettinen P, Myrvold HE, et al; Nordic GORD Study Group. Seven-year follow-up of a randomized clinical trial comparing proton-pump inhibition with surgical therapy for reflux oesophagitis. Br J Surg 2007; 94:198–203.
- Lundell L, Attwood S, Ell C, et al; LOTUS trial collaborators. Comparing laparoscopic antireflux surgery with esomeprazole in the management of patients with chronic gastro-oesophageal reflux disease: a 3-year interim analysis of the LOTUS trial. Gut 2008; 57:1207–1213.
- Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 2011; 33:601–606.
- Broeders JA, Bredenoord AJ, Hazebroek EJ, Broeders IA, Gooszen HG, Smout AJ. Effects of anti-reflux surgery on weakly acidic reflux and belching. Gut 2011; 60:435–441.
- American Gastroenterological Association medical position statement: guidelines on the use of esophageal pH recording. Gastroenterology 1996; 110:1981.
- el-Serag HB, Sonnenberg A. Comorbid occurrence of laryngeal or pulmonary disease with esophagitis in United States military veterans. Gastroenterology 1997; 113:755–760.
- Kiljander TO, Laitinen JO. The prevalence of gastroesophageal reflux disease in adult asthmatics. Chest 2004; 126:1490–1494.
- Chang AB, Lasserson TJ, Kiljander TO, Connor FL, Gaffney JT, Garske LA. Systematic review and meta-analysis of randomised controlled trials of gastro-oesophageal reflux interventions for chronic cough associated with gastro-oesophageal reflux. BMJ 2006; 332:11–17.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Iqbal M, Batch AJ, Spychal RT, Cooper BT. Outcome of surgical fundoplication for extraesophageal (atypical) manifestations of gastroesophageal reflux disease in adults: a systematic review. J Laparoendosc Adv Surg Tech A 2008; 18:789–796.
- Sontag SJ, O’Connell S, Khandelwal S, et al. Asthmatics with gastroesophageal reflux: long term results of a randomized trial of medical and surgical antireflux therapies. Am J Gastroenterol 2003; 98:987–999.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- Johnson DA, Benjamin SB, Vakil NB, et al. Esomeprazole once daily for 6 months is effective therapy for maintaining healed erosive esophagitis and for controlling gastroesophageal reflux disease symptoms: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Gastroenterol 2001; 96:27–34.
- Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987; 92:118–124.
- Westhoff B, Brotze S, Weston A, et al. The frequency of Barrett’s esophagus in high-risk patients with chronic GERD. Gastrointest Endosc 2005; 61:226–231.
- Rosenthal R, Peterli R, Guenin MO, von Flüe M, Ackermann C. Laparoscopic antireflux surgery: long-term outcomes and quality of life. J Laparoendosc Adv Surg Tech A 2006; 16:557–561.
- Broeders JA, Draaisma WA, Bredenoord AJ, Smout AJ, Broeders IA, Gooszen HG. Long-term outcome of Nissen fundoplication in non-erosive and erosive gastro-oesophageal reflux disease. Br J Surg 2010; 97:845–352.
- Parrilla P, Martínez de Haro LF, Ortiz A, et al. Long-term results of a randomized prospective study comparing medical and surgical treatment of Barrett’s esophagus. Ann Surg 2003; 237:291–298.
- Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol 2003; 98:2390–2394.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and antireflux medication use after antireflux surgery. Clin Gastroenterol Hepatol 2006; 4:299–305.
- Lord RV, Kaminski A, Oberg S, et al. Absence of gastroesophageal reflux disease in a majority of patients taking acid suppression medications after Nissen fundoplication. J Gastrointest Surg 2002; 6:3–9.
KEY POINTS
- If a PPI in twice-daily doses fails to relieve GERD symptoms, a pH study combined with multichannel intraluminal impedance testing can help in deciding whether to try surgery.
- Antireflux surgery can be considered for erosive esophagitis that does not resolve with drug therapy, for volume regurgitation (particularly if it occurs at night or if there is a risk of aspiration), and for patients who need lifelong treatment for reflux but have had a serious adverse event related to PPI therapy.
- Studies are needed to directly compare medical and surgical therapy in patients with extraesophageal manifestations of GERD and refractory symptoms, a difficult group of patients.
- Drugs that inhibit transient relaxation of the lower esophageal sphincter are under investigation, as are minimally invasive procedures to manipulate the physical barrier to reflux.
Purpuric lesion on the elbow
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
In reply: Essential tremor, beta-blockers, calcium channel blockers
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
Essential tremor, beta-blockers, and calcium channel blockers
To the Editor: In their thorough review of essential tremor,1Drs. Abboud, Ahmed, and Fernandez make a statement that needs clarification. In their list of absolute contraindications to propranolol (Inderal), the authors include “concurrent use of a calcium channel blocker.” This warning applies only to the nondihydropyridine calcium channel blockers, which are diltiazem (Cardizem) and verapamil (Calan). These two medications slow the heart rate and generally should not be combined with beta-blockers such as propranolol unless the patient requires this combination to control tachycardia. Most calcium channel blockers are dihydropyridines, which include amlodipine (Norvasc), nifedipine (Procardia), felodipine (Plendil), nisoldipine (Sular), isradipine (DynaCirc CR), and nicardipine (Cardene). These agents do not slow the heart rate significantly and therefore can be used freely in combination with propranolol. Of course, the dose of the calcium channel blocker may need to be decreased because of the antihypertensive effect of propranolol.
- Abboud H, Ahmed A, Fernandez HH. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011; 78:821–828.
To the Editor: In their thorough review of essential tremor,1Drs. Abboud, Ahmed, and Fernandez make a statement that needs clarification. In their list of absolute contraindications to propranolol (Inderal), the authors include “concurrent use of a calcium channel blocker.” This warning applies only to the nondihydropyridine calcium channel blockers, which are diltiazem (Cardizem) and verapamil (Calan). These two medications slow the heart rate and generally should not be combined with beta-blockers such as propranolol unless the patient requires this combination to control tachycardia. Most calcium channel blockers are dihydropyridines, which include amlodipine (Norvasc), nifedipine (Procardia), felodipine (Plendil), nisoldipine (Sular), isradipine (DynaCirc CR), and nicardipine (Cardene). These agents do not slow the heart rate significantly and therefore can be used freely in combination with propranolol. Of course, the dose of the calcium channel blocker may need to be decreased because of the antihypertensive effect of propranolol.
To the Editor: In their thorough review of essential tremor,1Drs. Abboud, Ahmed, and Fernandez make a statement that needs clarification. In their list of absolute contraindications to propranolol (Inderal), the authors include “concurrent use of a calcium channel blocker.” This warning applies only to the nondihydropyridine calcium channel blockers, which are diltiazem (Cardizem) and verapamil (Calan). These two medications slow the heart rate and generally should not be combined with beta-blockers such as propranolol unless the patient requires this combination to control tachycardia. Most calcium channel blockers are dihydropyridines, which include amlodipine (Norvasc), nifedipine (Procardia), felodipine (Plendil), nisoldipine (Sular), isradipine (DynaCirc CR), and nicardipine (Cardene). These agents do not slow the heart rate significantly and therefore can be used freely in combination with propranolol. Of course, the dose of the calcium channel blocker may need to be decreased because of the antihypertensive effect of propranolol.
- Abboud H, Ahmed A, Fernandez HH. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011; 78:821–828.
- Abboud H, Ahmed A, Fernandez HH. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011; 78:821–828.