User login
Bowel-Associated Dermatosis-Arthritis Syndrome in a Patient With Crohn Disease
To the Editor:
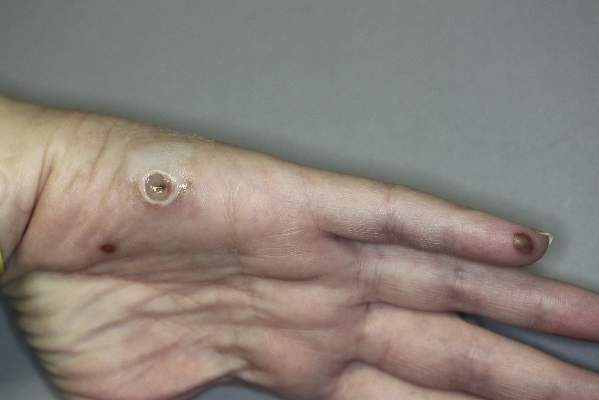
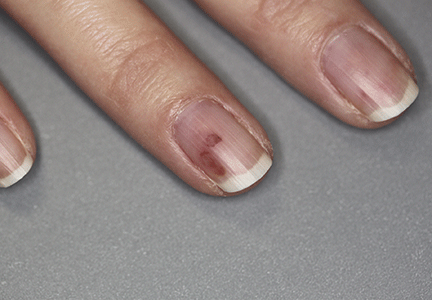
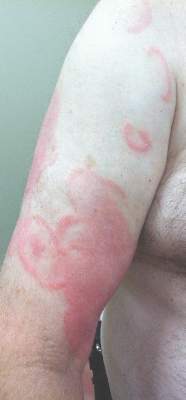
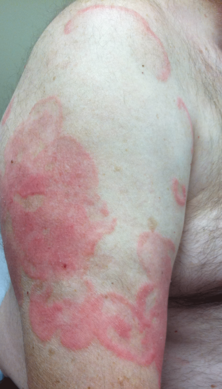
A 42-year-old woman with Crohn disease of 10 years’ duration presented to the clinic with a chief concern of nonpruritic pustular lesions on the bilateral arms. Physical examination revealed several pustules on the arms with secondary excoriation. She also had a warm tender nodule on the left upper shin and subungual hemorrhages under the fingernails (Figure 1). The patient had previously undergone infliximab therapy, which was discontinued 10 months prior to presentation in anticipation of a partial colectomy and temporary ileostomy that was performed 8 months prior to presentation. She recently had developed bilateral, radiating, sharp lower extremity pain extending from the feet to the hips over the last 2 weeks and swelling of the bilateral legs that impaired her ability to ambulate. Additionally, she had recently traveled to Colorado and a Lyme disease workup was initiated at an outside hospital in Colorado; however, the results were pending. The outside hospital also performed a spinal tap that was negative. At our clinic, biopsies were performed on the shin nodule and a right palmar pustule (Figure 2). There was clinical suspicion of erythema nodosum and subcorneal pustular dermatosis or a vesiculopustular skin manifestation of the patient’s Crohn disease. The patient was switched from generic doxycycline to a brand name variant 150 mg every night at bedtime for 2 weeks. She subsequently was admitted to the inpatient rheumatology service for a complete systemic workup.

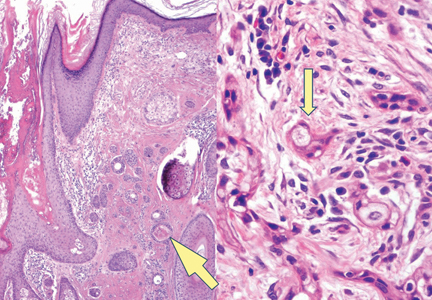
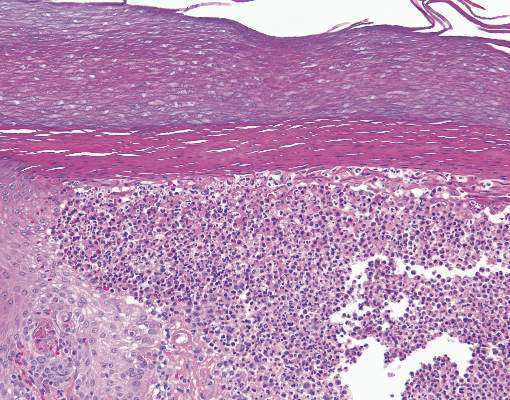
The punch biopsy of the left upper shin demonstrated operative hemorrhage and periadnexal lymphocytic inflammation without evidence of fungal or bacterial elements by Gram or Gomori methenamine-silver stain. Clinically, the diagnosis was most likely erythema nodosum, though insufficient hypodermis was present to make the diagnosis with pathology. The shave biopsy of the right medial palm was nondiagnostic but showed a transected pustule with no bacterial or fungal elements by Gram or Gomori methenamine-silver stain (Figure 3). Given the clinical context, the likely pathologic diagnosis was vesiculopustular Crohn disease.
Our patient was started on an empiric steroid trial with rapid improvement of the arthralgia and rash. The presumed diagnosis was a Crohn disease flare and the patient was discharged on an 8-week steroid taper. Three weeks later at a follow-up appointment, the patient’s skin lesions had nearly resolved. The swelling of the legs and feet had substantially decreased, but the joint pain, primarily in the ankles, persisted.
Routine laboratory studies showed a hemoglobin level of 11.6 g/dL (reference range, 12–15 g/dL), white blood cell count of 9.1 K/μL (reference range, 4.5–11.0 K/μL), C-reactive protein level of 20.15 mg/dL (reference range, <1.0 mg/dL), and an antinuclear antibody titer of 160 (<80). Serology for Lyme disease was negative. Serum chemistries were all within reference range and an echocardiogram was normal.
Up to one-third of patients with inflammatory bowel disease (IBD) experience extraintestinal manifestations of their condition. Of these patients, nearly one-third will develop cutaneous manifestations.1 The most common skin diseases associated with IBD are pyoderma gangrenosum and erythema nodosum.2 The differential diagnoses considered in this unique case included early pyoderma gangrenosum, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), and vesiculopustular Crohn disease. Vesiculopustular Crohn disease is a rare component of IBD and also can be present in bowel-associated dermatosis-arthritis syndrome (BADAS). In BADAS, symptoms often include arthritis and systemic symptoms such as fever and malaise. The skin manifestations typically involve the arms and trunk. It often is seen after intestinal bypass surgery but also can be present in patients with gastrointestinal diseases such as IBD.3 Due to its early association with bypass surgery, BADAS previously was referred to as bowel bypass syndrome but has since been seen in relation to other intestinal surgeries and IBD.4 Patients with BADAS often present with episodes of fever, fatigue, and malaise, in addition to arthralgia and cutaneous eruptions. Cases of BADAS related to IBD instead of bypass surgery often can be less severe in nature. Unlike many of these previously reported cases, our patient’s joint pain primarily was in the knees and ankles, whereas typical cases of BADAS cause upper extremity (ie, shoulder, elbow) arthralgia. Our patient occasionally experienced upper extremity pain, but it was less frequent and less severe than the knee and ankle pain. The vesiculopustular lesions in BADAS usually begin as 3- to 10-mm painful macules that then develop into aseptic pustular lesions. These manifestations arise on the upper arms and chest or trunk and can be accompanied by erythema nodosum on the legs.4
It has been hypothesized that BADAS occurs as an immune reaction to bacterial overgrowth in the bowel from IBD, infection, or surgery. The reaction is in response to a bacterial antigen and manifests cutaneously.5 This same pathogenesis is thought to cause various other manifestations of Crohn disease such as erythema nodosum. Bacteria that incite this immune response include Bacteroides fragilis, Escherichia coli, and Streptococcus.
Resolution of both vesiculopustular Crohn disease and of BADAS often occurs with treatment of the underlying IBD but also can be improved with steroids and antibiotics. However, response to antibiotics often is variable.5,6 The mainstay for treatment remains steroids and management of underlying bowel disease.
Bowel-associated dermatosis-arthritis syndrome often is overlooked when compiling differential diagnoses for neutrophilic dermatoses but should be considered in patients with bowel disease or recent surgery. Because the syndrome can be recurrent, early diagnosis can help to prevent and treat relapsing courses of BADAS.
- Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J. 2005;81:580-585.
- Havemann BD. A pustular skin rash in a woman with 2 weeks of diarrhea. MedGenMed. 2005;7:11.
- Bolognia JL, Jorizzo J, Rapini RP. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2008.
- Huang B, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13.
- Truchuelo MT, Alcántara J, Vano-Galván S, et al. Bowel associated dermatosis-arthritis syndrome: another cutaneous manifestation of inflammatory intestinal disease. Int J Dermatol. 2013;52:1596-1598.
- Ashok D, Kiely P. Bowel associated dermatosis-arthritis syndrome: a case report. J Med Case Rep. 2007;1:81.
To the Editor:
A 42-year-old woman with Crohn disease of 10 years’ duration presented to the clinic with a chief concern of nonpruritic pustular lesions on the bilateral arms. Physical examination revealed several pustules on the arms with secondary excoriation. She also had a warm tender nodule on the left upper shin and subungual hemorrhages under the fingernails (Figure 1). The patient had previously undergone infliximab therapy, which was discontinued 10 months prior to presentation in anticipation of a partial colectomy and temporary ileostomy that was performed 8 months prior to presentation. She recently had developed bilateral, radiating, sharp lower extremity pain extending from the feet to the hips over the last 2 weeks and swelling of the bilateral legs that impaired her ability to ambulate. Additionally, she had recently traveled to Colorado and a Lyme disease workup was initiated at an outside hospital in Colorado; however, the results were pending. The outside hospital also performed a spinal tap that was negative. At our clinic, biopsies were performed on the shin nodule and a right palmar pustule (Figure 2). There was clinical suspicion of erythema nodosum and subcorneal pustular dermatosis or a vesiculopustular skin manifestation of the patient’s Crohn disease. The patient was switched from generic doxycycline to a brand name variant 150 mg every night at bedtime for 2 weeks. She subsequently was admitted to the inpatient rheumatology service for a complete systemic workup.
The punch biopsy of the left upper shin demonstrated operative hemorrhage and periadnexal lymphocytic inflammation without evidence of fungal or bacterial elements by Gram or Gomori methenamine-silver stain. Clinically, the diagnosis was most likely erythema nodosum, though insufficient hypodermis was present to make the diagnosis with pathology. The shave biopsy of the right medial palm was nondiagnostic but showed a transected pustule with no bacterial or fungal elements by Gram or Gomori methenamine-silver stain (Figure 3). Given the clinical context, the likely pathologic diagnosis was vesiculopustular Crohn disease.
Our patient was started on an empiric steroid trial with rapid improvement of the arthralgia and rash. The presumed diagnosis was a Crohn disease flare and the patient was discharged on an 8-week steroid taper. Three weeks later at a follow-up appointment, the patient’s skin lesions had nearly resolved. The swelling of the legs and feet had substantially decreased, but the joint pain, primarily in the ankles, persisted.
Routine laboratory studies showed a hemoglobin level of 11.6 g/dL (reference range, 12–15 g/dL), white blood cell count of 9.1 K/μL (reference range, 4.5–11.0 K/μL), C-reactive protein level of 20.15 mg/dL (reference range, <1.0 mg/dL), and an antinuclear antibody titer of 160 (<80). Serology for Lyme disease was negative. Serum chemistries were all within reference range and an echocardiogram was normal.
Up to one-third of patients with inflammatory bowel disease (IBD) experience extraintestinal manifestations of their condition. Of these patients, nearly one-third will develop cutaneous manifestations.1 The most common skin diseases associated with IBD are pyoderma gangrenosum and erythema nodosum.2 The differential diagnoses considered in this unique case included early pyoderma gangrenosum, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), and vesiculopustular Crohn disease. Vesiculopustular Crohn disease is a rare component of IBD and also can be present in bowel-associated dermatosis-arthritis syndrome (BADAS). In BADAS, symptoms often include arthritis and systemic symptoms such as fever and malaise. The skin manifestations typically involve the arms and trunk. It often is seen after intestinal bypass surgery but also can be present in patients with gastrointestinal diseases such as IBD.3 Due to its early association with bypass surgery, BADAS previously was referred to as bowel bypass syndrome but has since been seen in relation to other intestinal surgeries and IBD.4 Patients with BADAS often present with episodes of fever, fatigue, and malaise, in addition to arthralgia and cutaneous eruptions. Cases of BADAS related to IBD instead of bypass surgery often can be less severe in nature. Unlike many of these previously reported cases, our patient’s joint pain primarily was in the knees and ankles, whereas typical cases of BADAS cause upper extremity (ie, shoulder, elbow) arthralgia. Our patient occasionally experienced upper extremity pain, but it was less frequent and less severe than the knee and ankle pain. The vesiculopustular lesions in BADAS usually begin as 3- to 10-mm painful macules that then develop into aseptic pustular lesions. These manifestations arise on the upper arms and chest or trunk and can be accompanied by erythema nodosum on the legs.4
It has been hypothesized that BADAS occurs as an immune reaction to bacterial overgrowth in the bowel from IBD, infection, or surgery. The reaction is in response to a bacterial antigen and manifests cutaneously.5 This same pathogenesis is thought to cause various other manifestations of Crohn disease such as erythema nodosum. Bacteria that incite this immune response include Bacteroides fragilis, Escherichia coli, and Streptococcus.
Resolution of both vesiculopustular Crohn disease and of BADAS often occurs with treatment of the underlying IBD but also can be improved with steroids and antibiotics. However, response to antibiotics often is variable.5,6 The mainstay for treatment remains steroids and management of underlying bowel disease.
Bowel-associated dermatosis-arthritis syndrome often is overlooked when compiling differential diagnoses for neutrophilic dermatoses but should be considered in patients with bowel disease or recent surgery. Because the syndrome can be recurrent, early diagnosis can help to prevent and treat relapsing courses of BADAS.
To the Editor:
A 42-year-old woman with Crohn disease of 10 years’ duration presented to the clinic with a chief concern of nonpruritic pustular lesions on the bilateral arms. Physical examination revealed several pustules on the arms with secondary excoriation. She also had a warm tender nodule on the left upper shin and subungual hemorrhages under the fingernails (Figure 1). The patient had previously undergone infliximab therapy, which was discontinued 10 months prior to presentation in anticipation of a partial colectomy and temporary ileostomy that was performed 8 months prior to presentation. She recently had developed bilateral, radiating, sharp lower extremity pain extending from the feet to the hips over the last 2 weeks and swelling of the bilateral legs that impaired her ability to ambulate. Additionally, she had recently traveled to Colorado and a Lyme disease workup was initiated at an outside hospital in Colorado; however, the results were pending. The outside hospital also performed a spinal tap that was negative. At our clinic, biopsies were performed on the shin nodule and a right palmar pustule (Figure 2). There was clinical suspicion of erythema nodosum and subcorneal pustular dermatosis or a vesiculopustular skin manifestation of the patient’s Crohn disease. The patient was switched from generic doxycycline to a brand name variant 150 mg every night at bedtime for 2 weeks. She subsequently was admitted to the inpatient rheumatology service for a complete systemic workup.
The punch biopsy of the left upper shin demonstrated operative hemorrhage and periadnexal lymphocytic inflammation without evidence of fungal or bacterial elements by Gram or Gomori methenamine-silver stain. Clinically, the diagnosis was most likely erythema nodosum, though insufficient hypodermis was present to make the diagnosis with pathology. The shave biopsy of the right medial palm was nondiagnostic but showed a transected pustule with no bacterial or fungal elements by Gram or Gomori methenamine-silver stain (Figure 3). Given the clinical context, the likely pathologic diagnosis was vesiculopustular Crohn disease.
Our patient was started on an empiric steroid trial with rapid improvement of the arthralgia and rash. The presumed diagnosis was a Crohn disease flare and the patient was discharged on an 8-week steroid taper. Three weeks later at a follow-up appointment, the patient’s skin lesions had nearly resolved. The swelling of the legs and feet had substantially decreased, but the joint pain, primarily in the ankles, persisted.
Routine laboratory studies showed a hemoglobin level of 11.6 g/dL (reference range, 12–15 g/dL), white blood cell count of 9.1 K/μL (reference range, 4.5–11.0 K/μL), C-reactive protein level of 20.15 mg/dL (reference range, <1.0 mg/dL), and an antinuclear antibody titer of 160 (<80). Serology for Lyme disease was negative. Serum chemistries were all within reference range and an echocardiogram was normal.
Up to one-third of patients with inflammatory bowel disease (IBD) experience extraintestinal manifestations of their condition. Of these patients, nearly one-third will develop cutaneous manifestations.1 The most common skin diseases associated with IBD are pyoderma gangrenosum and erythema nodosum.2 The differential diagnoses considered in this unique case included early pyoderma gangrenosum, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), and vesiculopustular Crohn disease. Vesiculopustular Crohn disease is a rare component of IBD and also can be present in bowel-associated dermatosis-arthritis syndrome (BADAS). In BADAS, symptoms often include arthritis and systemic symptoms such as fever and malaise. The skin manifestations typically involve the arms and trunk. It often is seen after intestinal bypass surgery but also can be present in patients with gastrointestinal diseases such as IBD.3 Due to its early association with bypass surgery, BADAS previously was referred to as bowel bypass syndrome but has since been seen in relation to other intestinal surgeries and IBD.4 Patients with BADAS often present with episodes of fever, fatigue, and malaise, in addition to arthralgia and cutaneous eruptions. Cases of BADAS related to IBD instead of bypass surgery often can be less severe in nature. Unlike many of these previously reported cases, our patient’s joint pain primarily was in the knees and ankles, whereas typical cases of BADAS cause upper extremity (ie, shoulder, elbow) arthralgia. Our patient occasionally experienced upper extremity pain, but it was less frequent and less severe than the knee and ankle pain. The vesiculopustular lesions in BADAS usually begin as 3- to 10-mm painful macules that then develop into aseptic pustular lesions. These manifestations arise on the upper arms and chest or trunk and can be accompanied by erythema nodosum on the legs.4
It has been hypothesized that BADAS occurs as an immune reaction to bacterial overgrowth in the bowel from IBD, infection, or surgery. The reaction is in response to a bacterial antigen and manifests cutaneously.5 This same pathogenesis is thought to cause various other manifestations of Crohn disease such as erythema nodosum. Bacteria that incite this immune response include Bacteroides fragilis, Escherichia coli, and Streptococcus.
Resolution of both vesiculopustular Crohn disease and of BADAS often occurs with treatment of the underlying IBD but also can be improved with steroids and antibiotics. However, response to antibiotics often is variable.5,6 The mainstay for treatment remains steroids and management of underlying bowel disease.
Bowel-associated dermatosis-arthritis syndrome often is overlooked when compiling differential diagnoses for neutrophilic dermatoses but should be considered in patients with bowel disease or recent surgery. Because the syndrome can be recurrent, early diagnosis can help to prevent and treat relapsing courses of BADAS.
- Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J. 2005;81:580-585.
- Havemann BD. A pustular skin rash in a woman with 2 weeks of diarrhea. MedGenMed. 2005;7:11.
- Bolognia JL, Jorizzo J, Rapini RP. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2008.
- Huang B, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13.
- Truchuelo MT, Alcántara J, Vano-Galván S, et al. Bowel associated dermatosis-arthritis syndrome: another cutaneous manifestation of inflammatory intestinal disease. Int J Dermatol. 2013;52:1596-1598.
- Ashok D, Kiely P. Bowel associated dermatosis-arthritis syndrome: a case report. J Med Case Rep. 2007;1:81.
- Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J. 2005;81:580-585.
- Havemann BD. A pustular skin rash in a woman with 2 weeks of diarrhea. MedGenMed. 2005;7:11.
- Bolognia JL, Jorizzo J, Rapini RP. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2008.
- Huang B, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13.
- Truchuelo MT, Alcántara J, Vano-Galván S, et al. Bowel associated dermatosis-arthritis syndrome: another cutaneous manifestation of inflammatory intestinal disease. Int J Dermatol. 2013;52:1596-1598.
- Ashok D, Kiely P. Bowel associated dermatosis-arthritis syndrome: a case report. J Med Case Rep. 2007;1:81.
What Is Your Diagnosis? Mycosis Fungoides
The Diagnosis: Mycosis Fungoides
Physical examination revealed erythematous polycyclic and arcuate plaques with fine overlying scale on the right arm and shoulder (Figure 1). Mild wrinkling and telangiectasias were noted on the skin surrounding the lesions. Laboratory tests showed normal values for antinuclear antibodies, anti–Sjögren syndrome–related antigen A, and anti–Sjögren syndrome–related antigen B.
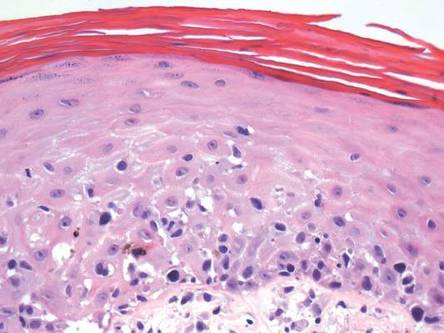
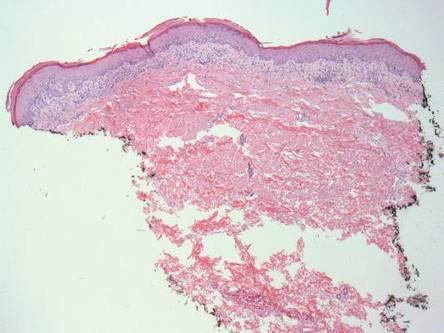
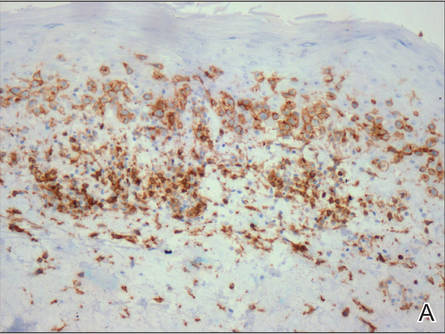
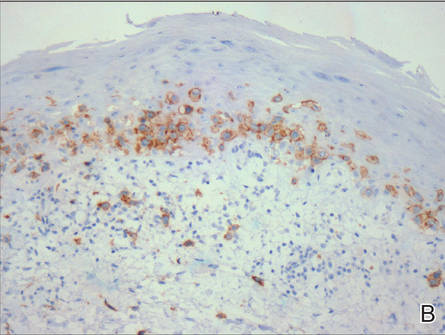
A skin biopsy of a plaque on the right upper arm showed enlarged pleomorphic lymphocytes arranged along the basal layer and in focal collections within the epidermis (Figure 2). Within the dermis were wiry bundles of collagen, a sparse superficial and patchy infiltrate of lymphocytes, and scattered large mononuclear cells (Figure 3). Immunoperoxidase staining revealed large intraepidermal lymphocytes positive for CD4 (Figure 4A) and CD5. Notably, these lymphocytes also stained positive for CD30 (Figure 4B). Staining for CD8, CD1a, CD56, and anaplastic lymphoma kinase was negative, with aberrant loss of CD3. The morphology and pattern of immunoreactivity supported the diagnosis of mycosis fungoides (MF).
Mycosis fungoides is the most common form of cutaneous T-cell lymphoma.1 Its progression is classified in 3 stages: (1) early (patch) stage, (2) plaque stage, and (3) tumor stage. Conclusive diagnosis of early stage MF often is difficult due to its clinical features that are similar to more common benign dermatoses (eg, atopic dermatitis, psoriasis, lichen planus), leading to shortcomings in determining prognosis and selecting an appropriate treatment regimen. With this diagnositic difficulty in mind, guidelines have been created to aid in the diagnosis of early stage MF.2
Clinical features consistent with early stage MF include multiple erythematous, well-demarcated lesions with varying shapes that typically are greater than 5 cm in diameter.2 Lesions usually are flat or thinly elevated and may exhibit slight scaling. As was noted in our patient, poikiloderma of the surrounding skin is fairly specific for early stage MF, as it is not a feature associated with common clinical mimics of MF (eg, atopic dermatitis, psoriasis, lichen planus). The distribution of skin lesions in non–sun-exposed areas is common. The eruption is persistent, though it may wax and wane in severity.2
| 
| |
|
|
Histopathologic examination is necessary to confirm a diagnosis of MF. Typically, early stage MF is marked by enlarged T lymphocytes within the epidermis as well as the papillary and superficial reticular dermis. Cerebriform nuclei are a key finding in the diagnosis of MF. Lymphocytes frequently are arranged linearly along the basal layer of the epidermis. Within the epidermis, clusters of atypical lymphocytes (Pautrier microabscesses) without spongiosis are uncommon but are a characteristic finding of MF if present.1 Papillary dermal fibrosis also may be evident.2
| 
| |
Figure 4. Large intraepidermal lymphocytes were highlighted on CD4 (A) and CD30 immunostaining (B)(original magnification ×200 and ×200). | ||
Immunostaining typically reveals positivity for CD3 and CD4, as well as for lymphocyte antigens CD2 and CD5.1 CD30 positivity in early stage MF rarely has been reported in the literature.3,4 Such cases appear histologically similarly to CD30‒negative cases in other respects. One study showed that the presence of CD30-positive lymphocytes does not alter the clinical course of MF.3 Another study found that, while epidermal CD30-postive lymphocytes had no prognostic relevance, an increased percentage of dermal CD30-positive cells was linked to a higher stage at diagnosis and worse overall prognosis.5 Pathogenesis underlying CD30 positivity in early MF is unknown. It is important to note that CD30-positive cells commonly are seen in lymphomatoid papulosis and anaplastic large cell lymphoma, as well as a variety of nonneoplastic conditions.3,6,7
- Smoller BR. Mycosis fungoides: what do/do not we know? J Cutan Pathol. 2008;35(suppl 2):35-39.
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
- Wu H, Telang GH, Lessin SR, et al. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212-216.
- Ohtani T, Kikuchi K, Koizumi H, et al. A case of CD30+ large-cell transformation in a patient with unilesional patch-stage mycosis fungoides. Int J Dermatol. 2009;48:623-626.
- Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860-1868.
- Resnik KS, Kutzner H. Of lymphocytes and cutaneous epithelium: keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am J Dermatopathol. 2010;32:314-315.
- Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
The Diagnosis: Mycosis Fungoides
Physical examination revealed erythematous polycyclic and arcuate plaques with fine overlying scale on the right arm and shoulder (Figure 1). Mild wrinkling and telangiectasias were noted on the skin surrounding the lesions. Laboratory tests showed normal values for antinuclear antibodies, anti–Sjögren syndrome–related antigen A, and anti–Sjögren syndrome–related antigen B.
A skin biopsy of a plaque on the right upper arm showed enlarged pleomorphic lymphocytes arranged along the basal layer and in focal collections within the epidermis (Figure 2). Within the dermis were wiry bundles of collagen, a sparse superficial and patchy infiltrate of lymphocytes, and scattered large mononuclear cells (Figure 3). Immunoperoxidase staining revealed large intraepidermal lymphocytes positive for CD4 (Figure 4A) and CD5. Notably, these lymphocytes also stained positive for CD30 (Figure 4B). Staining for CD8, CD1a, CD56, and anaplastic lymphoma kinase was negative, with aberrant loss of CD3. The morphology and pattern of immunoreactivity supported the diagnosis of mycosis fungoides (MF).
Mycosis fungoides is the most common form of cutaneous T-cell lymphoma.1 Its progression is classified in 3 stages: (1) early (patch) stage, (2) plaque stage, and (3) tumor stage. Conclusive diagnosis of early stage MF often is difficult due to its clinical features that are similar to more common benign dermatoses (eg, atopic dermatitis, psoriasis, lichen planus), leading to shortcomings in determining prognosis and selecting an appropriate treatment regimen. With this diagnositic difficulty in mind, guidelines have been created to aid in the diagnosis of early stage MF.2
Clinical features consistent with early stage MF include multiple erythematous, well-demarcated lesions with varying shapes that typically are greater than 5 cm in diameter.2 Lesions usually are flat or thinly elevated and may exhibit slight scaling. As was noted in our patient, poikiloderma of the surrounding skin is fairly specific for early stage MF, as it is not a feature associated with common clinical mimics of MF (eg, atopic dermatitis, psoriasis, lichen planus). The distribution of skin lesions in non–sun-exposed areas is common. The eruption is persistent, though it may wax and wane in severity.2
|
|
| |
|
|
Histopathologic examination is necessary to confirm a diagnosis of MF. Typically, early stage MF is marked by enlarged T lymphocytes within the epidermis as well as the papillary and superficial reticular dermis. Cerebriform nuclei are a key finding in the diagnosis of MF. Lymphocytes frequently are arranged linearly along the basal layer of the epidermis. Within the epidermis, clusters of atypical lymphocytes (Pautrier microabscesses) without spongiosis are uncommon but are a characteristic finding of MF if present.1 Papillary dermal fibrosis also may be evident.2
|
|
| |
Figure 4. Large intraepidermal lymphocytes were highlighted on CD4 (A) and CD30 immunostaining (B)(original magnification ×200 and ×200). | ||
Immunostaining typically reveals positivity for CD3 and CD4, as well as for lymphocyte antigens CD2 and CD5.1 CD30 positivity in early stage MF rarely has been reported in the literature.3,4 Such cases appear histologically similarly to CD30‒negative cases in other respects. One study showed that the presence of CD30-positive lymphocytes does not alter the clinical course of MF.3 Another study found that, while epidermal CD30-postive lymphocytes had no prognostic relevance, an increased percentage of dermal CD30-positive cells was linked to a higher stage at diagnosis and worse overall prognosis.5 Pathogenesis underlying CD30 positivity in early MF is unknown. It is important to note that CD30-positive cells commonly are seen in lymphomatoid papulosis and anaplastic large cell lymphoma, as well as a variety of nonneoplastic conditions.3,6,7
The Diagnosis: Mycosis Fungoides
Physical examination revealed erythematous polycyclic and arcuate plaques with fine overlying scale on the right arm and shoulder (Figure 1). Mild wrinkling and telangiectasias were noted on the skin surrounding the lesions. Laboratory tests showed normal values for antinuclear antibodies, anti–Sjögren syndrome–related antigen A, and anti–Sjögren syndrome–related antigen B.
A skin biopsy of a plaque on the right upper arm showed enlarged pleomorphic lymphocytes arranged along the basal layer and in focal collections within the epidermis (Figure 2). Within the dermis were wiry bundles of collagen, a sparse superficial and patchy infiltrate of lymphocytes, and scattered large mononuclear cells (Figure 3). Immunoperoxidase staining revealed large intraepidermal lymphocytes positive for CD4 (Figure 4A) and CD5. Notably, these lymphocytes also stained positive for CD30 (Figure 4B). Staining for CD8, CD1a, CD56, and anaplastic lymphoma kinase was negative, with aberrant loss of CD3. The morphology and pattern of immunoreactivity supported the diagnosis of mycosis fungoides (MF).
Mycosis fungoides is the most common form of cutaneous T-cell lymphoma.1 Its progression is classified in 3 stages: (1) early (patch) stage, (2) plaque stage, and (3) tumor stage. Conclusive diagnosis of early stage MF often is difficult due to its clinical features that are similar to more common benign dermatoses (eg, atopic dermatitis, psoriasis, lichen planus), leading to shortcomings in determining prognosis and selecting an appropriate treatment regimen. With this diagnositic difficulty in mind, guidelines have been created to aid in the diagnosis of early stage MF.2
Clinical features consistent with early stage MF include multiple erythematous, well-demarcated lesions with varying shapes that typically are greater than 5 cm in diameter.2 Lesions usually are flat or thinly elevated and may exhibit slight scaling. As was noted in our patient, poikiloderma of the surrounding skin is fairly specific for early stage MF, as it is not a feature associated with common clinical mimics of MF (eg, atopic dermatitis, psoriasis, lichen planus). The distribution of skin lesions in non–sun-exposed areas is common. The eruption is persistent, though it may wax and wane in severity.2
|
|
| |
|
|
Histopathologic examination is necessary to confirm a diagnosis of MF. Typically, early stage MF is marked by enlarged T lymphocytes within the epidermis as well as the papillary and superficial reticular dermis. Cerebriform nuclei are a key finding in the diagnosis of MF. Lymphocytes frequently are arranged linearly along the basal layer of the epidermis. Within the epidermis, clusters of atypical lymphocytes (Pautrier microabscesses) without spongiosis are uncommon but are a characteristic finding of MF if present.1 Papillary dermal fibrosis also may be evident.2
|
|
| |
Figure 4. Large intraepidermal lymphocytes were highlighted on CD4 (A) and CD30 immunostaining (B)(original magnification ×200 and ×200). | ||
Immunostaining typically reveals positivity for CD3 and CD4, as well as for lymphocyte antigens CD2 and CD5.1 CD30 positivity in early stage MF rarely has been reported in the literature.3,4 Such cases appear histologically similarly to CD30‒negative cases in other respects. One study showed that the presence of CD30-positive lymphocytes does not alter the clinical course of MF.3 Another study found that, while epidermal CD30-postive lymphocytes had no prognostic relevance, an increased percentage of dermal CD30-positive cells was linked to a higher stage at diagnosis and worse overall prognosis.5 Pathogenesis underlying CD30 positivity in early MF is unknown. It is important to note that CD30-positive cells commonly are seen in lymphomatoid papulosis and anaplastic large cell lymphoma, as well as a variety of nonneoplastic conditions.3,6,7
- Smoller BR. Mycosis fungoides: what do/do not we know? J Cutan Pathol. 2008;35(suppl 2):35-39.
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
- Wu H, Telang GH, Lessin SR, et al. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212-216.
- Ohtani T, Kikuchi K, Koizumi H, et al. A case of CD30+ large-cell transformation in a patient with unilesional patch-stage mycosis fungoides. Int J Dermatol. 2009;48:623-626.
- Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860-1868.
- Resnik KS, Kutzner H. Of lymphocytes and cutaneous epithelium: keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am J Dermatopathol. 2010;32:314-315.
- Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
- Smoller BR. Mycosis fungoides: what do/do not we know? J Cutan Pathol. 2008;35(suppl 2):35-39.
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
- Wu H, Telang GH, Lessin SR, et al. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212-216.
- Ohtani T, Kikuchi K, Koizumi H, et al. A case of CD30+ large-cell transformation in a patient with unilesional patch-stage mycosis fungoides. Int J Dermatol. 2009;48:623-626.
- Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860-1868.
- Resnik KS, Kutzner H. Of lymphocytes and cutaneous epithelium: keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am J Dermatopathol. 2010;32:314-315.
- Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
An otherwise healthy 62-year-old man presented for evaluation of multiple scaly erythematous plaques on the right upper arm and shoulder of 10 years’ duration. The patient reported a burning sensation but no exacerbation of the lesions upon sun exposure. He previously had been treated for a presumed clinical diagnosis of erythema annulare centrifugum but experienced only modest improvement with topical corticosteroids and tacrolimus ointment 0.1%. Previous trials of systemic antifungals also yielded minimal benefit.
Secondary Syphilis
Syphilis often is referred to as the “great imitator” due to the protean presentations of secondary-stage disease, the most common of which are skin manifestations.1 Secondary syphilis typically begins 3 to 10 weeks after initial exposure due to systemic dissemination of Treponema pallidum, and although presentations can vary widely, the classic presentation includes nonspecific generalized symptoms (eg, fever, malaise, lymphadenopathy), variable skin findings (eg, nonpruritic papulosquamous eruption), and mucosal ulcerations or plaques.1 Early and accurate diagnosis of syphilis is critical to avoid the morbidity associated with advanced disease.
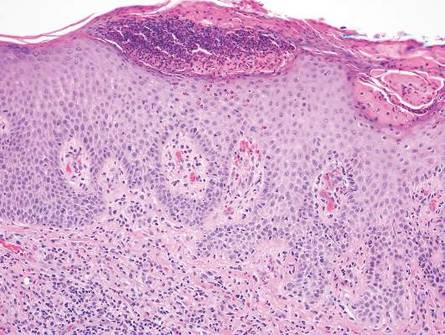
The classic histopathologic appearance of secondary syphilis is characterized by psoriasiform epidermal changes; a dermal inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells in a lichenoid and/or superficial and deep perivascular distribution (Figure 1); and endothelial swelling of dermal blood vessels.1 The presence of plasma cells in the infiltrate (Figure 2) is particularly useful for differentiating secondary syphilis from other clinicopathological mimickers, but this finding is not always present. Silver-based histochemical stains (eg, Warthin-Starry silver stain) can be used to high-light T pallidum organisms; however, histochemical staining is plagued by low diagnostic sensitivity for identifying the causative organism, making immunohistochemical and/or serologic testing the preferred method for confirming the diagnosis.1

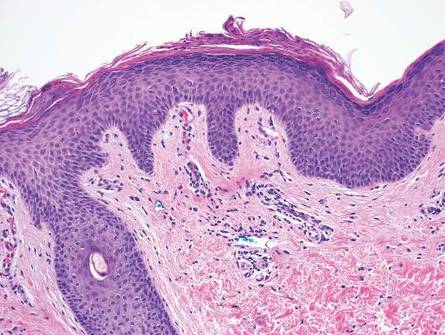
Arthropod assault is characterized by a superficial and deep perivascular lymphocytic inflammatory infiltrate with a variable number of polymorphonuclear cells.2 Overlying spongiosis or focal epidermal necrosis and increased eosinophils are typical of arthropod assault (Figure 3).2 The infiltrate seen following insect bites is classically described as wedge-shaped, although recent literature has disputed the sensitivity of this finding, identifying adnexal structure involvement as an alternative sensitive marker for identifying insect bites.2
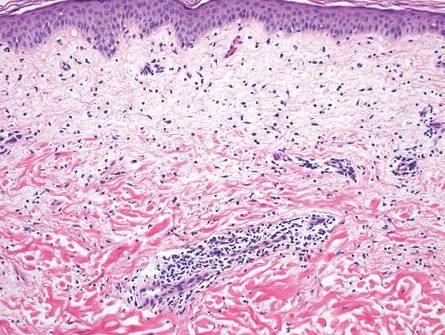
Chronic cutaneous lupus erythematosus demonstrates a spectrum of histopathologic changes depending on the age of the lesion biopsied; however, characteristic histopathologic features typically include variable epidermal atrophy or acanthosis with basal layer vacuolar degeneration, basement membrane thickening, follicular plugging, superficial and deep perivascular and periappendageal lymphocytic inflammation, and dermal mucin deposition (Figure 4).4
Fixed drug eruption histopathologically presents as an interface tissue reaction–associated single-cell necrosis to broader areas of epidermal necrosis, as well as superficial to mid-dermal lymphocytic infiltrate. Unlike secondary syphilis, a fixed drug eruption is characterized by prominent melanin pigment incontinence and eosinophils (Figure 5).5

Similar to secondary syphilis, pityriasis lichenoides et varioliformis acuta (PLEVA) demonstrates variable psoriasiform epidermal hyperplasia with a lichenoid and perivascular lymphocytic infiltrate. Other findings in PLEVA include parakeratosis, variable epidermal necrosis, and prominent exocytosis of lymphocytes. Unlike typical secondary syphilis, PLEVA often is associated with lymphocytic vasculitis, consisting of the invasion of vessel walls by lymphocytes with extravasation of erythrocytes and an absence of conspicuous plasma cells (Figure 6).6
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;3:595-599.
- Miteva M, Elsner P, Ziemer M. A histopathologic study of arthropod bite reactions in 20 patients highlights relevant adnexal involvement. J Cutan Pathol. 2009;36:26-33.
- Winkelmann RK, Reizner GT. Diffuse dermal neutrophilia in urticarial. Human Pathol. 1988;19:389-393.
- Sepehr A, Wenson S, Tahan SR. Histopathologic manifestations of systemic diseases: the example of cutaneous lupus erythematosus. J Cutan Pathol. 2010;37 (suppl 1):112-124.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727.
- Fernandes NF, Rozdeba PJ, Schwartz RA, et al. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49:257-261.
Syphilis often is referred to as the “great imitator” due to the protean presentations of secondary-stage disease, the most common of which are skin manifestations.1 Secondary syphilis typically begins 3 to 10 weeks after initial exposure due to systemic dissemination of Treponema pallidum, and although presentations can vary widely, the classic presentation includes nonspecific generalized symptoms (eg, fever, malaise, lymphadenopathy), variable skin findings (eg, nonpruritic papulosquamous eruption), and mucosal ulcerations or plaques.1 Early and accurate diagnosis of syphilis is critical to avoid the morbidity associated with advanced disease.
The classic histopathologic appearance of secondary syphilis is characterized by psoriasiform epidermal changes; a dermal inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells in a lichenoid and/or superficial and deep perivascular distribution (Figure 1); and endothelial swelling of dermal blood vessels.1 The presence of plasma cells in the infiltrate (Figure 2) is particularly useful for differentiating secondary syphilis from other clinicopathological mimickers, but this finding is not always present. Silver-based histochemical stains (eg, Warthin-Starry silver stain) can be used to high-light T pallidum organisms; however, histochemical staining is plagued by low diagnostic sensitivity for identifying the causative organism, making immunohistochemical and/or serologic testing the preferred method for confirming the diagnosis.1
Arthropod assault is characterized by a superficial and deep perivascular lymphocytic inflammatory infiltrate with a variable number of polymorphonuclear cells.2 Overlying spongiosis or focal epidermal necrosis and increased eosinophils are typical of arthropod assault (Figure 3).2 The infiltrate seen following insect bites is classically described as wedge-shaped, although recent literature has disputed the sensitivity of this finding, identifying adnexal structure involvement as an alternative sensitive marker for identifying insect bites.2
Chronic cutaneous lupus erythematosus demonstrates a spectrum of histopathologic changes depending on the age of the lesion biopsied; however, characteristic histopathologic features typically include variable epidermal atrophy or acanthosis with basal layer vacuolar degeneration, basement membrane thickening, follicular plugging, superficial and deep perivascular and periappendageal lymphocytic inflammation, and dermal mucin deposition (Figure 4).4
Fixed drug eruption histopathologically presents as an interface tissue reaction–associated single-cell necrosis to broader areas of epidermal necrosis, as well as superficial to mid-dermal lymphocytic infiltrate. Unlike secondary syphilis, a fixed drug eruption is characterized by prominent melanin pigment incontinence and eosinophils (Figure 5).5
Similar to secondary syphilis, pityriasis lichenoides et varioliformis acuta (PLEVA) demonstrates variable psoriasiform epidermal hyperplasia with a lichenoid and perivascular lymphocytic infiltrate. Other findings in PLEVA include parakeratosis, variable epidermal necrosis, and prominent exocytosis of lymphocytes. Unlike typical secondary syphilis, PLEVA often is associated with lymphocytic vasculitis, consisting of the invasion of vessel walls by lymphocytes with extravasation of erythrocytes and an absence of conspicuous plasma cells (Figure 6).6
Syphilis often is referred to as the “great imitator” due to the protean presentations of secondary-stage disease, the most common of which are skin manifestations.1 Secondary syphilis typically begins 3 to 10 weeks after initial exposure due to systemic dissemination of Treponema pallidum, and although presentations can vary widely, the classic presentation includes nonspecific generalized symptoms (eg, fever, malaise, lymphadenopathy), variable skin findings (eg, nonpruritic papulosquamous eruption), and mucosal ulcerations or plaques.1 Early and accurate diagnosis of syphilis is critical to avoid the morbidity associated with advanced disease.
The classic histopathologic appearance of secondary syphilis is characterized by psoriasiform epidermal changes; a dermal inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells in a lichenoid and/or superficial and deep perivascular distribution (Figure 1); and endothelial swelling of dermal blood vessels.1 The presence of plasma cells in the infiltrate (Figure 2) is particularly useful for differentiating secondary syphilis from other clinicopathological mimickers, but this finding is not always present. Silver-based histochemical stains (eg, Warthin-Starry silver stain) can be used to high-light T pallidum organisms; however, histochemical staining is plagued by low diagnostic sensitivity for identifying the causative organism, making immunohistochemical and/or serologic testing the preferred method for confirming the diagnosis.1
Arthropod assault is characterized by a superficial and deep perivascular lymphocytic inflammatory infiltrate with a variable number of polymorphonuclear cells.2 Overlying spongiosis or focal epidermal necrosis and increased eosinophils are typical of arthropod assault (Figure 3).2 The infiltrate seen following insect bites is classically described as wedge-shaped, although recent literature has disputed the sensitivity of this finding, identifying adnexal structure involvement as an alternative sensitive marker for identifying insect bites.2
Chronic cutaneous lupus erythematosus demonstrates a spectrum of histopathologic changes depending on the age of the lesion biopsied; however, characteristic histopathologic features typically include variable epidermal atrophy or acanthosis with basal layer vacuolar degeneration, basement membrane thickening, follicular plugging, superficial and deep perivascular and periappendageal lymphocytic inflammation, and dermal mucin deposition (Figure 4).4
Fixed drug eruption histopathologically presents as an interface tissue reaction–associated single-cell necrosis to broader areas of epidermal necrosis, as well as superficial to mid-dermal lymphocytic infiltrate. Unlike secondary syphilis, a fixed drug eruption is characterized by prominent melanin pigment incontinence and eosinophils (Figure 5).5
Similar to secondary syphilis, pityriasis lichenoides et varioliformis acuta (PLEVA) demonstrates variable psoriasiform epidermal hyperplasia with a lichenoid and perivascular lymphocytic infiltrate. Other findings in PLEVA include parakeratosis, variable epidermal necrosis, and prominent exocytosis of lymphocytes. Unlike typical secondary syphilis, PLEVA often is associated with lymphocytic vasculitis, consisting of the invasion of vessel walls by lymphocytes with extravasation of erythrocytes and an absence of conspicuous plasma cells (Figure 6).6
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;3:595-599.
- Miteva M, Elsner P, Ziemer M. A histopathologic study of arthropod bite reactions in 20 patients highlights relevant adnexal involvement. J Cutan Pathol. 2009;36:26-33.
- Winkelmann RK, Reizner GT. Diffuse dermal neutrophilia in urticarial. Human Pathol. 1988;19:389-393.
- Sepehr A, Wenson S, Tahan SR. Histopathologic manifestations of systemic diseases: the example of cutaneous lupus erythematosus. J Cutan Pathol. 2010;37 (suppl 1):112-124.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727.
- Fernandes NF, Rozdeba PJ, Schwartz RA, et al. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49:257-261.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;3:595-599.
- Miteva M, Elsner P, Ziemer M. A histopathologic study of arthropod bite reactions in 20 patients highlights relevant adnexal involvement. J Cutan Pathol. 2009;36:26-33.
- Winkelmann RK, Reizner GT. Diffuse dermal neutrophilia in urticarial. Human Pathol. 1988;19:389-393.
- Sepehr A, Wenson S, Tahan SR. Histopathologic manifestations of systemic diseases: the example of cutaneous lupus erythematosus. J Cutan Pathol. 2010;37 (suppl 1):112-124.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727.
- Fernandes NF, Rozdeba PJ, Schwartz RA, et al. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49:257-261.
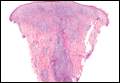
Erythematous Scaly Papules on the Shins and Calves
The Diagnosis: Hyperkeratosis Lenticularis Perstans
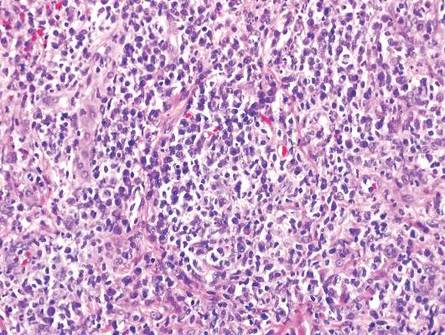
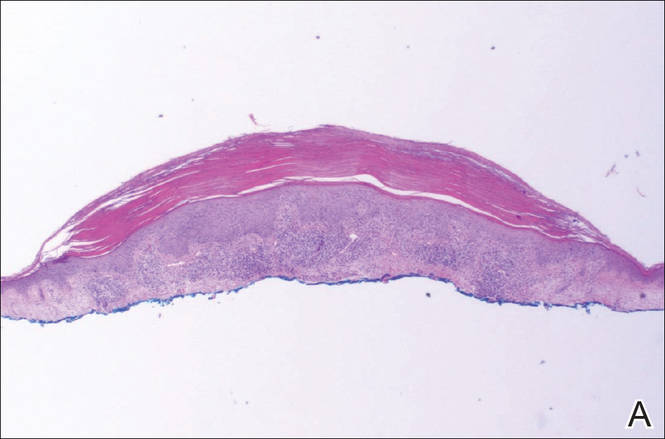
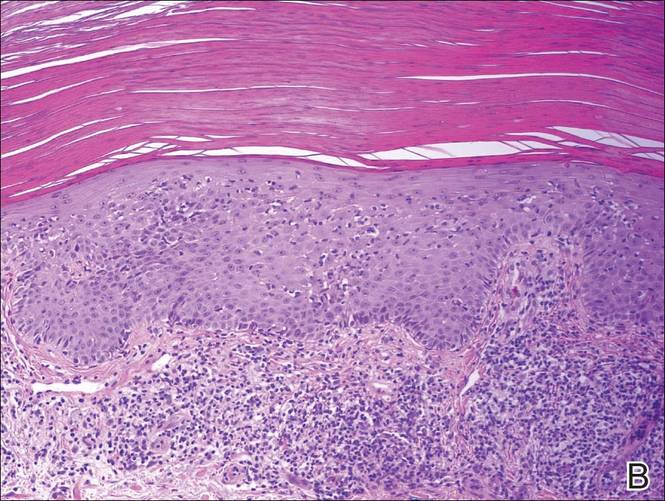
A shave biopsy of a lesion on the right leg was performed. Histopathology revealed a discrete papule with overlying compact hyperkeratosis. There was parakeratosis with an absent granular layer and a lichenoid lymphocytic infiltrate within the papillary dermis (Figure). Given the clinical context, these changes were consistent with a diagnosis of hyperkeratosis lenticularis perstans (HLP), also known as Flegel disease.
The patient was started on tretinoin cream 0.1% nightly for 3 months and triamcinolone ointment 0.1% as needed for pruritus but showed no clinical response. Given the benign nature of the condition and because the lesions were asymptomatic, additional treatment options were not pursued.
Originally described by Flegel1 in 1958, HLP is a rare skin disorder commonly seen in white individuals with onset in the fourth or fifth decades of life.1,2 While most cases are sporadic,3-6 HLP also has been associated with autosomal dominant inheritance.7-10
Patients with HLP typically present with multiple 1- to 5-mm reddish-brown, hyperkeratotic, scaly papules that reveal a moist, erythematous base with pinpoint bleeding upon removal of the scale. Lesions usually are distributed symmetrically and most commonly present on the extensor surfaces of the lower legs and dorsal feet.1,2,7 Lesions also may appear on the extensor surfaces of the arms, pinna, periocular region, antecubital and popliteal fossae, and oral mucosa and also may present as pits on the palms and soles.2,4,7,8 Furthermore, unilateral and localized variants of HLP have been described.11,12 Hyperkeratosis lenticularis perstans usually is asymptomatic but can present with mild pruritus or burning.3,5,13
The etiology and pathogenesis of HLP are unknown. Exposure to UV light has been implicated as an inciting factor14; however, reports of spontaneous resolution in the summer13 and upon treatment with psoralen plus UVA therapy15 make the role of UV light unclear. Furthermore, investigators disagree as to whether the primary pathogenic event in HLP is an inflammatory process or one of abnormal keratinization.1,3,7,10 Fernandez-Flores and Manjon16 suggested HLP is an inflammatory process with periods of exacerbations and remissions after finding mounds of parakeratosis with neutrophils arranged in different strata in the stratum corneum.
Histologically, compact hyperkeratosis usually is noted, often with associated parakeratosis, epidermal atrophy with thinning or absence of the granular layer, and a bandlike lymphohistiocytic infiltrate in the papillary dermis.1-3 Histopathologic differences between recent-onset versus longstanding lesions have been found, with old lesions lacking an inflammatory infiltrate.3 Furthermore, new lesions often show abnormalities in quantity and/or morphology of membrane-coating granules, also known as Odland bodies, in keratinocytes on electron microscopy,3,10,17 while old lesions do not.3 Odland bodies are involved in normal desquamation, leading some to speculate on their role in HLP.10 Currently, it is unclear whether abnormalities in these organelles cause the retention hyperkeratosis seen in HLP or if such abnormalities are a secondary phenomenon.3,17
There are questionable associations between HLP and diabetes mellitus type 2, hyperthyroidism, basal and squamous cell carcinomas of the skin, and gastrointestinal malignancy.4,9,18 Our patient had a history of basal cell carcinoma on the face, diet-controlled diabetes mellitus, and hypothyroidism. Given the high prevalence of these diseases in the general population, however, it is difficult to ascertain whether a true association with HLP exists.
While HLP can slowly progress to involve additional body sites, it is overall a benign condition that does not require treatment. Therapeutic options are based on case reports, with no single treatment showing a consistent response. From review of the literature, therapies that have been most effective include dermabrasion, excision,19 topical 5-fluorouracil,2,17,20 and oral retinoids.8 Hyperkeratosis lenticularis perstans generally is resistant to topical steroids, retinoids, and vitamin D3 analogs, although success with betamethasone dipropionate,5 isotretinoin
gel 0.05%,11 and calcipotriol have been reported.6 A case of HLP with clinical response to psoralen plus UVA therapy also has been described.15
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt. 1958;9:363-364.
- Pearson LH, Smith JG, Chalker DK. Hyperkeratosis lenticularis perstans (Flegel’s disease). J Am Acad Dermatol. 1987;16:190-195.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel’s disease). Am J Dermatopathol. 2006;28:122-126.
- Fernández-Crehuet P, Rodríguez-Rey E, Ríos-Martín JJ, et al. Hyperkeratosis lenticularis perstans, or Flegel disease, with palmoplantar involvement. Actas Dermosifiliogr. 2009;100:157-159.
- Sterneberg-Vos H, van Marion AM, Frank J, et al. Hyperkeratosis lenticularis perstans (Flegel’s disease)—successful treatment with topical corticosteroids. Int J Dermatol. 2008;47:38-41.
- Bayramgürler D, Apaydin R, Dökmeci S, et al. Flegel’s disease: treatment with topical calcipotriol. Clin Exp Dermatol. 2002;27:161-162.
- Price ML, Jones EW, MacDonald DM. A clinicopathological study of Flegel’s disease (hyperkeratosis lenticularis perstans). Br J Dermatol. 1987;116:681-691.
- Krishnan A, Kar S. Photoletter to the editor: hyperkeratosis lenticularis perstans (Flegel’s disease) with unusual clinical presentation. response to isotretinoin therapy. J Dermatol Case Rep. 2012;6:93-95.
- Beveridge GW, Langlands AO. Familial hyperkeratosis lenticularis perstans associated with tumours of the skin. Br J Dermatol. 1973;88:453-458.
- Frenk E, Tapernoux B. Hyperkeratosis lenticularis perstans (Flegel): a biological model for keratinization occurring in the absence of Odland bodies? Dermatologica. 1976;153:253-262.
- Miranda-Romero A, Sánchez Sambucety P, Bajo del Pozo C, et al. Unilateral hyperkeratosis lenticularis perstans (Flegel's disease). J Am Acad Dermatol. 1998;39:655-657.
- Gutiérrez MC, Hasson A, Arias MD, et al. Localized hyperkeratosis lenticularis perstans (Flegel's disease). Cutis. 1991;48:201-204.
- Fathy S, Azadeh B. Hyperkeratosis lenticularis perstans. Int J Dermatol. 1988;27:120-121.
- Rosdahl I, Rosen K. Hyperkeratosis lenticularis perstans: report on two cases. Acta Derm Venerol. 1985;65:562-564.
- Cooper SM, George S. Flegel's disease treated with psoralen ultraviolet A. Br J Dermatol. 2000;142:340-342.
- Fernandez-Flores A, Manjon JA. Morphological evidence of periodical exacerbation of hyperkeratosis lenticularis perstans. Acta Dermatovenerol Croat. 2009;17:16-19.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Ishibashi A, Tsuboi R, Fujita K. Familial hyperkeratosis lenticularis perstans. associated with cancers of the digestive organs. J Dermatol. 1984;11:407-409.
- Cunha Filho RR, Almeida Jr HL. Hyperkeratosis lenticularis perstans. An Bras Dermatol. 2011;86(4 suppl 1):S76-S77.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)—lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
The Diagnosis: Hyperkeratosis Lenticularis Perstans
A shave biopsy of a lesion on the right leg was performed. Histopathology revealed a discrete papule with overlying compact hyperkeratosis. There was parakeratosis with an absent granular layer and a lichenoid lymphocytic infiltrate within the papillary dermis (Figure). Given the clinical context, these changes were consistent with a diagnosis of hyperkeratosis lenticularis perstans (HLP), also known as Flegel disease.
The patient was started on tretinoin cream 0.1% nightly for 3 months and triamcinolone ointment 0.1% as needed for pruritus but showed no clinical response. Given the benign nature of the condition and because the lesions were asymptomatic, additional treatment options were not pursued.
Originally described by Flegel1 in 1958, HLP is a rare skin disorder commonly seen in white individuals with onset in the fourth or fifth decades of life.1,2 While most cases are sporadic,3-6 HLP also has been associated with autosomal dominant inheritance.7-10
Patients with HLP typically present with multiple 1- to 5-mm reddish-brown, hyperkeratotic, scaly papules that reveal a moist, erythematous base with pinpoint bleeding upon removal of the scale. Lesions usually are distributed symmetrically and most commonly present on the extensor surfaces of the lower legs and dorsal feet.1,2,7 Lesions also may appear on the extensor surfaces of the arms, pinna, periocular region, antecubital and popliteal fossae, and oral mucosa and also may present as pits on the palms and soles.2,4,7,8 Furthermore, unilateral and localized variants of HLP have been described.11,12 Hyperkeratosis lenticularis perstans usually is asymptomatic but can present with mild pruritus or burning.3,5,13
The etiology and pathogenesis of HLP are unknown. Exposure to UV light has been implicated as an inciting factor14; however, reports of spontaneous resolution in the summer13 and upon treatment with psoralen plus UVA therapy15 make the role of UV light unclear. Furthermore, investigators disagree as to whether the primary pathogenic event in HLP is an inflammatory process or one of abnormal keratinization.1,3,7,10 Fernandez-Flores and Manjon16 suggested HLP is an inflammatory process with periods of exacerbations and remissions after finding mounds of parakeratosis with neutrophils arranged in different strata in the stratum corneum.
Histologically, compact hyperkeratosis usually is noted, often with associated parakeratosis, epidermal atrophy with thinning or absence of the granular layer, and a bandlike lymphohistiocytic infiltrate in the papillary dermis.1-3 Histopathologic differences between recent-onset versus longstanding lesions have been found, with old lesions lacking an inflammatory infiltrate.3 Furthermore, new lesions often show abnormalities in quantity and/or morphology of membrane-coating granules, also known as Odland bodies, in keratinocytes on electron microscopy,3,10,17 while old lesions do not.3 Odland bodies are involved in normal desquamation, leading some to speculate on their role in HLP.10 Currently, it is unclear whether abnormalities in these organelles cause the retention hyperkeratosis seen in HLP or if such abnormalities are a secondary phenomenon.3,17
There are questionable associations between HLP and diabetes mellitus type 2, hyperthyroidism, basal and squamous cell carcinomas of the skin, and gastrointestinal malignancy.4,9,18 Our patient had a history of basal cell carcinoma on the face, diet-controlled diabetes mellitus, and hypothyroidism. Given the high prevalence of these diseases in the general population, however, it is difficult to ascertain whether a true association with HLP exists.
While HLP can slowly progress to involve additional body sites, it is overall a benign condition that does not require treatment. Therapeutic options are based on case reports, with no single treatment showing a consistent response. From review of the literature, therapies that have been most effective include dermabrasion, excision,19 topical 5-fluorouracil,2,17,20 and oral retinoids.8 Hyperkeratosis lenticularis perstans generally is resistant to topical steroids, retinoids, and vitamin D3 analogs, although success with betamethasone dipropionate,5 isotretinoin
gel 0.05%,11 and calcipotriol have been reported.6 A case of HLP with clinical response to psoralen plus UVA therapy also has been described.15
The Diagnosis: Hyperkeratosis Lenticularis Perstans
A shave biopsy of a lesion on the right leg was performed. Histopathology revealed a discrete papule with overlying compact hyperkeratosis. There was parakeratosis with an absent granular layer and a lichenoid lymphocytic infiltrate within the papillary dermis (Figure). Given the clinical context, these changes were consistent with a diagnosis of hyperkeratosis lenticularis perstans (HLP), also known as Flegel disease.
The patient was started on tretinoin cream 0.1% nightly for 3 months and triamcinolone ointment 0.1% as needed for pruritus but showed no clinical response. Given the benign nature of the condition and because the lesions were asymptomatic, additional treatment options were not pursued.
Originally described by Flegel1 in 1958, HLP is a rare skin disorder commonly seen in white individuals with onset in the fourth or fifth decades of life.1,2 While most cases are sporadic,3-6 HLP also has been associated with autosomal dominant inheritance.7-10
Patients with HLP typically present with multiple 1- to 5-mm reddish-brown, hyperkeratotic, scaly papules that reveal a moist, erythematous base with pinpoint bleeding upon removal of the scale. Lesions usually are distributed symmetrically and most commonly present on the extensor surfaces of the lower legs and dorsal feet.1,2,7 Lesions also may appear on the extensor surfaces of the arms, pinna, periocular region, antecubital and popliteal fossae, and oral mucosa and also may present as pits on the palms and soles.2,4,7,8 Furthermore, unilateral and localized variants of HLP have been described.11,12 Hyperkeratosis lenticularis perstans usually is asymptomatic but can present with mild pruritus or burning.3,5,13
The etiology and pathogenesis of HLP are unknown. Exposure to UV light has been implicated as an inciting factor14; however, reports of spontaneous resolution in the summer13 and upon treatment with psoralen plus UVA therapy15 make the role of UV light unclear. Furthermore, investigators disagree as to whether the primary pathogenic event in HLP is an inflammatory process or one of abnormal keratinization.1,3,7,10 Fernandez-Flores and Manjon16 suggested HLP is an inflammatory process with periods of exacerbations and remissions after finding mounds of parakeratosis with neutrophils arranged in different strata in the stratum corneum.
Histologically, compact hyperkeratosis usually is noted, often with associated parakeratosis, epidermal atrophy with thinning or absence of the granular layer, and a bandlike lymphohistiocytic infiltrate in the papillary dermis.1-3 Histopathologic differences between recent-onset versus longstanding lesions have been found, with old lesions lacking an inflammatory infiltrate.3 Furthermore, new lesions often show abnormalities in quantity and/or morphology of membrane-coating granules, also known as Odland bodies, in keratinocytes on electron microscopy,3,10,17 while old lesions do not.3 Odland bodies are involved in normal desquamation, leading some to speculate on their role in HLP.10 Currently, it is unclear whether abnormalities in these organelles cause the retention hyperkeratosis seen in HLP or if such abnormalities are a secondary phenomenon.3,17
There are questionable associations between HLP and diabetes mellitus type 2, hyperthyroidism, basal and squamous cell carcinomas of the skin, and gastrointestinal malignancy.4,9,18 Our patient had a history of basal cell carcinoma on the face, diet-controlled diabetes mellitus, and hypothyroidism. Given the high prevalence of these diseases in the general population, however, it is difficult to ascertain whether a true association with HLP exists.
While HLP can slowly progress to involve additional body sites, it is overall a benign condition that does not require treatment. Therapeutic options are based on case reports, with no single treatment showing a consistent response. From review of the literature, therapies that have been most effective include dermabrasion, excision,19 topical 5-fluorouracil,2,17,20 and oral retinoids.8 Hyperkeratosis lenticularis perstans generally is resistant to topical steroids, retinoids, and vitamin D3 analogs, although success with betamethasone dipropionate,5 isotretinoin
gel 0.05%,11 and calcipotriol have been reported.6 A case of HLP with clinical response to psoralen plus UVA therapy also has been described.15
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt. 1958;9:363-364.
- Pearson LH, Smith JG, Chalker DK. Hyperkeratosis lenticularis perstans (Flegel’s disease). J Am Acad Dermatol. 1987;16:190-195.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel’s disease). Am J Dermatopathol. 2006;28:122-126.
- Fernández-Crehuet P, Rodríguez-Rey E, Ríos-Martín JJ, et al. Hyperkeratosis lenticularis perstans, or Flegel disease, with palmoplantar involvement. Actas Dermosifiliogr. 2009;100:157-159.
- Sterneberg-Vos H, van Marion AM, Frank J, et al. Hyperkeratosis lenticularis perstans (Flegel’s disease)—successful treatment with topical corticosteroids. Int J Dermatol. 2008;47:38-41.
- Bayramgürler D, Apaydin R, Dökmeci S, et al. Flegel’s disease: treatment with topical calcipotriol. Clin Exp Dermatol. 2002;27:161-162.
- Price ML, Jones EW, MacDonald DM. A clinicopathological study of Flegel’s disease (hyperkeratosis lenticularis perstans). Br J Dermatol. 1987;116:681-691.
- Krishnan A, Kar S. Photoletter to the editor: hyperkeratosis lenticularis perstans (Flegel’s disease) with unusual clinical presentation. response to isotretinoin therapy. J Dermatol Case Rep. 2012;6:93-95.
- Beveridge GW, Langlands AO. Familial hyperkeratosis lenticularis perstans associated with tumours of the skin. Br J Dermatol. 1973;88:453-458.
- Frenk E, Tapernoux B. Hyperkeratosis lenticularis perstans (Flegel): a biological model for keratinization occurring in the absence of Odland bodies? Dermatologica. 1976;153:253-262.
- Miranda-Romero A, Sánchez Sambucety P, Bajo del Pozo C, et al. Unilateral hyperkeratosis lenticularis perstans (Flegel's disease). J Am Acad Dermatol. 1998;39:655-657.
- Gutiérrez MC, Hasson A, Arias MD, et al. Localized hyperkeratosis lenticularis perstans (Flegel's disease). Cutis. 1991;48:201-204.
- Fathy S, Azadeh B. Hyperkeratosis lenticularis perstans. Int J Dermatol. 1988;27:120-121.
- Rosdahl I, Rosen K. Hyperkeratosis lenticularis perstans: report on two cases. Acta Derm Venerol. 1985;65:562-564.
- Cooper SM, George S. Flegel's disease treated with psoralen ultraviolet A. Br J Dermatol. 2000;142:340-342.
- Fernandez-Flores A, Manjon JA. Morphological evidence of periodical exacerbation of hyperkeratosis lenticularis perstans. Acta Dermatovenerol Croat. 2009;17:16-19.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Ishibashi A, Tsuboi R, Fujita K. Familial hyperkeratosis lenticularis perstans. associated with cancers of the digestive organs. J Dermatol. 1984;11:407-409.
- Cunha Filho RR, Almeida Jr HL. Hyperkeratosis lenticularis perstans. An Bras Dermatol. 2011;86(4 suppl 1):S76-S77.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)—lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt. 1958;9:363-364.
- Pearson LH, Smith JG, Chalker DK. Hyperkeratosis lenticularis perstans (Flegel’s disease). J Am Acad Dermatol. 1987;16:190-195.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel’s disease). Am J Dermatopathol. 2006;28:122-126.
- Fernández-Crehuet P, Rodríguez-Rey E, Ríos-Martín JJ, et al. Hyperkeratosis lenticularis perstans, or Flegel disease, with palmoplantar involvement. Actas Dermosifiliogr. 2009;100:157-159.
- Sterneberg-Vos H, van Marion AM, Frank J, et al. Hyperkeratosis lenticularis perstans (Flegel’s disease)—successful treatment with topical corticosteroids. Int J Dermatol. 2008;47:38-41.
- Bayramgürler D, Apaydin R, Dökmeci S, et al. Flegel’s disease: treatment with topical calcipotriol. Clin Exp Dermatol. 2002;27:161-162.
- Price ML, Jones EW, MacDonald DM. A clinicopathological study of Flegel’s disease (hyperkeratosis lenticularis perstans). Br J Dermatol. 1987;116:681-691.
- Krishnan A, Kar S. Photoletter to the editor: hyperkeratosis lenticularis perstans (Flegel’s disease) with unusual clinical presentation. response to isotretinoin therapy. J Dermatol Case Rep. 2012;6:93-95.
- Beveridge GW, Langlands AO. Familial hyperkeratosis lenticularis perstans associated with tumours of the skin. Br J Dermatol. 1973;88:453-458.
- Frenk E, Tapernoux B. Hyperkeratosis lenticularis perstans (Flegel): a biological model for keratinization occurring in the absence of Odland bodies? Dermatologica. 1976;153:253-262.
- Miranda-Romero A, Sánchez Sambucety P, Bajo del Pozo C, et al. Unilateral hyperkeratosis lenticularis perstans (Flegel's disease). J Am Acad Dermatol. 1998;39:655-657.
- Gutiérrez MC, Hasson A, Arias MD, et al. Localized hyperkeratosis lenticularis perstans (Flegel's disease). Cutis. 1991;48:201-204.
- Fathy S, Azadeh B. Hyperkeratosis lenticularis perstans. Int J Dermatol. 1988;27:120-121.
- Rosdahl I, Rosen K. Hyperkeratosis lenticularis perstans: report on two cases. Acta Derm Venerol. 1985;65:562-564.
- Cooper SM, George S. Flegel's disease treated with psoralen ultraviolet A. Br J Dermatol. 2000;142:340-342.
- Fernandez-Flores A, Manjon JA. Morphological evidence of periodical exacerbation of hyperkeratosis lenticularis perstans. Acta Dermatovenerol Croat. 2009;17:16-19.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Ishibashi A, Tsuboi R, Fujita K. Familial hyperkeratosis lenticularis perstans. associated with cancers of the digestive organs. J Dermatol. 1984;11:407-409.
- Cunha Filho RR, Almeida Jr HL. Hyperkeratosis lenticularis perstans. An Bras Dermatol. 2011;86(4 suppl 1):S76-S77.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)—lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.

Cutaneous Leishmaniasis
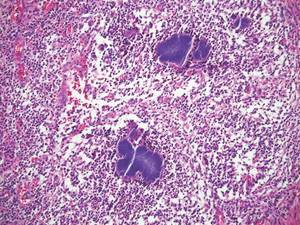
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
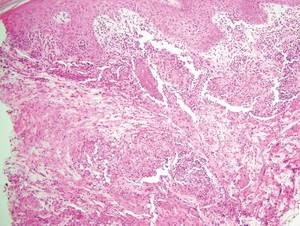

Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
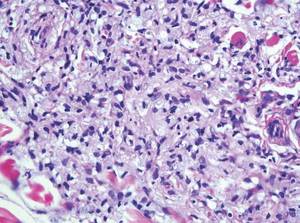
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
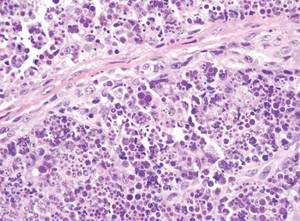
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4

Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4
Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4
Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
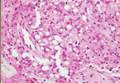
Scaly Plaque With Pustules and Anonychia on the Middle Finger
The Diagnosis: Acrodermatitis Continua of Hallopeau
Acrodermatitis continua of Hallopeau (ACH) is considered to be a form of acropustular psoriasis that presents as a sterile, pustular eruption initially affecting the fingertips and/or toes.1 The slow-growing pustules typically progress locally and can lead to onychodystrophy and/or osteolysis of the underlying bone.2,3 Most commonly affecting adult women, ACH often begins following local trauma to or infection of a single digit.4 As the disease progresses proximally, the small pustules burst, leaving a shiny, erythematous surface on which new pustules can develop. These pustules have a tendency to amalgamate, leading to the characteristic clinical finding of lakes of pus. Pustules frequently appear on the nail matrix and nail bed presenting as severe onychodystrophy and ultimately anonychia.5,6 Rarely, ACH can be associated with generalized pustular psoriasis as well as conjunctivitis, balanitis, and fissuring or annulus migrans of the tongue.2,7
Diagnosis can be established based on clinical findings, biopsy, and bacterial and fungal cultures revealing sterile pustules.8,9 Histologic findings are similar to those seen in pustular psoriasis, demonstrating subcorneal neutrophilic pustules, Munro microabscesses, and dilated blood vessels with lymphocytic infiltrate in the papillary dermis.10
Due to the refractory nature of the disease, there are no recommended guidelines for treatment of ACH. Most successful treatment regimens consist of topical psoriasis medications combined with systemic psoriatic therapies such as cyclosporine, methotrexate, acitretin, or biologic therapy.8,11-16 Our patient achieved satisfactory clinical improvement with clobetasol propionate ointment 0.05% twice daily alternating with calcipotriene cream 0.005% twice daily.
- Suchanek J. Relation of Hallopeau’s acrodermatitis continua to psoriasis. Przegl Dermatol. 1951;1:165-181.
- Adam BA, Loh CL. Acropustulosis (acrodermatitis continua) with resorption of terminal phalanges. Med J Malaysia. 1972;27:30-32.
- Mrowietz U. Pustular eruptions of palms and soles. In: Wolff K, Goldsmith LS, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:215-218.
- Yerushalmi J, Grunwald MH, Hallel-Halevy D, et al. Chronic pustular eruption of the thumbs. diagnosis: acrodermatitis continue of Hallopeau (ACH). Arch Dermatol. 2000:136:925-930.
- Granelli U. Impetigo herpetiformis; acrodermatitis continue of Hallopeau and pustular psoriasis; etiology and pathogenesis and differential diagnosis. Minerva Dermatol. 1956;31:120-126.
- Mobini N, Toussaint S, Kamino H. Noninfectious erythematous, papular, and squamous diseases. In: Elder DE, Elenitsas R, Johnson B, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005:174-210.
- Radcliff-Crocker H. Diseases of the Skin: Their Descriptions, Pathology, Diagnosis and Treatment. Philadelphia, PA: P. Blakiston, Son, & Co; 1888.
- Sehgal VN, Verma P, Sharma S, et al. Review: acrodermatitis continua of Hallopeau: evolution of treatment options. Int J Dermatol. 2011;50:1195-1211.
- Post CF, Hopper ME. Dermatitis repens: a report of two cases with bacteriologic studies. AMA Arc Derm Syphilol. 1951;63:220-223.
- Sehgal VN, Sharma S. The significance of Gram’s stain smear, potassium hydroxide mount, culture and microscopic pathology in the diagnosis of acrodermatitis continua of Hallopeau. Skinmed. 2011;9:260-261.
- Mosser G, Pillekamp H, Peter RU. Suppurative acrodermatitis continua of Hallopeau. a differential diagnosis of paronychia. Dtsch Med Wochenschr. 1998;123:386-390.
- Piquero-Casals J, Fonseca de Mello AP, Dal Coleto C, et al. Using oral tetracycline and topical betamethasone valerate to treat acrodermatitis continua of Hallopeau. Cutis. 2002;70:106-108.
- Tsuji T, Nishimura M. Topically administered fluorouracil in acrodermatitis continua of Hallopeau. Arch Dermatol. 1991;127:27-28.
- Van de Kerkhof PCM. In vivo effects of vitamin D3 analogs. J Dermatolog Treat. 1998;(suppl 3):S25-S29.
- Kokelj F, Plozzer C, Trevisan G. Uselessness of topical calcipotriol as monotherapy for acrodermatitis continua of Hallopeau. Acta Derm Venereol. 2001;81:153.
- Schneider LA, Hinrichs R, Scharffetter-Kochanek K. Phototherapy and photochemotherapy. Clin Dermatol. 2008;26:464-476.
The Diagnosis: Acrodermatitis Continua of Hallopeau
Acrodermatitis continua of Hallopeau (ACH) is considered to be a form of acropustular psoriasis that presents as a sterile, pustular eruption initially affecting the fingertips and/or toes.1 The slow-growing pustules typically progress locally and can lead to onychodystrophy and/or osteolysis of the underlying bone.2,3 Most commonly affecting adult women, ACH often begins following local trauma to or infection of a single digit.4 As the disease progresses proximally, the small pustules burst, leaving a shiny, erythematous surface on which new pustules can develop. These pustules have a tendency to amalgamate, leading to the characteristic clinical finding of lakes of pus. Pustules frequently appear on the nail matrix and nail bed presenting as severe onychodystrophy and ultimately anonychia.5,6 Rarely, ACH can be associated with generalized pustular psoriasis as well as conjunctivitis, balanitis, and fissuring or annulus migrans of the tongue.2,7
Diagnosis can be established based on clinical findings, biopsy, and bacterial and fungal cultures revealing sterile pustules.8,9 Histologic findings are similar to those seen in pustular psoriasis, demonstrating subcorneal neutrophilic pustules, Munro microabscesses, and dilated blood vessels with lymphocytic infiltrate in the papillary dermis.10
Due to the refractory nature of the disease, there are no recommended guidelines for treatment of ACH. Most successful treatment regimens consist of topical psoriasis medications combined with systemic psoriatic therapies such as cyclosporine, methotrexate, acitretin, or biologic therapy.8,11-16 Our patient achieved satisfactory clinical improvement with clobetasol propionate ointment 0.05% twice daily alternating with calcipotriene cream 0.005% twice daily.
The Diagnosis: Acrodermatitis Continua of Hallopeau
Acrodermatitis continua of Hallopeau (ACH) is considered to be a form of acropustular psoriasis that presents as a sterile, pustular eruption initially affecting the fingertips and/or toes.1 The slow-growing pustules typically progress locally and can lead to onychodystrophy and/or osteolysis of the underlying bone.2,3 Most commonly affecting adult women, ACH often begins following local trauma to or infection of a single digit.4 As the disease progresses proximally, the small pustules burst, leaving a shiny, erythematous surface on which new pustules can develop. These pustules have a tendency to amalgamate, leading to the characteristic clinical finding of lakes of pus. Pustules frequently appear on the nail matrix and nail bed presenting as severe onychodystrophy and ultimately anonychia.5,6 Rarely, ACH can be associated with generalized pustular psoriasis as well as conjunctivitis, balanitis, and fissuring or annulus migrans of the tongue.2,7
Diagnosis can be established based on clinical findings, biopsy, and bacterial and fungal cultures revealing sterile pustules.8,9 Histologic findings are similar to those seen in pustular psoriasis, demonstrating subcorneal neutrophilic pustules, Munro microabscesses, and dilated blood vessels with lymphocytic infiltrate in the papillary dermis.10
Due to the refractory nature of the disease, there are no recommended guidelines for treatment of ACH. Most successful treatment regimens consist of topical psoriasis medications combined with systemic psoriatic therapies such as cyclosporine, methotrexate, acitretin, or biologic therapy.8,11-16 Our patient achieved satisfactory clinical improvement with clobetasol propionate ointment 0.05% twice daily alternating with calcipotriene cream 0.005% twice daily.
- Suchanek J. Relation of Hallopeau’s acrodermatitis continua to psoriasis. Przegl Dermatol. 1951;1:165-181.
- Adam BA, Loh CL. Acropustulosis (acrodermatitis continua) with resorption of terminal phalanges. Med J Malaysia. 1972;27:30-32.
- Mrowietz U. Pustular eruptions of palms and soles. In: Wolff K, Goldsmith LS, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:215-218.
- Yerushalmi J, Grunwald MH, Hallel-Halevy D, et al. Chronic pustular eruption of the thumbs. diagnosis: acrodermatitis continue of Hallopeau (ACH). Arch Dermatol. 2000:136:925-930.
- Granelli U. Impetigo herpetiformis; acrodermatitis continue of Hallopeau and pustular psoriasis; etiology and pathogenesis and differential diagnosis. Minerva Dermatol. 1956;31:120-126.
- Mobini N, Toussaint S, Kamino H. Noninfectious erythematous, papular, and squamous diseases. In: Elder DE, Elenitsas R, Johnson B, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005:174-210.
- Radcliff-Crocker H. Diseases of the Skin: Their Descriptions, Pathology, Diagnosis and Treatment. Philadelphia, PA: P. Blakiston, Son, & Co; 1888.
- Sehgal VN, Verma P, Sharma S, et al. Review: acrodermatitis continua of Hallopeau: evolution of treatment options. Int J Dermatol. 2011;50:1195-1211.
- Post CF, Hopper ME. Dermatitis repens: a report of two cases with bacteriologic studies. AMA Arc Derm Syphilol. 1951;63:220-223.
- Sehgal VN, Sharma S. The significance of Gram’s stain smear, potassium hydroxide mount, culture and microscopic pathology in the diagnosis of acrodermatitis continua of Hallopeau. Skinmed. 2011;9:260-261.
- Mosser G, Pillekamp H, Peter RU. Suppurative acrodermatitis continua of Hallopeau. a differential diagnosis of paronychia. Dtsch Med Wochenschr. 1998;123:386-390.
- Piquero-Casals J, Fonseca de Mello AP, Dal Coleto C, et al. Using oral tetracycline and topical betamethasone valerate to treat acrodermatitis continua of Hallopeau. Cutis. 2002;70:106-108.
- Tsuji T, Nishimura M. Topically administered fluorouracil in acrodermatitis continua of Hallopeau. Arch Dermatol. 1991;127:27-28.
- Van de Kerkhof PCM. In vivo effects of vitamin D3 analogs. J Dermatolog Treat. 1998;(suppl 3):S25-S29.
- Kokelj F, Plozzer C, Trevisan G. Uselessness of topical calcipotriol as monotherapy for acrodermatitis continua of Hallopeau. Acta Derm Venereol. 2001;81:153.
- Schneider LA, Hinrichs R, Scharffetter-Kochanek K. Phototherapy and photochemotherapy. Clin Dermatol. 2008;26:464-476.
- Suchanek J. Relation of Hallopeau’s acrodermatitis continua to psoriasis. Przegl Dermatol. 1951;1:165-181.
- Adam BA, Loh CL. Acropustulosis (acrodermatitis continua) with resorption of terminal phalanges. Med J Malaysia. 1972;27:30-32.
- Mrowietz U. Pustular eruptions of palms and soles. In: Wolff K, Goldsmith LS, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:215-218.
- Yerushalmi J, Grunwald MH, Hallel-Halevy D, et al. Chronic pustular eruption of the thumbs. diagnosis: acrodermatitis continue of Hallopeau (ACH). Arch Dermatol. 2000:136:925-930.
- Granelli U. Impetigo herpetiformis; acrodermatitis continue of Hallopeau and pustular psoriasis; etiology and pathogenesis and differential diagnosis. Minerva Dermatol. 1956;31:120-126.
- Mobini N, Toussaint S, Kamino H. Noninfectious erythematous, papular, and squamous diseases. In: Elder DE, Elenitsas R, Johnson B, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005:174-210.
- Radcliff-Crocker H. Diseases of the Skin: Their Descriptions, Pathology, Diagnosis and Treatment. Philadelphia, PA: P. Blakiston, Son, & Co; 1888.
- Sehgal VN, Verma P, Sharma S, et al. Review: acrodermatitis continua of Hallopeau: evolution of treatment options. Int J Dermatol. 2011;50:1195-1211.
- Post CF, Hopper ME. Dermatitis repens: a report of two cases with bacteriologic studies. AMA Arc Derm Syphilol. 1951;63:220-223.
- Sehgal VN, Sharma S. The significance of Gram’s stain smear, potassium hydroxide mount, culture and microscopic pathology in the diagnosis of acrodermatitis continua of Hallopeau. Skinmed. 2011;9:260-261.
- Mosser G, Pillekamp H, Peter RU. Suppurative acrodermatitis continua of Hallopeau. a differential diagnosis of paronychia. Dtsch Med Wochenschr. 1998;123:386-390.
- Piquero-Casals J, Fonseca de Mello AP, Dal Coleto C, et al. Using oral tetracycline and topical betamethasone valerate to treat acrodermatitis continua of Hallopeau. Cutis. 2002;70:106-108.
- Tsuji T, Nishimura M. Topically administered fluorouracil in acrodermatitis continua of Hallopeau. Arch Dermatol. 1991;127:27-28.
- Van de Kerkhof PCM. In vivo effects of vitamin D3 analogs. J Dermatolog Treat. 1998;(suppl 3):S25-S29.
- Kokelj F, Plozzer C, Trevisan G. Uselessness of topical calcipotriol as monotherapy for acrodermatitis continua of Hallopeau. Acta Derm Venereol. 2001;81:153.
- Schneider LA, Hinrichs R, Scharffetter-Kochanek K. Phototherapy and photochemotherapy. Clin Dermatol. 2008;26:464-476.
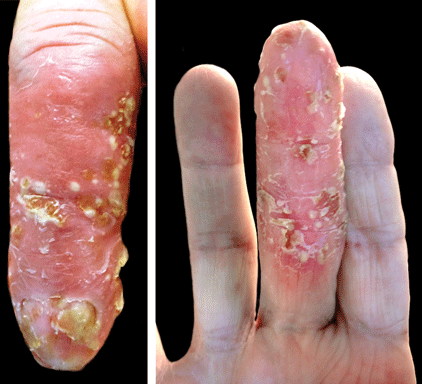
A 69-year-old man presented to our dermatology clinic with a persistent rash on the right middle finger of 5 years’ duration (left). Physical examination revealed a well-demarcated scaly plaque with pustules and anonychia localized to the right middle finger (right). Fungal and bacterial cultures revealed sterile pustules. The patient was successfully treated with an occluded superpotent topical steroid alternating with a topical vitamin D analogue.
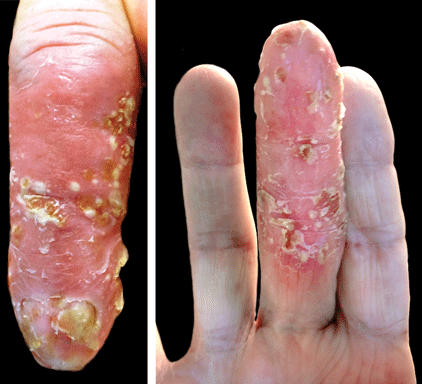
Sessile Pink Plaque on the Lower Back
The Diagnosis: Eccrine Poroma
A shave biopsy of the lesion was performed for definitive diagnosis and demonstrated a well-circumscribed tumor with cords and broad columns composed of uniform basaloid cells extending into the dermis and in areas connecting to the overlying epidermis (Figure). There also were small ducts and cysts admixed in the tumor columns that were embedded in a tumor stroma rich in blood vessels. A diagnosis of eccrine poroma was made based on these characteristic histologic features.
|
Biopsy revealed a basaloid tumor originating from the epidermis and extending into the dermis (A)(H&E, original magnification ×4). On higher magnification, ducts were evident amongst the tumor cells and a vascular rich stroma was revealed (B)(H&E, original magnification ×10). |
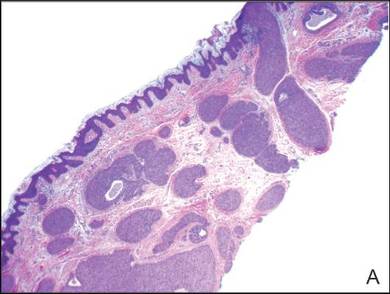

First described by Pinkus et al1 in 1956, eccrine poroma is a benign neoplasm of cells from the intraepidermal ductal portion of the eccrine sweat gland. Eccrine poroma (along with hidroacanthoma simplex, dermal duct tumor, and poroid hidradenoma) is one of the poroid neoplasms, which account for approximately 10% of all primary sweat gland tumors.2 Eccrine poroma usually is seen in patients over 40 years of age without any predilection for race or sex.
Characteristically, eccrine poromas clinically manifest as solitary, firm, sharply demarcated papules or nodules that may be sessile or pedunculated and rarely exceed 2 cm in diameter. This entity classically presents on acral, non–hair-bearing areas (eg, palms and soles). Eccrine poromas have a wide range of clinical appearances that can lead to broad differential diagnoses3 and have been described as flesh-colored,3 pink to red,4 purple,5 and pigmented3,4 papules or nodules depending on features such as blood vessel proliferation and pigment deposition.
Eccrine poromas also have been reported on hair-bearing areas of the body, including the head,3 neck,3,6 chest,4,6 hip,7 and pubic area,8 despite the paucity of eccrine glands in these areas on the body. These findings suggest that these neoplasms may not be purely eccrine in origin. The wide range of clinical presentations of eccrine poromas has prompted investigation into further classification and delineation of this neoplasm.3 The occurrence of eccrine poromas on areas of the skin known to have few eccrine glands suggests that eccrine poromas may not be purely comprised of eccrine ducts and instead may be of apocrine origin.3,9,10 Histologic features of eccrine poromas that suggest apocrine origination include sebaceous and follicular differentiation (eg, folliculocentric distribution), the association with the follicular infundibulum, and the presence of follicular germ cells.3,9,10 Thus, apocrine gland involvement in eccrine poromas may account for their appearance in anatomic areas that do not have high concentrations of eccrine glands, such as the trunk and pubic area.
Based on these findings, eccrine poromas may therefore be of eccrine and/or apocrine origin; however, the nomenclature of this neoplasm remains confusing and possibly misleading, as the term eccrine poroma continues to be accepted even in instances in which the differentiation appears to be largely apocrine. The terms poroma and eccrine poroma often are used interchangeably, which contributes to the confusion by failing to acknowledge the possibility of apocrine influence and possibly causing the clinician to exclude eccrine poromas from the differential diagnosis in areas that do not have high concentrations of eccrine glands.
Because of their high degree of clinical variability, characteristic acral location, and misleading nomenclature, eccrine poromas often are mistakenly confused with a long list of other cutaneous neoplasms, including hemangiomas, pyogenic granulomas, melanocytic nevi, warts, cysts, and other adnexal neoplasms.3 In our case, the lesion was abnormally large and was clinically concerning for an unusual sebaceous nevus. Its location on the lower back is not commonly noted and should remind the clinician of the possibility of apocrine differentiation. Clinicians should be aware of the wide phenotypic diversity of eccrine poromas, and therefore they should consider this diagnosis in their differential diagnosis for solitary papules or nodules occurring in any anatomic area.
- Pinkus H, Rogin JR, Goldman P. Eccrine poroma: tumors exhibiting features of the epidermal sweat duct unit. Arch Dermatol. 1956;74:511-521.
- Pylyser K, Dewolf-Peeters C, Marlen K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167:243-249.
- Moore TO, Orman HL, Orman SK, et al. Poromas of the head and neck. J Am Acad Dermatol. 2001;44:48-52.
- Agarwal S, Kumar B, Sharma N. Nodule on the chest. eccrine poroma. Indian J Dermatol Venereol Leprol. 2009;75:639.
- Ackerman AB, Abenoza P. Neoplasms With Eccrine Differentiation. Philadelphia, PA: Lea & Febinger; 1990:113-185.
- Okun M, Ansell H. Eccrine poroma. report of three cases, two with an unusual location. Arch Dermatol. 1963;88:561-566.
- Sarma DP, Zaman SU, Santos EE, et al. Poroma of the hip and buttock. Dermatol Online J. 2009;15:10.
- Altamura D, Piccolo D, Lozzi GP, et al. Eccrine poroma in an unusual site: a clinical and dermoscopic simulator of amelanotic melanoma. J Am Acad Dermatol. 2005;53:539-541.
- Groben PA, Hitchcock MG, Leshin B, et al. Apocrine poroma, a distinctive case in a patient with nevoid BCC. Am J Dermatopathol. 1992;21:31-33.
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9.
The Diagnosis: Eccrine Poroma
A shave biopsy of the lesion was performed for definitive diagnosis and demonstrated a well-circumscribed tumor with cords and broad columns composed of uniform basaloid cells extending into the dermis and in areas connecting to the overlying epidermis (Figure). There also were small ducts and cysts admixed in the tumor columns that were embedded in a tumor stroma rich in blood vessels. A diagnosis of eccrine poroma was made based on these characteristic histologic features.
|
Biopsy revealed a basaloid tumor originating from the epidermis and extending into the dermis (A)(H&E, original magnification ×4). On higher magnification, ducts were evident amongst the tumor cells and a vascular rich stroma was revealed (B)(H&E, original magnification ×10). |
First described by Pinkus et al1 in 1956, eccrine poroma is a benign neoplasm of cells from the intraepidermal ductal portion of the eccrine sweat gland. Eccrine poroma (along with hidroacanthoma simplex, dermal duct tumor, and poroid hidradenoma) is one of the poroid neoplasms, which account for approximately 10% of all primary sweat gland tumors.2 Eccrine poroma usually is seen in patients over 40 years of age without any predilection for race or sex.
Characteristically, eccrine poromas clinically manifest as solitary, firm, sharply demarcated papules or nodules that may be sessile or pedunculated and rarely exceed 2 cm in diameter. This entity classically presents on acral, non–hair-bearing areas (eg, palms and soles). Eccrine poromas have a wide range of clinical appearances that can lead to broad differential diagnoses3 and have been described as flesh-colored,3 pink to red,4 purple,5 and pigmented3,4 papules or nodules depending on features such as blood vessel proliferation and pigment deposition.
Eccrine poromas also have been reported on hair-bearing areas of the body, including the head,3 neck,3,6 chest,4,6 hip,7 and pubic area,8 despite the paucity of eccrine glands in these areas on the body. These findings suggest that these neoplasms may not be purely eccrine in origin. The wide range of clinical presentations of eccrine poromas has prompted investigation into further classification and delineation of this neoplasm.3 The occurrence of eccrine poromas on areas of the skin known to have few eccrine glands suggests that eccrine poromas may not be purely comprised of eccrine ducts and instead may be of apocrine origin.3,9,10 Histologic features of eccrine poromas that suggest apocrine origination include sebaceous and follicular differentiation (eg, folliculocentric distribution), the association with the follicular infundibulum, and the presence of follicular germ cells.3,9,10 Thus, apocrine gland involvement in eccrine poromas may account for their appearance in anatomic areas that do not have high concentrations of eccrine glands, such as the trunk and pubic area.
Based on these findings, eccrine poromas may therefore be of eccrine and/or apocrine origin; however, the nomenclature of this neoplasm remains confusing and possibly misleading, as the term eccrine poroma continues to be accepted even in instances in which the differentiation appears to be largely apocrine. The terms poroma and eccrine poroma often are used interchangeably, which contributes to the confusion by failing to acknowledge the possibility of apocrine influence and possibly causing the clinician to exclude eccrine poromas from the differential diagnosis in areas that do not have high concentrations of eccrine glands.
Because of their high degree of clinical variability, characteristic acral location, and misleading nomenclature, eccrine poromas often are mistakenly confused with a long list of other cutaneous neoplasms, including hemangiomas, pyogenic granulomas, melanocytic nevi, warts, cysts, and other adnexal neoplasms.3 In our case, the lesion was abnormally large and was clinically concerning for an unusual sebaceous nevus. Its location on the lower back is not commonly noted and should remind the clinician of the possibility of apocrine differentiation. Clinicians should be aware of the wide phenotypic diversity of eccrine poromas, and therefore they should consider this diagnosis in their differential diagnosis for solitary papules or nodules occurring in any anatomic area.
The Diagnosis: Eccrine Poroma
A shave biopsy of the lesion was performed for definitive diagnosis and demonstrated a well-circumscribed tumor with cords and broad columns composed of uniform basaloid cells extending into the dermis and in areas connecting to the overlying epidermis (Figure). There also were small ducts and cysts admixed in the tumor columns that were embedded in a tumor stroma rich in blood vessels. A diagnosis of eccrine poroma was made based on these characteristic histologic features.
|
Biopsy revealed a basaloid tumor originating from the epidermis and extending into the dermis (A)(H&E, original magnification ×4). On higher magnification, ducts were evident amongst the tumor cells and a vascular rich stroma was revealed (B)(H&E, original magnification ×10). |
First described by Pinkus et al1 in 1956, eccrine poroma is a benign neoplasm of cells from the intraepidermal ductal portion of the eccrine sweat gland. Eccrine poroma (along with hidroacanthoma simplex, dermal duct tumor, and poroid hidradenoma) is one of the poroid neoplasms, which account for approximately 10% of all primary sweat gland tumors.2 Eccrine poroma usually is seen in patients over 40 years of age without any predilection for race or sex.
Characteristically, eccrine poromas clinically manifest as solitary, firm, sharply demarcated papules or nodules that may be sessile or pedunculated and rarely exceed 2 cm in diameter. This entity classically presents on acral, non–hair-bearing areas (eg, palms and soles). Eccrine poromas have a wide range of clinical appearances that can lead to broad differential diagnoses3 and have been described as flesh-colored,3 pink to red,4 purple,5 and pigmented3,4 papules or nodules depending on features such as blood vessel proliferation and pigment deposition.
Eccrine poromas also have been reported on hair-bearing areas of the body, including the head,3 neck,3,6 chest,4,6 hip,7 and pubic area,8 despite the paucity of eccrine glands in these areas on the body. These findings suggest that these neoplasms may not be purely eccrine in origin. The wide range of clinical presentations of eccrine poromas has prompted investigation into further classification and delineation of this neoplasm.3 The occurrence of eccrine poromas on areas of the skin known to have few eccrine glands suggests that eccrine poromas may not be purely comprised of eccrine ducts and instead may be of apocrine origin.3,9,10 Histologic features of eccrine poromas that suggest apocrine origination include sebaceous and follicular differentiation (eg, folliculocentric distribution), the association with the follicular infundibulum, and the presence of follicular germ cells.3,9,10 Thus, apocrine gland involvement in eccrine poromas may account for their appearance in anatomic areas that do not have high concentrations of eccrine glands, such as the trunk and pubic area.
Based on these findings, eccrine poromas may therefore be of eccrine and/or apocrine origin; however, the nomenclature of this neoplasm remains confusing and possibly misleading, as the term eccrine poroma continues to be accepted even in instances in which the differentiation appears to be largely apocrine. The terms poroma and eccrine poroma often are used interchangeably, which contributes to the confusion by failing to acknowledge the possibility of apocrine influence and possibly causing the clinician to exclude eccrine poromas from the differential diagnosis in areas that do not have high concentrations of eccrine glands.
Because of their high degree of clinical variability, characteristic acral location, and misleading nomenclature, eccrine poromas often are mistakenly confused with a long list of other cutaneous neoplasms, including hemangiomas, pyogenic granulomas, melanocytic nevi, warts, cysts, and other adnexal neoplasms.3 In our case, the lesion was abnormally large and was clinically concerning for an unusual sebaceous nevus. Its location on the lower back is not commonly noted and should remind the clinician of the possibility of apocrine differentiation. Clinicians should be aware of the wide phenotypic diversity of eccrine poromas, and therefore they should consider this diagnosis in their differential diagnosis for solitary papules or nodules occurring in any anatomic area.
- Pinkus H, Rogin JR, Goldman P. Eccrine poroma: tumors exhibiting features of the epidermal sweat duct unit. Arch Dermatol. 1956;74:511-521.
- Pylyser K, Dewolf-Peeters C, Marlen K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167:243-249.
- Moore TO, Orman HL, Orman SK, et al. Poromas of the head and neck. J Am Acad Dermatol. 2001;44:48-52.
- Agarwal S, Kumar B, Sharma N. Nodule on the chest. eccrine poroma. Indian J Dermatol Venereol Leprol. 2009;75:639.
- Ackerman AB, Abenoza P. Neoplasms With Eccrine Differentiation. Philadelphia, PA: Lea & Febinger; 1990:113-185.
- Okun M, Ansell H. Eccrine poroma. report of three cases, two with an unusual location. Arch Dermatol. 1963;88:561-566.
- Sarma DP, Zaman SU, Santos EE, et al. Poroma of the hip and buttock. Dermatol Online J. 2009;15:10.
- Altamura D, Piccolo D, Lozzi GP, et al. Eccrine poroma in an unusual site: a clinical and dermoscopic simulator of amelanotic melanoma. J Am Acad Dermatol. 2005;53:539-541.
- Groben PA, Hitchcock MG, Leshin B, et al. Apocrine poroma, a distinctive case in a patient with nevoid BCC. Am J Dermatopathol. 1992;21:31-33.
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9.
- Pinkus H, Rogin JR, Goldman P. Eccrine poroma: tumors exhibiting features of the epidermal sweat duct unit. Arch Dermatol. 1956;74:511-521.
- Pylyser K, Dewolf-Peeters C, Marlen K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167:243-249.
- Moore TO, Orman HL, Orman SK, et al. Poromas of the head and neck. J Am Acad Dermatol. 2001;44:48-52.
- Agarwal S, Kumar B, Sharma N. Nodule on the chest. eccrine poroma. Indian J Dermatol Venereol Leprol. 2009;75:639.
- Ackerman AB, Abenoza P. Neoplasms With Eccrine Differentiation. Philadelphia, PA: Lea & Febinger; 1990:113-185.
- Okun M, Ansell H. Eccrine poroma. report of three cases, two with an unusual location. Arch Dermatol. 1963;88:561-566.
- Sarma DP, Zaman SU, Santos EE, et al. Poroma of the hip and buttock. Dermatol Online J. 2009;15:10.
- Altamura D, Piccolo D, Lozzi GP, et al. Eccrine poroma in an unusual site: a clinical and dermoscopic simulator of amelanotic melanoma. J Am Acad Dermatol. 2005;53:539-541.
- Groben PA, Hitchcock MG, Leshin B, et al. Apocrine poroma, a distinctive case in a patient with nevoid BCC. Am J Dermatopathol. 1992;21:31-33.
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9.
A 47-year-old man presented with an asymptomatic, 2.5×1.5-cm, sessile pink plaque with a coalescing papular texture on the lower back of 30 years’ duration. The patient reported that 2 papillated papules with peripheral rims of dark crust had developed in the center of the lesion over the past 6 months. His personal and family histories were unremarkable.
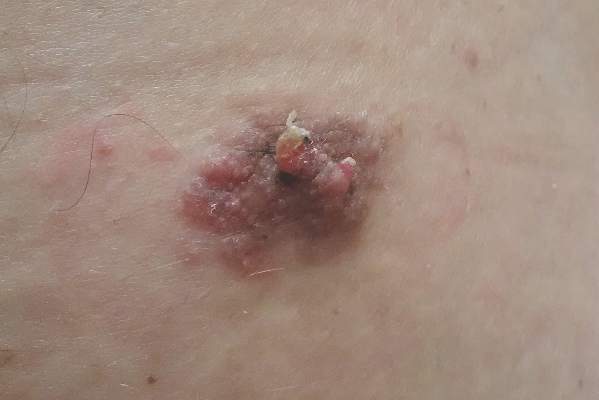
Bullous Henoch-Schönlein Purpura in Children
Henoch-Schönlein purpura (HSP) is a systemic, small vessel vasculitis affecting the skin, joints, gastrointestinal tract, and kidneys. It usually is self-limited, but relapses can be seen in one-third of cases.1 The classic cutaneous presentation includes palpable purpura localized to the legs and buttocks. Painful hemorrhagic bullae are uncommonly observed in childhood HSP and often could lead to a diagnostic dilemma. We report the case of a patient who presented with atypical features of painful hemorrhagic bullae and provide a review of the literature.
Case Report
An otherwise healthy 14-year-old adolescent girl presented to the hospital with painful ulcerative lesions covering the arms, legs, lower abdomen, and buttocks of 3 weeks’ duration. The rash first appeared on the ankles and spread in an ascending fashion, starting with bullous formation that was accompanied by joint pain, especially in the ankles and elbows. No abdominal pain was reported. The patient attributed the lesions to prolonged cold exposure followed by a hot bath. She had tried naproxen without any improvement of pain. She was afebrile with normal blood pressure.
On physical examination, numerous petechiae, palpable purpura, hemorrhagic bullae, and ulcers with surrounding erythematous to violaceous induration as well as central necrosis were noted on the arms, legs (Figure 1), abdomen, and buttocks. The palms, soles, trunk, and face were spared.
Laboratory values on admission revealed leukocytosis (17,500/μL [reference range, 4500–11,000/μL]), elevated erythrocyte sedimentation rate (42 mm/h [reference range, 0–20 mm/h]), elevated C-reactive protein (15.59 mg/L [reference range, 0.08–3.1 mg/L]), elevated C3 (174 mg/dL [reference range, 75–135 mg/dL]), normal C4 (32 mg/dL [reference range, 3–75 mg/dL]), normal blood urea nitrogen (13 mg/dL [reference range, 8–23 mg/dL]), and normal creatinine (0.72 mg/dL [reference range, 0.6–1.2 mg/dL]). Urinalysis showed microscopic hematuria and trace proteinuria. Platelet count was normal.
Diagnostic considerations included HSP, drug-induced leukocytoclastic vasculitis, and bullous pyoderma gangrenosum. The patient was started on oral prednisone 80 mg once daily. Additionally, oral doxycycline 100 mg twice daily was added for prevention of secondary bacterial infections and for anti-inflammatory effects. All nonsteroidal anti-inflammatory drugs were avoided. A commercial ointment containing 8-hydroxyquinoline sulfate 0.3% and triamcinolone acetonide ointment 0.1% were used to minimize skin irritation. Morphine, oxycodone-acetaminophen, and pregabalin followed by gabapentin were used for pain control. Hydrotherapy also was used for the treatment of skin lesions.
Two skin punch biopsies were performed at different stages. Biopsy of an early palpable purpuric lesion showed small vessel leukocytoclastic vasculitis with perivascular IgA on direct immunofluorescence. A second biopsy from a more hemorrhagic lesion performed 96 hours after admission to the hospital showed subepidermal vesicles with partial epidermal necrosis, confluent neutrophilic infiltrate in the papillary dermis, and small vessel vasculitis (Figures 2 and 3). Gram, periodic acid–Schiff, and acid-fast bacilli staining and cultures were negative. With continued treatment for 7 days, the clinical appearance of the lesions improved. On the tenth day of hospitalization, oral dapsone 25 mg once daily was initiated with the goal of weaning the patient off the prednisone as tolerated. She was discharged on prednisone (60 mg once daily) after 14 days of hospitalization. Dapsone also was continued.
| 
| |
Figure 2. Biopsy of a subepidermal bulla revealed neutrophilic inflammation within bullous space and evidence of dermal hemorrhage (H&E, original magnification ×100). | Figure 3. Leukocytoclastic vasculitis on biopsy (H&E, original magnification ×400). |
At 4-week follow-up, the lesions showed healing with mild residual pigmentation. The patient’s blood pressure and serum urea and creatinine levels were normal but the proteinuria was persistent, so the patient was started on oral lisinopril 5 mg once daily. Tapering of steroids over several months was initiated and the dose of dapsone was increased to 50 mg daily. Follow-up with a nephrologist was arranged to monitor renal function. She continued on lisinopril 5 mg once daily for treatment of nonnephrotic-range proteinuria, which was detected at 6 months following discharge.
Comment
The presence of atypical symptoms such as bullae and painful lesions in patients with suspected HSP can complicate the diagnosis. Initially, one of the top diagnostic considerations in our patient was bullous pyoderma gangrenosum, a neutrophilic dermatosis that typically presents with painful ulcerative lesions and inflammatory bullae. Other causes of bullae in children include erythema multiforme, toxic epidermal necrolysis, epidermolysis bullosa, bullous mastocytosis, pemphigus, bullous pemphigoid, dermatitis herpetiformis, linear IgA dermatosis, bullous impetigo, gangrenous cellulitis, and Vibrio vulnificus infection. However, the clinical symptoms of joint pain and hematuria/proteinuria in our patient as well as the punch biopsy findings pointed toward HSP as the most likely diagnosis.
Although bullous lesions are relatively common in adult-onset HSP (16%–60% of patients), they are very rare in pediatric patients (2% of patients).2-4 We performed a PubMed search of articles indexed for MEDLINE for bullous Henoch-Schönlein purpura in childhood using the search term Henoch-Schönlein purpura and bullous. The Table provides a summary of our search results from the English-language literature.5-22
Bullae often develop on several parts of the body but are more commonly observed on the legs.17 Pathergy and edema have been implicated in the pathogenesis, as these findings have been observed in sites such as malleoli and legs, respectively.12 Matrix metalloproteinases secreted in polymorphonuclear neutrophils have been found to be elevated in blister fluid and can cause bullae formation via degrading collagen in the basement membrane.9 Corticosteroids, by virtue of their inhibition of proinflammatory transcription factors (eg, nuclear factor κβ, intranuclear activator protein 1) and decreasing metalloproteinase levels, may be efficacious in bullous HSP. Although there is no consensus, corticosteroid therapy seems to be efficacious in treating the bullae, according to several reports.17-22
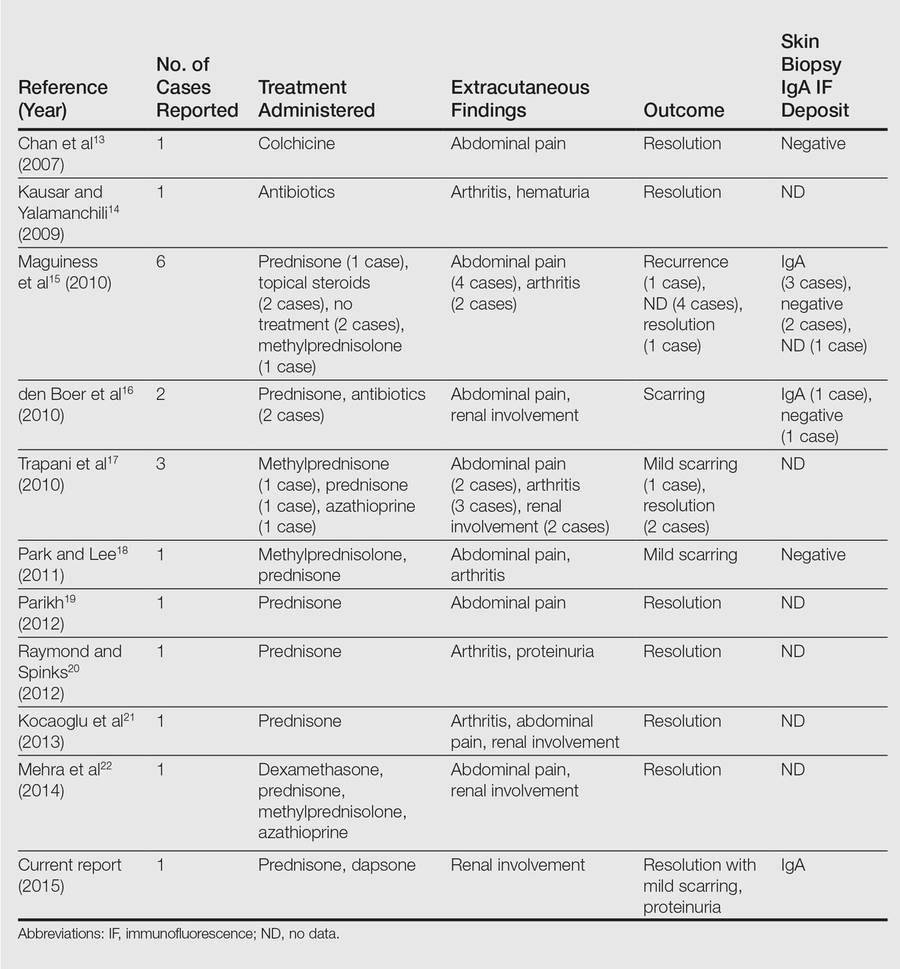
The use of glucocorticoids in bullous HSP in childhood remains controversial. Studies report shortening of the duration of abdominal pain, reducing risk of intussusception, decreasing recurrence risk, and reducing the risk of renal involvement with use of steroids in HSP.23-25 The use of systemic steroids has been described in children with bullous HSP to reduce the severity of HSP-related bullae and its associated painful ulcers and necrosis.16,21,25,26 The duration of steroid use ranged from a short burst to a prolonged course of weaning over weeks. Azathioprine also has been used in conjunction with methylprednisolone, prednisone, and dexamethasone.17,22 Because of its anti-IgA antioxidant antineutrophil effects, dapsone has been shown to be effective in the treatment of cutaneous HSP.27 In our patient, we used dapsone to help in weaning the patient off the prednisone. Based on our review of the literature, few cases of bullous HSP in children have reported remission without drug therapy. IgA was not found in all the reported cases in which a skin biopsy was done. As shown by the comparison of the 2 biopsies in our patient, biopsying an early lesion within 48 hours of appearance is essential to make a diagnosis because the biopsy of the older lesion could not rule out bullous pyoderma gangrenosum. Immunoreactants (IgA, C3) are destroyed within 48 hours and might lead to false-negative results on immunofluorescence in old and necrotic lesions.28,29 Most reported cases of bullous HSP showed resolution, but few resulted in scarring and/or pigmentation.10,17,18 Henoch-Schönlein purpura usually is self-limited but relapses can be seen in one-third of cases.1 One of the reported cases of bullous HSP showed recurrence of lesions.15 One of the cases showed persistent hematuria.8 Our patient also was started on lisinopril for persistent proteinuria.
1. Saulsbury FT. Henoch-Schönlein purpura in children. report of 100 patients and the review of literature. Medicine. 1999;78:395-409.
2. Cream JJ, Gumpel JM, Peachey RD. Schönlein-Henoch purpura in the adult. a study of 77 adults with anaphylactoid or Schönlein-Henoch purpura. Q J Med. 1970;39:461-484.
3. Tancrede-Bohin E, Ochonisky S, Vignon-Pennamen MD, et al. Schönlein-Henoch purpura in adult patients. predictive factors for IgA glomerulonephritis in a retrospective study of 57 cases. Arch Dermatol. 1997;133:438-442.
4. Abdel-Al YK, Hejazi Z, Majeed HA. Henoch Schönlein purpura in Arab children. analysis of 52 cases. Trop Geogr Med. 1990;42:52-57.
5. Garland JS, Chusid MJ. Henoch-Schöenlein purpura: association with unusual vesicular lesions. Wis Med J. 1985;84:21-23.
6. Crosby DL, Feldman SD. A pruritic vesicular eruption. Henoch-Schönlein purpura. Arch Dermatol. 1990;126:1497-1498.
7. Wananukul S, Pongprasit P, Korkij W. Henoch-Schönlein purpura presenting as hemorrhagic vesicles and bullae: case report and literature review. Pediatr Dermatol. 1995;12:314-317.
8. Saulsbury FT. Hemorrhagic bullous lesions in Henoch-Schönlein purpura. Pediatr Dermatol. 1998;15:357-359.
9. Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiological role of gelatinase (MMP-9). Dermatology. 1998;197:62-64.
10. Liu PM, Bong CN, Chen HH, et al. Henoch-Schönlein purpura with hemorrhagic bullae in children: report of two cases. J Microbiol Immunol Infect. 2004;37:375-378.
11. Ishii Y, Takizawa T, Arakawa H, et al. Hemorrhagic bullous lesions in Henoch-Schönlein purpura. Pediatr Int. 2005;47:694-697.
12. Leung AK, Robson WL. Hemorrhagic bullous lesions in a child with Henoch-Schönlein purpura. Pediatr Dermatol. 2006;23:139-141.
13. Chan K, Han N, Tang W, et al. Lesions in Henoch-Schönlein purpura. Pediatr Dermatol. 2007;24: 325-326.
14. Kausar S, Yalamanchili A. Management of haemorrhagic bullous lesions in Henoch-Schonlein purpura: is there any consensus? J Dermatolog Treat. 2009;20:88-90.
15. Maguiness S, Balma-Mena A, Pope E, et al. Bullous Henoch-Schönlein purpura in children: a report of 6 cases and review of the literature. Clin Pediatr. 2010;49: 1033-1037.
16. den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch-Schönlein purpura as indication to start systemic prednisone. Acta Paediatr. 2010;99:781-783.
17. Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature. Rheumatol Int. 2010;30:1355-1359.
18. Park SE, Lee JH. Haemorrhagic bullous lesions in a 3-year-old girl with Henoch-Schönlein purpura. Acta Paediatr. 2011;100:e283-e284.
19. Parikh K. 14-year-old boy with bullous lesions. Pediatr Ann. 2012;41:275-277.
20. Raymond M, Spinks J. Bullous Henoch Schönlein purpura. Arch Dis Child. 2012;97:617.
21. Kocaoglu C, Ozturk R, Unlu Y, et al. Successful treatment of hemorrhagic bullous Henoch-Schönlein purpura with oral corticosteroid: a case report [published online ahead of print April 16, 2013]. Case Rep Pediatr. 2013;2013:680208.
22. Mehra S, Suri D, Dogra S, et al. Hemorrhagic bullous lesions in a girl with Henoch Schönlein purpura. Indian J Pediatr. 2014;81:210-211.
23. Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schönlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr. 2006;149:241-247.
24. Weiss PF, Klink AJ, Localio R, et al. Corticosteroids may improve clinical outcomes during hospitalization for Henoch-Schönlein purpura. Pediatrics. 2010;126:674-681.
25. Rosato L, Chehade H, Cachat F. Re: steroids in haemorrhagic bullous Henoch-Schönlein purpura. Acta Paediatr. 2011;100:319-320.
26. Park SJ, Kim JH, Ha TS, et al. The role of corticosteroid in hemorrhagic bullous Henoch Schönlein purpura. Acta Paediatr. 2011;100:e3-e4.
27. Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
28. Davin JC, Weening JJ. Diagnosis of Henoch-Schönlein purpura: renal or skin biopsy? Pediatr Nephrol. 2003;18:1201-1203.
29. González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48: 1157-1165.
Henoch-Schönlein purpura (HSP) is a systemic, small vessel vasculitis affecting the skin, joints, gastrointestinal tract, and kidneys. It usually is self-limited, but relapses can be seen in one-third of cases.1 The classic cutaneous presentation includes palpable purpura localized to the legs and buttocks. Painful hemorrhagic bullae are uncommonly observed in childhood HSP and often could lead to a diagnostic dilemma. We report the case of a patient who presented with atypical features of painful hemorrhagic bullae and provide a review of the literature.
Case Report
An otherwise healthy 14-year-old adolescent girl presented to the hospital with painful ulcerative lesions covering the arms, legs, lower abdomen, and buttocks of 3 weeks’ duration. The rash first appeared on the ankles and spread in an ascending fashion, starting with bullous formation that was accompanied by joint pain, especially in the ankles and elbows. No abdominal pain was reported. The patient attributed the lesions to prolonged cold exposure followed by a hot bath. She had tried naproxen without any improvement of pain. She was afebrile with normal blood pressure.
On physical examination, numerous petechiae, palpable purpura, hemorrhagic bullae, and ulcers with surrounding erythematous to violaceous induration as well as central necrosis were noted on the arms, legs (Figure 1), abdomen, and buttocks. The palms, soles, trunk, and face were spared.
Laboratory values on admission revealed leukocytosis (17,500/μL [reference range, 4500–11,000/μL]), elevated erythrocyte sedimentation rate (42 mm/h [reference range, 0–20 mm/h]), elevated C-reactive protein (15.59 mg/L [reference range, 0.08–3.1 mg/L]), elevated C3 (174 mg/dL [reference range, 75–135 mg/dL]), normal C4 (32 mg/dL [reference range, 3–75 mg/dL]), normal blood urea nitrogen (13 mg/dL [reference range, 8–23 mg/dL]), and normal creatinine (0.72 mg/dL [reference range, 0.6–1.2 mg/dL]). Urinalysis showed microscopic hematuria and trace proteinuria. Platelet count was normal.
Diagnostic considerations included HSP, drug-induced leukocytoclastic vasculitis, and bullous pyoderma gangrenosum. The patient was started on oral prednisone 80 mg once daily. Additionally, oral doxycycline 100 mg twice daily was added for prevention of secondary bacterial infections and for anti-inflammatory effects. All nonsteroidal anti-inflammatory drugs were avoided. A commercial ointment containing 8-hydroxyquinoline sulfate 0.3% and triamcinolone acetonide ointment 0.1% were used to minimize skin irritation. Morphine, oxycodone-acetaminophen, and pregabalin followed by gabapentin were used for pain control. Hydrotherapy also was used for the treatment of skin lesions.
Two skin punch biopsies were performed at different stages. Biopsy of an early palpable purpuric lesion showed small vessel leukocytoclastic vasculitis with perivascular IgA on direct immunofluorescence. A second biopsy from a more hemorrhagic lesion performed 96 hours after admission to the hospital showed subepidermal vesicles with partial epidermal necrosis, confluent neutrophilic infiltrate in the papillary dermis, and small vessel vasculitis (Figures 2 and 3). Gram, periodic acid–Schiff, and acid-fast bacilli staining and cultures were negative. With continued treatment for 7 days, the clinical appearance of the lesions improved. On the tenth day of hospitalization, oral dapsone 25 mg once daily was initiated with the goal of weaning the patient off the prednisone as tolerated. She was discharged on prednisone (60 mg once daily) after 14 days of hospitalization. Dapsone also was continued.
|
|
| |
Figure 2. Biopsy of a subepidermal bulla revealed neutrophilic inflammation within bullous space and evidence of dermal hemorrhage (H&E, original magnification ×100). | Figure 3. Leukocytoclastic vasculitis on biopsy (H&E, original magnification ×400). |
At 4-week follow-up, the lesions showed healing with mild residual pigmentation. The patient’s blood pressure and serum urea and creatinine levels were normal but the proteinuria was persistent, so the patient was started on oral lisinopril 5 mg once daily. Tapering of steroids over several months was initiated and the dose of dapsone was increased to 50 mg daily. Follow-up with a nephrologist was arranged to monitor renal function. She continued on lisinopril 5 mg once daily for treatment of nonnephrotic-range proteinuria, which was detected at 6 months following discharge.
Comment
The presence of atypical symptoms such as bullae and painful lesions in patients with suspected HSP can complicate the diagnosis. Initially, one of the top diagnostic considerations in our patient was bullous pyoderma gangrenosum, a neutrophilic dermatosis that typically presents with painful ulcerative lesions and inflammatory bullae. Other causes of bullae in children include erythema multiforme, toxic epidermal necrolysis, epidermolysis bullosa, bullous mastocytosis, pemphigus, bullous pemphigoid, dermatitis herpetiformis, linear IgA dermatosis, bullous impetigo, gangrenous cellulitis, and Vibrio vulnificus infection. However, the clinical symptoms of joint pain and hematuria/proteinuria in our patient as well as the punch biopsy findings pointed toward HSP as the most likely diagnosis.
Although bullous lesions are relatively common in adult-onset HSP (16%–60% of patients), they are very rare in pediatric patients (2% of patients).2-4 We performed a PubMed search of articles indexed for MEDLINE for bullous Henoch-Schönlein purpura in childhood using the search term Henoch-Schönlein purpura and bullous. The Table provides a summary of our search results from the English-language literature.5-22
Bullae often develop on several parts of the body but are more commonly observed on the legs.17 Pathergy and edema have been implicated in the pathogenesis, as these findings have been observed in sites such as malleoli and legs, respectively.12 Matrix metalloproteinases secreted in polymorphonuclear neutrophils have been found to be elevated in blister fluid and can cause bullae formation via degrading collagen in the basement membrane.9 Corticosteroids, by virtue of their inhibition of proinflammatory transcription factors (eg, nuclear factor κβ, intranuclear activator protein 1) and decreasing metalloproteinase levels, may be efficacious in bullous HSP. Although there is no consensus, corticosteroid therapy seems to be efficacious in treating the bullae, according to several reports.17-22
The use of glucocorticoids in bullous HSP in childhood remains controversial. Studies report shortening of the duration of abdominal pain, reducing risk of intussusception, decreasing recurrence risk, and reducing the risk of renal involvement with use of steroids in HSP.23-25 The use of systemic steroids has been described in children with bullous HSP to reduce the severity of HSP-related bullae and its associated painful ulcers and necrosis.16,21,25,26 The duration of steroid use ranged from a short burst to a prolonged course of weaning over weeks. Azathioprine also has been used in conjunction with methylprednisolone, prednisone, and dexamethasone.17,22 Because of its anti-IgA antioxidant antineutrophil effects, dapsone has been shown to be effective in the treatment of cutaneous HSP.27 In our patient, we used dapsone to help in weaning the patient off the prednisone. Based on our review of the literature, few cases of bullous HSP in children have reported remission without drug therapy. IgA was not found in all the reported cases in which a skin biopsy was done. As shown by the comparison of the 2 biopsies in our patient, biopsying an early lesion within 48 hours of appearance is essential to make a diagnosis because the biopsy of the older lesion could not rule out bullous pyoderma gangrenosum. Immunoreactants (IgA, C3) are destroyed within 48 hours and might lead to false-negative results on immunofluorescence in old and necrotic lesions.28,29 Most reported cases of bullous HSP showed resolution, but few resulted in scarring and/or pigmentation.10,17,18 Henoch-Schönlein purpura usually is self-limited but relapses can be seen in one-third of cases.1 One of the reported cases of bullous HSP showed recurrence of lesions.15 One of the cases showed persistent hematuria.8 Our patient also was started on lisinopril for persistent proteinuria.
Henoch-Schönlein purpura (HSP) is a systemic, small vessel vasculitis affecting the skin, joints, gastrointestinal tract, and kidneys. It usually is self-limited, but relapses can be seen in one-third of cases.1 The classic cutaneous presentation includes palpable purpura localized to the legs and buttocks. Painful hemorrhagic bullae are uncommonly observed in childhood HSP and often could lead to a diagnostic dilemma. We report the case of a patient who presented with atypical features of painful hemorrhagic bullae and provide a review of the literature.
Case Report
An otherwise healthy 14-year-old adolescent girl presented to the hospital with painful ulcerative lesions covering the arms, legs, lower abdomen, and buttocks of 3 weeks’ duration. The rash first appeared on the ankles and spread in an ascending fashion, starting with bullous formation that was accompanied by joint pain, especially in the ankles and elbows. No abdominal pain was reported. The patient attributed the lesions to prolonged cold exposure followed by a hot bath. She had tried naproxen without any improvement of pain. She was afebrile with normal blood pressure.
On physical examination, numerous petechiae, palpable purpura, hemorrhagic bullae, and ulcers with surrounding erythematous to violaceous induration as well as central necrosis were noted on the arms, legs (Figure 1), abdomen, and buttocks. The palms, soles, trunk, and face were spared.
Laboratory values on admission revealed leukocytosis (17,500/μL [reference range, 4500–11,000/μL]), elevated erythrocyte sedimentation rate (42 mm/h [reference range, 0–20 mm/h]), elevated C-reactive protein (15.59 mg/L [reference range, 0.08–3.1 mg/L]), elevated C3 (174 mg/dL [reference range, 75–135 mg/dL]), normal C4 (32 mg/dL [reference range, 3–75 mg/dL]), normal blood urea nitrogen (13 mg/dL [reference range, 8–23 mg/dL]), and normal creatinine (0.72 mg/dL [reference range, 0.6–1.2 mg/dL]). Urinalysis showed microscopic hematuria and trace proteinuria. Platelet count was normal.
Diagnostic considerations included HSP, drug-induced leukocytoclastic vasculitis, and bullous pyoderma gangrenosum. The patient was started on oral prednisone 80 mg once daily. Additionally, oral doxycycline 100 mg twice daily was added for prevention of secondary bacterial infections and for anti-inflammatory effects. All nonsteroidal anti-inflammatory drugs were avoided. A commercial ointment containing 8-hydroxyquinoline sulfate 0.3% and triamcinolone acetonide ointment 0.1% were used to minimize skin irritation. Morphine, oxycodone-acetaminophen, and pregabalin followed by gabapentin were used for pain control. Hydrotherapy also was used for the treatment of skin lesions.
Two skin punch biopsies were performed at different stages. Biopsy of an early palpable purpuric lesion showed small vessel leukocytoclastic vasculitis with perivascular IgA on direct immunofluorescence. A second biopsy from a more hemorrhagic lesion performed 96 hours after admission to the hospital showed subepidermal vesicles with partial epidermal necrosis, confluent neutrophilic infiltrate in the papillary dermis, and small vessel vasculitis (Figures 2 and 3). Gram, periodic acid–Schiff, and acid-fast bacilli staining and cultures were negative. With continued treatment for 7 days, the clinical appearance of the lesions improved. On the tenth day of hospitalization, oral dapsone 25 mg once daily was initiated with the goal of weaning the patient off the prednisone as tolerated. She was discharged on prednisone (60 mg once daily) after 14 days of hospitalization. Dapsone also was continued.
|
|
| |
Figure 2. Biopsy of a subepidermal bulla revealed neutrophilic inflammation within bullous space and evidence of dermal hemorrhage (H&E, original magnification ×100). | Figure 3. Leukocytoclastic vasculitis on biopsy (H&E, original magnification ×400). |
At 4-week follow-up, the lesions showed healing with mild residual pigmentation. The patient’s blood pressure and serum urea and creatinine levels were normal but the proteinuria was persistent, so the patient was started on oral lisinopril 5 mg once daily. Tapering of steroids over several months was initiated and the dose of dapsone was increased to 50 mg daily. Follow-up with a nephrologist was arranged to monitor renal function. She continued on lisinopril 5 mg once daily for treatment of nonnephrotic-range proteinuria, which was detected at 6 months following discharge.
Comment
The presence of atypical symptoms such as bullae and painful lesions in patients with suspected HSP can complicate the diagnosis. Initially, one of the top diagnostic considerations in our patient was bullous pyoderma gangrenosum, a neutrophilic dermatosis that typically presents with painful ulcerative lesions and inflammatory bullae. Other causes of bullae in children include erythema multiforme, toxic epidermal necrolysis, epidermolysis bullosa, bullous mastocytosis, pemphigus, bullous pemphigoid, dermatitis herpetiformis, linear IgA dermatosis, bullous impetigo, gangrenous cellulitis, and Vibrio vulnificus infection. However, the clinical symptoms of joint pain and hematuria/proteinuria in our patient as well as the punch biopsy findings pointed toward HSP as the most likely diagnosis.
Although bullous lesions are relatively common in adult-onset HSP (16%–60% of patients), they are very rare in pediatric patients (2% of patients).2-4 We performed a PubMed search of articles indexed for MEDLINE for bullous Henoch-Schönlein purpura in childhood using the search term Henoch-Schönlein purpura and bullous. The Table provides a summary of our search results from the English-language literature.5-22
Bullae often develop on several parts of the body but are more commonly observed on the legs.17 Pathergy and edema have been implicated in the pathogenesis, as these findings have been observed in sites such as malleoli and legs, respectively.12 Matrix metalloproteinases secreted in polymorphonuclear neutrophils have been found to be elevated in blister fluid and can cause bullae formation via degrading collagen in the basement membrane.9 Corticosteroids, by virtue of their inhibition of proinflammatory transcription factors (eg, nuclear factor κβ, intranuclear activator protein 1) and decreasing metalloproteinase levels, may be efficacious in bullous HSP. Although there is no consensus, corticosteroid therapy seems to be efficacious in treating the bullae, according to several reports.17-22
The use of glucocorticoids in bullous HSP in childhood remains controversial. Studies report shortening of the duration of abdominal pain, reducing risk of intussusception, decreasing recurrence risk, and reducing the risk of renal involvement with use of steroids in HSP.23-25 The use of systemic steroids has been described in children with bullous HSP to reduce the severity of HSP-related bullae and its associated painful ulcers and necrosis.16,21,25,26 The duration of steroid use ranged from a short burst to a prolonged course of weaning over weeks. Azathioprine also has been used in conjunction with methylprednisolone, prednisone, and dexamethasone.17,22 Because of its anti-IgA antioxidant antineutrophil effects, dapsone has been shown to be effective in the treatment of cutaneous HSP.27 In our patient, we used dapsone to help in weaning the patient off the prednisone. Based on our review of the literature, few cases of bullous HSP in children have reported remission without drug therapy. IgA was not found in all the reported cases in which a skin biopsy was done. As shown by the comparison of the 2 biopsies in our patient, biopsying an early lesion within 48 hours of appearance is essential to make a diagnosis because the biopsy of the older lesion could not rule out bullous pyoderma gangrenosum. Immunoreactants (IgA, C3) are destroyed within 48 hours and might lead to false-negative results on immunofluorescence in old and necrotic lesions.28,29 Most reported cases of bullous HSP showed resolution, but few resulted in scarring and/or pigmentation.10,17,18 Henoch-Schönlein purpura usually is self-limited but relapses can be seen in one-third of cases.1 One of the reported cases of bullous HSP showed recurrence of lesions.15 One of the cases showed persistent hematuria.8 Our patient also was started on lisinopril for persistent proteinuria.
1. Saulsbury FT. Henoch-Schönlein purpura in children. report of 100 patients and the review of literature. Medicine. 1999;78:395-409.
2. Cream JJ, Gumpel JM, Peachey RD. Schönlein-Henoch purpura in the adult. a study of 77 adults with anaphylactoid or Schönlein-Henoch purpura. Q J Med. 1970;39:461-484.
3. Tancrede-Bohin E, Ochonisky S, Vignon-Pennamen MD, et al. Schönlein-Henoch purpura in adult patients. predictive factors for IgA glomerulonephritis in a retrospective study of 57 cases. Arch Dermatol. 1997;133:438-442.
4. Abdel-Al YK, Hejazi Z, Majeed HA. Henoch Schönlein purpura in Arab children. analysis of 52 cases. Trop Geogr Med. 1990;42:52-57.
5. Garland JS, Chusid MJ. Henoch-Schöenlein purpura: association with unusual vesicular lesions. Wis Med J. 1985;84:21-23.
6. Crosby DL, Feldman SD. A pruritic vesicular eruption. Henoch-Schönlein purpura. Arch Dermatol. 1990;126:1497-1498.
7. Wananukul S, Pongprasit P, Korkij W. Henoch-Schönlein purpura presenting as hemorrhagic vesicles and bullae: case report and literature review. Pediatr Dermatol. 1995;12:314-317.
8. Saulsbury FT. Hemorrhagic bullous lesions in Henoch-Schönlein purpura. Pediatr Dermatol. 1998;15:357-359.
9. Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiological role of gelatinase (MMP-9). Dermatology. 1998;197:62-64.
10. Liu PM, Bong CN, Chen HH, et al. Henoch-Schönlein purpura with hemorrhagic bullae in children: report of two cases. J Microbiol Immunol Infect. 2004;37:375-378.
11. Ishii Y, Takizawa T, Arakawa H, et al. Hemorrhagic bullous lesions in Henoch-Schönlein purpura. Pediatr Int. 2005;47:694-697.
12. Leung AK, Robson WL. Hemorrhagic bullous lesions in a child with Henoch-Schönlein purpura. Pediatr Dermatol. 2006;23:139-141.
13. Chan K, Han N, Tang W, et al. Lesions in Henoch-Schönlein purpura. Pediatr Dermatol. 2007;24: 325-326.
14. Kausar S, Yalamanchili A. Management of haemorrhagic bullous lesions in Henoch-Schonlein purpura: is there any consensus? J Dermatolog Treat. 2009;20:88-90.
15. Maguiness S, Balma-Mena A, Pope E, et al. Bullous Henoch-Schönlein purpura in children: a report of 6 cases and review of the literature. Clin Pediatr. 2010;49: 1033-1037.
16. den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch-Schönlein purpura as indication to start systemic prednisone. Acta Paediatr. 2010;99:781-783.
17. Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature. Rheumatol Int. 2010;30:1355-1359.
18. Park SE, Lee JH. Haemorrhagic bullous lesions in a 3-year-old girl with Henoch-Schönlein purpura. Acta Paediatr. 2011;100:e283-e284.
19. Parikh K. 14-year-old boy with bullous lesions. Pediatr Ann. 2012;41:275-277.
20. Raymond M, Spinks J. Bullous Henoch Schönlein purpura. Arch Dis Child. 2012;97:617.
21. Kocaoglu C, Ozturk R, Unlu Y, et al. Successful treatment of hemorrhagic bullous Henoch-Schönlein purpura with oral corticosteroid: a case report [published online ahead of print April 16, 2013]. Case Rep Pediatr. 2013;2013:680208.
22. Mehra S, Suri D, Dogra S, et al. Hemorrhagic bullous lesions in a girl with Henoch Schönlein purpura. Indian J Pediatr. 2014;81:210-211.
23. Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schönlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr. 2006;149:241-247.
24. Weiss PF, Klink AJ, Localio R, et al. Corticosteroids may improve clinical outcomes during hospitalization for Henoch-Schönlein purpura. Pediatrics. 2010;126:674-681.
25. Rosato L, Chehade H, Cachat F. Re: steroids in haemorrhagic bullous Henoch-Schönlein purpura. Acta Paediatr. 2011;100:319-320.
26. Park SJ, Kim JH, Ha TS, et al. The role of corticosteroid in hemorrhagic bullous Henoch Schönlein purpura. Acta Paediatr. 2011;100:e3-e4.
27. Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
28. Davin JC, Weening JJ. Diagnosis of Henoch-Schönlein purpura: renal or skin biopsy? Pediatr Nephrol. 2003;18:1201-1203.
29. González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48: 1157-1165.
1. Saulsbury FT. Henoch-Schönlein purpura in children. report of 100 patients and the review of literature. Medicine. 1999;78:395-409.
2. Cream JJ, Gumpel JM, Peachey RD. Schönlein-Henoch purpura in the adult. a study of 77 adults with anaphylactoid or Schönlein-Henoch purpura. Q J Med. 1970;39:461-484.
3. Tancrede-Bohin E, Ochonisky S, Vignon-Pennamen MD, et al. Schönlein-Henoch purpura in adult patients. predictive factors for IgA glomerulonephritis in a retrospective study of 57 cases. Arch Dermatol. 1997;133:438-442.
4. Abdel-Al YK, Hejazi Z, Majeed HA. Henoch Schönlein purpura in Arab children. analysis of 52 cases. Trop Geogr Med. 1990;42:52-57.
5. Garland JS, Chusid MJ. Henoch-Schöenlein purpura: association with unusual vesicular lesions. Wis Med J. 1985;84:21-23.
6. Crosby DL, Feldman SD. A pruritic vesicular eruption. Henoch-Schönlein purpura. Arch Dermatol. 1990;126:1497-1498.
7. Wananukul S, Pongprasit P, Korkij W. Henoch-Schönlein purpura presenting as hemorrhagic vesicles and bullae: case report and literature review. Pediatr Dermatol. 1995;12:314-317.
8. Saulsbury FT. Hemorrhagic bullous lesions in Henoch-Schönlein purpura. Pediatr Dermatol. 1998;15:357-359.
9. Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiological role of gelatinase (MMP-9). Dermatology. 1998;197:62-64.
10. Liu PM, Bong CN, Chen HH, et al. Henoch-Schönlein purpura with hemorrhagic bullae in children: report of two cases. J Microbiol Immunol Infect. 2004;37:375-378.
11. Ishii Y, Takizawa T, Arakawa H, et al. Hemorrhagic bullous lesions in Henoch-Schönlein purpura. Pediatr Int. 2005;47:694-697.
12. Leung AK, Robson WL. Hemorrhagic bullous lesions in a child with Henoch-Schönlein purpura. Pediatr Dermatol. 2006;23:139-141.
13. Chan K, Han N, Tang W, et al. Lesions in Henoch-Schönlein purpura. Pediatr Dermatol. 2007;24: 325-326.
14. Kausar S, Yalamanchili A. Management of haemorrhagic bullous lesions in Henoch-Schonlein purpura: is there any consensus? J Dermatolog Treat. 2009;20:88-90.
15. Maguiness S, Balma-Mena A, Pope E, et al. Bullous Henoch-Schönlein purpura in children: a report of 6 cases and review of the literature. Clin Pediatr. 2010;49: 1033-1037.
16. den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch-Schönlein purpura as indication to start systemic prednisone. Acta Paediatr. 2010;99:781-783.
17. Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature. Rheumatol Int. 2010;30:1355-1359.
18. Park SE, Lee JH. Haemorrhagic bullous lesions in a 3-year-old girl with Henoch-Schönlein purpura. Acta Paediatr. 2011;100:e283-e284.
19. Parikh K. 14-year-old boy with bullous lesions. Pediatr Ann. 2012;41:275-277.
20. Raymond M, Spinks J. Bullous Henoch Schönlein purpura. Arch Dis Child. 2012;97:617.
21. Kocaoglu C, Ozturk R, Unlu Y, et al. Successful treatment of hemorrhagic bullous Henoch-Schönlein purpura with oral corticosteroid: a case report [published online ahead of print April 16, 2013]. Case Rep Pediatr. 2013;2013:680208.
22. Mehra S, Suri D, Dogra S, et al. Hemorrhagic bullous lesions in a girl with Henoch Schönlein purpura. Indian J Pediatr. 2014;81:210-211.
23. Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schönlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr. 2006;149:241-247.
24. Weiss PF, Klink AJ, Localio R, et al. Corticosteroids may improve clinical outcomes during hospitalization for Henoch-Schönlein purpura. Pediatrics. 2010;126:674-681.
25. Rosato L, Chehade H, Cachat F. Re: steroids in haemorrhagic bullous Henoch-Schönlein purpura. Acta Paediatr. 2011;100:319-320.
26. Park SJ, Kim JH, Ha TS, et al. The role of corticosteroid in hemorrhagic bullous Henoch Schönlein purpura. Acta Paediatr. 2011;100:e3-e4.
27. Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
28. Davin JC, Weening JJ. Diagnosis of Henoch-Schönlein purpura: renal or skin biopsy? Pediatr Nephrol. 2003;18:1201-1203.
29. González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48: 1157-1165.
Practice Points
- The presence of painful hemorrhagic bullae is an uncommon presentation in pediatric patients with Henoch-Schönlein purpura (HSP) and can be a diagnostic challenge.
- Presence of joint pain, abdominal pain, or nephritis could corroborate the diagnosis.
- Early biopsy of the lesion within 48 hours of appearance is important for diagnosis. Presence of IgA deposits on immunofluorescence may aid in diagnosis.
- This finding of bullae in HSP does not seem to have any prognostic significance. Because of the rarity of incidence, there is no consensus on management. Supportive therapy and/or corticosteroids might be effective.
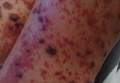
Chromoblastomycosis
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
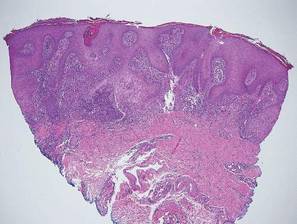

Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
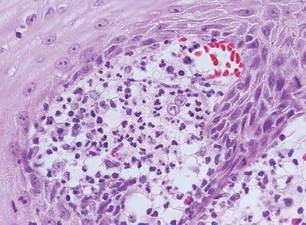
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
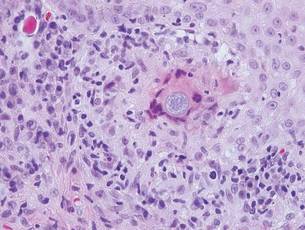
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
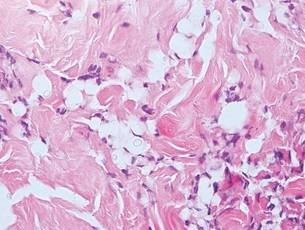
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
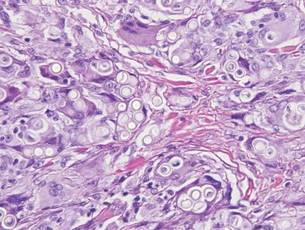
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Syringoid Eccrine Carcinoma
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
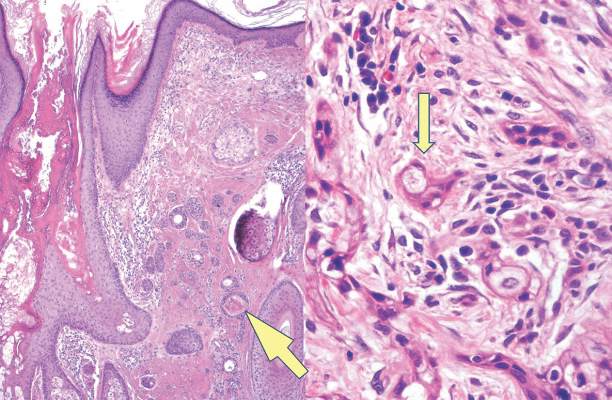
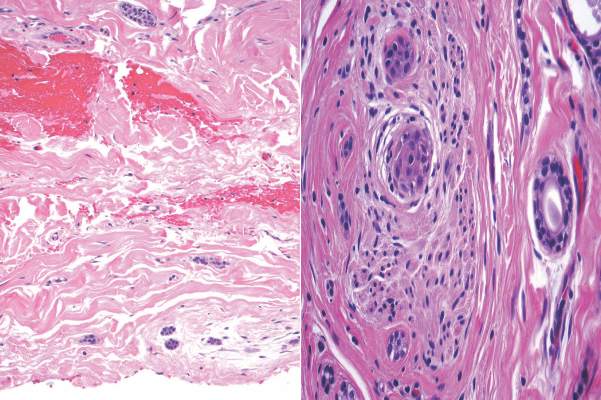
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
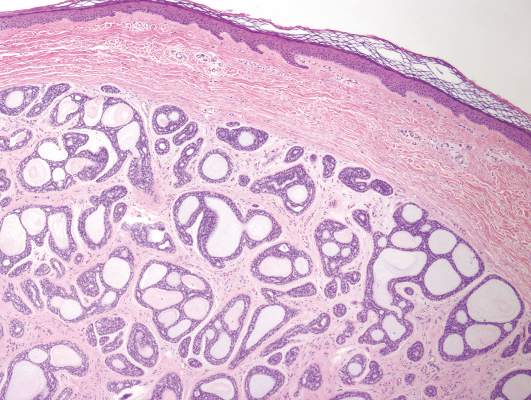
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
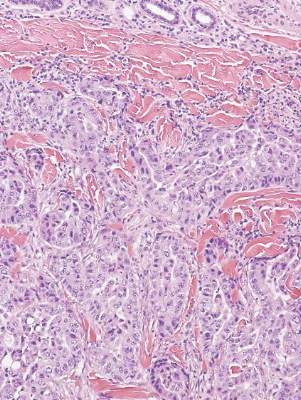
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
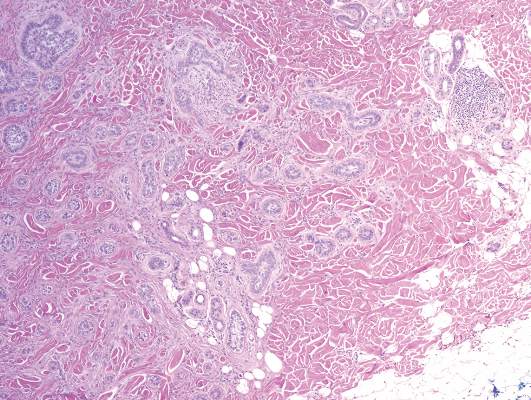
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
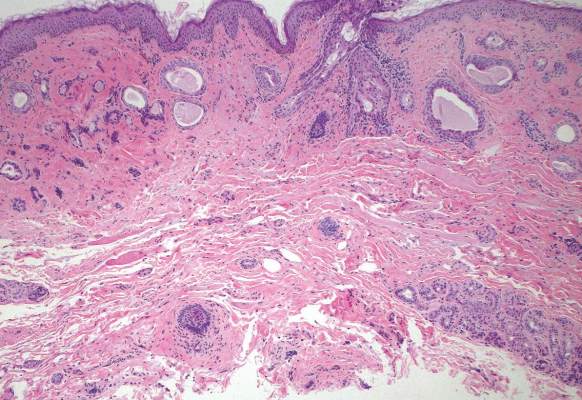
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.