User login
Idelalisib efficacy remains rock steady in relapsed CLL
MILAN – The benefits of combining idelalisib with rituximab remained unchanged in heavily pretreated, relapsed chronic lymphocytic leukemia unsuitable for chemotherapy in the second interim analysis of a phase III trial and extension study.
With 63% of events reported in Study 116, the primary endpoint of progression-free survival was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab and idelalisib.
These data were identical to those observed in the first interim analysis, save for an almost imperceptible shift in the hazard ratio from 0.15 to 0.18, with the same P value of less than .0001.

In the poor-prognosis subsets of patients with the 17p deletion and/or TP53 mutations or unmutated immunoglobulin heavy chain variable disease, median progression-free survival had also yet to be reached with the combination, compared with medians of 4.0 months (HR, 0.16) and 5.5 months (HR, 0.14) with single-agent rituximab, Dr. Steven Coutre said at the annual congress of the European Hematology Association.
As previously reported by this publication, Study 116 was stopped early after the first interim analysis and 50% of events reported due to overwhelming efficacy. The results also prompted the Food & Drug Administration to grant idelalisib, an oral phosphoinositide 3–kinase-delta inhibitor, breakthrough therapy designation for relapsed chronic lymphocytic leukemia (CLL).
In the second interim analysis, the addition of idelalisib to rituximab significantly increased the overall response rate from 15% to 77% (Odds ratio 17.3; P less than .0001) and number of patients achieving at least a 50% reduction in lymph nodes from 6% to 92% (OR, 165.5; P less than .0001), said Dr. Coutre, professor of medicine at Stanford (Calif.) University.
Median overall survival had yet to be reached at 18 months follow-up, but was significantly longer in the idelalisib arm (HR, 0.28; P = .003).
Adverse events were quite common in either arm, but "importantly, the combination demonstrated an acceptable and manageable safety profile," he said.
As previously seen, diarrhea, colitis, and grade 3/4 transaminase elevations were more common with idelalisib, but infusion site reactions were less frequent.
The 110 patients in the idelalisib arm, as compared with 108 controls, had more grade 3 or higher adverse events (AEs)(64% and 52%) and serious AEs (49% and 38%), but fewer AEs leading to treatment discontinuation (10% vs. 12%) or death (3% vs. 11%).
During a discussion of the results, the issue of the study’s comparator arm was raised once again. Dr. Coutre observed that while he is not a "big fan of single-agent rituximab" because "it is not particularly effective, certainly not in targeting the bone marrow, the reality in the United States is that it is probably the single most used regimen in this population ... so I think it is a very valid comparison."
Recruitment is currently underway for a phase II study, the first to evaluate idelalisib alone or combined with rituximab in untreated CLL and small lymphocytic lymphoma. Idelalisib has shown single-agent activity in relapsed or refractory CLL or small lymphocytic lymphoma, but Dr. Coutre said in an interview that "It is premature to consider idelalisib for front-line therapy. There are ongoing trials for this indication."
The study was supported by Gilead Sciences. Dr. Coutre reported personal fees from Gilead for a 1-day advisory board.
MILAN – The benefits of combining idelalisib with rituximab remained unchanged in heavily pretreated, relapsed chronic lymphocytic leukemia unsuitable for chemotherapy in the second interim analysis of a phase III trial and extension study.
With 63% of events reported in Study 116, the primary endpoint of progression-free survival was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab and idelalisib.
These data were identical to those observed in the first interim analysis, save for an almost imperceptible shift in the hazard ratio from 0.15 to 0.18, with the same P value of less than .0001.
In the poor-prognosis subsets of patients with the 17p deletion and/or TP53 mutations or unmutated immunoglobulin heavy chain variable disease, median progression-free survival had also yet to be reached with the combination, compared with medians of 4.0 months (HR, 0.16) and 5.5 months (HR, 0.14) with single-agent rituximab, Dr. Steven Coutre said at the annual congress of the European Hematology Association.
As previously reported by this publication, Study 116 was stopped early after the first interim analysis and 50% of events reported due to overwhelming efficacy. The results also prompted the Food & Drug Administration to grant idelalisib, an oral phosphoinositide 3–kinase-delta inhibitor, breakthrough therapy designation for relapsed chronic lymphocytic leukemia (CLL).
In the second interim analysis, the addition of idelalisib to rituximab significantly increased the overall response rate from 15% to 77% (Odds ratio 17.3; P less than .0001) and number of patients achieving at least a 50% reduction in lymph nodes from 6% to 92% (OR, 165.5; P less than .0001), said Dr. Coutre, professor of medicine at Stanford (Calif.) University.
Median overall survival had yet to be reached at 18 months follow-up, but was significantly longer in the idelalisib arm (HR, 0.28; P = .003).
Adverse events were quite common in either arm, but "importantly, the combination demonstrated an acceptable and manageable safety profile," he said.
As previously seen, diarrhea, colitis, and grade 3/4 transaminase elevations were more common with idelalisib, but infusion site reactions were less frequent.
The 110 patients in the idelalisib arm, as compared with 108 controls, had more grade 3 or higher adverse events (AEs)(64% and 52%) and serious AEs (49% and 38%), but fewer AEs leading to treatment discontinuation (10% vs. 12%) or death (3% vs. 11%).
During a discussion of the results, the issue of the study’s comparator arm was raised once again. Dr. Coutre observed that while he is not a "big fan of single-agent rituximab" because "it is not particularly effective, certainly not in targeting the bone marrow, the reality in the United States is that it is probably the single most used regimen in this population ... so I think it is a very valid comparison."
Recruitment is currently underway for a phase II study, the first to evaluate idelalisib alone or combined with rituximab in untreated CLL and small lymphocytic lymphoma. Idelalisib has shown single-agent activity in relapsed or refractory CLL or small lymphocytic lymphoma, but Dr. Coutre said in an interview that "It is premature to consider idelalisib for front-line therapy. There are ongoing trials for this indication."
The study was supported by Gilead Sciences. Dr. Coutre reported personal fees from Gilead for a 1-day advisory board.
MILAN – The benefits of combining idelalisib with rituximab remained unchanged in heavily pretreated, relapsed chronic lymphocytic leukemia unsuitable for chemotherapy in the second interim analysis of a phase III trial and extension study.
With 63% of events reported in Study 116, the primary endpoint of progression-free survival was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab and idelalisib.
These data were identical to those observed in the first interim analysis, save for an almost imperceptible shift in the hazard ratio from 0.15 to 0.18, with the same P value of less than .0001.
In the poor-prognosis subsets of patients with the 17p deletion and/or TP53 mutations or unmutated immunoglobulin heavy chain variable disease, median progression-free survival had also yet to be reached with the combination, compared with medians of 4.0 months (HR, 0.16) and 5.5 months (HR, 0.14) with single-agent rituximab, Dr. Steven Coutre said at the annual congress of the European Hematology Association.
As previously reported by this publication, Study 116 was stopped early after the first interim analysis and 50% of events reported due to overwhelming efficacy. The results also prompted the Food & Drug Administration to grant idelalisib, an oral phosphoinositide 3–kinase-delta inhibitor, breakthrough therapy designation for relapsed chronic lymphocytic leukemia (CLL).
In the second interim analysis, the addition of idelalisib to rituximab significantly increased the overall response rate from 15% to 77% (Odds ratio 17.3; P less than .0001) and number of patients achieving at least a 50% reduction in lymph nodes from 6% to 92% (OR, 165.5; P less than .0001), said Dr. Coutre, professor of medicine at Stanford (Calif.) University.
Median overall survival had yet to be reached at 18 months follow-up, but was significantly longer in the idelalisib arm (HR, 0.28; P = .003).
Adverse events were quite common in either arm, but "importantly, the combination demonstrated an acceptable and manageable safety profile," he said.
As previously seen, diarrhea, colitis, and grade 3/4 transaminase elevations were more common with idelalisib, but infusion site reactions were less frequent.
The 110 patients in the idelalisib arm, as compared with 108 controls, had more grade 3 or higher adverse events (AEs)(64% and 52%) and serious AEs (49% and 38%), but fewer AEs leading to treatment discontinuation (10% vs. 12%) or death (3% vs. 11%).
During a discussion of the results, the issue of the study’s comparator arm was raised once again. Dr. Coutre observed that while he is not a "big fan of single-agent rituximab" because "it is not particularly effective, certainly not in targeting the bone marrow, the reality in the United States is that it is probably the single most used regimen in this population ... so I think it is a very valid comparison."
Recruitment is currently underway for a phase II study, the first to evaluate idelalisib alone or combined with rituximab in untreated CLL and small lymphocytic lymphoma. Idelalisib has shown single-agent activity in relapsed or refractory CLL or small lymphocytic lymphoma, but Dr. Coutre said in an interview that "It is premature to consider idelalisib for front-line therapy. There are ongoing trials for this indication."
The study was supported by Gilead Sciences. Dr. Coutre reported personal fees from Gilead for a 1-day advisory board.
AT THE EHA CONGRESS
Major finding: Idelalisib and rituximab significantly increased progression-free survival (HR, 0.18; P less than .0001).
Data source: A prospective phase III study in 220 patients with relapsed CLL.
Key clinical point: Adding idelalisib to rituximab improves overall response rate, progression-free survival, and overall survival in heavily pretreated relapsed CLL.
Disclosures: The study was supported by Gilead Sciences. Dr. Coutre reported personal fees from Gilead for a 1-day advisory board.

Ibrutinib holds CLL at bay in majority of patients at 30 months
CHICAGO – Ibrutinib continues showing strength in a large majority of patients with chronic lymphocytic leukemia, including those with poor-prognosis chromosomal deletions, a study showed.
Three-year follow-up data of patients treated with ibrutinib (Imbruvica) monotherapy in a phase II trial and extension study showed 30-month overall survival (OS) rates of 96.6% for patients who received ibrutinib after an initial diagnosis, and 79.9% for patients with relapsed or refractory disease. Median overall survival has not been reached for either group, reported Dr. Susan M. O’Brien of the University of Texas M.D. Anderson Cancer Center, Houston.
Among patients with the poor-prognosis chromosome 17p deletion (del17p), 30-month OS was 65.9%, with the median OS not yet reached, Dr. O’Brien reported at the annual meeting of the American Society of Clinical Oncology.

"We achieved very rapid and quite durable responses with single-agent ibrutinib. We have not reached a median duration of response," she said.
The best investigator-assessed responses were 87% among 31 treatment-naive patients, and 90% among those with relapsed/refractory disease, some of whom had received four prior lines of therapy.
"I think one example of how this data will change clinical practice was the progression-free survival based on cytogenetics," commented invited discussant Dr. Nicole Lamanna of Columbia University Medical Center, New York.
The 30-month progression-free survival (PFS) by cytogenetics (as determined by fluorescence in situ hybridization) was 45.9% for patients with del17p, 74.2% for patients with the 11q deletion (del11q), and 89.0% for patients with neither deletion. The median PFS was 28.1 months among patients with del17p, but has not been reached in patients in the other two cytogenetic groups.
"With some of the other regimens that have been presented in patients with 17p, clearly a median PFS of this length has really not been achieved," she said.
In a separate study, also presented at ASCO 2014, investigators reported the results of the first interim analysis of a phase III randomized trial, which showed that 1-year overall survival for patients with previously treated chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) assigned to receive ibrutinib was 90%, compared with 81% for patients assigned to ofatumumab (Arzerra).
Independent Review
Ibrutinib is an oral inhibitor of Bruton’s tyrosine kinase, an enzyme essential for B-cell receptor signaling and adhesion that is present in many types of B-cell malignancies. Based on the results of phase II studies, ibrutinib received Food and Drug Administration approval for relapsed mantle cell lymphoma in November 2013, and for CLL in February 2014.
Dr. O’Brien and colleagues reviewed data on 31 patients aged 65 and over with newly diagnosed, untreated CLL/SLL, and 101 patients with relapsed/refractory CLL/SLL. The relapsed/refractory group included patients with high-risk CLL/SLL, defined as disease progression within 24 months of starting a chemoimmunotherapy regimen, or as a failure to respond.
The investigators initially tested ibrutinib at 420 and 840 mg daily doses, but soon discovered that there were no differences in efficacy or toxicity between the two doses. Dr. O’Brien therefore reported combined data on patients treated at each dose level.
For patients with relapsed/refractory disease, the median number of prior therapies was 4 (range, 1-12). All patients in this group had received chemotherapy, and more than 90% had received a nucleoside analogue, an alkylating agent, and anti–CD20-based therapy.
In all, 6% of treatment-naive patients and 34% of relapsed/refractory patients had del17p. Del11q was seen in 3% and 35% of patients, respectively.
At the most recent analysis, 25 of 31 treatment-naive patients (81%) and 42% of relapsed/refractory patients had been on ibrutinib for 2-3 years. Only one patient in the treatment-naive group and 19 in the relapsed/refractory group discontinued therapy because of disease progression.
Of the treatment-naive patients with responses, 13% had a complete response (CR), 3% had a near CR, 65% had a partial response (PR), and 6% had a PR with lymphocytosis. Of the relapsed/refractory group, 6% had a CR, 80% had a PR, and 4% had a PR with lymphocytosis.
The responses tend to improve over time, with a median time to first response of 1.9 months, and a median time to best response of 7.3 months.
Adverse events of grade 3 or greater occurred in 19 treatment-naive patients and 83 relapsed/refractory patients. Of these events, 7 (23%) in treatment-naive patients and 32 (32%) in relapsed/refractory patients were deemed to be ibrutinib related. One patient in the treatment-naive group and 11 in the relapsed/refractory group died during the study.
"If we look now at the adverse events over time, we see that in fact they tend to go down, and so – this is important – we’re not seeing a late, surprising side effect with 2 or 3 years of follow-up in terms of things that we didn’t see when we first started the patient on study," Dr. O’Brien said.
The most common side effects were hypertension and pneumonia.
The PCYC-1002 and -1003 studies were supported by Pharmacyclics. Dr. O’Brien disclosed receiving research funding from the company. Dr. Lamanna disclosed consulting/advising for Celgene, and receiving research funding from several companies, but not Pharmacyclics.
CHICAGO – Ibrutinib continues showing strength in a large majority of patients with chronic lymphocytic leukemia, including those with poor-prognosis chromosomal deletions, a study showed.
Three-year follow-up data of patients treated with ibrutinib (Imbruvica) monotherapy in a phase II trial and extension study showed 30-month overall survival (OS) rates of 96.6% for patients who received ibrutinib after an initial diagnosis, and 79.9% for patients with relapsed or refractory disease. Median overall survival has not been reached for either group, reported Dr. Susan M. O’Brien of the University of Texas M.D. Anderson Cancer Center, Houston.
Among patients with the poor-prognosis chromosome 17p deletion (del17p), 30-month OS was 65.9%, with the median OS not yet reached, Dr. O’Brien reported at the annual meeting of the American Society of Clinical Oncology.
"We achieved very rapid and quite durable responses with single-agent ibrutinib. We have not reached a median duration of response," she said.
The best investigator-assessed responses were 87% among 31 treatment-naive patients, and 90% among those with relapsed/refractory disease, some of whom had received four prior lines of therapy.
"I think one example of how this data will change clinical practice was the progression-free survival based on cytogenetics," commented invited discussant Dr. Nicole Lamanna of Columbia University Medical Center, New York.
The 30-month progression-free survival (PFS) by cytogenetics (as determined by fluorescence in situ hybridization) was 45.9% for patients with del17p, 74.2% for patients with the 11q deletion (del11q), and 89.0% for patients with neither deletion. The median PFS was 28.1 months among patients with del17p, but has not been reached in patients in the other two cytogenetic groups.
"With some of the other regimens that have been presented in patients with 17p, clearly a median PFS of this length has really not been achieved," she said.
In a separate study, also presented at ASCO 2014, investigators reported the results of the first interim analysis of a phase III randomized trial, which showed that 1-year overall survival for patients with previously treated chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) assigned to receive ibrutinib was 90%, compared with 81% for patients assigned to ofatumumab (Arzerra).
Independent Review
Ibrutinib is an oral inhibitor of Bruton’s tyrosine kinase, an enzyme essential for B-cell receptor signaling and adhesion that is present in many types of B-cell malignancies. Based on the results of phase II studies, ibrutinib received Food and Drug Administration approval for relapsed mantle cell lymphoma in November 2013, and for CLL in February 2014.
Dr. O’Brien and colleagues reviewed data on 31 patients aged 65 and over with newly diagnosed, untreated CLL/SLL, and 101 patients with relapsed/refractory CLL/SLL. The relapsed/refractory group included patients with high-risk CLL/SLL, defined as disease progression within 24 months of starting a chemoimmunotherapy regimen, or as a failure to respond.
The investigators initially tested ibrutinib at 420 and 840 mg daily doses, but soon discovered that there were no differences in efficacy or toxicity between the two doses. Dr. O’Brien therefore reported combined data on patients treated at each dose level.
For patients with relapsed/refractory disease, the median number of prior therapies was 4 (range, 1-12). All patients in this group had received chemotherapy, and more than 90% had received a nucleoside analogue, an alkylating agent, and anti–CD20-based therapy.
In all, 6% of treatment-naive patients and 34% of relapsed/refractory patients had del17p. Del11q was seen in 3% and 35% of patients, respectively.
At the most recent analysis, 25 of 31 treatment-naive patients (81%) and 42% of relapsed/refractory patients had been on ibrutinib for 2-3 years. Only one patient in the treatment-naive group and 19 in the relapsed/refractory group discontinued therapy because of disease progression.
Of the treatment-naive patients with responses, 13% had a complete response (CR), 3% had a near CR, 65% had a partial response (PR), and 6% had a PR with lymphocytosis. Of the relapsed/refractory group, 6% had a CR, 80% had a PR, and 4% had a PR with lymphocytosis.
The responses tend to improve over time, with a median time to first response of 1.9 months, and a median time to best response of 7.3 months.
Adverse events of grade 3 or greater occurred in 19 treatment-naive patients and 83 relapsed/refractory patients. Of these events, 7 (23%) in treatment-naive patients and 32 (32%) in relapsed/refractory patients were deemed to be ibrutinib related. One patient in the treatment-naive group and 11 in the relapsed/refractory group died during the study.
"If we look now at the adverse events over time, we see that in fact they tend to go down, and so – this is important – we’re not seeing a late, surprising side effect with 2 or 3 years of follow-up in terms of things that we didn’t see when we first started the patient on study," Dr. O’Brien said.
The most common side effects were hypertension and pneumonia.
The PCYC-1002 and -1003 studies were supported by Pharmacyclics. Dr. O’Brien disclosed receiving research funding from the company. Dr. Lamanna disclosed consulting/advising for Celgene, and receiving research funding from several companies, but not Pharmacyclics.
CHICAGO – Ibrutinib continues showing strength in a large majority of patients with chronic lymphocytic leukemia, including those with poor-prognosis chromosomal deletions, a study showed.
Three-year follow-up data of patients treated with ibrutinib (Imbruvica) monotherapy in a phase II trial and extension study showed 30-month overall survival (OS) rates of 96.6% for patients who received ibrutinib after an initial diagnosis, and 79.9% for patients with relapsed or refractory disease. Median overall survival has not been reached for either group, reported Dr. Susan M. O’Brien of the University of Texas M.D. Anderson Cancer Center, Houston.
Among patients with the poor-prognosis chromosome 17p deletion (del17p), 30-month OS was 65.9%, with the median OS not yet reached, Dr. O’Brien reported at the annual meeting of the American Society of Clinical Oncology.
"We achieved very rapid and quite durable responses with single-agent ibrutinib. We have not reached a median duration of response," she said.
The best investigator-assessed responses were 87% among 31 treatment-naive patients, and 90% among those with relapsed/refractory disease, some of whom had received four prior lines of therapy.
"I think one example of how this data will change clinical practice was the progression-free survival based on cytogenetics," commented invited discussant Dr. Nicole Lamanna of Columbia University Medical Center, New York.
The 30-month progression-free survival (PFS) by cytogenetics (as determined by fluorescence in situ hybridization) was 45.9% for patients with del17p, 74.2% for patients with the 11q deletion (del11q), and 89.0% for patients with neither deletion. The median PFS was 28.1 months among patients with del17p, but has not been reached in patients in the other two cytogenetic groups.
"With some of the other regimens that have been presented in patients with 17p, clearly a median PFS of this length has really not been achieved," she said.
In a separate study, also presented at ASCO 2014, investigators reported the results of the first interim analysis of a phase III randomized trial, which showed that 1-year overall survival for patients with previously treated chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) assigned to receive ibrutinib was 90%, compared with 81% for patients assigned to ofatumumab (Arzerra).
Independent Review
Ibrutinib is an oral inhibitor of Bruton’s tyrosine kinase, an enzyme essential for B-cell receptor signaling and adhesion that is present in many types of B-cell malignancies. Based on the results of phase II studies, ibrutinib received Food and Drug Administration approval for relapsed mantle cell lymphoma in November 2013, and for CLL in February 2014.
Dr. O’Brien and colleagues reviewed data on 31 patients aged 65 and over with newly diagnosed, untreated CLL/SLL, and 101 patients with relapsed/refractory CLL/SLL. The relapsed/refractory group included patients with high-risk CLL/SLL, defined as disease progression within 24 months of starting a chemoimmunotherapy regimen, or as a failure to respond.
The investigators initially tested ibrutinib at 420 and 840 mg daily doses, but soon discovered that there were no differences in efficacy or toxicity between the two doses. Dr. O’Brien therefore reported combined data on patients treated at each dose level.
For patients with relapsed/refractory disease, the median number of prior therapies was 4 (range, 1-12). All patients in this group had received chemotherapy, and more than 90% had received a nucleoside analogue, an alkylating agent, and anti–CD20-based therapy.
In all, 6% of treatment-naive patients and 34% of relapsed/refractory patients had del17p. Del11q was seen in 3% and 35% of patients, respectively.
At the most recent analysis, 25 of 31 treatment-naive patients (81%) and 42% of relapsed/refractory patients had been on ibrutinib for 2-3 years. Only one patient in the treatment-naive group and 19 in the relapsed/refractory group discontinued therapy because of disease progression.
Of the treatment-naive patients with responses, 13% had a complete response (CR), 3% had a near CR, 65% had a partial response (PR), and 6% had a PR with lymphocytosis. Of the relapsed/refractory group, 6% had a CR, 80% had a PR, and 4% had a PR with lymphocytosis.
The responses tend to improve over time, with a median time to first response of 1.9 months, and a median time to best response of 7.3 months.
Adverse events of grade 3 or greater occurred in 19 treatment-naive patients and 83 relapsed/refractory patients. Of these events, 7 (23%) in treatment-naive patients and 32 (32%) in relapsed/refractory patients were deemed to be ibrutinib related. One patient in the treatment-naive group and 11 in the relapsed/refractory group died during the study.
"If we look now at the adverse events over time, we see that in fact they tend to go down, and so – this is important – we’re not seeing a late, surprising side effect with 2 or 3 years of follow-up in terms of things that we didn’t see when we first started the patient on study," Dr. O’Brien said.
The most common side effects were hypertension and pneumonia.
The PCYC-1002 and -1003 studies were supported by Pharmacyclics. Dr. O’Brien disclosed receiving research funding from the company. Dr. Lamanna disclosed consulting/advising for Celgene, and receiving research funding from several companies, but not Pharmacyclics.
AT THE ASCO ANNUAL MEETING 2014
Major finding: At 30 months of follow-up, overall survival with ibrutinib monotherapy was 96.6% among patients with newly diagnosed chronic lymphocytic leukemia, and 79.9% for patients with relapsed/refractory CLL.
Data source: Long-term follow-up of 132 patients in a phase II safety and efficacy trial and extension study.
Disclosures: The PCYC-1002 and -1003 studies were supported by Pharmacyclics. Dr. O’Brien disclosed receiving research funding from the company. Dr. Lamanna disclosed consulting/advising for Celgene, and receiving research funding from several companies, but not Pharmacyclics.

VIDEO: Ibrutinib boosts survival of relapsed/refractory CLL
CHICAGO – For the first time an oral drug, ibrutinib, has been shown to significantly improve both progression-free and overall survival of patients with relapsed or refractory chronic lymphocytic leukemia, compared with a systemic agent.
We interviewed lead author Dr. John Byrd about the first interim analysis from RESONATE, a phase III randomized trial, comparing ibrutinib with ofatumumab for the treatment of relapsed or refractory chronic lymphocytic leukemia or small lymphocytic lymphoma at the annual meeting of the American Society of Clinical Oncology. The analysis showed that 1-year overall survival for patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma assigned to receive ibrutinib (Imbruvica) was 90%, compared with 81% for patients assigned to ofatumumab (Arzerra).
Dr. Byrd is professor of medicine at the Ohio State University Comprehensive Cancer Center, Columbus. The study was supported by Pharmacyclics. Dr. Byrd disclosed receiving research funding and serving as an unpaid advisor to the company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
CHICAGO – For the first time an oral drug, ibrutinib, has been shown to significantly improve both progression-free and overall survival of patients with relapsed or refractory chronic lymphocytic leukemia, compared with a systemic agent.
We interviewed lead author Dr. John Byrd about the first interim analysis from RESONATE, a phase III randomized trial, comparing ibrutinib with ofatumumab for the treatment of relapsed or refractory chronic lymphocytic leukemia or small lymphocytic lymphoma at the annual meeting of the American Society of Clinical Oncology. The analysis showed that 1-year overall survival for patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma assigned to receive ibrutinib (Imbruvica) was 90%, compared with 81% for patients assigned to ofatumumab (Arzerra).
Dr. Byrd is professor of medicine at the Ohio State University Comprehensive Cancer Center, Columbus. The study was supported by Pharmacyclics. Dr. Byrd disclosed receiving research funding and serving as an unpaid advisor to the company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
CHICAGO – For the first time an oral drug, ibrutinib, has been shown to significantly improve both progression-free and overall survival of patients with relapsed or refractory chronic lymphocytic leukemia, compared with a systemic agent.
We interviewed lead author Dr. John Byrd about the first interim analysis from RESONATE, a phase III randomized trial, comparing ibrutinib with ofatumumab for the treatment of relapsed or refractory chronic lymphocytic leukemia or small lymphocytic lymphoma at the annual meeting of the American Society of Clinical Oncology. The analysis showed that 1-year overall survival for patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma assigned to receive ibrutinib (Imbruvica) was 90%, compared with 81% for patients assigned to ofatumumab (Arzerra).
Dr. Byrd is professor of medicine at the Ohio State University Comprehensive Cancer Center, Columbus. The study was supported by Pharmacyclics. Dr. Byrd disclosed receiving research funding and serving as an unpaid advisor to the company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE ASCO ANNUAL MEETING 2014

Drugs approved in 2013
In 2013, the Food and Drug Administration approved 27 new molecular entities (i.e., drugs) for human use. Because of their indications, it is unlikely that four will be used in pregnancy or lactation, so they are not discussed here. The four agents are ospemifene (Osphena), an estrogen agonist/antagonist used for severe dyspareunia; [223Ra]radium dichloride (Xofigo), for late-stage metastatic prostate cancer; conjugated estrogens/bazedoxifene (Duavee) for hot flashes associated with menopause and to prevent osteoporosis; and flutemetamol F-18 injection (Vizamyl), a radioactive diagnostic agent to aid in the evaluation of Alzheimer’s disease and dementia.
There are two other drugs that are unlikely to be used in pregnancy: macitentan (Opsumit) and riociguat (Adempas). These drugs are oral vasodilators indicated for the treatment of pulmonary hypertension. Both are teratogenic in rats and rabbits, but there are no reports of their use in human pregnancy. For female patients of reproductive potential, they are only available through restricted programs. Pregnancy must be excluded before starting therapy, monthly during treatment, and for 1 month after treatment is stopped.
The remaining 21 agents can be classified into the following categories: anticonvulsant (1), antidepressant (1), antidiabetics (2), antineoplastics (7), antihyperlipidemic (1), anti-infectives (4), diagnostics (2), immunologic (1), and respiratory (2). It is important to note that, except for two drugs (fluticasone in a combination product and dimethyl fumarate), there is no reported human pregnancy experience for these agents. Moreover, all probably cross the placenta to the embryo and/or the fetus, at least in some part of pregnancy.
Eslicarbazepine (Aptiom) is indicated as adjunctive treatment of partial-onset seizures. Developmental toxicity was observed in three animals: teratogenicity (mice), embryolethality (rats), and fetal growth restriction (rabbits). The no-effect dose was not found in two species, and was less than the human dose based on body surface area in the third. If a pregnant woman is taking this drug, she should be encouraged to enroll in the North American Antiepileptic Drug Pregnancy Registry by calling 888-233-2334.
Vortioxetine (Brintellix) is indicated for the treatment of major depressive disorder. The drug was not teratogenic in animals but did cause developmental delays in one species. Although the antidepressant mechanism is not fully understood, it appears to be related to the inhibition of the reuptake of serotonin (5-hydroxytryptamine). If so, vortioxetine would be closely related to the drugs in the selective serotonin reuptake inhibitor (SSRI) class: citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac), fluvoxamine (Luvox), paroxetine (Paxil), sertraline (Zoloft), and vilazodone (Viibryd). The relationship could be important because the use of SSRIs or serotonin norepinephrine reuptake inhibitors (SNRIs) close to birth is related to significant toxicity in the newborn.
There are two new antidiabetic agents for the treatment of type 2 diabetes. Alogliptin (Nesina), a dipeptidyl peptidase–4 inhibitor, is in the same pharmacologic class as linagliptin (Tradjenta), saxagliptin (Onglyza), and sitagliptin (Januvia). Canagliflozin (Invokana) is a sodium-glucose cotransporter 2 inhibitor, the first drug in this class to be approved. The animal data for alogliptin suggest low risk, whereas canagliflozin caused renal toxicity in rats at exposures corresponding to the late second and third trimester in humans. Insulin remains the treatment of choice for pregnant diabetics because tight control of glucose levels is beneficial for the mother, embryo-fetus, and newborn.
The seven new antineoplastic agents are ado-trastuzumab emtansine (Kadcyla) for HER2-positive breast cancer; afatinib (Gilotrif) for non–small cell lung cancer; dabrafenib (Tafinlar) for unresectable or metastatic melanoma; ibrutinib (Imbruvica) for mantle cell lymphoma or chronic lymphocytic leukemia; obinutuzumab (Gazyva) for chronic lymphocytic leukemia; pomalidomide (Pomalyst) for multiple myeloma; and trametinib (Mekinist) for unresectable or metastatic melanoma. Only pomalidomide is contraindicated in pregnancy. Although obinutuzumab did not cause teratogenicity in monkeys, its use in the latter portion of pregnancy resulted in newborn depletion of B cells that took up to 6 months after birth to restore. Moreover, it is used in combination with chlorambucil, a known teratogen. The animal data suggest risk in the other five agents. Nevertheless, the maternal condition should determine whether any of these antineoplastics are used in a pregnant woman.
Mipomersen sodium (Kynamro) is given subcutaneously once a week as an adjunct to lipid-lowering medications. The drug caused embryo toxicity in one of three animal species.
Among the four anti-infectives are two oral agents for the treatment of chronic hepatitis C virus infection: simeprevir (Olysio) and sofosbuvir (Sovaldi). Because both agents are recommended to be combined with peginterferon alfa and ribavirin, they are classified as contraindicated in pregnancy. However, when used alone, the animal data suggest that sofosbuvir was low risk, whereas simeprevir might have higher risk.
Luliconazole (Luzu), an azole antifungal, is a cream used for the treatment of tinea pedis, tinea cruris, and tinea corporis. Systemic absorption is minimal. The animal data suggest low risk, but there are no human pregnancy reports. Nevertheless, topical use is probably compatible in pregnancy, as are the other topical azole antifungals in this pharmacologic class: clotrimazole (Lotrimin), econazole (Spectazole), ketoconazole (Kuric), miconazole (Micatin), oxiconazole (Oxistat), sertaconazole (Ertaczo), and sulconazole (Exelderm).
Dolutegravir (Tivicay) is an HIV-1 integrase strand transfer inhibitor given in combination with other antiretroviral drugs. The animal data suggest low risk. If indicated, the drug should not be withheld because of pregnancy.
Gadoterate meglumine (Dotarem), a gadolinium-based contrast agent, is indicated to detect and visualize areas with disruption of the blood brain barrier and/or abnormal vascularity. No developmental toxicity was observed in pregnant animals. Closely related diagnostic agents are gadobenate dimeglumine (MultiHance), gadodiamide (Omniscan), gadofosveset (Ablavar), gadopentetate dimeglumine (Magnevist), gadoteridol (Prohance), and gadoversetamide (OptiMARK). Although the animal data for these agents show risk, no harm has been reported in human pregnancies. However, the available human data are very limited, and the risk magnitude for embryo-fetal harm remains unknown.
Technetium (99mTc) tilmanocept (Lymphoseek) is a radioactive diagnostic agent used in patients with breast cancer or melanoma. The active ingredient is technetium (99mTc). Animal reproduction studies have not been conducted. 99mTc is probably compatible in pregnancy (see Drugs in Pregnancy and Lactation, 10th ed.; Philadelphia: Lippincott, Williams and Wilkins, 2014:1317-8; to be released in August), but the risk of the tilmanocept moiety is unknown.
The immunologic agent dimethyl fumarate (Tecfidera) is indicated for the treatment of patients with relapsing forms of multiple sclerosis. The drug caused developmental toxicity (embryolethality, impaired growth, and birth defects) in animals during all portions of pregnancy. In clinical trials, there were 38 exposed pregnancies with the following outcomes: 22 live births, 3 spontaneous abortions, 9 elective abortions, 3 ongoing pregnancies, and 1 lost to follow-up (CNS Drugs 2014;28:89-94). A pregnancy registry has been established, and patients should be encouraged to enroll by calling 800-456-2255.
Two new respiratory combination products were approved in 2013, both for chronic obstructive pulmonary disease: fluticasone/vilanterol (Breo Ellipta) and umeclidinium/vilanterol (Anoro Ellipta). Inhaled fluticasone, a corticosteroid, is compatible in pregnancy (see Drugs in Pregnancy and Lactation, 9th ed.; Philadelphia: Lippincott, Williams and Wilkins; 2011:599-601). Vilanterol is a long-acting beta2-adrenergic agonist that is probably compatible in pregnancy. The absolute bioavailability of inhaled fluticasone and vilanterol in nonpregnant adults was about 15% and 27%, respectively. The animal data for the combination or when given individually suggest low risk in pregnancy. Umeclidinium is a long-acting anticholinergic. It also is absorbed from the lung, but the amount was not specified by the manufacturer. The animal data for umeclidinium suggest low risk.
There are no reports of the above drugs being used during breastfeeding, but excretion into breast milk should be expected. The effect of these exposures on a nursing infant is unknown. However, if a mother is taking one of these drugs and breastfeeding, her infant should be monitored for adverse effects, especially those that are the most common (typically listed on the first page of the package insert) in patients taking the drug. Close monitoring is particularly important during the first 2 postpartum months. A 2003 study found that most adverse reactions in nursing infants occurred within that time period (Clin. Pediatr. 2003;42:325-40).
Mr. Briggs is a pharmacist clinical specialist at the outpatient clinics of Memorial Care Center for Women at Miller Children’s Hospital in Long Beach, Calif.; clinical professor of pharmacy at the University of California, San Francisco; and adjunct professor of pharmacy at the University of Southern California, Los Angeles, and Washington State University, Spokane. He also is coauthor of "Drugs in Pregnancy and Lactation," and coeditor of "Diseases, Complications, and Drug Therapy in Obstetrics." He had no other relevant financial disclosures. Contact him at [email protected].
In 2013, the Food and Drug Administration approved 27 new molecular entities (i.e., drugs) for human use. Because of their indications, it is unlikely that four will be used in pregnancy or lactation, so they are not discussed here. The four agents are ospemifene (Osphena), an estrogen agonist/antagonist used for severe dyspareunia; [223Ra]radium dichloride (Xofigo), for late-stage metastatic prostate cancer; conjugated estrogens/bazedoxifene (Duavee) for hot flashes associated with menopause and to prevent osteoporosis; and flutemetamol F-18 injection (Vizamyl), a radioactive diagnostic agent to aid in the evaluation of Alzheimer’s disease and dementia.
There are two other drugs that are unlikely to be used in pregnancy: macitentan (Opsumit) and riociguat (Adempas). These drugs are oral vasodilators indicated for the treatment of pulmonary hypertension. Both are teratogenic in rats and rabbits, but there are no reports of their use in human pregnancy. For female patients of reproductive potential, they are only available through restricted programs. Pregnancy must be excluded before starting therapy, monthly during treatment, and for 1 month after treatment is stopped.
The remaining 21 agents can be classified into the following categories: anticonvulsant (1), antidepressant (1), antidiabetics (2), antineoplastics (7), antihyperlipidemic (1), anti-infectives (4), diagnostics (2), immunologic (1), and respiratory (2). It is important to note that, except for two drugs (fluticasone in a combination product and dimethyl fumarate), there is no reported human pregnancy experience for these agents. Moreover, all probably cross the placenta to the embryo and/or the fetus, at least in some part of pregnancy.
Eslicarbazepine (Aptiom) is indicated as adjunctive treatment of partial-onset seizures. Developmental toxicity was observed in three animals: teratogenicity (mice), embryolethality (rats), and fetal growth restriction (rabbits). The no-effect dose was not found in two species, and was less than the human dose based on body surface area in the third. If a pregnant woman is taking this drug, she should be encouraged to enroll in the North American Antiepileptic Drug Pregnancy Registry by calling 888-233-2334.
Vortioxetine (Brintellix) is indicated for the treatment of major depressive disorder. The drug was not teratogenic in animals but did cause developmental delays in one species. Although the antidepressant mechanism is not fully understood, it appears to be related to the inhibition of the reuptake of serotonin (5-hydroxytryptamine). If so, vortioxetine would be closely related to the drugs in the selective serotonin reuptake inhibitor (SSRI) class: citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac), fluvoxamine (Luvox), paroxetine (Paxil), sertraline (Zoloft), and vilazodone (Viibryd). The relationship could be important because the use of SSRIs or serotonin norepinephrine reuptake inhibitors (SNRIs) close to birth is related to significant toxicity in the newborn.
There are two new antidiabetic agents for the treatment of type 2 diabetes. Alogliptin (Nesina), a dipeptidyl peptidase–4 inhibitor, is in the same pharmacologic class as linagliptin (Tradjenta), saxagliptin (Onglyza), and sitagliptin (Januvia). Canagliflozin (Invokana) is a sodium-glucose cotransporter 2 inhibitor, the first drug in this class to be approved. The animal data for alogliptin suggest low risk, whereas canagliflozin caused renal toxicity in rats at exposures corresponding to the late second and third trimester in humans. Insulin remains the treatment of choice for pregnant diabetics because tight control of glucose levels is beneficial for the mother, embryo-fetus, and newborn.
The seven new antineoplastic agents are ado-trastuzumab emtansine (Kadcyla) for HER2-positive breast cancer; afatinib (Gilotrif) for non–small cell lung cancer; dabrafenib (Tafinlar) for unresectable or metastatic melanoma; ibrutinib (Imbruvica) for mantle cell lymphoma or chronic lymphocytic leukemia; obinutuzumab (Gazyva) for chronic lymphocytic leukemia; pomalidomide (Pomalyst) for multiple myeloma; and trametinib (Mekinist) for unresectable or metastatic melanoma. Only pomalidomide is contraindicated in pregnancy. Although obinutuzumab did not cause teratogenicity in monkeys, its use in the latter portion of pregnancy resulted in newborn depletion of B cells that took up to 6 months after birth to restore. Moreover, it is used in combination with chlorambucil, a known teratogen. The animal data suggest risk in the other five agents. Nevertheless, the maternal condition should determine whether any of these antineoplastics are used in a pregnant woman.
Mipomersen sodium (Kynamro) is given subcutaneously once a week as an adjunct to lipid-lowering medications. The drug caused embryo toxicity in one of three animal species.
Among the four anti-infectives are two oral agents for the treatment of chronic hepatitis C virus infection: simeprevir (Olysio) and sofosbuvir (Sovaldi). Because both agents are recommended to be combined with peginterferon alfa and ribavirin, they are classified as contraindicated in pregnancy. However, when used alone, the animal data suggest that sofosbuvir was low risk, whereas simeprevir might have higher risk.
Luliconazole (Luzu), an azole antifungal, is a cream used for the treatment of tinea pedis, tinea cruris, and tinea corporis. Systemic absorption is minimal. The animal data suggest low risk, but there are no human pregnancy reports. Nevertheless, topical use is probably compatible in pregnancy, as are the other topical azole antifungals in this pharmacologic class: clotrimazole (Lotrimin), econazole (Spectazole), ketoconazole (Kuric), miconazole (Micatin), oxiconazole (Oxistat), sertaconazole (Ertaczo), and sulconazole (Exelderm).
Dolutegravir (Tivicay) is an HIV-1 integrase strand transfer inhibitor given in combination with other antiretroviral drugs. The animal data suggest low risk. If indicated, the drug should not be withheld because of pregnancy.
Gadoterate meglumine (Dotarem), a gadolinium-based contrast agent, is indicated to detect and visualize areas with disruption of the blood brain barrier and/or abnormal vascularity. No developmental toxicity was observed in pregnant animals. Closely related diagnostic agents are gadobenate dimeglumine (MultiHance), gadodiamide (Omniscan), gadofosveset (Ablavar), gadopentetate dimeglumine (Magnevist), gadoteridol (Prohance), and gadoversetamide (OptiMARK). Although the animal data for these agents show risk, no harm has been reported in human pregnancies. However, the available human data are very limited, and the risk magnitude for embryo-fetal harm remains unknown.
Technetium (99mTc) tilmanocept (Lymphoseek) is a radioactive diagnostic agent used in patients with breast cancer or melanoma. The active ingredient is technetium (99mTc). Animal reproduction studies have not been conducted. 99mTc is probably compatible in pregnancy (see Drugs in Pregnancy and Lactation, 10th ed.; Philadelphia: Lippincott, Williams and Wilkins, 2014:1317-8; to be released in August), but the risk of the tilmanocept moiety is unknown.
The immunologic agent dimethyl fumarate (Tecfidera) is indicated for the treatment of patients with relapsing forms of multiple sclerosis. The drug caused developmental toxicity (embryolethality, impaired growth, and birth defects) in animals during all portions of pregnancy. In clinical trials, there were 38 exposed pregnancies with the following outcomes: 22 live births, 3 spontaneous abortions, 9 elective abortions, 3 ongoing pregnancies, and 1 lost to follow-up (CNS Drugs 2014;28:89-94). A pregnancy registry has been established, and patients should be encouraged to enroll by calling 800-456-2255.
Two new respiratory combination products were approved in 2013, both for chronic obstructive pulmonary disease: fluticasone/vilanterol (Breo Ellipta) and umeclidinium/vilanterol (Anoro Ellipta). Inhaled fluticasone, a corticosteroid, is compatible in pregnancy (see Drugs in Pregnancy and Lactation, 9th ed.; Philadelphia: Lippincott, Williams and Wilkins; 2011:599-601). Vilanterol is a long-acting beta2-adrenergic agonist that is probably compatible in pregnancy. The absolute bioavailability of inhaled fluticasone and vilanterol in nonpregnant adults was about 15% and 27%, respectively. The animal data for the combination or when given individually suggest low risk in pregnancy. Umeclidinium is a long-acting anticholinergic. It also is absorbed from the lung, but the amount was not specified by the manufacturer. The animal data for umeclidinium suggest low risk.
There are no reports of the above drugs being used during breastfeeding, but excretion into breast milk should be expected. The effect of these exposures on a nursing infant is unknown. However, if a mother is taking one of these drugs and breastfeeding, her infant should be monitored for adverse effects, especially those that are the most common (typically listed on the first page of the package insert) in patients taking the drug. Close monitoring is particularly important during the first 2 postpartum months. A 2003 study found that most adverse reactions in nursing infants occurred within that time period (Clin. Pediatr. 2003;42:325-40).
Mr. Briggs is a pharmacist clinical specialist at the outpatient clinics of Memorial Care Center for Women at Miller Children’s Hospital in Long Beach, Calif.; clinical professor of pharmacy at the University of California, San Francisco; and adjunct professor of pharmacy at the University of Southern California, Los Angeles, and Washington State University, Spokane. He also is coauthor of "Drugs in Pregnancy and Lactation," and coeditor of "Diseases, Complications, and Drug Therapy in Obstetrics." He had no other relevant financial disclosures. Contact him at [email protected].
In 2013, the Food and Drug Administration approved 27 new molecular entities (i.e., drugs) for human use. Because of their indications, it is unlikely that four will be used in pregnancy or lactation, so they are not discussed here. The four agents are ospemifene (Osphena), an estrogen agonist/antagonist used for severe dyspareunia; [223Ra]radium dichloride (Xofigo), for late-stage metastatic prostate cancer; conjugated estrogens/bazedoxifene (Duavee) for hot flashes associated with menopause and to prevent osteoporosis; and flutemetamol F-18 injection (Vizamyl), a radioactive diagnostic agent to aid in the evaluation of Alzheimer’s disease and dementia.
There are two other drugs that are unlikely to be used in pregnancy: macitentan (Opsumit) and riociguat (Adempas). These drugs are oral vasodilators indicated for the treatment of pulmonary hypertension. Both are teratogenic in rats and rabbits, but there are no reports of their use in human pregnancy. For female patients of reproductive potential, they are only available through restricted programs. Pregnancy must be excluded before starting therapy, monthly during treatment, and for 1 month after treatment is stopped.
The remaining 21 agents can be classified into the following categories: anticonvulsant (1), antidepressant (1), antidiabetics (2), antineoplastics (7), antihyperlipidemic (1), anti-infectives (4), diagnostics (2), immunologic (1), and respiratory (2). It is important to note that, except for two drugs (fluticasone in a combination product and dimethyl fumarate), there is no reported human pregnancy experience for these agents. Moreover, all probably cross the placenta to the embryo and/or the fetus, at least in some part of pregnancy.
Eslicarbazepine (Aptiom) is indicated as adjunctive treatment of partial-onset seizures. Developmental toxicity was observed in three animals: teratogenicity (mice), embryolethality (rats), and fetal growth restriction (rabbits). The no-effect dose was not found in two species, and was less than the human dose based on body surface area in the third. If a pregnant woman is taking this drug, she should be encouraged to enroll in the North American Antiepileptic Drug Pregnancy Registry by calling 888-233-2334.
Vortioxetine (Brintellix) is indicated for the treatment of major depressive disorder. The drug was not teratogenic in animals but did cause developmental delays in one species. Although the antidepressant mechanism is not fully understood, it appears to be related to the inhibition of the reuptake of serotonin (5-hydroxytryptamine). If so, vortioxetine would be closely related to the drugs in the selective serotonin reuptake inhibitor (SSRI) class: citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac), fluvoxamine (Luvox), paroxetine (Paxil), sertraline (Zoloft), and vilazodone (Viibryd). The relationship could be important because the use of SSRIs or serotonin norepinephrine reuptake inhibitors (SNRIs) close to birth is related to significant toxicity in the newborn.
There are two new antidiabetic agents for the treatment of type 2 diabetes. Alogliptin (Nesina), a dipeptidyl peptidase–4 inhibitor, is in the same pharmacologic class as linagliptin (Tradjenta), saxagliptin (Onglyza), and sitagliptin (Januvia). Canagliflozin (Invokana) is a sodium-glucose cotransporter 2 inhibitor, the first drug in this class to be approved. The animal data for alogliptin suggest low risk, whereas canagliflozin caused renal toxicity in rats at exposures corresponding to the late second and third trimester in humans. Insulin remains the treatment of choice for pregnant diabetics because tight control of glucose levels is beneficial for the mother, embryo-fetus, and newborn.
The seven new antineoplastic agents are ado-trastuzumab emtansine (Kadcyla) for HER2-positive breast cancer; afatinib (Gilotrif) for non–small cell lung cancer; dabrafenib (Tafinlar) for unresectable or metastatic melanoma; ibrutinib (Imbruvica) for mantle cell lymphoma or chronic lymphocytic leukemia; obinutuzumab (Gazyva) for chronic lymphocytic leukemia; pomalidomide (Pomalyst) for multiple myeloma; and trametinib (Mekinist) for unresectable or metastatic melanoma. Only pomalidomide is contraindicated in pregnancy. Although obinutuzumab did not cause teratogenicity in monkeys, its use in the latter portion of pregnancy resulted in newborn depletion of B cells that took up to 6 months after birth to restore. Moreover, it is used in combination with chlorambucil, a known teratogen. The animal data suggest risk in the other five agents. Nevertheless, the maternal condition should determine whether any of these antineoplastics are used in a pregnant woman.
Mipomersen sodium (Kynamro) is given subcutaneously once a week as an adjunct to lipid-lowering medications. The drug caused embryo toxicity in one of three animal species.
Among the four anti-infectives are two oral agents for the treatment of chronic hepatitis C virus infection: simeprevir (Olysio) and sofosbuvir (Sovaldi). Because both agents are recommended to be combined with peginterferon alfa and ribavirin, they are classified as contraindicated in pregnancy. However, when used alone, the animal data suggest that sofosbuvir was low risk, whereas simeprevir might have higher risk.
Luliconazole (Luzu), an azole antifungal, is a cream used for the treatment of tinea pedis, tinea cruris, and tinea corporis. Systemic absorption is minimal. The animal data suggest low risk, but there are no human pregnancy reports. Nevertheless, topical use is probably compatible in pregnancy, as are the other topical azole antifungals in this pharmacologic class: clotrimazole (Lotrimin), econazole (Spectazole), ketoconazole (Kuric), miconazole (Micatin), oxiconazole (Oxistat), sertaconazole (Ertaczo), and sulconazole (Exelderm).
Dolutegravir (Tivicay) is an HIV-1 integrase strand transfer inhibitor given in combination with other antiretroviral drugs. The animal data suggest low risk. If indicated, the drug should not be withheld because of pregnancy.
Gadoterate meglumine (Dotarem), a gadolinium-based contrast agent, is indicated to detect and visualize areas with disruption of the blood brain barrier and/or abnormal vascularity. No developmental toxicity was observed in pregnant animals. Closely related diagnostic agents are gadobenate dimeglumine (MultiHance), gadodiamide (Omniscan), gadofosveset (Ablavar), gadopentetate dimeglumine (Magnevist), gadoteridol (Prohance), and gadoversetamide (OptiMARK). Although the animal data for these agents show risk, no harm has been reported in human pregnancies. However, the available human data are very limited, and the risk magnitude for embryo-fetal harm remains unknown.
Technetium (99mTc) tilmanocept (Lymphoseek) is a radioactive diagnostic agent used in patients with breast cancer or melanoma. The active ingredient is technetium (99mTc). Animal reproduction studies have not been conducted. 99mTc is probably compatible in pregnancy (see Drugs in Pregnancy and Lactation, 10th ed.; Philadelphia: Lippincott, Williams and Wilkins, 2014:1317-8; to be released in August), but the risk of the tilmanocept moiety is unknown.
The immunologic agent dimethyl fumarate (Tecfidera) is indicated for the treatment of patients with relapsing forms of multiple sclerosis. The drug caused developmental toxicity (embryolethality, impaired growth, and birth defects) in animals during all portions of pregnancy. In clinical trials, there were 38 exposed pregnancies with the following outcomes: 22 live births, 3 spontaneous abortions, 9 elective abortions, 3 ongoing pregnancies, and 1 lost to follow-up (CNS Drugs 2014;28:89-94). A pregnancy registry has been established, and patients should be encouraged to enroll by calling 800-456-2255.
Two new respiratory combination products were approved in 2013, both for chronic obstructive pulmonary disease: fluticasone/vilanterol (Breo Ellipta) and umeclidinium/vilanterol (Anoro Ellipta). Inhaled fluticasone, a corticosteroid, is compatible in pregnancy (see Drugs in Pregnancy and Lactation, 9th ed.; Philadelphia: Lippincott, Williams and Wilkins; 2011:599-601). Vilanterol is a long-acting beta2-adrenergic agonist that is probably compatible in pregnancy. The absolute bioavailability of inhaled fluticasone and vilanterol in nonpregnant adults was about 15% and 27%, respectively. The animal data for the combination or when given individually suggest low risk in pregnancy. Umeclidinium is a long-acting anticholinergic. It also is absorbed from the lung, but the amount was not specified by the manufacturer. The animal data for umeclidinium suggest low risk.
There are no reports of the above drugs being used during breastfeeding, but excretion into breast milk should be expected. The effect of these exposures on a nursing infant is unknown. However, if a mother is taking one of these drugs and breastfeeding, her infant should be monitored for adverse effects, especially those that are the most common (typically listed on the first page of the package insert) in patients taking the drug. Close monitoring is particularly important during the first 2 postpartum months. A 2003 study found that most adverse reactions in nursing infants occurred within that time period (Clin. Pediatr. 2003;42:325-40).
Mr. Briggs is a pharmacist clinical specialist at the outpatient clinics of Memorial Care Center for Women at Miller Children’s Hospital in Long Beach, Calif.; clinical professor of pharmacy at the University of California, San Francisco; and adjunct professor of pharmacy at the University of Southern California, Los Angeles, and Washington State University, Spokane. He also is coauthor of "Drugs in Pregnancy and Lactation," and coeditor of "Diseases, Complications, and Drug Therapy in Obstetrics." He had no other relevant financial disclosures. Contact him at [email protected].

FDA approves ofatumumab for chronic lymphocytic leukemia
The Food and Drug Administration has approved ofatumumab (Arzerra injection, for intravenous infusion) in combination with chlorambucil, for previously untreated patients with chronic lymphocytic leukemia for whom fludarabine-based therapy is considered inappropriate.
Ofatumumab, an anti-CD20-directed monoclonal antibody, manufactured by GlaxoSmithKline, was originally given an accelerated approval in 2009. As a condition of that approval, the company was required to conduct further studies. The trial used as the basis of the April 17 approval fulfilled that postmarketing requirement, and accelerated approval was converted to regular approval, said the FDA.
That multicenter, randomized, open-label trial compared ofatumumab in combination with chlorambucil to chlorambucil alone. Patients received an intravenous infusion of ofatumumab on the following schedule: 300 mg on cycle 1, day 1; 1,000 mg on cycle 1, day 8; and 1,000 mg on day 1 of all subsequent 28-day cycles. In both arms, chlorambucil was given at a dose of 10 mg/meter orally on days 1 to 7, every 28 days. Ofatumumab patients were premedicated with acetaminophen, an antihistamine, and a glucocorticoid.
Median progression-free survival was 22.4 months for patients receiving the combination, compared with 13.1 months for those who were given chlorambucil alone. Progression-free survival was assessed by a blinded independent review committee using the 2008 International Workshop on Chronic Lymphocytic Leukemia update of the National Cancer Institute Working Group guidelines.
The most common adverse reactions of the ofatumumab-chlorambucil combination were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain. Sixty-seven percent of patients who received ofatumumab experienced one or more symptoms of infusion reaction; 10% experienced a grade 3 or greater infusion reaction.
In September, the FDA added a black box warning to ofatumumab’s label on the risk of reactivation of hepatitis B virus infection in patients with prior infection.
For previously untreated chronic lymphocytic leukemia, the recommended dose and schedule is 300 mg on day 1; followed 1 week later by 1,000 mg on day 8; followed by 1,000 mg on day 1 of subsequent 28-day cycles for a minimum of 3 cycles until best response or a maximum of 12 cycles, according to the FDA.
On Twitter @aliciaault
The Food and Drug Administration has approved ofatumumab (Arzerra injection, for intravenous infusion) in combination with chlorambucil, for previously untreated patients with chronic lymphocytic leukemia for whom fludarabine-based therapy is considered inappropriate.
Ofatumumab, an anti-CD20-directed monoclonal antibody, manufactured by GlaxoSmithKline, was originally given an accelerated approval in 2009. As a condition of that approval, the company was required to conduct further studies. The trial used as the basis of the April 17 approval fulfilled that postmarketing requirement, and accelerated approval was converted to regular approval, said the FDA.
That multicenter, randomized, open-label trial compared ofatumumab in combination with chlorambucil to chlorambucil alone. Patients received an intravenous infusion of ofatumumab on the following schedule: 300 mg on cycle 1, day 1; 1,000 mg on cycle 1, day 8; and 1,000 mg on day 1 of all subsequent 28-day cycles. In both arms, chlorambucil was given at a dose of 10 mg/meter orally on days 1 to 7, every 28 days. Ofatumumab patients were premedicated with acetaminophen, an antihistamine, and a glucocorticoid.
Median progression-free survival was 22.4 months for patients receiving the combination, compared with 13.1 months for those who were given chlorambucil alone. Progression-free survival was assessed by a blinded independent review committee using the 2008 International Workshop on Chronic Lymphocytic Leukemia update of the National Cancer Institute Working Group guidelines.
The most common adverse reactions of the ofatumumab-chlorambucil combination were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain. Sixty-seven percent of patients who received ofatumumab experienced one or more symptoms of infusion reaction; 10% experienced a grade 3 or greater infusion reaction.
In September, the FDA added a black box warning to ofatumumab’s label on the risk of reactivation of hepatitis B virus infection in patients with prior infection.
For previously untreated chronic lymphocytic leukemia, the recommended dose and schedule is 300 mg on day 1; followed 1 week later by 1,000 mg on day 8; followed by 1,000 mg on day 1 of subsequent 28-day cycles for a minimum of 3 cycles until best response or a maximum of 12 cycles, according to the FDA.
On Twitter @aliciaault
The Food and Drug Administration has approved ofatumumab (Arzerra injection, for intravenous infusion) in combination with chlorambucil, for previously untreated patients with chronic lymphocytic leukemia for whom fludarabine-based therapy is considered inappropriate.
Ofatumumab, an anti-CD20-directed monoclonal antibody, manufactured by GlaxoSmithKline, was originally given an accelerated approval in 2009. As a condition of that approval, the company was required to conduct further studies. The trial used as the basis of the April 17 approval fulfilled that postmarketing requirement, and accelerated approval was converted to regular approval, said the FDA.
That multicenter, randomized, open-label trial compared ofatumumab in combination with chlorambucil to chlorambucil alone. Patients received an intravenous infusion of ofatumumab on the following schedule: 300 mg on cycle 1, day 1; 1,000 mg on cycle 1, day 8; and 1,000 mg on day 1 of all subsequent 28-day cycles. In both arms, chlorambucil was given at a dose of 10 mg/meter orally on days 1 to 7, every 28 days. Ofatumumab patients were premedicated with acetaminophen, an antihistamine, and a glucocorticoid.
Median progression-free survival was 22.4 months for patients receiving the combination, compared with 13.1 months for those who were given chlorambucil alone. Progression-free survival was assessed by a blinded independent review committee using the 2008 International Workshop on Chronic Lymphocytic Leukemia update of the National Cancer Institute Working Group guidelines.
The most common adverse reactions of the ofatumumab-chlorambucil combination were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain. Sixty-seven percent of patients who received ofatumumab experienced one or more symptoms of infusion reaction; 10% experienced a grade 3 or greater infusion reaction.
In September, the FDA added a black box warning to ofatumumab’s label on the risk of reactivation of hepatitis B virus infection in patients with prior infection.
For previously untreated chronic lymphocytic leukemia, the recommended dose and schedule is 300 mg on day 1; followed 1 week later by 1,000 mg on day 8; followed by 1,000 mg on day 1 of subsequent 28-day cycles for a minimum of 3 cycles until best response or a maximum of 12 cycles, according to the FDA.
On Twitter @aliciaault
Persistent lymphocytosis with ibrutinib does not indicate early relapse
Persistent lymphocytosis lasting more than 12 months in patients with chronic lymphocytic leukemia undergoing treatment with ibrutinib is not associated with a greater likelihood of early relapse.
A prospective observational study in 85 relapsed or refractory patients with chronic lymphocytic leukemia treated with ibrutinib showed lymphocytosis occurred in 77% in patients, persisting at 12 months in 20% of patients.
Dr. Jennifer A. Woyach of Ohio State University, Columbus, and her colleagues found no significant differences in progression-free survival between patients who responded to treatment but showed persistent lymphocytosis, and those with a partial or complete response to treatment without lymphocytosis.
They also found no significant differences in gene expression profiles in persistent lymphocytosis, suggesting that the lymphocytosis most likely represented the movement of quiescent cells (Blood 2014;123:1810-17).
Two authors were unpaid consultants for, three were paid consultants for, and two were employees of Pharmacyclics. One author received research funding from Janssen. The study was funded by the Four Winds Foundation, the Leukemia and Lymphoma Society, and several other organizations. Ibrutinib was provided by Pharmacyclics for in vitro experiments.
As more treatments targeting B-cell receptors became available, it was important for physicians to understand that lymphocytosis associated with this treatment was not indicative of treatment resistance or disease aggressiveness. The study by Woyach et al. provides the proof of principle that prolonged lymphocytosis produced by ibrutinib is composed of quiescent leukemic cells and provides a biological rationale in support of the current revisions of CLL [chronic lymphocytic leukemia] response criteria.
Dr. Davide Rossi and Dr. Gianluca Gaidano of the Amedeo Avogadro University of Eastern Piedmont, Alessandria, Italy, provided these comments in an editorial accompanying Dr. Woyach’s study (Blood 2014;123:1772-4). They declared that had no conflicts of interest.
Dr. Jennifer A. Woyach,
As more treatments targeting B-cell receptors became available, it was important for physicians to understand that lymphocytosis associated with this treatment was not indicative of treatment resistance or disease aggressiveness. The study by Woyach et al. provides the proof of principle that prolonged lymphocytosis produced by ibrutinib is composed of quiescent leukemic cells and provides a biological rationale in support of the current revisions of CLL [chronic lymphocytic leukemia] response criteria.
Dr. Davide Rossi and Dr. Gianluca Gaidano of the Amedeo Avogadro University of Eastern Piedmont, Alessandria, Italy, provided these comments in an editorial accompanying Dr. Woyach’s study (Blood 2014;123:1772-4). They declared that had no conflicts of interest.
As more treatments targeting B-cell receptors became available, it was important for physicians to understand that lymphocytosis associated with this treatment was not indicative of treatment resistance or disease aggressiveness. The study by Woyach et al. provides the proof of principle that prolonged lymphocytosis produced by ibrutinib is composed of quiescent leukemic cells and provides a biological rationale in support of the current revisions of CLL [chronic lymphocytic leukemia] response criteria.
Dr. Davide Rossi and Dr. Gianluca Gaidano of the Amedeo Avogadro University of Eastern Piedmont, Alessandria, Italy, provided these comments in an editorial accompanying Dr. Woyach’s study (Blood 2014;123:1772-4). They declared that had no conflicts of interest.
Persistent lymphocytosis lasting more than 12 months in patients with chronic lymphocytic leukemia undergoing treatment with ibrutinib is not associated with a greater likelihood of early relapse.
A prospective observational study in 85 relapsed or refractory patients with chronic lymphocytic leukemia treated with ibrutinib showed lymphocytosis occurred in 77% in patients, persisting at 12 months in 20% of patients.
Dr. Jennifer A. Woyach of Ohio State University, Columbus, and her colleagues found no significant differences in progression-free survival between patients who responded to treatment but showed persistent lymphocytosis, and those with a partial or complete response to treatment without lymphocytosis.
They also found no significant differences in gene expression profiles in persistent lymphocytosis, suggesting that the lymphocytosis most likely represented the movement of quiescent cells (Blood 2014;123:1810-17).
Two authors were unpaid consultants for, three were paid consultants for, and two were employees of Pharmacyclics. One author received research funding from Janssen. The study was funded by the Four Winds Foundation, the Leukemia and Lymphoma Society, and several other organizations. Ibrutinib was provided by Pharmacyclics for in vitro experiments.
Persistent lymphocytosis lasting more than 12 months in patients with chronic lymphocytic leukemia undergoing treatment with ibrutinib is not associated with a greater likelihood of early relapse.
A prospective observational study in 85 relapsed or refractory patients with chronic lymphocytic leukemia treated with ibrutinib showed lymphocytosis occurred in 77% in patients, persisting at 12 months in 20% of patients.
Dr. Jennifer A. Woyach of Ohio State University, Columbus, and her colleagues found no significant differences in progression-free survival between patients who responded to treatment but showed persistent lymphocytosis, and those with a partial or complete response to treatment without lymphocytosis.
They also found no significant differences in gene expression profiles in persistent lymphocytosis, suggesting that the lymphocytosis most likely represented the movement of quiescent cells (Blood 2014;123:1810-17).
Two authors were unpaid consultants for, three were paid consultants for, and two were employees of Pharmacyclics. One author received research funding from Janssen. The study was funded by the Four Winds Foundation, the Leukemia and Lymphoma Society, and several other organizations. Ibrutinib was provided by Pharmacyclics for in vitro experiments.
Dr. Jennifer A. Woyach,
Dr. Jennifer A. Woyach,
FROM BLOOD
Major finding: Patients with chronic lymphocytic leukemia who experienced persistent lymphocytosis associated with ibrutinib treatment did not have a greater likelihood of early relapse.
Data source: Prospective observational cohort study in 85 patients with relapsed or refractory chronic lymphocytic leukemia undergoing treatment with ibrutinib.
Disclosures: Two authors were unpaid consultants and three were paid consultants for Pharmacyclics, and two authors were employees of the company. One author received research funding from Janssen. The study was funded by the Four Winds Foundation, the Leukemia and Lymphoma Society, and several other organizations. Ibrutinib was provided by Pharmacyclics for in vitro experiments.
Idelalisib and rituximab extends survival in CLL patients
Adding idelalisib to rituximab significantly extends survival in patients with heavily pretreated relapsed chronic lymphocytic leukemia unsuitable for chemotherapy, according to published results from Study 116.
The primary endpoint of median progression-free survival (PFS) was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab plus idelalisib (hazard ratio, 0.15; P less than .0001).
The PFS advantage was seen across all subgroups including patients with the 17p deletion and TP53 mutations, Dr. Richard R. Furman reported at the annual meeting of the American Society of Hematology in December. The final results of the study were published online January 22 in the New England Journal of Medicine.
Overall survival was not reached in either group, but was also significantly longer with idelalisib (HR, 0.28; P = .018).
The investigators evenly randomized 220 patients to eight infusions of rituximab over 24 weeks plus either idelalisib 150 mg or placebo twice daily until disease progression. Rituximab was dosed at 375 mg/m2 in week 1, 500 mg/m2 every 2 weeks for four doses, followed by 500 mg/m2 every 4 weeks for three more doses.
Adding idelalisib to rituximab significantly extends survival in patients with heavily pretreated relapsed chronic lymphocytic leukemia unsuitable for chemotherapy, according to published results from Study 116.
The primary endpoint of median progression-free survival (PFS) was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab plus idelalisib (hazard ratio, 0.15; P less than .0001).
The PFS advantage was seen across all subgroups including patients with the 17p deletion and TP53 mutations, Dr. Richard R. Furman reported at the annual meeting of the American Society of Hematology in December. The final results of the study were published online January 22 in the New England Journal of Medicine.
Overall survival was not reached in either group, but was also significantly longer with idelalisib (HR, 0.28; P = .018).
The investigators evenly randomized 220 patients to eight infusions of rituximab over 24 weeks plus either idelalisib 150 mg or placebo twice daily until disease progression. Rituximab was dosed at 375 mg/m2 in week 1, 500 mg/m2 every 2 weeks for four doses, followed by 500 mg/m2 every 4 weeks for three more doses.
Adding idelalisib to rituximab significantly extends survival in patients with heavily pretreated relapsed chronic lymphocytic leukemia unsuitable for chemotherapy, according to published results from Study 116.
The primary endpoint of median progression-free survival (PFS) was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab plus idelalisib (hazard ratio, 0.15; P less than .0001).
The PFS advantage was seen across all subgroups including patients with the 17p deletion and TP53 mutations, Dr. Richard R. Furman reported at the annual meeting of the American Society of Hematology in December. The final results of the study were published online January 22 in the New England Journal of Medicine.
Overall survival was not reached in either group, but was also significantly longer with idelalisib (HR, 0.28; P = .018).
The investigators evenly randomized 220 patients to eight infusions of rituximab over 24 weeks plus either idelalisib 150 mg or placebo twice daily until disease progression. Rituximab was dosed at 375 mg/m2 in week 1, 500 mg/m2 every 2 weeks for four doses, followed by 500 mg/m2 every 4 weeks for three more doses.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
ONO-4059 makes waves in heavily pretreated CLL
NEW ORLEANS – Early data suggest that the second-generation oral BTK inhibitor ONO-4059 may give ibrutinib a run for its money in chronic lymphocytic leukemia.
The response rate to ONO-4059 monotherapy was 89% overall and 71% in those with the deleterious 17p deletion among 18 heavily pretreated patients with relapsed/refractory or high-risk CLL in a phase I, dose-escalation study.
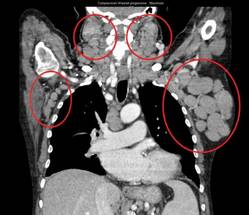
Patients had already received a median of three prior therapies, including rituximab (84%) and fludarabine (95%), and had no higher priority therapy available to them, said Dr. Gilles Salles of Hospices Civils de Lyon (France), Universite Claude Bernard Lyon.
All patients had improved hemoglobin and platelet counts after 3 months on treatment and rapid reductions in lymph node size within the first 28-day cycle. Tumor burden was reduced by 50% for most patients, and all but one patient experienced a response that was detectable on a CT scan.
"This was true whatever their FISH status or 17p or 11q deletion status," Dr. Salles said at the annual meeting of the American Society of Hematology.
ONO-4059 is a highly selective Bruton’s tyrosine kinase (BTK) inhibitor with antitumor activity in several preclinical models.
No patients had received prior treatment with a P13 kinase or a BTK inhibitor, including ibrutinib (Imbruvica), which recently gained accelerated approval for previously treated mantle cell lymphoma.
ONO-4059 was given at daily doses ranging from 20 mg to 320 mg for up to 6 months, with the option of additional dosing up to 2 years. Sustained BTK inhibition was established at doses of 40 mg and higher.
Overall, the best response was a partial response in 14 patients, as well as two partial responses with lymphocytosis and one stable disease, he said. No complete responses occurred.
One patient progressed roughly 1 month after showing an initial response and complete disappearance of all palpable disease on physical exam. Richter’s syndrome was suspected.
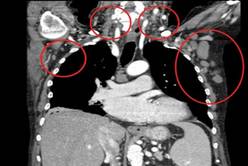
"It’s very promising efficacy in this highly pretreated population," Dr. Salles said.
Patients with relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma, especially the ABC subtype, also appear sensitive to ONO-4059. Overall response rates were 43% and 75%, respectively, including three complete responses reported from the phase I study in a separate poster presentation at the meeting.
ONO-4059 had a favorable safety profile with a single dose-limiting toxicity observed in a patient who had Waldenstrom’s macroglobulinemia, was on the 320-mg dose, and was intolerant to all prior therapies. The maximum tolerated dose has not yet been reached.
The majority of adverse events in the CLL patients were grades 1 and 2. There were no clinically significant bleeding events or bruising, and there was a low incidence of diarrhea and rash, Dr. Salles said.
ONO-4059–related grade 3-4 events were independent of dose and included one grade 3 neutropenia at 20 mg and two grade 4 events at 20 mg and 320 mg. Four serious adverse events (febrile neutropenia, pyrexia, rash, and neutropenia) occurred in three patients, all of whom are still in the study and showing good clinical response, Dr. Salles said. Of the 30 patients dosed to date, 22 remain in the study.
No other trials are firmly planned, and pharmacokinetics/pharmacodynamics data continue to be explored in order to assess a phase II dosage, he said in an interview.
Dr. Salles reported consulting for and receiving honoraria from Roche. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
NEW ORLEANS – Early data suggest that the second-generation oral BTK inhibitor ONO-4059 may give ibrutinib a run for its money in chronic lymphocytic leukemia.
The response rate to ONO-4059 monotherapy was 89% overall and 71% in those with the deleterious 17p deletion among 18 heavily pretreated patients with relapsed/refractory or high-risk CLL in a phase I, dose-escalation study.
Patients had already received a median of three prior therapies, including rituximab (84%) and fludarabine (95%), and had no higher priority therapy available to them, said Dr. Gilles Salles of Hospices Civils de Lyon (France), Universite Claude Bernard Lyon.
All patients had improved hemoglobin and platelet counts after 3 months on treatment and rapid reductions in lymph node size within the first 28-day cycle. Tumor burden was reduced by 50% for most patients, and all but one patient experienced a response that was detectable on a CT scan.
"This was true whatever their FISH status or 17p or 11q deletion status," Dr. Salles said at the annual meeting of the American Society of Hematology.
ONO-4059 is a highly selective Bruton’s tyrosine kinase (BTK) inhibitor with antitumor activity in several preclinical models.
No patients had received prior treatment with a P13 kinase or a BTK inhibitor, including ibrutinib (Imbruvica), which recently gained accelerated approval for previously treated mantle cell lymphoma.
ONO-4059 was given at daily doses ranging from 20 mg to 320 mg for up to 6 months, with the option of additional dosing up to 2 years. Sustained BTK inhibition was established at doses of 40 mg and higher.
Overall, the best response was a partial response in 14 patients, as well as two partial responses with lymphocytosis and one stable disease, he said. No complete responses occurred.
One patient progressed roughly 1 month after showing an initial response and complete disappearance of all palpable disease on physical exam. Richter’s syndrome was suspected.
"It’s very promising efficacy in this highly pretreated population," Dr. Salles said.
Patients with relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma, especially the ABC subtype, also appear sensitive to ONO-4059. Overall response rates were 43% and 75%, respectively, including three complete responses reported from the phase I study in a separate poster presentation at the meeting.
ONO-4059 had a favorable safety profile with a single dose-limiting toxicity observed in a patient who had Waldenstrom’s macroglobulinemia, was on the 320-mg dose, and was intolerant to all prior therapies. The maximum tolerated dose has not yet been reached.
The majority of adverse events in the CLL patients were grades 1 and 2. There were no clinically significant bleeding events or bruising, and there was a low incidence of diarrhea and rash, Dr. Salles said.
ONO-4059–related grade 3-4 events were independent of dose and included one grade 3 neutropenia at 20 mg and two grade 4 events at 20 mg and 320 mg. Four serious adverse events (febrile neutropenia, pyrexia, rash, and neutropenia) occurred in three patients, all of whom are still in the study and showing good clinical response, Dr. Salles said. Of the 30 patients dosed to date, 22 remain in the study.
No other trials are firmly planned, and pharmacokinetics/pharmacodynamics data continue to be explored in order to assess a phase II dosage, he said in an interview.
Dr. Salles reported consulting for and receiving honoraria from Roche. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
NEW ORLEANS – Early data suggest that the second-generation oral BTK inhibitor ONO-4059 may give ibrutinib a run for its money in chronic lymphocytic leukemia.
The response rate to ONO-4059 monotherapy was 89% overall and 71% in those with the deleterious 17p deletion among 18 heavily pretreated patients with relapsed/refractory or high-risk CLL in a phase I, dose-escalation study.
Patients had already received a median of three prior therapies, including rituximab (84%) and fludarabine (95%), and had no higher priority therapy available to them, said Dr. Gilles Salles of Hospices Civils de Lyon (France), Universite Claude Bernard Lyon.
All patients had improved hemoglobin and platelet counts after 3 months on treatment and rapid reductions in lymph node size within the first 28-day cycle. Tumor burden was reduced by 50% for most patients, and all but one patient experienced a response that was detectable on a CT scan.
"This was true whatever their FISH status or 17p or 11q deletion status," Dr. Salles said at the annual meeting of the American Society of Hematology.
ONO-4059 is a highly selective Bruton’s tyrosine kinase (BTK) inhibitor with antitumor activity in several preclinical models.
No patients had received prior treatment with a P13 kinase or a BTK inhibitor, including ibrutinib (Imbruvica), which recently gained accelerated approval for previously treated mantle cell lymphoma.
ONO-4059 was given at daily doses ranging from 20 mg to 320 mg for up to 6 months, with the option of additional dosing up to 2 years. Sustained BTK inhibition was established at doses of 40 mg and higher.
Overall, the best response was a partial response in 14 patients, as well as two partial responses with lymphocytosis and one stable disease, he said. No complete responses occurred.
One patient progressed roughly 1 month after showing an initial response and complete disappearance of all palpable disease on physical exam. Richter’s syndrome was suspected.
"It’s very promising efficacy in this highly pretreated population," Dr. Salles said.
Patients with relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma, especially the ABC subtype, also appear sensitive to ONO-4059. Overall response rates were 43% and 75%, respectively, including three complete responses reported from the phase I study in a separate poster presentation at the meeting.
ONO-4059 had a favorable safety profile with a single dose-limiting toxicity observed in a patient who had Waldenstrom’s macroglobulinemia, was on the 320-mg dose, and was intolerant to all prior therapies. The maximum tolerated dose has not yet been reached.
The majority of adverse events in the CLL patients were grades 1 and 2. There were no clinically significant bleeding events or bruising, and there was a low incidence of diarrhea and rash, Dr. Salles said.
ONO-4059–related grade 3-4 events were independent of dose and included one grade 3 neutropenia at 20 mg and two grade 4 events at 20 mg and 320 mg. Four serious adverse events (febrile neutropenia, pyrexia, rash, and neutropenia) occurred in three patients, all of whom are still in the study and showing good clinical response, Dr. Salles said. Of the 30 patients dosed to date, 22 remain in the study.
No other trials are firmly planned, and pharmacokinetics/pharmacodynamics data continue to be explored in order to assess a phase II dosage, he said in an interview.
Dr. Salles reported consulting for and receiving honoraria from Roche. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
AT ASH 2013
Major finding: The response rate was 89% overall and 71% for patients with 17p deletion.
Data source: A prospective, phase I dose-escalation study in 18 patients with relapsed/refractory or high-risk CLL.
Disclosures: Dr. Salles reported honoraria from Janssen, Gilead, and Celgene. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
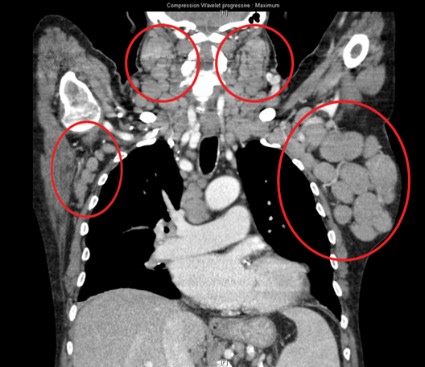
FDA provides steps to obtain ponatinib following suspension
The Food and Drug Administration has provided information on how health care professionals can ensure that patients who have benefitted from the leukemia drug ponatinib continue to have access to the treatment.
Less than a week after the agency announced that marketing and sales of the drug had been suspended because of the risk of life-threatening blood clots and severe narrowing of blood vessels associated with treatment, the FDA posted instructions on how to obtain emergency access to ponatinib through an Investigational New Drug (IND) application. This process entails contacting the FDA to obtain an emergency IND number for each patient who would benefit from continuing treatment and providing the manufacturer with that number to get ponatinib (Iclusig).
Health care professionals can obtain INDs for multiple patients during one phone call and should contact the agency at least 48-72 hours before the drug is needed, according to an FDA announcement.
The statement noted that health care professionals "may continue to use Iclusig for patients who they determine are responding to the drug and for whom the potential benefits outweigh the risks."
Ponatinib is a kinase inhibitor marketed by ARIAD Pharmaceuticals. It was approved in December 2012 for treating chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia in adults.
Serious adverse events associated with ponatinib should be reported online to the FDA or by phone at 800-332-0178. Information about the IND program is available online. The FDA’s emergency IND telephone number is 301-796-7550 (between 8 a.m. and 4:30 p.m. EST, weekdays and 866-300-4374 after 4:30 p.m. EST. More information on how to obtain an IND for patients on ponatinib is available at http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
The Food and Drug Administration has provided information on how health care professionals can ensure that patients who have benefitted from the leukemia drug ponatinib continue to have access to the treatment.
Less than a week after the agency announced that marketing and sales of the drug had been suspended because of the risk of life-threatening blood clots and severe narrowing of blood vessels associated with treatment, the FDA posted instructions on how to obtain emergency access to ponatinib through an Investigational New Drug (IND) application. This process entails contacting the FDA to obtain an emergency IND number for each patient who would benefit from continuing treatment and providing the manufacturer with that number to get ponatinib (Iclusig).
Health care professionals can obtain INDs for multiple patients during one phone call and should contact the agency at least 48-72 hours before the drug is needed, according to an FDA announcement.
The statement noted that health care professionals "may continue to use Iclusig for patients who they determine are responding to the drug and for whom the potential benefits outweigh the risks."
Ponatinib is a kinase inhibitor marketed by ARIAD Pharmaceuticals. It was approved in December 2012 for treating chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia in adults.
Serious adverse events associated with ponatinib should be reported online to the FDA or by phone at 800-332-0178. Information about the IND program is available online. The FDA’s emergency IND telephone number is 301-796-7550 (between 8 a.m. and 4:30 p.m. EST, weekdays and 866-300-4374 after 4:30 p.m. EST. More information on how to obtain an IND for patients on ponatinib is available at http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
The Food and Drug Administration has provided information on how health care professionals can ensure that patients who have benefitted from the leukemia drug ponatinib continue to have access to the treatment.
Less than a week after the agency announced that marketing and sales of the drug had been suspended because of the risk of life-threatening blood clots and severe narrowing of blood vessels associated with treatment, the FDA posted instructions on how to obtain emergency access to ponatinib through an Investigational New Drug (IND) application. This process entails contacting the FDA to obtain an emergency IND number for each patient who would benefit from continuing treatment and providing the manufacturer with that number to get ponatinib (Iclusig).
Health care professionals can obtain INDs for multiple patients during one phone call and should contact the agency at least 48-72 hours before the drug is needed, according to an FDA announcement.
The statement noted that health care professionals "may continue to use Iclusig for patients who they determine are responding to the drug and for whom the potential benefits outweigh the risks."
Ponatinib is a kinase inhibitor marketed by ARIAD Pharmaceuticals. It was approved in December 2012 for treating chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia in adults.
Serious adverse events associated with ponatinib should be reported online to the FDA or by phone at 800-332-0178. Information about the IND program is available online. The FDA’s emergency IND telephone number is 301-796-7550 (between 8 a.m. and 4:30 p.m. EST, weekdays and 866-300-4374 after 4:30 p.m. EST. More information on how to obtain an IND for patients on ponatinib is available at http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.