User login
Preserving fertility in female cancer patients: A snapshot of the options
In the last few decades, the survival rates have improved in many of the malignancies that affect young adults. This progress has made fertility preservation and quality of life after cancer treatment important, most of all in survivors of childhood cancers.
Men who are about to undergo cancer treatment can bank their sperm, but as yet no analogous noninvasive option is available for women. The most studied methods are often invasive and require the woman to take large doses of hormones. They may also necessitate a delay in starting cancer treatment.
Which method of fertility preservation a woman should choose depends on several factors, including the type of disease, the treatment required, the age of the patient, whether she has a long-term partner, and whether treatment can be delayed.
Chemotherapy and radiotherapy have well-known deleterious effects on female reproductive function. Many studies have shown that acute loss of growing follicles within the ovary and resultant premature ovarian failure often follow chemotherapy. This ovarian damage has long-term consequences, such as shortened reproductive life span and hormone deficiency.1
Fertility preservation requires a team effort. It should be managed by an oncology center that has built a close collaboration between oncologists, fertility specialists, psychologists, and primary care physicians to allow early discussion and to offer a full range of options to these patients.
The aim of this paper is to discuss the current options for preserving fertility in female cancer patients who have to undergo gonadotoxic cancer treatment.
FEMALE FERTILITY AND ASSESSMENT OF OVARIAN RESERVE
At birth, baby girls have about 1 million primordial ovarian follicles, which is the most they will ever have. By the time they reach menarche, this number has declined to 180,000, and at menopause only about 1,000 remain.2
Throughout a woman’s reproductive life, the number of oocytes remaining—both primordial follicles and the relatively small number of maturing, growing follicles—is called her ovarian reserve. When cancer is diagnosed, we need to assess the patient’s ovarian reserve to direct the discussion about the need for fertility preservation.
In search of markers of ovarian reserve
Fertility experts have been looking for clinical biomarkers that could give us an estimate of the number of nongrowing follicles and, consequently, of the ovarian reserve.
All methods used for the assessment of ovarian reserve actually provide an indirect determination of the remaining pool of oocytes. In clinical practice, a high blood level of follicle-stimulating hormone (FSH) on cycle day 3 (>15 mU/mL) or a low level of anti-Müllerian hormone (AMH) (<1 ng/mL) is generally associated with a low ovarian reserve.
The serum FSH level is the marker most commonly used, but it varies widely at different times in the menstrual cycle.
The FSH test is usually performed on day 3 of the menstrual cycle, when the estrogen level is expected to be low due to negative feedback. The FSH result needs to be combined with the estradiol level, especially in those patients with irregular menstruation or in cases of amenorrhea. A random FSH test is considered valid if the detected estrogen level is low. Women undergoing in vitro fertilization with a day 3 FSH lower than 15 mIU/mL are more likely to conceive than women with a higher FSH level.
AMH and antral follicles. Recently, two other variables have been introduced in clinical practice: the AMH concentration and the antral follicle count (the number of small antral follicles within the ovary) as assessed by transvaginal ultrasonography.
AMH is released by the granulosa cells of small, growing follicles. Because its level is much more stable over the menstrual cycle than the FSH level, it can be measured on any day of the cycle.3 In cancer survivors, AMH is particularly useful in demonstrating the degree of ovarian tissue damage induced by radiotherapy and chemotherapy and in evaluating the ovarian reserve.4
A reduced number of antral follicles causes a diminished AMH production, which becomes undetectable with menopause. The AMH level is strongly associated with the basal antral follicle count. Various threshold values (0.2–1.26 ng/mL) have been used to identify women with low ovarian reserve.
HOW CANCER TREATMENT DAMAGES THE OVARIES
Advances in surgery, radiotherapy, and chemotherapy have significantly improved the prognosis for young cancer patients. However, cancer treatments often result in ovarian dysfunction, and premature menopause and irreversible sterility are the most dramatic outcomes. The resulting low estrogen levels, in addition to their physiologic consequences, also worsen quality of life through psychological effects, which can as well influence the patient’s compliance with treatment.
Chemotherapy: Alkylating agents are the most gonadotoxic
The mechanism by which chemotherapy affects ovarian function is poorly understood. Histologically, chemotherapeutic drugs could lead to ovarian atrophy and stromal fibrosis, to depletion of the primordial follicle stockpile, and to reduced ovarian weight, resulting in ovarian dysfunction.5
The patient's age correlates with the probability of ovarian damage or, inversely, ovarian resistance to chemotherapy. Young women have more primordial oocytes, and after chemotherapy they face a sharp reduction of their ovarian reserve. Still, younger patients show a lower rate of ovarian toxicity than older women.6
The type and the cumulative dose of cytotoxic agents used are other important variables.7 There are six main classes of chemotherapeutic drugs: alkylating agents, platinum derivatives, antibiotics, antimetabolites, plant alkaloids, and taxanes. They all affect ovarian function, but alkylating agents are the most gonadotoxic.
Alkylating agents covalently bind an alkyl group to the DNA molecule and inhibit it from replicating. In the ovaries they directly injure primordial oocytes and deplete follicles. 8 They also seriously damage the ovarian vasculature so that the follicles cannot grow.9 Their destructive effect on the primordial follicles is dose-dependent and varies with the age and developmental maturity of the patient at the time of the therapy, with older women more likely to be left infertile afterward.10
Cyclophosphamide is an alkylating agent often used to treat severe manifestations of autoimmune diseases such as systemic lupus erythematosus, BehÇet disease, steroid-resistant glomerulonephritis, inflammatory bowel disease, pemphigus vulgaris, and others. Because it can lead to premature ovarian failure and infertility, women receiving cyclophosphamide for autoimmune conditions may also need treatment to preserve fertility.11
Radiotherapy
Ovarian follicles are remarkably vulnerable to damage from ionizing radiation, which results in ovarian atrophy and reduced follicle stores.1
The risk of radiotherapy-induced infertility is closely related to the patient’s age and developmental maturity at the time of therapy and to dose fractionation and the extent of the irradiation field. Every patient has a different sensitivity to radiation damage that is probably genetically predetermined, but age seems to be the most important variable. Wo and Viswanathan12 used a mathematical model devised by Wallace et al13 to show that the older the patient, the lower the radiation dose necessary to impair ovarian function.
The irradiation field is another aspect to consider. Pelvic radiation can be necessary in rectal cancer, cervical cancer, and lymphoma. In these cases, surgically moving the ovaries to a region outside the radiation field could be an option to minimize radiotherapy-induced ovarian damage.14
Radiotherapy can also damage the uterus. Pelvic irradiation can reduce uterine volume, alter uterine elasticity through myometrial fibrosis, and modify vascularization and endometrial thickness.15,16 These alterations are closely correlated with the total radiation dose, the site of irradiation, and the patient’s age at the time of the treatment, the prepubertal uterus being more susceptible to damage. 15,17 Larsen et al15 found that girls who received uterine irradiation before puberty had lower uterine volumes in adulthood than girls who received chemotherapy alone or radiation to other parts of the body, and that the younger the patient at the time of radiotherapy, the smaller the uterus later. These effects could result in adverse pregnancy outcomes.
Furthermore, radiation could also damage the uterine vasculature. Holm et al16 used ultrasonography to evaluate the effect of total-body irradiation and found that uterine volume and uterine blood flow were both impaired.15
All these possible alterations could lead to a reduced uterine response to cytotrophoblast invasion and to decreased fetoplacental blood flow, which could impair embryonic and fetal growth. In that case, a surrogate pregnancy would be the only method to achieve parenthood using the couple’s gametes.
OPTIONS FOR FERTILITY PRESERVATION
Assisted reproductive technology
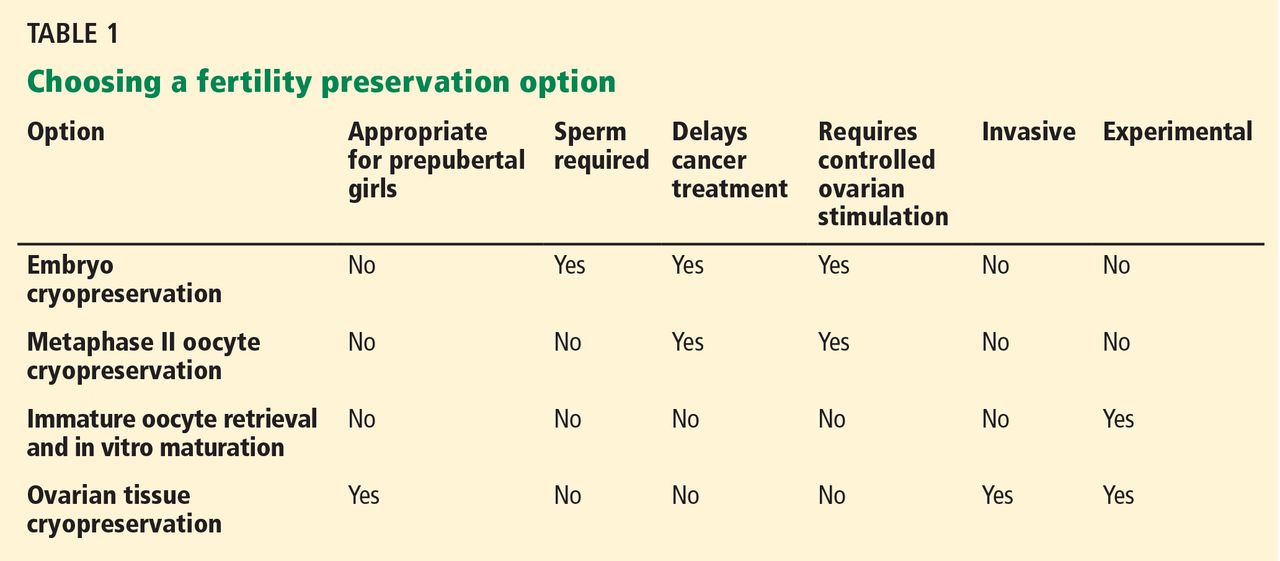
Young women diagnosed with an oncologic disease may wish to use assisted reproductive technology (Table 1) to keep open the possibility of having children at a later date. One approach is to harvest oocytes, fertilize them in vitro, and deep-freeze (cryopreserve) the resulting embryos to be thawed and implanted later. Alternatively, oocytes can be frozen directly, although success rates are lower with this method. And another approach is to obtain and cryopreserve ovarian tissue. If none of these is possible, oocytes may be obtained from a donor.
Controlled ovarian stimulation
If oocytes are to be harvested, a number of them should be harvested at one time.
There is not an optimal number of oocytes that should be retrieved, but cryopreservation of a large number of oocytes allows us to perform multiple attempts at in vitro fertilization, improving the chances of pregnancy. The fertilization rate (defined as the total number of zygotes at the 2-pronucleus stage divided by the number of fertilized oocytes) with intracytoplasmic sperm injection is 70% to 80%. On average, for every 10 eggs, 7 to 8 eggs will normally be fertilized. The implantation rate (defined as the total number of pregnancies divided by the total number of embryo transferred) with intracytoplasmic sperm injection is 40% to 50%—ie, only half of the transferred embryos will successfully implant and result in a pregnancy.
So that more than one ripe egg can be obtained at a time, the patient must undergo a regimen of controlled ovarian stimulation to achieve multifollicular growth. Stimulation protocols are based on giving pituitary hormones, ie, analogues of gonadotropin-releasing hormone (GnRH) (both agonists and antagonists), followed by recombinant FSH or human menopausal gonadotropin to promote follicular development. A single dose of human chorionic gonadotropin (hCG) is given to induce ovulation when the lead follicles have reached 18 to 20 mm in size.18
Many oncologists consider controlled ovarian stimulation dangerous for cancer patients because it takes time and thus delays cancer treatment. Furthermore, the regimen boosts the levels of circulating estrogens, which could be harmful in patients with hormone-dependent tumors.19
Oocytes can also be retrieved during unstimulated cycles. This avoids increasing estrogen concentrations above the physiologic level, but no more than a single oocyte is collected per cycle. The patient could undergo multiple oocyte harvestings, one per cycle, but this would delay her cancer treatment even more—by months—which is not recommended.
Serious efforts to minimize estrogen production during controlled ovarian stimulation have been made, although further studies are needed. Research is under way to develop appropriate ovarian stimulation protocols based on drugs with antiestrogenic effects.
Tamoxifen, an antagonist of the estrogen receptor, is widely used in breast cancer treatment. 20 It can also be given during controlled ovarian stimulation because it promotes follicular growth and induces ovulation.21 Oktay et al22 found that the embryo yield was 2.5 times higher in women with breast cancer treated with tamoxifen than in a retrospective control group consisting of breast cancer patients attempting natural-cycle oocyte retrieval.
Letrozole, an aromatase inhibitor, is also commonly used in treating breast and ovarian cancer. Aromatase is an enzyme that catalyzes the conversion of androgenic precursors to estrogens, and it is found in many tissues, including granulosa cells. Several studies report the use of letrozole, alone or in combination with low doses of recombinant FSH, in ovarian stimulation protocols in cancer patients, with positive clinical outcomes.23
Tamoxifen or letrozole, combined with recombinant FSH, is an attractive option in a controlled ovarian stimulation protocol for cancer patients,24 although further investigation is needed.
Cryopreservation methods
Two main protocols are currently used to freeze oocytes, embryos, and ovarian tissue: slow freezing and vitrification.
In the slow-freezing method, cryoprotectant agents are used to draw water out of the cells, raising intracellular viscosity without (or with minimal) intracellular ice crystal formation, while the sample is cooled slowly in a controlled manner. These cryoprotectant chemicals lower the freezing point of the solution, allowing greater cellular dehydration during the slow freezing. They also protect the plasmatic cell membrane by changing its physical state from liquid to partially dry. To avoid excessive deformation that could damage the cytoplasmic structures, cryoprotectant agents are added in successive stages.25
Vitrification is a newer method that uses higher concentrations of cryoprotectants and flash freezing. The instant drop in temperature converts this highly concentrated solution from an aqueous state to a semisolid, amorphous state that does not contain ice crystals, which are the main cause of damage during the freezing process.26 Since most cryoprotectant agents are extremely toxic, it is necessary to minimize the time that oocytes, embryos, and ovarian tissue are exposed to them.
Cryopreservation of embryos
According to the American Society of Clinical Oncology and the American Society of Reproductive Medicine, embryo freezing is the most established method for fertility preservation, with tangible and widely reported success.27 In normal practice, oocytes are retrieved after a controlled ovarian stimulation and then fertilized in vitro. Then they are treated with cryoprotectant agents, frozen, and stored. Upon demand, embryos can be thawed and implanted.
The Society for Assisted Reproductive Technology reports that the current live birth rate per transfer using thawed embryos from nondonor oocytes in US women under age 35 is 38.7%; at age 35 to 37 it is 35.1%.28 In women with cancer, storing as many embryos as possible could help improve embryo survival and the implantation rate. The optimal time to perform embryo cryopreservation is still being discussed,29 but, as in women without cancer, it is commonly done 3 to 5 days after fertilization.
In the past few years, the use of vitrification has greatly increased, as the post-thawing survival rates and pregnancy rates are higher with this method than with slow freezing.30
Despite its success, embryo cryopreservation has important drawbacks. First, the patient must be of reproductive age and have a partner or accept the use of donor sperm. Second, most patients undergo controlled ovarian stimulation before oocyte retrieval, causing a delay in starting cancer treatment, which is not acceptable in many cases. Moreover, the high serum estrogen levels caused by ovarian stimulation may be contraindicated in women with estrogen-sensitive malignancies.19
Oocyte cryopreservation
Cryopreservation of oocytes avoids the need for sperm and, thus, may be offered to more patients than embryo cryopreservation. In addition, it may circumvent ethical or legal considerations associated with embryo freezing, such as ownership of reproductive material.
However, oocytes are more difficult to cryopreserve than embryos. Indeed, ice crystals frequently form inside and outside the cells, damaging the cell membrane and the meiotic spindle. In routine practice, mature oocytes in metaphase II are used for cryopreservation. Metaphase II oocytes are large cells that contain a delicate meiotic spindle. Moreover, their cytoplasm contains a higher proportion of water than other cells, which could affect their viability after freezing and thawing due to ice crystal formation. In addition, cryopreservation could be responsible for hardening of the zona pellucida (through diffuse thickening of the cell membrane), adversely affecting fertilization rates.31
Significant improvements were achieved in fertilization of cryopreserved oocytes with intracytoplasmic sperm injection. Nevertheless, there are still concerns regarding oocyte cryopreservation. Further studies are needed to determine the risk of aneuploidy caused by damage to the meiotic spindle after oocyte cryopreservation. The potentially detrimental effects of high cryopreservant concentrations used in vitrification also need to be investigated.
In vitro maturation
A novel fertility preservation strategy involves collecting immature oocytes from primordial follicles in unstimulated cycles and then letting them mature in vitro.
To date, immature oocytes retrieved at the germinal vesicle stage can be cryopreserved with vitrification followed by in vitro maturation after thawing, but several studies have demonstrated that better results are obtained when vitrification follows the in vitro maturation process.32
Compared with mature oocytes, immature oocytes are less susceptible to damage during cryopreservation and thus have a better chance of surviving freezing and thawing, thanks to some peculiar characteristics: they are small, have few organelles, lack a zona pellucida, have low metabolic activity, and are in a state of relative quiescence.33 Controlled ovarian stimulation is not necessary, so this procedure preserves fertility without delaying the start of cancer treatment.
Patients are usually evaluated with transvaginal ultrasonography in the early follicular phase of the menstrual cycle (between day 2 and day 4) to count and measure the antral follicles. Immature oocytes are collected when the leading follicle has reached 10 to 12 mm in size and 36 hours after a subcutaneous injection of hCG.34 The retrieved oocytes are then incubated for 24 to 48 hours in a special medium supplemented with FSH and luteinizing hormone (LH). Immature oocytes can also be collected in the luteal phase.
This option could be considered when cancer treatment cannot be delayed for conventional follicular-phase retrieval35 or in case of a premature LH surge during ovarian stimulation. 36 It should be offered to patients facing infertility related to cancer treatment only after appropriate counseling and as a part of a clinical study.
Ovarian tissue cryopreservation
Cryopreservation of ovarian tissue is an experimental but highly promising technique for preserving fertility. Like oocyte cryopreservation, it avoids the need for hormonal stimulation and the need to delay cancer treatment. It may also be the only possible option for prepubertal girls, as well as for women who cannot postpone their cancer treatment. Although it is still experimental, it has obtained progressively better results: after having ovarian tissue preserved, thawed, and subsequently reimplanted in the same position or in a different part of the body, some patients transiently resumed having menstrual cycles and endocrine activity and in a very few cases achieved pregnancy.37
Ovarian tissue for cryopreservation is usually taken via laparoscopic surgery, unless the patient has to undergo open laparotomy for another indication. Laparoscopic surgery offers significant advantages, such as the possibility of performing it on short notice without delaying oncologic therapy. Considering that women at age 30 have about 35 primordial follicles per square millimeter of ovarian tissue, 5 cubic fragments 5 mm wide may be sufficient to obtain more than 4,000 primordial follicles.38 In cases in which complete ovariectomy is necessary, it is possible to remove and cryopreserve fragments of normal ovarian tissue located at the margins of the surgical specimen. Ovarian tissue withdrawal can also be performed in pediatric patients and during other surgical procedures.
The most studied method of cryopreserving ovarian tissue is slow freezing, but the use of vitrification is increasing. This technique was initially carried out in order to preserve the largest number of primordial follicles, but in recent years the possibility of cryopreserving the whole ovary with or without its vascular pedicle has also been studied. Martinez-Madrid et al39 described a protocol of cryopreserving the entire ovary with its stem and found it possible to reach a follicular survival rate of 75%, preserving vessels and stromal structure.39
Currently, the most promising approach seems to be the transplantation of the ovarian tissue back into the donor, ie, autotransplantation. This avoids the need for immunosuppression.
The location can be either orthotopic or heterotopic. In orthotopic transplantation the tissue is placed back in its original location. In theory, the patient could then become pregnant in the usual way if the rest of the reproductive system is not damaged.
In heterotopic transplantation the tissue is placed in a different location, usually easily accessible, like the forearm or the subcutaneous abdominal area. Heterotopic transplants have been shown to be able to restore ovarian function, but not to give pregnancies after oocyte collection.40 Indeed, the pregnancies obtained after transplantation came from autografts of ovarian cortex in orthotopic sites like the fossa ovarica or the remnant ovary.
With autotransplantation, there is a high risk of transmission of metastatic cancer cells. Blood-bone cancers such as leukemia and lymphomas are likely to be associated with the highest risk of ovarian metastasis through transplantation of thawed cryopreserved ovarian tissue. Neuroblastoma and breast cancer are associated with a moderate risk of metastasis to the ovaries. Ovarian involvement is extremely rare in Wilms tumor, lymphomas (except for Burkitt lymphoma), osteosarcomas, Ewing sarcoma, and extragenital rhabdomyosarcoma. Squamous cell cervical cancer metastasizes to the ovaries in fewer than 0.2% of cases, even in the most advanced stages. Histologic evaluation of ovarian samples before transplantation has been proposed to prevent cancer transmission, although it is not possible to completely abolish this risk.41,42 This jeopardy could potentially be eliminated by in vitro maturation of immature oocytes collected from cryopreserved ovarian tissue.
Despite significant advances, to date there have been fewer than 20 babies born worldwide through this method.43
Oocyte donation
Assisted reproduction techniques also include in vitro fertilization using a sperm sample from the partner and oocytes from a donor. The embryos obtained are then transferred, saving the woman from ovarian stimulation without any delay in starting the cancer treatment. Although this method has a high success rate, it inevitably raises personal considerations.
OVARIAN TRANSPOSITION
When a woman of childbearing age needs radiation treatment for a pelvic malignancy, transposition of the ovaries above the pelvic brim outside the radiation field (oophoropexy) should be considered before starting therapy. It is indicated in patients diagnosed with malignancies that require pelvic radiation but not the removal of the ovaries. It can be performed during surgical treatment of the tumor or as a separate laparoscopic procedure. The radiation dose that the transposed ovaries receive is considerably less than that in ovaries left in place.
Laparoscopic ovarian transposition is highly effective. However, the risk involved in the surgical procedure should not be underestimated. The most important complications are vascular injury, infarction of the fallopian tube, and ovarian cyst formation.44
PHARMACOLOGIC PROTECTION
Some drugs induce a state of ovarian quiescence similar to menopause. Can they be used during chemotherapy to protect the ovaries, allowing restoration of normal ovarian function and natural fertility after cancer treatment and preventing premature ovarian failure?
GnRH analogues slow the cellular activity of the gonads, in theory making them less sensitive to damage by cytotoxic agents. Initially, the release of gonadotropins is stimulated (flare-up effect), but after 10 to 15 days pituitary GnRH receptors are down-regulated by internalization of receptors. Since chemotherapy affects mainly actively dividing cells such as mature ovarian follicles, the use of the analogues is based on the assumption that by reducing FSH levels, follicles will remain quiescent, decreasing their sensitivity to the gonadotoxic effect of chemotherapy.
In a randomized study of 281 patients with early breast cancer, Del Mastro et al45 reported a reduction in the occurrence of early menopause in those treated with a GnRH analogue during chemotherapy after 1 year of follow-up. However, the debate regarding the effect of GnRH analogues on the fertility of cancer patients is still open and needs further investigation.
In recent years, research has focused on imatinib, a new, potentially protective drug,46 but a lot of work still needs to be done. To date, the use of GnRH analogues is not recommended outside of clinical studies, and it should be offered only after careful counseling about other options to preserve fertility.
- Meirow D, Biederman H, Anderson RA, Wallace WH. Toxicity of chemotherapy and radiation on female reproduction. Clin Obstet Gynecol 2010; 53:727–739.
- Wallace WH, Kelsey TW. Human ovarian reserve from conception to the menopause. PLoS One 2010; 5:e8772.
- van Disseldorp J, Lambalk CB, Kwee J, et al. Comparison of inter- and intra-cycle variability of anti-Mullerian hormone and antral follicle counts. Hum Reprod 2010; 25:221–227.
- Lie Fong S, Laven JS, Hakvoort-Cammel FG, et al. Assessment of ovarian reserve in adult childhood cancer survivors using anti-Müllerian hormone. Hum Reprod 2009; 24:982–990.
- Oktem O, Oktay K. Quantitative assessment of the impact of chemotherapy on ovarian follicle reserve and stromal function. Cancer 2007; 110:2222–2229.
- Partridge AH, Ruddy KJ, Gelber S, et al. Ovarian reserve in women who remain premenopausal after chemotherapy for early stage breast cancer. Fertil Steril 2010; 94:638–644.
- Lee SJ, Schover LR, Partridge AH, et al; American Society of Clinical Oncology. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24:2917–2931.
- Familiari G, Caggiati A, Nottola SA, Ermini M, Di Benedetto MR, Motta PM. Ultrastructure of human ovarian primordial follicles after combination chemotherapy for Hodgkin’s disease. Hum Reprod 1993; 8:2080–2087.
- Wulff C, Wilson H, Wiegand SJ, Rudge JS, Fraser HM. Prevention of thecal angiogenesis, antral follicular growth, and ovulation in the primate by treatment with vascular endothelial growth factor Trap R1R2. Endocrinology 2002; 143:2797–2807.
- Aubard Y, Piver P, Pech JC, Galinat S, Teissier MP. Ovarian tissue cryopreservation and gynecologic oncology: a review. Eur J Obstet Gynecol Reprod Biol 2001; 97:5–14.
- Elizur SE, Chian RC, Pineau CA, et al. Fertility preservation treatment for young women with autoimmune diseases facing treatment with gonadotoxic agents. Rheumatology (Oxford) 2008; 47:1506–1509.
- Wo JY, Viswanathan AN. Impact of radiotherapy on fertility, pregnancy, and neonatal outcomes in female cancer patients. Int J Radiat Oncol Biol Phys 2009; 73:1304–1312.
- Wallace WH, Thomson AB, Saran F, et al. Predicting age of ovarian failure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys 2005; 62:738–744.
- Gareer W, Gad Z, Gareer H. Needle oophoropexy: a new simple technique for ovarian transposition prior to pelvic irradiation. Surg Endosc 2011; 25:2241–2246.
- Larsen EC, Schmiegelow K, Rechnitzer C, Loft A, Müller J, Andersen AN. Radiotherapy at a young age reduces uterine volume of childhood cancer survivors. Acta Obstet Gynecol Scand 2004; 83:96–102.
- Holm K, Nysom K, Brocks V, Hertz H, Jacobsen N, Müller J. Ultrasound B-mode changes in the uterus and ovaries and Doppler changes in the uterus after total body irradiation and allogeneic bone marrow transplantation in childhood. Bone Marrow Transplant 1999; 23:259–263.
- Critchley HO, Wallace WH, Shalet SM, Mamtora H, Higginson J, Anderson DC. Abdominal irradiation in childhood; the potential for pregnancy. Br J Obstet Gynaecol 1992; 99:392–394.
- Goldberg JM, Falcone T, Attaran M. In vitro fertilization update. Cleve Clin J Med 2007; 74:329–338.
- Prest SJ, May FE, Westley BR. The estrogen-regulated protein, TFF1, stimulates migration of human breast cancer cells. FASEB J 2002; 16:592–594.
- Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1992; 339:71–85.
- Klopper A, Hall M. New synthetic agent for the induction of ovulation: preliminary trials in women. Br Med J 1971; 1:152–154.
- Oktay K, Buyuk E, Davis O, Yermakova I, Veeck L, Rosenwaks Z. Fertility preservation in breast cancer patients: IVF and embryo cryopreservation after ovarian stimulation with tamoxifen. Hum Reprod 2003; 18:90–95.
- Oktay K, Buyuk E, Libertella N, Akar M, Rosenwaks Z. Fertility preservation in breast cancer patients: a prospective controlled comparison of ovarian stimulation with tamoxifen and letrozole for embryo cryopreservation. J Clin Oncol 2005; 23:4347–4353.
- Oktay K. Further evidence on the safety and success of ovarian stimulation with letrozole and tamoxifen in breast cancer patients undergoing in vitro fertilization to cryopreserve their embryos for fertility preservation. J Clin Oncol 2005; 23:3858–3859.
- Gosden R. Cryopreservation: a cold look at technology for fertility preservation. Fertil Steril 2011; 96:264–268.
- Chen SU, Chien CL, Wu MY, et al. Novel direct cover vitrification for cryopreservation of ovarian tissues increases follicle viability and pregnancy capability in mice. Hum Reprod 2006; 21:2794–2800.
- Lee SJ, Schover LR, Partridge AH, et al; American Society of Clinical Oncology. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24:2917–2931.
- Society for Assisted Reproductive Technology (SART). http://www.sart.org. Accessed January 3, 2013.
- Granne I, Child T, Hartshorne G; British Fertility Society. Embryo cryopreservation: evidence for practice. Hum Fertil (Camb) 2008; 11:159–172.
- Loutradi KE, Kolibianakis EM, Venetis CA, et al. Cryopreservation of human embryos by vitrification or slow freezing: a systematic review and meta-analysis. Fertil Steril 2008; 90:186–193.
- Ko CS, Ding DC, Chu TW, et al. Changes to the meiotic spindle and zona pellucida of mature mouse oocytes following different cryopreservation methods. Anim Reprod Sci 2008; 105:272–282.
- Cao Y, Xing Q, Zhang ZG, et al. Cryopreservation of immature and in vitro-matured human oocytes by vitrification. Reprod Biomed Online 2009; 19:369–373.
- Toth TL, Baka SG, Veeck LL, Jones HW, Muasher S, Lanzendorf SE. Fertilization and in vitro development of cryopreserved human prophase I oocytes. Fertil Steril 1994; 61:891–894.
- Chian RC, Gülekli B, Buckett WM, Tan SL. Priming with human chorionic gonadotropin before retrieval of immature oocytes in women with infertility due to the polycystic ovary syndrome. N Engl J Med 1999; 341: 1624,1626.
- Maman E, Meirow D, Brengauz M, Raanani H, Dor J, Hourvitz A. Luteal phase oocyte retrieval and in vitro maturation is an optional procedure for urgent fertility preservation. Fertil Steril 2011; 95:64–67.
- Oktay K, Demirtas E, Son WY, Lostritto K, Chian RC, Tan SL. In vitro maturation of germinal vesicle oocytes recovered after premature luteinizing hormone surge: description of a novel approach to fertility preservation. Fertil Steril 2008; 89:228.e19–e22.
- Donnez J, Dolmans MM, Demylle D, et al. Livebirth after orthotopic transplantation of cryopreserved ovarian tissue. Lancet 2004; 364:1405–1410.
- Martinez-Madrid B, Dolmans MM, Van Langendonckt A, Defrère S, Donnez J. Freeze-thawing intact human ovary with its vascular pedicle with a passive cooling device. Fertil Steril 2004; 82:1390–1394.
- Martinez-Madrid B, Camboni A, Dolmans MM, Nottola S, Van Langendonckt A, Donnez J. Apoptosis and ultrastructural assessment after cryopreservation of whole human ovaries with their vascular pedicle. Fertil Steril 2007; 87:1153–1165.
- Oktay K, Economos K, Kan M, Rucinski J, Veeck L, Rosenwaks Z. Endocrine function and oocyte retrieval after autologous transplantation of ovarian cortical strips to the forearm. JAMA 2001; 286:1490–1493.
- Practice Committee of American Society for Reproductive Medicine. Ovarian tissue and oocyte cryopreservation. Fertil Steril 2008; 90(suppl 5):S241–S246.
- Oktay K. Ovarian tissue cryopreservation and transplantation: preliminary findings and implications for cancer patients. Hum Reprod Update 2001; 7:526–534.
- Donnez J, Silber S, Andersen CY, et al. Children born after autotransplantation of cryopreserved ovarian tissue. a review of 13 live births. Ann Med 2011; 43:437–450.
- Terenziani M, Piva L, Meazza C, Gandola L, Cefalo G, Merola M. Oophoropexy: a relevant role in preservation of ovarian function after pelvic irradiation. Fertil Steril 2009; 91:935.e15–e16.
- Del Mastro L, Boni L, Michelotti A, et al. Effect of the gonadotropin-releasing hormone analogue triptorelin on the occurrence of chemotherapy-induced early menopause in premenopausal women with breast cancer: a randomized trial. JAMA 2011; 306:269–276.
- Gonfloni S, Di Tella L, Caldarola S, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 2009; 15:1179–1185.
In the last few decades, the survival rates have improved in many of the malignancies that affect young adults. This progress has made fertility preservation and quality of life after cancer treatment important, most of all in survivors of childhood cancers.
Men who are about to undergo cancer treatment can bank their sperm, but as yet no analogous noninvasive option is available for women. The most studied methods are often invasive and require the woman to take large doses of hormones. They may also necessitate a delay in starting cancer treatment.
Which method of fertility preservation a woman should choose depends on several factors, including the type of disease, the treatment required, the age of the patient, whether she has a long-term partner, and whether treatment can be delayed.
Chemotherapy and radiotherapy have well-known deleterious effects on female reproductive function. Many studies have shown that acute loss of growing follicles within the ovary and resultant premature ovarian failure often follow chemotherapy. This ovarian damage has long-term consequences, such as shortened reproductive life span and hormone deficiency.1
Fertility preservation requires a team effort. It should be managed by an oncology center that has built a close collaboration between oncologists, fertility specialists, psychologists, and primary care physicians to allow early discussion and to offer a full range of options to these patients.
The aim of this paper is to discuss the current options for preserving fertility in female cancer patients who have to undergo gonadotoxic cancer treatment.
FEMALE FERTILITY AND ASSESSMENT OF OVARIAN RESERVE
At birth, baby girls have about 1 million primordial ovarian follicles, which is the most they will ever have. By the time they reach menarche, this number has declined to 180,000, and at menopause only about 1,000 remain.2
Throughout a woman’s reproductive life, the number of oocytes remaining—both primordial follicles and the relatively small number of maturing, growing follicles—is called her ovarian reserve. When cancer is diagnosed, we need to assess the patient’s ovarian reserve to direct the discussion about the need for fertility preservation.
In search of markers of ovarian reserve
Fertility experts have been looking for clinical biomarkers that could give us an estimate of the number of nongrowing follicles and, consequently, of the ovarian reserve.
All methods used for the assessment of ovarian reserve actually provide an indirect determination of the remaining pool of oocytes. In clinical practice, a high blood level of follicle-stimulating hormone (FSH) on cycle day 3 (>15 mU/mL) or a low level of anti-Müllerian hormone (AMH) (<1 ng/mL) is generally associated with a low ovarian reserve.
The serum FSH level is the marker most commonly used, but it varies widely at different times in the menstrual cycle.
The FSH test is usually performed on day 3 of the menstrual cycle, when the estrogen level is expected to be low due to negative feedback. The FSH result needs to be combined with the estradiol level, especially in those patients with irregular menstruation or in cases of amenorrhea. A random FSH test is considered valid if the detected estrogen level is low. Women undergoing in vitro fertilization with a day 3 FSH lower than 15 mIU/mL are more likely to conceive than women with a higher FSH level.
AMH and antral follicles. Recently, two other variables have been introduced in clinical practice: the AMH concentration and the antral follicle count (the number of small antral follicles within the ovary) as assessed by transvaginal ultrasonography.
AMH is released by the granulosa cells of small, growing follicles. Because its level is much more stable over the menstrual cycle than the FSH level, it can be measured on any day of the cycle.3 In cancer survivors, AMH is particularly useful in demonstrating the degree of ovarian tissue damage induced by radiotherapy and chemotherapy and in evaluating the ovarian reserve.4
A reduced number of antral follicles causes a diminished AMH production, which becomes undetectable with menopause. The AMH level is strongly associated with the basal antral follicle count. Various threshold values (0.2–1.26 ng/mL) have been used to identify women with low ovarian reserve.
HOW CANCER TREATMENT DAMAGES THE OVARIES
Advances in surgery, radiotherapy, and chemotherapy have significantly improved the prognosis for young cancer patients. However, cancer treatments often result in ovarian dysfunction, and premature menopause and irreversible sterility are the most dramatic outcomes. The resulting low estrogen levels, in addition to their physiologic consequences, also worsen quality of life through psychological effects, which can as well influence the patient’s compliance with treatment.
Chemotherapy: Alkylating agents are the most gonadotoxic
The mechanism by which chemotherapy affects ovarian function is poorly understood. Histologically, chemotherapeutic drugs could lead to ovarian atrophy and stromal fibrosis, to depletion of the primordial follicle stockpile, and to reduced ovarian weight, resulting in ovarian dysfunction.5
The patient's age correlates with the probability of ovarian damage or, inversely, ovarian resistance to chemotherapy. Young women have more primordial oocytes, and after chemotherapy they face a sharp reduction of their ovarian reserve. Still, younger patients show a lower rate of ovarian toxicity than older women.6
The type and the cumulative dose of cytotoxic agents used are other important variables.7 There are six main classes of chemotherapeutic drugs: alkylating agents, platinum derivatives, antibiotics, antimetabolites, plant alkaloids, and taxanes. They all affect ovarian function, but alkylating agents are the most gonadotoxic.
Alkylating agents covalently bind an alkyl group to the DNA molecule and inhibit it from replicating. In the ovaries they directly injure primordial oocytes and deplete follicles. 8 They also seriously damage the ovarian vasculature so that the follicles cannot grow.9 Their destructive effect on the primordial follicles is dose-dependent and varies with the age and developmental maturity of the patient at the time of the therapy, with older women more likely to be left infertile afterward.10
Cyclophosphamide is an alkylating agent often used to treat severe manifestations of autoimmune diseases such as systemic lupus erythematosus, BehÇet disease, steroid-resistant glomerulonephritis, inflammatory bowel disease, pemphigus vulgaris, and others. Because it can lead to premature ovarian failure and infertility, women receiving cyclophosphamide for autoimmune conditions may also need treatment to preserve fertility.11
Radiotherapy
Ovarian follicles are remarkably vulnerable to damage from ionizing radiation, which results in ovarian atrophy and reduced follicle stores.1
The risk of radiotherapy-induced infertility is closely related to the patient’s age and developmental maturity at the time of therapy and to dose fractionation and the extent of the irradiation field. Every patient has a different sensitivity to radiation damage that is probably genetically predetermined, but age seems to be the most important variable. Wo and Viswanathan12 used a mathematical model devised by Wallace et al13 to show that the older the patient, the lower the radiation dose necessary to impair ovarian function.
The irradiation field is another aspect to consider. Pelvic radiation can be necessary in rectal cancer, cervical cancer, and lymphoma. In these cases, surgically moving the ovaries to a region outside the radiation field could be an option to minimize radiotherapy-induced ovarian damage.14
Radiotherapy can also damage the uterus. Pelvic irradiation can reduce uterine volume, alter uterine elasticity through myometrial fibrosis, and modify vascularization and endometrial thickness.15,16 These alterations are closely correlated with the total radiation dose, the site of irradiation, and the patient’s age at the time of the treatment, the prepubertal uterus being more susceptible to damage. 15,17 Larsen et al15 found that girls who received uterine irradiation before puberty had lower uterine volumes in adulthood than girls who received chemotherapy alone or radiation to other parts of the body, and that the younger the patient at the time of radiotherapy, the smaller the uterus later. These effects could result in adverse pregnancy outcomes.
Furthermore, radiation could also damage the uterine vasculature. Holm et al16 used ultrasonography to evaluate the effect of total-body irradiation and found that uterine volume and uterine blood flow were both impaired.15
All these possible alterations could lead to a reduced uterine response to cytotrophoblast invasion and to decreased fetoplacental blood flow, which could impair embryonic and fetal growth. In that case, a surrogate pregnancy would be the only method to achieve parenthood using the couple’s gametes.
OPTIONS FOR FERTILITY PRESERVATION
Assisted reproductive technology
Young women diagnosed with an oncologic disease may wish to use assisted reproductive technology (Table 1) to keep open the possibility of having children at a later date. One approach is to harvest oocytes, fertilize them in vitro, and deep-freeze (cryopreserve) the resulting embryos to be thawed and implanted later. Alternatively, oocytes can be frozen directly, although success rates are lower with this method. And another approach is to obtain and cryopreserve ovarian tissue. If none of these is possible, oocytes may be obtained from a donor.
Controlled ovarian stimulation
If oocytes are to be harvested, a number of them should be harvested at one time.
There is not an optimal number of oocytes that should be retrieved, but cryopreservation of a large number of oocytes allows us to perform multiple attempts at in vitro fertilization, improving the chances of pregnancy. The fertilization rate (defined as the total number of zygotes at the 2-pronucleus stage divided by the number of fertilized oocytes) with intracytoplasmic sperm injection is 70% to 80%. On average, for every 10 eggs, 7 to 8 eggs will normally be fertilized. The implantation rate (defined as the total number of pregnancies divided by the total number of embryo transferred) with intracytoplasmic sperm injection is 40% to 50%—ie, only half of the transferred embryos will successfully implant and result in a pregnancy.
So that more than one ripe egg can be obtained at a time, the patient must undergo a regimen of controlled ovarian stimulation to achieve multifollicular growth. Stimulation protocols are based on giving pituitary hormones, ie, analogues of gonadotropin-releasing hormone (GnRH) (both agonists and antagonists), followed by recombinant FSH or human menopausal gonadotropin to promote follicular development. A single dose of human chorionic gonadotropin (hCG) is given to induce ovulation when the lead follicles have reached 18 to 20 mm in size.18
Many oncologists consider controlled ovarian stimulation dangerous for cancer patients because it takes time and thus delays cancer treatment. Furthermore, the regimen boosts the levels of circulating estrogens, which could be harmful in patients with hormone-dependent tumors.19
Oocytes can also be retrieved during unstimulated cycles. This avoids increasing estrogen concentrations above the physiologic level, but no more than a single oocyte is collected per cycle. The patient could undergo multiple oocyte harvestings, one per cycle, but this would delay her cancer treatment even more—by months—which is not recommended.
Serious efforts to minimize estrogen production during controlled ovarian stimulation have been made, although further studies are needed. Research is under way to develop appropriate ovarian stimulation protocols based on drugs with antiestrogenic effects.
Tamoxifen, an antagonist of the estrogen receptor, is widely used in breast cancer treatment. 20 It can also be given during controlled ovarian stimulation because it promotes follicular growth and induces ovulation.21 Oktay et al22 found that the embryo yield was 2.5 times higher in women with breast cancer treated with tamoxifen than in a retrospective control group consisting of breast cancer patients attempting natural-cycle oocyte retrieval.
Letrozole, an aromatase inhibitor, is also commonly used in treating breast and ovarian cancer. Aromatase is an enzyme that catalyzes the conversion of androgenic precursors to estrogens, and it is found in many tissues, including granulosa cells. Several studies report the use of letrozole, alone or in combination with low doses of recombinant FSH, in ovarian stimulation protocols in cancer patients, with positive clinical outcomes.23
Tamoxifen or letrozole, combined with recombinant FSH, is an attractive option in a controlled ovarian stimulation protocol for cancer patients,24 although further investigation is needed.
Cryopreservation methods
Two main protocols are currently used to freeze oocytes, embryos, and ovarian tissue: slow freezing and vitrification.
In the slow-freezing method, cryoprotectant agents are used to draw water out of the cells, raising intracellular viscosity without (or with minimal) intracellular ice crystal formation, while the sample is cooled slowly in a controlled manner. These cryoprotectant chemicals lower the freezing point of the solution, allowing greater cellular dehydration during the slow freezing. They also protect the plasmatic cell membrane by changing its physical state from liquid to partially dry. To avoid excessive deformation that could damage the cytoplasmic structures, cryoprotectant agents are added in successive stages.25
Vitrification is a newer method that uses higher concentrations of cryoprotectants and flash freezing. The instant drop in temperature converts this highly concentrated solution from an aqueous state to a semisolid, amorphous state that does not contain ice crystals, which are the main cause of damage during the freezing process.26 Since most cryoprotectant agents are extremely toxic, it is necessary to minimize the time that oocytes, embryos, and ovarian tissue are exposed to them.
Cryopreservation of embryos
According to the American Society of Clinical Oncology and the American Society of Reproductive Medicine, embryo freezing is the most established method for fertility preservation, with tangible and widely reported success.27 In normal practice, oocytes are retrieved after a controlled ovarian stimulation and then fertilized in vitro. Then they are treated with cryoprotectant agents, frozen, and stored. Upon demand, embryos can be thawed and implanted.
The Society for Assisted Reproductive Technology reports that the current live birth rate per transfer using thawed embryos from nondonor oocytes in US women under age 35 is 38.7%; at age 35 to 37 it is 35.1%.28 In women with cancer, storing as many embryos as possible could help improve embryo survival and the implantation rate. The optimal time to perform embryo cryopreservation is still being discussed,29 but, as in women without cancer, it is commonly done 3 to 5 days after fertilization.
In the past few years, the use of vitrification has greatly increased, as the post-thawing survival rates and pregnancy rates are higher with this method than with slow freezing.30
Despite its success, embryo cryopreservation has important drawbacks. First, the patient must be of reproductive age and have a partner or accept the use of donor sperm. Second, most patients undergo controlled ovarian stimulation before oocyte retrieval, causing a delay in starting cancer treatment, which is not acceptable in many cases. Moreover, the high serum estrogen levels caused by ovarian stimulation may be contraindicated in women with estrogen-sensitive malignancies.19
Oocyte cryopreservation
Cryopreservation of oocytes avoids the need for sperm and, thus, may be offered to more patients than embryo cryopreservation. In addition, it may circumvent ethical or legal considerations associated with embryo freezing, such as ownership of reproductive material.
However, oocytes are more difficult to cryopreserve than embryos. Indeed, ice crystals frequently form inside and outside the cells, damaging the cell membrane and the meiotic spindle. In routine practice, mature oocytes in metaphase II are used for cryopreservation. Metaphase II oocytes are large cells that contain a delicate meiotic spindle. Moreover, their cytoplasm contains a higher proportion of water than other cells, which could affect their viability after freezing and thawing due to ice crystal formation. In addition, cryopreservation could be responsible for hardening of the zona pellucida (through diffuse thickening of the cell membrane), adversely affecting fertilization rates.31
Significant improvements were achieved in fertilization of cryopreserved oocytes with intracytoplasmic sperm injection. Nevertheless, there are still concerns regarding oocyte cryopreservation. Further studies are needed to determine the risk of aneuploidy caused by damage to the meiotic spindle after oocyte cryopreservation. The potentially detrimental effects of high cryopreservant concentrations used in vitrification also need to be investigated.
In vitro maturation
A novel fertility preservation strategy involves collecting immature oocytes from primordial follicles in unstimulated cycles and then letting them mature in vitro.
To date, immature oocytes retrieved at the germinal vesicle stage can be cryopreserved with vitrification followed by in vitro maturation after thawing, but several studies have demonstrated that better results are obtained when vitrification follows the in vitro maturation process.32
Compared with mature oocytes, immature oocytes are less susceptible to damage during cryopreservation and thus have a better chance of surviving freezing and thawing, thanks to some peculiar characteristics: they are small, have few organelles, lack a zona pellucida, have low metabolic activity, and are in a state of relative quiescence.33 Controlled ovarian stimulation is not necessary, so this procedure preserves fertility without delaying the start of cancer treatment.
Patients are usually evaluated with transvaginal ultrasonography in the early follicular phase of the menstrual cycle (between day 2 and day 4) to count and measure the antral follicles. Immature oocytes are collected when the leading follicle has reached 10 to 12 mm in size and 36 hours after a subcutaneous injection of hCG.34 The retrieved oocytes are then incubated for 24 to 48 hours in a special medium supplemented with FSH and luteinizing hormone (LH). Immature oocytes can also be collected in the luteal phase.
This option could be considered when cancer treatment cannot be delayed for conventional follicular-phase retrieval35 or in case of a premature LH surge during ovarian stimulation. 36 It should be offered to patients facing infertility related to cancer treatment only after appropriate counseling and as a part of a clinical study.
Ovarian tissue cryopreservation
Cryopreservation of ovarian tissue is an experimental but highly promising technique for preserving fertility. Like oocyte cryopreservation, it avoids the need for hormonal stimulation and the need to delay cancer treatment. It may also be the only possible option for prepubertal girls, as well as for women who cannot postpone their cancer treatment. Although it is still experimental, it has obtained progressively better results: after having ovarian tissue preserved, thawed, and subsequently reimplanted in the same position or in a different part of the body, some patients transiently resumed having menstrual cycles and endocrine activity and in a very few cases achieved pregnancy.37
Ovarian tissue for cryopreservation is usually taken via laparoscopic surgery, unless the patient has to undergo open laparotomy for another indication. Laparoscopic surgery offers significant advantages, such as the possibility of performing it on short notice without delaying oncologic therapy. Considering that women at age 30 have about 35 primordial follicles per square millimeter of ovarian tissue, 5 cubic fragments 5 mm wide may be sufficient to obtain more than 4,000 primordial follicles.38 In cases in which complete ovariectomy is necessary, it is possible to remove and cryopreserve fragments of normal ovarian tissue located at the margins of the surgical specimen. Ovarian tissue withdrawal can also be performed in pediatric patients and during other surgical procedures.
The most studied method of cryopreserving ovarian tissue is slow freezing, but the use of vitrification is increasing. This technique was initially carried out in order to preserve the largest number of primordial follicles, but in recent years the possibility of cryopreserving the whole ovary with or without its vascular pedicle has also been studied. Martinez-Madrid et al39 described a protocol of cryopreserving the entire ovary with its stem and found it possible to reach a follicular survival rate of 75%, preserving vessels and stromal structure.39
Currently, the most promising approach seems to be the transplantation of the ovarian tissue back into the donor, ie, autotransplantation. This avoids the need for immunosuppression.
The location can be either orthotopic or heterotopic. In orthotopic transplantation the tissue is placed back in its original location. In theory, the patient could then become pregnant in the usual way if the rest of the reproductive system is not damaged.
In heterotopic transplantation the tissue is placed in a different location, usually easily accessible, like the forearm or the subcutaneous abdominal area. Heterotopic transplants have been shown to be able to restore ovarian function, but not to give pregnancies after oocyte collection.40 Indeed, the pregnancies obtained after transplantation came from autografts of ovarian cortex in orthotopic sites like the fossa ovarica or the remnant ovary.
With autotransplantation, there is a high risk of transmission of metastatic cancer cells. Blood-bone cancers such as leukemia and lymphomas are likely to be associated with the highest risk of ovarian metastasis through transplantation of thawed cryopreserved ovarian tissue. Neuroblastoma and breast cancer are associated with a moderate risk of metastasis to the ovaries. Ovarian involvement is extremely rare in Wilms tumor, lymphomas (except for Burkitt lymphoma), osteosarcomas, Ewing sarcoma, and extragenital rhabdomyosarcoma. Squamous cell cervical cancer metastasizes to the ovaries in fewer than 0.2% of cases, even in the most advanced stages. Histologic evaluation of ovarian samples before transplantation has been proposed to prevent cancer transmission, although it is not possible to completely abolish this risk.41,42 This jeopardy could potentially be eliminated by in vitro maturation of immature oocytes collected from cryopreserved ovarian tissue.
Despite significant advances, to date there have been fewer than 20 babies born worldwide through this method.43
Oocyte donation
Assisted reproduction techniques also include in vitro fertilization using a sperm sample from the partner and oocytes from a donor. The embryos obtained are then transferred, saving the woman from ovarian stimulation without any delay in starting the cancer treatment. Although this method has a high success rate, it inevitably raises personal considerations.
OVARIAN TRANSPOSITION
When a woman of childbearing age needs radiation treatment for a pelvic malignancy, transposition of the ovaries above the pelvic brim outside the radiation field (oophoropexy) should be considered before starting therapy. It is indicated in patients diagnosed with malignancies that require pelvic radiation but not the removal of the ovaries. It can be performed during surgical treatment of the tumor or as a separate laparoscopic procedure. The radiation dose that the transposed ovaries receive is considerably less than that in ovaries left in place.
Laparoscopic ovarian transposition is highly effective. However, the risk involved in the surgical procedure should not be underestimated. The most important complications are vascular injury, infarction of the fallopian tube, and ovarian cyst formation.44
PHARMACOLOGIC PROTECTION
Some drugs induce a state of ovarian quiescence similar to menopause. Can they be used during chemotherapy to protect the ovaries, allowing restoration of normal ovarian function and natural fertility after cancer treatment and preventing premature ovarian failure?
GnRH analogues slow the cellular activity of the gonads, in theory making them less sensitive to damage by cytotoxic agents. Initially, the release of gonadotropins is stimulated (flare-up effect), but after 10 to 15 days pituitary GnRH receptors are down-regulated by internalization of receptors. Since chemotherapy affects mainly actively dividing cells such as mature ovarian follicles, the use of the analogues is based on the assumption that by reducing FSH levels, follicles will remain quiescent, decreasing their sensitivity to the gonadotoxic effect of chemotherapy.
In a randomized study of 281 patients with early breast cancer, Del Mastro et al45 reported a reduction in the occurrence of early menopause in those treated with a GnRH analogue during chemotherapy after 1 year of follow-up. However, the debate regarding the effect of GnRH analogues on the fertility of cancer patients is still open and needs further investigation.
In recent years, research has focused on imatinib, a new, potentially protective drug,46 but a lot of work still needs to be done. To date, the use of GnRH analogues is not recommended outside of clinical studies, and it should be offered only after careful counseling about other options to preserve fertility.
In the last few decades, the survival rates have improved in many of the malignancies that affect young adults. This progress has made fertility preservation and quality of life after cancer treatment important, most of all in survivors of childhood cancers.
Men who are about to undergo cancer treatment can bank their sperm, but as yet no analogous noninvasive option is available for women. The most studied methods are often invasive and require the woman to take large doses of hormones. They may also necessitate a delay in starting cancer treatment.
Which method of fertility preservation a woman should choose depends on several factors, including the type of disease, the treatment required, the age of the patient, whether she has a long-term partner, and whether treatment can be delayed.
Chemotherapy and radiotherapy have well-known deleterious effects on female reproductive function. Many studies have shown that acute loss of growing follicles within the ovary and resultant premature ovarian failure often follow chemotherapy. This ovarian damage has long-term consequences, such as shortened reproductive life span and hormone deficiency.1
Fertility preservation requires a team effort. It should be managed by an oncology center that has built a close collaboration between oncologists, fertility specialists, psychologists, and primary care physicians to allow early discussion and to offer a full range of options to these patients.
The aim of this paper is to discuss the current options for preserving fertility in female cancer patients who have to undergo gonadotoxic cancer treatment.
FEMALE FERTILITY AND ASSESSMENT OF OVARIAN RESERVE
At birth, baby girls have about 1 million primordial ovarian follicles, which is the most they will ever have. By the time they reach menarche, this number has declined to 180,000, and at menopause only about 1,000 remain.2
Throughout a woman’s reproductive life, the number of oocytes remaining—both primordial follicles and the relatively small number of maturing, growing follicles—is called her ovarian reserve. When cancer is diagnosed, we need to assess the patient’s ovarian reserve to direct the discussion about the need for fertility preservation.
In search of markers of ovarian reserve
Fertility experts have been looking for clinical biomarkers that could give us an estimate of the number of nongrowing follicles and, consequently, of the ovarian reserve.
All methods used for the assessment of ovarian reserve actually provide an indirect determination of the remaining pool of oocytes. In clinical practice, a high blood level of follicle-stimulating hormone (FSH) on cycle day 3 (>15 mU/mL) or a low level of anti-Müllerian hormone (AMH) (<1 ng/mL) is generally associated with a low ovarian reserve.
The serum FSH level is the marker most commonly used, but it varies widely at different times in the menstrual cycle.
The FSH test is usually performed on day 3 of the menstrual cycle, when the estrogen level is expected to be low due to negative feedback. The FSH result needs to be combined with the estradiol level, especially in those patients with irregular menstruation or in cases of amenorrhea. A random FSH test is considered valid if the detected estrogen level is low. Women undergoing in vitro fertilization with a day 3 FSH lower than 15 mIU/mL are more likely to conceive than women with a higher FSH level.
AMH and antral follicles. Recently, two other variables have been introduced in clinical practice: the AMH concentration and the antral follicle count (the number of small antral follicles within the ovary) as assessed by transvaginal ultrasonography.
AMH is released by the granulosa cells of small, growing follicles. Because its level is much more stable over the menstrual cycle than the FSH level, it can be measured on any day of the cycle.3 In cancer survivors, AMH is particularly useful in demonstrating the degree of ovarian tissue damage induced by radiotherapy and chemotherapy and in evaluating the ovarian reserve.4
A reduced number of antral follicles causes a diminished AMH production, which becomes undetectable with menopause. The AMH level is strongly associated with the basal antral follicle count. Various threshold values (0.2–1.26 ng/mL) have been used to identify women with low ovarian reserve.
HOW CANCER TREATMENT DAMAGES THE OVARIES
Advances in surgery, radiotherapy, and chemotherapy have significantly improved the prognosis for young cancer patients. However, cancer treatments often result in ovarian dysfunction, and premature menopause and irreversible sterility are the most dramatic outcomes. The resulting low estrogen levels, in addition to their physiologic consequences, also worsen quality of life through psychological effects, which can as well influence the patient’s compliance with treatment.
Chemotherapy: Alkylating agents are the most gonadotoxic
The mechanism by which chemotherapy affects ovarian function is poorly understood. Histologically, chemotherapeutic drugs could lead to ovarian atrophy and stromal fibrosis, to depletion of the primordial follicle stockpile, and to reduced ovarian weight, resulting in ovarian dysfunction.5
The patient's age correlates with the probability of ovarian damage or, inversely, ovarian resistance to chemotherapy. Young women have more primordial oocytes, and after chemotherapy they face a sharp reduction of their ovarian reserve. Still, younger patients show a lower rate of ovarian toxicity than older women.6
The type and the cumulative dose of cytotoxic agents used are other important variables.7 There are six main classes of chemotherapeutic drugs: alkylating agents, platinum derivatives, antibiotics, antimetabolites, plant alkaloids, and taxanes. They all affect ovarian function, but alkylating agents are the most gonadotoxic.
Alkylating agents covalently bind an alkyl group to the DNA molecule and inhibit it from replicating. In the ovaries they directly injure primordial oocytes and deplete follicles. 8 They also seriously damage the ovarian vasculature so that the follicles cannot grow.9 Their destructive effect on the primordial follicles is dose-dependent and varies with the age and developmental maturity of the patient at the time of the therapy, with older women more likely to be left infertile afterward.10
Cyclophosphamide is an alkylating agent often used to treat severe manifestations of autoimmune diseases such as systemic lupus erythematosus, BehÇet disease, steroid-resistant glomerulonephritis, inflammatory bowel disease, pemphigus vulgaris, and others. Because it can lead to premature ovarian failure and infertility, women receiving cyclophosphamide for autoimmune conditions may also need treatment to preserve fertility.11
Radiotherapy
Ovarian follicles are remarkably vulnerable to damage from ionizing radiation, which results in ovarian atrophy and reduced follicle stores.1
The risk of radiotherapy-induced infertility is closely related to the patient’s age and developmental maturity at the time of therapy and to dose fractionation and the extent of the irradiation field. Every patient has a different sensitivity to radiation damage that is probably genetically predetermined, but age seems to be the most important variable. Wo and Viswanathan12 used a mathematical model devised by Wallace et al13 to show that the older the patient, the lower the radiation dose necessary to impair ovarian function.
The irradiation field is another aspect to consider. Pelvic radiation can be necessary in rectal cancer, cervical cancer, and lymphoma. In these cases, surgically moving the ovaries to a region outside the radiation field could be an option to minimize radiotherapy-induced ovarian damage.14
Radiotherapy can also damage the uterus. Pelvic irradiation can reduce uterine volume, alter uterine elasticity through myometrial fibrosis, and modify vascularization and endometrial thickness.15,16 These alterations are closely correlated with the total radiation dose, the site of irradiation, and the patient’s age at the time of the treatment, the prepubertal uterus being more susceptible to damage. 15,17 Larsen et al15 found that girls who received uterine irradiation before puberty had lower uterine volumes in adulthood than girls who received chemotherapy alone or radiation to other parts of the body, and that the younger the patient at the time of radiotherapy, the smaller the uterus later. These effects could result in adverse pregnancy outcomes.
Furthermore, radiation could also damage the uterine vasculature. Holm et al16 used ultrasonography to evaluate the effect of total-body irradiation and found that uterine volume and uterine blood flow were both impaired.15
All these possible alterations could lead to a reduced uterine response to cytotrophoblast invasion and to decreased fetoplacental blood flow, which could impair embryonic and fetal growth. In that case, a surrogate pregnancy would be the only method to achieve parenthood using the couple’s gametes.
OPTIONS FOR FERTILITY PRESERVATION
Assisted reproductive technology
Young women diagnosed with an oncologic disease may wish to use assisted reproductive technology (Table 1) to keep open the possibility of having children at a later date. One approach is to harvest oocytes, fertilize them in vitro, and deep-freeze (cryopreserve) the resulting embryos to be thawed and implanted later. Alternatively, oocytes can be frozen directly, although success rates are lower with this method. And another approach is to obtain and cryopreserve ovarian tissue. If none of these is possible, oocytes may be obtained from a donor.
Controlled ovarian stimulation
If oocytes are to be harvested, a number of them should be harvested at one time.
There is not an optimal number of oocytes that should be retrieved, but cryopreservation of a large number of oocytes allows us to perform multiple attempts at in vitro fertilization, improving the chances of pregnancy. The fertilization rate (defined as the total number of zygotes at the 2-pronucleus stage divided by the number of fertilized oocytes) with intracytoplasmic sperm injection is 70% to 80%. On average, for every 10 eggs, 7 to 8 eggs will normally be fertilized. The implantation rate (defined as the total number of pregnancies divided by the total number of embryo transferred) with intracytoplasmic sperm injection is 40% to 50%—ie, only half of the transferred embryos will successfully implant and result in a pregnancy.
So that more than one ripe egg can be obtained at a time, the patient must undergo a regimen of controlled ovarian stimulation to achieve multifollicular growth. Stimulation protocols are based on giving pituitary hormones, ie, analogues of gonadotropin-releasing hormone (GnRH) (both agonists and antagonists), followed by recombinant FSH or human menopausal gonadotropin to promote follicular development. A single dose of human chorionic gonadotropin (hCG) is given to induce ovulation when the lead follicles have reached 18 to 20 mm in size.18
Many oncologists consider controlled ovarian stimulation dangerous for cancer patients because it takes time and thus delays cancer treatment. Furthermore, the regimen boosts the levels of circulating estrogens, which could be harmful in patients with hormone-dependent tumors.19
Oocytes can also be retrieved during unstimulated cycles. This avoids increasing estrogen concentrations above the physiologic level, but no more than a single oocyte is collected per cycle. The patient could undergo multiple oocyte harvestings, one per cycle, but this would delay her cancer treatment even more—by months—which is not recommended.
Serious efforts to minimize estrogen production during controlled ovarian stimulation have been made, although further studies are needed. Research is under way to develop appropriate ovarian stimulation protocols based on drugs with antiestrogenic effects.
Tamoxifen, an antagonist of the estrogen receptor, is widely used in breast cancer treatment. 20 It can also be given during controlled ovarian stimulation because it promotes follicular growth and induces ovulation.21 Oktay et al22 found that the embryo yield was 2.5 times higher in women with breast cancer treated with tamoxifen than in a retrospective control group consisting of breast cancer patients attempting natural-cycle oocyte retrieval.
Letrozole, an aromatase inhibitor, is also commonly used in treating breast and ovarian cancer. Aromatase is an enzyme that catalyzes the conversion of androgenic precursors to estrogens, and it is found in many tissues, including granulosa cells. Several studies report the use of letrozole, alone or in combination with low doses of recombinant FSH, in ovarian stimulation protocols in cancer patients, with positive clinical outcomes.23
Tamoxifen or letrozole, combined with recombinant FSH, is an attractive option in a controlled ovarian stimulation protocol for cancer patients,24 although further investigation is needed.
Cryopreservation methods
Two main protocols are currently used to freeze oocytes, embryos, and ovarian tissue: slow freezing and vitrification.
In the slow-freezing method, cryoprotectant agents are used to draw water out of the cells, raising intracellular viscosity without (or with minimal) intracellular ice crystal formation, while the sample is cooled slowly in a controlled manner. These cryoprotectant chemicals lower the freezing point of the solution, allowing greater cellular dehydration during the slow freezing. They also protect the plasmatic cell membrane by changing its physical state from liquid to partially dry. To avoid excessive deformation that could damage the cytoplasmic structures, cryoprotectant agents are added in successive stages.25
Vitrification is a newer method that uses higher concentrations of cryoprotectants and flash freezing. The instant drop in temperature converts this highly concentrated solution from an aqueous state to a semisolid, amorphous state that does not contain ice crystals, which are the main cause of damage during the freezing process.26 Since most cryoprotectant agents are extremely toxic, it is necessary to minimize the time that oocytes, embryos, and ovarian tissue are exposed to them.
Cryopreservation of embryos
According to the American Society of Clinical Oncology and the American Society of Reproductive Medicine, embryo freezing is the most established method for fertility preservation, with tangible and widely reported success.27 In normal practice, oocytes are retrieved after a controlled ovarian stimulation and then fertilized in vitro. Then they are treated with cryoprotectant agents, frozen, and stored. Upon demand, embryos can be thawed and implanted.
The Society for Assisted Reproductive Technology reports that the current live birth rate per transfer using thawed embryos from nondonor oocytes in US women under age 35 is 38.7%; at age 35 to 37 it is 35.1%.28 In women with cancer, storing as many embryos as possible could help improve embryo survival and the implantation rate. The optimal time to perform embryo cryopreservation is still being discussed,29 but, as in women without cancer, it is commonly done 3 to 5 days after fertilization.
In the past few years, the use of vitrification has greatly increased, as the post-thawing survival rates and pregnancy rates are higher with this method than with slow freezing.30
Despite its success, embryo cryopreservation has important drawbacks. First, the patient must be of reproductive age and have a partner or accept the use of donor sperm. Second, most patients undergo controlled ovarian stimulation before oocyte retrieval, causing a delay in starting cancer treatment, which is not acceptable in many cases. Moreover, the high serum estrogen levels caused by ovarian stimulation may be contraindicated in women with estrogen-sensitive malignancies.19
Oocyte cryopreservation
Cryopreservation of oocytes avoids the need for sperm and, thus, may be offered to more patients than embryo cryopreservation. In addition, it may circumvent ethical or legal considerations associated with embryo freezing, such as ownership of reproductive material.
However, oocytes are more difficult to cryopreserve than embryos. Indeed, ice crystals frequently form inside and outside the cells, damaging the cell membrane and the meiotic spindle. In routine practice, mature oocytes in metaphase II are used for cryopreservation. Metaphase II oocytes are large cells that contain a delicate meiotic spindle. Moreover, their cytoplasm contains a higher proportion of water than other cells, which could affect their viability after freezing and thawing due to ice crystal formation. In addition, cryopreservation could be responsible for hardening of the zona pellucida (through diffuse thickening of the cell membrane), adversely affecting fertilization rates.31
Significant improvements were achieved in fertilization of cryopreserved oocytes with intracytoplasmic sperm injection. Nevertheless, there are still concerns regarding oocyte cryopreservation. Further studies are needed to determine the risk of aneuploidy caused by damage to the meiotic spindle after oocyte cryopreservation. The potentially detrimental effects of high cryopreservant concentrations used in vitrification also need to be investigated.
In vitro maturation
A novel fertility preservation strategy involves collecting immature oocytes from primordial follicles in unstimulated cycles and then letting them mature in vitro.
To date, immature oocytes retrieved at the germinal vesicle stage can be cryopreserved with vitrification followed by in vitro maturation after thawing, but several studies have demonstrated that better results are obtained when vitrification follows the in vitro maturation process.32
Compared with mature oocytes, immature oocytes are less susceptible to damage during cryopreservation and thus have a better chance of surviving freezing and thawing, thanks to some peculiar characteristics: they are small, have few organelles, lack a zona pellucida, have low metabolic activity, and are in a state of relative quiescence.33 Controlled ovarian stimulation is not necessary, so this procedure preserves fertility without delaying the start of cancer treatment.
Patients are usually evaluated with transvaginal ultrasonography in the early follicular phase of the menstrual cycle (between day 2 and day 4) to count and measure the antral follicles. Immature oocytes are collected when the leading follicle has reached 10 to 12 mm in size and 36 hours after a subcutaneous injection of hCG.34 The retrieved oocytes are then incubated for 24 to 48 hours in a special medium supplemented with FSH and luteinizing hormone (LH). Immature oocytes can also be collected in the luteal phase.
This option could be considered when cancer treatment cannot be delayed for conventional follicular-phase retrieval35 or in case of a premature LH surge during ovarian stimulation. 36 It should be offered to patients facing infertility related to cancer treatment only after appropriate counseling and as a part of a clinical study.
Ovarian tissue cryopreservation
Cryopreservation of ovarian tissue is an experimental but highly promising technique for preserving fertility. Like oocyte cryopreservation, it avoids the need for hormonal stimulation and the need to delay cancer treatment. It may also be the only possible option for prepubertal girls, as well as for women who cannot postpone their cancer treatment. Although it is still experimental, it has obtained progressively better results: after having ovarian tissue preserved, thawed, and subsequently reimplanted in the same position or in a different part of the body, some patients transiently resumed having menstrual cycles and endocrine activity and in a very few cases achieved pregnancy.37
Ovarian tissue for cryopreservation is usually taken via laparoscopic surgery, unless the patient has to undergo open laparotomy for another indication. Laparoscopic surgery offers significant advantages, such as the possibility of performing it on short notice without delaying oncologic therapy. Considering that women at age 30 have about 35 primordial follicles per square millimeter of ovarian tissue, 5 cubic fragments 5 mm wide may be sufficient to obtain more than 4,000 primordial follicles.38 In cases in which complete ovariectomy is necessary, it is possible to remove and cryopreserve fragments of normal ovarian tissue located at the margins of the surgical specimen. Ovarian tissue withdrawal can also be performed in pediatric patients and during other surgical procedures.
The most studied method of cryopreserving ovarian tissue is slow freezing, but the use of vitrification is increasing. This technique was initially carried out in order to preserve the largest number of primordial follicles, but in recent years the possibility of cryopreserving the whole ovary with or without its vascular pedicle has also been studied. Martinez-Madrid et al39 described a protocol of cryopreserving the entire ovary with its stem and found it possible to reach a follicular survival rate of 75%, preserving vessels and stromal structure.39
Currently, the most promising approach seems to be the transplantation of the ovarian tissue back into the donor, ie, autotransplantation. This avoids the need for immunosuppression.
The location can be either orthotopic or heterotopic. In orthotopic transplantation the tissue is placed back in its original location. In theory, the patient could then become pregnant in the usual way if the rest of the reproductive system is not damaged.
In heterotopic transplantation the tissue is placed in a different location, usually easily accessible, like the forearm or the subcutaneous abdominal area. Heterotopic transplants have been shown to be able to restore ovarian function, but not to give pregnancies after oocyte collection.40 Indeed, the pregnancies obtained after transplantation came from autografts of ovarian cortex in orthotopic sites like the fossa ovarica or the remnant ovary.
With autotransplantation, there is a high risk of transmission of metastatic cancer cells. Blood-bone cancers such as leukemia and lymphomas are likely to be associated with the highest risk of ovarian metastasis through transplantation of thawed cryopreserved ovarian tissue. Neuroblastoma and breast cancer are associated with a moderate risk of metastasis to the ovaries. Ovarian involvement is extremely rare in Wilms tumor, lymphomas (except for Burkitt lymphoma), osteosarcomas, Ewing sarcoma, and extragenital rhabdomyosarcoma. Squamous cell cervical cancer metastasizes to the ovaries in fewer than 0.2% of cases, even in the most advanced stages. Histologic evaluation of ovarian samples before transplantation has been proposed to prevent cancer transmission, although it is not possible to completely abolish this risk.41,42 This jeopardy could potentially be eliminated by in vitro maturation of immature oocytes collected from cryopreserved ovarian tissue.
Despite significant advances, to date there have been fewer than 20 babies born worldwide through this method.43
Oocyte donation
Assisted reproduction techniques also include in vitro fertilization using a sperm sample from the partner and oocytes from a donor. The embryos obtained are then transferred, saving the woman from ovarian stimulation without any delay in starting the cancer treatment. Although this method has a high success rate, it inevitably raises personal considerations.
OVARIAN TRANSPOSITION
When a woman of childbearing age needs radiation treatment for a pelvic malignancy, transposition of the ovaries above the pelvic brim outside the radiation field (oophoropexy) should be considered before starting therapy. It is indicated in patients diagnosed with malignancies that require pelvic radiation but not the removal of the ovaries. It can be performed during surgical treatment of the tumor or as a separate laparoscopic procedure. The radiation dose that the transposed ovaries receive is considerably less than that in ovaries left in place.
Laparoscopic ovarian transposition is highly effective. However, the risk involved in the surgical procedure should not be underestimated. The most important complications are vascular injury, infarction of the fallopian tube, and ovarian cyst formation.44
PHARMACOLOGIC PROTECTION
Some drugs induce a state of ovarian quiescence similar to menopause. Can they be used during chemotherapy to protect the ovaries, allowing restoration of normal ovarian function and natural fertility after cancer treatment and preventing premature ovarian failure?
GnRH analogues slow the cellular activity of the gonads, in theory making them less sensitive to damage by cytotoxic agents. Initially, the release of gonadotropins is stimulated (flare-up effect), but after 10 to 15 days pituitary GnRH receptors are down-regulated by internalization of receptors. Since chemotherapy affects mainly actively dividing cells such as mature ovarian follicles, the use of the analogues is based on the assumption that by reducing FSH levels, follicles will remain quiescent, decreasing their sensitivity to the gonadotoxic effect of chemotherapy.
In a randomized study of 281 patients with early breast cancer, Del Mastro et al45 reported a reduction in the occurrence of early menopause in those treated with a GnRH analogue during chemotherapy after 1 year of follow-up. However, the debate regarding the effect of GnRH analogues on the fertility of cancer patients is still open and needs further investigation.
In recent years, research has focused on imatinib, a new, potentially protective drug,46 but a lot of work still needs to be done. To date, the use of GnRH analogues is not recommended outside of clinical studies, and it should be offered only after careful counseling about other options to preserve fertility.
- Meirow D, Biederman H, Anderson RA, Wallace WH. Toxicity of chemotherapy and radiation on female reproduction. Clin Obstet Gynecol 2010; 53:727–739.
- Wallace WH, Kelsey TW. Human ovarian reserve from conception to the menopause. PLoS One 2010; 5:e8772.
- van Disseldorp J, Lambalk CB, Kwee J, et al. Comparison of inter- and intra-cycle variability of anti-Mullerian hormone and antral follicle counts. Hum Reprod 2010; 25:221–227.
- Lie Fong S, Laven JS, Hakvoort-Cammel FG, et al. Assessment of ovarian reserve in adult childhood cancer survivors using anti-Müllerian hormone. Hum Reprod 2009; 24:982–990.
- Oktem O, Oktay K. Quantitative assessment of the impact of chemotherapy on ovarian follicle reserve and stromal function. Cancer 2007; 110:2222–2229.
- Partridge AH, Ruddy KJ, Gelber S, et al. Ovarian reserve in women who remain premenopausal after chemotherapy for early stage breast cancer. Fertil Steril 2010; 94:638–644.
- Lee SJ, Schover LR, Partridge AH, et al; American Society of Clinical Oncology. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24:2917–2931.
- Familiari G, Caggiati A, Nottola SA, Ermini M, Di Benedetto MR, Motta PM. Ultrastructure of human ovarian primordial follicles after combination chemotherapy for Hodgkin’s disease. Hum Reprod 1993; 8:2080–2087.
- Wulff C, Wilson H, Wiegand SJ, Rudge JS, Fraser HM. Prevention of thecal angiogenesis, antral follicular growth, and ovulation in the primate by treatment with vascular endothelial growth factor Trap R1R2. Endocrinology 2002; 143:2797–2807.
- Aubard Y, Piver P, Pech JC, Galinat S, Teissier MP. Ovarian tissue cryopreservation and gynecologic oncology: a review. Eur J Obstet Gynecol Reprod Biol 2001; 97:5–14.
- Elizur SE, Chian RC, Pineau CA, et al. Fertility preservation treatment for young women with autoimmune diseases facing treatment with gonadotoxic agents. Rheumatology (Oxford) 2008; 47:1506–1509.
- Wo JY, Viswanathan AN. Impact of radiotherapy on fertility, pregnancy, and neonatal outcomes in female cancer patients. Int J Radiat Oncol Biol Phys 2009; 73:1304–1312.
- Wallace WH, Thomson AB, Saran F, et al. Predicting age of ovarian failure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys 2005; 62:738–744.
- Gareer W, Gad Z, Gareer H. Needle oophoropexy: a new simple technique for ovarian transposition prior to pelvic irradiation. Surg Endosc 2011; 25:2241–2246.
- Larsen EC, Schmiegelow K, Rechnitzer C, Loft A, Müller J, Andersen AN. Radiotherapy at a young age reduces uterine volume of childhood cancer survivors. Acta Obstet Gynecol Scand 2004; 83:96–102.
- Holm K, Nysom K, Brocks V, Hertz H, Jacobsen N, Müller J. Ultrasound B-mode changes in the uterus and ovaries and Doppler changes in the uterus after total body irradiation and allogeneic bone marrow transplantation in childhood. Bone Marrow Transplant 1999; 23:259–263.
- Critchley HO, Wallace WH, Shalet SM, Mamtora H, Higginson J, Anderson DC. Abdominal irradiation in childhood; the potential for pregnancy. Br J Obstet Gynaecol 1992; 99:392–394.
- Goldberg JM, Falcone T, Attaran M. In vitro fertilization update. Cleve Clin J Med 2007; 74:329–338.
- Prest SJ, May FE, Westley BR. The estrogen-regulated protein, TFF1, stimulates migration of human breast cancer cells. FASEB J 2002; 16:592–594.
- Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1992; 339:71–85.
- Klopper A, Hall M. New synthetic agent for the induction of ovulation: preliminary trials in women. Br Med J 1971; 1:152–154.
- Oktay K, Buyuk E, Davis O, Yermakova I, Veeck L, Rosenwaks Z. Fertility preservation in breast cancer patients: IVF and embryo cryopreservation after ovarian stimulation with tamoxifen. Hum Reprod 2003; 18:90–95.
- Oktay K, Buyuk E, Libertella N, Akar M, Rosenwaks Z. Fertility preservation in breast cancer patients: a prospective controlled comparison of ovarian stimulation with tamoxifen and letrozole for embryo cryopreservation. J Clin Oncol 2005; 23:4347–4353.
- Oktay K. Further evidence on the safety and success of ovarian stimulation with letrozole and tamoxifen in breast cancer patients undergoing in vitro fertilization to cryopreserve their embryos for fertility preservation. J Clin Oncol 2005; 23:3858–3859.
- Gosden R. Cryopreservation: a cold look at technology for fertility preservation. Fertil Steril 2011; 96:264–268.
- Chen SU, Chien CL, Wu MY, et al. Novel direct cover vitrification for cryopreservation of ovarian tissues increases follicle viability and pregnancy capability in mice. Hum Reprod 2006; 21:2794–2800.
- Lee SJ, Schover LR, Partridge AH, et al; American Society of Clinical Oncology. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24:2917–2931.
- Society for Assisted Reproductive Technology (SART). http://www.sart.org. Accessed January 3, 2013.
- Granne I, Child T, Hartshorne G; British Fertility Society. Embryo cryopreservation: evidence for practice. Hum Fertil (Camb) 2008; 11:159–172.
- Loutradi KE, Kolibianakis EM, Venetis CA, et al. Cryopreservation of human embryos by vitrification or slow freezing: a systematic review and meta-analysis. Fertil Steril 2008; 90:186–193.
- Ko CS, Ding DC, Chu TW, et al. Changes to the meiotic spindle and zona pellucida of mature mouse oocytes following different cryopreservation methods. Anim Reprod Sci 2008; 105:272–282.
- Cao Y, Xing Q, Zhang ZG, et al. Cryopreservation of immature and in vitro-matured human oocytes by vitrification. Reprod Biomed Online 2009; 19:369–373.
- Toth TL, Baka SG, Veeck LL, Jones HW, Muasher S, Lanzendorf SE. Fertilization and in vitro development of cryopreserved human prophase I oocytes. Fertil Steril 1994; 61:891–894.
- Chian RC, Gülekli B, Buckett WM, Tan SL. Priming with human chorionic gonadotropin before retrieval of immature oocytes in women with infertility due to the polycystic ovary syndrome. N Engl J Med 1999; 341: 1624,1626.
- Maman E, Meirow D, Brengauz M, Raanani H, Dor J, Hourvitz A. Luteal phase oocyte retrieval and in vitro maturation is an optional procedure for urgent fertility preservation. Fertil Steril 2011; 95:64–67.
- Oktay K, Demirtas E, Son WY, Lostritto K, Chian RC, Tan SL. In vitro maturation of germinal vesicle oocytes recovered after premature luteinizing hormone surge: description of a novel approach to fertility preservation. Fertil Steril 2008; 89:228.e19–e22.
- Donnez J, Dolmans MM, Demylle D, et al. Livebirth after orthotopic transplantation of cryopreserved ovarian tissue. Lancet 2004; 364:1405–1410.
- Martinez-Madrid B, Dolmans MM, Van Langendonckt A, Defrère S, Donnez J. Freeze-thawing intact human ovary with its vascular pedicle with a passive cooling device. Fertil Steril 2004; 82:1390–1394.
- Martinez-Madrid B, Camboni A, Dolmans MM, Nottola S, Van Langendonckt A, Donnez J. Apoptosis and ultrastructural assessment after cryopreservation of whole human ovaries with their vascular pedicle. Fertil Steril 2007; 87:1153–1165.
- Oktay K, Economos K, Kan M, Rucinski J, Veeck L, Rosenwaks Z. Endocrine function and oocyte retrieval after autologous transplantation of ovarian cortical strips to the forearm. JAMA 2001; 286:1490–1493.
- Practice Committee of American Society for Reproductive Medicine. Ovarian tissue and oocyte cryopreservation. Fertil Steril 2008; 90(suppl 5):S241–S246.
- Oktay K. Ovarian tissue cryopreservation and transplantation: preliminary findings and implications for cancer patients. Hum Reprod Update 2001; 7:526–534.
- Donnez J, Silber S, Andersen CY, et al. Children born after autotransplantation of cryopreserved ovarian tissue. a review of 13 live births. Ann Med 2011; 43:437–450.
- Terenziani M, Piva L, Meazza C, Gandola L, Cefalo G, Merola M. Oophoropexy: a relevant role in preservation of ovarian function after pelvic irradiation. Fertil Steril 2009; 91:935.e15–e16.
- Del Mastro L, Boni L, Michelotti A, et al. Effect of the gonadotropin-releasing hormone analogue triptorelin on the occurrence of chemotherapy-induced early menopause in premenopausal women with breast cancer: a randomized trial. JAMA 2011; 306:269–276.
- Gonfloni S, Di Tella L, Caldarola S, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 2009; 15:1179–1185.
- Meirow D, Biederman H, Anderson RA, Wallace WH. Toxicity of chemotherapy and radiation on female reproduction. Clin Obstet Gynecol 2010; 53:727–739.
- Wallace WH, Kelsey TW. Human ovarian reserve from conception to the menopause. PLoS One 2010; 5:e8772.
- van Disseldorp J, Lambalk CB, Kwee J, et al. Comparison of inter- and intra-cycle variability of anti-Mullerian hormone and antral follicle counts. Hum Reprod 2010; 25:221–227.
- Lie Fong S, Laven JS, Hakvoort-Cammel FG, et al. Assessment of ovarian reserve in adult childhood cancer survivors using anti-Müllerian hormone. Hum Reprod 2009; 24:982–990.
- Oktem O, Oktay K. Quantitative assessment of the impact of chemotherapy on ovarian follicle reserve and stromal function. Cancer 2007; 110:2222–2229.
- Partridge AH, Ruddy KJ, Gelber S, et al. Ovarian reserve in women who remain premenopausal after chemotherapy for early stage breast cancer. Fertil Steril 2010; 94:638–644.
- Lee SJ, Schover LR, Partridge AH, et al; American Society of Clinical Oncology. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24:2917–2931.
- Familiari G, Caggiati A, Nottola SA, Ermini M, Di Benedetto MR, Motta PM. Ultrastructure of human ovarian primordial follicles after combination chemotherapy for Hodgkin’s disease. Hum Reprod 1993; 8:2080–2087.
- Wulff C, Wilson H, Wiegand SJ, Rudge JS, Fraser HM. Prevention of thecal angiogenesis, antral follicular growth, and ovulation in the primate by treatment with vascular endothelial growth factor Trap R1R2. Endocrinology 2002; 143:2797–2807.
- Aubard Y, Piver P, Pech JC, Galinat S, Teissier MP. Ovarian tissue cryopreservation and gynecologic oncology: a review. Eur J Obstet Gynecol Reprod Biol 2001; 97:5–14.
- Elizur SE, Chian RC, Pineau CA, et al. Fertility preservation treatment for young women with autoimmune diseases facing treatment with gonadotoxic agents. Rheumatology (Oxford) 2008; 47:1506–1509.
- Wo JY, Viswanathan AN. Impact of radiotherapy on fertility, pregnancy, and neonatal outcomes in female cancer patients. Int J Radiat Oncol Biol Phys 2009; 73:1304–1312.
- Wallace WH, Thomson AB, Saran F, et al. Predicting age of ovarian failure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys 2005; 62:738–744.
- Gareer W, Gad Z, Gareer H. Needle oophoropexy: a new simple technique for ovarian transposition prior to pelvic irradiation. Surg Endosc 2011; 25:2241–2246.
- Larsen EC, Schmiegelow K, Rechnitzer C, Loft A, Müller J, Andersen AN. Radiotherapy at a young age reduces uterine volume of childhood cancer survivors. Acta Obstet Gynecol Scand 2004; 83:96–102.
- Holm K, Nysom K, Brocks V, Hertz H, Jacobsen N, Müller J. Ultrasound B-mode changes in the uterus and ovaries and Doppler changes in the uterus after total body irradiation and allogeneic bone marrow transplantation in childhood. Bone Marrow Transplant 1999; 23:259–263.
- Critchley HO, Wallace WH, Shalet SM, Mamtora H, Higginson J, Anderson DC. Abdominal irradiation in childhood; the potential for pregnancy. Br J Obstet Gynaecol 1992; 99:392–394.
- Goldberg JM, Falcone T, Attaran M. In vitro fertilization update. Cleve Clin J Med 2007; 74:329–338.
- Prest SJ, May FE, Westley BR. The estrogen-regulated protein, TFF1, stimulates migration of human breast cancer cells. FASEB J 2002; 16:592–594.
- Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1992; 339:71–85.
- Klopper A, Hall M. New synthetic agent for the induction of ovulation: preliminary trials in women. Br Med J 1971; 1:152–154.
- Oktay K, Buyuk E, Davis O, Yermakova I, Veeck L, Rosenwaks Z. Fertility preservation in breast cancer patients: IVF and embryo cryopreservation after ovarian stimulation with tamoxifen. Hum Reprod 2003; 18:90–95.
- Oktay K, Buyuk E, Libertella N, Akar M, Rosenwaks Z. Fertility preservation in breast cancer patients: a prospective controlled comparison of ovarian stimulation with tamoxifen and letrozole for embryo cryopreservation. J Clin Oncol 2005; 23:4347–4353.
- Oktay K. Further evidence on the safety and success of ovarian stimulation with letrozole and tamoxifen in breast cancer patients undergoing in vitro fertilization to cryopreserve their embryos for fertility preservation. J Clin Oncol 2005; 23:3858–3859.
- Gosden R. Cryopreservation: a cold look at technology for fertility preservation. Fertil Steril 2011; 96:264–268.
- Chen SU, Chien CL, Wu MY, et al. Novel direct cover vitrification for cryopreservation of ovarian tissues increases follicle viability and pregnancy capability in mice. Hum Reprod 2006; 21:2794–2800.
- Lee SJ, Schover LR, Partridge AH, et al; American Society of Clinical Oncology. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24:2917–2931.
- Society for Assisted Reproductive Technology (SART). http://www.sart.org. Accessed January 3, 2013.
- Granne I, Child T, Hartshorne G; British Fertility Society. Embryo cryopreservation: evidence for practice. Hum Fertil (Camb) 2008; 11:159–172.
- Loutradi KE, Kolibianakis EM, Venetis CA, et al. Cryopreservation of human embryos by vitrification or slow freezing: a systematic review and meta-analysis. Fertil Steril 2008; 90:186–193.
- Ko CS, Ding DC, Chu TW, et al. Changes to the meiotic spindle and zona pellucida of mature mouse oocytes following different cryopreservation methods. Anim Reprod Sci 2008; 105:272–282.
- Cao Y, Xing Q, Zhang ZG, et al. Cryopreservation of immature and in vitro-matured human oocytes by vitrification. Reprod Biomed Online 2009; 19:369–373.
- Toth TL, Baka SG, Veeck LL, Jones HW, Muasher S, Lanzendorf SE. Fertilization and in vitro development of cryopreserved human prophase I oocytes. Fertil Steril 1994; 61:891–894.
- Chian RC, Gülekli B, Buckett WM, Tan SL. Priming with human chorionic gonadotropin before retrieval of immature oocytes in women with infertility due to the polycystic ovary syndrome. N Engl J Med 1999; 341: 1624,1626.
- Maman E, Meirow D, Brengauz M, Raanani H, Dor J, Hourvitz A. Luteal phase oocyte retrieval and in vitro maturation is an optional procedure for urgent fertility preservation. Fertil Steril 2011; 95:64–67.
- Oktay K, Demirtas E, Son WY, Lostritto K, Chian RC, Tan SL. In vitro maturation of germinal vesicle oocytes recovered after premature luteinizing hormone surge: description of a novel approach to fertility preservation. Fertil Steril 2008; 89:228.e19–e22.
- Donnez J, Dolmans MM, Demylle D, et al. Livebirth after orthotopic transplantation of cryopreserved ovarian tissue. Lancet 2004; 364:1405–1410.
- Martinez-Madrid B, Dolmans MM, Van Langendonckt A, Defrère S, Donnez J. Freeze-thawing intact human ovary with its vascular pedicle with a passive cooling device. Fertil Steril 2004; 82:1390–1394.
- Martinez-Madrid B, Camboni A, Dolmans MM, Nottola S, Van Langendonckt A, Donnez J. Apoptosis and ultrastructural assessment after cryopreservation of whole human ovaries with their vascular pedicle. Fertil Steril 2007; 87:1153–1165.
- Oktay K, Economos K, Kan M, Rucinski J, Veeck L, Rosenwaks Z. Endocrine function and oocyte retrieval after autologous transplantation of ovarian cortical strips to the forearm. JAMA 2001; 286:1490–1493.
- Practice Committee of American Society for Reproductive Medicine. Ovarian tissue and oocyte cryopreservation. Fertil Steril 2008; 90(suppl 5):S241–S246.
- Oktay K. Ovarian tissue cryopreservation and transplantation: preliminary findings and implications for cancer patients. Hum Reprod Update 2001; 7:526–534.
- Donnez J, Silber S, Andersen CY, et al. Children born after autotransplantation of cryopreserved ovarian tissue. a review of 13 live births. Ann Med 2011; 43:437–450.
- Terenziani M, Piva L, Meazza C, Gandola L, Cefalo G, Merola M. Oophoropexy: a relevant role in preservation of ovarian function after pelvic irradiation. Fertil Steril 2009; 91:935.e15–e16.
- Del Mastro L, Boni L, Michelotti A, et al. Effect of the gonadotropin-releasing hormone analogue triptorelin on the occurrence of chemotherapy-induced early menopause in premenopausal women with breast cancer: a randomized trial. JAMA 2011; 306:269–276.
- Gonfloni S, Di Tella L, Caldarola S, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 2009; 15:1179–1185.
KEY POINTS
- Chemotherapy and radiotherapy are toxic to the ovaries, although the damage can be attenuated in some cases.
- The standard option for preserving fertility has been oocyte retrieval after controlled ovarian stimulation, followed by in vitro fertilization and subsequent cryopreservation of the resulting embryos.
- Unfortunately, controlled ovarian stimulation takes time, may delay needed cancer therapy, and may worsen estrogen-dependent cancers. Alternatives are being explored.
- Cryopreservation of unfertilized oocytes is an option for women who do not have a partner, although oocytes are more susceptible to damage during freezing than embryos are.
Recognizing and managing hereditary angioedema
Hereditary angioedema due to deficiency of C1 inhibitor is a rare autosomal dominant disease that can be life-threatening. It affects about 1 in 50,000 people,1 or about 6,000 people in the United States. There are no known differences in prevalence by ethnicity or sex. A form of hereditary angioedema with normal C1 inhibitor levels has also recently been identified.
Despite a growing awareness of hereditary angioedema in the medical community, repeated surveys have found an average gap of 10 years between the first appearance of symptoms and the correct diagnosis. In view of the risk of morbidity and death, recognizing this disease sooner is critical.
This article will discuss how to recognize hereditary angioedema and how to differentiate it from other forms of recurring angioedema. We will also review its acute and long-term management, with special attention to new therapies and clinical challenges.
EPISODES OF SWELLING WITHOUT HIVES
Hereditary angioedema involves recurrent episodes of nonpruritic, nonpitting, subcutaneous and submucosal edema that can affect the face, tongue, larynx, trunk, extremities, bowels, or genitals. Attacks typically follow a predictable course: swelling that increases slowly and continuously for 24 hours and then gradually subsides over the next 48 to 72 hours. Attacks that involve the oropharynx, larynx, or abdomen carry the highest risk of morbidity and death.1
The frequency and severity of attacks are highly variable and unpredictable. A few patients have no attacks, a few have two attacks per week, and most fall in between.
Hives suggests an allergic or idiopathic rather than hereditary cause and will not be discussed here in detail. A history of angioedema that was rapidly aborted by antihistamines, corticosteroids, or epinephrine also suggests an allergic rather than hereditary cause.
UNCHECKED BRADYKININ PRODUCTION
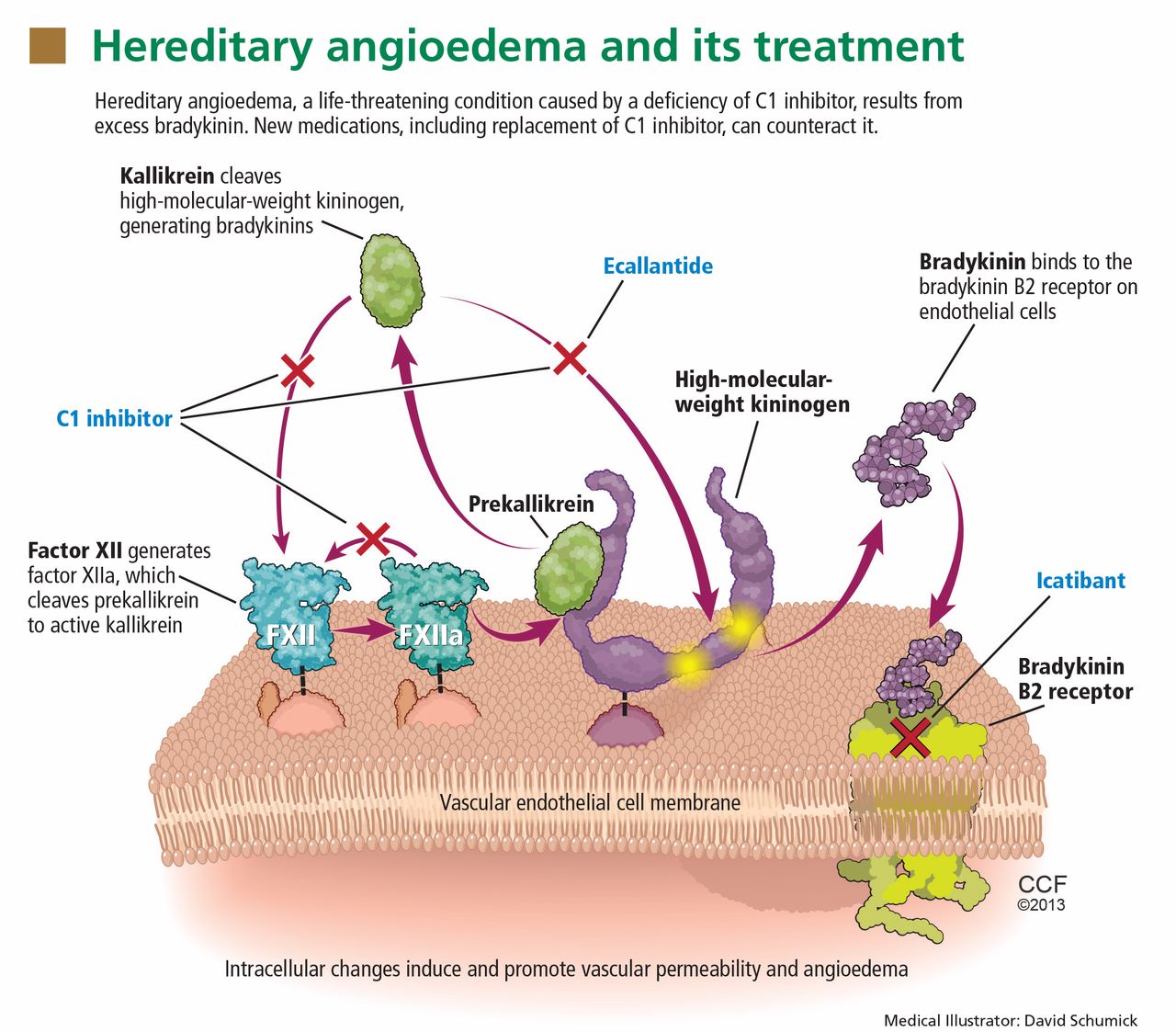
Substantial evidence indicates that hereditary angioedema results from extravasation of plasma into deeper cutaneous or mucosal compartments as a result of overproduction of the vasoactive mediator bradykinin (Figure 1).
Activated factor XII cleaves plasma prekallikrein to generate active plasma kallikrein (which, in turn, activates more factor XII).2 Once generated, plasma kallikrein cleaves high-molecular-weight kininogen, releasing bradykinin. Bradykinin binds to the B2 bradykinin receptor on endothelial cells, increasing the permeability of the endothelium.
Normally, C1 inhibitor helps control bradykinin production by inhibiting plasma kallikrein and activated factor XII. Without enough C1 inhibitor, the contact system is uninhibited and results in bradykinin being inappropriately generated.
Because the attacks of hereditary angioedema involve excessive bradykinin, they do not respond to the usual treatments for anaphylaxis and allergic angioedema (which involve mast cell degranulation), such as antihistamines, corticosteroids, and epinephrine.
TWO TYPES OF HEREDITARY ANGIOEDEMA
Figure 2 shows the evaluation of patients with suspected hereditary angioedema.
Hereditary angioedema due to C1 inhibitor deficiency
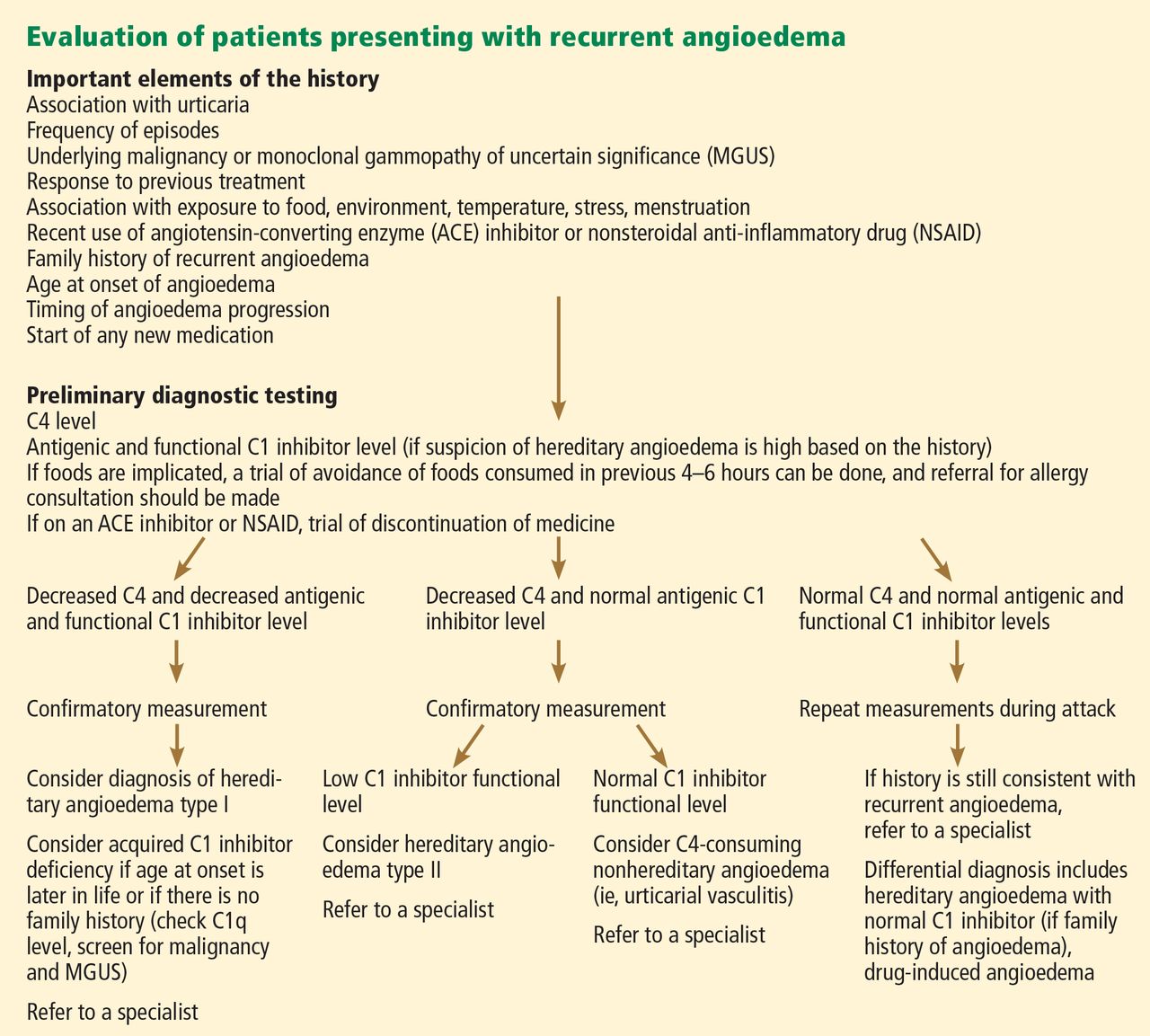
The classic forms of hereditary angioedema (types I and II) involve loss-of-function mutations in SERPING1—the gene that encodes for C1 inhibitor—resulting in low levels of functional C1 inhibitor.3 The mutation is inherited in an autosomal dominant pattern; however, in about 25% of cases, it appears to arise spontaneously,4 so a family history is not required for diagnosis.
Although C1 inhibitor deficiency is present from birth, the clinical disease most commonly presents for the first time when the patient is of school age. Half of patients have their first episode in the first decade of life, and another one-third first develop symptoms over the next 10 years.5
Clinically, types I and II are indistinguishable. Type I, accounting for 85% of cases,1 results from low production of C1 inhibitor. Laboratory studies reveal low antigenic and functional levels of C1 inhibitor.
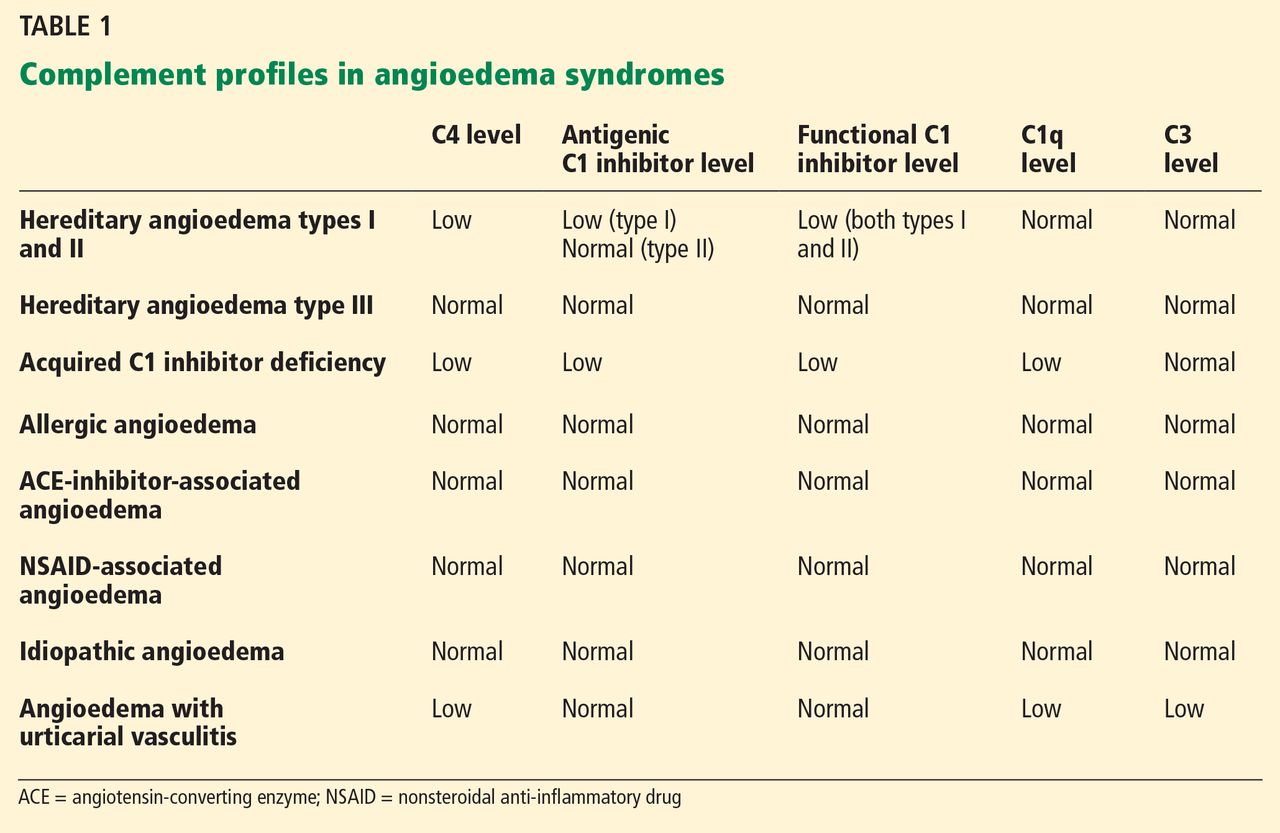
In type II, the mutant C1 inhibitor protein is present but dysfunctional and unable to inhibit target proteases. On laboratory testing, the functional level of C1 inhibitor is low but its antigenic level is normal (Table 1). Function can be tested by either chromogenic assay or enzyme-linked immunosorbent assay; the former is preferred because it is more sensitive.6
Because C1 inhibitor deficiency results in chronic activation of the complement system, patients with type I or II disease usually have low C4 levels regardless of disease activity, making measuring C4 the most economical screening test. When suspicion for hereditary angioedema is high, based on the presentation and family and clinical history, measuring antigenic and functional C1 inhibitor levels and C4 simultaneously is more efficient.
Hereditary angioedema with normal C1 inhibitor levels
Hereditary angioedema with normal C1 inhibitor levels is also inherited in an autosomal dominant pattern. It is often estrogen-sensitive, making it more severe in women. Symptoms tend to develop slightly later in life than in type I or II disease.7
Angioedema with normal C1 inhibitor levels has been associated with factor XII mutations in a minority of cases, but most patients do not have a specific laboratory abnormality. Because there is no specific laboratory profile, the diagnosis is based on clinical criteria. Hereditary angioedema with normal C1 inhibitor levels should be considered in patients who have recurrent angioedema, normal C4, normal antigenic and functional C1 inhibitor levels, a lack of response to high-dose antihistamines, and either a family history of angioedema without hives or a known factor XII mutation.7 However, other forms of angioedema (allergic, drug-induced, and idiopathic) should also be considered, as C4 and C1 inhibitor levels are normal in these forms as well.
DIFFERENTIAL DIAGNOSIS: OTHER TYPES OF ANGIOEDEMA
Acquired C1 inhibitor deficiency
Symptoms of acquired C1 inhibitor deficiency resemble those of hereditary angioedema but typically do not emerge until the fourth decade of life or later, and patients have no family history of the condition. It is often associated with other diseases, most commonly B-cell lymphoproliferative disorders, which cause uncontrolled complement activation and consumption of C1 inhibitor.
In some patients, autoantibodies to C1 inhibitor develop, greatly reducing its effectiveness and resulting in enhanced consumption. The autoantibody is often associated with a monoclonal gammopathy of unknown significance. The presence of a C1 inhibitor autoantibody does not preclude the presence of an underlying disorder, and vice versa.
Laboratory studies reveal low C4, low C1-inhibitor antigenic and functional levels, and usually a low C1q level owing to consumption of complement. Autoantibodies to C1 inhibitor can be detected by laboratory testing.
Because of the association with autoimmune disease and malignant disorders (especially B-cell malignancy), a patient diagnosed with acquired C1 inhibitor deficiency should be further evaluated for underlying conditions.
Allergic angioedema
Allergic angioedema results from preformed antigen-specific immunoglobulin E (IgE) antibodies that stimulate mast cells to degranulate when patients are exposed to a particular allergen—most commonly food, insect venom, latex, or drugs. IgE-mediated histamine release causes swelling, as histamine is a potent vasodilator.
Symptoms often begin within 2 hours of exposure to the allergen and generally include concurrent urticaria and swelling that last less than 24 hours. Unlike in hereditary angioedema, the swelling responds to antihistamines and corticosteroids. When very severe, these symptoms may also be accompanied by bronchoconstriction and gastrointestinal symptoms, especially if the allergen is ingested.
Histamine-mediated angioedema may also be associated with exercise as part of a syndrome called exercise-induced anaphylaxis or angioedema.
Drug-induced angioedema
Drug-induced angioedema is typically associated with angiotensin-converting enzyme (ACE) inhibitors or nonsteroidal anti-inflammatory drugs (NSAIDs).
Angioedema associated with ACE inhibitors is estimated to affect 0.1% to 6% of patients taking these medications, with African Americans being at significantly higher risk. Although 25% of affected patients develop symptoms of angioedema within the first month of taking the drugs, some tolerate them for as long as 10 years before the first episode.9 The swelling is not allergic or histamine-related. ACE normally degrades bradykinin; therefore, inhibiting ACE leads to accumulation of bradykinin. Because all ACE inhibitors have this effect, this class of drug should be discontinued in any patient who develops isolated angioedema.
NSAID-induced angioedema is often accompanied by other symptoms, including urticaria, rhinitis, cough, hoarseness, or breathlessness.10 The mechanism of NSAID-induced angioedema involves cyclooxygenase (COX) 1 (and to a lesser extent COX-2) inhibition. All NSAIDs (and aspirin) should be avoided in patients with recurrent angioedema. Specific COX-2 inhibitors, while theoretically capable of causing angioedema by the same mechanism, are generally well tolerated in patients who have had COX-1 inhibitor reactions.
Idiopathic angioedema
If no clear cause of recurrent angioedema (at least three episodes in a year) can be found, it is labeled idiopathic.11 Some patients with idiopathic angioedema fail to benefit from high doses of antihistamines, suggesting that the cause is bradykinin-mediated.
CLINICAL MANIFESTATIONS OF HEREDITARY ANGIOEDEMA
Attacks may start at one site and progress to involve additional sites.
Prodromal symptoms may begin up to several days before an attack and include tingling, warmth, burning, or itching at the affected site; increased fatigue or malaise; nausea, abdominal distention, or gassiness; or increased hunger, particularly before an abdominal attack.5 The most characteristic prodromal symptom is erythema marginatum—a raised, serpiginous, nonpruritic rash on the trunk, arms, and legs but often sparing the face.
Abdominal attacks are easily confused with acute abdomen
Almost half of attacks involve the abdomen, and almost all patients with type I or II disease experience at least one such attack.12 Symptoms can include severe abdominal pain, nausea, vomiting, and diarrhea. Abdominal attacks account for many emergency department visits, hospitalizations, and surgical procedures for acute abdomen; about one-third of patients with undiagnosed hereditary angioedema undergo an unnecessary surgery during an abdominal attack. Angioedema of the gastrointestinal tract can result in enough plasma extravasation and vasodilation to cause hypovolemic shock.
Eradicating Helicobacter pylori infection may alleviate abdominal attacks.13
Attacks of the extremities can be painful and disabling
Attacks of the extremities affect 96% of patients12 and can be very disfiguring and disabling. Driving or using the phone is often difficult when the hands are affected. When feet are involved, walking and standing become painful. While these symptoms rarely result in a lengthy hospitalization, they interfere with work and school and require immediate medical attention because they can progress to other parts of the body.
Laryngeal attacks are life-threatening
About half of patients with hereditary angioedema have an attack of laryngeal edema at some point in their lives.12 If not effectively managed, laryngeal angioedema can progress to asphyxiation. A survey of family history in 58 patients with hereditary angioedema suggested a 40% incidence of asphyxiation in untreated laryngeal attacks, and 25% to 30% of patients are estimated to have died of laryngeal edema before effective treatment became available.14
Symptoms of a laryngeal attack include change in voice, hoarseness, trouble swallowing, shortness of breath, and wheezing. Physicians must recognize these symptoms quickly and give effective treatment early in the attack to prevent morbidity and death.
Establishing an airway can be life-saving in the absence of effective therapy, but extensive swelling of the upper airway can make intubation extremely difficult.
Genitourinary attacks also occur
Attacks involving the scrotum and labia have been reported in up to two-thirds of patients with hereditary angioedema at some point in their lives. Attacks involving the bladder and kidneys have also been reported but are less common, affecting about 5% of patients.12 Genitourinary attacks may be triggered by local trauma, such as horseback riding or sexual intercourse, although no trigger may be evident.
MANAGING ACUTE ATTACKS
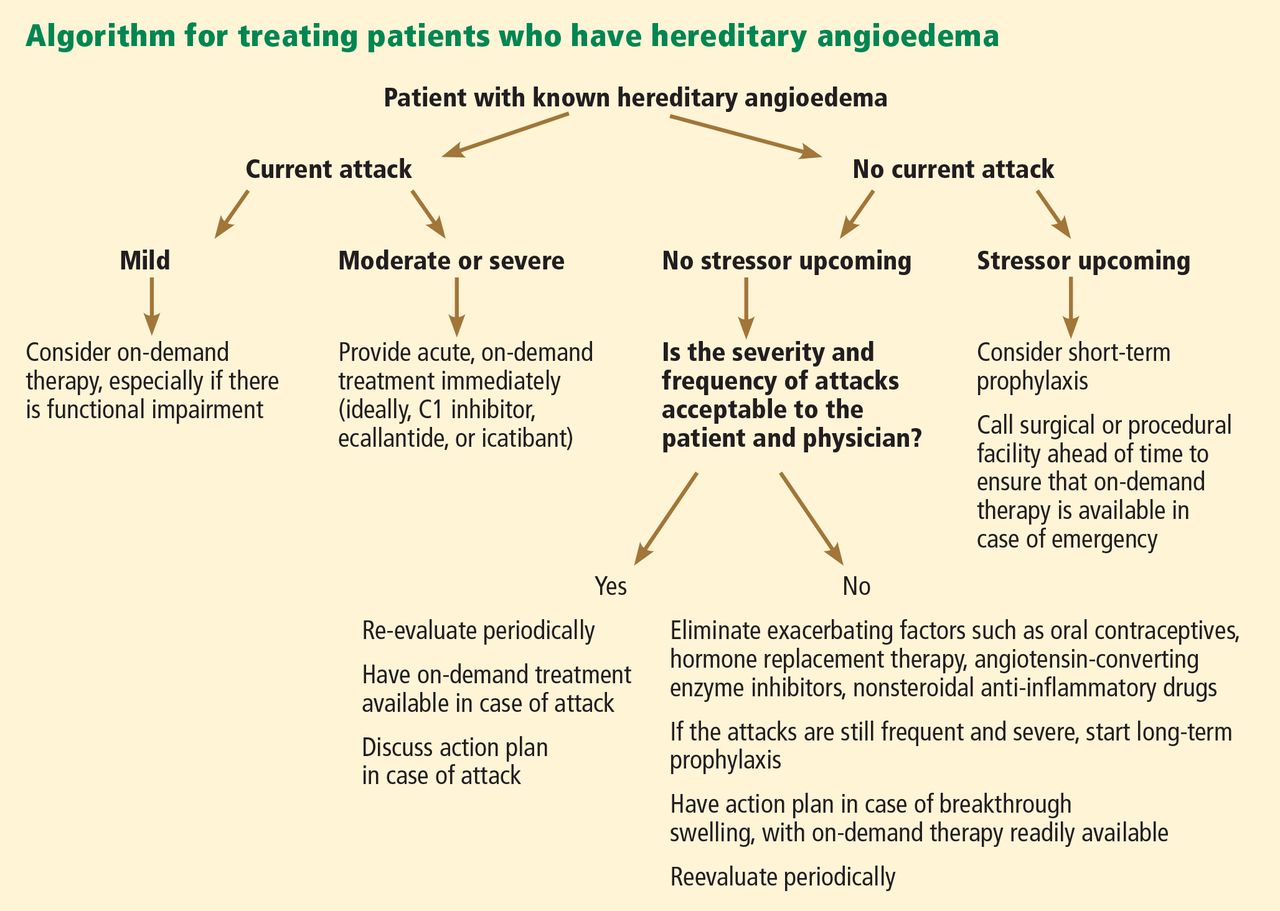
The goals of treatment are to alleviate acute exacerbations with on-demand treatment and to reduce the number of attacks with prophylaxis. Therapy should be individualized to each patient’s needs. Treatments have advanced greatly in the last several years, and new medications for treating acute attacks and preventing attacks have shown great promise (Figure 3, Table 2).
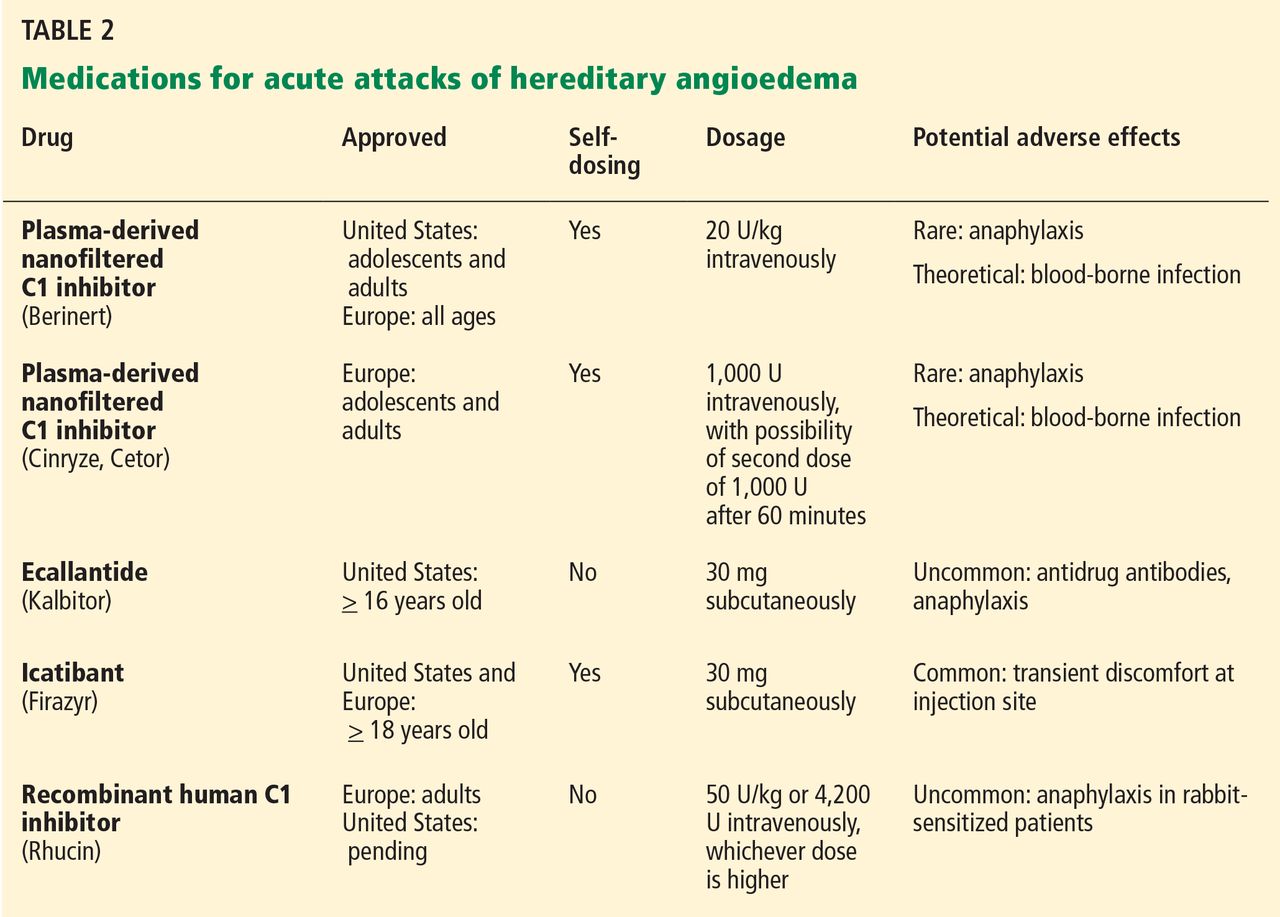
Patients tend to have recurrent symptoms interspersed with periods of health, suggesting that attacks ought to have identifiable triggers, although in most, no trigger is evident. The most commonly identified are local trauma (including medical and dental procedures), emotional stress, and acute infection. Disease severity may be worsened by menstruation, estrogen-containing oral contraceptives, hormone replacement therapy, ACE inhibitors, and NSAIDs.
It is critical that attacks be treated with an effective medication as soon as possible. Consensus guidelines state that all patients with hereditary angioedema due to C1 inhibitor deficiency, even if they are still asymptomatic, should have access to at least one of the drugs approved for on-demand treatment.15 The guidelines further state that whenever possible, “patients should have the on-demand medicine to treat acute attacks at home and should be trained to self-administer these medicines.”15
Plasma-derived C1 inhibitors
Several plasma-derived C1 inhibitors are available (Cinryze, Berinert, Cetor). They are prepared from fractionated plasma obtained from donors, then pasteurized and nanofiltered.
Berinert and Cinryze were each found to be superior to placebo in double-blind, placebo-controlled trials: attacks usually resolved 30 to 60 minutes after intravenous injection.16,17 Berinert 20 U/kg is associated with the onset of symptom relief as early as half an hour after administration, compared with 1.5 hours with placebo. Early use (at the onset of symptoms) of a plasma-derived C1 inhibitor in a low dose (500 U) can also be effective.18,19 Efficacy appears to be consistent at all sites of attack involvement, including laryngeal edema. Safety and efficacy have been demonstrated during pregnancy and lactation and in young children and babies.20
Plasma-derived C1 inhibitors can be self-administered. The safety and efficacy of self-administration (under physician supervision) were demonstrated in a study of Cinryze and Cetor, in which attack duration, pain medication use, and graded attack severity were significantly less with self-administered therapy than with therapy in the clinic.21
A concern about plasma-derived products is the possibility of blood-borne infection, but this has not been confirmed by experience.22
Recombinant human C1 inhibitor
A recombinant human C1 inhibitor (Rhucin) has been studied in two randomized placebo-controlled trials. Although this product has a shorter half-life than the plasma-derived C1 inhibitors (3 vs more than 24 hours), the two are equipotent: 1 U of recombinant human C1 inhibitor is equivalent to 1 U of plasma-derived C1 inhibitor. Because the supply of recombinant human C1 inhibitor is elastic, dosing has been higher, which may provide more efficacy.23 Similar to plasma-derived C1 inhibitor products, the recombinant human C1 inhibitor resulted in more rapid symptom relief than with saline (66 vs 122 minutes) and in a shorter time to minimal symptoms (247−266 vs 1,210 minutes).24
Allergy is of concern: in one study, a healthy volunteer with undisclosed rabbit allergy experienced an allergic reaction. Patients should be screened by a skin-prick test or serum testing for specific IgE to rabbit epithelium before being prescribed recombinant human C1 inhibitor. No data are available for use during pregnancy or breastfeeding.
Ecallantide
Ecallantide (Kalbitor) is a selective inhibitor of plasma kallikrein that is given in three subcutaneous injections. Ecallantide 30 mg was found superior to placebo during acute attacks.25,26
Ecallantide is well tolerated, with the most common adverse effects being headache, nausea, fatigue, diarrhea, and local injection-site reactions. Antibodies to ecallantide can be found in patients with increasing drug exposure but do not appear to correlate with adverse events. Hypersensitivity reactions have been observed in 2% to 3% of patients receiving repeated doses. Because of anaphylaxis risk, ecallantide must be administered by a health care professional.
Icatibant
Icatibant (Firazyr) is a bradykinin receptor-2 antagonist that is given in a single subcutaneous injection. Icatibant 30 mg significantly shortened time to symptom relief and time to almost complete resolution compared with placebo.27,28 Icatibant’s main adverse effect is transient local pain, swelling, and erythema at the injection site. Icatibant can be self-administered by patients.
Fresh-frozen plasma
Fresh-frozen plasma contains C1 inhibitor and was used before the newer products became available. Several noncontrolled studies reported benefit of its use in acute attacks.29 However, its use is controversial because it also contains contact-system proteins that could provide additional substrate for the generation of bradykinin, which could exacerbate attacks in some patients.1 This may be particularly dangerous in patients presenting with laryngeal edema: in such a situation, the physician should be ready to treat a sudden exacerbation with intubation. The risk of acquiring a blood-borne pathogen is also higher than with plasma-derived C1 inhibitor.
PROPHYLACTIC MANAGEMENT
Short-term and long-term prophylaxis have important roles in preventing attacks (Table 3).
Short-term prophylaxis before an anticipated attack

Short-term prophylaxis is used for patients whose disease is generally well controlled but who anticipate exposure to a potentially exacerbating situation, such as an invasive medical, surgical, or dental procedure. (Routine dental cleanings are generally considered safe and do not require prophylaxis.)
Prophylactic treatments include:
- Plasma-derived C1 inhibitor, 500 to 1,500 U 1 hour before the provoking event
- High-dose 17-alpha alkylated (attenuated) androgens (eg, danazol [Danocrine] 200 mg orally 3 times daily) for 5 to 10 days before the provoking event
- Fresh-frozen plasma, 2 U 1 to 12 hours before the event.1
Yet even with short-term prophylaxis, on-demand treatment should be available.
Long-term prophylaxis
While many patients can be managed with on-demand treatment only, other patients (reflecting the severity of their attacks, as well as their individual needs) may benefit from a combination of on-demand treatment plus long-term prophylaxis. Several options are available (Table 3).
17-alpha alkylated androgens. Patients treated with danazol 600 mg/day were attack-free 90% of the time during a 28-day period compared with only 2.2% of the time in placebo-treated patients.30 Use of anabolic androgens, however, is limited by their adverse effects, including weight gain, virilization, menstrual irregularities, headaches, depression, dyslipidemia, liver enzyme elevation, liver adenomas, and hepatocellular carcinoma. Arterial hypertension occurs in about 25% of treated patients.
Because adverse effects are dose-dependent, treatment should be empirically titrated to find the minimal effective dose, generally recommended to be no more than 200 mg per day of danazol or the equivalent.15
Contraindications include use by women during pregnancy or lactation and by children until growth is complete.
Regular follow-up is recommended every 6 months, with monitoring of liver enzymes, lipids, complete blood counts, alpha fetoprotein, and urinalysis. Abdominal ultrasonography (every 6 months if receiving 100 mg/day or more of danazol, every 12 months if less than 100 mg/day) is advisable for early diagnosis of liver tumors.
Antifibrinolytic drugs. Tranexamic acid (Lysteda) and aminocaproic acid (Amicar) have been found to be effective in reducing the number of attacks of hereditary angioedema compared with placebo but are considered to be less reliable than androgens. These drugs have been used in patients who do not tolerate anabolic androgens, and in children and pregnant women. Tranexamic acid is given at a dose of 20 to 50 mg/kg/day divided into two or three doses per day. The therapeutic dose of aminocaproic acid is 1 g orally three to four times per day.31 Patients with a personal or family history of thromboembolic disease may be at greater risk of venous or arterial thrombosis, but this has not occurred in clinical studies.
Plasma-derived C1 inhibitors. In a 24-week crossover study in 22 patients with hereditary angioedema, Cinryze 1,000 U every 3 to 4 days reduced the rate of attacks by 50% while also reducing their severity and duration.17 An open-label extension study in 146 patients for almost 3 years documented a 90% reduction in attack frequency with no evidence of tachyphylaxis.32
New treatments are costlier
The newer on-demand and prophylactic drugs are substantially costlier than the older alternatives (androgens, antifibrinolytics, and fresh-frozen plasma); however, they have a substantially better benefit-to-risk ratio. Furthermore, the costs of care for an attack requiring emergency treatment are also high. Hereditary angioedema patients are often young, otherwise healthy, and capable of leading normal productive lives. While formal pharmacoeconomic studies of the optimal use of these newer drugs have not yet been done, it is important that the use of these drugs be well justified. Ideally, physicians who prescribe these drugs should be knowledgeable in the management of hereditary angioedema.
SPECIAL CHALLENGES IN WOMEN
Women with hereditary angioedema have more frequent attacks and generally a more severe disease course than men.12 Optimizing care for women is challenging because hormonal changes often cause the disease to flare up in menarche, pregnancy, lactation, and menopause. Women also have a higher rate of discontinuing long-term androgen therapy because of side effects, including virilization and menstrual irregularities. Spironolactone (Aldactone) 100 to 200 mg daily can be used to control hirsutism.33
Contraception
Because estrogen can trigger attacks, progesterone-only formulations, intrauterine devices, or barrier methods are recommended for contraception.33 Progesterone-only pills are preferred and improve symptoms in more than 60% of women. Etonogestrel, another alternative, is available as an implant (Implanon) or vaginal ring (Nuvaring). Intrauterine devices are generally well tolerated, and no prophylaxis is needed during placement. The progesterone-eluting intrauterine device (Mirena) could be beneficial.34
Pregnancy and lactation
Pregnancy and lactation pose particular challenges. Anabolic androgens are contraindicated during pregnancy as well as during breastfeeding because they can be passed on in breast milk. Women receiving androgen prophylaxis should understand that they can still ovulate and need contraception if they are sexually active.34 Patients on attenuated androgens who desire pregnancy should discontinue them 2 months before trying to conceive.
Changes in attack patterns can be unpredictable during pregnancy. Attacks tend to be more severe during the first trimester and more frequent during the third. Due to its safety and efficacy, plasma-derived C1 inhibitor has become the treatment of choice for on-demand or prophylactic treatment during pregnancy and lactation. Antifibrinolytics are considered only when plasma-derived C1 inhibitor is not available.31 Ecallantide and icatibant have not been studied in pregnancy. If neither plasma-derived C1 inhibitor nor antifibrinolytics are available, fresh-frozen plasma or solvent-and-detergent-treated plasma can be used.
Short-term prophylaxis should be considered before amniocentesis, chorionic villous sampling, and dilation and curettage. Delivery should take place in a facility with rapid access to plasma-derived C1 inhibitor as well as consultants in obstetrics, anesthesiology, and perinatology. Although plasma-derived C1 inhibitor should be available at all times during labor and delivery, its prophylactic use is not required unless labor and delivery are particularly traumatic, the underlying hereditary angioedema is very severe, or if forceps, vacuum delivery, or cesarian section is performed. Close monitoring is recommended for at least 72 hours after routine vaginal delivery and for 1 week after cesarian section.
CONCLUSION
The goals of hereditary angioedema treatment are to alleviate morbidity and mortality associated with the disease and to improve the patient’s quality of life. Achieving these goals requires timely diagnosis, patient education, and careful selection of therapeutic modalities that are individualized to the needs of that patient. Treatments have advanced greatly in the last 4 years, and new medications for both the acute and chronic symptoms of hereditary angioedema have shown great promise.
Acknowledgment: K.T. is funded by National Institutes of Health grant T32 AI 07469.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359:1027–1036.
- Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol 2010; 126:918–925.
- Davis AE. C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988; 6:595–628.
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106:1147–1154.
- Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am 2006; 26:739–751.
- Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods 2008; 338:14–20.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol 2010; 6:15.
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 2012; 33:S145–S156.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Busse PJ. Angioedema: differential diagnosis and treatment. Allergy Asthma Proc 2011; 32(suppl 1):S3–S11.
- Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc 2009; 30:506–511.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119:267–274.
- Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 2001; 358:1695–1696.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 2001; 161:714–718.
- Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–157.
- Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009; 124:801–808.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010; 363:513–522.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005; 45:1774–1784.
- Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion 2009; 49:1987–1995.
- Farkas H, Varga L, Széplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics 2007; 120:e713–e722.
- Tourangeau LM, Castaldo AJ, Davis DK, Koziol J, Christiansen SC, Zuraw BL. Safety and efficacy of physician-supervised self-managed C1 inhibitor replacement therapy. Int Arch Allergy Immunol 2012; 157:417–424.
- De Serres J, Gröner A, Lindner J. Safety and efficacy of pasteurized C1 inhibitor concentrate (Berinert P) in hereditary angioedema: a review. Transfus Apher Sci 2003; 29:247–254.
- Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012; 67:123–130.
- Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010; 126:821–827.e14.
- Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010; 363:523–531.
- Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 2010; 104:523–529.
- Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010; 363:532–541.
- Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol 2011; 107:529–537.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98:383–388.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med 1976; 295:1444–1448.
- Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol 2010; 6:23.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med 2012: Epub ahead of print.
- Caballero T, Farkas H, Bouillet L, et al; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129:308–320.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol 2008; 199:484.e1–e4.
Hereditary angioedema due to deficiency of C1 inhibitor is a rare autosomal dominant disease that can be life-threatening. It affects about 1 in 50,000 people,1 or about 6,000 people in the United States. There are no known differences in prevalence by ethnicity or sex. A form of hereditary angioedema with normal C1 inhibitor levels has also recently been identified.
Despite a growing awareness of hereditary angioedema in the medical community, repeated surveys have found an average gap of 10 years between the first appearance of symptoms and the correct diagnosis. In view of the risk of morbidity and death, recognizing this disease sooner is critical.
This article will discuss how to recognize hereditary angioedema and how to differentiate it from other forms of recurring angioedema. We will also review its acute and long-term management, with special attention to new therapies and clinical challenges.
EPISODES OF SWELLING WITHOUT HIVES
Hereditary angioedema involves recurrent episodes of nonpruritic, nonpitting, subcutaneous and submucosal edema that can affect the face, tongue, larynx, trunk, extremities, bowels, or genitals. Attacks typically follow a predictable course: swelling that increases slowly and continuously for 24 hours and then gradually subsides over the next 48 to 72 hours. Attacks that involve the oropharynx, larynx, or abdomen carry the highest risk of morbidity and death.1
The frequency and severity of attacks are highly variable and unpredictable. A few patients have no attacks, a few have two attacks per week, and most fall in between.
Hives suggests an allergic or idiopathic rather than hereditary cause and will not be discussed here in detail. A history of angioedema that was rapidly aborted by antihistamines, corticosteroids, or epinephrine also suggests an allergic rather than hereditary cause.
UNCHECKED BRADYKININ PRODUCTION
Substantial evidence indicates that hereditary angioedema results from extravasation of plasma into deeper cutaneous or mucosal compartments as a result of overproduction of the vasoactive mediator bradykinin (Figure 1).
Activated factor XII cleaves plasma prekallikrein to generate active plasma kallikrein (which, in turn, activates more factor XII).2 Once generated, plasma kallikrein cleaves high-molecular-weight kininogen, releasing bradykinin. Bradykinin binds to the B2 bradykinin receptor on endothelial cells, increasing the permeability of the endothelium.
Normally, C1 inhibitor helps control bradykinin production by inhibiting plasma kallikrein and activated factor XII. Without enough C1 inhibitor, the contact system is uninhibited and results in bradykinin being inappropriately generated.
Because the attacks of hereditary angioedema involve excessive bradykinin, they do not respond to the usual treatments for anaphylaxis and allergic angioedema (which involve mast cell degranulation), such as antihistamines, corticosteroids, and epinephrine.
TWO TYPES OF HEREDITARY ANGIOEDEMA
Figure 2 shows the evaluation of patients with suspected hereditary angioedema.
Hereditary angioedema due to C1 inhibitor deficiency
The classic forms of hereditary angioedema (types I and II) involve loss-of-function mutations in SERPING1—the gene that encodes for C1 inhibitor—resulting in low levels of functional C1 inhibitor.3 The mutation is inherited in an autosomal dominant pattern; however, in about 25% of cases, it appears to arise spontaneously,4 so a family history is not required for diagnosis.
Although C1 inhibitor deficiency is present from birth, the clinical disease most commonly presents for the first time when the patient is of school age. Half of patients have their first episode in the first decade of life, and another one-third first develop symptoms over the next 10 years.5
Clinically, types I and II are indistinguishable. Type I, accounting for 85% of cases,1 results from low production of C1 inhibitor. Laboratory studies reveal low antigenic and functional levels of C1 inhibitor.
In type II, the mutant C1 inhibitor protein is present but dysfunctional and unable to inhibit target proteases. On laboratory testing, the functional level of C1 inhibitor is low but its antigenic level is normal (Table 1). Function can be tested by either chromogenic assay or enzyme-linked immunosorbent assay; the former is preferred because it is more sensitive.6
Because C1 inhibitor deficiency results in chronic activation of the complement system, patients with type I or II disease usually have low C4 levels regardless of disease activity, making measuring C4 the most economical screening test. When suspicion for hereditary angioedema is high, based on the presentation and family and clinical history, measuring antigenic and functional C1 inhibitor levels and C4 simultaneously is more efficient.
Hereditary angioedema with normal C1 inhibitor levels
Hereditary angioedema with normal C1 inhibitor levels is also inherited in an autosomal dominant pattern. It is often estrogen-sensitive, making it more severe in women. Symptoms tend to develop slightly later in life than in type I or II disease.7
Angioedema with normal C1 inhibitor levels has been associated with factor XII mutations in a minority of cases, but most patients do not have a specific laboratory abnormality. Because there is no specific laboratory profile, the diagnosis is based on clinical criteria. Hereditary angioedema with normal C1 inhibitor levels should be considered in patients who have recurrent angioedema, normal C4, normal antigenic and functional C1 inhibitor levels, a lack of response to high-dose antihistamines, and either a family history of angioedema without hives or a known factor XII mutation.7 However, other forms of angioedema (allergic, drug-induced, and idiopathic) should also be considered, as C4 and C1 inhibitor levels are normal in these forms as well.
DIFFERENTIAL DIAGNOSIS: OTHER TYPES OF ANGIOEDEMA
Acquired C1 inhibitor deficiency
Symptoms of acquired C1 inhibitor deficiency resemble those of hereditary angioedema but typically do not emerge until the fourth decade of life or later, and patients have no family history of the condition. It is often associated with other diseases, most commonly B-cell lymphoproliferative disorders, which cause uncontrolled complement activation and consumption of C1 inhibitor.
In some patients, autoantibodies to C1 inhibitor develop, greatly reducing its effectiveness and resulting in enhanced consumption. The autoantibody is often associated with a monoclonal gammopathy of unknown significance. The presence of a C1 inhibitor autoantibody does not preclude the presence of an underlying disorder, and vice versa.
Laboratory studies reveal low C4, low C1-inhibitor antigenic and functional levels, and usually a low C1q level owing to consumption of complement. Autoantibodies to C1 inhibitor can be detected by laboratory testing.
Because of the association with autoimmune disease and malignant disorders (especially B-cell malignancy), a patient diagnosed with acquired C1 inhibitor deficiency should be further evaluated for underlying conditions.
Allergic angioedema
Allergic angioedema results from preformed antigen-specific immunoglobulin E (IgE) antibodies that stimulate mast cells to degranulate when patients are exposed to a particular allergen—most commonly food, insect venom, latex, or drugs. IgE-mediated histamine release causes swelling, as histamine is a potent vasodilator.
Symptoms often begin within 2 hours of exposure to the allergen and generally include concurrent urticaria and swelling that last less than 24 hours. Unlike in hereditary angioedema, the swelling responds to antihistamines and corticosteroids. When very severe, these symptoms may also be accompanied by bronchoconstriction and gastrointestinal symptoms, especially if the allergen is ingested.
Histamine-mediated angioedema may also be associated with exercise as part of a syndrome called exercise-induced anaphylaxis or angioedema.
Drug-induced angioedema
Drug-induced angioedema is typically associated with angiotensin-converting enzyme (ACE) inhibitors or nonsteroidal anti-inflammatory drugs (NSAIDs).
Angioedema associated with ACE inhibitors is estimated to affect 0.1% to 6% of patients taking these medications, with African Americans being at significantly higher risk. Although 25% of affected patients develop symptoms of angioedema within the first month of taking the drugs, some tolerate them for as long as 10 years before the first episode.9 The swelling is not allergic or histamine-related. ACE normally degrades bradykinin; therefore, inhibiting ACE leads to accumulation of bradykinin. Because all ACE inhibitors have this effect, this class of drug should be discontinued in any patient who develops isolated angioedema.
NSAID-induced angioedema is often accompanied by other symptoms, including urticaria, rhinitis, cough, hoarseness, or breathlessness.10 The mechanism of NSAID-induced angioedema involves cyclooxygenase (COX) 1 (and to a lesser extent COX-2) inhibition. All NSAIDs (and aspirin) should be avoided in patients with recurrent angioedema. Specific COX-2 inhibitors, while theoretically capable of causing angioedema by the same mechanism, are generally well tolerated in patients who have had COX-1 inhibitor reactions.
Idiopathic angioedema
If no clear cause of recurrent angioedema (at least three episodes in a year) can be found, it is labeled idiopathic.11 Some patients with idiopathic angioedema fail to benefit from high doses of antihistamines, suggesting that the cause is bradykinin-mediated.
CLINICAL MANIFESTATIONS OF HEREDITARY ANGIOEDEMA
Attacks may start at one site and progress to involve additional sites.
Prodromal symptoms may begin up to several days before an attack and include tingling, warmth, burning, or itching at the affected site; increased fatigue or malaise; nausea, abdominal distention, or gassiness; or increased hunger, particularly before an abdominal attack.5 The most characteristic prodromal symptom is erythema marginatum—a raised, serpiginous, nonpruritic rash on the trunk, arms, and legs but often sparing the face.
Abdominal attacks are easily confused with acute abdomen
Almost half of attacks involve the abdomen, and almost all patients with type I or II disease experience at least one such attack.12 Symptoms can include severe abdominal pain, nausea, vomiting, and diarrhea. Abdominal attacks account for many emergency department visits, hospitalizations, and surgical procedures for acute abdomen; about one-third of patients with undiagnosed hereditary angioedema undergo an unnecessary surgery during an abdominal attack. Angioedema of the gastrointestinal tract can result in enough plasma extravasation and vasodilation to cause hypovolemic shock.
Eradicating Helicobacter pylori infection may alleviate abdominal attacks.13
Attacks of the extremities can be painful and disabling
Attacks of the extremities affect 96% of patients12 and can be very disfiguring and disabling. Driving or using the phone is often difficult when the hands are affected. When feet are involved, walking and standing become painful. While these symptoms rarely result in a lengthy hospitalization, they interfere with work and school and require immediate medical attention because they can progress to other parts of the body.
Laryngeal attacks are life-threatening
About half of patients with hereditary angioedema have an attack of laryngeal edema at some point in their lives.12 If not effectively managed, laryngeal angioedema can progress to asphyxiation. A survey of family history in 58 patients with hereditary angioedema suggested a 40% incidence of asphyxiation in untreated laryngeal attacks, and 25% to 30% of patients are estimated to have died of laryngeal edema before effective treatment became available.14
Symptoms of a laryngeal attack include change in voice, hoarseness, trouble swallowing, shortness of breath, and wheezing. Physicians must recognize these symptoms quickly and give effective treatment early in the attack to prevent morbidity and death.
Establishing an airway can be life-saving in the absence of effective therapy, but extensive swelling of the upper airway can make intubation extremely difficult.
Genitourinary attacks also occur
Attacks involving the scrotum and labia have been reported in up to two-thirds of patients with hereditary angioedema at some point in their lives. Attacks involving the bladder and kidneys have also been reported but are less common, affecting about 5% of patients.12 Genitourinary attacks may be triggered by local trauma, such as horseback riding or sexual intercourse, although no trigger may be evident.
MANAGING ACUTE ATTACKS
The goals of treatment are to alleviate acute exacerbations with on-demand treatment and to reduce the number of attacks with prophylaxis. Therapy should be individualized to each patient’s needs. Treatments have advanced greatly in the last several years, and new medications for treating acute attacks and preventing attacks have shown great promise (Figure 3, Table 2).
Patients tend to have recurrent symptoms interspersed with periods of health, suggesting that attacks ought to have identifiable triggers, although in most, no trigger is evident. The most commonly identified are local trauma (including medical and dental procedures), emotional stress, and acute infection. Disease severity may be worsened by menstruation, estrogen-containing oral contraceptives, hormone replacement therapy, ACE inhibitors, and NSAIDs.
It is critical that attacks be treated with an effective medication as soon as possible. Consensus guidelines state that all patients with hereditary angioedema due to C1 inhibitor deficiency, even if they are still asymptomatic, should have access to at least one of the drugs approved for on-demand treatment.15 The guidelines further state that whenever possible, “patients should have the on-demand medicine to treat acute attacks at home and should be trained to self-administer these medicines.”15
Plasma-derived C1 inhibitors
Several plasma-derived C1 inhibitors are available (Cinryze, Berinert, Cetor). They are prepared from fractionated plasma obtained from donors, then pasteurized and nanofiltered.
Berinert and Cinryze were each found to be superior to placebo in double-blind, placebo-controlled trials: attacks usually resolved 30 to 60 minutes after intravenous injection.16,17 Berinert 20 U/kg is associated with the onset of symptom relief as early as half an hour after administration, compared with 1.5 hours with placebo. Early use (at the onset of symptoms) of a plasma-derived C1 inhibitor in a low dose (500 U) can also be effective.18,19 Efficacy appears to be consistent at all sites of attack involvement, including laryngeal edema. Safety and efficacy have been demonstrated during pregnancy and lactation and in young children and babies.20
Plasma-derived C1 inhibitors can be self-administered. The safety and efficacy of self-administration (under physician supervision) were demonstrated in a study of Cinryze and Cetor, in which attack duration, pain medication use, and graded attack severity were significantly less with self-administered therapy than with therapy in the clinic.21
A concern about plasma-derived products is the possibility of blood-borne infection, but this has not been confirmed by experience.22
Recombinant human C1 inhibitor
A recombinant human C1 inhibitor (Rhucin) has been studied in two randomized placebo-controlled trials. Although this product has a shorter half-life than the plasma-derived C1 inhibitors (3 vs more than 24 hours), the two are equipotent: 1 U of recombinant human C1 inhibitor is equivalent to 1 U of plasma-derived C1 inhibitor. Because the supply of recombinant human C1 inhibitor is elastic, dosing has been higher, which may provide more efficacy.23 Similar to plasma-derived C1 inhibitor products, the recombinant human C1 inhibitor resulted in more rapid symptom relief than with saline (66 vs 122 minutes) and in a shorter time to minimal symptoms (247−266 vs 1,210 minutes).24
Allergy is of concern: in one study, a healthy volunteer with undisclosed rabbit allergy experienced an allergic reaction. Patients should be screened by a skin-prick test or serum testing for specific IgE to rabbit epithelium before being prescribed recombinant human C1 inhibitor. No data are available for use during pregnancy or breastfeeding.
Ecallantide
Ecallantide (Kalbitor) is a selective inhibitor of plasma kallikrein that is given in three subcutaneous injections. Ecallantide 30 mg was found superior to placebo during acute attacks.25,26
Ecallantide is well tolerated, with the most common adverse effects being headache, nausea, fatigue, diarrhea, and local injection-site reactions. Antibodies to ecallantide can be found in patients with increasing drug exposure but do not appear to correlate with adverse events. Hypersensitivity reactions have been observed in 2% to 3% of patients receiving repeated doses. Because of anaphylaxis risk, ecallantide must be administered by a health care professional.
Icatibant
Icatibant (Firazyr) is a bradykinin receptor-2 antagonist that is given in a single subcutaneous injection. Icatibant 30 mg significantly shortened time to symptom relief and time to almost complete resolution compared with placebo.27,28 Icatibant’s main adverse effect is transient local pain, swelling, and erythema at the injection site. Icatibant can be self-administered by patients.
Fresh-frozen plasma
Fresh-frozen plasma contains C1 inhibitor and was used before the newer products became available. Several noncontrolled studies reported benefit of its use in acute attacks.29 However, its use is controversial because it also contains contact-system proteins that could provide additional substrate for the generation of bradykinin, which could exacerbate attacks in some patients.1 This may be particularly dangerous in patients presenting with laryngeal edema: in such a situation, the physician should be ready to treat a sudden exacerbation with intubation. The risk of acquiring a blood-borne pathogen is also higher than with plasma-derived C1 inhibitor.
PROPHYLACTIC MANAGEMENT
Short-term and long-term prophylaxis have important roles in preventing attacks (Table 3).
Short-term prophylaxis before an anticipated attack
Short-term prophylaxis is used for patients whose disease is generally well controlled but who anticipate exposure to a potentially exacerbating situation, such as an invasive medical, surgical, or dental procedure. (Routine dental cleanings are generally considered safe and do not require prophylaxis.)
Prophylactic treatments include:
- Plasma-derived C1 inhibitor, 500 to 1,500 U 1 hour before the provoking event
- High-dose 17-alpha alkylated (attenuated) androgens (eg, danazol [Danocrine] 200 mg orally 3 times daily) for 5 to 10 days before the provoking event
- Fresh-frozen plasma, 2 U 1 to 12 hours before the event.1
Yet even with short-term prophylaxis, on-demand treatment should be available.
Long-term prophylaxis
While many patients can be managed with on-demand treatment only, other patients (reflecting the severity of their attacks, as well as their individual needs) may benefit from a combination of on-demand treatment plus long-term prophylaxis. Several options are available (Table 3).
17-alpha alkylated androgens. Patients treated with danazol 600 mg/day were attack-free 90% of the time during a 28-day period compared with only 2.2% of the time in placebo-treated patients.30 Use of anabolic androgens, however, is limited by their adverse effects, including weight gain, virilization, menstrual irregularities, headaches, depression, dyslipidemia, liver enzyme elevation, liver adenomas, and hepatocellular carcinoma. Arterial hypertension occurs in about 25% of treated patients.
Because adverse effects are dose-dependent, treatment should be empirically titrated to find the minimal effective dose, generally recommended to be no more than 200 mg per day of danazol or the equivalent.15
Contraindications include use by women during pregnancy or lactation and by children until growth is complete.
Regular follow-up is recommended every 6 months, with monitoring of liver enzymes, lipids, complete blood counts, alpha fetoprotein, and urinalysis. Abdominal ultrasonography (every 6 months if receiving 100 mg/day or more of danazol, every 12 months if less than 100 mg/day) is advisable for early diagnosis of liver tumors.
Antifibrinolytic drugs. Tranexamic acid (Lysteda) and aminocaproic acid (Amicar) have been found to be effective in reducing the number of attacks of hereditary angioedema compared with placebo but are considered to be less reliable than androgens. These drugs have been used in patients who do not tolerate anabolic androgens, and in children and pregnant women. Tranexamic acid is given at a dose of 20 to 50 mg/kg/day divided into two or three doses per day. The therapeutic dose of aminocaproic acid is 1 g orally three to four times per day.31 Patients with a personal or family history of thromboembolic disease may be at greater risk of venous or arterial thrombosis, but this has not occurred in clinical studies.
Plasma-derived C1 inhibitors. In a 24-week crossover study in 22 patients with hereditary angioedema, Cinryze 1,000 U every 3 to 4 days reduced the rate of attacks by 50% while also reducing their severity and duration.17 An open-label extension study in 146 patients for almost 3 years documented a 90% reduction in attack frequency with no evidence of tachyphylaxis.32
New treatments are costlier
The newer on-demand and prophylactic drugs are substantially costlier than the older alternatives (androgens, antifibrinolytics, and fresh-frozen plasma); however, they have a substantially better benefit-to-risk ratio. Furthermore, the costs of care for an attack requiring emergency treatment are also high. Hereditary angioedema patients are often young, otherwise healthy, and capable of leading normal productive lives. While formal pharmacoeconomic studies of the optimal use of these newer drugs have not yet been done, it is important that the use of these drugs be well justified. Ideally, physicians who prescribe these drugs should be knowledgeable in the management of hereditary angioedema.
SPECIAL CHALLENGES IN WOMEN
Women with hereditary angioedema have more frequent attacks and generally a more severe disease course than men.12 Optimizing care for women is challenging because hormonal changes often cause the disease to flare up in menarche, pregnancy, lactation, and menopause. Women also have a higher rate of discontinuing long-term androgen therapy because of side effects, including virilization and menstrual irregularities. Spironolactone (Aldactone) 100 to 200 mg daily can be used to control hirsutism.33
Contraception
Because estrogen can trigger attacks, progesterone-only formulations, intrauterine devices, or barrier methods are recommended for contraception.33 Progesterone-only pills are preferred and improve symptoms in more than 60% of women. Etonogestrel, another alternative, is available as an implant (Implanon) or vaginal ring (Nuvaring). Intrauterine devices are generally well tolerated, and no prophylaxis is needed during placement. The progesterone-eluting intrauterine device (Mirena) could be beneficial.34
Pregnancy and lactation
Pregnancy and lactation pose particular challenges. Anabolic androgens are contraindicated during pregnancy as well as during breastfeeding because they can be passed on in breast milk. Women receiving androgen prophylaxis should understand that they can still ovulate and need contraception if they are sexually active.34 Patients on attenuated androgens who desire pregnancy should discontinue them 2 months before trying to conceive.
Changes in attack patterns can be unpredictable during pregnancy. Attacks tend to be more severe during the first trimester and more frequent during the third. Due to its safety and efficacy, plasma-derived C1 inhibitor has become the treatment of choice for on-demand or prophylactic treatment during pregnancy and lactation. Antifibrinolytics are considered only when plasma-derived C1 inhibitor is not available.31 Ecallantide and icatibant have not been studied in pregnancy. If neither plasma-derived C1 inhibitor nor antifibrinolytics are available, fresh-frozen plasma or solvent-and-detergent-treated plasma can be used.
Short-term prophylaxis should be considered before amniocentesis, chorionic villous sampling, and dilation and curettage. Delivery should take place in a facility with rapid access to plasma-derived C1 inhibitor as well as consultants in obstetrics, anesthesiology, and perinatology. Although plasma-derived C1 inhibitor should be available at all times during labor and delivery, its prophylactic use is not required unless labor and delivery are particularly traumatic, the underlying hereditary angioedema is very severe, or if forceps, vacuum delivery, or cesarian section is performed. Close monitoring is recommended for at least 72 hours after routine vaginal delivery and for 1 week after cesarian section.
CONCLUSION
The goals of hereditary angioedema treatment are to alleviate morbidity and mortality associated with the disease and to improve the patient’s quality of life. Achieving these goals requires timely diagnosis, patient education, and careful selection of therapeutic modalities that are individualized to the needs of that patient. Treatments have advanced greatly in the last 4 years, and new medications for both the acute and chronic symptoms of hereditary angioedema have shown great promise.
Acknowledgment: K.T. is funded by National Institutes of Health grant T32 AI 07469.
Hereditary angioedema due to deficiency of C1 inhibitor is a rare autosomal dominant disease that can be life-threatening. It affects about 1 in 50,000 people,1 or about 6,000 people in the United States. There are no known differences in prevalence by ethnicity or sex. A form of hereditary angioedema with normal C1 inhibitor levels has also recently been identified.
Despite a growing awareness of hereditary angioedema in the medical community, repeated surveys have found an average gap of 10 years between the first appearance of symptoms and the correct diagnosis. In view of the risk of morbidity and death, recognizing this disease sooner is critical.
This article will discuss how to recognize hereditary angioedema and how to differentiate it from other forms of recurring angioedema. We will also review its acute and long-term management, with special attention to new therapies and clinical challenges.
EPISODES OF SWELLING WITHOUT HIVES
Hereditary angioedema involves recurrent episodes of nonpruritic, nonpitting, subcutaneous and submucosal edema that can affect the face, tongue, larynx, trunk, extremities, bowels, or genitals. Attacks typically follow a predictable course: swelling that increases slowly and continuously for 24 hours and then gradually subsides over the next 48 to 72 hours. Attacks that involve the oropharynx, larynx, or abdomen carry the highest risk of morbidity and death.1
The frequency and severity of attacks are highly variable and unpredictable. A few patients have no attacks, a few have two attacks per week, and most fall in between.
Hives suggests an allergic or idiopathic rather than hereditary cause and will not be discussed here in detail. A history of angioedema that was rapidly aborted by antihistamines, corticosteroids, or epinephrine also suggests an allergic rather than hereditary cause.
UNCHECKED BRADYKININ PRODUCTION
Substantial evidence indicates that hereditary angioedema results from extravasation of plasma into deeper cutaneous or mucosal compartments as a result of overproduction of the vasoactive mediator bradykinin (Figure 1).
Activated factor XII cleaves plasma prekallikrein to generate active plasma kallikrein (which, in turn, activates more factor XII).2 Once generated, plasma kallikrein cleaves high-molecular-weight kininogen, releasing bradykinin. Bradykinin binds to the B2 bradykinin receptor on endothelial cells, increasing the permeability of the endothelium.
Normally, C1 inhibitor helps control bradykinin production by inhibiting plasma kallikrein and activated factor XII. Without enough C1 inhibitor, the contact system is uninhibited and results in bradykinin being inappropriately generated.
Because the attacks of hereditary angioedema involve excessive bradykinin, they do not respond to the usual treatments for anaphylaxis and allergic angioedema (which involve mast cell degranulation), such as antihistamines, corticosteroids, and epinephrine.
TWO TYPES OF HEREDITARY ANGIOEDEMA
Figure 2 shows the evaluation of patients with suspected hereditary angioedema.
Hereditary angioedema due to C1 inhibitor deficiency
The classic forms of hereditary angioedema (types I and II) involve loss-of-function mutations in SERPING1—the gene that encodes for C1 inhibitor—resulting in low levels of functional C1 inhibitor.3 The mutation is inherited in an autosomal dominant pattern; however, in about 25% of cases, it appears to arise spontaneously,4 so a family history is not required for diagnosis.
Although C1 inhibitor deficiency is present from birth, the clinical disease most commonly presents for the first time when the patient is of school age. Half of patients have their first episode in the first decade of life, and another one-third first develop symptoms over the next 10 years.5
Clinically, types I and II are indistinguishable. Type I, accounting for 85% of cases,1 results from low production of C1 inhibitor. Laboratory studies reveal low antigenic and functional levels of C1 inhibitor.
In type II, the mutant C1 inhibitor protein is present but dysfunctional and unable to inhibit target proteases. On laboratory testing, the functional level of C1 inhibitor is low but its antigenic level is normal (Table 1). Function can be tested by either chromogenic assay or enzyme-linked immunosorbent assay; the former is preferred because it is more sensitive.6
Because C1 inhibitor deficiency results in chronic activation of the complement system, patients with type I or II disease usually have low C4 levels regardless of disease activity, making measuring C4 the most economical screening test. When suspicion for hereditary angioedema is high, based on the presentation and family and clinical history, measuring antigenic and functional C1 inhibitor levels and C4 simultaneously is more efficient.
Hereditary angioedema with normal C1 inhibitor levels
Hereditary angioedema with normal C1 inhibitor levels is also inherited in an autosomal dominant pattern. It is often estrogen-sensitive, making it more severe in women. Symptoms tend to develop slightly later in life than in type I or II disease.7
Angioedema with normal C1 inhibitor levels has been associated with factor XII mutations in a minority of cases, but most patients do not have a specific laboratory abnormality. Because there is no specific laboratory profile, the diagnosis is based on clinical criteria. Hereditary angioedema with normal C1 inhibitor levels should be considered in patients who have recurrent angioedema, normal C4, normal antigenic and functional C1 inhibitor levels, a lack of response to high-dose antihistamines, and either a family history of angioedema without hives or a known factor XII mutation.7 However, other forms of angioedema (allergic, drug-induced, and idiopathic) should also be considered, as C4 and C1 inhibitor levels are normal in these forms as well.
DIFFERENTIAL DIAGNOSIS: OTHER TYPES OF ANGIOEDEMA
Acquired C1 inhibitor deficiency
Symptoms of acquired C1 inhibitor deficiency resemble those of hereditary angioedema but typically do not emerge until the fourth decade of life or later, and patients have no family history of the condition. It is often associated with other diseases, most commonly B-cell lymphoproliferative disorders, which cause uncontrolled complement activation and consumption of C1 inhibitor.
In some patients, autoantibodies to C1 inhibitor develop, greatly reducing its effectiveness and resulting in enhanced consumption. The autoantibody is often associated with a monoclonal gammopathy of unknown significance. The presence of a C1 inhibitor autoantibody does not preclude the presence of an underlying disorder, and vice versa.
Laboratory studies reveal low C4, low C1-inhibitor antigenic and functional levels, and usually a low C1q level owing to consumption of complement. Autoantibodies to C1 inhibitor can be detected by laboratory testing.
Because of the association with autoimmune disease and malignant disorders (especially B-cell malignancy), a patient diagnosed with acquired C1 inhibitor deficiency should be further evaluated for underlying conditions.
Allergic angioedema
Allergic angioedema results from preformed antigen-specific immunoglobulin E (IgE) antibodies that stimulate mast cells to degranulate when patients are exposed to a particular allergen—most commonly food, insect venom, latex, or drugs. IgE-mediated histamine release causes swelling, as histamine is a potent vasodilator.
Symptoms often begin within 2 hours of exposure to the allergen and generally include concurrent urticaria and swelling that last less than 24 hours. Unlike in hereditary angioedema, the swelling responds to antihistamines and corticosteroids. When very severe, these symptoms may also be accompanied by bronchoconstriction and gastrointestinal symptoms, especially if the allergen is ingested.
Histamine-mediated angioedema may also be associated with exercise as part of a syndrome called exercise-induced anaphylaxis or angioedema.
Drug-induced angioedema
Drug-induced angioedema is typically associated with angiotensin-converting enzyme (ACE) inhibitors or nonsteroidal anti-inflammatory drugs (NSAIDs).
Angioedema associated with ACE inhibitors is estimated to affect 0.1% to 6% of patients taking these medications, with African Americans being at significantly higher risk. Although 25% of affected patients develop symptoms of angioedema within the first month of taking the drugs, some tolerate them for as long as 10 years before the first episode.9 The swelling is not allergic or histamine-related. ACE normally degrades bradykinin; therefore, inhibiting ACE leads to accumulation of bradykinin. Because all ACE inhibitors have this effect, this class of drug should be discontinued in any patient who develops isolated angioedema.
NSAID-induced angioedema is often accompanied by other symptoms, including urticaria, rhinitis, cough, hoarseness, or breathlessness.10 The mechanism of NSAID-induced angioedema involves cyclooxygenase (COX) 1 (and to a lesser extent COX-2) inhibition. All NSAIDs (and aspirin) should be avoided in patients with recurrent angioedema. Specific COX-2 inhibitors, while theoretically capable of causing angioedema by the same mechanism, are generally well tolerated in patients who have had COX-1 inhibitor reactions.
Idiopathic angioedema
If no clear cause of recurrent angioedema (at least three episodes in a year) can be found, it is labeled idiopathic.11 Some patients with idiopathic angioedema fail to benefit from high doses of antihistamines, suggesting that the cause is bradykinin-mediated.
CLINICAL MANIFESTATIONS OF HEREDITARY ANGIOEDEMA
Attacks may start at one site and progress to involve additional sites.
Prodromal symptoms may begin up to several days before an attack and include tingling, warmth, burning, or itching at the affected site; increased fatigue or malaise; nausea, abdominal distention, or gassiness; or increased hunger, particularly before an abdominal attack.5 The most characteristic prodromal symptom is erythema marginatum—a raised, serpiginous, nonpruritic rash on the trunk, arms, and legs but often sparing the face.
Abdominal attacks are easily confused with acute abdomen
Almost half of attacks involve the abdomen, and almost all patients with type I or II disease experience at least one such attack.12 Symptoms can include severe abdominal pain, nausea, vomiting, and diarrhea. Abdominal attacks account for many emergency department visits, hospitalizations, and surgical procedures for acute abdomen; about one-third of patients with undiagnosed hereditary angioedema undergo an unnecessary surgery during an abdominal attack. Angioedema of the gastrointestinal tract can result in enough plasma extravasation and vasodilation to cause hypovolemic shock.
Eradicating Helicobacter pylori infection may alleviate abdominal attacks.13
Attacks of the extremities can be painful and disabling
Attacks of the extremities affect 96% of patients12 and can be very disfiguring and disabling. Driving or using the phone is often difficult when the hands are affected. When feet are involved, walking and standing become painful. While these symptoms rarely result in a lengthy hospitalization, they interfere with work and school and require immediate medical attention because they can progress to other parts of the body.
Laryngeal attacks are life-threatening
About half of patients with hereditary angioedema have an attack of laryngeal edema at some point in their lives.12 If not effectively managed, laryngeal angioedema can progress to asphyxiation. A survey of family history in 58 patients with hereditary angioedema suggested a 40% incidence of asphyxiation in untreated laryngeal attacks, and 25% to 30% of patients are estimated to have died of laryngeal edema before effective treatment became available.14
Symptoms of a laryngeal attack include change in voice, hoarseness, trouble swallowing, shortness of breath, and wheezing. Physicians must recognize these symptoms quickly and give effective treatment early in the attack to prevent morbidity and death.
Establishing an airway can be life-saving in the absence of effective therapy, but extensive swelling of the upper airway can make intubation extremely difficult.
Genitourinary attacks also occur
Attacks involving the scrotum and labia have been reported in up to two-thirds of patients with hereditary angioedema at some point in their lives. Attacks involving the bladder and kidneys have also been reported but are less common, affecting about 5% of patients.12 Genitourinary attacks may be triggered by local trauma, such as horseback riding or sexual intercourse, although no trigger may be evident.
MANAGING ACUTE ATTACKS
The goals of treatment are to alleviate acute exacerbations with on-demand treatment and to reduce the number of attacks with prophylaxis. Therapy should be individualized to each patient’s needs. Treatments have advanced greatly in the last several years, and new medications for treating acute attacks and preventing attacks have shown great promise (Figure 3, Table 2).
Patients tend to have recurrent symptoms interspersed with periods of health, suggesting that attacks ought to have identifiable triggers, although in most, no trigger is evident. The most commonly identified are local trauma (including medical and dental procedures), emotional stress, and acute infection. Disease severity may be worsened by menstruation, estrogen-containing oral contraceptives, hormone replacement therapy, ACE inhibitors, and NSAIDs.
It is critical that attacks be treated with an effective medication as soon as possible. Consensus guidelines state that all patients with hereditary angioedema due to C1 inhibitor deficiency, even if they are still asymptomatic, should have access to at least one of the drugs approved for on-demand treatment.15 The guidelines further state that whenever possible, “patients should have the on-demand medicine to treat acute attacks at home and should be trained to self-administer these medicines.”15
Plasma-derived C1 inhibitors
Several plasma-derived C1 inhibitors are available (Cinryze, Berinert, Cetor). They are prepared from fractionated plasma obtained from donors, then pasteurized and nanofiltered.
Berinert and Cinryze were each found to be superior to placebo in double-blind, placebo-controlled trials: attacks usually resolved 30 to 60 minutes after intravenous injection.16,17 Berinert 20 U/kg is associated with the onset of symptom relief as early as half an hour after administration, compared with 1.5 hours with placebo. Early use (at the onset of symptoms) of a plasma-derived C1 inhibitor in a low dose (500 U) can also be effective.18,19 Efficacy appears to be consistent at all sites of attack involvement, including laryngeal edema. Safety and efficacy have been demonstrated during pregnancy and lactation and in young children and babies.20
Plasma-derived C1 inhibitors can be self-administered. The safety and efficacy of self-administration (under physician supervision) were demonstrated in a study of Cinryze and Cetor, in which attack duration, pain medication use, and graded attack severity were significantly less with self-administered therapy than with therapy in the clinic.21
A concern about plasma-derived products is the possibility of blood-borne infection, but this has not been confirmed by experience.22
Recombinant human C1 inhibitor
A recombinant human C1 inhibitor (Rhucin) has been studied in two randomized placebo-controlled trials. Although this product has a shorter half-life than the plasma-derived C1 inhibitors (3 vs more than 24 hours), the two are equipotent: 1 U of recombinant human C1 inhibitor is equivalent to 1 U of plasma-derived C1 inhibitor. Because the supply of recombinant human C1 inhibitor is elastic, dosing has been higher, which may provide more efficacy.23 Similar to plasma-derived C1 inhibitor products, the recombinant human C1 inhibitor resulted in more rapid symptom relief than with saline (66 vs 122 minutes) and in a shorter time to minimal symptoms (247−266 vs 1,210 minutes).24
Allergy is of concern: in one study, a healthy volunteer with undisclosed rabbit allergy experienced an allergic reaction. Patients should be screened by a skin-prick test or serum testing for specific IgE to rabbit epithelium before being prescribed recombinant human C1 inhibitor. No data are available for use during pregnancy or breastfeeding.
Ecallantide
Ecallantide (Kalbitor) is a selective inhibitor of plasma kallikrein that is given in three subcutaneous injections. Ecallantide 30 mg was found superior to placebo during acute attacks.25,26
Ecallantide is well tolerated, with the most common adverse effects being headache, nausea, fatigue, diarrhea, and local injection-site reactions. Antibodies to ecallantide can be found in patients with increasing drug exposure but do not appear to correlate with adverse events. Hypersensitivity reactions have been observed in 2% to 3% of patients receiving repeated doses. Because of anaphylaxis risk, ecallantide must be administered by a health care professional.
Icatibant
Icatibant (Firazyr) is a bradykinin receptor-2 antagonist that is given in a single subcutaneous injection. Icatibant 30 mg significantly shortened time to symptom relief and time to almost complete resolution compared with placebo.27,28 Icatibant’s main adverse effect is transient local pain, swelling, and erythema at the injection site. Icatibant can be self-administered by patients.
Fresh-frozen plasma
Fresh-frozen plasma contains C1 inhibitor and was used before the newer products became available. Several noncontrolled studies reported benefit of its use in acute attacks.29 However, its use is controversial because it also contains contact-system proteins that could provide additional substrate for the generation of bradykinin, which could exacerbate attacks in some patients.1 This may be particularly dangerous in patients presenting with laryngeal edema: in such a situation, the physician should be ready to treat a sudden exacerbation with intubation. The risk of acquiring a blood-borne pathogen is also higher than with plasma-derived C1 inhibitor.
PROPHYLACTIC MANAGEMENT
Short-term and long-term prophylaxis have important roles in preventing attacks (Table 3).
Short-term prophylaxis before an anticipated attack
Short-term prophylaxis is used for patients whose disease is generally well controlled but who anticipate exposure to a potentially exacerbating situation, such as an invasive medical, surgical, or dental procedure. (Routine dental cleanings are generally considered safe and do not require prophylaxis.)
Prophylactic treatments include:
- Plasma-derived C1 inhibitor, 500 to 1,500 U 1 hour before the provoking event
- High-dose 17-alpha alkylated (attenuated) androgens (eg, danazol [Danocrine] 200 mg orally 3 times daily) for 5 to 10 days before the provoking event
- Fresh-frozen plasma, 2 U 1 to 12 hours before the event.1
Yet even with short-term prophylaxis, on-demand treatment should be available.
Long-term prophylaxis
While many patients can be managed with on-demand treatment only, other patients (reflecting the severity of their attacks, as well as their individual needs) may benefit from a combination of on-demand treatment plus long-term prophylaxis. Several options are available (Table 3).
17-alpha alkylated androgens. Patients treated with danazol 600 mg/day were attack-free 90% of the time during a 28-day period compared with only 2.2% of the time in placebo-treated patients.30 Use of anabolic androgens, however, is limited by their adverse effects, including weight gain, virilization, menstrual irregularities, headaches, depression, dyslipidemia, liver enzyme elevation, liver adenomas, and hepatocellular carcinoma. Arterial hypertension occurs in about 25% of treated patients.
Because adverse effects are dose-dependent, treatment should be empirically titrated to find the minimal effective dose, generally recommended to be no more than 200 mg per day of danazol or the equivalent.15
Contraindications include use by women during pregnancy or lactation and by children until growth is complete.
Regular follow-up is recommended every 6 months, with monitoring of liver enzymes, lipids, complete blood counts, alpha fetoprotein, and urinalysis. Abdominal ultrasonography (every 6 months if receiving 100 mg/day or more of danazol, every 12 months if less than 100 mg/day) is advisable for early diagnosis of liver tumors.
Antifibrinolytic drugs. Tranexamic acid (Lysteda) and aminocaproic acid (Amicar) have been found to be effective in reducing the number of attacks of hereditary angioedema compared with placebo but are considered to be less reliable than androgens. These drugs have been used in patients who do not tolerate anabolic androgens, and in children and pregnant women. Tranexamic acid is given at a dose of 20 to 50 mg/kg/day divided into two or three doses per day. The therapeutic dose of aminocaproic acid is 1 g orally three to four times per day.31 Patients with a personal or family history of thromboembolic disease may be at greater risk of venous or arterial thrombosis, but this has not occurred in clinical studies.
Plasma-derived C1 inhibitors. In a 24-week crossover study in 22 patients with hereditary angioedema, Cinryze 1,000 U every 3 to 4 days reduced the rate of attacks by 50% while also reducing their severity and duration.17 An open-label extension study in 146 patients for almost 3 years documented a 90% reduction in attack frequency with no evidence of tachyphylaxis.32
New treatments are costlier
The newer on-demand and prophylactic drugs are substantially costlier than the older alternatives (androgens, antifibrinolytics, and fresh-frozen plasma); however, they have a substantially better benefit-to-risk ratio. Furthermore, the costs of care for an attack requiring emergency treatment are also high. Hereditary angioedema patients are often young, otherwise healthy, and capable of leading normal productive lives. While formal pharmacoeconomic studies of the optimal use of these newer drugs have not yet been done, it is important that the use of these drugs be well justified. Ideally, physicians who prescribe these drugs should be knowledgeable in the management of hereditary angioedema.
SPECIAL CHALLENGES IN WOMEN
Women with hereditary angioedema have more frequent attacks and generally a more severe disease course than men.12 Optimizing care for women is challenging because hormonal changes often cause the disease to flare up in menarche, pregnancy, lactation, and menopause. Women also have a higher rate of discontinuing long-term androgen therapy because of side effects, including virilization and menstrual irregularities. Spironolactone (Aldactone) 100 to 200 mg daily can be used to control hirsutism.33
Contraception
Because estrogen can trigger attacks, progesterone-only formulations, intrauterine devices, or barrier methods are recommended for contraception.33 Progesterone-only pills are preferred and improve symptoms in more than 60% of women. Etonogestrel, another alternative, is available as an implant (Implanon) or vaginal ring (Nuvaring). Intrauterine devices are generally well tolerated, and no prophylaxis is needed during placement. The progesterone-eluting intrauterine device (Mirena) could be beneficial.34
Pregnancy and lactation
Pregnancy and lactation pose particular challenges. Anabolic androgens are contraindicated during pregnancy as well as during breastfeeding because they can be passed on in breast milk. Women receiving androgen prophylaxis should understand that they can still ovulate and need contraception if they are sexually active.34 Patients on attenuated androgens who desire pregnancy should discontinue them 2 months before trying to conceive.
Changes in attack patterns can be unpredictable during pregnancy. Attacks tend to be more severe during the first trimester and more frequent during the third. Due to its safety and efficacy, plasma-derived C1 inhibitor has become the treatment of choice for on-demand or prophylactic treatment during pregnancy and lactation. Antifibrinolytics are considered only when plasma-derived C1 inhibitor is not available.31 Ecallantide and icatibant have not been studied in pregnancy. If neither plasma-derived C1 inhibitor nor antifibrinolytics are available, fresh-frozen plasma or solvent-and-detergent-treated plasma can be used.
Short-term prophylaxis should be considered before amniocentesis, chorionic villous sampling, and dilation and curettage. Delivery should take place in a facility with rapid access to plasma-derived C1 inhibitor as well as consultants in obstetrics, anesthesiology, and perinatology. Although plasma-derived C1 inhibitor should be available at all times during labor and delivery, its prophylactic use is not required unless labor and delivery are particularly traumatic, the underlying hereditary angioedema is very severe, or if forceps, vacuum delivery, or cesarian section is performed. Close monitoring is recommended for at least 72 hours after routine vaginal delivery and for 1 week after cesarian section.
CONCLUSION
The goals of hereditary angioedema treatment are to alleviate morbidity and mortality associated with the disease and to improve the patient’s quality of life. Achieving these goals requires timely diagnosis, patient education, and careful selection of therapeutic modalities that are individualized to the needs of that patient. Treatments have advanced greatly in the last 4 years, and new medications for both the acute and chronic symptoms of hereditary angioedema have shown great promise.
Acknowledgment: K.T. is funded by National Institutes of Health grant T32 AI 07469.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359:1027–1036.
- Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol 2010; 126:918–925.
- Davis AE. C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988; 6:595–628.
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106:1147–1154.
- Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am 2006; 26:739–751.
- Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods 2008; 338:14–20.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol 2010; 6:15.
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 2012; 33:S145–S156.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Busse PJ. Angioedema: differential diagnosis and treatment. Allergy Asthma Proc 2011; 32(suppl 1):S3–S11.
- Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc 2009; 30:506–511.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119:267–274.
- Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 2001; 358:1695–1696.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 2001; 161:714–718.
- Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–157.
- Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009; 124:801–808.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010; 363:513–522.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005; 45:1774–1784.
- Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion 2009; 49:1987–1995.
- Farkas H, Varga L, Széplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics 2007; 120:e713–e722.
- Tourangeau LM, Castaldo AJ, Davis DK, Koziol J, Christiansen SC, Zuraw BL. Safety and efficacy of physician-supervised self-managed C1 inhibitor replacement therapy. Int Arch Allergy Immunol 2012; 157:417–424.
- De Serres J, Gröner A, Lindner J. Safety and efficacy of pasteurized C1 inhibitor concentrate (Berinert P) in hereditary angioedema: a review. Transfus Apher Sci 2003; 29:247–254.
- Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012; 67:123–130.
- Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010; 126:821–827.e14.
- Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010; 363:523–531.
- Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 2010; 104:523–529.
- Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010; 363:532–541.
- Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol 2011; 107:529–537.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98:383–388.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med 1976; 295:1444–1448.
- Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol 2010; 6:23.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med 2012: Epub ahead of print.
- Caballero T, Farkas H, Bouillet L, et al; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129:308–320.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol 2008; 199:484.e1–e4.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359:1027–1036.
- Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol 2010; 126:918–925.
- Davis AE. C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988; 6:595–628.
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106:1147–1154.
- Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am 2006; 26:739–751.
- Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods 2008; 338:14–20.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol 2010; 6:15.
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 2012; 33:S145–S156.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Busse PJ. Angioedema: differential diagnosis and treatment. Allergy Asthma Proc 2011; 32(suppl 1):S3–S11.
- Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc 2009; 30:506–511.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119:267–274.
- Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 2001; 358:1695–1696.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 2001; 161:714–718.
- Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–157.
- Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009; 124:801–808.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010; 363:513–522.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005; 45:1774–1784.
- Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion 2009; 49:1987–1995.
- Farkas H, Varga L, Széplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics 2007; 120:e713–e722.
- Tourangeau LM, Castaldo AJ, Davis DK, Koziol J, Christiansen SC, Zuraw BL. Safety and efficacy of physician-supervised self-managed C1 inhibitor replacement therapy. Int Arch Allergy Immunol 2012; 157:417–424.
- De Serres J, Gröner A, Lindner J. Safety and efficacy of pasteurized C1 inhibitor concentrate (Berinert P) in hereditary angioedema: a review. Transfus Apher Sci 2003; 29:247–254.
- Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012; 67:123–130.
- Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010; 126:821–827.e14.
- Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010; 363:523–531.
- Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 2010; 104:523–529.
- Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010; 363:532–541.
- Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol 2011; 107:529–537.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98:383–388.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med 1976; 295:1444–1448.
- Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol 2010; 6:23.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med 2012: Epub ahead of print.
- Caballero T, Farkas H, Bouillet L, et al; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129:308–320.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol 2008; 199:484.e1–e4.
KEY POINTS
- Swelling in the airways is life-threatening and requires rapid treatment.
- Almost half of attacks involve the abdomen, and abdominal attacks account for many emergency department visits, hospitalizations, and unnecessary surgical procedures for acute abdomen.
- Acute attacks can be managed with plasma-derived or recombinant human preparations of C1 inhibitor (which is the deficient factor in this condition), ecallantide (a specific plasma kallikrein inhibitor), or icatibant (a B2 bradykinin receptor antagonist).
- Short-term prophylaxis may be used before events that could provoke attacks (eg, dental work or surgery). Long-term prophylaxis may be used in patients who have frequent or severe attacks or require more stringent control of their disease. Plasma-derived C1 inhibitor is both safe and effective when used as prophylaxis. Attenuated androgens are effective but associated with many adverse effects.
Child’s brain damage blamed on late cesarean … and more
A MOTHER WANTED A HOME BIRTH with a midwife. When complications arose and labor stopped progressing, the midwife called an ambulance. The emergency department (ED) physician ordered an urgent cesarean delivery, but the procedure did not begin for another 2 hours. The child was born with brain damage, multiple physical and mental disabilities, complex seizure disorder, and cerebral palsy.
PARENTS’ CLAIM The child’s injuries occurred because cesarean delivery was delayed for 2 hours. Based on fetal heart-rate monitoring, the injuries most likely occurred in the last 18 minutes before birth, and were probably caused by compression of the umbilical cord. An earlier cesarean delivery would have avoided the injuries.
DEFENDANTS’ DEFENSE All of the injuries occurred prior to the mother’s arrival at the hospital, while she was under the care of the midwife. Fetal distress was present for an hour before the ambulance was called. When the mother arrived at the ED, she was an unknown patient, as the midwife did not have a collaborating physician. While the ED physician determined that a cesarean delivery was required, it was not considered an emergency. The mother was taken to the OR as soon as possible. Fetal monitoring strips at the hospital were reassuring.
VERDICT A $55 million Maryland verdict was returned against the hospital, including $26 million in noneconomic damages. After the court reduced noneconomic damages and future lost wages awards, the net verdict was $28 million.
ARDS after hysterectomy
A MORBIDLY OBESE WOMAN underwent a hysterectomy. The asthmatic, 38-year-old patient vomited after surgery. A pulmonologist undertook her care and determined that she had acute respiratory distress syndrome (ARDS). He prescribed the administration of oxygen. When she vomited again during the early morning hours of the second postsurgical day, he ordered intubation and went to the hospital immediately, but the patient quickly deteriorated. She died from cardiac arrest.
ESTATE’S CLAIM The patient’s death was due to failure to diagnose and treat ARDS in a timely manner. A bronchoscopy and frequent radiographs should have been performed. If the patient had been intubated earlier and steps had been taken to reduce the risk of vomiting, she would have had a better chance of survival. She should have been transferred to another facility when ARDS was diagnosed.
DEFENDANTS’ DEFENSE A bronchoscopy was not necessary. ARDS was diagnosed and treated in a timely manner. She was too unstable to transfer to another hospital.
VERDICT The hospital reached a confidential settlement, and the claim against the anesthesiologist was dismissed. The trial proceeded against the pulmonologist and his group. A New York defense verdict was returned.
Mother’s HELLP syndrome missed; fetus dies
DURING HER PREGNANCY, a 23-year-old woman was monitored for hypertension by her ObGyn and nurse midwife. At her 36-week prenatal visit, she was found to have preeclampsia, including proteinuria. She was sent directly to the ED, where the baby was monitored and laboratory tests were ordered by a nurse and nurse midwife. After 2 hours, she was told she had a urinary tract infection and discharged. Three days later, she returned to the ED in critical condition; she had suffered an intrauterine fetal demise.
PARENTS’ CLAIM Lab results showed critical values and confirmed that the patient had developed HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome. The ED nurse and nurse midwife were negligent in their treatment: They never read the lab results or reported the results to the patient or an ObGyn.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $950,000 Virginia settlement was reached.

A PREGNANT WOMAN WAS AWAITING TRIAL in County jail when she went into preterm labor. She was taken to the ED but released 2 hours later, although she was dilated 2–3 cm and having contractions. She was returned to her locked cell and not monitored—no deputy or nurse was within sight or sound of the patient. Her water broke and contractions increased. Despite her screams, and those of other inmates, a nurse didn’t arrive for 2 hours, when the baby’s head was crowning. EMS services were called and the baby was delivered in the jail cell. The child had no heartbeat or respiration. Mother and baby were transported to the hospital, where the child was resuscitated. She has severe mental impairment and cerebral palsy.
There is no documentation that the mother received any prenatal or postpartum care in jail. The mother is now serving a life sentence after a conviction for felony murder, kidnapping, and conspiracy.
CHILD’S CLAIM The case was brought on behalf of the child, and claimed that deliberate indifference and the failure to provide medical attention caused the child’s impairments.
DEFENDANTS’ DEFENSE The County claimed qualified immunity as a government entity and argued that, when the child was injured, she was still a fetus, and therefore not protected by the Constitution and civil rights laws.
VERDICT The US Circuit Court of Appeals rejected the County’s argument that the child was not protected by the Constitution. An $8 million Michigan settlement was reached.
Dermoid cyst still present after wrong-site surgery
A DERMOID CYST WAS DETECTED on the left ovary of a 28-year-old woman during prenatal ultrasonography (US). A year later, US confirmed the dermoid cyst, and the patient underwent outpatient cystectomy.
At the first postsurgical visit, the patient reported right pelvic pain. When she called the ObGyn’s office a few days later to again report right pelvic pain, her call was not returned.
She then went to the ED, where testing determined that the ObGyn had performed a right salpingo-oophorectomy and that her left ovary and cyst were still intact. She again attempted to contact the ObGyn, without response.
PATIENT’S CLAIM The ObGyn performed wrong-site surgery. The patient was not informed of the error during a postsurgical visit, nor were her attempts at contacting the physician returned. Still at risk for malignancy, she is facing a second surgical procedure to remove the cyst. Her fertility is diminished due to the surgical error, and she suffers anxiety and mental stress as a result of the situation.
At first, the ObGyn refused to provide medical records to the patient’s lawyer. When the records were obtained and compared with records obtained from another physician who treated the patient, it was evident that the ObGyn had altered the records to state that the patient had complained of right-side pain.
PHYSICIAN’S DEFENSE There was no negligence. The patient was properly treated for right-sided pain. The records were not altered.
VERDICT A $1.42 million Maryland verdict was returned. The state cap on noneconomic damages will reduce the verdict to $680,000.
Sponge left behind after vacuum-assisted closure
A WOMAN WENT TO THE ED with abdominal pain. It was determined that she had an abdominal abscess, and a surgeon assumed her care. After surgically draining the abdominal abscess, the surgeon placed a large black sponge into the abdominal cavity and then used vacuum-assisted closure. The patient was discharged 6 days later. She continued to receive treatment for a surgical-site infection that failed to heal. Two weeks later, the patient was readmitted to the hospital for exploratory surgery. The surgeon found and removed the sponge.
PATIENT’S CLAIM The surgeon was negligent for leaving the surgical sponge in the patient’s abdomen. She claimed pain, scarring, wound necrosis, infection, and the need for additional hospitalizations due to retention of the sponge.
PHYSICIAN’S DEFENSE A settlement was reached during the trial.
VERDICT A confidential Florida settlement was reached.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
A MOTHER WANTED A HOME BIRTH with a midwife. When complications arose and labor stopped progressing, the midwife called an ambulance. The emergency department (ED) physician ordered an urgent cesarean delivery, but the procedure did not begin for another 2 hours. The child was born with brain damage, multiple physical and mental disabilities, complex seizure disorder, and cerebral palsy.
PARENTS’ CLAIM The child’s injuries occurred because cesarean delivery was delayed for 2 hours. Based on fetal heart-rate monitoring, the injuries most likely occurred in the last 18 minutes before birth, and were probably caused by compression of the umbilical cord. An earlier cesarean delivery would have avoided the injuries.
DEFENDANTS’ DEFENSE All of the injuries occurred prior to the mother’s arrival at the hospital, while she was under the care of the midwife. Fetal distress was present for an hour before the ambulance was called. When the mother arrived at the ED, she was an unknown patient, as the midwife did not have a collaborating physician. While the ED physician determined that a cesarean delivery was required, it was not considered an emergency. The mother was taken to the OR as soon as possible. Fetal monitoring strips at the hospital were reassuring.
VERDICT A $55 million Maryland verdict was returned against the hospital, including $26 million in noneconomic damages. After the court reduced noneconomic damages and future lost wages awards, the net verdict was $28 million.
ARDS after hysterectomy
A MORBIDLY OBESE WOMAN underwent a hysterectomy. The asthmatic, 38-year-old patient vomited after surgery. A pulmonologist undertook her care and determined that she had acute respiratory distress syndrome (ARDS). He prescribed the administration of oxygen. When she vomited again during the early morning hours of the second postsurgical day, he ordered intubation and went to the hospital immediately, but the patient quickly deteriorated. She died from cardiac arrest.
ESTATE’S CLAIM The patient’s death was due to failure to diagnose and treat ARDS in a timely manner. A bronchoscopy and frequent radiographs should have been performed. If the patient had been intubated earlier and steps had been taken to reduce the risk of vomiting, she would have had a better chance of survival. She should have been transferred to another facility when ARDS was diagnosed.
DEFENDANTS’ DEFENSE A bronchoscopy was not necessary. ARDS was diagnosed and treated in a timely manner. She was too unstable to transfer to another hospital.
VERDICT The hospital reached a confidential settlement, and the claim against the anesthesiologist was dismissed. The trial proceeded against the pulmonologist and his group. A New York defense verdict was returned.
Mother’s HELLP syndrome missed; fetus dies
DURING HER PREGNANCY, a 23-year-old woman was monitored for hypertension by her ObGyn and nurse midwife. At her 36-week prenatal visit, she was found to have preeclampsia, including proteinuria. She was sent directly to the ED, where the baby was monitored and laboratory tests were ordered by a nurse and nurse midwife. After 2 hours, she was told she had a urinary tract infection and discharged. Three days later, she returned to the ED in critical condition; she had suffered an intrauterine fetal demise.
PARENTS’ CLAIM Lab results showed critical values and confirmed that the patient had developed HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome. The ED nurse and nurse midwife were negligent in their treatment: They never read the lab results or reported the results to the patient or an ObGyn.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $950,000 Virginia settlement was reached.
A PREGNANT WOMAN WAS AWAITING TRIAL in County jail when she went into preterm labor. She was taken to the ED but released 2 hours later, although she was dilated 2–3 cm and having contractions. She was returned to her locked cell and not monitored—no deputy or nurse was within sight or sound of the patient. Her water broke and contractions increased. Despite her screams, and those of other inmates, a nurse didn’t arrive for 2 hours, when the baby’s head was crowning. EMS services were called and the baby was delivered in the jail cell. The child had no heartbeat or respiration. Mother and baby were transported to the hospital, where the child was resuscitated. She has severe mental impairment and cerebral palsy.
There is no documentation that the mother received any prenatal or postpartum care in jail. The mother is now serving a life sentence after a conviction for felony murder, kidnapping, and conspiracy.
CHILD’S CLAIM The case was brought on behalf of the child, and claimed that deliberate indifference and the failure to provide medical attention caused the child’s impairments.
DEFENDANTS’ DEFENSE The County claimed qualified immunity as a government entity and argued that, when the child was injured, she was still a fetus, and therefore not protected by the Constitution and civil rights laws.
VERDICT The US Circuit Court of Appeals rejected the County’s argument that the child was not protected by the Constitution. An $8 million Michigan settlement was reached.
Dermoid cyst still present after wrong-site surgery
A DERMOID CYST WAS DETECTED on the left ovary of a 28-year-old woman during prenatal ultrasonography (US). A year later, US confirmed the dermoid cyst, and the patient underwent outpatient cystectomy.
At the first postsurgical visit, the patient reported right pelvic pain. When she called the ObGyn’s office a few days later to again report right pelvic pain, her call was not returned.
She then went to the ED, where testing determined that the ObGyn had performed a right salpingo-oophorectomy and that her left ovary and cyst were still intact. She again attempted to contact the ObGyn, without response.
PATIENT’S CLAIM The ObGyn performed wrong-site surgery. The patient was not informed of the error during a postsurgical visit, nor were her attempts at contacting the physician returned. Still at risk for malignancy, she is facing a second surgical procedure to remove the cyst. Her fertility is diminished due to the surgical error, and she suffers anxiety and mental stress as a result of the situation.
At first, the ObGyn refused to provide medical records to the patient’s lawyer. When the records were obtained and compared with records obtained from another physician who treated the patient, it was evident that the ObGyn had altered the records to state that the patient had complained of right-side pain.
PHYSICIAN’S DEFENSE There was no negligence. The patient was properly treated for right-sided pain. The records were not altered.
VERDICT A $1.42 million Maryland verdict was returned. The state cap on noneconomic damages will reduce the verdict to $680,000.
Sponge left behind after vacuum-assisted closure
A WOMAN WENT TO THE ED with abdominal pain. It was determined that she had an abdominal abscess, and a surgeon assumed her care. After surgically draining the abdominal abscess, the surgeon placed a large black sponge into the abdominal cavity and then used vacuum-assisted closure. The patient was discharged 6 days later. She continued to receive treatment for a surgical-site infection that failed to heal. Two weeks later, the patient was readmitted to the hospital for exploratory surgery. The surgeon found and removed the sponge.
PATIENT’S CLAIM The surgeon was negligent for leaving the surgical sponge in the patient’s abdomen. She claimed pain, scarring, wound necrosis, infection, and the need for additional hospitalizations due to retention of the sponge.
PHYSICIAN’S DEFENSE A settlement was reached during the trial.
VERDICT A confidential Florida settlement was reached.
A MOTHER WANTED A HOME BIRTH with a midwife. When complications arose and labor stopped progressing, the midwife called an ambulance. The emergency department (ED) physician ordered an urgent cesarean delivery, but the procedure did not begin for another 2 hours. The child was born with brain damage, multiple physical and mental disabilities, complex seizure disorder, and cerebral palsy.
PARENTS’ CLAIM The child’s injuries occurred because cesarean delivery was delayed for 2 hours. Based on fetal heart-rate monitoring, the injuries most likely occurred in the last 18 minutes before birth, and were probably caused by compression of the umbilical cord. An earlier cesarean delivery would have avoided the injuries.
DEFENDANTS’ DEFENSE All of the injuries occurred prior to the mother’s arrival at the hospital, while she was under the care of the midwife. Fetal distress was present for an hour before the ambulance was called. When the mother arrived at the ED, she was an unknown patient, as the midwife did not have a collaborating physician. While the ED physician determined that a cesarean delivery was required, it was not considered an emergency. The mother was taken to the OR as soon as possible. Fetal monitoring strips at the hospital were reassuring.
VERDICT A $55 million Maryland verdict was returned against the hospital, including $26 million in noneconomic damages. After the court reduced noneconomic damages and future lost wages awards, the net verdict was $28 million.
ARDS after hysterectomy
A MORBIDLY OBESE WOMAN underwent a hysterectomy. The asthmatic, 38-year-old patient vomited after surgery. A pulmonologist undertook her care and determined that she had acute respiratory distress syndrome (ARDS). He prescribed the administration of oxygen. When she vomited again during the early morning hours of the second postsurgical day, he ordered intubation and went to the hospital immediately, but the patient quickly deteriorated. She died from cardiac arrest.
ESTATE’S CLAIM The patient’s death was due to failure to diagnose and treat ARDS in a timely manner. A bronchoscopy and frequent radiographs should have been performed. If the patient had been intubated earlier and steps had been taken to reduce the risk of vomiting, she would have had a better chance of survival. She should have been transferred to another facility when ARDS was diagnosed.
DEFENDANTS’ DEFENSE A bronchoscopy was not necessary. ARDS was diagnosed and treated in a timely manner. She was too unstable to transfer to another hospital.
VERDICT The hospital reached a confidential settlement, and the claim against the anesthesiologist was dismissed. The trial proceeded against the pulmonologist and his group. A New York defense verdict was returned.
Mother’s HELLP syndrome missed; fetus dies
DURING HER PREGNANCY, a 23-year-old woman was monitored for hypertension by her ObGyn and nurse midwife. At her 36-week prenatal visit, she was found to have preeclampsia, including proteinuria. She was sent directly to the ED, where the baby was monitored and laboratory tests were ordered by a nurse and nurse midwife. After 2 hours, she was told she had a urinary tract infection and discharged. Three days later, she returned to the ED in critical condition; she had suffered an intrauterine fetal demise.
PARENTS’ CLAIM Lab results showed critical values and confirmed that the patient had developed HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome. The ED nurse and nurse midwife were negligent in their treatment: They never read the lab results or reported the results to the patient or an ObGyn.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $950,000 Virginia settlement was reached.
A PREGNANT WOMAN WAS AWAITING TRIAL in County jail when she went into preterm labor. She was taken to the ED but released 2 hours later, although she was dilated 2–3 cm and having contractions. She was returned to her locked cell and not monitored—no deputy or nurse was within sight or sound of the patient. Her water broke and contractions increased. Despite her screams, and those of other inmates, a nurse didn’t arrive for 2 hours, when the baby’s head was crowning. EMS services were called and the baby was delivered in the jail cell. The child had no heartbeat or respiration. Mother and baby were transported to the hospital, where the child was resuscitated. She has severe mental impairment and cerebral palsy.
There is no documentation that the mother received any prenatal or postpartum care in jail. The mother is now serving a life sentence after a conviction for felony murder, kidnapping, and conspiracy.
CHILD’S CLAIM The case was brought on behalf of the child, and claimed that deliberate indifference and the failure to provide medical attention caused the child’s impairments.
DEFENDANTS’ DEFENSE The County claimed qualified immunity as a government entity and argued that, when the child was injured, she was still a fetus, and therefore not protected by the Constitution and civil rights laws.
VERDICT The US Circuit Court of Appeals rejected the County’s argument that the child was not protected by the Constitution. An $8 million Michigan settlement was reached.
Dermoid cyst still present after wrong-site surgery
A DERMOID CYST WAS DETECTED on the left ovary of a 28-year-old woman during prenatal ultrasonography (US). A year later, US confirmed the dermoid cyst, and the patient underwent outpatient cystectomy.
At the first postsurgical visit, the patient reported right pelvic pain. When she called the ObGyn’s office a few days later to again report right pelvic pain, her call was not returned.
She then went to the ED, where testing determined that the ObGyn had performed a right salpingo-oophorectomy and that her left ovary and cyst were still intact. She again attempted to contact the ObGyn, without response.
PATIENT’S CLAIM The ObGyn performed wrong-site surgery. The patient was not informed of the error during a postsurgical visit, nor were her attempts at contacting the physician returned. Still at risk for malignancy, she is facing a second surgical procedure to remove the cyst. Her fertility is diminished due to the surgical error, and she suffers anxiety and mental stress as a result of the situation.
At first, the ObGyn refused to provide medical records to the patient’s lawyer. When the records were obtained and compared with records obtained from another physician who treated the patient, it was evident that the ObGyn had altered the records to state that the patient had complained of right-side pain.
PHYSICIAN’S DEFENSE There was no negligence. The patient was properly treated for right-sided pain. The records were not altered.
VERDICT A $1.42 million Maryland verdict was returned. The state cap on noneconomic damages will reduce the verdict to $680,000.
Sponge left behind after vacuum-assisted closure
A WOMAN WENT TO THE ED with abdominal pain. It was determined that she had an abdominal abscess, and a surgeon assumed her care. After surgically draining the abdominal abscess, the surgeon placed a large black sponge into the abdominal cavity and then used vacuum-assisted closure. The patient was discharged 6 days later. She continued to receive treatment for a surgical-site infection that failed to heal. Two weeks later, the patient was readmitted to the hospital for exploratory surgery. The surgeon found and removed the sponge.
PATIENT’S CLAIM The surgeon was negligent for leaving the surgical sponge in the patient’s abdomen. She claimed pain, scarring, wound necrosis, infection, and the need for additional hospitalizations due to retention of the sponge.
PHYSICIAN’S DEFENSE A settlement was reached during the trial.
VERDICT A confidential Florida settlement was reached.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Four pillars of a successful practice: 2. Attract new patients
Pillar 1: Keep your current patients happy (March 2013)
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 3: Obtain and maintain physician referrals (June 2013)
Pillar 4: Motivate your staff (August 2013)
External marketing is nothing more than making potential patients aware of your service and areas of expertise. The public truly does not mind marketing, as long as it believes you are communicating useful information and providing value. Nevertheless, such marketing—getting the word out to the public and potential referring physicians—takes some physicians out of their comfort zone. Some doctors think that marketing is synonymous with advertising.
The truth is, you can make the public aware of your services and expertise in an ethical and professional fashion without spending large amounts of money on advertising or hiring an expensive consultant.
How?
The essence of external marketing is writing, speaking, and making use of the Internet. In this article, I review simple, inexpensive techniques to increase your visibility among your peers and in your community. These techniques do not require additional staff or anything more than minimal assistance from your hospital’s public relations and marketing departments and the creation of a few PowerPoint slides that will hold the attention of your audience. A future article will describe Internet marketing strategies.

Try your hand at public speaking
Few of us are natural-born orators, but if you get started on the speaking circuit and acquire effective skills, you’ll be amazed at the demand for your presentations and the commensurate number of new patients filling your appointment book. When you take your message to the podium, audiences have an opportunity not only to learn more about your medical topic and how it applies to their health and wellness, but also to interact with you before and after the presentation.
Most of us have been asked to give a presentation to a lay audience at some time or another. How many of us have set off with a PowerPoint presentation from a pharmaceutical company that contains information far too technical for a nonmedical audience? Is it any wonder that so few talks motivate new patients to call our practices?
How to get invited to speak at local events
Even if you have a knack for public speaking, you still need to generate invitations for speaking engagements. I systematically contact meeting planners at various churches, service organizations like the Junior League, women’s book clubs, and patient advocacy groups, such as the American Cancer Society and American Diabetes Association. A list of these organizations and clubs can be obtained from the Chamber of Commerce in your community.
When I began public speaking, I created a public relations packet and sent it to meeting planners in the community. The packet contained a brief biography that outlined my credentials, listed organizations or groups to which I have given talks in the past, and provided a few testimonials from previous audience members. I also included a fact sheet (see the box on this page) and several articles on the topic to be covered. The articles were written by me for local outlets or written by others for publication in national magazines or other lay publications.
After I delivered a talk, I hung around to answer questions. I also made sure to have plenty of business cards to hand out, as well as my practice brochure and articles that pertained to the topic I had just presented.
Overactive bladder: You don’t have to depend on Depends!
Overactive bladder is a common disorder that affects millions of American women and men. Most people who have this condition suffer in silence and do not seek help from a health-care professional. The good news: Most sufferers can be helped.
Overactive bladder:
- affects 33 million American men and women
- can result in reclusive behavior
- can be a source of tremendous embarrassment
- can cause recurrent urinary tract infections
- hinders workplace interactions
- limits personal mobility
- can cause skin infections
- may lead to falls and fractures
- may lead to nursing home institutionalization
- is expensive—economic costs exceeded $35 billion in 2008.
Help is available. No one needs to depend on Depends!
If you would like additional information on this topic, or you are interested in having Dr. Neil Baum speak to your group about overactive bladder and other urologic problems, please call (504) 891-8454 or write to Dr. Baum at [email protected].
Don’t overlook support groups and group appointments
Conducting a support group is an excellent way to target a specific diagnosis or disease state. If you can identify women who have a chronic problem, such as pelvic pain, incontinence, or endometriosis, and invite them to a meeting, you’ll find that they appreciate your interest and expertise and often become patients in your practice. Women who attend these meetings get to know who you are, what you do, and where to find you.
Start by organizing your current patients. I have discovered that it is easiest to start with patients in your own practice when organizing these meetings. These women know others with similar problems and soon invite them to your group.
How to start a support group
Choose a date for your meeting. Keep the following in mind:
- Select a date 2 or 3 months in the future. Decide on several possible alternative dates as well. Don’t choose a date near a major holiday. Because I practice in New Orleans, for example, I would never pick a date a week before or after Mardi Gras.
- Tuesday and Wednesday evenings are the best nights of the week. Most people do not schedule social engagements during the middle of the week.
- If your target audience is senior citizens, they may not be able to attend or drive at night. A Saturday morning or weekday afternoon meeting might be better for them.
- At the meeting, provide a sign-in sheet to record the names and email addresses of all who attend. You can use this list to contact attendees later through an online newsletter.
Within 1 week after your support group presentation, send a follow-up email and appropriate additional information to attendees on your sign-in sheet. The letter should thank them for attending and let them know you are available to answer any questions. You can then add their names to your database and contact them periodically when new treatments or diagnostic techniques become available.
Ethnic communities require special attention
With so many different ethnicities in many US metropolitan areas, you may have an opportunity to attract new patients from these groups. If possible, try to learn to speak the language of the ethnic group you primarily serve—you will have an advantage in attracting foreign-born immigrants if you can speak their language. Alternatively, you can serve their needs by having someone on staff who can translate for you.
Be aware, however, that professional medical interpreters recommend employing a trained medical professional to manage the translation. Without specific training in the language and familiarity with the nuances of translating during a medical examination, diagnostic cues and treatment recommendations may be missed or misinterpreted.
Some translation services specialize in medical translation. You can contact the service and request a translator in nearly any language, including Vietnamese, Russian, Serbian, and Afrikaans, and they will arrange for a translator to arrive at a designated time. The fees are reasonable, and using such a service ensures that you can communicate with patients when neither you nor a staffer speaks the language.
It is still a good idea for you to learn some basic vocabulary, such as greetings, farewells, and the names of body parts. Not only will this make diagnoses more efficient, it will make your patients feel welcome.
Provide translations of your educational materials for patients who are more comfortable with a language besides English. If these materials are not already available from pharmaceutical or medical manufacturing companies, have the most frequently used information translated. The nearest university or college might be a good resource. The language departments at these institutions often can refer you to people who do translations on a freelance basis.
Be sure to add information to your Web site and other social media that makes it clear that you accept patients who speak other languages.
Consider writing articles for lay publications
How many referrals or new patients do you get from articles you have written for professional journals?
There is a good chance that your answer is the same as mine: “None.”
My CV lists nearly 175 articles that have been published in peer-reviewed professional journals, but I have not seen a single referral or new patient as a result. However, I have written several hundred articles for local newspapers and magazines that have generated hundreds of new patient visits to my practice.
Become a media resource: Write, be proactive, be responsive
By writing articles for the local press, you can easily become a media resource. Reporters and editors will notice your pieces. Often they will contact you for articles or ask you for quotations to be included in articles they are writing. If you are responsive, they will keep you in their database as an expert to call on whenever your specialty is in the news.
You can promote this transition yourself. When Whoopi Goldberg shared her experience with urinary incontinence on the television talk show The View, I contacted my local paper, the Times-Picayune, and offered to provide information about the problems of incontinence and overactive bladder and how an outpatient evaluation can often lead to cure of this disease.
What should you write about?
Topics of interest to lay readers in your community undoubtedly include wellness, menopause, cancer prevention, female sexual dysfunction, and vaginal rejuvenation. You can create an interesting article about new procedures, new treatments, a unique case with an excellent result, or the use of new technologies, such as new in-office procedures for permanent contraception.
Like medical skills, writing skills can be learned and polished. The more you do it, the better you get. The better you get, the more women you will attract to your practice.
Use your Web site to attract new patients
For most ObGyns, the majority of patients they serve come from within their community. A clinician’s service area usually encompasses no more than three to five zip codes or a 25- to 50-mile radius. All of us enjoy seeing a patient who has traveled more than 100 miles to see us for a gynecologic problem. Imagine the excitement when a patient from 1,000, 5,000, or even 10,000 miles away contacts your office for an appointment. This is exactly what a Web site can do for you and your practice. (Note: In a future article, I will focus on Internet marketing.)
Blogging offers an opportunity to engage potential patients
If you have a Web site, then you’ve already taken the most critical step toward marketing your practice in an increasingly Internet-savvy age. Today’s patients rely on the Internet for personal health information; they also expect a level of interaction and communication from their clinician on the Web. That’s because popular social media platforms, like Facebook and Twitter, are growing rapidly, enabling patients to use a variety of social media resources for support, education, and treatment decisions. A static Web site that consists only of your practice name, staff biographies, your office address and phone numbers, and a map to guide patients to your practice won’t cut it any longer in terms of patient expectations.
Health-care practitioners are just beginning to embrace social media—Facebook, Twitter, YouTube, and blogging—as an important component of their Internet marketing strategy. Blogging is easy, quick, and free. In many cases, a blog already is integrated with the rest of your professionally designed Web site. To get started, you just need to contribute content to the blog.
Although a blog won’t deliver an instant return on investment, it can, with time, build awareness of your practice and help promote your services to existing and potential patients. Blogs are driven by content, and a blog tied to your practice gives you the freedom to write and publish content that is unique to you and your practice. Written effectively, blogs present the perfect opportunity to interact with your patients while promoting your services.
Blogs also can improve your search engine ranking significantly. By adding new content to your blog on a regular basis, you ensure that search engines “crawl” your site more often. More important, blogs make it possible to dually publish content on other social media sites, functioning as the nucleus of your social media maintenance. Regular posts to your blog can be synced with your Facebook and Twitter accounts for seamless social networking.
Choose a snappy headline
Few patients will read a blog post with a headline that doesn’t entice them in some way. A compelling headline is essential to get your visitor to read the rest of the article and revisit your blog for new posts in the future.
Think of your blog title as a billboard. Consider that you are trying to attract the attention of drivers who have only a few seconds to look at your signage. The same is true for the title of your blog. Visitors often read the title and make a decision about whether to read the rest of the content. For example, an article entitled “Evaluation and treatment of urinary incontinence” probably would not get the eyeballs to stick, compared with a headline like “You don’t have to depend on Depends!” Doctors tend to think conservatively and may generate bland titles more suitable for a medical journal. I suggest that you think more like a tabloid journalist to attract readers to your blog.
Keep blog posts lay-friendly
Because patients will be reading your blog, remember to write for them and not for your colleagues. Be conversational and avoid overusing medical terminology that your readers won’t appreciate or understand. Try to target your writing to the 10th grade level so that you attract both educated and less educated readers. Some blog sites evaluate your writing to determine its grade level and will assist you in keeping your material understandable by most readers.
For example, Writing Sample Analyzer uses syllable counts and sentence length to determine the average grade level of your material (http://sarahktyler.com/code/sam ple.php). And the Readability Calculator at http://www.online-utility.org/english/readability_test_and_improve.jsp is also useful. In general, these tools penalize writers for polysyllabic words and long, complex sentences. Your writing will score higher when you use simpler diction and write short sentences.
Educate, rather than advertise.
Blogs should be used to support your online marketing efforts and provide patients with important information about your practice and services. A blog is not designed to be an advertising tool. Using it as such a tool will cause readers to lose interest fast. If you think education first, your material will be attractive to readers and they may call your office for an appointment.
Some organizational pointers:
- Avoid lengthy blog posts; they can lose reader interest. Pages with a lot of white space are easier to scan and more likely to keep patients reading. Say enough to get your point across, but don’t lose your readers’ attention with irrelevant information.
- Include subheadings and bullet points every few paragraphs so readers can quickly browse your post for the information they want.
Provide fresh, unique content that is new and interesting. Offer advice and tips for improved health, and inform patients about new technology and treatments that are specific to your practice. For example, if you offer a noninvasive approach to a medical problem using a procedure that is new in your community, write a post on this topic and include a testimonial from one of your treated patients. This strategy is very effective at generating new patients.
Don’t let your content get stale
Post to your blog regularly, providing new and updated content. Once you develop an audience, keep them coming back by adhering to a schedule. Every update you make to your blog counts as fresh content—a significant factor search engines use to rank Web sites. I suggest that you consider blogging at a minimum of once a week.
We are in the age of social media. The social media train is leaving the station, and you better get on board. The easiest way to start is by creating and posting regularly on your blog site.
External marketing to attract new patients to your ObGyn practice basically consists of writing and speaking. If you want to market outside your practice, you need to think about putting your writing and speaking skills into action. So, speak up and get your pen or computer working!
We want to hear from you! Tell us what you think.
CLICK HERE to access recent articles on managing your ObGyn practice.

Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
Pillar 1: Keep your current patients happy (March 2013)
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 3: Obtain and maintain physician referrals (June 2013)
Pillar 4: Motivate your staff (August 2013)
External marketing is nothing more than making potential patients aware of your service and areas of expertise. The public truly does not mind marketing, as long as it believes you are communicating useful information and providing value. Nevertheless, such marketing—getting the word out to the public and potential referring physicians—takes some physicians out of their comfort zone. Some doctors think that marketing is synonymous with advertising.
The truth is, you can make the public aware of your services and expertise in an ethical and professional fashion without spending large amounts of money on advertising or hiring an expensive consultant.
How?
The essence of external marketing is writing, speaking, and making use of the Internet. In this article, I review simple, inexpensive techniques to increase your visibility among your peers and in your community. These techniques do not require additional staff or anything more than minimal assistance from your hospital’s public relations and marketing departments and the creation of a few PowerPoint slides that will hold the attention of your audience. A future article will describe Internet marketing strategies.
Try your hand at public speaking
Few of us are natural-born orators, but if you get started on the speaking circuit and acquire effective skills, you’ll be amazed at the demand for your presentations and the commensurate number of new patients filling your appointment book. When you take your message to the podium, audiences have an opportunity not only to learn more about your medical topic and how it applies to their health and wellness, but also to interact with you before and after the presentation.
Most of us have been asked to give a presentation to a lay audience at some time or another. How many of us have set off with a PowerPoint presentation from a pharmaceutical company that contains information far too technical for a nonmedical audience? Is it any wonder that so few talks motivate new patients to call our practices?
How to get invited to speak at local events
Even if you have a knack for public speaking, you still need to generate invitations for speaking engagements. I systematically contact meeting planners at various churches, service organizations like the Junior League, women’s book clubs, and patient advocacy groups, such as the American Cancer Society and American Diabetes Association. A list of these organizations and clubs can be obtained from the Chamber of Commerce in your community.
When I began public speaking, I created a public relations packet and sent it to meeting planners in the community. The packet contained a brief biography that outlined my credentials, listed organizations or groups to which I have given talks in the past, and provided a few testimonials from previous audience members. I also included a fact sheet (see the box on this page) and several articles on the topic to be covered. The articles were written by me for local outlets or written by others for publication in national magazines or other lay publications.
After I delivered a talk, I hung around to answer questions. I also made sure to have plenty of business cards to hand out, as well as my practice brochure and articles that pertained to the topic I had just presented.
Overactive bladder: You don’t have to depend on Depends!
Overactive bladder is a common disorder that affects millions of American women and men. Most people who have this condition suffer in silence and do not seek help from a health-care professional. The good news: Most sufferers can be helped.
Overactive bladder:
- affects 33 million American men and women
- can result in reclusive behavior
- can be a source of tremendous embarrassment
- can cause recurrent urinary tract infections
- hinders workplace interactions
- limits personal mobility
- can cause skin infections
- may lead to falls and fractures
- may lead to nursing home institutionalization
- is expensive—economic costs exceeded $35 billion in 2008.
Help is available. No one needs to depend on Depends!
If you would like additional information on this topic, or you are interested in having Dr. Neil Baum speak to your group about overactive bladder and other urologic problems, please call (504) 891-8454 or write to Dr. Baum at [email protected].
Don’t overlook support groups and group appointments
Conducting a support group is an excellent way to target a specific diagnosis or disease state. If you can identify women who have a chronic problem, such as pelvic pain, incontinence, or endometriosis, and invite them to a meeting, you’ll find that they appreciate your interest and expertise and often become patients in your practice. Women who attend these meetings get to know who you are, what you do, and where to find you.
Start by organizing your current patients. I have discovered that it is easiest to start with patients in your own practice when organizing these meetings. These women know others with similar problems and soon invite them to your group.
How to start a support group
Choose a date for your meeting. Keep the following in mind:
- Select a date 2 or 3 months in the future. Decide on several possible alternative dates as well. Don’t choose a date near a major holiday. Because I practice in New Orleans, for example, I would never pick a date a week before or after Mardi Gras.
- Tuesday and Wednesday evenings are the best nights of the week. Most people do not schedule social engagements during the middle of the week.
- If your target audience is senior citizens, they may not be able to attend or drive at night. A Saturday morning or weekday afternoon meeting might be better for them.
- At the meeting, provide a sign-in sheet to record the names and email addresses of all who attend. You can use this list to contact attendees later through an online newsletter.
Within 1 week after your support group presentation, send a follow-up email and appropriate additional information to attendees on your sign-in sheet. The letter should thank them for attending and let them know you are available to answer any questions. You can then add their names to your database and contact them periodically when new treatments or diagnostic techniques become available.
Ethnic communities require special attention
With so many different ethnicities in many US metropolitan areas, you may have an opportunity to attract new patients from these groups. If possible, try to learn to speak the language of the ethnic group you primarily serve—you will have an advantage in attracting foreign-born immigrants if you can speak their language. Alternatively, you can serve their needs by having someone on staff who can translate for you.
Be aware, however, that professional medical interpreters recommend employing a trained medical professional to manage the translation. Without specific training in the language and familiarity with the nuances of translating during a medical examination, diagnostic cues and treatment recommendations may be missed or misinterpreted.
Some translation services specialize in medical translation. You can contact the service and request a translator in nearly any language, including Vietnamese, Russian, Serbian, and Afrikaans, and they will arrange for a translator to arrive at a designated time. The fees are reasonable, and using such a service ensures that you can communicate with patients when neither you nor a staffer speaks the language.
It is still a good idea for you to learn some basic vocabulary, such as greetings, farewells, and the names of body parts. Not only will this make diagnoses more efficient, it will make your patients feel welcome.
Provide translations of your educational materials for patients who are more comfortable with a language besides English. If these materials are not already available from pharmaceutical or medical manufacturing companies, have the most frequently used information translated. The nearest university or college might be a good resource. The language departments at these institutions often can refer you to people who do translations on a freelance basis.
Be sure to add information to your Web site and other social media that makes it clear that you accept patients who speak other languages.
Consider writing articles for lay publications
How many referrals or new patients do you get from articles you have written for professional journals?
There is a good chance that your answer is the same as mine: “None.”
My CV lists nearly 175 articles that have been published in peer-reviewed professional journals, but I have not seen a single referral or new patient as a result. However, I have written several hundred articles for local newspapers and magazines that have generated hundreds of new patient visits to my practice.
Become a media resource: Write, be proactive, be responsive
By writing articles for the local press, you can easily become a media resource. Reporters and editors will notice your pieces. Often they will contact you for articles or ask you for quotations to be included in articles they are writing. If you are responsive, they will keep you in their database as an expert to call on whenever your specialty is in the news.
You can promote this transition yourself. When Whoopi Goldberg shared her experience with urinary incontinence on the television talk show The View, I contacted my local paper, the Times-Picayune, and offered to provide information about the problems of incontinence and overactive bladder and how an outpatient evaluation can often lead to cure of this disease.
What should you write about?
Topics of interest to lay readers in your community undoubtedly include wellness, menopause, cancer prevention, female sexual dysfunction, and vaginal rejuvenation. You can create an interesting article about new procedures, new treatments, a unique case with an excellent result, or the use of new technologies, such as new in-office procedures for permanent contraception.
Like medical skills, writing skills can be learned and polished. The more you do it, the better you get. The better you get, the more women you will attract to your practice.
Use your Web site to attract new patients
For most ObGyns, the majority of patients they serve come from within their community. A clinician’s service area usually encompasses no more than three to five zip codes or a 25- to 50-mile radius. All of us enjoy seeing a patient who has traveled more than 100 miles to see us for a gynecologic problem. Imagine the excitement when a patient from 1,000, 5,000, or even 10,000 miles away contacts your office for an appointment. This is exactly what a Web site can do for you and your practice. (Note: In a future article, I will focus on Internet marketing.)
Blogging offers an opportunity to engage potential patients
If you have a Web site, then you’ve already taken the most critical step toward marketing your practice in an increasingly Internet-savvy age. Today’s patients rely on the Internet for personal health information; they also expect a level of interaction and communication from their clinician on the Web. That’s because popular social media platforms, like Facebook and Twitter, are growing rapidly, enabling patients to use a variety of social media resources for support, education, and treatment decisions. A static Web site that consists only of your practice name, staff biographies, your office address and phone numbers, and a map to guide patients to your practice won’t cut it any longer in terms of patient expectations.
Health-care practitioners are just beginning to embrace social media—Facebook, Twitter, YouTube, and blogging—as an important component of their Internet marketing strategy. Blogging is easy, quick, and free. In many cases, a blog already is integrated with the rest of your professionally designed Web site. To get started, you just need to contribute content to the blog.
Although a blog won’t deliver an instant return on investment, it can, with time, build awareness of your practice and help promote your services to existing and potential patients. Blogs are driven by content, and a blog tied to your practice gives you the freedom to write and publish content that is unique to you and your practice. Written effectively, blogs present the perfect opportunity to interact with your patients while promoting your services.
Blogs also can improve your search engine ranking significantly. By adding new content to your blog on a regular basis, you ensure that search engines “crawl” your site more often. More important, blogs make it possible to dually publish content on other social media sites, functioning as the nucleus of your social media maintenance. Regular posts to your blog can be synced with your Facebook and Twitter accounts for seamless social networking.
Choose a snappy headline
Few patients will read a blog post with a headline that doesn’t entice them in some way. A compelling headline is essential to get your visitor to read the rest of the article and revisit your blog for new posts in the future.
Think of your blog title as a billboard. Consider that you are trying to attract the attention of drivers who have only a few seconds to look at your signage. The same is true for the title of your blog. Visitors often read the title and make a decision about whether to read the rest of the content. For example, an article entitled “Evaluation and treatment of urinary incontinence” probably would not get the eyeballs to stick, compared with a headline like “You don’t have to depend on Depends!” Doctors tend to think conservatively and may generate bland titles more suitable for a medical journal. I suggest that you think more like a tabloid journalist to attract readers to your blog.
Keep blog posts lay-friendly
Because patients will be reading your blog, remember to write for them and not for your colleagues. Be conversational and avoid overusing medical terminology that your readers won’t appreciate or understand. Try to target your writing to the 10th grade level so that you attract both educated and less educated readers. Some blog sites evaluate your writing to determine its grade level and will assist you in keeping your material understandable by most readers.
For example, Writing Sample Analyzer uses syllable counts and sentence length to determine the average grade level of your material (http://sarahktyler.com/code/sam ple.php). And the Readability Calculator at http://www.online-utility.org/english/readability_test_and_improve.jsp is also useful. In general, these tools penalize writers for polysyllabic words and long, complex sentences. Your writing will score higher when you use simpler diction and write short sentences.
Educate, rather than advertise.
Blogs should be used to support your online marketing efforts and provide patients with important information about your practice and services. A blog is not designed to be an advertising tool. Using it as such a tool will cause readers to lose interest fast. If you think education first, your material will be attractive to readers and they may call your office for an appointment.
Some organizational pointers:
- Avoid lengthy blog posts; they can lose reader interest. Pages with a lot of white space are easier to scan and more likely to keep patients reading. Say enough to get your point across, but don’t lose your readers’ attention with irrelevant information.
- Include subheadings and bullet points every few paragraphs so readers can quickly browse your post for the information they want.
Provide fresh, unique content that is new and interesting. Offer advice and tips for improved health, and inform patients about new technology and treatments that are specific to your practice. For example, if you offer a noninvasive approach to a medical problem using a procedure that is new in your community, write a post on this topic and include a testimonial from one of your treated patients. This strategy is very effective at generating new patients.
Don’t let your content get stale
Post to your blog regularly, providing new and updated content. Once you develop an audience, keep them coming back by adhering to a schedule. Every update you make to your blog counts as fresh content—a significant factor search engines use to rank Web sites. I suggest that you consider blogging at a minimum of once a week.
We are in the age of social media. The social media train is leaving the station, and you better get on board. The easiest way to start is by creating and posting regularly on your blog site.
External marketing to attract new patients to your ObGyn practice basically consists of writing and speaking. If you want to market outside your practice, you need to think about putting your writing and speaking skills into action. So, speak up and get your pen or computer working!
We want to hear from you! Tell us what you think.
CLICK HERE to access recent articles on managing your ObGyn practice.
Pillar 1: Keep your current patients happy (March 2013)
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 3: Obtain and maintain physician referrals (June 2013)
Pillar 4: Motivate your staff (August 2013)
External marketing is nothing more than making potential patients aware of your service and areas of expertise. The public truly does not mind marketing, as long as it believes you are communicating useful information and providing value. Nevertheless, such marketing—getting the word out to the public and potential referring physicians—takes some physicians out of their comfort zone. Some doctors think that marketing is synonymous with advertising.
The truth is, you can make the public aware of your services and expertise in an ethical and professional fashion without spending large amounts of money on advertising or hiring an expensive consultant.
How?
The essence of external marketing is writing, speaking, and making use of the Internet. In this article, I review simple, inexpensive techniques to increase your visibility among your peers and in your community. These techniques do not require additional staff or anything more than minimal assistance from your hospital’s public relations and marketing departments and the creation of a few PowerPoint slides that will hold the attention of your audience. A future article will describe Internet marketing strategies.
Try your hand at public speaking
Few of us are natural-born orators, but if you get started on the speaking circuit and acquire effective skills, you’ll be amazed at the demand for your presentations and the commensurate number of new patients filling your appointment book. When you take your message to the podium, audiences have an opportunity not only to learn more about your medical topic and how it applies to their health and wellness, but also to interact with you before and after the presentation.
Most of us have been asked to give a presentation to a lay audience at some time or another. How many of us have set off with a PowerPoint presentation from a pharmaceutical company that contains information far too technical for a nonmedical audience? Is it any wonder that so few talks motivate new patients to call our practices?
How to get invited to speak at local events
Even if you have a knack for public speaking, you still need to generate invitations for speaking engagements. I systematically contact meeting planners at various churches, service organizations like the Junior League, women’s book clubs, and patient advocacy groups, such as the American Cancer Society and American Diabetes Association. A list of these organizations and clubs can be obtained from the Chamber of Commerce in your community.
When I began public speaking, I created a public relations packet and sent it to meeting planners in the community. The packet contained a brief biography that outlined my credentials, listed organizations or groups to which I have given talks in the past, and provided a few testimonials from previous audience members. I also included a fact sheet (see the box on this page) and several articles on the topic to be covered. The articles were written by me for local outlets or written by others for publication in national magazines or other lay publications.
After I delivered a talk, I hung around to answer questions. I also made sure to have plenty of business cards to hand out, as well as my practice brochure and articles that pertained to the topic I had just presented.
Overactive bladder: You don’t have to depend on Depends!
Overactive bladder is a common disorder that affects millions of American women and men. Most people who have this condition suffer in silence and do not seek help from a health-care professional. The good news: Most sufferers can be helped.
Overactive bladder:
- affects 33 million American men and women
- can result in reclusive behavior
- can be a source of tremendous embarrassment
- can cause recurrent urinary tract infections
- hinders workplace interactions
- limits personal mobility
- can cause skin infections
- may lead to falls and fractures
- may lead to nursing home institutionalization
- is expensive—economic costs exceeded $35 billion in 2008.
Help is available. No one needs to depend on Depends!
If you would like additional information on this topic, or you are interested in having Dr. Neil Baum speak to your group about overactive bladder and other urologic problems, please call (504) 891-8454 or write to Dr. Baum at [email protected].
Don’t overlook support groups and group appointments
Conducting a support group is an excellent way to target a specific diagnosis or disease state. If you can identify women who have a chronic problem, such as pelvic pain, incontinence, or endometriosis, and invite them to a meeting, you’ll find that they appreciate your interest and expertise and often become patients in your practice. Women who attend these meetings get to know who you are, what you do, and where to find you.
Start by organizing your current patients. I have discovered that it is easiest to start with patients in your own practice when organizing these meetings. These women know others with similar problems and soon invite them to your group.
How to start a support group
Choose a date for your meeting. Keep the following in mind:
- Select a date 2 or 3 months in the future. Decide on several possible alternative dates as well. Don’t choose a date near a major holiday. Because I practice in New Orleans, for example, I would never pick a date a week before or after Mardi Gras.
- Tuesday and Wednesday evenings are the best nights of the week. Most people do not schedule social engagements during the middle of the week.
- If your target audience is senior citizens, they may not be able to attend or drive at night. A Saturday morning or weekday afternoon meeting might be better for them.
- At the meeting, provide a sign-in sheet to record the names and email addresses of all who attend. You can use this list to contact attendees later through an online newsletter.
Within 1 week after your support group presentation, send a follow-up email and appropriate additional information to attendees on your sign-in sheet. The letter should thank them for attending and let them know you are available to answer any questions. You can then add their names to your database and contact them periodically when new treatments or diagnostic techniques become available.
Ethnic communities require special attention
With so many different ethnicities in many US metropolitan areas, you may have an opportunity to attract new patients from these groups. If possible, try to learn to speak the language of the ethnic group you primarily serve—you will have an advantage in attracting foreign-born immigrants if you can speak their language. Alternatively, you can serve their needs by having someone on staff who can translate for you.
Be aware, however, that professional medical interpreters recommend employing a trained medical professional to manage the translation. Without specific training in the language and familiarity with the nuances of translating during a medical examination, diagnostic cues and treatment recommendations may be missed or misinterpreted.
Some translation services specialize in medical translation. You can contact the service and request a translator in nearly any language, including Vietnamese, Russian, Serbian, and Afrikaans, and they will arrange for a translator to arrive at a designated time. The fees are reasonable, and using such a service ensures that you can communicate with patients when neither you nor a staffer speaks the language.
It is still a good idea for you to learn some basic vocabulary, such as greetings, farewells, and the names of body parts. Not only will this make diagnoses more efficient, it will make your patients feel welcome.
Provide translations of your educational materials for patients who are more comfortable with a language besides English. If these materials are not already available from pharmaceutical or medical manufacturing companies, have the most frequently used information translated. The nearest university or college might be a good resource. The language departments at these institutions often can refer you to people who do translations on a freelance basis.
Be sure to add information to your Web site and other social media that makes it clear that you accept patients who speak other languages.
Consider writing articles for lay publications
How many referrals or new patients do you get from articles you have written for professional journals?
There is a good chance that your answer is the same as mine: “None.”
My CV lists nearly 175 articles that have been published in peer-reviewed professional journals, but I have not seen a single referral or new patient as a result. However, I have written several hundred articles for local newspapers and magazines that have generated hundreds of new patient visits to my practice.
Become a media resource: Write, be proactive, be responsive
By writing articles for the local press, you can easily become a media resource. Reporters and editors will notice your pieces. Often they will contact you for articles or ask you for quotations to be included in articles they are writing. If you are responsive, they will keep you in their database as an expert to call on whenever your specialty is in the news.
You can promote this transition yourself. When Whoopi Goldberg shared her experience with urinary incontinence on the television talk show The View, I contacted my local paper, the Times-Picayune, and offered to provide information about the problems of incontinence and overactive bladder and how an outpatient evaluation can often lead to cure of this disease.
What should you write about?
Topics of interest to lay readers in your community undoubtedly include wellness, menopause, cancer prevention, female sexual dysfunction, and vaginal rejuvenation. You can create an interesting article about new procedures, new treatments, a unique case with an excellent result, or the use of new technologies, such as new in-office procedures for permanent contraception.
Like medical skills, writing skills can be learned and polished. The more you do it, the better you get. The better you get, the more women you will attract to your practice.
Use your Web site to attract new patients
For most ObGyns, the majority of patients they serve come from within their community. A clinician’s service area usually encompasses no more than three to five zip codes or a 25- to 50-mile radius. All of us enjoy seeing a patient who has traveled more than 100 miles to see us for a gynecologic problem. Imagine the excitement when a patient from 1,000, 5,000, or even 10,000 miles away contacts your office for an appointment. This is exactly what a Web site can do for you and your practice. (Note: In a future article, I will focus on Internet marketing.)
Blogging offers an opportunity to engage potential patients
If you have a Web site, then you’ve already taken the most critical step toward marketing your practice in an increasingly Internet-savvy age. Today’s patients rely on the Internet for personal health information; they also expect a level of interaction and communication from their clinician on the Web. That’s because popular social media platforms, like Facebook and Twitter, are growing rapidly, enabling patients to use a variety of social media resources for support, education, and treatment decisions. A static Web site that consists only of your practice name, staff biographies, your office address and phone numbers, and a map to guide patients to your practice won’t cut it any longer in terms of patient expectations.
Health-care practitioners are just beginning to embrace social media—Facebook, Twitter, YouTube, and blogging—as an important component of their Internet marketing strategy. Blogging is easy, quick, and free. In many cases, a blog already is integrated with the rest of your professionally designed Web site. To get started, you just need to contribute content to the blog.
Although a blog won’t deliver an instant return on investment, it can, with time, build awareness of your practice and help promote your services to existing and potential patients. Blogs are driven by content, and a blog tied to your practice gives you the freedom to write and publish content that is unique to you and your practice. Written effectively, blogs present the perfect opportunity to interact with your patients while promoting your services.
Blogs also can improve your search engine ranking significantly. By adding new content to your blog on a regular basis, you ensure that search engines “crawl” your site more often. More important, blogs make it possible to dually publish content on other social media sites, functioning as the nucleus of your social media maintenance. Regular posts to your blog can be synced with your Facebook and Twitter accounts for seamless social networking.
Choose a snappy headline
Few patients will read a blog post with a headline that doesn’t entice them in some way. A compelling headline is essential to get your visitor to read the rest of the article and revisit your blog for new posts in the future.
Think of your blog title as a billboard. Consider that you are trying to attract the attention of drivers who have only a few seconds to look at your signage. The same is true for the title of your blog. Visitors often read the title and make a decision about whether to read the rest of the content. For example, an article entitled “Evaluation and treatment of urinary incontinence” probably would not get the eyeballs to stick, compared with a headline like “You don’t have to depend on Depends!” Doctors tend to think conservatively and may generate bland titles more suitable for a medical journal. I suggest that you think more like a tabloid journalist to attract readers to your blog.
Keep blog posts lay-friendly
Because patients will be reading your blog, remember to write for them and not for your colleagues. Be conversational and avoid overusing medical terminology that your readers won’t appreciate or understand. Try to target your writing to the 10th grade level so that you attract both educated and less educated readers. Some blog sites evaluate your writing to determine its grade level and will assist you in keeping your material understandable by most readers.
For example, Writing Sample Analyzer uses syllable counts and sentence length to determine the average grade level of your material (http://sarahktyler.com/code/sam ple.php). And the Readability Calculator at http://www.online-utility.org/english/readability_test_and_improve.jsp is also useful. In general, these tools penalize writers for polysyllabic words and long, complex sentences. Your writing will score higher when you use simpler diction and write short sentences.
Educate, rather than advertise.
Blogs should be used to support your online marketing efforts and provide patients with important information about your practice and services. A blog is not designed to be an advertising tool. Using it as such a tool will cause readers to lose interest fast. If you think education first, your material will be attractive to readers and they may call your office for an appointment.
Some organizational pointers:
- Avoid lengthy blog posts; they can lose reader interest. Pages with a lot of white space are easier to scan and more likely to keep patients reading. Say enough to get your point across, but don’t lose your readers’ attention with irrelevant information.
- Include subheadings and bullet points every few paragraphs so readers can quickly browse your post for the information they want.
Provide fresh, unique content that is new and interesting. Offer advice and tips for improved health, and inform patients about new technology and treatments that are specific to your practice. For example, if you offer a noninvasive approach to a medical problem using a procedure that is new in your community, write a post on this topic and include a testimonial from one of your treated patients. This strategy is very effective at generating new patients.
Don’t let your content get stale
Post to your blog regularly, providing new and updated content. Once you develop an audience, keep them coming back by adhering to a schedule. Every update you make to your blog counts as fresh content—a significant factor search engines use to rank Web sites. I suggest that you consider blogging at a minimum of once a week.
We are in the age of social media. The social media train is leaving the station, and you better get on board. The easiest way to start is by creating and posting regularly on your blog site.
External marketing to attract new patients to your ObGyn practice basically consists of writing and speaking. If you want to market outside your practice, you need to think about putting your writing and speaking skills into action. So, speak up and get your pen or computer working!
We want to hear from you! Tell us what you think.
CLICK HERE to access recent articles on managing your ObGyn practice.
Evaluating psychotic patients' risk of violence: A practical guide
When evaluating a patient’s risk of violence, the presence of psychosis is a crucial concern. Douglas et al1 found that psychosis was the most important predictor of violent behavior in an analysis of 204 studies examining the relationship between psychopathology and aggression. Clinicians need to be familiar with aspects of persecutory delusions and command auditory hallucinations that are associated with an increased risk of aggression because accurately assessing patients who are experiencing these 2 symptoms is an important part of a comprehensive violence risk assessment.
This article highlights the importance of investigating persecutory delusions and command auditory hallucinations when evaluating a psychotic patient’s risk for violence. We provide specific questions to ask to help gauge risk associated with these 2 symptoms.
Evaluating persecutory delusions
Do persecutory delusions increase the risk that a person will behave violently? Research examining delusions’ contribution to violent behavior does not provide a clear answer. Earlier studies suggested that persecutory delusions were associated with an increased risk of aggression.2 Delusions noted to increase the risk of violence were characterized by threat/control-override (TCO) symptoms. TCO symptoms are beliefs that one is being threatened (eg, being followed or poisoned) or is losing control to an external source (eg, one’s mind is dominated by forces beyond his or her control).3 Similarly, using data from the Epidemiologic Catchment Area surveys, Swanson et al4 found that patients who reported TCO symptoms were approximately twice as likely to engage in assaultive behavior compared with patients with other psychotic symptoms.
In contrast, the MacArthur Study of Mental Disorder and Violence5,6 showed that the presence of delusions did not predict higher rates of violence among recently discharged psychiatric patients. In particular, researchers did not find a relationship between the presence of TCO delusions and violent behavior. In a study comparing male criminal offenders with schizophrenia found not guilty by reason of insanity with matched non-offending schizophrenia patients, Stompe et al7 found no significant association between TCO symptoms and severity of violent behavior; prevalence of TCO symptoms did not differ between the 2 groups. However, nondelusional suspiciousness—such as misperceiving others’ behavior as indicating hostile intent—was associated with subsequent violence.6
Nederlof et al8 conducted a cross-sectional multicenter study to further examine whether TCO symptoms are related to aggressive behavior. Their study included 124 patients (88% men) who had paranoid schizophrenia (70%), “other forms” of schizophrenia (16%), schizoaffective disorder (3%), delusional disorder (1%), and psychosis not otherwise specified (10%). To measure TCO symptoms in a more detailed manner than in previous research, these researchers developed the Threat/Control-Override Questionnaire (TCOQ), a 14-item, self-report scale. The 7 threat items specific to the TCOQ are:8
- I am under the control of an external force that determines my actions.
- Other people have tried to poison me or to do me harm.
- Someone has deliberately tried to make me ill.
- Other people have been secretly plotting to ruin me.
- Someone has had evil intentions against me.
- I have the thought that I was being followed for a special reason.
- People have tried to drive me insane.
The 7 control-override items on the TCOQ are:8
- Other people control my way of movements.
- Other people can insert thoughts into my head.
- My thoughts are dominated by an external force.
- I have the feeling that other people can determine my thoughts.
- Other people can insert thoughts into my mind.
- I have the feeling that other people have control over me.
- My life is being determined by something or someone except for myself.
Nederlof et al8 determined that TCO symptoms were a significant correlate of aggression in their study sample. When the 2 domains of TCO symptoms were evaluated separately, only threat symptoms made a significant contribution to aggressive behavior. These researchers suggested that varying methods of measuring TCO symptoms may underlie previous studies’ seemingly contradictory findings.8 These recent findings indicate that the debate regarding the contribution of TCO symptoms, particularly threat symptoms, to future violence remains active.
Appelbaum et al9 used the MacArthur-Maudsley Delusions Assessment Schedule to examine the contribution of non-content-related delusional material to violence in interviews with 328 delusional hospitalized psychiatric patients. The 7 dimensions of the MacArthur-Maudsley Delusions Assessment Schedule are:
- Conviction—the degree of certainty about the delusional belief
- Negative affect—whether the delusional belief makes the patient unhappy, frightened, anxious, or angry
- Action—the extent to which the patient’s actions are motivated by the delusional belief
- Inaction—whether the patient has refrained from any action as a result of the delusional belief
- Preoccupation—the extent to which the patient indicates his or her thoughts focus exclusively on the delusion
- Pervasiveness—the degree to which the delusional belief penetrates all aspects of the patient’s experiences
- Fluidity—the degree to which the delusional belief changed frequently during the interview.
Patients with persecutory delusions had significantly higher scores on “action” and “negative affect” dimensions, indicating that those with persecutory delusions may be more likely to react in response to the dysphoric aspects of their symptoms.9 Subsequent research has demonstrated that patients who suffer from persecutory delusions and negative affect are more likely to act on their delusions2,10 and to act violently11 than patients without these symptoms.
When evaluating a patient who experiences persecutory delusions, inquire if he or she has employed “safety actions.” These are specific behaviors—such as avoiding a perceived persecutor or escaping a fearful situation—the individual has employed with the intention of minimizing a misperceived threat. In a study of 100 patients with persecutory delusions, 96% reported using safety behaviors in the past month.12 In this study, individuals with a history of violence reported a greater use of safety behaviors.
Table 1 lists 10 questions to ask patients to explore persecutory delusions and associated risk factors for aggression.
Table 1
Evaluating persecutory delusions: 10 questions
| 1. | Who or what do you believe wants to harm you? |
| 2. | How is this person attempting to harm you? (Ask about specific threat/control-override beliefs) |
| 3. | How certain are you that this is happening? |
| 4. | Is there anything that could convince you that this isn’t true? |
| 5. | How does your belief make you feel (eg, unhappy, frightened, anxious, or angry)? |
| 6. | Have you thought about any actions to take as a result of these beliefs? If so, what? |
| 7. | Have you taken any action as a result of your beliefs? If so, what specific actions? |
| 8. | Has your concern about being harmed stopped you from doing any action that you would normally do? Have you changed your routine in any way? |
| 9. | How much time do you spend thinking about this each day? |
| 10. | In what ways have these beliefs impacted your life? |
Assessing auditory hallucinations
A careful inquiry about hallucinations can help determine whether their presence increases a patient’s risk of committing a violent act. Command hallucinations provide some type of directive to the patient. Approximately 50% of hallucinating psychiatric patients experience command hallucinations.13 Most command hallucinations are nonviolent, and patients are more likely to obey nonviolent instructions than violent commands.14
Research on factors associated with a patient acting on harmful command hallucinations has been mixed. In a review of 7 controlled studies, no study demonstrated a positive relationship between command hallucinations and violence, and 1 found an inverse relationship.15 In contrast, in a study of 103 psychiatric inpatients, McNiel et al16 found 30% reported having command hallucinations to harm others during the past year and 22% reported they complied with such commands. These researchers concluded that compared with those without command hallucinations, patients in their study who experienced command hallucinations to harm others were more than twice as likely to be violent.
Much of the literature examining the relationship between a patient’s actions and command hallucinations has examined the patient’s response to all command hallucinations, without delineating factors specific to violent commands. Seven factors are associated with acting on command hallucinations:13
- the presence of coexisting delusions17
- having delusions that relate to the hallucination18
- knowing the voice’s identity18
- believing the voices to be real19
- believing that the voices are benevolent20
- having few coping strategies to deal with the voices17
- not feeling in control over the voices.20
These factors also have been found to indicate increased compliance with acting on violent command hallucinations.18,20 Studies that have examined compliance specific to harmful command hallucinations provide additional guidance when evaluating the patient’s risk of harm. Aspects relevant to increased compliance to violent command hallucinations include a belief that the voice is powerful,13,21 a patient’s sense of personal superiority,21 a belief that command hallucinations benefit the patient,13 delusions that were congruent with the action described,13 and hallucinations that generate negative emotions such as anger, anxiety, and sadness.11
Table 2 lists 10 questions to ask to further investigate general command auditory hallucinations and violent command auditory hallucinations.
Table 2
Evaluating command auditory hallucinations: 10 questions
| 1. | What are the voices telling you to do? |
| 2. | Do you have any thoughts or beliefs that are associated with what you are hearing? If so, what are they? |
| 3. | Do you know the voice’s identity? If so, who is it? |
| 4. | How convinced are you that these voices are real? |
| 5. | Are these voices wishing you well or do you think that they wish you harm? |
| 6. | Have you done anything to help make the voices go away? If so, what? |
| 7. | Do you feel you have control of the voices or do you feel they control you? |
| 8. | Do you believe the voice is powerful? |
| 9. | How do the voices make you feel? |
| 10. | Have you ever done what the voice has told you to do? If so, describe what you did. |
Related Resources
- MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study.http://macarthur.virginia.edu/risk.html.
- Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies [published online February 13, 2013]. PLoS One. 2013;8(2):e55942. doi: 10.1371/journal.pone.0055942.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Douglas KS, Guy LS, Hart SD. Psychosis as a risk factor for violence to others: a meta-analysis. Psychol Bull. 2009;135(5):679-706.
2. Wessely S, Buchanan A, Reed A, et al. Acting on delusions. I: Prevalence. Br J Psychiatry. 1993;163:69-76.
3. Link BG, Stueve A. Evidence bearing on mental illness as a possible cause of violent behavior. Epidemiol Rev. 1995;17(1):172-181.
4. Swanson JW, Borum R, Swartz MS, et al. Psychotic symptoms and disorders and the risk of violent behaviour in the community. Crim Behav Ment Health. 1996;6(4):309-329.
5. MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study. http://macarthur.virginia.edu/risk.html. Published April 2001. Accessed March 21 2013.
6. Monahan J, Steadman HJ, Silver E, et al. Rethinking risk assessment: the MacArthur study of mental disorder and violence. New York, NY: Oxford University Press, Inc.; 2001.
7. Stompe T, Ortwein-Swoboda G, Schanda H. Schizophrenia delusional symptoms, and violence: the threat/control override concept reexamined. Schizophr Bull. 2004;30(1):31-44.
8. Nederlof AF, Muris P, Hovens JE. Threat/control-override symptoms and emotional reactions to positive symptoms as correlates of aggressive behavior in psychotic patients. J Nerv Ment Dis. 2011;199(5):342-347.
9. Appelbaum PS, Robbins PC, Roth LH. Dimensional approach to delusions: comparison across types and diagnoses. Am J Psychiatry. 1999;156(12):1938-1943.
10. Buchanan A, Reed A, Wessely S, et al. Acting on delusions. II: The phenomenological correlates of acting on delusions. Br J Psychiatry. 1993;163:77-81.
11. Cheung P, Schweitzer I, Crowley K, et al. Violence in schizophrenia: role of hallucinations and delusions. Schizophr Res. 1997;26(2-3):181-190.
12. Freeman D, Garety PA, Kuipers E, et al. Acting on persecutory delusions: the importance of safety seeking. Behav Res Ther. 2007;45(1):89-99.
13. Shawyer F, MacKinnon A, Farhall J, et al. Command hallucinations and violence: implications for detention and treatment. Psychiatr Psychol Law. 2003;10(1):97-107.
14. Chadwick P, Birchwood M. The omnipotence of voices. A cognitive approach to auditory hallucinations. Br J Psychiatry. 1994;164(2):190-201.
15. Rudnick A. Relation between command hallucinations and dangerous behavior. J Am Acad Psychiatry Law. 1999;27(2):253-257.
16. McNiel DE, Eisner JP, Binder RL. The relationship between command hallucinations and violence. Psychiatr Serv. 2000;51(10):1288-1292.
17. Mackinnon A, Copolov DL, Trauer T. Factors associated with compliance and resistance to command hallucinations. J Nerv Ment Dis. 2004;192(5):357-362.
18. Junginger J. Predicting compliance with command hallucinations. Am J Psychiatry. 1990;147(2):245-247.
19. Erkwoh R, Willmes K, Eming-Erdmann A, et al. Command hallucinations: who obeys and who resists when? Psychopathology. 2002;35(5):272-279.
20. Beck-Sander A, Birchwood M, Chadwick P. Acting on command hallucinations: a cognitive approach. Br J Clin Psychol. 1997;36(pt 1):139-148.
21. Fox JRE, Gray NS, Lewis H. Factors determining compliance with command hallucinations with violent content: the role of social rank perceived power of the voice and voice malevolence. J Forens Psychiatry Psychol. 2004;15(3):511-531.
When evaluating a patient’s risk of violence, the presence of psychosis is a crucial concern. Douglas et al1 found that psychosis was the most important predictor of violent behavior in an analysis of 204 studies examining the relationship between psychopathology and aggression. Clinicians need to be familiar with aspects of persecutory delusions and command auditory hallucinations that are associated with an increased risk of aggression because accurately assessing patients who are experiencing these 2 symptoms is an important part of a comprehensive violence risk assessment.
This article highlights the importance of investigating persecutory delusions and command auditory hallucinations when evaluating a psychotic patient’s risk for violence. We provide specific questions to ask to help gauge risk associated with these 2 symptoms.
Evaluating persecutory delusions
Do persecutory delusions increase the risk that a person will behave violently? Research examining delusions’ contribution to violent behavior does not provide a clear answer. Earlier studies suggested that persecutory delusions were associated with an increased risk of aggression.2 Delusions noted to increase the risk of violence were characterized by threat/control-override (TCO) symptoms. TCO symptoms are beliefs that one is being threatened (eg, being followed or poisoned) or is losing control to an external source (eg, one’s mind is dominated by forces beyond his or her control).3 Similarly, using data from the Epidemiologic Catchment Area surveys, Swanson et al4 found that patients who reported TCO symptoms were approximately twice as likely to engage in assaultive behavior compared with patients with other psychotic symptoms.
In contrast, the MacArthur Study of Mental Disorder and Violence5,6 showed that the presence of delusions did not predict higher rates of violence among recently discharged psychiatric patients. In particular, researchers did not find a relationship between the presence of TCO delusions and violent behavior. In a study comparing male criminal offenders with schizophrenia found not guilty by reason of insanity with matched non-offending schizophrenia patients, Stompe et al7 found no significant association between TCO symptoms and severity of violent behavior; prevalence of TCO symptoms did not differ between the 2 groups. However, nondelusional suspiciousness—such as misperceiving others’ behavior as indicating hostile intent—was associated with subsequent violence.6
Nederlof et al8 conducted a cross-sectional multicenter study to further examine whether TCO symptoms are related to aggressive behavior. Their study included 124 patients (88% men) who had paranoid schizophrenia (70%), “other forms” of schizophrenia (16%), schizoaffective disorder (3%), delusional disorder (1%), and psychosis not otherwise specified (10%). To measure TCO symptoms in a more detailed manner than in previous research, these researchers developed the Threat/Control-Override Questionnaire (TCOQ), a 14-item, self-report scale. The 7 threat items specific to the TCOQ are:8
- I am under the control of an external force that determines my actions.
- Other people have tried to poison me or to do me harm.
- Someone has deliberately tried to make me ill.
- Other people have been secretly plotting to ruin me.
- Someone has had evil intentions against me.
- I have the thought that I was being followed for a special reason.
- People have tried to drive me insane.
The 7 control-override items on the TCOQ are:8
- Other people control my way of movements.
- Other people can insert thoughts into my head.
- My thoughts are dominated by an external force.
- I have the feeling that other people can determine my thoughts.
- Other people can insert thoughts into my mind.
- I have the feeling that other people have control over me.
- My life is being determined by something or someone except for myself.
Nederlof et al8 determined that TCO symptoms were a significant correlate of aggression in their study sample. When the 2 domains of TCO symptoms were evaluated separately, only threat symptoms made a significant contribution to aggressive behavior. These researchers suggested that varying methods of measuring TCO symptoms may underlie previous studies’ seemingly contradictory findings.8 These recent findings indicate that the debate regarding the contribution of TCO symptoms, particularly threat symptoms, to future violence remains active.
Appelbaum et al9 used the MacArthur-Maudsley Delusions Assessment Schedule to examine the contribution of non-content-related delusional material to violence in interviews with 328 delusional hospitalized psychiatric patients. The 7 dimensions of the MacArthur-Maudsley Delusions Assessment Schedule are:
- Conviction—the degree of certainty about the delusional belief
- Negative affect—whether the delusional belief makes the patient unhappy, frightened, anxious, or angry
- Action—the extent to which the patient’s actions are motivated by the delusional belief
- Inaction—whether the patient has refrained from any action as a result of the delusional belief
- Preoccupation—the extent to which the patient indicates his or her thoughts focus exclusively on the delusion
- Pervasiveness—the degree to which the delusional belief penetrates all aspects of the patient’s experiences
- Fluidity—the degree to which the delusional belief changed frequently during the interview.
Patients with persecutory delusions had significantly higher scores on “action” and “negative affect” dimensions, indicating that those with persecutory delusions may be more likely to react in response to the dysphoric aspects of their symptoms.9 Subsequent research has demonstrated that patients who suffer from persecutory delusions and negative affect are more likely to act on their delusions2,10 and to act violently11 than patients without these symptoms.
When evaluating a patient who experiences persecutory delusions, inquire if he or she has employed “safety actions.” These are specific behaviors—such as avoiding a perceived persecutor or escaping a fearful situation—the individual has employed with the intention of minimizing a misperceived threat. In a study of 100 patients with persecutory delusions, 96% reported using safety behaviors in the past month.12 In this study, individuals with a history of violence reported a greater use of safety behaviors.
Table 1 lists 10 questions to ask patients to explore persecutory delusions and associated risk factors for aggression.
Table 1
Evaluating persecutory delusions: 10 questions
| 1. | Who or what do you believe wants to harm you? |
| 2. | How is this person attempting to harm you? (Ask about specific threat/control-override beliefs) |
| 3. | How certain are you that this is happening? |
| 4. | Is there anything that could convince you that this isn’t true? |
| 5. | How does your belief make you feel (eg, unhappy, frightened, anxious, or angry)? |
| 6. | Have you thought about any actions to take as a result of these beliefs? If so, what? |
| 7. | Have you taken any action as a result of your beliefs? If so, what specific actions? |
| 8. | Has your concern about being harmed stopped you from doing any action that you would normally do? Have you changed your routine in any way? |
| 9. | How much time do you spend thinking about this each day? |
| 10. | In what ways have these beliefs impacted your life? |
Assessing auditory hallucinations
A careful inquiry about hallucinations can help determine whether their presence increases a patient’s risk of committing a violent act. Command hallucinations provide some type of directive to the patient. Approximately 50% of hallucinating psychiatric patients experience command hallucinations.13 Most command hallucinations are nonviolent, and patients are more likely to obey nonviolent instructions than violent commands.14
Research on factors associated with a patient acting on harmful command hallucinations has been mixed. In a review of 7 controlled studies, no study demonstrated a positive relationship between command hallucinations and violence, and 1 found an inverse relationship.15 In contrast, in a study of 103 psychiatric inpatients, McNiel et al16 found 30% reported having command hallucinations to harm others during the past year and 22% reported they complied with such commands. These researchers concluded that compared with those without command hallucinations, patients in their study who experienced command hallucinations to harm others were more than twice as likely to be violent.
Much of the literature examining the relationship between a patient’s actions and command hallucinations has examined the patient’s response to all command hallucinations, without delineating factors specific to violent commands. Seven factors are associated with acting on command hallucinations:13
- the presence of coexisting delusions17
- having delusions that relate to the hallucination18
- knowing the voice’s identity18
- believing the voices to be real19
- believing that the voices are benevolent20
- having few coping strategies to deal with the voices17
- not feeling in control over the voices.20
These factors also have been found to indicate increased compliance with acting on violent command hallucinations.18,20 Studies that have examined compliance specific to harmful command hallucinations provide additional guidance when evaluating the patient’s risk of harm. Aspects relevant to increased compliance to violent command hallucinations include a belief that the voice is powerful,13,21 a patient’s sense of personal superiority,21 a belief that command hallucinations benefit the patient,13 delusions that were congruent with the action described,13 and hallucinations that generate negative emotions such as anger, anxiety, and sadness.11
Table 2 lists 10 questions to ask to further investigate general command auditory hallucinations and violent command auditory hallucinations.
Table 2
Evaluating command auditory hallucinations: 10 questions
| 1. | What are the voices telling you to do? |
| 2. | Do you have any thoughts or beliefs that are associated with what you are hearing? If so, what are they? |
| 3. | Do you know the voice’s identity? If so, who is it? |
| 4. | How convinced are you that these voices are real? |
| 5. | Are these voices wishing you well or do you think that they wish you harm? |
| 6. | Have you done anything to help make the voices go away? If so, what? |
| 7. | Do you feel you have control of the voices or do you feel they control you? |
| 8. | Do you believe the voice is powerful? |
| 9. | How do the voices make you feel? |
| 10. | Have you ever done what the voice has told you to do? If so, describe what you did. |
Related Resources
- MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study.http://macarthur.virginia.edu/risk.html.
- Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies [published online February 13, 2013]. PLoS One. 2013;8(2):e55942. doi: 10.1371/journal.pone.0055942.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
When evaluating a patient’s risk of violence, the presence of psychosis is a crucial concern. Douglas et al1 found that psychosis was the most important predictor of violent behavior in an analysis of 204 studies examining the relationship between psychopathology and aggression. Clinicians need to be familiar with aspects of persecutory delusions and command auditory hallucinations that are associated with an increased risk of aggression because accurately assessing patients who are experiencing these 2 symptoms is an important part of a comprehensive violence risk assessment.
This article highlights the importance of investigating persecutory delusions and command auditory hallucinations when evaluating a psychotic patient’s risk for violence. We provide specific questions to ask to help gauge risk associated with these 2 symptoms.
Evaluating persecutory delusions
Do persecutory delusions increase the risk that a person will behave violently? Research examining delusions’ contribution to violent behavior does not provide a clear answer. Earlier studies suggested that persecutory delusions were associated with an increased risk of aggression.2 Delusions noted to increase the risk of violence were characterized by threat/control-override (TCO) symptoms. TCO symptoms are beliefs that one is being threatened (eg, being followed or poisoned) or is losing control to an external source (eg, one’s mind is dominated by forces beyond his or her control).3 Similarly, using data from the Epidemiologic Catchment Area surveys, Swanson et al4 found that patients who reported TCO symptoms were approximately twice as likely to engage in assaultive behavior compared with patients with other psychotic symptoms.
In contrast, the MacArthur Study of Mental Disorder and Violence5,6 showed that the presence of delusions did not predict higher rates of violence among recently discharged psychiatric patients. In particular, researchers did not find a relationship between the presence of TCO delusions and violent behavior. In a study comparing male criminal offenders with schizophrenia found not guilty by reason of insanity with matched non-offending schizophrenia patients, Stompe et al7 found no significant association between TCO symptoms and severity of violent behavior; prevalence of TCO symptoms did not differ between the 2 groups. However, nondelusional suspiciousness—such as misperceiving others’ behavior as indicating hostile intent—was associated with subsequent violence.6
Nederlof et al8 conducted a cross-sectional multicenter study to further examine whether TCO symptoms are related to aggressive behavior. Their study included 124 patients (88% men) who had paranoid schizophrenia (70%), “other forms” of schizophrenia (16%), schizoaffective disorder (3%), delusional disorder (1%), and psychosis not otherwise specified (10%). To measure TCO symptoms in a more detailed manner than in previous research, these researchers developed the Threat/Control-Override Questionnaire (TCOQ), a 14-item, self-report scale. The 7 threat items specific to the TCOQ are:8
- I am under the control of an external force that determines my actions.
- Other people have tried to poison me or to do me harm.
- Someone has deliberately tried to make me ill.
- Other people have been secretly plotting to ruin me.
- Someone has had evil intentions against me.
- I have the thought that I was being followed for a special reason.
- People have tried to drive me insane.
The 7 control-override items on the TCOQ are:8
- Other people control my way of movements.
- Other people can insert thoughts into my head.
- My thoughts are dominated by an external force.
- I have the feeling that other people can determine my thoughts.
- Other people can insert thoughts into my mind.
- I have the feeling that other people have control over me.
- My life is being determined by something or someone except for myself.
Nederlof et al8 determined that TCO symptoms were a significant correlate of aggression in their study sample. When the 2 domains of TCO symptoms were evaluated separately, only threat symptoms made a significant contribution to aggressive behavior. These researchers suggested that varying methods of measuring TCO symptoms may underlie previous studies’ seemingly contradictory findings.8 These recent findings indicate that the debate regarding the contribution of TCO symptoms, particularly threat symptoms, to future violence remains active.
Appelbaum et al9 used the MacArthur-Maudsley Delusions Assessment Schedule to examine the contribution of non-content-related delusional material to violence in interviews with 328 delusional hospitalized psychiatric patients. The 7 dimensions of the MacArthur-Maudsley Delusions Assessment Schedule are:
- Conviction—the degree of certainty about the delusional belief
- Negative affect—whether the delusional belief makes the patient unhappy, frightened, anxious, or angry
- Action—the extent to which the patient’s actions are motivated by the delusional belief
- Inaction—whether the patient has refrained from any action as a result of the delusional belief
- Preoccupation—the extent to which the patient indicates his or her thoughts focus exclusively on the delusion
- Pervasiveness—the degree to which the delusional belief penetrates all aspects of the patient’s experiences
- Fluidity—the degree to which the delusional belief changed frequently during the interview.
Patients with persecutory delusions had significantly higher scores on “action” and “negative affect” dimensions, indicating that those with persecutory delusions may be more likely to react in response to the dysphoric aspects of their symptoms.9 Subsequent research has demonstrated that patients who suffer from persecutory delusions and negative affect are more likely to act on their delusions2,10 and to act violently11 than patients without these symptoms.
When evaluating a patient who experiences persecutory delusions, inquire if he or she has employed “safety actions.” These are specific behaviors—such as avoiding a perceived persecutor or escaping a fearful situation—the individual has employed with the intention of minimizing a misperceived threat. In a study of 100 patients with persecutory delusions, 96% reported using safety behaviors in the past month.12 In this study, individuals with a history of violence reported a greater use of safety behaviors.
Table 1 lists 10 questions to ask patients to explore persecutory delusions and associated risk factors for aggression.
Table 1
Evaluating persecutory delusions: 10 questions
| 1. | Who or what do you believe wants to harm you? |
| 2. | How is this person attempting to harm you? (Ask about specific threat/control-override beliefs) |
| 3. | How certain are you that this is happening? |
| 4. | Is there anything that could convince you that this isn’t true? |
| 5. | How does your belief make you feel (eg, unhappy, frightened, anxious, or angry)? |
| 6. | Have you thought about any actions to take as a result of these beliefs? If so, what? |
| 7. | Have you taken any action as a result of your beliefs? If so, what specific actions? |
| 8. | Has your concern about being harmed stopped you from doing any action that you would normally do? Have you changed your routine in any way? |
| 9. | How much time do you spend thinking about this each day? |
| 10. | In what ways have these beliefs impacted your life? |
Assessing auditory hallucinations
A careful inquiry about hallucinations can help determine whether their presence increases a patient’s risk of committing a violent act. Command hallucinations provide some type of directive to the patient. Approximately 50% of hallucinating psychiatric patients experience command hallucinations.13 Most command hallucinations are nonviolent, and patients are more likely to obey nonviolent instructions than violent commands.14
Research on factors associated with a patient acting on harmful command hallucinations has been mixed. In a review of 7 controlled studies, no study demonstrated a positive relationship between command hallucinations and violence, and 1 found an inverse relationship.15 In contrast, in a study of 103 psychiatric inpatients, McNiel et al16 found 30% reported having command hallucinations to harm others during the past year and 22% reported they complied with such commands. These researchers concluded that compared with those without command hallucinations, patients in their study who experienced command hallucinations to harm others were more than twice as likely to be violent.
Much of the literature examining the relationship between a patient’s actions and command hallucinations has examined the patient’s response to all command hallucinations, without delineating factors specific to violent commands. Seven factors are associated with acting on command hallucinations:13
- the presence of coexisting delusions17
- having delusions that relate to the hallucination18
- knowing the voice’s identity18
- believing the voices to be real19
- believing that the voices are benevolent20
- having few coping strategies to deal with the voices17
- not feeling in control over the voices.20
These factors also have been found to indicate increased compliance with acting on violent command hallucinations.18,20 Studies that have examined compliance specific to harmful command hallucinations provide additional guidance when evaluating the patient’s risk of harm. Aspects relevant to increased compliance to violent command hallucinations include a belief that the voice is powerful,13,21 a patient’s sense of personal superiority,21 a belief that command hallucinations benefit the patient,13 delusions that were congruent with the action described,13 and hallucinations that generate negative emotions such as anger, anxiety, and sadness.11
Table 2 lists 10 questions to ask to further investigate general command auditory hallucinations and violent command auditory hallucinations.
Table 2
Evaluating command auditory hallucinations: 10 questions
| 1. | What are the voices telling you to do? |
| 2. | Do you have any thoughts or beliefs that are associated with what you are hearing? If so, what are they? |
| 3. | Do you know the voice’s identity? If so, who is it? |
| 4. | How convinced are you that these voices are real? |
| 5. | Are these voices wishing you well or do you think that they wish you harm? |
| 6. | Have you done anything to help make the voices go away? If so, what? |
| 7. | Do you feel you have control of the voices or do you feel they control you? |
| 8. | Do you believe the voice is powerful? |
| 9. | How do the voices make you feel? |
| 10. | Have you ever done what the voice has told you to do? If so, describe what you did. |
Related Resources
- MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study.http://macarthur.virginia.edu/risk.html.
- Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies [published online February 13, 2013]. PLoS One. 2013;8(2):e55942. doi: 10.1371/journal.pone.0055942.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Douglas KS, Guy LS, Hart SD. Psychosis as a risk factor for violence to others: a meta-analysis. Psychol Bull. 2009;135(5):679-706.
2. Wessely S, Buchanan A, Reed A, et al. Acting on delusions. I: Prevalence. Br J Psychiatry. 1993;163:69-76.
3. Link BG, Stueve A. Evidence bearing on mental illness as a possible cause of violent behavior. Epidemiol Rev. 1995;17(1):172-181.
4. Swanson JW, Borum R, Swartz MS, et al. Psychotic symptoms and disorders and the risk of violent behaviour in the community. Crim Behav Ment Health. 1996;6(4):309-329.
5. MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study. http://macarthur.virginia.edu/risk.html. Published April 2001. Accessed March 21 2013.
6. Monahan J, Steadman HJ, Silver E, et al. Rethinking risk assessment: the MacArthur study of mental disorder and violence. New York, NY: Oxford University Press, Inc.; 2001.
7. Stompe T, Ortwein-Swoboda G, Schanda H. Schizophrenia delusional symptoms, and violence: the threat/control override concept reexamined. Schizophr Bull. 2004;30(1):31-44.
8. Nederlof AF, Muris P, Hovens JE. Threat/control-override symptoms and emotional reactions to positive symptoms as correlates of aggressive behavior in psychotic patients. J Nerv Ment Dis. 2011;199(5):342-347.
9. Appelbaum PS, Robbins PC, Roth LH. Dimensional approach to delusions: comparison across types and diagnoses. Am J Psychiatry. 1999;156(12):1938-1943.
10. Buchanan A, Reed A, Wessely S, et al. Acting on delusions. II: The phenomenological correlates of acting on delusions. Br J Psychiatry. 1993;163:77-81.
11. Cheung P, Schweitzer I, Crowley K, et al. Violence in schizophrenia: role of hallucinations and delusions. Schizophr Res. 1997;26(2-3):181-190.
12. Freeman D, Garety PA, Kuipers E, et al. Acting on persecutory delusions: the importance of safety seeking. Behav Res Ther. 2007;45(1):89-99.
13. Shawyer F, MacKinnon A, Farhall J, et al. Command hallucinations and violence: implications for detention and treatment. Psychiatr Psychol Law. 2003;10(1):97-107.
14. Chadwick P, Birchwood M. The omnipotence of voices. A cognitive approach to auditory hallucinations. Br J Psychiatry. 1994;164(2):190-201.
15. Rudnick A. Relation between command hallucinations and dangerous behavior. J Am Acad Psychiatry Law. 1999;27(2):253-257.
16. McNiel DE, Eisner JP, Binder RL. The relationship between command hallucinations and violence. Psychiatr Serv. 2000;51(10):1288-1292.
17. Mackinnon A, Copolov DL, Trauer T. Factors associated with compliance and resistance to command hallucinations. J Nerv Ment Dis. 2004;192(5):357-362.
18. Junginger J. Predicting compliance with command hallucinations. Am J Psychiatry. 1990;147(2):245-247.
19. Erkwoh R, Willmes K, Eming-Erdmann A, et al. Command hallucinations: who obeys and who resists when? Psychopathology. 2002;35(5):272-279.
20. Beck-Sander A, Birchwood M, Chadwick P. Acting on command hallucinations: a cognitive approach. Br J Clin Psychol. 1997;36(pt 1):139-148.
21. Fox JRE, Gray NS, Lewis H. Factors determining compliance with command hallucinations with violent content: the role of social rank perceived power of the voice and voice malevolence. J Forens Psychiatry Psychol. 2004;15(3):511-531.
1. Douglas KS, Guy LS, Hart SD. Psychosis as a risk factor for violence to others: a meta-analysis. Psychol Bull. 2009;135(5):679-706.
2. Wessely S, Buchanan A, Reed A, et al. Acting on delusions. I: Prevalence. Br J Psychiatry. 1993;163:69-76.
3. Link BG, Stueve A. Evidence bearing on mental illness as a possible cause of violent behavior. Epidemiol Rev. 1995;17(1):172-181.
4. Swanson JW, Borum R, Swartz MS, et al. Psychotic symptoms and disorders and the risk of violent behaviour in the community. Crim Behav Ment Health. 1996;6(4):309-329.
5. MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study. http://macarthur.virginia.edu/risk.html. Published April 2001. Accessed March 21 2013.
6. Monahan J, Steadman HJ, Silver E, et al. Rethinking risk assessment: the MacArthur study of mental disorder and violence. New York, NY: Oxford University Press, Inc.; 2001.
7. Stompe T, Ortwein-Swoboda G, Schanda H. Schizophrenia delusional symptoms, and violence: the threat/control override concept reexamined. Schizophr Bull. 2004;30(1):31-44.
8. Nederlof AF, Muris P, Hovens JE. Threat/control-override symptoms and emotional reactions to positive symptoms as correlates of aggressive behavior in psychotic patients. J Nerv Ment Dis. 2011;199(5):342-347.
9. Appelbaum PS, Robbins PC, Roth LH. Dimensional approach to delusions: comparison across types and diagnoses. Am J Psychiatry. 1999;156(12):1938-1943.
10. Buchanan A, Reed A, Wessely S, et al. Acting on delusions. II: The phenomenological correlates of acting on delusions. Br J Psychiatry. 1993;163:77-81.
11. Cheung P, Schweitzer I, Crowley K, et al. Violence in schizophrenia: role of hallucinations and delusions. Schizophr Res. 1997;26(2-3):181-190.
12. Freeman D, Garety PA, Kuipers E, et al. Acting on persecutory delusions: the importance of safety seeking. Behav Res Ther. 2007;45(1):89-99.
13. Shawyer F, MacKinnon A, Farhall J, et al. Command hallucinations and violence: implications for detention and treatment. Psychiatr Psychol Law. 2003;10(1):97-107.
14. Chadwick P, Birchwood M. The omnipotence of voices. A cognitive approach to auditory hallucinations. Br J Psychiatry. 1994;164(2):190-201.
15. Rudnick A. Relation between command hallucinations and dangerous behavior. J Am Acad Psychiatry Law. 1999;27(2):253-257.
16. McNiel DE, Eisner JP, Binder RL. The relationship between command hallucinations and violence. Psychiatr Serv. 2000;51(10):1288-1292.
17. Mackinnon A, Copolov DL, Trauer T. Factors associated with compliance and resistance to command hallucinations. J Nerv Ment Dis. 2004;192(5):357-362.
18. Junginger J. Predicting compliance with command hallucinations. Am J Psychiatry. 1990;147(2):245-247.
19. Erkwoh R, Willmes K, Eming-Erdmann A, et al. Command hallucinations: who obeys and who resists when? Psychopathology. 2002;35(5):272-279.
20. Beck-Sander A, Birchwood M, Chadwick P. Acting on command hallucinations: a cognitive approach. Br J Clin Psychol. 1997;36(pt 1):139-148.
21. Fox JRE, Gray NS, Lewis H. Factors determining compliance with command hallucinations with violent content: the role of social rank perceived power of the voice and voice malevolence. J Forens Psychiatry Psychol. 2004;15(3):511-531.

Treating a patient who has ‘everything’
Patients who endorse multiple psychiatric symptoms and meet criteria for several DSM diagnoses pose diagnostic and therapeutic challenges. In community samples, approximately 40% of patients with a DSM diagnosis have >1 illness, and comorbidity is more frequent in clinical trials.1 We highlight things to consider when managing a patient who has “everything.”
Endorsing ‘everything’ means something in itself. Patients with borderline personality disorder often present with myriad, disparate diagnoses and urgent requests for care.2 Also consider primary or secondary gain, particularly if the patient’s descriptions of symptoms are unusual. Saying “yes” to every question or endorsing highly unusual symptoms described by the interviewer may represent suggestibility related to catatonia or confabulation.
Focus on the most impairing symptom. This may help put other symptoms in context and focus treatment.
Find a common goal. If you can’t pick a simple symptom, move on to helping the patient identify his or her goals by asking questions such as, “Four weeks from now, what would you like to be doing?” Picking an achievable, measurable goal may be therapeutic.
Are the symptoms valid? Examine individual symptoms for validity using the SAFER criteria (Table).3
Table
SAFER criteria for symptom validity
| State vs trait: has the symptom lasted <12 weeks? |
| Assessable: can the symptom be measured? |
| Face validity: does the symptom clearly affect the patient’s behavior and functioning? |
| Ecological validity: is the symptom valid with our knowledge of its occurrence? |
| Rule of the 3Ps: is the symptom Persistent; Pathologically disruptive and different than usual; and Pervasive across normal domains? |
| Source: Reference 3 |
Multiple diagnoses may be in play, but start by treating one. Many patients meet criteria for multiple diagnoses. There is little evidence about which diagnosis should be treated first. Use your judgment in picking “the best first step” and treat accordingly.
Resist polypharmacy. Target specific symptoms or goals until a clear diagnostic picture emerges.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kessler RC, Chiu WT, Demler O, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
2. Gunderson JG. Borderline personality disorder: ontogeny of a diagnosis. Am J Psychiatry. 2009;166(5):530-539.
3. Targum SD, Pollack MH, Fava M. Redefining affective disorders: relevance for drug development. CNS Neurosci Ther. 2008;14(1):2-9.
Patients who endorse multiple psychiatric symptoms and meet criteria for several DSM diagnoses pose diagnostic and therapeutic challenges. In community samples, approximately 40% of patients with a DSM diagnosis have >1 illness, and comorbidity is more frequent in clinical trials.1 We highlight things to consider when managing a patient who has “everything.”
Endorsing ‘everything’ means something in itself. Patients with borderline personality disorder often present with myriad, disparate diagnoses and urgent requests for care.2 Also consider primary or secondary gain, particularly if the patient’s descriptions of symptoms are unusual. Saying “yes” to every question or endorsing highly unusual symptoms described by the interviewer may represent suggestibility related to catatonia or confabulation.
Focus on the most impairing symptom. This may help put other symptoms in context and focus treatment.
Find a common goal. If you can’t pick a simple symptom, move on to helping the patient identify his or her goals by asking questions such as, “Four weeks from now, what would you like to be doing?” Picking an achievable, measurable goal may be therapeutic.
Are the symptoms valid? Examine individual symptoms for validity using the SAFER criteria (Table).3
Table
SAFER criteria for symptom validity
| State vs trait: has the symptom lasted <12 weeks? |
| Assessable: can the symptom be measured? |
| Face validity: does the symptom clearly affect the patient’s behavior and functioning? |
| Ecological validity: is the symptom valid with our knowledge of its occurrence? |
| Rule of the 3Ps: is the symptom Persistent; Pathologically disruptive and different than usual; and Pervasive across normal domains? |
| Source: Reference 3 |
Multiple diagnoses may be in play, but start by treating one. Many patients meet criteria for multiple diagnoses. There is little evidence about which diagnosis should be treated first. Use your judgment in picking “the best first step” and treat accordingly.
Resist polypharmacy. Target specific symptoms or goals until a clear diagnostic picture emerges.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Patients who endorse multiple psychiatric symptoms and meet criteria for several DSM diagnoses pose diagnostic and therapeutic challenges. In community samples, approximately 40% of patients with a DSM diagnosis have >1 illness, and comorbidity is more frequent in clinical trials.1 We highlight things to consider when managing a patient who has “everything.”
Endorsing ‘everything’ means something in itself. Patients with borderline personality disorder often present with myriad, disparate diagnoses and urgent requests for care.2 Also consider primary or secondary gain, particularly if the patient’s descriptions of symptoms are unusual. Saying “yes” to every question or endorsing highly unusual symptoms described by the interviewer may represent suggestibility related to catatonia or confabulation.
Focus on the most impairing symptom. This may help put other symptoms in context and focus treatment.
Find a common goal. If you can’t pick a simple symptom, move on to helping the patient identify his or her goals by asking questions such as, “Four weeks from now, what would you like to be doing?” Picking an achievable, measurable goal may be therapeutic.
Are the symptoms valid? Examine individual symptoms for validity using the SAFER criteria (Table).3
Table
SAFER criteria for symptom validity
| State vs trait: has the symptom lasted <12 weeks? |
| Assessable: can the symptom be measured? |
| Face validity: does the symptom clearly affect the patient’s behavior and functioning? |
| Ecological validity: is the symptom valid with our knowledge of its occurrence? |
| Rule of the 3Ps: is the symptom Persistent; Pathologically disruptive and different than usual; and Pervasive across normal domains? |
| Source: Reference 3 |
Multiple diagnoses may be in play, but start by treating one. Many patients meet criteria for multiple diagnoses. There is little evidence about which diagnosis should be treated first. Use your judgment in picking “the best first step” and treat accordingly.
Resist polypharmacy. Target specific symptoms or goals until a clear diagnostic picture emerges.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kessler RC, Chiu WT, Demler O, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
2. Gunderson JG. Borderline personality disorder: ontogeny of a diagnosis. Am J Psychiatry. 2009;166(5):530-539.
3. Targum SD, Pollack MH, Fava M. Redefining affective disorders: relevance for drug development. CNS Neurosci Ther. 2008;14(1):2-9.
1. Kessler RC, Chiu WT, Demler O, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
2. Gunderson JG. Borderline personality disorder: ontogeny of a diagnosis. Am J Psychiatry. 2009;166(5):530-539.
3. Targum SD, Pollack MH, Fava M. Redefining affective disorders: relevance for drug development. CNS Neurosci Ther. 2008;14(1):2-9.
Depressive recurrence on antidepressant treatment (DRAT): 4 next-step options
“Poop-out” and “tachyphylaxis” are terms used to describe loss of antidepressant response after initial benefit, but both descriptors are problematic. Poop-out carries a mildly offensive aura that conveys a lack of seriousness about the patient’s suffering. Tachyphylaxis is a pharmacologic term describing rapid or acute reduction in response to a drug after administration; it is not appropriate for antidepressant loss of response, which typically occurs months to years after treatment initiation.1
Depressive recurrence on antidepressant treatment (DRAT) is a better term to describe recurrence of a major depressive episode despite sustained treatment with an antidepressant that had induced remission. Maintenance studies of antidepressant treatment indicate DRAT occurs in approximately 10% of patients at 6 months and 20% at 2 years.2
Despite this high prevalence, there is little clinical trial data on which to base treatment decisions for patients who experience DRAT. There are 4 options:
1. Raise the dose. In small studies, doubling the dose of fluoxetine or duloxetine led to regaining response in approximately 60% of patients.3 However, these studies lacked a placebo comparison arm, so the specific benefit derived from the dose increase is unknown. Improvement with a dose increase is somewhat at odds with the known mechanism of action of selective serotonin reuptake inhibitors (SSRIs), which are thought to have a flat dose-response curve. That is, once an SSRI blocks approximately 80% of the serotonin transporters, further dose increases produce minimal further blockade and, presumably, little clinical benefit via this mechanism.4
2. Switch medication. No studies have examined switching antidepressants after DRAT. Switching to a medication with a different mechanism may be justified based on the results of treatment-resistant depression (TRD) trials, in which patients who failed to respond to an initial medication—typically an SSRI—improved after switching to an antidepressant from a different class. However, the biology of DRAT may differ from that of SSRI nonresponse. Unlike many patients with TRD, patients who experience DRAT while taking an SSRI have demonstrated previous response to serotonergic modulation.
3. Augmentation. Similar to switching, this approach has not been studied specifically for DRAT. This approach is derived from trials in which patients who did not attain response after treatment with a single antidepressant had a second medication added. Again, the biology of these patients may differ from those with DRAT, who at one point did remit with antidepressant treatment.
4. Psychotherapy. This addition is a low-risk option for patients who previously have not received evidence-based psychotherapy for depression.
Clinicians have scant evidence on which to base their decisions for this common and important problem. Dose increase after DRAT represents the best supported, simplest, and perhaps least costly next step in treatment.
Disclosure
Dr. Dunlop receives grant or research support from Bristol-Myers Squibb, Forest Pharmaceuticals, and GlaxoSmithKline, and is a consultant to MedAvante and Roche.
1. Rothschild AJ, Dunlop BW, Dunner DL, et al. Assessing rates and predictors of tachyphylaxis during the prevention of recurrent episodes of depression with venlafaxine ER for two years (PREVENT) study. Psychopharmacol Bull. 2009;42(3):5-20.
2. Hansen R, Gaynes B, Thieda P, et al. Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv. 2008;59(10):1121-1130.
3. Schmidt ME, Fava M, Zhang S, et al. Treatment approaches to major depressive disorder relapse. Part 1: dose increase. Psychother Psychosom. 2002;71(4):190-194.
4. Meyer JH, Wilson AA, Sagrati S, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry. 2004;161(5):826-835.
“Poop-out” and “tachyphylaxis” are terms used to describe loss of antidepressant response after initial benefit, but both descriptors are problematic. Poop-out carries a mildly offensive aura that conveys a lack of seriousness about the patient’s suffering. Tachyphylaxis is a pharmacologic term describing rapid or acute reduction in response to a drug after administration; it is not appropriate for antidepressant loss of response, which typically occurs months to years after treatment initiation.1
Depressive recurrence on antidepressant treatment (DRAT) is a better term to describe recurrence of a major depressive episode despite sustained treatment with an antidepressant that had induced remission. Maintenance studies of antidepressant treatment indicate DRAT occurs in approximately 10% of patients at 6 months and 20% at 2 years.2
Despite this high prevalence, there is little clinical trial data on which to base treatment decisions for patients who experience DRAT. There are 4 options:
1. Raise the dose. In small studies, doubling the dose of fluoxetine or duloxetine led to regaining response in approximately 60% of patients.3 However, these studies lacked a placebo comparison arm, so the specific benefit derived from the dose increase is unknown. Improvement with a dose increase is somewhat at odds with the known mechanism of action of selective serotonin reuptake inhibitors (SSRIs), which are thought to have a flat dose-response curve. That is, once an SSRI blocks approximately 80% of the serotonin transporters, further dose increases produce minimal further blockade and, presumably, little clinical benefit via this mechanism.4
2. Switch medication. No studies have examined switching antidepressants after DRAT. Switching to a medication with a different mechanism may be justified based on the results of treatment-resistant depression (TRD) trials, in which patients who failed to respond to an initial medication—typically an SSRI—improved after switching to an antidepressant from a different class. However, the biology of DRAT may differ from that of SSRI nonresponse. Unlike many patients with TRD, patients who experience DRAT while taking an SSRI have demonstrated previous response to serotonergic modulation.
3. Augmentation. Similar to switching, this approach has not been studied specifically for DRAT. This approach is derived from trials in which patients who did not attain response after treatment with a single antidepressant had a second medication added. Again, the biology of these patients may differ from those with DRAT, who at one point did remit with antidepressant treatment.
4. Psychotherapy. This addition is a low-risk option for patients who previously have not received evidence-based psychotherapy for depression.
Clinicians have scant evidence on which to base their decisions for this common and important problem. Dose increase after DRAT represents the best supported, simplest, and perhaps least costly next step in treatment.
Disclosure
Dr. Dunlop receives grant or research support from Bristol-Myers Squibb, Forest Pharmaceuticals, and GlaxoSmithKline, and is a consultant to MedAvante and Roche.
“Poop-out” and “tachyphylaxis” are terms used to describe loss of antidepressant response after initial benefit, but both descriptors are problematic. Poop-out carries a mildly offensive aura that conveys a lack of seriousness about the patient’s suffering. Tachyphylaxis is a pharmacologic term describing rapid or acute reduction in response to a drug after administration; it is not appropriate for antidepressant loss of response, which typically occurs months to years after treatment initiation.1
Depressive recurrence on antidepressant treatment (DRAT) is a better term to describe recurrence of a major depressive episode despite sustained treatment with an antidepressant that had induced remission. Maintenance studies of antidepressant treatment indicate DRAT occurs in approximately 10% of patients at 6 months and 20% at 2 years.2
Despite this high prevalence, there is little clinical trial data on which to base treatment decisions for patients who experience DRAT. There are 4 options:
1. Raise the dose. In small studies, doubling the dose of fluoxetine or duloxetine led to regaining response in approximately 60% of patients.3 However, these studies lacked a placebo comparison arm, so the specific benefit derived from the dose increase is unknown. Improvement with a dose increase is somewhat at odds with the known mechanism of action of selective serotonin reuptake inhibitors (SSRIs), which are thought to have a flat dose-response curve. That is, once an SSRI blocks approximately 80% of the serotonin transporters, further dose increases produce minimal further blockade and, presumably, little clinical benefit via this mechanism.4
2. Switch medication. No studies have examined switching antidepressants after DRAT. Switching to a medication with a different mechanism may be justified based on the results of treatment-resistant depression (TRD) trials, in which patients who failed to respond to an initial medication—typically an SSRI—improved after switching to an antidepressant from a different class. However, the biology of DRAT may differ from that of SSRI nonresponse. Unlike many patients with TRD, patients who experience DRAT while taking an SSRI have demonstrated previous response to serotonergic modulation.
3. Augmentation. Similar to switching, this approach has not been studied specifically for DRAT. This approach is derived from trials in which patients who did not attain response after treatment with a single antidepressant had a second medication added. Again, the biology of these patients may differ from those with DRAT, who at one point did remit with antidepressant treatment.
4. Psychotherapy. This addition is a low-risk option for patients who previously have not received evidence-based psychotherapy for depression.
Clinicians have scant evidence on which to base their decisions for this common and important problem. Dose increase after DRAT represents the best supported, simplest, and perhaps least costly next step in treatment.
Disclosure
Dr. Dunlop receives grant or research support from Bristol-Myers Squibb, Forest Pharmaceuticals, and GlaxoSmithKline, and is a consultant to MedAvante and Roche.
1. Rothschild AJ, Dunlop BW, Dunner DL, et al. Assessing rates and predictors of tachyphylaxis during the prevention of recurrent episodes of depression with venlafaxine ER for two years (PREVENT) study. Psychopharmacol Bull. 2009;42(3):5-20.
2. Hansen R, Gaynes B, Thieda P, et al. Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv. 2008;59(10):1121-1130.
3. Schmidt ME, Fava M, Zhang S, et al. Treatment approaches to major depressive disorder relapse. Part 1: dose increase. Psychother Psychosom. 2002;71(4):190-194.
4. Meyer JH, Wilson AA, Sagrati S, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry. 2004;161(5):826-835.
1. Rothschild AJ, Dunlop BW, Dunner DL, et al. Assessing rates and predictors of tachyphylaxis during the prevention of recurrent episodes of depression with venlafaxine ER for two years (PREVENT) study. Psychopharmacol Bull. 2009;42(3):5-20.
2. Hansen R, Gaynes B, Thieda P, et al. Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv. 2008;59(10):1121-1130.
3. Schmidt ME, Fava M, Zhang S, et al. Treatment approaches to major depressive disorder relapse. Part 1: dose increase. Psychother Psychosom. 2002;71(4):190-194.
4. Meyer JH, Wilson AA, Sagrati S, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry. 2004;161(5):826-835.
Long-acting injectable aripiprazole for adult schizophrenia
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
Suicide, depression, and CYP2D6: How are they linked?
Genetic variations in drug-metabolizing enzymes dramatically affect drug pharmacokinetics and can result in clinically relevant differences in drug efficacy or toxicity. Cytochrome P450 (CYP) enzymes such as CYP2D6 are involved in metabolism of antidepressants, including selective serotonin reuptake inhibitors (SSRIs), which often are a first-line choice for patients with major depressive disorder (MDD).1,2 CYP2D6 is a highly polymorphic gene with 75 allelic variants (CYP2D6*1 to *75) and >30 additional subvariants.3 These variants are associated with phenotypes where CYP2D6 activity is increased, reduced, or lost, which can increase the risk of adverse drug reactions, decrease efficacy, and possibly influence a patient’s suicide risk.
In this article, we review the pharmacogenetics of CYP2D6 and discuss a possible relationship between CYP2D6 genotype and suicidal events during antidepressant treatment for MDD.
CYP2D6: Many variants
CYP450 enzymes are a group of 57 proteins, each coded by a different gene. Five subfamilies in the CYP450 family metabolize most drugs: CYP1A2, CYP3A4, CYP2C19, CYP2E1, and CYP2D6.4
Researchers discovered CYP2D6 in studies of nonpsychotropics (Box).5-9 CYP2D6 is widely expressed in many tissues, with dominant expression in the liver. Although CYP2D6 accounts for 2% of the total CYP450 liver enzyme content, it mediates metabolism in 25% to 30% of drugs in common clinical use and has a major influence on the biotransformation of SSRIs (Table).10
I the late 1970s, 2 groups of researchers noted unexpected serious adverse reactions in studies of debrisoquine,5 a sympatholytic antihypertensive drug, and sparteine,6 an antiarrhythmic and oxytocic alkaloid drug. They observed that 5% to 10% of patients were unable to efficiently metabolize debrisoquine and sparteine and went on to define a genetic polymorphism responsible for these metabolic differences. They also observed that metabolism of antidepressants, antipsychotics, and beta blockers also was defective in these patients.
Further investigations established that the enzyme responsible for debrisoquine metabolism was a cytochrome P450 (CYP) enzyme that is now termed CYP2D6.7 In addition to biochemical evidence, the colocalization of sparteine oxidation deficiency and of the CYP2D6 locus at chromosome 22q13.1 confirmed CYP2D6 as the target gene of the debrisoquine/sparteine polymorphism.8,9
Table
CYP450 enzymes involved in biotransformation of SSRIs
| SSRI | Enzymes involved in biotransformation |
|---|---|
| Citalopram | CYP2C19, CYP2D6, CYP3A4 |
| Escitalopram | CYP2C19, CYP2D6, CYP3A4 |
| Fluoxetine | CYP2D6, CYP2C9, CYP2C19, CYP3A4 |
| Fluvoxamine | CYP1A2, CYP2D6 |
| Paroxetine | CYP2D6, CYP3A4 |
| Sertraline | CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
| CYP: cytochrome P450; SSRI: selective serotonin reuptake inhibitors Source: Reference 10 | |
Approximately 100 polymorphic CYP2D6 alleles (variants) have been identified.3 These alleles are active, resulting in normal CYP2D6 enzyme activity, or inactive, leading to decreased enzyme activity. Genotyping for most common CYP2D6 alleles in ethnically defined populations can predict poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultra-rapid metabolizers (UMs) with high accuracy.11 PMs are compound heterozygous for inactivating alleles or homozygous for an inactivating variant. IMs carry one functional allele and one nonfunctional allele but may demonstrate a range of enzyme activity levels. EMs have 2 functional gene copies and UMs have >2 functional genes from gene duplication, resulting in ultra-rapid metabolism.
Suicide and CYP2D6 status
The widespread use of antidepressants appears to have led to significant decline in suicide rates in many countries.12 Based on an investigation of suicide mortality in 27 countries from 1980 to 2000, Ludwig and Marcotte12 found that faster growth in SSRI sales per capita was associated with larger declines in suicide rates. This finding was not confounded by other suicide risk factors such as unemployment, sex, age, or divorce rate.12 Countries such as Germany, Austria, Estonia, Switzerland, Sweden, Denmark, Hungary, and Slovenia—which had the highest suicide rate in the world 20 years ago (20 to 46 per 100,000 per year)—have had impressive declines in suicide rates (24% to 57% in the last 2 decades) with a marked (6- to 8-fold) increase in SSRI prescriptions during the same period.13-15 On the other hand, a few countries, such as Portugal and Spain, have experienced dramatic increases (58% and 86%, respectively) in the suicide rate with a similar increase in SSRI prescribing during the same 20-year period.16
A review of the distribution of CYP2D6 genotype among countries indicates a south/north gradient of CYP2D6 gene duplications, which indicate UM status.16 The proportion of UMs increases by almost 2-fold in southern European countries (8.4% and 7% to 10% for Portugal and Spain, respectively) compared with northern European countries (1% to 2% and 3.6% for Sweden and Germany, respectively); this south/north trend extends to Africa.17 The prevalence of CYP2D6 UMs is lower in northern countries, where increased anti-depressant use appears to have reduced suicide rates, and higher in southern countries, where suicide rates increased despite higher antidepressant use.
Case reports and observational studies18-21 suggest that compared with other CYP2D6 phenotypes, UMs may need to take higher doses of antidepressants to achieve therapeutic response. In a case report, Bertilsson et al18 described 2 patients who were UMs and required high doses of nortriptyline and clomipramine to obtain appropriate plasma drug concentrations. Baumann et al19 described a depressed patient with CYP2D6 gene duplication who required higher-than-usual doses of clomipramine. Rau et al20 found a 3-fold increase in the frequency of UMs in a group of 16 depressed German patients who did not respond to SSRIs or serotonin–norepinephrine reuptake inhibitors, both of which are metabolized by CYP2D6. Kawanishi et al21 found a significantly greater prevalence of UMs among 81 Nordic patients who did not respond to SSRIs compared with the general population.
Because suicidality may be caused by inadequately treated depressive illness, MDD patients who are UMs may be more likely to commit suicide because of suboptimal antidepressant levels. In a 2010 Swedish study, Zackrisson et al22 found that compared with those who died of other causes, significantly more individuals who committed suicide had >2 active CYP2D6 genes. Stingl et al23 found that among 285 depressed German patients, UMs had an elevated risk of having a high suicidality score compared with individuals with other genotypes, after adjusting for sex, baseline score on the Hamilton Depression Rating Scale (after excluding item 3 for suicidality), and number of previous depressive episodes. Other researchers found that patients with eating disorders who are UMs have a greater risk of suicidal behavior.24 Although none of these 3 studies specified if these patients were treated with antidepressants, the association between CYP2D6 gene duplication and suicide risk suggests CYP2D6’s role in suicide risk might not be related solely to antidepressant metabolism.
Effects on serotonin, dopamine
CYP2D6 is expressed in the brain and localized primarily in large principle cells of the hippocampus and Purkinje cells of the cerebellum, with no expression in other brain regions such as glial cells.25 This heterogeneous expression among brain regions and cell types indicates that in addition to its role in metabolizing drugs, CYP2D6 might influence neurotransmitter levels. In vitro and in vivo animal studies suggest that CYP2D6 plays a role in biotransformation of serotonin and dopamine.26,27
Serotonin is likely to play a causal role in the pathophysiology of depression, and depressed patients have abnormalities in serotonin activity.28 Serotonin is generated primarily from the transformation of tryptophan by tryptophan decarboxylase and tryptamine 5-hydroxylase.29 Yu et al27 found that CYP2D6 may be an additional pathway to regenerate serotonin through O-demethylation from 5-methoxytryptamine, but it is unclear what proportion of the physiologic pool of serotonin in synaptic nerve terminals is generated through the CYP2D6 pathway. However, this discovery provides a mechanistic basis of CYP2D6 involvement in the endogenous serotonin balance and by extension, in serotonergic physiology and neuropsychiatric disorders such as depression.30 Because SSRIs target the serotonergic pathway, baseline levels of serotonin and all related components of this pathway—including CYP2D6—are likely to help determine a patient’s response to SSRIs.
Dopamine also is generated from tyramine through CYP2D6,31 and distribution of CYP2D6 in the brain follows that of dopamine nerve terminals.32 The serotonergic system has strong anatomical and functional interaction with the dopaminergic system,33 and imbalance between serotonin and dopamine activity is thought to give rise to behavioral changes,2 which play an important role in the development of anxiety and impulsivity.
CYP2D6 in clinical practice
Although research into a possible link between CYP2D6 status and suicide risk in depressed patients treated with antidepressants is ongoing, at present this connection is speculative. More studies are warranted to reveal the exact role of CYP2D6 in response to SSRI treatment and suicide risk.
Knowledge of this potential association can help clinicians keep CYP450 genotyping in mind when prescribing antidepressants to depressed patients. The FDA has approved a pharmacogenetic test to analyze polymorphisms of CYP2D6 and CYP2C19.34 The results of such testing might guide pharmacotherapy for depressed patients, including medication selection and dosing. For example, a patient who is a PM might be started at a lower antidepressant dosage to avoid potential adverse drug effects, whereas it might be appropriate to prescribe a higher starting dose for a UM patient to achieve an effective drug concentration.
Related Resources
- Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, et al. CYP2D6 and the severity of suicide attempts. Pharmacogenomics. 2012;13(2):179-184.
- Blasco-Fontecilla H, Peñas-Lledó E, Vaquero-Lorenzo C, et al. CYP2D6 polymorphism and mental and personality disorders in suicide attempters [published online February 11, 2013]. J Pers Disord. doi: 10.1521/pedi_2013_27_080.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Marwah Shahid and Ijlal Yazdani for their assistance with this article.
1. Meyer UA, Amrein R, Balant LP, et al. Antidepressants and drug-metabolizing enzymes—expert group report. Acta Psychiatr Scand. 1996;93(2):71-79.
2. Kroemer HK, Eichelbaum M. “It’s the genes stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56(26):2285-2298.
3. The Human Cytochrome P450 (CYP) Allele Nomenclature Database. CYP2D6 allele nomenclature. http://www.cypalleles.ki.se/cyp2d6.htm. Accessed February 25, 2013.
4. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab. 2002;3(1):13-37.
5. Mahgoub A, Idle JR, Dring LG, et al. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2(8038):584-586.
6. Eichelbaum M, Spannbrucker N, Steincke B, et al. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16(3):183-187.
7. Distlerath LM, Reilly PE, Martin MV, et al. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260(15):9057-9067.
8. Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P-450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23(4):455-458.
9. Gonzalez FJ, Vilbois F, Hardwick JP, et al. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Geonomics. 1988;2(2):174-179.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
11. Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405(6788):857-865.
12. Ludwig J, Marcotte DE. Anti-depressants suicide, and drug regulation. J Policy Anal Manage. 2005;24(2):249-272.
13. Isacsson G. Suicide prevention—a medical breakthrough? Acta Psychiatr Scand. 2000;102(2):113-117.
14. Rihmer Z. Can better recognition and treatment of depression reduce suicide rates? A brief review. Eur Psychiatry. 2001;16(7):406-409.
15. Rihmer Z. Decreasing national suicide rates—fact or fiction? World J Biol Psychiatry. 2004;5(1):55-56.
16. Rihmer Z, Akiskal H. Do antidepressants t(h)reat(en) depressives? Toward a clinically judicious formulation of the antidepressant-suicidality FDA advisory in light of declining national suicide statistics from many countries. J Affect Disord. 2006;94(1-3):3-13.
17. Correia C, Santos P, Coutinho AM, et al. Characterization of pharmacogenetically relevant CYP2D6 and ABCB1 gene polymorphisms in a Portuguese population sample. Cell Biochem Funct. 2009;27(4):251-255.
18. Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341(8836):63.-
19. Baumann P, Broly F, Kosel M, et al. Ultrarapid metabolism of clomipramine in a therapy-resistant depressive patient, as confirmed by CYP2 D6 genotyping. Pharmacopsychiatry. 1998;31(2):72.-
20. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants-a pilot study. Clin Pharmacol Ther. 2004;75(5):386-393.
21. Kawanishi C, Lundgren S, Agren H, et al. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol. 2004;59(11):803-807.
22. Zackrisson AL, Lindblom B, Ahlner J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin Pharmacol Ther. 2010;88(3):354-359.
23. Stingl JC, Viviani R. CYP2D6 in the brain: impact on suicidality. Clin Pharmacol Ther. 2011;89(3):352-353.
24. Peñas-Lledó EM, Dorado P, Agüera Z, et al. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol Psychiatry. 2011;16(7):691-692.
25. Siegle I, Fritz P, Eckhardt K, et al. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11(3):237-245.
26. Eichelbaum M. In search of endogenous CYP2D6 substrates. Pharmacogenetics. 2003;13(6):305-306.
27. Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36(2):243-277.
28. Cowen PJ. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci. 2008;29(9):433-436.
29. Kang S, Kang K, Lee K, et al. Characterization of tryptamine 5-hydroxylase and serotonin synthesis in rice plants. Plant Cell Rep. 2007;26(11):2009-2015.
30. Yu AM, Idle JR, Herraiz T, et al. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13(6):307-319.
31. Hiroi T, Imaoka S, Funae Y. Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun. 1998;249(3):838-843.
32. Niznik HB, Tyndale RF, Sallee FR, et al. The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch Biochem Biophys. 1990;276(2):424-432.
33. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153(4):466-476.
34. Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9(3):119-127.
Genetic variations in drug-metabolizing enzymes dramatically affect drug pharmacokinetics and can result in clinically relevant differences in drug efficacy or toxicity. Cytochrome P450 (CYP) enzymes such as CYP2D6 are involved in metabolism of antidepressants, including selective serotonin reuptake inhibitors (SSRIs), which often are a first-line choice for patients with major depressive disorder (MDD).1,2 CYP2D6 is a highly polymorphic gene with 75 allelic variants (CYP2D6*1 to *75) and >30 additional subvariants.3 These variants are associated with phenotypes where CYP2D6 activity is increased, reduced, or lost, which can increase the risk of adverse drug reactions, decrease efficacy, and possibly influence a patient’s suicide risk.
In this article, we review the pharmacogenetics of CYP2D6 and discuss a possible relationship between CYP2D6 genotype and suicidal events during antidepressant treatment for MDD.
CYP2D6: Many variants
CYP450 enzymes are a group of 57 proteins, each coded by a different gene. Five subfamilies in the CYP450 family metabolize most drugs: CYP1A2, CYP3A4, CYP2C19, CYP2E1, and CYP2D6.4
Researchers discovered CYP2D6 in studies of nonpsychotropics (Box).5-9 CYP2D6 is widely expressed in many tissues, with dominant expression in the liver. Although CYP2D6 accounts for 2% of the total CYP450 liver enzyme content, it mediates metabolism in 25% to 30% of drugs in common clinical use and has a major influence on the biotransformation of SSRIs (Table).10
I the late 1970s, 2 groups of researchers noted unexpected serious adverse reactions in studies of debrisoquine,5 a sympatholytic antihypertensive drug, and sparteine,6 an antiarrhythmic and oxytocic alkaloid drug. They observed that 5% to 10% of patients were unable to efficiently metabolize debrisoquine and sparteine and went on to define a genetic polymorphism responsible for these metabolic differences. They also observed that metabolism of antidepressants, antipsychotics, and beta blockers also was defective in these patients.
Further investigations established that the enzyme responsible for debrisoquine metabolism was a cytochrome P450 (CYP) enzyme that is now termed CYP2D6.7 In addition to biochemical evidence, the colocalization of sparteine oxidation deficiency and of the CYP2D6 locus at chromosome 22q13.1 confirmed CYP2D6 as the target gene of the debrisoquine/sparteine polymorphism.8,9
Table
CYP450 enzymes involved in biotransformation of SSRIs
| SSRI | Enzymes involved in biotransformation |
|---|---|
| Citalopram | CYP2C19, CYP2D6, CYP3A4 |
| Escitalopram | CYP2C19, CYP2D6, CYP3A4 |
| Fluoxetine | CYP2D6, CYP2C9, CYP2C19, CYP3A4 |
| Fluvoxamine | CYP1A2, CYP2D6 |
| Paroxetine | CYP2D6, CYP3A4 |
| Sertraline | CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
| CYP: cytochrome P450; SSRI: selective serotonin reuptake inhibitors Source: Reference 10 | |
Approximately 100 polymorphic CYP2D6 alleles (variants) have been identified.3 These alleles are active, resulting in normal CYP2D6 enzyme activity, or inactive, leading to decreased enzyme activity. Genotyping for most common CYP2D6 alleles in ethnically defined populations can predict poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultra-rapid metabolizers (UMs) with high accuracy.11 PMs are compound heterozygous for inactivating alleles or homozygous for an inactivating variant. IMs carry one functional allele and one nonfunctional allele but may demonstrate a range of enzyme activity levels. EMs have 2 functional gene copies and UMs have >2 functional genes from gene duplication, resulting in ultra-rapid metabolism.
Suicide and CYP2D6 status
The widespread use of antidepressants appears to have led to significant decline in suicide rates in many countries.12 Based on an investigation of suicide mortality in 27 countries from 1980 to 2000, Ludwig and Marcotte12 found that faster growth in SSRI sales per capita was associated with larger declines in suicide rates. This finding was not confounded by other suicide risk factors such as unemployment, sex, age, or divorce rate.12 Countries such as Germany, Austria, Estonia, Switzerland, Sweden, Denmark, Hungary, and Slovenia—which had the highest suicide rate in the world 20 years ago (20 to 46 per 100,000 per year)—have had impressive declines in suicide rates (24% to 57% in the last 2 decades) with a marked (6- to 8-fold) increase in SSRI prescriptions during the same period.13-15 On the other hand, a few countries, such as Portugal and Spain, have experienced dramatic increases (58% and 86%, respectively) in the suicide rate with a similar increase in SSRI prescribing during the same 20-year period.16
A review of the distribution of CYP2D6 genotype among countries indicates a south/north gradient of CYP2D6 gene duplications, which indicate UM status.16 The proportion of UMs increases by almost 2-fold in southern European countries (8.4% and 7% to 10% for Portugal and Spain, respectively) compared with northern European countries (1% to 2% and 3.6% for Sweden and Germany, respectively); this south/north trend extends to Africa.17 The prevalence of CYP2D6 UMs is lower in northern countries, where increased anti-depressant use appears to have reduced suicide rates, and higher in southern countries, where suicide rates increased despite higher antidepressant use.
Case reports and observational studies18-21 suggest that compared with other CYP2D6 phenotypes, UMs may need to take higher doses of antidepressants to achieve therapeutic response. In a case report, Bertilsson et al18 described 2 patients who were UMs and required high doses of nortriptyline and clomipramine to obtain appropriate plasma drug concentrations. Baumann et al19 described a depressed patient with CYP2D6 gene duplication who required higher-than-usual doses of clomipramine. Rau et al20 found a 3-fold increase in the frequency of UMs in a group of 16 depressed German patients who did not respond to SSRIs or serotonin–norepinephrine reuptake inhibitors, both of which are metabolized by CYP2D6. Kawanishi et al21 found a significantly greater prevalence of UMs among 81 Nordic patients who did not respond to SSRIs compared with the general population.
Because suicidality may be caused by inadequately treated depressive illness, MDD patients who are UMs may be more likely to commit suicide because of suboptimal antidepressant levels. In a 2010 Swedish study, Zackrisson et al22 found that compared with those who died of other causes, significantly more individuals who committed suicide had >2 active CYP2D6 genes. Stingl et al23 found that among 285 depressed German patients, UMs had an elevated risk of having a high suicidality score compared with individuals with other genotypes, after adjusting for sex, baseline score on the Hamilton Depression Rating Scale (after excluding item 3 for suicidality), and number of previous depressive episodes. Other researchers found that patients with eating disorders who are UMs have a greater risk of suicidal behavior.24 Although none of these 3 studies specified if these patients were treated with antidepressants, the association between CYP2D6 gene duplication and suicide risk suggests CYP2D6’s role in suicide risk might not be related solely to antidepressant metabolism.
Effects on serotonin, dopamine
CYP2D6 is expressed in the brain and localized primarily in large principle cells of the hippocampus and Purkinje cells of the cerebellum, with no expression in other brain regions such as glial cells.25 This heterogeneous expression among brain regions and cell types indicates that in addition to its role in metabolizing drugs, CYP2D6 might influence neurotransmitter levels. In vitro and in vivo animal studies suggest that CYP2D6 plays a role in biotransformation of serotonin and dopamine.26,27
Serotonin is likely to play a causal role in the pathophysiology of depression, and depressed patients have abnormalities in serotonin activity.28 Serotonin is generated primarily from the transformation of tryptophan by tryptophan decarboxylase and tryptamine 5-hydroxylase.29 Yu et al27 found that CYP2D6 may be an additional pathway to regenerate serotonin through O-demethylation from 5-methoxytryptamine, but it is unclear what proportion of the physiologic pool of serotonin in synaptic nerve terminals is generated through the CYP2D6 pathway. However, this discovery provides a mechanistic basis of CYP2D6 involvement in the endogenous serotonin balance and by extension, in serotonergic physiology and neuropsychiatric disorders such as depression.30 Because SSRIs target the serotonergic pathway, baseline levels of serotonin and all related components of this pathway—including CYP2D6—are likely to help determine a patient’s response to SSRIs.
Dopamine also is generated from tyramine through CYP2D6,31 and distribution of CYP2D6 in the brain follows that of dopamine nerve terminals.32 The serotonergic system has strong anatomical and functional interaction with the dopaminergic system,33 and imbalance between serotonin and dopamine activity is thought to give rise to behavioral changes,2 which play an important role in the development of anxiety and impulsivity.
CYP2D6 in clinical practice
Although research into a possible link between CYP2D6 status and suicide risk in depressed patients treated with antidepressants is ongoing, at present this connection is speculative. More studies are warranted to reveal the exact role of CYP2D6 in response to SSRI treatment and suicide risk.
Knowledge of this potential association can help clinicians keep CYP450 genotyping in mind when prescribing antidepressants to depressed patients. The FDA has approved a pharmacogenetic test to analyze polymorphisms of CYP2D6 and CYP2C19.34 The results of such testing might guide pharmacotherapy for depressed patients, including medication selection and dosing. For example, a patient who is a PM might be started at a lower antidepressant dosage to avoid potential adverse drug effects, whereas it might be appropriate to prescribe a higher starting dose for a UM patient to achieve an effective drug concentration.
Related Resources
- Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, et al. CYP2D6 and the severity of suicide attempts. Pharmacogenomics. 2012;13(2):179-184.
- Blasco-Fontecilla H, Peñas-Lledó E, Vaquero-Lorenzo C, et al. CYP2D6 polymorphism and mental and personality disorders in suicide attempters [published online February 11, 2013]. J Pers Disord. doi: 10.1521/pedi_2013_27_080.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Marwah Shahid and Ijlal Yazdani for their assistance with this article.
Genetic variations in drug-metabolizing enzymes dramatically affect drug pharmacokinetics and can result in clinically relevant differences in drug efficacy or toxicity. Cytochrome P450 (CYP) enzymes such as CYP2D6 are involved in metabolism of antidepressants, including selective serotonin reuptake inhibitors (SSRIs), which often are a first-line choice for patients with major depressive disorder (MDD).1,2 CYP2D6 is a highly polymorphic gene with 75 allelic variants (CYP2D6*1 to *75) and >30 additional subvariants.3 These variants are associated with phenotypes where CYP2D6 activity is increased, reduced, or lost, which can increase the risk of adverse drug reactions, decrease efficacy, and possibly influence a patient’s suicide risk.
In this article, we review the pharmacogenetics of CYP2D6 and discuss a possible relationship between CYP2D6 genotype and suicidal events during antidepressant treatment for MDD.
CYP2D6: Many variants
CYP450 enzymes are a group of 57 proteins, each coded by a different gene. Five subfamilies in the CYP450 family metabolize most drugs: CYP1A2, CYP3A4, CYP2C19, CYP2E1, and CYP2D6.4
Researchers discovered CYP2D6 in studies of nonpsychotropics (Box).5-9 CYP2D6 is widely expressed in many tissues, with dominant expression in the liver. Although CYP2D6 accounts for 2% of the total CYP450 liver enzyme content, it mediates metabolism in 25% to 30% of drugs in common clinical use and has a major influence on the biotransformation of SSRIs (Table).10
I the late 1970s, 2 groups of researchers noted unexpected serious adverse reactions in studies of debrisoquine,5 a sympatholytic antihypertensive drug, and sparteine,6 an antiarrhythmic and oxytocic alkaloid drug. They observed that 5% to 10% of patients were unable to efficiently metabolize debrisoquine and sparteine and went on to define a genetic polymorphism responsible for these metabolic differences. They also observed that metabolism of antidepressants, antipsychotics, and beta blockers also was defective in these patients.
Further investigations established that the enzyme responsible for debrisoquine metabolism was a cytochrome P450 (CYP) enzyme that is now termed CYP2D6.7 In addition to biochemical evidence, the colocalization of sparteine oxidation deficiency and of the CYP2D6 locus at chromosome 22q13.1 confirmed CYP2D6 as the target gene of the debrisoquine/sparteine polymorphism.8,9
Table
CYP450 enzymes involved in biotransformation of SSRIs
| SSRI | Enzymes involved in biotransformation |
|---|---|
| Citalopram | CYP2C19, CYP2D6, CYP3A4 |
| Escitalopram | CYP2C19, CYP2D6, CYP3A4 |
| Fluoxetine | CYP2D6, CYP2C9, CYP2C19, CYP3A4 |
| Fluvoxamine | CYP1A2, CYP2D6 |
| Paroxetine | CYP2D6, CYP3A4 |
| Sertraline | CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
| CYP: cytochrome P450; SSRI: selective serotonin reuptake inhibitors Source: Reference 10 | |
Approximately 100 polymorphic CYP2D6 alleles (variants) have been identified.3 These alleles are active, resulting in normal CYP2D6 enzyme activity, or inactive, leading to decreased enzyme activity. Genotyping for most common CYP2D6 alleles in ethnically defined populations can predict poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultra-rapid metabolizers (UMs) with high accuracy.11 PMs are compound heterozygous for inactivating alleles or homozygous for an inactivating variant. IMs carry one functional allele and one nonfunctional allele but may demonstrate a range of enzyme activity levels. EMs have 2 functional gene copies and UMs have >2 functional genes from gene duplication, resulting in ultra-rapid metabolism.
Suicide and CYP2D6 status
The widespread use of antidepressants appears to have led to significant decline in suicide rates in many countries.12 Based on an investigation of suicide mortality in 27 countries from 1980 to 2000, Ludwig and Marcotte12 found that faster growth in SSRI sales per capita was associated with larger declines in suicide rates. This finding was not confounded by other suicide risk factors such as unemployment, sex, age, or divorce rate.12 Countries such as Germany, Austria, Estonia, Switzerland, Sweden, Denmark, Hungary, and Slovenia—which had the highest suicide rate in the world 20 years ago (20 to 46 per 100,000 per year)—have had impressive declines in suicide rates (24% to 57% in the last 2 decades) with a marked (6- to 8-fold) increase in SSRI prescriptions during the same period.13-15 On the other hand, a few countries, such as Portugal and Spain, have experienced dramatic increases (58% and 86%, respectively) in the suicide rate with a similar increase in SSRI prescribing during the same 20-year period.16
A review of the distribution of CYP2D6 genotype among countries indicates a south/north gradient of CYP2D6 gene duplications, which indicate UM status.16 The proportion of UMs increases by almost 2-fold in southern European countries (8.4% and 7% to 10% for Portugal and Spain, respectively) compared with northern European countries (1% to 2% and 3.6% for Sweden and Germany, respectively); this south/north trend extends to Africa.17 The prevalence of CYP2D6 UMs is lower in northern countries, where increased anti-depressant use appears to have reduced suicide rates, and higher in southern countries, where suicide rates increased despite higher antidepressant use.
Case reports and observational studies18-21 suggest that compared with other CYP2D6 phenotypes, UMs may need to take higher doses of antidepressants to achieve therapeutic response. In a case report, Bertilsson et al18 described 2 patients who were UMs and required high doses of nortriptyline and clomipramine to obtain appropriate plasma drug concentrations. Baumann et al19 described a depressed patient with CYP2D6 gene duplication who required higher-than-usual doses of clomipramine. Rau et al20 found a 3-fold increase in the frequency of UMs in a group of 16 depressed German patients who did not respond to SSRIs or serotonin–norepinephrine reuptake inhibitors, both of which are metabolized by CYP2D6. Kawanishi et al21 found a significantly greater prevalence of UMs among 81 Nordic patients who did not respond to SSRIs compared with the general population.
Because suicidality may be caused by inadequately treated depressive illness, MDD patients who are UMs may be more likely to commit suicide because of suboptimal antidepressant levels. In a 2010 Swedish study, Zackrisson et al22 found that compared with those who died of other causes, significantly more individuals who committed suicide had >2 active CYP2D6 genes. Stingl et al23 found that among 285 depressed German patients, UMs had an elevated risk of having a high suicidality score compared with individuals with other genotypes, after adjusting for sex, baseline score on the Hamilton Depression Rating Scale (after excluding item 3 for suicidality), and number of previous depressive episodes. Other researchers found that patients with eating disorders who are UMs have a greater risk of suicidal behavior.24 Although none of these 3 studies specified if these patients were treated with antidepressants, the association between CYP2D6 gene duplication and suicide risk suggests CYP2D6’s role in suicide risk might not be related solely to antidepressant metabolism.
Effects on serotonin, dopamine
CYP2D6 is expressed in the brain and localized primarily in large principle cells of the hippocampus and Purkinje cells of the cerebellum, with no expression in other brain regions such as glial cells.25 This heterogeneous expression among brain regions and cell types indicates that in addition to its role in metabolizing drugs, CYP2D6 might influence neurotransmitter levels. In vitro and in vivo animal studies suggest that CYP2D6 plays a role in biotransformation of serotonin and dopamine.26,27
Serotonin is likely to play a causal role in the pathophysiology of depression, and depressed patients have abnormalities in serotonin activity.28 Serotonin is generated primarily from the transformation of tryptophan by tryptophan decarboxylase and tryptamine 5-hydroxylase.29 Yu et al27 found that CYP2D6 may be an additional pathway to regenerate serotonin through O-demethylation from 5-methoxytryptamine, but it is unclear what proportion of the physiologic pool of serotonin in synaptic nerve terminals is generated through the CYP2D6 pathway. However, this discovery provides a mechanistic basis of CYP2D6 involvement in the endogenous serotonin balance and by extension, in serotonergic physiology and neuropsychiatric disorders such as depression.30 Because SSRIs target the serotonergic pathway, baseline levels of serotonin and all related components of this pathway—including CYP2D6—are likely to help determine a patient’s response to SSRIs.
Dopamine also is generated from tyramine through CYP2D6,31 and distribution of CYP2D6 in the brain follows that of dopamine nerve terminals.32 The serotonergic system has strong anatomical and functional interaction with the dopaminergic system,33 and imbalance between serotonin and dopamine activity is thought to give rise to behavioral changes,2 which play an important role in the development of anxiety and impulsivity.
CYP2D6 in clinical practice
Although research into a possible link between CYP2D6 status and suicide risk in depressed patients treated with antidepressants is ongoing, at present this connection is speculative. More studies are warranted to reveal the exact role of CYP2D6 in response to SSRI treatment and suicide risk.
Knowledge of this potential association can help clinicians keep CYP450 genotyping in mind when prescribing antidepressants to depressed patients. The FDA has approved a pharmacogenetic test to analyze polymorphisms of CYP2D6 and CYP2C19.34 The results of such testing might guide pharmacotherapy for depressed patients, including medication selection and dosing. For example, a patient who is a PM might be started at a lower antidepressant dosage to avoid potential adverse drug effects, whereas it might be appropriate to prescribe a higher starting dose for a UM patient to achieve an effective drug concentration.
Related Resources
- Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, et al. CYP2D6 and the severity of suicide attempts. Pharmacogenomics. 2012;13(2):179-184.
- Blasco-Fontecilla H, Peñas-Lledó E, Vaquero-Lorenzo C, et al. CYP2D6 polymorphism and mental and personality disorders in suicide attempters [published online February 11, 2013]. J Pers Disord. doi: 10.1521/pedi_2013_27_080.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Marwah Shahid and Ijlal Yazdani for their assistance with this article.
1. Meyer UA, Amrein R, Balant LP, et al. Antidepressants and drug-metabolizing enzymes—expert group report. Acta Psychiatr Scand. 1996;93(2):71-79.
2. Kroemer HK, Eichelbaum M. “It’s the genes stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56(26):2285-2298.
3. The Human Cytochrome P450 (CYP) Allele Nomenclature Database. CYP2D6 allele nomenclature. http://www.cypalleles.ki.se/cyp2d6.htm. Accessed February 25, 2013.
4. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab. 2002;3(1):13-37.
5. Mahgoub A, Idle JR, Dring LG, et al. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2(8038):584-586.
6. Eichelbaum M, Spannbrucker N, Steincke B, et al. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16(3):183-187.
7. Distlerath LM, Reilly PE, Martin MV, et al. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260(15):9057-9067.
8. Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P-450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23(4):455-458.
9. Gonzalez FJ, Vilbois F, Hardwick JP, et al. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Geonomics. 1988;2(2):174-179.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
11. Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405(6788):857-865.
12. Ludwig J, Marcotte DE. Anti-depressants suicide, and drug regulation. J Policy Anal Manage. 2005;24(2):249-272.
13. Isacsson G. Suicide prevention—a medical breakthrough? Acta Psychiatr Scand. 2000;102(2):113-117.
14. Rihmer Z. Can better recognition and treatment of depression reduce suicide rates? A brief review. Eur Psychiatry. 2001;16(7):406-409.
15. Rihmer Z. Decreasing national suicide rates—fact or fiction? World J Biol Psychiatry. 2004;5(1):55-56.
16. Rihmer Z, Akiskal H. Do antidepressants t(h)reat(en) depressives? Toward a clinically judicious formulation of the antidepressant-suicidality FDA advisory in light of declining national suicide statistics from many countries. J Affect Disord. 2006;94(1-3):3-13.
17. Correia C, Santos P, Coutinho AM, et al. Characterization of pharmacogenetically relevant CYP2D6 and ABCB1 gene polymorphisms in a Portuguese population sample. Cell Biochem Funct. 2009;27(4):251-255.
18. Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341(8836):63.-
19. Baumann P, Broly F, Kosel M, et al. Ultrarapid metabolism of clomipramine in a therapy-resistant depressive patient, as confirmed by CYP2 D6 genotyping. Pharmacopsychiatry. 1998;31(2):72.-
20. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants-a pilot study. Clin Pharmacol Ther. 2004;75(5):386-393.
21. Kawanishi C, Lundgren S, Agren H, et al. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol. 2004;59(11):803-807.
22. Zackrisson AL, Lindblom B, Ahlner J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin Pharmacol Ther. 2010;88(3):354-359.
23. Stingl JC, Viviani R. CYP2D6 in the brain: impact on suicidality. Clin Pharmacol Ther. 2011;89(3):352-353.
24. Peñas-Lledó EM, Dorado P, Agüera Z, et al. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol Psychiatry. 2011;16(7):691-692.
25. Siegle I, Fritz P, Eckhardt K, et al. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11(3):237-245.
26. Eichelbaum M. In search of endogenous CYP2D6 substrates. Pharmacogenetics. 2003;13(6):305-306.
27. Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36(2):243-277.
28. Cowen PJ. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci. 2008;29(9):433-436.
29. Kang S, Kang K, Lee K, et al. Characterization of tryptamine 5-hydroxylase and serotonin synthesis in rice plants. Plant Cell Rep. 2007;26(11):2009-2015.
30. Yu AM, Idle JR, Herraiz T, et al. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13(6):307-319.
31. Hiroi T, Imaoka S, Funae Y. Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun. 1998;249(3):838-843.
32. Niznik HB, Tyndale RF, Sallee FR, et al. The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch Biochem Biophys. 1990;276(2):424-432.
33. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153(4):466-476.
34. Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9(3):119-127.
1. Meyer UA, Amrein R, Balant LP, et al. Antidepressants and drug-metabolizing enzymes—expert group report. Acta Psychiatr Scand. 1996;93(2):71-79.
2. Kroemer HK, Eichelbaum M. “It’s the genes stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56(26):2285-2298.
3. The Human Cytochrome P450 (CYP) Allele Nomenclature Database. CYP2D6 allele nomenclature. http://www.cypalleles.ki.se/cyp2d6.htm. Accessed February 25, 2013.
4. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab. 2002;3(1):13-37.
5. Mahgoub A, Idle JR, Dring LG, et al. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2(8038):584-586.
6. Eichelbaum M, Spannbrucker N, Steincke B, et al. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16(3):183-187.
7. Distlerath LM, Reilly PE, Martin MV, et al. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260(15):9057-9067.
8. Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P-450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23(4):455-458.
9. Gonzalez FJ, Vilbois F, Hardwick JP, et al. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Geonomics. 1988;2(2):174-179.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
11. Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405(6788):857-865.
12. Ludwig J, Marcotte DE. Anti-depressants suicide, and drug regulation. J Policy Anal Manage. 2005;24(2):249-272.
13. Isacsson G. Suicide prevention—a medical breakthrough? Acta Psychiatr Scand. 2000;102(2):113-117.
14. Rihmer Z. Can better recognition and treatment of depression reduce suicide rates? A brief review. Eur Psychiatry. 2001;16(7):406-409.
15. Rihmer Z. Decreasing national suicide rates—fact or fiction? World J Biol Psychiatry. 2004;5(1):55-56.
16. Rihmer Z, Akiskal H. Do antidepressants t(h)reat(en) depressives? Toward a clinically judicious formulation of the antidepressant-suicidality FDA advisory in light of declining national suicide statistics from many countries. J Affect Disord. 2006;94(1-3):3-13.
17. Correia C, Santos P, Coutinho AM, et al. Characterization of pharmacogenetically relevant CYP2D6 and ABCB1 gene polymorphisms in a Portuguese population sample. Cell Biochem Funct. 2009;27(4):251-255.
18. Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341(8836):63.-
19. Baumann P, Broly F, Kosel M, et al. Ultrarapid metabolism of clomipramine in a therapy-resistant depressive patient, as confirmed by CYP2 D6 genotyping. Pharmacopsychiatry. 1998;31(2):72.-
20. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants-a pilot study. Clin Pharmacol Ther. 2004;75(5):386-393.
21. Kawanishi C, Lundgren S, Agren H, et al. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol. 2004;59(11):803-807.
22. Zackrisson AL, Lindblom B, Ahlner J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin Pharmacol Ther. 2010;88(3):354-359.
23. Stingl JC, Viviani R. CYP2D6 in the brain: impact on suicidality. Clin Pharmacol Ther. 2011;89(3):352-353.
24. Peñas-Lledó EM, Dorado P, Agüera Z, et al. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol Psychiatry. 2011;16(7):691-692.
25. Siegle I, Fritz P, Eckhardt K, et al. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11(3):237-245.
26. Eichelbaum M. In search of endogenous CYP2D6 substrates. Pharmacogenetics. 2003;13(6):305-306.
27. Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36(2):243-277.
28. Cowen PJ. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci. 2008;29(9):433-436.
29. Kang S, Kang K, Lee K, et al. Characterization of tryptamine 5-hydroxylase and serotonin synthesis in rice plants. Plant Cell Rep. 2007;26(11):2009-2015.
30. Yu AM, Idle JR, Herraiz T, et al. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13(6):307-319.
31. Hiroi T, Imaoka S, Funae Y. Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun. 1998;249(3):838-843.
32. Niznik HB, Tyndale RF, Sallee FR, et al. The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch Biochem Biophys. 1990;276(2):424-432.
33. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153(4):466-476.
34. Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9(3):119-127.

Hospital Value‐Based Purchasing
The Centers for Medicaid and Medicare Services' (CMS) Hospital Inpatient Value‐Based Purchasing (VBP) Program, which was signed into law as part of the Patient Protection and Affordable Care Act of 2010, aims to incentivize inpatient providers to deliver high‐value, as opposed to high‐volume, healthcare.[1] Beginning on October 1, 2012, the start of the 2013 fiscal year (FY), hospitals participating in the VBP program became eligible for a variety of performance‐based incentive payments from CMS. These payments are based on an acute care hospital's ability to meet performance measurements in 6 care domains: (1) patient safety, (2) care coordination, (3) clinical processes and outcomes, (4) population or community health, (5) efficiency and cost reduction, and (6) patient‐ and caregiver‐centered experience.[2] The VBP program's ultimate purpose is to enable CMS to improve the health of Medicare beneficiaries by purchasing better care for them at a lower cost. These 3 characteristics of careimproved health, improved care, and lower costsare the foundation of CMS' conception of value.[1, 2] They are closely related to an economic conception of value, which is the difference between an intervention's benefit and its cost.
Although in principle not a new idea, the formal mandate of hospitals to provide high‐value healthcare through financial incentives marks an important change in Medicare and Medicaid policy. In this opportune review of VBP, we first discuss the relevant historical changes in the reimbursement environment of US hospitals that have set the stage for VBP. We then describe the structure of CMS' VBP program, with a focus on which facilities are eligible to participate in the program, the specific outcomes measured and incentivized, how rewards and penalties are allocated, and how the program will be funded. In an effort to anticipate some of the issues that lie ahead, we then highlight a number of potential challenges to the success of VBP, and discuss how VBP will impact the delivery and reimbursement of inpatient care services. We conclude by examining how the VBP program is likely to evolve over time.
HISTORICAL CONTEXT FOR VBP
Over the last decade, CMS has embarked on a number of initiatives to incentivize the provision of higher‐quality and more cost‐effective care. For example, in 2003, CMS implemented a national pay‐for‐performance (P4P) pilot project called the Premier Hospital Quality Incentive Demonstration (HQID).[3, 4] HQID, which ran for 6 years, tracked and rewarded the performance of 216 hospitals in 6 healthcare service domains: (1) acute myocardial infarction (AMI), (2) congestive heart failure (CHF), (3) pneumonia, (4) coronary artery bypass graft surgery, (5) hip and knee replacement surgery, and (6) perioperative management of surgical patients (including prevention of surgical site infections).[4] CMS then introduced its Hospital Compare Web site in 2005 to facilitate public reporting of hospital‐level quality outcomes.[3, 5] This Web site provides the public with access to data on hospital performance across a wide array of measures of process quality, clinical outcomes, spending, and resource utilization.[5] Next, in October 2008, CMS stopped reimbursing hospitals for a number of costly and common hospital‐acquired complications, including hospital‐acquired bloodstream infections and urinary tract infections, patient falls, and pressure ulcers.[3, 6] VBP is the latest and most comprehensive step that CMS has taken in its decade‐long effort to shift from volume to value‐based compensation for inpatient care.
Although CMS appears fully invested in using performance incentives to increase healthcare value, existing evidence of the effects of P4P on patient outcomes remains quite mixed.[7] On one hand, an analysis of an inpatient P4P program sponsored by the United Kingdom's National Health Service's (NHS) suggests that P4P may improve quality and save lives; indeed, hospitals that participated in the NHS P4P program significantly reduced inpatient mortality from pneumonia, saving an estimated 890 lives.[8] Additional empirical work suggests that the HQID was also associated with early improvements in healthcare quality.[9] However, a subsequent long‐term analysis found that participation in HQID had no discernible effect on 30‐day mortality rates.[10] Moreover, a meta‐analysis of P4P incentives for individual practitioners found few methodologically robust studies of P4P for clinicians and concluded that P4P's effects on individual practice patterns and outcomes remain largely uncertain.[11]
VBP: STRUCTURE AND DESIGN
This section reviews the structure of the VBP program. We describe current VBP eligibility criteria and sources of funding for the program, how hospitals participating in VBP are evaluated, and how VBP incentives for FY 2013 have been calculated.
Hospital Eligibility for VBP
All acute care hospitals in the United States (excluding Maryland) that are not psychiatric hospitals, rehabilitation hospitals, long‐term care facilities, children's hospitals, or cancer hospitals are eligible to participate in VBP in FY 2013 (full eligibility criteria is outlined in Table 1). For FY 2013, CMS chose to incentivize measures in just 2 care domains: (1) clinical processes of care and (2) patient experience of care. To be eligible for VBP in FY 2013, a hospital must report at least 10 cases each in at least 4 of 12 measures included in the clinical processes of care domain (Table 2), and/or must have at least 100 completed Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS). Designed and validated by CMS, the HCAHPS survey provides hospitals with a standardized instrument for gathering information about patient satisfaction with, and perspectives on, their hospital care.[12] HCAHPS will be used to assess 8 patient experience of care measures (Table 3).
|
| Inclusion criteria |
| Acute care hospital |
| Located in all 50 US states or District of Columbia (excluding Maryland) |
| Has at least 10 cases in at least 4 of 12 clinical process of care measures and/or at least 100 completed HCAHPS surveys |
| Exclusion criteria |
| Psychiatric, rehabilitation, long‐term care, children's or cancer hospital |
| Does not participate in Hospital Inpatient Quality Reporting Program during the VBP performance period |
| Cited by the Secretary of HHS for significant patient safety violations during performance period |
| Hospital does not meet minimum reporting requirements for number of cases, process measures, and surveys needed to participate in VBP |
| Disease Process | Process of Care Measure |
|---|---|
| |
| Acute myocardial infarction | Fibrinolytic therapy received within 30 minutes of hospital arrival |
| Primary percutaneous coronary intervention received within 90 minutes of hospital arrival | |
| Heart failure | Discharge instructions provided |
| Pneumonia | Blood cultures performed in the emergency department prior to initial antibiotic received in hospital |
| Initial antibiotic selection for community‐acquired pneumonia in immunocompetent patient | |
| Healthcare‐associated infections | Prophylactic antibiotic received within 1 hour prior to surgical incision |
| Prophylactic antibiotic selection for surgical patients | |
| Prophylactic antibiotics discontinued within 24 hours after surgery ends | |
| Cardiac surgery patients with controlled 6:00 am postoperative serum glucose | |
| Surgeries | Surgery patients on ‐blocker prior to arrival that received ‐blocker during perioperative period |
| Surgery patients with recommended venous thromboembolism prophylaxis ordered | |
| Surgery patients who received appropriate venous thromboembolism prophylaxis within 24 hours prior to surgery to 24 hours after surgery | |
| Communication with nurses |
| Communication with doctors |
| Responsiveness of hospital staff |
| Pain management |
| Communication about medicines |
| Cleanliness and quietness of hospital environment |
| Discharge information |
| Overall rating of hospital |
Participation in the program is mandatory for eligible hospitals, and CMS estimates that more than 3000 facilities across the United States will participate in FY 2013. Roughly $850 million dollars in VBP incentives will be paid out to these participating hospitals in FY 2013. The program is being financed through a 1% across‐the‐board reduction in FY 2013 diagnosis‐related group (DRG)‐based inpatient payments to participating hospitals. On December 20, 2012, CMS publically announced FY 2013 VBP incentives for all participating hospitals. Each hospital's incentive is retroactive and based on its performance between July 1, 2011 and March 31, 2012.
All data used for calculating VBP incentives is reported to CMS through its Hospital Inpatient Quality Reporting (Hospital IQR) Program, a national program instituted in 2003 that rewards hospitals for reporting designated quality measures. As of 2007, approximately 95% of eligible US hospitals were using the Hospital IQR program.[1] Measures evaluated via chart abstracts and surveys reflect a hospital's performance for its entire patient population, whereas measures assessed with claims data reflect hospital performance only for Medicare patients.
Evaluation of Hospitals
In FY 2013, hospital VBP incentive payments will be based entirely on performance in 2 domains: (1) clinical processes of care (weighted 70%) and (2) patient experience of care (weighted 30%). For each domain, CMS will evaluate each hospital's improvement over time as well as achievement compared to other hospitals in the VBP program. By assessing and rewarding both achievement and improvement, CMS will ensure that lower‐performing hospitals will still be rewarded for making substantial improvements in quality. To evaluate the first metricimprovement over timeCMS will compare a hospital's performance during a given reporting period with its baseline performance 2 years prior to this block of time. A hospital receives improvement points for improving its performance over time. To assess the second metricachievement compared to other hospitals in the VBP programCMS will compare each hospital's performance during a reporting period with the baseline performance (eg, performance 2 years prior to reporting period) of all other hospitals in the VBP program. A hospital is awarded achievement points if its performance exceeds the 50th percentile of all hospitals during the baseline performance period. Improvement scores range from 0 to 9, whereas achievement scores range from 0 to 10. The greater of a hospital's improvement and achievement scores on each VBP measure are used to calculate each hospital's total earned clinical care domain score and total earned HCAHPS base score. Hospitals that lack baseline performance data, which is required to assess improvement, will be evaluated solely on the basis of achievement points.[1] The total earned clinical care domain score is multiplied by 70% to reach the clinical care domain's contribution to a hospital's total performance score.
Each hospital's total patient experience domain, or HCAHPS performance, score consists of 2 components: a total earned HCAHPS base score as described above and a consistency score. The consistency score evaluates the reliability of a hospital's performance across all 8 patient experience of care measures (Table 3). If a hospital is above the 50th percentile of all hospital scores during the baseline period on all 8 measures, then it receives 100% of its consistency points. If a hospital is at the 0 percentile for a given measure, then it receives 0 consistency points for all measures. This provision promotes consistency by harshly penalizing hospitals with extremely poor performance on any 1 specific measure. If 1 or more measures are between the 0 and 50th percentiles, then it will receive a consistency score that takes into account how many measures were below the 50th percentile and their distance from this threshold. Each hospital's total HCAHPS performance score (the sum of total earned HCAHPS base points and consistency points) is then multiplied by 30% to arrive at the patient experience of care domain's contribution to a hospital's total performance score.
Importantly, CMS excluded from its VBP initiative 10 clinical process measures reported in the Hospital IQR Program because they are topped out; that is, almost all hospitals already perform them at very high rates (Table 4). Examples of these topped out process measures include administration of aspirin to all patients with AMI on arrival at the hospital; counseling of patients with AMI, CHF, and pneumonia about smoking cessation; and prescribing angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers to patients with CHF and left ventricular dysfunction.[1]
| Disease Process | Measure |
|---|---|
| |
| Acute myocardial infarction | Aspirin administered on arrival to the emergency department |
| ACEI or ARB prescribed on discharge | |
| Patient counseled about smoking cessation | |
| ‐Blocker prescribed on discharge | |
| Aspirin prescribed at discharge | |
| Heart failure | Patient counseled about smoking cessation |
| Evaluation of left ventricular systolic function | |
| ACEI or ARB prescribed for left ventricular systolic dysfunction | |
| Pneumonia | Patient counseled about smoking cessation |
| Surgical Care Improvement Project | Surgery patients with appropriate hair removal |
Calculation of VBP Incentives and Public Reporting
A hospital's total performance score for FY 2013 is equal to the sum of 70% of its clinical care domain score and 30% of its total HCAHPS performance score. This total performance score is entered into a linear mathematical formula to calculate each hospital's incentive payment. CMS projects that VBP will lead to a net increase in Medicare payments for one‐half of hospitals and a net decrease in payments for the other half of participating facilities.[1]
In December 2012, CMS publicly disclosed information about the initial performance of each hospital in the VBP program. Reported information included: (1) hospital performance for each applicable performance measure, (2) hospital performance by disease condition or procedure, and (3) hospital's total performance score. Initial analyses of this performance data revealed that 1557 hospitals will receive bonus payments under VBP in FY 2013, whereas 1427 hospitals will lose money under this program. Treasure Valley Hospital, a 10‐bed physician‐owned hospital in Boise, Idaho, will receive a 0.83% increase in Medicare payments, the largest payment increase under VBP in 2013. Conversely, Auburn Community Hospital in upstate New York, will suffer the most severe payment reduction: 0.9% per Medicare admission. The penalty will cost Auburn Hospital about $100,000, which is slightly more than 0.1% of its yearly $85 million operating budget.[13] For almost two‐thirds of participating hospitals, FY 2013 Medicare payments will change by <0.25%.[13] Additional information about VBP payments for FY 2013, including the number of hospitals who received VBP incentives and the size and range of these payments, is now accessible to the public through CMS' Hospital Compare Web site (
CHALLENGES OF VBP
As the Medicare VBP program evolves, and hospitals confront ever‐larger financial incentives to deliver high‐value as opposed to high‐volume care, it will be important to recognize limitations of the VBP program as they arise. Here we briefly discuss several conceptual and implementation challenges that physicians and policymakers should consider when assessing the merits of VBP in promoting high‐quality healthcare.
Rigorous and Continuous Evaluation of VBP Programs
The main premise of using VBP to incentivize hospitals to deliver high‐quality cost‐effective care is that the process measures used to determine hospital quality do impact patient outcomes. However, it is already well established that improvements in measures of process quality are not always associated with improvements in patient outcomes.[14, 15, 16] Moreover, incentivizing specific process measures encourages hospitals to shift resources away from other aspects of care delivery, which may have ambiguous, or even deleterious, effects on patient outcomes. Although incentives ideally push hospitals to shift resources away from low‐quality care toward high‐quality care, in practice this is not always the case. Hospital resources may instead be drawn away from areas that are not yet incented by VBP, but for which improvements in quality of care are desperately needed. The same empirical focus behind using VBP to incentivize hospitals to improve patient outcomes efficiently should be used to evaluate whether VBP is continually meeting its stated goals: reducing overall patient morbidity and mortality and improving patient satisfaction at ideally lower cost. The experience of the US education system with public policies designed to improve student testing performance may serve as a cautionary example here. Such policies, which provide financial rewards to schools whose students perform well on standardized tests, can indeed raise testing performance. However, these policies also lead educators to teach to the test, and to neglect important topics that are not tested on standardized exams.[17]
Prioritization of Process Measures
As payment incentives for VBP currently stand, process measures are weighted equally regardless of the clinical benefits they generate and the resources required to achieve improvements in process quality. For instance, 2 process measures, continuing home ‐blocker medications for patients with coronary artery disease undergoing surgery and early percutaneous coronary intervention for patients with AMI, may be weighted equally as process measures although both their clinical benefits and the costs of implementation are very different. Some hospitals responding to VBP incentives may choose to invest in areas where their ability to earn VBP incentive payments is high and the costs of improvement are low, although those areas may not be where interventions are most needed because clinical outcomes could be most improved. Recognizing that process measures have heterogeneous benefits and costs of implementation is important when prioritizing their reimbursement in VBP.
Measuring Improvements in Hospital Quality
Tying hospital financial compensation to hospital quality implies that measures of hospital quality should be robust. To incentivize hospitals to improve quality not only relative to other hospitals but to themselves in the past, the VBP program has established a baseline performance for each hospital. Each hospital is compared to its baseline performance in subsequent evaluation periods. Thus, properly measuring a hospital's baseline performance is important. During a given baseline period, some hospitals may have better or worse outcomes than their steady state due to random variation alone. Some hospitals deemed to have a low baseline will experience improvements in quality that are not related to active efforts to improve quality but through chance alone. Similarly, some hospitals deemed to have a high baseline will experience reductions in quality through chance. Of course, neither of these changes should be subject to differences in reimbursement because they do not reflect actual organizational changes made by the hospitals. The VBP program has made significant efforts to address this issue by requiring participating hospitals to have a large enough sample of cases such that estimated rates of process quality adherence meet a reliability threshold (ie, are likely to be consistent over time rather than vary substantially through chance alone). However, not all process measures exhibit high reliability, particularly those for which adverse events are rare (eg, foreign objects retained after surgery, air embolisms, and blood incompatibility). Ultimately, CMS's decision to balance the need for statistically reliable data with the goal of including as many hospitals as possible in the VBP program will require ongoing reevaluation of this issue.
Choosing Hospital Comparators Appropriately
In the current VBP program, hospitals will be evaluated in part by how they compare to hospitals nationally. However, studies of regional variation in healthcare have demonstrated large variations in practice patterns across the United States,[18, 19, 20] raising the question of whether hospitals should, at least initially, be compared to hospitals in the same geographic area. Although the ultimate goal of VBP should be to hold hospitals to a national standard, local practice patterns are not easily modified within 1‐ to 2‐year timeframes. Initially comparing hospitals to a national rather than local standard may unfairly penalize hospitals that are relative underperformers nationally but overperformers regionally. Although CMS's policy to reward improvement within hospitals over time mitigates issues arising from a cross‐sectional comparison of hospitals, the issue still remains if many hospitals within a region not only underperform relative to other hospitals nationally but also fail to demonstrate improvement. More broadly, this issue extends to differences across hospitals in factors that impact their ability to meet VBP goals. These factors may include, for example, hospital size, profitability, patient case and insurance mix, and presence of an electronic medical record. Comparing hospitals with vastly different abilities to achieve VBP goals and improve quickly may amount to inequitable policy.
Continual Evaluation of Topped‐Out Measures
Process measures that are met at high rates at nearly all hospitals are not used in evaluations by CMS for VBP. An assumption underlying CMS' decision to not reward hospitals for achieving these topped‐out measures is that once physicians and hospitals make cognitive and system‐level improvements that improve process quality, these gains will persist after the incentive is removed. Thus, CMS hopes and anticipates that although performance incentives will make it easier for well‐meaning physicians to learn to do the right thing, doctors will continue to do the right things for patients after these incentives are removed.[21, 22] Although this assumption may generally be accurate, it is important to continue to evaluate whether measures that are currently topped out continue to remain adequately performed, because rewarding new quality measures will necessarily lead hospitals to reallocate resources away from other clinical activities. Although we hope that the continued public reporting of topped‐out measures will prevent declines in performance on these measures, policy makers and clinicians should be aware that the lack of financial incentives for topped‐out measures may result in declines in quality. To this point, an analysis of 35 Kaiser Permanente facilities from 1997 to 2007 demonstrated that the removal of financial incentives for diabetic retinopathy and cervical cancer screening was associated with subsequent declines in performance of 3% and 1.6% per year, respectively.[23]
Will VBP Incentives Be Large Enough to Change Practice Patterns?
The VBP Program's ability to influence change depends, at least in part, on how the incentives offered under this program compare to the magnitude of the investments that hospitals must make to achieve a given reward. In general, larger incentives are necessary to motivate more significant changes in behavior or to influence organizations to invest the resources needed to achieve change. The incentives offered under VBP in FY 2013 are quite modest. Almost two‐thirds of participating hospitals will see their FY 2013 Medicare revenues change by <0.25%, roughly $125,000 at most.[13, 24] Although these incentives may motivate hospitals that can improve performance and achievement with very modest investments, they may have little impact on organizations that need to make significant upfront investments in care processes to achieve sustainable improvements in care quality. As CMS increases the size of VBP incentives over the next 2 to 4 years, it will also hold hospitals accountable for a broader and increasingly complex set of outcomes. Improving these outcomes may require investments in areas such as information technology and process improvement that far surpass the VBP incentive reward.
Moreover, prior research suggests that financial incentives like those available under VBP may contribute only slightly to performance improvements when public reporting already exists. For example, in a 2‐year study of 613 US hospitals implementing pay‐for‐performance plus public reporting or public reporting only, pay for performance plus public reporting was associated with only a 2.6% to 4.1% increase in a composite measure of quality when compared to hospitals with public reporting only.[9] Similarly, a study of 54 hospitals participating in the CMS pay for performance pilot initiative found no significant improvement in quality of care or outcomes for AMI when compared to 446 control hospitals.[25] A long‐term analysis of pay for performance in the Medicare Premier Hospital Quality Incentive Demonstration found that participation in the program had no discernible effect on 30‐day mortality rates.[10] Finally, a study of physician medical groups contracting with a large network healthcare maintenance organization found that the implementation of pay for performance did not result in major before and after improvements in clinical quality compared to a control group of medical groups.[26]
High‐Value Care Is Not Always Low‐Cost Care
Not surprisingly, the clinical process measures included in CMS' hospital VBP program evaluate a select and relatively small group of high‐value and low‐cost interventions (eg, appropriate administration of antibiotics and tight control of serum glucose in surgical patients). However, an important body of work has demonstrated that high‐cost care (eg, intensive inpatient hospital care for common acute medical conditions) may also be highly valuable in terms of improving survival.[20, 27, 28, 29, 30] As the hospital VBP program evolves, its overseers will need to consider whether to include additional incentives for high‐value high‐cost healthcare services. Such considerations will likely become increasingly salient as healthcare delivery organizations move toward capitated delivery models. In particular, the VBP program's Medicare Spending Per Beneficiary measure, which quantifies inpatient and subsequent outpatient spending per beneficiary after a given hospitalization episode, will need to distinguish between higher‐spending hospitals that provide highly effective care (eg, care that reduces mortality and readmissions) and facilities that provide less‐effective care.
FUTURE OF VBP
Although the future of VBP is unknown, CMS is likely to modify the program in a number of ways over the next 3 to 5 years. First, CMS will likely expand the breadth and focus of incentivized measures in the VBP program. In FY 2014, for example, CMS is adding a set of 3, 30‐day mortality outcome measures to VBP: 30‐day risk‐adjusted mortality for AMI, CHF, and pneumonia.[1] A hospital's performance with respect to these outcomes will represent 25% of its total performance score in 2014, whereas the clinical process of care and patient experience of care domains will account for 45% and 30% of this score, respectively. In 2015, patient experience and outcome measures will account for 30% each in a hospital's performance score, whereas process and efficiency measures will each account for 20% of this score, respectively. The composition of this performance score evidences a shift away from rewarding process‐based measures and toward incentivizing measures of clinical outcomes and patient satisfaction, the latter of which may be highly subjective and more representative of a hospital's catchment population than of a hospital's care itself.[31] Additional measures in the domains of patient safety, care coordination, population and community health, emergency room wait times, and cost control may also be added to the VBP program in FY 2015 to FY 2017. Furthermore, CMS will continue to reevaluate the appropriateness of measures that are already included in VBP and will stop incentivizing measures that have become topped out, or are no longer supported by the National Quality Forum.[1, 13]
Second, CMS has established an annual gradual increase of 0.25% in the percentage of each hospital's inpatient DRG‐based payment that is at stake under VBP. In FY 2014, for example, participating hospitals will be required to contribute 1.25% of inpatient DRG payments to the VBP program. This percentage is likely to increase to 2% or more by 2017.[1, 32]
Third, expansions of the VBP program complement a number of other quality improvement efforts overseen by CMS, including the Hospital Readmissions Reduction Program. Effective for discharges beginning on October 1, 2012, hospitals with excess readmissions for AMI, CHF, and pneumonia are at risk for reimbursement reductions for all Medicare admissions in proportion to the rate of excess rehospitalizations. Some of the same concerns about the hospital VBP program outlined above have also been raised for this program, namely, whether readmission penalties will be large enough to impact hospital behavior, whether readmissions are even preventable,[33, 34] and whether adjustments in hospital‐level policies will reduce admissions that are known to be heavily influenced by patient economic and social factors that are outside of a hospital's control.[35, 36] Despite the limitations of VBP and the challenges that lie ahead, there is optimism that rewarding hospitals that provide high‐value rather than high‐volume care will not only improve outcomes of hospitalized patients in the United States, but will potentially be able to do so at a lower cost. Encouraging hospitals to improve their quality of care may also have important spillover effects on other healthcare domains. For example, hospitals that adopt systems to ensure prompt delivery of antibiotics to patients with pneumonia may also observe positive spillover effects with the prompt antibiotic management of other acute infectious illnesses that are not covered by VBP. VBP may have spillover effects on medical malpractice liability and defensive medicine as well. Indeed, financial incentives to practice higher‐quality evidenced‐based care may reduce medical malpractice liability and defensive medicine.
The government's ultimate goal in implementing VBP is to identify a broad and clinically relevant set of outcome measures that can be used to incentivize hospitals to deliver high‐quality as opposed to high‐volume healthcare. The first wave of outcome measures has already been instituted. It remains to be seen whether the incentive rewards of Medicare's hospital VBP program will be large enough that hospitals feel compelled to improve and compete for them.
- Centers for Medicare and Medicaid Services. Hospital Value‐Based Purchasing Web site. 2013. Available at: http://www.cms.gov/Medicare/Quality‐Initiatives‐Patient‐Assessment‐Instruments/hospital‐value‐based‐purchasing/index.html. Accessed March 4, 2013.
- , . Value‐based purchasing—national programs to move from volume to value. N Engl J Med. 2012;367:292–295.
- , . Hospital value‐based purchasing: will Medicare's new policy exacerbate disparities? Circ Cardiovasc Qual Outcomes. 2012;5:148–149.
- Centers for Medicare and Medicaid Services. CMS/premier hospital quality incentive demonstration (QHID). 2013. Available at: https://www.premierinc.com/quality‐safety/tools‐services/p4p/hqi/faqs.jsp. Accessed March 5, 2013.
- Centers for Medicare and Medicaid Services. Hospital Compare Web site. 2013. Available at: http://www.medicare.gov/hospitalcompare. Accessed March 4, 2013.
- , , . “Never events”: not every hospital‐acquired infection is preventable. Clin Infect Dis. 2009;49:743–746.
- . Will pay for performance improve quality of care? The answer is in the details. N Engl J Med. 2012;367:1852–1853.
- , , , , , . Reduced mortality with hospital pay for performance in England. N Engl J Med. 2012;367:1821–1828.
- , , , et al. Public reporting and pay for performance in hospital quality improvement. N Engl J Med. 2007;356:486–496.
- , , , . The long‐term effect of premier pay for performance on patient outcomes. N Engl J Med. 2012;366:1606–1615.
- , , , , . Does performance‐based remuneration for individual health care practitioners affect patient care?: a systematic review. Ann Intern Med. 2012;157:889–899.
- Centers for Medicare and Medicaid Services. Hospital Consumer Assessment Of Healthcare Providers and Systems Web site. 2013. Available at: http://www.hcahpsonline.org. Accessed March 5, 2013.
- . Medicare discloses hospitals' bonuses, penalties based on quality. Kaiser Health News. December 20, 2012. Available at: http://www.kaiserhealthnews.org/stories/2012/december/21/medicare‐hospitals‐value‐based‐purchasing.aspx?referrer=search. Accessed March 26, 2013.
- , , , . Hospital quality and intensity of spending: is there an association? Health Aff (Millwood). 2009;28:w566–w572.
- , , , et al. Association between performance measures and clinical outcomes for patients hospitalized with heart failure. JAMA. 2007;297:61–70.
- , , . The advantages and disadvantages of process‐based measures of health care quality. Int J Qual Health Care. 2001;13:469–474.
- . Accountability, incentives and behavior: the impact of high‐stakes testing in the Chicago public schools. J Public Econ. 2005;89:761–796.
- , , , , , . The implications of regional variations in Medicare spending. Part 1: the content, quality, and accessibility of care. Ann Intern Med. 2003;138:273–287.
- . Medical care—is more always better? N Engl J Med. 2003;349:1665–1667.
- , , . Hospital spending and inpatient mortality: evidence from California: an observational study. Ann Intern Med. 2011;154:160–167.
- . Making it easy to do it right. N Engl J Med. 2001;345:991–993.
- , , , . A high rate of compliance with neonatal intensive care unit transfusion guidelines persists even after a program to improve transfusion guideline compliance ended. Transfusion. 2011;51:2519–2520.
- , , , et al. The impact of removing financial incentives from clinical quality indicators: longitudinal analysis of four Kaiser Permanente indicators. BMJ. 2010;340:c1898.
- , . Medicare's new hospital value‐based purchasing program is likely to have only a small impact on hospital payments. Health Aff (Millwood). 2012;31:1932–1940.
- , , , et al. Pay for performance, quality of care, and outcomes in acute myocardial infarction. JAMA. 2007;297:2373–2380.
- , , . Can you get what you pay for? Pay‐for‐performance and the quality of healthcare providers. Rand J Econ. 2010;41:64–91.
- , , , . Spending and mortality in US acute care hospitals. Am J Manag Care. 2013;19:e46–e54.
- , , , , , . Development and validation of hospital “end‐of‐life” treatment intensity measures. Med Care. 2009;47:1098–1105.
- , , , et al. Looking forward, looking back: assessing variations in hospital resource use and outcomes for elderly patients with heart failure. Circ Cardiovasc Qual Outcomes. 2009;2:548–557.
- , , , et al. Association of hospital spending intensity with mortality and readmission rates in Ontario hospitals. JAMA. 2012;307:1037–1045.
- , , . Patient satisfaction with hospital care: effects of demographic and institutional characteristics. Med Care. 2000;38:325–334.
- , , . Linking performance with payment: implementing the Physician Value‐Based Payment Modifier. JAMA. 2012;308:2089–2090.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183:E391–E402.
- , , , et al. Incidence of potentially avoidable urgent readmissions and their relation to all‐cause urgent readmissions. CMAJ. 2011;183:E1067–E1072.
- , . Thirty‐day readmissions—truth and consequences. N Engl J Med. 2012;366:1366–1369.
- , , . Thirty‐day readmission rates for Medicare beneficiaries by race and site of care. JAMA. 2011;305:675–681.
The Centers for Medicaid and Medicare Services' (CMS) Hospital Inpatient Value‐Based Purchasing (VBP) Program, which was signed into law as part of the Patient Protection and Affordable Care Act of 2010, aims to incentivize inpatient providers to deliver high‐value, as opposed to high‐volume, healthcare.[1] Beginning on October 1, 2012, the start of the 2013 fiscal year (FY), hospitals participating in the VBP program became eligible for a variety of performance‐based incentive payments from CMS. These payments are based on an acute care hospital's ability to meet performance measurements in 6 care domains: (1) patient safety, (2) care coordination, (3) clinical processes and outcomes, (4) population or community health, (5) efficiency and cost reduction, and (6) patient‐ and caregiver‐centered experience.[2] The VBP program's ultimate purpose is to enable CMS to improve the health of Medicare beneficiaries by purchasing better care for them at a lower cost. These 3 characteristics of careimproved health, improved care, and lower costsare the foundation of CMS' conception of value.[1, 2] They are closely related to an economic conception of value, which is the difference between an intervention's benefit and its cost.
Although in principle not a new idea, the formal mandate of hospitals to provide high‐value healthcare through financial incentives marks an important change in Medicare and Medicaid policy. In this opportune review of VBP, we first discuss the relevant historical changes in the reimbursement environment of US hospitals that have set the stage for VBP. We then describe the structure of CMS' VBP program, with a focus on which facilities are eligible to participate in the program, the specific outcomes measured and incentivized, how rewards and penalties are allocated, and how the program will be funded. In an effort to anticipate some of the issues that lie ahead, we then highlight a number of potential challenges to the success of VBP, and discuss how VBP will impact the delivery and reimbursement of inpatient care services. We conclude by examining how the VBP program is likely to evolve over time.
HISTORICAL CONTEXT FOR VBP
Over the last decade, CMS has embarked on a number of initiatives to incentivize the provision of higher‐quality and more cost‐effective care. For example, in 2003, CMS implemented a national pay‐for‐performance (P4P) pilot project called the Premier Hospital Quality Incentive Demonstration (HQID).[3, 4] HQID, which ran for 6 years, tracked and rewarded the performance of 216 hospitals in 6 healthcare service domains: (1) acute myocardial infarction (AMI), (2) congestive heart failure (CHF), (3) pneumonia, (4) coronary artery bypass graft surgery, (5) hip and knee replacement surgery, and (6) perioperative management of surgical patients (including prevention of surgical site infections).[4] CMS then introduced its Hospital Compare Web site in 2005 to facilitate public reporting of hospital‐level quality outcomes.[3, 5] This Web site provides the public with access to data on hospital performance across a wide array of measures of process quality, clinical outcomes, spending, and resource utilization.[5] Next, in October 2008, CMS stopped reimbursing hospitals for a number of costly and common hospital‐acquired complications, including hospital‐acquired bloodstream infections and urinary tract infections, patient falls, and pressure ulcers.[3, 6] VBP is the latest and most comprehensive step that CMS has taken in its decade‐long effort to shift from volume to value‐based compensation for inpatient care.
Although CMS appears fully invested in using performance incentives to increase healthcare value, existing evidence of the effects of P4P on patient outcomes remains quite mixed.[7] On one hand, an analysis of an inpatient P4P program sponsored by the United Kingdom's National Health Service's (NHS) suggests that P4P may improve quality and save lives; indeed, hospitals that participated in the NHS P4P program significantly reduced inpatient mortality from pneumonia, saving an estimated 890 lives.[8] Additional empirical work suggests that the HQID was also associated with early improvements in healthcare quality.[9] However, a subsequent long‐term analysis found that participation in HQID had no discernible effect on 30‐day mortality rates.[10] Moreover, a meta‐analysis of P4P incentives for individual practitioners found few methodologically robust studies of P4P for clinicians and concluded that P4P's effects on individual practice patterns and outcomes remain largely uncertain.[11]
VBP: STRUCTURE AND DESIGN
This section reviews the structure of the VBP program. We describe current VBP eligibility criteria and sources of funding for the program, how hospitals participating in VBP are evaluated, and how VBP incentives for FY 2013 have been calculated.
Hospital Eligibility for VBP
All acute care hospitals in the United States (excluding Maryland) that are not psychiatric hospitals, rehabilitation hospitals, long‐term care facilities, children's hospitals, or cancer hospitals are eligible to participate in VBP in FY 2013 (full eligibility criteria is outlined in Table 1). For FY 2013, CMS chose to incentivize measures in just 2 care domains: (1) clinical processes of care and (2) patient experience of care. To be eligible for VBP in FY 2013, a hospital must report at least 10 cases each in at least 4 of 12 measures included in the clinical processes of care domain (Table 2), and/or must have at least 100 completed Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS). Designed and validated by CMS, the HCAHPS survey provides hospitals with a standardized instrument for gathering information about patient satisfaction with, and perspectives on, their hospital care.[12] HCAHPS will be used to assess 8 patient experience of care measures (Table 3).
|
| Inclusion criteria |
| Acute care hospital |
| Located in all 50 US states or District of Columbia (excluding Maryland) |
| Has at least 10 cases in at least 4 of 12 clinical process of care measures and/or at least 100 completed HCAHPS surveys |
| Exclusion criteria |
| Psychiatric, rehabilitation, long‐term care, children's or cancer hospital |
| Does not participate in Hospital Inpatient Quality Reporting Program during the VBP performance period |
| Cited by the Secretary of HHS for significant patient safety violations during performance period |
| Hospital does not meet minimum reporting requirements for number of cases, process measures, and surveys needed to participate in VBP |
| Disease Process | Process of Care Measure |
|---|---|
| |
| Acute myocardial infarction | Fibrinolytic therapy received within 30 minutes of hospital arrival |
| Primary percutaneous coronary intervention received within 90 minutes of hospital arrival | |
| Heart failure | Discharge instructions provided |
| Pneumonia | Blood cultures performed in the emergency department prior to initial antibiotic received in hospital |
| Initial antibiotic selection for community‐acquired pneumonia in immunocompetent patient | |
| Healthcare‐associated infections | Prophylactic antibiotic received within 1 hour prior to surgical incision |
| Prophylactic antibiotic selection for surgical patients | |
| Prophylactic antibiotics discontinued within 24 hours after surgery ends | |
| Cardiac surgery patients with controlled 6:00 am postoperative serum glucose | |
| Surgeries | Surgery patients on ‐blocker prior to arrival that received ‐blocker during perioperative period |
| Surgery patients with recommended venous thromboembolism prophylaxis ordered | |
| Surgery patients who received appropriate venous thromboembolism prophylaxis within 24 hours prior to surgery to 24 hours after surgery | |
| Communication with nurses |
| Communication with doctors |
| Responsiveness of hospital staff |
| Pain management |
| Communication about medicines |
| Cleanliness and quietness of hospital environment |
| Discharge information |
| Overall rating of hospital |
Participation in the program is mandatory for eligible hospitals, and CMS estimates that more than 3000 facilities across the United States will participate in FY 2013. Roughly $850 million dollars in VBP incentives will be paid out to these participating hospitals in FY 2013. The program is being financed through a 1% across‐the‐board reduction in FY 2013 diagnosis‐related group (DRG)‐based inpatient payments to participating hospitals. On December 20, 2012, CMS publically announced FY 2013 VBP incentives for all participating hospitals. Each hospital's incentive is retroactive and based on its performance between July 1, 2011 and March 31, 2012.
All data used for calculating VBP incentives is reported to CMS through its Hospital Inpatient Quality Reporting (Hospital IQR) Program, a national program instituted in 2003 that rewards hospitals for reporting designated quality measures. As of 2007, approximately 95% of eligible US hospitals were using the Hospital IQR program.[1] Measures evaluated via chart abstracts and surveys reflect a hospital's performance for its entire patient population, whereas measures assessed with claims data reflect hospital performance only for Medicare patients.
Evaluation of Hospitals
In FY 2013, hospital VBP incentive payments will be based entirely on performance in 2 domains: (1) clinical processes of care (weighted 70%) and (2) patient experience of care (weighted 30%). For each domain, CMS will evaluate each hospital's improvement over time as well as achievement compared to other hospitals in the VBP program. By assessing and rewarding both achievement and improvement, CMS will ensure that lower‐performing hospitals will still be rewarded for making substantial improvements in quality. To evaluate the first metricimprovement over timeCMS will compare a hospital's performance during a given reporting period with its baseline performance 2 years prior to this block of time. A hospital receives improvement points for improving its performance over time. To assess the second metricachievement compared to other hospitals in the VBP programCMS will compare each hospital's performance during a reporting period with the baseline performance (eg, performance 2 years prior to reporting period) of all other hospitals in the VBP program. A hospital is awarded achievement points if its performance exceeds the 50th percentile of all hospitals during the baseline performance period. Improvement scores range from 0 to 9, whereas achievement scores range from 0 to 10. The greater of a hospital's improvement and achievement scores on each VBP measure are used to calculate each hospital's total earned clinical care domain score and total earned HCAHPS base score. Hospitals that lack baseline performance data, which is required to assess improvement, will be evaluated solely on the basis of achievement points.[1] The total earned clinical care domain score is multiplied by 70% to reach the clinical care domain's contribution to a hospital's total performance score.
Each hospital's total patient experience domain, or HCAHPS performance, score consists of 2 components: a total earned HCAHPS base score as described above and a consistency score. The consistency score evaluates the reliability of a hospital's performance across all 8 patient experience of care measures (Table 3). If a hospital is above the 50th percentile of all hospital scores during the baseline period on all 8 measures, then it receives 100% of its consistency points. If a hospital is at the 0 percentile for a given measure, then it receives 0 consistency points for all measures. This provision promotes consistency by harshly penalizing hospitals with extremely poor performance on any 1 specific measure. If 1 or more measures are between the 0 and 50th percentiles, then it will receive a consistency score that takes into account how many measures were below the 50th percentile and their distance from this threshold. Each hospital's total HCAHPS performance score (the sum of total earned HCAHPS base points and consistency points) is then multiplied by 30% to arrive at the patient experience of care domain's contribution to a hospital's total performance score.
Importantly, CMS excluded from its VBP initiative 10 clinical process measures reported in the Hospital IQR Program because they are topped out; that is, almost all hospitals already perform them at very high rates (Table 4). Examples of these topped out process measures include administration of aspirin to all patients with AMI on arrival at the hospital; counseling of patients with AMI, CHF, and pneumonia about smoking cessation; and prescribing angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers to patients with CHF and left ventricular dysfunction.[1]
| Disease Process | Measure |
|---|---|
| |
| Acute myocardial infarction | Aspirin administered on arrival to the emergency department |
| ACEI or ARB prescribed on discharge | |
| Patient counseled about smoking cessation | |
| ‐Blocker prescribed on discharge | |
| Aspirin prescribed at discharge | |
| Heart failure | Patient counseled about smoking cessation |
| Evaluation of left ventricular systolic function | |
| ACEI or ARB prescribed for left ventricular systolic dysfunction | |
| Pneumonia | Patient counseled about smoking cessation |
| Surgical Care Improvement Project | Surgery patients with appropriate hair removal |
Calculation of VBP Incentives and Public Reporting
A hospital's total performance score for FY 2013 is equal to the sum of 70% of its clinical care domain score and 30% of its total HCAHPS performance score. This total performance score is entered into a linear mathematical formula to calculate each hospital's incentive payment. CMS projects that VBP will lead to a net increase in Medicare payments for one‐half of hospitals and a net decrease in payments for the other half of participating facilities.[1]
In December 2012, CMS publicly disclosed information about the initial performance of each hospital in the VBP program. Reported information included: (1) hospital performance for each applicable performance measure, (2) hospital performance by disease condition or procedure, and (3) hospital's total performance score. Initial analyses of this performance data revealed that 1557 hospitals will receive bonus payments under VBP in FY 2013, whereas 1427 hospitals will lose money under this program. Treasure Valley Hospital, a 10‐bed physician‐owned hospital in Boise, Idaho, will receive a 0.83% increase in Medicare payments, the largest payment increase under VBP in 2013. Conversely, Auburn Community Hospital in upstate New York, will suffer the most severe payment reduction: 0.9% per Medicare admission. The penalty will cost Auburn Hospital about $100,000, which is slightly more than 0.1% of its yearly $85 million operating budget.[13] For almost two‐thirds of participating hospitals, FY 2013 Medicare payments will change by <0.25%.[13] Additional information about VBP payments for FY 2013, including the number of hospitals who received VBP incentives and the size and range of these payments, is now accessible to the public through CMS' Hospital Compare Web site (
CHALLENGES OF VBP
As the Medicare VBP program evolves, and hospitals confront ever‐larger financial incentives to deliver high‐value as opposed to high‐volume care, it will be important to recognize limitations of the VBP program as they arise. Here we briefly discuss several conceptual and implementation challenges that physicians and policymakers should consider when assessing the merits of VBP in promoting high‐quality healthcare.
Rigorous and Continuous Evaluation of VBP Programs
The main premise of using VBP to incentivize hospitals to deliver high‐quality cost‐effective care is that the process measures used to determine hospital quality do impact patient outcomes. However, it is already well established that improvements in measures of process quality are not always associated with improvements in patient outcomes.[14, 15, 16] Moreover, incentivizing specific process measures encourages hospitals to shift resources away from other aspects of care delivery, which may have ambiguous, or even deleterious, effects on patient outcomes. Although incentives ideally push hospitals to shift resources away from low‐quality care toward high‐quality care, in practice this is not always the case. Hospital resources may instead be drawn away from areas that are not yet incented by VBP, but for which improvements in quality of care are desperately needed. The same empirical focus behind using VBP to incentivize hospitals to improve patient outcomes efficiently should be used to evaluate whether VBP is continually meeting its stated goals: reducing overall patient morbidity and mortality and improving patient satisfaction at ideally lower cost. The experience of the US education system with public policies designed to improve student testing performance may serve as a cautionary example here. Such policies, which provide financial rewards to schools whose students perform well on standardized tests, can indeed raise testing performance. However, these policies also lead educators to teach to the test, and to neglect important topics that are not tested on standardized exams.[17]
Prioritization of Process Measures
As payment incentives for VBP currently stand, process measures are weighted equally regardless of the clinical benefits they generate and the resources required to achieve improvements in process quality. For instance, 2 process measures, continuing home ‐blocker medications for patients with coronary artery disease undergoing surgery and early percutaneous coronary intervention for patients with AMI, may be weighted equally as process measures although both their clinical benefits and the costs of implementation are very different. Some hospitals responding to VBP incentives may choose to invest in areas where their ability to earn VBP incentive payments is high and the costs of improvement are low, although those areas may not be where interventions are most needed because clinical outcomes could be most improved. Recognizing that process measures have heterogeneous benefits and costs of implementation is important when prioritizing their reimbursement in VBP.
Measuring Improvements in Hospital Quality
Tying hospital financial compensation to hospital quality implies that measures of hospital quality should be robust. To incentivize hospitals to improve quality not only relative to other hospitals but to themselves in the past, the VBP program has established a baseline performance for each hospital. Each hospital is compared to its baseline performance in subsequent evaluation periods. Thus, properly measuring a hospital's baseline performance is important. During a given baseline period, some hospitals may have better or worse outcomes than their steady state due to random variation alone. Some hospitals deemed to have a low baseline will experience improvements in quality that are not related to active efforts to improve quality but through chance alone. Similarly, some hospitals deemed to have a high baseline will experience reductions in quality through chance. Of course, neither of these changes should be subject to differences in reimbursement because they do not reflect actual organizational changes made by the hospitals. The VBP program has made significant efforts to address this issue by requiring participating hospitals to have a large enough sample of cases such that estimated rates of process quality adherence meet a reliability threshold (ie, are likely to be consistent over time rather than vary substantially through chance alone). However, not all process measures exhibit high reliability, particularly those for which adverse events are rare (eg, foreign objects retained after surgery, air embolisms, and blood incompatibility). Ultimately, CMS's decision to balance the need for statistically reliable data with the goal of including as many hospitals as possible in the VBP program will require ongoing reevaluation of this issue.
Choosing Hospital Comparators Appropriately
In the current VBP program, hospitals will be evaluated in part by how they compare to hospitals nationally. However, studies of regional variation in healthcare have demonstrated large variations in practice patterns across the United States,[18, 19, 20] raising the question of whether hospitals should, at least initially, be compared to hospitals in the same geographic area. Although the ultimate goal of VBP should be to hold hospitals to a national standard, local practice patterns are not easily modified within 1‐ to 2‐year timeframes. Initially comparing hospitals to a national rather than local standard may unfairly penalize hospitals that are relative underperformers nationally but overperformers regionally. Although CMS's policy to reward improvement within hospitals over time mitigates issues arising from a cross‐sectional comparison of hospitals, the issue still remains if many hospitals within a region not only underperform relative to other hospitals nationally but also fail to demonstrate improvement. More broadly, this issue extends to differences across hospitals in factors that impact their ability to meet VBP goals. These factors may include, for example, hospital size, profitability, patient case and insurance mix, and presence of an electronic medical record. Comparing hospitals with vastly different abilities to achieve VBP goals and improve quickly may amount to inequitable policy.
Continual Evaluation of Topped‐Out Measures
Process measures that are met at high rates at nearly all hospitals are not used in evaluations by CMS for VBP. An assumption underlying CMS' decision to not reward hospitals for achieving these topped‐out measures is that once physicians and hospitals make cognitive and system‐level improvements that improve process quality, these gains will persist after the incentive is removed. Thus, CMS hopes and anticipates that although performance incentives will make it easier for well‐meaning physicians to learn to do the right thing, doctors will continue to do the right things for patients after these incentives are removed.[21, 22] Although this assumption may generally be accurate, it is important to continue to evaluate whether measures that are currently topped out continue to remain adequately performed, because rewarding new quality measures will necessarily lead hospitals to reallocate resources away from other clinical activities. Although we hope that the continued public reporting of topped‐out measures will prevent declines in performance on these measures, policy makers and clinicians should be aware that the lack of financial incentives for topped‐out measures may result in declines in quality. To this point, an analysis of 35 Kaiser Permanente facilities from 1997 to 2007 demonstrated that the removal of financial incentives for diabetic retinopathy and cervical cancer screening was associated with subsequent declines in performance of 3% and 1.6% per year, respectively.[23]
Will VBP Incentives Be Large Enough to Change Practice Patterns?
The VBP Program's ability to influence change depends, at least in part, on how the incentives offered under this program compare to the magnitude of the investments that hospitals must make to achieve a given reward. In general, larger incentives are necessary to motivate more significant changes in behavior or to influence organizations to invest the resources needed to achieve change. The incentives offered under VBP in FY 2013 are quite modest. Almost two‐thirds of participating hospitals will see their FY 2013 Medicare revenues change by <0.25%, roughly $125,000 at most.[13, 24] Although these incentives may motivate hospitals that can improve performance and achievement with very modest investments, they may have little impact on organizations that need to make significant upfront investments in care processes to achieve sustainable improvements in care quality. As CMS increases the size of VBP incentives over the next 2 to 4 years, it will also hold hospitals accountable for a broader and increasingly complex set of outcomes. Improving these outcomes may require investments in areas such as information technology and process improvement that far surpass the VBP incentive reward.
Moreover, prior research suggests that financial incentives like those available under VBP may contribute only slightly to performance improvements when public reporting already exists. For example, in a 2‐year study of 613 US hospitals implementing pay‐for‐performance plus public reporting or public reporting only, pay for performance plus public reporting was associated with only a 2.6% to 4.1% increase in a composite measure of quality when compared to hospitals with public reporting only.[9] Similarly, a study of 54 hospitals participating in the CMS pay for performance pilot initiative found no significant improvement in quality of care or outcomes for AMI when compared to 446 control hospitals.[25] A long‐term analysis of pay for performance in the Medicare Premier Hospital Quality Incentive Demonstration found that participation in the program had no discernible effect on 30‐day mortality rates.[10] Finally, a study of physician medical groups contracting with a large network healthcare maintenance organization found that the implementation of pay for performance did not result in major before and after improvements in clinical quality compared to a control group of medical groups.[26]
High‐Value Care Is Not Always Low‐Cost Care
Not surprisingly, the clinical process measures included in CMS' hospital VBP program evaluate a select and relatively small group of high‐value and low‐cost interventions (eg, appropriate administration of antibiotics and tight control of serum glucose in surgical patients). However, an important body of work has demonstrated that high‐cost care (eg, intensive inpatient hospital care for common acute medical conditions) may also be highly valuable in terms of improving survival.[20, 27, 28, 29, 30] As the hospital VBP program evolves, its overseers will need to consider whether to include additional incentives for high‐value high‐cost healthcare services. Such considerations will likely become increasingly salient as healthcare delivery organizations move toward capitated delivery models. In particular, the VBP program's Medicare Spending Per Beneficiary measure, which quantifies inpatient and subsequent outpatient spending per beneficiary after a given hospitalization episode, will need to distinguish between higher‐spending hospitals that provide highly effective care (eg, care that reduces mortality and readmissions) and facilities that provide less‐effective care.
FUTURE OF VBP
Although the future of VBP is unknown, CMS is likely to modify the program in a number of ways over the next 3 to 5 years. First, CMS will likely expand the breadth and focus of incentivized measures in the VBP program. In FY 2014, for example, CMS is adding a set of 3, 30‐day mortality outcome measures to VBP: 30‐day risk‐adjusted mortality for AMI, CHF, and pneumonia.[1] A hospital's performance with respect to these outcomes will represent 25% of its total performance score in 2014, whereas the clinical process of care and patient experience of care domains will account for 45% and 30% of this score, respectively. In 2015, patient experience and outcome measures will account for 30% each in a hospital's performance score, whereas process and efficiency measures will each account for 20% of this score, respectively. The composition of this performance score evidences a shift away from rewarding process‐based measures and toward incentivizing measures of clinical outcomes and patient satisfaction, the latter of which may be highly subjective and more representative of a hospital's catchment population than of a hospital's care itself.[31] Additional measures in the domains of patient safety, care coordination, population and community health, emergency room wait times, and cost control may also be added to the VBP program in FY 2015 to FY 2017. Furthermore, CMS will continue to reevaluate the appropriateness of measures that are already included in VBP and will stop incentivizing measures that have become topped out, or are no longer supported by the National Quality Forum.[1, 13]
Second, CMS has established an annual gradual increase of 0.25% in the percentage of each hospital's inpatient DRG‐based payment that is at stake under VBP. In FY 2014, for example, participating hospitals will be required to contribute 1.25% of inpatient DRG payments to the VBP program. This percentage is likely to increase to 2% or more by 2017.[1, 32]
Third, expansions of the VBP program complement a number of other quality improvement efforts overseen by CMS, including the Hospital Readmissions Reduction Program. Effective for discharges beginning on October 1, 2012, hospitals with excess readmissions for AMI, CHF, and pneumonia are at risk for reimbursement reductions for all Medicare admissions in proportion to the rate of excess rehospitalizations. Some of the same concerns about the hospital VBP program outlined above have also been raised for this program, namely, whether readmission penalties will be large enough to impact hospital behavior, whether readmissions are even preventable,[33, 34] and whether adjustments in hospital‐level policies will reduce admissions that are known to be heavily influenced by patient economic and social factors that are outside of a hospital's control.[35, 36] Despite the limitations of VBP and the challenges that lie ahead, there is optimism that rewarding hospitals that provide high‐value rather than high‐volume care will not only improve outcomes of hospitalized patients in the United States, but will potentially be able to do so at a lower cost. Encouraging hospitals to improve their quality of care may also have important spillover effects on other healthcare domains. For example, hospitals that adopt systems to ensure prompt delivery of antibiotics to patients with pneumonia may also observe positive spillover effects with the prompt antibiotic management of other acute infectious illnesses that are not covered by VBP. VBP may have spillover effects on medical malpractice liability and defensive medicine as well. Indeed, financial incentives to practice higher‐quality evidenced‐based care may reduce medical malpractice liability and defensive medicine.
The government's ultimate goal in implementing VBP is to identify a broad and clinically relevant set of outcome measures that can be used to incentivize hospitals to deliver high‐quality as opposed to high‐volume healthcare. The first wave of outcome measures has already been instituted. It remains to be seen whether the incentive rewards of Medicare's hospital VBP program will be large enough that hospitals feel compelled to improve and compete for them.
The Centers for Medicaid and Medicare Services' (CMS) Hospital Inpatient Value‐Based Purchasing (VBP) Program, which was signed into law as part of the Patient Protection and Affordable Care Act of 2010, aims to incentivize inpatient providers to deliver high‐value, as opposed to high‐volume, healthcare.[1] Beginning on October 1, 2012, the start of the 2013 fiscal year (FY), hospitals participating in the VBP program became eligible for a variety of performance‐based incentive payments from CMS. These payments are based on an acute care hospital's ability to meet performance measurements in 6 care domains: (1) patient safety, (2) care coordination, (3) clinical processes and outcomes, (4) population or community health, (5) efficiency and cost reduction, and (6) patient‐ and caregiver‐centered experience.[2] The VBP program's ultimate purpose is to enable CMS to improve the health of Medicare beneficiaries by purchasing better care for them at a lower cost. These 3 characteristics of careimproved health, improved care, and lower costsare the foundation of CMS' conception of value.[1, 2] They are closely related to an economic conception of value, which is the difference between an intervention's benefit and its cost.
Although in principle not a new idea, the formal mandate of hospitals to provide high‐value healthcare through financial incentives marks an important change in Medicare and Medicaid policy. In this opportune review of VBP, we first discuss the relevant historical changes in the reimbursement environment of US hospitals that have set the stage for VBP. We then describe the structure of CMS' VBP program, with a focus on which facilities are eligible to participate in the program, the specific outcomes measured and incentivized, how rewards and penalties are allocated, and how the program will be funded. In an effort to anticipate some of the issues that lie ahead, we then highlight a number of potential challenges to the success of VBP, and discuss how VBP will impact the delivery and reimbursement of inpatient care services. We conclude by examining how the VBP program is likely to evolve over time.
HISTORICAL CONTEXT FOR VBP
Over the last decade, CMS has embarked on a number of initiatives to incentivize the provision of higher‐quality and more cost‐effective care. For example, in 2003, CMS implemented a national pay‐for‐performance (P4P) pilot project called the Premier Hospital Quality Incentive Demonstration (HQID).[3, 4] HQID, which ran for 6 years, tracked and rewarded the performance of 216 hospitals in 6 healthcare service domains: (1) acute myocardial infarction (AMI), (2) congestive heart failure (CHF), (3) pneumonia, (4) coronary artery bypass graft surgery, (5) hip and knee replacement surgery, and (6) perioperative management of surgical patients (including prevention of surgical site infections).[4] CMS then introduced its Hospital Compare Web site in 2005 to facilitate public reporting of hospital‐level quality outcomes.[3, 5] This Web site provides the public with access to data on hospital performance across a wide array of measures of process quality, clinical outcomes, spending, and resource utilization.[5] Next, in October 2008, CMS stopped reimbursing hospitals for a number of costly and common hospital‐acquired complications, including hospital‐acquired bloodstream infections and urinary tract infections, patient falls, and pressure ulcers.[3, 6] VBP is the latest and most comprehensive step that CMS has taken in its decade‐long effort to shift from volume to value‐based compensation for inpatient care.
Although CMS appears fully invested in using performance incentives to increase healthcare value, existing evidence of the effects of P4P on patient outcomes remains quite mixed.[7] On one hand, an analysis of an inpatient P4P program sponsored by the United Kingdom's National Health Service's (NHS) suggests that P4P may improve quality and save lives; indeed, hospitals that participated in the NHS P4P program significantly reduced inpatient mortality from pneumonia, saving an estimated 890 lives.[8] Additional empirical work suggests that the HQID was also associated with early improvements in healthcare quality.[9] However, a subsequent long‐term analysis found that participation in HQID had no discernible effect on 30‐day mortality rates.[10] Moreover, a meta‐analysis of P4P incentives for individual practitioners found few methodologically robust studies of P4P for clinicians and concluded that P4P's effects on individual practice patterns and outcomes remain largely uncertain.[11]
VBP: STRUCTURE AND DESIGN
This section reviews the structure of the VBP program. We describe current VBP eligibility criteria and sources of funding for the program, how hospitals participating in VBP are evaluated, and how VBP incentives for FY 2013 have been calculated.
Hospital Eligibility for VBP
All acute care hospitals in the United States (excluding Maryland) that are not psychiatric hospitals, rehabilitation hospitals, long‐term care facilities, children's hospitals, or cancer hospitals are eligible to participate in VBP in FY 2013 (full eligibility criteria is outlined in Table 1). For FY 2013, CMS chose to incentivize measures in just 2 care domains: (1) clinical processes of care and (2) patient experience of care. To be eligible for VBP in FY 2013, a hospital must report at least 10 cases each in at least 4 of 12 measures included in the clinical processes of care domain (Table 2), and/or must have at least 100 completed Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS). Designed and validated by CMS, the HCAHPS survey provides hospitals with a standardized instrument for gathering information about patient satisfaction with, and perspectives on, their hospital care.[12] HCAHPS will be used to assess 8 patient experience of care measures (Table 3).
|
| Inclusion criteria |
| Acute care hospital |
| Located in all 50 US states or District of Columbia (excluding Maryland) |
| Has at least 10 cases in at least 4 of 12 clinical process of care measures and/or at least 100 completed HCAHPS surveys |
| Exclusion criteria |
| Psychiatric, rehabilitation, long‐term care, children's or cancer hospital |
| Does not participate in Hospital Inpatient Quality Reporting Program during the VBP performance period |
| Cited by the Secretary of HHS for significant patient safety violations during performance period |
| Hospital does not meet minimum reporting requirements for number of cases, process measures, and surveys needed to participate in VBP |
| Disease Process | Process of Care Measure |
|---|---|
| |
| Acute myocardial infarction | Fibrinolytic therapy received within 30 minutes of hospital arrival |
| Primary percutaneous coronary intervention received within 90 minutes of hospital arrival | |
| Heart failure | Discharge instructions provided |
| Pneumonia | Blood cultures performed in the emergency department prior to initial antibiotic received in hospital |
| Initial antibiotic selection for community‐acquired pneumonia in immunocompetent patient | |
| Healthcare‐associated infections | Prophylactic antibiotic received within 1 hour prior to surgical incision |
| Prophylactic antibiotic selection for surgical patients | |
| Prophylactic antibiotics discontinued within 24 hours after surgery ends | |
| Cardiac surgery patients with controlled 6:00 am postoperative serum glucose | |
| Surgeries | Surgery patients on ‐blocker prior to arrival that received ‐blocker during perioperative period |
| Surgery patients with recommended venous thromboembolism prophylaxis ordered | |
| Surgery patients who received appropriate venous thromboembolism prophylaxis within 24 hours prior to surgery to 24 hours after surgery | |
| Communication with nurses |
| Communication with doctors |
| Responsiveness of hospital staff |
| Pain management |
| Communication about medicines |
| Cleanliness and quietness of hospital environment |
| Discharge information |
| Overall rating of hospital |
Participation in the program is mandatory for eligible hospitals, and CMS estimates that more than 3000 facilities across the United States will participate in FY 2013. Roughly $850 million dollars in VBP incentives will be paid out to these participating hospitals in FY 2013. The program is being financed through a 1% across‐the‐board reduction in FY 2013 diagnosis‐related group (DRG)‐based inpatient payments to participating hospitals. On December 20, 2012, CMS publically announced FY 2013 VBP incentives for all participating hospitals. Each hospital's incentive is retroactive and based on its performance between July 1, 2011 and March 31, 2012.
All data used for calculating VBP incentives is reported to CMS through its Hospital Inpatient Quality Reporting (Hospital IQR) Program, a national program instituted in 2003 that rewards hospitals for reporting designated quality measures. As of 2007, approximately 95% of eligible US hospitals were using the Hospital IQR program.[1] Measures evaluated via chart abstracts and surveys reflect a hospital's performance for its entire patient population, whereas measures assessed with claims data reflect hospital performance only for Medicare patients.
Evaluation of Hospitals
In FY 2013, hospital VBP incentive payments will be based entirely on performance in 2 domains: (1) clinical processes of care (weighted 70%) and (2) patient experience of care (weighted 30%). For each domain, CMS will evaluate each hospital's improvement over time as well as achievement compared to other hospitals in the VBP program. By assessing and rewarding both achievement and improvement, CMS will ensure that lower‐performing hospitals will still be rewarded for making substantial improvements in quality. To evaluate the first metricimprovement over timeCMS will compare a hospital's performance during a given reporting period with its baseline performance 2 years prior to this block of time. A hospital receives improvement points for improving its performance over time. To assess the second metricachievement compared to other hospitals in the VBP programCMS will compare each hospital's performance during a reporting period with the baseline performance (eg, performance 2 years prior to reporting period) of all other hospitals in the VBP program. A hospital is awarded achievement points if its performance exceeds the 50th percentile of all hospitals during the baseline performance period. Improvement scores range from 0 to 9, whereas achievement scores range from 0 to 10. The greater of a hospital's improvement and achievement scores on each VBP measure are used to calculate each hospital's total earned clinical care domain score and total earned HCAHPS base score. Hospitals that lack baseline performance data, which is required to assess improvement, will be evaluated solely on the basis of achievement points.[1] The total earned clinical care domain score is multiplied by 70% to reach the clinical care domain's contribution to a hospital's total performance score.
Each hospital's total patient experience domain, or HCAHPS performance, score consists of 2 components: a total earned HCAHPS base score as described above and a consistency score. The consistency score evaluates the reliability of a hospital's performance across all 8 patient experience of care measures (Table 3). If a hospital is above the 50th percentile of all hospital scores during the baseline period on all 8 measures, then it receives 100% of its consistency points. If a hospital is at the 0 percentile for a given measure, then it receives 0 consistency points for all measures. This provision promotes consistency by harshly penalizing hospitals with extremely poor performance on any 1 specific measure. If 1 or more measures are between the 0 and 50th percentiles, then it will receive a consistency score that takes into account how many measures were below the 50th percentile and their distance from this threshold. Each hospital's total HCAHPS performance score (the sum of total earned HCAHPS base points and consistency points) is then multiplied by 30% to arrive at the patient experience of care domain's contribution to a hospital's total performance score.
Importantly, CMS excluded from its VBP initiative 10 clinical process measures reported in the Hospital IQR Program because they are topped out; that is, almost all hospitals already perform them at very high rates (Table 4). Examples of these topped out process measures include administration of aspirin to all patients with AMI on arrival at the hospital; counseling of patients with AMI, CHF, and pneumonia about smoking cessation; and prescribing angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers to patients with CHF and left ventricular dysfunction.[1]
| Disease Process | Measure |
|---|---|
| |
| Acute myocardial infarction | Aspirin administered on arrival to the emergency department |
| ACEI or ARB prescribed on discharge | |
| Patient counseled about smoking cessation | |
| ‐Blocker prescribed on discharge | |
| Aspirin prescribed at discharge | |
| Heart failure | Patient counseled about smoking cessation |
| Evaluation of left ventricular systolic function | |
| ACEI or ARB prescribed for left ventricular systolic dysfunction | |
| Pneumonia | Patient counseled about smoking cessation |
| Surgical Care Improvement Project | Surgery patients with appropriate hair removal |
Calculation of VBP Incentives and Public Reporting
A hospital's total performance score for FY 2013 is equal to the sum of 70% of its clinical care domain score and 30% of its total HCAHPS performance score. This total performance score is entered into a linear mathematical formula to calculate each hospital's incentive payment. CMS projects that VBP will lead to a net increase in Medicare payments for one‐half of hospitals and a net decrease in payments for the other half of participating facilities.[1]
In December 2012, CMS publicly disclosed information about the initial performance of each hospital in the VBP program. Reported information included: (1) hospital performance for each applicable performance measure, (2) hospital performance by disease condition or procedure, and (3) hospital's total performance score. Initial analyses of this performance data revealed that 1557 hospitals will receive bonus payments under VBP in FY 2013, whereas 1427 hospitals will lose money under this program. Treasure Valley Hospital, a 10‐bed physician‐owned hospital in Boise, Idaho, will receive a 0.83% increase in Medicare payments, the largest payment increase under VBP in 2013. Conversely, Auburn Community Hospital in upstate New York, will suffer the most severe payment reduction: 0.9% per Medicare admission. The penalty will cost Auburn Hospital about $100,000, which is slightly more than 0.1% of its yearly $85 million operating budget.[13] For almost two‐thirds of participating hospitals, FY 2013 Medicare payments will change by <0.25%.[13] Additional information about VBP payments for FY 2013, including the number of hospitals who received VBP incentives and the size and range of these payments, is now accessible to the public through CMS' Hospital Compare Web site (
CHALLENGES OF VBP
As the Medicare VBP program evolves, and hospitals confront ever‐larger financial incentives to deliver high‐value as opposed to high‐volume care, it will be important to recognize limitations of the VBP program as they arise. Here we briefly discuss several conceptual and implementation challenges that physicians and policymakers should consider when assessing the merits of VBP in promoting high‐quality healthcare.
Rigorous and Continuous Evaluation of VBP Programs
The main premise of using VBP to incentivize hospitals to deliver high‐quality cost‐effective care is that the process measures used to determine hospital quality do impact patient outcomes. However, it is already well established that improvements in measures of process quality are not always associated with improvements in patient outcomes.[14, 15, 16] Moreover, incentivizing specific process measures encourages hospitals to shift resources away from other aspects of care delivery, which may have ambiguous, or even deleterious, effects on patient outcomes. Although incentives ideally push hospitals to shift resources away from low‐quality care toward high‐quality care, in practice this is not always the case. Hospital resources may instead be drawn away from areas that are not yet incented by VBP, but for which improvements in quality of care are desperately needed. The same empirical focus behind using VBP to incentivize hospitals to improve patient outcomes efficiently should be used to evaluate whether VBP is continually meeting its stated goals: reducing overall patient morbidity and mortality and improving patient satisfaction at ideally lower cost. The experience of the US education system with public policies designed to improve student testing performance may serve as a cautionary example here. Such policies, which provide financial rewards to schools whose students perform well on standardized tests, can indeed raise testing performance. However, these policies also lead educators to teach to the test, and to neglect important topics that are not tested on standardized exams.[17]
Prioritization of Process Measures
As payment incentives for VBP currently stand, process measures are weighted equally regardless of the clinical benefits they generate and the resources required to achieve improvements in process quality. For instance, 2 process measures, continuing home ‐blocker medications for patients with coronary artery disease undergoing surgery and early percutaneous coronary intervention for patients with AMI, may be weighted equally as process measures although both their clinical benefits and the costs of implementation are very different. Some hospitals responding to VBP incentives may choose to invest in areas where their ability to earn VBP incentive payments is high and the costs of improvement are low, although those areas may not be where interventions are most needed because clinical outcomes could be most improved. Recognizing that process measures have heterogeneous benefits and costs of implementation is important when prioritizing their reimbursement in VBP.
Measuring Improvements in Hospital Quality
Tying hospital financial compensation to hospital quality implies that measures of hospital quality should be robust. To incentivize hospitals to improve quality not only relative to other hospitals but to themselves in the past, the VBP program has established a baseline performance for each hospital. Each hospital is compared to its baseline performance in subsequent evaluation periods. Thus, properly measuring a hospital's baseline performance is important. During a given baseline period, some hospitals may have better or worse outcomes than their steady state due to random variation alone. Some hospitals deemed to have a low baseline will experience improvements in quality that are not related to active efforts to improve quality but through chance alone. Similarly, some hospitals deemed to have a high baseline will experience reductions in quality through chance. Of course, neither of these changes should be subject to differences in reimbursement because they do not reflect actual organizational changes made by the hospitals. The VBP program has made significant efforts to address this issue by requiring participating hospitals to have a large enough sample of cases such that estimated rates of process quality adherence meet a reliability threshold (ie, are likely to be consistent over time rather than vary substantially through chance alone). However, not all process measures exhibit high reliability, particularly those for which adverse events are rare (eg, foreign objects retained after surgery, air embolisms, and blood incompatibility). Ultimately, CMS's decision to balance the need for statistically reliable data with the goal of including as many hospitals as possible in the VBP program will require ongoing reevaluation of this issue.
Choosing Hospital Comparators Appropriately
In the current VBP program, hospitals will be evaluated in part by how they compare to hospitals nationally. However, studies of regional variation in healthcare have demonstrated large variations in practice patterns across the United States,[18, 19, 20] raising the question of whether hospitals should, at least initially, be compared to hospitals in the same geographic area. Although the ultimate goal of VBP should be to hold hospitals to a national standard, local practice patterns are not easily modified within 1‐ to 2‐year timeframes. Initially comparing hospitals to a national rather than local standard may unfairly penalize hospitals that are relative underperformers nationally but overperformers regionally. Although CMS's policy to reward improvement within hospitals over time mitigates issues arising from a cross‐sectional comparison of hospitals, the issue still remains if many hospitals within a region not only underperform relative to other hospitals nationally but also fail to demonstrate improvement. More broadly, this issue extends to differences across hospitals in factors that impact their ability to meet VBP goals. These factors may include, for example, hospital size, profitability, patient case and insurance mix, and presence of an electronic medical record. Comparing hospitals with vastly different abilities to achieve VBP goals and improve quickly may amount to inequitable policy.
Continual Evaluation of Topped‐Out Measures
Process measures that are met at high rates at nearly all hospitals are not used in evaluations by CMS for VBP. An assumption underlying CMS' decision to not reward hospitals for achieving these topped‐out measures is that once physicians and hospitals make cognitive and system‐level improvements that improve process quality, these gains will persist after the incentive is removed. Thus, CMS hopes and anticipates that although performance incentives will make it easier for well‐meaning physicians to learn to do the right thing, doctors will continue to do the right things for patients after these incentives are removed.[21, 22] Although this assumption may generally be accurate, it is important to continue to evaluate whether measures that are currently topped out continue to remain adequately performed, because rewarding new quality measures will necessarily lead hospitals to reallocate resources away from other clinical activities. Although we hope that the continued public reporting of topped‐out measures will prevent declines in performance on these measures, policy makers and clinicians should be aware that the lack of financial incentives for topped‐out measures may result in declines in quality. To this point, an analysis of 35 Kaiser Permanente facilities from 1997 to 2007 demonstrated that the removal of financial incentives for diabetic retinopathy and cervical cancer screening was associated with subsequent declines in performance of 3% and 1.6% per year, respectively.[23]
Will VBP Incentives Be Large Enough to Change Practice Patterns?
The VBP Program's ability to influence change depends, at least in part, on how the incentives offered under this program compare to the magnitude of the investments that hospitals must make to achieve a given reward. In general, larger incentives are necessary to motivate more significant changes in behavior or to influence organizations to invest the resources needed to achieve change. The incentives offered under VBP in FY 2013 are quite modest. Almost two‐thirds of participating hospitals will see their FY 2013 Medicare revenues change by <0.25%, roughly $125,000 at most.[13, 24] Although these incentives may motivate hospitals that can improve performance and achievement with very modest investments, they may have little impact on organizations that need to make significant upfront investments in care processes to achieve sustainable improvements in care quality. As CMS increases the size of VBP incentives over the next 2 to 4 years, it will also hold hospitals accountable for a broader and increasingly complex set of outcomes. Improving these outcomes may require investments in areas such as information technology and process improvement that far surpass the VBP incentive reward.
Moreover, prior research suggests that financial incentives like those available under VBP may contribute only slightly to performance improvements when public reporting already exists. For example, in a 2‐year study of 613 US hospitals implementing pay‐for‐performance plus public reporting or public reporting only, pay for performance plus public reporting was associated with only a 2.6% to 4.1% increase in a composite measure of quality when compared to hospitals with public reporting only.[9] Similarly, a study of 54 hospitals participating in the CMS pay for performance pilot initiative found no significant improvement in quality of care or outcomes for AMI when compared to 446 control hospitals.[25] A long‐term analysis of pay for performance in the Medicare Premier Hospital Quality Incentive Demonstration found that participation in the program had no discernible effect on 30‐day mortality rates.[10] Finally, a study of physician medical groups contracting with a large network healthcare maintenance organization found that the implementation of pay for performance did not result in major before and after improvements in clinical quality compared to a control group of medical groups.[26]
High‐Value Care Is Not Always Low‐Cost Care
Not surprisingly, the clinical process measures included in CMS' hospital VBP program evaluate a select and relatively small group of high‐value and low‐cost interventions (eg, appropriate administration of antibiotics and tight control of serum glucose in surgical patients). However, an important body of work has demonstrated that high‐cost care (eg, intensive inpatient hospital care for common acute medical conditions) may also be highly valuable in terms of improving survival.[20, 27, 28, 29, 30] As the hospital VBP program evolves, its overseers will need to consider whether to include additional incentives for high‐value high‐cost healthcare services. Such considerations will likely become increasingly salient as healthcare delivery organizations move toward capitated delivery models. In particular, the VBP program's Medicare Spending Per Beneficiary measure, which quantifies inpatient and subsequent outpatient spending per beneficiary after a given hospitalization episode, will need to distinguish between higher‐spending hospitals that provide highly effective care (eg, care that reduces mortality and readmissions) and facilities that provide less‐effective care.
FUTURE OF VBP
Although the future of VBP is unknown, CMS is likely to modify the program in a number of ways over the next 3 to 5 years. First, CMS will likely expand the breadth and focus of incentivized measures in the VBP program. In FY 2014, for example, CMS is adding a set of 3, 30‐day mortality outcome measures to VBP: 30‐day risk‐adjusted mortality for AMI, CHF, and pneumonia.[1] A hospital's performance with respect to these outcomes will represent 25% of its total performance score in 2014, whereas the clinical process of care and patient experience of care domains will account for 45% and 30% of this score, respectively. In 2015, patient experience and outcome measures will account for 30% each in a hospital's performance score, whereas process and efficiency measures will each account for 20% of this score, respectively. The composition of this performance score evidences a shift away from rewarding process‐based measures and toward incentivizing measures of clinical outcomes and patient satisfaction, the latter of which may be highly subjective and more representative of a hospital's catchment population than of a hospital's care itself.[31] Additional measures in the domains of patient safety, care coordination, population and community health, emergency room wait times, and cost control may also be added to the VBP program in FY 2015 to FY 2017. Furthermore, CMS will continue to reevaluate the appropriateness of measures that are already included in VBP and will stop incentivizing measures that have become topped out, or are no longer supported by the National Quality Forum.[1, 13]
Second, CMS has established an annual gradual increase of 0.25% in the percentage of each hospital's inpatient DRG‐based payment that is at stake under VBP. In FY 2014, for example, participating hospitals will be required to contribute 1.25% of inpatient DRG payments to the VBP program. This percentage is likely to increase to 2% or more by 2017.[1, 32]
Third, expansions of the VBP program complement a number of other quality improvement efforts overseen by CMS, including the Hospital Readmissions Reduction Program. Effective for discharges beginning on October 1, 2012, hospitals with excess readmissions for AMI, CHF, and pneumonia are at risk for reimbursement reductions for all Medicare admissions in proportion to the rate of excess rehospitalizations. Some of the same concerns about the hospital VBP program outlined above have also been raised for this program, namely, whether readmission penalties will be large enough to impact hospital behavior, whether readmissions are even preventable,[33, 34] and whether adjustments in hospital‐level policies will reduce admissions that are known to be heavily influenced by patient economic and social factors that are outside of a hospital's control.[35, 36] Despite the limitations of VBP and the challenges that lie ahead, there is optimism that rewarding hospitals that provide high‐value rather than high‐volume care will not only improve outcomes of hospitalized patients in the United States, but will potentially be able to do so at a lower cost. Encouraging hospitals to improve their quality of care may also have important spillover effects on other healthcare domains. For example, hospitals that adopt systems to ensure prompt delivery of antibiotics to patients with pneumonia may also observe positive spillover effects with the prompt antibiotic management of other acute infectious illnesses that are not covered by VBP. VBP may have spillover effects on medical malpractice liability and defensive medicine as well. Indeed, financial incentives to practice higher‐quality evidenced‐based care may reduce medical malpractice liability and defensive medicine.
The government's ultimate goal in implementing VBP is to identify a broad and clinically relevant set of outcome measures that can be used to incentivize hospitals to deliver high‐quality as opposed to high‐volume healthcare. The first wave of outcome measures has already been instituted. It remains to be seen whether the incentive rewards of Medicare's hospital VBP program will be large enough that hospitals feel compelled to improve and compete for them.
- Centers for Medicare and Medicaid Services. Hospital Value‐Based Purchasing Web site. 2013. Available at: http://www.cms.gov/Medicare/Quality‐Initiatives‐Patient‐Assessment‐Instruments/hospital‐value‐based‐purchasing/index.html. Accessed March 4, 2013.
- , . Value‐based purchasing—national programs to move from volume to value. N Engl J Med. 2012;367:292–295.
- , . Hospital value‐based purchasing: will Medicare's new policy exacerbate disparities? Circ Cardiovasc Qual Outcomes. 2012;5:148–149.
- Centers for Medicare and Medicaid Services. CMS/premier hospital quality incentive demonstration (QHID). 2013. Available at: https://www.premierinc.com/quality‐safety/tools‐services/p4p/hqi/faqs.jsp. Accessed March 5, 2013.
- Centers for Medicare and Medicaid Services. Hospital Compare Web site. 2013. Available at: http://www.medicare.gov/hospitalcompare. Accessed March 4, 2013.
- , , . “Never events”: not every hospital‐acquired infection is preventable. Clin Infect Dis. 2009;49:743–746.
- . Will pay for performance improve quality of care? The answer is in the details. N Engl J Med. 2012;367:1852–1853.
- , , , , , . Reduced mortality with hospital pay for performance in England. N Engl J Med. 2012;367:1821–1828.
- , , , et al. Public reporting and pay for performance in hospital quality improvement. N Engl J Med. 2007;356:486–496.
- , , , . The long‐term effect of premier pay for performance on patient outcomes. N Engl J Med. 2012;366:1606–1615.
- , , , , . Does performance‐based remuneration for individual health care practitioners affect patient care?: a systematic review. Ann Intern Med. 2012;157:889–899.
- Centers for Medicare and Medicaid Services. Hospital Consumer Assessment Of Healthcare Providers and Systems Web site. 2013. Available at: http://www.hcahpsonline.org. Accessed March 5, 2013.
- . Medicare discloses hospitals' bonuses, penalties based on quality. Kaiser Health News. December 20, 2012. Available at: http://www.kaiserhealthnews.org/stories/2012/december/21/medicare‐hospitals‐value‐based‐purchasing.aspx?referrer=search. Accessed March 26, 2013.
- , , , . Hospital quality and intensity of spending: is there an association? Health Aff (Millwood). 2009;28:w566–w572.
- , , , et al. Association between performance measures and clinical outcomes for patients hospitalized with heart failure. JAMA. 2007;297:61–70.
- , , . The advantages and disadvantages of process‐based measures of health care quality. Int J Qual Health Care. 2001;13:469–474.
- . Accountability, incentives and behavior: the impact of high‐stakes testing in the Chicago public schools. J Public Econ. 2005;89:761–796.
- , , , , , . The implications of regional variations in Medicare spending. Part 1: the content, quality, and accessibility of care. Ann Intern Med. 2003;138:273–287.
- . Medical care—is more always better? N Engl J Med. 2003;349:1665–1667.
- , , . Hospital spending and inpatient mortality: evidence from California: an observational study. Ann Intern Med. 2011;154:160–167.
- . Making it easy to do it right. N Engl J Med. 2001;345:991–993.
- , , , . A high rate of compliance with neonatal intensive care unit transfusion guidelines persists even after a program to improve transfusion guideline compliance ended. Transfusion. 2011;51:2519–2520.
- , , , et al. The impact of removing financial incentives from clinical quality indicators: longitudinal analysis of four Kaiser Permanente indicators. BMJ. 2010;340:c1898.
- , . Medicare's new hospital value‐based purchasing program is likely to have only a small impact on hospital payments. Health Aff (Millwood). 2012;31:1932–1940.
- , , , et al. Pay for performance, quality of care, and outcomes in acute myocardial infarction. JAMA. 2007;297:2373–2380.
- , , . Can you get what you pay for? Pay‐for‐performance and the quality of healthcare providers. Rand J Econ. 2010;41:64–91.
- , , , . Spending and mortality in US acute care hospitals. Am J Manag Care. 2013;19:e46–e54.
- , , , , , . Development and validation of hospital “end‐of‐life” treatment intensity measures. Med Care. 2009;47:1098–1105.
- , , , et al. Looking forward, looking back: assessing variations in hospital resource use and outcomes for elderly patients with heart failure. Circ Cardiovasc Qual Outcomes. 2009;2:548–557.
- , , , et al. Association of hospital spending intensity with mortality and readmission rates in Ontario hospitals. JAMA. 2012;307:1037–1045.
- , , . Patient satisfaction with hospital care: effects of demographic and institutional characteristics. Med Care. 2000;38:325–334.
- , , . Linking performance with payment: implementing the Physician Value‐Based Payment Modifier. JAMA. 2012;308:2089–2090.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183:E391–E402.
- , , , et al. Incidence of potentially avoidable urgent readmissions and their relation to all‐cause urgent readmissions. CMAJ. 2011;183:E1067–E1072.
- , . Thirty‐day readmissions—truth and consequences. N Engl J Med. 2012;366:1366–1369.
- , , . Thirty‐day readmission rates for Medicare beneficiaries by race and site of care. JAMA. 2011;305:675–681.
- Centers for Medicare and Medicaid Services. Hospital Value‐Based Purchasing Web site. 2013. Available at: http://www.cms.gov/Medicare/Quality‐Initiatives‐Patient‐Assessment‐Instruments/hospital‐value‐based‐purchasing/index.html. Accessed March 4, 2013.
- , . Value‐based purchasing—national programs to move from volume to value. N Engl J Med. 2012;367:292–295.
- , . Hospital value‐based purchasing: will Medicare's new policy exacerbate disparities? Circ Cardiovasc Qual Outcomes. 2012;5:148–149.
- Centers for Medicare and Medicaid Services. CMS/premier hospital quality incentive demonstration (QHID). 2013. Available at: https://www.premierinc.com/quality‐safety/tools‐services/p4p/hqi/faqs.jsp. Accessed March 5, 2013.
- Centers for Medicare and Medicaid Services. Hospital Compare Web site. 2013. Available at: http://www.medicare.gov/hospitalcompare. Accessed March 4, 2013.
- , , . “Never events”: not every hospital‐acquired infection is preventable. Clin Infect Dis. 2009;49:743–746.
- . Will pay for performance improve quality of care? The answer is in the details. N Engl J Med. 2012;367:1852–1853.
- , , , , , . Reduced mortality with hospital pay for performance in England. N Engl J Med. 2012;367:1821–1828.
- , , , et al. Public reporting and pay for performance in hospital quality improvement. N Engl J Med. 2007;356:486–496.
- , , , . The long‐term effect of premier pay for performance on patient outcomes. N Engl J Med. 2012;366:1606–1615.
- , , , , . Does performance‐based remuneration for individual health care practitioners affect patient care?: a systematic review. Ann Intern Med. 2012;157:889–899.
- Centers for Medicare and Medicaid Services. Hospital Consumer Assessment Of Healthcare Providers and Systems Web site. 2013. Available at: http://www.hcahpsonline.org. Accessed March 5, 2013.
- . Medicare discloses hospitals' bonuses, penalties based on quality. Kaiser Health News. December 20, 2012. Available at: http://www.kaiserhealthnews.org/stories/2012/december/21/medicare‐hospitals‐value‐based‐purchasing.aspx?referrer=search. Accessed March 26, 2013.
- , , , . Hospital quality and intensity of spending: is there an association? Health Aff (Millwood). 2009;28:w566–w572.
- , , , et al. Association between performance measures and clinical outcomes for patients hospitalized with heart failure. JAMA. 2007;297:61–70.
- , , . The advantages and disadvantages of process‐based measures of health care quality. Int J Qual Health Care. 2001;13:469–474.
- . Accountability, incentives and behavior: the impact of high‐stakes testing in the Chicago public schools. J Public Econ. 2005;89:761–796.
- , , , , , . The implications of regional variations in Medicare spending. Part 1: the content, quality, and accessibility of care. Ann Intern Med. 2003;138:273–287.
- . Medical care—is more always better? N Engl J Med. 2003;349:1665–1667.
- , , . Hospital spending and inpatient mortality: evidence from California: an observational study. Ann Intern Med. 2011;154:160–167.
- . Making it easy to do it right. N Engl J Med. 2001;345:991–993.
- , , , . A high rate of compliance with neonatal intensive care unit transfusion guidelines persists even after a program to improve transfusion guideline compliance ended. Transfusion. 2011;51:2519–2520.
- , , , et al. The impact of removing financial incentives from clinical quality indicators: longitudinal analysis of four Kaiser Permanente indicators. BMJ. 2010;340:c1898.
- , . Medicare's new hospital value‐based purchasing program is likely to have only a small impact on hospital payments. Health Aff (Millwood). 2012;31:1932–1940.
- , , , et al. Pay for performance, quality of care, and outcomes in acute myocardial infarction. JAMA. 2007;297:2373–2380.
- , , . Can you get what you pay for? Pay‐for‐performance and the quality of healthcare providers. Rand J Econ. 2010;41:64–91.
- , , , . Spending and mortality in US acute care hospitals. Am J Manag Care. 2013;19:e46–e54.
- , , , , , . Development and validation of hospital “end‐of‐life” treatment intensity measures. Med Care. 2009;47:1098–1105.
- , , , et al. Looking forward, looking back: assessing variations in hospital resource use and outcomes for elderly patients with heart failure. Circ Cardiovasc Qual Outcomes. 2009;2:548–557.
- , , , et al. Association of hospital spending intensity with mortality and readmission rates in Ontario hospitals. JAMA. 2012;307:1037–1045.
- , , . Patient satisfaction with hospital care: effects of demographic and institutional characteristics. Med Care. 2000;38:325–334.
- , , . Linking performance with payment: implementing the Physician Value‐Based Payment Modifier. JAMA. 2012;308:2089–2090.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183:E391–E402.
- , , , et al. Incidence of potentially avoidable urgent readmissions and their relation to all‐cause urgent readmissions. CMAJ. 2011;183:E1067–E1072.
- , . Thirty‐day readmissions—truth and consequences. N Engl J Med. 2012;366:1366–1369.
- , , . Thirty‐day readmission rates for Medicare beneficiaries by race and site of care. JAMA. 2011;305:675–681.