User login
Poor oral hygiene in the mentally ill: Be aware of the problem, and intervene
Poor oral health is common among mentally ill people and is related to inadequate nutrition, poor self-care, substance abuse, and medication side effects.1 Poor oral hygiene is a significant problem because it results in dental pathology that has an adverse influence on the whole body.
Compared with the general population, mentally ill patients are 3 times more likely to have their teeth removed.2 In a survey of mentally ill adults, 92% were found to have tooth decay—of which 23% were untreated and 40% smoked tobacco.3 Approximately 9% have periodontal disease, which most often occurs in those who smoke cigarettes.4
Lifestyle contributors
Drug abuse facilitates dental diseases, as evidenced by the high rate of caries among methamphetamine users.5 The drug induces xerostomia, encouraging users to drink sweetened beverages; this, combined with limited oral care, results in profound dental decay (“meth mouth”). Oral cocaine users often exhibit dental erosions or abrasions, gingival lacerations or necrosis, and mucosal lesions. Smoking Cannabis is associated with an increased rate of gingivitis, alveolar bone loss, leukoplakia, and oral papilloma or other cancers.5 Heroin users are at increased risk of tooth decay, periodontal disease, and oral infection.5
Alcohol consumption increases the risk of oral cancer. Long-term alcohol use suppresses bone marrow function, causing leukopenia and resulting in immunosuppression and an increased incidence of dental infections.6 Excessive alcohol consumption also can cause thrombocytopenia and bleeding, which can complicate dental procedures.
Smoking cigarettes increases the incidence of periodontal disease, especially necrotizing gingivitis and candidiasis.7 Ninety percent of patients with schizophrenia smoke—compared with up to 70% of patients with other psychiatric disorders, and 19% of the general population.7,8 Physiologic aspects of schizophrenia reinforce the smoking habit.7
Somatic ailments. Psychiatric disorders are strongly associated with diabetes, obesity, hypertension, stroke, heart disease, and arthritis, all of which contribute to oral pathology. Older age, greater dysfunction, longer duration of illness, and smoking are predictors of adverse dental outcomes.
Anxiety, depression, stress—all of these these disorders increase the circulating level of cortisol, thus raising the risk that periodontal disease will progress.9 Periodontitis increases the risk of stroke and heart attack by accelerating atherosclerotic plaque formation.10 Depression, anxiety, and substance abuse can lead to temporomandibular disorders that cause pain and restrict jaw movement.11 Stressed patients may experience muscle tension and bruxism, which can lead to temporomandibular joint discomfort.
Eating disorders. Patients who induce vomiting may exhibit enamel erosions (especially on the anterior maxillary teeth), increased tooth hypersensitivity, decay, and wear on dental restorative work.
Atypical odontalgia, characterized by chronic, burning pain in teeth and gums, is associated with depression and anxiety.11 Misdiagnosis can result in extractions or procedures without an appropriate indication and failure to alleviate the pain.
Medication side effects. Xerostomia can increase the risk for caries, periodontal disease, and oral infections such as candidiasis, glossitis, stomatitis, and parotitis.9 Extrapyramidal side effects (tardive dyskinesia, dystonia) may cause tooth damage and make managing dentures difficult.6
What to tell patients, and what you can do for them
Encourage your patients to reduce their sugar intake, brush and floss regularly, and work to stop smoking or ingesting substances of abuse. Teach appropriate hygiene and nutrition, which reduces the risk of dental caries, infection, and related problems. Recommend periodic oral health screening and how to secure such dental care.
From your position of familiarity with patients’ psychopharmacotherapy, make an effort to personalize and adjust their regimens when dental disease is present to address concerns about oral health that can be caused by medication side effects.
A multidisciplinary approach with patient advocacy, involving you and the patient’s dentist and primary care physician, facilitates health care and works to offer the patient access to global medical services.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mental Illness Fellowship of Australia Inc. Overview of the oral health of people affected by mental illness. http:// www.wfmh.com/links/external-contacts/mental-illness-fellowship-of-australia. Accessed June 18, 2014.
2. Kisely S, Quek LH, Pais J, et al. Advanced dental disease in people with severe mental illness: systematic review and meta-analysis. Br J Psychiatry. 2011;199(3):187-193.
3. Dental caries (tooth decay) in adults (age 20 to 64). National Institute of Dental and Craniofacial Research. http:// www.nidcr.nih.gov/DataStatistics/FindDataByTopic/ DentalCaries/DentalCariesAdults20to64.htm. Updated January 6, 2014. Accessed June 18, 2014.
4. Peridontal disease in adults (age 20 to 64). National Institute of Dental and Craniofacial Research. http://www.nidcr. nih.gov/DataStatistics/FindDataByTopic/GumDisease/ PeriodontaldiseaseAdults20to64.htm. Updated January 6, 2014. Accessed June 18, 2014.
5. Maloney WJ. The significance of illicit drug use to dental practice. http://www.webmedcentral.com/wmcpdf/ Article_WMC00455.pdf. Published July 28, 2010. Accessed June 18, 2014.
6. Oral health care for people with mental problems: guidelines and recommendations. British Society for Disability and Oral Health. http://www.bsdh.org.uk/guidelines/ mental.pdf. Updated January 2000. Accessed June 18, 2014.
7. Lohr JB, Flynn K. Smoking and schizophrenia. Schizophr Res. 1992;8(2):93-102.
8. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years–United States, 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
9. Yoffee L. The link between oral health and medical illness. http://www.everydayhealth.com/dental-health/oral-conditions/oral-health-and-other-diseases.aspx. Updated November 9, 2012. Accessed June 18, 2014.
10. Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease: the heart of the matter. J Am Dent Assoc. 2006;137(suppl 2):14S-20S; quiz 38S.
11. Mental illness and the dental patient. American Dental Hygienists’ Association. http://www.adha.org/ce-course-10. Accessed June 18, 2014.
Poor oral health is common among mentally ill people and is related to inadequate nutrition, poor self-care, substance abuse, and medication side effects.1 Poor oral hygiene is a significant problem because it results in dental pathology that has an adverse influence on the whole body.
Compared with the general population, mentally ill patients are 3 times more likely to have their teeth removed.2 In a survey of mentally ill adults, 92% were found to have tooth decay—of which 23% were untreated and 40% smoked tobacco.3 Approximately 9% have periodontal disease, which most often occurs in those who smoke cigarettes.4
Lifestyle contributors
Drug abuse facilitates dental diseases, as evidenced by the high rate of caries among methamphetamine users.5 The drug induces xerostomia, encouraging users to drink sweetened beverages; this, combined with limited oral care, results in profound dental decay (“meth mouth”). Oral cocaine users often exhibit dental erosions or abrasions, gingival lacerations or necrosis, and mucosal lesions. Smoking Cannabis is associated with an increased rate of gingivitis, alveolar bone loss, leukoplakia, and oral papilloma or other cancers.5 Heroin users are at increased risk of tooth decay, periodontal disease, and oral infection.5
Alcohol consumption increases the risk of oral cancer. Long-term alcohol use suppresses bone marrow function, causing leukopenia and resulting in immunosuppression and an increased incidence of dental infections.6 Excessive alcohol consumption also can cause thrombocytopenia and bleeding, which can complicate dental procedures.
Smoking cigarettes increases the incidence of periodontal disease, especially necrotizing gingivitis and candidiasis.7 Ninety percent of patients with schizophrenia smoke—compared with up to 70% of patients with other psychiatric disorders, and 19% of the general population.7,8 Physiologic aspects of schizophrenia reinforce the smoking habit.7
Somatic ailments. Psychiatric disorders are strongly associated with diabetes, obesity, hypertension, stroke, heart disease, and arthritis, all of which contribute to oral pathology. Older age, greater dysfunction, longer duration of illness, and smoking are predictors of adverse dental outcomes.
Anxiety, depression, stress—all of these these disorders increase the circulating level of cortisol, thus raising the risk that periodontal disease will progress.9 Periodontitis increases the risk of stroke and heart attack by accelerating atherosclerotic plaque formation.10 Depression, anxiety, and substance abuse can lead to temporomandibular disorders that cause pain and restrict jaw movement.11 Stressed patients may experience muscle tension and bruxism, which can lead to temporomandibular joint discomfort.
Eating disorders. Patients who induce vomiting may exhibit enamel erosions (especially on the anterior maxillary teeth), increased tooth hypersensitivity, decay, and wear on dental restorative work.
Atypical odontalgia, characterized by chronic, burning pain in teeth and gums, is associated with depression and anxiety.11 Misdiagnosis can result in extractions or procedures without an appropriate indication and failure to alleviate the pain.
Medication side effects. Xerostomia can increase the risk for caries, periodontal disease, and oral infections such as candidiasis, glossitis, stomatitis, and parotitis.9 Extrapyramidal side effects (tardive dyskinesia, dystonia) may cause tooth damage and make managing dentures difficult.6
What to tell patients, and what you can do for them
Encourage your patients to reduce their sugar intake, brush and floss regularly, and work to stop smoking or ingesting substances of abuse. Teach appropriate hygiene and nutrition, which reduces the risk of dental caries, infection, and related problems. Recommend periodic oral health screening and how to secure such dental care.
From your position of familiarity with patients’ psychopharmacotherapy, make an effort to personalize and adjust their regimens when dental disease is present to address concerns about oral health that can be caused by medication side effects.
A multidisciplinary approach with patient advocacy, involving you and the patient’s dentist and primary care physician, facilitates health care and works to offer the patient access to global medical services.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Poor oral health is common among mentally ill people and is related to inadequate nutrition, poor self-care, substance abuse, and medication side effects.1 Poor oral hygiene is a significant problem because it results in dental pathology that has an adverse influence on the whole body.
Compared with the general population, mentally ill patients are 3 times more likely to have their teeth removed.2 In a survey of mentally ill adults, 92% were found to have tooth decay—of which 23% were untreated and 40% smoked tobacco.3 Approximately 9% have periodontal disease, which most often occurs in those who smoke cigarettes.4
Lifestyle contributors
Drug abuse facilitates dental diseases, as evidenced by the high rate of caries among methamphetamine users.5 The drug induces xerostomia, encouraging users to drink sweetened beverages; this, combined with limited oral care, results in profound dental decay (“meth mouth”). Oral cocaine users often exhibit dental erosions or abrasions, gingival lacerations or necrosis, and mucosal lesions. Smoking Cannabis is associated with an increased rate of gingivitis, alveolar bone loss, leukoplakia, and oral papilloma or other cancers.5 Heroin users are at increased risk of tooth decay, periodontal disease, and oral infection.5
Alcohol consumption increases the risk of oral cancer. Long-term alcohol use suppresses bone marrow function, causing leukopenia and resulting in immunosuppression and an increased incidence of dental infections.6 Excessive alcohol consumption also can cause thrombocytopenia and bleeding, which can complicate dental procedures.
Smoking cigarettes increases the incidence of periodontal disease, especially necrotizing gingivitis and candidiasis.7 Ninety percent of patients with schizophrenia smoke—compared with up to 70% of patients with other psychiatric disorders, and 19% of the general population.7,8 Physiologic aspects of schizophrenia reinforce the smoking habit.7
Somatic ailments. Psychiatric disorders are strongly associated with diabetes, obesity, hypertension, stroke, heart disease, and arthritis, all of which contribute to oral pathology. Older age, greater dysfunction, longer duration of illness, and smoking are predictors of adverse dental outcomes.
Anxiety, depression, stress—all of these these disorders increase the circulating level of cortisol, thus raising the risk that periodontal disease will progress.9 Periodontitis increases the risk of stroke and heart attack by accelerating atherosclerotic plaque formation.10 Depression, anxiety, and substance abuse can lead to temporomandibular disorders that cause pain and restrict jaw movement.11 Stressed patients may experience muscle tension and bruxism, which can lead to temporomandibular joint discomfort.
Eating disorders. Patients who induce vomiting may exhibit enamel erosions (especially on the anterior maxillary teeth), increased tooth hypersensitivity, decay, and wear on dental restorative work.
Atypical odontalgia, characterized by chronic, burning pain in teeth and gums, is associated with depression and anxiety.11 Misdiagnosis can result in extractions or procedures without an appropriate indication and failure to alleviate the pain.
Medication side effects. Xerostomia can increase the risk for caries, periodontal disease, and oral infections such as candidiasis, glossitis, stomatitis, and parotitis.9 Extrapyramidal side effects (tardive dyskinesia, dystonia) may cause tooth damage and make managing dentures difficult.6
What to tell patients, and what you can do for them
Encourage your patients to reduce their sugar intake, brush and floss regularly, and work to stop smoking or ingesting substances of abuse. Teach appropriate hygiene and nutrition, which reduces the risk of dental caries, infection, and related problems. Recommend periodic oral health screening and how to secure such dental care.
From your position of familiarity with patients’ psychopharmacotherapy, make an effort to personalize and adjust their regimens when dental disease is present to address concerns about oral health that can be caused by medication side effects.
A multidisciplinary approach with patient advocacy, involving you and the patient’s dentist and primary care physician, facilitates health care and works to offer the patient access to global medical services.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mental Illness Fellowship of Australia Inc. Overview of the oral health of people affected by mental illness. http:// www.wfmh.com/links/external-contacts/mental-illness-fellowship-of-australia. Accessed June 18, 2014.
2. Kisely S, Quek LH, Pais J, et al. Advanced dental disease in people with severe mental illness: systematic review and meta-analysis. Br J Psychiatry. 2011;199(3):187-193.
3. Dental caries (tooth decay) in adults (age 20 to 64). National Institute of Dental and Craniofacial Research. http:// www.nidcr.nih.gov/DataStatistics/FindDataByTopic/ DentalCaries/DentalCariesAdults20to64.htm. Updated January 6, 2014. Accessed June 18, 2014.
4. Peridontal disease in adults (age 20 to 64). National Institute of Dental and Craniofacial Research. http://www.nidcr. nih.gov/DataStatistics/FindDataByTopic/GumDisease/ PeriodontaldiseaseAdults20to64.htm. Updated January 6, 2014. Accessed June 18, 2014.
5. Maloney WJ. The significance of illicit drug use to dental practice. http://www.webmedcentral.com/wmcpdf/ Article_WMC00455.pdf. Published July 28, 2010. Accessed June 18, 2014.
6. Oral health care for people with mental problems: guidelines and recommendations. British Society for Disability and Oral Health. http://www.bsdh.org.uk/guidelines/ mental.pdf. Updated January 2000. Accessed June 18, 2014.
7. Lohr JB, Flynn K. Smoking and schizophrenia. Schizophr Res. 1992;8(2):93-102.
8. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years–United States, 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
9. Yoffee L. The link between oral health and medical illness. http://www.everydayhealth.com/dental-health/oral-conditions/oral-health-and-other-diseases.aspx. Updated November 9, 2012. Accessed June 18, 2014.
10. Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease: the heart of the matter. J Am Dent Assoc. 2006;137(suppl 2):14S-20S; quiz 38S.
11. Mental illness and the dental patient. American Dental Hygienists’ Association. http://www.adha.org/ce-course-10. Accessed June 18, 2014.
1. Mental Illness Fellowship of Australia Inc. Overview of the oral health of people affected by mental illness. http:// www.wfmh.com/links/external-contacts/mental-illness-fellowship-of-australia. Accessed June 18, 2014.
2. Kisely S, Quek LH, Pais J, et al. Advanced dental disease in people with severe mental illness: systematic review and meta-analysis. Br J Psychiatry. 2011;199(3):187-193.
3. Dental caries (tooth decay) in adults (age 20 to 64). National Institute of Dental and Craniofacial Research. http:// www.nidcr.nih.gov/DataStatistics/FindDataByTopic/ DentalCaries/DentalCariesAdults20to64.htm. Updated January 6, 2014. Accessed June 18, 2014.
4. Peridontal disease in adults (age 20 to 64). National Institute of Dental and Craniofacial Research. http://www.nidcr. nih.gov/DataStatistics/FindDataByTopic/GumDisease/ PeriodontaldiseaseAdults20to64.htm. Updated January 6, 2014. Accessed June 18, 2014.
5. Maloney WJ. The significance of illicit drug use to dental practice. http://www.webmedcentral.com/wmcpdf/ Article_WMC00455.pdf. Published July 28, 2010. Accessed June 18, 2014.
6. Oral health care for people with mental problems: guidelines and recommendations. British Society for Disability and Oral Health. http://www.bsdh.org.uk/guidelines/ mental.pdf. Updated January 2000. Accessed June 18, 2014.
7. Lohr JB, Flynn K. Smoking and schizophrenia. Schizophr Res. 1992;8(2):93-102.
8. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years–United States, 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
9. Yoffee L. The link between oral health and medical illness. http://www.everydayhealth.com/dental-health/oral-conditions/oral-health-and-other-diseases.aspx. Updated November 9, 2012. Accessed June 18, 2014.
10. Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease: the heart of the matter. J Am Dent Assoc. 2006;137(suppl 2):14S-20S; quiz 38S.
11. Mental illness and the dental patient. American Dental Hygienists’ Association. http://www.adha.org/ce-course-10. Accessed June 18, 2014.
Prescribing for the pregnant patient
Primum non nocere: First, do no harm—a principle taught across the world to all medical students. It reminds the health care provider to consider the possible harm that any intervention might produce. Never is it more relevant in the mind of a clinician than when prescribing a medication for a pregnant woman. We are, after all, brought up in a society averse to medical risk.
When managing a pregnant patient, should the baby be the highest priority, whatever the mother may face? Or to take the extreme opposite position, should the mother be treated with the best possible options and the baby ignored?
And what about the views of the patient? There is a widespread cultural belief about the vulnerability of the mother and fetus during pregnancy. Therefore, when faced with the decision of whether to use a medication or not, what is the best recourse for the pregnant patient? Should she be the “good mother” and avoid all risk to the baby, or should she be the “responsible mother” who follows medical advice and takes treatment as recommended?
In truth, the path to safe management of a pregnant patient is rarely so dichotomous. In most cases, what is best for the mother is also best for the baby. However, caring for a pregnant or lactating woman can be challenging for clinicians facing insufficient information regarding medication safety, overestimation of the risk of medication by both the patient and the care provider, and increasing litigation costs.
This article provides key principles to guide clinicians caring for pregnant patients, as we find ourselves increasingly dependent on pharmacotherapy. It also includes sources of information clinicians can turn to when they need additional pregnancy safety data about a certain drug and when they want advice about conditions commonly seen in pregnancy and medications that can be justifiably used in those circumstances.
KEY CONCEPTS FOR PRESCRIBING IN PREGNANCY
The following concepts are key to prescribing for a pregnant patient:
No protective barrier exists between the maternal and fetal environments
The placenta contains a semipermeable membrane that selectively allows some substances to pass from the maternal to the fetal blood and excludes others. However, it is not really a “protective mechanism” when it comes to medications. Assume that the fetus will have exposure, at least to some degree.
In general, drugs that are lipophilic, of a low molecular weight, or not ionized at physiologic pH cross the placenta more efficiently than others. Heparin and insulin are notable exceptions to the rule that most drugs cross the placenta. They do not.
The gestational stage may determine the effect of a medication on the fetus
In animals and in humans, exposure of the embryo or fetus to a teratogen may produce a permanent abnormality of structure or function.
First-trimester exposures are most worrisome for structural malformations. However, fetal neurologic and behavioral development, fetal survival, and function of specific organs can be affected even after the first trimester. For example, while first-trimester exposure to angiotensin-converting enzyme inhibitors has been linked to a slight increase in congenital heart defects, exposure in the second or third trimester can result in fetal oligohydramnios, neonatal anuria, pulmonary hypoplasia, intrauterine growth restriction, and fetal death.
Physiologic changes of pregnancy affect the pharmacokinetics of medications
Pregnancy is associated with increased plasma volume, increased glomerular filtration rate, and dilutional hypoalbuminemia, which can all affect the bioavailability of medications. Absorption of oral agents also may be affected by slowed gastric motility in pregnancy.
Although these physiologic alterations do not routinely warrant a change in drug dosage, they may be important considerations when choosing an appropriate agent. For example, medications taken in multiple doses per day are more likely to have a sustained effect than once-daily medications, which would be rapidly cleared in a pregnant patient.
Sole reliance on the FDA pregnancy safety category may be inadequate

To help clinicians prescribe medications for pregnant women, the US Food and Drug Administration (FDA) assigns medications to one of five categories of risk (A, B, C, D, or X) (Table 1). Unfortunately, this classification system has several shortcomings:
- The categories are often seen as a grading system in which the risk increases from the lowest in category A to highest in category X, and the safety information in the accompanying narrative is not always appreciated by prescribers.
- Clinicians incorrectly assume that drugs in a particular category carry a similar risk. However, 65% to 70% of all medications are in category C. This category includes medications with adverse animal data or no animal data at all. In addition, adverse animal data may vary in severity from decreased fetal weight to major structural malformation and fetal loss, indicating a difference in expected risk.
- Most of the data on medication safety in pregnancy comes from animal studies, case reports, case series, case-control studies, or pregnancy registries, and each of these sources has significant limitations.
- The categories do not distinguish between supporting data from animal studies and human studies. For instance, a category-B drug may have animal studies that show no risk but no adequate human studies, or may have animal studies showing risk but human studies that do not.
Looking at the pregnancy risk classifications used in the United States (ie, the FDA system), Australia, and Sweden, researchers compared the classification of 236 drugs between the three systems and found that only one in four drugs was similarly classified into the same risk category. This discrepancy further brings into question the usefulness and reliability of these classifications.1
Finally, none of the classification systems tells us the potential harm from withholding a medication in pregnancy.
RESOURCES TO ASSESS MEDICATION SAFETY IN PREGNANCY
The FDA has proposed changes in the labeling of medications related to pregnancy and lactation.2 The proposed changes would eliminate the current categories and instead require a summary of the risks, the effects of the drug on the fetus, and clinical considerations for use during pregnancy. In addition, labeling would include a description of the medication’s effects on milk production, the amount of drug present in milk, and possible effects on the infant.
Until such changes are in place, what other resources can a busy clinician turn to for support?
The official drug labeling (or the package insert), also published in the Physicians’ Desk Reference, is one source of information, but it rarely provides up-to-date information about teratogenic risks in human pregnancies.
Several online databases review, summarize, and periodically update information from the peer-reviewed medical literature.3–7 The REPRORISK system4–7 maintained by Micromedex (Greenwood Village, CO) provides access to several databases that contain information about a wide range of individual medications: REPROTEXT, REPROTOX,5 Shepard’s Catalog of Teratogenic Agents,7 and the Teratogen Information System (TERIS).4 Online access and a smartphone “app” for these databases are available for a subscription fee. Summaries for individual medications can be ordered directly from TERIS, also for a fee. Several other resources are available in textbook format.8–10
In addition, health care providers can obtain information from or can refer pregnant and breastfeeding patients to a teratology information service for information and counseling about medication exposures. MotherToBaby,11 a service of the nonprofit Organization of Teratology Information Specialists, provides fact sheets, free phone consultation, risk assessment, and counseling by trained teratogen information specialists about environmental exposures, including prescription and over-the-counter medications and dietary and herbal supplements. Counselors from these services gather and synthesize information about exposures from the databases mentioned above, from the peer-reviewed medical literature, from drug manufacturers, and from other sources.

With the advent of electronic medical records and computerized provider order entry, clinical decision support systems hold promise as an additional resource for safe prescribing in pregnancy.
Fortunately, the list of teratogenic medications that are absolutely contraindicated in pregnancy remains small (Table 2).12,13
THE FOUR-QUESTION APPROACH TO CARING FOR THE PREGNANT PATIENT
Is the symptom self-limited or amenable to nonpharmacologic management?
It has been said that we live in a culture where every symptom warrants a pill. If this is true, there can be no better time for reevaluating this practice than during pregnancy.
Many of the medications most commonly used in pregnancy are for upper-respiratory-tract infections, headache, or psychological distress. Pregnancy is the ideal time to educate patients about the limited effectiveness of most cough-and-cold remedies and the inappropriateness of antibiotics for colds and viral bronchitis. It is also an ideal time for a trial of lifestyle modifications, relaxation, and biofeedback for a chronic headache problem. For cases of mild to moderate depression, it may be worth considering treatment with psychotherapy rather than medications.
Offering patients the option of no treatment or nonpharmacologic treatment for self-limited symptoms is an option worth considering.
How do the patient’s (and your) values and understanding affect the decision?
Is the patient willing to take medication? What are her beliefs with regard to her problem and how it should be managed in pregnancy?
Women and clinicians bring many worries and prejudices to the use of medications in pregnancy. The experiences of the patient and her family and friends may present huge obstacles to needed medication use in pregnancy. Misinformation in the media and from family members, friends, and other health care providers are other obstacles. The only way to deal with this issue is to ask your patient directly about her fears and concerns regarding each prescription written.
Clinicians also need to address fears or prejudices they themselves may have about medication safety in pregnancy. These may arise from a single bad experience in caring for a pregnant woman, discomfort with uncertainty, or a belief that pregnant women should avoid any and all risks of exposures, even when the mother’s condition warrants pharmacologic treatment.
Being informed, both scientifically and about one’s own biases or tendencies, is an essential foundation for rational prescribing in pregnancy.
Is the problem affected by pregnancy, and how?
Pregnancy can affect many medical conditions, and in different ways. Conditions such as asthma, migraine headache, and cardiac arrhythmia are exacerbated in pregnancy, placing the mother and fetus at increased risk of morbidity. Conditions such as Graves disease and hypertension may improve as pregnancy progresses, and medications often can be withdrawn as the patient progresses further along in gestation.
Understanding the effect of pregnancy on a particular problem may help the clinician to make an informed decision about medication use in pregnancy.
How does the problem affect pregnancy?
Considering the risk of untreated disease to the pregnancy may help in decision-making.
Many medical conditions can negatively affect the development of the fetus. A glaring example is diabetes mellitus, with poor glycemic control being linked to congenital malformations, spontaneous abortion, and fetal demise. Chronic conditions with periodic exacerbations such as asthma or epilepsy place the fetus at increased risk during a flare-up.
Therefore, for chronic conditions, continuing maintenance therapy is best. Preconception counseling in such cases is crucial, so that a drug with adequate safety data can be substituted before pregnancy. In this way, any risk to the mother or the embryo from exacerbation of disease as such adjustments are made is avoided.
For conditions arising de novo in pregnancy, the underlying principle remains the same. Is the risk of pharmacotherapy more than the risk of untreated disease? Invariably, the answer to this question supports medication use, and an educated provider will be able to choose a treatment that is justifiable in most circumstances.
CHOOSING A MEDICATION

Fetal well-being depends on maternal well-being. It therefore helps to think of medication use in pregnancy as “justified or not” rather than “safe or not.” Table 3 lists some conditions commonly seen in pregnancy, selected drugs of choice that can be safely used for treating those conditions, and alternates that may be justified in some circumstances.5,6,14–18
GOOD PRACTICES WHEN PRESCRIBING IN PREGNANCY
Prescribing in pregnancy will be most successful when both the patient and the prescribing physician consider the fetal benefit gained from optimizing maternal health. Good prescribing practices to ensure optimum therapeutic benefit when caring for a pregnant patient are to:
- Involve the patient in decision-making. Recognize her concerns, worries, and preferences regarding her illness and its treatment.
- Inform the patient of the risk of an untreated medical condition, weighed against the risk of medication.
- Choose medications with the most available safety data. Let the patient know what resources you have referred to in choosing the medication.
- It is advisable to perform a search each time a prescription is written for a pregnant or lactating woman.
- When possible, have the discussion in the preconception period.
- Consider the dynamic physiology of gestation. Choose the right drug for the right trimester.
- Discuss the plan with the patient and other providers.
- Define clear criteria for when to discontinue the treatment.
- Addis A, Sharabi S, Bonati M. Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 2000; 23:245–253.
- US Food and Drug Administration (FDA). Pregnancy and lactation labeling. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm. Accessed April 4, 2014.
- Lagoy CT, Joshi N, Cragan JD, Rasmussen SA. Medication use during pregnancy and lactation: an urgent call for public health action. J Womens Health (Larchmt) 2005; 14:104–109.
- Clinical Teratology Website. University of Washington. http://depts.washington.edu/terisweb/teris/. Accessed April 4, 2014.
- REPROTOX, An Online Reproductive Toxicology Resource. Reproductive Toxicology Center. www.reprotox.org. Accessed April 4, 2014.
- REPRORISK. Micromedex, Inc. www.micromedex.com/products/reprorisk. Accessed April 4, 2014.
- Shepard TH. Catalog of teratogenic agents. 13th ed. Baltimore, MD: Johns Hopkins University Press; 2010.
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: A reference guide to fetal and neonatal risk. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Koren G. Medication safety in pregnancy and breastfeeding. McGraw-Hill Professional Publishing; 2007.
- Friedman JM, Polifka JE. Teratogenic effects of drugs: A resource for clinicians (TERIS). Baltimore, MD: Johns Hopkins University Press; 2000.
- MotherToBaby. www.mothertobaby.org. Accessed April 4, 2014.
- Dunlop AL, Gardiner PM, Shellhaas CS, Menard MK, McDiarmid MA. The clinical content of preconception care: the use of medications and supplements among women of reproductive age. Am J Obstet Gynecol 2008; 199(suppl 2):S367–S372.
- Ciarkowski SL, Stalburg CM. Medication safety in obstetrics and gynecology. Clin Obstet Gynecol 2010; 53:482–499.
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med 1998; 338:1128–1137.
- Lambert K, Holt RI. The use of insulin analogues in pregnancy. Diabetes Obes Metab 2013; 15:888–900.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Nagler M, Haslauer M, Wuillemin WA. Fondaparinux—data on efficacy and safety in special situations. Thromb Res 2012; 129:407–417.
- Kweder SL, Powrie RO. Prescribing in pregnancy: a practical approach. In:Powrie RO, Greene M, Camann W, editors. De Swiet’s Medical disorders in Obstetric Practice. 5th ed. Hoboken, NJ: Wiley-Blackwell; 2010:633–640.
Primum non nocere: First, do no harm—a principle taught across the world to all medical students. It reminds the health care provider to consider the possible harm that any intervention might produce. Never is it more relevant in the mind of a clinician than when prescribing a medication for a pregnant woman. We are, after all, brought up in a society averse to medical risk.
When managing a pregnant patient, should the baby be the highest priority, whatever the mother may face? Or to take the extreme opposite position, should the mother be treated with the best possible options and the baby ignored?
And what about the views of the patient? There is a widespread cultural belief about the vulnerability of the mother and fetus during pregnancy. Therefore, when faced with the decision of whether to use a medication or not, what is the best recourse for the pregnant patient? Should she be the “good mother” and avoid all risk to the baby, or should she be the “responsible mother” who follows medical advice and takes treatment as recommended?
In truth, the path to safe management of a pregnant patient is rarely so dichotomous. In most cases, what is best for the mother is also best for the baby. However, caring for a pregnant or lactating woman can be challenging for clinicians facing insufficient information regarding medication safety, overestimation of the risk of medication by both the patient and the care provider, and increasing litigation costs.
This article provides key principles to guide clinicians caring for pregnant patients, as we find ourselves increasingly dependent on pharmacotherapy. It also includes sources of information clinicians can turn to when they need additional pregnancy safety data about a certain drug and when they want advice about conditions commonly seen in pregnancy and medications that can be justifiably used in those circumstances.
KEY CONCEPTS FOR PRESCRIBING IN PREGNANCY
The following concepts are key to prescribing for a pregnant patient:
No protective barrier exists between the maternal and fetal environments
The placenta contains a semipermeable membrane that selectively allows some substances to pass from the maternal to the fetal blood and excludes others. However, it is not really a “protective mechanism” when it comes to medications. Assume that the fetus will have exposure, at least to some degree.
In general, drugs that are lipophilic, of a low molecular weight, or not ionized at physiologic pH cross the placenta more efficiently than others. Heparin and insulin are notable exceptions to the rule that most drugs cross the placenta. They do not.
The gestational stage may determine the effect of a medication on the fetus
In animals and in humans, exposure of the embryo or fetus to a teratogen may produce a permanent abnormality of structure or function.
First-trimester exposures are most worrisome for structural malformations. However, fetal neurologic and behavioral development, fetal survival, and function of specific organs can be affected even after the first trimester. For example, while first-trimester exposure to angiotensin-converting enzyme inhibitors has been linked to a slight increase in congenital heart defects, exposure in the second or third trimester can result in fetal oligohydramnios, neonatal anuria, pulmonary hypoplasia, intrauterine growth restriction, and fetal death.
Physiologic changes of pregnancy affect the pharmacokinetics of medications
Pregnancy is associated with increased plasma volume, increased glomerular filtration rate, and dilutional hypoalbuminemia, which can all affect the bioavailability of medications. Absorption of oral agents also may be affected by slowed gastric motility in pregnancy.
Although these physiologic alterations do not routinely warrant a change in drug dosage, they may be important considerations when choosing an appropriate agent. For example, medications taken in multiple doses per day are more likely to have a sustained effect than once-daily medications, which would be rapidly cleared in a pregnant patient.
Sole reliance on the FDA pregnancy safety category may be inadequate
To help clinicians prescribe medications for pregnant women, the US Food and Drug Administration (FDA) assigns medications to one of five categories of risk (A, B, C, D, or X) (Table 1). Unfortunately, this classification system has several shortcomings:
- The categories are often seen as a grading system in which the risk increases from the lowest in category A to highest in category X, and the safety information in the accompanying narrative is not always appreciated by prescribers.
- Clinicians incorrectly assume that drugs in a particular category carry a similar risk. However, 65% to 70% of all medications are in category C. This category includes medications with adverse animal data or no animal data at all. In addition, adverse animal data may vary in severity from decreased fetal weight to major structural malformation and fetal loss, indicating a difference in expected risk.
- Most of the data on medication safety in pregnancy comes from animal studies, case reports, case series, case-control studies, or pregnancy registries, and each of these sources has significant limitations.
- The categories do not distinguish between supporting data from animal studies and human studies. For instance, a category-B drug may have animal studies that show no risk but no adequate human studies, or may have animal studies showing risk but human studies that do not.
Looking at the pregnancy risk classifications used in the United States (ie, the FDA system), Australia, and Sweden, researchers compared the classification of 236 drugs between the three systems and found that only one in four drugs was similarly classified into the same risk category. This discrepancy further brings into question the usefulness and reliability of these classifications.1
Finally, none of the classification systems tells us the potential harm from withholding a medication in pregnancy.
RESOURCES TO ASSESS MEDICATION SAFETY IN PREGNANCY
The FDA has proposed changes in the labeling of medications related to pregnancy and lactation.2 The proposed changes would eliminate the current categories and instead require a summary of the risks, the effects of the drug on the fetus, and clinical considerations for use during pregnancy. In addition, labeling would include a description of the medication’s effects on milk production, the amount of drug present in milk, and possible effects on the infant.
Until such changes are in place, what other resources can a busy clinician turn to for support?
The official drug labeling (or the package insert), also published in the Physicians’ Desk Reference, is one source of information, but it rarely provides up-to-date information about teratogenic risks in human pregnancies.
Several online databases review, summarize, and periodically update information from the peer-reviewed medical literature.3–7 The REPRORISK system4–7 maintained by Micromedex (Greenwood Village, CO) provides access to several databases that contain information about a wide range of individual medications: REPROTEXT, REPROTOX,5 Shepard’s Catalog of Teratogenic Agents,7 and the Teratogen Information System (TERIS).4 Online access and a smartphone “app” for these databases are available for a subscription fee. Summaries for individual medications can be ordered directly from TERIS, also for a fee. Several other resources are available in textbook format.8–10
In addition, health care providers can obtain information from or can refer pregnant and breastfeeding patients to a teratology information service for information and counseling about medication exposures. MotherToBaby,11 a service of the nonprofit Organization of Teratology Information Specialists, provides fact sheets, free phone consultation, risk assessment, and counseling by trained teratogen information specialists about environmental exposures, including prescription and over-the-counter medications and dietary and herbal supplements. Counselors from these services gather and synthesize information about exposures from the databases mentioned above, from the peer-reviewed medical literature, from drug manufacturers, and from other sources.
With the advent of electronic medical records and computerized provider order entry, clinical decision support systems hold promise as an additional resource for safe prescribing in pregnancy.
Fortunately, the list of teratogenic medications that are absolutely contraindicated in pregnancy remains small (Table 2).12,13
THE FOUR-QUESTION APPROACH TO CARING FOR THE PREGNANT PATIENT
Is the symptom self-limited or amenable to nonpharmacologic management?
It has been said that we live in a culture where every symptom warrants a pill. If this is true, there can be no better time for reevaluating this practice than during pregnancy.
Many of the medications most commonly used in pregnancy are for upper-respiratory-tract infections, headache, or psychological distress. Pregnancy is the ideal time to educate patients about the limited effectiveness of most cough-and-cold remedies and the inappropriateness of antibiotics for colds and viral bronchitis. It is also an ideal time for a trial of lifestyle modifications, relaxation, and biofeedback for a chronic headache problem. For cases of mild to moderate depression, it may be worth considering treatment with psychotherapy rather than medications.
Offering patients the option of no treatment or nonpharmacologic treatment for self-limited symptoms is an option worth considering.
How do the patient’s (and your) values and understanding affect the decision?
Is the patient willing to take medication? What are her beliefs with regard to her problem and how it should be managed in pregnancy?
Women and clinicians bring many worries and prejudices to the use of medications in pregnancy. The experiences of the patient and her family and friends may present huge obstacles to needed medication use in pregnancy. Misinformation in the media and from family members, friends, and other health care providers are other obstacles. The only way to deal with this issue is to ask your patient directly about her fears and concerns regarding each prescription written.
Clinicians also need to address fears or prejudices they themselves may have about medication safety in pregnancy. These may arise from a single bad experience in caring for a pregnant woman, discomfort with uncertainty, or a belief that pregnant women should avoid any and all risks of exposures, even when the mother’s condition warrants pharmacologic treatment.
Being informed, both scientifically and about one’s own biases or tendencies, is an essential foundation for rational prescribing in pregnancy.
Is the problem affected by pregnancy, and how?
Pregnancy can affect many medical conditions, and in different ways. Conditions such as asthma, migraine headache, and cardiac arrhythmia are exacerbated in pregnancy, placing the mother and fetus at increased risk of morbidity. Conditions such as Graves disease and hypertension may improve as pregnancy progresses, and medications often can be withdrawn as the patient progresses further along in gestation.
Understanding the effect of pregnancy on a particular problem may help the clinician to make an informed decision about medication use in pregnancy.
How does the problem affect pregnancy?
Considering the risk of untreated disease to the pregnancy may help in decision-making.
Many medical conditions can negatively affect the development of the fetus. A glaring example is diabetes mellitus, with poor glycemic control being linked to congenital malformations, spontaneous abortion, and fetal demise. Chronic conditions with periodic exacerbations such as asthma or epilepsy place the fetus at increased risk during a flare-up.
Therefore, for chronic conditions, continuing maintenance therapy is best. Preconception counseling in such cases is crucial, so that a drug with adequate safety data can be substituted before pregnancy. In this way, any risk to the mother or the embryo from exacerbation of disease as such adjustments are made is avoided.
For conditions arising de novo in pregnancy, the underlying principle remains the same. Is the risk of pharmacotherapy more than the risk of untreated disease? Invariably, the answer to this question supports medication use, and an educated provider will be able to choose a treatment that is justifiable in most circumstances.
CHOOSING A MEDICATION
Fetal well-being depends on maternal well-being. It therefore helps to think of medication use in pregnancy as “justified or not” rather than “safe or not.” Table 3 lists some conditions commonly seen in pregnancy, selected drugs of choice that can be safely used for treating those conditions, and alternates that may be justified in some circumstances.5,6,14–18
GOOD PRACTICES WHEN PRESCRIBING IN PREGNANCY
Prescribing in pregnancy will be most successful when both the patient and the prescribing physician consider the fetal benefit gained from optimizing maternal health. Good prescribing practices to ensure optimum therapeutic benefit when caring for a pregnant patient are to:
- Involve the patient in decision-making. Recognize her concerns, worries, and preferences regarding her illness and its treatment.
- Inform the patient of the risk of an untreated medical condition, weighed against the risk of medication.
- Choose medications with the most available safety data. Let the patient know what resources you have referred to in choosing the medication.
- It is advisable to perform a search each time a prescription is written for a pregnant or lactating woman.
- When possible, have the discussion in the preconception period.
- Consider the dynamic physiology of gestation. Choose the right drug for the right trimester.
- Discuss the plan with the patient and other providers.
- Define clear criteria for when to discontinue the treatment.
Primum non nocere: First, do no harm—a principle taught across the world to all medical students. It reminds the health care provider to consider the possible harm that any intervention might produce. Never is it more relevant in the mind of a clinician than when prescribing a medication for a pregnant woman. We are, after all, brought up in a society averse to medical risk.
When managing a pregnant patient, should the baby be the highest priority, whatever the mother may face? Or to take the extreme opposite position, should the mother be treated with the best possible options and the baby ignored?
And what about the views of the patient? There is a widespread cultural belief about the vulnerability of the mother and fetus during pregnancy. Therefore, when faced with the decision of whether to use a medication or not, what is the best recourse for the pregnant patient? Should she be the “good mother” and avoid all risk to the baby, or should she be the “responsible mother” who follows medical advice and takes treatment as recommended?
In truth, the path to safe management of a pregnant patient is rarely so dichotomous. In most cases, what is best for the mother is also best for the baby. However, caring for a pregnant or lactating woman can be challenging for clinicians facing insufficient information regarding medication safety, overestimation of the risk of medication by both the patient and the care provider, and increasing litigation costs.
This article provides key principles to guide clinicians caring for pregnant patients, as we find ourselves increasingly dependent on pharmacotherapy. It also includes sources of information clinicians can turn to when they need additional pregnancy safety data about a certain drug and when they want advice about conditions commonly seen in pregnancy and medications that can be justifiably used in those circumstances.
KEY CONCEPTS FOR PRESCRIBING IN PREGNANCY
The following concepts are key to prescribing for a pregnant patient:
No protective barrier exists between the maternal and fetal environments
The placenta contains a semipermeable membrane that selectively allows some substances to pass from the maternal to the fetal blood and excludes others. However, it is not really a “protective mechanism” when it comes to medications. Assume that the fetus will have exposure, at least to some degree.
In general, drugs that are lipophilic, of a low molecular weight, or not ionized at physiologic pH cross the placenta more efficiently than others. Heparin and insulin are notable exceptions to the rule that most drugs cross the placenta. They do not.
The gestational stage may determine the effect of a medication on the fetus
In animals and in humans, exposure of the embryo or fetus to a teratogen may produce a permanent abnormality of structure or function.
First-trimester exposures are most worrisome for structural malformations. However, fetal neurologic and behavioral development, fetal survival, and function of specific organs can be affected even after the first trimester. For example, while first-trimester exposure to angiotensin-converting enzyme inhibitors has been linked to a slight increase in congenital heart defects, exposure in the second or third trimester can result in fetal oligohydramnios, neonatal anuria, pulmonary hypoplasia, intrauterine growth restriction, and fetal death.
Physiologic changes of pregnancy affect the pharmacokinetics of medications
Pregnancy is associated with increased plasma volume, increased glomerular filtration rate, and dilutional hypoalbuminemia, which can all affect the bioavailability of medications. Absorption of oral agents also may be affected by slowed gastric motility in pregnancy.
Although these physiologic alterations do not routinely warrant a change in drug dosage, they may be important considerations when choosing an appropriate agent. For example, medications taken in multiple doses per day are more likely to have a sustained effect than once-daily medications, which would be rapidly cleared in a pregnant patient.
Sole reliance on the FDA pregnancy safety category may be inadequate
To help clinicians prescribe medications for pregnant women, the US Food and Drug Administration (FDA) assigns medications to one of five categories of risk (A, B, C, D, or X) (Table 1). Unfortunately, this classification system has several shortcomings:
- The categories are often seen as a grading system in which the risk increases from the lowest in category A to highest in category X, and the safety information in the accompanying narrative is not always appreciated by prescribers.
- Clinicians incorrectly assume that drugs in a particular category carry a similar risk. However, 65% to 70% of all medications are in category C. This category includes medications with adverse animal data or no animal data at all. In addition, adverse animal data may vary in severity from decreased fetal weight to major structural malformation and fetal loss, indicating a difference in expected risk.
- Most of the data on medication safety in pregnancy comes from animal studies, case reports, case series, case-control studies, or pregnancy registries, and each of these sources has significant limitations.
- The categories do not distinguish between supporting data from animal studies and human studies. For instance, a category-B drug may have animal studies that show no risk but no adequate human studies, or may have animal studies showing risk but human studies that do not.
Looking at the pregnancy risk classifications used in the United States (ie, the FDA system), Australia, and Sweden, researchers compared the classification of 236 drugs between the three systems and found that only one in four drugs was similarly classified into the same risk category. This discrepancy further brings into question the usefulness and reliability of these classifications.1
Finally, none of the classification systems tells us the potential harm from withholding a medication in pregnancy.
RESOURCES TO ASSESS MEDICATION SAFETY IN PREGNANCY
The FDA has proposed changes in the labeling of medications related to pregnancy and lactation.2 The proposed changes would eliminate the current categories and instead require a summary of the risks, the effects of the drug on the fetus, and clinical considerations for use during pregnancy. In addition, labeling would include a description of the medication’s effects on milk production, the amount of drug present in milk, and possible effects on the infant.
Until such changes are in place, what other resources can a busy clinician turn to for support?
The official drug labeling (or the package insert), also published in the Physicians’ Desk Reference, is one source of information, but it rarely provides up-to-date information about teratogenic risks in human pregnancies.
Several online databases review, summarize, and periodically update information from the peer-reviewed medical literature.3–7 The REPRORISK system4–7 maintained by Micromedex (Greenwood Village, CO) provides access to several databases that contain information about a wide range of individual medications: REPROTEXT, REPROTOX,5 Shepard’s Catalog of Teratogenic Agents,7 and the Teratogen Information System (TERIS).4 Online access and a smartphone “app” for these databases are available for a subscription fee. Summaries for individual medications can be ordered directly from TERIS, also for a fee. Several other resources are available in textbook format.8–10
In addition, health care providers can obtain information from or can refer pregnant and breastfeeding patients to a teratology information service for information and counseling about medication exposures. MotherToBaby,11 a service of the nonprofit Organization of Teratology Information Specialists, provides fact sheets, free phone consultation, risk assessment, and counseling by trained teratogen information specialists about environmental exposures, including prescription and over-the-counter medications and dietary and herbal supplements. Counselors from these services gather and synthesize information about exposures from the databases mentioned above, from the peer-reviewed medical literature, from drug manufacturers, and from other sources.
With the advent of electronic medical records and computerized provider order entry, clinical decision support systems hold promise as an additional resource for safe prescribing in pregnancy.
Fortunately, the list of teratogenic medications that are absolutely contraindicated in pregnancy remains small (Table 2).12,13
THE FOUR-QUESTION APPROACH TO CARING FOR THE PREGNANT PATIENT
Is the symptom self-limited or amenable to nonpharmacologic management?
It has been said that we live in a culture where every symptom warrants a pill. If this is true, there can be no better time for reevaluating this practice than during pregnancy.
Many of the medications most commonly used in pregnancy are for upper-respiratory-tract infections, headache, or psychological distress. Pregnancy is the ideal time to educate patients about the limited effectiveness of most cough-and-cold remedies and the inappropriateness of antibiotics for colds and viral bronchitis. It is also an ideal time for a trial of lifestyle modifications, relaxation, and biofeedback for a chronic headache problem. For cases of mild to moderate depression, it may be worth considering treatment with psychotherapy rather than medications.
Offering patients the option of no treatment or nonpharmacologic treatment for self-limited symptoms is an option worth considering.
How do the patient’s (and your) values and understanding affect the decision?
Is the patient willing to take medication? What are her beliefs with regard to her problem and how it should be managed in pregnancy?
Women and clinicians bring many worries and prejudices to the use of medications in pregnancy. The experiences of the patient and her family and friends may present huge obstacles to needed medication use in pregnancy. Misinformation in the media and from family members, friends, and other health care providers are other obstacles. The only way to deal with this issue is to ask your patient directly about her fears and concerns regarding each prescription written.
Clinicians also need to address fears or prejudices they themselves may have about medication safety in pregnancy. These may arise from a single bad experience in caring for a pregnant woman, discomfort with uncertainty, or a belief that pregnant women should avoid any and all risks of exposures, even when the mother’s condition warrants pharmacologic treatment.
Being informed, both scientifically and about one’s own biases or tendencies, is an essential foundation for rational prescribing in pregnancy.
Is the problem affected by pregnancy, and how?
Pregnancy can affect many medical conditions, and in different ways. Conditions such as asthma, migraine headache, and cardiac arrhythmia are exacerbated in pregnancy, placing the mother and fetus at increased risk of morbidity. Conditions such as Graves disease and hypertension may improve as pregnancy progresses, and medications often can be withdrawn as the patient progresses further along in gestation.
Understanding the effect of pregnancy on a particular problem may help the clinician to make an informed decision about medication use in pregnancy.
How does the problem affect pregnancy?
Considering the risk of untreated disease to the pregnancy may help in decision-making.
Many medical conditions can negatively affect the development of the fetus. A glaring example is diabetes mellitus, with poor glycemic control being linked to congenital malformations, spontaneous abortion, and fetal demise. Chronic conditions with periodic exacerbations such as asthma or epilepsy place the fetus at increased risk during a flare-up.
Therefore, for chronic conditions, continuing maintenance therapy is best. Preconception counseling in such cases is crucial, so that a drug with adequate safety data can be substituted before pregnancy. In this way, any risk to the mother or the embryo from exacerbation of disease as such adjustments are made is avoided.
For conditions arising de novo in pregnancy, the underlying principle remains the same. Is the risk of pharmacotherapy more than the risk of untreated disease? Invariably, the answer to this question supports medication use, and an educated provider will be able to choose a treatment that is justifiable in most circumstances.
CHOOSING A MEDICATION
Fetal well-being depends on maternal well-being. It therefore helps to think of medication use in pregnancy as “justified or not” rather than “safe or not.” Table 3 lists some conditions commonly seen in pregnancy, selected drugs of choice that can be safely used for treating those conditions, and alternates that may be justified in some circumstances.5,6,14–18
GOOD PRACTICES WHEN PRESCRIBING IN PREGNANCY
Prescribing in pregnancy will be most successful when both the patient and the prescribing physician consider the fetal benefit gained from optimizing maternal health. Good prescribing practices to ensure optimum therapeutic benefit when caring for a pregnant patient are to:
- Involve the patient in decision-making. Recognize her concerns, worries, and preferences regarding her illness and its treatment.
- Inform the patient of the risk of an untreated medical condition, weighed against the risk of medication.
- Choose medications with the most available safety data. Let the patient know what resources you have referred to in choosing the medication.
- It is advisable to perform a search each time a prescription is written for a pregnant or lactating woman.
- When possible, have the discussion in the preconception period.
- Consider the dynamic physiology of gestation. Choose the right drug for the right trimester.
- Discuss the plan with the patient and other providers.
- Define clear criteria for when to discontinue the treatment.
- Addis A, Sharabi S, Bonati M. Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 2000; 23:245–253.
- US Food and Drug Administration (FDA). Pregnancy and lactation labeling. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm. Accessed April 4, 2014.
- Lagoy CT, Joshi N, Cragan JD, Rasmussen SA. Medication use during pregnancy and lactation: an urgent call for public health action. J Womens Health (Larchmt) 2005; 14:104–109.
- Clinical Teratology Website. University of Washington. http://depts.washington.edu/terisweb/teris/. Accessed April 4, 2014.
- REPROTOX, An Online Reproductive Toxicology Resource. Reproductive Toxicology Center. www.reprotox.org. Accessed April 4, 2014.
- REPRORISK. Micromedex, Inc. www.micromedex.com/products/reprorisk. Accessed April 4, 2014.
- Shepard TH. Catalog of teratogenic agents. 13th ed. Baltimore, MD: Johns Hopkins University Press; 2010.
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: A reference guide to fetal and neonatal risk. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Koren G. Medication safety in pregnancy and breastfeeding. McGraw-Hill Professional Publishing; 2007.
- Friedman JM, Polifka JE. Teratogenic effects of drugs: A resource for clinicians (TERIS). Baltimore, MD: Johns Hopkins University Press; 2000.
- MotherToBaby. www.mothertobaby.org. Accessed April 4, 2014.
- Dunlop AL, Gardiner PM, Shellhaas CS, Menard MK, McDiarmid MA. The clinical content of preconception care: the use of medications and supplements among women of reproductive age. Am J Obstet Gynecol 2008; 199(suppl 2):S367–S372.
- Ciarkowski SL, Stalburg CM. Medication safety in obstetrics and gynecology. Clin Obstet Gynecol 2010; 53:482–499.
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med 1998; 338:1128–1137.
- Lambert K, Holt RI. The use of insulin analogues in pregnancy. Diabetes Obes Metab 2013; 15:888–900.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Nagler M, Haslauer M, Wuillemin WA. Fondaparinux—data on efficacy and safety in special situations. Thromb Res 2012; 129:407–417.
- Kweder SL, Powrie RO. Prescribing in pregnancy: a practical approach. In:Powrie RO, Greene M, Camann W, editors. De Swiet’s Medical disorders in Obstetric Practice. 5th ed. Hoboken, NJ: Wiley-Blackwell; 2010:633–640.
- Addis A, Sharabi S, Bonati M. Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 2000; 23:245–253.
- US Food and Drug Administration (FDA). Pregnancy and lactation labeling. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm. Accessed April 4, 2014.
- Lagoy CT, Joshi N, Cragan JD, Rasmussen SA. Medication use during pregnancy and lactation: an urgent call for public health action. J Womens Health (Larchmt) 2005; 14:104–109.
- Clinical Teratology Website. University of Washington. http://depts.washington.edu/terisweb/teris/. Accessed April 4, 2014.
- REPROTOX, An Online Reproductive Toxicology Resource. Reproductive Toxicology Center. www.reprotox.org. Accessed April 4, 2014.
- REPRORISK. Micromedex, Inc. www.micromedex.com/products/reprorisk. Accessed April 4, 2014.
- Shepard TH. Catalog of teratogenic agents. 13th ed. Baltimore, MD: Johns Hopkins University Press; 2010.
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: A reference guide to fetal and neonatal risk. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Koren G. Medication safety in pregnancy and breastfeeding. McGraw-Hill Professional Publishing; 2007.
- Friedman JM, Polifka JE. Teratogenic effects of drugs: A resource for clinicians (TERIS). Baltimore, MD: Johns Hopkins University Press; 2000.
- MotherToBaby. www.mothertobaby.org. Accessed April 4, 2014.
- Dunlop AL, Gardiner PM, Shellhaas CS, Menard MK, McDiarmid MA. The clinical content of preconception care: the use of medications and supplements among women of reproductive age. Am J Obstet Gynecol 2008; 199(suppl 2):S367–S372.
- Ciarkowski SL, Stalburg CM. Medication safety in obstetrics and gynecology. Clin Obstet Gynecol 2010; 53:482–499.
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med 1998; 338:1128–1137.
- Lambert K, Holt RI. The use of insulin analogues in pregnancy. Diabetes Obes Metab 2013; 15:888–900.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Nagler M, Haslauer M, Wuillemin WA. Fondaparinux—data on efficacy and safety in special situations. Thromb Res 2012; 129:407–417.
- Kweder SL, Powrie RO. Prescribing in pregnancy: a practical approach. In:Powrie RO, Greene M, Camann W, editors. De Swiet’s Medical disorders in Obstetric Practice. 5th ed. Hoboken, NJ: Wiley-Blackwell; 2010:633–640.
KEY POINTS
- There is no protective physiologic barrier between the maternal and fetal environments.
- The gestational stage may determine the effect of a medication on the fetus.
- The physiologic changes of pregnancy affect the pharmacokinetics of medications.
- Sole reliance on the US Food and Drug Administration’s pregnancy safety category may be inadequate.
- Key questions: Is the problem self-limited or amenable to nonpharmacologic management? How do the patient’s (and provider’s) presumptions affect decisions about this medication in pregnancy? How does pregnancy affect the problem, and how does the problem affect pregnancy?
Chronic obstructive pulmonary disease: An update for the primary physician
Chronic obstructive pulmonary disease (COPD) has seen several changes in its assessment and treatment in recent years, reflecting advances in our understanding of this common and serious disease.
This review updates busy practitioners on the major advances, including new assessment tools and new therapies.
COMMON AND INCREASING
COPD is the third leading cause of death in the United States, behind heart disease and cancer,1 and of the top five (the others being stroke and accidents), it is the only one that increased in incidence between 2007 and 2010.2 The 11th leading cause of disability-adjusted life years worldwide in 2002, COPD is projected to become the seventh by the year 2030.3
CHARACTERIZED BY OBSTRUCTION
COPD is characterized by persistent and progressive airflow obstruction associated with chronic airway inflammation in response to noxious particles and gases. Disease of the small airways (inflammation, mucus plugging, and fibrosis) and parenchymal destruction (emphysema) limit the flow of air.
COPD is diagnosed by spirometry—specifically, a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) of less than 0.7 after a bronchodilator is given. The severity of airflow limitation is revealed by the FEV1 as a percent of the predicted value.
Cigarette smoking is the major cause of COPD, but the prevalence of COPD is 6.6% in people who have never smoked, and one-fourth of COPD patients in the United States have never smoked.4
GOLDEN GOALS: FEWER SYMPTOMS, LOWER RISK
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) periodically issues evidence-based statements on how to prevent and treat COPD.
In its 2013 update,5 GOLD suggested two goals: improving symptoms and reducing the risk of death, exacerbations, progression of disease, and treatment-related adverse effects. The latter goal—reducing risk—is relatively new.
Exacerbations are acute inflammatory events superimposed on chronic inflammation. The inflammation is often brought on by infection6 and increases the risk of death7 and the risk of a faster decline in lung function.8
Exacerbations may characterize a phenotype of COPD. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) analyzed the frequency of COPD exacerbations and associated factors in 2,138 patients with COPD over a period of 3 years.9 Although patients with more severe obstruction tended to have more exacerbations, some patients appeared susceptible to exacerbations irrespective of the severity of obstruction. The best predictor of exacerbations was a history of exacerbations.
HOW DO I ASSESS A PATIENT WITH COPD ON PRESENTATION?
Markers of airflow obstruction such as the FEV1 do not correlate strongly with exertional capacity and health status in patients with COPD.10,11
The BODE index (body mass index, obstruction, dyspnea score, and exercise oximetry) takes into account the multidimensional nature of COPD. It performs better than the FEV1 in predicting the risk of death.12 The propensity for exacerbations and comorbidities further modulates outcome.
Assessing symptoms
The modified British Medical Research Council (mMRC) dyspnea scale, based on work by Fletcher in 1952,13 has five grades, numbered 0 through 4:
- Grade 0—Breathless with strenuous exercise only
- Grade 1—Breathless when hurrying on level ground or walking up a slight hill
- Grade 2—Walks slower than people of the same age on level ground because of shortness of breath or has to stop when walking at own pace on level ground
- Grade 3—Stops for breath after walking about 100 yards or after a few minutes on level ground
- Grade 4—Too breathless to leave the house or breathless when dressing or undressing.
Grade 2 or higher separates symptomatic from asymptomatic COPD.
The COPD Assessment Test (CAT) (www.catestonline.org) is a proprietary questionnaire. Patients use a 6-point scale (numbered 0 though 5) to rate eight symptoms (cough, mucus production, chest tightness, shortness of breath on exertion, limitations in home activities, lack of confidence leaving the home, poor sleep, and lack of energy). A total score of 10 or higher is abnormal.
Four GOLD groups

The new GOLD guidelines (Table 1)5 define four groups of patients according to their severity of airflow obstruction, symptoms, and exacerbation history:
- Group A—fewer symptoms, low risk: Fewer symptoms (“less symptoms,” as worded in the guidelines) means a CAT score less than 10 or an mMRC grade less than 2; “low risk” means no more than one exacerbation per year and an FEV1 of at least 50%
- Group B—more symptoms, low risk: “More symptoms” means a CAT score of 10 or more or an mMRC grade of 2 or more
- Group C—fewer symptoms, high risk: “High risk” means two or more exacerbations per year or an FEV1 less than 50%
- Group D—more symptoms, high risk.
Thus, a patient with an FEV1 of 60% (moderate airflow limitation) who has had one exacerbation during the past year and a CAT score of 8 would be in group A. In contrast, a patient who has an FEV1 of 40% (severe airflow limitation), no history of exacerbations, and a CAT score of 20 would be in group D.
Updated GOLD guidelines suggest utilizing a stepwise approach to treatment, akin to asthma management guidelines, based on patient grouping.5
How accurate is the new GOLD system?
Although practical and suited for use in primary care, the new GOLD system is arbitrary and has not been thoroughly studied, and may therefore need refinement.
Lange et al14 compared the new GOLD system with the previous one in 6,628 patients with COPD. As anticipated, the new system was better at predicting exacerbations, as it incorporates a history of exacerbations in stratification. The presence of symptoms (as determined by an mMRC grade ≥ 2) was a marker of mortality risk that distinguished group A from group B, and group C from group D. Surprisingly, the rate of death was higher in group B (more symptoms, low risk) than in group C (fewer symptoms, high risk).
Notably, most patients in group C qualified for this group because of the severity of airflow obstruction, not because of a history of exacerbations. Therefore, patients whose symptoms are out of proportion to the severity of obstruction may be at higher risk of death, possibly because of comorbidities such as cardiovascular disease.15 Patients who qualified for groups C and D by having both a history of frequent exacerbations (≥ 2 per year) and symptoms rather than either one alone had a higher risk of death in 3 years.
Similarly, the symptom-assessment tool that is used—ie, the mMRC grade or the CAT score—also makes a difference.
The Health-Related Quality of Life in COPD in Europe Study16 retrospectively analyzed data from 1,817 patients to determine whether the cutoff points for symptoms as assessed by mMRC grade and CAT score were equivalent. Although the mMRC grade correlated well with overall health status, the cutoff mMRC grade of 2 or higher did not correspond to a CAT score of 10 or higher, classifying patients with health status impairment as asymptomatic (mean weighted kappa 0.626). The two tools agreed much better when the cutoff was set at an mMRC grade of 1 or higher (mean weighted kappa 0.792).16
Although assessment schemes continue to evolve as data accumulate, we believe the new system is a welcome initiative that reflects the changing notions of COPD.
Comorbidities matter
Another shift is the recognition that certain comorbidities increase the risk of death. In 1,664 patients with COPD who were followed for 51 months, 12 distinct comorbidities were associated with a higher risk of death after multivariate analysis.17
The COTE index (COPD-Specific Comorbidity Test) is based on these findings. It awards points as follows:
- 6 points for cancer of the lung, esophagus, pancreas, or breast, or for anxiety
- 2 points for all other cancers, liver cirrhosis, atrial fibrillation or flutter, diabetes with neuropathy, or pulmonary fibrosis
- 1 point for congestive heart failure, gastric or duodenal ulcer, or coronary artery disease.
A COTE index score of 4 or higher was associated with a risk of death 2.2 times higher in each quartile of the BODE index.
We strongly recommend being aware of comorbidities in COPD patients, particularly when symptoms are out of proportion to the severity of obstruction.
SHOULD I USE ANTIBIOTICS TO TREAT ALL COPD EXACERBATIONS?
Infections are thought to cause more than 80% of acute exacerbations of COPD.
Anthonisen et al,18 in a landmark trial, found broad-spectrum antibiotics to be most helpful if the patient had at least two of the three cardinal symptoms of COPD exacerbation (ie, shortness of breath, increase in sputum volume, and sputum purulence). Antibiotics decreased the rate of treatment failure and led to a more rapid clinical resolution of exacerbation. However, they did not help patients who had milder exacerbations.
Antibiotics may nevertheless have a role in ambulatory patients with mild to moderate COPD who present with exacerbations characterized by one or more cardinal symptoms.
Llor et al,19 in a multicenter randomized double-blind placebo-controlled trial in Spain, concluded that amoxicillin clavulanate (Augmentin) led to higher clinical cure rates and longer time to the next exacerbation in these patients. Most of the benefit was in patients with more symptoms, consistent with the results of the study by Anthonisen et al.18
There is also strong evidence to support the use of antibiotics in addition to systemic corticosteroids in hospitalized patients with acute exacerbations of COPD. A 7-day course of doxycycline (Vibramycin) added to a standard regimen of corticosteroids was associated with higher rates of clinical and microbiological cure on day 10 of the exacerbation.20 In a large retrospective cohort study in 84,621 hospitalized patients with COPD exacerbations, fewer of those who received antibiotics needed mechanical ventilation, died, or were readmitted.21 Although sicker patients received antibiotics more frequently, their mortality rate was lower than in those who did not receive antibiotics, who were presumably less sick.
A meta-analysis confirmed the salutary effect of antibiotics in inpatients and particularly those admitted to the intensive care unit.22 Mortality rates and hospital length of stay were not affected in patients who were not in intensive care.
Biomarkers such as procalcitonin might help reduce the unnecessary use of antibiotics. Stolz et al23 conducted a randomized controlled trial in which they based the decision to give antibiotics on a threshold procalcitonin level of at least 1 μg/L in hospitalized patients with COPD exacerbation. The rate of antibiotic use was reduced by more than 40% in the procalcitonin group without any difference in clinical outcomes, 6-month exacerbation rate, or rehospitalization compared with controls. Nonstandardized procalcitonin assays are a possible barrier to the widespread adoption of this threshold.
Comment. In general, we recommend antibiotics for hospitalized patients with COPD exacerbation and look forward to confirmatory data that support the use of biomarkers. For outpatients, we find the Anthonisen criteria useful for decision-making at the point of care.
ARE THERE ANY NEW INTERVENTIONS TO PREVENT COPD EXACERBATIONS?
Macrolides
Macrolides have a proven role in managing chronic suppurative respiratory diseases such as cystic fibrosis24 and diffuse panbronchiolitis.25 Since they are beneficial at lower doses than those used to treat infection, the mechanism may be anti-inflammatory rather than antimicrobial.
Albert et al26 assigned 1,142 patients who had had a COPD exacerbation within a year before enrollment or who were on home oxygen therapy to receive azithromycin (Zithromax) 250 mg daily or placebo.25 The azithromycin group had fewer acute exacerbations (hazard ratio 0.73, 95% CI 0.63–0.84, P < .001), and more patients in the azithromycin group achieved clinically significant improvements in quality of life, ie, a reduction in the St. George’s Respiratory Questionnaire (SGRQ) score of at least 4 points (43% vs 36%, P = .03). Adverse events that were more common in the azithromycin group were hearing loss (25% vs 20%) and macrolide-resistant strains in nasopharyngeal secretions (81% vs 41%). In subgroup analysis, the benefit in terms of reducing exacerbations was greater in patients over age 65, patients on home oxygen, and patients with moderate or severe obstruction compared with those with very severe obstruction.

Comment. Macrolides are a valuable addition to the agents available for preventing COPD exacerbation (Table 2), but their role is still uncertain. Potential topics of research are whether these drugs have a role in patients already on preventive regimens, whether they would have a greater effect in distinct patient populations (eg, patients who have two or more exacerbations per year), and whether their broader use would lead to a change in the resident flora in the community.
Clinicians should exercise caution in the use of azithromycin in light of recent concern about associated cardiac morbidity and death. All patients should undergo electrocardiography to assess the QTc interval before starting treatment, as in the trial by Albert et al.26
Phosphodiesterase inhibitors

Roflumilast (Daliresp) is an oral phosphodiesterase 4 inhibitor approved for treating exacerbations and symptoms of chronic bronchitis in patients with severe COPD (Table 3). Phosphodiesterase 4, one of the 11 isoforms of the enzyme, is found in immune and inflammatory cells and promotes inflammatory responses. Roflumilast has anti-inflammatory properties but no acute bronchodilatory effect.27 Several phase 3 trials found the compound to have beneficial effects.
Calverley et al28 performed two placebo-controlled double-blind trials in outpatients with the clinical diagnosis of COPD who had chronic cough; increased sputum production; at least one recorded exacerbation requiring corticosteroids or hospitalization, or both; and an FEV1 of 50% or less. Patients were randomized to receive roflumilast 500 μg once a day (n = 1,537) or placebo (n = 1,554) for 1 year. The rate of moderate to severe exacerbations was 1.17 per year with roflumilast vs 1.37 with placebo (P < .0003). Adverse events were significantly more common with roflumilast and were related to the known side effects of the drug, namely, diarrhea, weight loss, decreased appetite, and nausea.
Fabbri et al29 performed two other placebo-controlled double-blind multicenter trials, studying the combinations of roflumilast with salmeterol (Serevent) and roflumilast with tiotropium (Spiriva) compared with placebo in 1,676 patients with COPD who had post-bronchodilator FEV1 values of 40% to 70% of predicted. The mean prebronchodilator FEV1 improved by 49 mL (P < .0001) in the salmeterol-plus-roflumilast trial and by 80 mL (P < .0001) in the tiotropium-plus-roflumilast trial compared with placebo. Fewer patients on roflumilast had exacerbations of any severity in both trials (risk ratio 0.82, P = .0419 and risk ratio 0.75, P = .0169, respectively).
No trial has yet addressed whether roflumilast is better than the combination of a long-acting muscarinic antagonist and a beta agonist, or whether roflumilast can be substituted for inhaled corticosteroids in a new triple-therapy combination. Clinicians should also be aware of psychiatric side effects of roflumilast, which include depression and, possibly, suicide.
ARE THERE ANY NEW BRONCHODILATORS FOR PATIENTS WITH COPD?
Long-acting muscarinic antagonists

Reversible airflow obstruction and mucus secretion are determined by the vagal cholinergic tone in patients with COPD.30 Antagonism of cholinergic (muscarinic) receptors results in bronchodilation and reduction in mucus production. Consequently, inhaled anticholinergic agents are the first-line therapy for COPD (Table 4).
Tiotropium bromide is a long-acting antimuscarinic approved in 2002 by the US Food and Drug Administration (FDA). The UPLIFT trial (Understanding Potential Long-Term Impacts on Function With Tiotropium)31 enrolled 5,993 patients with a mean FEV1 of 48% of predicted. Over a 4-year follow-up, significant improvements in mean FEV1 values (ranging from 87 mL to 103 mL before bronchodilation and 47 mL to 65 mL after bronchodilation, P < .001) in the tiotropium group were observed compared with placebo. The rate of the primary end point—the rate of decline in mean FEV1—was not different between tiotropium and placebo. However, there were important salutary effects in multiple clinical end points in the tiotropium group. Health-related quality of life as measured by the SGRQ improved in a clinically significant manner (> 4 points) in favor of tiotropium in a higher proportion of patients (45% vs 36%, P < .001). Tiotropium reduced the number of exacerbations per patient year (0.73 ± 0.02 vs 0.85 ± 0.02, RR = 0.86 (95% CI 0.81–0.91), P < .001) and the risk of respiratory failure (RR = 0.67, 95% CI 0.51–0.89). There were no significant differences in the risk of myocardial infarction, stroke, or pneumonia.
Aclidinium bromide (Tudorza Pressair) is a long-acting antimuscarinic recently approved by the FDA. Compared with tiotropium, it has a slightly faster onset of action and a considerably shorter half-life (29 hours vs 64 hours).32,33 Its dosage is 400 μg twice daily by inhalation. It provides sustained bronchodilation over 24 hours and may have a favorable side-effect profile, because it undergoes rapid hydrolysis in human plasma.34
ACCORD COPD I35 and ATTAIN,36 two phase 3 trials in patients with moderate-to severe COPD, found that twice-daily aclidinium was associated with statistically and clinically significant (> 100 mL) improvements in trough and peak FEV1 compared with placebo. Health status (assessed by SGRQ) and dyspnea (assessed by transitional dyspnea index) also improved significantly. However, improvements beyond minimum clinically significant thresholds were achieved only with 400 μg twice-daily dosing.
To date, no study has evaluated the impact of aclidinium on COPD exacerbation as a primary end point. Fewer moderate to severe exacerbations were reported in an earlier 52-week study of once-daily aclidinium (ACCLAIM COPD II) but not in ACCLAIM COPD I.37
Aclidinium may offer an advantage over tiotropium in patients who have nocturnal symptoms. Twice-daily aclidinium 400 μg was associated with superior FEV1 area-under-the-curve values compared with placebo and tiotropium, the difference mostly owing to improved nocturnal profile.38
Long-acting beta-2 agonists
Stimulation of airway beta-2 receptors relaxes smooth muscles and consequently dilates bronchioles via a cyclic adenosine monophosphate-dependent pathway.39
Short-acting beta-2 agonists such as albuterol and terbutaline have long been used as rescue medications for obstructive lung disease. Long-acting beta-2 agonists provide sustained bronchodilation and are therefore more efficacious as maintenance medications. Salmeterol, formoterol (Foradil), and arformoterol (Brovana) are long-acting beta-2 agonists in clinical use that are taken twice daily.
Clinical studies indicate that use of long-acting beta-2 agonists leads to significant improvements in FEV1,40–42 dynamic hyperinflation, exercise tolerance,43,44 and dyspnea.45,46 These drugs have also been associated with significant improvements in health-related quality of life and in the frequency of exacerbations.47–49
In patients with asthma, long-acting beta agonists may increase the risk of death.50 In contrast, in patients with COPD, they appear to offer a survival advantage when used in combination with inhaled corticosteroids,51 and some argue that this benefit is entirely from the long-acting beta agonist (a 17% reduction in mortality) rather than the inhaled corticosteroid (0% reduction in mortality).52

Indacaterol (Arcapta), approved in July 2011, is the first once-daily beta agonist or “ultra-long-acting” beta agonist (Table 5). Possibly because it has a high affinity for the lipid raft domain of the cell membrane where beta-2 receptors are coupled to second messengers,53 the drug has a 24-hour duration of action.
In patients with COPD, inhaled indacaterol 150 μg once daily improved airflow obstruction and health status as measured by SGRQ compared with salmeterol 50 μg twice daily and placebo.54 At the higher dose of 300 μg daily, the 52-week INVOLVE trial55 demonstrated early and more sustained improvement in FEV1 compared with placebo and formoterol. In this study, a lower exacerbation rate than with placebo was also noted. The drug has also shown equivalent bronchodilator efficacy at 150 μg and 300 μg daily dosing compared with tiotropium.56
The benefits of a longer-acting bronchodilator such as indacaterol are likely mediated by smoothing out airway bronchomotor tone over 24 hours without the dips seen with shorter-acting agents and by improvement of the FEV1 trough before the subsequent dose is due, aptly named “pharmacologic stenting.”57 Once-daily dosing should also foster better adherence. The safety profile appears excellent with no increase in cardiovascular or cerebrovascular events compared with placebo.58
The FDA approved the 75-μg daily dose instead of the higher doses used in the studies mentioned above. This decision was based on the observation that there appeared to be a flattened dose-response in patients with more severe COPD, with no further improvement in trough FEV1 at higher doses.59
DOES VITAMIN D SUPPLEMENTATION HAVE A ROLE IN COPD MANAGEMENT?
Vitamin D is vital for calcium and phosphate metabolism and bone health. Low vitamin D levels are associated with diminished leg strength and falls in the elderly.60 Osteoporosis, preventable with vitamin D and calcium supplementation, is linked to thoracic vertebral fracture and consequent reduced lung function.61,62

Patients with COPD are at higher risk of vitamin D deficiency, and more so if they also are obese, have advanced airflow obstruction, are depressed, or smoke.62 Therefore, there are sound reasons to look for vitamin D deficiency in patients with COPD and to treat it if the 25-hydroxyvitamin D level is less than 10 ng/ mL (Table 6).
Vitamin D may also have antimicrobial and immunomodulatory effects.63 Since COPD exacerbations are frequently caused by infection, it was hypothesized that vitamin D supplementation might reduce the rate of exacerbations.
In a study in 182 patients with moderate to very severe COPD and a history of recent exacerbations, high-dose vitamin D supplementation (100,000 IU) was given every 4 weeks for 1 year.64 There were no differences in the time to first exacerbation, in the rate of exacerbation, hospitalization, or death, or in quality of life between the placebo and intervention groups. However, subgroup analysis indicated that, in those with severe vitamin D deficiency at baseline, the exacerbation rate was reduced by more than 40%.
Comment. We recommend screening for vitamin D deficiency in patients with COPD. Supplementation is appropriate in those with low levels, but data indicate no role in those with normal levels.
WHAT ARE THE NONPHARMACOLOGIC APPROACHES TO COPD TREATMENT?
Noninvasive positive-pressure ventilation

Nocturnal noninvasive positive-pressure ventilation may be beneficial in patients with severe COPD, daytime hypercapnia, and nocturnal hypoventilation, particularly if higher inspiratory pressures are selected (Table 7).65,66
For instance, a randomized controlled trial of noninvasive positive-pressure ventilation plus long-term oxygen therapy compared with long-term oxygen therapy alone in hypercapnic COPD demonstrated a survival benefit in favor of ventilation (hazard ratio 0.6).67
In another randomized trial,68 settings that aimed to maximally reduce Paco2 (mean inspiratory positive airway pressure 29 cm H2O with a backup rate of 17.5/min) were compared with low-intensity positive airway pressure (mean inspiratory positive airway pressure 14 cm H2O, backup rate 8/min). The high inspiratory pressures increased the daily use of ventilation by 3.6 hours per day and improved exercise-related dyspnea, daytime Paco2, FEV1, vital capacity, and health-related quality of life66 without disrupting sleep quality.68
Caveats are that acclimation to the high pressures was achieved in the hospital, and the high pressures were associated with a significant increase in air leaks.66
Comments. Whether high-pressure noninvasive positive-pressure ventilation can be routinely implemented and adopted in the outpatient setting, and whether it is associated with a survival advantage remains to be determined. The advantages of noninvasive positive-pressure ventilation in the setting of hypercapnic COPD appear to augment those of pulmonary rehabilitation, with improved quality of life, gas exchange, and exercise tolerance, and a slower decline of lung function.69
Pulmonary rehabilitation

Pulmonary rehabilitation is a multidisciplinary approach to managing COPD (Table 8).
Patients participate in three to five supervised sessions per week, each lasting 3 to 4 hours, for 6 to 12 weeks. Less-frequent sessions may not be effective. For instance, in a randomized trial, exercising twice a week was not enough.70 Additionally, a program lasting longer than 12 weeks produced more sustained benefits than shorter programs.71
A key component is an exercise protocol centered on the lower extremities (walking, cycling, treadmill), with progressive exercise intensity to a target of about 60% to 80% of the maximal exercise tolerance,72 though more modest targets of about 50% can also be beneficial.73
Exercise should be tailored to the desired outcome. For instance, training of the upper arms may help with activities of daily living. In one study, unsupported (against gravity) arm training improved upper-extremity function more than supported arm training (by ergometer).74 Ventilatory muscle training is less common, as most randomized trials have not shown conclusive evidence of benefit. Current guidelines do not recommend routine inspiratory muscle training.71
Even though indices of pulmonary function do not improve after an exercise program, randomized trials have shown that pulmonary rehabilitation improves exercise capacity, dyspnea, and health-related quality of life; improves cost-effectiveness of health care utilization; and provides psychosocial benefits that often exceed those of other therapies. Although there is no significant evidence of whether pulmonary rehabilitation improves survival in patients with COPD,71 an observational study documented improvements in BODE scores as well as a reduction in respiratory mortality rates in patients undergoing pulmonary rehabilitation.75
A limitation of pulmonary rehabilitation is that endurance and psychological and cognitive function decline significantly if exercise is not maintained. However, the role of a maintenance program is uncertain, with long-term benefits considered modest.71
Lung-volume reduction surgery

Lung-volume reduction consists of surgical wedge resections of emphysematous areas of the lung (Table 9).
The National Emphysema Treatment Trial76 randomized 1,218 patients to undergo either lung-volume reduction surgery or maximal medical therapy. Surgery improved survival, quality of life, and dyspnea in patients with upper-lobe emphysema and a low exercise capacity (corresponding to < 40 watts for men or < 25 watts for women in the maximal power achieved on cycle ergometry). While conferring no survival benefit in patients with upper-lobe-predominant emphysema and high exercise capacity, this surgery is likely to improve exercise capacity and quality of life in this subset of patients.
Importantly, the procedure is associated with a lower survival rate in patients with an FEV1 lower than 20%, homogeneous emphysema, a diffusing capacity of the lung for carbon monoxide lower than 20%, non-upper-lobe emphysema, or high baseline exercise capacity.
The proposed mechanisms of improvement of lung function include placing the diaphragm in a position with better mechanical advantage, reducing overall lung volume, better size-matching between the lungs and chest cavity, and restoring elastic recoil.76,77
Ongoing trials aim to replicate the success of lung-volume reduction using nonsurgical bronchoscopic techniques with one-way valves, coils, biologic sealants, thermal ablation, and airway stents.
- Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. National Vital Statistics Reports 2010; 59:1–52. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf. Accessed April 10, 2014.
- Murphy SL, Xu J, Kochanek KD. Deaths: preliminary data from 2010. National Vital Statistics Reports 2012; 60:1–51. http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_04.pdf. Accessed April 10, 2014.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442.
- Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest 2005; 128:1239–1244.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed April 10, 2014.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173:1114–1121.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119(suppl 1):21–31.
- Jones PW. Issues concerning health-related quality of life in COPD. Chest 1995; 107(suppl):187S–193S.
- Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:1005–1012.
- Fletcher CM. The clinical diagnosis of pulmonary emphysema—an experimental study. J Royal Soc Med 1952; 45:577–584.
- Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med 2012; 186:975–981.
- Hurst J. Phenotype-based care in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:935–936.
- Jones PW, Adamek L, Nadeau G, Banik N. Comparisons of health status scores with MRC grades in COPD: implications for the GOLD 2011 classification. Eur Respir J 2013; 42:647–654.
- Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:155–161.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Llor C, Moragas A, Hernández S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Rothberg MB, Pekow PS, Lahti M, Brody O, Skiest DJ, Lindenauer PK. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA 2010; 303:2035–2042.
- Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012; 12:CD010257.
- Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131:9–19.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002; 57:212–216.
- Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. COPD 2010; 7:141–153.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Gross NJ, Skorodin MS. Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 1984; 129:856–870.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Cazzola M. Aclidinium bromide, a novel long-acting muscarinic M3 antagonist for the treatment of COPD. Curr Opin Investig Drugs 2009; 10:482–490.
- Gavaldà A, Miralpeix M, Ramos I, et al. Characterization of aclidinium bromide, a novel inhaled muscarinic antagonist, with long duration of action and a favorable pharmacological profile. J Pharmacol Exp Ther 2009; 331:740–751.
- Alagha K, Bourdin A, Tummino C, Chanez P. An update on the efficacy and safety of aclidinium bromide in patients with COPD. Ther Adv Respir Dis 2011; 5:19–28.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012; 9:90–101.
- Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012; 40:830–836.
- Jones PW, Rennard SI, Agusti A, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res 2011; 12:55.
- Fuhr R, Magnussen H, Sarem K, et al. Efficacy of aclidinium bromide 400 μg twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012; 141:745–752.
- Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 2010; 11:149.
- Mahler DA, Donohue JF, Barbee RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999; 115:957–965.
- Campbell M, Eliraz A, Johansson G, et al. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Hanrahan JP, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Neder JA, Fuld JP, Overend T, et al. Effects of formoterol on exercise tolerance in severely disabled patients with COPD. Respir Med 2007; 101:2056–2064.
- O’Donnell DE, Voduc N, Fitzpatrick M, Webb KA. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:86–94.
- Rennard SI, Anderson W, ZuWallack R, et al. Use of a long-acting inhaled beta 2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:1087–1092.
- Schultze-Werninghaus G. Multicenter 1-year trial on formoterol, a new long-acting beta 2-agonist, in chronic obstructive airway disease. Lung 1990; 168(suppl):83–89.
- Rodrigo GJ, Nannini LJ, Rodríguez-Roisin R. Safety of long-acting beta-agonists in stable COPD: a systematic review. Chest 2008; 133:1079–1087.
- Baker WL, Baker EL, Coleman CI. Pharmacologic treatments for chronic obstructive pulmonary disease: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2009; 29:891–905.
- Rodrigo GJ, Castro-Rodriguez JA, Plaza V. Safety and efficacy of combined long-acting beta-agonists and inhaled corticosteroids vs long-acting beta-agonists monotherapy for stable COPD: a systematic review. Chest 2009; 136:1029–1038.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008; 31:927–933.
- Lombardi D, Cuenoud B, Krämer SD. Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur J Pharm Sci 2009; 38:533–547.
- Kornmann O, Dahl R, Centanni S, et al; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators. Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J 2011; 37:273–279.
- Dahl R, Chung KF, Buhl R, et al; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators. Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 2010; 65:473–479.
- Donohue JF, Fogarty C, Lötvall J, et al; INHANCE Study Investigators. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010; 182:155–162.
- Beeh KM, Beier J. The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther 2010; 27:150–159.
- Worth H, Chung KF, Felser JM, Hu H, Rueegg P. Cardio- and cerebrovascular safety of indacaterol vs formoterol, salmeterol, tiotropium and placebo in COPD. Respir Med 2011; 105:571–579.
- Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol--the FDA’s review. N Engl J Med 2011; 365:2247–2249.
- Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA 2004; 291:1999–2006.
- Leech JA, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141:68–71.
- Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of vitamin D. PLoS One 2012; 7:e38934.
- Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M. Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med 2009; 179:630–636.
- Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2012; 156:105–114.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Dreher M, Storre JH, Schmoor C, Windisch W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: a randomised crossover trial. Thorax 2010; 65:303–308.
- McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Dreher M, Ekkernkamp E, Walterspacher S, et al. Noninvasive ventilation in COPD: impact of inspiratory pressure levels on sleep quality. Chest 2011; 140:939–945.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Ringbaek TJ, Broendum E, Hemmingsen L, et al. Rehabilitation of patients with chronic obstructive pulmonary disease. Exercise twice a week is not sufficient! Respir Med 2000; 94:150–154.
- Ries AL, Bauldoff GS, Carlin BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(suppl):4S–42S.
- Vallet G, Ahmaïdi S, Serres I, et al. Comparison of two training programmes in chronic airway limitation patients: standardized versus individualized protocols. Eur Respir J 1997; 10:114–122.
- Vogiatzis I, Williamson AF, Miles J, Taylor IK. Physiological response to moderate exercise workloads in a pulmonary rehabilitation program in patients with varying degrees of airflow obstruction. Chest 1999; 116:1200–1207.
- Martinez FJ, Vogel PD, Dupont DN, Stanopoulos I, Gray A, Beamis JF. Supported arm exercise vs unsupported arm exercise in the rehabilitation of patients with severe chronic airflow obstruction. Chest 1993; 103:1397–1402.
- Cote CG, Celli BR. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26:630–636.
- Fishman A, Martinez F, Naunheim K, et al; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348:2059–2073.
- Naunheim KS, Wood DE, Mohsenifar Z, et al; National Emphysema Treatment Trial Research Group. Long-term follow-up of patients receiving lung-volume-reduction surgery versus medical therapy for severe emphysema by the National Emphysema Treatment Trial Research Group. Ann Thorac Surg 2006; 82:431–443.
Chronic obstructive pulmonary disease (COPD) has seen several changes in its assessment and treatment in recent years, reflecting advances in our understanding of this common and serious disease.
This review updates busy practitioners on the major advances, including new assessment tools and new therapies.
COMMON AND INCREASING
COPD is the third leading cause of death in the United States, behind heart disease and cancer,1 and of the top five (the others being stroke and accidents), it is the only one that increased in incidence between 2007 and 2010.2 The 11th leading cause of disability-adjusted life years worldwide in 2002, COPD is projected to become the seventh by the year 2030.3
CHARACTERIZED BY OBSTRUCTION
COPD is characterized by persistent and progressive airflow obstruction associated with chronic airway inflammation in response to noxious particles and gases. Disease of the small airways (inflammation, mucus plugging, and fibrosis) and parenchymal destruction (emphysema) limit the flow of air.
COPD is diagnosed by spirometry—specifically, a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) of less than 0.7 after a bronchodilator is given. The severity of airflow limitation is revealed by the FEV1 as a percent of the predicted value.
Cigarette smoking is the major cause of COPD, but the prevalence of COPD is 6.6% in people who have never smoked, and one-fourth of COPD patients in the United States have never smoked.4
GOLDEN GOALS: FEWER SYMPTOMS, LOWER RISK
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) periodically issues evidence-based statements on how to prevent and treat COPD.
In its 2013 update,5 GOLD suggested two goals: improving symptoms and reducing the risk of death, exacerbations, progression of disease, and treatment-related adverse effects. The latter goal—reducing risk—is relatively new.
Exacerbations are acute inflammatory events superimposed on chronic inflammation. The inflammation is often brought on by infection6 and increases the risk of death7 and the risk of a faster decline in lung function.8
Exacerbations may characterize a phenotype of COPD. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) analyzed the frequency of COPD exacerbations and associated factors in 2,138 patients with COPD over a period of 3 years.9 Although patients with more severe obstruction tended to have more exacerbations, some patients appeared susceptible to exacerbations irrespective of the severity of obstruction. The best predictor of exacerbations was a history of exacerbations.
HOW DO I ASSESS A PATIENT WITH COPD ON PRESENTATION?
Markers of airflow obstruction such as the FEV1 do not correlate strongly with exertional capacity and health status in patients with COPD.10,11
The BODE index (body mass index, obstruction, dyspnea score, and exercise oximetry) takes into account the multidimensional nature of COPD. It performs better than the FEV1 in predicting the risk of death.12 The propensity for exacerbations and comorbidities further modulates outcome.
Assessing symptoms
The modified British Medical Research Council (mMRC) dyspnea scale, based on work by Fletcher in 1952,13 has five grades, numbered 0 through 4:
- Grade 0—Breathless with strenuous exercise only
- Grade 1—Breathless when hurrying on level ground or walking up a slight hill
- Grade 2—Walks slower than people of the same age on level ground because of shortness of breath or has to stop when walking at own pace on level ground
- Grade 3—Stops for breath after walking about 100 yards or after a few minutes on level ground
- Grade 4—Too breathless to leave the house or breathless when dressing or undressing.
Grade 2 or higher separates symptomatic from asymptomatic COPD.
The COPD Assessment Test (CAT) (www.catestonline.org) is a proprietary questionnaire. Patients use a 6-point scale (numbered 0 though 5) to rate eight symptoms (cough, mucus production, chest tightness, shortness of breath on exertion, limitations in home activities, lack of confidence leaving the home, poor sleep, and lack of energy). A total score of 10 or higher is abnormal.
Four GOLD groups
The new GOLD guidelines (Table 1)5 define four groups of patients according to their severity of airflow obstruction, symptoms, and exacerbation history:
- Group A—fewer symptoms, low risk: Fewer symptoms (“less symptoms,” as worded in the guidelines) means a CAT score less than 10 or an mMRC grade less than 2; “low risk” means no more than one exacerbation per year and an FEV1 of at least 50%
- Group B—more symptoms, low risk: “More symptoms” means a CAT score of 10 or more or an mMRC grade of 2 or more
- Group C—fewer symptoms, high risk: “High risk” means two or more exacerbations per year or an FEV1 less than 50%
- Group D—more symptoms, high risk.
Thus, a patient with an FEV1 of 60% (moderate airflow limitation) who has had one exacerbation during the past year and a CAT score of 8 would be in group A. In contrast, a patient who has an FEV1 of 40% (severe airflow limitation), no history of exacerbations, and a CAT score of 20 would be in group D.
Updated GOLD guidelines suggest utilizing a stepwise approach to treatment, akin to asthma management guidelines, based on patient grouping.5
How accurate is the new GOLD system?
Although practical and suited for use in primary care, the new GOLD system is arbitrary and has not been thoroughly studied, and may therefore need refinement.
Lange et al14 compared the new GOLD system with the previous one in 6,628 patients with COPD. As anticipated, the new system was better at predicting exacerbations, as it incorporates a history of exacerbations in stratification. The presence of symptoms (as determined by an mMRC grade ≥ 2) was a marker of mortality risk that distinguished group A from group B, and group C from group D. Surprisingly, the rate of death was higher in group B (more symptoms, low risk) than in group C (fewer symptoms, high risk).
Notably, most patients in group C qualified for this group because of the severity of airflow obstruction, not because of a history of exacerbations. Therefore, patients whose symptoms are out of proportion to the severity of obstruction may be at higher risk of death, possibly because of comorbidities such as cardiovascular disease.15 Patients who qualified for groups C and D by having both a history of frequent exacerbations (≥ 2 per year) and symptoms rather than either one alone had a higher risk of death in 3 years.
Similarly, the symptom-assessment tool that is used—ie, the mMRC grade or the CAT score—also makes a difference.
The Health-Related Quality of Life in COPD in Europe Study16 retrospectively analyzed data from 1,817 patients to determine whether the cutoff points for symptoms as assessed by mMRC grade and CAT score were equivalent. Although the mMRC grade correlated well with overall health status, the cutoff mMRC grade of 2 or higher did not correspond to a CAT score of 10 or higher, classifying patients with health status impairment as asymptomatic (mean weighted kappa 0.626). The two tools agreed much better when the cutoff was set at an mMRC grade of 1 or higher (mean weighted kappa 0.792).16
Although assessment schemes continue to evolve as data accumulate, we believe the new system is a welcome initiative that reflects the changing notions of COPD.
Comorbidities matter
Another shift is the recognition that certain comorbidities increase the risk of death. In 1,664 patients with COPD who were followed for 51 months, 12 distinct comorbidities were associated with a higher risk of death after multivariate analysis.17
The COTE index (COPD-Specific Comorbidity Test) is based on these findings. It awards points as follows:
- 6 points for cancer of the lung, esophagus, pancreas, or breast, or for anxiety
- 2 points for all other cancers, liver cirrhosis, atrial fibrillation or flutter, diabetes with neuropathy, or pulmonary fibrosis
- 1 point for congestive heart failure, gastric or duodenal ulcer, or coronary artery disease.
A COTE index score of 4 or higher was associated with a risk of death 2.2 times higher in each quartile of the BODE index.
We strongly recommend being aware of comorbidities in COPD patients, particularly when symptoms are out of proportion to the severity of obstruction.
SHOULD I USE ANTIBIOTICS TO TREAT ALL COPD EXACERBATIONS?
Infections are thought to cause more than 80% of acute exacerbations of COPD.
Anthonisen et al,18 in a landmark trial, found broad-spectrum antibiotics to be most helpful if the patient had at least two of the three cardinal symptoms of COPD exacerbation (ie, shortness of breath, increase in sputum volume, and sputum purulence). Antibiotics decreased the rate of treatment failure and led to a more rapid clinical resolution of exacerbation. However, they did not help patients who had milder exacerbations.
Antibiotics may nevertheless have a role in ambulatory patients with mild to moderate COPD who present with exacerbations characterized by one or more cardinal symptoms.
Llor et al,19 in a multicenter randomized double-blind placebo-controlled trial in Spain, concluded that amoxicillin clavulanate (Augmentin) led to higher clinical cure rates and longer time to the next exacerbation in these patients. Most of the benefit was in patients with more symptoms, consistent with the results of the study by Anthonisen et al.18
There is also strong evidence to support the use of antibiotics in addition to systemic corticosteroids in hospitalized patients with acute exacerbations of COPD. A 7-day course of doxycycline (Vibramycin) added to a standard regimen of corticosteroids was associated with higher rates of clinical and microbiological cure on day 10 of the exacerbation.20 In a large retrospective cohort study in 84,621 hospitalized patients with COPD exacerbations, fewer of those who received antibiotics needed mechanical ventilation, died, or were readmitted.21 Although sicker patients received antibiotics more frequently, their mortality rate was lower than in those who did not receive antibiotics, who were presumably less sick.
A meta-analysis confirmed the salutary effect of antibiotics in inpatients and particularly those admitted to the intensive care unit.22 Mortality rates and hospital length of stay were not affected in patients who were not in intensive care.
Biomarkers such as procalcitonin might help reduce the unnecessary use of antibiotics. Stolz et al23 conducted a randomized controlled trial in which they based the decision to give antibiotics on a threshold procalcitonin level of at least 1 μg/L in hospitalized patients with COPD exacerbation. The rate of antibiotic use was reduced by more than 40% in the procalcitonin group without any difference in clinical outcomes, 6-month exacerbation rate, or rehospitalization compared with controls. Nonstandardized procalcitonin assays are a possible barrier to the widespread adoption of this threshold.
Comment. In general, we recommend antibiotics for hospitalized patients with COPD exacerbation and look forward to confirmatory data that support the use of biomarkers. For outpatients, we find the Anthonisen criteria useful for decision-making at the point of care.
ARE THERE ANY NEW INTERVENTIONS TO PREVENT COPD EXACERBATIONS?
Macrolides
Macrolides have a proven role in managing chronic suppurative respiratory diseases such as cystic fibrosis24 and diffuse panbronchiolitis.25 Since they are beneficial at lower doses than those used to treat infection, the mechanism may be anti-inflammatory rather than antimicrobial.
Albert et al26 assigned 1,142 patients who had had a COPD exacerbation within a year before enrollment or who were on home oxygen therapy to receive azithromycin (Zithromax) 250 mg daily or placebo.25 The azithromycin group had fewer acute exacerbations (hazard ratio 0.73, 95% CI 0.63–0.84, P < .001), and more patients in the azithromycin group achieved clinically significant improvements in quality of life, ie, a reduction in the St. George’s Respiratory Questionnaire (SGRQ) score of at least 4 points (43% vs 36%, P = .03). Adverse events that were more common in the azithromycin group were hearing loss (25% vs 20%) and macrolide-resistant strains in nasopharyngeal secretions (81% vs 41%). In subgroup analysis, the benefit in terms of reducing exacerbations was greater in patients over age 65, patients on home oxygen, and patients with moderate or severe obstruction compared with those with very severe obstruction.
Comment. Macrolides are a valuable addition to the agents available for preventing COPD exacerbation (Table 2), but their role is still uncertain. Potential topics of research are whether these drugs have a role in patients already on preventive regimens, whether they would have a greater effect in distinct patient populations (eg, patients who have two or more exacerbations per year), and whether their broader use would lead to a change in the resident flora in the community.
Clinicians should exercise caution in the use of azithromycin in light of recent concern about associated cardiac morbidity and death. All patients should undergo electrocardiography to assess the QTc interval before starting treatment, as in the trial by Albert et al.26
Phosphodiesterase inhibitors
Roflumilast (Daliresp) is an oral phosphodiesterase 4 inhibitor approved for treating exacerbations and symptoms of chronic bronchitis in patients with severe COPD (Table 3). Phosphodiesterase 4, one of the 11 isoforms of the enzyme, is found in immune and inflammatory cells and promotes inflammatory responses. Roflumilast has anti-inflammatory properties but no acute bronchodilatory effect.27 Several phase 3 trials found the compound to have beneficial effects.
Calverley et al28 performed two placebo-controlled double-blind trials in outpatients with the clinical diagnosis of COPD who had chronic cough; increased sputum production; at least one recorded exacerbation requiring corticosteroids or hospitalization, or both; and an FEV1 of 50% or less. Patients were randomized to receive roflumilast 500 μg once a day (n = 1,537) or placebo (n = 1,554) for 1 year. The rate of moderate to severe exacerbations was 1.17 per year with roflumilast vs 1.37 with placebo (P < .0003). Adverse events were significantly more common with roflumilast and were related to the known side effects of the drug, namely, diarrhea, weight loss, decreased appetite, and nausea.
Fabbri et al29 performed two other placebo-controlled double-blind multicenter trials, studying the combinations of roflumilast with salmeterol (Serevent) and roflumilast with tiotropium (Spiriva) compared with placebo in 1,676 patients with COPD who had post-bronchodilator FEV1 values of 40% to 70% of predicted. The mean prebronchodilator FEV1 improved by 49 mL (P < .0001) in the salmeterol-plus-roflumilast trial and by 80 mL (P < .0001) in the tiotropium-plus-roflumilast trial compared with placebo. Fewer patients on roflumilast had exacerbations of any severity in both trials (risk ratio 0.82, P = .0419 and risk ratio 0.75, P = .0169, respectively).
No trial has yet addressed whether roflumilast is better than the combination of a long-acting muscarinic antagonist and a beta agonist, or whether roflumilast can be substituted for inhaled corticosteroids in a new triple-therapy combination. Clinicians should also be aware of psychiatric side effects of roflumilast, which include depression and, possibly, suicide.
ARE THERE ANY NEW BRONCHODILATORS FOR PATIENTS WITH COPD?
Long-acting muscarinic antagonists
Reversible airflow obstruction and mucus secretion are determined by the vagal cholinergic tone in patients with COPD.30 Antagonism of cholinergic (muscarinic) receptors results in bronchodilation and reduction in mucus production. Consequently, inhaled anticholinergic agents are the first-line therapy for COPD (Table 4).
Tiotropium bromide is a long-acting antimuscarinic approved in 2002 by the US Food and Drug Administration (FDA). The UPLIFT trial (Understanding Potential Long-Term Impacts on Function With Tiotropium)31 enrolled 5,993 patients with a mean FEV1 of 48% of predicted. Over a 4-year follow-up, significant improvements in mean FEV1 values (ranging from 87 mL to 103 mL before bronchodilation and 47 mL to 65 mL after bronchodilation, P < .001) in the tiotropium group were observed compared with placebo. The rate of the primary end point—the rate of decline in mean FEV1—was not different between tiotropium and placebo. However, there were important salutary effects in multiple clinical end points in the tiotropium group. Health-related quality of life as measured by the SGRQ improved in a clinically significant manner (> 4 points) in favor of tiotropium in a higher proportion of patients (45% vs 36%, P < .001). Tiotropium reduced the number of exacerbations per patient year (0.73 ± 0.02 vs 0.85 ± 0.02, RR = 0.86 (95% CI 0.81–0.91), P < .001) and the risk of respiratory failure (RR = 0.67, 95% CI 0.51–0.89). There were no significant differences in the risk of myocardial infarction, stroke, or pneumonia.
Aclidinium bromide (Tudorza Pressair) is a long-acting antimuscarinic recently approved by the FDA. Compared with tiotropium, it has a slightly faster onset of action and a considerably shorter half-life (29 hours vs 64 hours).32,33 Its dosage is 400 μg twice daily by inhalation. It provides sustained bronchodilation over 24 hours and may have a favorable side-effect profile, because it undergoes rapid hydrolysis in human plasma.34
ACCORD COPD I35 and ATTAIN,36 two phase 3 trials in patients with moderate-to severe COPD, found that twice-daily aclidinium was associated with statistically and clinically significant (> 100 mL) improvements in trough and peak FEV1 compared with placebo. Health status (assessed by SGRQ) and dyspnea (assessed by transitional dyspnea index) also improved significantly. However, improvements beyond minimum clinically significant thresholds were achieved only with 400 μg twice-daily dosing.
To date, no study has evaluated the impact of aclidinium on COPD exacerbation as a primary end point. Fewer moderate to severe exacerbations were reported in an earlier 52-week study of once-daily aclidinium (ACCLAIM COPD II) but not in ACCLAIM COPD I.37
Aclidinium may offer an advantage over tiotropium in patients who have nocturnal symptoms. Twice-daily aclidinium 400 μg was associated with superior FEV1 area-under-the-curve values compared with placebo and tiotropium, the difference mostly owing to improved nocturnal profile.38
Long-acting beta-2 agonists
Stimulation of airway beta-2 receptors relaxes smooth muscles and consequently dilates bronchioles via a cyclic adenosine monophosphate-dependent pathway.39
Short-acting beta-2 agonists such as albuterol and terbutaline have long been used as rescue medications for obstructive lung disease. Long-acting beta-2 agonists provide sustained bronchodilation and are therefore more efficacious as maintenance medications. Salmeterol, formoterol (Foradil), and arformoterol (Brovana) are long-acting beta-2 agonists in clinical use that are taken twice daily.
Clinical studies indicate that use of long-acting beta-2 agonists leads to significant improvements in FEV1,40–42 dynamic hyperinflation, exercise tolerance,43,44 and dyspnea.45,46 These drugs have also been associated with significant improvements in health-related quality of life and in the frequency of exacerbations.47–49
In patients with asthma, long-acting beta agonists may increase the risk of death.50 In contrast, in patients with COPD, they appear to offer a survival advantage when used in combination with inhaled corticosteroids,51 and some argue that this benefit is entirely from the long-acting beta agonist (a 17% reduction in mortality) rather than the inhaled corticosteroid (0% reduction in mortality).52
Indacaterol (Arcapta), approved in July 2011, is the first once-daily beta agonist or “ultra-long-acting” beta agonist (Table 5). Possibly because it has a high affinity for the lipid raft domain of the cell membrane where beta-2 receptors are coupled to second messengers,53 the drug has a 24-hour duration of action.
In patients with COPD, inhaled indacaterol 150 μg once daily improved airflow obstruction and health status as measured by SGRQ compared with salmeterol 50 μg twice daily and placebo.54 At the higher dose of 300 μg daily, the 52-week INVOLVE trial55 demonstrated early and more sustained improvement in FEV1 compared with placebo and formoterol. In this study, a lower exacerbation rate than with placebo was also noted. The drug has also shown equivalent bronchodilator efficacy at 150 μg and 300 μg daily dosing compared with tiotropium.56
The benefits of a longer-acting bronchodilator such as indacaterol are likely mediated by smoothing out airway bronchomotor tone over 24 hours without the dips seen with shorter-acting agents and by improvement of the FEV1 trough before the subsequent dose is due, aptly named “pharmacologic stenting.”57 Once-daily dosing should also foster better adherence. The safety profile appears excellent with no increase in cardiovascular or cerebrovascular events compared with placebo.58
The FDA approved the 75-μg daily dose instead of the higher doses used in the studies mentioned above. This decision was based on the observation that there appeared to be a flattened dose-response in patients with more severe COPD, with no further improvement in trough FEV1 at higher doses.59
DOES VITAMIN D SUPPLEMENTATION HAVE A ROLE IN COPD MANAGEMENT?
Vitamin D is vital for calcium and phosphate metabolism and bone health. Low vitamin D levels are associated with diminished leg strength and falls in the elderly.60 Osteoporosis, preventable with vitamin D and calcium supplementation, is linked to thoracic vertebral fracture and consequent reduced lung function.61,62
Patients with COPD are at higher risk of vitamin D deficiency, and more so if they also are obese, have advanced airflow obstruction, are depressed, or smoke.62 Therefore, there are sound reasons to look for vitamin D deficiency in patients with COPD and to treat it if the 25-hydroxyvitamin D level is less than 10 ng/ mL (Table 6).
Vitamin D may also have antimicrobial and immunomodulatory effects.63 Since COPD exacerbations are frequently caused by infection, it was hypothesized that vitamin D supplementation might reduce the rate of exacerbations.
In a study in 182 patients with moderate to very severe COPD and a history of recent exacerbations, high-dose vitamin D supplementation (100,000 IU) was given every 4 weeks for 1 year.64 There were no differences in the time to first exacerbation, in the rate of exacerbation, hospitalization, or death, or in quality of life between the placebo and intervention groups. However, subgroup analysis indicated that, in those with severe vitamin D deficiency at baseline, the exacerbation rate was reduced by more than 40%.
Comment. We recommend screening for vitamin D deficiency in patients with COPD. Supplementation is appropriate in those with low levels, but data indicate no role in those with normal levels.
WHAT ARE THE NONPHARMACOLOGIC APPROACHES TO COPD TREATMENT?
Noninvasive positive-pressure ventilation
Nocturnal noninvasive positive-pressure ventilation may be beneficial in patients with severe COPD, daytime hypercapnia, and nocturnal hypoventilation, particularly if higher inspiratory pressures are selected (Table 7).65,66
For instance, a randomized controlled trial of noninvasive positive-pressure ventilation plus long-term oxygen therapy compared with long-term oxygen therapy alone in hypercapnic COPD demonstrated a survival benefit in favor of ventilation (hazard ratio 0.6).67
In another randomized trial,68 settings that aimed to maximally reduce Paco2 (mean inspiratory positive airway pressure 29 cm H2O with a backup rate of 17.5/min) were compared with low-intensity positive airway pressure (mean inspiratory positive airway pressure 14 cm H2O, backup rate 8/min). The high inspiratory pressures increased the daily use of ventilation by 3.6 hours per day and improved exercise-related dyspnea, daytime Paco2, FEV1, vital capacity, and health-related quality of life66 without disrupting sleep quality.68
Caveats are that acclimation to the high pressures was achieved in the hospital, and the high pressures were associated with a significant increase in air leaks.66
Comments. Whether high-pressure noninvasive positive-pressure ventilation can be routinely implemented and adopted in the outpatient setting, and whether it is associated with a survival advantage remains to be determined. The advantages of noninvasive positive-pressure ventilation in the setting of hypercapnic COPD appear to augment those of pulmonary rehabilitation, with improved quality of life, gas exchange, and exercise tolerance, and a slower decline of lung function.69
Pulmonary rehabilitation
Pulmonary rehabilitation is a multidisciplinary approach to managing COPD (Table 8).
Patients participate in three to five supervised sessions per week, each lasting 3 to 4 hours, for 6 to 12 weeks. Less-frequent sessions may not be effective. For instance, in a randomized trial, exercising twice a week was not enough.70 Additionally, a program lasting longer than 12 weeks produced more sustained benefits than shorter programs.71
A key component is an exercise protocol centered on the lower extremities (walking, cycling, treadmill), with progressive exercise intensity to a target of about 60% to 80% of the maximal exercise tolerance,72 though more modest targets of about 50% can also be beneficial.73
Exercise should be tailored to the desired outcome. For instance, training of the upper arms may help with activities of daily living. In one study, unsupported (against gravity) arm training improved upper-extremity function more than supported arm training (by ergometer).74 Ventilatory muscle training is less common, as most randomized trials have not shown conclusive evidence of benefit. Current guidelines do not recommend routine inspiratory muscle training.71
Even though indices of pulmonary function do not improve after an exercise program, randomized trials have shown that pulmonary rehabilitation improves exercise capacity, dyspnea, and health-related quality of life; improves cost-effectiveness of health care utilization; and provides psychosocial benefits that often exceed those of other therapies. Although there is no significant evidence of whether pulmonary rehabilitation improves survival in patients with COPD,71 an observational study documented improvements in BODE scores as well as a reduction in respiratory mortality rates in patients undergoing pulmonary rehabilitation.75
A limitation of pulmonary rehabilitation is that endurance and psychological and cognitive function decline significantly if exercise is not maintained. However, the role of a maintenance program is uncertain, with long-term benefits considered modest.71
Lung-volume reduction surgery
Lung-volume reduction consists of surgical wedge resections of emphysematous areas of the lung (Table 9).
The National Emphysema Treatment Trial76 randomized 1,218 patients to undergo either lung-volume reduction surgery or maximal medical therapy. Surgery improved survival, quality of life, and dyspnea in patients with upper-lobe emphysema and a low exercise capacity (corresponding to < 40 watts for men or < 25 watts for women in the maximal power achieved on cycle ergometry). While conferring no survival benefit in patients with upper-lobe-predominant emphysema and high exercise capacity, this surgery is likely to improve exercise capacity and quality of life in this subset of patients.
Importantly, the procedure is associated with a lower survival rate in patients with an FEV1 lower than 20%, homogeneous emphysema, a diffusing capacity of the lung for carbon monoxide lower than 20%, non-upper-lobe emphysema, or high baseline exercise capacity.
The proposed mechanisms of improvement of lung function include placing the diaphragm in a position with better mechanical advantage, reducing overall lung volume, better size-matching between the lungs and chest cavity, and restoring elastic recoil.76,77
Ongoing trials aim to replicate the success of lung-volume reduction using nonsurgical bronchoscopic techniques with one-way valves, coils, biologic sealants, thermal ablation, and airway stents.
Chronic obstructive pulmonary disease (COPD) has seen several changes in its assessment and treatment in recent years, reflecting advances in our understanding of this common and serious disease.
This review updates busy practitioners on the major advances, including new assessment tools and new therapies.
COMMON AND INCREASING
COPD is the third leading cause of death in the United States, behind heart disease and cancer,1 and of the top five (the others being stroke and accidents), it is the only one that increased in incidence between 2007 and 2010.2 The 11th leading cause of disability-adjusted life years worldwide in 2002, COPD is projected to become the seventh by the year 2030.3
CHARACTERIZED BY OBSTRUCTION
COPD is characterized by persistent and progressive airflow obstruction associated with chronic airway inflammation in response to noxious particles and gases. Disease of the small airways (inflammation, mucus plugging, and fibrosis) and parenchymal destruction (emphysema) limit the flow of air.
COPD is diagnosed by spirometry—specifically, a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) of less than 0.7 after a bronchodilator is given. The severity of airflow limitation is revealed by the FEV1 as a percent of the predicted value.
Cigarette smoking is the major cause of COPD, but the prevalence of COPD is 6.6% in people who have never smoked, and one-fourth of COPD patients in the United States have never smoked.4
GOLDEN GOALS: FEWER SYMPTOMS, LOWER RISK
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) periodically issues evidence-based statements on how to prevent and treat COPD.
In its 2013 update,5 GOLD suggested two goals: improving symptoms and reducing the risk of death, exacerbations, progression of disease, and treatment-related adverse effects. The latter goal—reducing risk—is relatively new.
Exacerbations are acute inflammatory events superimposed on chronic inflammation. The inflammation is often brought on by infection6 and increases the risk of death7 and the risk of a faster decline in lung function.8
Exacerbations may characterize a phenotype of COPD. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) analyzed the frequency of COPD exacerbations and associated factors in 2,138 patients with COPD over a period of 3 years.9 Although patients with more severe obstruction tended to have more exacerbations, some patients appeared susceptible to exacerbations irrespective of the severity of obstruction. The best predictor of exacerbations was a history of exacerbations.
HOW DO I ASSESS A PATIENT WITH COPD ON PRESENTATION?
Markers of airflow obstruction such as the FEV1 do not correlate strongly with exertional capacity and health status in patients with COPD.10,11
The BODE index (body mass index, obstruction, dyspnea score, and exercise oximetry) takes into account the multidimensional nature of COPD. It performs better than the FEV1 in predicting the risk of death.12 The propensity for exacerbations and comorbidities further modulates outcome.
Assessing symptoms
The modified British Medical Research Council (mMRC) dyspnea scale, based on work by Fletcher in 1952,13 has five grades, numbered 0 through 4:
- Grade 0—Breathless with strenuous exercise only
- Grade 1—Breathless when hurrying on level ground or walking up a slight hill
- Grade 2—Walks slower than people of the same age on level ground because of shortness of breath or has to stop when walking at own pace on level ground
- Grade 3—Stops for breath after walking about 100 yards or after a few minutes on level ground
- Grade 4—Too breathless to leave the house or breathless when dressing or undressing.
Grade 2 or higher separates symptomatic from asymptomatic COPD.
The COPD Assessment Test (CAT) (www.catestonline.org) is a proprietary questionnaire. Patients use a 6-point scale (numbered 0 though 5) to rate eight symptoms (cough, mucus production, chest tightness, shortness of breath on exertion, limitations in home activities, lack of confidence leaving the home, poor sleep, and lack of energy). A total score of 10 or higher is abnormal.
Four GOLD groups
The new GOLD guidelines (Table 1)5 define four groups of patients according to their severity of airflow obstruction, symptoms, and exacerbation history:
- Group A—fewer symptoms, low risk: Fewer symptoms (“less symptoms,” as worded in the guidelines) means a CAT score less than 10 or an mMRC grade less than 2; “low risk” means no more than one exacerbation per year and an FEV1 of at least 50%
- Group B—more symptoms, low risk: “More symptoms” means a CAT score of 10 or more or an mMRC grade of 2 or more
- Group C—fewer symptoms, high risk: “High risk” means two or more exacerbations per year or an FEV1 less than 50%
- Group D—more symptoms, high risk.
Thus, a patient with an FEV1 of 60% (moderate airflow limitation) who has had one exacerbation during the past year and a CAT score of 8 would be in group A. In contrast, a patient who has an FEV1 of 40% (severe airflow limitation), no history of exacerbations, and a CAT score of 20 would be in group D.
Updated GOLD guidelines suggest utilizing a stepwise approach to treatment, akin to asthma management guidelines, based on patient grouping.5
How accurate is the new GOLD system?
Although practical and suited for use in primary care, the new GOLD system is arbitrary and has not been thoroughly studied, and may therefore need refinement.
Lange et al14 compared the new GOLD system with the previous one in 6,628 patients with COPD. As anticipated, the new system was better at predicting exacerbations, as it incorporates a history of exacerbations in stratification. The presence of symptoms (as determined by an mMRC grade ≥ 2) was a marker of mortality risk that distinguished group A from group B, and group C from group D. Surprisingly, the rate of death was higher in group B (more symptoms, low risk) than in group C (fewer symptoms, high risk).
Notably, most patients in group C qualified for this group because of the severity of airflow obstruction, not because of a history of exacerbations. Therefore, patients whose symptoms are out of proportion to the severity of obstruction may be at higher risk of death, possibly because of comorbidities such as cardiovascular disease.15 Patients who qualified for groups C and D by having both a history of frequent exacerbations (≥ 2 per year) and symptoms rather than either one alone had a higher risk of death in 3 years.
Similarly, the symptom-assessment tool that is used—ie, the mMRC grade or the CAT score—also makes a difference.
The Health-Related Quality of Life in COPD in Europe Study16 retrospectively analyzed data from 1,817 patients to determine whether the cutoff points for symptoms as assessed by mMRC grade and CAT score were equivalent. Although the mMRC grade correlated well with overall health status, the cutoff mMRC grade of 2 or higher did not correspond to a CAT score of 10 or higher, classifying patients with health status impairment as asymptomatic (mean weighted kappa 0.626). The two tools agreed much better when the cutoff was set at an mMRC grade of 1 or higher (mean weighted kappa 0.792).16
Although assessment schemes continue to evolve as data accumulate, we believe the new system is a welcome initiative that reflects the changing notions of COPD.
Comorbidities matter
Another shift is the recognition that certain comorbidities increase the risk of death. In 1,664 patients with COPD who were followed for 51 months, 12 distinct comorbidities were associated with a higher risk of death after multivariate analysis.17
The COTE index (COPD-Specific Comorbidity Test) is based on these findings. It awards points as follows:
- 6 points for cancer of the lung, esophagus, pancreas, or breast, or for anxiety
- 2 points for all other cancers, liver cirrhosis, atrial fibrillation or flutter, diabetes with neuropathy, or pulmonary fibrosis
- 1 point for congestive heart failure, gastric or duodenal ulcer, or coronary artery disease.
A COTE index score of 4 or higher was associated with a risk of death 2.2 times higher in each quartile of the BODE index.
We strongly recommend being aware of comorbidities in COPD patients, particularly when symptoms are out of proportion to the severity of obstruction.
SHOULD I USE ANTIBIOTICS TO TREAT ALL COPD EXACERBATIONS?
Infections are thought to cause more than 80% of acute exacerbations of COPD.
Anthonisen et al,18 in a landmark trial, found broad-spectrum antibiotics to be most helpful if the patient had at least two of the three cardinal symptoms of COPD exacerbation (ie, shortness of breath, increase in sputum volume, and sputum purulence). Antibiotics decreased the rate of treatment failure and led to a more rapid clinical resolution of exacerbation. However, they did not help patients who had milder exacerbations.
Antibiotics may nevertheless have a role in ambulatory patients with mild to moderate COPD who present with exacerbations characterized by one or more cardinal symptoms.
Llor et al,19 in a multicenter randomized double-blind placebo-controlled trial in Spain, concluded that amoxicillin clavulanate (Augmentin) led to higher clinical cure rates and longer time to the next exacerbation in these patients. Most of the benefit was in patients with more symptoms, consistent with the results of the study by Anthonisen et al.18
There is also strong evidence to support the use of antibiotics in addition to systemic corticosteroids in hospitalized patients with acute exacerbations of COPD. A 7-day course of doxycycline (Vibramycin) added to a standard regimen of corticosteroids was associated with higher rates of clinical and microbiological cure on day 10 of the exacerbation.20 In a large retrospective cohort study in 84,621 hospitalized patients with COPD exacerbations, fewer of those who received antibiotics needed mechanical ventilation, died, or were readmitted.21 Although sicker patients received antibiotics more frequently, their mortality rate was lower than in those who did not receive antibiotics, who were presumably less sick.
A meta-analysis confirmed the salutary effect of antibiotics in inpatients and particularly those admitted to the intensive care unit.22 Mortality rates and hospital length of stay were not affected in patients who were not in intensive care.
Biomarkers such as procalcitonin might help reduce the unnecessary use of antibiotics. Stolz et al23 conducted a randomized controlled trial in which they based the decision to give antibiotics on a threshold procalcitonin level of at least 1 μg/L in hospitalized patients with COPD exacerbation. The rate of antibiotic use was reduced by more than 40% in the procalcitonin group without any difference in clinical outcomes, 6-month exacerbation rate, or rehospitalization compared with controls. Nonstandardized procalcitonin assays are a possible barrier to the widespread adoption of this threshold.
Comment. In general, we recommend antibiotics for hospitalized patients with COPD exacerbation and look forward to confirmatory data that support the use of biomarkers. For outpatients, we find the Anthonisen criteria useful for decision-making at the point of care.
ARE THERE ANY NEW INTERVENTIONS TO PREVENT COPD EXACERBATIONS?
Macrolides
Macrolides have a proven role in managing chronic suppurative respiratory diseases such as cystic fibrosis24 and diffuse panbronchiolitis.25 Since they are beneficial at lower doses than those used to treat infection, the mechanism may be anti-inflammatory rather than antimicrobial.
Albert et al26 assigned 1,142 patients who had had a COPD exacerbation within a year before enrollment or who were on home oxygen therapy to receive azithromycin (Zithromax) 250 mg daily or placebo.25 The azithromycin group had fewer acute exacerbations (hazard ratio 0.73, 95% CI 0.63–0.84, P < .001), and more patients in the azithromycin group achieved clinically significant improvements in quality of life, ie, a reduction in the St. George’s Respiratory Questionnaire (SGRQ) score of at least 4 points (43% vs 36%, P = .03). Adverse events that were more common in the azithromycin group were hearing loss (25% vs 20%) and macrolide-resistant strains in nasopharyngeal secretions (81% vs 41%). In subgroup analysis, the benefit in terms of reducing exacerbations was greater in patients over age 65, patients on home oxygen, and patients with moderate or severe obstruction compared with those with very severe obstruction.
Comment. Macrolides are a valuable addition to the agents available for preventing COPD exacerbation (Table 2), but their role is still uncertain. Potential topics of research are whether these drugs have a role in patients already on preventive regimens, whether they would have a greater effect in distinct patient populations (eg, patients who have two or more exacerbations per year), and whether their broader use would lead to a change in the resident flora in the community.
Clinicians should exercise caution in the use of azithromycin in light of recent concern about associated cardiac morbidity and death. All patients should undergo electrocardiography to assess the QTc interval before starting treatment, as in the trial by Albert et al.26
Phosphodiesterase inhibitors
Roflumilast (Daliresp) is an oral phosphodiesterase 4 inhibitor approved for treating exacerbations and symptoms of chronic bronchitis in patients with severe COPD (Table 3). Phosphodiesterase 4, one of the 11 isoforms of the enzyme, is found in immune and inflammatory cells and promotes inflammatory responses. Roflumilast has anti-inflammatory properties but no acute bronchodilatory effect.27 Several phase 3 trials found the compound to have beneficial effects.
Calverley et al28 performed two placebo-controlled double-blind trials in outpatients with the clinical diagnosis of COPD who had chronic cough; increased sputum production; at least one recorded exacerbation requiring corticosteroids or hospitalization, or both; and an FEV1 of 50% or less. Patients were randomized to receive roflumilast 500 μg once a day (n = 1,537) or placebo (n = 1,554) for 1 year. The rate of moderate to severe exacerbations was 1.17 per year with roflumilast vs 1.37 with placebo (P < .0003). Adverse events were significantly more common with roflumilast and were related to the known side effects of the drug, namely, diarrhea, weight loss, decreased appetite, and nausea.
Fabbri et al29 performed two other placebo-controlled double-blind multicenter trials, studying the combinations of roflumilast with salmeterol (Serevent) and roflumilast with tiotropium (Spiriva) compared with placebo in 1,676 patients with COPD who had post-bronchodilator FEV1 values of 40% to 70% of predicted. The mean prebronchodilator FEV1 improved by 49 mL (P < .0001) in the salmeterol-plus-roflumilast trial and by 80 mL (P < .0001) in the tiotropium-plus-roflumilast trial compared with placebo. Fewer patients on roflumilast had exacerbations of any severity in both trials (risk ratio 0.82, P = .0419 and risk ratio 0.75, P = .0169, respectively).
No trial has yet addressed whether roflumilast is better than the combination of a long-acting muscarinic antagonist and a beta agonist, or whether roflumilast can be substituted for inhaled corticosteroids in a new triple-therapy combination. Clinicians should also be aware of psychiatric side effects of roflumilast, which include depression and, possibly, suicide.
ARE THERE ANY NEW BRONCHODILATORS FOR PATIENTS WITH COPD?
Long-acting muscarinic antagonists
Reversible airflow obstruction and mucus secretion are determined by the vagal cholinergic tone in patients with COPD.30 Antagonism of cholinergic (muscarinic) receptors results in bronchodilation and reduction in mucus production. Consequently, inhaled anticholinergic agents are the first-line therapy for COPD (Table 4).
Tiotropium bromide is a long-acting antimuscarinic approved in 2002 by the US Food and Drug Administration (FDA). The UPLIFT trial (Understanding Potential Long-Term Impacts on Function With Tiotropium)31 enrolled 5,993 patients with a mean FEV1 of 48% of predicted. Over a 4-year follow-up, significant improvements in mean FEV1 values (ranging from 87 mL to 103 mL before bronchodilation and 47 mL to 65 mL after bronchodilation, P < .001) in the tiotropium group were observed compared with placebo. The rate of the primary end point—the rate of decline in mean FEV1—was not different between tiotropium and placebo. However, there were important salutary effects in multiple clinical end points in the tiotropium group. Health-related quality of life as measured by the SGRQ improved in a clinically significant manner (> 4 points) in favor of tiotropium in a higher proportion of patients (45% vs 36%, P < .001). Tiotropium reduced the number of exacerbations per patient year (0.73 ± 0.02 vs 0.85 ± 0.02, RR = 0.86 (95% CI 0.81–0.91), P < .001) and the risk of respiratory failure (RR = 0.67, 95% CI 0.51–0.89). There were no significant differences in the risk of myocardial infarction, stroke, or pneumonia.
Aclidinium bromide (Tudorza Pressair) is a long-acting antimuscarinic recently approved by the FDA. Compared with tiotropium, it has a slightly faster onset of action and a considerably shorter half-life (29 hours vs 64 hours).32,33 Its dosage is 400 μg twice daily by inhalation. It provides sustained bronchodilation over 24 hours and may have a favorable side-effect profile, because it undergoes rapid hydrolysis in human plasma.34
ACCORD COPD I35 and ATTAIN,36 two phase 3 trials in patients with moderate-to severe COPD, found that twice-daily aclidinium was associated with statistically and clinically significant (> 100 mL) improvements in trough and peak FEV1 compared with placebo. Health status (assessed by SGRQ) and dyspnea (assessed by transitional dyspnea index) also improved significantly. However, improvements beyond minimum clinically significant thresholds were achieved only with 400 μg twice-daily dosing.
To date, no study has evaluated the impact of aclidinium on COPD exacerbation as a primary end point. Fewer moderate to severe exacerbations were reported in an earlier 52-week study of once-daily aclidinium (ACCLAIM COPD II) but not in ACCLAIM COPD I.37
Aclidinium may offer an advantage over tiotropium in patients who have nocturnal symptoms. Twice-daily aclidinium 400 μg was associated with superior FEV1 area-under-the-curve values compared with placebo and tiotropium, the difference mostly owing to improved nocturnal profile.38
Long-acting beta-2 agonists
Stimulation of airway beta-2 receptors relaxes smooth muscles and consequently dilates bronchioles via a cyclic adenosine monophosphate-dependent pathway.39
Short-acting beta-2 agonists such as albuterol and terbutaline have long been used as rescue medications for obstructive lung disease. Long-acting beta-2 agonists provide sustained bronchodilation and are therefore more efficacious as maintenance medications. Salmeterol, formoterol (Foradil), and arformoterol (Brovana) are long-acting beta-2 agonists in clinical use that are taken twice daily.
Clinical studies indicate that use of long-acting beta-2 agonists leads to significant improvements in FEV1,40–42 dynamic hyperinflation, exercise tolerance,43,44 and dyspnea.45,46 These drugs have also been associated with significant improvements in health-related quality of life and in the frequency of exacerbations.47–49
In patients with asthma, long-acting beta agonists may increase the risk of death.50 In contrast, in patients with COPD, they appear to offer a survival advantage when used in combination with inhaled corticosteroids,51 and some argue that this benefit is entirely from the long-acting beta agonist (a 17% reduction in mortality) rather than the inhaled corticosteroid (0% reduction in mortality).52
Indacaterol (Arcapta), approved in July 2011, is the first once-daily beta agonist or “ultra-long-acting” beta agonist (Table 5). Possibly because it has a high affinity for the lipid raft domain of the cell membrane where beta-2 receptors are coupled to second messengers,53 the drug has a 24-hour duration of action.
In patients with COPD, inhaled indacaterol 150 μg once daily improved airflow obstruction and health status as measured by SGRQ compared with salmeterol 50 μg twice daily and placebo.54 At the higher dose of 300 μg daily, the 52-week INVOLVE trial55 demonstrated early and more sustained improvement in FEV1 compared with placebo and formoterol. In this study, a lower exacerbation rate than with placebo was also noted. The drug has also shown equivalent bronchodilator efficacy at 150 μg and 300 μg daily dosing compared with tiotropium.56
The benefits of a longer-acting bronchodilator such as indacaterol are likely mediated by smoothing out airway bronchomotor tone over 24 hours without the dips seen with shorter-acting agents and by improvement of the FEV1 trough before the subsequent dose is due, aptly named “pharmacologic stenting.”57 Once-daily dosing should also foster better adherence. The safety profile appears excellent with no increase in cardiovascular or cerebrovascular events compared with placebo.58
The FDA approved the 75-μg daily dose instead of the higher doses used in the studies mentioned above. This decision was based on the observation that there appeared to be a flattened dose-response in patients with more severe COPD, with no further improvement in trough FEV1 at higher doses.59
DOES VITAMIN D SUPPLEMENTATION HAVE A ROLE IN COPD MANAGEMENT?
Vitamin D is vital for calcium and phosphate metabolism and bone health. Low vitamin D levels are associated with diminished leg strength and falls in the elderly.60 Osteoporosis, preventable with vitamin D and calcium supplementation, is linked to thoracic vertebral fracture and consequent reduced lung function.61,62
Patients with COPD are at higher risk of vitamin D deficiency, and more so if they also are obese, have advanced airflow obstruction, are depressed, or smoke.62 Therefore, there are sound reasons to look for vitamin D deficiency in patients with COPD and to treat it if the 25-hydroxyvitamin D level is less than 10 ng/ mL (Table 6).
Vitamin D may also have antimicrobial and immunomodulatory effects.63 Since COPD exacerbations are frequently caused by infection, it was hypothesized that vitamin D supplementation might reduce the rate of exacerbations.
In a study in 182 patients with moderate to very severe COPD and a history of recent exacerbations, high-dose vitamin D supplementation (100,000 IU) was given every 4 weeks for 1 year.64 There were no differences in the time to first exacerbation, in the rate of exacerbation, hospitalization, or death, or in quality of life between the placebo and intervention groups. However, subgroup analysis indicated that, in those with severe vitamin D deficiency at baseline, the exacerbation rate was reduced by more than 40%.
Comment. We recommend screening for vitamin D deficiency in patients with COPD. Supplementation is appropriate in those with low levels, but data indicate no role in those with normal levels.
WHAT ARE THE NONPHARMACOLOGIC APPROACHES TO COPD TREATMENT?
Noninvasive positive-pressure ventilation
Nocturnal noninvasive positive-pressure ventilation may be beneficial in patients with severe COPD, daytime hypercapnia, and nocturnal hypoventilation, particularly if higher inspiratory pressures are selected (Table 7).65,66
For instance, a randomized controlled trial of noninvasive positive-pressure ventilation plus long-term oxygen therapy compared with long-term oxygen therapy alone in hypercapnic COPD demonstrated a survival benefit in favor of ventilation (hazard ratio 0.6).67
In another randomized trial,68 settings that aimed to maximally reduce Paco2 (mean inspiratory positive airway pressure 29 cm H2O with a backup rate of 17.5/min) were compared with low-intensity positive airway pressure (mean inspiratory positive airway pressure 14 cm H2O, backup rate 8/min). The high inspiratory pressures increased the daily use of ventilation by 3.6 hours per day and improved exercise-related dyspnea, daytime Paco2, FEV1, vital capacity, and health-related quality of life66 without disrupting sleep quality.68
Caveats are that acclimation to the high pressures was achieved in the hospital, and the high pressures were associated with a significant increase in air leaks.66
Comments. Whether high-pressure noninvasive positive-pressure ventilation can be routinely implemented and adopted in the outpatient setting, and whether it is associated with a survival advantage remains to be determined. The advantages of noninvasive positive-pressure ventilation in the setting of hypercapnic COPD appear to augment those of pulmonary rehabilitation, with improved quality of life, gas exchange, and exercise tolerance, and a slower decline of lung function.69
Pulmonary rehabilitation
Pulmonary rehabilitation is a multidisciplinary approach to managing COPD (Table 8).
Patients participate in three to five supervised sessions per week, each lasting 3 to 4 hours, for 6 to 12 weeks. Less-frequent sessions may not be effective. For instance, in a randomized trial, exercising twice a week was not enough.70 Additionally, a program lasting longer than 12 weeks produced more sustained benefits than shorter programs.71
A key component is an exercise protocol centered on the lower extremities (walking, cycling, treadmill), with progressive exercise intensity to a target of about 60% to 80% of the maximal exercise tolerance,72 though more modest targets of about 50% can also be beneficial.73
Exercise should be tailored to the desired outcome. For instance, training of the upper arms may help with activities of daily living. In one study, unsupported (against gravity) arm training improved upper-extremity function more than supported arm training (by ergometer).74 Ventilatory muscle training is less common, as most randomized trials have not shown conclusive evidence of benefit. Current guidelines do not recommend routine inspiratory muscle training.71
Even though indices of pulmonary function do not improve after an exercise program, randomized trials have shown that pulmonary rehabilitation improves exercise capacity, dyspnea, and health-related quality of life; improves cost-effectiveness of health care utilization; and provides psychosocial benefits that often exceed those of other therapies. Although there is no significant evidence of whether pulmonary rehabilitation improves survival in patients with COPD,71 an observational study documented improvements in BODE scores as well as a reduction in respiratory mortality rates in patients undergoing pulmonary rehabilitation.75
A limitation of pulmonary rehabilitation is that endurance and psychological and cognitive function decline significantly if exercise is not maintained. However, the role of a maintenance program is uncertain, with long-term benefits considered modest.71
Lung-volume reduction surgery
Lung-volume reduction consists of surgical wedge resections of emphysematous areas of the lung (Table 9).
The National Emphysema Treatment Trial76 randomized 1,218 patients to undergo either lung-volume reduction surgery or maximal medical therapy. Surgery improved survival, quality of life, and dyspnea in patients with upper-lobe emphysema and a low exercise capacity (corresponding to < 40 watts for men or < 25 watts for women in the maximal power achieved on cycle ergometry). While conferring no survival benefit in patients with upper-lobe-predominant emphysema and high exercise capacity, this surgery is likely to improve exercise capacity and quality of life in this subset of patients.
Importantly, the procedure is associated with a lower survival rate in patients with an FEV1 lower than 20%, homogeneous emphysema, a diffusing capacity of the lung for carbon monoxide lower than 20%, non-upper-lobe emphysema, or high baseline exercise capacity.
The proposed mechanisms of improvement of lung function include placing the diaphragm in a position with better mechanical advantage, reducing overall lung volume, better size-matching between the lungs and chest cavity, and restoring elastic recoil.76,77
Ongoing trials aim to replicate the success of lung-volume reduction using nonsurgical bronchoscopic techniques with one-way valves, coils, biologic sealants, thermal ablation, and airway stents.
- Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. National Vital Statistics Reports 2010; 59:1–52. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf. Accessed April 10, 2014.
- Murphy SL, Xu J, Kochanek KD. Deaths: preliminary data from 2010. National Vital Statistics Reports 2012; 60:1–51. http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_04.pdf. Accessed April 10, 2014.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442.
- Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest 2005; 128:1239–1244.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed April 10, 2014.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173:1114–1121.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119(suppl 1):21–31.
- Jones PW. Issues concerning health-related quality of life in COPD. Chest 1995; 107(suppl):187S–193S.
- Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:1005–1012.
- Fletcher CM. The clinical diagnosis of pulmonary emphysema—an experimental study. J Royal Soc Med 1952; 45:577–584.
- Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med 2012; 186:975–981.
- Hurst J. Phenotype-based care in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:935–936.
- Jones PW, Adamek L, Nadeau G, Banik N. Comparisons of health status scores with MRC grades in COPD: implications for the GOLD 2011 classification. Eur Respir J 2013; 42:647–654.
- Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:155–161.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Llor C, Moragas A, Hernández S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Rothberg MB, Pekow PS, Lahti M, Brody O, Skiest DJ, Lindenauer PK. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA 2010; 303:2035–2042.
- Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012; 12:CD010257.
- Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131:9–19.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002; 57:212–216.
- Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. COPD 2010; 7:141–153.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Gross NJ, Skorodin MS. Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 1984; 129:856–870.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Cazzola M. Aclidinium bromide, a novel long-acting muscarinic M3 antagonist for the treatment of COPD. Curr Opin Investig Drugs 2009; 10:482–490.
- Gavaldà A, Miralpeix M, Ramos I, et al. Characterization of aclidinium bromide, a novel inhaled muscarinic antagonist, with long duration of action and a favorable pharmacological profile. J Pharmacol Exp Ther 2009; 331:740–751.
- Alagha K, Bourdin A, Tummino C, Chanez P. An update on the efficacy and safety of aclidinium bromide in patients with COPD. Ther Adv Respir Dis 2011; 5:19–28.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012; 9:90–101.
- Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012; 40:830–836.
- Jones PW, Rennard SI, Agusti A, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res 2011; 12:55.
- Fuhr R, Magnussen H, Sarem K, et al. Efficacy of aclidinium bromide 400 μg twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012; 141:745–752.
- Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 2010; 11:149.
- Mahler DA, Donohue JF, Barbee RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999; 115:957–965.
- Campbell M, Eliraz A, Johansson G, et al. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Hanrahan JP, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Neder JA, Fuld JP, Overend T, et al. Effects of formoterol on exercise tolerance in severely disabled patients with COPD. Respir Med 2007; 101:2056–2064.
- O’Donnell DE, Voduc N, Fitzpatrick M, Webb KA. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:86–94.
- Rennard SI, Anderson W, ZuWallack R, et al. Use of a long-acting inhaled beta 2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:1087–1092.
- Schultze-Werninghaus G. Multicenter 1-year trial on formoterol, a new long-acting beta 2-agonist, in chronic obstructive airway disease. Lung 1990; 168(suppl):83–89.
- Rodrigo GJ, Nannini LJ, Rodríguez-Roisin R. Safety of long-acting beta-agonists in stable COPD: a systematic review. Chest 2008; 133:1079–1087.
- Baker WL, Baker EL, Coleman CI. Pharmacologic treatments for chronic obstructive pulmonary disease: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2009; 29:891–905.
- Rodrigo GJ, Castro-Rodriguez JA, Plaza V. Safety and efficacy of combined long-acting beta-agonists and inhaled corticosteroids vs long-acting beta-agonists monotherapy for stable COPD: a systematic review. Chest 2009; 136:1029–1038.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008; 31:927–933.
- Lombardi D, Cuenoud B, Krämer SD. Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur J Pharm Sci 2009; 38:533–547.
- Kornmann O, Dahl R, Centanni S, et al; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators. Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J 2011; 37:273–279.
- Dahl R, Chung KF, Buhl R, et al; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators. Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 2010; 65:473–479.
- Donohue JF, Fogarty C, Lötvall J, et al; INHANCE Study Investigators. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010; 182:155–162.
- Beeh KM, Beier J. The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther 2010; 27:150–159.
- Worth H, Chung KF, Felser JM, Hu H, Rueegg P. Cardio- and cerebrovascular safety of indacaterol vs formoterol, salmeterol, tiotropium and placebo in COPD. Respir Med 2011; 105:571–579.
- Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol--the FDA’s review. N Engl J Med 2011; 365:2247–2249.
- Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA 2004; 291:1999–2006.
- Leech JA, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141:68–71.
- Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of vitamin D. PLoS One 2012; 7:e38934.
- Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M. Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med 2009; 179:630–636.
- Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2012; 156:105–114.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Dreher M, Storre JH, Schmoor C, Windisch W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: a randomised crossover trial. Thorax 2010; 65:303–308.
- McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Dreher M, Ekkernkamp E, Walterspacher S, et al. Noninvasive ventilation in COPD: impact of inspiratory pressure levels on sleep quality. Chest 2011; 140:939–945.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Ringbaek TJ, Broendum E, Hemmingsen L, et al. Rehabilitation of patients with chronic obstructive pulmonary disease. Exercise twice a week is not sufficient! Respir Med 2000; 94:150–154.
- Ries AL, Bauldoff GS, Carlin BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(suppl):4S–42S.
- Vallet G, Ahmaïdi S, Serres I, et al. Comparison of two training programmes in chronic airway limitation patients: standardized versus individualized protocols. Eur Respir J 1997; 10:114–122.
- Vogiatzis I, Williamson AF, Miles J, Taylor IK. Physiological response to moderate exercise workloads in a pulmonary rehabilitation program in patients with varying degrees of airflow obstruction. Chest 1999; 116:1200–1207.
- Martinez FJ, Vogel PD, Dupont DN, Stanopoulos I, Gray A, Beamis JF. Supported arm exercise vs unsupported arm exercise in the rehabilitation of patients with severe chronic airflow obstruction. Chest 1993; 103:1397–1402.
- Cote CG, Celli BR. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26:630–636.
- Fishman A, Martinez F, Naunheim K, et al; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348:2059–2073.
- Naunheim KS, Wood DE, Mohsenifar Z, et al; National Emphysema Treatment Trial Research Group. Long-term follow-up of patients receiving lung-volume-reduction surgery versus medical therapy for severe emphysema by the National Emphysema Treatment Trial Research Group. Ann Thorac Surg 2006; 82:431–443.
- Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. National Vital Statistics Reports 2010; 59:1–52. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf. Accessed April 10, 2014.
- Murphy SL, Xu J, Kochanek KD. Deaths: preliminary data from 2010. National Vital Statistics Reports 2012; 60:1–51. http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_04.pdf. Accessed April 10, 2014.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442.
- Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest 2005; 128:1239–1244.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed April 10, 2014.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173:1114–1121.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119(suppl 1):21–31.
- Jones PW. Issues concerning health-related quality of life in COPD. Chest 1995; 107(suppl):187S–193S.
- Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:1005–1012.
- Fletcher CM. The clinical diagnosis of pulmonary emphysema—an experimental study. J Royal Soc Med 1952; 45:577–584.
- Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med 2012; 186:975–981.
- Hurst J. Phenotype-based care in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:935–936.
- Jones PW, Adamek L, Nadeau G, Banik N. Comparisons of health status scores with MRC grades in COPD: implications for the GOLD 2011 classification. Eur Respir J 2013; 42:647–654.
- Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:155–161.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Llor C, Moragas A, Hernández S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Rothberg MB, Pekow PS, Lahti M, Brody O, Skiest DJ, Lindenauer PK. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA 2010; 303:2035–2042.
- Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012; 12:CD010257.
- Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131:9–19.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002; 57:212–216.
- Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. COPD 2010; 7:141–153.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Gross NJ, Skorodin MS. Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 1984; 129:856–870.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Cazzola M. Aclidinium bromide, a novel long-acting muscarinic M3 antagonist for the treatment of COPD. Curr Opin Investig Drugs 2009; 10:482–490.
- Gavaldà A, Miralpeix M, Ramos I, et al. Characterization of aclidinium bromide, a novel inhaled muscarinic antagonist, with long duration of action and a favorable pharmacological profile. J Pharmacol Exp Ther 2009; 331:740–751.
- Alagha K, Bourdin A, Tummino C, Chanez P. An update on the efficacy and safety of aclidinium bromide in patients with COPD. Ther Adv Respir Dis 2011; 5:19–28.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012; 9:90–101.
- Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012; 40:830–836.
- Jones PW, Rennard SI, Agusti A, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res 2011; 12:55.
- Fuhr R, Magnussen H, Sarem K, et al. Efficacy of aclidinium bromide 400 μg twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012; 141:745–752.
- Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 2010; 11:149.
- Mahler DA, Donohue JF, Barbee RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999; 115:957–965.
- Campbell M, Eliraz A, Johansson G, et al. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Hanrahan JP, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Neder JA, Fuld JP, Overend T, et al. Effects of formoterol on exercise tolerance in severely disabled patients with COPD. Respir Med 2007; 101:2056–2064.
- O’Donnell DE, Voduc N, Fitzpatrick M, Webb KA. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:86–94.
- Rennard SI, Anderson W, ZuWallack R, et al. Use of a long-acting inhaled beta 2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:1087–1092.
- Schultze-Werninghaus G. Multicenter 1-year trial on formoterol, a new long-acting beta 2-agonist, in chronic obstructive airway disease. Lung 1990; 168(suppl):83–89.
- Rodrigo GJ, Nannini LJ, Rodríguez-Roisin R. Safety of long-acting beta-agonists in stable COPD: a systematic review. Chest 2008; 133:1079–1087.
- Baker WL, Baker EL, Coleman CI. Pharmacologic treatments for chronic obstructive pulmonary disease: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2009; 29:891–905.
- Rodrigo GJ, Castro-Rodriguez JA, Plaza V. Safety and efficacy of combined long-acting beta-agonists and inhaled corticosteroids vs long-acting beta-agonists monotherapy for stable COPD: a systematic review. Chest 2009; 136:1029–1038.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008; 31:927–933.
- Lombardi D, Cuenoud B, Krämer SD. Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur J Pharm Sci 2009; 38:533–547.
- Kornmann O, Dahl R, Centanni S, et al; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators. Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J 2011; 37:273–279.
- Dahl R, Chung KF, Buhl R, et al; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators. Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 2010; 65:473–479.
- Donohue JF, Fogarty C, Lötvall J, et al; INHANCE Study Investigators. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010; 182:155–162.
- Beeh KM, Beier J. The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther 2010; 27:150–159.
- Worth H, Chung KF, Felser JM, Hu H, Rueegg P. Cardio- and cerebrovascular safety of indacaterol vs formoterol, salmeterol, tiotropium and placebo in COPD. Respir Med 2011; 105:571–579.
- Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol--the FDA’s review. N Engl J Med 2011; 365:2247–2249.
- Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA 2004; 291:1999–2006.
- Leech JA, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141:68–71.
- Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of vitamin D. PLoS One 2012; 7:e38934.
- Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M. Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med 2009; 179:630–636.
- Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2012; 156:105–114.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Dreher M, Storre JH, Schmoor C, Windisch W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: a randomised crossover trial. Thorax 2010; 65:303–308.
- McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Dreher M, Ekkernkamp E, Walterspacher S, et al. Noninvasive ventilation in COPD: impact of inspiratory pressure levels on sleep quality. Chest 2011; 140:939–945.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Ringbaek TJ, Broendum E, Hemmingsen L, et al. Rehabilitation of patients with chronic obstructive pulmonary disease. Exercise twice a week is not sufficient! Respir Med 2000; 94:150–154.
- Ries AL, Bauldoff GS, Carlin BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(suppl):4S–42S.
- Vallet G, Ahmaïdi S, Serres I, et al. Comparison of two training programmes in chronic airway limitation patients: standardized versus individualized protocols. Eur Respir J 1997; 10:114–122.
- Vogiatzis I, Williamson AF, Miles J, Taylor IK. Physiological response to moderate exercise workloads in a pulmonary rehabilitation program in patients with varying degrees of airflow obstruction. Chest 1999; 116:1200–1207.
- Martinez FJ, Vogel PD, Dupont DN, Stanopoulos I, Gray A, Beamis JF. Supported arm exercise vs unsupported arm exercise in the rehabilitation of patients with severe chronic airflow obstruction. Chest 1993; 103:1397–1402.
- Cote CG, Celli BR. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26:630–636.
- Fishman A, Martinez F, Naunheim K, et al; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348:2059–2073.
- Naunheim KS, Wood DE, Mohsenifar Z, et al; National Emphysema Treatment Trial Research Group. Long-term follow-up of patients receiving lung-volume-reduction surgery versus medical therapy for severe emphysema by the National Emphysema Treatment Trial Research Group. Ann Thorac Surg 2006; 82:431–443.
KEY POINTS
- A new COPD classification scheme is based on severity, symptoms, and exacerbations.
- Azithromycin 250 mg daily prevents exacerbations of COPD in those at high risk.
- Long-acting muscarinic antagonists such as aclidinium and tiotropium are first-line therapy.
- Relatively new options include roflumilast, an oral phosphodiesterase inhibitor, and indacaterol, an ultra-long-acting beta agonist that is taken once daily.
- Nondrug interventions include pulmonary rehabilitation, vitamin D supplementation, noninvasive positive-pressure ventilation, and lung-volume reduction surgery.
Frontotemporal dementia and its variants: What to look for
Frontotemporal dementia (FTD) is a neurologic disease that affects the frontal and the temporal lobes of the cerebral cortex.1 This disorder is observed most often in people between age 45 to 65, but also can manifest in younger or older persons.1 The cause varies among a range of pathologies affecting the anterior portions of the brain.2
Presentations
FTD presents with changes in personality, social skills, ability to concentrate, motivation, reasoning, and language abnormality.3 Memory loss is less prominent in this condition compared with other dementias; therefore, identification may be a diagnostic challenge. FTD can be misdiagnosed as a psychiatric illness or not recognized because social symptoms dominate over cognitive dysfunction. As the disease progresses, patients may become increasingly unable to plan or organize activities of daily living, behave appropriately, and react normally in social interactions.1
FTD has 3 diagnostic variants1-4:
Behavioral variant. Known as Pick disease or the “frontal variant,”1,2 this type of FTD manifests as changes in personality, improper behavior in social settings, personal neglect, or impulsivity, such as shoplifting or hypersexuality.
Primary progressive aphasia. Two types of language dysfunction are observed in FTD:
• Semantic dementia (SD)3: Left-sided SD presents with “meaningless speech” or “word substitutions” (eg, “chair” instead of “table”). Right-sided SD, however, is characterized by forgetting the faces of familiar people or objects.
• Primary nonfluent aphasia3: Language fluency is compromised. Persons with such language dysfunction cannot produce words easily, and their speech is stumbling and nonfluent.
FTD with motor neuron disease.4 The most common type of motor neuron disease associated with FTD is amyotrophic lateral sclerosis. Afflicted patients exhibit muscle weakness, spasms, and rigidity. This leads to difficulty in swallowing or breathing because the diaphragm and pharynx are paralyzed. Other diseases associated with FTD include corticobasal degeneration and progressive supranuclear palsy.
Diagnosis
In DSM-5, FTD has been renamed “frontotemporal lobar degeneration” under the category of “Major and Mild Neurocognitive Disorders.”5 The workup begins with a history, physical examination, and mental status assessment. Physical signs can include frontal-release, primitive reflexes. Early in the disease course, a palmomental reflex often is observed; later, as disease progress, the rooting reflex or palmar grasp may become apparent.1,5
Diagnosing FTD requires recognizing its symptoms and ruling out conditions such as Alzheimer’s disease, depression, and schizophrenia.6 Laboratory studies may help identify other conditions. Brain imaging, such as MRI, can depict frontotemporal pathology and rule in or exclude other diseases.3,5
Psychometric testing can evaluate memory or cognitive ability, which might be unremarkable during the initial phases of FTD.4 Further psychological assessments may provide objective verification of frontal lobe deficiencies in social skills or activities of daily living.3 Positron emission tomography and single-photon emission computed tomography may demonstrate areas of decreased activity or hypoperfusion in frontal and temporal lobes.7
Interventions
Treatment of FTD is limited to symptomatic therapy8; there are no specific, approved countermeasures available. Comorbid conditions, such as diabetes mellitus or hypertension, should be treated medically. Social interventions such as day care, increased supervision, and emotional support from the family can be effective adjuvants.2
Disclosures
The authors report no financial relationship whose products are mentioned in this article or with manufacturers of competing products.
1. Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140-143.
2. Frontotemporal degeneration. The Association for Frontotemporal Degeneration. http://www.theaftd.org/ frontotemporal-degeneration/ftd-overview. Accessed April 24, 2014.
3. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546-1554.
4. Clark CM, Forman MS. Frontotemporal lobar degeneration with motor neuron disease: a clinical and pathological spectrum. Arch Neurol. 2006;63(4):489-490.
5. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013:614-618.
6. Frontotemporal dementia diagnosis. UCSF Medical Center. http://www.ucsfhealth.org/conditions/frontotemporal_ dementia/diagnosis.html. Accessed April 24, 2014.
7. McMurtray AM, Chen AK, Shapira JS, et al. Variations in regional SPECT hypoperfusion and clinical features in frontotemporal dementia. Neurology. 2006;66(4):517-522.
8. Miller BL, Lee SE. Frontotemporal dementia: treatment. Up To Date. http://www.uptodate.com/contents/frontotemporal-dementia-treatment?source=search_result&search=frontote mporal+dementia+treatment&selectedTitle=1~150. Updated December 30, 2013. Accessed April 24, 2014.
Frontotemporal dementia (FTD) is a neurologic disease that affects the frontal and the temporal lobes of the cerebral cortex.1 This disorder is observed most often in people between age 45 to 65, but also can manifest in younger or older persons.1 The cause varies among a range of pathologies affecting the anterior portions of the brain.2
Presentations
FTD presents with changes in personality, social skills, ability to concentrate, motivation, reasoning, and language abnormality.3 Memory loss is less prominent in this condition compared with other dementias; therefore, identification may be a diagnostic challenge. FTD can be misdiagnosed as a psychiatric illness or not recognized because social symptoms dominate over cognitive dysfunction. As the disease progresses, patients may become increasingly unable to plan or organize activities of daily living, behave appropriately, and react normally in social interactions.1
FTD has 3 diagnostic variants1-4:
Behavioral variant. Known as Pick disease or the “frontal variant,”1,2 this type of FTD manifests as changes in personality, improper behavior in social settings, personal neglect, or impulsivity, such as shoplifting or hypersexuality.
Primary progressive aphasia. Two types of language dysfunction are observed in FTD:
• Semantic dementia (SD)3: Left-sided SD presents with “meaningless speech” or “word substitutions” (eg, “chair” instead of “table”). Right-sided SD, however, is characterized by forgetting the faces of familiar people or objects.
• Primary nonfluent aphasia3: Language fluency is compromised. Persons with such language dysfunction cannot produce words easily, and their speech is stumbling and nonfluent.
FTD with motor neuron disease.4 The most common type of motor neuron disease associated with FTD is amyotrophic lateral sclerosis. Afflicted patients exhibit muscle weakness, spasms, and rigidity. This leads to difficulty in swallowing or breathing because the diaphragm and pharynx are paralyzed. Other diseases associated with FTD include corticobasal degeneration and progressive supranuclear palsy.
Diagnosis
In DSM-5, FTD has been renamed “frontotemporal lobar degeneration” under the category of “Major and Mild Neurocognitive Disorders.”5 The workup begins with a history, physical examination, and mental status assessment. Physical signs can include frontal-release, primitive reflexes. Early in the disease course, a palmomental reflex often is observed; later, as disease progress, the rooting reflex or palmar grasp may become apparent.1,5
Diagnosing FTD requires recognizing its symptoms and ruling out conditions such as Alzheimer’s disease, depression, and schizophrenia.6 Laboratory studies may help identify other conditions. Brain imaging, such as MRI, can depict frontotemporal pathology and rule in or exclude other diseases.3,5
Psychometric testing can evaluate memory or cognitive ability, which might be unremarkable during the initial phases of FTD.4 Further psychological assessments may provide objective verification of frontal lobe deficiencies in social skills or activities of daily living.3 Positron emission tomography and single-photon emission computed tomography may demonstrate areas of decreased activity or hypoperfusion in frontal and temporal lobes.7
Interventions
Treatment of FTD is limited to symptomatic therapy8; there are no specific, approved countermeasures available. Comorbid conditions, such as diabetes mellitus or hypertension, should be treated medically. Social interventions such as day care, increased supervision, and emotional support from the family can be effective adjuvants.2
Disclosures
The authors report no financial relationship whose products are mentioned in this article or with manufacturers of competing products.
Frontotemporal dementia (FTD) is a neurologic disease that affects the frontal and the temporal lobes of the cerebral cortex.1 This disorder is observed most often in people between age 45 to 65, but also can manifest in younger or older persons.1 The cause varies among a range of pathologies affecting the anterior portions of the brain.2
Presentations
FTD presents with changes in personality, social skills, ability to concentrate, motivation, reasoning, and language abnormality.3 Memory loss is less prominent in this condition compared with other dementias; therefore, identification may be a diagnostic challenge. FTD can be misdiagnosed as a psychiatric illness or not recognized because social symptoms dominate over cognitive dysfunction. As the disease progresses, patients may become increasingly unable to plan or organize activities of daily living, behave appropriately, and react normally in social interactions.1
FTD has 3 diagnostic variants1-4:
Behavioral variant. Known as Pick disease or the “frontal variant,”1,2 this type of FTD manifests as changes in personality, improper behavior in social settings, personal neglect, or impulsivity, such as shoplifting or hypersexuality.
Primary progressive aphasia. Two types of language dysfunction are observed in FTD:
• Semantic dementia (SD)3: Left-sided SD presents with “meaningless speech” or “word substitutions” (eg, “chair” instead of “table”). Right-sided SD, however, is characterized by forgetting the faces of familiar people or objects.
• Primary nonfluent aphasia3: Language fluency is compromised. Persons with such language dysfunction cannot produce words easily, and their speech is stumbling and nonfluent.
FTD with motor neuron disease.4 The most common type of motor neuron disease associated with FTD is amyotrophic lateral sclerosis. Afflicted patients exhibit muscle weakness, spasms, and rigidity. This leads to difficulty in swallowing or breathing because the diaphragm and pharynx are paralyzed. Other diseases associated with FTD include corticobasal degeneration and progressive supranuclear palsy.
Diagnosis
In DSM-5, FTD has been renamed “frontotemporal lobar degeneration” under the category of “Major and Mild Neurocognitive Disorders.”5 The workup begins with a history, physical examination, and mental status assessment. Physical signs can include frontal-release, primitive reflexes. Early in the disease course, a palmomental reflex often is observed; later, as disease progress, the rooting reflex or palmar grasp may become apparent.1,5
Diagnosing FTD requires recognizing its symptoms and ruling out conditions such as Alzheimer’s disease, depression, and schizophrenia.6 Laboratory studies may help identify other conditions. Brain imaging, such as MRI, can depict frontotemporal pathology and rule in or exclude other diseases.3,5
Psychometric testing can evaluate memory or cognitive ability, which might be unremarkable during the initial phases of FTD.4 Further psychological assessments may provide objective verification of frontal lobe deficiencies in social skills or activities of daily living.3 Positron emission tomography and single-photon emission computed tomography may demonstrate areas of decreased activity or hypoperfusion in frontal and temporal lobes.7
Interventions
Treatment of FTD is limited to symptomatic therapy8; there are no specific, approved countermeasures available. Comorbid conditions, such as diabetes mellitus or hypertension, should be treated medically. Social interventions such as day care, increased supervision, and emotional support from the family can be effective adjuvants.2
Disclosures
The authors report no financial relationship whose products are mentioned in this article or with manufacturers of competing products.
1. Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140-143.
2. Frontotemporal degeneration. The Association for Frontotemporal Degeneration. http://www.theaftd.org/ frontotemporal-degeneration/ftd-overview. Accessed April 24, 2014.
3. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546-1554.
4. Clark CM, Forman MS. Frontotemporal lobar degeneration with motor neuron disease: a clinical and pathological spectrum. Arch Neurol. 2006;63(4):489-490.
5. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013:614-618.
6. Frontotemporal dementia diagnosis. UCSF Medical Center. http://www.ucsfhealth.org/conditions/frontotemporal_ dementia/diagnosis.html. Accessed April 24, 2014.
7. McMurtray AM, Chen AK, Shapira JS, et al. Variations in regional SPECT hypoperfusion and clinical features in frontotemporal dementia. Neurology. 2006;66(4):517-522.
8. Miller BL, Lee SE. Frontotemporal dementia: treatment. Up To Date. http://www.uptodate.com/contents/frontotemporal-dementia-treatment?source=search_result&search=frontote mporal+dementia+treatment&selectedTitle=1~150. Updated December 30, 2013. Accessed April 24, 2014.
1. Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140-143.
2. Frontotemporal degeneration. The Association for Frontotemporal Degeneration. http://www.theaftd.org/ frontotemporal-degeneration/ftd-overview. Accessed April 24, 2014.
3. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546-1554.
4. Clark CM, Forman MS. Frontotemporal lobar degeneration with motor neuron disease: a clinical and pathological spectrum. Arch Neurol. 2006;63(4):489-490.
5. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013:614-618.
6. Frontotemporal dementia diagnosis. UCSF Medical Center. http://www.ucsfhealth.org/conditions/frontotemporal_ dementia/diagnosis.html. Accessed April 24, 2014.
7. McMurtray AM, Chen AK, Shapira JS, et al. Variations in regional SPECT hypoperfusion and clinical features in frontotemporal dementia. Neurology. 2006;66(4):517-522.
8. Miller BL, Lee SE. Frontotemporal dementia: treatment. Up To Date. http://www.uptodate.com/contents/frontotemporal-dementia-treatment?source=search_result&search=frontote mporal+dementia+treatment&selectedTitle=1~150. Updated December 30, 2013. Accessed April 24, 2014.
Clozapine: Talking about risks, benefits, and alternatives with patients
Clozapine is a life-saving medication for many patients with schizophrenia, including those who have a schizophrenia spectrum disorder with suicidality or treatment-resistant disease, but clinicians’ discomfort with managing its risk profile has led to it being underutilized. Clinicians who are prepared to discuss the risks and benefits of clozapine—and alternatives, including no treatment—with patients may encounter less reluctance when they recommend a time-limited trial of the drug.
Risks
Clinicians need to be aware of both 1) serious adverse effects that can occur when clozapine needs to be interrupted or discontinued (Table)1 and 2) common side effects associated with continued use that can be managed without stopping the drug.2 Common side effects that patients may experience as treatment is initiated include sedation, orthostatic hypotension, constipation, drooling, tachycardia, and metabolic side effects such as weight gain, diabetes, and hyperlipidemia, which are problematic in the long term. 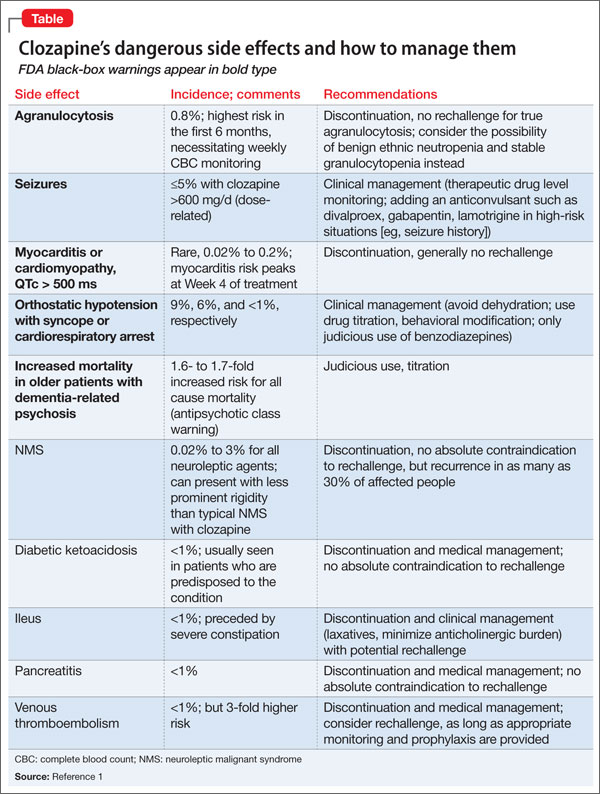
Reassure patients that frequent monitoring of metabolic metrics (including baseline HbA1C, lipid panel, waist circumference, and body mass index, as well as weight monitoring at each visit and metabolic laboratory monitoring every 3 to 6 months thereafter) should be expected, along with early intervention (eg, adding metformin) as appropriate. Constipation is common and can lead to serious, large bowel ileus. Ask about drooling, which can be treated by reducing the dosage or adding glycopyrrolate.
Extrapyramidal symptoms (EPS) including parkinsonism, dystonia, akathisia are uncommon (clozapine was the first “atypical” antipsychotic for this reason), but neuroleptic malignant syndrome (NMS) can occur. Although tardive dyskinesia (TD) is a small risk, clozapine will improve established TD in many patients once they are switched to clozapine. Blood dyscrasias include granulocytopenia and the rare risk of agranulocytosis which are monitored by means of a prescribing registry. Myocarditis and pancreatitis are likely idiosyncratic immune-related side effects that are unique to clozapine among antipsychotics. Other dangerous side effects include a dosage-related risk of seizure, severe hyperglycemia, and diabetic ketoacidosis.
Benefits
Clozapine is FDA-approved for treatment-resistant schizophrenia and for schizophrenia spectrum disorders with recurrent suicidality. Clozapine can be the best antipsychotic for patients who are sensitive to EPS and for those with TD. Antipsychotic efficacy often can be determined in a 2 to 3 month time-limited trial, although, in practice, you might need to wait 6 to 12 months to observe how well clozapine’s benefits have accrued.
Alternatives
Not using the most effective antipsychotic, or using no antipsychotic when one is indicated, often results in unstable psychiatric illness, which increases the risk of adverse outcomes (eg, suicide, accidents). Unstable psychiatric disease also complicates treatment of medical problems. An 11-year follow-up study in Finland of patients with schizophrenia showed a lower all-cause mortality with clozapine than with other antipsychotics, all of which collectively were associated with lower mortality compared with no antipsychotic use.3 Clozapine also is associated with the lowest discontinuation rate of any antipsychotic, which suggests that patients perceive its risk-benefit ratio favorably. Last, patients who might benefit from clozapine, but do not receive it, often will receive polypharmacy, which poses its own risks.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6): 603-613.
2. Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing; 2012.
3. Tiihonen J, Löngqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009; 374(9690):620-627.
Clozapine is a life-saving medication for many patients with schizophrenia, including those who have a schizophrenia spectrum disorder with suicidality or treatment-resistant disease, but clinicians’ discomfort with managing its risk profile has led to it being underutilized. Clinicians who are prepared to discuss the risks and benefits of clozapine—and alternatives, including no treatment—with patients may encounter less reluctance when they recommend a time-limited trial of the drug.
Risks
Clinicians need to be aware of both 1) serious adverse effects that can occur when clozapine needs to be interrupted or discontinued (Table)1 and 2) common side effects associated with continued use that can be managed without stopping the drug.2 Common side effects that patients may experience as treatment is initiated include sedation, orthostatic hypotension, constipation, drooling, tachycardia, and metabolic side effects such as weight gain, diabetes, and hyperlipidemia, which are problematic in the long term.
Reassure patients that frequent monitoring of metabolic metrics (including baseline HbA1C, lipid panel, waist circumference, and body mass index, as well as weight monitoring at each visit and metabolic laboratory monitoring every 3 to 6 months thereafter) should be expected, along with early intervention (eg, adding metformin) as appropriate. Constipation is common and can lead to serious, large bowel ileus. Ask about drooling, which can be treated by reducing the dosage or adding glycopyrrolate.
Extrapyramidal symptoms (EPS) including parkinsonism, dystonia, akathisia are uncommon (clozapine was the first “atypical” antipsychotic for this reason), but neuroleptic malignant syndrome (NMS) can occur. Although tardive dyskinesia (TD) is a small risk, clozapine will improve established TD in many patients once they are switched to clozapine. Blood dyscrasias include granulocytopenia and the rare risk of agranulocytosis which are monitored by means of a prescribing registry. Myocarditis and pancreatitis are likely idiosyncratic immune-related side effects that are unique to clozapine among antipsychotics. Other dangerous side effects include a dosage-related risk of seizure, severe hyperglycemia, and diabetic ketoacidosis.
Benefits
Clozapine is FDA-approved for treatment-resistant schizophrenia and for schizophrenia spectrum disorders with recurrent suicidality. Clozapine can be the best antipsychotic for patients who are sensitive to EPS and for those with TD. Antipsychotic efficacy often can be determined in a 2 to 3 month time-limited trial, although, in practice, you might need to wait 6 to 12 months to observe how well clozapine’s benefits have accrued.
Alternatives
Not using the most effective antipsychotic, or using no antipsychotic when one is indicated, often results in unstable psychiatric illness, which increases the risk of adverse outcomes (eg, suicide, accidents). Unstable psychiatric disease also complicates treatment of medical problems. An 11-year follow-up study in Finland of patients with schizophrenia showed a lower all-cause mortality with clozapine than with other antipsychotics, all of which collectively were associated with lower mortality compared with no antipsychotic use.3 Clozapine also is associated with the lowest discontinuation rate of any antipsychotic, which suggests that patients perceive its risk-benefit ratio favorably. Last, patients who might benefit from clozapine, but do not receive it, often will receive polypharmacy, which poses its own risks.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Clozapine is a life-saving medication for many patients with schizophrenia, including those who have a schizophrenia spectrum disorder with suicidality or treatment-resistant disease, but clinicians’ discomfort with managing its risk profile has led to it being underutilized. Clinicians who are prepared to discuss the risks and benefits of clozapine—and alternatives, including no treatment—with patients may encounter less reluctance when they recommend a time-limited trial of the drug.
Risks
Clinicians need to be aware of both 1) serious adverse effects that can occur when clozapine needs to be interrupted or discontinued (Table)1 and 2) common side effects associated with continued use that can be managed without stopping the drug.2 Common side effects that patients may experience as treatment is initiated include sedation, orthostatic hypotension, constipation, drooling, tachycardia, and metabolic side effects such as weight gain, diabetes, and hyperlipidemia, which are problematic in the long term.
Reassure patients that frequent monitoring of metabolic metrics (including baseline HbA1C, lipid panel, waist circumference, and body mass index, as well as weight monitoring at each visit and metabolic laboratory monitoring every 3 to 6 months thereafter) should be expected, along with early intervention (eg, adding metformin) as appropriate. Constipation is common and can lead to serious, large bowel ileus. Ask about drooling, which can be treated by reducing the dosage or adding glycopyrrolate.
Extrapyramidal symptoms (EPS) including parkinsonism, dystonia, akathisia are uncommon (clozapine was the first “atypical” antipsychotic for this reason), but neuroleptic malignant syndrome (NMS) can occur. Although tardive dyskinesia (TD) is a small risk, clozapine will improve established TD in many patients once they are switched to clozapine. Blood dyscrasias include granulocytopenia and the rare risk of agranulocytosis which are monitored by means of a prescribing registry. Myocarditis and pancreatitis are likely idiosyncratic immune-related side effects that are unique to clozapine among antipsychotics. Other dangerous side effects include a dosage-related risk of seizure, severe hyperglycemia, and diabetic ketoacidosis.
Benefits
Clozapine is FDA-approved for treatment-resistant schizophrenia and for schizophrenia spectrum disorders with recurrent suicidality. Clozapine can be the best antipsychotic for patients who are sensitive to EPS and for those with TD. Antipsychotic efficacy often can be determined in a 2 to 3 month time-limited trial, although, in practice, you might need to wait 6 to 12 months to observe how well clozapine’s benefits have accrued.
Alternatives
Not using the most effective antipsychotic, or using no antipsychotic when one is indicated, often results in unstable psychiatric illness, which increases the risk of adverse outcomes (eg, suicide, accidents). Unstable psychiatric disease also complicates treatment of medical problems. An 11-year follow-up study in Finland of patients with schizophrenia showed a lower all-cause mortality with clozapine than with other antipsychotics, all of which collectively were associated with lower mortality compared with no antipsychotic use.3 Clozapine also is associated with the lowest discontinuation rate of any antipsychotic, which suggests that patients perceive its risk-benefit ratio favorably. Last, patients who might benefit from clozapine, but do not receive it, often will receive polypharmacy, which poses its own risks.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6): 603-613.
2. Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing; 2012.
3. Tiihonen J, Löngqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009; 374(9690):620-627.
1. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6): 603-613.
2. Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing; 2012.
3. Tiihonen J, Löngqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009; 374(9690):620-627.
Using CBT effectively for treating depression and anxiety
Fewer than 20% of people seeking help for depression and anxiety disorders receive cognitive-behavioral therapy (CBT), the most established evidence-based psychotherapeutic treatment.1 Efforts are being made to increase access to CBT,2 but a substantial barrier remains: therapist training is a strong predictor of treatment outcome, and many therapists offering CBT services are not sufficiently trained to deliver multiple manual-based interventions with adequate fidelity to the model. Proposed solutions to this barrier include:
• abbreviated versions of CBT training for practitioners in primary care and community settings
• culturally adapted CBT training for community health workers3
• Internet-based CBT and telemedicine (telephone and video conferencing)2
• mobile phone applications that use text messaging, social support, and physiological monitoring as adjuncts to clinical practice or stand-alone interventions.4
New models of CBT also are emerging, including transdiagnostic CBT and metacognitive approaches (mindfulness-based cognitive therapy and acceptance and commitment therapy), and several new foci for exposure therapy.
In light of these ongoing modulations, this article is intended to help clinicians make informed decisions about CBT when selecting treatment for patients with depressive and anxiety disorders (Box5 ). We review the evidence of CBT’s efficacy for acute-phase treatment and relapse prevention; explain the common elements considered essential to CBT practice; describe CBT adaptations for specific anxiety disorders; and provide an overview of recent advances in conceptualizing and adapting CBT.

Efficacy for mood and anxiety disorders
Depression. Dozens of randomized controlled trials (RCT) and other studies support CBT’s efficacy in treating major depressive disorder (MDD). For acute treatment:
• CBT is more effective in producing remission when compared with no treatment, treatment as usual, or nonspecific psychotherapy.
• For mild to moderate depression, CBT is equivalent to antidepressant medication in terms of response and remission rates.
• Combining antidepressant therapy with CBT increases treatment adherence.6
Less well known may be that a successful response to CBT in the acute phase may have a protective effect against depression recurrences. A 2013 meta-analysis that totaled 506 individuals with depressive disorders found a trend toward significantly lower relapse rates when CBT was discontinued after acute therapy, compared with antidepressant therapy that continued beyond the acute phase.7
Anxiety. Among psychotherapies, CBT’s superior efficacy for anxiety disorders is well-established. CBT and its specific-disorder adaptations are considered first-line treatment.8
CBT’s essential elements
CBT focuses on distorted cognitions about the self, the world, and the future, and on behaviors that lead to or maintain symptoms.
Cognitive interventions seek to identify thoughts and beliefs that trigger emotional and behavioral reactions. A person with social anxiety disorder, for example, might believe that people will notice if he makes even a minor social mistake and then reject him, which will make him feel worthless. CBT can help him subject these beliefs to rational analysis and develop more adaptive beliefs, such as: “It is not certain that I will behave so badly that people would notice, but if that happened, the likelihood of being outright rejected is probably low. If—in the worst-case scenario—I was rejected, I am not worthless; I’m just a fallible human being.”
CBT’s behavioral component can be conceptualized as behavioral activation (BA), a structured approach to help the patient:
• increase behaviors and experiences that are rewarding
• overcome barriers to engaging in these new behaviors
• and decrease behaviors that maintain symptoms.
BA can be a useful intervention for individuals with depression characterized by lack of engagement or capacity for pleasurable experiences. During pregnancy and the postpartum period, for example, a woman undergoes physical, social, and environmental changes that might gradually deprive her of sources of pleasure and other reinforcing activities. BA would focus on developing creative solutions to regain access to or create new opportunities for rewarding experiences and to avoid behaviors (such as social withdrawal or physical activity restriction) that perpetuate depressed mood.
Common elements. Cognitive and behavioral interventions focus on problem solving, individualized case conceptualization (Figure 1), and collaborative empiricism.9
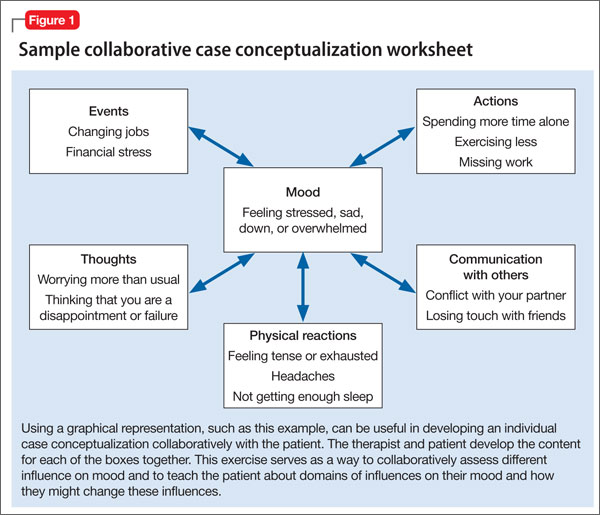
Individualized case conceptualization lays the foundation for the course of CBT, and may be thought of as a map for therapy. Case conceptualization brings in several domains of assessment including symptoms and diagnosis, the patient’s strengths, formative experiences (including biopsychosocial aspects), contextual factors, and cognitive factors that influence diagnosis and treatment, such as automatic thoughts or schemas. The case formulation leads to a working hypothesis about the optimal course and focus of CBT.
Collaborative empiricism is the way in which the patient and therapist work together to continually refine this working hypothesis. The pair works together to investigate the hypotheses and all aspects of the therapeutic relationship.
Although no specific technique defines CBT, a common practice is to educate a person about interrelationships between behaviors/activities, thoughts, and mood. A mood activity log (Figure 2) can illuminate links between moods and activities and be useful with targeting interventions. For a person with social anxiety, for example, a mood activity log could assist in developing a hierarchy of feared social situations and avoidance intensity. Systematic exposure therapy would follow, beginning with the least frightening/intense situation, accompanied by teaching new coping skills (such as relaxation strategies).
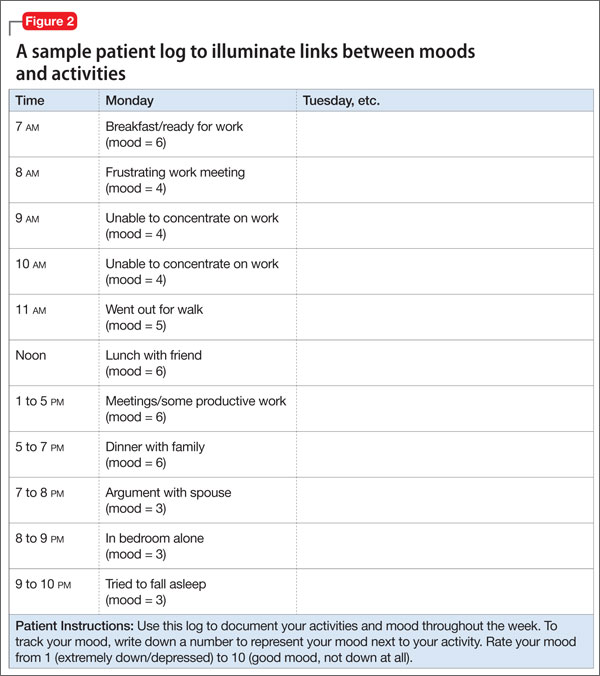
CBT adaptations for anxiety disorders
Elements of CBT have been adapted for a variety of anxiety disorders, based on specific symptoms and features (Table).10-15
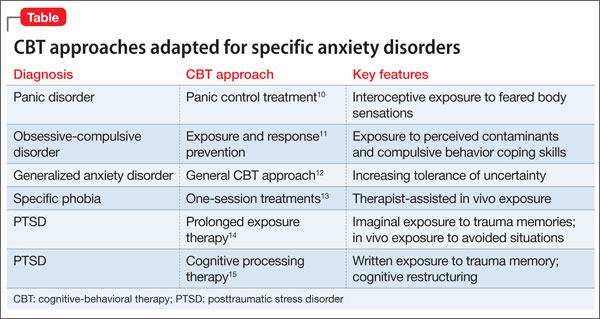
Panic disorder. Panic control treatment is considered the first-line intervention for panic disorder’s defining features: spontaneous panic attacks, worry about future occurrence of attacks, and perceived catastrophic consequences (such as heart attack, fainting).10 This CBT adaptation includes:
• patient education about the nature of panic
• breathing retraining to foster exposure to feared bodily sensations and avoided activities and places
• cognitive restructuring of danger-related thoughts (such as “I’m going to faint,” or “It would be catastrophic if I did”).
Obsessive-compulsive disorder. Exposure and response prevention (ERP) is the first-line treatment for obsessive-compulsive disorder (OCD).11 In traditional therapist-guided ERP, patients expose themselves to perceived contaminants while refraining from inappropriate compulsive behaviors (such as hand washing).
Cognitive interventions also can be an effective treatment of obsessions, without patients having to engage in exposure to their horrific thoughts and images.16 Consider, for example, a new mother who upon seeing the kitchen knife has the intrusive thought, “What if I stabbed my baby?” Instead of the traditional exposure approach for OCD (ie, having her vividly imagine stabbing her baby until her anxiety level subsided), the cognitive intervention would be to educate her about the normalcy of intrusive thoughts, particularly in the postpartum period.
Generalized anxiety disorder. CBT for generalized anxiety disorder (GAD) targets patients’ overestimation of the likelihood of negative events and the belief that these events, should they occur, would be catastrophic and render them unable to cope.12
Motivational interviewing (MI) appears to be a useful adjunct to precede traditional CBT, particularly for severe worriers.17 MI attempts to help individuals with GAD recognize their ambivalence about giving up worry. This technique acknowledges and validates perceived benefits of worry (eg, “It helps me prepare for the worst, so I won’t be emotionally devastated if it happens”), but also explores how worry is destructive.
Emerging CBT models for anxiety disorders
Metacognitive treatment. Evidence, such as presented by Dobson,18 suggests that the field of CBT is shifting towards a metacognitive model of change and treatment. A metacognitive approach goes beyond changing thinking and emphasizes thoughts about thoughts and experiences. Examples include mindfulness-based cognitive therapy (MBCT) and acceptance and commitment therapy (ACT).
MBCT typically consists of an 8-week program of 2-hour sessions each week and 1 full-day retreat. MBCT is modeled after Kabat-Zinn’s widely disseminated and empirically supported mindfulness based stress reduction course.19 MBCT was developed as a relapse prevention program for patients who had recovered from depression. Unlike traditional cognitive therapy for depression that targets changing the content of automatic thoughts and core beliefs, in MBCT patients are aware of negative automatic thoughts and find ways to change their relationship with these thoughts, learning that thoughts are not facts. This process mainly is carried out by practicing mindfulness meditation exercises. Importantly, MBCT goes beyond mindful acceptance of negative thoughts and teaches patients mindful acceptance of all internal experiences.
A fundamental difference between ACT and traditional CBT is the approach to cognitions.20 Although CBT focuses on changing the content of maladaptive thoughts, such as “I am a worthless person,” ACT focuses on changing the function of thoughts. ACT strives to help patients to accept their internal experiences—whether unwanted thoughts, feelings, bodily sensations, or memories—while committing themselves to pursuing their life goals and values. Strategies aim to help patients step back from their thoughts and observe them as just thoughts. The patient who thinks, “I am worthless” would be instructed to practice saying “I am having the thought I am worthless.” Therefore the thought no longer controls the person’s behavior.
These approaches train the patient to keenly observe distressing thoughts and experiences—not necessarily with the goal of changing them but to accept them and act in a way that is consistent with his (her) goals and values. A meta-analysis of 39 studies found mindfulness-based therapy effective in improving symptoms in participants with anxiety and mood disorders.21 Similarly, ACT has demonstrated efficacy with mixed anxiety disorders.22
Transdiagnostic CBT. Recent research18 suggests that mood and anxiety disorders may have more commonalities than differences in underlying biological and psychological traits. Because the symptoms of anxiety and depressive disorders tend to overlap, and their rate of comorbidity may be as high as 55%,23 so-called transdiagnostic treatments have been developed. Transdiagnostic treatments target impairing symptoms that cut across different diagnoses. For example, patients with depression, anxiety, or substance abuse might share a common difficulty with regulating and coping with negative emotions.
In a preliminary comparison trial,24 46 patients with social anxiety disorder, panic disorder, or GAD were randomly assigned to transdiagnostic CBT (n = 23) or diagnosis-specific CBT (n = 23). Treatments were based on widely used manuals and offered in 2-hour group sessions across 12 weeks. Transdiagnostic CBT was found to be as effective as specific CBT protocols in terms of symptom improvement. Participants attended an average of 8.46 sessions, with similar attendance in each protocol. Fourteen participants (30%) discontinued treatment, similar to attrition rates reported in other trials of transdiagnostic and diagnosis-specific CBT.
Transdiagnostic treatments may facilitate the dissemination of empirically supported treatments because therapists would not be required to have training and supervision to competency in delivering multiple manuals for specific anxiety disorders. This could be attractive to busy practitioners with limited time to learn new treatments.
Bottom Line
Efficacy of cognitive-behavioral therapy (CBT) for depression and anxiety is well established. Although no specific technique defines CBT, a common practice is to educate an individual about interrelationships between behaviors/activities, thoughts, and mood. CBT techniques can be customized to treat specific anxiety disorders, such as panic disorder, obsessive-compulsive disorder, and generalized anxiety disorder.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Collins K, Westra H, Dozois D, et al. Gaps in accessing treatment for anxiety and depressions: challenges for the delivery of care. Clin Psychol Rev. 2004;24(5):583-616.
2. Foa EB, Gillihan SJ, Bryant RA. Challenges and successes in dissemination of evidence-based treatments for posttraumatic stress: lessons learned from prolonged exposure therapy for PTSD. Psychological Science in the Public Interest. 2013;14(2):65-111.
3. Rahman A, Malik A, Sikander S, et al. Cognitive behaviour therapy-based intervention by community health workers for mothers with depression and their infants in rural Pakistan: a cluster-randomised controlled trial. Lancet. 2008;372(9642):902-909.
4. Aguilera A, Muench F. There’s an app for that: information technology applications for cognitive behavioral practitioners. Behavior Therapist. 2012;35(4):65-73.
5. Dimidjian S, Hollon SD, Dobson KS, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the acute treatment of adults with major depression. J Consult Clin Psychol. 2006; 74(4):658-670.
6. Hollon SD, Jarrett RB, Nierenberg AA, et al. Psychotherapy and medication in the treatment of adult and geriatric depression: which monotherapy or combined treatment? J Clin Psychiatry. 2005;66(4):455-468.
7. Cuijpers P, Hollon SD, van Straten A, et al. Does cognitive behaviour therapy have an enduring effect that is superior to keeping patients on continuation pharmacotherapy? A meta-analysis. BMJ Open. 2013;3(4):1-8.
8. Stewart R, Chambless D. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77(4): 595-606.
9. Wright JH, Basco MR, Thase M. Learning cognitive behavior therapy: an illustrated guide. Arlington, VA: American Psychiatric Publishing; 2006.
10. Barlow DH, Craske MG. Mastery of your anxiety and panic. 4th ed. New York, NY: Oxford University Press, Inc.; 2007.
11. Foa EB, Yadin E, Lichner TK. Exposure and response prevention for obsessive-compulsive disorder: therapist guide. New York, NY: Oxford University Press, Inc.; 2012.
12. Dugas MJ, Robichaud M. Cognitive-behavioral treatment for generalized anxiety disorder. New York, NY: Routledge; 2007.
13. Zlomke K, Davis TE. One-session treatment of specific phobias: a detailed description and review of treatment efficacy. Behav Ther. 2008;39(3):207-223.
14. Foa EB, Hembree E, Rothbaum B. Prolonged exposure therapy for PTSD: emotional processing of traumatic experiences. Therapist guide. New York, NY: Oxford University Press, Inc.; 2007.
15. Resick PA, Schnicke MK. Cognitive processing therapy for rape victims. London, United Kingdom: Sage Publications; 1996.
16. Whittal ML, Robichaud M, Woody SR. Cognitive treatment of obsessions: enhancing dissemination with video components. Cognitive and Behavioral Practice. 2010;17(1):1-8.
17. Westra H, Arkowitz H, Dozois D. Adding a motivational interviewing pretreatment to cognitive behavioral therapy for generalized anxiety disorder: a preliminary randomized controlled trial. J Anxiety Disord. 2009;23(2): 1106-1117.
18. Dobson KS. The science of CBT: toward a metacognitive model of change? Behav Ther. 2013;44(2):224-227.
19. Kabat-Zinn J. Full catastrophe living. Using the wisdom of your body and mind to face stress, pain, and illness. Revised edition. New York, NY: Bantam Books; 2013.
20. Hayes SC, Strosahl KD. Acceptance and commitment therapy. The process and practice of mindful change. 2nd ed. New York, NY: The Guilford Press; 2012.
21. Hofmann S, Sawyer A, Witt A, et al. The effect of mindfulness-based therapy on anxiety and depression: a meta-analytic review. J Consult Clin Psychol. 2010;78(2): 169-183.
22. Arch J, Eifert G, Davies C, et al. Randomized clinical trial of cognitive behavioral therapy (CBT) versus acceptance and commitment therapy (ACT) for mixed anxiety disorders. J Consult Clin Psychol. 2012;80(5):750-765.
23. Brown TA, Campbell LA, Lehman CL, et al. Current and lifetime comorbidity of the DSM-IV anxiety and mood disorders in a large clinical sample. J Abnorm Psychol. 2001;110(4):585-599.
24. Norton P, Barrera T. Transdiagnostic versus diagnosis-specific CBT for anxiety disorders: a preliminary randomized controlled noninferiority trial. Depress Anxiety. 2012;29(10):874-882.
Fewer than 20% of people seeking help for depression and anxiety disorders receive cognitive-behavioral therapy (CBT), the most established evidence-based psychotherapeutic treatment.1 Efforts are being made to increase access to CBT,2 but a substantial barrier remains: therapist training is a strong predictor of treatment outcome, and many therapists offering CBT services are not sufficiently trained to deliver multiple manual-based interventions with adequate fidelity to the model. Proposed solutions to this barrier include:
• abbreviated versions of CBT training for practitioners in primary care and community settings
• culturally adapted CBT training for community health workers3
• Internet-based CBT and telemedicine (telephone and video conferencing)2
• mobile phone applications that use text messaging, social support, and physiological monitoring as adjuncts to clinical practice or stand-alone interventions.4
New models of CBT also are emerging, including transdiagnostic CBT and metacognitive approaches (mindfulness-based cognitive therapy and acceptance and commitment therapy), and several new foci for exposure therapy.
In light of these ongoing modulations, this article is intended to help clinicians make informed decisions about CBT when selecting treatment for patients with depressive and anxiety disorders (Box5 ). We review the evidence of CBT’s efficacy for acute-phase treatment and relapse prevention; explain the common elements considered essential to CBT practice; describe CBT adaptations for specific anxiety disorders; and provide an overview of recent advances in conceptualizing and adapting CBT.
Efficacy for mood and anxiety disorders
Depression. Dozens of randomized controlled trials (RCT) and other studies support CBT’s efficacy in treating major depressive disorder (MDD). For acute treatment:
• CBT is more effective in producing remission when compared with no treatment, treatment as usual, or nonspecific psychotherapy.
• For mild to moderate depression, CBT is equivalent to antidepressant medication in terms of response and remission rates.
• Combining antidepressant therapy with CBT increases treatment adherence.6
Less well known may be that a successful response to CBT in the acute phase may have a protective effect against depression recurrences. A 2013 meta-analysis that totaled 506 individuals with depressive disorders found a trend toward significantly lower relapse rates when CBT was discontinued after acute therapy, compared with antidepressant therapy that continued beyond the acute phase.7
Anxiety. Among psychotherapies, CBT’s superior efficacy for anxiety disorders is well-established. CBT and its specific-disorder adaptations are considered first-line treatment.8
CBT’s essential elements
CBT focuses on distorted cognitions about the self, the world, and the future, and on behaviors that lead to or maintain symptoms.
Cognitive interventions seek to identify thoughts and beliefs that trigger emotional and behavioral reactions. A person with social anxiety disorder, for example, might believe that people will notice if he makes even a minor social mistake and then reject him, which will make him feel worthless. CBT can help him subject these beliefs to rational analysis and develop more adaptive beliefs, such as: “It is not certain that I will behave so badly that people would notice, but if that happened, the likelihood of being outright rejected is probably low. If—in the worst-case scenario—I was rejected, I am not worthless; I’m just a fallible human being.”
CBT’s behavioral component can be conceptualized as behavioral activation (BA), a structured approach to help the patient:
• increase behaviors and experiences that are rewarding
• overcome barriers to engaging in these new behaviors
• and decrease behaviors that maintain symptoms.
BA can be a useful intervention for individuals with depression characterized by lack of engagement or capacity for pleasurable experiences. During pregnancy and the postpartum period, for example, a woman undergoes physical, social, and environmental changes that might gradually deprive her of sources of pleasure and other reinforcing activities. BA would focus on developing creative solutions to regain access to or create new opportunities for rewarding experiences and to avoid behaviors (such as social withdrawal or physical activity restriction) that perpetuate depressed mood.
Common elements. Cognitive and behavioral interventions focus on problem solving, individualized case conceptualization (Figure 1), and collaborative empiricism.9
Individualized case conceptualization lays the foundation for the course of CBT, and may be thought of as a map for therapy. Case conceptualization brings in several domains of assessment including symptoms and diagnosis, the patient’s strengths, formative experiences (including biopsychosocial aspects), contextual factors, and cognitive factors that influence diagnosis and treatment, such as automatic thoughts or schemas. The case formulation leads to a working hypothesis about the optimal course and focus of CBT.
Collaborative empiricism is the way in which the patient and therapist work together to continually refine this working hypothesis. The pair works together to investigate the hypotheses and all aspects of the therapeutic relationship.
Although no specific technique defines CBT, a common practice is to educate a person about interrelationships between behaviors/activities, thoughts, and mood. A mood activity log (Figure 2) can illuminate links between moods and activities and be useful with targeting interventions. For a person with social anxiety, for example, a mood activity log could assist in developing a hierarchy of feared social situations and avoidance intensity. Systematic exposure therapy would follow, beginning with the least frightening/intense situation, accompanied by teaching new coping skills (such as relaxation strategies).
CBT adaptations for anxiety disorders
Elements of CBT have been adapted for a variety of anxiety disorders, based on specific symptoms and features (Table).10-15
Panic disorder. Panic control treatment is considered the first-line intervention for panic disorder’s defining features: spontaneous panic attacks, worry about future occurrence of attacks, and perceived catastrophic consequences (such as heart attack, fainting).10 This CBT adaptation includes:
• patient education about the nature of panic
• breathing retraining to foster exposure to feared bodily sensations and avoided activities and places
• cognitive restructuring of danger-related thoughts (such as “I’m going to faint,” or “It would be catastrophic if I did”).
Obsessive-compulsive disorder. Exposure and response prevention (ERP) is the first-line treatment for obsessive-compulsive disorder (OCD).11 In traditional therapist-guided ERP, patients expose themselves to perceived contaminants while refraining from inappropriate compulsive behaviors (such as hand washing).
Cognitive interventions also can be an effective treatment of obsessions, without patients having to engage in exposure to their horrific thoughts and images.16 Consider, for example, a new mother who upon seeing the kitchen knife has the intrusive thought, “What if I stabbed my baby?” Instead of the traditional exposure approach for OCD (ie, having her vividly imagine stabbing her baby until her anxiety level subsided), the cognitive intervention would be to educate her about the normalcy of intrusive thoughts, particularly in the postpartum period.
Generalized anxiety disorder. CBT for generalized anxiety disorder (GAD) targets patients’ overestimation of the likelihood of negative events and the belief that these events, should they occur, would be catastrophic and render them unable to cope.12
Motivational interviewing (MI) appears to be a useful adjunct to precede traditional CBT, particularly for severe worriers.17 MI attempts to help individuals with GAD recognize their ambivalence about giving up worry. This technique acknowledges and validates perceived benefits of worry (eg, “It helps me prepare for the worst, so I won’t be emotionally devastated if it happens”), but also explores how worry is destructive.
Emerging CBT models for anxiety disorders
Metacognitive treatment. Evidence, such as presented by Dobson,18 suggests that the field of CBT is shifting towards a metacognitive model of change and treatment. A metacognitive approach goes beyond changing thinking and emphasizes thoughts about thoughts and experiences. Examples include mindfulness-based cognitive therapy (MBCT) and acceptance and commitment therapy (ACT).
MBCT typically consists of an 8-week program of 2-hour sessions each week and 1 full-day retreat. MBCT is modeled after Kabat-Zinn’s widely disseminated and empirically supported mindfulness based stress reduction course.19 MBCT was developed as a relapse prevention program for patients who had recovered from depression. Unlike traditional cognitive therapy for depression that targets changing the content of automatic thoughts and core beliefs, in MBCT patients are aware of negative automatic thoughts and find ways to change their relationship with these thoughts, learning that thoughts are not facts. This process mainly is carried out by practicing mindfulness meditation exercises. Importantly, MBCT goes beyond mindful acceptance of negative thoughts and teaches patients mindful acceptance of all internal experiences.
A fundamental difference between ACT and traditional CBT is the approach to cognitions.20 Although CBT focuses on changing the content of maladaptive thoughts, such as “I am a worthless person,” ACT focuses on changing the function of thoughts. ACT strives to help patients to accept their internal experiences—whether unwanted thoughts, feelings, bodily sensations, or memories—while committing themselves to pursuing their life goals and values. Strategies aim to help patients step back from their thoughts and observe them as just thoughts. The patient who thinks, “I am worthless” would be instructed to practice saying “I am having the thought I am worthless.” Therefore the thought no longer controls the person’s behavior.
These approaches train the patient to keenly observe distressing thoughts and experiences—not necessarily with the goal of changing them but to accept them and act in a way that is consistent with his (her) goals and values. A meta-analysis of 39 studies found mindfulness-based therapy effective in improving symptoms in participants with anxiety and mood disorders.21 Similarly, ACT has demonstrated efficacy with mixed anxiety disorders.22
Transdiagnostic CBT. Recent research18 suggests that mood and anxiety disorders may have more commonalities than differences in underlying biological and psychological traits. Because the symptoms of anxiety and depressive disorders tend to overlap, and their rate of comorbidity may be as high as 55%,23 so-called transdiagnostic treatments have been developed. Transdiagnostic treatments target impairing symptoms that cut across different diagnoses. For example, patients with depression, anxiety, or substance abuse might share a common difficulty with regulating and coping with negative emotions.
In a preliminary comparison trial,24 46 patients with social anxiety disorder, panic disorder, or GAD were randomly assigned to transdiagnostic CBT (n = 23) or diagnosis-specific CBT (n = 23). Treatments were based on widely used manuals and offered in 2-hour group sessions across 12 weeks. Transdiagnostic CBT was found to be as effective as specific CBT protocols in terms of symptom improvement. Participants attended an average of 8.46 sessions, with similar attendance in each protocol. Fourteen participants (30%) discontinued treatment, similar to attrition rates reported in other trials of transdiagnostic and diagnosis-specific CBT.
Transdiagnostic treatments may facilitate the dissemination of empirically supported treatments because therapists would not be required to have training and supervision to competency in delivering multiple manuals for specific anxiety disorders. This could be attractive to busy practitioners with limited time to learn new treatments.
Bottom Line
Efficacy of cognitive-behavioral therapy (CBT) for depression and anxiety is well established. Although no specific technique defines CBT, a common practice is to educate an individual about interrelationships between behaviors/activities, thoughts, and mood. CBT techniques can be customized to treat specific anxiety disorders, such as panic disorder, obsessive-compulsive disorder, and generalized anxiety disorder.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Fewer than 20% of people seeking help for depression and anxiety disorders receive cognitive-behavioral therapy (CBT), the most established evidence-based psychotherapeutic treatment.1 Efforts are being made to increase access to CBT,2 but a substantial barrier remains: therapist training is a strong predictor of treatment outcome, and many therapists offering CBT services are not sufficiently trained to deliver multiple manual-based interventions with adequate fidelity to the model. Proposed solutions to this barrier include:
• abbreviated versions of CBT training for practitioners in primary care and community settings
• culturally adapted CBT training for community health workers3
• Internet-based CBT and telemedicine (telephone and video conferencing)2
• mobile phone applications that use text messaging, social support, and physiological monitoring as adjuncts to clinical practice or stand-alone interventions.4
New models of CBT also are emerging, including transdiagnostic CBT and metacognitive approaches (mindfulness-based cognitive therapy and acceptance and commitment therapy), and several new foci for exposure therapy.
In light of these ongoing modulations, this article is intended to help clinicians make informed decisions about CBT when selecting treatment for patients with depressive and anxiety disorders (Box5 ). We review the evidence of CBT’s efficacy for acute-phase treatment and relapse prevention; explain the common elements considered essential to CBT practice; describe CBT adaptations for specific anxiety disorders; and provide an overview of recent advances in conceptualizing and adapting CBT.
Efficacy for mood and anxiety disorders
Depression. Dozens of randomized controlled trials (RCT) and other studies support CBT’s efficacy in treating major depressive disorder (MDD). For acute treatment:
• CBT is more effective in producing remission when compared with no treatment, treatment as usual, or nonspecific psychotherapy.
• For mild to moderate depression, CBT is equivalent to antidepressant medication in terms of response and remission rates.
• Combining antidepressant therapy with CBT increases treatment adherence.6
Less well known may be that a successful response to CBT in the acute phase may have a protective effect against depression recurrences. A 2013 meta-analysis that totaled 506 individuals with depressive disorders found a trend toward significantly lower relapse rates when CBT was discontinued after acute therapy, compared with antidepressant therapy that continued beyond the acute phase.7
Anxiety. Among psychotherapies, CBT’s superior efficacy for anxiety disorders is well-established. CBT and its specific-disorder adaptations are considered first-line treatment.8
CBT’s essential elements
CBT focuses on distorted cognitions about the self, the world, and the future, and on behaviors that lead to or maintain symptoms.
Cognitive interventions seek to identify thoughts and beliefs that trigger emotional and behavioral reactions. A person with social anxiety disorder, for example, might believe that people will notice if he makes even a minor social mistake and then reject him, which will make him feel worthless. CBT can help him subject these beliefs to rational analysis and develop more adaptive beliefs, such as: “It is not certain that I will behave so badly that people would notice, but if that happened, the likelihood of being outright rejected is probably low. If—in the worst-case scenario—I was rejected, I am not worthless; I’m just a fallible human being.”
CBT’s behavioral component can be conceptualized as behavioral activation (BA), a structured approach to help the patient:
• increase behaviors and experiences that are rewarding
• overcome barriers to engaging in these new behaviors
• and decrease behaviors that maintain symptoms.
BA can be a useful intervention for individuals with depression characterized by lack of engagement or capacity for pleasurable experiences. During pregnancy and the postpartum period, for example, a woman undergoes physical, social, and environmental changes that might gradually deprive her of sources of pleasure and other reinforcing activities. BA would focus on developing creative solutions to regain access to or create new opportunities for rewarding experiences and to avoid behaviors (such as social withdrawal or physical activity restriction) that perpetuate depressed mood.
Common elements. Cognitive and behavioral interventions focus on problem solving, individualized case conceptualization (Figure 1), and collaborative empiricism.9
Individualized case conceptualization lays the foundation for the course of CBT, and may be thought of as a map for therapy. Case conceptualization brings in several domains of assessment including symptoms and diagnosis, the patient’s strengths, formative experiences (including biopsychosocial aspects), contextual factors, and cognitive factors that influence diagnosis and treatment, such as automatic thoughts or schemas. The case formulation leads to a working hypothesis about the optimal course and focus of CBT.
Collaborative empiricism is the way in which the patient and therapist work together to continually refine this working hypothesis. The pair works together to investigate the hypotheses and all aspects of the therapeutic relationship.
Although no specific technique defines CBT, a common practice is to educate a person about interrelationships between behaviors/activities, thoughts, and mood. A mood activity log (Figure 2) can illuminate links between moods and activities and be useful with targeting interventions. For a person with social anxiety, for example, a mood activity log could assist in developing a hierarchy of feared social situations and avoidance intensity. Systematic exposure therapy would follow, beginning with the least frightening/intense situation, accompanied by teaching new coping skills (such as relaxation strategies).
CBT adaptations for anxiety disorders
Elements of CBT have been adapted for a variety of anxiety disorders, based on specific symptoms and features (Table).10-15
Panic disorder. Panic control treatment is considered the first-line intervention for panic disorder’s defining features: spontaneous panic attacks, worry about future occurrence of attacks, and perceived catastrophic consequences (such as heart attack, fainting).10 This CBT adaptation includes:
• patient education about the nature of panic
• breathing retraining to foster exposure to feared bodily sensations and avoided activities and places
• cognitive restructuring of danger-related thoughts (such as “I’m going to faint,” or “It would be catastrophic if I did”).
Obsessive-compulsive disorder. Exposure and response prevention (ERP) is the first-line treatment for obsessive-compulsive disorder (OCD).11 In traditional therapist-guided ERP, patients expose themselves to perceived contaminants while refraining from inappropriate compulsive behaviors (such as hand washing).
Cognitive interventions also can be an effective treatment of obsessions, without patients having to engage in exposure to their horrific thoughts and images.16 Consider, for example, a new mother who upon seeing the kitchen knife has the intrusive thought, “What if I stabbed my baby?” Instead of the traditional exposure approach for OCD (ie, having her vividly imagine stabbing her baby until her anxiety level subsided), the cognitive intervention would be to educate her about the normalcy of intrusive thoughts, particularly in the postpartum period.
Generalized anxiety disorder. CBT for generalized anxiety disorder (GAD) targets patients’ overestimation of the likelihood of negative events and the belief that these events, should they occur, would be catastrophic and render them unable to cope.12
Motivational interviewing (MI) appears to be a useful adjunct to precede traditional CBT, particularly for severe worriers.17 MI attempts to help individuals with GAD recognize their ambivalence about giving up worry. This technique acknowledges and validates perceived benefits of worry (eg, “It helps me prepare for the worst, so I won’t be emotionally devastated if it happens”), but also explores how worry is destructive.
Emerging CBT models for anxiety disorders
Metacognitive treatment. Evidence, such as presented by Dobson,18 suggests that the field of CBT is shifting towards a metacognitive model of change and treatment. A metacognitive approach goes beyond changing thinking and emphasizes thoughts about thoughts and experiences. Examples include mindfulness-based cognitive therapy (MBCT) and acceptance and commitment therapy (ACT).
MBCT typically consists of an 8-week program of 2-hour sessions each week and 1 full-day retreat. MBCT is modeled after Kabat-Zinn’s widely disseminated and empirically supported mindfulness based stress reduction course.19 MBCT was developed as a relapse prevention program for patients who had recovered from depression. Unlike traditional cognitive therapy for depression that targets changing the content of automatic thoughts and core beliefs, in MBCT patients are aware of negative automatic thoughts and find ways to change their relationship with these thoughts, learning that thoughts are not facts. This process mainly is carried out by practicing mindfulness meditation exercises. Importantly, MBCT goes beyond mindful acceptance of negative thoughts and teaches patients mindful acceptance of all internal experiences.
A fundamental difference between ACT and traditional CBT is the approach to cognitions.20 Although CBT focuses on changing the content of maladaptive thoughts, such as “I am a worthless person,” ACT focuses on changing the function of thoughts. ACT strives to help patients to accept their internal experiences—whether unwanted thoughts, feelings, bodily sensations, or memories—while committing themselves to pursuing their life goals and values. Strategies aim to help patients step back from their thoughts and observe them as just thoughts. The patient who thinks, “I am worthless” would be instructed to practice saying “I am having the thought I am worthless.” Therefore the thought no longer controls the person’s behavior.
These approaches train the patient to keenly observe distressing thoughts and experiences—not necessarily with the goal of changing them but to accept them and act in a way that is consistent with his (her) goals and values. A meta-analysis of 39 studies found mindfulness-based therapy effective in improving symptoms in participants with anxiety and mood disorders.21 Similarly, ACT has demonstrated efficacy with mixed anxiety disorders.22
Transdiagnostic CBT. Recent research18 suggests that mood and anxiety disorders may have more commonalities than differences in underlying biological and psychological traits. Because the symptoms of anxiety and depressive disorders tend to overlap, and their rate of comorbidity may be as high as 55%,23 so-called transdiagnostic treatments have been developed. Transdiagnostic treatments target impairing symptoms that cut across different diagnoses. For example, patients with depression, anxiety, or substance abuse might share a common difficulty with regulating and coping with negative emotions.
In a preliminary comparison trial,24 46 patients with social anxiety disorder, panic disorder, or GAD were randomly assigned to transdiagnostic CBT (n = 23) or diagnosis-specific CBT (n = 23). Treatments were based on widely used manuals and offered in 2-hour group sessions across 12 weeks. Transdiagnostic CBT was found to be as effective as specific CBT protocols in terms of symptom improvement. Participants attended an average of 8.46 sessions, with similar attendance in each protocol. Fourteen participants (30%) discontinued treatment, similar to attrition rates reported in other trials of transdiagnostic and diagnosis-specific CBT.
Transdiagnostic treatments may facilitate the dissemination of empirically supported treatments because therapists would not be required to have training and supervision to competency in delivering multiple manuals for specific anxiety disorders. This could be attractive to busy practitioners with limited time to learn new treatments.
Bottom Line
Efficacy of cognitive-behavioral therapy (CBT) for depression and anxiety is well established. Although no specific technique defines CBT, a common practice is to educate an individual about interrelationships between behaviors/activities, thoughts, and mood. CBT techniques can be customized to treat specific anxiety disorders, such as panic disorder, obsessive-compulsive disorder, and generalized anxiety disorder.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Collins K, Westra H, Dozois D, et al. Gaps in accessing treatment for anxiety and depressions: challenges for the delivery of care. Clin Psychol Rev. 2004;24(5):583-616.
2. Foa EB, Gillihan SJ, Bryant RA. Challenges and successes in dissemination of evidence-based treatments for posttraumatic stress: lessons learned from prolonged exposure therapy for PTSD. Psychological Science in the Public Interest. 2013;14(2):65-111.
3. Rahman A, Malik A, Sikander S, et al. Cognitive behaviour therapy-based intervention by community health workers for mothers with depression and their infants in rural Pakistan: a cluster-randomised controlled trial. Lancet. 2008;372(9642):902-909.
4. Aguilera A, Muench F. There’s an app for that: information technology applications for cognitive behavioral practitioners. Behavior Therapist. 2012;35(4):65-73.
5. Dimidjian S, Hollon SD, Dobson KS, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the acute treatment of adults with major depression. J Consult Clin Psychol. 2006; 74(4):658-670.
6. Hollon SD, Jarrett RB, Nierenberg AA, et al. Psychotherapy and medication in the treatment of adult and geriatric depression: which monotherapy or combined treatment? J Clin Psychiatry. 2005;66(4):455-468.
7. Cuijpers P, Hollon SD, van Straten A, et al. Does cognitive behaviour therapy have an enduring effect that is superior to keeping patients on continuation pharmacotherapy? A meta-analysis. BMJ Open. 2013;3(4):1-8.
8. Stewart R, Chambless D. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77(4): 595-606.
9. Wright JH, Basco MR, Thase M. Learning cognitive behavior therapy: an illustrated guide. Arlington, VA: American Psychiatric Publishing; 2006.
10. Barlow DH, Craske MG. Mastery of your anxiety and panic. 4th ed. New York, NY: Oxford University Press, Inc.; 2007.
11. Foa EB, Yadin E, Lichner TK. Exposure and response prevention for obsessive-compulsive disorder: therapist guide. New York, NY: Oxford University Press, Inc.; 2012.
12. Dugas MJ, Robichaud M. Cognitive-behavioral treatment for generalized anxiety disorder. New York, NY: Routledge; 2007.
13. Zlomke K, Davis TE. One-session treatment of specific phobias: a detailed description and review of treatment efficacy. Behav Ther. 2008;39(3):207-223.
14. Foa EB, Hembree E, Rothbaum B. Prolonged exposure therapy for PTSD: emotional processing of traumatic experiences. Therapist guide. New York, NY: Oxford University Press, Inc.; 2007.
15. Resick PA, Schnicke MK. Cognitive processing therapy for rape victims. London, United Kingdom: Sage Publications; 1996.
16. Whittal ML, Robichaud M, Woody SR. Cognitive treatment of obsessions: enhancing dissemination with video components. Cognitive and Behavioral Practice. 2010;17(1):1-8.
17. Westra H, Arkowitz H, Dozois D. Adding a motivational interviewing pretreatment to cognitive behavioral therapy for generalized anxiety disorder: a preliminary randomized controlled trial. J Anxiety Disord. 2009;23(2): 1106-1117.
18. Dobson KS. The science of CBT: toward a metacognitive model of change? Behav Ther. 2013;44(2):224-227.
19. Kabat-Zinn J. Full catastrophe living. Using the wisdom of your body and mind to face stress, pain, and illness. Revised edition. New York, NY: Bantam Books; 2013.
20. Hayes SC, Strosahl KD. Acceptance and commitment therapy. The process and practice of mindful change. 2nd ed. New York, NY: The Guilford Press; 2012.
21. Hofmann S, Sawyer A, Witt A, et al. The effect of mindfulness-based therapy on anxiety and depression: a meta-analytic review. J Consult Clin Psychol. 2010;78(2): 169-183.
22. Arch J, Eifert G, Davies C, et al. Randomized clinical trial of cognitive behavioral therapy (CBT) versus acceptance and commitment therapy (ACT) for mixed anxiety disorders. J Consult Clin Psychol. 2012;80(5):750-765.
23. Brown TA, Campbell LA, Lehman CL, et al. Current and lifetime comorbidity of the DSM-IV anxiety and mood disorders in a large clinical sample. J Abnorm Psychol. 2001;110(4):585-599.
24. Norton P, Barrera T. Transdiagnostic versus diagnosis-specific CBT for anxiety disorders: a preliminary randomized controlled noninferiority trial. Depress Anxiety. 2012;29(10):874-882.
1. Collins K, Westra H, Dozois D, et al. Gaps in accessing treatment for anxiety and depressions: challenges for the delivery of care. Clin Psychol Rev. 2004;24(5):583-616.
2. Foa EB, Gillihan SJ, Bryant RA. Challenges and successes in dissemination of evidence-based treatments for posttraumatic stress: lessons learned from prolonged exposure therapy for PTSD. Psychological Science in the Public Interest. 2013;14(2):65-111.
3. Rahman A, Malik A, Sikander S, et al. Cognitive behaviour therapy-based intervention by community health workers for mothers with depression and their infants in rural Pakistan: a cluster-randomised controlled trial. Lancet. 2008;372(9642):902-909.
4. Aguilera A, Muench F. There’s an app for that: information technology applications for cognitive behavioral practitioners. Behavior Therapist. 2012;35(4):65-73.
5. Dimidjian S, Hollon SD, Dobson KS, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the acute treatment of adults with major depression. J Consult Clin Psychol. 2006; 74(4):658-670.
6. Hollon SD, Jarrett RB, Nierenberg AA, et al. Psychotherapy and medication in the treatment of adult and geriatric depression: which monotherapy or combined treatment? J Clin Psychiatry. 2005;66(4):455-468.
7. Cuijpers P, Hollon SD, van Straten A, et al. Does cognitive behaviour therapy have an enduring effect that is superior to keeping patients on continuation pharmacotherapy? A meta-analysis. BMJ Open. 2013;3(4):1-8.
8. Stewart R, Chambless D. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77(4): 595-606.
9. Wright JH, Basco MR, Thase M. Learning cognitive behavior therapy: an illustrated guide. Arlington, VA: American Psychiatric Publishing; 2006.
10. Barlow DH, Craske MG. Mastery of your anxiety and panic. 4th ed. New York, NY: Oxford University Press, Inc.; 2007.
11. Foa EB, Yadin E, Lichner TK. Exposure and response prevention for obsessive-compulsive disorder: therapist guide. New York, NY: Oxford University Press, Inc.; 2012.
12. Dugas MJ, Robichaud M. Cognitive-behavioral treatment for generalized anxiety disorder. New York, NY: Routledge; 2007.
13. Zlomke K, Davis TE. One-session treatment of specific phobias: a detailed description and review of treatment efficacy. Behav Ther. 2008;39(3):207-223.
14. Foa EB, Hembree E, Rothbaum B. Prolonged exposure therapy for PTSD: emotional processing of traumatic experiences. Therapist guide. New York, NY: Oxford University Press, Inc.; 2007.
15. Resick PA, Schnicke MK. Cognitive processing therapy for rape victims. London, United Kingdom: Sage Publications; 1996.
16. Whittal ML, Robichaud M, Woody SR. Cognitive treatment of obsessions: enhancing dissemination with video components. Cognitive and Behavioral Practice. 2010;17(1):1-8.
17. Westra H, Arkowitz H, Dozois D. Adding a motivational interviewing pretreatment to cognitive behavioral therapy for generalized anxiety disorder: a preliminary randomized controlled trial. J Anxiety Disord. 2009;23(2): 1106-1117.
18. Dobson KS. The science of CBT: toward a metacognitive model of change? Behav Ther. 2013;44(2):224-227.
19. Kabat-Zinn J. Full catastrophe living. Using the wisdom of your body and mind to face stress, pain, and illness. Revised edition. New York, NY: Bantam Books; 2013.
20. Hayes SC, Strosahl KD. Acceptance and commitment therapy. The process and practice of mindful change. 2nd ed. New York, NY: The Guilford Press; 2012.
21. Hofmann S, Sawyer A, Witt A, et al. The effect of mindfulness-based therapy on anxiety and depression: a meta-analytic review. J Consult Clin Psychol. 2010;78(2): 169-183.
22. Arch J, Eifert G, Davies C, et al. Randomized clinical trial of cognitive behavioral therapy (CBT) versus acceptance and commitment therapy (ACT) for mixed anxiety disorders. J Consult Clin Psychol. 2012;80(5):750-765.
23. Brown TA, Campbell LA, Lehman CL, et al. Current and lifetime comorbidity of the DSM-IV anxiety and mood disorders in a large clinical sample. J Abnorm Psychol. 2001;110(4):585-599.
24. Norton P, Barrera T. Transdiagnostic versus diagnosis-specific CBT for anxiety disorders: a preliminary randomized controlled noninferiority trial. Depress Anxiety. 2012;29(10):874-882.

Do biomarkers for Alzheimer’s disease have utility in everyday practice?
Guidelines for diagnosing Alzheimer’s disease (AD) are undergoing the first major changes since they were developed 30 years ago. The National Institute on Aging (NIA) and the Alzheimer’s Association (AA) have established workgroups to revise guidelines that were written in 1984.1
One of the major changes to these new guidelines is mention of research on biomarkers for diagnosing and monitoring progression of dementia in AD. This is an exciting and provocative development, but the questions practitioners who diagnose and treat AD should be asking are whether such biomarkers have utility in clinical practice today, or whether their application is a distant promise of continuing research.
Principles put forward in the guidelines
The new AD guidelines set forth in 3 major papers by the workgroups created by the NIA and AA include a change in nomenclature of AD.2 The workgroups have sought to define AD with specific stages that include:
• a preclinical/prodromal phase, in which the pathophysiology responsible for future cognitive changes is ongoing but lacks clinical manifestations3
• mild cognitive impairment, now considered a distinct entity from dementia and diagnosed when a person has early signs of AD; manifestations of impaired cognition in early disease are not significant enough to affect daily functioning.4
These newly formulated stages of AD rely on clinical judgment, and AD remains a clinical diagnosis. However, the new diagnostic guidelines include the use of biomarkers to measure disease progression.
Biomarkers of normal biologic function and pathology
The Biomarkers Definitions Working Group defines a biomarker as:
… a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.5
These characteristics include imaging studies and body fluids, such as serum and cerebrospinal fluid (CSF).
In AD, biomarkers are meant to measure the pathogenic processes of:
• accumulation and deposition of amyloid β _protein (Aβ42) plaques
• neuronal degeneration characterized by an increase in phosphorylated tau protein and neurofibrillary tangles.6
The purpose of these biomarkers is to identify ongoing disease and help the clinician stage patients who display a spectrum of symptoms.
Four classes of biomarkers (Table7)have been identified for use in the diagnosis of, and research on, AD:
• neuroimaging
• CSF
• serum
• genetic markers.

Neuroimaging
The basic purpose of CT and MRI of the head in the workup of cognitive impairment is to rule out a lesion in the brain, such as a tumor or hemorrhage, as the cause of, or contributor to, the impairment. Several neuroimaging studies are available to aid in diagnosing AD and distinguishing it from other causes of dementia, including:
• Fludeoxyglucose (FDG) positron-emission tomography (PET) scanning
• MRI
• Florbetapir F 18 Injection for PET.
FDG PET identifies areas of the brain in which glucose metabolism is decreased. This finding is thought to represent synaptic dysfunction.8 The true clinical utility of FDG PET appears to be as an aid in distinguishing cases of AD from frontotemporal dementia, by identifying regions of metabolic dysfunction.9 (Note: Medicare will reimburse for FDG PET only if 1) the patient has met diagnostic criteria for both AD and frontotemporal dementia for at least 6 months and 2) the cause of symptoms is uncertain.10)
FDG PET also can be useful in patients with mild cognitive impairment by identifying hypometabolism in the temporal and parietal regions of the brain years before clinical AD develops.In addition to FDG, 2 other imaging probes—Pittsburgh compound and 2-(1-{6-[(2-[fluorine-18]fluoroethyl)(methyl) amino]-2-naphthyl}-ethylidene) malononitrile (more commonly, FDDNP)—have been used with PET as research tools to demonstrate evidence of AD.11
MRI has been used to measure hippocampal atrophy and cortical thinning that occurs as a patient progresses from normal cognitive function or mild cognitive impairment to full dementia.5 The degree of atrophy has not been well correlated with the degree of functional impairment.
Florbetapir F 18 Injection was approved by the FDA in October 2013, under the brand name AMYViD, for measuring the quantity of Aβ42 deposition in the brain. When injected, this radiopharmaceutical binds to Aβ42 and can be detected on PET.12 Use criteria for AMYViD PET recently were developed13; the technique is indicated as an additional diagnostic tool for ruling out AD.
A negative AMYViD scan indicates sparse or no Aβ42 plaques, and is inconsistent with AD. However, a positive AMYViD scan does not establish a diagnosis of AD or other cognitive disorder.14 This lack of specificity decreases the potential utility of the scan in clinical practice.
Use of AMYViD PET in general practice also is constrained by cost, which varies by location, based on the fee for the PET scan ($1,000 to $3,000)15; to that, add the cost of a dose of AMYViD ($1,600, wholesale).16 The technique is not reimbursable, and the total out-of-pocket expense can be as much as $5,000—making an AMYViD PET prohibitive.
Cerebrospinal fluid markers
CSF biomarkers used in the evaluation of AD are Aβ42, t-tau protein, and p-tau protein.6,17 It is generally thought that the level of Aβ42 in CSF decreases in AD—indicative of Aβ42 being deposited in the brain.8 Tau proteins are elevated in CSF as neurons are destroyed. P-tau is associated with the neurofibrillary tangles of AD; its presence in CSF is thought to represent an increase in those tangles. The combination of a low level of Aβ42 and an elevated level of p-tau in CSF is considered the signature CSF biomarker of AD.6
Serum markers
The search for reliable serum biomarkers of AD is the area of greatest research interest because a blood test is a less invasive form of screening. Regrettably, the utility of serum biomarkers for clinical practice has not been established.
Aβ42 can be measured in serum, but levels do not correlate well with CSF levels.18 Other serum markers that have been evaluated for clinical utility include measures of lipid metabolism, oxidation, and inflammation. With none of these is there clear correlation between the level of protein and AD.18
Fourth front: Genetics
Several alleles are associated with AD. Mutations in amyloid precursor protein, presenilin 1, and presenilin 2 have been shown to cause a change in the processing of Aβ42 and thus lead to AD.19 These mutations are inherited in an autosomal-dominant fashion and are detected in early-onset (age <65) AD.
Mutations in apolipoprotein 4-β4 also has been the subject of much research; this allele usually is associated with increased risk of the more common, later-onset AD.20 Some evidence suggests that apolipoprotein 4-β4 carriers who develop AD might be at risk of earlier onset of symptoms, compared to noncarriers,21 but the clinical significance of that increased risk has not been established.
What utility do biomarkers have?
As we said at the beginning of this article, the question that clinicians should be asking is: “What is the current clinical utility of these sophisticated biomarkers and genetic testing?”
The answer is “little utility.” Diagnosing AD is a clinical enterprise, with, as we’ve outlined, specific and narrow exceptions.
Recently, researchers demonstrated biomarker evidence of AD before symptom onset in patients who have known autosomal-dominant gene mutations for AD.19 There is no evidence, however, that these biomarkers are useful for screening the general population to identify people who 1) are at risk of, or who have, AD and 2) do not have AD.
That being said, CSF and imaging biomarkers of AD are being used in clinical settings in some European countries to aid investigation of cognitive decline.
In conclusion
Here are key points to take away from this discussion of biomarkers of AD:
• The utility of these biomarkers today is in research—although some of them might, on occasion, be useful to distinguish dementia caused by AD from other dementias.
• The ultimate goal of research is to uncover a serum biomarker that can identify patients in the preclinical/prodromal stage of AD, so that disease-modifying therapies and preventive measures can be initiated before symptoms manifest.
• Science is a long way from making this goal a reality, but recent changes in the diagnostic criteria for AD will encourage research in this area of study.
Bottom Line
Researchers are working to uncover biomarkers that will identify patients in the preclinical or prodromal stage of Alzheimer’s disease, but diagnosis remains clinical. Recent changes to diagnostic criteria will encourage research in this area.
Related Resources
• Blennow K, Dubois B, Fagan AM, et al. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease [published online May 5, 2014]. Alzheimers Dement. doi: 10.1016/j.jalz.2014.02.004.
• Chase A. Alzheimer disease: Advances in imaging of AD biomarkers could aid early diagnosis. Nat Rev Neurol. 2014;10(5):239.
• De Riva V, Galloni E, Marcon M, et al. Analysis of combined CSF biomarkers in AD diagnosis. Clin Lab. 2014;60(4):629-634.
• Kristofikova Z, Ricny J, Kolarova M, et al. Interactions between amyloid-β and tau in cerebrospinal fluid of people with mild cognitive impairment and Alzheimer’s disease [published online March 26, 2014]. J Alzheimers Dis. doi: 10.3233/ JAD-132393.
Drug Brand Name
Florbetapir F 18 Injection • AMYViD
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):257-262.
2. McKhann GM, Knopman DS. Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
3. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
4. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):270-279.
5. Cummings JL. Biomarkers in Alzheimer’s disease– perspectives for the future. US Neurology. 2010;6(1):23-27.
6. Sperling R, Keith J. Biomarkers of Alzheimer disease: current and future applications to diagnostic criteria. Continuum (Minneap Minn). 2013;19(2 Dementia):325-338.
7. Craig-Shapiro R, Fagan AM, Holtzman DM. Biomarkers of Alzheimer’s disease. Neurobiol Dis. 2009;35(2):128-140.
8. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119-128.
9. Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130(pt 10):2616-2635.
10. National Coverage Determination (NCD) for FDG PET for Dementia and Neurodegenerative Diseases (220.6.13). Centers for Medicare and Medicaid Services. http://www. cms.gov/medicare-coverage-database/details/ncd-details. aspx?NCDId=288&ncdver=3&bc=BAABAAAAAAAA&. Accessed May 9, 2014.
11. Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161-172. 12. Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir- PET for imaging beta-amyloid pathology. JAMA. 2011;305(3): 275-283.
13. Johnson KA, Minoshima S, Bohnen NI, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid Imaging Task Force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimers Dement. 2013;9(4):e106-e109.
14. AMYViD [package insert]. Indianapolis, IN: Eli Lilly & Co; 2012.
15. First guidelines published for brain amyloid imaging in Alzheimer’s. Alzheimer’s Association. http://www.alz.org/ news_and_events_60578.asp. Published January 28, 2013. Accessed May 9, 2014.
16. Zakaib GD. FDA approves Amyvid for clinical use. Alzforum. http://www.alzforum.org/news/research-news/ fda-approves-amyvid-clinical-use. Published April 9, 2012. Accessed May 16, 2014.
17. Skillbäck T, Zetterberg H, Blennow K, et al. Cerebrospinal fluid biomarkers for Alzheimer disease and subcortical axonal damage in 5,542 clinical samples. Alzheimers Res Ther. 2013;5(5):47.
18. Irizarry MC. Biomarkers of Alzheimer disease in plasma. NeuroRx. 2004;1(2):226-234.
19. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795-804.
20. Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics. 2007;39(1):17-23.
21. Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53-77.
Guidelines for diagnosing Alzheimer’s disease (AD) are undergoing the first major changes since they were developed 30 years ago. The National Institute on Aging (NIA) and the Alzheimer’s Association (AA) have established workgroups to revise guidelines that were written in 1984.1
One of the major changes to these new guidelines is mention of research on biomarkers for diagnosing and monitoring progression of dementia in AD. This is an exciting and provocative development, but the questions practitioners who diagnose and treat AD should be asking are whether such biomarkers have utility in clinical practice today, or whether their application is a distant promise of continuing research.
Principles put forward in the guidelines
The new AD guidelines set forth in 3 major papers by the workgroups created by the NIA and AA include a change in nomenclature of AD.2 The workgroups have sought to define AD with specific stages that include:
• a preclinical/prodromal phase, in which the pathophysiology responsible for future cognitive changes is ongoing but lacks clinical manifestations3
• mild cognitive impairment, now considered a distinct entity from dementia and diagnosed when a person has early signs of AD; manifestations of impaired cognition in early disease are not significant enough to affect daily functioning.4
These newly formulated stages of AD rely on clinical judgment, and AD remains a clinical diagnosis. However, the new diagnostic guidelines include the use of biomarkers to measure disease progression.
Biomarkers of normal biologic function and pathology
The Biomarkers Definitions Working Group defines a biomarker as:
… a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.5
These characteristics include imaging studies and body fluids, such as serum and cerebrospinal fluid (CSF).
In AD, biomarkers are meant to measure the pathogenic processes of:
• accumulation and deposition of amyloid β _protein (Aβ42) plaques
• neuronal degeneration characterized by an increase in phosphorylated tau protein and neurofibrillary tangles.6
The purpose of these biomarkers is to identify ongoing disease and help the clinician stage patients who display a spectrum of symptoms.
Four classes of biomarkers (Table7)have been identified for use in the diagnosis of, and research on, AD:
• neuroimaging
• CSF
• serum
• genetic markers.
Neuroimaging
The basic purpose of CT and MRI of the head in the workup of cognitive impairment is to rule out a lesion in the brain, such as a tumor or hemorrhage, as the cause of, or contributor to, the impairment. Several neuroimaging studies are available to aid in diagnosing AD and distinguishing it from other causes of dementia, including:
• Fludeoxyglucose (FDG) positron-emission tomography (PET) scanning
• MRI
• Florbetapir F 18 Injection for PET.
FDG PET identifies areas of the brain in which glucose metabolism is decreased. This finding is thought to represent synaptic dysfunction.8 The true clinical utility of FDG PET appears to be as an aid in distinguishing cases of AD from frontotemporal dementia, by identifying regions of metabolic dysfunction.9 (Note: Medicare will reimburse for FDG PET only if 1) the patient has met diagnostic criteria for both AD and frontotemporal dementia for at least 6 months and 2) the cause of symptoms is uncertain.10)
FDG PET also can be useful in patients with mild cognitive impairment by identifying hypometabolism in the temporal and parietal regions of the brain years before clinical AD develops.In addition to FDG, 2 other imaging probes—Pittsburgh compound and 2-(1-{6-[(2-[fluorine-18]fluoroethyl)(methyl) amino]-2-naphthyl}-ethylidene) malononitrile (more commonly, FDDNP)—have been used with PET as research tools to demonstrate evidence of AD.11
MRI has been used to measure hippocampal atrophy and cortical thinning that occurs as a patient progresses from normal cognitive function or mild cognitive impairment to full dementia.5 The degree of atrophy has not been well correlated with the degree of functional impairment.
Florbetapir F 18 Injection was approved by the FDA in October 2013, under the brand name AMYViD, for measuring the quantity of Aβ42 deposition in the brain. When injected, this radiopharmaceutical binds to Aβ42 and can be detected on PET.12 Use criteria for AMYViD PET recently were developed13; the technique is indicated as an additional diagnostic tool for ruling out AD.
A negative AMYViD scan indicates sparse or no Aβ42 plaques, and is inconsistent with AD. However, a positive AMYViD scan does not establish a diagnosis of AD or other cognitive disorder.14 This lack of specificity decreases the potential utility of the scan in clinical practice.
Use of AMYViD PET in general practice also is constrained by cost, which varies by location, based on the fee for the PET scan ($1,000 to $3,000)15; to that, add the cost of a dose of AMYViD ($1,600, wholesale).16 The technique is not reimbursable, and the total out-of-pocket expense can be as much as $5,000—making an AMYViD PET prohibitive.
Cerebrospinal fluid markers
CSF biomarkers used in the evaluation of AD are Aβ42, t-tau protein, and p-tau protein.6,17 It is generally thought that the level of Aβ42 in CSF decreases in AD—indicative of Aβ42 being deposited in the brain.8 Tau proteins are elevated in CSF as neurons are destroyed. P-tau is associated with the neurofibrillary tangles of AD; its presence in CSF is thought to represent an increase in those tangles. The combination of a low level of Aβ42 and an elevated level of p-tau in CSF is considered the signature CSF biomarker of AD.6
Serum markers
The search for reliable serum biomarkers of AD is the area of greatest research interest because a blood test is a less invasive form of screening. Regrettably, the utility of serum biomarkers for clinical practice has not been established.
Aβ42 can be measured in serum, but levels do not correlate well with CSF levels.18 Other serum markers that have been evaluated for clinical utility include measures of lipid metabolism, oxidation, and inflammation. With none of these is there clear correlation between the level of protein and AD.18
Fourth front: Genetics
Several alleles are associated with AD. Mutations in amyloid precursor protein, presenilin 1, and presenilin 2 have been shown to cause a change in the processing of Aβ42 and thus lead to AD.19 These mutations are inherited in an autosomal-dominant fashion and are detected in early-onset (age <65) AD.
Mutations in apolipoprotein 4-β4 also has been the subject of much research; this allele usually is associated with increased risk of the more common, later-onset AD.20 Some evidence suggests that apolipoprotein 4-β4 carriers who develop AD might be at risk of earlier onset of symptoms, compared to noncarriers,21 but the clinical significance of that increased risk has not been established.
What utility do biomarkers have?
As we said at the beginning of this article, the question that clinicians should be asking is: “What is the current clinical utility of these sophisticated biomarkers and genetic testing?”
The answer is “little utility.” Diagnosing AD is a clinical enterprise, with, as we’ve outlined, specific and narrow exceptions.
Recently, researchers demonstrated biomarker evidence of AD before symptom onset in patients who have known autosomal-dominant gene mutations for AD.19 There is no evidence, however, that these biomarkers are useful for screening the general population to identify people who 1) are at risk of, or who have, AD and 2) do not have AD.
That being said, CSF and imaging biomarkers of AD are being used in clinical settings in some European countries to aid investigation of cognitive decline.
In conclusion
Here are key points to take away from this discussion of biomarkers of AD:
• The utility of these biomarkers today is in research—although some of them might, on occasion, be useful to distinguish dementia caused by AD from other dementias.
• The ultimate goal of research is to uncover a serum biomarker that can identify patients in the preclinical/prodromal stage of AD, so that disease-modifying therapies and preventive measures can be initiated before symptoms manifest.
• Science is a long way from making this goal a reality, but recent changes in the diagnostic criteria for AD will encourage research in this area of study.
Bottom Line
Researchers are working to uncover biomarkers that will identify patients in the preclinical or prodromal stage of Alzheimer’s disease, but diagnosis remains clinical. Recent changes to diagnostic criteria will encourage research in this area.
Related Resources
• Blennow K, Dubois B, Fagan AM, et al. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease [published online May 5, 2014]. Alzheimers Dement. doi: 10.1016/j.jalz.2014.02.004.
• Chase A. Alzheimer disease: Advances in imaging of AD biomarkers could aid early diagnosis. Nat Rev Neurol. 2014;10(5):239.
• De Riva V, Galloni E, Marcon M, et al. Analysis of combined CSF biomarkers in AD diagnosis. Clin Lab. 2014;60(4):629-634.
• Kristofikova Z, Ricny J, Kolarova M, et al. Interactions between amyloid-β and tau in cerebrospinal fluid of people with mild cognitive impairment and Alzheimer’s disease [published online March 26, 2014]. J Alzheimers Dis. doi: 10.3233/ JAD-132393.
Drug Brand Name
Florbetapir F 18 Injection • AMYViD
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Guidelines for diagnosing Alzheimer’s disease (AD) are undergoing the first major changes since they were developed 30 years ago. The National Institute on Aging (NIA) and the Alzheimer’s Association (AA) have established workgroups to revise guidelines that were written in 1984.1
One of the major changes to these new guidelines is mention of research on biomarkers for diagnosing and monitoring progression of dementia in AD. This is an exciting and provocative development, but the questions practitioners who diagnose and treat AD should be asking are whether such biomarkers have utility in clinical practice today, or whether their application is a distant promise of continuing research.
Principles put forward in the guidelines
The new AD guidelines set forth in 3 major papers by the workgroups created by the NIA and AA include a change in nomenclature of AD.2 The workgroups have sought to define AD with specific stages that include:
• a preclinical/prodromal phase, in which the pathophysiology responsible for future cognitive changes is ongoing but lacks clinical manifestations3
• mild cognitive impairment, now considered a distinct entity from dementia and diagnosed when a person has early signs of AD; manifestations of impaired cognition in early disease are not significant enough to affect daily functioning.4
These newly formulated stages of AD rely on clinical judgment, and AD remains a clinical diagnosis. However, the new diagnostic guidelines include the use of biomarkers to measure disease progression.
Biomarkers of normal biologic function and pathology
The Biomarkers Definitions Working Group defines a biomarker as:
… a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.5
These characteristics include imaging studies and body fluids, such as serum and cerebrospinal fluid (CSF).
In AD, biomarkers are meant to measure the pathogenic processes of:
• accumulation and deposition of amyloid β _protein (Aβ42) plaques
• neuronal degeneration characterized by an increase in phosphorylated tau protein and neurofibrillary tangles.6
The purpose of these biomarkers is to identify ongoing disease and help the clinician stage patients who display a spectrum of symptoms.
Four classes of biomarkers (Table7)have been identified for use in the diagnosis of, and research on, AD:
• neuroimaging
• CSF
• serum
• genetic markers.
Neuroimaging
The basic purpose of CT and MRI of the head in the workup of cognitive impairment is to rule out a lesion in the brain, such as a tumor or hemorrhage, as the cause of, or contributor to, the impairment. Several neuroimaging studies are available to aid in diagnosing AD and distinguishing it from other causes of dementia, including:
• Fludeoxyglucose (FDG) positron-emission tomography (PET) scanning
• MRI
• Florbetapir F 18 Injection for PET.
FDG PET identifies areas of the brain in which glucose metabolism is decreased. This finding is thought to represent synaptic dysfunction.8 The true clinical utility of FDG PET appears to be as an aid in distinguishing cases of AD from frontotemporal dementia, by identifying regions of metabolic dysfunction.9 (Note: Medicare will reimburse for FDG PET only if 1) the patient has met diagnostic criteria for both AD and frontotemporal dementia for at least 6 months and 2) the cause of symptoms is uncertain.10)
FDG PET also can be useful in patients with mild cognitive impairment by identifying hypometabolism in the temporal and parietal regions of the brain years before clinical AD develops.In addition to FDG, 2 other imaging probes—Pittsburgh compound and 2-(1-{6-[(2-[fluorine-18]fluoroethyl)(methyl) amino]-2-naphthyl}-ethylidene) malononitrile (more commonly, FDDNP)—have been used with PET as research tools to demonstrate evidence of AD.11
MRI has been used to measure hippocampal atrophy and cortical thinning that occurs as a patient progresses from normal cognitive function or mild cognitive impairment to full dementia.5 The degree of atrophy has not been well correlated with the degree of functional impairment.
Florbetapir F 18 Injection was approved by the FDA in October 2013, under the brand name AMYViD, for measuring the quantity of Aβ42 deposition in the brain. When injected, this radiopharmaceutical binds to Aβ42 and can be detected on PET.12 Use criteria for AMYViD PET recently were developed13; the technique is indicated as an additional diagnostic tool for ruling out AD.
A negative AMYViD scan indicates sparse or no Aβ42 plaques, and is inconsistent with AD. However, a positive AMYViD scan does not establish a diagnosis of AD or other cognitive disorder.14 This lack of specificity decreases the potential utility of the scan in clinical practice.
Use of AMYViD PET in general practice also is constrained by cost, which varies by location, based on the fee for the PET scan ($1,000 to $3,000)15; to that, add the cost of a dose of AMYViD ($1,600, wholesale).16 The technique is not reimbursable, and the total out-of-pocket expense can be as much as $5,000—making an AMYViD PET prohibitive.
Cerebrospinal fluid markers
CSF biomarkers used in the evaluation of AD are Aβ42, t-tau protein, and p-tau protein.6,17 It is generally thought that the level of Aβ42 in CSF decreases in AD—indicative of Aβ42 being deposited in the brain.8 Tau proteins are elevated in CSF as neurons are destroyed. P-tau is associated with the neurofibrillary tangles of AD; its presence in CSF is thought to represent an increase in those tangles. The combination of a low level of Aβ42 and an elevated level of p-tau in CSF is considered the signature CSF biomarker of AD.6
Serum markers
The search for reliable serum biomarkers of AD is the area of greatest research interest because a blood test is a less invasive form of screening. Regrettably, the utility of serum biomarkers for clinical practice has not been established.
Aβ42 can be measured in serum, but levels do not correlate well with CSF levels.18 Other serum markers that have been evaluated for clinical utility include measures of lipid metabolism, oxidation, and inflammation. With none of these is there clear correlation between the level of protein and AD.18
Fourth front: Genetics
Several alleles are associated with AD. Mutations in amyloid precursor protein, presenilin 1, and presenilin 2 have been shown to cause a change in the processing of Aβ42 and thus lead to AD.19 These mutations are inherited in an autosomal-dominant fashion and are detected in early-onset (age <65) AD.
Mutations in apolipoprotein 4-β4 also has been the subject of much research; this allele usually is associated with increased risk of the more common, later-onset AD.20 Some evidence suggests that apolipoprotein 4-β4 carriers who develop AD might be at risk of earlier onset of symptoms, compared to noncarriers,21 but the clinical significance of that increased risk has not been established.
What utility do biomarkers have?
As we said at the beginning of this article, the question that clinicians should be asking is: “What is the current clinical utility of these sophisticated biomarkers and genetic testing?”
The answer is “little utility.” Diagnosing AD is a clinical enterprise, with, as we’ve outlined, specific and narrow exceptions.
Recently, researchers demonstrated biomarker evidence of AD before symptom onset in patients who have known autosomal-dominant gene mutations for AD.19 There is no evidence, however, that these biomarkers are useful for screening the general population to identify people who 1) are at risk of, or who have, AD and 2) do not have AD.
That being said, CSF and imaging biomarkers of AD are being used in clinical settings in some European countries to aid investigation of cognitive decline.
In conclusion
Here are key points to take away from this discussion of biomarkers of AD:
• The utility of these biomarkers today is in research—although some of them might, on occasion, be useful to distinguish dementia caused by AD from other dementias.
• The ultimate goal of research is to uncover a serum biomarker that can identify patients in the preclinical/prodromal stage of AD, so that disease-modifying therapies and preventive measures can be initiated before symptoms manifest.
• Science is a long way from making this goal a reality, but recent changes in the diagnostic criteria for AD will encourage research in this area of study.
Bottom Line
Researchers are working to uncover biomarkers that will identify patients in the preclinical or prodromal stage of Alzheimer’s disease, but diagnosis remains clinical. Recent changes to diagnostic criteria will encourage research in this area.
Related Resources
• Blennow K, Dubois B, Fagan AM, et al. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease [published online May 5, 2014]. Alzheimers Dement. doi: 10.1016/j.jalz.2014.02.004.
• Chase A. Alzheimer disease: Advances in imaging of AD biomarkers could aid early diagnosis. Nat Rev Neurol. 2014;10(5):239.
• De Riva V, Galloni E, Marcon M, et al. Analysis of combined CSF biomarkers in AD diagnosis. Clin Lab. 2014;60(4):629-634.
• Kristofikova Z, Ricny J, Kolarova M, et al. Interactions between amyloid-β and tau in cerebrospinal fluid of people with mild cognitive impairment and Alzheimer’s disease [published online March 26, 2014]. J Alzheimers Dis. doi: 10.3233/ JAD-132393.
Drug Brand Name
Florbetapir F 18 Injection • AMYViD
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):257-262.
2. McKhann GM, Knopman DS. Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
3. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
4. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):270-279.
5. Cummings JL. Biomarkers in Alzheimer’s disease– perspectives for the future. US Neurology. 2010;6(1):23-27.
6. Sperling R, Keith J. Biomarkers of Alzheimer disease: current and future applications to diagnostic criteria. Continuum (Minneap Minn). 2013;19(2 Dementia):325-338.
7. Craig-Shapiro R, Fagan AM, Holtzman DM. Biomarkers of Alzheimer’s disease. Neurobiol Dis. 2009;35(2):128-140.
8. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119-128.
9. Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130(pt 10):2616-2635.
10. National Coverage Determination (NCD) for FDG PET for Dementia and Neurodegenerative Diseases (220.6.13). Centers for Medicare and Medicaid Services. http://www. cms.gov/medicare-coverage-database/details/ncd-details. aspx?NCDId=288&ncdver=3&bc=BAABAAAAAAAA&. Accessed May 9, 2014.
11. Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161-172. 12. Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir- PET for imaging beta-amyloid pathology. JAMA. 2011;305(3): 275-283.
13. Johnson KA, Minoshima S, Bohnen NI, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid Imaging Task Force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimers Dement. 2013;9(4):e106-e109.
14. AMYViD [package insert]. Indianapolis, IN: Eli Lilly & Co; 2012.
15. First guidelines published for brain amyloid imaging in Alzheimer’s. Alzheimer’s Association. http://www.alz.org/ news_and_events_60578.asp. Published January 28, 2013. Accessed May 9, 2014.
16. Zakaib GD. FDA approves Amyvid for clinical use. Alzforum. http://www.alzforum.org/news/research-news/ fda-approves-amyvid-clinical-use. Published April 9, 2012. Accessed May 16, 2014.
17. Skillbäck T, Zetterberg H, Blennow K, et al. Cerebrospinal fluid biomarkers for Alzheimer disease and subcortical axonal damage in 5,542 clinical samples. Alzheimers Res Ther. 2013;5(5):47.
18. Irizarry MC. Biomarkers of Alzheimer disease in plasma. NeuroRx. 2004;1(2):226-234.
19. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795-804.
20. Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics. 2007;39(1):17-23.
21. Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53-77.
1. Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):257-262.
2. McKhann GM, Knopman DS. Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
3. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
4. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):270-279.
5. Cummings JL. Biomarkers in Alzheimer’s disease– perspectives for the future. US Neurology. 2010;6(1):23-27.
6. Sperling R, Keith J. Biomarkers of Alzheimer disease: current and future applications to diagnostic criteria. Continuum (Minneap Minn). 2013;19(2 Dementia):325-338.
7. Craig-Shapiro R, Fagan AM, Holtzman DM. Biomarkers of Alzheimer’s disease. Neurobiol Dis. 2009;35(2):128-140.
8. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119-128.
9. Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130(pt 10):2616-2635.
10. National Coverage Determination (NCD) for FDG PET for Dementia and Neurodegenerative Diseases (220.6.13). Centers for Medicare and Medicaid Services. http://www. cms.gov/medicare-coverage-database/details/ncd-details. aspx?NCDId=288&ncdver=3&bc=BAABAAAAAAAA&. Accessed May 9, 2014.
11. Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161-172. 12. Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir- PET for imaging beta-amyloid pathology. JAMA. 2011;305(3): 275-283.
13. Johnson KA, Minoshima S, Bohnen NI, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid Imaging Task Force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimers Dement. 2013;9(4):e106-e109.
14. AMYViD [package insert]. Indianapolis, IN: Eli Lilly & Co; 2012.
15. First guidelines published for brain amyloid imaging in Alzheimer’s. Alzheimer’s Association. http://www.alz.org/ news_and_events_60578.asp. Published January 28, 2013. Accessed May 9, 2014.
16. Zakaib GD. FDA approves Amyvid for clinical use. Alzforum. http://www.alzforum.org/news/research-news/ fda-approves-amyvid-clinical-use. Published April 9, 2012. Accessed May 16, 2014.
17. Skillbäck T, Zetterberg H, Blennow K, et al. Cerebrospinal fluid biomarkers for Alzheimer disease and subcortical axonal damage in 5,542 clinical samples. Alzheimers Res Ther. 2013;5(5):47.
18. Irizarry MC. Biomarkers of Alzheimer disease in plasma. NeuroRx. 2004;1(2):226-234.
19. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795-804.
20. Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics. 2007;39(1):17-23.
21. Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53-77.

Lithium for bipolar disorder: A re-emerging treatment for mood instability
Lithium is among the most effective therapies for bipolar disorder (BD), and enthusiasm for this simple molecule is waxing. The history of lithium is fascinating,1 and recent considerations include that this element, the third on the periodic table, has few, if any, industry champions. The recent renaissance is caused by a groundswell of appreciation for the clinical efficacy of lithium and an increasing number of providers who are willing to manage patients with lithium.
Target: Bipolar disorder
The target illness for lithium is BD, a spectrum of mood disorders with characteristic features of unstable mood and affect. Shifts in mood include recurrent episodes of mania, which are pathologically energized states with misguided volition and behavior with intoxicating euphoria (or irritability).2 Psychomotor activity is elevated and out of character; speech and body movements are revved up, with a diminished need for sleep. The social, personal, and vocational consequences often are disastrous.
The most common mood state of BD is depression. Depressive episodes consist of pathologically compromised energy and volition with a slowing of bodily functions, most prominently cognition and concentration; a pervasive depressed or sad mood is common but not always present. Presence of mixed states, when features of depression and mania are present simultaneously, is one of the many challenges of treating BD; an elevated volitional or energized state may occur with a depressed, dysphoric mood.
Evidence for lithium
Efficacy studies of lithium have focused on managing mood disorders, treating mania and depression, and prevention or maintenance care.3 Most were performed during the 1970s and 1980s,3 but recent studies have been comparing lithium with other mood stabilizers4-7 and searching for a genetic basis for lithium response.8-10 Other researchers have examined the use of lithium to prevent suicide.11 Some have suggested a neuroprotective effect of lithium, which may have profound implications for neuropsychiatry if valid.12-14 Results of additional studies, which are at different stages of completion, will clarify lithium use,15,16 and characterize the genetic makeup of individuals who respond to lithium.17 The primary evidence for lithium, however, is for maintenance treatment of BD and for preventing manic and depressive episodes.
Biochemistry and physiology of lithium. The biochemical and physiological effects of lithium are complex, wide-ranging, and likely to affect hundreds, if not thousands, of genes and gene products. The mechanisms of action remain a focus of academic pursuit (for a review of hypotheses related to these mechanisms see Goodwin and Jamison2 and Can et al18) Lithium is involved in cell signaling pathways that involve complex molecular mechanisms of inter- and intracellular communication19; some neural receptors are down-regulated20 and others show inhibition,21 which is thought to be a mechanism of lithium. The hypothesized neuroprotective effect of lithium22 may be mediated through an increased level of brain-derived neurotrophic factor in brain tissue.14 Recently, investigators using induced pluripotent stem cell derived neurons have shown that patterns of calcium-related cell signaling in bipolar neurons are affected specifically by lithium in the culture media.23 There likely are many mechanisms through which lithium’s effects are mediated, including a series of dynamic pathways that vary over time and in reaction to the internal and external environments of the cell and person.
The lithium renaissance
In the past decade, there has been an increase in interest and use of lithium because clinicians recognize its efficacy and advantages and can monitor serum levels and gauge the patient’s response and side effects24 against the lithium level. This is important because balancing effi cacy and side effects depends on the serum level. Efficacy often is not immediate, although side effects may emerge early. All systems of the body may show effects that could be related to lithium use. It is helpful to be aware of the side effects in chronological order, because some immediate effects may be associated with starting at higher dosages (Table 1). Common side effects in the short term include:
• GI distress, such as nausea, vomiting, diarrhea, and abdominal discomfort
• a fine neurologic tremor, which may be seen with accentuation upon deliberate movement
• prominent thirst with polyuria
• drowsiness and clouded thinking, which can be upsetting to the patient and family.
In the longer term, adverse effects on kidney and thyroid function are common. Management must include monitoring of the serum level.
Lithium is FDA-approved for acute and maintenance treatment of mania in BD. There are reports that discuss most variants of mood disorders, including BD I, BD II, unipolar depression, rapid cycling, and even alcohol abuse.25-29 Lithium could help manage mood dysregulation in the context of temperament and personality.30 There is evidence that lithium has an antidepressant effect31-33 and has shown efficacy as an adjunctive treatment for depression.31-33 There are data that suggest that lithium, with its neuroprotective mechanisms, may prevent progression of mild cognitive impairment.34
Is there an ideal lithium candidate?
Mood instability is the characteristic feature of a lithium responder. The instability may be over the course of the day, such as a dysregulated temperament that often is associated with DSM-IV personality categories, shorter-term fluctuations (within days with BD II), or in the context of episodic shifts of mood states over weeks and months, which are characteristic of BD I. The hallmark of mood instability is fluctuation from depression to elevated mood states and charged emotions with increased energy.
The patient considered ideal for lithium treatment has BD I with recurrent severe euphoric manic episodes, absence of significant comorbid disorders such as substance abuse, and a family history of lithium response. However, any patient with a clinically significant and unstable mood disorder, regardless of the DSM diagnosis, should be considered for lithium treatment.
When considering a lithium trial for a patient with significant mood instability, it is critical to establish the target symptoms and behavior that will help you gauge the efficacy of the intervention. Measurement-based care utilizes clinician and self-report instruments to provide data on the illness course and response to intervention. Commonly used clinician driven assessments include the Young Mania Rating Scale35 and the Quick Inventory of Depressive Symptoms,36 while the self-report assessments are the Patient Health Questionnaire37 and the Altman Self- Rating Mania Scale.38
During acute mania or depression, lithium often is used in combination with another medications such as an antipsychotic or antidepressant. Used in the outpatient and non-acute setting, lithium may be an “add-on” or monotherapy for preventing recurrence of episodes. Response in early acute manic symptoms are predictive of later response and remission.39
Dosing strategies
An initial problem with lithium is side effects that emerge when beginning treatment, which may discourage the patient and family from using this agent. Starting with 150 mg/d for the first 2 or 3 doses is unlikely to produce any adverse effects and can show the patient that there is a high likelihood that he will be able to tolerate the medication. Gradual titration over several days—or even weeks—to the target dosage and serum levels will enhance patient compliance. Rate of dosage increase is best guided by tolerance to the medication. The general consensus is that lithium is most effective at levels of 0.6 to 0.8 mEq/L,40 although a lower level (0.5 mEq/L) over a 2-year period also can be effective.41 Lithium may be used in to treat acute mania at higher serum levels (0.8 to 1.2 mEq/L), however, the acute phase often requires urgent management, usually with an antipsychotic.
Emerging consensus
Although there is a need to gather and analyze longer observational periods to clarify the clinical and biological characteristics of persons who respond to lithium, there are several points of consensus. Management will be guided by patient characteristics such as age, comorbidities, and other therapies. Most studies that address the effect of lithium level focus on high vs low serum levels. There are 3 categories of lithium serum levels, low (<0.6 mEq/L), mid-range (0.6 to 0.8 mEq/L), and high (>0.8 mEq/L), each has risk-benefit considerations.
The LiTMUS study42 compared low-level lithium augmentation with optimized personal treatment without lithium. Both groups had similar outcomes but the lithium-treated group had significantly lower use of atypical antipsychotics. This may be important when considering the long-term risk of the metabolic syndrome because the tolerability and side-effect profile of lithium at lower levels is more favorable than that of atypical antipsychotics. As lithium levels increase, there seems to be concomitant increase in efficacy and side effects. Many patients will benefit with low-level lithium use; yet clearly some individuals require higher dosages for effective maintenance therapy.
Dosing and monitoring. In patients age >50 or those with comorbid medical conditions, use a lower level of lithium (<0.6 mEq/L). Most individuals with BD likely will benefit from the mid-range level strategy (0.6 to 0.8 mEq/L); however, there will be those who require a higher level. When beginning lithium, start at a low dosage (150 mg/d) and increase as tolerated to the desired serum level. With acute mania, temporary use of an antipsychotic will be required.
There are no tests available to determine whether a patient will do well at any of these lithium serum levels. Breakthrough mania in an adherent patient with a serum lithium level of 0.7 mEq/L indicates the need to obtain a higher lithium level. A major deficit in lithium research is the lack of long-term data (>5 years) on outcomes, clinical and biological features with lithium levels because of a lack of pharmaceutical company support.3,17 Monitoring mood symptoms using detailed mood charts, whether clinician-administered or self-reported, is an effective way to monitor outcomes, provided the clinician uses the same scales or methods to record a patient’s moods. If a patient wants to discontinue lithium, taper the drug over an extended period (months) to minimize the likelihood of emerging manic or depressive episodes related to drug discontinuation.
Managing side effects
Consider lithium’s side effects in the context of their short-, intermediate-, and long-term presence (Table 2). Gradually increasing the lithium dosage often will prevent side effects that manifest in the short term. If side effects emerge at low dosages, proceed slowly with lithium and manage symptoms with other medications. When a patient shows a change in side effects, obtain lithium and electrolytes levels; a change in mental status with confusion will require an acute lithium level. 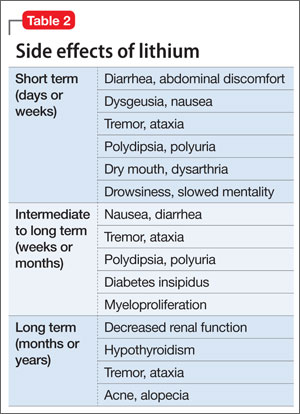
A diary of symptoms or clinically relevant matters such as fluid intake or frequency of GI- or neurological-related events will help the clinician monitor the frequency and severity of side effects. The patient and clinician should not be discouraged by emerging side effects in the short term, because they may dissipate or become minimally intrusive.
Several strategies can alleviate immediate GI effects, such as dosing with meals, enteric-coated formulations, multiple dose strategies, and short-term use of antidiarrheal medicine as needed. Side effects that disrupt a patient’s fluid and electrolyte balance (diabetes insipidus) to the point of clouding mental status will require discontinuing the medication until mental status improves, then reconsideration of the treatment regime, which will include managing diabetes insipidus with amiloride. Managing side effects may require consultation with specialty services. Likewise, some patients might experience neurologic side effects, such as profound tremor, that interferes with their ability to function. However, many side effects can be managed symptomatically with practical strategies (eg, a sugar-free lozenge for dry mouth or dysgeusia). Consider lower lithium dosages and serum levels because patients may experience benefits with lower therapeutic levels.
Long-term side effects include decreased renal function, hypothyroidism, persistent tremor, and dermatologic effects of acne and alopecia. Monitor renal and thyroid function annually in stable patients and more frequently when making changes in the treatment plan.
Before discontinuing lithium, consider discussing the medical issues with a specialist who has experience with complications of lithium.
Bottom Line
Lithium is an effective and under used medication for managing bipolar disorder. Initial prejudices and side effects often deter patients and prescribers from proceeding with a therapeutic trial of lithium. Although the mid-range lithium level of 0.6 to 0.8 mEq/L is desirable, many patients will experience significant benefits with lower levels. Initial strategies include the use of low-dose preparations that are unlikely to have uncomfortable side effects.
Related Resources
• Andreasen A, Ellingrod VL. Lithium-induced diabetes insipidus: prevention and management. Current Psychiatry. 2013;12(7):42-45.
• Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646.
Drug Brand Names
Amiloride • Midamor Lithium • Eskalith, Lithobid
Disclosure
Dr. McInnis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11(11 suppl 2):4-9.
2. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007.
3. Burgess S, Geddes J, Hawton K, et al. Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev. 2001:CD003013.
4. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex maintenance study group. Arch Gen Psychiatry. 2000;57(5):481-489.
5. Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
6. Swann AC, Bowden CL, Calabrese JR, et al. Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology. 2002;26(4):530-536.
7. Tohen M, Chengappa KN, Suppes T, et al. Efficacy of olanzapine in combination with valproate or lithium in the treatment of mania in patients partially nonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry. 2002;59(1):62-69.
8. Perlis RH, Smoller JW, Ferreira MA, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009; 166(6):718-725.
9. Grof P, Duffy A, Cavazzoni P, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63(10): 942-947.
10. Duffy A, Alda M, Kutcher S, et al. A prospective study of the offspring of bipolar parents responsive and nonresponsive to lithium treatment. J Clin Psychiatry. 2002;63(12): 1171-1178.
11. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473.
12. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010; 62(1):50-60.
13. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
14. de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54-56.
15. Nierenberg AA, Sylvia LG, Leon AC, et al; LiTMUS Study Group. Lithium treatment–moderate dose use study (LiTMUS) for bipolar disorder: rationale and design. Clin Trials. 2009;6(6):637-648.
16. Sylvia LG, Reilly-Harrington NA, Leon AC, et al. Methods to limit attrition in longitudinal comparative effectiveness trials: lessons from the Lithium Treatment - Moderate dose Use Study (LiTMUS) for bipolar disorder. Clin Trials. 2012;9(1):94-101.
17. McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 2010;11(10):1439-1465.
18. Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy [published online February 15, 2014]. Pharmacol Biochem Behav. doi: 10.1016/j.pbb.2014.02.004.
19. Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1-21.
20. Devaki R, Shankar Rao S, Nadgir SM. The effect of lithium on the adrenoceptor-mediated second messenger system in the rat brain. J Psychiatry Neurosci. 2006;31(4):246-252.
21. Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1397-1411.
22. Hu LW, Kawamoto EM, Brietzke E, et al. The role of Wnt signaling and its interaction with diverse mechanisms of cellular apoptosis in the pathophysiology of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):11-17.
23. Chen HM, DeLong CJ, Bame M, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients [published online March 25, 2014]. Transl Psychiatry. doi:10.1038/tp.2014.12.
24. Jefferson JW. Lithium. In: Aronson JK, ed. Side effects of drugs annual, volume 26. Amsterdam, The Netherlands: Elsevier Science; 2003:19-29.
25. Baldessarini RJ, Tondo L, Floris G, et al. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord. 2000;61(2):13-22.
26. Baldessarini RJ, Tondo L, Hennen J, et al. Latency and episodes before treatment: response to lithium maintenance in bipolar I and II disorders. Bipolar Disord. 1999;1(2): 91-97.
27. Fieve RR, Kumbaraci T, Dunner DL. Lithium prophylaxis of depression in bipolar I, bipolar II, and unipolar patients. Am J Psychiatry. 1976;133(8):925-929.
28. Peck CC, Pond SM, Becker CE, et al. An evaluation of the effects of lithium in the treatment of chronic alcoholism. II. Assessment of the two-period crossover design. Alcohol Clin Exp Res. 1981;5(2):252-255.
29. Peselow ED, Dunner DL, Fieve RR, et al. Lithium prophylaxis of depression in unipolar, bipolar II, and cyclothymic patients. Am J Psychiatry. 1982;139(6):747-752.
30. Bellino S, Paradiso E, Bogetto F. Efficacy and tolerability of pharmacotherapies for borderline personality disorder. CNS Drugs. 2008;22(8):671-692.
31. Alevizos B, Alevizos E, Leonardou A, et al. Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatrike. 2012;23(2):143-148.
32. Goldberg JF, Sacks MH, Kocsis JH. Low-dose lithium augmentation of divalproex in geriatric mania. J Clin Psychiatry. 2000;61(4):304.
33. Saunders KE, Goodwin GM. New approaches in the treatment of bipolar depression. Curr Top Behav Neurosci. 2013;14:291-307.
34. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
35. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429-435.
36. Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82.
37. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
38. Altman EG, Hedeker D, Peterson JL, et al. The Altman Self- Rating Mania Scale. Biol Psychiatry. 1997;42(10):948-955.
39. Machado-Vieira R, Luckenbaugh DA, Soeiro-de-Souza MG, et al. Early improvement with lithium in classic mania and its association with later response. J Affect Disord. 2013;144(1-2):160-164.
40. Severus WE, Lipkovich IA, Licht RW, et al. In search of optimal lithium levels and olanzapine doses in the long-term treatment of bipolar I disorder. A post-hoc analysis of the maintenance study by Tohen et al. 2005. Eur Psychiatry. 2010;25(8):443-449.
41. Vestergaard P, Licht RW, Brodersen A, et al. Outcome of lithium prophylaxis: a prospective follow-up of affective disorder patients assigned to high and low serum lithium levels. Acta Psychiatr Scand. 1998;98(4):310-315.
42. Nierenberg AA, Friedman ES, Bowden CL, et al. Lithium treatment moderate-dose use study (LiTMUS) for bipolar disorder: a randomized comparative effectiveness trial of optimized personalized treatment with and without lithium. Am J Psychiatry. 2013;170(1):102-111.
Lithium is among the most effective therapies for bipolar disorder (BD), and enthusiasm for this simple molecule is waxing. The history of lithium is fascinating,1 and recent considerations include that this element, the third on the periodic table, has few, if any, industry champions. The recent renaissance is caused by a groundswell of appreciation for the clinical efficacy of lithium and an increasing number of providers who are willing to manage patients with lithium.
Target: Bipolar disorder
The target illness for lithium is BD, a spectrum of mood disorders with characteristic features of unstable mood and affect. Shifts in mood include recurrent episodes of mania, which are pathologically energized states with misguided volition and behavior with intoxicating euphoria (or irritability).2 Psychomotor activity is elevated and out of character; speech and body movements are revved up, with a diminished need for sleep. The social, personal, and vocational consequences often are disastrous.
The most common mood state of BD is depression. Depressive episodes consist of pathologically compromised energy and volition with a slowing of bodily functions, most prominently cognition and concentration; a pervasive depressed or sad mood is common but not always present. Presence of mixed states, when features of depression and mania are present simultaneously, is one of the many challenges of treating BD; an elevated volitional or energized state may occur with a depressed, dysphoric mood.
Evidence for lithium
Efficacy studies of lithium have focused on managing mood disorders, treating mania and depression, and prevention or maintenance care.3 Most were performed during the 1970s and 1980s,3 but recent studies have been comparing lithium with other mood stabilizers4-7 and searching for a genetic basis for lithium response.8-10 Other researchers have examined the use of lithium to prevent suicide.11 Some have suggested a neuroprotective effect of lithium, which may have profound implications for neuropsychiatry if valid.12-14 Results of additional studies, which are at different stages of completion, will clarify lithium use,15,16 and characterize the genetic makeup of individuals who respond to lithium.17 The primary evidence for lithium, however, is for maintenance treatment of BD and for preventing manic and depressive episodes.
Biochemistry and physiology of lithium. The biochemical and physiological effects of lithium are complex, wide-ranging, and likely to affect hundreds, if not thousands, of genes and gene products. The mechanisms of action remain a focus of academic pursuit (for a review of hypotheses related to these mechanisms see Goodwin and Jamison2 and Can et al18) Lithium is involved in cell signaling pathways that involve complex molecular mechanisms of inter- and intracellular communication19; some neural receptors are down-regulated20 and others show inhibition,21 which is thought to be a mechanism of lithium. The hypothesized neuroprotective effect of lithium22 may be mediated through an increased level of brain-derived neurotrophic factor in brain tissue.14 Recently, investigators using induced pluripotent stem cell derived neurons have shown that patterns of calcium-related cell signaling in bipolar neurons are affected specifically by lithium in the culture media.23 There likely are many mechanisms through which lithium’s effects are mediated, including a series of dynamic pathways that vary over time and in reaction to the internal and external environments of the cell and person.
The lithium renaissance
In the past decade, there has been an increase in interest and use of lithium because clinicians recognize its efficacy and advantages and can monitor serum levels and gauge the patient’s response and side effects24 against the lithium level. This is important because balancing effi cacy and side effects depends on the serum level. Efficacy often is not immediate, although side effects may emerge early. All systems of the body may show effects that could be related to lithium use. It is helpful to be aware of the side effects in chronological order, because some immediate effects may be associated with starting at higher dosages (Table 1). Common side effects in the short term include:
• GI distress, such as nausea, vomiting, diarrhea, and abdominal discomfort
• a fine neurologic tremor, which may be seen with accentuation upon deliberate movement
• prominent thirst with polyuria
• drowsiness and clouded thinking, which can be upsetting to the patient and family.
In the longer term, adverse effects on kidney and thyroid function are common. Management must include monitoring of the serum level.
Lithium is FDA-approved for acute and maintenance treatment of mania in BD. There are reports that discuss most variants of mood disorders, including BD I, BD II, unipolar depression, rapid cycling, and even alcohol abuse.25-29 Lithium could help manage mood dysregulation in the context of temperament and personality.30 There is evidence that lithium has an antidepressant effect31-33 and has shown efficacy as an adjunctive treatment for depression.31-33 There are data that suggest that lithium, with its neuroprotective mechanisms, may prevent progression of mild cognitive impairment.34
Is there an ideal lithium candidate?
Mood instability is the characteristic feature of a lithium responder. The instability may be over the course of the day, such as a dysregulated temperament that often is associated with DSM-IV personality categories, shorter-term fluctuations (within days with BD II), or in the context of episodic shifts of mood states over weeks and months, which are characteristic of BD I. The hallmark of mood instability is fluctuation from depression to elevated mood states and charged emotions with increased energy.
The patient considered ideal for lithium treatment has BD I with recurrent severe euphoric manic episodes, absence of significant comorbid disorders such as substance abuse, and a family history of lithium response. However, any patient with a clinically significant and unstable mood disorder, regardless of the DSM diagnosis, should be considered for lithium treatment.
When considering a lithium trial for a patient with significant mood instability, it is critical to establish the target symptoms and behavior that will help you gauge the efficacy of the intervention. Measurement-based care utilizes clinician and self-report instruments to provide data on the illness course and response to intervention. Commonly used clinician driven assessments include the Young Mania Rating Scale35 and the Quick Inventory of Depressive Symptoms,36 while the self-report assessments are the Patient Health Questionnaire37 and the Altman Self- Rating Mania Scale.38
During acute mania or depression, lithium often is used in combination with another medications such as an antipsychotic or antidepressant. Used in the outpatient and non-acute setting, lithium may be an “add-on” or monotherapy for preventing recurrence of episodes. Response in early acute manic symptoms are predictive of later response and remission.39
Dosing strategies
An initial problem with lithium is side effects that emerge when beginning treatment, which may discourage the patient and family from using this agent. Starting with 150 mg/d for the first 2 or 3 doses is unlikely to produce any adverse effects and can show the patient that there is a high likelihood that he will be able to tolerate the medication. Gradual titration over several days—or even weeks—to the target dosage and serum levels will enhance patient compliance. Rate of dosage increase is best guided by tolerance to the medication. The general consensus is that lithium is most effective at levels of 0.6 to 0.8 mEq/L,40 although a lower level (0.5 mEq/L) over a 2-year period also can be effective.41 Lithium may be used in to treat acute mania at higher serum levels (0.8 to 1.2 mEq/L), however, the acute phase often requires urgent management, usually with an antipsychotic.
Emerging consensus
Although there is a need to gather and analyze longer observational periods to clarify the clinical and biological characteristics of persons who respond to lithium, there are several points of consensus. Management will be guided by patient characteristics such as age, comorbidities, and other therapies. Most studies that address the effect of lithium level focus on high vs low serum levels. There are 3 categories of lithium serum levels, low (<0.6 mEq/L), mid-range (0.6 to 0.8 mEq/L), and high (>0.8 mEq/L), each has risk-benefit considerations.
The LiTMUS study42 compared low-level lithium augmentation with optimized personal treatment without lithium. Both groups had similar outcomes but the lithium-treated group had significantly lower use of atypical antipsychotics. This may be important when considering the long-term risk of the metabolic syndrome because the tolerability and side-effect profile of lithium at lower levels is more favorable than that of atypical antipsychotics. As lithium levels increase, there seems to be concomitant increase in efficacy and side effects. Many patients will benefit with low-level lithium use; yet clearly some individuals require higher dosages for effective maintenance therapy.
Dosing and monitoring. In patients age >50 or those with comorbid medical conditions, use a lower level of lithium (<0.6 mEq/L). Most individuals with BD likely will benefit from the mid-range level strategy (0.6 to 0.8 mEq/L); however, there will be those who require a higher level. When beginning lithium, start at a low dosage (150 mg/d) and increase as tolerated to the desired serum level. With acute mania, temporary use of an antipsychotic will be required.
There are no tests available to determine whether a patient will do well at any of these lithium serum levels. Breakthrough mania in an adherent patient with a serum lithium level of 0.7 mEq/L indicates the need to obtain a higher lithium level. A major deficit in lithium research is the lack of long-term data (>5 years) on outcomes, clinical and biological features with lithium levels because of a lack of pharmaceutical company support.3,17 Monitoring mood symptoms using detailed mood charts, whether clinician-administered or self-reported, is an effective way to monitor outcomes, provided the clinician uses the same scales or methods to record a patient’s moods. If a patient wants to discontinue lithium, taper the drug over an extended period (months) to minimize the likelihood of emerging manic or depressive episodes related to drug discontinuation.
Managing side effects
Consider lithium’s side effects in the context of their short-, intermediate-, and long-term presence (Table 2). Gradually increasing the lithium dosage often will prevent side effects that manifest in the short term. If side effects emerge at low dosages, proceed slowly with lithium and manage symptoms with other medications. When a patient shows a change in side effects, obtain lithium and electrolytes levels; a change in mental status with confusion will require an acute lithium level.
A diary of symptoms or clinically relevant matters such as fluid intake or frequency of GI- or neurological-related events will help the clinician monitor the frequency and severity of side effects. The patient and clinician should not be discouraged by emerging side effects in the short term, because they may dissipate or become minimally intrusive.
Several strategies can alleviate immediate GI effects, such as dosing with meals, enteric-coated formulations, multiple dose strategies, and short-term use of antidiarrheal medicine as needed. Side effects that disrupt a patient’s fluid and electrolyte balance (diabetes insipidus) to the point of clouding mental status will require discontinuing the medication until mental status improves, then reconsideration of the treatment regime, which will include managing diabetes insipidus with amiloride. Managing side effects may require consultation with specialty services. Likewise, some patients might experience neurologic side effects, such as profound tremor, that interferes with their ability to function. However, many side effects can be managed symptomatically with practical strategies (eg, a sugar-free lozenge for dry mouth or dysgeusia). Consider lower lithium dosages and serum levels because patients may experience benefits with lower therapeutic levels.
Long-term side effects include decreased renal function, hypothyroidism, persistent tremor, and dermatologic effects of acne and alopecia. Monitor renal and thyroid function annually in stable patients and more frequently when making changes in the treatment plan.
Before discontinuing lithium, consider discussing the medical issues with a specialist who has experience with complications of lithium.
Bottom Line
Lithium is an effective and under used medication for managing bipolar disorder. Initial prejudices and side effects often deter patients and prescribers from proceeding with a therapeutic trial of lithium. Although the mid-range lithium level of 0.6 to 0.8 mEq/L is desirable, many patients will experience significant benefits with lower levels. Initial strategies include the use of low-dose preparations that are unlikely to have uncomfortable side effects.
Related Resources
• Andreasen A, Ellingrod VL. Lithium-induced diabetes insipidus: prevention and management. Current Psychiatry. 2013;12(7):42-45.
• Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646.
Drug Brand Names
Amiloride • Midamor Lithium • Eskalith, Lithobid
Disclosure
Dr. McInnis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Lithium is among the most effective therapies for bipolar disorder (BD), and enthusiasm for this simple molecule is waxing. The history of lithium is fascinating,1 and recent considerations include that this element, the third on the periodic table, has few, if any, industry champions. The recent renaissance is caused by a groundswell of appreciation for the clinical efficacy of lithium and an increasing number of providers who are willing to manage patients with lithium.
Target: Bipolar disorder
The target illness for lithium is BD, a spectrum of mood disorders with characteristic features of unstable mood and affect. Shifts in mood include recurrent episodes of mania, which are pathologically energized states with misguided volition and behavior with intoxicating euphoria (or irritability).2 Psychomotor activity is elevated and out of character; speech and body movements are revved up, with a diminished need for sleep. The social, personal, and vocational consequences often are disastrous.
The most common mood state of BD is depression. Depressive episodes consist of pathologically compromised energy and volition with a slowing of bodily functions, most prominently cognition and concentration; a pervasive depressed or sad mood is common but not always present. Presence of mixed states, when features of depression and mania are present simultaneously, is one of the many challenges of treating BD; an elevated volitional or energized state may occur with a depressed, dysphoric mood.
Evidence for lithium
Efficacy studies of lithium have focused on managing mood disorders, treating mania and depression, and prevention or maintenance care.3 Most were performed during the 1970s and 1980s,3 but recent studies have been comparing lithium with other mood stabilizers4-7 and searching for a genetic basis for lithium response.8-10 Other researchers have examined the use of lithium to prevent suicide.11 Some have suggested a neuroprotective effect of lithium, which may have profound implications for neuropsychiatry if valid.12-14 Results of additional studies, which are at different stages of completion, will clarify lithium use,15,16 and characterize the genetic makeup of individuals who respond to lithium.17 The primary evidence for lithium, however, is for maintenance treatment of BD and for preventing manic and depressive episodes.
Biochemistry and physiology of lithium. The biochemical and physiological effects of lithium are complex, wide-ranging, and likely to affect hundreds, if not thousands, of genes and gene products. The mechanisms of action remain a focus of academic pursuit (for a review of hypotheses related to these mechanisms see Goodwin and Jamison2 and Can et al18) Lithium is involved in cell signaling pathways that involve complex molecular mechanisms of inter- and intracellular communication19; some neural receptors are down-regulated20 and others show inhibition,21 which is thought to be a mechanism of lithium. The hypothesized neuroprotective effect of lithium22 may be mediated through an increased level of brain-derived neurotrophic factor in brain tissue.14 Recently, investigators using induced pluripotent stem cell derived neurons have shown that patterns of calcium-related cell signaling in bipolar neurons are affected specifically by lithium in the culture media.23 There likely are many mechanisms through which lithium’s effects are mediated, including a series of dynamic pathways that vary over time and in reaction to the internal and external environments of the cell and person.
The lithium renaissance
In the past decade, there has been an increase in interest and use of lithium because clinicians recognize its efficacy and advantages and can monitor serum levels and gauge the patient’s response and side effects24 against the lithium level. This is important because balancing effi cacy and side effects depends on the serum level. Efficacy often is not immediate, although side effects may emerge early. All systems of the body may show effects that could be related to lithium use. It is helpful to be aware of the side effects in chronological order, because some immediate effects may be associated with starting at higher dosages (Table 1). Common side effects in the short term include:
• GI distress, such as nausea, vomiting, diarrhea, and abdominal discomfort
• a fine neurologic tremor, which may be seen with accentuation upon deliberate movement
• prominent thirst with polyuria
• drowsiness and clouded thinking, which can be upsetting to the patient and family.
In the longer term, adverse effects on kidney and thyroid function are common. Management must include monitoring of the serum level.
Lithium is FDA-approved for acute and maintenance treatment of mania in BD. There are reports that discuss most variants of mood disorders, including BD I, BD II, unipolar depression, rapid cycling, and even alcohol abuse.25-29 Lithium could help manage mood dysregulation in the context of temperament and personality.30 There is evidence that lithium has an antidepressant effect31-33 and has shown efficacy as an adjunctive treatment for depression.31-33 There are data that suggest that lithium, with its neuroprotective mechanisms, may prevent progression of mild cognitive impairment.34
Is there an ideal lithium candidate?
Mood instability is the characteristic feature of a lithium responder. The instability may be over the course of the day, such as a dysregulated temperament that often is associated with DSM-IV personality categories, shorter-term fluctuations (within days with BD II), or in the context of episodic shifts of mood states over weeks and months, which are characteristic of BD I. The hallmark of mood instability is fluctuation from depression to elevated mood states and charged emotions with increased energy.
The patient considered ideal for lithium treatment has BD I with recurrent severe euphoric manic episodes, absence of significant comorbid disorders such as substance abuse, and a family history of lithium response. However, any patient with a clinically significant and unstable mood disorder, regardless of the DSM diagnosis, should be considered for lithium treatment.
When considering a lithium trial for a patient with significant mood instability, it is critical to establish the target symptoms and behavior that will help you gauge the efficacy of the intervention. Measurement-based care utilizes clinician and self-report instruments to provide data on the illness course and response to intervention. Commonly used clinician driven assessments include the Young Mania Rating Scale35 and the Quick Inventory of Depressive Symptoms,36 while the self-report assessments are the Patient Health Questionnaire37 and the Altman Self- Rating Mania Scale.38
During acute mania or depression, lithium often is used in combination with another medications such as an antipsychotic or antidepressant. Used in the outpatient and non-acute setting, lithium may be an “add-on” or monotherapy for preventing recurrence of episodes. Response in early acute manic symptoms are predictive of later response and remission.39
Dosing strategies
An initial problem with lithium is side effects that emerge when beginning treatment, which may discourage the patient and family from using this agent. Starting with 150 mg/d for the first 2 or 3 doses is unlikely to produce any adverse effects and can show the patient that there is a high likelihood that he will be able to tolerate the medication. Gradual titration over several days—or even weeks—to the target dosage and serum levels will enhance patient compliance. Rate of dosage increase is best guided by tolerance to the medication. The general consensus is that lithium is most effective at levels of 0.6 to 0.8 mEq/L,40 although a lower level (0.5 mEq/L) over a 2-year period also can be effective.41 Lithium may be used in to treat acute mania at higher serum levels (0.8 to 1.2 mEq/L), however, the acute phase often requires urgent management, usually with an antipsychotic.
Emerging consensus
Although there is a need to gather and analyze longer observational periods to clarify the clinical and biological characteristics of persons who respond to lithium, there are several points of consensus. Management will be guided by patient characteristics such as age, comorbidities, and other therapies. Most studies that address the effect of lithium level focus on high vs low serum levels. There are 3 categories of lithium serum levels, low (<0.6 mEq/L), mid-range (0.6 to 0.8 mEq/L), and high (>0.8 mEq/L), each has risk-benefit considerations.
The LiTMUS study42 compared low-level lithium augmentation with optimized personal treatment without lithium. Both groups had similar outcomes but the lithium-treated group had significantly lower use of atypical antipsychotics. This may be important when considering the long-term risk of the metabolic syndrome because the tolerability and side-effect profile of lithium at lower levels is more favorable than that of atypical antipsychotics. As lithium levels increase, there seems to be concomitant increase in efficacy and side effects. Many patients will benefit with low-level lithium use; yet clearly some individuals require higher dosages for effective maintenance therapy.
Dosing and monitoring. In patients age >50 or those with comorbid medical conditions, use a lower level of lithium (<0.6 mEq/L). Most individuals with BD likely will benefit from the mid-range level strategy (0.6 to 0.8 mEq/L); however, there will be those who require a higher level. When beginning lithium, start at a low dosage (150 mg/d) and increase as tolerated to the desired serum level. With acute mania, temporary use of an antipsychotic will be required.
There are no tests available to determine whether a patient will do well at any of these lithium serum levels. Breakthrough mania in an adherent patient with a serum lithium level of 0.7 mEq/L indicates the need to obtain a higher lithium level. A major deficit in lithium research is the lack of long-term data (>5 years) on outcomes, clinical and biological features with lithium levels because of a lack of pharmaceutical company support.3,17 Monitoring mood symptoms using detailed mood charts, whether clinician-administered or self-reported, is an effective way to monitor outcomes, provided the clinician uses the same scales or methods to record a patient’s moods. If a patient wants to discontinue lithium, taper the drug over an extended period (months) to minimize the likelihood of emerging manic or depressive episodes related to drug discontinuation.
Managing side effects
Consider lithium’s side effects in the context of their short-, intermediate-, and long-term presence (Table 2). Gradually increasing the lithium dosage often will prevent side effects that manifest in the short term. If side effects emerge at low dosages, proceed slowly with lithium and manage symptoms with other medications. When a patient shows a change in side effects, obtain lithium and electrolytes levels; a change in mental status with confusion will require an acute lithium level.
A diary of symptoms or clinically relevant matters such as fluid intake or frequency of GI- or neurological-related events will help the clinician monitor the frequency and severity of side effects. The patient and clinician should not be discouraged by emerging side effects in the short term, because they may dissipate or become minimally intrusive.
Several strategies can alleviate immediate GI effects, such as dosing with meals, enteric-coated formulations, multiple dose strategies, and short-term use of antidiarrheal medicine as needed. Side effects that disrupt a patient’s fluid and electrolyte balance (diabetes insipidus) to the point of clouding mental status will require discontinuing the medication until mental status improves, then reconsideration of the treatment regime, which will include managing diabetes insipidus with amiloride. Managing side effects may require consultation with specialty services. Likewise, some patients might experience neurologic side effects, such as profound tremor, that interferes with their ability to function. However, many side effects can be managed symptomatically with practical strategies (eg, a sugar-free lozenge for dry mouth or dysgeusia). Consider lower lithium dosages and serum levels because patients may experience benefits with lower therapeutic levels.
Long-term side effects include decreased renal function, hypothyroidism, persistent tremor, and dermatologic effects of acne and alopecia. Monitor renal and thyroid function annually in stable patients and more frequently when making changes in the treatment plan.
Before discontinuing lithium, consider discussing the medical issues with a specialist who has experience with complications of lithium.
Bottom Line
Lithium is an effective and under used medication for managing bipolar disorder. Initial prejudices and side effects often deter patients and prescribers from proceeding with a therapeutic trial of lithium. Although the mid-range lithium level of 0.6 to 0.8 mEq/L is desirable, many patients will experience significant benefits with lower levels. Initial strategies include the use of low-dose preparations that are unlikely to have uncomfortable side effects.
Related Resources
• Andreasen A, Ellingrod VL. Lithium-induced diabetes insipidus: prevention and management. Current Psychiatry. 2013;12(7):42-45.
• Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646.
Drug Brand Names
Amiloride • Midamor Lithium • Eskalith, Lithobid
Disclosure
Dr. McInnis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11(11 suppl 2):4-9.
2. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007.
3. Burgess S, Geddes J, Hawton K, et al. Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev. 2001:CD003013.
4. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex maintenance study group. Arch Gen Psychiatry. 2000;57(5):481-489.
5. Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
6. Swann AC, Bowden CL, Calabrese JR, et al. Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology. 2002;26(4):530-536.
7. Tohen M, Chengappa KN, Suppes T, et al. Efficacy of olanzapine in combination with valproate or lithium in the treatment of mania in patients partially nonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry. 2002;59(1):62-69.
8. Perlis RH, Smoller JW, Ferreira MA, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009; 166(6):718-725.
9. Grof P, Duffy A, Cavazzoni P, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63(10): 942-947.
10. Duffy A, Alda M, Kutcher S, et al. A prospective study of the offspring of bipolar parents responsive and nonresponsive to lithium treatment. J Clin Psychiatry. 2002;63(12): 1171-1178.
11. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473.
12. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010; 62(1):50-60.
13. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
14. de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54-56.
15. Nierenberg AA, Sylvia LG, Leon AC, et al; LiTMUS Study Group. Lithium treatment–moderate dose use study (LiTMUS) for bipolar disorder: rationale and design. Clin Trials. 2009;6(6):637-648.
16. Sylvia LG, Reilly-Harrington NA, Leon AC, et al. Methods to limit attrition in longitudinal comparative effectiveness trials: lessons from the Lithium Treatment - Moderate dose Use Study (LiTMUS) for bipolar disorder. Clin Trials. 2012;9(1):94-101.
17. McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 2010;11(10):1439-1465.
18. Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy [published online February 15, 2014]. Pharmacol Biochem Behav. doi: 10.1016/j.pbb.2014.02.004.
19. Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1-21.
20. Devaki R, Shankar Rao S, Nadgir SM. The effect of lithium on the adrenoceptor-mediated second messenger system in the rat brain. J Psychiatry Neurosci. 2006;31(4):246-252.
21. Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1397-1411.
22. Hu LW, Kawamoto EM, Brietzke E, et al. The role of Wnt signaling and its interaction with diverse mechanisms of cellular apoptosis in the pathophysiology of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):11-17.
23. Chen HM, DeLong CJ, Bame M, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients [published online March 25, 2014]. Transl Psychiatry. doi:10.1038/tp.2014.12.
24. Jefferson JW. Lithium. In: Aronson JK, ed. Side effects of drugs annual, volume 26. Amsterdam, The Netherlands: Elsevier Science; 2003:19-29.
25. Baldessarini RJ, Tondo L, Floris G, et al. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord. 2000;61(2):13-22.
26. Baldessarini RJ, Tondo L, Hennen J, et al. Latency and episodes before treatment: response to lithium maintenance in bipolar I and II disorders. Bipolar Disord. 1999;1(2): 91-97.
27. Fieve RR, Kumbaraci T, Dunner DL. Lithium prophylaxis of depression in bipolar I, bipolar II, and unipolar patients. Am J Psychiatry. 1976;133(8):925-929.
28. Peck CC, Pond SM, Becker CE, et al. An evaluation of the effects of lithium in the treatment of chronic alcoholism. II. Assessment of the two-period crossover design. Alcohol Clin Exp Res. 1981;5(2):252-255.
29. Peselow ED, Dunner DL, Fieve RR, et al. Lithium prophylaxis of depression in unipolar, bipolar II, and cyclothymic patients. Am J Psychiatry. 1982;139(6):747-752.
30. Bellino S, Paradiso E, Bogetto F. Efficacy and tolerability of pharmacotherapies for borderline personality disorder. CNS Drugs. 2008;22(8):671-692.
31. Alevizos B, Alevizos E, Leonardou A, et al. Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatrike. 2012;23(2):143-148.
32. Goldberg JF, Sacks MH, Kocsis JH. Low-dose lithium augmentation of divalproex in geriatric mania. J Clin Psychiatry. 2000;61(4):304.
33. Saunders KE, Goodwin GM. New approaches in the treatment of bipolar depression. Curr Top Behav Neurosci. 2013;14:291-307.
34. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
35. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429-435.
36. Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82.
37. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
38. Altman EG, Hedeker D, Peterson JL, et al. The Altman Self- Rating Mania Scale. Biol Psychiatry. 1997;42(10):948-955.
39. Machado-Vieira R, Luckenbaugh DA, Soeiro-de-Souza MG, et al. Early improvement with lithium in classic mania and its association with later response. J Affect Disord. 2013;144(1-2):160-164.
40. Severus WE, Lipkovich IA, Licht RW, et al. In search of optimal lithium levels and olanzapine doses in the long-term treatment of bipolar I disorder. A post-hoc analysis of the maintenance study by Tohen et al. 2005. Eur Psychiatry. 2010;25(8):443-449.
41. Vestergaard P, Licht RW, Brodersen A, et al. Outcome of lithium prophylaxis: a prospective follow-up of affective disorder patients assigned to high and low serum lithium levels. Acta Psychiatr Scand. 1998;98(4):310-315.
42. Nierenberg AA, Friedman ES, Bowden CL, et al. Lithium treatment moderate-dose use study (LiTMUS) for bipolar disorder: a randomized comparative effectiveness trial of optimized personalized treatment with and without lithium. Am J Psychiatry. 2013;170(1):102-111.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11(11 suppl 2):4-9.
2. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007.
3. Burgess S, Geddes J, Hawton K, et al. Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev. 2001:CD003013.
4. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex maintenance study group. Arch Gen Psychiatry. 2000;57(5):481-489.
5. Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
6. Swann AC, Bowden CL, Calabrese JR, et al. Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology. 2002;26(4):530-536.
7. Tohen M, Chengappa KN, Suppes T, et al. Efficacy of olanzapine in combination with valproate or lithium in the treatment of mania in patients partially nonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry. 2002;59(1):62-69.
8. Perlis RH, Smoller JW, Ferreira MA, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009; 166(6):718-725.
9. Grof P, Duffy A, Cavazzoni P, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63(10): 942-947.
10. Duffy A, Alda M, Kutcher S, et al. A prospective study of the offspring of bipolar parents responsive and nonresponsive to lithium treatment. J Clin Psychiatry. 2002;63(12): 1171-1178.
11. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473.
12. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010; 62(1):50-60.
13. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
14. de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54-56.
15. Nierenberg AA, Sylvia LG, Leon AC, et al; LiTMUS Study Group. Lithium treatment–moderate dose use study (LiTMUS) for bipolar disorder: rationale and design. Clin Trials. 2009;6(6):637-648.
16. Sylvia LG, Reilly-Harrington NA, Leon AC, et al. Methods to limit attrition in longitudinal comparative effectiveness trials: lessons from the Lithium Treatment - Moderate dose Use Study (LiTMUS) for bipolar disorder. Clin Trials. 2012;9(1):94-101.
17. McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 2010;11(10):1439-1465.
18. Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy [published online February 15, 2014]. Pharmacol Biochem Behav. doi: 10.1016/j.pbb.2014.02.004.
19. Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1-21.
20. Devaki R, Shankar Rao S, Nadgir SM. The effect of lithium on the adrenoceptor-mediated second messenger system in the rat brain. J Psychiatry Neurosci. 2006;31(4):246-252.
21. Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1397-1411.
22. Hu LW, Kawamoto EM, Brietzke E, et al. The role of Wnt signaling and its interaction with diverse mechanisms of cellular apoptosis in the pathophysiology of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):11-17.
23. Chen HM, DeLong CJ, Bame M, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients [published online March 25, 2014]. Transl Psychiatry. doi:10.1038/tp.2014.12.
24. Jefferson JW. Lithium. In: Aronson JK, ed. Side effects of drugs annual, volume 26. Amsterdam, The Netherlands: Elsevier Science; 2003:19-29.
25. Baldessarini RJ, Tondo L, Floris G, et al. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord. 2000;61(2):13-22.
26. Baldessarini RJ, Tondo L, Hennen J, et al. Latency and episodes before treatment: response to lithium maintenance in bipolar I and II disorders. Bipolar Disord. 1999;1(2): 91-97.
27. Fieve RR, Kumbaraci T, Dunner DL. Lithium prophylaxis of depression in bipolar I, bipolar II, and unipolar patients. Am J Psychiatry. 1976;133(8):925-929.
28. Peck CC, Pond SM, Becker CE, et al. An evaluation of the effects of lithium in the treatment of chronic alcoholism. II. Assessment of the two-period crossover design. Alcohol Clin Exp Res. 1981;5(2):252-255.
29. Peselow ED, Dunner DL, Fieve RR, et al. Lithium prophylaxis of depression in unipolar, bipolar II, and cyclothymic patients. Am J Psychiatry. 1982;139(6):747-752.
30. Bellino S, Paradiso E, Bogetto F. Efficacy and tolerability of pharmacotherapies for borderline personality disorder. CNS Drugs. 2008;22(8):671-692.
31. Alevizos B, Alevizos E, Leonardou A, et al. Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatrike. 2012;23(2):143-148.
32. Goldberg JF, Sacks MH, Kocsis JH. Low-dose lithium augmentation of divalproex in geriatric mania. J Clin Psychiatry. 2000;61(4):304.
33. Saunders KE, Goodwin GM. New approaches in the treatment of bipolar depression. Curr Top Behav Neurosci. 2013;14:291-307.
34. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
35. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429-435.
36. Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82.
37. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
38. Altman EG, Hedeker D, Peterson JL, et al. The Altman Self- Rating Mania Scale. Biol Psychiatry. 1997;42(10):948-955.
39. Machado-Vieira R, Luckenbaugh DA, Soeiro-de-Souza MG, et al. Early improvement with lithium in classic mania and its association with later response. J Affect Disord. 2013;144(1-2):160-164.
40. Severus WE, Lipkovich IA, Licht RW, et al. In search of optimal lithium levels and olanzapine doses in the long-term treatment of bipolar I disorder. A post-hoc analysis of the maintenance study by Tohen et al. 2005. Eur Psychiatry. 2010;25(8):443-449.
41. Vestergaard P, Licht RW, Brodersen A, et al. Outcome of lithium prophylaxis: a prospective follow-up of affective disorder patients assigned to high and low serum lithium levels. Acta Psychiatr Scand. 1998;98(4):310-315.
42. Nierenberg AA, Friedman ES, Bowden CL, et al. Lithium treatment moderate-dose use study (LiTMUS) for bipolar disorder: a randomized comparative effectiveness trial of optimized personalized treatment with and without lithium. Am J Psychiatry. 2013;170(1):102-111.

Woman loses both legs after salpingectomy: $64.3M award

Woman loses both legs after salpingectomy: $64.3M award
Due to an ectopic pregnancy, a 29-year-old woman underwent laparoscopic salpingectomy in October 2009. A resident supervised by Dr. A (gynecologist) performed the surgery. Although the patient reported abdominal pain and was febrile, Dr. B (gynecologist) discharged her on postsurgical day 2.
The next day, she returned to the emergency department (ED) with abdominal swelling and pain. Dr. C (ED physician), Dr. D (gynecologist), and Dr. E (general surgeon) examined her. Dr. D began conservative treatment for bowel obstruction. Two days later she was in septic shock. Dr. E repaired a 5-mm injury to the sigmoid colon and created a colostomy. The patient was placed in a medically induced coma for 3 weeks. She experienced cardiac arrest 3 times during her 73-day ICU stay. She underwent skin grafts, and suffered hearing loss as a result of antibiotic treatment. Due to gangrene, both legs were amputated below the knee.
At the trial’s conclusion in January 2014, the colostomy had not been reversed. She has difficulty caring for her daughter and has not worked since the initial operation.
PATIENT’S CLAIM The resident, who injured the colon and did not detect the injury during surgery, was improperly supervised by Dr. A. Hospital staff did not communicate the patient’s problem reports to the physicians. Dr. B should not have discharged her after surgery; based on her reported symptoms, additional testing was warranted. Drs. C, D, and E did not react to the patient’s pain reports in a timely manner, nor treat the resulting sepsis aggressively enough, leading to gangrene.
DEFENDANTS’ DEFENSE The patient’s colon injury was diagnosed and treated in a timely manner, but her condition deteriorated rapidly. The physicians acted responsibly based on the available information; a computed tomography scan did not show the colon injury. The injury likely occurred after the procedure due to an underlying bowel condition and is a known risk of the procedure. The colostomy can be reversed. Their efforts saved her life.
VERDICT The patient and Dr. E negotiated a $2.3 million settlement. A $62 million New York verdict was returned. The jury found the hospital 40% liable; Dr. A 30% liable; Dr. B 20% liable; and Dr. D 10% liable. Claims were dropped against the resident and Dr. C.
Related article: Oophorectomy or salpingectomy—which makes more sense? William H. Parker, MD (March 2014)
PARENTS REQUESTED EARLIER CESAREAN: CHILD HAS CP
A woman was in labor for 2 full days before her ObGyn performed a cesarean delivery. The child was born with abnormal Apgar scores and had seizures. Imaging studies revealed brain damage. She received a diagnosis of cerebral palsy.
PARENTS’ CLAIM The parents first requested cesarean delivery early on the second day, but the ObGyn allowed labor to progress. When the fetal heart-rate monitor showed signs of fetal distress 3 hours later, the parents made a second request; the ObGyn continued with vaginal delivery. The child was ultimately born by cesarean delivery. Her brain damage was caused by lack of oxygen from failure to perform an earlier cesarean delivery.
DEFENDANTS’ DEFENSE The case was settled during the trial.
VERDICT A $4.25 million Massachusetts settlement was reached.
BLADDER INJURED DURING CESAREAN DELIVERY
A 33-year-old woman gave birth via cesarean delivery performed by her ObGyn. During the procedure, the patient’s bladder was lacerated and the injury was immediately repaired. The patient reports occasional urinary incontinence and pain.
PATIENT’S CLAIM The ObGyn should have anticipated that the bladder would be shifted because of the patient’s previous cesarean delivery.
PHYSICIAN’S DEFENSE The injury is a known risk of the procedure. The patient had developed adhesions that caused the bladder to become displaced. She does not suffer permanent residual effects from the injury.
VERDICT A $125,000 New York verdict was returned.
Related article: 10 practical, evidence-based recommendations for improving maternal outcomes of cesarean delivery. Baha M. Sibai (March 2012)
PARENTS REQUESTED SPECIFIC GENETIC TESTING, BUT CHILD IS BORN WITH RARE CHROMOSOMAL CONDITION: $50M VERDICT
Parents sought prenatal genetic testing to determine if their fetus had a specific genetic condition because the father carries a rare chromosomal abnormality called an unbalanced chromosome translocation. This defect can only be identified if the laboratory is told precisely where to look for the specific translocation; it is not detected on routine prenatal genetic testing. After testing, the parents were told that the fetus did not have the chromosomal abnormality.
The child was born with the condition for which testing was sought, resulting in severe physical and cognitive impairments and multiple physical abnormalities. He will require 24-hour care for life.
PARENTS’ CLAIM Testing failed to identify the condition; the couple had decided to terminate the pregnancy if the child was affected. Due to budget cuts in the maternal-fetal medicine clinic, the medical center borrowed a genetic counselor from another hospital one day a week. The parents told the genetic counselor of the family’s history of the defect and explained that the laboratory’s procedures require the referring center to obtain and share the necessary detailed information with the lab. The lab was apparently notified that the couple had a family history of the defect, but the genetic counselor did not transmit specific information to the lab, and lab personnel did not appropriately follow-up.
DEFENDANTS’ DEFENSE The medical center blamed the laboratory: the lab’s standard procedures state that the lab should call the referring center to obtain the necessary detailed information if it was not provided; the lab employee who handled the specimen did not do so. The lab claimed that the genetic counselor did not transmit the specific information to the lab.
The laboratory disputed the child’s need for 24/7 care, maintaining that he could live in a group home with only occasional nursing care.
VERDICT A $50 million Washington verdict was returned against the medical center and laboratory; each defendant will pay $25 million.
Related article: Noninvasive prenatal testing: Where we are and where we’re going. Lee P. Shulman, MD (Commentary; May 2014)
NECROTIZING FASCIITIS AFTER SURGERY
A 57-year-old woman underwent surgery to repair vaginal vault prolapse, rectocele, and enterocele, performed by her gynecologist. Several days after discharge, the patient returned to the hospital with an infection in her leg that had evolved into necrotizing fasciitis. She underwent five fasciotomies and was hospitalized for 3 weeks.
PATIENT’S CLAIM The gynecologist should have administered prophylactic antibiotics before, during, and after surgery. The patient has massive scarring of her leg.
PHYSICIAN’S DEFENSE The infection was not a result of failing to administer antibiotics. The patient failed to seek timely treatment of symptoms that developed after surgery.
VERDICT A $400,000 New York verdict was returned but reduced because the jury found the patient 49% at fault.
OXYTOCIN BLAMED FOR CHILD’S CP
A mother had bariatric surgery 12 months before becoming pregnant, and she smoked during pregnancy. She developed placental insufficiency and labor was induced shortly after she reached 37 weeks’ gestation.
During delivery, the mother was given oxytocin to increase the frequency and strength of contractions. Nurses repeatedly stopped the oxytocin in response to decelerations in the fetal heart rate, but physicians ordered the oxytocin resumed, even after fetal heart-rate monitoring showed fetal distress.
Three days after birth, the child was transferred to another hospital, and was found to have cerebral palsy and other injuries. At age 5, the child is nonverbal, cannot walk, and requires a feeding tube.
PARENTS’ CLAIM Oxytocin should have been stopped and a cesarean delivery performed when fetal distress was first noted.
DEFENDANTS’ DEFENSE There was no need for cesarean delivery. Apgar scores, blood gases, and fetal presentation indicated that the injury occurred prior to labor.
VERDICT A $6 million Texas settlement was reached during the trial.
Related article: Q: Following cesarean delivery, what is the optimal oxytocin infusion duration to prevent postpartum bleeding? Robert L. Barbieri, MD (Editorial; April 2014)
MOTHER DISCHARGED DESPITE SEVERE ABDOMINAL PAIN
A woman had prenatal care at different locations. Her history included two cesarean deliveries.
Reporting severe abdominal pain, she was taken from a homeless shelter to an ED by ambulance. The mother was uncertain of the fetus’ gestational age; a 4th-year obstetric resident determined by physical examination that the pregnancy was at 36.5 weeks. The resident discussed the case with the attending ObGyn, who said to discharge the mother if her pain was gone. After 11 hours, the mother was returned to the shelter.
The mother returned to the ED 12 hours later. Thirty-five minutes after fetal distress was identified, an emergency cesarean delivery was performed. At birth, the child was found to be at 38 to 39 weeks’ gestation. He received a diagnosis of severe hypoxic ischemic encephalopathy and was transferred to a children’s hospital for brain cooling.
The child lives in a long-term care facility and is dependent on a ventilator and gastronomy tube.
PARENT’S CLAIM The mother should not have been discharged after the first visit. A cesarean delivery should have been performed at that time. The attending ObGyn never saw the mother.
DEFENDANTS’ DEFENSE The mother should have given her correct due date, which was in her prenatal records based on previous ultrasonograpy. The first discharge was proper, as the pain had improved. The homeless shelter should have called an ambulance earlier for the second admission.
VERDICT A $7.5 million California settlement was reached, plus payment of medical expenses exceeding $300,000.
Timing of child’s injury disputed
Vaginal birth after cesarean (VBAC) had been planned. After reporting to her ObGyn that she was in labor, a mother went to the ED.
During the next few hours, hospital staff called the ObGyn twice to report that fetal monitor strips indicated tachycardia. The ObGyn then spoke to the mother by phone and told her that cesarean delivery was necessary but could wait for him to get to the hospital. After the ObGyn arrived, he removed the fetal heart-rate monitor to prepare the mother’s abdomen; cesarean delivery occurred 15 minutes later.
The child has spastic dystonic quadriplegia and requires 24-hour care.
PARENT’S CLAIM The ObGyn should have come to the hospital and performed cesarean delivery when he was first notified that the fetus was tachycardic. The baby suffered an hypoxic ischemic event in the 15-minute period between when the monitor was removed and birth, causing hypoxic ischemic encephalopathy.
PHYSICIAN’S DEFENSE There was no indication of a need for earlier delivery. The brain injury occurred prior to labor and delivery.
VERDICT The hospital settled for a confidential amount before the trial. An Illinois defense verdict was returned for the ObGyn.
Were mammograms properly interpreted?
After reporting a lump in her breast, a 39-year-old woman underwent mammography in 2008 and 2009. Two different radiologists reported their findings as negative for cancer.
In 2010, the patient was found to have breast cancer. She underwent a mastectomy, chemotherapy, and radiation therapy, and was given a 75%–80% chance of 5-year survival.
PATIENT’S CLAIM The ObGyn failed to follow-up on the patient’s reports of a breast lump. The radiologists did not correctly interpret the 2008 and 2009 mammograms. If cancer had been detected earlier, treatment would have been less extreme.
PHYSICIANS’ DEFENSE The ObGyn claimed that he would have felt a lump if it was present. The first radiologist claimed that the 2008 mammography report was correct, noting that the patient’s cancer was a lobular carcinoma that does not always show on mammography or in patients with dense breasts, which this patient has.
VERDICT A directed verdict was granted to the radiologist who interpreted the 2009 mammography, as the results were lost. An Ohio defense verdict was returned for the ObGyn and the other radiologist.
Related article: Does screening mammography save lives? Janelle Yates, Senior Editor (April 2014)
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
TELL US WHAT YOU THINK! Drop us a line and let us know what you think about this or other current articles, which topics you'd like to see covered in future issues, and what challenges you face in daily practice.
Tell us what you think by emailing us at: [email protected] Please include your name, city and state.
Stay in touch! Your feedback is important to us!
Woman loses both legs after salpingectomy: $64.3M award
Due to an ectopic pregnancy, a 29-year-old woman underwent laparoscopic salpingectomy in October 2009. A resident supervised by Dr. A (gynecologist) performed the surgery. Although the patient reported abdominal pain and was febrile, Dr. B (gynecologist) discharged her on postsurgical day 2.
The next day, she returned to the emergency department (ED) with abdominal swelling and pain. Dr. C (ED physician), Dr. D (gynecologist), and Dr. E (general surgeon) examined her. Dr. D began conservative treatment for bowel obstruction. Two days later she was in septic shock. Dr. E repaired a 5-mm injury to the sigmoid colon and created a colostomy. The patient was placed in a medically induced coma for 3 weeks. She experienced cardiac arrest 3 times during her 73-day ICU stay. She underwent skin grafts, and suffered hearing loss as a result of antibiotic treatment. Due to gangrene, both legs were amputated below the knee.
At the trial’s conclusion in January 2014, the colostomy had not been reversed. She has difficulty caring for her daughter and has not worked since the initial operation.
PATIENT’S CLAIM The resident, who injured the colon and did not detect the injury during surgery, was improperly supervised by Dr. A. Hospital staff did not communicate the patient’s problem reports to the physicians. Dr. B should not have discharged her after surgery; based on her reported symptoms, additional testing was warranted. Drs. C, D, and E did not react to the patient’s pain reports in a timely manner, nor treat the resulting sepsis aggressively enough, leading to gangrene.
DEFENDANTS’ DEFENSE The patient’s colon injury was diagnosed and treated in a timely manner, but her condition deteriorated rapidly. The physicians acted responsibly based on the available information; a computed tomography scan did not show the colon injury. The injury likely occurred after the procedure due to an underlying bowel condition and is a known risk of the procedure. The colostomy can be reversed. Their efforts saved her life.
VERDICT The patient and Dr. E negotiated a $2.3 million settlement. A $62 million New York verdict was returned. The jury found the hospital 40% liable; Dr. A 30% liable; Dr. B 20% liable; and Dr. D 10% liable. Claims were dropped against the resident and Dr. C.
Related article: Oophorectomy or salpingectomy—which makes more sense? William H. Parker, MD (March 2014)
PARENTS REQUESTED EARLIER CESAREAN: CHILD HAS CP
A woman was in labor for 2 full days before her ObGyn performed a cesarean delivery. The child was born with abnormal Apgar scores and had seizures. Imaging studies revealed brain damage. She received a diagnosis of cerebral palsy.
PARENTS’ CLAIM The parents first requested cesarean delivery early on the second day, but the ObGyn allowed labor to progress. When the fetal heart-rate monitor showed signs of fetal distress 3 hours later, the parents made a second request; the ObGyn continued with vaginal delivery. The child was ultimately born by cesarean delivery. Her brain damage was caused by lack of oxygen from failure to perform an earlier cesarean delivery.
DEFENDANTS’ DEFENSE The case was settled during the trial.
VERDICT A $4.25 million Massachusetts settlement was reached.
BLADDER INJURED DURING CESAREAN DELIVERY
A 33-year-old woman gave birth via cesarean delivery performed by her ObGyn. During the procedure, the patient’s bladder was lacerated and the injury was immediately repaired. The patient reports occasional urinary incontinence and pain.
PATIENT’S CLAIM The ObGyn should have anticipated that the bladder would be shifted because of the patient’s previous cesarean delivery.
PHYSICIAN’S DEFENSE The injury is a known risk of the procedure. The patient had developed adhesions that caused the bladder to become displaced. She does not suffer permanent residual effects from the injury.
VERDICT A $125,000 New York verdict was returned.
Related article: 10 practical, evidence-based recommendations for improving maternal outcomes of cesarean delivery. Baha M. Sibai (March 2012)
PARENTS REQUESTED SPECIFIC GENETIC TESTING, BUT CHILD IS BORN WITH RARE CHROMOSOMAL CONDITION: $50M VERDICT
Parents sought prenatal genetic testing to determine if their fetus had a specific genetic condition because the father carries a rare chromosomal abnormality called an unbalanced chromosome translocation. This defect can only be identified if the laboratory is told precisely where to look for the specific translocation; it is not detected on routine prenatal genetic testing. After testing, the parents were told that the fetus did not have the chromosomal abnormality.
The child was born with the condition for which testing was sought, resulting in severe physical and cognitive impairments and multiple physical abnormalities. He will require 24-hour care for life.
PARENTS’ CLAIM Testing failed to identify the condition; the couple had decided to terminate the pregnancy if the child was affected. Due to budget cuts in the maternal-fetal medicine clinic, the medical center borrowed a genetic counselor from another hospital one day a week. The parents told the genetic counselor of the family’s history of the defect and explained that the laboratory’s procedures require the referring center to obtain and share the necessary detailed information with the lab. The lab was apparently notified that the couple had a family history of the defect, but the genetic counselor did not transmit specific information to the lab, and lab personnel did not appropriately follow-up.
DEFENDANTS’ DEFENSE The medical center blamed the laboratory: the lab’s standard procedures state that the lab should call the referring center to obtain the necessary detailed information if it was not provided; the lab employee who handled the specimen did not do so. The lab claimed that the genetic counselor did not transmit the specific information to the lab.
The laboratory disputed the child’s need for 24/7 care, maintaining that he could live in a group home with only occasional nursing care.
VERDICT A $50 million Washington verdict was returned against the medical center and laboratory; each defendant will pay $25 million.
Related article: Noninvasive prenatal testing: Where we are and where we’re going. Lee P. Shulman, MD (Commentary; May 2014)
NECROTIZING FASCIITIS AFTER SURGERY
A 57-year-old woman underwent surgery to repair vaginal vault prolapse, rectocele, and enterocele, performed by her gynecologist. Several days after discharge, the patient returned to the hospital with an infection in her leg that had evolved into necrotizing fasciitis. She underwent five fasciotomies and was hospitalized for 3 weeks.
PATIENT’S CLAIM The gynecologist should have administered prophylactic antibiotics before, during, and after surgery. The patient has massive scarring of her leg.
PHYSICIAN’S DEFENSE The infection was not a result of failing to administer antibiotics. The patient failed to seek timely treatment of symptoms that developed after surgery.
VERDICT A $400,000 New York verdict was returned but reduced because the jury found the patient 49% at fault.
OXYTOCIN BLAMED FOR CHILD’S CP
A mother had bariatric surgery 12 months before becoming pregnant, and she smoked during pregnancy. She developed placental insufficiency and labor was induced shortly after she reached 37 weeks’ gestation.
During delivery, the mother was given oxytocin to increase the frequency and strength of contractions. Nurses repeatedly stopped the oxytocin in response to decelerations in the fetal heart rate, but physicians ordered the oxytocin resumed, even after fetal heart-rate monitoring showed fetal distress.
Three days after birth, the child was transferred to another hospital, and was found to have cerebral palsy and other injuries. At age 5, the child is nonverbal, cannot walk, and requires a feeding tube.
PARENTS’ CLAIM Oxytocin should have been stopped and a cesarean delivery performed when fetal distress was first noted.
DEFENDANTS’ DEFENSE There was no need for cesarean delivery. Apgar scores, blood gases, and fetal presentation indicated that the injury occurred prior to labor.
VERDICT A $6 million Texas settlement was reached during the trial.
Related article: Q: Following cesarean delivery, what is the optimal oxytocin infusion duration to prevent postpartum bleeding? Robert L. Barbieri, MD (Editorial; April 2014)
MOTHER DISCHARGED DESPITE SEVERE ABDOMINAL PAIN
A woman had prenatal care at different locations. Her history included two cesarean deliveries.
Reporting severe abdominal pain, she was taken from a homeless shelter to an ED by ambulance. The mother was uncertain of the fetus’ gestational age; a 4th-year obstetric resident determined by physical examination that the pregnancy was at 36.5 weeks. The resident discussed the case with the attending ObGyn, who said to discharge the mother if her pain was gone. After 11 hours, the mother was returned to the shelter.
The mother returned to the ED 12 hours later. Thirty-five minutes after fetal distress was identified, an emergency cesarean delivery was performed. At birth, the child was found to be at 38 to 39 weeks’ gestation. He received a diagnosis of severe hypoxic ischemic encephalopathy and was transferred to a children’s hospital for brain cooling.
The child lives in a long-term care facility and is dependent on a ventilator and gastronomy tube.
PARENT’S CLAIM The mother should not have been discharged after the first visit. A cesarean delivery should have been performed at that time. The attending ObGyn never saw the mother.
DEFENDANTS’ DEFENSE The mother should have given her correct due date, which was in her prenatal records based on previous ultrasonograpy. The first discharge was proper, as the pain had improved. The homeless shelter should have called an ambulance earlier for the second admission.
VERDICT A $7.5 million California settlement was reached, plus payment of medical expenses exceeding $300,000.
Timing of child’s injury disputed
Vaginal birth after cesarean (VBAC) had been planned. After reporting to her ObGyn that she was in labor, a mother went to the ED.
During the next few hours, hospital staff called the ObGyn twice to report that fetal monitor strips indicated tachycardia. The ObGyn then spoke to the mother by phone and told her that cesarean delivery was necessary but could wait for him to get to the hospital. After the ObGyn arrived, he removed the fetal heart-rate monitor to prepare the mother’s abdomen; cesarean delivery occurred 15 minutes later.
The child has spastic dystonic quadriplegia and requires 24-hour care.
PARENT’S CLAIM The ObGyn should have come to the hospital and performed cesarean delivery when he was first notified that the fetus was tachycardic. The baby suffered an hypoxic ischemic event in the 15-minute period between when the monitor was removed and birth, causing hypoxic ischemic encephalopathy.
PHYSICIAN’S DEFENSE There was no indication of a need for earlier delivery. The brain injury occurred prior to labor and delivery.
VERDICT The hospital settled for a confidential amount before the trial. An Illinois defense verdict was returned for the ObGyn.
Were mammograms properly interpreted?
After reporting a lump in her breast, a 39-year-old woman underwent mammography in 2008 and 2009. Two different radiologists reported their findings as negative for cancer.
In 2010, the patient was found to have breast cancer. She underwent a mastectomy, chemotherapy, and radiation therapy, and was given a 75%–80% chance of 5-year survival.
PATIENT’S CLAIM The ObGyn failed to follow-up on the patient’s reports of a breast lump. The radiologists did not correctly interpret the 2008 and 2009 mammograms. If cancer had been detected earlier, treatment would have been less extreme.
PHYSICIANS’ DEFENSE The ObGyn claimed that he would have felt a lump if it was present. The first radiologist claimed that the 2008 mammography report was correct, noting that the patient’s cancer was a lobular carcinoma that does not always show on mammography or in patients with dense breasts, which this patient has.
VERDICT A directed verdict was granted to the radiologist who interpreted the 2009 mammography, as the results were lost. An Ohio defense verdict was returned for the ObGyn and the other radiologist.
Related article: Does screening mammography save lives? Janelle Yates, Senior Editor (April 2014)
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
TELL US WHAT YOU THINK! Drop us a line and let us know what you think about this or other current articles, which topics you'd like to see covered in future issues, and what challenges you face in daily practice.
Tell us what you think by emailing us at: [email protected] Please include your name, city and state.
Stay in touch! Your feedback is important to us!
Woman loses both legs after salpingectomy: $64.3M award
Due to an ectopic pregnancy, a 29-year-old woman underwent laparoscopic salpingectomy in October 2009. A resident supervised by Dr. A (gynecologist) performed the surgery. Although the patient reported abdominal pain and was febrile, Dr. B (gynecologist) discharged her on postsurgical day 2.
The next day, she returned to the emergency department (ED) with abdominal swelling and pain. Dr. C (ED physician), Dr. D (gynecologist), and Dr. E (general surgeon) examined her. Dr. D began conservative treatment for bowel obstruction. Two days later she was in septic shock. Dr. E repaired a 5-mm injury to the sigmoid colon and created a colostomy. The patient was placed in a medically induced coma for 3 weeks. She experienced cardiac arrest 3 times during her 73-day ICU stay. She underwent skin grafts, and suffered hearing loss as a result of antibiotic treatment. Due to gangrene, both legs were amputated below the knee.
At the trial’s conclusion in January 2014, the colostomy had not been reversed. She has difficulty caring for her daughter and has not worked since the initial operation.
PATIENT’S CLAIM The resident, who injured the colon and did not detect the injury during surgery, was improperly supervised by Dr. A. Hospital staff did not communicate the patient’s problem reports to the physicians. Dr. B should not have discharged her after surgery; based on her reported symptoms, additional testing was warranted. Drs. C, D, and E did not react to the patient’s pain reports in a timely manner, nor treat the resulting sepsis aggressively enough, leading to gangrene.
DEFENDANTS’ DEFENSE The patient’s colon injury was diagnosed and treated in a timely manner, but her condition deteriorated rapidly. The physicians acted responsibly based on the available information; a computed tomography scan did not show the colon injury. The injury likely occurred after the procedure due to an underlying bowel condition and is a known risk of the procedure. The colostomy can be reversed. Their efforts saved her life.
VERDICT The patient and Dr. E negotiated a $2.3 million settlement. A $62 million New York verdict was returned. The jury found the hospital 40% liable; Dr. A 30% liable; Dr. B 20% liable; and Dr. D 10% liable. Claims were dropped against the resident and Dr. C.
Related article: Oophorectomy or salpingectomy—which makes more sense? William H. Parker, MD (March 2014)
PARENTS REQUESTED EARLIER CESAREAN: CHILD HAS CP
A woman was in labor for 2 full days before her ObGyn performed a cesarean delivery. The child was born with abnormal Apgar scores and had seizures. Imaging studies revealed brain damage. She received a diagnosis of cerebral palsy.
PARENTS’ CLAIM The parents first requested cesarean delivery early on the second day, but the ObGyn allowed labor to progress. When the fetal heart-rate monitor showed signs of fetal distress 3 hours later, the parents made a second request; the ObGyn continued with vaginal delivery. The child was ultimately born by cesarean delivery. Her brain damage was caused by lack of oxygen from failure to perform an earlier cesarean delivery.
DEFENDANTS’ DEFENSE The case was settled during the trial.
VERDICT A $4.25 million Massachusetts settlement was reached.
BLADDER INJURED DURING CESAREAN DELIVERY
A 33-year-old woman gave birth via cesarean delivery performed by her ObGyn. During the procedure, the patient’s bladder was lacerated and the injury was immediately repaired. The patient reports occasional urinary incontinence and pain.
PATIENT’S CLAIM The ObGyn should have anticipated that the bladder would be shifted because of the patient’s previous cesarean delivery.
PHYSICIAN’S DEFENSE The injury is a known risk of the procedure. The patient had developed adhesions that caused the bladder to become displaced. She does not suffer permanent residual effects from the injury.
VERDICT A $125,000 New York verdict was returned.
Related article: 10 practical, evidence-based recommendations for improving maternal outcomes of cesarean delivery. Baha M. Sibai (March 2012)
PARENTS REQUESTED SPECIFIC GENETIC TESTING, BUT CHILD IS BORN WITH RARE CHROMOSOMAL CONDITION: $50M VERDICT
Parents sought prenatal genetic testing to determine if their fetus had a specific genetic condition because the father carries a rare chromosomal abnormality called an unbalanced chromosome translocation. This defect can only be identified if the laboratory is told precisely where to look for the specific translocation; it is not detected on routine prenatal genetic testing. After testing, the parents were told that the fetus did not have the chromosomal abnormality.
The child was born with the condition for which testing was sought, resulting in severe physical and cognitive impairments and multiple physical abnormalities. He will require 24-hour care for life.
PARENTS’ CLAIM Testing failed to identify the condition; the couple had decided to terminate the pregnancy if the child was affected. Due to budget cuts in the maternal-fetal medicine clinic, the medical center borrowed a genetic counselor from another hospital one day a week. The parents told the genetic counselor of the family’s history of the defect and explained that the laboratory’s procedures require the referring center to obtain and share the necessary detailed information with the lab. The lab was apparently notified that the couple had a family history of the defect, but the genetic counselor did not transmit specific information to the lab, and lab personnel did not appropriately follow-up.
DEFENDANTS’ DEFENSE The medical center blamed the laboratory: the lab’s standard procedures state that the lab should call the referring center to obtain the necessary detailed information if it was not provided; the lab employee who handled the specimen did not do so. The lab claimed that the genetic counselor did not transmit the specific information to the lab.
The laboratory disputed the child’s need for 24/7 care, maintaining that he could live in a group home with only occasional nursing care.
VERDICT A $50 million Washington verdict was returned against the medical center and laboratory; each defendant will pay $25 million.
Related article: Noninvasive prenatal testing: Where we are and where we’re going. Lee P. Shulman, MD (Commentary; May 2014)
NECROTIZING FASCIITIS AFTER SURGERY
A 57-year-old woman underwent surgery to repair vaginal vault prolapse, rectocele, and enterocele, performed by her gynecologist. Several days after discharge, the patient returned to the hospital with an infection in her leg that had evolved into necrotizing fasciitis. She underwent five fasciotomies and was hospitalized for 3 weeks.
PATIENT’S CLAIM The gynecologist should have administered prophylactic antibiotics before, during, and after surgery. The patient has massive scarring of her leg.
PHYSICIAN’S DEFENSE The infection was not a result of failing to administer antibiotics. The patient failed to seek timely treatment of symptoms that developed after surgery.
VERDICT A $400,000 New York verdict was returned but reduced because the jury found the patient 49% at fault.
OXYTOCIN BLAMED FOR CHILD’S CP
A mother had bariatric surgery 12 months before becoming pregnant, and she smoked during pregnancy. She developed placental insufficiency and labor was induced shortly after she reached 37 weeks’ gestation.
During delivery, the mother was given oxytocin to increase the frequency and strength of contractions. Nurses repeatedly stopped the oxytocin in response to decelerations in the fetal heart rate, but physicians ordered the oxytocin resumed, even after fetal heart-rate monitoring showed fetal distress.
Three days after birth, the child was transferred to another hospital, and was found to have cerebral palsy and other injuries. At age 5, the child is nonverbal, cannot walk, and requires a feeding tube.
PARENTS’ CLAIM Oxytocin should have been stopped and a cesarean delivery performed when fetal distress was first noted.
DEFENDANTS’ DEFENSE There was no need for cesarean delivery. Apgar scores, blood gases, and fetal presentation indicated that the injury occurred prior to labor.
VERDICT A $6 million Texas settlement was reached during the trial.
Related article: Q: Following cesarean delivery, what is the optimal oxytocin infusion duration to prevent postpartum bleeding? Robert L. Barbieri, MD (Editorial; April 2014)
MOTHER DISCHARGED DESPITE SEVERE ABDOMINAL PAIN
A woman had prenatal care at different locations. Her history included two cesarean deliveries.
Reporting severe abdominal pain, she was taken from a homeless shelter to an ED by ambulance. The mother was uncertain of the fetus’ gestational age; a 4th-year obstetric resident determined by physical examination that the pregnancy was at 36.5 weeks. The resident discussed the case with the attending ObGyn, who said to discharge the mother if her pain was gone. After 11 hours, the mother was returned to the shelter.
The mother returned to the ED 12 hours later. Thirty-five minutes after fetal distress was identified, an emergency cesarean delivery was performed. At birth, the child was found to be at 38 to 39 weeks’ gestation. He received a diagnosis of severe hypoxic ischemic encephalopathy and was transferred to a children’s hospital for brain cooling.
The child lives in a long-term care facility and is dependent on a ventilator and gastronomy tube.
PARENT’S CLAIM The mother should not have been discharged after the first visit. A cesarean delivery should have been performed at that time. The attending ObGyn never saw the mother.
DEFENDANTS’ DEFENSE The mother should have given her correct due date, which was in her prenatal records based on previous ultrasonograpy. The first discharge was proper, as the pain had improved. The homeless shelter should have called an ambulance earlier for the second admission.
VERDICT A $7.5 million California settlement was reached, plus payment of medical expenses exceeding $300,000.
Timing of child’s injury disputed
Vaginal birth after cesarean (VBAC) had been planned. After reporting to her ObGyn that she was in labor, a mother went to the ED.
During the next few hours, hospital staff called the ObGyn twice to report that fetal monitor strips indicated tachycardia. The ObGyn then spoke to the mother by phone and told her that cesarean delivery was necessary but could wait for him to get to the hospital. After the ObGyn arrived, he removed the fetal heart-rate monitor to prepare the mother’s abdomen; cesarean delivery occurred 15 minutes later.
The child has spastic dystonic quadriplegia and requires 24-hour care.
PARENT’S CLAIM The ObGyn should have come to the hospital and performed cesarean delivery when he was first notified that the fetus was tachycardic. The baby suffered an hypoxic ischemic event in the 15-minute period between when the monitor was removed and birth, causing hypoxic ischemic encephalopathy.
PHYSICIAN’S DEFENSE There was no indication of a need for earlier delivery. The brain injury occurred prior to labor and delivery.
VERDICT The hospital settled for a confidential amount before the trial. An Illinois defense verdict was returned for the ObGyn.
Were mammograms properly interpreted?
After reporting a lump in her breast, a 39-year-old woman underwent mammography in 2008 and 2009. Two different radiologists reported their findings as negative for cancer.
In 2010, the patient was found to have breast cancer. She underwent a mastectomy, chemotherapy, and radiation therapy, and was given a 75%–80% chance of 5-year survival.
PATIENT’S CLAIM The ObGyn failed to follow-up on the patient’s reports of a breast lump. The radiologists did not correctly interpret the 2008 and 2009 mammograms. If cancer had been detected earlier, treatment would have been less extreme.
PHYSICIANS’ DEFENSE The ObGyn claimed that he would have felt a lump if it was present. The first radiologist claimed that the 2008 mammography report was correct, noting that the patient’s cancer was a lobular carcinoma that does not always show on mammography or in patients with dense breasts, which this patient has.
VERDICT A directed verdict was granted to the radiologist who interpreted the 2009 mammography, as the results were lost. An Ohio defense verdict was returned for the ObGyn and the other radiologist.
Related article: Does screening mammography save lives? Janelle Yates, Senior Editor (April 2014)
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
TELL US WHAT YOU THINK! Drop us a line and let us know what you think about this or other current articles, which topics you'd like to see covered in future issues, and what challenges you face in daily practice.
Tell us what you think by emailing us at: [email protected] Please include your name, city and state.
Stay in touch! Your feedback is important to us!

The importance of hematologic, cytogenetic, and molecular testing and mutational analysis in chronic myeloid leukemia
The introduction of BCR-ABL1 tyrosine kinase inhibitors (TKIs) for treatment of chronic myeloid leukemia (CML) has made it possible for this cancer to be controlled in many patients for long periods with chronic medication and regular monitoring of disease status. Hematologic and cytogenetic testing, molecular monitoring, and BCR-ABL1 mutational analysis have become integral to the routine management of CML. The information that each type of test provides is essential to confirm a diagnosis, determine the disease stage, assess response to treatment, and monitor for signals of disease progression – all of which can be used to identify patients who might require further evaluation, closer follow-up, and additional intervention, and to guide clinical decisions.
Click on the PDF icon at the top of this introduction to read the full article.
The introduction of BCR-ABL1 tyrosine kinase inhibitors (TKIs) for treatment of chronic myeloid leukemia (CML) has made it possible for this cancer to be controlled in many patients for long periods with chronic medication and regular monitoring of disease status. Hematologic and cytogenetic testing, molecular monitoring, and BCR-ABL1 mutational analysis have become integral to the routine management of CML. The information that each type of test provides is essential to confirm a diagnosis, determine the disease stage, assess response to treatment, and monitor for signals of disease progression – all of which can be used to identify patients who might require further evaluation, closer follow-up, and additional intervention, and to guide clinical decisions.
Click on the PDF icon at the top of this introduction to read the full article.
The introduction of BCR-ABL1 tyrosine kinase inhibitors (TKIs) for treatment of chronic myeloid leukemia (CML) has made it possible for this cancer to be controlled in many patients for long periods with chronic medication and regular monitoring of disease status. Hematologic and cytogenetic testing, molecular monitoring, and BCR-ABL1 mutational analysis have become integral to the routine management of CML. The information that each type of test provides is essential to confirm a diagnosis, determine the disease stage, assess response to treatment, and monitor for signals of disease progression – all of which can be used to identify patients who might require further evaluation, closer follow-up, and additional intervention, and to guide clinical decisions.
Click on the PDF icon at the top of this introduction to read the full article.