User login
Limited treatment options exist for brittle nail syndrome
NEW ORLEANS – .
“The mainstay of treatment is irritant avoidance and moisturization,” Shari R. Lipner, MD, PhD, associate professor of clinical dermatology and director of the nail division at Weill Cornell Medicine, New York, said at the annual meeting of the American Academy of Dermatology. “This works well if patients are religious about doing it.”

Brittle nail syndrome affects about 20% of adults, she said, and is more common in females, particularly those older than age 50. Most cases are idiopathic, but some are secondary to dermatologic diseases including nail psoriasis and nail lichen planus, and systemic diseases such as hyperthyroidism and hypothyroidism. They are more common in patients in certain occupations such as carpentry. “The pathogenesis is poorly understood but is thought to be due to weakened intercellular keratinocyte bridges, decreased cholesterol sulphate in the nail plate, and reduced water content in the nail plate,” Dr. Lipner said.
Key clinical findings include onychoschizia (peeling of the nail plate), onychorrhexis (an increase in the longitudinal ridges and furrows, sometimes leading to splitting), and superficial granulation of keratin. Treatment involves general measures. “You want to treat the underlying cause and recommend that the patient avoid water and irritant exposure,” she said. Her general instructions for affected patients are to wear latex gloves for wet work and cotton gloves for dry work, avoid triclosan-based hand sanitizers, avoid nail cosmetics, minimize nail trauma, and foster moisturization.“It’s important to give these instructions verbally and in written form,” she said. “In our practice, we designed a QR code that links to our patient handout.”
According to Dr. Lipner, the promotion of vitamins and supplements such as biotin, vitamin D, amino acids, and chromium for treating brittle nail syndrome is rampant on the Internet and on social media, but no rigorously designed clinical trials have shown efficacy for any of them. “Very few people are deficient in biotin, except for those with inherited enzyme deficiencies,” and most people “can get all the biotin they need from a regular diet,” she said.
The initial rationale for using biotin for nails comes from the veterinary literature, she continued. In the 1940s, chickens with biotin deficiency developed fissures in their feet and parrot-like beaks. In the 1970s, pigs with biotin deficiency developed friable hooves, which was corrected with biotin supplementation. “By the 1980s it was standard practice to supplement the feet of pigs with biotin,” she said.
In a human trial from 1989, German researchers enrolled 71 patients with brittle nail syndrome who took oral biotin, 2.5 mg daily. Of the 45 patients evaluated, 41 (91%) showed improvement in firmness and hardness of the fingernails over the course of 5.5 months, but there was no good control group, Dr. Lipner said. In a follow-up study, the same German researchers used scanning electron microscopy to evaluate 22 patients with brittle nails who took oral biotin 2.5 mg daily and compared them with 10 patients with normal nails who did not take biotin. They found a 25% increase in nail plate thickness in the biotin group and onychoschizia resolved in 50% of patients who received biotin. “But again, there was no good control group,” Dr. Lipner said.
In a third study on the topic, researchers surveyed 46 patients who presented with onychorrhexis and/or onychoschizia on clinical exam and took 2.5 mg of biotin daily. Of the 35 survey respondents, 63% subjectively reported improvement in their nails at a mean of 2 months. “This is where we are today: There have been studies of only 80 patients that were done 25 years ago,” Dr. Lipner said. “That’s all of our evidence for biotin for the treatment of brittle nail syndrome.”
FDA warning about biotin
Additional cause for concern, she continued, is the safety communication issued by the FDA in 2017, stating that the use of biotin may interfere with certain lab tests such as thyroid tests and cardiac enzymes, in some cases leading to death. The safety communication was updated in 2019.
In 2018, Dr. Lipner and colleagues administered an anonymous survey to 447 patients at their clinic asking about their use of biotin supplements. Of the 447 patients, 34% reported current use of biotin. Among biotin users, 7% were aware of the FDA warning, 29% of respondents reported that it was recommended by either a primary care physician or a dermatologist, and 56% underwent laboratory testing while taking biotin. “It’s our duty to warn our patients about the evidence for biotin for treating brittle nails, and about this interference on laboratory tests,” Dr. Lipner said.
Other treatment options for brittle nail syndrome include two lacquers that are available by prescription. One contains hydroxypropyl chitosan, Equisetum arvense, and methylsulphonylmethane; the other contains 16% poly-ureaurethane, but has not been well studied. “These products can be very expensive if not covered by insurance,” Dr. Lipner said.
As an alternative, she recommends Nail Tek CITRA 2 Nail Strengthener, which is available for less than $10 from Walmart and other retailers.
Cyclosporine emulsion also has been studied for brittle nail syndrome, but results to date have been underwhelming. Dr. Lipner and colleagues are exploring the effect of platelet rich plasma for treating brittle nails on the premise that it will improve nail growth and promote healing, in a 16-week trial that has enrolled 10 patients and includes both a Physician Global Improvement Assessment (PGIA) and a Physician Global Assessment (PGA) score. “Our data is being analyzed by three independent nail experts, and we hope to report the findings next year,” she said.
Dr. Lipner reported having no disclosures relevant to her presentation.
NEW ORLEANS – .
“The mainstay of treatment is irritant avoidance and moisturization,” Shari R. Lipner, MD, PhD, associate professor of clinical dermatology and director of the nail division at Weill Cornell Medicine, New York, said at the annual meeting of the American Academy of Dermatology. “This works well if patients are religious about doing it.”
Brittle nail syndrome affects about 20% of adults, she said, and is more common in females, particularly those older than age 50. Most cases are idiopathic, but some are secondary to dermatologic diseases including nail psoriasis and nail lichen planus, and systemic diseases such as hyperthyroidism and hypothyroidism. They are more common in patients in certain occupations such as carpentry. “The pathogenesis is poorly understood but is thought to be due to weakened intercellular keratinocyte bridges, decreased cholesterol sulphate in the nail plate, and reduced water content in the nail plate,” Dr. Lipner said.
Key clinical findings include onychoschizia (peeling of the nail plate), onychorrhexis (an increase in the longitudinal ridges and furrows, sometimes leading to splitting), and superficial granulation of keratin. Treatment involves general measures. “You want to treat the underlying cause and recommend that the patient avoid water and irritant exposure,” she said. Her general instructions for affected patients are to wear latex gloves for wet work and cotton gloves for dry work, avoid triclosan-based hand sanitizers, avoid nail cosmetics, minimize nail trauma, and foster moisturization.“It’s important to give these instructions verbally and in written form,” she said. “In our practice, we designed a QR code that links to our patient handout.”
According to Dr. Lipner, the promotion of vitamins and supplements such as biotin, vitamin D, amino acids, and chromium for treating brittle nail syndrome is rampant on the Internet and on social media, but no rigorously designed clinical trials have shown efficacy for any of them. “Very few people are deficient in biotin, except for those with inherited enzyme deficiencies,” and most people “can get all the biotin they need from a regular diet,” she said.
The initial rationale for using biotin for nails comes from the veterinary literature, she continued. In the 1940s, chickens with biotin deficiency developed fissures in their feet and parrot-like beaks. In the 1970s, pigs with biotin deficiency developed friable hooves, which was corrected with biotin supplementation. “By the 1980s it was standard practice to supplement the feet of pigs with biotin,” she said.
In a human trial from 1989, German researchers enrolled 71 patients with brittle nail syndrome who took oral biotin, 2.5 mg daily. Of the 45 patients evaluated, 41 (91%) showed improvement in firmness and hardness of the fingernails over the course of 5.5 months, but there was no good control group, Dr. Lipner said. In a follow-up study, the same German researchers used scanning electron microscopy to evaluate 22 patients with brittle nails who took oral biotin 2.5 mg daily and compared them with 10 patients with normal nails who did not take biotin. They found a 25% increase in nail plate thickness in the biotin group and onychoschizia resolved in 50% of patients who received biotin. “But again, there was no good control group,” Dr. Lipner said.
In a third study on the topic, researchers surveyed 46 patients who presented with onychorrhexis and/or onychoschizia on clinical exam and took 2.5 mg of biotin daily. Of the 35 survey respondents, 63% subjectively reported improvement in their nails at a mean of 2 months. “This is where we are today: There have been studies of only 80 patients that were done 25 years ago,” Dr. Lipner said. “That’s all of our evidence for biotin for the treatment of brittle nail syndrome.”
FDA warning about biotin
Additional cause for concern, she continued, is the safety communication issued by the FDA in 2017, stating that the use of biotin may interfere with certain lab tests such as thyroid tests and cardiac enzymes, in some cases leading to death. The safety communication was updated in 2019.
In 2018, Dr. Lipner and colleagues administered an anonymous survey to 447 patients at their clinic asking about their use of biotin supplements. Of the 447 patients, 34% reported current use of biotin. Among biotin users, 7% were aware of the FDA warning, 29% of respondents reported that it was recommended by either a primary care physician or a dermatologist, and 56% underwent laboratory testing while taking biotin. “It’s our duty to warn our patients about the evidence for biotin for treating brittle nails, and about this interference on laboratory tests,” Dr. Lipner said.
Other treatment options for brittle nail syndrome include two lacquers that are available by prescription. One contains hydroxypropyl chitosan, Equisetum arvense, and methylsulphonylmethane; the other contains 16% poly-ureaurethane, but has not been well studied. “These products can be very expensive if not covered by insurance,” Dr. Lipner said.
As an alternative, she recommends Nail Tek CITRA 2 Nail Strengthener, which is available for less than $10 from Walmart and other retailers.
Cyclosporine emulsion also has been studied for brittle nail syndrome, but results to date have been underwhelming. Dr. Lipner and colleagues are exploring the effect of platelet rich plasma for treating brittle nails on the premise that it will improve nail growth and promote healing, in a 16-week trial that has enrolled 10 patients and includes both a Physician Global Improvement Assessment (PGIA) and a Physician Global Assessment (PGA) score. “Our data is being analyzed by three independent nail experts, and we hope to report the findings next year,” she said.
Dr. Lipner reported having no disclosures relevant to her presentation.
NEW ORLEANS – .
“The mainstay of treatment is irritant avoidance and moisturization,” Shari R. Lipner, MD, PhD, associate professor of clinical dermatology and director of the nail division at Weill Cornell Medicine, New York, said at the annual meeting of the American Academy of Dermatology. “This works well if patients are religious about doing it.”
Brittle nail syndrome affects about 20% of adults, she said, and is more common in females, particularly those older than age 50. Most cases are idiopathic, but some are secondary to dermatologic diseases including nail psoriasis and nail lichen planus, and systemic diseases such as hyperthyroidism and hypothyroidism. They are more common in patients in certain occupations such as carpentry. “The pathogenesis is poorly understood but is thought to be due to weakened intercellular keratinocyte bridges, decreased cholesterol sulphate in the nail plate, and reduced water content in the nail plate,” Dr. Lipner said.
Key clinical findings include onychoschizia (peeling of the nail plate), onychorrhexis (an increase in the longitudinal ridges and furrows, sometimes leading to splitting), and superficial granulation of keratin. Treatment involves general measures. “You want to treat the underlying cause and recommend that the patient avoid water and irritant exposure,” she said. Her general instructions for affected patients are to wear latex gloves for wet work and cotton gloves for dry work, avoid triclosan-based hand sanitizers, avoid nail cosmetics, minimize nail trauma, and foster moisturization.“It’s important to give these instructions verbally and in written form,” she said. “In our practice, we designed a QR code that links to our patient handout.”
According to Dr. Lipner, the promotion of vitamins and supplements such as biotin, vitamin D, amino acids, and chromium for treating brittle nail syndrome is rampant on the Internet and on social media, but no rigorously designed clinical trials have shown efficacy for any of them. “Very few people are deficient in biotin, except for those with inherited enzyme deficiencies,” and most people “can get all the biotin they need from a regular diet,” she said.
The initial rationale for using biotin for nails comes from the veterinary literature, she continued. In the 1940s, chickens with biotin deficiency developed fissures in their feet and parrot-like beaks. In the 1970s, pigs with biotin deficiency developed friable hooves, which was corrected with biotin supplementation. “By the 1980s it was standard practice to supplement the feet of pigs with biotin,” she said.
In a human trial from 1989, German researchers enrolled 71 patients with brittle nail syndrome who took oral biotin, 2.5 mg daily. Of the 45 patients evaluated, 41 (91%) showed improvement in firmness and hardness of the fingernails over the course of 5.5 months, but there was no good control group, Dr. Lipner said. In a follow-up study, the same German researchers used scanning electron microscopy to evaluate 22 patients with brittle nails who took oral biotin 2.5 mg daily and compared them with 10 patients with normal nails who did not take biotin. They found a 25% increase in nail plate thickness in the biotin group and onychoschizia resolved in 50% of patients who received biotin. “But again, there was no good control group,” Dr. Lipner said.
In a third study on the topic, researchers surveyed 46 patients who presented with onychorrhexis and/or onychoschizia on clinical exam and took 2.5 mg of biotin daily. Of the 35 survey respondents, 63% subjectively reported improvement in their nails at a mean of 2 months. “This is where we are today: There have been studies of only 80 patients that were done 25 years ago,” Dr. Lipner said. “That’s all of our evidence for biotin for the treatment of brittle nail syndrome.”
FDA warning about biotin
Additional cause for concern, she continued, is the safety communication issued by the FDA in 2017, stating that the use of biotin may interfere with certain lab tests such as thyroid tests and cardiac enzymes, in some cases leading to death. The safety communication was updated in 2019.
In 2018, Dr. Lipner and colleagues administered an anonymous survey to 447 patients at their clinic asking about their use of biotin supplements. Of the 447 patients, 34% reported current use of biotin. Among biotin users, 7% were aware of the FDA warning, 29% of respondents reported that it was recommended by either a primary care physician or a dermatologist, and 56% underwent laboratory testing while taking biotin. “It’s our duty to warn our patients about the evidence for biotin for treating brittle nails, and about this interference on laboratory tests,” Dr. Lipner said.
Other treatment options for brittle nail syndrome include two lacquers that are available by prescription. One contains hydroxypropyl chitosan, Equisetum arvense, and methylsulphonylmethane; the other contains 16% poly-ureaurethane, but has not been well studied. “These products can be very expensive if not covered by insurance,” Dr. Lipner said.
As an alternative, she recommends Nail Tek CITRA 2 Nail Strengthener, which is available for less than $10 from Walmart and other retailers.
Cyclosporine emulsion also has been studied for brittle nail syndrome, but results to date have been underwhelming. Dr. Lipner and colleagues are exploring the effect of platelet rich plasma for treating brittle nails on the premise that it will improve nail growth and promote healing, in a 16-week trial that has enrolled 10 patients and includes both a Physician Global Improvement Assessment (PGIA) and a Physician Global Assessment (PGA) score. “Our data is being analyzed by three independent nail experts, and we hope to report the findings next year,” she said.
Dr. Lipner reported having no disclosures relevant to her presentation.
AT AAD 2023
FDA Advisory panels consider easing isotretinoin requirements
for patients, pharmacies, and prescribers.
Isotretinoin, previously called Accutane, is marketed as Absorica, Absorica LD, Claravis, Amnesteem, Myorisan, and Zenatane.
In a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee, experts addressed ways to improve the modified iPLEDGE Risk Evaluation and Mitigation Strategy (iPLEDGE REMS) for isotretinoin that caused chaos after its rollout at the end of 2021.
In January 2022, problems were multiplying with the program for clinicians, pharmacists, and patients, causing extensive delays and prescription denials. In response, the FDA said it would continue to meet with the Isotretinoin Products Manufacturers Group (IPMG) to resolve problems.
March 28 was the first day of a 2-day meeting addressing what can be done to reduce burden with the iPLEDGE REMS while maintaining safety and preventing fetal exposure to the drug.
Key areas of concern
The meeting focused on several key areas.
The 19-day lockout period
The lockout is a current restriction for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the specified 7-day prescription window. Currently, those who miss the window must wait 19 days from the date of the first pregnancy test to take an additional pregnancy test to be eligible to receive the drug.
Lindsey Crist, PharmD, a risk management analyst for the FDA, who presented the FDA review committee’s analysis, acknowledged that the lockout period causes delays in treatment and adds frustration and costs.
She said it’s important to remember that the lockout applies only to the first prescription. “It’s intended as an additional layer of screening to detect pregnancy,” she said.
“At least 12 pregnancies have been identified during the 19-day lockout from March 2017–September of 2022,” she noted.
The FDA is looking to the advisory committee to provide recommendations on whether the lockout period should be changed.
Home testing
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory and home pregnancy tests have been allowed. The question now is whether home tests should continue to be allowed.
Ms. Crist said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” she said.
Ms. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Documenting counseling patients who cannot get pregnant
Currently, this documentation must be done monthly, primarily to counsel patients against drug sharing or giving blood. Proposed changes include extending the intervals for attestation or eliminating it to reduce burden on clinicians.
IPMG representative Gregory Wedin, PharmD, pharmacovigilance and risk management director for Upsher-Smith Laboratories, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while extending to 120 days would reduce burden on prescribers, it comes with risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Mr. Wedin said.
On March 29, the panel will hear more recommendations for and against modifications to iPLEDGE REMS and will vote on select modifications at the end of the meeting.
A version of this article first appeared on Medscape.com.
for patients, pharmacies, and prescribers.
Isotretinoin, previously called Accutane, is marketed as Absorica, Absorica LD, Claravis, Amnesteem, Myorisan, and Zenatane.
In a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee, experts addressed ways to improve the modified iPLEDGE Risk Evaluation and Mitigation Strategy (iPLEDGE REMS) for isotretinoin that caused chaos after its rollout at the end of 2021.
In January 2022, problems were multiplying with the program for clinicians, pharmacists, and patients, causing extensive delays and prescription denials. In response, the FDA said it would continue to meet with the Isotretinoin Products Manufacturers Group (IPMG) to resolve problems.
March 28 was the first day of a 2-day meeting addressing what can be done to reduce burden with the iPLEDGE REMS while maintaining safety and preventing fetal exposure to the drug.
Key areas of concern
The meeting focused on several key areas.
The 19-day lockout period
The lockout is a current restriction for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the specified 7-day prescription window. Currently, those who miss the window must wait 19 days from the date of the first pregnancy test to take an additional pregnancy test to be eligible to receive the drug.
Lindsey Crist, PharmD, a risk management analyst for the FDA, who presented the FDA review committee’s analysis, acknowledged that the lockout period causes delays in treatment and adds frustration and costs.
She said it’s important to remember that the lockout applies only to the first prescription. “It’s intended as an additional layer of screening to detect pregnancy,” she said.
“At least 12 pregnancies have been identified during the 19-day lockout from March 2017–September of 2022,” she noted.
The FDA is looking to the advisory committee to provide recommendations on whether the lockout period should be changed.
Home testing
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory and home pregnancy tests have been allowed. The question now is whether home tests should continue to be allowed.
Ms. Crist said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” she said.
Ms. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Documenting counseling patients who cannot get pregnant
Currently, this documentation must be done monthly, primarily to counsel patients against drug sharing or giving blood. Proposed changes include extending the intervals for attestation or eliminating it to reduce burden on clinicians.
IPMG representative Gregory Wedin, PharmD, pharmacovigilance and risk management director for Upsher-Smith Laboratories, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while extending to 120 days would reduce burden on prescribers, it comes with risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Mr. Wedin said.
On March 29, the panel will hear more recommendations for and against modifications to iPLEDGE REMS and will vote on select modifications at the end of the meeting.
A version of this article first appeared on Medscape.com.
for patients, pharmacies, and prescribers.
Isotretinoin, previously called Accutane, is marketed as Absorica, Absorica LD, Claravis, Amnesteem, Myorisan, and Zenatane.
In a joint meeting of the FDA’s Drug Safety and Risk Management Advisory Committee and Dermatologic and Ophthalmic Drugs Advisory Committee, experts addressed ways to improve the modified iPLEDGE Risk Evaluation and Mitigation Strategy (iPLEDGE REMS) for isotretinoin that caused chaos after its rollout at the end of 2021.
In January 2022, problems were multiplying with the program for clinicians, pharmacists, and patients, causing extensive delays and prescription denials. In response, the FDA said it would continue to meet with the Isotretinoin Products Manufacturers Group (IPMG) to resolve problems.
March 28 was the first day of a 2-day meeting addressing what can be done to reduce burden with the iPLEDGE REMS while maintaining safety and preventing fetal exposure to the drug.
Key areas of concern
The meeting focused on several key areas.
The 19-day lockout period
The lockout is a current restriction for patients who can become pregnant and do not pick up their first prescription of isotretinoin within the specified 7-day prescription window. Currently, those who miss the window must wait 19 days from the date of the first pregnancy test to take an additional pregnancy test to be eligible to receive the drug.
Lindsey Crist, PharmD, a risk management analyst for the FDA, who presented the FDA review committee’s analysis, acknowledged that the lockout period causes delays in treatment and adds frustration and costs.
She said it’s important to remember that the lockout applies only to the first prescription. “It’s intended as an additional layer of screening to detect pregnancy,” she said.
“At least 12 pregnancies have been identified during the 19-day lockout from March 2017–September of 2022,” she noted.
The FDA is looking to the advisory committee to provide recommendations on whether the lockout period should be changed.
Home testing
During the pandemic, iPLEDGE rules have been relaxed from having a pregnancy test done only at a Clinical Laboratory Improvement Amendments–certified laboratory and home pregnancy tests have been allowed. The question now is whether home tests should continue to be allowed.
Ms. Crist said that the FDA’s review committee recommends ending the allowance of home tests, citing insufficient data on use and the discovery of instances of falsification of pregnancy tests.
“One study at an academic medical center reviewed the medical records of 89 patients who used home pregnancy tests while taking isotretinoin during the public health emergency. It found that 15.7% submitted falsified pregnancy test results,” she said.
Ms. Crist added, however, that the review committee recommends allowing the tests to be done in a provider’s office as an alternative.
Documenting counseling patients who cannot get pregnant
Currently, this documentation must be done monthly, primarily to counsel patients against drug sharing or giving blood. Proposed changes include extending the intervals for attestation or eliminating it to reduce burden on clinicians.
IPMG representative Gregory Wedin, PharmD, pharmacovigilance and risk management director for Upsher-Smith Laboratories, said, “while we cannot support eliminating or extending the confirmation interval to a year, the [iPLEDGE] sponsors are agreeable [to] a 120-day confirmation interval.”
He said that while extending to 120 days would reduce burden on prescribers, it comes with risk in reducing oversight by a certified iPLEDGE prescriber and potentially increasing the risk for drug sharing.
“A patient may be more likely to share their drug with another person the further along with therapy they get as their condition improves,” Mr. Wedin said.
On March 29, the panel will hear more recommendations for and against modifications to iPLEDGE REMS and will vote on select modifications at the end of the meeting.
A version of this article first appeared on Medscape.com.
JAK inhibitor ivarmacitinib shows efficacy for atopic dermatitis in a pivotal trial
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.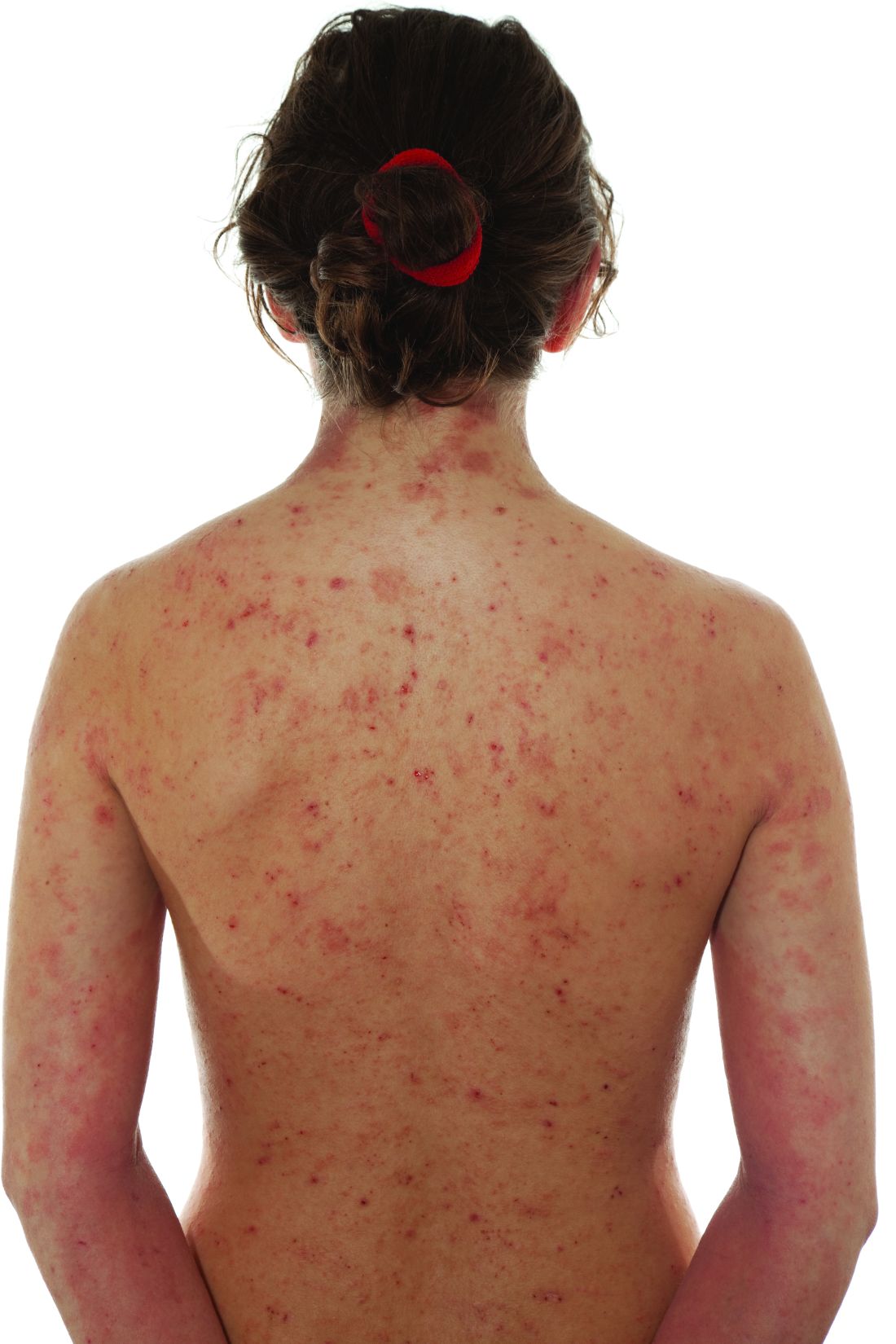
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
AT AAD 2023
COVID-19 potentially induced adult-onset IgA vasculitis
Plasma exchange successfully improved symptoms of immunoglobulin A vasculitis in an adult female patient who developed the condition after infection with COVID-19, according to a case report published in Cureus.
Immunoglobulin A (IgA) vasculitis can affect all ages, but is relatively rare in adults, and the etiology remains unclear, wrote Hassan Alwafi, MD, of Umm Al-Qura University, Makkah, Saudi Arabia, and colleagues.
COVID-19 has been associated with pulmonary and extrapulmonary complications, but , the authors wrote.
The authors described a case of a 41-year-old otherwise healthy Saudi Arabian woman who presented with an ascending rash on both lower extremities, along with arthralgia. Blood tests showed high blood urea nitrogen, creatinine, and inflammatory markers, and a negative immune panel. The patient had been infected with COVID-19 approximately 2 weeks before the onset of symptoms, but she was treated with supportive care and required no antiviral therapy of dexamethasone.
In addition, the patient’s urinalysis showed proteinuria and hematuria. After a kidney biopsy revealed additional abnormalities, the patient was started on intravenous methylprednisolone pulse therapy.
A few days after the initiation of therapy, the patient experienced nosebleeds and coughing up blood. After a chest x-ray showed bilateral pleural effusion, the patient was transferred to the ICU. The patient was started on intravenous piperacillin-tazobactam, and received two doses of intravenous immunoglobulin and plasma exchange after consultation with a nephrologist. Ultimately, the initial rash and other clinical symptoms improved, and the patient was discharged with a tapering schedule of oral prednisolone.
In this case, COVID-19 may have played a role in the development of IgA vasculitis, the authors said.
The authors also listed 21 cases of IgA vasculitis following COVID-19 infection, including 14 children and 7 adults. Of these, three cases had combined kidney and lung involvement, the two pediatric cases died from respiratory failure, while the adult case was successfully treated with steroid monotherapy.
“As COVID-19 is a novel disease and its pathogenic mechanism of causing IgA vasculitis is not well understood, every patient who is infected with or recently recovered from COVID-19 and presents with a skin rash or arthralgia should have baseline blood and urine tests done and should be treated promptly to avoid the emergence of irreversible consequences,” the authors wrote in their discussion.
Although case reports cannot prove a cause-and-effect link, the data from the cases in the current review suggest that COVID-19 infection may be an indirect trigger for IgA vasculitis, including cases associated with pulmonary renal syndrome, they said. However, more research is needed, especially on the efficacy of treatments in adults, they concluded.
The study received no outside funding. The researchers had no financial conflicts to disclose.
Plasma exchange successfully improved symptoms of immunoglobulin A vasculitis in an adult female patient who developed the condition after infection with COVID-19, according to a case report published in Cureus.
Immunoglobulin A (IgA) vasculitis can affect all ages, but is relatively rare in adults, and the etiology remains unclear, wrote Hassan Alwafi, MD, of Umm Al-Qura University, Makkah, Saudi Arabia, and colleagues.
COVID-19 has been associated with pulmonary and extrapulmonary complications, but , the authors wrote.
The authors described a case of a 41-year-old otherwise healthy Saudi Arabian woman who presented with an ascending rash on both lower extremities, along with arthralgia. Blood tests showed high blood urea nitrogen, creatinine, and inflammatory markers, and a negative immune panel. The patient had been infected with COVID-19 approximately 2 weeks before the onset of symptoms, but she was treated with supportive care and required no antiviral therapy of dexamethasone.
In addition, the patient’s urinalysis showed proteinuria and hematuria. After a kidney biopsy revealed additional abnormalities, the patient was started on intravenous methylprednisolone pulse therapy.
A few days after the initiation of therapy, the patient experienced nosebleeds and coughing up blood. After a chest x-ray showed bilateral pleural effusion, the patient was transferred to the ICU. The patient was started on intravenous piperacillin-tazobactam, and received two doses of intravenous immunoglobulin and plasma exchange after consultation with a nephrologist. Ultimately, the initial rash and other clinical symptoms improved, and the patient was discharged with a tapering schedule of oral prednisolone.
In this case, COVID-19 may have played a role in the development of IgA vasculitis, the authors said.
The authors also listed 21 cases of IgA vasculitis following COVID-19 infection, including 14 children and 7 adults. Of these, three cases had combined kidney and lung involvement, the two pediatric cases died from respiratory failure, while the adult case was successfully treated with steroid monotherapy.
“As COVID-19 is a novel disease and its pathogenic mechanism of causing IgA vasculitis is not well understood, every patient who is infected with or recently recovered from COVID-19 and presents with a skin rash or arthralgia should have baseline blood and urine tests done and should be treated promptly to avoid the emergence of irreversible consequences,” the authors wrote in their discussion.
Although case reports cannot prove a cause-and-effect link, the data from the cases in the current review suggest that COVID-19 infection may be an indirect trigger for IgA vasculitis, including cases associated with pulmonary renal syndrome, they said. However, more research is needed, especially on the efficacy of treatments in adults, they concluded.
The study received no outside funding. The researchers had no financial conflicts to disclose.
Plasma exchange successfully improved symptoms of immunoglobulin A vasculitis in an adult female patient who developed the condition after infection with COVID-19, according to a case report published in Cureus.
Immunoglobulin A (IgA) vasculitis can affect all ages, but is relatively rare in adults, and the etiology remains unclear, wrote Hassan Alwafi, MD, of Umm Al-Qura University, Makkah, Saudi Arabia, and colleagues.
COVID-19 has been associated with pulmonary and extrapulmonary complications, but , the authors wrote.
The authors described a case of a 41-year-old otherwise healthy Saudi Arabian woman who presented with an ascending rash on both lower extremities, along with arthralgia. Blood tests showed high blood urea nitrogen, creatinine, and inflammatory markers, and a negative immune panel. The patient had been infected with COVID-19 approximately 2 weeks before the onset of symptoms, but she was treated with supportive care and required no antiviral therapy of dexamethasone.
In addition, the patient’s urinalysis showed proteinuria and hematuria. After a kidney biopsy revealed additional abnormalities, the patient was started on intravenous methylprednisolone pulse therapy.
A few days after the initiation of therapy, the patient experienced nosebleeds and coughing up blood. After a chest x-ray showed bilateral pleural effusion, the patient was transferred to the ICU. The patient was started on intravenous piperacillin-tazobactam, and received two doses of intravenous immunoglobulin and plasma exchange after consultation with a nephrologist. Ultimately, the initial rash and other clinical symptoms improved, and the patient was discharged with a tapering schedule of oral prednisolone.
In this case, COVID-19 may have played a role in the development of IgA vasculitis, the authors said.
The authors also listed 21 cases of IgA vasculitis following COVID-19 infection, including 14 children and 7 adults. Of these, three cases had combined kidney and lung involvement, the two pediatric cases died from respiratory failure, while the adult case was successfully treated with steroid monotherapy.
“As COVID-19 is a novel disease and its pathogenic mechanism of causing IgA vasculitis is not well understood, every patient who is infected with or recently recovered from COVID-19 and presents with a skin rash or arthralgia should have baseline blood and urine tests done and should be treated promptly to avoid the emergence of irreversible consequences,” the authors wrote in their discussion.
Although case reports cannot prove a cause-and-effect link, the data from the cases in the current review suggest that COVID-19 infection may be an indirect trigger for IgA vasculitis, including cases associated with pulmonary renal syndrome, they said. However, more research is needed, especially on the efficacy of treatments in adults, they concluded.
The study received no outside funding. The researchers had no financial conflicts to disclose.
FROM CUREUS
Melatonin: A new way to reduce self-harm?
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CHILD PSYCHOLOGY AND PSYCHIATRY
COVID led to rise in pregnancy-related deaths: New research
The rise in deaths was most pronounced among Black mothers.
In 2021, 1,205 women died from pregnancy-related causes, making the year one of the worst for maternal mortality in U.S. history, according to newly released data from the Centers for Disease Control and Prevention. Maternal mortality is defined as occurring during pregnancy, at delivery, or soon after delivery.
COVID was the driver of the increased death rate, according to a study published in the journal Obstetrics & Gynecology. The researchers noted that unvaccinated pregnant people are more likely to get severe COVID, and that prenatal and postnatal care were disrupted during the early part of the pandemic. From July 2021 to March 2023, the rate of women being vaccinated before pregnancy has risen from 22% to 70%, CDC data show.
Maternal mortality rates jumped the most among Black women, who in 2021 had a maternal mortality rate of nearly 70 deaths per 100,000 live births, which was 2.6 times the rate for White women.
Existing risks based on a mother’s age also increased from 2020 to 2021. The maternal mortality rates by age in 2021 per 100,000 live births were:
- 20.4 for women under age 25.
- 31.3 for women ages 25 to 39.
- 138.5 for women ages 40 and older.
Iffath Abbasi Hoskins, MD, FACOG, president of the American College of Obstetricians and Gynecologists, called the situation “stunning” and “preventable.”
The findings “send a resounding message that maternal health and evidence-based efforts to eliminate racial health inequities need to be, and remain, a top public health priority,” Dr. Hoskins said in a statement.
“The COVID-19 pandemic had a dramatic and tragic effect on maternal death rates, but we cannot let that fact obscure that there was – and still is – already a maternal mortality crisis to compound,” she said.
A version of this article first appeared on WebMD.com.
The rise in deaths was most pronounced among Black mothers.
In 2021, 1,205 women died from pregnancy-related causes, making the year one of the worst for maternal mortality in U.S. history, according to newly released data from the Centers for Disease Control and Prevention. Maternal mortality is defined as occurring during pregnancy, at delivery, or soon after delivery.
COVID was the driver of the increased death rate, according to a study published in the journal Obstetrics & Gynecology. The researchers noted that unvaccinated pregnant people are more likely to get severe COVID, and that prenatal and postnatal care were disrupted during the early part of the pandemic. From July 2021 to March 2023, the rate of women being vaccinated before pregnancy has risen from 22% to 70%, CDC data show.
Maternal mortality rates jumped the most among Black women, who in 2021 had a maternal mortality rate of nearly 70 deaths per 100,000 live births, which was 2.6 times the rate for White women.
Existing risks based on a mother’s age also increased from 2020 to 2021. The maternal mortality rates by age in 2021 per 100,000 live births were:
- 20.4 for women under age 25.
- 31.3 for women ages 25 to 39.
- 138.5 for women ages 40 and older.
Iffath Abbasi Hoskins, MD, FACOG, president of the American College of Obstetricians and Gynecologists, called the situation “stunning” and “preventable.”
The findings “send a resounding message that maternal health and evidence-based efforts to eliminate racial health inequities need to be, and remain, a top public health priority,” Dr. Hoskins said in a statement.
“The COVID-19 pandemic had a dramatic and tragic effect on maternal death rates, but we cannot let that fact obscure that there was – and still is – already a maternal mortality crisis to compound,” she said.
A version of this article first appeared on WebMD.com.
The rise in deaths was most pronounced among Black mothers.
In 2021, 1,205 women died from pregnancy-related causes, making the year one of the worst for maternal mortality in U.S. history, according to newly released data from the Centers for Disease Control and Prevention. Maternal mortality is defined as occurring during pregnancy, at delivery, or soon after delivery.
COVID was the driver of the increased death rate, according to a study published in the journal Obstetrics & Gynecology. The researchers noted that unvaccinated pregnant people are more likely to get severe COVID, and that prenatal and postnatal care were disrupted during the early part of the pandemic. From July 2021 to March 2023, the rate of women being vaccinated before pregnancy has risen from 22% to 70%, CDC data show.
Maternal mortality rates jumped the most among Black women, who in 2021 had a maternal mortality rate of nearly 70 deaths per 100,000 live births, which was 2.6 times the rate for White women.
Existing risks based on a mother’s age also increased from 2020 to 2021. The maternal mortality rates by age in 2021 per 100,000 live births were:
- 20.4 for women under age 25.
- 31.3 for women ages 25 to 39.
- 138.5 for women ages 40 and older.
Iffath Abbasi Hoskins, MD, FACOG, president of the American College of Obstetricians and Gynecologists, called the situation “stunning” and “preventable.”
The findings “send a resounding message that maternal health and evidence-based efforts to eliminate racial health inequities need to be, and remain, a top public health priority,” Dr. Hoskins said in a statement.
“The COVID-19 pandemic had a dramatic and tragic effect on maternal death rates, but we cannot let that fact obscure that there was – and still is – already a maternal mortality crisis to compound,” she said.
A version of this article first appeared on WebMD.com.
Retinopathy ‘emerging decades earlier’ in kids with type 2 diabetes than in adults
Nearly one in four children diagnosed with type 2 diabetes for 5 years or more develop diabetic retinopathy, according to a new report.
The global prevalence of diabetic retinopathy in pediatric patients with type 2 diabetes is about 7%, which appears to increase with age.
“In our clinical practice, we have seen an increase in children presenting with type 2 diabetes over the past few years. These patients present with multiple simultaneous comorbidities and complications like hypertension, fatty liver, and other conditions,” senior author M. Constantine Samaan, MD, told this news organization.
“The exact scale of diabetes-related eye disease was not clear, and we decided to quantify it,” said Dr. Samaan, associate professor of pediatrics at McMaster University and pediatric endocrinologist at McMaster Children’s Hospital in Hamilton, Ont.
“What we found was that in pediatric patients with type 2 diabetes, diabetic retinopathy is present in 1 in 14 youth. The risk of retinopathy increased significantly 5 years after diagnosis to almost one in four,” he noted.
“While we acknowledged that the number of diabetic retinopathy cases was relatively small and there was heterogeneity in studies, we were surprised that retinopathy rates rose so fast in the first few years after diabetes diagnosis,” Dr. Samaan indicated.
The findings signal that the increase in the prevalence of diabetic retinopathy is emerging decades earlier among children compared with adults with type 2 diabetes, the authors wrote in their article published online in JAMA Network Open.
“While the guidelines for eye care in children with type 2 diabetes recommend screening at diagnosis and annually afterward, these recommendations are not followed in almost half of these patients,” Dr. Samaan said. “There is a need to ensure that patients get screened to try and prevent or delay retinopathy onset and progression.”
Analyzing prevalence rates
Diabetic retinopathy is the leading cause of blindness in patients with type 2 diabetes. Between 21% and 39% of adults have diabetic retinopathy at diagnosis, with rates subsequently increasing, the authors wrote.
Dr. Samaan and colleagues conducted a systematic review and meta-analysis to estimate the global prevalence of diabetic retinopathy in pediatric patients with type 2 diabetes. They included studies that had a study population of at least 10 participants diagnosed at age 21 and younger, an observational study design, and prevalence data on diabetic retinopathy.
Among the 29 studies included, 6 were cross-sectional, 13 had a retrospective cohort design, and 10 had a prospective cohort design. Patients were diagnosed between age 6.5 and 21 years, and the diabetes duration ranged from 0 to 15 years after diagnosis.
The overall global prevalence of diabetic retinopathy in 5,924 pediatric patients was 7.0%. Prevalence varied by study design, ranging from 1.1% in cross-sectional studies to 6.5% in prospective cohort studies and 11.3% in retrospective cohort studies.
In the nine studies that reported diabetic retinopathy classification based on criteria, the prevalence of minimal-to-moderate nonproliferative diabetic retinopathy was 11.2%, the prevalence of severe nonproliferative diabetic retinopathy was 2.6%, the prevalence of proliferative diabetic retinopathy was 2.4%, and the prevalence of macular edema was 3.1%.
In the five studies that reported diabetic retinopathy diagnosis using fundoscopy, the prevalence was 0.5%. In the four studies that used 7-field stereoscopic fundus photography, the prevalence was 13.6%.
In the pooled analysis of 27 studies, the prevalence of diabetic retinopathy was 1.8% less than 2.5 years after diabetes diagnosis but more than doubled to 5.1% in years 2.5 to 5 and jumped to 28.8% more than 5 years after diagnosis.
Differences by sex, ethnicity
“We were also surprised that there was very limited evidence to understand the sex and race differences in retinopathy risk,” said Dr. Samaan. “Further research is warranted, considering that more girls develop type 2 diabetes than boys, and the risk of type 2 diabetes is higher in some racial groups.”
In addition, older age, longer diabetes duration, and higher hypertension prevalence were associated with diabetic retinopathy prevalence. There were no associations with obesity prevalence or mean age at diabetes diagnosis. However, patients who developed diabetic retinopathy had a higher mean A1c level of 1.4% compared to those without retinopathy.
Dr. Samaan and colleagues are continuing to research the comorbidities and complications that children with type 2 diabetes face as well as mechanisms that drive diabetes outcomes among children and adolescents.
For now, the findings highlight the importance of retinopathy screening and personalized diabetes treatment to protect vision, Dr. Samaan reiterated.
No funding source for the study was reported. The authors have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Nearly one in four children diagnosed with type 2 diabetes for 5 years or more develop diabetic retinopathy, according to a new report.
The global prevalence of diabetic retinopathy in pediatric patients with type 2 diabetes is about 7%, which appears to increase with age.
“In our clinical practice, we have seen an increase in children presenting with type 2 diabetes over the past few years. These patients present with multiple simultaneous comorbidities and complications like hypertension, fatty liver, and other conditions,” senior author M. Constantine Samaan, MD, told this news organization.
“The exact scale of diabetes-related eye disease was not clear, and we decided to quantify it,” said Dr. Samaan, associate professor of pediatrics at McMaster University and pediatric endocrinologist at McMaster Children’s Hospital in Hamilton, Ont.
“What we found was that in pediatric patients with type 2 diabetes, diabetic retinopathy is present in 1 in 14 youth. The risk of retinopathy increased significantly 5 years after diagnosis to almost one in four,” he noted.
“While we acknowledged that the number of diabetic retinopathy cases was relatively small and there was heterogeneity in studies, we were surprised that retinopathy rates rose so fast in the first few years after diabetes diagnosis,” Dr. Samaan indicated.
The findings signal that the increase in the prevalence of diabetic retinopathy is emerging decades earlier among children compared with adults with type 2 diabetes, the authors wrote in their article published online in JAMA Network Open.
“While the guidelines for eye care in children with type 2 diabetes recommend screening at diagnosis and annually afterward, these recommendations are not followed in almost half of these patients,” Dr. Samaan said. “There is a need to ensure that patients get screened to try and prevent or delay retinopathy onset and progression.”
Analyzing prevalence rates
Diabetic retinopathy is the leading cause of blindness in patients with type 2 diabetes. Between 21% and 39% of adults have diabetic retinopathy at diagnosis, with rates subsequently increasing, the authors wrote.
Dr. Samaan and colleagues conducted a systematic review and meta-analysis to estimate the global prevalence of diabetic retinopathy in pediatric patients with type 2 diabetes. They included studies that had a study population of at least 10 participants diagnosed at age 21 and younger, an observational study design, and prevalence data on diabetic retinopathy.
Among the 29 studies included, 6 were cross-sectional, 13 had a retrospective cohort design, and 10 had a prospective cohort design. Patients were diagnosed between age 6.5 and 21 years, and the diabetes duration ranged from 0 to 15 years after diagnosis.
The overall global prevalence of diabetic retinopathy in 5,924 pediatric patients was 7.0%. Prevalence varied by study design, ranging from 1.1% in cross-sectional studies to 6.5% in prospective cohort studies and 11.3% in retrospective cohort studies.
In the nine studies that reported diabetic retinopathy classification based on criteria, the prevalence of minimal-to-moderate nonproliferative diabetic retinopathy was 11.2%, the prevalence of severe nonproliferative diabetic retinopathy was 2.6%, the prevalence of proliferative diabetic retinopathy was 2.4%, and the prevalence of macular edema was 3.1%.
In the five studies that reported diabetic retinopathy diagnosis using fundoscopy, the prevalence was 0.5%. In the four studies that used 7-field stereoscopic fundus photography, the prevalence was 13.6%.
In the pooled analysis of 27 studies, the prevalence of diabetic retinopathy was 1.8% less than 2.5 years after diabetes diagnosis but more than doubled to 5.1% in years 2.5 to 5 and jumped to 28.8% more than 5 years after diagnosis.
Differences by sex, ethnicity
“We were also surprised that there was very limited evidence to understand the sex and race differences in retinopathy risk,” said Dr. Samaan. “Further research is warranted, considering that more girls develop type 2 diabetes than boys, and the risk of type 2 diabetes is higher in some racial groups.”
In addition, older age, longer diabetes duration, and higher hypertension prevalence were associated with diabetic retinopathy prevalence. There were no associations with obesity prevalence or mean age at diabetes diagnosis. However, patients who developed diabetic retinopathy had a higher mean A1c level of 1.4% compared to those without retinopathy.
Dr. Samaan and colleagues are continuing to research the comorbidities and complications that children with type 2 diabetes face as well as mechanisms that drive diabetes outcomes among children and adolescents.
For now, the findings highlight the importance of retinopathy screening and personalized diabetes treatment to protect vision, Dr. Samaan reiterated.
No funding source for the study was reported. The authors have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Nearly one in four children diagnosed with type 2 diabetes for 5 years or more develop diabetic retinopathy, according to a new report.
The global prevalence of diabetic retinopathy in pediatric patients with type 2 diabetes is about 7%, which appears to increase with age.
“In our clinical practice, we have seen an increase in children presenting with type 2 diabetes over the past few years. These patients present with multiple simultaneous comorbidities and complications like hypertension, fatty liver, and other conditions,” senior author M. Constantine Samaan, MD, told this news organization.
“The exact scale of diabetes-related eye disease was not clear, and we decided to quantify it,” said Dr. Samaan, associate professor of pediatrics at McMaster University and pediatric endocrinologist at McMaster Children’s Hospital in Hamilton, Ont.
“What we found was that in pediatric patients with type 2 diabetes, diabetic retinopathy is present in 1 in 14 youth. The risk of retinopathy increased significantly 5 years after diagnosis to almost one in four,” he noted.
“While we acknowledged that the number of diabetic retinopathy cases was relatively small and there was heterogeneity in studies, we were surprised that retinopathy rates rose so fast in the first few years after diabetes diagnosis,” Dr. Samaan indicated.
The findings signal that the increase in the prevalence of diabetic retinopathy is emerging decades earlier among children compared with adults with type 2 diabetes, the authors wrote in their article published online in JAMA Network Open.
“While the guidelines for eye care in children with type 2 diabetes recommend screening at diagnosis and annually afterward, these recommendations are not followed in almost half of these patients,” Dr. Samaan said. “There is a need to ensure that patients get screened to try and prevent or delay retinopathy onset and progression.”
Analyzing prevalence rates
Diabetic retinopathy is the leading cause of blindness in patients with type 2 diabetes. Between 21% and 39% of adults have diabetic retinopathy at diagnosis, with rates subsequently increasing, the authors wrote.
Dr. Samaan and colleagues conducted a systematic review and meta-analysis to estimate the global prevalence of diabetic retinopathy in pediatric patients with type 2 diabetes. They included studies that had a study population of at least 10 participants diagnosed at age 21 and younger, an observational study design, and prevalence data on diabetic retinopathy.
Among the 29 studies included, 6 were cross-sectional, 13 had a retrospective cohort design, and 10 had a prospective cohort design. Patients were diagnosed between age 6.5 and 21 years, and the diabetes duration ranged from 0 to 15 years after diagnosis.
The overall global prevalence of diabetic retinopathy in 5,924 pediatric patients was 7.0%. Prevalence varied by study design, ranging from 1.1% in cross-sectional studies to 6.5% in prospective cohort studies and 11.3% in retrospective cohort studies.
In the nine studies that reported diabetic retinopathy classification based on criteria, the prevalence of minimal-to-moderate nonproliferative diabetic retinopathy was 11.2%, the prevalence of severe nonproliferative diabetic retinopathy was 2.6%, the prevalence of proliferative diabetic retinopathy was 2.4%, and the prevalence of macular edema was 3.1%.
In the five studies that reported diabetic retinopathy diagnosis using fundoscopy, the prevalence was 0.5%. In the four studies that used 7-field stereoscopic fundus photography, the prevalence was 13.6%.
In the pooled analysis of 27 studies, the prevalence of diabetic retinopathy was 1.8% less than 2.5 years after diabetes diagnosis but more than doubled to 5.1% in years 2.5 to 5 and jumped to 28.8% more than 5 years after diagnosis.
Differences by sex, ethnicity
“We were also surprised that there was very limited evidence to understand the sex and race differences in retinopathy risk,” said Dr. Samaan. “Further research is warranted, considering that more girls develop type 2 diabetes than boys, and the risk of type 2 diabetes is higher in some racial groups.”
In addition, older age, longer diabetes duration, and higher hypertension prevalence were associated with diabetic retinopathy prevalence. There were no associations with obesity prevalence or mean age at diabetes diagnosis. However, patients who developed diabetic retinopathy had a higher mean A1c level of 1.4% compared to those without retinopathy.
Dr. Samaan and colleagues are continuing to research the comorbidities and complications that children with type 2 diabetes face as well as mechanisms that drive diabetes outcomes among children and adolescents.
For now, the findings highlight the importance of retinopathy screening and personalized diabetes treatment to protect vision, Dr. Samaan reiterated.
No funding source for the study was reported. The authors have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
New state bill could protect docs prescribing abortion pills to out-of-state patients
California lawmakers are considering legislation to protect California physicians and pharmacists who prescribe abortion pills to out-of-state patients. The proposed law would shield health care providers who are legally performing their jobs in California from facing prosecution in another state or being extradited.
State Sen. Nancy Skinner, who introduced the bill, said the legislation is necessary in a fractured, post-Roe legal landscape where doctors in some states can face felony charges or civil penalties for providing reproductive health care. It’s part of a package of 17 new bills aiming to “strengthen California’s standing as a safe haven for abortion, contraception, and pregnancy care,” according to a press release.
“I’m trying to protect our healthcare practitioners so they can do their jobs, without fear,” Ms. Skinner said in a statement on March 24.
Most abortions are banned in 14 states after the Supreme Court overturned Roe v. Wade. Lawmakers in those states have established a variety of penalties for doctors, pharmacists, and other clinicians to provide abortion care or assist patients in obtaining abortions, including jail time, fines, and loss of professional licenses.
As a result, doctors in restrictive states have anguished over having to delay treatment for patients experiencing miscarriages, ectopic pregnancies, and other conditions until their lives are enough at risk to satisfy exceptions to state abortion laws.
“As a physician, I believe everyone deserves the care they need, regardless of where they live,” said Daniel Grossman, MD, a University of California, San Francisco, ob.gyn. professor who directs the university’s Advancing New Standards in Reproductive Health program.
“Since the fall of Roe v. Wade, patients are being forced to travel long distances – often over 500 miles – to access abortion care in a clinic. People should be able to access this essential care closer to home, including by telemedicine, which has been shown to be safe and effective. I am hopeful that SB 345 will provide additional legal protections that would allow California clinicians to help patients in other states,” he stated.
Other states, including New York, Vermont, New Jersey, Massachusetts, and Connecticut, have passed or are considering similar legislation to protect doctors using telemedicine to prescribe abortion medication to out-of-state patients. These laws come amid a growing push by some states and anti-abortion groups to severely restrict access to abortion pills.
Wyoming is the first state to explicitly ban the pills, although a judge on March 22 blocked that ban. And, in a closely watched case, a conservative federal judge could soon rule to ban sales of mifepristone, one of the medications in a two-pill regimen approved for abortions early in pregnancy.
California’s legislation protects clinicians from losing their California professional licenses if an out-of-state medical board takes action against them. It also allows clinicians to sue anyone who tries to legally interfere with the care they are providing.
It also covers California physicians prescribing contraceptives or gender-affirming care to out-of-state patients. At least 21 states are considering restrictions on gender-affirming care for minors and another 9 states have passed them, according to the advocacy group Human Rights Campaign. Courts have blocked the restrictions in some states.
“It’s understandable that states like California want to reassure their doctors ... that, if one of their patients is caught in one of those states and can’t get help locally, they can step up to help and feel safe in doing so,” said Matthew Wynia, MD, MPH, FACP, director of the Center for Bioethics and Humanities at the University of Colorado at Denver, Aurora.
“This is also a crazy development in terms of the law. It’s just one part of the legal mayhem that was predicted when the Supreme Court overturned Roe,” Dr. Wynia said of the growing number of bills protecting in-state doctors. These bills “will almost certainly end up being litigated over issues of interstate commerce, cross-state licensure and practice compacts, FDA regulations and authorities, and maybe more. It’s a huge mess, in which both doctors and patients are being hurt.”
A version of this article first appeared on Medscape.com.
California lawmakers are considering legislation to protect California physicians and pharmacists who prescribe abortion pills to out-of-state patients. The proposed law would shield health care providers who are legally performing their jobs in California from facing prosecution in another state or being extradited.
State Sen. Nancy Skinner, who introduced the bill, said the legislation is necessary in a fractured, post-Roe legal landscape where doctors in some states can face felony charges or civil penalties for providing reproductive health care. It’s part of a package of 17 new bills aiming to “strengthen California’s standing as a safe haven for abortion, contraception, and pregnancy care,” according to a press release.
“I’m trying to protect our healthcare practitioners so they can do their jobs, without fear,” Ms. Skinner said in a statement on March 24.
Most abortions are banned in 14 states after the Supreme Court overturned Roe v. Wade. Lawmakers in those states have established a variety of penalties for doctors, pharmacists, and other clinicians to provide abortion care or assist patients in obtaining abortions, including jail time, fines, and loss of professional licenses.
As a result, doctors in restrictive states have anguished over having to delay treatment for patients experiencing miscarriages, ectopic pregnancies, and other conditions until their lives are enough at risk to satisfy exceptions to state abortion laws.
“As a physician, I believe everyone deserves the care they need, regardless of where they live,” said Daniel Grossman, MD, a University of California, San Francisco, ob.gyn. professor who directs the university’s Advancing New Standards in Reproductive Health program.
“Since the fall of Roe v. Wade, patients are being forced to travel long distances – often over 500 miles – to access abortion care in a clinic. People should be able to access this essential care closer to home, including by telemedicine, which has been shown to be safe and effective. I am hopeful that SB 345 will provide additional legal protections that would allow California clinicians to help patients in other states,” he stated.
Other states, including New York, Vermont, New Jersey, Massachusetts, and Connecticut, have passed or are considering similar legislation to protect doctors using telemedicine to prescribe abortion medication to out-of-state patients. These laws come amid a growing push by some states and anti-abortion groups to severely restrict access to abortion pills.
Wyoming is the first state to explicitly ban the pills, although a judge on March 22 blocked that ban. And, in a closely watched case, a conservative federal judge could soon rule to ban sales of mifepristone, one of the medications in a two-pill regimen approved for abortions early in pregnancy.
California’s legislation protects clinicians from losing their California professional licenses if an out-of-state medical board takes action against them. It also allows clinicians to sue anyone who tries to legally interfere with the care they are providing.
It also covers California physicians prescribing contraceptives or gender-affirming care to out-of-state patients. At least 21 states are considering restrictions on gender-affirming care for minors and another 9 states have passed them, according to the advocacy group Human Rights Campaign. Courts have blocked the restrictions in some states.
“It’s understandable that states like California want to reassure their doctors ... that, if one of their patients is caught in one of those states and can’t get help locally, they can step up to help and feel safe in doing so,” said Matthew Wynia, MD, MPH, FACP, director of the Center for Bioethics and Humanities at the University of Colorado at Denver, Aurora.
“This is also a crazy development in terms of the law. It’s just one part of the legal mayhem that was predicted when the Supreme Court overturned Roe,” Dr. Wynia said of the growing number of bills protecting in-state doctors. These bills “will almost certainly end up being litigated over issues of interstate commerce, cross-state licensure and practice compacts, FDA regulations and authorities, and maybe more. It’s a huge mess, in which both doctors and patients are being hurt.”
A version of this article first appeared on Medscape.com.
California lawmakers are considering legislation to protect California physicians and pharmacists who prescribe abortion pills to out-of-state patients. The proposed law would shield health care providers who are legally performing their jobs in California from facing prosecution in another state or being extradited.
State Sen. Nancy Skinner, who introduced the bill, said the legislation is necessary in a fractured, post-Roe legal landscape where doctors in some states can face felony charges or civil penalties for providing reproductive health care. It’s part of a package of 17 new bills aiming to “strengthen California’s standing as a safe haven for abortion, contraception, and pregnancy care,” according to a press release.
“I’m trying to protect our healthcare practitioners so they can do their jobs, without fear,” Ms. Skinner said in a statement on March 24.
Most abortions are banned in 14 states after the Supreme Court overturned Roe v. Wade. Lawmakers in those states have established a variety of penalties for doctors, pharmacists, and other clinicians to provide abortion care or assist patients in obtaining abortions, including jail time, fines, and loss of professional licenses.
As a result, doctors in restrictive states have anguished over having to delay treatment for patients experiencing miscarriages, ectopic pregnancies, and other conditions until their lives are enough at risk to satisfy exceptions to state abortion laws.
“As a physician, I believe everyone deserves the care they need, regardless of where they live,” said Daniel Grossman, MD, a University of California, San Francisco, ob.gyn. professor who directs the university’s Advancing New Standards in Reproductive Health program.
“Since the fall of Roe v. Wade, patients are being forced to travel long distances – often over 500 miles – to access abortion care in a clinic. People should be able to access this essential care closer to home, including by telemedicine, which has been shown to be safe and effective. I am hopeful that SB 345 will provide additional legal protections that would allow California clinicians to help patients in other states,” he stated.
Other states, including New York, Vermont, New Jersey, Massachusetts, and Connecticut, have passed or are considering similar legislation to protect doctors using telemedicine to prescribe abortion medication to out-of-state patients. These laws come amid a growing push by some states and anti-abortion groups to severely restrict access to abortion pills.
Wyoming is the first state to explicitly ban the pills, although a judge on March 22 blocked that ban. And, in a closely watched case, a conservative federal judge could soon rule to ban sales of mifepristone, one of the medications in a two-pill regimen approved for abortions early in pregnancy.
California’s legislation protects clinicians from losing their California professional licenses if an out-of-state medical board takes action against them. It also allows clinicians to sue anyone who tries to legally interfere with the care they are providing.
It also covers California physicians prescribing contraceptives or gender-affirming care to out-of-state patients. At least 21 states are considering restrictions on gender-affirming care for minors and another 9 states have passed them, according to the advocacy group Human Rights Campaign. Courts have blocked the restrictions in some states.
“It’s understandable that states like California want to reassure their doctors ... that, if one of their patients is caught in one of those states and can’t get help locally, they can step up to help and feel safe in doing so,” said Matthew Wynia, MD, MPH, FACP, director of the Center for Bioethics and Humanities at the University of Colorado at Denver, Aurora.
“This is also a crazy development in terms of the law. It’s just one part of the legal mayhem that was predicted when the Supreme Court overturned Roe,” Dr. Wynia said of the growing number of bills protecting in-state doctors. These bills “will almost certainly end up being litigated over issues of interstate commerce, cross-state licensure and practice compacts, FDA regulations and authorities, and maybe more. It’s a huge mess, in which both doctors and patients are being hurt.”
A version of this article first appeared on Medscape.com.
DMARDs taper-to-discontinuation trial deemed inconclusive
The small size of a new study of the feasibility of tapering conventional synthetic disease-modifying antirheumatic drug (csDMARD) doses to half for patients with rheumatoid arthritis in remission, and then to zero, makes suspect the validity of its finding of no statistical difference between continuing half doses and stopping altogether, according to one rheumatologist’s analysis.
In the open-label, randomized trial of 56 patients, which was published as a research letter in JAMA, more patients in the group that discontinued csDMARDs experienced flares within 1 year than did the half-dose group, but this difference was not statistically significant.
Most patients in the drug-free group did not experience disease flares, the authors note.
“The results show that in this population, a majority of patients remained flare-free for at least a year after csDMARD discontinuation. This highlights a potential for drug-free remission in a subgroup of RA patients, and the data provide a basis for shared decision-making in this patient group. We know that tapering is a common question from patients and thus think that the data are especially clinically relevant,” first author Siri Lillegraven, MD, MPH, PhD, of the Center for Treatment of Rheumatic and Musculoskeletal Diseases at the Diakonhjemmet Hospital, Oslo, said in an interview.
While several studies have demonstrated that patients with RA can maintain remission on lower doses of medication, James O’Dell, MD, chief of the Division of Rheumatology at the University of Nebraska Medical Center, Omaha, urged caution in interpreting these results because the study was so small – just 56 patients. “Every analysis they did favored staying on treatment, but the confidence interval slightly crossed null, so they can’t say [that group] was superior,” Dr. O’Dell said in an interview. He was not involved in the research. “Had this study been double this size and they got the same results, they would have clearly shown that staying on medicines were superior,” he said.
Dr. Lillegraven acknowledged the impact of the trial’s small sample size. “This is a study with a limited study sample, and it is conceivable that a larger study might have shown a statistical difference between the groups,” she said.
In addition to the small number of patients in the study, Dr. O’Dell also noted that this study group was already a selected group of patients who had maintained remission on half-dose therapy for at least 1 year. Even then, “what they showed was that 39% of the patients who they discontinued [then] flared, compared with 17% when they didn’t taper [off medication],” he said. “That’s a pretty important clinical difference.”
While Dr. O’Dell thinks the study was too small to inform practice, he emphasized that tapering off full doses of medications can be beneficial for patients with RA that has been in remission for 6 months or longer. “It seems to take less medicines to keep somebody in remission than it did to get them there in the first place,” he said. “I come out strongly in favor of tapering medications in rheumatoid arthritis patients who are in remission, and that includes tapering and stopping biologics if patients are on conventional therapy,” he added, “but tapering patients off all of their conventional therapy is something that I think is a bridge too far.”
This trial was the second part of the ARCTIC REWIND study, which involved patients with RA that was in sustained remission, per their Disease Activity Score. In the first part of the trial, 160 participants from 10 hospitals in Norway were enrolled and were randomly assigned to either continue their standard csDMARD dosing or taper down to a half dose. Patients whose doses were tapered to a half dose and whose conditions were in remission for 1 year were eligible for the second half the study.
Of the 56 participants who were included, 26 discontinued csDMARD therapy, while 30 continued taking a half dose for 12 months of follow-up. Most patients in both groups had received methotrexate monotherapy (21 in the discontinuation group and 26 in the continued half-dosing group). Triple therapy (methotrexate, sulfasalazine, and hydroxychloroquine) was used by three patients in the discontinuation group and by two in the half-dose group. Two additional patients in the discontinuation group and two in the half-dose group took other mono/duo therapies. Clinic visits occurred every 4 months; visits were more frequent if there was an increase in disease activity. For patients who experienced a disease flare, full-dose csDMARD treatment was resumed.
Ten patients in the discontinuation group experienced flares during 1 year, compared with five patients in the half-dose group. The risk difference between the two groups was not statistically significant (RD, 21.5%; 95% CI, –3.4% to 49.7%). The median time to flare was 179 days in the discontinuation group and 133 days in the half-dose group.
Of those who experienced flares, 8 of 10 patients in the discontinuation group and 2 of 5 in the half-dose group regained remission when full-dose therapy was resumed.
The study was funded by the Research Council of Norway and the South-Eastern Norway Regional Health Authorities. Many of the authors disclosed financial ties to pharmaceutical companies. Dr. O’Dell disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The small size of a new study of the feasibility of tapering conventional synthetic disease-modifying antirheumatic drug (csDMARD) doses to half for patients with rheumatoid arthritis in remission, and then to zero, makes suspect the validity of its finding of no statistical difference between continuing half doses and stopping altogether, according to one rheumatologist’s analysis.
In the open-label, randomized trial of 56 patients, which was published as a research letter in JAMA, more patients in the group that discontinued csDMARDs experienced flares within 1 year than did the half-dose group, but this difference was not statistically significant.
Most patients in the drug-free group did not experience disease flares, the authors note.
“The results show that in this population, a majority of patients remained flare-free for at least a year after csDMARD discontinuation. This highlights a potential for drug-free remission in a subgroup of RA patients, and the data provide a basis for shared decision-making in this patient group. We know that tapering is a common question from patients and thus think that the data are especially clinically relevant,” first author Siri Lillegraven, MD, MPH, PhD, of the Center for Treatment of Rheumatic and Musculoskeletal Diseases at the Diakonhjemmet Hospital, Oslo, said in an interview.
While several studies have demonstrated that patients with RA can maintain remission on lower doses of medication, James O’Dell, MD, chief of the Division of Rheumatology at the University of Nebraska Medical Center, Omaha, urged caution in interpreting these results because the study was so small – just 56 patients. “Every analysis they did favored staying on treatment, but the confidence interval slightly crossed null, so they can’t say [that group] was superior,” Dr. O’Dell said in an interview. He was not involved in the research. “Had this study been double this size and they got the same results, they would have clearly shown that staying on medicines were superior,” he said.
Dr. Lillegraven acknowledged the impact of the trial’s small sample size. “This is a study with a limited study sample, and it is conceivable that a larger study might have shown a statistical difference between the groups,” she said.
In addition to the small number of patients in the study, Dr. O’Dell also noted that this study group was already a selected group of patients who had maintained remission on half-dose therapy for at least 1 year. Even then, “what they showed was that 39% of the patients who they discontinued [then] flared, compared with 17% when they didn’t taper [off medication],” he said. “That’s a pretty important clinical difference.”
While Dr. O’Dell thinks the study was too small to inform practice, he emphasized that tapering off full doses of medications can be beneficial for patients with RA that has been in remission for 6 months or longer. “It seems to take less medicines to keep somebody in remission than it did to get them there in the first place,” he said. “I come out strongly in favor of tapering medications in rheumatoid arthritis patients who are in remission, and that includes tapering and stopping biologics if patients are on conventional therapy,” he added, “but tapering patients off all of their conventional therapy is something that I think is a bridge too far.”
This trial was the second part of the ARCTIC REWIND study, which involved patients with RA that was in sustained remission, per their Disease Activity Score. In the first part of the trial, 160 participants from 10 hospitals in Norway were enrolled and were randomly assigned to either continue their standard csDMARD dosing or taper down to a half dose. Patients whose doses were tapered to a half dose and whose conditions were in remission for 1 year were eligible for the second half the study.
Of the 56 participants who were included, 26 discontinued csDMARD therapy, while 30 continued taking a half dose for 12 months of follow-up. Most patients in both groups had received methotrexate monotherapy (21 in the discontinuation group and 26 in the continued half-dosing group). Triple therapy (methotrexate, sulfasalazine, and hydroxychloroquine) was used by three patients in the discontinuation group and by two in the half-dose group. Two additional patients in the discontinuation group and two in the half-dose group took other mono/duo therapies. Clinic visits occurred every 4 months; visits were more frequent if there was an increase in disease activity. For patients who experienced a disease flare, full-dose csDMARD treatment was resumed.
Ten patients in the discontinuation group experienced flares during 1 year, compared with five patients in the half-dose group. The risk difference between the two groups was not statistically significant (RD, 21.5%; 95% CI, –3.4% to 49.7%). The median time to flare was 179 days in the discontinuation group and 133 days in the half-dose group.
Of those who experienced flares, 8 of 10 patients in the discontinuation group and 2 of 5 in the half-dose group regained remission when full-dose therapy was resumed.
The study was funded by the Research Council of Norway and the South-Eastern Norway Regional Health Authorities. Many of the authors disclosed financial ties to pharmaceutical companies. Dr. O’Dell disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The small size of a new study of the feasibility of tapering conventional synthetic disease-modifying antirheumatic drug (csDMARD) doses to half for patients with rheumatoid arthritis in remission, and then to zero, makes suspect the validity of its finding of no statistical difference between continuing half doses and stopping altogether, according to one rheumatologist’s analysis.
In the open-label, randomized trial of 56 patients, which was published as a research letter in JAMA, more patients in the group that discontinued csDMARDs experienced flares within 1 year than did the half-dose group, but this difference was not statistically significant.
Most patients in the drug-free group did not experience disease flares, the authors note.
“The results show that in this population, a majority of patients remained flare-free for at least a year after csDMARD discontinuation. This highlights a potential for drug-free remission in a subgroup of RA patients, and the data provide a basis for shared decision-making in this patient group. We know that tapering is a common question from patients and thus think that the data are especially clinically relevant,” first author Siri Lillegraven, MD, MPH, PhD, of the Center for Treatment of Rheumatic and Musculoskeletal Diseases at the Diakonhjemmet Hospital, Oslo, said in an interview.
While several studies have demonstrated that patients with RA can maintain remission on lower doses of medication, James O’Dell, MD, chief of the Division of Rheumatology at the University of Nebraska Medical Center, Omaha, urged caution in interpreting these results because the study was so small – just 56 patients. “Every analysis they did favored staying on treatment, but the confidence interval slightly crossed null, so they can’t say [that group] was superior,” Dr. O’Dell said in an interview. He was not involved in the research. “Had this study been double this size and they got the same results, they would have clearly shown that staying on medicines were superior,” he said.
Dr. Lillegraven acknowledged the impact of the trial’s small sample size. “This is a study with a limited study sample, and it is conceivable that a larger study might have shown a statistical difference between the groups,” she said.
In addition to the small number of patients in the study, Dr. O’Dell also noted that this study group was already a selected group of patients who had maintained remission on half-dose therapy for at least 1 year. Even then, “what they showed was that 39% of the patients who they discontinued [then] flared, compared with 17% when they didn’t taper [off medication],” he said. “That’s a pretty important clinical difference.”
While Dr. O’Dell thinks the study was too small to inform practice, he emphasized that tapering off full doses of medications can be beneficial for patients with RA that has been in remission for 6 months or longer. “It seems to take less medicines to keep somebody in remission than it did to get them there in the first place,” he said. “I come out strongly in favor of tapering medications in rheumatoid arthritis patients who are in remission, and that includes tapering and stopping biologics if patients are on conventional therapy,” he added, “but tapering patients off all of their conventional therapy is something that I think is a bridge too far.”
This trial was the second part of the ARCTIC REWIND study, which involved patients with RA that was in sustained remission, per their Disease Activity Score. In the first part of the trial, 160 participants from 10 hospitals in Norway were enrolled and were randomly assigned to either continue their standard csDMARD dosing or taper down to a half dose. Patients whose doses were tapered to a half dose and whose conditions were in remission for 1 year were eligible for the second half the study.
Of the 56 participants who were included, 26 discontinued csDMARD therapy, while 30 continued taking a half dose for 12 months of follow-up. Most patients in both groups had received methotrexate monotherapy (21 in the discontinuation group and 26 in the continued half-dosing group). Triple therapy (methotrexate, sulfasalazine, and hydroxychloroquine) was used by three patients in the discontinuation group and by two in the half-dose group. Two additional patients in the discontinuation group and two in the half-dose group took other mono/duo therapies. Clinic visits occurred every 4 months; visits were more frequent if there was an increase in disease activity. For patients who experienced a disease flare, full-dose csDMARD treatment was resumed.
Ten patients in the discontinuation group experienced flares during 1 year, compared with five patients in the half-dose group. The risk difference between the two groups was not statistically significant (RD, 21.5%; 95% CI, –3.4% to 49.7%). The median time to flare was 179 days in the discontinuation group and 133 days in the half-dose group.
Of those who experienced flares, 8 of 10 patients in the discontinuation group and 2 of 5 in the half-dose group regained remission when full-dose therapy was resumed.
The study was funded by the Research Council of Norway and the South-Eastern Norway Regional Health Authorities. Many of the authors disclosed financial ties to pharmaceutical companies. Dr. O’Dell disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM JAMA
Safety, efficacy of analgesics for low back pain ‘uncertain’
Higher-quality randomized controlled trials of head-to-head comparisons are needed, study investigator Michael A. Wewege, PhD candidate, research fellow, University of New South Wales and Neuroscience Research Australia, Sydney, said in an interview.
“Until then, doctors should use caution when prescribing analgesic medicines for adults with nonspecific acute low back pain. They should use this new evidence in line with their own expertise and the patient sitting in front of them when making any decision about a medication,” he added.
The findings were published online in the BMJ.
Poor quality evidence
Analgesics such as ibuprofen, acetaminophen, and codeine are widely used to treat nonspecific low-back pain, which is defined as pain lasting less than 6 weeks, but evidence for the comparative efficacy of these agents is limited.
To fill this knowledge gap, the researchers conducted a systematic review and analysis of controlled trials comparing analgesics with another analgesic, placebo, or no treatment in patients with acute, nonspecific low back pain.
The review involved 98 randomized controlled trials that included 15,134 adults (49% women) aged 30-60 years with pain duration ranging from 24 hours to 21 days. The median baseline pain intensity was 65 on a pain scale of 0-100.
Of the included trials, 39% were placebo controlled, 67% masked both participants and clinicians, and 41% reported industry sponsorship.
The studies compared an analgesic medicine with another analgesic, placebo, or no treatment comprised of usual care or being placed on a wait list.
Study medications, which had to be approved in the United States, Europe, or Australia, included nonsteroidal anti-inflammatory drugs, paracetamol, opioids, anticonvulsants, antidepressants, muscle relaxants, and corticosteroids.
These drugs were administered systemically as a single drug or in combination formulations, at any dose.
Researchers used a network meta-analysis, which combines direct and indirect information across a network of randomized clinical trials to estimate the comparative effectiveness of multiple treatments.
The primary outcomes were reductions in low back pain intensity (measured with a visual analogue scale), numerical rating scale or another ordinal scale, and safety as indicated by the number of participants who had any adverse event.
Investigators found several medications were associated with large reductions in pain intensity, compared with placebo, though with low or very low confidence.
Low or very low confidence was found for reduced pain intensity after treatment with tolperisone (mean difference, −26.1; 95% confidence interval, −34.0 to −18.2), aceclofenac plus tizanidine (mean difference, −26.1; 95% CI, −38.5 to −13.6), pregabalin (mean difference, −24.7; 95% CI, −34.6 to −14.7), and 14 other medicines, compared with placebo, the researchers report.
In addition, they found low or very low confidence for no difference between the effects of several of these medications.
Increased adverse events had moderate to very low confidence with tramadol (risk ratio, 2.6; 95% CI, 1.5-4.5), paracetamol plus sustained release tramadol (RR, 2.4; 95% CI, 1.5-3.8), baclofen (RR, 2.3; 95% CI, 1.5-3.4), and paracetamol plus tramadol (RR, 2.1; 95% CI, 1.3-3.4), compared with placebo, the investigators add.
“These medicines could increase the risk of adverse events, compared with other medicines with moderate to low confidence. Moderate to low confidence was also noted for secondary outcomes and secondary analysis of medicine classes,” the researchers note.
The review suggested 14 additional comparisons favored the treatment over placebo, all with very low confidence except for one with low confidence.
In the 68 trials that included the number of participants reporting an adverse event, there was moderate confidence for increased adverse events with the opioid tramadol (RR, 2.6; 95% CI, 1.5-4.5), paracetamol plus sustained release tramadol (RR, 2.4; 95% CI, 1.5-3.8), paracetamol plus tramadol (RR, 2.1; 95% CI, 1.3-3.4), and low confidence for baclofen (RR, 2.3; 1.5-3.4), compared with placebo.
The review also uncovered moderate to low confidence for secondary outcomes, which included low back-specific function, serious adverse events, and acceptability (number of participants who dropped out).
Unexpected findings
The new results were somewhat unexpected, said Mr. Wewege.
“When we set out to do this review, we envisioned the evidence would be a lot more comprehensive. We didn’t think it would be so disconnected and there would be so few trials looking at the different comparisons that would lead us to have low confidence in most of the findings.”
Various factors contributed to this low confidence, he said. One was the risk of bias – about 90% of trials had some concerns or high risk of bias. Another factor was the heterogeneity in effect estimates.
Most of the evidence is based on studies comparing different analgesics to placebo, Mr. Wewege noted. The lack of head-to-head drug comparisons is because “the easiest way to get a drug approved is just to demonstrate it’s better than placebo,” he said.
In addition to these new findings, clinicians should consider a medication’s availability, their own expertise, and patient preferences when selecting an analgesic, said Mr. Wewege. He noted most patients with acute low back pain get better within a few weeks without any intervention.
“Patients should be reassured that things will heal naturally and that they are not going to be in pain forever,” he said.
Determining optimal treatment is key
Chris Gilligan, MD, associate chief medical officer, Brigham and Women’s Hospital, and associate professor of anesthesia, Harvard Medical School, both in Boston, said determining which medications are optimal is “key,” as acute low back pain is very common and analgesics are used frequently.
The new review does provide information on which medications have the strongest evidence for pain reduction, said Dr. Gilligan. “On the one hand, it directionally points you towards certain medications, and even certain classes of medication, for comparative effectiveness.”
However, he said, the confidence for this effectiveness is low or very low, “so I wouldn’t overweight it.”
The data on adverse effects, where the confidence is mostly moderate to low, might have more of an influence on prescribing, he said.
“For example, there’s some indication tramadol may be more closely associated with adverse events in patients with acute low back pain and that would add to our caution about using tramadol; it’s not that we would never use it, but [we]would take that into account.”
Dr. Gilligan agrees clinicians should be cautious about prescribing analgesics for low back pain. One reason for being conservative in terms of treatments, he noted, is that “acute low back pain has a very favorable natural history.”
While clinical practice guidelines recommend nonpharmacologic therapies as first- and second-line treatment for acute, nonspecific low back pain, Dr. Gilligan noted that as with drugs, evidence for nondrug therapies also has low or very low confidence.
The study received funding from a 2020 Exercise Physiology Research (Consumables) Grant from the University of New South Wales, which was used to obtain translations of studies published in languages other than English.
Mr. Wewege was supported by a Postgraduate Scholarship from the National Health and Medical Research Council of Australia, a School of Medical Sciences Top-Up Scholarship from the University of New South Wales, and a PhD Supplementary Scholarship from Neuroscience Research Australia. Dr. Gilligan reports that he conducts clinical trials with companies and groups, including the National Institutes of Health related to medications, devices, and procedures for pain.
A version of this article first appeared on Medscape.com.
Higher-quality randomized controlled trials of head-to-head comparisons are needed, study investigator Michael A. Wewege, PhD candidate, research fellow, University of New South Wales and Neuroscience Research Australia, Sydney, said in an interview.
“Until then, doctors should use caution when prescribing analgesic medicines for adults with nonspecific acute low back pain. They should use this new evidence in line with their own expertise and the patient sitting in front of them when making any decision about a medication,” he added.
The findings were published online in the BMJ.
Poor quality evidence
Analgesics such as ibuprofen, acetaminophen, and codeine are widely used to treat nonspecific low-back pain, which is defined as pain lasting less than 6 weeks, but evidence for the comparative efficacy of these agents is limited.
To fill this knowledge gap, the researchers conducted a systematic review and analysis of controlled trials comparing analgesics with another analgesic, placebo, or no treatment in patients with acute, nonspecific low back pain.
The review involved 98 randomized controlled trials that included 15,134 adults (49% women) aged 30-60 years with pain duration ranging from 24 hours to 21 days. The median baseline pain intensity was 65 on a pain scale of 0-100.
Of the included trials, 39% were placebo controlled, 67% masked both participants and clinicians, and 41% reported industry sponsorship.
The studies compared an analgesic medicine with another analgesic, placebo, or no treatment comprised of usual care or being placed on a wait list.
Study medications, which had to be approved in the United States, Europe, or Australia, included nonsteroidal anti-inflammatory drugs, paracetamol, opioids, anticonvulsants, antidepressants, muscle relaxants, and corticosteroids.
These drugs were administered systemically as a single drug or in combination formulations, at any dose.
Researchers used a network meta-analysis, which combines direct and indirect information across a network of randomized clinical trials to estimate the comparative effectiveness of multiple treatments.
The primary outcomes were reductions in low back pain intensity (measured with a visual analogue scale), numerical rating scale or another ordinal scale, and safety as indicated by the number of participants who had any adverse event.
Investigators found several medications were associated with large reductions in pain intensity, compared with placebo, though with low or very low confidence.
Low or very low confidence was found for reduced pain intensity after treatment with tolperisone (mean difference, −26.1; 95% confidence interval, −34.0 to −18.2), aceclofenac plus tizanidine (mean difference, −26.1; 95% CI, −38.5 to −13.6), pregabalin (mean difference, −24.7; 95% CI, −34.6 to −14.7), and 14 other medicines, compared with placebo, the researchers report.
In addition, they found low or very low confidence for no difference between the effects of several of these medications.
Increased adverse events had moderate to very low confidence with tramadol (risk ratio, 2.6; 95% CI, 1.5-4.5), paracetamol plus sustained release tramadol (RR, 2.4; 95% CI, 1.5-3.8), baclofen (RR, 2.3; 95% CI, 1.5-3.4), and paracetamol plus tramadol (RR, 2.1; 95% CI, 1.3-3.4), compared with placebo, the investigators add.
“These medicines could increase the risk of adverse events, compared with other medicines with moderate to low confidence. Moderate to low confidence was also noted for secondary outcomes and secondary analysis of medicine classes,” the researchers note.
The review suggested 14 additional comparisons favored the treatment over placebo, all with very low confidence except for one with low confidence.
In the 68 trials that included the number of participants reporting an adverse event, there was moderate confidence for increased adverse events with the opioid tramadol (RR, 2.6; 95% CI, 1.5-4.5), paracetamol plus sustained release tramadol (RR, 2.4; 95% CI, 1.5-3.8), paracetamol plus tramadol (RR, 2.1; 95% CI, 1.3-3.4), and low confidence for baclofen (RR, 2.3; 1.5-3.4), compared with placebo.
The review also uncovered moderate to low confidence for secondary outcomes, which included low back-specific function, serious adverse events, and acceptability (number of participants who dropped out).
Unexpected findings
The new results were somewhat unexpected, said Mr. Wewege.
“When we set out to do this review, we envisioned the evidence would be a lot more comprehensive. We didn’t think it would be so disconnected and there would be so few trials looking at the different comparisons that would lead us to have low confidence in most of the findings.”
Various factors contributed to this low confidence, he said. One was the risk of bias – about 90% of trials had some concerns or high risk of bias. Another factor was the heterogeneity in effect estimates.
Most of the evidence is based on studies comparing different analgesics to placebo, Mr. Wewege noted. The lack of head-to-head drug comparisons is because “the easiest way to get a drug approved is just to demonstrate it’s better than placebo,” he said.
In addition to these new findings, clinicians should consider a medication’s availability, their own expertise, and patient preferences when selecting an analgesic, said Mr. Wewege. He noted most patients with acute low back pain get better within a few weeks without any intervention.
“Patients should be reassured that things will heal naturally and that they are not going to be in pain forever,” he said.
Determining optimal treatment is key
Chris Gilligan, MD, associate chief medical officer, Brigham and Women’s Hospital, and associate professor of anesthesia, Harvard Medical School, both in Boston, said determining which medications are optimal is “key,” as acute low back pain is very common and analgesics are used frequently.
The new review does provide information on which medications have the strongest evidence for pain reduction, said Dr. Gilligan. “On the one hand, it directionally points you towards certain medications, and even certain classes of medication, for comparative effectiveness.”
However, he said, the confidence for this effectiveness is low or very low, “so I wouldn’t overweight it.”
The data on adverse effects, where the confidence is mostly moderate to low, might have more of an influence on prescribing, he said.
“For example, there’s some indication tramadol may be more closely associated with adverse events in patients with acute low back pain and that would add to our caution about using tramadol; it’s not that we would never use it, but [we]would take that into account.”
Dr. Gilligan agrees clinicians should be cautious about prescribing analgesics for low back pain. One reason for being conservative in terms of treatments, he noted, is that “acute low back pain has a very favorable natural history.”
While clinical practice guidelines recommend nonpharmacologic therapies as first- and second-line treatment for acute, nonspecific low back pain, Dr. Gilligan noted that as with drugs, evidence for nondrug therapies also has low or very low confidence.
The study received funding from a 2020 Exercise Physiology Research (Consumables) Grant from the University of New South Wales, which was used to obtain translations of studies published in languages other than English.
Mr. Wewege was supported by a Postgraduate Scholarship from the National Health and Medical Research Council of Australia, a School of Medical Sciences Top-Up Scholarship from the University of New South Wales, and a PhD Supplementary Scholarship from Neuroscience Research Australia. Dr. Gilligan reports that he conducts clinical trials with companies and groups, including the National Institutes of Health related to medications, devices, and procedures for pain.
A version of this article first appeared on Medscape.com.
Higher-quality randomized controlled trials of head-to-head comparisons are needed, study investigator Michael A. Wewege, PhD candidate, research fellow, University of New South Wales and Neuroscience Research Australia, Sydney, said in an interview.
“Until then, doctors should use caution when prescribing analgesic medicines for adults with nonspecific acute low back pain. They should use this new evidence in line with their own expertise and the patient sitting in front of them when making any decision about a medication,” he added.
The findings were published online in the BMJ.
Poor quality evidence
Analgesics such as ibuprofen, acetaminophen, and codeine are widely used to treat nonspecific low-back pain, which is defined as pain lasting less than 6 weeks, but evidence for the comparative efficacy of these agents is limited.
To fill this knowledge gap, the researchers conducted a systematic review and analysis of controlled trials comparing analgesics with another analgesic, placebo, or no treatment in patients with acute, nonspecific low back pain.
The review involved 98 randomized controlled trials that included 15,134 adults (49% women) aged 30-60 years with pain duration ranging from 24 hours to 21 days. The median baseline pain intensity was 65 on a pain scale of 0-100.
Of the included trials, 39% were placebo controlled, 67% masked both participants and clinicians, and 41% reported industry sponsorship.
The studies compared an analgesic medicine with another analgesic, placebo, or no treatment comprised of usual care or being placed on a wait list.
Study medications, which had to be approved in the United States, Europe, or Australia, included nonsteroidal anti-inflammatory drugs, paracetamol, opioids, anticonvulsants, antidepressants, muscle relaxants, and corticosteroids.
These drugs were administered systemically as a single drug or in combination formulations, at any dose.
Researchers used a network meta-analysis, which combines direct and indirect information across a network of randomized clinical trials to estimate the comparative effectiveness of multiple treatments.
The primary outcomes were reductions in low back pain intensity (measured with a visual analogue scale), numerical rating scale or another ordinal scale, and safety as indicated by the number of participants who had any adverse event.
Investigators found several medications were associated with large reductions in pain intensity, compared with placebo, though with low or very low confidence.
Low or very low confidence was found for reduced pain intensity after treatment with tolperisone (mean difference, −26.1; 95% confidence interval, −34.0 to −18.2), aceclofenac plus tizanidine (mean difference, −26.1; 95% CI, −38.5 to −13.6), pregabalin (mean difference, −24.7; 95% CI, −34.6 to −14.7), and 14 other medicines, compared with placebo, the researchers report.
In addition, they found low or very low confidence for no difference between the effects of several of these medications.
Increased adverse events had moderate to very low confidence with tramadol (risk ratio, 2.6; 95% CI, 1.5-4.5), paracetamol plus sustained release tramadol (RR, 2.4; 95% CI, 1.5-3.8), baclofen (RR, 2.3; 95% CI, 1.5-3.4), and paracetamol plus tramadol (RR, 2.1; 95% CI, 1.3-3.4), compared with placebo, the investigators add.
“These medicines could increase the risk of adverse events, compared with other medicines with moderate to low confidence. Moderate to low confidence was also noted for secondary outcomes and secondary analysis of medicine classes,” the researchers note.
The review suggested 14 additional comparisons favored the treatment over placebo, all with very low confidence except for one with low confidence.
In the 68 trials that included the number of participants reporting an adverse event, there was moderate confidence for increased adverse events with the opioid tramadol (RR, 2.6; 95% CI, 1.5-4.5), paracetamol plus sustained release tramadol (RR, 2.4; 95% CI, 1.5-3.8), paracetamol plus tramadol (RR, 2.1; 95% CI, 1.3-3.4), and low confidence for baclofen (RR, 2.3; 1.5-3.4), compared with placebo.
The review also uncovered moderate to low confidence for secondary outcomes, which included low back-specific function, serious adverse events, and acceptability (number of participants who dropped out).
Unexpected findings
The new results were somewhat unexpected, said Mr. Wewege.
“When we set out to do this review, we envisioned the evidence would be a lot more comprehensive. We didn’t think it would be so disconnected and there would be so few trials looking at the different comparisons that would lead us to have low confidence in most of the findings.”
Various factors contributed to this low confidence, he said. One was the risk of bias – about 90% of trials had some concerns or high risk of bias. Another factor was the heterogeneity in effect estimates.
Most of the evidence is based on studies comparing different analgesics to placebo, Mr. Wewege noted. The lack of head-to-head drug comparisons is because “the easiest way to get a drug approved is just to demonstrate it’s better than placebo,” he said.
In addition to these new findings, clinicians should consider a medication’s availability, their own expertise, and patient preferences when selecting an analgesic, said Mr. Wewege. He noted most patients with acute low back pain get better within a few weeks without any intervention.
“Patients should be reassured that things will heal naturally and that they are not going to be in pain forever,” he said.
Determining optimal treatment is key
Chris Gilligan, MD, associate chief medical officer, Brigham and Women’s Hospital, and associate professor of anesthesia, Harvard Medical School, both in Boston, said determining which medications are optimal is “key,” as acute low back pain is very common and analgesics are used frequently.
The new review does provide information on which medications have the strongest evidence for pain reduction, said Dr. Gilligan. “On the one hand, it directionally points you towards certain medications, and even certain classes of medication, for comparative effectiveness.”
However, he said, the confidence for this effectiveness is low or very low, “so I wouldn’t overweight it.”
The data on adverse effects, where the confidence is mostly moderate to low, might have more of an influence on prescribing, he said.
“For example, there’s some indication tramadol may be more closely associated with adverse events in patients with acute low back pain and that would add to our caution about using tramadol; it’s not that we would never use it, but [we]would take that into account.”
Dr. Gilligan agrees clinicians should be cautious about prescribing analgesics for low back pain. One reason for being conservative in terms of treatments, he noted, is that “acute low back pain has a very favorable natural history.”
While clinical practice guidelines recommend nonpharmacologic therapies as first- and second-line treatment for acute, nonspecific low back pain, Dr. Gilligan noted that as with drugs, evidence for nondrug therapies also has low or very low confidence.
The study received funding from a 2020 Exercise Physiology Research (Consumables) Grant from the University of New South Wales, which was used to obtain translations of studies published in languages other than English.
Mr. Wewege was supported by a Postgraduate Scholarship from the National Health and Medical Research Council of Australia, a School of Medical Sciences Top-Up Scholarship from the University of New South Wales, and a PhD Supplementary Scholarship from Neuroscience Research Australia. Dr. Gilligan reports that he conducts clinical trials with companies and groups, including the National Institutes of Health related to medications, devices, and procedures for pain.
A version of this article first appeared on Medscape.com.
FROM BMJ