User login
CDC issues new pneumococcal vaccine recommendations for adults
The recommendations, voted on by the CDC’s Advisory Committee on Immunization Practices (ACIP) in October and made final in January with publication in the agency’s Morbidity and Mortality Weekly Report (MMWR), call for use of the 15-valent pneumococcal conjugate vaccine (PCV15; Vaxneuvance, Merck Sharp & Dohme) or 20-valent PCV (PREVNAR20; Wyeth Pharmaceuticals).
The recommendations apply to PCV-naive adults in the United States who are either aged 65 years or older, or who are aged 19-64 years and have underlying conditions such as diabetes, chronic heart or liver disease, or HIV, and have not previously received a PCV or whose previous vaccination history is unknown.
If the PCV15 vaccine is used, a subsequent dose of the 23-valent pneumococcal polysaccharide vaccine (PPSV23; Pneumovax23, Merck Sharp & Dohme) should be provided, typically at least 1 year later, under the recommendations.
As reported by this news organization, PCV15 and PREVNAR20 received approval from the Food and Drug Administration last July.
Those approvals provided an impetus for the revised recommendations, “offer[ing] an opportunity to review the existing recommendations and available data,” Miwako Kobayashi, MD, first author of the MMWR report and a medical epidemiologist with the National Center for Immunization and Respiratory Diseases, CDC, in Atlanta, said in an interview.
“As part of that process, ACIP strived to simplify the recommendations,” she said.
The previous recommendations called for the PCV13 vaccine and the PPSV23 and had varying conditions (depending on certain age and risk groups) that added complexity to the process. Under the new approach, the same recommendation applies regardless of specific medical conditions or other risk factors.
“With the simplified recommendation for adults 19 through 64, we expect coverage may increase among this population,” Dr. Kobayashi said.
Compared with the PCV13 vaccine, PREVNAR20 protects against seven additional serotypes involved in cases of invasive pneumococcal disease (IPD) and pneumonia, which are responsible for up to 40% of all cases of pneumococcal disease and related deaths in the United States.
While the PREVNAR20 includes five more pneumococcal serotypes than PCV15, the
CDC does not recommend one over the other, Dr. Kobayashi noted.
More than 90% of cases of adult IPD involve older adults and adults with chronic medical conditions or immunocompromising conditions, cerebrospinal fluid leaks, or cochlear implants, the MMWR report notes.
Commenting on the recommendations, Amit A. Shah, MD, a geriatrician with the Mayo Clinic in Phoenix, Ariz., underscored the need for clinicians to be proactive in recommending the vaccines to those patients.
“Despite only needing one vaccine dose after turning 65 to be considered vaccinated, only about 70% of people in this group have received any pneumococcal vaccination,” he said in an interview. “This percentage has not increased much over the past several years.”
The new approach should help change that, he said.
“These new recommendations are a significant simplification from the prior confusing and challenging-to-implement recommendations from 2019,” Dr. Shah explained.
Among the 2019 recommendations was a stipulation for “shared decision-making” with PCV13, and a conversation that often only complicated matters, he noted.
“Patients and providers alike had confusion about this since it was not a clear-cut ‘yes, give it’ or ‘no, do not give it any longer’ recommendation.”
“Now that this new recommendation will require no extra time for a discussion in the clinic, and just a simple ‘it’s time for your pneumonia shot’ offer, this may become more feasible,” Dr. Shah added. “In addition, removal of the shared decision-making stipulation allows for this immunization to be easily protocolized in the clinic, similar to automatic offers to the flu vaccine for patients each year.”
According to the CDC, pneumococcal pneumonia causes an estimated 150,000 hospitalizations each year in the United States, while pneumococcal meningitis and bacteremia killed approximately 3,250 people in the United States in 2019.
“Clinicians are patients’ most trusted resource when it comes to vaccine recommendations,” Dr. Kobayashi said. “We encourage all clinicians to recommend pneumococcal vaccines when indicated.”
Dr. Kobayashi and Dr. Shah have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The recommendations, voted on by the CDC’s Advisory Committee on Immunization Practices (ACIP) in October and made final in January with publication in the agency’s Morbidity and Mortality Weekly Report (MMWR), call for use of the 15-valent pneumococcal conjugate vaccine (PCV15; Vaxneuvance, Merck Sharp & Dohme) or 20-valent PCV (PREVNAR20; Wyeth Pharmaceuticals).
The recommendations apply to PCV-naive adults in the United States who are either aged 65 years or older, or who are aged 19-64 years and have underlying conditions such as diabetes, chronic heart or liver disease, or HIV, and have not previously received a PCV or whose previous vaccination history is unknown.
If the PCV15 vaccine is used, a subsequent dose of the 23-valent pneumococcal polysaccharide vaccine (PPSV23; Pneumovax23, Merck Sharp & Dohme) should be provided, typically at least 1 year later, under the recommendations.
As reported by this news organization, PCV15 and PREVNAR20 received approval from the Food and Drug Administration last July.
Those approvals provided an impetus for the revised recommendations, “offer[ing] an opportunity to review the existing recommendations and available data,” Miwako Kobayashi, MD, first author of the MMWR report and a medical epidemiologist with the National Center for Immunization and Respiratory Diseases, CDC, in Atlanta, said in an interview.
“As part of that process, ACIP strived to simplify the recommendations,” she said.
The previous recommendations called for the PCV13 vaccine and the PPSV23 and had varying conditions (depending on certain age and risk groups) that added complexity to the process. Under the new approach, the same recommendation applies regardless of specific medical conditions or other risk factors.
“With the simplified recommendation for adults 19 through 64, we expect coverage may increase among this population,” Dr. Kobayashi said.
Compared with the PCV13 vaccine, PREVNAR20 protects against seven additional serotypes involved in cases of invasive pneumococcal disease (IPD) and pneumonia, which are responsible for up to 40% of all cases of pneumococcal disease and related deaths in the United States.
While the PREVNAR20 includes five more pneumococcal serotypes than PCV15, the
CDC does not recommend one over the other, Dr. Kobayashi noted.
More than 90% of cases of adult IPD involve older adults and adults with chronic medical conditions or immunocompromising conditions, cerebrospinal fluid leaks, or cochlear implants, the MMWR report notes.
Commenting on the recommendations, Amit A. Shah, MD, a geriatrician with the Mayo Clinic in Phoenix, Ariz., underscored the need for clinicians to be proactive in recommending the vaccines to those patients.
“Despite only needing one vaccine dose after turning 65 to be considered vaccinated, only about 70% of people in this group have received any pneumococcal vaccination,” he said in an interview. “This percentage has not increased much over the past several years.”
The new approach should help change that, he said.
“These new recommendations are a significant simplification from the prior confusing and challenging-to-implement recommendations from 2019,” Dr. Shah explained.
Among the 2019 recommendations was a stipulation for “shared decision-making” with PCV13, and a conversation that often only complicated matters, he noted.
“Patients and providers alike had confusion about this since it was not a clear-cut ‘yes, give it’ or ‘no, do not give it any longer’ recommendation.”
“Now that this new recommendation will require no extra time for a discussion in the clinic, and just a simple ‘it’s time for your pneumonia shot’ offer, this may become more feasible,” Dr. Shah added. “In addition, removal of the shared decision-making stipulation allows for this immunization to be easily protocolized in the clinic, similar to automatic offers to the flu vaccine for patients each year.”
According to the CDC, pneumococcal pneumonia causes an estimated 150,000 hospitalizations each year in the United States, while pneumococcal meningitis and bacteremia killed approximately 3,250 people in the United States in 2019.
“Clinicians are patients’ most trusted resource when it comes to vaccine recommendations,” Dr. Kobayashi said. “We encourage all clinicians to recommend pneumococcal vaccines when indicated.”
Dr. Kobayashi and Dr. Shah have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The recommendations, voted on by the CDC’s Advisory Committee on Immunization Practices (ACIP) in October and made final in January with publication in the agency’s Morbidity and Mortality Weekly Report (MMWR), call for use of the 15-valent pneumococcal conjugate vaccine (PCV15; Vaxneuvance, Merck Sharp & Dohme) or 20-valent PCV (PREVNAR20; Wyeth Pharmaceuticals).
The recommendations apply to PCV-naive adults in the United States who are either aged 65 years or older, or who are aged 19-64 years and have underlying conditions such as diabetes, chronic heart or liver disease, or HIV, and have not previously received a PCV or whose previous vaccination history is unknown.
If the PCV15 vaccine is used, a subsequent dose of the 23-valent pneumococcal polysaccharide vaccine (PPSV23; Pneumovax23, Merck Sharp & Dohme) should be provided, typically at least 1 year later, under the recommendations.
As reported by this news organization, PCV15 and PREVNAR20 received approval from the Food and Drug Administration last July.
Those approvals provided an impetus for the revised recommendations, “offer[ing] an opportunity to review the existing recommendations and available data,” Miwako Kobayashi, MD, first author of the MMWR report and a medical epidemiologist with the National Center for Immunization and Respiratory Diseases, CDC, in Atlanta, said in an interview.
“As part of that process, ACIP strived to simplify the recommendations,” she said.
The previous recommendations called for the PCV13 vaccine and the PPSV23 and had varying conditions (depending on certain age and risk groups) that added complexity to the process. Under the new approach, the same recommendation applies regardless of specific medical conditions or other risk factors.
“With the simplified recommendation for adults 19 through 64, we expect coverage may increase among this population,” Dr. Kobayashi said.
Compared with the PCV13 vaccine, PREVNAR20 protects against seven additional serotypes involved in cases of invasive pneumococcal disease (IPD) and pneumonia, which are responsible for up to 40% of all cases of pneumococcal disease and related deaths in the United States.
While the PREVNAR20 includes five more pneumococcal serotypes than PCV15, the
CDC does not recommend one over the other, Dr. Kobayashi noted.
More than 90% of cases of adult IPD involve older adults and adults with chronic medical conditions or immunocompromising conditions, cerebrospinal fluid leaks, or cochlear implants, the MMWR report notes.
Commenting on the recommendations, Amit A. Shah, MD, a geriatrician with the Mayo Clinic in Phoenix, Ariz., underscored the need for clinicians to be proactive in recommending the vaccines to those patients.
“Despite only needing one vaccine dose after turning 65 to be considered vaccinated, only about 70% of people in this group have received any pneumococcal vaccination,” he said in an interview. “This percentage has not increased much over the past several years.”
The new approach should help change that, he said.
“These new recommendations are a significant simplification from the prior confusing and challenging-to-implement recommendations from 2019,” Dr. Shah explained.
Among the 2019 recommendations was a stipulation for “shared decision-making” with PCV13, and a conversation that often only complicated matters, he noted.
“Patients and providers alike had confusion about this since it was not a clear-cut ‘yes, give it’ or ‘no, do not give it any longer’ recommendation.”
“Now that this new recommendation will require no extra time for a discussion in the clinic, and just a simple ‘it’s time for your pneumonia shot’ offer, this may become more feasible,” Dr. Shah added. “In addition, removal of the shared decision-making stipulation allows for this immunization to be easily protocolized in the clinic, similar to automatic offers to the flu vaccine for patients each year.”
According to the CDC, pneumococcal pneumonia causes an estimated 150,000 hospitalizations each year in the United States, while pneumococcal meningitis and bacteremia killed approximately 3,250 people in the United States in 2019.
“Clinicians are patients’ most trusted resource when it comes to vaccine recommendations,” Dr. Kobayashi said. “We encourage all clinicians to recommend pneumococcal vaccines when indicated.”
Dr. Kobayashi and Dr. Shah have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE MMWR
Immunocompromised patients should receive fourth COVID shot: CDC
The Centers for Disease Control and Prevention contacted pharmacies on Jan. 26 to reinforce the message that people with moderate to severe immune suppression should receive a fourth COVID-19 vaccine, according to Kaiser Health News.
The conference call came a day after the news outlet reported that immunocompromised people were being turned away by pharmacies. White House officials also emphasized on Jan. 26 that immunocompromised people should receive an additional shot.
During the call, the CDC “reiterated the recommendations, running through case examples,” Mitchel Rothholz, RPh, MBA, chief of governance and state affiliates for the American Pharmacists Association, told KHN.
While on the call, Mr. Rothholz asked for a “prepared document” with the CDC’s recommendations “so we can clearly and consistently communicate the message.” The CDC officials on the call said they would create a document but “don’t know how long that will take,” Mr. Rothholz told KHN.
The CDC recommends an additional shot -– or a fourth shot – for those who have weak immune systems, which makes them more at risk for severe COVID-19 and death. About 7 million American adults are considered immunocompromised, KHN reported, which includes people who have certain medical conditions that impair their immune response or who take immune-suppressing drugs because of organ transplants, cancer, or autoimmune diseases.
The CDC first recommended fourth shots for immunocompromised people in October. This month, the CDC shortened the time for booster shots from 6 months to 5 months, and some immunocompromised people who are due for another shot have begun to seek them. The agency has been educating pharmacists and other health providers since then, a CDC spokesperson told KHN.
While patients don’t need to provide proof that they are immunocompromised, according to the CDC, some have been turned away, KHN reported.
To improve communication with the public, large pharmacies could issue news releases and update their websites “explicitly stating that they are offering fourth doses” to immunocompromised people, Ameet Kini, MD, a professor of pathology and laboratory medicine at Loyola University Medical Center in Chicago, told KHN.
Pharmacies should also update their patient portals and provide “clear guidance for their pharmacists,” he said.
A version of this article first appeared on WebMD.com.
The Centers for Disease Control and Prevention contacted pharmacies on Jan. 26 to reinforce the message that people with moderate to severe immune suppression should receive a fourth COVID-19 vaccine, according to Kaiser Health News.
The conference call came a day after the news outlet reported that immunocompromised people were being turned away by pharmacies. White House officials also emphasized on Jan. 26 that immunocompromised people should receive an additional shot.
During the call, the CDC “reiterated the recommendations, running through case examples,” Mitchel Rothholz, RPh, MBA, chief of governance and state affiliates for the American Pharmacists Association, told KHN.
While on the call, Mr. Rothholz asked for a “prepared document” with the CDC’s recommendations “so we can clearly and consistently communicate the message.” The CDC officials on the call said they would create a document but “don’t know how long that will take,” Mr. Rothholz told KHN.
The CDC recommends an additional shot -– or a fourth shot – for those who have weak immune systems, which makes them more at risk for severe COVID-19 and death. About 7 million American adults are considered immunocompromised, KHN reported, which includes people who have certain medical conditions that impair their immune response or who take immune-suppressing drugs because of organ transplants, cancer, or autoimmune diseases.
The CDC first recommended fourth shots for immunocompromised people in October. This month, the CDC shortened the time for booster shots from 6 months to 5 months, and some immunocompromised people who are due for another shot have begun to seek them. The agency has been educating pharmacists and other health providers since then, a CDC spokesperson told KHN.
While patients don’t need to provide proof that they are immunocompromised, according to the CDC, some have been turned away, KHN reported.
To improve communication with the public, large pharmacies could issue news releases and update their websites “explicitly stating that they are offering fourth doses” to immunocompromised people, Ameet Kini, MD, a professor of pathology and laboratory medicine at Loyola University Medical Center in Chicago, told KHN.
Pharmacies should also update their patient portals and provide “clear guidance for their pharmacists,” he said.
A version of this article first appeared on WebMD.com.
The Centers for Disease Control and Prevention contacted pharmacies on Jan. 26 to reinforce the message that people with moderate to severe immune suppression should receive a fourth COVID-19 vaccine, according to Kaiser Health News.
The conference call came a day after the news outlet reported that immunocompromised people were being turned away by pharmacies. White House officials also emphasized on Jan. 26 that immunocompromised people should receive an additional shot.
During the call, the CDC “reiterated the recommendations, running through case examples,” Mitchel Rothholz, RPh, MBA, chief of governance and state affiliates for the American Pharmacists Association, told KHN.
While on the call, Mr. Rothholz asked for a “prepared document” with the CDC’s recommendations “so we can clearly and consistently communicate the message.” The CDC officials on the call said they would create a document but “don’t know how long that will take,” Mr. Rothholz told KHN.
The CDC recommends an additional shot -– or a fourth shot – for those who have weak immune systems, which makes them more at risk for severe COVID-19 and death. About 7 million American adults are considered immunocompromised, KHN reported, which includes people who have certain medical conditions that impair their immune response or who take immune-suppressing drugs because of organ transplants, cancer, or autoimmune diseases.
The CDC first recommended fourth shots for immunocompromised people in October. This month, the CDC shortened the time for booster shots from 6 months to 5 months, and some immunocompromised people who are due for another shot have begun to seek them. The agency has been educating pharmacists and other health providers since then, a CDC spokesperson told KHN.
While patients don’t need to provide proof that they are immunocompromised, according to the CDC, some have been turned away, KHN reported.
To improve communication with the public, large pharmacies could issue news releases and update their websites “explicitly stating that they are offering fourth doses” to immunocompromised people, Ameet Kini, MD, a professor of pathology and laboratory medicine at Loyola University Medical Center in Chicago, told KHN.
Pharmacies should also update their patient portals and provide “clear guidance for their pharmacists,” he said.
A version of this article first appeared on WebMD.com.
FDA approves risankizumab (Skyrizi) for psoriatic arthritis
The Food and Drug Administration on Jan. 21 approved risankizumab-rzaa (Skyrizi) for a second indication – treating adults with active psoriatic arthritis (PsA) – making it the second anti–interleukin-23 monoclonal antibody available to treat PsA, according to an announcement from manufacturer AbbVie.
The agency previously approved risankizumab in April 2019 for adults with moderate to severe plaque psoriasis.
The dosing regimen for PsA is the same as it is for patients with moderate to severe plaque psoriasis: a single 150-mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4), and it can be administered alone or in combination with disease-modifying antirheumatic drugs (DMARDs).
Two phase 3 trials, KEEPsAKE 1 and KEEPsAKE 2, were the basis for the approval. These two trials tested the biologic agent in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or nonbiologic DMARDs. Fulfillment of the trials’ primary endpoint of at least a 20% improvement in American College of Rheumatology response criteria at 24 weeks occurred in 51.3%-57.3% of patients, compared with 26.5%-33.5% of placebo-treated patients.
Those on risankizumab also achieved significantly higher rates of ACR50 and ACR70 responses than those on placebo. In addition, patients with preexisting dactylitis and enthesitis experienced improvements in these PsA manifestations. Risankizumab was also associated with an improvement in physical function at 24 weeks on the Health Assessment Questionnaire–Disability Index, bettering placebo by a mean difference of 0.16-0.20 points in the two trials. A significantly higher percentage of patients who had psoriatic skin lesions experienced at least 90% improvement with risankizumab on the Psoriasis Area and Severity Index, compared with placebo.
AbbVie said that the safety profile of risankizumab in patients with PsA has been generally consistent with its effects in patients with plaque psoriasis.
The KEEPsAKE 1 and KEEPsAKE 2 studies are ongoing, and patients in the long-term extensions of the trials remain blinded to the original randomized allocation for the duration of the studies.
Phase 3 trials of risankizumab are also ongoing in patients with Crohn’s disease and ulcerative colitis.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on Jan. 21 approved risankizumab-rzaa (Skyrizi) for a second indication – treating adults with active psoriatic arthritis (PsA) – making it the second anti–interleukin-23 monoclonal antibody available to treat PsA, according to an announcement from manufacturer AbbVie.
The agency previously approved risankizumab in April 2019 for adults with moderate to severe plaque psoriasis.
The dosing regimen for PsA is the same as it is for patients with moderate to severe plaque psoriasis: a single 150-mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4), and it can be administered alone or in combination with disease-modifying antirheumatic drugs (DMARDs).
Two phase 3 trials, KEEPsAKE 1 and KEEPsAKE 2, were the basis for the approval. These two trials tested the biologic agent in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or nonbiologic DMARDs. Fulfillment of the trials’ primary endpoint of at least a 20% improvement in American College of Rheumatology response criteria at 24 weeks occurred in 51.3%-57.3% of patients, compared with 26.5%-33.5% of placebo-treated patients.
Those on risankizumab also achieved significantly higher rates of ACR50 and ACR70 responses than those on placebo. In addition, patients with preexisting dactylitis and enthesitis experienced improvements in these PsA manifestations. Risankizumab was also associated with an improvement in physical function at 24 weeks on the Health Assessment Questionnaire–Disability Index, bettering placebo by a mean difference of 0.16-0.20 points in the two trials. A significantly higher percentage of patients who had psoriatic skin lesions experienced at least 90% improvement with risankizumab on the Psoriasis Area and Severity Index, compared with placebo.
AbbVie said that the safety profile of risankizumab in patients with PsA has been generally consistent with its effects in patients with plaque psoriasis.
The KEEPsAKE 1 and KEEPsAKE 2 studies are ongoing, and patients in the long-term extensions of the trials remain blinded to the original randomized allocation for the duration of the studies.
Phase 3 trials of risankizumab are also ongoing in patients with Crohn’s disease and ulcerative colitis.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on Jan. 21 approved risankizumab-rzaa (Skyrizi) for a second indication – treating adults with active psoriatic arthritis (PsA) – making it the second anti–interleukin-23 monoclonal antibody available to treat PsA, according to an announcement from manufacturer AbbVie.
The agency previously approved risankizumab in April 2019 for adults with moderate to severe plaque psoriasis.
The dosing regimen for PsA is the same as it is for patients with moderate to severe plaque psoriasis: a single 150-mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4), and it can be administered alone or in combination with disease-modifying antirheumatic drugs (DMARDs).
Two phase 3 trials, KEEPsAKE 1 and KEEPsAKE 2, were the basis for the approval. These two trials tested the biologic agent in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or nonbiologic DMARDs. Fulfillment of the trials’ primary endpoint of at least a 20% improvement in American College of Rheumatology response criteria at 24 weeks occurred in 51.3%-57.3% of patients, compared with 26.5%-33.5% of placebo-treated patients.
Those on risankizumab also achieved significantly higher rates of ACR50 and ACR70 responses than those on placebo. In addition, patients with preexisting dactylitis and enthesitis experienced improvements in these PsA manifestations. Risankizumab was also associated with an improvement in physical function at 24 weeks on the Health Assessment Questionnaire–Disability Index, bettering placebo by a mean difference of 0.16-0.20 points in the two trials. A significantly higher percentage of patients who had psoriatic skin lesions experienced at least 90% improvement with risankizumab on the Psoriasis Area and Severity Index, compared with placebo.
AbbVie said that the safety profile of risankizumab in patients with PsA has been generally consistent with its effects in patients with plaque psoriasis.
The KEEPsAKE 1 and KEEPsAKE 2 studies are ongoing, and patients in the long-term extensions of the trials remain blinded to the original randomized allocation for the duration of the studies.
Phase 3 trials of risankizumab are also ongoing in patients with Crohn’s disease and ulcerative colitis.
A version of this article first appeared on Medscape.com.
Children and COVID: United States passes 10 million total cases
Weekly COVID-19 cases in children topped 1 million for the first time as the cumulative count surpassed 10 million since the start of the pandemic, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
the AAP and CHA said in their weekly COVID report. Those 10.6 million child cases represent 18.4% of all cases, and the latest 1.15 million represented 25.5% of all cases for the week.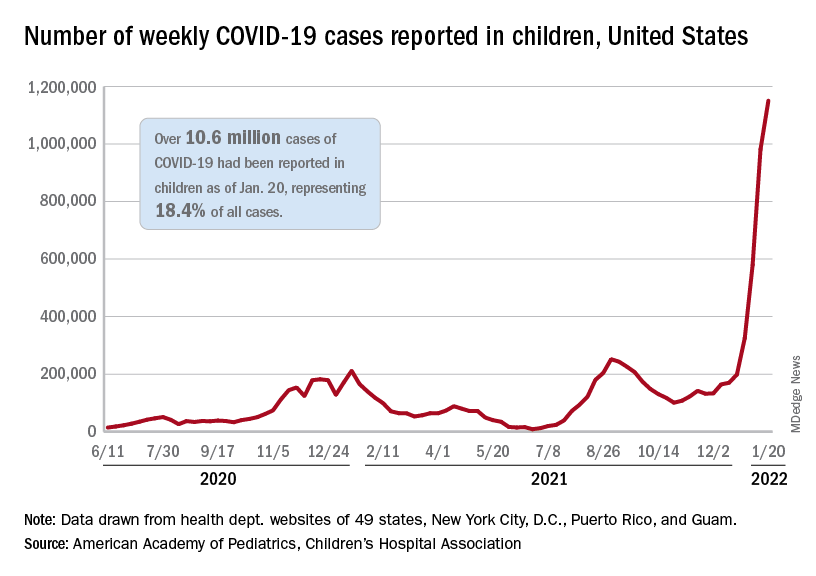
Regionally, the South had the most cases with over 380,000 for the week of Jan. 14-20, while the West was next with close to 350,000, followed by the Midwest and then the East. Among the states, the largest percent increases – on the order of 30% – came in New England (Massachusetts, Rhode Island, and Vermont), as well as Virginia and California, the AAP and CHA said.
Examining all those cases by vaccination status shows an obvious difference between the Omicron and Delta variants: The fully vaccinated have been hit much harder than before. For the week ending Dec. 25, 2021, the incidence of COVID-19 in children aged 12-17 years was 704 per 100,000 among those were unvaccinated and 384 per 100,000 in those who were fully vaccinated. During the Delta surge in the summer of 2021, the peak rates were 938 (unvaccinated) and 79 (vaccinated), the Centers for Disease Control and Prevention said.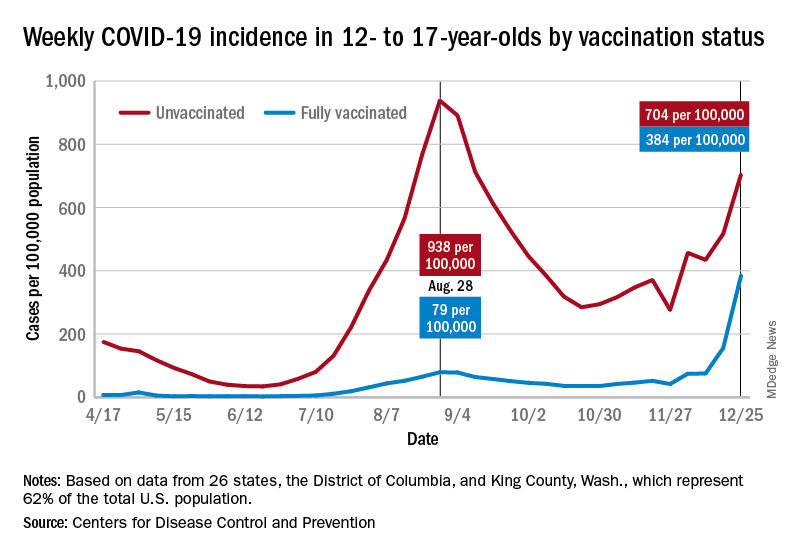
Hospitalizations are also at record levels, but two separate CDC databases seem to show a decline in child admissions over the last available week or so of data, which follows the trend among all ages. The peak among children aged 0-17 years came on Jan. 15, when the rate of new admissions reached 1.25 per 100,000, based on reporting to the CDC from 5,265 hospitals nationwide.
The second database, the COVID-19–Associated Hospitalization Surveillance Network (COVID-NET), indicates that children aged 0-4 years had the highest admission rate, 14.5 per 100,000, for the week ending Jan. 8, compared with 5.5 per 100,000 for 12- to 17-year-olds and 2.3 per 100,000 for those aged 5-11 years. COVID-NET covers almost 100 counties in 10 states, along with 4 entire states, and represents about 10% of the U.S. population.
Vaccinations rose briefly in late December and into January to meet the Omicron surge, but the numbers for the latest week show a return to their earlier levels. In children aged 5-11 years, new vaccinations went from 381,000 for the week of Dec. 20-26 to 524,000 for Jan. 3-9, but fell to just 260,000 during Jan. 17-23. The response was a little later for those aged 12-17, with the big week coming Jan. 10-16, but there was still a 38% drop for Jan. 17-23, according to the CDC’s COVID Data Tracker.
Currently, 29.3% of all 5- to 11-year-olds have received at least one dose of the COVID vaccine, and an even 20.0% are fully vaccinated. For children aged 12-17, the corresponding figures are 65.8% and 55.1%, the CDC said.
Statewide vaccination rates vary from Vermont’s high of 61% for those aged 5-11 to 12% for Alabama, Louisiana, and Mississippi, while Hawaii has the highest rate for 12- to 17-year-olds at 92% and Wyoming has the lowest at 39%, the AAP reported.
Weekly COVID-19 cases in children topped 1 million for the first time as the cumulative count surpassed 10 million since the start of the pandemic, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
the AAP and CHA said in their weekly COVID report. Those 10.6 million child cases represent 18.4% of all cases, and the latest 1.15 million represented 25.5% of all cases for the week.
Regionally, the South had the most cases with over 380,000 for the week of Jan. 14-20, while the West was next with close to 350,000, followed by the Midwest and then the East. Among the states, the largest percent increases – on the order of 30% – came in New England (Massachusetts, Rhode Island, and Vermont), as well as Virginia and California, the AAP and CHA said.
Examining all those cases by vaccination status shows an obvious difference between the Omicron and Delta variants: The fully vaccinated have been hit much harder than before. For the week ending Dec. 25, 2021, the incidence of COVID-19 in children aged 12-17 years was 704 per 100,000 among those were unvaccinated and 384 per 100,000 in those who were fully vaccinated. During the Delta surge in the summer of 2021, the peak rates were 938 (unvaccinated) and 79 (vaccinated), the Centers for Disease Control and Prevention said.
Hospitalizations are also at record levels, but two separate CDC databases seem to show a decline in child admissions over the last available week or so of data, which follows the trend among all ages. The peak among children aged 0-17 years came on Jan. 15, when the rate of new admissions reached 1.25 per 100,000, based on reporting to the CDC from 5,265 hospitals nationwide.
The second database, the COVID-19–Associated Hospitalization Surveillance Network (COVID-NET), indicates that children aged 0-4 years had the highest admission rate, 14.5 per 100,000, for the week ending Jan. 8, compared with 5.5 per 100,000 for 12- to 17-year-olds and 2.3 per 100,000 for those aged 5-11 years. COVID-NET covers almost 100 counties in 10 states, along with 4 entire states, and represents about 10% of the U.S. population.
Vaccinations rose briefly in late December and into January to meet the Omicron surge, but the numbers for the latest week show a return to their earlier levels. In children aged 5-11 years, new vaccinations went from 381,000 for the week of Dec. 20-26 to 524,000 for Jan. 3-9, but fell to just 260,000 during Jan. 17-23. The response was a little later for those aged 12-17, with the big week coming Jan. 10-16, but there was still a 38% drop for Jan. 17-23, according to the CDC’s COVID Data Tracker.
Currently, 29.3% of all 5- to 11-year-olds have received at least one dose of the COVID vaccine, and an even 20.0% are fully vaccinated. For children aged 12-17, the corresponding figures are 65.8% and 55.1%, the CDC said.
Statewide vaccination rates vary from Vermont’s high of 61% for those aged 5-11 to 12% for Alabama, Louisiana, and Mississippi, while Hawaii has the highest rate for 12- to 17-year-olds at 92% and Wyoming has the lowest at 39%, the AAP reported.
Weekly COVID-19 cases in children topped 1 million for the first time as the cumulative count surpassed 10 million since the start of the pandemic, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
the AAP and CHA said in their weekly COVID report. Those 10.6 million child cases represent 18.4% of all cases, and the latest 1.15 million represented 25.5% of all cases for the week.
Regionally, the South had the most cases with over 380,000 for the week of Jan. 14-20, while the West was next with close to 350,000, followed by the Midwest and then the East. Among the states, the largest percent increases – on the order of 30% – came in New England (Massachusetts, Rhode Island, and Vermont), as well as Virginia and California, the AAP and CHA said.
Examining all those cases by vaccination status shows an obvious difference between the Omicron and Delta variants: The fully vaccinated have been hit much harder than before. For the week ending Dec. 25, 2021, the incidence of COVID-19 in children aged 12-17 years was 704 per 100,000 among those were unvaccinated and 384 per 100,000 in those who were fully vaccinated. During the Delta surge in the summer of 2021, the peak rates were 938 (unvaccinated) and 79 (vaccinated), the Centers for Disease Control and Prevention said.
Hospitalizations are also at record levels, but two separate CDC databases seem to show a decline in child admissions over the last available week or so of data, which follows the trend among all ages. The peak among children aged 0-17 years came on Jan. 15, when the rate of new admissions reached 1.25 per 100,000, based on reporting to the CDC from 5,265 hospitals nationwide.
The second database, the COVID-19–Associated Hospitalization Surveillance Network (COVID-NET), indicates that children aged 0-4 years had the highest admission rate, 14.5 per 100,000, for the week ending Jan. 8, compared with 5.5 per 100,000 for 12- to 17-year-olds and 2.3 per 100,000 for those aged 5-11 years. COVID-NET covers almost 100 counties in 10 states, along with 4 entire states, and represents about 10% of the U.S. population.
Vaccinations rose briefly in late December and into January to meet the Omicron surge, but the numbers for the latest week show a return to their earlier levels. In children aged 5-11 years, new vaccinations went from 381,000 for the week of Dec. 20-26 to 524,000 for Jan. 3-9, but fell to just 260,000 during Jan. 17-23. The response was a little later for those aged 12-17, with the big week coming Jan. 10-16, but there was still a 38% drop for Jan. 17-23, according to the CDC’s COVID Data Tracker.
Currently, 29.3% of all 5- to 11-year-olds have received at least one dose of the COVID vaccine, and an even 20.0% are fully vaccinated. For children aged 12-17, the corresponding figures are 65.8% and 55.1%, the CDC said.
Statewide vaccination rates vary from Vermont’s high of 61% for those aged 5-11 to 12% for Alabama, Louisiana, and Mississippi, while Hawaii has the highest rate for 12- to 17-year-olds at 92% and Wyoming has the lowest at 39%, the AAP reported.
Children and COVID: U.S. sees almost 1 million new cases
Another record week for COVID-19 brought almost 1 million new cases to the children of the United States, according to new data from the American Academy of Pediatrics and the Children’s Hospital Association.
The pre-Omicron high for new cases in a week – 252,000 during the Delta surge of the late summer and early fall – has been surpassed each of the last 3 weeks and now stands at 981,000 (Jan. 7-13), according to the AAP/CHA weekly COVID-19 report. Over the 3-week stretch from Dec. 17 to Jan. 13, weekly cases increased by 394%.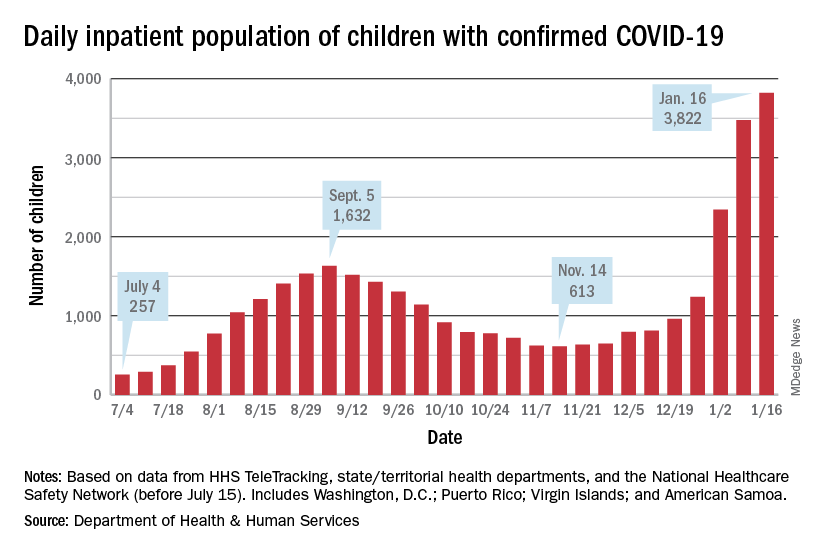
Hospitalizations also climbed to new heights, as daily admissions reached 1.23 per 100,000 children on Jan. 14, an increase of 547% since Nov. 30, when the rate was 0.19 per 100,000. Before Omicron, the highest rate for children was 0.47 per 100,000, based on data from the Centers for Disease Control and Prevention.
The inpatient population count, meanwhile, has followed suit. On Jan. 16, there were 3,822 children hospitalized in pediatric inpatient beds with laboratory-confirmed COVID-19, which is 523% higher than the 613 children who were hospitalized on Nov. 14, according to the Department of Health & Human Services. In the last week, though, the population was up by just 10%.
The one thing that has not surged in the last few weeks is vaccination. Among children aged 5-11 years, the weekly count of those who have received at least one dose dropped by 34% over the last 5 weeks, falling from 527,000 for Dec.11-17 to 347,000 during Jan. 8-14, the CDC said on the COVID Data Tracker, which also noted that just 18.4% of this age group is fully vaccinated.
The situation was reversed in children aged 12-15, who were up by 36% over that same time, but their numbers were much smaller: 78,000 for the week of Dec. 11-17 and 106,000 for Jan. 8-14. Those aged 16-17 were up by just 4% over that 5-week span, the CDC data show.
Over the course of the entire pandemic, almost 9.5 million cases of COVID-19 in children have been reported, and children represent 17.8% of all cases reported in 49 states (excluding New York but including New York City), the District of Columbia, Puerto Rico, and Guam, the AAP and CHA said in their report.
Three states (Alabama, Nebraska, and Texas) stopped public reporting over the summer, but many states count individuals up to age 19 as children, and others (South Carolina, Tennessee, and West Virginia) go up to age 20, the AAP and CHA noted. The CDC, by comparison, puts the number of cases for those aged 0-17 at 8.3 million, but that estimate is based on only 51 million of the nearly 67 million U.S. cases as of Jan. 18.
Another record week for COVID-19 brought almost 1 million new cases to the children of the United States, according to new data from the American Academy of Pediatrics and the Children’s Hospital Association.
The pre-Omicron high for new cases in a week – 252,000 during the Delta surge of the late summer and early fall – has been surpassed each of the last 3 weeks and now stands at 981,000 (Jan. 7-13), according to the AAP/CHA weekly COVID-19 report. Over the 3-week stretch from Dec. 17 to Jan. 13, weekly cases increased by 394%.
Hospitalizations also climbed to new heights, as daily admissions reached 1.23 per 100,000 children on Jan. 14, an increase of 547% since Nov. 30, when the rate was 0.19 per 100,000. Before Omicron, the highest rate for children was 0.47 per 100,000, based on data from the Centers for Disease Control and Prevention.
The inpatient population count, meanwhile, has followed suit. On Jan. 16, there were 3,822 children hospitalized in pediatric inpatient beds with laboratory-confirmed COVID-19, which is 523% higher than the 613 children who were hospitalized on Nov. 14, according to the Department of Health & Human Services. In the last week, though, the population was up by just 10%.
The one thing that has not surged in the last few weeks is vaccination. Among children aged 5-11 years, the weekly count of those who have received at least one dose dropped by 34% over the last 5 weeks, falling from 527,000 for Dec.11-17 to 347,000 during Jan. 8-14, the CDC said on the COVID Data Tracker, which also noted that just 18.4% of this age group is fully vaccinated.
The situation was reversed in children aged 12-15, who were up by 36% over that same time, but their numbers were much smaller: 78,000 for the week of Dec. 11-17 and 106,000 for Jan. 8-14. Those aged 16-17 were up by just 4% over that 5-week span, the CDC data show.
Over the course of the entire pandemic, almost 9.5 million cases of COVID-19 in children have been reported, and children represent 17.8% of all cases reported in 49 states (excluding New York but including New York City), the District of Columbia, Puerto Rico, and Guam, the AAP and CHA said in their report.
Three states (Alabama, Nebraska, and Texas) stopped public reporting over the summer, but many states count individuals up to age 19 as children, and others (South Carolina, Tennessee, and West Virginia) go up to age 20, the AAP and CHA noted. The CDC, by comparison, puts the number of cases for those aged 0-17 at 8.3 million, but that estimate is based on only 51 million of the nearly 67 million U.S. cases as of Jan. 18.
Another record week for COVID-19 brought almost 1 million new cases to the children of the United States, according to new data from the American Academy of Pediatrics and the Children’s Hospital Association.
The pre-Omicron high for new cases in a week – 252,000 during the Delta surge of the late summer and early fall – has been surpassed each of the last 3 weeks and now stands at 981,000 (Jan. 7-13), according to the AAP/CHA weekly COVID-19 report. Over the 3-week stretch from Dec. 17 to Jan. 13, weekly cases increased by 394%.
Hospitalizations also climbed to new heights, as daily admissions reached 1.23 per 100,000 children on Jan. 14, an increase of 547% since Nov. 30, when the rate was 0.19 per 100,000. Before Omicron, the highest rate for children was 0.47 per 100,000, based on data from the Centers for Disease Control and Prevention.
The inpatient population count, meanwhile, has followed suit. On Jan. 16, there were 3,822 children hospitalized in pediatric inpatient beds with laboratory-confirmed COVID-19, which is 523% higher than the 613 children who were hospitalized on Nov. 14, according to the Department of Health & Human Services. In the last week, though, the population was up by just 10%.
The one thing that has not surged in the last few weeks is vaccination. Among children aged 5-11 years, the weekly count of those who have received at least one dose dropped by 34% over the last 5 weeks, falling from 527,000 for Dec.11-17 to 347,000 during Jan. 8-14, the CDC said on the COVID Data Tracker, which also noted that just 18.4% of this age group is fully vaccinated.
The situation was reversed in children aged 12-15, who were up by 36% over that same time, but their numbers were much smaller: 78,000 for the week of Dec. 11-17 and 106,000 for Jan. 8-14. Those aged 16-17 were up by just 4% over that 5-week span, the CDC data show.
Over the course of the entire pandemic, almost 9.5 million cases of COVID-19 in children have been reported, and children represent 17.8% of all cases reported in 49 states (excluding New York but including New York City), the District of Columbia, Puerto Rico, and Guam, the AAP and CHA said in their report.
Three states (Alabama, Nebraska, and Texas) stopped public reporting over the summer, but many states count individuals up to age 19 as children, and others (South Carolina, Tennessee, and West Virginia) go up to age 20, the AAP and CHA noted. The CDC, by comparison, puts the number of cases for those aged 0-17 at 8.3 million, but that estimate is based on only 51 million of the nearly 67 million U.S. cases as of Jan. 18.
FDA approves two JAK-1 inhibitors for moderate to severe atopic dermatitis
The available for this indication in the United States.
“It’s big news because a few years ago we didn’t have any systemic treatments that are safer than the classical immunosuppressants like cyclosporine and methotrexate,” Emma Guttman-Yassky, MD, PhD, Waldman professor and system chair of dermatology at the Icahn School of Medicine at Mount Sinai in New York, told this news organization commenting on upadacitinib’s approval.
“The only oral approved drug for AD up to now was oral prednisone, which has terrible safety concerns. This is basically the first oral medication that we can provide our patients for long-term use.”
Upadacitinib
The approval of upadacitinib (Rinvoq), marketed by AbbVie, for moderate to severe AD in patients ages 12 and older, comes on the heels of findings from three pivotal phase 3 studies involving more than 2,500 adults and children 12 years of age and older with moderate to severe AD: Measure Up 1 and 2, led by Dr. Guttman-Yassky, which evaluated upadacitinib compared with placebo, and AD UP, which compared upadacitinib along with topical corticosteroids, compared with placebo.
Across the three studies, upadacitinib – both 15 mg and 30 mg once daily monotherapy – met all primary and secondary endpoints at week 16, with some patients achieving higher levels of skin clearance based on the Eczema Area and Severity Index 90 (EASI-90) and EASI-100.
“I always say that patients with AD need options,” Dr. Guttman-Yassky said. “We need biologics. We need oral medications. Not everybody likes an injectable. The plus of the class of JAK inhibitors in general is the quick onset of action.” Many patients in her clinic are maintained on upadacitinib more than two years later “and are super happy,” she said. “Many of them failed cyclosporine and other immunosuppressants such as methotrexate and prednisone.”
She predicted that health insurance companies will find coverage cost-effective “because it sets a new bar for efficacy, and because many patients have failed other treatments.”
Abrocitinib
Abrocitinib (Cibinqo), marketed by Pfizer, was approved for adults with moderate to severe AD. The approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The recommended doses are 100 mg and 200 mg, with the 200 mg dose recommended for patients who are not responding to the 100 mg dose.
The labeling of abrocitinib and upadacitinib include a boxed warning for JAK inhibitors, regarding the risk of serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Dr. Guttman-Yassky has served as a principal investigator for AbbVie and has received consulting fees from the company.
The available for this indication in the United States.
“It’s big news because a few years ago we didn’t have any systemic treatments that are safer than the classical immunosuppressants like cyclosporine and methotrexate,” Emma Guttman-Yassky, MD, PhD, Waldman professor and system chair of dermatology at the Icahn School of Medicine at Mount Sinai in New York, told this news organization commenting on upadacitinib’s approval.
“The only oral approved drug for AD up to now was oral prednisone, which has terrible safety concerns. This is basically the first oral medication that we can provide our patients for long-term use.”
Upadacitinib
The approval of upadacitinib (Rinvoq), marketed by AbbVie, for moderate to severe AD in patients ages 12 and older, comes on the heels of findings from three pivotal phase 3 studies involving more than 2,500 adults and children 12 years of age and older with moderate to severe AD: Measure Up 1 and 2, led by Dr. Guttman-Yassky, which evaluated upadacitinib compared with placebo, and AD UP, which compared upadacitinib along with topical corticosteroids, compared with placebo.
Across the three studies, upadacitinib – both 15 mg and 30 mg once daily monotherapy – met all primary and secondary endpoints at week 16, with some patients achieving higher levels of skin clearance based on the Eczema Area and Severity Index 90 (EASI-90) and EASI-100.
“I always say that patients with AD need options,” Dr. Guttman-Yassky said. “We need biologics. We need oral medications. Not everybody likes an injectable. The plus of the class of JAK inhibitors in general is the quick onset of action.” Many patients in her clinic are maintained on upadacitinib more than two years later “and are super happy,” she said. “Many of them failed cyclosporine and other immunosuppressants such as methotrexate and prednisone.”
She predicted that health insurance companies will find coverage cost-effective “because it sets a new bar for efficacy, and because many patients have failed other treatments.”
Abrocitinib
Abrocitinib (Cibinqo), marketed by Pfizer, was approved for adults with moderate to severe AD. The approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The recommended doses are 100 mg and 200 mg, with the 200 mg dose recommended for patients who are not responding to the 100 mg dose.
The labeling of abrocitinib and upadacitinib include a boxed warning for JAK inhibitors, regarding the risk of serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Dr. Guttman-Yassky has served as a principal investigator for AbbVie and has received consulting fees from the company.
The available for this indication in the United States.
“It’s big news because a few years ago we didn’t have any systemic treatments that are safer than the classical immunosuppressants like cyclosporine and methotrexate,” Emma Guttman-Yassky, MD, PhD, Waldman professor and system chair of dermatology at the Icahn School of Medicine at Mount Sinai in New York, told this news organization commenting on upadacitinib’s approval.
“The only oral approved drug for AD up to now was oral prednisone, which has terrible safety concerns. This is basically the first oral medication that we can provide our patients for long-term use.”
Upadacitinib
The approval of upadacitinib (Rinvoq), marketed by AbbVie, for moderate to severe AD in patients ages 12 and older, comes on the heels of findings from three pivotal phase 3 studies involving more than 2,500 adults and children 12 years of age and older with moderate to severe AD: Measure Up 1 and 2, led by Dr. Guttman-Yassky, which evaluated upadacitinib compared with placebo, and AD UP, which compared upadacitinib along with topical corticosteroids, compared with placebo.
Across the three studies, upadacitinib – both 15 mg and 30 mg once daily monotherapy – met all primary and secondary endpoints at week 16, with some patients achieving higher levels of skin clearance based on the Eczema Area and Severity Index 90 (EASI-90) and EASI-100.
“I always say that patients with AD need options,” Dr. Guttman-Yassky said. “We need biologics. We need oral medications. Not everybody likes an injectable. The plus of the class of JAK inhibitors in general is the quick onset of action.” Many patients in her clinic are maintained on upadacitinib more than two years later “and are super happy,” she said. “Many of them failed cyclosporine and other immunosuppressants such as methotrexate and prednisone.”
She predicted that health insurance companies will find coverage cost-effective “because it sets a new bar for efficacy, and because many patients have failed other treatments.”
Abrocitinib
Abrocitinib (Cibinqo), marketed by Pfizer, was approved for adults with moderate to severe AD. The approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The recommended doses are 100 mg and 200 mg, with the 200 mg dose recommended for patients who are not responding to the 100 mg dose.
The labeling of abrocitinib and upadacitinib include a boxed warning for JAK inhibitors, regarding the risk of serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Dr. Guttman-Yassky has served as a principal investigator for AbbVie and has received consulting fees from the company.
ACIP releases new dengue vaccine recommendations
The vaccine is only to be used for children aged 9-16 who live in endemic areas and who have evidence with a specific diagnostic test of prior dengue infection.
Dengue is a mosquito-borne virus found throughout the world, primarily in tropical or subtropical climates. Cases had steadily been increasing to 5.2 million in 2019, and the geographic distribution of cases is broadening with climate change and urbanization. About half of the world’s population is now at risk.
The dengue virus has four serotypes. The first infection may be mild or asymptomatic, but the second one can be life-threatening because of a phenomenon called antibody-dependent enhancement.
The lead author of the new recommendations is Gabriela Paz-Bailey, MD, PhD, division of vector-borne diseases, dengue branch, CDC. She told this news organization that, during the second infection, when there are “low levels of antibodies from that first infection, the antibodies help the virus get inside the cells. There the virus is not killed, and that results in increased viral load, and then that can result in more severe disease and the plasma leakage” syndrome, which can lead to shock, severe bleeding, and organ failure. The death rate for severe dengue is up to 13%.
Previous infection with Zika virus, common in the same areas where dengue is endemic, can also increase the risk for symptomatic and severe dengue for subsequent infections.
In the United States, Puerto Rico is the main focus of control efforts because 95% of domestic dengue cases originate there – almost 30,000 cases between 2010 and 2020, with 11,000 cases and 4,000 hospitalizations occurring in children between the ages of 10 and 19.
Because Aedes aegypti, the primary mosquito vector transmitting dengue, is resistant to all commonly used insecticides in Puerto Rico, preventive efforts have shifted from insecticides to vaccination.
Antibody tests prevaccination
The main concern with the Sanofi’s dengue vaccine is that it could act as an asymptomatic primary dengue infection, in effect priming the body for a severe reaction from antibody-dependent enhancement with a subsequent infection. That is why it’s critical that the vaccine only be given to children with evidence of prior disease.
Dr. Paz-Bailey said: “The CDC came up with recommendations of what the performance of the test used for prevaccination screening should be. And it was 98% specificity and 75% sensitivity. ... But no test by itself was found to have a specificity of 98%, and this is why we’re recommending the two-test algorithm,” in which two different assays are run off the same blood sample, drawn at a prevaccination visit.
If the child has evidence of prior dengue, they can proceed with vaccination to protect against recurrent infection. Dengvaxia is given as a series of three shots over 6 months. Vaccine efficacy is 82% – so not everyone is protected, and additionally, that protection declines over time.
There is concern that it will be difficult to achieve compliance with such a complex regimen. Dr. Paz-Bailey said, “But I think that the trust in vaccines that is highly prevalent for [Puerto] Rico and trusting the health care system, and sort of the importance that is assigned to dengue by providers and by parents because of previous outbreaks and previous experiences is going to help us.” She added, “I think that the COVID experience has been very revealing. And what we have learned is that Puerto Rico has a very strong health care system, a very strong network of vaccine providers. ... Coverage for COVID vaccine is higher than in other parts of the U.S.”
One of the interesting things about dengue is that the first infection can range from asymptomatic to life-threatening. The second infection is generally worse because of this antibody-dependent enhancement phenomenon. Eng Eong Ooi, MD, PhD, professor of microbiology and immunology, National University of Singapore, told this news organization, “After you have two infections, you seem to be protected quite well against the remaining two [serotypes]. The vaccine serves as another episode of infection in those who had prior dengue, so then any natural infections after the vaccination in the seropositive become like the outcome of a third or fourth infection.”
Vaccination alone will not solve dengue. Dr. Ooi said, “There’s not one method that would fully control dengue. You need both vaccines as well as control measures, whether it’s Wolbachia or something else. At the same time, I think we need antiviral drugs, because hitting this virus in just one part of its life cycle wouldn’t make a huge, lasting impact.” Dr. Ooi added that as “the spread of the virus and the population immunity drops, you’re actually now more vulnerable to dengue outbreaks when they do get introduced. So, suppressing transmission alone isn’t the answer. You also have to keep herd immunity levels high. So if we can reduce the virus transmission by controlling either mosquito population or transmission and at the same time vaccinate to keep the immunity levels high, then I think we have a chance of controlling dengue.”
Dr. Paz-Bailey concluded: “I do want to emphasize that we are excited about having these tools, because for years and years, we have had really limited options to prevent and control dengue. It’s an important addition to have the vaccine be approved to be used within the U.S., and it’s going to pave the road for future vaccines.”
Dr. Paz-Bailey and Dr. Ooi reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The vaccine is only to be used for children aged 9-16 who live in endemic areas and who have evidence with a specific diagnostic test of prior dengue infection.
Dengue is a mosquito-borne virus found throughout the world, primarily in tropical or subtropical climates. Cases had steadily been increasing to 5.2 million in 2019, and the geographic distribution of cases is broadening with climate change and urbanization. About half of the world’s population is now at risk.
The dengue virus has four serotypes. The first infection may be mild or asymptomatic, but the second one can be life-threatening because of a phenomenon called antibody-dependent enhancement.
The lead author of the new recommendations is Gabriela Paz-Bailey, MD, PhD, division of vector-borne diseases, dengue branch, CDC. She told this news organization that, during the second infection, when there are “low levels of antibodies from that first infection, the antibodies help the virus get inside the cells. There the virus is not killed, and that results in increased viral load, and then that can result in more severe disease and the plasma leakage” syndrome, which can lead to shock, severe bleeding, and organ failure. The death rate for severe dengue is up to 13%.
Previous infection with Zika virus, common in the same areas where dengue is endemic, can also increase the risk for symptomatic and severe dengue for subsequent infections.
In the United States, Puerto Rico is the main focus of control efforts because 95% of domestic dengue cases originate there – almost 30,000 cases between 2010 and 2020, with 11,000 cases and 4,000 hospitalizations occurring in children between the ages of 10 and 19.
Because Aedes aegypti, the primary mosquito vector transmitting dengue, is resistant to all commonly used insecticides in Puerto Rico, preventive efforts have shifted from insecticides to vaccination.
Antibody tests prevaccination
The main concern with the Sanofi’s dengue vaccine is that it could act as an asymptomatic primary dengue infection, in effect priming the body for a severe reaction from antibody-dependent enhancement with a subsequent infection. That is why it’s critical that the vaccine only be given to children with evidence of prior disease.
Dr. Paz-Bailey said: “The CDC came up with recommendations of what the performance of the test used for prevaccination screening should be. And it was 98% specificity and 75% sensitivity. ... But no test by itself was found to have a specificity of 98%, and this is why we’re recommending the two-test algorithm,” in which two different assays are run off the same blood sample, drawn at a prevaccination visit.
If the child has evidence of prior dengue, they can proceed with vaccination to protect against recurrent infection. Dengvaxia is given as a series of three shots over 6 months. Vaccine efficacy is 82% – so not everyone is protected, and additionally, that protection declines over time.
There is concern that it will be difficult to achieve compliance with such a complex regimen. Dr. Paz-Bailey said, “But I think that the trust in vaccines that is highly prevalent for [Puerto] Rico and trusting the health care system, and sort of the importance that is assigned to dengue by providers and by parents because of previous outbreaks and previous experiences is going to help us.” She added, “I think that the COVID experience has been very revealing. And what we have learned is that Puerto Rico has a very strong health care system, a very strong network of vaccine providers. ... Coverage for COVID vaccine is higher than in other parts of the U.S.”
One of the interesting things about dengue is that the first infection can range from asymptomatic to life-threatening. The second infection is generally worse because of this antibody-dependent enhancement phenomenon. Eng Eong Ooi, MD, PhD, professor of microbiology and immunology, National University of Singapore, told this news organization, “After you have two infections, you seem to be protected quite well against the remaining two [serotypes]. The vaccine serves as another episode of infection in those who had prior dengue, so then any natural infections after the vaccination in the seropositive become like the outcome of a third or fourth infection.”
Vaccination alone will not solve dengue. Dr. Ooi said, “There’s not one method that would fully control dengue. You need both vaccines as well as control measures, whether it’s Wolbachia or something else. At the same time, I think we need antiviral drugs, because hitting this virus in just one part of its life cycle wouldn’t make a huge, lasting impact.” Dr. Ooi added that as “the spread of the virus and the population immunity drops, you’re actually now more vulnerable to dengue outbreaks when they do get introduced. So, suppressing transmission alone isn’t the answer. You also have to keep herd immunity levels high. So if we can reduce the virus transmission by controlling either mosquito population or transmission and at the same time vaccinate to keep the immunity levels high, then I think we have a chance of controlling dengue.”
Dr. Paz-Bailey concluded: “I do want to emphasize that we are excited about having these tools, because for years and years, we have had really limited options to prevent and control dengue. It’s an important addition to have the vaccine be approved to be used within the U.S., and it’s going to pave the road for future vaccines.”
Dr. Paz-Bailey and Dr. Ooi reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The vaccine is only to be used for children aged 9-16 who live in endemic areas and who have evidence with a specific diagnostic test of prior dengue infection.
Dengue is a mosquito-borne virus found throughout the world, primarily in tropical or subtropical climates. Cases had steadily been increasing to 5.2 million in 2019, and the geographic distribution of cases is broadening with climate change and urbanization. About half of the world’s population is now at risk.
The dengue virus has four serotypes. The first infection may be mild or asymptomatic, but the second one can be life-threatening because of a phenomenon called antibody-dependent enhancement.
The lead author of the new recommendations is Gabriela Paz-Bailey, MD, PhD, division of vector-borne diseases, dengue branch, CDC. She told this news organization that, during the second infection, when there are “low levels of antibodies from that first infection, the antibodies help the virus get inside the cells. There the virus is not killed, and that results in increased viral load, and then that can result in more severe disease and the plasma leakage” syndrome, which can lead to shock, severe bleeding, and organ failure. The death rate for severe dengue is up to 13%.
Previous infection with Zika virus, common in the same areas where dengue is endemic, can also increase the risk for symptomatic and severe dengue for subsequent infections.
In the United States, Puerto Rico is the main focus of control efforts because 95% of domestic dengue cases originate there – almost 30,000 cases between 2010 and 2020, with 11,000 cases and 4,000 hospitalizations occurring in children between the ages of 10 and 19.
Because Aedes aegypti, the primary mosquito vector transmitting dengue, is resistant to all commonly used insecticides in Puerto Rico, preventive efforts have shifted from insecticides to vaccination.
Antibody tests prevaccination
The main concern with the Sanofi’s dengue vaccine is that it could act as an asymptomatic primary dengue infection, in effect priming the body for a severe reaction from antibody-dependent enhancement with a subsequent infection. That is why it’s critical that the vaccine only be given to children with evidence of prior disease.
Dr. Paz-Bailey said: “The CDC came up with recommendations of what the performance of the test used for prevaccination screening should be. And it was 98% specificity and 75% sensitivity. ... But no test by itself was found to have a specificity of 98%, and this is why we’re recommending the two-test algorithm,” in which two different assays are run off the same blood sample, drawn at a prevaccination visit.
If the child has evidence of prior dengue, they can proceed with vaccination to protect against recurrent infection. Dengvaxia is given as a series of three shots over 6 months. Vaccine efficacy is 82% – so not everyone is protected, and additionally, that protection declines over time.
There is concern that it will be difficult to achieve compliance with such a complex regimen. Dr. Paz-Bailey said, “But I think that the trust in vaccines that is highly prevalent for [Puerto] Rico and trusting the health care system, and sort of the importance that is assigned to dengue by providers and by parents because of previous outbreaks and previous experiences is going to help us.” She added, “I think that the COVID experience has been very revealing. And what we have learned is that Puerto Rico has a very strong health care system, a very strong network of vaccine providers. ... Coverage for COVID vaccine is higher than in other parts of the U.S.”
One of the interesting things about dengue is that the first infection can range from asymptomatic to life-threatening. The second infection is generally worse because of this antibody-dependent enhancement phenomenon. Eng Eong Ooi, MD, PhD, professor of microbiology and immunology, National University of Singapore, told this news organization, “After you have two infections, you seem to be protected quite well against the remaining two [serotypes]. The vaccine serves as another episode of infection in those who had prior dengue, so then any natural infections after the vaccination in the seropositive become like the outcome of a third or fourth infection.”
Vaccination alone will not solve dengue. Dr. Ooi said, “There’s not one method that would fully control dengue. You need both vaccines as well as control measures, whether it’s Wolbachia or something else. At the same time, I think we need antiviral drugs, because hitting this virus in just one part of its life cycle wouldn’t make a huge, lasting impact.” Dr. Ooi added that as “the spread of the virus and the population immunity drops, you’re actually now more vulnerable to dengue outbreaks when they do get introduced. So, suppressing transmission alone isn’t the answer. You also have to keep herd immunity levels high. So if we can reduce the virus transmission by controlling either mosquito population or transmission and at the same time vaccinate to keep the immunity levels high, then I think we have a chance of controlling dengue.”
Dr. Paz-Bailey concluded: “I do want to emphasize that we are excited about having these tools, because for years and years, we have had really limited options to prevent and control dengue. It’s an important addition to have the vaccine be approved to be used within the U.S., and it’s going to pave the road for future vaccines.”
Dr. Paz-Bailey and Dr. Ooi reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM MMWR RECOMMENDATIONS AND REPORTS
CDC to update mask recommendations as Omicron spreads
Director Rochelle Walensky, MD, said on Jan. 12.
“We are preparing an update to the info on our mask website to best reflect the options that are available to people and the different levels of protection different masks provide, and we want to provide Americans the best and most updated information to choose what mask is going to be right for them,” she said at a White House news briefing.
While the higher-quality masks provide better protection, they can be uncomfortable to wear, expensive, and harder to find. That’s why Dr. Walensky added an important caveat.
“Any mask is better than no mask, and we do encourage all Americans to wear a well-fitting mask to protect themselves and prevent the spread of COVID-19. That recommendation is not going to change,” she said.
“Most importantly, the best mask that you wear is the one you will wear and the one you can keep on all day long and tolerate in public indoor settings.”
Meanwhile, the World Health Organization was more focused on vaccines.
WHO officials stressed on Jan. 12 that global vaccine distribution is first priority in defeating the highly contagious Omicron variant, as well as other variants that may evolve.
The WHO’s Technical Advisory Group on COVID-19 Vaccine Composition – a group of experts assessing how COVID-19 vaccines perform against Omicron and other emerging variants – says there is an “urgent need” for broader access to vaccines, along with reviewing and updating current vaccines as needed to ensure protection.
The WHO also disputed the idea that COVID-19 could become endemic in one largely vaccinated nation, while the rest of the world remains unprotected.
“It is up to us how this pandemic unfolds,” Maria Van Kerkhove, PhD, the WHO’s technical lead on COVID-19 response, said at a news briefing.
The WHO has a goal of vaccinating 70% of the population of every country by the middle of the year.
But right now, 90 countries have yet to reach 40% vaccination rates, and 36 of those countries have less than 10% of their populations vaccinated, according to WHO Director General Tedros Adhanom Ghebreyesus, PhD.
A staggering 85% of the African population has not received a first dose.
But progress is being made, Dr. Ghebreyesus said at the briefing.
The WHO said there were over 15 million COVID-19 cases reported last week – the most ever in a single week – and this is likely an underestimate.
The Omicron variant, first identified in South Africa 2 months ago and now found on all seven continents, is “rapidly replacing Delta in almost all countries,” Dr. Ghebreyesus said.
Dr. Walensky said this week’s U.S. daily average COVID-19 case count was 751,000, an increase of 47% from last week. The average daily hospital admissions this week is 19,800, an increase of 33%. Deaths are up 40%, reaching 1,600 per day.
But she also reported new data that supports other research showing Omicron may produce less severe disease. Kaiser Permanente Southern California released a study on Jan. 11 showing that, compared with Delta infections, Omicron was associated with a 53% reduction in hospitalizations, a 74% reduction in intensive care unit admissions, and a 91% lower risk of death.
In the study, no patients with Omicron required mechanical ventilation. The strain now accounts for 98% of cases nationwide.
But Dr. Walensky warned the lower disease severity is not enough to make up for the sheer number of cases that continue to overwhelm hospital systems.
“While we are seeing early evidence that Omicron is less severe than Delta and that those infected are less likely to require hospitalization, it’s important to note that Omicron continues to be much more transmissible than Delta,” she said. “The sudden rise in cases due to Omicron is resulting in unprecedented daily case counts, sickness, absenteeism, and strains on our health care system.”
A version of this article first appeared on WebMD.com.
Director Rochelle Walensky, MD, said on Jan. 12.
“We are preparing an update to the info on our mask website to best reflect the options that are available to people and the different levels of protection different masks provide, and we want to provide Americans the best and most updated information to choose what mask is going to be right for them,” she said at a White House news briefing.
While the higher-quality masks provide better protection, they can be uncomfortable to wear, expensive, and harder to find. That’s why Dr. Walensky added an important caveat.
“Any mask is better than no mask, and we do encourage all Americans to wear a well-fitting mask to protect themselves and prevent the spread of COVID-19. That recommendation is not going to change,” she said.
“Most importantly, the best mask that you wear is the one you will wear and the one you can keep on all day long and tolerate in public indoor settings.”
Meanwhile, the World Health Organization was more focused on vaccines.
WHO officials stressed on Jan. 12 that global vaccine distribution is first priority in defeating the highly contagious Omicron variant, as well as other variants that may evolve.
The WHO’s Technical Advisory Group on COVID-19 Vaccine Composition – a group of experts assessing how COVID-19 vaccines perform against Omicron and other emerging variants – says there is an “urgent need” for broader access to vaccines, along with reviewing and updating current vaccines as needed to ensure protection.
The WHO also disputed the idea that COVID-19 could become endemic in one largely vaccinated nation, while the rest of the world remains unprotected.
“It is up to us how this pandemic unfolds,” Maria Van Kerkhove, PhD, the WHO’s technical lead on COVID-19 response, said at a news briefing.
The WHO has a goal of vaccinating 70% of the population of every country by the middle of the year.
But right now, 90 countries have yet to reach 40% vaccination rates, and 36 of those countries have less than 10% of their populations vaccinated, according to WHO Director General Tedros Adhanom Ghebreyesus, PhD.
A staggering 85% of the African population has not received a first dose.
But progress is being made, Dr. Ghebreyesus said at the briefing.
The WHO said there were over 15 million COVID-19 cases reported last week – the most ever in a single week – and this is likely an underestimate.
The Omicron variant, first identified in South Africa 2 months ago and now found on all seven continents, is “rapidly replacing Delta in almost all countries,” Dr. Ghebreyesus said.
Dr. Walensky said this week’s U.S. daily average COVID-19 case count was 751,000, an increase of 47% from last week. The average daily hospital admissions this week is 19,800, an increase of 33%. Deaths are up 40%, reaching 1,600 per day.
But she also reported new data that supports other research showing Omicron may produce less severe disease. Kaiser Permanente Southern California released a study on Jan. 11 showing that, compared with Delta infections, Omicron was associated with a 53% reduction in hospitalizations, a 74% reduction in intensive care unit admissions, and a 91% lower risk of death.
In the study, no patients with Omicron required mechanical ventilation. The strain now accounts for 98% of cases nationwide.
But Dr. Walensky warned the lower disease severity is not enough to make up for the sheer number of cases that continue to overwhelm hospital systems.
“While we are seeing early evidence that Omicron is less severe than Delta and that those infected are less likely to require hospitalization, it’s important to note that Omicron continues to be much more transmissible than Delta,” she said. “The sudden rise in cases due to Omicron is resulting in unprecedented daily case counts, sickness, absenteeism, and strains on our health care system.”
A version of this article first appeared on WebMD.com.
Director Rochelle Walensky, MD, said on Jan. 12.
“We are preparing an update to the info on our mask website to best reflect the options that are available to people and the different levels of protection different masks provide, and we want to provide Americans the best and most updated information to choose what mask is going to be right for them,” she said at a White House news briefing.
While the higher-quality masks provide better protection, they can be uncomfortable to wear, expensive, and harder to find. That’s why Dr. Walensky added an important caveat.
“Any mask is better than no mask, and we do encourage all Americans to wear a well-fitting mask to protect themselves and prevent the spread of COVID-19. That recommendation is not going to change,” she said.
“Most importantly, the best mask that you wear is the one you will wear and the one you can keep on all day long and tolerate in public indoor settings.”
Meanwhile, the World Health Organization was more focused on vaccines.
WHO officials stressed on Jan. 12 that global vaccine distribution is first priority in defeating the highly contagious Omicron variant, as well as other variants that may evolve.
The WHO’s Technical Advisory Group on COVID-19 Vaccine Composition – a group of experts assessing how COVID-19 vaccines perform against Omicron and other emerging variants – says there is an “urgent need” for broader access to vaccines, along with reviewing and updating current vaccines as needed to ensure protection.
The WHO also disputed the idea that COVID-19 could become endemic in one largely vaccinated nation, while the rest of the world remains unprotected.
“It is up to us how this pandemic unfolds,” Maria Van Kerkhove, PhD, the WHO’s technical lead on COVID-19 response, said at a news briefing.
The WHO has a goal of vaccinating 70% of the population of every country by the middle of the year.
But right now, 90 countries have yet to reach 40% vaccination rates, and 36 of those countries have less than 10% of their populations vaccinated, according to WHO Director General Tedros Adhanom Ghebreyesus, PhD.
A staggering 85% of the African population has not received a first dose.
But progress is being made, Dr. Ghebreyesus said at the briefing.
The WHO said there were over 15 million COVID-19 cases reported last week – the most ever in a single week – and this is likely an underestimate.
The Omicron variant, first identified in South Africa 2 months ago and now found on all seven continents, is “rapidly replacing Delta in almost all countries,” Dr. Ghebreyesus said.
Dr. Walensky said this week’s U.S. daily average COVID-19 case count was 751,000, an increase of 47% from last week. The average daily hospital admissions this week is 19,800, an increase of 33%. Deaths are up 40%, reaching 1,600 per day.
But she also reported new data that supports other research showing Omicron may produce less severe disease. Kaiser Permanente Southern California released a study on Jan. 11 showing that, compared with Delta infections, Omicron was associated with a 53% reduction in hospitalizations, a 74% reduction in intensive care unit admissions, and a 91% lower risk of death.
In the study, no patients with Omicron required mechanical ventilation. The strain now accounts for 98% of cases nationwide.
But Dr. Walensky warned the lower disease severity is not enough to make up for the sheer number of cases that continue to overwhelm hospital systems.
“While we are seeing early evidence that Omicron is less severe than Delta and that those infected are less likely to require hospitalization, it’s important to note that Omicron continues to be much more transmissible than Delta,” she said. “The sudden rise in cases due to Omicron is resulting in unprecedented daily case counts, sickness, absenteeism, and strains on our health care system.”
A version of this article first appeared on WebMD.com.
Children and COVID: New cases and hospital admissions skyrocket
, according to the American Academy of Pediatrics and the Children’s Hospital Association.
The total for the week of Dec. 31 to Jan. 6 – the highest since the pandemic began – was an increase of 78% over the previous week (325,000) and 192% higher than just 2 weeks before (199,000), the AAP and CHA said in their weekly COVID-19 report. No region of the country was spared, as all four saw at least 50,000 more cases than the week before, but the increase was largest in the West and smallest in the Midwest.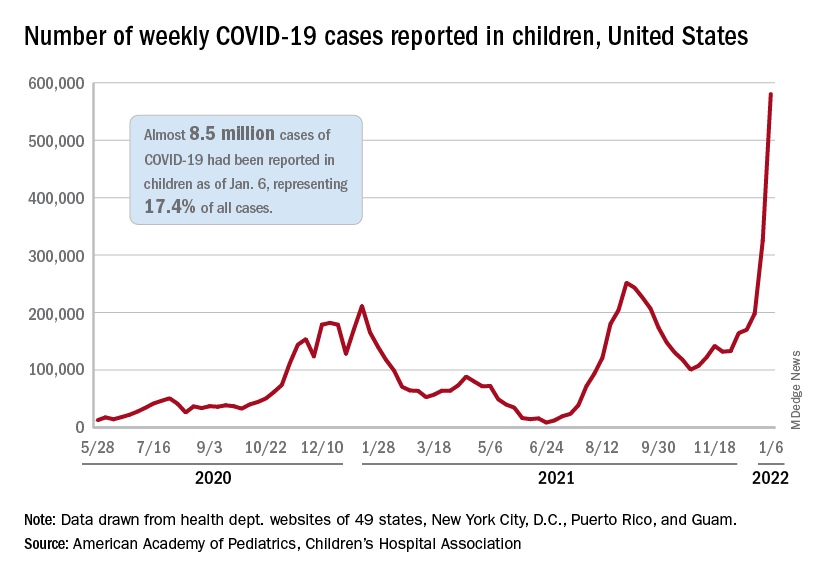
“Nearly 8.5 million children have tested positive for COVID-19 since the onset of the pandemic; nearly 11% of these cases have been added in the past 2 weeks,” the AAP said.
The situation is the same for hospitalizations. On Dec. 15, the daily rate of new admissions for children aged 0-17 years was 0.26 per 100,000, and by Jan. 7 it had more than quadrupled to 1.15 per 100,000, the Centers for Disease Control and Prevention reported. Before Omicron, the highest rate was 0.47 per 100,000 on Sept. 4, 2021.
The number of children occupying inpatient beds who had laboratory-confirmed COVID-19 went from 2,343 on Jan. 2 to 3,476 on Jan. 9, a jump of more than 48% in just 1 week. Texas had more hospitalized children (392) than any other state on Jan. 9, with California (339) and New York (313) the only other states over 300, according to data from the Department of Health & Human Services.
For vaccinations. however, the situation is definitely not the same. The number of children added to the ranks of those with at least one dose of COVID-19 vaccine was down in early 2022 (Jan. 3-9) for both 5- to 11-year-olds (–8.2%) and 16- to 17-year-olds (–12.2%) but higher among those aged 12-15 (12.2%), compared with the previous week (Dec. 27 to Jan. 2), the CDC said on its COVID Data Tracker.
Cumulative figures show that 26.3% of all children aged 5-11 had received at least one dose of vaccine and 17.2% were fully vaccinated as of Jan. 10, compared with 62.2% and 52.0% of 12- to 15-year-olds and 68.5% and 58.1% of those aged 16-17. Altogether, over 23.8 million children in those three age groups have received at least one dose and almost 18.6 million are fully vaccinated, the CDC said.
, according to the American Academy of Pediatrics and the Children’s Hospital Association.
The total for the week of Dec. 31 to Jan. 6 – the highest since the pandemic began – was an increase of 78% over the previous week (325,000) and 192% higher than just 2 weeks before (199,000), the AAP and CHA said in their weekly COVID-19 report. No region of the country was spared, as all four saw at least 50,000 more cases than the week before, but the increase was largest in the West and smallest in the Midwest.
“Nearly 8.5 million children have tested positive for COVID-19 since the onset of the pandemic; nearly 11% of these cases have been added in the past 2 weeks,” the AAP said.
The situation is the same for hospitalizations. On Dec. 15, the daily rate of new admissions for children aged 0-17 years was 0.26 per 100,000, and by Jan. 7 it had more than quadrupled to 1.15 per 100,000, the Centers for Disease Control and Prevention reported. Before Omicron, the highest rate was 0.47 per 100,000 on Sept. 4, 2021.
The number of children occupying inpatient beds who had laboratory-confirmed COVID-19 went from 2,343 on Jan. 2 to 3,476 on Jan. 9, a jump of more than 48% in just 1 week. Texas had more hospitalized children (392) than any other state on Jan. 9, with California (339) and New York (313) the only other states over 300, according to data from the Department of Health & Human Services.
For vaccinations. however, the situation is definitely not the same. The number of children added to the ranks of those with at least one dose of COVID-19 vaccine was down in early 2022 (Jan. 3-9) for both 5- to 11-year-olds (–8.2%) and 16- to 17-year-olds (–12.2%) but higher among those aged 12-15 (12.2%), compared with the previous week (Dec. 27 to Jan. 2), the CDC said on its COVID Data Tracker.
Cumulative figures show that 26.3% of all children aged 5-11 had received at least one dose of vaccine and 17.2% were fully vaccinated as of Jan. 10, compared with 62.2% and 52.0% of 12- to 15-year-olds and 68.5% and 58.1% of those aged 16-17. Altogether, over 23.8 million children in those three age groups have received at least one dose and almost 18.6 million are fully vaccinated, the CDC said.
, according to the American Academy of Pediatrics and the Children’s Hospital Association.
The total for the week of Dec. 31 to Jan. 6 – the highest since the pandemic began – was an increase of 78% over the previous week (325,000) and 192% higher than just 2 weeks before (199,000), the AAP and CHA said in their weekly COVID-19 report. No region of the country was spared, as all four saw at least 50,000 more cases than the week before, but the increase was largest in the West and smallest in the Midwest.
“Nearly 8.5 million children have tested positive for COVID-19 since the onset of the pandemic; nearly 11% of these cases have been added in the past 2 weeks,” the AAP said.
The situation is the same for hospitalizations. On Dec. 15, the daily rate of new admissions for children aged 0-17 years was 0.26 per 100,000, and by Jan. 7 it had more than quadrupled to 1.15 per 100,000, the Centers for Disease Control and Prevention reported. Before Omicron, the highest rate was 0.47 per 100,000 on Sept. 4, 2021.
The number of children occupying inpatient beds who had laboratory-confirmed COVID-19 went from 2,343 on Jan. 2 to 3,476 on Jan. 9, a jump of more than 48% in just 1 week. Texas had more hospitalized children (392) than any other state on Jan. 9, with California (339) and New York (313) the only other states over 300, according to data from the Department of Health & Human Services.
For vaccinations. however, the situation is definitely not the same. The number of children added to the ranks of those with at least one dose of COVID-19 vaccine was down in early 2022 (Jan. 3-9) for both 5- to 11-year-olds (–8.2%) and 16- to 17-year-olds (–12.2%) but higher among those aged 12-15 (12.2%), compared with the previous week (Dec. 27 to Jan. 2), the CDC said on its COVID Data Tracker.
Cumulative figures show that 26.3% of all children aged 5-11 had received at least one dose of vaccine and 17.2% were fully vaccinated as of Jan. 10, compared with 62.2% and 52.0% of 12- to 15-year-olds and 68.5% and 58.1% of those aged 16-17. Altogether, over 23.8 million children in those three age groups have received at least one dose and almost 18.6 million are fully vaccinated, the CDC said.
FDA OKs new adult insomnia med
The Food and Drug Administration has approved the dual orexin receptor antagonist daridorexant (Quviviq) for the treatment of insomnia in adults, the drug’s manufacturer, Idorsia, has announced.
The FDA’s decision was based partly on a phase 3 trial of adults with moderate to severe insomnia who were randomly assigned to receive 25 or 50 mg of daridorexant or matching placebo. Daridorexant was associated with dose-dependent improvements in wake after sleep onset, total sleep time, and latency to persistent sleep.
Whereas the overall results are very positive, the improvements in daytime functioning are especially “exciting,” Thomas Roth, PhD, director of the Sleep Disorders and Research Center at Henry Ford Hospital in Detroit, said in an interview.
“That’s sort of a big deal. For me, that’s the biggest deal there is,” said Dr. Roth, who was a consultant on the design of the phase 3 trial and on the interpretation of the data.
The drug will be available in doses of 25 mg and 50 mg, and the FDA has recommended that it be classified as a controlled substance. After it is scheduled by the Drug Enforcement Administration, daridorexant is expected to be made available in May.
Favorable safety profile
Insomnia is a common disorder characterized by difficulty falling asleep or staying asleep and by early-morning awakenings. Patients with insomnia often report fatigue, irritability, and difficulty with concentration. The condition can also result in significant problems with work and social activities, thus contributing to anxiety or depression.
As with other dual orexin receptor antagonists, daridorexant competitively binds with both orexin receptors in the lateral hypothalamus to block the activity of orexin in a reversible way. This approach decreases the downstream action of the wake-promoting neurotransmitters that are overactive in patients with insomnia.
The phase 3 trial measured daytime functioning using the new Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ), a patient-reported outcome instrument. Daridorexant was associated with significant improvements in daytime function, particularly in sleepiness and mood.
Previous trials of other dual orexin receptor antagonists did not use the IDSIQ as an outcome, so it is not possible to compare daridorexant with those drugs in this respect, Dr. Roth noted. Researchers also have not conducted head-to-head trials of the drug with other dual orexin receptor antagonists.
Daridorexant also had a favorable safety profile and was not associated with rebound insomnia or withdrawal effects. The most common adverse events were headache and somnolence or fatigue.
“They had no effect on sleep stage distribution [and] they had no significant effects on sleep and breathing in people with mild to moderate sleep apnea,” said Dr. Roth, who presented the phase 3 findings at SLEEP 2020.
In addition to serving as a consultant for Idorsia on the trial design and interpretation of results, Dr. Roth has also served as a consultant for other companies that develop sleep agents.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the dual orexin receptor antagonist daridorexant (Quviviq) for the treatment of insomnia in adults, the drug’s manufacturer, Idorsia, has announced.
The FDA’s decision was based partly on a phase 3 trial of adults with moderate to severe insomnia who were randomly assigned to receive 25 or 50 mg of daridorexant or matching placebo. Daridorexant was associated with dose-dependent improvements in wake after sleep onset, total sleep time, and latency to persistent sleep.
Whereas the overall results are very positive, the improvements in daytime functioning are especially “exciting,” Thomas Roth, PhD, director of the Sleep Disorders and Research Center at Henry Ford Hospital in Detroit, said in an interview.
“That’s sort of a big deal. For me, that’s the biggest deal there is,” said Dr. Roth, who was a consultant on the design of the phase 3 trial and on the interpretation of the data.
The drug will be available in doses of 25 mg and 50 mg, and the FDA has recommended that it be classified as a controlled substance. After it is scheduled by the Drug Enforcement Administration, daridorexant is expected to be made available in May.
Favorable safety profile
Insomnia is a common disorder characterized by difficulty falling asleep or staying asleep and by early-morning awakenings. Patients with insomnia often report fatigue, irritability, and difficulty with concentration. The condition can also result in significant problems with work and social activities, thus contributing to anxiety or depression.
As with other dual orexin receptor antagonists, daridorexant competitively binds with both orexin receptors in the lateral hypothalamus to block the activity of orexin in a reversible way. This approach decreases the downstream action of the wake-promoting neurotransmitters that are overactive in patients with insomnia.
The phase 3 trial measured daytime functioning using the new Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ), a patient-reported outcome instrument. Daridorexant was associated with significant improvements in daytime function, particularly in sleepiness and mood.
Previous trials of other dual orexin receptor antagonists did not use the IDSIQ as an outcome, so it is not possible to compare daridorexant with those drugs in this respect, Dr. Roth noted. Researchers also have not conducted head-to-head trials of the drug with other dual orexin receptor antagonists.
Daridorexant also had a favorable safety profile and was not associated with rebound insomnia or withdrawal effects. The most common adverse events were headache and somnolence or fatigue.
“They had no effect on sleep stage distribution [and] they had no significant effects on sleep and breathing in people with mild to moderate sleep apnea,” said Dr. Roth, who presented the phase 3 findings at SLEEP 2020.
In addition to serving as a consultant for Idorsia on the trial design and interpretation of results, Dr. Roth has also served as a consultant for other companies that develop sleep agents.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the dual orexin receptor antagonist daridorexant (Quviviq) for the treatment of insomnia in adults, the drug’s manufacturer, Idorsia, has announced.
The FDA’s decision was based partly on a phase 3 trial of adults with moderate to severe insomnia who were randomly assigned to receive 25 or 50 mg of daridorexant or matching placebo. Daridorexant was associated with dose-dependent improvements in wake after sleep onset, total sleep time, and latency to persistent sleep.
Whereas the overall results are very positive, the improvements in daytime functioning are especially “exciting,” Thomas Roth, PhD, director of the Sleep Disorders and Research Center at Henry Ford Hospital in Detroit, said in an interview.
“That’s sort of a big deal. For me, that’s the biggest deal there is,” said Dr. Roth, who was a consultant on the design of the phase 3 trial and on the interpretation of the data.
The drug will be available in doses of 25 mg and 50 mg, and the FDA has recommended that it be classified as a controlled substance. After it is scheduled by the Drug Enforcement Administration, daridorexant is expected to be made available in May.
Favorable safety profile
Insomnia is a common disorder characterized by difficulty falling asleep or staying asleep and by early-morning awakenings. Patients with insomnia often report fatigue, irritability, and difficulty with concentration. The condition can also result in significant problems with work and social activities, thus contributing to anxiety or depression.
As with other dual orexin receptor antagonists, daridorexant competitively binds with both orexin receptors in the lateral hypothalamus to block the activity of orexin in a reversible way. This approach decreases the downstream action of the wake-promoting neurotransmitters that are overactive in patients with insomnia.
The phase 3 trial measured daytime functioning using the new Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ), a patient-reported outcome instrument. Daridorexant was associated with significant improvements in daytime function, particularly in sleepiness and mood.
Previous trials of other dual orexin receptor antagonists did not use the IDSIQ as an outcome, so it is not possible to compare daridorexant with those drugs in this respect, Dr. Roth noted. Researchers also have not conducted head-to-head trials of the drug with other dual orexin receptor antagonists.
Daridorexant also had a favorable safety profile and was not associated with rebound insomnia or withdrawal effects. The most common adverse events were headache and somnolence or fatigue.
“They had no effect on sleep stage distribution [and] they had no significant effects on sleep and breathing in people with mild to moderate sleep apnea,” said Dr. Roth, who presented the phase 3 findings at SLEEP 2020.
In addition to serving as a consultant for Idorsia on the trial design and interpretation of results, Dr. Roth has also served as a consultant for other companies that develop sleep agents.
A version of this article first appeared on Medscape.com.